UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| | | | | |
| ☑ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 20202023
| | | | | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from __________to__________
Commission file number 0-22705
NEUROCRINE BIOSCIENCES, INC.
(Exact name of registrant as specified in its charter)
| | | | | | | | | | | | | | |
| Delaware | | 33-0525145 |
(State or other jurisdiction of
incorporation or organization) | | (I.R.S. Employer
Identification No.) |
| | | | |
| 12780 El Camino Real, | San Diego, | California | | 92130 |
| (Address of principal executive offices) | | (Zip Code) |
(858) 617-7600
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | | | | | | | |
| Common Stock, $0.001 par value | | NBIX | | Nasdaq Global Select Market |
| (Title of each class) | | (Trading Symbol) | | (Name of each exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☑ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☑
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☑ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Large accelerated filer | ☑ | | Accelerated filer | ☐ | | Non-accelerated filer | ☐ | | Smaller reporting company | ☐ | | Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☑ No ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☑
The aggregate market value of registrant’s common stock held by non-affiliates of the registrant, computed by reference to the closing price as of the last business day of the registrant’s most recently completed second fiscal quarter, June 30, 2020,2023, was approximately $11,231,617,436.$7.9 billion.
As of January 29, 2021, 93,943,645February 5, 2024, 99,507,490 shares of the registrant’s common stock were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement relating to the registrant’s annual meeting of stockholders to be filed pursuant to Regulation 14A within 120 days following the end of the registrant’s fiscal year ended December 31, 20202023 are incorporated by reference into Part III of this Form 10-K.
TABLE OF CONTENTS
| | | | | | | | |
| | | Page |
| | |
| | | |
| Item 1. | | |
| Item 1A. | | |
| Item 1B. | | |
| Item 1C. | | |
| Item 2. | | |
| Item 3. | | |
| Item 4. | | |
| | | |
| | |
| | | |
| Item 5. | | |
Item 6. | Selected Financial Data | 34 |
| Item 7. | | |
| Item 7A. | | |
| Item 8. | | |
| Item 9. | | |
| Item 9A. | | |
| Item 9B. | | |
| Item 9C. | | |
| | | |
| | |
| | | |
| Item 10. | | |
| Item 11. | | |
| Item 12. | | |
| Item 13. | | |
| Item 14. | | |
| | | |
| | |
| | | |
| Item 15. | | |
NEUROCRINE, the Neurocrine logo, INGREZZA,® the INGREZZA logo, and ONGENTYS® other Neurocrine Biosciences trademarks are registered trademarksthe property of Neurocrine Biosciences, Inc. ALKINDI, EFMODY, and other Diurnal trademarks are the property of Diurnal Limited, a Neurocrine Biosciences company. Any other brand names or trademarks appearing in this Annual Report that are not the property of Neurocrine Biosciences, Inc. are the property of their respective holders.
PART I
Forward-Looking Statements
This Annual Report on Form 10-K and the information incorporated herein by reference contain forward-looking statements that involve a number of risks and uncertainties. Although our forward-looking statements reflect the good faith judgment of our management, these statements can only be based on facts and factors currently known by us. Consequently, these forward-looking statements are inherently subject to risks and uncertainties, and actual results and outcomes may differ materially from results and outcomes discussed in the forward-looking statements.
Forward-looking statements can be identified by the use of forward-looking words such as “believes,” “expects,” “hopes,” “may,” “will,” “plan,” “intends,” “estimates,” “could,” “should,” “would,” “continue,” “seeks,” “pro forma,” or “anticipates,” or other similar words (including their use in the negative), or by discussions of future matters such as the development of new products, technology enhancements, possible changes in legislation and other statements that are not historical. These statements include but are not limited to statements under the captions “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business,” as well as other sections in this report. You should be aware that the occurrence of any of the events discussed under the heading in Part I titled “Item 1A. Risk Factors” and elsewhere in this report could substantially harm our business, results of operations and financial condition and that if any of these events occurs, the trading price of our common stock could decline and you could lose all or a part of the value of your shares of our common stock.
The cautionary statements made in this report are intended to be applicable to all related forward-looking statements wherever they may appear in this report. We urge you not to place undue reliance on these forward-looking statements, which speak only as of the date of this report. Except as required by law, we assume no obligation to update our forward-looking statements, even if new information becomes available in the future.
Item 1. Business
Overview
We areNeurocrine Biosciences is a neuroscience-focused, biopharmaceutical company dedicatedwith a simple purpose: to discovering, developing and delivering life-changing treatmentsrelieve suffering for people with serious, challenging and under-addressed neurological, endocrine and psychiatric disorders. Our diverse portfolio includes United States Food and Drug Administration, or FDA, approved treatments for tardive dyskinesia, Parkinson’s disease, endometriosis*, uterine fibroids* and clinical programs in multiple therapeutic areas.great needs, but few options. For nearly three decades, we have specialized in targetingapplied our unique insight into neuroscience and interrupting disease-causing mechanisms involving the interconnected pathwaysinterconnections between brain and body systems to advance medicines for the treatment of under-addressed neurological, neuroendocrine and neuropsychiatric disorders and we will continue to relentlessly pursue medicines to ease the nervousburden of debilitating diseases and endocrine systems. (*in collaboration with AbbVie Inc.)
Product Pipeline
Exclusive and Partnered Commercial Products
The following table summarizes our exclusive and partnered commercial products and is followed by detailed descriptions of each product:
INGREZZA (valbenazine)disorders.
We launched INGREZZA in the U.S. in May 2017 after receiving FDA approval for INGREZZA as the first FDA-approvedU.S. Food and Drug Administration (FDA)-approved drug for the treatment of tardive dyskinesia and in April 2017.August 2023 for the treatment of chorea associated with Huntington's disease. INGREZZA provides a once-daily dosing treatment option for tardive dyskinesia and has two dosing options (40 mg and 80 mg capsules), with a recommended dose of 40 mg taken for the first seven days of treatment for tardive dyskinesia and fourteen days for chorea associated with Huntington’s disease, and an option to take 40 mg, 60 mg, or 80 mg thereafter, depending on the patient’s dosing needs.
In February 2021, Mitsubishi Tanabe Pharmaceutical Company, or MTPC, reported positive top-line results from the J-KINECT Phase III study, designed to evaluate the efficacy and safety of valbenazine in tardive dyskinesia. Detailed results from this trial will be presented at a future medical conference. With positive data in hand, a marketing authorization with the Ministry of Health and Welfare is planned for 2021 in Japan. In addition, MTPC submitted filings for marketing authorizations in South Korea, Thailand, Singapore, Indonesia, and Malaysia in 2020.
Tardive dyskinesia is defined by hyperkinetic involuntary movements which arise after months or years of treatment with dopamine receptor blocking agents, such as antipsychotics used for treating schizophrenia, bipolar disorder and depression, and certain treatments for nausea, vomiting and gastric emptying in patients with gastroparesis. While the prevalence rates of tardive dyskinesia can vary greatly in accordance with the population being studied, it is estimated that over 500 thousand individuals are2023, INGREZZA helped more people affected by tardive dyskinesia than ever before, reflecting higher prescription demand driven by increased commercial activities, including the continued investment in our branded direct-to-consumer INGREZZA advertising campaign and benefit from the U.S. alone (Kantar Health).
expansion of our sales force completed in April 2022. Going forward, key elements of our commercial strategy include maximizing the opportunity in INGREZZA through consistent and effective commercial execution, continued development of valbenazine as the best-in-class treatment for new patient populations and to lead the evolving understanding of VMAT2 biology and its role in disease. INGREZZA net product sales totaled $993.1 million, $752.9 million$1.8 billion for 2023, $1.4 billion for 2022 and $409.6 million$1.1 billion for 2020, 20192021 and 2018, respectively, and represented the significant majorityaccounted for approximately 99% of our total net product sales for 20202023.
Our internal research and all ofdevelopment efforts are focused on innovative therapies with clear and defined clinical and regulatory paths to approval. From time to time, we supplement our net product sales for 2019internal research and 2018.development efforts by in-licensing the rights to certain clinical development programs or by acquiring businesses that synergize with and allow us to capitalize on our existing development and commercial capabilities.
ONGENTYS (opicapone)
We launched ONGENTYSCommercial Products
| | | | | | | | | | | |
| Product | Indication | | Major Markets |
| | | |
| Tardive Dyskinesia | | U.S., Japan, Select Asian Markets (1) |
| Chorea Associated with Huntington’s Disease | |
| | |
| | | |
| Adrenal Insufficiency | | U.S., United Kingdom, EU4 (2)(3) |
| | |
| | | |
| Classic Congenital Adrenal Hyperplasia | | United Kingdom, EU4 (3) |
| | |
| | | |
| Endometriosis | | U.S. (4) |
| | |
| | | |
| Uterine Fibroids | | U.S. (4) |
| | |
(1) INGREZZA is marketed as DYSVAL® (valbenazine) in Japan and REMLEAS® (valbenazine) in other select Asian markets, where Mitsubishi Tanabe Pharma Corporation retains commercialization rights.
(2) ALKINDI is marketed as ALKINDI SPRINKLE® (hydrocortisone) in the U.S. in September 2020, after receiving FDA approval for ONGENTYS as an adjunctive therapy to levodopa/DOPA decarboxylase inhibitors in adult Parkinson's disease patients in April 2020. We acquired, where Eton Pharmaceuticals, Inc. retains commercialization rights.
(3) The EU4 market is made up of the U.S.following countries: Germany, France, Italy and CanadaSpain.
(4) AbbVie Inc. retains global commercialization rights to ONGENTYS from BIAL – Portela & Ca, S.A., or BIAL, in the first quarterelagolix.
Marketing and Distribution
Our specialty sales force consists of 2017.
ONGENTYS is a novel, once-daily, peripherally acting, highly selective Catechol-O-methyltransferase, or COMT, inhibitor utilized as an adjunct therapy to levodopa/carbidopa in patients with Parkinson’s disease experiencing motor fluctuations. COMT inhibitors are utilized to prolong the duration of effect of levodopa, the primary treatment option for Parkinson’s disease patients, during periods of the day where the effects of levodopa wear off and motor symptoms worsen, also referred to as “off-time.” Parkinson’s disease is a chronic and progressive movement disorder that affects approximately 1 million individuals in the U.S. alone.
ORILISSA (elagolix)
AbbVie Inc., or AbbVie, launched ORILISSA400 experienced sales professionals located in the U.S. and Canada in Augustis divided into three dedicated sales teams focused on psychiatry, neurology and November 2018, respectively, after receiving FDA and Health Canada approval for ORILISSA for the management of moderate to severe endometriosis pain in women in July and October 2018, respectively. Discovered and developed through Phase II clinical studies by us, we out-licensed the global rights to elagolix to AbbVie in 2010.long-term care.
The World Endometriosis Research Foundation estimates that there are over 170 million women worldwide who suffer from endometriosis, including approximately 7.5 million womenFor INGREZZA, our customers in the U.S. alone.consist of a limited network of specialty pharmacy providers that deliver INGREZZA to patients by mail, wholesale distributors that distribute INGREZZA primarily to certain specialty pharmacies, and specialty distributors that distribute INGREZZA primarily to closed-door pharmacies and government facilities. We rely on third-party service providers to perform a variety of functions related to the packaging, storage and distribution of INGREZZA.
ORIAHNN (elagolix, estradiol,
Manufacturing and norethindrone acetate; elagolix)Supply
AbbVie launched ORIAHNN in the U.S. in June 2020, after receiving FDA approval for ORIAHNN as the first FDA-approved non-surgical, oral medication optionWe currently rely on, and intend to continue to rely on, third-party manufacturers for the managementproduction of heavy menstrual bleeding associatedINGREZZA and our product candidates. Raw materials, active pharmaceutical ingredients (API) and other supplies required for the production of INGREZZA and our product candidates are sourced from various third-party manufacturers and suppliers in quantities adequate to meet our needs. Continuing adequate supply of such raw materials and API is assured through our long-term commercial supply and manufacturing agreements with uterine fibroidsmultiple manufacturers and our continued focus on the expansion and diversification of our third-party manufacturing relationships.
We believe our outsourced manufacturing strategy enables us to direct our financial resources to the maximization of our opportunity with INGREZZA, investment in pre-menopausal womenour internal research and development programs and expansion of our clinical pipeline through business development opportunities.
Our third-party manufacturers, suppliers and service providers may be subject to routine current Good Manufacturing Practice (cGMP) inspections by the FDA or comparable agencies in May 2020.other jurisdictions. We out-licensed the global rights to elagolix to AbbVie in 2010.
Uterine fibroids are benign hormonally responsive tumors that form in the walldepend on our third-party partners and our quality system oversight of the uterusthem for continued compliance with a prevalence rate of at least 25% (American College of ObstetricianscGMP requirements and Gynecologists) and are a leading indication for hysterectomy in the U.S., with approximately 250,000 hysterectomies performed each year related to uterine fibroids (Whiteman et al AJOG 2008, 198, e1).applicable foreign standards.
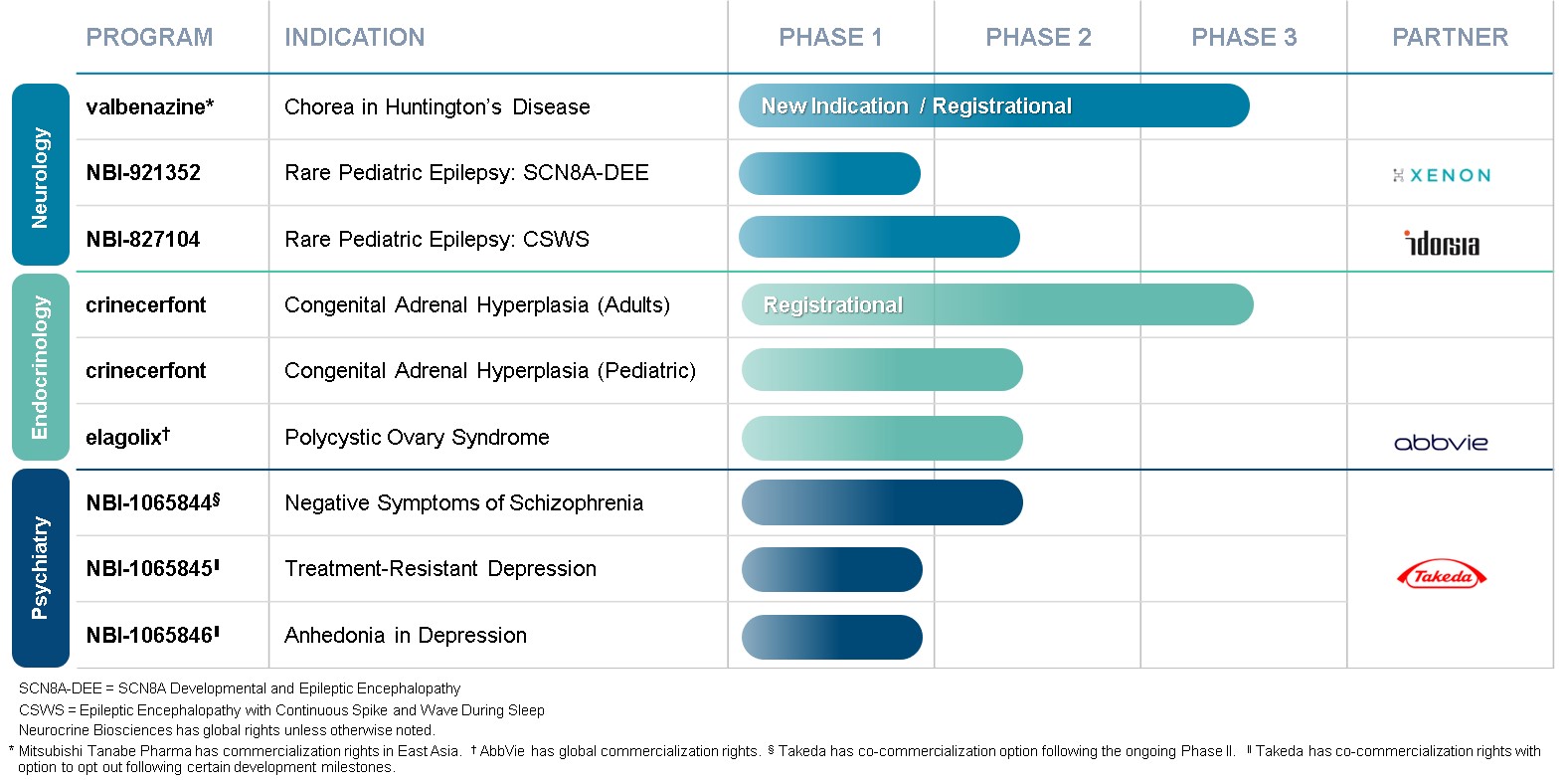
Clinical Development PipelinePrograms
The following table summarizeshighlights our current clinical development pipelineprograms and is followed by detailed descriptionsthe current phase of each program:
development for such programs.
_________________________
* Mitsubishi Tanabe Pharma Corporation retains commercialization rights in Japan and other select Asian markets.
† Heptares Therapeutics Limited retains commercialization rights in Japan, where Neurocrine Biosciences retains the right to opt in to a 50:50 profit sharing arrangement upon certain development events.
(1) This program was in-licensed from Heptares Therapeutics Limited.
(2) This program was in-licensed from Idorsia Pharmaceuticals Ltd.
(3) This program was in-licensed from Xenon Pharmaceuticals Inc.
(4) This program was in-licensed from Sanofi S.A.
(5) This program was in-licensed from Takeda Pharmaceutical Company Limited
Neurocrine Biosciences retains global rights unless otherwise noted.
Neurology
valbenazine – VMAT2 Inhibitor | | | | | |
| Program | Indication |
Valbenazine. Valbenazine is a highly selective VMAT2 inhibitor. VMAT2 is a protein concentrated in the human brain that is essential for the transmission of nerve impulses between neurons. VMAT2 is primarily responsible for packaging and transporting monoamines (dopamine, norepinephrine, serotonin and histamine) in neurons. Specifically, dopamine enables neurotransmission among nerve cells that are involved in voluntary and involuntary motor control. | Dyskinetic Cerebral Palsy. Dyskinetic cerebral palsy is a non-progressive, permanent disorder marked by involuntary movement and is a result of damage to the fetal or infant brain’s basal ganglia. The basal ganglia are responsible for submitting messages to the body to help coordinate and control movements. When damaged, voluntary movements are compromised, resulting in involuntary and abnormal movements. It affects development and movement and has long term effects on patients’ quality of life. The long-term outlook for patients with dyskinetic cerebral palsy will depend upon the severity of the brain damage and how well the treatment works. Dyskinetic cerebral palsy affects up to 15% of the estimated 500,000 to 1 million people affected by cerebral palsy in the U.S. |
NBI-921352. NBI-921352 is a potent, highly selective Nav1.6 sodium channel inhibitor being developed to treat pediatric patients with SCN8A-DEE and other potential indications. We acquired the global rights to NBI-921352 in December 2019. | SCN8A Developmental and Epileptic Encephalopathy Syndrome, or SCN8A-DEE. SCN8A-DEE is a rare, extremely severe, single-gene epilepsy caused by mutations in the SCN8A gene that activates Nav1.6, the most highly expressed sodium channel in the excitatory pathways of the central nervous system. Children born with SCN8A-DEE typically start experiencing seizures between birth and 18 months of age, and most have multiple seizures per day. Other symptoms include learning difficulties, muscle spasms, low or high muscle tone, poor coordination, developmental delay and features similar to autism. As SCN8a mutations were discovered only recently, prevalence estimates will be determined in the future as awareness of and access to genetic surveillance increases. NBI-921352 has been granted orphan drug and rare pediatric disease designations for the treatment of SCN8A-DEE in the U.S. |
VMAT2 is a protein concentratedValbenazine in the human brain that is essential for the transmission of nerve impulses between neurons. VMAT2 is primarily responsible for packagingPediatrics and transporting monoamines (dopamine, norepinephrine, serotonin and histamine) in neurons. Specifically, dopamine enables neurotransmission among nerve cells that are involved in voluntary and involuntary motor control. Disease states such as tardive dyskinesia, Tourette syndrome, Huntington’s chorea, schizophrenia, and tardive dystonia are characterized in part by a hyperdopaminergic state in the brain, and modulation of neuronal dopamine levels may provide symptomatic benefits for patientsAdults with these conditions, among others.
Dyskinetic Cerebral Palsy.We are currently conducting the KINECT-HD study, ahave an ongoing Phase III,3 randomized, placebo-controlled, double-blind, multi-center Phase IIIplacebo-controlled clinical study to evaluate the efficacy, safety and tolerability of valbenazine for the treatment of choreadyskinetic cerebral palsy in 120 patientspediatrics and adults (aged 6 to 70 years).
NBI-921352 in Pediatrics and Adolescents with Huntington’s disease, or HD, withSCN8A-DEE. We have ongoing the KAYAKTM study, a Phase III top-line data expected in2 randomized, double-blind, placebo-controlled clinical study to evaluate the fourth quarter of 2021.
HD is a hereditary progressive neurodegenerative disorder, in which neurons within the brain break down, resulting in motor, cognitive and psychiatric symptoms. Symptoms generally appear between the ages of 30 to 50 and worsen over a 10 to 25-year period. Many patients with HD experience chorea, a troublesome involuntary movement disorder, in which patients develop abnormal, abrupt or irregular movements. Chorea can affect various body parts, and interfere with speech, swallowing, posture and gait. HD is estimated to affect approximately 30,000 adults in the U.S., with more than 200,000 at risk of inheriting the disease (NORD).
NBI-921352 (XEN901) – Nav1.6 Sodium Channel Inhibitor
NBI-921352 is a potent, highly selective Nav1.6 sodium channel inhibitor being developed to treat pediatric patients with SCN8A-DEE and other potential indications, including adult focal epilepsy.
Theefficacy, safety tolerability and pharmacokinetics of NBI-921352 have been evaluatedas adjunctive therapy for seizures in a randomized, double-blind, placebo-controlled Phase I study using a powder-in-capsule formulation of NBI-921352 in healthy adult subjects.
Xenon has developed a pediatric-specific, granule formulation of NBI-921352, and completed juvenile toxicology studiesadolescents (aged 12 to support pediatric development activities.
In October 2020, the FDA requested additional non-clinical data to support the IND we submitted in August 2020 in support of a Phase II clinical study for NBI-921352 in patients21 years) with SCN8A-DEE. Based on feedback received inIn January 2021, we plan to initiate a Phase II clinical study in adolescent patients (aged 12 years and older) with SCN8A-DEE in the third quarter of 2021, and2022, the study protocol will bewas amended to include younger pediatric patientspediatrics (aged 2-112 to 11 years) with SCN8A-DEE as soon as the FDA has reviewed and approved additional non-clinical information. We are also advancing clinical plans to initiate a Phase II clinical study of NBI-921352 for the treatment of adult focal epilepsy in 2021. In addition, in October 2020, we announced the FDA granted us Rare Pediatric Disease Designation for NBI-921352 for the treatment of SCN8A-DEE.
SCN8A-DEE is a rare, extremely severe, single-gene epilepsy caused by mutations in the SCN8A gene that activates Nav1.6, the most highly expressed sodium channel in the excitatory pathways of the central nervous system, or CNS. Children born with SCN8A-DEE typically start experiencing seizures between birth and 18 months of age, and most have multiple seizures per day. Other symptoms include learning difficulties, muscle spasms, low or high muscle tone, poor coordination, developmental delay, and features similar to autism. An estimated 10% of people with SCN8A are reported to have experienced sudden unexpected death in epilepsy. The prevalence of SCN8A-DEE is estimated to be 1% of all developmental and epileptic encephalopathies (Larsen et al, Neurology 2015, 84, 480). As SCN8A mutations were discovered only recently (i.e., in 2012), the number of SCN8A-DEE cases is expected to increase as awareness of and access to genetic surveillance increases. SCN8A-DEE is generally refractory to anti-epilepsy treatments.
We are developing NBI-921352 with Xenon Pharmaceuticals Inc., or Xenon, as part of a strategic collaboration announced in December 2019.
NBI-827104 (ACT-709478) – T-type Calcium Channel Blocker
We acquired the global rights to NBI-827104 from Idorsia Pharmaceuticals Ltd., or Idorsia, in May 2020. NBI-827104 is a potent, selective, orally active and brain penetrating T-type calcium channel blocker, being developed for the treatment of a rare pediatric epilepsy and other potential indications, including essential tremor.
In November 2020, we initiated a Phase II clinical study for NBI-827104 in a rare pediatric epileptic encephalopathy known as Continuous Spike and Wave During Sleep, or CSWS. CSWS typically impacts children initially between the ages of two and four years old and manifests itself via a variety of seizure types, including atypical absence seizures, generalized tonic-clonic seizures and focal seizures that usually occur during sleep. In addition, children with CSWS often present with cognitive, behavioral and developmental regression or delay. Due to the differentiated mechanism of action of this molecule, when compared to non-selective calcium channel inhibitors, treatment with NBI-827104 could lead to an enhanced benefit risk profile for patients with this rare pediatric form of epilepsy. In parallel we are advancing clinical plans to initiate a Proof of Concept clinical study of NBI-827104 for the treatment of essential tremor in 2021.
Endocrinology
crinecerfont (NBI-74788) – CRF1 Antagonist
Crinecerfont is a potent, selective, orally active, corticotropin-releasing factor1, or CRF1, receptor antagonist as demonstrated in a range of in vitro and in vivo assays. CRF1, is a hypothalamic hormone released directly into the hypophyseal portal vasculature which acts on the CRF1 receptor, a G protein-coupled receptor, or GPCR, in the anterior pituitary to stimulate the release of adrenocorticotropin hormone, or ACTH. The primary role of ACTH is the stimulation of the synthesis and release of adrenal steroids, including cortisol. Cortisol from the adrenals have a negative feedback role at the level of the hypothalamus that decreases CRF1 release as well as at the level of the pituitary to inhibit the release of ACTH. This tight control loop is known as the hypothalamic-pituitary-adrenal axis. Blockade of CRF1 receptors at the pituitary has been shown to decrease the release of ACTH, which in turn decreases the production of adrenal steroids including androgens, and potentially the symptoms associated with classic CAH. Lower ACTH levels would also reduce the amount of exogenous corticosteroid necessary for classic CAH patients to thrive avoiding the side-effects currently associated with excessive steroid therapy.
Classic CAH is a group of autosomal recessive genetic disorders that affects approximately 30 thousandNeuroendocrinology
| | | | | |
| Program | Indication |
Crinecerfont. Crinecerfont is an investigational, oral, selective corticotropin-releasing factor type 1 (CRF1) receptor antagonist being developed to reduce and control excess adrenal androgens through a steroid-independent mechanism for the treatment of classic congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency (21-OHD). Crinecerfont has received orphan drug designation in the U.S. from the FDA and in the European Union (EU) from the European Medicines Agency (EMA). Crinecerfont has also received Breakthrough Therapy designation in the U.S. from the FDA for the treatment of CAH due to 21-OHD in adults and pediatrics. | Classic Congenital Adrenal Hyperplasia. CAH is a genetic disorder that causes little to no cortisol production and increased secretion of adrenocorticotropic hormone (ACTH) and androgens. In approximately 75% of cases, the adrenal glands cannot produce aldosterone, which can result in salt wasting adrenal crisis, causing extreme weakness, low blood pressure, shock, and even death. There are currently no non-steroidal FDA-approved treatments for CAH. CAH affects up to an estimated 30,000 people in the U.S. and 50,000 people in Europe. |
EFMODY. EFMODY is a modified-release preparation of hydrocortisone that mimics the physiological circadian rhythm of cortisol and has been specifically designed for patients with diseases of cortisol deficiency, such as CAH and adrenal insufficiency. | Classic Congenital Adrenal Hyperplasia. |
Adrenal Insufficiency. Adrenal insufficiency is a rare condition caused by inadequate production of steroid hormones in the cortex of the adrenal glands. Adrenal insufficiency can result in severe fatigue and, if left untreated, adrenal crisis that may be life threatening. |
Crinecerfont in the U.S. and approximately 50 thousand people in the EU, and results in an enzyme deficiency altering the production of adrenal steroids. Because of this deficiency, the adrenal glands have little to no cortisol biosynthesis resulting in a potentially life-threatening condition. If left untreated, classic CAH can result in salt wasting, dehydration, and eventually death. EvenAdults with cortisol replacement, persistent elevation of ACTHCAH.In September 2023, we announced positive top-line data from the pituitary gland results in excessive androgen levels leading to virilization of females including precocious puberty, menstrual irregularity, short stature, hirsutism, acne and fertility problems.
Corticosteroids are the current standard of care for classic CAH and are used chronically to both correct the endogenous cortisol deficiency and to reduce the excessive ACTH levels and androgen excess. However, the dose and duration of steroid use required to suppress ACTH is well above the normal physiological level of cortisol; resulting in metabolic syndrome, bone loss, growth impairment, and Cushing’s syndrome as common and serious side effects. We have been granted orphan drug designation for crinecerfont in the treatment of classic CAH in the U.S. and the EU.
In June 2020, positive data from a completed Phase II, open-label, pharmacokinetic/pharmacodynamic3 CAHtalyst™ clinical study of crinecerfont in adults with CAH due to 21-OHD. The Phase 3 adult patientsstudy met its primary endpoint at Week 24, demonstrating that treatment with classic CAH, which assessedcrinecerfont resulted in a statistically significant percent reduction in daily glucocorticoid (GC) dose versus placebo while maintaining androgen control (p-value <0.0001). The study also met important key pharmacodynamic biomarkers including ACTH, 17-hydroxyprogesterone (17-OHP), androgen and cortisol levels collected the morning following bedtime dosing on Day 1 and Day 14, demonstrated meaningful reductions in elevated ACTH and 17-hydroxyprogesterone (17-OHP) levels (by 54% to 75%) at all doses studied, togethersecondary endpoints, with a dose-relatedstatistically significant decrease in androstenedione (A4) levels, ranging from 21% to 64%at Week 4 versus placebo (p-value <0.0001). At Week 24, approximately 63% of patients on crinecerfont achieved a reduction to a physiologic GC dose versus approximately 18% on placebo (p-value <0.0001). The data from the highest dosePhase 3 adult study, including data from the open-label treatment period, will support New Drug Application (NDA) submission to the FDA in the second quarter of 2024.
Crinecerfont in Pediatrics with CAH.In October 2023, we announced positive top-line data from the Phase 3 CAHtalyst™ clinical study of crinecerfont (100 mg twice daily), 75% of patients showedin pediatrics (aged 2 to 17 years) with CAH due to 21-OHD. The Phase 3 pediatric study met its primary endpoint, demonstrating that treatment with crinecerfont resulted in a response ofstatistically significant decrease in serum androstenedione from baseline at least 50%Week 4 versus placebo following a GC stable period (p = 0.0002). Consistent with the results from the Phase 3 adult study, crinecerfont treatment led to a statistically significant percent reduction from baseline in daily GC dose while maintaining androgen control at Week 28 versus placebo (p < 0.0001). Approximately 30% of participants receiving crinecerfont achieved a reduction to a physiologic GC dose while maintaining androgen control compared to 0% of participants receiving placebo. The study also met the other key secondary endpoint demonstrating a statistically significant decrease in serum 17-hydroxyprogesterone from baseline at Week 4 versus placebo (p < 0.0001). The data from the Phase 3 pediatric study, including data from the open-label treatment period, will support NDA submission to the FDA in the second quarter of 2024.
EFMODY in Adolescents and Adults with CAH. We have an ongoing Phase 2 randomized, double-blind, active-controlled clinical study to evaluate the efficacy, safety and tolerability of twice-daily EFMODY compared with twice-daily Cortef® (immediate-release hydrocortisone tablets) in adolescents and adults (aged 16 years and older) with CAH. We anticipate having top-line data for eachthis clinical study in the first half of 2024.
EFMODY in Adults with Adrenal Insufficiency. We have ongoing the three hormone markers at day 14. Treatment with crinecerfont was well tolerated with a favorable safety profile with no related serious adverse events reported. Adverse events reported in two or more participants included headache, upper respiratory tract infection, fatigue, contusion, insomnia and nausea.
In July 2020, we initiated the CAHtalystCHAMPAIN study, a global registrational Phase III,2 randomized, double-blind, double-dummy, two-way crossover clinical study to evaluate the efficacy, safety and tolerability of twice-daily EFMODY compared with once-daily Plenadren® (modified-release hydrocortisone tablets) in adults with primary adrenal insufficiency. We anticipate having top-line data for this clinical study in the first half of 2024.
Neuropsychiatry | | | | | |
| Program | Indication |
Valbenazine. Valbenazine is a highly selective VMAT2 inhibitor. VMAT2 is a protein concentrated in the human brain that is essential for the transmission of nerve impulses between neurons. VMAT2 is primarily responsible for packaging and transporting monoamines (dopamine, norepinephrine, serotonin and histamine) in neurons. Specifically, dopamine enables neurotransmission among nerve cells that are involved in voluntary and involuntary motor control. | Schizophrenia. Schizophrenia is a spectrum of serious neuropsychiatric brain diseases in which people interpret reality abnormally. Schizophrenia may result in some combination of hallucinations, delusions and extremely disordered thinking and behavior that impairs daily life. People with schizophrenia typically require lifelong treatment. Early treatment may help improve long-term prognosis and get symptoms under control before serious complications develop. Schizophrenia affects an estimated 3.5 million people in the U.S. All currently approved antipsychotic medications are believed to work through direct action on monoaminergic receptors, with approximately 40% of patients reporting negative side effects and approximately 30% not benefiting adequately from these medications. |
NBI-1117568. NBI-1117568 is a potential first-in-class muscarinic M4 receptor agonist with the potential to be developed for the treatment of schizophrenia. As a selective M4 orthosteric agonist, NBI-1117568 offers the potential for an improved safety profile without the need for combination therapy to ameliorate off-target effects or for cooperativity with acetylcholine. Muscarinic receptors are central to brain function and validated as drug targets in psychosis and cognitive disorders. We acquired the global rights to NBI-1117568 in December 2021. |
Luvadaxistat. Luvadaxistat is a potential first-in-class D-Amino Acid Oxidase (DAAO) inhibitor with the potential to be developed for the treatment of cognitive impairment associated with schizophrenia. We acquired the global rights to luvadaxistat in June 2020. | Cognitive Impairment Associated with Schizophrenia, or CIAS. CIAS, which may include deficits in attention, working memory and executive function, has a negative impact on patients’ quality of life and ability to function. Although cognitive symptoms in schizophrenia are well characterized, no formal diagnostic criteria exist. Furthermore, no pharmacological agents are approved to treat the condition, and no marketed therapy tested to date has established clear, meaningful efficacy, which underscores the difficulty of drug development in this arena and accentuates the unmet need for proven treatment options. Approximately 80% of the estimated 3.5 million people affected by schizophrenia in the U.S. experience clinically relevant cognitive impairment. |
NBI-1065845. NBI-1065845 is a potential first-in-class Alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazole Propionic Acid (AMPA) potentiator with the potential to be developed for the treatment of inadequate response to treatment in major depressive disorder. We acquired the global rights to NBI-1065845 in June 2020. NBI-1065845 is currently designated as a 50:50 profit-share product with Takeda Pharmaceutical Company Limited, which retains a one-time opt-out right to convert the designation to a royalty-bearing product. | Major Depressive Disorder. Major depressive disorder is one of the leading causes of disability and is characterized by a persistently depressed mood or loss of interest in daily activities that is present most of the day in addition to other symptoms that can impact normal daily functioning, relationships and overall quality of life. Treatments range from selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors, atypical antipsychotics, tricyclic antidepressants and psychotherapies, among others. Approximately 30% of the more than 16 million people affected by the disorder in the U.S. do not adequately respond to treatment. |
Valbenazine in Adolescents and Adults with Schizophrenia.We have an ongoing Phase 3 randomized, double-blind, placebo-controlled clinical study to evaluate the efficacy, safety and efficacytolerability of crinecerfontvalbenazine when administered orally once daily as adjunctive treatment in 165 adult patientsadolescents and adults (aged 13 years and older) with classic CAH, followed byschizophrenia who have had an open-label treatment period.inadequate response to antipsychotics.
In July 2019, we initiated aNBI-1117568 in Adults with Schizophrenia. We have an ongoing Phase IIa proof-of-concept, pharmacokinetic/pharmacodynamic2 multi-center, randomized, double-blind, placebo-controlled, multi-arm, multi-stage clinical study to evaluate the efficacy, safety and tolerability of crinecerfontNBI-1117568 in pediatric patientsadults with classic CAH.schizophrenia who are experiencing an acute exacerbation or relapse of symptoms. We plan to initiate a single global registrational Phase IIIanticipate having top-line data for this clinical study for crinecerfont in pediatric patients with CAH in 2021.
elagolix – GnRH Antagonist
The gonadotropin-releasing hormone, or GnRH, is the endogenous peptide that binds to the GnRH receptor and stimulates the secretion of the pituitary hormones that are responsible for sex steroid production and normal reproductive function. Researchers have found that chronic administration of GnRH agonists, after initial stimulation, reversibly shuts down this transmitter pathway and is clinically useful in treating hormone-dependent diseases such as Polycystic Ovary Syndrome, orPCOS.
AbbVie initiated a Phase II clinical study of elagolix in patients with PCOS in mid-2019. The study is designed to evaluate whether there is a potential impact on disordered hormonal dynamics in women with PCOS. We out-licensed the global rights to elagolix to AbbVie in 2010.
PCOS is one of the most common hormonal disorders among women of reproductive age, affecting approximately 3.5 million women in the U.S. PCOS occurs when the ovaries or adrenal glands produce more male hormones (androgens) than normal. Women with PCOS experience irregular menstrual periods, infertility, pelvic pain, weight gain, acne and excess hair growth on the face, chest, stomach and thighs. There is no cure for PCOS, and treatment options are limited. If left untreated, PCOS can lead to certain cancers, diabetes and coronary artery disease.second half of 2024.
Psychiatry
Luvadaxistat in Adults with CIAS.We acquiredhave ongoing the global rightsERUDITE™ study, a Phase 2 randomized, double-blind, parallel, placebo-controlled clinical study to developevaluate the efficacy, safety, tolerability and commercialize NBI-1065844 (TAK-831), NBI-1065845 (TAK-653) and NBI-1065846 (TAK-041) from Takeda Pharmaceutical Company Limited, or Takeda,pharmacokinetics of luvadaxistat when administered orally once daily as adjunctive treatment in June 2020.
NBI-1065844 (TAK-831) – DAAO Inhibitor
NBI-1065844 is a potential first-in-class D-Amino Acid Oxidase, or DAAO, inhibitor that has completed multiple Phase I clinical studies and is currently in on-going Phase II clinical studies, including the Phase II INTERACT proof-of-conceptadults with CIAS. We anticipate having top-line data for this clinical study in negative symptomsthe second half of schizophrenia,2024.
NBI-1065845 in Adults with Inadequate Response to Treatment in Major Depressive Disorder.We have ongoing the SAVITRI™ study, a Phase II2 randomized, double-blind, placebo-controlled clinical study to evaluate the efficacy and safety of NBI-1065845 as adjunctive treatment in adults with inadequate response to treatment in major depressive disorder. We anticipate having top-line data expectedfor this clinical study in the first quarterhalf of 2021.
According to the World Health Organization, or WHO, 20 million people across the globe are affected by schizophrenia. In the U.S., the prevalence of schizophrenia is estimated to be approximately 0.6% of the population. The negative symptoms associated with schizophrenia describe a lessening or absence of behaviors and functions related to motivation and interest, or verbal and emotional expression. There are currently no approved treatment options in the U.S. for patients with predominant negative symptoms of schizophrenia.
NBI-1065844 is currently designated as a royalty-bearing product for Takeda. Takeda retains a one-time opt-in right for a 50:50 profit share arrangement upon achievement of a certain development event.
NBI-1065845 (TAK-653) – AMPA Potentiator
NBI-1065845 is a potential first-in-class Alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazole Propionic Acid, or AMPA, potentiator with the potential to be developed for treatment-resistant depression. NBI-1065845 has completed multiple Phase I clinical studies. We plan to initiate a Phase II clinical study of NBI-1065845 in treatment-resistant depression in 2021.
According to the WHO, major depressive disorder, or MDD, is one of the leading causes of disability. While there are a number of marketed treatments for MDD, approximately 1/3 of patients do not benefit from them. There is a significant need to develop new therapies with improved, faster onset of efficacy that are well tolerated.
NBI-1065845 is currently designated as a 50:50 profit share product with Takeda. Takeda retains a one-time opt-out right to convert the designation to a royalty-bearing product dependent on a certain development event.
NBI-1065846 (TAK-041) – G Protein-Coupled Receptor 139 Agonist
NBI-1065846 is a potential first-in-class G Protein-Coupled Receptor 139, or GPR139, agonist with the potential to be developed for the treatment of anhedonia in depression. NBI-1065846 has completed multiple Phase I clinical studies. We plan to initiate a Phase II clinical study of NBI-1065846 in anhedonia in 2021.
Anhedonia is a psychological condition characterized by the inability to experience pleasure. In patients with depression, anhedonia often does not improve with current treatments and predicts lack of functional improvement.
NBI-1065846 is currently designated as a 50:50 profit share product with Takeda. Takeda retains a one-time opt-out right to convert the designation to a royalty-bearing product dependent on a certain development event.
Research Programs
We invest in research and development in order to address diseases and disorders of the central nervous and endocrine systems, which include therapeutic categories ranging from hypothalamic-pituitary-adrenal disorders to stress-related disorders and neurological/neuropsychiatric diseases. CNS and endocrinology drug therapies are among the largest therapeutic categories, accounting for over $110 billion in drug sales in the U.S. alone according to IQVIA (2018).
Business Strategy
Our mission is to improve the lives of patients living with serious and under-addressed neurological, neuro-endocrinology and psychiatry related diseases and disorders. The following are the key elements of our business strategy:
Commercializing Our Product Portfolio. In April 2017, we received approval from the FDA for INGREZZA for the treatment of tardive dyskinesia. In April 2020, we received FDA approval for ONGENTYS as an adjunctive therapy to levodopa/DOPA decarboxylase inhibitors in adult Parkinson's disease patients. We market INGREZZA and ONGENTYS in the U.S. The commercial launch of INGREZZA occurred in May 2017 and ONGENTYS occurred in September 2020. We have built a specialty sales force in the U.S. of approximately 250experienced sales professionals. This specialty sales force focuses on promotion to physicians, primarily psychiatrists and neurologists.Ourcommercial team is comprised of experienced professionals in marketing, access and reimbursement, managed markets, market research, commercial operations, and sales force planning and management. In addition, our commercial infrastructure includes capabilities in manufacturing, medical affairs, quality control and compliance. We intend to retain commercial rights to certain products, including INGREZZA, that we can effectively and efficiently develop, secure regulatory approval and commercialize, which includes products with a concentrated prescriber base and well-defined patient population that can be accessed with an efficient patient and prescriber outreach program.
Advancing Life-Changing Discoveries in Neurology, Neuro-Endocrinology and Psychiatry. We believe that by continuing to advance and extend our product pipeline, we can mitigate some of the clinical development risks associated with drug development. We currently have multiple programs in various stages of research and development, including symptomatic disease modifying and curative treatments. We take a portfolio approach to managing our pipeline that balances the size of the market opportunities with clear and defined clinical and regulatory paths to approval. By doing so, we focus our internal development resources on innovative therapies with improved probabilities of technical and commercial success.
Discovering Novel Medicines to Address Unmet Patient Needs. We seek to identify and validate new medicines on novel targets for internal development or collaboration. We believe the creativity and productivity of our discovery research group will continue to be a critical component for our ongoing success.
Acquiring Rights to Commercial Products, Drug Development Candidates and Technologies. We plan to continue to selectively acquire rights to programs at all stages of development and commercial products to take advantage of our drug development and commercial capabilities.
Corporate Collaborations and Strategic Alliances
One of our business strategies is to utilize strategic alliances to enhance our development and commercialization capabilities. The following is a summary of our significant collaborations/alliances:
Takeda. In June 2020, we entered into an exclusive license agreement with Takeda, which became effective in July 2020, to develop and commercialize certain compounds in Takeda’s early to mid-stage psychiatry pipeline. Specifically, Takeda granted us an exclusive license to the following seven assets: (i) NBI-1065844 (TAK-831) for schizophrenia, (ii) NBI-1065845 (TAK-653) for treatment-resistant depression, (iii) NBI-1065846 (TAK-041) for anhedonia (which together with the NBI-1065845 are referred to as the Phase II Ready Assets), and (iv) four non-clinical stage assets, or the Non-Clinical Assets.
NBI-1065844 is deemed a royalty-bearing product under the license agreement pursuant to which we will be responsible for all costs and expenses associated with the development, manufacture, and commercialization of such asset, subject to certain exceptions, and Takeda will be eligible to receive development and commercial milestones and royalties with respect to such asset, or a Royalty-Bearing Product, and Takeda will retain the right to opt-in to a profit sharing arrangement pursuant to which we and Takeda will equally share in the operating profits and losses related to such asset, subject to certain exceptions, in lieu of receiving milestones and royalties, or a Profit-Share Product. Subject to specified conditions, Takeda may elect to exercise such opt-in right for NBI-1065844 before we initiate a Phase III clinical trial. Each of the Phase II Ready Assets is deemed a Profit-Share Product and Takeda will retain the right to opt-out of the profit-sharing arrangement for such asset pursuant to which such asset would become a Royalty-Bearing Product. Takeda may elect to exercise such opt-out rights with respect to a Phase II
Ready Asset immediately following the completion of the second Phase II clinical trial for such Phase II Ready Asset. In addition, under certain circumstances related to the development and commercialization activities to be performed by us, Takeda may elect to opt-out of the profit-sharing arrangement for a Profit-Share Product before the initiation of a Phase III clinical trial for such product.
Each of the Non-Clinical Assets will be Royalty-Bearing Products pursuant to which we will be responsible for all costs and expenses associated with the development, manufacture, and commercialization of such assets, subject to certain exceptions.
Unless earlier terminated, the license agreement will continue on a licensed product-by-licensed product and country-by-country basis until the date on which, (i) for any Royalty-Bearing Product, the royalty term has expired in such country; and (ii) for any Profit-Share Product, for so long as we continue to develop, manufacture, or commercialize such licensed product.We may terminate the license agreement for convenience in its entirety or in one or more (but not all) of the United States, Japan, the European Union, and the United Kingdom, or the Major Markets, on 6 months’ written notice to Takeda (i) with respect to all licensed products prior to the first commercial sale of the first licensed product for which first commercial sale occurs, or (ii) with respect to all licensed products in one or more given target classes, as defined in the agreement, prior to the first commercial sale of the first licensed product in such target class(es) for which first commercial sale occurs. We may terminate the license agreement for convenience in its entirety or in one or more (but not all) of the Major Markets on 12 months’ written notice to Takeda (i) with respect to all licensed products following the first commercial sale of the first licensed product for which first commercial sale occurs, or (ii) with respect to all licensed products in one or more given target classes following the first commercial sale of the first licensed product in such target class(es) for which first commercial sale. Takeda may terminate the license agreement, subject to specified conditions, (i) if we challenge the validity or enforceability of certain Takeda intellectual property rights or (ii) on a target class-by-target class basis, in the event that we do not conduct any material development or commercialization activities with respect to any licensed product within such target class for a specified continuous period. Subject to a cure period, either party may terminate the license agreement in the event of any material breach, solely with respect to the target class of a licensed product to which such material breach relates, or in its entirety in the event of any material breach that relates to all licensed products.
Idorsia. We acquired the global rights to NBI-827104 from Idorsia in May 2020. NBI-827104 is a potent, selective, orally active and brain penetrating T-type calcium channel blocker, being developed for the treatment of a rare pediatric epilepsy and other potential indications, including essential tremor. The agreement also included a research collaboration to discover and identify additional novel T-type calcium channel blockers as development candidates. Under the terms of the agreement, we are responsible for all manufacturing, development and commercialization costs of any collaboration product. We may terminate the collaboration and licensing agreement, in its entirety or with respect to a particular compound or development candidate, by providing 90 days’ written notice to Idorsia. Further, in the event a party commits a material breach and fails to cure such material breach within 90 days after receiving written notice thereof, the non-breaching party may terminate the agreement in its entirety immediately upon written notice to the breaching party.
Xenon. In December 2019, we entered into a license and collaboration agreement with Xenon to identify, research, and develop sodium channel inhibitors, including clinical candidate NBI-921352 and three preclinical candidates, which compounds we will have the exclusive right to further develop and commercialize under the terms and conditions set forth in the agreement.
We will be solely responsible, at our sole cost and expense, for all development and manufacturing of the compounds and any pharmaceutical product that contains a compound, subject to Xenon’s right to elect to co-fund the development of one product in a major indication and thus receive a mid-single digit percentage increase in royalties owed on the net sales of such product in the U.S. If Xenon exercises such option, the parties will share equally all reasonable and documented costs and expenses incurred in connection with the development of such product in the applicable indication, except costs and expenses that are solely related to the development of such product for regulatory approval outside the U.S.
Unless earlier terminated, the term of the license and collaboration agreement will continue on a product-by-product and country-by-country basis until the expiration of the royalty term for such product in such country. Upon the expiration of the royalty term for a particular product and country, the exclusive license granted by Xenon to us with
respect to such product and country will become fully paid, royalty free, perpetual, and irrevocable. We may terminate the license and collaboration agreement by providing at least 90 days’ written notice, provided that such unilateral termination will not be effective for certain products until we have used commercially reasonable efforts to complete certain specified clinical studies. Either party may terminate the agreement in the event of a material breach in whole or in part, subject to specified conditions.
Voyager.We entered into a collaboration and license agreement with Voyager, a clinical-stage gene therapy company, which became effective in March 2019. The agreement is focused on the development and commercialization of four programs using Voyager’s proprietary gene therapy platform. The four programs consist of the following: NBIb-1817 for Parkinson’s disease, the Friedreich’s ataxia program and two undisclosed programs.
Pursuant to development plans agreed to by us and Voyager, unless Voyager exercises its co-development and co-commercialization rights as provided for in the agreement, we will be responsible for all development costs. Further, upon the occurrence of a specified event for each program, we will assume responsibility for the development, manufacturing, and commercialization activities of such program.
On February 2, 2021, we notified Voyager of our termination of the NBIb-1817 for Parkinson’s disease program. The effective date of this termination will be August 2, 2021. The termination does not apply to any other development program other than NBIb-1817 for Parkinson’s disease, and our collaboration and license agreement with Voyager will otherwise continue in effect. With respect to the other programs, we may terminate the collaboration and license agreement with Voyager upon 180 days written notice to Voyager prior to the first commercial sale of any collaboration product or upon 1 year after the date of notice if such notice is provided after the first commercial sale of any collaboration product. Unless terminated earlier, the agreement will continue in effect until the expiration of the last to expire royalty term with respect to any collaboration product or the last expiration or termination of any exercised co-development and co-commercialization rights by Voyager as provided for in the agreement.
BIAL.We acquired the U.S. and Canada rights to ONGENTYS from BIAL in the first quarter of 2017. We launched ONGENTYS in the U.S. in September 2020, after receiving FDA approval for ONGENTYS as an adjunctive therapy to levodopa/DOPA decarboxylase inhibitors in adult Parkinson's disease patients in April 2020.
Under the terms of the agreement, we are responsible for the commercialization of ONGENTYS in the U.S. and Canada. Further, werely on BIAL for the commercial supply of ONGENTYS. Upon our written request prior to the estimated expiration of the term of a licensed product, the parties shall negotiate a good faith continuation of BIAL’s supply of such licensed product after the term. After the term, and if BIAL is not supplying a certain licensed product, we shall pay BIAL a trademark royalty based on the net sales of such licensed product.
Upon commercialization of ONGENTYS, we determined certain annual sales forecasts. In the event we fail to meet the minimum sales requirements for a particular year, we would be obligated to pay BIAL an amount equal to the difference between the actual net sales and minimum sales requirements for such year.
Unless earlier terminated, the agreement will continue on a licensed product-by-product and country-by-country basis until a generic product in respect of such licensed product under the agreement is sold in a country and sales of such generic product are greater than a specified percentage of total sales of such licensed product in such country.
Either party may terminate the agreement if the other party materially breaches the agreement and does not cure the breach within a specified notice period, or upon the other party’s insolvency. BIAL may terminate the agreement if we fail to use commercially reasonable efforts to submit a new drug application, or NDA, for a licensed product by a specified date, in the event we fail to meet the minimum sales requirements for any two years, or under certain circumstances involving a change of control of Neurocrine Biosciences. Under certain circumstances where BIAL elects to terminate the agreement in connection with a change of control of Neurocrine Biosciences, BIAL would be obligated to pay us a termination fee. We may terminate the agreement at any time for any reason upon nine months’ written notice to BIAL.
MTPC. In March 2015, we entered into a collaboration and license agreement with MTPC for the development and commercialization of INGREZZA for movement disorders in Japan and other select Asian markets. Under the terms of the agreement, MTPC is responsible for all development, marketing and commercialization costs in Japan and other select Asian markets, with the exception of a single Huntington’s chorea study to be performed by us. We will
be entitled to a percentage of sales of INGREZZA in Japan and other select Asian markets for the longer of ten years or the life of the related patent rights. MTPC may terminate the agreement at its discretion upon 180 days’ written notice to us. In such event, all INGREZZA product rights for Japan and other select Asian markets would revert to us.
AbbVie. In June 2010, we entered into an exclusive worldwide collaboration with AbbVie to develop and commercialize elagolix and all next-generation GnRH antagonists, or collectively the GnRH Compounds, for women’s and men’s health.
AbbVie received approval of ORILISSA for the management of moderate to severe endometriosis pain in women from the FDA in July 2018 and Health Canada in October 2018. In May 2020, AbbVie received approval from the FDA for ORIAHNN for the management of heavy menstrual bleeding associated with uterine fibroids in pre-menopausal women.
Under the terms of the agreement, AbbVie is responsible for all third-party development, marketing, and commercialization costs. We are entitled to a percentage of worldwide sales of GnRH Compounds for the longer of ten years or the life of the related patent rights. AbbVie may terminate the collaboration at its discretion upon 180 days’ written notice to us.
Intellectual Property
We actively seek to protect our products, and product candidates, and related inventions and improvements that we consider important to our business. We own a portfolio of U.SU.S. and non-U.S.ex-U.S. patents and patent applications, and have also licensed rights to a number of U.S. and non-U.S.ex-U.S. patents and patent applications. Our owned and licensed patents and patent applications cover or relate to our products and product candidates, including certain formulations, useduses to treat particular conditions, and methods of administration, drug delivery technologies and delivery profiles, and methods of manufacturing.
We own or have licensed rights toBelow is a description of the following U.S. and ex-U.S. patents relating to INGREZZA and our other products and product candidates in our pipeline (in addition to non-U.S. patents and certain patents covering our early-stage product candidates):crinecerfont:
•INGREZZA, our highly selective VMAT2 inhibitor approved in the U.S. for the treatment of tardive dyskinesia and of chorea associated with Huntington’s disease, is covered by eight22 issued, FDA Orange Book-listed U.S. patents that are listed in the FDA’s Orange Book andwhich are set to expire between 2027 and 2037. There is also a potential patent2040. Patent term extension corresponding to regulatory approval delay of up to an additional two years552 days has been received for U.S. Patent No. 8,039,627, which is currently set to expirenow expires in 20292031 and is the earliest patent coveringcovers valbenazine, the active pharmaceutical ingredient contained in INGREZZA. In Japan and in certain other East Asian markets, we are actively pursuing most of the patents corresponding to those listed in the FDA’s Orange Book entry for INGREZZA. In 2023, we entered into settlement agreements resolving all patent litigation brought by us against the companies that filed ANDAs seeking approval to market generic versions of INGREZZA, and all cases have been dismissed. Pursuant to the terms of the respective settlement agreements, such companies have the right to sell generic versions of INGREZZA in the U.S. beginning March 1, 2038, or earlier under certain circumstances. Refer to Note 13 to the consolidated financial statements for a more detailed description of these matters.
