UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the Fiscal Year Ended December 31, 20202023
or
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
FOR THE TRANSITION PERIOD FROM TO
Commission file number 000-19319
Vertex Pharmaceuticals Incorporated
(Exact name of registrant as specified in its charter)
Massachusetts
(State or other jurisdiction of incorporation or organization)
50 Northern Avenue, Boston, Massachusetts
(Address of principal executive offices)
04-3039129
(I.R.S. Employer Identification No.)
02210
(Zip Code)
Registrant’s telephone number, including area code (617) 341-6100
Securities registered pursuant to Section 12(b) of the Exchange Act:
| | | | | | | | | | | | | | |
| Title of Each Class | Trading Symbol | Name of Each Exchange on Which Registered |
| Common Stock, $0.01 Par Value Per Share | VRTX | The Nasdaq Global Select Market |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act (Check one):
Large accelerated filer ☒ Accelerated filer ☐ Non-accelerated filer ☐ Smaller reporting company ☐ Emerging growth company ☐
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant based on the closing price on June 30, 20202023 (the last business day of the registrant’s most recently completed second fiscal quarter of 2020)2023) was $74.8$90.7 billion.
As of January 31, 2021,February 9, 2024, the registrant had 259,960,062258,307,816 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement for the 20212024 Annual Meeting of Shareholders, which we expect to hold on May 19, 2021,15, 2024, are incorporated by reference into Part III of this Annual Report on Form 10-K.
VERTEX PHARMACEUTICALS INCORPORATED
ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
“We,Vertex,” “us,“we,” “Vertex”“us” and the “Company”“our” as used in this Annual Report on Form 10-K refer to Vertex Pharmaceuticals Incorporated, a Massachusetts corporation, and its subsidiaries.
“Vertex,VERTEX®,” “KALYDECO®,” “ORKAMBI®,” “SYMDEKO®,” “SYMKEVI®,” and “TRIKAFTA®,” “KAFTRIO®,” and “CASGEVY™” are registered trademarks of Vertex. The trademark for “KAFTRIO” is pending in the United States and registered in the European Union. Other brands, names and trademarks contained in this Annual Report on Form 10-K are the property of their respective owners.
We use the brand name for our products when we refer to the product that has been approved and with respect to the indications on the approved label. Otherwise, including in discussions of our cystic fibrosis, sickle cell disease, and beta thalassemia development programs, we refer to our compoundsproduct candidates by their scientific (or generic) name or VX developmental designation.
This Annual Report on Form 10-K contains forward-looking statements. Words such as “anticipates,” “may,” “forecasts,” “expects,” “intends,” “plans,” “potentially,” “believes,” “seeks,” “estimates,” variations of such words and similar expressions are intended to identify such forward-looking statements, although not all forward-looking statements contain these identifying words. Please refer to “Special Note Regarding Forward-Looking Statements” set forth in Part I, Item 1A, for a discussion of our forward-looking statements and the related risks and uncertainties of such statements.
PART I
ITEM 1.BUSINESS
OVERVIEW
We are a global biotechnology company that invests in scientific innovation to create transformative medicines for people with serious diseases, with a focus on specialty markets.
Cystic Fibrosis We have four approved medicines that treat the underlying cause of cystic fibrosis (“CF”), a life-threatening genetic disease, and one approved therapy that treats severe sickle cell disease (“SCD”) and transfusion dependent beta thalassemia (“TDT”), life shortening inherited blood disorders. Our pipeline includes clinical-stage programs in CF, sickle cell disease, beta thalassemia, acute and neuropathic pain, APOL1-mediated kidney disease, type 1 diabetes, myotonic dystrophy type 1 and alpha-1 antitrypsin deficiency.
Our goal in CF is to develop treatment regimens that will provide benefits to all people with cystic fibrosis, or CF and will enhance the benefits currently provided to people taking our medicines. Our marketed medicines that treat people with CF are TRIKAFTA/KAFTRIO (elexacaftor/tezacaftor/ivacaftor and ivacaftor), SYMDEKO/SYMKEVI (tezacaftor/ivacaftor and ivacaftor), ORKAMBI (lumacaftor/ivacaftor) and KALYDECO (ivacaftor). Our triple combination regimen, TRIKAFTA/KAFTRIO, was approved in 2019 in the United States, or U.S., and in 2020 in the European Union, or E.U. Collectively, our four marketed CF medicines are approvedbeing used to treat the majoritynearly three quarters of the approximately 83,00092,000 people with CF in North America, Europe and Australia.
Through label expansions, approval of new medicines, and expanded reimbursement, we are focused on increasing the number of people with CF who are eligible and able to receive our medicines. We are evaluating our current medicines including our triple combination regimen, in additional patient populations, including younger children, with the goal of having small molecule treatments for upall people who have at least one mutation in their cystic fibrosis transmembrane conductance regulator (“CFTR”) gene that is responsive to 90%our CFTR modulators. We have completed Phase 3 development of a triple combination of vanzacaftor/tezacaftor/deutivacaftor, which has demonstrated the potential to provide additional clinical benefits, as well as once-daily dosing, to people with CF. CF who have at least one mutation in their CFTR gene that is responsive to CFTR modulators. This regimen also carries a lower royalty burden. We plan to submit global regulatory filings for this new triple combination regimen by mid-2024. In addition, we have initiated the multiple ascending dose portion of the Phase 1/2 clinical trial of VX-522, an investigational messenger ribonucleic acid (“mRNA”) therapeutic we are developing in collaboration with Moderna, Inc. (“Moderna”). We expect to share data from this clinical trial in late 2024 or early 2025. VX-522 has the potential to benefit the more than 5,000 people with CF in North America, Europe and Australia who do not make full-length CFTR protein and cannot benefit from CFTR modulators. In addition, we are continuing our research and development of CFTR modulators, with the aim of developing best-in-class medicines that can help more patients achieve carrier levels of CFTR function, and we are investigating additional potential treatments for people with CF who do not make full-length CFTR protein and cannot benefit from CFTR modulators.
In SCD and TDT, our goal is to eliminate vaso-occlusive crises (“VOCs”) (as well as vaso-occlusive organ damage) and transfusion dependence, respectively. Our marketed therapy is CASGEVY (exagamglogene autotemcel, or “exa-cel”), an ex-vivo, non-viral CRISPR/Cas9 gene-edited cell therapy, which has been approved in the United States (“U.S.”), the European Union (“E.U.”), the United Kingdom (“U.K.”), the Kingdom of Saudi Arabia (“Saudi Arabia”), and the Kingdom of Bahrain (“Bahrain”) for treatment of SCD and TDT. We estimate approximately 35,000 people with severe SCD or TDT could be eligible for CASGEVY in the U.S. and Europe, with additional eligible people in Saudi Arabia and Bahrain. In connection with our serial innovation approach, we are progressing preclinical assets for gentler conditioning for CASGEVY, which could broaden the eligible patient population, and we are investigating small molecules for the potential treatment of SCD and TDT.
In January 2024, we announced positive results from our Phase 3 clinical trials evaluating VX-548, a non-opioid investigational NaV 1.8 inhibitor, for the treatment of moderate-to-severe acute pain. We plan to submit for regulatory approval of VX-548 in moderate-to-severe acute pain in the U.S. by mid-2024. In addition, in December 2023, we announced positive results from the Phase 2 clinical trial evaluating VX-548 for the treatment of diabetic peripheral neuropathy (“DPN”), a type of peripheral neuropathic pain. We expect to advance VX-548 into pivotal development in DPN in 2024.
The following chart represents our clinical-stage programs, and select pre-clinical programs:
Beyond CF, SCD, TDT and pain, we are advancing programs across multiple disease areas and modalities, including:
•APOL1-Mediated Kidney Disease. We are pursuing genetic therapiesevaluating inaxaplin, formerly known as VX-147, our investigational small molecule for the treatment of APOL1-mediated kidney disease (“AMKD”) in a Phase 2/3 clinical trial. We expect to select a dose and move to the Phase 3 portion of the clinical trial in the first quarter of 2024.
•Type 1 Diabetes. We are evaluating VX-880, an investigational allogeneic stem-cell derived, fully differentiated islet cell therapy, for the treatment of type 1 diabetes (“T1D”) in a Phase 1/2 clinical trial in which patients also receive immunosuppressive therapy to protect the islet cells from immune rejection. We have completed enrollment in Part C of this clinical trial. VX-880 is on a protocol-specified pause, pending review of the totality of the data by the independent data monitoring committee and global regulators. Our second clinical program in T1D, VX-264, in which the implanted islet cells are encapsulated in an immunoprotective device, is ongoing. We have completed Part A of this Phase 1/2 clinical trial and we have initiated Part B in multiple centers and countries.
•Myotonic Dystrophy Type 1. We are exploring multiple approaches to address the remaining 10% of peopleunderlying causal biology for myotonic dystrophy type 1 (“DM1”), including small molecules. We initiated a Phase 1/2 clinical trial evaluating VX-670, an oligonucleotide-based approach that we have in-licensed from Entrada Therapeutics, Inc. (“Entrada”). The clinical trial is active and enrolling in Canada and will initiate in the U.K. in the near term.
•Alpha-1 Antitrypsin Deficiency. We continue to enroll and dose healthy volunteers in Phase 1 clinical trials for VX-634 and VX-668, our next-wave investigational molecules with CF.significantly improved potency and drug-like properties as compared to our previous alpha-1 antitrypsin (“AAT”) correctors.
Research•In addition to the programs listed above, we have additional research programs aimed at diseases that fit our research and Developmentdevelopment strategy and follow-on programs in our existing disease areas in accord with our serial innovation approach.
Our goalcore strategy is to identifydiscover and develop innovative medicines by combining transformative advances in the understanding of human disease and in the science of therapeutics to dramatically advance human health. That strategy focuses on validated targets that address causal human biology, predictive lab assays and clinical biomarkers, rapid paths to registration and approval, and product candidates that hold the potential for transformative patient benefit. Our research and early development strategyapproach includes advancing multiple compounds or therapies from each program into early clinical trials and evaluatingto obtain patient data that can inform selection of the resulting data tomost promising therapies for later stage development as well as inform drugour ongoing discovery and development efforts. We aim to rapidly follow our first-in-class therapies that achieve proof-of-concept with the goal of bringingpotential best-in-class therapiescandidates. We plan to patients. Thiscontinue investing to advance our strategy, fostering scientific innovation by identifying additional product candidates through internal research efforts, and investing in business development transactions to access
emerging technologies, products and product candidates.
Our serial innovation approach is intended to increase the likelihood of successfully bringing transformative medicines to patients.
Small Molecule Programs
Alpha-1 Antitrypsin Deficiency. Wepatients and to provide durable clinical and commercial success. Our CF medicines are focused on identifyingthe exemplar of this strategy, as we continue to reach more people with CF than ever before through label expansions, approvals of new medicines and developing multiple drug candidates with the potential to increase the levels of functional alpha-1 antitrypsin, or AAT, in the blood, to address the lung and liver manifestations of AAT deficiency. Enrollment is ongoing in a Phase 2 proof-of-concept trialexpanded reimbursement. In addition, we have obtained historic approvals for VX-864, an investigational small molecule correctorCASGEVY for the treatment of AAT deficiency. We expect data from this clinical trial in the first half of 2021.
APOL1-Mediated Kidney Diseases. We are evaluating inhibitors of APOL1 function to reduce levels of protein in the urine, or proteinuria, in people with serious kidney diseases, including focal segmental glomerulosclerosis, or FSGS, and other APOL1-mediated kidney diseases. In 2020, we initiated a Phase 2 proof-of-concept clinical trial designed to evaluate the reduction in proteinuria in people with APOL1-mediated FSGS after treatment with VX-147. Enrollment in this clinical trial is ongoing and we expect data in 2021.
Pain. We believe that NaV1.8 inhibitors have the potential to provide an effective non-opioid treatment for pain. We are advancing a portfolio of NaV1.8 inhibitors through pre-clinical and early clinical development.
Cell and Genetic Therapies
Sickle cell disease and transfusion-dependent beta thalassemia. We are co-developing CTX001, an investigational CRISPR/Cas9-based gene-editing therapy for sickle cell disease, or SCD and transfusion-dependent beta thalassemia, or TDT, with CRISPR Therapeutics AG, or CRISPR. Enrollment and dosing are ongoing in two Phase 1/2 clinical trials to evaluate CTX001 as a potential one-time curative therapy for people with severe SCD and TDT. In December 2020, we announced positive interim data from 10 people treated with CTX001 and that a total of thirteen people with TDT and seven people with severe SCD have been dosed with CTX001.are working towards broad access for eligible patients to this potentially curative treatment option. We expectcontinue to complete enrollmentadvance our broad and diverse pipeline and prepare for potential near-term global commercial launches in both clinical trials in 2021.
Type 1 Diabetes. In 2019, we acquired Semma Therapeutics, Inc., or Semma,new disease areas, and established preclinical cell therapy programs for type 1 diabetes, or T1D. We are pursuing two programs for the transplant of functional islets into patients: transplantation of islet cells alone, using immunosuppression to protect the implanted cells, and implantation of the islet cells inside a novel immunoprotective device. The FDA has clearedfurther strengthen our Investigational New Drug Application, or IND, forfinancial profile.
VX-880, the first program (transplantation of islet cells alone), and we expect to initiate a Phase 1/2 clinical trial evaluating VX-880 in the first half of 2021.
Duchenne muscular dystrophy, or DMD, and myotonic dystrophy type 1, or DM1. In 2019, we acquired Exonics Therapeutics, Inc., or Exonics, and expanded our collaboration with CRISPR enabling the establishment of preclinical genetic therapy programs for DMD and DM1.
We plan to continue investing in our research and development programs and fostering scientific innovation by continuing to identify additional drug candidates through our internal research efforts and investing in business development transactions to access emerging technologies, drugs and drug candidates.
CYSTIC FIBROSISMARKETED PRODUCTS
BackgroundInformation regarding our marketed products, including information regarding the disease area, initial approval and age group for which the therapy is approved, are set forth in the table below.
| | | | | | | | |
| Disease | Initial Approval | Eligible Age Group(1) |
| Cystic Fibrosis |
| 2019 | 2 years of age and older |
| 2020 | 2 years of age and older |
| 2018 | 6 years of age and older |
| 2018 | 6 years of age and older |
| 2015 | 1 year of age and older |
| 2012 | 1 month of age and older |
| Sickle Cell Disease and Transfusion-Dependent Beta Thalassemia |
| 2023 | 12 years of age and older |
(1) Specifies the youngest eligible age group in any major market.
CF
Our CF medicines are collectively being used by nearly three quarters of the approximately 92,000 people with CF in North America, Europe, and Australia. CF is a life-shortening genetic disease caused by a defective or missing CFTR protein resulting from mutations in the CFTR gene. To develop CF, children must inherit two defective CFTR genes, which are referred to as alleles; one allele is inherited from each parent. The vast majority of patients with CF carry at least one of the two most prevalent mutations, the F508del mutation and the G551D mutation. The F508del mutation results in a defect in the CFTR protein in which the CFTR protein does not reach the surface of the cells in sufficient quantities and does not adequately transport chloride ions. The G551D mutation results in a defect in the CFTR protein in which the defective protein reaches the surface of a cell but does not adequately transport chloride ions across the cell membrane.
The absence of working CFTR proteinsprotein results in poor flow of salt and water into and out of cells in a number of organs, including the lungs. As a result, thick, sticky mucus builds up and blocks the passages in many organs, leading to a variety of symptoms. In particular, mucus builds up and clogs the airways in the lungs, causing chronic lung infections and progressive lung damage. CFTR potentiators, such as ivacaftor, and VX-561 increase the probability that the CFTR protein channels open on the cell surface, increasing the flow of salt and water into and out of the cell. Our CFTR correctors, such as lumacaftor, tezacaftor, and elexacaftor, helpincrease the proper protein processing and folding of mutant CFTR proteins, reach the cell surface.such that a larger amount of
functional CFTR protein reaches the cell surface. Our Medicines
Our medicines, TRIKAFTA/KAFTRIO, SYMDEKO/SYMKEVI, ORKAMBICFTR regimens target the underlying cause of disease and KALYDECO, are collectively approvedhave been shown to treat the majority ofimprove CFTR protein function in people with CF, and as such have been shown to provide transformative benefit for people living with CF.
Our CF medicines are used by patients in North America, Europeover 60 countries, and Australia. OurTRIKAFTA/KAFTRIO is now approved medicines, including information regarding the indication and age groups for which the medicine is approved, are set forthreimbursed or accessible in the table below.
| | | | | | | | | | | | | | |
Product | Scientific Name | Region/Initial Approval | Indication | Eligible Age Group |
| elexacaftor/tezacaftor/ivacaftor and ivacaftor | U.S.
(2019) | People with CF with (i) at least one F508del mutation, or (ii) another mutation that is responsive to elexacaftor/tezacaftor/ivacaftor and ivacaftor
| 12 years of age and older |
| elexacaftor/tezacaftor/ivacaftor and ivacaftor | E.U. (2020) | People with CF with (i) at least one F508del mutation and one minimal function mutation, or (ii) two F508del mutations
| 12 years of age and older |
| tezacaftor/ivacaftor and ivacaftor | U.S.
(2018) | People with CF (i) homozygous for the F508del mutation or (ii) with at least one mutation that is responsive to tezacaftor/ivacaftor
| 6 years of age and older |
| tezacaftor/ivacaftor | E.U.
(2018) | People with CF (i) homozygous for the F508del mutation or (ii) with one copy of the F508del mutation and one copy of certain mutations that result in residual CFTR activity
| 6 years of age and older |
| lumacaftor/ivacaftor | U.S.
(2015) | People with CF homozygous for the F508del mutation
| 2 years of age and older |
lumacaftor/ivacaftor | E.U.
(2015) | People with CF homozygous for the F508del mutation
| 2 years of age and older |
| ivacaftor | U.S.
(2012) | People with CF with G551D and other specified mutations
| 4 months of age and older |
ivacaftor | E.U.
(2012) | People with CF with G551D and other specified mutations
| 4 months of age and older |
more than 40 of these countries. In addition to the E.U. and the U.S., we market our products in additional countries, including the United Kingdom,U.K., Australia, Switzerland, Israel,Canada, Brazil and Canada. Currently, our medicines treat almost half of the people with CF in these geographies.Switzerland. We continuously seekcontinue to increase the number of patients eligible and able to receive our current medicines through label expansions approval of new medicines and expanded reimbursement. Since the beginning of 2020, activities in support of these efforts include:
TRIKAFTA/KAFTRIO
•In August, the European Commission granted marketing authorization for KAFTRIO to treat people with CF 12 years of age and older with one F508del mutation and one minimal function mutation, or two F508del mutations.
•The FDA expanded the eligibility for TRIKAFTA to include people with CF 12 years of age and older with certain mutations that are responsive to TRIKAFTA based on in vitro data.
•In January 2021, the FDA accepted our supplemental New Drug Application, or sNDA, for TRIKAFTA for the treatment of children 6 to 11 years of age with at least one F508del mutation or have certain mutations that are responsive to TRIKAFTA based on in vitro data. The FDA granted Priority Review of the sNDA.
•Swissmedic, the Swiss Agency for Therapeutic Products, granted marketing authorization and a reimbursement agreement was reached for TRIKAFTA in Switzerland for the treatment of people with CF 12 years of age and older who have two copies of the F508del mutation, or one F508del mutation and one minimal function mutation.
•Health Canada accepted for Priority Review a New Drug Submission for TRIKAFTA for the treatment of people with CF 12 years of age and older.
SYMDEKO/SYMKEVI
•The European Commission approved SYMKEVI for the treatment of people with CF 6 years of age and older with two copies of the F508del mutation, or one F508del mutation and certain residual function mutations.
•The FDA approved SYMDEKO for additional responsive mutations in people with CF 6 years of age and older.
ORKAMBI
•We entered into a reimbursement agreement with the Swiss government for ORKAMBI for the treatment of people with CF 2 years of age and older, and for SYMDEKO for the treatment of people 12 years of age and older in Switzerland.
KALYDECO
•The FDA approved KALYDECO for treatment of infants with CF four months of age and older who have at least one mutation in their CFTR gene that is responsive to KALYDECO.
•The European Commission approved KALYDECO for treatment of infants with CF four months of age and older who have the R117H mutation or certain gating mutations.
•The FDA approved KALYDECO for treatment of infants with CF four months of age and older with additional responsive mutations.
CF PIPELINE
•VX-561, a CFTR potentiator we acquired from Concert Pharmaceuticals, Inc., and VX-121, a CFTR corrector, are being evaluated in Phase 2 clinical development.
•We continue to identify and develop additional CFTR modulators with the goal of achieving carrier levels of CFTR activity for the 90% of people with CF who respond to CFTR modulators.
•We continue to research genetic therapies, such as messenger ribonucleic acid, or mRNA, and gene-editing approaches, to treat the remaining 10% of people who do not make CFTR protein and, as a result, are not eligible for CFTR modulators.
•We extended our collaboration with Moderna, Inc., or Moderna, aimed at the discovery and development of mRNA therapeutics for the treatment of CF. In addition, we entered into a new collaboration with Moderna for the discovery and development of lipid nanoparticles and mRNAs that can deliver gene-editing therapies to lung cells for the treatment of CF.
RESEARCH AND DEVELOPMENT PROGRAMS
We invest in research and development in order to discover and develop transformative medicines for people with serious diseases with a focus on specialty markets. Our strategy is to combine transformative advances in the understanding of human disease and the science of therapeutics in order to discover and develop new medicines. Our approach to drug discovery has been validated through our success in moving novel small molecule drug candidates into clinical trials and obtaining marketing approvals for TRIKAFTA/KAFTRIO, KALYDECO, ORKAMBI and SYMDEKO/SYMKEVI for the treatment of CF and INCIVEK (telaprevir) for the treatment of hepatitis C infection. In addition, we have achieved clinical proof of concept for Nav1.8 inhibition in the treatment of three different pain models, and for gene-editing of BCL11A for the treatment of beta thalassemia and SCD.
We continue to research and develop small molecule drug candidates for the treatment of serious diseases, including CF, AAT deficiency, APOL1-mediated kidney diseases, and pain. Our research and development approach includes advancing multiple small molecules into clinical trials, pursuing multiple modalities and evaluating clinical and non-clinical data to inform drug discovery and development, with the goal of bringing best-in-class therapies to patients.
Over the last several years, we have expanded our capabilities to include additional innovative therapeutic approaches with a focus on cell and genetic therapies, which have the potential to treat, and in some cases, cure diseases by addressing the underlying cause of the disease. We have expanded our capabilities by increasing our internal investment in cell and genetic therapies, including plans to establish a new research and development site in Boston that will focus primarily on cell and genetic therapies. In addition, we have made several significant investments in external innovation, including:
•our collaboration with CRISPR to access and develop therapeutics based on the CRISPR gene-editing technology;
•our establishment of cell therapy programs for T1D through our acquisition of Semma;
•our establishment of genetic therapy programs for DMD and DM1, through our acquisition of Exonics;
•our collaboration with Moderna for the discovery and development of lipid nanoparticles and mRNAs that can deliver gene-editing therapies; and
•our collaboration with Affinia Therapeutics, Inc., or Affinia, to engineer novel adeno-associated virus (AAV) capsids to deliver gene therapies.
The experience we gained developing medicines for CF and our analysis of research and development programs conducted by other companies in our industry have shaped a disciplined strategy that guides our investments in research and development and external innovation that focuses on:
• transformative treatments for life-threatening diseases with a high unmet medical need;
• targets validated as playing a causal role in the human biology of a disease;
•innovative therapeutic approaches to addressing those targets;
•biological assays and clinical biomarkers that we believe will be predictive of clinical responses; and
• efficient clinical and regulatory paths to bring new medicines to patients.
To augment our internal programs, we plan to continue acquiring businesses and technologies and collaborating with biopharmaceutical and technology companies, leading academic research institutions, government laboratories, foundations and other organizations to advance research in our areas of therapeutic interest as well as to access technologies needed to execute on our strategy. We have established such relationships with organizations around the world and intend to extend and leverage that experience to further our research efforts to discover transformational medicines for serious diseases. We will continue to identify and evaluate potential acquisitions and collaborations that may be similar to or different from the transactions that we have engaged in previously.
Small Molecule Programs
Alpha-1 Antitrypsin Deficiency
AAT deficiency is caused by mutations in the SERPINA1 gene that encodes the AAT protein. People who inherit two mutant SERPINA1 alleles (one from each parent) develop AAT deficiency. Most people who develop AAT deficiency have two copies of the mutant Z allele. The mutations result in a defect in the AAT protein in which the protein does not fold correctly. This folding defect causes the AAT protein to accumulate in the liver (where it is produced at high levels), which can cause liver damage. As a result, the protein fails to reach other organs in adequate quantity and function, particularly in the lungs, where its normal role is to protect them from the digestive effects of certain proteases. The unchecked activity of these proteases can cause auto-digestion of lung tissue and may lead to emphysema or chronic pulmonary obstructive disease, and lung infections over time. Currently, there is no cure or treatment that targets the underlying cause of the disease in both the liver and the lung. Available treatments are aimed at transiently increasing levels of AAT in the blood but have no effect
in the liver. Patients living with AAT deficiency typically experience recurring hospital visits and a shortened life expectancy.
We seek to develop medicines that treat the underlying cause of AAT deficiency. In the laboratory, we have discovered multiple small molecule correctors that restore folding of the mutant AAT protein, with the potential to affect both the liver and lung diseases caused by AAT deficiency, and we are focused on identifying and developing multiple drug candidates with the potential to correct the misfolded protein. In 2020, we advanced two Phase 2 proof-of-concept clinical trials evaluating two investigational oral small molecule correctors, VX-814 and VX-864, for the treatment of people with AAT deficiency who have two copies of the Z mutation. In October 2020, we discontinued development of VX-814 based on the safety and pharmacokinetic profile observed in the clinical trial. Enrollment is ongoing in the clinical trial evaluating VX-864, and we expect data from this clinical trial in the first half of 2021. In addition, we continue to discover and develop additional molecules with the potential to correct AAT deficiency.
APOL1-Mediated Kidney Diseases
Inherited mutations in the APOL1 gene play a causal role in the biology of FSGS as well as other kidney diseases. FSGS is a rare disease that attacks the kidney’s filtering units, causing leakage of protein into the urine followed by deterioration in kidney function, scarring, and, ultimately, permanent kidney damage. FSGS is a leading cause of nephrotic syndrome in children and kidney failure in adults. We are evaluating multiple novel small molecules that inhibit the function of APOL1 protein with the potential to treat APOL1-mediated FSGS. In 2020, we initiated a Phase 2 proof-of-concept clinical trial for VX-147, our first investigational oral small molecule medicine for the treatment of FSGS and other serious kidney diseases. Enrollment is ongoing in this Phase 2 clinical trial and we expect data from the trial in 2021.
Pain
Pain can develop from a variety of pathophysiological and psychological conditions. Patients with pain can suffer from acute pain (for example, following surgery or an injury), neuropathic pain (when there is damage to a nerve), and musculoskeletal pain. Current treatments may not work well and can cause significant side effects. In addition, there is the potential for addiction and the practice of over- and mis-utilization, as well as underutilization of current pain medicines.
Vertex has discovered multiple inhibitors of the voltage-gated sodium channel 1.8, or NaV1.8, as potential treatments for pain. Consistent with our research strategy, the Nav1.8 channel is a validated target for pain based both on inherited mutations that cause pain syndromes as well as our own clinical trial data. Specifically, we have obtained positive results from three separate Phase 2 clinical trials evaluating VX-150, a NaV1.8 inhibitor, in patients with three different pain conditions: acute post-surgical, chronic neuropathic and chronic musculoskeletal pain. We continue to focus our research and development efforts on discovering, developing and advancing a portfolio of multiple inhibitors of NaV1.8 as potential treatments for pain.
Cell and Genetic Therapies
Sickle Cell Disease and Transfusion-Dependent Beta Thalassemia
CASGEVY, our therapy for SCD and beta thalassemiaTDT, was recently approved in the U.S., the E.U., the U.K. Saudi Arabia, and Bahrain. SCD and TDT are hemoglobinopathies, a group of inherited blood disorders that result from gene mutations that alter hemoglobin, a protein in red blood cells that delivers oxygen throughout the body. We estimate approximately 35,000 people with severe SCD or TDT could be eligible for CASGEVY in the U.S. and Europe, with additional eligible people in Saudi Arabia and Bahrain.
SCD is caused by the change of a single amino acid in the hemoglobinβ-hemoglobin gene that causes red cells to change shape in settings of low oxygen. These sickled cells block blood flow and can lead to severe pain, organ damage, and shortened life span. Treatment is typically focused on relieving pain and minimizing organ damage, requiring medication and, for some patients, monthly blood transfusions and frequent hospital visits. We believe there are approximately 25,000 patients with severe SCD in the U.S. and E.U.
Beta thalassemia is caused by loss-of-function mutations in hemoglobinthe β-hemoglobin gene that lead to severe anemia in patients, which causes fatigue and shortness of breath. In infants, beta thalassemia causes failure to thrive, jaundice, and feeding problems. Complications of beta thalassemia can lead to an enlarged spleen, liver and/or heart, misshapen bones and delayed puberty. Treatment for beta thalassemia varies depending on the disease severity for each patient. Patients with TDT, the most severe form of the disease, require regular blood transfusions, as frequently as every two to four weeks. Repeated blood transfusions eventually cause an unhealthy buildup of iron in the patient, leading to organ damage. We believe that there are approximately 7,000 patients with TDT in the U.S. and E.U.
In collaboration with CRISPR, we are co-developing CTX001, an investigationalCASGEVY, our ex-vivo, non-viral CRISPR/Cas9-based gene-editing therapy, was developed for the treatment of severe SCD and TDT. Our therapeutic approach involves isolatingTDT, with our collaborator, CRISPR Therapeutics AG (“CRISPR”). Patients first undergo a treatment at an authorized treatment center (an “ATC”) that mobilizes a population of hematopoietic stem and progenitor cells or HSPCs, which give rise to red blood cells, from a patient, treating those cells ex vivo with CRISPR/Cas9 in order to modify the erythroid-specific enhancer in the BCL11A gene, and reintroducing the edited cells back into the patient. This approach has the potential to increase levels of fetal hemoglobin in erythrocytes and reduce or eliminate symptoms associated with disease.
We and CRISPR are investigating CTX001 in two Phase 1/2 open-label clinical trials designed to assess the safety and efficacy of a single dose of CTX001 in patients ages 12 to 35 with TDT (CLIMB THAL-111) and severe SCD (CLIMB SCD-121), respectively.Patients enrolled in the clinical trial first undergo a treatment which mobilizes a population of HSPCs(“HSPC”) from the bone marrow into the bloodstream. Blood cells are collected from the patient’s bloodstream and senttransferred to a manufacturing facility where the HSPCs are purified and CRISPR/Cas9 gene-editing is performed. The gene editing procedure results in a precise and specific gene edit in a non-coding intron of the BCL11A gene. After the HSPC cells are returned to the patient and engraft, the gene edit results in significant increases in the levels of fetal hemoglobin erythrocytes, thereby reducing or eliminating symptoms associated with disease. Following manufacturing, the edited cells, now called CTX001,CASGEVY, are senttransferred back to the clinical site.ATC. Patients are preconditioned with a myeloablative conditioning treatment that ablates their bone marrow prior to infusion of CTX001.CASGEVY. Efficacy data presented to date support the profile of CASGEVY as a potential one-time functional cure for people with severe SCD and TDT.
Our global launch strategy for CASGEVY is focused on disease education and awareness for patients, caregivers, health care professionals, payors, and policymakers, as well as engagement with the scientific and medical community regarding CASGEVY clinical data. Our strategy is also focused on activation of ATCs to ensure their readiness to treat patients and establishing a globally-enabled manufacturing and supply chain to meet patient demand. In addition, our approach concentrates on achieving access for patients through reimbursement agreements with governments and commercial payors, as well as through early access programs where applicable.
RESEARCH AND DEVELOPMENT PROGRAMS
We invest in research and development to discover and develop transformative medicines for people with serious diseases, with a focus on specialty markets. Our research strategy is to combine transformative advances in the understanding of human disease and in the science of therapeutics to dramatically advance human health. We focus on:
•disease areas with known causal human biology;
•targets validated by causal human biology;
•predictive lab assays and clinical biomarkers;
•potential for transformative benefit regardless of modality; and
•efficient path to registration and approval.
Our development-stage product candidates for the treatment of the serious diseases on which we are focused, include CF, SCD, TDT, acute and neuropathic pain, AMKD, T1D, DM1, and AAT deficiency (“AATD”). In December 2020,pursuit of serial innovation, our research and development approach includes advancing multiple candidates into clinical trials and pursuing multiple modalities with the goal of bringing first-in-class and/or best-in-class therapies to patients.
Our research and development strategy has been validated through our success in moving novel product candidates into clinical trials and obtaining marketing approvals for TRIKAFTA/KAFTRIO, KALYDECO, ORKAMBI, and SYMDEKO/SYMKEVI for the treatment of CF, and CASGEVY for the treatment of SCD and TDT. Our approach to drug discovery has been further validated by our successful demonstration of clinical proof-of-concept in four additional disease areas: in acute and neuropathic pain with our NaV1.8 inhibitors, in AMKD with inaxaplin, and in T1D with a stem cell-derived islet cell therapy.
Over the last several years, this strategy has led us to expand our capabilities to include additional innovative therapeutic modalities with a focus on cell and genetic therapies, which have the potential to treat, and in some cases, cure diseases by addressing the underlying cause of the disease. CASGEVY, recently approved in multiple geographies, including the U.S., is one such example. We continue to make significant internal investments in cell and genetic therapies. These investments include the development of a Boston-based campus for research and current Good Manufacturing Practices (“cGMP”) clinical manufacturing capabilities dedicated to our portfolio of cell and genetic therapy technologies and teams.
To augment our internal programs, we acquire businesses and technologies and collaborate with biopharmaceutical and technology companies, leading academic research institutions, government laboratories, foundations and other organizations to advance research in our disease areas of interest, as well as to access technologies needed to execute on our strategy. Our internal and external innovation approaches are based on the same strategy, which enables us to effectively integrate and execute on new internal capabilities as we invest in external innovation. Our investments in external innovation include our collaboration with CRISPR, which resulted in the successful development and approval of CASGEVY and the establishment and advancement of other pipeline programs, including our T1D program through our acquisition of Semma Therapeutics, Inc. (“Semma”), our mRNA therapeutic, VX-522, for treatment of CF through our collaboration with Moderna, and our intracellular therapeutics for DM1, including VX-670, through our collaboration with Entrada.
CF
We have completed the Phase 3 global program evaluating a once-daily investigational triple combination of vanzacaftor/tezacaftor/deutivacaftor. Our Phase 3 program consisted of two 52-week randomized, controlled clinical trials, SKYLINE 102 and SKYLINE 103, which evaluated the safety and efficacy of the new combination relative to TRIKAFTA in approximately 950 people with CF 12 years of age and older, and the single arm RIDGELINE trial, evaluating vanzacaftor/tezacaftor/deutivacaftor in children with CF 6 to 11 years of age. In February 2024, we announced positive interim data from 10 people treated with CTX001 andthis Phase 3 program. The data from these trials demonstrate that 20this triple combination provides additional benefit beyond TRIKAFTA for people with severe hemoglobinopathiesCF who have been dosedthe F508del mutation on at least one allele that is CFTR modulator responsive. This new triple combination regimen was safe and well-tolerated in all three clinical trials. We expect to submit global regulatory filings for this triple combination by mid-2024, including a New Drug Application (“NDA”) to the U.S. Food and Drug Administration (“FDA”), using a priority review voucher, and Marketing Authorization Applications to the EMA and Health Canada, for people with CTX001CF 6 years of age and older. We estimate that approximately 90% of people with CF could benefit from vanzacaftor/tezacaftor/deutivacaftor. We continue to identify and develop additional CFTR modulators with the goal of developing best-in-class medicines that can help more patients who respond to CFTR modulators achieve carrier levels of CFTR activity.
In order to treat people with CF who do not make full-length CFTR protein, and as a result, cannot benefit from our CFTR modulators, we are researching and developing genetic therapies, such as mRNA, and gene-editing approaches to CF. In collaboration with Moderna, we are developing VX-522, a CF mRNA therapeutic designed to treat the underlying cause of CF for these people by enabling cells in the ongoinglungs to produce functional CFTR protein. At the end of 2023, we completed dosing in the single ascending dose part of the Phase 1/2 clinical trials. All seventrial for VX-522 in people with CF, and we initiated the
multiple ascending dose part of the clinical trial. We expect to share data from this clinical trial in late 2024 or early 2025.
Sickle Cell Disease and Transfusion-Dependent Beta Thalassemia
Two global Phase 3 clinical trials evaluating CASGEVY in people 5 to 11 years of age with severe SCD (the CLIMB SCD-151 clinical trial) and TDT were transfusion independent at last follow-up(the CLIMB THAL-141 clinical trial) are ongoing. We have completed enrollment in these two clinical trials.
In connection with our serial innovation approach, we are working on preclinical assets for myeloablative conditioning agents with improved tolerability profiles which could be used in connection with treatment with CASGEVY, significantly broadening the eligible SCD and TDT patient population. In addition, we also are investigating small molecules for the potential treatment of SCD and TDT.
Pain
Pain can be debilitating and develop from a variety of conditions. Patients with pain can be categorized as suffering from one of three types of pain: acute pain, chronic neuropathic pain (caused primarily by damage or dysfunction of peripheral nerves), or chronic musculoskeletal pain (caused primarily by damage to muscle, joints or bone). Acute pain usually resolves in days or weeks (for example, following surgery or an injury), while chronic pain generally lasts greater than three months due to unresolved or ongoing damage to tissues. Currently, some available treatments have limited efficacy, others cause significant side effects and/or carry the risk of addiction. Because of these challenges, over- and under-utilization, as well as mis-utilization, of current pain medicines may occur.
The sodium channels NaV1.8 and NaV1.7 play important roles in the physiology of pain. We have discovered multiple selective small molecule inhibitors of NaV1.8 as potential treatments for pain. We obtained pharmacological validation of NaV1.8 inhibition with a first generation NaV1.8 inhibitor in all three clinical pain types: acute pain, chronic neuropathic pain, and chronic musculoskeletal pain.
In acute pain, we completed two randomized, placebo-controlled Phase 3 clinical trials for our lead compound, VX-548, evaluating patients with moderate-to-severe acute pain following abdominoplasty or bunionectomy surgery. We also completed a single-arm clinical trial evaluating the safety and effectiveness of VX-548 in multiple other types of moderate-to-severe acute pain. In January 2024, we announced positive results from these clinical trials. The clinical trials demonstrated that treatment with VX-548 led to statistically significant improvement on the primary endpoint of the time-weighted sum of the pain intensity difference compared to placebo, as well as clinically meaningful reduction in pain from baseline on the numeric pain rating scale after abdominoplasty surgery or bunionectomy surgery. VX-548 was safe and well tolerated in all three Phase 3 studies. We also announced our plans to submit an NDA to the FDA by mid-2024, with the goal of securing a broad label for the treatment of moderate-to-severe acute pain.
In neuropathic pain, in December 2023, we announced results from a Phase 2 clinical trial evaluating VX-548 in people with SCD were freeDPN, a common form of vaso-occlusive crisesperipheral neuropathic pain. The clinical trial demonstrated that treatment with VX-548 led to a statistically significant and clinically meaningful reduction in the primary endpoint of change from CTX001 infusion throughbaseline in the last follow-up. Enrollmentweekly average of daily pain intensity on a numeric pain rating scale at week 12. VX-548 was generally well-tolerated at all doses tested in the clinical trial. We also announced our plans to advance VX-548 for the treatment of DPN into pivotal development, with the ultimate goal of securing a broad label for the treatment of peripheral neuropathic pain.
In support of this broad peripheral neuropathic pain indication we seek, in December 2023, we initiated a second Phase 2 clinical trial in lumbosacral radiculopathy, which is pain caused by impairment or injury to nerve roots in the area of the lumbar spine, another type of peripheral neuropathic pain. There are no approved medicines in the U.S. that are labeled for the treatment of peripheral neuropathic pain.
In keeping with our serial innovation approach, we have also completed a Phase 1 clinical trial evaluating an oral formulation of VX-993, a next generation NaV1.8 inhibitor, and dosing are ongoing, and completion of enrollment for bothwe anticipate initiating Phase 2 clinical trials is expectedevaluating VX-993 for the treatment of moderate-to-severe acute pain and for the treatment of peripheral neuropathic pain in 2021.2024. We also anticipate initiating a Phase 1 clinical trial evaluating an intravenous formulation of VX-993 in 2024. Additionally, we are advancing multiple NaV1.8 inhibitors and NaV1.7 inhibitors through research and earlier stages of development for pain.
APOL1-Mediated Kidney Disease
Inherited mutations in the APOL1 gene play a causal role in the biology of severe proteinuric kidney diseases referred to as AMKD. In AMKD, the kidney’s filtering units known as the glomeruli, and within them the cells known as podocytes, are damaged, leading to leakage of protein into the urine, deterioration in kidney function, scarring, and, ultimately, permanent kidney damage. Patients with proteinuria who inherited two copies of the APOL1 mutations demonstrate rapid progression to end-stage kidney disease. Some patients with AMKD have the histological finding of focal segmental glomerulosclerosis (“FSGS”) and co-morbidities such as hypertension. We are evaluating multiple novel small molecules that inhibit the function of the mutant APOL1 protein with the potential to treat APOL1-mediated kidney disease.
In a Phase 2 proof-of-concept clinical trial, patients with APOL1-mediated FSGS treated with inaxaplin on top of standard of care achieved a statistically significant, substantial, and clinically meaningful reduction of proteinuria. In this clinical trial, inaxaplin was well tolerated by patients. Based on this positive Phase 2 data, we initiated pivotal development of inaxaplin in a single Phase 2/3 adaptive clinical trial in patients with AMKD in 2022. Enrollment in the Phase 2B portion of the Phase 2/3 clinical trial has completed. We expect to select a dose and begin the Phase 3 portion of the clinical trial in the first quarter of 2024.
Type 1 Diabetes
T1D is a chronic metabolic disorder caused by an absence ofinsufficient insulin secretion by the beta cells in the pancreas. In patients with T1D, the person’s own immune system attacks the insulin-producing islet cells of the pancreas are destroyed by the person’s own immune system, resulting in a complete lack of insulin. While insulin therapy allows patients to live for decades with the disease, challenges of insulin therapy include inadequate control of blood sugar (both hyper- and hypo-glycemia), a substantial burden of care on patients and families, and long-term vascular complications.
In 2019, we acquired Semma and established programs to develop cell-basedWe are developing non-autologous (allogeneic) fully differentiated, stem-cell derived islet cell therapies designed to replace insulin-producing islet cells that are destroyed in people with T1D. T1D, with the goal of delivering a functional cure. We are pursuing twothree programs for the transplant of functional islets into patients: transplantation of islet cells alone usingfollowing immunosuppression to protect the implanted cells, and implantation of the islet cells inside a novel immunoprotective device, and development of hypoimmune islet cells to optimize protection of the implanted islet cells from the immune system.
VX-880, our first program, is a stem cell-derived, allogeneic, fully differentiated, insulin-producing islet cell replacement therapy, using standard immunosuppression to protect the implanted cells. The Phase 1/2 study is designed as a sequential, multi-part clinical trial to evaluate the safety and efficacy of VX-880. In Part A, the first two patients received half the target dose of VX-880 cells and the results demonstrated proof-of-concept that VX-880 can restore glucose-regulated insulin production and improve glycemic control. In Part B, patients received the full target dose. We have completed enrollment in Part C of the clinical trial, with concurrent dosing at the full target dose, with trial sites in the U.S., Canada, Norway, the U.K., Italy, Germany, Switzerland, the Netherlands and France. Data presented to date from the VX-880 clinical trial continue to demonstrate unprecedented efficacy and curative potential. Safety data is consistent with the immunosuppressives, the perioperative period, and past medical history. We have placed the clinical trial on a protocol-specified pause, pending review of the totality of the data by an independent data monitoring committee and global regulators, following two patient deaths both of which were determined by the trial investigators to be unrelated to VX-880.
In our second program, we are evaluating VX-264, in which the allogeneic stem cell-derived, fully differentiated, insulin-producing islet cells are encapsulated in an immunoprotective device. The FDA has cleared our IND for VX-880, the first program (transplantation of islet cells alone),device that is implanted in patients. We are enrolling and we expect to initiatedosing patients with VX-264 in a Phase 1/2 clinical trial evaluating VX-880that is a sequential, multi-part study to evaluate the safety, tolerability and efficacy of VX-264. Part A of the study dosed patients with a partial dose of cells/devices and a stagger between patients, and Part B will dose patients with a full target dose and a stagger between patients before moving to concurrent, full dosing in Part C. The clinical trial is enrolling patients in the first halfU.S., Canada, the U.K., Switzerland, Italy, Germany, and the Netherlands, with additional global sites to be activated in the coming months. We have completed Part A of 2021. Thisthe clinical trial will involve an infusion ofand Part B has been initiated in multiple centers and countries.
In our third program, research is directed toward developing hypoimmune cells by gene-editing the same stem cells used in the VX-880 and VX-264 programs prior to differentiation into fully differentiated functional islet cells, and chronic administration of concomitant immunosuppressive therapy,islets in order to protectcloak them from the islet cells from immune rejection.system. This program continues to advance in pre-clinical development.
Myotonic Dystrophy Type 1
DMD and DM1 areis an inherited diseasesdisease that resultresults in the weakening and breakdowndestruction of skeletal muscles over time. In 2019,We have an internal small molecule program for DM1 and, in 2022, we acquired Exonics and expanded ourestablished a collaboration with CRISPR establishing preclinical programs to develop gene-editing therapies for DMD and DM1. We areEntrada focused on advancing gene-editing therapies aimed at treatingenabling efficient intracellular delivery of an oligonucleotide, which we believe will address the underlying causepathophysiology and help restore normal cell function. Our Phase 1/2 clinical trial evaluating the first candidate, VX-670, in patients with DM1 has been initiated in Canada and will initiate in the U.K. in the near term.
Alpha-1 Antitrypsin Deficiency
AATD is a severe disease of DMDthe liver and lung, caused by restoring expressioninherited mutations in the SERPINA1 gene that encodes the AAT protein. We have discovered multiple small molecule correctors that restore folding of near-full length dystrophinthe mutant Z-AAT protein (the most common mutant form of SERPINA1 in AATD patients), leading to increased production of functional AAT protein. We continue to enroll and dose subjects in DM1 by addressingPhase 1 clinical trials evaluating VX-634 and VX-668, the repeat expansion that causes the disease. Our collaborationnext-wave of investigational small molecule AAT correctors with Affinia enables access to a novel library of AAV capsids to supportsignificantly improved potency and drug-like properties compared with our ongoing research and development efforts in genetic therapies, including DMD and DM1.previous AATD correctors.
COMMERCIALIZATION OF OUR MEDICINES
Commercial Organization
Our commercial organization focuses on supporting salesthe appropriate use of TRIKAFTA/KAFTRIO, SYMDEKO/SYMKEVI, ORKAMBI, KALYDECO and KALYDECOCASGEVY in the markets where these products have been approved. Our sales and marketing organizations are responsible for promoting products to health care providers, ensuring our products are distributed effectively, ensuring the safe use of our products, and obtaining reimbursement for our products from third-party payors, including governmental organizations in the U.S. and ex-U.S. markets.
We market our products through personal interactions with physicians and allied health care professionals. In parallel, our government affairs and public policy group advocates for policies that promote life sciences innovation and increase awareness of the diseases on which we are focusing with state and federal legislatures, government agencies, public health officials and other policymakers.
Commercialization of CF Medicines
In the U.S., we primarily sell our CF products to a limited number of specialty pharmacy and specialty distributors. In international markets, we sell our CF products primarily through distributor arrangements and to retail pharmacies, as well as to hospitals and clinics, many of which are government-owned or supported.
Our U.S. field-based CF commercial team is comprised of a small number of individuals to support commercialization of our medicines for CF. We focus our CF marketing efforts in the U.S. on a relatively small number of physicians and health
care professionals who write most of the prescriptions for CF medicines. Many of these physicians and health care professionals are located at a limited number of accredited centers in the U.S. focused on the treatment of CF. In international markets, we or our distributors have small sales forces that support KALYDECO,TRIKAFTA/KAFTRIO, SYMDEKO/SYMKEVI, ORKAMBI SYMDEKO/SYMKEVI and TRIKAFTA/KAFTRIOKALYDECO in jurisdictions where these products are approved.
We market our products through personal interactions with physicians and allied health care professionals. In addition, our government affairs and public policy group advocates for policies that promote life sciences innovation and increase awareness of the diseases on which we are focusing with state and federal legislatures, government agencies, public health officials and other policymakers. We also have established programs in the U.S. that provide our CF products to qualified uninsured or underinsured patients at no charge or at a reduced charge, based on specific eligibility criteria.
Commercialization of CASGEVY
We have begun the commercial launch of CASGEVY, following approvals in the U.S., the E.U., the U.K., Saudi Arabia, and Bahrain. We expect to obtain additional regulatory approvals for CASGEVY in Canada and Switzerland in 2024. We estimate that there are approximately 35,000 people with severe SCD and TDT in the U.S. and Europe, with additional eligible people in Saudi Arabia and Bahrain.
As CASGEVY is the first CRISPR-based therapy to be approved, our global launch strategy is focused on educating physicians, patients and caregivers, payors, and policymakers about the significant disease burden of SCD and TDT and the availability of CASGEVY as a treatment option. In addition, we are concentrating on communications with the scientific and
medical community regarding the clinical profile of CASGEVY and data demonstrating its safety and efficacy. We have also established a global manufacturing network to ensure adequate production and supply of CASGEVY at a commercial scale. Given the geographic concentration of patients, we expect to reach the majority of patients through our specialty commercial model, which is comprised of a small number of field-based individuals. Because the administration of CASGEVY requires specialized experience in stem cell transplantation, we are engaging with experienced hospitals to establish a network of approximately 75 authorized treatment centers (each, an “ATC”) across the U.S., Europe and the Middle East. We have activated 12 ATCs in the U.S., three ATCs in Europe, and one ATC in Saudi Arabia, and we are working to activate additional centers.
Our teams are actively engaged with key treatment centers, policymakers and third-party payors to ensure that patients who may be eligible for CASGEVY have access to this potentially curative therapy. We are seeking broad access with government and commercial payors for CASGEVY, both in and outside the U.S. In the U.S., we are working to develop medical policies and identify reimbursement pathways for people with SCD or TDT through both government and commercial payors. In the U.K., we also have begun engagement with the National Institute for Health and Care Excellence (“NICE”), which is the start of the process for seeking government reimbursement of CASGEVY. In the E.U., we are working with national health authorities to achieve access and reimbursement. In France, the National Authority for Health (“HAS”) approved our request for the implementation of an early access program (“EAP”) for the use of CASGEVY to treat eligible people with TDT, and we are pursuing a similar program for SCD. In the Middle East, we have established a local presence in the region and are working with local healthcare authorities in Saudi Arabia and Bahrain to achieve access and reimbursement.
Near-Term Launch Opportunities
We are also preparing for near-term potential launches for two product candidates: one for our triple combination of vanzacaftor/tezacaftor/deutivacaftor for the treatment of CF and one for VX-548 for the treatment of acute pain.
CF
We believe that the triple combination of vanzacaftor/tezacaftor/deutivacaftor will help more people with CF achieve carrier levels of CFTR function and in doing so, lead to improved clinical outcomes for patients and reinforce our established position as the leading company in CF. In addition, this new triple combination is a once-daily regimen and has a lower royalty burden. We believe the new triple combination will present an opportunity for three types of patients: (i) those who are currently on a CFTR modulator who may want to switch to the new triple combination, (ii) those patients who have not yet been initiated on a CFTR modulator, and (iii) those who have discontinued from a CFTR modulator. We expect to support the launch of the vanzacaftor/tezacaftor/deutivacaftor triple combination with our existing commercial infrastructure.
Acute Pain
We believe the innovation of VX-548 could provide a transformative option for acute pain, based on the suboptimal benefit risk profiles of existing agents, such as the adverse effects and addictive potential of opioids, and the benefit risk profile of VX-548 as established in the results from our Phase 3 clinical trials.
For our near-term opportunity in acute pain, we are focused on the estimated 80 million patients in the U.S. who are prescribed a medicine for their moderate-to-severe acute pain each year. Currently, more than two-thirds of patients receive acute pain prescriptions either during a hospital or ambulatory surgery center visit or at discharge. As hospital-driven prescribing is concentrated amongst approximately 2,000 hospitals and 200 integrated delivery networks, we believe we can reach a large portion of people treated for acute pain with a specialty sales force. We have made significant progress in building our organization to support the launch of VX-548 and are currently hiring additional key teams that will support the use of VX-548 at hospitals and other settings in the U.S. We plan to complete the buildout of our field team in 2024. In addition, we are working with policymakers and institutions that establish practice guidelines to support the appropriate use of non-opioid pain medications, including avoiding meaningful financial barriers to patients accessing such medicines relative to generic opioids.
Reimbursement of Approved Therapies
Sales of our products depend, to a large degree, on the extent to which our products will be reimbursed by third-party payors, such as government health programs, commercial insurance, and managed health care organizations. Increasingly, these third-party payors are becoming stricter in the ways they evaluate and reimburse medical products and services.
Additionally, the containment of health care costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could limit our revenues. Decisions by third-party payors to not cover a product could reduce physician usage of the product.
Our CF medicines are broadly reimbursed by third-party payors in the U.S., including the federal government. We participate in the Medicaid Drug Rebate program, Medicare, and other governmental pricing programs. Medicaid is a joint federal and state program that is administered by the states for low-income and disabled beneficiaries. Under the Medicaid Drug Rebate program, we are required to pay a rebate to each state Medicaid program for our covered outpatient drugs, which include inpatient drugs reimbursed separately from a bundled payment rate that are dispensed to Medicaid beneficiaries. Medicaid rebates are based on pricing data reported by us on a monthly and quarterly basis to the Centers for Medicare & Medicaid Services (“CMS”), the federal agency that administers the Medicaid and Medicare programs.
Any company that participates in the Medicaid Drug Rebate program also must participate in the 340B drug pricing program (the “340B program”), and the Federal Supply Schedule (“FSS”) pricing program. The 340B program, which is administered by the Health Resources and Services Administration, requires participating companies to agree to charge statutorily defined covered entities no more than the 340B “ceiling price” for covered outpatient drugs. The 340B ceiling price is calculated using a statutory formula, which is based on pricing data calculated under the Medicaid Drug Rebate Program. The FSS pricing program, which is administered by the Department of Veterans Affairs (“VA”), also requires participating companies to extend discounted prices to the VA, Department of Defense, Coast Guard, and Public Health Service. Similar to the 340B program, FSS prices are calculated utilizing pricing data reported by us to the VA on a quarterly and annual basis.
The Medicare program includes “Part A” that generally covers certain hospital services for eligible beneficiaries. In general, Part A covers inpatient hospital services, skilled nursing, and hospice care. Most individuals are enrolled in Medicare Part A upon reaching age 65 (although other individuals qualify for Part A, including those receiving services for end stage renal disease). Prescription Drug, Improvement,drugs that are used as part of an inpatient hospital stay will be covered by Medicare Part A, and Modernization Actthese products typically are paid as part of 2003,a bundled or the MMA, established thecomposite rate (e.g., diagnosis related group).
The Medicare Part D program to provideprovides a voluntary prescription drug benefit to Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities, which provide coverage of outpatient prescription drugs.drugs such as our CF medicines. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, including CF, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutics committee. GovernmentU.S. government payment for some of the costs of prescription drugs may increase demand for products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan likely will be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private
Private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. AnyAs a result, any reduction in payment that results from the MMAPart D reimbursement may result in a similar reduction in payments from non-governmental payors.payors for our products. Additionally, private payors, including health maintenance organizations and pharmacy benefit managers in the U.S., are adopting more aggressive utilization management techniques, and are increasingly applying restrictive plan designs that can impact patients and manufacturers and they continue to push for significant discounts and rebates from manufacturers. As a consequence, these payors may not cover or adequately reimburse for use of our products or may do so at levels that disadvantage them relative to competitive products.
The U.S. government has shown significant interest in implementing cost-containment programs for medicines and has enacted reforms at the state and federal level designed to, among other things, modify prescription drug reimbursement amounts and methodologies, and otherwise control health care costs. For example, the American Recovery and Reinvestment Act of 2009 provided funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research was to be developed by the Department of Health and Human Services, or HHS, the Agency for Healthcare Research and Quality and the National Institutes of Health, and periodic reports on the status of the research and related expenditures were to be made to the U.S. Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of our products. In the future, it is possible that comparative effectiveness research demonstrating benefits of a competitor’s product could adversely affect the
sales of our products. If third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Patient Protection and Affordable Care Act or ACA,(“ACA”) was enacted in March 2010 and was designed to expand coverage for the uninsured while at the same time containing overall health care costs. With regard to pharmaceutical products, among other things, the ACA was designed to expand and increase industry rebates for drugs covered under Medicaid programs, impose an annual fee on branded pharmaceutical manufacturers, subject biological products to potential competition by lower-cost biosimilars, and make changes to the coverage requirements under the Medicare Part D program. Additionally, in August 2022, the Inflation Reduction Act (“IRA”) was enacted, establishing a Medicare Drug Price Negotiation Program, a Medicare inflationary rebate, and a redesign of the Part D benefit structure. Certain orphan drugs, including our CF medicines, are excluded from the IRA negotiation program. We anticipate that the U.S. government will continue to engage in activities seeking to address drug pricing and reimbursement. Furthermore, certain states have enacted laws establishing Prescription Drug Affordability Boards (“PDABs”). Some state PDABs, including those in Colorado, Maryland, Washington, and Minnesota, either have the authority or have defined a pathway pursuant to which they may be granted the authority to establish upper payment limits for prescription drugs. For example, in 2023, the Colorado PDAB selected five drugs for an affordability review, including TRIKAFTA, although it ultimately concluded that TRIKAFTA is not unaffordable, and thus not eligible for an upper payment limit.
In Europe and other foreign jurisdictions, the success of our products depends largely on obtaining and maintaining government reimbursement, because many patients are generally unable to access prescription pharmaceutical products that are not
reimbursed by their governments. Negotiating reimbursement agreements in foreign countries can delay the commercialization of a pharmaceutical product and can result in a reimbursement price that is lower than the net price that companies can obtain for the product in the U.S.
In some countries, such as Germany, commercial sales of a new product may begin while thepricing and reimbursement rate that a company will receive isterms are under discussion. In other countries, a company must complete reimbursement negotiations prior to the commencement of commercial supply of the pharmaceutical product. The requirements governing drug pricing vary widely from country to country.country-by-country and region-by-region. For example, the member states of the E.U. can restrict the range of drugs for which their national health insurance systems provide reimbursement and can control the prices of prescription drugs. In addition, many ex-U.S. government payerspayors require companies to provide health economic assessments of products, which are evaluated by government agencies set up for this purpose. A member state may approve a specific price for the drug, or it may instead adopt a system of direct or indirect controls on the total amount of money that a company may receive for supply of a drug. Countries also may consider increasing mandatory discounts over time in an attempt to manage increased demands on healthcare budgets. Reimbursement discussions in foreign countries often result in a reimbursement price that is lower than the net price that companies can obtain for the product in the U.S. In addition, reimbursement discussions may take a significant period of time resulting in commercialization delays. Reimbursement for our products cannot be assured. In addition, it is possible thatassured because a country or region may only provide for reimbursement on terms that we do not deem adequate. Additionally,Further, many ex-U.S. governments have introduced or are in the process of introducing legislation focusing on cost containment measures in the pharmaceutical industry. The impact of these laws where finalized, the final form of laws under consideration, and their relevant practical application, are unknown at this time, but may lead to lower prices, paybacks, or other forms of discounts or special taxes.
Our CF medicines are used by patients in more than 60 countries, and TRIKAFTA/KAFTRIO is reimbursed or accessible in more than 40 of these countries outside the U.S. We expect to continue to focus significant resources to maintain reimbursement discussionsand obtain expanded reimbursement for our CF medicines and pipeline therapies in ex-U.S. markets.
In SCD and TDT, CASGEVY is approved in the U.S., the E.U., the U.K., Saudi Arabia, and Bahrain, and we are actively engaged with relevant stakeholders to obtain broad access and reimbursement with government and commercial payors for CASGEVY. In the U.S., federal and private payors are taking different approaches to reimbursement of novel cell and genetic therapy products, including providing separate reimbursement outside of the bundled payment. We have been working with key government and commercial payors and policymakers in the U.S. to support access to CASGEVY, in part through an understanding of the burden of the diseases and the ability of CASGEVY to treat SCD and TDT effectively. Currently in the U.S., across commercial and government payors, all eligible CASGEVY patients have case-by-case coverage through single case agreements. We continue to make progress with payors, having identified potential reimbursement pathways and are engaging in targeted, innovative contract solutions. For example, we have signed an agreement with Synergie Medication Collective, which covers approximately 100 million people, to provide access to CASGEVY. Within Medicaid, we have engaged all of the Medicaid administrators in all 50 U.S. states, focused on the 25 states with the highest prevalence of SCD patients, and have confirmed pathways to reimbursement in nearly all 25 of those priority states. In addition, in February 2023, the Biden Administration demonstrated its commitment to paying for cell and gene therapies in
diseases such as SCD through the CMS program known as The Cell and Gene Therapy Access Model (“CGT Access Model”). The CGT Access Model was designed to provide an opportunity to accelerate and enhance broad Medicaid access for eligible patients across all 50 U.S. states by allowing state Medicaid agencies to delegate authority to CMS to coordinate and facilitate outcomes-based payment arrangements (“OBAs”) with cell and gene therapy manufacturers, such as ours.
In addition to payor administration for CASGEVY, hospitals will also seek reimbursement for the administration of CASGEVY, which requires procedures for the collection of cells from patients, followed by other procedures before and after infusion of the gene therapy. The same may be true for other pipeline cell and gene therapies. The manner and level at which reimbursement is provided for these services also are important. An inadequate reimbursement for such services may adversely affect ATCs and physicians’ decisions to recommend any product for which we obtain approval in the future and our ability to market or sell the related cell or genetic therapy. The potential treatment center network at launch and growth of such network could also impact uptake and necessitate out-of-state access for some beneficiaries if an ATC is not available within their home state, which could result in further underpayment from out-of-state Medicaid programs.
For CASGEVY in ex-U.S. markets, may take a significant period of time.we anticipate some early access programs initially, such as the recently approved EAP for TDT in France, and in parallel we are pursuing long-term reimbursement arrangements. We have begun engagement with payors in the U.K., the E.U., Saudi Arabia and Bahrain.
STRATEGIC TRANSACTIONS AND COLLABORATIONS
As part of our business strategy, we seek to license or acquire drugs, drugtechnologies, products, product candidates, and businesses that are aligned with our corporate and other technologies that have the potential toresearch and development strategies and complement and advance our ongoing research and development efforts. In addition, we establish business relationships with collaborators to support our research activities and to lead or support development and/or commercialization of certain drugproduct candidates. We expect to continue to identify and evaluate potential acquisitions, licenses and collaborations that may be similar or different from the transactions that we have engaged in previously.
Strategic Transactions
Acquisitions•In 2017, we enhanced our CF portfolio through our acquisition of certain CF assets, including deutivacaftor, from Concert Pharmaceuticals Inc. We recently announced positive results from our Phase 3 clinical trials evaluating our new, once-daily investigational triple combination therapy, vanzacaftor/tezacaftor/deutivacaftor, for people with CF 6 years of age and older.
•In 2019, we acquiredestablished our T1D program through our acquisition of Semma, a privately-heldprivately held company focused on the use of stem cell-derived human islets as a potentially curative treatment for T1D. Our acquisition of Semma advanced our cell therapy capabilities and supports the development of transformative therapiesWe are evaluating VX-880 for T1D. In connection with the acquisition, we acquired all of the outstanding equity of Semma for approximately $950.0 million in cash.
In 2019, we acquired Exonics, a privately held company focused on creating transformative gene-editing therapies to repair mutations that cause DMD and other severe neuromuscular diseases, including DM1. Our acquisition of Exonics enhanced our gene-editing capabilities and supports the potential developmenttreatment of novel therapies for DMDT1D in a clinical trial and DM1.have completed enrollment in Part C of that trial. In connection withaddition, our second clinical program in T1D, VX-264, in which the acquisition, we acquired allimplanted islet cells are encapsulated in an immunoprotective device, is ongoing. We have completed dosing in Part A of the outstanding equity of Exonics for an upfront payment of approximately $245.0 million plus customary working capital adjustments in cash,that trial and certain potential future payments based primarily upon the successful achievement of specified development and regulatory milestones for the DMD and DM1 programs.Part B is underway.
Collaboration and Licensing Arrangements
Joint Development and Commercialization Agreement with CRISPR
In December 2017, we entered into a joint development and commercialization agreement or JDA,(“Original JDCA”) with CRISPR, pursuant to which we are co-developing and preparing to co-commercialize CTX001co-commercializing CASGEVY for TDTSCD and SCD. This JDA wasTDT. We entered into the Original JDCA following our exercise of an option to co-develop and co-commercialize the hemoglobinopathies program that was contained in the collaboration agreement that we entered into with CRISPR in 2015.
TheIn April 2021, we and CRISPR amended and restated the Original JDCA (the “A&R JDCA”). Pursuant to the A&R JDCA, the parties agreed to, among other things, (a) adjust the governance structure for the collaboration and adjust the responsibilities of each party thereunder; (b) adjust the allocation of net profits and net losses as applicable,between the parties; and (c) exclusively license (subject to CRISPR’s reserved rights to conduct certain activities) certain intellectual property rights to us relating to the products that may be researched, developed, manufactured and commercialized under such agreement.
Pursuant to the A&R JDCA, we lead global development, manufacturing and commercialization of CASGEVY, with support from CRISPR. Subject to the terms and conditions of the A&R JDCA, we have the right to conduct all research,
development, manufacturing, and commercialization activities relating to the product candidates and products under the A&R JDCA (including CASGEVY) throughout the world, subject to CRISPR’s reserved right to conduct certain activities.
In connection with the A&R JDCA, we made a $900.0 million upfront payment to CRISPR in the second quarter of 2021. CRISPR earned an additional one-time $200.0 million milestone payment upon receipt of marketing approval of CASGEVY from the FDA.
We and CRISPR shared equally all expenses incurred under the JDA willOriginal JDCA. On July 1, 2021, with respect to CASGEVY, the net profits and net losses incurred pursuant to the A&R JDCA began to be allocated 60% to us and 40% to CRISPR, subject to certain adjustments, while all other product candidates and products continue to have net profits and net losses shared equally bybetween the parties. Under the JDA, CRISPR will be responsible for commercialization activities in the U.S. and we will be responsible for commercialization activities outside of the U.S. There is a joint committee to provide high-level oversight and decision-making regarding the activities covered by the JDA. The committee contains an equal number of representatives from us and CRISPR.
Either party canmay terminate the JDAA&R JDCA upon the other party’s material breach, subject to specified notice and cure provisions, or, in our case, in the event that CRISPR becomes subject to specified bankruptcy, winding up, or similar
circumstances. Either party may terminate the JDAA&R JDCA in the event the other party commences or participates in any action or proceeding challenging the validity or enforceability of any patent that is licensed to such challenging party pursuant to the JDA.A&R JDCA. We also have the right to terminate the JDAA&R JDCA for convenience at any time after giving prior written notice. If circumstances arise pursuant to which a party would have the right to terminate the JDAA&R JDCA on account of an uncured material breach, such party may elect to keep the JDAA&R JDCA in effect and cause such breaching party to be treated as if it had exercised its opt-out rights with respect to the products associated with such uncured material breach and the royalties payable to the breaching party would be reduced by a specified percentage.
Either party may opt of out of the development of a product candidate under the JDAA&R JDCA after predetermined points in the development of the product candidate, on a candidate-by-candidate basis. In the event of such opt-out, the party opting-out will no longer share in the net profits and net losses associated with such product candidate and, instead, the opting out party will be entitled to high single to mid- teenmid-teen percentage royalties on the net sales of such product, if commercialized.
In-License Agreements
We have entered into various agreements pursuant to which we have obtained access to technologies from third parties and are conducting research and development activities with collaborators. Pursuant to these arrangements, we have obtained development and commercialization rights to resulting drugproduct candidates. Depending on the terms of the arrangements, we may be responsible for the costs of research activities, required to make upfront payments and/or milestone payments upon the achievement of certain research, development, and developmentcommercial objectives, and/or pay royalties on future sales, if any, of commercial products resulting from the collaboration. Our current in-license agreements include:
•Affinia Therapeutics, Inc. In 2020, we entered into a collaboration with Affinia to gain access to a novel library of AAV capsids to support on our ongoing research and development efforts in genetic therapies, including DMD, DM1 and CF.
•Arbor Biotechnologies, Inc. In 2018, we entered into a collaboration with Arbor Biotechnologies, pursuant to which we are focusing on the discovery of novel proteins, including DNA endonucleases, to advance the development of new gene-editing therapies.
•CRISPR Therapeutics AG. In 2015, we entered into a collaborationaddition to our arrangement with CRISPR for the discovery and development of potential new treatments aimed at the underlying genetic causes of human diseases using CRISPR-Cas9 gene-editing technology. As described above, we currently are co-developing CTX001 for the treatment of SCD and beta thalassemia and, if successful, have agreed to co-commercialize CTX001. In addition, we have exercised options to exclusively license treatments for specific targets, including CF, that were subject to the research program.program under the collaboration agreement we entered into with CRISPR in 2015. In 2019, we obtained exclusive worldwide rights to CRISPR’s intellectual property for DMDDuchenne muscular dystrophy (“DMD”) and DM1 gene-editing products through a new agreement with CRISPR.
•Kymera Therapeutics, Inc. In 2019,2023, we entered into a collaboration with Kymera Therapeuticsobtained non-exclusive rights to CRISPR’s intellectual property for the research and development of small molecule protein degraders. Under the collaboration, Kymera Therapeutics conducts research activities in multiple targets, and upon designation ofhypoimmune gene-edited cell therapies for T1D through a clinical development candidate for a target, we have the option to exclusively license molecules against the target.new agreement with CRISPR.
•Moderna, Inc. In 2016, we entered into a collaboration with Moderna pursuant to which we are seeking to identifyfor the identification and developdevelopment of mRNA therapeutics encoding CFTR for the treatment of CF. In 2020,December 2022, the FDA cleared our Investigational New Drug Application (“IND”) for VX-522, an mRNA therapeutic we entered intoare developing with Moderna pursuant to this collaboration. At the end of 2023, we completed dosing in the single ascending dose part of the Phase 1/2 clinical trial for VX-522 in people with CF, and we initiated the multiple ascending dose part of the clinical trial.
•Entrada Therapeutics, Inc. In 2022, we established a new strategic collaboration with Moderna aimed at the discovery and developmentEntrada focused on enabling efficient intracellular delivery of lipid nanoparticles and mRNAs that can deliver gene-editing therapies to lung cellsan oligonucleotide. This collaboration includes ENTR-701, now known as VX-670, an investigational candidate for the treatment of CF.
•Skyhawk Therapeutics, Inc. In December 2020, we entered into a collaboration with Skyhawk Therapeutics, forDM1 that is in clinical development. The clinical trial evaluating VX-670 is active and enrolling in Canada and will initiate in the discovery and development of novel small molecules that modulate RNA splicing forU.K. in the treatment of serious diseases.
•Other Arrangements. In 2019, we entered into collaborations with Molecular Templates, Inc. and Ribometrix, Inc. In 2018, we entered into agreements with Genomics plc, Merck KGaA, Darmstadt, Germany, and X-Chem, Inc. in order to support our research and development efforts.near term.
Out-license Agreements
We have entered into various agreements pursuant to which we have out-licensed rights to certain drugproduct candidates to third-party collaborators. Pursuant to these out-license arrangements, our collaborators are responsible for all costs related to the continued development of such drugproduct candidates and obtain development and commercialization rights to these drugproduct candidates. Depending on the terms of the arrangements, our collaborators may be required to make upfront payments, milestone payments upon the achievement of certain research and development objectives and/or pay royalties on future sales, if any, of commercial products licensed under the agreement. Our current out-license agreements include a Strategic Collaboration and License Agreement with Merck KGaA, Darmstadt, Germany, that we entered into in 2017, pursuant to which we granted an exclusive worldwide license to research, develop and commercialize four oncology research and development programs.
Cystic Fibrosis Foundation Therapeutics Incorporated
In 2004, we entered into a collaboration agreement with the Cystic Fibrosis Foundation, or CFF, as successor in interest to the Cystic Fibrosis Foundation Therapeutics, Inc., to support research and development activities. Pursuant to the collaboration agreement, as amended, we have agreed to pay tiered royalties ranging from single digits to sub-teens on covered compounds first synthesized and/or tested during a research term on or before February 28, 2014, including KALYDECO (ivacaftor), ORKAMBI (lumacaftor in combination with ivacaftor)ivacaftor, lumacaftor and SYMDEKO/SYMKEVI (tezacaftor in combination with ivacaftor)tezacaftor and royalties ranging from low-single digits to mid-single digits on potential net sales of certain compounds first synthesized and/or tested between March 1, 2014 and August 31, 2016, including elexacaftor. We do not have any royalty obligations on compounds first synthesized and tested on or after September 1, 2016. For combination products, such as ORKAMBI, SYMDEKO/SYMKEVI and TRIKAFTA/KAFTRIO, (elexacaftor, tezacaftor, and ivacaftor), sales are allocated equally to each of the active pharmaceutical ingredients in the combination product.
INTELLECTUAL PROPERTY
Patents and other proprietaryintellectual property rights such as trademarks, trade secrets, and copyrights are critical to our business. We actively seek protection for our products and proprietary information by means of U.S. and foreign patents, trademarks, and copyrights, as appropriate. In addition, we rely upon trade secret protection and contractual arrangements to protect certain of our proprietary information and products.
Patents provide a period of exclusivity that can make it more difficult for competitors to market and use our technology. We own and control patents and pending patent applications that relate to compounds, formulations, synthetic routes, intermediates, devices, treatment of diseases, synthetic routes, intermediates and other inventions.
To protect our intellectual property, we typically apply for patents several years before a product receives marketing approval. Under current law, a patent expires 20 years from its first effective filing date. Since the drug development process may last for many years, there may be a period of time in which we have an issued patent but not marketing approval to sell the drug. To compensate for patent term lost while a product is in clinical trials and undergoing review for marketing approval, we may be able to apply for patent term extensions or supplementary protection certificates (“SPCs”) in some countries. In addition to patent protection, we have received regulatory exclusivity from U.S. and European regulatory agencies for the active pharmaceutical and biological agents and, where applicable, their approved orphan indications for a certain time period. Regulatory exclusivity runs concurrently with patent exclusivity and provides complementary protection.protection for our products.
WeFor our approved commercial products, and those in development, we own or hold exclusive and non-exclusive licenses to several hundred patents inaround the world. In the U.S. Upon approval of, once an NDA, or a supplement thereto, NDA sponsorsis approved we are required to list with the FDA each U.S. patent with claims that cover the applicant’sour product or a method of using the product. Each ofThe FDA publishes the patents listed bywe list in a book referred to as the NDA sponsor is published in the FDA’s Orange Book. We have tenthirteen issued U.S. patents listed in the Orange Book that cover the active pharmaceutical ingredients in KALYDECO, its marketed formulations, and/or its approved indication. We have 1920 issued U.S. patents listed in the Orange Book that cover the active pharmaceutical ingredients in ORKAMBI, its marketed formulations, and/or its approved indication. We have 2124 issued U.S. patents listed in the Orange Book that cover the active pharmaceutical ingredients in SYMDEKO, its marketed formulation, and/or its approved indication. We have 2128 issued U.S. patents listed in the Orange Book that cover the active pharmaceutical ingredients in TRIKAFTA, its marketed formulation, and/or its approved indication.
Products approved by the FDA under a Biologics Licensing Application (“BLA”), including CASGEVY, receive 12 years of regulatory exclusivity in the U.S. from a product’s approval date. Additionally, we have licenses to dozens of issued U.S. patents that cover CASGEVY, its approved indication, and/or its manufacture. Products approved by the FDA under a
BLA are not subject to the Orange Book patent listing requirement.
The table below sets forth the year of projected expiration for the basic product patentspatent covering each of our approved products. For products that are combinations of two or more active ingredients, the table lists the projected expiration of the
latest expiring patent covering any of the active pharmaceutical ingredients (lumacaftor for ORKAMBI, tezacaftor for SYMDEKO/SYMKEVI and elexacaftor for TRIKAFTA/KAFTRIO). PatentUnless otherwise noted, patent term extensions, supplementary protection certificates, and pediatric exclusivity periods are not reflected in the expiration dates listed in the table below and may extend protection. In some instances, we also own later-expiring patents and applications relating to solid forms, formulations, methods of manufacture, or the use of these drugs in the treatment of particular diseases or conditions. In some cases, however, such patents may not protect our drug from generic competition after the expiration of the basic patent.
| | Product | Product | Projected Expiration of U.S. Patent | Projected Status of European Patent | Product | Expiration Year
of U.S. Basic Product Patent | Expiration Year
of European Basic Product Patent |
| KALYDECO | KALYDECO | 2027 | 2025 1 | KALYDECO | 2027 | 2027 1 |
| ORKAMBI | ORKAMBI | 2030 | 2026 2 | ORKAMBI | 2030 | 2030 1 |
| SYMDEKO/SYMKEVI | SYMDEKO/SYMKEVI | 2027 | 2028 3 | SYMDEKO/SYMKEVI | 2027 | 2033 1 |
| TRIKAFTA/KAFTRIO | TRIKAFTA/KAFTRIO | 2037 | TRIKAFTA/KAFTRIO | 2037 |
| CASGEVY | | CASGEVY | 2035 2 | 2034 2, 3 |
1 CertainExpiration date reflects SPCs granted in the five major European countries have granted supplementary protection certificates for KALYDECO, which expire in 2027.markets (France, Germany, Italy, Spain and the U.K.).
2 Certain European countries have granted supplementary protection certificatesExpiration year reflects the expiration of regulatory exclusivity, which expires later than the basic product patent for ORKAMBI, which expirethis product in 2030.this market.
