UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
|
| |
| x | Annual report pursuant to Section 13 or 15 (d) of the Securities Exchange Act of 1934 |
For the fiscal year ended December 31, 20162018
or
|
| |
| ¨ | Transition report pursuant to Section 13 or 15 (d) of the Securities Exchange Act of 1934 |
For the transition period from to
Commission file number: 0-27756
ALEXION PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
|
| |
| Delaware | 13-3648318 |
| (State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) |
100 College Street, New Haven, Connecticut 06510121 Seaport Boulevard, Boston Massachusetts 02210
(Address of Principal Executive Offices) (Zip Code)
475-230-2596
(Registrant’s telephone number, including area code)
|
| | |
| Securities registered pursuant to Section 12(b) of the Act: | | Common Stock, par value $0.0001 |
| | | |
Name of each exchange on which registered: The NASDAQNasdaq Stock Market LLC |
| Securities registered pursuant to Section 12(g) of the Act: None |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨x No x¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer” andfiler,” “smaller reporting company”company,” and "emerging growth company" in Rule 12b-2 of the Exchange Act. Check One:
Large accelerated filer x Accelerated filer ¨ Non-accelerated filer ¨ (Do not check if a smaller reporting company)
Smaller reporting company¨ Emerging growth company ¨
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the Common Stock held by non-affiliates of the registrant, based upon the last sale price of the Common Stock reported on The NASDAQNasdaq Stock Market LLC on June 30, 2016,29, 2018, was $25,314,108,813.$26,514,235,288.(1)
The number of shares of Common Stock outstanding as of February 13, 2017January 31, 2019 was 224,613,750.223,469,381.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Definitive Proxy Statement to be used in connection with its 2019 Annual Meeting of Stockholders currently anticipated to be held on May 10, 2017,14, 2019, are incorporated by reference into Part III of this report.
(1) Excludes 7,417,8979,186,789 shares of common stock held by directors, and executive officers and their respective affiliates at June 30, 2016.29, 2018. Exclusion of shares held by any person should not be construed to indicate that such person possesses the power, directly or indirectly, to direct or cause the direction of the management or policies of the registrant, or that such person is controlled by or under common control with the registrant.
Alexion Pharmaceuticals, Inc.
Table of Contents
|
| | |
PART I | | Page |
| PART I | | |
| Item 1. | | |
| Item 1A. | | |
| Item 1B. | | |
| Item 2. | | |
| Item 3. | | |
| Item 4. | | |
| | | |
| PART II | | |
| Item 5. | | |
| Item 6. | | |
| Item 7. | | |
| Item 7.A | | |
| Item 8. | | |
| Item 9. | | |
| Item 9A. | | |
| Item 9A(T). | | |
| Item 9B. | | |
| | | |
| PART III | | |
| Item 10. | | |
| Item 11. | | |
| Item 12. | | |
| Item 13. | | |
| Item 14. | | |
| | | |
| PART IV | | |
| Item 15. | | |
| Item 16. | | |
| | | |
| |
PART I
Unless the context requires otherwise, references in this report to “Alexion”,“Alexion,” the “Company”, “we”,“Company,” “we,” “our” or “us” refer to Alexion Pharmaceuticals, Inc. and its subsidiaries.
Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements. Words such as “anticipates,” “may,” “forecasts,” “expects,” “intends,” “plans,” “believes,” “seeks,” “estimates,” variations of such words and similar expressions are intended to identify such forward-looking statements, although not all forward-looking statements contain these identifying words. Forward-looking statements are not guarantees of future performance and are subject to certain risks, uncertainties, and assumptions that have been made pursuantare difficult to the provisions of the Private Securities Litigation Reform Act of 1995.predict; therefore, actual results may differ materially from those expressed or forecasted in any such statements. Such forward-looking statements are based on current expectations, estimates and projections about our industry, management’smanagement's beliefs, and certain assumptions made by our management, and may include, but are not limited to, statements regarding regarding:
the potential benefits and commercial potential of Soliris®UTLOMIRIS™, Strensiq®SOLIRIS®, STRENSIQ® and Kanuma®KANUMA® for approved indications and any expanded uses, timing and effect of sales of our products in various markets worldwide, pricing for our products, level of insurance coverage and reimbursement for our products, level of future product sales and collections, timing regarding development and regulatory approvals for additional indications or in additional territories, the medical and commercial potentialterritories;
plans for clinical trials (and proof of additional indications for Soliris, failure to satisfactorily address the issues raised by the U.S. Food and Drug Administration (FDA) in the March 2013 Warning Letter and Form 483s issued by the FDA, costs, expenses and capital requirements, cash outflows, cash from operations, status of reimbursement, price approval and funding processes in various countries worldwide, progress in developing interest about our products and our product candidates in the patient, physician and payer communities, the safety and efficacy of our products and our product candidates, estimates of the potential markets and estimated commercialization dates for our products and our product candidates around the world, sales and marketing plans, any changes in the current or anticipated market demand or medical need for our products or our product candidates,concept trials), status of our ongoing clinical trials for eculizumab, asfotase alfa, sebelipase alfa and our other product candidates, commencement dates for new clinical trials, clinical trial results and evaluation of our clinical trial results by regulatory agencies, agencies;
potential benefits offered by product candidates, including improved dosing intervals;
the medical and commercial potential of additional indications for our products;
the expected timing for the completion and/or regulatory approval of our facilities and facilities of our third-party manufacturers;
future expansion of our commercial organization;
future governmental and regulatory decisions regarding pricing (and discounts) and the adoption, implementation and interpretation of healthcare laws and regulations (and the impact on our business);
plans and prospects for future regulatory approval of products and product candidates;
competitors, potential competitors and future competitive products (including biosimilars);
plans to grow our product pipeline (and diversify our business, including through acquisitions) and anticipated benefits to the Company;
future objective to expand business and sales;
future plans to retain earnings and not pay dividends;
expected decisions to appeal certain litigation and intellectual property decisions;
expectations to realize the carrying value of product inventory;
impact of accounting standards;
future costs, operating expenses (including research and development, sales, general and administrative and restructuring expenses) and capital requirements, capital investment, sufficiency of cash to fund operations, the sufficiency of our existing capital resources and projected cash needs, price approval and funding processes in various countries;
anticipated future milestone, contingent and royalty payments (and expected impact on liquidity);
timing and anticipated amounts of future tax payments and benefits, as well as timing of conclusion of tax audits;
collection of accounts receivable;
the safety and efficacy of our products and our product candidates;
the adequacy of our pharmacovigilance and drug safety reporting processes, prospects for regulatory approval of our products and our product candidates, need for additional research and testing, processes;
the uncertainties involved in the drug development process and manufacturing, manufacturing;
performance and reliance on third party service providers, providers;
our future research and development activities, plans for acquired programs, our ability to develop and commercialize products with our collaborators, assessmentcollaborators;
periods of competitorspatent, regulatory and potential competitors,market exclusivity for our products;
the scope of our intellectual property and the outcome of any challenges andor opposition proceedings to our intellectual property, assertion or potential assertion by third parties that the manufacture, use or sale of our products infringes their intellectual property, property; and
estimates of the capacity of manufacturing and other service facilities to support our business, operations, products and ourproduct candidates.
Such risks and uncertainties include, but are not limited to, increased competition, actions by regulatory agencies, product candidates potential costs resulting from product liability or other third party claims, the sufficiency of our existing capital resources and projected cash needs,not receiving regulatory approvals, the possibility that expected tax benefits will not be realized, assessment of impact of recent accounting pronouncements, potential declines in sovereign credit ratings or sovereign defaults in countries where we sell our products, delay of collection or reduction in reimbursement due to adverse economic conditions or changes in government and private insurer regulations and approaches to reimbursement, uncertainties surrounding legal proceedings, company investigations and government investigations, including our Securities and Exchange Commission (SEC) and U.S. Department of Justice (DOJ) investigations, the securities fraud class action litigation filed in December 2016, the investigation by our Audit and Finance Committee announced November 2016 (the Audit Committee Investigation), and the inquiry by the U.S. Attorney’sAttorney's Office for the District of Massachusetts requesting documents relating generally to our support of patient assistance programs, the investigation of our Brazilian operations by Brazilian authorities, the investigation by the MHLW in Japan, risks related to potential disruptions to our business as a result of the leadership changes and transition announced in December 2016, the risk that hiring a new CEO may take longer than anticipated, the short and long-term effects of other government healthcare measures, and the effect of shifting foreign exchange rates. Words suchrates, as “anticipates,” “expects,” “intends,” “plans,” “believes,” “seeks,” “estimates,” variations of such words and similar expressions are intended to identify such forward-looking statements, although not all forward-looking statements contain these identifying words. These statements are not guarantees of future performance and are subject to certain risks, uncertainties, and assumptions that are difficult to predict; therefore, actual results may differ materially fromwell as those expressed or forecasted in any such forward-looking statements. Such risks and uncertainties include, but are not limited to, those discussed later in this report under the section entitled “Risk Factors”.Factors.” Unless required by law, we undertake no obligation to update publicly any forward-looking statements, whether because of new information, future events or otherwise. However, readers should carefully review the risk factors set forth in this and other reports or documents we file from time to time with the SEC.
Note Regarding Trademarks
We have proprietary rights to a number of registered and unregistered trademarks that we believe are important to our business, including but not limited to: Alexion Pharmaceuticals, Inc., Alexion, ULTOMIRIS, SOLIRIS, STRENSIQ and KANUMA. We have, in certain cases, omitted the ®, © and ™ designations for these and other trademarks used in this Annual Report on Form 10-K. Nevertheless, all rights to such trademarks are reserved. These and other trademarks referenced in this Annual Report on Form 10-K are the property of their respective owners.
Item 1. BUSINESS.
(dollars and shares in millions)
Overview
We areAlexion is a global biopharmaceutical company focused on serving patients with devastating and ultra-rare disordersfamilies affected by rare diseases through the innovation, development and commercialization of life-transforming therapeutic products.life-changing therapies.
In ourWe are the global leader in complement franchise, Soliris® isinhibition and have developed and commercialize the only two approved complement inhibitors to treat patients with paroxysmal nocturnal hemoglobinuria (PNH), as well as the first and only therapeutic approved for patients with either paroxysmal nocturnal hemoglobinuria (PNH), a life-threatening and ultra-rare genetic blood disorder, orcomplement inhibitor to treat atypical hemolytic uremic syndrome (aHUS), a and anti-acetylcholine receptor (AchR) antibody-positive generalized myasthenia gravis (gMG). In addition, Alexion has two highly innovative enzyme replacement therapies for patients with life-threatening and ultra-rare genetic disease. PNHmetabolic disorders, hypophosphatasia (HPP) and aHUS result from chronic uncontrolled activation oflysosomal acid lipase deficiency (LAL-D).
As the leader in complement biology for over 20 years, Alexion focuses its research efforts on novel molecules and targets in the complement componentcascade, and its development efforts on the core therapeutic areas of the immune system.
In ourhematology, nephrology, neurology, and metabolic franchise, we commercialize Strensiq® for the treatment of patients with Hypophosphatasia (HPP) and Kanuma® for the treatment of patients with Lysosomal Acid Lipase Deficiency (LAL-D). HPP is an ultra-rare genetic disease characterized by defective bone mineralization that can lead to deformity of bones and other skeletal abnormalities. LAL-D is a serious, life threatening ultra-rare disease in which genetic mutations result in decreased activity of the Lysosomal Acid Lipase (LAL) enzyme leading to marked accumulation of lipids in vital organs, blood vessels and other tissues.
We are also evaluating additional potential indications for eculizumab in other severe and devastating diseases in which uncontrolled complement activation is the underlying mechanism, and we are progressing in various stages of development with additional product candidates as potential treatments for patients with devastating and ultra-rare disorders.
We were incorporated in 1992. In June 2015, we acquired all1992 under the laws of the outstanding sharesState of common stock of Synageva BioPharma Corp. (Synageva), a publicly-held clinical-stage biotechnology company. The acquisition furthered our objective to develop and commercialize life-transforming therapies for patients with devastating and ultra-rare diseases.Delaware.
Products and Development Programs
We focus our productproducts and development programs on life-transforming therapeutics for devastating and ultra-rarerare diseases for which we believe the current treatments are either non-existent or inadequate. We have developed or are developing innovative products for the following indications:
|
| |
| Paroxysmal Nocturnal Hemoglobinuria (PNH) | PNH is a debilitating and life-threatening, ultra-rare genetic blood disorder defined by chronic uncontrolled complement activation leading to the destruction of red blood cells (hemolysis). Chronic hemolysis in patients with PNH may be associated with life-threatening thromboses, recurrent pain, kidney disease, disabling fatigue, impaired quality of life, severe anemia, pulmonary hypertension, shortness of breath and intermittent episodes of dark-colored urine (hemoglobinuria). |
|
| |
| Atypical Hemolytic Uremic Syndrome (aHUS) | aHUS is a severe and life-threatening, ultra-rare genetic disease characterized by chronic uncontrolled complement activation and thrombotic microangiopathy (TMA), the formation of blood clots in small blood vessels throughout the body, causing a reduction in platelet count (thrombocytopenia) and life-threatening damage to the kidney, brain, heart and other vital organs. |
|
| |
| Generalized Myasthenia Gravis (gMG) | Myasthenia Gravis (MG) is a debilitating, complement-mediated neuromuscular disease in which patients suffer profound muscle weakness throughout the body, resulting in slurred speech, impaired swallowing and choking, double vision, upper and lower extremity weakness, disabling fatigue, shortness of breath due to respiratory muscle weakness and episodes of respiratory failure.
|
|
| |
| Hypophosphatasia (HPP) | HPP is an ultra-rare genetic and progressive metabolic disease in which patients experience devastating effects on multiple systems of the body, leading to debilitating or life-threatening complications. HPP is characterized by defective bone mineralization that can lead to deformity of bones and other skeletal abnormalities, as well as systemic complications such as profound muscle weakness, seizures, pain, and respiratory failure leading to premature death in infants.
|
|
| |
| Lysosomal Acid Lipase Deficiency (LAL Deficiency or LAL-D) | LAL-D is a serious, life-threatening ultra-rare disease associated with premature mortality and significant morbidity. LAL-D is a chronic disease in which genetic mutations result in decreased activity of the LAL enzyme that leads to marked accumulation of lipids in vital organs, blood vessels, and other tissues, resulting in progressive and systemic organ damage including hepatic fibrosis, cirrhosis, liver failure, accelerated atherosclerosis, cardiovascular disease, and other devastating consequences.
|
|
| |
| Relapsing Neuromyelitis Optica Spectrum Disorder (NMOSD) | Relapsing NMOSD is a severe and ultra-rare autoimmune disease of the central nervous system that primarily affects the optic nerves and the spinal cord. Each relapse of the disorder results in a stepwise accumulation of disability, including blindness and paralysis, and sometimes premature death. |
|
| |
| Wilson Disease | Wilson disease is a rare disorder that can lead to severe liver disease, including cirrhosis and acute liver failure, as well as debilitating neurological morbidities such as impaired movement, gait, speech, swallowing, and psychiatric disorders. |
|
| |
| Warm Autoimmune Hemolytic Anemia (WAIHA) | WAIHA is a rare autoimmune disorder caused by pathogenic Immunoglobulin G (IgG) antibodies that react with and cause the premature destruction of red blood cells at normal body temperature. The disease is often characterized by profound, and potentially life-threatening anemia and other acute complications, including severe and life-threatening hemolysis, severe weakness, enlarged spleen and/or liver, rapid heart rate (tachycardia), chest pain, heart failure and fainting (syncope). |
Marketed Products
Our marketed products include the following:
|
| | | | |
| Product | | DevelopmentTherapeutic Area | | Approved Indication |
Soliris (eculizumab) | | Hematology | | Paroxysmal Nocturnal Hemoglobinuria (PNH) |
| | | Hematology | PNH |
|
|
| | Hematology | PNH |
| | Hematology/Nephrology | | Atypical Hemolytic Uremic Syndrome (aHUS)aHUS |
Strensiq (asfotase alfa) | Neurology | gMG
|
| | Metabolic Disorders | | Hypophosphatasia (HPP)HPP |
Kanuma (sebelipase alfa) | | Metabolic Disorders | | Lysosomal Acid Lipase Deficiency (LAL-D)LAL-D |
Soliris (eculizumab)
SolirisULTOMIRIS (ALXN1210/ravulizumab-cwvz)
ULTOMIRIS is designed to inhibit a specific aspect of the complement component of the immune system and thereby treat inflammation associated with chronic disorders in several therapeutic areas, including hematology, nephrology neurology and transplant rejection. Solirisneurology. As of the date hereof, ULTOMIRIS has only been approved as a therapy in the US for adult patients with PNH. ULTOMIRIS is a humanized monoclonal antibody that effectively blocks terminal complement activity at the doses currently prescribed. The initial indicationULTOMIRIS is the first and only long-acting C5 inhibitor that provides immediate and complete inhibition for whicheight weeks.
In December 2018, ULTOMIRIS was approved by the U.S. Food and Drug Administration (FDA) as a new treatment option for adult patients living with PNH.
In June 2018, we received approval for Soliris is PNH.
Paroxysmal Nocturnal Hemoglobinuria (PNH)
PNH issubmitted a debilitating and life-threatening, ultra-rare genetic blood disorder defined by chronic uncontrolled complement activation leadingMarketing Authorization Application (MAA) to the destructionEuropean Medicines Agency (EMA) for approval of red blood cells (hemolysis). The chronic hemolysis inULTOMIRIS for the treatment of patients with PNH may beand in July 2018 the MAA was accepted for review in the European Union (EU). In September 2018, we also filed an application with the Japan Pharmaceuticals and Medical Devices (PDMA) for the approval of ULTOMIRIS for patients with PNH.
ULTOMIRIS has received Orphan Drug Designation (ODD) for the treatment of patients with PNH in the U.S., EU and Japan.
SOLIRIS (eculizumab)
SOLIRIS is designed to inhibit a specific aspect of the complement component of the immune system and thereby treat inflammation associated with life-threatening thromboses, recurrent pain, kidney disease, disabling fatigue, impaired quality of life, severe anemia, pulmonary hypertension, shortness of breathchronic disorders in several therapeutic areas, including hematology, nephrology and intermittent episodes of dark-colored urine (hemoglobinuria). We continue to work with researchers to expandneurology. SOLIRIS is a humanized monoclonal antibody that effectively blocks terminal complement activity at the base of knowledge in PNH and the utility of Soliris to treat patients with PNH. Solirisdoses currently prescribed.
SOLIRIS is approved for the treatment of PNH in the United States (U.S.)U.S., Europe, Japan and in several other territories.countries. We are sponsoring a multinational registry to gather information regarding the natural history of patients with PNH and the longer term outcomes during SolirisSOLIRIS treatment. In addition, SolirisSOLIRIS has been granted orphan drug designation for the treatment of PNH in the U.S., Europe, Japan and several other territories.countries.
Atypical Hemolytic Uremic Syndrome (aHUS)
aHUS is a severe and life-threatening, ultra-rare genetic disease characterized by chronic uncontrolled complement activation and thrombotic microangiopathy (TMA), the formation of blood clots in small blood vessels throughout the body, causing a reduction in platelet count (thrombocytopenia) and life-threatening damage to the kidney, brain, heart and other vital
organs. SolirisSOLIRIS is approved for the treatment of pediatric and adult patients with aHUS in the U.S., Europe, Japan and Japan.in several other countries. We are sponsoring a multinational registry to gather information regarding the natural history of patients with aHUS and the longer termlonger-term outcomes during SolirisSOLIRIS treatment. In addition, the FDA and European Commission (EC) have granted SolirisSOLIRIS orphan drug designation for the treatment of patients with aHUS.
Strensiq
In 2017, the FDA and EC approved SOLIRIS for the treatment of refractory gMG in adults who are anti-acetylcholine receptor (AChR) antibody-positive. Additionally, in 2017 the Ministry of Health, Labour and Welfare (MHLW) in Japan approved SOLIRIS as a treatment for patients with gMG who are AChR antibody-positive and whose symptoms are difficult to control with high-dose intravenous immunoglobulin therapy or plasmapheresis (PLEX). SOLIRIS has received orphan drug designation for the treatment of patients with MG in the U.S. and Europe, and for the treatment of patients with refractory gMG, a subset of MG, in Japan.
STRENSIQ (asfotase alfa)
Hypophosphatasia (HPP)
HPP is an ultra-rare genetic and progressive metabolic disease in which patients experience devastating effects on multiple systems of the body, leading to debilitating or life-threatening complications. HPP is characterized by defective bone mineralization that can lead to deformity of bones and other skeletal abnormalities, as well as systemic complications such as profound muscle weakness, seizures, pain, and respiratory failure leading to premature death in infants.
Strensiq,STRENSIQ, a targeted enzyme replacement therapy, is the first and only approved therapy for patients with HPP and is designed to directly address underlying causes of HPP by aiming to restore the genetically defective metabolic process, thereby preventing or reversing the severe and potentially life-threatening complications in patients with HPP. In 2015,STRENSIQ is approved in the FDA approved StrensiqU.S. for patients with
perinatal-, infantile- and juvenile-onset HPP, the EC granted marketing authorization for StrensiqEurope for the treatment of patients with pediatric-onset HPP, and Japan’s Ministry of Health Labour and Welfare (MHLW) approved StrensiqJapan for the treatment of patients with HPP. We are sponsoring a multinational registry to gather information regarding the natural history of patients with HPP and the longer-term outcomes during StrensiqSTRENSIQ treatment.
KanumaKANUMA (sebelipase alfa)
Lysosomal Acid Lipase Deficiency (LAL Deficiency or LAL-D)
LAL-D is a serious, life-threatening ultra-rare disease associated with premature mortality and significant morbidity. LAL-D is a chronic disease in which genetic mutations result in decreased activity of the LAL enzyme that leads to marked accumulation of lipids in vital organs, blood vessels, and other tissues, resulting in progressive and systemic organ damage including hepatic fibrosis, cirrhosis, liver failure, accelerated atherosclerosis, cardiovascular disease, and other devastating consequences.
Kanuma,KANUMA, a recombinant form of the human LAL enzyme, is the only enzyme-replacement therapy that is approved for the treatment for patients with LAL-D. In 2015,KANUMA is approved in the FDA approved KanumaU.S. for the treatment of patients with LAL-D, and the EC granted marketing authorization of KanumaEurope for long-term enzyme replacement therapy in patients of all ages with LAL-D. On March 28, 2016, we announced that the MHLW approved KanumaLAL-D, and Japan for the treatment of patients of all ages in Japan with LAL-D. We are sponsoring a multinational registry to gather information regarding the natural history of patients with LAL-D and the longer termlonger-term outcomes during KanumaKANUMA treatment.
Clinical Development Programs
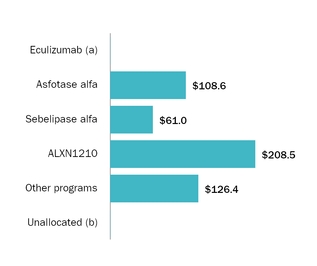
Our programs, including investigator sponsoredongoing clinical development programs include the following:
|
| | | | | | |
| Product | | Development Area | Indication | IndicationPhase I | Phase II | Phase III | Filed |
ULTOMIRIS (ALXN1210/ravulizumab-cwvz)
(Intravenous) | Hematology/Nephrology | aHUS | | Development Stage | l | |
Soliris (eculizumab) | | | |
Neurology
| gMG | | Refractory Generalized Myasthenia Gravis (gMG) | l | | Phase III |
| | | | | Relapsing Neuromyelitis Optica Spectrum Disorder (NMOSD)
| | Phase III |
ULTOMIRIS (ALXN1210/ravulizumab-cwvz)
(Subcutaneous) | Hematology/Nephrology | PNH/aHUS | | | Transplantl | | Antibody Mediated Rejection (AMR) Presensitized Renal Transplant - Deceased Donor | | Phase II |
cPMP (ALXN1101) | | Metabolic Disorders
| | Molybdenum Cofactor Deficiency (MoCD )Type A | | Phase II / III |
SBC-103 | | Metabolic Disorders
| | Mucopolysaccharidoses IIIB
(MPS IIIB)
| | Phase I / II |
ALXN1210 (IV) | | Next Generation Complement Inhibitor | | Paroxysmal Nocturnal Hemoglobinuria (PNH) | | Phase III |
| | | | | Atypical Hemolytic Uremic Syndrome (aHUS) | | Phase III |
ALXN1210 ALXN1810 (Subcutaneous) | | Next Generation Subcutaneous Complement Inhibitor | | l | | | |
| | | | Phase I | | |
| SOLIRIS (eculizumab) | Neurology | NMOSD | | | | l |
| | | | |
ALXN1840 (WTX101) | Metabolic Disorders | Wilson disease | | | l | |
| | | | | | |
ALXN1830 (SYNT001) | Hematology | WAIHA | | l | | |
Soliris (eculizumab)
NeurologyRefractory Generalized Myasthenia Gravis (gMG)ULTOMIRIS (ALXN1210/ravulizumab-cwvz)
Refractory gMGALXN1210 (ravulizumab-cwvz) is an ultra-rare segmentinnovative, long-acting C5 inhibitor discovered and developed by Alexion that works by inhibiting the C5 protein in the terminal complement cascade. In clinical studies, ALXN1210 demonstrated rapid, complete, and sustained reduction of Myasthenia Gravis,free C5 levels for eight weeks.
Intravenous (IV)
Enrollment was completed in late May 2018 in a debilitating, complement-mediated neuromuscular diseasePhase III, single arm, multicenter study to evaluate the safety and efficacy of ALXN1210 administered by IV infusion every 8 weeks to adult patients with aHUS who have never been treated with a complement inhibitor. In January 2019, we announced the results of the Phase
III study with ALXN1210 meeting its primary objective in whichcomplement inhibitor-naïve patients suffer profound muscle weakness throughoutwith aHUS. In the body, resultinginitial 26 week treatment period in slurred speech, impaired swallowingthis study, 53.6 percent of patients demonstrated complete thrombotic microangiopathy (TMA) response. A second Phase III, single arm, multicenter study to evaluate the safety, efficacy, pharmacokinetics (PK), and choking, double vision, upperpharmaco-dynamics (PD) of ALXN1210 administered by IV infusion every 8 weeks in pediatric patients (including adolescents) with aHUS who have never been treated with a complement inhibitor (inhibitor-naïve patients) is ongoing.
Alexion plans to initiate a Phase III study with ALXN1210 administered by IV infusion every 8 weeks to adult patients for the treatment of gMG in 2019.
In addition to aHUS and lower extremity weakness, disabling fatigue, shortnessgMG, Alexion plans to initiate clinical studies of breath dueALXN1210 in NMOSD. In addition, in 2019 we also plan to respiratory muscle weaknessinitiate proof of concept trials for ALXN1210 as a therapy for Amyotrophic Lateral Sclerosis (ALS), and episodesPrimary Progressive Multiple Sclerosis (PPMS).
Subcutaneous (SC) Delivery
In late 2018, Alexion initiated a single, PK-based Phase III study of respiratory failure. TheALXN1210 delivered subcutaneously once per week to PNH patients to support regulatory approval submissions in both PNH and aHUS.
In October 2017, the FDA EC and MHLW have granted orphan drug designation to the subcutaneous formulation of ALXN1210 for eculizumab as athe treatment of aHUS.
ALXN1810 Subcutaneous (SC) Delivery
ALXN1810 combines ALXN1210 with recombinant human hyaluronidase enzyme (rHuPH20) from Halozyme Therapeutics, Inc. to potentially further extend the dosing interval for patientsALXN1210 SC to once every two weeks or once per month. A SC healthy volunteer study with refractory gMG.ALXN1810 was initiated in August 2018.
SOLIRIS (eculizumab)
In June 2016,September 2018, we announced toplinethe results of the Phase III REGAIN trial of eculizumab for the treatment of refractory gMG. The primary efficacy endpoint of change from baseline in Myasthenia Gravis-Activities of Daily Living Profile (MG-ADL) total score, a patient-reported assessment, at week 26, did not reach statistical significance (p=0.0698) as measured by a worst-rank analysis. The totality of data reviewed to date, including the first three secondary endpoints and a series of prospectively defined sensitivity analyses, shows early and sustained substantial improvements over 26 weeks for patients treated with eculizumab compared to placebo. The safety of eculizumab in this study was consistent with the Soliris labels. Additional data from the Phase III study was presented in July 2016. The data showed that 18 of 22 pre-defined endpoints and pre-specified analyses in the study, based on the primary and five secondary endpoints, achieved p-values below 0.05.
In January 2017, we announced that we filed for regulatory approval for eculizumab in refractory gMG in both the U.S. and Europe. These marketing applications were based on the comprehensive data from the Phase III REGAIN trial.
Relapsing Neuromyelitis Optica Spectrum Disorder (NMOSD)
Relapsing NMOSD is a severe and ultra-rare autoimmune disease of the central nervous system (CNS) that primarily affects the optic nerves and spinal cord. The disease leads to severe weakness, paralysis, respiratory failure, loss of bowel and bladder function, blindness and premature death. Enrollment and dosing are ongoing in a global, randomized, double-blind, placebo-controlled trialstudy to evaluate eculizumab as a treatment for patients with relapsing NMOSD. The study met its primary endpoint of time to first adjudicated on-trial relapse, demonstrating that treatment with eculizumab reduced the risk of NMOSD relapse by 94.2 percent compared to placebo. At 48 weeks, 97.9 percent of patients receiving eculizumab were free of relapse compared to 63.2 percent of patients receiving placebo. Eculizumab had a safety profile consistent with that seen in previous clinical studies. The FDA, EC, and MHLW have each granted orphan designation for eculizumab as a treatment for patients with relapsing NMOSD.
Transplant
Antibody Mediated Rejection (AMR) in Presensitized Kidney Transplant Patients
AMR is the term used to describe a type of transplant rejection that occurs when the recipient has antibodiesIn December 2018, we submitted our requests for regulatory approval to the donor organ. Enrollment in a multi-national, multi-center controlled clinical trial of eculizumab in presensitized kidney transplant patients at elevated risk for AMR who received kidneys from deceased organ donors was completed in March 2013FDA and patient follow-upour MAA in the trialEU for eculizumab for the potential treatment of NMOSD.
ALXN1840 (WTX101)
ALXN1840 (WTX101), an innovative product candidate that addresses the underlying cause of Wilson disease, is continuing. In September 2013, researchers presented positive preliminary dataa first-in-class oral copper-binding agent with a unique mechanism of action and ability to access and bind copper from serum and promote its removal from the eculizumab deceased-donor AMR kidney transplant study.liver.
ALXN1840 is in Phase III development as a treatment for Wilson disease. In May 2015, new data from the Phase II single-arm deceased-donor transplant trial of eculizumab in prevention of acute AMR was presented and was consistent with previous positive reports.
cPMP (ALXN1101)
Molybdenum Cofactor Deficiency (MoCD) Disease Type A (MoCD Type A)
MoCD Type A is an ultra-rare metabolic disorder characterized by severe and rapidly progressive neurologic damage and death in newborns. MoCD Type A results from a genetic deficiency in cyclic Pyranopterin Monophosphate (cPMP), a molecule that enables the function of certain enzymes and the absence of which allows neurotoxic sulfite to accumulateaddition, ALXN1840 has received Fast Track designation in the brain. To date, there is no approved therapy available for MoCD Type A. There has been some early clinical experience with the recombinant cPMP replacement therapy in a small number of children with MoCD Type A,U.S. and we have completed enrollment in a natural history study in patients with MoCD Type A. cPMP received Breakthrough TherapyOrphan Drug Designation from the FDA for the treatment of patientsWilson disease in the U.S. and EU.
ALXN1830 (SYNT001)
ALXN1830 (SYNT001) is a humanized monoclonal antibody that is designed to inhibit the interaction of the neonatal Fc receptor (FcRn) with MoCD Type A. Evaluation of our synthetic form of cPMP replacement therapyIgG and IgG immune complexes and has the potential to improve treatment in a number of rare IgG-mediated diseases. ALXN1830 (SYNT001) is currently being evaluated in Phase I healthy volunteer study is complete. In addition, we completed enrollment in a multi-center, multinational open-label clinical trial of synthetic cPMP1b/2a studies in patients with MoCD Type A switched from treatment with recombinant cPMP. Enrollment is ongoing in the Phase II/III pivotal open-label, single-arm trial of ALXN1101 for treatment-naïve neonates with MoCD Type A.
SBC-103
Mucopolysaccharidosis IIIB (MPS IIIB)
MPS IIIB is an ultra-rare, devastatingwarm autoimmune hemolytic anemia (WAIHA) and life-threatening disease which typically presents in children during the first few years of life. Genetic mutations result in decreased activity of the alpha-N-acetyl-glucosaminidase (NAGLU) enzyme, which leads to a buildup of abnormal amounts of heparan sulfate (HS) in the brain and throughout the body. Over time, this unrelenting systemic accumulation of HS causes progressive and severe cognitive decline, behavioral problems, speech loss, increasing loss of mobility, and premature death. Current treatments are palliative for the behavioral problems, sleep disturbances, seizures, and other complications, and these treatments do not address the root cause of MPS IIIB or stop disease progression.
SBC-103, a recombinant form of natural human NAGLU is designed to replace the missing (or deficient) NAGLU enzyme. SBC-103 was granted orphan drug designation by the FDA and by the EC. It received Fast Track designation by the FDA. The first-in-human trial of patients with MPS IIIB is ongoing. In March 2016, researchers presented 24-week results from this study that showed a 26.2 percent mean reduction in heparan sulfate in cerebrospinal fluid at the highest dose studied (3mg/kg every other week) in a Phase I/II study at six months. In July 2016, researchers presented preliminary results on brain MRI and neurocognitive assessments performed after 24 weeks of dosing suggesting preliminary evidence of potential for dose-dependent disease stabilization in patients treated with 0.3, 1, or 3mg/kg every other week of doses of SBC-103. Planned dose escalation of SBC-103 is now ongoing in this trial. In February 2017, the Board of Directors of Alexion made the decision to reduce our investment in SBC-103. The current Phase I/II clinical trial will not be expanded and no new patients will be added to the trial. Patients currently enrolled in the trial will continue to receive therapy.
ALXN1210
ALXN1210 is a highly innovative, longer-acting anti-C5 antibody discovered and developed by Alexion that inhibits terminal complement. In early studies, ALXN1210 demonstrated rapid, complete, and sustained reduction of free C5 levels. Alexion has completed enrollment in two ongoing clinical studies of ALXN1210 in patients with PNH-a Phase 1/2 dose-escalating studypemphigus vulgaris (PV) or pemphigus foliaceus (PF). In 2019, Alexion plans to initiate two pivotal trials, one in WAIHA and an open-label, multi-dose Phase II study that is also evaluating longer dosing intervals beyond 8 weeks.one in gMG.
Paroxysmal Nocturnal Hemoglobinuria (PNH)
In June 2016, we announced interim data from a Phase I/II study in patients with PNH showing that once-monthly dosing of ALXN1210 achieved rapid and sustained reductions in hemolysis, as measured by mean levels of lactate dehydrogenase (LDH), in 100 percent of treated patients. Chronic hemolysis in patients with PNH may be associated with life-threatening thromboses, recurrent pain, kidney disease, disabling fatigue, impaired quality of life, severe anemia, pulmonary hypertension, shortness of breath and intermittent episodes of dark-colored urine (hemoglobinuria). Researchers also reported that, at the time of analysis, 80 percent of patients who required at least 1 blood transfusion in the 12 months prior to treatment with ALXN1210 did not require transfusions while on treatment with ALXN1210. Furthermore, in December 2016, we reported new data from this same ongoing study that showed rapid and sustained reductions LDH in patients with PNH treated with once-monthly dosing. Patients also had improvements in Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue score from baseline, with patients in the higher-dose cohort achieving a two-fold greater improvement compared with the lower-dose cohort. In addition, we have completed enrollment and treatment is ongoing in an open-label, multi-dose Phase II study of ALXN1210 in patients with PNH designed to measure reductions in hemolysis and safety in several dosing cohorts and intervals evaluating monthly and longer dosing intervals. We have initiated a Phase III open-label, multinational, active-controlled study of ALXN1210 compared to eculizumab (Soliris) in adult patients with PNH who have never been treated with a complement inhibitor. The study is evaluating ALXN1210 administered intravenously every eight weeks. Patient enrollment is ongoing in this trial.
In June 2016 and January 2017, the EC and the FDA, respectively, granted orphan drug designation to ALXN1210, for the treatment of patients with PNH.
Atypical Hemolytic Uremic Syndrome (aHUS)Manufacturing
We initiated a Phase III open-label, single arm, multicenter study of ALXN1210 in adolescent and adult patients with aHUS who have never been treated with a complement inhibitor. In patients with aHUS, complement-mediated TMA leads to life-threatening damage to the kidney, brain, heart and other vital organs. The study will evaluate ALXN1210 administered intravenously every eight weeks. Patient recruitment will initiate in 2017 on this trial.
Subcutaneous (SC) Delivery
We have completed enrollment in a Phase I study in healthy volunteers to evaluate ALXN1210 delivered subcutaneously.
Manufacturing
We currently rely onutilize both internal manufacturing facilities and third party contract manufacturers to supply clinical and commercial quantities of our products and product candidates. Our internal manufacturing capability includes our Ireland facilities, a fill/finish facility in Athlone and a packaging facility in Dublin, as well as facilities in Massachusetts and Georgia. Third party contract manufacturers, including Lonza Group AG and its affiliates (Lonza), to supply clinical and commercial quantities of our commercial products and product candidates. Our internal manufacturing facilities include our Ireland manufacturing facilities, our Rhode Island manufacturing facility (ARIMF), and facilities in Massachusetts and Georgia. We also utilize third party contract manufacturers forprovide bulk drug substance as well as other manufacturing services includinglike purification, product filling, finishing, packaging, and labeling.
We have various agreements with Lonza through 2028,2029, with remaining total non-cancellable commitments of approximately $1,148.$1,084.6. If we terminate certain supply agreements with Lonza without cause, we will be required to pay for product scheduled for manufacture under our arrangements. Under an existing arrangement, with Lonza, we also pay Lonza a royalty on sales of SolirisSOLIRIS that was previously manufactured at ARIMFthe Alexion Rhode Island Manufacturing Facility (ARIMF) and a payment with respect to sales of SolirisSOLIRIS manufactured at Lonza facilities. During 2015, we entered into a new supply agreement withThe ARIMF site was sold in 2018. Lonza whereby Lonza will constructis in the process of qualifying a new manufacturing facility dedicated to Alexion manufacturing at one of its existing facilities.products and commitments entered into under this arrangement are included in the non-cancellable commitments amount noted above.
In addition, we have non-cancellable commitments of approximately $27$104.1 through 20192020 with other third party manufacturers.
InMarch 2013, we received a Warning Letter (Warning Letter) from the FDA regarding compliance with current Good Manufacturing Practices (cGMP) at ARIMF. The Warning Letter followed receipt of a Form 483 Inspectional Observations by the FDA in connection with an FDA inspection that concluded in August 2012. The observations relate to commercial and clinical manufacture of Soliris at ARIMF. We responded to the Warning Letter in a letter to the FDA dated in April 2013. As previously disclosed, the FDA issued Form 483s in August 2014 and August 2015 relating to observations at ARIMF and the inspectional observations from the August 2014 and 2015 Form 483s have since been closed out by the FDA. During July 2016, the FDA completed a routine inspection at ARIMF and have since confirmed receipt of our responses to the inspectional observations included in the Form 483 received during that inspection. We continue to manufacture products, including Soliris, at ARIMF, and we anticipate that the supply of Soliris to patients will not be interrupted as a result of the inspectional observations. While the resolution of the issues raised in the Warning Letter is difficult to predict, we do not currently believe a
loss related to this matter is probable or that the potential magnitude of such loss or range of loss, if any, can be reasonably estimated.
In April 2014, we purchased a fill/finish facility in Athlone, Ireland. After regulatory approvals, the facility willIreland, which has been refurbished to become our first company-owned fill/finish facility for our commercial and clinical products.facility. In July 2016, we announced plans to construct a new biologics manufacturing facility at this site, whichthe construction of this facility is expected to be completed by 2018.on-going and, based on current expectations, we anticipate this facility will receive regulatory approval in 2020.
In May 2015, we announced plans to construct a new biologics manufacturing facility on our existing property in Dublin, Ireland, which is expected to be completed bythe construction of this facility has commenced and, based on current
expectations, we anticipate this facility will receive regulatory approval in 2020.
Sales and Marketing
We have established a commercial organization to support current and future sales of our products in the U.S., Europe, Japan, Latin America, Asia Pacific countries, and other territories. Our sales force is small compared to that ofthose for other drugspharmaceutical companies with similar revenues; however, we believe that a relatively smaller sales force is appropriate to effectively market our products due to the incidence and prevalence of rare diseases. If we receive regulatory approval in new territories or for new products or indications, we may expand our own commercial organizations in such territories and market and sell our products through our own sales force in these territories. However, we evaluate each jurisdiction on a country-by-country basis, and, in certain territories, we promote our products in collaboration with marketing partners or rely on relationships with one or more companies with established distribution systems and direct sales forces in certain countries. In addition, we have recently announced that, in an effort to align the structure of our commercial organization with our re-focused corporate strategy and to realize operational efficiencies, certain portions of our international commercial operations will transition to a new operating model in which sales and marketing efforts in the designated countries will rely to a greater extent on third-party entities and alliances to promote and sell our products, and our direct sales presence will decrease in these regions (as we focus our direct sales resources on those regions where it can have a more cost-effective impact).
Customers
Our customers are primarily comprised of distributors, pharmacies, hospitals, hospital buying groups, and other healthcare providers. In some cases, we may also sell our products to governments and government agencies.
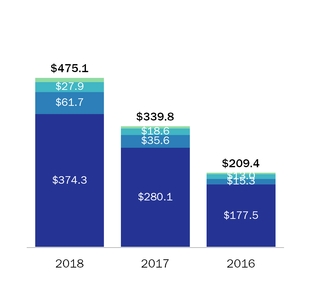
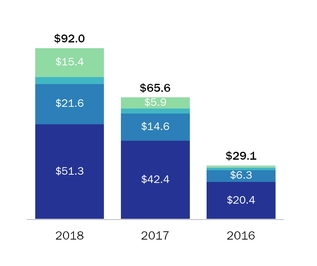
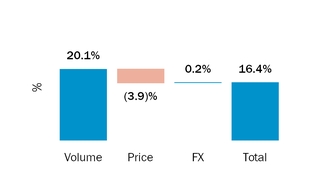
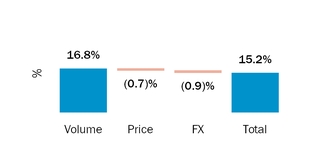
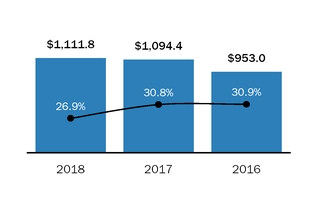
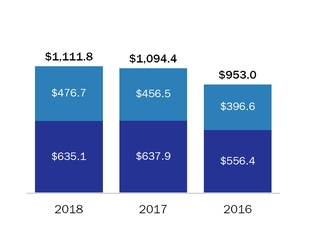
During 2016 and 2015, sales to our largest customerFor the year ended December 31, 2018, four customers accounted for 16% and 18% respectively,50.3% of netour product sales, with these individual customers ranging from 10.0% to 16.4% of product sales. For the year ended December 31, 2017, three customers accounted for 37.0% of our product sales, with these individual customers ranging from 10.8% to 15.0% of product sales. For the year ended December 31, 2016, three customers accounted for 36.7% of our product sales, with these individual customers ranging from 10.0% to 16.0% of product sales.
Because of factors such as the pricing of our products, the limited number of patients, the short period from product sale to patient use and the lack of contractual return rights, customers often carry limited inventory. We also monitor inventory within our sales
channels to determine whether deferrals are appropriate based on factors such as inventory levels compared to demand, contractual terms, financial strength of distributors and our ability to estimate returns.
Please also see “Management’s“Management’s Discussion and Analysis – Net Product Sales,” and Note 1819 “Segment Information” of the Consolidated Financial Statements included in this Annual Report on Form 10-K, for financial information aboutby geographic areas.
Intellectual Property Rights and Market Exclusivity
Patents and other intellectual property rights protect our investment in discovering, developing and marketing our products, and are therefore important to our business. We own or license a number ofrights to many patents in the U.S. and foreign countries that cover our products and investigational compounds. We also file and prosecute many patent applications covering new technologies and inventions that we believe are or may become meaningful to our business. In addition to patents, we rely on trade secrets, know-how, trademarks, regulatory exclusivity and other forms of intellectual property.property and regulatory exclusivity. Our intellectual property rights have material value and we act to protect them.
In the biopharmaceutical industry, two forms of intellectual property generally determine the period of a product’s market exclusivity: patentPatent rights and regulatory forms of exclusivity. Duringprotections are the two principal considerations that determine the period of market exclusivity an innovative product generally realizes mostfor our products. It is during the period of itsmarket exclusivity that our products have their greatest commercial value.
Patents provide the owner with a right to exclude others from practicing an invention.invention for a defined period of time. In our business, patents may cover the active ingredients, uses, formulations, doses, administrations, delivery mechanisms, manufacturing processes and other aspects of a product. The period of patent protection for any given product may depend on the expiration date of various patents and may differ from country to country according to the type of patents, the scope of coverage and the remedies for infringement available in a country.
Most of our products and investigational compounds are protected by patents with varying terms that depend on the type of patent and its filing date. However, Because a significant portion of a biopharmaceutical product’s patent lifeprotection can elapse during the time it takes to developcourse of developing and obtainobtaining regulatory approval of the product. As compensation for such delayproduct, certain countries willprovide compensatory mechanisms to extend a patent’s term, subject to a number of factors and caps.patent terms for the biopharmaceutical products.
Regulatory forms of exclusivityprotections are another source of valuableexclusive rights that can contribute toward market exclusivity for an innovative biopharmaceutical product.our products. Many developed countries provide such non-patent incentives to develop medicines. In the U.S., Europe and Japan, for instance, regulatory intellectual property rights provide incentives to develop medicines for rare diseases, or orphan drugs, and medicines for pediatric patients. ThoseFor example, countries and others also provide data protection for a
period of time after the approval of a new drug, during which regulatory agencies may not rely on the innovator’s data to approve a biosimilar or generic copy. Some countries provide additional incentives to develop medicines for rare diseases, or orphan drugs, and medicines for pediatric patients. Regulatory forms of exclusivityprotections can work in
conjunction with patents to strengthen market exclusivity, and in countries where patent protection has expired or does not exist, regulatory forms of exclusivityprotections can extend a product’s market exclusivity period. Different forms of regulatory protection are described in the section of this Annual Report on Form 10-K titled Government Regulation. For information regarding lawsuits alleging that ULTOMIRIS infringes patents held by a third party, see Note 11 “Commitments and Contingencies” to the notes to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K.
SolirisSOLIRIS Exclusivity
With respect to Soliris,SOLIRIS, we own an issued U.S. patent that covers the eculizumab composition of matter andthat will expire in 2021, taking into account patent term extension. SolirisWe also own other issued U.S. patents that cover the composition, use and formulation of eculizumab, that expire in 2027. SOLIRIS is also protected in the U.S. by regulatory data exclusivity until 2019 and bythat will expire in March 2019. SOLIRIS also benefits from orphan drug exclusivity for treating gMG until 2024 (orphan drug exclusivity for SOLIRIS for treating PNH and aHUS until 2018.previously expired). In Europe we have supplementary protection certificates that extend rights associated with a composition of matter patent until 2020 in certain countries. SolirisSOLIRIS is also protected in Europe by orphan drug exclusivity until 2019 for PNH, through late 2023 for aHUS and until 2027 for gMG. In Japan we own issued patents that cover the eculizumab composition of matter and will expire in 2019 and 2027. SOLIRIS is also protected in Japan by orphan drug exclusivity until 2020 for PNH, until 2023 for aHUS.aHUS and until 2027 for gMG. In addition to the foregoing patent and regulatory protections, we own other patents and pending patent applications that are directed to various aspects of eculizumab and which may provide additional protection for Soliris.SOLIRIS in the U.S., Europe, Japan and other countries.
StrensiqULTOMIRIS Exclusivity
With respect to Strensiq,ULTOMIRIS, we own issued U.S. patents that cover the composition, use and formulation of ravulizumab which will expire in 2035. ULTOMIRIS is also protected in the U.S. by regulatory data exclusivity until 2030 and we have applied for orphan drug exclusivity for treating PNH, which, if granted, would protect that indication through 2025. Although ULTOMIRIS is not yet approved for any indication in Europe or Japan, it is also protected in those regions by patents that cover ravulizumab which will expire in 2035. If ULTOMIRIS is approved in Europe or Japan, we also expect regulatory protections to apply in those regions. In addition to the foregoing patent and regulatory protections, we own other patents and pending patent applications that are directed to various aspects of ravulizumab and which may provide additional protection
for ULTOMIRIS in the U.S., Europe, Japan and other countries.
STRENSIQ Exclusivity
With respect to STRENSIQ, we own an issued U.S. patent that covers the asfotase alfa composition of matter andthat will expire in 2026. We have applied for an extension of thethis U.S. patent term. StrensiqSTRENSIQ is also protected in the U.S. by orphan drug exclusivity until 2022 and by regulatory data exclusivity until 2027. In Europe, we own two issued patents that cover the asfotase alfa composition of matter and will expire in 2025 and 2028. We have applied for supplementary protection certificates in the European countries. StrensiqSTRENSIQ is also protected in Europe by orphan drug exclusivity and regulatory data exclusivity until 2025. In other countries we own corresponding patents that will expire between 2025 and 2028, not including possible extensions.
KanumaKANUMA Exclusivity
With respect to Kanuma,KANUMA, we own issued patents in the U.S., Europe and other countries that cover methods of using the product to treat LAL-D and will expire in 2031. TheWe maintained the European patent is under challenge in an administrative opposition proceeding.proceeding that was favorably resolved in 2017. An exclusively licensed composition of matter patent also protects KanumaKANUMA in certain European countries until it expires in 2021, though we also applied for supplementary protection certificates in those countries. In the U.S. Kanuma, KANUMA also is protected by orphan drug exclusivity until 2022 and by regulatory data exclusivity until 2027. In Europe it is protected by orphan drug exclusivity and regulatory data exclusivity until 2025.
Soliris, Strensiq, and Kanuma Regulatory ProtectionInvestigational Compounds
As noted above, for each of Soliris, Strensiq and Kanuma we rely on regulatory forms of exclusivity such as data protection and orphan drug protection to support the product’s market exclusivity. Specific aspects of the laws governing regulatory exclusivity vary by country, but most forms of regulatory exclusivity do not prevent competitive products from gaining regulatory approval on the basis of the competitor’s own safety and efficacy data, even when the competitive product is a biosimilar or generic copy. In certain countries, however, orphan drugs can obtain a period of exclusivity during which no competitive product containing the same drug may be approved for the same orphan indication.
We also own U.S. and foreign patents and patent applications that protect our investigational compounds and product candidates. At present, it iswe do not knownknow whether any such investigational compound or product candidate will be approved for human use and sale.sale.
License and Collaboration Agreements
From time to time, we enter into arrangements with third parties, including collaboration and licensing arrangements, for the development, manufacture and commercialization of products and product candidates. These strategic alliances are intended to strengthen and advance our R&D capabilities and diversify our product pipeline to support the growth of our marketed product base. The arrangements, which generally provide Alexion with rights to specialized technology and intellectual property for the development of potential product candidates, often require non-refundable, upfront license fees, development, regulatory and commercial milestones, as well as royalty payments on commercial sales.
Importance of Intellectual Property Exclusivities and Rights
The pharmaceutical industry places considerable importance on obtaining and enforcing patent (including licensed patents), trade secret and other intellectual property protection for new therapies, technologies, products, services and processes. Our success therefore depends, in part, on our ability to obtain and enforce our patents (including licensed patents) and other intellectual property rights necessary to protect our current and future products, to obtain and preserve our trade secrets and other confidential intellectual property and to avoid or neutralize intellectual property threats from third parties. The existence of patents does not guarantee our right to practice the patented technology or commercialize the patented product. Litigation, oppositions, inter partes reviews or other proceedings are, have been and may in the future be necessary in some instances to determine the validity and scope of certain of our patents, regulatory exclusivities or other proprietary rights, and in other instances to determine the validity, scope or non-infringement of certain patent rights claimed by third parties to be pertinent to the manufacture, use or sale of our products. We may also face challenges to our patents, regulatory exclusivities and other proprietary rights covering our products by manufacturers of biosimilars. For additional information, see Item 1A “Risk Factors - Risks Related to Intellectual Property” elsewhere in this Annual Report on Form 10-K (including a recent European Patent Office ruling to revoke a previously issued patent relating to the formulation of SOLIRIS).
Government Regulation
Drug Development and Approval in the United States
The preclinical studies and clinical testing, manufacture, labeling, storage, record keeping, advertising, promotion, pharmacovigilance reporting, export, and marketing, among other things, of our products and product candidates, , including Soliris, StrensiqULTOMIRIS, SOLIRIS, STRENSIQ and Kanuma,KANUMA, are subject to extensive regulation by governmental authorities in the US,U.S., the European Union (EU)EU, Japan and other territories. In the U.S., pharmaceutical products are regulated by the FDA under the Federal Food, Drug, and Cosmetic Act and other laws, including, in the case of biologics, the Public Health Service Act. Our threefour approved products are regulated by the FDA as biologics. Biologics require the submission of a Biologics License Application (BLA) and approval by the FDA prior to being marketed in the U.S. In the case of Kanuma,KANUMA, which is derived from egg whites from select hens, we also submitted a New Animal Drug Application (NADA) for approval by the FDA. Manufacturers of biologics and drugs derived from animal origin may also be subject to state regulation. Failure to comply with FDA and state requirements, both before and after product approval, may subject us and/
or our partners, contract manufacturers, and suppliers to administrative or judicial sanctions, including FDA refusal to approve applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, fines and/or criminal prosecution.
The process for obtaining regulatory approval to market a biologic is expensive, often takes many years, and can vary substantially based on the type, complexity, and novelty of the product candidates involved. The steps required before a biologic may be approved for marketing of an indication in the U.S. generally include:
(1) preclinical laboratory tests and animal tests;
(2) submission to the FDA of an investigational new drug (IND) application for human clinical testing, which must become effective before human clinical trials may commence;
(3) adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its intended use;
(4) submission to the FDA of a BLA or supplemental BLA;
(5) FDA pre-approval inspection of the manufacturing sites identified in the BLA; and
(6) FDA review and approval of the BLA or supplemental BLA.
Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as toxicological and pharmacological animal studies to assess the potential safety and efficacy of the product candidate. Preclinical safety tests intended for submission to FDA must be conducted in compliance with FDA’s Good Laboratory Practice (GLP) regulations and the U.S. Department of Agriculture’s Animal Welfare Act. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND application which must become effective before human clinical trials may be commenced. The IND will automatically become effective 30 days after receipt by the FDA, unless the FDA, before that time, raises concerns about the drug candidate or the conduct of the trials as outlined in the IND. The IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed. We cannot assure you that submission of an IND will result in FDA authorization to commence clinical trials or that once commenced, other concerns will not arise.arise that will prevent the trials from moving forward. FDA may stop the clinical trials by placing them on “clinical hold” because of concerns about the safety of the product being tested, or for other reasons.
Clinical trials involve the administration of the investigational product to healthy volunteers or to patients, under the supervision of qualified principal investigators. The conduct of clinical trials is subject to
extensive regulation, including compliance with the FDA’s bioresearch monitoring regulations and Good Clinical Practice (GCP) requirements, which establish standards for conducting, recording data from, and reporting the results of clinical trials, and are intended to assure that the data and reported results are credible and accurate, and that the rights, safety, and well-being of study participants are protected. Clinical trials must be conducted in accordance with protocols that detail the objectives of the study, the criteria for determining subject eligibility, the dosing plan, patient monitoring requirements, timely reporting of adverse events, and other elements necessary to ensure patient safety, and any efficacy criteria to be evaluated. Each protocol must be submitted to FDA as part of the IND; further, each clinical study at each clinical site must be reviewed and approved by an independent institutional review board, prior to the recruitment of subjects. The institutional review board’s role is to protect the rights and welfare of human subjects involved in clinical studies by evaluating, among other things, the potential risks and benefits to subjects, processes for obtaining informed consent, monitoring of data to ensure subject safety, and provisions to protect the subjects’ privacy. Foreign studies conducted under an IND application must meet the same requirements that apply to studies being conducted in the U.S. Data from a foreign study not conducted under an IND may be submitted in support of a BLA if the study was conducted in accordance with GCP and FDA is able to validate the data.
Clinical trials are typically conducted in three sequential phases, but the phases may overlap and different trials may be initiated with the same drug candidate within the same phase of development in similar or differing patient populations. Phase I studies may be conducted in a limited number of patients, but are usually conducted in healthy volunteer subjects. The drug is usually tested for safety and, as appropriate, for absorption, metabolism, distribution, excretion, pharmaco-dynamics and pharmaco-kinetics. Phase II usually involves studies in a larger, but still limited patient population to evaluate preliminarily the efficacy of the drug candidate for specific, targeted indications; to determine dosage tolerance and optimal dosage; and to identify possible short-term adverse effects and safety risks.
Phase III trials are undertaken to gather additional information to evaluate the product’s overall risk-benefit profile, and to provide a basis for physician labeling. Phase III trials evaluate clinical efficacy of a specific endpoint and test further for safety within an expanded patient population at geographically dispersed clinical study sites. Phase I, Phase II or Phase III testing might not be completed successfully within any specific time period, if at all, with respect to any of our product candidates. Results from one trial are not necessarily predictive of results from later trials. Furthermore, the FDA, sponsor or institutional review board may suspend
clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
We must register each controlled clinical trial, other than Phase I trials, on a website administered by National Institutes of Health (NIH) (http://clinicaltrials.gov). Registration must occur not later than 21 days after the first patient is enrolled, and the submission must include descriptive information (e.g., a summary in lay terms of the study design, type and desired outcome), recruitment information (e.g., target number of participants and whether healthy volunteers are accepted), location and contact information, and other administrative data (e.g., FDA identification numbers). Within one year of a trial’s completion, information about the trial including characteristics of the patient sample, primary and secondary outcomes, trial results written in lay and technical terms, and the full trial protocol must be submitted to the FDA.NIH. The results information is posted to the website unless the drug has not yet been approved, in which case the FDANIH posts the information shortly after approval. A BLA, BLA supplement, and certain other submissions to the FDA require certification of compliance with these clinical trials database requirements. There are proposals to expand these registration requirements to additional studies.
The results of the preclinical studies and clinical trials, together with other detailed information, including information on the manufacture and composition of the product and proposed labeling for the product, are submitted to the FDA as part of a BLA requesting approval to market the product candidate for a proposed indication. Under the Prescription Drug User Fee Act, as amended, the fees payable to the FDA for reviewing a BLA, as well as annual fees for commercial manufacturing establishments and for approved products, can be substantial. The BLA review fee alone can exceed $2$2.0 subject to certain limited deferrals, waivers and reductions that may be available. Each BLA submitted to the FDA for approval is typically reviewed for administrative completeness and reviewability within sixty days following submission of the application. If the FDA finds the BLA sufficiently complete, the FDA will “file” the BLA, thus triggering a full review of the application. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission. FDA performance goals provide for action on an applicationwithin 12 months of submission. The FDA, however, may not approve a drug within these established goals and its review goals are subject to change from time to time because the review process is often significantly extended by FDA requests for additional information or clarification. As part of its review, the FDA may refer the BLA to an advisory committee composed of outside experts for evaluation and a recommendation as to whether the application should be approved. Although the FDA is not bound by the recommendation of an advisory committee, the agency usually has followed such recommendations.
Further, the outcome of the review, even if generally favorable, may not be an actual approval but instead a “complete response letter” communicating the FDA’s decision not to approve the application, outlining the deficiencies in the BLA, and identifying what information and/or data (including additional pre-clinical or clinical data) is required before the application can be approved. Even if such additional information and data are submitted, the FDA may decide that the BLA still does not meet the standards for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we do.
Before approving a BLA, the FDA typically will inspect the facilities at which the product is manufactured and will not approve the product unless the facilities comply with the FDA’s cGMPcurrent Good Manufacturer Practice (cGMP) requirements. The FDA may deny approval of a BLA if applicable statutory or regulatory criteria are not satisfied, or may require additional testing or information, which can delay the approval process. FDA approval of any applicationBLA may include many delays and requests for additional information or never be granted. If a product is approved, the approval will impose limitations on the indicated uses for which the product may be marketed, may require that warning statements be included in the product labeling, and may require that additional studies be conducted following approval as a condition of the approval. FDA also may impose restrictions and conditions on product distribution, prescribing or dispensing in the form of a Risk Evaluation and Mitigation StrategiesStrategy (REMS), or otherwise limit the scope of any approval. A REMS may
include various elements, ranging from a medication guide to limitations on who may prescribe or dispense the drug, depending on what the FDA considers necessary for the safe use of the drug. To market a product for other indicated uses, or to make certain manufacturing or other changes, requires FDA review and approval of a BLA Supplementsupplement or new BLA and the payment of applicable review fees. Further post-marketing testing and surveillance to monitor the safety or efficacy of a product may be required. In addition, new government requirements may be established that could delay or prevent regulatory approval of our product candidates under development.
In 2010, the Biologics Price Competition and Innovation Act (BPCIA) was enacted, creating a statutory pathway for licensure, or approval, of biological products that are biosimilar to, and possibly interchangeable with, reference biological products licensed under the Public Health Service Act. The objectives of the BPCIA are conceptually similar to those of the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the “Hatch-Waxman Act”, which established abbreviated pathways for the approval of small molecule drug products. Under the BPCIA, innovator manufacturers of original reference biological products are granted 12 years of exclusive use before
biosimilar versions of such products can be licensed for marketing in the U.S. This means that the FDA may not approve an application for a biosimilar version of a reference biological product until 12 years after the date of approval of the reference biological product (with a potential six-month extension of exclusivity if certain pediatric studies are conducted and the results reported to FDA), although a biosimilar application may be submitted four years after the date of licensure of the reference biological product. Additionally, the BPCIA establishes procedures by which the biosimilar applicant must provide information about its application and product to the reference product sponsor, and by which information about potentially relevant patents is shared and litigation over patents may proceed in advance of approval. The BPCIA also provides a period of exclusivity for the first biosimilar to be determined by the FDA to be interchangeable with the reference product.
FDA has released numerous guidance documents interpreting the BPCIA in recent years. These guidance documents, among other things, elaborate on the definition of a biosimilar as a biological product that is highly similar to an already approved biological product, notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biosimilar and the approved biological product in terms of the safety, purity, and potency. More recently,The FDA has also released final guidance documents on the assignment of nonproprietary, clearly distinguishable nonproprietary product names for both biologic and biosimilar products, labeling for biosimilar products, and interchangeability.questions and answers on issues involving biosimilar development, as well as draft guidance on interchangeability and evaluation of analytical similarity.
The FDA approved the first biosimilar product under the BPCIA in 2015, and theas of December 2018, sixteen (16) biosimilar products have been approved in total. The agency continues to refine the procedures and standards it will apply in implementing this approval pathway. In July 2018, the FDA issued a Biosimilars Action Plan, asserting its intent to take steps to facilitate biosimilars competition. We anticipate that the contours of the BPCIA will continue to be defined as the statute is implemented over a period of years. This likely will be accomplished by a variety of means, including FDA issuance of guidance documents, proposed regulations, and decisions in the course of considering specific applications. The approval of a biologic product biosimilar to one of our products, including SOLIRIS, could have a material impact on our business because it may be significantly less costly to bring to market and may be priced significantly lower than our products.
Both before and after the FDA approves a product, the manufacturer and the holder or holders of the BLA, and in the case of Kanuma,KANUMA, the NADA, for the product are subject to comprehensive regulatory oversight. If ongoing regulatory requirements are not satisfied or if
safety problems occur after the product reaches the market, the FDA may at any time withdraw its approval or take actions that would suspend marketing. For example, quality control and manufacturing procedures must conform, on an ongoing basis, to cGMP requirements, and the FDA periodically subjects manufacturing facilities to unannounced inspections to assess compliance with cGMP. Failure to comply with applicable cGMP requirements and other conditions of product approval may lead the FDA to take regulatory action, including fines, recalls, civil penalties, injunctions, suspension of manufacturing operations, operating restrictions, withdrawal of FDA approval, seizure or recall of products, and criminal prosecution. Accordingly, manufacturers must continue to spend time, money, and effort to maintain cGMP compliance.
The FDA and other federal regulatory agencies also closely regulate the promotion of drugs and biologics through, among other things, standards and regulations for direct-to-consumer advertising, communications regarding unapproved uses, industry-sponsored scientific and educational activities, and promotional activities involving the Internet and social media. A product cannot be commercially promoted before it is approved. After approval, product promotion can include only those claims relating to safety and effectiveness that are consistent with the labeling approved by the FDA. Healthcare providers are permitted to prescribe drugs and biologics for “usesuses not approved by the FDA and therefore not described in the product’s labeling - because the FDA does not regulate the practice of medicine. However, FDA regulations impose stringent restrictions on manufacturers’ communications regarding such uses. Broadly speaking, a manufacturer may not promote a drug or biologic for an unapproved use, but may engage in non-promotional, balanced communication regarding such uses under certain conditions. Failure to comply with applicable FDA requirements and restrictions in this area may subject a company to adverse publicity and enforcement action by the FDA, the Department of Justice, or the Office of the Inspector General (OIG) of the
Department of Health and Human Services (HHS), as well as state authorities. Noncompliance could subject a company to a range of penalties that could have a significant commercial impact, including civil and criminal fines and agreements that materially restrict the manner in which a company promotes or distributes drug or biologic products.
Orphan Drug Designation in the U.S., the EU and Other Foreign Jurisdictions
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs and biological products intended to treat a “rare disease or condition,” which generally is a disease or condition that affects fewer than two hundred thousand individuals in the U.S. Orphan drug designation must be requested before submitting a BLA or supplemental BLA. AfterIf the FDA grants
orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are publicly disclosed by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. If a product which has an orphan drug designation subsequently receives the first FDA approval for that drug or biologic for the indication for which it has such designation, the product is entitled to an orphan exclusivity period, in which the FDA may not approve any other applications to market the same drug or biologic for the same indication for seven years, except in limited circumstances, such as where the sponsor of a different version of the product is able to demonstrate that its product is clinically superior to the approved orphan drug product. This exclusivity does not prevent a competitor from obtaining approval to market a different product that treats the same disease or condition or the same product to treat a different disease or condition. The FDA can revoke a product’s orphan drug exclusivity under certain circumstances, including when the holder of the approved orphan drug application is unable to assure the availability of sufficient quantities of the drug to meet patient needs. A sponsor of a product application that has received an orphan drug designation is also granted tax incentives for clinical research undertaken to support the application. In addition, the FDA will typically coordinate with the sponsor on research study design for an orphan drug and may exercise its discretion to grant marketing approval on the basis of more limited product safety and efficacy data than would ordinarily be required.
MedicinalIn the EU, medicinal products: (a) that are used to treat or prevent life-threatening or chronically debilitating conditions that affect no more than five in ten thousand people in the EU;EU when the application is made; or (b) that are used to treat or prevent life-threatening or chronically debilitating conditions and that, for economic reasons, would be unlikely to be developed without incentives; and (c) where no satisfactory method of diagnosis, prevention or treatment of the condition concerned exists, or, if such a method exists, the medicinal product would be of significant benefit to those affected by the condition, may be granted an orphan designation in the EU.designation. The application for orphan designation must be submitted to the EMA and approved before an application is made for marketing authorization for the product. Once authorized, orphan medicinal products are entitled to up to ten years of market exclusivity.exclusivity (which may be extended for an additional two years if pediatric data have been produced in accordance with an agreed pediatric investigational plan). During this ten year period, with a limited number of exceptions, neither the competent authorities of the EU member states,Member States, the EMA, or the EC are permitted to accept applications or grant marketing authorization for other similar medicinal products with the same therapeutic indication. However, marketing authorization may be granted to a similar
medicinal product with the same orphan indication during the ten year period with the consent of the marketing authorization holder for the original orphan medicinal product or if the manufacturer of the original orphan medicinal product is unable to supply sufficient quantities. Marketing authorization may also be granted to a similar medicinal product with the same orphan indication if this latter product is safer, more effectiveefficacious or otherwise clinically superior to the original orphan medicinal product. The period of market exclusivity may, in addition, be reduced to six years if it can be demonstrated on the basis of available evidence that the originalcriteria for orphan designation are no longer met or if the orphan medicinal product is sufficiently profitable not to justify maintenance of market exclusivity.
SolirisULTOMIRIS has received orphan drug designation for the treatment of patients with PNH in the U.S., EU and Japan, and for the subcutaneous treatment of patients with aHUS in the U.S. SOLIRIS has received orphan drug designation for (a) the treatment of PNH and aHUS in the U.S., the EU, and in several other territories; (b) the prevention of delayed graft function in renal transplant patients in the U.S.; (c) the treatment of patients with myasthenia gravisgMG in the U.S., Japan, and the EU; and (d) the prevention of graft rejection and delayed graft rejection following solid organ transplantation in the EU.EU and (e) and for the treatment of NMOSD in the U.S., EU, and Japan. In 2008, StrensiqSTRENSIQ received orphan drug designation for the treatment of patients with HPP in the U.S. and the EU, and in Japan in November 2014. Furthermore, in 2010, KanumaKANUMA received orphan drug designation for the treatment of LAL-D in the U.S. and the EU. OrphanAs noted above, orphan drug designation provides certain regulatory and filing fee advantages, including market exclusivity, except in limited circumstances, for several years after approval.
Breakthrough Designation in the U.S.
Congress has created the Breakthrough Therapy designation program under which the FDA may grant Breakthrough Therapy status to a drug intended for the treatment of a serious condition when preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint over existing therapies. The Breakthrough Therapy designation, which may be requested by a sponsor when filing or amending an IND, is intended to facilitate and expedite the development and FDA review of a product candidate. Specifically, the Breakthrough Therapy designation may entitle the sponsor to more frequent meetings with FDA during drug development, intensive guidance on clinical trial design, and expedited FDA review by a cross-disciplinary team comprised of senior managers. The designation does not guarantee a faster
development or review time as compared to other drugs however, nor does it assure that the drug will obtain ultimate marketing approval by the FDA. Once granted, the FDA may withdraw this
designation at any time.time if subsequent data no longer support the breakthrough therapy designation. We have received Breakthrough Therapy designations for StrensiqSTRENSIQ for HPP in perinatal-, infant-, and juvenile-onset patients; and for KanumaKANUMA in the treatment of LAL-D presenting in infants; and for cyclic Pyranopterin Monophosphate, intended to treat Molybdenum Cofactor Deficiency Type A. Because the Breakthrough Therapy designation program is relatively new, itinfants. It is difficult for us to predict the impact that these designations will have on the development and FDA review of our products.
21st Century Cures Act (the Cures Act)
In December 2016, Congress passed the Cures Act which included a number of provisions designed to speed development of innovative therapies, provide funding authorization to the NIH, and provide funding for certain oncology-directed research. Because the FDA is still working to implement many aspects of the Cures Act, has only recently been enacted, its potential affecteffect on our business remains unclear with the exception of a provision requiring that we post our policies on the availability of expanded access programs for individuals. In addition, the Cures Act includes requiring the FDA to assess and publish guidance on the use of novel clinical trial designs, the use of real world evidence in applications, the availability of summary level review for supplemental applications for certain indications, and the qualification of drug development tools. Because these provisions allow the FDA to spend several years developing these policies, the effect on us could be delayed. At this time, we cannot anticipate what effect these future policies may have on our business.
The Cures Act also authorizes $1,800$1,800.0 in funding for “cancer moonshot”the “Cancer Moonshot” initiative (the Initiative) to be run by the NIH. The Cancer Moonshot Initiative’s strategic goals encourage inter-agency cooperation and fund research and innovation to catalyze new scientific breakthroughs, bring new therapies to patients, and strengthen prevention and diagnosis. The Initiative aims to stimulate drug development through the creation of a public-private partnership with 20 to 30 pharmaceutical and biotechnology companies to expedite cancer researchers’ access to investigational agents and approved drugs. This partnership is designed to permit researchers to obtain drugs and other technologies from a preapproved “formulary” list without having to negotiate with each company for individual research projects. We will monitor these developments but cannot currently assess how the Initiative may impact our businessbusiness.
Foreign Regulation of Drug Development and Approval
In addition to regulations in the U.S., we are subject to a variety of foreign regulatory requirements including those governing human clinical trials, marketing approval, and post-marketing regulation for drugs. The foreign regulatory approval process includes all of the risks associated with FDA approval set forth above, as well as additional country-specific regulations. Whether or not we obtain FDA approval for a product, we must
obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. Approval by one regulatory authority does not ensure approval by regulatory authorities in other jurisdictions. The approval process varies from country to country, can involve additional testing beyond that required by FDA, and may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing, promotion, and reimbursement vary greatly from country to country.
Under the EU regulatory system, we may submit applications for marketing authorizations either under a centralized, decentralized, or mutual recognition marketing authorization procedure. The centralized procedure provides for the grant of a single marketing authorization for a medicinal product by the EC on the basis of a positive opinion by the EMA.EMA and is mandatory for certain categories of medicinal products, such as orphan medicinal products. A centralized marketing authorization is valid for all EU member statesMember States and three of the four EFTA States (Iceland, Liechtenstein and Norway).European Economic Area states. The decentralized procedure and the mutual recognition procedure apply between EU member states.Member States. The decentralized marketing authorization procedure involves the submission of an application for marketing authorization to the competent authority of all EU member states in which the product is to be marketed. One national competent authority, selected by the applicant, assesses the application for marketing authorization. The competent authorities of the other EU member statesMember States are subsequently required to grant marketing authorization for their territory on the basis of this assessment, except where grounds of potential serious risk to public health require this authorization to be refused. The mutual recognition procedure provides for mutual recognition of marketing authorizations delivered by the national competent authorities of EU member statesMember States by the competent authorities of other EU member states.Member States. The holder of a national marketing authorization may submit an application to the competent authority of aan EU member state requesting that this authority recognize the marketing authorization delivered by the competent authority of another EU member state for the same medicinal product. The EC may agree upon recommendation of the EMA to grant for medicines designated as orphan medicines a (i) conditional marketing authorization in the interest of public health under certain conditions; namely that unmet medical needs will be fulfilled, the benefit-risk balance of the product is positive, the benefit to public health of the medicinal product’s immediate availability on the market outweighs the risks due to need for further data and it is likely that the applicant will be able to provide comprehensive data; or (ii) marketing authorization under “exceptional circumstances” when the applicant can show that it is unable to provide
comprehensive data on the efficacy and safety under normal conditions of use and subject to specific procedures being introduced. This may arise in particular when the intended indications are very rare, in the present state of scientific knowledge, it is not possible to provide comprehensive information, or when generating data may be contrary to generally accepted ethical principles.
Similarly to the U.S., both marketing authorization holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA and the competent authorities of the individual EU member statesMember States both before and after grant of the manufacturing and marketing authorizations. This includes control of compliance by the entities withcompanies within the EU legal framework (i.e., GCP, GLP, cGMP and pharmacovigilance rules, which govern quality control of the manufacturing process and require documentation policies and procedures.procedures). We
and our third party manufacturers are required under regulations to ensure that all of our processes, methods, and equipment are compliant with cGMP.GCP, GLP, cGMP and pharmacovigilance rules. The EMA and national competent authorities may arrange inspections to ensure that we adhere to these principles and regulations. Any adverse findings from such inspections, depending on their severity, may result in significant delays in obtaining a marketing authorization, may impose penalties or may result in other action by regulatory authorities.
Failure by us or by any of our third party partners, including suppliers, manufacturers, and distributors to comply with EU laws and the related national laws of individual EU member statesMember States governing the conduct of clinical trials, manufacturing approval, marketing authorization of medicinal products, pre-approval promotion of products, reporting of adverse health events, both before and after grant of marketing authorization, and marketingmarketing/promotion of such products following grant of authorization may result in administrative, civil, or criminal penalties. These penalties could include delays in or refusal to authorize the conduct of clinical trials or to grant marketing authorization, product withdrawals and recalls, product seizures, suspension, or variation of the marketing authorization, total or partial suspension of production, distribution, manufacturing, or clinical trials, operating restrictions, injunctions, suspension of licenses, fines, and criminal penalties.
The EU has had an established regulatory pathway for biosimilars since 2005 and has approved several biosimilar products. In addition, in February 2017 the EMA will launchlaunched a pilot project with the aim of providing scientific advice to companies for the development of new biosimilar products.
The approval of a biosimilar of one of our products marketed in the EU could have a material impact on our business. The biosimilar may be less costly to bring to
market, may be priced significantly lower than our products, and result in a reduction in the pricing and reimbursement of our products.
Pharmaceutical Pricing and Reimbursement
Sales of pharmaceutical products depend in significant part on the extent of coverage and reimbursement from third party payers, including government programs includingsuch as Medicare and Medicaid in the U.S., and other third party payers.U.S, as well as private health insurers. Third party payers are sensitive to the cost of drugs and are increasingly seeking to implement cost containment measures to control, restrict access to, or influence the purchase of drugs, biologicals,biologics, and other health care products and services. GovernmentsFor example, governments may regulate reimbursement, pricing, and coverage of products in order to control costs or to affect utilization levels of use of certain products. PrivateIn addition, private health insurance plans may restrict coverage of some products, such as by using payerdrug formularies under which only selectedselect drugs or uses of select drugs are covered, through the implementation of variable co-paymentspatient co-payment obligations that make non-preferred drugs that are not preferred by the payer more expensive for patients, and by employing utilization management controls, such as requirements for prior authorization or prior failure on another type of treatment.treatment before the insurer will cover and reimburse a particular therapy. Payers may especially impose these obstacles to coverage for higher-priced drugs such as those we sell. Consequently, all of our products may be subject to payer-driven restrictions, rendering patients responsible for a higher percentage of the total cost of drugs in the outpatient setting. This can lower the demand for our products if the increased patient cost-sharing obligations are more than theypatients can afford.
Medicare is a U.S. federal government insurance program that covers individuals aged 65 years or older, as well as individuals of any age with certain disabilities, and individuals with End-Stage Renal Disease. The primaryend-stage renal disease. Our products are primarily reimbursed by Medicare programs that may affect reimbursement for Soliris areunder Medicare Part B, which generally covers physician services and outpatient care, including some outpatient prescription drugs under limited conditions, and Medicare Part D, which provides a voluntaryan outpatient prescription drug benefit.benefit for Medicare beneficiaries.
Generally speaking, Medicare Part B provides limited coverage of certain outpatient drugs and biologicalsbiologics that are reasonable and necessary for diagnosis or treatment of an illness or injury. Under Part B, reimbursement for most drugs is based on a fixed percentage above the applicable product’s average sales price (ASP). Manufacturers calculate ASP based on a statutory formula and must report ASP information to the Centers for Medicare and Medicaid Services (CMS), the federal agency within HHS that administers Medicare and the Medicaid Drug Rebate Program, on a quarterly basis. Under the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, Medicare
pays physicians and suppliers ASP + 6.0% for most Part B-covered drugs and biologics (Medicare payments for ULTOMIRIS will also be based on the ASP formula commencing with the period two quarters after approval). Medicare payment for separately payable Part B drugs reimbursed through the hospital outpatient prospective payment system is generally under the discretion of CMS, meaning it can be changed without legislative action from Congress. The current reimbursement rate for most separately payable Part B drugs and biologicalsused in both the hospital outpatient department setting and the physician office setting is ASP + 6%. The rate for the physician clinic setting is set by statute, but CMS has the authority to adjust the rate for the hospital outpatient setting onis ASP plus 6.0%. One exception, however, is that, effective January 1, 2018, Medicare pays 340B hospital covered entities ASP minus 22.5% for separately payable Part-B covered drugs and biologics that were purchased under the 340B Program in an annual basis. This reimbursement rate may decreaseoutpatient clinic setting, as discussed further below. In addition, the sequester that is currently in the future.place through 2027, reduces payments providers receive for Part B-Covered drugs by 1.6%, which results in a net payment equivalent to ASP plus 4.3%. The sequester affects other Medicare payments and is also discussed in more detail below. In both settings (i.e., physician office and hospital outpatient), the amount of reimbursement is updated quarterly based on the manufacturer’s submission of new ASP information.
Medicare Part D is aan outpatient prescription drug benefit available to all Medicare beneficiaries. It is a voluntary benefit that is implemented through private insurance plans under contractual arrangements withbetween the plans and the federal government. Similar to pharmaceutical coverage through private health insurance, Part D plans develop formularies, impose utilization controls (such as prior authorization, step therapy, and quantity limits), and negotiate discounts from drug manufacturers. Medicare Part D coverage is available through private plans, andBecause of this, the list of prescription drugs covered by Part D plans varies by plan. However, with limited exceptions, individual plans are required by statute to cover certain therapeutic categories and classes of drugs or biologicalsbiologics and to have at least two drugs in each unique therapeutic category or class, with certain exceptions.class.
Our products can also be provided under Medicare Parts A and C (Medicare Advantage). Medicare Part A generally covers inpatient hospital benefits. Hospitals typically receive a single payment for an inpatient stay depending on the Medicare Severity Diagnosis Related Group (MS-DRG) to which the inpatient stay is assigned. The MS-DRG for a hospital inpatient stay varies based on the patient’s condition. Hospitals generally do not receive separate payment for drugs and biologicalsbiologics administered to patients during an inpatient hospital stay. As a result, hospitals may not have a financial incentive to utilize our products for inpatients.inpatients where lower cost alternative therapies are available. Finally, Medicare beneficiaries can receive their Part A, B, and D benefits through a Medicare Advantage organization plan that is administered by a private insurance company pursuant to Medicare Part C. Similar to private health
insurance plan, Medicare Advantage organization plans negotiate discounts with health care providers and implement utilization controls, including, most notably, step therapy for Part B drugs beginning January 1, 2019.
Beginning April 1, 2013, the Budget Control Act of 2011, Pub. L. No. 112-25, as amended, by the American Taxpayer Relief Act of 2012, Pub. L. 112-240, requiredrequires Medicare payments for all items and services, including drugs and biologicals,biologics, to be reduced by 2%up to 2.0% under sequestration (i.e., automatic spending reductions)reductions, calculated each year by the Office of Management and Budget). Subsequent legislation extended the 2%2.0% reduction, on average, to 2025.2027. This 2%2.0% reduction in Medicare payments affects all Parts of the Medicare program and could impact sales of our products. Additional sequestration orders could also be triggered, potentially resulting in up to a 4% reduction in Medicare payments.
Pursuant to the Medicaid Drug Rebate Statute (42 U.S.C. § 1396r-8(a)(1)), we are required to participate in the Medicaid Drug Rebate Program in order for federal payment to be available for our products under Medicaid and Medicare Part B. Medicaid is a government health insurance program for eligible low-income adults, children, families, pregnant women, and people with certain disabilities. It is jointly funded by the federal and state governments, and it is administered by individual states within parameters established by the federal government. CoverageAs a result, coverage and reimbursement requirements for drugs and biologics thus variesvary by state. DrugsFor example, drugs and biologics may be covered under the medical or pharmacy benefit. Statebenefit, and state Medicaid programs may impose different utilization management controls, such as prior authorization, step therapy, or quantity limits on drugs and biologics.biologics, subject to federal limitations for such controls. But all states must generally provide coverage and reimbursement for a manufacturer’s covered outpatient drugs, as that term is defined by applicable law, if a manufacturer participates in the Medicaid also includesDrug Rebate Program.
Under the Medicaid Drug Rebate Program, under which we are required to, among other things, pay a rebate to each state Medicaid program for quantities of our products utilized on an outpatient basis (with some exceptions) that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program asprogram. Medicaid Drug Rebate Program Rebates are calculated using a condition of having federal funds being made available to the states for our products under Medicaidstatutory formula, state-reported utilization data, and Medicare Part B. Those rebatespricing data that are based on pricing datacalculated and reported by us on a monthly and quarterly basis to CMS. These data include the average manufacturer price and, in the case of innovator products, the best price for each product we sell.drug. As further described below under “U.S. Healthcare Reform and Other U.S. Healthcare Laws,” the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the PPACA), made significant changes to the Medicaid Drug Rebate Program that could negatively impact our results of operations.
Federal law requires that any company that participates
In addition to participating in the Medicaid Drug Rebate Program, alsofederal law requires manufacturers like us to participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B drug pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities only include a varietyhealth care organizations that have certain federal designations or receive funding from specific federal programs, including Federally Qualified Health Centers, Ryan White HIV/AIDS Program grantees, and certain types of community healthhospitals and specialized clinics, and other entities that receive health services grants from the Public Health Service, as well as certain hospitals that serve a disproportionate share of low-income patients. PPACA expanded the 340B program to include additional types of covered entities: certain children’s hospitals, certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, each as defined by PPACA. However, “orphan drugs” i.e., those designated under section 526 of the FDCA, such as each of our products that have received market authorization are exempted from the ceiling price requirements for these newly-eligible entities when used for the rare disease or condition for which they received an orphan designation. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program. ChangesProgram, and in general, products subject to the Medicaid Drug Rebate Program are also subject to the 340B ceiling price calculation and discount requirement. Any changes to the definition of Medicaid average manufacturer price and the Medicaid rebate amount under PPACA and CMS’s issuance of final regulations implementing those changes also could affect our 340B ceiling price calculation for our products and could negatively impact our results of operations. As described below under “U.S. Healthcare Reform and Other U.S. Healthcare Laws,” PPACA expandedIn addition, after multiple delays, the final rule implementing civil monetary penalties against manufacturers for instances of overcharging 340B program to include additional types of covered entities but exempts “orphan drugs”-those designated under section 526 ofbecame effective on January 1, 2019. Accordingly, we could be subject to such penalties if the FDCA, such as Soliris from the ceiling price requirementsgovernment finds that we knowingly and intentionally overcharged a 340B covered entity.
Federal law requires that for these newly-eligible entities.
In ordera company to be eligible to have ourits products paid for with federal funds under the Medicaid and Medicare Part B programs andas well as to be purchased by certain federal agencies and grantees, weit also must participate in the Department of Veterans Affairs (VA) Federal Supply Schedule (FSS) pricing program. To participate, we are required to enter into an FSS contract and other agreements with the VA for our products, which qualify as “covered drugs.” Under these agreements, we must make our products available to the “Big Four” federal agencies the VA, the Department of Defense (DoD), the Public Health Service (including the Indian Health Service), and the Coast Guard at pricing that is capped pursuant to a statutory
federal ceiling price, or FSS, pricing program, established byFCP, formula set forth in Section 603 of the Veterans Health Care Act of 1992. Under this program, we are obligated to make our innovator “covered drugs” available for procurement on an FSS contract and charge a price to four federal agencies, Department of Veterans Affairs, Department of Defense, Public Health Service and Coast Guard that is no higher than the statutory Federal Ceiling Price, or FCP.1992 (VHCA). The FCP is based on thea weighted average non-federal average manufacturer price or Non-FAMP,(Non-FAMP), which we calculate andmanufacturers are required to report to the Department of Veterans Affairs on a quarterly and annual basis.basis to the VA. Pursuant to the VHCA, knowing provision of false information in connection with a Non-FAMP filing can subject a manufacturer to a penalty for each item of false information and could result in other potential liability as well, including liability under the False Claims Act (which is discussed in more detail below).
FSS contracts are federal procurement contracts that include standard government terms and conditions, separate pricing for each product, and extensive disclosure and certification requirements. All items on FSS contracts are subject to a standard FSS contract clause that requires FSS contract price reductions under certain circumstances where pricing is reduced to an agreed “tracking customer.” Further, in addition to the “Big Four” agencies, all other federal agencies and some non-federal entities are authorized to purchase off FSS contracts. FSS contractors are permitted to charge FSS purchasers other than the Big Four agencies “negotiated pricing” for covered drugs that is not capped by the FCP; instead, such pricing is negotiated based on a mandatory disclosure of the contractor’s commercial “most favored customer” pricing. We also participate inoffer dual pricing on our FSS contract.