•ONGENTYS,Crinecerfont, a highly selective COMT inhibitor for Parkinson’s disease, is covered by nine issued U.S. patents that are listed in the FDA’s Orange Book and set to expire between 2026 to 2035 (not including a potential patent term extension of up to an additional four years for one of these patents).
•ORILISSA, our small molecule GnRHCRF1 receptor antagonist for the treatment of endometriosis pain, is covered by eight issued U.S. patents that are listed in the FDA’s Orange Book and are set to expire between 2021 to 2036 (not including a potential patent term extension of up to an additional five years for one of the patents currently set to expire either in 2021 or 2024).
•ORIAHNN, containing our small molecule GnRH antagonist for the treatment of menstrual bleeding associated with uterine fibroids, is covered by six issued U.S. patents that are listed in the FDA’s Orange Book and are set to expire between 2021 to 2024 (not including a potential patent term extension of up to an additional five years for one of the patents).
•Valbenazine, our highly selective VMAT2 inhibitor under further clinical development for the treatment of choreaCAH in Huntington’s disease, is covered by at least six of the issued U.S. patents that are listed in the FDA’s Orange Book entry for INGREZZAadults and are set to expire between 2027 and 2036 . There is also a potential patent term extension of up to an additional two years for U.S. Patent No. 8,039,627, which is currently set to expire in 2029.
•Crinecerfont, our CRF1 antagonist for the treatment of CAH,children, is covered by U.S. Patent No.Nos. 10,905,690, which expires in 2035 (not including a potential11,311,544, and 11,730,739, among other patents and pending patent term extension of up to an additional five years).
•NBI-1065844, a DAAO inhibitor for the treatment of negative symptoms of schizophrenia, is covered by U.S. Patent No. 9,290,456, among others, which expires in 2032 (not including a potential patent term extension of up to an additional five years).
•NBI-827104, an inhibitor of T-type calcium channels for the treatment of CSWS epilepsy, is covered by U.S. Patent No. US 9,932,314, among others, which expires in 2035 (not including a potential patent term extension of up to an additional five years).
•NBI-921352, an inhibitor of the Nav1.6 voltage-gated sodium channel for the treatment of SCN8A-DEE epilepsy, is covered by U.S. Patent No. US 10,246,453, among others, which expires in 2037 (not including a potential patent term extension of up to an additional five years).
•Elagolix, our small molecule GnRH antagonist under further development for the treatment of polycystic ovary syndrome, is covered by six of the issued U.S. patents that are listed in the FDA’s Orange Book entry for ORILISSA and areapplications, set to expire between 2021 to 20242035 and 2044 (not including aany potential patent term extension of up to an additional five years for one of the patents)extensions).
•NBI-1065845, a positive allosteric modulator of AMPA for the treatment of treatment-resistant depression is covered by U.S. Patent No. 8,778,934, among others, which expires in 2031 (not including a potential patent term extension of upWe also own, or have licensed rights to, an additional five years).
•NBI-1065846, a GPR139 agonist for the treatment of anhedonia in depression, is covered by U.S. Patent No. 9,556,130, among others, which expires in 2035 (not including a potential patent term extension of up to an additional five years).
patents covering our other products and earlier stage product candidates. In addition to the potential patent term extensions referenced above, the products and product candidates in our pipeline may be subject to additional terms of exclusivity that we mightmay obtain by future patent issuances.
Separately, the U.S., the European Union, or EU, and Japan alleach provide data and marketing exclusivity for new medicinal compounds. If this protection is available, no competitor may use the original applicant’s data as the basis of a generic marketing application during the period of data and marketing exclusivity, which is measured from the date of marketing approval by the FDA or corresponding foreign regulatory authority. This period of exclusivity is generally five years in the U.S., six years in Japan and ten10 years in the EU, except that for biologics, thisthe period of exclusivity in the U.S. is twelve12 years under the Biologics Price Competition and Innovation Act. In addition, if granted orphan drug designation, certain of our product candidates, including, for example, crinecerfont, may also be eligible for marketmarketing exclusivity in the U.S. and EU for seven years and ten years, respectively.EU for 10 years.
ManufacturingRefer to Part I, Item 1A. Risk Factors for a discussion of the challenges we may face in obtaining or maintaining patent and/or trade secret protection and Supply
We currently rely on, and intendNote 13 to continue to rely on, third-party manufacturers for the production of INGREZZA and our product candidates. Raw materials, active pharmaceutical ingredients, consolidated financial statements for API, and other supplies required for the production of INGREZZA and our product candidates are procured from various third-party manufacturers and suppliers in quantities adequate to meet our needs. Continuing adequate supply of such raw materials and API is assured through our long-term commercial supply and manufacturing agreements with multiple manufacturers and our continued focus on the expansion and diversificationa description of our third-party manufacturing relationships. In addition, under the terms of our agreement with BIAL, we rely on BIAL and its supplierslegal proceedings related to supply all drug product for the commercialization of ONGENTYS.
We believe our outsource manufacturing strategy enables us to direct our financial resources to the maximization of our opportunities with INGREZZA and ONGENTYS, investment in our internal R&D programs and expansion of our clinical pipeline through business development opportunities.
Our third-party manufacturers, suppliers and service providers may be subject to routine current Good Manufacturing Practice, or cGMP, inspections by the FDA or comparable agencies in other jurisdictions. We depend on our third-party partners for continued compliance with cGMP requirements and applicable foreign standards.intellectual property matters.
Marketing, SalesCompetition
The biotechnology and Distribution
Our sales forcepharmaceutical industries are subject to rapid and intense technological change. We face, and will continue to face, competition in the U.S. consistsdevelopment and marketing of approximately 250 experienced sales professionals focused on educating health care professionals,our products and product candidates from academic institutions, government agencies, research institutions and biotechnology and pharmaceutical companies. Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approval and marketing than we do.
Competition may also arise from, among other things, other drug development technologies, methods of preventing or reducing the incidence of disease, including psychiatristsvaccines, and neurologists, who treat patientsnew small molecule or other classes of therapeutic agents. Such developments by others (including the development of generic equivalents) may render our product candidates or technologies obsolete or noncompetitive.
•INGREZZA competes with AUSTEDO® (deutetrabenazine), marketed by Teva Pharmaceuticals Industries, for the treatment of tardive dyskinesia in adults and Parkinson’schorea associated with Huntington's disease. A once-daily dosing of AUSTEDO (AUSTEDO XR) was introduced in February 2023. Additionally, there are a number of commercially available medicines used to treat tardive dyskinesia off-label, such as XENAZINE® (tetrabenazine) and generic equivalents, and various antipsychotic medications (e.g., clozapine), anticholinergics, benzodiazepines (off-label), and botulinum toxin. In addition, there are several programs in clinical development by other companies targeting Huntington's disease.
•ORILISSA and ORIAHNN each compete with several FDA-approved products for the treatment of endometriosis, uterine fibroids, infertility and central precocious puberty. Additionally, there is also competition from surgical intervention, including hysterectomies and ablations. Separate from these options, there are many programs in clinical development which serve as potential future competition. Lastly, there are numerous medicines used to treat the symptoms of disease (vs. endometriosis or uterine fibroids directly) which may also serve as competition: oral contraceptives, NSAIDs and other pain medications, including opioids.
•For INGREZZA, our customers inCAH, high doses of corticosteroids are the current standard of care to both correct the endogenous cortisol deficiency as well as reduce the excessive ACTH levels. In the U.S. consist of a limited network of specialty pharmacy providers that deliver INGREZZA to patientsalone, there are more than two dozen companies manufacturing steroid-based products. In addition, there are several programs in clinical development by mail and a specialty distributor that distributes INGREZZA primarily to closed-door pharmacies and government facilities. For ONGENTYS, our customers in the U.S. consist primarily of wholesale distributors. We rely on third-party service providers to performother companies targeting CAH with a variety of functions relatedapproaches including gene therapy.
•Our investigational treatments for potential use in epilepsy may in the future compete with numerous approved anti-seizure medications and development-stage programs being pursued by several other companies. Commonly used anti-seizure medications include phenytoin, levetiracetam, brivaracetam, cenobamate, carbamazepine, clobazam, lamotrigine, valproate, oxcarbazepine, topiramate, lacosamide, perampanel and cannabidiol, among others. There are currently no FDA-approved treatments specifically indicated for the early infantile epileptic encephalopathy SCN8A-DEE; however, a number of different anti-seizure medications are currently used in these patient populations.
•Our investigational treatments for potential use in schizophrenia, anhedonia and depression may in the future compete with several development-stage programs being pursued by other companies. Currently, there are no FDA-approved treatments specifically indicated for anhedonia or CIAS; however, there are a number of different anti-psychotic medications currently used in these patient populations.
•Our investigational treatments for potential use in neurology, neuroendocrinology and neuropsychiatry may in the future compete with numerous approved products and development-stage programs being pursued by several other companies.
Collaboration and License Agreements
Refer to Note 2 to the packaging, storageconsolidated financial statements for more information on our significant collaboration and distribution of INGREZZA and ONGENTYS.license agreements.
Government Regulation
Our business activities are subject to extensive regulation by the U.S. and other countries. Regulation by government authorities in the U.S. and foreign countries is a significant factor in the development, manufacture, distribution, tracking, marketing and sale of our proposed products and in our ongoing research and product development activities. All of our products in development will require regulatory approval by government agencies prior to commercialization. The process of obtaining these approvals and the subsequent compliance with appropriate federal and state statutes and regulations require the expenditure of substantial time and financial resources.
In addition, federal and state healthcare laws, and equivalent supranational and foreign laws, restrict business practices in the pharmaceutical industry. These laws include, without limitation, federal, state and stateforeign fraud and abuse laws, false claims laws, data privacy and security laws, as well as transparency laws and industry codes of conduct regarding payments or other items of value provided to healthcare providers. We have a comprehensive compliance program designed to ensure our business practices remain compliant.
The U.S. federal Anti-Kickback Statute and equivalent foreign laws makes it illegal for any person or entity to knowingly and willfully, directly or indirectly, solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, lease of any good, facility, item or service for which payment may be made under programs such as a federal healthcare program, such as Medicare or Medicaid.Medicaid in the U.S.
Federal and equivalent foreign civil and criminal false claims laws and the federal civil monetary penalties law and equivalent foreign laws, which prohibit among other things, any person or entity from knowingly presenting, or causing to be presented, for payment to, or approval by, federal programs, including Medicare and Medicaid, claims for items or services, including drugs, that are false or fraudulent or not provided as claimed and knowingly making, or causing to be made, a false record or to avoid or decrease an obligation to pay money to the federal government.
The Health Insurance Portability and Accountability Act of 1996 or HIPAA,(HIPAA) created additional federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services.services and equivalent foreign laws.
In addition, weWe may be subject to HIPPA,HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 or HITECH,(HITECH) and their privacy and security regulations, which impose certain obligations, including the adoption of administrative, physical and technical safeguards to protect individually identifiable health information on covered entities subject to HIPPAHIPAA (i.e., health plans, healthcare clearinghouses and certain healthcare providers) and their business associates that perform certain services for or on their behalf involving the use or disclosure of individually identifiable health information as wewell as their covered subcontractors.
The federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies to report annually to the Centers for Medicare and& Medicaid Services or CMS,(CMS) information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), other healthcare professionals (such as physician assistants and nurse practitioners) and teaching hospitals, and applicable entities to reportas well as information regarding ownership and investment interests held by the physicians and their immediate family members. Beginning in 2022, such obligations will include payments and other transfers of value provided in the previous year to certain other healthcare professionals, including physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified nurse anesthetists, and certified nurse-midwives.
Also, many states have similar healthcare statutes or regulations that may be broader in scope and may apply regardless of payor. Additionally, to the extent that our product is sold in a foreign country, we may be subject to similar foreign laws.
The U.S. Foreign Corrupt Practices Act (FCPA) prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. It is illegal to pay, offer to pay or authorize the payment of anything of value to any foreign government official, government staff member, political party, or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. The FCPA also imposes accounting standards and requirements on publicly traded U.S. corporations and their foreign affiliates, which are intended to prevent the diversion of corporate funds to the payment of bribes and other improper payments. Similar laws exist in other countries, such as the United Kingdom (UK) or in EU member states, that restrict improper payments to public and private parties. Many countries have laws prohibiting these types of payments within the respective country. In addition to these anti-corruption laws, we are subject to import and export control laws, tariffs, trade barriers, economic sanctions, and regulatory limitations on our ability to operate in certain foreign markets.
Failure to comply with these laws, where applicable, can result in significant penalties, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgement, imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal and equivalent foreign healthcare programs, and additional reporting requirements and regulatory oversight, any of which could adversely affect our ability to operate our business and our results of operations.
Development and Marketing Approval for Products
Products. Preclinical studies generally are conducted in laboratory animals to evaluate the potential safety and efficacy of a product. Drug developers submit the results of preclinical studies to the FDA as a part of an investigational new drug or IND, application (IND) and to equivalent foreign authorities before clinical trials can begin in humans. Typically, clinical evaluation involves a time consuming and costly three-phasemulti-phase process.
| | | | | | | | |
Phase I1 | | Clinical trials are conducted with a small number of subjects to determine the early safety profile, maximum tolerated dose and pharmacokinetic properties of the product in human volunteers and for certain products, such as gene therapies,or in patients with the target disease. |
| | |
Phase II2 | | Clinical trials are conducted with groups of patients afflicted with a specific disease in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. |
| | |
Phase III3 | | Larger, multi-center, comparative clinical trials are conducted with patients afflicted with a specific disease in order to determine safety and efficacy as primary support for regulatory approval by the FDA, the European Commission, or equivalent foreign authorities, to market a product candidate for a specific disease. |
The FDA closely monitors the progress of each of the three phases of clinical trials that are conducted in the U.S. and may, at its discretion, re-evaluate, alter, suspend or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. Institutional Review Boards, Institutional Ethics Committees and Data Safety Monitoring Boards also closely monitor the conduct of our trials and may also place holds on our clinical trials or recommend that we voluntarily do so. Clinical trials conducted in foreign countries are also subject to oversight by regulatory authorities in those countries.
Once Phase III3 trials are completed, drug developers submit the results of preclinical studies and clinical trials to the FDA in the form of an NDA or a biologics licensenew drug application or BLA,(NDA) for approval to commence commercial sales. In most cases, the submission of an NDA or BLA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act or PDUFA,(PDUFA), the FDA has a goal of ten10 months from the date of filing of a standard NDA for a new molecular entity to review and act on the submission. The FDA generally has a six-month review goal of priority NDAs.
In addition, under the Pediatric Research Equity Act of 2003 as amended and reauthorized, certain applications or supplements to an application must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults or full or partial waivers from the pediatric data requirements.
The FDA also may require submission of a risk evaluation and mitigation strategy to ensure that the benefits of the drug outweigh its risks. The risk evaluation and mitigation strategy could include medication guides, physician communication plans, assessment plans and/or additional elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA conducts a preliminary review of all NDAs and BLAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an application for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA or BLA to determine, among other things, whether the drug is safe and effective for its intended use and whether the facility in which it is
manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, or BLA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with current Good Manufacturing PracticecGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, or BLA, the FDA may inspect one or more clinical trial sites to assure compliance with Good Clinical Practice (GCP) requirements.
After evaluating the NDA or BLA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the application and may require additional clinical or preclinical testing in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase IV4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a risk evaluation and mitigation strategy, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
We will also have to complete an approval process similar to that in the U.S. in order to commercialize our product candidates in each foreign country. The approval procedure and the time required for approval vary from country to country and may involve additional testing. Foreign approvals may not be granted on a timely basis, or at all. In addition, regulatory approval of prices is required in most countries other than the U.S., except for a certain limited number of drugs sold to certain Medicare beneficiaries beginning in 2023. The resulting prices may not be sufficient to generate an acceptable return to us or our corporate collaborators.
In the EU, there are currently two potential tracks for seeking marketing approval for a product not authorized in any EU member state: a decentralized procedure and a centralized procedure. In the decentralized procedure, identical applications for marketing authorization are submitted simultaneously to the national regulatory agencies. Regulatory review is led by one member state (the reference-member state), and its assessment—based on safety, quality and efficacy—is reviewed and approved (assuming there are no concerns that the product poses a serious risk to public health) by the other member states from which the applicant is seeking approval (the concerned-member states). The decentralized procedure leads to a series of single national approvals in all relevant countries. In the centralized procedure, which is required of all products derived from biotechnology, a company submits a single marketing authorization application to the European Medicines Agency, or EMA, which conducts an evaluation of the dossier, drawing upon its scientific resources across Europe. If the drug product is proven to fulfill the requirements for quality, safety and efficacy, the EMA’s Committee for Medicinal Products of Human Use, or CHMP, adopts a positive opinion, which is transmitted to the European Commission for final decision on grant of the marketing authorization. While the EC generally follows the CHMP’s opinion, it is not bound to do so. Subsequent commercialization is enabled by country-by-country reimbursement approval.
Orphan Drug Designation
Designation. Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the U.S., or if it affects more than 200,000, there is no reasonable expectation that sales of the drug in the U.S. will be sufficient to offset the costs of developing and making the drug available in the U.S. Orphan drug designation must be requested before submitting an NDA or BLA.NDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If the FDA approves a sponsor’s marketing application for a designated orphan drug for use in the rare disease or condition for which it was designated, the sponsor is eligible for a seven-year period of marketing exclusivity, during which the FDA may not approve another sponsor’s marketing application for a drug with the same active moiety and intended for the same use or indication as the approved orphan drug, except in limited circumstances, such as if a subsequent sponsor demonstrates its product is clinically superior. During a sponsor’s orphan drug exclusivity period, competitors, however, may receive approval for drugs with different active moieties for the same indication as the approved orphan drug, or for drugs with the same active moiety as the approved orphan drug, but for different indications. Orphan drug exclusivity could block the approval of one of our products for seven years if a competitor obtains approval for a drug with the same active moiety intended for the same indication before we do, unless we are able to demonstrate that grounds for withdrawal of the orphan drug exclusivity exist, or that our product is clinically superior. Further, if a designated orphan drug receives marketing approval for an indication broader than the rare disease or condition for which it received orphan drug designation, it may not be entitled to exclusivity.
Legislation similar to the Orphan Drug Act has been enacted in other countries outside of the United States, including the EU. The orphan legislation in the EU is available for therapies addressing conditions that affect five or fewer out of 10,000 persons, are life‑threatening or chronically debilitating conditions and for which no satisfactory treatment is authorized. The market exclusivity period is for ten years, although that period can be reduced to six years if, at the end of the fifth year, available evidence establishes that the product does not justify maintenance of market exclusivity.
Post-Approval Requirements
Requirements. Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual program user fee requirements for any marketed products, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA or BLA.NDA. For example, the FDA may require post-marketing testing, including Phase IV4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with current Good Manufacturing Practices, or cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess
new safety risks; or imposition of distribution or other restrictions under a risk evaluation and mitigation strategy program.restrictions. Other potential consequences include, among other things:
•restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
•fines, warning letters or holds on post-approval clinical trials;
•refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals;
•product seizure or detention, or refusal to permit the import or export of products; or
•injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indicationsindication(s) and in accordance with the provisions of the approved label. However, companies may share truthful and not misleading information that is otherwise consistent with a product’s FDA approved labeling. The FDA and other agencies actively enforce the laws and regulations prohibiting pre-approval promotion of investigational drugs, as well as the promotion of off-label uses of approved drugs, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. The FDA does not regulate behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off-label use of their products.
Additional Regulation for Gene Therapy Products
In addition to the regulations discussed above, there are a number of standards that apply to gene therapy. FDA has issued various guidance documents regarding gene therapies, which outline factors that FDA will consider at each of the stages of development and relate to, among other things: the proper preclinical assessment of gene therapies; the chemistry, manufacturing and controls information that should be included in an IND application; the proper design of tests to measure product potency in support of an IND or BLA; and measures to observe delayed adverse effects in subjects who have been exposed to investigational gene therapies when the risk of such effects is high. For instance, FDA usually recommends that sponsors observe all surviving subjects who receive treatment using gene therapies that are based on adeno-associated virus vectors in clinical trials for potential gene therapy-related delayed adverse events for a minimum five-year period, followed by 10 years of annual queries, either in person or by questionnaire. FDA does not require the long-term tracking to be complete prior to its review of the BLA.
In addition to FDA oversight and oversight by institutional review boards, gene therapy clinical trials are also subject to review and oversight by an institutional biosafety committee, or IBC, a local institutional committee that reviews and oversees research utilizing recombinant or synthetic nucleic acid molecules at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment.
Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we obtain regulatory approval. In the U.S. and markets in other countries, sales of any products for which we receive regulatory approval will depend, in part, on the extent to which third-party payors provide coverage and establish adequate reimbursement levels for such drug products.
In the U.S., third-party payors include federal and state healthcare programs, government authorities, private managed care providers, private health insurers and other organizations. No uniform policy for coverage and reimbursement exists in the United States,U.S., and coverage and reimbursement can differ significantly from payor to payor. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates, but also have their own methods and approval process apart from Medicare determinations. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our drug products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
instance or applied consistently.Third-party payors are increasingly challenging the price, examining the medical necessity and reviewing the cost-effectiveness of medical drug products and medical services, in addition to questioning their safety, efficacy and efficacy.clinical appropriateness. Such payors may limit coverage to specific drug products on an approved list, also known as a formulary, which might not include all of the FDA-approved drugs for a particular indication. We may need to conduct expensive pharmaco-economic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Nonetheless, our products or product candidates, including INGREZZA, may not be considered medically necessary or cost-effective.
Moreover, the process for determining whether a third-party payor will provide coverage for a drug product may be separate from the process for setting the price of a drug product or for establishing the reimbursement rate that such a payor will pay for the drug product. A payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a drug product does not assure that other payors will also provide coverage for the drug product. Adequate third-party payor reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
The marketability of any product or product candidates for which we or our collaborators receive regulatory approval for commercial sale may suffer if third-party payors fail to provide coverage and adequate reimbursement. In addition, emphasis on managed care in the U.S. has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party payor reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Healthcare Reform Measures
The U.S. and some foreign jurisdictions have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. In the U.S., the pharmaceutical industry and the cost of prescription drugs has been a continuous focus of these efforts and has been significantly affected by major legislative initiatives.
Most recently, in August 2022, President Biden signed into law the Inflation Reduction Act of 2022 (IRA), which, among other things, (1) directs the Secretary of the U.S. Department of Health and Human Services (HHS) to negotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare, (2) redesigns the Medicare Part D prescription drug benefit to lower patient out-of-pocket costs and increase manufacturer liability and (3) requires drug manufacturers to pay rebates on drugs whose prices increase greater than the rate of inflation. The IRA also extends enhanced subsidies for individuals purchasing health insurance coverage in the ACA marketplaces through plan year 2025 and eliminates the “donut hole” under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost to $2,000 through a newly established manufacturer discount program. These provisions take effect progressively starting in 2023. On August 29, 2023, HHS announced the list of the first 10 drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is currently subject to legal challenges. It is currently unclear how the IRA will be implemented; however, it is likely to have a significant impact on the pharmaceutical industry and prescription drug pricing.
While the IRA targets high-expenditure drugs that have been on the market for several years without generic or biosimilar competition, we expect to qualify for the small biotech manufacturer exemption that is set to expire in 2029. However, the qualification for this exemption is subject to various requirements and there is no assurance that we will continue to qualify for this exemption in the future. Further, the loss of this exemption or the potential loss of this exemption, including as a result of a potential acquisition or strategic transaction, could have an adverse impact on our business.
The most significant prior revisions to federal law governing the pharmaceutical industry and prescription drug pricing were enacted through the March 2010 Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the ACA). Thislaw was intended to broaden access to health insurance by reducing the number of uninsured persons, reducing or constraining the growth of healthcare spending, enhancing remedies against fraud and abuse, adding transparency requirements for the healthcare and health insurance industries, imposing taxes and fees on the health industry and imposing additional health policy reforms.
We expect that these health reform measures may result in more rigorous coverage criteria and lower reimbursement for prescription drugs, as well as result in additional downward pressure on any price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private third-party payors.
Other significant legislative changes impacting the pharmaceutical industry and prescription drug pricing have been adopted since the ACA was enacted. These changes include, among others, aggregate reductions to Medicare payments to providers of up to 2% per fiscal year pursuant to the Budget Control Act of 2011, which began in 2013 and, due to subsequent legislative amendments, including the Investment and Jobs Act, will remain in effect through 2032.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to examine and/or control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. For example, on January 5, 2024, the FDA approved Florida’s Section 804 Importation Program (SIP) proposal to import certain drugs from Canada for specific state healthcare programs. It is unclear how this program will be implemented, including which drugs will be chosen, and whether it will be subject to legal challenges in the United States or Canada. Other states have also submitted SIP proposals that are pending review by the FDA. Any such approved importation plans, when implemented, may result in lower drug prices for products covered by those programs. Further, certain states through legislation have created a state prescription drug affordability board (PDAB) to help control costs of drugs for that state. The functions of the PDABs vary by state, and may include among others, negotiating the price the state pays for certain drugs, recommending or setting upper limits on drug prices, performing drug affordability reviews, and advising state lawmakers on additional ways to reduce the state’s drug spending. It is possible that the actions taken by the PDABs may result in lower prices for certain drug products sold in their states.
Proposed Healthcare Reform Measures
The U.S. and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the U.S. and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. In the U.S., the pharmaceutical industry has been a particular focus of these efforts and has beenmay be significantly affected by major legislative initiatives.
By wayWe are currently unable to predict what other additional legislation or regulation, if any, relating to the healthcare industry may be enacted in the future or what effect recently enacted federal legislation or any such additional legislation or regulation would have on our business.
Regulation and Procedures Governing Approval of example,Medicinal Products in March 2010, the Patient ProtectionEU
To market any product outside of the U.S., a company must also comply with numerous and Affordable Care Act, as amendedvarying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of products. Whether or not it obtains FDA approval for a product, an applicant will need to obtain the necessary approvals by the Health Carecomparable foreign regulatory authorities before it can initiate clinical trials or marketing of the product in those countries or jurisdictions. Specifically, the process governing approval of medicinal products in the EU generally aligns with the requirements in the U.S. It entails satisfactory completion of pharmaceutical development, nonclinical studies and Education Reconciliation Actadequate and well-controlled clinical trials to establish the safety and efficacy of 2010, collectively the ACA, was signedmedicinal product for each proposed indication.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement may vary from country to country. In all cases, the clinical trials must be conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki. Medicines used in clinical trials must be manufactured in accordance with cGMP and in a GMP licensed facility, which can be subject to GMP inspections.
Clinical Trials in the EU. In the EU, the Clinical Trials Regulation (EU) No 536/2014 (CTR) entered into law, which intendedapplication on January 31, 2022 repealing and replacing the former Clinical Trials Directive 2001/20 (CTD). The regulation introduces a streamlined application procedure via a single entry point, the “EU portal”, the Clinical Trials Information System (CTIS); a single set of documents to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraudbe prepared and abuse, add transparency requirementssubmitted for the healthcare and health insurance industries, impose taxes and fees on the health industry and impose additional health policy reforms.
We expect that the ACA,application as well as other healthcare reform measures that maysimplified reporting procedures for clinical trial sponsors. A harmonized procedure for the assessment of applications for clinical trials has been introduced and is divided into two parts.
The extent to which on-going clinical trials will be adopted ingoverned by the future, may result in more rigorous coverage criteria and lower reimbursement, and in additional downward pressureCTR will depend on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private third-party payor.
There remain legal and political challenges to certain aspectsduration of the ACA. Since January 2017,individual clinical trial. For clinical trials in relation to which an application for approval was made on the Trump administration signed several executive orders and other directives designed to delay, circumvent, or loosen certain requirements mandated by the ACA. Concurrently, Congress considered legislation that would repeal or repeal and replace all or partbasis of the ACA. Legislation enacted in 2017, informally titledCTD before January 31, 2023, the Tax Cuts and Jobs Act, includesCTD will continue to apply on a provisiontransitional basis until January 31, 2025. By that repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage fordate, all or part of a year that is commonly referred to as the “individual mandate”. In addition, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminated the health insurer tax. On December 14, 2018, a Texas U.S. District Court Judge ruled that the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as part of the Tax Cuts and Jobs Act. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit ruled that the individual mandate was unconstitutional and remanded the case backongoing trials will become subject to the District Court to determine whether the remaining provisions of the ACACTR. The CTR will apply to clinical trials from an earlier date if the related clinical trial application was made on the basis of the CTR or if the clinical trial has already transitioned to the CTR framework before January 31, 2025.
Marketing Authorizations. In the EU, medicinal products can only be commercialized after a related marketing authorization (MA) has been granted. To obtain an MA for a product in the EU, an applicant must submit a marketing authorization application (MAA) either under a centralized procedure administered by the EMA or one of the procedures administered by the competent authorities of EU Member States (decentralized procedure, national procedure or mutual recognition procedure). An MA may be granted only to an applicant established in the EU.
The centralized procedure provides for the grant of a single MA by the European Commission that is valid throughout the European Economic Area (which is comprised of the 27 EU Member States plus Norway, Iceland and Liechtenstein). Pursuant to Regulation (EC) No 726/2004, the centralized procedure is compulsory for specific products, including for (i) medicinal products derived from biotechnological processes, (ii) products designated as orphan medicinal products, (iii) advanced therapy medicinal products (ATMPs), and (iv) products with a new active substance indicated for the treatment of HIV/AIDS, cancer, neurodegenerative diseases, diabetes, auto-immune and other immune dysfunctions and viral diseases. For products with a new active substance indicated for the treatment of other diseases and products that are invalid as well. Thehighly innovative or for which a centralized process is in the interest of patients, authorization through the centralized procedure is optional on related approval.
U.S. Supreme CourtAccelerated assessment may be granted by the EMA’s Committee for Medicinal Products for Human Use (CHMP) in exceptional cases, when a medicinal product targeting an unmet medical need is currently reviewing this case, althoughexpected to be of major interest from the point of view of public health and, in particular, from the viewpoint of therapeutic innovation. If the CHMP accepts a request for accelerated assessment, the time limit of 210 days will be reduced to 150 days (excluding clock stops). The CHMP can, however, revert to the standard time limit for the centralized procedure if it considers that it is unclearno longer appropriate to conduct an accelerated assessment.
An MA has, in principle, an initial validity of five years. The MA may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the EU Member State in which the original MA was granted. The European Commission or the competent authorities of the EU Member States may decide on justified grounds relating to pharmacovigilance, to proceed with one further five-year renewal period for the MA. Once subsequently definitively renewed, the MA shall be valid for an unlimited period. Any authorization which is not followed by the actual placing of the medicinal product on the EU market (for a centralized MA) or on the market of the authorizing EU Member State within three years after authorization ceases to be valid (the so-called sunset clause).
Upon receiving an MA, innovative medicinal products are generally entitled to receive eight years of data exclusivity and 10 years of market exclusivity. Data exclusivity, if granted, prevents regulatory authorities in the EU from referencing the innovator’s data to assess a generic application or biosimilar application for eight years from the date of authorization of the innovative product, after which a generic or biosimilar MAA can be submitted, and the innovator’s data may be referenced. The market exclusivity period prevents a successful generic or biosimilar applicant from commercializing its product in the EU until 10 years have elapsed from the initial MA of the reference product in the EU. The overall ten-year period may, occasionally, be extended for a further year to a maximum of 11 years if, during the first eight years of those ten years, the MA holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
Orphan Designation and related Exclusivity in the EU. In the EU, Regulation (EC) No. 141 provides that a medicinal product can be designated as an orphan medicinal product by the European Commission if its sponsor can establish that: (i) the product is intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions; (ii) either (a) such conditions affect not more than five in 10,000 persons in the EU when a decisionthe application is made, or (b) the product without the benefits derived from orphan status, would not generate sufficient return in the EU to justify the necessary investment in developing the medicinal product; and (iii) there exists no satisfactory authorized method of diagnosis, prevention, or treatment of the condition that has been authorized in the EU, or even if such method exists, the product will be made. Itof significant benefit to those affected by that condition.
Upon grant of a marketing authorization, orphan medicinal products are entitled to a ten-year period of market exclusivity for the approved therapeutic indication, which means that the EMA cannot accept another marketing authorization application or accept an application to extend for a similar product and the European Commission cannot grant a marketing authorization for the same indication for a period of ten years. The period of market exclusivity is extended by two years for orphan medicinal products that have also unclear how such litigation will impactcomplied with an agreed PIP. The period of market exclusivity may, however, be reduced to six years if, at the ACA.end of the fifth year, it is established that the product no longer meets the criteria on the basis of which it received orphan medicinal product destination.
Other legislative changes have been proposed and adopted sincePost-Authorization Obligations in the ACA was enacted. These changes include, among others, aggregate reductionsEU. Where an MA is granted in relation to Medicare paymentsa medicinal product in the EU, the holder of the MA is required to providerscomply with a range of up to 2% per fiscal year pursuantregulatory requirements applicable to the Budget Control Actmanufacturing, marketing, promotion and sale of 2011, which began in 2013 and will remain in effect through 2030, except for a temporary suspension from May 1, 2020 through May 31, 2021 duemedicinal products. Similar to the COVID-19 pandemic, unless additional Congressional action is taken.
Also, there has been heightened governmental scrutiny recently over pharmaceutical pricing practices in lightU.S., both MA holders and manufacturers of the rising cost of prescription drugs and biologics. Such scrutiny has resulted in several Congressional hearings and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. Both the U.S. House of Representatives and the Senate Finance Committee passed legislation in 2019 to reform pharmaceutical pricing in a variety of meaningful ways, and we expect legislative efforts to reform drug pricing to continue in 2021.
At the federal level, the Trump administration’s budget proposal for fiscal year 2021 included a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. Additionally, the Trump administration previously released a “Blueprint” to lower drug prices and reduce out-of-pocket costs of drugs. On July 24, 2020 and September 13, 2020, the Trump administration announced several executive orders related to drug pricing that sought to implement several of the administration’s proposals. As a result, the FDA released a final rule on September 24, 2020, effective November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. This rule is undergoing legal challenge.
Further, on November 20, 2020, HHS finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers. This rule has been stayed until 2023 while pending litigation is heard in the courts.
On November 20, 2020, CMS issued an interim final rule implementing President Trump’s Most Favored Nation executive order, which would tie Medicare Part B payments for certain physician-administered drugs to the lowest price paid in other economically advanced countries, effective January 1, 2021. On December 28, 2020, the United States District Court in Northern California issued a nationwide preliminary injunction against implementation of the interim final rule. Two additional lawsuits in other jurisdictions are challenging the legality of this rule.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Further, it is possible that additional governmental action is taken in response to the COVID-19 pandemic.
Competition
The biotechnology and pharmaceutical industriesmedicinal products are subject to rapidcomprehensive regulatory oversight by the EMA, the European Commission and/or the competent regulatory authorities of the individual EU Member States. The holder of an MA must establish and intense technological change. We face,maintain a pharmacovigilance system and will continue to face, competition in the developmentappoint an individual qualified person for pharmacovigilance who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and marketingsubmission of our products and product candidates from academic institutions, government agencies, research institutions and biotechnology and pharmaceutical companies. Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approval and marketing than we do.
Competition may also arise from, among other things, new drug development technologies, new or improved treatment options for preventing or reducing the incidence of disease in diseases our products treat and new smallperiodic safety update reports (PSURs).
molecule orIn the EU, the advertising and promotion of medicinal products are subject to both EU and EU Member States’ laws governing promotion of medicinal products, interactions with physicians and other classeshealthcare professionals, misleading and comparative advertising and unfair commercial practices. General requirements for advertising and promotion of therapeutic agents. Such developmentsmedicinal products, such as direct-to-consumer advertising of prescription medicinal products are established in EU law. However, the details are governed by competitors could reduce or eliminateregulations in individual EU Member States and can differ from one country to another.
Brexit and the use of ourRegulatory Framework in the UK. The UK’s withdrawal from the EU on January 31, 2020, commonly referred to as Brexit, has changed the regulatory relationship between the UK and the EU. The Medicines and Healthcare products or may limitRegulatory Agency (MHRA) is now the utilityUK’s standalone regulator for medicinal products and application of ongoingmedical devices. Great Britain (England, Scotland and Wales) is now a third country to the EU. Northern Ireland continues to follow the EU regulatory rules.
The UK regulatory framework in relation to clinical trials for our product candidates.
Additional information about the competition that our marketed products face is set forth below.
Tardive Dyskinesia. INGREZZA competes with AUSTEDO (deutetrabenazine), which was approvedgoverned by the FDAMedicines for Human Use (Clinical Trials) Regulations 2004, as amended, which is derived from the CTD, as implemented into UK national law through secondary legislation. In October 2023, the MHRA announced a new Notification Scheme for clinical trials which enables a more streamlined and risk-proportionate approach to initial clinical trial applications for Phase 4 and low-risk Phase 3 clinical trial applications.
Marketing authorizations in the UK are governed by the Human Medicines Regulations (SI 2012/1916), as amended. This legislation includes procedures to prioritize access to new medicines that will benefit patients, including a 150-day assessment route, a rolling review procedure and the International Recognition Procedures (IRP) which entered into application on January 1, 2024. Since January 1, 2024, the MHRA may rely on the IRP when reviewing certain types of marketing authorization applications. There is no pre-marketing authorization orphan designation for medicinal products in the UK. Instead, the MHRA reviews applications for orphan designation in parallel to the corresponding marketing authorization application. The criteria are essentially the same as those in the EU but have been tailored for the treatment of tardive dyskinesia in adults in August 2017 and is marketed by Teva Pharmaceutical Industries, and several clinical development-stage programs targeting tardive dyskinesia and related movement disorders. Additionally, there are a number of commercially available medicines used to treat tardive dyskinesia off-label, such as Xenazine® (tetrabenazine) and generic equivalents, and various antipsychotic medications (e.g., clozapine), anticholinergics, benzodiazepines (off-label), and botulinum toxin.
Parkinson’s Disease. ONGENTYS competes with two other FDA-approved COMT inhibitors and their generic equivalents. Additionally, there are a number of alternative adjunctive treatment options (FDA-approved and in clinical development) for Parkinson’s patients which compete with ONGENTYS, including various L-dopa preparations, dopamine agonists, MAO-B inhibitors and others. In terms of potential future competition, there are several programs in late-stage clinical development.
Endometriosis and Uterine Fibroids. ORILISSA and ORIAHNN each compete with several FDA-approved products for the treatment of endometriosis, uterine fibroids, infertility, and central precocious puberty. Additionally, there is also competition from surgical intervention, including hysterectomies and ablations. Separate from these options, there are many programs in clinical development which serve as potential future competition. Lastly, there are numerous medicines used to treat the symptoms of disease (vs. endometriosis or uterine fibroids directly) which may also serve as competition: oral contraceptives, NSAIDs and other pain medications including opioids.
Congenital Adrenal Hyperplasia. For CAH, high doses of corticosteroids are the current standard of care to both correct the endogenous cortisol deficiency as well as reduce the excessive ACTH levels. In the U.S. alone, there are more than two dozen companies manufacturing steroid-based products. In addition, there are several clinical development-stage programs targeting CAH and several companies developing medicinal treatments for CAH.
Epilepsy. Our investigational treatments for potential use in epilepsy may in the future compete with numerous approved anti-seizure medications, or ASMs, and development-stage programs being pursued by several other companies. Commonly used ASMs, among others, include phenytoin, levetiracetam, brivaracetam, cenobamate, carbamazepine, clobazam, lamotrigine, valproate, oxcarbazepine, topiramate, lacosamide, perampanel and cannabidiol. There are currently no FDA-approved treatments specifically indicated for the early infantile epileptic encephalopathies SCN8A-DEE and EE-CSWS; however, a number of different ASMs are currently used in these patient populations.
Schizophrenia. The investigational treatment NBI-1065844 for the negative symptoms of schizophrenia may in the future compete with off-label antipsychotic and antidepressant medicines, including cariprazine, clozapine, fluoxetine, citalopram, sertraline, and amisulpride. In addition, there are several development-stage programs being pursued by other companies, including pimavanserin, roluperidone, RO6889450 and sodium benzoate. Currently, there are no-FDA approved treatments specifically indicated for the negative symptoms of schizophrenia.
Other. Our investigational treatments for potential use in endocrinology, neurology, and psychiatry, as well as our investigational gene therapies, may in the future compete with numerous approved products and development-stage programs being pursued by several other companies.market.
Human Capital
Our Employees. We have grown to a team of over 845more than 1,400 employees as of December 31, 2020, all of whom were2023, primarily employed in the U.S. Our highly qualified and experienced team, which includes scientists, physicians and professionals across sales, marketing, manufacturing, regulatory, finance and other importantessential functions are critical to our success. We also leverage temporary workers to provide flexibility for our business needs. During 2020,2023, we added 191approximately 200 new employees to our team.
We expect to continue to add additional employees in 20212024 with a focus on expanding our expertise and bandwidth in clinical and preclinical research and development.development organization. We continually evaluate our business needs and opportunities
and balance in house expertise and capacityin-house with external expertise and capacity. Currently, we rely on third-party contract manufacturers.
Our Culture. The success of our human capital management investments is evidenced by our low employee turnover, a number which is regularly reviewed by our Board of Directors as part of their oversight of our human capital strategy. In recognition of our efforts, in 2020,2023, we were named to theranked #8 in Fortune Best Small and Medium Workplaces 2020 list ranking Number 8 across the country. We were also named a Great Place to Work Certified company and were recognized on Great Place to Work’s Best Workplace for Parents 2020 list.in BiopharmaTM.
Employee Engagement, Talent Development & Benefits. We believe that our future success largely depends upon our continued ability to attract and retain highly skilled employees. We provide our employees with competitive salaries and bonuses, opportunities for equity ownership, development programs that enable continued learning and growth and a robust employment package that promotes well-being across all aspects of their lives, including health care,healthcare, retirement planning and paid time off. As part of our promotion and retention efforts, we also invest in ongoing leadership development through programs as well as offer tuition reimbursement. In addition, we regularly conduct an employee surveysurveys to gauge employee engagement and identify areas of focus.
Diversity & Inclusion. Much of our success is rooted in the diversity of our teams and our commitment to inclusion. We value diversity at all levels and continue to focus on extending our diversity and inclusion initiatives across our entire workforce. We believe that our business benefits from the different perspectives a diverse workforce brings, and we pride ourselves on having a strong, inclusive and positive culture based on our shared mission and values.
Corporate Information
We were originally incorporated in California in January 1992 and reincorporated in Delaware in May 1996. Our principal executive offices are located at 12780 El Camino Real, San Diego, California 92130. Our telephone number is (858) 617-7600.
Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to reports filed pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended, are available free of charge on our website at www.neurocrine.com, as soon as reasonably practicable after such reports are available on the Securities and Exchange Commission or SEC,(SEC) website at www.sec.gov. Additionally, copies of our Annual Report will be made available, free of charge, upon written request. Information found on, or accessible through, our website is not a part of, and is not incorporated into, this Annual Report on Form 10-K.
Item 1A. Risk Factors
The following information sets forth risk factors that could cause our actual results to differ materially from those contained in forward-looking statements we have made in this Annual Report on Form 10-K and those we may make from time to time. If any of the following risks actually occur, our business, operating results, prospects or financial condition could be harmed. Additional risks not presently known to us, or that we currently deem immaterial, may also affect our business operations.
Summary Risk Factors
We face risks and uncertainties related to our business, many of which are beyond our control. In particular, risks associated with our business include:
•We may not be able to continue to successfully commercialize INGREZZA ONGENTYS,or any of our other products, or any of our product candidates if they are approved in the future.
•If physicians and patients do not continue to accept INGREZZA or do not accept ONGENTYS,any of our other products, or our sales and marketing efforts are not effective, we may not generate sufficient revenue.
•GovernmentalEnacted healthcare reform, drug pricing measures and third-party payors may impose sales and pharmaceutical pricing controls onother recent legislative initiatives, including the Inflation Reduction Act of 2022, could adversely affect our products or limit coverage and/or reimbursement for our products that could limit our product revenues and delay sustained profitability.business.
•Our business could be adversely affected by the effects of health pandemics or epidemics, includingwhich could also cause significant disruption in the COVID-19 pandemic, in regions where we or third parties on which we rely have significant sales and marketing efforts or manufacturing facilities, concentrations of clinical trial sites or other business operations, or materially affect our operations, and at our clinical trial sites, as well as the business or operations of ourthird-party manufacturers, CROscontract research organizations (CROs), or other third parties withupon whom we conduct business.rely.
•We face intense competition, and if we are unable to compete effectively, the demand for our products may be reduced.
•Because the development of our product candidates is subject to a substantial degree of technological uncertainty, we may not succeed in developing any of our product candidates.
•Our clinical studiestrials may be delayed for safety or other reasons, or fail to demonstrate the safety and efficacy of our product candidates, which could prevent or significantly delay their regulatory approval. For example, the FDA has placed a clinical hold on the RESTORE-1 study, a Phase II, randomized, placebo-surgery controlled, double-blind, multi-center clinical study of NBIb-1817 in Parkinson’s disease patients, following our submission of an IND safety report related to the observation of MRI abnormalities in some study participants. The clinical implications of this observation are currently unknown and are being evaluated. On February 2, 2021, we notified Voyager of our termination of the NBIb-1817 for Parkinson’s disease program. The effective date of the termination will be August 2, 2021.
•We depend on our current collaborators for the development and commercialization of several of our products and product candidates and may need to enter into future collaborations to develop and commercialize certain of our product candidates.
•Use of our approved products or those of our collaborators could be associated with side effects or adverse events.
•We face intense competition,have increased the size of our organization and ifwill need to continue to increase the size of our organization. We may encounter difficulties with managing our growth, which could adversely affect our results of operations.
•If we are unable to compete effectively,retain and recruit qualified scientists and other employees or if any of our key senior executives discontinues his or her employment with us, it may delay our development efforts or impact our commercialization of INGREZZA or any of our other products, or any product candidate approved by the demand for our products may be reduced.FDA in the future.
•We currently have no manufacturing capabilities. If third-party manufacturers of INGREZZA ONGENTYSor any of our other products, or any of our product candidates fail to devote sufficient time and resources to our concerns, or if their performance is substandard, our clinical trials and product introductions may be delayed, and our costs may rise.
•We currently depend on a limited number of third-party suppliers. The loss of these suppliers, or delays or problems in the supply of INGREZZA or ONGENTYS,any of our other products, could materially and adversely affect our ability to successfully commercialize INGREZZA or ONGENTYS.
•If we are unable to retain and recruit qualified scientists or if any of our key senior executives discontinues his or her employment with us, it may delay our development efforts or impact our commercialization of INGREZZA, ONGENTYS or any product candidate approved by the FDA.other products.
•We license some of our core technologies and drug candidates from third parties. If we default on any of our obligations under those licenses, or violate the terms of these licenses, we could lose our rights to those technologies and drug candidates or be forced to pay damages.
•If we are unable to protect our intellectual property, our competitors could develop and market products based on our discoveries, which may reduce demand for our products.
•Government and third-party payors may impose sales and pharmaceutical pricing controls on our products, or limit coverage and/or reimbursement for our products or impose policies and/or make decisions that regarding the status of our products that could limit our product revenues and delay sustained profitability.
•Our indebtedness and liabilities could limit the cash flow available for our operations, expose us to risks that could adversely affect our business, financial condition and results of operations.
•We have a history of losses and expect to increase our expenses for the foreseeable future, and we may not be able to sustain profitability.
•We have recently increased the size of our organization and will need to continue to increase the size of our organization. We may encounter difficulties with managing our growth, which could adversely affect our results of operations.
•Our customers are concentrated and therefore the loss of a significant customer may harm our business.
•We may need additional capital in the future. If we cannot raise additional funding, we may be unable to completefund our business plan and our future research, development, of our product candidates or establish commercial and manufacturing capabilities in the future.
•Health care reform measures and other recent legislative initiatives could adversely affect our business.
•If we are unable to protect our intellectual property, our competitors could develop and market products based on our discoveries, which may reduce demand for our products.efforts.
Risks Related to Our Company
We may not be able to continue to successfully commercialize INGREZZA ONGENTYS,or any of our other products, or any of our product candidates if they are approved in the future.
Our ability to produce INGREZZA revenues consistent with expectations ultimately depends on our ability to continue to successfully commercialize INGREZZA and secure adequate third-party reimbursement. Our experience in marketing and selling pharmaceutical products began with INGREZZA’s approval in 2017, when we hired our sales force and established our distribution and reimbursement capabilities, all of which are necessary to successfully commercialize our current and future products. We have continued to invest in our commercial infrastructure and distribution capabilities, including the expansion of our specialty sales force, which we announced in the past four years, including our sales force expansionthird quarter of 2021 and completed in late 2018.April 2022. While our team members and consultants have experience marketing and selling pharmaceutical products, we may face difficulties related to managing the rapid growth of our personnel and infrastructure, and there can be no guarantee that we will be able to maintain the personnel, systems, arrangements and capabilities necessary to continue to successfully commercialize INGREZZA or to successfully commercialize ONGENTYSany of our other products, or any product candidate approved by the FDA, or equivalent foreign authorities, in the future.
In addition, our business has been and may continue to be adversely affected by the effects of health pandemics or epidemics, includingepidemics. In parts of the ongoing COVID-19 pandemic. Mostcountry, some hospitals, community mental health facilities, and other healthcare facilities continue to have implemented policies that limit access of our sales representatives, medical affairs personnel and patients to such facilities. Due to these closures and our work from home decisions, our field force is currently functioning utilizing digital and telephonic engagement tools and tactics,In addition, many healthcare practitioners have adopted telehealth for patient interactions, which may be less effective than our ordinary sales and marketing and medical education programs. The ultimate impact the ability of the COVID-19 pandemic, including any lasting effects on the way we conduct our business, is highly uncertainhealthcare practitioner to screen for and subject to change. If we fail to maintain successful marketing, sales and reimbursement capabilities, our product revenues may suffer.diagnose tardive dyskinesia or chorea associated with Huntington's disease.
If physicians and patients do not continue to accept INGREZZA or do not accept ONGENTYSany of our other products, or our sales and marketing efforts are not effective, we may not generate sufficient revenue.
The commercial success of INGREZZA or ONGENTYSany of our other products will depend upon the acceptance of those products as safe and effective by the medical community and patients.
The market acceptance of INGREZZA or ONGENTYSany of our other products could be affected by a number of factors, including:
•the timing of receipt of marketing approvals for additional indications;
•the safety and efficacy of the products;
•the pricing of our products;
•the availability of healthcare payor coverage and adequate reimbursement for the products;
•public perception regarding any products we may develop;
•the success of existing competitor products addressing our target markets or the emergence of equivalent or superior products; and
•the cost-effectiveness of the products.
If the medical community, patients and patientspayors do not continue to accept our products as being safe, effective, superior and/or cost-effective, we may not generate sufficient revenue.
GovernmentalGovernment and third-party payors may impose sales and pharmaceutical pricing controls on our products or limit coverage and/or reimbursement for our products or impose policies and/or make decisions regarding the status of our productsthat could limit our product revenues and delay sustained profitability.
Our ability to continue to commercialize INGREZZA successfully or to commercialize ONGENTYS,any of our other products will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available. The continuing efforts of government and third-party payors to contain or reduce the costs of health care
healthcare and the price of prescription drugs through various means may reduceimpact our potential revenues. These payors’ efforts could decrease the price that we receive for any products we may develop and sell in the future.
Assuming we obtain coverage for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. Patients who are prescribed medications for the treatment of their conditions, and their prescribing physicians, generally rely on third-party payors to reimburse all or part of the costs associated with their prescription drugs. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the out-of-pocket cost of our products. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available regardless of whether they are approved by the FDA for that particular use. Coverage decisions by payors for our competitors' products may also impact coverage for our products.
Government authorities and other third-party payors are developing increasingly sophisticated methods of controlling healthcare costs, such as by limiting coverage and the amount of reimbursement for particular medications. Further, no uniform policy requirement for coverage and reimbursement for drug products exists among third-party payors in the U.S. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. In addition, communications from government officials, media outlets, and others regarding health carehealthcare costs and pharmaceutical pricing could have a negative impact on our stock price, even if such communications do not ultimately impact coverage or reimbursement decisions for our products.
There may also be significant delays in obtaining coverage and reimbursement for newly approved drugs or indications, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. In addition, we could also be subject to amendments in our rebate agreements with pharmaceutical benefit managers that require us to pay larger rebate amounts or modify our formulary position, which could have a material adverse effect on our business. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future. In addition, gene therapy treatments,For example, government authorities could make a decision that adversely impacts the status of one of our products, which could impact the eligibility and/or the amount of government reimbursement for that product.
As a pharmaceutical manufacturer, we are developing pursuantsubject to our collaborationvarious federal statutes and license agreement with Voyager, face additional uncertaintyregulations requiring the reporting of price data and the subsequent provision of concessions to certain purchasers/payors, including state Medicaid programs. Federal agencies issue guidance to manufacturers related to pricingthe interpretation of laws and reimbursement. As an example, there are a limited numberregulations, and this guidance has changed and may change or be updated over time. In interpreting these laws, regulations and guidance, manufacturers may make reasonable assumptions to fill gaps, and these reasonable assumptions may need to be updated upon issuance of gene therapy products currently approved for coverage and reimbursement by the Centers for Medicare & Medicaid Services, or CMS.additional agency guidance.
If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may notbe unable to successfully commercialize INGREZZA ONGENTYSor any of our other products, or any other product candidate for which we obtain marketing approval.approval in the future. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition. Further, a majority of our current revenue is derived from federal healthcare program payors, including Medicare and Medicaid. Thus, changes in government reimbursement policies, government negotiation of the price of any of products, reductions in payments and/or our suspension or exclusion from participation in federal healthcare programs could have a material adverse effect on our business.
Our business could be adversely affected by the effects of health pandemics or epidemics, includingFurther, during the COVID-19 pandemic, the use of physician telehealth services rapidly increased, fueled by an unprecedented expansion of coverage and reimbursement for telehealth services across public and private insurers. The limitations that telehealth places on the ability to conduct a thorough physical examination may impact the ability of providers to screen for movement disorders, leading to fewer patients being diagnosed and/or treated.
Outside the United States, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. The EU provides options for EU Member States to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. An EU Member State may approve a specific price for the medicinal product, it may refuse to reimburse a product at the price set by the manufacturer or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market.
To obtain reimbursement for our products in regions wheresome European countries, including some EU Member States, we or third partiesmay be required to compile additional data comparing the cost-effectiveness of our products to other available therapies. The Health Technology Assessment (HTA) of medicinal products is becoming an increasingly common part of the pricing and reimbursement procedures in some EU Member States, including those representing the larger markets. The HTA process is the procedure to assess therapeutic, economic and societal impact of a given medicinal product in the national healthcare systems of the individual country. The outcome of an HTA will often influence the pricing and reimbursement status granted to these medicinal products by the competent authorities of individual EU Member States. The extent to which pricing and reimbursement decisions are influenced by the HTA of the specific medicinal product currently varies between EU Member States. In December 2021, Regulation No 2021/2282 on HTA, amending Directive 2011/24/EU, was adopted in the EU. This regulation, which entered into force in January 2022 will apply as of January 2025. The regulation will permit EU Member States to use common HTA tools, methodologies, and procedures across the EU to identify promising technologies early, and continuing voluntary cooperation in other areas. Individual EU Member States will continue to be responsible for assessing non-clinical (e.g., economic, social, ethical) aspects of health technologies, and making decisions on pricing and reimbursement. If we are unable to maintain favorable pricing and reimbursement status in EU Member States for product candidates that we may successfully develop and for which we rely have significant salesmay obtain regulatory approval, any anticipated revenue from and marketing efforts or manufacturing facilities, concentrationsgrowth prospects for those products in the EU could be negatively affected.