3Certain European countries have granted supplementary protection certificates for SYMKEVI, Product is approved in Great Britain with regulatory exclusivity until November 2033, which expire in 2033.is later than the expiration of the basic product patent.
In addition to protecting our marketed products, we actively monitor and file patent applications in the U.S. and in foreign countries on inventions relating to our pipeline. For example, we also own and/or control U.S. and foreign patents and/or we have patent applications relating to the following:
•CTX001Vanzacaftor, deutivacaftor, and other potential gene-editing approaches for treating hemoglobinopathies.
•VX-864 and other compounds being studied for the potential treatment of AAT deficiency.
•VX-147 and other compounds being studied for the potential treatment of APOL1-mediated kidney diseases.
•CF potentiators and correctors and many other related compounds, and the use of those compounds to treatfor the treatment CF.
•VX-522 and other mRNA-based approaches for treating CF.
•VX-548 and other compounds being studied for the potential treatment of pain.
•Inaxaplin and other compounds being studied for the potential treatment of AMKD.
•VX-880, VX-264, and other cell-based approaches for treating T1D.
•VX-634, VX-668, and other compounds being studied for the potential treatment of AATD.
•VX-670 for the treatment of DM1.
•Other pre-clinical and clinical candidates and the use of such candidates to treat specified diseases.
•The manufacture, pharmaceutical compositions, related solid forms, formulations, dosing regimens, and methods of use of many of the above compounds.
We and CRISPR intend to rely upon a combination of rights, including patent rights, trade secret protection, and regulatory exclusivities to protect CTX001.CASGEVY. CRISPR has licensed certain rights to a worldwide patent portfolio that covers various aspects of the CRISPR/Cas9 editing platform technology including, for example, compositions of matter and methods of use, including their use in targeting or cutting DNA, from Dr. Emmanuelle Charpentier. In addition to Dr. Charpentier, this patent portfolio has named inventors who assigned their rights to the Regents of the University of California or the University of Vienna, to whom we refer, together with Dr. Charpentier, as the CVC Group. CRISPR has non-exclusive or co-exclusive rights to the patent rights that protect the core CRISPR/Cas9 gene-editing technology. For example, certain third parties, including competitors, have reported obtaining a license to rights in this patent portfolio in certain fields. In addition, patents and patent applications in this patent portfolio are the subject of adversarial proceedings in the U.S., Europe, and other
jurisdictions, including proceedings between the CVC and the Broad Institute in the U.S. Patent and Trademark Office or USPTO.(the “USPTO”), between the CVC Group and, separately, Sigma-Aldrich, Co. LLC (“Sigma-Aldrich”), ToolGen, Inc. (“ToolGen”), and the Broad Institute, Harvard University, and Massachusetts Institute of Technology (collectively, “Broad”). To date, both the CVC Group and the Broad have obtained granted patents that purport to cover aspects of CRISPR/Cas9 editing platform technology. The patents and patent applications within the patent portfolios of the CVC patent portfolio and theGroup, Broad, patent portfolioSigma-Aldrich and/or ToolGen are, or may in the future be, involved in proceedings similar to interferences or priority disputes in Europe or other foreign jurisdictions. In December 2023, we entered into an agreement with Editas Medicine, Inc. (“Editas”), providing us a non-exclusive sublicense to certain patents relating to CRISPR/Cas9 technology, owned by Broad and Harvard, which are licensed to Editas. In addition to the patent portfolioportfolios licensed from Dr. Charpentier, Broad, and Harvard, we own patents and/or patent applications relating to the composition, manufacture, and use of CTX001.CASGEVY.
From time to time, we enter into exclusive and non-exclusive license agreements for proprietary third-party technology used in connection with our research activities. These license agreements typically provide for the payment by us of a license fee but may also include terms providing for milestone payments or royalties for the development and/or commercialization of our drug products arising from the related research.
We cannot be certain that issued patents we own or license will be enforceable or provide adequate protection or that pending patent applications will result in issued patents. The existence of patents does not guarantee our right to practice the patented technology or commercialize the patented product. Litigation, interferences, oppositions, inter partes reviews,
administrative challenges or other similar types of proceedings may be necessary in some instances to determine the validity and scope of certain patents, regulatory exclusivities or other proprietary rights, and in other instances to determine the validity, scope or non-infringement of intellectual property rights that may be claimed by third parties to be pertinent to the manufacture, use or sale of our products.
MANUFACTURING
As we market and sell our approved products and advance our drugproduct candidates through clinical development toward commercialization, we continue to build and maintain our supply chain and quality assurance resources. We rely on internal capabilities and a global network of third parties to manufacture and distribute our product candidates for clinical trials, as well as our products for commercial sale and post-approval clinical trials and to manufacture and distribute our drug candidates for clinical trials. In addition to establishing supply chains for each new approved product, we need tomust adapt our supply chain for existing products to include additional formulations that are often required in order to treat younger patients or to increase scale of production for existing products. We are focused on ensuring the stability of the supply chains for our current products, including KALYDECO, ORKAMBI, SYMDECO/SYMKEVI, TRIKAFTA/KAFTRIO, CASGEVY, and for our pipeline programs. In addition, we are focused on identifying and ensuring the optimalefficient manufacturing and delivery requirementsprocesses for the cell and genetic therapies we are developing.developing, including our stem cell therapy program for T1D.
We have established our own manufacturing capabilities in Boston, which we use for clinical trial and commercial supplies, including certain manufacturing steps related to our commercial supply of TRIKAFTA/KAFTRIO. We expect that we willto continue to rely on third parties to meet our commercial supply needs, including for TRIKAFTA/KAFTRIO and pipeline programs, and a significant portion of our clinical supply needs for the foreseeable future. We have established our own small-scale manufacturing capabilities in Boston, which we use for clinical trial and commercial supplies, and are evaluatingcontinue to evaluate additional manufacturing capacity for our current and future products.products, including products with near-term launch potential (e.g., the triple combination of vanzacaftor/tezacaftor/deutivacaftor for CF and VX-548 for pain).
Our supply chain for sourcing raw materials and manufacturing drug product ready for distributionour products, including obtaining all necessary supplies, is a multi-step global endeavor. In general, these raw materials and other necessary supplies are available from multiple sources. Third-party contract manufacturers, including some in China, perform different parts of our manufacturing process. Contract manufacturers may supply us with raw materials, convert these raw materials into drug substance and/or convert the drug substance or product into final dosage form. In addition, third parties assist us with packaging, warehousing, and global distribution of our products.
Establishing and managing this global supply chain for each of our drugsproducts and drugproduct candidates requires a significant financial commitment and the creation and maintenance of numerous third-party contractual relationships. To ensure the stability of our supply chains, we aim to develop alternatives for each step of our manufacturing process at the time of, or shortly after, marketing approval. Therefore, at any point in time, we may have a limited number of single source manufacturers for certain steps in our manufacturing processes, particularly for recently launched products.
In order to manufacture our commercial CF products, we utilize both continuous manufacturing technology as well as batch manufacturing processes. While continuous process manufacturing has been used in many industries, we believe that we are the first company to obtain FDA approval for a fully-continuous drug product manufacturing process.
We have developed systems and processes to track, monitor, and oversee our and our third-party manufacturers’
activities, including a quality assurance program intended to ensure that our third-party manufacturers comply with current Good Manufacturing Practices, or cGMP. We devote substantial time, resources and effort in the areas of production, quality control, and quality assurance to maintain cGMP compliance. We regularly evaluate the performance of our third-party manufacturers with the objective of confirming their continuing capabilities to meet our needs efficiently and economically. Manufacturing facilities, both foreign and domestic, are subject to inspections by or under the authority of the FDA and other U.S. and foreign government authorities. Although we actively engage with regulatory authorities, the timing of inspections and regulatory approvals for each of these facilities may be delayed for a number of reasons.
The manufacturing processes for cell and genetic therapies are more complex than those required for small molecule drugs and require different systems, equipment, facilities, and expertise. Additionally, we are unable to rely onutilize a single process for all of our cell and genetic therapies; they must be customized for each program and therapy. Although we have been building expertise in these areas, which was augmented through our acquisitions of ExonicsWe are investing and Semma, we will needplan to continue to expandinvest significant resources in expanding and strengthenstrengthening our manufacturing supplies, infrastructure and capabilities, such as cGMP clinical manufacturing, both independently and/orand through a third-party network,networks, in an effort to successfully develop and commercialize our cell and genetic therapies. We are focused on evaluating and securing potentialhave secured relationships with third parties globally for CASGEVY, and are identifying and evaluating various third parties that wouldwill enable us to expand and strengthen such capabilities to support our current and future cell and genetic therapy programs. We expect to make significant investment in our manufacturing capabilities and partnerships for our genetic and cell-based therapy programs, in order to continue to advance and, in the future, commercialize these programs.
We and CRISPR rely on third-party manufacturers to produce or process cell culture reagents, gene-editing components, such as Cas9 protein and guide RNA molecules for clinical trials and commercial supply of CASGEVY, and to generate gene-edited cells to supply CTX001 for clinical trials. If approved, we expect toCASGEVY. We continue to rely on third-party manufacturers for commercial supply of CTX001.CASGEVY. The current manufacturing process for CTX001CASGEVY involves a number of steps prior to the final infusion of drug product into patients. Following mobilization and collection of blood cells from the patient, at the clinical site, cells are transferred to a manufacturing site where HSPCs are purified and CRISPR/Cas9 gene-editing is performed. The edited cellular product, called CTX001,CASGEVY, is frozen and transported back to the clinical siteATC where it is stored prior to infusion into the patient. Each step must be completed successfully, and in a timely manner, requiring coordination between CRISPR, Vertex, clinical sites,us, ATCs, third-party manufacturers and shipping vendors. To increase production to commercial levels, Vertexwe are making significant investments to secure additional capacity and CRISPR will need to coordinate manufacturing, testing, and logistics activities at a larger scale across multiple facilities into serve the geographies in which CTX001we expect to treat patients with CASGEVY. In addition to clinical data establishing the safety and efficacy of CASGEVY, regulatory approval of the processes and facilities used to manufacture and test critical gene-editing components is approved. Approval willrequired.
We have established our own manufacturing capabilities in the Boston area to support our T1D program. In addition, to further expand our capabilities in cell therapy manufacturing, in June 2023, we announced a strategic agreement with Lonza to support the manufacture of our portfolio of investigational allogeneic stem cell-derived, fully differentiated, insulin-producing islet cell therapies. We expect to rely on inspection and approvalthird parties to meet significant portions of these facilities by global health authorities.our future commercial supply needs for our T1D program.
COMPETITION
The pharmaceutical industry is characterized by extensive research efforts, rapid technological progress, and intense competition. There are many public and private companies, including pharmaceutical companies and biotechnology companies, engaged in developing products for the indications our drugs are approved to treat and the therapeutic areas we are targeting with our research and development activities. Potential competitors also include academic institutions, government agencies, other public and private research organizations and charitable venture philanthropy organizations that conduct research, seek patent protection and/or establish collaborative arrangements for research, development, manufacturing and commercialization. Mergers and acquisitions in the pharmaceutical, biotechnology and gene therapy industries may result in a larger concentration of resources among a smaller number of our competitors. Some of our competitors may have substantially greater financial, technical, marketing and human resources than we do.
We believe that competition in our industry is based on, among other factors, innovative research, the effective and rapid development of drugproduct candidates, the ability to market and obtain reimbursement for products and the ability to establish effective patent protection. We face competition based on the safety and efficacy of our product and drugproduct candidates, the timing and scope of regulatory approvals, the availability and cost of supply, marketing and sales capabilities, reimbursement coverage, price, patent protection and other factors. Our competitors may develop or commercialize more effective, safer, or more affordable products than we are able to develop or commercialize or obtain more effective patent protection. As a result, our competitors may commercialize products more rapidly or effectively than we do, which would adversely affect our
competitive position, the likelihood that our drugproduct candidates, if approved, would achieve and maintain market acceptance and our ability to generate meaningful revenues from our products. Future competitive products may render our products, or future products, obsolete or noncompetitive. Another key element of remaining competitive in our industry is recruiting and retaining leading scientific, technical and management personnel to conduct our research activities and advance our development programs, including with the commercial expertise to effectively market our products.
Cystic Fibrosis
A number of companies are seeking to identify and develop drugproduct candidates for the treatment of CF, including CFTR modulators and other therapies intended to address the underlying causes of CF.
AbbVie, Inc., or AbbVie, has indicated that it plans to develop a triple combination CFTR modulator therapy comprised of a potentiator and correctors. Currently, AbbVie is evaluating the combination of a potentiator and a corrector in a Phase 2 clinical trial. In March 2020, AbbVie disclosed plans to file an IND application with the FDA for another corrector in the second quarter of 2020. In addition, Proteostasis Therapeutics, Inc. was developing potential CFTR modulator therapies prior to its acquisition by Yumanity Therapeutics, Inc., or Yumanity. (“Yumanity”). Following the merger, Yumanity hasout-licensed the CF program to Fair Therapeutics. In August 2023, HIT-CF Europe, a research project from CF-Europe, a federation of European patient advocacy groups, announced plans to divest itsinitiate a Phase 3 trial of a combination of Fair Therapeutics’ CFTR modulators in European CF program.patients with rare mutations of the CFTR gene. Sionna Therapeutics has at least two CFTR modulator in Phase 1 development, and several CFTR modulators in preclinical development.
Other therapeutic approaches include addressing CF utilizing nucleic acid therapies, and read-through agents, which are compounds that allow expression of a functional CFTR protein and are relevant for the more than 5,000 people with CF who cannot make full-length protein.CFTR protein and cannot benefit from CFTR modulators. Nucleic acid therapies are under development by companies such as Translate Bio, Arcturus Therapeutics Holdings, Inc., ReCode Therapeutics, Inc., Krystal Biotech, Inc., Spirovant Sciences, Inc. andBoehringer Ingelheim International, GmbH, 4D Molecular Therapeutics, Inc. Translate Bio is evaluating its mRNA therapy in a proof of concept Phase 1/2 clinical trial. Eloxx Pharmaceuticals, Inc. is evaluating a read-through therapy for nonsense CFTR mutations in two Phase 2 clinical trials.
Our success in rapidly developing and commercializing our products may increase the resources that our competitors allocate to the development of these potential treatments for CF. In addition, clinical trials conducted by our competitors could take place simultaneously with our own trials and may slow down our pace of development if we are unable to recruit sufficient clinical trial subjects. If one or more competing therapies are successfully developed as a treatment for people with CF, our revenues from our current products and/or additional CF products, if then approved, could face significant competitive pressure.
Sickle Cell and Beta Thalassemia
There are multiple approved small molecule and biologic treatments for SCD and beta thalassemia, including products from Novartis International AG (“Novartis”), Pfizer Inc. (“Pfizer”), and Bristol Myers Squibb together with Merck & Co. In addition, bluebird bio, Inc. (“bluebird”), obtained FDA approval of its gene therapy, Zynteglo (betibeglogene autotemcel) in August 2022 for the treatment of patients with beta thalassemia who require regular red blood cell transfusions. In late 2023, bluebird also obtained FDA approval for its gene therapy for SCD, Lyfgenia (lovotibeglogene autotemcel).
Editas is also investigating a CRISPR gene-editing asset that is currently in clinical trials. In addition, various companies and private academic/medical institutes are developing gene therapy or gene-editing candidates for the treatment of SCD or beta thalassemia utilizing CRISPR technology, lentiviral vectors, zinc finger nuclease technology, or transcription activator-like effector nuclease, gene correction, base, or prime editing.
Pipeline
In recent years, we have committed significant research resources to, and made significant investments in, our pipeline of potential new therapies for AAT deficiency, APOL1-mediated kidney diseases, TDT, SCD,pain, AMKD, T1D, muscular dystrophies, T1DAATD and other diseases. We plan
Pain
The acute pain market largely consists of conventional analgesics, including opioids, non-steroidal anti-inflammatory drugs, acetaminophen and local anesthetics, low-cost generics, and reformulations aiming to continue investingprovide safer, more tolerable, or more convenient therapies. Peripheral neuropathic pain is a type of chronic pain caused by injury or dysfunction of peripheral nerves. The peripheral neuropathic pain market largely consists of generic anticonvulsants and anti-depressant drugs.
Several companies are pursuing clinical development of novel mechanisms of action for acute and chronic pain indications, including a selective NaV1.8 inhibitor in a Phase 1 clinical trial by Orion Corporation. Several additional
NaV1.8/1.7 inhibitors are in preclinical development for pain, including programs by Merck and Grünenthal.
Additional Programs
Certain of our pipeline, including expanding beyond small molecule therapiesother product candidates face competition from many pharmaceutical and intobiotechnology companies. For example, other pharmaceutical and biotechnology companies are actively engaged in the discoveryresearch and development of products for T1D, including strategies to prevent the destruction of beta cells, to protect beta cell function, or to replace missing beta cells, as well as other cell therapy approaches such as immune evasive technologies to hide the cell from the immune system, micro- and gene therapies. For example, we remain focused on our ongoing evaluation of CTX001, an investigational CRISPR/Cas9-based gene-editingmacro-encapsulation technologies that potentially require no immunosuppression, and islets cell in combination with immunosuppression. In addition to CellTrans, Inc.’s Lantidra, the first FDA-approved cadaveric islet therapy for treatment of SCD and TDT currently in clinical development.
There are multiple approved treatments for TDT and SCD, including products from Novartis International AG, or Novartis, Global Blood Therapeutics, Inc. and Bristol Myers Squibb together with Acceleron Pharma, Inc. Bluebird Bio, Inc., or Bluebird, has a gene therapy, Zynteglo (Lentiglobin), approved by the European Medicines Agency, or EMA, for the treatment of certain TDT genotypesT1D, other companies developing cell therapies for T1D include Novo Nordisk A/S, Eli Lilly & Co., Sernova Corp, Sana Biotechnology, Inc., Seraxis, Inc., and in clinical development for SCD. In addition, variousEvotec A.G. The T1D standard of care of exogenous insulin injections continues to progress toward an artificial pancreas as multiple companies and private academic/medical institutes are developing gene therapy or gene-editing candidatesnovel insulin formulations, advanced pumps and glucose sensors, and closed loop systems.
There are no approved therapies targeting AMKD. People with chronic kidney disease (“CKD”) take angiotensin-converting enzyme inhibitors and angiotensin receptor blockers to treat hypertension; steroids and immunosuppressants to reduce proteinuria; and SGLT2 inhibitors to reduce the risk of CKD progression. These CKD treatments may reduce proteinuria, but do not stop the rapid disease progression seen in AMKD patients. Several companies have early programs developing APOL1-targeted assets for patients with AMKD, including AstraZeneca, Maze Therapeutics, and OmniAb. Eli Lilly and Company’s Janus kinase inhibitor (baricitinib) is being investigated in AMKD patients in a Phase 2 study by Duke University.
Several new therapies are under development specifically for the treatment of SCD or TDT utilizing CRISPR technology, lenti-viral vectors, zinc finger nuclease technology, or base editing.patients with FSGS. We are not aware of any of these therapies having a mechanism of action targeting FSGS patients with two APOL1 mutations. Travere’s sparsentan is currently under regulatory review while Dimerix’s QYTOVRA is in a pivotal study.
Many other pharmaceutical and biotechnology companies are also investing resources for the discovery and development of small molecules gene therapies and cell and gene therapies to treat the same diseasesdisease areas for which we are developing therapies.therapies in our pipeline. If any of these competitors develop or successfully commercialize products involving therapies competitive with our pipeline therapies, the potential return on our investment in those pipeline therapies could be impacted.
GOVERNMENT REGULATION
Our operations and activities are subject to extensive regulation by numerous government authorities in the U.S., the E.U.Europe and other countries. In the U.S., the E.U.Europe and other countries, drugsour products are subject to rigorous regulations governing thetheir testing, manufacture, labeling, storage, record keeping, approval, and advertising and promotion of our products.promotion. As a result of these regulations, product development and product approval processes are very expensive and time consuming. The regulatory requirements applicable to drug and biologic development, approval, and marketing are subject to change. In addition, regulations and administrative guidance often are revised or reinterpreted by the agencies in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or FDA or comparable ex-U.S. regulations, guidance or interpretations will change.
United States Government Regulation
New Drug Application and Biologics License Application Approval Processes
The process required by the FDA before a drug or biologic may be marketed in the U.S. generally involves the following:
•completion of preclinical laboratory tests, animal studies and formulation studies conducted according to Good Laboratory Practices or GLP,(“GLP”), and other applicable regulations;
•submission to the FDA of an IND, application, which must become effective before clinical trials in the U.S. may begin;
•performance of adequate and well-controlled clinical trials according to Good Clinical Practices or GCP,(“GCP”), and other clinical trial-related regulations to establish the safety and efficacy of the proposed drug for its intended use;
•submission to the FDA of an NDA or a New Drug Application, or an NDA;BLA;
•satisfactory completion of ana pre-approval FDA inspection of the manufacturing facility or facilities at which the product will be produced to assess compliance with cGMP; and
•FDA review and approval of the NDA.
Once a drug candidateor biologic is identified for development, it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal pharmacology and toxicology studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND, which seeks FDA approval to test the drug candidateor biologic in humans. Preclinical or nonclinical testing typically continues even after the IND is submitted.
If the FDA accepts the IND, the drug candidateor biologic can then be studied in human clinical trials to determine if the drugproduct candidate is safe and effective. These clinicalClinical trials involve three separate phases that often overlap, can take many years and are expensive. These three phases, which are subject to considerable regulation, are as follows:
•Phase 1. The drug or biologic initially is introduced into a limited number of healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution and elimination. In the case of some drug candidatesdrugs or biologics for severe or life-threatening diseases, such as cancer, especially when the drug candidateor biologic may be inherently too toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
•Phase 2. Clinical trials are next initiated in a limited patient population with the specified disease or condition the drug or biologic is intended to treat to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the drug or biologic candidate for specific targeted diseasesthe disease or condition it is intended to treat and to determine dosage tolerance and optimal dosage.
•Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk-benefit ratio of the drug candidateor biologic and provide an adequate basis for regulatory approval and product labeling.
It is possible that Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within any specified period, if at all. The FDA or the sponsor may, at any time during the initial 30-day IND review period or while clinical trials are ongoing under the IND, impose a partial or complete clinical hold or suspend a clinical trial at any time for a variety of reasons, including a finding that the healthy volunteers or patients are being exposed to an unacceptable health risk. All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCP. Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently in other situations, includingand the occurrence of serious adverse events.events must also be reported. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health for public dissemination on the www.clinicaltrials.gov website.
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and are commonly intended to generate additional safety data regarding use of the product in a clinical setting. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA or BLA.
The results of drug or biologic development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug candidate,or biologic, proposed labeling and other relevant information are submitted to the FDA as part of an NDA or BLA requesting approval to market the drug candidate.or biologic. The FDA reviews each NDA or BLA submitted to ensure that it is sufficiently complete for substantive review before it accepts it for filing. It may request additional information rather than accept an NDA or BLA for filing.
Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA reviews an NDA or BLA to determine, among other things, whether a drug candidateor biologic is safe and effective for its intended use and whether its manufacturing is cGMP-compliant to assure and preserve the drug candidate’sor biologic’s identity, strength, quality and purity. The FDA may refer the NDA or BLA to an advisory committee for review and recommendation as to whether the NDA or BLA should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA or BLA, the FDA will inspect the facility or facilities
where the drug candidateor biologic is manufactured and tested. Additionally, before approving an NDA or BLA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP requirements.
The FDA may require, as a condition of approval, restricted distribution and use, enhanced labeling, special packaging or labeling, expedited reporting of certain adverse events, pre-approval of promotional materials, restrictions on direct-to-consumer advertising or commitments to conduct additional research post-approval. The FDA will issue a complete response letter if the agency decides not to approve the NDA or BLA in its present form.
Biologics License Application Process
Certain of our drug candidates may be regulated by the FDA under the Food, Drug, and Cosmetic Act, or FDCA, and the Public Health Service Act as biologics. Biologics can present special safety, efficacy and manufacturing challenges that may differ from those present in the regulation of small molecule drugs. As such, while similar to the NDA review process described above, in lieu of filing an NDA, biologics require the submission of a Biologics License Application, or BLA, and approval of such BLA by the FDA prior to being marketed in the U.S.
Expedited Review and Approval
The FDA has developed a number of distinct approaches to make new drugs or biologics available as rapidly as possible in cases where there is no available treatment or there are advantages over existing treatments.
The FDA may grant “accelerated approval” to products that have been studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit to patients over existing treatments. For accelerated approval, the product must have an effect on a surrogate endpoint or an intermediate clinical endpoint that is considered reasonably likely to predict the clinical benefit of a drug, such as an effect on irreversible morbidity and mortality. When approval is based on surrogate endpoints or clinical endpoints other than survival or morbidity, the sponsor will be required to conduct additional post-approval clinical studies to verify and describe the clinical benefit. These studies are known as “confirmatory trials.” Approval of a drug may be withdrawn, or the labeled indication of the drug changed if these trials fail to verify clinical benefit or do not demonstrate sufficient clinical benefit to justify the risks associated with the drug.drug or biologic.
The FDA may grant “fast track” status to products that treat serious diseases or conditions and demonstrate the potential to address an unmet medical need. Fast track is a process designed to facilitate the development and expedite the review of such products by providing, among other things, more frequent meetings with the FDA to discuss the product’s development plan and rolling review, which allows submission of individually completed sections of an NDA or BLA for FDA review before the entire submission is completed. Fast track status does not ensure that a product will be developed more quickly or receive FDA approval.
“Breakthrough Therapy” designation is a process designed to expedite the development and review of drugs or biologics that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug or biologic may demonstrate substantial improvement over available therapy on aone or more clinically significant endpoint. For drugs and biologics that have been designated asendpoints. Breakthrough Therapies,Therapy designation provides all of the benefits of fast-track designation in addition to robust FDA-sponsor interaction and communication canto help to identify the most efficient and expeditious path for clinical development while minimizing the number of patients placed in ineffective control regimens.
“Regenerative Medicine Advanced Therapy,” or RMAT,(“RMAT”) designation is a process created by the 21st Century Cures Act in December 2016. A product is eligible for RMAT designation if it is a regenerative medicine therapy that is intended to treat, modify, reverse or cure a serious or life-threatening disease or condition, and if preliminary clinical evidence indicates that the product has the potential to address unmet medical needs for such disease or condition. The benefits of RMAT designation include the benefits available to breakthrough therapies, including potential eligibility for priority review and accelerated approval based on surrogate or intermediate endpoints.
The FDA may grant “priority review” status to productsa product that, if approved, would provide significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions. Priority review is intended to reduce the time it takes for the FDA to review an NDA or BLA, with the goal to take action on the application within six months from when the application is filed, compared to ten months for a standard review.
Manufacturing Quality Control
Among the conditions for NDA or BLA approval is the requirement that the prospective manufacturer’s quality control and manufacturing procedures continually conform with cGMP. In complying with cGMP, manufacturersManufacturers must devote substantial time, money and effort in the areas of production, quality control, and quality assurance to maintain cGMP compliance. Material changes in manufacturing equipment, location, or process, may result in additional regulatory review and approval. The FDA, and other regulatory agencies, conduct periodic visits to inspect equipment, facilities, and processes following the initial approval of a product. If a manufacturing facility is not in substantial compliance with the applicable regulations and requirements imposed when the product was approved, regulatory or judicial enforcement action may be taken,initiated, which may include a warning
letter, suspension of manufacturing, product seizure, or an injunction against shipment of products from the facility and/or recall of products previously shipped. We rely, and expect to continue to rely, on third parties for the production of our products. Future FDA, state, and foreign inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt manufacture or distribution of our products or require substantial resources to correct.
Post-approval Requirements
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory standardsrequirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or complete withdrawal of the product from the market. In addition, under the FDCA the sponsor of an approved drug in the U.S. may not promote that drug for unapproved, or off-label, uses, although a physician may prescribe a drug for an off-label use in accordance with the practice of medicine. AfterThe FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. Further, after approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. In addition, the FDA may require testing, including Phase 4 trials, and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs.
The FDA typically advises that patients treated with gene therapy undergo follow-up observations for potential adverse events for a 15-year period. As part of the FDA approval of CASGEVY for the treatment of SCD and TDT, we are required to conduct two post-marketing requirement (“PMR”) safety studies to assess the long-term risk of hematologic malignancies and off-target genome editing effects by CRISPR/Cas9. Both PMR safety studies are consistent with the FDA’s guidance for industry on Long Term Follow-up After Administration of Human Gene Therapy Products.
Products manufacturedwe manufacture or distributed by usdistribute pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things:
• record-keeping requirements;
• reporting of adverse experiences with the product;
• providing the FDA with updated safety and efficacy information;
• drug sampling and distribution requirements;
• notifying the FDA and gaining its approval of specified manufacturing or labeling changes;
• complying with certain electronic records and signature requirements; and
• complying with FDA promotion and advertising requirements.
Failure to comply with the applicable U.S. requirements at any time during the drug or biologic development process, approval process or after approval, may subject us or our collaborators to administrative or judicial sanctions, any of which could have a material adverse effect on us. These sanctions could include:
• restrictions on marketing or manufacturing of the product;
• safety alerts, Dear Healthcare Provider letters, press releases, or other communications containing warnings or other safety information about the product;
• refusal to approve or delay in review of pending applications;
• withdrawal of an approval or the implementation of limitations on a previously approved indication for use;
• imposition of a clinical hold, a risk evaluation and mitigation and evaluation strategy (“REMS”) or other safety-related limitations;
• warning letters or “untitled letters”;letters;”
• product seizures;seizures, recalls, or detentions, or refusal to permit the import or export of products;
• total or partial suspension of production or distribution;
• consent decrees, corporate integrity agreements, debarment or exclusion from federal healthcare programs; or
• injunctions, fines, disgorgement, refusals of government contracts, or civil or criminal penalties.
United States Patent Term Restoration and Regulatory Exclusivity
Upon approval, products may be entitled to certain kinds of exclusivity under applicable intellectual property and regulatory regimes. The Drug Price Competition and Patent Term Restoration Act of 1984 (commonly known as the Hatch-Waxman Act) permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. The length of the patent extension is roughly based on 50 percent of the period of time from the filing of an IND for a compound to the submission of the NDA for such compound, plus 100 percent of the time period from NDA submission to regulatory approval. The extension, however, cannot exceed five years and the patent term remaining after regulatory approval cannot exceed 14 years.
If the FDA approves a drug product that contains an active ingredienta new chemical entity not previously approved, the product is typically entitled to five years of non-patent regulatory exclusivity. Other products may be entitled to three years of exclusivity if approval was based on the FDA’s reliance on new clinical studies essential to approval submitted by the NDA applicant. If
the NDA applicant studies the product for use by children, the FDA may grant pediatric exclusivity, which extends by 180 days each existing exclusivity (patent and regulatory) related to the product.
Biologics are also entitled to exclusivity under the Biologics Price Competition and Innovation Act (the “BPCIA”), which was passed as Title VII to the ACA. The law provides a pathway for approval of biosimilars followingproducts that are biosimilar to or interchangeable with an FDA-licensed reference biological product. Under the expiration ofBPCIA, a reference biological product is granted 12 years of data exclusivity, the period of time during which an innovator’s clinical data cannot be used by other companies, from the time of first licensure of the product, and an application for a biosimilar product may not be submitted to the innovator biologic andFDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a potential additional 180 day-extension termbiosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. The BPCIA also created certain exclusivity periods for conducting pediatric studies.biosimilars approved as interchangeable products. At this juncture, it is unclear whether products deemed “interchangeable” by the FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy law. Biologics are also eligible for orphan drug exclusivity, as discussed below. The law also includes an extensive process for the innovator biologic and biosimilar manufacturer to litigate patent infringement, validity, and enforceability prior to the approval of the biosimilar. There have been ongoing federal legislative and administrative efforts as well as judicial challenges seeking to repeal, modify or invalidate some or all of the provisions of the ACA. While none of those efforts have focused on changes to the provisions of the ACA related to the biosimilar regulatory framework, if the ACA is repealed, substantially modified, or invalidated, it is unclear what, if any, impact such action would have on biosimilar regulation.
If the NDA or BLA applicant studies the product for use by children, the FDA may grant pediatric exclusivity, which extends by 180 days each existing exclusivity (patent and regulatory) related to the product.
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drug candidatesdrugs or biologics intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 people in the U.S.
If a drug candidateor biologic that has orphan drug designation subsequently receives the first FDA approval for that drug or biologic for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indicationdisease or condition for seven years following marketing approval, except in certain very limited circumstances, such as if the later product is shown to be clinically superior to the orphan product. Orphan drug exclusivity, however, also could block the approval of our drug candidatesproducts for seven years if a competitor first obtains approval of the same productdrug, as defined by the FDA, or if our drug candidate is determined to be contained within the competitor’s product for the same indicationdisease or disease.condition for which we were seeking approval. KALYDECO, ORKAMBI, SYMDEKO, TRIKAFTA, and TRIKAFTACASGEVY have been granted orphan drug exclusivity by the FDA.
We may pursue orphan drug designation for certain of our future product candidates. Even if we obtain orphan drug designation for a product candidate, we may not be the first to obtain marketing approval for the product candidate for any particular orphan indication due to the uncertainties associated with developing novel therapies. Further, even if we obtain orphan drug exclusivity for a product candidate, that exclusivity may not effectively protect the product from competition
because orphan drug exclusivity does not prevent different drugs from being approved for the same condition. Moreover, orphan drug exclusivity may not prevent the approval of another sponsor’s product that is considered to be the same drug for a different disease or condition, even where such product could be used off-label for the indication that is protected by orphan drug exclusivity. Even after an orphan drug is approved, regulators may subsequently approve the same drug made by another manufacturer for the same condition if the regulator concludes that the later drug is safer, more effective, or makes a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a drug or biologic nor gives the drug or biologic any advantage in the regulatory review or approval process.
Foreign Regulation
We conduct clinical trials and market our products in numerous jurisdictions outside the U.S. Most of these jurisdictions have clinical trial, product approval and post-approval regulatory processes that are similar in principle to those in the U.S. Thus, whether or not we obtain FDA approval for a drugproduct candidate, we must obtain approval by the comparable regulatory authorities of foreign countries or economic areas, such as the E.U., before we can commence clinical trials or market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Under the E.U. regulatory systems,system, a company may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for orphan medicines, medicines produced by biotechnology, orand those medicines intended to treat AIDS, cancer, neurodegenerative disorders, or diabetes, and optional for those medicines that are highly innovative, provides for the grant of a single marketing authorization that is valid for all E.U. member states. In addition to the centralized procedure, Europethe E.U. also has a nationalized procedure, which requires a separate application to and approval determination by each country; a decentralized procedure, whereby applicants submit identical applications to several countries and receive simultaneous approval; and a mutual recognition procedure, where applicants submit an application to one country for review and other countries may accept or reject the initial decision.
Additionally, our European headquarters and European research facility are located in the U.K. Despite the U.K. formally withdrawing from the E.U. on January 31, 2020, a number of E.U. regulations were retained in U.K. law. The U.K. government has communicated an intent to remove or replace some of these E.U. provisions, which may increase some regulatory divergence between the U.K. and the E.U. Given the uncertainty as to what changes may be incorporated into U.K. law, it is unclear if any such changes could adversely affect our business, financial condition, and operating results.
Other Regulations
Pharmaceutical companies are also subject to various laws pertaining to healthcare “fraud and abuse,” including anti-kickbackthe federal Anti-Kickback Statute (“AKS”), the False Claims Act (“FCA”), and false claims laws. Anti-kickbackother state and federal laws and regulations. In the U.S., the Anti-Kickback Statute generally makemakes it illegal to knowingly and willfully solicit, offer, receive or pay any remuneration in return for or to induce the referral of business, including the purchase or prescription of a particular drug that is reimbursed by a state or federal health care program. False claims laws prohibitThe FCA prohibits knowingly and willingly presenting, or causing to be presented for payment to third-party payors (including Medicare and Medicaid), any claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed or claims for medically unnecessary items or services. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, as well as by the possibility of exclusion from federal healthcare programs (including Medicare and Medicaid). Liability under the false claims lawsFCA may also arise when a violation of certain laws or regulations related to the underlying products (e.g., violations regarding improper promotional activity, manufacturing regulations, or unlawful
payments) contributes to the submission of a false claim. If we were subject to allegations concerning, or convicted of violating, these laws, our business could be harmed.
Laws and regulations also have been enacted by the federal government and various states to regulate the sales and marketing practices of pharmaceutical manufacturers. The laws and regulations generally limit financial interactions between manufacturers and health care providers, require manufacturers to adopt certain compliance standards or require disclosure to the government and public of such interactions. The laws include U.S. federal and state “sunshine” provisions. The federal sunshine provisions apply to pharmaceutical manufacturers with products reimbursed under certain government programs and require those manufacturers to disclose annually to the federal government (for re-disclosure to the public) certain payments and other transfers of value made to physicians, physicians assistants, advanced practice registered nurses, and teaching hospitals and, beginning with disclosures in 2022, to certain non-physician practitioners.hospitals. State laws may also require disclosure of pharmaceutical pricing information and marketing expenditures.
Many of these laws and regulations contain requirements that are subject to interpretation. Outside the U.S., other countries have implemented requirements for disclosure of financial interactions with healthcare providers and additional countries may consider or implement such laws.
We are subject to various federal and foreign laws that govern our international business practices with respect to payments to government officials. Those laws include the U.S. Foreign Corrupt Practices Act or FCPA,(“FCPA”), which prohibits U.S. companies and their representatives from paying, offering to pay, promising, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate for the purpose of obtaining or retaining business or to otherwise obtain favorable treatment or influence a person working in an official capacity. In many countries, the health care professionals we regularly interact with may meet the FCPA’s definition of a foreign government official. We are also subject to U.K. Bribery Act 2010 or (“the Bribery Act,Act”), which proscribes giving and receiving bribes in the public and private sectors, bribing a foreign public official, and failing to have adequate procedures to prevent employees and other agents from giving bribes. U.S. companies that conduct business in the United Kingdom, or U.K., generally will be subject to the Bribery Act.
We are subject to federal laws, including the Medicaid Drug Rebate Program, the 340 program, and the FSS pricing program, that require pharmaceutical manufacturers to report certain calculated product prices to the government or provide certain discounts or rebates to government authorities or private entities, often as a condition of reimbursement under government healthcare programs.
Our collection and use of personal data as part of our business activities is subject to various privacy and data security laws and regulations, including oversight by various regulatory or other governmental bodies, in the U.S., E.U., U.K., Canada, Australia, Brazil and other jurisdictions. Such laws and regulations have the potential to affect our business materially, continue to evolve and increasingly are being enforced.
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations, and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import, export and use and disposal of hazardous or potentially hazardous substances are or may be applicable to our activities. In addition, as we expand our pipeline and contemplate different approaches that may incorporate the use of medical devices, such approaches may necessitate compliance with regulatory laws applicable to medical devices, including those governing the testing, manufacture, approval, distribution, and marketing of medical devices. Furthermore, the extent of government regulation, which might result from future legislation or administrative action, cannot accurately be predicted.
We have a global corporate compliance program designed to actively identify, prevent, and mitigate healthcare fraud and abuse risk through, among other things, the implementation of compliance policies and systems and through the promotion of a culture of compliance. We expect towill continue to devote substantial resources to maintain, administerenhance and expand theour corporate compliance program as necessary to help us manage and mitigate our evolving compliance risk environment as our business grows and expands globally. WeEven with these measures, however, we cannot be certain, however, that our compliance program will ensureguarantee compliance with the various complex laws and regulations to which we are subject now or in the future.
EMPLOYEES AND HUMAN CAPITAL MANAGEMENT
As of December 31, 2020,2023, we had approximately 3,4005,400 employees. Of these employees, approximately 2,8004,400 were based in the U.S. and approximately 6001,000 were based outside the U.S. OurNone of our U.S. employees are not covered by a collective bargaining agreement, except for aagreement. A small number of employees outside the U.S. are covered by such agreements due to local law or industry requirements. We consider our relations with our employees to be good. We continue to face intense competition for our personnel from our competitors and other companies throughout our industry and from universities and research institutions. Over the last several years, the challenges in recruiting and retaining employees across the biotechnology industry have increased substantially due to current industry job market dynamics.
We rely on skilled, experienced, and innovative employees to conduct the operations of our company. The biotechnology industry is very competitive, and recruiting and retaining such employees is important to the continued success of our business. We are committed to building an outstanding, committed, and passionate team, at Vertex, and we focus on a culture that values inclusion, diversity, and equity.equity for all employees. We believe that each employee brings unique perspectives and strengths, and by embracing these strengths, we can do our best work for patients. We focus on recruiting, retaining, and
developing employees from a diverse range of backgrounds to conduct our research, development, commercial, and commercialother business activities.
Our executive management team reflects our commitment to inclusion, diversity, inclusion and equity begins with our executive management team: fourequity: five of ourthe ten members are women and/or from diverse racial and ethnic groups.underrepresented communities. On our Board of Directors, sixfour of our teneleven members are women and/orand four members are from a diverse racial and ethnic group.underrepresented communities. As of December 31, 2020,2023, women represented 53%54.5% of our global workforce and 38%40.0% of our leadership (VP and above). As of December 31, 2020, 34%2023, 40.5% of our U.S. workforce, and 18%20.5% of our U.S. leadership (VP and above), were from diverseunderrepresented ethnic and racial and ethnic groups.
The leader of ourOur inclusion, diversity, inclusion, and equity strategy and efforts isare important to our culture. We view diversity through a Vice Presidentbroad lens that encompasses culture, backgrounds, experiences, and worldview to foster creativity and innovation. Our initiatives include learning, resources, and forums that promote inclusion, diversity, and equity in our human resources group. Additionally, ourworkplaces; efforts to develop a diverse pipeline of talent from early career through leadership; four global employee resource networks that promote connectivity and collaboration across levels and functions, and engage colleagues in personal and professional development opportunities, including mentoring, community outreach, and cultural awareness activities.activities; and investments to support efforts to address racism and social injustice in our surrounding communities.
To promote our employees’ continued well-being and development, we offer a variety of inclusive benefits and opportunities. We offer comprehensive work-life benefits, including health, dental, and income protection, such as life insurance and retirement savings programs. In 2020,programs and we enhanced and expanded ourthose employee benefits in response to the COVID-19 pandemic.benefits. For example, we increased company-wide personal time off, provided resourcescontinued to enable employees to work from home,promote and introduced and expandedexpand mental wellness tools, and enhanced child/elder care benefits for all employees. We continually review and augment our programs to include benefits such as expanded parental bonding, increased support for family planning, and gender affirming benefits. Our management has continuedcontinues to assess and respond to the evolving needs of our workforce throughout the pandemic.workforce.
In addition, we provide our employees with career development and advancement opportunities, including job rotations, mentoring, and managerial training. We also are committed to identifying and developing our next generation of leaders and have developed programs focused on manager excellence, talent and succession for critical roles in our organization.
Succession Planning
In 2020, we successfully executed a leadership succession plan with the transition of Dr. Kewalramani to the role of Chief Executive Officer and our former Chief Executive Officer, Dr. Leiden, to the role of Executive Chairman. This transition was the culmination of a multi-year planning process led by our independent directors.
OTHER MATTERS
Financial Information and Significant Customers
The Company operatesWe operate in one segment, pharmaceuticals. Financial information about our revenue by product and significant customers is set forth in Note Q, “Segment Information,” to our consolidated financial statements included in this Annual Report on Form 10-K.
Information Available on the Internet
Our internet address is www.vrtx.com. Our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K, and all amendments to those reports, are available to you free of charge through the “Investors-SEC“Investors/Financial Information/SEC Filings” section of our website as soon as reasonably practicable after those materials have been electronically filed with, or furnished to, the Securities and Exchange Commission.
Corporate Information
Vertex was incorporated in Massachusetts in 1989, and our principal executive offices are located at 50 Northern Avenue Boston, Massachusetts 02210.
INFORMATION ABOUT OUR EXECUTIVE OFFICERS
The names, ages and positions held by our executive officers are as follows:
| | | | | | | | | | | |
| Name | Age | Position |
| Reshma Kewalramani, M.D. | 4851 | Chief Executive Officer and President |
| Jeffrey M. Leiden, M.D., Ph.D. | 6568 | Executive Chairman |
| David Altshuler, M.D., Ph.D. | 5659 | Executive Vice President, Global Research and Chief Scientific Officer |
| Stuart A. Arbuckle | 5558 | Executive Vice President and Chief CommercialOperating Officer |
| E. Morrow Atkinson, III, Ph.D. | 58 | Executive Vice President, Chief Technical Operations Officer, Head of Biopharmaceutical Science and Manufacturing Operations |
| Jonathan Biller, J.D. | 60 | Executive Vice President, Chief Legal Officer |
| Carmen Bozic, M.D. | 5861 | Executive Vice President, Global Medicines Development and Medical Affairs, and Chief Medical Officer |
Michael Parini, J.D. | 46 | Executive Vice President, Chief Administrative, Legal and Business Development Officer |
| Amit K. Sachdev, J.D. | 5356 | Executive Vice President, Chief Patient Officer |
Bastiano Sanna, Ph.D. | 46 | Executive Vice President, Chief of Cell and Genetic TherapiesExternal Affairs Officer |
| Ourania “Nia” Tatsis, Ph.D. | 5154 | Executive Vice President and Chief Regulatory and Quality Officer |
| Charles F. Wagner, Jr. | 5255 | Executive Vice President and Chief Financial Officer |
Paul M. SilvaKristen C. Ambrose, CPA | 5447 | Senior Vice President and Chief Accounting Officer |
Dr. Kewalramani has been our Chief Executive Officer and President since April 2020 and a member of our Board of Directors since February 2020. Dr. Kewalramani was our Executive Vice President and Chief Medical Officer from April 2018 through April 2020. She was our Senior Vice President, Late Development from February 2017 until April 2018. From August 2004 to January 2017, she served in roles of increasing responsibility at Amgen Inc., most recently as Vice President Global Clinical Development, Nephrology & Metabolic Therapeutic Area and as Vice President,Head of U.S. Medical Organization. From 2014 through 2019, Dr. Kewalramani was the industry representative to the FDA’s Endocrine and Metabolic Drug Advisory Committee. Dr. Kewalramani also has served on the board of Ginkgo Bioworks since September 2021. She completed her internship and residency in Internal Medicine at the Massachusetts General Hospital and her fellowship in Nephrology at the Massachusetts General Hospital and Brigham and Women’s Hospital combined program. Dr. Kewalramani holds a B.A. from Boston University and an M.D. from Boston University School of Medicine. Dr. Kewalramani also completed the General Management Program at Harvard Business School and is an alumnusalumna of the school.
Dr. Leiden becameis our Executive Chairman, a position he has held since in April 2020. He was our Chief Executive Officer and President from 2012 through March 2020. He has been a member of our Board of Directors since July 2009, the Chairman of our Board of Directors since May 2012, and served as our lead independent director from October 2010 through December 2011. Dr. Leiden was a Managing Director at Clarus Ventures, a life sciences venture capital firm, from 2006 through January 2012. Dr. Leiden was President and Chief Operating Officer of Abbott Laboratories, Pharmaceuticals Products Group, and a member of the Board of Directors of Abbott Laboratories from 2001 to 2006. From 1987 to 2000, Dr. Leiden held several academic appointments, including the Rawson Professor of Medicine and Pathology and Chief of Cardiology and Director of the Cardiovascular Research Institute at the University of Chicago, the Elkan R. Blout Professor of Biological Sciences at the Harvard School of Public Health, and Professor of Medicine at Harvard Medical School. He is an elected member of both the American Academy of Arts and Sciences and the Institute of Medicine of the National Academy of Sciences. Dr. Leiden serves asis a directormember of the Board of Directors of the Massachusetts Mutual Life Insurance Company, ana private insurance company.company, since 2015. Dr. Leiden is the Executive Chairman of the Board of Directors of Odyssey Therapeutics, a private biotechnology company, since 2022, and is the Chairman of the Board of Directors of Casana, a private healthcare technology company, since 2021. Dr. Leiden was a director and the non-executive Vice Chairman of the board of Shire plc, a specialty biopharmaceutical company, from 2006 to January 2012, and a director of Quest Diagnostics, a publicly traded medical diagnostics company, from December 2014 to May 2019.2019, and the Chairman of Revolution Healthcare Acquisition Corp., a special purpose acquisition corporation, from April 2021 to December 2022. Dr. Leiden received his M.D., Ph.D. and B.A. degrees from the University of Chicago.
Dr. Altshuler has been our Executive Vice President, Global Research and Chief Scientific Officer since January 2015 and was a member of our Board of Directors from May 2012 through December 2014. Dr. Altshuler was one of four founding members of the Broad Institute, a research collaboration of Harvard University and the Massachusetts Institute of Technology, The Whitehead Institute and the Harvard Hospitals. He served as the Director of the Institute’s Program in
Medical and Population Genetics from 2003 through December 2014 and as the Institute’s Deputy Director and Chief Academic Officer from 2009 through December 2014. Dr. Altshuler joined the faculty at Harvard Medical School and the Massachusetts General Hospital in 2000 and held the academic rank of Professor of Genetics and Medicine from 2008 through December 2014. He served as Adjunct Professor of Biology at MIT from 2012 through December 2014. Dr. Altshuler earned a B.S. from MIT, a Ph.D. from Harvard University and an M.D. from Harvard Medical School. Dr. Altshuler completed his clinical training in Internal Medicine, and in Endocrinology, Diabetes and Metabolism, at the Massachusetts General Hospital.
Mr. Arbuckle is our Executive Vice President, Chief CommercialOperating Officer, a position he has held since July 2021. Previously, Mr. Arbuckle served as Executive Vice President, Chief Commercial and Operations Officer from March 2021 to July 2021, and as our Executive Vice President, Chief Commercial Officer from September 2012.2012 to February 2021. Prior to joining us, Mr. Arbuckle held multiple commercial leadership roles at Amgen, Inc. from July 2004 through August 2012. Mr. Arbuckle has worked in the biopharmaceuticals industry since 1986, including more than 15 years at GlaxoSmithKline plc, where he held sales and marketing roles of increasing responsibility for medicines aimed at treating respiratory, metabolic, musculoskeletal, cardiovascular and other diseases. He served as a member of the Board of Directors of Cerulean Pharma, Inc. from June 2015 through July 2017 and has served as a member of the Board of Directors of ImmunoGen, Inc. since January 2018 and of Rhythm Pharmaceuticals Inc. since July 2019. Mr. Arbuckle holds a BSc in pharmacologyPharmacology and physiologyPhysiology from the University of Leeds.
Dr. Atkinson has been our Executive Vice President, Chief Technical Operations Officer, Head of Biopharmaceutical Sciences and Manufacturing Operations since August 2023. He previously served as our Senior Vice President, Head of Commercial Manufacturing and Supply Chain since July 2020. Prior to joining us, Dr. Atkinson served in various roles at Bristol-Myers Squibb Co., including as Senior Vice President, Global Manufacturing Operations from September 2019 to June 2020; Vice President and Integration Leader, Corporate Cell Therapy and Global Development and Manufacturing from January 2019 to September 2019; Vice President, Internal Manufacturing, Biologics from June 2017 to January 2019; and Vice President, Biologics Development and Clinical Manufacturing from 2012 to June 2017. Before Bristol-Myers Squibb, he held various roles at Cook Pharmica, LLC (now owned by Catalent, Inc.) and Eli Lilly & Co. Dr. Atkinson has served as a member of the Board of Directors of 89bio, Inc., a public biopharmaceutical company, since February 2022. Dr. Atkinson holds a B.S. in Biology from Indiana University and a Ph.D. in Biological Sciences from Stanford University.
Mr. Biller has been our Executive Vice President, Chief Legal Officer since September 2022. From November 2019 until he joined us, Mr. Biller served in several executive roles at Agios Pharmaceuticals, Inc., including Chief Legal Officer and, most recently, Chief Financial Officer and Head of Corporate Affairs. Prior to Agios, he served as Executive Vice President, General Counsel at Celgene from July 2018 to November 2019, where he was responsible for their global legal function, and served as Senior Vice President, Tax and Treasury from 2011 to June 2018. Prior to Celgene, Mr. Biller was General Counsel, Chief Tax Officer and Secretary at Bunge Limited, a global publicly traded agriculture and food company. Earlier in his career he held various leadership roles at Alcon, Inc. and was a partner at Hopkins & Sutter and Foley & Lardner. Mr. Biller holds a B.A. from Brown University and a J.D. from Yale Law School.
Dr. Bozic is our Executive Vice President, Global Medicines Development and Medical Affairs, a position she has held since October 2019, and she has been our Chief Medical Officer since April 2020. She was our Senior Vice President and Head of Global Clinical Development from May 2019 to October 2019. Prior to joining Vertex,us, Dr. Bozic spent more than 20 years at Biogen Inc., a biotechnology company focused on neurological diseases, most recently as Senior Vice President of Global Development and Portfolio Transformation from 2015 to May 2019 and as Senior Vice President of Clinical and Safety Sciences from 2013 to 2015. Dr. Bozic has served as the industry representative to the FDA’s Risk Communication Advisory Committee, and was a member of PhRMA’s Clinical and Preclinical Development Committee and the Board of Managers at BioMotiv. She is a member of the Clinical Advisory Board at Akili Interactive. She received her M.D., C.M., completed her residency, and was Chief Resident in Internal Medicine at McGill University. She completed her fellowship in Pulmonary and Critical Care Medicine at Brigham and Women’s Hospital, and was an Associate Physician at Beth Israel Deaconess Medical Center and Harvard Medical School before joining the biopharmaceutical industry.
Mr. Parini is our Executive Vice President, Chief Administrative, Legal and Business Development Officer, a role he has held since March 2020. From January 2017 to March 2020, he was our Executive Vice President, Chief Legal and Administrative Officer. From January 2016 to January 2017, he was our Executive Vice President and Chief Legal Officer. From 2004 until he joined Vertex, Mr. Parini served in various roles of increasing responsibility at Pfizer Inc., a pharmaceutical company, most recently as Senior Vice President and Associate General Counsel. Prior to Pfizer, Mr. Parini was an attorney at Akin, Gump, Strauss, Hauer & Feld, L.L.P. Mr. Parini holds a B.A. from Georgetown University and a J.D. from the Georgetown University Law Center.
Mr. Sachdev is our Executive Vice President, Chief Patient and External Affairs Officer, a role he has held since July 2023. From October 2019.2019 to July 2023, he was our Executive Vice President, Chief Patient Officer. In addition, Mr. Sachdev has served in the role of Chief of Staff to the CEO since April 2020. He served as our Executive Vice President and Chief Regulatory Officer from January 2017 until September 2019, and as our Executive Vice President, Policy, Access and Value from October 2014 through December 2016. In 2010, he established our first international commercial operations in Canada.
In 2007, he joined us as a Senior Vice President, and has ledto establish our government affairs and public policy activities, as well as our patient advocacy programs. Prior to joining us, Mr. Sachdev served as Executive Vice President, Health, of the Biotechnology Industry Organization (BIO) and was the Deputy Commissioner for Policy at the FDA, where he also served in several other senior positions. Prior to the FDA, Mr. Sachdev served as Majority Counsel to the Committee on Energy and Commerce in the United StatesU.S. House of Representatives and practiced law at the Chemical Manufacturers Association,American Chemistry Council, and subsequently at the law firm of Ropes & Gray LLP. He has served as a member of the Board of Directors of Eiger BioPharmaceuticals since MayApril 2019. Mr. Sachdev holds a B.S from Carnegie Mellon University and a J.D. from Emory University School of Law.
Dr. Sanna has been our Executive Vice President, Chief of Cell and Genetic Therapies since February 2020. From October 2019 to February 2020, he was President of Semma Therapeutics, Inc., a private biotechnology company that Vertex acquired in October 2019. Prior to the acquisition, Dr. Sanna was the Chief Executive Officer and President of Semma from May 2018 until October 2019. Dr. Sanna was Chief Operating Officer at Magenta Therapeutics from May 2016 through April 2018. He served on the leadership team of the Novartis Cell and Gene Therapy Unit as the Global Program Head of Stem Cell Transplant and early programs from 2014 through 2016. Dr. Sanna served as Global Head of Strategic Planning and Portfolio Management at the Novartis Institutes for BioMedical Research from 2010 through 2014. Dr. Sanna has served as a member of the Board of Directors of Adicet Bio, Inc., a biotechnology company since December 2020. Dr. Sanna received a Ph.D. in Biotechnology from the University of Sassari.
Dr. Tatsis has beenis our Executive Vice President, Chief Regulatory and Quality Officer, a position she has held since August 2020. ShePreviously, she was our Senior Vice President and Chief Regulatory Officer from October 2019 to August 2020. She served as2020, and our Senior Vice President, Global Regulatory Affairs from September 2017 to October 2019. Prior to joining Vertex,us, Dr. Tatsis held positions of increasing responsibility at several pharmaceutical companies, including Sanofi, Stemnion, Pfizer, and Wyeth. Most
recently, from 2014 to 2017, she was Vice President, Head of Global Regulatory Affairs, at the Sanofi Genzyme Business Unit focused on Inflammation/Immunology, Rare Disease, Multiple Sclerosis, Ophthalmology, Neurology, and Oncology/Immuno-Oncology. Dr. Tatsis also worked as an associate staff scientist and research fellow in Immunology and Vaccine Development at the Wistar Institute and completed a post-doctoral research fellowship in Immunology at Thomas Jefferson University. She received her Ph.D. in Cell and Molecular Biology from the University of Vermont and holds a B.S. in Biology from Temple University.
Mr. Wagner has beenis our Executive Vice President and Chief Financial Officer, a position he has held since April 2019. Prior to joining Vertex,us, Mr. Wagner was Chief Financial Officer and Executive Vice President, Finance, of Ortho Clinical Diagnostics, a Carlyle Group portfolio company, from June 2015 to March 2019. In that role, he led the finance, accounting, tax, treasury, global informationfinancial systems, lender relations, and acquisitions and divestiture groups, as well asand also had shared leadership overfor several enterprise-wide projects. From July 2012 to June 2015, Mr. Wagner served as Executive Vice President, Chief Financial Officer of Bruker Corporation, a scientific instruments manufacturer. Prior to that, Mr. Wagner served as Chief Financial Officer for Progress Software Corporation, a provider of enterprise software, and Millipore Corporation, a global provider of products and services in the life science tools market. Mr. Wagner served as a director and chairman of the Audit Committee of Good Start Genetics, Inc., a molecular diagnostics company, from April 2014 to August 2017 and served as a director and member of the Audit Committee of Bruker Corporation from August 2010 to June 2012. He has served as a member of the Board of Directors of The TJX Companies, Inc., a public department store company, since September 2023. Mr. Wagner holds a B.S. in accountingAccounting from Boston College and a M.B.A from Harvard Business School.
Mr. SilvaMs. Ambrose is our Senior Vice President, Chief Accounting Officer, a position heshe has held since April 2011. Mr. Silva alsoMay 2021. Ms. Ambrose previously served as our interim Chief Financial OfficerSenior Vice President, Accounting, Tax, Treasury, Strategic Sourcing and Corporate Services since March 2021. From February 2003 until she joined us, Ms. Ambrose held roles of increasing responsibility at Boston Scientific Corporation, a medical device company, most recently as Vice President of Finance and Controller of the Global Endoscopy Division from JanuaryJuly 2019 to April 2019. Mr. Silva joined us in August 2007March 2021 and as Senior Director, Accounting Operations and was our Vice President and Corporate Controllerof Global Internal Audit from September 2008 through April 2011.February 2017 to June 2019. Prior to joining us, he was the Vice President, Internal ReportingBoston Scientific Corporation, Ms. Ambrose served as an accountant at Iron Mountain Incorporated from July 2006 until August 2007 and a consultant to Iron Mountain’s finance department from April 2005 until July 2006. He was the Finance Director of the Bioscience Technologies Division of Thermo Electron Corporation from 2002 to April 2005. Mr. Silva holds aErnst & Young LLP. She received her B.S. in accountingCommerce from Assumption College.
ITEM 1A. RISK FACTORS
Investing in our common stock involves a high degree of risk, and you should carefully consider the risks and uncertainties described below in addition to the other information included or incorporated by reference in this Annual Report on Form 10-K. If any of the following risks or uncertainties actually occurs, our business, financial condition or results of operations would likely suffer, possibly materially. In that case, the trading price of our common stock could decline.
SUMMARY OF RISK FACTORS
Our business is subject to numerous risks and uncertainties, discussed in more detail in the following section. These risks include, among others, the following key risks:
Risks Related to Our Business
•AllWe invest significant resources in the research, development, manufacturing and supply of therapies for serious diseases, and if we are unable to successfully develop and commercialize additional products, our business could be materially harmed.
•Over the last several years all of our product revenues and the vast majority of our total revenues arewere derived from sales of medicines for the treatment of CF.our CF medicines. If we are unable to continue to increase revenues from sales of our CF medicines, our business would be materially harmed and the market price of our common stock would likely decline.
•WeIf we are investing significant resourcesnot successful in the researchcommercializing CASGEVY, our revenue growth could be limited and development of therapies for serious diseases other than CF, and ifour business could be materially harmed.
•If we are unable to successfully develop, obtain approval, and commercialize one or more of these therapies,treatments for acute and neuropathic pain, our business could be materially harmed.
•If our competitors bring drugsproducts with superior product profiles to market, our drugsproducts may not be competitive, and our revenues could decline.
•If we discover safety issues with any of our products or if we fail to comply with continuing U.S. and applicable foreign regulations, commercialization efforts for the product could be negatively affected, the approved product could lose its approval or sales could be suspended, and our business could be materially harmed.
•If physicians and patients do not accept our medicines,products, or if patients do not remain on treatment or comply with their prescribed dosing regimen, our product revenues would be materially harmed in future periods.
•Cell and genetic therapies face increased scrutiny from the public and medical communities and commercial success will depend, in part, upon the acceptance of those communities.
Risks Related to Pricing of Our Products
•Government and other third-party payors seek to contain costs of health care through legislative and other means. If they fail to provide coverage and adequate reimbursement rates for our products, our revenues will be harmed.
•We may experience pricing pressure on our products, which could reduce our revenues and future profitability.
•Current health care laws and regulations in the U.S. and future legislative or regulatory reforms to the U.S. health care system may affect our ability to commercialize our marketed products profitably.
•We have experienced challenges commercializing products outside of the U.S,U.S., and our future revenues will be dependent on our ability to obtain adequate reimbursement for our products.products in ex-U.S. markets.
•We have limited experience developing cellInsurance coverage and genetic therapies and could experience challenges with these programs, which could result in delays or prevent the development, manufacturing and commercializationreimbursement of our cell andor genetic therapies.therapies is uncertain.
Risks Related to Development and Clinical Testing of Our Products and DrugProduct Candidates
•Our drugproduct candidates remain subject to clinical testing and regulatory approval. Ourapproval, and our future success is dependent on our ability to successfully develop additional drugproduct candidates for both CF and non-CF indications.
•If we are unable to obtain or are delayed in obtaining regulatory approval, we willmay incur additional costs, experience delays in commercialization, or be unable to commercialize our drugproduct candidates.
•If clinical trials are prolonged or delayed, our development timelines for the affected development program could be extended, our costs to develop the drugproduct candidate could increase and the competitive position of the drugproduct candidate could be adversely affected.
•Difficulty in enrolling patients could delay or prevent clinical trials of our product candidates, and ultimately delay or prevent regulatory approval.
•Enrollment for clinical trials for our cell and gene therapies may face additional and unique challenges and adverse developments associated with these clinical trials could result in action by regulatory bodies, including revised requirements for approval.
Risks Related to Government Regulation
•If regulatory authorities interpret any of our conduct, including our marketing practices, as being in violation of applicable health care laws, including fraud and abuse laws, laws prohibiting off-label promotion, disclosure laws or other similar laws, we may be subject to civil or criminal penalties.
•If we fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate Program or other governmental pricing programs in the U.S., we could be subject to additional reimbursement requirements, penalties, sanctions, and fines whichthat could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
•If our processes and systems are not compliant with regulatory requirements, we could be subject to restrictions on marketing our products or could be delayed in submitting regulatory filings seeking approvals for our drugproduct candidates.
•We are subjectThe regulatory approval process for our cell and genetic therapies involves additional consultations with regulatory agencies, costs, and potentially longer timelines as compared to various and evolving laws and regulations governing the privacy and security of personal data, and our failure to comply could adversely affect our business, result in fines and/or criminal penalties, and damage our reputation.those for small molecules.
Risks Related to Business Development Activities
•Our ability to execute on our long-term strategy depends in part on our ability to engage in transactions and collaborations with other entities that add to our pipeline or provide us with new commercial opportunities.
•We may not realize the anticipated benefits of acquisitions of businesses or technologies, and the integration following any such acquisition may disrupt our business and management.
•We face risks in connection with existing and future collaborations with respect to the development, manufacture and commercialization of our products and drug candidates.
•We may not be able to attract collaborators or external funding for the development and commercialization of certain of our drug candidates.
Risks Related to Third-PartySupply, Manufacturing and Reliance on Third Parties
•We depend on third-party manufacturers and our internal capabilities to manufacture our products and the materials we require for our clinical trials. We rely on third party logistics providers to manage our shipments globally.We may not be able to maintain theseour third-party relationships and could experience supply disruptions outside of our control.
•We rely on third parties to conduct pre-clinical work, clinical trials and other activities, and those third parties may not perform satisfactorily, including failing to meet established deadlines for the completion of such studies and/or trials or failing to satisfy regulatory requirements.
Risks Related to Business Development Activities
•Our ability to execute on our long-term strategy depends in part on our ability to engage in transactions and collaborations with other entities that add to our pipeline or provide us with new commercial opportunities.
•We face risks in connection with existing and future collaborations with respect to the development, manufacture and commercialization of our products and product candidates.
•We may not realize the anticipated benefits of existing or future acquisitions of businesses or technologies, and the integration following any such acquisition may disrupt our business and management.
Risks Related to Intellectual Property
•If our patents do not protect our drugs orproducts and our drugsproducts infringe third-party patents, we could be subject to litigation which could result in injunctions preventing us from selling out products, substantial damages, or circumvention of our products or substantial liabilities.patents by third parties.
•Uncertainty over intellectual property in the pharmaceutical and biotechnology industry has been the source of litigation and other disputes which isthat are inherently costly and unpredictable.
•We may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Risks Related to Our Operations
•RisksIf we fail to scale our operations to accommodate growth, our business may suffer.
•A variety of risks associated with operating in foreign countries could materially adversely affect our business.
•We are subject to risks associated with the global COVID-19 pandemic.
•If we fail to attract and retain skilled employees, our business could be materially harmed.
•Our business faces potential risks relatingA breakdown or breach of our information technology systems could subject us to liability or interrupt the United Kingdom’s withdrawal from the European Union.operation of our business.
Risks Related to Financial Results and Holding Our Common Stock
•Our stock price may fluctuate.
•Changeseffective tax rate fluctuates, and changes in tax laws, regulations and treaties, unfavorable resolution to the tax positions we have taken or exposure to additional income tax liabilities could affecthave a material impact on our future taxable income.
Risks Related to Our Business
AllWe invest significant resources in the research, development, manufacturing and supply of therapies for serious diseases, and if we are unable to successfully develop and commercialize additional products, our business could be materially harmed.
We invest significant resources in the research and development of therapies for serious diseases, including CF, SCD, TDT, acute and neuropathic pain, AMKD, T1D, DM1, and AATD. Product development is highly uncertain and expensive, and we may experience unforeseen delays, including regulatory and commercialization delays. Product candidates that may appear promising in the early phases of research and development may fail to reach commercial success for many reasons, including the failure to demonstrate acceptable clinical trial results or obtain marketing approval, the inability to manufacture or commercialize the product candidate on economically feasible terms, or the appearance of safety issues.
When we receive marketing approval for a pipeline product, we cannot be sure that we will obtain market acceptance or adequate reimbursement levels from third-party payors or foreign governments for such product. Additionally, many of the therapies that we are developing in our pipeline target rare diseases that affect a limited number of patients. There can be no guarantee that we will effectively identify patients that are eligible for enrollment in our clinical trials or treatment with our product candidates. Even if we do successfully identify eligible patients, the number of patients that our product candidates are able to treat may turn out to be lower than we expect or new patients may become increasingly difficult to identify, each of which may adversely affect our revenues and materially harm our business. If we are not able to successfully develop and commercialize additional products our business could be materially harmed.
Over the last several years all of our product revenues and the vast majority of our total revenues arewere derived from sales of medicines for the treatment of CF.our CF medicines. If we are unable to continue to increase revenues from sales of our CF medicines, our business would be materially harmed and the market price of our common stock would likely decline.
OurSubstantially all of our net product revenues and the vast majority of our total revenues arehave been derived from the sale of our CF medicines.medicines over the last several years. As a result, our future successbusiness is dependent upon our ability to sustain and increase revenues from sales of our CF medicines. This will require usWe seek to continue to gainincrease our CF product revenue through serial innovation, including the potential approval and reimbursement forof our vanzacaftor/tezacaftor/deutivacaftor triple combination, therapydevelopment and commercialization CF medicines in ex-U.S. markets and successfully develop and commercialize our triple combination therapy for younger children with CF.CF and through securing additional approvals and reimbursements for our CF medicines in ex-U.S. markets.
Our concentrated source of revenues presents a number of risks to our business, including:
•that one or more competing therapies may successfully be developed successfully as a treatment for people with CF;
•that reimbursement policies of payors and other third parties may make it difficult to obtain reimbursement or reduce the net price we receive for our products;
•that we may experience manufacturing or supply disruptions for our CF medicines; and
•that we may experience adverse developments with respect to development or commercialization of our CF medicines and/or CF drug candidates.medicines.
If any of the above risks were to materialize, if we are otherwise unable to increase revenues from sales of our CF medicines, or if we do not meet the expectations of investors or public equity market analysts, our business would be materially harmed and our ability to fund our operations could be adversely affected. For example, if
If we are unable to increase revenues from sales ofnot successful in commercializing CASGEVY, our CF medicines,revenue growth could be limited and our ability to fund our research and development programsbusiness could be materially harmed.
We recently obtained approval for CASGEVY for the discoverytreatment of people 12 years and development or acquisition of new products would be harmed, which would limit our ability to diversify our revenue baseolder with SCD and our stock price would likely be adversely affected.
TDT in the U.S., the E.U., the U.K., Saudi Arabia, and Bahrain. We are investinginvested significant resources in the research and development of therapiesCASGEVY. While we have previously successfully commercialized several small molecule drugs, we have limited experience with the commercialization of cell and genetic therapies. Manufacturing and commercialization of CASGEVY is subject to similar risks and uncertainties as small molecules. In addition:
•the manufacturing process for serious diseasesCASGEVY is more complex than the manufacturing processes for our CF medicines and we may encounter difficulties in the production of CASGEVY and ensuring that the product meets required
specifications;
•there are multiple steps along the CASGEVY patient treatment journey, many of which involve significant clinical complexities performed by third parties, including the collection of blood cells from patients, transfer of those cells to and from a manufacturing facility, and other than CF,procedures either before or after delivery of CASGEVY;
•the commercial success of CASGEVYwill depend in part on the medical community, patients, governments, and ifthird-party or governmental payors accepting and providing adequate reimbursement of CASGEVY products, and recognizing the applicable medicine as medically useful, cost-effective, ethical, and safe; and
•market acceptance will be dependent in part on the prevalence and severity of side effects associated with the procedure by which CASGEVY is administered, the prevalence and severity of any side effects resulting from the myeloablative preconditioning regime.
If we are not successful in commercializing CASGEVY, our revenue growth could be limited and our business could be materially harmed.
If we are unable to successfully develop, obtain approval and commercialize one or more of these therapies,treatments for acute and neuropathic pain, our business could be materially harmed.
We are investing significant resources inbelieve that a portion of the research and development of medicines for serious diseases including AAT deficiency, APOL1-mediated kidney diseases, pain, beta thalassemia, SCD, T1D, DMD and DM1. Some of these programs have progressed into clinical trials, while others are still in pre-clinical development. Product developmentvalue attributed to our company by investors is highly uncertain and expensive, and product candidates that may appear promising in the early phases of research and development may fail to reach commercial success for many reasons, including the failure to demonstrate acceptable clinical trial results or obtain marketing approval, the inability to manufacture or commercialize the product candidate on economically feasible terms, or the appearance of safety issues. For example, in October 2020, we discontinued development of VX-814, a drug candidate for the treatment of AAT, based on our potential treatments for acute and neuropathic pain, including VX-548. We have completed the safetyPhase 3 development program for VX-548 in acute pain and pharmacokinetic profile observedwe are planning to submit an NDA to the FDA by mid-2024. We are planning to initiate a Phase 3 development program for VX-548 in aneuropathic pain based on positive Phase 2 clinical trial.results we received in the fourth quarter of 2023.
EvenObtaining approval for VX-548 is uncertain process and we may not be successful. If we do not obtain approval of VX-548, our business may be materially harmed. VX-548, if approved, may not gain or maintain market acceptance among physicians and patients or other members of the medical community. In addition to the risks normally associated with launching a new branded product, VX-548 will need to compete in an acute pain market that largely consists of low-cost generic drugs, including opioids, non-steroidal anti-inflammatory drugs, acetaminophen and local anesthetics. Similarly, if we gain marketingare successful in developing and obtaining approval for one or more pipeline products, we cannot be sure that weVX-548 in neuropathic pain, VX-548 will obtain market acceptance or adequate reimbursement levelsface competition from third-party payors or foreign governments for such products. Additionally, many of the therapies thatgeneric anticonvulsant and antidepressant drugs. If we are developing in our pipeline target rare diseases that affect a limited number of patients. There can be no guarantee that we will effectively identify patients that are eligible for enrollment in our clinical trials or treatment with our drug candidates. Even if we do successfully identify eligible patients, the number of patients that our drug candidates arenot able to treat may turn out to be lower than we expect or new patients may become increasingly difficult to identify, each of which may adversely affectsuccessfully develop, obtain approval for and commercialize treatments for acute and neuropathic pain, our future net product revenues and cash flows will be adversely affected and our business could be materially harm our business. For these and other reasons, we may never be successful in expanding our pipeline and future revenue may continue to depend on sales of our CF medicines.
If our competitors bring drugsproducts with superior product profiles to market, our drugsproducts may not be competitive, and our revenues could decline.
A number of companies are seeking to identify and develop drugproduct candidates for the treatment of CF, SCD, TDT, pain, and other therapeutic areas we are targeting with our research and development activities. Our success in rapidly developing and commercializing our CF medicines may increase the resources that our competitors allocate to the development of potential competitive treatments. If one or more competing therapies are successfully developed as a treatment for people with CF, SCD, TDT, pain or any of the other diseasesdisease areas we are currently targeting in our pipeline, our products and our net product revenues could face competitive pressures. If one or more competing therapies prove to be superior to our then existingthen-existing products and/or drugproduct candidates, our business could be materially adversely affected.
In addition, our business faces competition from major pharmaceutical companies possessing substantially greater financial resources than we possess. We also face competitionpossess, as well as from numerous smaller public and private companies, academic institutions, government agencies, public and private research organizations, and charitable venture philanthropy organizations that conduct research, seek patent protection, and/or establish collaborative arrangements for research, development, manufacturing, and commercialization.
Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies also may prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Our products and any drugsproducts that we develop in the future may not be able to compete effectively with marketed drugs
therapies or new drugstherapies that may be developed by competitors. The risk of competition is particularly important to our company because substantially all of our revenues as well as our most advanced drug candidates are related to the treatment of people with CF. There are many other companies developing drugsproducts for the same patient populations that we are pursuing. In order toTo compete successfully in these areas, we must demonstrate improved safety, efficacy and/or tolerability, ease of manufacturing, and gain and maintain market acceptance over competing drugs.products.
If we discover safety issues with any of our products or if we fail to comply with continuing U.S. and applicable foreign regulations, commercialization efforts for the product could be negatively affected, the approved product could lose its approval or sales could be suspended, and our business could be materially harmed.
Our products are subject to continuing regulatory oversight, including the review of additional safety information. DrugsProducts are more widely used by patients once approval has been obtained and therefore side effects and other problems may be observed after approval that were not seen or anticipated, or were not as prevalent or severe, during pre-approval clinical trials or nonclinical studies. The subsequent discovery of previously unknown or underestimated problems with a product could negatively affect commercial sales of the product, result in restrictions on the product or lead to the withdrawal of the product from the market. ThreeEach of our commercial products are combination products, and each of ourCF products shares at least one active pharmaceutical ingredient with another of our products. As a result, if any of our CF products were to experience safety issues, our other CF products may be adversely affected. In SCD and TDT, as part of the FDA approval for CASGEVY, we are required to conduct two post-marketing requirement safety studies to assess the long-term risk of hematologic malignancies and off-target genome editing effects by CRISPR/Cas9. Negative or ambiguous results from these studies could also have a significant impact on our ability to commercialize CASGEVY. The reporting of adverse safety events involving our products or public speculation about such events could cause our stock price to decline or experience periods of volatility. Our business also may be materially harmed by impaired sales of our products, denial or withdrawal of regulatory approvals, non-renewal of conditional regulatory approvals, required label changes or additional clinical trials, reputational harm, or government investigations or lawsuits brought against us.
In addition, ourOur products are subject to ongoing regulatory requirements governing the testing, manufacturing, labeling, packaging, storage, advertising, promotion, sale, distribution, import, export, recordkeeping, and submission of safety and other post-market information. We and our third-party manufacturers must comply with cGMP and other applicable regulations governing the manufacturing and distribution of our products. Regulatory authorities periodically inspect our drug manufacturing facilities, and those of our third-party manufacturers, to evaluate compliance with cGMP and other regulatory requirements.
If we or our collaborators, or third-parties acting on our behalf, fail to comply with applicable continuing regulatory requirements, we or our collaborators may be subject to fines, suspension or withdrawal of regulatory approvals for specific
products, product recalls and seizures, operating restrictions and/or criminal prosecutions, any of which could have a material adverse effect on our business, reputation, financial condition, and results of operations.
If physicians and patients do not accept our medicines,products, or if patients do not remain on treatment or comply with their prescribed dosing regimen, our product revenues would be materially harmed in future periods.
