441 Ninth Avenue, 14th Floor Title of each class Name of each exchange on which registered Common Stock, par value $0.001 per share OVID The Nasdaq Stock Market LLC x o o Large Accelerated Filer o Accelerated Filer Non-accelerated Filer x Smaller Reporting Company x Emerging growth company o Portions of the registrant’s definitive proxy statement for its trademarks and trade names referred to, including logos, artwork and other visual displays, may appear without the ® or potential small molecule neurotherapeutics. the long-term future of treating genetic epilepsies and neurological disorders. Accordingly, we engage in appropriately scaled, early-stage research programs with certain collaborators, including Gensaic, Inc., formerly M13 Therapeutics, Inc. (“Gensaic”), a next-generation gene therapy developer. In June 2023, the Healx for OV329 Patent Rights. Patents, and, upon commercialization of any such products, will be required to pay to Northwestern a tiered royalty on net sales of such products by the Company, its affiliates or sublicensees, at percentages in the low to mid-single-digits, subject to standard reductions and offsets. Our royalty obligations continue on a product-by-product and country-by-country basis until the later of the expiration of the last-to-expire valid claim in a licensed patent covering the applicable product in such country and As it relates to OV888 (GV101), we believe Neurelis Inc. is our most direct competitor. application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the CMS Innovation Center which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether the models will be utilized in any health reform measures in the future. Further, on December 7, 2023, the Biden administration announced an initiative to control the price of prescription drugs through the use of march-in rights under the Bayh-Dole Act. On December 8, 2023, the National Institute of Standards and Technology published for comment a Draft Interagency Guidance Framework for Considering the Exercise of March-In Rights which for the first time includes the price of a product as one factor an agency can use when deciding to exercise march-in rights. While march-in rights have not previously been exercised, it is uncertain if that will continue under the new framework. As of December 31, have received LEED Platinum certification. business strategy. limited. ability to generate revenue from drug sales depends heavily on our, or any current or future collaborators’, success in the following areas, including but not limited to: the United States. Failure to obtain regulatory approval in the United States or other jurisdictions would prevent us from commercializing and marketing our current and future drug candidates. are a number of large pharmaceutical and biotechnology companies that currently market and sell drugs or are pursuing the development of drug candidates for the treatment of the indications that we are pursuing. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization. Pursuant to the Ligand Agreement, Ligand will receive a 13% portion of the royalties and milestones owed to us pursuant to the RLT Agreement. amount of reimbursement to be provided for any drug candidates that we develop will be made on a payor-by-payor basis. One third-party payor’s determination to provide coverage for a drug does not assure that other payors will also provide coverage, and adequate reimbursement, for the drug. Additionally, a third-party payor’s decision to provide coverage for a therapy does not imply that an adequate reimbursement rate will be approved. Each third-party payor determines whether or not it will provide coverage for a therapy, what amount it will pay the manufacturer for the therapy, and on what tier of its formulary it will be placed. The position on a third-party payor’s list of covered drugs, or formulary, generally determines the co-payment that a patient will need to make to obtain the therapy and can strongly influence the adoption of such therapy by patients and physicians. Patients who are prescribed treatments for their conditions and providers prescribing such services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use our drugs unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our drugs. System (“MIPS”). Under both APMs and MIPS, performance data collected each performance year will affect Medicare payments in later years, including potentially reducing payments. research and development partnerships or similar agreements. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, consulting agreements or other similar agreements with our advisors, employees, third-party contractors and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, including our trade secrets. Despite the contractual provisions employed when working with third parties, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure could have an adverse effect on our business and results of operations. our results of operations. Additionally, we do not currently maintain “key person” life insurance on the lives of our executives or any of our employees. levels of persistence, sophistication and intensity, and are also being conducted by sophisticated and organized groups and individuals with a wide range of motives (including, but not limited to, industrial espionage) and expertise, including organized criminal groups, “hacktivists,” nation states and others. a ransomware attack, but we may be unwilling or unable to make such payments due to, for example, applicable laws or regulations prohibiting such payments. obligations. security laws, we may be bound by other contractual obligations related to data privacy and security, and our efforts to comply with such obligations may not be successful. We also publish privacy policies, marketing materials, and other statements regarding data privacy and security and if these policies, materials, or statements are found to be deficient, lacking in transparency, deceptive, unfair, or misrepresentative of our practices, we may be subject to investigation, enforcement actions by regulators, or other adverse consequences. If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common stock. report on internal control over financial reporting issued by our independent registered public accounting firm in this Annual Report on Form 10-K for the fiscal year ended December 31, 2023. dilutive. business. disagree. current management by making it more difficult for stockholders to replace members of our board of directors. Among other things, these provisions: General Corporation Law (the “DGCL”), which may discourage, delay or prevent someone from acquiring us or merging with us whether or not it is desired by or beneficial to our stockholders. Under the DGCL, a corporation may not, in general, engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other things, the board of directors has approved the transaction. Any provision of our amended and restated certificate of incorporation or amended and restated bylaws or Delaware law that has the effect of delaying or deterring a change of control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock and could also affect the price that some investors are willing to pay for our common stock. Sales of a substantial number of shares of our common stock in the public market could cause the market price of our common stock to drop significantly. intellectual property rights; complying with applicable regulatory requirements. 2022 Year Ended Year Ended December 31, December 31, 2021 2020 Change $ (in thousands) Revenue: License and other revenue $ 12,383 $ 12,617 $ (234 ) License revenue - related party 196,000 - 196,000 Total revenue 208,383 12,617 195,766 Operating expenses: Research and development 46,940 63,417 (16,477 ) General and administrative 37,234 30,631 6,603 Total operating expenses 84,174 94,048 (9,874 ) Income (loss) from operations 124,209 (81,431 ) 205,640 Other (expense) income, net (46 ) 395 (441 ) Income (loss) before provision for income taxes 124,163 (81,036 ) 205,199 Provision for income taxes 1,329 - 1,329 Net income (loss) $ 122,834 $ (81,036 ) $ 203,871 Year Ended Year Ended December 31, December 31, 2021 2020 Change $ (in thousands) Preclinical and development expenses $ 30,386 $ 43,612 $ (13,226 ) Payroll and payroll-related expenses 13,454 15,439 (1,985 ) Other expenses 3,101 4,366 (1,265 ) Total research and development $ 46,940 $ 63,417 $ (16,476 ) Year Ended Year Ended December 31, December 31, 2021 2020 Change $ (in thousands) Payroll and payroll-related expenses $ 14,008 $ 13,390 $ 618 Legal and professional fees 17,071 13,824 3,247 General office expenses 6,154 3,417 2,737 Total general and administrative $ 37,234 $ 30,631 $ 6,603 unrealized loss on long-term equity investments. issuance of this Annual Report on Form 10-K. negatively affect our See “Risk Factors” for additional risks associated with our capital requirements. Year Ended Year Ended December 31, December 31, 2021 2020 (in thousands) Net cash provided by (used in): Operating activities $ 118,612 $ (51,584 ) Investing activities (1,821 ) 34,648 Financing activities 904 47,072 Net increase in cash, cash equivalents, and restricted cash $ 117,694 $ 30,137 changes. in U.S. treasury funds. employee stock purchase plan. and Policies royalty agreements. reduced disclosure obligations regarding executive compensation arrangements; and 2023. year ended December 31, 2023. NOTE has not declared any dividends. No dividends on the common stock shall be declared and paid unless dividends on the provides for the grant of performance On January 1, 2024, an additional 3,534,600 shares were reserved for issuance under the 2017 Plan. months, respectively under the 2017 Plan and 6 months under the 2014 Plan.. stock prices. awards recognized. $9.3 million and $11.5 million as of December 31, The Company’s stock-based compensation expense was recognized in operating expenses as follows: For the Year Ended December 31, 2021 2020 Research and development $ 1,679,183 $ 2,779,758 General and administrative 3,375,274 4,745,596 Total $ 5,054,457 $ 7,525,354 For the Year Ended December 31, 2021 2020 Stock options $ 4,970,670 $ 7,372,381 Employee Stock Purchase Plan 83,787 152,973 Total $ 5,054,457 $ 7,525,354 For the Year Ended December 31, 2021 2020 Weighted Weighted Volatility 84.38 % 81.48 % Expected term in years 6.03 5.88 Dividend rate 0.00 % 0.00 % Risk-free interest rate 0.90 % 0.58 % Fair value of option on grant date $ 2.50 $ 2.62 The fair value of nonemployee options granted and remeasured during the years ended December 31, For the Year Ended December 31, 2021 2020 Weighted Weighted Volatility 80.43 % 74.37 % Expected term in years 6.23 5.64 Dividend rate 0.00 % 0.00 % Risk-free interest rate 1.03 % 2.32 % Fair value of option on grant date $ 2.49 $ 1.18 Weighted Weighted Average Average Remaining Aggregate Number of Exercise Contractual Intrinsic Shares Price Life in Years Value Options Outstanding at December 31, 2019 7,405,295 $ 5.82 8.01 $ 4,488,930 Vested and exercisable at December 31, 2019 3,296,547 $ 7.93 6.53 $ 356,443 Granted 3,583,160 3.93 9.68 Exercised (165,384 ) 1.82 Forfeited (415,526 ) 5.00 Options Outstanding December 31, 2020 10,403,420 $ 5.26 7.59 $ 652,438 Vested and exercisable at December 31, 2020 5,395,658 $ 6.45 6.13 $ 445,599 Granted 2,045,913 3.53 9.47 Exercised (288,525 ) 2.36 Forfeited (1,384,050 ) 4.08 Options Outstanding December 31, 2021 10,776,758 $ 4.97 6.07 $ 2,389,890 Vested and exercisable at December 31, 2021 6,188,200 $ 5.98 4.63 $ 1,531,907 last ownership change. The tax effects of temporary differences that gave rise to significant portions of the deferred tax assets and liabilities were as follows: December 31, 2021 2020 Deferred tax assets/liabilities: Net operating loss carryovers 49,086,998 $ 74,064,673 Research and development tax credits 2,422,331 5,458,004 Share-based compensation 4,663,578 4,978,370 Accrued compensation (28,243 ) 514,316 Depreciation (51,920 ) (29,760 ) Charitable contributions 87,672 87,120 Intangible assets 7,121,484 4,742,031 Total gross deferred tax assets/liabilities 63,301,900 89,814,754 Valuation allowance (63,301,900 ) (89,814,754 ) Net deferred tax assets (liabilities) $ 0 $ 0 December 31, 2021 2020 Federal income tax benefit at statutory rate 21.00 (21.00 ) State income tax, net of federal benefit (1) 1.05 6.15 Permanent items 0.53 0.75 Change in valuation allowance (21.39 ) 16.72 Research and development tax credits (1.05 ) (2.64 ) Other 0.93 0.02 Effective income tax (benefit) expense rate 1.07 % 0.00 % (1) Inclusive of $5.8 million deferred tax benefit due to change in apportionment Patent Rights. The Company is developing OV329 under this agreement. contingencies are expensed as incurred. The Company is not currently involved in any legal matters arising in the normal course of business. AGREEMENTS year ended December 31, 2023. During the year ended December 31, The For the Year Ended December 31, 2021 2020 Net income (loss) $ 122,834,584 $ (81,035,576 ) Net income attributable to participating securities (2,997,344 ) - Net income (loss) attributable to common stockholders $ 119,839,261 $ (81,035,576 ) For the Year Ended December 31, 2021 2020 Basic and Diluted Net income (loss) attributable to common stockholders $ 119,839,261 $ (81,035,576 ) Weighted average common shares outstanding used in 67,479,403 58,451,293 Weighted average common shares outstanding used in 68,067,992 58,451,293 Net income (loss) per share, basic $ 1.78 $ (1.39 ) Net income (loss) per share, diluted $ 1.76 $ (1.39 ) ☒x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 19342021OR☐o TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934Delaware 2834 46-5270895 Delaware283446-52708951460 Broadway, Suite 15021, New York10036(646) 10001Indicatendicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ Noo ☒No ☐ o No ☒xYesx ☒ No ☐Yesx ☒ No ☐☐☒☐☒☒☒oYes ☐ No ¨☒If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ¨☐o No ☒x2021,2023, the last day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the Common Stock held by non-affiliates of the registrant was approximately $217.2$191.1 million based on the closing price of the registrant’s common stock on June 30, 2021.2023. The calculation excludes shares of the registrant’s common stock held by current executive officers, directors and stockholders that the registrant has concluded are affiliates of the registrant. This determination of affiliate status is not a determination for other purposes.10, 2022,5, 2024, there were 70,373,16270,709,857 shares of common stock outstanding.20222024 Annual Meeting of Stockholders, which the registrant intends to file pursuant to Regulation 14A with the Securities and Exchange Commission not later than 120 days after the registrant’s fiscal year ended December 31, 2021,2023, are incorporated by reference into Part III of this Annual Report on Form 10-K.istatementsour expectations regarding government and third-party payor coverage and reimbursement;COVID-19 pandemic and its effectsimpact of geopolitical tensions, including war or the perception that hostilities may be imminent, adverse global economic conditions, terrorism, natural disasters or public health crises on our operations, access to capital, research and development and clinical trials and potential disruption in the operations and business of third-party manufacturers, contract research organizations, or CROs, other service providers,third parties and collaborators with whom we conduct business;our expectations regarding government and third-party payor coverage and reimbursement;•our ability to compete in the markets we serve;•the impact of government laws and regulations;•developments relating to our competitors and our industry; and•subsidiaries.subsidiary. This Annual Report on Form 10-K also contains references to our trademarks and to trademarks belonging to other entities. Solely for convenience,1TM™ symbols, but such references are not intended to indicate, in any