In addition, pursuant to regulations issued by the Tricare Retail Pharmacy program, established byDoD to implement Section 703 of the National Defense Authorization Act for FYFiscal Year 2008, and related regulations,each of our covered drugs is listed on an agreement with the Defense Health Agency (DHA) under which we pay quarterly rebates on utilization of innovatorhave agreed to honor the “Big Four” pricing for our products thatwhen they are dispensed throughto TRICARE beneficiaries by TRICARE retail network pharmacies. More specifically, we have agreed to provide rebates (or refunds) on such utilization. Companies are required to enter into a DHA Agreement for “covered drug” products in order for the Tricare Retail Pharmacy networkcovered drug to Tricare beneficiaries.be eligible for DoD formulary inclusion and available to TRICARE beneficiaries without preauthorization. The rebates are calculated asformula for determining the rebate is established in the regulations and our DHA agreement and is based on the difference between Annualthe annual Non-FAMP and FCP.the FCP (as described above, these price points are required to be calculated by us under the VHCA).
As noted in the foregoing, pricing and rebate calculations vary among products and programs. The calculations can be very complex and are often subject to interpretation by us, governmental or regulatory agencies and the courts. We cannot assure you that our submissions will not be found by CMS or other governmental agencies to be incomplete or incorrect. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for
amounts previously estimated or paid. For example, if we become aware that certain Medicaid Drug Rebate Program price reporting for a prior quarter was incorrect, or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected data for a period not to exceed twelve quarters from the quarter in which the data originally were due, and CMS may consider restatements for earlier periods as well depending on the circumstance. Such restatements and recalculations increase our costs for complying with the laws and regulations governing the Medicaid Drug Rebate Program. Any corrections to our Medicaid rebate calculations could result in an increase or decrease in our rebate liability for past quarters, depending on the nature of the correction. Price recalculations also may affect the ceiling price at which we are required to offer our products to certain covered entities under the 340B drug pricing program.
Any failure to comply with these price reporting and rebate payment obligations could negatively impact our financial results. Civil monetary penalties can be applied if we are found to have knowingly submitted any false price information to the government, if we are found to have made a misrepresentation in the reporting of our average sales price, or if we fail to submit the required price data on a timely basis. Such conduct also could be grounds for CMS to terminate our Medicaid drug rebate agreement, in which case federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs, as well as provide a basis for other potential liability under other federal laws such as the False Claims Act.
Payers also are increasingly considering new metrics as the basis for reimbursement rates, such as ASP, average manufacturer price, and actual acquisition cost. The existing data for reimbursement based on these metrics is relatively limited, although certain states have begun to survey acquisition cost data for the purpose of setting Medicaid reimbursement rates. CMS surveys and publishes retail community pharmacy acquisition cost information in the form of National Average Drug Acquisition Cost files to provide state Medicaid agencies with a basis of comparison for their own reimbursement and pricing methodologies and rates. It may be difficult to project the impact of these evolving reimbursement mechanics on the willingness of payers to cover our products.
Federal law requires that for a company to be eligible to have its products paid for with federal funds under the Medicaid and Medicare Part B programs as well as to be purchased by certain federal agencies and grantees, it also must participateFurther, in the Department of Veterans Affairs (VA) Federal Supply Schedule (FSS)U.S., there is increased focus on drug pricing, program. To participate, we are required to enter into an FSS contract with the VA, under which we must make our innovator “covered drugs” available to the “Big Four” federal
agencies - the VA, the Department of Defense (DoD) the Public Health Service, and the Coast Guard - atPresident, policy officials (including the FDA) and lawmakers have expressed a clear interest in efforts to reduce prices for drugs and biologics, further increase transparency around prices and price increases, lower out-of-pocket costs for consumers, and decrease spending on drugs by government programs. In addition, members of Congress have launched an investigation into the pricing that is capped pursuant to a statutory federal ceiling price, or FCP, formula set forth in Section 603practices of the Veterans Health Care Act of 1992 (VHCA). The FCP is based on a weighted average non-federal average manufacturer price (Non-FAMP) which manufacturers are required to report on a quarterly and annual basis to the VA. If a company misstates Non-FAMPs or FCPs it must restate these figures. Pursuant to the VHCA, knowing provision of false informationprescription drug industry (and hearings may be held in 2019 in connection with this investigation). We expect
regulatory changes and continued Congressional investigations and negative media attention in the coming months with respect to drugs reimbursed by federal healthcare programs, like ours, which could have a Non-FAMP filing can subject a manufacturer to penalties of one hundred seventy eight thousand dollars for each item of false information.
FSS contracts are federal procurement contracts that include standard government terms and conditions, separate pricing for each product, and extensive disclosure and certification requirements. All items on FSS contracts are subject to a standard FSS contract clause that requires FSS contract price reductions under certain circumstances where pricing is reduced to an agreed “tracking customer.” Further, in addition to the “Big Four” agencies, all other federal agencies and some non-federal entities are authorized to access FSS contracts. FSS contractors are permitted to charge FSS purchasers other than the Big Four agencies “negotiated pricing” for covered drugs that is not capped by the FCP; instead, such pricing is negotiated based on a mandatory disclosure of the contractor’s commercial “most favored customer” pricing. We offer dual pricingnegative impact on our FSS contract.
In addition, pursuant to regulations issued by the DoD TRICARE Management Activity, now the Defense Health Agency, to implement Section 703 of the National Defense Authorization Act for Fiscal Year 2008, each of our covered drugs is listed on a Section 703 Agreement under which we have agreed to pay rebates on covered drug prescriptions dispensed to TRICARE beneficiaries by TRICARE network retail pharmacies. Companies are required to list their innovator products on Section 703 Agreements in order for those products to be eligible for DoD formulary inclusion. The formula for determining the rebate is established in the regulations and our Section 703 Agreement and is based on the difference between the annual Non-FAMP and the FCP (as described above, these price points are required to be calculated by us under the VHCA).operations.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. Moreover, the requirements governing drug pricing and reimbursement vary widely from country to country. For example, in the EU, the sole legal instrument at the EU level governing the pricing and reimbursement of medicinal products is Council Directive 89/105/EEC (the Price Transparency Directive). The aim of the Price Transparency Directive is to ensure that pricing and reimbursement mechanisms established in EU member statesMember States are transparent and objective, do not hinder the free movement and trade of medicinal products in the EU and do not hinder, prevent or distort competition on the market. The Price Transparency Directive does not, however, provide any guidance concerning the specific criteria on the basis of which pricing and reimbursement decisions are to be made in individual EU member states.Member States. Neither does it have any direct consequence for pricing or levels of reimbursement in individual EU member states. TheMember States. Pricing of prescription only medicinal products is a national prerogative. Therefore the relevant national authorities of the individual EU member statesMember States are free to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices and/or reimbursement of medicinal products for human use. Some individual EU member statesMember States adopt policies according to which a specific price or level of reimbursement is approved for the medicinal product. Other EU member statesMember States adopt a system of reference pricing, basing the price or reimbursement level in their territory either, on the pricing and reimbursement levels in other countries, or on the pricing and reimbursement levels of medicinal products intended for the same therapeutic indication. Furthermore, some EU member statesMember States impose direct or indirect controls on the profitability of the company placing the medicinal product on the market.
Health Technology Assessment (HTA) of medicinal products is becoming an increasingly common part of the pricing and reimbursement procedures in some EU member states.Member States. These countries include the United Kingdom, France, Germany and Sweden. The HTA process in the EU member statesMember States is governed by the national laws of these countries. HTA is the procedure according to which the assessment of the public health impact, therapeutic impact and the economic and societal impact of the use of a given medicinal product in the national healthcare systems of the individual country is conducted. HTA generally focuses on the clinical efficacy and effectiveness, safety, cost, and cost-effectiveness of individual medicinal products as well
as their potential implications for the national healthcare system. Those elements of medicinal products are compared with other treatment options available on the market.
The outcome of HTA may influence the pricing and reimbursement status for specific medicinal products within individual EU member states.Member States. The extent to which pricing and reimbursement decisions are influenced by the HTA of a specific medicinal product vary between the EU member states.Member States.
In 2011, Directive 2011/24/EU was adopted at the EU level. This Directive concerns the application of patients’ rights in cross-border healthcare. The Directive is intended to establish rules for facilitating access to safe and high-quality cross-border healthcare in the EU. Pursuant to Directive 2011/24/EU, a voluntary network of national authorities or bodies responsible for HTA in the individual EU Member States was established. The purpose of the network is to facilitate and support the exchange of scientific information concerning HTAs. This could lead to harmonization of the criteria taken into account in the conduct of
HTA between EU member statesMember States in pricing and reimbursement decisions and negatively impact price in at least some EU member states.Member States.
On a continuous basis, we engage with appropriate authorities in individual countries on the operational, reimbursement, price approval and funding processes that are separately required in each country.
Fraud and Abuse
Pharmaceutical companies participating in federal healthcare programs like Medicare or Medicaid are subject to various U.S. federal and state laws pertaining to healthcare “fraud and abuse,” including anti-kickback and false claims laws. Violations of U.S. federal and state fraud and abuse laws may be punishable by criminal, civil and administrative sanctions, including fines, damages, civil monetary penalties and exclusion from participation in federal healthcare programs (including Medicare and Medicaid). Applicable U.S. statutes, include, but are not limited to, the following:
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully soliciting, offering, receiving, or paying any remuneration, directly or indirectly, in cash or in kind, to induce or reward purchasing, ordering or arranging for or recommending the purchase or order of any item or service for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. Liability may be established without a person or entity having actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it. This statute has been interpreted to apply broadly to arrangements between pharmaceutical manufacturers on the one hand
and individuals such as prescribers, patients, purchasers and formulary managers on the other. In addition, PPACA amended the Social Security Act to provide that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statuteAnti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.Act (which is discussed below). A conviction for violation of the Anti-kickbackAnti-Kickback Statute results in criminal fines and requires mandatory exclusion from participation in federal health care programs. Although there are a number of statutory exemptionsexceptions and regulatory safe harbors protectingto the federal Anti-Kickback Statute that protect certain common, activitiesindustry practices from prosecution, the exemptionsexceptions and safe harbors are drawn narrowly, and those activitiesarrangements may be subject to scrutiny or penalty if they do not qualify forfully satisfy all elements of an exemptionavailable exception or safe harbor. The discount safe harbor is currently the subject of possible reform. Any changes to the discount safe harbor may cause us to review our arrangements and pricing strategies with payers.
The federal civil False Claims Act (FCA) prohibits,imposes civil penalties against individuals or entities for, among other things, knowingly presenting, or causing to be presented, claims for payment ofto the government funds that are false or fraudulent, or knowingly making, using or causing to be made or used a false record or statement material to such a false or fraudulent claim, or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. This statute also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery. False Claims ActFCA liability is potentially significant in the healthcare industry because the statute provides for treble damages and mandatory penalties of five thousand to eleven thousand dollars per false claim or statement (and tenone hundred eighty-one to twenty-two thousand to twenty thousandthree hundred sixty-three dollars per false claim or statement for penalties assessed after August 1, 2016 forJanuary 29, 2018, with respect to violations occurring after November 2, 2015)2015 (and penalties of five thousand five hundred to eleven thousand dollars with respect to violations occurring before that date). Government enforcement agencies and private whistleblowers have investigated pharmaceutical companies for or asserted liability under the FCA for a variety of alleged inappropriate promotional and marketing activities, such as providingincluding those involving the provision of free product or other items of value to customers, certain financial arrangements with
healthcare providers, misstated government pricing information, and purported “off-label” promotion of products, among other things.
Under the expectation that the customers would bill federal programs for the product; providing consulting fees and other benefitscriminal statute on false statements relating to physicianshealth care matters, it is a crime to induce them to prescribe products; engaging in promotion for “off-label” uses; and submitting inflated best price information to the Medicaid Rebate Program.
The federal False Statements Statute prohibits knowingly and willfully falsifying, concealing,falsify, conceal, or coveringcover up a material fact, or makingmake any materially false, fictitious, or fraudulent statementstatements or representation,representations, or makingmake or usinguse any materially false writing or document knowing the same to contain any materially false, fictitious, or fraudulent statement or entry in connection with the delivery of or payment for federally funded healthcare benefits, items, or services.
Under the Health Insurance Portability and Accountability Act of 1996 (HIPAA) criminal federal health care fraud statute, it is a crime to knowingly and willfully execute, or attempt to execute, a scheme or artifice to defraud any health care benefit program or to obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any health care benefit program, in connection with the delivery of or payment for health care benefits, items, or services.
The federal Civil Monetary Penalties Law authorizes the imposition of substantial civil monetary penalties against an entity, such as a pharmaceutical manufacturer, that engages in activities including, among others (1) knowingly presenting, or causing to be presented, a claim for services not provided as claimed or that is otherwise false or fraudulent in any way; (2) arranging for or contracting with an individual or entity that is excluded from participation in federal healthcare programs to provide items or services reimbursable by a federal healthcare program; (3) violations of the federal Anti-Kickback Statute; or (4) failing to report and return a known overpayment.
The majority of states also have statutes similar to the federal anti-kickback lawAnti-Kickback Statute and false claims lawsFCA that apply to items and services reimbursed under Medicaid and other state health care programs, or, in several states, apply regardless of the payer.
The federal OpenPhysician Payments programSunshine Act requires manufacturers“applicable manufacturers” of products, including biologics, for which payment is available under Medicare, Medicaid or the State Children’s Health Insurance Program, among others, to track and report annually to the federal government (for disclosure to the public) certain payments and other transfers of value madethey make to ”covered recipients.” The term covered recipients includes physicians, teaching
hospitals, and, teaching hospitals.for reports submitted on or after January 1, 2022, physician assistances, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse-midwives. In addition, several U.S. states and localities have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports, with the state, and/or make periodic public disclosures on sales, marketing, pricing, clinical trials, and other activities. Other state laws prohibit certain marketing-related activities including the provision of gifts, meals or other items to certain healthcare providers.providers, and restrict the ability of manufacturers to offer co-pay support to patients for certain prescription drugs. Some states and cities require identification or licensing of state representatives. In addition, several recently passed state laws require disclosures related to state agencies and/or commercial purchasers with respect to certain price increases that exceed a certain level as identified in the relevant statutes. Many of these laws and regulations contain ambiguous requirements that government officials have not yet clarified. Given the lack of clarity in the laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent federal and state laws and regulations.
Sanctions under federal and state fraud and abuse laws may include significant criminal, civil, monetaryand administrative penalties, including damages, fines, imprisonment, and exclusion of a manufacturer’s products from reimbursement under government programs, monetary damages, criminal fines, and imprisonment.programs. Any of the foregoing would be expected to have a negative impact on our business which may be material.
Federal and state authorities are continuing to devote significant attention and resources to enforcement of fraud and abuse laws within the pharmaceutical industry, and private individuals have been active in alleging violations of the law and bringing suits on behalf of the government under the FCA. For example, federal enforcement agencies recently have investigated certain pharmaceutical companies’ product and patient assistance programs, including manufacturer reimbursement support services, relationships with specialty pharmacies, and grants to independent charitable foundations. If we, our vendors, or donation recipients are deemed to fail to comply with relevant laws, regulations or evolving government guidance in the operation of these programs, we could be subject to damages, fines, penalties or other criminal, civil or administrative sanctions or enforcement actions. We cannot ensure that our compliance controls, policies and procedures will be sufficient to protect against acts of our employees, business partners or vendors that may violate the laws or regulations of the jurisdictions
in which we operate. In December 2016, we received a subpoena from the U.S. Attorney’s Office (USAO) for the District of Massachusetts relating generally to our support of 501(c)(3) organizations that provide financial assistance to Medicare patients, Alexion’s provision of free drug to Medicare patients and Alexion’s related compliance policies and training materials. SomePlease see the discussion below in the “Risk Factors” section and Note 11 “Commitments and Contingencies” to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K for additional details regarding this investigation. Similar investigations of these investigationsother pharmaceutical companies have resulted in significant civil and criminal settlements. Efforts to ensure that our business arrangements continue to comply with applicable healthcare laws and regulations could be costly.
Outside the U.S., other countries have implemented similar laws and regulations relating to fraud and abuse in the sale of pharmaceutical products and requirements for disclosure of financial interactions with healthcare providers and additional countries may consider or implement such laws.
U.S. Healthcare Reform and Other U.S. and International Healthcare Laws
PPACA was adopted in the U.S. in March 2010. This law substantially changes the way healthcare is financed in the U.S. by both governmental and private insurers, in the U.S., and significantly impacts the pharmaceutical industry. PPACA contains a number of provisions that have and are expected to impact our business and operations. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under the health insurance exchanges, expansion of the 340B program, expansion of state Medicaid programs, and fraud and abuse and enforcement. These changes will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program.
PPACA contains several provisions that have or could potentially have an impact on our business. PPACA made significant changes to the Medicaid Drug Rebate Program. Effective March 23, 2010, rebate liability expanded from fee-for-service Medicaid utilization to include the utilization of Medicaid managed care organizations as well. With regard to the amount of the rebates owed, PPACA increased the minimum Medicaid rebate percentage from 15.1% to 23.1% of the average manufacturer price for most innovator products; changed the calculation of the rebate for certain innovator products that qualify as line extensions of existing drugs; and capped the total rebate amount for innovator drugs at 100%100.0% of the average manufacturer
price. In addition, PPACA and subsequent legislation changed the definition of average manufacturer price. In early 2016, CMS issued final regulations to implement the changes to the Medicaid Drug Rebate Program under PPACA, which became effective on April 1, 2016. Finally, PPACA requires pharmaceutical manufacturers of branded prescription drugs to pay a branded prescription drug fee to the federal government. Each individual pharmaceutical manufacturer pays a prorated share of the aggregate branded prescription drug fee of $4,000paid by all covered entities ($2,800 in 2017 (and set to increase in2019 and each ensuing years)year), based on, the dollar value ofamong other things, its applicable branded prescription drug sales to certain federal programs identified in the law. Sales of “orphan drugs” are excluded from this fee. “Orphan drugs” are specifically defined for purposes of the fee. For each indication approved by the FDA for the drug, such indication must have been designated as orphan by the FDA under section 526 of the FDCA, an orphan drug tax credit under section 45C of the Internal Revenue Code of 1986 (Internal Revenue Code) must have been claimed with respect to such indication, and such tax credit must not have been disallowed by the Internal Revenue Service.Service (IRS). Finally, the FDA must not have approved the drug for any indication other than an
orphan indication for which a section 45C orphan drug tax credit was claimed (and not disallowed). LegislativeIn early 2016, CMS issued a final regulation to implement the changes to the Medicaid Drug Rebate Program under PPACA, also remain possiblewhich became effective on April 1, 2016. The issuance of the final regulation, as well as any other regulations and appear likelycoverage expansion by various governmental agencies relating to the Medicaid Drug Rebate Program, has increased and will continue to increase our costs and the complexity of compliance, has been and will continue to be time-consuming to implement, and could have a material adverse effect on our results of operations, particularly if CMS challenges the approach we take in our implementation of the 115th U.S. Congress and under the Trump Administration.final rule.
Additional provisions of PPACA may negatively affect manufacturer’s revenues in the future. For example, as part of PPACA’s provisions closing a coverage gap that currently exists in the Medicare Part Dprescription drug program (commonly known as the “donut hole”), manufacturers of branded prescription drugs and biologics are required to provide a 50%50.0% discount on branded prescription drugs and biologics dispensed to beneficiaries within this donut hole. This discount was recently increased to 70.0%, beginning January 1, 2019, by the Bipartisan Budget Act of 2018.
As noted above, PPACA also expanded the Public Health Service’s 340B drug pricing discount program.program by including additional types of covered entities. The 340B pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. PPACA expanded the 340B program to include additional types of covered entities: certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, eachentities as defined by PPACA.described above. PPACA exempts “orphan drugs”-those designated under section 526 of the
FDCA, such as our products-fromproducts, from the ceiling pricepricing requirements for these newly-eligible covered entities.
Finally, numerousMoreover, certain legislative changes to and regulatory changes under PPACA have occurred under the Trump Administration. For example, the Tax Cuts and Jobs Act enacted in 2017 eliminated the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code, commonly referred to as the “individual mandate,” beginning in 2019. In December 2018, a federal district court in Texas ruled the individual mandate was unconstitutional and could not be severed from the PPACA. As a result, the court ruled the remaining provisions of the PPACA were also invalid, though the court declined to issue a preliminary injunction with respect to the PPACA. However, it remains unclear whether the court’s ruling will be upheld by appellate courts. In addition, further legislative changes to and regulatory changes under PPACA remain possible.
Privacy, Data Protection and Information Security
Numerous international, federal, and state laws, including state security breach notification and information security laws, state health information privacy laws, and federal and state consumer protection laws govern the collection, use, and disclosure of personal information. In addition, most healthcare providers who prescribe and dispense our products and research institutions with whom we collaborate for our sponsored clinical trials are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996 (HIPAA),HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (HITECH), and its implementing regulations. Although we are neither a “covered entity” nor a “business associate” undernot directly subject to HIPAA and these privacy and security requirements do not applyother than with respect to us, the regulations may affect our interactions with healthcare providers, health plans, and research institutions from whom we obtain patient health information. Further,providing certain employee benefits, we could be potentially subject to criminal penalties if we, our affiliates, or our agents knowingly obtain or disclose individually identifiable health information frommaintained by a HIPAA covered entity in a manner that is not authorized or permitted by HIPAA HIPAA. In addition, in December 2018, HHS issued cybersecurity guidance for all healthcare organizations that addresses organizations’ enterprise-level information security generally, including individually identifiable health information. Failure to comply with current and future laws and regulations could result in governmental enforcement actions (including the imposition of significant penalties), criminal and civil liability for our Company and our officers and directors, and/or for aidingadverse publicity that negatively affects our business. Further, the EU’s General Data Protection Regulation (GDPR) and abettingimplementing laws in the violationEU member states govern the collection and processing of HIPAA.EU residents’ personal data and, among other requirements, imposes certain consent and data access rights. Such laws may impact our ability to conduct clinical trials that involve EU personal data and engage in other activities that require the processing of EU personal data. Outside of the U.S.
and the EU, there are numerous other jurisdictions that have their own privacy and information security laws, and new laws and regulations are being considered and/or enacted globally, which may affect our ability to collect, process, and store their residents’ personal data. Two such examples are the California Consumer Privacy Act of 2018 and the Brazilian Data Protection Law, which both go into effect in early 2020, and may impact our collection and use of personal information related to their jurisdictions.
Moreover, we rely on our and third-party provided information technology systems and applications to support our operations and to maintain and process company information including personal information, confidential business information and proprietary information. If these information technology systems are subject to cybersecurity attacks, or are otherwise compromised, due to cyberattacks, human error or malfeasance, system errors or otherwise, it may adversely impact our business, disrupt our operations, or lead to the loss, theft, destruction, corruption or compromise of company information and personal information. Such information technology or security events could also lead to legal liability, regulatory investigations or actions, loss of business, negative media coverage, and reputational damage. While we maintain an information security program with technical controls to mitigate these risks and training to educate and prepare our employees, the healthcare sector continues to see a high frequency of cyberattacks and threat actors that continue to become more sophisticated and better resourced, and our systems and the information maintained within those systems remain potentially vulnerable to data security incidents. Moreover, losses from such events may not be completely covered by insurance coverage. Finally, as cyber threats continue to evolve and privacy and cybersecurity laws and regulations continue to develop, we may need to invest additional resources to implement new compliance measures, strengthen our information security posture, or respond to cyber threats and incidents.
Other Regulations
We are also subject to the U.S. Foreign Corrupt Practices Act (FCPA), the U.K. Bribery Act (U.K. Bribery Act), and other anti-corruption laws and regulations pertaining to our financial relationships and interactions with foreign government officials. The FCPA prohibits U.S. companies and their employees, officers, and representatives from paying, offering to pay, promising, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate to obtain or retain business or to otherwise seek favorable treatment. In many countries in which we operate or sell our products, the healthcare professionals with whom we interact may be deemed to be foreign government officials for
purposes of the FCPA. The U.K. Bribery Act, which applies to any company incorporated or doing business in the UK, prohibits giving, offering, or promising bribes in the public and private sectors, bribing a foreign public official or private person, and failing to have adequate procedures to prevent bribery amongst employees and other agents. Penalties under the U.K. Bribery Act include potentially unlimited fines for companies and criminal sanctions for corporate officers under certain circumstances. Liability in relation to breaches of the U.K. Bribery Act is strict. This means that it is not necessary to demonstrate elements of a corrupt state of mind. However, a defense of having in place adequate procedures designed to prevent bribery is available.
Recent years have seen a substantial increase in anti-bribery law enforcement activity by U.S. regulators, with more frequent and aggressive investigations and enforcement proceedings by both the DOJ and the SEC, increased enforcement activity by non-U.S. regulators, and increases in criminal and civil proceedings brought against companies and individuals. Increasing regulatory scrutinyIn May 2015, we received a subpoena in connection with an investigation by the Enforcement Division of the promotionalSEC requesting information related to our grant-making activities and compliance with the FCPA in various countries. In addition, in October 2015, Alexion received a request from the DOJ for the voluntary production of pharmaceutical companiesdocuments and other information pertaining to Alexion’s compliance with the FCPA. For information concerning this investigation see Note 11 “Commitments and Contingencies” to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K and, with respect to the the risks associated with the investigation, see our Risk Factors, including "Our business and operations may be materially adversely affected by government investigations."
The EU also has been observed in a number of EU member states.
Similarimposes strict restrictions are imposed on the promotion and marketing of drug products in the EU, where a large portion of our non-U.S. business is conducted, and other territories. Increasing regulatory scrutiny of the promotional activities of pharmaceutical companies also has been observed in a number of EU Member States. Laws in the EU, including in the individual EU member states,Member States, require promotional materials and advertising for drug products to comply with the product’s Summary of Product Characteristics (SmPC), which is approved by the competent authorities. Promotion of a medicinal product which does not comply with the SmPC is considered to constitute off-label promotion. The off-label promotion of medicinal products is prohibited in the EU and in other territories. The promotion of medicinal products that are not subject to a marketing authorization is also considered to constitute off-label promotion and is prohibited in the EU. Laws in the EU, including in the individual EU member states,Member States, also prohibit the direct-to-consumer advertising of prescription-only medicinal products. Violations of the rules governing the promotion of medicinal products in
the EU and in other territories could be penalized by administrative measures, fines and imprisonment.
Under the new Clinical Trial Regulation there is an obligation to publish clinical trial within a certain timeframe. A breach of this obligation would constitute non-compliance with an EU Regulation and may be met with penalties set by each Member State, including civil and criminal liability.
Japan and other countries in which we operate also have strict regulations and requirements regarding the promotion of pharmaceutical products.
Interactions between pharmaceutical companies and physicians are also governed by strict laws, regulations, industry self-regulation codes of conduct and physicians’ codes of professional conduct in the individual EU member states.Member States. The provision of any inducements to physicians to prescribe, recommend, endorse, order, purchase, supply, use or administer a medicinal product is prohibited. A number of EU member statesMember States have introduced additional rules requiring pharmaceutical companies to publicly disclose their interactions with physicians and to obtain approval from employers, professional organizations and/or competent authorities before entering into agreements with physicians. These rules have been supplemented by provisions of related industry codes, including the EFPIA Disclosure Code on Disclosure of Transfers of Value from Pharmaceutical Companies to Healthcare Professionals and Healthcare Organizations and related codes developed at national level in individual EU member states.Member States. Additional countries may consider or implement similar laws and regulations. Violations of these rules could lead to reputational risk, public reprimands, and/or the imposition of fines or imprisonment.
Our present and future business has been and will continue to be subject to various other laws and regulations. Laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances, including radioactive compounds, used in connection with our research work are or may be applicable to our activities. We cannot predict the impact of government regulation, which may result from future legislation or administrative action, on our business.
Competition
Soliris
Competition
ULTOMIRIS and SOLIRIS are currently the only approved therapies for the treatment of PNH (although several companies are currently evaluating other complement inhibitors for the treatment of PNH in clinical trials). SOLIRIS is currently the only approved therapy for the treatment of aHUS, and the only approved complement inhibitor therapy for the treatment of AChR antibody-positive gMG (although similar to PNH, there are companies evaluating other complement inhibitors in both aHUS and aHUS.gMG clinical trials). We have recently announced the results of our SOLIRIS Phase III PREVENT trial in patients with anti-aquaporin-4 (AQP4) auto antibody-positive NMOSD. Based on these results, we submitted applications for marketing authorization for SOLIRIS as a treatment for NMOSD in the US and the E.U. and expect additional marketing authorization applications to be submitted in other jurisdictions in the future. While we are unable to assess our competitive position with respect to potential NMOSD competitors, as we have not yet received regulatory approval in any jurisdiction, we are aware that others companies are also developing and testing therapies for NMOSD. We are also in advanced clinical studies of SolirisULTOMIRIS and SOLIRIS for the treatment of other indications, and we believe there are currently no competitors for the patient segments we target. Strensiqtarget with respect to these products. STRENSIQ is currently the only product approved for the treatment of HPP and KanumaKANUMA is the only product approved for the treatment of LAL-D. Many pharmaceutical and biotech companies have publicly announced intention to establish or develop rare disease programs that may be competitive with ours. We also experience competition in drug development from universities and other research institutions, and pharmaceutical companies compete with us to attract universities and academic research institutions as drug development partners, including for licensing their proprietary technology. Some of these entities may have:
•greater financial and other resources;
•larger research and development staffs;
•lower labor costs; and/or
•more extensive marketing and manufacturing organizations.
Many of these companies and organizations have significant experience in preclinical testing, human clinical trials, product manufacturing, marketing, sales and distribution and other regulatory approval and commercial procedures. They may also have a greater number of significant patents and greater legal resources to seek remedies for cases of alleged infringement of their patents by us to block, delay or compromise our own drug development process.
We compete with large pharmaceutical companies that produce and market synthetic compounds and with specialized biotechnology firms in the United States,
Europe and in other countries and regions, as well as a growing number of large pharmaceutical companies that are developing biotechnology products. A number of biotechnology and pharmaceutical companies are developing new products for the treatment of the same diseases being targeted by us. Other companies have initiated clinical studies for the treatment of PNH, aHUS, AMR, MG and NMOSD, and we are aware of companies that are planning to initiate studies for diseases we are also targeting. In the future, our products may alsoaddition, we are aware that companies are conducting clinical trials for biosimilars of SOLIRIS and we expect to compete with biosimilars.biosimilars in the future.
Several biotechnology and pharmaceutical companies have programs to develop complement inhibitor therapies or have publicly announced their intentions to develop drugs which target the inflammatory effects of complement in the immune system or have had programs to develop complement inhibitor therapies. SolirisSOLIRIS is the only therapy that has demonstrated to be safe and effective in two clinical indications by regulators in many jurisdictions around the world.
Employees
As of December 31, 2016,2018, we had 3,1212,656 full-time, world-wide employees, of which 1,247954 were engaged in research, product development, manufacturing, and clinical development, 1,2401,299 in sales and marketing, and 634403 in administration, human resources, information technology and finance. Our U.S. employees are not represented by any collective bargaining unit, and we regard the relationships with all our employees as satisfactory.
EXECUTIVE OFFICERS OF THE COMPANY
The executive officers of the Company and their respective ages and positions as of February 13, 20176, 2019 are as follows:
|
| | | |
| Name | Age | Position with Alexion | Age |
David R. BrennanLudwig Hantson, Ph.D. | 62 |
| Interim Chief Executive Officer | 56 |
DavidPaul J. Anderson, M.B.A. | 67 |
Clancy | Executive Vice President, and Chief Financial Officer | 57 |
Clare Carmichael | 57 |
Ellen Chiniara, J.D. | Executive Vice President, General Counsel and Corporate Secretary | 60 |
| Indrani Franchini, J.D. | Executive Vice President, Chief Compliance Officer | 47 |
| Brian Goff | Executive Vice President, Chief Commercial Officer | 49 |
| Anne-Marie Law | Executive Vice President, Chief Human Resources Officer | 51 |
Martin Mackay, Ph.D. | 60 |
John Orloff, M.D. | Executive Vice President, and Global Head of Research and Development | 61 |
|
John B. Moriarty, J.D. | 49 |
|
| | Ludwig N. Hantson, Ph.D., is Chief Executive Officer of Alexion. Dr. Hantson is an accomplished healthcare executive with more than 30 years of experience in the biopharmaceutical industry.
Prior to joining Alexion in March 2017, Dr. Hantson was President and Chief Executive Officer of Baxalta and also served on the company’s Board of Directors. He led Baxalta’s successful spin-off as a public company from Baxter in July 2015 where he was President of Baxter BioScience. Dr. Hantson joined Baxter in May 2010 and established the BioScience division as one of the most innovative specialty and rare disease companies by building a robust pipeline of 25 new product candidates, and launching 13 new products.
Dr. Hantson held several leadership roles during his decade-long tenure at Novartis from 2001-2010, including CEO of Pharma North America, CEO of Europe, and President of Pharma Canada. Prior to Novartis, he spent 13 years with Johnson & Johnson in roles of increasing responsibility in marketing, and research and development. Mr. Hantson serves on the Board of Directors of Hologic Inc., which is a medical technology company.
Dr. Hantson received his Ph.D. in motor rehabilitation and physical therapy, master’s degree in physical education, and a certification in high secondary education, all from the University of Louvain in Belgium.
|
|
|
| | |
| | Paul J. Clancyis Executive Vice President, Chief Financial Officer of Alexion. Mr. Clancy is responsible for global financial management, treasury, internal audit, corporate strategy, business development, investor relations, information technology, and security activities.
Prior to joining Alexion in July 2017, Mr. Clancy served as the Executive Vice President, Finance and Chief Financial Officer and a member of the Executive Committee of Biogen, where he led the financial performance of the company. Prior to joining Biogen, Mr. Clancy spent 13 years at PepsiCo, serving in a range of finance, strategy and general management positions. Mr. Clancy serves on the Board of Directors of the biopharmaceutical companies Agios Pharmaceuticals, Inc. and Incyte Corporation.
Mr. Clancy holds an MBA from Columbia University and a Bachelor of Science in Finance from Babson College.
|
|
|
| | |
| | Ellen Chiniara is Executive Vice President, General Counsel and Corporate Secretary of Alexion. In this role, she is responsible for overseeing all global legal matters for the Company.
Prior to joining Alexion in January 2018, Ms. Chiniara was Senior Vice President and General Counsel of Alere Inc., a point-of-care diagnostics company, from October 2006 to October 2017 where she was responsible for all legal matters and, from June 2014 to October 2017 she had oversight of compliance and government affairs matters. She managed the legal aspects of the company’s numerous acquisitions and dispositions and was also the executive sponsor of Alere’s corporate social responsibility efforts.
Prior to joining Alere, Ms. Chiniara served as Associate General Counsel for Serono’s Neurology division from 2002 to 2006. Earlier in her career, Ms. Chiniara was a partner at the law firm Hale and Dorr LLP (now Wilmer Cutler Pickering Hale and Dorr LLP).
Ms. Chiniara received her J.D. from Stanford University’s School of Law and her Bachelor's Degree from Bryn Mawr College. She also was a graduate fellow at Yale University in Slavic Languages.
|
Julie O’Neill, M.B.A. |
|
| 50 |
|
| | Indrani Franchini, J.D., is Executive Vice President, Chief Compliance Officer of Alexion. Ms. Franchini is responsible for leading Alexion’s global compliance program and co-leads the Global OperationsCorporate Compliance Committee. Ms. Franchini has extensive experience developing and building the infrastructure and company-wide standards for global compliance programs. Prior to joining Alexion in June 2017, Ms. Franchini served as Chief Compliance Officer at Hess Corporation (a leading independent energy company) from June 2012 to July 2017. She previously spent nearly ten years with Pfizer overseeing all compliance elements for the development, marketing, and promotion of its global business. Earlier in her career, Ms. Franchini served as an attorney with Milbank, Tweed, Hadley & McCloy in the firm’s New York and Tokyo offices.
Ms. Franchini earned her J.D. from the University of Michigan Law School and a Bachelor of Arts from Princeton University. In addition, she spent a year as a Fulbright Fellow at the Kyushu University Graduation School in Fukuoka, Japan. |
Carsten Thiel, Ph.D. |
|
| 53 |
|
| | Brian Goff is Executive Vice President, Chief Commercial Officer of Alexion. Mr. Goff leads commercial operations globally with responsibility for country operations in each of Alexion’s affiliates in North America, EMEA, Japan, Asia Pacific, and Latin America.
Mr. Goff is a proven global biopharmaceutical executive with a 25-year track record of consistently delivering sustainable growth through multiple business cycles. He has deep expertise in commercial operations across multiple therapeutic areas, as well as broad expertise managing global cross-functional teams, including R&D, Medical Affairs, Manufacturing and Quality with a number of industry-leading biopharmaceutical companies.
Prior to joining Alexion in June 2017, Mr. Goff was Chief Operating Officer and a Member of the Board of Directors of Neurovance Inc. from December 2016 until its acquisition by Otsuka Pharmaceuticals in March 2017. Prior to joining Neurovance, Mr. Goff served as Baxalta’s Executive Vice President & President — Hematology Division from January 2015 to July 2016. He previously served with Baxter Healthcare Corporation as Global Hemophilia Franchise Head from June 2012 to December 2014. Earlier in his career, Mr. Goff held positions of increasing responsibility in sales and marketing roles with Novartis Pharmaceuticals, and the pharmaceutical division of Johnson & Johnson.
Mr. Goff has a MBA from the Wharton School at the University of Pennsylvania and a Bachelor of Arts from Skidmore College. |
|
|
| | |
| | Anne-Marie Lawis Executive Vice President, Chief Human Resources Officer of Alexion. She is responsible for Human Resources on a global basis, with the goal of continuing to build the organization capabilities to advance Alexion’s strategy. Ms. Law brings more than 25 years of experience at global corporations to the organization. Prior to joining Alexion in June 2017, she served as Chief Human Resources Officer at Hyatt Hotels Corporation from October 2016 to May 2017, where she was responsible for building the strategy to support the company’s 100,000 employees worldwide, and designing talent systems to create world class leadership and customer connectivity capabilities. She previously served as Executive Vice President and Chief Commercial OfficerHead of Human Resources for Baxalta Incorporated from April 2009 to December 2014, and held various senior human resources positions at McKesson Corporation, including the Specialty Health Division, VeriSign, and Xilinx, Inc.
Ms. Law is a graduate of Leicester University with a degree in Art History in the United Kingdom and the National College of Ireland, Dublin. |
Edward Miller, J.D. | 52 |
| Senior Vice President and Global Chief Compliance Officer |
Heidi L. Wagner, J.D. | 52 |
| Senior Vice President, Global Governmental Affairs |
David R. Brennan has been a member of the Board of Directors since July 2014 and as Interim Chief Executive Officer since December 2016. Prior to joining Alexion, Mr. Brennan held various positions of increasing responsibility at AstraZeneca PLC, from 1999-2012, including Chief Executive Officer and Executive Director, Executive Vice President of North America, and Senior Vice President of Commercialization and Portfolio Management. Mr. Brennan began his career in 1975 at Merck and Co. Inc., where he held various sales and general manager positions. Mr. Brennan currently serves on the Board of Directors of Innocoll, Inc. and Insmed Incorporated, and previously served on the Board of Directors of AstraZeneca PLC, Reed Elsevier PLC and the Pharmaceutical Research & Manufacturers of America (PhRMA). Mr. Brennan received a Bachelor of Arts in Business Administration from Gettysburg College, where he is a member of the Board of Trustees.
David J. Anderson, M.B.A. has been with Alexion since December 2016, serving as Executive Vice President and Chief Financial Officer. Prior to joining Alexion, Mr. Anderson served as Senior Vice President and Chief Financial Officer of Honeywell International from 2003-2014, where he was responsible for all corporate finance activities including accounting, treasury, tax, audit, investments, financial planning and acquisitions, and was integral to the reshaping of the company’s business portfolio. Prior to joining Honeywell, Mr. Anderson was Senior Vice President and Chief Financial Officer of ITT Industries, as well as Newport News Shipbuilding. Previously, he also held senior financial positions with RJR Nabisco and the Quaker Oats Company. Mr. Anderson serves on the Boards of several public companies, including Cardinal Health, a Fortune 20 leader in healthcare products and services. Mr. Anderson received a Bachelor of Science in Economics from Indiana University and a Masters of Business Administration from the University of Chicago (Booth School of Business).
Clare Carmichael has been with Alexion since August 2011 and has served as Executive Vice President and Chief Human Resources Officer since September 2014. From August 2011 to September 2014, Ms. Carmichael served as Senior Vice President and Chief Human Resources Officer. Prior to joining Alexion, Ms. Carmichael served as Senior Vice President, Global Human Resources at Watson Pharmaceuticals, Inc., from August 2008 to March 2011, where she established and executed global HR strategies. From December 2005 to August 2008, Ms. Carmichael held various human resources positions of increasing responsibility at Schering-Plough Corporation, including Vice President of Global Human Resources at the Schering-Plough Research Institute. From December 2003 to December 2005, Ms. Carmichael was Vice President of Human Resources at Eyetech Pharmaceuticals, Inc. Prior to Eyetech, she held various positions of increasing responsibility in human resources at Pharmacia Corporation. Ms. Carmichael received a Bachelor of Arts in Psychology from Rider University.
Martin Mackay, Ph.D. has been Executive Vice President, Global Head of Research & Development since joining Alexion in May 2013. Prior to joining Alexion, Dr. Mackay served as President, Research and Development at AstraZeneca from June 2010 to February 2012, where he led all R&D functions worldwide, including discovery research, clinical development, regulatory affairs and key related R&D functions. From April 1995 to May 2010, he held various positions of increasing responsibility at Pfizer, including President, Head of Pfizer Pharmatherapeutics, R&D, where he oversaw all aspects of small molecule discovery and development across multiple therapeutic areas. Dr. Mackay has also worked in the CIBA organization, now Novartis, and held positions within academia. Dr. Mackay received a Microbiology First Class Honors Degree from Heriot-Watt University, Scotland, and a Ph.D. in Molecular Genetics from the University of Edinburgh, Scotland.
John B. Moriarty, J.D. has been with Alexion since December 2012 and has served as Executive Vice President and General Counsel since September 2014. From December 2012 to September 2014, Mr. Moriarty served as Senior Vice President and General Counsel. From December 2010 to December 2012, Mr. Moriarty served as General Counsel and Chief Legal Officer at
Elan Corporation plc, an Irish public limited company traded on the New York and Irish Stock Exchanges, and also served as a member of Elan’s Executive Management team. Prior to assuming the role of General Counsel, Mr. Moriarty served as Senior Vice President of Law, Litigation and Commercial Operations at Elan from December 2008 to December 2010. From 2002 to 2008, Mr. Moriarty held various positions with Amgen, Inc., including Executive Director and Associate General Counsel, Global Commercial Operations - Amgen Oncology and Senior Counsel, Complex Litigation, Products Liability and Government Investigations. Between 1994 and 2002, Mr. Moriarty served in various capacities in private practice focused on healthcare and as a healthcare fraud prosecutor in the U.S. Attorney’s Office and the Virginia Attorney General’s Office. Mr. Moriarty received his Bachelor’s of Arts, with distinction, from the University of Virginia and his J.D., cum laude, from the University of Georgia School of Law.
Julie O’Neill, M.B.A. has been with Alexion since February 2014 and has served as Executive Vice President of Global Operations since January 2015. From January 2014 to January 2015, Ms. O’Neill was Senior Vice President Global Manufacturing Operations and General Manager of Alexion Pharma International Trading. Prior to joining Alexion, Ms. O’Neill served in various leadership positions at Gilead Sciences from February 1997 to February 2014 including Vice President of Operations and General Manager of Ireland from 2011 to 2014. Prior to Gilead Sciences, Ms. O’Neill held leadership positions at Burnil Pharmacies and Helsinn Birex Pharmaceuticals. She is the Chairperson for the National Standards Authority of Ireland and is a member of the Boards of the National Institute for Bioprocessing Research & Training and the American Chamber of Commerce, Ireland. Ms. O’Neill received a Bachelor of Science in Pharmacy from University of Dublin, Trinity College and a Masters of Business Administration from University College Dublin (Smurfit School of Business).
Carsten Thiel, Ph.D. has been with Alexion since September 2014 and has served as Chief Commercial Officer since September 2015. From January 2015 to September 2015, Mr. Thiel served as Senior Vice President EMEA and Asia Pacific and from September 2014 to January 2015, Mr. Thiel was Senior Vice President EMEA and Australasia-Canada. Prior to joining Alexion, Mr. Thiel served in various senior leadership positions at Amgen from 2002 to 2014, including Vice President, Head of Europe, General Manager, Germany, General Manager, CEE and Head of the Oncology Franchise in Europe. Prior to Amgen, Mr. Thiel held several sales and marketing leadership roles across Europe at Roche. Mr. Thiel has a Master Degree in Biochemistry from the University of Marburg, Germany, and a Ph.D. in Molecular Biology and Biochemistry from the Max Planck Institute, Germany.
Edward Miller, J.D. has been Senior Vice President and Global Chief Compliance Officer since joining Alexion in September 2014. Prior to joining Alexion, Mr. Miller served in various compliance and legal leadership positions at Boehringer Ingelheim from 2000 to August 2014, including Vice President, Associate General Counsel, Global Head of Litigation and Government Investigations; Vice President and Acting Global Compliance Officer and Vice President, Chief Compliance Officer and Head of Litigation. Prior to Boehringer Ingelheim, Mr. Miller was a Senior Trial Attorney at the DOJ in Washington, D.C. Mr. Miller received a Bachelor of Arts from Princeton University and his J.D. from Rutgers University School of Law.
Heidi L. Wagner, J.D., has been with Alexion since September 2009 and has served as Senior Vice President, Global Governmental Affairs since September 2012. From September 2009 to September 2012, Ms. Wagner served as Vice President, Global Government Affairs. Prior to joining Alexion, Ms. Wagner was the Sr. Director of Governmental Affairs for Genentech, and also consulted for a variety of health plans, biopharmaceutical and other healthcare-related companies. Ms. Wagner received a Bachelor of Science in Journalism and Mass Communication from the University of Colorado in Boulder, and her J.D. from the George Mason University School of Law in Virginia. |
| | |
| | John Orloff, M.D., is Executive Vice President, Head of Research & Development of Alexion. Dr. Orloff is focused on strengthening Alexion’s clinical pipeline and research programs, enhancing research and development productivity, overseeing regulatory and medical affairs, and supporting business development. Dr. Orloff has 20 years of experience in the biopharmaceutical industry and deep expertise spanning various stages of clinical and non-clinical development, including developing medicines for rare diseases. Prior to joining Alexion in June 2017, Dr. Orloff served as Executive Vice President, Head of Research & Development at Novelion from November 2016 to May 2017, where he currently sits on the Board of Directors. From July 2015 to July 2016, he served with Baxalta as Global Head of R&D and Chief Scientific Officer, where he advanced the company’s pipeline and oversaw regulatory approval of 10 unique products and two devices. He also held executive R&D roles with Baxter International from July 2014 to June 2015, Merck Serono from January 2014 to May 2014, Novartis from April 2003 to October 2013 and Merck Research Laboratories. Prior to joining the biopharmaceutical industry in 1997, Dr. Orloff was with the Yale School of Medicine for seven years. Dr. Orloff received a Bachelor of Arts from Dartmouth College, and a M.D. from the University of Vermont College of Medicine. He completed his medical training at the University of Pittsburgh Medical Center and Yale University School of Medicine. |
|
|
Available Information
Our internet website address is http://www.alexion.com. Through our website, we make available, free of charge, our Annual Reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, any amendments to those reports, proxy and registration statements, and all of our insider Section 16 reports, as soon as reasonably practicable after such material is electronically filed with, or furnished to, the SEC. These SEC reports can be accessed through the “Investors” section of our website. The information found on our website (or that may be accessed through links on our website) is not part of this or any other report we file with, or furnish to, the SEC. Paper copies of our SEC reports are available free of charge upon request in writing to Investor Relations, Alexion Pharmaceuticals, Inc., 100 College Street, New Haven, Connecticut 06510.121 Seaport Boulevard, Boston Massachusetts 02210. In addition, any document we file may be inspected, without charge, at the SEC’s public reference room at 100 F Street NE, Washington, DC 20549, orviewed at the SEC’s internet address at http://www.sec.gov.www.sec.gov. (This website address is not intended to function as a hyperlink, and the information contained in the SEC’s website is not intended to be a part of this filing). Information related
The company intends to use its website http://www.alexion.com as a means of disclosing material non-public information and for complying with its disclosure obligations under SEC Regulation FD. Such disclosures will be included on the operationcompany’s website under the heading “Investors”. Accordingly, investors should monitor such portions of the SEC’scompany’s website, in addition to following the company’s press releases, SEC filings and public reference room may be obtained by calling the SEC at 800-SEC-0330 (800-732-0330).conference calls and webcasts.
Item 1A. Risk Factors.
(amounts in millions, except percentages)
You should carefully consider the following risk factors before you decide to invest in Alexion securities and our business, because these risk factorsthe risks described below may have a significantmaterial impact on our business, operating results, financial condition, and cash flows. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business operations. If any of the following risks actually occurs, our business, financial condition and results of operations could be materially and adversely affected.
Risks Related to Our Products and Product Candidates
We depend heavily on the success of, and revenue from, Soliris.
Since 2007, our lead product,revenue has depended primarily on the sales of Soliris. IfUnless we are able to develop or acquire new products and technologies, successfully commercialize ULTOMIRIS as described in the following risk factor, and/or materially increase sales of Strensiq and Kanuma (two of our other currently approved products), we will remain dependent on sales of Soliris are adversely affected, our business may be materially harmed.
Currently, our ability to generate revenues depends primarily on the commercial success of Soliris and whether physicians, patients and healthcare payers view Soliris as therapeutically effective and safe relative to cost. Since we launched Soliris in the U.S. in 2007, substantially alla source of our revenue has been attributed to sales of Soliris. In 2015, we received marketing approval in the U.S., the EU and Japan, of our second marketed product, Strensiq, for the treatment of HPP. We also received marketing approval in 2015 in the United States and the EU for our third product, Kanuma, for the treatment of LAL-D. However, we anticipate that Soliris product sales will continue to contribute a significant percentage of our total revenue over the next several years.revenue.
The commercial success of SolirisSOLIRIS and our ability to generate revenuesrevenue depends on several factors, as discussed in greater detail below, includingincluding: the safety and efficacy of Soliris,SOLIRIS; coverage or reimbursement by government or third-party payers for SOLIRIS; pricing for SOLIRIS; the analysis by doctors and patients of the cost of SOLIRIS relative to the perceived benefits; manufacturing and uninterrupted supply,supply; the introduction of and success of competing products by competitors (including novel products and biosimilars to SOLIRIS); the size of patient populations and the number of patients diagnosed who may be treated with Soliris, adverseSOLIRIS; the impact of legal, administrative, regulatory or legislative developments,developments; and our ability to develop, registerobtain regulatory approval for and commercialize SolirisSOLIRIS for new indications.
While SOLIRIS has been studied for indications beyond PNH, aHUS and gMG (which are the current approved indications of SOLIRIS), there is no guarantee that we can obtain regulatory approval or achieve any commercial sales of SOLIRIS for other indications. Despite positive topline results from the Phase 3 PREVENT study of SOLIRIS in patients with anti-aquaporin-4 (AQP4) auto antibody-positive neuromyelitis optica spectrum disorder (NMOSD), we may not be able to obtain regulatory approval to sell SOLIRIS as a treatment for NMOSD due to the failure to meet applicable regulatory requirements. Additionally, even if we obtain regulatory approval, physicians and patients
may not accept SOLIRIS as a treatment for NMOSD or payers may not be willing to pay for or reimburse the costs of SOLIRIS as a therapy for NMOSD.
If we are not able to maintain revenues from sales of Soliris,SOLIRIS, or our SOLIRIS revenues do not grow as anticipated,decrease, our operating results would be negatively impacted and our ability to fund research and development programs for the discovery and commercialization or acquisition of operationsnew products would be harmed, which would limit our ability to diversify our revenue base and our stock price could be adversely affected.
If PNH patients do not switch from SOLIRIS to ULTOMIRIS or ULTOMIRIS does not gain market acceptance, our future operating results may be adversely impacted.
In December 2018, ULTOMIRIS was approved by the FDA for use in the U.S. for adult patients with PNH (and applications for approval of ULTOMIRIS are under review by the European Medicines Agency (EMA) and the Ministry of Health, Labour and Welfare (MHLW) in Japan for patients with PNH).
One of our principal business objectives is to facilitate the conversion of PNH patients from SOLIRIS to ULTOMIRIS. While clinical trials demonstrated that ULTOMIRIS is non-inferior to SOLIRIS at an 8 week dosing interval (compared to a 2 week dosing interval for SOLIRIS), existing PNH patients taking SOLIRIS and their physicians may decline to switch to ULTOMIRIS for many reasons including: reluctance to try a new therapy, lack of clinical evidence that ULTOMIRIS is superior to SOLIRIS, no (or limited) reimbursement by government or third-party payers (including as a result of SOLIRIS being available as an alternative therapy), or our inability to manufacture quantities necessary to meet demands.
If we achieve our goal of promptly facilitating the conversion of current PNH patients from SOLIRIS to ULTOMIRIS, we anticipate that revenue from SOLIRIS, which accounted for approximately $3,563.0, or 86.3%, of our revenues in 2018, will decline as we move patients to ULTOMIRIS. We have established a price for ULTOMIRIS in the U.S. that, on an annual basis, represents an approximate 10% discount to the cost of current labeled maintenance therapy for SOLIRIS for adult PNH patients of average weight. However, this represents an approximate 10% premium to the cost of SOLIRIS in a patient’s first year of switching due to the loading doses required.
We may not obtain marketing approval for ULTOMIRIS as a treatment for PNH in any jurisdictions beyond the U.S. or for any indications beyond PNH.
There is no guarantee that the EMA or the MHLW (or any other regulatory authority) will promptly approve the use of ULTOMIRIS in PNH patients or that they will approve the use of ULTOMIRIS in PNH patients at all. We believe that the EU and Japan may be important
potential markets for ULTOMIRIS and if we are not able to sell ULTOMIRIS in these geographies, our business may be adversely impacted.
Subject to successful completion of clinical trials, we intend to pursue marketing approval for ULTOMIRIS in the U.S., the EU, Japan and other jurisdictions for indications in addition to PNH and, potentially, other delivery mechanisms. The FDA, the EMA or the MHLW could reject our applications for indications beyond PNH (and the EMA and the MHLW could reject our application for ULTOMIRIS for PNH) or for a subcutaneous delivery mechanism for many reasons, including due to a finding of inadequate safety, tolerability, potency or efficacy profiles. Additionally, these and other regulatory agencies may request that we provide additional safety or efficacy data, which may require significant additional time and expense to generate prior to a decision on approval.
If ULTOMIRIS is not approved for use in PNH patients in the EU or Japan (or other jurisdictions) or for any other indications or for subcutaneous administration in the U.S., the EU, Japan or elsewhere or if any such approval is delayed, our future business and results of operations may be harmed. In the event of any of the foregoing, while we would continue to sell SOLIRIS in the jurisdictions and for the indications authorized by the appropriate authorities, certain of the patents and regulatory exclusivities related to SOLIRIS expire earlier than patents and regulatory exclusivities we hold on ULTOMIRIS, which may allow competitors to enter those markets at an earlier date utilizing SOLIRIS or biosimilar technology.
Our future commercial success depends on gaining regulatory approval for new products and obtaining approvals for existing products for new indications.
We have invested, and continue to invest, significant amounts in acquiring new products and technologies and advancing our existing product candidates and technologies. Our long-term success and revenue growth will depend upon the successful identification, acquisition (including licenses from third parties), development and commercialization of new products and technologies, from our research and development activities, including those licensed or acquired from third parties and approval of additional indications for our existing products.products and products under development. Product development (including products acquired in connection with acquisitions) is very expensive, takes significant time to obtain regulatory approval and involves a high degree of risk. Only a small number of research and development programs result in the commercialization of a product. The process for obtaining regulatory approval to market a biologic is expensive, often takes many years, and can vary substantially based on the type, complexity, andthe novelty of the product candidates involved. involved and the indications to be treated. Further, success in early clinical trials, which may lead to further investment in a product candidate by us, may not result in success in later stage
trials. In addition, our recent acquisitions have focused on new technologies with which we have very limited experience, including antibody therapeutics targeting the neonatal Fc receptor, which may make the development, approval and commercialization of such potential products challenging.
Our ability to maintain or grow revenues wouldmay be adversely affected if we are delayed or unable to successfully develop the products in our pipeline, including Solirisif we are unable to gain approval for SOLIRIS and ULTOMIRIS for additional indications and in new jurisdictions, obtain marketing approval for StrensiqSTRENSIQ and KanumaKANUMA in additional territories, obtain approval for additional delivery systems for our therapies (such as subcutaneous administration) or acquire or license products and technologies from third parties.
We dedicate significant resources to the worldwide development, manufacture and commercialization of our products. We cannot guarantee that any marketing application for our product candidates will be approved or maintained in any country where we seek marketing authorization. If we do not obtain regulatory approval of new products or additional indications for existing products or additional delivery systems, or are significantly delayed or limited in doing so, our revenue growth willmay be adversely affected, we may experience surplus inventory, we may be required to write down certain assets, our business may be materially harmed and we may need to significantly curtail operations.
Because the targetWe develop therapies for rare diseases with limited patient populations of Kanuma and Stensiq are small andthat have not been definitively determined, we must be ableand our success will depend on our ability to successfully identify patients in order to maintain growth.the disease areas we target.
KanumaThe therapies that we have developed and Stensiqthat are in our product pipeline target diseases that have a limited number of patients and for which, in many cases, there are either no or limited diagnostics tools. For example, KANUMA and STRENSIQ are currently approved to treat ultra-rare diseases with small patient populations that have not been definitively determined. Our development pipeline programs that may be the basis for future revenue growth also focus on rare (and ultra-rare) diseases for which there are a very limited number of patients. The lack of diagnostic tools, coupled with the fact that there is frequently limited awareness among certain health care providers concerning the rare diseases we treat, often means that a proper diagnosis can, and frequently does, take years to identify (or an appropriate diagnosis may never be made for certain patients). As a result, we may not be able to grow our revenues (even as we introduce new products or as existing products are approved for additional indications). There can be no guarantee that any of our programs will be effective at identifying patients, and even if we can identify patients that our therapies can help, the number of patients in the United States, Japan and Europe and elsewherethat our therapies treat may turn out to be lower than expected,we expect, may not be otherwise amenable to treatment with Kanumaour products (such as KANUMA and Stensiq,STRENSIQ), or new patients may become increasingly difficult to identify, all of which wouldmay adversely affect our results of operations and our business. In addition, even in instances where we do
add patients, the number may be less than the number of patients that discontinue use of the applicable product in a given period resulting in a net loss of patients and potentially decreased revenue.
We may not be able to gain or maintain market acceptance of our products among the medical community, patients or payers, which could prevent us from maintaining profitability or growth.
Our products may not gain or maintain market acceptance among physicians, patients, healthcare payers and others. Although we have received regulatory approval for certain of our products in certain territories, such approvals do not guarantee future revenue. We cannot predict whether physicians, other healthcare providers, government agencies or private insurers will determine or continue to accept that our products are safe and therapeutically effective and that the benefits are meaningful relative to the cost. Nor can we predict whether patients, physicians or payers will continue use of SOLIRIS or elect to convert to ULTOMIRIS in the U.S. (or other jurisdictions if and when approved for use by the appropriate regulatory authorities) or alternative treatments that may become available. Physicians’ willingness to prescribe, and patients’ willingness to accept, our products, depends on many factors, including:
prevalence and severity of adverse side effects in both clinical trials and commercial use;
the timing of the market introduction of competitive drugs and biosimilars;
demonstrated clinical safety and efficacy compared to other drugs;
perceived cost-effectiveness and/or evaluations in HTAs;
pricing and availability of reimbursement from third-party payers, including governmental entities;
convenience and ease of administration;
effectiveness of our marketing strategy;
publicity concerning our products and our other product candidates (and those of competitive products); and
availability of alternative treatments.
The likelihood of physicians to prescribe SOLIRIS for patients with aHUS (and ULTOMIRIS, if approved for use by aHUS patients) may also depend on how quickly SOLIRIS can be delivered to the hospital or clinic and our distribution methods may not be sufficient to satisfy this need. In addition, we are aware that some healthcare providers have determined not to continue SOLIRIS treatment for some patients with aHUS. While SOLIRIS as a treatment for aHUS is recommended by some regulatory authorities to be used for the duration of a patient’s lifetime, we are aware that some healthcare providers prescribe SOLIRIS for aHUS for a shorter time
period and, in some cases, may prescribe SOLIRIS for aHUS in emergency or acute situations only. Decisions such as this by aHUS patients and healthcare providers to use our products for a period that is less than the remaining lifetime of the patient or in only acute circumstances can cause our SOLIRIS revenues, and revenues for our other products, to fluctuate and past sales of our products may not be indicative of future sales for such products.
If our products fail to achieve or maintain market acceptance among the medical community or patients in a particular country, we may not be able to market and sell our products successfully in such country, which may limit our ability to generate revenue and could harm our overall business.
If our products harm patients, or are perceived to harm patients even when such harm is unrelated to our products, our regulatory approvals could be revoked or otherwise negatively impacted and we could be subject to costly and damaging product liability claims.
The testing, manufacturing, marketing and sale of biologics for use in humans may cause harm to patients, which exposes us to product liability risks and regulatory penalties.
Our products and our product candidates treat patients with rare diseases and, as a result, we generally are able to test our products in only a small number of patients. As more patients use our products, including more children and adolescents, new risks and side effects may be discovered, the rate of known risks or side effects may increase, and risks previously viewed as less significant could be determined to be significant. Previously unknown risks and adverse effects may also be discovered in connection with unapproved uses of our products, which may include administration of our products under acute emergency conditions, such as the Enterohemorrhagic E. coli health crisis in Europe, primarily Germany, which began in May 2011. Under pharmacovigilance guidelines, we are required to timely report any adverse events any patient using our products experiences and any clinical evaluations of outcomes in the post-marketing setting are required to be reported to appropriate regulatory agencies in accordance with relevant regulations, as a result any potential adverse events will be promptly brought to the attention of regulators that may likely require prompt remedial action (and any failure to report these adverse events or report such events in a timely manner may result in penalties being imposed by regulators). In the event any new risks or adverse effects discovered as new patients are treated for approved indications, or as our products are studied in or used by patients for other indications, regulatory authorities may delay or revoke their approvals, we may be required to conduct additional clinical trials and safety studies, make changes in labeling, reformulate our products or make changes and obtain new approvals for our and our suppliers’
manufacturing facilities. If we experience any of the foregoing actions, it may harm our reputation and, particularly given that we rely on a very limited number of products for our revenue, our business and results of operations could be materially and adversely impacted. Further, any investigation into the circumstances surrounding an adverse event may be costly and time consuming (even if it is ultimately determined that the adverse event is not the result of the use of our product) or the investigation may not be sufficiently conclusive to prevent a regulatory authority from taking one of the foregoing actions against us.
In addition, many patients who use our products are already very ill and may suffer adverse events, including death, during treatment for reasons that may or may not be related to our products. Also, there are risks associated with our products; for example, use of C5 Inhibitors, such as SOLIRIS and ULTOMIRIS, is associated with an increased risk for certain types of infection, including meningococcal infection. In certain cases, a physician may not have the opportunity to timely vaccinate a patient in the event of an acute emergency episode, such as in a patient presenting with aHUS, which could result in the patient using SOLIRIS or ULTOMIRIS experiencing a life-threatening meningococcal infection (and even in certain cases in which a vaccination can be delivered to the patient, it may not, eliminate all risk of meningococcal infection). Patients using our products and product candidates have died or suffered potentially life-threatening conditions either during or after ending their treatments, and these include patients who have died while participating in a clinical trial (for example, four patients died during the ULTOMIRIS Phase III clinical trial for aHUS, although none of these were considered related to the treatment with ULTOMIRIS). We may be sued by patients who are harmed during the course of using our products, whether as a prescribed therapy, during a clinical trial, during an investigator initiated study, or otherwise. Any such product liability lawsuit or injury claim, which could include class actions, could harm our reputation among patients, physicians, payers and others and require us to pay substantial amounts of money to injured patients, and even if successfully defended, could have a material adverse effect on our business, financial condition or results of operations due to the expense of defending any such claim. While we do have product liability insurance, it may not cover all potential types of liabilities or may not cover certain liabilities completely. Moreover, we may not be able to maintain our insurance on acceptable terms, or at all.
We anticipate that we may face increased competition from companies that will enter into the markets we currently serve and as our product pipeline expands into markets that are currently served by other companies.
We expect that the business environment in which we operate will become increasingly competitive.
Currently, certain of our products are the only approved therapy for the indication they treat. For example, SOLIRIS and ULTOMIRIS (in the U.S.) are the only approved treatments of PNH. In the future, we expect that SOLIRIS and ULTOMIRIS may compete with new, novel drugs and pharmaceuticals currently in development. For example, several companies are developing and engaged in clinical trials for therapies to treat PNH, aHUS, and gMG. If SOLIRIS is approved for treatment of NMOSD, we expect there may be competition in that market as well. Since other companies are also operating clinical trials in this disease state. Additionally, other pharmaceutical companies have publicly stated that they are developing and intend to commercialize a SOLIRIS biosimilar and these biosimilars may be commercially available in the future. STRENSIQ and KANUMA may also experience competition in the future. We are also aware of companies that are planning to initiate studies for diseases that we are also targeting with our product pipeline. Our revenues could be negatively affected if patients or potential patients enroll in our clinical trials or clinical trials of other companies with respect to diseases that we also target with approved therapies.
Other pharmaceutical companies have publicly announced intentions to establish or develop rare disease programs and may introduce products that compete with ours (or products that are in our pipeline). These and other companies, many of which have significantly greater financial, technical and marketing resources than us, may commercialize products that are cheaper, more effective, safer, have less frequent dosing schedules, or easier to administer than our products. Our current and future competitors may develop products that are more broadly accepted or may receive patent protection that dominates, blocks or adversely affects our product development or business. These competitive products, including any biosimilars approved under alternative regulatory pathways, may significantly reduce both the price that we receive for such marketed products and the volume of products that we sell, which may negatively impact our revenues and profitability. Given that a significant portion of our 2018 revenue was attributable to SOLIRIS, one or more competitive products or biosimilar could have a significant impact on our entire business. In addition, we experience competition in drug development from universities and other research institutions, and pharmaceutical companies compete with us to attract universities and academic research institutions as drug development partners, including for licensing their proprietary technology. If our competitors successfully enter into such arrangements with academic institutions, we may be precluded from pursuing those unique opportunities and may not be able to find equivalent opportunities elsewhere.
If a company announces successful clinical trial results for a product that may be competitive with one
of our products or product candidates, receives marketing approval of a competitive product, or gets to the market before we do with a competitive product, our business may be harmed or our stock price may decline.
Risks Related to Pricing and Reimbursement
Sales of our products depend on reimbursement by government health administration authorities, private health insurers and other organizations.organizations, each of which are subject to pressures to contain costs. If we are unable to obtain, or maintain at anticipated levels, reimbursement for and access to our products, or coverage is reduced, our pricing may be adversely affected or our product sales, results of operations or financial condition could be harmed.
We may not be able to sell our products on a profitable basis or our profitability may be reduced if we are required to sell our products at lower than anticipated prices or reimbursement is unavailable or limited in scope or amount. Our products are significantly more expensive than traditional drug treatments and almost all patients require some form of third party coverage to afford their cost. We depend, to a significant extent, on governmental payers, such as Medicare and Medicaid in the U.S. or country specific governmental organizations in foreign countries, andand/or private third-party payers to defraypay all or a portion of the cost of our productsproducts. There is also a significant trend in the health care industry by public and private payers to patients. These entities may refuse to provide coverage and reimbursement, determine to provide a lower level of coverage and reimbursement than anticipated,contain or reduce previously approved levelstheir costs. As a result, payers have in the past (i) decreased the portion of coveragecosts they will cover, (ii) ceased providing adequate payment for our products or (iii) not covered our products at all, each of which payers may continue to do in the future (or other payers who have not taken such actions in the past may do so in the future). Any of the foregoing may have an adverse impact on our revenue and reimbursement,results of operations.
Our ability to set the price for our products varies significantly from country to country, including in the form of higher mandatory rebates or modified pricing terms.
In certainthose countries where we sell or are seeking or may seek to commercialize our products, pricing, coverage, and level of reimbursement or funding of prescription drugs are subject to governmental control. We may be unable to timely or successfully negotiate coverage, pricing and reimbursement on terms that are favorable to us (or at all), or such coverage, pricing and reimbursement may differ in separate regions in the same country. In some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. As discussed above in the subsection entitled “Pharmaceutical Pricing and Reimbursement,” the requirements governing drugmarketed, which could delay market entry (or, if pricing vary widely from country to country, which may include a combination of distinct potential payers, including private insurance and governmental payers as well as a HTA assessment of medicinal products for pricing and reimbursement methodologies. Therefore,is not approved, we may not successfully concludebe unable to sell at all in a country where we have received regulatory approval for a product). In addition, authorities in some countries impose additional obligations, such as HTAs, which assess how well a pharmaceutical works in relation to its cost. Additionally, U.S. payers are increasingly considering new metrics as the necessary processes and commercializebasis for reimbursement rates. If our products in every,do not meet or even most countries in which we seek to sellsurpass these metrics, including any HTAs and other metrics imposed on our products.
A significant reduction inproducts, these payers may not reimburse for use of our products or may reduce the amountrate of reimbursement or pricing for our products and as a result we expect revenue from such product may decrease. We may also, in onesome
cases, elect to reduce prices or more countries may reduce our profitability and adversely affect our financial condition. Certainreimbursement with third parties which we believe provides value in the long term.
Further, certain countries establish pricing and reimbursement amounts by reference to the price of the same or similar products in other countries. Therefore, if coverage or the level of reimbursement is limited in one or more countries, we may be unable to obtain or maintain anticipated pricing or reimbursement in other countries or in new markets. In Canada, for example, the Patented Medicine Prices Review Board (PMPRB) issued a decision in an administrative pricing matter that we had excessively priced SOLIRIS in a manner inconsistent with the Canadian pricing rules and guidelines and ordered that the price be decreased to no higher than the lowest price in seven comparator countries (we filed an application for judicial review of the PMPRB’s decision in the Federal Court of Canada, and a hearing on the matter was held in November 2018, but we are unable to determine the outcome of this review at this time since the court has not yet issued its opinion). In addition, the current or new territories. InU.S. presidential administration recently unveiled a number of proposals, among these was a recommendation to move from the current U.S. pricing and reimbursement regime to one that would establish pharmaceutical pricing by reference to a target price derived from the international price index (such a change may be expected to result in significant savings for the government for purchases of certain pharmaceuticals). If the U.S., which accounted for a significant portion of our revenue in 2018, were to move to a pricing system based on the EU member states, and elsewhere, there have been, andinternational price index (or similar model) that were to apply to our products, we expect there will continue to be, efforts to control and reduce healthcare costs. Inthat our revenues for sales in the U.S. for example, the(or any other country adopting such a price of drugs has come under intense scrutiny by the U.S. Congress. Third party payers decide which drugs they will pay forindex) may decrease, and establish reimbursement and co-payment levels. Government and other third-party payers are increasingly challenging the prices charged for healthcare products, examiningsuch decrease may be material in amount.
Due to the cost effectiveness of drugs in addition to their safety and efficacy, and limiting or attempting to limit both coverage and the level of reimbursement for prescription drugs. See additional discussion below under the headings “Changes in healthcare law and implementing regulations, including those based on recently enacted legislation, as well as changes in healthcare policy and government initiatives that affect coverage and reimbursement of drug products may impact our business in ways that we cannot currently predict and these changes could adversely affect our business and financial condition” and “The credit and financial market conditions may aggravate certain risks affecting our business.”
Thetherapies, any potential increase in the number of patients receiving Solirisour products (for example, we expect there may be increases in sales of SOLIRIS for patients with NMOSD, if approved by regulatory authorities for that indication), may cause third-party payers to modify, limit or limiteliminate coverage or reimbursement for Soliris for the treatmentour products because they may require an allocation of PNH, aHUS, or both indications. To the extent we are successful in developing Soliris for indications other than PNH and aHUS,a greater percentage of the potential increase in the numberfinancial resources of patients receiving Soliris may cause third-party payers to refuseany public or limit coverage or reimbursementprivate payer for Soliris for the treatment of PNH, aHUS or for any other approved indication, or provide a lower level of coverage or reimbursement than anticipated or currently in effect.our products.
As discussed above in the subsection entitled “Pharmaceutical Pricing and Reimbursement,”Further, health insurance programs may restrictutilize coverage of someincentives and obstacles to discourage beneficiaries from using higher priced products by using payersuch as ours, including:
establishing formularies under which only selected drugs are covered,covered;
utilizing variable co-payments that make drugs that are not preferred by the payer more expensive for patients,patients; and by using utilization
utilizing management controls, such as requirements for prior authorization or failure first on another type of treatment. Payers
Any of these actions may especially impose these obstacles to coverage for higher-priced drugs, and consequentlysubject our products may be subject to payer-driven restrictions. Additionally, U.S. payers are increasingly considering new metrics as the basis for reimbursement rates.
In countries where patients have access to insurance, their insurance co-payment amounts or other benefit limits may represent a barrier to obtaining or continuing Soliris. Weuse of our products or adoption of new treatment options, such as ULTOMIRIS. The continuation of the use of these types of limits or barriers by insurers or the imposition of similar limitations or barriers in the future may have an adverse impact on our revenue and results of operations. In some cases, we have financially supported non-profit organizations that assist patients in accessing treatment for PNH and aHUS, including Soliris.SOLIRIS, among other therapies. Such organizations assist patients whose insurance coverage imposes prohibitive co-payment amounts or other expensive financial obligations. Such organizations’ ability to provide assistance to patients is dependent on funding from external sources, and we cannot guarantee that such funding will be provided at adequate levels, if at all. We have also provided our products without charge to patients who have no insurance (or limited insurance) coverage for drugs through related charitable purposes. We are not able to predict the financial impact of the support we may provide for these and other charitable purposes; however, substantial support could have a material adverse effect on our profitability in the future.
As third-party payers attempt to contain health care costs they are demanding price discounts or rebates and limiting both the types and variety of drugs that they may cover and the amounts that they will pay for drugs. As a result, they may not cover or provide adequate payment to patients for our products or they may demand discounts or rebates from us, which may be material.
Our commercial success depends on obtaining and maintaining pricing for our products, which is directly tied to reimbursement for our products at anticipated levels reimbursement for our products. It may beis difficult to project the impact of evolving reimbursement mechanics on the willingness of payers to cover our products.products, but we expect pharmaceutical pricing to continue to be subject to intense payer, political and societal pressures on a global basis. If we are unable to obtain or maintain coverage for our products, or coverage is reduced or eliminated in one or more countries or if the U.S. (or other countries) were to move to an international price index for our products, our pricing, may be affected or our product sales, results of operations or financial condition could be harmed.
Risks Related to Business Operations
We may not be ablerely on a limited number of facilities to maintain market acceptance ofproduce our products among the medical community or patients, or gain market acceptance of our products in the future, which could prevent us from maintaining profitability or growth.
We cannot be certain that our products will maintain market acceptance in a particular country among physicians, patients, healthcare payers, and others. Although we have received regulatory approval of our products in certain territories, such approvals do not guarantee future revenue. We cannot predict whether physicians, other healthcare providers, government agencies or private insurers will determine or continue to accept that our products are safe and therapeutically effective relative to their cost. Physicians’ willingness to prescribe, and patients’ willingness to accept, our products, depends on many factors, including prevalence and severity of adverse side effects in both clinical trials and commercial use, the timing of the market introduction of competitive drugs, lower demonstrated clinical safety and efficacy compared to other drugs, perceived lack of cost-effectiveness, pricing and lack of availability of reimbursement from third-party payers, convenience and ease of administration, effectiveness of our marketing strategy, publicity concerning the product, our other product candidates and availability of alternative treatments, including bone marrow transplant as an alternative treatment for PNH. The likelihood of physicians to prescribe Soliris for patients with aHUS may also depend on how quickly Soliris can be delivered to the hospital or clinic and our distribution methods may not be sufficient to satisfy this need. In addition, we are aware that medical doctors have determined not to continue Soliris treatment for some patients with aHUS.
If our products fail to achieve or maintain market acceptance among the medical community or patients in a particular country, we may not be able to market and sell our products successfully in such country, which would limit our ability to generate revenue and could harm our overall business.
Manufacturingmanufacturing issues at our facilities or the facilities of our third party service providers could cause product shortages, stop or delay commercialization of our products, disrupt or delay our clinical trials or regulatory approvals, and adversely affect our business.business.
The majority of our products and product candidates are biologics, which cannot be manufactured synthetically and must be produced from biologic sources. As a result, the production of biologic therapeutics that meet all product specification and regulatory requirements is particularly complex. Even slight deviations at any point in the production process may lead to production failures or recalls. For example, in 2013 and 2014 we undertook a voluntary recall of SOLIRIS due to the presence of visible particles in a limited number of vials. In addition, because the production process involves the use of materials that are derived from biological sources, the process can be affected by contaminants that could impact those biological micro-organisms. Therefore, the manufacture of our products and our product candidates is highly regulated, complex and difficult, requiring a multi-step controlled process and, as noted above, even minor technical problems or deviations could result in significant defects or failures. Wefailures and regulatory action against us. These manufacturing challenges are coupled with the fact that we have limited experience manufacturing commercial quantities of StrensiqULTOMIRIS, STRENSIQ and Kanuma. OnlyKANUMA (so we may have limited previous experience resolving any issues in connection with the manufacture of these products and it may take significant time to remediate or we may be unable to solve any manufacturing problems) and we rely on a smalllimited number of companies have the ability and capacityfacilities to manufacture our products for our development, clinical and commercialization needs. Dueneeds, some of which we own and some of which are owned by third parties.
If we and/or our third party suppliers fail to meet the highly technical requirements of manufacturing our biologic products and theour strict quality and control specifications, we and our third party providers(or they) may be unable to manufacture or supply our products despiteproducts. We depend on our third party manufacturers to perform effectively on a timely basis and their efforts. Failureto comply with regulatory requirements and meet our product specifications. If they are unable to do so, our contractual rights to address any failures and right to recover damages are limited. Our failure or the failure of our third-party manufacturers to produce sufficient quantities of our products and product candidates could result in lost revenue, diminish our profitability, delay the development of our product candidates, delay regulatory approval, result in the rejection of our product candidates or result in supply shortages for our patients, which may lead to lawsuits,
loss of revenue or could accelerate introduction of competing products to the market.
TheAs noted above, the manufacture of our products and product candidates is at high risk of product loss due to contamination, equipment malfunctions, human error or raw material shortages. Deviations from established manufacturing processes couldshortages, which may result in reduced production yields, product defects and other supply disruptions. If microbial, viral or other contaminations are discovered in our products or manufacturing facilities, or the facilities of our third party manufacturers, we or our third party manufacturers may need to close our or their manufacturing facilities for an extended period of time to investigate and remediate the contaminant. The occurrence of any such event could adversely affect our ability to satisfy demand for any of our products, which could materially and adversely affect our operating results.
Many additional factors could cause production interruptions at our facilities or at the facilities of our third party providers, including natural disasters, labor disputes, acts of terrorism or war. The occurrence of any such event could adversely affect our ability to satisfy demand for Soliris, which could materially and adversely affect our operating results.
We expect that the demand for Soliris will increase. We mayIf we underestimate demand for SolirisULTOMIRIS, SOLIRIS or any of our products, or experience product interruptions at Alexion’s internal manufacturing facilities or a facility of a third party provider, including as a result of risks and uncertainties described in this report.Annual Report on Form 10-K, we may not be able to increase our revenues and alternative therapies may gain greater market acceptance.
We also face external factors, many of which are beyond our control, that could cause production interruptions at our facilities or at the facilities of our third party providers, including natural disasters, labor disputes, acts of terrorism or war.
The risks to our business of any manufacturing stops or interruptions (whether the result of internal or external factors) are amplified because we rely on a limited number of facilities to produce our products and product candidates. For example, each of our products is manufactured at only one to two facilities. Sales of SOLIRIS, which accounted for 86.3% of our revenue for the fiscal year ended December 31, 2018, in the U.S., the EU, Japan and certain other territories were manufactured exclusively by Lonza at its facilities in Singapore and Spain. Manufacturing SOLIRIS for commercial sale in certain other territories may only be performed at a single facility in some cases until such time as we have received the required regulatory approval for an additional facility, if ever. We expect that we will continue to rely on a very limited number of manufacturing facilities in the future for all of our products, including ULTOMIRIS.
We and our third party providers are required to maintain compliance with cGMP and other stringent operation and manufacturing requirements and are subject to inspections by the FDA and comparable agencies in other jurisdictions to confirm such compliance. Governmental authorities will generally not permit products manufactured at a facility that is not registered by the applicable government agency to enter into the country and such products may be returned for failure to comply with such regulation, which may decrease or delay sales and result in the loss of inventory. Any delay, interruption or other issues that
arise in the manufacture, fill-finish, packaging or storage of our products as a result of a failure of our facilities or the facilities or operations of third parties to pass any regulatory agency inspection or comply with on-going operating regulations could significantly impair our ability to supply our products and product candidates. Significant noncompliance could also result in the imposition of monetary penalties or other civil or criminal sanctions and damage our reputation.
Our efforts to bring more of our manufacturing operations under our control present additional challenges. We rely on onehave completed the build-out of a fill-finish facility in Ireland to two facilitiessupport global drug product manufacture or vial fill finish of SOLIRIS and certain of our other clinical and commercial products. We also completed construction of a facility in Dublin, Ireland in the fourth quarter of 2015, which is comprised of laboratories, packaging and warehousing operations and we intend to make significant further investment in this facility for the manufacture each of our products. We are authorizedalso constructing new biologics manufacturing facilities at both sites. Despite the significant investment we have made in these facilities and operations, we cannot guarantee that we will be able to sell Solirissuccessfully and timely complete the construction of the biologics facilities or the appropriate validation processes or obtain the necessary regulatory approvals for these and other facilities, or that is manufactured by Lonzawe will be able to perform the intended manufacturing and supply chain services at ARIMF in the U.S., the EU, Japan and certain other territories. However, manufacturing Soliristhese facilities for commercial
sale in certain other territories may only be performed at a single facility in some cases until or clinical use. Prior to such time, as we have received the required regulatory approval for an additional facility, if ever, however in certain territories only a single manufacturing facility may be registered and we will continue to rely on a singlethird parties for these services.
If we experience any manufacturing facility in such instances. We will continueissues, we may be unable to depend entirely on one facility to manufacture Soliris for commercial sale in such other territories until that time. We also depend entirely on one facility to manufacture Strensiqtimely identify alternative manufacturers, and on one facility for the purification of Kanuma for commercial sale. Regarding Kanuma, we rely on two animal facilities to produce the starting material, and a single manufacturing facility to manufacture the drug product.
We depend on a very limited number of third party providers for supply chain services with respect to our clinical and commercial product requirements, including product filling, finishing, packaging, and labeling. Our third party providers operate as independent entities and we do not have control over any third party provider’s compliance with our internal or external specifications or the rules and regulations of regulatory agencies, including the FDA, competent authorities of the EU member states, or any other applicable regulations or standards.
Any difficulties or delays in our third party manufacturing, or any failure of our third party providers to comply with our internal and external specifications or any applicable rules, regulations and standards could increase our costs, constrain our ability to satisfy demand for our products from customers, cause us to lose revenue or incur penalties for failure to deliver product, make us postpone or cancel clinical trials, or cause our products to be recalled or withdrawn, such as the voluntary recalls that we initiated in 2013 and 2014 due to the presence of visible particles in a limited number of vials in specific lots. Even if we are able to find alternatives theytimely identify alternative manufacturers, such alternative manufactures may ultimatelynot be insufficient forable to satisfy our needs.requirements. No guarantee can be made that regulators will approve additional third party providers in a timely manner or at all, or that any third party providers will be able to perform services for sufficient product volumes for any country or territory. Further, due to the nature of the current market for third-party commercial manufacturing, many arrangements require substantial penalty payments by the customer for failure to use the manufacturing capacity for which it contracted. Penalty payments under these agreements typically decrease over the life of the agreement, and may be substantial initially and de minimis or non-existent in the final period. The payment of a substantial penalty could harm our financial condition.condition and may restrict our ability to transition to internal manufacturing or manufacturing by other third parties. In addition, the terms and conditions to engage an additional third party manufacturer may not be as favorable to us as our current arrangements and may likely reduce the profit on the sales of any products to which they relate.
It can take longer than five yearsIn addition, KANUMA is a transgenic product and the facilities on which we rely to build and validate a new manufacturing facility and it can take longer than three years to qualify and validate a new contract manufacturer. We have completedproduce raw material for KANUMA are the build-out of a fill-finish facilityonly animal facilities in Ireland to support global drug product manufacture or vial fill finish of Soliris and Alexion’s other clinical and commercial products. We cannot guaranteethe world that this facility will receiveproduce the necessary global regulatory approvalsegg whites from transgenic
chickens. Natural disasters, disease, such as exotic Newcastle disease or avian influenza, or other catastrophic events could have a significant impact on the supply of unpurified KANUMA, or destroy our animal operations altogether. If our animal operations are disrupted, it may be extremely difficult to set up another animal facility to supply the unpurified KANUMA.
Any adverse developments affecting our manufacturing operations or the operations of our third-party providers could result in a timely mannerproduct shortage of clinical or commercial requirements, withdrawal of our product candidates or any approved products, shipment delays, lot failures or recalls. We may also have to write-off inventory and we will continueincur other charges and expenses for products that fail to meet specifications, undertake costly remediation efforts or seek more costly manufacturing alternatives. Each of these could have an adverse material impact on our business individually or in the aggregate. Such manufacturing issues could increase our cost of goods, cause us to lose revenue, reduce our profitability or damage our reputation.
We rely on appropriate third parties to supplementa limited number of providers for our fill finish operations until that time. We also completed construction of a new facility in Dublin, Ireland in the fourth quarter of 2015, which is comprised of laboratories, packagingraw materials and warehousing operations and we intend to make significant further investment in this facility for the manufacture our products. We cannot guarantee that we will be able to successfully and timely complete the appropriate validation processes or obtain the necessary regulatory approvals, or that we will be able to perform the intended supply chain services, at either of these facilities for commercial orwhich could result in our being unable to continue to successfully commercialize our products and our product candidates (if approved) and to advance our clinical use.pipeline.
Certain of the raw materials required in the manufacture and the formulation of our products are derived from biological sources. Such raw materials are difficult to procure and may be subject to contamination or recall. Access to and supply of sufficient quantities of raw materials which meet the technical specifications for the production process is challenging, and often limited to single-source suppliers. Finding an alternative supplier could take a significant amount of time and involve significant expense due to the nature of the products and the need to obtain regulatory approvals. The failure of these single-source suppliers to supply adequate quantities of raw materials for the production process in a timely manner may impact our ability to produce sufficient quantities of our products for clinical or commercial requirements. A material shortage, contamination, recall, or restriction on the use of certain biologically derived substances or any raw material used in the manufacture of our products could adversely impact or disrupt manufacturing.manufacturing and materially limit our ability to generate revenues.
We also depend on a very limited number of third party providers for supply chain services with respect to our clinical and commercial product requirements, including product filling, finishing, packaging and labeling.