Our medicinesapproved products may not gain or maintain market acceptance among physicians and patients.patients or other members of the medical community. Effectively marketing our drugsproducts and any of our drugproduct candidates or investigational therapies, if approved, requires substantial efforts, both prior to launch and after approval. Physicians may elect not to prescribe our drugsproducts or recommend our cell or genetic therapies, and patients may elect not to take them or receive them or they may discontinue use of our drugsproducts after initiation of treatment, for a variety of reasons including:
•prevalence and severity of adverse side effects;
•lack of reimbursement availability from third-party payors, including governmental entities;
•lower demonstrated efficacy, safety and/or tolerability compared to alternative treatment methods;
•lack of cost-effectiveness;
•a decision to wait for the approval of other therapies in development that have significant perceived advantages over our drug;product;
•convenience and ease of administration;
•limitations or warnings contained in the labeling;
•the timing of market introduction of our product as well as competitive products;
•other potential advantages of alternative treatment methods; and
•inadequate sales, marketing and/or distribution support, including as a result of limitations or restrictions resulting from COVID-19.support.
If our medicines fail to achieve or maintain market acceptance, we may not be able to generate significant revenues in future periods.
Cell and genetic therapies face increased scrutiny from the public and medical communities and commercial success will depend, in part, upon the acceptance of those communities.
We face uncertainty as to whether cell and gene therapy treatments will gain the acceptance of the public or the medical community. The commercial success of cell and gene therapy treatments, including CASGEVY, will depend, in part, on the acceptance of physicians, patients, and third-party payors of gene therapy products in general, and our product candidates in particular, as medically necessary, cost-effective and safe. In particular, our success will depend upon physicians prescribing our therapies in lieu of existing treatments they are already familiar with and for which greater clinical data may be available. Moreover, physicians and patients may delay acceptance of cell and gene therapies until the therapies have been on the market for a certain amount of time. In addition, medical centers, including ATCs, that administer procedures accompanying treatment could experience capacity constraints, and these centers are subject to competing priorities that could delay patient access to procedures associated with cell and gene therapy products. Negative public opinion or more restrictive government regulations may delay or impair the successful commercialization of, and demand for, cell and gene therapies.
Risks Related to Pricing of Our Products
Government and other third-party payors seek to contain costs of health care through legislative and other means. If they fail to provide coverage and adequate reimbursement rates for our products, our revenues will be harmed.
Sales of our products depend in part upon the availability of reimbursement from third-party payors. Third-party payors include government health programs such as Medicare and Medicaid in the U.S. and the national health care systems in ex-U.S. markets, managed care providers, private health insurers and other organizations. The trend in the health care industry is cost containment, and efforts of third-party payors to contain or reduce health care costs may adversely affect our ability to establish or maintain appropriate prices for our products or any drugs that we may develop and commercialize.
In most ex-U.S. markets, the pricing and reimbursement of therapeutic and other pharmaceutical products is subject to governmental control, and government authorities are making greater efforts to limit or regulate the price of drug products. In the U.S., there have been, and we expect that there will continue to be, a number of federal and state proposals to implement governmental controls that are similar to those that currently exist in Europe. For example, the ACA required manufacturers of Medicare Part D brand name drugs to provide discounts on those drugs to Medicare Part D beneficiaries during the coverage gap; increased the rebates paid by pharmaceutical companies to state Medicaid programs on drugs covered by Medicaid; and imposed an annual fee, which increases annually, on sales by branded pharmaceutical manufacturers.
There also has been an increase Additionally, private payors, including health maintenance organizations and pharmacy benefit managers in legislation and regulations related to drug pricing and drug pricing transparency. In the U.S., various states, including Nevada, Maryland, Louisiana, New York, California, Washington, Massachusetts, Connecticut, Utah, Minnesotaare adopting more aggressive utilization management techniques and Oregon, have passed legislation requiring companies to disclose extensive information relating to drug prices, drug price increases,are increasingly applying restrictive plan designs that can impact patients and spending on research, development,manufacturers, and marketing. Although it is not clear what states will do with the collected information, some laws were designed to obtain additional product discounts. We maythey continue to see more state action requiring additional disclosurespush for significant discounts and rebates from manufacturers.
Additionally, on August 16, 2022, the IRA was enacted. Among other things, the IRA establishes a Drug Price Negotiation Program, under which the government may negotiate maximum fair prices for certain drugs covered by Medicare that do not have generic or biosimilar competition. The first set of maximum fair prices will be effective in 2026. Certain products are excluded from the negotiation program including drugs that have a single orphan drug designation and that are not approved for any other actions. In addition, we could see increased federal activity relatedorphan or non-orphan diseases or conditions. We cannot predict with certainty whether there will be future legislative changes to the scope of these exclusions. The law also requires manufacturers to pay a rebate to Medicare if the price of a Medicare drug pricing(under both Part B and transparency requiring disclosures or other actions insteadPart D) increases faster than the rate of orinflation. The law also redesigns the Part D benefit. The current Coverage Gap Discount Program, which requires manufacturers to provide a 70% discount on brand drugs and biologics during the coverage gap phase, will be eliminated after the 2024 plan year. Starting in addition2025, manufacturers of brand drugs and biologics will be required to state requirements. Similar initiatives are also occurring in, or being considered by, some ofprovide a 10% discount during the ex-U.S. markets, including Italy and Brazil.initial
Complyingphase and a 20% discount during the catastrophic phase of the Part D benefit. The IRA continues a trend in the U.S. toward reducing drug prices and limiting spending by the federal health care programs on drugs. We cannot predict how CMS will interpret the IRA or how the provisions of the law will affect our business once fully implemented, but it is possible that these changes or other legislative updates will have an adverse impact on our revenue. The IRA also requires the Secretary of the Department of Health and Human Services (the “Secretary”) to issue program guidance on numerous areas associated with these lawsimplementation of the law’s requirements, including for drug price negotiation and inflation rebates. We cannot know what form this program guidance would take or how it would affect our business.
It is expensivepossible the U.S. Congress or administration may take further actions to address health care costs and requires significant personnelaccess to medicine, and operational resourcesspecifically address coverage and detersreimbursement of cell and gene therapies. For example, in October 2022, President Biden issued an Executive Order directing the Center for Medicare and Medicaid Innovation (“CMMI”), to consider new healthcare payment and delivery models that would lower drug costs and promote access to innovative drug therapies for Medicare and Medicaid beneficiaries. In February 2023, the Secretary submitted a report to the White House describing three models that the Secretary selected for testing. Among the selected models is a Cell & Gene Therapy Access Model, under which CMS would structure and coordinate multi-state Medicaid outcomes-based agreements between participating states and manufacturers. The report also directs CMS to consider potential Medicare fee-for-service options to support cell and gene therapy access and affordability. In October 2023, CMMI further announced that it will move the start-date for the Cell & Gene Therapy Access Model from 2026 to 2025. On January 30, 2024,CMMI released additional information about the Cell & Gene Therapy Access Model, including the initial focus on our business. Additionally, any additionalcell and gene therapies for sickle cell disease. CMS intends to negotiate outcomes-based agreements with manufacturers between May 2024 and November 2024. In addition to the supplemental rebate negotiated under the outcomes-based agreement, participating manufacturers would be required discounts would adversely affectto cover certain fertility preservation services and supports for ancillary services (e.g., travel, case management, behavioral health services). CMS is requesting that states submit an optional, non-binding letter of intent by April 2024. States may begin participating in the pricing of,Cell & Gene Therapy Access Model on a rolling-basis, between January 2025 and revenues from, our products. Finally, while we seek to comply with all statutory and regulatory requirements, we face increased enforcement activity by the U.S. federal government, state governments, and private payors against pharmaceutical and biotechnology companies for pricing and reimbursement-related issues.
Recently, there also has been rulemaking related to importation of prescription drugs from Canada as well as guidance related to importation of prescription drugs from other foreign countries. HHS also has issued a regulation seeking to establish a model for reference pricing of certain physician-administered drugs. While the recent regulation does not apply to our current medicines, it could affect future medicines. Additionally, in 2020, the Trump Administration issued several executive orders relating to drug pricing which were intended to broadly impact the pharmaceutical industry. Likewise, HHS recently issued a final regulation adopting changes to anti-kickback laws for rebates offered to pharmacy benefit managers. We expect such government scrutiny over drug pricing, reimbursement, and distribution to increase. Potential future government regulation of drug prices or reimbursement creates uncertainties about our portfolio and could have a material adverse effect on our operations.January 2026.
Third-party payors throughout the world also have been attempting to control drug spending through various other actions, and this is expected to be an area of intensified focus for all payors in light of the global economic pressures, including due to the COVID-19 pandemic.pressures. In reimbursement negotiations, many payors are demandingrequesting price discounts and caps on total expenditures and limiting both the types and variety of drugs that they will cover if they are not able to secure them. Some payors restrict reimbursement to certain patient groups or by indication. As part of these negotiations, many ex-U.S. government payerspayors also are requiring companies to establish product cost-effectiveness as a condition of reimbursement and companies’ data-backed explanations are assessed by government agencies set up for this purpose.reimbursement. These cost-effectiveness reviews may not account foroverlook many of the benefits provided by innovative medicines, and for the most part, have not taken into account the specific circumstances of products that treat rare diseases. This has led to conclusions that certain medicines, including our products in certain jurisdictions, are not cost-effective. As a result, certain countries have declined to reimburse, or delayed their reimbursement of, some of our products. Although not mandated in the U.S., various organizations have started advocating for cost-effectiveness analyses in the U.S. as well as value-based contracting in which the amount of reimbursement for a product is based on patient outcomes and other clinical or economic metrics related to the performance of such product. If U.S. payors were to adopt such assessments and make negative coverage determinations or utilize value-based contracts that result in penalties to, or lower rates of, reimbursement, it could adversely affect our product revenues. Our business would be materially adversely affected if we are not able to obtain or maintain coverage and reimbursement of our products from third-party payors on a broad, timely, or satisfactory basis, or if such coverage is subject to overly broad or restrictive utilization management controls.
The U.S. government, individual states and some foreign jurisdictions also have been aggressively pursuing legislative and regulatory reforms that could affect our ability to sell products. For example, in the U.S., there have been ongoing federal legislative and administrative efforts to repeal, substantially modify or invalidate some or all of the provisions of the ACA. Various portions of the ACA are subject to legal challenges in various jurisdictions, including the U.S. Supreme Court, which could affect coverage and payment for medicines. Other reforms include the Bipartisan Budget Act of 2018, which contained various provisions that affect coverage and reimbursement of drugs, including an increase in the discount that manufacturers of Medicare Part D brand name drugs must provide to Medicare Part D beneficiaries during the coverage gap from 50% to 70%. There are a number of additional bills pending in Congress that would affect drug pricing in the Medicare and Medicaid programs. Additional healthcare reform efforts have sought to address issues related to the COVID-19 pandemic, including an expansion of telehealth coverage under Medicare and accelerated or advanced Medicare payments to healthcare providers. Adoption of new healthcare reform legislation at the federal or state level could affect demand for, or pricing of, our products or product candidates if approved for sale. We cannot, however, predict the ultimate content, timing or effect of any healthcare reform legislation or action, or its impact on us, including increased compliance requirements and costs, all of which may adversely affect our future business, operations and financial results.
The increasing availability and use of innovative specialty pharmaceuticals for rare diseases, combined with their relative higher cost as compared to other types of pharmaceutical products, is generating significant third-party payor interest in developing cost-containment strategies targeted to this sector. Government regulations in both U.S. and ex-U.S. markets could further limit the prices that can be charged for our products and may limit our commercial opportunity. The increasing use of cost-effectiveness assessments in markets around the world and the financial challenges faced by many governments may lead to significant adverse effects on our business.
We may experience pricing pressure on our products, which could reduce our revenues and future profitability.
There also has been an increase in state legislation and regulations related to drug pricing and drug pricing transparency. In the U.S., various states, including Nevada, Maryland, Louisiana, New York, California, Washington, Massachusetts, New Jersey, Connecticut, Vermont, New Hampshire, Utah, Minnesota, Oregon, Colorado, New Mexico, Virginia, Maine, Texas, North Dakota, West Virginia, Florida, and New Jersey have passed legislation requiring companies to disclose extensive
information relating to drug prices, drug price increases, and spending on research, development, and marketing, among other things. Although it is not always clear what states will do with the collected information, some laws were designed to obtain additional product discounts. Additionally, certain states have enacted laws establishing PDABs. Some state PDABs either have the authority or have defined a pathway where they may be granted the authority to establish upper payment limits for prescription drugs, including Colorado, Maryland, Washington, and Minnesota. Under the Washington law, the PDAB cannot select for an affordability review drugs that are solely for the treatment of an orphan-designated disease or condition. In August 2023, the Colorado PDAB selected five drugs for an affordability review, including TRIKAFTA; in December, it found TRIKAFTA to be not unaffordable, and thus not eligible for an upper payment limit. We cannot, however, predict whether future reviews by the Colorado PDAB, or any legislationother PDAB, will come to the same conclusion about TRIKAFTA or regulatory changesany of our other therapies, or relaxationthe amount of laws that restrict importsany potential upper payment limit. We may continue to see more state action requiring additional disclosures or other actions. Additional state actions, including the importation of drugs from other countries, revisionsalso may affect the availability and accessibility of our medicines. For example, on January 5, 2024, the FDA authorized Florida’s Agency for Health Care Administration’s drug importation program under section 804 of the Federal Food, Drug, and Cosmetic Act, which eventually would allow Florida to reimbursementimport certain prescription drugs from Canada. The importation of drugs from Canada or pharmaceuticals under government programsother countries that potentially could compete with our medicines could create increased pressure on our revenue and profitability. In addition, we could see increased federal activity related to drug pricing and transparency requiring disclosures or general budget controlother actions instead of, or in addition to, state requirements. Similar initiatives also could reduceare occurring in, or being considered by, some of our ex-U.S. markets, including Italy and Brazil.
Complying with these laws can be expensive and requires significant personnel and operational resources. Furthermore, any additional required discounts would adversely affect the net price we receive forpricing of, and revenues from, our products. Finally, while we seek to comply with all statutory and regulatory requirements, we face increased enforcement activity by the U.S. federal government, state governments, and private payors against pharmaceutical and biotechnology companies for pricing and reimbursement-related issues as well as inquiries from the U.S. Congress.
Other federal activities seeking to specifically address drug pricing and reimbursement include:
•rulemaking related to importation of prescription drugs from Canada, as well as guidance related to importation of prescription drugs from other foreign countries;
•attempts to establish reference pricing for certain physician-administered drugs;
•executive orders relating to drug pricing that are intended to broadly impact the pharmaceutical industry;
•changes to the federal anti-kickback statute safe harbors that eliminate anti-kickback statute discount safe harbor protection for certain manufacturer rebate arrangements; and
•legislation relating to drug pricing, including enhanced transparency measures into drug pricing.
We expect government scrutiny over drug pricing, reimbursement, and distribution to continue. Potential future government regulation of drug prices or reimbursement creates uncertainties about our portfolio and could have a material adverse effect on our operations. Moreover, antitrust and/or competition laws are increasingly being used to scrutinize pricing on high-value medicines. Defending against an antitrust or competition claim can be expensive and requires significant personnel and operational resources, may ultimately lead to a reduction in the prices of our products, and can ultimately result a material adverse effect on profitability and our business overall. Additionally, governmental efforts to pursue compulsory licensing, including the Biden Administration’s proposed framework to pursue so-called “march in” rights, could affect our pricing strategy and result in an adverse impact on our revenue.
Current health care laws and regulations in the U.S. and future legislative or regulatory reforms to the U.S. health care system may affect our ability to commercialize our marketed products profitably.
The U.S. government, individual states and some foreign jurisdictions also have been aggressively pursuing legislative and regulatory reforms that could affect our ability to sell products. For example, in the U.S., there have been federal legislative and administrative efforts to repeal, substantially modify, or invalidate some or all of the provisions of the ACA, which could affect coverage and payment for medicines. The federal government additionally has proposed and enacted legislation leading to aggregate reductions of Medicare payments to providers, which ultimately could affect utilization of medicines.
Other reforms include the Bipartisan Budget Act of 2018, which contained various provisions that affect coverage and reimbursement of drugs, including an increase in the discount that manufacturers of Medicare Part D brand name drugs must provide to Medicare Part D beneficiaries during the coverage gap from 50% to 70%. Under the IRA, the coverage gap phase and the associated coverage gap discount program will be eliminated after the 2024 plan year. Starting in 2025, there will be a new Part D manufacturer discount program, which requires a 10% discount in the initial phase and a 20% discount in the catastrophic phase of the benefit. The IRA also authorizes the government to negotiate maximum fair prices for certain Medicare drugs. It also establishes mandatory rebates for Part B and Part D drugs with prices that increase faster than inflation. These new laws or any other similar laws introduced in the future may result in additional reductions in Medicare and other health care funding, which could negatively affect our customers and accordingly, our financial operations. Moreover, payment methodologies may be subject to changes in health care legislation and regulatory initiatives. For example, CMS may develop new payment and delivery models, such as bundled payment models.
Adoption of new health care reform legislation at the federal or state level could affect demand for, or pricing of, our products or product candidates if approved for sale. We cannot, however, predict the ultimate content, timing, or effect of any health care reform legislation or action, or its impact on us, including increased compliance requirements and costs, all of which may adversely affect our future business, operations, and financial results.
We have experienced challenges commercializing products outside of the U.S., and our future revenues will be dependent on our ability to obtain adequate reimbursement for our products.products in ex-U.S. markets.
In most ex-U.S. markets, the pricing and reimbursement of therapeutic and other pharmaceutical products is subject to governmental control. Given recent global economic pressures, including due to the COVID-19 pandemic,control and geopolitical uncertainty, government authorities throughout the world are increasingly attemptingmaking greater efforts to limit or regulate the price of drug products. The reimbursement process in ex-U.S. markets can take a significant time to conclude and reimbursement decisions are made on a country-by-countrycountry by country or region by region basis. Further, many ex-U.S. governments are introducing new legislation focusing on cost containment measures in the pharmaceutical industry. The final form of these laws and the relevant practical application is unknown at this time, but may lead to lower prices, paybacks or other forms of discounts or special taxes.
Our medicines treat life-threatening conditions and address relatively small patient populations, and our research and development programs are primarily focused on developing medicines to treat similar diseases. Particular attention is being paid by payors, includingBoth government and private payors toare targeting these types of high-cost medicines, and countries are increasinglytherapies, in some cases refusing to reimburse costly medicines.pay for them. We have experienced challenges in obtaining timely reimbursement for our products in various countries outside the U.S. We continue to experience such challenges in some countries. For example, we obtained reimbursement for ORKAMBI and SYMKEVI in England in the fourth quarter of 2019, four years after ORKAMBI’s initial approval in 2015. Our future product revenues, including from TRIKAFTA/KAFTRIO, depend on, among other things, our ability to completemaintain reimbursement discussions in ex-U.S. markets for our products. There is no assurance that coverage and reimbursement will be available outside of the U.S. for our fourfive approved medicines or any future medicine, and, even if it is available, whether the timing or the level of reimbursement will be sufficient to allow us to market our medicines. Adverse pricing limitations or a delay in obtaining coverage and reimbursement would decrease our future net product revenues and harm our business.
We have limited experience developing cellInsurance coverage and genetic therapies and could experience challenges with these programs, which could result in delays or prevent the development, manufacturing and commercialization of our cell and genetic therapies.
We are investing significant resources in the research, development and manufacturing of cell and genetic therapies. While we have previously successfully developed, manufactured and commercialized several small molecule drugs, we have limited experience with the development, manufacture and commercialization of cell and genetic therapies. Development, manufacturing and commercializationreimbursement of cell and genetic therapies are subject to the same risks and uncertainties as development, manufacturing and commercializing small molecules. In addition:
•the manufacturing processes for cell and genetic therapies are different and more complex than the manufacturing processes required for small molecule drugs, and require different systems, equipment, facilities and expertise to develop and maintain;
•we may encounter difficulties in the production of our cell and genetic therapies and ensuring that the product meets required specifications;
•there have been a limited number of regulatory approvals for genetic therapies to date, the regulatory requirements governing genetic therapies continue to evolve, and regulatory positions and interpretations can change or lead to delays or significant unexpected costs with respect to our genetic therapy programs;
•the commercial success of cell or genetic therapies, including CTX001, if approved, will depend in part on the medical community, patients, and third-party or governmental payers accepting cell or genetic therapy products in general, and the applicable medicine as medically useful, cost-effective, and safe; and
•market acceptance will be dependent in part on the prevalence and severity of side effects associated with the procedure by which the cell or genetic therapy is administered, including, with respect to CTX001, if approved, the prevalence and severity of any side effects resulting from the myeloablative preconditioning regimen.
For programs addressing rare genetic diseases with small patient populations, we may not be able to identify, recruit and enroll a sufficient number of patients, or those with required or desired characteristics, to complete our clinical studies in an adequate and timely manner. Additionally, patients may be unwilling to participate in our clinical trials because of concerns that cell and genetic therapies are unsafe or unethical, negative publicity from adverse events in the biotechnology or gene therapy industries or for other reasons, including competitive clinical studies for similar patient populations.
In order to develop and commercialize any future cell or genetic therapies, we will need to incur substantial expenditures to develop, contract for, or otherwise arrange for the necessary manufacturing capabilities. Additionally, the manufacture of cell and genetic therapies requires significant expertise. Even with the relevant experience and expertise, manufacturers of cell and genetic therapy products often encounter difficulties in production, including difficulties with production costs and yields, quality control, and compliance with federal, state and foreign regulations. We cannot make any assurances that these problems will not occur, or that we will be able to resolve or address problems that occur in a timely manner, or at all.
To the extent we develop capabilities internally, there are many risks that could result in delays and additional costs, including the need to hire and train qualified employees and obtain access to necessary equipment and third-party technology. To the extent we partner with third parties to manufacture our cell or genetic therapies, any complexity in the manufacture of our products and product candidates may require lengthy technology transfers.uncertain.
There also is significant uncertainty related to the insurance coverage and reimbursement of cell or genetic therapy products, including gene therapies that are potential one-time treatments.treatments (e.g., CASGEVY). It is difficult to predict what third party payors, including U.S. or ex-U.S. governments or private insurance companies, will decide with respect to reimbursement for CASGEVY and the other novel cell and genetic therapies like the ones in our pipeline. Additionally, reimbursement rates for cell and genetic therapies approved before ours could create an adverse environment for reimbursement of any therapies we ultimately commercialize. The administration of our products may require procedures for the collection of cells from patients, followed by other procedures either before or after delivery of the cell or genetic therapy. The manner and level at which reimbursement is provided for these services also is important. An inadequateInadequate reimbursement for such services may adversely affect physician decisiondiscourage physicians from recommending decisions to recommend any product for which we obtain approval in the future and impair our ability to market or sell them.
Given there are only a few approved cell and genetic therapy products, it also is difficult to determine how long it will take or reasonably estimate the costs to develop, manufacture and commercializeassociated cell or genetic therapies. In addition, our cell-based therapies include approaches involving devices, which are subject to additional regulatory requirements. If we are unable to successfully develop, manufacture or commercialize such therapies on a timely or profitable basis, or at all, we may not realize benefits or generate cash flows based on our investments in these programs and our business, financial condition, results of operations and our stock price would likely be adversely affected.therapy.
We are dependent upon a small number of customers for a significant portion of our revenue, and the loss of, or significant reduction in sales to, these customers would adversely affect our results of operations.
In the U.S., we sell our products principally to a limited number of specialty pharmacy and specialty distributors, which subsequently resell our products to patients and health care providers. Internationally, we sell our products primarily to a limited number of specialty distributors and retail chains, as well as hospitals and clinics. We expect this significant customer concentration to continue for the foreseeable future. Our ability to generate and grow sales of our CF medicines will depend significantly on the extent to which these specialty distributors and specialty pharmacies are able to provide adequate distribution of our products to patients and healthcare providers. The loss of any large customer, a significant reduction in sales we make to them, any cancellation of orders they have made with us, or any failure to pay for the products we have shipped to them could adversely affect our business, financial condition and results of operations.
Risks Related to Development and Clinical Testing of Our Products and DrugProduct Candidates
Our drugproduct candidates remain subject to clinical testing and regulatory approval. Ourapproval, and our future success is dependent on our ability to successfully develop additional drugproduct candidates for both CF and non-CF indications.
Our business depends upon the successful development and commercialization of drugproduct candidates. These drugproduct candidates are in various stages of development and must satisfy rigorous standards of safety and efficacy before they can be
approved for sale by the FDA or comparable foreign regulatory authorities. To satisfy these standards, we must allocate resources among our various development programs and must engage in expensive and lengthy testing of our drugproduct candidates. Discovery and development efforts for new pharmaceutical and biological products, including new combination therapies, are resource-intensive and may take 10 to 15 years or longer for each drugproduct candidate. It is impossible to predict when or if any of our product candidates will prove effective and safe in humans or will receive regulatory approval. Despite our efforts, our drugproduct candidates may not:
•offer therapeutic or other improvement over existing competitive therapies;
•show the level of safety and efficacy, including the level of statistical significance, required by the FDA or other regulatory authorities for approval of a drug candidate;
• meet applicable regulatory standards;
•be capable of being produced in commercial quantities at acceptable costs; or
•if approved for commercial sale, be successfully marketed as pharmaceutical or biological products.
We have recently completed and/or have ongoing or planned clinical trials for several of our drugproduct candidates. The strength of our product portfolio and pipeline will depend in large part upon the outcomes of these clinical trials, including clinical trialsthose evaluating our triple combination therapyTRIKAFTA/KAFTRIO and vanzacaftor/tezacaftor/deutivacaftor in younger children with CF, VX-522 in CF, VX-548 in neuropathic pain, and VX-880 and VX-264 in T1D. Failure to advance product candidates through clinical development could impair our clinical trials of potential medicinesability to treat other diseases. ultimately commercialize products, which could materially harm our business and long-term prospects.
Results of our clinical trials and findings from our nonclinical studies, including toxicology findings in nonclinical studies conducted concurrently with clinical trials, could lead to abrupt changes in our development activities, including the possible cessation of development activities associated with a particular drugproduct candidate or program. For example, in October 2020,2023, we discontinued development of VX-814,decided not to progress VX-864, a drug candidate for the treatment of AAT, based on the safety and pharmacokinetic profile observedAATD, into further development due to non-serious rash events in the clinical trial.some patients.
Moreover, clinical data are often susceptible to varying interpretations, and many companies that have believed their drugproduct candidates performed satisfactorily in clinical trials have nonetheless failed to obtain marketing approval of their drugproduct candidate. Furthermore, results from our clinical trials may not meet the level of statistical significance or otherwise provide the level of evidence or safety and efficacy required by the FDA or other regulatory authorities for approval of a drugproduct candidate. Finally, clinical trials are expensive and require significant operational resources to implement and maintain.
Many companies in the pharmaceutical and biotechnology industries, including our company, have suffered significant setbacks in later-stage clinical trials even after achieving promising results in earlier-stage clinical trials. For example, the results from completed preclinical studies and clinical trials may not be replicated in later clinical trials, and ongoing clinical trials for our drugproduct candidates may not be predictive of the results we may obtain in later-stage clinical trials or of the likelihood of approval of a drugproduct candidate for commercial sale.
In addition, from time to time, we report interim, topline, and preliminary data from our clinical trials.trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change. Interim or preliminary data from a clinical trial may not be predictive of final results from the clinical trial. Failuretrial and are subject to advance drug candidates throughthe risk that one or more of the clinical development could impairoutcomes may materially change as patient enrollment and treatment continues and more patient data become available or as patients from our clinical trials continue other treatments for their disease. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution until the final data are available. If the interim, topline, or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to ultimatelyobtain approval for, and commercialize, products,our product candidates may be harmed, which could materially harm our business, operating results, prospects or financial condition.
The ability of third parties to review and/or analyze data from our clinical trials, including as a result of government disclosure, also may increase the risk of commercial confidentiality breaches and long-term prospects.result in enhanced scrutiny of our clinical trial results. For example, Clinical Trial Regulation (EU) No. 536/2014, and the EMA policy on publication of clinical data
for medicinal products for human use, both permit the EMA to publish clinical information submitted in marketing authorization applications. Third party review and scrutiny could result in public misconceptions regarding our drugs and product candidates. These publications could also result in the disclosure of information to our competitors that we might otherwise deem confidential, which could harm our business.
If we are unable to obtain or are delayed in obtaining regulatory approval, we willmay incur additional costs, experience delays in commercialization, or be unable to commercialize our drugproduct candidates.
The time required to complete clinical trials and to satisfy the FDA and other countries’ regulatory review processes is uncertain and typically takes many years. Our analysis of data obtained from nonclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. We also may encounter unanticipated delays or increased costs due to government regulation from future legislation or administrative action or changes in governmental policy during the period of drug development, clinical trials and governmental regulatory review.
We may seek a Fast Track, Priority Review, Breakthrough Therapy, and/or RMAT designation for some of our drugproduct candidates. DrugProduct candidates that receive one or more of these designations may be eligible for, among other things, a priority regulatory review. Each of these designations is within the discretion of the FDA. Accordingly, even if we believe one of our drugproduct candidates meets the criteria for Fast Track, Priority Review, Breakthrough Therapy and/or RMAT designation, the FDA may disagree and instead determine not to make such designation. The receipt of one or more of these designations for a drugproduct candidate does not guarantee a faster development process, review or approval compared to drugsproducts developed or considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our drugsproducts or drugproduct candidates qualifies for Fast Track, Priority Review, Breakthrough Therapy and/or RMAT designation, the FDA may later decide to withdraw such designation if it determines that the drugproduct or drugproduct candidate no longer meets the conditions for qualification.
Any failure to obtain regulatory approvals for a drugproduct candidate would prevent us from commercializing that drugproduct candidate. Any delay in obtaining required regulatory approvals could materially adversely affect our ability to successfully commercialize a drugproduct candidate. Furthermore, any regulatory approval to market a drugproduct may be subject to limitations that we do not expect on the indicated uses for which we may market the drug.product. Any such limitations could reduce the size or demand of the market for the drug.
We also are subject to numerous foreign regulatory requirements governing the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. Non-U.S. jurisdictions have different approval
procedures than those required by the FDA, and these jurisdictions may impose additional testing requirements for our drugproduct candidates. The foreign regulatory approval process includes all of the risks associated with the FDA approval process described above, as well as risks attributable to the satisfaction of foreign requirements. Approval by the FDA does not ensure approval by regulatory authorities outside the U.S. and approval by a foreign regulatory authority does not ensure approval by the FDA. In addition, although the FDA may accept data from clinical trials conducted outside the U.S., acceptance of this data is subject to conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The trial population also must adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. In addition, while these clinical trials are subject to applicable local laws, FDA acceptance of the data will depend on its determination that the trials also complied with all applicable U.S. laws and regulations. If the FDA does not accept the data from any trial that we conduct outside the U.S., it would likely result in the need for additional trials, which would be costly and time-consuming and delay or permanently halt our development of the applicable drugproduct candidate.
If clinical trials are prolonged or delayed, our development timelines for the affected development program could be extended, our costs to develop the drugproduct candidate could increase and the competitive position of the drugproduct candidate could be adversely affected.
We cannot predict whether or not we will encounter problems with any of our completed, ongoing or planned clinical trials that will cause us or regulatory authorities to delay or suspend clinical trials, or delay the analysis of data from our completed or ongoing clinical trials. Among the factors that could delay our development programs are:
•ongoing discussions with the FDA or comparable foreign authorities regarding the scope or design of our clinical trials and the number of clinical trials we must conduct;
•failure or delay in reaching agreement on acceptable terms with prospective contract research organizations (“CROs”) and clinical trial sites;
•failure to add or delay in adding a sufficient number of clinical trial sites and obtaining institutional review board or independent ethics committee approval at each clinical trial site;
•suspension or termination of clinical trials of product candidates for various reasons, including non-compliance with regulatory requirements;
•clinical trial sites deviating from clinical trial protocol or dropping out of a clinical trial;
•delays in enrolling volunteers or patients into clinical trials, including as a result of low numbers of patients that meet the eligibility criteria for the trial;
•a lower than anticipated retention rate of volunteers or patients in clinical trials;
•the need to repeat clinical trials as a result of unfavorable or inconclusive results, unforeseen complications in testing or clinical investigator error;
•inadequate supply or deficient quality of drugproduct candidate materials or other materials necessary for the conduct of our clinical trials;
•unfavorable FDA or foreign regulatory authority inspection and review of a manufacturing facility that supplied clinical trial materials or its relevant manufacturing records or a clinical trial site or records of any clinical or preclinical investigation;
•unfavorable or inconclusive scientific results from clinical trials;
•serious and unexpected drug-relatedtreatment-related side-effects experienced by participants in our clinical trials or by participants in clinical trials being conducted by our competitors to evaluate drugproduct candidates with similar mechanisms of action or structures to therapies that we are developing;
•favorable results in testing of our competitors’ drugproduct candidates, or FDA or foreign regulatory authority approval of our competitors’ drugproduct candidates; or
•action by the FDA or a foreign regulatory authority to place a clinical hold or partial clinical hold on a trial or compound or deeming the clinical trial conduct as problematic.
For planning purposes, we estimate the timing of the accomplishment of various scientific, clinical, regulatory, and other product development goals, which we sometimes refer to as milestones. These milestones may include the commencement or completion of scientific studies and clinical trials and the submission of regulatory filings. From time to time, we publicly announce the expected timing of some of these milestones. All of these milestones are based on a variety of assumptions. The actual timing of these milestones can vary dramatically compared to our estimates, in many cases for reasons beyond our control. If we do not meet these milestones as publicly announced, the commercialization of our products may be delayed and the credibility of our estimates may be adversely affected and, as a result, our stock price may decline.
Difficulty in enrolling patients could delay or prevent clinical trials of our product candidates, and ultimately delay or prevent regulatory approval.
Our ability to enroll patients in our clinical trials in sufficient numbers and on a timely basis is subject to a number of factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites, the availability of effective treatments for the relevant disease, the number of other clinical trials ongoing and competing for patients in the same indication, the eligibility criteria for the clinical trial, and the ongoing COVID-19 pandemic. In addition, patients may drop out of our clinical trials or may be lost to follow-up medical evaluation after treatment ends, and this could impair the validity or statistical significance of the trials.factors. Clinical trials are expensive and require significant operational
resources. Delays in patient enrollment or unforeseen drop-out rates may result in increased costs and longer development times. The enrollment of patients further depends on many factors, including:
•the proximity of patients to clinical trial sites;
•the size of the patient population, the nature of the protocol, and the design of the clinical trial;
•our ability to recruit clinical trial investigators with the appropriate competencies and experience;
•the number of other clinical trials ongoing and competing for patients in the same indication;
•our ability to obtain and maintain patient consents;
•reporting of the preliminary results of any of our clinical trials;
•the availability of effective treatments for the relevant disease and eligibility criteria for the clinical trial;
•the risk that patients enrolled in clinical trials will drop out of the clinical trials before clinical trial completion; and
•factors we may not be able to control, such as current or potential pandemics that may limit patients, principal investigators or staff or clinical site availability.
We, our collaborators, the FDA, or other applicable regulatory authorities may suspend clinical trials of a drugproduct candidate at any time if we or they believe the healthy volunteers or patients participating in such clinical trials are being exposed to unacceptable health risks or for other reasons. Any such suspension could materially adversely affect the development of a particular drugproduct candidate and our business.
Enrollment for clinical trials for our cell and gene therapies may face additional and unique challenges and adverse developments associated with these clinical trials could result in action by regulatory bodies, including revised requirements for approval.
For cell and genetic therapy programs addressing rare genetic diseases with small patient populations, we may not be able to identify, recruit and enroll a sufficient number of patients, or those with required or desired characteristics, to complete our clinical studies in an adequate and timely manner. Additionally, patients may be unwilling to participate in our clinical trials because of concerns that cell and genetic therapies are unsafe or unethical, negative publicity from adverse safety events in the biotechnology or gene therapy industries, or for other reasons, including competitive clinical studies for similar patient populations. Moreover, adverse developments in clinical trials conducted by others of cell and genetic therapy products or products created using similar technology, or adverse public perception of the field of cell and genetic therapies, may cause the FDA and other regulatory bodies to revise the requirements for approval of any cell or genetic therapy product candidates we may develop or limit the use of products utilizing technologies such as ours, either of which could materially harm our business.
Risks Related to Government Regulation
If regulatory authorities interpret any of our conduct, including our marketing practices, as being in violation of applicable health care laws, including fraud and abuse laws, laws prohibiting off-label promotion, disclosure laws or other similar laws, we may be subject to civil or criminal penalties.
We are subject to health care fraud and abuse laws, such as the federal False Claims ActFCA and anti-kickback laws, which prohibit off-label product promotionthe AKS, and other similar laws and regulations both in the U.S. and in non-U.S. markets.
The federal anti-kickback lawIn the U.S., the Federal Anti-Kickback Statute prohibits knowingly and willfully offering, paying, soliciting, receiving or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the ordering, furnishing, arranging for or recommending of an item or service that is reimbursable, in whole or in part, by a federal health care program, such as Medicare or Medicaid. Because of the broad scope of the prohibition, most financial interactions between pharmaceutical manufacturers and prescribers, purchasers, third party payors and patients would be subject to the statute. Although there are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution, the exceptions and safe harbors are narrow. Financialfinancial interactions must therefore be structured carefully to qualify for protection or otherwise withstand scrutiny.
Federal false claims laws, including the FCA, prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. Pharmaceutical companies have been prosecuted under these laws for a variety of alleged promotional and marketing activities, such as providing free product to customers with the expectation that the customers would bill federal programs for the product; reporting to pricing services inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in promotion for uses that the FDA has not approved, known as “off-label” uses, that caused claims to be submitted to Medicaid for those off-label uses; submitting inflated “best price” information to
the Medicaid Rebate Program; and certain manufacturing-related violations. The scope of this and other laws may expand in ways that make compliance more difficult and expensive.
The FDA and other regulatory agencies closely regulate the post-approval marketing and promotion of products to ensure that they are marketed only for the approved indications and in accordance with the provisions of the approved labeling. Although physicians are generally permitted, based on their medical judgment, to prescribe products for indications other than those approved by the FDA,applicable regulatory agency, manufacturers are prohibited from promoting their products for such off-label uses. We market our products to eligible people with CF, SCD, and TDT for whom the applicable product has been approved and provide promotional materials and traininginformational programs to physicians regarding the use of each product in these patient populations. These eligible people do not represent all people with CF.CF, SCD, and TDT. If the FDAa regulatory agency determines that our promotional materials, training or other activities constitute off-label promotion, it could request that we modify our training or promotional materials or other activities, conduct corrective advertising, or subject us to regulatory enforcement actions, includingsuch as the issuance of a warning or untitled letter, injunction, seizure, civil finefines and criminal penalties. It also is possible that other federal, state, or foreign enforcement authorities might take action if they believe that the alleged improper promotion led to the submission and payment of claims for an off-label use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. Even if it is later determined we were not in violation of these laws, we may be faced with negative publicity, incur significant expenses defending our actions, and have to divert significant management resources from other matters.
In the U.S., federal and state laws regulate financial interactions between pharmaceutical manufacturers and healthcare providers, require disclosure to government authorities and the public of such interactions, and mandate the adoption of compliance standards or programs. For example, the so-called federal “sunshine law” requires pharmaceutical manufacturers to report annually to the Centers for Medicare & Medicaid Services, or CMS payments or other transfers of value made by that entity to physicians, physicians assistants, advanced practice registered nurses, and teaching hospitals (and additional categories of health care practitioners beginning with reports submitted on or after January 1, 2022).hospitals. We also have similar reporting obligations with respect to financial interactions throughout the E.U. We expended significant efforts to establish, and are continuing to devote significant resources to
maintain and enhance, systems and processes in order to comply with these regulations. Requirements to track and disclose financial interactions with health care providers and organizations increase government and public scrutiny of these financial interactions. Failure to comply with the reporting requirements could result in significant civil monetary penalties.
The sales and marketing practices of our industry have been the subject of increased scrutiny from government authorities in the U.S. and other countries in which we market our products, and we believe that this trend will continue. Many of these laws have not been fully interpreted by the government authorities or the courts, and their provisions are subject to a variety of interpretations. While we have a corporate compliance program which, together with our policies and procedures, is designed to actively identify, prevent and mitigate risk through the implementation of compliance policies and systems and the promotion of a culture of compliance, if we are found not to be in full compliance with these laws and regulations, our business could be materially harmed. We may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from federal health care programs and/or the curtailment or restructuring of our operations. Even if we successfully defend against government challenge, responding to the challenge may cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
If we fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate Program or other governmental pricing programs in the U.S., we could be subject to additional reimbursement requirements, penalties, sanctions, and fines whichthat could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We participate in the Medicaid Drug Rebate Program, the 340B Drug Pricing Program,program, and a number of other federal and state government pricing programs in the U.S. in order to obtain coverage for our products by certain government health care programs. These programs would generally require us to pay rebates or provide discounts to certain government payors or private purchasers or government payers in connection with our products when dispensed to beneficiaries of these programs. In some cases, such as with the Medicaid Drug Rebate Program, the rebates are based on pricing and rebate calculations that we report on a monthly and quarterly basis to the government agencies that administer the programs. The terms, scope and complexity of these government pricing programs change frequently. For example, regulations finalized in December 2020 created an alternative Medicaid rebate formula for “line extensions” of oral solid dosage forms. Moreover, in December 2020, CMS finalized changes to Medicaid Drug Rebate Program pricing calculations regarding the provision of co-payment assistance to patients that may be impacted by so-called accumulator programs operated by private insurers or pharmacy benefit managers. The portion of this rule
dealing with manufacturer co-payment assistance was struck down by the U.S. District Court for the District of Columbia in May 2022 (and the deadline for an appeal has lapsed). In May 2023, CMS issued a proposed rule, which would withdraw the challenged accumulator adjustment regulations, consistent with the Court’s order. The rule also proposes significant changes, which, if finalized, could have an impact on our Medicaid rebate liability, impact our participation in the Medicaid Drug Rebate Program, and impose new reporting requirements.
Additionally, the expansion of the 340B Drug Discount Program through the ACA has increased the number of purchasers who are eligible for significant discounts on branded drugs. These and future changes to government pricing programs, laws, and regulations may have a material adverse impact on our revenue and operations.
We also may also have reimbursement obligations or be subject to penalties if we fail to provide timely and accurate information to the government, pay the correct rebates, or offer the correct discounted pricing. Changes to the price reporting or rebate requirements of these programs would affect our obligations to pay rebates or offer discounts. CMS recently proposed changesFor example, the removal of the current statutory 100% of Average Manufacturer Price per-unit cap on Medicaid rebate liability for single source and innovator multiple source drugs, effective as of January 1, 2024, under the American Rescue Plan Act of 2021 may affect the amount of rebates paid on prescription drugs under Medicaid and the prices that are required to be charged to covered entities under the Medicaid340B Drug Rebate calculationsDiscount Program. Additionally, the IRA requires manufacturers to address treatmentpay rebates for Medicare Part B and Part D drugs with prices that increase faster than the rate of value-based arrangements, accumulator adjustment programs implemented by payers, and new formulations of existing products.inflation. Responding to current and future changes to these and other Medicaid Drug Rebate Program requirements may increasereduce our costsnet revenues and the complexity of compliance, will be time-consuming, and could have a material adverse effect on our results of operations.
If our processes and systems are not compliant with regulatory requirements, we could be subject to restrictions on marketing our products or could be delayed in submitting regulatory filings seeking approvals for our drugproduct candidates.
We have a number of regulated processes and systems that are required both prior to and following approval of our drugs and drugproduct candidates. These processes and systems are subject to continual review and periodic inspection by the FDA and other regulatory bodies. In addition, the clinical research organizations and other third parties that we work with in our non-clinical studies and clinical trials and our oversight of such parties are subject to similar reviews and periodic inspection by the FDA and other regulatory bodies. If compliance issues are identified at any point in the development and approval process, we may experience delays in filing for regulatory approval for our drugproduct candidates, or delays in obtaining regulatory approval after filing, if at all. Any later discovery of previously unknown problems or safety issues with approved drugs or manufacturing processes, or failure to comply with regulatory requirements, may result in restrictions on such drugs or manufacturing processes, withdrawal of drugs from the market, the imposition of civil or criminal penalties or a refusal by the FDA and/or other regulatory bodies to approve pending applications for marketing approval of new drugs or supplements to approved applications, any of which could have a material adverse effect on our business. In addition, we are party to agreements that transfer responsibility for complying with specified regulatory requirements, such as filing and maintenance of marketing authorizations and safety reporting or compliance with manufacturing requirements, to our collaborators and third-party manufacturers. If our collaborators or third-party manufacturers do not fulfill these regulatory obligations, any drugs for which we or they obtain approval may be subject to later restrictions on manufacturing or sale, which could have a material adverse effect on our business.
The regulatory approval process for our cell or genetic therapies involves additional consultations with regulatory agencies, costs, and potentially longer timelines as compared to those for small molecules.
As we advance our cell and genetic therapy product candidates, we will be required to consult with various regulatory authorities, and we must comply with all applicable laws, rules, and regulations, which may change from time to time, including during the course of development of our cell and genetic therapy product candidates. If we fail to do so, we may be required to delay or discontinue the clinical development of certain of our cell and genetic therapy product candidates. These additional processes may result in a review and approval process that is longer than we otherwise would have expected. Even if we comply with applicable laws, rules, and regulations, and even if we maintain close coordination with the applicable regulatory authorities with oversight over our cell and genetic therapy product candidates, our development programs may experience delays or fail to succeed. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential cell or genetic therapy product to market would materially adversely affect our business, financial condition, results of operations and prospects.
The regulatory approval process and clinical trial requirements for cell and genetic therapies can be more expensive and take longer than for other, better known or more extensively studied product candidates, and regulatory requirements
governing cell and genetic therapy products have changed frequently and may continue to change in the future. For example, the FDA established the Office of Tissues and Advanced Therapies within its Center for Biologics Evaluation and Research (“CBER”) to consolidate the review of cell therapies and related products, and the Cellular, Tissue and Gene Therapies Advisory Committee to advise CBER on its review. These and other regulatory review agencies, committees and advisory groups, and the requirements and guidelines they promulgate, may lengthen the regulatory review process, require us to perform additional preclinical studies or clinical trials, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of these treatment candidates or lead to significant post-approval limitations or restrictions.
We are subject to various and evolving laws and regulations governing the privacy and security of personal data, and our failure to comply could adversely affect our business, result in fines and/or criminal penalties, and damage our reputation.
We are subject to data privacy and security laws and regulations in various jurisdictions that apply to the collection, storage, use, sharing, and security of personal data, including health information, and impose significant compliance obligations. In addition, numerous other federal and state laws, including state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use, disclosure and security of personal information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues with the potential to affect our business.
For example, the E.U. General Data Protection Regulation or GDPR,(“GDPR”) went into effect in 2018 and has imposed new obligations on us with respect to our processing of personal data and the cross-border transfer of such data, including higher standards of obtaining consent, more robust transparency requirements, data breach notification requirements, requirements for contractual language with our data processors, and stronger individual data rights. Different E.U. member states have interpreted the GDPR differently and many have imposed additional requirements, which add to the complexity of processing personal data in the E.U. The GDPR also imposes strict rules on the transfer of personal data to countries outside the E.U., including the U.S. and the U.K., and permits data protection authorities to impose large penalties for violations of the GDPR. The GDPR rules related to cross border data transfers continue to evolve based on E.U. court decisions and regulator guidance, which presents certain practical challenges to compliance. Regulators also continue to focus enforcement efforts on behavioral advertising and other online tracking technologies commonly used by companies. Compliance with these evolving rules is challenging, as country specific guidance and rules are continually changing and limited alternatives currently exist in the market. Compliance with the GDPR is a rigorous and time-intensive process that may increase our cost of doing business or require us to change our business practices, and despite those efforts, there is a risk that we may be subject to fines and penalties, litigation, and reputational harm in connection with any activities falling within the scope of the GDPR.
In the U.S., California has passed the California Consumer Privacy Act (the “CCPA”), which went into effect on January 1, 2020. In November 2020, California also passed the California Privacy Rights Act (the “CPRA”), which expands and builds upon the consumer privacy rights of the CCPA. The CPRA came into effect January 1, 2023. Certain other states have also enacted legislation governing the protection of personal data and several other states and the federal government are actively considering similar proposed legislation. A number of other states have also introduced privacy legislation, governing the protection of personalwith some focusing specifically on health data. Additionally, Brazil passed the General Data Protection Law, or LGPD, which went into effect in August 2020. While we continue to address the implications of the new data privacy regulations, data privacy remains an evolving landscape at both the domestic and international level, with new regulations coming into effect and continued legal challenges. Each law is also subject to various interpretations by courts and regulatory agencies, creating even more uncertainty. While we have a global privacy program that addresses such laws and regulations, our efforts to comply with the evolving data protection rules may be unsuccessful.
We must devote significant resources to understanding and complying with the changing landscape in this area. Failure to comply with data protection laws may expose us to risk of enforcement actions taken by data protection authorities, private rights of action in some jurisdictions, and potential significant penalties if we are found to be non-compliant. Failure to comply with the GDPR and applicable national data protection laws of European Economic Area member states could lead to fines of up to €20,000,000 or up to 4% of the total worldwide annual revenue of the preceding financial year, whichever is higher. Some of these laws and regulations also carry the possibility of criminal sanctions. For example, while we are not directly subject to the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act or HIPAA,(“HIPAA”), we could be subject to penalties, including criminal penalties if we knowingly obtain or disclose individually identifiable health information from a HIPAA-covered health care provider or
research institution that has not complied with HIPAA’s requirements for disclosing such information. In addition, the commercialization of cell and gene therapies requires the collection and processing of a greater amount of personal data than traditional therapies, potentially increasing risk. Furthermore, the number of government investigations related to data security incidents and privacy violations, with a specific focus on online data sharing, continue to increase and government investigations typically require significant resources and generate negative publicity, which could harm our business and our reputation.
The COVID-19 pandemic has added further complexity to the processing of personal data. For example, safety measures and government health regulations intended to protect our employees, contractors, and other visitors to our sites may require the collection of certain personal data. Although we are focused on ensuring that personal data is properly protected, our efforts may be unsuccessful and we could unintentionally be subject to unauthorized access or disclosure of such personal data.
Clinical Trial Regulation (EU) No. 536/2014, or the Clinical Trial Regulation, and the EMA policy on publication of clinical data for medicinal products for human use both permit the EMA to publish clinical information submitted in MAAs. This provision of the Clinical Trial Regulation is expected to be effective by the end of 2021. The ability of third parties to review and/or analyze data from our clinical trials may increase the risk of commercial confidentiality breaches and result in enhanced scrutiny of our clinical trial results. Such scrutiny could result in public misconceptions regarding our drugs and drug candidates. These publications could also result in the disclosure of information to our competitors that we might otherwise deem confidential, which could harm our business.
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
Our research and development efforts involve the regulated use of hazardous materials, chemicals, and various controlled and radioactive compounds. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state, federal and foreign regulations, the risk of loss of, or accidental contamination or injury from, these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We also are subject to numerous environmental, health, and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens, and the handling of biohazardous materials. Although we maintain workers’ compensation insurance to cover us for costs we may incur due to injuries to our employees resulting from the use of these materials, this insurance may not provide adequate coverage against potential liabilities. We maintain insurance to cover pollution conditions or other extraordinary or unanticipated events relating to our use and disposal of hazardous materials that we believe is appropriate based on the small amount of hazardous materials we generate. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of these laws or regulations.
Risks Related to Business Development Activities
Our ability to execute on our long-term strategy depends in part on our ability to engage in transactions and collaborations with other entities that add to our pipeline or provide us with new commercial opportunities.
In order to achieve our long-term business objectives, we seek to license or acquire drugs, drug candidates and other technologies that have the potential to complement our ongoing research and development efforts, access emerging technologies and license or acquire pipeline assets. These transactions may be similar to prior transactions or may involve larger transactions or later-stage assets. We have faced and will continue to face significant competition for the acquisition of rights to these types of drugs, drug candidates and other technologies from a variety of other companies, many of which have significantly more financial resources and experience in business development activities than we have. In addition, non-profit organizations may be willing to provide capital to the companies that control additional drugs, drug candidates or technologies, which may provide incentives for companies to advance these drugs, drug candidates or technologies independently. Also, the cost of acquiring, in-licensing or otherwise obtaining rights to such drugs, drug candidates or other technologies has grown dramatically in recent years and may be at levels that we cannot afford or that we believe are not justified by market potential. As a result, we may not be able to acquire, in-license or otherwise obtain rights to additional drugs, drug candidates or other technologies on acceptable terms or at all.
We may not realize the anticipated benefits of acquisitions of businesses or technologies, and the integration following any such acquisition may disrupt our business and management.
It is challenging to effectively integrate businesses and technologies that we acquire, including the acquisitions of Semma and Exonics and the exclusive licenses that we have acquired from CRISPR and Moderna, and we may not realize the benefits anticipated from such transactions. Achieving the anticipated benefits of any transaction and successfully integrating acquired businesses or technologies involves a number of risks, including:
• failure to successfully develop and commercialize the acquired drugs, drug candidates or technologies or to achieve other strategic objectives;
• delays or inability to progress preclinical programs into clinical development or unfavorable data from clinical trials evaluating the acquired or licensed drug or drug candidates;
• difficulty in integrating the drugs, drug candidates, technologies, business operations and personnel of an acquired asset or company;
• disruption of our ongoing business and distraction of our management and employees from daily operations or other opportunities and challenges;
•the potential loss of key employees of an acquired company;
• entry into markets in which we have no or limited direct prior experience or where competitors in such markets have stronger market positions;
• potential failure of the due diligence processes to identify significant problems, liabilities or challenges of an acquired company, or acquired or licensed drug, drug candidate or technology, including but not limited to, problems, liabilities or challenges with respect to intellectual property, clinical or non-clinical data, safety, accounting practices, employee, or third-party relations and other known and unknown liabilities;
• liability for activities of the acquired company or licensor before the acquisition or license, including intellectual property infringement claims, violations of laws, commercial disputes, tax liabilities, and other known and unknown liabilities;
• exposure to litigation or other claims in connection with, or inheritance of claims or litigation risk as a result of an acquisition or license, including but not limited to, claims from terminated employees, customers, former equity holders or other third parties; and
• difficulties in the integration of the acquired company’s departments, systems, including accounting, human resource and other administrative systems, technologies, books and records, and procedures, as well as in maintaining uniform standards, controls, including internal control over financial reporting required by the Sarbanes-Oxley Act of 2002 and related procedures and policies.
Acquisitions, licensing arrangements and other strategic transactions are inherently risky, and ultimately, if we do not complete an announced acquisition, collaboration or strategic transaction or integrate an acquired or licensed asset, business or technology successfully and in a timely manner, we may not realize the anticipated benefits of the strategic transaction.
We may later incur impairment charges related to assets acquired in any such transaction. For example, we entered into a strategic collaboration and license agreement with Parion Sciences, Inc. to develop ENaC inhibitors in 2015 and incurred an impairment charge related to this collaboration in 2017. Even if we achieve the long-term benefits associated with our strategic transactions, our expenses and short-term costs may increase materially and adversely affect our liquidity and short-term net income. Future strategic transactions could result in potentially dilutive issuances of equity securities, the incurrence of debt, the creation of contingent liabilities, impairment expenses related to goodwill, or impairment or amortization expenses related to other intangible assets, all of which could harm our financial condition.
We face risks in connection with existing and future collaborations with respect to the development, manufacture and commercialization of our products and drug candidates.
The risks that we face in connection with our current collaborations, including CRISPR, and any future collaborations, include the following:
• Our collaborators may change the focus of their development and commercialization efforts or may have insufficient resources or expertise to effectively develop, manufacture or commercialize our drug candidates.
•The ability of some of our therapies to reach their potential could be limited if collaborators are unable to effectively develop, manufacture or commercialize these therapies or drug candidates or decrease or fail to increase development or commercialization efforts related to those therapies or drug candidates. Our collaboration agreements allocate development, manufacturing and commercialization responsibilities between us and our collaborators and provide our collaborators with a level of discretion in determining the amount and timing of efforts and resources that they will apply to these collaborations.
•Our collaborators may have limited experience in developing, manufacturing and commercializing therapies, either generally, or in the specific therapeutic area. For example, CRISPR, which is responsible for leading commercialization of CTX001 in the U.S., has no prior experience commercializing a therapy and is in the process of establishing the capabilities that would be required to commercialize CTX001 in the U.S.
• Collaboration agreements may have the effect of limiting the areas of research and development that we may pursue, either alone or in collaboration with third parties.
• Collaborators may develop and commercialize, either alone or with others, drugs that are similar to or competitive with the drugs or drug candidates that are the subject of their collaborations with us.
• Disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or termination of the research, development or commercialization of drug candidates, might lead to additional responsibilities or costs for us with respect to drug candidates, or might result in litigation or arbitration. Any such disagreements would divert management attention and resources and would be time-consuming and expensive.
• Collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation.
• Collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability.
• Investigations and/or compliance or enforcement actions against a collaborator, which may expose us to indirect liability as a result of our partnership with such collaborator.
• Our collaboration agreements are subject to termination under various circumstances.
Additionally, if a collaborator were to be involved in a business combination with a third party, it might de-emphasize or terminate the development or commercialization of any drug candidate licensed to it by us. If one of our collaborators terminates its agreement with us, we may find it more difficult to attract new collaborators and our perception in the business and financial communities could be harmed.
We may not be able to attract collaborators or external funding for the development and commercialization of certain of our drug candidates.
As part of our ongoing strategy, we may seek additional collaborative arrangements or external funding for certain of our development programs and/or seek to expand existing collaborations to cover additional commercialization and/or development activities. We have a number of research programs and clinical development programs, some of which are being developed in collaboration with a third party. For example, we are co-developing CTX001, an investigational CRISPR/Cas9-based gene-editing therapy for SCD and TDT with our collaborator, CRISPR. At any time, we may determine that in order to continue development of a drug candidate or program or successfully commercialize a drug we need to identify a collaborator or amend or expand an existing collaboration. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA, EMA or other regulatory authorities, the potential market for the subject drug candidate, the costs and complexities of manufacturing and delivering such drug candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of the applicable intellectual property, which can exist if there is a challenge to such ownership without regard to the merits of the challenge, and industry and market conditions generally. Potentially, and depending on the circumstances, we may desire that a collaborator either agree to fund portions of a drug development program led by us, or agree to provide all of the funding and directly lead the development and commercialization of a program. No assurance can be given that any efforts we make to seek additional collaborative arrangements will be successfully completed on a timely basis or at all. If we elect to fund and undertake development or commercialization activities on our own, we may need to obtain additional expertise and additional capital, which may not be available to us on acceptable terms or at all. If we are unable to enter into acceptable collaborative relationships, one or more of our development programs could be delayed or terminated and the possibility of our receiving a return on our investment in the program could be impaired.
Risks Related to Third-PartySupply, Manufacturing and Reliance on Third Parties
We depend on third-party manufacturers and our internal capabilities to manufacture our products and the materials we require for our clinical trials. We rely on third party logistics providers to manage our shipments globally. We may not be able to maintain theseour third-party relationships and could experience supply disruptions outside of our control.
We rely on a worldwide network of third-party manufacturers and our internal capabilities, including our own manufacturing facilities in Boston, to manufacture our drugs for commercial use and our drugproduct candidates for clinical trials. Astrials as well as our medicines for commercial use. While we have developed internal capabilities to supply product candidates for use in our clinical trials as well as our products for commercial sale, a resultmajority of the manufacturing steps needed to produce our medicines, product candidates, and drug products are performed through a third-party manufacturing network. The manufacture of our relianceproducts and product candidates can be complex, which may require lengthy technology transfers between us and the third parties on these third-party manufacturerswhich we rely. We expect that we will continue to rely on third parties to meet our commercial supply needs and suppliers, wea significant portion of our clinical supply needs for the foreseeable future.
We could be subject to significant supply interruptions as a result of disruptions outside ofto third party or our control.internal manufacturing capabilities. Our supply chain for sourcing raw materials and manufacturing drug product ready for distribution, including obtaining necessary supplies, is a multi-step international endeavor. Third-party contract manufacturers, including some in China, perform different parts of our manufacturing process. Contract manufacturers may supply us with raw materials, convert these raw materials into drug substance and/or convert the drug substance into final dosage form. Third parties are used for packaging, warehousing and distribution of products. In
The manufacturing and logistics for cell and genetic therapies are highly complex, short lead time operations that require partnership with an extensive network of third parties also willto deliver product. These manufacturing and logistics operations require significant investment by us to secure capacity at third parties with expertise to meet our requirements. Even with the relevant experience and expertise, manufacturers of cell and genetic therapy products often encounter difficulties in production, including difficulties with production costs and yields, quality control, and compliance with federal, state and
foreign regulations. There are many risks that could result in delays and additional costs, including the need to hire and train qualified employees and obtain access to necessary equipment and third-party technology. This capacity may be usedlimited by the number of other clinical trials and commercial manufacturing ongoing for other companies seeking similar support.
If third parties are unwilling or unable to meet our requirements, we could experience supply disruptions outside of our control. Additionally, manufacturing facilities, both manufactureforeign and deliverdomestic, are subject to inspections by the FDA and other U.S. and foreign government authorities. Although we actively engage with regulatory authorities, the timing of regulatory approvals for each of these facilities may be delayed for a variety of reasons. We may experience supply disruptions if regulatory agencies are unable to inspect the manufacturing facilities on which we rely. In addition, we and the third parties with whom we engage are required to maintain compliance with quality regulations globally. An inability to maintain compliance with such regulations, including cGMP requirements, could cause significant disruptions to our therapies, which requires significant expertise.business and operations.
Establishing,Additionally, establishing, managing and expanding thisour global manufacturing and supply chain requires a significant financial commitment and the creation and maintenance of our numerous third-party contractual relationships. We may not be able to agree on contractual terms with third parties as needed for manufacturing of our products. Although we attempt to manage the business relationships with companies in our supply chain,partners, we could be subject to supply disruptions outside of our control. For example, we are collaborating with CRISPR on establishing the supply chain to support clinical trials and commercial supply for CTX001, if approved. As a result, we do not have independent control over the related supply operations and are reliant on CRISPR to adequately establish the corresponding supply chains.
Supply disruptions may result from a number of factors, including shortages in product raw materials, labor or technical difficulties, regulatory inspections or restrictions, shipping or customs delays, general global supply chain disruptions, or any other performance failure by us or any third-party manufacturer on which we rely. Additionally, unfavorable geopolitical events or situations could affect our ability to interact with or conduct business with specific vendors within our global supply network, or could prevent or delay the transportation of supplies or products to their planned destination. Any supplysuch disruptions could disrupt sales of our products and/or the timing or advancement of our clinical trials.
We require a supply for our medicines for commercial sale and a supply of our drug candidates for use in our clinical trials. While we have developed some internal capabilities, a majority of the manufacturing steps needed to produce our drug candidates and drug products are performed through a third-party manufacturing network. To ensure the stability of our supply chains, we aim to develop additional sources of manufacture for all steps of our manufacturing processes at the time of, or shortly after, marketing approval. Therefore, at any point in time, we may have a limited number of single source manufacturers for certain steps in our manufacturing processes, particularly for recently launched products.
If we or our third-party manufacturers become unable or unwilling to continue manufacturing product and we are not able to promptly identify another manufacturer, we could experience a disruption in the commercial supply of our then-marketed medicines, which would have a significant effect on patients, our business, and our product revenues. Similarly, a disruption in the clinical supply of drug productsproduct candidates could delay the completion of clinical trials and affect timelines for regulatory filings. ThereWe have a limited number of critical steps in our manufacturing process that are single sourced, including for commercialized products. To ensure the stability of our supply chains, we continue to develop alternative suppliers for our manufacturing processes. However, there can be no assurance that we will be able to establish and maintain additional manufacturers or capacity for all of our drugproduct candidates and drug products on a timely basis or at all.
In the course of providing its services, a contract manufacturer may develop process technology related to the manufacture of our products or drugproduct candidates that the manufacturer owns, either independently or jointly with us. This would increase our reliance on that manufacturer or require us to obtain a license from that manufacturer in order to have our products or drugproduct candidates manufactured by other suppliers utilizing the same process.
We rely on third parties to conduct pre-clinical work, clinical trials and other activities, and those third parties may not perform satisfactorily, including failing to meet established deadlines for the completion of such studies and/or trials or failing to satisfy regulatory requirements.
We rely on third parties such as contract research organizationsCROs to help manage certain pre-clinical work and our clinical trials and on medical institutions, clinical investigators, and clinical research organizations such as the Therapeutic Development Network, which is primarily funded by the CFF,Cystic Fibrosis Foundation, to assist in the design and review of, and to conduct our clinical trials, including enrolling qualified patients. In addition, we engage third party contractors to support numerous other research, commercial and administrative activities. Our reliance on these third parties for clinical development activities reduces our control over these activities but does not relieve us of our responsibilities. For example, we remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the clinical trial. Moreover, the FDA requires us to comply with standards, commonly referred to as good laboratory practices
and good clinical practices, for conducting, recording and reporting the results of pre-clinical and clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Such standards, particularly with respect to newer cell and genetic therapies, will continue to evolve and subject us and third parties to new or changing requirements.
If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be
required to replace them. Although we believe that there are a number of other third-party contractors we could engage to continue the activities, it may result in a delay of the affected clinical trial, drug development program or applicable activity. If clinical trials are not conducted in accordance with our contractual expectations or regulatory requirements, action by regulatory authorities might significantly and adversely affect the conduct or progress of these clinical trials or in specific circumstances might result in a requirement that a clinical trial be redone. Accordingly, our efforts to obtain regulatory approvals for and commercialize our drugproduct candidates could be delayed. In addition, failure of any third partythird-party contractor to conduct activities in accordance with our expectations, could adversely affect the relevant research, development, commercial or administrative activity.
Risks Related to Business Development Activities
Our ability to execute on our long-term strategy depends in part on our ability to engage in transactions and collaborations with other entities that add to our pipeline or provide us with new commercial opportunities.
To achieve our long-term business objectives, we seek to license or acquire products, product candidates and other technologies that have the potential to complement our ongoing research and development efforts, access emerging technologies and license or acquire pipeline assets. These transactions may be similar to prior transactions, may be structured differently than prior transactions, or may involve larger transactions or later-stage assets. We have faced and will continue to face significant competition for the acquisition of rights to these types of products, product candidates and other technologies from a variety of other companies, some of which have significantly more financial resources and experience in business development activities than we have. In addition, non-profit organizations may be willing to provide capital to the companies that control additional products, product candidates or technologies, which may provide incentives for companies to advance these products, product candidates or technologies independently. Also, the cost of acquiring, in-licensing or otherwise obtaining rights to such products, product candidates or other technologies has grown dramatically in recent years and may be at levels that we cannot afford or that we believe are not justified by market potential. As a result, we may not be able to acquire, in-license or otherwise obtain rights to additional products, product candidates or other technologies on acceptable terms or at all.
We face risks in connection with existing and future collaborations with respect to the development, manufacture and commercialization of our products and product candidates.
The risks that we face in connection with our current collaborations, including with CRISPR, Moderna, and Entrada, and any future collaborations, include the following:
•Collaborators may develop and commercialize, either alone or with others, drugs or therapies that are similar to or competitive with the products or product candidates that are the subject of their collaborations with us.
•Disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or termination of the research, development or commercialization of product candidates, might lead to additional responsibilities or costs for us with respect to product candidates, or might result in litigation or arbitration. Any such disagreements would divert management attention and resources and would be time-consuming and expensive.
•Collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation.
•Collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability.
•Investigations and/or compliance or enforcement actions against a collaborator, which may expose us to indirect liability as a result of our partnership with such collaborator.
If a collaborator were to be involved in a business combination with a third party, it might de-emphasize or terminate the development or commercialization of any product candidate licensed to it by us. If one of our collaborators terminates its agreement with us, we may find it more difficult to attract new collaborators and our perception in the business and financial communities could be harmed.
Moreover, as part of our ongoing strategy, we may seek additional collaborative arrangements for certain of our development programs and/or seek to expand existing collaborations to cover additional commercialization and/or development activities. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA, EMA or other regulatory authorities, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of the applicable intellectual property, which can exist if there is a challenge to such ownership without regard to the merits of the challenge, and industry and market conditions generally. No assurance can be given that any efforts we make to seek additional collaborative arrangements will be successfully completed on a timely basis or at all.
We may not realize the anticipated benefits of existing or future acquisitions of businesses or technologies, and the integration following any such acquisition may disrupt our business and management.
Effectively integrating acquired businesses, technologies and exclusive licenses is challenging. We may not realize the benefits anticipated from our external innovation transactions. Achieving the anticipated benefits of any transaction and successfully integrating acquired businesses or technologies involves a number of risks, including:
•failure to successfully develop and commercialize the acquired products, product candidates or technologies or to achieve other strategic objectives;
•delays or inability to progress preclinical programs into clinical development or unfavorable data from clinical trials evaluating the acquired or licensed product or product candidates;
•difficulty in integrating the products, product candidates, technologies, business operations and personnel of an acquired asset or company;
•disruption of our ongoing business and distraction of our management and employees from daily operations or other opportunities and challenges;
•the potential loss of key employees of an acquired company;
•entry into markets in which we have no or limited direct prior experience or where competitors in such markets have stronger market positions;
•potential failure of the due diligence processes to identify significant problems, liabilities or challenges of an acquired company, or acquired or licensed products, product candidate or technology, including problems, liabilities or challenges with respect to intellectual property, clinical or non-clinical data, safety, accounting practices, employee, or third-party relations and other known and unknown liabilities;
•liability for activities of the acquired company or licensor before the acquisition or license, including intellectual property infringement claims, violations of laws, commercial disputes, tax liabilities, and other known and unknown liabilities;
•exposure to litigation or other claims in connection with, or inheritance of claims or litigation risk as a result of an acquisition or license, including claims from terminated employees, customers, former equity holders or other third parties; and
•difficulties in the integration of the acquired company’s departments, systems, including accounting, human resource and other administrative systems, technologies, books and records, and procedures, as well as in maintaining uniform standards, controls, including internal control over financial reporting required by the Sarbanes-Oxley Act of 2002 and related procedures and policies.
Acquisitions, licensing arrangements and other strategic transactions are inherently risky, and ultimately, if we do not complete an announced acquisition, collaboration or strategic transaction or integrate an acquired or licensed asset, business or technology successfully and in a timely manner, we may not realize the anticipated benefits of the strategic transaction.
We may later incur impairment charges related to assets acquired in any such transaction. Even if we achieve the long-
term benefits associated with our strategic transactions, our expenses and short-term costs may increase materially and adversely affect our liquidity and short-term net income. Future strategic transactions could result in increased operating expenses, potentially dilutive issuances of equity securities, the incurrence of debt, the creation of contingent liabilities, impairment expenses related to goodwill, or impairment or amortization expenses related to other intangible assets, all of which could harm our financial condition.
Risks Related to Intellectual Property
If our patents do not protect our drugsproducts or our drugsproducts infringe third-party patents, we could be subject to litigation which could result in injunctions preventing us from selling our products, substantial damages, or substantial liabilities.circumvention of our patents by third parties.
We haveown and/or control numerous issued patents and pending patent applications in the U.S., as well as counterparts in other countries. Our success will depend, in significant part, on our ability to obtain and defend U.S. and foreign patents covering our drugs,products, their uses and our processes, to preserve our trade secrets and to operate without infringing the proprietary rights of third parties. We cannot be certain that any patents will issue from our pending patent applications or, even if patents issue or have issued, that the issued claims will provide us with adequate protection against competitive products or otherwise be commercially valuable.
Due to evolving legal standards relating to the patentability, validity and enforceability of patents covering pharmaceutical inventions and the scope of claims made under these patents, our ability to obtain, maintain and enforce patents is uncertain and involves complex legal and factual questions. Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents in the U.S. The Leahy-Smith America Invents Act, or the Leahy-Smith Act, includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted and may also affect patent litigation. The U.S. Patent Office developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, became effective in March 2013. The first to file provisions limit the rights of an inventor who is the first to invent an invention but is not the first to file an application claiming that invention. U.S. and foreign patent applications typically are maintained in confidence for a period of time after they initially are filed with the applicable patent office. Consequently, we cannot be certain that we were the first to invent, or the first to file patent applications on our products or drugproduct candidates or their use. If a third party alsothird-party has an earlier filed a U.S. patent application relating to our drugsproduct or drug candidates, their uses, or a similar invention, we may have to participate in legal or administrative proceedings to determine priority of invention. For applications governed by the Leahy-Smith Act, if a third-party has an earlier filed U.S. patent application relating to our drugs or drugproduct candidates, their uses, or a similar invention, we may be unable to obtain an issued patent from our application.
Due to evolving legal standards relating to the patentability, validity, and enforceability of patents covering pharmaceutical and biotechnological inventions and the scope of claims made under these patents, our ability to obtain, maintain and enforce patents is uncertain and involves complex legal and factual questions.
The issuance of a patent is not conclusive as to its inventorship, scope, validity, or enforceability. Our patents may be challenged by third parties and certain of our patents have been challenged in the past.challenged. This could result in the patent being deemed invalid, unenforceable or narrowed in scope, or the third party may circumvent any such issued patents.patents, including through compulsory licensing mechanisms. Also, our pending patent applications may not issue, and we may not receive any additional patents.
Our patents or patents we license might not contain claims that are sufficiently broad to prevent others from developing competing products. For instance, issued patents, or patents that may issue in the future, (i) relating to our small molecules may be limited to a particular molecule or molecules and may not cover similar molecules that have similar clinical properties, and (ii) relating to cell or genetic therapies may not cover similar technologies that would allow competitors to achieve similar results. Consequently, our competitors may independently develop competing products that do not infringe our patents or other intellectual property. In addition, CRISPR only has non-exclusive or co-exclusive rights to the patent rights that protect the core CRISPR/Cas9 gene-editing technology.
The laws of many foreign jurisdictions do not protect intellectual property rights to the same extent as in the U.S. and many companies in our segment of the pharmaceutical industry have encountered significant difficulties in protecting and defending such rights in foreign jurisdictions. If we encounter such difficulties in protecting or are otherwise precluded from effectively protecting our intellectual property rights in foreign jurisdictions, including through compulsory licensing, our business could be substantially harmed.
Because of the extensive time required for the discovery, development, testing and regulatory review of drugproduct candidates, it is possible that a patent may expire before a drugproduct candidate can be commercialized, or a patent may expire or remain in effect for only a short period following commercialization of such drugproduct candidate. This would result in a minimal or non-existent period of patent exclusivity. If our drugproduct candidates are not commercialized significantly ahead of the expiration date of any applicable patent, or if we have no patent protection on such drugproduct candidates, then, to the extent available we would rely on other forms of exclusivity, such as regulatorydata exclusivity provided by the FDCA and its counterpart agencies in various jurisdictions, and/or orphan drug exclusivity.
Uncertainty over intellectual property in the pharmaceutical and biotechnology industry has been the source of litigation and other disputes which isthat are inherently costly and unpredictable.
There is considerable uncertainty within our industry about the validity, scope, and enforceability of many issued patents in the U.S. and elsewhere in the world, and, to date, the law and practice remains in substantial flux both in the agencies that grant
patents and in the courts. We cannot currently determine the ultimate scope and validity of patents which may be granted to third parties in the future or which patents might be asserted as being infringed by the manufacture, use and sale of our products.
There has been, and we expect that there may continue to be, significant litigation in the pharmaceutical industry regarding patents and other intellectual property rights. Litigation, arbitrations, administrative proceedings, and other legal actions with private parties and governmental authorities concerning patents and other intellectual property rights may be protracted, expensive, and distracting to management. Competitors may sue us as a way of delaying the introduction of our drugsproducts or to remove our drugsproducts from the market. Any litigation, including litigation related to Abbreviated New Drug Applications or ANDA,(“ANDA”), litigation related to 505(b)(2) applications, interference proceedings to determine priority of inventions, derivations proceedings, inter partes review, oppositions to patents in foreign countries, litigation against our collaborators or similar actions, may be costly and time consuming and could harm our business. We expect that litigation may be necessary in some instances to determine the validity and scope of certain of our proprietary rights. Litigation may be necessary in other instances to determine the validity, scope or non-infringement of certain patent rights claimed by third parties to be pertinent to the manufacture, use or sale of our products. Ultimately, the outcome of such litigation could adversely affect the validity and scope of our patent or other proprietary rights, hinder our ability to manufacture and market our products, or result in the assessment of significant monetary damages against us that may exceed amounts, if any, accrued in our consolidated financial statements.
On July 24, 2020, we filed a lawsuit against Sun Pharmaceutical Industries Limited or Sun,(“Sun”) in the U.S. District Court for the District of Delaware alleging infringement of our U.S. Patent No. 10,646,481 or (“the ’481 patent.patent”). The lawsuit follows Vertex’sour receipt of a Notice Letter on June 11, 2020, advising that Sun had submitted an ANDA to the FDA seeking approval to manufacture and market a generic version of the 150 mg tablet of KALYDECO in the U.S. The Notice Letter indicated that Sun submitted a “Paragraph IV” certification to the FDA in which Sun assertedasserts that the ’481 patent is invalid or would not be infringed by Sun’s generic product. The ’481 patent, which expires inon August 13, 2029, was issued on May 12, 2020, and listed in the Orange Book with respect to the KALYDECO 150 mg tabletstablet on June 1, 2020. By letter dated June 5, 2023, Sun notified us that it had amended its ANDA to include a Paragraph IV certification with respect to our U.S. Patent No. 11,564,916 (“the ’916 patent”), which issued on January 31, 2023, is related to the ’481 patent and was listed in the FDA’s Orange Book on February 28, 2023. On June 16, 2023, we filed a lawsuit against Sun in the U.S. District Court for the District of Delaware alleging infringement of the ’916 patent. In December 2023, we settled the case against Sun. The terms of the settlement are confidential.