In light of clinical trial sites orthe fact that the UK has left the EU, Regulation No 2021/2282 on HTA will not apply in the UK. However, the MHRA is working with UK HTA bodies and other business operations, or materially affect our operations, and at our clinical trial sites, as wellnational organizations, such as the business or operationsScottish Medicines Consortium, the National Institute for Health and Care Excellence, and the All-Wales Medicines Strategy Group, to introduce new pathways supporting innovative approaches to the safe, timely and efficient development of our manufacturers, CROs or other third parties with whom we conduct business.medicinal products.
Our business could be adversely affected by the effects of health pandemics or epidemics in regions where we have concentrations of clinical trial sites or other business operations,Legislators, policymakers and could cause significant disruptionhealthcare insurance funds in the operations of third-party manufacturersEU and CROs upon whom we rely. As a result of the ongoing COVID-19 pandemic, weUK may experience disruptionscontinue to propose and implement cost-containing measures to keep healthcare costs down, particularly due to the financial strain that could severely impact our supply chain, ongoing and future clinical trials and commercialization of INGREZZA and ONGENTYS. For example, the COVID-19 pandemic has resultedplaced on national healthcare systems of European countries. These measures could include limitations on the prices we would be able to charge for product candidates that we may successfully develop and for which we may obtain regulatory approval or the level of reimbursement available for these products from governmental authorities or third-party payors. Further, an increasing number of EU and other foreign countries use prices for medicinal products established in increased travel restrictions andother countries as “reference prices” to help determine the shutdown or delay of business activities in various regions, including San Diego, California, where our headquarters are located. In response to state and local restrictions, we implemented work-from-home policies for all employees except certain key essential members involved in business-critical activities. The effectsprice of the stay at home orderproduct in their own territory. Consequently, a downward trend in prices of medicinal products in some countries could contribute to similar downward trends elsewhere.
We face intense competition, and if we are unable to compete effectively, the demand for our work-from-home policiesproducts may negatively impact productivity, disruptbe reduced.
The biotechnology and pharmaceutical industries are subject to rapid and intense technological change. We face, and will continue to face, competition in the development and marketing of our businessproducts and delayproduct candidates from academic institutions, government agencies, research institutions and biotechnology and pharmaceutical companies.
Competition may also arise from, among other things:
•other drug development technologies;
•methods of preventing or reducing the incidence of disease, including vaccines; and
•new small molecule or other classes of therapeutic agents.
Developments by others (including the development of generic equivalents) may render our product candidates or technologies obsolete or noncompetitive.
We are commercializing and performing research on or developing products for the treatment of several disorders including endometriosis, tardive dyskinesia, chorea associated with Huntington's disease, uterine fibroids, classic congenital adrenal hyperplasia, pain, Parkinson’s disease and other neurology, neuroendocrinology and neuropsychiatry-related diseases and disorders, and there are a number of competitors to our products and product candidates. If one or more of our competitors’ products or programs are successful (including the development of generic equivalents), the market for our products may be reduced or eliminated.
•INGREZZA competes with AUSTEDO® (deutetrabenazine), marketed by Teva Pharmaceuticals Industries, for the treatment of tardive dyskinesia in adults and chorea associated with Huntington's disease. A once-daily dosing of AUSTEDO (AUSTEDO XR) was introduced in February 2023. Additionally, there are a number of commercially available medicines used to treat tardive dyskinesia off-label, such as XENAZINE® (tetrabenazine) and generic equivalents, and various antipsychotic medications (e.g., clozapine), anticholinergics, benzodiazepines (off-label), and botulinum toxin. In addition, there are several programs in clinical development by other companies targeting Huntington's disease.
•ORILISSA and ORIAHNN each compete with several FDA-approved products for the treatment of endometriosis, uterine fibroids, infertility and central precocious puberty. Additionally, there is also competition from surgical intervention, including hysterectomies and ablations. Separate from these options, there are many programs in clinical development which serve as potential future competition. Lastly, there are numerous medicines used to treat the symptoms of disease (vs. endometriosis or uterine fibroids directly) which may also serve as competition: oral contraceptives, NSAIDs and other pain medications, including opioids.
•For CAH, high doses of corticosteroids are the current standard of care to both correct the endogenous cortisol deficiency as well as reduce the excessive ACTH levels. In the U.S. alone, there are more than two dozen companies manufacturing steroid-based products. In addition, there are several programs in clinical development by other companies targeting CAH.
•Our investigational treatments for potential use in epilepsy may in the future compete with numerous approved anti-seizure medications and development-stage programs being pursued by several other companies. Commonly used anti-seizure medications include phenytoin, levetiracetam, brivaracetam, cenobamate, carbamazepine, clobazam, lamotrigine, valproate, oxcarbazepine, topiramate, lacosamide, perampanel and cannabidiol, among others. There are currently no FDA-approved treatments specifically indicated for the early infantile epileptic encephalopathy SCN8A-DEE; however, a number of different anti-seizure medications are currently used in these patient populations.
•Our investigational treatments for potential use in schizophrenia, anhedonia and depression may in the future compete with several development-stage programs being pursued by other companies. Currently, there are no FDA-approved treatments specifically indicated for anhedonia or CIAS; however, there are a number of different anti-psychotic medications currently used in these patient populations.
•Our investigational treatments for potential use in neurology, neuroendocrinology and neuropsychiatry may in the future compete with numerous approved products and development-stage programs being pursued by several other companies.
Compared to us, many of our competitors and potential competitors have substantially greater:
•capital resources;
•sales and marketing experience;
•research and development resources, including personnel and technology;
•regulatory experience;
•preclinical study and clinical testing experience;
•manufacturing, marketing and distribution experience; and
•production facilities.
Moreover, increased competition in certain disorders or therapies may make it more difficult for us to recruit or enroll patients in our clinical programs and timelines, the magnitude of which willtrials for similar disorders or therapies.
depend, in part, on the length and severity of the restrictions and other limitations on our ability to conduct our business in the ordinary course. In addition, we may face several challenges or disruptions upon a return back to the workplace if and when the COVID-19 pandemic subsides, including re-integration challenges by our employees and distractions to management related to such transition. These and similar, and perhaps more severe, disruptions in our operations due to the COVID-19 pandemic could negatively impact our business, operating results and financial condition.
Quarantines, stay at home orders and other state and local restrictions, or the perception that such orders, shutdowns or other restrictions on the conduct of business operations could occur, related to COVID-19 or other infectious diseases, could impact personnel at third-party manufacturing facilities in the United States and other countries, or the availability or cost of materials, which would disrupt our supply chain.
In addition, clinical site initiation and patient enrollment may be delayed due to concerns for patient safety and prioritization of healthcare resources toward the COVID-19 pandemic. Some patients may not be able to comply with clinical trial protocols if quarantines impede patient movement or interrupt healthcare services. Similarly, our ability to recruit and retain patients, principal investigators and site staff (who as healthcare providers may have heightened exposure to COVID-19) may be hindered, which would adversely impact our clinical trial operations. For example, due to the impact of the COVID-19 pandemic, we initially paused enrollment of new patients in several of our clinical trials.Since then, we have begun enrolling patients again in the Phase III study of valbenazine for chorea in HD and the Phase IIa pediatric study of crinecerfont in CAH.However, increases in COVID-19 cases or hospitalizations in the future could cause us to again limit or suspend our patient enrollment and screening activities.
The spread of COVID-19, which has caused a broad impact globally, may materially affect us economically. While the potential economic impact brought by, and the duration of, the COVID-19 pandemic may be difficult to assess or predict, the pandemic is currently resulting in disruption of global financial markets. This disruption, if sustained or recurrent, could make it more difficult for us to access capital, which could in the future negatively affect our liquidity. In addition, a recession or market correction resulting from the spread of COVID-19 could materially affect our business and the value of our common stock.
The global COVID-19 pandemic continues to rapidly evolve. The ultimate impact of the COVID-19 pandemic or a similar health pandemic or epidemic is highly uncertain and subject to change. We do not yet know the full extent of potential delays or impacts on our business, our clinical trials, healthcare systems or the global economy as a whole. These effects could have a material impact on our operations.
Because the development of our product candidates is subject to a substantial degree of technological uncertainty, we may not succeed in developing any of our product candidates.
All of our product candidates are currently in research or clinical development with the exceptions of INGREZZA, which has been approved by the FDA for tardive dyskinesia, ONGENTYS, which has been approved by the FDA for Parkinson’s disease, ORILISSA (partnered with AbbVie), which has been approved by the FDA for the management of moderate to severe endometriosis pain in women, and ORIAHNN (partnered with AbbVie), which has been approved by the FDA for the management of heavy menstrual bleeding associated with uterine fibroids in pre-menopausal women. Only a small number of research and development programs ultimately result in commercially successful drugs. In addition, to date the FDA has granted regulatory approval for only a very limited number of gene therapy products and the clinical development of a gene therapy product may result in unforeseen adverse events. For example, the FDA has placed a clinical hold on the RESTORE-1 study, a Phase II, randomized, placebo-surgery controlled, double-blind, multi-center clinical study of NBIb-1817 in Parkinson’s disease patients, following our submission of an IND safety report related to the observation of MRI abnormalities in some study participants. The clinical implications of this observation are currently unknown and are being evaluated. On February 2, 2021, we notified Voyager of our termination of the NBIb-1817 for Parkinson’s disease program. The effective date of the termination will be August 2, 2021.
Potential products that appear to be promising at early stages of development may not reach the market for a number of reasons. These reasons include the possibilities that the potential products may:
•be found ineffective or cause harmful side effects during preclinical studies or clinical trials;
•fail to receive necessary regulatory approvals on a timely basis or at all;
•be precluded from commercialization by proprietary rights of third parties;
•be difficult to manufacture on a large scale; or
•be uneconomical to commercialize or fail to achieve market acceptance.
If any of our product candidates encounters any of these potential problems, we may never successfully market that product candidate.
Our clinical trials may be delayed for safety or other reasons or fail to demonstrate the safety and efficacy of our product candidates, which could prevent or significantly delay their regulatory approval.
Before obtaining regulatory approval for the sale of any of our potential products, we must subject these product candidates to extensive preclinical and clinical testing to demonstrate their safety and efficacy for humans. Clinical trials are expensive, time-consuming and may take years to complete and the outcomes are uncertain.
In connection with the clinical trials of our product candidates, we face the risks that:
•the FDA or similar foreign regulatory authority may not allow an IND application or foreign equivalent filings required to initiate human clinical studies for our drug candidates or the FDA or similar foreign regulatory authorities may require additional preclinical studies as a condition of the initiation of Phase I1 clinical studies, or additional clinical studies for progression from Phase I1 to Phase II,2, or Phase II2 to Phase III,3, or for NDA approval;
•the product candidate may not prove to be effective or as effective as other competing product candidates;
•we may discover that a product candidate may cause harmful side effects or results of required toxicology or other studies may not be acceptable to the FDA;FDA or similar foreign regulatory authorities;
•theclinical trial results may not replicate the results of earlier, smallerprevious trials;
•the FDA or similar foreign regulatory authorities may require use of new or experimental endpoints that may prove insensitive to treatment effects;
•we or the FDA or similar foreign regulatory authorities may suspend or vary the trials;
•the results may not be statistically significant;
•clinical site initiation or patient recruitment and enrollment may be slower or more difficult than expected;
•the FDA or similar foreign regulatory authorities may not accept the data from any trial or trial site outside of the US;U.S.;
•patients may drop out of the trials;
•unforeseen disruptions or delays may occur, caused by man-made or natural disasters or public health pandemics or epidemics or other business interruptions, including, for example, the COVID-19 pandemic;conflict between Russia and Ukraine and the conflict in the Middle East; and
•regulatory requirements may change.
These risks and uncertainties impact all of our clinical programs and any of the clinical, regulatory or operational events described above could change our planned clinical and regulatory activities. For example, due to the impact of the COVID-19 pandemic, we paused enrollment of new patients in several of our clinical trials,conflict between Russia and increases in COVID-19 cases or hospitalizations in the future could causeUkraine, together with sanctions imposed on Russia, caused us to further limit or suspend all planned clinical trial activities in Russia and Ukraine. As a result, our patient enrollmentplanned clinical development timelines for valbenazine and screening activities.luvadaxistat were significantly delayed while we identified and operationalized alternative clinical trial sites, which we have now done. Additionally, any of these events described above could result in suspension of a program and/or obviate any filings for necessary regulatory approvals.
In addition, late-stage clinical trials are often conducted with patients having the most advanced stages of disease. During the course of treatment, these patients can die or suffer other adverse medical effects for reasons that may not be related to the pharmaceutical agent being tested but which can nevertheless adversely affect clinical trial conduct, completion and results. Any failure or substantial delay in completing clinical trials for our product candidates may severely harm our business.
Even if the clinical trials are successfully completed, we cannot guarantee that the FDA or foreign regulatory authorities will interpret the results as we do, and more trials could be required before we submit our product candidates for approval. To the extent that the results of the trials are not satisfactory to the FDA or foreign regulatory authorities for support of a marketing application, approval of our product candidates may be significantly delayed, or we may be required to expend significant additional resources, which may not be available to us, to conduct additional trials in support of potential approval of our product candidates.
We depend on our current collaborators for the development and commercialization of several of our products and product candidates and may need to enter into future collaborations to develop and commercialize certain of our product candidates.
We depend on our current collaborators for the development and commercialization of several of our products and product candidates and may need to enter into future collaborations to develop and commercialize certain of our product candidates. For example, we collaborate withdepend on AbbVie for the manufacture and commercialization of two of our commercial products, ORILISSA and ORIAHNN and for the continued development of elagolix. We collaborate with MTPC for the commercialization of DYSVAL in Japan and for the continued development and commercialization of INGREZZAvalbenazine for movement disorders in Japan and other select Asian markets. We also rely on BIAL for the commercial supply of ONGENTYS. In addition, we collaborate withOur additional collaborators include Xenon for the development of NBI-921352,Pharmaceuticals, Inc., Idorsia for the development of NBI-827104Pharmaceuticals Ltd., Takeda Pharmaceutical Company Limited, Heptares Therapeutics Limited and Takeda for the development of NBI-1065844.Voyager Therapeutics, Inc.
Our current and future collaborations and licenses could subject us to a number of risks, including:
•strategic collaborators may sell, transfer or divest assets or programs related to our partnered product or product candidates;
•we may be required to undertake the expenditure of substantial operational, financial and management resources;
•we may be required to assume substantial actual or contingent liabilities;
•we may not be able to control the amount and timing of resources that our strategic collaborators devote to the development or commercialization of our products or product candidates;
•we may not be able to influence our strategic collaborator’s decisions regarding the development and collaboration of our partnered product and product candidates, and as a result, our collaboration partners may not pursue or prioritize the development and commercialization of those partnered products and product candidates in a manner that is in our best interest;
•strategic collaborators may select indications or design clinical trials in a way that may be less successful than if we were doing so;
•strategic collaborators may not conduct collaborative activities in a timely manner, provide insufficient funding, terminate a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new version of a product candidate for clinical testing;
•strategic collaborators may not pursue further development and commercialization of products resulting from the strategic collaboration arrangement or may elect to discontinue research and development programs;
•disagreements or disputes may arise between us and our strategic collaborators that result in delays or in costly litigation or arbitration that diverts management’s attention and consumes resources;
•strategic collaborators may experience financial difficulties;
•strategic collaborators may not properly maintain, enforce or defend our intellectual property rights or may use our proprietary information in a manner that could jeopardize or invalidate our proprietary information or expose us to potential litigation;
•we or strategic collaborators could terminate the arrangement (in whole or in part) or allow it to expire, which would delay the development and commercialization, andresult in disagreements or disputes or may increase the cost of developing and commercializing our products or product candidates; and
•strategic collaborators could develop, either alone or with others, products or product candidates that may compete with ours.
If any of these issues arise, it may delay and/or negatively impact the development and commercialization of drug candidates and, ultimately, our generation of product revenues.
We may not be able to successfully commercialize ONGENTYS.
In April 2020, we received FDA approval for ONGENTYS as an adjunctive therapy to levodopa/DOPA decarboxylase inhibitors in adult Parkinson’s disease patients, and in September 2020, we launched the commercial sale of ONGENTYS with our existing INGREZZA infrastructure. The successful commercialization of ONGENTYS is subject to many risks, and there are numerous examples of unsuccessful product launches and failures, including by pharmaceutical companies with more experience and resources than us. If we are unable to effectively train our employees and equip them with effective materials, including medical and sales literature to help them inform and educate health care practitioners about the benefits of ONGENTYS and its proper administration, our commercialization of ONGENTYS may not be successful. Even if we are successful in effectively training and equipping our sales force, there are many factors that could cause the commercialization of ONGENTYS to be unsuccessful, including a number of factors that are outside our control. Health care practitioners may not prescribe ONGENTYS and patients may be unwilling to use ONGENTYS if insurance coverage is not provided or reimbursement is inadequate. In addition, our ability to train our employees and effectively communicate with potential prescribers could be adversely affected by the effects of health pandemics or epidemics, including the ongoing COVID-19 pandemic.
Use of our approved products or those of our collaborators could be associated with side effects or adverse events.
As with most pharmaceutical products, use of our approved products or those of our collaborators could be associated with side effects or adverse events which can vary in severity (from minor adverse reactions to death) and frequency (infrequent or prevalent). Side effects or adverse events associated with the use of our products or those of our collaborators may be observed at any time, including after a product is commercialized, and reports of any such side effects or adverse events may negatively impact demand for our or our collaborators’ products or affect our or our collaborators’ ability to maintain regulatory approval for such products. Side effects or other safety issues associated with the use of our approved products or those of our collaborators could require us or our collaborators to modify or halt commercialization of these products or expose us to product liability lawsuits which will harm our business. We or our collaborators may be required by regulatory agencies to conduct additional studies regarding the safety and efficacy of our products which we have not planned or anticipated. Furthermore, there can be no assurance that we or our collaborators will resolve any issues related to any product related adverse events to the satisfaction of the FDA or any regulatory agency in a timely manner or ever, which could harm our business, prospects and financial condition.
Gene therapy treatments, which we are developing pursuant to our collaboration and license agreement with Voyager, may be perceived as unsafe or may result in unforeseen adverse events. Negative public opinion and increased regulatory scrutiny of gene therapy may adversely affect our ability to initiate or continue clinical development or obtain regulatory approvals for gene therapy product candidates or the commercialization of gene therapy products.
Gene therapy remains a novel technology, with few gene therapy products approved to date in the US. Public perception may be influenced by claims that gene therapy is unsafe, and gene therapy may not gain the acceptance of the public or the medical community. Even if we are able to successfully complete clinical development of a gene therapy product and obtain commercial approval, the success of our collaboration with Voyager will depend upon physicians who specialize in the treatment of genetic diseases targeted by gene therapy product candidates, prescribing treatments that involve the use of our product candidates in lieu of, or in addition to, existing treatments with which they are familiar and for which greater clinical data may be available. More restrictive government regulations, negative public opinion related to gene therapy products, or safety issues identified in our clinical trials may delay or impair the development and commercialization of our gene therapy product candidates or demand for any gene therapy products we develop.
The limited precedent for gene therapy approvals makes it difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for the product candidates we are developing through our collaboration with Voyager.
The FDA has limited experience in the review and approval of gene therapy products. The limited precedent for gene therapy approvals makes it difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for the product candidates we are developing through our collaboration with Voyager.
Regulatory requirements governing gene therapy products have changed frequently and may continue to change in the future. As a result, the regulatory review process may take longer or cost more than we anticipate, including requirements for additional preclinical studies or clinical trials, and delay or prevent approval and commercialization of our gene therapy product candidates we are developing through our collaboration with Voyager. While the FDA has issued draft guidance for the development of gene therapies and proposed rules that would streamline certain requirements to which gene therapies are currently subject, it remains to be seen as to whether such initiatives will ultimately increase the speed of drug development in gene therapies such as the product candidates we are developing through our collaboration with Voyager.
Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market could decrease our ability to generate sufficient product revenue, and our business, financial condition, results of operations and prospects would be harmed. If our gene therapy products are approved but fail to achieve market acceptance among physicians, patients, hospitals, third-party payors or others in the medical community, we will not be able to generate significant revenue.
We face intense competition, and if we are unable to compete effectively, the demand for our products may be reduced.
The biotechnology and pharmaceutical industries are subject to rapid and intense technological change. We face, and will continue to face, competition in the development and marketing of our products and product candidates from academic institutions, government agencies, research institutions and biotechnology and pharmaceutical companies.
Competition may also arise from, among other things:
•other drug development technologies;
•methods of preventing or reducing the incidence of disease, including vaccines; and
•new small molecule or other classes of therapeutic agents.
Developments by others may render our product candidates or technologies obsolete or noncompetitive.
We are commercializing and performing research on or developing products for the treatment of several disorders including endometriosis, tardive dyskinesia, uterine fibroids, essential tremor, classic congenital adrenal hyperplasia, pain, Parkinson’s disease, Friedreich’s ataxia, and other neurological and endocrine-related diseases and disorders, and there are a number of competitors to our products and product candidates. If one or more of our competitors’ products or programs are successful, the market for our products may be reduced or eliminated.
•With respect to INGREZZA for tardive dyskinesia, we compete with Teva Pharmaceutical Industries, which received FDA approval for AUSTEDO to treat tardive dyskinesia in August 2017, and several clinical development-stage programs targeting tardive dyskinesia and related movement disorders. Additionally, there are a number of commercially available medicines used to treat tardive dyskinesia off-label, such as Xenazine (tetrabenazine) and generic equivalents, and various antipsychotic medications (e.g., clozapine), anticholinergics, benzodiazepines (off-label), and botulinum toxin.
•In endometriosis, ORILISSA and ORIAHNN each compete with several FDA-approved products for the treatment of endometriosis, uterine fibroids, infertility, and central precocious puberty. Additionally, there is also competition from surgical intervention, including hysterectomies and ablations. Separate from these options, there are many programs in clinical development which serve as potential future competition. Lastly, there are numerous medicines used to treat the symptoms of disease (vs. endometriosis or uterine fibroids directly) which may also serve as competition: oral contraceptives, NSAIDs and other pain medications including opioids.
•With respect to ONGENTYS for Parkinson’s disease, there are currently two other FDA-approved COMT inhibitors. ONGENTYS competes directly with these two drugs and their generic equivalents. Additionally, there are a number of alternative adjunctive treatment options (FDA-approved and in clinical development) for Parkinson’s patients which compete with ONGENTYS, including various L-dopa preparations, dopamine agonists, MAO-B inhibitors and others. In terms of potential future competition, there are several programs in late-stage clinical development.
•As for CAH, high doses of corticosteroids are the current standard of care to both correct the endogenous cortisol deficiency as well as reduce the excessive ACTH levels. In the U.S. alone, there are more than two dozen companies manufacturing steroid-based products. Additionally, there are several clinical development-stage programs targeting CAH and several companies developing medicinal treatments for CAH.
•Our investigational treatments for potential use in epilepsy may in the future compete with numerous approved anti-seizure medications, or ASMs, and development-stage programs being pursued by several other companies. Commonly used ASMs, among others, include phenytoin, levetiracetam, brivaracetam, cenobamate, carbamazepine, clobazam, lamotrigine, valproate, oxcarbazepine, topiramate, lacosamide, perampanel and cannabidiol. There are currently no FDA-approved treatments specifically indicated for the early infantile epileptic encephalopathies SCN8A-DEE and EE-CSWS; however, a number of different ASMs are currently used in these patient populations.
•The investigational treatment NBI-1065844 for the negative symptoms of schizophrenia may in the future compete with off-label antipsychotic and antidepressant medicines, including ciraprazine, clozapine, fluoxetine, citalopram, sertraline, and amisulpride. In addition, there are several development-stage programs being pursued by other companies, including pimavanserin, roluperidone, RO6889450 and sodium benzoate. Currently, there are no-FDA approved treatments specifically indicated for the negative symptoms of schizophrenia.
•Our investigational treatments for potential use in endocrinology, neurology, and psychiatry, as well as our investigational gene therapies, may in the future compete with numerous approved products and development-stage programs being pursued by several other companies.
Compared to us, many of our competitors and potential competitors have substantially greater:
•capital resources;
•research and development resources, including personnel and technology;
•regulatory experience;
•preclinical study and clinical testing experience;
•manufacturing, marketing and distribution experience; and
•production facilities.
Moreover, increased competition in certain disorders or therapies may make it more difficult for us to recruit or enroll patients in our clinical trials for similar disorders or therapies.
We currently have no manufacturing capabilities. If third-party manufacturers of INGREZZA, ONGENTYS or any of our product candidates fail to devote sufficient time and resources to our concerns, or if their performance is substandard, our clinical trials and product introductions may be delayed, and our costs may rise.
We have in the past utilized, and intend to continue to utilize, third-party manufacturers to produce the drug compounds we use in our clinical trials and for the commercialization of our products. We have limited experience in manufacturing products for commercial purposes and do not currently have any manufacturing facilities. Establishing internal commercial manufacturing capabilities would require significant time and resources, and we may not be able to timely or successfully establish such capabilities. Consequently, we depend on, and will continue to depend on, several contract manufacturers for all production of products for development and commercial purposes, including INGREZZA and ONGENTYS. If we are unable to obtain or retain third-party manufacturers, we will not be able to develop or commercialize our products, including INGREZZA and ONGENTYS. The
manufacture of our products for clinical trials and commercial purposes is subject to specific FDA regulations, including current Good Manufacturing Practice regulations. Our third-party manufacturers, including BIAL and its suppliers, might not comply with FDA regulations relating to manufacturing our products for clinical trials and commercial purposes or other regulatory requirements now or in the future. In addition, the manufacture of gene therapy products, which will be necessary under our collaboration and license agreement with Voyager, is technically complex and necessitates substantial expertise and capital investment. Our reliance on contract manufacturers also exposes us to the following risks:
•contract manufacturers may encounter difficulties in achieving volume production, quality control or quality assurance, and also may experience shortages in qualified personnel. As a result, our contract manufacturers might not be able to meet our clinical schedules or adequately manufacture our products in commercial quantities when required;
•switching manufacturers may be difficult because the number of potential manufacturers is limited. It may be difficult or impossible for us to find a replacement manufacturer quickly on acceptable terms, or at all;
•our contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to successfully produce, store or distribute our products; and
•drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, the U.S. Drug Enforcement Administration, and other agencies to ensure strict compliance with cGMP and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards.
Our current dependence upon third parties for the manufacture of our products may reduce our profit margin, if any, on the sale of INGREZZA, ONGENTYS, or our future products and our ability to develop and deliver products on a timely and competitive basis.
We currently depend on a limited number of third-party suppliers. The loss of these suppliers, or delays or problems in the supply of INGREZZA or ONGENTYS, could materially and adversely affect our ability to successfully commercialize INGREZZA or ONGENTYS.
The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of process controls required to consistently produce the active pharmaceutical ingredients, or API, and the finished product in sufficient quantities while meeting detailed product specifications on a repeated basis. Manufacturers of pharmaceutical products may encounter difficulties in production, such as difficulties with production costs and yields, process controls, quality control and quality assurance, including testing of stability, impurities and impurity levels and other product specifications by validated test methods, compliance with strictly enforced U.S., state, and non-U.S. regulations, and disruptions caused by man-made or natural disasters or public health pandemics or epidemics or other business interruptions, including, for example, the COVID-19 pandemic. We depend on a limited number of suppliers for the production of INGREZZA and its API. If our third-party suppliers for INGREZZA encounter these or any other manufacturing, quality or compliance difficulties, we may be unable to meet commercial demand for INGREZZA, which could materially and adversely affect our ability to successfully commercialize INGREZZA. In addition, under the terms of our agreement with BIAL, although we are responsible for the management of all ONGENTYS commercialization activities, we rely on BIAL and its suppliers to supply all drug product for the commercialization of ONGENTYS. BIAL relies on third-party contract manufacturers to produce ONGENTYS. These contract manufacturers may encounter difficulties in achieving volume production, quality control, or quality assurance. As a result, these contract manufacturers may not be able to adequately produce ONGENTYS in commercial quantities when required, which may impact our ability to deliver ONGENTYS on a timely basis.
In addition, if our suppliers fail or refuse to supply us with INGREZZA or its API for any reason, it would take a significant amount of time and expense to qualify a new supplier. The FDA and similar international regulatory bodies must approve manufacturers of the active and inactive pharmaceutical ingredients and certain packaging materials used in pharmaceutical products. The loss of a supplier could require us to obtain regulatory clearance and to incur validation and other costs associated with the transfer of the API or product manufacturing processes. If there are delays in qualifying new suppliers or facilities or a new supplier is unable to meet FDA or a similar international regulatory body’s requirements for approval, there could be a shortage of INGREZZA, which could
materially and adversely affect our ability to successfully commercialize INGREZZA. If BIAL is unable or refuses to supply us with ONGENTYS drug product for any reason, or does not meet FDA or international regulators’ requirements for approval, we have limited opportunity to qualify a new supplier. This could materially and adversely affect our ability to successfully commercialize ONGENTYS.
The independent clinical investigators and contract research organizations that we rely upon to conduct our clinical trials may not be diligent, careful or timely, and may make mistakes, in the conduct of our trials.
We depend on independent clinical investigators and contract research organizations, or CROs, to conduct our clinical trials under their agreements with us. The investigators are not our employees, and we cannot control the amount or timing of resources that they devote to our programs. If our independent investigators fail to devote sufficient time and resources to our drug development programs, or if their performance is substandard, or not in compliance with Good Clinical Practices, it may delay or prevent the approval of our FDA applications and our introduction of new drugs. The CROs we contract with for execution of our clinical trials play a significant role in the conduct of the trials and the subsequent collection and analysis of data. Failure of the CROs to meet their obligations could adversely affect clinical development of our products. Moreover, these independent investigators and CROs may also have relationships with other commercial entities, some of which may compete with us. If independent investigators and CROs assist our competitors at our expense, it could harm our competitive position.
We do not and will not have access to all information regarding the products and product candidates we licensed to AbbVie.
We do not and will not have access to all information regarding elagolix, including potentially material information about commercialization plans, medical information strategies, clinical trial design and execution, safety reports from clinical trials, safety reports, regulatory affairs, process development, manufacturing and other areas known by AbbVie. In addition, we have confidentiality obligations under our agreement with AbbVie. Thus, our ability to keep our shareholders informed about the status of elagolix will be limited by the degree to which AbbVie keeps us informed and allows us to disclose such information to the public. If AbbVie fails to keep us informed about commercialization efforts related to elagolix, or the status of the clinical development or regulatory approval pathway of other product candidates licensed to it, we may make operational and/or investment decisions that we would not have made had we been fully informed, which may materially and adversely affect our business and operations.
We are subject to ongoing obligations and continued regulatory review for INGREZZA. Additionally, our other product candidates, if approved, could be subject to labeling and other post-marketing requirements and restrictions.
Regulatory approvals for any of our product candidates may be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase IV clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. For example, with respect to the FDA’s approval of INGREZZA for tardive dyskinesia in April 2017, we are subject to certain post-marketing requirements and commitments. In addition, with respect to INGREZZA, and any product candidate that the FDA or a comparable foreign regulatory authority approves, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current Good Manufacturing Practices for any clinical trials that we conduct post-approval. Failure to comply with these ongoing regulatory requirements, or later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, may result in, among other things:
•restrictions on the marketing or manufacturing of the product, changes in the product’s label, withdrawal of the product from the market, or voluntary or mandatory product recalls;
•fines, warning letters or holds on clinical trials;
•refusal by the FDA to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product license approvals;
•product seizure or detention, or refusal to permit the import or export of products; and
•product injunctions or the imposition of civil or criminal penalties.
The occurrence of any of these events may adversely affect our business, prospects and ability to achieve or sustain profitability on a sustained basis.
If we are unable to retain and recruit qualified scientists or if any of our key senior executives discontinues his or her employment with us, it may delay our development efforts or impact our commercialization of INGREZZA, ONGENTYS or any product candidate approved by the FDA.
We are highly dependent on the principal members of our management and scientific staff. The loss of any of these people could impede the achievement of our objectives, including the successful commercialization of INGREZZA, ONGENTYS or any product candidate approved by the FDA. Furthermore, recruiting and retaining qualified scientific personnel to perform research and development work in the future, along with personnel with experience marketing and selling pharmaceutical products, is critical to our success. We may be unable to attract and retain personnel on acceptable terms given the competition among biotechnology, pharmaceutical and health care companies, universities and non-profit research institutions for experienced scientists and individuals with experience marketing and selling pharmaceutical products. We may face particular retention challenges in light of the recent rapid growth in our personnel and infrastructure and the perceived impact of those changes upon our corporate culture. In addition, we rely on a significant number of consultants to assist us in formulating our research and development strategy and our commercialization strategy. Our consultants may have commitments to, or advisory or consulting agreements with, other entities that may limit their availability to us.
If the market opportunities for our products and product candidates are smaller than we believe they are, our revenues may be adversely affected, and our business may suffer.
Certain of the diseases that INGREZZA, ONGENTYS and our other product candidates are being developed to address are in underserved and underdiagnosed populations. Our projections of both the number of people who have these diseases, as well as the subset of people with these diseases who will seek treatment utilizing our products or product candidates, may not be accurate. If our estimates of the prevalence or number of patients potentially on therapy prove to be inaccurate, the market opportunities for INGREZZA, ONGENTYS and our other product candidates may be smaller than we believe they are, our prospects for generating expected revenue may be adversely affected and our business may suffer.
We license some of our core technologies and drug candidates from third parties. If we default on any of our obligations under those licenses, or violate the terms of these licenses, we could lose our rights to those technologies and drug candidates or be forced to pay damages.
We are dependent on licenses from third parties for some of our key technologies. These licenses typically subject us to various commercialization, reporting and other obligations. If we fail to comply with these obligations, we could lose important rights. If we were to default on our obligations under any of our licenses, we could lose some or all of our rights to develop, market and sell products covered by these licenses. For example, BIAL may terminate our license agreement, pursuant to which we have rights to commercialize ONGENTYS, if we fail to use commercially reasonable efforts to comply with specified obligations under the license agreement, or if we otherwise breach the license agreement. In addition, several of our collaboration and license agreements allow our licensors to terminate such agreements if we challenge the validity or enforceability of certain intellectual property rights or if we commit a material breach in whole or in part of the agreement and do not cure such breach within the agreed upon cure period. In addition, if we were to violate any of the terms of our licenses, we could become subject to damages. Likewise, if we were to lose our rights under a license to use proprietary research tools, it could adversely affect our existing collaborations or adversely affect our ability to form new collaborations. We also face the risk that our licensors could, for a number of reasons, lose patent protection or lose their rights to the technologies we have licensed, thereby impairing or extinguishing our rights under our licenses with them.
We have increased the size of our organization and will need to continue to increase the size of our organization. We may encounter difficulties with managing our growth, which could adversely affect our results of operations.
As of December 31, 2023, we had approximately 1,400 full-time employees. Although we have substantially increased the size of our organization, we may need to add additional qualified personnel and resources, especially with the recent increase in the size of our sales force. Our current infrastructure may be inadequate to support our development and commercialization efforts and expected growth. Future growth will impose significant added responsibilities on our organization, including the need to identify, recruit, maintain and integrate additional employees and implement and expand managerial, operational and financial systems and may be costly and take time away from running other aspects of our business, including development and commercialization of our product candidates. For example, we are in the process of implementing a new company-wide enterprise resource planning (ERP) system to streamline certain existing business, operational, and financial processes. This project has required and may continue to require investment of capital and human resources, the re-engineering of processes of our business, and the attention of many employees who would otherwise be focused on other aspects of our business. Any disruptions, delays, or deficiencies in the implementation or design of the ERP system could adversely affect the effectiveness of our internal control over financial reporting or our ability to accurately maintain our books and records, provide accurate, timely and reliable reports on our financial and operating results, or otherwise operate our business. Any of these consequences could have an adverse effect on our results of operations and financial condition.
Our future financial performance and our ability to commercialize INGREZZA and any of our other products, or any of our product candidates that receive regulatory approval in the future, will partially depend on our ability to manage any future growth effectively. In particular, as we commercialize INGREZZA, we will need to support the training and ongoing activities of our sales force and will likely need to continue to expand the size of our employee base for managerial, operational, financial and other resources. To that end, we must be able to successfully:
•manage our development efforts effectively;
•integrate additional management, administrative and manufacturing personnel;
•further develop our marketing and sales organization;
•compensate our employees on adequate terms in an increasingly competitive, inflationary market;
•attract and retain personnel; and
•maintain sufficient administrative, accounting and management information systems and controls.
We may not be able to accomplish these tasks or successfully manage our operations and, accordingly, may not achieve our research, development and commercialization goals. Our failure to accomplish any of these goals could harm our financial results and prospects.
If we are unable to retain and recruit qualified scientists and other employees or if any of our key senior executives discontinues his or her employment with us, it may delay our development efforts or impact our commercialization of INGREZZA or any of our other products, or any product candidate approved by the FDA in the future.
We are highly dependent on the principal members of our management, commercial and scientific staff. The loss of any of these people could impede the achievement of our objectives, including the successful commercialization of INGREZZA or any of our other products, or any product candidate approved by the FDA in the future. Furthermore, recruiting and retaining qualified scientific personnel to perform research and development work in the future, along with personnel with experience marketing and selling pharmaceutical products, is critical to our success. We may be unable to attract and retain personnel on acceptable terms given the competition among biotechnology, pharmaceutical and healthcare companies, universities and non-profit research institutions for experienced scientists and individuals with experience marketing and selling pharmaceutical products. We may face particular retention challenges in light of the recent rapid growth in our personnel and infrastructure and the perceived impact of those changes upon our corporate culture. In addition, we rely on a significant number of consultants to assist us in formulating our research and development strategy and our commercialization strategy. Our consultants may have commitments to, or advisory or consulting agreements with, other entities that may limit their availability to us.
We currently have no manufacturing capabilities. If third-party manufacturers of INGREZZA or any of our other products, or any of our product candidates fail to devote sufficient time and resources to our concerns, or if their performance is substandard, our clinical trials and product introductions may be delayed, and our costs may rise.
We have in the past utilized, and intend to continue to utilize, third-party manufacturers to produce the drug compounds we use in our clinical trials and for the commercialization of our products. We have limited experience in manufacturing products for commercial purposes and do not currently have any manufacturing facilities. Establishing internal commercial manufacturing capabilities would require significant time and resources, and we may not be able to timely or successfully establish such capabilities. Consequently, we depend on, and will continue to depend on, several contract manufacturers for all production of products for development and commercial purposes, including INGREZZA. If we are unable to obtain or retain third-party manufacturers, we will not be able to develop or commercialize our products, including INGREZZA. The manufacture of our products for clinical trials and commercial purposes is subject to specific FDA and equivalent foreign regulations, including current Good Manufacturing Practice regulations. Our third-party manufacturers might not comply with FDA or equivalent foreign regulations relating to manufacturing our products for clinical trials and commercial purposes or other regulatory requirements now or in the future. Our reliance on contract manufacturers also exposes us to the following risks:
•contract manufacturers may encounter difficulties in achieving volume production, quality control or quality assurance, and also may experience shortages in qualified personnel or materials and ingredients necessary to conduct their operations. As a result, our contract manufacturers might not be able to meet our clinical schedules or adequately manufacture our products in commercial quantities when required;
•switching manufacturers may be difficult because the number of potential manufacturers is limited. It may be difficult or impossible for us to find a replacement manufacturer quickly on acceptable terms, or at all;
•our contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to successfully produce, store or distribute our products; and
•drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, the U.S. Drug Enforcement Administration, equivalent foreign regulatory authorities, and other agencies to ensure strict compliance with cGMP and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards.
Our current dependence upon third parties for the manufacture of our products may reduce our profit margin, if any, on the sale of INGREZZA or any of our other products, or our future products and our ability to develop and deliver products on a timely and competitive basis.
We currently depend on a limited number of third-party suppliers. The loss of these suppliers, or delays or problems in the supply of INGREZZA or any of our other products, could materially and adversely affect our ability to successfully commercialize INGREZZA or any of our other products.
The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of process controls required to consistently produce the active pharmaceutical ingredients (API), the finished drug product and packaging in sufficient quantities while meeting detailed product specifications on a repeated basis. Manufacturers of pharmaceutical products may encounter difficulties in production, such as difficulties with production costs and yields, process controls, quality control and quality assurance, including testing of stability, impurities and impurity levels and other product specifications by validated test methods, compliance with strictly enforced U.S., state and non-U.S. regulations, and disruptions or delays caused by man-made or natural disasters, pandemics or epidemics, or other business interruptions. We depend on a limited number of suppliers for the production and packaging of INGREZZA and its API. If our third-party suppliers for INGREZZA encounter these or any other manufacturing, quality or compliance difficulties, we may be unable to meet commercial demand for INGREZZA, which could materially and adversely affect our ability to successfully commercialize INGREZZA.
In addition, if our suppliers fail or refuse to supply us with INGREZZA or its API for any reason, it would take a significant amount of time and expense to qualify a new supplier. The FDA and similar foreign regulatory authorities must approve manufacturers of the active and inactive pharmaceutical ingredients and certain packaging materials used in pharmaceutical products. The loss of a supplier could require us to obtain regulatory clearance and to incur validation and other costs associated with the transfer of the API or product manufacturing processes. If there are delays in qualifying new suppliers or facilities or if a new supplier is unable to meet FDA or a similar foreign regulatory authority’s requirements for approval, there could be a shortage of INGREZZA, which could materially and adversely affect our ability to successfully commercialize INGREZZA.
The independent clinical investigators and contract research organizations that we rely upon to conduct our clinical trials may not be diligent, careful or timely, or may make mistakes in the conduct of our trials.
We depend on independent clinical investigators and CROs to conduct our clinical trials under their agreements with us. The investigators are not our employees, and we cannot control the amount or timing of resources that they devote to our programs. If our independent investigators fail to devote sufficient time and resources to our drug development programs, or if their performance is substandard, or not in compliance with GCPs, it may delay or prevent the approval of our regulatory applications and our introduction of new treatments. The CROs we contract with for execution of our clinical trials play a significant role in the conduct of the trials and the subsequent collection and analysis of data. Failure of the CROs to meet their obligations could adversely affect clinical development of our products. Moreover, these independent investigators and CROs may also have relationships with other commercial entities, some of which may compete with us. If independent investigators and CROs assist our competitors at our expense, it could harm our competitive position.
We are subject to ongoing obligations and continued regulatory review for INGREZZA. Additionally, our other product candidates, if approved, could be subject to labeling and other post-marketing requirements and restrictions.
Regulatory approvals for any of our product candidates may be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. In addition, with respect to INGREZZA, and any product candidate that the FDA or a comparable foreign regulatory authority approves, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with GCPs for any clinical trials that we conduct post-approval. Failure to comply with these ongoing regulatory requirements, or later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, may result in, among other things:
•restrictions on the marketing or manufacturing of the product, changes in the product’s label, withdrawal of the product from the market, or voluntary or mandatory product recalls;
•fines, warning or untitled letters or holds on clinical trials;
•refusal by the FDA or similar foreign regulatory authorities to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product license approvals;
•adverse inspection findings or other activities that temporarily delay manufacture and distribution of our products;
•product seizure or detention, or refusal to permit the import or export of products; and
•product injunctions or the imposition of civil or criminal penalties.
The occurrence of any of these events may adversely affect our business, prospects and ability to achieve or sustain profitability on a sustained basis.
If the market opportunities for our products and product candidates are smaller than we believe they are, our expected revenues may be adversely affected, and our business may suffer.
Certain of the diseases that INGREZZA, crinecerfont, and our other product candidates are being developed to address are in underserved and underdiagnosed populations. Our projections of both the number of people who have these diseases, as well as the subset of people with these diseases who will seek treatment utilizing our products or product candidates, may not be accurate. If our estimates of the prevalence or number of patients potentially on therapy prove to be inaccurate, the market opportunities for INGREZZA, crinecerfont, and our other product candidates may be smaller than we believe they are, our prospects for generating expected revenue may be adversely affected and our business may suffer.
Because our operating results may vary significantly in future periods, our stock price may decline.
Our quarterly revenues, expenses and operating results have fluctuated in the past and are likely to fluctuate significantly in the future. Our financial results are unpredictable and may fluctuate, for among other reasons, due to seasonality and timing of customer purchases and commercial sales of INGREZZA, royalties from out-licensed products, the impact of Medicare Part D coverage, including redesign of the Part D benefit enacted as part of the Inflation Reduction Act, our achievement of product development objectives and milestones, clinical trial enrollment and expenses, research and development expenses and the timing and nature of contract manufacturing, contract research payments, fluctuations in our effective tax rate, and disruptions caused by man-made or natural disasters or public health pandemics or epidemics or other business interruptions, including, for example, the conflict between Russia and Ukraine, or in the Middle East. Because a majority of our costs are predetermined on an annual basis, due in part to our significant research and development costs, small declines in revenue could disproportionately affect financial results in a quarter. Thus, our future operating results and profitability may fluctuate from period to period, and even if we become profitable on a quarterly or annual basis, we may not be able to sustain or increase our profitability. Moreover, as our company and our market capitalization have grown, our financial performance has become increasingly subject to quarterly and annual comparisons with the expectations of securities analysts or investors. The failure of our financial results to meet these expectations, either in a single quarterly or annual period over a sustained period time, could cause our stock price to decline.
Our indebtedness and liabilities could limit the cash flow available for our operations, expose us to risks that could adversely affect our business, financial condition and results of operations.
In May 2017, we sold $517.5 million aggregate principal amount of 2.25% convertible senior notes due May 15, 2024, or the 2024 Notes. In November 2020, we entered into separate, privately negotiated transactions with certain
holders of the 2024 Notes to repurchase $136.2 million aggregate principal amount of the 2024 Notes for an aggregate repurchase price of $186.9 million in cash. AtIn 2022, we entered into separate, privately negotiated transactions with certain holders of the 2024 Notes to repurchase $210.8 million aggregate principal amount of the 2024 Notes for an aggregate repurchase price of $279.0 million in cash. As of December 31, 2020, $381.32023, $170.4 million aggregate principal amount of the 2024 Notes remained outstanding. We may also incur additional indebtedness to meet future financing needs. Our indebtedness could have significant negative consequences for our security holders and our business, results of operations and financial condition by, among other things:
•increasing our vulnerability to adverse economic and industry conditions;
•limiting our ability to obtain additional financing;
•requiring the dedication of a substantial portion of our cash flow from operations to service our indebtedness, which will reduce the amount of cash available for other purposes;
•limiting our flexibility to plan for, or react to, changes in our business;
•diluting the interests of our existing stockholders as a result of issuing shares of our common stock upon conversion of the 2024 Notes; and
•placing us at a possible competitive disadvantage with competitors that are less leveraged than us or have better access to capital.
Our business may not generate sufficient funds, and we may otherwise be unable to maintain sufficient cash reserves, to pay amounts due under the 2024 Notes and any additional indebtedness that we may incur. In addition, our cash needs may increase in the future. In addition, any future indebtedness that we may incur may contain financial and other restrictive covenants that limit our ability to operate our business, raise capital or make payments under our other indebtedness. If we fail to comply with these covenants or to make payments under our indebtedness when due, then we would be in default under that indebtedness, which could, in turn, result in that and our other indebtedness becoming immediately payable in full.
We have a history of losses and expect to increase our expenses for the foreseeable future, and we may not be able to sustain profitability.
Since our inception, we have incurred significant net losses and negative cash flow from operations. AtAs of December 31, 2020,2023, we had an accumulated deficit of $0.7 billion$157.1 million as a result of historical operating losses.
We received FDA approval for INGREZZA for tardive dyskinesia in April 2017 and for ONGENTYS for Parkinson’schorea associated with Huntington's disease in April 2020.August 2023. Our partner AbbVie received FDA approval for ORILISSA for endometriosis in July 2018 and for ORIAHNN for uterine fibroids in May 2020. Additionally, our partner MTPC received Japanese Ministry of Health, Labour and Welfare approval for DYSVAL for the treatment of tardive dyskinesia in March 2022. However, we have not yet obtained regulatory approvals for any other product candidates. Even if we continue to succeed in commercializing INGREZZA, or if we successfully commercialize ONGENTYS or are successful in developing and commercializing any of our other product candidates, we may not be able to sustain profitability. We also expect to continue to incur significant operating and capital expenditures as we:
•commercialize INGREZZA for tardive dyskinesia;
•commercialize ONGENTYS for Parkinson’sdyskinesia and chorea associated with Huntington's disease;
•seek regulatory approvals for our product candidates;candidates or for additional indications for our current products;
•develop, formulate, manufacture and commercialize our product candidates;
•in-license or acquire new product development opportunities;
•implement additional internal systems and infrastructure; and
•hire additional clinical, scientific, sales and marketing personnel.
We expect to increase our expenses and other investments in the coming years as we fund our operations in-licensing or acquisition opportunities, and capital expenditures. While we were profitable for the year ended December 31, 2020,Thus, our future operating results and profitability may fluctuate from period to period due to the factors described above, and we will need to generate significant revenues to achieve and maintain profitability and positive cash flow on a sustained basis. We may not be able to generate these revenues, and we may never achieve
profitability on a sustained basis in the future. Our failure to maintain or increase profitability on a sustained basis could negatively impact the market price of our common stock.
We have recently increased the size of our organization and will need to continue to increase the size of our organization. We may encounter difficulties with managing our growth, which could adversely affect our results of operations.
At December 31, 2020, we had approximately 845 full-time employees. Although we have substantially increased the size of our organization, we may need to add additional qualified personnel and resources, especially now that we have a commercial sales force. Our current infrastructure may be inadequate to support our development and commercialization efforts and expected growth. Future growth will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain and integrate additional employees, and may take time away from running other aspects of our business, including development and commercialization of our product candidates.
Our future financial performance and our ability to commercialize INGREZZA, ONGENTYS and any other product candidates that receive regulatory approval will depend, in part, on our ability to manage any future growth effectively. In particular, as we commercialize INGREZZA and ONGENTYS, we will need to support the training and ongoing activities of our sales force and will likely need to continue to expand the size of our employee base for managerial, operational, financial and other resources. To that end, we must be able to successfully:
•manage our development efforts effectively;
•integrate additional management, administrative and manufacturing personnel;
•further develop our marketing and sales organization; and
•maintain sufficient administrative, accounting and management information systems and controls.
We may not be able to accomplish these tasks or successfully manage our operations and, accordingly, may not achieve our research, development, and commercialization goals. Our failure to accomplish any of these goals could harm our financial results and prospects.
We may be subject to claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is commonplace in the biotechnology industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Because our operating results may vary significantly in future periods, our stock price may decline.
Our quarterly revenues, expenses and operating results have fluctuated in the past and are likely to fluctuate significantly in the future. Our financial results are unpredictable and may fluctuate, for among other reasons, due to seasonality and timing of customer purchases and commercial sales of INGREZZA, impact of the commercial launch of ONGENTYS and ORIAHNN, royalties from out-licensed products, the impact of Medicare Part D coverage, our achievement of product development objectives and milestones, clinical trial enrollment and expenses, research and development expenses and the timing and nature of contract manufacturing, contract research payments, fluctuations in our effective tax rate, and disruptions caused by man-made or natural disasters or public health pandemics or epidemics or other business interruptions, including, for example, the COVID-19 pandemic. A high portion of our costs are predetermined on an annual basis, due in part to our significant research and development costs. Thus, small declines in revenue could disproportionately affect financial results in a quarter. While we were profitable for the year ended December 31, 2020, our future operating results and profitability may fluctuate from period to period, and even if we become profitable on a quarterly or annual basis, we may not be able to sustain or increase our profitability. Moreover, as our company and our market capitalization have grown, our financial performance has become increasingly subject to quarterly and annual comparisons with the expectations of
securities analysts or investors. The failure of our financial results to meet these expectations, either in a single quarterly or annual period over a sustained period time, could cause our stock price to decline.
Changes in tax laws or regulations that are applied adversely to us or our customers may have a material adverse effect on our business, cash flows, financial condition or results of operations.
NewEffective January 1, 2022, legislation enacted in 2017, informally titled the Tax Cuts and Jobs Act of 2017 eliminated the option to deduct research and development expenses for tax purposes in the year incurred and requires taxpayers to capitalize and subsequently amortize such expenses over five years for research activities conducted in the U.S. and over 15 years for research activities conducted outside the U.S. Unless the U.S. Department of the Treasury issues regulations that narrow the application of this provision to a smaller subset of our research and development expenses or the provision is deferred, modified, or repealed by Congress, we expect a material decrease in our cash flows from operations and an offsetting similarly sized increase in our net deferred tax assets over these amortization periods. The actual impact of this provision will depend on multiple factors, including the amount of research and development expenses we will incur and whether we conduct our research and development activities inside or outside the U.S.
In addition, new income, sales, use, excise or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could adversely affect our business and financial condition. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, legislation enacted in 2017, informally titled the Tax Cuts and Jobs Act orof 2017, the TaxCoronavirus Aid, Relief, and Economic Security Act and the Inflation Reduction Act enacted many significant changes to the USU.S. tax laws. Future guidance from the Internal Revenue Service and other tax authorities with respect to the Tax Actsuch legislation may affect us, and certain aspects of the Tax Actsuch legislation could be repealed or modified in future legislation. For example, the Coronavirus Aid, Relief, and Economic Security Act, or the CARES Act, modified certain provisions of the Tax Act. In addition,Furthermore, it is uncertain if and to what extent various states will conform to the Tax Act or any newly enacted federal tax legislation. Changes in corporatelaws. Future tax rates, the realization of net deferred tax assets relating to our operations, the taxation of foreign earnings, and the deductibility of expenses under the Tax Act or future reform legislation could have a material impact on the value of our deferred tax assets, could result in significant one-time charges, and could increase our future USU.S. tax expense.
Our ability to use net operating loss carryforwards and certain other tax attributes may be limited.
Our net operating loss, or NOL, carryforwards generated in tax years ending on or prior to December 31, 2017, are only permitted to be carried forward for 20 years under applicable US tax law. Under the Tax Cut and Jobs Act, as modified by the CARES Act, our federal NOLs generated in tax years beginning after December 31, 2017, may be carried forward indefinitely, but the deductibility of such federal NOLs in tax years beginning after December 31, 2020, is limited to 80% of taxable income. It is uncertain if and to what extent various states will conform to the Tax Cut and Jobs Act or the CARES Act. In addition, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in its equity ownership over a three-year period, the corporation’s ability to use itscertain pre-change NOL carryforwardsfederal tax attributes such as research and other pre-changedevelopment tax attributescredits to offset its post-change income or taxes may be limited. WeBased on completed Section 382 analysis done annually, we do not believe we have experienced any previous ownership changes, but the determination is complex and there can be no assurance we are correct. Furthermore, we may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control.
As a result, our pre-2018 NOL carryforwards may expire prior to being used and our NOL carryforwards generated in tax years beginning after December 31, 2020, will be subject to a percentage limitation and, if we undergo an ownership change (or if we previously underwent such an ownership change), our ability to use all of our pre-change NOLs and other pre-change tax attributes (such as research tax credits) to offset our post-change income or taxes may be limited.