These third party raw material providers and supply chain service providers operate as independent entities and we do not exercise control over any such third party provider’s operations or their compliance with our internal or external specifications or the rules and
regulations of regulatory agencies, including the FDA, competent authorities of the EU Member States, or any other applicable regulations or standards. Any contractual remedies we may have under agreements with these parties may not protect us from the harm suffered by our business or our patients if they fail to provide material or perform services that meet our specifications. Due to the highly specialized nature of the services performed by these third parties, particularly the supply of our raw materials, we do not believe that we could quickly find replacement suppliers or service providers and, even if we were able to identify additional third parties, the terms of any such arrangement may not be favorable to us. In either of these cases, our revenue, results of operations, business and reputation may be harmed and we may not be able to provide the therapies that our patients require.
The success of our business may also depend on the security of our products while in the supply chain for delivery to patients, which, as noted above, is dependent on third-party providers. For example, if our products are not fully and adequately secured from unauthorized access by third parties, any of our products may be tampered with or contaminated. If our products were exposed to any tampering or contamination, or if they are not transported in accordance with the required specifications, our patients may be harmed through use of our products, and such harm may be severe. In addition, if the supply chain is not secure (or our distributors do not exercise control over our products while in their possession), we are also at risk for our products to be diverted to patients other than those who are the intended recipient or to patients who do not have a prescription to receive our therapies (or it may be used for treatment by physicians who have not completed the necessary REMs protocols in order to treat patients) or it may be sold by distributors, channels or other entities that are not authorized by Alexion to sell our products. In addition, an unauthorized distributor may not properly store or ship our products, thereby exposing patients to potential harm from use of the product that was not handled in accordance with our standards. In any of the foregoing were to happen, we could be subject to costly litigation, significant monetary penalties, harm to our reputation and investigation by regulatory authorities (and potentially subject to regulatory action, including recall, product withdrawals, suspensions and monetary penalties).
The sale and use of counterfeit versions of our products could result in significant harm to patients, reduced sales of our products and harm to our reputation.
We are aware that counterfeit versions of our products have been sold by entities that are not affiliated with Alexion using product packaging suggesting that the product was manufactured by Alexion. If unauthorized third parties illegally distribute and sell
counterfeit versions of our products, those products may not meet our very stringent product specifications (or the manufacturing, handling and distribution requirements for our products) and any patient that takes any counterfeit product may suffer serious adverse health consequences, including death. Our reputation and business could suffer harm as a result of counterfeit drugs sold under our brand name and could result in lost sales for us and decreased revenues.
If we are unable to establish and maintain effective sales, marketing and distribution capabilities or to enter into agreements with third parties to do so, we may be unable to successfully commercialize our products.
We currently market and sell our products in the U.S., the EU Japan and several other territories through a direct sales force. Most of our products are relatively new to the market (ULTOMIRIS for the treatment of PNH was approved by the FDA in December 2018, for example), and we have recently hired several senior members of our sales and commercial team. In addition, in order to gain greater efficiencies in our operations, we have begun to implement a plan pursuant to which certain portions of our international commercial operations will transition to a new operating model in which sales and marketing efforts in the designated countries will rely to a greater extent on third-parties to promote and sell our products, and our direct sales presence will decrease in these regions.
Due to the fact that many of our products are new to the market, we do not have significant experience in marketing and selling these productions to patients, healthcare providers and payers (for example, we are new to certain therapy areas, such as neurology (gMG), and our sales force has had very limited exposure in educating and targeting sales to patients and physicians in neurology practices). This challenge is coupled with the fact that many of our sales and marketing team are new to Alexion and we are transitioning to third parties to market and sell our product in certain countries. If we are unable to successfully market and sell our new products and to successfully sell our products in new therapy areas, as well as successfully implement the transition to third parties to distribute and market our products in certain countries, our business and sales may be harmed. One of our objectives is to expand our business and sales in the future. If we are unable to establish and/or expand our capabilities to sell, market and distribute our products in those jurisdictions where we will continue to rely on our direct sales force and, at the same time, effectively transition from a direct sales force model (or maintain such distributor capabilities in countries where we have already commenced commercial sales), we may not be able to successfully sell our products. In that event, we may not be able to maintain or increase revenues and achieve our goal of expanding our business. We cannot guarantee that we will be able to establish and maintain our own
capabilities or enter into and maintain any marketing or distribution agreements with third-party providers on acceptable terms, if at all, or that we will be able to manage the transition to distributors in the relevant jurisdictions that will not cause any interruption or disruption in our business and sales of our products.
Even if we hire the qualified sales and marketing personnel necessary to support our objectives, or enter into marketing and distribution agreements with third parties on acceptable terms, we may not hire such employees or enter into such agreements in an efficient manner or on a timely basis. We may not be able to forecast accurately the size and experience of the sales and marketing force and the scale of distribution capabilities necessary to successfully market and sell our products. Establishing and maintaining sales, marketing and distribution capabilities are competitive, expensive and time-consuming. In addition, as we launch new products, such as ULTOMIRIS for the treatment of PNH, and we move into new therapeutic areas (such as neurology), and, if and when, the products we acquire in connection with acquisitions and development agreements with third parties move closer to regulatory approval, we may have a larger product portfolio and address more therapeutic areas and the foregoing risks may continue to apply and may even increase. Our expenses associated with building up and maintaining the sales force and distribution capabilities around the world, and in transitioning from direct sales to third party marketers and distributors, may be disproportionate compared to the revenues we may be able to generate on sales or any savings or efficiencies we gain through use of such third-parties. We cannot guarantee that we will be successful in commercializing any of our products for the above referenced or other reasons.
Completion of proof of concept trials, preclinical studies or clinical trials does not guarantee advancement to the next phase of development or regulatory approval or successful commercialization.
Completion of preclinical studies or clinical trials does not guarantee that we will initiate additional studies or trials for our product candidates, if further studies or trials are initiated, what the scope and phase of the trial will be or that they will be completed, or if these further studies or trials are completed, that the design or results may provide a sufficient basis to apply for or receive regulatory approvals or to commercialize products. Results of clinical trials could be inconclusive, requiring additional or repeat trials. Data obtained from preclinical studies and clinical trials are subject to varying interpretations that could delay, limit or prevent regulatory approval. If the design or results achieved in our clinical trials are insufficient to proceed to further trials or to regulatory approval of our product candidates, we could be materially adversely affected. Failure of a clinical trial to achieve its pre-specified primary endpoint
generally increases the likelihood that additional studies or trials may be required if we determine to continue development of the product candidate, reduces the likelihood of timely development of and regulatory approval to market the product candidate, and may decrease the chances for successfully achieving the primary endpoint in scientifically similar indications.
We are currently planning and conducting several clinical trials of products and product candidates that we anticipate may be important to our goal of expanding our business and diversifying our product portfolio. These trials may not yield the anticipated results for a number of reasons. For example, the fact that we have obtained marketing authorization in the U.S. for ULTOMIRIS as a treatment for PNH does not mean that ULTOMIRIS will be approved as a treatment for aHUS, gMG and NMOSD or that any clinical trials may achieve its designated endpoints and prove to be safe and effective for use in patients with these indications. In addition, we are also conducting clinical trials in therapeutic areas with which we have limited experience (for example, in 2018 we acquired ALXN1840 (WTX101), a therapy for Wilson’s disease acquired from Wilson Therapeutics and are currently in Phase III clinical trials) and with technology platforms with which we also have limited experience (for example, in 2018 we acquired Syntimmune that develops humanized monoclonal antibody that inhibits the interaction of FcRn with Immunoglobulin G (IgG) and IgG immune complexes). Each of these clinical trials is subject to the risks highlighted in the preceding paragraph and the investments we have made in these technologies may not generate the expected returns if the clinical trials do not produce results that will meet the requirements of regulators and the needs of patients and their healthcare providers.
In addition, Kanumawe intend to further increase the number of products in our preclinical and early-stage clinical pipeline and the number of indications that our products address. For example, in 2019 we plan to initiate proof of concept clinical trials for ULTOMIRIS as a treatment for Amyotrophic Lateral Sclerosis (ALS) and Primary Progressive Multiple Sclerosis (PPMS). There is no guarantee that any proof of concept trial will provide sufficient evidence to advance our research beyond the proof of concept stage, and we may expend significant resources in an effort to establish proof of concept that ULTOMIRIS is a transgenic product. It is producedpotential therapy for ALS or PPMS or that any other product in development will meet the standard for proof of concept for other indications. In the event that a product does satisfactorily establish proof of concept, and it does advance into preclinical or clinical trials, such product may face the risks and challenges identified in the egg whitespreceding paragraph.
Our clinical studies may be costly and lengthy, and there are many reasons why drug testing could be delayed or terminated.
For human trials, patients must be recruited and each product candidate must be tested at various doses and formulations for each clinical indication. In addition, to ensure safety and effectiveness, the effects of genetically modified chickens who receive copiesdrugs often must be studied over a long period of time, especially for the chronic diseases that we are studying. Many of our programs focus on diseases with small patient populations making patient enrollment difficult. Insufficient patient enrollment in our clinical trials could delay or cause us to abandon a product development program. We may decide to abandon development of a product candidate or a study at any time due to unfavorable results or other reasons, including if there are concerns about patient safety. We may have to spend considerable resources repeating clinical trials or conducting additional trials, either of which may increase costs and delay revenue from those product candidates, if any. We may open clinical sites and enroll patients in countries where or for indications in which we have little experience.
We rely on a small number of clinical research organizations to carry out our clinical trial related activities, and one contract research organization (CRO) is responsible for many of our studies. We rely on such parties to accurately report their results. Our reliance on CROs may impact our ability to control the timing, conduct, expense and quality of our clinical trials. In addition, we may be responsible for any errors in clinical trials by a CRO as a result of the human lysosomal acid lipase geneperformance of services in connection with a clinical trial on our behalf. And regulatory agencies, in connection with a potential product or approval or as part of on-going monitoring, will review a CROs compliance with regulatory requirements relating to produce recombinant human lysosomal acid lipase. The facilitiesclinical trials and we may be subject to findings and regulatory action (including denial or delay of product approval) if a CRO fails to comply with regulations.
Additional factors that can cause delay, impairment or termination of our clinical trials or our product development efforts include:
delay or failure in obtaining institutional review board (IRB) approval or the approval of other reviewing entities to conduct a clinical trial at each site;
delay or failure in reaching agreement on acceptable terms with prospective CROs, and clinical trial sites, the terms of which we relycan be subject to produce raw materialextensive negotiation and may vary significantly among different CROs and trial sites;
withdrawal of clinical trial sites from our clinical trials as a result of changing standards of care or the ineligibility of a site to participate in our clinical trials;
clinical sites and investigators deviating from trial protocol, failing to conduct the trial in accordance with regulatory requirements, or dropping out of a trial;
delay or failure in having patients complete a trial or return for recombinant lysosomal acid lipase arepost-treatment follow-up;
long treatment time required to demonstrate effectiveness;
lack of sufficient supplies of the only animalproduct candidate;
disruption of operations at the clinical trial sites;
adverse medical events or side effects in treated patients;
failure of patients taking the placebo to continue to participate in our clinical trials;
insufficient clinical trial data to support safety and effectiveness of the product candidates;
lack of effectiveness or safety of the product candidate being tested;
inability to meet required specifications or to manufacture sufficient quantities of the product candidate for development or commercialization activities in a timely and cost-efficient manner;
decisions by regulatory authorities, the IRB, ethics committee, or us, or recommendation by a data safety monitoring board, to suspend or terminate clinical trials at any time for safety issues or for any other reason;
failure to obtain the necessary regulatory approvals for the product candidate or the approvals for the facilities in which such product candidate is manufactured; and
decisions by competent authorities, IRBs or ethics committees to demand variations in protocols or conduct of clinical trials.
We may not accurately forecast demand for our products, including our new products, or the worldconversion of PNH patients to ULTOMIRIS, which may cause our operating results to fluctuate,
Our quarterly revenues, expenses and net income (loss) may fluctuate, even significantly, due to certain risks, including those described in these “Risk Factors” as well as the timing of charges and expenses that produceswe may take and acquisitions (such as the Wilson Therapeutics and Syntimmune acquisitions). In the future, we may not generate sufficient revenues or control expenses to achieve our financial goals, including continued profitability. We may not be able to sustain or increase profitability on a quarterly or annual basis. You should not consider our financial performance, including our revenue growth, in recent periods as indicative of our future performance. Since
we have a limited sales and operating history with certain of our products (such as ULTOMIRIS as a treatment for PNH in the US) and for new indications of existing products (such as SOLIRIS as a treatment for gMG), we may not be able to accurately forecast demand for our products. STRENSIQ and KANUMA, also relatively new products, each received marketing approval in 2015, and both products treat rare diseases for which there was no existing therapy in a new therapeutic area. We have recently filed for regulatory approval for SOLIRIS as a treatment for NMOSD. Since approval of ULTOMIRIS as a treatment for PNH in the U.S. in December 2018, we have undertaken efforts to facilitate the conversion of PNH patients in the U.S. from SOLIRIS to ULTOMIRIS. Product demand and, in the case of conversion to ULTOMIRIS, product preference and conversion, is dependent on a number of factors, many of which are beyond our control. For these reasons, we may not be able to accurately forecast demand for our products.
We cannot guarantee that we will achieve our financial goals, including our ability to maintain profitability on a quarterly or annual basis in the future.
Our investors and investment analysts may have widely varying expectations that may be materially higher or lower than actual revenues and profits and if our revenues and profits are different from these expectations, our stock price may experience significant volatility. Our revenues and profits are also subject to foreign exchange rate fluctuations due to the global nature of our operations and our results of operations could be adversely affected due to unfavorable foreign exchange rates. Although we use derivative instruments to manage foreign currency risk, our efforts to reduce currency exchange losses may not be successful.
In addition, we have in the past provided, and expect to continue to provide, financial guidance for future periods and if our actual operating results fail to meet or exceed the guidance that we have previously provided to our investors, our stock price could drop suddenly and significantly.
As we attempt to expand our pipeline, obtain regulatory approval for new products, facilitate the conversion of PNH patients from SOLIRIS to ULTOMIRIS in the U.S., seek regulatory approval for existing products in new jurisdictions and approval of new indications for existing products (such as SOLIRIS as a treatment for NMOSD), we may have substantial expenses as we continue our research and development efforts, continue to conduct clinical trials and continue to develop and expand manufacturing, sales, marketing and distribution capabilities worldwide, some of which could be delayed, scaled-back or eliminated to achieve our financial objectives. These expenses may increase and such increases may exceed analyst and investor expectations.
If we fail to achieve the expected financial and operating benefits of our corporate restructurings, our business and financial results may be harmed.
We have undertaken corporate restructuring activities to re-align our global organization with our re-focused strategy, reduce costs, and realize operational efficiencies. We estimate that our most recent restructuring, which includes our transition in certain jurisdictions from a direct sales model to increased use of third parties, will result in a charge of up to $25.0 in 2019. These recent restructuring activities, including work force reductions, closing certain operational sites and our increased use of third parties in certain countries to market and distribute products (and rely less on a direct sales force), subject us to many risks, including loss of business continuity, unanticipated costs, and higher than usual employee turnover. In addition, we will not exercise the same degree of control over any third parties that we do over our direct sales force and the ability to direct the third party or provide incentives for such third party to sell our products may not be as strong as in the case of a direct sales force. The expected cost savings and operational efficiencies from the restructuring activities are based on assumptions and expectations that we believe were reasonable in our judgment at the time made but may not be achieved due to unforeseen difficulties and challenges that are beyond our control. If these assumptions and expectations are incorrect or if we experience delays or unforeseen events in realizing the benefits of the restructuring activities, our business operations and financial results may be harmed.
As we implement any restructurings, we must execute on our re-focused strategy, including growing and maximizing our rare disease business and pursuing disciplined business development to expand our pipeline. If we are unable to effectively execute with fewer human resources and/or attract, retain or motivate key employees, our business may be adversely affected.
If we fail to attract and retain highly qualified personnel, we may not be able to successfully develop, manufacture or commercialize our products or products candidates.
The success of our business is dependent in large part on our continued ability to attract and retain our senior management, and other highly qualified personnel in our scientific, clinical, manufacturing and commercial organizations. There is intense competition in the biopharmaceutical industry for these types of personnel. In March 2017, our Board appointed a new Chief Executive Officer (CEO) and we have experienced other recent significant management changes. In addition, since 2017, we have moved our global headquarters and undertaken company-wide restructurings with the goal of re-aligning our global organization with our re-focused strategy and to make our international operations more efficient and effective.
The relocation of our headquarters and restructurings have the potential to adversely impact our ability to recruit and/or retain key employees as well as to disrupt our business operations, financial conditions, programs, plans and strategies.
Our business is specialized and global and we must attract and retain highly qualified individuals across many geographies. We may not be able to continue to attract and retain the highly qualified personnel necessary to develop, manufacture and commercialize our products and product candidates. If we are unsuccessful in our recruitment and retention efforts, or if our recruitment efforts take longer than anticipated, our business may be harmed.
If we fail to satisfy our debt service obligations or obtain the capital necessary to fund our operations, we may be unable to commercialize our products or continue or complete our product development.
In June 2018, we amended and restated our credit facility to, among other things, increase the amount available under the revolving credit facility from $500.0 to $1,000.0 and extend the maturity date of the revolving credit facility and the term loan facility to June 7, 2023. As a result, we have significant debt service obligations. In addition to the obligations to make interest and principal payments under the facility throughout the term of the loans, any changes in interest rates related to this debt could significantly increase our annual interest expense and any hedging of this interest may not be effective to control expenses.
In addition, we have substantial contingent liabilities, including milestone and royalty obligations under acquisitions and strategic transactions, and we have been, and in the future may again be, engaged in disputes with certain counterparties regarding potential milestone and royalty obligations. Our increased indebtedness, including increased interest expense, together with our significant contingent liabilities, could, among other things:
•make us more vulnerable to economic or industry downturns and competitive pressures;
•make it difficult for us to make payments on our credit facilities and require us to use cash flow from operations to satisfy our debt obligations, which may reduce the availability of our cash flow for other purposes, including business development efforts, research and development and mergers and acquisitions;
•limit our ability to incur additional debt or access the capital markets; and
•limit our flexibility in planning for, or reacting to changes in, our business.
The Amended and Restated Credit Agreement requires us to comply with certain financial covenants and negative covenants, restricting or limiting our ability
and the ability of our subsidiaries to, among other things, incur additional indebtedness, grant liens, and engage in certain investment, acquisition and disposition transactions, subject to limited exceptions. If an event of default occurs, the interest rate may increase and the administrative agent may be entitled to take various actions, including the acceleration of amounts due under the Amended and Restated Credit Agreement. If the interest rate imposed under our Amended and Restated Credit Agreement were to increase as a result of a default, our expenses may increase and we may need to allocate additional funds to this interest expense (which may limit the use of these funds for other purposes, including growing our business or responding to changes in our business and industry). If some or all of the amounts outstanding under the Amended and Restated Credit Agreement were to be accelerated by the lenders, we may not have sufficient cash on hand to pay the amounts due, we may not be able to refinance such debt on terms acceptable to us (or at all) and we may be required to sell certain assets on terms that are unfavorable to us.
Our ability to satisfy our obligations under the Amended and Restated Credit Agreement and meet our debt service obligations and our royalty and milestone obligations will depend upon our future performance, which will be subject to financial, business and other factors affecting our operations, many of which are beyond our control.
We may not be able to access the capital and credit markets on terms that are favorable to us or at all.
We may need to raise additional capital to supplement our existing funds and cash generated from operations for working capital, capital expenditure and debt service requirements, and other business activities. Funding needs may shift and the amount of capital we may need depends on many factors, including, the cost of any acquisition or any new collaborative, licensing or other commercial relationships that we may establish, the time and cost necessary to build our manufacturing facilities or enhance our manufacturing operations, amounts we may need to pay in connection with the resolution of any government investigation or litigation matter (including any securities class action matter or any product liability claim), the cost of obtaining and maintaining the necessary egg whites from transgenic chickens. Natural disasters, disease,regulatory approvals for our manufacturing facilities, and the progress, timing and scope of our preclinical studies, clinical trials and product development and commercialization efforts. The capital and credit markets have experienced and may continue to experience extreme volatility and disruption. We may not receive additional funding when we need it or funding may only be available on unfavorable terms. If we cannot raise adequate funds to satisfy our working capital, capital requirements and debt repayment obligations (or royalty and milestone obligations), we may have to delay, scale-back or
eliminate certain research, development, manufacturing, acquisition or commercial activities or sell certain assets and technologies.
Our business involves environmental risks and potential exposure to environmental liabilities.
As a biopharmaceutical company, our business involves the use of certain hazardous materials in our research, development, manufacturing and other activities. We and our third party providers are subject to various federal, state and local and foreign environmental laws and regulations concerning the handling and disposal of non-hazardous and hazardous wastes, such as exotic Newcastle diseasemedical and biological wastes, and emissions and discharges into the environment, such as air, soils and water sources. We also are subject to laws and regulations that impose liability and clean-up responsibility for releases of hazardous substances into the environment and a current or avian influenza,previous owner or other catastrophic eventsoperator of property may be liable for the costs of remediating its property or locations, without regard to whether the owner or operator knew of or caused the contamination. Although we believe that our safety procedures for handling and disposing of hazardous materials comply with the laws and regulations established by state, federal and foreign regulations, the risk of loss of, or accidental contamination or injury from, these materials cannot be eliminated. If an accident or environmental discharge occurs, or if we discover contamination caused by prior owners and operators of properties we acquire, we could be liable for remediation obligations, damages and fines that could exceed our insurance coverage and financial resources. Such obligations and liabilities, which to date have not been material, could have a significantmaterial impact on our business and financial condition. Additionally, the supplycost of unpurified Kanuma,compliance with environmental and safety laws and regulations may increase in the future, and we may be required to dedicate more resources, including substantial financial resources, to comply with such laws and regulations or destroy Alexion’s animal operations altogether. Ifpurchase supplemental insurance coverage, which may not be available on acceptable terms or at all.
In order to meet one of our animal operationskey business objectives of advancing and rebuilding our product pipeline, we plan to expand our business and product offerings through acquisitions of businesses and technologies. Our efforts to identify opportunities or complete transactions that satisfy our strategic criteria may not be successful, and we may not realize the anticipated benefits of any completed acquisition or other strategic transaction.
As noted above, in 2018 a substantial portion of our total revenue was derived from SOLIRIS. We expect that there may be increased competition to SOLIRIS from, among other products and therapies, biosimilars, and we are disruptedstill in the very early stages of the launch of ULTOMIRIS in the U.S. for PNH and cannot guarantee that our efforts to facilitate the conversion of patients
from SOLIRIS to ULTOMIRIS or destroyed, itto have new PNH patients prescribed ULTOMIRIS will be extremelysuccessful (or that we will obtain clearance for ULTOMIRIS for PNH in the EU, Japan and other jurisdictions). As a result, we have identified rebuilding our product pipeline as a key strategic objective and, in order to achieve this objective, we expect to purchase businesses and acquire, co-develop or license technologies and products from third parties in the future. For example, in 2018, among other transactions, we completed acquisitions of Wilson Therapeutics and Syntimmune, Inc. We anticipate that we will regularly evaluate potential merger, acquisition, partnering and in-license opportunities in an effort to expand our pipeline or product offerings, and enhance our research platforms. Acquisitions of new businesses or products and in-licensing of new technologies and products may involve numerous risks, including:
•substantial cash expenditures;
•potentially dilutive issuance of equity securities and incurrence of debt;
•assumption of material liabilities in connection with the target or purchased technology, some of which may be difficult or impossible to set up another animal facilityidentify at the time of acquisition;
•difficulties in assimilating the operations of the acquired companies;
•failure of any acquired businesses or products or in-licensed products or technologies to supplyachieve the unpurified Kanuma. This wouldscientific, medical, commercial or other results we anticipate;
•diverting our management’s attention away from other business opportunities and concerns;
•the potential loss of our key employees or key employees of the acquired companies; and
•risks of entering disease areas and indications in which we have limited or no direct experience.
A substantial portion of our strategic efforts are focused on opportunities for rare disorders, but the availability of such opportunities is limited. We may not be able to identify opportunities that satisfy our strategic criteria or are acceptable to us or our stockholders. Several companies have publicly announced intentions to establish or develop rare disease programs and we may compete with these companies for the same opportunities. For these and other reasons, we may not be able to acquire the rights to additional product candidates or approved products on terms that we or our stockholders find acceptable, or at all. In such event, we may not be able to rebuild our pipeline and any future revenue may remain largely dependent on our existing products which, as noted above, may be subject to increasing competition from biosimilars and other competitive or novel therapies.
Even if we are able to successfully identify and complete acquisitions and other strategic transactions,
we may not be able to integrate or take full advantage of them. An acquisition or other strategic transaction may not result in short-term or long-term benefits to us. We may also incorrectly judge the value or worth of an acquired company or business or an acquired or in-licensed product, particularly if the acquired technology is preclinical trials or early-stage clinical trials.
To effectively manage our current and future potential growth, we must continue to effectively enhance and develop our global employee base and our operational and financial processes. Supporting our growth strategy may require significant capital expenditures and management resources, including investments in research, development, sales and marketing, manufacturing and other areas of our operations. The development or expansion of our business, any acquired business or any acquired or in-licensed products may require a substantial capital investment by us and we may likely incur substantial expenses in advancing acquired products to commercialization. We may not have the necessary funds for these capital expenditures and expenses or they might not be available to us on acceptable terms or at all. We may also seek to raise funds by incurring additional indebtedness and selling shares of our capital stock, which could dilute current stockholders’ ownership interest in our company, or securities convertible into our capital stock, which could dilute current stockholders’ ownership interest in us upon conversion.
We may incur impairment charges in the future for certain of our assets, including goodwill in connection with acquisitions, and such amounts may be material.
If the purchase price of a business acquisition exceeds the value of the assets (and liabilities) acquired, the acquirer must recognize goodwill in such amount. We may be required to recognize impairment charges for our goodwill and other intangible assets, and such charges may be material and have an adverse impact on our financial results in the period such charges are incurred.
As of December 31, 2018, the net carrying value of our goodwill and other intangible assets, net totaled $8,678.7. As required by GAAP, we periodically assess these assets to determine if there are indicators of impairment. We have recorded charges that include inventory write-downs for failed quality specifications or recalls, impairments with respect to investments and acquisitions, fixed assets and long-lived assets, outcomes of litigation and other legal or administrative proceedings, regulatory matters and tax matters, and payments in connection with acquisitions and other business development activities, such as milestone payments. The impairment of tangible and intangible assets may be triggered by developments both within and outside our control. Deteriorating economic
conditions, technological changes, disruptions to our business, inability to effectively integrate acquired businesses, unexpected significant changes or planned changes in the use of the assets, intensified competition, divestitures, market capitalization declines and other factors may impair our goodwill and other intangible assets. Any charges relating to such impairments could adversely affect our abilityresults of operations in the periods in which an impairment is recognized. As part of our standard quarterly procedures, we reviewed the KANUMA asset as of December 31, 2018 and determined that there were no indicators of impairment. We will continue to satisfy demandreview the related valuation and accounting of this asset in future quarters as new information becomes available to us. Changes to assumptions used in our net cash flow projections may result in impairment charges in subsequent periods. The net book value of the KANUMA intangible asset as of December 31, 2018 is $3,252.6.
Our business could be adversely affected by litigation, government investigations and enforcement actions.
We operate in many jurisdictions in a highly regulated industry and we could be subject to litigation, government investigation and enforcement actions on a variety of matters in the U.S. or foreign jurisdictions, including, without limitation, intellectual property, regulatory, product liability, environmental, whistleblower, Qui Tam, false claims, privacy, anti-kickback, anti-bribery, securities, commercial, employment and other claims and legal proceedings which may arise from conducting our business. See Note 11 “Commitments and Contingencies” to the footnotes to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K for Kanuma,information on our material legal proceedings. For example, in May 2015, we received a subpoena in connection with an investigation by the Enforcement Division of the SEC requesting information related to our grant-making activities and compliance with the FCPA in various countries. In addition, in October 2015, we received a request from the DOJ for the voluntary production of documents and other information pertaining to Alexion’s compliance with FCPA. The SEC and DOJ also seek information related to Alexion’s recalls of specific lots of Soliris and related securities disclosures. Alexion is cooperating with these investigations. The investigations have focused on operations in various countries, including Brazil, Colombia, Japan, Russia and Turkey, and Alexion's compliance with the FCPA and other applicable laws. Any determination that our operations or activities are not in compliance with existing laws or regulations, by the SEC or DOJ in the above referenced matter for example, could result in the imposition of fines, civil and criminal penalties, equitable remedies, including disgorgement, injunctive relief, exclusion from the federal healthcare programs, healthcare debarment, product recalls, reputational damage and modifications of our business
practices and/or other sanctions against us, and remediation of any such findings could have an adverse effect on our business operations. Legal proceedings, government investigations, including the SEC and DOJ investigations, and enforcement actions have been and we expect may continue to be expensive and time consuming. Any future litigation or investigation may also likely be expensive and time consuming.
The efficiency of our corporate structure depends on the application of the tax laws and regulations in the countries where we operate and we may have exposure to additional tax liabilities or our effective tax rate could increase, which could have a material impact on our results of operations and financial position.
As a company with international operations, we are subject to income taxes, as well as non-income based taxes, in both the U.S. and various foreign jurisdictions. Significant judgment is required in determining our worldwide tax liabilities. Although we believe our estimates are reasonable at the time made, the final taxes we owe may differ from the amounts recorded in our financial statements (and such differences may be material). If the IRS, or other taxing authority, disagrees with the positions we take, we could have additional tax liability, and this could have a material impact on our results of operations and financial position. Our effective tax rate could be adversely affected by changes in the mix of earnings in countries with different statutory tax rates, changes in the valuation of deferred tax assets and liabilities, changes in tax laws and regulations, changes in interpretations of tax laws, including pending tax law changes, changes in our manufacturing activities and changes in our future levels of research and development spending.
We have designed, and from time to time we modify, our corporate structure, the manner in which we develop and use our intellectual property, and our intercompany transactions between our affiliates in a way that is intended to enhance our operational and financial efficiency and increase our overall profitability. The application of the tax laws and regulations of various countries in which we operate and to our global operations is subject to interpretation. We also must operate our business in a manner consistent with our corporate structure to realize such efficiencies. The tax authorities of the countries in which we operate may challenge our methodologies for valuing developed technology or for transfer pricing or other operations. If tax authorities determine that the manner in which we operate results in our business not achieving the intended tax consequences, our effective tax rate could increase (and such increase may be material) and harm our financial position and results of operations. In addition, certain governments are considering and may adopt tax reform measures that significantly increase our worldwide tax liabilities. The Organization for Economic Co-operation and Development and other
government bodies have focused on issues related to the taxation of multinational corporations, including, in the area of “base erosion and profit shifting,” where payments are made from affiliates in jurisdictions with high tax rates to affiliates in jurisdictions with lower tax rates. It is possible that these reform measures could increase our effective tax rate (and such increase may be material) and harm our financial position and results of operations over the next several years.
Our sales and operations are subject to a variety of risks relating to the conduct of our international business.
We have increased our international presence, including in emerging markets. Our operations in foreign countries subject us to a variety of risks, including:
difficulties or the inability to obtain necessary foreign regulatory or reimbursement approvals of our products in a timely manner or at all;
political or economic determinations that adversely impact pricing or reimbursement policies;
economic problems or political instability;
fluctuations in currency exchange rates;
difficulties or inability to obtain financing in markets;
unexpected changes in tariffs, trade barriers and regulatory requirements;
customs and tax officials in foreign jurisdictions may disagree with the value we set when we or others import our products (including products that are donated for charitable purposes) and we may be required to pay additional duties or fines and such amounts may be substantial;
difficulties in establishing and enforcing contractual and intellectual property rights;
compliance with complex import and export control laws;
trade restrictions and restrictions on direct investments by foreign entities;
compliance with tax, employment and labor laws;
costs and difficulties in recruiting and retaining qualified managers and employees to manage and operate the business in local jurisdictions;
costs and difficulties in managing and monitoring international operations; and
longer payment cycles.
Additionally, our business and marketing methods are subject to the laws and regulations of the countries in which we operate, which may differ significantly from country to country and may conflict with U.S. laws and regulations. The FCPA and anti-bribery laws and regulations in the locations in which we operate our business are extensive and far-reaching, and we must maintain accurate records and control over the activities
of our distributors and third party service providers in countries where we operate. We have policies and procedures, and we are currently implementing an enhanced company-wide compliance program and effort, designed to help ensure that we and our representatives, including our employees and our vendors and distributors, comply with such laws, however we cannot guarantee that these policies and procedures will protect us against liability under the FCPA or other anti-bribery laws for actions taken by us or our representatives. Any determination that our operations or activities are not in compliance with existing laws or regulations, including the FCPA and the UK Anti-Bribery Act, could result in the imposition of fines, civil and criminal penalties, equitable remedies, including disgorgement, injunctive relief, and/or other sanctions against us, and remediation of such findings could have a material and adverse effect on our business operations. In addition, as our international operations expand, we are likely to become subject to new anti-corruption/anti-bribery laws or existing laws may govern our activities in new jurisdictions in which we operate. In addition, as we move from a direct sales force to third-party distributors and marketers in certain countries and regions, we may also have liability under the FCPA and anti-bribery laws and regulations for their actions. Although we can impose contractual restrictions on what they are authorized to do on our behalf, we will exercise only limited control over the actions of these third parties but may still face the same liabilities for their actions. Our failure, and the failure of others who we engage to act on our behalf, to comply, with the laws and regulations of the countries in which we operate, or will operate in the future, could materially harm our business.
Currency fluctuations and changes in exchange rates could adversely affect our revenue, increase our costs and negatively affect our profitability.
We conduct a substantial portion of our business in currencies other than the U.S. dollar. We are exposed to fluctuations in foreign currency exchange rates and such fluctuations affect our operating results.
Any adverse developments affecting our manufacturing operations or the operations The exposures result from portions of our third-party providers could resultrevenues, as well as the related receivables, and expenses that are denominated in currencies other than the U.S. dollar, including the Euro, Japanese Yen, British Pound, Canadian dollar and Turkish Lira. As the U.S. dollar strengthens against these foreign currencies, the relative value of sales made in the respective foreign currencies decrease. When the U.S. dollar weakens against these currencies, the relative value of such sales increase. We manage a product shortage of clinical or commercial requirements, withdrawalportion of our product candidates or any approved products, shipment delays, lot failures, or recalls.foreign currency transaction risk within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes. We mayenter into foreign exchange forward contracts to hedge exposures resulting from portions of our forecasted
revenues, including intercompany revenues that are denominated in currencies other than the U.S. dollar. The purpose of the revenue hedges is to reduce the volatility of exchange rate fluctuations on our operating results and to increase the visibility of the foreign exchange impact on forecasted revenues. Further, we enter into foreign exchange forward contracts, with durations of approximately 30 days, designed to limit the balance sheet exposure of monetary assets and liabilities. We enter into these hedges to reduce the impact of fluctuating exchange rates on our operating results. Gains and losses on these hedge transactions are designed to offset gains and losses on underlying balance sheet exposures. While we attempt to hedge certain currency risks, currency fluctuations between the U.S. dollar and the currencies in which we do business have, in the past, caused foreign currency transaction gains and losses and have also have to write-off inventory and incur other chargesimpacted the amounts of revenues and expenses for productscalculated in U.S. dollars and will do so in the future. Likewise, past currency fluctuations have at times resulted in foreign currency transaction gains, and there can be no assurance that fail to meet specifications, undertake costly remediation efforts or seek more costly manufacturingthese gains can be reproduced. Any significant foreign currency exchange rate fluctuations could adversely affect our financial condition and results of operations.
alternatives. Such manufacturing issues could increase our cost of goods, cause usRisks Related to lose revenue, reduce our profitability or damage our reputation.the Regulatory Environment
We operate in a highly regulated industry and if we or our third party providers fail to comply with U.S. and foreign regulations, we or our third party providers could lose our approvals to market our products or our product candidates, and our business wouldmay be seriously harmed.
We and our current and future partners,third party vendors, contract manufacturers, CROs, distributors and suppliers and logistic providers are subject to rigorous and extensive regulation by governmental authorities around the world, including the FDA, EMA, the competent authorities of the EU member states,Member States and the MHLW. These regulations, many of which are complex, relate to almost all aspects of our business, including GCP, GLP, cGMP and pharmacovigilance rules (for additional information on the regulations relating to our business, see “Business - Government Regulation” in Item 1 above in this Annual Report on Form 10-K). If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured (such as product contamination), or in the case of Kanuma,KANUMA, problems with animal operations, a regulatory agency may impose restrictions on that product, the manufacturing facility or us. For example, in March 2013, weWe have received a Warning Letter from the FDA relating to compliance with FDA’s cGMP requirements at ARIMF. We are working with the FDA to resolve the issues identified in the Warning Letter. Failureone of our facilities, which was remediated. If we had failed to address the FDA’s concerns may leador if we (or one of our third party contract manufacturers) were to receive
another Warning Letter in the future relating to cGMP or other applicable regulations, the FDA or other regulatory authorities tocould take regulatory action, including fines, civil penalties, recalls, seizure of product, suspension of manufacturing operations, operating restrictions, injunctions, suspension of clinical trials, withdrawal of FDA approval and/or criminal prosecution.
If we do not resolve outstanding concerns expressed by the FDA in the Warning Letter and the Form 483s to the satisfaction of the FDA, EMA or any other regulatory agency, or we or our third-party providers, including our product fill-finish providers, packagers and labelers, fail to comply fully with applicable regulations, then we may be required to initiate a recall or withdrawal of our products. LikeIn addition to our manufacturing operations and those of contract manufacturers’ manufacturing operations being subject to inspection and potential regulatory action for failure to comply with (among other regulations) cGMP, our animal operations willmay also be subject to FDA and U.S. Department of Agriculture, Animal and Plant Health Inspection Service (USDA APHIS) inspection to evaluate whether our animal husbandry, containment, personnel, and record keeping practices are sufficient to ensure safety and security of our transgenic chickens and animal products (e.g.(e.g., eggs, waste, etc.). Our animal operations may also be subject to inspection by the U.S. Department of Agriculture, Animal and Plant Health Inspection Service (USDA APHIS), the agency responsible for administering the Animal Welfare Act. Any failure to ensure safety and security of our transgenic chickens and/or animal products could result in regulatory action by the FDA or another regulatory body, including USDA APHIS.
TheFailure to comply with the laws and requirements, including statutes and regulations, administered by the FDA, the EC, the competent authorities of the EU Member States, the MHLW or other agencies, could result in:
a product recall;
a product withdrawal;
significant administrative and judicial sanctions, including, warning letters or untitled letters;
significant fines and other civil penalties;
suspension, variation or withdrawal of a previously granted approval for our products;
interruption of production;
operating restrictions, such as a shutdown of production facilities or production lines, or new manufacturing requirements;
suspension or termination of ongoing clinical trials;
delays in approving or refusal to approve our products including pending BLAs or BLA supplements for our products or a facility that manufactures our products;
seizing or detaining product;
requiring us or our partners to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance;
injunctions; and/or
criminal prosecution.
In addition, we are subject to antitrust regulations with respect to our acquisitions, as well as our interactions with other participants in the markets we serve. In addition, these antitrust laws are vigorously enforced in the U.S. and in other jurisdictions in which we operate.
Our product candidates require extensive clinical testing and regulatory approval and failure of to satisfy regulatory requirements to meet the appropriate safety and efficacy thresholds may prevent us from being able to market our products and limit our ability to grow our business and diversify our revenue.
We believe our future success may depend on our ability to develop and commercialize our product candidates and, to this end, we have recently acquired companies and technologies in an effort to expand our product pipeline. Our product candidates are in various stages of development and must satisfy the rigid safety and efficacy requirements of the FDA and other foreign regulatory agencies before they can be approved for sale to patients. To satisfy these standards, we must ensure, among other things, that we have appropriately established our protocol designs, obtained the necessary IRB approval, provide adequate patient enrollment rates, timely and appropriately report any adverse events and serious adverse events to the appropriate authorities and ensure compliance with cGCP. If we or our third-party clinical trial providers or third-party CROs do not successfully carry out these clinical activities, our clinical trials or the potential regulatory approval of a product candidate may be delayed or be unsuccessful.
If we discover safety or safety reporting issues with any of our approved products, or if we fail to comply with continuing U.S. and applicable foreign regulations, our revenue may decrease, an approved product could lose its marketing approval or sales could be suspended and our business could be materially harmed.
Following marketing approval of a pharmaceutical product, the safety profile of anysuch product continues to be closely monitored by the FDA and other foreign regulatory authorities after approval.authorities. Regulations continue to apply after product approval, and cover, among other things, testing, manufacturing, quality control, finishing, filling, labeling, advertising, promotion, risk mitigation, adverse event reporting requirements and export of biologics. For example, the risk managementREMS program established in 2007 upon the FDA’s approval of Soliris for the treatment of PNH was replaced with a Risk Evaluation and Mitigation Strategy (REMS) program, approvedSOLIRIS, most recently updated by the FDA in 2010, and further revised in December 2015, concerningrequires prescribing information regarding the level of fever needed to seek medical attention and reporting adverse events. Future changes to the SolirisSOLIRIS REMS (or similar requirements for other products) could be costly and burdensome to implement.
We are required to report any serious and unexpected adverse experiences and certain quality
problems with our products to the FDA, the EMA, the MHLW and other health agencies. We or any health agencyAdverse safety events involving our products may have to notify healthcare providersa negative impact on our business. Discovery of any such developments. Non-compliancesafety issues with safety reporting requirementsour products could result in product liability claims and could cause additional regulatory action that may include civil actionscrutiny and requirements for additional labeling or safety monitoring, withdrawal of products from the market and the imposition of fines or criminal penalties. In addition, governmental authorities are making greater amounts of safety information directly available to the public through periodic safety update reports, patient registries and other reporting requirements. The reporting of adverse safety events may also damage physician, patient and/or investor confidence in our products and our reputation. Any adverse events in connection with the use of our products could result in liabilities, loss of revenues, material write-offs of inventory, material impairments of intangible assets, goodwill and fixed assets, material restructuring charges and other adverse impacts on our results of operations.
Regulatory agencies periodically inspect our pharmacovigilance processes, including our adverse event reporting.processes. If these regulatory agencies determine that we or other parties whom we do not control that perform services on our behalf, including clinical trial investigators, have not complied with the applicable reporting or other pharmacovigilance requirements, we may become subject to additional inspections, warning letters or other enforcement actions, including monetary fines, marketing authorization withdrawal and other penalties.
As a condition of approval for marketing our products, governmental authorities may require us to conduct additional studies. In connection with the approval of SolirisSOLIRIS in the U.S., EU and Japan, for the treatment of PNH, we agreed to establish a PNH Registry, monitor immunogenicity, monitor compliance with vaccination requirements, and determine the effects of anticoagulant withdrawal among PNH patients receiving eculizumab, and, specifically in Japan, we agreed to conduct a trial in a limited number of Japanese PNH patients to evaluate the safety of a meningococcal vaccine. In connection with the approval of SolirisSOLIRIS in the U.S. for the treatment of aHUS, we agreed to establish an aHUS Registry and complete additional human clinical studies in adult and pediatric patients. Furthermore, in connection with the approval of StrensiqSTRENSIQ in the U.S., we agreed to conduct a prospective observational study in treated patients to assess the long-term safety of StrensiqSTRENSIQ therapy and to develop complementary assays. Similarly, in connection with the approval of KanumaKANUMA in the U.S., we have agreed to conduct a long-term observational study of treated patients, either as a standalone study or as a component of the existing LAL Registry. In the EU, in connection with the grant of authorization for Strensiq,STRENSIQ, we agreed to conduct a multicenter, randomized, open-label, Phase 2a study of StrensiqSTRENSIQ in patients with HPP
and to extend the studies ENB-008-10 and ENB-009-10 to provide efficacy data in patients 13 to 18 years of age. We also agreed to set up an observational, longitudinal, prospective, long-term registry of patients with HPP to collect information on the epidemiology of the disease, including clinical outcomes and quality of life, and to evaluate safety and effectiveness data in patients treated with Strensiq. age, which we have commenced.
In the U.S., the FDA can also propose to withdraw
approval for a product if it determines that such additional studies are inadequate or if new clinical data or information shows that a product is not safe for use in an approved indication.
Failure to comply with the lawsIn addition, similar or more stringent post-approval requirements and requirements, including statutes and regulations, administeredobligations may be imposed by the FDA the EC, the competent authorities of the EU member states, the MHLW and/or other regulatory agencies including without limitation, failures or delays in resolving the concerns raised by the FDA in the Warning Letter, could result in:
a product recall;
a product withdrawal;
significant administrative and judicial sanctions, including, warning letters or untitled letters;
significant fines and other civil penalties;
suspension, variation or withdrawal of a previously granted approval for Soliris;
interruption of production;
operating restrictions, such as a shutdown of production facilities or production lines, or new manufacturing requirements;
suspension of ongoing clinical trials;
delays in approving or refusal to approve our products including pending BLAs or BLA supplements for our products or a facility that manufactures our products;
seizing or detaining product;
requiring us or our partners to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance;
injunctions; and/or
criminal prosecution.
If the use of our products harms people, or is perceived to harm patients even when such harm is unrelatedwith respect to our future products our regulatory approvals could be revoked(such as ULTOMIRIS or otherwise negatively impacted and we could be subject to costly and damaging product liability claims.
The testing, manufacturing, marketing and sale of drugs for use in humans exposes us to product liability risks. Side effects and other problems from using our products could (1) lessen the frequency with which physicians decide to prescribe our products, (2) encourage physicians to stop prescribing our products to their patients who previously had been prescribed our products, (3) cause serious adverse events and give rise to product liability claims against us, and (4) result in our need to withdraw or recall our products from the marketplace. Some of these risks are unknown at this time.
Our products and our product candidates treat patients with ultra-rare diseases. We generally test our products in only a small number of patients. For example, the FDA marketing approvalSOLIRIS for the treatment of patients with aHUS was based on two prospective studies in a total of 37 adult and adolescent patients, together with a retrospective study that included 19 pediatric patients. As more patients use our products, including more children and adolescents, new risks and side effects may be discovered, the rate of known risks or side effects may increase, and risks previously viewed as less significant could be determined to be significant. Previously unknown risks and adverse effects may also be discovered in connection with unapproved uses of our products, which may include administration of our products under acute emergency conditions, such as the Enterohemorrhagic E. coli health crisis in Europe, primarily Germany, which began in May 2011. We do not promote, or in any way support or encourage the promotion of our products for unapproved uses in violation of applicable law, but physicians are permitted to use products for unapproved purposes and we are aware of such uses of Soliris. In addition, we are studying and expect to continue to study Soliris in diseases other than PNH and aHUS in controlled clinical settings, and independent investigators are doing so as well. In the event of any new risks or adverse effects discovered as new patients are treated for approved indications, or as our products are studied in or used by patients for other indications, regulatory authorities may delay or revoke their approvals, we may be required to conduct additional clinical trials and safety studies, make changes in labeling, reformulate our products or make changes and obtain new approvals for our and our suppliers’ manufacturing facilities. We may also experience a significant drop in potential sales, experience harm to our reputation and the reputation of our products in the marketplace or become subject to lawsuits, including class actions. Any of these results could decrease or prevent any sales or substantially increase the costs and expenses of commercializing and marketing our products.
We may be sued by people who use our products, whether as a prescribed therapy, during a clinical trial, during an investigator initiated study, or otherwise. Many patients who use our products are already very ill. Any informed consents or waivers obtained from people who enroll in our trials or use our products may not protect us from liability or litigation. Our product liability insurance may not cover all potential types of liabilities or may not cover certain liabilities completely. Moreover, we may not be able to maintain our insurance on acceptable terms. In addition, negative publicity relating to the use of our products or a product candidate, or to a product liability claim, may make it more difficult, or impossible, for us to market and sell. As a result of these factors, a product liability claim, evenNMOSD, if successfully defended, could have a material adverse effect on our business, financial condition or results of operations.
Patients who use our products already often have severe and advanced stages of disease and known as well as unknown significant pre-existing and potentially life-threatening health risks. During the course of treatment, patients may suffer adverse events, including death, for reasons that may or may not be related to our products. Some patients treated with our products, including patients who have participated in our clinical trials, have died or suffered potentially life-threatening diseases either during or after ending their treatments. Patients who delay or miss a dose or discontinue treatment may also experience complications, including death. Such events could subject us to costly litigation, require us to pay substantial amounts of money to injured patients, delay, negatively impact or end our opportunity to receive or maintain regulatory approval to market our products, or require us to suspend or abandon our commercialization efforts. Even in a circumstance in which we do not believe that an adverse event is related to our products, the investigation into the circumstance may be time consuming or inconclusive. These investigations may interrupt our sales efforts, delay our regulatory approval process in other countries, or impact and limit the type of regulatory approvals that our products receive or maintain.
For example, use of C5 Inhibitors, such as Soliris, is associated with an increased risk for certain types of infection, including meningococcal infection. Under controlled settings, patients in our eculizumab trials all receive vaccination against meningococcal infection prior to first administration of Soliris and patients who are prescribed Soliris in most countries are required by prescribing guidelines to be vaccinated prior to receiving their first dose. A physician may not have the opportunity to timely vaccinate a patient in the event of an acute emergency episode, such as in a patient presenting with aHUS or during the health crisis that began in May 2011 in Europe, principally in Germany, due to the epidemic of infections from Enterohemorrhagic E. coli. Vaccination does not, however, eliminate all risk of meningococcal infection. Additionally, in some countries there may not be any vaccine approved for general use or approved for use by the FDA and such agencies). Compliance with these post-approval requirements could result in infantsincreased cost and children. Some patients treated with Soliris who had been vaccinated have nonetheless experienced meningococcal infection, including patients who have suffered serious illness or death. Each such incident is required to be reported to appropriate regulatory agencies in accordance with relevant regulations.
Clinical evaluations of outcomes in the post-marketing setting are required to be reported to appropriate regulatory agencies in accordance with relevant regulations. Determination of significant complications associated with the delay or discontinuation ofexpense and decrease our products could have a material adverse effect on our ability to sell our products.
Ifoperating margins and, if we are unable to establish and maintain effective sales, marketing and distribution capabilities, or to enter into agreementscomply with third parties to do so, we will be unable to successfully commercialize our products.
We are marketing and selling our products ourselves in the U.S., Europe, Japan and several other territories. Strensiq and Kanuma were approved in 2015, are in the early stages of commercial launch and are the second and third new product launches in Alexion’s history. If we are unable to establish and/or expand our capabilities to sell, market and distribute our products, either through our own capabilities or by entering into agreements with others, or to maintain such capabilities in countries where we have already commenced commercial sales, we will not be able to successfully sell our products. In that event, we will not be able to generate significant revenues. We cannot guarantee that we will be able to establish and maintain our own capabilities or enter into and maintain any marketing or distribution agreements with third-party providers on acceptable terms, if at all. Even if we hire the qualified sales and marketing personnel we need to support our objectives, or enter into marketing and distribution agreements with third parties on acceptable terms, we may not do so in an efficient manner or on a timely basis. We may not be able to correctly judge the size and experience of the sales and marketing force and the scale of distribution capabilities necessary to successfully market and sell our products. Establishing and maintaining sales, marketing and distribution capabilities are competitive, expensive and time-consuming. Our expenses associated with building up and maintaining the sales force and distribution capabilities around the world may be disproportionate compared to the revenuesthese requirements, we may be ablesubject to generate on sales. We cannot guarantee that we will be successfulregulatory action by the applicable regulatory agency and the penalties may include fines and product withdrawals or restrictions in commercializing anythe use of our products.a product.
If we fail to comply with applicable healthcare laws orand regulations, including those related to healthcare fraud and abuse, we may be subject to investigations and civil or criminal penalties and our business could be adversely affected.
In addition to FDA and related regulatory requirements, weWe are subject to healthcare “fraud and abuse” laws, such as the federal False Claims Act (FCA),FCA, the anti-kickback provisions of the federal Social Security Act, laws prohibiting off-label product promotion and other related federal and state laws and regulations. As discussed above in the subsection entitled “Fraud and Abuse,” the
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving any remuneration, directly or indirectly, in cash or in kind to induce, or reward the purchasing, leasing, ordering or arranging for or recommending the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid, or other federal healthcare programs. Liability may be established without a person or entity having actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it. A conviction for violation of the Anti-kickback Statute requires mandatory exclusion from participation in federal healthcare programs. The majority of states also have statutes similar to the federal Anti-Kickback Statute and false claims laws that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. We seek to comply with the anti-kickback laws and with the available statutory exemptions and safe harbors. However, our practices may not in all cases fit within the safe harbors, and our practices may therefore be subject to scrutiny on
a case-by-case basis. As discussed above in the subsection entitled “Fraud and Abuse,” theThe FCA prohibits any person from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds, or knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim. Pharmaceutical companies have been
investigated and have reached substantial financial settlements with the Federal government under the FCA for a variety of alleged promotional and marketing activities, such as allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees and other benefits to physicians to induce them to prescribe products; engaging in promotion of pharmaceuticals for uses that the FDA has not approved, or “off-label” uses; and submitting inflated best price information to the Medicaid Rebate Program.
We seek to comply with the Anti-Kickback Statute and FCA laws, including operating within any available safe harbors, but we cannot assure that our compliance program, policies and procedures will always protect Alexionus from acts committed by its employees or third-party distributors or service providers.
Other related federal and state laws and regulations that may affect our ability to operate include, among others, the federal False Statements Statute, the federal Civil Monetary Penalties Law, HIPAA, the federal Open Payments program, state anti-kickback and false claims acts, and state and local disclosure requirements and marketing restrictions. Additional information about the scope of these requirements and potential penalties is provided under “Government Regulation - Fraud and Abuse” included above in Item 1 in this Annual Report on Form 10-K.
In recent years, legislation has been adopted at the federal, state and local level requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports or make periodic public disclosures on sales, marketing, pricing, clinical trials, health care provider payments and other activities. For example, as part of the PPACA, the federal government enacted the Open Payments (commonly known as the Sunshine Act) provisions. Open Payments requires pharmaceutical manufacturers to report annually to CMS payments or other transfers of value made by that entity to physicians and teaching hospitals. We also now have similar reporting obligations throughout the EU. Failure to comply with the reporting requirements may result in significant civil monetary penalties.
Violations of U.S. federal and state fraud and abuse laws (and comparable laws in foreign jurisdictions) may result in criminal, civil and administrative sanctions, including fines, damages, civil monetary penalties (which may be material in amount) and exclusion from federal healthcare programs (including Medicare and Medicaid). Any action initiated against us for violation of these laws, even if we successfully defend against it, could require the expenditure of significant resources and generate negative publicity, which could materially adversely affect our ability to operate our business and our financial results.
Although physicians in
Finally, the U.S. are permitted to, basedFDA, the EU and EU Member States and the MHLW impose restrictions on their medical judgment, prescribethe promotion and marketing of drug products and prohibit pharmaceutical manufacturers from promoting products for indications other than those cleared or approved by regulatory authorities or for use in manner that is not consistent with the FDA, manufacturersproduct label approved by regulatory agencies, or off-label promotion. In certain instances, physicians are, prohibited from promotinghowever, in their medical judgment permitted to use products for unapproved purposes and we are aware of such off-label uses. Inuses of SOLIRIS. For information regarding a recent MHLW inquiry focused on our communication efforts regarding the U.S., we marketproper use of SOLIRIS in Japan for aHUS, see Note 11 “Commitments and Contingencies” to our products for their approved uses.consolidated financial statements included elsewhere in this Annual Report on Form 10-K. Although we believe our marketing materials and training programs for physicians do not constitute improper promotion, the FDA, the U.S. Department of Justice Department, or(DOJ), other federal or state government agencies, the EU, EU Member States or the MHLW may disagree. If the FDA or other government agencies determineany governmental authority determines that our promotional materials, training or other activities constitute improper promotion of any of our products, it could request that we modify our training or promotional materials or other activities or subject us to regulatory enforcement actions, including the issuance of a warning letter, product withdrawal or recall, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal or state enforcement authorities might take action if they believe that the alleged improper promotion led to the submission and payment of claims for an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false or fraudulent claims for payment of government funds.
As discussed above in the subsection entitled “Other Regulations,” the EU imposes similar strict restrictions on the promotionThe sales and marketing practices of drug products. The off-label promotionthe pharmaceutical industry have been the subject of medicinal products is prohibited inincreased scrutiny from authorities such as the EUDOJ, and in other territories. The promotionwe expect that this trend may continue and may increase. If the government or the courts determine that we breached any of medicinal products that are notthese sales and marketing laws, we may be subject to a marketing authorization ispenalties identified above. Any action against us for violation of these laws, even if we successfully defend against them, also prohibited incould cause us to incur significant legal expenses, harm our reputation and divert our management’s attention from the EU. Violationsoperation of the rules governing the promotion of medicinal products in the EUour business.
Our business and in other territories couldoperations may be penalizedmaterially adversely affected by administrative measures, fines and imprisonment.government investigations.
As discussed above in the subsection entitled “Other Regulations,” weWe are subject to the FCPA, the U.K. Bribery Act and other anti-corruption laws and regulations that generally prohibit companies and their intermediaries from making improper payments to government officials and/or other persons for the purpose of obtaining or retaining business and we operate in countries that are recognized as having a greater potential for
governmental and commercial corruption. WeWhile we have enhanced our compliance and training programs, we cannot assure that our compliance program, policies and procedures will always protect Alexionus from acts committed by its employees or third-party distributors or service providers.third-parties acting on our behalf.
In May 2015, we received a subpoena in connection with an investigation by the Enforcement Division of the SEC requesting information related to our grant-making activities and compliance with the FCPA in various countries. The SEC also seeks information related to Alexion’s recalls of specific lots of Soliris and related securities disclosures. In addition, in October 2015, Alexionwe received a request from the DOJ for the voluntary production of documents and other information pertaining to Alexion’sour compliance with the FCPAFCPA. The SEC and inDOJ also sought information related to our recalls of specific lots of SOLIRIS and related securities disclosures. In December 2016, we received a subpoena from the USAOU.S. Attorney’s Office for the District of Massachusetts requesting documents relating generally to our support of certain 501(c)(3) organizations (as described below). We understand that provide financial assistance to Medicare patients, Alexion’s provisionthe U.S. Attorney's Office is coordinating its inquiry with the Office of free drug to Medicare patientsInspector General (OIG) of the U.S. Department of Health and Alexion’s related compliance policiesHuman Services. In May 2017, Brazilian authorities seized records and training materials. Alexion isdata from our Sao Paulo, Brazil offices as part of an investigation being conducted into our Brazilian operations. In October 2018, the MHLW conducted an inspection of our Japanese operations. We are cooperating with these investigations. At this time, Alexion iswe are unable to predict the duration, scope or outcome of these investigations.
Any determination that our operations or activities are not, or were not, in compliance with existing U.S. or foreign laws or regulations, including by the SEC or DOJ pursuant to its investigation of our compliance with the FCPA and other matters, could result in the imposition of a broad range of civil and criminal sanctions against Alexionus and certain of our directors, officers and/or employees, including injunctive relief, disgorgement, substantial fines or penalties, imprisonment, and other legal or equitable sanctions.sanctions, including exclusion from Medicare, Medicaid, and other governmental healthcare programs. Any attempts to resolve some or all of these matters may not be successful. If we were to engage in settlement discussions with respect to any current or future investigation or litigation (and we may accrue amounts due to the nature of such discussions), but the matter is not settled, the ultimate resolution may result in monetary or other penalties materially stricter or greater than the terms or amounts that we proposed in discussions (or the amount that we accrued for such matter during negotiations). For example, in connection with the investigation by the U.S. Attorney's Office for the District of Massachusetts relating generally to our support of Patient Services, Inc. (PSI) and National Organization for Rare Disorders (NORD), 501(c)(3) organizations that provide financial assistance to Medicare patients taking drugs sold by Alexion (among other matters) we have accrued approximately $13.0 in the fourth quarter of 2018 as a result of our agreement
in principle to settle this investigation (but there is no guarantee that the steps necessary to conclusively resolve this matter will be successful or that the settlement terms will be finalized (and, if not completed, our liability in connection with this matter may exceed $13.0)). Additionally, remediation of any such findings resulting from these and any future investigations could have an adverse effect on our business operations, and we could experience interruptions of business, harm to our reputation, debarment from government contracts, loss of supplier, vendor or other third-party relationships, and necessary licenses and permits could be terminated. Other internal or government investigations or legal or regulatory proceedings, including lawsuits brought by private litigants, may also follow as a consequence. Cooperating with and responding to the SEC and the DOJrequests for information in connection with its investigation of our FCPA practices and other matters,these ongoing investigations, as well as responding to any future U.S., state or foreign governmental investigation or whistleblower lawsuit, has resulted and could continue to result in substantial expenses, and could divert management’s attention from other business concerns and could have a material adverse effect on our business and financial condition and growth prospects.
Changes in healthcare laws and implementing regulations, as well as changes in healthcare policy, may affect coverage and reimbursement of our products in ways that we cannot currently predict and these changes could adversely affect our business and financial condition.
Completion In the U.S., there have been a number of preclinical studies or clinical trials does not guarantee advancementlegislative and regulatory initiatives focused on containing the cost of healthcare. The PPACA substantially changed the way healthcare is financed by both governmental and private insurers in the U.S., and significantly impacts the pharmaceutical industry. The PPACA contains a number of provisions that are expected to impact our business and operations, in some cases in ways we cannot currently predict. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under health insurance exchanges, expansion of the 340B program, expansion of state Medicaid programs, fraud and abuse enforcement and rules governing the approval of biosimilar products (and allowing biosimilars access to the next phasemarket in accordance with the FDA’s Biosimilars Action Plan). These changes may impact existing government healthcare programs and may result in the development of development.new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program. In 2016, CMS implemented changes to the Medicaid Drug Rebate Program under the PPACA. Moreover, in the future, Congress could enact legislation that further increases Medicaid drug rebates or other costs and charges associated with participating in the
Completion
Medicaid Drug Rebate Program. The issuance of preclinical studiesregulations and coverage expansion by various governmental agencies relating to the Medicaid Drug Rebate Program has and may continue to increase our costs and the complexity of compliance, has been and may be time-consuming, and could have a material adverse effect on our results of operations.
Similar efforts to those in the United States, and in some cases even more aggressive efforts, are being taken by governments to control the costs of pharmaceutical drugs in countries outside the U.S. In these markets outside the U.S., the pricing and reimbursement of pharmaceutical products is subject to direct or clinical trials doesindirect governmental control and such government authorities are increasingly attempting to limit or regulate the price of drug products and due to their control over pricing are able to move quickly to implement pricing changes.
We may face uncertainties as a result of federal and administrative efforts to repeal, substantially modify or invalidate some or all of the provisions of the PPACA. There is no assurance that the PPACA, as currently enacted or as amended in the future, will not guaranteeadversely affect our business and financial results, and we cannot predict how future federal or state legislative or administrative changes relating to healthcare reform may affect our business.
The current presidential administration has also indicated an intent to address prescription drug pricing and recent Congressional hearings have brought increased public attention to the costs of prescription drugs. These actions and the uncertainty about the future of the PPACA and healthcare laws may put downward pressure on pharmaceutical pricing and increase our regulatory burdens and operating costs.
State governments have sought to put in place limits and caps on pharmaceutical prices and have also requested rebates for certain pharmaceuticals. Attempts to decrease prices of pharmaceuticals products may lead to increased use of managed care organizations by Medicaid programs which could lead to managed care organizations influencing prescription decisions for beneficiaries and a corresponding limitation on prices and reimbursement for our products.
Governments in countries where we operate have adopted or have also shown significant interest in pursuing legislative initiatives to reduce costs of healthcare. We expect that we will initiate additional studiesthe implementation of current laws and policies, the amendment of those laws and policies in the future, as well as the adoption of new laws and policies, could have a material adverse effect on our industry generally and on our ability to maintain or trials forincrease our product sales or successfully commercialize our product candidates, or could limit or eliminate our future spending on development projects. The announcement or adoption of regulatory or legislative proposals could delay or prevent our entry
into new markets, affect our reimbursement or sales in the markets where we are already selling our products and materially harm our business, financial condition and results of operations.
If we fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate Program, Medicare, or other governmental pricing programs, we could be subject to additional reimbursement requirements, penalties, sanctions and fines which could have a material adverse effect on our business, financial condition, results of operations and prospects.
We participate in and have certain price reporting obligations to the Medicaid Drug Rebate Program and we have obligations to report the average sales price under the Medicare program. Under the Medicaid Drug Rebate Program, we are required to pay a rebate to each state Medicaid program for quantities of our products that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our products under Medicaid and Medicare Part B. Those rebates are based on pricing data reported by us on a monthly and quarterly basis to CMS. Any failure to comply with these price reporting and rebate payment obligations could negatively impact our financial results.
Pricing and rebate calculations vary among products and programs. The calculations, including those in connection with the Medicaid Drug Rebate Program and 340B drug pricing program (as described further below) are complex and are often subject to interpretation by us, governmental or regulatory agencies and the courts. We cannot assure you that our submissions will not be found by CMS or other applicable government authorities to be incomplete or incorrect. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. For example, if further studieswe become aware that our reporting to CMS for a prior quarter was incorrect, or trials are initiated what the scope and phasehas changed as a result of recalculation of the trial willpricing data, we are obligated to resubmit the corrected data for a period not to exceed twelve quarters from the quarter in which the data originally were due, and CMS may request or require restatements for earlier periods as well. Such restatements and recalculations increase our costs for complying with the laws and regulations governing these programs, including the Medicaid Drug Rebate Program. Any corrections to our rebate calculations could result in an overage or underage in our rebate liability for past quarters, depending on the nature of the correction. Price recalculations also may affect the ceiling price at which we are required to offer our products to certain covered entities under the 340B pricing program.
We are liable for errors associated with our submission of pricing data. In addition to retroactive rebates and the potential for 340B program refunds,
civil monetary penalties can be applied if we are found to have knowingly submitted any false pricing information to the government, if we are found to have made a misrepresentation in the reporting of our average sales price, or if we fail to submit the required pricing data on a timely basis. Such conduct also could be grounds for CMS to terminate our Medicaid drug rebate agreement, pursuant to which we participate in the Medicaid program. In the event that they willCMS terminates our rebate agreement, federal payments may not be completed,available under Medicaid or Medicare Part B for our covered outpatient drugs. If a governmental authority, such as CMS, were to take any of the foregoing actions, our business and results of operations may be negatively impacted.
The Public Health Service’s 340B drug pricing program, and other comparable government and payer regulations, may have a negative impact on the price we can charge for our products and result in a decrease in revenues.
Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. The 340B pricing program is described in Pharmaceutical Pricing and Reimbursement in Item 1 Business in this Annual Report on Form 10-K. The 340B ceiling price is calculated using a statutory formula, which is based on, among other prices, the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program. We are a participant in the 340B drug pricing program and are, for the applicable covered entities, subject to the price ceiling. Any changes to the 340B drug pricing program, including:
the method of calculating the 340B ceiling price for our products (such as the pricing regulations that have been further delayed until July 2019);
any expansion of the entities that qualify as covered entities; and
any requirement that participating manufacturers agree to provide 340B discounted pricing on drugs used in an inpatient settings;
could have a material and negative impact our revenue and results of operations.
In addition, the agreement that manufacturers must sign to participate in the 340B pricing program obligates a manufacturer to offer the 340B price to covered entities if these further studiesthe manufacturer makes the drug
available to any other purchaser at any price and to report to the government the ceiling prices for its drugs.
Beyond the Public Health Service’s 340B drug pricing program, federal law requires that a company must participate in the Department of Veterans Affairs Federal Supply Schedule (FFS) pricing program to be eligible to have its products paid for with federal funds. If we overcharge the government in connection with our FSS contract or trialsSection 703 Agreement, whether due to a misstated FCP or otherwise, we are completed, thatrequired to refund the design or results will provide a sufficient basisdifference to apply for or receive regulatory approvals the government. Failure to make necessary disclosures and/or to commercialize products. Resultsidentify contract overcharges can result in allegations against us under the FCA and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, may be expensive, and could have a material adverse effect on our business, financial condition, results of clinical trialsoperations and growth prospects.
We may be subject to numerous and varying privacy and security laws, and our failure to comply could be inconclusive, requiring additional or repeat trials. Data obtained from preclinical studiesresult in penalties and clinical trialsreputational damage.
We are subject to laws and regulations covering data privacy and the protection of personal information including health information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business. In the U.S., numerous federal and state laws and regulations, including state security breach notification laws, state health information privacy laws, and federal and state consumer protection laws, govern the collection, use, disclosure, and protection of personal information. Each of these laws is subject to varying interpretations by courts and government agencies, creating complex compliance issues for us. If we fail to comply with applicable laws and regulations we could be subject to penalties or sanctions, including criminal penalties if we knowingly obtain or disclose individually identifiable health information from a covered entity in a manner that is not authorized or permitted by HIPAA.
Numerous other countries have, or are developing, laws governing the collection, use and transmission of personal information as well. EU Member States and other jurisdictions have adopted data protection laws and regulations, which impose significant compliance obligations. For example, the EC adopted the EU Data Protection Directive, as implemented into national laws by the EU Member States, which imposes strict obligations and restrictions on the ability to collect, analyze, and transfer personal data, including health data from clinical trials and adverse event reporting. Data protection authorities from different EU Member States have interpreted the privacy laws differently, which adds to the complexity of processing personal data in the EU, and guidance on implementation and
compliance practices are often updated or otherwise revised. Any failure to comply with the rules arising from the EU Data Protection Directive and related national laws of EU Member States could delay, limitlead to government enforcement actions and significant penalties against us, and adversely impact our operating results.
In May 2016, the EU formally adopted the General Data Protection Regulation, which applies in all EU Member States and went into effect on May 25, 2018 and replaced the EU Data Protection Directive on that date. The regulation introduces new data protection requirements in the EU and substantial fines for breaches of the data protection rules. It increases our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with the new EU data protection rules.
Security breaches, cyber-attacks or prevent regulatory approval.other disruptions could expose us to liability and affect our business and reputation.
We are increasingly dependent on our information technology systems and infrastructure for our business. We collect, store and transmit sensitive information including intellectual property, proprietary business information and personal information in connection with business operations. The secure maintenance of this information is critical to our operations and business strategy. Some of this information could be an attractive target of criminal attack by third parties with a wide range of motives and expertise, including organized criminal groups, “hacktivists,” patient groups, disgruntled current or former employees and others. Cyber-attacks are of ever-increasing levels of sophistication, and despite our security measures, our information technology and infrastructure may be vulnerable to such attacks or may be breached, including due to employee error or malfeasance. We have implemented information security measures to protect patients’ personal information against the risk of inappropriate and unauthorized external use and disclosure. However, despite these measures, and due to the ever changing information cyber-threat landscape, we may be subject to data breaches through cyber-attacks. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. If our systems become compromised, we may not promptly discover the design or results achievedintrusion. Like other companies in our clinical trials are insufficientindustry, we have experienced attacks to proceedour data and systems, including malware and computer viruses. If our systems failed or were breached or disrupted, we could lose product sales, and suffer reputational damage and loss of customer confidence. Such incidents may result in notification obligations to further trialsaffected individuals and government agencies, legal claims or proceedings, and liability under foreign, federal and state laws that protect the privacy and security of personal information. Any one of these events could
cause our business to be materially harmed and our results of operations may be adversely impacted.
Negative public opinion and increased regulatory approvalscrutiny of recombinant and transgenic products, genetically modified products and genetically modified animals generally may damage public perception of our product candidates, our company could be materiallycurrent and future products or adversely affected. Failure of a clinical trial to achieve its pre-specified primary endpoint, such as the Phase III Soliris trial for gMG that we announced in June 2016, generally increases the likelihood that additional studies or trials will be required if we determine to continue development of the product candidate, reduces the likelihood of timely development of and regulatory approval to market the product candidate, and may decrease the chances for successfully achieving the primary endpoint in scientifically similar indications.
Our clinical studies may be costly and lengthy, and there are many reasons why drug testing could be delayed or terminated.
For human trials, patients must be recruited and each product candidate must be tested at various doses and formulations for each clinical indication. In addition, to ensure safety and effectiveness, the effect of drugs often must be studied over a long period of time, especially for the chronic diseases that we are studying. Many of our programs focus on diseases with small patient populations making patient enrollment difficult. Insufficient patient enrollment in our clinical trials could delay or cause us to abandon a product development program. We may decide to abandon development of a product candidate or a study at any time due to unfavorable results or other reasons, or we may have to spend considerable resources repeating clinical trials or conducting additional trials, either of which would increase costs and delay any revenue from those product candidates, if any. We may open clinical sites and enroll patients in countries where we have little experience. We rely on a small number of clinical research organizations to carry out our clinical trial related activities, and one CRO is responsible for many of our studies. We rely on such parties to accurately report their results. Our reliance on CROs may impactaffect our ability to controlconduct our business and obtain regulatory approvals we may seek.
KANUMA is a transgenic product produced in the timing, conduct, expenseegg whites of genetically modified chickens who receive copies of the human lysosomal acid lipase gene to produce recombinant human lysosomal acid lipase. The success of KANUMA may depend in part on public attitudes of the use of genetic engineering. Public attitudes may be influenced by claims and qualityperceptions that these types of activities or products are unsafe, and our clinical trials.
Additional factors that can cause delay, impairmentproducts may not gain sufficient acceptance by, or terminationfall out of our clinical trials or our product development efforts include:
delay or failure in obtaining institutional review board (IRB), approvalfavor with, the public or the approval of other reviewing entitiesmedical community. Negative public attitudes to genetic engineering activities in general could result in more restrictive legislation or regulations and could impede our ability to conduct aour business, delay preclinical or clinical trial at each site;
delaystudies, or failure in reaching agreement on acceptable terms with prospective contract research organizations (CROs), and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
withdrawal of clinical trial sitesotherwise prevent us from commercializing our clinical trials as a result of changing standards of care or the ineligibility of a site to participate in our clinical trials;
clinical sites and investigators deviating from trial protocol, failing to conduct the trial in accordance with regulatory requirements, or dropping out of a trial;
slow patient enrollment, including, for example, due to the rarity of the disease being studied;
delay or failure in having patients complete a trial or return for post-treatment follow-up;
long treatment time required to demonstrate effectiveness;
lack of sufficient supplies of the product candidate;
disruption of operations at the clinical trial sites;
adverse medical events or side effects in treated patients, and the threat of legal claims and litigation alleging injuries;
failure of patients taking the placebo to continue to participate in our clinical trials;
insufficient clinical trial data to support effectiveness of the product candidates;
lack of effectiveness or safety of the product candidate being tested;
lack of sufficient funds;
inability to meet required specifications or to manufacture sufficient quantities of the product candidate for development or commercialization activities in a timely and cost-efficient manner;
decisions by regulatory authorities, the IRB, ethics committee, or us, or recommendation by a data safety monitoring board, to suspend or terminate clinical trials at any time for safety issues or for any other reason;
failure to obtain the necessary regulatory approvals for the product candidate or the approvals for the facilities in which such product candidate is manufactured; and
decisions by competent authorities, IRBs or ethics committees to demand variations in protocols or conduct of clinical trials.
product.
Risks Related to Intellectual Property
If we cannot obtain new patents, maintain our existing patents and protect the confidentiality and proprietary nature of our trade secrets and other intellectual property, our business and competitive position willmay be harmed.
Our success will dependdepends in part on our ability to obtain and maintain patent and regulatory protections for our products and investigational compounds, to preserve our trade secrets and other proprietary rights, to operate without infringing the proprietary rights of third parties and to prevent third parties from circumventing our rights. Due to the time and expense of bringing new products through development and regulatory approval to the marketplace, there is particular importance in obtaining patent and trade secret protection for significant new technologies, products and processes.
We have and may in the future obtain patents or the right to practice patents through ownership or license. Our patent applications may not result in the issue of patents in the U.S. or other countries. Our patentsIn addition, a patent may be issued in one country, but a counterpart patent may not be issued in another country. For example, we have applied for a certain patent in the EU that would provide protection for the composition of matter for SOLIRIS through 2027, and while a similar patent was granted in the U.S., the European patent application remains under examination by the European Patent Office and a hearing on it scheduled for February 2019 has been delayed until later in the year. Even if a patent is issued, that is not conclusive as to
inventorship, scope, validity or enforceability and therefore that patent may not afford adequate (or any) protection for our products. Third parties may challenge our patents, and have challenged our patents in the past.past and, in some cases have been successful in such challenges. For example, on January 21, 2019, the Opposition Division of the European Patent Office determined, following multi-party opposition proceedings, to revoke our European patent No. 2359834, which relates to the formulation of SOLIRIS. If any of our patents are narrowed, invalidated, revoked or become unenforceable, competitors may develop and market products similar to ours that do not conflict with or infringe our patents rights, which could have a material adverse effect on our financial condition. We may also finance and collaborate in research conducted by government organizations, hospitals, universities or other educational or research institutions. Such research partners may be unwilling to grant us exclusive rights to technology or products developed through such collaborations. There is also a risk that disputes may arise as to the rights to technology or products developed in collaboration with other parties. Our products and product candidates are expensive and time-consuming to test and develop. Even if we obtain and maintain patents, our business may be significantly harmed if the patents are not broad enough to protect our products from copycat products.
Significant legal questions exist concerning the extent and scope of patent protection for biopharmaceutical products and processes in the U.S. and elsewhere. Accordingly, there is no certainty that patent applications owned or licensed by us will issue as patents, or that our issued patents will afford meaningful protection against competitors. Once issued, patents are subject to challenge through both administrative and judicial proceedings in the U.S. and other countries. Such proceedings include re-examinations, inter partes reviews, post-grant reviews and interference proceedings before the U.S. Patent and Trademark Office, as well as opposition proceedings before the European Patent Office and other non-U.S. patent offices. Certain countries have laws that provide stronger bases for challenging third party patent rights than are available to challenge patents in other countries. Therefore, we may be able to defend our patents against a third party claim in one country but counterpart patents may be invalidated in other countries and we may be able to invalidate a third-party patent in one country but not invalidate its counterpart patents in other countries. Litigation may be required to enforce, defend or obtain our patent and other intellectual property rights. Any administrative proceeding or litigation could require a significant commitment of our resources and, depending on outcome, could adversely affect the scope, validity or enforceability of certain of our patent or other proprietary rights.
In addition, our business requires using sensitive technology, techniques and proprietary compounds that we protect as trade secrets. However, we may also rely heavily on collaboration with, or discuss the potential for collaboration with, suppliers, outside scientists and other biopharmaceutical companies.companies, including in connection with development efforts such as those with Complement Pharma and Dicerna. Collaboration and discussion of potential collaboration present a strong risk of exposing our trade secrets. If our trade secrets were exposed, it wouldwe may lose the protection and potential exclusive rights afforded by trade secret law, and such exposure may likely help our competitors and allow them to access technology without restriction and adversely affect our business prospects.
IfIf we are found to be infringing onthird party patents, owned by others, we may be forced to pay damages to the patent owner and/or obtain a license to continue the manufacture, sale or development of our products. If we cannot obtain a license, we may be prevented from the manufacture, sale or development of our products or product candidates, which wouldmay adversely affect our business.
Parts of our technology, techniques, proprietary compounds and potential product candidates, including those which are or may be in-licensed, may be found to infringe patents owned by or granted to others. We previously reported that certainhave and may in the future receive notices claiming our products infringe third party patents and third parties filedhave and may in the future file civil lawsuits against us claiming infringement of their intellectual property rights. Each of those matters was resolved. However, additionalMost recently, in late-2018, Chugai Pharmaceutical Co., Ltd. filed suits in the U.S. and Japan alleging that ULTOMIRIS infringes a U.S. and two Japanese patents, respectively, held by Chugai (these suits are still in the early stages). Additional third parties may claim that the manufacture, use or sale of our products or product candidates infringes patents owned or granted to such third parties. We have in the past received, and may in the future receive, notices from third parties claiming that their patents may be infringed by the development, manufacture or sale of our products or product candidates. We are aware of patents owned by third parties that might be claimed by such third parties to be infringed by the development and commercialization of our products or investigational compounds. In respect to some of these patents, we have obtained licenses, or expect to obtain licenses. However, with regard to other patents, we have determined in our judgment that:
| |
▪ | our products and investigational compounds do not infringe the patents; |
| |
▪ | the patents are not valid or enforceable; and/or |
| |
▪ | we have identified and are testing various alternatives that should not infringe the patents and which should permit continued development and commercialization of our products and investigational compounds. |
our products and investigational compounds do not infringe the patents;
the patents are not valid or enforceable; and/or
we have identified and are testing various alternatives that should not infringe the patents and which should permit continued development and commercialization of our products and investigational compounds.
Any holder of these patents or other patents covering similar technology could sue us for damages
and seek to prevent us from manufacturing, selling or developing our products. LegalIntellectual property disputes, such as those initiated by Chugai, can be costly and time consuming to defend. Prior to launch of a new product (or an existing product for a new indication), for various reasons, a patent owner may not be able to assert its patent rights so it is likely that any potential challenges to our products may be made after a product has been commercialized and not while the product is in development, in clinical trials or during the regulatory review process. If we cannot successfully defend against any future actions or conflicts, if they arise, we may incur substantial legal costs and may be liable for damages, be required to obtain costly licenses or needbe forced to stop manufacturing, using or selling our products, which
would may adversely affect our business. We may seek to obtain a license prior to or during legal actions in order to reduce the risks in connection with product launches (or at a later time) and to reduce further costs and the risk of a court determination that our technology, techniques, proprietary compounds or potential product infringescandidates infringe the third party’s patents. A required license may be costly or may not be available on acceptable terms, if at all. A costly license, or inability to obtain a necessary license, could have a material adverse effect on our business. In addition, even if we obtained a license, it would likely be non-exclusive and any competitive advantage resulting from the licensed technology may be of limited value and the same technology could be utilized by competitors.
There can be no assurance thatIn some instances, we wouldbelieve we may prevail in a patent infringement actionaction. There can, however, be no assurance that the court will agree with our position or that they will decide this or any other infringement case in our favor. Nor can we wouldbe certain that, if we do not prevail in litigation, that we may be able to obtain a license to any third-party patent on commercially reasonable terms or any terms at all; successfully develop non-infringing alternatives on a timely basis;basis (or at all); or license alternative non-infringing technology, if any exists, on commercially reasonable terms.terms (or at all). Any impediment to our ability to manufacture, use or sell approved forms of our products or our product candidates could have a material adverse effect on our business and prospects.
It is possible that we could lose market exclusivity for a product earlier than expected, which wouldmay harm our competitive position.position.
In our industry, much of an innovative product’s commercial value is realized while it has market exclusivity. When market exclusivity expires and biosimilar or generic versions of the product are approved and marketed, there can be substantial decline in the innovative product’s sales.sales.
Market exclusivity for our products is based upon patent rights and certain regulatory forms of exclusivity.protection. The scope of our product patent rights vary from country
to country and areis dependent on the availability of meaningful legal remedies in each country. The failure to obtain patent and other intellectual property rights, or limitations on the use, or loss of such rights, could be material to our business. In some countries, patent protections for our products may not exist because certain countries did not historically offer the right to obtain specific types of patents or we did not file patents in those markets. Also, the patent environment is unpredictable and the validity and enforceability of patents cannot be predicted with certainty. Absent relevant patent protection for a product, once regulatory exclusivity periods expire, biosimilar or generic versions of the product can be approved and marketed. Even prior to the expiration of regulatory exclusivity, a competitor could seek to obtain marketing approval by submitting its own clinical trial data.
The market exclusivity of our products may be impacted by competitive products that are either innovative or biosimilar or generic copies. In our industry, the potential for biosimilar challenges has been an increasing risk to product market exclusivity. U.S. law includes an approval pathway for biosimilar versions of innovative biological products. Under the pathway, the FDA may approve products that are similar to (but not generic copies of) innovative biologics on the basis of less extensive data than is required for a full biologic license application. After an innovator has marketed its product for four years, other manufacturers may apply for approval of a biosimilar version of the innovator product. However, qualified innovative biological products will receive 12 years of regulatory market exclusivity meaning that(i.e., the FDA may not actually approve a biosimilar version untilproduct cannot be approved before 12 years after the innovative product received its approval.biological product). The law also provides a mechanism for innovators to enforce their patents that protect their products and for biosimilar applicants to challenge the patents. Such litigation may begin as early as four years after the innovative biological product is first approved by the FDA. Pathways for biosimilar products also exist in many other markets, including Europe, Japan and Japan.
Risks RelatedRussia. Other companies are developing and advancing SOLIRIS biosimilar programs, including conducting clinical trials. Competition, including from biosimilars approved for marketing, may likely result in a decrease in prices, increased promotion efforts and lower margins for our products. In addition, approval of a biosimilar that is substitutable for one of our products may increase the risk of accelerated market penetration by that biosimilar. Further, if patients or healthcare providers do not believe that ULTOMIRIS provides a compelling profile for patient conversion from SOLIRIS, a SOLIRIS biosimilar may not only be expected to Our Operations
We have identified a material weaknessand negative impact on our SOLIRIS revenues and margins (which accounted for a significant percentage of our revenue in our internal control over financial reporting. If we2018), it may also have a material impact on ULTOMIRIS revenue and margins and the ability of ULTOMIRIS to gain market acceptance.
Our other products are unable to remediate this material weaknesses, or if we experience additional material weaknesses or deficiencies inalso at risk from biosimilars. Other than SOLIRIS for the future or otherwise fail to maintain an effective systemtreatment of internal controls, we may not be able to accurately or timely report our financial condition or results of operations, which may adversely affect investor confidence in usgMG and SOLIRIS and ULTOMIRIS as a result, the value of our common stock.
Current management concluded and the Audit Committee concurred that there was a material weakness in the Company’s internal controls over financial reporting because we did not maintain an effective control environment as senior management failed to set an appropriate “Tone at the Top.” A “material weakness” is defined as a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. As further described in Item 9A “Controls and Procedures”, the aforementioned material weakness arose from actions identified during the Audit Committee Investigation which found that senior management applied pressure on personnel to use pull-in sales to meet targets, and such pressure was particularly significant during the fourth quarter 2015. The Audit Committee Investigation also found that certain Company personnel engaged in inappropriate business conduct to realize pull-in sales, as a result of pressure from senior management.
As further described in Item 9A “Controls and Procedures-Remediation Plan and Activities”, we have undertaken steps to improve our internal controls over financial reporting. However, there can be no assurance that we will be successful in making the improvements necessary to remediate the material weakness identified by management, that we will do so in a timely manner, or that we will not identify additional control deficiencies or material weaknesses in the future. If we are unable to
successfully remediate our existing or any future material weaknesses in our internal control over financial reporting, the accuracy and timing of our financial reporting may be adversely affected, we may be unable to maintain compliance with securities laws and NASDAQ listing requirements regarding the timely filing of periodic reports, investors may lose confidence in our financial reporting and our stock price may decline.
We may not accurately forecast demandtreatment for our products, including our new products, which may cause our operating results to fluctuate, and we cannot guarantee that we will achieve our financial goals, including our ability to maintain profitability on a quarterly or annual basis in the future.
We have maintained profitability on a quarterly basis since the quarter ended June 30, 2008 and on an annual basis beginning with the year ended December 31, 2008. Our quarterly revenues, expenses and net income (loss) may fluctuate, even significantly, due to the risks described in these “Risk Factors” as well as the timing of charges and expenses that we may take. We believe that we formulate our annual operating budgets with reasonable assumptions and targets, however we may not generate sufficient revenues or control expenses to achieve our financial goals, including continued profitability. We may not be able to sustain or increase profitability on a quarterly or annual basis. You should not consider our financial performance, including our revenue growth, in recent periods as indicative of our future performance. We may not accurately forecast demand for our products, especially Strensiq and Kanuma. Strensiq and Kanuma are in the early stages of commercial launch havingPNH, each received marketing approval in 2015, and both products treat rare diseases for which there was no existing therapy in a new therapeutic area. Product demand is dependent on a number of factors. Our investors may have widely varying expectations that may be materially higher or lower than actual revenues and if our revenues are different from these expectations, our stock price may experience significant volatility. Our revenues are also subject to foreign exchange rate fluctuations due to the global nature of our operations and our results of operations could be adversely affected due to unfavorable foreign exchange rates. Although we use derivative instruments to manage foreign currency risk, our efforts to reduce currency exchange losses may not be successful.