On July 13, 2021, we filed a lawsuit against Lupin Limited and Lupin Pharmaceuticals, Inc. (collectively, “Lupin”) in the U.S. District Court for the District of Delaware alleging infringement of the ’481 patent. The lawsuit follows our receipt of a Notice Letter on June 2, 2021, advising that Lupin had submitted an ANDA to the FDA seeking approval to manufacture and market a generic version of the 150 mg tablet of KALYDECO in the U.S. The Notice Letter indicated that Lupin submitted a “Paragraph IV” certification to the FDA in which Lupin asserts that the ’481 patent is invalid or would not be infringed by Lupin’s generic product. By letter dated April 25, 2023, Lupin notified us that it had amended its ANDA to include a Paragraph IV certification with respect to the ’916 patent. On May 26, 2023, we filed a lawsuit against Lupin in the U.S. District Court for the District of Delaware alleging infringement of the ’916 patent. In November 2023, we settled the case against Lupin. The terms of the settlement agreement are confidential.
On June 2, 2022, we filed a lawsuit against Aurobindo Pharma Limited (“Aurobindo”) in the U.S. District Court for the District of Delaware alleging infringement of the ’481 patent. The lawsuit follows our receipt of a Notice Letter on April 21, 2022, advising that Aurobindo had submitted an ANDA to the FDA seeking approval to manufacture and market a generic version of the 150 mg tablet of KALYDECO in the U.S. The Notice Letter indicated that Aurobindo submitted a “Paragraph IV” certification to the FDA in which Aurobindo asserts that the ’481 patent is invalid or would not be infringed by Aurobindo’s generic product. By letter dated April 12, 2023, Aurobindo notified us that it had amended its ANDA to include a Paragraph IV certification with respect to the ’916 patent. On May 26, 2023, we filed a lawsuit against Aurobindo in the U.S. District Court for the District of Delaware alleging infringement of the ’916 patent. In November 2023, we settled the case against Aurobindo. The terms of the settlement agreement are confidential.
On July 22, 2022, we filed a lawsuit against Lupin in the U.S. District Court for the District of Delaware alleging infringement of the ’481 patent and U.S. Patent Nos. 8,883,206 (“the ’206 patent”), 10,272,046 (“the ’046 patent”), and 11,147,770 (“the ’770 patent”). The lawsuit follows our receipt of a Notice Letter on June 9, 2022, advising that Lupin had submitted an ANDA to the FDA seeking approval to manufacture and market a generic version of KALYDECO granules in
the U.S. The Notice Letter indicated that Lupin submitted a “Paragraph IV” certification to the FDA in which Lupin asserts that the ’481 patent, the ’206 patent, and the ’046 patent are invalid or would not be infringed by Lupin’s generic product. By letter dated April 25, 2023, Lupin notified us that it had amended its ANDA to include a Paragraph IV certification with respect to the ’916 patent. On May 26, 2023, we filed a lawsuit against Lupin in the U.S. District Court for the District of Delaware alleging infringement of the ’916 patent. On October 11, 2023, U.S. Patent No. 11,752,106 (the “’106 patent”) was listed in the Orange Book as covering KALYDECO granules. We have not yet received notification that Lupin has submitted a “Paragraph IV” certification for the ’106 patent. Other than the ’770 patent, which was listed in the Orange Book on April 14, 2022, Lupin does not appear to challenge our other U.S. patents covering KALYDECO. Vertex intendsKALYDECO granules, the last of which expires on August 5, 2027. Therefore, regardless of the outcome of the litigation, Lupin cannot receive final approval of its ANDA before that date. A scheduling order has been entered by the court and trial is set for April 2025. We intend to vigorously enforce itsour intellectual property rights relating to KALYDECO includinggranules and the ’481, patent.’206, ’046, ’770, ’916, and ’106 patents.
CRISPR has licensed certain rights to a worldwide patent portfolio that covers various aspects of the CRISPR/Cas9 editing platform technology including, for example, compositions of matter and methods of use including their use in targeting or cutting DNA from Dr. Emmanuelle Charpentier, one of the named inventors of this patent portfolio. The patent portfolio also has named inventors who assigned their rights either to the Regents of the University of California or the University of Vienna, to whom we refer, together with Dr. Charpentier, as the CVC Group. For example, in connection with their collaboration, Novartis and Intellia Therapeutics, Inc. have reportedly obtained a license to this patent portfolio in certain fields. Both the CVC Group and Broad have obtained granted patents that purport to cover aspects of CRISPR/Cas9 editing platform technology. Patents and patent applications in this patent portfolio have been the subject of numerous contentious proceedings in the U.S., Europe, and other jurisdictions, including interference proceedings in the USPTO between the CVC Group and (separately) Broad, Sigma-Aldrich and ToolGen. On February 28, 2021, the USPTO issued a decision in Interference No. 106, 115, concluding that Broad Institute in the USPTO. Decisions rendered to date in these proceedings may be subject to appeal. To date, both the CVC and the Broad have obtained granted patents that purport to cover aspectsinvented certain applications of CRISPR/Cas9 editing platform technology. The patents and patent
applications withintechnology in eukaryotic cells before the CVC patent portfolioGroup. The CVC Group has appealed the decision to the U.S. Court of Appeals for the Federal Circuit. If the decision is upheld on appeal (including a potential subsequent appeal to the Supreme Court), Broad would maintain its granted patents directed to those applications CRISPR/Cas9 technology in eukaryotic cells, and the BroadCVC Group’s pending patent portfolio are, or may in the future be, involved in proceedings similarapplications directed to interferences or priority disputes in Europe or other foreign jurisdictions.that subject matter would not proceed to grant. We can give no assurances to the ultimate outcome of these proceedings or the disputedisputes between the CVC Group and Broad.Broad, Sigma-Aldrich and ToolGen. In December 2023, we entered into an agreement with Editas, providing us a non-exclusive sublicense to certain patents relating to CRISPR/Cas9 technology owned by Broad and Harvard, which are licensed to Editas.
In addition to the Broad, other third parties have filed patent applications claiming CRISPR/Cas9-related inventions and may allege that they invented one or more of the inventions claimed by the CVC Group. Thus, the USPTO may, in the future, declare an interference between certain CVC Group patent applications and one or more patent applications. The Broad, as well as other thirdThird parties could seek to assert itstheir patents, if issued, patents against us based on our CRISPR/Cas9-based activities, including commercialization. Defense of these claims, regardless of their merit, could involve substantial litigation expense and could result in a substantial diversion of management and other employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible or require substantial time and monetary expenditure. In that event, we could be unable to further develop and commercialize CTX001CASGEVY or other products that we may develop using the CRISPR/Cas9 technology we license from CRISPR.
To the extent that valid present or future third-party patents or other intellectual property rights cover our drugs, drugproducts, product candidates or technologies, we or our strategic collaborators may seek licenses or other agreements from the holders of such rights in order to avoid or settle legal claims. Such licenses may not be available on acceptable terms, which may hinder our ability to, or prevent us from being able to, manufacture and market our drugs.products. Payments under any licenses that we are able to obtain would reduce our profits derived from the covered products.
We may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that these employees or we have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. Litigation may be necessary to defend against these claims.
In addition, while it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own. Our and their assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to management.
Risks Related To Our Operations
RisksIf we fail to scale our operations to accommodate growth, our business may suffer.
We have expanded and are continuing to expand our global operations and capabilities, which has placed, and will continue to place, significant demands on our management and our operational, research and development and financial infrastructure. To effectively manage our business, we need to continue to adapt as our business grows in scale and complexity across multiple disease states, modalities, and geographies, including by:
•implement and clearly communicating our corporate-wide strategies;
•enhancing our operational and financial infrastructure, including expansion of our controls over data, records and information;
•enhancing our operational, administrative, financial and management processes, including our cross-functional decision-making processes and our budget prioritization systems;
•effectively growing, training and managing our global employee base; and
•expanding our compliance and legal resources.
A variety of risks associated with operating in foreign countries could materially adversely affect our business.
We have expanded our international operations over the past several years in order to market our CF medicines and expand our research and development capabilities. New laws and industry codes in the E.U. and elsewhere have expanded transparency requirements regarding payments and transfers of value to healthcare professionals, requirements surrounding patient-level clinical trial data, the protection of personal data and increased sanctions for violations. Collectively, our expansion and these new requirements are adding to our compliance costs and potentially exposes us to sanctions in the event of an infringement or failure to report in these jurisdictions. In addition, a significant portion of our commercial supply chain, including sourcing of raw materials and manufacturing, is located in China and the E.U. Consequently, we are, and will continue to be, subject to risks related to operating in foreign countries, including risks relating to intellectual property protections and business interruptions. These risks are increased with respect to countries such as China that have substantially different local laws and business practices and weaker protections for intellectual property. Risks associated with operating a global biotechnology company include:
•differing regulatory requirements for drug approvals and regulation of approved drugs in foreign countries;
• varying reimbursement regimes and difficulties or the inability to obtain reimbursement for our products in foreign countries in a timely manner;
•differing patient treatment infrastructures, particularly since our business is focused on the treatment of serious diseases that affect relatively smaller numbers of patients and are typically prescribed by specialist physicians;
•collectability of accounts receivable;
•changes in tariffs, trade barriers, and regulatory requirements, the risks of which appear to have increased in the current political environment;
•economic weakness, including recession and inflation, or political instability in particular foreign economies and markets;
•differing levels of enforcement and/or recognition of contractual and intellectual property rights;
•complying with local laws and regulations, which can change significantly over time;
•foreign taxes, including withholding of payroll taxes;
•foreign currency fluctuations, which could result in reduced revenues or increased operating expenses, and other obligations incident to doing business or operating in another country;
•workforce uncertainty in countries where labor unrest is more common than in the U.S.;
•reliance on third-party vendors, distributors and suppliers;
•import and export licensing requirements, tariffs, and other trade and travel restrictions;
•global or regional public health emergencies that could affect our operations or business;
•production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
•business interruptions resulting from geo-political actions, including war and terrorism.
Our revenues are subject to foreign exchange rate fluctuations due to the global nature of our operations. Although we have foreign currency forward contracts to hedge certain forecasted product revenues denominated in foreign currencies, our efforts to reduce currency exchange losses may not be successful. As a result, currency fluctuations among our reporting currency, the U.S. dollar, and the currencies in which we do business will affect our operating results, often in unpredictable ways.
In addition, our international operations are subject to regulation under U.S. law. For example, the FCPA prohibits U.S. companies and their representatives from offering, promising, authorizing or making payments to foreign officials for the purpose of obtaining or retaining business abroad. In many countries, the health care professionals we regularly interact with may meet the definition of a foreign government official for purposes of the FCPA. We also are subject to import/export control laws. Failure to comply with domestic or foreign laws could result in various adverse consequences, including the possible delay in approval or refusal to approve a product, recalls, seizures, withdrawal of an approved product from the market, the imposition of civil or criminal sanctions, the prosecution of executives overseeing our international operations and corresponding bad publicity and negative perception of our company in foreign countries.
We are subject to risks associated with the global COVID-19 pandemic.
The COVID-19 pandemic has broadly affected the global economy, resulted in significant travel and work restrictions in many regions and has put a significant strain on healthcare resources. COVID-19 has had, and we expect it will continue to have, an impact on our operations, an impact on the operations of our collaborators, third-party contractors and other entities, including governments, governmental agencies and payors, with which we interact, and an impact on the people with CF who take our medicines. To date, the most significant effect on our business operations has been the requirement that a majority of our employees work remotely. We have re-initiated enrollment and dosing in all of our ongoing clinical trials and initiated new clinical trials despite some temporary pauses to enrollment and dosing early in the pandemic.
We continue to monitor local COVID-19 trends and government guidance for each of our site locations and are utilizing a phased, site-specific approach to assess employee access to our sites. Currently, all of our research and manufacturing sites are open to essential employees. There can be no assurance that our sites will remain open, when additional employees will gain access to our sites or whether we will be required to pause enrollment and dosing at clinical trial sites. Any site closure or pause of a clinical trial could harm our operations and delay the development of our product candidates. In addition, even if sites or clinical trials are open for enrollment, COVID-19 may nevertheless impact clinical trial enrollment or participation, for example due to suspension of in-person procedures required for enrollment, government shut-down orders, or decreased patient willingness to participate compared to pre-COVID-19 pandemic levels. COVID-19 may also impact uptake of our medicines generally and patient retention in clinical trials, potentially resulting in higher drop-out rates or missed visits, which may negatively affect the strength of our clinical trial data.
In the future, the economic impacts of the COVID-19 pandemic could affect our business directly or indirectly, including potentially affecting the net prices for our products through changes in our payor mix as a result of increased unemployment in the U.S. or increased pressure on healthcare costs in the U.S. and around the world. The effects on our research, development, manufacturing and commercialization activities, including the virtual launch of KAFTRIO in the E.U., will be dependent on, among other things, the severity and duration of the COVID-19 pandemic and any worsening of the global economic environment as a result thereof, as well as the impact of the pandemic on our third-party manufacturers, suppliers, distributors, subcontractors and customers. While the ultimate impact of COVID-19 on our business is highly uncertain, any negative impacts that materialize could materially adversely affect our operations, financial performance and stock price. Any negative impacts of COVID-19, alone or in combination with others, could exacerbate other risk factors discussed herein. The full extent to which the COVID-19 pandemic will negatively affect our operations, financial performance and stock price will depend on future developments that are highly uncertain and cannot be predicted, including the scope and duration of the pandemic and actions taken by governmental authorities and other third parties in response to the pandemic.
If we fail to manage our operations effectively, our business may suffer.
We have expanded and are continuing to expand our global operations and capabilities, which has placed, and will continue to place, significant demands on our management and our operational, research and development and financial infrastructure. To effectively manage our business, we need to:
•implement and clearly communicate our corporate-wide strategies;
•enhance our operational and financial infrastructure, including our controls over records and information;
•enhance our operational, financial and management processes, including our cross-functional decision-making processes and our budget prioritization systems;
•train and manage our global employee base; and
•enhance our compliance and legal resources.
If we fail to attract and retain skilled employees, our business could be materially harmed.
Due to the highly technical nature of our drug discovery and development activities, we require the services of highly qualified and trained scientists who have the skills necessary to conduct these activities. In addition, we need to attract and retain employees with experience in development, marketing and commercialization of medicines.medicines and therapies, including cell and genetic therapies. We have entered into employment agreements with some executives and provide stock-related compensation benefits to all of our key employees that vest over time and therefore induce them to remain with us.us and have entered into employment agreements with some executives. However, the employment agreements can be terminated by the executive on relatively short notice. The value to employees of stock-related benefits that vest over time - such as restricted stock units and stock options - can be significantly affected by movements in our stock price and business performance, and may, at any point in time, be insufficient to counteract more lucrative offers from other companies. We face intense competition for our personnel from our competitors and other companies throughout our industry, especially with respect to employees with expertise in cell or genetic therapies. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. Moreover, the growth of local biotechnology companies and the expansion of major pharmaceutical companies into the Boston area has increased competition for the available pool of skilled employees, especially in technical fields. The high cost of living can make it difficult to attract employees from other parts of the country to our Massachusetts headquarters.global headquarters in Boston and our international headquarters in London. Current job market dynamics, caused in part by the effects of COVID-19 and other macro-level events, with many employers unable to fill existing openings at all levels of their organizations, could result in significant increases to our costs to recruit and retain employees. Challenges could adversely affect our operations and financial results if we do not have sufficient staff to perform necessary functions. In addition, the
available pool of skilled employees would be further reduced if immigration laws change in a manner that increases restrictions on immigration. Our ability to continue to commercialize our products and achieve our
research and development objectives depends on our ability to respond effectively to these demands. If we are unable to hire and retain qualified personnel, there could be a material adverse effect on our business.
OurA breakdown or breach of our information technology systems could subject us to liability or interrupt the operation of our business.
We maintain and rely extensively on information technology systems and network infrastructures for the effective operation of our business. In the course of our business, faces potentialwe collect, store, and transmit confidential information (including personal information and intellectual property), and it is critical that we do so in a secure manner to maintain the confidentiality, integrity, and availability of such confidential information. A disruption, infiltration, or failure of our information technology systems or any of our data centers as a result of software or hardware malfunctions, computer viruses, cyber-attacks, employee theft or misuse, power disruptions, natural disasters, floods or accidents could cause breaches of data security and loss of critical data, which in turn could materially adversely affect our business and subject us to both private and governmental causes of action. While we have implemented security measures to minimize these risks relating to our data and information technology systems and have adopted a business continuity plan to deal with a disruption to our information technology systems, there can be no assurance that our efforts to protect our data and information systems will prevent breakdowns or breaches in our systems that could adversely affect our business. In addition, we maintain cyber liability insurance, however, this insurance may not be sufficient to cover the United Kingdom’s withdrawalfinancial, legal, business or reputational losses that may result from an interruption or breach of our systems and those of critical third parties.
Cyber-attacks are increasing in their frequency, sophistication, and intensity, and are becoming increasingly difficult to detect. They are often carried out by well-resourced and skilled parties, including nation states, organized crime groups, “hacktivists” and employees or contractors acting carelessly or with malicious intent. Cyber-attacks include deployment of harmful malware and key loggers, ransomware, denial-of-service attacks, malicious websites, the European Union.
Our European headquartersuse of social engineering, and European research facility are locatedother means to affect the confidentiality, integrity and availability of our technology systems and data. Cyber-attacks also include manufacturing, hardware or software supply chain attacks, which could cause a delay in the U.K.,manufacturing of products or products produced for contract manufacturing or lead to a data privacy or security breach. Our key business partners face similar risks, and aany security breach of their systems could adversely affect our security. In addition, our increased use of cloud technologies heightens these third party and other operational risks, and any failure by cloud or other technology service providers to adequately safeguard their systems and prevent cyber-attacks could disrupt our operations and result in misappropriation, corruption, or loss of confidential or propriety information. A significant portion of our ex-U.S. net product revenues are derived from sales inworkforce continues to leverage hybrid work. Risk of cyber-attack is increased with employees working remotely. Remote work increases the U.K. On January 31, 2020, the U.K. formally withdrew from the E.U., also knownrisk we may be vulnerable to cybersecurity-related events such as Brexit. The U.K.phishing attacks and the E.U. negotiated a detailed post-Brexit Trade and Cooperating Agreement which went into effect on January 1, 2021. As of January 1, 2021, E.U. Treaties, E.U. free movement rights and the general principals of E.U. law no longer apply in relation to the U.K. By virtue of the E.U. (Withdrawal) Act 2018, E.U. relations will continue to apply in U.K. domestic law to the extent that they are not modified of revoked by regulations under that Act. Brexit could lead to legal uncertainty and potentially divergent national laws and regulations as the U.K. determines which E.U. laws to replace or replicate. Given the lack of comparable precedent, it is unclear what financial, trade, regulatory and legal implications the withdrawal of the U.K. from the E.U. would have and how such withdrawal would affect us. Any of these effects of Brexit, among others, could adversely affect our business, financial condition and operating results.other security threats.
Our business has a substantial risk of product liability claims and other litigation liability.
We are or may be involved in various legal proceedings, including securities/shareholder matters and claims related to product liability, intellectual property, employment law, competition law, data privacy, and breach of contract. Such proceedings may involve claims for, or the possibility of, damages or fines and penalties involving substantial amounts of money or other relief, including but not limited to civil or criminal fines and penalties. If any of these legal proceedings were to result in an adverse outcome, it could have a material adverse effect on our business.
With respect to product liability and clinical trial risks, in the ordinary course of business we are subject to liability claims and lawsuits, including potential class actions, alleging that our products or drugproduct candidates have caused, or could cause, serious adverse events or other injury. We have product liability insurance and clinical trial insurance in amounts that we believe are adequate to cover this risk. However, our insurance may not provide adequate coverage against all potential liabilities. If a claim is brought against us, we might be required to pay legal and other expenses to defend the claim, as well as pay uncovered damage awards resulting from a claim brought successfully against us and these damages could be significant and have a material adverse effect on our financial condition. Furthermore, whether or not we are ultimately successful in defending any such claims, we might be required to direct significant financial and managerial resources to such defense and adverse publicity is likely to result.
A breakdown or breach of our information technology systems could subject us to liability or interrupt the operation of our business.
We maintain and rely extensively on information technology systems and network infrastructures for the effective operation of our business. In the course of our business, we collect, store and transmit confidential information (including personal information and intellectual property), and it is critical that we do so in a secure manner to maintain the confidentiality and integrity of such confidential information. The size and complexity of our information technology and information security systems makes such systems potentially vulnerable to service interruptions and to security breaches. A disruption, infiltration or failure of our information technology systems or any of our data centers as a result of software or hardware malfunctions, computer viruses, cyber-attacks, employee theft or misuse, power disruptions, natural disasters, floods or accidents could cause breaches of data security and loss of critical data, which in turn could materially adversely affect our business and subject us to both private and governmental causes of action. While we have implemented security measures in an attempt to minimize these risks to our data and information technology systems and have adopted a business continuity plan to deal with a disruption to our information technology systems, cyber-attacks are increasing in their frequency, sophistication and intensity, and have become increasingly difficult to detect. There can be no assurance that our efforts to protect our data and information systems will prevent breakdowns or breaches in our systems that could adversely affect our business. In addition, our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyber-attacks or other related liabilities.
Risk of cyber-attack is increased with the majority of employees working remotely during the ongoing COVID-19 pandemic. During this time, there is an increased risk that we may be vulnerable to cybersecurity-related events such as phishing attacks and other security threats as a result of our employees, third party vendors and collaborators working remotely from non-corporate managed networks.
If our facilities were to experience a catastrophic loss, our operations would be seriously harmed.
Most of our operations, including our research and development activities, are conducted in a limited number of facilities. If any of our major facilities were to experience a catastrophic loss, due to an earthquake, severe storms, fire or similar event, our operations could be seriously harmed. For example, our corporate headquarters, as well as additional leased space that we use for certain logistical and laboratory operations and manufacturing, are located in a flood zone along the Massachusetts coast. We have adopted a business continuity planplans to address most crises. However, if we are unable to fully implement our business continuity plans, we may experience delays in recovery of data and/or an inability to perform vital corporate functions, which could result in a significant disruption in our research, development, manufacturing and/or commercial activities, large expenses to repair or replace the facility and/or the loss of critical data, which could have a material adverse effect on our business.
The use of social media platforms and artificial intelligence tools presents risks and challenges.
Social media is being used by third parties to communicate about our products and drugproduct candidates and the diseases our therapies are designed to treat. We believe that members of the CF communitycommunities supporting serious diseases may be more active on social media asas compared to other patient populations due to the demographics of thisthose patient population.populations. Social media practices in the pharmaceutical and biotechnology industries are evolving, which creates uncertainty and risk of noncompliance with regulations applicable to our business. For example, patients may use social media platforms to comment on the effectiveness of, or adverse experiences with, a drugproduct or a drugproduct candidate, which could result in reporting obligations. In addition, our employees may engage on social media in ways that may not comply with legal or regulatory requirements, which may give rise to liability, lead to the loss of trade secrets and other intellectual property, or result in public disclosure of protected personal information. There is a risk of inappropriate disclosure of sensitive information or negative or inaccurate posts or comments about us on any social networking website. Negative sentiment about us or our business shared over social media, or misinformation disseminated from fraudulent accounts impersonating our employees or our business, or otherwise, could harm our business and reputation, whether or not it is based in fact. Certain data protection regulations, such as the GDPR, apply to personal data contained on social media. If any of these events were to occur or we otherwise fail to comply with applicable regulations, we could incur liability, face regulatory actions or incur harm to our business, including damage to our reputation. Similar risks relating to inappropriate disclosure of sensitive information or inaccurate information appearing in the public domain may also apply from our employees engaging with and use of new artificial intelligence tools, such as ChatGPT.
Risks Related to Financial Results and Holding Our Common Stock
Our stock price may fluctuate.
Market prices for securities of companies such as ours are highly volatile. From January 1, 20202023 to December 31, 2020,2023, our common stock traded between $197.47$282.21 and $306.08$413.00 per share. The market for our stock, like that of other companies in the biotechnology industry, has experienced significant price and volume fluctuations. The future market price of our securities could be significantly and adversely affected by factors such as:
•the information contained in our quarterly earnings releases, including updates regarding our commercialized products or our product candidates, our net product revenues and operating expenses for completed periods and financial guidance regarding future periods;
•announcements of FDA actions with respect to our therapies or those of our competitors, or regulatory filings for our therapies or those of our competitors, or announcements of interim or final results of clinical trials or nonclinical studies relating to our therapies or those of our competitors;
•announcements we make or commentary by public equity analysts with respect to clinical development of the product candidates in our pain program;
•developments in domestic and international governmental policy or regulation, for example, relating to drug pricing or intellectual property rights;and tax reform;
•technological innovations or the introduction of new drugs by our competitors;
•government regulatory action;
•public concern as to the safety of drugs developed by us or our competitors;
•developments in patent or other intellectual property rights or announcements relating to these matters;
•information disclosed by third parties regarding our business or products;
•developments relating specifically to other companies and market conditions for pharmaceutical and biotechnology stocks or stocks in general;
• business development, capital structuring or financing activities; and
•general worldwide or national economic, political and capital market conditions, including as a result of the ongoing COVID-19 pandemic.inflation and rapid fluctuations in interest rates.
Following periods of volatility in the market price of a company’s securities, stockholder derivative lawsuits and securities class action litigation are common. Such litigation, if instituted against us or our officers and directors, could result in substantial costs and a diversion of management’s attention and resources.
Our effective tax rate fluctuates, and changes in tax laws, regulations and treaties, unfavorable resolution to the tax positions we have taken or exposure to additional income tax liabilities could have a material impact on our future taxable income.
Our effective tax rate is derived from a combination of applicable tax rates in the various places that we operate globally. Our effective tax rate may be different than experienced in the past due to numerous factors, including changes in the mix of our profitability from country to country, tax authority examinations/audits of our tax filings, adjustments to the value of our uncertain tax positions, changes in accounting for income taxes, and changes in tax laws or modifications of treaties in various jurisdictions. Any of these factors could cause us to experience an effective tax rate that is significantly different from previous periods or our current expectations.
On December 12, 2022, E.U. member states reached an agreement to implement the minimum tax component (“Pillar Two”) of the Organization for Economic Co-operation and Development’s (the “OECD’s”), global international tax reform initiative with effective dates of January 1, 2024 and 2025. On July 17, 2023, the OECD published Administrative Guidance proposing certain safe harbors that effectively extend certain effective dates to January 1, 2027. E.U. member states need to adopt this Administrative Guidance in their local Pillar Two legislation for such safe harbors to apply. In addition, the U.K. has independently released draft legislation to introduce the OECD’s Pillar Two reforms into U.K. law. We are continuing to evaluate the potential impact on future periods of the Pillar Two guidance, pending legislative adoption by individual countries, including those in which we do business.
We are subject to ongoing tax audits in various jurisdictions, and local tax authorities may disagree with certain positions we have taken and assess additional taxes. We regularly assess the probable outcomes of these audits to determine the appropriateness of our tax provision, and we have established contingency reserves for material tax exposures. However, there can be no assurance that we will accurately predict the outcomes of these disputes or other tax audits or that issues raised by tax authorities will be resolved at a financial cost that does not exceed our related reserves and the actual outcomes of these disputes and other tax audits could have a material impact on our results of operations or financial condition.
Our quarterly operating results are subject to significant fluctuation.
Our operating results have fluctuated from quarter to quarter in the past, and we expect that they will continue to do so in the future. Our revenues are primarily dependent on the levelamount of net product revenues from sales of our CF medicines. Our total net product revenues could vary on a quarterly basis based on, among other factors, the timing of orders from our significant customers. Additional factors that have caused quarterly fluctuations to our operating results in recent years include variable amounts of revenues,revenues; expenses related to businessresulting from our significant investments in research and development, activities,acquired in-process research and development, and commercialization activities; changes in the fair value of our strategic investments, impairment charges,derivative instruments and contingent consideration liabilities; charges for excess and obsolete inventories, changes in the fair value of derivative instrumentsinterest income, interest expenses; and the consolidation or deconsolidation of variable interest entities.our provision for income taxes. Our revenues also are subject to foreign exchange rate fluctuations due to the global nature of our operations. Although we have foreign currency forward contracts to hedge forecasted product revenues denominated in foreign currencies, our efforts to reduce currency exchange losses may not be successful. As a result, currency fluctuations among our reporting currency, the U.S. dollar, and the currencies in which we
do business may affect our operating results, often in unpredictable ways. Our quarterly results also could be materially affected by significant charges, which may or may not be similar to charges we have experienced in the past. Most of our operating expenses relate to our research and development activities, do not vary directly with the amount of revenues and are difficult to adjust in the short term. As a result, if revenues in a particular quarter are below expectations, we are unlikely to reduce operating expenses proportionately for that quarter. These examples are only illustrative and other risks, including those discussed in these “Risk Factors,” could also cause fluctuations in our reported financial results. Our operating results during any one period do not necessarily suggest the results of future periods.
We expect that results from our clinical development activities and the clinical development activities of our competitors will continue to be released periodically, and may result in significant volatility in the price of our common stock.
Any new information regarding our products and drugproduct candidates, or competitive products or potentially competitive drugproduct candidates, can substantially affect investors’ perceptions regarding our future prospects. We, our collaborators, and our competitors periodically provide updates regarding drug and therapy development programs, typically through press releases, conference calls and presentations at medical conferences. These periodic updates often include interim or final results from clinical trials conducted by us or our competitors and/or information about our or our competitors’ expectations regarding regulatory filings and submissions as well as future clinical development of our products or drugproduct candidates, competitive products or potentially competitive drugproduct candidates. The timing of the release of information by us regarding our drug and therapy development programs is often beyond our control and is influenced by the timing of receipt of data from our clinical trials and by the general preference among pharmaceutical companies to disclose clinical data during medical conferences. In addition, the information disclosed about our clinical trials, or our competitors’ clinical trials, may be based on interim rather than final data that may involve interpretation difficulties and may in any event not accurately predict final results. The release of such information may result in volatility in the price of our common stock.
Changes in tax laws, regulations and treaties could affect our future taxable income.
We are subject to taxation in numerous countries, states and other jurisdictions. As a result, our effective tax rate is derived from a combination of applicable tax rates in the various places that we operate globally. Our effective tax rate may be different than experienced in the past due to numerous factors, including changes in the mix of our profitability from country to country, the results of tax authority examinations/audits of our tax filings, adjustments to the value of our uncertain tax positions, changes in accounting for income taxes and changes in tax laws or modifications of treaties in various jurisdictions. For example, changes to the U.S. tax code are anticipated under the new administration. Any of these factors could cause us to experience an effective tax rate that is significantly different from previous periods or our current expectations.
We continue to assess the impact of various tax reform proposals and modifications to existing tax treaties in all jurisdictions where we have operations to determine the potential effect on our business and any assumptions we have made about our future taxable income. We cannot predict whether any specific proposals will be enacted, the terms of any such proposals or what effect, if any, such proposals would have on our business if they were to be enacted.
Recommendations from the Organization for Economic Co-operation and Development that are part of the base erosion and profit shifting, or BEPS, framework could result in changes in tax laws in countries where we do business and adversely affect our provision for income taxes and our current rate. If these recommendations (or other changes in law) were adopted by the countries in which we do business, it could adversely affect our provision for income tax and our current rate.
General Risk Factors
We may need to raise additional capital that may not be available.
We may need to raise additional capital in the future. Any potential public offering, private placement or debt financing may or may not be similar to the transactions that we entered into in the past. Any debt financing may be on terms that, among other things, include conversion features that could result in dilution to our then-existing security holders and restrict our ability to pay interest and dividends—although we do not intend to pay dividends for the foreseeable future. Any equity financings would result in dilution to our then-existing security holders. If adequate funds are not available on acceptable terms, or at all, we may be required to curtail significantly or discontinue one or more of our research, drug discovery or development programs, including clinical trials, incur significant cash exit costs, or attempt to obtain funds through arrangements with collaborators or others that may require us to relinquish rights to certain of our technologies, drugs or drug candidates. Based on many factors, including general economic conditions, additional financing may not be available on acceptable terms, if at all.
Future indebtedness could materially and adversely affect our financial condition, and the terms of our credit agreements impose restrictions on our business, reducing our operational flexibility and creating default risks.
In 2019,July 2022, we entered into a credit agreement providing for a $500$500.0 million revolving facility.credit facility and terminated an existing $500.0 million credit agreement entered into in 2019. In September 2020, we2022, our $2.0 billion credit agreement that was entered into a secondin 2020 expired in accordance with its terms. Subject to certain conditions, our current credit agreement providing for a $2.0 billion revolving facility. Each of the credit agreements provides that subject to the satisfaction of certain conditions, we may request the borrowing capacity be increased by an additional $500.0 million.million for a total of $1.0 billion. If we borrow under our current credit agreementsagreement or any future credit agreement,agreements, such indebtedness could have important consequences to our business, including increasing our vulnerability to general adverse financial, business, economic and industry conditions, as well as other factors that are beyond our control. The credit agreements requireagreement requires that we comply with certain financial covenants, including (i) a consolidated leverage ratio covenant and (ii) a consolidated interest coverage ratio covenant, in each case to be measured on a quarterly basis.covenant. Further, the credit agreements includeagreement includes negative covenants, subject to exceptions, restricting or limiting our ability and the ability of our subsidiaries to, among other things, incur additional indebtedness, grant liens, engage in certain investment, acquisition and disposition transactions, pay dividends, repurchase capital stock and enter into transactions with affiliates. As a result, we may be restricted from engaging in business activities that may otherwise improve our business. Failure to comply with the covenants could result in an event of default that could trigger acceleration of our indebtedness, which would require us to repay all amounts owingowed under the credit agreements and/or our finance leases and could have a material adverse effect on our business. Additionally, our obligations under the credit agreementsagreement are unconditionally guaranteed by certain of our domestic subsidiaries. If we incur additional indebtedness, the risks related to our business and our ability to service or repay our indebtedness would increase.
Issuances of additional shares of our common stock could cause the price of our common stock to decline.
As of December 31, 2020,2023, we had 259.9257.7 million shares of common stock issued and outstanding. As of December 31, 2020,2023, we also had 3.0 million unvested restricted stock units (“RSUs”), 1.2 million unvested performance stock units (“PSUs”), and outstanding options to purchase 4.21.9 million shares of common stock with a weighted-average exercise price of $140.47$151.37 per share.
The majority of our unvested RSUs are likely to vest based on our employees’ continued employment. The number of PSUs that vest is dependent on a potential range of shares issuable pursuant to certain financial and non-financial milestones, and our employees’ continued employment. Outstanding vested options are likely to be exercised if the market price of our common stock exceeds the applicable exercise price, and, inprice. In the future, we expect to issue a limited number of additional equity awards options
to directors and employees. our directors.
In addition, we may issue additional common stock or restricted securities in the future as part of financing activities or business development activities and any such issuances may have a dilutive effect on our then-existing shareholders. Sales of substantial amounts of our common stock in the open market, or the availability of such shares for sale, could adversely affect the price of our common stock. The issuance of restricted common stock or common stock upon exercise of any outstanding options would be dilutive, and may cause the market price for a share of our common stock to decline.
There can be no assurance that we will repurchase shares of common stock or that we will repurchase shares at favorable prices.
In November 2020,February 2023, our Board of Directors authorizedapproved a share repurchase program (the “Share Repurchase Program”) pursuant to which we are authorized to repurchase up to $500 million$3.0 billion of our common stock by December 31, 2022. As of December 31, 2020, we had repurchased $75.1 million of common stock under the share repurchase program and had remaining available $424.9 millionfrom time to repurchase additional shares pursuant to this program.
time through open market or privately negotiated transactions. Our stock repurchases will depend upon, among other factors, market conditions, our cash balances and potential future capital requirements, results of operations, financial condition, and other factors that we may deem relevant. We can provide no assurance that we will repurchase stock at favorable prices, if at all.
We have adopted anti-takeover provisions in our articles of organization and by-laws and are subject to Massachusetts corporate laws that may frustrate any attempt to remove or replace members of our current managementboard or to effectuate acertain types of business combinationcombinations involving Vertex.us.
Our corporate charter and by-law provisionsProvisions of our articles of organization, by-laws and Massachusetts state laws may frustrate any attempt to remove or replace members of our current Board of Directors and may discourage certain types of transactionsbusiness combinations involving an actual or potential change of control of Vertex that might be beneficial to us or our security holders.us. Our by-laws grantallow the directors a rightBoard of Directors to adjourn annualany meetings of shareholders and certain provisions of our by-laws may be amended only with an 80% shareholder vote.prior to the time the meeting has been convened. We may issue shares of any class or series of preferred stock in the future without shareholder approval and upon such terms as our Board of Directors may determine. The rights of the holders of common stock will be subject to, and may be adversely affected by, the rights of the holders of any class or series of preferred stock that may be issued in the future. Massachusetts state law also prohibits us from engaging in specified business combinations with an interested stockholder, subject to certain exceptions, unless the combination is approved or consummated in a prescribed manner, and prohibitsplaces restrictions on voting by any shareholder who acquires 20% or more of ourthe aggregate shareholder voting stockpower without shareholder approval.approval by non-interested shareholders. As a result, shareholders or other parties may find it more difficult to remove or replace our current management.directors or to effectuate certain types of business combinations involving us.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K, including the descriptions of our Business set forth in Part I, Item 1, our Risk Factors set forth in Part I, Item 1A, and our Management’s Discussion and Analysis of Financial Condition and Results of Operations set forth in Part II, Item 7, contains forward-looking statements. Forward-looking statements are not purely historical and may be accompanied by words such as “anticipates,” “may,” “forecasts,” “expects,” “intends,” “plans,” “potentially,” “believes,” “seeks,” “estimates,” and other words and terms of similar meaning. Such statements may relate to:
•our expectations regarding the amount of, timing of, and trends with respect to our financial performance, including revenues, costs and expenses, and other gains and losses;
•our expectations regarding clinical trials, including expectations for patient enrollment, development timelines, the expected timing of data from our ongoing and planned clinical trials, and regulatory authority filings and other submissions for our therapies;
•our ability to maintain and obtain adequate reimbursement for our medicines in ex-U.S. marketsproducts and product candidates, our ability to launch, commercialize and market our medicinesproducts or any of our other therapies for which we obtain regulatory approval;approval, including CASGEVY, and our ability to obtain label expansions for existing therapies;
•our expectations regarding our ability to continue to grow our CF business by increasing the number of people with CF eligible and able to receive our medicines and providing improved treatment options for people who are already eligible for one of our medicines;
•the data that will be generated by ongoing and planned clinical trials and the ability to use that data to advance compounds, continue development or support regulatory filings;filings, including from the multiple ascending dose portion of the Phase 1/2 clinical trial of VX-522, the durable efficacy and effectiveness of CASGEVY as one-time functional cure for people with SCD and TDT, and the benefit risk profile supporting VX-548 as a transformative option for acute pain as compared to existing agents, and that our triple combinations of vanzacaftor/tezacaftor/deutivacaftor will provide additional clinical benefits to people with CF who have at least one mutation in their CFTR;
•our beliefs and plans with respect to the potential near-term launch of our triple combinations of vanzacaftor/tezacaftor/deutivacaftor for treatment of CF and for VX-548 for the treatment of acute pain;
•our beliefs regarding the support provided by clinical trials and preclinical and nonclinical studies of our therapies for further investigation, clinical trials or potential use as a treatment;
•our plans to continue investing in our research and development programs, including anticipated timelines for our programs, and our strategy to develop our pipeline programs, alone or with third party-collaborators;
•our beliefs regarding the approximate patient populations for each of ourthe disease areas;areas on which we focus;
•the potential benefits and therapeutic scope of our acquisitions and collaborations;collaborations, including our acquisition of ViaCyte and its potential to accelerate development of our stem-cell based T1D programs, and our collaboration with CRISPR for their gene-editing technology to accelerate the development of our hypoimmune cell therapies for T1D;
•potential business development activities, including the identification of potential collaborative partners or acquisition targets;
•the establishment, development and maintenance of collaborative relationships, including potential milestone payments or other obligations;
52•our ability to expand and protect our intellectual property portfolio and otherwise maintain exclusive rights to products;
• potential business development activities, including the identification of potential collaborative partners or acquisition targets;
• potential fluctuations in foreign currency exchange rates;rates and the effectiveness of our foreign currency management program;
•our expectations regarding the amount of cash to generated by operations, our cash balance and expected generation and interest income;
•our expectations regarding our provision for or benefit from income taxes and the utilization of our deferred tax assets, including the impact of the Coronavirus Aid, Relief and Economic Security Act;assets;
•our ability to use our research programs to identify and develop new drugproduct candidates to address serious diseases and significant unmet medical needs;
•the effectiveness of our governance, plans and strategy with respect to managing cybersecurity risks and other threats to our information technology systems;
•our plans to expand, strengthen, and invest in our global supply chains and manufacturing infrastructure and capabilities;capabilities, including for cell and gene therapies;
•our ability to attract and retain skilled personnel;
•our expectations regardinginvolving governmental cost containment and other regulatory efforts;
•our expectations surrounding the effect of the COVID-19 pandemic on, among other things,competitive landscape facing our financial performance, liquidity, businessproducts and operations, including manufacturing, supply chain, researchproduct candidates; and development activities and pipeline programs; and
•our liquidity and our expectations regarding the possibility of raising additional capital.
Forward-looking statements are subject to certain risks, uncertainties, or other factors that are difficult to predict and could cause actual events or results to differ materially from those indicated in any such statements. These risks, uncertainties, and other factors include, but are not limited to, those described in our Risk Factors, set forth in Part I, Item 1A, and elsewhere in this report and those described from time to time in our future reports filed with the Securities and Exchange Commission.
Any such forward-looking statements are made on the basis of our views and assumptions as of the date of the filing and are not estimates of future performance. Except as required by law, we undertake no obligation to publicly update any forward-looking statements. The reader is cautioned not to place undue reliance on any such statements.
ITEM 1B.UNRESOLVED STAFF COMMENTS
We did not receive any written comments from the Securities and Exchange Commission prior to the date 180 days before the end of the fiscal year ended December 31, 20202023 regarding our filings under the Securities Exchange Act of 1934, as amended, that have not been resolved.
ITEM 1C.CYBERSECURITY
Risk Management and Strategy
We recognize the critical importance of developing, implementing, and maintaining robust cybersecurity measures to maintain the security, confidentiality, integrity, and availability of our business systems and confidential information, including personal information and intellectual property. Our cybersecurity program includes systems and processes for assessing, identifying and managing material risks from cybersecurity threats and include maintenance and monitoring of information security policies aligned with global regulatory controls; user and employee awareness of cyber policies and practices; information systems configuration management; third-party risk management systems; identity and information asset protection; infrastructure security systems; and cyber threat operations with continuous monitoring and threat hunting. This program includes processes to oversee and identify material risks from cybersecurity threats associated with our use of third-party service providers. We also engage a range of third-party experts in connection with various development, implementation, and maintenance activities related to our cybersecurity program.
Our cybersecurity program is integrated into our overall risk management systems, including our annual enterprise risk management program, internal audit program, business continuity and crisis management programs, third-party risk management program, insurance risk management program, and employee compliance programs. As part of our overall risk management program, we maintain a global insurance portfolio with comprehensive cyber coverage. Our Chief Information Security Officer (“CISO”) and the Information Security function advises, consults with, or provides input to each of these programs to ensure that material risks from cybersecurity threats are appropriately assessed, identified, and managed.
As of the date of this report, there have been no cybersecurity threats that have materially affected or are reasonably likely to materially affect our business, operations, or financial condition.
Governance
While our board of directors has oversight responsibility for risk management generally, the Audit and Finance Committee (“Audit Committee”) is specifically responsible for overseeing our cybersecurity risk management program to ensure that cybersecurity risks are identified, assessed, managed, and monitored. Our CISO provides periodic updates to the Audit Committee in this regard, and covers the state of our cybersecurity program, supported by key performance indicators across the range of cybersecurity functions related to risk management and governance, identity and information asset protection, core security and endpoint security, and cyber threat operations. These updates include descriptions of cybersecurity incidents of interest, including those associated with our third-party service providers; the board will be informed promptly of material risks from cybersecurity threats.
We strive to create a culture of cybersecurity resilience and awareness and believe that cybersecurity is the responsibility of every employee and contractor. At the same time, primary responsibility for assessing, monitoring, and managing our cybersecurity risks lies with our CISO, Michael Daly. Mr. Daly has more than 35 years of experience in security and information systems and spent 25 years with Raytheon Technologies, most recently as Chief Technology Officer of Cybersecurity, Special Missions, Training & Services. Mr. Daly supported the U.S. President's National Security Telecommunications Advisory Committee for more than 20 years, is a member of the Massachusetts Cybersecurity Strategy Council, and is Chair of the Kogod Cybersecurity Governance Center at American University. Formerly, he served on the Rhode Island Homeland Security Advisory Board and was a member of various commercial cyber product councils.
Mr. Daly oversees a team of skilled cybersecurity professionals who have Certified Information Systems Security Professional (“CISSP”) credentials, Global Information Assurance Certification from the SANS Institute, and other security and network certifications. The cybersecurity team monitors and evaluates our cybersecurity posture and performance on an ongoing basis, including through regular vulnerability scans, penetration tests, and threat intelligence feeds. The cybersecurity team uses various tools and methodologies to manage cybersecurity risk that are tested on a regular cadence,
and assesses and evaluates cybersecurity incidents, escalating certain cybersecurity incidents to Mr. Daly according to protocol. Mr. Daly is continually informed regarding the performance of the cybersecurity program, as well as the latest developments in cybersecurity, including potential threats and innovative risk management techniques.
ITEM 2.PROPERTIES
Corporate Headquarters
We lease approximately 1.1 million square feet of office and laboratory space at our corporate headquarters in Boston, Massachusetts in two buildings pursuant to two leases that we entered into in May 2011. These leases commenced in December 2013 and will extend until December 2028. We have an option to extend the term of the leases for an additional ten years.
Additional United States and Worldwide Locations
In addition to our corporate headquarters, we lease an aggregate of approximately 678,000840,000 square feet of space globally. This space includes logistical, laboratory, commercial and manufacturing operations, as well as laboratory and office space to support our research and development organizations. We also own approximately 213,000 square feet at our continuous manufacturing facility in Massachusetts.
ITEM 3.LEGAL PROCEEDINGS
We are not currently subject to any material legal proceedings.
ITEM 4.MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5.MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is traded on The Nasdaq Global Select Market under the symbol “VRTX.”
Shareholders
As of January 31, 2021,February 9, 2024, there were 115106 holders of record of our common stock.
Performance Graph
Our performance graph includes the NASDAQ Biotechnology Index, which we believe is a comparable index consisting of companies with similar industry classifications, and which we plan to use in our future performance graphs.
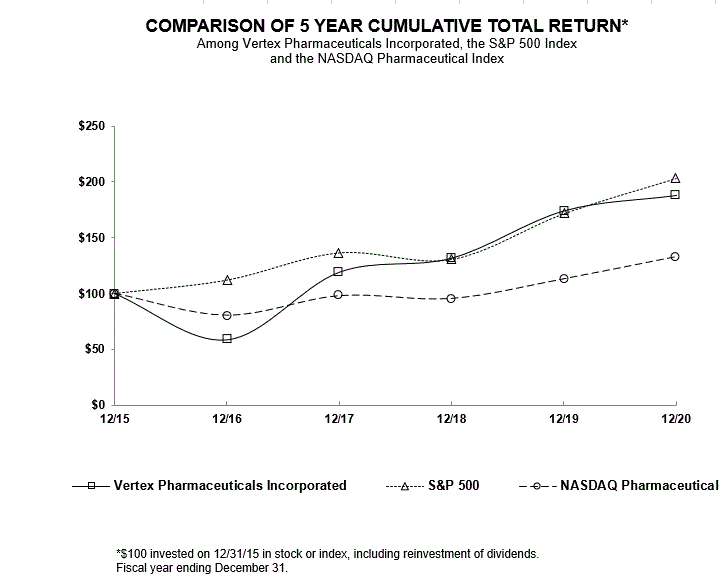
Dividends
We currently expect thathave never paid any future earnings will be retained for use in our business. Any future determination to declare cash dividends will be subject to the discretion of our board of directors and applicable law and will depend on various factors, including our results of operations, financial condition, prospects and any other factors deemed relevant by our board of directors. In addition, our credit agreement limits our ability to pay cash dividends on our common stock.
stock, and we do not anticipate paying any in the foreseeable future.Issuer Repurchases of Equity Securities
In July 2019,February 2023, our Board of Directors approved a share repurchase program (the “2019 Share“Share Repurchase Program”), pursuant to which we were authorized to repurchase up to $500.0 million of our common stock between August 1, 2019 and December 31, 2020. As of December 31, 2020, we had repurchased the entire $500.0 million of common stock that was authorized under the 2019 Share Repurchase Program.
In November 2020, our Board of Directors approved a new share repurchase program (the “2020 Share Repurchase Program”), pursuant to which we are authorized to repurchase up to $500.0 million$3.0 billion of our common stock by December 31, 2022. As of December 31, 2020, there was a total of $424.9 million remaining for repurchases under the 2020stock. Our Share Repurchase Program.
Program does not have an expiration date and can be discontinued at any time. The table set forth below shows repurchases of securities by us during the three months ended December 31, 20202023 under our 2019 Share Repurchase Program and our 2020 Share Repurchase Program.
| | | | | | | | | | | | | | |
| Period | Total Number
of Shares Purchased | Average Price
Paid per Share | Total Number of Shares
Purchased as Part of
Publicly Announced
Plans or Programs (3) | Approximate dollar value of
Shares that May Yet be Purchased Under
the Plans or Programs (3) |
| Oct. 1, 2020 to Oct. 31, 2020 (1) | 252,375 | | $ | 221.90 | | 252,375 | | $ | — | |
| Nov. 1, 2020 to Nov. 30, 2020 (2) | 345,897 | | $ | 217.08 | | 345,897 | | $ | 424,912,410 | |
| Dec. 1, 2020 to Dec. 31, 2020 (2) | — | | $ | — | | — | | $ | 424,912,410 | |
| Total | 598,272 | | $ | 219.12 | | 598,272 | | $ | 424,912,410 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Period | Total Number
of Shares Purchased | | Average Price
Paid per Share | | Total Number of Shares Purchased as Part of Publicly Announced Programs (1) | | Approximate Dollar Value of Shares that May Yet Be Purchased Under the Programs (1) |
| Oct. 1, 2023 to Oct. 31, 2023 | 179,000 | | | $ | 360.95 | | | 179,000 | | | $ | 2,651,316,977 | |
| Nov. 1, 2023 to Nov. 30, 2023 | 172,552 | | | $ | 362.50 | | | 172,552 | | | $ | 2,588,767,489 | |
| Dec. 1, 2023 to Dec. 31, 2023 | 46,464 | | | $ | 352.39 | | | 46,464 | | | $ | 2,572,394,027 | |
| Total | 398,016 | | | $ | 360.62 | | | 398,016 | | | $ | 2,572,394,027 | |
(1)Shares purchased and approximate dollar value of shares that may yet be purchased under our 2019 Share Repurchase Program.
(2)Shares purchased and approximate dollar value of shares that may yet be purchased under our 2020 Share Repurchase Program.
(3)Under our 2020 Share Repurchase Program, we are authorized to purchase shares from time to time through open market or privately negotiated transactions. Such purchases may be made pursuant to Rule 10b5-1 plans or other means as determined by our management and in accordance with the requirements of the Securities and Exchange Commission. The approximate dollar value of shares that may yet be repurchased is based solely on shares that may be repurchased under the share repurchase program and excludes any shares that may be repurchased under our employee equity programs.
ITEM 6.[RESERVED]
ITEM 6.SELECTED FINANCIAL DATA
The following unaudited selected consolidated financial data are derived from our audited consolidated financial statements. These data should be read in conjunction with our audited consolidated financial statements and related notes that are included elsewhere in this Annual Report on Form 10-K and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in Item 7.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2020 | | 2019 | | 2018 | | 2017 | | 2016 |
| Consolidated Statements of Operations Data: | (in thousands, except per share amounts) |
| Product revenues, net | $ | 6,202,783 | | | $ | 4,160,726 | | | $ | 3,038,325 | | | $ | 2,165,480 | | | $ | 1,683,632 | |
| Collaborative and royalty revenues | 2,900 | | | 2,095 | | | 9,272 | | | 323,172 | | | 18,545 | |
| Total revenues | 6,205,683 | | | 4,162,821 | | | 3,047,597 | | | 2,488,652 | | | 1,702,177 | |
| Total costs and expenses (1) | 3,349,393 | | | 2,965,255 | | | 2,412,447 | | | 2,365,409 | | | 1,692,241 | |
| Provision for (benefit from) income taxes (2) | 405,151 | | | 218,109 | | | (1,486,862) | | | (107,324) | | | 16,665 | |
| Net income (loss) attributable to Vertex | $ | 2,711,647 | | | $ | 1,176,810 | | | $ | 2,096,896 | | | $ | 263,484 | | | $ | (112,052) | |
| Diluted income (loss) per share attributable to Vertex common shareholders | $ | 10.29 | | | $ | 4.51 | | | $ | 8.09 | | | $ | 1.04 | | | $ | (0.46) | |
| Shares used in per diluted share calculations | 263,396 | | | 260,673 | | | 259,185 | | | 253,225 | | | 244,685 | |
| | | | | | | | | |
| As of December 31, |
| 2020 | | 2019 | | 2018 | | 2017 | | 2016 |
| Consolidated Balance Sheet Data: | (in thousands) |
| Cash, cash equivalents and marketable securities | $ | 6,658,897 | | | $ | 3,808,294 | | | $ | 3,168,242 | | | $ | 2,088,666 | | | $ | 1,434,557 | |
| Deferred tax assets (2) | 882,779 | | | 1,190,815 | | | 1,499,672 | | | — | | | — | |
| Total assets | 11,751,808 | | | 8,318,465 | | | 6,245,898 | | | 3,546,014 | | | 2,896,787 | |
| Total current liabilities | 1,877,533 | | | 1,334,827 | | | 1,120,290 | | | 807,260 | | | 792,537 | |
| Long-term finance leases | 539,042 | | | 538,576 | | | 581,550 | | | 583,902 | | | 521,335 | |
| Other long-term liabilities | 648,418 | | | 359,818 | | | 108,853 | | | 112,546 | | | 244,724 | |
| Total shareholders’ equity | 8,686,815 | | | 6,085,244 | | | 4,435.203 | | | 2,042,306 | | | 1,338,191 | |
(1) Total costs and expenses included (i) in 2017, an intangible asset impairment charge of $255.3 million, and (ii) in 2020, 2019, 2018 and 2017, collaborative license and asset acquisition expenses of $184.6 million, $318.3 million, $111.9 million and $168.7 million, respectively. See Note B, “Collaborative Arrangements.”
(2) In 2018, we released the valuation allowance on the majority of our net operating losses and other deferred tax assets resulting in a benefit from income taxes of $1.56 billion in the fourth quarter of 2018 and we recorded a $1.50 billion deferred tax asset on our consolidated balance sheet as of December 31, 2018. In 2020 and 2019, we began recording a provision for income taxes on our pre-tax income approximating statutory rates. In 2020, our provision for income taxes included discrete tax benefits associated with the $209.0 million transfer of intellectual property rights to the U.K., the write-off of a long-term intercompany receivable, and an increase in the U.K.’s corporate tax rate. See Note O, “Income Taxes.” In 2017, we recorded a benefit from income taxes related to the impairment of an intangible asset.
ITEM 7.MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Our discussion and analysis of our financial condition and results of operations for 2023 as compared to 2022 are discussed below. For a discussion of our financial condition and results of operations for 2022 as compared to 2021, please refer to Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our 2022 Annual Report on Form 10-K, except as set forth below.
OVERVIEW
We investare a global biotechnology company that invests in scientific innovation to create transformative medicines for people with serious diseases, with a focus on specialty markets. We have four approved medicines tothat treat the underlying cause of cystic fibrosis or CF,(“CF”), a life-threatening genetic disease, and we are focused on increasing the number of patients eligibleone approved therapy that treats severe sickle cell disease (“SCD”) and able to receive our current medicines through label expansions, approval of new medicinestransfusion dependent beta thalassemia (“TDT”), life shortening inherited blood disorders. Our pipeline includes clinical-stage programs in CF, sickle cell disease, beta thalassemia, acute and expanded reimbursement. We are broadening our pipeline into additionalneuropathic pain, APOL1-mediated kidney disease, areas through internal research effortstype 1 diabetes, myotonic dystrophy type 1 and accessing external innovation through business development transactions.alpha-1 antitrypsin deficiency.
Our triple combination regimen, TRIKAFTA/KAFTRIO (elexacaftor/tezacaftor/ivacaftor and ivacaftor), was approved in 2019 in the United States or (“U.S.,”) and in 2020 in the European Union or (“E.U.”). Collectively, our four medicines are approvedbeing used to treat the majoritynearly three quarters of the approximately 83,00092,000 people with CF in North America, Europe, and Australia. We are evaluating our CF medicines including our triple combination regimen, in additional patient populations, including younger children, with the goal of having small molecule treatments for upall people who have at least one mutation in their cystic fibrosis transmembrane conductance regulator (“CFTR”) gene that is responsive to 90% of people with CF.our CFTR modulators. We also are also pursuing genetic therapies to address the remaining 10% of people with CF.
Beyond CF, our small molecule programs include programs focused on developing treatments for alpha-1 antitrypsin, or AAT, deficiency, APOL1-mediated kidney diseases and pain. We are also focused on developing cellmessenger ribonucleic acid (“mRNA”) and genetic therapies for various diseases in our pipeline. We are evaluating CTX001, a genetic therapy,people with CF who do not make full-length CFTR protein and, as a potentialresult, cannot benefit from our current CF medicines.
CASGEVY (exagamglogene autotemcel or “exa-cel”), an ex-vivo, non-viral CRISPR/Cas9 gene-edited cell therapy, was recently approved in the U.S., the E.U., the United Kingdom (“U.K.”), the Kingdom of Saudi Arabia (“Saudi Arabia”), and the Kingdom of Bahrain (“Bahrain”) for the treatment for sickle cell disease, orof people 12 years of age and older with SCD and transfusion-dependent beta thalassemia,TDT. We estimate approximately 35,000 people with severe SCD or TDT could be eligible for CASGEVY in Phase 1/2 clinical trialsthe U.S. and Europe, with additional people in collaboration with CRISPR Therapeutics AG, or CRISPR.Saudi Arabia and Bahrain. In T1D,addition, we are pursuing two programspreparing for the transplantnear-term launches of functional islets into patients: transplantation of islet cells alone, using immunosuppression to protect the implanted cells,potential new products in CF and implantation of the islet cells inside a novel immunoprotective device. In 2020, we continued to advance our cell and genetic therapy pipeline programs through internal research efforts and investing in business development transactions to access emerging technologies.acute pain.
Financial Highlights
Revenues
In 2020, our net product revenues continued to increase due to the approval of TRIKAFTA in late 2019 and uptake of our medicines in ex-U.S. markets following the approval of KAFTRIO and completion of several significant reimbursement agreements.
Expenses
Our total R&D and SG&A expenses increased from $2.41 billion in 2019 to $2.60 billion in 2020. In 2020, cost of sales was approximately 12% of our net product revenues.
| | | | | |
| Revenues | In 2023, our net product revenues increased to $9.9 billion as compared to $8.9 billion in 2022. The increase was primarily due to the continued strong uptake of TRIKAFTA/KAFTRIO in ex-U.S. markets and label extensions in younger age groups, and the continued performance of TRIKAFTA in the U.S., following the launch of TRIKAFTA in children with CF 2 to 5 years of age. |
| Expenses | Our total research and development (“R&D”), acquired in-process research and development (“AIPR&D”), and selling, general and administrative (“SG&A”) expenses increased to $4.8 billion in 2023 as compared to $3.6 billion in 2022. The increase was primarily due to increased AIPR&D, the progression of several product candidates in mid- to late-stage clinical development and costs to support global launches. Cost of sales was 13% and 12% of our net product revenues in 2023 and 2022, respectively. |
| Cash | Our total cash, cash equivalents and marketable securities increased to $13.7 billion as of December 31, 2023 as compared to $10.9 billion as of December 31, 2022 primarily due to our income from operations driven by our net product revenues, and interest income, partially offset by our income tax payments and repurchases of our common stock. |
Note: Charts above may not add due to rounding.
Business Updates
Marketed Products
Cystic Fibrosis
We expect to continue to grow our CF business with (i) continued uptake by patients in countries where we are early in our launch, such as those with recently achieved reimbursement agreements, (ii) label expansions, including into younger patient groups, and (iii) growth in the number of people living with CF. Recent progress in activities supporting continued uptake and label expansions is included below:
•The U.S. Food and Drug Administration (“FDA”), the European Commission, the Medicines and Healthcare Products Regulatory Agency (“MHRA”), and Health Canada approved TRIKAFTA/KAFTRIO for the treatment of children with CF 2 to 5 years of age who have at least one F508del mutation in the CFTR gene.
•The European Medicines Agency (“EMA”) validated the Marketing Authorization Application (“MAA”) extension for KAFTRIO in combination with ivacaftor to include people with CF who have a rare mutation in the CFTR gene that is responsive based on clinical and/or in vitro data, including the N1303K mutation. We plan to submit regulatory filings for these mutations in Australia, Brazil, Canada, New Zealand and Switzerland, and we plan to submit for regulatory approval of a subset of these mutations not currently included in the U.S. TRIKAFTA label to the FDA.
•The FDA and the European Commission approved the use of ORKAMBI for children with CF 12 to less than 24 months of age who are homozygous for the F508del mutation in the CFTR gene.
•The FDA and MHRA approved KALYDECO in children with CF from 1 month to less than 4 months of age.
Sickle Cell Disease and Beta Thalassemia
•CASGEVY is now approved in the U.S., the E.U., the U.K., Saudi Arabia, and Bahrain for people 12 years of age and older with SCD or TDT.
•The French National Authority for Health (“HAS”) approved our request for the implementation of an early access program (“EAP”) for the use of CASGEVY to treat eligible people with TDT from 12 to 35 years of age. We are also pursuing an EAP submission for SCD in France and we expect to receive the outcome of this decision in the coming months.
•Our regulatory submission for CASGEVY in both SCD and TDT is currently under review in Switzerland. We expect to submit for regulatory approval of CASGEVY in Canada in the first half of 2024.
•We have activated 12 authorized treatment centers (“ATCs”) in the U.S. and three ATCs in Europe. We are aiming to activate approximately 50 ATCs in the U.S. and 25 in Europe. We also have activated one of two planned ATCs in Saudi Arabia.
•We entered into an agreement with Synergie Medication Collective, a medication contracting organization, which covers approximately 100 million people, to provide access to CASGEVY.
Potential Near-Term Launch Opportunities
We are preparing for the following near-term launches of potential new products:
Vanzacaftor/tezacaftor/deutivacaftor in CF
•We completed the pivotal SKYLINE 102 and SKYLINE 103 clinical trials, which evaluate the efficacy and safety of our new once-daily investigational triple combination vanzacaftor/tezacaftor/deutivacaftor relative to TRIKAFTA in people with CF 12 years of age and older, and the RIDGELINE clinical trial of vanzacaftor/tezacaftor/deutivacaftor in children with CF 6 to 11 years of age, at the end of 2023.
•In February 2024, we announced positive data from this Phase 3 program evaluating the new triple combination regimen. We expect to submit global regulatory filings by mid-2024, including a New Drug Application (“NDA”) to the FDA, using a priority review voucher, and MAAs to the EMA and Health Canada.
VX-548 in Acute Pain
•We completed the pivotal program evaluating our lead compound, VX-548, for the treatment of moderate-to-severe acute pain, which included one randomized controlled Phase 3 pivotal trial in abdominoplasty, one randomized, controlled Phase 3 clinical trial in bunionectomy, and one single-arm safety and effectiveness clinical trial. In January 2024, we announced positive results from these clinical trials and we announced our plans to submit an NDA to the FDA for VX-548 in moderate-to-severe acute pain by mid-2024.
•In the U.S., VX-548 has been granted Breakthrough Therapy and Fast Track designations for moderate-to-severe acute pain.
Pipeline
We continue to advance a diversified pipeline of potentially transformative medicines for serious diseases utilizing a range of modalities. Recent and anticipated progress in activities supporting these efforts is included below:
Cystic Fibrosis
•In collaboration with Moderna, we are developing VX-522, a CFTR mRNA therapeutic for the treatment of people with CF who do not produce full-length CFTR protein. We completed dosing in the single ascending dose part of the clinical trial for VX-522 and initiated the multiple ascending dose part of the clinical trial in late 2023. We expect to share data from this clinical trial in late 2024 or early 2025. In the U.S., the FDA has granted Fast Track designation for VX-522.
•We are investigating a portfolio of other small molecules targeting the underlying cause of CF with the aim of achieving carrier levels of CFTR function. We also are investigating additional potential treatments for people with CF who do not make full-length CFTR protein and cannot benefit from CFTR modulators.
Sickle Cell Disease and Beta Thalassemia
•We have completed enrollment in two global Phase 3 clinical trials evaluating CASGEVY in children 5 to 11 years of age with SCD or TDT.
•We continue to work on preclinical assets for myeloablative conditioning agents that would have milder side-effects and could be used in connection with CASGEVY, which could broaden the eligible patient population.
Peripheral Neuropathic Pain
•We have completed the Phase 2 dose-ranging clinical trial evaluating VX-548 in patients with diabetic peripheral neuropathy, a common form of chronic peripheral neuropathic pain, and announced positive results from this clinical trial. We plan to meet with regulators in the first quarter of 2024 and then we expect to advance VX-548 for the treatment of diabetic peripheral neuropathy into pivotal development.
•We initiated a second Phase 2 clinical trial evaluating VX-548 in patients with peripheral neuropathic pain in December 2023. This clinical trial will evaluate VX-548 in patients with lumbosacral radiculopathy, a second type of peripheral neuropathic pain. Screening, enrollment and dosing are underway in this clinical trial.
•We expect to initiate a Phase 2 clinical trial evaluating the oral formulation of VX-993, a next generation NaV1.8 inhibitor, for the treatment of peripheral neuropathic pain in 2024.
Acute Pain
•We have completed a Phase 1 clinical trial evaluating an oral formulation of VX-993, a next generation NaV1.8 inhibitor. We expect to initiate a Phase 2 clinical trial evaluating the oral formulation of VX-993 for the treatment of moderate-to-severe acute pain in the second half of 2024.
•We anticipate completing IND-enabling studies and filing an Investigational New Drug Application (“IND”) for an intravenous formulation of VX-993 for the treatment of moderate-to-severe acute pain in 2024.
•We are advancing multiple NaV1.7 inhibitors, including in combination with NaV1.8 inhibitors, through research and earlier stages of development for both acute and peripheral neuropathic pain.
APOL1-Mediated Kidney Disease
•Inaxaplin is our small molecule for the treatment of APOL1-mediated kidney disease (“AMKD”), including APOL1-mediated focal segmental glomerulosclerosis (“FSGS”). We completed enrollment in the Phase 2B dose-ranging portion of the pivotal program for inaxaplin, a single Phase 2/3 clinical trial in patients with AMKD. We expect to select a dose and begin the Phase 3 portion of the clinical trial in the first quarter of 2024.
•The FDA grantedBreakthrough Therapy designation to inaxaplin for APOL1-mediated FSGS and the EMA granted Orphan Drug and PRIME designations to inaxaplin for AMKD.
Type 1 Diabetes
•VX-880 is an allogeneic stem cell-derived, fully differentiated, insulin-producing islet cell replacement therapy, using standard immunosuppression to protect the implanted cells. We are evaluating VX-880 as a potential treatment for type 1 diabetes (“T1D”) in a sequential, three-part Phase 1/2 clinical trial. We have completed enrollment in Part C of the clinical trial. We have placed the clinical trial on a protocol-specified pause, pending review of the totality of the data by an independent data monitoring committee and global regulators, following two patient deaths, both unrelated to VX-880.
•Our second Phase 1/2 program in T1D evaluates VX-264, which encapsulates the same VX-880 cells in a novel device designed to eliminate the need for immunosuppression. This trial is a sequential, multi-part study to evaluate the safety, tolerability and efficacy of VX-264. We have completed Part A of the clinical trial and Part B has been initiated in multiple centers and countries.
•Our hypoimmune islet cell program uses CRISPR/Cas9 technology to gene-edit the same allogeneic stem cell-derived, fully differentiated islets used in the VX-880 and VX-264 programs. The goal is to cloak the cells from the immune system to explore another possible path to eliminate the need for immunosuppressive therapy. This program continues to progress through the research stage.
Myotonic Dystrophy Type 1
•We have established a collaboration with Entrada Therapeutics, Inc. (“Entrada”) to address the functional impact of the causal mutation in the gene that causes myotonic dystrophy type 1 (“DM1”), which includes VX-670. VX-670 is designed to enable efficient intracellular delivery of an oligonucleotide, which we believe will address the underlying pathophysiology and restore normal cell function.
•We initiated a Phase 1/2 clinical trial evaluating VX-670 in patients with DM1. The clinical trial is active and enrolling in Canada and will initiate in the U.K. in the near term.
Alpha-1 Antitrypsin Deficiency
•We are working to address the underlying genetic cause of alpha-1 antitrypsin (“AAT”) deficiency (“AATD”). We are developing novel small molecule correctors of Z-AAT protein folding, with the goal of enabling the secretion of functional AAT into the blood and addressing both the lung and the liver aspects of AATD. We continue to enroll and dose healthy volunteers in Phase 1 clinical trials evaluating VX-634 and VX-668, our next-wave investigational molecule AAT correctors with significantly improved potency and drug-like properties as compared to the first-generation AATD correctors.
Our Business Environment
In 2023, our net product revenues came from the sale of our medicines for the treatment of CF. Our CF strategy involves continuing to develop and obtain approval and reimbursement for treatment regimens that will provide benefits to all people with CF and increasing the number of people with CF eligible and able to receive our medicines, including through label expansions, expanded reimbursement, and providing improved treatment options for people whothe development of new medicines. We are already eligible for one ofadvancing our medicines. Since the beginning of 2020, we have made important progress in activities supporting these efforts.
•In August, the European Commission granted marketing authorization for KAFTRIO to treat people with CF 12 years of age and older with one F508del mutation and one minimal function mutation, or two F508del mutations.
•The FDA expanded the eligibility for TRIKAFTA to include people with CF 12 years of age and older with certain mutations that are responsive to TRIKAFTA based on in vitro data. SYMDEKO and KALYDECO also received approvals to include additional responsive mutations in people with CF 6 years of age and older and 4 months of age and older, respectively.
•In January 2021, the FDA accepted our supplemental New Drug Application, or sNDA, for TRIKAFTA for the treatment of children with CF 6 to 11 years of age with at least one F508del mutation or have certain mutations that are responsive to TRIKAFTA based on in vitro data. The FDA granted Priority Review of the sNDA.
•The European Commission approved the label extension of SYMKEVI in combination with KALYDECO for the treatment of children with CF 6 years of age and older with two F508del mutations or one F508del mutation and certain residual function mutations.
•The FDA approved KALYDECO for treatment of infants with CF four months of age and older who have at least one mutation in their CFTR gene that is responsive to KALYDECO.
•The European Commission approved KALYDECO for treatment of infants with CF four months of age and older who have the R117H mutation or certain gating mutations.
Pipeline
We continue to advance a broad pipeline of potentially transformative small molecule, cell and genetic therapies aimed at treating serious diseases. Since the beginning of 2020, we have made important progress in activities supporting these efforts.
Beta Thalassemia and Sickle Cell Disease
•In December, we and our collaborator, CRISPR, announced positive interim data from 10 people with TDT or SCD treated with CTX001 and that 20 people with severe hemoglobinopathies have been dosed with CTX001 in the ongoing Phase 1/2 clinical trials. Enrollment and dosing are ongoing, and completion of enrollment in both clinical trials is expected in 2021.
Alpha-1 Antitrypsin Deficiency
•Enrollment is ongoing in a Phase 2 proof-of-concept clinical trial for the corrector VX-864. We expect data from this clinical trial in the first half of 2021.
•We discontinued development of VX-814, our first corrector, based on the safety and pharmacokinetic profile of VX-814 observed in a Phase 2 clinical trial.
APOL1-Mediated Kidney Diseases
•Enrollment is ongoing in a Phase 2 proof-of-concept clinical trial designed to evaluate the reduction of proteinuria in people with APOL1-mediated FSGS after treatment with VX-147. We expect data from this clinical trial in 2021.
Type 1 Diabetes
•We are developing a cell therapy designed to replace insulin-producing islet cells in patients with T1D. We are pursuing two programs for the transplant of these functional islets into patients: transplantation of islet cells alone,
using immunosuppression to protect the implanted cells, and implantation of the islet cells inside a novel immunoprotective device.
•In January 2021, the FDA cleared our IND for VX-880, the islet cells alone program. We expect to initiate a Phase 1/2 clinical trial evaluating this program in the first half of 2021. This clinical trial will involve an infusion of fully differentiated, functional islet cells, and chronic administration of concomitant immunosuppressive therapy, to protect the islet cells from immune rejection.
Investment in External Innovation
•We entered into a collaboration with Skyhawk Therapeutics, Inc., or Skyhawk, for the discovery and development of novel small molecules that modulate RNA splicingproduct candidates for the treatment of serious diseases.
•We entered into a new collaboration with Moderna, Inc.diseases outside of CF, including CASGEVY, which recently received marketing approvals in the U.S., or Moderna, aimed at the discoveryE.U., the U.K., Saudi Arabia, and development of lipid nanoparticles and mRNAs that can deliver gene-editing therapies to lung cellsBahrain for the treatment of CF.
•SCD and TDT. We entered into a collaborationanticipate broad access with Affinia Therapeutics, Inc., or Affinia, to gain access to a novel library of AAV capsids to support on our ongoing researchgovernment and development efforts in genetic therapies, including DMD, DM1 and CF.
COVID-19
We continue to monitor the impacts of the COVID-19 global pandemic on our business. COVID-19 has not affected our supply chain or the demandcommercial payors for our medicines, and we believe that we will be able to continue to supply all of our approved medicines to patients globally. We have adjusted our business operations in response to COVID-19, with a majority of our employees continuing to work remotely. We continue to monitor local COVID-19 trends and government guidance for each of our site locations, and are utilizing a phased, site-specific approach to assess employee access to our sites. Currently, all of our research and manufacturing sites are open to essential employees. To provide a safe working environment for our on-site employees, we have, among other things, limited employee numbers at our open sites and increased safety measures, including at home and on-site testingCASGEVY in the U.S., enhanced cleaning and sanitation protocols, required usewe anticipate early access programs initially, such as the recently approved early access program for TDT in France, and are pursuing long-term reimbursement agreements outside of personal protective equipment for all on-site employees, hand sanitation stations throughout our open sites and implementation of various social distancing measures while on-site.the U.S.
Research
We continue to invest in our research programs and foster scientific innovation in order to identify and develop transformative medicines. Our strategy is to combine transformative advances in the understanding of causal human diseasebiology and the science of therapeutics in order to identifydiscover and develop innovative medicines. This approach includes advancing multiple compounds or therapies from each program, spanning multiple modalities, into early clinical trials to obtain patient data that can inform selection of the most promising therapies for later-stage development, as well as to inform discovery and development efforts. We aim to rapidly follow our first-in-class therapies that achieve proof-of-concept with potential best-in-class candidates to provide durable clinical and commercial success.
In pursuit of new medicines.product candidates and therapies in specialty markets, we invest in research and development. We believe that pursuing innovative approaches to treatresearch in diverse diseases of great unmet needareas allows us to balance the risks inherent in drugproduct development and may provide drugproduct candidates that will form our pipeline in future years. To supplement our internal research programs, we acquire technologies and programs and collaborate with biopharmaceutical and technology companies, leading academic research institutions, government laboratories, foundations and other organizations, as needed, to advance research in our areas of therapeutic interest and to access technologies needed to execute on our strategy.
Drug Discovery and Development
Discovery and development of a new pharmaceutical or biological product is a difficult and lengthy process that requires significant financial resources along with extensive technical and regulatory expertise. Potential drug candidates are subjected to rigorous evaluations, driven in part by stringent regulatory considerations, designed to generate information concerning efficacy, side effects, proper dosage levels and a variety of other physical and chemical characteristics that are important in determining whether a drug candidate should be approved for marketing as a pharmaceutical product. Most chemical compounds that are investigated asAcross the industry, most potential drug candidatesor biological products never progress into development, and most drug candidatesproducts that do advance into development never receive marketing approval. Because ourOur investments in drugproduct candidates are subject to considerable risks, werisks. We closely monitor the results of our discovery, research, clinical trials and nonclinical studies and frequently evaluate our drugproduct development programs in light of new data and scientific, business and commercial insights, with the objective of balancing risk and potential. This process can result in rapid changes in focus and priorities as new information becomes available and as we gain additional understanding of our ongoing programs and potential new programs, as well as those of our competitors. For example, in October 2020,
Our business also requires ensuring appropriate manufacturing and reimbursement of our products. As we discontinuedadvance our product candidates through clinical development toward commercialization and market and sell our approved products, we build and maintain our supply chain and quality assurance resources. We rely on a global network of VX-814, athird parties and our internal capabilities to manufacture and distribute our products for commercial sale and post-approval clinical trials and to
drug candidatemanufacture and distribute our product candidates for clinical trials. In addition to establishing supply chains for each new approved product, we adapt our supply chain for existing products to include additional formulations or to increase scale of production for existing products as needed. The processes for cell and genetic therapies can be more complex than those required for small molecule drugs and require additional investments in different systems, equipment, facilities and expertise. We are focused on ensuring the treatment of AAT, based on the safety and pharmacokinetic profile of VX-814 observed in a Phase 2 clinical trial.
If we believe that data from a completed registration program support approval of a drug candidate, we submit an NDA or BLA to the FDA requesting approval to market the drug candidate in the U.S. and seek analogous approvals from comparable regulatory authorities in jurisdictions outside the U.S. To obtain approval, we must, among other things, demonstrate with evidence gathered in nonclinical studies and well-controlled clinical trials that the drug candidate is safe and effective for the disease it is intended to treat and that the manufacturing facilities, processes and controls for the manufacturestability of the drug candidate are adequate. The FDA and ex-U.S. regulatory authorities have substantial discretion in deciding whether or not a drug candidate should be granted approval based on the benefits and risks of the drug candidate in the treatment of a particular disease, and could delay, limit or deny regulatory approval. If regulatory delays are significant or regulatory approval is limited or denied altogether,supply chains for our financial results and the commercial prospects for the drug candidate involved will be harmed.