Similar provisions of state tax law may also apply to limit our use of accumulated state tax attributes.attributes, including net operating loss (NOL) carryforwards. In addition, at the state level, there may be periods during which the use of NOLs or credits is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. For example, California passed legislation imposing limits on the usability of California state NOLs and certain tax credits in tax years beginning after 2019 and before 2023. As a result, we may be unable to use all or a material portion of our NOLs, research and development credits, and other tax attributes, which could adversely affect our future cash flows.
Our effective tax rate may fluctuate, and we may incur obligations in tax jurisdictions in excess of accrued amounts.
Our effective tax rate is derived from a combination of applicable tax rates in the various places that we operate. In preparing our financial statements, we estimate the amount of tax that will become payable in each of such places.place. Nevertheless, our effective tax rate may be different than experienced in the past due to numerous factors, including the impact of stock-based compensation, changes in the mix of our profitability from statejurisdiction to state,jurisdiction, the results of examinations and audits of our tax filings, our inability to secure or sustain acceptable agreements with tax authorities, changes in accounting for income taxes and changes in tax laws. Any of these factors could cause us to experience an effective tax rate significantly different from previous periods or our current expectations and may result in tax obligations in excess of amounts accrued in our financial statements.
In addition, on December 31, 2020, we determined, based on our facts and circumstances, that it was more likely than not that a substantial portion of our deferred tax assets would be realized and, as a result, substantially all of our valuation allowance against our deferred tax assets was released. Therefore, beginning in 2021, we expect to commence recording income tax expense at an estimated tax rate that will likely approximate statutory tax rates, which would result in a significant reduction in our net income and net income per share.
The price of our common stock is volatile.
The market prices for securities of biotechnology and pharmaceutical companies historically have been highly volatile, and the market for these securities has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. The COVID-19 pandemic, for example, has negatively affected the stock market and investor sentiment and has resulted in significant volatility.volatility, as has the applicability of the Medicare drug price negotiation provisions in the Inflation Reduction Act. Furthermore, especially as we and our market capitalization have grown, the price of our common stock has been increasingly affected by quarterly and annual comparisons with the valuations and recommendations of the analysts who cover our business. If our results do not meet these analysts’ forecasts, the expectations of our investors or the financial guidance we provide to investors in any period, which is based on assumptions that may be incorrect or that may change from quarter to quarter, the market price of our common stock could decline. Over the course of the last twelve12 months, the price of our common stock has ranged from approximately $72$89 per share to approximately $136$143 per share.
The market price of our common stock may fluctuate in response to many factors, including:
•sales of INGREZZA and ORILISSA;
•impact of the commercial launch of ONGENTYS and ORIAHNN;
•the status and cost of our post-marketing commitments for INGREZZA and ONGENTYS;other products;
•the results of our clinical trials;
•reports of safety issues related to INGREZZA, ONGENTYS, ORILISSA, ORIAHNN, DYSVAL, or ORIAHNN;any of our other products;
•developments concerning new and existing collaboration agreements;
•announcements of technological innovations or new therapeutic products by us or others;others, including our competitors;
•general economic and market conditions, including economic and market conditions affecting the biotechnology industry;
•developments in patent or other proprietary rights;
•developments related to the FDA;FDA, CMS and foreign regulatory agencies;
•government regulation, including the Inflation Reduction Act;
•future sales of our common stock by us or our stockholders;
•comments by securities analysts;
•additions or departures of key personnel;
•fluctuations in our operating results;
•potential litigation matters;
•government regulation;
•government and third-party payor coverage and reimbursement;
•failure of any of our product candidates, if approved, to achieve commercial success;
•disruptions caused by man-made or natural disasters, or public health pandemics or epidemics or other business interruptions, including, for example, the COVID-19 pandemic;pandemic and the conflict between Russia and Ukraine; and
•public concern as to the safety of our drugs.
In addition, we are a member of the S&P MidCap 400 index. If we cease to be represented in the S&P MidCap 400 index, or other indexes or indexed products, as a result of our market capitalization falling below the threshold for inclusion in the index, certain institutional shareholders may, due to their internal policies and investment guidelines, be required to sell their shareholdings. Such sales may result in further negative pressure on our stock price and, when combined with reduced trading volume and liquidity, could adversely affect the value of your investment and your ability to sell your shares.
Our customers are concentrated and therefore the loss of a significant customer may harm our business.
We have entered into agreements for the distribution of INGREZZA with a limited number of specialty pharmacy providers and a specialty distributor,distributors, and all of our product sales of INGREZZA are to these customers. TwoFour of these customers
represented approximately 86%91% of our total product revenuesales for the year ended December 31, 20202023 and a significant majorityapproximately 98% of our accounts receivable balance atas of December 31, 2020.2023. If any of these significant customers becomes subject to bankruptcy, is unable to pay us for our products or is acquired by a company that wants to terminate the relationship with us, or if we otherwise lose any of these significant customers, our revenue, results of operations and cash flows would be adversely affected. Even if we replace the loss of a significant customer, we cannot predict with certainty that such transition would not result in a decline in our revenue, results of operations and cash flows.
We may need additional capital in the future. If we cannot raise additional funding, we may be unable to completefund our business plan and our future research, development, of our product candidates or establish commercial and manufacturing capabilities in the future.efforts.
We may require additionalOur future funding to continue our researchrequirements will depend on many factors and development programs, to conduct preclinical studies and clinical trials, for operating expenses, to pursue regulatory approvals for our product candidates, for the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims, if any, and the cost of product in-licensing and any possible acquisitions. In addition, we may requireneed to raise additional fundingcapital to establishfund our business plan and our future research, development, commercial and manufacturing and marketing capabilities in the future. We believe that our existing capital resources, together with investment income, and future payments due under our strategic alliances, will be sufficient to satisfy our current and projected funding requirements for at least the next twelve months. However, these resources might be insufficient to conduct research and development programs, the cost of product in-taking and possible acquisitions, fully commercialize products and operate the company to the full extent currently planned. If we cannot obtain adequate funds, we may be required to curtail significantly our commercial plans or one or more of our research and development programs or obtain funds through additional arrangements with corporate collaborators or others that may require us to relinquish rights to some of our technologies or product candidates.efforts.
Our future capital requirements will depend on many factors, including:
•the commercial success of INGREZZA, ONGENTYS, ORILISSA, ORIAHNN, DYSVAL, and/or ORIAHNN;any of our other products;
•debt serviceservices obligations on the 2024 Notes;
•continued scientific progress in our R&D and clinical development programs;
•the magnitude and complexity of our research and development programs;
•progress with preclinical testing and clinical trials;
•the time and costs involved in obtaining regulatory approvals;
•the costscost involved in filing and pursuing patent applications, enforcing patent claims, or engaging in interference proceedings or other patent litigation;
•costs associated with securing adequate coverage and reimbursement for our products;
•competing technological and market developments;
•the establishment of additional strategic alliances;
•developments related to any future litigation;
•the cost of commercialization activities and arrangements, including advertising campaigns;
•the cost of manufacturing of our product candidates;
•the impact of the COVID-19 pandemic or a future pandemic or epidemic on our business; and
•the cost of any strategic alliances, collaborations, product in-licensing, and any possibleor acquisitions.
We intend to seek additional funding through strategic alliances and may seek additional funding through public or private sales of our securities, including equity securities. In addition, during the second quarter of 2017, we issued the 2024 Notes and we have previously financed capital purchases and may continue to pursue opportunities to obtain additional debt financing in the future. In November 2020, we entered into separate, privately negotiated transactions with certain holders of the 2024 Notes to repurchase $136.2 million aggregate principal amount of the 2024 Notes for an aggregate repurchase price of $186.9 million in cash. AtIn 2022, we entered into separate, privately negotiated transactions with certain holders of the 2024 Notes to repurchase $210.8 million aggregate principal amount of the
2024 Notes for an aggregate repurchase price of $279.0 million in cash. As of December 31, 2020, $381.32023, $170.4 million aggregate principal amount of the 2024 Notes remained outstanding. Additional equity or debt financing might not be available on reasonable terms, if at all. In addition, disruptions due to the COVID-19 pandemic could make it more difficult for us to access capital. Any additional equity financings will be dilutive to our stockholders and any additional debt financings may involve operating covenants that restrict our business.
Compliance with changing regulation of corporate governance and public disclosure may result in additional expenses.
Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Dodd-Frank Wall Street Reform and Consumer Protection Act, new SEC regulations and Nasdaq rules, are creating uncertainty for companies such as ours. These laws, regulations and standards are subject to varying interpretations in some cases due to their lack of specificity, and as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies, which could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We are committed to maintaining high standards of corporate governance and public disclosure. As a result, our efforts to comply with evolving laws, regulations and standards have resulted in, and are likely to continue to result in, increased selling, general and administrative expenses and management time related to compliance activities. If we fail to comply with these laws, regulations and standards, our reputation may be harmed and we might be subject to sanctions or investigation by regulatory authorities, such as the SEC. Any such action could adversely affect our financial results and the market price of our common stock.
Increasing use of social media could give rise to liability and result in harm to our business.
Our employees are increasingly utilizing social media tools and our website as a means of communication. Despite our efforts to monitor social media communications, there is risk that the unauthorized use of social media by our employees to communicate about our products or business, or any inadvertent disclosure of material, nonpublic information through these means, may result in violations of applicable laws and regulations, which may give rise to liability and result in harm to our business. In addition, there is also risk of inappropriate disclosure of sensitive information, which could result in significant legal and financial exposure and reputational damages that could potentially have a material adverse impact on our business, financial condition and results of operations. Furthermore, negative posts or comments about us or our products on social media could seriously damage our reputation, brand image and goodwill.
We may be subject to claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is commonplace in the biotechnology industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Our business could be adversely affected by the effects of health pandemics or epidemics, which could also cause significant disruption in the operations of third-party manufacturers, CROs, or other third parties upon whom we rely.
Our business could be adversely affected by the effects of health pandemics or epidemics, which could also cause significant disruption in the operations of third-party manufacturers, CROs and other third parties upon whom we rely. As a result, we may experience disruptions that could severely impact our supply chain, ongoing and future clinical trials and commercialization of INGREZZA or any of our other products. In response to the COVID-19 pandemic, we implemented a remote work model for all employees except certain key essential members involved in business-critical activities. Our employees have resumed in-person interactions and have returned to the office under flexible work guidelines. However, a remote work model may nevertheless need to be reinstated at some point in the future. The effects of a remote and flexible work model may negatively impact productivity, disrupt our business and delay our clinical programs and timelines, the magnitude of which will depend on our ability to conduct our business in the ordinary course. Remote work may also create increased risks to our information technology systems and data, as more of our employees utilize network connections, computers and devices outside our premises or network, including working at home, while in transit and in public locations. In addition, we may face several challenges or disruptions upon a return back to the workplace, including re-integration challenges by our employees and distractions to management related to such transition. These and similar, and perhaps more severe, disruptions in our operations could negatively impact our business, operating results and financial condition.
In addition, clinical site initiation and patient enrollment may be delayed due to concerns for patient safety. Some patients may not be able to comply with clinical trial protocols and our ability to recruit and retain patients, principal investigators and site staff may be hindered, which would adversely impact our clinical trial operations.
The ultimate effects of health pandemics or epidemics is highly uncertain and subject to change and these effects could have a material impact on our operations, or the operations of third parties on whom we rely.
Risks Related to Our Industry
Health careIf we are unable to protect our intellectual property, our competitors could develop and market products based on our discoveries, which may reduce demand for our products.
Our success will depend on our ability to, among other things:
•obtain patent protection for our products;
•preserve our trade secrets;
•prevent third parties from infringing upon our proprietary rights; and
•operate without infringing upon the proprietary rights of others, both in the U.S. and internationally.
Because of the substantial length of time and expense associated with bringing new products through the development and regulatory approval processes in order to reach the marketplace, the pharmaceutical industry places considerable importance on obtaining patent and trade secret protection for new technologies, products and processes. Accordingly, we intend to seek patent protection for our proprietary technology and compounds. However, we face the risk that we may not obtain any of these patents and that the breadth of claims we obtain, if any, may not provide adequate protection of our proprietary technology or compounds. Additionally, if our employees, commercial collaborators or consultants use generative artificial intelligence (AI) technologies to develop our proprietary technology and compounds, it may impact our ability to obtain or successfully defend certain intellectual property rights.
We also rely upon unpatented trade secrets and improvements, unpatented know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, through confidentiality agreements with our commercial collaborators, employees and consultants. We also have invention or patent assignment agreements with our employees and some, but not all, of our commercial collaborators and consultants. However, if our employees, commercial collaborators or consultants breach these agreements, we may not have adequate remedies for any such breach, and our trade secrets may otherwise become known or independently discovered by our competitors.
In addition, although we own a number of patents, the issuance of a patent is not conclusive as to its validity or enforceability, and third parties may challenge the validity or enforceability of our patents. We cannot assure you how much protection, if any, will be given to our patents if we attempt to enforce them and they are challenged in court or in other proceedings. It is possible that a competitor may successfully challenge our patents or that challenges will result in limitations of their coverage. Moreover, competitors may infringe our patents or successfully avoid them through design innovation. In addition, potential competitors have in the past and may in the future file an abbreviated new drug application (ANDA) with the FDA seeking approval to market a generic version of our products, or our competitors’ products, before the expiration of the patents covering our products or our competitors’ products, as applicable. To prevent infringement or unauthorized use, we have in the past and may in the future need to file infringement claims, which are expensive and time-consuming. Refer to Note 13 to the consolidated financial statements for a description of our legal proceedings related to intellectual property matters. In addition, in an infringement proceeding a court may decide that a patent of ours or a patent of a competitor is not valid or is unenforceable or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover its technology. Derivation proceedings declared by the U.S. Patent and Trademark Office may be necessary to determine the priority of inventions with respect to our patent applications (or those of our licensors) or a patent of a competitor. Litigation or derivation proceedings may fail and, even if successful, may result in substantial costs and be a distraction to management. Litigation or derivation proceedings, including proceedings of a competitor, may also result in a competitor entering the marketplace faster than expected. We cannot assure you that we will be able to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the U.S.
Enacted healthcare reform, drug pricing measures and other recent legislative initiatives could adversely affect our business.
The business and financial condition of pharmaceutical and biotechnology companies are affected by the efforts of governmentalgovernment and third-party payors to contain or reduce the costs of health carehealthcare and to lower drug prices. In the U.S., comprehensive health care reformdrug pricing legislation was enacted by the Federal government and we expect that there will continue to be a number of federal and state proposals to implementimplements, for the first time, government control over the pricing of prescription pharmaceuticals. In addition, increasing emphasis on reducing the cost of health care in the U.S. will continue to put pressure on the rate of adoption and pricing ofcertain prescription pharmaceuticals. Moreover, in some foreign jurisdictions, pricing of prescription pharmaceuticals is alreadyalso subject to government control. Additionally, other federal and state legislationlaws impose obligations on manufacturers of pharmaceutical products, among others, related to product trackingdisclosure of new drug products introduced to the market and tracing. Amongincreases in drug prices above a specified threshold.
For example, in August 2022, President Biden signed into law the Inflation Reduction Act of 2022, or the IRA, which, among other things: (1) directs the Secretary of the HHS to negotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare; (2) redesigns the Medicare Part D prescription drug benefit to lower patient out-of-pocket costs and increase manufacturer liability; and (3) requires drug manufacturers to pay rebates on drugs whose prices increase greater than the rate of inflation. The IRA also extends enhanced subsidies for individuals purchasing health insurance coverage in the ACA marketplaces through plan year 2025 and beginning in 2025, eliminates the “donut hole” under the Medicare Part D program and creates a new, permanent cap on beneficiary out-of-pocket spending, in addition to a newly established manufacturer discount program. The IRA permits HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years. HHS has issued and updated and will continue to issue and update guidance as these programs are implemented. These provisions take effect progressively starting in 2023. On August 29, 2023, HHS announced the list of the first 10 drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is currently subject to legal challenges. It is currently uncertain how the IRA will be implemented over time; however, it is likely to have a significant impact on the pharmaceutical industry and prescription drug pricing.
While the IRA drug price negotiation program targets high-expenditure drugs that have been on the market for several years without generic or biosimilar competition, we believe we will qualify for the small biotech exception from negotiation that is set to expire in 2029. However, the qualification for this exception is subject to various requirements and there is no assurance that we will continue to qualify for this exemption in the future. Further, the loss of this newexception or the potential loss of this exception, including as a result of a potential acquisition or strategic transaction, could have an adverse impact on our business.
Prior to the IRA’s enactment, the most significant recent federal legislation manufacturers are required to provide certain information regardingimpacting the drug product provided to individuals and entities to which product ownership is transferred, label drug product with a product identifier, and keep certain records regarding distribution of the drug product. Further, under this new legislation, manufacturers will have drug product investigation, quarantine, disposition, notification and purchaser license verification responsibilities related to counterfeit, diverted, stolen, and intentionally adulterated products, as well as products that are the subject of fraudulent transactions or which are otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death.
Additionally,pharmaceutical industry occurred in March 2010, Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectivelywhen the ACA was signed into law, whichlaw. The ACA was intended to broaden access to health insurance and reduce the number of uninsured individuals, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add transparency requirements for the healthcare and health insurance industries, impose taxes and fees
on the health industry and impose additional health policy reforms. Among the provisions of the ACA of importance to our potential drug candidates are:
•an annual, nondeductible fee on any entity that manufactures, or imports, specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs;
•an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for branded and generic drugs, respectively;
•a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected;
•extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations;
•expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability;
•a new Medicare Part D coverage gap discount program, in which manufacturers must now agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D;
•expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; and
•a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research.
There remain legal and political challenges to certain aspects of the ACA. Since January 2017, several executive orders and other directives designed to delay, circumvent, or loosen certain requirements mandated by the ACA have been put into place. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. Legislation enacted in 2017, informally titled the Tax Cuts and Jobs Act includes a provision that repealed, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. In addition, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminated the health insurer tax. On December 14, 2018, a U.S. District Court Judge in Texas ruled that the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as part of the Tax Cuts and Jobs Act. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit ruled that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. On March 2, 2020, the United States Supreme Court is currently reviewing this case, although it is unclear when a decision will be made. It is also unclear how such litigation will impact the ACA and our business.
Other legislative changes have been proposed and adopted since the ACA was enacted. These changes include aggregate reductions to Medicare payments to providers of up to 2% per fiscal year pursuant to the Budget Control Act of 2011, which began in 2013 and, due to subsequent legislative amendments to the statute, including the Bipartisan BudgetInfrastructure Investment and Jobs Act and Consolidated Appropriations Act of 2018,2023, will remain in effect through 2030, except for a temporary suspension from May 1, 2020 through May 31, 2021 due to the COVID-19 pandemic, unless additional Congressional action is taken.until 2032. The American Taxpayer Relief Act of 2012, among other things, further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Additional changes that may affect our business include the expansion of new programs such as Medicare payment for performance initiatives for physicians under the Medicare Access and CHIP Reauthorization Act of 2015, which
ended the use of the statutory formula, also referred to as the Sustainable Growth Rate, for clinician payment and established a quality payment incentive program, also referred to as the Quality Payment Program. This program provides clinicians with two ways to participate, including through the Advanced Alternative Payment Models, or APMs, and the Merit-based Incentive Payment System, or MIPS. In November 2019, CMS issued a final rule finalizing the changes to the Quality Payment Program. At this time, it remains unclear how the introduction of the Quality Payment Program will impact overall physician reimbursement.
Also, there has been heightened governmental scrutiny recently over pharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. Such scrutiny has resulted in several recent Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. At the federal level, the Trump administration’s budget proposal for the 2021 fiscal year includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. Further, the Trump administration released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. On July 24, 2020 and September 13, 2020, the Trump administration announced several executive orders related to prescription drug pricing that attempt to implement several of the administration’s proposals. As a result, the FDA released a final rule on September 24, 2020, effective November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. Further, on November 20, 2020, the U.S. Department of Health and Human Services, or HHS, finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers. On November 20, 2020, CMS issued an interim final rule implementing President Trump’s Most Favored Nation executive order, which would tie Medicare Part B payments for certain physician-administered drugs to the lowest price paid in other economically advanced countries, effective January 1, 2021. On December 28, 2020, the U.S. District Court in Northern California issued a nationwide preliminary injunction against implementation of the interim final rule. It is unclear whether the Biden administration will work to reverse these measures or pursue similar policy initiatives. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
We expect that For example, on January 5, 2024, the ACA, as well as otherFDA approved Florida’s SIP proposal to import certain drugs from Canada for specific state healthcare reform measures that mayprograms. It is unclear how this program will be adoptedimplemented, including which drugs will be chosen, and whether it will be subject to legal challenges in the future,United States or Canada. Other states have also submitted SIP proposals that are pending review by the FDA. Any such approved importation plans, when implemented, may result in more rigorous coverage criterialower drug prices for products covered by those programs. Further, certain states through legislation have created a state PDAB to help control costs of drugs for that state. The functions of the PDABs vary by state, and lower reimbursement, and in additional downward pressuremay include among other things, recommending or setting upper limits on the price that we receivethe state pays for any approved product. In particular, itcertain drugs, performing drug affordability reviews, and advising state lawmakers on additional ways to reduce the state’s drug spending. It is possible that additional governmental action isthe actions taken in response toby the COVID-19 pandemic. Any reduction in reimbursement from Medicare or other government-funded programsPDABs may result in a similar reductionlower prices for certain drug products sold in payments from private payors. their in states.
The implementation of these cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain sustained profitability or commercialize our drugs.drugs, particularly since the majority of our current revenue is derived from federal healthcare programs, including Medicare and Medicaid.
Proposed healthcare reform, drug pricing measures and other prospective legislative initiatives could adversely affect our business.
We expect that there will continue to be a number of federal and state proposals to implement additional government controls over the pricing of prescription pharmaceuticals. In addition, increasing emphasis on reducing the cost of healthcare in the U.S. will continue to put pressure on the pricing and reimbursement of prescription pharmaceuticals. For example, in response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the Center for Medicare and Medicaid Innovation which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether the models will be utilized in any health reform measures in the future.
In addition, certain jurisdictions outside of the U.S., including the EU, have instituted price ceilings on specific products and therapies, as described further in the risk factor titled “Government and third-party payors may impose sales and pharmaceutical pricing controls on our products or limit coverage and/or reimbursement for our products or impose policies and/or make decisions regarding the status of our products that could limit our product revenues and delay sustained profitability.”
We are currently unable to predict what other additional legislation or regulation, if any, relating to the health carehealthcare industry may be enacted in the future or what effect recently enacted federal or equivalent foreign legislation or any such additional legislation or regulation would have on our business. The pendency or approval of such proposals or reforms could result in a decrease in our stock price or limit our ability to raise capital or to enter into collaboration agreements for the further development and commercialization of our programs and products.
Any relationships with healthcare professionals, principal investigators, consultants, customers (actual and potential) and third-party payors in connection with our current and future business activities are and will continue to be subject, directly or indirectly, to federal and state healthcare laws. If we are unable to comply, or have not fully complied, with such laws, we could face penalties, contractual damages, reputational harm, diminished profits and future earnings and curtailment or restructuring of our operations.
Our business operations and activities may be directly, or indirectly, subject to various federal and state healthcare laws, including without limitation, fraud and abuse laws, false claims laws, data privacy and security laws, as well as transparency laws regarding payments or other items of value provided to healthcare providers. These laws may restrict or prohibit a wide range of business activities, including, but not limited to, research, manufacturing, distribution, pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. These laws may impact, among other things, our current activities with principal investigators and research subjects, as well as current and future sales, marketing, patient co-payment assistance and education programs.
Such laws include:
•the federal Anti-Kickback Statute which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid;
•the federal civil and criminal false claims laws, including the federal civil False Claims Act, and civil monetary penalties laws,Civil Monetary Penalties Laws, which impose criminal and civil penalties against individuals or entities for, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government;
•HIPAA, which imposes criminal and civil liability for, among other things, executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters;
•HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act,HITECH and its implementing regulations, which also imposes obligations, including mandatory contractual terms, on covered entities, including certain healthcare providers, health plans and healthcare clearinghouses, as well as their business associates and their covered subcontractors, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
•the federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to CMS information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), other healthcare professionals (such as physician assistants and nurse practitioners) and teaching hospitals, and applicable manufacturers and applicable group purchasing organizations to report annually to CMS ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding its relationships with physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetistsmembers; and certified nurse midwives during the previous year; and
•analogous state, local and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third party payors, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures or drug pricing; state laws that require disclosure of price increases above certain identified thresholds as well as of new commercial launches in the state; state laws that create Prescription Drug Price Affordability Boards to review or attempt to cap drug spending; state and local laws that require the registration of pharmaceutical sales representatives; state and local “drug take
back” laws and regulations; and state and foreign laws governing the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Efforts to ensure that our business arrangements will comply with applicable healthcare laws may involve substantial costs. While our interactions with healthcare professionals, including our speaker programs and other arrangements such as our contributions to patient assistance programs, have been structured to comply with these laws and related guidance, it is possible that governmental and enforcement authorities will conclude that our business practices, or a rogue employee’s activities, may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws. For example, we maintain a patient assistance program to help eligible patients afford our products. These and other types of programs have become the subject of governmental scrutiny, and numerous organizations, including pharmaceutical manufacturers, have been subject to litigation, enforcement actions and settlements related to their patient assistance programs. If our operations or activities are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to, without limitation, significant civil, criminal and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, contractual damages, reputational harm, diminished profits and future earnings and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate.
In addition, any sales of our product once commercialized outside the U.S. will also likely subject us to foreign equivalents of the healthcare laws mentioned above, among other foreign laws.
We could face liability if a regulatory authority determines that we are promoting INGREZZA ONGENTYS or any of our product candidates that receives regulatory approval, for “off-label” uses.
A company may not promote “off-label” uses for its drug products. An off-label use is the use of a product for an indication that is not described in the product’s FDA-approved label in the U.S. or for uses in other jurisdictions that differ from those approved by the applicable regulatory agencies. Physicians, on the other hand, may prescribe products for off-label uses. Although the FDA and other regulatory agencies do not regulate a physician’s choice of drug treatment made in the physician’s independent medical judgment, they do restrict promotional communications from companies or their sales force with respect to off-label uses of products for which marketing clearance has not been issued. However, companies may share truthful and not misleading information that is otherwise consistent with a product’s FDA approved labeling. A company that is found to have promoted off-label use of its product may be subject to significant liability, including civil and criminal sanctions. We intend to comply with the requirements and restrictions of the FDA and other regulatory agencies with respect to our promotion of our products, including INGREZZA and ONGENTYS, but we cannot be sure that the FDA or other regulatory agencies will agree that we have not violated their restrictions. As a result, we may be subject to criminal and civil liability. In addition, our management’s attention could be diverted to handle any such alleged violations.
If the FDA or any other governmental agency, including equivalent foreign authorities, initiates an enforcement action against us, or if we are the subject of a qui tam suit brought by a private plaintiff on behalf of the government, and it is determined that we violated prohibitions relating to the promotion of products for unapproved uses, we could be subject to substantial civil or criminal fines or damage awards and other sanctions such as consent decrees and corporate integrity agreements pursuant to which our activities would be subject to ongoing scrutiny and monitoring to ensure compliance with applicable laws and regulations. Any such fines, awards or other sanctions would have an adverse effect on our revenue, business, financial prospects and reputation.
If we are unable to protect our intellectual property, our competitors could develop and market products based on our discoveries, which may reduce demand for our products.
Our success will depend on our ability to, among other things:
•obtain patent protection for our products;
•preserve our trade secrets;
•prevent third parties from infringing upon our proprietary rights; and
•operate without infringing upon the proprietary rights of others, both in the U.S. and internationally.
Because of the substantial length of time and expense associated with bringing new products through the development and regulatory approval processes in order to reach the marketplace, the pharmaceutical industry places
considerable importance on obtaining patent and trade secret protection for new technologies, products and processes. Accordingly,If our information technology systems, those third parties upon which we intendrely, or our data is or were compromised, we could experience adverse impacts resulting from such compromise, including, but not limited to, seek patent protection forinterruptions to our proprietary technology and compounds. However, we face the riskoperations such as our clinical trials, claims that we breached our data protection obligations, harm to our reputation, regulatory investigations or actions, litigation, fines and penalties, and a loss of customers or sales.
We are increasingly dependent on information technology systems and infrastructure, including mobile technologies, to operate our business. In the ordinary course of our business, we and the third parties upon which we rely, collect, receive, store, process, generate, disclose, make accessible, protect, dispose of, transmit, use, safeguard, share and transfer, or collectively, process, confidential and sensitive electronic information on our networks and in our data centers. This information includes, among other things, de-identified or pseudonymous sensitive personal data (including health data), our intellectual property and proprietary information, the confidential information of our collaborators and licensees, and the personal data of our employees. It is important to our operations and business strategy that this electronic information remains secure and is perceived to be secure. The size and complexity of our information technology systems, and those of third-party vendors with whom we contract, and the volume of data we retain, make such systems potentially vulnerable to a variety of evolving threats, including but not limited to social-engineering attacks (including through deep fakes, which may be increasingly more difficult to identify as fake, and phishing attacks), malicious code, malware (such as malicious code, adware, and command and control (C2)), denial-of-service attacks, credential harvesting, personnel misconduct or error, ransomware attacks, supply-chain attacks, software bugs, server malfunctions, software or hardware failures, loss of data or other information technology assets, attacks enhanced or facilitated by AI, telecommunications failures, and other similar threats. Cyber-attacks, malicious internet-based activity, online and offline fraud, and other similar activities threaten the confidentiality, integrity, and availability of our sensitive information and information technology systems, and those of the third parties upon which we rely. Such threats continue to rise, are increasingly difficult to detect, and come from a variety of sources, including traditional computer “hackers,” threat actors, “hacktivists,” organized criminal threat actors, personnel (such as through theft or misuse), sophisticated nation states, and nation-state-supported actors (also referred to as APTs). Some actors now engage and are expected to continue to engage in cyber-attacks, including without limitation nation-state actors for geopolitical reasons and in conjunction with military conflicts and defense activities. During times of war and other major conflicts, we and the third parties upon which we rely may be vulnerable to a heightened risk of these attacks, including retaliatory cyber-attacks, which could materially disrupt our systems and operations, as well as our ability to conduct clinical trials. Ransomware attacks are also becoming increasingly prevalent and severe, and can lead to significant interruptions in our operations (including our ability to conduct clinical trials), loss of sensitive data (including related to our clinical trials) and income, reputational harm, and diversion of funds. To alleviate the financial, operational and reputational impact of a ransomware attack, it may be preferable to make extortion payments, but we may be unwilling or unable to do so (including, for example, if applicable laws or regulations prohibit such payments). Similarly, supply chain attacks have increased in frequency and severity, and we cannot guarantee that third parties in our supply chain have not been compromised or that they do not contain exploitable defects, vulnerabilities, or bugs that could result in a breach of or disruption to our information technology systems and infrastructure or the information technology systems and infrastructure of third parties that support our operations. Remote work has become more common and has increased risks to our information technology systems and data, as more of our employees work from home, utilizing network connections, computers and devices outside our premises, including at home, while in transit or in public locations.
Additionally, natural disasters, public health pandemics or epidemics, terrorism, war and geopolitical conflicts, and telecommunication and electrical failures may result in damage to or the interruption or impairment of key business processes, or the loss or corruption of confidential information, including intellectual property, proprietary business information and personal data.
Future or past business transactions (such as acquisitions or integrations) could expose us to additional cybersecurity risks and vulnerabilities, as our systems could be negatively affected by vulnerabilities present in acquired or integrated entities’ systems and technologies. Furthermore, we may discover security issues that were not found during due diligence of such acquired or integrated entities, and it may be difficult to integrate companies into our information technology environment and security program.
As cyber threats continue to evolve, we may be required to expend significant additional resources to continue to modify or enhance our protective measures or to investigate and remediate any information security vulnerabilities or modify our business activities (including our clinical trial activities) to try to protect against security incidents.
We take steps designed to detect, mitigate, and remediate vulnerabilities in our information security systems (such as our hardware and/or software, including that of third parties upon which we rely). We may not, obtain anyhowever, detect and remediate all such vulnerabilities including on a timely basis. Further, we may experience delays in developing and deploying remedial measures and patches designed to address identified vulnerabilities. Vulnerabilities could be exploited and result in a security incident.
We rely on third-party service providers and technologies to operate critical business systems to process sensitive information in a variety of these patentscontexts, including, without limitation, cloud-based infrastructure, data center facilities, encryption and that the breadth of claims we obtain, if any, may not provide adequate protection of our proprietaryauthentication technology, or compounds.
employee email and other functions. We also rely upon unpatented trade secretson third-party service providers to provide other products, services, parts, or otherwise to operate our business, including clinical trial sites and improvements, unpatented know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, through confidentiality agreements with our commercial collaborators, employeesinvestigators, contractors, manufacturers, suppliers and consultants. We also have invention or patent assignment agreements with our employeesOur ability to monitor these third parties’ information security practices is limited, and some, but not all, of our commercial collaborators and consultants. However, if our employees, commercial collaborators or consultants breach these agreements, wethird parties may not have adequate remedies forinformation security measures in place. If our third-party service providers or CROs experience a security incident or other interruption, we could experience adverse consequences. In addition, supply-chain attacks have increased in frequency and severity, and we cannot guarantee that third parties’ infrastructure in our supply chain or our third-party partners’ supply chains have not been compromised or otherwise subject to a security incident. While we may be entitled to damages if our third-party service providers fail to satisfy their privacy or security-related obligations to us, any award may be insufficient to cover our damages, or we may be unable to recover such breach,award.
Although to our knowledge we, or the third parties upon who we rely, have not experienced a security incident or disruption to date that is material to us, we and our trade secretsvendors have been, either directly or indirectly, the target of cybersecurity incidents and expect them to continue. While we have implemented security measures designed to protect our data security and information technology systems, such measures may otherwise become knownnot prevent such events. Furthermore, while we have implemented and are planning to implement redundancies designed to avoid interruptions to our operations, not all potential events can be anticipated and interruptions to our operations could lead to decreased productivity.
If we (or a third party upon whom we rely) experience a security incident, ransomware attack or independently discovered byare perceived to have experienced a security incident, we may experience adverse consequences. Such consequences may include: government enforcement actions (for example, investigations, fines, penalties, audits and inspections); additional reporting requirements and/or oversight; restrictions on processing sensitive information (including personal data); litigation (including class claims); indemnification obligations; negative publicity; reputational harm (including but not limited todamage to our competitors.patient, partner, or employee relationships); monetary fund diversions; diversion of management’s attention; interruptions in our operations (including availability of data, loss of connectivity to our network or internet); financial loss (including decreased productivity resulting from interruptions in our operations); and other similar harms. Similarly, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. In addition, theft of our intellectual property or proprietary business information could require substantial expenditures to remedy. Applicable data privacy and security obligations may also require us to notify relevant stakeholders, including affected individuals, customers, regulators, and investors, of security incidents. Such disclosures are costly, and the disclosure or the failure to comply with such requirements could lead to adverse consequences.
Our contracts, with for example third parties or CROs, may not contain limitations of liability, and even where they do, there can be no assurance that limitations of liability in our contracts are sufficient to protect us from liabilities, damages, or claims related to our data privacy and security obligations. We also cannot be sure that our insurance coverage will be adequate or sufficient to protect us from or to mitigate liabilities arising out of our privacy and security practices, that such coverage will continue to be available on commercially reasonable terms or at all, or that such coverage will pay future claims.
In addition although we ownto experiencing a number of patents, the issuance of a patent is not conclusive as to its validity or enforceability, andsecurity incident, third parties may challenge the validitygather, collect, or enforceabilityinfer sensitive information about us from public sources, data brokers, or other means that reveals competitively sensitive details about our organization and could be used to undermine our competitive advantage or market position. Additionally, our sensitive information could be leaked, disclosed, or revealed as a result of our patents. We cannot assure you how much protection, if any, will be given to our patents if we attempt to enforce them and they are challenged in court or in other proceedings. It is possible that a competitor may successfully challengeconnection with our patentsemployees’, personnel’s, or that challenges will result in limitationsvendors’ potential use of their coverage. Moreover, competitors may infringe our patents or successfully avoid them through design innovation. To prevent infringement or unauthorized use, we may need to file infringement claims, which are expensive and time-consuming. In addition, in an infringement proceeding a court may decide that a patent of ours is not valid or is unenforceable or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover its technology. Interference proceedings declared by the U.S. Patent and Trademark Office may be necessary to determine the priority of inventions with respect to our patent applications or those of our licensors. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and be a distraction to management. We cannot assure you that we will be able to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the U.S.generative AI technologies.
If we fail to obtain or maintain orphan drug designation or other regulatory exclusivity for some of our product candidates, our competitive position would be harmed.
In addition to any patent protection, we rely on forms of regulatory exclusivity to protect our products such as orphan drug designation. A product candidate that receives orphan drug designation can benefit from a streamlined regulatory process as well as potential commercial benefits following approval. Currently, this designation provides market exclusivity in the U.S. and the EU for seven years and tenEU for 10 years respectively, if a product is the first such product approved for such orphan indication. This market exclusivity does not, however, pertain to indications other than those for which the drug was specifically designated in the approval, nor does it prevent other types of drugs from receiving orphan designations or approvals in these same indications. Further, even after an orphan drug is approved, the FDA can subsequently approve the same drug for the same condition if the FDA concludes that the new drug is clinically superior to the orphan product or a market shortage occurs.
In the EU, orphan exclusivity may be reduced to six years if the drug no longer satisfies the original designation criteria or can be lost altogether if the marketing authorization holder consents to a second orphan drug application or cannot supply enough drug, or when a second applicant demonstrates its drug is “clinically superior” to the original orphan drug.
If we do not have adequate patent protection for our products, then the relative importance of obtaining regulatory exclusivity is even greater. We may not be successful obtaining orphan drug designations for any indications and, even if we succeed, such product candidates with such orphan drug designations may fail to achieve FDA approval. Even if a product candidate with orphan drug designation may receive marketing approval from the FDA, it may fail to result in or maintain orphan drug exclusivity upon approval, which would harm our competitive position.
The technologies we use in our research as well as the drug targets we select may infringe the patents or violate the proprietary rights of third parties.
We cannot assure you that third parties will not assert patent or other intellectual property infringement claims against us or our collaborators with respect to technologies used in potential products. If a patent infringement suit were brought against us or our collaborators, we or our collaborators could be forced to stop or delay developing, manufacturing or selling potential products that are claimed to infringe a third party’s intellectual property unless that party grants us or our collaborators rights to use its intellectual property. In such cases, we could be required to obtain licenses to patents or proprietary rights of others in order to continue to commercialize our products. However, we may not be able to obtain any licenses required under any patents or proprietary rights of third parties
on acceptable terms, or at all. Even if our collaborators or we were able to obtain rights to the third party’s intellectual property, these rights may be non-exclusive, thereby giving our competitors access to the same intellectual property. Ultimately, we may be unable to commercialize some of our potential products or may have to cease some of our business operations as a result of patent infringement claims, which could severely harm our business.
Our business operations may subject us to disputes, claims and lawsuits, which may be costly and time-consuming and could materially and adversely impact our financial position and results of operations.
From time to time, we may become involved in disputes, claims and lawsuits relating to our business operations. In particular, we may face claims related to the safety of our products, intellectual property matters, employment matters, tax matters, commercial disputes, competition, sales and marketing practices, environmental matters, personal injury, insurance coverage and acquisition or divestiture-related matters. Any dispute, claim or lawsuit may divert management’s attention away from our business, we may incur significant expenses in addressing or defending any dispute, claim or lawsuit, and we may be required to pay damage awards or settlements or become subject to equitable remedies that could adversely affect our operations and financial results. For example, we recently settled various intellectual property litigation matters against potential competitors related to INGREZZA. Refer to Note 13 to the consolidated financial statements for a more detailed description of these matters.
Litigation related to these disputes may be costly and time-consuming and could materially and adversely impact our financial position and results of operations if resolved against us. In addition, the uncertainty associated with litigation could lead to increased volatility in our stock price.
Our employees, independent contractors, principal investigators, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees and independent contractors, such as principal investigators, consultants, commercial partners and vendors, or by employees of our commercial partners could include failures to comply with FDA regulations, to provide accurate information to the FDA, to comply with manufacturing standards we have established, to comply with federal and state healthcare fraud and abuse laws, to report financial information or data accurately, to maintain the confidentiality of our trade secrets or the trade secrets of our commercial partners, or to disclose unauthorized activities to us. In particular, sales, marketing and other business arrangements in the healthcare industry are subject to extensive laws intended to prevent fraud, kickbacks, self-dealing and other abusive practices. Employee and independent contractor misconduct could also involve the improper use of individually identifiable information, including, without limitation, information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. Any action against our employees, independent contractors, principal investigators, consultants, commercial partners or vendors for violations of these laws could result in significant civil, criminal and administrative penalties, fines and imprisonment.
We face potential product liability exposure far in excess of our insurance coverage.
The use of any of our potential products in clinical trials, and the sale of any approved products, including INGREZZA, and ONGENTYS, may expose us to liability claims. These claims might be made directly by consumers, health carehealthcare providers, pharmaceutical companies or others selling our products. We have product liability insurance coverage for both our clinical trials in the amount of $45.0 million per occurrence and $45.0 million in the aggregate. In addition, we have product liability insuranceas well as related to the sale of INGREZZA and ONGENTYS in the amount of $45.0 million per occurrence and $45.0 million in the aggregate.amounts consistent with customary industry practices. However, our insurance may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability from any current or future clinical trials or approved products. A successful product liability claim, or series of claims, brought against us would decrease our cash reserves and could cause our stock price to fall. Furthermore, regardless of the eventual outcome of a product liability claim, any product liability claim against us may decrease demand for our approved products, including INGREZZA, and ONGENTYS, damage our reputation, result in regulatory investigations that could require costly recalls or product modifications, cause clinical trial participants to withdrawal, result in costs to defend the related litigation, decrease our revenue, and divert management’s attention from managing our business.
Our activities involve hazardous materials, and we may be liable for any resulting contamination or injuries.
Our research activities involve the controlled use of hazardous materials. We cannot eliminate the risk of accidental contamination or injury from these materials. If an accident occurs, a court may hold us liable for any resulting damages, which may harm our results of operations and cause us to use a substantial portion of our cash reserves, which would force us to seek additional financing.
Cyber security breaches and other disruptions could compromise our information, including the theft of our intellectual property, and could expose us to liability, which would cause our business and reputation to suffer.
We are increasingly dependentsubject to stringent and changing obligations related to data privacy and information security. Our actual or perceived failure to comply with such obligations could have a material adverse effect on information technology systems and infrastructure, including mobile technologies, to operate our business. reputation, business, financial condition or results of operations.
In the ordinary course of our business, we collect and storeprocess confidential and sensitive electronic information, including personal data, proprietary and confidential business data, trade secrets, intellectual property, data we collect about clinical trial participants in connection with clinical trials, and sensitive third-party data, on our networks and in our data centers. ThisWe are subject to numerous federal, state, local and foreign laws, orders, codes, regulations and regulatory guidance regarding privacy, data protection, information includes, among other things, our intellectual property and proprietary information, the confidential information of our collaborators and licensees,security and the personally identifiableprocessing of personal information (including clinical trial data), the number and scope of which are expanding, changing, subject to differing applications and interpretations, and may be inconsistent among jurisdictions. Our data processing activities may also subject us to other data privacy and security obligations, such as industry standards, external and internal privacy and security policies, contracts and other obligations that govern the processing of data by us and by third parties on our employees. It is important to our operations and business strategy that this electronic information remains secure and is perceived to be secure. The size andbehalf.
complexityLaws regarding privacy, data protection, information security and the processing of personal data are becoming increasingly common in the U.S. at both the federal and state level. Additionally, in the past few years, numerous U.S. states—including California, Virginia, Colorado, Connecticut, and Utah—have enacted comprehensive privacy laws that impose certain obligations on covered businesses, including providing specific disclosures in privacy notices and affording residents with certain rights concerning their personal data. As applicable, such rights may include the right to access, correct, or delete certain personal data, and to opt-out of certain data processing activities, such as targeted advertising, profiling, and automated decision-making. The exercise of these rights may impact our business and ability to provide our products and services. Certain states also impose stricter requirements for processing certain personal data, including sensitive information, technology systems,such as conducting data privacy impact assessments. These state laws allow for statutory fines for noncompliance. For example, the California Consumer Privacy Act, as amended by the California Privacy Rights Act of 2020 (CPRA) (collectively, CCPA), requires businesses to provide specific disclosures in privacy notices, and thosehonor requests of third-party vendorsCalifornia residents to exercise certain privacy rights. The CCPA allows for fines for noncompliance (up to $7,500 per intentional violation). Although some U.S. comprehensive privacy laws and the CCPA exempt some data processed in the context of clinical trials, these laws may increase compliance costs and potential liability with respect to other personal data we may maintain about California residents. Other states have also enacted data privacy laws and we expect more jurisdictions to pass similar laws in the future. These developments may further complicate compliance efforts, and may increase legal risk and compliance costs for us and the third parties upon whom we contract,rely.
Additionally, HIPAA, as amended by HITECH, imposes specific requirements relating to the privacy, security, and transmission of individually identifiable health information.
Laws in Europe regarding privacy, data protection, information security and the volumeprocessing of data we retain, make such systems potentially vulnerable to breakdown, malicious intrusion, security breaches and other cyber-attacks. Additionally, natural disasters, public health pandemics or epidemics (including, for example, the COVID-19 pandemic), terrorism, war and telecommunication and electrical failures may result in damage to or the interruption or impairment of key business processes, or the loss or corruption of confidential information, including intellectual property, proprietary business information and personal information. Information security risks have significantly increased in recent years in part due to the proliferation of new technologies and the increased sophistication and activities of organized crime, hackers, terrorists and other external parties, including foreign private parties and state actors. A security breach or privacy violation that leads to disclosure or modification of or prevents access to personally identifiable information or other protected information could harm our reputation, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, require us to verify the correctness of database contents and otherwise subject us to liability under laws and regulations that protect personal data resulting in increased costs or loss of revenue. Similarly, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval effortshave also been significantly reformed and significantly increase our costs to recover or reproduce the data. Additionally, theft of our intellectual property or proprietary business information could require substantial expenditures to remedy. If we are unable to prevent such security breaches or privacy violations or implement satisfactory remedial measures, our operations could be disrupted, and we may suffer loss of reputation, financial loss and other regulatory penalties because of lost or misappropriated information. In addition, these breaches and other inappropriate access can be difficult to detect, and any delay in identifying them may lead to increased harm of the type described above. Moreover, the prevalent use of mobile devices that access confidential information increases the risk of data security breaches, which could lead to the loss of confidential information, trade secrets or other intellectual property. As cyber threats continue to evolve, we may be required to expend significant additional resources to continue to modify or enhance our protective measures or to investigate and remediate any information security vulnerabilities. While we have implemented security measures to protect our data security and information technology systems, such measures may not prevent such events. Significant disruptions of our information technology systems or breaches of data security could have a material adverse effect on our business, financial condition and results of operations.
Compliance with evolving U.S. and global privacy and data security requirements could result in additional costs and liabilities to us or inhibit our ability to collect and process data globally, and the failure to comply with such requirements could have a material adverse effect on our business, financial condition or results of operations.
The regulatory framework for the collection, use, safeguarding, sharing, transfer and other processing of information worldwide is rapidly evolving and is likely to remain uncertain for the foreseeable future.undergo reform. For example, the EU’s General Data Protection Regulation or(EU GDPR) and the UK’s GDPR imposes(UK GDPR) (collectively, GDPR) impose strict obligations onrequirements for processing the processing of personal data including personal health data,of individuals located, respectively, within the European Economic Area (EEA) and the free movement of such data.UK. The GDPR applies to any company established in the EU as well as any company outside the EU that processes personal data in connection with the offering of goods or services to individuals in the EU or the monitoring of their behavior. The GDPR enhancesprovides for enhanced data protection obligations for processors and controllers of personal data, including, for example, obligations relating to: processing health and other sensitive data; obtaining consent of individuals; providing notice to individuals regarding data processing activities; responding to data subject requests; taking certain measures when engaging third-party processors; notifying data subjects and regulators of data breaches; and implementing safeguards to protect the security and confidentiality of personal data; and transferring personal data to countries outside the EU, including the U.S.data. The GDPR imposesimpose substantial fines for breaches of data protection requirements, whichrequirements. For example, under the GDPR, such fines can be up to four percent of global revenue or 20 million euros under the EU GDPR / 17.5 million pounds sterling under the UK GDPR, whichever is greater in either case, and it also confers aallow for private rightlitigation related to processing of action onpersonal data brought by classes of data subjects for breaches of dataor consumer protection requirements.organizations authorized at law to represent their interests. The GDPR and other changes in laws or regulations associated with the enhanced protection of certain types of sensitive data, such as EU regulations governing clinical trial data and other healthcare data, could require us to change our business practices or lead to government enforcement actions, private litigation or significant penalties against us and could have a material adverse effect on our business, financial condition or results of operations.
Additionally,We may be subject to additional foreign data laws. For example, in Canada, the California Consumer PrivacyPersonal Information Protection and Electronic Documents Act (PIPEDA) and various related provincial laws, as well as Canada’s Anti-Spam Legislation (CASL), may apply to our operations. As another example, the General Data Protection Law, Lei Geral de Proteção de Dados Pessoais (LGPD) (Law No. 13,709/2018), may apply to our operations. The LGPD broadly regulates processing personal data of individuals in Brazil and imposes compliance obligations and penalties comparable to those of the EU GDPR. We also target customers in Asia and may be subject to new and emerging data privacy regimes in Asia, including Japan’s Act on the Protection of Personal Information and Singapore’s Personal Data Protection Act.
In the ordinary course of business, we may transfer personal data from Europe and other jurisdictions to the U.S. or CCPA, which went into effect in 2020, created new individual privacy rights for California consumers (as that word is broadly definedother countries. Certain jurisdictions have enacted data localization laws and cross-border personal data transfers laws. For example, countries in the law)EEA and placesthe UK have significantly restricted the transfer of personal data to the U.S. and other countries, whose privacy laws it generally believes are inadequate. Although there are currently various mechanisms that may be used to transfer personal data from the EEA and UK to the U.S. in compliance with law, such as the EEA standard contractual clauses, the UK’s International Data Transfer Agreement / Addendum, and the EU-U.S. Data Privacy Framework and the UK extension thereto (which allows for transfers for to relevant U.S.-based organizations who self-certify compliance and participate in the Framework), these mechanisms are subject to legal challenges, and there is no assurance that we can satisfy or rely on these measures to lawfully transfer personal data to the U.S. If we cannot implement a valid compliance mechanism for cross-border personal data transfers or if the requirements for a legally-compliant transfer are too onerous, we may face increased exposure to regulatory actions, substantial fines and injunctions against processing or transferring personal data from Europe or elsewhere. The inability to import personal data to the U.S. may significantly and negatively impact our business operations, including by limiting our ability to conduct clinical trial activities in Europe and elsewhere; limiting our ability to collaborate with parties subject to European and other data protection laws or requiring us to increase our personal data processing capabilities in Europe and/or elsewhere at significant expense. Other jurisdictions may adopt similarly stringent interpretations of their data localization and cross-border data transfer laws. Additionally, companies that transfer personal data out of the EEA and UK to other jurisdictions, particularly to the United States, are subject to increased scrutiny from regulators, individual litigants, and activist groups. Some European regulators have ordered certain companies to suspend or permanently cease certain transfers out of Europe for allegedly violating the GDPR’s cross-border data transfer limitations.
Our employees and personnel may use generative AI technologies to perform some of their work, and the disclosure and use of personal information data in generative AI technologies is subject to various privacy laws and other privacy obligations. Governments have passed and are likely to pass additional laws regulating generative AI. Our use of this technology could result in additional compliance costs, regulatory investigations and actions, and consumer lawsuits. Furthermore, any use of generative AI to develop our proprietary technology and compounds may also impact our ability to obtain or successfully defend certain intellectual property rights. If we are unable to use generative AI, it could make our business less efficient and result in competitive disadvantages.
In addition to data privacy and security laws, we may contractually be subject to industry standards adopted by industry groups and, we are, or may become subject to such obligations on entities handlingin the future. We are also bound by contractual obligations related to data privacy and security, and our efforts to comply with such obligations may not be successful. We publish privacy policies, marketing materials and other statements regarding data privacy and security. If these policies, materials or statements are found to be deficient, lacking in transparency, deceptive, unfair, or misrepresentative of our practices, we may be subject to investigation, enforcement actions by regulators or other adverse consequences.
Our obligations related to data privacy and security (and consumers’ data privacy expectations) are quickly changing in an increasingly stringent fashion and creating uncertainty. These obligations may be subject to differing applications and interpretations, which may be inconsistent among jurisdictions or in conflict. Preparing for and complying with these obligations requires us to devote significant resources (including, without limitation, financial and time-related resources). These obligations may necessitate changes to our information technologies, systems and practices and those of any third parties that process personal data of consumers or households. For example, theon our behalf. In addition, these obligations may even require us to change our business model.
CCPA requires covered companiesAlthough we endeavor to provide additional disclosurescomply with all applicable data privacy and security obligations, we may at times fail (or be perceived to California consumers, and provideshave failed) to do so. Moreover, despite our efforts, our personnel or third-parties upon whom we rely may fail to comply such consumers with new rights, such as the ability to opt out of certain disclosures of personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for data breachesobligations that is expected to increase data breach litigation. The CCPA may increaseimpacts our compliance costsposture. If we fail, or are perceived to have failed, to address or comply with data privacy and security obligations, we could face significant consequences. These consequences may include, but are not limited to, government enforcement actions, litigation(including class claims), additional reporting requirements and/or oversight, bans on processing personal data, imprisonment of company officials, and orders to destroy or not use personal data. In particular, plaintiffs have become increasingly more active in bringing privacy-related claims against companies, including class claims and mass arbitration demands. Some of these claims allow for the recovery of statutory damages on a per violation basis, and, if viable, carry the potential liability. Some observersfor monumental statutory damages, depending on the volume of data and the number of violations. Any of these events could have noted that the CCPA could mark the beginninga material adverse effect on our reputation, business, financial condition or results of a trend toward more stringent privacy legislation in the U.S., which could increase our potential liability and adversely affect our business.operations.
Item 1B. Unresolved Staff Comments
None.
Item 1C. Cybersecurity
Risk Management and Strategy.We rely on information technology and data to operate our business and develop, market, and deliver our therapies to our customers. We have implemented and maintain various information security processes designed to identify, assess and manage material risks from cybersecurity threats to critical computer networks, third party hosted services, communications systems, hardware, lab equipment, software, and our critical data includes confidential, personal, proprietary, and sensitive data (collectively “Information Assets”). Accordingly, we maintain certain risk assessment processes intended to identify cybersecurity threats, determine their likelihood of occurring, and assess potential material impact to our business. Based on our assessment, we implement and maintain risk management processes designed to protect the confidentiality, integrity, and availability of our Information Assets and mitigate harm to our business.
The Company’s general risk management program is designed to manage identified material risks, which would include material cybersecurity risks.
We engage in processes designed to identify such threats by, among other things, monitoring the threat environment using manual and automated tools, subscribing to reports and services that identify cybersecurity threats, analyzing reports of threats and actors, conducting scans of the threat environment, evaluating our and our industry’s risk profile, evaluating threats reported to us, coordinating with law enforcement concerning threats, conducting threat assessments for internal and external threats, and conducting vulnerability assessments to identify vulnerabilities.