way, that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other companies.2committedthat is dedicated to developing medicines that transformmeaningfully improving the lives of people affected by certain epilepsies and seizure-related disorders.brain conditions with seizure symptoms. We believe that addressing these epilepsiesdisorders represents a substantial scientific, medical and seizure-related disorders represent an attractive area for drug development ascommercial opportunity. Over the last decade, scientific understanding of the underlying biology of neuronal hyperexcitability and the related pathophysiology of epilepsy and many neurological disorders has grown meaningfullyimproved. This understanding of disease, coupled with advances in preclinical research tools, is improving the predictive potential of translational research, and thereby, may increase the probability of successful clinical development of anti-seizure medicines (“ASMs”). Emerging science also indicates that addressing the underlying causes of hyperexcitability may have therapeutic applications in broad neurological disease well beyond epilepsy.few100 years, and today represents a substantial number of epilepsy patients continue to experience breakthrough seizures that can cause enduring damage to the brain. Individuals who suffer from rare forms of refractory epilepsies may experience persistent seizure rates ranging from 50 - 90%. The seizures they suffer can have a devastating impact both upon patients and their families, by triggering permanent motor, cognitive and developmental delays, as well as epileptogenesis, which is a cascade of seizures begetting more seizures. Some patients with developmental epileptic encephalopathies experience even greater rates of refractory seizures that are resistant to drug therapy.commercial opportunity.scientific advancements have set the stage for a potential era of neurotherapeutics, which we believe will be led by ASMs.aim to identify, discover and develop novel therapeutics based onbelieve that major developments in the rapid increase in scientific understanding of the rolebiology of geneticsthese diseases means that key areas of unmet need, including many epilepsies and key biological pathways relevantseizure disorders, are now addressable and offer significant medical potential. Our team has proven expertise in understanding MoAs that underlie seizures and shaping potential therapies to diseasestreat rare epilepsies and disorders with seizures. Specifically, we have built a pipeline focused on treating the extrinsic or intrinsic causes of neuronal hyperexcitability. This know-how affords us an ability to build Ovid in a manner focused on delivering successive, novel medicines for epilepsies and seizure-related neurological conditions.brain. development and subsequent repurchase of our rights to soticlestat by Takeda. In 2017, we in-licensed a 50% stake in soticlestat for $26.0 million, and further invested $57.0 million in designing and executing soticlestat’s early and mid-stage clinical trials. In 2021, following encouraging Phase 2 findings, which we delivered six months ahead of schedule, we entered into a Royalty, License and Termination Agreement (“RLT Agreement”) through which we sold back our rights to soticlestat to Takeda. The RLT Agreement provided us with $196.0 million paid in Q1 2021 and, if soticlestat is approved and successfully commercialized, we are eligible to receive up to $660.0 million in sales and regulatory milestone payments and low double-digits up to 20% of potential net sales-based tiered royalty payments. The RLT Agreement provides us with a potential stream of non-dilutive capital. The funds received from this transaction have enabled us to invest in and secure what we believe is a world-leading pipeline during a period when we believe the cost of capital would have been unduly expensive. In 2023, we sold a 13% stake in the royalty, regulatory and commercial milestone payments that we are eligible to receive under the RLT Agreement to Ligand Pharmaceuticals Incorporated (“Ligand”) for $30.0 million.knowledgesubject-matter expertise in seizures and neurological conditions. This includes epileptologists, physicians, academic scientists, commercial and biopharmaceutical industry leaders. In total, we have five individuals with M.D. degrees and 13 professionals with Ph.D. degrees specializing in the sciences. Our operational and commercial leaders have extensive experience fostering market access and sales for leading neurological medicines. In total, our team’s collective professional experience has involved the successful development or commercial launch of raremore than 25 CNS medicines, including several epilepsy products.diseases, howconditions, including seizures and epilepsies.themthe heterogeneous causes of seizures. Accordingly, our pipeline seeks to curate and how to identify the clinically meaningful endpoints required for developmentdevelop a unique set of compounds that can be effective by itself and/or can provide the necessary intervention to impact multiple mechanisms. We believe this approach has the potential to provide the basis of a modern differentiated and leading epilepsy franchise. Core tenets of our approach include a focus on:these conditions. As a resultsynthetic methodology, formulation technology, generative artificial intelligence and biology target research have unlocked more opportunities for innovative and creative medicines. The compounds we seek to develop are brain-permeable and are designed to modulate specific biological targets. We seek to take advantage of this knowledge,the versatility small molecules have to offer in terms of manufacturing, chemistry, and dosing to potentially deliver novel and next-generation medicines that will make meaningful improvements in the lives of patients.have developedpursue directly or indirectly modulate hyperexcitability. These targets are associated with regulating inherent intracellular excitatory/inhibitory balance within neurons.programstractable seizure types. Similarly, many ASMs were later proven to have clinical efficacy in other neurological conditions. Simply put, effective therapeutic outcomes in highly pharmaco-resistant indications often bodes well for treating broader neuropathologies.demonstratedmedicines that are well tolerated and exhibit few DDIs. In addition, we strive to impact the broader symptomatology of disease, including for example, the measurement of behavior and communication change. model by progressing compounds through to late-stage development. Our strategy focuses initially on evaluating our investigational medicines for pathways and targets impacting rare diseases, andbrain conditions. If effective in rare disease, our intent is to explore expanding on that may also have therapeutic relevance forsuccess to broader conditions of the brain. We are executing on our strategybrain for which the MoA may hold therapeutic relevance. Accordingly, in the future, we expect to build this pipeline by discovering, in-licensing and collaborating with leading biopharmaceutical companies and academic institutions.Our Focus: Epilepsies and Rare Neurological DisordersNeurological disorders have a devastating impact on patients and their families. Patients suffering from rare disorders of the brain often require extensive care, and go largely underserved by impactful therapeutics. We believe that there are at least 100 neurological disorders, epileptic encephalopathies and other related rare neurological disorders that we may be able to target. Theseextend our knowledge and drug programs to other neurological conditions. This approach is supported by research indicating that more than ten ASMs are used in treatment of other (non-epilepsy) neurological diseases today.characterized by seizures and degenerationeither uniquely (1) expressed in the developmentCNS, such as: CH24 and functioningKCC2 co-transporters or (2) over-expressed in a pathological state, such as ROCK and GABA-AT.brain. Due to a historical overwhelming preferencemost difficult organs in the body to treat, in part due to the challenge of penetrating the BBB. Ovid’s drug industry to develop medicines for broader neurological indications, manydevelopment programs include potential small molecule therapies that demonstrate penetration of these disorders have no approved therapies. As a result, recent scientific advancements have been overlooked, which we believe presents us with an opportunity to pursue these indications. These reasons include:•Potential to affect seizures and disease progression. We are focusing primarily on pediatric disorders that are typically characterized by epilepsy-related symptoms and seizures. Today, it is estimated that as many as 30-40% of patients treated for epilepsies continue to experience seizures; signifying that additional effective treatment options are needed. We believe that our therapeutic candidates may be able to meaningfully reduce harmful seizures, address symptoms, and potentially alter the progression of disease, especially if they can be administered early in life.BBB.High penetrance linking genetic defect to disorder pathology.Clinically translatable preclinical models Some rare neurological disorders have a genetic origin and typically have a strong correlation, or penetrance, between the presence of a gene and the manifestation of the corresponding disease pathology. As a result, we believe in those cases we can develop drug candidates that will be efficacious in patients with a given genetic profile.•Predictive genetic and other models.. Recent advances in genetics enable us to employ predictive in vitro and in vivo genetic models of certain ofspecific brain diseases. We believe these disorders. Thesepredictive models will allow us to evaluate and observe a drug candidate’s potential activity prior to initiation of clinicalhuman trials. ThroughApplying these models, we believe we will be able to select the most relevant clinicalindication and seizure endpoints for our trialsstudies and increase the potential for clinical success.Overlapping pathophysiologyClear primary endpoints and symptoms.scales Neurological. We primarily focus on disorders that are often characterized by a numberepilepsy-related symptoms and seizures. Many seizure types afford clear observable endpoints and biomarkers that help capture and measure evidence of overlappingthe clinical impact of our drug candidates. Our team of development experts has extensive experience designing scales to measure other symptoms that are common among seizure-disorders, such as seizures, sleep disturbances,cognitive declines, movement deficiencies and behavioral manifestations. We believe these commonalities will enable us to employ clinical endpoints that may be translatable from one disorder to another,These skills support our desire and ability to develop drugsmedicines that may provide a clinical benefit across multiple indications.aspects of patient health.EarlyTrial design enables early observation of proof-of-concept. By employing clinicalsurrogate biomarkers and endpoints that are highly relevant and are designed to detect meaningful clinical benefits, we anticipate that many of our studies may provide early proof-of-conceptPOC in clinical development.development, thereby directing the use of our capital to projects with higher probability of later-stage success.theseepilepsies and brain disorders have increasing access to diagnostics and genetic testing. Additionally, many are avid users of social media, tothrough which they learn new insights about thetheir conditions and share relevant information and experiences. We conduct patient disease community outreach and activities on digital platforms to efficiently identify new patients for our clinical trials, raise disease awareness and help connect with patients and caregivers.The Ovid StrategyWe believe that the sciencemedicine underlying discoveryDisciplined Business Developmentof new drugsefforts in collaboration with external leaders in the field and academic collaborators; and (2) business development activities to partner assets that have promising potential in and outside our chosen therapeutic areas.the brain has changed fundamentally over the last decade.laboratory facilities. We identify compounds with what we believe that major developmentsis untapped value sitting in other organizations’ pipelines and look to in-license or enter into collaborative agreements to secure such assets and advance clinical development. This strategy directs our efforts where we excel at creating value, which is specifically shaping translational and clinical-stage development in our understandingtherapeutic area. An integral part of our process is establishing collaborations with academic research centers to support translational expertise for our programs, including the biologyStephen Moss Lab of these diseases means3that key areasNeuropharmacology at Tufts University.unmet need are now addressable and offer tremendous medical and economic opportunity.our compounds in non-core indications or extend regional market rights outside the United States. We believe that we now have a deep understandingare well positioned to execute on our business development strategy due to the extensive experience and networks of the mechanismsour management team. Collectively, our senior management has transacted hundreds of in-licensing deals and collaborations.underlie seizureswe hope will create new possibilities and many brain disorders. This know-how offers an opportunity to build our company in a manner deeply focused on delivering novel medicines to address these medically important needs. We plan to focus our efforts in the areas ofmore good days for people living with rare epilepsies and then progressively, as science and medicine evolve, we expect to apply our knowledge to larger indications.Our strategy is to pursue drug discovery and development for epilepsies and rare central nervous system (CNS) disorders in a manner that is scientifically driven, patient focused and is coupled with an integrated and discipled approach to research, clinical development, and business development. Our team has significant experience and understanding of these epilepsies, seizure related disorders, and rare neurological conditions, and we continue build insight into the way the different molecular mechanisms and pathways underlying these disorders impact the symptoms patients suffer. We have set out to be a leader in the field, and have developed a differentiated pipeline containing three novel mechanisms of action to target different causes of epilepsies and seizures. Our team’s knowledge of epilepsy disease biology and pathology, now contributes to our pursuit of additional relevant genetic targets and molecular pathways that are the cause of seizures. We are pursuing therapeutic assets for epilepsies, seizure related disorders, and rare neurological disorders and seek to leverage those learnings in accelerated development programs. If successfully developed and marketed in rare conditions, we intend to explore our assets for broader neurologic indications. Our cohesive focus in epilepsies and seizures reinforces our belief that we have the potential to develop and produce multiple novel medicines, and thereby succeed in our mission.Scientifically DrivenWe take a scientifically driven approach to identify promising drug candidates for our pipeline. We are building our portfolio based on the existence of known biological rationales, including a focus on epilepsies, seizure-related disorders and neurological disorders that are associated with validated targets, genetic linkages and clear endpoints, such as seizures, for study in clinical trials. We use our deep understanding of neurology and epilepsies to identify differentiated mechanisms of action for initial drug candidates. As we advance our drug candidates into and through the clinical evaluation, we are building on the emerging body of scientific and