Regulatory Compliance
Our marketing of pharmaceuticalcurrent products, is subject to extensive and complex laws and regulations. We have a corporate compliance program designed to actively identify, prevent and mitigate risk through the implementation of compliance policies and systems and through the promotion of a culture of compliance. Among other laws, regulations and standards, we are subject to various U.S. federal and state laws, and comparable laws in other jurisdictions, pertaining to health care fraud and abuse, including anti-kickback and false claims laws, and laws prohibiting the promotion of drugs for unapproved or off-label uses. Anti-kickback laws generally make it illegal for a prescription drug manufacturer to knowingly and willfully solicit, offer, receive or pay any remuneration in return for or to induce the referral of business, including the purchase or prescription of a particular drug that is reimbursed by a state or federal health care program. False claims laws prohibit anyone from knowingly or willfully presenting for payment to third-party payors, including Medicare and Medicaid, claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. We are subject to laws and regulations that regulate the sales and marketing practices of pharmaceutical manufacturers, as well as laws such as the U.S. Foreign Corrupt Practices Act, which governfor our international business practices with respect to payments to government officials. In addition, we are subject to various data protection and privacy laws and regulations in the U.S., E.U., U.K., Canada, Australia and other jurisdictions. We expect to continue to devote substantial resources to maintain, administer and expand these compliance programs globally.
Reimbursementpipeline programs.
Sales of our products depend, to a large degree, on the extent to which our products are reimbursed by third-party payors, such as government health programs, commercial insurance and managed health care organizations. Reimbursement for our products, including our potential pipeline therapies, cannot be assured and may take significant periods of time to obtain. We dedicate substantial management and other resources in order to obtain and maintain appropriate levels of reimbursement for our products from third-party payors, including governmental organizations in the U.S. and ex-U.S. markets.
In the U.S., we have worked successfully with third partythird-party payors in order to promptly obtain appropriate levels of reimbursement for our CF medicines.medicines and are currently working with U.S. government and commercial payors with respect to CASGEVY. We plan to continue to engage in discussions with numerous commercial insurers and managed health care organizations, along with government health programs that are typically managed by authorities in the individual states, to ensure that payors recognize the significant benefits that our medicinestherapies provide by treating the underlying causeand provide patients with appropriate levels of CF and continue to provide access to our medicines.
In Europemedicines and othertherapies now and in the future. We cannot, however, predict how recent changes in the law, including through the Inflation Reduction Act of 2022 and passage of state laws (e.g., transparency laws and prescription drug affordability boards), will affect our ability to negotiate successfully with third-party payors and distribute our products. Similarly, in ex-U.S. markets, we seek government reimbursement for our medicines on a country-by-country basis.or region-by-region basis, as required. This is necessary for each new medicine, as well as for label expansions for our current medicines. We successfully obtained reimbursement for KALYDECO in each significantare beginning to work with ex-U.S. market within two years of approval, but experienced significant challenges in obtaining reimbursement for ORKAMBI in certain ex-U.S. markets. With the completion of reimbursement discussions in England and France in 2019, we have reimbursement for ORKAMBI or SYMKEVI in most of our significant ex-U.S. markets. In addition, in several ex-U.S. markets, including England, Ireland, Denmark and Australia, our reimbursement agreements include innovative arrangements that provide a pathwaypayors with respect to access and rapid reimbursement for certain future CF medicines. For example, our existing reimbursement agreements in England, Ireland, and Denmark have been expanded to include KAFTRIO.CASGEVY. We expect to continue to focus significant resources to obtain appropriateexpand and maintain reimbursement for our productsCF medicines, CASGEVY and, ultimately, pipeline therapies, in U.S. and ex-U.S. markets.
Strategic Transactions
Acquisitions
As part of our business strategy, we seek to acquire drugs, drugtechnologies, products, product candidates and other technologies and businesses that have the potential toare aligned with our corporate and research and development strategies and complement and advance our ongoing research and development efforts. We have engaged in a number of acquisitions of privately held biotechnology companies over the last several years. In 2019, we invested significantly in business development transactions designed to augment our pipeline, including the acquisition ofacquired Semma Therapeutics, Inc., or Semma, a privately-held company focused on the use of stem cell-derived human islets as a potentially curative treatment forpursuant to which we established our T1D program, and Exonics Therapeutics, Inc., or Exonics, a privately-held company focused on creating transformativewhich enhanced our gene-editing therapiescapabilities to repair mutations that cause DMD and other severe neuromuscular diseases, including DM1.such as DM1 and Duchenne muscular dystrophy. In 2022, we acquired ViaCyte, Inc. (“ViaCyte”), which had intellectual property, tools, technologies and assets with the Semma acquisition, we paid approximately $950.0 million in cashpotential to Semma equity holders. In the Exonics acquisition, we paid approximately $245.0 million upfront to Exonics equity holders and agreed to additional payments based upon successful achievement of specifiedaccelerate development and regulatory milestones. We expect to continue to identify and evaluate potential acquisitions and may include larger transactions or later stage assets.
Both of our 2019 acquisitions wereT1D programs. Our acquisition of ViaCyte, for $315.0 million, was accounted for as a business combinations.combination. As of the acquisition date, for each transaction, the cash payments, as well as the fair value of contingent consideration for Exonics, werepayment was allocated primarily to goodwill and the fair value of severalan in-process research and development assetsasset.
In 2023 and 2022, we also acquired programs that we acquired. The fair valuewere accounted for as asset acquisitions resulting in $47.5 million and $60.0 million of contingent consideration related to Exonics was recorded as a liability and continues to be adjusted on a quarterly basis. As a result, these acquisitions are primarily reflected in additional assets and liabilities on our consolidated balance sheet. Operating expenses incurred by Exonics and Semma after the acquisition dates and specific expenses associated with the acquisitions are reflected in our consolidated statement of operations.AIPR&D, respectively.
Please refer toour critical accounting policies, “Acquisitions,“Acquisitions,” for further information regarding the significant judgments and estimates related to our acquisitions.
Collaboration and LicensingIn-Licensing Arrangements
We enter into arrangements with third parties, including collaboration and licensing arrangements, for the development, manufacture and commercialization of drugs, drugproducts, product candidates, and other technologies that have the potential to complement our ongoing research and development efforts. We expect to continue to identify and evaluate collaboration and licensing opportunities that may be similar to or different from the collaborations and licenses that we have engaged in previously.
In-License Agreements
We have entered into collaborations with biotechnology and pharmaceutical companies in order to acquire rights or to license drug candidates or technologies that enhance our pipeline and/or our research capabilities. Over the last several years, we entered into collaboration agreements with a number of companies, including Affinia, Arbor Biotechnologies,CRISPR, Entrada, and Moderna, Inc., CRISPR, Kymera Therapeutics, Inc., Moderna, Molecular Templates, Inc. and Skyhawk. Generally, when we in-license a technology or drugproduct candidate, we make upfront payments to the collaborator, assume the costs of the program and/or agree to make contingent payments, which could consist of milestone, royalty and option payments. Most of these collaboration payments are expensed as AIPR&D, including, in 2023, our total payments of $242.6 million to Entrada and our total upfront and milestone payments of $170.0 million to CRISPR related to T1D, because they are primarily attributable to acquired in-process research and development expenses; however,for which there is no
alternative future use. However, depending on many factors, including the structure of the collaboration, the stage of development of the acquired technology, the significance of the in-licensed drugproduct candidate to the collaborator’s operations and the other activities in which our collaborators are engaged, the accounting for these transactions can vary significantly. OurWe expect to continue to identify and evaluate collaboration and licensing opportunities that may be similar to or different from the collaborations and licenses that we have engaged in previously.
Joint Development and Commercialization Agreement with CRISPR
In 2017, we entered into a joint development and commercialization agreement (the “Original JDCA”) with CRISPR, pursuant to which we are developing and commercializing CASGEVY for SCD and TDT. The Original JDCA was entered into following our exercise of an option to co-develop and co-commercialize the hemoglobinopathies program that was contained in a collaboration agreement that we entered into with CRISPR in 2015.
In April 2021, we and CRISPR entered into an amendment and restatement of the Original JDCA (the “A&R JDCA”). In June 2021, we made a $900.0 million upfront payment to CRISPR in connection with the closing of the transactions contemplated by the A&R JDCA. We concluded that we did not have any alternative future use for the acquired in-process research and development expensesand recorded this upfront payment to AIPR&D. Under the terms of the A&R JDCA, we lead worldwide development, manufacturing, and commercialization of CASGEVY. As of July 1, 2021, 60% of the net profits and net losses for CASGEVY are allocated to us and 40% of the net profits and net losses for CASGEVY are allocated to CRISPR, subject to certain adjustments. We concluded that the Original JDCA and the A&R JDCA are cost-sharing arrangements, which result in the net impact of the arrangements, excluding amounts recorded to AIPR&D, being recorded in “Research and development expenses” or “Selling, general and administrative expenses.” in our consolidated statements of income.
CASGEVY was approved by the FDA in December 2023 for the treatment of SCD. Upon this approval, we made a $200.0 million milestone payment to CRISPR, which we recorded within “Other intangible assets, net” on our consolidated balance sheet as of December 31, 2023. We also recorded an immaterial amount of intangible asset amortization expense to “Cost of sales” in the fourth quarter of 2023 related to this intangible asset. Subsequent to receiving marketing approval for CASGEVY, we continue to lead the research and development activities under the A&R JDCA, subject to CRISPR’s reserved right to conduct certain activities. We are reimbursed by CRISPR for its 40% share of these research and development activities, subject to certain adjustments, and we continue to record this reimbursement from CRISPR within “Research and development expenses.” We also share with CRISPR 40% of the net commercial profits or losses incurred with respect to CASGEVY, subject to certain adjustments. In 2023, the net commercial loss incurred with respect to CASGEVY was not material to our consolidated statement of income.
Acquired In-Process Research and Development Expenses
In 2023 and 2022, our AIPR&D included $184.6$527.1 million in 2020, $318.3and $115.5 million, in 2019 and $111.9 million in 2018respectively, related to upfront, andcontingent milestone, or other payments pursuant to our collaboration agreements.
Out-License Agreements
We also have out-licensed internally-developed programs to collaborators who are leadingbusiness development transactions, including the developmentcollaborations, licenses of these programs. These out-license arrangements include our agreement with Merck KGaA, Darmstadt, Germany, which licensed oncology researchthird-party technologies, and development programs from us in early 2017. Pursuant to these out-licensing arrangements, our collaborators are responsible for the research, development and commercialization costs associated with these programs, and we are entitled to receive contingent milestone and/or royalty payments. As a result, we do not expect to incur significant expenses in connection with these programs and have the potential for future collaborative and royalty revenues resulting from these programs.
asset acquisitions described above. Please refer to Note B, “Collaborative“Collaboration, License and Other Arrangements,” for further information regarding our collaboration, in-license agreements and out-license agreements.
Strategic Investments
In connection with our business development activities, we have periodically made equity investments in our collaborators. As of December 31, 2020, we held strategic equity investments in several public companies and certain private companies, and we plan to make additional strategic equity investments in the future. While we invest the majority of our cash, cash equivalents and marketable securities in instruments that meet specific credit quality standards and limit our exposure to any one issue or type of instrument, our strategic investments are maintained and managed separately from our other cash, cash equivalents and marketable securities. Any changes in the fair value of equity investments with readily determinable fair values (including publicly traded securities) are recorded to other income (expense), net in our consolidated statement of operations.
In 2020, 2019 and 2018, we recorded within other income (expense), net gains of $311.9 million, $197.6 million and $2.6 million, respectively, related to changes in the fair value of our strategic investments and from sales of certain investments. As of December 31, 2020, the fair value of our investments in publicly traded companies was $195.8 million. To the extent that we continue to hold strategic investments, particularly strategic investments in publicly traded companies, we will record other income (expense) related to these strategic investments on a quarterly basis. Due to the increased volatility of the global markets, including as a result of COVID-19, and the high volatility of stocks in the biotechnology industry, we expect the value of these strategic investments to fluctuate and that the increases or decreases in the fair value of these strategic investments will continue to have material impacts on our net income (expense) and our profitability on a quarterly and/or annual basis.asset acquisitions.
RESULTS OF OPERATIONS
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | 2020/2019
Comparison | | 2019/2018
Comparison |
| | | | | | | Increase/(Decrease) | | Increase/(Decrease) |
| 2020 | | 2019 | | 2018 | | $ | | % | | $ | | % |
| (in thousands, except percentages and per share amounts) |
| Revenues | $ | 6,205,683 | | | $ | 4,162,821 | | | $ | 3,047,597 | | | $ | 2,042,862 | | | 49 | % | | $ | 1,115,224 | | | 37 | % |
| Operating costs and expenses | 3,349,393 | | | 2,965,255 | | | 2,412,447 | | | 384,138 | | | 13 | % | | 552,808 | | | 23 | % |
| Income from operations | 2,856,290 | | | 1,197,566 | | | 635,150 | | | 1,658,724 | | | 139 | % | | 562,416 | | | 89 | % |
| Other non-operating income (expense), net | 260,508 | | | 197,353 | | | (25,116) | | | 63,155 | | | 32 | % | | ** | | ** |
| Provision for (benefit from) income taxes | 405,151 | | | 218,109 | | | (1,486,862) | | | 187,042 | | | 86 | % | | ** | | ** |
| Net income attributable to Vertex | $ | 2,711,647 | | | $ | 1,176,810 | | | $ | 2,096,896 | | | $ | 1,534,837 | | | 130 | % | | $ | (920,086) | | | ** |
| | | | | | | | | | | | | |
| Net income per diluted share attributable to Vertex common shareholders | $ | 10.29 | | | $ | 4.51 | | | $ | 8.09 | | | | | |
| Diluted shares used in per share calculations | 263,396 | | | 260,673 | | | 259,185 | | | | | | | | | |
| | | | | | | | | ** Not meaningful |
Net Income Attributable to Vertex
Our net income attributable to Vertex increased to $2.71 billion in 2020 as compared to $1.18 billion in 2019 primarily due to increased revenues and increased other income (expense) related to our strategic investments partially offset by increased operating costs and expenses and an increased provision for income taxes. The increased revenues were primarily due to the U.S. approval of TRIKAFTA in the fourth quarter of 2019, E.U. approval of KAFTRIO in the third quarter of 2020 and continued uptake of our medicines in ex-U.S. markets. The increased operating costs and expenses were primarily due to increased cost of sales consistent with increased net product revenues, increased investment in research and development and increased sales, general and administrative expenses to support our business.
Net income attributable to Vertex in 2018 included a one-time non-cash benefit from income taxes of $1.56 billion resulting from our release of our valuation allowance. Net income attributable to Vertex decreased in 2019 as compared to 2018 as a result of this one-time tax benefit and increased operating costs and expenses. The increases in operating costs and expenses were primarily due to increased cost of sales due to increased net product revenues and increased research expenses associated with our business development activities. These decreases in our net income in 2019 as compared to 2018 were partially offset by increased net product revenues and increased gains recorded to other income (expense) related to our strategic investments.
Earnings Per Share
In 2020, 2019, and 2018, net income attributable to Vertex was $10.29, $4.51 and $8.09, respectively, per diluted share. In 2018, the benefit from income taxes as a result of the release of our valuation allowance increased net income attributable to Vertex by $6.03 per diluted share.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2023 | | % Change | | 2022 | | % Change | | 2021 | | | | |
| (in millions, except percentages and per share amounts) | | | | |
| Revenues | $ | 9,869.2 | | | 11% | | $ | 8,930.7 | | | 18% | | $ | 7,574.4 | | | | | |
| Operating costs and expenses | 6,037.2 | | | 31% | | 4,623.3 | | | (4)% | | 4,792.3 | | | | | |
| Income from operations | 3,832.0 | | | (11)% | | 4,307.4 | | | 55% | | 2,782.1 | | | | | |
| Other non-operating income (expense), net | 547.8 | | | ** | | (75.0) | | | 45% | | (51.7) | | | | | |
| Provision for income taxes | 760.2 | | | (16)% | | 910.4 | | | 134% | | 388.3 | | | | | |
| Net income | $ | 3,619.6 | | | 9% | | $ | 3,322.0 | | | 42% | | $ | 2,342.1 | | | | | |
| | | | | | | | | | | | | |
| Net income per diluted common share | $ | 13.89 | | | | | $ | 12.82 | | | | | $ | 9.01 | | | |
| Diluted shares used in per share calculations | 260.5 | | | | | 259.1 | | | | | 259.9 | | | | | |
| | | | | | | ** Not meaningful | | |
Revenues
| | 2023 | | | 2023 | | % Change | | 2022 | | % Change | | 2021 |
| (in millions, except percentages) | | | (in millions, except percentages) |
| TRIKAFTA/KAFTRIO | |
| KALYDECO | |
| ORKAMBI | |
| SYMDEKO/SYMKEVI | |
| Product revenues, net | |
| Other revenues | |
| Total revenues | |
| | 2020/2019 Comparison | | 2019/2018 Comparison |
| Increase/(Decrease) | | Increase/(Decrease) |
| 2020 | | 2019 | | 2018 | | $ | | % | | $ | | % |
| (in thousands, except percentages) |
| Product revenues, net | $ | 6,202,783 | | | $ | 4,160,726 | | | $ | 3,038,325 | | | $ | 2,042,057 | | | 49 | % | | $ | 1,122,401 | | | 37 | % |
| Collaborative and royalty revenues | 2,900 | | | 2,095 | | | 9,272 | | | 805 | | | 38 | % | | (7,177) | | | (77) | % |
| Total revenues | $ | 6,205,683 | | | $ | 4,162,821 | | | $ | 3,047,597 | | | $ | 2,042,862 | | | 49 | % | | $ | 1,115,224 | | | 37 | % |
| | | | | | | ** Not meaningful | |
| | | | | | | ** Not meaningful | |
| | | | | | | ** Not meaningful | |
Product Revenues, Net
| | | | | | | | | | | | | | | | | |
| 2020 | | 2019 | | 2018 |
| (in thousands) |
| TRIKAFTA/KAFTRIO | $ | 3,863,824 | | | $ | 420,105 | | | $ | — | |
| SYMDEKO/SYMKEVI | 628,577 | | | 1,417,668 | | | 768,657 | |
| ORKAMBI | 907,512 | | | 1,331,891 | | | 1,262,166 | |
| KALYDECO | 802,870 | | | 991,062 | | | 1,007,502 | |
| Product revenues, net | $ | 6,202,783 | | | $ | 4,160,726 | | | $ | 3,038,325 | |
In 2020,2023, our net product revenues increased by $2.04 billion$938.5 million, or 11%, as compared to 2019. In 2019, our net product revenues increased by $1.12 billion as compared to 2018.2022. The increase in total net product revenues in 2020 was primarily due to the continued strong uptake of TRIKAFTA/KAFTRIO in ex-U.S. markets and label extensions in younger age groups, and the continued performance of TRIKAFTA in the U.S., following the launch of TRIKAFTA in the U.S in the fourth quarterchildren with CF 2 to 5 years of 2019 and KAFTRIO in the E.U. in the third quarter of 2020.age. Decreases in revenues for our products other productsthan TRIKAFTA/KAFTRIO were primarily the result of patients in the U.S. switching from these medicines to TRIKAFTA, partially offset by label expansions and expanded access to our medicines in ex-U.S. markets. The increase in totalTRIKAFTA/KAFTRIO.
Our net product revenues in 2019 was primarily due tofrom the increasing number of patients being treated with SYMDEKO/SYMKEVI, the October 2019 approval of TRIKAFTA in the U.S., label expansions for KALYDECO and ORKAMBI and expanded access to our medicines in ex-U.S. markets. In 2020, 2019 and 2018, our net product revenues included product revenues of $1.4 billion, $1.1 billion and $682.4 million, respectively, from ex-U.S. markets.markets were as follows:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2023 | | % Change | | 2022 | | % Change | | 2021 |
| (in millions, except percentages) |
| United States | $ | 6,040.4 | | | 6% | | $ | 5,699.3 | | | 8% | | $ | 5,287.3 | |
| ex-U.S. | 3,828.8 | | | 18% | | 3,231.4 | | | 41% | | 2,286.1 | |
| Product revenues, net | $ | 9,869.2 | | | 11% | | $ | 8,930.7 | | | 18% | | $ | 7,573.4 | |
We expect that our CF net product revenues will increase in 2021 due to increasing numbers of people being treated with our medicines2024 as a result of the continued uptakeperformance of TRIKAFTA, the approval of TRIKAFTA/KAFTRIO, by the European Commission, label expansions into younger age groups for our previously approved products, and expanded access to our medicines.
Upon reaching an agreement with the French government for ORKAMBI Starting in the fourth quarter of 2019, including the final amount2023, we have received approval for ORKAMBI distributed through early access programs, we recognized an adjustment to increase net product revenues related to prior period shipments of ORKAMBI distributed through early access programs of $155.8 million. Please refer to “Critical Accounting Policies - Revenue Recognition” below for a discussion of our accounting treatment for our early access program for ORKAMBI in France.
Collaborative and Royalty Revenues
Our collaborative and royalty revenues were $2.9 million, $2.1 million and $9.3 million in 2020, 2019 and 2018, respectively. Our collaborative revenues have historically fluctuated significantly from one period to another and may continue to fluctuateCASGEVY in the future. Our future royalty revenues willU.S., the E.U., the U.K., Saudi Arabia, and Bahrain for the treatment of SCD and TDT. We expect 2024 to be dependent on if, and when, our collaborators are ablea foundational year for CASGEVY, as we lay the groundwork for the first patients to successfully develop drug candidates that we have out-licensed to them.initiate this multi-month treatment.
Operating Costs and Expenses
| | 2020/2019 Comparison | | 2019/2018 Comparison |
| Increase/(Decrease) | | Increase/(Decrease) |
| 2020 | | 2019 | | 2018 | | $ | | % | | $ | | % |
| (in thousands, except percentages) |
| 2023 | | | 2023 | | % Change | | 2022 | | % Change | | 2021 |
| (in millions, except percentages) | | | (in millions, except percentages) |
| Cost of sales | Cost of sales | $ | 736,300 | | | $ | 547,758 | | | $ | 409,539 | | | $ | 188,542 | | | 34 | % | | $ | 138,219 | | | 34 | % |
| Research and development expenses | Research and development expenses | 1,829,537 | | | 1,754,540 | | | 1,416,476 | | | 74,997 | | | 4 | % | | 338,064 | | | 24 | % |
| Sales, general and administrative expenses | 770,456 | | | 658,498 | | | 557,616 | | | 111,958 | | | 17 | % | | 100,882 | | | 18 | % |
| Acquired in-process research and development expenses | |
| Selling, general and administrative expenses | |
| Change in fair value of contingent consideration | Change in fair value of contingent consideration | 13,100 | | | 4,459 | | | — | | | 8,641 | | | 194 | % | | 4,459 | | | ** |
| Restructuring income | — | | | — | | | (184) | | | — | | | ** | | 184 | | | ** |
| Intangible asset impairment charge | — | | | — | | | 29,000 | | | — | | | ** | | (29,000) | | | ** |
| Total costs and expenses | Total costs and expenses | $ | 3,349,393 | | | $ | 2,965,255 | | | $ | 2,412,447 | | | $ | 384,138 | | | 13 | % | | $ | 552,808 | | | 23 | % |
| | ** Not meaningful |
| | | | | | | ** Not meaningful | |
| | | | | | | ** Not meaningful | |
| | | | | | | ** Not meaningful | |
Beginning in 2022, we separately classify upfront, contingent milestone, or other payments pursuant to our business development transactions, including collaborations, licenses of third-party technologies, and asset acquisitions as “Acquired in-process research and development expenses,” in our consolidated statements of income in cases where such acquired assets do not have an alternative future use. To conform prior periods to our current presentation, we have reclassified $1.1 billion from “Research and development expenses” to AIPR&D for 2021.
Cost of Sales
Our cost of sales primarily consists of the third-party royalties payable on our net sales of our products as well as the cost of producing inventories that corresponded to product revenues for the reporting period.inventories. Pursuant to our agreement with the Cystic Fibrosis Foundation, or CFF, our tiered third-party royalties on sales of TRIKAFTA/KAFTRIO, SYMDEKO/SYMKEVI, KALYDECO, and ORKAMBI, calculated as a percentage of net sales, range from the single digits to the sub-teens, with lower royalties on sales of TRIKAFTA/KAFTRIO slightly lower than for our other products. Over the last several years, our cost of sales has been increasing due to increased net product revenues. Our cost of sales as a percentage of our net product revenues was approximately13% and 12%, 13%, in 2023 and 13% for 2020, 2019 and 2018,2022, respectively. In 2021,2024, we expect our total cost of sales, including as a percentage of our net product revenues, will increase due to expected increases in our CF net product revenues and our cost of sales as a percentage of total net product revenues will be similar to our cost of sales as a percentage of total net product revenues in 2020.costs associated with CASGEVY.
Research and Development Expenses
| | 2020/2019 Comparison | | 2019/2018 Comparison |
| Increase/(Decrease) | | Increase/(Decrease) |
| 2020 | | 2019 | | 2018 | | $ | | % | | $ | | % |
| (in thousands, except percentages) |
| 2023 | | | 2023 | | % Change | | 2022 | | % Change | | 2021 |
| (in millions, except percentages) | | | (in millions, except percentages) |
| Research expenses | Research expenses | $ | 636,759 | | | $ | 732,772 | | | $ | 438,360 | | | $ | (96,013) | | | (13) | % | | $ | 294,412 | | | 67 | % |
| Development expenses | Development expenses | 1,192,778 | | | 1,021,768 | | | 978,116 | | | 171,010 | | | 17 | % | | 43,652 | | | 4 | % |
| Total research and development expenses | Total research and development expenses | $ | 1,829,537 | | | $ | 1,754,540 | | | $ | 1,416,476 | | | $ | 74,997 | | | 4 | % | | $ | 338,064 | | | 24 | % |
Our research and development expenses include internal and external costs incurred for research and development of our drugsproducts and drug candidates and expenses related to certain technology that we acquire or license through business development transactions.product candidates. We do not assign our internal costs, such as salary and benefits, stock-based compensation expense, laboratory supplies and other direct expenses and infrastructure costs, to individual drugsproducts or drugproduct candidates, because the employees within our research and development groups typically are deployed across multiple research and development programs. These internal costs are significantly greater than ourWe assign external costs, such as the costs of services provided to us by clinical research organizations and other outsourced research which we allocate by individual program. Our internal costs are greater than our external costs. All research and development costs for our drugsproducts and drugproduct candidates are expensed as incurred.
Over the past three years, we have incurred $5.0$9.4 billion in total research and development and AIPR&D expenses associated with drugproduct discovery and development. The successful development of our drugproduct candidates is highly uncertain and subject to a number of risks. In addition, the duration of clinical trials may vary substantially according to the type, complexity and novelty of the drugproduct candidate and the disease indication being targeted. The FDA and comparable agencies in foreign countries impose substantial requirements on the introduction of therapeutic pharmaceutical products, typically requiring lengthy and detailed laboratory and clinical testing procedures, sampling activities and other costly and time-consuming procedures. Data obtained from nonclinical and clinical activities at any step in the testing process may be adverse and lead to discontinuation or redirection of development activities. Data obtained from these activities also are susceptible to varying interpretations,
which could delay, limit or prevent regulatory approval. The duration and cost of
discovery, nonclinical studies and clinical trials may vary significantly over the life of a project and are difficult to predict. Therefore, accurate and meaningful estimates of the ultimate costs to bring our drugproduct candidates to market are not available.
In 2020, 2019 and 2018, costs related to our CF programs represented the largest portion of our development costs. Any estimates regarding development and regulatory timelines for our drugproduct candidates are highly subjective and subject to change. Until we have data from Phase 3 clinical trials, we cannot make a meaningful estimate regarding when, or if, a clinical development program will generate revenues and cash flows.
Research Expenses
| | 2020/2019 Comparison | | 2019/2018 Comparison |
| Increase/(Decrease) | | Increase/(Decrease) |
| 2020 | | 2019 | | 2018 | | $ | | % | | $ | | % |
| (in thousands, except percentages) |
| 2023 | | | 2023 | | Change % | | 2022 | | Change % | | 2021 |
| (in millions, except percentages) | | | (in millions, except percentages) |
| Research Expenses: | Research Expenses: | |
| Salary and benefits | |
| Salary and benefits | |
| Salary and benefits | Salary and benefits | $ | 129,835 | | | $ | 134,642 | | | $ | 87,773 | | | $ | (4,807) | | | (4) | % | | $ | 46,869 | | | 53 | % |
| Stock-based compensation expense | Stock-based compensation expense | 85,609 | | | 69,417 | | | 62,925 | | | 16,192 | | | 23 | % | | 6,492 | | | 10 | % |
| Outsourced services and other direct expenses | Outsourced services and other direct expenses | 116,182 | | | 116,575 | | | 89,355 | | | (393) | | | — | % | | 27,220 | | | 30 | % |
| Collaborative payments | 184,600 | | | 307,828 | | | 111,600 | | | (123,228) | | | (40) | % | | 196,228 | | | 176 | % |
| Intangible asset impairment charge | |
| Infrastructure costs | Infrastructure costs | 120,533 | | | 104,310 | | | 86,707 | | | 16,223 | | | 16 | % | | 17,603 | | | 20 | % |
| Total research expenses | Total research expenses | $ | 636,759 | | | $ | 732,772 | | | $ | 438,360 | | | $ | (96,013) | | | (13) | % | | $ | 294,412 | | | 67 | % |
| | | | | | | ** Not meaningful | |
| | | | | ** Not meaningful | |
| | | | | ** Not meaningful | |
We expect to continue to invest in our research programs with a focus on identifying drug candidates with the goal of creating transformative medicines for serious diseases. Our total research expenses have historically fluctuated, and are expected to continue to fluctuate, from one period to another due to upfront and milestone payments related to our business development activities that are reflected in the preceding table as collaborative payments. Our research expenses excluding these collaborative payments, have been increasing over the last several years as we have invested in our pipeline and expanded our cell and genetic therapies capabilities.therapy capabilities, resulting in increased headcount, outside services and other direct expenses, and infrastructure costs associated with our research facilities. We expect to continue to invest in our research programs with a focus on creating transformative medicines for serious diseases.
Development Expenses
| | 2020/2019 Comparison | | 2019/2018 Comparison |
| Increase/(Decrease) | | Increase/(Decrease) |
| 2020 | | 2019 | | 2018 | | $ | | % | | $ | | % |
| (in thousands, except percentages) |
| 2023 | | | 2023 | | Change % | | 2022 | | Change % | | 2021 |
| (in millions, except percentages) | | | (in millions, except percentages) |
| Development Expenses: | Development Expenses: | |
| Salary and benefits | |
| Salary and benefits | |
| Salary and benefits | Salary and benefits | $ | 295,744 | | | $ | 249,860 | | | $ | 220,128 | | | $ | 45,884 | | | 18 | % | | $ | 29,732 | | | 14 | % |
| Stock-based compensation expense | Stock-based compensation expense | 177,081 | | | 155,141 | | | 140,187 | | | 21,940 | | | 14 | % | | 14,954 | | | 11 | % |
| Outsourced services and other direct expenses | Outsourced services and other direct expenses | 512,157 | | | 425,149 | | | 471,338 | | | 87,008 | | | 20 | % | | (46,189) | | | (10) | % |
| Collaborative payments | — | | | 10,440 | | | 250 | | | (10,440) | | | ** | | 10,190 | | | ** |
| Infrastructure costs | Infrastructure costs | 207,796 | | | 181,178 | | | 146,213 | | | 26,618 | | | 15 | % | | 34,965 | | | 24 | % |
| Total development expenses | Total development expenses | $ | 1,192,778 | | | $ | 1,021,768 | | | $ | 978,116 | | | $ | 171,010 | | | 17 | % | | $ | 43,652 | | | 4 | % |
| | | ** Not meaningful |
Our development expenses increased by $171.0$543.7 million, or 17%28%, in 20202023 as compared to 2019 and increased by $43.7 million, or 4%, in 2019 as compared to 2018,2022, primarily due to increased expensescosts to support clinical trials and drug supply associated with our advancing pipeline programs, including pain and T1D, and our stock-based compensation expense. We are investing significantly in headcount, leveraging outsourced services, and in infrastructure to support these programs. Our stock-based compensation expense increased in 2023 as compared to 2022, primarily due increased headcount eligible for our employee stock programs and the timing of expense recognition related to our diversifying pipeline, including clinical trials, headcount and infrastructure costs.certain performance-based restricted stock units. We expect our development expenses to continue to increase in 2021 as a result of2024 due to our advancing pipeline.pipeline programs, including our T1D program.
Acquired In-process Research and Development Expenses
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2023 | | % Change | | 2022 | | % Change | | 2021 |
| (in millions, except percentages) |
| Acquired in-process research and development expenses | $ | 527.1 | | | 356% | | $ | 115.5 | | | (90)% | | $ | 1,113.3 | |
AIPR&D in 2023 was primarily related to our $225.1 million upfront payment to Entrada, $100.0 million upfront
Sales,payment and $70.0 million T1D research milestone to CRISPR, $47.5 million acquisition of a novel GPCR program from Septerna, and various other payments. AIPR&D in 2022 was primarily related to the $60.0 million payment to acquire Catalyst’s complement portfolio, a $25.0 million upfront payment pursuant to our license agreement with Verve, and various other payments. Our AIPR&D has historically fluctuated, and is expected to continue to fluctuate, from one period to another due to upfront, contingent milestone, and other payments pursuant to our existing and future business development transactions, including collaborations, licenses of third-party technologies, and asset acquisitions.
AIPR&D in 2021 primarily related to the $900.0 million upfront payment we made to CRISPR in connection with the A&R JDCA.
Selling, General and Administrative Expenses
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | 2020/2019 Comparison | | 2019/2018 Comparison |
| | | | | | | Increase/(Decrease) | | Increase/(Decrease) |
| 2020 | | 2019 | | 2018 | | $ | | % | | $ | | % |
| (in thousands, except percentages) |
| Sales, general and administrative expenses | $ | 770,456 | | | $ | 658,498 | | | $ | 557,616 | | | $ | 111,958 | | | 17 | % | | $ | 100,882 | | | 18 | % |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 2023 | | % Change | | 2022 | | % Change | | 2021 |
| (in millions, except percentages) |
| Selling, general and administrative expenses | $ | 1,136.6 | | | 20% | | $ | 944.7 | | | 12% | | $ | 840.1 | |
Sales,Selling, general and administrative expenses increased by 17%20% in 20202023 as compared to 2019, and by 18% in 2019 as compared to 2018,2022, primarily due to increased global support for our medicines, including incrementalthe continued investment to support the launchcommercialization of our triple combination regimenmedicines, including increased commercial headcount, outsourced services to support our pipeline product candidates and increased support for our CF pipeline products and other disease areas.stock-based compensation expense. We expect our sales,selling, general and administrative expenses to continue to increase in 2021.2024 as we progress our efforts in preparation for the potential commercialization of VX-548 and launch CASGEVY.
Contingent Consideration
In 2020 and 2019, the increase in2023, the fair value of our contingent consideration potentially payable to Exonics’ former equity holders was $13.1decreased by $51.6 million and $4.5 million, respectively, primarily due to changesour determination that additional pre-clinical studies of the delivery system for our gene-editing components for Duchenne muscular dystrophy (“DMD”) will be required. In 2022, the fair value of our contingent consideration decreased by $57.5 million, primarily as a result of a revision to the scope of certain gene-editing programs in market interest rates. There were no similar amounts in 2018.the second quarter of 2022. In future periods, we expect the fair value of contingent consideration to increase or decrease based on, among other things, our estimates of the probability of achieving and the timing of these contingent development and regulatory milestone payments, as well as the time value of money and changes in market interest rates.
Intangible Asset Impairment Charge
In 2018, we recorded a $29.0 million impairment charge related to VX-210 that was licensed from BioAxone Biosciences, Inc., or BioAxone, in 2014. This charge was attributable to non-controlling interest on our consolidated statement of operations because we consolidated BioAxone as a variable interest entity, or VIE, until December 31, 2018. There were no corresponding intangible asset impairment charges in 2020 or 2019.
Other Non-Operating Income (Expense), Net
Interest Income
Interest income increased from $38.4to $614.7 million in 2018 to $63.7 million in 2019 and decreased to $22.2 million in 2020. The increase in our interest income in 20192023, as compared to 2018 was$144.6 million in 2022, primarily due to increases in our cash equivalents and available-for-sale debt securities and prevailing market interest rates. The decrease in our interest income in 2020 as compared to 2019 was primarily due to a decrease in prevailingincreased market interest rates despite continued increases in ourand increased cash equivalents and available-for-sale debt securities. Our future interest income will beis dependent on the amount of, and prevailing market interest rates on, our outstanding cash equivalents and available-for-sale debt securities.
Interest Expense
Interest expense was $58.2$44.1 million in 2020, $58.52023, as compared to $54.8 million in 2019 and $72.5 million in 2018.2022. The majority of our interest expense in these periods was related to imputed interest expense associated with our leased corporate headquarters in Boston and our research site in San Diego. On January 1, 2019, we adopted ASC 842, Leases, which resulted in a reduction in our imputed interest expense associated with these leases in 2020 and 2019. Our future interest expense will be dependent on whether, and to what extent, we borrow amounts under our credit facilities.Boston.
Other Income (Expense), Net
In 2020 and 2019, we recordedOther income (expense), net other incomewas expenses of $296.4$22.8 million and $192.2$164.8 million respectively,in 2023 and 2022, respectively. These amounts related primarily related to net unrealized gains or losses resulting from changes in the fair value or sales of certain of our strategic investments. In 2018,equity investments, which consist of investments in our collaborators that may be public or private companies. To the extent that we recorded net other expense of $0.8 million. Wecontinue to hold strategic equity investments in publicly traded companies, we expect that due to the volatility of the stock price of biotechnology companies, our other income (expense), net will fluctuate in future periods based on increases or decreases in the fair value of our strategic equity investments. As of December 31, 2023, these investments had a fair value of $46.0 million. We discuss these potential future fluctuations further in Item 7a. Quantitative and Qualitative Disclosures About Market Risk.
Noncontrolling Interest (VIEs)
In 2018, our $9.8 million net loss attributable to noncontrolling interest reflects BioAxone’s net loss for the reporting period. We deconsolidated BioAxone from our consolidated financial statements as of December 31, 2018 and did not consolidate any VIEs into our consolidated financial statements in 2020 or 2019.
Income Taxes
Our effective tax rate fluctuates from year to year due to the global nature of our operations. The factors that most significantly impact our effective tax rate include changes in tax laws, variability in the allocation of our taxable earnings among multiple jurisdictions, the amount and characterization of our research and development expenses, the levels of certain deductions and credits, adjustments to the value of our uncertain tax positions, acquisitions and third-party collaboration and licensing transactions.
Our provision for income taxes was $405.2$760.2 million for 20202023 and $218.1$910.4 million for 2019.2022. Our effective tax rate of 13%17.4% for 20202023 was lower than the U.S. statutory rate primarily due to (i) discretea benefit from a research and development tax benefits associated with the $209.0 million transfer of intellectual property rights to the U.K., the write-off of a long-term intercompany receivable,credit study that was completed in 2023 and an increase in the U.K.’s corporate tax rate; and (ii) excess tax benefits related to stock-based compensation. The impact of these items wascompensation, partially offset by a U.S. deemed dividend. changes in uncertain tax positions.
Our effective tax rate of 16%21.5% for 20192022 was lowerhigher than the U.S. statutory rate primarily due to an increase in our uncertain tax positions associated with intercompany transfer pricing matters partially offset by excess tax benefits related to stock-based compensation, tax credits, and research and developmentchanges in our estimated prior-year tax credits.
In 2018, we recorded a benefit from income taxes of $1.5 billion because we released our valuation allowance on the majority of our net operating losses and other deferred tax assets in the fourth quarter of 2018, resulting in a non-cash credit to net income of $1.56 billion. Starting in 2019, we began recording a provision for income taxes on our pre-tax income using an effective tax rate approximating statutory rates. Due to our ability to offset our pre-tax income against previously benefited net operating losses, the majority of our tax provision in 2020 and 2019 represented a non-cash expense. We utilized substantially all of our remaining previously benefited U.S. net operating losses in 2020. As a result, a larger portion of our tax provision will represent a cash tax payable in future periods.liabilities.
LIQUIDITY AND CAPITAL RESOURCES
The following table summarizes the components of our financial condition as of December 31, 20202023 and 2019:2022:
| | 2023 | | | 2023 | | 2022 | | % Change |
| (in millions, except percentages) | | | (in millions, except percentages) |
| Cash, cash equivalents and marketable securities: | |
| Cash and cash equivalents | |
| Cash and cash equivalents | |
| Cash and cash equivalents | |
| Marketable securities | |
| Marketable securities | |
| Marketable securities | |
| Long-term marketable securities | |
| Long-term marketable securities | |
| Long-term marketable securities | |
| Total cash, cash equivalents and marketable securities | |
| Total cash, cash equivalents and marketable securities | |
| Total cash, cash equivalents and marketable securities | | $ | 13,716.1 | | | $ | 10,890.7 | | | 26% |
| | Increase/(Decrease) |
| 2020 | | 2019 | | $ | | % |
| (in thousands, except percentages) |
| Cash, cash equivalents and marketable securities | $ | 6,658,897 | | | $ | 3,808,294 | | | $ | 2,850,603 | | | 75 | % |
| Working Capital: | Working Capital: | |
| Working Capital: | |
| Working Capital: | |
| Total current assets | |
| Total current assets | |
| Total current assets | Total current assets | $ | 8,133,379 | | | $ | 4,822,829 | | | $ | 3,310,550 | | | 69 | % | $ | 14,144.2 | | | $ | | $ | 13,234.8 | | | 7% | | 7% |
| Total current liabilities | Total current liabilities | (1,877,533) | | | (1,334,827) | | | (542,706) | | | (41) | % | Total current liabilities | (3,547.4) | | | (2,742.1) | | (2,742.1) | | | 29% | | 29% |
| Total working capital | Total working capital | $ | 6,255,846 | | | $ | 3,488,002 | | | $ | 2,767,844 | | | 79 | % | Total working capital | $ | 10,596.8 | | | $ | | $ | 10,492.7 | | | 1% | | 1% |
|
As of December 31, 2020,Working Capital
Our total working capital was $6.3 billion, which represented an increase of $2.8 billion from $3.5$10.6 billion as of December 31, 2019. The increase in2023 was similar to our total working capital of $10.5 billion as of December 31, 2022 primarily due to $3.8 billion of income from operations mostly offset by net purchases of long-term marketable securities of $2.4 billion, income tax payments and repurchases of our common stock of $427.6 million.
Cash Flows
| | | | | | | | | | | | | | | | | | | | | |
| 2023 | | | | 2022 | | | | 2021 |
| (in millions) |
| Net cash provided by (used in): | | | | | | | | | |
| Operating activities | $ | 3,537.3 | | | | | $ | 4,129.9 | | | | | $ | 2,643.5 | |
| Investing activities | $ | (3,141.7) | | | | | $ | (321.1) | | | | | $ | (340.9) | |
| Financing activities | $ | (562.2) | | | | | $ | (67.7) | | | | | $ | (1,478.0) | |
Operating Activities
Cash provided by operating activities was $3.5 billion in 20202023 as compared to $4.1 billion in 2022. The largest driver of
the decrease in cash provided by operating activities in 2023 as compared to 2022 was an increase in cash paid for income taxes.
Investing Activities
Cash used in investing activities was $3.1 billion and $321.1 million in 2023 and 2022, respectively. In 2023, our investing activities primarily related to $3.3 billionnet purchases of cash provided by operations partially offset by $539.1available-for-sale debt securities of $2.9 billion. In 2022, our investing activities included a net payment of $295.9 million of cash used to repurchase our common stock pursuant our share repurchase programsacquire ViaCyte and $204.7 million in purchases of property and equipment, partially offset by net sales and maturities of $259.8available-for-sale debt securities of $227.3 million.
Financing Activities
Cash used in financing activities was $562.2 million and $67.7 million in 2023 and 2022, respectively. In 2023, our financing activities primarily related to repurchases of our common stock pursuant to our share repurchase program and payments related to our employee stock benefit plans. In 2022, the largest portions of our financing activities were payments related to our employee stock benefit plans and payments on finance leases.
Sources and Uses of Liquidity
As of December 31, 2020,2023, we had current cash, cash equivalents, and marketable securities of $6.7$11.2 billion, which represented an increase of $2.9 billion$439.8 million from $3.8$10.8 billion as of December 31, 2019.2022. We intend to rely on our existing cash, cash equivalents and current marketable securities together with cash flows from product sales as our primary source of liquidity.
We may borrow up to a total of $2.5 billion pursuant to two revolving credit facilities. We may repay and reborrow amounts under these revolving credit agreements without penalty. Subject to certain conditions, we may request that the borrowing capacity for each of the credit agreements be increased by an additional $500.0 million, for a total of $3.5 billion collectively. Other possible sources of future liquidity include commercial debt, public and private offerings of our equity and debt securities, strategic sales of assets or businesses and financial transactions. Negative covenants in our credit agreement may prohibit or limit our ability to access these sources of liquidity. As of December 31, 2020, we were in compliance with these covenants.
Future Capital Requirements
We have significant future capital requirements including:
•significant expected operating expenses to conduct research and development activities and to operate our organization; and
•substantial facility and finance lease obligations.
In addition:
•We have entered into certain collaboration agreements with third parties that include the funding of certain research, development and commercialization efforts. Certain of our business development transactions, including collaborations and acquisitions, include the potential for future milestone and royalty payments by us upon the achievement of pre-established developmental and regulatory targets and/or commercial targets. We may enter into additional business development transactions, including acquisitions, collaborations and equity investments, that require additional capital.
•To the extent we borrow amounts under the credit agreements we entered into in 2020 and 2019, we would be required to repay any outstanding principal amounts in 2022 or 2024, respectively.
•As of December 31, 2020, $424.9 million remained available to fund repurchases under the 2020 Share Repurchase Program that we announced in November 2020.
We expect that cash flows from our productsproduct sales together with our current cash, cash equivalents and current marketable securities will be sufficient to fund our operations for at least the next twelve months and do not expect COVID-19 to have an adverse effect on our liquidity.months. The adequacy of our available funds to meet our future operating and capital requirements will depend on many factors, including the amounts ofour future revenues generated by our products,product sales, and the potential introduction of one or more of our other drugproduct candidates to the market, the level of our business development activities and the number, breadth, cost and prospects of our research and development programs.
Credit Facilities & Financing Strategy
We may borrow up to a total of $500.0 million pursuant to a revolving credit facility that we entered into in July 2022 and could repay and reborrow amounts under this revolving credit agreement without penalty. Subject to certain conditions, we could request that the borrowing capacity be increased by an additional $500.0 million, for a total of $1.0 billion. Negative covenants in our credit agreement could prohibit or limit our ability to access this source of liquidity. As of December 31, 2023, the facility was undrawn, and we were in compliance with these covenants.
We may also raise additional capital by borrowing under credit agreements, through public offerings or private placements of our securities, or securing new collaborative agreements or other methods of financing. We will continue to manage our capital structure and will consider all financing opportunities, whenever they may occur, that could strengthen our long-term liquidity profile. There can be no assurance that any such financing opportunities will be available on acceptable terms, if at all.
Future Capital Requirements
We have significant future capital requirements, including:
•Cash that we pay for income taxes.
CONTRACTUAL COMMITMENTS AND OBLIGATIONS•Royalties we pay related to sales of our CF products.
The following table sets forth our commitments•Facility, operating and finance lease obligations as of December 31, 2020:described below.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Payments Due by Period |
| 2021 | | 2022-2023 | | 2024-2025 | | 2026 and later | | Total |
| (in thousands) |
| Fan Pier Leases | $ | 66,540 | | | $ | 145,177 | | | $ | 155,942 | | | $ | 233,913 | | | $ | 601,572 | |
| Finance leases, excluding Fan Pier Leases | 18,930 | | | 33,028 | | | 30,913 | | | 182,542 | | | 265,413 | |
| Operating leases | 15,266 | | | 68,782 | | | 65,677 | | | 320,684 | | | 470,409 | |
| Research and development costs | 66,955 | | | 1,724 | | | — | | | — | | | 68,679 | |
| Total contractual commitments and obligations | $ | 167,691 | | | $ | 248,711 | | | $ | 252,532 | | | $ | 737,139 | | | $ | 1,406,073 | |
Leases
We lease two buildings that are located at Fan Pier in Boston, Massachusetts. We commenced lease payments on these two buildings in December 2013 and the initial lease periods end in December 2028. We also lease office and laboratoryIn addition, other potential significant future capital requirements may include:
space in San Diego, California. We commenced lease payments for this building in the second quarter of 2019 pursuant to an initial 16 year lease term. The future minimum rental payments that we are obligated to pay related to the San Diego building are included in “Finance leases, excluding Fan Pier Leases,” which also reflects leases of equipment and a land lease. The remainder of our real estate leases are reflected in “Operating leases” in the table above, including office and laboratory space at our Cell and Genetic Therapies facility near our corporate headquarters. Base rent payments will commence for this building in the fourth quarter of 2021 pursuant to an initial 15 year lease term.
Research and Development Costs
“Research and development costs” included in the table above primarily relate to pharmaceutical materials to be utilized in our clinical trials and research costs related to our advancing pipeline. The amounts reflected in “Research and development costs” do not include certain payments we anticipate making to clinical research organizations, or CROs, because these contracts are cancelable, at our option, with notice. However, we historically have not cancelled such contracts. As of December 31, 2020, we had accrued $42.8 million related to these contracts for costs incurred for services provided through December 31, 2020, and we have approximately $237.6 million in cancelable future commitments based on existing contracts as of December 31, 2020. These amounts reflect planned expenditures based on existing contracts and do not reflect any future modifications to, or terminations of, existing contracts or anticipated or potential new contracts.
Collaborative Arrangements and Asset Acquisitions
•We have entered into certain researchbusiness development-related and development collaborationstrategic agreements with third parties and acquired certain assets that include the funding of certain research, development, manufacturing and commercialization efforts withefforts. Certain of our transactions, including collaborations, licensing arrangements, and asset acquisitions, include the potential for future milestone and royalty payments by us upon the achievement of pre-established developmental and regulatory targets and/or commercial targets. Other transactions include the potential for future lease-related expenses and other costs. Our obligation to fund these research and development and commercialization efforts and to pay these potential milestones, expenses and royalties is contingent upon continued involvement in the programs and/or the lack of any adverse events that could cause the discontinuance of the programs. These payments,their discontinuance. We may enter into additional business development transactions and strategic agreements, including acquisitions, collaborations, licensing arrangements and equity investments, which are not included in the contractual obligations table above, include:
•CFF: We pay royalties, which are included in cost of sales, to CFF on sales of our CF products.require additional capital.
•Research and Development Milestones: The majority ofTo the extent we borrow amounts under our in-license agreements and our acquisitions have milestone and royalty payments payable by us upon the successful achievement of pre-established developmental, regulatory and/or commercial targets or net sales. Contingent payments under these agreements become due and payable only upon achievement of certain milestones.existing credit agreement, we would be required to repay any outstanding principal amounts in 2027.
Tax-related Obligations
We exclude liabilities pertaining to uncertain tax positions from our summary of contractual obligations as we cannot make a reliable estimate of the period of cash settlement with the respective taxing authorities. •As of December 31, 2020,2023, we had $2.6 billion remaining authorization available under our liabilities associated with uncertain tax positions were $86.6 million.$3.0 billion Share Repurchase Program that our Board of Directors approved in February 2023. We expect to fund repurchases of our common stock through a combination of cash on hand and cash generated by operations. This program does not have an expiration date and can be discontinued at any time.
Other Funding CommitmentsAdditional information on several of our future capital requirements is provided below.
Research and Development Costs
At any point in time, we have several ongoing clinical trials at various stages of clinical development. Our clinical trial costs are dependent on, among other things, our research activities advancing to later-stage clinical development as well as the size, number, and length of our clinical trials.
Leases
Finance Leases
Our table detailing contractual commitmentscorporate headquarters is in two buildings that we lease at Fan Pier in Boston, Massachusetts. We commenced lease payments on these buildings in 2013 and obligations does not include severance payment obligationsthe initial lease periods end in December 2028. We also lease office and laboratory space in San Diego, California. We commenced lease payments for this building in 2019 pursuant to certainan initial 16-year lease term. We account for each of our executive officers in the event of a not-for-cause employment termination under existing employment contracts. these buildings as finance leases.
Operating Leases
We will provide information regarding these obligations annually in our proxy statementaccount for our annual meetingremaining real estate leases as operating leases, including office and laboratory space at the Jeffrey Leiden Center for Cell and Genetic Therapies near our corporate headquarters. Base rent payments commenced in 2021 pursuant to an initial 15-year lease term for this building.
Our total future minimum lease payments for our finance and operating leases for each of shareholders.the next five years and in total are included in Note L, “Leases.” The total future undiscounted minimum lease payments were $583.0 million and $436.1 million related to our finance and operating leases, respectively, as of December 31, 2023.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
Our discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements prepared in accordance with generally accepted accounting principles in the U.S. The preparation of these financial statements requires us to make certain estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reported periods. These items are monitored and analyzed by management for changes in facts and circumstances, and material changes in these estimates could occur in the future. Changes in estimates are reflected in reported results for the period in which the change occurs. We base our estimates on historical experience and various other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from our estimates if past experience or other assumptions do not turn out to be substantially accurate.
We believe that our application of the following accounting policies, each of which requires significant judgments and estimates on the part of management, are the most critical to aid in fully understanding and evaluating our reported financial results:
•revenue recognition;
•acquisitions, including intangible assets, goodwill and contingent consideration; and
•income taxes.
Our accounting policies, including the ones discussed below, are more fully described in the Notes to our consolidated financial statements, including Note A, “Nature of Business and Accounting Policies,Policies.” included in this Annual Report on Form 10-K.
Revenue Recognition
CF Product Revenues, Net
We generate CF product revenues from sales in the U.S. and in international markets. We sell our CF products principally to a limited number of specialty pharmacy and specialty distributors in the U.S., which account for the largest portion of our total revenues, and make international sales primarily to specialty distributors and retail chains, as well as hospitals and clinics, many of which are government-owned or supported customers, collectively, our customers.revenues. Our customers in the U.S. subsequently resell our products to patients and health care providers. We contract with government agencies so that our products will be eligible for purchase by, or partial or full reimbursement from, such third-party payors. We make international sales primarily through distributor arrangements and to retail pharmacies, as well as to hospitals and clinics, many of which are government-owned or supported customers. We recognize net CF product revenues from sales of our products when our customers obtain control of our products, which typically occurs upon delivery to our customers. Revenues from our CF product sales are recorded at the net sales price, or “transactiontransaction price,” which requires us to make several significant estimates regarding the net sales price.
The most significant estimate we are required to make for our CF product revenues is related to government and private payor rebates, chargebacks, discounts and fees, collectively rebates. The valuevalues of the rebates provided to third-party payors per course of treatment vary significantly and are based on government-mandated discounts and our arrangements with other third-party payors. In order toTo estimate our total rebates, we estimate the percentage of prescriptions that will be covered by each third-party payor, which is referred to as the payor mix. We track available information regarding changes, if any, to the payor mix for our products, to our contractual terms with third-party payors and to applicable governmental programs and regulations and levels of our products in the distribution channel. We adjust our estimated rebates based onupon new information as it becomes available, including information regarding actual rebates for our products, as it becomes available.products. Claims by third-party payors for rebates are submitted to us significantly after the related sales, potentially resulting in adjustments in the period in which the new information becomes known. Our credits to revenue related to prior period sales, excluding the adjustment to the transaction price for ORKAMBI distributed through early access programs in France in 2019, have not been significant (typically less than 1% of gross product revenues) and primarily related to U.S. rebates.
The following table summarizes activity related to our CF product revenue accruals for rebates (including our refund liability to the French government related to ORKAMBI distributed through early access programs in France as described below) for the three years ended December 31, 2020:2023, 2022 and 2021:
| | | | | |
| (in thousands)millions) |
Balance as of December 31, 20172020 | $ | 112,215775.6 | |
Provision related to 20182021 sales and the adoption of ASC 606 | 2,126.1 | | 684,299 |
| Adjustments related to prior year(s) sales | (22,099)(27.6) | |
| Credits/payments made | (229,361)(2,035.5) | |
Balance as of December 31, 20182021 | $ | 545,054838.6 | |
Provision related to 20192022 sales | 2,977.2 | | 655,980 |
| Adjustments related to prior year(s) sales | (95,480)(10.4) | |
| Credits/payments made | (469,832)(2,514.0) | |
Balance as of December 31, 20192022 | $ | 635,7221,291.4 | |
Provision related to 20202023 sales | 3,481.4 | | 1,284,068 |
| Adjustments related to prior year(s) sales | 631 (6.5) | |
| Credits/payments made | (1,144,832)(3,064.7) | |
Balance as of December 31, 20202023 | $ | 775,5891,701.6 | |
We have also entered into annual contracts with government-owned and supported customers in international markets that limit the amount of annual reimbursement we can receive.receive for our CF products. Upon exceeding the annual reimbursement amount provided by the customer’s contract with us, products are provided free of charge, which is a material right. WeIf we estimate that the annual reimbursement amount under a contract will be exceeded for an annual period, we defer a portion of the consideration received, which includes upfront payments and fees, for shipments made up to the annual reimbursement limit as “Other current liabilities.” TheOnce the annual reimbursement limit has been reached, we recognize the deferred amount is recognized as revenue when we ship the free products are shipped. In order toproducts. To estimate the portion of the consideration received to recognizebe recognized as revenue and the portion of the amount to defer,be deferred, we rely on our forecast of the number of units we will distribute during the applicable annual period in each international market in which our contracts with government-owned and supported customers limit the amount of annual reimbursement we can receive. Our forecasts are based on, among other things, our historical experience.
The preceding estimates and judgments materially affect our recognition of net CF product revenues. Changes in our estimates of net CF product revenues could have a material effect on net CF product revenues recorded in the period in which we determine that change occurs.
French Early Access ProgramsAcquisitions
In 2015,As part of our business strategy, we began distributing ORKAMBI through early access programsseek to acquire products, product candidates and other technologies and businesses that are aligned with our corporate and research and development strategies and complement and advance our ongoing research and development efforts. If we determine that substantially all the fair value associated with an acquisition is concentrated in France and remained engaged in reimbursement discussions with the French government for ORKAMBI, including ORKAMBI distributed through early access programs, until November 2019, when we reached an agreement with the French government. From the time we began distributing ORKAMBI through early access programs in France, we expected that the difference between the amounts collected based on the invoiced amounta single asset, and the final amountacquisition does not constitute a business, we account for ORKAMBI distributed through these programs would be returnedit as an asset acquisition. For an asset acquisition involving rights to the French government. Our refund liabilityintellectual property related to in-process research and development that is not yet associated with a product that has achieved regulatory approval, we generally record our upfront payment to AIPR&D, because there is no alternative future use for the early access programsasset that was acquired. For example, we accounted for our A&R JDCA with CRISPR in France was classified2021 as an asset acquisition of rights to in-process research and development for which we did not have any alternative future use and recorded the associated $900.0 million upfront payment to AIPR&D.
If the fair value that we acquired in “Accrued expenses” on our consolidated balance sheets.
Froman acquisition is distributed among more than one asset, and the first quarter of 2018 through the third quarter of 2019,acquisition constitutes a business, we recognized net product revenuesaccount for ORKAMBI sales in France under the early access programs based onit as a transaction price that reflected our estimate of consideration we expected to retain that would not be subject to a significant reversal in amounts recognized, which resulted in revenue representing a portion of the invoiced amount.
Upon reaching an agreement with the French government for ORKAMBI, including the final amount for ORKAMBI distributed through early access programs in France in the fourth quarter of 2019, we updated the transaction price related to ORKAMBI distributed through early access programs and recognized net product revenues of $155.8 million related to these shipments, which occurred from 2015 through the date of our agreement with the French government, because the final amount for these shipments exceeded our previous estimate.
Acquisitionsbusiness combination.
We are required to make several significant judgments and estimates in order to calculate the purchase price for our business combinations and then allocate it to the assets that we have acquired and the liabilities that we have assumed on our consolidated balance sheet. The most significant judgments and estimates relate to the fair value of the in-process research and development assets and contingent consideration liabilities related to these business combinations. Based on these judgments and estimates, the fair value of the goodwill that we record as a result of these business combinations may be material. Once recorded, these assets are subject to quarterly impairment analysis
In-process Research and our contingent consideration liability is adjusted quarterly, which requires similar judgments and estimates.
Development Intangible Assets
In 2019,As of each of December 31, 2023 and 2022, we recordedhad $603.6 million of in-process research and development assets related to our acquisitions of Exonics and Semma totaling $400.0 million on our consolidated balance sheet which remainedwithin “Other intangible assets, net.” Most recently, we recorded a $216.6 million in-process research and development intangible asset related to our acquisition of ViaCyte on our consolidated balance sheet as of December 31, 2020. Each of thesein 2022. We characterize in-process research and development assets is accounted for as an indefinite-lived intangible asset and is maintained on our consolidated balance sheetsheets as indefinite-lived intangible assets until either the project underlying it is completed or the asset becomes impaired.impaired and is subsequently written-off. We test our in-process research and development intangible assets for impairment on an annual basis, and more frequently if indicators are present or changes in circumstances suggest that impairment may exist. When we determine that an indefinite-lived intangible asset has become impaired or we abandon athe associated research and development project, we write down the carrying value of the related intangible asset to its fair value and record an impairment charge in the period in which the impairment occurs. In 2018,For example, we recorded a full$13.0 million impairment charge of $29.0 million foran in-process research and development intangible asset to “Research and development expenses” in 2022 due to a decision to revise the scope of certain acquired gene-editing programs. If one of our product candidates achieves regulatory approval, the in-process research and development asset that had previously been recorded on our consolidated balance sheets related to our collaborationintangible assets associated with BioAxone.the product candidate become finite-lived intangible assets as described below.
To determine the fair value of our in-process research and development assets, we utilizehave utilized either the multi-period excess earnings methodor the relief from royalty methods of the income approach, whichapproach. Each method requires us to make estimates ofestimate the probability of technical and regulatory success, development cost assumptions, revenue projections and growth rates, commercial cost estimates and appropriate discount rates. These assumptions require significant management judgment and reasonable changes intax rates.
The multi-period excess earnings method also requires us to estimate development and commercial costs. The relief from royalty method also requires us to estimate the assumptions can cause material changes to the fair valueafter-tax royalty savings expected from ownership of the intangible assets. Due to the early stage of Exonics and Semma’s programs, these significant assumptions could be affected by future economic and market conditions.asset that we acquired.
Contingent Consideration
As of December 31, 20202023 and 2019,2022, we had $189.6$77.4 million and $176.5$129.0 million, respectively, of liabilities on our consolidated balance sheetsheets attributable to the fair value of the contingent development and regulatory payments that we may owe to Exonics’ former equity holders upon the achievement of certain events. Our acquisition of Semma in 2019 did not include similar contingent payments; therefore, we are not required to record contingent consideration liabilities related to our acquisition of Semma.events associated with research programs focused on DMD and other severe neuromuscular diseases, including DM1.
We record an increase or a decrease in the fair value of the contingent consideration liabilityliabilities on our consolidated balance sheet and in our consolidated statement of operationsincome on a quarterly basis. We determine the fair value of our contingent consideration liabilityliabilities using a probability weighted discounted cash flow method of the income approach, which requires us to make estimates of the timing of regulatory and commercial milestone achievement and the corresponding estimated probability of technical and regulatory success rates. SignificantWe use significant judgment is used in determining the appropriateness of these assumptions during each reporting period. Reasonable changes in these assumptions can cause material changes to the fair value of our contingent consideration liability.liabilities. Due to the early stage of Exonics’ DMD and DM1 programs, these significant assumptions could be affected by future economic and market conditions.
In November 2023, we determined that additional pre-clinical studies of the delivery system for our gene-editing components for DMD will be required. As a result, we revised our estimates regarding the timing of the development and regulatory milestones and the probability of achieving those milestones, which reduced the estimated fair value of our contingent consideration as of December 31, 2023.
Goodwill
In 2020,As of December 31, 2023 and 2022, we had goodwill of $1.1 billion on our consolidated balance sheets. During 2023, we did not have any business combinations; therefore,combinations. In 2022, we did not record any additionalrecorded $85.8 million of goodwill on our consolidated balance sheet. In 2019, we recorded goodwill of $554.6 million and $397.1 millionsheet related to our acquisitionsacquisition of Semma and Exonics, respectively.ViaCyte. Goodwill reflects the difference between the fair value of the consideration transferred and the fair value of the net assets acquired. Thus, the goodwill that we record is dependent on the significant judgments and estimates inherent in these fair values. We have one reporting unit for goodwill reporting purposes. We evaluate our goodwill for impairment on an annual basis, and more frequently if indicators are present or changes in circumstances suggest that impairment may exist. We have not identified any goodwill impairment to date.
Finite-lived Intangible Assets
As of December 31, 2023, we had $236.3 million of finite-lived intangible assets on our consolidated balance sheet within “Other intangible assets, net.” These finite-lived intangible assets, which were recorded during 2023, relate to $208.0 million of CASGEVY regulatory approval milestones, including $200.0 million we paid to CRISPR pursuant to the A&R JDCA, and $30.0 million for a non-exclusive sublicense of developed technology related to CASGEVY.
We amortize our finite-lived intangible assets using the straight-line method within “Cost of sales” over the remaining estimated life of the assets beginning in the period in which regulatory approval is achieved or the assets are acquired and continuing through the period that we no longer have either exclusive rights to market the products associated with the assets or in-license rights to the intellectual property underlying the assets. We test finite-lived intangible assets for impairment if indicators are present or changes in circumstances suggest that the carrying value of an asset may not be recoverable. If we determine that the carrying value of a finite-lived intangible asset may not be recoverable, we compare the carrying value of the asset to the undiscounted cash flows that we expect the asset to generate. When we determine that a finite-lived intangible asset has become impaired, we write down the carrying value of the asset to its fair value of our in-process research and development assets and contingent consideration liabilities.record an impairment charge in the period in which the impairment occurs.
Income Taxes
We utilize the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement carrying amounts and tax basis of assets and liabilities using enacted tax rates in effect for years in which the temporary differences are expected to reverse. If our estimate
of the tax effect of reversing temporary differences is (i) not reflective of actual outcomes, (ii) modified to reflect new
developments or interpretations of the tax law, or (iii) revised to incorporate new accounting principles, or changes in the expected timing or manner of the reversal, our results of operations could be materially impacted.
We are engaged in research and development activities and incurred significant net operating losses for a number of years before recently becoming profitable. Accordingly, we did not report any tax benefits relating to our net operating loss carryforwards and income tax credit carryforwards that were available for utilization in future periods because we maintained a valuation allowance on the majority of our net operating losses and other deferred tax assets until December 31, 2018. We released the valuation allowance on the majority of our net operating losses and other deferred tax assets resulting in a non-cash benefit from income taxes of $1.56 billion in the fourth quarter of 2018.
We provide a valuation allowance when it is more likely than not that deferred tax assets will not be realized. On a periodic basis, we reassess our valuation allowances on our deferred tax assets, weighing positive and negative evidence to assess the recoverability of the deferred tax assets. In the fourth quarter of 2018, we reassessed our valuation allowances and considered positive evidence including significant cumulative consolidated and U.S. income over the three years ended December 31, 2018, revenue growth, clinical program progression, including the advancement and clinical trial data from our triple combination regimens, and expectations regarding future profitability, and negative evidence, including potential impact of competition on our projections and cumulative losses in the jurisdictions. After assessing both the positive evidence and the negative evidence, we released the valuation allowance on the majority of our net operating losses and other deferred tax assets as of December 31, 2018.
Significant judgment is required in making these assessments to maintain or reverseadjust our valuation allowances and, to the extent our future expectations change we would have to assess the recoverability of these deferred tax assets at that time. The determinationAs of December 31, 2023, we maintained a valuation allowance of $266.6 million related primarily to releaseU.S. state tax attributes.
We record liabilities related to uncertain tax positions by prescribing a minimum recognition threshold and measurement attribute for the majorityfinancial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. We adjust our valuation allowances increasedliability to reflect any subsequent changes in the relevant facts and circumstances surrounding the uncertain positions. We are subject to tax laws and audits in multiple jurisdictions and significant judgment is required in making this assessment. Consequently, we regularly re-evaluate uncertain tax positions and consider various factors, including changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, and changes in facts or circumstances related to a tax position. As of December 31, 2023, our net income by $1.56 billion, or $6.03 per share in 2018.liability for uncertain tax positions was $615.9 million.
RECENT ACCOUNTING PRONOUNCEMENTS
Refer to Note A, “Nature of Business and Accounting Policies,” in the accompanying notes to the consolidated financial statements for a discussion of recent accounting pronouncements and new accounting pronouncements adopted during 2020.2023.
ITEM 7A.QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk
Financial Instruments
As part of our investment portfolio, we own financial instruments that are sensitive to market risks. The investment portfolio is used to preserve our capital. None of these market risk-sensitivecapital, provide adequate liquidity and earn returns commensurate with our risk appetite. We invest in instruments are held for trading purposes. We do not have derivative financial instrumentsthat meet the credit quality standards outlined in our investment portfolio.
Interest Rate Risk
We invest our cash in a varietypolicy, which also limits the amount of financialcredit exposure to any one issue or type of instrument. These instruments principallyprimarily include securities issued by the U.S. government and its agencies, investment-grade corporate bonds and commercial paper, and money market funds. These investments are denominated in U.S. Dollars. Dollars and none are held for trading purposes.
All of our interest-bearing securities are subject to interest rate risk and could declinechange in value if interest rates fluctuate, including potential fluctuations as a result of COVID-19.fluctuate. Substantially all of our investment portfolio consists of marketable securities with active secondary or resale markets to help ensure portfolio liquidity, and we have implemented guidelines limiting the term-to-maturity of our investment instruments. Since we account for these securities as available-for-sale, no gains or losses are realized due to changes in the fair value of our investments unless we sell our investments prior to maturity or incur a credit loss. Due to the conservative nature of these instruments, we do not believe that we havethe fair value of our investments has a material exposure to interest rate risk. If
While we are exposed to global interest rates were to increase or decrease by 1%, the fair value ofrate fluctuations, our investment portfolio would increase or decreaseis most affected by an immaterial amount.fluctuations in U.S. interest rates, which affect the interest earned on our cash, cash equivalents and marketable securities.
WeCredit Agreement
In 2022, we entered into a $500.0 million unsecured revolving credit agreement in each of 2020 and 2019.facility (“credit agreement”). Loans under thesethis credit agreementsagreement bear interest, at our option, at either a base rate or a Eurocurrency rate, in each caseSecured Overnight Financing Rate (“SOFR”), plus an applicable margin based on our consolidated leverage ratio (the ratio of our total consolidated funded indebtedness to our consolidated EBITDA for the most recently completed four fiscal quarter period). Pursuant to theour credit agreement, that we entered into in 2019, the applicable margin on base rate loans ranges from 0.125%0.000% to 0.500% and the applicable margin on EurocurrencySOFR loans ranges from 1.125%1.000% to 1.500%. Pursuant to the credit agreement that we entered into in 2020, the applicable margin on base rate loans ranges from 0.500% to 0.875% and the applicable margin on Eurocurrency loans ranges from 1.500% to 1.875%. We do not believe that changes in interest rates related to eitherour credit agreement would have a material effect on our consolidated financial statements. As
of December 31, 2020,2023, we had no principal or interest outstanding under either of our existing credit facilities.facility. A portion of our “Interest expense” in 20212024 will be dependent on whether, and to what extent, we borrow amounts under these existing facilities.this facility.
Foreign Exchange Market Risk
As a result of our foreign operations, we face significant exposure to movements in foreign currency exchange rates, primarily the Euro and British Pound against the U.S. Dollar.dollar. Fluctuations in the global markets, including as a resultamounts of COVID-19,our foreign revenues and fluctuations in foreign currency exchange rates, may have a positive or negative effect on our foreign exchange rate exposure. The current exposures arise primarily from cash, accounts receivable, intercompany receivables and payables, payables, and accruals, and inventories. Both positive and negative effects to our net revenues from international product sales from movements in exchange rates are partially mitigated by the natural, opposite effect that exchange rates have on our international operating costs and expenses.
We have a foreign currency management program, which is separate from our investment policy and portfolio, with the objective of reducing the effect of exchange rate fluctuations on our operating results and forecasted revenues and expenses denominated in foreign currencies. We currently have cash flow hedges for the Euro, British Pound, Canadian Dollar, Swiss Franc and Australian Dollar related to a portion of our forecasted product revenues that qualify for hedge accounting treatment under U.S. GAAP. We do not seek hedge accounting treatment for our foreign currency forward contracts related to monetary assets and liabilities that impact our operating results. As of December 31, 2020,2023, we held foreign exchange forward contracts that were designated as cash flow hedges with notional amounts totaling $1.1$2.4 billion representing a net liability of $63.5$31.9 million recorded on our consolidated balance sheet.
Although not predictive in nature, we believe a hypothetical 10% threshold reflects a reasonably possible near-term change in exchange rates. Assuming thatIf the December 31, 20202023 exchange rates were to change by a hypothetical 10%, the fair value recorded on our consolidated balance sheet related to our foreign exchange forward contracts that were designated as cash flow hedges as of December 31, 20202023 would change by approximately $109.2$239.2 million. However, since these contracts hedge a specific portion of our forecasted product revenues denominated in certain foreign currencies, any change in the fair value of these contracts is recorded in “Accumulated other comprehensive loss”(loss) income” on our consolidated balance sheetsheets and is
reclassified to earnings in the same periods during which the underlying product revenues affect earnings. Therefore, any change in the fair value of these contracts that would result from a hypothetical 10% change in exchange rates would be entirely offset by the change in value associated with the underlying hedged product revenues resulting in no impact on our future anticipated earnings and cash flows with respect to the hedged portion of our forecasted product revenues.
Equity Price Risk
Information required by this section is incorporated by reference from the discussionWe hold strategic equity investments in certain public and private companies, and we expect to make additional strategic equity investments in the “Strategic Investments” sectionfuture. In 2023 and 2022, we recorded net losses of this Part II, Item 7, “Management’s Discussion$0.6 million and Analysis$149.1 million, respectively, to “Other Income (Expense), Net” in our consolidated statements of Financial Condition and Resultsincome to reflect changes in the fair value of Operations.”equity investments with readily determinable fair values (including publicly traded securities). The fair value of our equity investments in publicly traded companies was less than $50.0 million as of December 31, 2023.
To the extent that we continue to hold strategic equity investments in publicly traded companies, we expect that due to the volatility of the stock price of biotechnology companies, our “Other Income (Expense), Net” will fluctuate in future periods based on increases or decreases in the fair value of our strategic equity investments.
ITEM 8.FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information required by this Item 8 is contained on pages F-1 through F-49F-46 of this Annual Report on Form 10-K.
ITEM 9.CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable.
ITEM 9A.CONTROLS AND PROCEDURES
(1) Evaluation of Disclosure Controls and Procedures. The Company’sOur chief executive officer and chief financial officer, after evaluating the effectiveness of the Company’sour disclosure controls and procedures (as defined in Rule 13a-15(e) and Rule 15d-15(e) promulgated under the Securities Exchange Act of 1934, as amended) as of the end of the period covered by this Annual Report on Form 10-K, have concluded that, based on such evaluation, the Company’sour disclosure controls and procedures were effective. In designing and evaluating the disclosure controls and procedures, the Company’s management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and the Company’s management necessarily was required to apply itsour judgment in evaluating the cost-benefit relationship of possible controls and procedures.
(2) Management’s Annual Report on Internal Control Over Financial Reporting. The management of the CompanyManagement is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) and Rule 15d-15(f) promulgated under the Securities Exchange Act of 1934, as amended, as a process designed by, or under the supervision of, the Company’sour principal executive and principal financial officers and effected by the Company’sour board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. The Company’sOur internal control over financial reporting includesinclude those policies and procedures that:
• pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company;our assets;
• provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company;our directors; and
• provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’sour assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s managementManagement assessed the effectiveness of the Company’sour internal control over financial reporting as of December 31, 2020.2023. In making this assessment, itwe used the criteria set forth in the Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO)(“COSO”). Based on itsour assessment, the Company’s management has concluded that, as of December 31, 2020, the Company’s2023, our internal control over financial reporting is effective based on those criteria.
The Company’sOur independent registered public accounting firm, Ernst & Young LLP, issued an attestation report on the Company’sour internal control over financial reporting. See Section 4 below.
(3) Changes in Internal Controls. During the quarter ended December 31, 2020,2023, there were no changes in the Company’sour internal control over financial reporting that materially affected, or are reasonably likely to materially affect, the Company’sour internal control over financial reporting.
(4)
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of Vertex Pharmaceuticals Incorporated
Opinion on Internal Control overOver Financial Reporting
We have audited Vertex Pharmaceuticals Incorporated’s internal control over financial reporting as of December 31, 2020,2023, based on criteria established in Internal Control-IntegratedControl–Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Vertex Pharmaceuticals Incorporated (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2020,2023, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the 20202023 consolidated financial statements of the Company and our report dated February 11, 2021,15, 2024, expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control overOver Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Boston, Massachusetts
February 11, 202115, 2024
ITEM 9B. OTHER INFORMATION
Michael Parini, oneRule 10b5-1 Trading Plans
Our policy governing transactions in our securities by our directors, officers, and employees permits our officers, directors and employees to enter into trading plans complying with Rule 10b5-1 under the Securities Exchange Act of 1934, as amended, which plans are intended to satisfy the affirmative defense conditions of Rule 10b5-1 (each, a “Trading Plan”). None of our current executive officers or directors entered into a Trading Plan in the fourth quarter of 2023.
Other Information
Dr. Bastiano Sanna has informed us that hestepped down from his role as Executive Vice President and Chief of Cell and Genetic Therapies for personal reasons. Dr. Sanna will be leavingcontinue to provide advisory services in his position at our company effective March 1, 2021.
Stuart Arbuckle, one of our current executive officers, will assume additional responsibilities and be appointednew role as a consultant to the position of EVP and Chief Commercial and Operations Officer, effective March 1, 2021.company.