We rely on a multidisciplinary team (including from our information security function, management, and third party service providers, as described further below) to assess how identified cybersecurity threats could impact our business. These assessments may leverage, among other processes, industry tools and metrics designed to assist in the assessment of risks from such cybersecurity threats.
Depending on the environment, we implement and maintain various technical, physical and organizational measures designed to manage and mitigate material risks from cybersecurity threats to our Information Assets. The cybersecurity risk management and mitigation measures we implement for certain of our Information Assets include: policies and procedures designed to address cybersecurity threats, including an incident response plan, vulnerability management policy, and disaster recovery/business continuity plans; incident detection and response tools; internal and/or external audits to assess our exposure to cybersecurity threats, environment, compliance with risk mitigation procedures, and effectiveness of relevant controls; documented risk assessments; implementation of security standards/certifications; credit and background checks on our and/or third parties’ personnel; encryption of data; network security controls; threat modeling; data segregation; physical and electronic access controls; physical security; asset management, tracking and disposal; systems monitoring; vendor risk management program; employee security training; penetration testing; red/blue team exercises; cyber insurance; dedicated cybersecurity staff/officer.
We work with third parties from time to time that assist us from time to time to identify, assess, and manage cybersecurity risks, including professional services firms, threat intelligence service providers, cybersecurity consultants, cybersecurity software providers, managed cybersecurity service providers, and penetration testing.
To operate our business, we utilize certain third-party service providers to perform a variety of functions, such as outsourced business critical functions, clinical research, professional services, SaaS platforms, managed services, property management, cloud-based infrastructure, data center facilities, content delivery, encryption and authentication technology, corporate productivity services, and other functions. We have certain vendor management processes designed to help to manage cybersecurity risks associated with our use of certain of these providers. Depending on the nature of the services provided, the sensitivity and quantity of information processed, and the identity of the service provider, our vendor management process may include reviewing the cybersecurity practices of such provider, contractually imposing obligations on the provider related to the services they provide and/or the information they process, conducting security assessments, conducting on-site inspections, requiring their completion of written questionnaires regarding their services and data handling practices, and conducting periodic re-assessments during their engagement.
For a description of the risks from cybersecurity threats that may materially affect us and how they may do so, refer to Part I, Item 1A. Risk Factors for additional information about cybersecurity-related risks.
Governance.Our cybersecurity risk assessment and management processes are implemented and maintained by certain Company management, including a Chief Information Officer, who reports to the CFO. Management is also responsible for hiring appropriate personnel, integrating cybersecurity considerations into the company’s overall risk management strategy, and for communicating key priorities to employees, as well as for approving budgets, helping prepare for cybersecurity incidents, approving cybersecurity processes, and reviewing security assessments and other security-related reports. Our cybersecurity incident response and vulnerability management processes involve management, who participates in our disclosure controls and procedures.
Our cybersecurity incident response and vulnerability management processes are designed to escalate certain cybersecurity incidents and vulnerabilities to members of management depending on the circumstances, including work with the company’s incident response team to help the company mitigate and remediate cybersecurity incidents of which they are notified. In addition, the company’s incident response processes include reporting to the Audit committee of the board of directors for certain cybersecurity incidents.
Management is involved with the Company’s efforts to prevent, detect, and mitigate cybersecurity incidents by overseeing preparation of cybersecurity policies and procedures, testing of incident response plans, engagement of vendors to conduct penetration tests. Management participates in cybersecurity incident response efforts by being a member of the incident response team and helping direct the company’s response to cybersecurity incidents.
Our board of directors addresses the Company’s cybersecurity risk management as part of its general oversight function. The board of directors’ audit committee is responsible for overseeing the company’s cybersecurity risk management processes, including oversight and mitigation of risks from cybersecurity threats. The audit committee also has access to various reports, summaries or presentations related to cybersecurity threats, risk, and mitigation.
Item 2. Properties
We lease ourOur corporate headquarters which are located in San Diego, California, and consist of 141 thousand square feet of laboratory and office space located at 12780 El Camino Real, 88 thousand square feet of office space located at 12790 El Camino Real, 46 thousand square feet of laboratory space located at 10420 Wateridge Circle, and 45 thousand square feet of office space located at 12777 High Bluff Drive.
California. We believe that our property and equipment are generally well maintained, in good operating condition and suitable for the conduct of our business.
Details of our leased facilities, which include our corporate headquarters and consist of office space and research and development laboratories, follow.
| | | | | | | | | | | | | | |
| Address | | Type | | Square Feet |
| 12780 El Camino Real, San Diego, California | | Office Space, Research and Development Laboratories | | 141,000 | |
| 6027 Edgewood Bend Court, San Diego, California | | Office Space | | 124,000 | |
| 6029 Edgewood Bend Court, San Diego, California | | Office Space | | 110,000 | |
| 12790 El Camino Real, San Diego, California | | Office Space | | 88,000 | |
| 10420 Wateridge Circle, San Diego, California | | Research and Development Laboratories | | 46,000 | |
| 12777 High Bluff Drive, San Diego, California | | Office Space | | 45,000 | |
| 12770 El Camino Real, San Diego, California | | Office Space | | 26,000 | |
On February 8, 2022, we entered into a lease agreement for a four-building campus facility to be constructed in San Diego, California, including a six-year option for the construction of a fifth building. This campus facility, comprised of office space and research and development laboratories, will serve as our new corporate headquarters.
The construction of the campus facility is phased. The first phase of construction relating to office space was completed in December 2023. As we begin to occupy our new campus facility, we will sublease certain of our existing leased premises when we determine there is excess leased capacity.
Item 3. Legal Proceedings
From time to time in the normal courseFor a description of business, we may be subject to various legal matters such as threatened or pending claims or proceedings. We are not currently a party to any materialour legal proceedings, or claims, nor are we aware of any pending or threatened litigation or claims that could have a material adverse effect on our business, operating results, cash flows orrefer to Note 13 to the consolidated financial condition should such litigation or claim be resolved unfavorably.statements, which is incorporated herein by reference.
Item 4. Mine Safety Disclosures
None.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock is traded on the Nasdaq Global Select Market under the symbol “NBIX”.
At January 29, 2021,As of February 5, 2024, there were approximately 4743 stockholders of record of our common stock. We have not paid any cash dividends on our common stock since inception and do not anticipate paying cash dividends in the foreseeable future.
Information about our equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report on Form 10-K.
Recent Sales of Unregistered Securities and Issuer Purchases of Equity Securities
There were no unregistered sales of our equity securities during 2020. In addition,and we did not repurchase any of our equity securities during 2020.2023.
Stock Performance Graph and Cumulative Total Return*
The following graph below showspresents the cumulative total stockholder return assuming the investment of $100 on December 31, 20152018 (and the reinvestment of dividends thereafter) in each of (i) Neurocrine Biosciences, Inc.’s common stock, (ii) the Nasdaq Composite Index and (iii) the Nasdaq Biotechnology Index. The comparisons in the graph below are based upon historical data and are not indicative of, or intended to forecast, future performance of our common stock or Indexes.
* The material in this section is not “soliciting material”, is not deemed “filed” with the Securities and Exchange Commission or SEC, and is not to be incorporated by reference into any of our SEC filings whether made before or after the date hereof and irrespective of any general incorporation language in any such SEC filing except to the extent we specifically incorporate this section by reference.
Item 6. Selected Financial Data
The following selected financial data have been derived from our audited financial statements. The information set forth below is not necessarily indicative of our results of future operations and should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the financial statements and notes thereto appearing elsewhere in this Annual Report on Form 10-K.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| (in millions, except per share data) | 2020 | | 2019 | | 2018 | | 2017 | | 2016 |
| Consolidated Statements of Operations Data |
| Revenues: | | | | | | | | | |
| Product sales, net | $ | 994.1 | | | $ | 752.9 | | | $ | 409.6 | | | $ | 116.6 | | | $ | — | |
| Collaboration revenue | 51.8 | | | 35.2 | | | 41.6 | | | 45.0 | | | 15.0 | |
| Total revenues | 1,045.9 | | | 788.1 | | | 451.2 | | | 161.6 | | | 15.0 | |
| Operating expenses: | | | | | | | | | |
| Cost of sales | 10.1 | | | 7.4 | | | 4.9 | | | 1.3 | | | — | |
| Research and development | 275.0 | | | 200.0 | | | 155.8 | | | 91.8 | | | 94.3 | |
| Acquired in-process research and development | 164.5 | | | 154.3 | | | 4.8 | | | 30.0 | | | — | |
| Selling, general and administrative | 433.3 | | | 354.1 | | | 248.9 | | | 169.9 | | | 68.1 | |
| Total operating expenses | 882.9 | | | 715.8 | | | 414.4 | | | 293.0 | | | 162.4 | |
| Operating income (loss) | 163.0 | | | 72.3 | | | 36.8 | | | (131.4) | | | (147.4) | |
| Other (expense) income: | | | | | | | | | |
| Interest expense | (32.8) | | | (32.0) | | | (30.5) | | | (19.5) | | | — | |
| Unrealized loss on restricted equity securities | (17.7) | | | (13.0) | | | — | | | — | | | — | |
| Loss on extinguishment of convertible senior notes | (18.4) | | | — | | | — | | | — | | | — | |
| Investment income and other, net | 12.6 | | | 19.2 | | | 15.5 | | | 8.3 | | | 6.3 | |
| Total other (expense) income, net | (56.3) | | | (25.8) | | | (15.0) | | | (11.2) | | | 6.3 | |
| Income (loss) before (benefit from) provision for income taxes | 106.7 | | | 46.5 | | | 21.8 | | | (142.5) | | | (141.1) | |
| (Benefit from) provision for income taxes | (300.6) | | | 9.5 | | | 0.7 | | | — | | | — | |
| Net income (loss) | $ | 407.3 | | | $ | 37.0 | | | $ | 21.1 | | | $ | (142.5) | | | $ | (141.1) | |
| Net income (loss) per share, basic | $ | 4.38 | | | $ | 0.40 | | | $ | 0.23 | | | $ | (1.62) | | | $ | (1.63) | |
| Net income (loss) per share, diluted | $ | 4.16 | | | $ | 0.39 | | | $ | 0.22 | | | $ | (1.62) | | | $ | (1.63) | |
| Weighted average common shares outstanding: | | | | | | | | | |
| Basic | 93.1 | | | 91.6 | | | 90.2 | | | 88.1 | | | 86.7 | |
| Diluted | 97.8 | | | 95.7 | | | 95.4 | | | 88.1 | | | 86.7 | |
| | | | | | | | | |
| Consolidated Balance Sheets Data | | | | | | | | | |
| Cash, cash equivalents and debt securities available-for-sale | $ | 1,028.1 | | | $ | 970.2 | | | $ | 866.9 | | | $ | 763.3 | | | $ | 350.8 | |
| Working capital | $ | 829.7 | | | $ | 265.7 | | | $ | 649.5 | | | $ | 500.5 | | | $ | 280.0 | |
| Total assets | $ | 1,734.7 | | | $ | 1,306.0 | | | $ | 993.2 | | | $ | 817.6 | | | $ | 365.1 | |
| Convertible senior notes | $ | 317.9 | | | $ | 408.8 | | | $ | 388.5 | | | $ | 369.6 | | | $ | — | |
| Accumulated deficit | $ | (725.4) | | | $ | (1,132.7) | | | $ | (1,177.8) | | | $ | (1,198.9) | | | $ | (1,056.3) | |
| Total stockholders’ equity | $ | 1,126.2 | | | $ | 636.9 | | | $ | 480.8 | | | $ | 372.1 | | | $ | 314.9 | |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations section contains forward-looking statements pertaining to, among other things, the commercialization of our product and product candidates, the expected continuation of our collaborative agreements, the receiptprogress, timing, results or implications of research and development payments thereunder, the future achievement of various milestones in product development and the receipt of payments related thereto, the potential receipt of royalty payments, preclinical testing and clinical trials of potential products,and other development activities, our plans and timing with respect to seeking regulatory approvals, the period of time that our existing capital resources will meet our funding requirements, and our financial results of operations. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of various risks and uncertainties, including those set forth in this Annual Report on Form 10-K under the heading “Item 1A. Risk Factors.” See “Forward-Looking Statements” in Part I of this Annual Report on Form 10-K.
Overview
We areNeurocrine Biosciences is a neuroscience-focused, biopharmaceutical company with a simple purpose: to relieve suffering for people with great needs, but few options. We are dedicated to discovering developing and deliveringdeveloping life-changing treatments for peoplepatients with serious, challenging and under-addressed neurological, endocrineneuroendocrine and psychiatricneuropsychiatric disorders. OurThe Company’s diverse portfolio includes United StatesU.S. Food and Drug Administration or FDA,(FDA) approved treatments for tardive dyskinesia, Parkinson’schorea associated with Huntington's disease, endometriosis*adrenal insufficiency, and endometriosis and uterine fibroids in collaboration with AbbVie Inc. (AbbVie), uterine fibroids*a European Medicines Agency (EMA) approved treatment for classic congenital adrenal hyperplasia (CAH) and clinicala diversified portfolio of advanced clinical-stage programs in multiple therapeutic areas. For nearly three decades, we have specialized in targeting and interrupting disease-causing mechanisms involving the interconnected pathways of the nervous and endocrine systems. (*in collaboration with AbbVie Inc.)
We launched INGREZZA® (valbenazine) in the U.S. with our specialty sales force in May 2017, after receiving FDA approval for INGREZZA as the first FDA-approved drug for the treatment of tardive dyskinesia in April 2017. In September 2020, we launched ONGENTYS® (opicapone) inMay 2017 and for the U.S. leveraging our existing INGREZZA commercial infrastructure after receiving FDA approval for ONGENTYS for Parkinson'streatment of adults with chorea associated with Huntington's disease in April 2020.August 2023. INGREZZA net product sales represent the significant majoritytotaled $1.8 billion for 2023 and accounted for approximately 99% of our total net product sales.sales for 2023.
Our partner AbbVie Inc., or AbbVie,Mitsubishi Tanabe Pharma Corporation (MTPC) launched ORILISSADYSVAL®(elagolix) in the U.S. and Canada in August and November 2018, respectively, after receiving FDA and Health Canada approval for ORILISSA for endometriosis in July and October 2018, respectively. In June 2020, AbbVie launched ORIAHNNTM (elagolix, estradiol,(valbenazine) in Japan for the treatment of tardive dyskinesia in June 2022 and norethindrone acetate; elagolix)subsequently in the U.S. after receiving FDA approval for ORIAHNN for uterine fibroids in May 2020.other select Asian markets, where it is marketed as REMLEAS® (valbenazine). We receive royalties at tiered percentage rates on anyMTPC net sales of ORILISSA and ORIAHNN.
In addition, we have a rapidly expanding pipeline of potential treatments and gene therapies for diseases such as Huntington’s disease, or HD, Parkinson’s disease, epilepsy, congenital adrenal hyperplasia, or CAH, schizophrenia and depression. Refer to Part I, Item 1, “Business” for more information about our exclusive and partnered commercial products, clinical development pipeline and research programs.
Highlights:
•INGREZZA net product sales for 2020 increased $240.2 million, or 31.9%, to $993.1 million, primarily reflecting strong refill and persistency rates for existing INGREZZA patients.valbenazine.
•Our partner AbbVie launched ORILISSAWe launched ONGENTYS® (elagolix tablets) in the U.S. in September 2020, after receiving FDA approval for ONGENTYS as an adjunctive therapy to levodopa/DOPA decarboxylase inhibitors in adult Parkinson's disease patients in April 2020.
•AbbVie launched ORIAHNN in the U.S. in June 2020, after receiving FDA approval for ORIAHNN as the first FDA-approved non-surgical, oral medication option for the management of heavy menstrual bleeding associated with uterine fibroids in pre-menopausal women in May 2020. We recognized a $30.0 million event-based milestone as revenue in the second quarter of 2020.
•Completed strategic partnerships with Idorsia Pharmaceuticals Ltd, or Idorsia, and Takeda Pharmaceutical Company Limited, or Takeda, to expand clinical pipeline for epilepsy and psychiatry disorders.Recognized
in-process research and development, or IPR&D, expense for 2020 of $164.5 million, related to upfront payments.
•Total debt outstanding decreased by $136.2 million to $381.3 million after repurchase of approximately 26% of our debt outstanding in December 2020. The total aggregate repurchase price of $186.9 million was paid in cash and resulted in an $18.4 million loss.
•At December 31, 2020, in part because we achieved three years of cumulative pretax income, management determined that there is sufficient positive evidence to conclude that it is more likely than not that deferred tax assets of $319.4 million are realizable. We therefore reduced the valuation allowance accordingly.
Pipeline Highlights:
•Crinecerfont (NBI-74788): In July 2020, we initiated the CAHtalyst study, a global registrational Phase III, randomized, double-blind, placebo-controlled clinical study to evaluate the safety and efficacy of crinecerfont in 165 adult patients with classic CAH, followed by an open-label treatment period.
•NBI-827104 (ACT-709478): In November 2020, we initiated a Phase II clinical study for NBI-827104 in a rare pediatric epileptic encephalopathy known as Continuous Spike and Wave During Sleep.
•INGREZZA: In February 2021, Mitsubishi Tanabe Pharmaceutical Company, or MTPC, reported positive top-line results from the J-KINECT Phase III study, designed to evaluate the efficacy and safety of valbenazine in tardive dyskinesia. Detailed results from this trial will be presented at a future medical conference. With positive data in hand, a marketing authorization with the Ministry of Health and Welfare is planned for 2021 in Japan. In addition, MTPC submitted filings for marketing authorizations in South Korea, Thailand, Singapore, Indonesia, and Malaysia in 2020.
•NBIb-1817 (VY-AADC): On February 2, 2021, we notified Voyager Therapeutics, Inc., or Voyager, of our termination of the NBIb-1817 for Parkinson’s disease program. The effective date of this termination will be August 2, 2021. The termination does not apply to any other development program other than NBIb-1817 for Parkinson’s disease, and our collaboration and license agreement with Voyager will otherwise continue in effect.
COVID-19
The global COVID-19 pandemic has dramatically changed the ways in which we live and interact with one another. While we adapt to this new shared reality, our mission remains unchanged: to discover and develop life-changing treatments for people with serious, challenging and under-addressed disorders.
While we are unable to reliably estimate the duration or extent of any potential business disruption or financial impact during this time, including any impacts on INGREZZA product sales or R&D expense, we remain committed to (1) prioritizing the safety, health and well-being of patients, their caregivers, healthcare providers and our employees; (2) ensuring patients with tardive dyskinesia are well supported and have continued uninterrupted access to INGREZZA, for which we currently do not expect any supply disruption; and (3) advancing ongoing clinical studies. As part of this commitment, we implemented a “Work from Home Policy” in early March 2020 for employees not involved in business-critical activities. For employees involved in business-critical activities, we implemented safety measures designed to comply with federal, state and local guidelines.
Due to the impact of COVID-19, we initially paused enrollment of new patients in several of our clinical trials. Beginning in the third quarter of 2020, we began enrolling patients in our HD and CAH studies. To date, we have not experienced any interruption of our supply of drug products needed to support our ongoing clinical studies, but we expect that completion and data readouts for several of our ongoing and planned studies will be delayed.
We continue to believe that existing funds, cash generated from operations, and existing sources of and access to financing are adequate to satisfy our needs for working capital, capital expenditures, debt service requirements and other business development initiatives that we plan to strategically pursue. However, should the COVID-19 pandemic and any associated recession or depression continue for a prolonged period, our results of operations, financial condition, liquidity and cash flows could be materially impacted by lower revenues and profitability and a lower likelihood of effectively and efficiently developing new medicines.
Results of Operations
Revenues
The following table presents revenues by category.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2020 | | 2019 | | 2018 |
| INGREZZA product sales, net | $ | 993.1 | | | $ | 752.9 | | | $ | 409.6 | |
| ONGENTYS product sales, net | 1.0 | | | — | | | — | |
| Collaboration revenues | 51.8 | | | 35.2 | | | 41.6 | |
| Total revenues | $ | 1,045.9 | | | $ | 788.1 | | | $ | 451.2 | |
Product Sales, net.Net product sales were $994.1 million for 2020, $752.9 million for 2019 and $409.6 million for 2018.
Collaboration Revenues.Collaboration revenues reflect the achievement of certain event-based milestones, royalties earned at tiered percentage rates on any net sales of ORILISSA and ORIAHNN and license fees earned under our collaboration agreements with AbbVie and MTPC.
In the second quarter of 2020, we recognized a $30.0 million event-based milestone as revenue upon FDA-approval of AbbVie’s ORIAHNN for uterine fibroids. In the third quarter of 2019, we recognized a $20.0 million event-based milestone as revenue upon the FDA’s acceptance of AbbVie’s new drug application, or NDA, submission of elagolix for uterine fibroids. In the third quarter of 2018, we recognized a $40.0 million event-based milestone as revenue upon FDA-approval of AbbVie’s ORILISSA for the treatment of moderate to severe pain associated with endometriosis.endometriosis in August 2018 and ORIAHNN® (elagolix, estradiol and norethindrone acetate capsules and elagolix capsules) in the U.S. for the treatment of heavy menstrual bleeding due to uterine fibroids in June 2020. We receive royalties at tiered percentage rates on AbbVie net sales of elagolix.
Business Highlights
•INGREZZA net product sales for 2023 increased $0.4 billion, or 28.6%, to $1.8 billion, reflecting higher prescription demand and increased commercial activities, including continued investment in our branded direct-to-consumer INGREZZA advertising campaign and benefit from the expansion of our sales force completed in April 2022.
•In the fourth quarter of 2023, we announced that all patent litigation brought by Neurocrine Biosciences against the companies that filed an Abbreviated New Drug Application (ANDA) to the FDA seeking approval to market generic versions of INGREZZA prior to the expiration of the Orange Book listed patents have been resolved. Pursuant to the terms of the respective settlement agreements, such companies have the right to sell generic versions of INGREZZA in the U.S. beginning March 1, 2038, or earlier under certain circumstances.
Pipeline Highlights
•Announced positive top-line data from the Phase 3 clinical studies of crinecerfont in adults and pediatrics with CAH. Crinecerfont subsequently received Breakthrough Therapy designation from the FDA for the treatment of CAH. Data from the Phase 3 studies will support a New Drug Application (NDA) submission to the FDA in the second quarter of 2024.
•Expanded strategic partnership with Voyager Therapeutics Inc. (Voyager) to advance multiple gene therapy programs, each enabled by Voyager's next-generation TRACERTM capsids, for the treatment of neurological diseases. Upfront fee associated with the agreement totaled $175.0 million, including an equity investment valued at $31.3 million on the transaction date, with the remaining $143.9 million of the purchase price, which includes the applicable transaction costs, expensed as in-process research and development in 2023.
•In the third quarter of 2023, we announced the FDA accepted the NDA for INGREZZA oral granules, a new sprinkle formulation of INGREZZA capsules for oral administration. The agency set a Prescription Drug User Fee Act target action date of April 30, 2024.
•In the third quarter of 2023, the FDA approved INGREZZA for the treatment of adults with chorea associated with Huntington's disease.
•In the fourth quarter of 2023, we announced the Phase 2 clinical studies of NBI-921352 in focal onset seizures and NBI-1065846 for anhedonia in major depressive disorder (MDD) did not meet their primary endpoints. No further development of NBI-921352 in focal onset seizures or NBI-1065846 for anhedonia in MDD is planned at this time.
Results of Operations
Revenues
Net Product Sales by Sales Product.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| INGREZZA | $ | 1,836.0 | | | $ | 1,427.8 | | | $ | 1,081.9 | |
| Other | 24.6 | | | 13.1 | | | 8.2 | |
| Total net product sales | $ | 1,860.6 | | | $ | 1,440.9 | | | $ | 1,090.1 | |
The increases in total net product sales from 2021 to 2022 and from 2022 to 2023 were primarily driven by increased INGREZZA net product sales on higher prescription demand and increased commercial activities, including continued investment in our branded direct-to-consumer INGREZZA advertising campaign and benefit from the expansion of our sales force completed in April 2022.
Collaboration Revenues by Category.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Royalties | $ | 21.2 | | | $ | 22.3 | | | $ | 22.3 | |
| Milestones | — | | | 20.0 | | | 15.0 | |
| Collaboration and other | 5.3 | | | 5.5 | | | 6.1 | |
| Total collaboration revenue | $ | 26.5 | | | $ | 47.8 | | | $ | 43.4 | |
Royalties reflect revenue earned on AbbVie net sales of elagolix for all periods presented and MTPC net sales of valbenazine beginning in June 2022.
For ORILISSA and ORIAHNN, we recognized royalty2022, total collaboration revenue also reflected the achievement of $19.2a $20.0 million milestone in connection with MTPC's first commercial sale of DYSVAL in Japan.
For 2021, total collaboration revenue also reflected the achievement of a $15.0 million milestone in connection with MTPC's marketing authorization application submission for 2020, $14.3 millionvalbenazine for 2019 and $1.6 million for 2018.the treatment of tardive dyskinesia in Japan.
Operating Expenses
Cost of Sales. CostRevenues.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Cost of revenues | $ | 39.7 | | | $ | 23.2 | | | $ | 14.3 | |
For 2023 compared to 2022, the increase in cost of salesrevenues was $10.1 million for 2020, $7.4 million for 2019primarily driven by increased INGREZZA and $4.9 million for 2018, primarily reflecting a higher annual volume of INGREZZAother net product sales, since commercial launchincreased amortization costs related to intangible assets, increased reserves for ONGENTYS inventory obsolescence in April 2017.connection with the termination of our license agreement with BIAL, and increased manufacturing costs in connection with our supply of valbenazine drug product under our collaboration with MTPC.
For 2022 compared to 2021, the increase in cost of revenues was primarily driven by increased INGREZZA net product sales.
Research and Development. Development by Category.
We support our drug discovery and development efforts through the commitment of significant resources to discovery, R&Dresearch and development programs, and business development opportunities.
Costs are reflected in the applicable development stage based upon the program status when incurred. Therefore, the same program could be reflected in different development stages in the same reporting period. For several of our programs, the R&Dresearch and development activities are part of our collaborative and other relationships.arrangements.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Late stage | $ | 106.1 | | | $ | 68.7 | | | $ | 55.7 | |
| Early stage | 107.4 | | | 81.1 | | | 43.9 | |
| Research and discovery | 96.5 | | | 63.7 | | | 50.5 | |
| Milestones | 0.8 | | | 42.7 | | | 5.4 | |
| Payroll and benefits | 206.7 | | | 163.8 | | | 129.1 | |
| Facilities and other | 47.5 | | | 43.8 | | | 43.5 | |
| Research and development | $ | 565.0 | | | $ | 463.8 | | | $ | 328.1 | |
Late stage consistsStage.Consists of costs incurred related tofor product candidates in Phase II2 registrational studies and onwards. all subsequent activities.
The increases in late stage expenses from 2021 to 2022 and from 2022 to 2023 primarily reflected increased investment in the Phase 3 programs for crinecerfont in CAH and valbenazine in schizophrenia and Phase 2 program for EFMODY in CAH.
Early stage consistsStage.Consists of costs incurred related tofor product candidates in post-investigationalafter the approval of an investigational new drug application or IND,by the applicable regulatory agency through Phase II2 non-registrational studies.
For 2023 compared to 2022, the increase in early stage expenses primarily reflected increased investment in the Phase 2 program for NBI-1117568 in schizophrenia and other advancing Phase 2 programs in psychiatry, partially offset by decreased spend on early stage programs in epilepsy.
For 2022 compared to 2021, the increase in early stage expenses primarily reflected increased investment in advancing Phase 2 programs in epilepsy and psychiatry.
Research and Discovery.Consists of expenses incurred prior to the approval of an investigational new drug application by the applicable regulatory agency.
For 2023 compared to 2022, the increase in research and discovery consistsexpenses primarily reflected increased investment in preclinical development programs including muscarinic agonists, gene therapies, and second generation VMAT2 inhibitors.
For 2022 compared to 2021, the increase in research and discovery expenses reflected increased investment in preclinical development programs including psychiatry, epilepsy, and gene therapies .
Milestones. Consist of pre-IND costs. Milestonedevelopment and regulatory milestone expenses reflect payments madeincurred in connection with our collaborative arrangements.
In 2022, we recognized milestone expenses of $30.0 million in connection with the FDA's acceptance of the investigational new drug application for NBI-1117568 in schizophrenia, $7.3 million in connection with the FDA's acceptance of the amended KAYAKTM study protocol, and other relationships. $5.0 million in connection with the approval of the clinical trial application for NBI-1070770 in major depressive disorder.
In 2021, we recognized milestone expense of $5.4 million in connection with the regulatory approval of the clinical trial application in Europe for NBI-921352 in epilepsy.
Payroll and benefits consistsBenefits. Consists of costs incurred for salaries and wages, payroll taxes, benefits and share-basedstock-based compensation associated with employees involved in ongoing R&Dresearch and development activities. Share-basedStock-based compensation may fluctuate from period to period based on factors that are not within our control, such as our stock price on the dates share-basedstock-based grants are issued.
For 2023 compared to 2022, the increase in payroll and benefits expenses primarily reflected higher headcount and an increase of $10.3 million in non-cash stock-based compensation expense primarily driven by an incremental charge related to a change in equity grant agreement terms.
For 2022 compared to 2021, the increase in payroll and benefits expenses primarily reflected higher headcount, including an increase of $9.3 million in non-cash stock-based compensation expense driven by an August 2021 equity grant of approximately 0.5 million restricted stock units to our full-time employees other than our executive officers and performance-based restricted stock units to our executive officers for which attainment of the performance-based criteria was achieved in 2022.
Facilities and other consistsOther.Consists of indirect costs incurred in supportfor the benefit of overall R&D activities and non-specific programs, including activities that benefit multiple programs, such as management costs, as well asincluding depreciation, information technology, and other facility-based expenses. These costs are not allocatedexpenses, such as rent expense.
Acquired In-Process Research and Development, or IPR&D.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Acquired in-process research and development | $ | 143.9 | | | $ | — | | | $ | 105.3 | |
In 2023, we recognized $143.9 million of IPR&D expense in connection with our payment of the upfront fee pursuant to our expanded strategic partnership with Voyager.
In 2021, we recognized $105.3 million of IPR&D expense, of which $100.3 million was in connection with our payment of the upfront fee pursuant to our collaboration with Heptares Therapeutics Limited.
Selling, General and Administrative, or SG&A.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Selling, general and administrative | $ | 887.6 | | | $ | 752.7 | | | $ | 583.3 | |
For 2023 compared to 2022, the increase in SG&A expenses was primarily driven by increased investment in our commercial initiatives, including our branded direct-to-consumer INGREZZA advertising campaign and deployment of our expanded salesforce completed in April 2022, and increased payroll and benefits expenses on higher headcount and an increase of $10.9 million in non-cash stock-based compensation expense primarily driven by an incremental charge related to a specific program or stage.change in equity grant agreement terms.
For 2022 compared to 2021, the increase in SG&A expenses was primarily driven by increased investment in our commercial initiatives and increased payroll and benefits expenses on higher headcount and an increase of $29.6 million in non-cash stock-based compensation expense driven by an August 2021 equity grant of approximately 0.5 million restricted stock units to our full-time employees other than our executive officers and performance-based restricted stock units to our executive officers for which attainment of the performance-based criteria was achieved in 2022.
Other Income (Expense), Net.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Interest expense | $ | (4.6) | | | $ | (7.1) | | | $ | (25.8) | |
| Unrealized gain on equity securities | 28.4 | | | 30.8 | | | 20.9 | |
| Loss on extinguishment of convertible senior notes | — | | | (70.0) | | | — | |
| Investment income and other, net | 57.4 | | | 11.2 | | | 3.8 | |
| Total other income (expense), net | $ | 81.2 | | | $ | (35.1) | | | $ | (1.1) | |
The following table presents R&D expense by category:
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2020 | | 2019 | | 2018 |
| Late stage | $ | 55.1 | | | $ | 43.7 | | | $ | 14.2 | |
| Early stage | 30.2 | | | 25.3 | | | 41.7 | |
| Research and discovery | 43.3 | | | 24.6 | | | 17.0 | |
| Milestone payments | 20.0 | | | 10.0 | | | 10.0 | |
| Payroll and benefits | 95.4 | | | 71.3 | | | 62.0 | |
| Facilities and other | 31.0 | | | 25.1 | | | 10.9 | |
| Total R&D expense | $ | 275.0 | | | $ | 200.0 | | | $ | 155.8 | |
R&D expense was $275.0 million for 2020, $200.0 million for 2019change in other income (expense), net from 2021 to 2022 and $155.8 million for 2018. The increase in R&D expense wasfrom 2022 to 2023 primarily the result of increased investment to support advancing our expanded clinical portfolio and increased personnel expenses on higher headcount.
Acquired In-Process Research and Development. IPR&D expense was $164.5 million for 2020, $154.3 million for 2019 and $4.8 million for 2018. For 2020, we recorded IPR&D expense of $46.0 million and $118.5 millionreflected debt extinguishment charges in connection with the paymentsrepurchase of the upfront fees pursuant to our collaborations with Idorsia and Takeda, respectively. For 2019, we recorded IPR&D expense of $118.1 million and $36.2 millionconvertible senior notes in connection with the payments of the upfront fees pursuant to our collaborations with Voyager and Xenon Pharmaceuticals, Inc., or Xenon. For 2018, we recorded IPR&D expense of $4.8 million in connection with payment of the upfront fee to Jnana to obtain access to Jnana’s proprietary drug discovery platform.
Selling, General and Administrative. Selling, general and administrative, or SG&A, expense was $433.3 million for 2020, $354.1 million for 2019 and $248.9 million for 2018. The increase in SG&A expense from 2019 to 2020 was primarily due to increased personnel expenses on higher headcount and continued investment in INGREZZA marketing. The increase in SG&A expense from 2018 to 2019 was primarily due to the sales force expansion completed2022, periodic fluctuations in the third quarterfair values of 2018, the national launch of a patient-focused disease state awareness campaign, Talk About TD,our equity security investments, increased interest income on our debt security investments and an increase in the Branded Pharmaceutical Drug Fee expense.
Other Expense
Otherdecreased interest expense net, was $56.3 million for 2020, $25.8 million for 2019 and $15.0 million for 2018. Periodic fluctuationson lower total debt outstanding. The change in other expense, net primarily reflect unrealized losses recognizedfrom 2021 to adjust our equity investments in Voyager and Xenon Pharmaceuticals Inc.2022 also reflected decreased interest expense due to fair value. For 2020, other expense, net, also reflects an $18.4 million lossthe adoption of ASU 2020-06 on debt extinguishment recognized for the partial repurchase of the 2024 Notes in November 2020.January 1, 2022.
(Benefit from) Provision for Income TaxesTaxes.
Our | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Provision for income taxes | $ | 82.4 | | | $ | 59.4 | | | $ | 11.8 | |
For 2023, the effective tax rate varied from the federal and state statutory rates primarily due to credits generated for research activities, certain nondeductible expenses, the impact of changes in the state effective rate, and losses incurred in foreign jurisdictions for which no tax benefit was recorded as management cannot conclude that it is more likely than not that the tax benefit of such losses will be realized in the future.
For 2022, the effective tax rate varied from income taxes was $300.6 millionthe federal and state statutory rates primarily due to credits generated for 2020, comparedresearch activities and certain nondeductible expenses, including the premium paid on the repurchase of our convertible senior notes in 2022.
For 2021, the effective tax rate varied from the federal and state statutory rates primarily due to excess tax benefits associated with stock-based compensation and credits generated for research activities. In the first quarter of 2021, we began recording a provision for income taxes of $9.5 million for 2019using an effective tax rate that approximated federal and $0.7 million for 2018. The benefit fromstate statutory rates.
Net Income.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Net income | $ | 249.7 | | | $ | 154.5 | | | $ | 89.6 | |
For 2023 compared to 2022, the increase in net income taxes for 2020 included a $296.3 million benefit related toprimarily reflected increased INGREZZA net product sales, decreased debt extinguishment charges in connection with the release of substantially allrepurchase of our valuation allowance againstconvertible senior notes in 2022, and decreased milestone expenses in connection with our deferred tax assets on December 31, 2020. The decision to release the valuation allowance was made after we determined that it was more likely than not the deferred tax assets, including net operating lossescollaborations, partially offset by increased upfront payments in connection with our expanded strategic partnership with Voyager and tax credits, would be realized, and was based on the evaluation and weighting of both positive and negative evidence, such as our achievement of a cumulative three-year income position at December 31, 2020, as well as our consideration of forecasts of future operating results and utilization of net operating losses and tax credits prior to their expiration. The provision for income taxes for 2019 and 2018 reflected estimated current state income taxes for both periods. At December 31, 2019 and 2018, we had full valuation allowances against our net deferred tax assets as realization was uncertain. Our tax expense for 2020, 2019 and 2018 varied from the statutory tax rate primarily due to changesincreased investment in our valuation allowances,commercial initiatives and expanded clinical portfolio.
For 2022 compared to 2021, the increase in net of other permanent book/tax differences, tax credits generated and impacts of changes in tax laws.
Net Income
Net income was $407.3 million, or $4.16 diluted earnings per share, for 2020, $37.0 million, or $0.39 diluted earnings per share, for 2019 and $21.1 million, or $0.22 diluted earnings per share, for 2018. The change from 2019
to 2020 was primarily the result ofreflected increased INGREZZA net product sales and a non-cash tax benefitlower upfront payments for asset acquisitions, partially offset by increased debt extinguishment charges in connection with the repurchase of $296.3 million relatedour convertible senior notes in 2022 and increased investment in our commercial initiatives and expanded clinical portfolio.
Liquidity and Capital Resources
Sources of Liquidity
We believe that our existing capital resources, funds generated by anticipated INGREZZA net product sales and investment income will be sufficient to satisfy our current and projected funding requirements for at least the release of substantiallynext 12 months. However, we cannot guarantee that our existing capital resources and anticipated revenues will be sufficient to conduct and complete all of our valuation allowance against our deferred tax assetsresearch and development programs or commercialization activities as planned. We may seek to access the public or private equity markets whenever conditions are favorable or pursue opportunities to obtain additional debt financing in the future. We may also seek additional funding through strategic alliances or other financing mechanisms. However, we cannot provide assurance that adequate funding will be available on December 31, 2020, offset by $164.5 million of IPR&Dterms acceptable to us, if at all.
Information Regarding Our Financial Condition.
| | | | | | | | | | | |
| December 31, |
| (in millions) | 2023 | | 2022 |
| Total cash, cash equivalents and marketable securities | $ | 1,719.1 | | | $ | 1,288.7 | |
| Working Capital: | | | |
| Total current assets | $ | 1,607.0 | | | $ | 1,453.5 | |
| Less total current liabilities | 654.8 | | | 537.7 | |
| Total working capital | $ | 952.2 | | | $ | 915.8 | |
Information Regarding Our Cash Flows.
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Cash flows from operating activities | $ | 389.9 | | | $ | 339.4 | | | $ | 256.5 | |
| Cash flows from investing activities | (467.1) | | | (177.1) | | | (130.2) | |
| Cash flows from financing activities | 65.3 | | | (234.3) | | | 27.4 | |
| Effect of exchange rate changes on cash and cash equivalents | 0.3 | | | (1.3) | | | — | |
| Change in cash, cash equivalents and restricted cash | $ | (11.6) | | | $ | (73.3) | | | $ | 153.7 | |
Cash Flows from Operating Activities.
For 2023 compared to 2022, the change in cash flows from operating activities primarily reflected increased INGREZZA net product sales and lower milestone payments in connection with our collaborations, partially offset by higher upfront payments in connection with Idorsiaour expanded strategic partnership with Voyager and Takeda, ongoing support forincreased investment in our commercial initiatives and expanded clinical portfolio.
For 2022 compared to 2021, the commercial launch of INGREZZA for tardive dyskinesia and progression of our clinical pipeline. The change in cash flows from 2018 to 2019 wasoperating activities primarily the result ofreflected increased INGREZZA net product sales and lower upfront payments for asset acquisitions, partially offset by $154.3 million of IPR&Dincreased investment in connection with our collaborations with Voyagercommercial initiatives and Xenon, ongoing support for the commercial launch of INGREZZA for tardive dyskinesia and progression of ourexpanded clinical pipeline.
Liquidity and Capital Resources
Cash, cash equivalents and debt securities available-for-sale totaled $1.0 billion and $970.2 million at December 31, 2020 and 2019, respectively.
Net cash provided by operating activities was $228.5 million for 2020, $147.0 million for 2019 and $101.4 million for 2018. Theportfolio. In addition, we experienced an increase in positive cash flow from 2019 to 2020 was primarily due toaccounts receivable driven by increased INGREZZA net product sales partially offset by incremental INGREZZA investment and progressionon extended customer payment terms attributed to the expansion of our clinical pipeline. Thedistribution network at the end of 2021 and an increase in positive cash flow from 2018 to 2019 was primarily due toaccrued liabilities driven by increased revenue-related reserves for discounts and allowances on higher INGREZZA net product sales partially offset by incremental INGREZZA investment and upfront paymentsthe timing of $154.3 million in connection with our collaborations with Voyager and Xenon.payments.
Net cash provided by investing activities was $4.1 million for 2020, compared with net cash used in investing activities of $211.1 million for 2019 and $242.9 million for 2018. Cash Flows from Investing Activities.
Periodic fluctuations in cash flows from investing activities primarily reflectfor all periods presented reflected timing differences inrelated to our purchases, sales, and maturities of debt securities available-for-salesecurity investments and changes in our portfolio-mix. Net
For 2023, cash used inflows from investing activities also reflected a $31.3 million equity investment in Voyager.
For 2022, cash flows from investing activities also reflected the acquisition of Diurnal Group plc for 2019 also reflects equity investments of $54.7$42.7 million in Voyagercash, which is net of cash acquired, and $14.2a $7.7 million equity investment in Xenon Pharmaceuticals Inc.
Cash Flows from Financing Activities.
Cash flows from financing activities for all periods presented reflected proceeds from issuances of our common stock.
For 2022, cash flows from financing activities also reflected the repurchase of $210.8 million aggregate principal amount of our convertible senior notes for an aggregate repurchase price of $279.0 million in Xenon.cash.
Net cash used
Material Cash Requirements
In the pharmaceutical industry, it can take a significant amount of time and capital resources to successfully complete all stages of research and development and commercialize a product candidate, which ultimate length of time and spend required cannot be accurately estimated as it varies substantially according to the type, complexity, novelty and intended use of a product candidate.
The funding necessary to execute our business strategies is subject to numerous uncertainties and we may be required to make substantial expenditures if unforeseen difficulties arise in financingcertain areas of our business. In particular, our future capital requirements will depend on many factors, including:
•the commercial success of INGREZZA, ORILISSA, ORIAHNN and/or DYSVAL;
•continued scientific progress in our research and clinical development programs;
•the magnitude and complexity of our research and development programs;
•progress with preclinical testing and clinical trials;
•the time and costs involved in obtaining regulatory approvals;
•the cost of commercialization activities and arrangements, including our advertising campaigns;
•the cost of manufacturing of our product candidates;
•the costs involved in filing and pursuing patent applications, enforcing patent claims, or engaging in interference proceedings or other patent litigation;
•competing technological and market developments; and
•developments related to any future litigation.
In addition to the foregoing factors, we have significant future capital requirements, including:
External Business Developments.In addition to our independent efforts to develop and market products, we may enter into collaboration and license agreements or acquire businesses from time-to-time to enhance our drug development and commercial capabilities. With respect to our existing collaboration and license agreements, we may be required to make potential future payments of up to $17.0 billion upon the achievement of certain event-based milestones.
Refer to Note 2 to the consolidated financial statements for more information on our significant collaboration and license agreements.
Leases.Our operating leases that have commenced have terms that expire beginning 2025 through 2036 and consist of office space and research and development laboratories, including our corporate headquarters.
On February 8, 2022, we entered into a lease agreement for a four-building campus facility to be constructed in San Diego, California, including a six-year option for the construction of a fifth building. This campus facility, comprised of office space and research and development laboratories, will serve as our new corporate headquarters.
The construction of the campus facility is phased. The first phase of construction relating to office space was $157.8completed in December 2023. As we begin to occupy our new campus facility, we will sublease certain of our existing leased premises when we determine there is excess leased capacity.
Refer to Note 11 to the consolidated financial statements for more information on our leases, including a presentation of our approximate future minimum lease payments under non-cancelable operating leases.
Convertible Senior Notes.On May 2, 2017, we completed a private placement of $517.5 million forin aggregate principal amount of 2.25% fixed-rated convertible senior notes due May 15, 2024 (the 2024 Notes). In 2020, primarily reflecting our repurchase ofwe repurchased $136.2 million aggregate principal amount of the 2024 Notes for an aggregate repurchase price of $186.9 million in cash in November 2020, compared to net cash provided by financing activities of $32.4cash. In 2022, we repurchased $210.8 million for 2019 and $29.5 million in 2018. For 2019 and 2018, periodic fluctuations in cash flows from financing activities reflect proceeds from issuances of our common stock.
Shelf Registration Statement. In February 2017, we filed an automatic shelf registration statement which immediately became effective by rule of the Securities and Exchange Commission, or SEC. We sold no securities under this shelf registration statement in 2020, 2019 or 2018.
Convertible Senior Notes. In May 2017, we completed a private placement of $517.5 million in aggregate principal amount of 2.25% convertible senior notes scheduled to mature on May 15, 2024, unless earlier converted, redeemed, or repurchased. We may not redeem the 2024 Notes prior to May 15, 2021. On or after this date, atfor an aggregate repurchase price of $279.0 million in cash. As of December 31, 2023, $170.4 million aggregate principal amount of the 2024 Notes remained outstanding.
At our election, we may redeem all or any portion of the 2024 Notes under certain circumstances. In addition, holders of the 2024 Notes may convert the 2024 Notes at any time until the close of business on the scheduled trading day immediately preceding May 15, 2024. Upon conversion, holders will receive the principal amount of their 2024 Notes and any conversion premium, calculated based on the per share volume-weighted average price for each of the 30 consecutive trading days during the observation period (as more fully described in the 2017 Indenture), in cash. Unless earlier converted, redeemed, or repurchased, we would be required to pay interest of $1.9 million in 2024 and pay the aggregate principal amount outstanding of $170.4 million upon maturity of the 2024 Notes.
The 2024 Notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, the issuance of other indebtedness or the issuance or repurchase of securities by us. There are customary events of default with respect to the 2024 Notes, including that upon certain events of default, 100% of the principal and accrued and unpaid interest on the 2024 Notes will automaticallywould become due and payable. Amounts
Refer to Note 5 to the consolidated financial statements for more information on the 2024 Notes and related interest in the table above assume that the 2024 Notes will be held until maturity. In November 2020, we entered into separate, privately negotiated transactions with certain holders of the 2024 Notes to repurchase $136.2 million aggregate principal amount of the 2024 Notes for an aggregate repurchase price of $186.9 million in cash. At December 31, 2020, $381.3 million aggregate principal amount of the 2024 Notes remained outstanding.Notes.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations is based upon financial statements that we have prepared in accordance with accounting principles generally accepted in the United States of America or GAAP.(GAAP). The preparation of these financial statements requires management to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses, and related disclosures. On an on-going basis, we evaluate these estimates, including those related to revenue recognition and share-based compensation.recognition. Estimates are based on historical experience, information received from third parties and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about
the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. Historically, revisions to our estimates have not resulted in a material change to the financial statements.
The items in our financial statements requiring significant estimates and judgments are as follows:
Product Sales, Net.Reserves for Government Rebates. RevenuesWe recognize revenues from product sales are recordedof INGREZZA net of reserves established for applicable discounts and allowances that are offered within contracts with our customers, payors, and other third parties. The transaction price, which includes variable consideration reflecting the impact of discounts and allowances, may be subject to constraint and is included in the net sales price only to the extentSuch reserves include estimates for government rebates that it is probable that a significant reversal of the amount of the cumulative revenue recognized will not occur in a future period. Actual amounts may ultimately differ from our estimates. If actual results vary, we adjust these estimates, which could have an effect on earnings in the period of adjustment.
Government Rebates. We are obligated to pay rebates for mandated discounts including under the Medicaid Drug Rebate Program.Program and Medicare Part D. The liability for such rebates consists of invoices received for claims from prior quarters that remain unpaid, or for which an invoice has not been received, and estimated rebates for the current applicable reporting period, whichperiod. Such estimates require us to project the magnitude of our sales that will be subject to such rebates and are primarily based on actual historical rebates by state, estimated payor mix, state and federal regulations, and relatedrelevant contractual terms. Estimated rebates are recordedterms, as supplemented by management’s judgement. There is a reduction of revenuesignificant time-lag in the period the relatedour receiving rebate notices from each state (generally, several months or longer after a sale is recognized.recognized). To date, actual government rebates have not differed materially from our estimates.
Share-Based Compensation. For purposes of calculating share-based compensation, we estimate the fair value of share-based compensation awards using a Black-Scholes option-pricing model. The determination of the fair value of share-based compensation awards utilizing the Black-Scholes model is affected by our stock price and a number of assumptions, including but not limited to expected stock price volatility over the term of the awards and the expected term of stock options. Our stock options have characteristics significantly different from those of traded options, and changes in the assumptions can materially affect the fair value estimates. For example, an increase in the underlying stock price results in a significant increase in the Black-Scholes option-pricing. The fair value of performance-based restricted stock units, or PRSUs, is estimated based on the closing sale price of our common stock on the date of grant. Expense recognition for PRSUs commences when attainment of the associated performance-based criteria is determined to be probable.
If factors change and we employ different assumptions, share-based compensation expense may differ significantly from what we have recorded in the past. If there is a difference between the assumptions used in determining share-based compensation expense and the actual factors which become known over time, we may change the input factors used in determining share-based compensation expense for future grants. These changes, if any, may materially impact our results of operations in the period such changes are made. For actual forfeitures, we recognize the adjustment to compensation expense in the period the forfeitures occur.
Income Taxes. Our income tax benefit (provision)provision is computed under the asset and liability method. Significant estimates are required in determining our income tax benefit (provision).provision. Some of these estimates are based on interpretations of existing tax laws or regulations. We recognize deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts (temporary differences) at enacted tax rates in effect for the years in which the differences are expected to reverse. A valuation allowance is established for deferred tax assets for which it is more likely than not that some portion or all of the deferred tax assets, including net operating losses and tax credits, will not be realized. We periodically re-assess the need for a valuation allowance against our deferred tax assets based on various factors including our historical earnings experience by taxing jurisdiction, and forecasts of future operating results and utilization of net operating losses and tax credits prior to their expiration. Significant judgment is required in making this assessment and, to the extent that a reversal of any portion of our valuation allowance against our deferred tax assets is deemed appropriate, a tax benefit will be recognized against our income tax provision in the period of such reversal. Prior to 2020, we recorded a valuation allowance that fully offset our deferred tax assets. On December 31, 2020, based on our evaluation of various factors, such as our achievement of a cumulative three-year income position as of December 31, 2020, as well as our consideration of forecasts of future operating results and utilization of net operating losses and tax credits prior to their expiration, we released substantially all of our valuation allowance against our deferred tax assets and recorded
a corresponding income tax benefit. We continue to maintain a valuation allowance against our California state deferred tax assets.
Additional Information
Refer to Note 1 to the consolidated financial statements for information on accounting pronouncements that have impacted or are expected to materially impact our consolidated financial condition, results of operations, or cash flows.
Factors That May Affect Future Financial Condition and Liquidity
The funding necessary to execute our business strategies is subject to numerous uncertainties, which may adversely affect our liquidity and capital resources. Marketing of approved pharmaceuticals and completion of clinical trials may take several years or more, but the length of time generally varies substantially according to the type, complexity, novelty and intended use of a product candidate. It is also important to note that if a clinical candidate is identified, the further development of that candidate can be halted or abandoned at any time due to a number of factors. These factors include, but are not limited to, funding constraints, safety or a change in market demand.
The nature and efforts required to develop our product candidates into commercially viable products include research to identify a clinical candidate, preclinical development, clinical testing, FDA approval and commercialization. In the pharmaceutical industry, total R&D spend for a drug candidate that successfully completes all stages of R&D and is commercialized may exceed $2 billion. Further, it can take in excess of ten years to complete all stages of R&D for a drug candidate.
We test our potential product candidates in numerous preclinical studies to identify disease indications for which our product candidates may show efficacy. We may conduct multiple clinical trials to cover a variety of indications for each product candidate. As we obtain results from trials, we may elect to discontinue clinical trials for certain product candidates or for certain indications in order to focus our resources on more promising product candidates or indications. The duration and the cost of clinical trials may vary significantly over the life of a project as a result of differences arising during the clinical trial protocol, including, among others, the following:
•we or the FDA or similar foreign regulatory authorities may suspend the trials;
•we may discover that a product candidate may cause harmful side effects;
•patient recruitment and enrollment may be slower or more difficult than expected; and
•patients may drop out of the trials.
For each of our programs, we periodically assess the scientific progress and merits of the programs to determine if continued R&D is economically viable. Certain of our programs have been terminated due to the lack of scientific progress and lack of prospects for ultimate commercialization. Because of the uncertainties associated with R&D of these programs, we may not be successful in achieving commercialization. As such, the ultimate timeline and costs to commercialize a product cannot be accurately estimated.
Our in-license, research and clinical development agreements are generally cancellable with written notice within 180 days or less. We may be required to pay up to $8.5 billion in milestone payments, plus sales royalties, in the event that all scientific research, development and commercialization milestones under these agreements are achieved.
Other than INGREZZA, which has been FDA-approved for the treatment of tardive dyskinesia; ONGENTYS, which has been FDA-approved as an adjunctive therapy to levodopa/DOPA decarboxylase inhibitors in adult Parkinson’s disease patients; ORILISSA (partnered with AbbVie), which has been FDA-approved for the management of moderate to severe endometriosis pain in women; and ORIAHNN (partnered with AbbVie), which has been FDA-approved for the management of heavy menstrual bleeding associated with uterine fibroids in pre-menopausal women, our product candidates have not yet achieved FDA regulatory approval, which is required before we can market them as therapeutic products in the U.S. In order to proceed to subsequent clinical trial stages and to ultimately achieve regulatory approval, the FDA must conclude that our clinical data establish safety and efficacy. We must satisfy the requirements of similar regulatory authorities in foreign countries in order to market products in those countries. The results from preclinical testing and early clinical trials may not be predictive of results in later
clinical trials. It is possible for a candidate to show promising results in clinical trials, but subsequently fail to establish sufficient safety and efficacy data necessary to obtain regulatory approvals.
As a result of the uncertainties discussed above, among others, the duration and completion costs of our R&D projects, clinical trials, and post-marketing studies are difficult to estimate and are subject to considerable variation. Our inability to complete our R&D projects in a timely manner or our failure to enter into collaborative agreements, when appropriate, could significantly increase our capital requirements and could adversely impact our liquidity. These uncertainties could force us to seek additional, external sources of financing from time to time in order to continue with our business strategy. Our inability to raise additional capital, or to do so on terms reasonably acceptable to us, would jeopardize the future success of our business.
We currently have limited experience in marketing and selling pharmaceutical products. If we fail to maintain successful marketing, sales, and reimbursement capabilities, or fail to enter into successful arrangements with third parties, our product revenues may suffer. We also may be required to make further substantial expenditures if unforeseen difficulties arise in other areas of our business. In particular, our future capital requirements will depend on many factors, including:
•the commercial success of INGREZZA, ONGENTYS, ORILISSA, and/or ORIAHNN;
•debt service obligations on the 2024 Notes;
•continued scientific progress in our R&D and clinical development programs;
•the magnitude and complexity of our research and development programs;
•progress with preclinical testing and clinical trials;
•the time and costs involved in obtaining regulatory approvals;
•the costs involved in filing and pursuing patent applications, enforcing patent claims, or engaging in interference proceedings or other patent litigation;
•competing technological and market developments;
•the establishment of additional strategic alliances;
•developments related to any future litigation;
•the impact of the COVID-19 pandemic on our business;
•the cost of commercialization activities and arrangements, including manufacturing of our product candidates; and
•the cost of product in-licensing and any possible acquisitions.