clinical insights developed by us and others in the biopharmaceutical industry to target important disease pathways of the brain. As we evaluate data from previous and ongoing preclinical studies and clinical trials, we intend to refine and improve our scientific approach and apply these insights to continue to build our current and prospective pipeline programs and clinical trials.In particular, our approach is driven by the following scientific principles:•pursue epilepsy-related disorders and rare neurological conditions for which significant therapeutic need persists•identify the genetic origin of the disorder, when possible;•target biological pathways or genes for which proof-of-concept has been established via in vitro or animal models;•focus on the mechanisms that cause the pathology of the disorder that generate the symptoms that we can target; and which may hold relevance for development of therapeutics for broader neurologic conditions in the future;•develop understanding of gene expression and link to pathophysiology;•target optimal mechanism of action for drug candidates; and•seek clear endpoints and biomarkers, if possible, to help demonstrate evidence of the clinical impact of our drug candidates.Patient FocusedWe are developing product candidatesbrain conditions. For example, we believe havethat our therapeutic candidates may be able to meaningfully reduce harmful seizures, mitigate burdensome symptoms, and potentially slow down the potential to transform the livesunderlying progression of individuals affected by epilepsies and related neurological disorders. disease.rare condition that carries serious morbiditiesrisk of morbidity and requires extensive and specialized involvement from the patients’ families, caregivers, and physicians and from patient advocacy groups.efficient clinical trialspatients,patient communities, caregivers, families, disease foundations and key opinion leaders, to better understand the history of these disorders, raise awareness, identify patients and facilitate enrollment of clinical trials;patientsthe patient communities and their physicians and caregivers; and4Research and Development Coupled with Business Developmenthave builtare focused on delivering long-term value for shareholders. Our financial strategy seeks to apply our capital in a broadfocused manner to advance a differentiated pipeline of potential drug candidatesneurotherapeutics, which we believe may generate multiple value-creating milestones from data and ultimately, commercial sales.treat epilepsies, seizure related disorders, and related neurological disorders. Our current pipeline isfund Ovid's operations into the resultfirst half of two complementary efforts: 1) internal research and development efforts2026. To achieve this cash runway, in collaboration with external leadersOctober 2023, we sold 13% of our interest in the fieldmilestones and academic collaborators; and 2) focused business development to in-license or partner assets. Central to our internal process has been collaborations that we form with world-leading academic research centers such as Columbia University and the University of Connecticut to augment target discovery and early biology. Overall, our goal is to develop a cohesive and diversified pipeline with multiple, novel mechanisms that leverage our strong capabilities in this area and provide a sustainable pipeline for years to come.We are building a specialized, scalable and robust infrastructure that we believe will make us a leader in our chosen area and potentially a partner of choice for leading biopharmaceutical companies that wish to pursue valuable drug candidates or research platforms in epilepsies and seizures. This infrastructure spans the critical domains of discovery, delivery and development. We believeroyalties that we are particularly well positioned eligibleexecutereceive from soticlestat's potential approval and commercialization to Ligand for $30.0 million. We retain 87% interest in soticlestat's regulatory and commercial milestones and royalties on sales if the drug is successfully approved and commercialized by Takeda. These future payments may contribute to funding our operations and business development strategy becauseactivities. For additional information, see the description of both the extensive network of our management teamLigand Agreement below under the heading “License and experience delivering valued partnerships with companies including Takeda, LundbeckCollaboration Agreements – 2023 Ligand Pharmaceuticals Milestones and AstraZeneca.OurRoyalties Monetization.”two drug candidates (gaboxadol and soticlestat) from pre-clinical proof of concept and either through pivotal trials or to the initiation of pivotal trials, whether by us or our collaborators.POC into human clinical trials. Today, we are one of the few epilepsy-focused biopharma companies withthat has researched and developed three noveldistinct MoAs to target seizures and one of the only clinical development programs to evaluate a highly selective ROCK2 inhibitor for neurological disease. We believe this pipeline of potential first-in-class or potential best-in-class mechanisms differentiates us and provides the basis of action for epilepsy within its pipeline. This is critical given that as many as 30 - 40 percentan attractive franchise of patients treated for epilepsy continue to experience seizures.the status and mechanism of action of our drug candidates: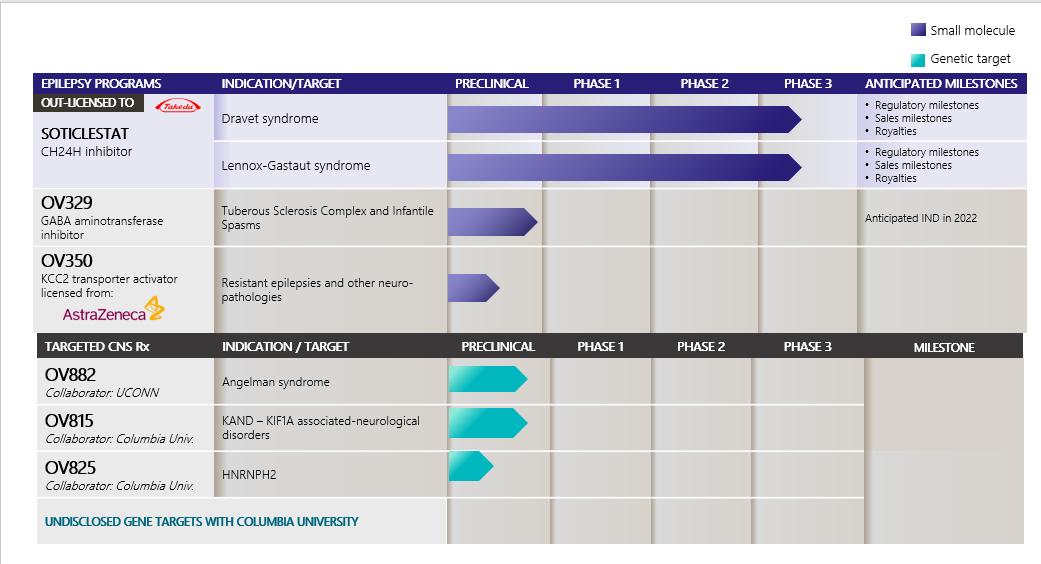
Internal ProgramsOV329OV329 (GABA aminotransferase inhibitor) iscandidate programs and their development status, MoA, and anticipated near-term milestones:
next-generation GABA aminotransferasenovel CH24 inhibitor being developed by us for the potential treatment of seizures associatedpatients with Tuberous Sclerosis Complexresistant epilepsies, following our role in its successful early and Infantile Spasms. We expect to file an IND for OV329 in 2022. OV329 functions by substantially reducing the activity of GABA aminotransferase (GABA-AT), a key enzyme responsible for the degradation of the brain’s major inhibitory neurotransmitter, GABA. OV329 leads to increased concentrations of GABA by inhibiting its metabolism. Given that epilepsy is characterized by excessive neuronal excitation, the increased levels of GABA may suppress this excitatory signaling and may reduce seizures. If successful, we anticipate OV329 could be used to treat seizures associated with Tuberous Sclerosis Complex and Infantile Spasms and we are currently assessing this approach in the pre-clinical setting. If the5safety and efficacy profile of the product is appropriate we will consider lifecycle management in epilepsy in other areas. In December 2016, we entered into a license agreement with Northwestern University, or Northwestern, pursuant to which Northwestern granted us an exclusive, worldwide license to patent rights in certain inventions, or the Northwestern Patent Rights, which relate to a specific compound and related methods of use for such compound, along with certain Know-How related to the practice of the inventions claimed in the Northwestern Patents.OV350OV350 is a small molecule that directly activates the KCC2 transporter, which is an important channel in seizure control. Ovid licensed OV350 and a library of associated compounds (previously AZ8333) from AstraZeneca in December 2021 to complement our growing pipeline of potential medicines with novel mechanisms of action for treating epilepsies.mid-stage development program. We believe itsoticlestat has the potential to bebecome a first-in-class direct activatormedicine targeting the metabolism of cholesterol in the KCC2 channel. Extensive academic literature indicates thatbrain. It has been shown to gradually reduce inflammation in the KCC2 transporter is an essential channel to maintaining homeostasis within neurons, thereby contributing to seizure control. In vivo studies illustrated that KCC2 activity leads to reduced seizure sensitivity and seizure-induced mortality. Pre-clinical mechanistic studies have also demonstrated that OV350 wasbrain as well tolerated and did not induce sedation.as indirectly acting on the N-methyl D-aspartate pathway. We believe OV350that this dual mechanism plays an important role in modulating excitatory signals involved in epilepsy, and thereby suppressing seizures.potential to be developed for multiple epilepsies and other CNS indications. Our initial efforts are focused on optimizing multiple potential formulations for OV350 and conducting animal models to inform our development plans. While we plan to develop the compound and seek initial approvals in rare epilepsies, if successful, and if the safety and efficacy profile of the product candidate is appropriate, we will consider lifecycle management in larger epilepsy indications.OV882OV882 is a short hairpin RNA (shRNA-551)RLT Agreement, we are evaluating aseligible to receive regulatory and commercial milestones payments of up to $660.0 million, in addition to potential net sales based tiered royalties of up to 20%. In 2023, we sold a potential disease-modifying gene therapy for Angelman syndrome. The most common cause of Angelman syndrome is the loss of functional UBE3A protein due to a defect13% stake in the maternal copyroyalty, regulatory and commercial milestone payments that we are eligible to receive under the RLT Agreement to Ligand for $30.0 million. Royalty payments are eligible on net sales across all regions and all future indications. The future milestones payments do not include an initial upfront payment that we received from Takeda in March 2021 of the UBE3A gene. Our aim is to develop a disease-modifying noncoding RNA vector that reduces expression of UBE3A-antisense and restores UBE3A expression via the paternal gene copy. We are$196.0 million. early stages of our research with OV882, but benefit from robust natural history and baseline data for a prior clinical trial we conducted with gaboxadol in Angelman's syndrome. From these prior programs, Ovid has deep knowledge of the Angelman's condition and retains ownership of baseline data from clinical trials with approximately 100 Angelman's patients. These proprietary insights will aid our OV882 program, which is being conducted in collaboration with the University of Connecticut School of Medicine.OV815 and OV825 - Columbia University ProgramIn June 2020, we began a strategic research collaboration with Columbia University Irving Medical Center, or Columbia, to advance genetic based therapies for a range of rare neurological conditions, complementary of our pipeline. This collaboration provides us with the potential to expand our future drug development portfolio and impact individuals living with rare genetic neurological conditions by working closely with the Precision Medicine Resource in the Irving Institute at Columbia. Our first research program with Columbia is OV815 and focuses on the kinesin-family of proteins and KIF1A-associated neurological disorder (KAND), under this research and translational development alliance, Columbia will align its expertise in rare disease genetics and deep clinical understanding of rare neurological diseases with our discovery, translational, and clinical development expertise in neurodevelopmental disorders and rare epilepsies.Soticlestat: Continued interest via Out-Licensing Agreement with Takeda PharmaceuticalsBackgroundIn January 2017 we entered intoas a license and collaboration agreement withbetween us and Takeda orfor rare epilepsies. Under this original agreement, Ovid held a 50% ownership stake in soticlestat and Takeda retained the Takeda collaboration agreement, to develop and commercialize soticlestat, which culminated inremaining 50%. Following a successful Phase 2 development program. Onprogram led by us, in March 29, 2021, we entered into a royalty, license and termination agreement, or the Takeda License and Termination Agreement, with Takeda.RLT Agreement. Under the terms of the Takeda License and TerminationRLT Agreement, we terminated the Takedaour original collaboration agreement with Takeda, and Takeda subsequently secured an exclusive license and intellectual property rights to repurchase our 50% global share in soticlestat. Takeda secured an exclusive license of soticlestat and our relevant intellectual property rights inIn exchange, for an upfront payment, development and commercial milestone payments, and royalties. Takeda has since assumed all responsibility for, and costs of, both development and commercialization of soticlestat.In March 2021, we received an upfront payment of $196 million$196.0 million. In addition, if soticlestat achieves regulatory approval, and is successfully commercialized, we are eligible to receive up to an additional $660$660.0 million in development, regulatory and sales milestones. In addition, if soticlestat achieves regulatory approval, we will receivecommercial milestone payments and potential tiered royalties on net sales of soticlestat at percentages ranging from the low double-digits up to 20%, subject to standard reductions in certain circumstances. Royalties are payable on a country-by-country and product-by-product basis during the period beginning on the date of the first commercial sale of such product in such country and ending on the later to occur of the expiration of patent rights covering the product in such country andcountry.specified anniversaryresult of such first commercial sale. The Takeda collaborationthis agreement, will remain in effect until Takeda’s cessation of commercialization of soticlestat.6Following the out-licensing of soticlestat to Takeda, Takeda initiated two pivotal, phase three programs for soticlestat, which are referred to as SKYWAY for LGS and SKYLINE for DS. SKYWAY applies the use of Major Motor Drop Seizures as an endpoint which it believes will more accurately reflect soticlestat's effectiveness in reducing seizures in LGS. It is anticipated that Takeda will have data from these Phase 3 programs in the first half of 2023 and will seek regulatory decisions by March 2024. Takeda has reported it is preparing for a multi-regional launch of soticlestat. The United States Food and Drug Administration, or the FDA, has granted orphan drug designation for soticlestat and China has granted it Breakthrough Status for the treatment of DS and LGS.Soticlestat - MechanismSoticlestat is a potential first-in-class inhibitor of cholesterol 24 hydroxylase (CH24H), which may modulate the excitatory signals involved in epilepsy, and thereby suppress seizures. The investigational soticlestat program began as a joint development collaboration between Ovid and Takeda for rare epilepsies. Following a successful Phase 2 development program led by us, we entered into a royalty, license and termination agreement with Takeda, or the Takeda License and Termination Agreement, in March 2021 under which Takeda secured all of the global rights from Ovid to develop and commercialize soticlestat for the treatment of developmental and epileptic encephalopathies, including Dravet syndrome (DS)DS and Lennox Gastaux syndrome (LGS).LGS. In addition, Takeda has assumed responsibility for all development and commercialization costs associated with soticlestat. We have no ongoing costs or obligations.Takeda’s continuedthat the ROCK2 signaling pathway may be hyperactivated in multiple neurological diseases, including disorders involving vascular structures and nerve myelination diseases that can result in seizures, spasms and a variety of other symptoms. Despite this link, there has been limited clinical development of soticlestatROCK2 inhibitors due to challenges penetrating the BBB and the inherent challenge of avoiding inhibition of rho-associated coiled-coil-containing protein kinase 1, which can have unwanted side effects.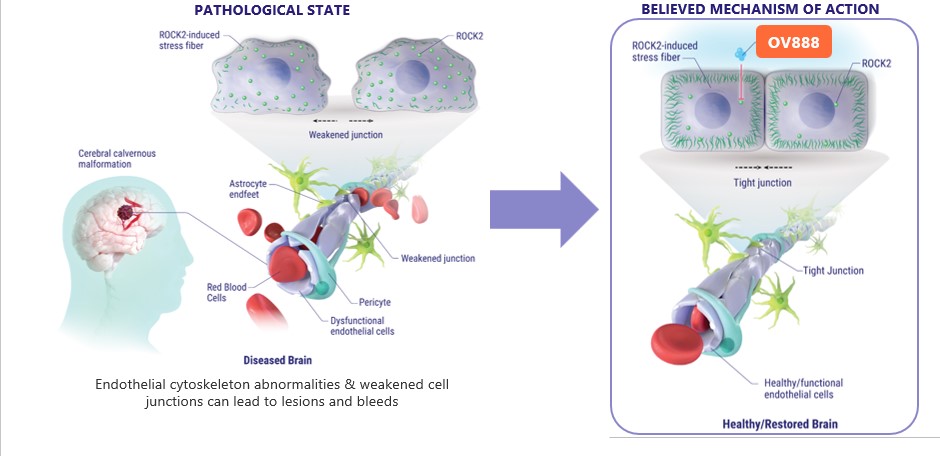
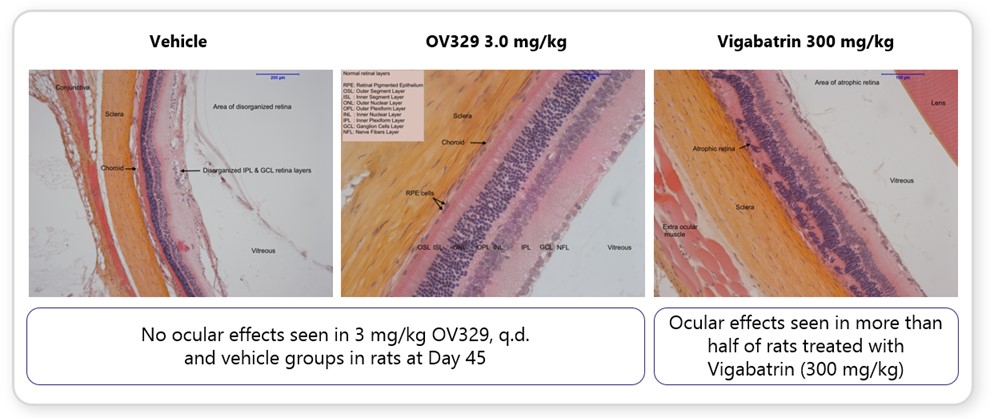