Additional information regarding Mr. Arbuckle and Mr. Parini is provided in Part I, Item 1 of this Annual Report on Form 10-K.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS.
None.
PART III
Portions of our definitive Proxy Statement for the 20212024 Annual Meeting of Shareholders or 2021(“2024 Proxy Statement,Statement”) are incorporated by reference into this Part III of our Annual Report on Form 10-K.
ITEM 10.DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information regarding directors required by this Item 10 will be included in our 20212024 Proxy Statement and is incorporated herein by reference. We expect this information to be provided under “Election of Directors,” “Corporate Governance and Risk Management,” “Shareholder Proposals for the 20212025 Annual Meeting and Nominations for Director,” “Delinquent Section 16(a) Reports” and “Code of Conduct.” The information regarding executive officers required by this Item 10 is included in Part I of this Annual Report on Form 10-K.
ITEM 11.EXECUTIVE COMPENSATION
The information required by this Item 11 will be included in the 20212024 Proxy Statement and is incorporated herein by reference. We expect this information to be provided under “Compensation Committee Interlocks and Insider Participation,” “Compensation Discussion and Analysis,” “Compensation and Equity Tables,” “Director Compensation,” “Management Development and Compensation Committee Report” and/or “Corporate Governance and Risk Management.”
ITEM 12.SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item 12 will be included in the 20212024 Proxy Statement and is incorporated herein by reference. We expect this information to be provided under “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information.”
ITEM 13.CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item 13 will be included in the 20212024 Proxy Statement and is incorporated herein by reference. We expect this information to be provided under “Election of Directors,” “Corporate Governance and Risk Management,” and “Audit and Finance Committee.”
ITEM 14.PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this Item 14 will be included in the 20212024 Proxy Statement and is incorporated herein by reference. We expect this information to be provided under “Ratification of the Appointment of Independent Registered Public Accounting Firm.”
PART IV
ITEM 15.EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a)(1) The Financial Statements required to be filed by Items 8 and 15(c) of Form 10-K, and filed herewith, are as follows:
| | | | | |
| Page Number in
|
| Report of Independent Registered Public Accounting Firm (PCAOB ID: 42) | |
Consolidated Statements of OperationsIncome for the years ended December 31, 2020, 20192023, 2022 and 20182021 | |
Consolidated Statements of Comprehensive Income for the years ended December 31, 2020, 20192023, 2022 and 20182021 | |
Consolidated Balance Sheets as of December 31, 20202023 and 20192022 | |
Consolidated Statements of Shareholders’ Equity and Noncontrolling Interest for the years ended December 31, 2020, 20192023, 2022 and 20182021 | |
Consolidated Statements of Cash Flows for the years ended December 31, 2020, 20192023, 2022 and 20182021 | |
| Notes to Consolidated Financial Statements | |
(a)(2) Financial Statement Schedules have been omitted because they are either not applicable or the required information is included in the consolidated financial statements or notes thereto listed in (a)(1) above.
(a)(3) Exhibits.
The following is a list of exhibits filed as part of this Annual Report on Form 10-K.
| | | | | | | | | | | | | | | | | |
| Exhibit Number | Exhibit Description | Filed with
| Incorporated by
Reference herein
| Filing Date/
Period Covered | SEC File/Reg. Number |
| Plan of Acquisition | | | |
| 2.1 | | | 10-Q
(Exhibit 2.1) | October 31, 2019 | 000-19319 |
2.2 | Agreement and Plan of Merger, dated as of June 6, 2019, among Vertex Pharmaceuticals Incorporated, VXP Merger Sub, Inc., Exonics Therapeutics, Inc. and Shareholder Representative Services LLC, solely in its Capacity as Shareholders’ Representative, as amended by the Amendment to Agreement and Plan of Merger, dated as of June 12, 2019, among Vertex Pharmaceuticals Incorporated, VXP Merger Sub, Inc., Exonics Therapeutics, Inc. and Shareholder Representative Services LLC, solely in its Capacity as Shareholders’ Representative.† | | 10-Q
(Exhibit (Exhibit 10.1) | August 1, 2019 | 000-19319 |
| Governance Documents | | | |
| 3.1 | | | 10-Q
(Exhibit 3.1) | July 26, 2018 | 000-19319 |
| 3.2 | | | 10-Q10-K
(Exhibit 3.2) | May 1, 2020February 10, 2023 | 000-19319 |
| Stock Certificate | | | |
| 4.1 | | | 10-K
(Exhibit 4.1) | February 15, 2018 | 000-19319 |
| 4.2 | | | 10-K (Exhibit
(Exhibit 4.2) | February 13, 202010, 2023 | 000-19319 |
| Collaboration Agreement | | | |
| 10.1 | | | 10-Q/A10-Q
(Exhibit 10.2)10.1) | August 19, 2011November 3, 2021 | 000-19319 |
| 10.2 | | | 10-K10-Q
(Exhibit 10.9)10.2) | March 16, 2006November 3, 2021 | 000-19319 |
| 10.3 | | | 10-Q/A
(Exhibit 10.6) | August 19, 2011 | 000-19319 |
| | | | | | | | | | | | | | | | | |
Exhibit Number | Exhibit Description | Filed with
this report | Incorporated by
Reference herein
from—Form
or Schedule | Filing Date/
Period Covered | SEC File/Reg.
Number
|
| 10.4 | | | 10-Q
(Exhibit 10.3) | August 9, 2011November 3, 2021 | 000-19319 |
| | | | | | | | | | | | | | | | | |
| Exhibit Number | Exhibit Description | Filed with this report | Incorporated by
Reference herein from—Form or Schedule | Filing Date/
Period Covered | SEC File/Reg. Number |
| 10.5 | | | 10-K10-Q
(Exhibit 10.05)10.4) | February 23, 2017November 3, 2021 | 000-19319 |
| 10.6 | Amended and Restated Joint Development and Commercialization Agreement, dated December 12, 2017,April 16, 2021, between Vertex Pharmaceuticals Incorporated, Vertex Pharmaceuticals (Europe) Limited and CRISPR Therapeutics AG, CRISPR Therapeutics Limited, CRISPR Therapeutics, Inc., TRACR Hematology Ltd.† | | 10-Q
(Exhibit 10.1) | July 30, 2021 | 000-19319 |
| 10.7 | | X | | | |
| Leases | | | | | |
10.710.8 | | | 10-Q
(Exhibit 10.4)10.2) | August 9, 2011July 30, 2021 | 000-19319 |
10.810.9 | | | 10-Q
(Exhibit 10.5)10.3) | August 9, 2011July 30, 2021 | 000-19319 |
| Financing Agreements |
10.910.10 | | | 10-Q
(Exhibit 10.1) | October 31, 2019 | 000-19319 |
10.10 | | X | | | |
10.11 | | | 10-Q (Exhibit 10.1) | October 30, 2020August 5, 2022 | 000-19319 |
| Equity Plans | | | |
10.1210.11 | | | 10-Q
(Exhibit 10.1) | October 25, 2018 | 000-19319 |
10.13 | | | 8-K
(Exhibit 10.2) | May 15, 2006 | 000-19319 |
10.1410.12 | | | 10-K
(Exhibit 10.20) | February 13, 2015 | 000-19319 |
10.1510.13 | | | DEF 14A
(Appendix A) | April 26, 20197, 2022 | 000-19319 |
10.1610.14 | | | 10-K
(Exhibit 10.17) | February 13, 2015 | 000-19319 |
10.17 | | | 10-K
(Exhibit 10.18) | February 13, 2015 | 000-19319 |
10.1810.15 | | | 10-K
(Exhibit 10.25) | February 16, 2016 | 000-19319 |
10.1910.16 | | | 10-K
(Exhibit 10.19) | February 13, 2015 | 000-19319 |
10.2010.17 | | | 10-K (Exhibit
(Exhibit 10.17) | February 13, 2020 | 000-19319 |
10.2110.18 | | | 10-K
(Exhibit 10.27) | February 16, 2016 | 000-19319 |
10.2210.19 | | | DEF 14A
(Appendix B) | April 26, 2019 | 000-19319 |
| Agreements with Executive Officers and Directors | | | |
10.2310.20 | | | 8-K
(Exhibit 10.1) | April 1, 2020 | 000-19319 |
10.2410.21 | | | 10-K
(Exhibit 10.24) | February 9, 2022 | 000-19319 |
| 10.22 | | | 10-K
(Exhibit 10.23) | February 10, 2023 | 000-19319 |
| 10.23 | | | 10-K
(Exhibit 10.35) | February 22, 2012 | 000-19319 |
10.2510.24 | | | 8-K
(Exhibit 10.1) | July 25, 2019 | 000-19319 |
10.2610.25 | | | 8-K
(Exhibit 10.2) | July 25, 2019 | 000-19319 |
| | | | | | | | | | | | | | | | | |
Exhibit Number | Exhibit Description | Filed with
this report | Incorporated by
Reference herein
from—Form
or Schedule | Filing Date/
Period Covered | SEC File/Reg.
Number
|
10.2710.26 | | | 10-Q
(Exhibit 10.1) | November 6, 2012 | 000-19319 |
10.2810.27 | | | 10-Q
(Exhibit 10.2) | November 6, 2012 | 000-19319 |
10.2910.28 | | | 10-K
(Exhibit 10.34) | February 16, 2016 | 000-19319 |
| | | | | | | | | | | | | | | | | |
| Exhibit Number | Exhibit Description | Filed with this report | Incorporated by
Reference herein from—Form or Schedule | Filing Date/
Period Covered | SEC File/Reg. Number |
10.3010.29 | | | 10-K
(Exhibit 10.35) | February 16, 2016 | 000-19319 |
10.31 | | | 10-K
(Exhibit 10.40) | February 23, 2017 | 000-19319 |
10.32 | | | 10-K
(Exhibit 10.41) | February 23, 2017 | 000-19319 |
10.3310.30 | | | 10-K
(Exhibit (Exhibit 10.42) | February 23, 2017 | 000-19319 |
10.3410.31 | | | 10-K
(Exhibit (Exhibit 10.43) | February 23, 2017 | 000-19319 |
10.3510.32 | | | 10-Q
(Exhibit 10.1) | May 1, 2019 | 000-19319 |
10.3610.33 | | | 10-Q
(Exhibit 10.2) | May 1, 2019 | 000-19319 |
10.3710.34 | | | 10-K (Exhibit 10.35)
(Exhibit 10.36) | February 13, 20209, 2022 | 000-19319 |
10.3810.35 | | | 10-Q (Exhibit 10.13)10-K
(Exhibit 10.37) | November 4, 2009February 9, 2022 | 000-19319 |
10.39 | | | 10-K (Exhibit 10.66) | February 17, 2009 | 000-19319 |
10.4010.36 | | | 10-K
(Exhibit 10.46)X | | February 15, 2018 | 000-19319 |
10.4110.37 | | | X10-K
(Exhibit 10.39) | February 9, 2022 | | 000-19319 |
| Subsidiaries | | | | |
| 21.1 | | X | | | |
| Consent | | | | |
| 23.1 | | X | | | |
| Certifications | | | | |
| 31.1 | | X | | | |
| 31.2 | | X | | | |
32.132 | | X | | | |
| Clawback Policy | | | | |
| 97.1 | | X | | | |
| 101.INS | XBRL Instance | X | | | |
| 101.SCH | XBRL Taxonomy Extension Schema | X | | | |
| 101.CAL | XBRL Taxonomy Extension Calculation | X | | | |
| 101.LAB | XBRL Taxonomy Extension Labels | X | | | |
| 101.PRE | XBRL Taxonomy Extension Presentation | X | | | |
| 101.DEF | XBRL Taxonomy Extension Definition | X | | | |
| 104 | Cover Page Interactive Data File––the cover page interactive data file does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | X | | | |
| | | | | |
| * | Management contract, compensatory plan or agreement. |
| † | Confidential portions of this document have been redacted according to the applicable rules. |
* Management contract, compensatory plan or agreement.
† Confidential portions of this document have been redacted according to the applicable rules.
ITEM 16.FORM 10-K SUMMARY
Not applicable.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | | |
| Vertex Pharmaceuticals Incorporated |
| | |
February 11, 202115, 2024 | By: | /s/ Reshma Kewalramani |
| | Reshma Kewalramani Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Name | | | | Title | | | | | Date | | |
| | |
| /s/ Reshma Kewalramani | | |
| Reshma Kewalramani | President, Chief Executive Officer and Director (Principal Executive Officer) | February 11, 202115, 2024 |
| | |
| /s/ Charles F. Wagner, Jr. | | |
| Charles F. Wagner, Jr. | Executive Vice President and Chief Financial Officer (Principal Financial Officer) | February 11, 202115, 2024 |
| | |
/s/ Paul M. SilvaKristen C. Ambrose | | |
Paul M. SilvaKristen C. Ambrose | Senior Vice President and Chief Accounting Officer (Principal Accounting Officer) | February 11, 202115, 2024 |
| | |
| /s/Jeffrey M. Leiden | | |
| Jeffrey M. Leiden | Executive Chairman | February 11, 202115, 2024 |
| | |
| /s/ Sangeeta N. Bhatia | | |
| Sangeeta N. Bhatia | Director | February 11, 202115, 2024 |
| | |
| /s/ Lloyd Carney | | |
| Lloyd Carney | Director | February 11, 202115, 2024 |
| | |
| /s/ Alan Garber | | |
| Alan Garber | Director | February 11, 202115, 2024 |
| | |
| /s/ Terrence C. Kearney | | |
| Terrence C. Kearney | Director | February 11, 202115, 2024 |
| | | | | | | | | | | | | |
/s/ Yuchun LeeMichel Lagarde | | |
Yuchun LeeMichel Lagarde | Director | February 11, 202115, 2024 |
| | |
/s/ Margaret G. McGlynn | | |
Margaret G. McGlynn | Director | February 11, 2021 |
| | | | | | | | | | | | | |
| /s/ Diana McKenzie | | |
| Diana McKenzie | Director | February 11, 202115, 2024 |
| | |
| /s/ Nancy A. Thornberry | | |
| Nancy A. Thornberry | Director | February 15, 2024 |
| | | | | | | | | | | | | |
| /s/ Bruce I. Sachs | | |
| Bruce I. Sachs | Director | February 11, 202115, 2024 |
| | |
| /s/ Suketu Upadhyay | | |
| Suketu Upadhyay | Director | February 15, 2024 |
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of Vertex Pharmaceuticals Incorporated
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Vertex Pharmaceuticals Incorporated (the Company) as of December 31, 20202023 and 2019,2022, the related consolidated statements of operations,income, comprehensive income, shareholders’ equity and noncontrolling interest, and cash flows for each of the three years in the period ended December 31, 2020,2023, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 20202023 and 2019,2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2020,2023, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2020,2023, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated February 11, 2021,15, 2024, expressed an unqualified opinion thereon.
Adoption of New Accounting Standard
ASU No. 2016-02
As discussed in Note A to the consolidated financial statements, the Company changed its method for lease accounting as a result of the adoption of ASU No. 2016-02, Leases (Topic 842), and the related amendments effective January 1, 2019.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
| | | | | | | | |
| | Revenue recognition - Payor Mix Impact on Measuring Variable ConsiderationMedicaid Drug Rebate Program in the U.S. |
| Description of the Matter | | As discussed in Note A to the Company’s consolidated financial statements, the Company recordsrecognizes revenue from product sales at thebased on amounts due from customers net sales price, or “transaction price,”of allowances for variable consideration, which requires the Company to make several significant estimates regarding the net sales price.include, among others, rebates mandated by law under Medicaid and other government pricing programs. The most significant estimates relate to government and private payor rebates, chargebacks, discounts and fees, collectively rebates. DueThe Company includes an estimate of variable consideration in its transaction price at the time of sale, when control of the product transfers to the delay in receiptcustomer. The Company estimates its Medicaid and other government pricing accruals based on monthly sales, historical experience of claims submitted by third-party payors, the Company estimatesvarious states and jurisdictions, historical rebate rates and estimated lag time of the percentage of prescriptions that will be covered by each third-party payor, which is referred to as the payor mix.rebate invoices. Rebate accruals inclusive of estimated amounts due for claims not yet received or processed as part of the Company’s Medicaid program are recorded within accrued expenses on the Company’s consolidated balance sheet. Auditing the measurement ofallowances for rebates owed pursuant to the Company’s net product revenuesMedicaid Drug Rebate Program in the U.S. was especially complex and judgmental due to the significant estimation required in determining certain assumptions including the levels of expected utilization of these rebates based on the amount of consideration that will be collected netproduct sold to eligible patients, as well as the complexity of estimatesthe government mandated calculations. The allowances for payor rebates. In particular,rebates owed pursuant to the net sales price is affected byMedicaid Drug Rebate Program in the U.S. are sensitive to these significant assumptions in payor behavior such as changes in payor mix, payor collections, current customer contractual requirements, and experience with ultimate collection from third-party payors.calculations. |
| How We Addressed the Matter in Our Audit | | We obtained an understanding, evaluated the design and tested the operating effectiveness of controls over the Company’s revenue recognition process, including controls over the underlying assumptions and inputs used by management to estimate amounts due to third-party payors and the completeness and accuracymanagement’s review of the data used in the estimates.allowances for Medicaid rebates. We also tested the Company’s controls to assess the completeness and accuracy of the current and historical data that supports the estimate.Medicaid estimate, significant assumptions related to the inputs utilized as well as management’s review of the application of the government pricing regulations. Our audit procedures to test the Company’s recognition of net product revenuesallowances for rebates owed pursuant to the Medicaid Drug Rebate Program in the U.S., included, among others, assessingthe following: we performed audit procedures to assess the methodology used to determine the estimate and testingtested the significant assumptions andas well as the underlying data used by the Company in its analysis, which included historical claims data. To assess the payor mix assumptions we tested contracted rates, historical claims and payment data and related trends, and other relevant factors.analysis. We also assessed the historical accuracy of the Company’s estimates of Medicaid rebates by comparing assumptions to historical trends and evaluating the change from prior periods. We further tested the completeness and accuracy of the underlying data used in the Company’s calculations through reconciliation to third-party payorinvoices, claims data and actual cash payments. In addition, we involved our government pricing specialists to assist in evaluating management’s methodology and calculations used in the measurement of certain estimated rebates. |
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2005.
Boston, Massachusetts
February 11, 202115, 2024
VERTEX PHARMACEUTICALS INCORPORATED
Consolidated Statements of OperationsIncome
(in thousands,millions, except per share amounts)
| | Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Year Ended December 31, | | | Year Ended December 31, |
| 2023 | | | 2023 | | 2022 | | 2021 |
| Revenues: | Revenues: | | | | | |
| Product revenues, net | Product revenues, net | $ | 6,202,783 | | | $ | 4,160,726 | | | $ | 3,038,325 | |
| Collaborative and royalty revenues | 2,900 | | | 2,095 | | | 9,272 | |
| Product revenues, net | |
| Product revenues, net | |
| Other revenues | |
| Total revenues | Total revenues | 6,205,683 | | | 4,162,821 | | | 3,047,597 | |
| Costs and expenses: | Costs and expenses: | |
| Cost of sales | Cost of sales | 736,300 | | | 547,758 | | | 409,539 | |
| Cost of sales | |
| Cost of sales | |
| Research and development expenses | Research and development expenses | 1,829,537 | | | 1,754,540 | | | 1,416,476 | |
| Sales, general and administrative expenses | 770,456 | | | 658,498 | | | 557,616 | |
| Acquired in-process research and development expenses | |
| Selling, general and administrative expenses | |
| Change in fair value of contingent consideration | Change in fair value of contingent consideration | 13,100 | | | 4,459 | | | 0 | |
| Restructuring income | 0 | | | 0 | | | (184) | |
| Intangible asset impairment charge | 0 | | | 0 | | | 29,000 | |
| Total costs and expenses | Total costs and expenses | 3,349,393 | | | 2,965,255 | | | 2,412,447 | |
| Income from operations | Income from operations | 2,856,290 | | | 1,197,566 | | | 635,150 | |
| Interest income | Interest income | 22,239 | | | 63,678 | | | 38,352 | |
| Interest expense | Interest expense | (58,151) | | | (58,502) | | | (72,471) | |
| Other income (expense), net | 296,420 | | | 192,177 | | | (790) | |
| Income before provision for (benefit from) income taxes | 3,116,798 | | | 1,394,919 | | | 600,241 | |
| Provision for (benefit from) income taxes | 405,151 | | | 218,109 | | | (1,486,862) | |
| Other (expense) income, net | |
| Income before provision for income taxes | |
| Provision for income taxes | |
| Net income | Net income | 2,711,647 | | | 1,176,810 | | | 2,087,103 | |
| Loss attributable to noncontrolling interest | 0 | | | 0 | | | 9,793 | |
| Net income attributable to Vertex | $ | 2,711,647 | | | $ | 1,176,810 | | | $ | 2,096,896 | |
| | Amounts per share attributable to Vertex common shareholders: | |
| Net income: | |
| Net income per common share: | |
| Net income per common share: | |
| Net income per common share: | |
| Basic | |
| Basic | |
| Basic | Basic | $ | 10.44 | | | $ | 4.58 | | | $ | 8.24 | |
| Diluted | Diluted | $ | 10.29 | | | $ | 4.51 | | | $ | 8.09 | |
| Shares used in per share calculations: | Shares used in per share calculations: | |
| Basic | Basic | 259,841 | | | 256,728 | | | 254,292 | |
| Basic | |
| Basic | |
| Diluted | Diluted | 263,396 | | | 260,673 | | | 259,185 | |
Please refer to Note A, “Nature of Business and Accounting Policies,” for an explanation of amounts reclassified from “Research and development expenses” to “Acquired in-process research and development expenses” for 2021.
The accompanying notes are an integral part of thethese consolidated financial statements.
VERTEX PHARMACEUTICALS INCORPORATED
Consolidated Statements of Comprehensive Income
(in thousands)millions)
| | | | | | | | | | | | | | | | | |
| Year ended December 31, |
| 2020 | | 2019 | | 2018 |
| Net income | $ | 2,711,647 | | | $ | 1,176,810 | | | $ | 2,087,103 | |
| Changes in other comprehensive income: | | | | | |
| Unrealized holding (losses) gains on marketable securities, net | (169) | | | 1,039 | | | 58 | |
| Unrealized (losses) gains on foreign currency forward contracts, net of tax of $14.3 million, $7.0 million and $(7.1) million, respectively | (51,555) | | | (14,003) | | | 27,438 | |
| Foreign currency translation adjustment | (14,783) | | | 10,332 | | | 8,855 | |
| Total other comprehensive (loss) income | (66,507) | | | (2,632) | | | 36,351 | |
| Comprehensive income | 2,645,140 | | | 1,174,178 | | | 2,123,454 | |
| Comprehensive loss attributable to noncontrolling interest | 0 | | | 0 | | | 9,793 | |
| Comprehensive income attributable to Vertex | $ | 2,645,140 | | | $ | 1,174,178 | | | $ | 2,133,247 | |
| | | | | | | | | | | | | | | | | |
| Year ended December 31, |
| 2023 | | 2022 | | 2021 |
| Net income | $ | 3,619.6 | | | $ | 3,322.0 | | | $ | 2,342.1 | |
| Other comprehensive (loss) income: | | | | | |
| Unrealized holding gains (losses) on marketable securities, net of tax of $(2.7), zero and zero, respectively | 9.7 | | | 0.4 | | | (0.8) | |
| Unrealized (losses) gains on foreign currency forward contracts, net of tax of $14.0, $1.0 and $(21.8), respectively | (50.9) | | | (4.1) | | | 83.2 | |
| Foreign currency translation adjustment | 26.1 | | | (11.4) | | | 2.0 | |
| Total other comprehensive (loss) income | (15.1) | | | (15.1) | | | 84.4 | |
| Comprehensive income | $ | 3,604.5 | | | $ | 3,306.9 | | | $ | 2,426.5 | |
The accompanying notes are an integral part of thethese consolidated financial statements.
VERTEX PHARMACEUTICALS INCORPORATED
Consolidated Balance Sheets
(in thousands, exceptmillions, except share and per share amounts)data)
| | December 31, |
| 2020 | | 2019 |
| December 31, | | | December 31, |
| 2023 | | | 2023 | | 2022 |
| Assets | Assets | | | |
| Current assets: | Current assets: | |
| Current assets: | |
| Current assets: | |
| Cash and cash equivalents | |
| Cash and cash equivalents | |
| Cash and cash equivalents | Cash and cash equivalents | $ | 5,988,187 | | | $ | 3,109,322 | |
| Marketable securities | Marketable securities | 670,710 | | | 698,972 | |
| Accounts receivable, net | Accounts receivable, net | 885,352 | | | 633,518 | |
| Inventories | Inventories | 280,777 | | | 167,502 | |
| Prepaid expenses and other current assets | Prepaid expenses and other current assets | 308,353 | | | 213,515 | |
| Total current assets | Total current assets | 8,133,379 | | | 4,822,829 | |
| Property and equipment, net | Property and equipment, net | 958,534 | | | 745,080 | |
| Goodwill | Goodwill | 1,002,158 | | | 1,002,158 | |
| Intangible assets | 400,000 | | | 400,000 | |
| Other intangible assets, net | |
| Deferred tax assets | Deferred tax assets | 882,779 | | | 1,190,815 | |
| Operating lease assets | Operating lease assets | 325,564 | | | 88,202 | |
| Long-term marketable securities | |
| Other assets | Other assets | 49,394 | | | 69,381 | |
| Total assets | Total assets | $ | 11,751,808 | | | $ | 8,318,465 | |
| Liabilities and Shareholders’ Equity | Liabilities and Shareholders’ Equity | | | |
| Current liabilities: | Current liabilities: | |
| Current liabilities: | |
| Current liabilities: | |
| Accounts payable | |
| Accounts payable | |
| Accounts payable | Accounts payable | $ | 155,139 | | | $ | 87,610 | |
| Accrued expenses | Accrued expenses | 1,404,971 | | | 1,116,912 | |
| Other current liabilities | Other current liabilities | 317,423 | | | 130,305 | |
| Total current liabilities | Total current liabilities | 1,877,533 | | | 1,334,827 | |
| Long-term finance lease liabilities | Long-term finance lease liabilities | 539,042 | | | 538,576 | |
| Long-term operating lease liabilities | Long-term operating lease liabilities | 350,463 | | | 84,292 | |
| Long-term contingent consideration | 189,600 | | | 176,500 | |
| Other long-term liabilities | Other long-term liabilities | 108,355 | | | 99,026 | |
| Total liabilities | Total liabilities | 3,064,993 | | | 2,233,221 | |
| Commitments and contingencies | Commitments and contingencies | 0 | | | 0 | |
| Shareholders’ equity: | Shareholders’ equity: | |
| Preferred stock, $0.01 par value; 1,000 shares authorized; NaN issued and outstanding | 0 | | | 0 | |
| Common stock, $0.01 par value; 500,000 shares authorized, 259,890 and 258,993 shares issued and outstanding, respectively | 2,599 | | | 2,589 | |
| Preferred stock, $0.01 par value; 1,000,000 shares authorized; none issued and outstanding | |
| Preferred stock, $0.01 par value; 1,000,000 shares authorized; none issued and outstanding | |
| Preferred stock, $0.01 par value; 1,000,000 shares authorized; none issued and outstanding | |
| Common stock, $0.01 par value; 500,000,000 shares authorized, 257,695,221 and 257,011,628 shares issued and outstanding, respectively | |
| Additional paid-in capital | Additional paid-in capital | 7,894,027 | | | 7,937,606 | |
| Accumulated other comprehensive loss | (68,480) | | | (1,973) | |
| Retained earnings (accumulated deficit) | 858,669 | | | (1,852,978) | |
| Accumulated other comprehensive (loss) income | |
| Retained earnings | |
| Total shareholders’ equity | Total shareholders’ equity | 8,686,815 | | | 6,085,244 | |
| Total liabilities and shareholders’ equity | Total liabilities and shareholders’ equity | $ | 11,751,808 | | | $ | 8,318,465 | |
The accompanying notes are an integral part of thethese consolidated financial statements.
VERTEX PHARMACEUTICALS INCORPORATED
Consolidated Statements of Shareholders’ Equity and Noncontrolling Interest
(in thousands)millions)
| | Common Stock | | Additional
Paid-in Capital | | Accumulated
Other
Comprehensive Income (Loss) | | Retained Earnings (Accumulated Deficit) | | Total Vertex
Shareholders’ Equity | | Noncontrolling
Interest | | Total
Shareholders’ Equity |
| Shares | | Amount | |
| Balance, December 31, 2017 | 253,253 | | | $ | 2,512 | | | $ | 7,157,362 | | | $ | (11,572) | | | $ | (5,119,723) | | | $ | 2,028,579 | | | $ | 13,727 | | | $ | 2,042,306 | |
| Cumulative effect adjustment for adoption of new accounting guidance | — | | | — | | | — | | | (24,120) | | | 33,349 | | | 9,229 | | | — | | | 9,229 | |
| Common Stock | | | Common Stock | | Additional
Paid-in Capital | | Accumulated
Other
Comprehensive Income (Loss) | | Retained Earnings | | Total
Shareholders’ Equity |
| Shares | |
| Balance at December 31, 2020 | |
| Balance at December 31, 2020 | |
| Balance at December 31, 2020 | |
| Other comprehensive income, net of tax | Other comprehensive income, net of tax | — | | | — | | | — | | | 36,351 | | | — | | | 36,351 | | | — | | | 36,351 | |
| Net income (loss) | — | | | — | | | — | | | — | | | 2,096,896 | | | 2,096,896 | | | (9,793) | | | 2,087,103 | |
| Net income | |
| Repurchases of common stock | Repurchases of common stock | (2,094) | | | (21) | | | (350,022) | | | — | | | — | | | (350,043) | | | — | | | (350,043) | |
| Common stock withheld for employee tax obligations | |
| Issuance of common stock under benefit plans | Issuance of common stock under benefit plans | 4,013 | | | 55 | | | 288,480 | | | — | | | — | | | 288,535 | | | — | | | 288,535 | |
| Stock-based compensation expense | Stock-based compensation expense | — | | | — | | | 325,656 | | | — | | | — | | | 325,656 | | | — | | | 325,656 | |
| VIE noncontrolling interest upon deconsolidation | — | | | — | | | — | | | — | | | — | | | — | | | (3,540) | | | (3,540) | |
| Other VIE activity | — | | | — | | | — | | | — | | | — | | | — | | | (394) | | | (394) | |
| Balance, December 31, 2018 | 255,172 | | | $ | 2,546 | | | $ | 7,421,476 | | | $ | 659 | | | $ | (2,989,478) | | | $ | 4,435,203 | | | $ | 0 | | | $ | 4,435,203 | |
Cumulative effect adjustment for adoption of new accounting guidance | — | | | — | | | — | | | — | | | (40,310) | | | (40,310) | | | — | | | (40,310) | |
| Balance at December 31, 2021 | |
| Other comprehensive loss, net of tax | |
| Net income | |
| | Common stock withheld for employee tax obligations | |
| Common stock withheld for employee tax obligations | |
| Common stock withheld for employee tax obligations | |
| Issuance of common stock under benefit plans | |
| Stock-based compensation expense | |
| Balance at December 31, 2022 | |
| Other comprehensive loss, net of tax | Other comprehensive loss, net of tax | — | | | — | | | — | | | (2,632) | | | — | | | (2,632) | | | — | | | (2,632) | |
| Net income | Net income | — | | | — | | | — | | | — | | | 1,176,810 | | | 1,176,810 | | | — | | | 1,176,810 | |
| Repurchases of common stock | Repurchases of common stock | (1,046) | | | (10) | | | (186,010) | | | — | | | — | | | (186,020) | | | — | | | (186,020) | |
Common stock withheld for employee tax obligations | Common stock withheld for employee tax obligations | (28) | | | — | | | (5,995) | | | — | | | — | | | (5,995) | | | — | | | (5,995) | |
| Issuance of common stock under benefit plans | Issuance of common stock under benefit plans | 4,895 | | | 53 | | | 345,926 | | | — | | | — | | | 345,979 | | | — | | | 345,979 | |
| Stock-based compensation expense | Stock-based compensation expense | — | | | — | | | 362,209 | | | — | | | — | | | 362,209 | | | — | | | 362,209 | |
| Balance, December 31, 2019 | 258,993 | | | $ | 2,589 | | | $ | 7,937,606 | | | $ | (1,973) | | | $ | (1,852,978) | | | $ | 6,085,244 | | | $ | 0 | | | $ | 6,085,244 | |
| Other comprehensive loss, net of tax | — | | | — | | | — | | | (66,507) | | | — | | | (66,507) | | | — | | | (66,507) | |
| Net income | — | | | — | | | — | | | — | | | 2,711,647 | | | 2,711,647 | |
| — | |
| 2,711,647 | |
| Repurchases of common stock | (2,405) | | | (24) | | | (539,112) | | | — | | | — | | | (539,136) | | | — | | | (539,136) | |
Common stock withheld for employee tax obligations | (804) | | | (8) | | | (200,263) | | | — | | | — | | | (200,271) | | | — | | | (200,271) | |
| Issuance of common stock under benefit plans | 4,106 | | | 42 | | | 262,726 | | | — | | | — | | | 262,768 | | | — | | | 262,768 | |
| Stock-based compensation expense | — | | | — | | | 433,070 | | | — | | | — | | | 433,070 | | | — | | | 433,070 | |
| Balance, December 31, 2020 | 259,890 | | | $ | 2,599 | | | $ | 7,894,027 | | | $ | (68,480) | | | $ | 858,669 | | | $ | 8,686,815 | | | $ | 0 | | | $ | 8,686,815 | |
| Balance at December 31, 2023 | |
The accompanying notes are an integral part of thethese consolidated financial statements.
VERTEX PHARMACEUTICALS INCORPORATED
Consolidated Statements of Cash Flows
(in thousands)millions)
| | Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Year Ended December 31, | | | Year Ended December 31, |
| 2023 | | | 2023 | | 2022 | | 2021 |
| Cash flows from operating activities: | Cash flows from operating activities: | | | | | |
| Net income | |
| Net income | |
| Net income | Net income | $ | 2,711,647 | | | $ | 1,176,810 | | | $ | 2,087,103 | |
| Adjustments to reconcile net income to net cash provided by operating activities: | Adjustments to reconcile net income to net cash provided by operating activities: | |
| Stock-based compensation expense | Stock-based compensation expense | 429,461 | | | 360,489 | | | 325,047 | |
| Depreciation expense | 109,515 | | | 106,941 | | | 72,420 | |
| Deferred income taxes (including benefit from valuation allowance release in 2018) | 277,341 | | | 167,387 | | | (1,512,325) | |
| Gains on equity securities | (311,937) | | | (197,597) | | | (2,558) | |
| Increase in fair value of contingent consideration | 13,100 | | | 4,459 | | | 0 | |
| Intangible asset impairment charge | 0 | | | 0 | | | 29,000 | |
| Stock-based compensation expense | |
| Stock-based compensation expense | |
| Depreciation and amortization expenses | |
| Deferred income taxes | |
| Losses (gains) on equity securities | |
| Decrease in fair value of contingent consideration | |
| Other non-cash items, net | Other non-cash items, net | 78,832 | | | 16,942 | | | 33,579 | |
| Changes in operating assets and liabilities: | Changes in operating assets and liabilities: | |
| Accounts receivable, net | |
| Accounts receivable, net | |
| Accounts receivable, net | Accounts receivable, net | (223,444) | | | (225,587) | | | (108,152) | |
| Inventories | Inventories | (132,014) | | | (64,047) | | | (31,965) | |
| Prepaid expenses and other assets | Prepaid expenses and other assets | (297,562) | | | 35,440 | | | 16,684 | |
| Accounts payable | Accounts payable | 51,276 | | | (22,785) | | | 36,554 | |
| Accrued expenses | Accrued expenses | 122,198 | | | 172,881 | | | 302,755 | |
| Other liabilities | Other liabilities | 425,092 | | | 37,997 | | | 22,144 | |
| Net cash provided by operating activities | Net cash provided by operating activities | 3,253,505 | | | 1,569,330 | | | 1,270,286 | |
| Cash flows from investing activities: | Cash flows from investing activities: | |
| Payments to acquire businesses, net of cash acquired | 0 | | | (1,154,212) | | | 0 | |
| Purchases of available-for-sale debt securities | Purchases of available-for-sale debt securities | (431,396) | | | (537,196) | | | (431,918) | |
| Maturities of available-for-sale debt securities | 372,342 | | | 475,924 | | | 431,576 | |
| Purchases of available-for-sale debt securities | |
| Purchases of available-for-sale debt securities | |
| Sales and maturities of available-for-sale debt securities | |
| Purchases of property and equipment | |
| Sale of equity securities | Sale of equity securities | 437,567 | | | 94,936 | | | 0 | |
| Purchases of property and equipment | (259,798) | | | (75,451) | | | (95,449) | |
| Investment in equity securities | (19,327) | | | (39,319) | | | (83,471) | |
| Investment in note receivable | 0 | | | 0 | | | (15,000) | |
| Decrease in restricted cash due to deconsolidation of VIE | — | | | — | | | (7,896) | |
| Net cash provided by (used in) investing activities | 99,388 | | | (1,235,318) | | | (202,158) | |
| Investment in equity securities and notes receivable | |
| Payments related to finite-lived intangible assets | |
| Payment to acquire ViaCyte, net of cash acquired | |
| Net cash used in investing activities | |
| Cash flows from financing activities: | Cash flows from financing activities: | |
| Issuances of common stock under benefit plans | |
| Issuances of common stock under benefit plans | |
| Issuances of common stock under benefit plans | Issuances of common stock under benefit plans | 264,946 | | | 343,244 | | | 289,293 | |
| Repurchases of common stock | Repurchases of common stock | (539,136) | | | (186,020) | | | (350,043) | |
| Payments in connection with common stock withheld for employee tax obligations | Payments in connection with common stock withheld for employee tax obligations | (200,271) | | | (5,995) | | | 0 | |
| Payments on finance leases * | (42,275) | | | (39,185) | | | (33,388) | |
| Proceeds from finance leases * | 13,251 | | | 10,046 | | | 20,840 | |
| Other financing activities | (1,796) | | | 4,683 | | | 2,079 | |
| Net cash (used in) provided by financing activities | (505,281) | | | 126,773 | | | (71,219) | |
| Payments on finance leases | |
| Proceeds from finance leases and other financing activities | |
| Net cash used in financing activities | |
| Effect of changes in exchange rates on cash | Effect of changes in exchange rates on cash | 20,552 | | | 1,643 | | | (6,182) | |
| Net increase in cash, cash equivalents and restricted cash | 2,868,164 | | | 462,428 | | | 990,727 | |
| Net (decrease) increase in cash, cash equivalents and restricted cash | |
| Cash, cash equivalents and restricted cash—beginning of period | Cash, cash equivalents and restricted cash—beginning of period | 3,120,681 | | | 2,658,253 | | | 1,667,526 | |
| Cash, cash equivalents and restricted cash—end of period | Cash, cash equivalents and restricted cash—end of period | $ | 5,988,845 | | | $ | 3,120,681 | | | $ | 2,658,253 | |
| | Supplemental disclosure of cash flow information: | Supplemental disclosure of cash flow information: | |
| Supplemental disclosure of cash flow information: | |
| Supplemental disclosure of cash flow information: | |
| Cash paid for income taxes | |
| Cash paid for income taxes | |
| Cash paid for income taxes | |
| Cash paid for interest | Cash paid for interest | $ | 54,520 | | | $ | 55,554 | | | $ | 66,458 | |
| Cash paid for income taxes | $ | 191,776 | | | $ | 24,730 | | | $ | 12,402 | |
| Capitalization of costs related to construction financing lease obligation * | $ | 0 | | | $ | 0 | | | $ | 3,389 | |
| Issuances of common stock from employee benefit plans receivable | $ | 642 | | | $ | 2,820 | | | $ | 86 | |
| * For the year ended December 31, 2018, amounts are related to the Company’s capital leases and construction financing lease obligations pursuant to ASC 840, Leases, which was applicable until December 31, 2018. | | Net payments due to CRISPR Therapeutics related to finite-lived intangible assets | |
The accompanying notes are an integral part of thethese consolidated financial statements.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements
A.Nature of Business and Accounting Policies
Business
Vertex Pharmaceuticals Incorporated (“Vertex”Vertex,” “we,” “us” or the “Company”“our”) is a global biotechnology company that invests in scientific innovation to create transformative medicines for people with serious diseases. The Company’s business is focuseddiseases, with a focus on developing and commercializing therapies forspecialty markets. We have four approved medicines that treat the treatmentunderlying cause of cystic fibrosis (“CF”), a life-threatening genetic disease, and advancing researchone approved therapy that treats severe sickle cell disease (“SCD”) and developmenttransfusion dependent beta thalassemia (“TDT”), life shortening inherited blood disorders. Our pipeline includes clinical-stage programs in other indications. The Company’sCF, sickle cell disease, beta thalassemia, acute and neuropathic pain, APOL1-mediated kidney disease, type 1 diabetes, myotonic dystrophy type 1 and alpha-1 antitrypsin deficiency.
Our marketed productsCF medicines are TRIKAFTA/KAFTRIO (elexacaftor/tezacaftor/ivacaftor and ivacaftor), SYMDEKO/SYMKEVI (tezacaftor in combination with(tezacaftor/ivacaftor and ivacaftor), ORKAMBI (lumacaftor in combination with (lumacaftor/ivacaftor) and KALYDECO (ivacaftor).
Starting in the fourth quarter of 2023, we have received approval to market CASGEVY (exagamglogene autotemcel or “exa-cel”) for the treatment of SCD and TDT in the United States (“U.S.”), which are approved to treat people with CF who have specific mutations in their cystic fibrosis transmembrane conductance regulatorthe European Union, the United Kingdom (“CFTR”U.K.”) gene.
As, the Kingdom of December 31, 2020,Saudi Arabia, and the Company had cash, cash equivalents and marketable securitiesKingdom of $6.7 billion. The Company expects that cash flows from the sales of its products, together with its cash, cash equivalents and marketable securities, will be sufficient to fund its operations for at least the next twelve months.
The Company is subject to risks common to companies in its industry including, but not limited to, the dependence on revenues from its CF products, competition, uncertainty about clinical trial outcomes and regulatory approvals, uncertainties relating to pharmaceutical pricing and reimbursement, uncertainty related to international expansion, uncertain protection of proprietary technology, the need to comply with government regulations, share price volatility, dependence on collaborative relationships and potential product liability.Bahrain.
Basis of Presentation
The accompanying consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of AmericaU.S. (“U.S. GAAP”), reflect the operations of (i) the Company, (ii) its wholly-owned subsidiariesVertex and (iii) a consolidated variable interest entity (“VIE”). In 2018, the Company deconsolidated BioAxone Biosciences, Inc. (“BioAxone”), a VIE the Company had consolidated since 2014. As of December 31, 2020 and 2019, the Company did not have any consolidated VIEs.our wholly owned subsidiaries. All material intercompany balances and transactions have been eliminated. The Company operatesWe operate in 1one segment, pharmaceuticals. Please refer to Note Q, “Segment Information,” for enterprise-wide disclosures regarding the Company’sour revenues, major customers and long-lived assets by geographic area. The Company has
In 2022, we began to separately classify upfront, contingent milestone, and other payments pursuant to our business development transactions, including collaborations, licenses of third-party technologies, and asset acquisitions as “Acquired in-process research and development expenses” in our consolidated statements of income in cases where such acquired assets do not have an alternative future use. To conform prior periods to current presentation, we reclassified certain items$1.1 billion from the prior year’s consolidated financial statements“Research and development expenses” to conform“Acquired in-process research and development expenses” for 2021. Please refer to the current year’s presentation.Note B, “Collaboration, License and Other Arrangements,” for further information on these transactions.
Use of Estimates
The preparation of consolidated financial statements in accordance with U.S. GAAP requires managementus to make certain estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of theour consolidated financial statements, and the amounts of revenues and expenses during the reported periods. Significant estimates in these consolidated financial statements have been made in connection with (i) determining the transaction price of revenues, (ii) accounting for acquisitions, including intangible assets, goodwill and contingent consideration and (iii) evaluating deferred tax asset valuation allowances and the provision for income taxes. The Company bases itsWe base our estimates on historical experience and various other assumptions, including in certain circumstances future projections that management believeswe believe to be reasonable under the circumstances. Actual results could differ from those estimates. Changes in estimates are reflected in reported results in the period in which they become known.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Revenue Recognition
The Company recognizesWe recognize revenue when a customer obtains control of promised goods or services. The Company recordsWe record the amount of revenue that reflects the consideration that it expectswe expect to receive in exchange for those goods or services. The Company appliesWe apply the following five-step model in order to determine this amount: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations, including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) the Company satisfieswe satisfy each performance obligation.
The CompanyWe only appliesapply the five-step model to contracts when it is probable that itwe will collect the consideration to which it iswe are entitled in exchange for the goods or services that it transferswe transfer to the customer. Once a contract is determined to be within the
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
scope of Accounting Standards Codification (“ASC”) 606, Revenue from Contracts with Customers (“ASC 606”) at contract inception, the Company reviewswe review the contract to determine which performance obligations itwe must deliver and which of these performance obligations are distinct. The Company recognizesWe recognize as revenue the amount of the transaction price that is allocated to each performance obligation when that performance obligation is satisfied or as it is satisfied. Generally, the Company’sour performance obligations are transferred to customers at a point in time, typically upon delivery.
Product Revenues, Net
The Company sells its products principally to a limited number of specialty pharmacy and specialty distributors in the United States (“U.S.”), which account for the largest portion of its total revenues, and makes international sales primarily to specialty distributors and retail chains, as well as hospitals and clinics, many of which are government-owned or supported (collectively, its “Customers”). The Company’s Customers in the U.S. subsequently resell the products to patients and health care providers. The Company recognizes net product revenues from sales when the Customers obtain control of the Company’s products, which typically occurs upon delivery to the Customer. The Company’s payment terms are approximately 30 days in the U.S. and consistent with prevailing practice in international markets.
Revenues from product sales are recorded at the net sales price, or “transaction price,” which includes estimates of variable consideration that result from (a) invoice discounts for prompt payment and distribution fees, (b) government and private payor rebates, chargebacks, discounts and fees and (c) other incentives for certain indirect customers, including costs of co-pay assistance programs for patients, as well as other incentives for certain indirect customers.patients. Reserves are established for the estimates of variable consideration based on the amounts earned or to be claimed on the related sales. The reserves are classified as reductions to “Accounts receivable, net” if payable to a Customercustomer or “Accrued expenses” if payable to a third-party. Where appropriate, the Company utilizeswe utilize the expected value method to determine the appropriate amount for estimates of variable consideration based on factors such as the Company’sour historical experience, current contractual and statutory requirements, specific known market events and trends, industry data and forecasted customer buying and payment patterns. The amount of variable consideration that is included in the transaction price may be constrained and is included in our net product revenues only to the extent that it is probable that a significant reversal in the amount of the cumulative revenue recognized will not occur in a future period. Actual amounts of consideration ultimately received may differ from the Company’sour estimates. If actual results vary from the Company’sour estimates, the Company adjustswe adjust these estimates, which would affect net product revenue and earnings in the period such variances become known.
Invoice Discounts and Distribution Fees: The Company generally providesIn the U.S., we may provide invoice discounts on product sales to its Customersour customers for prompt payment and payspay fees for distribution services, such as fees for certain data that Customerscustomers provide to the Company. The Company estimatesus. We estimate that, based on itsour experience, its Customersour customers will earn these discounts and fees, and deductsdeduct the full amount of these discounts and fees from itsour gross product revenues and accounts receivable at the time such revenues are recognized.
Rebates, Chargebacks, Discounts and Fees: The Company contractsWe contract with government agencies (its(our “Third-party Payors”) so that products will be eligible for purchase by, or partial or full reimbursement from, such Third-party Payors. The Company estimatesWe estimate the rebates, chargebacks, discounts and fees itwe will provide to Third-party Payors and deductsdeduct these estimated amounts from itsour gross product revenues at the time the revenues are recognized. For each product, the Company estimateswe estimate the aggregate rebates, chargebacks and discounts that itwe will provide to Third-party Payors based upon (i) the Company’sour contracts with these Third-party Payors, (ii) the government-mandated discounts and fees
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
applicable to government-funded programs, (iii) information obtained from the Company’s Customersour customers and other third-party data regarding the payor mix for such product and (iv) historical experience.
Other Incentives: Other incentives that the Company offerswe offer include co-pay mitigation rebates provided bythat we provide in the CompanyU.S. to commercially insured patients who have coverage and who reside in states that permit co-pay mitigation programs. Based upon the terms of the Company’sour co-pay mitigation programs, the Company estimateswe estimate average co-pay mitigation amounts for each of itsour products in order to establish appropriate accruals.
The Company makesWe make significant estimates and judgments that materially affect itsour recognition of net product revenues. The Company adjusts itsWe adjust our estimated rebates, chargebacks and discounts based on new information, including information regarding actual rebates, chargebacks and discounts for itsour products, as it becomes available. Claims by third-party payors for rebates, chargebacks and discounts frequently are submitted to the Companyus significantly after the related sales, potentially resulting in adjustments in the period in which the new information becomes known. The Company’sOur credits to product revenue related to prior period sales have not been significant and primarily related to rebates and discounts.
The Company excludesWe exclude taxes collected from Customerscustomers relating to product sales and remitted to governmental authorities from revenues.
CF Product Revenues
We sell our CF products principally to a limited number of specialty pharmacy and specialty distributors in the U.S.,
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
which account for the largest portion of our total revenues. Our customers in the U.S. subsequently resell the products to patients and health care providers. We make international sales primarily to specialty distributors and retail pharmacies, as well as hospitals and clinics, many of which are government-owned or supported. As noted above, we recognize net product revenues from sales when our customers obtain control of our products, which typically occurs upon delivery to our CF customers. Our payment terms are approximately 30 days in the U.S. and consistent with prevailing practice in international markets.
CASGEVY Product Revenues
We expect to sell CASGEVY principally to a limited number of specialty distributors or directly to authorized hospitals and clinics in markets where a specialty distributor is not utilized. Control is expected to transfer to our CASGEVY customers, resulting in revenue recognition, upon infusion of this gene-editing therapy into our patients.
Contract Liabilities
The Company recordedWe had contract liabilities of $191.5$170.3 million and $62.3$159.6 million as of December 31, 20202023 and 2019,2022, respectively, related to annual contracts with government-owned and supported customers in international markets that limit the amount of annual reimbursement the Companywe can receive.receive for our CF products. Upon exceeding the annual reimbursement amount provided by the customer’s contract with us, our CF products are provided free of charge, which is a material right. These contracts include upfront payments and fees. The Company defersIf we estimate that we will exceed the annual reimbursement amount under a contract, we defer a portion of the consideration received for shipments made up to the annual reimbursement limit as a portion of “Other current liabilities.” TheOnce the reimbursement limit has been reached, we recognize the deferred amount is recognized as revenue when we ship the free products are shipped. The Company’sproducts. Our CF product revenue contracts include performance obligations that are one year or less.
The Company’sOur contract liabilities at the end of each fiscal year relate to contracts with CF annual reimbursement limits in international markets in which the annual period associated with the contract is not the same as the Company’sour fiscal year. In these markets the Company recognizeswe recognize revenues related to performance obligations satisfied in previous years; however, these revenues do not relate to any performance obligations that were satisfied more than 12 months prior to the beginning of the current year. During the years ended December 31, 2020, 20192023, 2022 and 2018, the Company2021, we recorded $62.3$159.6 million, $24.9$171.7 million and $1.7$191.5 million, respectively, of CF product revenues that were recorded as contract liabilities at the beginning of the year.
French Early Access Programs
In 2015, the Company began distributing ORKAMBI through early access programs in France and remained engaged in reimbursement discussions with the French government until November 2019, when the Company reached an agreement with the French government for ORKAMBI, including ORKAMBI distributed through early access programs. From the time the Company began distributing ORKAMBI through early access programs in France, it expected the difference between the amounts collected based on the invoiced amount and the final amount for ORKAMBI distributed through early access programs would be returned to the French government. As a result, the Company had classified a refund liability related to the early access programs in France within “Accrued expenses” on its consolidated balance sheets.
From the first quarter of 2018 through the third quarter of 2019, the Company recognized net product revenues for ORKAMBI sales in France under the early access programs based on a transaction price that reflected the Company’s estimate of consideration it expected to retain that would not be subject to a significant reversal in amounts recognized. When determining if variable consideration should be constrained, the Company considers whether there are factors outside its control that could result in a significant reversal of revenue. In making these assessments, the Company considers the likelihood and magnitude of a potential reversal of revenue.
Upon reaching an agreement with the French government for ORKAMBI, including ORKAMBI distributed through early access programs in November 2019, the Company updated the transaction price to reflect the final amount for ORKAMBI distributed through early access programs. As a result, the Company recognized net product revenues of
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
$155.8 million related to prior period ORKAMBI early access program sales in the fourth quarter of 2019 because the updated transaction price for ORKAMBI distributed through these programs exceeded the Company’s previous estimate of the consideration it expected to retain that would not be subject to a significant reversal in amounts recognized. The Company paid the final amount due to the French government in 2020.
Collaborative and Royalty Revenues
The Company has not recorded significant collaborative and royalty revenues during the three years ended December 31, 2020; however, in future periods, it may recognize collaborative revenues generated through collaborative research, development and/or commercialization agreements. The terms of these agreements typically include payment to the Company related to one or more of the following: nonrefundable, upfront license fees; development and commercial milestones; funding of research and/or development activities; and royalties on net sales of licensed products. Revenue is recognized upon satisfaction of a performance obligation by transferring control of a good or service to the collaborator.
For each collaborative research, development and/or commercialization agreement that results in revenue, the Company identifies all material performance obligations, which may include a license to intellectual property and know-how, research and development activities and/or transition activities. In order to determine the transaction price, in addition to any upfront payment, the Company estimates the amount of variable consideration at the outset of the contract either utilizing the expected value or most likely amount method, depending on the facts and circumstances relative to the contract. The Company constrains (reduces) the estimate of variable consideration such that it is probable that a significant reversal of previously recognized revenue will not occur throughout the life of the contract. When determining if variable consideration should be constrained, management considers whether there are factors outside the Company’s control that could result in a significant reversal of revenue. In making these assessments, the Company considers the likelihood and magnitude of a potential reversal of revenue. These estimates are re-assessed each reporting period as required.
Once the estimated transaction price is established, amounts are allocated to the performance obligations that have been identified. The transaction price is generally allocated to each separate performance obligation on a relative standalone selling price basis. In order to account for these agreements, the Company must develop assumptions that require judgment to determine the standalone selling price, which may include (i) the probability of obtaining marketing approval for the drug candidate, (ii) estimates regarding the timing of and the expected costs to develop and commercialize the drug candidate, (iii) estimates of future cash flows from potential product sales with respect to the drug candidate and (iv) appropriate discount and tax rates. Standalone selling prices used to perform the initial allocation are not updated after contract inception. The Company does not include a financing component to its estimated transaction price at contract inception unless it estimates that certain performance obligations will not be satisfied within one year.
Upfront License Fees: If a license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in an arrangement, the Company recognizes revenue from the related nonrefundable, upfront license fees based on the relative standalone selling price prescribed to the license compared to the total selling price of the arrangement. The revenue is recognized when the license is transferred to the collaborator and the collaborator is able to use and benefit from the license. For licenses that are not distinct from other obligations identified in the arrangement, the Company utilizes judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time. If the combined performance obligation is satisfied over time, the Company applies an appropriate method of measuring progress for purposes of recognizing revenue from nonrefundable, upfront license fees. The Company evaluates the measure of progress each reporting period and, if necessary, adjusts the measure of performance and related revenue recognition.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Development and Regulatory Milestone Payments: Depending on facts and circumstances, the Company may conclude that it is appropriate to include certain milestones in the estimated transaction price or that it is appropriate to fully constrain the milestones. A milestone payment is included in the transaction price in the reporting period that the Company concludes that it is probable that recording revenue in the period will not result in a significant reversal in amounts recognized in future periods. This may result in revenues from certain milestones and a corresponding contract asset being recorded in a reporting period before the milestone is achieved. Milestone payments that have not been included in the transaction price to date are fully constrained until the Company concludes that their achievement is probable and that recognition of the related revenue will not result in a significant reversal in amounts recognized in future periods. The Company re-evaluates the probability of achievement of such development milestones and any related constraint each reporting period and adjusts its estimate of the overall transaction price, including the amount of collaborative revenue that it has recorded, if necessary.
Research and Development Activities/Transition Services: If the Company is entitled to reimbursement from its collaborators for specified research and development expenses, it accounts for the related services that it provides as separate performance obligations if it determines that these services represent a material right. The Company also determines whether the reimbursement of research and development expenses should be accounted for as collaborative revenues or an offset to research and development expenses in accordance with the provisions of gross or net revenue presentation. The Company recognizes the corresponding revenues or records the corresponding offset to research and development expenses as it satisfies the related performance obligations.
Sales-based Milestone and Royalty Payments: The Company’s collaborators may be required to pay the Company sales-based milestones or royalties on future sales of commercial products. The Company recognizes revenues related to sales-based milestone and royalties upon the later to occur of (i) achievement of the collaborator’s underlying sales or (ii) satisfaction of any performance obligation(s) related to these sales, in each case assuming the license to the Company’s intellectual property is deemed to be the predominant item to which the sales-based milestones and/or royalties relate.
Concentration of Credit Risk
Financial instruments that potentially subject the Companyus to concentration of credit risk consist principally of money market fundscash equivalents and marketable securities. The Company placesWe place these investments with highly rated financial institutions, and, by policy, limitslimit the amountsamount of credit exposure to any one financial institution. These amounts at times may exceed federally insured limits. The CompanyWe also maintainsmaintain a foreign currency hedging program that includes foreign currency forward contracts with several counterparties. The Company hasWe have not experienced any credit losses related to these financial instruments and doesdo not believe it iswe are exposed to any significant credit risk related to these instruments.
The CompanyWe are also is subject to credit risk from itsour accounts receivable related to itsour product sales and collaborators. The Company evaluatesWe evaluate the creditworthiness of each of itsour customers and hashave determined that all of itsour material customers are creditworthy. To date, the Company haswe have not experienced significant losses with respect to the collection of itsour accounts receivable. The Company believesWe believe that itsour allowances, which are not significant to itsour consolidated financial statements, are adequate at December 31, 2020.2023. Please refer to Note Q, “Segment Information,” for further information.
Cash and Cash Equivalents
The Company considersWe consider all highly liquid investments with original maturities of three months or less at the date of purchase to be cash equivalents.
Marketable Securities
As of December 31, 2020, the Company’s2023, our marketable securities consisted of investments in available-for-sale debt securities and corporate equity securities with readily determinable fair values. The Company classifiesWe classify marketable securities available to fundwith current operationsmaturities of less than one year as current assets on itsour consolidated balance sheets. MarketableThe remainder of our marketable securities are classified as long-term assets within “Long-term marketable securities” on theour consolidated balance sheets if (i) they have been in an unrealized loss position for longer than one year and (ii) the Company has the ability and intent to hold them (a) until the carryingsheets. The fair value is recovered and (b) such holdingof
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
period may be longer than one year. The Company’s marketable securities are stated at fair value. The fair value of these securities is based on quoted prices for identical or similar assets.
The Company recordsWe record unrealized gains (losses) on available-for-sale debt securities as a component of “Accumulated other comprehensive loss,(loss) income,” which is a separate component of shareholders’ equity on itsour consolidated balance sheet,sheets, until such gains and losses are realized. Realized gains and losses, if any, are determined using the specific identification method.
The Company records changes in the fair value of its investments in corporate equity securities to “Other income (expense), net” in its consolidated statements of operations. Realized gains and losses, which are also included in “Other income (expense), net,” are determined on an original weighted-average cost basis.
The Company adopted Accounting Standards Update (“ASU”) 2016-13, Financial Instruments - Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments (“ASU 2016-13”) as of January 1, 2020, which did not have a significant impact on its consolidated financial statements. For available-for-sale debt securities in unrealized loss positions, ASU 2016-13 requires the Companywe are required to assess whether to record an allowance for credit losses using an expected loss model, which replaces the incurred loss model required under the previous guidance.model. A credit loss is limited to the amount by which the amortized cost of an investment exceeds its fair value. A previously recognized credit loss may be decreased in subsequent periods if the Company’sour estimate of fair value for the investment increases. To determine whether to record a credit loss, the Company considerswe consider issuer specific credit ratings and historical losses as well as current economic conditions and itsour expectations for future economic conditions.
We record changes in the fair value of our investments in corporate equity securities to “Other (expense) income, net” in our consolidated statements of income. Realized gains and losses, which are also included in “Other (expense) income, net,” are determined on an original weighted-average cost basis.
Accounts Receivable
The Company deductsWe deduct invoice discounts for prompt payment and fees for distribution services from itsour accounts receivable based on itsour experience that the Company’s Customersour customers will earn these discounts and fees. The Company’sOur estimates for itsour allowance for credit losses, which has not been significant to date, is determined based on existing contractual payment terms, historical payment patterns, current economic conditions and the Company’sour expectation for future economic conditions.
Stock-based Compensation Expense
The Company expensesWe expense the fair value of employee restricted stock units and other forms of stock-based employee compensation over the associated employee service period on a straight-line basis. Stock-based compensation expense is determined based on the fair value of the award at the grant date and is adjusted each period to reflect actual forfeitures and the outcomes of certain performance conditions.
For awards with performance conditions in which the award does not vest unless the performance condition is met, the Company recognizeswe recognize expense if, and to the extent that, the Company estimateswe estimate that achievement of the performance condition is probable. If the Company concludeswe conclude that vesting is probable, it recognizeswe recognize expense from the date it reacheswe reach this conclusion through the estimated vesting date.
The Company providesWe provide to employees who have rendered a certain number of years of service to the CompanyVertex and meet certain age requirements, partial or full acceleration of vesting of these equity awards, subject to certain conditions including a notification period, upon a termination of employment other than for cause. Approximately 5% of the Company’sour employees were eligible for partial or full acceleration of any of their equity awards as of December 31, 2020. The Company recognizes2023. We recognize stock-based compensation expense related to these awards over a service period reflecting qualified employees’ eligibility for partial or full acceleration of vesting.
Please refer to Note N, “Stock-based Compensation Expense,” for tables displaying our stock-based compensation expense by type of award and by line item within our consolidated statements of income.
Research and Development Expenses
The Company expenses as incurred all research and development expenses, including amounts funded by research and development collaborations. The Company capitalizes nonrefundable advance payments made by the Company for research and development activities and expenses the payments as the related goods are delivered or the related services are performed.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Research and development expenses are comprised of costs incurred by the Companywe incur in performing research and development activities, including salary and benefits; stock-based compensation expense; outsourced services and other direct expenses, including clinical trial, and pharmaceutical development and drug supply costs; collaborative payments;intangible asset impairment charges; and infrastructure costs, including facilities costs and depreciation expense. We recognize research and development expenses as incurred. We capitalize nonrefundable advance payments we make for research and development activities and expense the payments as the related goods are delivered or the related services are performed.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Acquired In-process Research and Development Expenses
Our research and development activities include upfront, contingent milestone, and other payments pursuant to our business development transactions, including collaborations, licenses of third-party technologies, and asset acquisitions. In-process research and development that is acquired in a transaction that does not qualify as a business combination under U.S. GAAP and that does not have an alternative future use is recorded to “Acquired in-process research and development expenses” in our consolidated statements of income in the period in which it is acquired.
Inventories
The Company values itsWe value our inventories at the lower-of-cost or net realizable value. The Company determinesWe determine the cost of itsour inventories, which includesinclude amounts related to materials and manufacturing overhead, on a first-in, first-out basis. The Company performsWe perform an assessment of the recoverability of our capitalized inventory during each reporting period and writeswrite down any excess and obsolete inventories to their net realizable value in the period in which the impairment is first identified. Shipping and handling costs incurred for inventory purchases are capitalized and recorded upon sale in “Cost of sales” in theour consolidated statements of operations.income. Shipping and handling costs incurred for product shipments are recorded as incurred in “Cost of sales” in theour consolidated statements of operations.income.
The Company capitalizesWe capitalize inventories produced in preparation for initiating sales of a drugproduct candidate when the related drugproduct candidate is considered to have a high likelihood of regulatory approval and the related costs are expected to be recoverable through sales of the inventories. In determining whether or not to capitalize such inventories, the Company evaluates,we evaluate, among other factors, information regarding the drugproduct candidate’s safety and efficacy, the status of regulatory submissions and communications with regulatory authorities and the outlook for commercial sales, including the existence of current or anticipated competitive drugs and the availability of reimbursement. In addition, the Company evaluateswe evaluate risks associated with manufacturing the drugproduct candidate and the remaining shelf-life of the inventories.
Property and Equipment
Property and equipment are recorded at cost, net of accumulated depreciation. Depreciation expense is recorded using the straight-line method over the estimated useful life of the related asset generally as follows:
| | | | | | | | |
| Description | | Estimated Useful Life |
| Buildings and improvements | | 15 to 40 years |
FurnitureLaboratory equipment, other equipment and equipmentfurniture | | 7 to 10 years |
| Leasehold improvements; assets under finance leases | | The shorter of the useful life of the assets or the estimated remaining term of the associated lease |
| Computers and software | | 3 to 5 years |
Maintenance and repairs to an asset that do not improve or extend its life are charged to operations.expensed as incurred. When assets are retired or otherwise disposed of, the assets and related accumulated depreciation are eliminated from the accounts and any resulting gain or loss is reflected in the Company’sour consolidated statements of operations. The Company performsincome. We perform an assessment of the fair value of the assets if indicators of impairment are identified during a reporting period and recordsrecord the assets at the lower of the net book value or the fair value of the assets.
The Company capitalizes internalWe capitalize costs incurred to develop software for internal use during the application development stage. Amortization of capitalized internally developed software costs is recorded in depreciation expensestage, which are depreciated over the useful life of the related asset.
Cloud Computing Service Contracts
We classify costs incurred to implement cloud computing service contracts as “Other assets” on our consolidated balance sheets. Amortization is recorded over the noncancellable term of the cloud computing service contract, plus any optional renewal periods that are reasonably certain to be exercised.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Leases
The Company adopted ASU 2016-02, Leases (Topic 842) (“ASC 842”), as of January 1, 2019. Under ASC 842, the Company determinesWe determine whether thean arrangement contains a lease at the inception of an arrangement.inception. If a lease is identified in an arrangement, the Company recognizeswe recognize a right-of-use asset and liability on itsour consolidated balance sheet and determinesdetermine whether the lease should be classified as a finance or operating lease. The Company doesWe do not recognize assets or liabilities for leases with lease terms of less than 12 months.
A lease qualifies as a finance lease if any of the following criteria are met at the inception of the lease: (i) there is a transfer of ownership of the leased asset to the CompanyVertex by the end of the lease term, (ii) the Company holdswe hold an option to purchase the leased asset that it iswe are reasonably certain to exercise, (iii) the lease term is for a major part of the remaining
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
economic life of the leased asset, (iv) the present value of the sum of lease payments equals or exceeds substantially all of the fair value of the leased asset, or (v) the nature of the leased asset is specialized to the point that it is expected to provide the lessor no alternative use at the end of the lease term. All other leases are recorded as operating leases.
Finance and operating lease assets and liabilities are recognized at the lease commencement date based on the present value of the lease payments over the lease term using the discount rate implicit in the lease. If the rate implicit is not readily determinable, the Company utilizes itswe utilize our incremental borrowing rate at the lease commencement date. Operating lease assets are further adjusted for prepaid or accrued lease payments. Operating lease payments are expensed using the straight-line method as an operating expense over the lease term. Finance lease assets are amortized to depreciation expense using the straight-line method over the shorter of the useful life of the related asset or the lease term. Finance lease payments are bifurcated into (i) a portion that is recorded as imputed interest expense and (ii) a portion that reduces the finance liability associated with the lease.
The Company doesWe do not separate lease and non-lease components for our real estate leases when determining which lease payments to include in the calculation of itsour lease assets and liabilities. Variable lease payments are expensed as incurred. If a lease includes an option to extend or terminate the lease, the Company reflectswe reflect the option in the lease term if it is reasonably certain itwe will exercise the option.
Finance leases are recorded in “Property and equipment, net,” “Other current liabilities” and “Long-term finance lease liabilities”liabilities,” and operating leases are recorded in “Operating lease assets,” “Other current liabilities” and “Long-term operating lease liabilities” on the Company’sour consolidated balance sheet.
In 2018, prior to the adoption of ASC 842 on January 1, 2019, the Company applied build-to-suit accounting and was the deemed owner of its leased corporate headquarters in Boston and research site in San Diego, for which it was recognizing depreciation expense over the buildings’ useful lives and imputed interest on the corresponding construction financing lease obligations.sheets.
Income Taxes
Our provision for income taxes is accounted for under the asset and liability method and includes federal, state, local and foreign taxes.
Deferred tax assets and liabilities are recognized for the expectedestimated future tax consequences of temporary differences between the financial statement carrying amounts and the income tax bases of assets and liabilities. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the year in which the temporary differences are expected to be recovered or settled. A valuation allowance is applied against any net deferred tax asset if, based on the available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. On a periodic basis, the Company reassesseswe reassess the valuation allowance on itsour deferred income tax assets weighing positive and negative evidence to assess the recoverability of itsour deferred tax assets. The Company includes,We include, among other things, itsour recent financial performance and itsour future projections in this periodic assessment.
The Company recordsWe record liabilities related to uncertain tax positions by prescribing a minimum recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. The Company does not believe any suchWe evaluate our uncertain tax positions currently pending will haveon a material adverse effect on its consolidated financial statements.
Variable Interest Entities
The Company reviews each collaboration agreement pursuantquarterly basis and consider various factors, including, but not limited to, which it licenses assets owned bychanges in tax law, the measurement of tax positions taken or expected to be taken in our tax returns, and changes in facts or circumstances related to a collaborator in ordertax position. We adjust our liabilities to determine whether or not it has a variable interest via the license agreement with the collaborator and if the variable interest is a variable interestreflect any subsequent changes in the collaboratorrelevant facts and circumstances surrounding the uncertain positions. We accrue interest and penalties related to unrecognized tax benefits as a whole. In assessing whether the Company has a variable interest in the collaborator as a whole, the Company considers and makes judgments regarding the purpose and designcomponent of our “Provision for income taxes.”
As part of the entity, the valueU.S. Tax Cut and Jobs Act of the licensed assets2017, we are subject to the collaborator, the value of the collaborator’s total assets and the significant activities of the collaborator. If the Company has a variable interest in the collaborator as a whole, the Company assesses whether or not the Company is the primary beneficiary of that VIE based on a number of factors, including (i)territorial tax system, under which party has the power to direct the activities that most significantly affect the VIE’s economic performance, (ii) the parties’ contractual rights and responsibilities pursuant to the collaboration agreement and (iii) which party has the obligation to absorb losses of or the right to receive benefits from the VIE that could be significant to the VIE. If the Company determines it is the primary beneficiary of a VIE at the onset of the collaboration agreement, the collaboration is treated as a business combination and the Company consolidates the financial statements of the VIE into the Company’s consolidated financial statements. On a quarterly basis,we must
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
establish an accounting policy to provide for tax on Global Intangible Low Taxed Income (“GILTI”) earned by certain foreign subsidiaries. We have elected to treat the Company evaluatesimpact of GILTI as a current tax expense in our “Provision for income taxes.”
Variable Interest Entities
We review each agreement pursuant to which we license technologies owned by a third party to determine whether it continues to beor not we have a variable interest via the license agreement with the third party and if the variable interest is a variable interest in the third party as a whole and whether or not we are the primary beneficiary of anythat variable interest entity (“VIE”). If we determine we are the primary beneficiary of a VIE at the onset of a license agreement, it is treated as a business combination and we consolidate the financial statements of the VIE into our consolidated VIEs. If the Company determines that it isfinancial statements until we are no longer the primary beneficiary of athe consolidated VIE, or no longer hashave a variable interest in the VIE, it deconsolidates the VIE in the period that the determination is made.VIE. As of December 31, 2023 and 2022, we did not have any consolidated VIEs.
Fair Value of In-process Research and Development Assets and Contingent PaymentsConsideration
The present-value models the Company uses to estimate the fair values of in-process research and development assets and contingent payments pursuant to collaborations and acquisitions incorporate significant assumptions.
The Company’s discounted cash flow models pertaining to in-process research and development assets include: (i) assumptions regarding the probability of obtaining marketing approval for a drug candidate; (ii) the timing of and the expected costs to develop and commercialize a drug candidate; (iii) estimates of future cash flows from potential product sales with respect to a drug candidate; and (iv) appropriate discount and tax rates.
The Company bases itsWe base our estimates of the probability of achieving the milestones relevant to the fair value of contingent payments which could include milestone, royalty and option payments, on industry data.data and our knowledge of the programs and viability of the programs. Estimates included in the discounted cash flow models pertaining to contingent payments also include: (i) estimateestimates regarding the timing of the relevant development and commercial milestones and royalties, and (ii) and appropriate discount rates. We record any increases or decreases in the fair value of our contingent payments to “Change in fair value of contingent consideration” in our consolidated statements of income. We record our contingent consideration liabilities at fair value on our consolidated balance sheets as “Other current liabilities” or “Other long-term liabilities” depending on when we estimate we will pay them. Please refer to Note D, “Fair Value Measurements,” for further information.
In-process Research and Development Assets
The Company recordsWe record the fair value of in-process research and development assets as of the transaction date of a business combination. Each of thesecombination on our consolidated balance sheets as “Other intangible assets, is accounted fornet.” These assets are used in research and development activities but have not yet reached technological feasibility, which occurs when we complete the research and development efforts by obtaining regulatory approval to market an underlying product candidate. We characterize in-process research and development assets on our consolidated balance sheets as an indefinite-lived intangible asset and is maintained on the Company’s consolidated balance sheetassets until either the project underlying it is completedthey achieve regulatory approval and become finite-lived intangible assets, or the asset becomesassets are impaired. IfUpon completion of the asset becomes impaired or is abandoned,associated research and development efforts, we will determine the remaining estimated life of the marketed product and begin amortizing the carrying value of the related intangible assetassets over this period. If the assets become impaired or are abandoned, the carrying value is written down to its fair value, and we record an impairment charge is recorded in the period in which the impairment occurs. If a project is completed, the carrying value of the related intangible asset is amortized as a part of “Cost of sales” over the remaining estimated life of the asset beginning in the period in which the project is completed. In-processWe test in-process research and development assets are tested for impairment on an annual basis as of October 1, and more frequently if indicators are present or changes in circumstances suggest that impairment may exist.
In-processThe fair value of our in-process research and development assets is determined using either the multi-period excess earnings or the relief from royalty methods of the income approach. Each method requires us to make: (i) assumptions regarding the probability of obtaining marketing approval for a product candidate; (ii) estimates of future cash flows from potential product sales with respect to a product candidate; and (iii) appropriate discount and tax rates. The multi-period excess earnings method also requires us to estimate the timing of and the expected costs to develop and commercialize a product candidate. The relief from royalty method also requires us to estimate the after-tax royalty savings expected from ownership of a product candidate that is acquired in a transaction that does not qualifywe acquired.
Finite-lived Intangible Assets
We record finite-lived intangible assets at cost, net of accumulated amortization, on our consolidated balance sheets as a business combination under U.S. GAAP“Other intangible assets, net.” Each of these assets relates to our marketed products and that does not have an alternative future use is recorded to “Researchmay include, among other things, completed research and development expenses”projects that were previously reflected on our consolidated balance sheets as in-process research and development assets, or rights to developed technology associated with in-licenses, regulatory approval milestones due to our collaborators, or other payments. We amortize our finite-lived intangible assets using the straight-line method within “Cost of sales” over the remaining estimated life of the assets beginning in the period in which itregulatory
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
approval is acquired.achieved or the assets are acquired and continuing through the period that we no longer have either exclusive rights to market the products associated with the assets or in-license rights to the intellectual property underlying the assets. We test our finite-lived intangible assets for impairment if indicators are present or changes in circumstances suggest that the carrying value of the assets may not be recoverable. If we determine that the carrying value of a finite-lived intangible asset may not be recoverable, we compare the carrying value of the asset to the undiscounted cash flows that we expect the asset to generate. When we determine that a finite-lived intangible asset has become impaired, we write down the carrying value of the asset to its fair value and record an impairment charge in the period in which the impairment occurs.
Goodwill
The difference between the purchase price and the fair value of assets acquired and liabilities assumed in a business combination is allocated to goodwill. Goodwill is evaluated for impairment on an annual basis as of October 1, and more frequently if indicators are present or changes in circumstances suggest that impairment may exist. As noted in Basis of Presentation above, the Company has 1we have one operating segment, pharmaceuticals, which is itsour only reporting unit.
Deconsolidation
Upon the occurrence of certain events and on a regular basis, the Company evaluates whether it no longer has a controlling interest in its subsidiaries, including consolidated VIEs. If the Company determines it no longer has a controlling interest, the subsidiary is deconsolidated. The Company records a gain or loss on deconsolidation based on the difference on the deconsolidation date between (i) the aggregate of (a) the fair value of any consideration received, (b) the fair value of any retained noncontrolling investment in the former subsidiary and (c) the carrying amount of any noncontrolling interest in the subsidiary being deconsolidated, less (ii) the carrying amount of the former subsidiary’s assets and liabilities.
Discontinued Operations
The Company assesses whether a deconsolidation is required to be presented as discontinued operations in its consolidated financial statements on the deconsolidation date. This assessment is based on whether or not the deconsolidation
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
represents a strategic shift that has or will have a major effect on the Company’s operations or financial results. If the Company determines that a deconsolidation requires presentation as a discontinued operation on the deconsolidation date, or at any point during the one year period following such date, it will present the former subsidiary as a discontinued operation in current and comparative period financial statements.
Embedded Derivatives
Embedded derivatives are required to be bifurcated from the host instruments and recorded at fair value if the derivatives are not clearly and closely related to the host instruments on the date of issuance. The Company did not have any material embedded derivatives that required bifurcation recorded on its consolidated balance sheets as of December 31, 2020 and 2019, respectively.