We have significant debt service obligations as a result of the debt we incurred to finance the acquisition of Synageva. Changes in interest rates related to this debt could significantly increase our annual interest expense. As we advance our most robust pipeline in our history and launch our second and third products worldwide, we will have substantial expenses as we continue our research and development efforts, continue to conduct clinical trials and continue to develop manufacturing, sales, marketing and distribution capabilities worldwide, some of which could be delayed, scaled-back or eliminated to achieve our financial objectives.
We have also recorded, or may be required to record, charges that include inventory write-downs for failed quality specifications or recalls, impairments with respect to investments, fixed assets and long-lived assets, outcomes of litigation and other legal or administrative proceedings, regulatory matters and tax matters, and payments in connection with acquisitions and other business development activities, such as milestone payments.
Each of our products is currently the only approved drug for the disease(s) the product treats. If a competitive product is approved for sale, including a biosimilar or generic product, our market share and our revenues could decline, particularly if the competitive product is perceived to be more effective or is less expensive than our product.
We operate in a highly competitive environment. Soliris is currently the only approved therapy for the treatment of PNH and aHUS. We are in advanced clinical studies of Soliris for the treatment of other diseases, and there are currently no approved drugs for any of these other diseases. Strensiq is currently the only product approved to treat HPP and Kanuma is the only product approved to treat LAL-D. In the future, Soliris may compete with new drugs currently in development, and Strensiq and Kanuma may also experience competition. Other companies have initiated clinical studies for the treatment of PNH and NMO, and we are aware of companies that are planning to initiate studies for diseases that we are also targeting. Our revenues could be negatively affected if patients or potential patients enroll in our clinical trials or clinical trials of other companies with respect to diseases that we also target with approved therapies.
Pharmaceutical companies have publicly announced intentions to establish or develop rare disease programs and these companies may introduce products that are competitive with ours. These and other companies, many of which have significantly greater financial, technical and marketing resources than us, may commercialize products that are cheaper, more effective, safer, or easier to administer than our products. In the future, our products may also compete with biosimilars or generics. We experience competition in drug development from universities and other research institutions, and pharmaceutical companies compete with us to attract universities and academic research institutions as drug development partners, including for licensing their proprietary technology. If our competitors successfully enter into such arrangements with academic institutions, we will be precluded from pursuing those unique opportunities and may not be able to find equivalent opportunities elsewhere.
If a company announces successful clinical trial results for a product that may be competitive with one of our products or product candidates, receives marketing approval of a competitive product, or gets to the market before we do with a competitive product, our business may be harmed or our stock price may decline.
If we fail to attract and retain highly qualified personnel, we may not be able to successfully develop, manufacture or commercialize our products or products candidates.
The success of our business is dependent in large part on our continued ability to attract and retain our senior management, and other highly qualified personnel in our scientific, clinical, manufacturing and commercial organizations. There is intense competition in the biopharmaceutical industry for these types of personnel. In December 2016, our Board appointed an interim CEO, and a search for a new CEO is ongoing. Our business is specialized and global and we must attract and retain highly qualified individuals across many geographies. We may not be able to continue to attract and retain the highly qualified personnel necessary for developing, manufacturing and commercializing our products and product candidates. If we are unsuccessful in our recruitment and retention efforts, or if our recruitment efforts take longer than anticipated, our business may be harmed.
If we fail to satisfy our debt service obligations or obtain the capital necessary to fund our operations, we may be unable to commercialize our products or continue or complete our product development.
In June 2015, we acquired Synageva and used a substantial portion of our cash on hand and incurred significant debt under the terms of a senior secured credit facility to finance the acquisition. In addition, we have substantial contingent liabilities, including milestone and royalty obligations under earlier acquisitions and strategic transactions. Our increased indebtedness, including increased interest expense, together with our significant contingent liabilities, could, among other things:
make us more vulnerable to economic or industry downturns and competitive pressures;
make it difficult for us to make payments on the credit facilities and require us to use cash flow from operations to satisfy our debt obligations, which would reduce the availability of our cash flow for other purposes, including business development efforts, research and development and mergers and acquisitions;
limit our ability to incur additional debt or access the capital markets; and
limit our flexibility in planning for, or reacting to changes in, our business.
The Credit Agreement requires us to comply with certain financial covenants on a quarterly basis and includes negative covenants, subject to exceptions, restricting or limiting our ability and the ability of our subsidiaries to, among other things, incur additional indebtedness, grant liens, and engage in certain investment, acquisition and disposition transactions. If an event of default occurs, the interest rate would increase and the administrative agent would be entitled to take various actions, including the acceleration of amounts due under the Credit Agreement.
Our ability to satisfy our obligations under the Credit Agreement and meet our debt service obligations will depend upon our future performance, which will be subject to financial, business and other factors affecting our operations, many of which are beyond our control.
We may not be able to access the capital and credit markets on terms that are favorable to us.
We may need to raise additional capital to supplement our existing funds and cash generated from operations for working capital, capital expenditure and debt service requirements, and other business activities. Funding needs may shift and the amount of capital we may need depends on many factors, including, the cost of any acquisition or any new collaborative, licensing or other commercial relationships that we may establish, the time and cost necessary to build our manufacturing facilities or enhance our manufacturing operations, the cost of obtaining and maintaining the necessary regulatory approvals for our manufacturing facilities, and the progress, timing and scope of our preclinical studies and clinical trials. The capital and credit markets have experienced extreme volatility and disruption. We may not receive additional funding when we need it or funding may only be available on unfavorable terms. If we cannot raise adequate funds to satisfy our capital requirements, we may have to delay, scale-back or eliminate certain research, development, manufacturing or commercial activities.
Our business involves environmental risks and potential exposure to environmental liabilities.
As a biopharmaceutical company, our business involves the use of certain hazardous materials in our research, development, manufacturing, and other activities. We and our third party providers are subject to various federal, state and local environmental laws and regulations concerning the handling and disposal of non-hazardous and hazardous wastes, such as medical and biological wastes, and emissions and discharges into the environment, such as air, soils and water sources. We also are subject to laws and regulations that impose liability and clean-up responsibility for releases of hazardous substances into the environment and a current or previous owner or operator of property may be liable for the costs of remediating its property or locations, without regard to whether the owner or operator knew of or caused the contamination. If an accident or environmental discharge occurs, or if we discover contamination caused by prior owners and operators of properties we acquire, we could be liable for remediation obligations, damages and fines that could exceed our insurance coverage and
financial resources. Such obligations and liabilities, which to date have not been material, could have a material impact on our business and financial condition. Additionally, the cost of compliance with environmental and safety laws and regulations may increase in the future, and we may be required dedicate more resources to comply with such developments or purchase supplemental insurance coverage.
We are seeking to expand our business through strategic initiatives. Our efforts to identify opportunities or complete transactions that satisfy our strategic criteria may not be successful, and we not realize the anticipated benefits of any completed acquisition or other strategic transaction.
Our business strategy includes expanding our products and capabilities. We regularly evaluate potential merger, acquisition, partnering and in-license opportunities that we expect will expand our pipeline or product offerings, and enhance our research platforms. Acquisitions of new businesses or products and in-licensing of new products may involve numerous risks, including:
substantial cash expenditures;
potentially dilutive issuance of equity securities;
incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition;
difficulties in assimilating the operations of the acquired companies;
failure of any acquired businesses or products or in-licensed products to achieve the scientific, medical, commercial or other results anticipated;
diverting our management’s attention away from other business concerns;
the potential loss of our key employees or key employees of the acquired companies; and
risks of entering markets in which we have limited or no direct experience.
A substantial portion of our strategic efforts are focused on opportunities for rare disorders and life-saving therapies, but the availability of such opportunities is limited. We may not be able to identify opportunities that satisfy our strategic criteria or are acceptable to us or our stockholders. Several companies have publicly announced intentions to establish or develop rare disease programs and we may compete with these companies for the same opportunities. For these and other reasons, we may not be able to acquire the rights to additional product candidates or approved products on terms that we or our stockholders find acceptable, or at all.
Even if we are able to successfully identify and complete acquisitions and other strategic transactions, we may not be able to integrate them or take full advantage of them. An acquisition or other strategic transaction may not result in short-term or long-term benefits to us. We may also incorrectly judge the value or worth of an acquired company or business or an acquired or in-licensed product.
To effectively manage our current and future potential growth, we must continue to effectively enhance and develop our global employee base, and our operational and financial processes. Supporting our growth strategy will require significant capital expenditures and management resources, including investments in research, development, sales and marketing, manufacturing and other areas of our operations. The development or expansion of our business, any acquired business or any acquired or in-licensed products may require a substantial capital investment by us. We may not have these necessary funds or they might not be available to us on acceptable terms or at all. We may also seek to raise funds by selling shares of our capital stock, which could dilute current stockholders’ ownership interest in our company, or securities convertible into our capital stock, which could dilute current stockholders’ ownership interest in our company upon conversion.
We may be required to recognize impairment charges for our goodwill and other intangible assets.
As of December 31, 2016, the net carrying value of our goodwill and other intangible assets totaled $9,340. As required by generally accepted accounting principles, we periodically assess these assets to determine if they are impaired. Impairment of intangible assets may be triggered by developments both within and outside our control. Deteriorating economic conditions, technological changes, disruptions to our business, inability to effectively integrate acquired businesses, unexpected significant changes or planned changes in use of the assets, intensified competition, divestitures, market capitalization declines and other factors may impair our goodwill and other intangible assets. For example, in the fourth quarter 2016, we recorded an impairment charge of $85 related to SBC-103 as discussed below in our “Results of Operations.” Any charges relating to such impairments could adversely affect our results of operations in the periods in which an impairment is recognized.
Our business could be affected by litigation, government investigations and enforcement actions.
We operate in many jurisdictions in a highly regulated industry and we could be subject to litigation, government investigation and enforcement actions on a variety of matters in the U.S. or foreign jurisdictions, including, without limitation, intellectual property, regulatory, product liability, environmental, whistleblower, Qui Tam, false claims, privacy, anti-kickback, anti-bribery, securities, commercial, employment, and other claims and legal proceedings which may arise from conducting our business. As previously disclosed, in May 2015, we received a subpoena in connection with an investigation by the Enforcement Division of the SEC requesting information related to our grant-making activities and compliance with the FCPA in various countries. The SEC also seeks information related to Alexion’s recalls of specific lots of Soliris and related securities disclosures. In addition, in October 2015, Alexion received a request from the DOJ for the voluntary production of documents and other information pertaining to Alexion’s compliance with the FCPA and in December 2016, we received a subpoena from the USAO for the District of Massachusetts requesting documents relating generally to our support of 501(c)(3) organizations that provide financial assistance to Medicare patients, Alexion’s provision of free drug to Medicare patients and Alexion’s related compliance policies and training materials . Further, securities fraud class action litigation has been filed against the Company and individual executive officers, and we could also become subject to legal proceedings and government investigations relating to matters addressed in the Audit Committee Investigation. Legal proceedings, government investigations, including the SEC and DOJ investigations, and enforcement actions can be expensive and time consuming. An adverse outcome could result in significant damages awards, fines, penalties, exclusion from the federal healthcare programs, healthcare debarment, injunctive relief, product recalls, reputational damage and modifications of our business practices, which could have a material adverse effect on our business and results of operations.
The intended efficiency of our corporate structure depends on the application of the tax laws and regulations in the countries where we operate and we may have exposure to additional tax liabilities or our effective tax rate could change, which could have a material impact on our results of operations and financial position.
As a company with international operations, we are subject to income taxes, as well as non-income based taxes, in both the U.S. and various foreign jurisdictions. Significant judgment is required in determining our worldwide tax liabilities. Although we believe our estimates are reasonable, the ultimate outcome with respect to the taxes we owe may differ from the amounts recorded in our financial statements. If the Internal Revenue Service, or other taxing authority, disagrees with the positions we take, we could have additional tax liability, and this could have a material impact on our results of operations and financial position. Our effective tax rate could be adversely affected by changes in the mix of earnings in countries with different statutory tax rates, changes in the valuation of deferred tax assets and liabilities, changes in tax laws and regulations, changes in interpretations of tax laws, including pending tax law changes, changes in our manufacturing activities and changes in our future levels of research and development spending.
We have designed our corporate structure, the manner in which we develop and use our intellectual property, and our intercompany transactions between our affiliates in a way that is intended to enhance our operational and financial efficiency and increase our overall profitability. The application of the tax laws and regulations of various countries in which we operate and to our global operations is subject to interpretation. We also must operate our business in a manner consistent with our corporate structure to realize such efficiencies. The tax authorities of the countries in which we operate may challenge our methodologies for valuing developed technology or for transfer pricing. If tax authorities determine that the manner in which we operate results in our business not achieving the intended tax consequences, our effective tax rate could increase and harm our financial position and results of operations.
In addition, the U.S. Federal government and other U.S. State and foreign governments are considering and may adopt tax reform measures that significantly increase our worldwide tax liabilities. The U.S. Congress, the Organization for Economic Co-operation and Development and other government agencies in countries where we and our affiliates operate have focused on issues related to the taxation of multinational corporations, including, for example, in the area of “base erosion and profit shifting,” where payments are made between affiliates from a jurisdiction with high tax rates to a jurisdiction with lower tax rates. We established operations in Ireland in 2013 and Ireland tax authorities announced changes to the treatment of non-resident Irish entities. The changes are not expected to impact existing non-resident Irish entities, such as ours, until after December 31, 2020. These changes and other prospective changes in the U.S. and other countries in which we and our affiliates operate could increase our effective tax rate, and harm our financial position and results of operations.
Our sales and operations are subject to a variety of risks relating to the conduct and expansion of our international business.
We continue to increase our international presence, including in emerging markets. Our operations in foreign countries subject us to a variety of risks, including:
difficulties or the inability to obtain necessary foreign regulatory or reimbursement approvals of our products in a timely manner;
political or economic determinations that adversely impact pricing or reimbursement policies;
economic problems or political instability;
fluctuations in currency exchange rates;
difficulties or inability to obtain financing in markets;
unexpected changes in tariffs, trade barriers and regulatory requirements;
difficulties enforcing contractual and intellectual property rights;
compliance with complex import and export control laws;
trade restrictions and restrictions on direct investments by foreign entities;
compliance with tax, employment and labor laws;
costs and difficulties in recruiting and retaining qualified managers and employees to manage and operate the business in local jurisdictions;
costs and difficulties in managing and monitoring international operations; and
longer payment cycles.
Additionally, our business and marketing methods are subject to the laws and regulations of the countries in which we operate, which may differ significantly from country to country and may conflict with U.S. laws and regulations. The FCPA and anti-bribery laws and regulations are extensive and far-reaching, and we must maintain accurate records and control over the activities of our distributors and third party service providers in countries where we operate. We have policies and procedures designed to help ensure that we and our representatives, including our employees, comply with such laws, however we cannot guarantee that these policies and procedures will protect us against liability under the FCPA or other anti-bribery laws for actions taken by our representatives. Although we conducted due diligence of Synageva’s operations prior to the acquisition, we may discover or identify deficiencies or non-compliance with such laws as we complete the integration of the Synageva business and conduct operations. Failure to comply with the laws and regulations of the countries in which we operate could materially harm our business.
Currency fluctuations and changes in exchange rates could adversely affect our revenue growth, increase our costs and negatively affect our profitability.
We conduct a substantial portion of our business in currencies other than the U.S. dollar. We are exposed to fluctuations in foreign currency exchange rates and fluctuations in foreign currency exchange rates affect our operating results. The exposures result from portions of our revenues, as well as the related receivables, and expenses that are denominated in currencies other than the U.S. dollar, including the Euro, Japanese Yen, British Pound, Swiss Franc, and Russian Ruble. As the U.S. dollar strengthens against these foreign currencies, the relative value of sales made in the respective foreign currencies decrease. When the U.S. dollar weakens against these currencies, the relative value of such sales increase. We manage our foreign currency transaction risk within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes. We enter into foreign exchange forward contracts to hedge exposures resulting from portions of our forecasted revenues, including intercompany revenues, that are denominated in currencies other than the U.S. dollar. The purpose of the hedges of revenue is to reduce the volatility of exchange rate fluctuations on our operating results and to increase the visibility of the foreign exchange impact on forecasted revenues. Further, we enter into foreign exchange forward contracts, with durations of approximately 30 days, designed to limit the balance sheet exposure of monetary assets and liabilities. We enter into these hedges to reduce the impact of fluctuating exchange rates on our operating results. Gains and losses on these hedge transactions are designed to offset gains and losses on underlying balance sheet exposures. While we attempt to hedge certain currency risks, currency fluctuations between the U.S. dollar and the currencies in which we do business have, in the past, caused foreign currency transaction gains and losses and have also impacted the amounts of revenues and expenses calculated in U.S. dollars and will do so in the future. Likewise, past currency fluctuations have at times resulted in foreign currency transaction gains, and there can be no assurance that these gains can be reproduced. Any significant foreign currency exchange rate fluctuations could adversely affect our financial condition and results of operations.
Changes in healthcare laws and implementing regulations, as well as changes in healthcare policy, may affect coverage and reimbursement of our products in ways that we cannot currently predict and these changes could adversely affect our business and financial condition.
In the U.S., there have been a number of legislative and regulatory initiatives focused on containing the cost of healthcare. The Patient Protection and Affordable Care Act (PPACA) was enacted in the U.S. in March 2010. This law substantially changes the way healthcare is financed by both governmental and private insurers in the U.S., and significantly impacts the pharmaceutical industry. PPACA contains a number of provisions that are expected to impact our business and operations, in some cases in ways we cannot currently predict. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under health insurance exchanges, expansion of the 340B program, expansion of state Medicaid programs, fraud and abuse enforcement and rules governing the approval of biosimilar products. These changes will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to
the physician quality reporting system and feedback program. In early 2016, CMS issued final regulations to implement the changes to the Medicaid Drug Rebate Program under PPACA. These regulations became effective on April 1, 2016. Moreover, in the future, Congress could enact legislation that further increases Medicaid drug rebates or other costs and charges associated with participating in the Medicaid Drug Rebate Program. Legislative changes to the PPACA also remain possible and appear likely in the 115th U.S. Congress under the Trump Administration. The issuance of regulations and coverage expansion by various governmental agencies relating to the Medicaid Drug Rebate Program has and will continue to increase our costs and the complexity of compliance, has been and will be time-consuming, and could have a material adverse effect on our results of operations.
Governments in countries where we operate have adopted or have shown significant interest in pursuing legislative initiatives to reduce costs of healthcare. We expect that the implementation of current laws and policies, the amendment of those laws and policies in the future, as well as the adoption of new laws and policies, could have a material adverse effect on our industry generally and on our ability to maintain or increase our product sales or successfully commercialize our product candidates, or could limit or eliminate our future spending on development projects. In many cases, these government initiatives, even if enacted into law, are subject to future rulemaking by regulatory agencies. Although we have evaluated these government initiatives and the impact on our business, we cannot know with certainty whether any such law, rule or regulation will adversely affect coverage and reimbursement of our products, or to what extent, until such laws, rules and regulations are promulgated, implemented and enforced, which could sometimes take many years. The announcement or adoption of regulatory or legislative proposals could delay or prevent our entry into new markets, affect our reimbursement or sales in the markets where we are already selling our products and materially harm our business, financial condition and results of operations.
If we fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate Program, Medicare, or other governmental pricing programs, we could be subject to additional reimbursement requirements, penalties, sanctions and fines which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
Pricing and rebate calculations vary among products and programs. The calculations are complex and are often subject to interpretation by us, governmental or regulatory agencies and the courts. We cannot assure you that our submissions will not be found by CMS to be incomplete or incorrect. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. The Medicaid rebate amount is computed each quarter based on our submission to CMS of our current average manufacturer price and best price for the quarter. If we become aware that our reporting for a prior quarter was incorrect, or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected data for a period not to exceed twelve quarters from the quarter in which the data originally were due, and CMS may request or require restatements for earlier periods as well. Such restatements and recalculations increase our costs for complying with the laws and regulations governing the Medicaid Drug Rebate Program. Any corrections to our rebate calculations could result in an overage or underage in our rebate liability for past quarters, depending on the nature of the correction. Price recalculations also may affect the ceiling price at which we are required to offer our products to certain covered entities, such as safety-net providers, under the 340B drug discount program.
We are liable for errors associated with our submission of pricing data. In addition to retroactive rebates and the potential for 340B program refunds, if we are found to have knowingly submitted false average manufacturer price, ASP, or best price information to the government, we may be liable for civil monetary penalties in the amount of one hundred seventy-eight thousand dollars per item of false information. If we are found to have made a misrepresentation in the reporting of our ASP, the Medicare statute provides for civil monetary penalties of up to thirteen thousand dollars for each misrepresentation for each day in which the misrepresentation was applied. Our failure to submit monthly/quarterly average manufacturer price, ASP, and best price data on a timely basis could result in a civil monetary penalty of eighteen thousand dollars per day for each day the information is late beyond the due date. Such failure also could be grounds for CMS to terminate our Medicaid drug rebate agreement, pursuant to which we participate in the Medicaid program. In the event that CMS terminates our rebate agreement, federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs. A final regulation that has been published but is not yet effective would impose a civil monetary penalty of up to five thousand dollars for each instance of knowingly and intentionally charging a 340B covered entity more than the 340B ceiling price.
As discussed above in the subsection entitled “Pharmaceutical Pricing and Reimbursement,” federal law requires that a company must participate in the FSS pricing program to be eligible to have its products paid for with federal funds. If we overcharge the government in connection with our FSS contract or Section 703 Agreement, whether due to a misstated FCP or otherwise, we are required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the FCA and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We may be subject to numerous and varying privacy and security laws, and our failure to comply could result in penalties and reputational damage.
We are subject to laws and regulations covering data privacy and the protection of personal information including health information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business. In the U.S., we may be subject to state security breach notification laws, state health information privacy laws and federal and state consumer protections laws which impose requirements for the collection, use, disclosure and transmission of personal information. Each of these laws are subject to varying interpretations by courts and government agencies, creating complex compliance issues for us. If we fail to comply with applicable laws and regulations we could be subject to penalties or sanctions, including criminal penalties if we knowingly obtain individually identifiable health information from a covered entity in a manner that is not authorized or permitted by the federal Health Insurance Portability and Accountability Act of 1996, as amended (HIPAA) or for aiding and abetting the violation of HIPAA.
Numerous other countries have, or are developing, laws governing the collection, use and transmission of personal information as well. EU member states and other jurisdictions have adopted data protection laws and regulations, which impose significant compliance obligations. For example, the EC adopted the EU Data Protection Directive, as implemented into national laws by the EU member states, which imposed strict obligations and restrictions on the ability to collect, analyze, and transfer personal data, including health data from clinical trials and adverse event reporting. Data protection authorities from different EU member states have interpreted the privacy laws differently, which adds to the complexity of processing personal data in the EU, and guidance on implementation and compliance practices are often updated or otherwise revised. Any failure to comply with the rules arising from the EU Data Protection Directive and related national laws of EU member states could lead to government enforcement actions and significant penalties against us, and adversely impact our operating results.
In May 2016, the EU formally adopted the General Data Protection Regulation, which will apply to all EU member states from May 25, 2018 and will replace the current EU Data Protection Directive on that date. The regulation introduces new data protection requirements in the EU and substantial fines for breaches of the data protection rules. It will increase our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with the new EU data protection rules.
Security breaches, cyber-attacks, or other disruptions could expose us to liability and affect our business and reputation.
We are increasingly dependent on our information technology systems and infrastructure for our business. We collect, store, and transmit sensitive information including intellectual property, proprietary business information and personal information in connection with business operations. The secure maintenance of this information is critical to our operations and business strategy. Some of this information could be an attractive target of criminal attack by third parties with a wide range of motives and expertise, including organized criminal groups, “hactivists,” patient groups, disgruntled current or former employees, and others. Cyber-attacks are of ever-increasing levels of sophistication, and despite our security measures, our information technology and infrastructure may be vulnerable to such attacks or may be breached, including due to employee error or malfeasance. We have implemented information security measures to protect patients’ personal information against the risk of inappropriate and unauthorized external use and disclosure. However, despite these measures, and due to the ever changing information cyber-threat landscape, we may be subject to data breaches through cyber-attacks. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. If our systems become compromised, we may not promptly discover the intrusion. Like other companies in our industry, we have experienced attacks to our data and systems, including malware and computer viruses. If our systems failed or were breached or disrupted, we could lose product sales, and suffer reputational damage and loss of customer confidence. Such incidents would result in notification obligations to affected individuals and government agencies, legal claims or proceedings, and liability under federal and state laws that protect the privacy and security of personal information. Any one of these events could cause our business to be materially harmed and our results of operations would be adversely impacted.
Negative public opinion and increased regulatory scrutiny of recombinant and transgenic products, genetically modified products, and genetically modified animals generally may damage public perception of our current and future products or adversely affect our ability to conduct our business and obtain regulatory approvals we may seek.
Kanuma is a transgenic product produced in the egg whites of genetically modified chickens who receive copies of the human lysosomal acid lipase gene to produce recombinant human lysosomal acid lipase. The success of Kanuma will depend in part on public attitudes of the use of genetic engineering. Public attitudes may be influenced by claims and perceptions that these types of activities or products are unsafe, and our products may not gain sufficient acceptance by, or fall out of favor with, the public or the medical community. Negative public attitudes to genetic engineering activities in general could result in more restrictive legislation or regulations and could impede our ability to conduct our business, delay preclinical or clinical studies, or otherwise prevent us from commercializing our product.
Risks Related to Our Common Stock
Our stock price is extremely volatile.
The trading price of our common stock has been extremely volatile and may continue to be volatile in the future. Many factors could have an impact on our stock price, including fluctuations in our or our competitors’ operating results, clinical trial results or adverse events associated with our products, product development by us or our competitors, changes in laws, including healthcare, tax or intellectual property laws, intellectual property developments, changes in reimbursement or drug pricing, the existence or outcome of litigation or government proceedings, including the SEC/DOJ investigation failure to resolve, delays in resolving or other developments with respect toand the issues raised in the Warning Letter,Chugai lawsuits alleging patent infringement, acquisitions or other strategic transactions, and the perceptions of our investors that we are not performing or meeting expectations. The trading price of the common stock of many biopharmaceutical companies, including ours, has experienced extreme price and volume fluctuations, which have at times been unrelated to the operating performance of the companies whose stocks were affected.
Anti-takeover provisions in our charter and bylaws and under Delaware law could make a third-party acquisition of us difficult and may frustrate any attempt to remove or replace our current management.
Our corporate charter and by-law provisions may discourage certain types of transactions involving an actual or potential change of control that might be beneficial to us or our stockholders. Our bylaws provide that special meetings of our stockholders may be called only by the Chairman of the Board of Directors, the President, the Secretary, or a majority of the Board of Directors, or upon the written request of stockholders who together own of record 25%25.0% of the outstanding stock of all classes entitled to vote at such meeting. Our bylaws also specify that the authorized number of directors may be changed only by resolution of the boardBoard of directors.Directors. Our charter does not include a provision for cumulative voting for directors, which may have enabled a minority stockholder holding a sufficient percentage of a class of shares to elect one or more directors. Under our charter, our boardBoard of directorsDirectors has the authority, without further action by stockholders, to designate up to 5five million shares of preferred stock in one or more series. The rights of the holders of common
stock will be subject to, and may be adversely affected by, the rights of the holders of any class or series of preferred stock that may be issued in the future.
Because we are a Delaware corporation, the anti-takeover provisions of Delaware law could make it more difficult for a third party to acquire control of us, even if the change in control wouldmay be beneficial to stockholders. We are subject to the provisions of Section 203 of the Delaware General Laws, which prohibits a person who owns in excess of 15%15.0% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15%15.0% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
| |
| Item 1B. | UNRESOLVED STAFF COMMENTS. |
None.
We conduct our primary operations at the owned and leased facilities described below.
| | | Location | | Operations Conducted | | Approximate Square Feet | | Lease Expiration Dates | | Operations Conducted | | Approximate Square Feet | | Lease Expiration Dates |
| Boston, Massachusetts | | | Corporate headquarters and executive, sales, research and development offices | | 150,000 | | 2031 |
| New Haven, Connecticut | | Corporate headquarters and executive, sales, research and development offices | | 514,000 |
| | 2030 | | Research and process development laboratories, clinical supply and quality, enterprise business services | | 263,000 | | 2030 |
| Dublin, Ireland | | Global supply chain, distribution, and administration offices | | 160,000 |
| | Owned | | Global supply chain, distribution, and administration offices | | 160,000 | | Owned |
| Athlone, Ireland | | Commercial, research and development manufacturing | | 80,000 |
| | Owned | | Commercial, research and development manufacturing | | 80,000 | | Owned |
| Lexington, Massachusetts | | Research and development offices | | 81,000 |
| | 2019 | |
| Bogart, Georgia | | Commercial, research and development manufacturing | | 70,000 |
| | Owned | | Commercial, research and development manufacturing | | 70,000 | | Owned |
| Smithfield, Rhode Island | | Commercial, research and development manufacturing | | 67,000 |
| | Owned | |
| Zurich, Switzerland | | Regional executive and sales offices | | 69,000 |
| | 2025 | | Regional executive and sales offices | | 40,000 | | 2025 |
We believe that our administrative office space is adequate to meet our needs for the foreseeable future. We also believe that our research and development facilities and our manufacturing facilities, together with third party manufacturing facilities, will be adequate for our on-going activities. In addition to the locations above, we also lease space in other U.S. locations and in foreign countries to support our operations as a global organization.
In May 2015,April 2014, we announced plans to constructpurchased a new bulk biologics manufacturingfill/finish facility on our existing property in DublinAthlone, Ireland, which is expectedhas been refurbished to be completed by 2020.
become our first company-owned fill/finish facility. In July 2016, we announced plans to construct a new biologics manufacturing facility at this site, the construction of this facility is on-going and, based on current expectations, we anticipate this facility will receive regulatory approval in 2020.
In May 2015, we announced plans to construct a new biologics manufacturing facility on our existing property in Athlone,Dublin, Ireland, which is expected to be completed by 2018.the construction of this facility has commenced and, based on current expectations, we anticipate this facility will receive regulatory approval in 2020.
In the fourth quarter of 2018, we amended the New Haven lease agreement significantly reducing our rented square footage in the building beginning in 2019 through the expiration of the lease.
| |
| Item 3. | LEGAL PROCEEDINGS. |
In May 2015, we received
For a subpoena in connection with an investigation by the Enforcement Divisiondiscussion of the SEC requesting information related tolegal matters as of December 31, 2018, see Note 11, “Commitments and Contingencies,” Contingent Liabilities, within our grant-making activities and compliance with the FCPA in various countries. In addition, in October 2015, we received a request from the DOJ for the voluntary production of documents and other information pertaining to Alexion’s compliance with FCPA. The SEC and DOJ also seek information related to Alexion’s recalls of specific lots of Soliris and related securities disclosures. Alexion is cooperating with these investigations. At this time, Alexion is unable to predict the duration, scope or outcome of these investigations. While it is possible that a loss related to these matters may be incurred, given the ongoing nature of these investigations, management cannot reasonably estimate the potential magnitude of such loss or range of loss, if any.
Several securities class action lawsuits have been filed against the Company and former officers in federal district court alleging violations of Sections 10(b) and 20(a) of the Securities Exchange Act of 1934, 15 U.S.C. § 78j(b), and Rule 10b-5, promulgated thereunder, alleging that defendants made misstatements and/or omissions concerning the Company’s sales of Soliris.
On November 17, 2016, a shareholder filed a putative class action in the U.S. District Court for the Southern District of New York. While the litigation was in the early stages, and before defendants had respondednotes to the complaint, on December 30, 2016 plaintiffs filed a notice of voluntary dismissal and dismissed all claims without prejudice. This case is now closed.
On December 29, 2016, a second shareholder filed a putative class action against the Company and certain former employees in the U.S. District Court for the District of Connecticut, alleging that defendants made misrepresentations and omissions about Soliris between February 10, 2014 and December 9, 2016. On January 17, 2017, three parties filed motions to be named lead plaintiffconsolidated financial statements included in this action. BriefingAnnual Report on these motionsForm 10-K, which is ongoing. The litigation is in the early stages, and defendants have not yet responded to the complaint. Given the early stages ofincorporated into this litigation, management does not currently believe that a loss related to this matter is probable or that the potential magnitude of such loss or range of loss, if any, can be reasonably estimated. item by reference.
In December 2016, we received a subpoena from the USAO for the District of Massachusetts requesting documents relating generally to our support of 501(c)(3) organizations that provide financial assistance to Medicare patients taking drugs sold by Alexion, Alexion’s provision of free drug to Medicare patients, and Alexion compliance policies and training materials concerning the anti-kickback statute or payments to any 501(c)(3) organization that provides financial assistance to Medicare patients. Other companies have disclosed similar inquiries. We are cooperating with this inquiry.
| |
| Item 4. | MINE SAFETY DISCLOSURES. |
Not applicable.
PART II
| |
| Item 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES. |
Our common stock is quoted on The NASDAQNasdaq Stock Market, LLC under the symbol “ALXN.” The following table sets forth the range of high and low sales prices for our common stock on The NASDAQ Stock Market, LLC for the periods indicated since January 1, 2015.
|
| | | | | | | |
| Fiscal 2015 | High | | Low |
| First Quarter | | | |
| (January 1, 2015 to March 31, 2015) | $ | 193.27 |
| | $ | 171.08 |
|
| Second Quarter | | | |
| (April 1, 2015 to June 30, 2015) | $ | 191.00 |
| | $ | 150.06 |
|
| Third Quarter | | | |
| (July 1, 2015 to September 30, 2015) | $ | 208.88 |
| | $ | 142.02 |
|
| Fourth Quarter | | | |
| (October 1, 2015 to December 31, 2015) | $ | 193.45 |
| | $ | 150.69 |
|
| Fiscal 2016 | | | |
| First Quarter | | | |
| (January 1, 2016 to March 31, 2016) | $ | 187.59 |
| | $ | 124.16 |
|
| Second Quarter | | | |
| (April 1, 2016 to June 30, 2016) | $ | 162.00 |
| | $ | 110.56 |
|
| Third Quarter | | | |
| (July 1, 2016 to September 30, 2016) | $ | 138.40 |
| | $ | 115.84 |
|
| Fourth Quarter | | | |
| (October 1, 2016 to December 31, 2016) | $ | 145.42 |
| | $ | 109.12 |
|
As of February 8, 2017,January 28, 2019, we had approximately 11194 stockholders of record of our common stock and an estimated 185,102210,846 beneficial owners. The closing sale price of our common stock on February 8, 2017January 28, 2019 was $126.37$119.21 per share.
DIVIDEND POLICY
We have never paid cash dividends. We do not expect to declare or pay any cash dividends on our common stock in the near future. We intend to retain all earnings, if any, to invest in our operations. The payment of future dividends is within the discretion of our boardBoard of directorsDirectors and will depend upon our future earnings, if any, our capital requirements, financial condition and other relevant factors. In addition, restrictive covenants under our amended and restated credit agreement prohibit or limit the payment of cash dividends if we are not in compliance with certain covenants.
ISSUER PURCHASES OF EQUITY SECURITIES (amounts in millions except per share amounts)
The following table summarizes our common stock repurchase activity during the fourth quarter of 2016:2018: |
| | | | | | | | | | | | |
| Period | Total Number of Shares Purchased | | Average Price Paid per Share | | Total Number of Shares Purchased as Part of Publicly Announced Programs | | Maximum Dollar Value of Shares that May Yet Be Purchased Under the Programs |
| October 1-31, 2016 | 0.25 |
| | $ | 122.74 |
| | 0.25 |
| | 325 |
|
| November 1-30, 2016 | — |
| | — |
| | — |
| | 325 |
|
| December 1-31, 2016 | — |
| | — |
| | — |
| | 325 |
|
| Total | 0.25 |
| | $ | 122.74 |
| | 0.25 |
| |
|
|
| | | | | | | | | | | | |
| Period | Total Number of Shares Purchased | | Average Price Paid per Share | | Total Number of Shares Purchased as Part of Publicly Announced Programs | | Maximum Dollar Value of Shares that May Yet Be Purchased Under the Programs |
| October 1-31, 2018 | — |
| | $ | — |
| | — |
| | 451.5 |
|
| November 1-30, 2018 | — |
| | — |
| | — |
| | 451.5 |
|
| December 1-31, 2018 | — |
| | — |
| | — |
| | 451.5 |
|
| Total | — |
| | $ | — |
| | — |
| |
|
In November 2012, our Board of Directors authorized a share repurchase program. The repurchase program does not have an expiration date and we are not obligated to acquire a particular number of shares. In May 2015, our Board of Directors increased the authorization of shares up to $1,000 for future purchases under the repurchase program. In February 2017, our Board of Directors increased the authorization of shares upamount that we are authorized to $1,000 forexpend on future purchasesrepurchases to $1,000 under the repurchase program, which superseded all prior repurchase programs. As of February 16, 2017,6, 2019, there is a total of $1,000$451.5 remaining for repurchases under the repurchase program.
EQUITY COMPENSATION PLAN INFORMATION(amounts in millions except per share amounts)
| | | Plan Category | Number of shares of common stock to be issued upon exercise of outstanding options (1) | | Weighted- average exercise price of outstanding options | | Weighted- average term to expiration of options outstanding (years) | | Number of shares of common stock remaining available for future issuance under equity compensation plans (2) | Number of shares of common stock to be issued upon exercise of outstanding options (1) | | Weighted- average exercise price of outstanding options | | Weighted- average term to expiration of options outstanding (years) | | Number of shares of common stock remaining available for future issuance under equity compensation plans (2) |
| Equity compensation plans approved by stockholders | 6 |
| | $ | 116.65 |
| | 6.02 |
| | 7 |
| 3.6 | | $119.68 | | 4.74 | | 17.7 |
| Equity compensation plans not approved by stockholders | — |
| | $ | — |
| | — |
| | — |
| — | | $— | | — | | — |
| |
| (1) | Reflects number of shares of common stock to be issued upon exercise of outstanding options under all our equity compensation plans, including our 2017 Incentive Plan. Does not include 3.7 of outstanding restricted stock units, including performance-based restricted stock units, that were issued under the 2017 Incentive plan and the previous Amended and Restated 2004 Incentive Plan. Does not include 3 restricted shares outstanding that were issued under the Amended and Restated 2004 Incentive Plan. |
| |
| (2) | Of these shares, 617.0 remain available for future issuance under the Amended and Restated 20042017 Incentive Plan and 10.7 remain available under the 2015 Employee Stock Purchase Plan. |
The outstanding options and restricted sharesstock units are not transferable for consideration and do not have dividend equivalent rights attached.
THE COMPANY’S STOCK PERFORMANCE
The following graph compares cumulative total return of the Company’s Common Stockcommon stock with the cumulative total return of (i) the NASDAQNasdaq Stock Market-United States, and (ii) the NASDAQNasdaq Biotechnology Index. The graph assumes (a) $100 was invested on December 31, 20112013 in each of the Company’s Common Stock,common stock, the stocks comprising the NASDAQNasdaq Stock Market-United States and the stocks comprising the NASDAQNasdaq Biotechnology Index, and (b) the reinvestment of dividends. The comparisons shown in the graph are based on historical data and the stock price performance shown in the graph is not necessarily indicative of, or intended to forecast, future performance of our stock.
CUMULATIVE TOTAL RETURN
|
| | | | | | | | | | | | | | | | | |
| | 12/11 | | 12/12 | | 12/13 | | 12/14 | | 12/15 | | 12/16 |
| Alexion Pharmaceuticals, Inc. | 100.00 |
| | 131.10 |
| | 185.85 |
| | 258.78 |
| | 266.78 |
| | 171.12 |
|
| NASDAQ Composite | 100.00 |
| | 116.41 |
| | 165.47 |
| | 188.69 |
| | 200.32 |
| | 216.54 |
|
| NASDAQ Biotechnology | 100.00 |
| | 134.68 |
| | 232.37 |
| | 307.67 |
| | 328.76 |
| | 262.08 |
|
|
| | | | | | | | | | | |
| | 12/13 | | 12/14 | | 12/15 | | 12/16 | | 12/17 | | 12/18 |
| Alexion Pharmaceuticals, Inc. | 100.00 | | 139.24 | | 143.55 | | 92.07 | | 90.00 | | 73.27 |
| Nasdaq Composite | 100.00 | | 114.62 | | 122.81 | | 133.19 | | 172.11 | | 165.84 |
| Nasdaq Biotechnology | 100.00 | | 131.71 | | 140.56 | | 112.25 | | 133.67 | | 121.24 |
| |
| Item 6. | SELECTED FINANCIAL DATA. DATA. |
(amounts in millions, except per share amounts)
The following selected financial data is derived from, and should be read in conjunction with, the financial statements,Consolidated Financial Statements, including the notes thereto, and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report on Form 10-K.
(amounts in millions, except per share amounts)
| | Consolidated Statements of Operations Data: | | | Year Ended December 31, | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 | | 2013 | | 2012 | 2018 |
| 2017 |
| 2016 |
| 2015 |
| 2014 |
Net product sales (1) | $ | 3,082 |
| | $ | 2,603 |
| | $ | 2,234 |
| | $ | 1,551 |
| | $ | 1,134 |
| $ | 4,130.1 |
| | $ | 3,549.5 |
| | $ | 3,081.7 |
| | $ | 2,602.5 |
| | $ | 2,233.7 |
|
| Other revenue | 2 |
| | 1 |
| | — |
| | — |
| | — |
| 1.1 |
| | 1.6 |
| | 2.4 |
| | 1.5 |
| | — |
|
| Total revenues | 3,084 |
| | 2,604 |
| | 2,234 |
| | 1,551 |
| | 1,134 |
| 4,131.2 |
| | 3,551.1 |
| | 3,084.1 |
| | 2,604.0 |
| | 2,233.7 |
|
| Cost of sales: | | | | | | | | | | |
| Cost of sales | 258 |
| | 233 |
| | 174 |
| | 168 |
| | 126 |
| |
| Change in contingent liability from intellectual property settlements | — |
| | — |
| | — |
| | 9 |
| | (53 | ) | |
| Total cost of sales | 258 |
| | 233 |
| | 174 |
| | 177 |
| | 73 |
| |
Cost of sales (2) | | 374.3 |
| | 454.2 |
| | 258.3 |
| | 233.1 |
| | 173.9 |
|
| Operating expenses: | | | | | | | | | | | | | | | | | | |
| Research and development | 757 |
| | 709 |
| | 514 |
| | 317 |
| | 223 |
| 730.4 |
| | 878.4 |
| | 757.2 |
| | 709.5 |
| | 513.8 |
|
| Selling, general and administrative | 954 |
| | 863 |
| | 630 |
| | 490 |
| | 385 |
| 1,111.8 |
| | 1,094.4 |
| | 953.0 |
| | 862.6 |
| | 630.2 |
|
Amortization of purchased intangible assets (2) | 322 |
| | 117 |
| | — |
| | — |
| | — |
| |
Acquired in-process research and development (3) | | 1,183.0 |
| | — |
| | — |
| | — |
| | — |
|
Amortization of purchased intangible assets (4) | | 320.1 |
| | 320.1 |
| | 322.2 |
| | 116.6 |
| | — |
|
| Change in fair value of contingent consideration | 36 |
| | 64 |
| | 20 |
| | 4 |
| | 7 |
| 116.5 |
| | 41.0 |
| | 35.7 |
| | 64.2 |
| | 20.3 |
|
| Acquisition-related costs | 2 |
| | 39 |
| | — |
| | 1 |
| | 16 |
| — |
| | — |
| | 2.3 |
| | 39.2 |
| | — |
|
| Restructuring expenses | 3 |
| | 42 |
| | 15 |
| | — |
| | — |
| |
Restructuring expenses (2) | | 25.5 |
| | 104.6 |
| | 3.0 |
| | 42.1 |
| | 15.3 |
|
| Impairment of intangible assets | 85 |
| | — |
| | 12 |
| | 34 |
| | 26 |
| — |
| | 31.0 |
| | 85.0 |
| | — |
| | 11.5 |
|
| Total operating expenses | 2,159 |
| | 1,834 |
| | 1,191 |
| | 846 |
| | 657 |
| 3,487.3 |
| | 2,469.5 |
| | 2,158.4 |
| | 1,834.2 |
| | 1,191.1 |
|
| Operating income | 667 |
| | 537 |
| | 869 |
| | 528 |
| | 404 |
| 269.6 |
| | 627.4 |
| | 667.4 |
| | 536.7 |
| | 868.7 |
|
| Other income (expense) | (91 | ) | | (39 | ) | | 3 |
| | (2 | ) | | (6 | ) | |
Other (expense) income (5) | | (27.4 | ) | | (79.6 | ) | | (91.2 | ) | | (38.6 | ) | | 3.4 |
|
| Income before income taxes | 576 |
| | 498 |
| | 872 |
| | 526 |
| | 398 |
| 242.2 |
| | 547.8 |
| | 576.2 |
| | 498.1 |
| | 872.1 |
|
Income tax expense (3) (4) | 177 |
| | 354 |
| | 215 |
| | 273 |
| | 143 |
| |
Income tax expense (6) (7) (8) | | 164.6 |
| | 104.5 |
| | 176.8 |
| | 353.7 |
| | 215.2 |
|
| Net income | $ | 399 |
| | $ | 144 |
| | $ | 657 |
| | $ | 253 |
| | $ | 255 |
| $ | 77.6 |
| | $ | 443.3 |
| | $ | 399.4 |
| | $ | 144.4 |
| | $ | 656.9 |
|
| Earnings per common share | | | | | | | | | | | | | | | | | | |
| Basic | $ | 1.78 |
| | $ | 0.68 |
| | $ | 3.32 |
| | $ | 1.29 |
| | $ | 1.34 |
| $ | 0.35 |
| | $ | 1.98 |
| | $ | 1.78 |
| | $ | 0.68 |
| | $ | 3.32 |
|
| Diluted | $ | 1.76 |
| | $ | 0.67 |
| | $ | 3.26 |
| | $ | 1.27 |
| | $ | 1.28 |
| $ | 0.35 |
| | $ | 1.97 |
| | $ | 1.76 |
| | $ | 0.67 |
| | $ | 3.26 |
|
| Shares used in computing earnings per common share | | | | | | | | | | | | | | | | | | |
| Basic | 224 |
| | 213 |
| | 198 |
| | 196 |
| | 190 |
| 222.7 |
| | 223.9 |
| | 224.3 |
| | 213.4 |
| | 198.1 |
|
| Diluted | 227 |
| | 216 |
| | 202 |
| | 200 |
| | 199 |
| 224.5 |
| | 225.4 |
| | 226.3 |
| | 215.9 |
| | 201.6 |
|
| | Consolidated Balance Sheet Data: | | | As of December 31, | As of December 31, |
| | 2016 | | 2015 | | 2014 | | 2013 | | 2012 | 2018 | | 2017 | | 2016 | | 2015 | | 2014 |
| Cash, cash equivalents and marketable securities | $ | 1,293 |
| | $ | 1,385 |
| | $ | 1,962 |
| | $ | 1,515 |
| | $ | 990 |
| $ | 1,563.8 |
| | $ | 1,474.1 |
| | $ | 1,293.4 |
| | $ | 1,385.0 |
| | $ | 1,961.6 |
|
Total assets (5)(9) | 13,253 |
| | 13,097 |
| | 4,202 |
| | 3,318 |
| | 2,614 |
| 13,931.9 |
| | 13,583.3 |
| | 13,253.3 |
| | 13,097.9 |
| | 4,202.0 |
|
Long-term debt (current and noncurrent) (6) | 3,055 |
| | 3,420 |
| | 58 |
| | 113 |
| | 149 |
| |
Long-term debt (current and noncurrent) (10) | | 2,595.5 |
| | 2,888.1 |
| | 3,055.1 |
| | 3,420.9 |
| | 57.5 |
|
| Contingent consideration (current and noncurrent) | 153 |
| | 177 |
| | 163 |
| | 143 |
| | 142 |
| 280.8 |
| | 168.9 |
| | 152.9 |
| | 177.2 |
| | 163.0 |
|
| Facility lease obligation (current and noncurrent) | 243 |
| | 151 |
| | 107 |
| | 32 |
| | — |
| 372.2 |
| | 353.3 |
| | 243.4 |
| | 151.3 |
| | 107.1 |
|
Total stockholders’ equity (7) | 8,694 |
| | 8,259 |
| | 3,303 |
| | 2,383 |
| | 1,971 |
| |
Total stockholders’ equity (11) | | 9,165.3 |
| | 8,893.1 |
| | 8,693.8 |
| | 8,258.6 |
| | 3,302.0 |
|
In addition to the following notes, see “Item 7. Management’s“Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the Consolidated Financial Statements and accompanying notes and previously filed Annual Reports on Form 10-K for further information regarding our consolidated results of operations and financial position for periods reported therein.
(1) In March 2014, we entered into an agreement with the French government which positively impacted prospective reimbursement of SolirisSOLIRIS and also provided for reimbursement for shipments made in years prior to January 1, 2014. As a result of the agreement, in 2014 we recognized $88$87.8 of net product sales from SolirisSOLIRIS in France relating to years prior to January 1, 2014.
(2) In 2017, we committed to an operational plan to re-align the global organization with its refocused corporate strategy. As a result of this re-alignment, in 2017, we recorded additional asset related charges of $152.1 associated with the planned closure of the ARIMF facility to cost of sales (which facility was subsequently sold in 2018). These charges primarily relate to accelerated depreciation and the impairment of manufacturing assets. Additionally, the re-alignment in 2017 resulted in restructuring expenses of $104.6, primarily related to employee separation costs.
(3) In the second quarter 2018, we completed the acquisition of Wilson Therapeutics AB (publ). We acquired in-process research and development related to WTX101, an early Phase III asset in development for the treatment of Wilson Disease. Due to the stage of development of this asset, the value of this asset of $803.7 was expensed during 2018. In the fourth quarter of 2018 we completed the acquisition of Syntimmune, Inc. We acquired in-process research and development related to SYNT001, which is in Phase 1b/2a trials and in development for the treatment of Immunoglobulin G and IgG-mediated autoimmune diseases. Due to the stage of development of this asset, the value of this asset of $379.3 was expensed during 2018.
(4) In the third quarter 2015, we received regulatory approval for StrensiqSTRENSIQ and Kanuma.KANUMA. As a result, we began amortizing intangible assets associated with StrensiqSTRENSIQ and Kanuma.KANUMA.
(3)(5) We recognized an unrealized gain of $44.4 on our Moderna Therapeutics equity investment in 2018. Additionally, in 2016, we incurred a full year of interest expense on our credit facility entered into in 2015.
(6) We recognized tax (benefit) expense of $(56.5) and $45.8 in 2018 and 2017, respectively, as a result of the Tax Cuts and Jobs Act. In 2017, we recorded certain impacts of the Tax Act on a provisional basis. As of December 22, 2018, our accounting for the impact of the Tax Act was complete. See Note 12, “Income Taxes” for additional information.
(7) In 2016, we recognized deferred tax expense of $119.3 associated with the distribution of earnings from our captive foreign partnership.
(8) In connection with the integration of the Synageva business with and into the Alexion business, we incurred a one-time tax expense of $316$315.6 in the third quarter 2015. This tax expense is attributable to the change in our deferred tax liability for the outside basis difference resulting from the movement of assets into our captive foreign partnership.
(4)(9) In 2013, we recognized tax expense of approximately $96 resulting from the centralization of our global supply chain and technical operations2015, in Ireland.
(5) In connection with the acquisition of Synageva, we acquired $4,236$4,236.0 of intangible assets and $4,783$4,783.4 of goodwill.
(6)(10) In 2015, in connection with the acquisition of Synageva, we borrowed $3,500$3,500.0 under our term loan under a new credit facility. This credit facility was amended and restated in June 2018.
(7)(11) In 2015, in connection with the acquisition of Synageva, we issued $4,918$4,917.8 of common stock to former Synageva stockholders.
| |
| Item 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.(amounts in millions, except percentages and per share data) |
(amounts in millions, except percentages and per share data)
In addition to historical information, this report contains forward-looking statements that involve risks and uncertainties which may cause our actual results to differ materially from plans and results discussed in forward-looking statements. We encourage you to review the risks and uncertainties, discussed in the section entitled item 1A “Risk Factors”, and the “Note Regarding Forward-Looking Statements”, included at the beginning of this Annual Report on Form 10-K. The risks and uncertainties can cause actual results to differ significantly from those forecast in forward-looking statements or implied in historical results and trends.
The following discussion should be read in conjunction with our consolidated financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K.
Overview
We areAlexion is a global biopharmaceutical company focused on serving patients with devastating and ultra-rare disordersfamilies affected by rare diseases through the innovation, development and commercialization of life-transforminglife-changing therapies.
We are the global leader in complement inhibition and have developed and commercialize the only two approved complement inhibitors to treat patients with paroxysmal nocturnal hemoglobinuria (PNH), as well as the first and only approved complement inhibitor to treat atypical hemolytic uremic syndrome (aHUS) and anti-acetylcholine receptor (AchR) antibody-positive generalized myasthenia gravis (gMG). In addition, Alexion has two highly innovative enzyme replacement therapies for patients with life-threatening and ultra-rare metabolic disorders, hypophosphatasia (HPP) and lysosomal acid lipase deficiency (LAL-D).
As the leader in complement biology for over 20 years, Alexion focuses its research efforts on novel molecules and targets in the complement cascade, and its development efforts on the core therapeutic products.areas of hematology, nephrology, neurology, and metabolic disorders.
RecentDevelopments
In our complement franchise, Soliristhe fourth quarter 2018, we completed the acquisition of Syntimmune, Inc. (Syntimmune), a clinical-stage biotechnology company developing an antibody therapy targeting the neonatal Fc receptor (FcRn). The lead candidate from this acquisition, ALXN1830 (SYNT001), is a monoclonal antibody that inhibits the interaction of FcRn with Immunoglobulin G (IgG) and IgG immune complexes, and is being studied in Phase 1b/2a trials for the treatment of IgG-mediated autoimmune diseases. Under the terms of the agreement, Alexion
acquired Syntimmune for an upfront payment of $400.0, with the potential for additional milestone-dependent payments of up to $800.0, for a total value of up to $1,200.0.
In December 2018 ULTOMIRIS™ was approved by the FDA as a new treatment option for adult patients living with paroxysmal nocturnal hemoglobinuria (PNH). ULTOMIRIS is the first and only therapeutic approvedlong-acting C5 inhibitor that provides immediate and complete inhibition for eight weeks.
In January 2019, we submitted our filings to the FDA and the EU for marketing clearance for SOLIRIS as a potential treatment of NMOSD.
On January 21, 2019, the Opposition Division of the European Patent Office determined, following multi-party opposition proceedings, to revoke our European patent No. 2359834, which relates to the formulation of SOLIRIS. Subject to our review of the final written decision of the European Patent Office, we currently expect that we will appeal this decision. While any appeal is pending at the European Patent Office, the claims in the originally granted patent remain in force.
In January 2019, we announced the results of the Phase III study with ULTOMIRIS (ALXN1210) meeting its primary objective in complement inhibitor-naïve patients with either PNH,aHUS. In the initial 26 week treatment period in this study, 53.6 percent of patients demonstrated complete thrombotic microangiopathy (TMA) response.
In January 2019, we entered into a life-threatening and ultra-rare genetic blood disorder, or aHUS, a life-threatening and ultra-rare genetic disease. PNH and aHUS result from chronic uncontrolled activationcollaboration agreement with Caelum Biosciences (Caelum) to develop CAEL101 for light chain (AL) amyloidosis. Under the terms of the complement componentagreement, we acquired a minority equity interest in Caelum and an exclusive option to acquire the remaining equity in the company based on Phase II data, for pre-negotiated economics. We made an upfront payment of $30.0 and could be required to pay up to an additional $30.0 in contingent milestone-dependent option fees. The collaboration also provides for potential additional payments, in the immune system.
In our metabolic franchise, we commercialize Strensiqevent Alexion exercises the acquisition option, for the treatment of patients with HPPup to $500.0, which includes an upfront option exercise payment and Kanuma for the treatment of patients with LAL-D. HPP is an ultra-rare genetic disease characterized by defective bone mineralization that can lead to deformity of bonespotential regulatory and other skeletal abnormalities. LAL-D is a serious, life threatening ultra-rare disease in which geneticcommercial milestone payments.
mutations result in decreased activity of the LAL enzyme leading to marked accumulation of lipids in vital organs, blood vessels and other tissues.
We are also evaluating additional potential indications for eculizumab in other severe and devastating diseases in which uncontrolled complement activation is the underlying mechanism, and we are progressing in various stages of development with additional product candidates as potential treatments for patients with devastating and ultra-rare disorders.
Recent Developments
As previously reported, the Audit and Finance Committee of the Company’s Board of Directors (Audit Committee) commenced an investigation of allegations made by a former employee concerning the Company’s Soliris sales practices. The former employee alleged that certain of such practices resulted in certain customers placing orders for shipments of Soliris in an earlier fiscal quarter than the fiscal quarter they otherwise would have (referred to here as pull-in or advanced sales, and more fully described below). The former employee alleged that such practices were used by the Company in order to meet certain financial targets and at times involved inappropriate business conduct. The Audit Committee conducted its investigation (Audit Committee Investigation) with the assistance of outside counsel, forensic accountants and other accounting firm advisors. The Audit Committee Investigation is substantially complete, and no further investigative procedures are currently planned except as necessary to respond to regulatory inquiries, if any, or because of matters that arise in the ordinary course of the Company’s future business activities.
The Audit Committee concluded, based on the facts of the investigation that the Company’s previously issued financial results do not require restatement. In addition, the Audit Committee Investigation did not identify any instances of improper revenue recognition associated with pull-in sales, instances where Soliris orders were not placed by customers for patients in order to fulfill an actual need, or instances where Soliris was sold to build stock of unwanted product. However, management concluded and the Audit Committee concurred that there was a material weakness in the Company’s internal control over financial reporting because we did not maintain an effective control environment as senior management failed to set an appropriate “Tone at the Top.” Specifically, senior management failed to reinforce the need for compliance with the Company’s policies and procedures, which resulted in inappropriate business conduct. The Audit Committee Investigation found that senior management applied pressure on personnel to use pull-in sales to meet targets, and such pressure was particularly significant during the fourth quarter of 2015. The Audit Committee Investigation also found that certain Company personnel engaged in inappropriate business conduct to realize pull-in sales, as a result of pressure from senior management.
For purposes of this Annual Report on Form 10-K, “pull-in” or “advanced” sales are certain Soliris sales transactions, coordinated by Company personnel (primarily personnel in the customer operations department in their capacity as coordinators for the shipment of orders for customers) that increase revenue recognized in an earlier fiscal quarter than the one in which a sale otherwise would have occurred and result in a corresponding decrease in the revenue that will be recognized in the subsequent fiscal quarter. The Company is able to forecast the estimated date of certain shipments of Soliris due to customer order history, known infusion dates, or other similar data to support the operations of our business and patient needs. Pull-in sales may occur, for example, when a customer, as a result of encouragement by a Company employee, places an order for a patient earlier than the customer might otherwise place the order. Pull-in sales are not inherently problematic or impermissible, when in accordance with U.S. GAAP. The Audit Committee Investigation included a review of sales transactions for evidence of pull-in sales, the reasons for pull-in sales, whether such transactions were conducted in accordance with the Company’s policies and procedures, and whether revenue from pull-in sales was properly recognized in accordance with U.S. GAAP.
The Audit Committee Investigation concluded that revenue from the pull-in sales under review was appropriately recognized in the quarter in which such sales actually occurred and that there were no financial statement errors related to the pull-in sales. However, the Audit Committee Investigation found that certain revenue pulled into the fourth quarter of 2015 from the first quarter of 2016 was realized as the result of employee actions that involved inappropriate business conduct, including conduct that was inconsistent with, and in violation of Company policies and procedures. Pull-in sales during the fourth quarter of 2015 were estimated to be between approximately $10 to $17 and were significantly higher than for other quarters. Some portion of these estimated sales did not involve inappropriate business conduct. These estimated pull-in sales represented less than 1% of total revenue for 2015.
During the past two completed fiscal years and through the fourth quarter of 2016, but excluding the fourth quarter of 2015, pull-in sales were estimated to be between $1 to $7 in the aggregate, representing 0% - 1% of total revenue.
Although pull-in sales are not inherently problematic or impermissible, they must not be realized through violations of the Company’s policies and procedures and must be in accordance with U.S. GAAP. The conclusions concerning the material weakness in the Company’s internal controls over financial reporting are discussed further under Item 9A “Controls and Procedures” in this Form 10-K.
Critical Accounting Policies and the Use of Estimates
The significant accounting policies and basis of preparation of our consolidated financial statements are described in Note 1, “Business Overview and Summary of Significant Accounting Policies” of the Consolidated Financial Statements included in this Annual Report on Form 10-K. Under accounting principles generally accepted in the U.S., we are required to make estimates, judgments and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses and disclosure of contingent assets and liabilities in our financial statements. Actual results could differ from those estimates.estimates and such differences may be material.
We believe the judgments, estimates and assumptions associated with the following critical accounting policies have the greatest potential impact on our consolidated financial statements:
Revenue recognition;
Contingent liabilities;
Inventories;
Share-based compensation;
Valuation of goodwill, acquired intangible assets and in-process research and development (IPR&D);
Valuation of contingent consideration; and
Income taxes.
Revenue Recognition
Net Product SalesIn May 2014, the Financial Accounting Standards Board (FASB) issued a comprehensive new standard which amends revenue recognition principles. We adopted the new standard on January 1, 2018 by applying the modified retrospective method to all contracts that were not completed as of that date. Under the new guidance, revenue is recognized when a customer obtains control of promised goods or services, in an amount that reflects the consideration expected to be received in exchange for those goods or services. Revenue is recognized through a five-step process: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) a performance obligation is satisfied. The Company only applies the five-step model to contracts when it is probable that the Company will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer. At contract inception, the Company assesses the goods or services promised within each contract, and determines those that are performance obligations. Revenue is recognized for the applicable performance element when each distinct performance obligation is satisfied.
While results for reporting periods beginning after January 1, 2018 are presented under the new guidance,
prior period amounts are not adjusted and continue to be reported under the accounting standards in effect for the prior period. The accounting policy for revenue recognition for periods prior to January 1, 2018 is described in Note 1 of the Notes to the Consolidated Financial Statements included in our Annual Report on Form 10-K for the year ended December 31, 2017.
Nature of Products
Our principal source of revenue is product sales. WeOur contracts with customers generally contain a single performance obligation and we recognize revenue from product sales when persuasive evidencewe have satisfied our performance obligation by transferring control of an arrangement exists, titlethe product to our customers. Control of the product and associated risk of loss has passedgenerally transfers to the customer the price is fixed or determinable, collection from the customer is reasonably assured,upon delivery. In certain countries, we sell to distributors on a consignment basis and we have no further performance obligations. Depending on these criteria,record revenue is usually recorded upon receiptwhen control of the product bytransfers to the customer upon sale to the end customer, which is typically a hospital, physician’s office, private or government pharmacy or other healthcare facility. On a regular basis, we review revenue arrangements, such as distributor relationships, to determine whether changes in these criteria have an impact on revenue recognition. Amounts collected from customers and remitted to governmental authorities, such as value-added taxes (VAT) in foreign jurisdictions, are presented on a net basis in our consolidated statements of operations and do not impact net product sales.user.
Our customers are primarily comprised of distributors, pharmacies, hospitals, hospital buying groups, and other healthcare providers. In some cases, we may also sell product to governments and government agencies.
Because of factors such as the price of our products, the limited number of patients, the short period from product sale to patient infusion and the lack of contractual return rights, customers often carry limited inventory. We also monitor inventory within our sales channels to determine whether deferrals are appropriate based on factors such as inventory levels compared to demand, contractual terms, financial strength of distributors and our ability to estimate returns. In certain countries, exact quantities of inventory in the channel are not precisely known, requiring us to estimate these amounts. If actual amounts of inventory differ from these estimates, these adjustments could have an impact in the period in which these estimates change.
In addition to sales in countries where product isour products are commercially available, we have also recorded revenue on sales for patients receiving treatment through named-patient programs. The relevant authorities or institutions in those countries have agreed to reimburse for product sold on a named-patient basis where product hasour products have not received final approval for commercial sale.
We record estimated rebates payable under governmental programs, including Medicaid in the U.S. and other programs outside the U.S., as a reduction of revenueRevenue is recognized at the time of product sale. Our calculations relatedamount to these rebate accruals require analysis of historical claim patterns and estimates of customer mixwhich we expect to determine which sales will be subject to rebates and the amount of such rebates. We update our estimates and assumptions each period and record any necessary adjustments, which may have an impact on revenueentitled in the period in which the adjustment is made. Generally, the length of time between product sale and the processing and reporting of the rebates is three to six months.
We have entered into volume-based arrangements with governments in certain countries in which reimbursement is limited to a contractual amount. Under this type of arrangement, amounts billed in excess of the contractual limitation are repaid to these governments as a rebate. We estimate incremental discounts resulting from these contractual limitations, based on estimated sales during the limitation period, and we apply the discount percentage to product shipments as a reduction of revenue. Our calculations related to these arrangements require estimation of sales during the limitation period, and adjustments in these estimates may have an impact in the period in which these estimates change.
We have provided balances and activity in the rebates payable accountexchange for the years ended December 31, 2016, 2015sale of our products. This amount includes both fixed and 2014variable consideration and excludes amounts that are collected from customers and remitted to governmental authorities, such as follows:
|
| | | |
| | Rebates Payable |
| Balance at December 31, 2013 | $ | 124 |
|
| Current provisions relating to sales in current year | 63 |
|
| Adjustments relating to prior years | (87 | ) |
| Payments/credits relating to sales in current year | (34 | ) |
| Payments/credits relating to sales in prior years | (29 | ) |
| Balance at December 31, 2014 | $ | 37 |
|
| Current provisions relating to sales in current year | 90 |
|
| Adjustments relating to prior years | (2 | ) |
| Payments/credits relating to sales in current year | (43 | ) |
| Payments/credits relating to sales in prior years | (26 | ) |
| Balance at December 31, 2015 | $ | 56 |
|
| Current provisions relating to sales in current year | 115 |
|
| Adjustments relating to prior years | (2 | ) |
| Payments/credits relating to sales in current year | (50 | ) |
| Payments/credits relating to sales in prior years | (49 | ) |
| Balance at December 31, 2016 | $ | 70 |
|
In 2016 compared to 2015, current provisions relating to salesvalue-added taxes in the current year increased by $25 primarily due to increased unit volumes in the U.S.foreign jurisdictions. Shipping and Europe which were subject to rebates.
In 2015 compared to 2014, current provisions relating to sales in the current year increased by $27 primarily due to increased unit volumes in the U.S. and Europe which were subject to rebates.
In March 2014, we entered into an agreementhandling costs associated with the French government which positively impacts prospective reimbursementoutbound freight after control of Soliris and also provides for reimbursement for shipments in years prior to January 1, 2014. As a result of this agreement, in the first quarter 2014, we reduced the rebate payable and recognized $88 of net product sales from Soliris in France relating to years prior to January 1, 2014.
We record distribution and other fees paidhas transferred to our customers are accounted for as a reductionfulfillment cost and are included in operating expenses. The cost for any shipping and handling activities (including customs clearance activities) associated with transactions for which revenue has been recognized are accrued if not completed before the respective period end.
The timing between the recognition of revenue unless we receive an identifiablefor product sales and separate benefit for the considerationreceipt of payment is not significant. Our standard credit terms, which vary based on the country of sale, generally range from 30 to 120 days and we can reasonably estimate the fair valueall arrangements generally are payable within one year of the benefit received. If both conditions are met, we recordtransfer of the consideration paidproduct. We do not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between the transfer of the promised good to the customer as an operating expense. These costs are typically known at the timeand receipt of sale, resulting in minimal adjustments subsequent to the period of sale.payment will be one year or less.
We enter into foreign exchange forward contracts to hedge exposures resulting from portions of our forecasted revenues, including intercompany revenues, that are denominated in currencies other than the U.S. dollar. These hedges are designated as cash flow hedges upon inception. We record the effective portion of these cash flow hedges to revenue in the period in which the sale is made to an unrelated third party and the derivative contract is settled.
We evaluate the creditworthiness of customers on a regular basis. In certain European countries, sales by us are subject to payment terms that are statutorily determined. This is primarily the case in countries where the payer is government-owned or government-funded, which we consider to be creditworthy. The length of time from sale to receipt of payment in certain countries exceeds our credit terms. In countries in which collections from customers extend beyond normal payment terms, we seek to collect interest. We record interest on customer receivables as interest income when collected. For non-interest bearing receivables with an estimated payment beyond one year, we discount the accounts receivable to present value at the date of sale, with a corresponding adjustment to revenue. Subsequent adjustments for further declines in credit rating are recorded as bad debt expense as a component of selling, general and administrative expense. We also use judgments as to our ability to collect outstanding receivables and provide allowances for the portion of receivables if and when collection becomes doubtful, and we also assess on an ongoing basis whether collectibility is reasonably assuredprobable at the time of sale. As of December 31, 2018 and December 31, 2017, allowances on receivables were not material.
Variable Consideration
We pay distribution fees to our distributors and offer rebates and/or discounts, or enter into volume-based reimbursement arrangements with certain customers. We reduce the transaction price on our sales for these amounts. For variable amounts, we estimate the amount of consideration to which we expect to be entitled based on all available historic, current and forecast information. We primarily use the expected value method to estimate variable payments and, in limited circumstances, will apply the most likely method based on the type of variable consideration and what method better predicts the amount of consideration we expect to be entitled to. Consideration that is received from a customer that we expect will need to be refunded in the future is recorded as a refund liability to the customer within accrued expenses. Actual amounts of consideration ultimately received or refunded may differ from our estimates, and such difference may be material. If actual results in the future vary from our estimates, we adjust these estimates, which would affect net product sales and earnings in the period such variances become known, and such variances may be material.
Variability in the transaction price for our products pursuant to our contracts with customers primarily arises from the following:
Discounts and Rebates: We offer discounts and rebates to certain distributors and customers under our arrangements. In many cases, these amounts are fixed at the time of sale and the transaction price is reduced accordingly. We also provide for rebates under certain governmental programs, including Medicaid in the U.S. and other programs outside the U.S., which are payable based on actual claim data. We estimate these rebates based on an analysis of historical claim patterns and estimates of customer mix to determine which sales will be subject to rebates and the amount of such rebates. We update our estimates and assumptions each period and record any necessary adjustments, which may have
an impact on revenue in the period in which the adjustment is made (and such impact may be material). Generally, the length of time between product sale and the processing and reporting of the rebates is three to six months.
Volume-Based Arrangements: We have entered into volume-based arrangements with governments in certain countries and other customers in which reimbursement is limited to a contractual amount. Under this type of arrangement, amounts billed in excess of the contractual limitation are repaid to the customer as a rebate. We estimate incremental discounts resulting from these contractual limitations, based on forecasted sales during the limitation period, and we apply the discount percentage to product shipments as a reduction of revenue. Our calculations related to these arrangements require estimation of sales during the limitation period, and adjustments in these estimates may have a material impact in the period in which these estimates change.
We have provided balances and activity in the rebates payable account for the years ended December 31, 2018, 2017 and 2016 as follows:
|
| | | |
| | Rebates Payable |
| Balances, December 31, 2015 | $ | 55.6 |
|
| Current provisions relating to sales in current year | 114.6 |
|
| Adjustments relating to prior years | (1.7 | ) |
| Payments/credits relating to sales in current year | (50.3 | ) |
| Payments/credits relating to sales in prior years | (48.7 | ) |
| Balances, December 31, 2016 | $ | 69.5 |
|
| Current provisions relating to sales in current year | 193.8 |
|
| Adjustments relating to prior years | (4.5 | ) |
| Payments/credits relating to sales in current year | (97.4 | ) |
| Payments/credits relating to sales in prior years | (62.3 | ) |
| Balances, December 31, 2017 | $ | 99.1 |
|
| Current provisions relating to sales in current year | 235.4 |
|
| Adjustments relating to prior years | (2.4 | ) |
| Payments/credits relating to sales in current year | (119.3 | ) |
| Payments/credits relating to sales in prior years | (90.0 | ) |
| Balances, December 31, 2018 | $ | 122.8 |
|
Current provisions relating to sales in the current year increased by $41.6 in 2018 compared to 2017 and $79.2 in 2017 compared to 2016. The increase in 2018 was primarily due to increased unit volumes in the U.S. which were subject to rebates as well as increases in rebate rates in the U.S. on certain product sales. The increase in 2017 was attributable to increased unit volumes in the U.S. and Europe, which were subject to rebates, as well as to increases in rebate rates in certain geographical regions and on certain product sales as compared to the prior year.
Distribution & Other Fees: We pay distribution and other fees to certain customers in connection with the sales of our products. We record distribution and other
fees paid to our customers as a reduction of revenue, unless the payment is for a distinct good or service from the customer and we can reasonably estimate the fair value of the goods or services received. If both conditions are met, we record the consideration paid to the customer as an operating expense. These costs are typically known at the time of sale, resulting in minimal adjustments subsequent to the period of sale.
Product Returns: Our contracts with customers generally provide for returns only if the product is damaged or defective upon delivery. We assess our sales transactions and arrangements with customers and monitor inventory within our sales channels to determine whether a provision for returns is warranted and a resulting adjustment to the transaction price is necessary. This assessment is based on historical experience and assumptions as of the date of sale and changes in these estimates could have an impact in the period in which the change occurs (and such impact may be material). Because of factors such as the price of our products, the limited number of patients, the short period from product sale to patient infusion and limited contractual return rights, our customers often carry limited inventory.
The amount of variable consideration included in the transaction price is constrained by the amount that is probable will not result in a significant reversal of revenue. We consider our experience with similar transactions and expectations regarding the contract in estimating the amount of variable consideration to which we expect to be entitled, and determining whether the estimated variable consideration should be constrained. We do not have any material constraints on the variable consideration included within the transaction price of our current revenue arrangements.
We continue to monitor economic conditions, including volatility associated with international economies and the associated impacts on the financial markets and our business. For additional information related to our concentration of credit risk associated with certain international accounts receivable balances, refer to the “Financial Condition, Liquidity and Capital Resources” and “Quantitative and Qualitative Disclosures About Market Risk” sectionsections below.
Contingent liabilities
We are currently involved in various claims and legal proceedings. On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. If the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Because of uncertainties related to claims and litigation, accruals are based on our best estimates based on available information. On a periodic basis, as additional information becomes available, or based on specific events such as the outcome of litigation or settlement of claims (and our offers of settlement), we may reassess the potential liability related to these matters and may revise these estimates, which could result in a material adjustment to our operating results and liquidity.
Inventories
Inventories are stated at the lower of cost or estimatednet realizable value. We determine the cost of inventory onCost is determined in a standard cost basis, whichmanner that approximates average costs.
We capitalize inventory produced for commercial sale, which may include costs incurred for certain products awaiting regulatory approval. We capitalize inventory produced in preparation of product launches sufficient to support estimated initial market demand. Capitalization of such inventory begins when we have (i) obtained positive results in clinical trials that we believe are necessary to support regulatory approval, (ii) concluded that uncertainties regarding regulatory approval have been sufficiently reduced, and (iii) determined that the inventory has probable future economic benefit. In evaluating whether these conditions have been met, we consider clinical trial results for the underlying product candidate, results from meetings with regulatory authorities, and the compilation of the regulatory application. If we are aware of any material risks or contingencies outside of the standard regulatory review and approval process, or if there are any specific negative issues identified relating to the safety, efficacy, manufacturing, marketing or labeling of the product that would have a significant negative impact on its future economic benefits, the related inventory would not be capitalized.
Products that have been approved by the FDA or other regulatory authorities are also used in clinical programs to assess the safety and efficacy of the products for usage in diseases that have not been approved by the FDA or other regulatory authorities. The form of product utilized for both commercial and clinical programs is identical and, as a result, the inventory has an “alternative future use” as defined in authoritative guidance. Raw materials and purchased drug product associated with clinical development programs are included in inventory and charged to research and development expense when the product enters the
research and development process and no longer can be used for commercial purposes and, therefore, does not have an “alternative future use”.
For products which are under development and have not yet been approved by regulatory authorities, purchased drug product is charged to research and development expense upon delivery. Delivery occurs when the inventory passes quality inspection and ownership transfers to us. Nonrefundable advance payments for research and development activities, including production of purchased drug product, are deferred and capitalized until the goods are delivered. We also recognize expense for raw materials purchased for developmental purposes when the raw materials pass quality inspection, and we have an obligation to pay for the materials.
We analyze our inventory levels to identify inventory that may expire prior to sale, inventory that has a cost basis in excess of its estimated realizable value, or inventory in excess of expected sales requirements. Although the manufacturing of our product is subject to strict quality control, certain batches or units of product may no longer meet quality specifications or may expire, which would require adjustments to our inventory values. We also apply judgment related to the results of quality tests that we perform throughout the production process, as well as our understanding of regulatory guidelines, to determine if it is probable that inventory will be saleable. These quality tests are performed throughout the pre- and post-production process, and we continually gather information regarding product quality for periods after the manufacturing date. Our products currently have a maximum estimated life range of 36 to 48 months and, based on our sales forecasts, we expect to realize the carrying value of the product inventory. In the future, reduced demand, quality issues or excess supply beyond those anticipated by management may result in a material adjustment to inventory levels, which would be recorded as an increase to cost of sales.
The determination of whether or not inventory costs will be realizable requires estimates by our management. A critical input in this determination is future expected inventory requirements based on internal sales forecasts. We then compare these requirements to the expiry dates of inventory on hand. For inventories that are capitalized in preparation of product launch, we also consider the expected approval date in assessing realizability. To the extent that inventory is expected to expire prior to being sold, we will write down the value of inventory. If actual results differ from those estimates, additional inventory write-offs may be required.required, and such write-offs may be material.
Share-Based Compensation
We have two share-based compensation plans pursuant to which awards are currently being made: (i)
the Amended and Restated 20042017 Incentive Plan (2004(2017 Plan) and (ii) the 2015 Employee Stock Purchase Plan (ESPP). The 2017 Plan replaced the Amended & Restated 2004 Incentive Plan, effective May 10, 2017. Under the 20042017 Plan, restricted stock, restricted stock units, stock options and other stock-related awards may be granted to our directors, officers,
employees and consultants or advisors of the Company or any subsidiary. Under the ESPP, eligible employees can purchase shares of common stock at a discount semi-annually through payroll deductions. To date, share-based compensation issued under the plans consists of incentive and non-qualified stock options, restricted stock and restricted stock units, including restricted stock units with market and non-market performance conditions, and shares issued under our ESPP. Stock-related awards are also outstanding under other share-based compensation plans, but we have not granted awards under these plans since 2004.
Compensation expense for our share-based awards is recognized based on the estimated fair value of the awards on the grant date. Compensation expense reflects an estimate of the number of awards expected to vest and is primarily recognized on a straight-line basis over the requisite service period of the individual grants, which typically equals the vesting period. Compensation expense for awards with performance conditions is recognized using the graded-vesting method.
Our estimates of employee stock option values rely on estimates of factors we input into the Black-Scholes model. The key factors involve an estimate of future uncertain events. Significant assumptions include the use of historical volatility to determine the expected stock price volatility. We also estimate expected term until exercise and the reduction in the expense from expected forfeitures. We currently use historical exercise and cancellation patterns as our best estimate of future estimated life. Actual volatility and lives of options may be significantly different from our estimates.
For our non-market performance-based awards, we estimate the anticipated achievement of the performance targets, including forecasting the achievement of future financial targets. These estimates are revised periodically based on the probability of achieving the performance targets and adjustments are made throughout the performance period as necessary. We use payout simulation models to estimate the grant date fair value of market performance-based awards. The payout simulation models assume volatility of our common stock and the common stock of a comparator group of companies, as well as correlations of returns of the price of our common stock and the common stock prices of the comparator group.
The purchase price of common stock under our ESPP is equal to 85% of the lower of (i) the market value per share of the common stock on the first business day of an offering period or (ii) the market value per share
of the common stock on the purchase date. The fair value of the discounted purchases made under our ESPP is calculated using the Black-Scholes model. The fair value of the look-back provision plus the 15% discount is recognized as compensation expense over the 6 month purchase period.
If factors change or we employ different assumptions to value our stock-based awards, the share-based compensation expense that we record in future periods may differ materially from our prior recorded amounts.
Valuation of Goodwill, Acquired Intangible Assets and In-Process Research and Development (IPR&D)
We have recorded goodwill, acquired intangible assets and IPR&D related to our business combinations. When identifiable intangible assets, including IPR&D, are acquired, we determine the fair values of the assets as of the acquisition date. Discounted cash flow models are typically used in these valuations if quoted market prices are not available, and the models require the use of significant estimates and assumptions including but not limited to:
timing and costs to complete the in-process projects;
timing and probability of success of clinical events or regulatory approvals;
estimated future cash flows from product sales resulting from completed products and in-process projects; and
discount rates.
We may also utilize a cost approach, which estimates the costs that would be incurred to replace the assets being purchased. Significant inputs into the cost approach include estimated rates of return on historical costs that a market participant would expect to pay for these assets.
Intangible assets with definite useful lives are amortized to their estimated residual values over their estimated useful lives and reviewed for impairment if certain events occur.
Intangible assets related to IPR&D projects are considered to be indefinite-lived until the completion or abandonment of the associated research and development efforts. During the period the assets are considered indefinite-lived, they will not be amortized but will be tested for impairment. Impairment testing is performed at least annually or when a triggering event occurs that could indicate a potential impairment. If and when development is complete, which generally occurs when regulatory approval to market a product is obtained, the associated assets are deemed finite-lived and are amortized over a period that best reflects the economic benefits provided by these assets.
If projects are not successfully developed, our sales and profitability may be adversely affected in future periods. Additionally, the value of the acquired intangible assets, including IPR&D, may become impaired if the underlying projects do not progress as we initially estimated. We believe that the assumptions used in developing our estimates of intangible asset values were reasonable at the time of the respective acquisitions. However, the underlying assumptions used to estimate expected project
sales, development costs, profitability, or the events associated with such projects, such as clinical results, may not occur as we estimated at the acquisition date.
Goodwill represents the excess of purchase price over fair value of net assets acquired in a business combination and is not amortized. Goodwill is subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. We are organized and operate as a single reporting unit and therefore the goodwill impairment test is performed using our overall market value, as determined by our traded share price, compared to our book value of net assets.
Valuation of Contingent Consideration
We record contingent consideration resulting from a business combination at its fair value on the acquisition date. We determine the fair value of the contingent consideration based primarily on the following factors:
timing and probability of success of clinical events or regulatory approvals;
| |
| �� | timing and probability of success of clinical events or regulatory approvals; |
timing and probability of success of meeting commercial milestones, such as estimated future sales levels of a specific compound; and
discount rates.
Our contingent consideration liabilities arose in connection with our business combinations. On a quarterly basis, we revalue these obligations and record increases or decreases in their fair value as an adjustment to operating earnings. Changes to contingent consideration obligations can result from adjustments to discount rates, accretion of the discount rates due to the passage of time, changes in our estimates of the likelihood or timing of achieving development or commercial milestones, changes in the probability of certain clinical events or changes in the assumed probability associated with regulatory approval.
The assumptions related to determining the value of contingent consideration include a significant amount of judgment, and any changes in the underlying estimates could have a material impact on the amount of contingent consideration expense recorded in any given period.
Income Taxes
We utilize the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement carrying amounts and tax basis of assets and liabilities using enacted tax rates in effect for years in which the temporary differences are expected to reverse.
On December 22, 2017, the Tax Cuts and Jobs Act (Tax Act) was enacted into law. The Tax Act decreased the U.S. statutory corporate tax rate for years beginning after December 31, 2017, and included other domestic and international tax provisions that affect the measurement of our deferred tax assets and liabilities. As a result, we revalued our deferred tax assets and liabilities as of December 31, 2017 and recorded a deferred tax benefit of $292.4. We recorded other impacts of the Tax Act on a provisional basis in 2017. As of December 22, 2018, our accounting for the impact of the Tax Act was complete. See Note 12, “Income Taxes” to our consolidated financial statements included elsewhere in this Annual Report on Form 10-K for additional information.
If our estimate of the tax effect of reversing temporary differences is not reflective of actual outcomes, is modified to reflect new developments or interpretations of the tax law, revised to incorporate new accounting principles, or changes in the expected timing or manner of the reversal our results of operations could be materially impacted. We provide a valuation allowance when it is more likely than not that deferred tax assets will not be realized. We recognize the benefit of an uncertain tax position that has been taken or we expect to take on income tax returns if such tax position is more likely than not to be sustained.
We follow the authoritative guidance regarding accounting for uncertainty in income taxes, which prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. These unrecognized tax benefits relate primarily to issues common among multinational corporations in our industry. We apply a variety of methodologies in making these estimates which include studies performed by independent economists, advice from industry and subject experts, evaluation of public actions taken by the Internal Revenue ServiceIRS and other taxing authorities, as well as our own industry experience. We provide estimates for unrecognized tax benefits which may be subject to material adjustments until matters are resolved with taxing authorities or statutes expire. If our estimates are not representative of actual outcomes, our results of operations could be materially impacted.
We continue to maintain a valuation allowance against certain deferred tax assets where realization is not certain. We periodically evaluate the likelihood of the realization of deferred tax assets and reduce the carrying amount of these deferred tax assets by a valuation allowance to the extent we believe a portion will not be realized. We consider many factors when assessing the likelihood of future realization of deferred
tax assets, including our recent cumulative earnings experience by taxing jurisdiction, expectations of future taxable income, carryforward periods available to us for tax reporting purposes, various income tax strategies and other relevant factors. Significant judgment is required in making this assessment and, to the extent future expectations change, we would assess the recoverability of our deferred tax assets at that time. If we determine that the deferred tax assets are not realizable in a future period, we would record material adjustments to income tax expense in that period.period, and such adjustments may be material.
New Accounting Pronouncements
In February 2016, the FASB issued a new standard that requires lessees to recognize leases on-balance sheet and disclose key information about leasing arrangements. The new standard establishes a right-of-use (ROU) model that requires a lessee to recognize a ROU asset and lease liability on the balance sheet for all leases with a term longer than 12 months. Leases will be classified as finance or operating, with classification affecting the pattern and classification of expense recognition in the income statement. The standard is effective on January 1, 2019, with early adoption permitted. We adopted the new standard on January 1, 2019 and use the effective date as our date of initial application. In July 2018, the FASB issued an update that provided an additional transition option that allows companies to continue applying the guidance under the lease standard in effect at that time in the comparative periods presented in the consolidated financial statements. Companies that elect this option would record a cumulative-effect adjustment to the opening balance of retained earnings on the date of adoption. We elected this optional transition method. We also elected the “package of practical expedients”, which permits us not to reassess under the new standard our prior conclusions about lease identification, lease classification and initial direct costs. We continue to evaluate other practical expedients available under the standard.
We have substantially completed our assessment of the standard as well as implementation of our leasing software, including data upload and test procedures. We continue to finalize our calculations, including our discount rate assumptions, related to the new standard. We are also continuing to establish new processes and internal controls that may be required to comply with the new lease accounting and disclosure requirements set by the new standard. We expect the impact of the standard adoption to decrease our assets, liabilities and retained earnings within our consolidated balance sheet. These decreases will result from the derecognition of our existing assets and financing obligations related to our build to suit leases offset by the recognition of new ROU assets and liabilities as a result of the leasing standard.