We believe that our existing capital resources, funds generated by anticipated INGREZZA net product sales and investment income will be sufficient to satisfy our current and projected funding requirements for at least the next twelve months. However, we cannot guarantee that our existing capital resources and anticipated revenues will be sufficient to conduct and complete all of our research and development programs or commercialization activities as planned.
The spread of COVID-19, which has caused a broad impact globally, may materially affect us economically. While the potential economic impact brought by, and the duration of, the COVID-19 pandemic may be difficult to assess or predict, the pandemic is currently resulting in disruption of global financial markets. This disruption, if sustained or recurrent, could make it more difficult for us to access capital, which could in the future negatively affect our liquidity. In addition, a recession or market correction resulting from the spread of COVID-19 could materially affect our business and the value of our common stock.
We may require additional funding to continue our research and product development programs, to conduct preclinical studies and clinical trials, for operating expenses, to pursue regulatory approvals for our product candidates, for the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims, if any, the cost of product in-licensing and any possible acquisitions, and we may require additional funding to establish manufacturing and marketing capabilities in the future. We may seek to access the public or private
equity markets whenever conditions are favorable. For example, we have an effective shelf registration statement on file with the SEC which allows us to issue an unlimited number of shares of our securities from time to time. In addition, we issued $517.5 million of convertible debt in May 2017 and we have previously financed capital purchases and may continue to pursue opportunities to obtain additional debt financing in the future. In November 2020, we entered into separate, privately negotiated transactions with certain holders of the 2024 Notes to repurchase $136.2 million aggregate principal amount of the 2024 Notes for an aggregate repurchase price of $186.9 million in cash. At December 31, 2020, $381.3 million aggregate principal amount of the 2024 Notes remained outstanding. We may also seek additional funding through strategic alliances or other financing mechanisms. We cannot assure you that adequate funding will be available on terms acceptable to us, if at all. In addition, COVID-19 pandemic is currently resulting in disruption of global financial markets. This disruption, if sustained or recurrent, could make it more difficult for us to access capital, which could in the future negatively affect our liquidity. Any additional equity financings will be dilutive to our stockholders and any additional debt may involve operating covenants that may restrict our business. If adequate funds are not available through these means, we may be required to curtail significantly one or more of our research or development programs or obtain funds through arrangements with collaborators or others. This may require us to relinquish rights to certain of our technologies, products or product candidates. To the extent that we are unable to obtain third-party funding for such expenses, we expect that increased expenses will result in increased cash flow losses from operations. We cannot assure you that we will successfully develop our products under development or that our approved products will generate revenues sufficient to enable us to earn a profit.
Contractual Obligations
The following table presents our contractual obligations at December 31, 2020.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| (in millions) | Total | | 2021 | | 2022 | | 2023 | | 2024 | | 2025 and
Thereafter |
2024 Notes and related interest (1) | $ | 411.5 | | | $ | 8.7 | | | $ | 8.6 | | | $ | 8.6 | | | $ | 385.6 | | | $ | — | |
Operating leases (2) | 159.6 | | | 12.3 | | | 14.9 | | | 15.5 | | | 16.0 | | | 100.9 | |
| Total contractual obligations | $ | 571.1 | | | $ | 21.0 | | | $ | 23.5 | | | $ | 24.1 | | | $ | 401.6 | | | $ | 100.9 | |
(1) In May 2017, we completed a private placement of $517.5 million in aggregate principal amount of 2.25% convertible senior notes scheduled to mature on May 15, 2024, unless earlier converted, redeemed, or repurchased. We may not redeem the 2024 Notes prior to May 15, 2021. On or after this date, at our election, we may redeem all, or any portion, of the 2024 Notes under certain circumstances. The 2024 Notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, the issuance of other indebtedness or the issuance or repurchase of securities by us. There are customary events of default with respect to the 2024 Notes, including that upon certain events of default, 100% of the principal and accrued and unpaid interest on the 2024 Notes will automatically become due and payable. Amounts for the 2024 Notes and related interest in the table above assume that the 2024 Notes will be held until maturity. In November 2020, we entered into separate, privately negotiated transactions with certain holders of the 2024 Notes to repurchase $136.2 million aggregate principal amount of the 2024 Notes for an aggregate repurchase price of $186.9 million in cash. At December 31, 2020, $381.3 million aggregate principal amount of the 2024 Notes remained outstanding.
(2) We lease our corporate headquarters, which consist of laboratory and office space located San Diego, California, under various operating lease agreements. In addition to minimum rental commitments, these operating leases may require us to pay additional amounts for taxes, insurance, maintenance and other operating expenses. The non-cancelable lease terms for these operating leases expire at various dates between 2025 and 2031 and do not include renewal options. Amounts for operating leases presented in the table above reflect future minimum rental commitments under non-cancelable operating leases as of December 31 for each of the periods presented.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Interest Rate Risk
We maintain a diversified investment portfolio consisting of low-risk, investment-grade debt securities with maturities of up to three years, including investments in commercial paper, securities of government-sponsored entities and corporate bonds that are exposedsubject to interest rate risk on our short-term investments.risk. The primary objective of our investment activities is to preserve principal while at the same time maximizing yields without significantly increasing risk. To achieve this objective, we invest in highly liquid and high-quality government and other debt securities. To minimize our exposure due to adverse shifts in interest rates, we invest in short-term securities and ensure that the maximum average maturity of our investments does not exceed twelve months.maintain liquidity. If a 1% unfavorable change in interest rates were to have occurred on December 31, 2020, this change2023, it would not have had a material effect on the fair value of our investment portfolio as of that date. Due to the short holding period of our investments, we have concluded that we do not have a material financial market risk exposure.
Item 8. Financial Statements and Supplementary Data
NEUROCRINE BIOSCIENCES, INC.
INDEX TO THE CONSOLIDATED FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting Firm
To the ShareholdersStockholders and the Board of Directors of Neurocrine Biosciences, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Neurocrine Biosciences, Inc. (the “Company”) as of December 31, 20202023 and 2019,2022, the related consolidated statements of income and comprehensive income, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2020,2023, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 20202023 and 2019,2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2020,2023, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2020,2023, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated February 5, 20219, 2024 expressed an unqualified opinion thereon.
Adoption of New Accounting Standard
ASU No. 2016-02
As discussed in Note 1 to the consolidated financial statements, the Company changed its method of accounting for leases effective January 1, 2019, due to the adoption of Accounting Standards Update (ASU) No. 2016-02, Leases (Topic 842), and the related amendments.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit MattersMatter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below providing a separate opinion on the critical audit matter or on the account or disclosure to which it relates.
| | | | | |
| Reserves for government rebates related to product sales |
| |
| Description of the Matter | The Company sells drugsproduct to specialty pharmacies and specialty distributors in the U.S.US (collectively, “customers”). As described in Note 1 to the consolidated financial statements, the Company recognizes revenues for sales of INGREZZA to its customers after deducting management’s estimates of reserves, including drug coverage gap rebates, it will provide under government rebate programs (“government rebates”). Estimated government rebates are presented within accounts payable and accrued liabilities on the consolidated balance sheets.sheet. |
| |
| Auditing the estimates of government rebates was complex and judgmental due to the level of uncertainty involved in management’s assumptions used in the measurement process. In particular, management was required to estimate for product that remains in the distribution channel at December 31, 2020,2023, the portion of product that is expected to be subject to a government rebate, and the applicable contractual government rebate percentage by forecasting the revenue, the payor type underlying the revenue and the applicable rebate amount applicable for the payor type. |
| | | | | |
| How We Addressed the Matter in Our Audit | We tested the Company’s internal controls over management’s process for estimating the portion of product that is expected to be subject to a government rebate for product that remains in the distribution channel at December 31, 2020, including2023. This included controls over management’s forecastreview of revenuesignificant assumptions and other inputs into the estimation of government rebates including the accuracy of data used in the calculation. |
| |
| To test management’s estimate of government rebate reserves our audit procedures included, among others, evaluating the methodologies used, testing the significant assumptions discussed above and testing the completeness and accuracy of the underlying data used by the Company in its analyses. Specifically, we compared the significant assumptions to third-party reports used by the Company to estimate product remaining in the distribution channelgovernment rebate reserves at December 31, 2020.2023. In addition, we compared the underlying government rebate percentages used in the Company’s analyses to those published by the applicable government entity. We assessed the historical accuracy of management’s rebate estimates, tested payments of rebates and performed a sensitivity analysis of significant assumptions to evaluate the changes in the rebate allowance that would result from changes in the assumptions. |
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 1992.
San Diego, California
February 5, 20219, 2024
NEUROCRINE BIOSCIENCES, INC.
CONSOLIDATED BALANCE SHEETS
| | | | December 31, | | December 31, |
| (in millions, except per share data) | (in millions, except per share data) | 2020 | | 2019 | (in millions, except per share data) | 2023 | | 2022 |
| Assets | Assets | | | |
| Current assets: | Current assets: | |
| Current assets: | |
| Current assets: | |
| Cash and cash equivalents | Cash and cash equivalents | $ | 187.1 | | | $ | 112.3 | |
| Debt securities available-for-sale (amortized cost $612.4 million at December 31, 2020 and $557.3 million at December 31, 2019) | 613.9 | | | 558.2 | |
| Accounts receivable | 157.1 | | | 126.6 | |
| Inventories | 28.0 | | | 17.3 | |
| Cash and cash equivalents | |
| Cash and cash equivalents | |
| Debt securities available-for-sale | |
| Accounts receivable, net | |
| Inventory, net | |
| Other current assets | Other current assets | 30.1 | | | 16.6 | |
| Total current assets | Total current assets | 1,016.2 | | | 831.0 | |
| Debt securities available-for-sale (amortized cost $226.7 million at December 31, 2020 and $299.3 million at December 31, 2019) | 227.1 | | | 299.7 | |
| Deferred tax assets | |
| Debt securities available-for-sale | |
| Right-of-use assets | Right-of-use assets | 82.8 | | | 74.3 | |
| Equity securities | Equity securities | 38.2 | | | 55.9 | |
| Property and equipment, net | Property and equipment, net | 44.6 | | | 41.9 | |
| Deferred tax assets | 319.4 | | | 0 | |
| Restricted cash | 3.2 | | | 3.2 | |
| Other long-term assets | 3.2 | | | 0 | |
| Intangible assets, net | |
| Other assets | |
| Total assets | Total assets | $ | 1,734.7 | | | $ | 1,306.0 | |
| | Liabilities and Stockholders’ Equity | Liabilities and Stockholders’ Equity | |
| Liabilities and Stockholders’ Equity | |
| Liabilities and Stockholders’ Equity | |
| Current liabilities: | Current liabilities: | |
| Current liabilities: | |
| Current liabilities: | |
| Accounts payable and accrued liabilities | |
| Accounts payable and accrued liabilities | |
| Accounts payable and accrued liabilities | Accounts payable and accrued liabilities | $ | 168.7 | | | $ | 141.3 | |
| Convertible senior notes | Convertible senior notes | 0 | | | 408.8 | |
| Other current liabilities | Other current liabilities | 17.8 | | | 15.2 | |
| Total current liabilities | Total current liabilities | 186.5 | | | 565.3 | |
| Convertible senior notes | 317.9 | | | 0 | |
| | Noncurrent operating lease liabilities | |
| Noncurrent operating lease liabilities | |
| Noncurrent operating lease liabilities | Noncurrent operating lease liabilities | 94.4 | | | 86.7 | |
| Other long-term liabilities | Other long-term liabilities | 9.7 | | | 17.1 | |
| Total liabilities | Total liabilities | 608.5 | | | 669.1 | |
| | Stockholders’ equity: | Stockholders’ equity: | |
| Preferred stock, $0.001 par value; 5.0 shares authorized; 0 shares issued and outstanding at December 31, 2020 and 2019 | 0 | | | 0 | |
| Common stock, $0.001 par value; 220.0 shares authorized; issued and outstanding shares were 93.5 million and 92.3 million at December 31, 2020 and 2019, respectively | 0.1 | | | 0.1 | |
| Stockholders’ equity: | |
| Stockholders’ equity: | |
| Preferred stock, $0.001 par value; 5.0 shares authorized; no shares issued and outstanding | |
| Preferred stock, $0.001 par value; 5.0 shares authorized; no shares issued and outstanding | |
| Preferred stock, $0.001 par value; 5.0 shares authorized; no shares issued and outstanding | |
| Common stock, $0.001 par value; 220.0 shares authorized; 98.7 and 96.5 shares issued and outstanding, respectively | |
| Additional paid-in capital | Additional paid-in capital | 1,849.7 | | | 1,768.1 | |
| Accumulated other comprehensive income | 1.8 | | | 1.4 | |
| Accumulated other comprehensive income (loss) | |
| Accumulated deficit | Accumulated deficit | (725.4) | | | (1,132.7) | |
| Total stockholders’ equity | Total stockholders’ equity | 1,126.2 | | | 636.9 | |
| Total liabilities and stockholders’ equity | Total liabilities and stockholders’ equity | $ | 1,734.7 | | | $ | 1,306.0 | |
See accompanying notes to consolidated financial statements.
NEUROCRINE BIOSCIENCES, INC.
CONSOLIDATED STATEMENTS INCOME
AND COMPREHENSIVE INCOME
| | Year Ended December 31, |
| Year Ended December 31, | | | Year Ended December 31, |
| (in millions, except per share data) | (in millions, except per share data) | 2020 | | 2019 | | 2018 | (in millions, except per share data) | 2023 | | 2022 | | 2021 |
| Revenues: | Revenues: | | | | | |
| Product sales, net | $ | 994.1 | | | $ | 752.9 | | | $ | 409.6 | |
| Net product sales | |
| Net product sales | |
| Net product sales | |
| Collaboration revenue | Collaboration revenue | 51.8 | | | 35.2 | | | 41.6 | |
| Total revenues | Total revenues | 1,045.9 | | | 788.1 | | | 451.2 | |
| Operating expenses: | Operating expenses: | |
| Cost of sales | 10.1 | | | 7.4 | | | 4.9 | |
| Cost of revenues | |
| Cost of revenues | |
| Cost of revenues | |
| Research and development | Research and development | 275.0 | | | 200.0 | | | 155.8 | |
| Acquired in-process research and development | Acquired in-process research and development | 164.5 | | | 154.3 | | | 4.8 | |
| Selling, general and administrative | Selling, general and administrative | 433.3 | | | 354.1 | | | 248.9 | |
| Total operating expenses | Total operating expenses | 882.9 | | | 715.8 | | | 414.4 | |
| Operating income | Operating income | 163.0 | | | 72.3 | | | 36.8 | |
| Other (expense) income: | |
| Other income (expense): | |
| Interest expense | Interest expense | (32.8) | | | (32.0) | | | (30.5) | |
| Unrealized loss on restricted equity securities | (17.7) | | | (13.0) | | | 0 | |
| Interest expense | |
| Interest expense | |
| Unrealized gain on equity securities | |
| Loss on extinguishment of convertible senior notes | Loss on extinguishment of convertible senior notes | (18.4) | | | 0 | | | 0 | |
| Investment income and other, net | Investment income and other, net | 12.6 | | | 19.2 | | | 15.5 | |
| Total other expense, net | (56.3) | | | (25.8) | | | (15.0) | |
| Income before (benefit from) provision for income taxes | 106.7 | | | 46.5 | | | 21.8 | |
| (Benefit from) provision for income taxes | (300.6) | | | 9.5 | | | 0.7 | |
| Total other income (expense), net | |
| Income before provision for income taxes | |
| Provision for income taxes | |
| Net income | Net income | 407.3 | | | 37.0 | | | 21.1 | |
| Unrealized gain (loss) on debt securities available-for-sale | 0.4 | | | 3.4 | | | (0.1) | |
| Foreign currency translation adjustments, net of tax | |
| Unrealized gain (loss) on debt securities available-for-sale, net of tax | |
| Comprehensive income | Comprehensive income | $ | 407.7 | | | $ | 40.4 | | | $ | 21.0 | |
| Net income per share, basic | $ | 4.38 | | | $ | 0.40 | | | $ | 0.23 | |
| Net income per share, diluted | $ | 4.16 | | | $ | 0.39 | | | $ | 0.22 | |
| Earnings per share, basic | |
| Earnings per share, diluted | |
| Weighted average common shares outstanding, basic | Weighted average common shares outstanding, basic | 93.1 | | | 91.6 | | | 90.2 | |
| Weighted average common shares outstanding, diluted | Weighted average common shares outstanding, diluted | 97.8 | | | 95.7 | | | 95.4 | |
See accompanying notes to consolidated financial statements.
NEUROCRINE BIOSCIENCES, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Common Stock | | Additional
Paid-In Capital | | Accumulated
Other
Comprehensive Income (Loss) | | Accumulated Deficit | | Total Stockholders’ Equity |
| (in millions) | Shares | | $ | | | | |
| Balances at December 31, 2017 | 88.8 | | | $ | 0.1 | | | $ | 1,572.8 | | | $ | (1.9) | | | $ | (1,198.8) | | | $ | 372.2 | |
| Net income | — | | | — | | | — | | | — | | | 21.1 | | | 21.1 | |
| Unrealized loss on debt securities available-for-sale | — | | | — | | | — | | | (0.1) | | | — | | (0.1) | |
| Share-based compensation expense | — | | | — | | | 58.1 | | | — | | | — | | 58.1 | |
| Issuance of common stock for vested restricted stock units | 0.4 | | | — | | | — | | | — | | | — | | — | |
| Issuance of common stock for stock option exercises | 1.6 | | | — | | | 29.5 | | | — | | | — | | 29.5 | |
| Balances at December 31, 2018 | 90.8 | | | $ | 0.1 | | | $ | 1,660.4 | | | $ | (2.0) | | | $ | (1,177.7) | | | $ | 480.8 | |
| Net income | — | | | — | | | — | | | — | | | 37.0 | | | 37.0 | |
| Unrealized gain on debt securities available-for sale | — | | | — | | | — | | | 3.4 | | | — | | | 3.4 | |
| Share-based compensation expense | — | | | — | | | 75.3 | | | — | | | — | | | 75.3 | |
| Cumulative-effect adjustment to equity due to adoption of ASU 2016-02 | — | | | — | | | — | | | — | | | 8.0 | | | 8.0 | |
| Issuance of common stock for vested restricted stock units | 0.4 | | | — | | | — | | | — | | | — | | | — | |
| Issuance of common stock for stock option exercises | 1.0 | | | — | | | 27.3 | | | — | | | — | | | 27.3 | |
| Issuance of common stock for employee stock purchase plan | 0.1 | | | — | | | 5.1 | | | — | | | — | | | 5.1 | |
| Balances at December 31, 2019 | 92.3 | | | $ | 0.1 | | | $ | 1,768.1 | | | $ | 1.4 | | | $ | (1,132.7) | | | $ | 636.9 | |
| Net income | — | | | — | | | — | | | — | | | 407.3 | | | 407.3 | |
| Unrealized gain on debt securities available-for-sale, net of tax | — | | | — | | | — | | | 0.4 | | | — | | | 0.4 | |
| Share-based compensation expense | — | | | — | | | 100.0 | | | — | | | — | | | 100.0 | |
| Equity component of repurchased convertible senior notes, net | — | | | — | | | (47.5) | | | — | | | — | | | (47.5) | |
| Issuance of common stock for vested restricted stock units | 0.5 | | | — | | | — | | | — | | | — | | | — | |
| Issuance of common stock for stock option exercises | 0.6 | | | — | | | 23.5 | | | — | | | — | | | 23.5 | |
| Issuance of common stock for employee stock purchase plan | 0.1 | | | — | | | 5.6 | | | — | | | — | | | 5.6 | |
| Balances at December 31, 2020 | 93.5 | | | $ | 0.1 | | | $ | 1,849.7 | | | $ | 1.8 | | | $ | (725.4) | | | $ | 1,126.2 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Common Stock | | Additional
Paid-In Capital | | Accumulated
Other
Comprehensive Income (Loss) | | Accumulated Deficit | | Total Stockholders’ Equity |
| (in millions) | Shares | | $ | | | | |
| Balances at December 31, 2020 | 93.5 | | | $ | 0.1 | | | $ | 1,849.7 | | | $ | 1.8 | | | $ | (725.4) | | | $ | 1,126.2 | |
| Net income | — | | | — | | | — | | | — | | | 89.6 | | | 89.6 | |
| Other comprehensive loss, net of tax | — | | | — | | | — | | | (3.5) | | | — | | (3.5) | |
| Stock-based compensation expense | — | | | — | | | 134.2 | | | — | | | — | | 134.2 | |
| Issuances of common stock under stock plans | 1.4 | | | — | | | 27.5 | | | — | | | — | | 27.5 | |
| Balances at December 31, 2021 | 94.9 | | | $ | 0.1 | | | $ | 2,011.4 | | | $ | (1.7) | | | $ | (635.8) | | | $ | 1,374.0 | |
| Net income | — | | | — | | | — | | | — | | | 154.5 | | | 154.5 | |
| Other comprehensive loss, net of tax | — | | | — | | | — | | | (6.2) | | | — | | | (6.2) | |
| Cumulative-effect adjustment due to adoption of ASU 2020-06 | — | | | — | | | (106.8) | | | — | | | 74.5 | | | (32.3) | |
| Stock-based compensation expense | — | | | — | | | 173.1 | | | — | | | — | | | 173.1 | |
| Issuances of common stock under stock plans | 1.6 | | | — | | | 44.7 | | | — | | | — | | | 44.7 | |
| Balances at December 31, 2022 | 96.5 | | | $ | 0.1 | | | $ | 2,122.4 | | | $ | (7.9) | | | $ | (406.8) | | | $ | 1,707.8 | |
| Net income | — | | | — | | | — | | | — | | | 249.7 | | | 249.7 | |
| Other comprehensive income, net of tax | — | | | — | | | — | | | 14.9 | | | — | | | 14.9 | |
| Stock-based compensation expense | — | | | — | | | 194.3 | | | — | | | — | | | 194.3 | |
| Issuances of common stock under stock plans | 2.2 | | | — | | | 65.3 | | | — | | | — | | | 65.3 | |
| Balances at December 31, 2023 | 98.7 | | | $ | 0.1 | | | $ | 2,382.0 | | | $ | 7.0 | | | $ | (157.1) | | | $ | 2,232.0 | |
See accompanying notes to consolidated financial statements.
NEUROCRINE BIOSCIENCES, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | Year Ended December 31, |
| Year Ended December 31, | | | Year Ended December 31, |
| (in millions) | (in millions) | 2020 | | 2019 | | 2018 | (in millions) | 2023 | | 2022 | | 2021 |
| Cash Flows from Operating Activities: | | | | | |
| Cash flows from operating activities: | |
| Net income | Net income | $ | 407.3 | | | $ | 37.0 | | | $ | 21.1 | |
| Reconciliation of net income to net cash provided by operating activities: | |
| Share-based compensation expense | 100.0 | | | 75.3 | | | 58.1 | |
| Net income | |
| Net income | |
| Adjustments to reconcile net income to net cash from operating activities: | |
| Stock-based compensation expense | |
| Stock-based compensation expense | |
| Stock-based compensation expense | |
| Depreciation | Depreciation | 8.6 | | | 7.4 | | | 4.0 | |
| (Accretion) amortization of (discount) premium on investments, net | |
| Amortization of debt discount | Amortization of debt discount | 20.0 | | | 18.9 | | | 17.6 | |
| Amortization of debt issuance costs | Amortization of debt issuance costs | 1.4 | | | 1.4 | | | 1.3 | |
| Change in fair value of equity securities | 17.7 | | | 13.0 | | | 0 | |
| Deferred income taxes (including benefit from valuation allowance release) | (310.7) | | | 0 | | | 0 | |
| Amortization of intangible assets | |
| Changes in fair value of equity securities | |
| Deferred income taxes | |
| Loss on extinguishment of convertible senior notes | Loss on extinguishment of convertible senior notes | 18.4 | | | 0 | | | 0 | |
| Other | Other | 3.7 | | | (1.2) | | | 1.0 | |
| Changes in operating assets and liabilities: | Changes in operating assets and liabilities: | |
| Accounts receivable | Accounts receivable | (30.5) | | | (69.2) | | | (25.1) | |
| Inventories | (10.7) | | | (6.4) | | | (3.5) | |
| Accounts receivable | |
| Accounts receivable | |
| Inventory | |
| Accounts payable and accrued liabilities | Accounts payable and accrued liabilities | 26.9 | | | 54.0 | | | 24.2 | |
| Other assets and liabilities, net | Other assets and liabilities, net | (23.6) | | | 16.8 | | | 2.7 | |
| Net cash provided by operating activities | 228.5 | | | 147.0 | | | 101.4 | |
| Cash flows from operating activities | |
| | Cash Flows from Investing Activities: | |
| Cash flows from investing activities: | |
| Cash flows from investing activities: | |
| Cash flows from investing activities: | |
| Purchases of debt securities available-for-sale | |
| Purchases of debt securities available-for-sale | |
| Purchases of debt securities available-for-sale | Purchases of debt securities available-for-sale | (735.5) | | | (797.2) | | | (545.9) | |
| Sales and maturities of debt securities available-for-sale | Sales and maturities of debt securities available-for-sale | 750.5 | | | 669.7 | | | 327.8 | |
| Acquisition of business, net of cash acquired | |
| Purchases of equity securities | Purchases of equity securities | 0 | | | (68.9) | | | 0 | |
| Purchases of property and equipment | (10.9) | | | (14.7) | | | (24.8) | |
| Net cash provided by (used in) investing activities | 4.1 | | | (211.1) | | | (242.9) | |
| Capital expenditures | |
| Cash flows from investing activities | |
| | Cash Flows from Financing Activities: | |
| Cash flows from financing activities: | |
| Cash flows from financing activities: | |
| Cash flows from financing activities: | |
| Issuances of common stock under benefit plans | Issuances of common stock under benefit plans | 29.1 | | | 32.4 | | | 29.5 | |
| Partial repurchase of convertible senior notes | (186.9) | | | 0 | | | 0 | |
| Net cash (used in) provided by financing activities | (157.8) | | | 32.4 | | | 29.5 | |
| Issuances of common stock under benefit plans | |
| Issuances of common stock under benefit plans | |
| Repurchases of convertible senior notes | |
| Cash flows from financing activities | |
| Effect of exchange rate changes on cash and cash equivalents | |
| Change in cash and cash equivalents and restricted cash | Change in cash and cash equivalents and restricted cash | 74.8 | | | (31.7) | | | (112.0) | |
| Cash and cash equivalents and restricted cash at beginning of period | 115.5 | | | 147.2 | | | 259.2 | |
| Cash and cash equivalents and restricted cash at end of period | $ | 190.3 | | | $ | 115.5 | | | $ | 147.2 | |
| Cash, cash equivalents and restricted cash at beginning of period | |
| Cash, cash equivalents and restricted cash at end of period | |
| | Supplemental Disclosure: | Supplemental Disclosure: | |
| Supplemental Disclosure: | |
| Supplemental Disclosure: | |
| Non-cash capital expenditures | Non-cash capital expenditures | $ | 1.4 | | | $ | 1.0 | | | $ | 2.3 | |
| Right-of-use assets acquired through operating leases | $ | 12.8 | | | $ | 77.1 | | | $ | 0 | |
| Non-cash capital expenditures | |
| Non-cash capital expenditures | |
| Right-of-use assets obtained in exchange for new operating lease liabilities | |
| Cash paid for interest | Cash paid for interest | $ | 11.6 | | | $ | 11.6 | | | $ | 11.6 | |
| Cash paid for income taxes | Cash paid for income taxes | $ | 15.3 | | | $ | 0.5 | | | $ | 0 | |
See accompanying notes to consolidated financial statements.
NEUROCRINE BIOSCIENCES, INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and Summary of Significant Accounting Policies
Business Activities.Organization and Business. Neurocrine Biosciences, Inc., or Neurocrine, and its subsidiaries (Neurocrine Biosciences, the Company, we, our or us, was incorporated in California in 1992 and reincorporated in Delaware in 1996. Neurocrine Continental, Inc.,us) is a Delaware corporation and a wholly owned subsidiary of Neurocrine. We also have 2 wholly-owned Irish subsidiaries, Neurocrine Therapeutics, Ltd. and Neurocrine Europe, Ltd. both of which were formed in December 2014 and are inactive.
We are a neuroscience-focused biopharmaceutical company dedicated tofocused on discovering, developing and delivering life-changing treatmentsinnovative therapies to help ease the burden of debilitating disorders and diseases.
We operate in a single business segment, which includes all activities related to the research, development and commercialization of pharmaceuticals for patients with serious, challengingthe treatment of neurological, neuroendocrine and under-addressed neurological, endocrineneuropsychiatric disorders and psychiatric disorders. Our diverse portfolio includes FDA-approved treatments for tardive dyskinesia, Parkinson’s disease, endometriosis*, uterine fibroids*reflects the way in which internally-reported financial information is regularly reviewed by our chief operating decision maker to analyze performance, make decisions and clinical programs in multiple therapeutic areas. For nearly three decades, we specialize in targeting and interrupting disease-causing mechanisms involving the interconnected pathways of the nervous and endocrine systems. (*in collaboration with AbbVie Inc.)allocate resources.
Principles of Consolidation. The consolidated financial statements include the accounts of Neurocrine Biosciences as well as our wholly owned subsidiaries. All intercompany transactions and balances have been eliminated in consolidation.
Use of Estimates. The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America or GAAP,(GAAP) requires management to make estimates and assumptions that affect the amounts reported in the financial statements and the accompanying notes. Actual results could differ from those estimates.
Industry Segment and Geographic Information. We operate in a single industry segment – the discovery, development and marketing of pharmaceuticals for the treatment of neurological, endocrine and psychiatric-based diseases and disorders. We had no foreign-based operations during any of the years presented.
Reclassifications. Certain amounts in prior year periods have been reclassified to conform with the presentation adopted in the current year.
Cash Equivalents. We consider all highly liquid investments that are readily convertible into cash without penalty and have an original maturity of three months or less at the time of purchase to be cash equivalents.
Accounts Receivable. Accounts receivable are recorded net of customer allowances for prompt payment discounts, chargebacks, and any allowance for doubtful accounts. Wecredit losses. Our estimate for the allowance for doubtful accountscredit losses, which has not been significant to date, is determined based on existing contractual payment terms, actual payment patterns of our customers, and individual customer circumstances. To date,
Our exposure to credit losses may increase if our customers are adversely affected by changes in healthcare laws, coverage and reimbursement, economic pressures or uncertainty associated with local or global economic recessions, or other customer-specific factors.
Inventory. Inventory is valued at the lower of cost or net realizable value. We determine the cost of inventory using the standard-cost method, which approximates actual cost based on the first-in, first-out method. We perform an allowance for doubtful accounts has not been material.assessment of the recoverability of our inventory on a quarterly basis and write down any excess and obsolete inventory to its net realizable value in the period in which the impairment is first identified. When future commercialization is considered probable and the future economic benefit is expected to be realized, based on management’s judgment, we capitalize pre-launch inventory costs prior to regulatory approval.
Debt Securities. Debt securities consist of investments in certificates of deposit, corporate debt securities and securities of government-sponsored entities. We classify debt securities as available-for-sale. Debt securities available-for-sale are recorded at fair value, with unrealized gains and losses included in other comprehensive income or loss, net of tax. We exclude accrued interest from both the fair value and amortized cost basis of debt securities. A debt security is placed on nonaccrual status at the time any principal or interest payments become 90 days delinquent. Interest accrued but not received for a debt security placed on nonaccrual status is reversed against interest income.
Interest income includes amortization of purchase premium or discount. Premiums and discounts on debt securities are amortized using the effective interest rate method. Gains and losses on sales of debt securities are recorded on the trade date in investment income and other, net, and determined using the specific identification method.
Allowance for Credit Losses. For debt securities available-for-sale in an unrealized loss position, we first assess whether we intend to sell, or it is more likely than not that we will be required to sell, the security before recovery of its amortized cost basis. If either of the criteria regarding intent or requirement to sell is met, the security’s amortized cost basis is written down to fair value through earnings. For debt securities available-for-sale that do not meet the aforementioned criteria, we evaluate whether the decline in fair value has resulted from credit losses or other factors. In making this assessment, we consider the extent to which fair value is less than amortized cost, any changes in interest rates, and any changes to the rating of the security by a rating agency, among other factors. If this
assessment indicates that a credit loss exists, the present value of cash flows expected to be collected from the security is compared to the amortized cost basis of the security. If the present value of cash flows expected to be collected is less than the amortized cost basis, a credit loss exists and an allowance for credit losses is recorded, limited by the amount that the fair value is less than the amortized cost basis. Any impairment that has not been recorded through an allowance for credit losses is recognized in other comprehensive income or loss, as applicable.
Accrued interest receivables on debt securities available-for-sale totaled $3.7were $11.2 million atand $4.7 million, respectively, as of December 31, 2020.2023 and 2022. We do not measure an allowance for credit losses for accrued interest receivables. For the purposes of identifying and measuring an impairment, accrued interest is excluded from both the fair value and amortized cost basis of the debt security. Uncollectible accrued interest receivables associated with an impaired debt security are reversed against interest income upon identification of the impairment. NaNNo accrued interest receivables were written off during 2020, 20192023, 2022 or 2018.2021.
Fair Value of Financial Instruments. We record cash equivalents, debt securities available-for-sale and equity securitiessecurity investments at fair value based on a fair value hierarchy that distinguishes between assumptions based on market data (observable inputs) and our own assumptions (unobservable inputs). The fair value hierarchy consists of the following three levels:
Level 1 – Quoted prices (unadjusted) in active markets for identical assets or liabilities.
Level 2 – Quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active or inputs that are observable, either directly or indirectly, for substantially the full term of the asset or liability.
Level 3 – Unobservable inputs that reflect our own assumptions about the assumptions that market participants would use in pricing the asset or liability when there is little, if any, market activity for the asset or liability at the measurement date.
Investments in debt securities available-for-sale are classified as Level 2 and carried at fair value. We estimate the fair value of debt securities available-for-sale by utilizing third-party pricing services. These pricing services utilize industry standard valuation models, including both income and market-based approaches, for which all significant inputs are observable, either directly or indirectly, to estimate fair value. Such inputs include market pricing based on real-time trade data for similar instruments, issuer credit spreads, benchmark yields, broker/dealer quotes and other observable inputs. We validate valuations obtained from third-party pricing services by understanding the models used, obtaining market values from other pricing sources and analyzing data in certain instances.
InvestmentsWe deem transfers between levels of the fair value hierarchy to have occurred at the end of the reporting period during which the event or change in circumstances that caused the transfer occurred.
Equity Investments. We account for certain equity securities of certain companies that areinvestments subject to holding period restrictions longer than one yearthe equity method of accounting, or through which we have the ability to exercise significant influence (but not control) over the operating and financial policies of an investee, under the fair value option. In assessing whether we exercise significant influence, we consider the nature and magnitude of such an investment, the voting and protective rights we hold, any participation in the governance of the investee and other relevant factors, such as the presence of a collaborative or other business relationship. Such investments in publicly traded companies are currently classified aswithin Level 31 of the fair value hierarchy and carried at fair value, using an option pricing valuation model. The most significant assumptions within the option pricing valuation model are the stock price volatility, which is based on the historical volatility of similar companies, and the discount for lack of marketability related to the term of the restrictions.
The carrying amounts of accounts receivable and accounts payable and accrued liabilities approximate their fair values due to their short-term maturities.
There were no transfers between levelswith any changes in the fair value hierarchy during 2020 or 2019.
Inventory. Inventory is valued at the lower of cost or net realizable value. We determine the cost of inventory using the standard-cost method, which approximates actual cost based on the first-in, first-out method. We assess the valuation of our inventory on a quarterly basis and adjust the value for excess and obsolete inventory to the extent management determines that the cost cannot be recovered based on estimates about future demand. Inventory costs resulting from these adjustments aresuch investments recognized as cost of sales in the period in which they are incurred. When future commercialization is considered probable and the future economic benefit is expected to be realized, based on management’s judgment, we capitalize pre-launch inventory costs prior to regulatory approval.earnings.
Property and Equipment. Property and equipment are stated at cost and depreciated over the estimated useful lives of the assets using the straight-line method. Equipment is depreciated over an average estimated useful life of three3 to seven7 years. Leasehold improvements are depreciated over the shorter of their estimated useful lives or the remaining lease term. Depreciation expense was $8.6$17.8 million for 2020, $7.42023, $15.1 million for 20192022 and $4.0$10.9 million for 2018.2021.
ImpairmentBusiness Combinations. Under the acquisition method of accounting, we allocate the fair value of the total consideration transferred to the tangible and identifiable intangible assets acquired and liabilities assumed based on their estimated fair values on the date of acquisition. These valuations require us to make estimates and assumptions, especially with respect to intangible assets. We record the excess consideration over the aggregate fair value of tangible and intangible assets, net of liabilities assumed, as goodwill. In addition, costs that we incur to complete the business combination, such as legal and other professional fees, are expensed as selling, general and administrative when incurred.
Goodwill, Intangible Assets and Other Long-Lived Assets.We review long-livedAssets acquired, including intangible assets and in-process research and development (IPR&D) and liabilities assumed are measured at fair value as of the acquisition date. Goodwill, which has an indefinite useful life, represents the excess of cost over fair value of the net assets acquired. Intangible assets acquired in a business combination that are used for IPR&D activities are considered indefinite lived until the completion or abandonment of the associated research and development efforts. Upon reaching the end of the relevant research and development project (i.e., upon commercialization), the IPR&D asset is amortized over its estimated useful life. If the relevant research and development project is abandoned, the IPR&D asset is expensed in the period of abandonment.
Goodwill and IPR&D are not amortized; however, they are reviewed for impairment whenever events or changes in circumstances indicate thatat least annually, as of October 1, and more frequently if an event occurs indicating the potential for impairment. Goodwill and IPR&D are considered to be impaired if the carrying value of the reporting unit or IPR&D asset exceeds its respective fair value.
We perform our goodwill impairment analysis at the reporting unit level, which aligns with our reporting structure and availability of discrete financial information. During the goodwill impairment review, we assess qualitative factors to determine whether it is more likely than not that the fair value of the reporting unit is less than the carrying amount, including goodwill. The qualitative factors include, but are not limited to, macroeconomic conditions, industry and market considerations, and our overall financial performance. If, after assessing the totality of these qualitative factors, we determine that it is not more likely than not that the fair value of the reporting unit is less than the carrying amount, then no additional assessment is deemed necessary. Otherwise, we proceed to compare the estimated fair value of the reporting unit with the carrying value, including goodwill. If the carrying amount of the reporting unit exceed the fair value, we record an assetimpairment loss based on the difference. We may not be recoverable.elect to bypass the qualitative assessment in a period and proceed to perform the quantitative goodwill impairment test.
Our identifiable intangible assets with a finite life are typically comprised of acquired product rights. The cost of identifiable intangible assets with finite lives is generally amortized on a straight-line basis over the assets’ respective estimated useful lives.
We perform regular reviews to determine if any event has occurred that may indicate that intangible assets with finite useful lives and other long-lived assets are potentially impaired. If indicators of impairment exist, wean impairment test is performed to assess the recoverability of the affected long-lived assets by determining whether the carrying valueamount of such assets can be recovered throughexceeds the undiscounted expected future operating cash flows. If the carrying amount isaffected assets are not recoverable, we measureestimate the amountfair value of anythe assets and record an impairment by comparingloss if the carrying value of the assets exceeds the fair value. Factors that may indicate potential impairment include a significant decline in our stock price and market capitalization compared to the net book value, significant changes in the ability of a particular asset to generate positive cash flows for our strategic business objectives, and the pattern of utilization of a particular asset.
Leases. We determine if an arrangement is a lease at contract inception. Right-of-use (ROU) assets represent our right to use an underlying asset for the lease term and operating lease liabilities represent our obligation to make lease payments arising from the lease. ROU assets and operating lease liabilities are recognized at the commencement date based on the present value of lease payments over the expected future cash flowslease term. When determining the lease term, we include options to extend or terminate the lease when it is reasonably certain that such options will be exercised.
As none of our operating leases provide an implicit rate, we use our incremental borrowing rate based on the information available at commencement date in determining the present value of lease payments. Our incremental borrowing rate is determined using a secured borrowing rate for the same currency and term as the associated lease.
The lease payments used to determine our ROU assets may include prepaid or accrued lease payments and any lease incentives received and are recognized in ROU assets in our consolidated balance sheets.
Our lease agreements may include both lease and non-lease components, which we account for as a single lease component when the payments are fixed. Variable payments included in lease agreements are expensed as incurred.
Our operating leases are reflected in ROU assets, noncurrent operating lease liabilities, and other current liabilities in our consolidated balance sheets. Lease expense for minimum lease payments is recognized on a straight-line basis over the lease term.
Foreign currency.Assets and liabilities are translated into the reporting currency using the exchange rates in effect on the consolidated balance sheet dates. Equity accounts are translated at historical rates, except for the change in retained earnings during the period, which is the result of the income statement translation process. Revenue and expense accounts are translated using the weighted average exchange rate during the period. The cumulative translation adjustments associated with the usenet assets of foreign subsidiaries are recorded in accumulated other comprehensive income (loss) in the asset.accompanying consolidated statements of stockholders’ equity.
Revenue Recognition. We recognize revenue when the customer obtains control of promised goods or services in an amount that reflects the consideration which we expect to receive in exchange for thosesuch goods or services. Revenue is recognized using a five-step model: (i) identify contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenues when (or as) we satisfy the performance obligation. We only apply the five-step model to contracts when it is probable that we will collect the consideration we are entitled to in exchange for the goods or services we transfer to the customer.
Net Product Sales, Net.Sales. In the U.S., our product sales, net consist of sales ofwe sell INGREZZA® (valbenazine) primarily to specialty pharmacy providers and a specialty distributor, and sales of ONGENTYS, primarily to wholesale distributors. We recognize net product sales net when the customer obtains control of our product, which occurs at a point in time, typically upon delivery of our product to the customer.
Revenues from product sales are recorded net of reserves established for applicable discounts and allowances that are offered within contracts with our customers, payors, and other third parties. Such estimates are based on information received from external sources (such as written or oral information obtained from our customers with respect to their period-end inventory levels and sales to end-users during the reporting period), as supplemented by management’s judgement. Our process for estimating reserves established for these variable consideration components does not differ materially from historical practices. The transaction price, which includes variable consideration reflecting the impact of discounts and allowances, may be subject to constraint and is included in the net sales price only to the extent that it is probable that a significant reversal of the amount of the cumulative revenue recognized will not occur in a future period. Actual amounts may ultimately differ from our estimates. If actual results vary, we adjust these estimates, which could have an effect on earnings in the period of adjustment.
Our significant categories of sales discounts and allowances are as follows:
Product Discounts. Product discounts are based on payment terms extended to our customers at the time of sale, which include incentives offered for prompt payment. We maintain a reserve for product discounts based on our historical experience, including the timing of customer payments. To date, actual product discounts have not differed materially from our estimates.
Government Rebates. We are obligated to pay rebates for mandated discounts including under the Medicaid Drug Rebate Program.Program and Medicare Part D. The liability for such rebates consists of invoices received for claims from prior quarters that remain unpaid, or for which an invoice has not been received, and estimated rebates for the current applicable reporting period. Such estimates are based on actual historical rebates by state, estimated payor mix, state and federal regulations, and relevant contractual terms, as supplemented by management’s judgement. Our rebate accrual calculations require us to project the magnitude of our sales by state, that will be subject to these rebates. There is a significant time-lag in our receiving rebate notices from each state (generally, several months or longer after a sale is recognized). Estimated rebates are recorded as a reduction of revenue in the period the related sale is recognized. To date, actual government rebates have not differed materially from our estimates.
Chargebacks. The difference between the list price, or the price at which we sell our products to our customers, and the contracted price, or the price at which our customers sell our products to qualified healthcare professionals, is charged back to us by our customers. In addition to actual chargebacks received, we maintain a reserve for chargebacks based on estimated contractual discounts on product inventory levels on-hand in our distribution channel. To date, actual chargebacks have not differed materially from our estimates.
Payor and Pharmacy Rebates. We are obligated to pay rebates as a percentage of sales under payor and pharmacy contracts. We estimate these rebates based on actual historical rebates, contractual rebate percentages, sales made through the payor channel, and purchases made by pharmacies. To date, actual payor and pharmacy rebates have not differed materially from our estimates.
Co-paymentPatient Financial Assistance. WeTo help patients afford our products, we offer financial assistance to qualified patients with prescription drug co-payments required by insurance.copay requirements. We accrue for copaypatient financial assistance based on estimated claims and the cost per claim we expect to receive associatedin connection with inventory that remains in the distribution channel at period end. To date, actual copaypatient financial assistance has not differed materially from our estimates.
Distributor and Other Fees. In connection with the sales of our products, we pay distributor and other fees, which are generally recorded as a reduction of revenue, to certain customers that provide us with inventory management, data, and/or distribution services. Costs associated with such services are expensed as selling, general and distribution services, which are generally recorded as a reduction of revenue. Toadministrative to the extent we can demonstrate a separable benefit and fair value for these services, we classify the associated costs in selling, generalsuch services. To date, actual distributor and administrative expenses. These costs are typically known at the time of sale, resulting in minimal adjustments subsequent to the period of sale.other fees have not differed materially from our estimates.
Product Returns. For INGREZZA, we offer our customers product return rights primarily limited to errors in shipment and damaged product. We do not permit returns of INGREZZA for expiring or expired product. Accordingly, we have limited return risk resulting from INGREZZA product sales and therefore do not record an associated returns allowance. For ONGENTYS, we offer our customers product return rights primarily limited to errors in shipment, damaged product, and expiring or expired product, provided it is within a specified period aroundof the product expiration date, as set forth in the associated distribution agreement. Once product is returned, it is destroyed. Where actual returns history is not available, we estimate the associateda returns allowance based on benchmarking data for similar products and industry experience. We record this estimateSuch estimates are recorded as a reduction of revenue in the period the related sale is recognized. Once product is returned, it is destroyed. To date, actual product returns have not differed materially from our estimates.
Collaboration Revenues. We have entered into collaboration and licensinglicense agreements under which we licenseout-license certain rights to our product candidates to third parties. The terms of these arrangements typically include payment to us offor one or more of the following: non-refundable, up-front license fees; development, regulatory, and/or commercial milestone payments; and royalties on net sales of licensedthe out-licensed products.
Licenses of Intellectual Property. If the license to our intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, we recognize revenue from non-refundable, up-front fees allocated to the license when the license is transferred to the customer and the customer is able to use, and benefit from, the license. For licenses that are bundled with other promises, we use judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligationit is satisfied over time or at a point in time and, if over time, the appropriate method of measuring progress for purposes of recognizing revenue from non-refundable, up-front fees. We evaluate the measure of progress each reporting period and, if necessary, adjust the measure of performance and related revenue recognition.
Milestone Payments.Milestones. At the inception of each arrangement that includes developmental,development, regulatory, and/or commercial milestone payments,milestones, we evaluate whether achieving the milestones is considered probable and estimate the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the value of the associated milestone is included in the transaction price. Milestone paymentsAmounts for milestones that are not within our control, such as approvals from regulators or where attainmentwhen achievement of thea specified event is dependent on the development activities of a third party or approvals from regulators, are not considered probable of being achieved until those approvals are received or thesuch specified event occurs. Revenue is recognized from the satisfaction of performance obligations in the amount billable to the customer.
Royalty Revenues.Royalties. For arrangements that include sales-based royalties, including milestone payments based on the level of sales, and under which the license is deemed to be the predominant item to which the royalties relate, we recognize revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied). Each quarterly period, sales-based royalties are recorded based on estimated quarterly net sales of the associated collaboration products. Differences between actual results and estimated amounts are adjusted for in the period in which they become known, which typically follows the quarterly period in which the estimate was made. To date, actual royalties received have not differed materially from our estimates.
Concentration of Credit Risk. Financial instruments that potentially subject us to concentration of credit risk consist primarily of cash and cash equivalents, accounts receivable and marketable securities.debt securities available-for-sale. We have established guidelines to limit our exposure to credit risk by diversifying our investment portfolio with low-risk investment-grade debt securities with maturities of up to three years and by placing our investments with high credit qualityhigh-credit-quality financial
institutions and maturities thatto maintain safety and liquidity. To date, we have not experienced any credit losses and do not believe we are exposed to any significant credit risk in relationconnection with these financial instruments.
We have entered into agreements for the distribution of INGREZZA with a limited number of specialty pharmacy providers and distributors and all of our product sales of INGREZZA are to these financial instruments.
We are also subject to credit risk from our accounts receivable related to our product sales. Our two largestcustomers. Four of these customers represented approximately 86%91% of our total product revenuessales for both 20202023 and 2019, and the significant majority of our accounts receivable balances at December 31, 2020 and 2019. For 2018, our three largest customers represented approximately 93% of our product revenue and substantially all98% of our accounts receivable balance atas of December 31, 2018. To date, we have not experienced any significant losses with respect to the collection of these accounts receivable.2023.
Cost of Sales.Revenues. Cost of salesrevenues includes third-party manufacturing, transportation, freight, and indirect overhead costs associated withprimarily for the manufacture and distribution of INGREZZA and ONGENTYS,drug product sold, manufacturing costs in connection with our supply of valbenazine drug product under our collaboration with Mitsubishi Tanabe Pharma Corporation, royalty fees on net sales of ORILISSA and ORIAHNN,elagolix, amortization of intangible assets, and adjustments for excess and obsolete inventory to the extent management determines that the cost cannot be recovered based on estimates about future demand.
Research and Development, Expenses.or R&D. R&D expenses primarily consist primarily of salaries, payroll taxes, employee benefits and share-based compensation charges for those individuals involved in ongoing R&D efforts; as well as scientific consulting fees, preclinical and clinical trial costs, payroll and benefits costs, including stock-based compensation associated with employees involved in R&D facilities costs, laboratory supplyactivities, certain facility-based costs, and depreciation of scientific equipment.costs associated with our collaborative arrangements, including event-based milestones. All such costs are charged toexpensed as R&D expense aswhen incurred. These expenses result from our independent R&D efforts, as well as efforts associated with collaborations, in-licenses and third-party funded research arrangements, including event based milestones.
Asset Acquisitions. We account for acquisitions of an asset or groupassets (or groups of assetsassets) that do not meet the definition of a business using the cost accumulation method, whereby the cost of the acquisition, including certain transaction costs, is allocated to the assets (or group of assets) acquired on the basis of their relative fair values.value(s) on the measurement date. No goodwill is recognized in an asset acquisition. Intangible assets that are acquired in an asset acquisition for use in R&D activities which have no alternative future use are expensed as in-process research and development, or IPR&D on the acquisition date. Future costs to develop these assets are recorded toexpensed as R&D expense as they arewhen incurred.
Advertising Expense. Advertising costs are expensed as selling, general and administrative when services are performed, or goods are delivered. We incurred advertising costs related to INGREZZA and ONGENTYS of $64.8incurred. Advertising expense was $159.9 million for 2020, $40.62023, $149.7 million for 20192022 and $20.5$139.8 million for 2018.2021.
Share-BasedStock-Based Compensation. We grant stock options to purchase our common stock to eligible employees and directors and also grant certain employees restricted stock units or RSUs,(RSUs) and performance-based restricted stock units or PRSUs.(PRSUs). Additionally, we allow employees to participate in an employee stock purchase plan or ESPP.(ESPP).
We estimate the fair value of stock options and shares to be issued under the ESPP using the Black-Scholes option-pricing model on the date of grant. Restricted stock unitsRSUs are valued based on the closing price of our common stock on the date of grant. The fair value of equity instruments expected to vest areis recognized and amortized on a straight-line basis over the requisite service period of the award, which is generally three to four years; however, certain provisions in our equity compensation plans provide for shorter vesting periods under certain circumstances. The fair value of shares to be issued under the ESPP areis recognized and amortized on a straight-line basis over the purchase period, which is generally six months. Additionally, we granted certain PRSUs that vest upon the achievement of certain predefined company-specific performance-based criteria. Expense related to these PRSUs is generally recognized ratably over the expected performance period once the predefined performance-based criteria for vesting becomes probable.
Income Taxes. Our income tax benefit (provision)provision is computed under the asset and liability method. Significant estimates are required in determining our income tax benefit (provision).provision. Some of these estimates are based on interpretations of existing tax laws or regulations. We recognize deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts (temporary differences) at enacted tax rates in effect for the years in which the differences are expected to reverse. A valuation allowance is established for deferred tax assets for which it is more likely than not that some portion or all of the deferred tax assets, including net operating losses and tax credits, will not be realized. We periodically re-assess the need for a valuation allowance against our deferred tax assets based on various factors including our historical earnings experience by
taxing jurisdiction, and forecasts of future operating results and utilization of net operating losses and tax credits prior to their expiration. Significant judgment is required in making this assessment and, to the extent that a reversal of any portion of our valuation allowance against our deferred tax assets is deemed appropriate, a tax benefit will be recognized against our income tax provision in the period of such reversal. Prior to 2020, we recorded a valuation allowance that fully offset our deferred tax assets. On December 31, 2020, based on our evaluation of various factors, such as our achievement of a cumulative three-year income position as of December 31, 2020, as well as our consideration of forecasts of future operating results and utilization of net operating losses and tax credits prior to their expiration, we released substantially all of our valuation allowance against our deferred tax assets and recorded a corresponding income tax benefit. Refer to Note 9 to the consolidated financial statements for more information.
We recognize tax benefits from uncertain tax positions only if it is more likely than not that the tax position will be sustained upon examination by the tax authorities based on the technical merits of the position. An adverse resolution of one or more of these uncertain tax positions in any period could have a material impact on the results of operations for that period.
Net IncomeEarnings Per Share. Basic net incomeearnings per share isare computed using the weighted average number of common shares outstanding during the period. Diluted net incomeearnings per share isare computed using the treasury stock and if-converted methods and reflect the weighted average number of common and potentially dilutive shares outstanding during the period, includingexcluding those which effect would be anti-dilutive.
In 2021, we entered into the potentially dilutive shares resulting fromFirst Supplemental Indenture to the conversion of the 2024 Notes and excluding the effect of stock options and restricted stock outstanding for periods when their effect is anti-dilutive, using the treasury stock method.
Convertible debt instruments that may be settled entirely or partly in cash (such as the 2024 Notes) may, in certain circumstances where the borrower has the ability and intent2017 Indenture, pursuant to settle in cash, be accounted for under the treasury stock method. We issued the 2024 Notes with a combination settlement feature, which we have the ability and intent to use upon conversion of the 2024 Notes,irrevocably elected to settle the principal amount of debt forthe 2.25% fixed-rate convertible senior notes due May 15, 2024 in cash upon conversion and the excess of the principal portionto settle any conversion premium in either cash or shares of our common stock. As a result, of the approximately 5.0 million shares underlying the 2024 Notes at December 31, 2020, only the shares required to settle the excess of the principal portion would beany conversion premium are considered dilutive under the treasury stockif-converted method. Further, approximately 0.2 million PRSUs werefor which the performance condition has not been achieved are excluded from the calculation of diluted net incomeearnings per share.