Hedging Activities
The Company recognizesWe recognize the fair value of hedging instruments that are designated and qualify as hedging instruments pursuant to U.S. GAAP, foreign currency forward contracts, as either assets or liabilities on theour consolidated balance sheets. Changes in the fair value of these instruments are recorded each period in “Accumulated other comprehensive loss”(loss) income” as unrealized gains and losses until the forecasted underlying transaction occurs. Unrealized gains and losses on these foreign currency forward contracts are included in “Prepaid expenses and other current assets” or “Other assets,” and “Other current liabilities” or “Other long-term liabilities,” respectively, on the Company’sour consolidated balance sheets depending on the remaining period until their contractual maturity. Realized gains and losses for the effective portion of such contracts are recognized in “Product revenues, net” in theour consolidated statement of operationsincome in the same period that it recognizeswe recognize the product revenues that were impacted by the hedged foreign exchange rate changes. The Company classifiesWe classify the cash flows from hedging instruments in the same category as the cash flows from the hedged items.
Certain of the Company’sour hedging instruments are subject to master netting arrangements to reduce the risk arising from such transactions with itsour counterparties. The Company presentsWe present unrealized gains and losses on itsour foreign currency forward contracts on a gross basis within itsour consolidated balance sheets.
The CompanyWe also entersenter into foreign currency forward contracts with contractual maturities of less than one month designed to mitigate the effect of changes in foreign exchange rates on monetary assets and liabilities including intercompany balances. These contracts are not designated as hedging instruments pursuant to U.S. GAAP. Realized gains and losses for such contracts are recognized in “Other (expense) income, (expense), net” in the consolidated statement of operations each period.
Restructuring Expenses
The Company records costs and liabilities associated with exit and disposal activities based on estimates of fair value in the period the liabilities are incurred. The Company’s exit and disposal activities have primarily been associated with the Company’s facilities, but also have included the termination of employees in some cases. The Company’s initial estimate of its liabilities for net ongoing costs associated with its facility obligations are recorded at fair value on the cease use date. On a quarterly basis, the Company evaluates and adjusts these liabilities as appropriate for changes in circumstances. Changes to the Company’s estimate of these liabilities are recorded as additional restructuring expenses (credits). These costs are included in “Restructuring expense (income)” on the Company’sour consolidated statements of operations.income each period.
Comprehensive Income
Comprehensive income consists of net income and other comprehensive income (loss), which includes foreign currency translation adjustments and unrealized gains and losses on foreign currency forward contracts and certain marketableour available-for-sale debt securities. For purposes of comprehensive income disclosures, the Company recordswe record provisions for or benefits from income taxes related to the unrealized gains and losses on foreign currency forward contracts and certain marketableour available-for-sale debt securities. The Company does notWe record provisions for or benefits from income taxes related to theour cumulative translation adjustment as the Company intends to permanently reinvestonly, for those undistributed earnings in itsour foreign subsidiaries.
VERTEX PHARMACEUTICALS INCORPORATED
Notessubsidiaries that we do not intend to Consolidated Financial Statements (Continued)
permanently reinvest.Foreign Currency Translation and Transactions
The majority of the Company’sour operations occur in entities that have the U.S. dollar denominated as their functional currency. The assets and liabilities of the Company’sour entities with functional currencies other than the U.S. dollar are translated into U.S. dollars at exchange rates of exchange in effect at the end of the year. Revenue and expense amounts for these entities are translated using the average exchange rates for the period. Net unrealized gains and lossesChanges resulting from foreign currency translation are included in “Accumulated other comprehensive loss.(loss) income.” Net foreign currency exchange transaction losses, which are included in “Other (expense) income, (expense), net” on the Company’sour consolidated statementstatements of operations,income, were $16.1$24.6 million, $5.2$15.1 million and $1.1$13.9 million for 2020, 20192023, 2022 and 2018,2021, respectively. These net foreign currency exchange losses are presented net of the impact of the foreign
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
currency forward contracts designed to mitigate their effect on our consolidated statements of income.
Share Repurchase Programs
Repurchases of our common stock are recorded as reductions to “Common Stock” and “Additional paid-in capital” pursuant to our established accounting policy. Repurchases in excess of the Company’s consolidated statement of operations.par value will be recorded as reductions to “Retained earnings” in the event that “Additional paid-in capital” is reduced to zero.
Net Income Per Share Attributable to Vertex Common Shareholders
Basic and diluted net income per share attributable to Vertex common shareholders are presented in conformity with the two-class method required for participating securities. Shares of unvested restricted stock granted under the Company’s Amended and Restated 2006 Stock and Option Plan had the non-forfeitable right to receive dividends on an equal basis with other outstanding common stock. As a result, these unvested shares of restricted stock were considered participating securities under the two-class method. In 2020 and 2019, the Company did not have a significant amount of restricted stock outstanding under this plan: therefore, the two-class method did not impact its basic and diluted net income per share attributable to Vertex common shareholders for the years ended December 31, 2020 and 2019.
Under the two-class method, earnings are allocated to (i) Vertex common shares, excluding unvested restricted stock, and (ii) participating securities, based on their respective weighted-average shares outstanding for the period. Potentially dilutive shares result from the assumed exercise of outstanding stock options (the proceeds of which are then assumed to have been used to repurchase outstanding stock using the treasury-stock method).Share
Basic net income per share attributable to Vertex common shareholdersshare is based upon the weighted-average number of common shares outstanding during the period, excluding restricted stock that had been issued but had not yet vested, if any.period. Diluted net income per common share attributable to Vertex common shareholdersutilizing the treasury-stock method is based upon the weighted-average number of common shares outstanding during the period plus additional weighted-average common equivalent shares outstanding during the period when the effect is dilutive. Potentially dilutive shares result from the assumed (i) vesting of restricted stock units and performance-based restricted stock units, and (ii) exercise of outstanding stock options. The proceeds of such vestings or exercises are assumed to have been used to repurchase outstanding stock using the treasury-stock method.
Recently Adopted and Issued Accounting Standards
Internal-Use SoftwareWe have not been required to adopt any accounting standards that had a significant impact on our consolidated financial statements in the three years ended December 31, 2023.
Recently Issued Accounting Standards
Segment Reporting
In 2018,2023, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures (“ASU 2018-15, Intangibles—Goodwill and Other—Internal-Use Software (Subtopic 350-40): Customer’s Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract (“ASU 2018-15”2023-07”), which clarifiesrequires public entities to disclose significant segment expenses and other segment items. ASU 2023-07 also requires public entities to provide in interim periods all disclosures about a reportable segment’s profit or loss and assets that are currently required annually. ASU 2023-07 becomes effective for the accounting for implementation costs in cloud computing arrangements. ASU 2018-15 became effectiveannual period starting on January 1, 2020. The2024, and for the interim periods starting on January 1, 2025. We are in the process of analyzing the impact that the adoption of ASU 2018-15 resulted in an insignificant amount of additional assets recorded2023-07 will have on the Company’s consolidated balance sheet.our segment disclosures.
Fair Value MeasurementIncome Tax Disclosures
In 2018,2023, the FASB issued ASU 2018-13,2023-09, Fair Value MeasurementIncome Taxes (Topic 820)740): Disclosure Framework-ChangesImprovements to the Disclosure Requirements for Fair Value MeasurementIncome Tax Disclosures (“ASU 2018-13”2023-09”), which modifiesrequires public entities to disclose in their rate reconciliation table additional categories of information about federal, state and foreign income taxes and to provide more details about the disclosure requirementsreconciling items in some categories if items meet a quantitative threshold. ASU 2023-09 becomes effective for fair value measurements. ASU 2018-13 became effectivethe annual period starting on January 1, 2020. The Company2025. We are in the process of analyzing the impact that the adoption of ASU 2018-13 resulted2023-09 will have on our income tax disclosures.
B.Collaboration, License and Other Arrangements
We have entered into numerous agreements with third parties to collaborate on research, development and commercialization programs, license technologies, or acquire assets. Our “Acquired in-process research and development expenses” included $527.1 million, $115.5 million and $1.1 billion in additional disclosures2023, 2022 and 2021, respectively, related to upfront, contingent milestone, or other payments pursuant to our business development transactions, including collaborations, licenses of third-party technologies, and asset acquisitions that qualify as in-process research and development.
In-license Agreements
We have entered into several in-license agreements to advance and obtain access to technologies and services related to our research and early-development activities. We are generally required to make an upfront payment upon execution of our license agreements; development, regulatory and commercialization milestones payments upon the Company’s Level 3 inputs. Please refer to Note D, “Fair Value Measurements,” for further information.
Credit Losses
In 2016, the FASB issued ASU 2016-13, which requires entities to record expected credit losses forachievement of certain financial instruments, including trade receivables, as an allowance that reflects the entity's current estimate of credit losses expected to be incurred. For available-for-sale debt securities in unrealized loss positions, ASU 2016-13 requires allowances to be
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
recorded instead of reducing the amortized cost of the investment. ASU 2016-13 became effective on January 1, 2020. The adoption of ASU 2016-13 did not have a significant impact on the Company’s consolidated financial statements.
Leases
On January 1, 2019, the Company adopted ASC 842 using the modified-retrospective method. Until December 31, 2018, the Company applied build-to-suit accounting and was the deemed owner of its leased corporate headquarters in Boston and research site in San Diego, for which it was recognizing depreciation expense over the buildings’ useful lives and imputed interest on the corresponding construction financing lease obligations. Under the amended guidance that became effective January 1, 2019, the Company accounts for these buildings as finance leases, resulting in increased depreciation expense over the respective lease terms of approximately 15 years, which are significantly shorter than the buildings’ useful lives of 40 years. The amended guidance also results in a reduction in imputed interest expense in the initial years of each finance lease term. As of January 1, 2019, the Company recorded a cumulative effect adjustment to increase its “Accumulated deficit” by $40.3 million related to the adjustments to its build-to-suit leases. Please refer to “Leases” above for further information.
Revenue Recognition
On January 1, 2018, the Company adopted ASC 606, using the modified retrospective adoption method for all contracts that were not completed as of the date of adoption.Based on the Company’s review of existing customer contracts as of January 1, 2018, the Company concluded that the only significant impact that the adoption of ASC 606 had on its consolidated financial statements related to shipments of ORKAMBI under early access programs in France. Prior to the adoption of ASC 606, the Company did not recognize revenue on the proceeds received from sales of ORKAMBI under early access programs in France because the price was not fixed or determinable based on the status of ongoing pricing discussions. As of January 1, 2018, the Company recorded a cumulative effect adjustment to its accumulated deficit of $8.3 million related to the adoption of ASC 606, which primarily represented the Company’s estimated amount of consideration it expected to retain related to these shipments that would not be subject to a significant reversal in amounts recognized, net of costs previously deferred related to these shipments.
Equity Investments
On January 1, 2018, the Company adopted ASU 2016-01, Recognition and Measurement of Financial Assets and Financial Liabilities (“ASU 2016-01”), using the required modified-retrospective adoption method. Under ASU 2016-01, equity investments (except those accounted for under the equity method of accounting or those that result in consolidation of an investee) are measured at fair value with changes in fair value recognized in net income. As of January 1, 2018, the Company held publicly traded equity investments and equity investments accounted for under the cost method. As a result, in 2018, the Company recorded a $25.1 million cumulative effect adjustment to “Accumulated deficit” related to its publicly traded equity investments equal to the unrealized gain, net of tax, that was recorded in “Accumulated other comprehensive loss” as of December 31, 2017.
Recently Issued Accounting Standards
Income Taxes
In 2019, the FASB issued ASU 2019-12, Income Taxes (Topic 740) (“ASU 2019-12”), which simplifies the accounting for income taxes. ASU 2019-12 was effective on January 1, 2021. The Company does not expect the adoption of ASU 2019-12 to have a significant impact on its consolidated financial statements.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
B.Collaborative Arrangements
The Company has entered into numerous agreements pursuant to which it collaborates with third parties on research, development and commercialization programs, including in-license and out-license agreements.
In-license Agreements
The Company has entered into a number of license agreements in order to advance and obtain access to technologies and services related to its research and early-development activities. The Company is generally required to make an upfront payment upon execution of the license agreement; development, regulatory and commercialization milestones payments upon the achievement of certain product research, development and commercialization objectives; and royalty payments on future sales, if any, of commercial products resulting from the collaboration.our collaborations.
Pursuant to the terms of itsour in-license agreements, the Company’sour collaborators typically lead the discovery efforts and the Company leadswe lead all preclinical, development and commercialization activities associated with the advancement of any drugproduct candidates and fundsfund all expenses.
The CompanyWe typically can terminate itsour in-license agreements by providing advance notice to its collaborators; the required length of notice is dependent on whether any product developed under the license agreement has received marketing approval. The Company’sour collaborators. Our license agreements may be terminated by either party for a material breach by the other, subject to notice and cure provisions. Unless earlier terminated, these license agreements generally remain in effect until the date on which the royalty term and all payment obligations with respect to all products in all countries have expired.
CRISPR Therapeutics AG
CRISPR-Cas9 Gene-editing Therapies Agreements
In 2015, the Companywe entered into a strategic collaboration, option and license agreement (the “CRISPR Agreement”) with CRISPR Therapeutics AG and its affiliates (“CRISPR”) to collaborate on the discovery and development of potential new treatments aimed at the underlying genetic causes of human diseases using CRISPR-Cas9 gene-editing technology. The CompanyWe had the exclusive right to license certain targets. In the fourth quarter of 2019, the Company paid an aggregate of $30.0 millionwe elected to exclusively license 3 CRISPR-Cas9-basedthree targets, including CF, pursuant to the CRISPR Agreement. The Company recorded the $30.0 million total option payment to “Research and development expenses” in the fourth quarter of 2019. For each of the 3three targets that the Companywe elected to license, CRISPR has the potential to receive up to an additional $410.0 million in development, regulatory and commercial milestones as well as royalties on resulting net product sales.
In 2017, the Companywe entered into a co-developmentjoint development and co-commercializationcommercialization agreement with CRISPR pursuant to the terms of the CRISPR Agreement (the “Original JDCA”), under which the Companywe and CRISPR arewere co-developing and willpreparing to co-commercialize CTX001 (the “CTX001 Co-Co Agreement”)CASGEVY for the treatment of hemoglobinopathies, including treatments for sickle cell diseaseSCD and beta thalassemia. As part ofTDT.
In 2021, we and CRISPR amended and restated the Original JDCA (the “A&R JDCA”), pursuant to which the parties agreed to, among other things, (a) adjust the governance structure for the collaboration and adjust the Companyresponsibilities of each party thereunder; (b) adjust the allocation of net profits and net losses between the parties; and (c) exclusively license (subject to CRISPR’s reserved rights to conduct certain activities) certain intellectual property rights to us relating to the products that may be researched, developed, manufactured and commercialized under such agreement. Pursuant to the A&R JDCA, we lead global development, manufacturing and commercialization of CASGEVY, with support from CRISPR, share equallyas described further below. We also conduct all research, development, costsmanufacturing and potential worldwide revenues relatedcommercialization activities relating to potential hemoglobinopathy treatments. The Companyother product candidates and products under the A&R JDCA throughout the world subject to CRISPR’s reserved right to conduct certain activities.
In connection with the A&R JDCA, we made a $900.0 million upfront payment to CRISPR in the second quarter of 2021. We concluded that the CTX001 Co-Co Agreement is a cost-sharing arrangement, which results in the net impact of the arrangement being recorded in “Research and development expenses” in its consolidated statements of operations. During the years ended December 31, 2020, 2019 and 2018, the net expense related to the CTX001 Co-Co Agreement was $50.6 million, $30.1 million and $19.7 million, respectively.
In July 2019, the Company entered into a separate strategic collaboration and license agreement (the “CRISPR DMD/DM1 Agreement”) with CRISPR. Pursuant to this agreement, the Company received an exclusive worldwide license to CRISPR’s existing and future intellectual property for Duchenne muscular dystrophy (“DMD”) and myotonic dystrophy type 1 (“DM1”) and the Company made an upfront payment of $175.0 million to CRISPR. The Company concluded that itwe did not have any alternative future use for the acquired in-process research and development and recorded thethis upfront payment to “Acquired in-process research and development expenses.” Prior to receiving marketing approval for CASGEVY, we accounted for the A&R JDCA as a cost-sharing arrangement, with costs incurred related to CASGEVY allocated 60% to us and 40% to CRISPR, subject to certain adjustments, and we recognized the impact of the arrangement as either “Research and development expenses” or “Selling, general and administrative expenses.” Prior to July 1, 2021, we and CRISPR shared equally all expenses incurred under the Original JDCA, which we also accounted for as a cost-sharing arrangement.
CASGEVY was approved by the U.S. Food and Drug Administration in December 2023 for the third quartertreatment of 2019.SCD. In 2020,connection with this approval, we made a $200.0 million milestone payment to CRISPR in January 2024, which we accrued to “Other current liabilities” and recorded within “Other intangible assets, net” on our consolidated balance sheet as of December 31, 2023. Please refer to Note J, “Goodwill and Other Intangible Assets,” for further information. Subsequent to receiving marketing approval for CASGEVY, we continue to lead the Company recorded $25.0 millionresearch and development activities under the A&R JDCA, subject to CRISPR’s reserved right to conduct certain activities. We are reimbursed by CRISPR for its 40% share of these research and development activities, subject to certain adjustments, and we continue to record this reimbursement from CRISPR within “Research and development expenses” related to a pre-clinical milestone earned byexpenses.” We also share with CRISPR under40% of the CRISPR DMD/DM1 Agreement. CRISPR has the potential to receive up to an additional $800.0 million in research, development, regulatory andnet commercial milestones for the DMD and DM1 programs as well as royalties on net product sales. CRISPR has the option to co-develop and co-commercialize all DM1 products globally and forego the milestones and royalties associated with theprofits or
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
DM1 program. The Company funds all expenses associatedlosses incurred with respect to CASGEVY, subject to certain adjustments. In 2023, the collaboration except for research costs for specified guide RNA research conducted by CRISPR, which the Company and CRISPR share equally.
Kymera Therapeutics Inc.net commercial loss incurred with respect to CASGEVY was not material to our consolidated statement of income.
In May 2019,2023, 2022 and 2021, we recognized the Companynet impact of the A&R JDCA and Original JDCA as “Research and development expenses” of $227.0 million, $194.2 million and $108.2 million, respectively, and “Selling, general and administrative expenses” of $94.9 million, $61.4 million and $20.8 million, respectively, within our consolidated statements of income.
CRISPR-Cas9 Gene-editing Hypoimmune Cell Therapies Agreement
In March 2023, we entered into a strategicnon-exclusive license agreement (the “CRISPR T1D Agreement”) for the use of CRISPR’s CRISPR-Cas9 gene-editing technology to accelerate the development of our hypoimmune cell therapies for type 1 diabetes (“T1D”). Pursuant to the CRISPR T1D Agreement, we made a $100.0 million upfront payment to CRISPR, and we determined that substantially all the fair value of our upfront payment was attributable to in-process research and development, collaboration agreement with Kymera Therapeutics Inc. (“Kymera”)for which there is no alternative future use, and that no substantive processes were acquired that would constitute a business. In the second quarter of 2023, we achieved a research milestone that resulted in a $70.0 million payment to advance small molecule protein degraders against multiple targets. Kymera’s proprietary platform technology is being applied inCRISPR. We recorded the collaboration activities in exchange for an upfront payment of $50.0 million. The Company hasand the exclusive rightresearch milestone, totaling $170.0 million, to license up“Acquired in-process research and development expenses” in 2023. CRISPR is eligible to 6 protein targets, for each of which Kymera may receive up to $170.0an additional $160.0 million in payments, includingresearch, development, regulatory and commercial milestones for any products that may result from the agreement, as well as royalties on resulting net product sales.
Entrada Therapeutics, Inc.
In addition to theFebruary 2023, we closed a strategic collaboration and license agreement (the “Entrada Agreement”) with Entrada Therapeutics, Inc. (“Entrada”) focused on discovering and developing intracellular therapeutics for myotonic dystrophy type 1 (“DM1”). Upon closing, we made an upfront payment the Companyof $225.1 million to Entrada, and purchased $20.0$24.9 million of Kymera’s preferred stock. The CompanyEntrada’s common stock in connection with the Entrada Agreement. We determined that substantially all the fair value of itsour upfront payment was attributable to in-process research and development, for which there is no alternative future use, and that no substantive processes were acquired that would constitute a business. In 2023, Entrada also achieved a research milestone, resulting in a $17.5 million payment to Entrada. We recorded the upfront and milestone payments totaling $242.6 million to “Acquired in-process research and development expenses” in 2023. We recorded the investment in Kymera’s preferredEntrada’s common stock which did not have a readily determinableat fair value approximated $20.0 million. The preferred stock convertedon our consolidated balance sheet within “Marketable securities.” Entrada is eligible to receive up to an additional $410.0 million in research, development, regulatory and commercial milestones for any products that may result from the Entrada Agreement, as well as royalties on resulting net product sales.
Verve Therapeutics, Inc.
In 2022, we entered into a strategic collaboration and license agreement with Verve Therapeutics, Inc. (“Verve”) focused on discovering and developing an in vivo gene-editing program for a liver disease. Under the terms of the agreement, we made a $25.0 million upfront payment to Verve and also purchased $35.0 million of Verve’s common stock when Kymera became a publicly traded company in 2020.
The Companyconnection with the agreement. Verve is also eligible to receive up to $66.0 million in success payments, up to an additional $340.0 million in development, regulatory and commercial milestones for any products that may result from the collaboration agreement, and royalties on net product sales. We determined that substantially all of the fair value of the Kymera collaboration agreementupfront payment was attributable to in-process research and development and no substantive processes were acquired that would constitute a business. The CompanyWe concluded that itthere is no alternative future use for the acquired in-process research and development and recorded the upfront payment to “Acquired in-process research and development expenses.” The investment in Verve’s common stock is recorded at fair value on our consolidated balance sheet within “Marketable securities.”
ApoLo1 Bio, LLC
In 2016, we entered into a strategic collaboration and license agreement with ApoLo1 Bio, LLC (“ApoLo1”) related to our drug discovery efforts in APOL1-mediated kidney disease. In 2021, based on positive results from a Phase 2 proof-of-concept study of VX-147 in patients with APOL1-mediated focal segmental glomerulosclerosis, we paid ApoLo1 a $15.0 million milestone and exercised our $60.0 million option to buy-out all future development milestones, regulatory milestones, and future royalties on net product sales. We recorded these payments to “Acquired in-process research and
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
development expenses” because we concluded that we did not have any alternative future use for the acquired in-process research and development and recorded the $50.0 million upfront payment to “Research and development expenses” in 2019.development.
Moderna, Inc.
In 2016, the Companywe entered into a strategic collaboration and licensing agreement with Moderna, Inc. (“Moderna”), pursuant to which the parties are seeking to identify and develop messenger ribonucleic acid (“mRNA”) therapeutics encoding cystic fibrosis transmembrane conductance regulator for the treatment of CF. Moderna is eligible to receive up to $270.0 million in development and regulatory milestones as well as royalties on net product sales related to this agreement.
In September 2020, the Companywe entered into a newsecond strategic collaboration and licensing agreement with Moderna (the “2020 Moderna Agreement”) aimed at the discovery and development of lipid nanoparticles and mRNAs that can deliver gene-editing therapies to lung cells for the treatment of CF. Pursuant to the 2020 Moderna Agreement, Moderna received an upfront payment of $75.0 million and is eligible to receive up to $380.0 million in development, regulatory and commercial milestones as well as royalties on net product sales. The Companysales related to this agreement.
Asset Acquisitions
Septerna, Inc. - Novel G Protein-coupled Receptor Program
In September 2023, pursuant to an asset purchase agreement, we acquired a novel G protein-coupled receptor (“GPCR”) program from Septerna, Inc. We determined that substantially all the fair value acquired is concentrated in the GPCR in-process research and development asset, which does not constitute a business, and for which we determined there is no alternative future use. As a result, we recorded $47.5 million to “Acquired in-process research and development expenses” in 2023.
Catalyst Biosciences, Inc. - Complement 3 Degrader Program
In 2022, pursuant to an asset purchase agreement, we acquired from Catalyst Biosciences, Inc.’s a portfolio of protease medicines that target the complement system and related intellectual property, including CB 2782-PEG, which is a pre-clinical complement component 3 degrader program for geographic atrophy in dry age-related macular degeneration. We determined that substantially all the fair value acquired is concentrated in the CB-2782 PEG in-process research and development assets, which do not constitute a business, and for which we determined there is no alternative future use. As a result, we recorded our $60.0 million upfront payment to “Acquired in-process research and development expenses.”
Additional In-License Agreements and Other Arrangements
In addition to the agreements described above, we recorded upfront, option and milestone payments totaling $67.0 million in 2023, $30.5 million in 2022 and $138.3 million in 2021 to “Acquired in-process research and development expenses” related to additional in-license agreements and other business development transactions that we do not consider to be individually significant to our consolidated financial statements. The most significant payments included within these amounts were upfront payments of $31.0 million to Mammoth Biosciences, Inc. (“Mammoth”) and $25.0 million to Arbor Biotechnologies, Inc. (“Arbor”) in 2021.
For Mammoth, Arbor, and several other in-license agreements that we entered into in 2021, 2022 and 2023 that are not individually significant to our consolidated financial statements, we determined that substantially all the fair value of the 2020 Moderna Agreementconsideration for each individual agreement was attributable to in-process research and development, for which we did not have any alternative future use, and no substantive processes were acquired that would constitute a business. The Company concluded that it did not have any alternative future use for the acquired in-process research and development andWe recorded the upfront paymentpayments for these agreements to “Research and development expenses” in the third quarter of 2020.
Other In-License Agreements
In addition to the collaborative arrangements described above, the Company has entered into additional in-license agreements that it does not consider to be individually significant to its consolidated financial statements. In addition to the payments described above, the Company recorded upfront, option and milestone payments totaling $84.6 million in 2020, $63.3 million in 2019 and $46.9 million in 2018 to “Research and development expenses” which included a $40.0 million upfront payment to Skyhawk Therapeutics, Inc. (“Skyhawk”) in 2020.
For Skyhawk and several other in-license agreements that are not individually significant to the Company’s consolidated financial statements, the Company determined that substantially all of the fair value of each individual agreement was attributable to“Acquired in-process research and development and no substantive processes were acquired that would constitute a business. The Company concluded that it did not have any alternative future use for the acquired in-process research and development associated with the agreements and recorded the related portion of the upfront payments to “Research and development expenses.” Please refer to Note D, “Fair Value Measurements,” and Note E, “Marketable Securities and Equity Investments,” for further information regarding the Company’sour investments in itsour collaborators.
Variable Interest Entities (VIEs)Editas Medicine, Inc.
The Company licensed rightsIn December 2023, we entered into a sublicense agreement (the “Editas Agreement”) with Editas Medicine, Inc. (“Editas”) to certain drug candidates from these third-party collaborators, which has resulted inobtain a non-exclusive license for Editas CRISPR/Cas9 gene-editing technology related to SCD and TDT, including CASGEVY. Pursuant to the consolidation of the third-parties’ financial statements into the Company’s consolidated financial statements as VIEs forEditas Agreement, we made a $50.0 million upfront payment to Editas and Editas is
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
eligible to receive an additional $50.0 million license payment upon the resolution of certain periodscontingencies related to Editas’ license to the technology. Editas is also eligible to receive commercial milestones based on certain annual CASGEVY sales thresholds. Pursuant to the A&R JDCA described above, CRISPR will reimburse us for 40%, or $20.0 million, of time. Most recently, the Company deconsolidatedupfront payment, and would also reimburse us for 40% of the financial statements of BioAxone from itsadditional contingent $50.0 million license payment, subject to certain adjustments. We recorded the net $30.0 million upfront payment related to our sublicense for Editas’ developed technology to “Other intangible assets, net” on our consolidated financial statementsbalance sheet as of December 31, 2018. The Company has not consolidated a VIE into its financial statements since December 31, 2018.
BioAxone Biosciences, Inc.
In 2014, the Company entered into a license2023. Please refer to Note J, “Goodwill and collaboration agreement (the “BioAxone Agreement”) with BioAxone, which resulted in the consolidation of BioAxone as a VIE.
In October 2018, the Company announced it would stop clinical development of VX-210 and terminate the Phase 2b clinical trial of VX-210 based on the recommendation of the clinical trial’s Data Safety Monitoring Board and the Company’s review of interim data. In December 2018, the Company notified BioAxone of its intent to terminate the BioAxone Agreement and executed a release that immediately allowed BioAxone to control development of its neurological programs other than VX-210 without the Company’s consent. As a result of this decision, the Company recorded a $29.0 million impairment charge related to VX-210 that was attributable to noncontrolling interest.
As a result, the Company deconsolidated BioAxone as of December 31, 2018 because it determined that it no longer was the primary beneficiary of BioAxone as it no longer had the power to direct the significant activities of BioAxone. The net impact of the deconsolidation was not material to the Company’s consolidated statement of operations. The Company’s net loss attributable to noncontrolling interestOther Intangible Assets,” for the year ended December 31, 2018 was $9.8 million.further information.
Out-license Agreements
The Company hasWe have entered into licensing agreements pursuant to which it haswe have out-licensed rights to certain drugproduct candidates to third-party collaborators. Pursuant to these out-license agreements, the Company’sour collaborators become responsible for all costs related to the continued development of such drugproduct candidates and obtain development and commercialization rights to these drugproduct candidates. Depending on the terms of the agreements, the Company’sour collaborators may be required to make upfront payments, milestone payments upon the achievement of certain product research and development objectives and may also be required to pay royalties on future sales, if any, of commercial products resulting from the collaboration. The termination provisions associated with these collaborations are generally the same as those described above related to the Company’sour in-license agreements.
Merck KGaA, Darmstadt, Germany
In January 2017, the Company entered into a strategic collaboration and license agreement (the “Oncology Agreement”) with Merck KGaA, Darmstadt, Germany (the “Licensee”). Pursuant to the Oncology Agreement, the Company granted the Licensee an exclusive worldwide license to research, develop and commercialize 4 oncology research and development programs including 2 clinical-stage programs targeting DNA damage repair: its ataxia telangiectasia and Rad3-related protein kinase inhibitor program, or ATR program, including VX-970 and VX-803, and its DNA-dependent protein kinase inhibitor program, or DNA-PK program, including VX-984. In addition, the Company granted the Licensee exclusive, worldwide rights to 2 pre-clinical programs.
The Oncology Agreement provided for an upfront payment from the Licensee to the Company of $230.0 million, which was recorded as “Collaborative and royalty revenues,” under the multiple element arrangement accounting guidance that was applicable in 2017. All of the Company’s activities related to the Oncology Agreement were substantially complete as of December 31, 2017.
In December 2018, the Company entered into an agreement with Merck KGaA, Darmstadt, Germany (the “DNA-PK Agreement”) whereby the Company licensed the two lead Vertex DNA-PK compounds from its DNA-PK program for use in the field of gene integration for six specific indications. In exchange for this exclusive worldwide license to research, develop and commercialize the DNA-PK program for the specified indications within the field of gene integration, the Company made an upfront payment of $65.0 million. Merck KGaA, Darmstadt, Germany has the potential to receive additional milestones, primarily related to approval and reimbursement in various markets, as well as royalties on net product sales.
The Company evaluated the DNA-PK Agreement and concluded it represents a modification of the Oncology Agreement pursuant to ASC 606. As of December 2018, when the Company entered into the DNA-PK Agreement, the Company had
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
completed its obligations under the Oncology Agreement, but the Oncology Agreement was an open contract pursuant to ASC 606 since the Company could receive future royalty payments from the commercialization of the licensed programs under the Oncology Agreement.
In applying ASC 606, the Company determined that the license granted under the DNA-PK Agreement is distinct from the license granted by the Company under the Oncology Agreement since the license to the two lead Vertex DNA-PK compounds is capable of being distinct as the Company is able to benefit from the license via its ability to internally develop and commercialize the two lead Vertex DNA-PK compounds in the six named indications in the field of gene-editing, and the license is not dependent on Merck KGaA, Darmstadt, Germany providing any specialized services to the Company. In addition, the license to the two lead Vertex DNA-PK compounds granted to the Company under the DNA-PK Agreement is distinct from the license granted by the Company under the Oncology Agreement as the rights conveyed in the licenses differ and both parties have the ability to commercially benefit from the licenses on their own. Furthermore, the consideration attributable to the license of the two lead Vertex DNA-PK compounds represents fair value. Therefore, the Company determined it should account for the DNA-PK Agreement as a separate agreement.
The Company determined that substantially all of the fair value of the DNA-PK Agreement was attributable to a single in-process research and development asset that did not constitute a business. The Company concluded that it did not have any alternative future use for the acquired in-process research and development and recorded the $65.0 million payment to “Research and development expenses” in 2018 accordingly.
Janssen Pharmaceuticals, Inc.
In 2014, the Company entered into an agreement with Janssen Pharmaceuticals, Inc. (“Janssen”). In the third quarter of 2020, Janssen exercised its right to terminate its exclusive worldwide license to develop and commercialize certain drug candidates for the treatment of influenza, based on Phase 3 clinical trial results for pimodivir.
Cystic Fibrosis Foundation
The Company has a research, development and commercialization agreement that was originallyIn 2004, we entered into in 2004a collaboration agreement with the Cystic Fibrosis Foundation, as successor in interest to the Cystic Fibrosis Foundation Therapeutics, Inc. This agreement was most recently amended in 2016., to support research and development activities. Pursuant to the collaboration agreement, as amended, the Companywe have agreed to pay royalties ranging from low-single digits to mid-single digits on potential sales of certain compounds first synthesized and/or tested between March 1, 2014 and August 31, 2016, including elexacaftor, and tiered royalties ranging from single digits to sub-teens on covered compounds first synthesized and/or tested during a research term on or before February 28, 2014, including KALYDECO (ivacaftor), ORKAMBI (lumacaftor in combination with ivacaftor)ivacaftor, lumacaftor and SYMDEKO/SYMKEVI (tezacaftor in combination with ivacaftor).tezacaftor and royalties ranging from low-single digits to mid-single digits on potential net sales of certain compounds first synthesized and/or tested between March 1, 2014 and August 31, 2016, including elexacaftor. We do not have any royalty obligations on compounds first synthesized and tested on or after September 1, 2016. For combination products, such as ORKAMBI, SYMDEKO/SYMKEVI and TRIKAFTA/KAFTRIO, (elexacaftor/tezacaftor/ivacaftor and ivacaftor), sales are allocated equally to each of the active pharmaceutical ingredients in the combination product. We record expenses related to these royalty obligations to “Cost of sales.”
C.Earnings Per Share
Basic net income per share attributable to Vertex common shareholders is based upon the weighted-average number of common shares outstanding during the period. Diluted net income per share attributable to Vertex common shareholders utilizing the treasury-stock method is based upon the weighted-average number of common shares outstanding during the period plus additional weighted-average common equivalent shares outstanding during the period when the effect is dilutive.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Earnings Per ShareThe following table sets forth the computation of basic and diluted net income per common share for the periods ended:
| | | | | | | | | | | | | | | | | |
| 2020 | | 2019 | | 2018 |
| (in thousands, except per share amounts) |
| Basic net income attributable to Vertex per common share calculation: | | | | | |
| Net income attributable to Vertex common shareholders | $ | 2,711,647 | | | $ | 1,176,810 | | | $ | 2,096,896 | |
| Less: Undistributed earnings allocated to participating securities | 0 | | | 0 | | | (501) | |
| Net income attributable to Vertex common shareholders—basic | $ | 2,711,647 | | | $ | 1,176,810 | | | $ | 2,096,395 | |
| | | | | |
| Basic weighted-average common shares outstanding | 259,841 | | | 256,728 | | | 254,292 | |
| Basic net income attributable to Vertex per common share | $ | 10.44 | | | $ | 4.58 | | | $ | 8.24 | |
| | | | | |
| Diluted net income attributable to Vertex per common share calculation: | | | | | |
| Net income attributable to Vertex common shareholders | $ | 2,711,647 | | | $ | 1,176,810 | | | $ | 2,096,896 | |
| Less: Undistributed earnings allocated to participating securities | 0 | | | 0 | | | (492) | |
| Net income attributable to Vertex common shareholders—diluted | $ | 2,711,647 | | | $ | 1,176,810 | | | $ | 2,096,404 | |
| | | | | |
| Weighted-average shares used to compute basic net income per common share | 259,841 | | | 256,728 | | | 254,292 | |
| Effect of potentially dilutive securities: | | | | | |
| Stock options | 1,801 | | | 2,231 | | | 2,913 | |
| Restricted stock units (including PSUs) and restricted stock | 1,741 | | | 1,700 | | | 1,963 | |
| Employee stock purchase program | 13 | | | 14 | | | 17 | |
| Weighted-average shares used to compute diluted net income per common share | 263,396 | | | 260,673 | | | 259,185 | |
| Diluted net income attributable to Vertex per common share | $ | 10.29 | | | $ | 4.51 | | | $ | 8.09 | |
| | | | | | | | | | | | | | | | | |
| 2023 | | 2022 | | 2021 |
| (in millions, except per share amounts) |
| Net income | $ | 3,619.6 | | | $ | 3,322.0 | | | $ | 2,342.1 | |
| | | | | |
| Basic weighted-average common shares outstanding | 257.7 | | | 256.1 | | | 257.7 | |
| Effect of potentially dilutive securities: | | | | | |
| Restricted stock units (including PSUs) | 1.6 | | | 1.6 | | | 1.1 | |
| Stock options | 1.2 | | | 1.4 | | | 1.1 | |
| Employee stock purchase program | 0.0 | | | 0.0 | | | 0.0 | |
| Diluted weighted-average common shares outstanding | 260.5 | | | 259.1 | | | 259.9 | |
| | | | | |
| Basic net income per common share | $ | 14.05 | | | $ | 12.97 | | | $ | 9.09 | |
| Diluted net income per common share | $ | 13.89 | | | $ | 12.82 | | | $ | 9.01 | |
The CompanyWe did not include the securities in the following table in the computation of the diluted net income per share attributable to Vertex common shareholders calculationsshare because the effect would have been anti-dilutive during each period.period:
| | 2020 | | 2019 | | 2018 |
| (in thousands) |
| 2023 | | | 2023 | | 2022 | | 2021 |
| (in millions) | | | (in millions) |
| Unvested restricted stock units (including PSUs) | |
| Stock options | Stock options | 312 | | | 2,833 | | | 2,217 | |
| Unvested restricted stock units (including PSUs) and restricted stock | 257 | | | 6 | | | 5 | |
D.Fair Value Measurements
The fair value of the Company’s financial assets and liabilities reflects the Company’s estimate of amounts that it would have received in connection with the sale of the assets or paid in connection with the transfer of the liabilities in an orderly transaction between market participants at the measurement date. In connection with measuring the fair value of its assets and liabilities, the Company seeks to maximize the use of observable inputs (market data obtained from sources independent from the Company) and to minimize the use of unobservable inputs (the Company’s assumptions about how market participants would price assets and liabilities). The following fair value hierarchy is used to classify assets and liabilities based on the observable inputs and unobservable inputs used in order to determine the fair value of the Company’sour financial assets and liabilities:
| | | | | |
| Level 1: | Quoted prices in active markets for identical assets or liabilities. An active market for an asset or liability is a market in which transactions for the asset or liability occur with sufficient frequency and volume to provide pricing information on an ongoing basis. |
| Level 2: | Observable inputs other than Level 1 inputs. Examples of Level 2 inputs include quoted prices in active markets for similar assets or liabilities and quoted prices for identical assets or liabilities in markets that are not active. |
| Level 3: | Unobservable inputs based on the Company’sour assessment of the assumptions that market participants would use in pricing the asset or liability. |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
The Company’sOur investment strategy is focused on capital preservation. The Company investsWe invest in instruments that meet the credit quality standards outlined in the Company’sour investment policy. This policy, which also limits the amount of credit exposure to any one issue or type of instrument. The Company maintainsWe maintain strategic equity investments separately from the investment policy that governs itsour other cash, cash equivalents and marketable securities as described in Note E, “Marketable Securities and Equity Investments.” Additionally, the Company utilizeswe utilize foreign currency forward contracts intended to mitigate the effect of changes in foreign exchange rates on itsour consolidated statementstatements of operations.income.
During the three years ended December 31, 2020, the Company did not record any other-than-temporary impairment charges related
VERTEX PHARMACEUTICALS INCORPORATED
Notes to its financial assets.Consolidated Financial Statements (Continued)
The following tables set forth the Company’sour financial assets and liabilities subject to fair value measurements by level within the fair value hierarchy (and does not include $2.8$3.3 billion and $2.3$3.1 billion of cash as of December 31, 20202023 and 2019,2022, respectively):
| | As of December 31, 2020 | | As of December 31, 2019 |
| | Fair Value Hierarchy | | | Fair Value Hierarchy |
| Total | | Level 1 | | Level 2 | | Level 3 | | Total | | Level 1 | | Level 2 | | Level 3 |
| (in thousands) |
| Financial instruments carried at fair value (asset position): | | | |
| As of December 31, 2023 | | | As of December 31, 2023 | | As of December 31, 2022 |
| | | Fair Value Hierarchy | | | | | Fair Value Hierarchy |
| Total | | | Total | | Level 1 | | Level 2 | | Level 3 | | Total | | Level 1 | | Level 2 | | Level 3 |
| (in millions) | | | (in millions) |
| Financial instruments carried at fair value (asset positions): | |
| Cash equivalents: | |
| Cash equivalents: | |
| Cash equivalents: | Cash equivalents: | | | |
| Money market funds | Money market funds | $ | 3,141,053 | | | $ | 3,141,053 | | | $ | 0 | | | $ | 0 | | | $ | 791,039 | | | $ | 791,039 | | | $ | 0 | | | $ | 0 | |
| Money market funds | |
| Money market funds | |
| Time deposits | |
| U.S. Treasury securities | |
| Government-sponsored enterprise securities | |
| Corporate debt securities | Corporate debt securities | 0 | | | 0 | | | 0 | | | 0 | | | 6,070 | | | 0 | | | 6,070 | | | 0 | |
| Commercial paper | Commercial paper | 0 | | | 0 | | | 0 | | | 0 | | | 29,472 | | | 0 | | | 29,472 | | | 0 | |
| Marketable securities: | Marketable securities: | | | |
| Corporate equity securities | Corporate equity securities | 195,781 | | | 15,650 | | | 180,131 | | | 0 | | | 282,084 | | | 261,797 | | | 20,287 | | | 0 | |
| Corporate equity securities | |
| Corporate equity securities | |
| U.S. Treasury securities | |
| Government-sponsored enterprise securities | Government-sponsored enterprise securities | 80,063 | | | 80,063 | | | 0 | | | 0 | | | 12,733 | | | 12,733 | | | 0 | | | 0 | |
| Asset-backed securities | |
| Certificates of deposit | |
| Corporate debt securities | Corporate debt securities | 231,598 | | | 0 | | | 231,598 | | | 0 | | | 301,799 | | | 0 | | | 301,799 | | | 0 | |
| Commercial paper | Commercial paper | 163,268 | | | 0 | | | 163,268 | | | 0 | | | 102,356 | | | 0 | | | 102,356 | | | 0 | |
| Prepaid expenses and other current assets: | Prepaid expenses and other current assets: | | | |
| Foreign currency forward contracts | Foreign currency forward contracts | 0 | | | 0 | | | 0 | | | 0 | | | 9,725 | | | 0 | | | 9,725 | | | 0 | |
| Foreign currency forward contracts | |
| Foreign currency forward contracts | |
| Other assets: | |
| Foreign currency forward contracts | |
| Foreign currency forward contracts | |
| Foreign currency forward contracts | |
| Total financial assets | Total financial assets | $ | 3,811,763 | | | $ | 3,236,766 | | | $ | 574,997 | | | $ | 0 | | | $ | 1,535,278 | | | $ | 1,065,569 | | | $ | 469,709 | | | $ | 0 | |
| | Financial instruments carried at fair value (liability position): | | | |
| Financial instruments carried at fair value (liability positions): | |
| Financial instruments carried at fair value (liability positions): | |
| Financial instruments carried at fair value (liability positions): | |
| Other current liabilities: | |
| Other current liabilities: | |
| Other current liabilities: | Other current liabilities: | | | |
| Foreign currency forward contracts | Foreign currency forward contracts | $ | (59,184) | | | $ | 0 | | | $ | (59,184) | | | $ | 0 | | | $ | (5,533) | | | $ | 0 | | | $ | (5,533) | | | $ | 0 | |
| Long-term contingent consideration | (189,600) | | | 0 | | | 0 | | | (189,600) | | | (176,500) | | | 0 | | | 0 | | | (176,500) | |
| Foreign currency forward contracts | |
| Foreign currency forward contracts | |
| Contingent consideration | |
| Other long-term liabilities: | Other long-term liabilities: | | | |
| Foreign currency forward contracts | Foreign currency forward contracts | (4,283) | | | 0 | | | (4,283) | | | 0 | | | (1,821) | | | 0 | | | (1,821) | | | 0 | |
| Foreign currency forward contracts | |
| Foreign currency forward contracts | |
| Contingent consideration | |
| Total financial liabilities | Total financial liabilities | $ | (253,067) | | | $ | 0 | | | $ | (63,467) | | | $ | (189,600) | | | $ | (183,854) | | | $ | 0 | | | $ | (7,354) | | | $ | (176,500) | |
Please refer to Note E, “Marketable Securities and Equity Investments,” for the carrying amount and related unrealized gains (losses) by type of investment.
Fair Value of Corporate Equity Securities
The Company maintains strategic investments in corporate equity securities separately from the investment policy that governs its other cash, cash equivalents and marketable securities. The Company classifies itsWe classify our investments in publicly traded companiescorporate equity securities as “Marketable securities” on itsour consolidated balance sheets. Generally, the Company’sour investments in the common stock of these publicly traded companies are valued based on Level 1 inputs because they have readily determinable fair values. However, certain of the Company’sour investments in publicly traded companies have been or continue to be valued based on Level 2 inputs due to transfer restrictions associated with these investments. During the year ended December 31, 2020, the Company transferred the fair value of one of its strategic investments in a publicly traded company from Level 2 to
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Level 1 upon the expiration of transfer restrictions associated with this investment. Please refer to Note E, “Marketable Securities and Equity Investments,” for further information on these investments.
As of December 31, 2023, one of our investments in publicly traded corporate equity securities was subject to a contractual sales restriction expiring partially in 2024 and partially in 2025 with a total fair value of $24.4 million. We
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
purchased this investment directly from the publicly traded company in the first quarter of 2023, and do not anticipate any circumstances that would cause this restriction to lapse prior to the periods listed above.
Fair Value of Contingent Consideration
In 2019, the Companywe acquired Exonics Therapeutics, Inc. (“Exonics”), a privately-heldprivately held company focused on creating transformative gene-editing therapies to repair mutations that cause DMDDuchenne muscular dystrophy (“DMD”) and other severe neuromuscular diseases, including DM1. The Company’sOur Level 3 contingent consideration liabilities are related to $678.3 million of development and regulatory milestones potentially payable to Exonics’ former Exonics equity holders. The Company bases itsWe base our estimates of the probability of achieving the milestones relevant to the fair value of contingent payments on industry data attributable to rare diseases.gene therapies and our knowledge of the progress and viability of the programs.
The following table represents a rollforward of the fair value of our contingent consideration liabilities:
| | | | | |
| Year Ended December 31, 2023 |
| (in millions) |
| Balance at December 31, 2022 | $ | 129.0 | |
| Decrease in fair value of contingent payments | (51.6) | |
| Balance at December 31, 2023 | $ | 77.4 | |
In November 2023, we determined that additional pre-clinical studies of the delivery system for our gene-editing components for DMD will be required, which resulted in a decrease in the fair value of our contingent consideration liabilities in the fourth quarter of 2023. The primary drivers of this decrease were reassessments of our estimates regarding the timing of the development and regulatory milestones and the probability of achieving the milestones. The discount rates used in the valuation model for contingent payments, which were between 0.4%4.7% and 1.9%4.9% as of December 31, 2020,2023, represent a measure of credit risk and market risk associated with settling the liabilities.
Significant judgment is used in determining the appropriateness of these assumptions at each reporting period. Due to the uncertainties associated with development and commercialization of drugproduct candidates in the pharmaceutical industry and the effects of changes in other assumptions including discount rates, the Company expects itswe expect our estimates regarding the fair value of contingent consideration to continue to change in the future, resulting in adjustments to the fair value of the Company’sour contingent consideration liabilities, and the effect of any such adjustments could be material.
The following table represents a rollforward of the fair value of the Company’s contingent consideration liabilities:
| | | | | |
| Year Ended December 31, 2020 |
| (in thousands) |
Balance at December 31, 2019 | $ | 176,500 | |
Increase in fair value of contingent payments | 13,100 | |
Balance at December 31, 2020 | $ | 189,600 | |
The “Increase in fair value of contingent payments” in the table above was primarily due to changes in market interest rates.
E.Marketable Securities and Equity Investments
A summary of the Company’s cash equivalents and marketable securities, which are recorded at fair value (and do not include $2.8 billion and $2.3 billion of cash as of December 31, 2020 and 2019, respectively), is shown below:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| As of December 31, 2020 | | As of December 31, 2019 |
| Amortized Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value | | Amortized Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value |
| (in thousands) |
| Cash equivalents: | | | | | | | | | | | | | | | |
| Money market funds | $ | 3,141,053 | | | $ | 0 | | | $ | 0 | | | $ | 3,141,053 | | | $ | 791,039 | | | $ | 0 | | | $ | 0 | | | $ | 791,039 | |
| Corporate debt securities | 0 | | | 0 | | | 0 | | | 0 | | | 6,070 | | | 0 | | | 0 | | | 6,070 | |
| Commercial paper | 0 | | | 0 | | | 0 | | | 0 | | | 29,470 | | | 3 | | | (1) | | | 29,472 | |
| Total cash equivalents | $ | 3,141,053 | | | $ | 0 | | | $ | 0 | | | $ | 3,141,053 | | | $ | 826,579 | | | $ | 3 | | | $ | (1) | | | $ | 826,581 | |
| Marketable securities: | | | | | | | | | | | | | | | |
| Government-sponsored enterprise securities | $ | 80,046 | | | $ | 17 | | | $ | 0 | | | $ | 80,063 | | | $ | 12,689 | | | $ | 44 | | | $ | 0 | | | $ | 12,733 | |
| Corporate debt securities | 231,263 | | | 377 | | | (42) | | | 231,598 | | | 301,458 | | | 391 | | | (50) | | | 301,799 | |
| Commercial paper | 163,286 | | | 19 | | | (37) | | | 163,268 | | | 102,240 | | | 121 | | | (5) | | | 102,356 | |
| Total marketable debt securities | 474,595 | | | 413 | | | (79) | | | 474,929 | | | 416,387 | | | 556 | | | (55) | | | 416,888 | |
| Corporate equity securities | 51,427 | | | 144,354 | | | 0 | | | 195,781 | | | 113,829 | | | 168,255 | | | 0 | | | 282,084 | |
| Total marketable securities | $ | 526,022 | | | $ | 144,767 | | | $ | (79) | | | $ | 670,710 | | | $ | 530,216 | | | $ | 168,811 | | | $ | (55) | | | $ | 698,972 | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Available-for-sale debt securities were classified on the Company’s consolidated balance sheets as follows at fair value:
| | | | | | | | | | | |
| December 31, |
| 2020 | | 2019 |
| (in thousands) |
| Cash and cash equivalents | $ | 3,141,053 | | | $ | 826,581 | |
| Marketable securities | 474,929 | | | 416,888 | |
| Total | $ | 3,615,982 | | | $ | 1,243,469 | |
Available-for-sale debt securities by contractual maturity were as follows:
| | | | | | | | | | | |
| December 31, |
| 2020 | | 2019 |
| (in thousands) |
| Matures within one year | $ | 3,526,185 | | | $ | 1,137,942 | |
| Matures after one year through five years | 89,797 | | | 105,527 | |
| Total | $ | 3,615,982 | | | $ | 1,243,469 | |
The Company has a limited number of available-for-sale debt securities in insignificant loss positions as of December 31, 2020, which it does not intend to sell and has concluded it will not be required to sell before recovery of the amortized costs for the investments at maturity. The Company did 0t record any charges for other-than-temporary declines in the fair value of available-for-sale debt securities or gross realized gains or losses in 2020, 2019 or 2018.
The Company records changes in the fair value of its investments in corporate equity securities to “Other income (expense), net” in the Company’s consolidated statements of operations. During the three years ended December 31, 2020, the Company’s net unrealized gains on corporate equity securities held at the conclusion of each period were as follows:
| | | | | | | | | | | | | | | | | |
| 2020 | | 2019 | | 2018 |
| (in thousands) |
| Net unrealized gains | $ | 136,167 | | | $ | 143,175 | | | $ | 2,558 | |
During the years ended December 31, 2020 and 2019, the Company sold the common stock of publicly traded companies, which were primarily sales of its investment in CRISPR, resulting in the following:
| | | | | | | | | | | | | |
| 2020 | | 2019 | | |
| (in thousands) |
| Proceeds received | $ | 437,567 | | | $ | 94,936 | | | |
| Weighted-average cost basis | $ | 103,332 | | | $ | 29,825 | | | |
During the year ended December 31, 2018, the Company did 0t sell any common stock of publicly traded companies.
As of December 31, 2020, the carrying value of the Company’s equity investments without readily determinable fair values, which are recorded in “Other assets” on its consolidated balance sheets, was $20.8 million.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
E.Marketable Securities and Equity Investments
A summary of our cash equivalents and marketable securities, which are recorded at fair value (and do not include $3.3 billion and $3.1 billion of cash as of December 31, 2023 and 2022, respectively), is shown below:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| As of December 31, 2023 | | As of December 31, 2022 |
| Amortized Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value | | Amortized Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value |
| (in millions) |
| Cash equivalents: | | | | | | | | | | | | | | | |
| Money market funds | $ | 5,328.4 | | | $ | — | | | $ | — | | | $ | 5,328.4 | | | $ | 5,162.6 | | | $ | — | | | $ | — | | | $ | 5,162.6 | |
| Time deposits | 1,450.0 | | | — | | | — | | | 1,450.0 | | | 2,000.0 | | | — | | | — | | | 2,000.0 | |
| U.S. Treasury securities | 68.9 | | | — | | | — | | | 68.9 | | | — | | | — | | | — | | | — | |
| Government-sponsored enterprise securities | 174.8 | | | — | | | — | | | 174.8 | | | — | | | — | | | — | | | — | |
| Corporate debt securities | 5.2 | | | — | | | — | | | 5.2 | | | 5.8 | | | — | | | — | | | 5.8 | |
| Commercial paper | 6.6 | | | — | | | — | | | 6.6 | | | 204.5 | | | — | | | — | | | 204.5 | |
| Total cash equivalents | $ | 7,033.9 | | | $ | — | | | $ | — | | | $ | 7,033.9 | | | $ | 7,372.9 | | | $ | — | | | $ | — | | | $ | 7,372.9 | |
| Marketable securities: | | | | | | | | | | | | | | | |
| U.S. Treasury securities | $ | 544.5 | | | $ | 3.0 | | | $ | (1.0) | | | $ | 546.5 | | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| Government-sponsored enterprise securities | 424.8 | | | 0.9 | | | (0.5) | | | 425.2 | | | 127.0 | | | 0.2 | | | (0.1) | | | 127.1 | |
| Asset-backed securities | 304.9 | | | 1.4 | | | (0.3) | | | 306.0 | | | — | | | — | | | — | | | — | |
| Certificates of deposit | 33.7 | | | — | | | — | | | 33.7 | | | — | | | — | | | — | | | — | |
| Corporate debt securities | 1,794.0 | | | 10.5 | | | (1.7) | | | 1,802.8 | | | 87.2 | | | — | | | (0.2) | | | 87.0 | |
| Commercial paper | 186.8 | | | 0.1 | | | (0.1) | | | 186.8 | | | 55.8 | | | — | | | — | | | 55.8 | |
| Total marketable debt securities | 3,288.7 | | | 15.9 | | | (3.6) | | | 3,301.0 | | | 270.0 | | | 0.2 | | | (0.3) | | | 269.9 | |
| Corporate equity securities | 72.1 | | | — | | | (26.1) | | | 46.0 | | | 104.4 | | | 30.9 | | | (18.5) | | | 116.8 | |
| Total marketable securities | $ | 3,360.8 | | | $ | 15.9 | | | $ | (29.7) | | | $ | 3,347.0 | | | $ | 374.4 | | | $ | 31.1 | | | $ | (18.8) | | | $ | 386.7 | |
Available-for-sale debt securities were classified on our consolidated balance sheets at fair value as follows:
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| (in millions) |
| Cash and cash equivalents | $ | 5,583.9 | | | $ | 5,372.9 | |
| Marketable securities | 803.2 | | | 157.7 | |
| Long-term marketable securities | 2,497.8 | | | 112.2 | |
| Total | $ | 8,884.9 | | | $ | 5,642.8 | |
Available-for-sale debt securities by contractual maturity were as follows:
| | | | | | | | | | | |
| December 31, |
| 2023 | | 2022 |
| (in millions) |
| Matures within one year | $ | 6,387.1 | | | $ | 5,530.6 | |
| Matures after one year through five years | 2,495.6 | | | 112.2 | |
| Matures after five years | 2.2 | | | — | |
| Total | $ | 8,884.9 | | | $ | 5,642.8 | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
We did not record any allowances for credit losses to adjust the fair value of available-for-sale debt securities in 2023, 2022 or 2021. Additionally, we did not record any realized gains or losses that were material to our consolidated statements of income in 2023, 2022 or 2021. As of December 31, 2023, we held available-for-sale debt securities with a total fair value of $1.5 billion that were in unrealized loss positions totaling $3.6 million; however, none of these investments had been in an unrealized loss position for greater than twelve months.
We record changes in the fair value of our investments in corporate equity securities to “Other (expense) income, net” in our consolidated statements of income. During the three years ended December 31, 2023, our net unrealized (losses) gains on corporate equity securities held at the conclusion of each period were as follows:
| | | | | | | | | | | | | | | | | |
| 2023 | | 2022 | | 2021 |
| (in millions) |
| Net unrealized (losses) gains | $ | (7.5) | | | $ | (149.1) | | | $ | 17.1 | |
In 2023, we received proceeds of $95.1 million related to the sale of the common stock of a publicly traded company, which had a total original cost basis of $57.3 million. In 2022 and 2021, we did not sell any common stock of publicly traded companies.
As of December 31, 2023, the carrying value of our equity investments without readily determinable fair values, which are recorded in “Other assets” on our consolidated balance sheets, was $98.6 million.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
F.Accumulated Other Comprehensive Income (Loss)
The following table summarizes the changes in accumulated other comprehensive income (loss) by component:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Unrealized Holding Gains (Losses), Net of Tax | | |
| Foreign Currency Translation Adjustment | | On Available-For-Sale Debt Securities | | On Equity Securities | | On Foreign Currency Forward Contracts | | Total |
| (in thousands) |
| Balance as of December 31, 2017 | $ | (21,031) | | | $ | (594) | | | $ | 25,069 | | | $ | (15,016) | | | $ | (11,572) | |
| Other comprehensive income before reclassifications | 8,855 | | | 58 | | | 0 | | | 25,664 | | | 34,577 | |
| Amounts reclassified from accumulated other comprehensive income (loss) | 0 | | | 0 | | | 0 | | | 1,774 | | | 1,774 | |
| Net current period other comprehensive income | 8,855 | | | 58 | | | 0 | | | 27,438 | | | 36,351 | |
| Amounts reclassified to accumulated deficit pursuant to adoption of new accounting standard | 949 | | | 0 | | | (25,069) | | | 0 | | | (24,120) | |
| Balance as of December 31, 2018 | $ | (11,227) | | | $ | (536) | | | $ | 0 | | | $ | 12,422 | | | $ | 659 | |
| Other comprehensive income before reclassifications | 10,332 | | | 1,039 | | | 0 | | | 11,513 | | | 22,884 | |
| Amounts reclassified from accumulated other comprehensive income (loss) | 0 | | | 0 | | | 0 | | | (25,516) | | | (25,516) | |
| Net current period other comprehensive income (loss) | 10,332 | | | 1,039 | | | 0 | | | (14,003) | | | (2,632) | |
| Balance as of December 31, 2019 | $ | (895) | | | $ | 503 | | | $ | 0 | | | $ | (1,581) | | | $ | (1,973) | |
| Other comprehensive loss before reclassifications | (14,783) | | | (169) | | | 0 | | | (54,467) | | | (69,419) | |
| Amounts reclassified from accumulated other comprehensive income (loss) | 0 | | | 0 | | | 0 | | | 2,912 | | | 2,912 | |
| Net current period other comprehensive loss | (14,783) | | | (169) | | | 0 | | | (51,555) | | | (66,507) | |
| Balance as of December 31, 2020 | $ | (15,678) | | | $ | 334 | | | $ | 0 | | | $ | (53,136) | | | $ | (68,480) | |
| | | | | | | | | | | | | | | | | | | | | | | |
| | | Unrealized Holding Gains (Losses), Net of Tax | | |
| Foreign Currency Translation Adjustment | | On Available-For-Sale Debt Securities | | On Foreign Currency Forward Contracts | | Total |
| (in millions) |
| Balance as of December 31, 2020 | $ | (15.6) | | | $ | 0.3 | | | $ | (53.2) | | | $ | (68.5) | |
| Other comprehensive income (loss) before reclassifications | 2.0 | | | (0.8) | | | 59.7 | | | 60.9 | |
| Amounts reclassified from accumulated other comprehensive income (loss) | — | | | — | | | 23.5 | | | 23.5 | |
| Net current period other comprehensive income (loss) | 2.0 | | | (0.8) | | | 83.2 | | | 84.4 | |
| Balance as of December 31, 2021 | $ | (13.6) | | | $ | (0.5) | | | $ | 30.0 | | | $ | 15.9 | |
| Other comprehensive (loss) income before reclassifications | (11.4) | | | (3.3) | | | 138.9 | | | 124.2 | |
| Amounts reclassified from accumulated other comprehensive income (loss) | — | | | 3.7 | | | (143.0) | | | (139.3) | |
| Net current period other comprehensive (loss) income | (11.4) | | | 0.4 | | | (4.1) | | | (15.1) | |
| Balance as of December 31, 2022 | $ | (25.0) | | | $ | (0.1) | | | $ | 25.9 | | | $ | 0.8 | |
| Other comprehensive income (loss) income before reclassifications | 26.1 | | | 9.7 | | | (27.2) | | | 8.6 | |
| Amounts reclassified from accumulated other comprehensive income (loss) | — | | | — | | | (23.7) | | | (23.7) | |
| Net current period other comprehensive income (loss) | 26.1 | | | 9.7 | | | (50.9) | | | (15.1) | |
| Balance as of December 31, 2023 | $ | 1.1 | | | $ | 9.6 | | | $ | (25.0) | | | $ | (14.3) | |
G.Hedging
Foreign currency forward contracts - Designated as hedging instruments
The Company maintainsWe maintain a hedging program intended to mitigate the effect of changes in foreign exchange rates for a portion of the Company’sour forecasted product revenues denominated in certain foreign currencies. The program includes foreign currency forward contracts that are designated as cash flow hedges under U.S. GAAP having contractual durations from one to eighteen months. The Company recognizesWe recognize realized gains and losses for the effective portion of such contracts in “Product revenues, net” in itsour consolidated statements of operationsincome in the same period that it recognizeswe recognize the product revenues that were impacted by the hedged foreign exchange rate changes.
The CompanyWe formally documentsdocument the relationship between foreign currency forward contracts (hedging instruments) and forecasted product revenues (hedged items), as well as the Company’sour risk management objective and strategy for undertaking various hedging activities, which includes matching all foreign currency forward contracts that are designated as cash flow hedges to forecasted transactions. The CompanyWe also formally assesses,assess, both at the hedge’s inception and on an ongoing basis, whether the foreign currency forward contracts are highly effective in offsetting changes in cash flows of hedged items on a prospective and retrospective basis. If the Companywe were to determine that a (i) foreign currency forward contract is not highly effective as a cash flow hedge, (ii) foreign currency forward contract has ceased to be a highly effective hedge or (iii) forecasted transaction is no longer probable of occurring, the Companywe would discontinue hedge accounting treatment prospectively. The Company measuresWe measure effectiveness based on the change in fair value of the forward contracts and the fair value of the hypothetical foreign currency forward
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
contracts with terms that match the critical terms of the risk being hedged. As of December 31, 2020,2023, all hedges were determined to be highly effective.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Prior to the adoption of ASU 2017-12 on January 1, 2019, the Company did not record any ineffectiveness related to its foreign currency forward contracts that were designated as hedging instruments in the year ended December 31, 2018. ASU 2017-12 eliminated the requirement to separately measure and report hedge ineffectiveness.
The Company considersWe consider the impact of itsour counterparties’ credit risk on the fair value of the foreign currency forward contracts. As of December 31, 20202023 and December 31, 2019,2022, credit risk did not change the fair value of the Company’sour foreign currency forward contracts.
The following table summarizes the notional amount in U.S. dollars of the Company’sour outstanding foreign currency forward contracts designated as cash flow hedges under U.S. GAAP:
| | As of December 31, |
| 2020 | | 2019 |
| As of December 31, | | | As of December 31, |
| 2023 | | | 2023 | | 2022 |
| Foreign Currency | Foreign Currency | (in thousands) | Foreign Currency | (in millions) |
| Euro | Euro | $ | 745,099 | | | $ | 501,197 | |
| Canadian dollar | |
| British pound sterling | British pound sterling | 160,427 | | | 87,032 | |
| Australian dollar | Australian dollar | 99,922 | | | 89,705 | |
| Canadian dollar | 86,468 | | | 50,452 | |
| Swiss Franc | |
| Total foreign currency forward contracts | Total foreign currency forward contracts | $ | 1,091,916 | | | $ | 728,386 | |
Foreign currency forward contracts - Not designated as hedging instruments
The CompanyWe also entersenter into foreign currency forward contracts with contractual maturities of less than one month, thatwhich are designed to mitigate the effect of changes in foreign exchange rates on monetary assets and liabilities, including intercompany balances. These contracts are not designated as hedging instruments under U.S. GAAP. The Company recognizesWe recognize realized gains and losses for such contracts in “Other (expense) income, (expense), net” in itsour consolidated statements of operationsincome each period. As of December 31, 2020, the notional amount of the Company’s2023, we did not have any outstanding foreign currency forward contracts where hedge accounting under U.S. GAAP iswas not applied was $198.9 million.applied.
During the three years ended December 31, 2020, the Company2023, we recognized the following related to foreign currency forward contactscontracts in itsour consolidated statements of operations:income:
| | December 31, |
| 2020 | | 2019 | | 2018 |
| (in thousands) |
| 2023 | | | 2023 | | 2022 | | 2021 |
| (in millions) | | | (in millions) |
| Designated as hedging instruments - Reclassified from AOCI | Designated as hedging instruments - Reclassified from AOCI | |
| Product revenues, net | Product revenues, net | $ | (3,714) | | | $ | 32,546 | | | $ | (1,252) | |
| Product revenues, net | |
| Product revenues, net | |
| Not designated as hedging instruments | Not designated as hedging instruments | |
| Other income (expense), net | $ | 22,113 | | | $ | (4,838) | | | $ | (623) | |
| Other (expense) income, net | |
| Other (expense) income, net | |
| Other (expense) income, net | |
| | Total reported in the Consolidated Statement of Operations | |
| Total reported in the Consolidated Statements of Income | |
| Total reported in the Consolidated Statements of Income | |
| Total reported in the Consolidated Statements of Income | |
| Product revenues, net | Product revenues, net | $ | 6,202,783 | | | $ | 4,160,726 | | | $ | 3,038,325 | |
| Other income (expense), net | $ | 296,420 | | | $ | 192,177 | | | $ | (790) | |
| Product revenues, net | |
| Product revenues, net | |
| Other (expense) income, net | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
The following table summarizes the fair value of the Company’sour outstanding foreign currency forward contracts designated as cash flow hedges under U.S. GAAP included on itsour consolidated balance sheets:
| As of December 31, 2020 | | As of December 31, 2023 | | As of December 31, 2023 |
| Assets | Assets | | Liabilities | Assets | | Liabilities |
| Classification | Classification | | Fair Value | | Classification | | Fair Value | Classification | | Fair Value | | Classification | | Fair Value |
(in thousands) | | (in millions) | | (in millions) |
| Prepaid expenses and other current assets | Prepaid expenses and other current assets | | $ | 0 | | | Other current liabilities | | $ | (59,184) | |
| Other assets | Other assets | | 0 | | | Other long-term liabilities | | (4,283) | |
| Total assets | Total assets | | $ | 0 | | | Total liabilities | | $ | (63,467) | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
| As of December 31, 2019 | | As of December 31, 2022 | | As of December 31, 2022 |
| Assets | Assets | | Liabilities | Assets | | Liabilities |
| Classification | Classification | | Fair Value | | Classification | | Fair Value | Classification | | Fair Value | | Classification | | Fair Value |
(in thousands) | | (in millions) | | (in millions) |
| Prepaid expenses and other current assets | Prepaid expenses and other current assets | | $ | 9,725 | | | Other current liabilities | | $ | (5,533) | |
| Other assets | Other assets | | 0 | | | Other long-term liabilities | | (1,821) | |
| Total assets | Total assets | | $ | 9,725 | | | Total liabilities | | $ | (7,354) | |
As of December 31, 2020,2023, we expect the Company expects amounts that are related to foreign exchange forward contracts designated as cash flow hedges under U.S. GAAP recorded in “Prepaid expenses and other current assets” and “Other current liabilities” to be reclassified to earnings within twelve months.
As discussed in “Note A, “Nature of Business and Accounting Policies,” we present the fair value of our foreign currency forward contracts on a gross basis within our consolidated balance sheets. The following table summarizes the potential effect of offsetting derivatives by type of financial instrument designated as cash flow hedges under U.S. GAAP on the Company’sour consolidated balance sheets:
| | As of December 31, 2020 |
| Gross Amounts Recognized | | Gross Amounts Offset | | Gross Amounts Presented | | Gross Amounts Not Offset | | Legal Offset |
| As of December 31, 2023 | | | As of December 31, 2023 |
| Gross Amounts Recognized | | | Gross Amounts Recognized | | Gross Amounts Offset | | Gross Amounts Presented | | Gross Amounts Not Offset | | Legal Offset |
| Foreign currency forward contracts | Foreign currency forward contracts | (in thousands) | Foreign currency forward contracts | (in millions) |
| Total assets | Total assets | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | |
| Total liabilities | Total liabilities | (63,467) | | | 0 | | | (63,467) | | | 0 | | | (63,467) | |
| | As of December 31, 2019 |
| Gross Amounts Recognized | | Gross Amounts Offset | | Gross Amounts Presented | | Gross Amounts Not Offset | | Legal Offset |
| As of December 31, 2022 | | | As of December 31, 2022 |
| Gross Amounts Recognized | | | Gross Amounts Recognized | | Gross Amounts Offset | | Gross Amounts Presented | | Gross Amounts Not Offset | | Legal Offset |
| Foreign currency forward contracts | Foreign currency forward contracts | (in thousands) | Foreign currency forward contracts | (in millions) |
| Total assets | Total assets | $ | 9,725 | | | $ | 0 | | | $ | 9,725 | | | $ | (7,354) | | | $ | 2,371 | |
| Total liabilities | Total liabilities | (7,354) | | | 0 | | | (7,354) | | | 7,354 | | | 0 | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
H.Inventories
“Inventories” consisted of the following:
| | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 |
| (in millions) |
| Raw materials | $ | 78.7 | | | $ | 38.1 | |
| Work-in-process | 525.1 | | | 260.7 | |
| Finished goods | 135.0 | | | 161.8 | |
| Total | $ | 738.8 | | | $ | 460.6 | |
H.I.InventoriesProperty and Equipment
Inventories“Property and equipment, net” consisted of the following:
| | | | | | | | | | | |
| As of December 31, |
| 2020 | | 2019 |
| (in thousands) |
| Raw materials | $ | 46,232 | | | $ | 26,247 | |
| Work-in-process | 161,324 | | | 107,021 | |
| Finished goods | 73,221 | | | 34,234 | |
| Total | $ | 280,777 | | | $ | 167,502 | |
| | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 |
| (in millions) |
| Buildings and improvements | $ | 928.6 | | | $ | 903.1 | |
| Laboratory equipment, other equipment and furniture | 579.1 | | | 476.5 | |
| Leasehold improvements | 474.6 | | | 410.9 | |
| Computers and software | 332.8 | | | 312.1 | |
| Land | 33.1 | | | 33.1 | |
| Total property and equipment, gross | 2,348.2 | | | 2,135.7 | |
| Less: accumulated depreciation | (1,188.9) | | | (1,027.3) | |
| Total property and equipment, net | $ | 1,159.3 | | | $ | 1,108.4 | |
We recorded depreciation expense of $167.8 million, $148.3 million and $125.6 million in 2023, 2022 and 2021, respectively, which includes our finance lease amortization.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
J.Goodwill and Other Intangible Assets
Intangible Assets
“Other intangible assets, net” consisted of the following:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of December 31, 2023 | | As of December 31, 2022 |
| Estimated Useful lives | | Gross Carrying Amount | | Accumulated Amortization | | Net Carrying Amount | | Gross Carrying Amount | | Accumulated Amortization | | Net Carrying Amount |
| | | (in millions, except useful lives) |
| Finite-lived intangible assets | 10 to 12 yrs. | | $ | 238.0 | | | $ | (1.7) | | | $ | 236.3 | | | $ | — | | | $ | — | | | $ | — | |
| In-process research and development | Indefinite | | 603.6 | | | — | | | $ | 603.6 | | | 603.6 | | | — | | | $ | 603.6 | |
| Total other intangible assets, net | | | $ | 841.6 | | | $ | (1.7) | | | $ | 839.9 | | | $ | 603.6 | | | $ | — | | | $ | 603.6 | |
As of December 31, 2023, finite-lived intangible assets totaled $238.0 million. Following the regulatory approval of CASGEVY in several markets we paid $208.0 million in regulatory approval milestones, including $200.0 million to CRISPR, as well as a net amount of $30.0 million related to the Editas Agreement. We recorded these amounts as finite-lived intangible assets and are amortizing each on a straight-line basis over the longer of the last underlying patents to expire or the period that we have exclusive rights to market CASGEVY. We recorded intangible asset amortization expense of $1.7 million to “Cost of sales” related to these assets in 2023. Please refer to Note B, “Collaboration, License and Other Arrangements,” for further information.
As of December 31, 2023, the estimated future amortization of our finite-lived intangible assets was as follows:
| | | | | | | | |
| Year | | Estimated Amortization Expense |
| | (in millions) |
| 2024 | | $ | 20.2 | |
| 2025 | | $ | 20.2 | |
| 2026 | | $ | 20.2 | |
| 2027 | | $ | 20.2 | |
| 2028 | | $ | 20.2 | |
In 2022, we recorded a $13.0 million impairment of an in-process research and development intangible asset to “Research and development expenses” due to a decision to revise the scope of certain acquired gene-editing programs.
Goodwill
As of December 31, 2023 and 2022, we had goodwill of $1.1 billion on our consolidated balance sheets.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
I.K.Property and EquipmentAdditional Balance Sheet Detail
Property“Prepaid expenses and equipment, netother current assets” consisted of the following:
| | | | | | | | | | | |
| As of December 31, |
| 2020 | | 2019 |
| (in thousands) |
| Buildings and improvements | $ | 876,091 | | | $ | 713,412 | |
| Furniture and equipment | 346,698 | | | 317,567 | |
| Leasehold improvements | 234,585 | | | 175,769 | |
| Computers and software | 258,612 | | | 230,872 | |
| Land | 33,128 | | | 0 | |
| Total property and equipment, gross | 1,749,114 | | | 1,437,620 | |
| Less: accumulated depreciation | (790,580) | | | (692,540) | |
| Total property and equipment, net | $ | 958,534 | | | $ | 745,080 | |
| | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 |
| (in millions) |
| Tax-related prepaid and receivables | $ | 429.0 | | | $ | 319.8 | |
| Prepaid expenses | 108.6 | | | 111.8 | |
| Fair value of cash flow hedges | 1.8 | | | 47.5 | |
| Other | 84.3 | | | 74.4 | |
| Total | $ | 623.7 | | | $ | 553.5 | |
The Company recorded depreciation expense“Accrued expenses” consisted of $109.5 million, $106.9 million and $72.4 million in 2020, 2019 and 2018, respectively. The Company’s finance lease amortization is included in depreciation expense.
In the third quarter of 2020, the Company purchased its continuous manufacturing facility located near its corporate headquarters in Boston, Massachusetts from its former landlord for $155.3 million in cash. The Company’s December 31, 2020 consolidated balance sheet reflects the following: (i) classification
| | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 |
| (in millions) |
| Product revenue accruals | $ | 1,716.4 | | | $ | 1,300.8 | |
| Payroll and benefits | 295.0 | | | 246.5 | |
| Research, development and commercial contract costs | 265.2 | | | 198.7 | |
| Royalty payable | 237.6 | | | 215.0 | |
| Tax related accruals | 99.5 | | | 123.3 | |
| Other | 41.6 | | | 42.4 | |
| Total | $ | 2,655.3 | | | $ | 2,126.7 | |
“Other current liabilities” consisted of the building within “Property and equipment, net” with a 40 year useful life, (ii) derecognition of the previously recorded insignificant right-of-use asset and operating lease liability for the facility and (iii) a finance lease for the land on which the facility is constructed.following:
J.Intangible Assets and Goodwill
Intangible Assets
As of December 31, 2020 and 2019, the Company had $400.0 million of in-process research and development intangible assets classified as “Intangible assets” on its consolidated balance sheet. In 2019, the Company recorded $387.0 million and $13.0 million of in-process research and development intangible assets related to its acquisitions of Semma Therapeutics, Inc. (“Semma”) and Exonics, respectively. In 2018, the Company recorded an intangible asset impairment charge of $29.0 million related to VX-210 that was licensed from BioAxone in 2014. Please refer to Note B, “Collaborative Arrangements,” for further information regarding the events and circumstances associated with this impairment charge.