New
In June 2016, the FASB issued a new standard intended to improve reporting requirements specific to loans, receivables and other financial instruments. The new standard requires that credit losses be reported based on expected losses compared to the current incurred loss model. The new standard also requires enhanced disclosure of credit risk associated with respective assets. The standard is effective for interim and annual periods beginning after December 15, 2019 with early adoption permitted. We are currently assessing the impact of this standard on our financial condition and results of operations.
In February 2018, the FASB issued a new standard that would permit entities to make a one time reclassification from accumulated other comprehensive income (AOCI) to retained earnings for the stranded tax effects resulting from the newly enacted corporate tax rates under the Tax Act, that was effective for the year ended December 31, 2017. The amount of the reclassification is calculated on the basis of the difference between the historical tax rate and newly enacted tax rate. The standard is effective for interim and annual periods beginning after December 15, 2018 with early adoption permitted. We are currently assessing the impact of this standard on our financial condition.
In August 2018, the FASB issued a new standard on a customer's accounting for implementation, set-up, and other upfront costs incurred in a cloud computing arrangement (CCA). Under the new guidance, customers will assess if a CCA includes a software license and if a CCA does include a software license, implementation and set-up costs will be accounted for consistent with existing internal-use software implementation guidance. Implementation costs associated with a CCA that does not include a software license would be expensed to operating expenses. The standard also provides classification guidance on these implementation costs as well as additional quantitative and qualitative disclosures. The standard is effective for public business entities for fiscal years beginning after December 15, 2019, and interim periods within those fiscal years. Early adoption is permitted, including adoption in any interim periods. Entities can choose to adopt the new guidance prospectively or retrospectively. We are currently assessing the impact this standard will have on our statement of financial condition and results of operations.
Recently Adopted Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (FASB)FASB issued a comprehensive new standard which amends revenue recognition principles and provides a single set of criteria for revenue recognition among all industries. The new standard provides a five stepfive-step framework whereby revenue is recognized when promised goods or services are transferred to a customer at
an amount that reflects the
consideration to which the entity expects to be entitled in exchange for those goods or services. The standard also requires enhanced disclosures pertaining to revenue recognition in both interim and annual periods. The standard is effective for interim and annual periods beginning after December 15, 2017 and allows for adoption using a full retrospective method, or a modified retrospective method. Entities may elect to early adoptWe adopted the standard for annual periods beginning after December 15, 2016. We currently anticipate adopting the standard using the modified retrospective method. We do not expect the implementation of this new standard to have a material impact on our financial position and results of operations.January 1, 2018.
In April 2015,January 2017, the FASB issued a new standard simplifyingthat clarifies the presentationdefinition of debt issuance costs. The new standard aligns the treatmenta business and determines when an integrated set of debt issuance costs with debt discountsassets and premiums andactivities is not a business. This framework requires debt issuance costs be presented as a direct deduction from the carrying amountthat if substantially all of the related debt.fair value of gross assets acquired or disposed of is concentrated in a single asset or group of similar identifiable assets, the assets would not represent a business. We adopted the provisionsnew standard on January 1, 2018 and applied the new guidance prospectively to transactions occurring after adoption. We anticipate that the adoption of this new standard will likely result in the first quarter 2016 and reclassified $9 of deferred financing costs from prepaid expenses and other current assetsmore transactions, to the current portion of long-term debt and $27 from other assets to long-term debt, less current portion in our consolidated balance sheetsextent that such transactions are undertaken by the Company, being accounted for as of December 31, 2015.asset acquisitions.
In April 2015, the FASB issued a new standard clarifying the accounting for a customer’s fees paid in a cloud computing arrangement. Under this standard, if a cloud computing arrangement includes a software license, the customer would account for the software license consistent with other software licenses. If a cloud computing arrangement does not include a software license, the customer would account for the arrangement as a service contract. We adopted the provisions of this standard in the first quarter 2016. The adoption did not have a material effect on our financial condition or results of operations.
In FebruaryJanuary 2016, the FASB issued a new standard that changes accounting for equity investments, financial liabilities under the fair value option, and presentation and disclosure requirements for financial instruments. In addition, the FASB clarified guidance related to the valuation allowance assessment when recognizing deferred tax assets resulting from unrealized losses on available-for-sale debt securities. Equity investments with readily determinable fair values will be measured at fair value with changes in fair value recognized in net income. Companies have the option to either measure equity investments without readily determinable fair values at fair value, or at cost adjusted for changes in observable prices minus impairment. We adopted the new standard on January 1, 2018, and elected to measure our existing equity investments without readily determinable fair values at cost adjusted for changes in observable prices minus impairment. In connection with the adoption of the new standard, we reclassified an immaterial amount of unrealized gains on equity securities from accumulated other comprehensive income to retained earnings. The guidance related to equity investments without readily determinable fair values was applied prospectively to equity investments that existed as of the date of adoption. We will assess equity investments without readily determinable fair values for observable price changes and impairment on a quarterly basis. See Note 7, “Other Investments,” to our consolidated financial statements included elsewhere in this Annual Report on Form 10-K for further details.
In March 2017, the FASB issued a new standard that improves the presentation of net periodic pension cost and net periodic post retirement benefit cost by requiring the bifurcation of net benefit cost. Under the new standard, the service cost component of net benefit cost will be presented with other employee costs in operating expenses, while other components will be reported separately in other income and expense. We adopted the new standard on January 1, 2018. The
adoption of this standard did not have a material impact on our consolidated statements of operations.
In November 2016, the FASB issued a new standard that clarifies how entities should present restricted cash in the rightsstatement of cash flows. Under the new standard, changes in total cash, inclusive of restricted cash, should be reflected in the statement of cash flows. As a result, transfers between cash and obligations arising from leasesrestricted cash will no longer be recognizedreflected as activity within the statement of cash flows. We adopted the new standard on January 1, 2018. The adoption of this standard did not have a material impact on our consolidated statements of cash flows.
In August 2017, the balance sheet by recordingFASB issued a right-of-use assetnew standard intended to improve and corresponding lease liability.simplify certain aspects of the
accounting for hedges. The new standard is intended to more closely align hedge accounting with companies’ risk management strategies, simplify the application of hedge accounting, and increase transparency as to the scope and results of hedging programs. It also requires qualitativeamends the presentation and quantitative disclosures to understand the amount, timing,disclosure requirements and uncertainty of cash flows arising from leases, as well as significant management estimates utilized.changes how companies assess effectiveness. The standard is effective for interim and annual periods beginning after December 15, 2018 and requires awith early adoption permitted. We early adopted the new standard in the second quarter 2018 using the modified retrospective adoption. We are currently assessing the impactmethod. The adoption of this standard did not have a material impact on our consolidated financial condition and results of operations.statements.
In March 2016, the FASB issued a new standard intended to simplify certain aspects of the accounting for employee share-based payments. We elected to early adopt this standard during the third quarter of 2016. One aspect of the standard requires an entity to recognize all excess tax benefits and deficiencies associated with stock-based compensation as a reduction or increase to tax expense in the income statement. Previously, such amounts were recognized in additional paid-in capital. This aspect of the new standard was adopted prospectively, and accordingly we recorded tax benefits of $10 within income tax expense for the year ended December 31, 2016. The amendments require recognition of excess tax benefits regardless of whether the benefit reduces taxes payable in the current period. As a result, $238 associated with previously unrecognized excess tax benefits was recorded as a deferred tax asset and an increase in retained earnings as of the beginning of 2016. Furthermore, the amendment requires that excess tax benefits be classified as an operating activity in the statement of cash flows instead of a financing activity. We elected to adopt this provision of the standard prospectively and thus, prior periods have not been adjusted. We have also elected to continue to estimate the impact of forfeitures when determining the amount of compensation cost to be recognized each period rather than account for forfeitures as they occur.
In October 2016 the FASB issued a new standard that eliminates the prohibition of immediate recognition of current and deferred income tax impacts for an intra-entity asset transfer other than inventory. Under the new standard, entities should recognize the income tax consequences on an intra-entity transfer of an asset other than inventory when the transfer occurs. This new standard will be effective for interim periods beginning after December 15, 2017 and requires a modified retrospective adoption through a cumulative-effect adjustment directly to retained earnings as of the beginning of the period of adoption. We are currently assessing the impact of this standard on our financial condition and results of operations.
Results of Operations
The following table sets forth consolidated statements of operations data for the periods indicated. This information has been derived from the consolidated financial statements included elsewhere in this Annual Report on Form 10-K.
| | | | Year Ended December 31, | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 | 2018 |
| 2017 |
| 2016 |
| Net product sales | $ | 3,082 |
| | $ | 2,603 |
| | $ | 2,234 |
| $ | 4,130.1 |
| | $ | 3,549.5 |
| | $ | 3,081.7 |
|
| Other revenue | 2 |
| | 1 |
| | — |
| 1.1 |
| | 1.6 |
| | 2.4 |
|
| Total revenues | 3,084 |
| | 2,604 |
| | 2,234 |
| 4,131.2 |
| | 3,551.1 |
| | 3,084.1 |
|
| Cost of sales | 258 |
| | 233 |
| | 174 |
| 374.3 |
| | 454.2 |
| | 258.3 |
|
| Operating expenses: | | | | | | | | | | |
| Research and development | 757 |
| | 709 |
| | 514 |
| 730.4 |
| | 878.4 |
| | 757.2 |
|
| Selling, general and administrative | 954 |
| | 863 |
| | 630 |
| 1,111.8 |
| | 1,094.4 |
| | 953.0 |
|
| Acquired in-process research and development | | 1,183.0 |
| | — |
| | — |
|
| Amortization of purchased intangible assets | 322 |
| | 117 |
| | — |
| 320.1 |
| | 320.1 |
| | 322.2 |
|
| Change in fair value of contingent consideration | 36 |
| | 64 |
| | 20 |
| 116.5 |
| | 41.0 |
| | 35.7 |
|
| Acquisition-related costs | 2 |
| | 39 |
| | — |
| — |
| | — |
| | 2.3 |
|
| Restructuring expenses | 3 |
| | 42 |
| | 15 |
| 25.5 |
| | 104.6 |
| | 3.0 |
|
| Impairment of intangible assets | 85 |
| | — |
| | 12 |
| — |
| | 31.0 |
| | 85.0 |
|
| Total operating expenses | 2,159 |
| | 1,834 |
| | 1,191 |
| 3,487.3 |
| | 2,469.5 |
| | 2,158.4 |
|
| Operating income | 667 |
| | 537 |
| | 869 |
| 269.6 |
| | 627.4 |
| | 667.4 |
|
| Other (expense) income | (91 | ) | | (39 | ) | | 3 |
| |
| Other expense | | (27.4 | ) | | (79.6 | ) | | (91.2 | ) |
| Income before income taxes | 576 |
| | 498 |
| | 872 |
| 242.2 |
| | 547.8 |
| | 576.2 |
|
| Income tax expense | 177 |
| | 354 |
| | 215 |
| 164.6 |
| | 104.5 |
| | 176.8 |
|
| Net income | $ | 399 |
| | $ | 144 |
| | $ | 657 |
| $ | 77.6 |
| | $ | 443.3 |
| | $ | 399.4 |
|
| Earnings per common share: | | | | | | | | | | |
| Basic | $ | 1.78 |
| | $ | 0.68 |
| | $ | 3.32 |
| $ | 0.35 |
| | $ | 1.98 |
| | $ | 1.78 |
|
| Diluted | $ | 1.76 |
| | $ | 0.67 |
| | $ | 3.26 |
| $ | 0.35 |
| | $ | 1.97 |
| | $ | 1.76 |
|
Comparison of the YearYears Ended December 31, 2018, 2017, and 2016 to the Year Ended December 31, 2015
Net Product Sales
Net product sales by product and significant geographic region are as follows:
|
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | % Change |
| | 2018 | | 2017 | | 2016 | | 2018 compared to 2017 | | 2017 compared to 2016 |
| SOLIRIS | | | | | | | | | |
| United States | $ | 1,588.4 |
| | $ | 1,235.0 |
| | $ | 1,058.5 |
| | 28.6 | % | | 16.7 | % |
| Europe | 1,036.7 |
| | 985.2 |
| | 939.7 |
| | 5.2 | % | | 4.8 | % |
| Asia Pacific | 382.0 |
| | 328.1 |
| | 303.8 |
| | 16.4 | % | | 8.0 | % |
| Rest of World | 555.9 |
| | 595.8 |
| | 541.2 |
| | (6.7 | )% | | 10.1 | % |
| | $ | 3,563.0 |
| | $ | 3,144.1 |
|
| $ | 2,843.2 |
| | 13.3 | % | | 10.6 | % |
| | | | | | | | | | |
| STRENSIQ | | | | | | | | | |
| United States | $ | 374.3 |
| | $ | 280.1 |
| | $ | 177.5 |
| | 33.6 | % | | 57.8 | % |
| Europe | 61.7 |
| | 35.6 |
| | 15.3 |
| | 73.3 | % | | 132.7 | % |
| Asia Pacific | 27.9 |
| | 18.6 |
| | 13.0 |
| | 50.0 | % | | 43.1 | % |
| Rest of World | 11.2 |
| | 5.5 |
| | 3.6 |
| | 103.6 | % | | 52.8 | % |
| | $ | 475.1 |
| | $ | 339.8 |
|
| $ | 209.4 |
| | 39.8 | % | | 62.3 | % |
| | | | | | | | | | |
| KANUMA | | | | | | | | | |
| United States | $ | 51.3 |
| | $ | 42.4 |
| | $ | 20.4 |
| | 21.0 | % | | 107.8 | % |
| Europe | 21.6 |
| | 14.6 |
| | 6.3 |
| | 47.9 | % | | 131.7 | % |
| Asia Pacific | 3.7 |
| | 2.7 |
| | 1.3 |
| | 37.0 | % | | 107.7 | % |
| Rest of World | 15.4 |
| | 5.9 |
| | 1.1 |
| | ** |
| | ** |
|
| | $ | 92.0 |
| | $ | 65.6 |
|
| $ | 29.1 |
| | 40.2 | % | | 125.4 | % |
| | | | | | | | | | |
| Total Net Product Sales | $ | 4,130.1 |
| | $ | 3,549.5 |
|
| $ | 3,081.7 |
| | 16.4 | % | | 15.2 | % |
** Percentages not meaningful
Net Product Sales (consolidated)
|
| | | | | | | | | | |
| | Year Ended December 31, |
| | 2016 | | 2015 | | % Change |
| Net product sales: | | | | | |
| United States | $ | 1,257 |
| | $ | 951 |
| | 32 | % |
| Europe | 961 |
| | 841 |
| | 14 | % |
| Asia Pacific | 318 |
| | 276 |
| | 15 | % |
| Rest of World | 546 |
| | 535 |
| | 2 | % |
| | $ | 3,082 |
| | $ | 2,603 |
| | 18 | % |
|
| | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
Net
SOLIRIS net product sales by product are as follows:
|
| | | | | | | | | | |
| | Year Ended December 31, |
| | 2016 | | 2015 | | % Change |
| Net product sales: | | | | | |
| Soliris | 2,843 |
| | 2,591 |
| | 10 | % |
| Strensiq | 210 |
| | 12 |
| | - |
|
| Kanuma | 29 |
| | — |
| | - |
|
| | $ | 3,082 |
| | $ | 2,603 |
| | 18 | % |
|
| | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
STRENSIQ net product sales
|
| | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
KANUMA net product sales
|
| | | |
| United States | | Asia Pacific |
| Europe | | Rest of World |
The components of the increase in net product sales for the year ended December 31, 20162018 as compared to 2017 are as follows:
|
| | |
| Year Ended December 31, |
| 2016 |
Components of change: | |
Price | (1 | )% |
Volume | 22 | % |
Foreign exchange | (3 | )% |
Total change in net product sales | 18 | % |
The increase in net product sales for fiscal year 20162018, as compared to the same period in 2015fiscal year 2017, was primarily due to an increase in unit volumes of 22%20.1%. This increase in unit volumes is primarily due to increased global demand globally for SolirisSOLIRIS therapy, forincluding sales to patients with PNH or aHUS andgMG, which received regulatory approval in the second half of 2017. Additional unit volume increases were due to increased sales of StrensiqSTRENSIQ and KanumaKANUMA during 2016.2018 as a result of our continuing efforts to identify and reach more patients with HPP and LAL-D globally.
The positive impact of volume onincrease in net product sales for fiscal year 2018, as compared to fiscal year 2017, was partially offset by price decreases of 3.9% due, in part, to a price change in Turkey resulting from a formalized reimbursement agreement, subsequent to marketing authorization, in the negative impact on foreign exchangethird quarter of 3%,2018. In addition, rebates in the U.S. and reimbursement agreements outside the U.S. for our metabolic products also contributed to this decrease in net product sales.
The components of the increase in revenues for the year ended December 31, 2016,2017 as compared to the same period in 2015. 2016 are as follows:
The negative impact on foreign exchange of $(74), or 3%, was dueincrease in net product sales for fiscal year 2017 as compared to changes in foreign currency exchange rates (inclusive of hedging activity) versus the U.S. dollar for thefiscal year ended December 31, 2015. The negative impact2016 was primarily due to the weakeningan increase in unit volumes of the Euro, Japanese Yen, Russian Ruble,16.8% due to increased demand globally for SOLIRIS therapy for patients with PNH and the British Pound. We recorded a gain in revenueaHUS and increased sales of $73STRENSIQ and $118 related to our foreign currency cash flow hedging program, for the years ended December 31, 2016 and 2015, respectively. We expect the strong dollar compared to other currencies to continue to have a negative impact on revenue intoKANUMA during 2017.
Cost of Sales
Cost of sales includes manufacturing costs, as well as actual and estimated royalty expenses associated with sales of our products.products, and amortization of licensing rights.
The following table summarizes cost of sales for the years ended December 31, 2018, 2017 and 2016:
|
| |
| Cost of Sales |
| ● | Cost of sales as a percentage of net product sales |
Cost of sales for the year ended December 31, 2018 and December 31, 2017 included asset related charges of $5.8 and $152.1, respectively, associated with the closure of the ARIMF facility announced in the third quarter of 2017 (this facility was sold in 2018). These charges primarily relate to accelerated depreciation and the impairment of manufacturing assets.
Exclusive of the items mentioned above, cost of sales as a percentage of net product sales were 8.9%, 8.5% and 8.4% for the years ended December 31, 2018, 2017 and 2016, and 2015:respectively.
|
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2016 | | 2015 | | Change |
| Cost of sales | $ | 258 |
| | $ | 233 |
| | $ | 25 |
|
| Cost of sales as a percentage of net product sales | 8 | % | | 9 | % | | (1 | )% |
Research and Development Expense
|
| |
| Research and Development Expense (R&D) |
| ● | R&D as a % of net product sales |
Our research and development expense includes personnel, facility and externaldirect costs associated with the research and development (R&D) of our product candidates, as well as product development costs. We groupFor additional information on our researchdevelopment programs, please refer to Product and development expenses into two major categories: external direct expenses and all other research and development (R&D) expenses.Development Programs in Item IBusiness of this Annual Report on Form 10-K.
External directR&D expenses are comprised of costs paid to outside parties for clinical development, product development and discovery research, as well as costs associated with certain strategic licensing agreements we have entered into with third parties. Clinical development costs are comprised of costs to conduct and manage clinical trials related to eculizumab, ALXN1210 and other product candidates, including ALX1210.candidates. Product development costs are those incurred in performing duties related to manufacturing development and regulatory functions, including manufacturing of material for clinical and research activities.activities, milestone expenses related to our licensing agreements and collaborations and other administrative costs incurred during product development. Discovery research costs are incurred in conducting laboratory studies and performing preclinical research for other uses of our products and other product candidates. Licensing agreement costscandidates and milestone expenses related to our licensing agreements and collaborations in the discovery stage. Upfront payments include upfront payments related to licenses and milestone payments made in connection with strategic licensing arrangements we have entered into with third parties.collaborations. Clinical development costs have been accumulated and allocated to each of our programs, while product development and discovery research costs have not been allocated.
AllFacilities and other R&D expenses consist of costs to compensate personnel, to maintain our facility,facilities and equipment, and overhead and similarother occupancy costs ofassociated with our research and development efforts. These costs relate to efforts on our clinical and preclinical products, our product development and our discovery research efforts. These costs have not been allocated directly to each program.
The following tablegraph provides information regarding research and development expenses:
|
| | | | | | | | | | | | | | |
| | Year Ended December 31, 2016 | | Year Ended December 31, 2015 | | $ Change | | % Change |
| Clinical development | $ | 208 |
| | $ | 155 |
| | $ | 53 |
| | 34 | % |
| Product development | 168 |
| | 120 |
| | 48 |
| | 40 | % |
| Licensing agreements | 10 |
| | 130 |
| | (120 | ) | | (92 | )% |
| Discovery research | 54 |
| | 44 |
| | 10 |
| | 23 | % |
| Total external direct expenses | 440 |
| | 449 |
| | (9 | ) | | (2 | )% |
| Payroll and benefits | 274 |
| | 219 |
| | 55 |
| | 25 | % |
| Facilities and other costs | 43 |
| | 41 |
| | 2 |
| | 5 | % |
| Total other R&D expenses | 317 |
| | 260 |
| | 57 |
| | 22 | % |
| Research and development expense | $ | 757 |
| | $ | 709 |
| | $ | 48 |
| | 7 | % |
|
| | | |
| Clinical Development | | Discovery |
| Product Development | | Payroll and Benefits |
| Upfront Payments | | Facilities and Other |
During the year ended December 31, 2016,2018, we incurred research and developmentR&D expenses of $757, an increase$730.4, a decrease of $48,$148.0, or 7%16.8%, versus the $709$878.4 incurred during the year ended December 31, 2015.2017. The increasedecrease was primarily related to the following:
IncreaseDecrease of $53$70.9 in externaldirect clinical development expenses related primarily to andecreases in various eculizamab clinical studies, offset by expansion of studies for ALXN1210, sebelipase alfa, and eculizumab (see table below).ALXN1210.
Increase of $48$13.0 in externaldirect product development expenses related primarily to an increase in costs associated with the manufacturing of material for increasedALXN1210 offset by a decrease in ALXN6000 clinical research activities (the ALXN6000 program has been discontinued).
Decrease of $22.2 in upfront payments due to the nature and clinical studies.timing of licensing and collaborations agreements executed in 2018 compared to 2017.
Decrease of $120$12.9 in licensing agreementdiscovery primarily related to de-prioritized preclinical arrangements with Moderna Therapeutics and Blueprint Medicines. We no longer conduct development efforts with these entities.
Decrease of $26.0 in payroll and benefits primarily related to headcount reductions resulting from restructuring activities initiated in 2017.
Decrease of $29.0 in facilities and other related expenses primarily related to decreased
facilities expenses primarily resulting from the impact of the 2017 restructuring.
During the year ended December 31, 2017, we incurred research and development expenses of $878.4, an increase of $121.2, or 16.0%, versus the $757.2 incurred during the year ended December 31, 2016. The increase was primarily related to the following:
Increase of $23.0 in direct clinical development expenses related primarily to an expansion of ALXN 1210 studies.
Increase of $36.2 in direct product development expenses related primarily to an increase in costs associated with the manufacturing of material for ALXN1210 and ALXN6000 clinical research activities.
Increase of $48.9 in upfront payments made in the firstfourth quarter 2015.of 2017 related to a collaboration and license agreement with Halozyme Therapeutics, Inc.
Increase of $10 i$16.9n discovery research expenses primarily related to increases in external research expenses associated with our collaboration agreements.
Increase of $55 in payroll and benefits expenserelated primarily to increased bonus performance and stock compensation expense.
Increase of $17.4 in facilities and other expenses related primarily to accelerated depreciation on assets that support R&D activities associated with the 2017 restructuring activities.
The following graph summarizes expenses related to the additional headcount acquired as part of the Synageva acquisition on June 22, 2015 and the continued global expansion of staff supporting our increasing number of clinical and development programs.programs:
The following tablegraph summarizes externalaccumulated direct expenses related to our clinical development programs. Please referprograms from January 1, 2006 to Item 1, “Business”,December 31, 2018:
(a) From 1992 through 2006, substantially all research and development expenses were related to two products, eculizumab and pexelizumab. We obtained approval in the U.S. for a descriptioneculizumab for PNH in 2007 and for aHUS in 2010, and we ceased development of each of these programs:pexelizumab in 2006.
(b) Unallocated costs shared across various development programs. |
| | | | | | | | | | | |
| | Year Ended December 31, 2016 | | Year Ended December 31, 2015 | | Accumulated Expenditures |
| External direct expenses | | | | | |
| Eculizumab | $ | 88 |
| | $ | 78 |
| | (a) |
| Asfotase alfa | 18 |
| | 22 |
| | $ | 85 |
|
| cPMP | 7 |
| | 8 |
| | 32 |
|
| ALXN1007 | 7 |
| | 14 |
| | 28 |
|
| Sebelipase alfa | 24 |
| | 5 |
| | 29 |
|
| ALXN1210 | 37 |
| | 8 |
| | 46 |
|
| SBC-103 | 9 |
| | 3 |
| | 12 |
|
| Other programs | 7 |
| | 10 |
| | 31 |
|
| Shared expenses | 11 |
| | 7 |
| | (b) |
| | $ | 208 |
| | $ | 155 |
| | $ | 263 |
|
| (a) From 1992 through 2006, substantially all research and development expenses were related to two products, eculizumab and pexelizumab. We obtained approval in the U.S. for eculizumab for PNH in 2007 and for aHUS in 2010, and we ceased development of pexelizumab in 2006. |
| (b) External costs shared across various development programs. |
The successful development of our drug candidates is uncertain and subject to a number of risks. We cannot guarantee that results of clinical trials will be favorable or sufficient to support regulatory approvals for any of our otherproduct development programs. We could decide to abandon development or be required to spend considerable resources not otherwise contemplated. For additional discussion regarding the risks and uncertainties regarding our development programs, please refer to Item 1A “Risk Factors” in this Annual Report on Form 10-K.
We expect our research and development expenses to increaseremain consistent as a percentage of sales in 2017 due2019 as compared to clinical development and manufacturing costs related to our expanding development programs. For additional information on these programs, please refer to “Product and Development Programs” in Item I “Business” of this Annual Report on Form 10-K.2018.
Selling, General and Administrative Expense
|
| |
| Selling General and Administrative Expense (SG&A) |
| ● | SG&A as a % of net product sales |
Our selling, general and administrative expense includes commercial and administrative personnel, corporate facility and external costs required to support the marketing and sales of our commercialized products. These selling, general and administrative costs include: corporate facility operating expenses and depreciation; marketing and sales operations in support of our products; human resources; finance, legal, information technology and support personnel expenses; and other corporate costs such as telecommunications, insurance, audit, government affairs and our global corporate compliance program.
The table below provides information regarding selling, general and administrative expense:
|
| | | | | | | | | | | | | | |
| | Year Ended December 31, 2016 | | Year Ended December 31, 2015 | | $ Change | | % Change |
| Salary, benefits and other labor expense | $ | 556 |
| | $ | 550 |
| | $ | 6 |
| | 1 | % |
| External selling, general and administrative expense | 398 |
| | 313 |
| | 85 |
| | 27 | % |
| Total selling, general and administrative expense | $ | 954 |
| | $ | 863 |
| | $ | 91 |
| | 11 | % |
|
| |
| Salary, benefits and other labor expense |
| External selling, general and administrative expense |
During the year ended December 31, 2016,2018, we incurred selling, general and administrative expenses of $954, $1,111.8, an increase of $91,$17.4, or 11%1.6%, versus the $863$1,094.4 incurred during the year ended December 31, 2015.2017. The increase was primarily related to the following:
Increase in external selling, general and administrative expenses of $85.$20.2. The increase was primarily due to an increase in legalprofessional services and asset related charges associated with previously announced restructuring programs. These increases were partially offset by decreased distribution expenses from investigations overseen by the Audit and Finance Committee relatingas compared to the SECsame period in 2017.
During the year ended December 31, 2017, we incurred selling, general and DOJadministrative expenses of $1,094.4, an increase of $141.4, or 14.8%, versus the $953.0 incurred during the year ended December 31, 2016. The increase was primarily related to the following:
Increase in salary, benefits and other labor expenses of $81.5, primarily related to increase of commercial activities to support the continued global launches of STRENSIQ and KANUMA and the launch of SOLIRIS for gMG. Employee related costs associated with executive leadership changes and incentive compensation also increased.
investigationsIncrease in external selling, general and administrative expenses of $59.9. The increase was primarily due to an increase in charitable contributions and additional professional services, offset in part by decreases in advertising and promotional cost as well as the Audit Committee Investigation that occurred in the fourth quartercompared to 2016. The increase was also attributabledue to additional facilities costs as a result of continuing growth of operations worldwide.asset impairment charges that were recorded in 2017 related to restructuring activities.
We expect our selling, general and administrative expenses to decrease as a percentage of sales in 2019 as compared to 2018.
Acquired In-Process Research and Development
For the year ended December 31, 2018 we recorded acquired in-process research and development (IPR&D) expense of $1,183.0. The increase in 2017, reflecting our continued growth as a commercial organization throughout the world.
Amortization of Purchase Intangible Assets
In the third quarter 2015, we received regulatory approval for Strensiq and Kanuma. As a result,acquired IPR&D for the year ended December 31, 2018, as compared to 2017 and 2016, is due to the Wilson Therapeutics acquisition completed in the second quarter of 2018 and 2015, we recorded amortizationthe Syntimmune acquisition
completed in the fourth quarter of 2018. The IPR&D assets associated with each of these acquisitions, which were the principal assets acquired in each transaction, had not reached technological feasibility and had no alternative future use as of the acquisition date and were therefore expensed in 2018.
Amortization of Purchased Intangible Assets
Amortization expense of $322associated with purchased intangible assets was $320.1, $320.1 and $117, respectively,$322.2 for the years ended December 31, 2018, 2017 and 2016, respectively. Amortization expense is primarily associated with intangible assets related to StrensiqSTRENSIQ and Kanuma.KANUMA.
Change in Fair Value of Contingent Consideration
For the years ended December 31, 20162018, 2017 and 2015,2016, the change in fair value of contingent consideration expense associated with our prior business combinations was $36$116.5, $41.0 and $64,$35.7, respectively. The change in the fair value of contingent consideration forwill fluctuate based on the timing of recognition of changes in the probability of achieving and the expected timing of milestone payments in connection with previous acquisitions.
In September 2018, we amended the terms of certain contingent milestone payments due under our prior merger agreement with Enobia Pharma Corp. (Enobia), dated December 28, 2011. The agreement removed our obligations with respect to a regulatory milestone and redistributed the contingent payment associated with this milestone to various sales
milestones. As a result of this agreement and the probability of achieving the various sales milestones, our contingent consideration liability increased by $48.7 in the third quarter 2018.
For the year ended December 31, 2018, changes in the fair value of contingent consideration expense primarily reflect the impact of the agreement with Enobia to amend milestones and changes in the expected timing of payments of contingent consideration, as well as the interest component of contingent consideration related to the passage of time.
Restructuring Expenses
For the years ended December 31, 20162018 and 2015 was primarily due2017, we recorded $25.5 and $104.6, respectively, in restructuring expenses. The charges for the year ended 2018 were mainly attributable to increases in the likelihood of payments for contingent consideration.
Acquisition-related Costs
For the years ended December 31, 2016 and 2015, acquisition-related costs associated with our business combinations included the following:
|
| | | | | | | |
| | Year Ended December 31, 2016 | | Year Ended December 31, 2015 |
Transaction costs (1) | $ | — |
| | $ | 27 |
|
| Integration costs | 2 |
| | 12 |
|
| | $ | 2 |
| | $ | 39 |
|
| | | | |
| (1) Transaction costs include investment advisory, legal, and accounting fees | | | |
Restructuring Expenses
In connection with the relocation of our corporate headquarters tofrom New Haven, Connecticut we entered into a lease termination agreement in December 2015 forto Boston, Massachusetts and other related costs and the previous corporate headquarters located in Cheshire, Connecticut. We recorded contract termination fees of $11 in restructuring expense in the fourth quarter of 2015.
In conjunction with the acquisition and integration of Synageva we recorded restructuring expense of $13 primarily related to employee costs during 2015. Synageva restructuring charges were not material for the year ended December 31, 2016.2017 were mainly attributable to employee separation costs in connection with the 2017 restructuring (as described below).
In the fourthfirst quarter 2014,of 2017, we announced plansinitiated a company-wide restructuring designed to relocatehelp position the Company for sustainable, long-term growth that we believe will further allow us to fulfill our European headquarters from Lausannemission of serving patients and families with rare diseases. The initial restructuring activities primarily focused on a reduction of the Company's global workforce. In September 2017, we committed to Zurich, Switzerland.an operational plan to re-align the global organization with its refocused corporate strategy. The re-alignment focused investments in priority growth areas to maximize leadership in complement and grow the rare disease business. The re-alignment also included the relocation of our Europeanthe Company's headquarters supports our operational needs based on growthto Boston, Massachusetts in 2018. Our New Haven, Connecticut site continues to support employees working in the European region. Asresearch and process development laboratories, the clinical supply and quality teams, nurse case management and a resultnumber of this action, we recordedimportant enterprise business services. The 2017 restructuring expenses of $15 related to employee costs inplan reduced the fourth quarter of 2014. DuringCompany's global workforce by approximately 20.0%. The restructuring achieved cost savings by focusing the years ended December 31, 2016 and 2015, we incurred additional restructuring costs of $4 and $18, respectively.development portfolio, simplifying
business structures and processes across the Company's global operations, and closing of multiple Alexion sites, including ARIMF and certain regional and country-based offices.
In the first quarter 2019, we have undertaken corporate restructuring activities to re-align our global organization with our re-focused strategy, reduce costs, and realize operational efficiencies. We expect to incur estimated expenses up to $25.0 associated with this recent restructuring by the end of 2019. For additional information on this 2019 corporate restructuring activity, see Item 1. “Business - Sales and Marketing” elsewhere in this Annual Report on Form 10-K.
Impairment of Intangible AssetAssets
During the fourth quarter of 2016, we reviewed SBC-103, an early stage clinical indefinite-lived intangible asset related to the Synageva acquisition as part of our annual impairment testing. The estimated fair value that can be obtained for this asset from a market participant in an arm’s length transaction is $31,was determined to be $31.0, which was lower than the carrying amount of the asset. As a result, in the fourth quarter 2016, we recognized an impairment charge of $85$85.0 to write-down this asset to fair value. In the second quarter 2017, due to clinical results, we recognized an impairment charge of $31.0 related to our SBC-103 acquired in-process research and development asset to write-down the asset to fair value, which was determined to be de minimis.
As of December 31, 2018, we reviewed the KANUMA asset for impairment and determined that there were no indicators of impairment. We will continue to review the related valuation and accounting of this asset in future quarters as new information becomes available to us. Changes to assumptions used in our net cash flow projections may result in impairment charges in subsequent periods. The net book value of the KANUMA intangible asset as of December 31, 2018 is $3,252.6.
Other Income and Expense(Expense)
The following table provides information regarding other income and expense:
|
| | | | | | | | | | | |
| | Year Ended December 31, 2016 | | Year Ended December 31, 2015 | | $
Change |
| Investment income | $ | 11 |
| | $ | 8 |
| | $ | 3 |
|
| Interest expense | (97 | ) | | (48 | ) | | (49 | ) |
| Foreign currency gain (loss) | (5 | ) | | 1 |
| | (6 | ) |
| Total other income (expense) | $ | (91 | ) | | $ | (39 | ) | | $ | (52 | ) |
|
| |
| Investment Income |
| Interest Expense |
| Other Income (expense) |
The increase in interest expense forFor the year ended December 31, 2016 as compared2018, we experienced an increase in investment income primarily due to the prior year was due to us borrowing $3,500 under a term loan facility in conjunction with the acquisitionrecognition of Synagevaunrealized gains of $44.4 on June 22, 2015. The increase was also attributable to increases in interest expense associated with our facility lease obligations.Moderna Therapeutics equity investment.
Income Taxes
During the year ended December 31, 2016, we recorded an income tax expense of $177 and an effective tax rate of 30.7%, compared to an income tax expense of $354 and an effective tax rate of 71.0% for the year ended December 31, 2015. The decrease in the effective tax rate is primarily attributable to the tax charge we recorded in 2015 related to the integration of Synageva assets into our captive foreign partnership. This one-time charge increased our effective tax rate in 2015 by approximately 63.0%. This decrease was partially offset by deferred tax expense we recognized in 2016 attributable to first quarter distributions from our foreign captive partnership. This distribution increased our 2016 effective tax rate by 20.7%. Exclusive of these charges, we expect to continue to benefit from a reduced tax rate compared to periods prior to January 1, 2014 as a result of centralizing our global supply chain and technical operations in Ireland in the fourth quarter 2013.
|
| |
| Tax Expense |
| ● | Effective Tax Rate |
The income tax expense for the years ended December 31, 2018, 2017 and 2016 is attributable to the U.S. federal, state and foreign income taxes on our profitable operations. Additionally,During the year ended December 31, 2018, we recorded an income tax expense of $164.6 and an effective tax rate of 68.0%, compared to an income tax expense of $104.5 and $176.8 and an effective tax rate of 19.1% and 30.7% for the years ended December 31, 2017 and 2016, respectively. The increase in the effective tax rate during 2018, from 19.1% for the year ended December 31, 2017 to 68.0% for the year ended December 31, 2018 was primarily attributable to the acquisitions of Syntimmune and Wilson Therapeutics. Absent successful clinical results and regulatory approval, there is no alternative future use for the in-process research assets we acquired in these acquisitions. Accordingly, the value of the assets acquired of $1,183.0 were expensed as acquired in-process research and development, for which no tax benefit has been recognized. The Syntimmune and Wilson Therapeutics acquisitions resulted in an increase in the effective tax rate of approximately 102.6%. This increase was partially offset by the decrease to the U.S. statutory rate and other related adjustments as a result of the Tax Act. These items resulted in a decrease of approximately 45.7%.
In December 2017, the Tax Act was enacted into law. The Tax Act decreased the U.S. federal corporate tax rate to 21.0%, imposed a minimum tax on foreign earnings and incorporated a one-time transition tax on previously unremitted foreign earnings. We incorporated the impact of the Tax Act in our results of operations or calculated provisional amounts for the tax effects of the Tax Act that could be reasonably estimated for the year ended December 31, 2017. We recorded adjustments to this provisional accounting during 2018, which
resulted in a decrease to tax expense of $56.5. We completed our accounting for the Tax Act in the fourth quarter 2018.
The Tax Act resulted in an increase to tax expense for the year ended December 31, 2017 of $45.8. This increase included a transition tax expense of $177.9 and deferred tax expense related to the new GILTI minimum tax of $165.4, partially offset by the $297.5 benefit of re-measuring balance sheet taxes to the new 21.0% US federal tax rate. The re-measurement benefit included $292.4 related to decreases to our net deferred tax liability and $5.1 related to decreases to income taxes payable. The deferred tax expense related to the GILTI minimum tax included incremental deferred tax of $236.9, net of a related $71.5 decrease for uncertain tax positions.
The decrease in the effective tax rate during 2017, from 30.7% for the year ended December 31, 2016 isto 19.1% for the impactyear ended December 31, 2017 was primarily attributable to the net increase to tax expense in 2017 of $45.8 attributable to the Tax Act, offset by decreases attributable to the deferred tax attributable to first quarter distributionscost of $119.3 associated with the distribution of earnings from our captive foreign partnership in 2016 and the conclusion of $119.
In the third quarter 2015, we contributed certain supply chain assets, commercial operation rightsIRS examination of our 2013 and intellectual property acquired2014 tax years in 2017. The impact of the Synageva acquisition toenactment of the Tax Act increased our captive foreign partnership. This contributioneffective tax rate in 2017 by 8.4%. The 2016 distribution of earnings increased our 2016 effective tax rate by 20.7%. Conclusion of the IRS examination resulted in a revaluationdecrease to our 2017 effective tax rate of our captive foreign partnership, an increase toapproximately 3.6% for the outside basis difference our U.S. parent company has in the captive foreign partnership, and a corresponding one-time deferred tax expense of $316. There was no cash tax payment associated with this deferred expense.year ended December 31, 2017.
We continue to maintain a valuation allowance against certain other deferred tax assets where realization is not certain. We periodically evaluate the likelihood of the realization ofrealizing deferred tax assets and reduce the carrying amount of these deferred tax assets by a valuation allowance to the extent we believe a portion will not be realized.
Comparison of the Year Ended December 31, 2015 to the Year Ended December 31, 2014
Net Product Sales
Net product sales by significant geographic region are as follows:
|
| | | | | | | | | | |
| | Year Ended December 31, |
| | 2015 | | 2014 | | % Change |
| Net product sales: | | | | | |
| United States | $ | 951 |
| | $ | 730 |
| | 30 | % |
| Europe (1) | 841 |
| | 836 |
| | 1 | % |
| Asia Pacific | 276 |
| | 244 |
| | 13 | % |
| Rest of World | 535 |
| | 424 |
| | 26 | % |
| | $ | 2,603 |
| | $ | 2,234 |
| | 17 | % |
Net product sales by product are as follows:
|
| | | | | | | | | | |
| | Year Ended December 31, |
| | 2015 | | 2014 | | % Change |
| Net product sales: | | | | | |
| Soliris (1) | $ | 2,591 |
| | $ | 2,234 |
| | 16 | % |
| Strensiq | 12 |
| | — |
| | N/A |
|
| Kanuma | — |
| | — |
| | N/A |
|
| | $ | 2,603 |
| | $ | 2,234 |
| | 17 | % |
| (1) In March 2014, we entered into an agreement with the French government which positively impacts prospective reimbursement of Soliris and also provides for reimbursement for shipments made in years prior to January 1, 2014. As a result of the agreement, in the first quarter of 2014, we recognized $88 of net product sales from Soliris in France relating to years prior to January 1, 2014. Exclusive of the $88, net product sales in Europe increased 12% for the year ended December 31, 2015 compared to the year ended December 31, 2014. |
The components of the increase in net product sales for the year ended December 31, 2015, exclusive of the $88 recognized related to prior years, are as follows:
|
| | |
| Year Ended December 31, |
| 2015 |
Components of change: | |
Price | — | % |
Volume | 29 | % |
Foreign exchange | (8 | )% |
Total change in net product sales | 21 | % |
The increase in net product sales for fiscal year 2015 as compared to the same period in 2014, was primarily due to an increase in unit volumes of 29% due to increased demand globally for Soliris therapy for patients with PNH or aHUS during the respective periods.
The positive impact of volume on net product sales was offset by the negative impact on foreign exchange of 8%, for the year ended December 31, 2015, as compared to the same period in 2014. The negative impact on foreign exchange of $165, or 8%, was due to changes in foreign currency exchange rates (inclusive of hedging activity) versus the U.S. dollar for the year ended December 31, 2014. The negative impact was primarily due to the weakening of the Euro, Japanese Yen and Russian Ruble. We recorded a gain in revenue of $118 and $19 related to our foreign currency cash flow hedging program, for the years ended December 31, 2015 and 2014, respectively.
Cost of Sales
Cost of sales includes manufacturing costs as well as actual and estimated royalty expenses associated with sales of Soliris.
The following table summarizes cost of sales for the year ended December 31, 2015 and 2014:
|
| | | | | | | | |
| | Year Ended December 31, |
| | 2015 | | 2014 | | Change |
| Cost of sales | 233 |
| | 174 |
| | 59 |
|
| Cost of sales as a percentage of net product sales | 9 | % | | 8 | % | | 1 | % |
We recorded an expense of $24 in the first quarter of 2015 associated with a portion of a single manufacturing campaign at a third party manufacturer for Strensiq. The cost was comprised of raw materials, internal overhead and external production costs. This expense did not impact the clinical supply of inventory or the commercial launch of Strensiq.
Exclusive of the item mentioned above, cost of sales as a percentage of net product sales was 8% for the years ended December 31, 2015 and 2014.
Research and Development Expense
Our research and development expense includes personnel, facility and external costs associated with the research and development of our product candidates, as well as product development costs. We group our research and development expenses into two major categories: external direct expenses and all other research and development (R&D) expenses.
External direct expenses are comprised of costs paid to outside parties for clinical development, product development and discovery research, as well as costs associated with strategic licensing agreements we have entered into with third parties. Clinical development costs are comprised of costs to conduct and manage clinical trials related to eculizumab and other product candidates. Product development costs are those incurred in performing duties related to manufacturing development and regulatory functions, including manufacturing of material for clinical and research activities. Discovery research costs are incurred in conducting laboratory studies and performing preclinical research for other uses of our products and other product candidates. Licensing agreement costs include upfront and milestone payments made in connection with strategic licensing arrangements we have entered into with third parties. Clinical development costs have been accumulated and allocated to each of our programs, while product development and discovery research costs have not been allocated.
All other R&D expenses consist of costs to compensate personnel, to maintain our facility, equipment and overhead and similar costs of our research and development efforts. These costs relate to efforts on our clinical and preclinical products, our product development and our discovery research efforts. These costs have not been allocated directly to each program.The following table provides information regarding research and development expenses:
|
| | | | | | | | | | | | | | |
| | Year Ended December 31, 2015 | | Year Ended December 31, 2014 | | $ Change | | % Change |
| Clinical development | $ | 155 |
| | $ | 116 |
| | $ | 39 |
| | 34 | % |
| Product development | 120 |
| | 58 |
| | 62 |
| | 107 | % |
| Licensing agreements | 130 |
| | 110 |
| | 20 |
| | 18 | % |
| Discovery research | 44 |
| | 14 |
| | 30 |
| | 214 | % |
| Total external direct expenses | 449 |
| | 298 |
| | 151 |
| | 51 | % |
| Payroll and benefits | 219 |
| | 191 |
| | 28 |
| | 15 | % |
| Facilities and other costs | 41 |
| | 25 |
| | 16 |
| | 64 | % |
| Total other R&D expenses | 260 |
| | 216 |
| | 44 |
| | 20 | % |
| Research and development expense | $ | 709 |
| | $ | 514 |
| | $ | 195 |
| | 38 | % |
During the year ended December 31, 2015, we incurred research and development expenses of $709, an increase of $195, or 38%, versus the $514 incurred during the year ended December 31, 2014. The increase was primarily related to the following:
Increase of $39 in external clinical development expenses related primarily to an expansion of studies for eculizumab, ALXN1007, ALXN1210, and other programs (see table below).
Increase of $62 in external product development expenses related primarily to an increase in costs associated with the manufacturing of material for increased clinical research activities and clinical studies.
Increase of $20 in licensing agreement expenses related to the achievement of additional license milestones.
Increase of $30 in discovery research expenses primarily related to increases in external research expenses associated with our Moderna agreement and other external research expenses.
Increase of $28 R&D payroll and benefit expense related to the additional headcount acquired as part of the Synageva acquisition in the second quarter 2015 and the continued global expansion of staff supporting our increasing number of clinical and development programs.
Increases of $16 in R&D facilities and other costs related to the additional R&D facilities as part of the Synageva acquisition in the second quarter 2015 and the additional costs associated with the continued expansion of global supply chain facilities and support services.
The following table summarizes external direct expenses related to our clinical development programs. Please refer to Item 1, “Business”, for a description of each of these programs:
|
| | | | | | | |
| | Year Ended December 31, 2015 | | Year Ended December 31, 2014 |
| External direct expenses | | | |
| Eculizumab | $ | 78 |
| | $ | 68 |
|
| Asfotase alfa | 22 |
| | 27 |
|
| cPMP | 8 |
| | 8 |
|
| ALXN1007 | 14 |
| | 3 |
|
| Sebelipase alfa | 5 |
| | — |
|
| ALXN1210 | 8 |
| | 1 |
|
| Other programs | 13 |
| | 3 |
|
| Unallocated | 7 |
| | 6 |
|
| | $ | 155 |
| | $ | 116 |
|
The successful development of our drug candidates is uncertain and subject to a number of risks. We cannot guarantee that results of clinical trials will be favorable or sufficient to support regulatory approvals for our other programs. We could decide to abandon development or be required to spend considerable resources not otherwise contemplated. For additional discussion regarding the risks and uncertainties regarding our development programs, please refer to Item 1A “Risk Factors” in this Annual Report on Form 10-K.
Selling, General and Administrative Expense
Our selling, general and administrative expense includes commercial and administrative personnel, corporate facility and external costs required to support the marketing and sales of our commercialized products. These selling, general and administrative costs include: corporate facility operating expenses and depreciation; marketing and sales operations in support of Soliris; human resources; finance, legal, information technology and support personnel expenses; and other corporate costs such as telecommunications, insurance, audit, government affairs and our global corporate compliance program.
The table below provides information regarding selling, general and administrative expense:
|
| | | | | | | | | | | |
| | Year Ended December 31, 2015 | | Year Ended December 31, 2014 | | $
Change |
| Salary, benefits and other labor expense | $ | 550 |
| | $ | 389 |
| | $ | 161 |
|
| External selling, general and administrative expense | 313 |
| | 241 |
| | 72 |
|
| Total selling, general and administrative expense | $ | 863 |
| | $ | 630 |
| | $ | 233 |
|
During the year ended December 31, 2015, we incurred selling, general and administrative expenses of $863, an increase of $233, or 37%, versus the $630 incurred during the year ended December 31, 2014. The increase was primarily related to the following:
Increase in salary, benefits and other labor expenses of $161. The increase was a result of increased staff costs related to commercial development activities and increases in payroll and benefits within our general and administrative functions to support our infrastructure growth as a global commercial entity. The increase was also attributable to additional global commercial staff costs due to our acquisition of Synageva in the second quarter 2015 and additional stock-based compensation expense of $30 related to the acceleration of Alexion stock awards for former Synageva employees.
Increase in external selling, general and administrative expenses of $72. The increase was primarily due to an increase in external marketing costs to support the global launches of Strensiq and Kanuma and professional services to support the continuing growth of the company.
Amortization of Purchase Intangible Assets
In the third quarter 2015, we received regulatory approval for Strensiq and Kanuma. As a result, for the year ended December 31, 2015, we recorded amortization expense of $117 associated with intangible assets related to Strensiq and Kanuma.
Acquisition-related Costs
For the years ended December 31, 2015 and 2014, acquisition-related costs associated with our business combinations included the following:
|
| | | | | | | |
| | Year Ended December 31, 2015 | | Year Ended December 31, 2014 |
Transaction costs (1) | $ | 27 |
| | $ | — |
|
| Integration costs | 12 |
| | — |
|
| | $ | 39 |
| | $ | — |
|
| | | | |
| (1) Transaction costs include investment advisory, legal, and accounting fees | | | |
The increase in acquisition related costs was due to the Synageva acquisition that occurred during 2015.
Change in Fair Value of Contingent Consideration
For the years ended December 31, 2015 and 2014, the change in fair value of contingent consideration expense associated with our prior business combinations was $64 and $20 respectively. The increase in the fair value of contingent consideration for the year ended December 31, 2015 as compared the prior year was primarily due to increases in the likelihood of payments for contingent consideration and a net decrease in discount rates.
Restructuring Expenses
In connection with the relocation of our corporate headquarters to New Haven, Connecticut, we entered into a lease termination agreement in December 2015 for the previous corporate headquarters located in Cheshire, Connecticut. We recorded contract termination fees of $11 in restructuring expense in the fourth quarter of 2015.
In conjunction with the acquisition and integration of Synageva we recorded restructuring expense of $13 primarily related to employee costs during 2015.
In the fourth quarter of 2014 we announced plans to move the European headquarters from Lausanne, Switzerland to Zurich, Switzerland resulting in restructuring expenses of $15. The relocation of the European headquarters supports our growing operational needs based on current business forecasts. During the year ended December 31, 2015, we incurred additional restructuring costs of $18.
Impairment of Intangible Asset
During the fourth quarter of 2014, we reviewed for impairment the value of the early stage, Phase II indefinite-lived intangible asset related to the Orphatec acquisition. We initiated such review as part of our annual impairment testing and increased costs associated with clinical trial studies. Although we will continue to develop this asset, the estimated fair value that can be obtained from a market participant in an arm’s length transaction was determined to be de minimis as of December 31, 2014. As a result, in the fourth quarter 2014, we recognized an impairment charge of $8 to write-down these assets to fair value.
Other Income and Expense
The following table provides information regarding other income and expense:
|
| | | | | | | | | | | |
| | Year Ended December 31, 2015 | | Year Ended December 31, 2014 | | $
Change |
| Investment income | $ | 8 |
| | $ | 8 |
| | $ | — |
|
| Interest expense | (48 | ) | | (3 | ) | | (45 | ) |
| Foreign currency loss | 1 |
| | (2 | ) | | 3 |
|
| Total other income (expense) | $ | (39 | ) | | $ | 3 |
| | $ | (42 | ) |
The increase in interest expense for the year ended December 31, 2015 as compared to the prior year was due to us borrowing $3,500 under a term loan facility in conjunction with the acquisition of Synageva.
Income Taxes
During the year ended December 31, 2015, we recorded an income tax expense of $354 and an effective tax rate of 71.0%, compared to an income tax expense of $215 and an effective tax rate of 24.7% for the year ended December 31, 2014. The increase in the effective tax rate is primarily attributable to the integration of Synageva assets into our captive foreign partnership.
This one-time charge increased our effective tax rate in 2015 by approximately 63.0%. Exclusive of such one-time charges, we expect to continue to benefit from a reduced tax rate compared to periods prior to January 1, 2014 as a result of centralizing our global supply chain and technical operations in Ireland in the fourth quarter 2013.
The income tax expense for 2015 is attributable to the U.S. federal, state and foreign income taxes on our profitable operations, as well as the tax impact associated with integration of the Synageva business with and into the Alexion business.
In the third quarter 2015, we contributed certain supply chain assets, commercial operation rights and intellectual property acquired in the Synageva acquisition to our captive foreign partnership. This contribution resulted in a revaluation of our captive foreign partnership, an increase to the outside basis difference our U.S. parent company has in the captive foreign partnership, and a corresponding one-time deferred tax expense of $316. There was no cash tax payment associated with this deferred expense.
The income tax expense for 2014 is attributable to the U.S. federal, state and foreign income taxes on our profitable operations. Additionally, included for the year ended December 31, 2014 is $2 of tax attributable to our agreement with the French government that provided reimbursement for shipments of Soliris made prior to January 1, 2014.
We continue to maintain a valuation allowance against certain other deferred tax assets where realization is not certain. We periodically evaluate the likelihood of the realization of deferred tax assets and reduce the carrying amount of these deferred tax assets by a valuation allowance to the extent we believe a portion will not be realized.
Financial Condition, Liquidity and Capital Resources
The following table summarizes the components of our financial condition as of December 31, 20162018 and 2015:2017:
|
| | | | | | | |
| | December 31, 2018 | | December 31, 2017 |
| Cash and cash equivalents | $ | 1,365.5 |
| | $ | 584.4 |
|
| Marketable securities | 198.3 |
| | 889.7 |
|
| Long-term debt (includes current portion & revolving credit facility) | 2,862.5 |
| | 2,906.3 |
|
| |
|
| | |
| Current assets | $ | 3,385.0 |
| | $ | 2,953.9 |
|
| Current liabilities | 1,174.0 |
| | 952.5 |
|
| Working capital | $ | 2,211.0 |
| | $ | 2,001.4 |
|
|
| | | | | | | | | | | |
| | December 31, 2016 | | December 31, 2015 | | $ Change |
| Cash and cash equivalents | $ | 966 |
| | $ | 1,010 |
| | $ | (44 | ) |
| Marketable securities | 327 |
| | 375 |
| | (48 | ) |
| Long-term debt (includes current portion) | 3,081 |
| | 3,456 |
| | (375 | ) |
| | | | | | |
| Current assets | $ | 2,578 |
| | $ | 2,416 |
| | $ | 162 |
|
| Current liabilities | 823 |
| | 709 |
| | 114 |
|
| Working capital | $ | 1,755 |
| | $ | 1,707 |
| | $ | 48 |
|
The aggregate decreaseincrease in cash and cash equivalents and marketable securities of $89.7 at December 31, 2018 as compared to December 31, 2017 was primarily attributable to cash generated from operations and net proceeds from the issuance of common stock under share-based compensation arrangements. Partially offsetting these increases was cash utilized to repurchase shares of common stock, principal payments on our term loan payments of contingent consideration,(in connection with entering into our Amended and Restated Credit Agreement in June 2018), and purchases of property, plant, and equipment. Partially offsetting these decreases was cash generated through operations.
Excluding the impact of any future asset acquisitions, we expect our annual operating expenses to decrease as a percentage of sales in 2019 as compared to 2018. We also expect continued growthreduced capital investment in our expenditures, particularly those related2019 as compared to research and product development, clinical trials, regulatory approvals, international expansion, commercialization of products and capital investment. However, we2018. We anticipate that cash generated from operations and our existing available cash, cash equivalents and marketable securities should provide us adequate resources to fund our operations as currently planned.planned for at least the next twelve months.
We have financed our operations and capital expenditures primarily through positive cash flows from operations. We expect to continue to be able to fund our operations, including principal and interest payments on our credit facilityAmended and Restated Credit Agreement and contingent payments from our acquisitions principally through our cash flows from operations. We may, from time to time, also seek additional funding through a combination of equity or debt financings or from other sources, if necessary for future acquisitions or other strategic purposes. New sources of financing through equity and/or debt financing(s) may not always be available on acceptable terms, or at all, and we may be required to obtain certain consents in connection with completing such financings.
Financial Instruments
Until required for use in the business, we may invest our cash reserves in money market funds, bank deposits, reverse repurchase agreements, and high-quality marketable debt securities in accordance with our investment policy. The stated objectives of our investment policy isare to preserve capital, provide liquidity consistent with forecasted cash flow requirements, maintain appropriate diversification and generate returns relative to these investment objectives and prevailing market conditions.
Financial instruments that potentially expose us to concentrations of credit risk are cash equivalents, marketable securities, accounts receivable and our derivative contracts. At December 31, 2016,2018, three customers accounted for 47%48.7% of the accounts receivable balance, with these individual customers accounting for 14%14.0% to 19%19.1% of the accounts
receivable balance. At December 31, 2015, three2017, four customers accounted for 51%57.7% of the accounts receivable balance, with these individual customers accounting for 14%10.2% to 22%18.9% of the accounts receivable balance.
For the year endedDecember 31, 2016, three2018, four customers accounted for 37%50.3% of our product sales, with these individual customers ranging from 10%10.0% to 16%16.4% of product sales. For the year ended December 31, 2015,2017, three customers accounted for 37.0% 38%of our product sales, with these individual customers ranging from 10%10.8% to 18%15.0% of product sales. For the year ended December 31, 2016, three customers accounted for 36.7% of our product sales, with these individual customers ranging from 10.0% to 16.0% of product sales.
We continue to monitor economic conditions, including volatility associated with international economies and the associated impacts on the financial markets and our business. A substantial portionSubstantially all of our accounts receivable due from these countries are due from or backed by sovereign or local governments,wholesale distributors, public hospitals and other government entities. We monitor the amountfinancial performance of non-sovereign accounts receivable isour customers so that we can appropriately respond to changes in their credit worthiness. We operate in certain jurisdictions where weakness in economic conditions can result in extended collection periods. We continue to monitor these conditions and assess their possible impact on our business. To date, we have not material. Althoughexperienced any significant losses with respect to collection of our accounts receivables from certain countries may extend beyond our credit terms, we do not expect any such delays to have a material impact on our financial condition or results of operations.receivable.
We manage our foreign currency transaction risk and interest rate risk within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes. As of December 31, 2016,2018, we havehad foreign exchange forward contracts with notional amounts totaling $2,389.$2,523.0. These outstanding foreign exchange forward contracts had a net fair value liability of $140,$18.9, of which $156$40.8 is included in other current assets and noncurrent assets and $16$21.9 is included in other current liabilities and noncurrent liabilities. As of December 31, 2016,2018, we havehad interest rate swap contracts with notional amounts totaling $656.$3,881.3. These outstanding interest rate swap contracts had a net fair value of $10,$2.0, of which $20.1 is included in other current assets and $18.1 is included in other current liabilities and noncurrent assets.liabilities. The counterparties to these contracts are large domestic and multinational commercial banks, and we believe the risk of nonperformance is not material.
At December 31, 2016,2018, our financial assets and liabilities were recorded at fair value. We have classified our financial assets and liabilities as Level 1, 2 or 3 within the fair value hierarchy. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Our Level 1 assets consist of mutual
fund investments and equity securities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, butfor substantially the full term of the financial instrument. Our Level 2 assets consist primarily of institutional money market funds, commercial paper, municipal bonds, reverse repurchase agreements, U.S. and foreign government-related debt, corporate debt securities, certificates of deposit, equity securities subject to holding period restrictions and derivative contracts. Our Level 2 liabilities consist also of derivative contracts. Level 3 inputs are unobservable inputs based on our own assumptions used to measure assets and liabilities at fair value. Our Level 3 liabilities consist of contingent consideration related to acquisitions.
Business Combinations and Contingent Consideration Obligations
TheAt December 31, 2018, the purchase agreements for our business combinations include contingent payments totaling up to $766$702.0 that will become payable if and when certain development and commercial milestones are achieved. Of these milestone amounts, $451$367.0 and $315$335.0 of the contingent payments relate to development and commercial milestones, respectively. We do not expect these amounts to have an impact on our liquidity in the near-term, and, during the next 12 months, we expect to make milestone payments of approximately $25$100.0 associated with our prior business combinations. As additional future payments become probable, we will evaluate methods of funding payments, which could be made from available cash and marketable securities, cash generated from operations or proceeds from other financing. the sale of equity securities or debt.
Asset Acquisitions and License Agreements
In the fourth quarter 2016,2018, Alexion acquired Syntimmune, a clinical-stage biotechnology company developing an antibody therapy targeting the criteria were metneonatal Fc receptor (FcRn), for an upfront payment of $400.0. Under the terms of the agreement, we could also be required to pay up to $800.0 upon the achievement of specified research, development, regulatory and commercial milestones.
In October 2018, we entered into a milestone collaboration agreement with Dicerna Pharmaceuticals, Inc. (Dicerna) that provides us with exclusive worldwide licenses and development and commercial rights for two preclinical RNA interference (RNAi) subcutaneously delivered molecules for complement-mediated diseases, as well as an exclusive option for other preclinical RNAi molecules for two additional targets within the complement pathway. In addition to the collaboration agreement, we made an equity investment in Dicerna. Under the terms of the agreements, we made an upfront
payment of $37.0 for the exclusive licenses and the equity investment. The market value of the equity investment was $10.3 as of the date of acquisition, which we recorded in other assets in our consolidated balance sheets. Due to the early stage of the assets we are licensing, we recorded expense for the upfront license payment of $26.7 during the fourth quarter 2018. In addition, as of December 31, 2018, we could also be required to pay up to approximately $625.0 for option exercise fees and amounts due upon the achievement of specified research, development, regulatory and commercial milestones, as well as royalties on commercial sales.
In December 2017, we entered into a collaboration and license agreement with Halozyme Therapeutics, Inc. that allows us to use drug-delivery technology in the development of subcutaneous formulations for our portfolio of products for up to four targets. Due to the early stage of the assets we are licensing, we recorded expense for the upfront payment of $40.0 during the fourth quarter 2017. In addition, as of December 31, 2018, we could be required to pay an additional $160.0 for each target developed, subject to achievement of specified development, regulatory and sales-based milestones, as well as royalties on commercial sales.
In addition, we have entered into other license agreements under which we may be required to pay up to an additional $137.2 if certain development, regulatory and commercial milestones are met.
We do not expect the payments associated with milestones under our acquisitionasset acquisitions and licensing agreements to have a significant impact on our liquidity in the near-term. During the next 12 months, we may make milestone payments related to these arrangements of Enobia Pharma Corp. In connection with this, $60 was paid in December 2016.approximately $255.0.
Financing Lease Obligations
In November 2012, we entered into a lease agreement for office and laboratory space to be constructed in New Haven, Connecticut. The term of the lease commenced in 2015 and will expire in 2030, with a renewal option of ten years. Although we do not legally own the premises, we are deemed to be the owner of the building due to the substantial improvements directly funded during the construction period based on applicable accounting guidance for build-to-suit leases. Accordingly, the landlord’s costs of constructing the facility during the construction period are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. Construction of the new facility was completed and the building was placed into service in the first quarter 2016. Associated with this arrangement we recognized interest expense of $13.3, $14.2, and $14.0 for the years ended December 31, 2018, 2017, and 2016, respectively. As of December 31, 20162018 and 2015,2017, our total facility lease
obligation was $136$133.5 and $133,$134.6, respectively, recorded within other current liabilities and facility lease obligation onin our consolidated balance sheets.
During the third quarter 2015, we entered into a newan agreement with Lonza Group AG and its affiliates (Lonza) whereby Lonza will construct a new manufacturing facility dedicated to Alexion at one of its existing facilities. As a result of our contractual right to full capacity of the new manufacturing facility, a portion of the payments under the agreement are considered to be lease payments and a portion as payment for the supply of inventory. Although we will not legally own the premises, we are deemed to be the owner of the manufacturing facility during the construction period based on applicable accounting guidance for build-to-suit leases due to our involvement during the construction period. Accordingly, the landlord’s costs of constructing the facility during the construction period are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. We expect the completion of the facility, including obtaining regulatory approval, to be in 2019. As of December 31, 20162018 and 2015,2017, we recorded a construction-in-process asset of $118$203.9 and $19,$180.6, respectively, and an offsetting facility lease obligation of $107$155.1 and $15,$159.1, respectively, within other current liabilities and facility lease obligation onin our consolidated balance sheets.
License Agreements
In March 2015,September 2017, we entered into a collaborationlease agreement with a third party that allows usfor approximately 150,000 square feet of office space to identify and optimize drug candidates. Alexion will have the exclusive worldwide rights to develop and commercialize products arising from the collaboration. Due to the early stagebe constructed in Boston, Massachusetts. Construction of the assets we are licensingfacility was completed and the building was placed into service in connection with the collaboration, we recorded expense forsecond quarter 2018. The term of the upfront paymentlease commenced upon the landlord's substantial completion of $15the facility during the firstsecond quarter 2015. In addition, as of December 31, 2016 we could be required2018 and will expire on the thirteenth anniversary of commencement, with an option to pay up to
an additional $249 if certain development, regulatory, and commercial milestones are met over time, as well as royalties on commercial sales.
In January 2015, we entered into a license agreement with a third party to obtain an exclusive research, development and commercial licenserenew for specific therapeutic molecules. Due to the early stage of these assets, we recorded expense for the upfront payment of $50 during the first quarter 2015. In addition, as of December 31, 2016 we could be required to pay up to an additional $822 if certain development, regulatory,10 years. Although we do not legally own the premises, due to our involvement during the construction period, we are deemed to be the owner of the portion of the building that we will lease based on applicable accounting guidance for build-to-suit leases. Accordingly, the landlord's costs of constructing the facility during the construction period are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. Interest expense recognized during 2018 was not material. As of December 31, 2018 and commercial milestones are met over time, as well as royalties2017, our total facility lease obligation was $83.6 and $59.6, respectively, recorded within facility lease obligation in our consolidated balance sheets.
Our facility lease obligations will be derecognized in 2019 upon adoption of the new lease accounting standard and will be replaced by ROU liabilities going forward. See Note 1 Business Overview and Summary of Significant Accounting Policies to our Consolidated Financial Statements included elsewhere in this Annual Report on commercial sales.Form 10-K for more information.
In December 2014, weLong-term Debt
On June 7, 2018, Alexion entered into an agreement with X-Chem Pharmaceuticals (X-Chem) that allows us to identify novel drug candidates from X-Chem’s proprietary drug discovery engine. Alexion will have the exclusive worldwide rights to developAmended and commercialize products arising from the collaboration in up to three program targets. Due to the early stage of these assets, we recorded expense for an upfront payment of $8. In addition, for each program target, for a maximum of three targets, we could be required to make additional payments upon the achievement of specified research, development and regulatory milestones up to $75, as well as royalties on commercial sales.
In January 2014, we entered into an agreement with Moderna Therapeutics, Inc. (Moderna) that allows us to purchase ten product options to develop and commercialize treatments for rare diseases with Moderna’s messenger RNA (mRNA) therapeutics platform. Alexion will lead the discovery, development and commercialization of the treatments produced through this broad, long-term strategic agreement, while Moderna will retain responsibility for the design and manufacture of the messenger RNA against selected targets. Due to the early stage of these assets, we recorded expense for an upfront payment of $100. We will also be responsible for funding research activities under the program. In addition, for each drug target, up to a maximum of ten targets, we could be required to make an option exercise payment of $15 and to pay up to an additional $120 with respect to a rare disease product and $400 with respect to a non-rare disease product in development and sales milestones if the specific milestones are met over time as well as royalties on commercial sales.
In addition, we have entered into other license agreements under which we would be required to pay up to an additional $415 if certain development, regulatory and commercial milestones are met.
Our license agreements include contingent payments that will become payable if and when certain development, regulatory and commercial milestones are achieved. We do not expect the payments associated with these milestones to have a significant impact on our liquidity in the near-term. During the next 12 months, we expect to make milestone payments related to our license agreements of approximately $51.
Long-term Debt
On June 22, 2015, Alexion entered into a credit agreementRestated Credit Agreement (the Credit Agreement) with a syndicateBank of banks, whichAmerica N.A. as administrative agent. The Credit Agreement amends and restates our credit agreement dated as of June 22, 2015 (the Prior Credit Agreement). The Credit Agreement provides for a $3,500$2,612.5 term loan facility and a $500$1,000.0 revolving facility. Borrowings under the term loan facility are payable in quarterly installments equal to 1.25% of the original loan amount, beginning December 31, 2015. Final repayment of the term loan and any draw down of revolving credit loans are due on June 22, 2020. In addition to borrowings in which prior notice is required, the revolving credit facility includes a sublimit of $100 in the form of letters of credit and borrowings on same-day notice, referred to as swingline loans, of up to $25. Borrowings can be used for working capital requirements, acquisitions and other general corporate purposes.
Under Beginning with the Credit Agreement,quarter ending June 30, 2019, we are required to deliver to the administrative agent, not later than 50 days after each fiscal quarter, our quarterly financial statements, and within 5 days thereafter, a compliance certificate. In November 2016, we obtained a waiver from the necessary lenders for this requirement and the due date for deliverymake amortization payments of 5.00% of the third quarter 2016 financial statements and compliance certificate was extended to January 18, 2017. The postingaggregate principal amount of the Third Quarter report on Form 10-Q on our website on January 4, 2017 satisfied the financial statement covenant, and we simultaneously delivered the required compliance certificate, as required by the lenders.
In connection with the acquisition of Synageva in June 2015, we borrowed $3,500 under the term loan facility and $200 under the revolving facility, and we used our available cash for the remaining cash consideration. In June 2015, we repaid the revolving facilityannually, payable in full. equal quarterly installments.
As of December 31, 2016,2018, we had $3,081$2,612.5 outstanding on the term loan.loan and $250.0 of borrowings outstanding under the revolving credit facility. The $250.0 of proceeds on the revolving credit facility was used to refinance amounts outstanding under the Prior Credit Agreement. As of December 31, 2016,2018, we had open letters of credit of $15, and$1.7 that offset our borrowing availability underon the revolving facility. In January 2019 we paid the outstanding revolving credit facility was $485.of $250.0 in full.
Manufacturing Obligations
We have supply agreements with Lonza through 2028 relating to the manufacture of SolirisSOLIRIS and Strensiq,STRENSIQ, which requires payments to Lonza at the inception of contract and upon the initiation and completion of product manufactured. On an ongoing basis, we evaluate our plans for future levels of manufacturing by Lonza, which depends upon our commercial requirements and the progress of our clinical development programs and the production levels of ARIMF.programs.
We have various agreements with Lonza, with remaining total non-cancellable commitments of approximately $1,148$1,084.6 through 2028.2029. Certain commitments may be canceled only in limited circumstances. If we terminate certain supply agreements
with Lonza without cause, we will be required to pay for product scheduled for manufacture under our arrangement. Under an existing arrangement with Lonza, we also pay Lonza a royalty on sales of SolirisSOLIRIS that was manufactured at ARIMF prior to its sale and a payment with respect to sales of SolirisSOLIRIS manufactured at Lonza facilities.
In addition to Lonza, we have non-cancellable commitments of approximately $27$104.1 through 20192020 with other third party manufacturers.
Taxes
We do not record U.S.have recorded tax expense on the undistributed earnings of our controlled foreign corporation (CFC) subsidiaries. These earnings relate to ongoing operations and were approximately $1,462 at December 31, 2016. We intend to reinvest these earnings permanently outside the U.S. or repatriate the earnings only when it is tax efficient to do so. Accordingly, we believe that U.S. tax on any earnings that might be repatriated would be substantially offset by realizing the benefit of tax attributes, such as U.S. foreign tax credits or by utilizing deficits in the foreign earnings and profits account.
During the fourth quarter of 2013, in connection with the centralization of our global supply chain and technical operations in Ireland, our U.S. parent company became a direct partner in a foreign partnership subsidiary. To the extent that our U.S. parent company receives its allocation of partnership taxable income, the amounts will be taxable in the U.S., and therefore the permanent reinvestment assertion will no longer apply.
We doCFC earnings may not have any present or anticipated future need for cash held by our CFCs, as cash generated in the U.S., as well as borrowings, are expected to be sufficient to meet U.S. liquidity needs for the foreseeable future. At December 31, 2016, approximately $445 of our cash and cash equivalents was held by foreign subsidiaries, a significant portion of which is required for liquidity needs of our foreign subsidiaries. These subsidiaries will settle any outstanding intercompany trade payables prior to having excess cash available which could be repatriated to our entities in the U.S. Whileas a dividend distribution due to limitations imposed by law, we intend to reinvest CFC earnings permanently outsidehave not recorded the related potential withholding, foreign local, and U.S. or repatriate the earnings only when it is tax efficient to do so, certain unforeseen future events could impact our permanent reinvestment assertion. Such events include acquisitions, corporate restructurings or tax law changes not currently contemplated.state income taxes.
Common Stock Repurchase Program
In November 2012, our Board of Directors authorized a share repurchase program. In May 2015, our Board of Directors increased the authorization to acquire shares with an aggregate value of up to $1,000 for future purchases under the repurchase program, which superseded all prior repurchase programs. The repurchase
program does not have an expiration date, and we are not obligated to acquire a particular number of shares. The repurchase program may be discontinued at any time at the Company’s discretion. We expect that cash generated from operations and our existing available cash and cash equivalents will be sufficient to fund any share repurchases.
Under the program, we repurchased 3 and 2 shares of our common stock at a cost of $430 and $328 during the years ended December 31, 2016 and 2015, respectively. As of December 31, 2016, there is a total of $325 remaining for repurchases under the program. The Company did not repurchase any shares during the pendency of the Synageva acquisition, and the Company began repurchasing shares again in the third quarter 2015.
In February 2017, our Board of Directors increased the authorizationamount that we are authorized to acquire shares with an aggregate value of upexpend on future repurchases to $1,000 for future purchases under the repurchase program, which superseded all prior repurchase programs. .Under the program, we repurchased 0.7 and 4.0 shares of our common stock at a cost of $85.0 and $463.6 during the years ended December 31, 2018 and 2017, respectively. As of February 16, 2017,December 31, 2018, there is a total of $1,000$451.5 remaining for repurchases under the repurchase program.
Cash Flows
The following summarizes our net change in cash and cash equivalents:
|
| | | | | | | | | | | |
| | Year Ended December 31, | | |
| | 2016 | | 2015 | | $ Change |
| Net cash provided by operating activities | $ | 1,086 |
| | $ | 675 |
| | $ | 411 |
|
| Net cash used in investing activities | (287 | ) | | (3,585 | ) | | 3,298 |
|
| Net cash provided by financing activities | (836 | ) | | 2,985 |
| | (3,821 | ) |
| Effect of exchange rate changes on cash | (7 | ) | | (9 | ) | | 2 |
|
| Net change in cash and cash equivalents | $ | (44 | ) | | $ | 66 |
| | $ | (110 | ) |
|
| | | | | | | | | | | |
| | Year Ended December 31, | | |
| | 2018 | | 2017 | | $ Change |
| Net cash provided by operating activities | $ | 426.0 |
| | $ | 1,115.6 |
| | $ | (689.6 | ) |
| Net cash provided by (used in) investing activities | 470.5 |
| | (918.3 | ) | | 1,388.8 |
|
| Net cash used in financing activities | (102.4 | ) | | (596.6 | ) | | 494.2 |
|
| Effect of exchange rate changes on cash and cash equivalents and restricted cash | (11.2 | ) | | 17.7 |
| | (28.9 | ) |
| Net change in cash and cash equivalents | $ | 782.9 |
|
| $ | (381.6 | ) | | $ | 1,164.5 |
|
Operating Activities
Cash flows provided by operations in 20162018 were $1,086$426.0 compared to $675$1,115.6 in 2015.2017. The increasedecrease in cash provided by operating activities was primarily due to an increase in gross margin on product salesthe acquisition of $454 resulting primarily from an increase in global demandWilson Therapeutics and Syntimmune and higher cash payments for Solirisrestructuring and the launch of Strensiq and Kanuma,incentive compensation, as well as athe impact of the timing of cash receipts and other payments for the year ended 2018 as compared to the same period in the prior year. This decrease in cash outflows relates to our licensing arrangements of $120. The increase in gross margin was partially offset by an increase in clinical development costs, interest expenseoperating income, excluding the impact of the IPR&D charge associated with the Wilson Therapeutics and selling, general and administrative expenses during 2016.
In 2017, we expect increases in cash flow from operations which will be highly dependent on sales levels, and the related cash collections from sales of our products. We also expect cash outflows of approximately $51 related to milestone payments on our license agreements.Syntimmune acquisitions.
Investing Activities
Cash used forprovided by (used in) investing activities in 20162018 was $287$470.5 compared to $3,585$(918.3) in 2015.2017. The decreaseincrease in cash usedprovided by investing activities was primarily dueattributable to the payment of $3,939 in 2015 related to the Synageva acquisition and net cash flows related to the purchaseproceeds and maturities of available-for-sale marketable securities, which resulted
in a net cash inflows of $51$690.8 in 20162018 compared to $640a net cash outflows of $(558.9) in 2015.2017.
We expectDuring 2018, we also had lower cash outlays associated with the purchase of property, plant and equipment of $213.0 as compared to continue to have$357.3 in 2017. The significant spending on property, plant and equipment in 2017 related primarily to the construction of our new biologics manufacturing facilities in Ireland.
Financing Activities
Cash flows (used in) provided byused in financing activities in 20162018 were $(836)$102.4 compared to $2,985$596.6 in 2015.2017. The decrease in cash used for financing activities was primarily due to the following:
Borrowingrepurchasing $378.6 less of $3,655,our common stock in 2018 than 2017. Additionally, cash used for financing activities decreased as a result of reduced net of issuance costs, underpayments on our credit facility, in connection with the acquisition of Synageva in June 2015.
Principal payments against theoutstanding credit facility of $375$43.8 in 2016, 2018,compared to $301$175.0 in 2015.2017.
Repurchased of common stock of $430 in 2016, compared to $328 in 2015.
Contractual Obligations
The following table summarizes our contractual obligations at December 31, 20162018 and the effect such obligations and commercial commitments are expected to have on our liquidity and cash flow in future fiscal years. These do not include potential milestone payments and assume non-termination of agreements.
These obligations, commitments and supporting arrangements represent payments based on current operating forecasts at December 31, 2018, which are subject to change:
| | | | Total | | Less than 1 Year | | 1-3 Years | | 3-5 Years | | More than 5 Years | Total | | Less than 1 Year | | 1-3 Years | | 3-5 Years | | More than 5 Years |
| Contractual obligations: | | | | | | | | | | | | | | | | | | |
Long-term debt | $ | 3,081 |
| | $ | — |
| | $ | 325 |
| | $ | 2,756 |
| | $ | — |
| $ | 2,862.5 |
| | $ | 348.0 |
| | $ | 261.2 |
| | $ | 2,253.3 |
| | $ | — |
|
Interest expense (1)(2) | 265 |
| | 79 |
| | 151 |
| | 35 |
| | — |
| 407.2 |
| | 81.8 |
| | 194.7 |
| | 130.7 |
| | — |
|
Facility lease obligation (2) | 225 |
| | 16 |
| | 31 |
| | 32 |
| | 146 |
| |
Facility lease obligations (3) | | 197.6 |
| | 13.7 |
| | 31.1 |
| | 32.2 |
| | 120.6 |
|
| Operating leases | 90 |
| | 21 |
| | 32 |
| | 13 |
| | 24 |
| 48.0 |
| | 14.1 |
| | 14.9 |
| | 7.4 |
| | 11.6 |
|
| Total contractual obligations | $ | 3,661 |
| | $ | 116 |
| | $ | 539 |
| | $ | 2,836 |
| | $ | 170 |
| $ | 3,515.3 |
| | $ | 457.6 |
| | $ | 501.9 |
| | $ | 2,423.6 |
| | $ | 132.2 |
|
| Commercial commitments: | | | | | | | | | | | | | | | | | | |
Clinical and manufacturing development (3)(4) | $ | 1,175 |
| | $ | 227 |
| | $ | 356 |
| | $ | 200 |
| | $ | 392 |
| $ | 1,188.7 |
| | $ | 271.8 |
| | $ | 317.5 |
| | $ | 227.4 |
| | $ | 372.0 |
|
| Total commercial commitments | $ | 1,175 |
| | $ | 227 |
| | $ | 356 |
| | $ | 200 |
| | $ | 392 |
| $ | 1,188.7 |
| | $ | 271.8 |
| | $ | 317.5 |
| | $ | 227.4 |
| | $ | 372.0 |
|
| | | | | | | | | | | | | | | | | | | |
(1) Interest on variable rate debt calculated based on interest rates at December 31, 2016. Interest that is fixed, associated to our interest rate swaps, is calculated based on the fixed interest swap rate at December 31, 2016 | |
(2) Facility lease obligation includes the lease agreement signed in November 2012, for office and laboratory space to be constructed in New Haven, Connecticut. Although we do not legally own the premises, we were deemed to be the owner of the building during the construction period based on applicable accounting guidance for build-to-suit leases due to our involvement during the construction period. Accordingly, the landlord’s costs of constructing the facility are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. | |
(3) Clinical and manufacturing development commitments include only non-cancellable commitments, including all Lonza agreements, at December 31, 2016. | |
(1) Includes our term loan and the $250.0 revolving credit facility balance. Our revolving credit facility is classified as a current liability and has been included in payments to be made within one year. In January 2019, we paid the revolving credit facility of $250.0 in full. | | (1) Includes our term loan and the $250.0 revolving credit facility balance. Our revolving credit facility is classified as a current liability and has been included in payments to be made within one year. In January 2019, we paid the revolving credit facility of $250.0 in full. |
(2) Interest on variable rate debt is calculated based on interest rates at December 31, 2018. Interest that is fixed, associated to our interest rate swaps, is calculated based on the fixed interest swap rate at December 31, 2018. | | (2) Interest on variable rate debt is calculated based on interest rates at December 31, 2018. Interest that is fixed, associated to our interest rate swaps, is calculated based on the fixed interest swap rate at December 31, 2018. |
(3) Facility lease obligations include the lease agreement signed in November 2012, for office and laboratory space in New Haven, Connecticut and the lease agreement signed in September 2017 for office space in Boston, Massachusetts. In the fourth quarter of 2018 we amended the New Haven lease agreement significantly reducing our leased square footage in the building beginning in 2019 through the expiration of the lease (in connection with this amendment, in the fourth quarter of 2018, we made a payment of $53.0 to a third party as in incentive to lease the released square footage). Although we do not legally own these premises, we were deemed to be the owner of the buildings during the construction period based on applicable accounting guidance for build-to-suit leases due to our involvement during the construction period. Accordingly, the landlord’s costs of constructing the facility are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. | | (3) Facility lease obligations include the lease agreement signed in November 2012, for office and laboratory space in New Haven, Connecticut and the lease agreement signed in September 2017 for office space in Boston, Massachusetts. In the fourth quarter of 2018 we amended the New Haven lease agreement significantly reducing our leased square footage in the building beginning in 2019 through the expiration of the lease (in connection with this amendment, in the fourth quarter of 2018, we made a payment of $53.0 to a third party as in incentive to lease the released square footage). Although we do not legally own these premises, we were deemed to be the owner of the buildings during the construction period based on applicable accounting guidance for build-to-suit leases due to our involvement during the construction period. Accordingly, the landlord’s costs of constructing the facility are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet. |
(4) Clinical and manufacturing development commitments include only non-cancellable commitments, including all Lonza agreements, at December 31, 2018. | | (4) Clinical and manufacturing development commitments include only non-cancellable commitments, including all Lonza agreements, at December 31, 2018. |
The contractual obligations table above does not include contingent royalties and other contingent contractual payments we may owe to third parties in the future because such payments are contingent on future sales of our products and the existence and scope of third party intellectual property rights and other factors described in Item 1A “Risk Factors”“Risk Factors” and Note 9 “Commitments11 “Commitments and Contingencies” ofContingencies” to the Consolidated Financial Statements included elsewhere in thethis Annual Report on Form 10-K.
The liability for unrecognized tax benefits related to various federal, state and foreign income tax matters of $139$92.7 at December 31, 20162018 was not included within the table above. The timing of the settlement of these amounts was not reasonably estimable at December 31, 2016.2018. We do not expect payment of amounts related to the unrecognized tax benefits within the next twelve months.
Contingent payments related to business acquisitions, completed in prior yearsasset acquisitions or license agreements
are not included within the table above, as the satisfaction of the contingent consideration obligations is uncertain at December 31, 2018 and, if satisfied, the timing of payment for these amounts was not reasonably estimable at December 31, 2016.2018. Contingent payments associated with these business combinations total up to $766$702.0 which will become payable if and when certain development and commercial milestones are achieved. During the next 12 months, we expect to make milestone payments of approximately $25$100.0 associated with our prior business combinations. License commitmentsCommitments related to asset acquisitions and license agreements include contingent payments that will become payable if and when certain development, regulatory and commercial milestones are achieved under which we would be required to pay additional amounts if certain development, regulatory and commercial milestones are met.achieved. During the next 12 months, we expect tomay make milestone payments related to our asset acquisitions and license agreements of approximately $51.$255.0.
Future obligations related to our defined benefit plans are not included within the table above, as the timing and amounts of these payments was not reasonably estimable as of December 31, 2016.2018. The total unfunded obligation on our defined benefit plans as of December 31, 20162018 was $20.$17.6. Our unfunded obligation can be impacted by changes in the laws and regulations, interest rates, investment returns, and other variables.
Credit Facilities
On June 7, 2018, we entered into an Amended and Restated Credit Agreement (the Credit Agreement), with Bank of America N.A. as administrative agent. The Credit Agreement amends and restates our agreement dated as of June 22, 2015 Alexion entered into a credit agreement (Credit(the Prior Agreement) with a syndicate of banks, which.
The Credit Agreement provides for a $3,500$1,000.0 revolving credit facility and a $2,612.5 term loan facility. The revolving credit facility and term loan facility and a $500 revolving credit facility maturing in five years. Borrowings undermature on June 7, 2023. Beginning with the term loanquarter ending June 30, 2019, we are payable in quarterly installments equalrequired to 1.25%make amortization payments of 5.00% of the original loanaggregate principal amount beginning December 31, 2015. Final repayment of the term loan and revolving credit loans are due on June 22, 2020. In addition to borrowingsfacility annually, payable in which prior notice is required, the revolving credit facility includes a sublimit of $100 in the form of letters of credit and borrowings on same-day notice, referred to as swingline loans, of up to $25. Borrowings can be used for working capital requirements, acquisitions and other general corporate purposes. With the consent of the lenders and the administrative agent, and subject to satisfaction of certain conditions, we may increase the term loan facility and/or the revolving credit facility in an amount that does not cause our consolidated net leverage ratio to exceed the maximum allowable amount.equal quarterly installments.
Under the Credit Agreement we may elect that the loansLoans under the Credit Agreement bear interest, at a rate per annum equal toour option, at either athe base rate or a Eurodollar rate, plus, in each case plus an applicable margin. TheUnder the Credit Agreement, the applicable margins on base rate loans range from 0.25% to 1.00% and the applicable margins on Eurodollar loans range from 1.25% to 2.00%, in each case depending uponbased on our consolidated net leverage ratio (as calculated in accordance with the Credit Agreement).