2. Collaboration and License Agreements
Heptares Therapeutics Limited, or Heptares. We entered into a collaboration and license agreement with Heptares, which became effective in December 2021, to develop and commercialize certain compounds containing sub-type selective muscarinic M1, M4, or dual M1/M4 receptor agonists, for which we have the exclusive rights to develop, manufacture and commercialize worldwide, excluding in Japan, where Heptares retains the rights to develop, manufacture, and commercialize all compounds comprised of M1 receptor agonists, subject to certain exceptions. With respect to such rights retained by Heptares, we retain the rights to opt in to profit sharing arrangements, pursuant to which we and Heptares will equally share in the operating profits and losses for such compounds in Japan. Subject to specified conditions, we may elect to exercise such opt-in rights with respect to each such compound either before initiation of the first proof of concept Phase 2 clinical trial for such compound or following our receipt from Heptares of the top-line data from such clinical trial for such compound. We are responsible for all development, manufacturing, and commercialization costs of any collaboration product.
In connection with the agreement, we paid Heptares $100.0 million upfront, which, including certain transaction-related costs, was expensed as IPR&D in 2021 as the performance condition haslicense had no foreseeable alternative future use. We accounted for the transaction as an asset acquisition as the set of acquired assets did not been achieved. constitute a business.
In loss periods, basic net loss per share and diluted net loss per share are identical becauseconnection with the otherwise dilutive potential common shares become anti-dilutive and are therefore excluded.FDA's acceptance of our investigational new drug application for NBI-1117568 for the treatment of schizophrenia in June 2022, we paid Heptares a milestone of $30.0 million, which was expensed as R&D in 2022.
Recently Adopted Accounting Pronouncements.
ASU 2016-13.On January 1, 2020, we adopted Accounting Standards Update, or ASU, 2016-13,Financial Instruments – Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments,usingUnder the modified retrospective transition method. For debt securities available-for-sale, the standard requires an investor to determine whether a decline in the fair value below the amortized cost basisterms of the investment is dueagreement, Heptares may be entitled to credit-related factors. Credit-related impairment is recognized as an allowance for credit lossreceive potential future payments of up to $2.6 billion upon the achievement of certain event-based milestones and would be entitled to receive royalties on the balance sheet with a corresponding adjustment to earnings. Credit losses are limited to the amount by which the investment’s amortized cost basis exceeds its fair value and may be subsequently reversed if conditions change. Any impairment that is not credit related is recognized in other comprehensive income or loss, as applicable,future net sales of applicable taxes.
The adoption of ASU 2016-13 did not result in a cumulative-effect adjustment to retained earnings. The comparative prior period information continues to be reported under the accounting standards in effect during those periods.
Recently Issued Accounting Pronouncements.
ASU 2019-12.In December 2019, the FASB issued ASU 2019-12,Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes, which simplifies the accounting for income taxes by removing certain exceptions to the general principles in Topic 740 and amends existing guidance to improve consistent application of Topic 740. ASU 2019-12 is effective for fiscal years beginning after December 15, 2020, including interim periods within those fiscal years, with early adoption permitted in any interim period for which financial statements have not yet been made available for issuance. We are currently evaluating the effect ASU 2019-12 will have on our condensed consolidated financial statements and related disclosures.
ASU 2020-06. In August 2020, the FASB issued ASU 2020-06,Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging – Contracts in Entity's Own Equity (Subtopic 815-40): Accountingcollaboration product.
Unless earlier terminated, the agreement will continue on a licensed product-by-licensed product and country-by-country basis until the date on which the royalty term for Convertible Instrumentssuch licensed product has expired in such country. On a licensed product-by-licensed product and Contracts in an Entity's Own Equity, which simplifiescountry-by-country basis, royalty payments would commence on the accounting for certain financial instruments with characteristicsfirst commercial sale of liabilitiesa licensed product and equity, including convertible instruments, and amends existing earnings-per-share, or EPS, guidance by requiring that an entity useterminate on the if-converted method when calculating diluted EPS for convertible instruments. ASU 2020-06 is effective for fiscal years beginning after December 15, 2021, including interim periods within those fiscal years, with early adoption permitted for fiscal years beginning after December 15, 2020, including interim periods within those fiscal years. We plan to adopt ASU 2020-06 effective January 1, 2022 and are currently evaluatinglater of (i) the effect ASU 2020-06 will have on our consolidated financial statements and related disclosures.
2. License and Collaboration Agreements
Under the termsexpiration of the last patent covering such licensed product in such country, (ii) a number of years from the first commercial sale of such licensed product in such country and (iii) the expiration of regulatory exclusivity for such licensed product in such country.
We may terminate the agreement in its entirety or with respect to one or more targets upon 180 days’ written notice to Heptares during the research collaboration term and upon 90 days’ written notice to Heptares following licensethe expiration of the research collaboration term. Following the expiration of the research collaboration term, Heptares may terminate the agreement on a target-by-target basis in the event that we do not conduct any material development activities outside of Japan with respect to a certain compound or licensed product within the applicable target class for a continuous period of not less than 365 days and collaboration agreements, wedo not commence any such activities within 120 days of receiving written notice. Either party may be requiredterminate the agreement, subject to make milestone payments upon achievementspecified conditions, (i) in the event of material breach by the other party, subject to a cure period, (ii) if the other party challenges the validity or enforceability of certain development and regulatory activities of upintellectual property rights, subject to $8.5 billion and pay royalties on future sales,a cure period, or (iii) if any, of commercial products resulting from these agreements.the other party becomes insolvent or takes certain actions related to insolvency.
Takeda Pharmaceutical Company Limited, or Takeda.. We In 2020, we entered into an exclusive license agreement with Takeda, Pharmaceutical Company Limited, or Takeda,pursuant to which became effective in July 2020,we acquired the exclusive rights to develop and commercialize certain early-to-mid stage psychiatry compounds, in Takeda’s early to mid-stage psychiatry pipeline. Specifically, Takeda granted us an exclusive license to the following seven assets: (i) NBI-1065844 (TAK-831) for schizophrenia, (ii)including luvadaxistat, NBI-1065845, (TAK-653) for treatment-resistant depression, (iii) NBI-1065846 (TAK-041) for anhedonia (which together with the NBI-1065845 are referred to as the Phase II Ready Assets), and (iv) four non-clinical stage assets, orcompounds. Luvadaxistat and the Non-Clinical Assets.
NBI-1065844 is deemedfour non-clinical stage compounds have each been designated as a royalty-bearing product under the license agreement pursuant to which we will beproduct. NBI-1065845 and NBI-1065846 are currently each designated as a profit-share product. We are responsible for all costs and expenses associated with themanufacturing, development, manufacture, and commercialization costs of such asset, subject to certain exceptions, and Takeda will be eligible to receive development and commercial milestones and royalties withany royalty-bearing product.
With respect to such asset, or a Royalty-Bearing Product,NBI-1065845 and Takeda will retain the right to opt-in to a profit sharing arrangement pursuant to whichNBI-1065846, we and Takeda will equally share in the operating profits and losses related to such asset, subject to certain exceptions, in lieu of receiving milestones and royalties, or a Profit-Share Product. Subject to specified conditions,losses. Takeda may elect to exercise such opt-in right for NBI-1065844 before we initiate a Phase III clinical trial. Each ofretains the Phase II Ready Assets is deemed a Profit-Share Product and Takeda will retain the rightrights to opt-out of the profit-sharing arrangement for such assetarrangements, pursuant to which Takeda would be entitled to receive potential future payments upon the achievement of certain event-based milestones with respect to such asset would become a Royalty-Bearing Product.compounds and receive royalties on the future net sales of such compounds (in lieu of equally sharing in the operating profits and losses). Takeda may elect to exercise such opt-out rights with respect to a Phase II Ready Assetright for such compound immediately following the completion of thea second Phase II2 clinical trial for such Phase II Ready Asset. In addition,compound, or, under certain circumstances related to the development and commercialization activities to be performed by us, Takeda may elect to opt-out of the profit-sharing arrangement for a Profit-Share Product before the initiation of a Phase III3 clinical trial for such product.compound.
Each of the Non-Clinical Assets will be Royalty-Bearing Products pursuant to which we will be responsible for all costs and expenses associated with the development, manufacture, and commercialization of such assets, subject to certain exceptions.
In connection with the agreement,approval of our clinical trial application for NBI-1070770 for the treatment of major depressive disorder in 2022, we paid Takeda $120.0a milestone of $5.0 million, upfront, which including certain transaction related costs, was expensed as in-process research and development, or IPRR&D in the third quarter of 2020. Pursuant to2022.
Under the terms of the agreement, Takeda may also be entitled to receive additionalpotential future payments of up to $1.9 billion upon the achievement of certain event-based milestones associated with Royalty-Bearing Products, as well asand would be entitled to receive royalties on the future net sales of Royalty-Bearing Products. any royalty-bearing product.
Unless earlier terminated, the agreement will continue on a licensed product-by-licensed product and country-by-country basis until the date on which, (i) for any royalty-bearing product, the royalty term has expired in such country; and (ii) for any profit-share product, for so long as we continue to develop, manufacture, or commercialize such licensed product.On a country-by-countrylicensed product-by-licensed product and product-by-productcountry-by-country basis, royalty payments would commence on the first commercial sale of a Royalty-Bearing Productroyalty-bearing product and terminate on the later of (i) the expiration of the last patent covering such Royalty-Bearing Productroyalty-bearing product in such country, (ii) a number of years from the first commercial sale of such Royalty-Bearing Productroyalty-bearing product in such country and (iii) the expiration of regulatory exclusivity for Royalty-Bearing Productsuch royalty-bearing product in such country.
We may terminate the agreement in its entirety or in one or more (but not all) of the U.S., Japan, the European Union (EU) and the United Kingdom (UK) (collectively, the major markets) upon six months’ written notice to Takeda (i) with respect to all licensed products prior to the first commercial sale of the first licensed product for which first commercial sale occurs, or (ii) with respect to all licensed products in one or more given target classes, as defined in the agreement, prior to the first commercial sale of the first licensed product in such target class for which first commercial sale occurs. We may terminate the agreement in its entirety or in one or more (but not all) of the major markets upon 12 months’ written notice to Takeda (i) with respect to all licensed products following the first commercial sale of the first licensed product for which first commercial sale occurs, or (ii) with respect to all licensed products in one or more given target classes following the first commercial sale of the first licensed product in such target class for which first commercial sale occurs. Takeda may terminate the agreement, subject to specified conditions, (i) if we challenge the validity or enforceability of certain Takeda intellectual property rights or (ii) on a target class-by-target class basis, in the event that we do not conduct any material development or commercialization activities with respect to any licensed product within such target class for a specified continuous period. Subject to a cure period, either party may terminate the agreement in the event of any material breach, solely with respect to the target class of a licensed product to which such material breach relates, or in its entirety in the event of any material breach that relates to all licensed products.
Idorsia Pharmaceuticals Ltd., or Idorsia. In May 2020, we entered into a collaboration and licensinglicense agreement with Idorsia, Pharmaceuticals Ltd, or Idorsia,pursuant to licensewhich we acquired the global rights to NBI-827104, (ACT-709478), a potent, selective, orally active and brain penetrating T-type calcium channel blocker in clinical development for the treatment of a rare pediatric epilepsy. The agreement also includes a research collaboration to discoverepilepsy and identify additional novel T-type calcium channel blockers as development candidates.
In connection with the exercise of the option, we paid Idorsia $45.0 million upfront, which we expensed as IPR&D in the second quarter of 2020. Further, as part of the research collaboration, we provided Idorsia with an incremental $7.2 million in funding, which we recorded as a prepaid asset and is being expensed over the two-year research collaboration term.
Pursuant to the terms of the agreement, upon the achievement of certain development and regulatory milestones, Idorsia may be entitled to receive additional payments of up to $365.0 million with respect to NBI-827104 and $620.0 million with respect to the development candidates. Idorsia may also be entitled to receive additional payments of up to $750.0 million upon the achievement of certain commercial milestones, as well as receive royalties on the future net sales of any collaboration product. Further, we will beother potential indications, including essential tremor. We are responsible for all manufacturing, development, and commercialization costs of any collaboration product.
Xenon Pharmaceuticals, Inc. In December 2019, we entered into a license and collaboration agreement with Xenon Pharmaceuticals Inc., or Xenon, to identify, research, and develop sodium channel inhibitors, including clinical candidate NBI-921352 (XEN901) and three preclinical candidates, which compounds we will have the exclusive right to further develop and commercialize under the terms and conditions set forth in the agreement.
We will be solely responsible, at our sole cost and expense, for all development and manufacturing of the compounds and any pharmaceutical product that contains a compound, subject to Xenon’s right to elect to co-fund the development of one product in a major indication and thus receive a mid-single digit percentage increase in royalties owed on the net sales of such product in the U.S. If Xenon exercises such option, the parties will share equally all reasonable and documented costs and expenses incurred in connection with the development of such product in the applicable indication, except costs and expenses that are solely related to the development of such product for regulatory approval outside the U.S.
In connection with the agreement, we paid Xenon $30.0 million upfront and purchased $20.0 million of Xenon’s common stock at $14.196 per share, representing approximately 1.4 million shares. Pursuant toUnder the terms of the agreement, XenonIdorsia may also be entitled to receive additionalpotential future payments of up to $1.7 billion upon the achievement of certain event-based milestones as well asand would be entitled to receive royalties on the future net sales of any collaboration product.
We may terminate the agreement, in its entirety or with respect to a particular compound or development candidate, upon 90 days’ written notice to Idorsia. Further, in the event a party commits a material breach and fails to cure such material breach within 90 days after receiving written notice thereof, the non-breaching party may terminate the agreement in its entirety immediately upon written notice to the breaching party.
Xenon Pharmaceuticals Inc., or Xenon. In 2019, we entered into a collaboration and license agreement with Xenon to identify, research and develop sodium channel inhibitors, including NBI-921352 and three preclinical candidates, which compounds we have the exclusive rights to develop and commercialize. We are responsible for all development and manufacturing costs of any collaboration product, subject to certain exceptions.
In connection with the agreement, we purchased approximately 1.4 million shares (at $14.196 per share) of Xenon common stock in 2019. The purchased shares were recorded at a fair value of $14.1 million after considering Xenon’s stock price and certain transfer restrictions that were applicable to the shares on the measurement date.
In connection with the regulatory approval of our clinical trial application in Europe for NBI-921352 for the treatment of focal onset seizures in adults in 2021, we paid Xenon a regulatory milestone of $10.0 million, including a purchase of approximately 0.3 million shares (at $19.9755 per share) of Xenon common stock. The purchased shares were recorded at a fair value of $4.6 million after considering Xenon’s stock price and certain transfer restrictions that were applicable to the shares on the measurement date. The remaining $5.4 million of the milestone payment was expensed as R&D in 2021.
In connection with the FDA's acceptance of our amended KAYAKTM study protocol in 2022, we paid Xenon a regulatory milestone of $15.0 million, including a purchase of approximately 0.3 million shares (at $31.855 per share) of Xenon common stock. The purchased shares were recorded at a fair value of $7.7 million after considering Xenon’s stock price on the measurement date. The remaining $7.3 million of the milestone payment was expensed as R&D in 2022.
Under the terms of the agreement, Xenon may be entitled to receive potential future payments of up to $1.7 billion upon the achievement of certain event-based milestones and would be entitled to receive royalties on the future net sales of any collaboration product. Xenon retains the right to elect to co-develop one product in a major indication, pursuant to which Xenon would receive a mid-single digit percentage increase in royalties earned on the future net sales of such product in the U.S. and we and Xenon would equally share in the development costs of such product in the applicable indication, except where such development costs relate solely to the regulatory approval of such product outside the U.S.
Unless earlier terminated, the agreement will continue on a licensed product-by-licensed product and country-by-country basis until the expiration of the royalty term for such product in such country. Upon the expiration of the royalty term for a particular licensed product and country, the license obtained by us with respect to such product and country will become fully paid, royalty free, perpetual and irrevocable. We may terminate the agreement upon 90 days’ written notice to Xenon, provided that such unilateral termination will not be effective for certain products until we have used commercially reasonable efforts to complete certain specified clinical studies. Either party may terminate the agreement in the event of a material breach in whole or in part, subject to specified conditions.
Voyager Therapeutics, Inc., or Voyager.
2019 Voyager Agreement. In 2019, we entered into a collaboration and license agreement with Voyager (the 2019 Voyager Agreement), pursuant to which we retain certain rights to develop and commercialize the Friedreich’s ataxia program and two undisclosed programs. We are responsible for all development and commercialization costs of any collaboration product under the 2019 Voyager Agreement, subject to certain co-development and co-commercialization rights retained by Voyager.
In connection with the 2019 Voyager Agreement, we purchased approximately 4.2 million shares (at $11.9625 per share) of Voyager common stock (the 2019 Purchased Shares), which are subject to certain transfer, beneficial ownership, and voting restrictions for a period of up to three years from the effective date of the 2023 Voyager Agreement (defined below). The 2019 Purchased Shares were recorded at a fair value of $54.7 million after considering Voyager’s stock price and certain transfer restrictions that were applicable to the shares on the measurement date.
Under the terms of the 2019 Voyager Agreement, Voyager may be entitled to receive potential future payments of up to $1.3 billion upon the achievement of certain event-based milestones and would be entitled to receive royalties on the future net sales of any collaboration product, subject to certain co-development and co-commercialization rights retained by Voyager.
Unless terminated earlier, the 2019 Voyager Agreement will continue in effect until the expiration of the last to expire royalty term with respect to any collaboration product under the agreement or the last expiration or termination of any exercised co-development and co-commercialization rights by Voyager as provided for in the 2019 Voyager Agreement. We may terminate the 2019 Voyager Agreement upon 180 days’ written notice to Voyager prior to the first commercial sale of any collaboration product under the 2019 Voyager Agreement or upon one year after the date of notice if such notice is provided after the first commercial sale of any collaboration product under the 2019 Voyager Agreement.
2023 Voyager Agreement. In 2023, we entered into a collaboration and license agreement with Voyager (the 2023 Voyager Agreement), pursuant to which we acquired the global rights to the gene therapy products directed to the gene that encodes glucosylceramidase beta 1 (GBA1) for the treatment of Parkinson's disease and other diseases associated with GBA1 (the GBA1 Program), and three gene therapy programs directed to rare central nervous system (CNS) targets, each enabled by Voyager's next-generation TRACERTM capsids. With respect to collaboration products subject to the GBA1 Program, we are responsible for all development and commercialization costs of any such products, including in the U.S., where Voyager retains certain co-development and co-commercialization rights. Voyager may elect to exercise such rights, pursuant to which we and Voyager would equally share in the operating profits and losses of such products in the U.S. (in lieu of Voyager being entitled to receive potential future payments of certain event-based milestones upon their achievement in the U.S. and receive royalties on the future net sales of such products in the U.S.), following Voyager’s receipt of the top-line data from a first Phase 1 clinical trial for each such product. Irrespective of Voyager’s election to exercise such rights, Voyager may be entitled to receive potential future payments upon the achievement of certain event-based milestones outside the U.S. and would be entitled to receive royalties on the future net sales of any such product outside the U.S. With respect to collaboration products subject to the three gene therapy programs directed to rare CNS targets, we are responsible for all development and commercialization costs for any such products.
In connection with the 2023 Voyager Agreement, we paid Voyager $175.0 million upfront, including a purchase of approximately 4.4 million shares (at $8.88 per share) of Voyager common stock (the 2023 Purchased Shares), which are subject to certain transfer, beneficial ownership, and voting restrictions for a period of up to three years from the effective date of the 2023 Voyager Agreement. In addition, as part of the collaboration, Jude Onyia, Ph.D., Chief Scientific Officer of Neurocrine, was appointed to Voyager's board of directors with an initial term expiring in 2024. Mr. Onyia (or another individual designated by us) will be nominated for election to Voyager's board of directors annually for a maximum duration of 10 years from the effective date of the 2023 Voyager Agreement. As a result, our strategic investment in Voyager became subject to the equity method of accounting, and Voyager became a related party under ASC 850, following our purchase of the 2023 Purchased Shares, after which, together with the 2019 Purchased Shares, we owned approximately 19.9% of the voting stock of Voyager. We elected the fair value option to account for our strategic investment in Voyager as we believe it creates greater transparency regarding the investment's fair value at future reporting dates. The 2023 Purchased Shares were recorded at a fair value of $31.3 million after considering Voyager’s stock price on the measurement date. The remaining $143.9 million of the purchase price, which includes certain transaction-related costs, was expensed as IPR&D in 2023 as the license had no foreseeable alternative future use. We accounted for the transaction as an asset acquisition as the set of acquired assets did not constitute a business. Our equityWe recognized unrealized gains of $15.5 million for 2023 and $14.5 million for 2022 and an unrealized loss of $8.7 million for 2021 on our strategic investment in Xenon was recorded at aVoyager. As of December 31, 2023, the fair value (Level 1) of $14.1 million after considering Xenon’s stock price on the date of closing and certain lock-up and voting provisions applicable to the acquired shares. The remaining $36.2 million of the purchase price, which includes the applicable transaction costs,our strategic investment in Voyager was expensed as IPR&D in the fourth quarter of 2019.$72.4 million.
Voyager Therapeutics, Inc.We entered into a collaboration and license agreement with Voyager Therapeutics, Inc., or Voyager, which became effective in March 2019, to develop and commercialize four programs using Voyager’s proprietary gene therapy platform. The four programs consist of the NBIb-1817 (VY-AADC) program for Parkinson’s disease, the Friedreich’s ataxia program and the rights to two undisclosed programs.
In connection with the agreement, we paid Voyager $115.0 million upfront and purchased $50.0 million of Voyager’s common stock at $11.9625 per share, representing approximately 4.2 million shares. Pursuant toUnder the terms of the agreement,2023 Voyager Agreement, Voyager may also be entitled to receive additionalpotential future payments of up to $1.7$6.1 billion upon the achievement of certain event-based milestones as well as receive royalties on the future net sales of any collaboration product.
Pursuant to development plans agreed to by us and Voyager, unless Voyager exercises its co-development and co-commercialization rights as provided for in the agreement, we will be responsible for all development costs. Further, upon the occurrence of a specified event for each program, we will assume responsibility for the development, manufacturing, and commercialization activities of such program.
We accounted for the transaction as an asset acquisition as the set of acquired assets did not constitute a business. Our equity investment in Voyager was recorded at a fair value of $54.7 million after considering Voyager’s stock price on the date of closing and certain lock-up and voting provisions applicable to the acquired shares. The remaining $113.1 million of the purchase price, which includes the applicable transaction costs, was expensed as in-process research and development, or IPR&D, in the first quarter of 2019.
In June 2019, we entered into an amendment to the collaboration and license agreement with Voyager. Under the terms of the amendment, we paid Voyager $5.0 million upfront to obtain rights outside the U.S. to the Friedreich’s ataxia program in connection with the early return of those rights to Voyager pursuant to a restructuring of Voyager’s gene therapy relationship with Sanofi Genzyme. The upfront payment was expensed as IPR&D in the second quarter of 2019.
On February 2, 2021, we notified Voyager of our termination of the NBIb-1817 for Parkinson’s disease program. The effective date of this termination will be August 2, 2021. The termination does not apply to any other development program other than NBIb-1817 for Parkinson’s disease, and our collaboration and license agreement with Voyager will otherwise continue in effect.
BIAL – Portela & Ca, S.A.We acquired the U.S. and Canada rights to ONGENTYS® from BIAL in the first quarter of 2017. We launched ONGENTYS in the U.S. in September 2020, after receiving FDA approval for ONGENTYS as an adjunctive therapy to levodopa/DOPA decarboxylase inhibitors in adult Parkinson's disease patients in April 2020. FDA approval for ONGENTYS for Parkinson’s disease resulted in a $20.0 million event-based payment to BIAL, which we expensed as R&D in the second quarter of 2020. We further recognized R&D expense of $10.0 million in each 2019 and 2018 in connection with BIAL’s achievement of certain regulatory event-based milestones related to then ongoing development of ONGENTYS. Pursuant to the terms of the agreement, BIAL may alsowould be entitled to receive additional payments of up to $75.0 million upon the achievement of certain event-based milestones.
Under the terms of the agreement, we are responsible for the commercialization of ONGENTYS in the U.S. and Canada. Further, we rely on BIAL for the commercial supply of ONGENTYS. Upon our written request prior to the estimated expiration of the term of a licensed product, the parties shall negotiate a good faith continuation of BIAL’s supply of such licensed product after the term. After the term, and if BIAL is not supplying a certain licensed product, we shall pay BIAL a trademark royalty based on the net sales of such licensed product.
Upon commercialization of ONGENTYS, we determined certain annual sales forecasts. In the event we fail to meet the minimum sales requirements for a particular year, we would be obligated to pay BIAL an amount equal to the difference between the actual net sales and minimum sales requirements for such year.
Mitsubishi Tanabe Pharma Corporation.In March 2015, we entered into a collaboration and license agreement with Mitsubishi Tanabe Pharma Corporation, or MTPC, for the development and commercialization of INGREZZA for movement disorders in Japan and other select Asian markets.
Since inception of the agreement, we have recognized revenue of $19.8 million associated with the delivery of a technology license and existing know-how and $15.0 million associated with the achievement of a certain event-based milestone. We further recognized revenue of $2.7 million in 2020 and $0.9 million in 2019 in connection with the ongoing KINECT-HD study, a placebo-controlled Phase III study of valbenazine in adult Huntington’s disease patients with chorea. In accordance with our continuing performance obligations, $6.7 million of the $30.0 million upfront payment received from MTPC is being deferred and will be recognized as revenue over the ongoing study period using an input method according to costs incurred to-date relative to estimated total costs associated with the study.
Pursuant to the terms of the agreement, we may also be entitled to receive additional payments of up to $70.0 million upon the achievement of certain event-based milestones, receive payments for the manufacture of certain pharmaceutical products, as well as receive royalties on the future net sales of any collaboration product, subject to certain co-development and co-commercialization rights retained by Voyager.
Unless terminated earlier, the 2023 Voyager Agreement will continue in select territories in Asia.
Undereffect until the termsexpiration of the last to expire royalty term with respect to any collaboration product under the 2023 Voyager Agreement or the last expiration or termination of any exercised co-development and co-commercialization rights by Voyager as provided for in the 2023 Voyager Agreement. We may terminate the 2023 Voyager Agreement upon 180 days’ written notice to Voyager prior to the first commercial sale of any collaboration product under the 2023 Voyager Agreement or upon one year after the date of notice if such notice is provided after the first commercial sale of any collaboration product under the 2023 Voyager Agreement.
BIAL – Portela & Ca, S.A., or BIAL. In 2017, we received from BIAL a license to commercialize and market ONGENTYS® (opicapone) in the U.S. and Canada. We launched ONGENTYS in the U.S. as an FDA-approved add-on treatment to levodopa/carbidopa in patients with Parkinson's disease experiencing motor fluctuations in 2020. In 2023, we provided BIAL with written notice of termination of the license agreement MTPC is responsibleto commercialize and market ONGENTYS in the U.S. and Canada, and recognized reserves for all third-party development, marketing, and commercialization costsONGENTYS inventory obsolescence totaling $5.2 million in cost of revenues in connection with the termination, which became effective in December 2023, as management determined the cost cannot be recovered.
Mitsubishi Tanabe Pharma Corporation, or MTPC.We out-licensed the rights to valbenazine in Japan and other select Asian markets to MTPC in 2015. In 2020, we entered into a commercial supply agreement with MTPC, pursuant to which we supply MTPC with valbenazine drug product for commercial use in such markets. MTPC is responsible for all development, manufacturing, and we would be entitled to a percentagecommercialization costs of sales of INGREZZAvalbenazine in such markets.
MTPC launched DYSVAL® (valbenazine) in Japan for the treatment of tardive dyskinesia in June 2022 and subsequently in other select Asian markets, where it is marketed as REMLEAS® (valbenazine). We receive royalties at tiered percentage rates on MTPC net sales of valbenazine. In connection with MTPC's first commercial sale of DYSVAL in Japan, we received a milestone payment of $20.0 million in 2022. ASC 606 provides a royalty exception for a sales-based or usage-based royalty promised in exchange for a license of intellectual property. Under the royalty exception, the milestone would be recognized as revenue only when the later of (1) the subsequent sale or usage occurs or (2) the performance obligation to which some or all of the sales-based or usage-based royalty has been allocated has been satisfied (or partially satisfied). As the milestone related to a license of intellectual property and was contingent upon MTPC’s first commercial sale of DYSVAL in Japan, the milestone was recognized as revenue in 2022.
Under the terms of our license agreement with MTPC, we may be entitled to receive potential future payments of up to $30.0 million upon the achievement of certain sales-based milestones and are entitled to receive royalties at tiered percentage rates on future MTPC net sales of valbenazine for the longer of ten10 years or the life of the related patent rights. Further,MTPC may terminate the collaboration effort between the partiesagreement upon 180 days’ written notice to advance INGREZZA towards commercialization in Japan and other select Asian markets is governed by joint steering and development committees with representatives from both parties.
us. In such event, all out-licensed product rights would revert to us.AbbVie Inc. In June 2010, we entered into an exclusive worldwide collaboration with AbbVie Inc., or AbbVie. We out-licensed the global rights to elagolix to AbbVie to developin 2010. AbbVie is responsible for all development and commercialize elagolix and all next-generation gonadotropin-releasing factor, or GnRH, antagonists and collectively, GnRH Compounds, for women’s and men’s health.commercialization costs of elagolix.
AbbVie received approval forlaunched ORILISSA® (elagolix tablets) in the U.S. for the managementtreatment of moderate to severe pain associated with endometriosis pain in women from the FDA in JulyAugust 2018 and Health CanadaORIAHNN® (elagolix, estradiol and norethindrone acetate capsules and elagolix capsules) in October 2018. In May 2020, AbbVie received FDA approval for ORIAHNNthe U.S. for the managementtreatment of heavy menstrual bleeding associated withdue to uterine fibroids in pre-menopausal women.June 2020. We recognized sales-basedreceive royalties at tiered percentage rates on AbbVie net sales of ORILISSAelagolix and ORIAHNN of $19.2 million in 2020, $14.3 million in 2019 and $1.6 million in 2018.
FDA approval for ORIAHNN for uterine fibroids resulted in the achievement of a $30.0 million event-based milestone, which we recognized as collaboration revenue in the second quarter of 2020. In 2019, we recognized collaborationelagolix royalty revenue of $20.0$16.7 million in connection with the FDA’s acceptance of AbbVie’s NDA submission for the approval of ORIAHNN2023, $21.2 million for uterine fibroids. In 2018, we recognized collaboration revenue of $40.02022 and $22.3 million in connection with the FDA’s approval for ORILISSA for endometriosis.2021.
Since inception of the agreement, we have recognized revenue of $75.0 million associated with the delivery of a technology license and existing know-how and $165.0 million associated with the achievement of certain event-based milestones. Pursuant toUnder the terms of theour license agreement with AbbVie, we may also be entitled to receive additionalpotential future payments of up to $366.0 million upon the achievement of certain event-based milestones.
Under the terms of the agreement, AbbVie is responsible for all third-party development, marketing,milestones and commercialization costs. We are entitled to areceive royalties at tiered percentage of worldwiderates on future AbbVie net sales of GnRH Compoundselagolix for the longer of ten10 years or the life of the related patent rights. AbbVie may terminate the agreement upon 180 days’ written notice to us. In such event, all out-licensed product rights would revert to us.
3. Debt Securities
The following table summarizespresents the amortized cost, unrealized gain and loss recognized in accumulated other comprehensive income (loss), allowance for credit losses, and fair value of debt securities available-for-sale, at December 31, 2020, aggregated by major security type and contractual maturity:maturity.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| (in millions) | Contractual
Maturity | | Amortized
Cost | | Unrealized
Gain | | Unrealized
Loss | | Allowance for Credit Losses | | Fair
Value |
| Commercial paper | Within 1 year | | $ | 82.2 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 82.2 | |
| Corporate debt securities | Within 1 year | | 299.3 | | | 1.4 | | | 0 | | | 0 | | | 300.7 | |
| Securities of government-sponsored entities | Within 1 year | | 230.9 | | | 0.1 | | | 0 | | | 0 | | | 231.0 | |
| | | $ | 612.4 | | | $ | 1.5 | | | $ | 0 | | | $ | 0 | | | $ | 613.9 | |
| | | | | | | | | | | |
| Corporate debt securities | 1 to 2 years | | $ | 144.8 | | | $ | 0.4 | | | $ | 0 | | | $ | 0 | | | $ | 145.2 | |
| Securities of government-sponsored entities | 1 to 2 years | | 81.9 | | | 0.1 | | | (0.1) | | | 0 | | | 81.9 | |
| | | $ | 226.7 | | | $ | 0.5 | | | $ | (0.1) | | | $ | 0 | | | $ | 227.1 | |
The following table summarizes the amortized cost, unrealized gain and loss recognized in accumulated other comprehensive income, and fair value of debt securities available-for-sale at December 31, 2019, aggregated by major security type and contractual maturity:
| | | | December 31,
2023 | | | | | December 31,
2023 | | December 31,
2022 |
| (in millions) | (in millions) | Contractual
Maturity | | Amortized
Cost | | Unrealized
Gain | | Unrealized
Loss | | Fair
Value | (in millions) | Contractual
Maturity | | Amortized
Cost | | Unrealized Gain | | Unrealized Loss | | Fair
Value | | Amortized
Cost | | Unrealized Gain | | Unrealized Loss | | Fair
Value |
| Commercial paper | Commercial paper | Within 1 year | | $ | 144.5 | | | $ | 0 | | | $ | 0 | | | $ | 144.5 | |
| Corporate debt securities | Corporate debt securities | Within 1 year | | 270.5 | | | 0.5 | | | 0 | | | 271.0 | |
| Securities of government-sponsored entities | Securities of government-sponsored entities | Within 1 year | | 142.3 | | | 0.4 | | | 0 | | | 142.7 | |
| $ | 557.3 | | | $ | 0.9 | | | $ | 0 | | | $ | 558.2 | |
| | | $ | |
| | Corporate debt securities | Corporate debt securities | 1 to 2 years | | $ | 250.5 | | | $ | 0.5 | | | $ | (0.1) | | | $ | 250.9 | |
| Corporate debt securities | |
| Corporate debt securities | |
| Securities of government-sponsored entities | Securities of government-sponsored entities | 1 to 2 years | | 48.8 | | | 0 | | | 0 | | | 48.8 | |
| $ | 299.3 | | | $ | 0.5 | | | $ | (0.1) | | | $ | 299.7 | |
| | | $ | |
The following table summarizes debt securities available-for-sale in an unrealized loss position for which an allowance for credit losses has not been recorded at December 31, 2020, aggregated by major security type and length of time in a continuous unrealized loss position:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Less Than 12 Months | | 12 Months or Longer | | Total |
| (in millions) | Fair
Value | | Unrealized
Loss | | Fair
Value | | Unrealized
Loss | | Fair
Value | | Unrealized
Loss |
| Securities of government-sponsored entities | $ | 95.0 | | | $ | (0.1) | | | $ | 0 | | | $ | 0 | | | $ | 95.0 | | | $ | (0.1) | |
At December 31, 2020, our security portfolio consisted of 148 securities related to investments in debt securities available-for-sale, of which 30 securities were in an unrealized loss position.
Our investments in corporate debt securities in an unrealized loss position at December 31, 2020 are of high credit quality (rated A or higher). Unrealized losses on theseour available-for-sale debt security investments were primarily due to changes in interest rates. WeThese investments are of high credit quality, and we do not intend to sell these investments and it is not more likely than not that we will be required to sell these investments before recovery of their amortized cost basis. No allowance for credit losses was recognized as of December 31, 2023 or 2022.
The following table summarizespresents debt securities available-for-sale that were in an unrealized loss position atas of December 31, 2019,2023, aggregated by major security type and length of time in a continuous loss position.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Less Than 12 Months | | 12 Months or Longer | | Total |
| (in millions) | Fair
Value | | Unrealized
Loss | | Fair
Value | | Unrealized
Loss | | Fair
Value | | Unrealized
Loss |
| | | | | | | | | | | |
| Corporate debt securities | $ | 265.1 | | | $ | (0.4) | | | $ | 183.8 | | | $ | (1.0) | | | $ | 448.9 | | | $ | (1.4) | |
| Securities of government-sponsored entities | $ | 214.6 | | | $ | (0.2) | | | $ | 16.7 | | | $ | (0.4) | | | $ | 231.3 | | | $ | (0.6) | |
The following table presents debt securities available-for-sale that were in an unrealized loss position:position as of December 31, 2022, aggregated by major security type and length of time in a continuous loss position.
| | Less Than 12 Months | | 12 Months or Longer | | Total |
| Less Than 12 Months | | | Less Than 12 Months | | 12 Months or Longer | | Total |
| (in millions) | (in millions) | Fair
Value | | Unrealized
Loss | | Fair
Value | | Unrealized
Loss | | Fair
Value | | Unrealized
Loss | (in millions) | Fair
Value | | Unrealized
Loss | | Fair
Value | | Unrealized
Loss | | Fair
Value | | Unrealized
Loss |
| Commercial paper | |
| Corporate debt securities | Corporate debt securities | $ | 186.1 | | | $ | (0.1) | | | $ | 0 | | | $ | 0 | | | $ | 186.1 | | | $ | (0.1) | |
| Securities of government-sponsored entities | |
4. Fair Value Measurements
Investments at December 31, 2020,The following table presents a summary of financial assets, which were measured at fair value on a recurring basis, consisted of the following:basis.
| | | | | | | | | | | | | | | | | | | | | | | |
| | | Fair Value Measurements Using |
| (in millions) | Fair Value | | Level 1 | | Level 2 | | Level 3 |
| Cash and cash equivalents: | | | | | | | |
| Cash and money market funds | $ | 187.1 | | | $ | 187.1 | | | $ | 0 | | | $ | 0 | |
| Total cash and cash equivalents | 187.1 | | | 187.1 | | | 0 | | | 0 | |
| Restricted cash: | | | | | | | |
| Certificates of deposit | 3.2 | | | 3.2 | | | 0 | | | 0 | |
| Total restricted cash | 3.2 | | | 3.2 | | | 0 | | | 0 | |
| Debt securities available-for-sale: | | | | | | | |
| Commercial paper | 82.2 | | | 0 | | | 82.2 | | | 0 | |
| Corporate debt securities | 445.9 | | | 0 | | | 445.9 | | | 0 | |
| Securities of government-sponsored entities | 312.9 | | | 0 | | | 312.9 | | | 0 | |
| Total debt securities available-for-sale | 841.0 | | | 0 | | | 841.0 | | | 0 | |
| Equity securities: | | | | | | | |
| Equity securities–biotechnology industry | 38.2 | | | 0 | | | 0 | | | 38.2 | |
| Total equity securities | 38.2 | | | 0 | | | 0 | | | 38.2 | |
| Total recurring fair value measurements | $ | 1,069.5 | | | $ | 190.3 | | | $ | 841.0 | | | $ | 38.2 | |
Investments at December 31, 2019, which were measured at fair value on a recurring basis, consisted of the following:
| | | | | | | | | | | | | | | | | | | | | | | |
| | | Fair Value Measurements Using |
| (in millions) | Fair Value | | Level 1 | | Level 2 | | Level 3 |
| Cash and cash equivalents: | | | | | | | |
| Cash and money market funds | $ | 112.3 | | | $ | 112.3 | | | $ | 0 | | | $ | 0 | |
| Total cash and cash equivalents | 112.3 | | | 112.3 | | | 0 | | | 0 | |
| Restricted cash: | | | | | | | |
| Certificates of deposit | 3.2 | | | 3.2 | | | 0 | | 0 |
| Total restricted cash | 3.2 | | | 3.2 | | | 0 | | 0 |
| Debt securities available-for-sale: | | | | | | | |
| Commercial paper | 144.5 | | | 0 | | 144.5 | | | 0 |
| Corporate debt securities | 521.9 | | | 0 | | 521.9 | | | 0 |
| Securities of government-sponsored entities | 191.5 | | | 0 | | 191.5 | | | 0 |
| Total debt securities available-for-sale | 857.9 | | | 0 | | 857.9 | | | 0 |
| Equity securities: | | | | | | | |
| Equity securities–biotechnology industry | 55.9 | | | 0 | | 0 | | 55.9 | |
| Total equity securities | 55.9 | | | 0 | | 0 | | 55.9 | |
| Total recurring fair value measurements | $ | 1,029.3 | | | $ | 115.5 | | | $ | 857.9 | | | $ | 55.9 | |
The following table presents a reconciliation of equity securities, which were measured at fair value on a recurring basis using significant unobservable inputs (Level 3):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions) | 2020 | | 2019 | | 2018 |
| Beginning balance | $ | 55.9 | | | $ | 0 | | | $ | 0 | |
| Purchases | 0 | | | 68.9 | | | 0 | |
| Unrealized loss included in earnings | (17.7) | | | (13.0) | | | 0 | |
| Ending balance | $ | 38.2 | | | $ | 55.9 | | | $ | 0 | |
At December 31, 2020, the discount for lack of marketability used in the valuation analysis of equity securities ranged from 15.0% to 34.0% (weighted average of 24.8%). The discount for lack of marketability was weighted by the relative fair value of the instruments. A significant increase (decrease) in the discount for lack of marketability in isolation would result in a significantly lower (higher) fair value measurement. Unrealized gains and losses on equity securities are included in other income (expense), net. | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| December 31,
2023 | | December 31,
2022 |
| Fair
Value | | Leveling | | Fair
Value | | Leveling |
| (in millions) | | Level 1 | | Level 2 | | | | | Level 1 | | Level 2 | | |
| Cash and money market funds | $ | 251.1 | | | $ | 251.1 | | | $ | — | | | | | $ | 262.9 | | | $ | 262.9 | | | $ | — | | | |
| Restricted cash | 8.0 | | | 8.0 | | | — | | | | | 7.8 | | | 7.8 | | | — | | | |
| Commercial paper | 53.5 | | | — | | | 53.5 | | | | | 156.0 | | | — | | | 156.0 | | | |
| Corporate debt securities | 867.2 | | | — | | | 867.2 | | | | | 548.4 | | | — | | | 548.4 | | | |
| Securities of government-sponsored entities | 547.3 | | | — | | | 547.3 | | | | | 321.4 | | | — | | | 321.4 | | | |
| Equity securities | 161.9 | | | 161.9 | | | — | | | | | 102.1 | | | 102.1 | | | — | | | |
| $ | 1,889.0 | | | $ | 421.0 | | | $ | 1,468.0 | | | | | $ | 1,398.6 | | | $ | 372.8 | | | $ | 1,025.8 | | | |
5. Convertible Senior Notes
On May 2, 2017, we completed a private placement of $517.5 million in aggregate principal amount of 2.25% fixed-rate convertible senior notes due May 15, 2024 or the(the 2024 Notes,Notes) and entered into an indenture agreement, or the 20242017 Indenture with respect to the 2024 Notes. TheInterest on the 2024 Notes accrue interest at a fixed rate of 2.25% per year, payable semiannually in arrearsis due semi-annually on May 15 and November 15 of each year, beginning on November 15, 2017. The 2024 Notes mature on May 15, 2024. The net proceeds from the issuanceyear.
In 2020, we repurchased $136.2 million aggregate principal amount of the 2024 Notes werefor an aggregate repurchase price of $186.9 million in cash. In 2022, we repurchased $210.8 million aggregate principal amount of the 2024 Notes for an aggregate repurchase price of $279.0 million in cash, which resulted in the recognition of a $70.0 million loss on extinguishment.
The following table presents a summary of the 2024 Notes as of December 31, 2023.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Principal
Amount | | Unamortized Issuance Costs | | Net Carrying
Amount | | Fair Value |
| (in millions) | | | | Amount | | Leveling |
| 2024 Notes | $ | 170.4 | | | $ | (0.3) | | | $ | 170.1 | | | $ | 295.7 | | | Level 2 |
The following table presents a summary of the 2024 Notes as of December 31, 2022.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Principal
Amount | | Unamortized Issuance Costs | | Net Carrying
Amount | | Fair Value |
| (in millions) | | | | Amount | | Leveling |
| 2024 Notes | $ | 170.4 | | | $ | (1.0) | | | $ | 169.4 | | | $ | 268.0 | | | Level 2 |
The following table presents a summary of the interest expense of the 2024 Notes.
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Coupon interest | $ | 3.9 | | | $ | 5.9 | | | $ | 8.5 | |
| Amortization of debt discount and issuance costs | 0.7 | | | 1.2 | | | 17.3 | |
| Total interest expense | $ | 4.6 | | | $ | 7.1 | | | $ | 25.8 | |
The initial conversion rate for the 2024 Notes, which is subject to adjustment in some events (as provided for in the 2017 Indenture), is 13.1711 shares of common stock per $1,000 principal amount and equivalent to an initial conversion price of approximately $502.8 million, after deducting commissions$75.92 per share, reflecting a conversion premium of approximately 42.5% above the closing price of $53.28 per share of our common stock on April 26, 2017.
We may redeem for cash all or part of the 2024 Notes if the last reported sale price (as defined in the 2017 Indenture) of our common stock has been at least 130% of the conversion price then in effect (equal to $98.70 as of December 31, 2023) for at least 20 trading days (whether or not consecutive) during any 30 consecutive trading-day period ending on, and including, the offering expenses payable by us.trading day immediately before the date which we provide notice of redemption.
Holders of the 2024 Notes may convert the 2024 Notes at any time prior to the close of business on the business day immediately preceding May 15, 2024, only under the following circumstances:
(i)during any calendar quarter (and only during such calendar quarter), if the last reported sale price of our common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than 130% of the conversion price (equal to $98.70 as of December 31, 2023) on each applicable trading day;
(ii)during the 5five business-day period immediately after any 5five consecutive trading-day period (the measurement period) in which the trading price (as defined in the 20242017 Indenture) per $1,000 principal amount of the
2024 Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price of our common stock and the conversion rate on each such trading day;
(iii)upon the occurrence of specified corporate events, including a merger or a sale of all or substantially all of our assets; or
(iv)if we call the 2024 Notes for redemption, until the close of business on the business day immediately preceding the redemption date.
On or after January 15, 2024, untilUntil the close of business on the scheduled trading day immediately preceding May 15, 2024, holders of the 2024 Notes may convert theirthe 2024 Notes at any time.
Upon On January 4, 2024, we provided notice to the holders of the 2024 Notes electing to settle all conversions of the 2024 Notes in cash. As such, upon conversion, holders will receive the principal amount of their 2024 Notes and any excess conversion value,premium, calculated based on the per share volume-weighted average price for each of the 30 consecutive trading days during the observation period (as more fully described in the 20242017 Indenture). For both the principal and excess conversion value, holders may receive cash, shares of our common stock or a combination of cash and shares of our common stock, at our option.
It is our intent and policy to settle conversions through combination settlement, which essentially involves repayment of an amount of cash equal to the “principal portion” and delivery of the “share amount”, in excess of the principal portion in shares of common stock or cash. In general, for each $1,000 in principal, the “principal portion” of cash upon settlement is defined as the lesser of $1,000, and the conversion value during the 25-day observation period as described in the notes. The conversion value is the sum of the daily conversion value which is the product of the effective conversion rate divided by 25 days and the daily volume-weighted average price, or VWAP, of our common stock. The “share amount” is the cumulative “daily share amount” during the observation period, which is calculated by dividing the daily VWAP into the difference between the daily conversion value (i.e., conversion rate x daily VWAP) and $1,000.
The initial conversion rate for the 2024 Notes is 13.1711 shares of common stock per $1,000 principal amount, which is equivalent to an initial conversion price of approximately $75.92 per share of our common stock. At the initial conversion rate, settlement of the 2024 Notes for shares of our common stock would approximate 5.0 million shares. The conversion rate will be subject to adjustment in some events but will not be adjusted for any accrued and unpaid interest. The initial conversion price of the 2024 Notes represented a premium of approximately 42.5% to the closing sale price of $53.28 per share of our common stock on the Nasdaq Global Select Market on April 26, 2017, the date that we priced the private offering of the 2024 Notes.
In the event of conversion, holders would forgo all future interest payments, any unpaid accrued interest and the possibility of further stock price appreciation. Upon the receipt of conversion requests, the settlement of the 2024 Notes will be paid pursuant to the terms of the 2024 Indenture. In the event that all of the 2024 Notes are converted, we would be required to repay the outstanding principal value and any conversion premium in any combination of cash and shares of its common stock (at our option).
We may not redeem the 2024 Notes prior to May 15, 2021. On or after May 15, 2021, we may redeem for cash all or part of the 2024 Notes if the last reported sale price (as defined in the 2024 Indenture) of our common stock has been at least 130% of the conversion price then in effect for at least 20 trading days (whether or not consecutive) during any 30 consecutive trading-day period ending on, and including, the trading day immediately before the date which we provide notice of redemption. The redemption price will equal the sum of (i) 100% of the principal amount of the 2024 Notes being redeemed, plus (ii) accrued and unpaid interest, including additional interest, if any, to, but excluding, the redemption date. No sinking fund is provided for the 2024 Notes.
If we undergo a fundamental change as(as defined in the 2024 Indenture,2017 Indenture), subject to certain conditions, holders of the 2024 Notes may require us to repurchase for cash all or part of their 2024 Notes at a repurchase price equal to 100% of the principal amount of the 2024 Notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date. In addition, if a ‘‘make-whole fundamental change’’change (as defined in the 20242017 Indenture) occurs prior to January 15, 2024, we will,would, in certain circumstances, increase the conversion rate for a holder who elects to convert their notes in connection with the make-whole fundamental change.
The 2024 Notes are our general unsecured obligations that rank senior in right of payment to all of our indebtedness that is expressly subordinated in right of payment to the 2024 Notes, and equal in right of payment to our unsecured indebtedness.
We are required to separately account for the liability and equity components of the 2024 Notes, as they may be settled entirely or partially in cash upon conversion in a manner that reflects our economic interest cost. The liability component of the instrument was valued in a manner that reflects the market interest rate for a similar nonconvertible instrument at the date of issuance. The initial carrying value of the liability component of $368.3 million was calculated using a 7.5% assumed borrowing rate. The equity component of $149.2 million, representing the conversion option, was determined by deducting the fair value of the liability component from the par value of the 2024 Notes and was recorded in additional paid-in capital on the consolidated balance sheet at the issuance date. That equity component is treated as a discount on the liability component of the 2024 Notes, which is amortized over the seven-year term of the 2024 Notes using the effective interest rate method. The equity component is not re-measured as long as it continues to meet the conditions for equity classification. At December 31, 2020, the remaining period over which the discount on the liability component will be amortized was approximately 3.4 years.
We allocated the total transaction costs of approximately $14.7 million related to the issuance of the 2024 Notes to the liability and equity components of the 2024 Notes based on their relative values. Transaction costs attributable to the liability component are amortized to interest expense over the seven-year term of the 2024 Notes, and transaction costs attributable to the equity component are netted with the equity component in stockholders’ equity.
The 2024 Notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, the issuance of other indebtedness or the issuance or repurchase of securities by us. The 20242017 Indenture contains customary events of default with respect to the 2024 Notes, including that upon certain events of default, 100% of the principal and accrued and unpaid interest on the 2024 Notes will automatically become due and payable.
In November 2020, we entered into separate, privately negotiated transactions with certain holders of
6. Goodwill and Intangible Assets
The following table presents the 2024 Notes to repurchase $136.2 million aggregate principal amount ofchanges in the 2024 Notes for an aggregate repurchase price of $186.9 million in cash. We accounted for the partial repurchase of the 2024 Notes as a debt extinguishment. As a result, we attributed $130.7 million of the aggregate repurchase price to the liability component based on the fair value of the liability component immediately before extinguishment. The fair value of the liability component was calculated at settlement using a discounted cash flow analysis with a discount rate of 3.37%, which was the market rate for similar notes that have no conversion rights. The difference of $56.3 million between the fair value of the aggregate consideration remitted to certain holders of the 2024 Notes and the fair value of the liability component was attributed to the reacquisition of the equity component and recognized as a reduction to additional paid-in capital. The carrying amount of the liability of $112.4 million at settlement was recognized as a reduction to convertible senior notes and resultedgoodwill. Goodwill is included in an $18.4 million loss on extinguishment.other assets in our consolidated balance sheets.
| | | | | |
| (in millions) | Amount |
| Balance as of December 31, 2021 | $ | — | |
| Goodwill recognized in connection with business combination | 5.2 | |
| Foreign currency translation adjustments | 0.2 | |
| Balance as of December 31, 2022 | 5.4 | |
| Foreign currency translation adjustments | 0.4 | |
| Balance as of December 31, 2023 | $ | 5.8 | |
The 2024 Notes,following table presents information relating to our recognized intangible assets as of December 31, 2023.
| | | | | | | | | | | | | | | | | | | | | | | |
| (dollars in millions) | Useful Life | | Gross Carrying Amount | | Accumulated Amortization | | Net
Carrying Amount |
| Developed product rights | 10 years | | $ | 35.9 | | | $ | 4.0 | | | $ | 31.9 | |
| Acquired IPR&D | Indefinite | | $ | 3.6 | | | $ | — | | | 3.6 | |
| Total intangible assets, net | | | | | | | $ | 35.5 | |
The following table presents approximate future annual amortization expense for our finite-lived intangible assets as of December 31, 2023.
| | | | | |
| (in millions) | Amount |
| Year ending December 31, 2024 | $ | 3.6 | |
| Year ending December 31, 2025 | $ | 3.6 | |
| Year ending December 31, 2026 | $ | 3.6 | |
| Year ending December 31, 2027 | $ | 3.6 | |
| Year ending December 31, 2028 | $ | 3.6 | |
| Thereafter | $ | 13.9 | |
7. Other Balance Sheet Details
Inventory, net, of discounts and deferred financing costs, consisted of the following:
| | | | | | | | | | | |
| | December 31, |
| (in millions) | 2020 | | 2019 |
| Principal | $ | 381.3 | | | $ | 517.5 | |
| Deferred financing costs | (4.0) | | | (6.9) | |
| Debt discount, net | (59.4) | | | (101.8) | |
| Net carrying amount | $ | 317.9 | | | $ | 408.8 | |
The 2024 Notes were recorded at the estimated value of a similar non-convertible instrument on the date of issuance and accretes to the face value of the 2024 Notes over their seven-year term. The fair value of the 2024 Notes, which was estimated utilizing market quotations from an over-the-counter trading market (Level 2), was $514.3 million and $596.8 million at December 31, 2020 and 2019, respectively. | | | | | | | | | | | |
| December 31, |
| (in millions) | 2023 | | 2022 |
| Raw materials | $ | 21.5 | | | $ | 12.0 | |
| Work in process | 9.7 | | | 5.6 | |
| Finished goods | 12.3 | | | 17.5 | |
| 43.5 | | | 35.1 | |
| Less inventory reserves | (5.2) | | | — | |
| Total inventory, net | $ | 38.3 | | | $ | 35.1 | |
6. Other Balance Sheet Details
Inventories consisted of the following:
| | | | | | | | | | | |
| December 31, |
| (in millions) | 2020 | | 2019 |
| Raw materials | $ | 16.6 | | | $ | 14.1 | |
| Work in process | 2.4 | | | 1.5 | |
| Finished goods | 9.0 | | | 1.7 | |
| Total inventories | $ | 28.0 | | | $ | 17.3 | |
Property and equipment, net, consisted of the following:
| | December 31, |
| December 31, | | | December 31, |
| (in millions) | (in millions) | 2020 | | 2019 | (in millions) | 2023 | | 2022 |
| Tenant improvements | Tenant improvements | $ | 29.5 | | | $ | 26.3 | |
| Scientific equipment | Scientific equipment | 39.2 | | | 33.5 | |
| Computer equipment | Computer equipment | 13.9 | | | 12.5 | |
| Furniture and fixtures | Furniture and fixtures | 3.7 | | | 3.2 | |
| 86.3 | | | 75.5 | |
| 153.8 | |
| Less accumulated depreciation | Less accumulated depreciation | (41.7) | | | (33.6) | |
| Total property and equipment, net | Total property and equipment, net | $ | 44.6 | | | $ | 41.9 | |
Accounts payable and accrued liabilities consisted of the following:
| | December 31, |
| December 31, | | | December 31, |
| (in millions) | (in millions) | 2020 | | 2019 | (in millions) | 2023 | | 2022 |
| Sales rebates and reserves | |
| Accrued employee related costs | Accrued employee related costs | $ | 38.2 | | | $ | 38.9 | |
| Revenue-related reserves for discounts and allowances | 34.6 | | | 30.6 | |
| Current branded prescription drug fee | |
| Accrued development costs | Accrued development costs | 32.9 | | | 25.5 | |
| Accrued Branded Prescription Drug Fee | 23.6 | | | 4.9 | |
| Current income taxes payable | |
| Accounts payable and other accrued liabilities | Accounts payable and other accrued liabilities | 39.4 | | | 41.4 | |
| Total accounts payable and accrued liabilities | Total accounts payable and accrued liabilities | $ | 168.7 | | | $ | 141.3 | |
Other long-term liabilities consisted of the following:
| | | | | | | | | | | |
| December 31, |
| (in millions) | 2023 | | 2022 |
| Noncurrent income taxes payable | $ | 96.0 | | | $ | 19.8 | |
| Noncurrent branded prescription drug fee | 10.3 | | | 9.9 | |
| Total other long-term liabilities | $ | 106.3 | | | $ | 29.7 | |
The following table providespresents a reconciliation of cash, and cash equivalents and restricted cash reported within the consolidated balance sheets that sum to the total of the same such amounts shown in the consolidated statements of cash flows.
| | December 31, |
| December 31, | | | December 31, |
| (in millions) | (in millions) | 2020 | | 2019 | (in millions) | 2023 | | 2022 |
| Cash and cash equivalents | Cash and cash equivalents | $ | 187.1 | | | $ | 112.3 | |
| Restricted cash | Restricted cash | 3.2 | | | 3.2 | |
| Total cash, cash equivalents and restricted cash | Total cash, cash equivalents and restricted cash | $ | 190.3 | | | $ | 115.5 | |
7. Net Income8. Earnings Per Share
Net incomeEarnings per share waswere calculated as follows:
| | Year Ended December 31, |
| Year Ended December 31, | | | Year Ended December 31, |
| (in millions, except per share data) | (in millions, except per share data) | 2020 | | 2019 | | 2018 | (in millions, except per share data) | 2023 | | 2022 | | 2021 |
| Net income - basic and diluted | Net income - basic and diluted | $ | 407.3 | | | $ | 37.0 | | | $ | 21.1 | |
| Weighted-average common shares outstanding: | Weighted-average common shares outstanding: | |
| Basic | Basic | 93.1 | | | 91.6 | | | 90.2 | |
| Effect of dilutive securities: | |
| | Stock options | 2.4 | | | 2.6 | | | 3.2 | |
| Restricted stock units | 0.5 | | | 0.4 | | | 0.6 | |
| 2024 Notes | 1.8 | | | 1.1 | | | 1.3 | |
| Basic | |
| Basic | |
| Effect of dilutive securities | | Effect of dilutive securities | 3.3 | | 3.1 | | 3.3 |
| Diluted | Diluted | 97.8 | | | 95.7 | | | 95.4 | |
| Net income per share: | | | | | |
| Earnings per share: | |
| Basic | |
| Basic | |
| Basic | Basic | $ | 4.38 | | | $ | 0.40 | | | $ | 0.23 | |
| Diluted | Diluted | $ | 4.16 | | | $ | 0.39 | | | $ | 0.22 | |
Shares which have been excluded from diluted per share amounts because their effect would have been anti-dilutive were 2.5 million, 2.1 million and 0.94.7 million for 2020, 20192023, 4.6 million for 2022 and 2018, respectively.4.1 million for 2021.