Goodwill
As of December 31, 2020 and December 31, 2019, goodwill of $1.00 billion was recorded on the Company’s consolidated balance sheet. During 2019, the Company recorded goodwill of $554.6 million and $397.1 million related to its acquisitions of Semma and Exonics, respectively.
| | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 |
| (in millions) |
| Milestone payment due to CRISPR | $ | 200.0 | | | $ | — | |
| Contract liabilities | 170.3 | | | 159.6 | |
| Finance lease liabilities | 50.6 | | | 40.8 | |
| Operating lease liabilities | 33.1 | | | 48.6 | |
| Other | 73.2 | | | 62.5 | |
| Total | $ | 527.2 | | | $ | 311.5 | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
K.Additional Balance Sheet Detail
Accrued expenses“Other long-term liabilities” consisted of the following:
| | | | | | | | | | | |
| As of December 31, |
| 2020 | | 2019 |
| (in thousands) |
| Product revenue accruals | $ | 781,903 | | | $ | 641,368 | |
| Payroll and benefits | 169,387 | | | 159,464 | |
| Research, development and commercial contract costs | 136,704 | | | 105,663 | |
| Royalty payable | 165,404 | | | 98,578 | |
| Tax related accruals | 104,173 | | | 72,293 | |
| Other | 47,400 | | | 39,546 | |
| Total | $ | 1,404,971 | | | $ | 1,116,912 | |
| | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 |
| (in millions) |
| Tax-related liabilities | $ | 681.4 | | | $ | 452.8 | |
| Contingent consideration | 77.4 | | | 114.4 | |
| Other | 118.9 | | | 118.6 | |
| Total | $ | 877.7 | | | $ | 685.8 | |
Other current liabilities consistedCash, Cash Equivalents and Restricted Cash Presented in Consolidated Statements of the following:
| | | | | | | | | | | |
| As of December 31, |
| 2020 | | 2019 |
| (in thousands) |
| Contract liabilities | $ | 191,519 | | | $ | 62,332 | |
| Fair value of cash flow hedges | 59,184 | | | 5,533 | |
| Finance lease liabilities | 42,434 | | | 38,794 | |
| Other | 24,286 | | | 23,646 | |
| Total | $ | 317,423 | | | $ | 130,305 | |
The cash, cash equivalents and restricted cash balances at the beginning and ending of each period presented in the Company’sour consolidated statements of cash flows consisted of the following:
| | As of December 31, |
| 2020 | | 2019 | | 2018 | | 2017 |
| (in thousands) |
| As of December 31, | | | As of December 31, |
| 2023 | | | 2023 | | 2022 | | 2021 | | 2020 |
| (in millions) | | | (in millions) |
| Cash and cash equivalents | Cash and cash equivalents | $ | 5,988,187 | | | $ | 3,109,322 | | | $ | 2,650,134 | | | $ | 1,665,412 | |
| Prepaid expenses and other current assets | Prepaid expenses and other current assets | 658 | | | 8,004 | | | 4,910 | | | 2,114 | |
| Other assets | 0 | | | 3,355 | | | 3,209 | | | 0 | |
| Cash, cash equivalents and restricted cash per consolidated statement of cash flows | $ | 5,988,845 | | | $ | 3,120,681 | | | $ | 2,658,253 | | | $ | 1,667,526 | |
| Cash, cash equivalents and restricted cash per consolidated statements of cash flows | |
The Company’sOur restricted cash, if any, is included in “Prepaid expenses and other current assets” and “Other assets” on itsour consolidated balance sheets.
Cloud Computing Service Contracts
As of December 31, 2023 and 2022, “Other assets” included $58.9 million and $40.2 million, respectively, related to costs incurred to implement cloud computing service contracts. We recorded amortization associated with cloud computing service contracts of $11.8 million in 2023. Our amortization associated with cloud computing service contracts was not material in 2022 or 2021.
L.Leases
Finance Leases
The Company’sOur finance lease assets and liabilities primarily relate to itsour corporate headquarters in Boston and research site in San Diego (the “Buildings”). These Buildings are classified as finance leases because the present value of the sum of the lease payments associated with the Buildings exceedsexceeded substantially all of the fair value of the Buildings. The CompanyBuildings at lease inception. We also hashave an outstanding finance leaseslease for equipment and land.
Pursuantland related to ASC 842, which was adopted on January 1, 2019, the Company’s finance lease assets are amortized to depreciation expense using the straight-line method over the remaining lease term; and the Company records imputed interest expense associated with its finance lease liabilities. In 2018, prior to the adoption of ASC 842, the Company applied build-to-
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
suit accounting and was deemed for accounting purposes to be the owner of the Buildings, for which it recognized depreciation expense over the Buildings 40 years useful lives and imputed interest expense on the corresponding financing lease obligations.a facility that we own.
Corporate Headquarters
In 2011, the Companywe entered into 2two lease agreements, pursuant to which the Company leaseswe lease approximately 1.1 million square feet of office and laboratory space in 2two buildings in Boston, Massachusetts for a term of 15 years. Base rent payments commenced in December 2013 and will continue through December 2028. The Company utilizesWe utilize this initial period as itsour lease term. The Company hasWe have an option to extend the lease term for an additional ten years.
San Diego Lease
In 2015, the Companywe entered into a lease agreement pursuant to which the Company leaseswe lease approximately 170,000 square feet of office and
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
laboratory space in San Diego, California for a term of 16 years. Base rent payments commenced in the second quarter of 2019 and will continue through May 2034. The Company utilizesWe utilize this initial period as itsour lease term. The Company hasWe have an option to extend the lease term for up to 2two additional five-year terms. The Company placed this building into service in the second quarter of 2018.
Operating Leases
The Company’sOur operating leases relate to itsour real estate leases that are not classified as finance leases.
Jeffrey Leiden Center for Cell and Genetic Therapies Lease
In 2019, the Companywe entered into an agreement to lease approximately 269,000 square feet of office and laboratory space near itsour corporate headquarters in Boston, Massachusetts. The lease agreement includes an initialMassachusetts for a term of 15 years plus a16 years. Base rent payments commenced in 2021 and will continue through November 2036. We utilize the initial period to install leasehold improvements, withas our lease term. We have an option to extend the lease term for up to 2two additional ten-year periods. Base rent payments will commence in the fourth quarter of 2021. The Company has utilized the initial period, which commenced in the third quarter of 2020 upon occupation of the building, as its lease term. As of December 31, 2020, the Company recorded a right-of-use asset of $253.5 million and an operating lease liability of $286.6 million related to the lease agreement on its consolidated balance sheet.
Please refer to the Company’sour accounting policy, Leases, in Note A, “Nature of Business and Accounting Policies,” for further information on the accounting treatment for the Company’sour finance and operating leases.
Aggregate Lease Information Related to the Application of ASC 842
The following information is disclosed in accordance with ASC 842, which became effective January 1, 2019. The components of lease cost recorded in the Company’sour consolidated statementstatements of operationsincome were as follows:
| | 2020 | | 2019 |
| | 2023 | | | 2023 | | 2022 | | 2021 |
| (in millions) | | | (in millions) |
| Operating lease cost | Operating lease cost | $ | 23,128 | | | $ | 11,972 | |
| Finance lease cost | Finance lease cost | |
| Amortization of leased assets | |
| Amortization of leased assets | |
| Amortization of leased assets | Amortization of leased assets | 51,223 | | | 49,778 | |
| Interest on lease liabilities | Interest on lease liabilities | 50,188 | | | 52,839 | |
| Variable lease cost | Variable lease cost | 30,776 | | | 27,997 | |
| Sublease income | Sublease income | (4,021) | | | (6,391) | |
| Net lease cost | Net lease cost | $ | 151,294 | | | $ | 136,195 | |
The Company’sOur variable lease cost during 20202023, 2022 and 20192021 primarily related to operating expenses, taxes and insurance associated with itsour finance leases. The Company’s sublease income during 2020 and 2019 primarily related to subleases for an insignificant portion of the Company’s corporate headquarters.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
The Company’sOur leases are included on itsour consolidated balance sheets as follows:
| | As of December 31, |
| 2020 | | 2019 |
| (in thousands) |
| As of December 31, | | | As of December 31, |
| 2023 | | | 2023 | | 2022 |
| (in millions) | | | (in millions) |
| Finance leases | Finance leases | |
| Property and equipment, net | |
| Property and equipment, net | |
| Property and equipment, net | Property and equipment, net | $ | 431,193 | | | $ | 445,336 | |
| Total finance lease assets | Total finance lease assets | $ | 431,193 | | | $ | 445,336 | |
| | Other current liabilities | |
| Other current liabilities | |
| Other current liabilities | Other current liabilities | $ | 42,434 | | | $ | 38,795 | |
| Long-term finance lease liabilities | Long-term finance lease liabilities | 539,042 | | | 538,576 | |
| Total finance lease liabilities | Total finance lease liabilities | $ | 581,476 | | | $ | 577,371 | |
| | Operating leases | Operating leases | |
| Operating leases | |
| Operating leases | |
| Operating lease assets | |
| Operating lease assets | |
| Operating lease assets | Operating lease assets | $ | 325,564 | | | $ | 88,202 | |
| Total operating lease assets | Total operating lease assets | $ | 325,564 | | | $ | 88,202 | |
| | Other current liabilities | Other current liabilities | $ | 10,521 | | | $ | 11,504 | |
| Other current liabilities | |
| Other current liabilities | |
| Long-term operating lease liabilities | Long-term operating lease liabilities | 350,463 | | | 84,292 | |
| Total operating lease liabilities | Total operating lease liabilities | $ | 360,984 | | | $ | 95,796 | |
Maturities of the Company’sour finance and operating lease liabilities as of December 31, 20202023 were as follows:
| | | | | | | | | | | | | | | | | | | | |
| Year | | Finance Leases | | Operating Leases | | Total |
| | (in thousands) |
| 2021 | | $ | 85,470 | | | $ | 15,266 | | | $ | 100,736 | |
| 2022 | | 90,020 | | | 34,119 | | | 124,139 | |
| 2023 | | 88,185 | | | 34,663 | | | 122,848 | |
| 2024 | | 93,434 | | | 34,113 | | | 127,547 | |
| 2025 | | 93,421 | | | 31,564 | | | 124,985 | |
| Thereafter | | 416,455 | | | 320,684 | | | 737,139 | |
| Total lease payments | | 866,985 | | | 470,409 | | | 1,337,394 | |
| Less: tenant allowance | | 0 | | | (36,051) | | | (36,051) | |
| Less: amount representing interest | | (285,509) | | | (73,374) | | | (358,883) | |
| Present value of lease liabilities | | $ | 581,476 | | | $ | 360,984 | | | $ | 942,460 | |
The weighted-average remaining lease terms and discount rates related to the Company’s leases were as follows:
| | | | | | | | | | | |
| As of December 31, |
| 2020 | | 2019 |
| Weighted-average remaining lease term (in years) | | | |
| Finance leases | 11.58 | | 9.74 |
| Operating leases | 14.10 | | 9.70 |
| | | |
| Weighted-average discount rate | | | |
| Finance leases | 8.36 | % | | 9.04 | % |
| Operating leases | 2.28 | % | | 3.75 | % |
| | | | | | | | | | | | | | | | | | | | |
| Year | | Finance Leases | | Operating Leases | | Total |
| | (in millions) |
| 2024 | | $ | 81.4 | | | $ | 42.1 | | | $ | 123.5 | |
| 2025 | | 89.0 | | | 41.4 | | | 130.4 | |
| 2026 | | 89.3 | | | 40.4 | | | 129.7 | |
| 2027 | | 89.7 | | | 36.5 | | | 126.2 | |
| 2028 | | 90.1 | | | 33.5 | | | 123.6 | |
| Thereafter | | 143.5 | | | 242.2 | | | 385.7 | |
| Total lease payments | | 583.0 | | | 436.1 | | | 1,019.1 | |
| Less: amount representing interest | | (156.3) | | | (54.4) | | | (210.7) | |
| Present value of lease liabilities | | $ | 426.7 | | | $ | 381.7 | | | $ | 808.4 | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
| | | | | | | | | | | |
| As of December 31, |
| 2023 | | 2022 |
| Weighted-average remaining lease term (in years) | | | |
| Finance leases | 10.06 | | 10.60 |
| Operating leases | 11.24 | | 11.57 |
| | | |
| Weighted-average discount rate | | | |
| Finance leases | 8.20 | % | | 8.36 | % |
| Operating leases | 2.42 | % | | 2.46 | % |
Supplemental cash flow information related to the Company’sour leases was as follows:
| | | | | | | | | | | |
| 2020 | | 2019 |
| (in thousands) |
| Cash paid for amounts included in the measurement of lease liabilities | | | |
| Operating cash flows from operating leases | $ | 16,254 | | | $ | 10,650 | |
| Operating cash flows from finance leases | $ | 48,908 | | | $ | 50,527 | |
| Financing cash flows from finance leases | $ | 42,275 | | | $ | 39,185 | |
| | | |
| Right-of-use assets obtained in exchange for lease obligations | | | |
| Operating leases * | $ | 293,604 | | | $ | 34,605 | |
| Finance leases | $ | 33,128 | | | $ | 0 | |
| * 2019 includes $33.7 million acquired in 2019 pursuant to the Company’s acquisitions of Semma and Exonics. | | |
Additional Lease Information Related to the Application of ASC 840
Rental expense was $17.3 million during the year ended December 31, 2018, which is disclosed in accordance with ASC 840, Leases (Topic 840), which was applicable during the year ended December 31, 2018. | | | | | | | | | | | | | | | | | |
| 2023 | | 2022 | | 2021 |
| (in millions) |
| Cash paid for amounts included in the measurement of lease liabilities | | | | | |
| Operating cash flows from operating leases | $ | 62.8 | | | $ | 50.8 | | | $ | 21.5 | |
| Operating cash flows from finance leases | $ | 38.4 | | | $ | 42.5 | | | $ | 46.2 | |
| Financing cash flows from finance leases | $ | 44.9 | | | $ | 85.5 | | | $ | 47.0 | |
| | | | | |
| Right-of-use assets obtained in exchange for lease obligations | | | | | |
| Operating leases | $ | 2.4 | | | $ | 58.6 | | | $ | 36.3 | |
| | | | | |
M.Common Stock, Preferred Stock and Equity Plans
Common Stock and Preferred Stock
The Company isWe are authorized to issue 500,000,000500.0 million shares of common stock. Holders of common stock are entitled to 1one vote per share. Holders of common stock are entitled to receive dividends, if and when declared by the Company’sour Board of Directors, and to share ratably in the Company’sour assets legally available for distribution to the Company’sour shareholders in the event of liquidation. Holders of common stock have no preemptive, subscription, redemption or conversion rights. The holders of common stock do not have cumulative voting rights.
The Company isWe are authorized to issue 1,000,0001.0 million shares of preferred stock in one or more series and to fix the powers, designations, preferences and relative participating, option or other rights thereof, including dividend rights, conversion rights, voting rights, redemption terms, liquidation preferences and the number of shares constituting any series, without any further vote or action by the Company’sour shareholders. As of December 31, 20202023 and 2019, the Company2022, we had 0no shares of preferred stock issued or outstanding.
Share Repurchase Programs
During 2018, the Company’sIn November 2020, our Board of Directors approved a share repurchase program, (the “2018 Share Repurchase Program”), pursuant to which the Company waswe repurchased $500.0 million of our common stock in 2020 and 2021. In 2021, we repurchased 2.0 million shares of our common stock under this program for an aggregate of $424.9 million.
In June 2021, our Board of Directors approved a share repurchase program, pursuant to which we were authorized to repurchase up to $500.0 million$1.5 billion of itsour common stock between February 1, 2018 andby December 31, 2019. During the years ended December 31, 2019 and 2018, the Company2022. In 2021, we repurchased 832,186 and 2,093,8915.3 million shares respectively, of itsour common stock under the 2018 Share Repurchase Programthis program for an aggregate of $150.0 million and $350.0 million, respectively, including commissions and fees. As of June 30, 2019,$1.0 billion. On December 31, 2022, the Company had repurchased the entire $500.0 million it was authorized to repurchase of its common stock under the 2018 Share Repurchase Program.
In July 2019, the Company’s Board of Directors approved a second share repurchase program (the “2019 Share Repurchase Program”), pursuant to which the Company was authorized to repurchase up to $500.0expired with $499.7 million of its common stock between August 1, 2019 and December 31, 2020. During the years ended December 31, 2020 and 2019, the Company repurchased 2,058,962 and 213,548 shares, respectively, of its common stock under the 2019 Share Repurchase Program for an aggregate of $464.0 million and $36.0 million, respectively, including commissions and fees. As of December 31, 2020, the Company had repurchased the entire $500.0 million it was authorized to repurchase of its common stock under the 2019 Share Repurchase Program.
In November 2020, the Company’s Board of Directors approved a third share repurchase program (the “2020 Share Repurchase Program”), pursuant to which the Company is authorized to repurchase up to $500.0 million of its common stockauthorization remaining.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
by December 31, 2022. In the fourth quarterFebruary 2023, our Board of 2020, the CompanyDirectors approved a share repurchase program, pursuant to which we are authorized to repurchase up to $3.0 billion of our common stock. This program does not have an expiration date and can be discontinued at any time. In 2023, we repurchased 345,8971.3 million shares of itsour common stock under the 2020 Share Repurchase Programthis program for an aggregate of $75.1 million including commissions and fees. The Company expects to fund further repurchases of its common stock through a combination of cash on hand and cash generated by operations.$427.6 million. As of December 31, 2020, there was a total of $424.9 million2023, we had $2.6 billion remaining for repurchases of its common stockauthorization under the 2020 Share Repurchase Program.
Under the 2020 Share Repurchase Program, the Company is authorized to purchase shares from time to time through open market or privately negotiated transactions. Such purchases are made pursuant to Rule 10b5-1 plans or other means as determined by the Company’s management and in accordance with the requirements of the SEC.this program.
Stock and Option Plans
The purpose of each of the Company’sour stock and option plans is to attract, retain and motivate itsour employees, consultants and directors. Awards granted under these plans can be nonstatutory stock options (“NSOs”), incentive stock options (“ISOs”), restricted stock units (“RSUs”) including performance-based RSUs (“PSUs”), restricted stock (“RSs”), or other equity-based awards, as specified in the individual plans.
Shares issued under all of the Company’sour plans are funded through the issuance of new shares. The following table contains information about the Company’sour equity plans:
| | As of December 31, 2020 |
| | | | | | As of December 31, 2023 | | | | | | | | As of December 31, 2023 |
| Title of Plan | Title of Plan | | Group Eligible | | Type of Award Granted | | Awards Outstanding | | Additional Awards Authorized for Grant | Title of Plan | | Group Eligible | | Type of Award Granted | | Awards Outstanding | | Additional Awards Authorized for Grant |
| | | | | | (in thousands) | | | | | | | | (in thousands) |
| 2013 Stock and Option Plan | 2013 Stock and Option Plan | | Employees, Non-employee Directors and Consultants | | NSO,
RS, RSU and PSU | | 7,231,859 | | | 13,385,321 | |
| 2006 Stock and Option Plan | 2006 Stock and Option Plan | | Employees, Non-employee Directors and Consultants | | NSO,
RS and RSU | | 383,818 | | | 0 | |
| Total | | 7,615,677 | | | 13,385,321 | |
| | | | Total | |
Restricted Stock Units (excluding PSUs)
The following table summarizes our restricted stock unit activity during the year ended December 31, 2023:
| | | | | | | | | | | |
| Restricted Stock Units (excluding PSUs) |
| Number of Shares | | Weighted-average
Grant-date Fair Value |
| (in thousands) | | (per share) |
| Unvested at December 31, 2022 | 2,913 | | | $ | 240.54 | |
| Granted | 1,574 | | | $ | 331.23 | |
| Vested | (1,349) | | | $ | 238.92 | |
| Cancelled | (176) | | | $ | 275.18 | |
| Unvested at December 31, 2023 | 2,962 | | | $ | 287.41 | |
The total fair value of restricted stock units that vested during 2023, 2022 and 2021 (measured based on the market price of our common stock on the date of vesting) was $433.4 million, $372.5 million and $281.1 million, respectively.
Performance-based RSUs (PSUs)
The potential range of shares issuable pursuant to our PSU awards range from 0% to 200% of the target shares based on financial and non-financial measures. For the majority of our PSU awards, 50% of PSUs that could be earned have a one-year performance period with the amount actually earned dependent upon our financial performance and with vesting of the earned shares in three equal installments over a three-year period. For these same PSU awards, the remaining 50% of PSUs that could be earned have a three-year performance period with the amount earned dependent upon the achievement of multiple clinical development milestones and with the earned shares cliff vesting at the end of the three-year performance period.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
The following table summarizes our PSU activity during the year ended December 31, 2023:
| | | | | | | | | | | |
| Performance-Based RSU |
| Number of Units | | Weighted-average
Grant-date Fair Value |
| (in thousands) | | (per share) |
| Unvested at December 31, 2022 (1) | 1,227 | | | $ | 226.75 | |
| Granted (2) | 581 | | | $ | 317.83 | |
| Vested | (545) | | | $ | 237.14 | |
| Cancelled | (68) | | | $ | 241.25 | |
| Unvested at December 31, 2023 | 1,195 | | | $ | 247.12 | |
| | | |
| (1) “Unvested” represents our PSUs at target to the extent performance has not been certified plus the actual number of shares that continue to be subject to service conditions for which the performance has been achieved and certified. |
| (2) “Granted” represents (i) the target number of shares issuable for grants during 2023 and (ii) any change in the number of shares issuable pursuant to outstanding PSUs based on performance certification during 2023. |
The total fair value of PSUs that vested during 2023, 2022 and 2021 (measured on the date of vesting) was $160.4 million, $98.7 million and $92.2 million, respectively.
Stock Options
All options granted under the Company’sour 2013 Stock and Option Plan (“2013 Plan”) and 2006 Stock and Option Plan (“2006 Plan”) were granted with an exercise price equal to the fair value of the underlying common stock on the date of grant. As of December 31, 2020, the stock and option plan under which the Company is2023, we are only authorized to make new equity awards is the Company’sunder our 2013 Plan. Under the 2013 Plan, no stock options can be awarded with an exercise price less than the fair market value on the date of grant. In 2019 and 2018, the Company’s shareholders approved increases in the number of shares authorized for issuance pursuant to the 2013 Stock and Option Plan of (i) 5,000,000 shares in 2019 and (ii) 8,000,000 shares in 2018.
During the three years ended December 31, 2020, grants to current employees and directors primarily had a grant date that was the same as the date the award was approved by the Company’s Board of Directors. During the three years ended December 31, 2020, for grants to new employees and directors, the date of grant for awards was the employee’s first day of employment or the date the director was elected to the Company’s Board of Directors. All options awarded under the Company’sour stock and option plans expire not more than 10 years from the grant date.
VERTEX PHARMACEUTICALS INCORPORATED
Notes In each of the three years ended December 31, 2023, we only granted stock options to Consolidated Financial Statements (Continued)
Stock Optionscertain of our non-employee directors on May 1.
The following table summarizes information related to the outstanding and exercisable options during the year ended December 31, 2020:2023:
| | | | | | | | | | | | | | | | | | | | | | | |
| Stock Options | | Weighted-average Exercise Price | | Weighted-average Remaining Contractual Life | | Aggregate Intrinsic Value |
| (in thousands) | | (per share) | | (in years) | | (in thousands) |
| Outstanding at December 31, 2019 | 6,272 | | | $ | 134.92 | | | | | |
| Granted | 23 | | | $ | 286.27 | | | | | |
| Exercised | (1,883) | | | $ | 121.42 | | | | | |
| Forfeited | (174) | | | $ | 165.39 | | | | | |
| Expired | 0 | | | $ | 0 | | | | | |
| Outstanding at December 31, 2020 | 4,238 | | | $ | 140.47 | | | 6.34 | | $ | 397,211 | |
| Exercisable at December 31, 2020 | 2,894�� | | | $ | 125.83 | | | 5.73 | | $ | 313,936 | |
| | | | | | | | | | | | | | | | | | | | | | | |
| Stock Options | | Weighted-average Exercise Price | | Weighted-average Remaining Contractual Life | | Aggregate Intrinsic Value |
| (in thousands) | | (per share) | | (in years) | | (in millions) |
| Outstanding at December 31, 2022 | 2,527 | | | $ | 146.11 | | | | | |
| Granted | 16 | | | $ | 342.73 | | | | | |
| Exercised | (602) | | | $ | 134.27 | | | | | |
| Forfeited | (1) | | | $ | 174.87 | | | | | |
| | | | | | | |
| Outstanding at December 31, 2023 | 1,940 | | | $ | 151.37 | | | 3.97 | | $ | 497.6 | |
| Exercisable at December 31, 2023 | 1,940 | | | $ | 151.37 | | | 3.97 | | $ | 497.6 | |
The aggregate intrinsic value in the table above represents the total pre-tax amount, net of exercise price, that would have been received by option holders if all option holders had exercised all options with an exercise price lower than the market price on the last business day of 2020,2023, which was $233.91$407.84 based on the average of the high and low price of the Company’sour common stock on that date.
The total intrinsic value (the amount by which the fair market value exceeded the exercise price) of stock options exercised during 2020, 20192023, 2022 and 20182021 was $255.0$128.4 million, $325.9$157.2 million and $258.2$43.0 million, respectively. The total cash we received by the Company as a result of employee stock option exercises during 2020, 20192023, 2022 and 20182021 was $228.2$80.8 million, $317.8$144.6 million and $263.4 million, respectively.
The following table summarizes information about stock options outstanding and exercisable at December 31, 2020:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Options Outstanding | | Options Exercisable |
| Range of Exercise Prices | | Number Outstanding | | Weighted-average Remaining Contractual Life | | Weighted-average Exercise Price | | Number Exercisable | | Weighted-average Exercise Price |
| | (in thousands) | | (in years) | | (per share) | | (in thousands) | | (per share) |
| $36.28–$40.00 | | 60 | | | 0.85 | | $ | 38.06 | | | 60 | | | $ | 38.06 | |
| $40.01–$60.00 | | 154 | | | 1.73 | | $ | 47.07 | | | 154 | | | $ | 47.07 | |
| $60.01–$80.00 | | 97 | | | 3.25 | | $ | 74.53 | | | 97 | | | $ | 74.54 | |
| $80.01–$100.00 | | 991 | | | 5.34 | | $ | 88.93 | | | 937 | | $ | 89.07 | |
| $100.01–$120.00 | | 127 | | | 4.07 | | $ | 109.24 | | | 127 | | | $ | 109.22 | |
| $120.01–$140.00 | | 271 | | | 4.71 | | $ | 129.49 | | | 270 | | | $ | 129.50 | |
| $140.01–$160.00 | | 715 | | | 7.05 | | $ | 155.49 | | | 415 | | | $ | 155.42 | |
| $160.01–$180.00 | | 578 | | | 7.47 | | $ | 168.40 | | | 312 | | | $ | 166.01 | |
| $180.01–$200.00 | | 1,222 | | | 7.85 | | $ | 185.31 | | | 499 | | | $ | 184.92 | |
| $200.01–$286.27 | | 23 | | | 9.42 | | $ | 286.27 | | | 23 | | | $ | 286.27 | |
| Total | | 4,238 | | | 6.34 | | $ | 140.47 | | | 2,894 | | | $ | 125.83 | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Restricted Stock Units (excluding PSUs) and Restricted Stockmillion, respectively.
The following table summarizes the restrictedinformation about stock unit and restricted stock activityoptions outstanding as of the Company during the year ended December 31, 2020:2023, which were all exercisable:
| | | | | | | | | | | | | | | | | | | | | | | |
| Restricted Stock Units (excluding PSUs) | | Restricted Stock |
| Number of Shares | | Weighted-average
Grant-date Fair Value | | Number of Units | | Weighted-average Grant-date Fair Value |
| (in thousands) | | (per share) | | (in thousands) | | (per share) |
| Unvested at December 31, 2019 | 3,131 | | | $ | 163.61 | | | 92 | | | $ | 91.97 | |
| Granted | 1,244 | | | $ | 258.45 | | | 0 | | | $ | 0 | |
| Vested | (1,443) | | | $ | 159.10 | | | (92) | | | $ | 91.97 | |
| Cancelled | (210) | | | $ | 193.84 | | | 0 | | | $ | 0 | |
| Unvested at December 31, 2020 | 2,722 | | | $ | 206.99 | | | 0 | | | $ | 0 | |
The total fair value of restricted stock units that vested during 2020, 2019 and 2018 (measured on the date of vesting) was $370.3 million, $178.2 million and $104.8 million, respectively. The total fair value of restricted stock that vested during 2020, 2019 and 2018 (measured on the date of vesting) was $21.4 million, $70.7 million and $114.5 million, respectively.
Performance-based RSUs (PSUs)
The potential range of shares issuable pursuant to the Company’s PSU awards range from 0% to 200% of the target shares based on financial and non-financial measures. NaN percent of PSUs that could be earned have a one-year performance period with the amount actually earned dependent upon the Company’s financial performance and with vesting of the earned shares in three equal installments over a three-year period. The remaining 50% of PSUs that could be earned have a three-year performance period with the amount actually earned dependent upon the achievement of multiple clinical development milestones and with the earned shares cliff vesting at the end of the three-year performance period.
The following table summarizes the PSU activity of the Company during the year ended December 31, 2020:
| | | | | | | | | | | |
| Performance-Based RSU |
| Number of Units | | Weighted-average
Grant-date
Fair Value |
| (in thousands) | | (per share) |
| Unvested at December 31, 2019 (1) | 734 | | | $ | 143.21 | |
| Granted (2) | 547 | | | $ | 241.38 | |
| Vested | (577) | | | $ | 114.11 | |
| Cancelled | (48) | | | $ | 194.46 | |
| Unvested at December 31, 2020 | 656 | | | $ | 202.06 | |
| | | |
| (1) “Unvested” represents the Company’s PSUs at target to the extent performance has not been certified plus the actual number of shares that continue to be subject to service conditions for which the performance has been achieved and certified. |
| (2) “Granted” represents (i) the target number of shares issuable for grants during 2020 and (ii) any change in the number of shares issuable pursuant to outstanding PSUs based on performance certification during 2020. |
The total fair value of PSUs that vested during 2020, 2019 and 2018 (measured on the date of vesting) was $138.5 million, $73.3 million and $23.2 million, respectively. | | | | | | | | | | | | | | | | | | | | |
| | Options Outstanding and Exercisable |
| Range of Exercise Prices | | Number Outstanding | | Weighted-average Remaining Contractual Life | | Weighted-average Exercise Price |
| | (in thousands) | | (in years) | | (per share) |
| $77.31–$100.00 | | 495 | | | 2.58 | | $ | 88.70 | |
| $100.01–$150.00 | | 170 | | | 1.74 | | $ | 123.38 | |
| $150.01–$200.00 | | 1,189 | | | 4.59 | | $ | 172.73 | |
| $200.01–$342.73 | | 86 | | | 7.69 | | $ | 272.22 | |
| Total | | 1,940 | | | 3.97 | | $ | 151.37 | |
Employee Stock Purchase Plan
The Company hasWe have an employee stock purchase plan (the “ESPP”). The ESPP permits eligible employees to enroll in a twelve-month offering period comprising 2two six-month purchase periods. Participants may purchase shares of the Company’sour common stock, through payroll deductions, at a price equal to 85% of the fair market value of the common stock on the first day of the applicable twelve-month offering period, or the last day of the applicable six-month purchase period,
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
whichever is lower. Purchase dates under the ESPP occur on or about May 14 and November 14 of each year. As of December 31, 2020,2023, there were 1,996,3211.3 million shares of common stock authorized for issuance pursuant to the ESPP.
In 2020,2023, the following shares were issued to employees under the ESPP:
| | | | | |
| Year Ended December 31, 2020 |
| (in thousands,
except per share amount)2023 |
| Number of shares (in thousands) | 203,055203 | |
| Average price paid per share | $ | 167.93259.42 | |
Employee Benefits
The Company hasWe have a 401(k) retirement plan (the “Vertex 401(k) Plan”) in which substantially all of itsour permanent U.S. employees are eligible to participate. Participants may contribute up to 60% of their annual compensation to the Vertex 401(k) Plan, subject to statutory limitations. The CompanyWe may declare discretionary matching contributions to the Vertex 401(k) Plan. The Company paysWe pay matching contributions in the form of cash. For the years ended December 31, 2020, 2019In ex-U.S. markets, we have similar benefit plans. In 2023, 2022 and 2018, the Company contributed2021, we recorded approximately $19.2$43.6 million, $15.8$36.4 million and $13.9$29.9 million of expense related to the plan,these plans, respectively.
N.Stock-based Compensation Expense
The Company recognizesWe recognize share-based payments to employees as compensation expense using the fair value method. The fair value of stock options and shares purchased pursuant to the ESPP is calculated using the Black-Scholes option pricing model. The fair value of restricted stock units, including PSUs, and restricted stock is based on the intrinsic value on the date of grant. The fair value of shares purchased pursuant to the ESPP and stock options is calculated using the Black-Scholes option pricing model. Stock-based compensation expense, measured at the grant date based on the fair value of the award, is typically recognized as expense ratably over the requisite service period.
The effect of stock-based compensation expense during the three years ended December 31, 2020 was as follows:
| | | | | | | | | | | | | | | | | |
| 2020 | | 2019 | | 2018 |
| (in thousands) |
| Stock-based compensation expense by line item: | | | | | |
| Cost of sales | $ | 5,579 | | | $ | 5,575 | | | $ | 4,543 | |
| Research and development expenses | 262,690 | | | 224,558 | | | 203,112 | |
| Sales, general and administrative expenses | 161,192 | | | 130,356 | | | 117,392 | |
| Total stock-based compensation expense included in costs and expenses | 429,461 | | | 360,489 | | | 325,047 | |
| Income tax effect | (146,959) | | | (124,225) | | | 0 | |
| Total stock-based compensation included in costs and expenses, net of tax | $ | 282,502 | | | $ | 236,264 | | | $ | 325,047 | |
The Company maintained a valuation allowance on the majority of its NOLs and other deferred tax assets until December 31, 2018. Therefore, there was no “Income tax effect” of stock-based compensation expense for the year ended December 31, 2018.
The stock-based compensation expense by type of award during the three years ended December 31, 2020 was as follows:
| | | | | | | | | | | | | | | | | |
| 2020 | | 2019 | | 2018 |
| (in thousands) |
| Stock-based compensation expense by type of award: | | | | | |
| Restricted stock units (including PSUs) and restricted stock | $ | 360,364 | | | $ | 254,276 | | | $ | 207,845 | |
| Stock options | 59,722 | | | 96,737 | | | 107,854 | |
| ESPP share issuances | 12,984 | | | 11,196 | | | 9,933 | |
| Stock-based compensation expense related to inventories | (3,609) | | | (1,720) | | | (585) | |
| Total stock-based compensation expense included in costs and expenses | $ | 429,461 | | | $ | 360,489 | | | $ | 325,047 | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
The Company capitalizesDuring the three years ended December 31, 2023, we recognized the following stock-based compensation expense:
| | | | | | | | | | | | | | | | | |
| 2023 | | 2022 | | 2021 |
| (in millions) |
| Stock-based compensation expense by type of award: | | | | | |
| Restricted stock units (including PSUs) | $ | 563.7 | | | $ | 456.1 | | | $ | 384.3 | |
| ESPP share issuances | 15.8 | | | 16.7 | | | 24.4 | |
| Stock options | 4.0 | | | 17.6 | | | 36.8 | |
| Stock-based compensation expense related to inventories | (2.3) | | | 0.9 | | | (4.1) | |
| Total stock-based compensation expense included in “Total costs and expenses” | $ | 581.2 | | | $ | 491.3 | | | $ | 441.4 | |
| | | | | |
| Stock-based compensation expense by line item: | | | | | |
| Cost of sales | $ | 7.5 | | | $ | 9.4 | | | $ | 6.3 | |
| Research and development expenses | 354.9 | | | 297.9 | | | 268.3 | |
| Selling, general and administrative expenses | 218.8 | | | 184.0 | | | 166.8 | |
| Total stock-based compensation expense included in “Total costs and expenses” | 581.2 | | | 491.3 | | | 441.4 | |
| Income tax effect | (167.5) | | | (144.1) | | | (82.9) | |
| Total stock-based compensation expense, net of tax | $ | 413.7 | | | $ | 347.2 | | | $ | 358.5 | |
We capitalize a portion of our stock-based compensation expense to inventories, all of which is attributable to employees who support the Company’s manufacturing operations for the Company’sof our products.
The following table sets forth the Company’sour unrecognized stock-based compensation expense as of December 31, 2020,2023, by type of award and the weighted-average period over which that expense is expectedwe expect to be recognized:recognize the expense:
| | | | | | | | | | | |
| As of December 31, 2020 |
| Unrecognized Expense | | Weighted-average Recognition Period |
| (in thousands) | | (in years) |
| Type of award: | | | |
| Restricted stock units (including PSUs) and restricted stock | $ | 401,100 | | | 1.88 |
| Stock options | $ | 62,392 | | | 1.77 |
| ESPP share issuances | $ | 11,333 | | | 0.59 |
Stock Options
In each of the three years ended December 31, 2020, the Company issued stock options to its non-employee directors. In 2019 and 2018, the Company issued stock options with service conditions, which were generally the vesting periods of the awards, to its employees. The Company uses the Black-Scholes option pricing model to estimate the fair value of stock options at the grant date. The Black-Scholes option pricing model uses the option exercise price as well as estimates and assumptions related to the expected price volatility of the Company’s stock, the rate of return on risk-free investments, the expected period during which the options will be outstanding, and the expected dividend yield for the Company’s stock to estimate the fair value of a stock option on the grant date. The options granted during 2020, 2019 and 2018 had a weighted-average grant-date fair value per share of $88.37, $61.32 and $60.83, respectively.
The fair value of each option granted during 2020, 2019 and 2018 was estimated on the date of grant using the Black-Scholes option pricing model with the following weighted-average assumptions:
| | | | | | | | | | | | | | | | | |
| 2020 | | 2019 | | 2018 |
| Stock options granted | 22,636 | | 1,520,743 | | 2,297,328 |
| | | | | |
| Expected stock price volatility | 35.87% | | 36.99% | | 40.50% |
| Risk-free interest rate | 0.43% | | 2.32% | | 2.61% |
| Expected term of options (in years) | 4.67 | | 4.27 | | 4.55 |
| Expected annual dividends | 0 | | 0 | | 0 |
The weighted-average valuation assumptions were determined as follows:
•Expected stock price volatility: Expected stock price volatility is calculated using the trailing one-month average of daily implied volatilities prior to the grant date. Implied volatility is based on options to purchase the Company’s stock with remaining terms of greater than one year that are regularly traded in the market.
•Risk-free interest rate: The Company bases the risk-free interest rate on the interest rate payable on U.S. Treasury securities in effect at the time of grant for a period that is commensurate with the assumed expected option term.
•Expected term of options: The expected term of options represents the period of time options are expected to be outstanding. The Company uses historical data to estimate employee exercise and post-vest termination behavior. The Company believes that all groups of employees exhibit similar exercise and post-vest termination behavior and therefore does not stratify employees into multiple groups in determining the expected term of options.
•Expected annual dividends: The estimate for annual dividends is $0.00 because the Company has not historically paid, and does not intend for the foreseeable future to pay, a dividend.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
| | | | | | | | | | | |
| As of December 31, 2023 |
| Unrecognized Expense | | Weighted-average Recognition Period |
| (in millions) | | (in years) |
| Type of award: | | | |
| Restricted stock units (including PSUs) | $ | 593.6 | | | 1.90 |
| ESPP share issuances | 11.9 | | | 0.61 |
| Total unrecognized stock-based compensation expense | $ | 605.5 | | | |
Restricted Stock Units and Performance-based Restricted Stock Units
The Company awardsWe award restricted stock units with service conditions, which are generally the vesting periods of the awards.
As described in Note M, “Common Stock, Preferred Stock and Equity Plans,” we grant the majority of December 31, 2020, the Company did not have any unvested restricted stock awards remaining, which it awarded until 2016.
The Company grantsour PSUs to certain members of senior management. Half of theOur financial-based PSUs contain financial goals as the performance metric and the other half contain non-financial goals. A target number of shares is established for each award, however the actual number of shares that are issued when an award vests may range from 0 to 200% of the target amount depending upon the level of achievement of the applicable performance metric. The financial-based PSUssenior management vest in three equal installments over a three-year period and are expensed ratably over that same period based upon an assessment of the likely level of achievement. TheOur non-financial based PSUs to senior management cliff vest at the end of the three-year performance period and are expensed on a straight-line basis over that same period based upon an assessment of the likely level of achievement.
Employee Stock Purchase Plan
The weighted-average fair value of each purchase right granted during 2020, 20192023, 2022 and 20182021 was $65.88, $47.79$90.91, $79.36 and $44.04,
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
$51.71, respectively. The following table reflects the weighted-average assumptions used in the Black-Scholes option pricing model for 2020, 20192023, 2022 and 2018:2021:
| | 2020 | | 2019 | | 2018 |
| 2023 | | | 2023 | | 2022 | | 2021 |
| Expected stock price volatility | Expected stock price volatility | 37.70% | | 33.43% | | 36.51% | Expected stock price volatility | 28.52% | | 33.55% | | 34.06% |
| Risk-free interest rate | Risk-free interest rate | 0.11% | | 2.08% | | 2.36% | Risk-free interest rate | 5.13% | | 4.05% | | 0.05% |
| Expected term (in years) | Expected term (in years) | 0.71 | | 0.74 | | 0.75 | Expected term (in years) | 0.71 | | 0.71 | | 0.69 |
| Expected annual dividends | Expected annual dividends | 0 | | 0 | | 0 | Expected annual dividends | — | | — |
The expectedStock Options
We issued stock price volatility for ESPP offerings is based on implied volatility. The Company basesoptions to our non-employee directors with total grant date fair values of $1.8 million in each of 2023, 2022 and 2021. We use the risk-free interest rate onBlack-Scholes option pricing model to estimate the interest rate payable on U.S. Treasury securities in effectfair value of stock options at the timegrant date. The options granted during 2023, 2022 and 2021 had a weighted-average grant-date fair value per share of grant for a period that is commensurate with the assumed expected term. The expected term represents purchases$114.92, $89.65 and purchase periods that take place within the offering period. The expected annual dividends estimate is $0.00 because the Company has not historically paid, and does not for the foreseeable future intend to pay, a dividend.$65.94, respectively.
O.Income Taxes
We are subject to U.S. federal, state, and foreign income taxes. The components of income before provision for (benefit from) income taxes during the three years ended December 31, 20202023, consisted of the following:
| | 2020 | | 2019 | | 2018 |
| (in thousands) |
| 2023 | | | 2023 | | 2022 | | 2021 |
| (in millions) | | | (in millions) |
| United States | United States | $ | 2,885,423 | | | $ | 1,263,379 | | | $ | 812,086 | |
| Foreign | Foreign | 231,375 | | | 131,540 | | | (211,845) | |
| Income before provision for (benefit from) income taxes | $ | 3,116,798 | | | $ | 1,394,919 | | | $ | 600,241 | |
| Income before provision for income taxes | |
The components of our provision for income taxes during the three years ended December 31, 2023, consisted of the following:
| | | | | | | | | | | | | | | | | |
| 2023 | | 2022 | | 2021 |
| (in millions) |
| Current taxes: | | | | | |
| Federal | $ | 900.4 | | | $ | 779.0 | | | $ | 374.9 | |
| State | 46.2 | | | 34.9 | | | 26.5 | |
| Foreign | 350.1 | | | 372.4 | | | 141.5 | |
| Total current taxes | 1,296.7 | | | 1,186.3 | | | 542.9 | |
| Deferred taxes: | | | | | |
| Federal | (569.9) | | | (404.0) | | | (36.9) | |
| State | (21.9) | | | (11.0) | | | (19.3) | |
| Foreign | 55.3 | | | 139.1 | | | (98.4) | |
| Total deferred taxes | (536.5) | | | (275.9) | | | (154.6) | |
| Provision for income taxes | $ | 760.2 | | | $ | 910.4 | | | $ | 388.3 | |
Unremitted Earnings
As of December 31, 2023, we do not consider a portion of the earnings of our foreign subsidiaries to be indefinitely reinvested. Upon repatriation of the non-indefinitely invested earnings in the form of distributions or otherwise, we could be
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
subject to immaterial U.S. federal withholding taxes payable to various foreign countries and income taxes in certain states. There are no material deferred taxes recorded on the excess of financial statement reporting over the tax basis of our investments in our foreign subsidiaries. Any permanently reinvested basis differences could reverse if we sell our foreign subsidiaries or various other events occur, none of which were considered probable as of December 31, 2023. The tax liabilities described above would not be material to our consolidated financial statements.
Effective Tax Rate Reconciliation
A reconciliation between the U.S. federal statutory rate of 21% and our effective tax rate was as follows:
| | | | | | | | | | | | | | | | | |
| 2023 | | 2022 | | 2021 |
| Federal statutory tax rate | 21.0 | % | | 21.0 | % | | 21.0 | % |
| State taxes, net of federal benefit | 0.3 | % | | 0.6 | % | | 0.8 | % |
| Foreign income tax rate differential | (0.6) | % | | (0.3) | % | | (0.3) | % |
| U.S. tax on foreign earnings, net of credits | 0.7 | % | | 1.9 | % | | 0.7 | % |
| Foreign derived intangible income deduction | (1.7) | % | | (1.4) | % | | (0.8) | % |
| Tax credits | (6.0) | % | | (2.2) | % | | (6.4) | % |
| Tax rate change | 0.0 | % | | 0.0 | % | | (3.5) | % |
| Stock compensation (benefit), shortfalls and cancellations | (0.8) | % | | (0.8) | % | | 0.0 | % |
| Uncertain tax positions | 3.4 | % | | 2.7 | % | | 2.0 | % |
| Other | 1.1 | % | | 0.0 | % | | 0.7 | % |
| Effective tax rate | 17.4 | % | | 21.5 | % | | 14.2 | % |
Our 17.4% effective tax rate for 2023 was lower than the U.S. statutory rate primarily due to a benefit from a research and development tax credit study that was completed in 2023 and excess tax benefits related to stock-based compensation, partially offset by changes in uncertain tax positions.
Our 21.5% effective tax rate for 2022 was higher than the U.S. statutory rate primarily due to an increase in our uncertain tax positions associated with intercompany transfer pricing matters offset by excess tax benefits related to stock-based compensation, tax credits and changes in our estimated prior-year tax liabilities.
Our 14.2% effective tax rate for 2021 was lower than the U.S. statutory rate primarily due to an increase in the U.K.’s corporate tax rate from 19% to 25%, which was enacted in 2021 and became effective in April 2023, and benefit from a research and development tax credit study that we completed in 2021.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
The components of the provision for (benefit from) income taxes during the three years ended December 31, 2020 consisted of the following:
| | | | | | | | | | | | | | | | | |
| 2020 | | 2019 | | 2018 |
| (in thousands) |
| Current taxes: | | | | | |
| Federal | $ | 71,377 | | | $ | 0 | | | $ | 772 | |
| Foreign | 37,582 | | | 37,194 | | | 15,600 | |
| State | 18,851 | | | 13,528 | | | 9,018 | |
| Total current taxes | 127,810 | | | 50,722 | | | 25,390 | |
| Deferred taxes: | | | | | |
| Federal | 510,220 | | | 184,312 | | | (1,105,053) | |
| Foreign | (239,579) | | | (24,797) | | | (364,919) | |
| State | 6,700 | | | 7,872 | | | (42,280) | |
| Total deferred taxes | 277,341 | | | 167,387 | | | (1,512,252) | |
| Provision for (benefit from) income taxes | $ | 405,151 | | | $ | 218,109 | | | $ | (1,486,862) | |
A reconciliation of the provision for (benefit from) income taxes as computed by applying the U.S. federal statutory rate of 21% for the three years ended December 31, 2020 to the provision for (benefit from) income taxes is as follows:
| | | | | | | | | | | | | | | | | |
| 2020 | | 2019 | | 2018 |
| (in thousands) |
| Income before provision for (benefit from) income taxes | $ | 3,116,798 | | | $ | 1,394,919 | | | $ | 600,241 | |
| | | | | |
| Expected provision for income taxes | 654,528 | | | 292,933 | | | 126,051 | |
| State taxes, net of federal benefit | 18,596 | | | 8,478 | | | 8,680 | |
| Foreign income tax rate differential | 5,433 | | | 6,178 | | | 23,427 | |
| Tax credits | (55,824) | | | (59,459) | | | (52,629) | |
| Benefit from income taxes attributable to valuation allowances | 17,384 | | | (2,672) | | | (1,563,169) | |
| Tax rate change | (37,688) | | | 0 | | | 0 | |
| Stock compensation (benefit), shortfalls and cancellations | (70,628) | | | (56,324) | | | (49,044) | |
| Officer’s compensation | 13,079 | | | 10,666 | | | 8,310 | |
| Long-term intercompany receivable write-off | (53,821) | | | 0 | | | 0 | |
| Deconsolidation of VIE | 0 | | | 0 | | | (9,390) | |
| Uncertain tax positions | 39,808 | | | 14,070 | | | 15,431 | |
| Inter-entity transfer of intellectual property rights | (209,000) | | | 0 | | | 0 | |
| U.S. deemed dividend | 71,727 | | | 79 | | | 64 | |
| Other | 11,557 | | | 4,160 | | | 5,407 | |
| Provision for (benefit from) income taxes | $ | 405,151 | | | $ | 218,109 | | | $ | (1,486,862) | |
The Company is subject to U.S. federal, state,Deferred Tax Assets and foreign income taxes. The Company’s provision for income taxes during the years ended December 31, 2020 and 2019, has increased compared to historical amounts due to the release of the Company’s valuation allowance on the majority of its net operating losses (“NOLs”) and other deferred tax assets as of December 31, 2018. Starting in 2019, the Company began recording a provision for income taxes approximating the statutory rates on its pre-tax income. The Company’s effective tax rate of 13% for 2020 was lower than the U.S. statutory rate primarily due to (i) discrete tax benefits associated with an intra-entity transfer of intellectual property rights to the United Kingdom (“U.K.”), the write-off of a long-term intercompany receivable, and an increase in the U.K.’s corporate tax rate; and (ii) excess tax benefits related to stock-based compensation. The impact of these items was partially offset by a U.S. deemed dividend. The Company’s effective tax rate of 16% for 2019 was lower than the U.S. statutory rate primarily due to excess tax benefits related to stock-based compensation and research and development tax credits. The Company utilized substantially all of its remaining previously benefited U.S. NOLs in 2020. As a result, a larger portion of the Company’s tax provision will represent a cash tax payable in future periods.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
In the second quarter of 2020, the Company completed an intra-entity transfer of intellectual property rights to the U.K., resulting in a deferred tax benefit of $209.0 million. The Company expects to be able to utilize the deferred tax asset resulting from the intra-entity transfer.
In 2018, the change in the “Benefit from income taxes attributable to valuation allowances” was primarily related to the release of the Company’s valuation allowances on the majority of its NOLs and other deferred tax assets related to the U.S. and the U.K. The Company released these valuation allowances after determining it was more likely than not that its deferred tax assets would be realized in the future based on positive evidence, including significant cumulative consolidated and U.S. income over the three years ended December 31, 2018, revenue growth, clinical trial data from the Company’s triple combination regimens, competitor clinical progress and expectations regarding future profitability, and negative evidence, including the potential impact of competition on the Company’s projections and cumulative losses in one of the jurisdictions.Liabilities
Deferred tax assets and liabilities are determined based on the difference between financial statement and tax bases using enacted tax rates in effect for the year in which the differences are expected to reverse. The components of the deferred taxes were as follows:
| | As of December 31, |
| 2020 | | 2019 |
| (in thousands) |
| As of December 31, | | | As of December 31, |
| 2023 | | | 2023 | | 2022 |
| (in millions) | | | (in millions) |
| Deferred tax assets: | Deferred tax assets: | |
| Net operating loss | $ | 140,563 | | | $ | 512,256 | |
| Tax credit carryforwards | |
| Tax credit carryforwards | |
| Tax credit carryforwards | Tax credit carryforwards | 406,120 | | | 549,543 | |
| Intangible assets | Intangible assets | 507,484 | | | 275,290 | |
| Deferred revenues | 20,962 | | | 18,833 | |
| Stock-based compensation | Stock-based compensation | 89,203 | | | 85,199 | |
| Accrued expenses | 47,326 | | | 44,367 | |
| Finance lease liabilities | Finance lease liabilities | 118,749 | | | 119,160 | |
| Operating lease assets | Operating lease assets | 65,046 | | | 13,114 | |
| R&D capitalization | |
| Other | Other | 987 | | | 8,596 | |
| Gross deferred tax assets | Gross deferred tax assets | 1,396,440 | | | 1,626,358 | |
| Valuation allowance | Valuation allowance | (213,750) | | | (205,192) | |
| Total deferred tax assets | Total deferred tax assets | 1,182,690 | | | 1,421,166 | |
| Deferred tax liabilities: | Deferred tax liabilities: | |
| Property and equipment | Property and equipment | (116,955) | | | (101,235) | |
| Property and equipment | |
| Property and equipment | |
| Acquired intangibles | Acquired intangibles | (87,025) | | | (87,160) | |
| Unrealized gain | (17,553) | | | (28,838) | |
| Operating lease liabilities | Operating lease liabilities | (63,310) | | | (13,118) | |
| Other | Other | (15,068) | | | 0 | |
| Total deferred tax liabilities | |
| Net deferred tax assets | Net deferred tax assets | $ | 882,779 | | | $ | 1,190,815 | |
On a periodic basis, the Company reassesseswe reassess the valuation allowance on itsour deferred income tax assets, weighing positive and negative evidence to assess the recoverability of theour deferred tax assets. As of December 31, 2020, the Company2023, we maintained a valuation allowance of $213.8$266.6 million related primarily to U.S. state and foreign tax attributes.
In addition to deferred tax assets and liabilities, we have recorded deferred charges related to intra-entity sales of inventory. As of December 31, 2023 and 2022, the total deferred charges were $185.8 million and $195.1 million, respectively.
As of December 31, 2020, the Company2023, we had NOLnet operating loss (“NOL”) carryforwards of $97.4$53.1 million, which are subject to annual utilization limitations and tax credits of $249.2 million for U.S. federal income tax purposes. AsIn 2027, our definite lived U.S. federal NOLs of December 31, 2020,$18.2 million will begin to expire, while the Companyremaining portion may be carried forward indefinitely. For U.S. state income tax purposes, we had NOL carryforwards of $708.2$525.5 million and tax creditscredit carryforwards of $179.1 million for U.S.$334.8 million. The state NOL and tax credit carryforwards begin to expire in 2024. For foreign income tax purposes.purposes, we had NOL carryforwards of $113.2 million and tax credit carryforwards of $20.6 million. The U.S. federal NOLsforeign NOL carryforwards may be carried forward indefinitely, with the exception of $29.1$38.2 million that will expire in 2041. The foreign tax credit carryforwards will begin to expire in 2030. The state NOL carryforwards2024.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Unrecognized Tax Benefits
Unrecognized tax benefits during the three years ended December 31, 2023 were as follows:
| | | | | | | | | | | | | | | | | |
| 2023 | | 2022 | | 2021 |
| (in millions) |
| Balance at beginning of the period | $ | 459.6 | | | $ | 147.2 | | | $ | 86.6 | |
| Increases related to current period tax positions | 116.0 | | | 128.3 | | | 42.0 | |
| Increases related to prior period tax positions | 62.5 | | | 205.3 | | | 19.9 | |
| Decreases related to prior period tax positions | (14.4) | | | (14.4) | | | — | |
| Statute of limitations expiration | (8.1) | | | (4.5) | | | (1.3) | |
| Foreign currency translation adjustment | 0.3 | | | (2.3) | | | — | |
| Balance at end of period | $ | 615.9 | | | $ | 459.6 | | | $ | 147.2 | |
During 2023, we increased our gross unrecognized tax benefits by $156.3 million, primarily associated with intercompany transfer pricing matters. This unrecognized tax benefit was recorded as a $3.7 million increase to our gross deferred tax assets and a $160.0 million gross tax credits expire at various dates through 2040liability.
During 2022, we increased our gross unrecognized tax benefits by $312.4 million, primarily associated with intercompany transfer pricing matters. This unrecognized tax benefit was recorded as a $29.7 million reduction to our gross deferred tax assets and may be used to offset future state incomea $282.7 million gross tax liabilities. liability.
As of December 31, 2020,2023, we have classified $47.9 million and $568.0 million of our unrecognized tax benefits as credits to “Deferred tax assets” and “Other long-term liabilities,” respectively, on our consolidated balance sheet.
Included in our unrecognized tax benefits as of December 31, 2023, 2022 and 2021, we had $288.7 million, $208.5 million and $129.5 million (net of the Company hadfederal benefit on state issues), respectively, of unrecognized tax benefits, which would affect our effective income tax rate if recognized.
We recognize potential interest and penalties related to unrecognized tax benefits in our provision for income taxes. In 2023 and 2022, we recognized total interest and penalty expenses of $84.9 million and $36.6 million, respectively. Our total interest and penalty expenses in 2021 was not material to our consolidated financial statements. As of December 31, 2023 and 2022, our accrual for interest and penalties was $124.1 million and $39.2 million, respectively.
The U.S. Internal Revenue Service and other local and foreign net operating loss carryforwardstax authorities routinely examine our tax returns, including intercompany transfer pricing, and it is reasonably possible that we will adjust the value of $521.6 million,our uncertain tax positions related these matters and other issues as we receive additional information from various taxing authorities, including $10.6 millionreaching settlements with such authorities. In the case of intercompany transfer pricing, it is reasonably possible that weretaxing authorities do not agree with each other on the reallocation of income or the valuation of intellectual property, in which case we could be subject to expirationdouble taxation, despite bilateral treaty agreements available to prevent this. During the year ended December 31, 2023, we came to settlement with the U.K.’s HM Revenue & Customs (“HMRC”) with respect to our tax positions for 2015 through 2020. Due to the nature of the adjustments, we will be asserting our rights under the U.S./U.K. Income Tax Convention pursuant to the mutual agreement procedures for the relief of double taxation for these matters.
As a result of various audit closures, settlements and statutes of limitations, we estimate that it is reasonably possible that our gross unrecognized tax benefits could decrease by up to $81.8 million in the next 12 months due to statute of limitations expirations.
We file U.S. federal income tax returns and income tax returns in various state, local and foreign jurisdictions. We have various income tax audits ongoing at various dates through 2040 and $511.0 millionany time throughout the world. Except for jurisdictions where we have NOLs or tax credit carryforwards, we are no longer subject to any tax assessment from tax authorities for years prior to 2014 in jurisdictions that had an indefinite carryforward period.have a material impact on our consolidated financial statements.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Unrecognized tax benefits during the three years ended December 31, 2020 were as follows:
| | | | | | | | | | | | | | | | | |
| 2020 | | 2019 | | 2018 |
| (in thousands) |
| Balance at beginning of the period | $ | 33,920 | | | $ | 19,549 | | | $ | 3,814 | |
| Increases related to current period tax positions | 26,653 | | | 14,407 | | | 9,704 | |
| Increases related to prior period tax positions | 26,732 | | | 598 | | | 6,031 | |
| Decreases related to prior period tax positions | 0 | | | (156) | | | 0 | |
| Settlement with tax authorities | 0 | | | (478) | | | 0 | |
| Statute of limitations expiration | (657) | | | 0 | | | 0 | |
| Balance at end of period | $ | 86,648 | | | $ | 33,920 | | | $ | 19,549 | |
As of December 31, 2020, the Company has classified $48.1 million and $38.5 million of its unrecognized tax benefits as credits to “Deferred tax assets” and “Accrued expenses,” respectively, on its consolidated balance sheet.
The Company has reviewed the tax positions taken, or to be taken, in its tax returns for all tax years currently open to examination by a taxing authority. Unrecognized tax benefits represent the aggregate tax effect of differences between tax return positions and the benefits recognized in the consolidated financial statements. As of December 31, 2020, 2019 and 2018, the Company had $75.8 million, $33.9 million and $19.5 million, respectively, of net unrecognized tax benefits, which would affect the Company’s tax rate if recognized. The Company does not expect that its unrecognized tax benefits will materially change within the next twelve months. The Company accrues interest and penalties related to unrecognized tax benefits as a component of its “Provision for (benefit from) income taxes.” The Company did not recognize any material interest or penalties related to uncertain tax positions during the three years ended December 31, 2020.
In March 2020, the Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) was signed into law. The CARES Act includes provisions relating to several aspects of corporate income taxes. The CARES Act did not have a significant impact on the Company’s provision for income taxes.
As of December 31, 2020, foreign earnings, which were not significant, have been retained by the Company’s foreign subsidiaries for indefinite reinvestment. Upon repatriation of those earnings, in the form of dividends or otherwise, the Company could be subject to withholding taxes payable to the various foreign countries.
The Company files U.S. federal income tax returns and income tax returns in various state, local and foreign jurisdictions. The Company is no longer subject to any tax assessment from an income tax examination in the U.S. or any other major taxing jurisdiction for years before 2011, except where the Company has NOLs or tax credit carryforwards that originate before 2011. The Company has various income tax audits ongoing at any time throughout the world.
P.Commitments and Contingencies
Revolving2022 Credit FacilitiesFacility
The CompanyIn July 2022, Vertex and certain of its subsidiaries have entered into 2 credit agreementsa $500.0 million unsecured revolving facility (the “Credit Agreements”Agreement”) with Bank of America, N.A., as administrative agent and the lenders referred to therein (the “Lenders”)., which matures on July 1, 2027. The Credit Agreements wereAgreement was not drawn upon at closing and the Company haswe have not drawn upon themit to date. Amounts drawn pursuant to the Credit Agreements,Agreement, if any, will be used for general corporate purposes. Subject to satisfaction of certain conditions, we may request that the borrowing capacity for the Credit Agreement be increased by an additional $500.0 million. Additionally, the Credit Agreement provides a sublimit of $100.0 million for letters of credit.
Any amounts borrowed under the Credit AgreementsAgreement will bear interest, at the Company’sour option, at either a base rate or a Eurocurrency rate,Secured Overnight Financing Rate (“SOFR”), in each case plus an applicable marginmargin. Under the Credit Agreement, the applicable margins on base rate loans range from 0.000% to 0.500% and the applicable margins on SOFR loans range from 1.000% to 1.500%, in each case based on the Company’sour consolidated leverage ratio (the ratio of the Company’sour total consolidated funded indebtedness to the Company’sour consolidated EBITDA for the most recently completed four fiscal quarter period).
In September 2019, the Company and certain of its subsidiaries entered into a $500.0 million unsecured revolving facility (the “2019 Credit Agreement”) with the Lenders, which matures on September 17, 2024. The 2019 Credit Agreement superseded the Company’s credit agreement entered into in 2016 with Bank of America, N.A serving in the same capacity. Under the 2019 Credit Agreement, the applicable margins on base rate loans range from 0.125% to 0.500% and the
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
applicable margins on Eurocurrency loans range from 1.125% to 1.500%. The 2019 Credit Agreement provides a sublimit of $50.0 million for letters of credit.
In September 2020, the Company and certain of its subsidiaries entered into a $2.0 billion unsecured revolving facility (the “2020 Credit Agreement”) with the Lenders, which matures on September 18, 2022. Under the 2020 Credit Agreement, the applicable margins on base rate loans range from 0.500% to 0.875% and the applicable margins on Eurocurrency loans range from 1.500% to 1.875%. The 2020 Credit Agreement does not support the issuance of letters of credit.
Subject to satisfaction of certain conditions, the Company may request that the borrowing capacity for each of the Credit Agreements be increased by an additional $500.0 million. Any amounts borrowed pursuant to the Credit AgreementsAgreement are guaranteed by certain of the Company’sour existing and future domestic subsidiaries, subject to certain exceptions.
The Credit Agreements containAgreement contains customary representations and warranties and affirmative and negative covenants, including a financial covenantscovenant to maintain (x) subject to certain limited exceptions, a consolidated leverage ratio of 3.50 to 1.00, subject to an increase to 4.00 to 1.00 following a material acquisition and (y) a consolidated interest coverage ratio of 2.50 to 1.00, in each case measured on a quarterly basis.acquisition. As of December 31, 2020, the Company was2023, we were in compliance with the covenants described above. The Credit AgreementsAgreement also containcontains customary events of default. In the case of a continuing event of default, the administrative agent would be entitled to exercise various remedies, including the acceleration of amounts due under outstanding loans.
Direct costs related to the Credit Agreements, whichAgreement are recorded over its term and were not material to the Company’sour financial statements, were deferred and recorded over the term of the Credit Agreements.statements.
Guaranties and Indemnifications
As permitted under Massachusetts law, the Company’sour Articles of Organization and By-laws provide that the Companywe will indemnify certain of itsour officers and directors for certain claims asserted against them in connection with their service as an officer or director. The maximum potential amount of future payments that the Companywe could be required to make under these indemnification provisions is unlimited. However, the Company haswe have purchased directors’ and officers’ liability insurance policies that could reduce itsour monetary exposure and enable itus to recover a portion of any future amounts paid. NaNNo indemnification claims currently are outstanding, and the Company believeswe believe the estimated fair value of these indemnification arrangements is minimal.
The CompanyWe customarily agreesagree in the ordinary course of itsour business to indemnification provisions in agreements with clinical trial investigators and sites in its drugour product development programs, sponsored research agreements with academic and not-for-profit institutions, various comparable agreements involving parties performing services for the Company,us, and itsour real estate leases. The CompanyWe also customarily agreesagree to certain indemnification provisions in itsour drug discovery, development and commercialization collaboration agreements. With respect to the Company’sour clinical trials and sponsored research agreements, these indemnification provisions typically apply to any claim asserted against the investigator or the investigator’s institution relating to personal injury or property damage, violations of law or certain breaches of the Company’sour contractual obligations arising out of the research or clinical testing of the Company’sour compounds or drugproduct candidates. With respect to lease agreements, the indemnification provisions typically apply to claims asserted against the landlord relating to personal injury or property damage caused by the Company,us, to violations of law by the Companyus or to certain breaches of the Company’sour contractual obligations. The indemnification provisions appearing in the Company’sour collaboration agreements are similar to those for the other agreements discussed above, but in addition provide some limited indemnification for itsour collaborator in the event of third-party claims alleging infringement of intellectual property rights. In each of the cases above, the indemnification obligation generally survives the termination of the agreement for some extended period, although the Company believeswe believe the obligation typically has the most relevance during the contract term and for a short period of time thereafter. The maximum potential amount of future payments that the Companywe could be required to make under these provisions is generally unlimited. The Company hasWe have purchased insurance policies covering personal injury, property damage and
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
general liability that reduce itsour exposure for indemnification and would enable itus in many cases to recover all or a portion of any future amounts paid. The Company hasWe have never paid any material amounts to defend lawsuits or settle claims related to these indemnification provisions. Accordingly, the Company believeswe believe the estimated fair value of these indemnification arrangements is minimal.
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Other Contingencies
The Company hasWe have certain contingent liabilities that arise in the ordinary course of itsour business activities. The Company accrues a reserveWe accrue for such contingent liabilities when it is probable that future expenditures will be made and such expenditures can be reasonably estimated. ThereOther than our contingent consideration liabilities discussed in Note D, “Fair Value Measurements,” there were 0no material contingent liabilities accrued as of December 31, 20202023 or 2019.2022.
Q.Segment Information
Segment reporting is prepared on the same basis that the Company’sour chief executive officer, who is the Company’sour chief operating decision maker, manages the business, makes operating decisions and assesses performance. The Company operatesWe operate in 1one segment, pharmaceuticals. Enterprise-wide disclosures about revenues, significant customers, and property and equipment, net by location are presented below.
Revenues by Product
Product revenues, net consisted of the following:
| | 2020 | | 2019 | | 2018 |
| (in thousands) |
| 2023 | | | 2023 | | 2022 | | 2021 |
| (in millions) | | | (in millions) |
| TRIKAFTA/KAFTRIO | TRIKAFTA/KAFTRIO | $ | 3,863,824 | | | $ | 420,105 | | | $ | 0 | |
| KALYDECO | |
| ORKAMBI | |
| SYMDEKO/SYMKEVI | SYMDEKO/SYMKEVI | 628,577 | | | 1,417,668 | | | 768,657 | |
| ORKAMBI | 907,512 | | | 1,331,891 | | | 1,262,166 | |
| KALYDECO | 802,870 | | | 991,062 | | | 1,007,502 | |
| Total product revenues, net | Total product revenues, net | $ | 6,202,783 | | | $ | 4,160,726 | | | $ | 3,038,325 | |
Product Revenues by Geographic Location
Net product revenues are attributed to countries based on the location of the customer. Collaborativecustomer and royalty revenues are attributed to countries based on the location of the Company’s subsidiary associated with the collaborative arrangement related to such revenues. Total revenues from external customers and collaborators by geographic region consisted of the following:
| | 2020 | | 2019 | | 2018 |
| (in thousands) |
| 2023 | | | 2023 | | 2022 | | 2021 |
| (in millions) | | | (in millions) |
| United States | United States | $ | 4,829,282 | | | $ | 3,062,555 | | | $ | 2,365,079 | |
| Outside of the United States | Outside of the United States | |
| Europe | Europe | 1,126,460 | | | 885,762 | | | 543,179 | |
| Europe | |
| Europe | |
| Other | Other | 249,941 | | | 214,504 | | | 139,339 | |
| Total revenues outside of the United States | 1,376,401 | | | 1,100,266 | | | 682,518 | |
| Total revenues | $ | 6,205,683 | | | $ | 4,162,821 | | | $ | 3,047,597 | |
| Total product revenues outside of the United States | |
| Total product revenues, net | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
Significant Customers
Gross product revenues and accounts receivable from each of the Company’sour customers who individually accounted for 10% or more of total gross product revenues and/or 10% or more of total accounts receivable consisted of the following:
| | Percent of Total Gross Product Revenues | | Percent of Accounts Receivable |
| Year Ended December 31, | | As of December 31, |
| 2020 | | 2019 | | 2018 | | 2020 | | 2019 |
| Percent of Total Gross Product Revenues | | | Percent of Total Gross Product Revenues | | Percent of Accounts Receivable |
| Year Ended December 31, | | | Year Ended December 31, | | As of December 31, |
| 2023 | | | 2023 | | 2022 | | 2021 | | 2023 | | 2022 |
| McKesson Corporation | McKesson Corporation | 20 | % | | 17 | % | | 14 | % | | 14 | % | | 22 | % | McKesson Corporation | 26 | % | | 25 | % | | 22 | % | | 23 | % | | 22 | % |
| Accredo/Curascript | 15 | % | | 14 | % | | 14 | % | | 10 | % | | 15 | % |
| Accredo Health Group, Inc. | | Accredo Health Group, Inc. | 11 | % | | 12 | % | | 12 | % | | <10% |
| Walgreen Co. | Walgreen Co. | 14 | % | | 15 | % | | 20 | % | | 10 | % | | 14 | % | Walgreen Co. | <10% | | 10 | % | | 10 | % | | <10% |
| Lloyds Pharmacy* | Lloyds Pharmacy* | <10% | | <10% | | <10% | | 19 | % | | <10% | Lloyds Pharmacy* | <10% | | <10% | | 10 | % | | 12 | % |
*A wholly-owned subsidiary of McKesson Corporation in the U.K. | | *A wholly owned subsidiary of McKesson Corporation in the U.K. until 2022. | | *A wholly owned subsidiary of McKesson Corporation in the U.K. until 2022. |
Long-lived Assets by Location
Long-lived assets by location consisted of the following:
| | As of December 31, |
| 2020 | | 2019 |
| (in thousands) |
| As of December 31, | | | As of December 31, |
| 2023 | | | 2023 | | 2022 |
| (in millions) | | | (in millions) |
| United States | United States | $ | 1,207,748 | | | $ | 768,572 | |
| Outside of the United States | Outside of the United States | |
| United Kingdom | |
| United Kingdom | |
| United Kingdom | United Kingdom | 61,462 | | | 57,383 | |
| Other | Other | 14,888 | | | 7,327 | |
| Total long-lived assets outside of the United States | Total long-lived assets outside of the United States | 76,350 | | | 64,710 | |
| Total long-lived assets | Total long-lived assets | $ | 1,284,098 | | | $ | 833,282 | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
R.Quarterly Financial Data (unaudited)
The following tables set forth the Company’s quarterly financial data for the two years ended December 31, 2020:
| | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended |
| March 31,
2020 | | June 30,
2020 | | September 30,
2020 | | December 31,
2020 |
| (in thousands, except per share amounts) |
| Revenues: | | | | | | | |
| Product revenues, net | $ | 1,515,107 | | | $ | 1,524,485 | | | $ | 1,536,271 | | | $ | 1,626,920 | |
| Collaborative and royalty revenues | 0 | | | 0 | | | 2,000 | | | 900 | |
| Total revenues | 1,515,107 | | | 1,524,485 | | | 1,538,271 | | | 1,627,820 | |
| Costs and expenses: | | | | | | | |
| Cost of sales | 162,497 | | | 184,520 | | | 186,182 | | | 203,101 | |
| Research and development expenses (1) | 448,528 | | | 420,928 | | | 493,497 | | | 466,584 | |
| Sales, general and administrative expenses | 182,258 | | | 191,804 | | | 184,551 | | | 211,843 | |
| Change in fair value of contingent consideration | 1,600 | | | 9,200 | | | 1,800 | | | 500 | |
| Total costs and expenses | 794,883 | | | 806,452 | | | 866,030 | | | 882,028 | |
| Income from operations | 720,224 | | | 718,033 | | | 672,241 | | | 745,792 | |
| Interest income | 12,576 | | | 4,243 | | | 3,100 | | | 2,320 | |
| Interest expense | (14,136) | | | (13,871) | | | (13,856) | | | (16,288) | |
| Other (expense) income, net (2) | (61,130) | | | 116,365 | | | 84,386 | | | 156,799 | |
| Income before provision for income taxes | 657,534 | | | 824,770 | | | 745,871 | | | 888,623 | |
| Provision for (benefit from) income taxes (3) | 54,781 | | | (12,500) | | | 78,437 | | | 284,433 | |
| Net income attributable to Vertex | $ | 602,753 | | | $ | 837,270 | | | $ | 667,434 | | | $ | 604,190 | |
| | | | | | | |
| Amounts per share attributable to Vertex common shareholders: | | | | | | | |
| Net income: | | | | | | | |
| Basic | $ | 2.32 | | | $ | 3.22 | | | $ | 2.56 | | | $ | 2.32 | |
| Diluted | $ | 2.29 | | | $ | 3.18 | | | $ | 2.53 | | | $ | 2.30 | |
| Shares used in per share calculations: | | | | | | | |
| Basic | 259,815 | | | 259,637 | | | 260,392 | | | 260,038 | |
| Diluted | 263,515 | | | 263,403 | | | 264,079 | | | 263,106 | |
VERTEX PHARMACEUTICALS INCORPORATED
Notes to Consolidated Financial Statements (Continued)
| | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended |
| March 31,
2019 | | June 30,
2019 | | September 30,
2019 | | December 31,
2019 |
| (in thousands, except per share amounts) |
| Revenues: | | | | | | | |
| Product revenues, net (4) | $ | 857,253 | | | $ | 940,380 | | | $ | 949,828 | | | $ | 1,413,265 | |
| Collaborative and royalty revenues | 1,182 | | | 913 | | | 0 | | | 0 | |
| Total revenues | 858,435 | | | 941,293 | | | 949,828 | | | 1,413,265 | |
| Costs and expenses: | | | | | | | |
| Cost of sales | 95,092 | | | 135,740 | | | 131,914 | | | 185,012 | |
| Research and development expenses (1) | 339,490 | | | 379,091 | | | 555,948 | | | 480,011 | |
| Sales, general and administrative expenses | 147,045 | | | 156,502 | | | 159,674 | | | 195,277 | |
| Change in fair value of contingent consideration | 0 | | | 0 | | | 2,959 | | | 1,500 | |
| Total costs and expenses | 581,627 | | | 671,333 | | | 850,495 | | | 861,800 | |
| Income from operations | 276,808 | | | 269,960 | | | 99,333 | | | 551,465 | |
| Interest income | 15,615 | | | 18,076 | | | 17,628 | | | 12,359 | |
| Interest expense | (14,868) | | | (14,837) | | | (14,548) | | | (14,249) | |
| Other income (expense), net (2) | 42,610 | | | 53,939 | | | (31,747) | | | 127,375 | |
| Income before provision for income taxes | 320,165 | | | 327,138 | | | 70,666 | | | 676,950 | |
| Provision for income taxes | 51,534 | | | 59,711 | | | 13,148 | | | 93,716 | |
| Net income attributable to Vertex | $ | 268,631 | | | $ | 267,427 | | | $ | 57,518 | | | $ | 583,234 | |
| | | | | | | |
| Amounts per share attributable to Vertex common shareholders: | | | | | | | |
| Net income: | | | | | | | |
| Basic | $ | 1.05 | | | $ | 1.04 | | | $ | 0.22 | | | $ | 2.26 | |
| Diluted | $ | 1.03 | | | $ | 1.03 | | | $ | 0.22 | | | $ | 2.23 | |
| Shares used in per share calculations: | | | | | | | |
| Basic | 255,695 | | | 256,154 | | | 256,946 | | | 258,003 | |
| Diluted | 260,175 | | | 259,822 | | | 260,473 | | | 262,108 | |
1.The Company incurred research and development expenses of $75.0 million related to its 2020 Moderna Agreement in the third quarter of 2020 and $175.0 million related to its CRISPR DMD/DM1 Agreement in the third quarter of 2019. See Note B, “Collaborative Arrangements.”
2.In 2020 and 2019, “Other income (expense), net” was primarily related to changes in the fair value of the Company’s investments in corporate equity securities, and from sales of certain corporate equity securities. See Note E, “Marketable Securities and Equity Investments.”
3.In the second quarter of 2020, the Company completed an intra-entity transfer of intellectual property rights to the U.K., resulting in a deferred tax benefit of $209.0 million. See Note O, “Income Taxes.”
4.In the fourth quarter of 2019, the Company updated its transaction price and recognized net product revenues of $155.8 million related to prior period ORKAMBI sales upon reaching a reimbursement agreement with the French government for ORKAMBI, including ORKAMBI distributed through early access programs. See Note A, “Nature of Business and Accounting Policies.”