Our obligations under the credit facilitiesCredit Agreement are guaranteed by certain of Alexion’sour foreign and domestic subsidiaries and secured by liens on certain of Alexion’s and itsour subsidiaries’ equity interests, subject to certain exceptions. Under the terms of the Credit Agreement, we must maintain a ratio of total net debt to EBITDA of 3.50 to 1.00 (subject to certain limited adjustments) and EBITDA to cash interest expense ratio of at least 3.50 to 1.00, in each case as calculated in accordance with the Credit Agreement.
The Credit Agreement requires us to comply withcontains certain financial covenants on a quarterly basis. Further, the Credit Agreement includes negative covenants, subject to exceptions, restricting or limiting our ability and the ability of our subsidiaries to, among other things, incur additional indebtedness, grant liens, and engage in certain investment, acquisition and disposition transactions. The Credit Agreement also contains customary representations and warranties, affirmative and negative covenants and events of default, including payment defaults, breachdefault. The negative covenants in the Credit Agreement restrict Alexion’s and its subsidiaries’ ability, subject to certain baskets and exceptions, to (among other things) incur liens or indebtedness, make investments, enter into mergers and other fundamental changes, make dispositions or pay dividends. The restriction on dividend payments includes an exception that permits us to pay dividends and make other restricted payments regardless of representations and warranties, covenant defaults and cross defaults. If an event of default occurs,dollar amount so long as, after giving pro forma effect thereto, we have consolidated net leverage ratio, as defined in the interest rate would increase and the administrative agent would be entitled Credit Agreement, within predefined ranges, subject
to take various actions, including the acceleration of amounts due under the loan.certain increases following designated material acquisitions.
Operating Leases
Our operating leases are principally for facilities and equipment. We currently lease office space in the U.S. and foreign countries to support our operations as a global organization.
We believe that our administrative office space is adequate to meet our needs for the foreseeable future. We also believe that our research and development facilities and our manufacturing facility,facilities, together with third party manufacturing facilities, will be adequate for our on-going activities.
In addition to the minimum rental commitments on our operating leases we may also be required to pay amounts for taxes, insurance, maintenance and other operating expenses.
Commercial Commitments
Our commercial commitments consist of research and development, license, operational, clinical development, and manufacturing cost commitments, along with anticipated supporting arrangements, subject to certain limitations and cancellation clauses. The timing and level of our commercial scale manufacturing costs, which may or may not be realized, are contingent upon the progress of our clinical development programs and our commercialization plans. Our commercial commitments are represented principally by our supply agreementagreements with Lonza described above. Our commitments with Lonza do not include amounts for estimated consumer price index, or CPI, adjustments.adjustments which we are obligated to pay to Lonza.
| |
Item 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK. |
Item 7A.QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
(amounts in millions, except percentages)
Interest Rate Risk
As of December 31, 2016,2018, we invested our cash in a variety of financial instruments, principally money market funds, corporate bonds, repurchase agreements, municipal bonds, commercial paper and government-related obligations. Most of our interest-bearing securities are subject to interest rate risk and could decline in value if interest rates fluctuate. Our investment portfolio is comprised of marketable debt securities of highly rated financial institutions and investment-grade debt instruments, and we have guidelines to limit the term-to-maturity of our investments. Based on the type of securities we hold, we do not believe a change in interest rates would have a material impact on our financial statements. If interest rates were to increase or decrease by 1%1.00%, the fair value of our investment portfolio would (decrease) increase by approximately $(4)$(0.5) and $4,$0.5, respectively.
InOn June 2015,7, 2018, we entered into an Amended and Restated Credit Agreement (the Credit Agreement), with Bank of America N.A. as administrative agent. The Credit Agreement amends and restates our agreement dated as of June 22, 2015 (the Prior Agreement). Loans under the Credit Agreement withbear interest, at a rate per annum equal toour option, at either athe base rate or a Eurodollar rate, plus, in each case plus an applicable margin. TheUnder the Credit Agreement, the applicable margins on base rate loans range from 0.25% to 1.00% and the applicable margins on Eurodollar loans range from 1.25% to 2.00%, in each case depending uponbased on our consolidated net leverage ratio (as calculated in accordance with the Credit Agreement).
Changes in interest rates related to the Credit Agreement could have a material effect on our financial statements.
To achieve a desired mix of floating and fixed interest rates on our term loan, we entered into twoa number of interest rate swap agreements in June 2016 that qualified for and are designated as cash flow hedges. The first agreementAs of December 31, 2018, we had a notional amountcash flow hedges with aggregate amounts of $3,281 and was effective from June 30, 2016 through December 30, 2016. This agreement hedged the contractual floating interest rateapproximately 87.0% of our term loan. As a result of this agreement, the interest rate for ourcurrent outstanding term loan was fixed at 0.535%, pluscovering periods over the borrowing spread, until December 30, 2016. The second agreement has a notional amount of $656 and is effective December 31, 2016 through December 31, 2019. The second agreement converts the floating rate on a portion of our term loan to a fixed rate of 0.98%, plus a borrowing spread, from December 31, 2016 through December 2019. The impact of a hypothetical increase or decrease in interest rates on the fair value of our interest rate swap contract would be offset by a change in the value of the underlying liability.next twelve months. If interest rates were to increase or decrease by 1%1.00%, annual interest expense, beginning in 2017,over the next year would increase or decrease by $24,$3.0, based on the unhedged portion of our outstanding term loan.loan as of December 31, 2018.
Foreign Exchange Market Risk
Our operations include activities in many countries outside the U.S., including countries in Europe, Latin America and Asia Pacific. As a result, our financial results are impacted by factors such as changes in foreign currency
exchange rates or weak economic conditions in the foreign markets where we operate. We have exposure to movements in foreign currency exchange rates, the most significant of which are the Euro and Japanese Yen, against the U.S. dollar. We are a net receiver of many foreign currencies, and our consolidated financial results benefit from a weaker U.S. dollar and are adversely impacted by a stronger U.S. dollar relative to foreign currencies in which we sell our product.products.
Our monetary exposures on our balance sheet arise primarily from cash, accounts receivable, intercompany receivables and payables denominated in foreign currencies. Approximately 51%49.0% of our net product sales were denominated in foreign currencies during 2016,2018, and our revenues are also exposed to fluctuations in the foreign currency exchange rates over time. In certain foreign countries, we may sell in U.S. dollar, but our customers may be impacted adversely inby fluctuations in foreign currency exchange rates which may also impact the timing and amount of our revenue.
Both positive and negative impacts to our international product sales from movements in foreign currency exchange rates are only partially mitigated by the natural, opposite impact that foreign currency exchange rates have on our international operating expenses. Additionally, we have operations based in Switzerland and Ireland,Europe and accordingly, our expenses are impacted by fluctuations in the value of the Swiss Franc and Euro against the U.S. dollar.
We currently have a derivative program in place to achieve the following: 1)(1) limit the foreign currency exposure of our monetary assets and liabilities on our balance sheet, using contracts with durations of approximately 90 daysup to 6 months and 2)(2) hedge a portion of our forecasted product sales (in some currencies), including intercompany sales, and certain forecasted expenses using contracts with durations of up
to 60 months. The objectivesobjective of this program areis to reduce the volatility of our operating results due to fluctuation of foreign exchange and to increase the visibility of the foreign exchange impact on forecasted revenues.exchange. This program utilizes foreign exchange forward contracts intended to reduce, not eliminate, the volatility of operating results due to fluctuations in foreign exchange rates.
As of December 31, 20162018 and 2015,2017, we held foreign exchange forward contracts with notional amounts totaling $2,389$2,523.0 and $2,536,$2,708.1, respectively. The decrease in outstanding foreign exchange forward contracts resulted primarily from increases in forecasted revenues and, for certain currencies, extended duration of hedges. As of December 31, 20162018 and 2015,2017, our outstanding foreign exchange forward contracts had a net fair value of $140$18.9 and $148,$(47.5), respectively.
We do not use derivative financial instruments for speculative trading purposes. The counterparties to these foreign exchange forward contracts are large domestic and multinational commercial banks. We believe the risk of counterparty nonperformance is not material.
Based on our foreign currency exchange rate exposures at December 31, 2016,2018, a hypothetical 10% adverse fluctuation in exchange rates would decrease the fair value of our foreign exchange forward contracts that are designated as cash flow hedges by approximately $162$101.5 at December 31, 2016.2018. The resulting loss on these forward contracts would be offset by the gain on the underlying transactions and therefore would have minimal impact on future anticipated earnings and cash flows. Similarly, adverse fluctuations in exchange rates that would decrease the fair value of our foreign exchange forward contracts that are not designated as hedge instruments would be offset by a positive impact of the underlying monetary assets and liabilities.
Credit Risk
As a result of our foreign operations, we are exposed to changes in the general economic conditions in the countries in which we conduct business. The majority of our receivables are due from wholesale distributors, public hospitals and other government entities. We monitor the financial performance and creditworthiness of our large customers so that we can properly assess and respond to changes in their credit profile. We continue to monitor these conditions, including the volatility associated with international economies and the relevant financial markets, and assess their possible impact on our business. Although collection of our accounts receivables from certain countries may extend beyond our standard credit terms, we do not expect any such delays to have a material impact on our financial condition or results of operationsoperations.
| |
| Item 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA. |
The consolidated financial statements and supplementary data of the Company required in this item are set forth beginning on page F-1.
| |
| Item 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None.
| |
| Item 9A. | CONTROLS AND PROCEDURES. |
Disclosure Controls and Procedures.Procedures
We have established disclosure controls and procedures to provide reasonable assurance that information is accumulated and communicated to our management, including our principal executive officer
and principal financial officer, as appropriate to allow timely decisions regarding required disclosure, and ensure that information required to be disclosed in the reports we file or submit under the Securities Exchange Act of 1934, as amended (Exchange Act) is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms.
Our management, with the participation of our Interim Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures, (asas defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, as of December 31, 2016.2018. Based on this evaluation, our Interim Chief Executive Officer and Chief Financial Officer concluded that as of December 31, 2018, our disclosure controls and procedures were not effective as of December 31, 2016, due toat the material weakness in internal control over financial reporting that was previously disclosed in our Form 10-Q filed on January 4, 2017 and described below, which was not remediated as of December 31, 2016.reasonable assurance level.
Management’s Report on Internal Control overOver Financial Reporting
Management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial
statements for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 20162018 based on the framework in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). A material weakness is a deficiency, or combination of deficiencies, inBased on that evaluation, management has concluded that the Company maintained an effective internal control over financial reporting such that there is a reasonable possibility that a material misstatementas of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis.December 31, 2018.
We did not maintain an effective control environment as our senior management failed to set an appropriate Tone at the Top. Specifically, senior management failed to reinforce the need for compliance with the Company’s policies and procedures, which resulted in inappropriate business conduct. This control deficiency did not result in a misstatement to the Company’s consolidated financial statements. However, this control deficiency could result in a misstatement to disclosures that would result in a material misstatement to our annual or interim consolidated financial statements that would not be prevented or detected. Accordingly, our management has determined that this control deficiency constitutes a material weakness.
The effectiveness of our internal control over financial reporting as of December 31, 20162018 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
Changes in Internal Control over Financial Reporting.Reporting
There has been no change in our internal control over financial reporting that occurred during the quarter ended December 31, 20162018 that has materially affected,
or is reasonably likely to materially affect, our internal control over financial reporting.
Remediation Plan and Activities
Management is engaged in remedial activities to address the material weakness described above. The remedial activities include the following:
•The Board of Directors has and will reinforce to key leadership the importance of setting appropriate Tone at the Top and of appropriate behavior with respect to the Company’s commitment to ethics and compliance programs in the performance of the Company’s mission, as well as adherence to the Company’s internal control over financial reporting framework;
•Members of senior management, with the participation and input of the Audit and Finance Committee and the Board of Directors, have and will increase communication with, and training of employees regarding:
▪Our commitment to ethical standards and the integrity of our business practices;
▪Requirements for compliance with applicable laws, our Code of Ethics and Business Conduct and other Company policies; and
▪Availability of and processes for reporting suspected violations of law or our Code of Ethics and Business Conduct.
•Revised financial reporting processes to ensure that all employees annually confirm compliance with the Company’s Code of Ethics and Business Conduct and that deviations are identified and timely remediated; and
•The Board of Directors, together with management, is evaluating certain Company practices and procedures, including those related to compensation, planning and forecasting, as well as the Company’s organizational structure, to determine which practices and procedures should be modified or terminated, and management is assessing roles and responsibilities to enhance controls and compliance.
In addition, on December 11, 2016, our Board of Directors oversaw a change in the Company’s senior leadership when it appointed a new Interim Chief Executive Officer and a new Chief Financial Officer following the departures of our former Chief Executive Officer and Chief Financial Officer, as well as other personnel changes.
The Company is committed to maintaining a strong internal control environment. Management believes the foregoing efforts will effectively remediate the material weakness. We will provide further updates to the Company’s Board on an ongoing basis with measurable milestones and responsibilities regarding the progress of our remediation efforts.
| |
| Item 9A(T). | CONTROLS AND PROCEDURES. |
Not applicable
| |
| Item 9B. | OTHER INFORMATION. |
None.
PART III
| |
| Item 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE. |
The information required by this item with respect to our executive officers is provided under the caption entitled “Executive Officers of the Company” in Part I of this Annual Report on Form 10-K and is incorporated by reference herein. The information required by this item with respect to our directors and our audit committee and audit committee financial expert will be set forth in our definitive Proxy Statement under the captions “General Information About the Board of Directors” and “Election of Directors”, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
SECTION 16(a) BENEFICIAL OWNERSHIP REPORTING COMPLIANCE
The information regarding compliance with Section 16(a) of the Securities Exchange Act of 1934 required by this Item will be set forth in our definitive Proxy Statement under the caption “Section 16(a) Beneficial Ownership Reporting Compliance”, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
CODE OF ETHICS
We have adopted the Alexion Pharmaceuticals, Inc. Code of Ethics and Business Conduct, or code of ethics, that applies to directors, officers and employees of Alexion and its subsidiaries and complies with the requirements of Item 406 of Regulation S-K and the listing standards of the NASDAQNasdaq Global Select Market. Our code of ethics is located on our website (http://ir.alexionpharm.com/corporate-governance.cfm)ir.alexion.com/index.php/corporate-governance). We amended the code of ethics in September 2015 and any future amendments or waivers to our code of ethics will be promptly disclosed on our website and as required by applicable laws, rules and regulations of the SEC and NASDAQ.
Nasdaq.
| |
Item 11. | EXECUTIVE COMPENSATION. |
Item 11.EXECUTIVE COMPENSATION.
The information required by this Item will be set forth in our definitive Proxy Statement, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
| |
| Item 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
The information required by this Item will be set forth in our definitive Proxy Statement, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
| |
| Item 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE. |
The information required by this Item will be set forth in our definitive Proxy Statement, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
| |
| Item 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES. |
The information required by this Item will be set forth in our definitive Proxy Statement under the caption “Independent Registered Public Accounting Firm”, to be filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and is incorporated herein by reference to our Proxy Statement.
PART IV
| |
| Item 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES. |
The financial statements required by this item are submitted in a separate section beginning on page F-1 of this report.
| |
| (2) | Financial Statement Schedules |
Schedules have been omitted because of the absence of conditions under which they are required or because the required information is included in the financial statements or notes thereto beginning on page F-1 of this report.
|
| | |
|
| Agreement and Plan of Merger by and among Alexion, TPCA Corporation, Taligen Therapeutics, Inc., each stockholder of Taligen that signed the Agreement as a seller of Series Bl Call Rights, and, only for the limited purposes described therein as Stockholders’ Representatives (and not in their individual capacities), Nick Galakatos, Ed Hurwitz and Timothy Mills, dated as of January 28, 2011.(1)+ |
| | |
|
| Agreement and Plan of Merger by and among Alexion, EMRD Corporation, Enobia Pharma Corp., and the Stockholder Representatives named therein, dated as of December 28, 2011.(2)+ |
| | |
|
| Amendment No. 1 to the Agreement and Plan of Merger, dated December 28, 2011, by and among Alexion, EMRD Corporation, Enobia Pharma Corp., and the Stockholder Representatives named therein, dated February 1, 2012.(3) |
| | |
|
| Agreement, dated as of September 7, 2018, by and between Alexion Pharma Holding Unlimited Company, Shareholder Representative Services LLC, Fonds de Solidarité des Travailleurs du Québec F.T.Q., Capital Régional e Coopératif Desjardins, CTI Life Sciences Fund, L.P., OrbiMed Private Investments III, LP and OrbiMed Associates III, LP (in connection with the Agreement and Plan of Merger, dated December 28, 2011 pursuant to which Alexion acquired Enobia Pharma Corp.)(4) |
| |
|
| Agreement and Plan of Reorganization, dated May 5, 2015, among Alexion Pharmaceuticals, Inc., Pulsar Merger Sub Inc., Galaxy Merger Sub LLC and Synageva BioPharma Corp. (4)
(5) |
| | |
|
| Agreement and Plan of Merger, dated as of September 25, 2018, by and among Alexion Pharmaceuticals, Inc., Syracuse Merger Sub, Inc., Syntimmune, Inc. and Shareholder Representative Services LLC,(4)+ |
| |
|
| Certificate of Incorporation, as amended.(5)(6) |
| | |
|
| Certificate of Amendment of the Certificate of Incorporation.(6) |
| |
3.3 |
| Bylaws, as amended.(7) |
| | |
4.1 |
| Specimen Common Stock Certificate.Bylaws, as amended.(8) |
| | |
10.1 |
| Consulting Agreement, by and between Alexion Pharmaceuticals, Inc. and Dr. Leonard Bell, dated April 1, 2015.Specimen Common Stock Certificate.(9) |
| | |
10.2 |
| Amendment to the April 1, 2015,Consulting Agreement by and between Alexion Pharmaceuticals, Inc. and Dr. Leonard Bell, dated September 21, 2016.(26) |
| |
10.3 |
| Letter Agreement, by and between Alexion Pharmaceuticals, Inc. and Dr. Leonard Bell, dated April 1, 2015.(9) |
| |
10.4 |
| Confidential Separation Agreement and Release by and between Vikas Sinha and Alexion Pharmaceuticals, Inc. dated December 11, 2016. |
| |
10.5 |
| Confidential Release and Separation Agreement by and between David Hallal and Alexion Pharmaceuticals, Inc. dated December 11, 2016. |
| |
10.6 |
| Employment Agreement, dated as of December 11, 2016,March 27, 2017, by and between David BrennanLudwig N. Hantson and Alexion Pharmaceuticals, Inc. (23)** |
| | |
10.7 |
| Employment Agreement, dated as of December 12, 2016,June 11, 2017, by and between DavidPaul J. AndersonClancy and Alexion Pharmaceuticals, Inc. |
| |
10.8 |
| Employment Agreement, dated February 26, 2016, by and between Alexion Pharmaceuticals, Inc. and Clare Carmichael.(27) (24)** |
| | |
10.9 |
| Employment Agreement, dated February 26, 2016,as of June 1, 2017, by and between Brian Goff and Alexion Pharmaceuticals, Inc. and Martin Mackay.(27)(26)** |
| | |
10.10 |
| Employment Agreement, dated February 26, 2016, by and between Alexion Pharmaceuticals, Inc. and John Moriarty.(27)** |
| |
10.11 |
| Form of Employment Agreement (Senior Vice Presidents).(10)** |
| | |
10.12 |
| Form of Amendment No. 1 to Employment Agreements (Senior Vice Presidents). (11)** |
| | |
|
| | |
10.13 |
| Form of Indemnification Agreement for Officers and Directors. (12) |
| | |
10.14 |
| Lease, dated November 15, 2012, between Alexion and WE Route 34, LLC.(14) |
| |
10.15 |
| Alexion’s 2000 Stock Option Plan, as amended.(15)(13)** |
| | |
10.16 |
| Alexion’s 1992 Outside Directors Stock Option Plan, as amended.(16)(14)** |
| | |
10.17 |
| Alexion’s Amended and Restated 2004 Incentive Plan.(17)(15)** |
| | |
10.18 |
| License Agreement dated March 27, 1996 between Alexion and Medical Research Council.(18)(16)+ |
| | |
10.19 |
| | |
|
| Master Manufacturing and Supply Agreement, dated December 16, 2014 between Alexion Pharma International Trading, Alexion Pharmaceuticals, Inc,Inc., Lonza Group AG, Lonza Biologics Tuas PTE LTD and Lonza Sales AG. (24)*
(22)+ |
| | |
10.20 |
| Form of 2004 Incentive Plan Stock Option Agreement for Directors.(20)(18)** |
| | |
10.21 |
| Form of 2004 Incentive Plan Stock Option Agreement for Executive Officers (Form A).(21)(19)** |
| | |
10.22 |
| Form of 2004 Incentive Plan Stock Option Agreement for Executive Officers (Form B).(21)(19)** |
10.23 |
| Form of 2004 Incentive Plan Restricted Stock Award Agreement for Executive Officers (Form A).(22)(20)** |
| | |
10.24 |
| Form of 2004 Incentive Plan Stock Option Agreement (Incentive Stock Options).(19)(17) |
| | |
10.25 |
| Form of 2004 Incentive Plan Stock Option Agreement (Nonqualified Stock Options).(19)(17) |
| | |
10.26 |
| Form of 2004 Incentive Plan Restricted Stock Award Agreement.(19)(17) |
| | |
10.27 |
| Form of 2004 Incentive Plan Restricted Stock Unit Award Agreement.(23)(21) |
| | |
10.28 |
| Form of 2004 Incentive Plan Stock Option Agreement for Participants in France.(19)(17)** |
| | |
10.29 |
| Form of 2004 Incentive Plan Restricted Stock Unit Agreement for Participants in France.(19)(17)** |
| | |
10.30 |
| Amended and Restated Credit Agreement, dated as of June 22, 2015,7, 2018, by and among Alexion Pharmaceuticals, Inc,Inc., as administrative borrower, the guarantors referred to therein,subsidiary borrowers party thereto, the lenders referred to thereinand other financial institutions party thereto and Bank of America, N.A., as administrative agent. (25)(27) |
| | |
|
| Alexion Pharmaceuticals, Inc. 2017 Incentive Plan (25)** |
| |
|
| Form of 2017 Incentive Plan Restricted Stock Unit Agreement.(26)** |
| |
|
| Form of 2017 Incentive Plan Nonqualified Stock Option Agreement.(26)** |
| |
|
| Form of 2017 Incentive Plan Performance Stock Unit Agreement (TSR.)(26)** |
| |
|
| Form of 2017 Incentive Plan Performance Stock Unit Agreement (R&D Units.)(26)** |
| |
|
| Alexion Pharmaceuticals, Inc. 2017 Incentive Plan Rules for Awards Granted to Participants in France.(26)**
|
| |
|
| Form of 2017 Incentive Plan Restricted Stock Unit Agreement for French Participants.(26)** |
| |
|
| Form of 2017 Incentive Plan Global Stock Option Agreement.(26)** |
| |
|
| Alexion Pharmaceuticals, Inc. Amended and Restated 2015 Employee Stock Purchase Plan.(4)** |
| |
|
| Form of 2017 Incentive Plan Restricted Stock Unit Agreement for Non-U.S. Participants.(26)** |
| |
|
| Subsidiaries of Alexion Pharmaceuticals, Inc. |
| | |
|
| Consent of PricewaterhouseCoopers LLP, an Independent Registered Public Accounting Firm |
| | |
|
| Certificate of Chief Executive Officer pursuant to Exchange Act Rules 13a-14 and 15d-14, as adopted pursuant to Section 302 Sarbanes Oxley Act of 2002. |
| | |
|
| Certificate of Chief Financial Officer pursuant to Exchange Act Rules 13a-14 and 15d-14, as adopted pursuant to Section 302 of Sarbanes Oxley Act of 2002. |
| | |
|
| Certificate of Chief Executive Officer pursuant to Section 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act. |
| | |
|
| Certificate of Chief Financial Officer pursuant to Section 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act. |
| | |
| 101 |
| The following materials from the Alexion Pharmaceuticals, Inc. Annual Report on Form 10-K for the year ended December 31, 20162018 formatted in eXtensible Business Reporting Language (XBRL): (i) the Consolidated Statements of Operations, (ii) the Consolidated Statements of Comprehensive Income, (iii) the Consolidated Balance Sheets, (iv) the Consolidated Statements of Changes in Stockholders’ Equity, (v) the Consolidated Statements of Cash Flows and (vi) related notes, tagged as blocks of text. |
_____________________
| |
| (1) | Incorporated by reference to our Report on Form 8-K, filed on February 3, 2011. |
| |
| (2) | Incorporated by reference to our Report on Form 8-K, filed on January 4, 2012. |
| |
| (3) | Incorporated by reference to our Report on Form 8-K, filed on February 7, 2012. |
| |
| (4) | Incorporated by reference to our Quarterly Report on Form 10-Q, for the quarter ended September 30, 2018. |
| |
| (5) | Incorporated by reference to our Report on Form 8-K, filed on May 6, 2015. |
| |
(5)(6) | Incorporated by reference to our Registration Statement on Form S-3 (Reg. No. 333-128085), filed on September 2, 2005. |
| |
(6)(7) | Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2011. |
| |
(7)(8) | Incorporated by reference to our Report on Form 8-K, filed on January 8, 2016. |
| |
(8)(9) | Incorporated by reference to our Registration Statement on Form S-1 (Reg. No. 333-00202). |
| |
(9) | Incorporated by reference to our Report on Form 8-K, filed April 7, 2015. |
| |
| (10) | Incorporated by reference to our Report on Form 8-K, filed on February 16, 2006. |
| |
| (11) | Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2009. |
| |
| (12) | Incorporated by reference to our Report on Form 8-K, filed on September 17, 2010. |
| |
| (13) | Incorporated by reference to our Registration Statement on Form S-3 (Reg. No. 333-36738) filed on May 10, 2000. |
| |
(14) | Incorporated by reference to our Quarterly Report on Form 10-Q for the quarter ended March 31, 2013. |
| |
(15) | Incorporated by reference to our quarterly report on Form 10-Q for the quarter ended January 31, 2004. |
| |
(16)(14) | Incorporated by reference to our Registration Statement on Form S-8 (Reg. No. 333-71879) filed on February 5, 1999. |
| |
(17)(15) | Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2013. |
| |
(18)(16) | Incorporated by reference to our Annual Report on Form 10-K/A for the fiscal year ended July 31, 1996. |
| |
(19)(17) | Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2008. |
| |
(20)(18) | Incorporated by reference to our reportReport on Form 8-K, filed on December 16, 2004. |
| |
(21)(19) | Incorporated by reference to our Quarterly Report on Form 10-Q for the quarter ended January 31, 2005. |
| |
(22)(20) | Incorporated by reference to our reportReport on Form 8-K, filed on March 14, 2005. |
| |
(23)(21) | Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2010. |
| |
(24)(22) | Incorporated by reference to our Report on Form 10-K for the fiscal year ended December 31, 2014. |
| |
(25) | Incorporated by reference to our report on Form 8-K, filed on June 23, 2015. |
| |
(26) | Incorporated by reference to our report on Form 8-K, filed on September 22, 2016. |
| |
(27)(23) | Incorporated by reference to our Quarterly Report on Form 10-Q for the quarter ended March 31, 2016.2017. |
| |
| (24) | Incorporated by reference to our Quarterly Report on Form 10-Q for the quarter ended June 30, 2017. |
| |
| (25) | Incorporated by reference to our Registration Statement on Form S-8 (Reg. No. 333-217905) filed on May 5, 2017. |
| |
| (26) | Incorporated by reference to our Annual Report on Form 10-K for the fiscal year ended December 31, 2017. |
| |
| (27) | Incorporated by reference to our Report on Form 8-K, filed on June 13, 2018. |
| |
| + | Confidential treatment was granted for portions of such exhibit. |
| |
* | Confidential treatment requested under 17 C.F.R. §§200.80(b)(4) and 24b-2. The confidential portions of this exhibit have been omitted and are marked accordingly. The confidential portions have been filed separately with the SEC pursuant to the confidential treatment request. |
** Indicates a management contract or compensatory plan or arrangement required to be filed pursuant to Item 15(b) of Form 10-K.
Item 15(b) Exhibits
See (a) (3) above.
Item 15(c) Financial Statement Schedules
See (a) (2) above.
Item 16 Form 10-K Summary
Not applicable.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
| | | |
| | ALEXION PHARMACEUTICALS, INC. |
| | | |
| | By: | /s/ Ludwig N. Hantson, Ph.D. |
| Date: | February 6, 2019 | | ALEXION PHARMACEUTICALS, INC.Ludwig N. Hantson, Ph.D.
Chief Executive Officer (principal executive officer) |
| | |
By: | /s/ David R. Brennan |
| | David R. Brennan
Interim Chief Executive Officer (principal executive officer)
Dated: February 16, 2017
|
| | |
| By: | /s/ DavidPaul J. Anderson Clancy |
| Date: | February 6, 2019 | | DavidPaul J. Anderson, Clancy
Executive Vice President and Chief Financial Officer
(principal financial officer) Dated: February 16, 2017
|
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
| | | |
/s/ David R. BrennanLudwig N. Hantson | | Interim Chief Executive Officer and Director (principal executive officer) | February 16, 20176, 2019 |
David R. BrennanLudwig N. Hantson | | | |
| | | | |
/s/ DavidPaul J. AndersonClancy | | Executive Vice President and Chief Financial Officer (principal financial officer) | February 16, 20176, 2019 |
DavidPaul J. AndersonClancy | | | |
| | | | |
| /s/ Daniel A. Bazarko | | Senior Vice President and Chief Accounting Officer (principal accounting officer) | February 16, 20176, 2019 |
| Daniel A. Bazarko, C.P.A. | | | |
| | | | |
/s/ Leonard BellDavid R. Brennan | | Chairman | February 16, 20176, 2019 |
Leonard Bell, M.D.David R. Brennan | | | |
| | | | |
| /s/ Felix J. Baker | | Director | February 16, 20176, 2019 |
| Felix J. Baker, Ph.D. | | | |
| | | |
/s/ M. Michele Burns | | Director | February 16, 2017 |
M. Michele Burns | | | |
| | | | |
| /s/ Christopher J. Coughlin | | Director | February 16, 20176, 2019 |
| Christopher J. Coughlin | | | |
| | | |
| /s/ Deborah Dunsire | | Director | February 6, 2019 |
| Deborah Dunsire, M.D. | | | |
| | | |
| /s/ Paul A. Friedman | | Director | February 6, 2019 |
| Paul A. Friedman, M.D. | | | |
| | | | |
| /s/ John T. Mollen | | Director | February 16, 20176, 2019 |
| John T. Mollen | | | |
| | | | |
/s/ R. Douglas NorbyFrancois Nader | | Director | February 16, 20176, 2019 |
R. Douglas NorbyFrancois Nader, M.D. | | | |
| | | | |
/s/ Alvin S. ParvenJudith A. Reinsdorf | | Director | February 16, 20176, 2019 |
Alvin S. ParvenJudith A. Reinsdorf, J.D. | | | |
| | | | |
| /s/ Andreas Rummelt | | Director | February 16, 20176, 2019 |
| Andreas Rummelt, Ph.D. | | | |
| | | | |
/s/ Ann M. Veneman | | Director | February 16, 2017 |
Ann M. Veneman | | | |
| | | |
Alexion Pharmaceuticals, Inc.
Contents
For the Years Ended December 31, 2016, 20152018, 2017 and 20142016
|
| |
| | Page(s) |
| |
| Consolidated Financial Statements | |
| |
| |
| |
| |
|
|
| |
Report of Independent Registered Public Accounting Firm
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Alexion Pharmaceuticals, Inc.
Opinions on the Financial Statements and Internal Control over Financial Reporting
In our opinion,We have audited the accompanying consolidated balance sheets of Alexion Pharmaceuticals, Inc. and its subsidiaries(the “Company”) as of December 31, 2018 and December 31, 2017 and the relatedconsolidatedstatements of operations, comprehensive income, changes in stockholders’ equity and cash flowsfor each of the three years in the period ended December 31, 2018, including the related notes (collectively referred to as the “consolidated financial statements”).We also have audited the Company's internal control over financial reporting as of December 31, 2018, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
In our opinion, the consolidatedfinancial statements referred to above present fairly, in all material respects, the financial position of Alexion Pharmaceuticals, Inc.and its subsidiariesthe Company as of December 31, 20162018 and 2015, December 31, 2017and the results of theirits operations and theirits cash flows for each of the three years in the period endedDecember 31, 2016 2018in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company did not maintain,maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016,2018, based on criteria established in Internal Control - Integrated Framework (2013)issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) becauseCOSO.a material weakness in internal control over financial reporting existed as of that date related to not maintaining an effective control environment as senior management failed to set an appropriate tone at the top. Specifically, senior management failed to reinforce the need
Basis for compliance with the Company’s policies and procedures, which resulted in inappropriate business conduct. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis. Opinions
The material weakness referred to above is described in Management’s Report on Internal Control over Financial Reporting appearing under Item 9A. We considered this material weakness in determining the nature, timing, and extent of audit tests applied in our audit of the 2016consolidatedfinancial statements, and our opinion regarding the effectiveness of the Company’s internal control over financial reporting does not affect our opinion on those consolidatedfinancial statements. The Company’sCompany's management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in management’s report referred to above.Management's Report on Internal Control over Financial Reporting under Item 9A. Our responsibility is to express opinions on thesethe Company’s consolidated financial statements and on the Company’sCompany's internal control over financial reporting based on our integrated audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States).PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence supportingregarding the amounts and disclosures in the consolidated financial statements, assessingstatements. Our audits also included evaluating the accounting principles used and significant estimates made by management, andas well as evaluating the overall presentation of the consolidated financial statement presentation.statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
As discussed in Note 1 to the consolidated financial statements, the Company changed the manner in which it accounts for the classificationDefinition and Limitations of debt issuance costs and the manner in which it accounts for certain elements of its employee share-based payments in 2016.
Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/PricewaterhouseCoopers LLP
Hartford, Connecticut
February 16, 20176, 2019
We have served as the Company’s auditor since 2002.
Alexion Pharmaceuticals, Inc.
Consolidated Balance Sheets
(amounts in millions, except per share amounts)
| | | | December 31, | December 31, |
| | 2016 | | 2015 | 2018 | | 2017 |
| Assets | | | |
|
|
|
| Current Assets: | | | |
|
|
|
| Cash and cash equivalents | $ | 966 |
| | $ | 1,010 |
| $ | 1,365.5 |
|
| $ | 584.4 |
|
| Marketable securities | 327 |
| | 375 |
| 198.3 |
|
| 889.7 |
|
| Trade accounts receivable, net | 650 |
| | 533 |
| 922.3 |
|
| 726.5 |
|
| Inventories | 375 |
| | 290 |
| 472.5 |
|
| 460.4 |
|
| Prepaid expenses and other current assets | 260 |
| | 208 |
| 426.4 |
|
| 292.9 |
|
| Total current assets | 2,578 |
| | 2,416 |
| 3,385.0 |
|
| 2,953.9 |
|
| Property, plant and equipment, net | 1,036 |
| | 697 |
| 1,471.5 |
|
| 1,325.4 |
|
| Intangible assets, net | 4,303 |
| | 4,708 |
| 3,641.3 |
|
| 3,954.4 |
|
| Goodwill | 5,037 |
| | 5,048 |
| 5,037.4 |
|
| 5,037.4 |
|
| Other assets | 299 |
| | 228 |
| 396.7 |
|
| 312.2 |
|
| Total assets | $ | 13,253 |
| | $ | 13,097 |
| $ | 13,931.9 |
|
| $ | 13,583.3 |
|
| Liabilities and Stockholders’ Equity | | | |
|
|
|
| Current Liabilities: | | | |
|
|
|
| Accounts payable | $ | 64 |
| | $ | 57 |
| |
| Accrued expenses | 508 |
| | 403 |
| |
| Deferred revenue | 37 |
| | 21 |
| |
| Accounts payable and accrued expenses | | $ | 698.2 |
|
| $ | 710.2 |
|
| Revolving credit facility | | 250.0 |
| | — |
|
| Current portion of long-term debt | 167 |
| | 166 |
| 93.8 |
|
| 167.4 |
|
| Current portion of contingent consideration | 24 |
| | 56 |
| 97.6 |
|
| — |
|
| Other current liabilities | 23 |
| | 6 |
| 34.4 |
|
| 74.9 |
|
| Total current liabilities | 823 |
| | 709 |
| 1,174.0 |
|
| 952.5 |
|
| Long-term debt, less current portion | 2,888 |
| | 3,254 |
| 2,501.7 |
|
| 2,720.7 |
|
| Contingent consideration | 129 |
| | 121 |
| 183.2 |
|
| 168.9 |
|
| Facility lease obligation | 233 |
| | 151 |
| |
| Facility lease obligations | | 361.0 |
|
| 342.9 |
|
| Deferred tax liabilities | 396 |
| | 529 |
| 391.1 |
|
| 365.0 |
|
| Other liabilities | 90 |
| | 74 |
| 155.6 |
|
| 140.2 |
|
| Total liabilities | 4,559 |
| | 4,838 |
| 4,766.6 |
|
| 4,690.2 |
|
| Commitments and contingencies (Note 10) |
|
|
| |
| Commitments and contingencies (Note 11) | |
|
|
|
| Stockholders’ Equity: | | | |
|
|
|
| Common stock, $.0001 par value; 290 shares authorized; 232 and 230 shares issued at December 31, 2016 and 2015, respectively | — |
| | — |
| |
| Common stock, $.0001 par value; 290.0 shares authorized; 236.2 and 234.3 shares issued at 2018 and 2017, respectively | | — |
|
| — |
|
| Additional paid-in capital | 7,957 |
| | 7,727 |
| 8,539.1 |
|
| 8,290.3 |
|
| Treasury stock, at cost, 8 and 5 shares at December 31, 2016 and 2015, respectively | (1,141 | ) | | (711 | ) | |
| Accumulated other comprehensive income | 60 |
| | 62 |
| |
| Treasury stock, at cost, 12.7 and 12.0 shares at 2018 and 2017, respectively | | (1,689.9 | ) |
| (1,604.9 | ) |
| Accumulated other comprehensive loss | | (9.7 | ) |
| (34.4 | ) |
| Retained earnings | 1,818 |
| | 1,181 |
| 2,325.8 |
|
| 2,242.1 |
|
| Total stockholders’ equity | 8,694 |
| | 8,259 |
| 9,165.3 |
|
| 8,893.1 |
|
| Total liabilities and stockholders’ equity | $ | 13,253 |
| | $ | 13,097 |
| $ | 13,931.9 |
|
| $ | 13,583.3 |
|
The accompanying notes are an integral part of these consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Consolidated Statements of Operations
(amounts in millions, except per share amounts)
| | | | Year Ended December 31, | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 | 2018 |
| 2017 |
| 2016 |
| Net product sales | $ | 3,082 |
| | $ | 2,603 |
| | $ | 2,234 |
| $ | 4,130.1 |
| | $ | 3,549.5 |
| | $ | 3,081.7 |
|
| Other revenue | 2 |
| | 1 |
| | — |
| 1.1 |
| | 1.6 |
| | 2.4 |
|
| Total revenues | 3,084 |
| | 2,604 |
| | 2,234 |
| 4,131.2 |
| | 3,551.1 |
| | 3,084.1 |
|
| Cost of sales | 258 |
| | 233 |
| | 174 |
| 374.3 |
| | 454.2 |
| | 258.3 |
|
| Operating expenses: |
| |
| |
|
| |
| |
|
| Research and development | 757 |
| | 709 |
| | 514 |
| 730.4 |
| | 878.4 |
| | 757.2 |
|
| Selling, general and administrative | 954 |
| | 863 |
| | 630 |
| 1,111.8 |
| | 1,094.4 |
| | 953.0 |
|
| Acquired in-process research and development | | 1,183.0 |
| | — |
| | — |
|
| Amortization of purchased intangible assets | 322 |
| | 117 |
| | — |
| 320.1 |
| | 320.1 |
| | 322.2 |
|
| Change in fair value of contingent consideration | 36 |
| | 64 |
| | 20 |
| 116.5 |
| | 41.0 |
| | 35.7 |
|
| Acquisition-related costs | 2 |
| | 39 |
| | — |
| — |
| | — |
| | 2.3 |
|
| Restructuring expenses | 3 |
| | 42 |
| | 15 |
| 25.5 |
| | 104.6 |
| | 3.0 |
|
| Impairment of intangible assets | 85 |
| | — |
| | 12 |
| — |
| | 31.0 |
| | 85.0 |
|
| Total operating expenses | 2,159 |
| | 1,834 |
| | 1,191 |
| 3,487.3 |
| | 2,469.5 |
| | 2,158.4 |
|
| Operating income | 667 |
| | 537 |
| | 869 |
| 269.6 |
| | 627.4 |
| | 667.4 |
|
| Other income and expense: |
| |
| |
|
| |
| |
|
| Investment income | 11 |
| | 8 |
| | 8 |
| 65.3 |
| | 18.5 |
| | 10.9 |
|
| Interest expense | (97 | ) | | (48 | ) | | (3 | ) | (98.2 | ) | | (98.4 | ) | | (96.9 | ) |
| Foreign currency gain (loss) | (5 | ) | | 1 |
| | (2 | ) | |
| Other income and (expense) | | 5.5 |
| | 0.3 |
| | (5.2 | ) |
| Income before income taxes | 576 |
| | 498 |
| | 872 |
| 242.2 |
| | 547.8 |
| | 576.2 |
|
| Income tax expense | 177 |
| | 354 |
| | 215 |
| 164.6 |
| | 104.5 |
| | 176.8 |
|
| Net income | $ | 399 |
| | $ | 144 |
| | $ | 657 |
| $ | 77.6 |
| | $ | 443.3 |
| | $ | 399.4 |
|
| Earnings per common share |
| |
| |
|
| |
| |
|
| Basic | $ | 1.78 |
| | $ | 0.68 |
| | $ | 3.32 |
| $ | 0.35 |
| | $ | 1.98 |
| | $ | 1.78 |
|
| Diluted | $ | 1.76 |
| | $ | 0.67 |
| | $ | 3.26 |
| $ | 0.35 |
| | $ | 1.97 |
| | $ | 1.76 |
|
| Shares used in computing earnings per common share |
| |
| |
|
| |
| |
|
| Basic | 224 |
| | 213 |
| | 198 |
| 222.7 |
| | 223.9 |
| | 224.3 |
|
| Diluted | 227 |
| | 216 |
| | 202 |
| 224.5 |
| | 225.4 |
| | 226.3 |
|
| | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Consolidated Statements of Comprehensive Income
(amounts in millions)
|
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 |
| Net income | $ | 399 |
| | $ | 144 |
| | $ | 657 |
|
| Other comprehensive (loss) income, net of tax: | | | | | |
| Foreign currency translation | (4 | ) | | (6 | ) | | (6 | ) |
| Unrealized losses on marketable securities | — |
| | (1 | ) | | — |
|
| Unrealized gains (losses) on pension obligation | 3 |
| | 7 |
| | (5 | ) |
| Unrealized (losses) gains on hedging activities, net of tax of $0, $6 and $45, respectively | (1 | ) | | 6 |
| | 91 |
|
| Other comprehensive (loss) income, net of tax | (2 | ) | | 6 |
| | 80 |
|
| Comprehensive income | $ | 397 |
| | $ | 150 |
| | $ | 737 |
|
|
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2018 |
| 2017 |
| 2016 |
| Net income | $ | 77.6 |
| | $ | 443.3 |
| | $ | 399.4 |
|
| Other comprehensive income (loss), net of tax: | | | | | |
| Foreign currency translation | (0.5 | ) | | 8.4 |
| | (4.3 | ) |
| Unrealized (losses) gains on debt securities | (0.5 | ) | | 0.6 |
| | 0.4 |
|
| Unrealized gains on pension obligation | 2.2 |
| | 1.9 |
| | 2.9 |
|
| Unrealized gains (losses) on hedging activities, net of tax of $7.3, $(59.0) and $(0.2), respectively | 23.5 |
| | (105.8 | ) | | (0.8 | ) |
| Other comprehensive income (loss), net of tax | 24.7 |
| | (94.9 | ) | | (1.8 | ) |
| Comprehensive income | $ | 102.3 |
| | $ | 348.4 |
| | $ | 397.6 |
|
The accompanying notes are an integral part of these consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Consolidated Statements of Changes in Stockholders’ Equity
(amounts in millions)
| | | | Common Stock | | Additional Paid-In Capital | | Treasury Stock at Cost | | Accumulated Other Comprehensive Income (Loss) | | Retained Earnings (Deficit) | | Total Stockholders’ Equity | Common Stock | | Additional Paid-In Capital | | Treasury Stock at Cost | | Accumulated Other Comprehensive Income (Loss) | | Retained Earnings | | Total Stockholders’ Equity |
| | Shares Issued | | Amount | | Shares | | Amount | | Shares Issued | | Amount | | Shares | | Amount | |
| Balances, December 31, 2013 | 198 |
| | $ | — |
| | $ | 2,107 |
| | 1 |
| | $ | (80 | ) | | $ | (24 | ) | | $ | 380 |
| | $ | 2,383 |
| |
| Repurchase of common stock | — |
| | — |
| | — |
| | 2 |
| | (303 | ) | | — |
| | — |
| | (303 | ) | |
| Issuance of common stock from exercise of options | 3 |
| | — |
| | 114 |
| | — |
| | — |
| | — |
| | — |
| | 114 |
| |
| Issuance of restricted common stock | 1 |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| |
| Excess tax benefit from stock options | — |
| | — |
| | 251 |
| | — |
| | — |
| | — |
| | — |
| | 251 |
| |
| Share-based compensation expense | — |
| | — |
| | 121 |
| | — |
| | — |
| | — |
| | — |
| | 121 |
| |
| Net income | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 657 |
| | 657 |
| |
| Other comprehensive income | — |
| | — |
| | — |
| | — |
| | — |
| | 80 |
| | — |
| | 80 |
| |
| Balances, December 31, 2014 | 202 |
| | $ | — |
| | $ | 2,593 |
| | 3 |
| | $ | (383 | ) | | $ | 56 |
| | $ | 1,037 |
| | $ | 3,303 |
| |
| Repurchase of common stock | — |
| | — |
| | — |
| | 2 |
| | (328 | ) | | — |
| | — |
| | (328 | ) | |
| Issuance of common stock, net of issuance costs of $4 | 26 |
| | — |
| | 4,914 |
| | — |
| | — |
| | — |
| | — |
| | 4,914 |
| |
| Issuance of common stock under stock option and stock purchase plans | 1 |
| | — |
| | 82 |
| | — |
| | — |
| | — |
| | — |
| | 82 |
| |
| Issuance of restricted common stock | 1 |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| |
| Excess tax benefit from stock options | — |
| | — |
| | (90 | ) | | — |
| | — |
| | — |
| | — |
| | (90 | ) | |
| Share-based compensation expense | — |
| | — |
| | 228 |
| | — |
| | — |
| | — |
| | — |
| | 228 |
| |
| Net income | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 144 |
| | 144 |
| |
| Other comprehensive income | — |
| | — |
| | — |
| | — |
| | — |
| | 6 |
| | — |
| | 6 |
| |
| Balances, December 31, 2015 | 230 |
| | $ | — |
| | $ | 7,727 |
| | 5 |
| | $ | (711 | ) | | $ | 62 |
| | $ | 1,181 |
| | $ | 8,259 |
| 230.5 |
| | $ | — |
| | $ | 7,726.6 |
| | 4.9 |
| | $ | (710.7 | ) | | $ | 62.3 |
| | $ | 1,180.4 |
| | $ | 8,258.6 |
|
| Repurchase of common stock | — |
| | — |
| | — |
| | 3 |
| | (430 | ) | | — |
| | — |
| | (430 | ) | — |
| | — |
| | — |
| | 3.1 |
| | (430.6 | ) | | — |
| | — |
| | (430.6 | ) |
| Issuance of common stock under stock option and stock purchase plans | 1 |
| | — |
| | 37 |
| | — |
| | — |
| | — |
| | — |
| | 37 |
| 0.6 |
| | — |
| | 37.1 |
| | — |
| | — |
| | — |
| | — |
| | 37.1 |
|
| Issuance of restricted common stock | 1 |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| 0.8 |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
|
| Excess tax benefit from stock options | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| |
| Share-based compensation expense | — |
| | — |
| | 193 |
| | — |
| | — |
| | — |
| | — |
| | 193 |
| — |
| | — |
| | 193.3 |
| | — |
| | — |
| | — |
| | — |
| | 193.3 |
|
| Net income | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 399 |
| | 399 |
| — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 399.4 |
| | 399.4 |
|
| Other comprehensive loss | — |
| | — |
| | — |
| | — |
| | — |
| | (2 | ) | | — |
| | (2 | ) | — |
| | — |
| | — |
| | — |
| | — |
| | (1.8 | ) | | — |
| | (1.8 | ) |
| Adoption of new share-based compensation guidance | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 238 |
| | 238 |
| — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 237.8 |
| | 237.8 |
|
| Balances, December 31, 2016 | $ | 232 |
| | $ | — |
| | $ | 7,957 |
| | $ | 8 |
| | $ | (1,141 | ) | | $ | 60 |
| | $ | 1,818 |
| | $ | 8,694 |
| 231.9 |
| | $ | — |
| | $ | 7,957.0 |
| | 8.0 |
| | $ | (1,141.3 | ) | | $ | 60.5 |
| | $ | 1,817.6 |
| | $ | 8,693.8 |
|
| Repurchase of common stock | | — |
| | — |
| | — |
| | 4.0 |
| | (463.6 | ) | | — |
| | — |
| | (463.6 | ) |
| Issuance of common stock under stock option and stock purchase plans | | 1.3 |
| | — |
| | 85.9 |
| | — |
| | — |
| | — |
| | — |
| | 85.9 |
|
| Issuance of restricted common stock | | 1.1 |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
|
| Share-based compensation expense | | — |
| | — |
| | 247.4 |
| | — |
| | — |
| | — |
| | — |
| | 247.4 |
|
| Net income | | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 443.3 |
| | 443.3 |
|
| Other comprehensive loss | | — |
| | — |
| | — |
| | — |
| | — |
| | (94.9 | ) | | — |
| | (94.9 | ) |
| Adoption of new intra-entity tax guidance | | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | (18.8 | ) | | (18.8 | ) |
| Balances, December 31, 2017 | | 234.3 |
| | $ | — |
| | $ | 8,290.3 |
| | 12.0 |
| | $ | (1,604.9 | ) | | $ | (34.4 | ) | | $ | 2,242.1 |
| | $ | 8,893.1 |
|
| Repurchase of common stock | | — |
| | — |
| | — |
| | 0.7 |
| | (85.0 | ) | | — |
| | — |
| | (85.0 | ) |
| Issuance of common stock under stock option and stock purchase plans | | 0.6 |
| | — |
| | 47.6 |
| | — |
| | — |
| | — |
| | — |
| | 47.6 |
|
| Issuance of restricted common stock | | 1.3 |
| | — |
| | (0.3 | ) | | — |
| | — |
| | — |
| | — |
| | (0.3 | ) |
| Share-based compensation expense | | — |
| | — |
| | 201.5 |
| | — |
| | — |
| | — |
| |
|
| | 201.5 |
|
| Net income | | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 77.6 |
| | 77.6 |
|
| Other comprehensive income | | — |
| | — |
| | — |
| | — |
| | — |
| | 24.7 |
| | — |
| | 24.7 |
|
| Adoption of new accounting standards | | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 6.1 |
| | 6.1 |
|
| Balances, December 31, 2018 | | 236.2 |
| | $ | — |
| | $ | 8,539.1 |
| | 12.7 |
| | $ | (1,689.9 | ) | | $ | (9.7 | ) | | $ | 2,325.8 |
| | $ | 9,165.3 |
|
The accompanying notes are an integral part of these consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(amounts in millions)
| | | | Year Ended December 31, | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 | 2018 | | 2017 | | 2016 |
| Cash flows from operating activities: | | | | | | | | | | |
| Net income | $ | 399 |
| | $ | 144 |
| | $ | 657 |
| $ | 77.6 |
| | $ | 443.3 |
| | $ | 399.4 |
|
| Adjustments to reconcile net income to net cash flows from operating activities: | | | | | | | | | | |
| Depreciation and amortization | 396 |
| | 167 |
| | 47 |
| 405.3 |
| | 496.7 |
| | 396.4 |
|
| Impairment of intangible assets | 85 |
| | — |
| | 12 |
| |
| Impairment of assets | | 13.5 |
| | 118.8 |
| | 85.0 |
|
| Change in fair value of contingent consideration | 36 |
| | 64 |
| | 20 |
| 116.5 |
| | 41.0 |
| | 35.7 |
|
| Payments of contingent consideration | | — |
| | (18.0 | ) | | — |
|
| Share-based compensation expense | 192 |
| | 227 |
| | 114 |
| 203.0 |
| | 243.1 |
| | 192.3 |
|
| Non-cash expense for acquired IPR&D | | 64.6 |
| | — |
| | — |
|
| Deferred taxes | 104 |
| | 395 |
| | (154 | ) | 32.9 |
| | (45.9 | ) | | 104.3 |
|
| Change in excess tax benefit from stock options | — |
| | 90 |
| | (251 | ) | |
| Unrealized foreign currency loss (gain) | | 4.8 |
| | (9.4 | ) | | 6.9 |
|
| Unrealized (gain) loss on forward contracts | | (15.8 | ) | | 11.1 |
| | (3.6 | ) |
| Unrealized gain on equity investments | | (40.2 | ) | | — |
| | — |
|
| Other | 10 |
| | 3 |
| | 37 |
| (2.0 | ) | | 5.4 |
| | 6.5 |
|
| Changes in operating assets and liabilities, excluding the effect of acquisitions: | | | | | | | | | | |
| Accounts receivable | (122 | ) | | (116 | ) | | (28 | ) | (208.8 | ) | | (55.2 | ) | | (122.1 | ) |
| Inventories | (84 | ) | | (88 | ) | | (67 | ) | (14.7 | ) | | (88.2 | ) | | (83.8 | ) |
| Prepaid expenses and other assets | (97 | ) | | (57 | ) | | (18 | ) | (155.6 | ) | | (137.2 | ) | | (97.5 | ) |
| Accounts payable, accrued expenses and other liabilities | 150 |
| | (116 | ) | | 265 |
| (55.1 | ) | | 110.1 |
| | 166.8 |
|
| Deferred revenue | 17 |
| | (38 | ) | | 6 |
| |
| Net cash provided by operating activities | 1,086 |
| | 675 |
| | 640 |
| 426.0 |
| | 1,115.6 |
| | 1,086.3 |
|
| Cash flows from investing activities: | | | | | | | | | | |
| Purchases of available-for-sale securities | (667 | ) | | (520 | ) | | (664 | ) | |
| Proceeds from maturity or sale of available-for-sale securities | 718 |
| | 1,160 |
| | 620 |
| |
| Purchases of trading securities | (8 | ) | | (15 | ) | | (3 | ) | |
| Proceeds from sale of trading securities | 4 |
| | 10 |
| | — |
| |
| Purchases of available-for-sale debt securities | | (782.7 | ) | | (1,648.8 | ) | | (667.1 | ) |
| Proceeds from maturity or sale of available-for-sale debt securities | | 1,473.5 |
| | 1,089.9 |
| | 717.8 |
|
| Purchases of mutual funds related to nonqualified deferred compensation plan | | (12.1 | ) | | (9.9 | ) | | (8.5 | ) |
| Proceeds from sale of mutual funds related to nonqualified deferred compensation plan | | 12.3 |
| | 7.7 |
| | 4.0 |
|
| Purchases of property, plant and equipment | (333 | ) | | (286 | ) | | (137 | ) | (213.0 | ) | | (357.3 | ) | | (332.7 | ) |
| Purchases of other investments | — |
| | — |
| | (38 | ) | (10.3 | ) | | — |
| | — |
|
| Payments for acquisitions of businesses, net of cash acquired | — |
| | (3,939 | ) | | — |
| |
| Other | (1 | ) | | 5 |
| | (1 | ) | 2.8 |
| | 0.1 |
| | (1.1 | ) |
| Net cash used in investing activities | (287 | ) | | (3,585 | ) | | (223 | ) | |
| Net cash provided by (used in) investing activities | | 470.5 |
| | (918.3 | ) | | (287.6 | ) |
| Cash flows from financing activities: | | | | | | | | | | |
| Debt issuance costs | — |
| | (45 | ) | | — |
| |
| Proceeds from revolving credit facility | — |
| | 200 |
| | — |
| 250.0 |
| | — |
| | — |
|
| Payments on revolving credit facility | — |
| | (200 | ) | | — |
| |
| Proceeds from term loan | — |
| | 3,500 |
| | — |
| |
| Payments on term loan | (375 | ) | | (101 | ) | | (55 | ) | (293.8 | ) | | (175.0 | ) | | (375.0 | ) |
| Equity issuance costs for shares issued in connection with acquisition of business | — |
| | (4 | ) | | — |
| |
| Change in excess tax benefit from stock options | — |
| | (90 | ) | | 251 |
| |
| Repurchase of common stock | (430 | ) | | (328 | ) | | (303 | ) | (85.0 | ) | | (463.6 | ) | | (430.6 | ) |
| Net proceeds from issuance of stock under share-based compensation arrangements | 37 |
| | 82 |
| | 114 |
| 47.3 |
| | 85.9 |
| | 37.1 |
|
| Payment of contingent consideration | (60 | ) | | (50 | ) | | — |
| |
| Proceeds from development-related grants | — |
| | 26 |
| | — |
| |
| Payments of contingent consideration | | — |
| | (7.0 | ) | | (60.0 | ) |
| Repayment of development-related grants | | — |
| | (26.0 | ) | | — |
|
| Other | (8 | ) | | (5 | ) | | — |
| (20.9 | ) | | (10.9 | ) | | (7.7 | ) |
| Net cash (used in) provided by financing activities | (836 | ) | | 2,985 |
| | 7 |
| |
| Effect of exchange rate changes on cash | (7 | ) | | (9 | ) | | (10 | ) | |
| Net change in cash and cash equivalents | (44 | ) | | 66 |
| | 414 |
| |
| Cash and cash equivalents at beginning of period | 1,010 |
| | 944 |
| | 530 |
| |
| Cash and cash equivalents at end of period | $ | 966 |
| | $ | 1,010 |
| | $ | 944 |
| |
| Net cash used in financing activities | | (102.4 | ) | | (596.6 | ) | | (836.2 | ) |
| Effect of exchange rate changes on cash and cash equivalents and restricted cash | | (11.2 | ) | | 17.7 |
| | (6.6 | ) |
| Net change in cash and cash equivalents and restricted cash | | 782.9 |
| | (381.6 | ) | | (44.1 | ) |
| Cash and cash equivalents and restricted cash at beginning of period | | 584.4 |
| | 966.0 |
| | 1,010.1 |
|
| Cash and cash equivalents and restricted cash at end of period | | $ | 1,367.3 |
| | $ | 584.4 |
| | $ | 966.0 |
|
| | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(amounts in millions)
| | | | Year Ended December 31, | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 | 2018 | | 2017 | | 2016 |
| | | | | | | | | | | |
| Supplemental cash flow disclosures: | | | | | | | | | | |
| Cash paid for interest (net of amounts capitalized) | $ | 80 |
| | $ | 41 |
| | $ | 2 |
| $ | 90.9 |
| | $ | 95.3 |
| | $ | 79.6 |
|
| Cash paid for income taxes | $ | 38 |
| | $ | 123 |
| | $ | 91 |
| $ | 163.9 |
| | $ | 162.1 |
| | $ | 37.7 |
|
| Supplemental cash flow disclosures from investing and financing activities: | | �� | | | | | | | | |
| Common stock issued in acquisition of business | $ | — |
| | $ | 4,918 |
| | $ | — |
| |
| Capitalization of construction costs related to facility lease obligations | $ | 103 |
| | $ | 41 |
| | $ | 75 |
| $ | 44.8 |
| | $ | 121.8 |
| | $ | 103.1 |
|
| Accrued expenses for purchases of property, plant and equipment | $ | 23 |
| | $ | 30 |
| | $ | 17 |
| |
| Accrued expenses for purchases of property, plant and equipment and intangible assets | | $ | 21.4 |
| | $ | 34.7 |
| | $ | 23.5 |
|
The accompanying notes are an integral part of these consolidated financial statements.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
| |
| 1. | Business Overview and Summary of Significant Accounting Policies |
Business
Alexion Pharmaceuticals, Inc. (Alexion, the Company, we, our or us) is a global biopharmaceutical company focused on serving patients with devastating and ultra-rare disordersfamilies affected by rare diseases through the innovation, development and commercialization of life-transforming therapeutic products.life-changing therapies.
In ourWe are the global leader in complement franchise, Soliris® isinhibition and have developed and commercialize the only two approved complement inhibitors to treat patients with paroxysmal nocturnal hemoglobinuria (PNH), as well as the first and only therapeutic approved for patients with either paroxysmal nocturnal hemoglobinuria (PNH), a life-threatening and ultra-rare genetic blood disorder, orcomplement inhibitor to treat atypical hemolytic uremic syndrome (aHUS), a life-threatening and ultra-rare genetic disease. PNH and aHUS areanti-acetylcholine receptor (AchR) antibody-positive generalized myasthenia gravis (gMG). In addition, Alexion has two disorders resulting from chronic uncontrolled activation of the complement component of the immune system.
In our metabolic franchise, we commercialize Strensiq® for the treatment of patients with Hypophosphatasia (HPP) and Kanuma® for the treatment of patients with Lysosomal Acid Lipase Deficiency (LAL-D). HPP is an ultra-rare genetic disease characterized by defective bone mineralization that can lead to deformity of bones and other skeletal abnormalities. LAL-D is a serious, life threatening ultra-rare disease in which genetic mutations result in decreased activity of the Lysosomal Acid Lipase (LAL)highly innovative enzyme leading to marked accumulation of lipids in vital organs, blood vessels and other tissues. We initiated sales of these products in the third quarter 2015.
We are also evaluating additional potential indications for eculizumab in other severe and devastating diseases in which uncontrolled complement activation is the underlying mechanism, and we are progressing in various stages of development with additional product candidates as potential treatments for patients with devastating and ultra-rare disorders.
In June 2015, we acquired all of the outstanding shares of common stock of Synageva BioPharma Corp. (Synageva), a publicly-held clinical-stage biotechnology company. The acquisition furthered our objective to develop and commercialize life-transformingreplacement therapies for patients with devastatinglife-threatening and ultra-rare diseases.metabolic disorders, hypophosphatasia (HPP) and lysosomal acid lipase deficiency (LAL-D).
As the leader in complement biology for over 20 years, Alexion focuses its research efforts on novel molecules and targets in the complement cascade, and its development efforts on the core therapeutic areas of hematology, nephrology, neurology, and metabolic disorders. We were incorporated in 1992 under the laws of the State of Delaware.
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements include the accounts of Alexion and its wholly-owned subsidiaries. All intercompany balances and transactions have been eliminated in consolidation. For each of our business combinations, all of the assets acquired and liabilities assumed were recorded at their respective fair values as of the date of acquisition, and their results of operations are included in the consolidated financial statements from the date of acquisition.
Dividend Policy
We have never paid a cash dividend on shares of our stock. We currently intend to retain our earnings to finance future operations and do not anticipate paying any cash dividends on our stock in the foreseeable future.
Critical Accounting Estimates
The preparation of our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S., requires us to make estimates, judgments and assumptions that may affect the reported amounts of assets, liabilities, revenues, expenses and related disclosure of contingent assets and liabilities in our financial statements. We believe the most complex judgments result primarily from the need to make estimates about the effects of matters that are inherently uncertain and are significant to our consolidated financial statements. We base our estimates on historical experience and on various other assumptions that we believe are reasonable, the results of which form the basis for making judgments about the carrying values of assets and liabilities. We evaluate our estimates, judgments and assumptions on an ongoing basis. Actual results may differ from these estimates under different assumptions or conditions.
The most significant areas involving estimates, judgments and assumptions used in the preparation of our consolidated financial statements are as follows:
Revenue recognition;
Contingent liabilities;
Inventories;
Share-based compensation;
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
Valuation of goodwill, acquired intangible assets and in-process research and development (IPR&D);
Valuation of contingent consideration; and
Income taxes.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
Foreign Currency Translation
The financial statements of our subsidiaries with functional currencies other than the U.S. dollar are translated into U.S. dollars using period-end exchange rates for assets and liabilities, historical exchange rates for stockholders’ equity and weighted average exchange rates for operating results. Translation gains and losses are included in accumulated other comprehensive income (loss), net of tax, in stockholders’ equity. Foreign currency transaction gains and losses are included in the results of operations in other income and expense.
Cash and Cash Equivalents
Cash and cash equivalents are stated at cost plus accrued interest, which approximates fair value, and include short-term highly liquid investments with original maturities of three months or less. As of December 31, 2018 and 2017, cash equivalents were comprised of money market funds, reverse repurchase agreements, and other debt securities with maturities less than 90 days from the date of purchase.
Fair Value of Financial Instruments
The carrying amounts reflected in the consolidated balance sheets for cash and cash equivalents, accounts receivable, other assets, accounts payable, accrued expenses and other liabilities approximate fair value due to their short-term maturities. Our marketable securities are valued based upon pricing of securities with similar investment characteristics and holdings. Our mutual fund investments and equity securities are valued based on quoted market prices in active markets with no valuation adjustment. Investments in equity securities of publicly traded companies which are subject to holding period restrictions are carried at fair value using an option pricing valuation model and observable market inputs such as the historical volatility of similar companies and risk-free interest rates. Our derivative financial instruments are measured at fair value using observable market inputs such as forward rates, interest rates, our own credit risk and our counterparties’ credit risks. Our debt obligations are carried at historical cost, which approximates fair value. Our contingent consideration liabilities related to our acquisitions are valued based on various estimates, including probability of success, estimated revenues, discount rates and amount of time until the conditions of the milestone payments are met.
Marketable Securities
We invest our excess cash balances in marketable securities of highly rated financial institutions and investment-grade debt instruments. We seek to diversify our investments and limit the amount of investment concentrations for individual institutions, maturities and investment types. We classify these marketable debt securities as available-for-sale and, accordingly, record such securities at fair value. We classify these marketable securities as current assets as these investments are intended to be available to the Company for use in funding current operations.
Unrealized gains and losses on our marketable debt securities that are deemed temporary are included in accumulated other comprehensive income (loss) as a separate component of stockholders’ equity. If any adjustment to fair value reflects a significant decline in the value of the security, we evaluate the extent to which the decline is determined to be other-than-temporary and would mark the security to market through a charge to our consolidated statement of operations. Credit losses are identified when we do not expect to receive cash flows sufficient to recover the amortized cost basis of a security. In the event of a credit loss, only the amount associated with the credit loss is recognized in operating results, with the amount of loss relating to other factors recorded in accumulated other comprehensive income (loss).
We sponsor a nonqualified deferred compensation plan which allows certain highly-compensated employees to elect to defer income to future periods. Participants in the plan earn a return on their deferrals based on several investments options, which mirror returns on underlying mutual fund investments. We choose to invest in the underlying mutual fund investments to offset the liability associated with our nonqualified deferred compensation plan. These securitiesmutual fund investments are classified as trading securitiesvalued at net asset value per share and are carried at fair value with gains and losses included in investment income. The changes in the underlying liability to the employee are recorded in operating expenses.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
Accounts Receivable
Our standard credit terms vary based on the country of sale and range from 30 to 120 days. days and all arrangements are payable within one year of the transfer of the product. Our consolidated average days’ sales outstanding ranges from 60 to 8070 days. We evaluate the creditworthiness of customers on a regular basis. In certain European countries, sales by us are subject to payment terms that are statutorily determined. This is primarily the case in countries where the payer is government-owned or government-funded, which we consider to be creditworthy. The length of time from sale to receipt of payment in certain countries exceeds our credit terms. In countries in which collections from
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
customers extend beyond normal payment terms, we seek to collect interest. We record interest on customer receivables as interest income when collected. For non-interest bearing receivables with an estimated payment beyond one year, we discount the accounts receivable to present value at the date of sale, with a corresponding adjustment to revenue. Subsequent adjustments for further declines in credit rating are recorded as bad debt expense as a component of selling, general and administrative expense. We also use judgments as to our ability to collect outstanding receivables and provide allowances for the portion of receivables if and when collection becomes doubtful, and we also assess on an ongoing basis whether collectibility is reasonably assuredprobable at the time of sale. As of December 31, 2018 and 2017, allowances on receivables were not material.
Concentration of Credit Risk
Financial instruments that potentially expose the Company to concentrations of credit risk are limited to cash equivalents, marketable securities, accounts receivable and our foreign exchange derivative contracts. We invest our cash reserves in money market funds or high-quality marketable debt securities in accordance with our investment policy. The stated objectives of our investment policy is to preserve capital, provide liquidity consistent with forecasted cash flow requirements, maintain appropriate diversification and generate returns relative to these investment objectives and prevailing market conditions.
At December 31, 2016,2018, three customers accounted for 47%48.7% of the accounts receivable balance, with these individual customers ranging from 14%14.0% to 19%19.1% of the accounts receivable balance. At December 31, 2015, three2017, four customers accounted for 51%57.7% of the accounts receivable balance, with these individual customers ranging from 14%10.2% to 22%18.9% of the accounts receivable balance.
For the year ended December 31, 2018, four customers accounted for 50.3% of our product sales, with these individual customers ranging from 10.0% to 16.4% of our product sales. For the year endedDecember 31, 2016,2017, three customers accounted for 37% of our product sales, with these individual customers ranging from 10%10.8% to 16%15.0% of our product sales. For the year endedDecember 31, 2015,2016, three customers accounted for 38%36.7% of our product sales, with these individual customers ranging from 10%10.0% to 18%16.0% of our product sales. No other customers accounted for more than 10%10.0% of accounts receivable or net product sales. David Anderson, Alexion’s Executive Vice President and Chief Financial Officer since December 2016, has been a member of the Board of Directors of Cardinal Health, Inc. since April 2014. Cardinal Health, Inc. and its affiliates provide product distribution and other services to Alexion in the United States.
As a result of our foreign operations, we are exposed to changes in the general economic conditions in the countries in which we conduct business. Substantially all of our accounts receivable due from these countries are due from or backed by sovereign or local governments, and the amount of non-sovereign accounts receivable is not material. We continue to monitor economic conditions, including volatility associated with international economies and the associated impacts on the financial markets and our business. AlthoughSubstantially all of our accounts receivable are due from wholesale distributors, public hospitals and other government entities. We monitor the financial performance of our customers so that we can appropriately respond to changes in their credit worthiness. We can operate in certain jurisdictions where weakness in economic conditions can result in extended collection periods. To date, we have not experienced any significant losses with respect to collection of our accounts receivables due from certain countries may extend beyond our standard credit terms, we do not expect any such delays to have a material impact on our financial condition or results of operations.receivable.
Inventories
Inventories are stated at the lower of cost or estimatednet realizable value. We determine the cost of inventory onCost is determined in a standard cost basis, whichmanner that approximates average costs.
The components of inventory are as follows:
| | | | December 31, | December 31, |
| | 2016 | | 2015 | 2018 | | 2017 |
| Raw materials | $ | 17 |
| | $ | 18 |
| $ | 31.4 |
| | $ | 4.7 |
|
| Work-in-process | 143 |
| | 180 |
| 90.4 |
| | 148.6 |
|
| Finished goods | 215 |
| | 92 |
| 350.7 |
| | 307.1 |
|
| | $ | 375 |
| | $ | 290 |
| $ | 472.5 |
| | $ | 460.4 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
Capitalization of Inventory Costs
We capitalize inventory produced for commercial sale, which may include costs incurred for certain products awaiting regulatory approval. We capitalize inventory produced in preparation of product launches sufficient to support estimated initial market demand. Capitalization of such inventory begins when we have (i) obtained positive results in clinical trials that we believe are necessary to support regulatory approval, (ii) concluded that uncertainties regarding regulatory approval have been sufficiently reduced, and (iii) determined that the inventory has probable future economic benefit. In evaluating whether these conditions have been met, we consider clinical trial results for the underlying product candidate, results from meetings with regulatory authorities, and the compilation of the regulatory application. If we are aware of any material risks or contingencies outside of the standard regulatory review and approval process, or if there are any specific negative issues identified relating to the safety, efficacy, manufacturing, marketing or labeling of the product that would have a significant negative impact on its future economic benefits, the related inventory would not be capitalized. We had no inventory capitalized for products awaiting regulatory approval as of December 31, 20162018 and 2015.2017.
Products that have been approved by the U.S. Food and Drug Administration (FDA) or other regulatory authorities are also used in clinical programs to assess the safety and efficacy of the products for usage in diseases that have not been approved by the FDA or other regulatory authorities. The form of the products utilized for both commercial and clinical programs is identical and, as a result, the inventory has an “alternative future use” as defined in authoritative guidance. Raw materials and purchased drug product associated with clinical development programs are included in inventory and charged to research and development expense when the product enters the research and development process and no longer can be used for commercial purposes and, therefore, does not have an “alternative future use”.
For products which are under development and have not yet been approved by regulatory authorities, purchased drug product is charged to research and development expense upon delivery. Delivery occurs when the inventory passes quality inspection and ownership transfers to us. Nonrefundable advance payments for research and development activities, including production of purchased drug product, are deferred and capitalized until the goods are delivered. We also recognize expense for raw materials purchased for developmental purposes when the raw materials pass quality inspection and we have an obligation to pay for the materials.
Inventory Write-Offs
We analyze our inventory levels to identify inventory that may expire prior to sale, inventory that has a cost basis in excess of its estimated realizable value, or inventory in excess of expected sales requirements. Although the manufacturing of our product is subject to strict quality control, certain batches or units of product may no longer meet quality specifications or may expire, which requires adjustments to our inventory values. We also apply judgment related to the results of quality tests that we perform throughout the production process, as well as our understanding of regulatory guidelines, to determine if it is probable that inventory will be saleable. These quality tests are performed throughout the pre-and post-production process, and we continually gather additional information regarding product quality for periods after the manufacture date. Our products currently have a maximum estimated life ranging from 36 to 48 months and, based on our sales forecasts, we expect to realize the carrying value of our inventory. In the future, reduced demand, quality issues or excess supply beyond those anticipated by management may result in a material adjustment to inventory levels, which would be recorded as an increase to cost of sales.
The determination of whether or not inventory costs will be realizable requires estimates by our management. A critical input in this determination is future expected inventory requirements based on internal sales forecasts. We then compare these requirements to the expiry dates of inventory on hand. For inventories that are capitalized in preparation of product launch, we also consider the expected approval date in assessing realizability. To the extent that inventory is expected to expire prior to being sold, we will write down the value of inventory.
Derivative Instruments
We record the fair value of derivative instruments as either assets or liabilities on the balance sheet. The accounting for gains and losses resulting from changes in fair value is dependent on the use of the derivative and whether it is designated and qualifies for hedge accounting.
All qualifying hedging activities are documented at the inception of the hedge and must meet the definition of highly effective in offsetting changes to future cash. The effectiveness of the qualifyingOn a quarterly basis, we perform an assessment to confirm that outstanding hedges remain highly effective and continue to qualify for hedge contract is assessed quarterly.accounting. We record the fair value of the qualifying hedges in other current assets, other assets, other current liabilities and other liabilities. Gains or losses resulting from changes in the fair value of qualifying hedges are recorded in other comprehensive income (loss) until the forecasted transaction occurs. When the forecasted transaction occurs, this amount is reclassified into revenue or interestAll unrealized
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
gains and losses on derivatives that are designated and qualify for hedge accounting are reported in other comprehensive income (loss) and recognized when the underlying hedged transaction affects earnings. When the forecasted transaction occurs, this amount is reclassified into the consolidated statement of operations and presented in the same financial statement line item as the hedged item.
expense, based on the nature of the derivative instrument. Any non-qualifying portion of theDerivative instruments for which hedge accounting is not applied are recorded at fair value in other current assets and other current liabilities. Unrealized gains orand losses resulting from changes in the fair value if any, isof these derivatives are reported in other income and expense.
Property, Plant and Equipment
Property, plant and equipment are stated at cost and are depreciated on a straight-line basis over the estimated useful lives of the assets. We estimate economic lives as follows:
Building and improvements—fifteen to thirty five years
Machinery and laboratory equipment—five to fifteen years
Computer hardware and software—three to seven years
Furniture and office equipment— five to ten years
Leasehold improvements and assets under capital lease arrangements are amortized over the lesser of the asset’s estimated useful life or the term of the respective lease. Maintenance costs are expensed as incurred.
Construction-in-progress reflects amounts incurred for property, plant, or equipment construction or improvements that have not been placed in service.
Assets Held for Sale
We classify assets as held for sale when the following criteria are met: i) management, having the authority to approve the action, commits to a plan to sell the asset, ii) the asset is available for immediate sale in its present condition subject only to terms that are usual and customary for sales of similar assets, iii) an active program to locate a buyer and other actions required to complete the plan to sell the asset have been initiated, iv) the sale of the asset is probable, and transfer of the asset is expected to qualify for recognition as a completed sale, within one year, v) the asset is being actively marketed for sale at a price that is reasonable in relation to its current fair value, and vi) actions required to complete the plan indicate that it is unlikely that significant changes to the plan will be made or that the plan will be withdrawn. Assets that are classified as held for sale are recorded at the lower of their carrying value or their fair value less the costs to sell.
In the third quarter 2017, we announced our intention to close the Alexion Rhode Island Manufacturing Facility (ARIMF). In the fourth quarter 2017, we met the criteria for assets held for sale and reclassified the ARIMF assets from property, plant and equipment to assets held for sale recorded within prepaid expenses and other current assets. We subsequently sold ARIMF during the third quarter of 2018.
Manufacturing Facilities
We capitalize costs incurred for the construction of facilities which support commercial manufacturing. We also capitalize costs related to validation activities which are directly attributable to preparing the facility for its intended use, including engineering runs and inventory production necessary to obtain approval of the facility from government regulators for the production of a commercially approved drug. When the facility is substantially complete and ready for its intended use and regulatory approval for commercial production has been received, we will place the asset in service.
The production of inventory for preparing the facility for its intended use requires two types of production: engineering runs which are used for testing purposes only and do not result in saleable inventory, and validation runs which are used for validating equipment and may result in saleable inventory. The costs associated with inventory produced during engineering runs and normal production losses during validation runs are capitalized to fixed assets and depreciated over the asset’s useful life. Saleable inventory produced during the validation process is initially treated as a fixed asset; however, upon regulatory approval, this inventory is reclassified to inventory and expensed in cost of goods sold as product is sold, or in research and development expenses as product is utilized in R&D activities. Abnormal production costs incurred during the validation process are expensed as incurred.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
Acquisitions
Business combinations are accounted for using the acquisition method of accounting. Under the acquisition method of accounting, the tangible and intangible assets acquired and the liabilities assumed are recorded as of the acquisition date at their respective fair values. We evaluate a business as an integrated set of activities and assets that is capable of being conducted and managed for the purpose of providing a return in the form of dividends, lower costs or other economic benefits and consists of inputs and substantive processes applied to those inputs that provide or have the ability to providecontribute to the creation of outputs. If substantially all of the fair value of gross assets acquired is concentrated in a single asset or group of similar identifiable assets, the assets do not represent a business. In an acquisition of a business, the excess of the fair value of the consideration transferred over the fair value of the net assets acquired is recorded as goodwill. In an acquisition
Acquisitions of netassets or group of assets that doesdo not constitutemeet the definition of a business noare accounted for as asset acquisitions using the cost accumulation method, whereby the cost of the acquisition, including certain transaction costs, is allocated to the assets acquired on the basis of relative fair values. No goodwill is recognized.recognized in an asset acquisition. Intangible assets that are acquired in an asset acquisition for use in research and development activities which have an alternative future use are capitalized as in-process research and development (IPR&D). Acquired IPR&D which has no alternative future use is recognized as research and development expense at acquisition. Contingent milestone payments associated with asset acquisitions are recognized when probable and estimable. These amounts are expensed to research and development if there is no alternative future use associated with the asset, or capitalized as an intangible asset if alternative future use of the asset exists.
Our consolidated financial statements include the results of operations of an acquired business after the completion of the acquisition.
Intangible Assets
Our intangible assets generally consist of licenses,licensing rights, patents, purchased technology, acquired IPR&D and acquired in-process research and development (IPR&D).other intangibles. Intangible assets with definite lives are amortized based on their pattern of economic benefit over their estimated useful lives and reviewed periodically for impairment.
Intangible assets related to IPR&D projects are considered to be indefinite-lived until the completion or abandonment of the associated research and development efforts. During the period the assets are considered indefinite-lived, they will not be amortized but will be tested for impairment. Impairment testing is performed at least annually or when a triggering event occurs that could indicate a potential impairment. If and when development is complete, which generally occurs when regulatory
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
approval to market a product is obtained, the associated assets are deemed finite-lived and are amortized over a period that best reflects the economic benefits provided by these assets.
Goodwill
Goodwill represents the excess of purchase price over fair value of net assets acquired in a business combination and is not amortized. Goodwill is subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment. We are organized and operate as a single reporting unit and therefore the goodwill impairment test is performed using our overall market value, as determined by our traded share price, compared to our book value of net assets.
Impairment of Long-Lived Assets
Our long-lived assets are primarily comprised of intangible assets and property, plant and equipment. We evaluate our finite-lived intangible assets and property, plant and equipment, for impairment whenever events or changes in circumstances indicate the carrying value of an asset or group of assets is not recoverable. If these circumstances exist, recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset group to future undiscounted net cash flows expected to be generated by the asset group. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the assets exceeds the fair value of the assets.
In addition, indefinite-lived intangible assets, comprised of IPR&D, are reviewed for impairment annually and whenever events or changes in circumstances indicate that it is more likely than not that the asset is impaired by comparing the fair value to the carrying value of the asset. In the second quarter 2017, we recognized an impairment charge of $31.0 related to our SBC-103 acquired in-process research and development asset due to clinical results.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
Other Investments
From time to time, we make strategic investments in equity securities of certain biotechnology companies. Our strategic investment portfolio may include equity securities in publicly traded companies, as well as investments in companies with securities that are not publicly traded and where fair value is not readily available. These investments are included in other assets in our consolidated balance sheets.
We have historically recorded our investments in securities that are not publicly traded at cost, less impairments. As of January 1, 2018, we continue to record these investments at cost, less impairments; however, we also adjust the investment for any changes resulting from an observable price change in an orderly transaction for identical or similar investments of the same issuer. We assess relevant transactions that occur on or before the balance sheet date to identify observable price changes, and we regularly monitor these investments to evaluate whether there is an indication that the investment is impaired, based on the implied value of recent company financings, public market prices of comparable companies, and general market conditions.
Our investments in equity securities in publicly traded companies which are unrestricted are regularly measured and carried at fair value and classified as Level 1 equity securities within the fair value hierarchy. Investments in publicly traded companies which are subject to holding period restrictions are carried at fair value using an option pricing valuation model and classified as Level 2 equity securities within the fair value hierarchy. The most significant assumptions within the option pricing valuation model are the term of the restrictions and the stock price volatility, which is based upon the historical volatility of similar companies. We also use a constant maturity risk-free interest rate to match the remaining term of the restrictions on such investments.
ContingentConsideration
We record contingent consideration resulting from a business combination at its fair value on the acquisition date. On a quarterly basis, we revalue these obligations and record increases or decreases in their fair value as an adjustment to operating earnings. Changes to contingent consideration obligations can result from adjustments to discount rates, accretion of the liability due to the passage of time, changes in our estimates of the likelihood or timing of achieving development or commercial milestones, changes in the probability of certain clinical events or changes in the assumed probability associated with regulatory approval.
ContingentLiabilities
We are currently involved in various claims and legal proceedings. On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. If the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Because of uncertainties related to claims and litigation, accruals are based on our best estimates based on available information. On a periodic basis, as additional information becomes available, or based on specific events such as the outcome of litigation or settlement of claims (and our offers of settlement), we may reassess the potential liability related to these matters and may revise these estimates.estimates (and these revisions may be material).
Treasury Stock
Treasury stock is accounted for using the cost method, with the purchase price of the common stock recorded separately as a deduction from stockholders’ equity.
Revenue Recognition
Our principal sourceIn May 2014, the FASB issued a comprehensive new standard which amends revenue recognition principles. We adopted the new standard on January 1, 2018 by applying the modified retrospective method to all contracts that were not completed as of that date. Under the new guidance, revenue is product sales. Werecognized when a customer obtains control of promised goods or services, in an amount that reflects the consideration expected to be received in exchange for those goods or services. Revenue is recognized through a five-step process: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue from product sales when persuasive evidence of an arrangement exists, title(or as) a performance obligation is satisfied. The Company only applies the five-step model to product and associated risk of loss has passedcontracts when it is probable that the Company will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer,customer. At contract inception, the price is fixedCompany assesses the goods or determinable, collection from the customer is reasonably assured,services promised within each contract, and we have no furtherdetermines those that are performance obligations. Depending on these criteria, revenue is usually recorded upon receipt of the product by the end customer, which is typically a hospital, physician’s office, private or government pharmacy or other healthcare facility. On a regular basis, we review revenue arrangements, such as distributor relationships, to determine whether changes in these criteria have an impact on revenue recognition. Amounts collected from customers and remitted to governmental authorities, such as value-added taxes (VAT) in foreign jurisdictions, are presented on a net basis in our consolidated statements of operations and do not impact net product sales.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
obligations. Revenue is recognized for the applicable performance element when each distinct performance obligation is satisfied.
While results for reporting periods beginning after January 1, 2018 are presented under the new guidance, prior period amounts are not adjusted and continue to be reported under the accounting standards in effect for the prior period. The adoption of the new standard did not significantly change our accounting policies.
Nature of Products
Our principal source of revenue is product sales. Our contracts with customers generally contain a single performance obligation and we recognize revenue from product sales when we have satisfied our performance obligation by transferring control of the product to our customers. Control of the product generally transfers to the customer upon delivery. In certain countries, we sell to distributors on a consignment basis and record revenue when control of the product transfers to the customer upon sale to the end user.
Our customers are primarily comprised of distributors, pharmacies, hospitals, hospital buying groups, and other healthcare providers. In some cases, we may also sell to governments and government agencies.
Because of factors such as the price of our products, the limited number of patients, the short period from product sale to patient infusion and the lack of contractual return rights, our customers often carry limited inventory. We also monitor inventory within our sales channels to determine whether deferrals are appropriate based on factors such as inventory levels compared to demand, contractual terms, financial strength of distributors and our ability to estimate returns. In some cases, exact quantities of inventory in the channel are not precisely known, requiring us to estimate these amounts. If actual amounts of inventory differ from these estimates, these adjustments could have an impact in the period in which these estimates change.
In addition to sales in countries where our products are commercially available, we have also recorded revenue on sales for patients receiving treatment through named-patient programs. The relevant authorities or institutions in those countries have agreed to reimburse for product sold on a named-patient basis where our products have not received final approval for commercial sale.
Revenue is recognized at the amount to which we expect to be entitled in exchange for the sale of our products. This amount includes both fixed and variable consideration and excludes amounts that are collected from customers and remitted to governmental authorities, such as value-added taxes in foreign jurisdictions. Shipping and handling costs associated with outbound freight after control of a product has transferred to our customers are accounted for as a fulfillment cost and are included in operating expenses. The cost for any shipping and handling activities (including customs clearance activities) associated with transactions for which revenue has been recognized are accrued if not completed before the respective period end.
The timing between the recognition of revenue for product sales and the receipt of payment is not significant. Our standard credit terms, which vary based on the country of sale, range from 30 to 120 days and all arrangements are payable within one year of the transfer of the product. We record estimateddo not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between the transfer of the promised good to the customer and receipt of payment will be one year or less.