becomebe therapeutic in refractory and drug-resistant epilepsies such as seizures associated with tuberous sclerosis and refractory status epilepticus (“SE”).first-in-classvalidated drug target for seizures. Specifically, it works by substantially reducing the activity of GABA-AT, a key enzyme responsible for the degradation of the brain’s major inhibitory neurotransmitter, GABA. OV329 leads to increased concentrations of GABA by inhibiting its metabolism. Given that epilepsy is characterized by excessive neuronal excitation, the increased levels of GABA may suppress this excitatory signaling and only-in-class compound targetingmay reduce seizures.metabolismoral formulation has the potential to provide (in comparison to vigabatrin) preferred (lower) dosing. We believe that these preclinical data suggests that OV329 could allow for seizure reduction efficacy and improved tolerability in comparison to existing therapeutics.cholesterolOV329 (see Figure 3 below). These findings in both chronic and acute seizure models provide additional confidence about the therapeutic potential of OV329 in humans.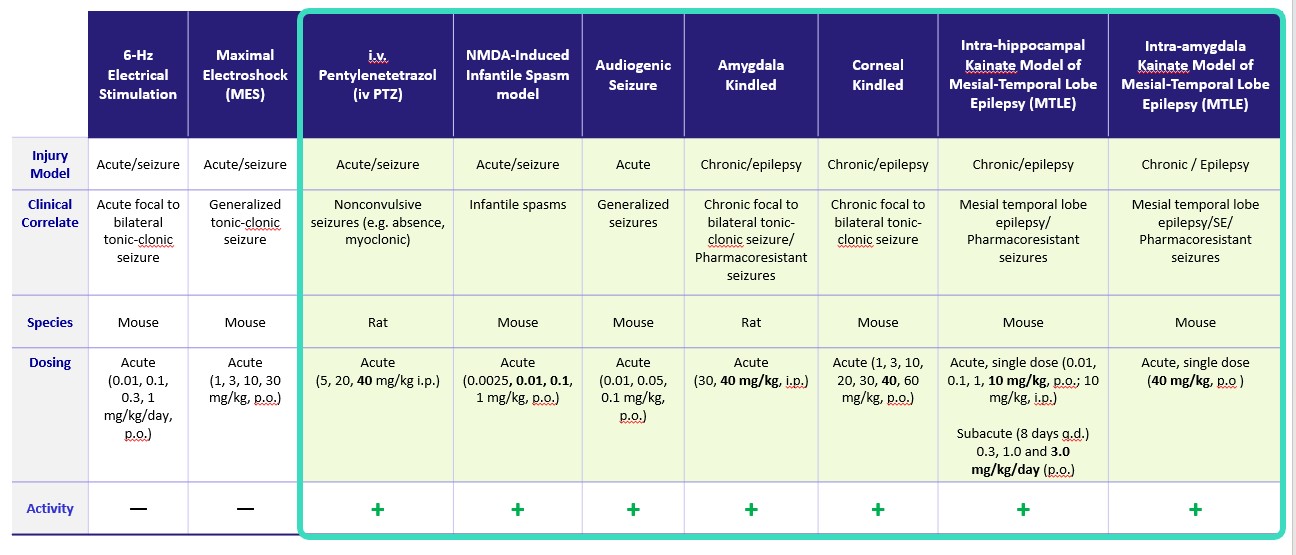
whichdamage and increased rates of mortality.
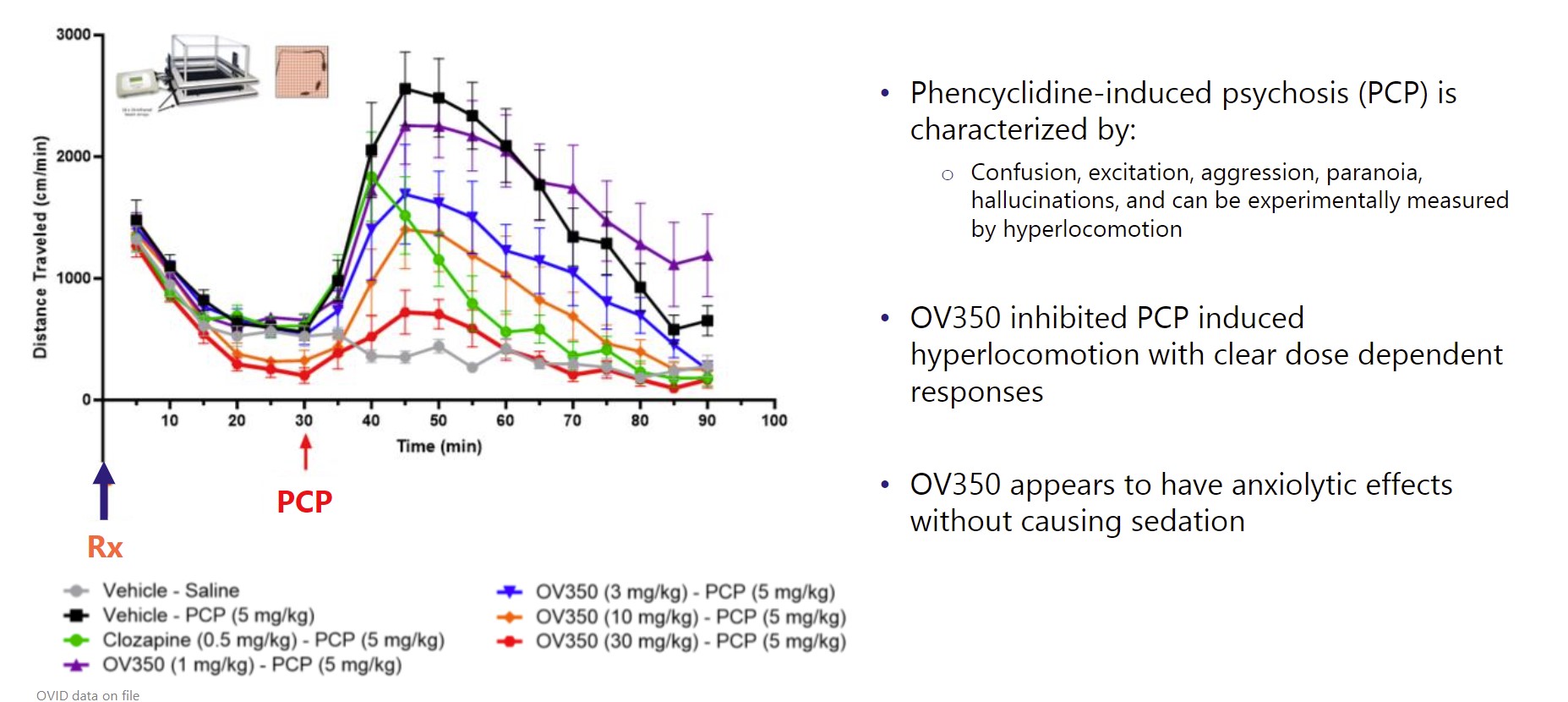
reduce inflammationexpire in 2041 excluding any potential regulatory extensions.playsgenetic medicine will play an important role in seizures.OnIn March 1, 2022, we entered into an exclusive patent license agreement with Marinus, Pharmaceuticals, Inc. or the (“Marinus License Agreement.Agreement”). Under the Marinus License Agreement, we granted Marinus an exclusive, non-transferable (except as expressly provided therein), royalty-bearing right and license under certain Ovid patents relating to ganaxolone to develop, make, have made, commercialize, promote, distribute, sell, offer for sale and import licensed products in the territory (which consistconsists of the United States, the European Economic Area, United Kingdom and Switzerland) for the treatment of CDKL5 deficiency disorders in humans.disorders. Following the potential date of regulatory approval by the FDA of the first licensed product in the territory, which was received on March 18, 2022, Marinus will,issued, at ourthe Company's option, (i) pay to us the sum of $1,500,000 in cash; or (ii) issue us 123,255 shares of Marinus common stock, par value $0.001 per share. The Marinus License Agreement also provides for payment of royalties from Marinus to us in single digitssingle-digits on net sales of each such licensed product sold.OnIn February 1, 2022, we entered into an exclusive license option agreement or the (“Healx License and Option Agreement,Agreement”) with Healx.Healx, Ltd (“Healx”). Under the terms of the Healx License and Option Agreement, Healx has secured a one-year option to investigate gaboxadol (OV101) as part of a potential combination therapy for Fragile X syndrome in a Phase 2A clinical trial, as welland as a treatment for other indications, for an upfront payment of $0.5 million, and fees to support prosecution and maintenance of our relevant intellectual property rights. In February 2023, we amended the Healx Agreement to extend the option period by three months. At the end of the one-year option period, Healx has the option to secure rights to an exclusive license under our relevant intellectual property rights, in exchange for an upfrontadditional payment of $2.0 million (“Option Fee”), development and commercial milestone payments, and low to mid-tier double digit royalties. Royalties are payable on a country-by-country and product-by-product basis during the period beginning on the date of the first commercial sale of such product in such country and ending on the later to occur of the expiration of patent rights covering the product in such country and a specified anniversary of such first commercial sale.will assumeAgreement was further amended to: (i) grant Healx a development license; (ii) grant a commercial license upon payment of the Option Fee to us; and (iii) restructure the timing of the Option Fee payable to us.both development andof gaboxadol in June 2023. Healx will also be responsible for all commercialization costs of gaboxadol following the exercisegrant of a commercial license upon payment of the option.Option Fee. We will retain the option to co-develop and co-commercialize the program with Healx or the (“Ovid Opt-In Right,Right”) at the end of a positive readout of clinical phase 2B, and, willin such case, would share net profits and losses in lieu of the milestones and royalty payments. We do not plan to conduct further trials of gaboxadol. The term of the Healx License and Option Agreement will continue until the later of (a) the expiration of all relevant royalty terms, or in the event that Healx does not exercise its option during the option period defined in the Healx License and Option Agreement, or the Option Period, the expiration of such period, or in the event that Healx does exercise its option during the Option Period, and we do not exercise the Ovid Opt-In Right during the period of time we have to opt-in, or the Opt-In Period, or the opt-in terms are otherwise terminated, upon the expiration of all payment obligations, or (c) in the event that Healx does exercise the Option during the Option Period, and we do exercise the Ovid Opt-In Right during the Opt-In Period, such time as neither Healx nor Ovid is continuing to exploit the gaboxadol. As part of the revised contractual obligations with Lundbeck, Ovid will owe Lundbeck a share of all milestone and royalty payments received from Healx, if we do not exercise the Ovid Opt-In Right. If we to exercise the Ovid Opt-In Right to co-develop and co-commercialize the program with Healx, we will owe a share of the net profit share to Lundbeck.7OnIn December 30, 2021, we entered into an exclusive license agreement or the (“AstraZeneca Exclusive License Agreement,Agreement”) with AstraZeneca. Under the terms of the AstraZeneca Exclusive License Agreement, we have obtained worldwide rights to a libraryportfolio of early-stage, small moleculesmolecule compounds targeting the KCC2 transporter, including our lead compound, OV350. In exchange offor an upfront payment of $5.0 million in cash and $7.3 million in shares of Ovidour common stock to AstraZeneca, we are responsible for using commercially reasonable efforts to carry out all future development and commercialization of KCC2 transporter activators in epilepsies and potentially other neuropathic conditions. We are obligated to pay AstraZeneca potential clinical development milestones of up to $8.0 million, regulatory milestones of up to $45.0 million and total commercial milestones of up to $150.0 million, as well as tiered royalty payments ranging from the single digitssingle-digits up to 10ten percent on net sales. At the time of proof of clinical efficacy, AstraZeneca will have the right of first negotiation to opt in to co-develop and co-commercialize KCC2 transporter activators with Ovid. The license option will continue until the expiration of all relevant royalty terms.License Agreement with H. Lundbeck A/SIn March 2015, we entered into a license agreement with Lundbeck, which we subsequently amended in May 2019, July 2020 and in February 2022. As part of the Lundbeck agreement, we obtained from Lundbeck an exclusive (subject to certain reserved non-commercial rights), worldwide license to develop, manufacture, and commercialize OV101, also known as gaboxadol, for the treatment of human disease. Under the agreement, we were responsible for conducting commercially reasonable efforts to carry out all future development and commercialization of OV101. We were also obligated to make certain manufacturing-related payments to Lundbeck, including for its preparation of a drug master file for OV101.In connection with the Lundbeck agreement, we issued 489,756 shares of our common stock to Lundbeck. We also agreed to pay to Lundbeck milestone payments up to an aggregate of $189.0 million upon the achievement of certain global development, regulatory and sales milestone events with gaboxadol. Pursuant to the Lundbeck agreement, the first milestone payment is $1.0 million, which is due upon the successful completion of the first Phase 3 trial for a product in which OV101 is an active ingredient. In addition, if we successfully developed and commercialized OV101, we would be obligated to pay to Lundbeck tiered royalties based on single-digit and low-double digit percentage of net sales of OV101, subject to certain reductions for generic product sales and for royalties paid for licenses to third party intellectual property. If Lundbeck manufactures OV101 compound for us after the expiration of the royalty term, we will pay to Lundbeck, in addition to the fully burdened cost of such manufacture, a low, single-digit manufacturing royalty on the net sales of OV101 manufactured by Lundbeck.We closed our OV101 (gaboxadol) program in Angelman syndrome in early 2021. On February 1, 2022, we entered into Amendment No. 3 to the Lundbeck agreement, or Amendment No. 3, to permit our performance under the Healx License and Option Agreement. Under the terms of Amendment No. 3, if Healx exercises its option, we will owe Lundbeck a share of all milestone and royalty payments received from Healx if we choose not to exercise the Ovid Opt-In Right. If we choose to exercise the Ovid Opt-In Right and to co-develop and co-commercialize the program with Healx, we will owe a share of the net profit share to Lundbeck.or the (“Northwestern Patent Rights,Rights”) which relate to a specific compound (OV329) and related methods of use for such compound, along with certain Know-Howknow-how related to the practice of the inventions claimed in the Northwestern Patents.uses. We have agreed that we will not use the Northwestern Patent Rights to develop any products for the treatment of cancer, but Northwestern may not grant rights in the technology to others for use inuses, other than cancer. We also have an option, exercisable during the term of the agreement to an exclusive license under certain intellectual property rights covering novel compounds with the same or similar mechanism of action as the primary compound that is the subject of the license agreement. Northwestern has retained the right, on behalf of itself and other non-profit institutions, to use the Northwestern Patent Rights and practice the inventions claimed therein for educational and research purposes and to publish information about the inventions covered by the Northwestern Patent Rights.agreement,Agreement, we paid an upfront non-creditable one-time license issuance fee of $75,000, and we are required to pay an annual license maintenance fee of $20,000, which will be creditable against any royalties payable to Northwestern following first commercial sale of licensed products under the agreement. We are responsible for all ongoing costs of filing, prosecuting and maintaining the Northwestern Patents,Patent Rights, but we also have the right to control such activities using our own patent counsel. In consideration for the rights granted to us under the Northwestern agreement, we are required to pay to Northwestern up to an aggregate of $5.3 million upon the achievement of certain development and regulatory milestones for the first product covered by the Northwestern810ten years following the first commercial sale of such product in such country. If Ovid sublicenses a Northwestern Patent Right, it will be obligated to pay to Northwestern a specified percentage of sublicense revenue received by us, ranging from the high single digitssingle-digits to the low-teens.agreementAgreement requires that we use commercially reasonable efforts to develop and commercialize at least one product that is covered by the Northwestern Patent Rights.agreementAgreement will remain in force until the expiration of our payment obligations thereunder. We have the right to terminate the agreement for any reason upon prior written notice or for an uncured material breach by Northwestern. Northwestern may terminate the agreement for our uncured material breach or insolvency.rare disorders of the brain.epilepsies and seizure-related disorders. In markets for which commercialization may be less capital efficient for us, we may selectively pursue strategic collaborations with third parties in order to maximize the commercial potential of our drug candidates.(which is set to be acquired(acquired by UCB in 2022), Jazz Pharmaceuticals Inc.plc (acquired by UCB in 2022), Sage Therapeutics, Inc., Marinus Pharmaceuticals, Inc., Mallinckrodt Pharmaceuticals,plc, SK Biopharmaceuticals Inc., StokeEpygenix Therapeutics, Inc., Ultragenyx, Roche, and PTCStoke Therapeutics, Inc. and Xenon Pharmaceuticals, Inc. are our most direct competitors with respect to soticlestat, OV329 and OV350.9 For example, the proprietary map of disease-relevant biological pathways underlying orphan disorders of the brain that we developed would not be appropriate for patent protection and, as a result, we rely on trade secrets to protect this aspect of our business. currently own issued U.S. patents directed to treatment of Angelman syndrome and Fragile X syndrome with OV101 that expire in 2035, excluding any regulatory extensions. We also have exclusively licensed a portfolio of issued U.S. and international patents from Lundbeck directed to polymorphic forms of OV101 and their preparation and these patents expire on dates ranging from 2025 to 2028. In addition, we have exclusively licensed from Lundbeck an issued U.S. patent that will expire in 2036 and a pending application directed to an OV101methods of manufacturing processes that, if issued, would have a statutory expiration in 2036.OV101. We have also filed, and own, multiple patent families directed to methods of treatment and formulations with OV101. Additional issued patentsSubsequently, we have licensed much of this portfolio to Healx under the Amended Healx Agreement.pending applications are directed to methods of treating neurodegenerative diseases and developmental disorders. We are seeking or will seekown, multiple patent protection for these inventions in numerous countries and regions including, among others, Europe, Australia, Canada, Mexico, Israel, Japan, China, and Korea.We licensed from Takeda a portfolio of U.S. and international patents and applicationsfamilies directed to the soticlestatsynthesis of OV329 and methods of treatment with OV329.these patents and applicationsif issued, will expire in 2032,2041, excluding any potential regulatory extensions.or the USPTO,(the “USPTO”) delay in issuing the patent as well as a portion of the term effectively lost as a result of the FDA regulatory review period. However, as to the FDA component, the restoration period cannot be longer than five years and the total patent term including the restoration period must not exceed 14 years following FDA approval. The duration of foreign patents varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective filing date. The actual protection afforded by a patent may vary on a product-by-product basis, from country to country and can depend upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.10or (“FDCA”) and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with applicable(NDAs)(“NDAs”) or Biologics License Applications (BLAs)(“BLAs”), withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.or (“GLP regulations.begin.or (“IRB”) at each clinical site before each trial may be initiated.or (“GCP”) requirements to establish the safety and efficacy of the proposed drug product for each indication.BLA.applicable.or (“cGMP”), requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and11or (“NIH”) for public dissemination on their www.clinicaltrials.gov website.or (“PDUFA”), guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to FDA because the FDA has approximately two months to make a “filing” decision.or (“PREA”) as amended and reauthorized, certain research must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.or (“REMS”) plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.12(ANDA)(“ANDA”) or