Note 8. Share-Based9. Stock-Based Compensation
2020 Equity Incentive Plan.In May 2011, we adopted2022, our stockholders approved an amendment of the 20112020 Equity Incentive Plan as(as so amended, or the 2011 Plan.Amended 2020 Plan). The 2011Amended 2020 Plan authorized 21 million shares of common stock for issuance and allowedprovides for the grant of stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, or RSUs, performance stock awards, performance-based restricted stock units, or PRSUs, and certain other awards. During 2020, the 2011 Plan was merged into the 2020 Plan (defined below). As a result, there were 0 shares of common stock remaining available for future grant under the 2011 Plan.
In May 2018, we adopted the 2018 Employee Stock Purchase Plan, or ESPP, pursuant to which 0.3 million shares of common stock are authorized for issuance. At December 31, 2020, 0.22023, 10.5 million shares of common stock remain available for future grant under the 2018 ESPP.
In May 2020, we adopted the 2020 Equity Incentive Plan, or theAmended 2020 Plan. The
Under the terms of the Amended 2020 Plan, authorized 3.3 millionthe number of shares of common stock available for issuance will be: (i) reduced by (a) one share for each share issued pursuant to an appreciation award (as defined in the Amended 2020 Plan) granted under the Amended 2020 Plan and allows(b) 2.13 shares for each share issued pursuant to a full value award (as defined in the grantAmended 2020 Plan) granted under the Amended 2020 Plan on or after May 18, 2022; and (ii) increased by (a) one share for each share subject to an appreciation award that becomes available again for issuance under the terms of stock options, stock appreciation rights, restricted stock awards, RSUs, performance stock awards, PRSUsthe Amended 2020 Plan and certain other awards.(b) 2.13 shares for each share subject to a full value award that becomes available again for issuance under the terms of the Amended 2020 Plan on or after May 18, 2022.
2011 Equity Incentive Plan.In May 2011, we adopted the 2011 Equity Incentive Plan (the 2011 Plan). The 2011 Plan was merged intoa stockholder-approved plan pursuant to which outstanding awards have been made, but from which no further awards can or will be made.
2018 Employee Stock Purchase Plan.In May 2021, our stockholders approved an amendment and restatement of the 20202018 Employee Stock Purchase Plan (as so amended and as a result, all remaining shares inrestated, the 2011 Plan were transferred into the 2020 Plan. AtAmended 2018 ESPP). As of December 31, 2020, 8.22023, 0.5 million shares of common stock remain available for future grantissuance under the 2020 Plan.Amended 2018 ESPP.
Share-BasedStock-Based Compensation Expense. The effect of share-basedstock-based compensation expense on our consolidated statements of income and comprehensive income by line-item follows:
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions) | 2020 | | 2019 | | 2018 |
| Selling, general and administrative expense | $ | 66.3 | | | $ | 49.5 | | | $ | 31.9 | |
| Research and development expense | 33.7 | | | 25.8 | | | 26.2 | |
| Total share-based compensation expense | $ | 100.0 | | | $ | 75.3 | | | $ | 58.1 | |
Share-based compensation expense by award-type follows:
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions) | 2020 | | 2019 | | 2018 |
| Stock options | $ | 47.5 | | | $ | 36.5 | | | $ | 35.4 | |
| RSUs | 44.2 | | | 30.5 | | | 21.9 | |
| PRSUs | 5.3 | | | 5.6 | | | 0 | |
| ESPP | 3.0 | | | 2.7 | | | 0.8 | |
| Total share-based compensation expense | $ | 100.0 | | | $ | 75.3 | | | $ | 58.1 | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Selling, general and administrative expense | $ | 126.3 | | | $ | 115.4 | | | $ | 85.8 | |
| Research and development expense | 68.0 | | | 57.7 | | | 48.4 | |
| Total stock-based compensation expense | $ | 194.3 | | | $ | 173.1 | | | $ | 134.2 | |
AtStock-based compensation expense by award-type follows:
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Stock options | $ | 91.6 | | | $ | 62.6 | | | $ | 60.5 | |
| RSUs | 93.4 | | | 86.4 | | | 62.5 | |
| PRSUs | 4.6 | | | 20.1 | | | 7.6 | |
| ESPP | 4.7 | | | 4.0 | | | 3.6 | |
| Total stock-based compensation expense | $ | 194.3 | | | $ | 173.1 | | | $ | 134.2 | |
As of December 31, 2020,2023, unrecognized share-basedstock-based compensation expense by award-type and the weighted-average period over which such expense is expected to be recognized, as applicable, werewas as follows:
| | | | | | | | | | | |
| (dollars in millions) | Unrecognized Expense | | Weighted-Average Recognition Period |
| Stock options | $ | 86.294.1 | | | 2.3 years |
| RSUs | $ | 99.5162.4 | | | 2.3 years |
| PRSUs | $ | 22.3 | | | |
Stock Options. Typically, stock options have a ten-year10-year term and vest over a three to four-year period. The exercise price of stock options granted is equal to the closing price of our common stock on the date of grant. We estimate the fair value of stock options using the Black-Scholes option-pricing model on the date of grant. The Black-Scholes option-pricing model incorporates various and highly sensitive assumptions including expected volatility, term and interest rates. The weighted-average grant-date fair values of stock options granted were $45.67, $41.74$45.19 for 2023, $32.05 for 2022 and $43.42$45.02 for 2020, 2019 and 2018, respectively.2021.
The fair value of each stock option granted was estimated on the date of grant using the Black-Scholes option-pricing valuation model with the following weighted-average assumptions:
| | Year Ended December 31, |
| 2020 | | 2019 | | 2018 |
| Year Ended December 31, | | | Year Ended December 31, |
| 2023 | | | 2023 | | 2022 | | 2021 |
| Risk-free interest rate | Risk-free interest rate | 1.4 | % | | 2.4 | % | | 2.5 | % | Risk-free interest rate | 3.9 | % | | 1.8 | % | | 0.6 | % |
| Expected volatility of common stock | Expected volatility of common stock | 48.5 | % | | 54.8 | % | | 59.5 | % | Expected volatility of common stock | 40.8 | % | | 42.6 | % | | 45.9 | % |
| Dividend yield | Dividend yield | 0.0 | % | | 0.0 | % | | 0.0 | % | Dividend yield | 0.0 | % | | 0.0 | % | | 0.0 | % |
| Expected option term | Expected option term | 5.3 years | | 5.4 years | | 4.7 years | Expected option term | 5.5 years | | 5.0 years | | 5.2 years |
The weighted-average valuation assumptions were determined as follows:
•The expected volatility of common stock is estimated based on the historical volatility of our common stock over the most recent period commensurate with the estimated expected term of our stock options.
•The expected option term is estimated based on historical experience as well as the status of the employee. For example, directors and officers have a longer expected option term than all other employees.
•The risk-free interest rate for periods within the contractual life of a stock option is based upon observed interest rates appropriate for the expected term of our employee stock options.
•We have not historically declared or paid dividends and do not intend to do so in the foreseeable future.
AThe following table presents summary of activity related to stock options follows:options.
| | (in millions, except weighted average data) | (in millions, except weighted average data) | Number of
Stock Options | | Weighted
Average
Exercise Price | | Weighted-Average Remaining Contractual Term | | Aggregate Intrinsic Value | (in millions, except weighted average data) | Number of
Stock Options | | Weighted
Average
Exercise Price | | Weighted-Average Remaining Contractual Term | | Aggregate Intrinsic Value |
| Outstanding at December 31, 2019 | 6.1 | | | $ | 52.62 | | | | | |
| Outstanding at December 31, 2022 | |
| Granted | |
| Granted | |
| Granted | Granted | 1.3 | | | $ | 103.44 | | |
| Exercised | Exercised | (0.6) | | | $ | 43.90 | | |
| Exercised | |
| Exercised | |
| Canceled | Canceled | 0 | | | $ | 0 | | |
| Outstanding at December 31, 2020 | 6.8 | | | $ | 62.98 | | | 6.4 years | | $ | 235.4 | |
| Exercisable at December 31, 2020 | 4.7 | | | $ | 49.80 | | | 5.5 years | | $ | 218.2 | |
| Canceled | |
| Canceled | |
| Outstanding at December 31, 2023 | |
| Outstanding at December 31, 2023 | |
| Outstanding at December 31, 2023 | |
| Exercisable at December 31, 2023 | |
The total intrinsic value of stock options exercised during 2020, 2019was $39.9 million for 2023, $39.7 million for 2022 and 2018 was $40.2$58.0 million $64.3 million and $117.0 million, respectively.for 2021. Cash received from stock option exercises during 2020, 2019was $55.5 million for 2023, $37.0 million for 2022 and 2018 was $23.5$20.7 million $27.3 million and $29.5 million, respectively.for 2021.
Restricted Stock Units. Typically, RSUs typically vest over a four-year period.period and may be subject to a deferred delivery arrangement at the election of eligible employees. The fair value of RSUs is based on the closing sale price of our common stock on the date of issuance. RSUs may be subject to a deferred delivery arrangement at the election of eligible employees.
A summary of activity related to RSUs follows:
| | | | | | | | | | | | | | | | | | | | | | | |
| (in millions, except weighted average data) | Number of
RSUs | | Weighted-Average Grant Date
Fair Value | | Weighted-Average Remaining Contractual Term | | Aggregate Intrinsic Value |
| Unvested at December 31, 2019 | 1.4 | | | $ | 74.77 | | | | | |
| Granted | 0.7 | | | $ | 102.92 | | | | | |
| Released | (0.5) | | | $ | 67.86 | | | | | |
| Canceled | (0.1) | | | $ | 84.95 | | | | | |
| Unvested at December 31, 2020 | 1.5 | | | $ | 89.60 | | | 1.3 years | | $ | 147.5 | |
The total fair value of RSUs that vested during 2020, 2019was $101.0 million for 2023, $72.4 million for 2022 and 2018 was $49.7$64.3 million $36.1 million and $35.5 million, respectively.for 2021.
The following table presents a summary of activity related to RSUs.
| | | | | | | | | | | | | | | | | | | | | | | |
| (in millions, except weighted average data) | Number of
RSUs | | Weighted-Average Grant Date
Fair Value | | Weighted-Average Remaining Contractual Term | | Aggregate Intrinsic Value |
| Unvested at December 31, 2022 | 2.3 | | | $ | 92.61 | | | | | |
| Granted | 1.1 | | | $ | 103.54 | | | | | |
| Released | (0.9) | | | $ | 93.46 | | | | | |
| Canceled | (0.1) | | | $ | 95.62 | | | | | |
| Unvested at December 31, 2023 | 2.4 | | | $ | 97.32 | | | 1.3 years | | $ | 312.5 | |
Performance-Based Restricted Stock Units. PRSUs vest based on the achievement of certain predefined Company-specific performance criteria. Any unvested PRSUs will expire if it is determined the related performance criteria and expirehas not been met during the applicable fourthree to five years from the grant date.four-year performance period. The fair value of PRSUs is estimated based on the closing sale price of our common stock on the date of grant. Expense recognition for PRSUs commences when attainment of the performance-based criteria is determined to be probable.
A summary of activity related to PRSUs follows:
| | | | | | | | | | | | | | | | | | | | | | | |
| (in millions, except weighted average data) | Number of
PRSUs | | Weighted-Average Grant Date
Fair Value | | Weighted-Average Remaining Contractual Term | | Aggregate Intrinsic Value |
| Unvested at December 31, 2019 | 0.3 | | | $ | 59.62 | | | | | |
| Granted | 0.2 | | | $ | 102.90 | | | | | |
| Released | (0.1) | | | $ | 82.04 | | | | | |
| Canceled | (0.2) | | | $ | 45.67 | | | | | |
| Unvested at December 31, 2020 | 0.2 | | | $ | 102.90 | | | 2.2 years | | $ | 15.8 | |
At December 31, 2020, unrecognized share-based compensation expense for PRSUs was $17.0 million. The total fair value of PRSUs that vested during 20202023 was $13.5$34.4 million. NaNNo PRSUs vested during 20192022 or 2018.2021.
The following table presents a summary of activity related to PRSUs.
| | | | | | | | | | | | | | | | | | | | | | | |
| (in millions, except weighted average data) | Number of
PRSUs | | Weighted-Average Grant Date
Fair Value | | Weighted-Average Remaining Contractual Term | | Aggregate Intrinsic Value |
| Unvested at December 31, 2022 | 0.5 | | | $ | 101.00 | | | | | |
| Granted | 0.3 | | | $ | 97.22 | | | | | |
| Released | (0.3) | | | $ | 98.43 | | | | | |
| Canceled | (0.2) | | | $ | 115.60 | | | | | |
| Unvested at December 31, 2023 | 0.3 | | | $ | 89.23 | | | 1.7 years | | $ | 33.0 | |
Employee Stock Purchase Plan. Under the Amended 2018 ESPP, eligible employees may purchase shares of our common stock at a discount semi-annually based on a percentage of their annual compensation. The discounted purchase price is equal to the lower of 85% of (i) the market value per share of the common stock on the first day of the offering period or (ii) the market value per share of common stock on the purchase date.
Note 9.10. Income Taxes
Components ofThe following table presents income tax expense forfrom continuing operations were as follows:before provision for income taxes for domestic and international operations.
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions) | 2020 | | 2019 | | 2018 |
| Current: | | | | | |
| Federal | $ | 0 | | | $ | 0 | | | $ | (0.1) | |
| State | 10.1 | | | 9.5 | | | 0.8 | |
| Total current taxes | 10.1 | | | 9.5 | | | 0.7 | |
| Deferred: | | | | | |
| Federal | (287.5) | | | 0 | | | 0 | |
| State | (23.2) | | | 0 | | | 0 | |
| Total deferred taxes | (310.7) | | | 0 | | | 0 | |
| (Benefit from) provision for income taxes | $ | (300.6) | | | $ | 9.5 | | | $ | 0.7 | |
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| U.S. | $ | 409.2 | | | $ | 218.0 | | | $ | 101.4 | |
| Foreign | (77.1) | | | (4.1) | | | — | |
| Income before provision for income taxes | $ | 332.1 | | | $ | 213.9 | | | $ | 101.4 | |
The following table presents the components of income tax expense (benefit) for continuing operations.
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions) | 2023 | | 2022 | | 2021 |
| Current: | | | | | |
| Federal | $ | 115.0 | | | $ | 17.1 | | | $ | — | |
| State | 28.1 | | | 20.3 | | | 6.3 | |
| Current income taxes | 143.1 | | | 37.4 | | | 6.3 | |
| Deferred: | | | | | |
| Federal | (45.2) | | | 27.5 | | | 5.9 | |
| State | (15.5) | | | (5.5) | | | (0.4) | |
| Deferred income taxes | (60.7) | | | 22.0 | | | 5.5 | |
| Provision for income taxes | $ | 82.4 | | | $ | 59.4 | | | $ | 11.8 | |
The provision for income taxes on earnings subject to income taxes differs from the statutory federal rate due to the following:
| | Year Ended December 31, |
| Year Ended December 31, | | | Year Ended December 31, |
| (in millions) | (in millions) | 2020 | | 2019 | | 2018 | (in millions) | 2023 | | 2022 | | 2021 |
| Federal income taxes at 21% for 2020, 2019, 2018 | $ | 22.4 | | | $ | 9.8 | | | $ | 4.6 | |
| Federal income taxes at 21% | |
| State income tax, net of federal benefit | State income tax, net of federal benefit | 5.5 | | | 4.0 | | | 0.4 | |
| Non-deductible expenses | 0.6 | | | 0.8 | | | 0.4 | |
| Branded prescription drug fee | Branded prescription drug fee | 4.9 | | | 3.7 | | | 0 | |
| Share-based compensation expense | (6.7) | | | (12.8) | | | (9.8) | |
| Loss on extinguishment of convertible senior notes | |
| Stock-based compensation expense | |
| Officer compensation | Officer compensation | 3.7 | | | 3.1 | | | 0.9 | |
| Change in tax rate | Change in tax rate | 3.3 | | | (4.1) | | | (0.2) | |
| Expired tax attributes | Expired tax attributes | 1.1 | | | 1.2 | | | 13.9 | |
| Research credits | Research credits | (39.0) | | | (10.4) | | | (13.5) | |
| Change in valuation allowance | Change in valuation allowance | (296.3) | | | 13.9 | | | 4.3 | |
| Other | Other | (0.1) | | | 0.3 | | | (0.3) | |
| (Benefit from) provision for income taxes | $ | (300.6) | | | $ | 9.5 | | | $ | 0.7 | |
| Provision for income taxes | |
SignificantThe following table presents the significant components of our deferred tax assets asassets.
| | | | | | | | | | | |
| | December 31, |
| (in millions) | 2023 | | 2022 |
| Deferred tax assets: | | | |
| Net operating losses | $ | 36.4 | | | $ | 27.4 | |
| Research and development credits | 55.3 | | | 108.9 | |
| Capitalized research and development | 178.7 | | | 91.1 | |
| Stock-based compensation expense | 52.7 | | | 45.9 | |
| Operating lease assets | 72.0 | | | 26.8 | |
| Intangible assets | 110.0 | | | 80.7 | |
| Other | 25.0 | | | 24.9 | |
| Total deferred tax assets | 530.1 | | | 405.7 | |
| Deferred tax liabilities: | | | |
| | | |
| Operating lease liabilities | (66.3) | | | (21.0) | |
| Other | (12.3) | | | (11.8) | |
| Total deferred tax liabilities | (78.6) | | | (32.8) | |
| Net of deferred tax assets and liabilities | 451.5 | | | 372.9 | |
| Valuation allowance | (88.9) | | | (67.0) | |
| Net deferred tax assets | $ | 362.6 | | | $ | 305.9 | |
As of December 31, 2020 and 2019 are listed below.
| | | | | | | | | | | |
| | December 31, |
| (in millions) | 2020 | | 2019 |
| Deferred tax assets: | | | |
| Net operating losses | $ | 111.4 | | | $ | 181.3 | |
| Research and development credits | 109.6 | | | 71.9 | |
| Capitalized research and development | 24.7 | | | 28.0 | |
| Share-based compensation expense | 29.8 | | | 22.9 | |
| Operating lease assets | 25.2 | | | 23.3 | |
| Intangible assets | 86.7 | | | 49.3 | |
| Other | 23.9 | | | 18.5 | |
| Total deferred tax assets | 411.3 | | | 395.2 | |
| Deferred tax liabilities: | | | |
| Convertible senior notes | (13.8) | | | (24.1) | |
| Operating lease liabilities | (19.9) | | | (18.2) | |
| Other | (8.4) | | | (6.9) | |
| Total deferred tax liabilities | (42.1) | | | (49.2) | |
| Net of deferred tax assets and liabilities | 369.2 | | | 346.0 | |
| Valuation allowance | (49.8) | | | (346.0) | |
| Net deferred tax assets | $ | 319.4 | | | $ | 0 | |
At December 31, 2020,2023, our deferred tax assets were primarily the result of federal net operating loss carry forwards, capitalized research costs, acquired intangible assets and tax credit carryforwards. AtAs of December 31, 20202023 and 2019,2022, we recorded a valuation allowance of $49.8$88.9 million and $346.0$67.0 million, respectively, against our gross deferred tax asset balance.
AtAs of each reporting date, management considers new evidence, both positive and negative, that could affect its assessment of the future realizability of our deferred tax assets. AtAs of December 31, 2020, in part because we achieved three years of cumulative pretax income,2023, management determined there iswas sufficient positive evidence to conclude that it is more likely than not deferred tax assets of $319.4$362.6 million are realizable. Accordingly, weThe recorded a net valuation release of $296.3 million on the basis of management’s assessment. The remaining valuation allowance of $49.8$88.9 million consistsconsisted primarily of state and foreign net operating loss carryforwards and state credit carryforwards for which management cannot conclude it is more likely than not to be realized. The release
As of the valuation allowance is reported under continuing operations as a benefit to income tax expense.
At December 31, 2020,2023, we had federalstate and stateforeign income tax net operating loss carryforwards of $518.2$286.0 million and $340.8$134.3 million, respectively. TheWe had no federal netincome tax operating losses will begin to expire in 2028, unless previously utilized.
loss carryforwards as of December 31, 2023. California net operating losses will begin to expire in 20282029 unless previously utilized and the net operating losses related to other states will begin to expire in 2026. Swiss net operating losses will begin to expire in 2030 unless previously utilized. UK net operating losses will carry forward indefinitely.
In addition,As of December 31, 2023, we have federal and Californiahad state R&D tax credit carryforwards of $92.1 million and $56.4 million, respectively. A portion of the federal R&D tax credit carryforwards expired in 2020. The remaining federal R&D tax credits will continue to expire beginning in 2021, unless previously utilized. The$85.6 million. California R&D tax credits carry forward indefinitely.indefinitely, while R&D tax credits related to other states will begin to expire in 2033 unless previously utilized.
Additionally, the future utilization of our net operating loss and R&D tax credit carryforwards to offset future taxable income may be subject to an annual limitation, pursuant to Internal Revenue Code Sections 382 and 383, as a result of ownership changes that could occur in the future. No ownership changes have occurred through December 31, 2020.2023.
The impact of an uncertain income tax position on the income tax return must be recognized at the largest amount that is more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained.
Our policy is toWe recognize interest orand penalties related to income tax matters in income tax expense. InterestWe had accruals for interest related to income tax matters of $3.1 million and $1.2 million, respectively, as of December 31, 2023 and 2022. We had accruals for penalties relates to income tax matters of $2.2 million and $0.4 million, respectively, as of December 31, 2023 and 2022. Accruals for interest and penalties related to income tax matters were not material for 2020, 2019 or 2018.as of December 31, 2021.
We are subject to taxation in the U.S. and various state and foreign jurisdictions. Our taxTax years for 2001 (federal)2020 for federal, inception for California, 2016 to 2020 for other significant state jurisdictions, and 2008 (California)2021 and forward for foreign are subject to examination by federal and state tax authorities due to the carryforward of unutilized net operating losses and R&D tax credits.
AThe following table presents a summary of activity related to unrecognized tax benefits follows:benefits.
| | Year Ended December 31, |
| Year Ended December 31, | | | Year Ended December 31, |
| (in millions) | (in millions) | 2020 | | 2019 | | 2018 | (in millions) | 2023 | | 2022 | | 2021 |
| Balance at January 1 | Balance at January 1 | $ | 63.9 | | | $ | 54.8 | | | $ | 37.4 | |
| (Decrease) increase related to prior year tax positions | (5.7) | | | 0.3 | | | 6.1 | |
| Increase related to prior year tax positions | |
| Increase related to current year tax positions | Increase related to current year tax positions | 3.9 | | | 9.5 | | | 11.7 | |
| Settlements related to prior year tax positions | (0.2) | | | 0 | | | 0 | |
| Decrease related to prior year tax positions | |
| Expiration of the statute of limitations for the assessment of taxes | Expiration of the statute of limitations for the assessment of taxes | (1.1) | | | (0.7) | | | (0.4) | |
| Balance at December 31 | Balance at December 31 | $ | 60.8 | | | $ | 63.9 | | | $ | 54.8 | |
We excluded those deferred tax assets that are not more-likely-than-not to be sustained under the technical meritsAs of the tax position. Such unrecognized tax benefits total $3.9 million for current year tax positions, as reflected in the table above.
At December 31, 2020,2023, we had $53.9$105.3 million of unrecognized tax benefits that, if recognized and realized, would affect the effective tax rate, subject to changes in the valuation allowance. We do not expect a significant change in our unrecognized tax benefits in the next twelve12 months.
Note 10.In 2021, the OECD announced an Inclusive Framework on Base Erosion and Profit Shifting including Pillar Two Model Rules defining the global minimum tax, which calls for the taxation of large multinational corporations at a minimum rate of 15%. Subsequently, multiple sets of administrative guidance have been issued. Many non-U.S. tax jurisdictions have either recently enacted legislation to adopt certain components of the Pillar Two Model Rules beginning in 2024 (including EU Member States) with the adoption of additional components in later years or announced their plans to enact legislation in future years. We are continuing to evaluate the impacts of enacted legislation and pending legislation to enact Pillar Two Model Rules in the non-U.S. tax jurisdictions we operate in.
11. Leases
We haveOur operating leases for ourthat have commenced have terms that expire beginning 2025 through 2036 and consist of office space and laboratory facilities,research and development laboratories, including our corporate headquarters, with terms that expire from 2025 through 2031. We have 2 options to extend the termheadquarters. Certain of the operatingthese lease agreements contain clauses for renewal at our corporate headquarters for a period of ten years each. However, asoption. As we were not reasonably certain to exercise eitherany of thosethese renewal options at lease commencement neither option wasof the associated leases, no such options were recognized as part of the associatedour ROU assets or operating lease right-of-use, or ROU, asset or liability. In connection with ourliabilities.
The following table presents supplemental operating lease information for operating leases that have commenced.
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| (in millions, except weighted average data) | 2023 | | 2022 | | 2021 |
| Operating lease cost | $ | 17.1 | | | $ | 16.3 | | | $ | 15.3 | |
| Sublease income | (0.7) | | | — | | | — | |
| Net operating lease cost | $ | 16.4 | | | $ | 16.3 | | | $ | 15.3 | |
| Cash paid for amounts included in the measurement of operating lease liabilities | $ | 17.9 | | | $ | 16.9 | | | $ | 12.6 | |
| | | | | |
| | | December 31,
2023 | | December 31,
2022 |
| Weighted average remaining lease term | | | 10.8 years | | 7.9 years |
| Weighted average discount rate | | | 5.1 | % | | 5.3 | % |
| Restricted cash related to letters of credit issued in lieu of cash security deposits | | | $ | 7.8 | | | $ | 7.8 | |
The following table presents approximate future non-cancelable minimum lease payments under operating leases and sublease income as of December 31, 2023.
| | | | | | | | | | | |
| (in millions) | Operating Leases (1) | | Sublease
Income |
| Year ending December 31, 2024 | $ | 33.0 | | | $ | (1.7) | |
| Year ending December 31, 2025 | 34.7 | | | (1.7) | |
| Year ending December 31, 2026 | 34.0 | | | (1.7) | |
| Year ending December 31, 2027 | 34.8 | | | (1.7) | |
| Year ending December 31, 2028 | 35.6 | | | (1.7) | |
| Thereafter | 211.4 | | | (4.3) | |
| Total operating lease payments (sublease income) | 383.5 | | | $ | (12.8) | |
| Less accreted interest | 93.2 | | | |
| Total operating lease liabilities | 290.3 | | | |
| Less current operating lease liabilities included in other current liabilities | 32.0 | | | |
| Noncurrent operating lease liabilities | $ | 258.3 | | | |
_________________________
(1) Amounts presented in lieu of cash security deposits, Wells Fargo Bank, N.A., issued letters of credit on our behalf, which are secured by deposits totaling $3.2 million.
Our operating lease cost was $10.1the table above exclude $15.4 million for 2020 and $8.12025, $23.6 million for 2019. Cash paid for amounts included in the measurement of lease liabilities was $8.62026, $24.3 million for 2020 and $7.72027, $25.1 million for 2019.
Our operating leases had a weighted-average remaining lease term2028 and $223.5 million thereafter of approximately 10.3 years and 11.2 years at December 31, 2020 and 2019, respectively, and a weighted-average discount rate of 5.6% and 5.8% at December 31, 2020 and 2019, respectively.
Approximateapproximate non-cancelable future minimum lease payments under operating leases were as follows:
| | | | | |
(in millions) | December 31,
2020 |
Year ending December 31, 2021 | $ | 10.7 | |
Year ending December 31, 2022 | 12.4 | |
Year ending December 31, 2023 | 12.7 | |
Year ending December 31, 2024 | 13.1 | |
Year ending December 31, 2025 | 13.5 | |
Thereafter | 77.5 | |
Total operating lease payments | 139.9 | |
Less accreted interest | 35.2 | |
Total operating lease liabilities | 104.7 | |
Less current operating lease liabilities | 10.3 | |
Noncurrent operating lease liabilities | $ | 94.4 | |
Note 1: Amounts presented in the table above exclude $19.7 million of non-cancelable future minimum lease payments for operating leases that have not yet commenced.
Note 2: CurrentNew Campus Facility.On February 8, 2022, we entered into a lease agreement for a four-building campus facility to be constructed in San Diego, California, including a six-year option for the construction of a fifth building. This campus facility, comprised of office space and research and development laboratories, will serve as our new corporate headquarters.
The construction of the campus facility is phased. We recognized ROU assets of $199.0 million and operating lease liabilities are includedof $189.8 million in other current liabilitiesassociation with the commencement of operating leases following the completion of the first phase of construction relating to office space in December 2023.
As we begin to occupy our new campus facility, we will sublease certain of our existing leased premises when we determine there is excess leased capacity. Certain of these subleases contain both lease and non-lease components. Sublease income is recognized as an offset to operating expense on a straight-line basis over the consolidated balance sheets.lease term. Income related to non-lease components is recognized in operating expenses as a reduction to costs we incur in relation to the primary lease.
Note 11.12. Retirement Plan
We have a 401(k) defined contribution savings plan or the 401(k) Plan. The 401(k) Plan is for the benefit of all qualifying employees and permits voluntary contributions by employees up to 60% of base salary limited by the IRS-imposed maximum. Employer contributions were $6.7 million, $4.9 million, and $1.8$12.5 million for 2020, 20192023, $10.3 million for 2022 and 2018, respectively.$8.1 million for 2021.
Note 12. Selected Quarterly Financial Data (Unaudited)
A summaryDuring 2021, 2022, and 2023, we received notices from (i) Teva Pharmaceuticals Development, Inc., (ii) Lupin Limited, (iii) Crystal Pharmaceutical (Suzhou) Co. Ltd., (iv) Sandoz Inc. and (v) Zydus Pharmaceuticals (USA) Inc. that each company had filed an abbreviated new drug application (ANDA) with the FDA seeking approval of a generic version of INGREZZA. These companies represented that their respective ANDAs each contained a Paragraph IV Patent Certification alleging that certain of our quarterly results follows:
| | | | | | | | | | | | | | | | | | | | | | | |
| (in millions, except per share data) | First
Quarter | | Second
Quarter | | Third
Quarter | | Fourth
Quarter |
| Year Ended December 31, 2020: | | | | | | | |
| Total revenues | $ | 237.1 | | | $ | 302.4 | | | $ | 258.5 | | | $ | 247.9 | |
Total operating expenses (1) | $ | 178.2 | | | $ | 225.8 | | | $ | 302.8 | | | $ | 176.1 | |
Net income (loss) (1) | $ | 37.4 | | | $ | 79.6 | | | $ | (57.6) | | | $ | 347.9 | |
Net income (loss) per share, basic (1) | $ | 0.40 | | | $ | 0.86 | | | $ | (0.62) | | | $ | 3.72 | |
Net income (loss) per share, diluted (1) | $ | 0.39 | | | $ | 0.81 | | | $ | (0.62) | | | $ | 3.58 | |
| Weighted average common shares outstanding, basic | 92.6 | | | 93.0 | | | 93.3 | | | 93.5 | |
| Weighted average common shares outstanding, diluted | 97.0 | | | 98.2 | | | 93.3 | | | 97.2 | |
| | | | | | | |
| Year Ended December 31, 2019: | | | | | | | |
| Total revenues | $ | 138.4 | | | $ | 183.5 | | | $ | 222.1 | | | $ | 244.1 | |
Total operating expenses (2) | $ | 239.4 | | | $ | 149.1 | | | $ | 132.0 | | | $ | 195.3 | |
Net (loss) income (2) | $ | (102.1) | | | $ | 51.3 | | | $ | 53.8 | | | $ | 34.0 | |
Net (loss) income per share, basic (2) | $ | (1.12) | | | $ | 0.56 | | | $ | 0.59 | | | $ | 0.37 | |
Net (loss) income per share, diluted (2) | $ | (1.12) | | | $ | 0.54 | | | $ | 0.56 | | | $ | 0.35 | |
| Weighted average common shares outstanding, basic | 91.1 | | | 91.4 | | | 91.9 | | | 92.2 | |
| Weighted average common shares outstanding, diluted | 91.1 | | | 94.8 | | | 96.1 | | | 97.2 | |
(1) In connection withpatents covering INGREZZA are invalid and/or will not be infringed by the paymentmanufacture, use or sale of the upfront fee pursuant to our collaboration and license agreement with Idorsia, we recorded a charge of $46.0 million, accountedmedicine for as IPR&D,which the ANDA was submitted.
We filed suit in the second quarterU.S. District Court for the District of 2020. In connection with the payment of the upfront fee pursuant to our collaboration with Takeda, we recorded Delaware during 2021, 2022 and 2023, against (i) Teva Pharmaceuticals, Inc., Teva Pharmaceuticals Development, Inc., Teva Pharmaceuticals USA, Inc. and Teva Pharmaceutical Industries Ltd. (entity dismissed), collectively, "Teva", (ii) Lupin Limited, Lupin Pharmaceuticals, Inc., Lupin Inc. and Lupin Atlantis Holdings S.A., collectively, “Lupin”, (iii) Crystal Pharmaceutical (Suzhou) Co., Ltd., Crystal Pharmatech Co., Ltd., collectively, “Crystal”, (iv) Sandoz Inc., Sandoz International GmbH (entity dismissed) and Sandoz AG (entity dismissed), collectively, “Sandoz” and (v) Zydus Pharmaceuticals (USA) Inc., Zydus Worldwide DMCC, Zydus Lifesciences Limited (formerly known as Cadila Healthcare Limited d/b/a charge of $118.5 million, accounted for as IPR&D,Zydus Cadila) and Zydus Healthcare (USA) LLC (entity dismissed), collectively, “Zydus”. We also filed suit in the third quarterU.S. District Court for the District of 2020.New Jersey during 2021, 2022 and 2023 against Zydus.
(2) In connection with the payment of the upfront fee pursuant to our collaboration and license agreement with Voyager, we recorded a charge of $113.1 million, accounted for as IPR&D, in the first quarter of 2019. In the second quarter of 2019,2023 we entered into an amendmentsettlement agreements resolving the foregoing litigation with each of (i) Sandoz and Crystal, collectively, the “Sandoz Parties”, (ii) Teva, (iii) Lupin and (iv) Zydus. Pursuant to the collaboration and license agreement with Voyager, pursuant to which we paid Voyager $5.0 million upfront, accounted for as IPR&D, to obtain outsideterms of the U.S. rights to the Friedreich’s ataxia program. In connectionrespective agreements with the paymentSandoz Parties, Teva, Lupin, and Zydus, each of Teva, the Sandoz Parties, Lupin, and Zydus has the right to sell generic versions of INGREZZA in the United States beginning March 1, 2038, or earlier under certain circumstances. The settlements with Teva, the Sandoz Parties, Lupin and Zydus resolve all patent litigation brought by us against the companies that filed ANDAs seeking approval to market generic INGREZZA, and all cases have been dismissed.
From time to time, we may also become subject to other legal proceedings or claims arising in the ordinary course of our business. We currently believe that none of the upfront fee pursuantclaims or actions pending against us is likely to our collaboration with Xenon, we recorded a charge of $36.2 million, accounted for as IPR&D,have, individually or in the fourth quarteraggregate, a material adverse effect on our business, financial condition or results of 2019.operations. Given the unpredictability inherent in litigation, however, we cannot predict the outcome of these matters.
Item 9. Changes and Disagreements with Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the timelines specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As required by SEC Rule 13a-15(b), we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the year covered by this report. Based on the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control Over Financial Reporting
Internal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
(1) Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
(2) Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and
(3) Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk. Management is responsible for establishing and maintaining adequate internal control over financial reporting for the company.
Management has used the framework set forth in the report entitled Internal Control-Integrated Framework (2013 framework) published by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), known as COSO, to evaluate the effectiveness of our internal control over financial reporting. Based on this assessment, management has concluded that our internal control over financial reporting was effective as of December 31, 2020.2023. Ernst & Young, LLP, our independent registered public accounting firm, has issued an attestation report on our internal control over financial reporting as of December 31, 2020,2023, which is included herein.
There has been no change in our internal control over financial reporting during our most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Report of Independent Registered Public Accounting Firm
To the ShareholdersStockholders and the Board of Directors of Neurocrine Biosciences, Inc.
Opinion on Internal Control overOver Financial Reporting
We have audited Neurocrine Biosciences, Inc.’s internal control over financial reporting as of December 31, 2020,2023, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), (the COSO criteria). In our opinion, Neurocrine Biosciences, Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2020,2023, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 20202023 and 2019,2022, the related consolidated statements of income and comprehensive income, stockholders‘ equity and cash flows for each of the three years in the period ended December 31, 2020,2023, and the related notes and our report dated February 5, 20219, 2024 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
San Diego, California
February 5, 20219, 2024
Item 9B. Other Information
During the period from October 1, 2023, to December 31, 2023, our executive officers and directors adopted or terminated contracts, instructions or written plans for the purchase or sale of our securities as noted below:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Name and Title | | Action | | Date | | Trading Arrangement | | Total Shares Authorized
to be Sold | | Expiration
Date |
| | | Rule 10b5-1* | | Non-Rule 10b5-1** | | |
| George Morrow | | Adopt | | 12/14/2023 | | X | | | | 40,000 | | | 11/15/2024 |
| (Director) | | | | | | | | | | | | |
| Eric Benevich | | Terminate (1) | | 11/30/2023 | | X | | | | 131,341 | | | 12/31/2023 |
| (Chief Commercial Officer) | | Adopt | | 11/29/2023 | | X | | | | 169,818 | | | 11/27/2024 |
| Ingrid Delaet | | Adopt | | 11/29/2023 | | X | | | | 30,000 | | | 9/7/2025 |
| (Chief Regulatory Officer) | | | | | | | | | | | | |
| Leslie Norwalk | | Adopt | | 11/28/2023 | | X | | | | 9,106 | | | 11/28/2024 |
| (Director) | | | | | | | | | | | | |
| Shalini Sharp | | Adopt | | 11/27/2023 | | X | | | | 1,106 | | | 5/31/2024 |
| (Director) | | | | | | | | | | | | |
| Richard Pops | | Adopt | | 11/21/2023 | | X | | | | 42,100 | | | 11/30/2024 |
| (Director) | | | | | | | | | | | | |
______________* Intended to satisfy the affirmative defense of Rule 10b5-1(c)
** Not intended to satisfy the affirmative defense of Rule 10b5-1(c)
(1) On November 30, 2023, Eric Benevich, Chief Commercial Officer, terminated a trading arrangement that was intended to satisfy the affirmative defense of Rule 10b5-1 (the “Benevich 10b5-1 Plan”). The Benevich 10b5-1 Plan was entered into on February 23, 2022, with an expiration date of December 31, 2023.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
None.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Information required by this item will be contained in our Definitive Proxy Statement for our 20212024 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A with the Securities and Exchange CommissionSEC within 120 days of December 31, 2020.2023. Such information is incorporated herein by reference.
We have adopted a code of ethics that applies to our Chief Executive Officer, Chief Financial Officer, and to all of our other officers, directors, employees and agents. The code of ethics is available at the Corporate Governance section of the Investors page on our website at www.neurocrine.com. We intend to disclose future amendments to, or waivers from, certain provisions of our code of ethics on the above website within four business days following the date of such amendment or waiver. Information found on, or accessible through, our website is not part of, and is not incorporated into, this Annual Report on Form 10-K.
Item 11. Executive Compensation
Information required by this item will be contained in our Definitive Proxy Statement for our 20212024 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A with the Securities and Exchange CommissionSEC within 120 days of December 31, 2020.2023. Such information is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Information required by this item will be contained in our Definitive Proxy Statement for our 20212024 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A with the Securities and Exchange CommissionSEC within 120 days of December 31, 2020.2023. Such information is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Information required by this item will be contained in our Definitive Proxy Statement for our 20212024 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A with the Securities and Exchange CommissionSEC within 120 days of December 31, 2020.2023. Such information is incorporated herein by reference.
Item 14. Principal Accounting Fees and Services
Information required by this item will be contained in our Definitive Proxy Statement for our 20212024 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A with the Securities and Exchange CommissionSEC within 120 days of December 31, 2020.2023. Such information is incorporated herein by reference.
PART IV
Item 15. Exhibits, Financial Statement Schedules
(a) Documents filed as part of this report.
1. List of Financial Statements. The following are included in Item 8 of this report:
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets as of December 31, 20202023 and 20192022
Consolidated Statements of Income and Comprehensive Income for the years ended December 31, 2020, 20192023, 2022 and 20182021
Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2020, 20192023, 2022 and 20182021
Consolidated Statements of Cash Flows for the years ended December 31, 2020, 20192023, 2022 and 20182021
Notes to the Consolidated Financial Statements (includes unaudited Selected Quarterly Financial Data)
2. List of all Financial Statement schedules. All schedules are omitted because they are not applicable, or the required information is shown in the Financial Statements or notes thereto.
3. List of Exhibits required by Item 601 of Regulation S-K. See part (b) below.
(b) Exhibits. The following exhibits are filed as part of, or incorporated by reference into, this report:
| | | | | | | | | | | | | | | | | |
| Exhibit | | | | | |
| | | | | | |
| 3.1 | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 3.1 of the Company’s Quarterly Report on Form 10-Q filed on November 5, 2018 | |
| | | | | |
| 3.2 | | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 3.2 of the Company’sCompany's Quarterly Report on Form 10-Q filed on November 5, 2018 | August 1, 2023 |
| | | | | | |
3.3 | | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 3.1 of the Company’s Current Report on Form 8-K filed on February 4, 2020 | |
| | | | | |
3.4 | | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 3.1 of the Company’s Current Report on Form 8-K filed on August 28, 2020 | |
| | | | | |
| 4.1 | | Description: | | | |
| | | Reference: | | Incorporated by reference to the Company’s Registration Statement on Form S-1 (Registration No. 333-03172) | |
| | | | | | |
| 4.2 | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 4.1 of the Company’s Current Report on Form 8-K filed on May 2, 2017 |
| | | | |
| 4.3 | | Description: | | |
| | Reference: | | Incorporated by reference to Exhibit 4.3 of the Company’s Annual Report on Form 10-K filed on February 11, 2022 |
| | | | | | |
4.34.4 | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 99.1 of the Company’s Current Report on Form 8-K filed on May 2, 2017 | |
| | | | | |
4.44.5 | | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 4.4 of the Company’s Annual Report on Form 10-K filed on February 7, 2020 | |
| | | | | | |
| 21.1 | | Description: | | | |
| | | | | | |
| 23.1 | | Description: | | | |
| | | | | | |
| | | | | | | | | | | | | | | | | |
| 31.1 | | Description: | | | |
| | | | | | |
| 31.2 | | Description: | | | |
| | | | | | |
| 32*** | | Description: | | | |
| | | | | |
| | | | | | | | | | | | | | |
97+ | | Description: | | |
| | | | |
| 101.INS | | Description: | | Inline XBRL Instance Document. – the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | |
| | | | | | |
| 101.SCH | | Description: | | Inline XBRL Taxonomy Extension Schema Document. | |
| | | | | | |
| 101.CAL | | Description: | | Inline XBRL Taxonomy Extension Calculation Linkbase Document. | |
| | | | | | |
| 101.DEF | | Description: | | Inline XBRL Taxonomy Extension Definition Linkbase Document. | |
| | | | | | |
| 101.LAB | | Description: | | Inline XBRL Taxonomy Extension Label Linkbase Document. | |
| | | | | | |
| 101.PRE | | Description: | | Inline XBRL Taxonomy Extension Presentation Linkbase Document. | |
| | | | | | |
| 104 | | Description: | | Cover Page Interactive Data File (formatted as Inline XBRL with applicable taxonomy extension information contained in Exhibit 101) | |
| | | | | |
Collaboration and License Agreements: | |
| | | | | |
| 10.1** | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.210.1 of the Company’sCompany's Quarterly Report on Form 10-Q filed on July 29, 2010May 5, 2021 | |
| | | | | | |
| 10.2** | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.2 of the Company’sCompany's Quarterly Report on Form 10-Q filed on October 31, 2011May 5, 2021 | |
| | | | | | |
| 10.3** | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.110.3 of the Company’sCompany's Quarterly Report on Form 10-Q filed on April 30, 2015May 5, 2021 | |
| | | | | | |
| 10.4* | | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 99.4 of the Company’s Current Report on Form 8-K filed on April 25, 2017 | |
| | | | | |
10.5* | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.5 of the Company’s Annual Report on Form 10-K filed on February 7, 2019 | |
| | | | | | |
10.610.5 | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.6 of the Company’s Annual Report on Form 10-K filed on February 7, 2019 | |
| | | | | | |
10.710.6 | | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 10.7 of the Company’s Annual Report on Form 10-K filed on February 7, 2019 | |
| | | | | |
10.8 | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q filed on July 29, 2019 | |
| | | | | |
10.9*10.7** | | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 10.3 of the Company’s Quarterly Report on Form 10-Q filed on August 3, 2020 |
| | | | |
| 10.8** | | Description: | | |
| | Reference: | | Incorporated by reference to Exhibit 10.10 of the Company’s Annual Report on Form 10-K filed on February 11, 2022 |
| | | | |
| 10.9** | | Description: | | |
| | Reference: | | Incorporated by reference to Exhibit 10.2 of the Company’s Quarterly Report on Form 10-Q filed on May 3, 2023 |
| | | | |
| 10.10 | | Description: | | |
| | Reference: | | Incorporated by reference to Exhibit 10.3 of the Company’s Quarterly Report on Form 10-Q filed on May 3, 2023 |
| | | | |
| | | | | | | | | | | | | | |
| 10.11 | | Description: | | | | | | | | | | | | | | | | |
| | Reference: | | | | | Incorporated by reference to Exhibit 10.4 of the Company’s Quarterly Report on Form 10-Q filed on May 3, 2023 |
Equity Plans and Related Agreements: | |
| | | | | | |
10.1010.12+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 99.1 of the Company’s Current Report on Form 8-K filed on May 30, 2018 | |
| | | | | |
10.1110.13+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 99.1 of the Company’s Current Report on Form 8-K filed on June 1, 2015 | |
| | | | | | |
10.1210.14+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.17 of the Company’s Annual Report on Form 10-K filed on February 13, 2018 | |
| | | | | | |
10.1310.15+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q filed on July 29, 2015 | |
| | | | | | |
10.1410.16+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 99.2 of the Company’s Current Report on Form 8-K filed on May 30, 2018 | |
| | | | | |
10.15+
| | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 10.110.3 of the Company’s Quarterly Report on Form 10-Q filed on August 3, 20204, 2022 | |
| | | | | |
10.1610.17+
| | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 10.210.17 of the Company’s Annual Report on Form 10-K filed on February 11, 2022 |
| | | | |
10.18+ | | Description: | | |
| | Reference: | | Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q filed on August 3, 20201, 2023 | |
| | | | | |
Agreements with Officers and Directors: | |
| | | | | | |
10.1710.19+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.3 of the Company’s Quarterly Report on Form 10-Q filed on August 3, 2007 | |
| | | | | | |
10.1810.20+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.32 of the Company’s Annual Report on Form 10-K filed on February 11, 2008 | |
| | | | | |
10.1910.21+
| | Description: | | | |
| | Reference: | | | | Incorporated by reference to Exhibit 10.1610.1 of the Company’s AnnualQuarterly Report on Form 10-K10-Q filed on February 6, 2020May 3, 2023 | |
| | | | | | |
10.2010.22+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.3 of the Company’s Annual Report on Form 10-K filed on February 14, 2017 | |
| | | | | | |
10.2110.23+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.26 of the Company’s Annual Report on Form 10-K filed on February 13, 2018 | |
| | | | | | |
10.2210.24+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q filed on November 1, 2017 | |
| | | | | |
| | | | | | | | | | | | | | | | | |
10.2310.25+
| | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.2 of the Company’s Quarterly Report on Form 10-Q filed on July 29, 2019 |
| | | | |
10.26+ | | Description: | | |
| | Reference: | | | | | Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q filed on August 4, 2022 |
Agreements Related to Real Property: | |
| | | | | | |
10.2410.27 | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 99.2 of the Company’s Current Report on Form 8-K filed on January 18, 2012 | |
| | | | | | |
10.2510.28 | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q filed on August 3, 2017 | |
| | | | | | |
10.2610.29 | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.3 of the Company’s Quarterly Report on Form 10-Q filed on November 1, 2017 | |
| | | | | | |
10.2710.30 | | Description: | | | |
| | Reference: | | Incorporated by reference to Exhibit 10.3 of the Company’s Current Report on Form 8-K filed on December 10, 2007; Exhibit 10.5 of the Company’s Annual Report on Form 10-K filed on February 9, 2015; and Exhibit 10.2 of the Company’s Quarterly Report on Form 10-Q filed on August 3, 2017 | |
| | | | | |
10.28 | | Description: | | | |
| | | Reference: | | Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q filed on November 4, 2019 |
| | | | |
| 10.31 | | Description: | | |
| | Reference: | | Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q filed on May 4, 2022 |
| | | | | | | | |
| + | | Management contract or compensatory plan or arrangement. |
| | |
| * | | Confidential treatment has been granted with respect to certain portions of the exhibit. |
| | |
| ** | | Certain portions of theinformation in this exhibit havehas been omitted because the omitted information is not material and would likely cause competitive harm if publicly disclosed.pursuant to Item 601 of Regulation S-K. |
| | |
| *** | | These certifications are being furnished solely to accompany this annual report pursuant to 18 U.S.C. Section 1350 and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934 and are not to be incorporated by reference into any filing of Neurocrine Biosciences, Inc., whether made before or after the date hereof, regardless of any general incorporation language in such filing. |
| | |
| | Except as specifically noted above, the Company’s Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K have a Commission File Number of 000-22705. |
(c) Financial Statement Schedules. See Item 15(a)(2) above.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | |
| NEUROCRINE BIOSCIENCES, INC. |
| (Registrant) |
| |
| By: | /s/ Kevin C. Gorman |
| Kevin C. Gorman |
| Chief Executive Officer |
| |
| Date: | February 5, 20219, 2024 |
| |
| By: | /s/ Matthew C. Abernethy |
| | Matthew C. Abernethy |
| | Chief Financial Officer |
| |
| Date: | February 5, 20219, 2024 |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Kevin C. Gorman and Matthew C. Abernethy, and each of them, as his or her true and lawful attorneys-in-fact and agents, with full power of substitution for him or her, and in his or her name in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power of authority to do and perform each and every act and thing requisite and necessary to be done therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, and any of them, his or her substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the Registrant and in the capacities indicated as of February 5, 2021:9, 2024:
| | | | | | | | |
| Signature | | Title |
| | |
/s/ Kevin C. Gorman | | Chief Executive Officer and Director |
| Kevin C. Gorman, Ph.D. | | (Principal Executive Officer) |
| | |
/s/ Matthew C. Abernethy | | Chief Financial Officer |
| Matthew C. Abernethy | | (Principal Financial and Accounting Officer) |
| | |
/s/ William H. Rastetter | | Chairman of the Board of Directors |
| William H. Rastetter, Ph.D. | | |
| | |
/s/ Gary A. Lyons | | Director |
| Gary A. Lyons | | |
| | |
/s/ Johanna Mercier | | Director |
| Johanna Mercier | | |
| | |
/s/ George J. Morrow | | Director |
| George J. Morrow | | |
| | |
/s/ Leslie V. Norwalk | | Director |
| Leslie V. Norwalk | | |
| | |
/s/ Christine A. Poon | | Director |
| Christine A. Poon | | |
| | |
/s/ Richard F. Pops | | Director |
| Richard F. Pops | | |
| | |
/s/ Shalini Sharp | | Director |
| Shalini Sharp | | |
| | |
/s/ Stephen A. Sherwin | | Director |
| Stephen A. Sherwin, M.D. | | |
| | |
/s/ Shalini Sharp
| | Director |
Shalini Sharp | | |