Variable Consideration
We pay distribution fees to our distributors and offer rebates payableand/or discounts, or enter into volume-based reimbursement arrangements with certain customers. We reduce the transaction price on our sales for these amounts. For variable amounts, we estimate the amount of consideration to which we expect to be entitled based on all available historic, current and forecast information. We primarily use the expected value method to estimate variable payments and, in limited circumstances, will apply the most likely method based on the type of variable consideration and what method better predicts the amount of consideration we expect to be entitled to. Consideration that is received from a customer that we expect will need to be refunded in the future is recorded as a refund liability to the customer within accrued expenses. Actual amounts of consideration ultimately received or refunded may differ from our estimates. If actual results in the future vary from our estimates, we adjust these estimates, which would affect net product sales and earnings in the period such variances become known.
Variability in the transaction price for our products pursuant to our contracts with customers primarily arises from the following:
Discounts and Rebates: We offer discounts and rebates to certain distributors and customers under our arrangements. In many cases, these amounts are fixed at the time of sale and the transaction price is reduced accordingly. We also provide for rebates under certain governmental programs, including Medicaid in the U.S. and other programs outside the U.S., as a reduction of revenue at the time of product sale. Our calculations related towhich are payable based on actual claim data. We estimate these rebate accruals requirerebates based on an analysis of historical claim patterns and estimates of customer mix to determine which sales will be subject to rebates and the amount of such rebates. We update our estimates and assumptions each period and record any necessary adjustments, which may have an impact on revenue in the period in which the
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
adjustment is made. Generally, the length of time between product sale and the processing and reporting of the rebates is three to six months.
Volume-Based Arrangements: We have entered into volume-based arrangements with governments in certain countries and other customers in which reimbursement is limited to a contractual amount. Under this type of arrangement, amounts billed in excess of the contractual limitation are repaid to these governmentsthe customer as a rebate. We estimate incremental discounts resulting from these contractual limitations, based on estimatedforecasted sales during the limitation period, and we apply the discount percentage to product shipments as a reduction of revenue. Our calculations related to these arrangements require estimation of sales during the limitation period, and adjustments in these estimates may have a material impact in the period in which these estimates change.
Distribution & Other Fees: We pay distribution and other fees to certain customers in connection with the sales of our products. We record distribution and other fees paid to our customers as a reduction of revenue, unless we receive an identifiable and separate benefitthe payment is for a distinct good or service from the considerationcustomer and we can reasonably estimate the fair value of the benefitgoods or services received. If both conditions are met, we record the consideration paid to the customer as an operating expense. These costs are typically known at the time of sale, resulting in minimal adjustments subsequent to the period of sale.
Product Returns: Our contracts with customers generally provide for returns only if the product is damaged or defective upon delivery. We enter into foreign exchange forward contractsassess our sales transactions and arrangements with customers and monitor inventory within our sales channels to hedge exposuresdetermine whether a provision for returns is warranted and a resulting from portionsadjustment to the transaction price is necessary. This assessment is based on historical experience and assumptions as of our forecasted revenues, including intercompany revenues, that are denominatedthe date of sale and changes in currencies other than the U.S. dollar. These hedges are designated as cash flow hedges upon inception. We record the effective portion of these cash flow hedges to revenueestimates could have an impact in the period in which the change occurs. Because of factors such as the price of our products, the limited number of patients, the short period from product sale to patient infusion and limited contractual return rights, our customers often carry limited inventory.
The amount of variable consideration included in the transaction price is madeconstrained by the amount that is probable will not result in a significant reversal of revenue. We consider our experience with similar transactions and expectations regarding the contract in estimating the amount of variable consideration to an unrelated third partywhich we expect to be entitled, and determining whether the derivativeestimated variable consideration should be constrained. We do not have any material constraints on the variable consideration included within the transaction price of our current revenue arrangements.
See Note 19 “Segment Information” for a summary of revenue from contracts with customers by product and geographical region.
Contract Balances and Receivables
Contract liabilities relate to consideration received and/or billed for goods that have not been delivered to the customer and for which the performance obligation has not yet been completed. These amounts are included within other current liabilities in the consolidated statements of operations.
The following table provides information about receivables and contract is settled.liabilities from our contracts with customers.
|
| | | | | | | |
| | December 31, 2018 | | December 31, 2017 |
| Receivables, which are included in "Trade accounts receivable, net" | $ | 922.3 |
| | $ | 726.5 |
|
| Contract liabilities, which are included in "Other current liabilities" | $ | 3.4 |
| | $ | 15.9 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
Research and Development Expenses
Research and development expenses are comprised of costs incurred in performing research and development activities including payroll and benefits, pre-clinical,preclinical, clinical trial and related clinical manufacturing costs, manufacturing development and scale-up costs, product development and regulatory costs, contract services and other outside contractor costs, research license fees, depreciation and amortization of lab facilities, and lab supplies. These costs are expensed as incurred. We accrue costs for clinical trial activities based upon estimates of the services received and related expenses incurred that have yet to be invoiced by the contract research organizations, clinical study sites, laboratories, consultants, or other clinical trial vendors that perform the activities.
Share-Based Compensation
We have two share-based compensation plans pursuant to which awards are currently being made: (i) the Amended and Restated 20042017 Incentive Plan (2004(2017 Plan) and (ii) the 2015 Employee Stock Purchase Plan (ESPP). The 2017 Plan replaced the Amended & Restated 2004 Incentive Plan (2004 Plan), effective May 10, 2017. Under the 20042017 Plan, restricted stock, restricted stock units, stock options and other stock-related awards may be granted to our directors, officers, employees and consultants or advisors of the Company or any subsidiary. Under the ESPP, eligible employees can purchase shares of common stock at a discount semi-annually through payroll deductions. To date, share-based compensation issued under the plans consists of incentive and non-qualified stock options, restricted stock and restricted stock units, including restricted stock units with market and non-market performance conditions, and shares issued under our ESPP.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
Compensation expense for our share-based awards is recognized based on the estimated fair value of the awards on the grant date. Compensation expense reflects an estimate of the number of awards expected to vest and is primarily recognized on a straight-line basis over the requisite service period of the individual grants, which typically equals the vesting period. Compensation expense for awards with performance conditions is recognized using the graded-vesting method.
Our estimates of employee stock option values rely on estimates of factors we input into the Black-Scholes model. The key factors involve an estimate of future uncertain events. Significant assumptions include the use of historical volatility to determine the expected stock price volatility. We also estimate expected term until exercise and the reduction in the expense from expected forfeitures. We currently use historical exercise and cancellation patterns as our best estimate of future estimated life.
For our non-market performance-based awards, we estimate the anticipated achievement of the performance targets, including forecasting the achievement of future financial targets. These estimates are revised periodically based on the probability of achieving the performance targets and adjustments are made throughout the performance period as necessary. We use payout simulation models to estimate the grant date fair value of market performance-based awards.awards with market-based performance conditions. The payout simulation models assume volatility of our common stock and the common stock of a comparator group of companies, as well as correlations of returns of the price of our common stock and the common stock prices of the comparator group.
The purchase price of common stock under our ESPP is equal to 85%85.0% of the lower of (i) the market value per share of the common stock on the first business day of an offering period or (ii) the market value per share of the common stock on the purchase date. The fair value of the discounted purchases made under our ESPP is calculated using the Black-Scholes model. The fair value of the look-back provision plus the 15%15.0% discount is recognized as compensation expense over the 6 month purchase period.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
Restructuring and restructuring related expenses
We record liabilities associated with one-time employee termination benefits and exit or disposal activities in the period in which the liability is incurred. One-time employee benefits are incurred when communicated to employees and where detailed action plans have been approved. Costs for one-time termination benefits in which the employee is required to render service until termination in order to receive benefits are recognized ratably over the service period. For existing benefit arrangements, employee termination costs are accrued when the exit or disposal cost are probable and estimable.
Restructuring related expenses include accelerated depreciation costs and impairment charges associated with assets impacted by a restructuring exit activity. Accelerated depreciation costs represent the difference between the depreciation expense recognized over the revised useful life of the asset, based upon the anticipated date an impacted site closure and the depreciation expense as determined using the useful life prior to the restructuring activities.
Earnings Per Common Share
Basic earnings per common share (EPS) is computed by dividing net income by the weighted-average number of shares of common stock outstanding. For purposes of calculating diluted EPS, the denominator reflects the potential dilution that could occur if stock options, unvested restricted stock units or other contracts to issue common stock were exercised or converted into common stock, using the treasury stock method.
The following table summarizes the calculation of basic and diluted EPS for years ended December 31, 2016, 20152018, 2017 and 2014:2016:
| | | | Year Ended December 31, | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 | 2018 | | 2017 | | 2016 |
| Net income used for basic and diluted calculation | $ | 399 |
| | $ | 144 |
| | $ | 657 |
| $ | 77.6 |
| | $ | 443.3 |
| | $ | 399.4 |
|
| Shares used in computing earnings per common share—basic | 224 |
| | 213 |
| | 198 |
| 222.7 |
| | 223.9 |
| | 224.3 |
|
| Weighted-average effect of dilutive securities: | | | | | | | | | | |
| Stock awards | 3 |
| | 3 |
| | 4 |
| 1.8 |
| | 1.5 |
| | 2.0 |
|
| Shares used in computing earnings per common share—diluted | 227 |
| | 216 |
| | 202 |
| 224.5 |
| | 225.4 |
| | 226.3 |
|
| Earnings per common share: | | | | | | | | | | |
| Basic | $ | 1.78 |
| | $ | 0.68 |
| | $ | 3.32 |
| $ | 0.35 |
| | $ | 1.98 |
| | $ | 1.78 |
|
| Diluted | $ | 1.76 |
| | $ | 0.67 |
| | $ | 3.26 |
| $ | 0.35 |
| | $ | 1.97 |
| | $ | 1.76 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
We exclude from EPS the weighted-average number of securities whose effect is anti-dilutive. Excluded from the calculation of EPS for the years ended December 31, 2016, 20152018, 2017 and 20142016 were 4, 2,2.8, 4.0, and 14.2 shares of common stock, respectively, because their effect is anti-dilutive.
Income Taxes
We utilize the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement carrying amounts and tax basis of assets and liabilities using enacted tax rates in effect for years in which the temporary differences are expected to reverse. We periodically evaluate the likelihood of the realization of deferred tax assets and reduce the carrying amount of these deferred tax assets by a valuation allowance when it is more likely than not that deferred tax assets will not be realized.
We recognize the benefit of an uncertain tax position that has been taken or we expect to take on income tax returns if such tax position is more likely than not to be sustained. The tax benefit recognized in the financial statements for a particular tax position is based on the largest benefit that is more likely than not to be realized. The amount of unrecognized tax benefits is adjusted, as appropriate, for changes in facts and circumstances, such as significant amendments to existing tax law, new regulations or interpretations by the taxing authorities, or new information obtained during a tax examination or resolution of an examination. We also accrued for potential interest and penalties related to unrecognized tax benefits as a component of tax expense.
In December 2017, the Tax Cuts and Jobs Act (Tax Act) was enacted into law. The Tax Act decreased the U.S. federal corporate tax rate to 21.0%, imposed a minimum tax on foreign earnings related to intangible assets (GILTI), a
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
one-time transition tax on previously unremitted foreign earnings, and modified the taxation of other income and expense items. With regard to the GILTI minimum tax, foreign earnings are reduced by the profit attributable to tangible assets and a deductible allowance of up to 50.0%, subject to annual limitations. We have elected to account for the impact of the minimum tax in deferred taxes.
Comprehensive Income
Comprehensive income is comprised of net income and other comprehensive income (loss). Other comprehensive income (loss) includes changes in equity that are excluded from net income, such as changes in pension liabilities, unrealized gains and losses on marketable debt securities, unrealized gains and losses on hedge contracts and foreign currency translation adjustments. Certain of these changes in equity are reflected net of tax.
Other Investments
We invest in companies with securities that are not publicly traded and where fair value is not readily available. Other investments include an investment in the preferred stock of the non-public entity Moderna Therapeutics, Inc. During 2014, we purchased $38 of preferred equity of Moderna. We recorded our investment at cost within other assets in our condensed consolidated balance sheets. We regularly monitor these investments to evaluate whether there has been an other-than-temporary decline in its fair value, based on the implied value of recent company financings, public market prices of comparable companies, and general market conditions. The carrying value of these investments was not impaired as of December 31, 2016.
Reclassifications and Adjustments
Certain items in the prior year’s consolidated financial statements have been reclassified to conform to the current presentation.
New Accounting Pronouncements
In February 2016, the FASB issued a new standard that requires lessees to recognize leases on-balance sheet and disclose key information about leasing arrangements. The new standard establishes a right-of-use (ROU) model that requires a lessee to recognize a ROU asset and lease liability on the balance sheet for all leases with a term longer than 12 months. Leases will be classified as finance or operating, with classification affecting the pattern and classification of expense recognition in the income statement. The standard is effective on January 1, 2019, with early adoption permitted. We adopted the new standard on January 1, 2019 and use the effective date as our date of initial application. In July 2018, the FASB issued an update that provided an additional transition option that allows companies to continue applying the guidance under the lease standard in effect at that time in the comparative periods presented in the consolidated financial statements. Companies that elect this option would record a cumulative-effect adjustment to the opening balance of retained earnings on the date of adoption. We elected this optional transition method. We also elected the “package of practical expedients”, which permits us not to reassess under the new standard our prior conclusions about lease identification, lease classification and initial direct costs. We continue to evaluate other practical expedients available under the standard.
We have substantially completed our assessment of the standard as well as implementation of our leasing software, including data upload and test procedures. We continue to finalize our calculations, including our discount rate assumptions, related to the new standard. We are also continuing to establish new processes and internal controls that may be required to comply with the new lease accounting and disclosure requirements set by the new standard. We expect the impact of the standard adoption to decrease our assets, liabilities and retained earnings within our consolidated balance sheet. These decreases will result from the derecognition of our existing assets and financing obligations related to our build to suit leases offset by the recognition of new ROU assets and liabilities as a result of the leasing standard.
In June 2016, the FASB issued a new standard intended to improve reporting requirements specific to loans, receivables and other financial instruments. The new standard requires that credit losses be reported based on expected losses compared to the current incurred loss model. The new standard also requires enhanced disclosure of credit risk associated with respective assets. The standard is effective for interim and annual periods beginning after December 15, 2019 with early adoption permitted. We are currently assessing the impact of this standard on our financial condition and results of operations.
In February 2018, the FASB issued a new standard that would permit entities to make a one time reclassification from accumulated other comprehensive income (AOCI) to retained earnings for the stranded tax effects resulting from the newly enacted corporate tax rates under the Tax Cuts and Jobs Act (the Tax Act), that was effective for the year ended December 31, 2017. The amount of the reclassification is calculated on the basis of the difference between the historical tax rate and newly enacted tax rate. The standard is effective for interim and annual periods beginning after December 15, 2018 with early adoption permitted. We are currently assessing the impact of this standard on our financial condition.
In August 2018, the FASB issued a new standard on a customer's accounting for implementation, set-up, and other upfront costs incurred in a cloud computing arrangement (CCA). Under the new guidance, customers will assess if a CCA includes a software license and if a CCA does include a software license, implementation and set-up costs will be accounted for consistent with existing internal-use software implementation guidance. Implementation costs
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
associated with a CCA that does not include a software license would be expensed to operating expenses. The standard also provides classification guidance on these implementation costs as well as additional quantitative and qualitative disclosures. The standard is effective for public business entities for fiscal years beginning after December 15, 2019, and interim periods within those fiscal years. Early adoption is permitted, including adoption in any interim periods. Entities can choose to adopt the new guidance prospectively or retrospectively. We are currently assessing the impact this standard will have on our statement of financial condition and results of operations.
Recently Adopted Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (FASB)FASB issued a comprehensive new standard which amends revenue recognition principles and provides a single set of criteria for revenue recognition among all industries. The new standard provides a five stepfive-step framework whereby revenue is recognized when promised goods or services are transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. We adopted the new standard on January 1, 2018.
In January 2017, the FASB issued a new standard that clarifies the definition of a business and determines when an integrated set of assets and activities is not a business. This framework requires that if substantially all of the fair value of gross assets acquired or disposed of is concentrated in a single asset or group of similar identifiable assets, the assets would not represent a business. We adopted the new standard on January 1, 2018 and applied the new guidance prospectively to transactions occurring after adoption. We anticipate that the adoption of this new standard will likely result in more transactions, to the extent that such transactions are undertaken by the Company, being accounted for as asset acquisitions.
In January 2016, the FASB issued a new standard that changes accounting for equity investments, financial liabilities under the fair value option, and presentation and disclosure requirements for financial instruments. In addition, the FASB clarified guidance related to the valuation allowance assessment when recognizing deferred tax assets resulting from unrealized losses on available-for-sale debt securities. Equity investments with readily determinable fair values will be measured at fair value with changes in fair value recognized in net income. Companies have the option to either measure equity investments without readily determinable fair values at fair value, or at cost adjusted for changes in observable prices minus impairment. We adopted the new standard on January 1, 2018, and elected to measure our existing equity investments without readily determinable fair values at cost adjusted for changes in observable prices minus impairment. In connection with the adoption of the new standard, we reclassified an immaterial amount of unrealized gains on equity securities from accumulated other comprehensive income to retained earnings. The guidance related to equity investments without readily determinable fair values was applied prospectively to equity investments that existed as of the date of adoption. We will assess equity investments without readily determinable fair values for observable price changes and impairment on a quarterly basis. Refer to Note 7, Other Investments, for further details.
In March 2017, the FASB issued a new standard that improves the presentation of net periodic pension cost and net periodic post retirement benefit cost by requiring the bifurcation of net benefit cost. Under the new standard, the service cost component of net benefit cost will be presented with other employee costs in operating expenses, while other components will be reported separately in other income and expense. We adopted the new standard on January 1, 2018. The adoption of this standard did not have a material impact on our consolidated statements of operations.
In November 2016, the FASB issued a new standard that clarifies how entities should present restricted cash in the statement of cash flows. Under the new standard, changes in total cash, inclusive of restricted cash, should be reflected in the statement of cash flows. As a result, transfers between cash and restricted cash will no longer be reflected as activity within the statement of cash flows. We adopted the new standard on January 1, 2018. The adoption of this standard did not have a material impact on our consolidated statements of cash flows.
In August 2017, the FASB issued a new standard intended to improve and simplify certain aspects of the accounting for hedges. The new standard is intended to more closely align hedge accounting with companies’ risk management strategies, simplify the application of hedge accounting, and increase transparency as to the scope and results of hedging programs. It also requires enhanced disclosures pertaining to revenue recognition in both interimamends the presentation and annual periods.disclosure requirements and changes how companies assess effectiveness. The standard is effective for interim and annual periods beginning after December 15, 2017 and allows for2018 with early adoption using a full retrospective method, or a modified retrospective method. Entities may elect topermitted. We early adoptadopted the new standard for annual periods beginning after December 15, 2016. We currently anticipate adoptingin the standardsecond quarter 2018 using the modified retrospective method. We do not expect the implementationThe adoption of this new standard todid not have a material impact on our consolidated financial position and results of operations.
In April 2015, the FASB issued a new standard simplifying the presentation of debt issuance costs. The new standard aligns the treatment of debt issuance costs with debt discounts and premiums and requires debt issuance costs be presented as a direct deduction from the carrying amount of the related debt. We adopted the provisions of this standard in the first quarterstatements.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
Impacts of the New Revenue Standard
2016 and reclassified $9 of deferred financing costs from prepaid expenses and other current assets to the current portion of long-term debt and $27 from other assets to long-term debt, less current portion in our consolidated balance sheets as of December 31, 2015.
In April 2015, the FASB issued a new standard clarifying the accounting for a customer’s fees paid in a cloud computing arrangement. Under this standard, if a cloud computing arrangement includes a software license, the customer would account for the software license consistent with other software licenses. If a cloud computing arrangement does not include a software license, the customer would account for the arrangement as a service contract. We adopted the provisionsnew revenue standard by applying the modified retrospective method to all contracts that were not completed as of this standard in the first quarter 2016. The adoption did not have a material effect on our financial condition or results of operations.
In February 2016, the FASB issued a new standard requiring that the rights and obligations arising from leases be recognized on the balance sheet by recording a right-of-use asset and corresponding lease liability. The new standard also requires qualitative and quantitative disclosures to understand the amount, timing, and uncertainty of cash flows arising from leases, as well as significant management estimates utilized. The standard is effectiveJanuary 1, 2018. Results for interim and annualreporting periods beginning after December 15,January 1, 2018 and requires a modified retrospective adoption. We are currently assessingpresented under the impact of this standard on our financial condition and results of operations.
In March 2016, the FASB issued a new standard, intendedwhile prior period amounts are not adjusted and continue to simplify certain aspects ofbe reported under the accounting standards in effect for employee share-based payments. We elected to early adopt this standard during the third quarter of 2016. One aspect of the standard requires an entity to recognize all excess tax benefits and deficiencies associated with stock-based compensation as a reduction or increase to tax expense in the income statement. Previously, such amounts were recognized in additional paid-in capital. This aspectprior period. Upon adoption of the new revenue recognition standard, was adopted prospectively,on January 1, 2018, we reduced our deferred revenue balance by $10.4, with an offsetting increase of $6.0 in retained earnings due to the cumulative impact of adopting this new standard.
The impact to net product sales and accordingly we recorded tax benefits of $10, withinnet income tax expense for the year ended December 31, 2016. The amendments require recognition2018 was an increase of excess tax benefits regardless of whether the benefit reduces taxes payable in the current period. As$5.3 and $4.8, respectively, as a result $238 associated with previously unrecognized excess tax benefits was recorded asof adopting the new standard. The new standard also resulted in a decrease of $17.9 in deferred tax assetrevenue and an increase of $10.8 in retained earnings as of the beginning of 2016. Furthermore, the amendment requires that excess tax benefits be classified as an operating activity in the statement of cash flows instead of a financing activity. We elected to adopt this provisionDecember 31, 2018. The adoption of the new revenue standard prospectively and thus, prior periodsdid not have not been adjusted. We have also elected to continue to estimatea material impact on any other balances within the impact of forfeitures when determining the amount of compensation cost to be recognized each period rather than account for forfeitures as they occur.
In October 2016 the FASB issued a new standard that eliminates the prohibition of immediate recognition of current and deferred income tax impacts for an intra-entity asset transfer other than inventory. Under the new standard, entities should recognize the income tax consequences on an intra-entity transfer of an asset other than inventory when the transfer occurs. This new standard will be effective for interim periods beginning after December 15, 2017 and requires a modified retrospective adoption through a cumulative-effect adjustment directly to retained earningsconsolidated financial statements as of and for the beginning of the period of adoption. We are currently assessing the impact of this standard on our financial condition and results of operations.year ended December 31, 2018.
2. Acquisitions
Wilson Therapeutics AB
On June 22, 2015,May 25, 2018, we completed the acquisition of Synageva,Wilson Therapeutics AB (publ), a biopharmaceutical company based in Stockholm, Sweden (Wilson Therapeutics) that develops a novel therapy for patients with rare copper-mediated disorders, pursuant to a recommended public cash offer of SEK 232 for each share of stock of Wilson Therapeutics. As a result of the acquisition, we added WTX101 (ALXN1840), a highly innovative drug candidate that is currently in the early stages of Phase III clinical trials for the treatment of patients with Wilson disease, to our clinical pipeline.
The acquisition of Wilson Therapeutics is accounted for as an asset acquisition, as substantially all of the fair value of the gross assets acquired is concentrated in a transaction accountedsingle asset, WTX101.
The following table summarizes the total consideration for under the acquisition methodand the value of accounting for business combinations. Under the acquisition method of accounting, the assets acquired and liabilities assumedassumed:
|
| | | |
| Consideration | |
| Cash paid for acquisition of Wilson Therapeutics outstanding shares | $ | 749.3 |
|
| Transaction costs | 15.1 |
|
| Total consideration | $ | 764.4 |
|
| | |
| Assets Acquired and Liabilities Assumed | |
| Cash | $ | 45.1 |
|
| In-process research & development | 803.7 |
|
| Employee related liabilities | (71.4 | ) |
| Other assets and liabilities | (13.0 | ) |
| Total net assets acquired | $ | 764.4 |
|
The acquired in-process research and development asset relates to WTX101. Due to the stage of development of this asset, significant risk remains and it is not yet probable that there is future economic benefit from Synagevathis asset. Absent successful clinical results and regulatory approval for the asset, there is no alternative future use associated with WTX101. Accordingly, the value of this asset of $803.7 was expensed during the year ended December 31, 2018.
Employee related liabilities include the value of outstanding employee equity incentive awards that were recorded as ofaccelerated in connection with the Wilson Therapeutics acquisition date at their respective fair values. Synageva’s results of operations arethat have been settled in cash. Also included in this amount are employer tax obligations associated with the consolidated financial statementsemployee equity incentive awards.
In connection with rights to WTX101 that were previously acquired by Wilson Therapeutics from the date of acquisition. The acquisition furthered our objectivethird parties, we could be required to developpay up to approximately $19.0 if certain development, regulatory and commercialize life-transforming therapies to an increasing number of patients with devastating and rare diseases. Synageva’s lead product candidate was Kanuma, an enzyme replacement therapy for patients suffering with LAL-D, a life-threatening, ultra-rare disease for which there were no approved treatments at the closing of the business combination.
We acquired all of the outstanding shares of common stock of Synageva for $4,565 in cash and 26 shares of common stock. We financed the cash consideration with existing cash and proceeds from our new credit facility described further in Note 8.
The aggregate consideration to acquire Synageva consisted of: |
| | | |
| Stock consideration | $ | 4,918 |
|
| Cash consideration | 4,565 |
|
| Total purchase price | $ | 9,483 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
Syntimmune, Inc.
In September 2018, we entered into a definitive agreement to acquire Syntimmune, Inc. (Syntimmune), a clinical-stage biotechnology company developing an antibody therapy targeting the neonatal Fc receptor (FcRn). Syntimmune’s lead candidate, SYNT001 (ALXN1830), is a monoclonal antibody that is designed to inhibit the interaction of FcRn with Immunoglobulin G (IgG) and IgG immune complexes, and is being studied in Phase 1b/2a trials for the treatment of IgG-mediated autoimmune diseases. The acquisition of Syntimmune closed in November 2018. Under the terms of the agreement, Alexion acquired Syntimmune for an upfront cash payment of $400.0, with the potential for additional milestone-dependent payments of up to $800.0, for a total value of up to $1,200.0.
The acquisition of Syntimmune is accounted for as an asset acquisition, as substantially all of the fair value of the gross assets acquired is concentrated in a single in-process research and development asset, SYNT001.
The following table summarizes the estimated fair valuestotal consideration for the acquisition and the value of the assets acquired and liabilities assumed:
|
| | | |
| Cash | $ | 626 |
|
| Inventory | 24 |
|
| In-process research and development (IPR&D) | 4,236 |
|
| Deferred tax liabilities, net | (160 | ) |
| Other assets and liabilities | (26 | ) |
| Net assets acquired | 4,700 |
|
| Goodwill | 4,783 |
|
| Total purchase price | $ | 9,483 |
|
|
| | | |
| Consideration | |
| Upfront payment for acquisition of Syntimmune outstanding shares | $ | 400.0 |
|
| Cash acquired | 4.2 |
|
| Working capital adjustment | 6.4 |
|
| Transaction costs | 0.9 |
|
| Total consideration | $ | 411.5 |
|
| | |
| Assets Acquired and Liabilities Assumed | |
| Cash | $ | 4.2 |
|
| In-process research & development | 379.3 |
|
| Deferred tax assets | 25.1 |
|
| Other assets and liabilities | 2.9 |
|
| Total net assets acquired | $ | 411.5 |
|
The fairacquired in-process research and development asset relates to SYNT001. Due to the stage of development of this asset, significant risk remains and it is not yet probable that there is future economic benefit from this asset. Absent successful clinical results and regulatory approval for the asset, there is no alternative future use associated with SYNT001. Accordingly, the value of the assets acquired and liabilities assumed were initially based upon preliminary calculations, and our estimates and assumptions were subject to change as we obtained additional information for our estimatesthis asset of $379.3 was expensed during the measurement period (up to one year from the acquisition date). During the year ended December 31, 2016, we recorded fair value adjustments of $11 primarily due to tax related items.
We acquired $24 of Kanuma inventory. The estimated fair value of work-in-process and finished goods inventory was determined utilizing the comparative sales method, based on the expected selling price of the inventory, adjusted for incremental costs to complete the manufacturing process and for direct selling efforts, as well as for a reasonable profit allowance. The estimated fair value of raw material inventory was valued at replacement cost, which is equal to the value a market participant would pay to acquire the inventory.
Intangible assets associated with IPR&D projects primarily relate to Kanuma. The estimated fair value of IPR&D assets of $4,236 was determined using the multi-period excess earnings method, a variation of the income approach. The multi-period excess earnings method estimates the value of an intangible asset equal to the present value of the incremental after-tax cash flows attributable to that intangible asset. The fair value using the multi-period excess earnings method was dependent on an estimated weighted average cost of capital for Synageva of 10%, which represents a rate of return that a market participant would expect for these assets.
The excess of purchase price over the fair value amounts of the assets acquired and liabilities assumed represents the goodwill amount resulting from the acquisition. The goodwill, which is not tax-deductible, has been recorded as a noncurrent asset and is not amortized, but is subject to an annual review for impairment. The goodwill represents future economic benefits arising from other assets acquired that could not be individually identified and separately recognized and expected synergies that are specific to our business and not available to market participants, including our unique ability to commercialize therapies for rare diseases, our existing relationships with specialty physicians who can identify patients with LAL-D, a global distribution network to facilitate drug delivery and other benefits that we believe will result from combining the operations of Synageva within our operations.
We recorded a net deferred tax liability of $160. This amount was primarily comprised of $603 of deferred tax liabilities related to the IPR&D and inventory acquired, offset by $443 of deferred tax assets related to net operating loss carryforwards (NOLs), tax credits, and other temporary differences, which we expect to utilize.
For the year ended December 31, 2015, we recorded $96 of pre-tax operating losses associated with the continuing operations of Synageva in our consolidated statements of operations.
Pro forma financial information (unaudited)
The following unaudited pro forma information presents the combined results of Alexion and Synageva as if the acquisition of Synageva had been completed on January 1, 2014, with adjustments to give effect to pro forma events that are directly attributable to the acquisition. The unaudited pro forma results do not reflect operating efficiencies or potential cost savings which may result from the consolidation of operations. Accordingly, the unaudited pro forma financial information is not necessarily indicative of the results of operations that would have had we completed the transaction on January 1, 2014.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
|
| | | | | | | |
| | Year Ended December 31, | | Year Ended December 31, |
| | 2015 | | 2014 |
| Pro forma revenues | $ | 2,606 |
| | $ | 2,240 |
|
| Pro forma net income | 21 |
| | 261 |
|
| Earnings per common share | | | |
| Basic | $ | 0.09 |
| | $ | 1.16 |
|
| Diluted | $ | 0.09 |
| | $ | 1.14 |
|
The unaudited pro forma consolidated results include the following pro forma adjustments related to non-recurring activity:
Alexion and Synageva expenses of $33 and $127, respectively, associated with the accelerated vesting of stock based compensation as a result of the acquisition were excluded from net income for the year ended December 31, 2015. These expenses were included in net income for the year ended December 31, 2014;
Alexion and Synageva acquisition-related and restructuring costs of $53 and $62, respectively, were excluded from income for the year ended December 31, 2015. These expenses were included in net income for the year ended December 31, 2014.
Acquisition-Related Costs
Acquisition-related costs associated with our business combinations for the years ended December 31, 2016, 2015 and 2014 include the following:
|
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 |
Transaction costs (1) | $ | — |
| | $ | 27 |
| | $ | — |
|
| Integration costs | 2 |
| | 12 |
| | — |
|
| | $ | 2 |
| | $ | 39 |
| | $ | — |
|
| | | | | | |
| (1) Transaction costs include investment advisory, legal, and accounting fees | | | | | |
| |
The acquisition of Synageva resulted in $13 of restructuring related charges for the year ended December 31, 2015. Synageva restructuring related charges were not material for the year ended December 31, 2016. See Note 17 for additional details.2018.
| |
| 3. | Property, Plant and Equipment, Net |
A summary of property, plant and equipment is as follows:
| |
| December 31, 2016 | | December 31, 2015 | December 31, 2018 | | December 31, 2017 |
| Land | $ | 10 |
| | $ | 9 |
| $ | 9.6 |
| | $ | 9.6 |
|
| Buildings and improvements | 450 |
| | 252 |
| 520.1 |
| | 427.9 |
|
| Machinery and laboratory equipment | 126 |
| | 92 |
| 161.7 |
| | 159.2 |
|
| Computer hardware and software | 123 |
| | 84 |
| 144.8 |
| | 141.5 |
|
| Furniture and office equipment | 25 |
| | 16 |
| 27.5 |
| | 23.8 |
|
| Construction-in-progress | 495 |
| | 420 |
| 827.1 |
| | 723.7 |
|
| 1,229 |
| | 873 |
| 1,690.8 |
| | 1,485.7 |
|
| Less: Accumulated depreciation and amortization | (193 | ) | | (176 | ) | (219.3 | ) | | (160.3 | ) |
| $ | 1,036 |
| | $ | 697 |
| $ | 1,471.5 |
| | $ | 1,325.4 |
|
Included in construction-in-progress at December 31, 20152017 was $227$64.1 of costs associated with the construction of our leased facility in New Haven, Connecticut. ThisBoston, Massachusetts. Construction of this facility was completed and the building was placed into service in 2016.the second quarter 2018. Additionally, there were costs of $118$203.9 and $19$180.6 as of December 31, 20162018 and 2015,2017, respectively, included within construction-in-process associated with the construction of a new Lonza manufacturing facility. Although we willdo not legally own these premises, we are deemed to be the owner of the buildings during
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
during the construction period based on applicable accounting guidance for build-to-suit leases, see Note 9, “Facility10, “Facility Lease Obligations”Obligations” for additional information.
In connection with the construction of the facility in New Haven, Connecticut, we entered into an agreement with the State of Connecticut Department of Economic and Community Development which provides for a forgivable loan and grants totaling $26 and tax credits of up to $25. The program requires that we meet certain criteria in order to prevent forfeiture or repayment of the loan, grants and credits, which include (i) maintaining corporate headquarters in Connecticut for ten years; (ii) satisfying minimum employment obligations; and (iii) minimum capital spending requirements. In the third quarter 2015, we received $26 for the forgivable loan and grants. In 2016, we satisfied the second and third criteria. The proceeds reduce the costs of our assets associated with the project. As of December 31, 2016, we have not received any tax credits associated with our agreement with the State of Connecticut.
Depreciation and amortization of property, plant and equipment was approximately $64, $44$77.9, $95.8 and $35$64.0 recorded within operating expenses on our consolidated statement of operations for the years ended December 31, 2018, 2017 and 2016, 2015respectively. Included within this amount for the years ended December 31, 2018 and 2014, respectively.2017 were charges related to the 2017 restructuring activities. See Note 18, “Restructuring and Related Expenses” for additional information.
At December 31, 20162018 and 2015,2017, computer software costs included in property, plant and equipment were $37$50.3 and $20,$58.2, respectively. Depreciation and amortization expense for capitalized computer software costs was $12, $10$17.4, $16.0 and $7$12.4 for the years ended December 31, 2018, 2017 and 2016, 2015respectively.
In January 2019, we adopted a new lease accounting standard. See Note 1 “Business Overview & Summary of Significant Accounting Policies” for an overview of the impact this standard will have on our property, plant and 2014, respectively.equipment balances in 2019.
| |
| 4. | Intangible Assets and Goodwill |
IntangibleThe following table summarizes the carrying amount of our intangible assets and goodwill, net of accumulated amortization, are as follows:amortization:
| | | | | | December 31, 2016 | | December 31, 2015 | | | December 31, 2018 | | December 31, 2017 |
| | Estimated
Life (years) | | Cost | | Accumulated
Amortization | | Net | | Cost | | Accumulated
Amortization | | Net | Estimated
Life (years) | | Cost | | Accumulated
Amortization | | Net | | Cost | | Accumulated
Amortization | | Net |
| Licenses | 6-8 | | $ | 29 |
| | $ | (29 | ) | | $ | — |
| | $ | 29 |
| | $ | (29 | ) | | $ | — |
| |
| Licensing Rights | | 5-8 | | $ | 39.0 |
| | $ | (29.3 | ) | | $ | 9.7 |
| | $ | 31.0 |
| | $ | (28.5 | ) | | $ | 2.5 |
|
| Patents | 7 | | 11 |
| | (11 | ) | | — |
| | 11 |
| | (11 | ) | | — |
| 7 | | 10.5 |
| | (10.5 | ) | | — |
| | 10.5 |
| | (10.5 | ) | | — |
|
| Purchased technology | 6-16 | | 4,711 |
| �� | (439 | ) | | 4,272 |
| | 4,709 |
| | (117 | ) | | 4,592 |
| 6-16 | | 4,710.5 |
| | (1,079.1 | ) | | 3,631.4 |
| | 4,710.5 |
| | (758.9 | ) | | 3,951.6 |
|
| Acquired IPR&D | Indefinite | | 31 |
| | — |
| | 31 |
| | 116 |
| | — |
| | 116 |
| |
| Other Intangibles | | 5 | | 0.4 |
| | (0.2 | ) | | 0.2 |
| | 0.4 |
| | (0.1 | ) | | 0.3 |
|
| Total | | $ | 4,782 |
| | $ | (479 | ) | | $ | 4,303 |
| | $ | 4,865 |
| | $ | (157 | ) | | $ | 4,708 |
| | $ | 4,760.4 |
| | $ | (1,119.1 | ) | | $ | 3,641.3 |
| | $ | 4,752.4 |
| | $ | (798.0 | ) | | $ | 3,954.4 |
|
| Goodwill | Indefinite | | $ | 5,040 |
| | $ | (3 | ) | | $ | 5,037 |
| | $ | 5,051 |
| | $ | (3 | ) | | $ | 5,048 |
| Indefinite | | $ | 5,040.3 |
| | $ | (2.9 | ) | | $ | 5,037.4 |
| | $ | 5,040.3 |
| | $ | (2.9 | ) | | $ | 5,037.4 |
|
Amortization expense was $322, $117$321.1, $320.2 and $11$322.2 for the years ended December 31, 2016, 20152018, 2017 and 2014,2016, respectively. Assuming no changes in the gross cost basis of intangible assets, the total estimated amortization expense for finite-lived intangible assets is $320approximately $322.0 for each of the years ending December 31, 20172019 through December 31, 2021.2023.
During the fourth quarter 2016, we reviewed SBC-103, an early stage clinical indefinite-lived intangible asset related to the Synageva acquisition as part of our annual impairment testing.
The amortized cost, gross unrealized holding gains, gross unrealized holding losses and fair value of this IPR&D asset was determined using the income approach and included significant unobservable (Level 3) inputs. These unobservable inputs included, among other things, expected development, regulatory and commercial time lines, risk-adjusted forecasted future cash flows to be generatedavailable-for-sale debt securities by this asset, contributory asset charges for other assets employed in this IPR&D project and the determinationtype of an appropriate discount rate based on a weighted cost of capital of 12% to be applied in calculating the present value of future cash flows. Based on our strategic portfolio evaluation, increases in development, regulatory and commercial time lines and updated cash flows, the estimated value that can be obtained for this asset from a market participant in an arm’s length transaction is $31, which was lower than the carrying amount of the asset. As a result, in the fourth quarter 2016, we recognized an impairment charge of $85 to write-down this asset to fair value. The impairment was recorded in operating expenses in our consolidated statement of operations for the year endedsecurity at December 31, 2016. In February2018 and December 31, 2017 the Board of Directors of Alexion made the decision to reduce our investment in SBC-103. The current Phase I/II clinical trial will not be expanded and no new patients will be added to the trial. Patients currently enrolled in the trial will continue to receive therapy. We will reassess the value of this asset on a go forward basis for indicators of impairment.were as follows:
|
| | | | | | | | | | | | | | | | |
| | | December 31, 2018 |
| | | Amortized Cost | | Gross Unrealized Holding Gains | | Gross Unrealized Holding Losses | | Fair Value |
| Commercial paper | | $ | 52.1 |
| | $ | — |
| | $ | — |
| | $ | 52.1 |
|
| Corporate bonds | | 122.9 |
| | — |
| | (0.1 | ) | | 122.8 |
|
| Other government related obligations: | | | | | | | | |
| U.S. | | 17.5 |
| | — |
| | — |
| | 17.5 |
|
| Bank certificates of deposit | | 33.2 |
| | — |
| | — |
| | 33.2 |
|
| Total available-for-sale debt securities | | $ | 225.7 |
| | $ | — |
| | $ | (0.1 | ) | | $ | 225.6 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
The following table summarizes the changes in the carrying amount of goodwill:
|
| | | |
| Balance at December 31, 2014 | $ | 254 |
|
| Goodwill resulting from the Synageva acquisition | 4,794 |
|
| Balance at December 31, 2015 | $ | 5,048 |
|
| Change in goodwill associated with prior acquisition | $ | (11 | ) |
| Balance at December 31, 2016 | $ | 5,037 |
|
The amortized cost, gross unrealized holding gains, gross unrealized holding losses and estimated fair value of available-for-sale investments by type of security at December 31, 2016 and December 31, 2015 were as follows:
|
| | | | | | | | | | | | | | | | |
| | | December 31, 2016 |
| | | Amortized Cost | | Gross Unrealized Holding Gains | | Gross Unrealized Holding Losses | | Fair Value |
| Commercial paper | | $ | 114 |
| | $ | — |
| | $ | — |
| | $ | 114 |
|
| Corporate bonds | | 124 |
| | — |
| | (1 | ) | | 123 |
|
| Municipal bonds | | 91 |
| | — |
| | — |
| | 91 |
|
| Other government related obligations: | | | | | | | | |
| U.S. | | 28 |
| | — |
| | — |
| | 28 |
|
| Foreign | | 73 |
| | — |
| | (1 | ) | | 72 |
|
| Bank certificates of deposit | | 5 |
| | — |
| | — |
| | 5 |
|
| Total available-for-sale debt securities | | $ | 435 |
| | $ | — |
| | $ | (2 | ) | | $ | 433 |
|
| Equity securities | | — |
| | 1 |
| | — |
| | 1 |
|
| Total available-for-sale securities | | $ | 435 |
| | $ | 1 |
| | $ | (2 | ) | | $ | 434 |
|
| | | | | December 31, 2015 | | December 31, 2017 |
| | | Amortized Cost | | Gross Unrealized Holding Gains | | Gross Unrealized Holding Losses | | Fair Value | | Amortized Cost | | Gross Unrealized Holding Gains | | Gross Unrealized Holding Losses | | Fair Value |
| Commercial paper | | $ | 254 |
| | $ | — |
| | $ | — |
| | $ | 254 |
| | $ | 16.0 |
| | $ | — |
| | $ | — |
| | $ | 16.0 |
|
| Repurchase agreements | | | 27.0 |
| | — |
| | — |
| | 27.0 |
|
| Corporate bonds | | 133 |
| | — |
| | — |
| | 133 |
| | 432.2 |
| | 0.5 |
| | (0.2 | ) | | 432.5 |
|
| Municipal bonds | | 87 |
| | — |
| | — |
| | 87 |
| |
| Other government related obligations: | | | | | | | | | | | | | | | | |
| U.S. | | 25 |
| | — |
| | — |
| | 25 |
| |
| Foreign | | 164 |
| | — |
| | (1 | ) | | 163 |
| | 426.3 |
| | 0.2 |
| | (0.2 | ) | | 426.3 |
|
| Bank certificates of deposit | | 27 |
| | — |
| | — |
| | 27 |
| | 11.8 |
| | — |
| | — |
| | 11.8 |
|
| Total available-for-sale securities | | $ | 690 |
| | $ | — |
| | $ | (1 | ) | | $ | 689 |
| |
| Total available-for-sale debt securities | | | $ | 913.3 |
| | $ | 0.7 |
| | $ | (0.4 | ) | | $ | 913.6 |
|
The aggregate fair value of available-for-sale debt securities in an unrealized loss position as of December 31, 20162018 and December 31, 20152017 was $265$128.7 and $294.$436.2, respectively. Investments that have been in a continuous unrealized loss position for more than 12twelve months werewas $12.0 as of December 31, 2017. We did not material.have any investments in a continuous unrealized loss position for more than twelve months as of December 31, 2018. As of December 31, 20162018 we believe that the cost basis of our available-for-sale investmentsdebt securities is recoverable.
The fair values of available-for-sale debt securities by classification in the consolidated balance sheet were as follows:
|
| | | | | | | |
| | December 31, 2016 | | December 31, 2015 |
| Cash and cash equivalents | $ | 120 |
| | $ | 323 |
|
| Marketable securities | 314 |
| | 366 |
|
| | $ | 434 |
| | $ | 689 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
|
| | | | | | | |
| | December 31, 2018 | | December 31, 2017 |
| Cash and cash equivalents | $ | 43.8 |
| | $ | 42.7 |
|
| Marketable securities | 181.8 |
| | 870.9 |
|
| | $ | 225.6 |
| | $ | 913.6 |
|
The fair values of available-for-sale debt securities atas of December 31, 2016,2018, by contractual maturity, are summarized as follows:
| | | | December 31, 2016 | December 31, 2018 |
| Due in one year or less | $ | 236 |
| $ | 211.5 |
|
| Due after one year through three years | 197 |
| 14.1 |
|
| Due after three years through five years | — |
| — |
|
| | $ | 433 |
| $ | 225.6 |
|
We sponsor a nonqualified deferred compensation plan which allows certain highly-compensated employees to elect to defer income to future periods. Participants in the plan earn a return on their deferrals based on several investment options, which mirror returns on underlying mutual fund investments. We choose to invest in the underlying mutual fund investments to offset the liability associated with our nonqualified deferred compensation plan. These mutual fund investments are valued at net asset value per share and are carried at fair value with gains and losses included in investment income. The changes in the underlying liability to the employee are recorded in operating expenses. As of December 31, 20162018 and December 31, 2015,2017, the fair value of our trading securitiesthese investments was $13$16.5 and $9.$18.5, respectively.
We utilize the specific identification method in computing realized gains and losses. Realized gains and losses on our available-for-sale and tradingmarketable securities were not material for the yearyears ended December 31, 20162018, 2017 and 2015.2016.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
| |
| 6. | Derivative Instruments and Hedging Activities |
We operate internationally and, in the normal course of business, are exposed to fluctuations in foreign currency exchange rates. The exposures result from portions of our revenues, as well as the related receivables, and expenses that are denominated in currencies other than the U.S. dollar, primarily the Euro and Japanese Yen. We are also exposed to fluctuations in interest rates on outstanding borrowings under our outstandingrevolving credit facility and term loan debt.facility. We manage these exposures within specified guidelines through the use of derivatives. All of our derivative instruments are utilized for risk management purposes, and we do not use derivatives for speculative trading purposes.
We enter into foreign exchange forward contracts, with durations of up to 60 months, to hedge exposures resulting from portions of our forecasted revenues, including intercompany revenues, and certain forecasted expenses that are denominated in currencies other than the U.S. dollar. The purpose of these hedges is to reduce the volatility of exchange rate fluctuations on our operating results and to increase the visibility of the foreign exchange impact on forecasted revenues.results. These hedges are designated as cash flow hedges upon contract inception. At As of December 31, 2016,2018, we had open revenue related foreign exchange forward contracts with notional amounts totaling $1,742$991.1 that qualified for hedge accounting.accounting with current contract maturities through December 2020. As of December 31, 2018, we had open expense related foreign exchange forward contracts with notional amounts totaling $20.3 that qualified for hedge accounting with contract maturities through September 2022.
To achieve a desired mix of floating and fixed interest rates on our term loan, we enteredenter into two interest rate swap agreements in June 2016 that qualifiedqualify for and are designated as cash flow hedges. These contracts convert the floating interest rate on a portion of our debt to a fixed rate, plus a borrowing spread.
The firstfollowing tables summarize the total interest rate swap contracts executed as of December 31, 2018:
|
| | | | |
| Type of Interest Rate Swap | Notional Amount | Effective Date | Termination Date | Fixed Interest Rate or Rate Range |
| Floating to Fixed | 2,031.3 | December 2016 - January 2018 | December 2018 - December 2019 | 0.98% - 1.62% |
| Floating to Fixed | 450.0 | December 2018 | December 2022 | 2.60% - 2.79% |
| Floating to Fixed | 300.0 | January 2019 | December 2019 | 2.08% |
| Floating to Fixed | 1,100.0 | December 2019 | December 2022 | 2.70% - 2.83% |
In January 2019, we entered into an additional interest rate swap agreement hadwith a notional amount of $3,281 and was$200.0 that is effective from June 30, 2016 through December 30, 2016. This agreement hedged the contractual floating interest rate of our term loan. As a result of this agreement, the interest rate for our term loan was fixed at 0.535%, plus the borrowing spread, until December 30, 2016. The second agreement has a notional amount of $656 and is effective December 31, 20162019 through December 31, 2019. The second agreement2022 and converts the floating rate on a portion of our term loan to a fixed rate of 0.98%2.37%, plus a borrowing spread, fromspread.
During the second quarter 2018, we adopted the new standard for accounting for hedges that is designed to simplify the application of hedge accounting and increase transparency as to the scope and results of hedging programs. The updated guidance no longer requires the separate measurement and reporting of hedge ineffectiveness. Following adoption, all unrealized gains and losses on derivatives that are designated and qualify for hedge accounting are reported in other comprehensive income (loss) and recognized in our consolidated statements of operations when the underlying hedged transaction affects earnings.
The amount of gains and losses recognized in the consolidated statements of operations for the years ended December 31, 2016 through December 2019. In the first quarter of2018, 2017, we entered into an additionaland 2016from foreign exchange and interest rate swap agreement with a notional amount of $300 that is effective from January 31, 2017 through December 31, 2018 that converts the floating rate on a portion of our term loan to a fixed rate of 1.29%, plus a borrowing spread, from January 2017 through December 2018.
The impact on accumulated other comprehensive income (AOCI) and earnings from derivative instrumentscontracts that qualified as cash flow hedges for the years ended December 31, 2016 and 2015were as follows:
|
| | | | | | | |
| | Year Ended December 31, |
| Foreign Exchange Contracts: | 2016 | | 2015 |
| Gain recognized in AOCI, net of tax | $ | 40 |
| | $ | 111 |
|
| Gain reclassified from AOCI to net product sales (effective portion), net of tax | $ | 47 |
| | $ | 103 |
|
| Gain reclassified from AOCI to other income and expense (ineffective portion), net of tax | $ | — |
| | $ | 2 |
|
| Interest Rate Contracts: | | | |
| Gain recognized in AOCI, net of tax | $ | 6 |
| | $ | — |
|
| Gain reclassified from AOCI to interest expense, net of tax | $ | — |
| | $ | — |
|
Assuming no change in foreign exchange rates or LIBOR-based interest rates from market rates at December 31, 2016, $78 of gains recognized in AOCI will be reclassified to revenue over the next 12 months. The amount of gains recognized in AOCI that will be reclassified to interest expense over the next 12 months is immaterial. |
| | | | | | | | | | | | | | | | | | | | | | | |
| | Year ended December 31, |
| | 2018 | | 2017 | | 2016 |
| | Net Product Sales | | Interest Expense | | Net Product Sales | | Interest Expense | | Net Product Sales | | Interest Expense |
| Financial Statement Line Item in which the Effects of Cash Flow Hedges are Recorded | $ | 4,130.1 |
| | $ | (98.2 | ) | | $ | 3,549.5 |
| | $ | (98.4 | ) | | $ | 3,081.7 |
| | $ | (96.9 | ) |
| Impact of cash flow hedging relationships: | | | | | | | | | | | |
| Foreign Exchange Forward Contracts | $ | (1.8 | ) | | $ | — |
| | $ | 28.9 |
| | $ | — |
| | $ | 73.0 |
| | $ | — |
|
| Interest Rate Swap Contracts | $ | — |
| | $ | 13.6 |
| | $ | — |
| | $ | (1.8 | ) | | $ | — |
| | $ | (0.2 | ) |
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
The impact on accumulated other comprehensive income (AOCI) and earnings from foreign exchange and interest rate swap contracts that qualified as cash flow hedges, for the years ended December 31, 2018, 2017, and 2016 were as follows:
|
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2018 | | 2017 | | 2016 |
| Foreign Exchange Contracts: | | | | | |
| Gain (loss) recognized in AOCI, net of tax | $ | 37.7 |
| | $ | (96.1 | ) | | $ | 40.2 |
|
| Gain (loss) reclassified from AOCI to net product sales (effective portion), net of tax | $ | (1.4 | ) | | $ | 18.7 |
| | $ | 47.3 |
|
| Interest Rate Contracts: | | | | | |
| Gain (loss) recognized in AOCI, net of tax | $ | (4.8 | ) | | $ | 7.9 |
| | $ | 6.2 |
|
| Gain (loss) reclassified from AOCI to interest expense, net of tax | $ | 10.8 |
| | $ | (1.1 | ) | | $ | (0.1 | ) |
Assuming no change in foreign exchange rates from market rates at December 31, 2018, $9.8 of gains recognized in AOCI will be reclassified to revenue over the next 12 months. Assuming no change in LIBOR-based interest rates from market rates at December 31, 2018, $19.3 of gains recognized in AOCI will be reclassified to interest expense over the next 12 months. Amounts recognized in AOCI for expense related foreign exchange forward contracts was immaterial as of December 31, 2018.
We enter into foreign exchange forward contracts, with durations of approximately 90 days,up to 6 months, designed to limit the balance sheet exposure of monetary assets and liabilities. We enter into these hedges to reduce the impact of fluctuating exchange rates on our operating results. Hedge accounting is not applied to these derivative instruments as gains and losses on these hedge transactions are designed to offset gains and losses on underlying balance sheet exposures. As of December 31, 2016,2018, the notional amount of foreign exchange contracts where hedge accounting is not applied was $647.$1,511.6.
We recognized a gain (loss) gain of $(5), $5$23.0, $(14.7) and $26,$(5.2), in other income and expense for the years ended December 31, 2016, 20152018, 2017 and 2014,2016, respectively, associated with the foreign exchange contracts not designated as hedging instruments. These amounts were largelypartially offset by gains or losses inon monetary assets and liabilities.
The following tables summarize the fair value of outstanding derivatives at December 31, 20162018 and 2015:2017:
|
| | | | | | | | | | | |
| | December 31, 2016 |
| | Asset Derivatives | | Liability Derivatives |
| | Balance Sheet
Location | | Fair
Value | | Balance Sheet
Location | | Fair
Value |
| Derivatives designated as hedging instruments: | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | $ | 80 |
| | Other current liabilities | | $ | 2 |
|
| Foreign exchange forward contracts | Other assets | | 59 |
| | Other liabilities | | 4 |
|
| Interest rate contracts | Prepaid expenses and other current assets | | — |
| | Other current liabilities | | — |
|
| Interest rate contracts | Other assets | | 10 |
| | Other liabilities | | — |
|
| Derivatives not designated as hedging instruments: | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | 17 |
| | Other current liabilities | | 10 |
|
| Total fair value of derivative instruments | | | $ | 166 |
| | | | $ | 16 |
|
| | | | December 31, 2015 | December 31, 2018 |
| | Asset Derivatives | | Liability Derivatives | Asset Derivatives | | Liability Derivatives |
| | Balance Sheet
Location | | Fair
Value | | Balance Sheet
Location | | Fair
Value | Balance Sheet
Location | | Fair
Value | | Balance Sheet
Location | | Fair
Value |
| Derivatives designated as hedging instruments: | | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | $ | 85 |
| | Other current liabilities | | $ | 1 |
| Prepaid expenses and other current assets | | $ | 16.9 |
| | Other current liabilities | | $ | 7.3 |
|
| Foreign exchange forward contracts | Other assets | | 66 |
| | Other liabilities | | 5 |
| Other assets | | 0.3 |
| | Other liabilities | | 3.1 |
|
| Interest rate contracts | | Prepaid expenses and other current assets | | 20.1 |
| | Other current liabilities | | 0.8 |
|
| Interest rate contracts | | Other assets | | — |
| | Other liabilities | | 17.3 |
|
| Derivatives not designated as hedging instruments: | | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | 7 |
| | Other current liabilities | | 4 |
| Prepaid expenses and other current assets | | 23.6 |
| | Other current liabilities | | 11.5 |
|
| Total fair value of derivative instruments | | $ | 158 |
| | $ | 10 |
| | $ | 60.9 |
| | $ | 40.0 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
|
| | | | | | | | | | | |
| | December 31, 2017 |
| | Asset Derivatives | | Liability Derivatives |
| | Balance Sheet
Location | | Fair
Value | | Balance Sheet
Location | | Fair
Value |
| Derivatives designated as hedging instruments: | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | $ | 12.9 |
| | Other current liabilities | | $ | 34.8 |
|
| Foreign exchange forward contracts | Other assets | | 4.1 |
| | Other liabilities | | 26.0 |
|
| Interest rate contracts | Prepaid expenses and other current assets | | 9.3 |
| | Other current liabilities | | — |
|
| Interest rate contracts | Other assets | | 12.5 |
| | Other liabilities | | — |
|
| Derivatives not designated as hedging instruments: | | | | | | | |
| Foreign exchange forward contracts | Prepaid expenses and other current assets | | 10.0 |
| | Other current liabilities | | 13.7 |
|
| Total fair value of derivative instruments | | | $ | 48.8 |
| | | | $ | 74.5 |
|
Although we do not offset derivative assets and liabilities within our condensed consolidated balance sheets, our International Swap and Derivatives Association agreements provide for net settlement of transactions that are due to or from the same counterparty upon early termination of the agreement due to an event of default or other termination event. The following tables summarize the potential effect on our consolidated balance sheets of offsetting our foreign exchange forward contracts and interest rate contracts subject to such provisions:
| | | December 31, 2016 | |
| December 31, 2018 | | December 31, 2018 |
| | | | | | | | | Gross Amounts Not Offset in the Consolidated Balance Sheet | | | | | | | | | | Gross Amounts Not Offset in the Consolidated Balance Sheet | | |
| Description | | Gross Amounts of Recognized Assets/Liabilities | | Gross Amounts Offset in the Consolidated Balance Sheet | | Net Amounts of Assets/Liabilities Presented in the Consolidated Balance Sheet | | Derivative Financial Instruments | | Cash Collateral Received (Pledged) | | Net Amount | | Gross Amounts of Recognized Assets/Liabilities | | Gross Amounts Offset in the Consolidated Balance Sheet | | Net Amounts of Assets/Liabilities Presented in the Consolidated Balance Sheet | | Derivative Financial Instruments | | Cash Collateral Received (Pledged) | | Net Amount |
| Derivative assets | | $ | 166 |
| | $ | — |
| | $ | 166 |
| | $ | (16 | ) | | $ | — |
| | $ | 150 |
| | $ | 60.9 |
| | $ | — |
| | $ | 60.9 |
| | $ | (30.2 | ) | | $ | — |
| | $ | 30.7 |
|
| Derivative liabilities | | (16 | ) | | — |
| | (16 | ) | | 16 |
| | — |
| | — |
| | $ | (40.0 | ) | | $ | — |
| | $ | (40.0 | ) | | $ | 30.2 |
| | $ | — |
| | $ | (9.8 | ) |
| | | December 31, 2015 | |
| December 31, 2017 | | December 31, 2017 |
| | | | | | | | | Gross Amounts Not Offset in the Consolidated Balance Sheet | | | | | | | | | | Gross Amounts Not Offset in the Consolidated Balance Sheet | | |
| Description | | Gross Amounts of Recognized Assets/Liabilities | | Gross Amounts Offset in the Consolidated Balance Sheet | | Net Amounts of Assets/Liabilities Presented in the Consolidated Balance Sheet | | Derivative Financial Instruments | | Cash Collateral Received (Pledged) | | Net Amount | | Gross Amounts of Recognized Assets/Liabilities | | Gross Amounts Offset in the Consolidated Balance Sheet | | Net Amounts of Assets/Liabilities Presented in the Consolidated Balance Sheet | | Derivative Financial Instruments | | Cash Collateral Received (Pledged) | | Net Amount |
| Derivative assets | | $ | 158 |
| | $ | — |
| | $ | 158 |
| | $ | (10 | ) | | $ | — |
| | $ | 148 |
| | $ | 48.8 |
| | $ | — |
| | $ | 48.8 |
| | $ | (26.3 | ) | | $ | — |
| | $ | 22.5 |
|
| Derivative liabilities | | (10 | ) | | — |
| | (10 | ) | | 10 |
| | — |
| | — |
| | $ | (74.5 | ) | | $ | — |
| | $ | (74.5 | ) | | $ | 26.3 |
| | $ | — |
| | $ | (48.2 | ) |
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
| |
| 7. | Accrued ExpensesOther Investments |
Accrued expenses consistOther investments include strategic equity investments in Moderna Therapeutics, Inc. (Moderna) and Dicerna Pharmaceuticals, Inc. (Dicerna).
Moderna
During 2014, we purchased $37.5 of preferred equity of Moderna, a nonpublic biotechnology company, which was recorded at cost. During the first quarter of 2018, Moderna announced the completion of a new round of financing. We considered this transaction and the rights of the
following: new shares issued in the new round, compared to the rights of the preferred equity that we hold, and concluded that Moderna’s new round of financing represented an observable price change in an orderly transaction for a similar investment. We further concluded, based on the respective rights of the stock and consideration of potential liquidity events, that the value of our preferred stock was equivalent to the value of the newly issued preferred stock. As a result, we recognized an unrealized gain of $100.8 in investment income during the first quarter 2018 to adjust our equity investment in Moderna to fair value as of the date of the observable price change, based on the per share price in Moderna's new round of financing. |
| | | | | | | |
| | December 31, 2016 | | December 31, 2015 |
| Royalties | $ | 20 |
| | $ | 30 |
|
| Payroll and employee benefits | 121 |
| | 115 |
|
| Taxes payable | 39 |
| | 12 |
|
| Rebates payable | 70 |
| | 56 |
|
| Clinical | 64 |
| | 57 |
|
| Manufacturing | 52 |
| | 19 |
|
| Other | 142 |
| | 114 |
|
| | $ | 508 |
| | $ | 403 |
|
The carrying value of this investment was $81.9 and $37.5 as of December 31, 2018 and 2017, respectively.
Dicerna
In October 2018, we purchased $10.3 of Dicerna common stock in connection with a collaboration agreement that we entered into with Dicerna, a publicly-traded biopharmaceutical company, see Note 11, “Commitments and Contingencies” for additional information on collaboration agreement. As our equity investment in Dicerna common stock has a readily determinable fair value, we are recording the investment at fair value. We have considered the effects of a six month holding period restriction and determined the impact on the fair value is immaterial. During the fourth quarter 2018, we recognized an unrealized loss of $1.4 in investment income to adjust our equity investment in Dicerna to fair value as of December 31, 2018.
The fair value of this investment was $8.9 as of December 31, 2018.
| |
| 8. | Accounts Payable and Accrued Expenses |
Accounts payable and accrued expenses consist of the following: |
| | | | | | | |
| | December 31, 2018 | | December 31, 2017 |
| Accounts Payable | $ | 74.4 |
| | $ | 70.8 |
|
| Royalties | 27.0 |
| | 22.5 |
|
| Payroll and employee benefits | 170.4 |
| | 149.9 |
|
| Taxes payable | 24.4 |
| | 30.7 |
|
| Rebates payable | 122.8 |
| | 99.1 |
|
| Clinical | 58.6 |
| | 79.1 |
|
| Manufacturing | 72.0 |
| | 41.1 |
|
| Accrued restructuring costs | 4.2 |
| | 58.2 |
|
| Other | 144.4 |
| | 158.8 |
|
| | $ | 698.2 |
| | $ | 710.2 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
On June 7, 2018, we entered into an Amended and Restated Credit Agreement (the Credit Agreement), with Bank of America, N.A. as Administrative Agent. The Credit Agreement amends and restates our credit agreement dated as of June 22, 2015 Alexion entered into a credit agreement (Credit(the Prior Credit Agreement) with a syndicate of banks, which.
The Credit Agreement provides for a $3,500$1,000.0 revolving credit facility and a $2,612.5 term loan facility. The revolving credit facility and the term loan facility and a $500 revolving credit facility maturing in five years. Borrowings undermature on June 7, 2023. Beginning with the term loanquarter ending June 30, 2019, we are payable in quarterly installments equalrequired to 1.25%make amortization payments of 5.00% of the original loanaggregate principal amount beginning December 31, 2015. Final repayment of the term loan and revolving credit loans are due on June 22, 2020. In addition to borrowingsfacility annually, payable in which prior notice is required, the revolving credit facility includes a sublimit of $100 in the form of letters of credit and borrowings on same-day notice, referred to as swingline loans, of up to $25. Borrowings can be used for working capital requirements, acquisitions and other general corporate purposes. With the consent of the lenders and the administrative agent, and subject to satisfaction of certain conditions, we may increase the term loan facility and/or the revolving credit facility in an amount that does not cause our consolidated net leverage ratio to exceed the maximum allowable amount.equal quarterly installments.
Under the Credit Agreement we may elect that the loansLoans under the Credit Agreement bear interest, at a rate per annum equal toour option, at either a base rate or a Eurodollar rate, plus, in each case plus an applicable margin. TheUnder the Credit Agreement, the applicable margins on base rate loans range from 0.25% to 1.00% and the applicable margins on Eurodollar loans range from 1.25%to 2.00%, in each case
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
depending upon based on our consolidated net leverage ratio (as calculated in accordance with the Credit Agreement). At December 31, 2016,2018, the interest rate on our outstanding loans under the Credit Agreement was 2.52%3.90%. Our obligations under the credit facilitiesCredit Agreement are guaranteed by certain of Alexion’sAlexion Pharmaceuticals, Inc.'s foreign and domestic subsidiaries and secured by liens on certain of Alexion’s and itsour subsidiaries’ equity interests, subject to certain exceptions. Under the terms of the Credit Agreement, we must maintain a ratio of total net debt to EBITDA of 3.50 to 1.00 (subject to certain limited adjustments) and EBITDA to cash interest expense ratio of at least 3.50 to 1.00, in each case as calculated in accordance with the Credit Agreement.
The Credit Agreement requires us to comply withcontains certain financial covenants on a quarterly basis. Under these financial covenants, we are required to deliver to the administrative agent, not later than 50 days after each fiscal quarter, our quarterly financial statements, and within 5 days thereafter, a compliance certificate. In November 2016, we obtained a waiver from the necessary lenders for this requirement and the due date for delivery of the third quarter 2016 financial statements and compliance certificate was extended to January 18, 2017. The posting of the Third Quarter report on Form 10-Q on our website on January 4, 2017 satisfied the financial statement covenant, and we simultaneously delivered the required compliance certificate, as required by the lenders.
Further, the Credit Agreement includes negative covenants, subject to exceptions, restricting or limiting our ability and the ability of our subsidiaries to, among other things, incur additional indebtedness, grant liens, and engage in certain investment, acquisition and disposition transactions. The Credit Agreement also contains customary representations and warranties, affirmative and negative covenants and events of default, including payment defaults, breachdefault. The negative covenants in the Credit Agreement restrict Alexion’s and its subsidiaries’ ability, subject to certain baskets and exceptions, to (among other things) incur liens or indebtedness, make investments, enter into mergers and other fundamental changes, make dispositions or pay dividends. The restriction on dividend payments includes an exception that permits us to pay dividends and make other restricted payments regardless of representations and warranties, covenant defaults and cross defaults. If an event of default occurs,dollar amount so long as, after giving pro forma effect thereto, we have a consolidated net leverage ratio, as defined in the interest rate would increase and the administrative agent would be entitledCredit Agreement, within predefined ranges, subject to take various actions, including the acceleration of amounts due under the loan.certain increases following designated material acquisitions.
In connection with entering into the Credit Agreement and the Prior Credit Agreement, we paid $45an aggregate of$53.1 in financing costs. Financing costs which are being amortized as interest expense over the life of the debt. Amortization expense associated with deferred financing costs for the years ended December 31, 2018, 2017, and 2016 was $8.0, $9.2, and 2015 was $10 and $6,$10.3, respectively. Amortization expense associated withRemaining unamortized deferred financing costs for the year endedas of December 31, 2014 was not material.
In connection with the acquisition of Synageva in June 2015, we borrowed $3,500 under the term loan facility2018 and $200 under the revolving facility, and we used our available cash for the remaining cash consideration. We made principal payments of $375 during the year ended December 31, 2016. At2017 were $20.8 and $21.0, respectively.
As of December 31, 2016,2018, we had $3,081$2,612.5 outstanding on the term loan and zero$250.0 of borrowings outstanding under the revolving credit facility. The $250.0 of proceeds on the revolving facility. Atcredit facility was used to refinance amounts outstanding under the Prior Credit Agreement. As of December 31, 2016,2018, we had open letters of credit of $15, and$1.7 that offset our borrowing availability underin the revolving facility. In January 2019 we paid the outstanding revolving credit facility was $485.of $250.0 in full.
The fair value of our long term debt, which is measured using Level 2 inputs of the fair value hierarchy, approximates book value.
The contractual maturities of our long-term debt obligations, including our revolving credit facility, due subsequent to December 31, 20162018 are as follows:
| | | Year | | |
| 2017 | $ | — |
| |
| 2018 | 150 |
| |
| 2019 | 175 |
| $ | 348.0 |
|
| 2020 | 2,756 |
| 130.6 |
|
| 2021 | | 130.6 |
|
| 2022 | | 130.6 |
|
| 2023 | | 2,122.7 |
|
Based upon our intent and ability
Alexion Pharmaceuticals, Inc.
Notes to make payments during 2017, we included $175 within current liabilities on our consolidated balance sheet as ofConsolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016 net of current deferred financing costs.
(amounts in millions except per share amounts)
| |
9.10. | Facility Lease Obligations |
New Haven Facility Lease Obligation
In November 2012, we entered into a lease agreement for office and laboratory space to be constructed in New Haven, Connecticut. The term of the lease commenced in 2015 and will expire in 2030, with a renewal option of 10ten years. Although we do not legally own the premises, we are deemed to be the owner of the building due to the substantial improvements directly funded by us during the construction period based on applicable accounting guidance for build-to-suit leases. Accordingly, the landlord’s costs of constructing the facility during the construction period are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheet.sheets.
Construction of the new facility was completed and the building was placed into service in the first quarter 2016. The imputed interest rate on this facility lease obligation as of December 31, 20162018 was approximately 11%. ForAssociated with this arrangement, we recognized interest expense of $13.3, $14.2, and $14.0 for the yearyears ended December 31, 2018, 2017, and 2016, and 2015, we recognized $14 and $5, respectively, of interest expense associated with this arrangement.respectively. As of December 31, 20162018 and 2015,2017, our total facility lease obligation was $136$133.5 and $133,$134.6, respectively, recorded within other current liabilities and facility lease obligation onin our consolidated balance sheets.
Alexion Pharmaceuticals, Inc.
NotesIn the fourth quarter of 2018 we amended the New Haven lease agreement significantly reducing our leased square footage in the building beginning in 2019 through the expiration of the lease. This amendment does not impact our previous conclusions that we are deemed the owner of the building for accounting purposes. In conjunction with this lease modification, during the fourth quarter of 2018 we made a payment of $53.0 to Consolidated Financial Statements
Fora third party as an incentive to lease the Years endedreleased square footage. This was capitalized within other assets in our consolidated balance sheets as of December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
2018.
Aggregate future minimum non-cancellable commitments under the New Haven facility lease obligation, as of December 31, 20162018 are as follows:
| | | Year | | |
| 2017 | $ | 16 |
| |
| 2018 | 15 |
| |
| 2019 | 16 |
| $ | 9.4 |
|
| 2020 | 16 |
| 8.8 |
|
| 2021 | 16 |
| 9.0 |
|
| 2022 | | 9.2 |
|
| 2023 | | 9.2 |
|
| Thereafter | 146 |
| 63.9 |
|
Lonza Facility Lease Obligation
During the third quarter 2015, we entered into a new agreement with Lonza Group AG and its affiliates (Lonza) whereby Lonza will construct a new manufacturing facility dedicated to Alexion at one of its existing facilities. The agreement requires us to make certain payments during the construction of the new manufacturing facility and annual payments for ten years thereafter. As a result of our contractual right to full capacity of the new manufacturing facility, a portion of the payments under the agreement are considered to be lease payments and a portion as payment for the supply of inventory. Although we will not legally own the premises, we are deemed to be the owner of the manufacturing facility during the construction period based on applicable accounting guidance for build-to-suit leases due to our involvement during the construction period. Accordingly, the landlord’s costs of constructing the facility during the construction period are required to be capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheets. We expect the completion of the facility, including obtaining regulatory approval, to be in 2019. As of December 31, 20162018 and 2015,2017, we recorded a construction-in-process asset of $118$203.9 and $19,$180.6, respectively, and an offsetting facility lease obligation of $107$155.1 and $15,$159.1, respectively, associated with the manufacturing facility.within other current liabilities and facility lease obligation on our consolidated balance sheets.
Payments to Lonza under the agreement are allocated to the purchases of inventory and the repayment of the facility lease obligation on a relative fair value basis. In 2016,2018, we incurred $58$73.8 of payments to Lonza under this agreement, of which $8$9.6 was applied against the outstanding facility lease obligation and $50$64.2 was recognized as a prepayment of inventory. See Note 1011 “Commitments and Contingencies” for minimum fixed payments due under Lonza agreements.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
Boston Facility Lease Obligation
In September 2017, we entered into a lease agreement for approximately 150,000 square feet of office space to be constructed in Boston, Massachusetts. Construction of the facility was completed and the building was placed into service in the second quarter 2018. The term of the lease commenced upon the landlord's substantial completion of the facility in the second quarter of 2018 and will expire on the thirteenth anniversary of commencement, with an option to renew for up to an additional ten years. Although we do not legally own the premises, due to our involvement during the construction period, we are deemed to be the owner of the portion of the building that we will lease based on applicable accounting guidance for build-to-suit leases. Accordingly, the landlord's costs of constructing the facility during the construction period were capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in our consolidated balance sheets.
Interest expense recognized during 2018 was not material. As of December 31, 2018 and December 31, 2017, our total facility lease obligation was $83.6 and $59.6, respectively, recorded within facility lease obligation in our consolidated balance sheets.
Aggregate future minimum non-cancellable commitments under the Boston facility lease obligation, as of December 31, 2018 are as follows:
|
| | | |
| Year | |
| 2019 | $ | 4.3 |
|
| 2020 | 6.6 |
|
| 2021 | 6.7 |
|
| 2022 | 6.8 |
|
| 2023 | 7.0 |
|
| Thereafter | 56.7 |
|
In January 2019, we adopted a new lease accounting standard. See Note 1 “Business Overview & Summary of Significant Accounting Policies” for an overview of the impact this standard is expected to have on our facility lease obligation balances in 2019.
| |
10.11. | Commitments and Contingencies |
Commitments
License Agreements
We have entered into a number of license agreements since our inception in order to advance and obtain technologies and services related to our business. License agreements generally provide forrequire us to pay an initial fee followed by milestone and royalty payments if certain conditions are met. Certain agreements call for future payments upon the attainment of agreed upon development and/or commercial milestones. These agreements may also require minimum royalty payments based on sales of products developed from the applicable technologies, if any.
In March 2015, we entered into an agreement with a third party that allowed us to exercise an option with another third party for exclusive, worldwide, perpetual license rights to a specialized technology and other intellectual property, and we simultaneously exercised the option. Due to the early stage of these assets, we recorded expense for the payments of $47 during the first quarter 2015.
In March 2015,October 2018, we entered into a collaboration agreement with a third partyDicerna that allowsprovides us with exclusive worldwide licenses and development and commercial rights for two preclinical RNA interference (RNAi) subcutaneously delivered molecules for complement-mediated diseases, as well as an exclusive option for other preclinical RNAi molecules for two additional targets within the complement pathway. In addition to identify and optimize drug candidates. Alexion will havethe collaboration agreement, we made an equity investment in Dicerna. Under the terms of the agreements, we made an upfront payment of $37.0 for the exclusive worldwide rights to developlicenses and commercialize products arising from the collaboration.equity investment. The market value of the equity investment was $10.3 as of the date of acquisition, which we recorded in other assets in our consolidated balance sheets. Due to the early stage of the assets we are licensing, in connection with the collaboration, we recorded expense for the upfront license payment of $15$26.7 as research and development expense during the firstfourth quarter 2015.2018. In addition, as of December 31, 2016,2018, we could also be required to pay up to an additional $249 if certainapproximately $625.0 for option exercise fees and amounts due upon the achievement of specified research, development, regulatory and commercial milestones, are met over time, as well as royalties on commercial sales.
In January 2015, we entered into a license agreement with a third party to obtain an exclusive research, development and commercial license for specific therapeutic molecules. Due to the early stage of these assets, we recorded expense for the upfront payment of $50 during the first quarter 2015. In addition, we could be required to pay up to an additional $822 if certain development, regulatory, and commercial milestones are met over time, as well as royalties on commercial sales.
In December 2014,2017, we entered into ana collaboration and license agreement with X-Chem Pharmaceuticals (X-Chem)Halozyme Therapeutics, Inc. that allows us to identify novel drug candidates from X-Chem’s proprietary drug discovery engine. Alexion will haveuse drug-delivery technology in the exclusive worldwide rightsdevelopment of subcutaneous formulations for our portfolio of products for up to four targets. Due to the early stage of the assets we are licensing, we recorded expense for the upfront payment of $40.0 during the fourth quarter 2017. In addition, as of December 31, 2018, we could be required to pay an additional
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
develop and commercialize products arising from the collaboration in up to three program targets. Due to the early stage of these assets, we recorded expense for an upfront payment of $8. In addition,$160.0 for each program target for a maximum of three targets, we could be requireddeveloped, subject to make additional payments upon the achievement of specified research, development, regulatory and regulatorysales-based milestones, up to $75, as well as royalties on commercial sales.
In January 2014,addition, as of December 31, 2018, we have entered into an agreement with Moderna Therapeutics, Inc. (Moderna) that allows us to purchase ten product options to develop and commercialize treatments for rare diseases with Moderna’s messenger RNA (mRNA) therapeutics platform. Alexion will lead the discovery, development and commercialization of the treatments produced through this broad, long-term strategic agreement, while Moderna will retain responsibility for the design and manufacture of the messenger RNA against selected targets. Due to the early stage of these assets,other license agreements under which we recorded expense for an upfront payment of $100. We will alsomay be responsible for funding research activities under the program. In addition, for each drug target, up to a maximum of ten targets, we could be required to make an option exercise payment of $15 and to pay up to an additional $120 with respect to a rare disease product$137.2 if certain development, regulatory and $400 with respect to a non-rare disease product in development and sales milestones if the specificcommercial milestones are met over time as well as royalties on commercial sales.met.
Manufacturing Agreements
We have various manufacturing development and license agreements to support our clinical and commercial product needs.
We rely on Lonza, a third party manufacturer, to produce a portion of commercial and clinical quantities of Solirisour commercial products and Strensiq.product candidates. We have various manufacturing and license agreements with Lonza, with remaining total non-cancellable future commitments of approximately $1,148.$1,084.6. If we terminate certain supply agreements with Lonza without cause, we will be required to pay for product scheduled for manufacture under our arrangement. Under an existing arrangement with Lonza, we also pay Lonza a royalty on sales of SolirisSOLIRIS that was manufactured at Alexion Rhode Island Manufacturing Facility (ARIMF)the ARIMF facility prior to its sale and a payment with respect to sales of SolirisSOLIRIS manufactured at Lonza facilities.
In addition to Lonza, as of December 31, 2018, we have non-cancellable commitments of $27approximately $104.1 through 2020 with other third party manufacturers.
Contingent Liabilities
We are currently involved in various claims, lawsuits and legal proceedings. On a quarterly basis, we review the status of each significant matter and assess its potential financial exposure. If the potential loss from any claim, asserted or unasserted, or legal proceeding is considered probable and the amount can be reasonably estimated, we accrue a liability for the estimated loss. Because of uncertainties related to claims and litigation, accruals are based on our best estimates based on information available information.at the time of the assessment. On a periodic basis, as additional information becomes available, or based on specific events such as the outcome of litigation or settlement of claims (and offers of settlement), we may reassess the potential liability related to these matters and may revise these estimates, which could result in a material adverse adjustment to our operating results. Costs associated with our involvement in legal proceedings are expensed as incurred. The outcome of any such proceedings, regardless of the merits, is inherently uncertain. If we were unable to prevail in any such proceedings, our consolidated financial position, results of operations, and future cash flows may be materially impacted.
We have in the past received, and may in the future receive, notices from third parties claiming that their patents may be infringed by the development, manufacture or sale of our products. Under the guidance of ASC 450, Contingencies, we record a royalty accrual based on our best estimate of the fair value percent of net sales of our products that we could be required to pay the owners of patents for technology used in the manufacture and sale of our products. A costly license, or inability to obtain a necessary license, could have a material adverse effect on our financial results.
In May 2015, we received a subpoena in connection with an investigation by the Enforcement Division of the U.S. Securities and Exchange Commission (SEC)SEC requesting information related to our grant-making activities and compliance with the Foreign Corrupt Practices Act (FCPA)FCPA in various countries. In addition, in October 2015, Alexionwe received a request from the U.S. Department of Justice (DOJ)DOJ for the voluntary production of documentdocuments and other information pertaining to Alexion’s compliance with FCPA. The SEC and DOJ also seek information related to Alexion’s recalls of specific lots of SolirisSOLIRIS and related securities disclosures. Alexion is cooperating with these investigations.
The investigations have focused on operations in various countries, including Brazil, Colombia, Japan, Russia and Turkey, and Alexion's compliance with the FCPA and other applicable laws.
At this time, Alexion is unable to predict the duration, scope or outcome of these investigations. While it is possible that a loss related to these matters may be incurred, given the ongoing nature of these investigations, management cannot reasonably estimate the potential magnitude of any such loss or range of loss, if any.
Several securities class action lawsuits have been filed againstor the Company and former officers in federal district court alleging violations of Sections 10(b) and 20(a)cost of the Securities Exchange Actongoing investigation. Any determination that our operations or activities are not or were not in compliance with existing laws or regulations could result in the imposition of 1934, 15 U.S.C. § 78j(b),fines, civil and Rule 10b-5, promulgated thereunder, alleging that defendants made misstatementscriminal penalties, equitable remedies, including disgorgement, injunctive relief, and/or omissions concerning the Company’s salesother sanctions against us, and remediation of Soliris. any such findings could have an adverse effect on our business operations.
Alexion is committed to strengthening its compliance program and is currently implementing a comprehensive company-wide transformation plan to enhance and remediate its business processes, structures, controls, training, talent and systems across Alexion’s global operations.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
On November 17, 2016, a shareholder filed a putative class action in the U.S. District Court for the Southern District of New York. While the litigation was in the early stages, and before defendants had responded to the complaint,As previously reported, on December 30, 2016 plaintiffs filed a notice of voluntary dismissal and dismissed all claims without prejudice. This case is now closed.
On December 29, 2016, a second shareholder filed a putative class action against the Company and certain former employees in the U.S. District Court for the District of Connecticut, alleging that defendants made misrepresentations and omissions about SolirisSOLIRIS. On April 12, 2017, the court appointed a lead plaintiff. On July 14, 2017, the lead plaintiff filed an amended putative class action complaint against the Company and seven current or former employees. The complaint alleges that defendants made misrepresentations and omissions about SOLIRIS, including alleged misrepresentations regarding sales practices, management changes, and related investigations, between February 10,January 30, 2014 and May 26, 2017, and that the Company's stock price dropped upon the purported disclosure of the misrepresentations. The plaintiffs seek to recover unspecified monetary relief, unspecified equitable and injunctive relief, interest, and attorneys’ fees and costs. Defendants moved to dismiss the amended complaint on September 12, 2017. Plaintiffs filed an opposition to defendants’ motion to dismiss on November 13, 2017, and defendants’ filed a reply brief in further support of their motion on December 9, 2016. On January 17, 2017, three parties filed motions28, 2017. Defendants’ motion to be named lead plaintiff in this action. Briefing on these motionsdismiss is ongoing. The litigation is innow fully briefed and pending before the early stages, and defendants have not yet responded to the complaint.court. Given the early stages of this litigation, management does not currently believe that a loss related to this matter is probable or thatan estimate of the potential magnitude of suchpossible loss or range of loss if any, cancannot be reasonably estimated. made at this time.
In December 2016, we received a subpoena from the U.S. Attorney’sAttorney's Office for the District of Massachusetts requesting documents relating generally to our support of Patient Services, Inc. (PSI) and National Organization for Rare Disorders (NORD), 501(c)(3) organizations that provide financial assistance to Medicare patients taking drugs sold by Alexion,Alexion; Alexion’s provision of free drug to Medicare patients,patients; and Alexion compliance policies and training materials concerning the anti-kickback statute or paymentsand information on donations to any 501(c)(3) organization that provides financial assistance to Medicare patients.PSI and NORD from 2010 through 2016. Other companies have disclosed similar inquiries. We are cooperating with this inquiry. We have been engaged in discussions with the DOJ about a potential resolution of this matter and, in December 2018, we reached an agreement in principle to resolve this matter by entering into a civil settlement agreement with the DOJ and the Office of Inspector General (OIG) of the U.S. Department of Health and Human Services. As part of the proposed resolution, Alexion will pay approximately $13.0 to the DOJ and OIG. While we have reached an agreement in principle, there can be no assurance that the steps necessary to conclusively resolve this matter will be successful or that the settlement terms will be finalized. We are unable to determine when a potential final settlement may be reached. Further, if we are unable to reach a final agreement based on the agreement in principle, we will not be able to predict when these matters will be resolved or what further action, if any, the government will take in connection with them.
InMarch 2013, May 2017, Brazilian authorities seized records and data from our Sao Paulo, Brazil offices as part of an investigation being conducted into Alexion’s Brazilian operations. We are cooperating with this inquiry.
In June 2017, we received a Warning Letter (Warning Letter) fromdemand to inspect certain of our books and records pursuant to Section 220 of the FDA regarding compliance with current Good Manufacturing Practices (cGMP) at ARIMF. The Warning Letter followed receiptGeneral Corporation Law of the State of Delaware on behalf of a Form 483 Inspectional Observationspurported stockholder. Among other things, the demand sought to determine whether to institute a derivative lawsuit against certain of the Company’s directors and officers in relation to the investigation by our Audit and Finance Committee announced in November 2016 and the investigations instituted by the FDA in connection with an FDA inspectionSEC, DOJ, U.S. Attorney’s Office for the District of Massachusetts, and Brazilian law enforcement officials that concluded in August 2012. The observations relate to commercial and clinical manufacture of Soliris at ARIMF.are described above. We have responded to the Warning Letter in a letter todemand. Given the FDA dated in April 2013. As previously disclosed, the FDA issued Form 483s in August 2014 and August 2015 related to observations at ARIMF and the inspectional observations from the August 2014 and 2015 Forms 483s have since been closed out by the FDA. During July 2016, the FDA completed a routine inspection at ARIMF and have since confirmed receiptearly stages of our responses to the inspectional observations included in the Form 483 received during that inspection. The observations are inspectional and do not represent a final FDA determination of compliance. We continue to manufacture products, including Soliris, in this facility. While the resolutionmatter, an estimate of the issues raised in the Warning Letter is difficult to predict, we do not currently believe a loss related to this matter is probable or that the potential magnitude of suchpossible loss or range of loss cannot be made at this time.