Biosimilar application, to market a drug or biologic with the same active moiety for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Orphan drug exclusivity does not prevent FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the application user fee. A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.13recalls.14statesstate and foreign governments in which we will conduct our business, including our clinical research, proposed sales, marketing and educational programs.or HIPAA,(“HIPPA”), as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), which governs the conduct of “covered entities,” including certain healthcare providers, health plans, and healthcare clearinghouses, as well as their respective “business associates”associates,” including their covered subcontractors, that create, receive, maintain or transmit individually identifiable health information for or on behalf of a covered entity, with respect to certain electronic healthcare transactions and protecting the security and privacy of protected health information; certain state laws governing the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; the federal Anti-Kickback Statute, which prohibits, among other things, individuals or entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; federal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent; federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; the Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics, and medical supplies to report annually to the U.S. Department of Health and Human Services (“HHS”) information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other healthcare professionals (such as physician assistants and nurse practitioners) and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members.15or the (“PPACA”), has substantially changed healthcare financing and delivery by both governmental and private insurers, and significantly impacted the pharmaceutical industry. The PPACA, among other things, established an annual, nondeductible fee on any entity that manufactures or imports certain specified branded prescription drugs and biologic agents, revised the methodology by which rebates owed by manufacturers to the state and federal government for covered outpatient drugs under the Medicaid Drug Rebate Program are calculated, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations, provided incentives to programs that increase the federal government’s comparative effectiveness research and created a licensure frame work for follow-on biologic products. Since its enactment there have been executive, judicial and Congressional challenges to certain aspects of the PPACA. For example, President Trump has signed several Executive Orders and other directives designed to delay the implementation of certain provisions of the PPACA or otherwise circumvent some of the requirements for health insurance mandated by the PPACA. Concurrently, Congress considered legislation to repeal or repeal and replace all or part of the PPACA. While Congress has not passed comprehensive repeal legislation, several bills affecting the implementation of certain taxes under the PPACA have been signed into law. The Tax Cuts and Jobs Act of 2017 includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the PPACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. On June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the PPACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. Thus, the PPACA will remain in effect in its current form. Prior to the U.S. Supreme Court ruling, ,Further, on January 28, 2021,August 16, 2022, President Biden issued an executive order to initiate a special enrollment periodsigned thepurposes of obtainingindividuals purchasing health insurance coverage in PPACA marketplaces through plan year 2025. The IRA also eliminates the PPACA marketplace. The executive order also instructed certain governmental agencies to review“donut hole” under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create barriers to obtaining access to health insurance coverage through Medicaid or the PPACA.creating a new manufacturer discount program. It is possible that the PPACA will be subject to judicial or Congressional challenges in the future. It is unclear how any such challenges and the healthcare reform measures of the Biden administration will impact the PPACA.through 2031until 2032 unless additional Congressional action is taken. However, COVID-19 relief support legislation suspended the 2% Medicare sequester from May 1, 2020 through March 31, 2022. Under current legislation, the actual reduction in Medicare payments will vary from 1% in 2022 to up to 3% in the final fiscal year of this sequester. Additionally, in January 2013, then President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Congress is also considering additional health reform measures. presidential executive orders and several recent presidential executive orders, Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. For example, in July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to Biden’s executive order, on September 9, 2021, the U.S. Department of Health and Human Services, or HHS released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. No legislation or administrative actions have been finalizedFurther, the IRA, among other things (i) directs HHS to implement these principles. We expectnegotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that additional federaloutpace inflation. These provisions started to take effect in fiscal year 2023 and state, as well as foreign, healthcare reform measureswill continue to take effect progressively thereafter, although may be subject to legal challenges. On August 29, 2023, HHS announced the list of the first ten drugs that will be adopted insubject to price negotiations, although the future, any of which could result in reduced demand for our products or additional pricing pressure. Further, itMedicare drug price negotiation program is possible that additional governmental actioncurrently subject to legal challenges. It is taken incurrently unclear how the IRA will be implemented but is likely to have a significant impact on the pharmaceutical industry. In response to the COVID-19 pandemic.162021,2023, we had 5740 full-time employees, the majority of whom were primarily engaged in research and development activities, including more than 10five individuals with M.D. degrees and 13 professionals with Ph.D. degrees withspecializing in the sciences. Many of these professionals have extensive epilepsy and neurology experience. In total, within our management team, we have colleagues who worked to shape the development or commercialization of a number of important marketed neurology and ASMs, including: Ztalmy, Fintepla, Brineura, Gilenya, Tysabri and Tecfidera.Ourresources objectives include,assets, including, among others: employee engagement, development and training, talent acquisition and retention, employee wellness, diversity, inclusion, and compensation, benefits and equity.applicable, identifying, recruiting, retaining, incentivizingare one-third of our board of directors. Approximately half of our organization is multicultural.integratingtheir courage and perseverance to overcome obstacles and operate with a sense of purpose and urgency on behalf of patients. Grounded in these guiding principles, we believe we have developed a collaborative environment where our existingcolleagues feel respected, valued, and additional employees. The principal purposes of ourcan contribute to their fullest potential.1460 Broadway,441 Ninth Avenue, 14th Floor, New York, New York. In June 2022, we formally instituted our hybrid work policies. In 2022, we executed what we believe was a smooth transition to a hybrid work environment while ensuring that ample resources, support and flexibility were available to our employees.on a monthly basis. In 2021, we entered into a 10-year lease of an office space at 441 Ninth Avenue, New York, New York. We anticipate using this space as our new principal executive offices and expect that our employees will move into those offices mid-year 2022.1460 Broadway, Suite 15021441 Ninth Avenue, 14th Floor, New York, New York 1003610001 and our telephone number is (646) 661-7661. Our corporate website address is www.ovidrx.com. Information contained on or accessible through our website is not a part of this Annual Report, and the inclusion of our website address in this Annual Report is an inactive textual reference only.or the SEC,(“SEC”) our annual reports on Form 10-K, Annualquarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act. We make available on our website at www.ovidrx.com under “Investors,” free of charge, copies of these reports as soon as reasonably practicable after filing or furnishing these reports with the SEC.17 In addition, such risks may be amplified by the COVID-19 pandemic and its potential impact on Ovid’s business and the global economy. for these or any other indications, or successfully develop any other drug candidates, or experience significant delays in doing so, our business will be harmed.time-consumingtime consuming and difficult to design and implement and involve uncertain outcomes. Further, we may encounter substantial delays in our clinical trials or we may fail to demonstrate safety and efficacy in our preclinical studies and clinical trials to the satisfaction of applicable regulatory authorities.Takeda License and TerminationRLT Agreement, we are entitled to receive royalty and milestone payments in connection with the development and commercialization of soticlestat. If Takeda fails to progress, delays or discontinues the development of soticlestat, we may not receive some or all of such payments, which would materially harm our business.18•COVID-19 could adversely impact our business, including our preclinical studies, clinical trials and access to capital.Historically, we have incurred significant operating losses andDue to a one-time, upfront payment of $196.0 million pursuant to the royalty, license and termination agreement (the “Takeda License and Termination Agreement”) with Takeda Pharmaceutical Company Limited (“Takeda”), our net incomeOur net loss was $122.8$52.3 million for the year ended December 31, 2021.2023. As of December 31, 2021,2023, we had an accumulated deficit of $171.4$277.9 million. We expect to continue to incur increasing operating losses for the foreseeable future. Since inception, we have devoted substantially all of our efforts to research and preclinical and clinical development of our drug candidates, as well as hiring employees and building our infrastructure.AllMost of our drug candidates are still in the preclinical testing stage. It could be several years, if ever, before we have a commercialized drug. We expect to continue to incur significant expenses and operating losses over the next several years, and the the net losseslosses we incur may fluctuate significantly from quarter to quarter and year to year. We anticipate that our expenses will increase substantially if, and as, we:experience further delays in our preclinical studies and clinical trials due to the ongoing COVID-19 pandemic;•19in April 2014, primarily due to research and development of our drug candidates, organizing and staffing our company, business planning, raising capital, and acquiring assets and undertaking the development of our drug candidates.assets. We have not yet demonstrated the ability to obtain marketing approvals, manufacture a commercial-scale drug or conduct sales and marketing activities necessary for successful commercialization. Consequently, any predictions about our future success or viability may not be as accurate as they could be if we had more experience developing drug candidates.2021,2023, our cash, and cash equivalents and marketable securities were $187.8$105.8 million and we had an accumulated deficit of $171.4$277.9 million. We believe that our existing cash, and cash equivalents and marketable securities will fund our current operating plans through at least 12 months from the filing of this Annual Report on Form 10-K. However, our operating plans may change because of many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings, third-party funding, marketing and distribution arrangements, as well as other collaborations, strategic alliances and licensing arrangements, or any combination of these approaches. TheCOVID-19 pandemicwar between Russia and Ukraine and war in Israel, are difficult to assess or predict, each of these events has already resulted in acaused significant disruption ofdisruptions to the global financial markets.markets and contributed to a general global economic slowdown. Furthermore, inflation rates have increased recently to levels not seen in decades. Increased inflation may result in increased operating costs (including labor costs) and may affect our operating budgets. In addition, the U.S. Federal Reserve has raised interest rates in response to concerns about inflation. High interest rates, especially if coupled with reduced government spending and volatility in financial markets, may further increase economic uncertainty and heighten these risks. If the disruption persistsdisruptions and deepens,slowdown deepen or persist, we could experience an inabilitymay not be able to access additional capital. If we do not raise additional capital on favorable terms, or at all, which could in sufficient amounts, or on terms acceptable to us, we may be prevented from pursuing developmentthe future negatively affect our financial condition and commercialization efforts, which will harm our business, operating results and prospects.Raising additional capital or acquiring or licensing assets by issuing equity or debt securities may cause dilution to our stockholders, and raising funds through lending and licensing arrangements may restrict our operations or require us to relinquish proprietary rights.Until such time as we can generate substantial revenue from drug sales, if ever, we expect to finance our cash needs through a combination of equity and debt financings, strategic alliances, and license and development agreements in connection with any collaborations. We do not have any committed external source of funds. To the extent that we issue additional equity securities, our stockholders may experience substantial dilution, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a stockholder. In addition, we may issue equity or debt securities as consideration for obtaining rights to additional compounds.20Debt and equity financings, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as redeemingpursue our shares, making investments, issuing additional equity, incurring additional debt, making capital expenditures, declaring dividends or placing limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could negatively impact our ability to conduct our business. If we raise additional capital through future collaborations, strategic alliances or third-party licensing arrangements, we may have to relinquish valuable rights to our intellectual property, future revenue streams, research programs or drug candidates, or grant licenses on terms that may not be favorable to us.incurred in the taxable year beginning after December 31, 2020 is limited to 80% of taxable income.20212023 and 2020,2022, we recorded no U.S. federal andor state income tax provisionsprovision, based on pre-tax losses of $1.3$52.3 million and zero, respectively, on pre-tax income of $124.2 million and pre-tax loss of $81.0$54.2 million, respectively. As of December 31, 2021,2023, we had available approximately $171.3$150.2 million of unused NOL carryforwards for U.S. federal and New York state income tax purposes, $12.6 million of unused NOL carryforwards for Massachusetts income tax purposes, $164.1 million of unused NOL carryforwards for New York income tax purposes, and approximately $163.9 million of unused NOL carryforwards for New York City income