On September 27, 2017, a hearing panel of the Canadian Patented Medicine Prices Review Board (PMPRB) issued a decision in a previously pending administrative pricing matter that we had excessively priced SOLIRIS in a manner inconsistent with the Canadian pricing rules and guidelines. In its decision, the PMPRB ordered Alexion to decrease the price of SOLIRIS to an upper limit based upon pricing in certain other countries, and to forfeit excess revenues for the period between 2009 and 2017. The amount of excess revenues was not determined to be a material amount. In October 2017, Alexion filed an application for judicial review of the PMPRB’s decision in the Federal Court of Canada. The hearing of that application for judicial review took place on November 15 and 16, 2018 but a decision on this matter has not yet been delivered by the Court. At this time, we cannot predict the outcome of these judicial review proceedings or any appeals that may follow and cannot reasonably estimate the amount of any additional forfeitures that will be required to be made or the potential impact to future SOLIRIS revenues in Canada relating to any potential future price reduction.
In October 2018, the Japanese Ministry of Health, Labour and Welfare (MHLW) conducted an administrative inspection of Alexion’s Japanese operations. The MHLW inquiry has been primarily focused on our communication efforts regarding the proper use of SOLIRIS in Japan for aHUS, among other matters. We have cooperated, and will continue to cooperate, with this inquiry. An estimate of the possible loss or range of loss, or what further action, if any, canthe MHLW will take in connection with this matter, cannot be reasonably estimated.made at this time.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
Chugai Pharmaceutical Co., Ltd. has filed two lawsuits against Alexion. The first was filed in November 2018 in the United States District Court for the District of Delaware against Alexion Pharmaceuticals, Inc. alleging that ULTOMIRIS infringes one U.S. patent held by Chugai Pharmaceutical Co., Ltd. The second lawsuit was filed in December 2018 in the Tokyo District Court against Alexion Pharma GK (a wholly-owned subsidiary of Alexion) in Japan and alleges that ULTOMIRIS infringes two Japanese patents held by Chugai Pharmaceutical Co., Ltd. Chugai’s complaints seek unspecified damages and certain injunctive relief. Alexion has filed an answer to the U.S. complaint that denies infringement. In addition, Alexion has raised defenses and has brought several counterclaims against Chugai Pharmaceutical Co., Ltd. that request a finding of non-infringement and patent invalidity. Alexion Pharma GK responded to Chugai at the Tokyo District Court and raised defenses against Chugai Pharmaceutical Co., Ltd. that request a finding of non-infringement and patent invalidity. Given the early stages of these litigations, an estimate of the possible loss or range of loss cannot be made at this time.
Operating Leases
As of December 31, 2016,2018, we have operating leases for office and laboratory space in U.S. and foreign locations to support our operations as a global organization.
Aggregate lease expense was $29, $28$24.4, $27.2 and $23$29.3 for the years ended December 31, 2016, 20152018, 2017 and 2014,2016, respectively. Lease expense is being recorded on a straight-line basis over the applicable lease terms.
Aggregate future minimum annual rental payments, for the next five years and thereafter under non-cancellable operating leases (including facilities and equipment) as of December 31, 20162018 are:
| | | Year | | |
| 2017 | $ | 21 |
| |
| 2018 | 19 |
| |
| 2019 | 13 |
| $ | 14.1 |
|
| 2020 | 7 |
| 9.3 |
|
| 2021 | 6 |
| 5.6 |
|
| 2022 | | 3.9 |
|
| 2023 | | 3.5 |
|
| Thereafter | 24 |
| 11.6 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
The income tax expense is based on income before income taxes as follows:
| | | | Year Ended December 31, | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 | 2018 |
| 2017 |
| 2016 |
| U.S. | $ | (165 | ) | | $ | (126 | ) | | $ | 222 |
| $ | (451.4 | ) | | $ | (43.9 | ) | | $ | (164.6 | ) |
| Non-U.S. | 741 |
| | 624 |
| | 650 |
| 693.6 |
| | 591.7 |
| | 740.8 |
|
| | $ | 576 |
| | $ | 498 |
| | $ | 872 |
| $ | 242.2 |
| | $ | 547.8 |
| | $ | 576.2 |
|
During the fourth quarter of 2013, in connection with the centralization of our global supply chain and technical operations in Ireland, our U.S. parent company became a direct partner in a captive foreign partnership. The partnership income, which is derived in foreign jurisdictions, is classified as “non-U.S. income” for purposes of financial reporting. Substantially all non-U.S. income for the years ended December 31, 2016 and 2015 relates to income from our captive foreign partnership.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
The components of the income tax expense are as follows:
|
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 |
| Domestic | | | | | |
| Current | $ | 4 |
| | $ | (88 | ) | | $ | 285 |
|
| Deferred | 107 |
| | 389 |
| | (112 | ) |
| | 111 |
| | 301 |
| | 173 |
|
| Foreign | | | | | |
| Current | 69 |
| | 49 |
| | 82 |
|
| Deferred | (3 | ) | | 4 |
| | (40 | ) |
| | 66 |
| | 53 |
| | 42 |
|
| Total | | | | | |
| Current | 73 |
| | (39 | ) | | 367 |
|
| Deferred | 104 |
| | 393 |
| | (152 | ) |
| | $ | 177 |
| | $ | 354 |
| | $ | 215 |
|
We continue to maintain a valuation allowance against certain deferred tax assets where realization is not certain. |
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2018 |
| 2017 |
| 2016 |
| Domestic | | | | | |
| Current | $ | 57.0 |
| | $ | 42.9 |
| | $ | 3.4 |
|
| Deferred | 49.5 |
| | 7.2 |
| | 107.6 |
|
| | 106.5 |
| | 50.1 |
| | 111.0 |
|
| Foreign | | | | | |
| Current | 74.7 |
| | 107.5 |
| | 69.1 |
|
| Deferred | (16.6 | ) | | (53.1 | ) | | (3.3 | ) |
| | 58.1 |
| | 54.4 |
| | 65.8 |
|
| Total | | | | | |
| Current | 131.7 |
| | 150.4 |
| | 72.5 |
|
| Deferred | 32.9 |
| | (45.9 | ) | | 104.3 |
|
| | $ | 164.6 |
| | $ | 104.5 |
| | $ | 176.8 |
|
We continue to pay cash taxes in U.S. Federal, various U.S. state, and foreign jurisdictions where we have utilized all of our tax attributes or have met the applicable limitation for attribute utilization.
At December 31, 2016, we have tax effected federal and state net operating loss carryforwards of $57 and $5, respectively. Our NOL’s expire between 2020 and 2036. We also have federal and state income tax credit carryforwards of $536 and $11, respectively. These income tax credits expire between 2019 and 2036.Effective Tax Rate
The provision (benefit) for income taxes differs from the U.S. federal statutory tax rate. The reconciliation of the statutory U.S. federal income tax rate to our effective income tax rate is as follows:
|
| | | | | | | | |
| | Year Ended December 31, |
| | 2018 |
| 2017 |
| 2016 |
| U.S. federal statutory tax rate | 21.0 | % | | 35.0 | % | | 35.0 | % |
| Benefit of foreign earnings | (71.2 | )% | | 60.1 | % | | (7.2 | )% |
| Tax credits | (17.0 | )% | | (10.7 | )% | | (6.0 | )% |
| Tax reserves | 12.1 | % | | (14.0 | )% | | 2.7 | % |
| Re-measurement of deferred taxes as a result of the Tax Act | — | % | | (53.4 | )% | | — | % |
| Acquired in-process research & development | 102.6 | % | | — | % | | — | % |
| U.S. state taxes | 14.2 | % | | 1.5 | % | | 4.1 | % |
| Other permanent differences | 6.3 | % | | 0.6 | % | | 2.1 | % |
| | | | | | |
| Effective Income Tax Rate | 68.0 | % | | 19.1 | % | | 30.7 | % |
In our reconciliation of our statutory U.S. federal income tax rate to our effective tax rate above, we have included a Benefit of foreign earnings amount which encapsulates the various tax impacts that result from our foreign derived income. As a result of U.S. Tax Reform, a substantial portion of our foreign earnings are subject to the GILTI minimum tax at an effective rate which is lower than the U.S. statutory tax rate of 21.0%. While we are also subject to tax in foreign jurisdictions locally, substantially all of these taxes are creditable against U.S. taxes imposed on foreign earnings. As a result, the effective tax rate on our foreign earnings is lower than the U.S. statutory rate.
In the year ended December 31, 2018, the Benefit of foreign earnings includes foreign local tax expense of $58.1, substantially all of which is offset by the benefit from U.S. foreign tax credits of $54.2, resulting in a net increase to the effective tax rate of 1.6%. We incurred U.S. tax expense on our foreign earnings of $206.1, which includes GILTI minimum tax. The U.S. tax on our foreign earnings reflects a benefit of $108.7 or 44.8%, primarily related to the Section 250(a) deduction, compared to the U.S. statutory rate. Also included in this component is a benefit of $67.7 from adjustments to 2018 provisional accounting for the Tax Act, which resulted in a decrease to our effective tax rate of approximately 28.0%.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
|
| | | | | | | | |
| | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 |
| U.S. federal statutory tax rate | 35.0 | % | | 35.0 | % | | 35.0 | % |
| State and local income taxes | 4.1 | % | | (0.8 | )% | | 0.9 | % |
| Foreign income tax rate differential | (33.8 | )% | | (32.5 | )% | | (16.5 | )% |
| Tax credits, net of nondeductible expenses | (6.0 | )% | | (7.6 | )% | | (2.5 | )% |
| Foreign income tax credits | (8.4 | )% | | (7.6 | )% | | (4.8 | )% |
| Foreign income subject to U.S. taxation | 26.6 | % | | 24.3 | % | | 15.8 | % |
| U.S. deferred taxes on foreign earnings | 16.5 | % | | 60.1 | % | | — | % |
| Other permanent differences | (3.3 | )% | | 0.1 | % | | (3.2 | )% |
| Effective income tax rate | 30.7 | % | | 71.0 | % | | 24.7 | % |
DuringIn the fourth quarteryear ended December 31, 2017, the Benefit of 2013, in connection withforeign earnings includes foreign local tax expense of $54.4 partially offset by the centralizationbenefit from U.S. foreign tax credits of our global supply chain and technical operations in Ireland, our U.S. parent company became a direct partner$33.2, resulting in a captive foreign partnership. Starting in 2014, a significant portionnet increase to the effective tax rate of 3.9%. We incurred transition tax imposed by the Tax Act of $177.9 and US deferred taxes related to the GILTI provisions of the non-U.S. income flows through the partnership and the portionTax Act of the partnership income that is attributable to our U.S. parent company’s ownership percentage is taxed$165.4. These Tax Act-related adjustments resulted in the U.S. The remainder of the non-U.S. income is taxed based on the tax rate enacted in the local foreign jurisdictions in which the income is earned.
We have operations in many foreign tax jurisdictions, which impose income taxes at different rates than the U.S. The impact of these rate differences is included in the foreign income tax rate differential that we disclose in our reconciliation of the U.S. statutory income tax ratean increase to our effective tax rate of approximately 62.7%. Additional U.S. tax imposed on our foreign earnings of $171.7 reflects a benefit of $35.4 or 6.5% compared to the U.S. statutory rate.
In the year ended December 31, 2016 the Benefit of foreign earnings includes foreign local tax expense of $42.8, more than offset by the benefit from U.S. foreign tax credits of $48.3, resulting in a net decrease to the effective tax rate of 0.9%. Additional U.S. tax imposed on our foreign earnings of $103.9 reflects a benefit of $155.4 or 27.0% compared to the U.S. statutory rate. Additionally,Also included in the foreign income tax rate differential line itemthis component is the impact of taxesto deferred tax attributable to intercompany transactionsdistributions from our captive foreign partnership of $119.3, which increased the effective tax rate by 20.7%.
The effective tax rate reconciliation includes the tax impact of acquisitions of IPR&D assets. Absent successful clinical results and regulatory approval, there is no alternative use for certain acquired IPR&D assets. An increase to the effective tax rate results when the value of such assets are expensed, and no tax benefit is recognized. In the year ended December 31, 2018, this component of the effective tax rate includes an increase to tax expense of $248.4 related to the acquired IPR&D costs for the acquisitions of Wilson Therapeutics and Syntimmune, which increased our effective tax rate by 69.7% and 32.9%, respectively.
In the year ended December 31, 2018, Other permanent differences includes tax expense of $21.1 or 8.7% related to nondeductible compensation and tax benefit of $10.9 or 4.5% related to Foreign-Derived Intangible Income.
In 2017, we concluded the IRS examination of our 2013 and 2014 tax years. Conclusion of the IRS examination resulted in a decrease to the amounttax reserves component of the 2017 effective tax rate of approximately $22, $24,3.6%.
The Tax Act
In December 2017, the Tax Cuts and $23Jobs Act (Tax Act) was enacted into law. The Tax Act decreased the US federal corporate tax rate to 21.0%, imposed a minimum tax on foreign earnings related to intangible assets (GILTI), a one-time transition tax on previously unremitted foreign earnings, and modified the taxation of other income and expense items. With regard to the GILTI minimum tax, foreign earnings are reduced by the profit attributable to tangible assets and a deductible allowance of up to 50.0%, subject to annual limitations. We have elected to account for the impact of the minimum tax in deferred taxes.
At December 31, 2017, the Tax Act resulted in an increase to tax expense and the effective tax rate of $45.8 and 8.4%, respectively:
| |
| (a) | Income tax expense increased $177.9 or 32.5% related to the transition tax on unremitted earnings imposed by the Tax Act. This increase includes foreign income subject to US tax of $195.6, partially offset by a related benefit of foreign tax credits of $17.7. |
| |
| (b) | The decrease to the U.S. federal tax rate resulted in a decrease to deferred tax expense of $292.4 or 53.4%. This decrease includes the $121.3 or 22.2% benefit of re-measuring domestic deferred taxes and an additional decrease attributable to re-measuring deferred taxes on foreign earnings of $171.1 or 31.2%. |
| |
| (c) | Other permanent differences includes a decrease to tax expense of $5.1 or 0.9% related to the re-measurement of income taxes payable as a result of changes in U.S. federal tax rates under the Tax Act. |
| |
| (d) | The enactment of the GILTI minimum tax increased US deferred taxes on foreign earnings $165.4 or 30.2%. This increase includes deferred tax expense related to the GILTI minimum tax of $236.9. This deferred expense is partially offset by a related decrease to deferred expense for the release of reserves for uncertain tax positions of $71.5. |
We calculated provisional amounts for 2016, 2015,the tax effects of the Tax Act that could be reasonably estimated, but not completed, in our results for the year ended December 31, 2017. As of the fourth quarter 2018 we had completed our analysis of all provisional estimates, and 2014, respectively.concluded as follows:
| |
| (a) | We calculated a reasonable estimate of the one-time transition tax on previously unremitted earnings, which resulted in an increase to U.S. Federal tax expense of $177.9 and an increase to taxes payable, net of tax credits, of $28.0 in the period ended December 31, 2017. Our initial accounting for the transition tax was not |
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
complete as of December 31, 2017 because there was uncertainty regarding the calculation of the amounts subject to the tax. We completed our analysis of the transition tax and related interpretive guidance during the third quarter 2018. No significant measurement period adjustment to our initial accounting was required.
| |
| (b) | We calculated a reasonable estimate of the impact of the GILTI minimum tax on deferred taxes, which resulted in an increase to U.S. Federal tax expense and the deferred tax liability of $236.9 in the period ended December 31, 2017. Our initial accounting for the minimum tax was incomplete because there was uncertainty regarding the calculation of the temporary differences subject to the minimum tax. We completed our analyses of these temporary differences and the expected timing and manner of their reversal during the fourth quarter 2018. We recorded measurement period adjustments during 2018 which resulted in a decrease to U.S. federal tax expense of $67.7. |
| |
| (c) | We calculated a reasonable estimate of the Tax Act’s limits on deductions for employee remuneration, including remuneration in kind, which resulted in an insignificant impact to tax expense, taxes payable, and deferred taxes in the period ended December 31, 2017. Our initial accounting for these limits was incomplete because there was uncertainty regarding the value of the deduction-limited remuneration. We completed our analysis of the relevant employee remuneration arrangements during the third quarter 2018. No measurement period adjustment to our initial accounting was required. |
| |
| (d) | We calculated a reasonable estimate of the impact of the Tax Act to U.S. state income taxes, which resulted in an increase to tax expense, taxes payable, and deferred taxes of $2.9, $2.2, and $0.7, respectively, in the period ended December 31, 2017. We interpreted the effect of the Tax Act's changes to federal law on each U.S. state's system of taxation as of the date of enactment. We completed additional analysis of the effect of modifications to federal deductions and income inclusions on U.S. state tax systems in the fourth quarter 2018. No measurement period adjustment to our initial accounting was required. |
| |
| (e) | We calculated the deferred tax liability related to our foreign captive partnership in the period ended December 31, 2017 consistent with our calculation in periods prior to enactment of the Tax Act. As a result, the deferred tax liability we recorded as of December 31, 2017 of $533.4 related to our foreign captive partnership was provisional. We completed additional analysis of the direct and indirect effects of the Tax Act during the fourth quarter 2018. We recorded measurement period adjustments during 2018 which resulted in an increase to U.S. state income tax expense and deferred taxes of $11.1. |
Deferred Taxes
Provisions have been made for deferred taxes based on the differences between the basis of the assets and liabilities for financial statement purposes and the basis of the assets and liabilities for tax purposes using currently enacted tax rates and regulations that will be in effect when the differences are expected to be recovered or settled. The components of the deferred tax assets and liabilities are as follows:
|
| | | | | | | |
| | December 31, | | December 31, |
| | 2016 | | 2015 |
| Deferred tax assets: | | | |
| Net operating losses | $ | 58 |
| | $ | 168 |
|
| Income tax credits | 537 |
| | 209 |
|
| Stock compensation | 89 |
| | 74 |
|
| Accruals and allowances | 91 |
| | 86 |
|
| Research and development expenses | 15 |
| | 19 |
|
| Accrued royalties | 23 |
| | 16 |
|
| | 813 |
| | 572 |
|
| Valuation allowance | (4 | ) | | (5 | ) |
| Total deferred tax assets | 809 |
| | 567 |
|
| Deferred tax liabilities: |
| |
|
| Depreciable assets | (95 | ) | | (83 | ) |
| Unrealized gains | (44 | ) | | (47 | ) |
| Investment in foreign partnership | (546 | ) | | (409 | ) |
| Intangible assets | (502 | ) | | (543 | ) |
| Total deferred tax liabilities | (1,187 | ) | | (1,082 | ) |
| Net deferred tax (liability) asset | $ | (378 | ) | | $ | (515 | ) |
In the second quarter of 2016, we adopted the new share-based compensation guidance. Under the prior guidance, the effect of certain windfall tax benefit deductions were not recognized in deferred tax. The new guidance fully incorporates the deferred tax impact of these deductions. As a result, we recorded an increase to the deferred tax asset for income tax credits for
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
|
| | | | | | | |
| | December 31, | | December 31, |
| | 2018 | | 2017 |
| Deferred tax assets: | | | |
| Net operating losses | $ | 41.8 |
| | $ | 4.9 |
|
| Income tax credits | 371.6 |
| | 442.5 |
|
| Stock compensation | 47.6 |
| | 66.6 |
|
| Accruals and allowances | 105.8 |
| | 97.7 |
|
| Unrealized losses | — |
| | 6.6 |
|
| Research and development expenses | 5.2 |
| | 7.2 |
|
| Accrued royalties | 89.1 |
| | 74.2 |
|
| | 661.1 |
| | 699.7 |
|
| Valuation allowance | (19.6 | ) | | (3.4 | ) |
| Total deferred tax assets | 641.5 |
| | 696.3 |
|
| Deferred tax liabilities: |
| |
|
| Depreciable assets | (88.7 | ) | | (75.2 | ) |
| Unrealized gains | (6.6 | ) | | — |
|
| Investment in foreign partnership | (566.6 | ) | | (607.9 | ) |
| Intangible assets | (268.8 | ) | | (285.6 | ) |
| Total deferred tax liabilities | (930.7 | ) | | (968.7 | ) |
| Net deferred tax (liability) asset | $ | (289.2 | ) | | $ | (272.4 | ) |
the period endedAt December 31, 2016. Consistent with this new guidance, deferred2018, we have tax balances for the period ended December 31, 2015effected federal and state net operating loss carryforwards of $20.0 and $11.6, respectively. Our net operating losses expire between 2022 and 2038. We also have not been restated.federal and state income tax credit carryforwards of $370.6 and $8.4, respectively. These income tax credits expire between 2025 and 2038.
The decreaseincrease in our net operating losses is due to the utilizationacquisition of Syntimmune and the recognition of historical Synageva net operating loss carryforwards.carryforwards which are subject to annual utilization limitations in accordance with Section 382 of the Internal Revenue Code. The increasedecrease in income tax credits is primarily attributable to the adoptionutilization of new share-based compensation guidance and the corresponding recognition of “windfall” tax benefits.Orphan Drug credits. The increasedecrease in our investment in foreign partnership deferred tax liability is due to 2016 distributions fromadjustments we recorded in 2018 to our provisional accounting for the Tax Act and the reversal of other component temporary differences. We continue to maintain a valuation allowance against certain deferred tax assets where realization is not certain.
Included in our investment in foreign partnership above is $(24.1) associated with GILTI minimum tax. Our accounting for the GILTI minimum tax on our captive foreign partnership.partnership as a result of the Tax Act was provisional at December 31, 2017. We completed our accounting for the Tax Act in the fourth quarter 2018.
Unrecognized Tax Benefits
We follow authoritative guidance regarding accounting for uncertainty in income taxes, which prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. The interpretation also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosures, and transition.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
The beginning and ending amounts of unrecognized tax benefits reconciles as follows:
| | | | 2016 | | 2015 | | 2014 | 2018 | | 2017 | | 2016 |
| Beginning of period balance | $ | 114 |
| | $ | 29 |
| | $ | 46 |
| $ | 60.9 |
| | $ | 138.9 |
| | $ | 113.9 |
|
| Increases for tax positions taken during a prior period | 3 |
| | 2 |
| | 1 |
| 9.1 |
| | 5.6 |
| | 3.4 |
|
| Decreases for tax positions taken during a prior period | (1 | ) | | — |
| | (2 | ) | (5.8 | ) | | (85.8 | ) | | (1.1 | ) |
| Increases for tax positions taken during the current period | 23 |
| | 85 |
| | 9 |
| 28.8 |
| | 19.3 |
| | 22.8 |
|
| Decreases for tax positions related to settlements | — |
| | (1 | ) | | (25 | ) | — |
| | (15.8 | ) | | — |
|
| Decreases for tax positions related to lapse of statute | — |
| | (1 | ) | | — |
| (0.3 | ) | | (1.3 | ) | | (0.1 | ) |
| | $ | 139 |
| | $ | 114 |
| | $ | 29 |
| $ | 92.7 |
| | $ | 60.9 |
| | $ | 138.9 |
|
The total amount of accrued interest and penalties was not significant as of December 31, 2016.2018. The total amount of tax benefit recorded during 2016, 2015,2018, 2017, and 20142016 which related to unrecognized tax benefits was $22, $83,$35.4, $27.1, and $17,$21.5, respectively. All of our unrecognized tax benefits, if recognized, would have a favorable impact on the effective tax rate.
It is reasonably possible that a portion of our unrecognized tax benefits could reverse within the next twelve months. Reversal of these amounts is contingent upon the completion of field audits by the taxing authorities in several jurisdictions, whether a tax adjustment is proposed, the nature and amount of any adjustment, and the administrative path to resolving the proposed adjustment. We cannot reasonably estimate the range of the potential change.
Tax Audits
We file federal and state income tax returns in the U.S. and in numerous foreign jurisdictions. The U.S. and foreign jurisdictions have statutes of limitations ranging from 3 to 56 years. However, the limitation period could be extended due to our tax attribute carryforward position in a number of our jurisdictions. The tax authorities generally have the ability to review income tax returns for periods where the limitation period has previously expired and can subsequently adjust tax attribute values.
The Internal Revenue Service (IRS) hasIn 2017, the IRS commenced an examination of our U.S. income tax returns for 2013 and 2014.2015. We anticipate this audit will conclude within the next twelve months. As of February 16, 2017, weWe have not been notified of any significant adjustments proposed adjustments by the IRS.
Undistributed Earnings
We do not record U.S.have recorded tax expense on the undistributed earnings of our controlled foreign corporation (CFC) subsidiaries. We intend to reinvest these earnings permanently outside the U.S. or repatriate the earnings only when it is tax efficient to do so. Accordingly, we believe that U.S. tax on any earnings that might be repatriated would be substantially offset by other tax attributes, such as foreign tax credits or deficits in the foreign earnings and profits account. At December 31, 2016, the cumulative amount of these earnings was approximately $1,462.
During the fourth quarter of 2013, in connection with the centralization of our global supply chain and technical operations in Ireland, our U.S. parent company became a direct partner in a captive foreign partnership. To the extent that our U.S. parent company receives its allocation of partnership income, the amounts willCFC earnings may not be taxable inrepatriated to the U.S. each year. The permanent reinvestment assertion is inapplicable to such earnings.
We do not have any present or anticipated future need for cash held by our CFCs, as cash generated in the U.S., as well as borrowings, are expected to be sufficient to meet U.S. liquidity needs for the foreseeable future.
It is not practicable to estimate the amount of additional taxes which might be payable on our CFCs’ undistributed earningsa dividend distribution due to a variety of factors, includinglimitations imposed by law, we have not recorded the timing, extentrelated potential withholding, foreign local, and nature of any repatriation. While our expectation is that all foreign undistributed earnings, other than our U.S. parent company’s share of the foreign partnership profits, are permanently
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
invested, there could be certain unforeseen future events that could impact our permanent reinvestment assertion. Such events include acquisitions, corporate restructuring or tax law changes not currently contemplated.state income taxes.
| |
12.13. | Share-based Compensation |
Amended and Restated 20042017 Incentive Plan
The 20042017 Plan was approved by our stockholders in May 20132017 and replaced the 2004 Plan effective May 10, 2017. The 2017 Plan is a broad based plan that provides for the grant of equity awards including restricted stock and restricted stock units (collectively referred to as Restricted Stock), incentive and non-qualified stock options, and other stock-related awards to our directors, officers, key employees and consultants, for up to a maximum of 48 shares.18.2 shares in addition to awards outstanding under the 2004 Incentive Plan on or after March 14, 2017 that are subsequently canceled, cash settled, expired, forfeited, or otherwise terminated without the delivery of such shares, subject to the limitations in the 2017 Plan. Stock options granted under the 20042017 Plan have a maximum contractual term of ten years from the date of grant, have an exercise price not less than the fair value of the stock on the grant date and generally vest over four years. Restricted stockStock awards also generally vest over four years, with performance-based restricted stock units having a three-year vesting period.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
Stock Options
A summary of the status of our stock option plansoptions at December 31, 2016,2018, and changes during the year then ended is presented in the table and narrative below:
|
| | | | | | | | | | | | |
| | Number of
shares | | Weighted
Average Exercise
Price | | Weighted
Average
Remaining
Contractual
Term (in years) | | Aggregate Intrinsic
Value |
| Outstanding at December 31, 2015 | 6 |
| | $ | 110.15 |
| | | | |
| Granted | 2 |
| | 139.71 |
| | | | |
| Exercised | (1 | ) | | 55.36 |
| | | | |
| Forfeited and canceled | (1 | ) | | 157.80 |
| | | | |
| Outstanding at December 31, 2016 | 6 |
| | $ | 116.65 |
| | 6.02 | | $ | 177 |
|
| Vested and unvested expected to vest at December 31, 2016 | 6 |
| | $ | 116.08 |
| | 5.97 | | $ | 177 |
|
| Exercisable at December 31, 2016 | 4 |
| | $ | 97.02 |
| | 4.59 | | $ | 175 |
|
|
| | | | | | | | | | | | |
| | Number of
shares | | Weighted
Average Exercise
Price | | Weighted
Average
Remaining
Contractual
Term (in years) | | Aggregate Intrinsic
Value |
| Outstanding at December 31, 2017 | 5.3 |
| | $ | 124.71 |
| | | | |
| Granted | — |
| | — |
| | | | |
| Exercised | (0.5 | ) | | 82.70 |
| | | | |
| Forfeited and canceled | (1.2 | ) | | 156.20 |
| | | | |
| Outstanding at December 31, 2018 | 3.6 |
| | $ | 119.68 |
| | 4.74 | | $ | 44.5 |
|
| Vested and unvested expected to vest at December 31, 2018 | 3.6 |
| | $ | 119.63 |
| | 4.73 | | $ | 44.5 |
|
| Exercisable at December 31, 2018 | 3.2 |
| | $ | 117.73 |
| | 4.38 | | $ | 44.5 |
|
Total intrinsic value of stock options exercised during the years ended December 31, 2016, 20152018, 2017 and 20142016 was $42, $168$27.5, $88.9 and $460,$41.7, respectively. We primarily utilize newly issued shares to satisfy the exercise of stock options. The total fair value of options vested during the years ended December 31, 2016, 20152018, 2017 and 20142016 was $58, $51$27.2, $61.5 and $36,$58.1, respectively.
TheWe did not grant any stock options during the year ended December 31, 2018. For the years ended December 31, 2017 and 2016, the fair value of options at the date of grant was estimated using the Black-Scholes model with the following ranges of weighted average assumptions:
| | | | December 31, | | December 31, | | December 31, | December 31, | | December 31, |
| | 2016 | | 2015 | | 2014 | 2017 |
| 2016 |
| Expected life in years | 3.82 - 6.29 | | 3.57 - 9.00 | | 3.64 - 5.30 | 4.07 - 4.29 | | 3.82 - 6.29 |
| Interest rate | 0.87% - 1.66% | | 0.84% - 2.17% | | 0.97% - 1.74% | 1.64% - 1.92% | | 0.87% - 1.66% |
| Volatility | 33.45% - 37.61% | | 33.35% - 38.13% | | 32.15% - 34.87% | 38.78% - 39.01% | | 33.45% - 37.61% |
| Dividend yield | — | | — | | — | — | | — |
The expected stock price volatility rates are based on historical volatilities of our common stock. The risk-free interest rates are based on the U.S. Treasury yield curve in effect at the time of grant for periods corresponding with the expected life of the option. The average expected life represents the weighted average period of time that options granted are expected to be outstanding. We have evaluated three distinct employee groups in determining the expected life assumptions, and we estimate the expected life of stock options based on historical experience of exercises, cancellations and forfeitures of our stock options.
The weighted average fair value at the date of grant for options granted during the years ended December 31, 2016, 20152017 and 20142016 was $41.46, $53.03$42.59 and $51.22$41.46 per option, respectively.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
Restricted Stock
A summary of the status of our nonvested Restricted Stock at December 31, 2018 and changes during the periodyear then ended is as follows:
| | | | Number of Shares | | Weighted Average Grant Date Fair Value | Number of Shares | | Weighted Average Grant Date Fair Value |
| Nonvested Restricted Stock at December 31, 2015 | 2 |
| | $ | 167.21 |
| |
| Nonvested Restricted Stock at December 31, 2017 | | 3.6 |
| | $ | 130.75 |
|
| Shares granted | 2 |
| | 133.35 |
| 2.1 |
| | 119.27 |
|
| Shares forfeited | — |
| | 165.23 |
| (0.7 | ) | | 126.78 |
|
| Shares vested | (1 | ) | | 155.03 |
| (1.3 | ) | | 135.17 |
|
| Nonvested Restricted Stock at December 31, 2016 | 3 |
| | $ | 149.48 |
| |
| Nonvested Restricted Stock at December 31, 2018 | | 3.7 |
| | $ | 123.25 |
|
The fair value of restricted stockRestricted Stock at the date of grant is based on the fair market value of the shares of common stock underlying the awards on the date of grant. The weighted average fair value at the date of grant for restricted stockRestricted Stock awards granted during the years ended December 31, 2016, 20152018, 2017 and 2014,2016, including restricted stock units with performance conditions, was $133.35, $184.09$119.27, $125.39 and $174.22$133.35 per share, respectively. The total weighted average grant date fair value of restricted stockRestricted Stock vested during the years ended December 31, 2016, 20152018, 2017 and 20142016 was $124, $135$181.7, $157.0 and $41,$124.4, respectively.
We also grant market-based performance awardsIncluded in the table above is 0.3 shares granted to senior management with market-based performance conditions which provide the recipient the right to receive restricted stock at the end of a three year performance period, based on pre-established market-based performance goals. We useused payout simulation models to estimate the grant date fair value of the awards.these awards at $123.25. Expense recognized for awards with market-based performance awardsconditions was not material$14.9 for the year ended December 31, 2018 and immaterial for the years ended December 31, 2016, 20152017 and 2014.2016.
Employee Stock Purchase Plan
During 2015, the Company adopted the ESPP under which employees can purchase shares of our common stock based on a percentage of their compensation subject to certain limits. The purchase price per share is equal to the lower of 85%85.0% of the fair market value of our common stock on the offering date or the purchase date with a six month look-back feature. Under the ESPP, up to 11.0 shares of common stock may be issued to eligible employees who elect to participate in the purchase plan. Shares issued and compensation expense recognized under the ESPP for the years ended December 31, 20162018, 2017 and 20152016 were not material.
Share-Based Compensation Expense
The following table summarizes the share-based compensation expense in the consolidated statements of operations: | | | | Year Ended December 31, | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 | 2018 | | 2017 | | 2016 |
| Cost of sales | $ | 11 |
| | $ | 7 |
| | $ | 4 |
| $ | 16.0 |
| | $ | 11.1 |
| | $ | 11.1 |
|
| Research and development | 57 |
| | 64 |
| | 36 |
| 57.5 |
| | 76.4 |
| | 57.6 |
|
| Selling, general and administrative | 124 |
| | 156 |
| | 74 |
| 129.5 |
| | 155.6 |
| | 123.6 |
|
| Total share-based compensation expense | 192 |
| | 227 |
| | 114 |
| 203.0 |
| | 243.1 |
| | 192.3 |
|
| Income tax effect | (70 | ) | | (84 | ) | | (42 | ) | (46.5 | ) | | (89.3 | ) | | (70.3 | ) |
| Total share-based compensation expense, net of tax | $ | 122 |
| | $ | 143 |
| | $ | 72 |
| $ | 156.5 |
| | $ | 153.8 |
| | $ | 122.0 |
|
Share-based compensation expense capitalized to inventory during the years ended December 31, 2018, 2017 and 2016 2015was $14.5, $15.4, and 2014 was $12, $8, and $10,$12.1, respectively.
As of December 31, 2016,2018, there was $356$312.7 of total unrecognized share-based compensation expense related to non-vested share-based compensation arrangements granted under the 2004 Plan.our share-based compensation plans. The expense is expected to be recognized over a weighted-average period of 2.711.70 years.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
| |
13.14. | Stockholders’ Equity |
Common Stock
In June 2015, in connection with our acquisition of Synageva, we issued 26 shares of common stock to former Synageva stockholders and employees. The fair value of the stock was $4,914, and we incurred $4 of issuance costs.
Share Repurchases
In November 2012, our Board of Directors authorized a share repurchase program. The repurchase program does not have an expiration date, and we are not obligated to acquire a particular number of shares. The repurchase program may be discontinued at any time at the Company’s discretion. In May 2015,February 2017, our Board of Directors increased the authorizationamount that we are authorized to acquire shares with an aggregate value of upexpend on future repurchases to $1,000 for future purchases under the repurchase program, which superseded all prior repurchase programs. The repurchase program does not have an expiration date. The repurchase program may be discontinued at any time at our discretion. Under the program, we repurchased 30.7 and 24.0 shares of our common stock at a cost of $430$85.0 and $328$463.6 during the years ended December 31, 20162018 and 2015,2017, respectively. The Company did not repurchase any shares during the pendency of the Synageva acquisition in the second quarter of 2015 and the Company began repurchasing shares again in the third quarter 2015.
In February 2017, our Board of Directors increased the authorization to acquire shares with an aggregate value of up to $1,000 for future purchases under the repurchase program, which superseded all prior repurchase programs. As of February 16, 2017,6, 2019, there is a total of $1,000$451.5 remaining for repurchases under the repurchase program.
| |
14.15. | Other Comprehensive Income and Accumulated Other Comprehensive Income |
The following table summarizes the changes in AOCI, by component, for the years ended December 31, 2016, 20152018, 2017 and 2014:2016:
| |
| Defined Benefit Pension Plans | | Unrealized Gains (Losses) from Marketable Securities | | Unrealized Gains (Losses) from Hedging Activities | | Foreign Currency Translation Adjustment | | Total Accumulated Other Comprehensive Income (Loss) | Defined Benefit Pension Plans | | Unrealized Gains (Losses) from Debt Securities | | Unrealized Gains (Losses) from Hedging Activities | | Foreign Currency Translation Adjustment | | Total Accumulated Other Comprehensive Income (Loss) |
| Balances, December 31, 2013 | $ | (12 | ) | | $ | — |
| | $ | (4 | ) | | $ | (8 | ) | | $ | (24 | ) | |
| Other comprehensive income before reclassifications | (6 | ) | | — |
| | 110 |
| | (6 | ) | | 98 |
| |
| Amounts reclassified from other comprehensive income | 1 |
| | — |
| | (19 | ) | | — |
| | (18 | ) | |
| Net other comprehensive income (loss) | (5 | ) | | — |
| | 91 |
| | (6 | ) | | 80 |
| |
| Balances, December 31, 2014 | $ | (17 | ) | | $ | — |
| | $ | 87 |
| | $ | (14 | ) | | $ | 56 |
| |
| Other comprehensive income before reclassifications | (2 | ) | | (1 | ) | | 111 |
| | (6 | ) | | 102 |
| |
| Amounts reclassified from other comprehensive income | 9 |
| | — |
| | (105 | ) | | — |
| | (96 | ) | |
| Net other comprehensive income (loss) | 7 |
| | (1 | ) | | 6 |
| | (6 | ) | | 6 |
| |
| Balances, December 31, 2015 | $ | (10 | ) | | $ | (1 | ) | | $ | 93 |
| | $ | (20 | ) | | $ | 62 |
| $ | (9.6 | ) | | $ | (0.8 | ) | | $ | 92.7 |
| | $ | (20.0 | ) | | $ | 62.3 |
|
| Other comprehensive income before reclassifications | 2 |
| | — |
| | 46 |
| | (4 | ) | | 44 |
| 2.6 |
| | 0.2 |
| | 46.4 |
| | (4.3 | ) | | 44.9 |
|
| Amounts reclassified from other comprehensive income | 1 |
| | — |
| | (47 | ) | | — |
| | (46 | ) | 0.3 |
| | 0.2 |
| | (47.2 | ) | | — |
| | (46.7 | ) |
| Net other comprehensive income (loss) | 3 |
| | — |
| | (1 | ) | | (4 | ) | | (2 | ) | 2.9 |
| | 0.4 |
| | (0.8 | ) | | (4.3 | ) | | (1.8 | ) |
| Balances, December 31, 2016 | $ | (7 | ) | | $ | (1 | ) | | $ | 92 |
| | $ | (24 | ) | | $ | 60 |
| $ | (6.7 | ) | | $ | (0.4 | ) | | $ | 91.9 |
| | $ | (24.3 | ) | | $ | 60.5 |
|
| Other comprehensive income before reclassifications | | 0.5 |
| | (0.2 | ) | | (88.2 | ) | | 8.4 |
| | (79.5 | ) |
| Amounts reclassified from other comprehensive income | | 1.4 |
| | 0.8 |
| | (17.6 | ) | | — |
| | (15.4 | ) |
| Net other comprehensive income (loss) | | 1.9 |
| | 0.6 |
| | (105.8 | ) | | 8.4 |
| | (94.9 | ) |
| Balances, December 31, 2017 | | $ | (4.8 | ) | | $ | 0.2 |
| | $ | (13.9 | ) | | $ | (15.9 | ) | | $ | (34.4 | ) |
| Other comprehensive income before reclassifications | | 1.5 |
| | 0.1 |
| | 32.9 |
| | (0.5 | ) | | 34.0 |
|
| Amounts reclassified from other comprehensive income | | 0.7 |
| | (0.6 | ) | | (9.4 | ) | | — |
| | (9.3 | ) |
| Net other comprehensive income (loss) | | 2.2 |
| | (0.5 | ) | | 23.5 |
| | (0.5 | ) | | 24.7 |
|
| Balances, December 31, 2018 | | $ | (2.6 | ) | | $ | (0.3 | ) | | $ | 9.6 |
| | $ | (16.4 | ) | | $ | (9.7 | ) |
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
The table below provides details regarding significant reclassifications from AOCI during the years ended December 31, 2016, 20152018, 2017 and 2014:2016: | | | Details about Accumulated Other Comprehensive Income Components | | Amount Reclassified From Accumulated Other Comprehensive Income during the year ended December 31, | | Affected Line Item in the Consolidated Statements of Operations | | Amount Reclassified From Accumulated Other Comprehensive Income during the year ended December 31, | | Affected Line Item in the Consolidated Statements of Operations |
| | 2016 | 2015 | 2014 | | | 2018 | 2017 | 2016 | |
| Unrealized Gains (Losses) on Hedging Activity | | | | | | | | |
| Effective portion of foreign exchange contracts | | $ | 73 |
| $ | 118 |
| $ | 19 |
| | Net product sales | | $ | (1.8 | ) | $ | 28.9 |
| $ | 73.0 |
| | Net product sales |
| Ineffective portion of foreign exchange contracts | | — |
| 2 |
| 3 |
| | Foreign currency gain (loss) | |
| Effective portion of interest rate swap contracts | | | 13.6 |
| (1.8 | ) | (0.2 | ) | | Interest expense |
| | | 73 |
| 120 |
| 22 |
| | | 11.8 |
| 27.1 |
| 72.8 |
| |
| | | (26 | ) | (15 | ) | (3 | ) | | Income tax expense | | (2.4 | ) | (9.5 | ) | (25.6 | ) | | Income tax expense |
| | | $ | 47 |
| $ | 105 |
| $ | 19 |
| | | $ | 9.4 |
| $ | 17.6 |
| $ | 47.2 |
| |
| Defined Benefit Pension Items | | | | | | | |
| Amortization of prior service costs and actuarial losses | | $ | (1 | ) | $ | (1 | ) | $ | (1 | ) | | (a) | | $ | (0.3 | ) | $ | (0.4 | ) | $ | (0.5 | ) | | (a) |
| Curtailment | | — |
| (10 | ) | — |
| | (a) | | (0.6 | ) | (1.8 | ) | — |
| | (a) |
| | | (1 | ) | (11 | ) | (1 | ) | | | (0.9 | ) | (2.2 | ) | (0.5 | ) | |
| | | — |
| 2 |
| — |
| | Income tax expense | | 0.2 |
| 0.8 |
| 0.2 |
| | Income tax expense |
| | | $ | (1 | ) | $ | (9 | ) | $ | (1 | ) | | | $ | (0.7 | ) | $ | (1.4 | ) | $ | (0.3 | ) | |
(a) This AOCI component is included in the computation of net periodic pension benefit cost (see Note 1617, Employee Benefit Plans, for additional details).
| |
15.16. | Fair Value Measurement |
Authoritative guidance establishes a valuation hierarchy for disclosure of the inputs to the valuation used to measure fair value. This hierarchy prioritizes the inputs into three broad levels as follows. Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities. Level 2 inputs are quoted prices for similar assets and liabilities in active markets or inputs that are observable for the asset or liability, either directly or indirectly through market corroboration, for substantially the full term of the financial instrument. Level 3 inputs are unobservable inputs based on our own assumptions used to measure assets and liabilities at fair value.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
The following tables present information about our assets and liabilities that are measured at fair value on a recurring basis as of December 31, 20162018 and 2015,2017, and indicate the fair value hierarchy of the valuation techniques we utilized to determine such fair value.
|
| | | | | | | | | | | | | | | | |
| | | Fair Value Measurement at
December 31, 2016 |
| Balance Sheet Classification | Type of Instrument | Total | | Level 1 | | Level 2 | | Level 3 |
| Cash equivalents | Money market funds | $ | 266 |
| | $ | — |
| | $ | 266 |
| | $ | — |
|
| Cash equivalents | Commercial paper | $ | 70 |
| | $ | — |
| | $ | 70 |
| | $ | — |
|
| Cash equivalents | Corporate bonds | $ | 10 |
| | $ | — |
| | $ | 10 |
| | $ | — |
|
| Cash equivalents | Municipal bonds | $ | 40 |
| | $ | — |
| | $ | 40 |
| | $ | — |
|
| Marketable securities | Mutual funds | $ | 13 |
| | $ | 13 |
| | $ | — |
| | $ | — |
|
| Marketable securities | Commercial paper | $ | 44 |
| | $ | — |
| | $ | 44 |
| | $ | — |
|
| Marketable securities | Corporate bonds | $ | 113 |
| | $ | — |
| | $ | 113 |
| | $ | — |
|
| Marketable securities | Municipal bonds | $ | 51 |
| | $ | — |
| | $ | 51 |
| | $ | — |
|
| Marketable securities | Other government-related obligations | $ | 100 |
| | $ | — |
| | $ | 100 |
| | $ | — |
|
| Marketable securities | Bank certificates of deposit | $ | 5 |
| | $ | — |
| | $ | 5 |
| | $ | — |
|
| Marketable securities | Equity securities | $ | 1 |
| | $ | 1 |
| | $ | — |
| | $ | — |
|
| Prepaid expenses and other current assets | Foreign exchange forward contracts | $ | 97 |
| | $ | — |
| | $ | 97 |
| | $ | — |
|
| Other assets | Foreign exchange forward contracts | $ | 59 |
| | $ | — |
| | $ | 59 |
| | $ | — |
|
| Other current liabilities | Foreign exchange forward contracts | $ | 12 |
| | $ | — |
| | $ | 12 |
| | $ | — |
|
| Other liabilities | Foreign exchange forward contracts | $ | 4 |
| | $ | — |
| | $ | 4 |
| | $ | — |
|
| Other assets | Interest rate contracts | $ | 10 |
| | $ | — |
| | 10 |
| | $ | — |
|
| Current portion of contingent consideration | Acquisition-related contingent consideration | $ | 24 |
| | $ | — |
| | $ | — |
| | $ | 24 |
|
| Contingent consideration | Acquisition-related contingent consideration | $ | 129 |
| | $ | — |
| | $ | — |
| | $ | 129 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
|
| | | | | | | | | | | | | | | | |
| | | Fair Value Measurement at
December 31, 2018 |
| Balance Sheet Classification | Type of Instrument | Total | | Level 1 | | Level 2 | | Level 3 |
| Cash equivalents | Money market funds | $ | 569.4 |
| | $ | — |
| | $ | 569.4 |
| | $ | — |
|
| Cash equivalents | Commercial paper | $ | 35.4 |
| | $ | — |
| | $ | 35.4 |
| | $ | — |
|
| Cash equivalents | Corporate bonds | $ | 0.2 |
| | $ | — |
| | $ | 0.2 |
| | $ | — |
|
| Cash equivalents | Other government-related obligations | $ | 8.2 |
| | $ | — |
| | $ | 8.2 |
| | $ | — |
|
| Marketable securities | Mutual funds | $ | 16.5 |
| | $ | 16.5 |
| | $ | — |
| | $ | — |
|
| Marketable securities | Commercial paper | $ | 16.7 |
| | $ | — |
| | $ | 16.7 |
| | $ | — |
|
| Marketable securities | Corporate bonds | $ | 122.6 |
| | $ | — |
| | $ | 122.6 |
| | $ | — |
|
| Marketable securities | Other government-related obligations | $ | 9.3 |
| | $ | — |
| | $ | 9.3 |
| | $ | — |
|
| Marketable securities | Bank certificates of deposit | $ | 33.2 |
| | $ | — |
| | $ | 33.2 |
| | $ | — |
|
| Other assets | Equity securities | $ | 90.8 |
| | $ | 8.9 |
| | $ | 81.9 |
| | $ | — |
|
| Prepaid expenses and other current assets | Foreign exchange forward contracts | $ | 40.5 |
| | $ | — |
| | $ | 40.5 |
| | $ | — |
|
| Other assets | Foreign exchange forward contracts | $ | 0.3 |
| | $ | — |
| | $ | 0.3 |
| | $ | — |
|
| Other current liabilities | Foreign exchange forward contracts | $ | 18.8 |
| | $ | — |
| | $ | 18.8 |
| | $ | — |
|
| Other liabilities | Foreign exchange forward contracts | $ | 3.1 |
| | $ | — |
| | $ | 3.1 |
| | $ | — |
|
| Prepaid expenses and other current assets | Interest rate contracts | $ | 20.1 |
| | $ | — |
| | $ | 20.1 |
| | $ | — |
|
| Other current liabilities | Interest rate contracts | $ | 0.8 |
| | $ | — |
| | $ | 0.8 |
| | $ | — |
|
| Other liabilities | Interest rate contracts | $ | 17.3 |
| | $ | — |
| | $ | 17.3 |
| | $ | — |
|
| Current portion of contingent consideration | Acquisition-related contingent consideration | $ | 97.6 |
| | $ | — |
| | $ | — |
| | $ | 97.6 |
|
| Contingent consideration | Acquisition-related contingent consideration | $ | 183.2 |
| | $ | — |
| | $ | — |
| | $ | 183.2 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
| | | | | Fair Value Measurement at
December 31, 2015 | | Fair Value Measurement at
December 31, 2017 |
| Balance Sheet Classification | Type of Instrument | Total | | Level 1 | | Level 2 | | Level 3 | Type of Instrument | Total | | Level 1 | | Level 2 | | Level 3 |
| Cash equivalents | Money market funds | $ | 180 |
| | $ | — |
| | $ | 180 |
| | $ | — |
| |
| Cash equivalents | Commercial paper | $ | 192 |
| | $ | — |
| | $ | 192 |
| | $ | — |
| |
| Cash equivalents | Corporate bonds | $ | 13 |
| | $ | — |
| | $ | 13 |
| | $ | — |
| Commercial paper | $ | 9.5 |
| | $ | — |
| | $ | 9.5 |
| | $ | — |
|
| Cash equivalents | Municipal bonds | $ | 60 |
| | $ | — |
| | $ | 60 |
| | $ | — |
| Reverse repurchase agreements | $ | 27.0 |
| | $ | — |
| | $ | 27.0 |
| | $ | — |
|
| Cash equivalents | Other government-related obligations | $ | 31 |
| | $ | — |
| | $ | 31 |
| | $ | — |
| Corporate bonds | $ | 1.2 |
| | $ | — |
| | $ | 1.2 |
| | $ | — |
|
| Cash equivalents | Bank certificates of deposit | $ | 27 |
| | $ | — |
| | $ | 27 |
| | $ | — |
| Other government-related obligations | $ | 5.0 |
| | $ | — |
| | $ | 5.0 |
| | $ | — |
|
| Marketable securities | Mutual funds | $ | 9 |
| | $ | 9 |
| | $ | — |
| | $ | — |
| Mutual funds | $ | 18.5 |
| | $ | 18.5 |
| | $ | — |
| | $ | — |
|
| Marketable securities | Commercial paper | $ | 62 |
| | $ | — |
| | $ | 62 |
| | $ | — |
| Commercial paper | $ | 6.5 |
| | $ | — |
| | $ | 6.5 |
| | $ | — |
|
| Marketable securities | Corporate bonds | $ | 120 |
| | $ | — |
| | $ | 120 |
| | $ | — |
| Corporate bonds | $ | 431.3 |
| | $ | — |
| | $ | 431.3 |
| | $ | — |
|
| Marketable securities | Municipal bonds | $ | 27 |
| | $ | — |
| | $ | 27 |
| | $ | — |
| Other government-related obligations | $ | 421.3 |
| | $ | — |
| | $ | 421.3 |
| | $ | — |
|
| Marketable securities | Other government-related obligations | $ | 157 |
| | $ | — |
| | $ | 157 |
| | $ | — |
| Bank certificates of deposit | $ | 11.8 |
| | $ | — |
| | $ | 11.8 |
| | $ | — |
|
| Marketable securities | | Equity securities | $ | 0.3 |
| | $ | 0.3 |
| | $ | — |
| | $ | — |
|
| Prepaid expenses and other current assets | Foreign exchange forward contracts | $ | 92 |
| | $ | — |
| | $ | 92 |
| | $ | — |
| Foreign exchange forward contracts | $ | 22.9 |
| | $ | — |
| | $ | 22.9 |
| | $ | — |
|
| Other assets | Foreign exchange forward contracts | $ | 66 |
| | $ | — |
| | $ | 66 |
| | $ | — |
| Foreign exchange forward contracts | $ | 4.1 |
| | $ | — |
| | $ | 4.1 |
| | $ | — |
|
| Other current liabilities | Foreign exchange forward contracts | $ | 5 |
| | $ | — |
| | $ | 5 |
| | $ | — |
| Foreign exchange forward contracts | $ | 48.5 |
| | $ | — |
| | $ | 48.5 |
| | $ | — |
|
| Other liabilities | Foreign exchange forward contracts | $ | 5 |
| | $ | — |
| | $ | 5 |
| | $ | — |
| Foreign exchange forward contracts | $ | 26.0 |
| | $ | — |
| | $ | 26.0 |
| | $ | — |
|
| Current portion of contingent consideration | Acquisition-related contingent consideration | $ | 56 |
| | $ | — |
| | $ | — |
| | $ | 56 |
| |
| Prepaid expenses and other current assets | | Interest rate contracts | $ | 9.3 |
| | $ | — |
| | $ | 9.3 |
| | $ | — |
|
| Other assets | | Interest rate contracts | $ | 12.5 |
| | $ | — |
| | $ | 12.5 |
| | $ | — |
|
| Contingent consideration | Acquisition-related contingent consideration | $ | 121 |
| | $ | — |
| | $ | — |
| | $ | 121 |
| Acquisition-related contingent consideration | $ | 168.9 |
| | $ | — |
| | $ | — |
| | $ | 168.9 |
|
There were no securities transferred between Level 1, 2 and 3 during the year ended December 31, 2016.2018.
Valuation Techniques
We classify mutual fund investments and equity securities, which are valued based on quoted market prices in active markets with no valuation adjustment, as Level 1 assets within the fair value hierarchy.
Cash equivalents and marketable securities classified as Level 2 within the valuation hierarchy consist of institutional money market funds, commercial paper, municipal bonds,reverse repurchase agreements, U.S. and foreign government-related debt, corporate debt securities and certificates of deposit. We estimate the fair values of these marketable securities by taking into consideration valuations obtained from third-party pricing sources. These pricing sources utilize industry standard valuation models, including both income and market-based approaches, for which all significant inputs are observable, either directly or indirectly, to estimate fair value. These inputs include market pricing based on real-time trade data for the same or similar securities, issuer credit spreads, benchmark yields, and other observable inputs. We validate the prices provided by our third-party pricing sources by understanding the models used, obtaining market values from other pricing sources and analyzing pricing data in certain instances.
Alexion Pharmaceuticals, Inc.
NotesOther investments in equity securities of publicly traded companies which are subject to Consolidated Financial Statements
Forholding period restrictions are carried at fair value using an option pricing valuation model and classified as Level 2 equity securities within the Years ended December 31, 2016, 2015fair value hierarchy. The most significant assumptions within the option pricing valuation model are the term of the restrictions and 2014
(amounts in millions except per share amounts)
the stock price volatility, which is based upon the historical volatility of similar companies. We also use a constant maturity risk-free interest rate to match the remaining term of the restrictions on such investments.
Our derivative assets and liabilities include foreign exchange and interest rate derivatives that are measured at fair value using observable market inputs such as forward rates, interest rates, our own credit risk as well as an
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
evaluation of our counterparties’ credit risks. Based on these inputs, the derivative assets and liabilities are classified within Level 2 of the valuation hierarchy.
Contingent consideration liabilities related to acquisitions are classified as Level 3 within the valuation hierarchy and are valued based on various estimates, including probability of success, discount rates and amount of time until the conditions of the milestone payments are met.
As of December 31, 2016,2018, there has not been any impact to the fair value of our derivative liabilities due to our own credit risk. Similarly, there has not been any significant adverse impact to our derivative assets based on our evaluation of our counterparties’ credit risks.
Contingent Consideration
In connection with prior acquisitions,business combinations, we may be required to pay future consideration that is contingent upon the achievement of specified development, regulatory approvals or sales-based milestone events. We determine the fair value of these obligations on the acquisition date using various estimates that are not observable in the market and represent a Level 3 measurement within the fair value hierarchy. The resulting probability-weighted cash flows were discounted using a cost of debt of 4.7%ranging from 4.2% to 5.1% for developmental milestones and a weighted average cost of capital ranging from 10%9.0% to 21%21.0% for sales-based milestones.
Each reporting period, we adjust the contingent consideration to fair value with changes in fair value recognized in operating earnings. Changes in fair values reflect new information about the probability and timing of meeting the conditions of the milestone payments. In the absence of new information, changes in fair value will only reflect the interest component of contingent consideration related to the passage of time.
EstimatedAs of December 31, 2018, estimated future contingent milestone payments related to prior business combinations range from zero if no milestone events are achieved, to a maximum of $766$702.0 if all development, regulatory and sales-based milestones are reached. As of December 31, 2016,2018, the fair value of acquisition-related contingent consideration was $153.$280.8. The following table represents a roll-forward of our acquisition-related contingent consideration:
| |
| December 31, 2016 | 2018 |
| Balance at beginning of period | $ | (177 | ) | $ | 168.9 |
|
| Milestone payments | 60 |
| |
| Amounts derecognized upon sale of asset | | (4.6 | ) |
| Changes in fair value | (36 | ) | 116.5 |
|
| Balance at end of period | $ | (153 | ) | $ | 280.8 |
|
In September 2018, we sold all our assets, rights and obligations related to the fourthALXN1101 program to a third party and, as a result, in the quarter 2016, the criteria was metended September 30, 2018, derecognized $4.6 of contingent consideration due under our prior purchase agreement with Orphatec Pharmaceuticals GmbH, dated February 8, 2011. The definitive agreement related to our sale of ALXN1101 provides for contingent consideration payments to Alexion upon the achievement of various regulatory and commercial milestones and other events, as well as royalties on commercial sales. The amount of contingent consideration related to these contingent payments is deemed to be fully constrained as of December 31, 2018, and therefore has not been included in the transaction price. During the third quarter 2018, we recognized an immaterial gain on the sale of ALXN1101 within operating income.
In September 2018, we amended the terms of certain contingent milestone payments due under our prior merger agreement with Enobia Pharma Corp., dated December 28, 2011. The agreement removed our obligations with respect to a regulatory milestone and redistributed the contingent payment associated with this milestone to various sales milestones. As a result of this agreement and the probability of achieving the various sales milestones, our acquisition of Enobia Pharma Corp. In connection with this, $60 was paidcontingent consideration liability increased by $48.7 in December 2016.the third quarter 2018.
| |
16.17. | Employee Benefit Plans |
Deferred Compensation Plan
We have a nonqualified deferred compensation plan which allows certain highly-compensated employees to make voluntary deferrals of up to 80% of their base salary and incentive bonuses. The plan is designed to work in conjunction with the 401(k) plan and provides for a total combined employer match of up to 6% of an employee’s eligible earnings,
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
up to the IRS annual 401(k) contribution limitations. Deferred compensation amounts under this plan as of December 31, 20162018 and 20152017 were $13$16.5 and $9,$18.5, respectively, and are included in other liabilities within the consolidated balance sheets. Employer matching contributions under the plan for the years ended December 31, 2016, 20152018, 2017 and 20142016 were not material.
Defined Contribution Plan
We have one qualified 401(k) plan covering all eligible employees. Under the plan, employees may contribute up to the statutory allowable amount for any calendar year. We make matching contributions equal to $1.00 for each dollar contributed up to the first 6% of an individual’s base salary and incentive cash bonus up to the annual IRS maximum. For the years ended December 31, 2016, 20152018, 2017 and 2014,2016, we recorded matching contributions of approximately $17, $11,$14.1, $15.9, and $9$16.7 respectively.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 2015 and 2014
(amounts in millions except per share amounts)
Defined Benefit Plans
We maintain defined benefit plans for employees in certain countries outside the U.S., including retirement benefit plans required by applicable local law. The plans are valued by independent actuaries using the projected unit credit method. The liabilities correspond to the projected benefit obligations of which the discounted net present value is calculated based on years of employment, expected salary increases, and pension adjustments.
In 20152018 and 2017 we recorded the impacts of a curtailment related to our Swiss plan as a result of a reduction of employees due to the relocation of our European headquartersrestructuring events as discussed in Note 17, “Restructuring”18, “Restructuring and Related Expenses”.
The following table sets forth the funded status and the amounts recognized for defined benefit plans, including the impacts of the 2015 curtailment:curtailments:
| | | | December 31, | December 31, |
| | 2016 | | 2015 | 2018 | | 2017 |
| Change in benefit obligation: | | | | | | |
| Projected benefit obligation, beginning of year | $ | 45 |
| | $ | 51 |
| $ | 43.4 |
| | $ | 48.4 |
|
| Prior service cost | — |
| | — |
| |
| Service cost | 8 |
| | 10 |
| 6.3 |
| | 7.8 |
|
| Interest cost | — |
| | 1 |
| |
| Change in assumptions | (1 | ) | | 2 |
| |
| Recognized actuarial net loss | — |
| | 4 |
| |
| Curtailment | — |
| | (25 | ) | (3.8 | ) | | (9.6 | ) |
| Plan amendment | (4 | ) | | — |
| |
| Foreign currency exchange rate changes | (3 | ) | | — |
| |
| Net transfers to (from) plan | — |
| | 2 |
| |
| Other | 3 |
| | — |
| (6.5 | ) | | (3.2 | ) |
| Projected benefit obligation, end of year | $ | 48 |
| | $ | 45 |
| $ | 39.4 |
| | $ | 43.4 |
|
| Accumulated benefit obligation, end of year | $ | 41 |
| | $ | 42 |
| $ | 36.1 |
| | $ | 39.4 |
|
| | | | December 31, | December 31, |
| | 2016 | | 2015 | 2018 |
| 2017 |
| Change in plan assets: | | | | | | |
| Fair value of plan assets, beginning of year | $ | 22 |
| | $ | 27 |
| $ | 24.2 |
| | $ | 28.2 |
|
| Return on plan assets | — |
| | — |
| |
| Employer contributions | 3 |
| | 4 |
| 3.0 |
| | 4.3 |
|
| Plan participants' contributions | 2 |
| | 2 |
| 1.3 |
| | 1.5 |
|
| Curtailment | — |
| | (13 | ) | (2.4 | ) | | (6.6 | ) |
| Foreign currency exchange rate changes | (1 | ) | | — |
| |
| Net transfers to (from) plan | — |
| | 2 |
| |
| Other | 2 |
| | — |
| (4.3 | ) | | (3.2 | ) |
| Fair value of plan assets, end of year | $ | 28 |
| | $ | 22 |
| $ | 21.8 |
| | $ | 24.2 |
|
| Funded status at end of year | $ | (20 | ) | | $ | (23 | ) | $ | (17.6 | ) | | $ | (19.2 | ) |
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
The Company measures the fair value of plan assets based on the prices that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The following table presents total plan assets by investment category as of December 31, 2016 and 2015 and the classification of each investment category within the fair value hierarchy with respect to the inputs used to measure fair value:
|
| | | | | | | | | | | | | |
| | December 31, 2016 | | December 31, 2015 |
| | Fair Value (Level 2) | | as % of total plan assets | | Fair Value (Level 2) | | as % of total plan assets |
| Cash and cash equivalents | $ | — |
| | — | % | | $ | — |
| | — | % |
| Equity security funds | 2 |
| | 7 | % | | 2 |
| | 9 | % |
| Debt security funds | 22 |
| | 79 | % | | 17 |
| | 77 | % |
| Real estate funds | 4 |
| | 14 | % | | 3 |
| | 14 | % |
| | $ | 28 |
| | 100 | % | | $ | 22 |
| | 100 | % |
All plan asset investments are classified as Level 2 within the fair value hierarchy and are valued utilizing observable prices for similar instruments and quoted prices for identical or similar instruments in markets that are not active. Plan assets are managed by an independent investment fiduciary and are primarily invested in debt and equity securities and real estate funds in order to maximize the overall return from investment income considering asset allocation limits as determined by pension law.
At December 31, 2016,2018, we have recorded a liability of $20$17.6 in other noncurrent liabilities and a chargean additional minimum liability of $2.6, net of tax, to accumulated other comprehensive income, net of tax, of $7 related to an additional minimum liability.income.
The following table provides the weighted average assumptions used to calculate net periodic benefit cost and the actuarial present value of projected benefit obligations:
| | | | December 31, | December 31, |
| | 2016 | | 2015 | 2018 | | 2017 |
| Weighted average assumptions - Net Periodic Benefit Cost: | | | | | | |
| Discount rate | 0.6 | % | | 1.4 | % | 0.8 | % | | 0.7 | % |
| Long term rate of return on assets | 3.0 | % | | 3.5 | % | 2.5 | % | | 3.0 | % |
| Rate of compensation increase | 1.4 | % | | 1.5 | % | 1.3 | % | | 1.4 | % |
| Weighted average assumptions - Projected Benefit Obligation: | | | | | | |
| Discount Rate | 0.7 | % | | 0.6 | % | 0.8 | % | | 0.8 | % |
| Rate of compensation increase | 1.4 | % | | 1.4 | % | 1.3 | % | | 1.3 | % |
The discount rates used to determine the net periodic benefit cost and projected benefit obligation represent the yield on high quality AA-rated corporate bonds for periods that match the duration of the benefit obligations.
The expected long-term rate of return on plan assets represents a weighted average of expected returns per asset category. The rate of return considers historical and estimated future risk free rates of return as well as risk premiums for the relevant investment categories.
The components of net periodic benefit cost are as follows:
| | | | Year Ended December 31, | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 | 2018 | | 2017 | | 2016 |
| Service cost | $ | 8 |
| | $ | 10 |
| | $ | 8 |
| $ | 6.3 |
| | $ | 7.8 |
| | $ | 8.2 |
|
| Interest cost | — |
| | 1 |
| | 1 |
| |
| Expected return on plan assets | — |
| | (1 | ) | | (1 | ) | |
| Employee contributions | (2 | ) | | (2 | ) | | (2 | ) | (1.3 | ) | | (1.5 | ) | | (1.6 | ) |
| Amortization of prior service costs | — |
| | — |
| | — |
| (0.3 | ) | | (0.4 | ) | | — |
|
| Curtailment | — |
| | (2 | ) | | — |
| (0.8 | ) | | (1.1 | ) | | — |
|
| Amortization and deferral of actuarial gain | 1 |
| | 1 |
| | 1 |
| 0.5 |
| | 0.8 |
| | 0.9 |
|
| Other | | (0.3 | ) | | (0.6 | ) | | (0.8 | ) |
| Total net periodic benefit cost | $ | 7 |
| | $ | 7 |
| | $ | 7 |
| $ | 4.1 |
| | $ | 5.0 |
| | $ | 6.7 |
|
During 2018, service costs were recorded to operating expenses while all other components of the net periodic benefit was recorded to other income and expense within our consolidated statement of operations.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
Other changes in plan assets and benefit obligations recognized in AOCI are as follows:
|
| | | |
| Amount included in AOCI - December 31, 2014 | $ | (17 | ) |
| Prior service cost | — |
|
| Net loss arising during the period | (4 | ) |
| Change in assumptions | (2 | ) |
| Amortization of net gain | 1 |
|
| Plan assets losses | (1 | ) |
| Curtailment | 10 |
|
| Foreign currency exchange rate changes | — |
|
| Taxes | 3 |
|
| Amount included in AOCI - December 31, 2015 | $ | (10 | ) |
| Prior service cost | — |
|
| Net loss arising during the period | — |
|
| Plan amendment | 4 |
|
| Change in assumptions | 1 |
|
| Amortization of net gain | 1 |
|
| Plan assets losses | — |
|
| Curtailment | — |
|
| Foreign currency exchange rate changes | — |
|
| Taxes | (1 | ) |
| Other | (2 | ) |
| Amount included in AOCI - December 31, 2016 | $ | (7 | ) |
|
| | | |
| Amount included in AOCI - December 31, 2016 | $ | (6.7 | ) |
| Prior service cost | (0.4 | ) |
| Amortization of net gain | 0.8 |
|
| Curtailment | 1.9 |
|
| Taxes | (0.6 | ) |
| Other | 0.2 |
|
| Amount included in AOCI - December 31, 2017 | $ | (4.8 | ) |
| Prior service cost | (0.3 | ) |
| Amortization of net gain | 0.6 |
|
| Curtailment | 0.6 |
|
| Taxes | (0.7 | ) |
| Other | 2.0 |
|
| Amount included in AOCI - December 31, 2018 | $ | (2.6 | ) |
We estimate that we will pay employer contributions of approximately $3$2.4 in 2017.2019. The expected future benefits to be paid in respect of the pension plans as of December 31, 20162018 were as follows:
| | | Year | | |
| 2017 | $ | 2 |
| |
| 2018 | 1 |
| |
| 2019 | 1 |
| $ | 1.9 |
|
| 2020 | 1 |
| 1.7 |
|
| 2021 | 1 |
| 1.7 |
|
| 2022 to 2026 | 5 |
| |
| 2022 | | 1.9 |
|
| 2023 | | 1.6 |
|
| 2024 to 2028 | | 9.1 |
|
| |
17.18. | Restructuring and Related Expenses |
In connectionthe first quarter of 2017, we initiated a company-wide restructuring designed to help position the Company for sustainable, long-term growth that we believe will further allow us to fulfill our mission of serving patients and families with rare diseases. The initial restructuring activities primarily focused on a reduction of the completionCompany's global workforce. In September 2017, we committed to an operational plan to re-align the global organization with its refocused corporate strategy. The re-alignment focuses investments in priority growth areas to maximize leadership in complement and grow the rare disease business. The re-alignment also included the relocation of our new corporatethe Company's headquarters locatedto Boston, Massachusetts which was completed in the second quarter of 2018. Our New Haven, Connecticut we entered into a lease termination agreement for the previous corporate headquarters located in Cheshire, Connecticut during December 2015. As a result of this action, we recorded restructuring expense of $11 for contract termination costssite continues to support employees working in the fourth quarterresearch and process development laboratories, the clinical supply and quality teams, nurse case management and a number of 2015.
In connection withimportant enterprise business services. The plan also reduced the acquisitionCompany's global workforce by approximately 20.0%. The restructuring is designed to result in cost savings by focusing the development portfolio, simplifying business structures and integrationprocesses across the Company's global operations, and closing of Synageva in 2015, we recorded restructuring expense of $13 primarily related to employee costs during 2015. Synageva restructuring charges were not material in 2016.
In the fourth quarter 2014, we announced plans to relocate our European headquarters from Lausanne to Zurich, Switzerland. The relocation of our European headquarters supports our operational needs based on growth in the European region. As a result of this action, we recorded restructuring expenses of $15 related to employee costs in the fourth quarter of 2014. During the years ended December 31, 2016multiple Alexion sites, including ARIMF and 2015, we incurred additional restructuring costs of $4certain regional and $18, respectively.country-based offices.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
The following table summarizes the total expenses recorded related to the restructuring activities by type of activity and the locations recognized within the consolidated statements of operations:
|
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | December 31, 2018 | | December 31, 2017 | | December 31, 2016 |
| | Employee Separation Costs | Asset-Related Charges | Other | Total | | Employee Separation Costs | Asset-Related Charges | Other | Total | | Employee Separation Costs | Asset-Related Charges | Other | Total |
| Cost of Sales | $ | — |
| $ | 5.8 |
| $ | — |
| $ | 5.8 |
| | $ | — |
| $ | 152.1 |
| $ | — |
| $ | 152.1 |
| | $ | — |
| $ | — |
| $ | — |
| $ | — |
|
| Research and Development | — |
| 0.1 |
| — |
| 0.1 |
| | — |
| 16.3 |
| — |
| 16.3 |
| | — |
| — |
| — |
| — |
|
| Selling , General and Administrative | — |
| 19.4 |
| — |
| 19.4 |
| | — |
| 10.9 |
| — |
| 10.9 |
| | — |
| — |
| — |
| — |
|
| Restructuring Expense | 4.6 |
| — |
| 20.9 |
| 25.5 |
| | 87.3 |
| — |
| 17.3 |
| 104.6 |
| | 3.0 |
| — |
|
|
| 3.0 |
|
| Other expense | — |
| — |
| (0.1 | ) | (0.1 | ) | | — |
| — |
| 2.6 |
| 2.6 |
| | — |
| — |
| — |
| — |
|
| | $ | 4.6 |
| $ | 25.3 |
| $ | 20.8 |
| $ | 50.7 |
| | $ | 87.3 |
| $ | 179.3 |
| $ | 19.9 |
| $ | 286.5 |
| | $ | 3.0 |
| $ | — |
| $ | — |
| $ | 3.0 |
|
Employee separation costs are associated with headcount reductions, as well as corporate employees not relocating to the Company's headquarters in 2018.
Asset-related charges consist of accelerated depreciation costs and asset impairment charges. Accelerated depreciation costs primarily relates to site closures, including ARIMF (which was sold to a third-party in 2018). Accelerated depreciation costs represent the difference between the depreciation expense recognized over the revised useful life of the asset, based upon the anticipated date the site closure and the depreciation expense as determined using the useful life prior to the restructuring activities. Asset impairment charges primarily related to manufacturing assets that will no longer be utilized due to the 2017 restructuring activities.
Other costs consist of contract termination expenses, relocation costs, and other costs incurred as a direct result of an exit plan.
The following table presents a reconciliation of the restructuring reserve recorded within accounts payable and accrued expenses on the Company’sCompany's consolidated balance sheets for the years ended December 31, 20162018 and 2015, respectively:2017:
|
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | December 31, 2016 | | December 31, 2015 |
| | Employee Separation Costs | | Contract Termination Costs | | Other Costs | | Total | | Employee Separation Costs | | Contract Termination Costs | | Other Costs | | Total |
| Liability, beginning of period | $ | 6 |
| | $ | 1 |
| | $ | — |
| | $ | 7 |
| | $ | 15 |
| | $ | — |
| | $ | — |
| | $ | 15 |
|
| Restructuring expenses | — |
| | 2 |
| | 1 |
| | 3 |
| | 22 |
| | 12 |
| | 4 |
| | 38 |
|
| Cash settlements | (5 | ) | | (4 | ) | | (1 | ) | | (10 | ) | | (35 | ) | | (11 | ) | | (4 | ) | | (50 | ) |
| Adjustments to previous estimates | (1 | ) | | 1 |
| | — |
| | — |
| | 4 |
| | — |
| | — |
| | 4 |
|
| Liability, end of period | $ | — |
| | $ | — |
| | $ | — |
| | $ | — |
| | $ | 6 |
| | $ | 1 |
| | $ | — |
| | $ | 7 |
|
|
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | December 31, 2018 | | December 31, 2017 |
| | Employee Separation Costs | | Asset-Related Charges | | Other | | Total | | Employee Separation Costs | | Asset-Related Charges | | Other | | Total |
| Liability, beginning of year | $ | 53.8 |
| | $ | — |
| | $ | 4.4 |
| | $ | 58.2 |
| | $ | 0.5 |
| | $ | — |
| | $ | 0.1 |
| | $ | 0.6 |
|
| Charges | 5.8 |
| | 25.3 |
| | 21.1 |
| | 52.2 |
| | 88.2 |
| | 179.3 |
| | 19.9 |
| | 287.4 |
|
| Settlements | (54.2 | ) | | — |
| | (25.2 | ) | | (79.4 | ) | | (34.0 | ) | | — |
| | (15.6 | ) | | (49.6 | ) |
| Adjustments to previous estimates | (1.2 | ) | | — |
| | (0.3 | ) | | (1.5 | ) | | (0.9 | ) | | — |
| | — |
| | (0.9 | ) |
| Non Cash Activity | — |
| | (25.3 | ) | | — |
| | (25.3 | ) | | — |
| | (179.3 | ) | | — |
| | (179.3 | ) |
| Liability, end of year | $ | 4.2 |
| | $ | — |
| | $ | — |
| | $ | 4.2 |
| | $ | 53.8 |
| | $ | — |
| | $ | 4.4 |
| | $ | 58.2 |
|
The restructuring reserve of $4.2 and $58.2 is recorded in accounts payable and accrued expenses on the Company's consolidated balance sheet as of December 31, 2018 and 2017, respectively.
As a result of the relocation of our corporate headquarters to Boston, Massachusetts, we were required to repay a forgivable loan and grant that were provided by the State of Connecticut Department of Economic Community Development in 2015 in connection with the construction of our current headquarters in New Haven, Connecticut. The loan and grant totaled $26.0 and were recognized, upon receipt, as a reduction in the cost of our New Haven facility-related fixed assets. As a result, the $26.0 repayment obligation was recorded in the third quarter 2017 with an offsetting increase in the carrying value of the related assets. We repaid this amount in the fourth quarter 2017.
In the first quarter 2019, we have undertaken corporate restructuring activities to re-align our global organization with our re-focused strategy, reduce costs, and realize operational efficiencies. We expect to incur estimated expenses up to $25.0 associated with this recent restructuring by the end of 2019.
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2018, 2017 and 2016
(amounts in millions except per share amounts)
| |
18.19. | Segment Information |
We operate in a single segment, focusing on serving patients with devastating and ultra-rare disordersaffected by rare diseases through the innovation, development and commercialization of life-transforming therapeutic products.life-changing therapies. Consistent with our operational structure, our chief operating decision maker manages and allocates resources at a global, consolidated level. Therefore, results of our operations are reported on a consolidated basis for purposes of segment reporting, consistent with our management reporting. Disclosures about net product sales and long-lived assets by geographic area are presented below.
Net product salesProduct Sales
Net product sales by product and geographic region are as follows:
|
| | | | | | | | | | | |
| | Year Ended December 31, |
| | 2016 | | 2015 | | 2014 |
| Net product sales: | | | | | |
| Soliris (1) | $ | 2,843 |
| | $ | 2,591 |
| | $ | 2,234 |
|
| Strensiq | 210 |
| | 12 |
| | — |
|
| Kanuma | 29 |
| | — |
| | — |
|
| | $ | 3,082 |
| | $ | 2,603 |
| | $ | 2,234 |
|
|
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | % Change |
| | 2018 |
| 2017 | | 2016 | | 2018 compared to 2017 | | 2017 compared to 2016 |
| SOLIRIS | | | | | | | | | |
| United States | $ | 1,588.4 |
| | $ | 1,235.0 |
| | $ | 1,058.5 |
| | 28.6 | % | | 16.7 | % |
| Europe | 1,036.7 |
| | 985.2 |
| | 939.7 |
| | 5.2 | % | | 4.8 | % |
| Asia Pacific | 382.0 |
| | 328.1 |
| | 303.8 |
| | 16.4 | % | | 8.0 | % |
| Rest of World | 555.9 |
| | 595.8 |
| | 541.2 |
| | (6.7 | )% | | 10.1 | % |
| | $ | 3,563.0 |
| | $ | 3,144.1 |
|
| $ | 2,843.2 |
| | 13.3 | % | | 10.6 | % |
| | | | | | | | | | |
| STRENSIQ | | | | | | | | | |
| United States | $ | 374.3 |
| | $ | 280.1 |
| | $ | 177.5 |
| | 33.6 | % | | 57.8 | % |
| Europe | 61.7 |
| | 35.6 |
| | 15.3 |
| | 73.3 | % | | 132.7 | % |
| Asia Pacific | 27.9 |
| | 18.6 |
| | 13.0 |
| | 50.0 | % | | 43.1 | % |
| Rest of World | 11.2 |
| | 5.5 |
| | 3.6 |
| | 103.6 | % | | 52.8 | % |
| | $ | 475.1 |
| | $ | 339.8 |
|
| $ | 209.4 |
| | 39.8 | % | | 62.3 | % |
| | | | | | | | | | |
| KANUMA | | | | | | | | | |
| United States | $ | 51.3 |
| | $ | 42.4 |
| | $ | 20.4 |
| | 21.0 | % | | 107.8 | % |
| Europe | 21.6 |
| | 14.6 |
| | 6.3 |
| | 47.9 | % | | 131.7 | % |
| Asia Pacific | 3.7 |
| | 2.7 |
| | 1.3 |
| | 37.0 | % | | 107.7 | % |
| Rest of World | 15.4 |
| | 5.9 |
| | 1.1 |
| | ** |
| | ** |
|
| | $ | 92.0 |
| | $ | 65.6 |
|
| $ | 29.1 |
| | 40.2 | % | | 125.4 | % |
| Total Net Product Sales | 4,130.1 |
| | $ | 3,549.5 |
|
| $ | 3,081.7 |
| | 16.4 | % | | 15.2 | % |
Geographical information** Percentages not meaningful
Long-Lived Assets
Long-lived assets consist of property, plant and equipment. |
| | | | | | | | | | | |
| | Year Ended December 31, |
| Net product sales: | 2016 | | 2015 | | 2014 |
| United States | $ | 1,257 |
| | $ | 951 |
| | $ | 730 |
|
| Europe (1) | 961 |
| | 841 |
| | 836 |
|
| Asia Pacific | 318 |
| | 276 |
| | 244 |
|
| Rest of World | 546 |
| | 535 |
| | 424 |
|
| | $ | 3,082 |
| | $ | 2,603 |
| | $ | 2,234 |
|
| (1) Included within the Soliris and Europe revenues for 2014 is a reimbursement of $88 for shipments made in years prior to January 1, 2014 as a result of an agreement with the French government. |
|
| | | | | | | |
| | December 31, |
| | 2018 | | 2017 |
| United States | $ | 468.3 |
| | $ | 455.9 |
|
| Europe | 1,001.1 |
| | 864.6 |
|
| Other | 2.1 |
| | 4.9 |
|
| | $ | 1,471.5 |
| | $ | 1,325.4 |
|
Alexion Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
For the Years ended December 31, 2016, 20152018, 2017 and 20142016
(amounts in millions except per share amounts)
|
| | | | | | | | | |
| | | | December 31, |
| Long-lived assets (2): | | | 2016 | | 2015 |
| United States | | | $ | 490 |
| | $ | 444 |
|
| Europe | | | 538 |
| | 248 |
|
| Other | | | 8 |
| | 5 |
|
| | | | $ | 1,036 |
| | $ | 697 |
|
| |
(2) | Long-lived assets consist of property, plant and equipment. |
| |
19.20. | Quarterly Financial Information (unaudited) |
The following condensed quarterly financial information is for the years ended December 31, 20162018 and 2015:2017:
| | | | March 31 | | June 30 | | September 30 | | December 31 | | March 31 | | June 30 | | September 30 | | December 31 | |
| 2016: | | | | | | | | | |
| 2018: | | | | | | | | | |
| Revenues | $ | 701 |
| | $ | 753 |
| | $ | 799 |
| | $ | 831 |
| | $ | 930.9 |
| | $ | 1,045.0 |
| | $ | 1,026.5 |
| | $ | 1,128.8 |
| |
| Cost of sales | 59 |
| | 60 |
| | 71 |
| | 68 |
| | 91.6 |
| | 95.3 |
| | 90.6 |
| | 96.8 |
| |
| Operating expenses | 476 |
| | 498 |
| | 549 |
| | 636 |
| (1) | 571.9 |
| | 1,349.8 |
| (1) | 577.3 |
| | 988.3 |
| (1) |
| Operating income | 166 |
| | 195 |
| | 179 |
| | 127 |
| | 267.4 |
| | (400.1 | ) | | 358.6 |
| | 43.7 |
| |
| Net income (loss) | $ | 92 |
| | $ | 120 |
| (2) | $ | 94 |
| | $ | 93 |
| | |
| Earnings (loss) per common share | | | | | | | | | |
| Net income | | $ | 249.1 |
| | $ | (457.4 | ) | | $ | 330.9 |
| | $ | (45.0 | ) | |
| Earnings per common share | | | | | | | | | |
| Basic | $ | 0.41 |
| | $ | 0.54 |
| (2) | $ | 0.42 |
| | $ | 0.41 |
| | $ | 1.12 |
| | $ | (2.05 | ) | | $ | 1.48 |
| | $ | (0.20 | ) | |
| Diluted | $ | 0.41 |
| | $ | 0.53 |
| (2) | $ | 0.42 |
| | $ | 0.41 |
| | $ | 1.11 |
| | $ | (2.05 | ) | | $ | 1.47 |
| | $ | (0.20 | ) | |
| | | | | | | | | | | | | | | | | |
| | March 31 | | June 30 | | September 30 | | December 31 | | March 31 | | June 30 | | September 30 | | December 31 | |
| 2015: | | | | | | | | | |
| 2017: | | | | | | | | | |
| Revenues | $ | 600 |
| | $ | 636 |
| | $ | 667 |
| | $ | 701 |
| | $ | 869.6 |
| | $ | 912.7 |
| | $ | 859.1 |
| | $ | 909.7 |
| |
| Cost of sales | 69 |
| | 52 |
| | 54 |
| | 58 |
| | 69.0 |
| | 83.6 |
| | 157.0 |
| (2) | 144.6 |
| (2) |
| Operating expenses | 427 |
| | 403 |
| | 458 |
| | 546 |
| | 588.6 |
| (3) | 602.4 |
| (4) | 622.0 |
| (3) | 656.5 |
| |
| Operating income | 104 |
| | 181 |
| | 155 |
| | 97 |
| | 212.0 |
| | 226.7 |
| | 80.1 |
| | 108.6 |
| |
| Net income (loss) | $ | 91 |
| | $ | 170 |
| | $ | (184 | ) | (3) | $ | 67 |
| | |
| Earnings (loss) per common share | | | | | | | | | |
| Net income | | $ | 170.1 |
| | $ | 165.2 |
| | $ | 78.0 |
| | $ | 30.0 |
| (5) |
| Earnings per common share | | | | | | | | | |
| Basic | $ | 0.46 |
| | $ | 0.84 |
| | $ | (0.81 | ) | | $ | 0.30 |
| | $ | 0.76 |
| | $ | 0.74 |
| | $ | 0.35 |
| | $ | 0.13 |
| |
| Diluted | $ | 0.45 |
| | $ | 0.83 |
| | $ | (0.81 | ) | | $ | 0.29 |
| | $ | 0.75 |
| | $ | 0.73 |
| | $ | 0.35 |
| | $ | 0.13 |
| |
(1) Included within operating expenses for the second and fourth quarter of 20162018 we recognized $803.7 and $379.3, respectively, of acquired in-process research and development expense related to our Wilson and Syntimmune acquisitions, respectively. See Note 2 “Acquisitions” for additional information.
(2) Included within cost of sales for the third and fourth quarters 2017 are $83.0 and $69.1, respectively, of restructuring related expenses associated with the planned closure of the ARIMF facility.
(3) Included within operating expenses for the first and third quarters 2017 are $23.8 and $79.4, respectively of restructuring and related expenses.
(4) Included within operating expenses for the second quarter 2017 is an impairment charge of $85$31.0, associated with an early stage clinical indefinite-lived intangible asset .asset.
(2) Included within net income(5) We recognized a tax (benefit) expense of $(56.5) and $45.8 in 2018 and 2017, respectively, as a result of the Tax Cuts and Jobs Act. In 2017, we recorded certain impacts of the Tax Act on a provisional basis. As of December 22, 2018, our accounting for the second quarter of 2016 are tax benefits of $5 resulting from our adoption of new share-based compensation guidance during the third quarter of 2016. This resulted in an increase in basic EPS and diluted EPS of $0.03 and $0.02, respectively.
(3) Included within net income for the third quarter of 2015 is a one-time tax expense of $316 resulting from our integrationimpact of the Synageva business with and into the Alexion business. This tax expense is attributable to the change in our deferred tax liabilityTax Act was complete. See Note 12, “Income Taxes” for the outside basis difference resulting from the movement of assets into our captive foreign partnership.additional information.
In January 2019, we entered into a collaboration agreement with Caelum Biosciences (Caelum) to develop CAEL101 for light chain (AL) amyloidosis. Under the terms of the agreement, we acquired a minority equity interest in Caelum and an exclusive option to acquire the remaining equity in the company based on Phase 2 data, for pre-negotiated economics. We made an upfront payment of $30.0 and could be required to pay up to an additional $30.0 in contingent milestone-dependent option fees. The collaboration also provides for potential additional payments, in the event Alexion exercises the acquisition option, for up to $500.0, which includes an upfront option exercise payment and potential regulatory and commercial milestone payments.