tax purposes, that may be applied against future taxable income. Our NOL carryforwards are significantly limited such that if we achieve profitability in future periods, we may not be able to utilize most of the NOL carryforwards, which could have a material adverse effect on cash flow and results of operations.efforts and all our drug candidates are in preclinical development.efforts. If we are unable to successfully develop, receive regulatory approval for and commercialize our drug candidates, for these or any other indications, or successfully develop any other drug candidates, or experience significant delays in doing so, our business will be harmed. very early in our development efforts and all of the drug candidates (including OV329 and OV350), for which we control developmental and commercial responsibility are still in preclinical development. For example, weefforts. We previously publicly announced we anticipate filing three investigational new drug (“IND”) applicationsINDs in three years, beginning in 2022,2022; however, we cannot guarantee success of pre-clinicalpreclinical development to achieve all such IND applications.INDs. Following IND acceptance, each of our drug candidates will need to be progressed through clinical development in order to achieve regulatory approval, and we will also need to address issues relating to manufacture and supply, which may involve building our own capacity and expertise. In order to commercialize any product that achieves regulatory approval, we will need to build a commercial organization or successfully outsource commercialization, all of which will require substantial investment and significant marketing efforts before we have the ability to generate any revenue from drug sales. We do not have any drugs that are approved for commercial sale, and we may never be able to develop or commercialize marketable drugs.21whether due to the impacts of the ongoing COVID-19 pandemic or otherwise, could have an adverse effect on our business, financial condition, results of operations and growth prospects.TAK-935,Takeda License and TerminationRLT Agreement. Without those funds, we may need to raise significant additional capital to pursue the development and commercialization of our current and future pipeline.22 For instance, our NEPTUNE trial of OV101, one of our former drug candidates, did not meet its primary endpoints despite earlier encouraging results from our phase 2 trial STARS, the first clinical trial evaluating efficacy of OV101 in patients with Angelman syndrome. We closed our OV101 program in Angelman syndrome in early 2021. The results from preclinical studies of our current and future drug candidates may not be predictive of the effects of these compounds in later stage clinical trials. If we do not observe favorable results in clinical trials of one of our drug candidates, we may decide to delay or abandon clinical development of that drug candidate. Any such delay or abandonment could harm our business, financial condition, results of operations and prospects.It is difficult to predict the time and cost of product candidate development and subsequently obtaining regulatory approval for our gene therapy candidates.Our future success depends in part on the successful development of our early-stage gene therapy product candidates. We may experience delays in developing a sustainable, reproducible, and scalable manufacturing process or transferring that process to internal and external commercial manufacturing sites, which may prevent us from initiating or completing our clinical trials or commercializing our product candidates on a timely or profitable basis, if at all.The regulatory approval process for novel gene therapy products such as ours can be more expensive and take longer than for other product types, which are better known or more extensively studied to date. Regulatory approaches and requirements for gene therapy products continue to evolve, and any changes could create significant delay and unpredictability for product development and approval as compared to technologies with which regulatory authorities have more substantial experience.Also, before a clinical trial can begin to enroll at a site, each clinical site's Institutional Review Board (“IRB”) and its Institutional Biosafety Committee will have to review the proposed clinical trial to assess appropriateness to conduct the clinical trial at that site. In addition, adverse events in clinical trials of gene therapy products conducted by others may cause the FDA or other regulatory authorities outside the U.S. to change the requirements for human research on or for approval of any of our product candidates.Negative public opinion and increased regulatory scrutiny of gene therapy and genetic research may damage public perception of our product candidates or adversely affect our ability to conduct our business or obtain marketing approvals for our product candidates.Public perception may be influenced by claims that gene therapy is unsafe, and gene therapy may not gain the acceptance of the public or the medical community. More restrictive government regulations or negative public opinion would have a negative effect on our business or financial condition and may delay or impair the development and commercialization of our product candidates or demand for any products we may develop. Trials using early versions of retroviral vectors, which integrate into, and thereby alter, the host cell’s DNA, have led to several well-publicized adverse events. The risk of serious adverse events remains a concern for gene therapy and we23cannot assure that it will not occur in any of our future clinical trials. In addition, there is the potential risk of delayed adverse events following exposure to gene therapy products due to persistent biological activity of the genetic material or other components of products used to carry the genetic material.Adverse events in trials or studies conducted by us or other parties, even if not ultimately attributable to our product candidates, and resulting publicity, could result in increased governmental regulation, unfavorable public perception, potential regulatory delays in the testing or approval of our product candidates, stricter labeling requirements for those product candidates that are approved and a decrease in demand for any such product candidates.a new drug application (“NDA”)an NDA or Biologics License Application (“BLA”)BLA for regulatory approval. We cannot predict with any certainty if or when we might submit an NDA or BLA for any of our product candidates or whether any such application will be approved by the FDA. Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous review and regulatory requirements by numerous government authorities in the United States and in other countries where we intend to test and market our product candidates. For instance, the FDA may not agree with our proposed endpoints for any future clinical trial of our product candidates, which may delay the commencement of such clinical trial.trials, whether as a result of the COVID-19 pandemic or otherwise;24geo-political actions,global geopolitical tensions, including the ongoing war between Russia and Ukraine and war in Israel, any other war or the perception that hostilities may be imminent, including the ongoing conflict in Ukraine and terrorism, or natural disasters andor public health epidemics.crises.Clinical site initiation and patient enrollment may be delayed due to prioritization of hospital resources toward the COVID-19 pandemic. Patients may not be able to comply with clinical trial protocols if quarantines impede patient movement or interrupt healthcare services. Similarly, our ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19, could be limited, which in turn could adversely impact our clinical trial operations. Additionally, we may experience interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel, quarantines or social distancing protocols imposed or recommended by federal or state governments, employers and others in connection with the ongoing COVID-19 pandemic. As a result of the COVID-19 pandemic, we have faced and may continue to face delays in meeting our anticipated timelines for our ongoing and planned preclinical studies and clinical trials.Angelman syndrome,CCM, have not been established, and accordingly we may have to develop new modalities or modify existing endpoints to measure efficacy, which may increase the time it takes for us to commence or complete clinical trials. In addition, we believe investigators in this area may be inexperienced in conducting trials in this area due to the current lack of drugs to treat these disorders, which may result in increased time and expense to train investigators and open clinical sites.Good Clinical Practice (“GCP”)GCP regulations, that we are exposing participants to unacceptable health risks, or if the FDA finds deficiencies in our IND applications or the conduct of these trials. Therefore, we cannot predict with any certainty the schedule for commencement25seizure relatedseizure-related disorders and we may choose to pursue drug candidates in other areas of interest including other disorders that we believe would be in the best interest of the Company and our stockholders. We plan to continuously review our strategies and modify as necessary based on attractive areas of interest and assets that we choose to pursue. We intend to develop our portfolio of drug candidates by in-licensing and entering into collaborations with leading biopharmaceutical companies or academic institutions for new drug candidates. Identifying new drug candidates requires substantial technical, financial and human resources, whether or not any drug candidates are ultimately identified. The COVID-19 pandemic could also impact our ability to do in-person due diligence, negotiations and other interactions to identify new opportunities. Even if we identify drug candidates that initially show promise, we may fail to in-license or acquire these assets and may also fail to successfully develop and commercialize such drug candidates for many reasons, including the following:seizure relatedseizure-related disorders and rare neurological disorders we are pursuing is small and has not been established with precision. If the actual number of patients with these disorders is smaller than we anticipate, we may encounter difficulties in enrolling patients in our clinical trials, thereby delaying or preventing development and approval of our drug candidates. Even once enrolled we may be unable to retain a sufficient number of patients to complete any of our trials. Patient enrollment and retention in clinical trials depends on many factors, including the size of the patient population, the nature of the trial protocol, the existing body of safety and efficacy data, the number and nature of competing treatments and ongoing clinical trials of competing therapies for the same indication, the proximity of patients to clinical sites and the eligibility criteria for the trial, any such enrollment issues could cause delays or prevent development and approval of our drug candidates. Because we are focused on addressing seizure relatedseizure-related disorders and rare neurological disorders, there are limited patient pools from which to draw in order to complete our clinical trials in a timely and cost-effective manner. Furthermore, our efforts to build relationships with patient communities may not succeed, which could result in delays in patient enrollment in our clinical trials. In addition, any negative results we may report in clinical trials26and OV935, two of our former drug candidates.candidate, and soticlestat. Clinical trials may not demonstrate any ocular safety benefits for OV329 relative to vigabatrin. If clinical experience indicates that any of our drug candidates causes adverse events or serious or life-threatening adverse events, the development of that drug candidate may fail or be delayed, or, if the drug candidate has received regulatory approval, such approval may be revoked, which would harm our business, prospects, operating results and financial condition.2728 Due to the ongoing COVID-19 pandemic, it is possible that we could experience delays in the timing of our interactions with regulatory authorities due to absenteeism by governmental employees, inability to conduct planned physical inspections related to regulatory approval, or the diversion of regulatory authority efforts and attention to approval of other therapeutics or other activities related to COVID-19, which could delay anticipated approval decisions and otherwise delay or limit our ability to make planned regulatory submissions or obtain new product approvals.actions,tensions, including the ongoing war (includingbetween Russia and Ukraine and the conflictwar in Ukraine) andIsrael, any other war or the perception that hostilities may be imminent, terrorism, or natural disasters andor public health pandemics, such as COVID-19.crises.29Takeda License and TerminationRLT Agreement, we are entitled to receive royalty and milestone payments in connection with the development and commercialization of soticlestat. If Takeda fails to progress or discontinues the development of soticlestat, we may not receive some or all of such payments, which would materially harm our business.Takeda License and TerminationRLT Agreement, pursuant to which Takeda secured rights to our 50% global share in soticlestat, which we had originally licensed from Takeda, and we granted to Takeda an exclusive worldwide license under our relevant intellectual property rights to develop and commercialize the investigational medicine soticlestat for the treatment of developmental and epileptic encephalopathies, including Dravet syndrome and Lennox-Gastaut syndrome. All rights in soticlestat are now owned by Takeda or exclusively licensed to Takeda by us. Following the closing date of the Takeda License and TerminationRLT Agreement, Takeda assumed all responsibility for, and costs of, both development and commercialization of soticlestat, and we will no longer have any financial obligation to Takeda under the original collaboration agreement, including for milestone payments or any future development and commercialization costs. Upon closing of the Takeda License and TerminationRLT Agreement, we received a one-time, upfront payment of $196.0 million and, if soticlestat is successfully developed, we will be eligible to receive up to an additional $660.0 million upon Takeda achieving specified regulatory and sales milestones. In addition, if soticlestat achieves regulatory approval, we will be entitled to receive tiered royalties at percentages ranging from the low double-digits up to 20% on sales of soticlestat. Royalties will be payable on a country-by-country and product-by-product basis during the period beginning on the date of the first commercial sale of suchTakeda License and TerminationRLT Agreement, Takeda now has sole discretion over the conduct of the development and commercialization of soticlestat. If for any reason, Takeda fails to progress, or elects to terminate the development of soticlestat as contemplated by the Takeda License and TerminationRLT Agreement, or if the development or commercialization of soticlestat is delayed or deprioritized by Takeda, we may not receive some or all of the royalty and milestone payments under such agreement. We are dependent upon Takeda’s progression of such development and the resulting payments to fund the regulatory development of our current and future drug candidates. If we are unable to find alternative sources of revenue, our inability to receive royalty or milestone payments under the Takeda License and TerminationRLT Agreement would negatively impact our business and results of operations.3031seizure relatedseizure-related disorders and we may choose to pursue drug candidates in other areas of interest including other disorders and diseases that we believe would be in the best interest of the Company and our stockholders. We plan to continuously review our strategies and modify as necessary based on attractive areas of interest and assets that we choose to pursue.Anti-KickbackAnti-3233Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively PPACA)PPACA was passed, which substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry.President Trump signed Executive Orders and other directives designed to delay the implementationof certain provisions of the PPACA or otherwise circumvent some of the requirements for health insurance mandated by the PPACA. Concurrently, Congress considered legislation to repeal or repeal and replace all or part of the PPACA. While Congress has not passed comprehensive repeal legislation, it has enacted laws that modify certain provisions of the PPACA such as removing penalties, effective January 1, 2019, for not complying with the PPACA’s individual mandate to carry health insurance, delaying the implementation of certain PPACA-mandated fees, and increasing the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D. Onon June 17, 2021 the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the PPACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. Thus,Further, there have been a number of health reform measures by the PPACA will remain in effect in its current form. Further, prior toBiden administration that have impacted the U.S. Supreme Court ruling, on January 28, 2021,PPACA. On August 16, 2022, President Biden issued an executive order that initiated a special enrollment periodsigned the IRA into law, which among other things, extends enhanced subsidies for purposes of obtainingindividuals purchasing health insurance coverage in PPACA marketplaces through plan year 2025. The IRA also eliminates the PPACA marketplace. The executive order also instructed certain governmental agencies to review“donut hole” under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create barriers to obtaining access to health insurance coverage through Medicaid or the PPACA.by creating a new manufacturer discount program. It is possible that the PPACA will be subject to judicial or Congressional challenges in the future. It is unclear how any such challenges and the healthcare reform measures of the Biden administration will impact the PPACA and our business.including the BBA, will remain in effect through 2031until 2032 unless additional Congressional action is taken. However, COVID-19 relief legislation suspended the 2% Medicare sequester from May 1, 2020 through March 31, 2022. Under current legislation, the actual reduction in Medicare payments will vary from 1% in 2022 to up to 3% in the final fiscal year of this sequester. The American Taxpayer Relief Act of 2012, among other things, further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Congress is also considering additional health reform measures.34In November 2019, CMS issued a final rule finalizingThis program provides clinicians with two ways to participate, including through the changes toAdvanced Alternative Payment Models (“APMs”) and the QualityMerit-based Incentive Payment Program. At this time, the full impact to overall physician reimbursement as a result of the introduction of the Quality Payment Program remains unclear.the Trump administration used several means to propose or implement drug pricing reform, including through federal budget proposals, executive orders and policy initiatives. For example, on July 24, 2020 and September 13, 2020, the Trump administration announced several executive orders related to prescription drug pricing that attempt to implement several of the administration’s proposals. Additionally, in July 2021, the Biden administration released an executive order, “Promoting Competition in the American Economy,” with multiple provisions aimed at prescription drugs. In response to Biden’s executive order, on September 9, 2021, the U.S. Department of Health and Human Services or HHS,(“HHS”) released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. No legislation or administrative actions have been finalizedFurther, the IRA, among other things (i) directs HHS to implement these principles.negotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation. These provisions take effect progressively starting in fiscal year 2023. On August 29, 2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is currently subject to legal challenges. Additionally, the Biden administration released an additional executive order on October 14, 2022, directing HHS to report on how the Center for Medicare and Medicaid Innovation can be further leveraged to test new models for lowering drug costs for Medicare and Medicaid beneficiaries. At the state level, legislatures have increasingly passed and implemented regulations designed to control pharmaceutical and biological product pricing, including pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. We expect that these and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved drug. For example, basedin response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a recent executive order,report outlining three new models for testing by the Centers for Medicare & Medicaid Services Innovation Center which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether the models will be utilized in any health reform measures in the future. Further, on December 7, 2023, the Biden administration expressed its intentannounced an initiative to pursue certain policy initiativescontrol the price of prescription drugs through the use of march-in rights under the Bayh-Dole Act. On December 8, 2023, the National Institute of Standards and Technology published for comment a Draft Interagency Guidance Framework for Considering the Exercise of March-In Rights which for the first time includes the price of a product as one factor an agency can use when deciding to reduce drug prices.exercise march-in rights. While march-in rights have not previously been exercised, it is uncertain if that will continue under the new framework. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our drugs.It is possible that additional governmental action is taken in response to the ongoing COVID-19 pandemic.3536(the “Leahy-Smith(“Leahy-Smith Act”) was signed into law. The Leahy-Smith Act includes a number of significant changes to United States patent law. These include provisions that affect the way patent applications are prosecuted and may also affect patent litigation. The United States Patent Office recently developed new regulations and procedures to governU.S. Patent and Trademark Office (the “USPTO”)USPTO or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or drugs and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize drugs without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future drug candidates.3738394041COVID-19 could adversely impact our business, including our preclinical studies, clinical trials and access to capital.The ongoing COVID-19 pandemic has resulted in travel and other restrictions in order to reduce the spread of the disease. In response to initial state and local orders across the country directing individuals to shelter at their places of residence, directing businesses and governmental agencies to cease non-essential operations at physical locations, prohibiting certain non-essential gatherings, and ordering cessation of non-essential travel, in March 2020 we implemented work-from-home policies for all employees. Our current plans to return to the office remain fluid as the pandemic persists and federal, state and local guidelines, rules and regulations regarding the conduct of in-person business operations continue to evolve. The effects of our work-from-home policies may negatively impact productivity, disrupt our business and delay our clinical programs and timelines, the magnitude of which will depend, in part, on the length and severity of any applicable restrictions and other limitations on our ability to conduct our business in the ordinary course. These and similar, and perhaps more severe, disruptions in our operations could negatively impact our business, operating results and financial condition.42Any new imposition of quarantines, shelter-in-place and similar government orders related to COVID-19 may adversely impact our business operations and the business operations of our CROs conducting our preclinical studies and clinical trials and our third-party manufacturing facilities in the United States and other countries. In particular, some of our third-party manufacturers which we use for the supply of materials for drug candidates or other materials necessary to manufacture product to conduct preclinical studies and clinical trials are located in countries affected by COVID-19, and should they experience disruptions, such as temporary closures or suspension of services, we would likely experience delays in advancing these tests and trials. Currently, we expect no material impact on the clinical supply of any of our drug candidates.In addition, our previous clinical trials were, and our future clinical trials may be, affected by the COVID-19 pandemic. Clinical site initiation and patient enrollment may be delayed due to prioritization of hospital resources toward the COVID-19 pandemic. Some patients may not be willing or able to comply with clinical trial protocols if quarantines impede patient movement or interrupt healthcare services. Similarly, our ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 and adversely impact our clinical trial operations. As a result of the COVID-19 pandemic, we have faced and may continue to face delays in meeting our anticipated timelines for our previous and planned clinical trials.The spread of COVID-19, which has caused a broad impact globally, may materially affect us economically. While the potential economic impact brought by, and the duration of, COVID-19 may be difficult to assess or predict, a sustained, widespread pandemic could result in significant disruption of global financial markets, reducing our ability to access capital, which could in the future negatively affect our liquidity. In addition, a recession or market correction resulting from the spread of COVID-19 could materially affect our business and the value of our common stock.The global COVID-19 pandemic continues to evolve. The extent to which the COVID-19 pandemic impacts our business, our clinical development and regulatory efforts will depend on future developments that are highly uncertain and cannot be predicted with confidence, such as the ultimate duration of the outbreak, the extent of sustained or new travel restrictions, social distancing requirements and business closures in the United States, Europe and other countries, and the effectiveness of actions taken in the United States, Europe and other countries to contain and treat the disease, including vaccination efforts. Accordingly, we do not yet know the full extent of potential delays or impacts on our business, our clinical and regulatory activities, healthcare systems or the global economy as a whole. However, these impacts could adversely affect our business, financial condition, results of operations and growth prospects.In addition, to the extent the ongoing COVID-19 pandemic adversely affects our business and results of operations, it may also have the effect of heightening many of the other risks and uncertainties described in this ‘‘Risk Factors’’ section.432021,2023, we had 5740 full-time employees. As our development and commercialization plans and strategies for our current pipeline of product candidates develop, we expect to need additional managerial, operational, sales, marketing, financial, legal and other resources. Our management may need to divert a disproportionate amount of its attention away from our day-to-day operations and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, operational inefficiencies, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of our current and potential future drug candidates. If our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate and grow revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance, our ability to commercialize drug candidates, develop a scalable infrastructure and compete effectively will depend, in part, on our ability to effectively manage any future growth.the work from homeour hybrid-work policies, we implemented due to COVID-19, information that is normally protected, including company confidential information, may be less secure. If actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could result in the imposition of significant fines or other sanctions, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, contractual damages, reputational harm, diminished profits and future earnings and curtailment of operations, any of which could adversely affect our ability to operate our business and our results of operations. Whether or not we are successful in defending against such actions or investigations, we could incur substantial costs, including legal fees, and divert the attention of management in defending ourselves against any of these claims or investigations.information, including intellectual property, proprietary business information, personal informationdata, and, other confidential information. It is criticalas a result, we and the third parties upon which we rely face a variety of evolving threats that we do so in a secure manner to maintain the confidentiality, integrity and availability of such sensitive information.could cause security incidents. We have also outsourced elements of our operations (including elements of our information technology infrastructure) to third parties, and as a result, we manage a number of third-party vendors who may or could have access to our computer networks or our confidential information.sensitive data. In addition, many of those third parties in turn subcontract or outsource some of their responsibilities to other third parties. While all information technology operations are inherently vulnerable to inadvertent or intentional security breaches, incidents, attacks and exposures, the accessibility and distributed nature of our information technology systems, and the sensitive informationdata stored on those systems, make such systems potentially vulnerable to unintentional or malicious, internal and external attacks on our technology environment. Furthermore, our ability to monitor the aforementioned third parties’ information security practices is limited, and these third parties may not have adequate information security measures in place. If our third-party service providers experience a security incident or other interruption, we could experience adverse consequences. While we may be entitled to damagesthe ongoing COVID-19 pandemic,our hybrid-work environment, we have enabled allmay be more vulnerable to cyberattacks as more of our employees utilize network connections, computers, and devices outside our premises or network, including working at home, while in transit and in public locations. Additionally, future or past business transactions (such as acquisitions or integrations) could expose us to work remotely, whichadditional cybersecurity risks and vulnerabilities, as our systems could be negatively affected by vulnerabilities present in acquired or integrated entities’ systems and technologies. Furthermore, we may make us more vulnerablediscover security issues that were not found during due diligence of such acquired or integrated entities, and it may be difficult to cyberattacks. integrate companies into our information technology environment and security program.AttacksWe take steps designed to detect, mitigate, and remediate vulnerabilities in our information systems (such as our hardware and/or software, including that of this naturethird parties upon which we rely); however we may not detect and remediate all such vulnerabilities on a timely basis. Further, we may experience delays in deploying remedial measures and patches designed to address identified vulnerabilities. Vulnerabilities could be exploited and result in a security incident.44In addition to the extraction of sensitive information, suchSuch attacks could include the deployment of harmful malware (including as a result of advanced persistent threat intrusions), ransomware attacks, denial-of-service attacks, credential stuffing and/or harvesting, social engineering (including through deep fakes, which may be increasingly more difficult to identify as fake, and phishing attacks), supply-chain attacks, software bugs, server malfunctions, software or hardware failures, loss of sensitive data or other information technology assets, adware, attacks enhanced or facilitated by artificial intelligence, telecommunications failures, earthquakes, fires, floods and other means to affect service reliability and threaten the confidentiality, integrity and availability of information.our information systems and sensitive data. In addition,particular, severe ransomware attacks are becoming increasingly prevalent and can lead to significant interruptions in our operations, ability to provide our products or services, loss of sensitive data and income, reputational harm, and diversion of funds. Extortion payments may alleviate the prevalent usenegative impact of mobile devices increases the risk of data security incidents.information,data, which could result in financial, legal, regulatory, business and reputational harm to us. In addition, information technology system disruptions, whether from attacks on our technology environment or from computer viruses, natural disasters, terrorism, war and telecommunication and electrical failures, could result in a material disruption of our development programs and our business operations. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data.There is no wayknowingsecurity incidents. Such disclosures are costly, and the disclosure or the failure to comply with certainty whethersuch requirements could lead to adverse consequences.any dataa security incidents that have not been discovered. While we have no reason to believe this to be the case, attackers have become very sophisticated in the way they conceal access to systems, and many companies that have been attacked are not aware that they have been attacked. Any event that leads to unauthorized access, use or disclosure of personal information,incident, including but not limited to a security incident involving personal information regarding our patients or employees, could disruptwe may experience adverse consequences, such as disruptions to our business, harm to our reputation, compel us to comply with applicable federalgovernment enforcement actions (for example, investigations, fines, penalties, audits, and inspections), additional reporting requirements, and/or state breach notification laws and foreign law equivalents,oversight, or we may otherwise be subject us to time consuming, distracting and expensive litigation, regulatory investigation and oversight, mandatory corrective action, require us to verify the correctness of database contents, or otherwise subject us to liability under laws, regulations and contractual obligations, including those that protect the privacy and security of personal information. This could result in increasedinformation, which could include personally identifiable information,data, may result in governmental investigations, enforcement actions, regulatory fines, litigation, or public statements against us by advocacy groups or others, and could cause third parties, including clinical sites, regulators or current and potential partners, to lose trust in us or we could be subject to claims by third parties that we have breached our privacy- or confidentiality-related obligations, which could materially and adversely affect our business and prospects. Moreover, data security incidents and other inappropriate access can be difficult to detect, and any delay in identifying them may lead to increased harm of the type described above.successfully prevent service interruptionsbe effective. Our contracts may not contain limitations of liability, and even where they do, there can be no assurance that limitations of liability in our contracts are sufficient to protect us from liabilities, damages, or claims related to our data privacy and security incidents.may beare subject to numerousstringent and varyingevolving privacy and security laws, regulations, contractual obligations, industry standards, policies, and other obligations, and our failure or perceived failure to comply with such obligations could result in regulatory investigations or actions, litigation (including class actions), fines and penalties, disruptions of our business operations, loss of revenue or profits, reputational damage and reputational damage.We areother adverse business consequences.including health information.and other sensitive data. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business. In the U.S.,United States, we may be subject to state security breach notification laws, state health information privacy laws and federal and state consumer protections laws which impose requirements for the collection, use, disclosure and transmission of personal information. Each of these laws is subject to varying interpretations by courts and government agencies, creating complex compliance issues for us. If we fail to comply with applicable laws and regulations, we could be subject to penalties or sanctions, including criminal penalties if we knowingly obtain individually identifiable health information from a covered entity in a manner that is not authorized or permitted by HIPAA or for aiding and abetting the violation of HIPAA.or GDPR,(“GDPR”), which applies to all EU member states as of May 25, 2018 and replaces the former EU Data Protection Directive. The regulation introduces new data protection requirements in the EU and imposes substantial fines for breaches of the data protection rules. The GDPR must be implemented into national laws by the EU member states imposes strict obligations and restrictions on the ability to collect, analyze, and transfer personal data, including health data from clinical trials and adverse event reporting. Data protection authorities from different EU member states have interpreted the privacy laws differently, which adds to the complexity of processing personal data in the EU, and guidance on implementation and compliance practices are often updated or otherwise revised. Any failure to comply with the rules arising from the GDPR and related national laws of EU member states could lead to government enforcement actions and45significant penalties against us, fines of up to 20 million Euros or 4% of annual global revenue, whichever is greater, and adversely impact our operating results. The GDPR will increase our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with EU data protection rules.legislation thatwhich has been dubbed the first “GDPR-like” law in the United States. In the past few years, numerous other U.S. states—including Virginia, Colorado, Connecticut, and Utah—have also enacted comprehensive privacy laws that impose certain obligations on covered businesses, including providing specific disclosures in privacy notices and affording residents with certain rights concerning their personal data. The CCPA gives California residents expanded rights to access, correct and delete their personal information, opt out of certain personal information sharing and receive detailed information about how their personal information is used by requiring covered companies to provide new disclosures to California consumers (as that term is broadly defined) and provide such consumers new ways to opt-out of certain sales of personal information. The CCPA provides for civil penalties forTheAlthough there are limited exemptions for clinical trial data under the CCPA (and the other similar state privacy laws), the CCPA and other similar laws may increaseimpact (possibly significantly) our compliance costsbusiness activities depending on how it is interpreted, should we become subject to the CCPA in the future.potential liability. an “emerging growth company” and a “smaller reporting company” and the reduced disclosure requirements applicable to such companies may make our common stock less attractive to investors.an emerging growth company (“EGC”),currently a “smaller reporting company” as defined in the Jumpstart Our Business StartupsSecurities Exchange Act of 20121934, as amended (the “JOBS“Exchange Act”). We will remain an EGC untilbe a smaller reporting company and may take advantage of the earlier of:scaled-back disclosures available to smaller reporting companies for so long as (i) the market value of our voting and non-voting ordinary shares held by non-affiliates is less than $250.0 million measured on the last business day of our second fiscal quarter or (ii) (a) our annual revenue is less than $100.0 million during the most recently completed fiscal year in which we have total annual gross revenuesand (b) the market value of $1.07 billion or more; (ii) December 31, 2022,our voting and non-voting ordinary shares held by non-affiliates is less than $700.0 million measured on the last business day of theour second fiscal year following the fifth anniversary of the date of the completion of our IPO; (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; or (iv) the date on whichquarter.deemedpermitted to comply with scaled-back disclosure obligations in our SEC filings compared to other issuers, including with respect to disclosure obligations regarding executive compensation in our periodic reports and proxy statements. We have elected to adopt the accommodations available to smaller reporting companies. Until we cease to be a large accelerated filer undersmaller reporting company, the rulesscaled-back disclosure in our SEC filings will result in less information about our company being available than for other public companies. If investors consider our common shares less attractive as a result of our election to use the scaled-back disclosure permitted for smaller reporting companies, there may be a less active trading market for our common shares and our share price may be more volatile.Securities and Exchange Commission (“SEC”). For so long as we remain an EGC, we are permitted and intendscaled-back disclosures available to rely on exemptions from certain disclosure requirements that are applicable to other publicsmaller reporting companies, that areincluding but not emerging growth companies. These exemptions include:•not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002 (“Section 404”);not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Boardreduced disclosure obligations regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the auditexecutive compensation arrangements; and the financial statements;disclosure;•reduced disclosure obligations regarding executive compensation arrangements; and•exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved.We currently take advantage of some, but not all, of the reduced regulatory and reporting requirements that are available to us so long as we qualify as an EGC. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock, and our stock price may be more volatile and may decline.In addition, the JOBS Act provides that an EGC may take advantage of an extended transition period for complying with new or revised accounting standards. This allows an EGC to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not an EGC.We are also a smaller reporting company as defined in the Exchange Act. We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies and will be able to take advantage of these scaled disclosures for so long as (i) our voting and non-voting common stock held by nonaffiliates is less than $250.0 million measured on the last business day of our second fiscal quarter or (ii) our annual revenue is less than $100.0 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is less than $700.0 million measured on the last business day of our second fiscal quarter.We will continue to incur increased costs as a result of operating as a public company, and our management will devote substantial time to new compliance initiatives.46As a public company, and particularly after we are no longer an EGC, we will incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act of 2002 and rules subsequently implemented by the SEC and The Nasdaq Stock Market LLC have imposed various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel devote a substantial amount of time to these and other compliance initiatives. Moreover, these rules and regulations will continue to increase our legal and financial compliance costs and will make some activities more time-consuming and costly.A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However,as long asthe year ended December 31, 2022, we remain an emerging growth company as defined in the JOBS Act, we intendwere required to take advantageprovide management’s attestation on internal controls pursuant to Section 404 of the exemption permitting us not to comply with theSarbanes-Oxley Act, and our independent registered public accounting firm was required to attest to the effectiveness of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act. However, as of the last business day of our second fiscal quarter of 2023, we determined that we requalify as a smaller reporting company and as a non-accelerated filer for the year ended December 31, 2023. We are therefore no longer required to include an attestation requirement.willin future periods may require that we incur substantial expense and expend significant management efforts. We currently do not have an internal audit group, and we willrely on experienced consultants to support this function. We may need to hire additional consultants or accounting and financial staff with appropriate public company experience and technical accounting knowledge and compile the system and process documentation necessaryin order to perform the evaluation needed tocontinually comply with Section 404. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting, once that firm begins its Section 404 reviews, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by The Nasdaq Stock Market LLC, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.including very recently in connection with the ongoing COVID-19 pandemic, which has resulted in decreased stock prices for many companies notwithstanding the lack of a fundamental change in their underlying business models or prospects. Broad market and industry factors, including potentially worsening economic conditions and other adverse effects or developments relating to thenew or ongoing COVID-19 pandemic,public health crises or other inflationary factors, may negatively affect the market price of our common stock, regardless of our actual operating performance. As a result of this volatility, you may lose all or part of your investment in our common stock since you might be unable to sell your shares at or above the price you paid for the shares. The market price for our common stock may be influenced by many factors, including:47For example, the COVID-19 pandemic resulted in widespread unemployment, economic slowdown and extreme volatility in the capital markets. Similarly, the current conflict between Ukraine and Russia hasGlobal geopolitical tensions have created extreme volatility in the global capital markets and isare expected to have further global economic consequences, including disruptions of the global supply chain and energy markets. Any such volatility and disruptions may have adverse consequences on us or the third parties on whom we rely. If the equity and credit markets deteriorate, including as a result of political unrest or war, it may make any necessary debt or equity financing more difficult to obtain in a timely manner or on favorable terms, more costly or more dilutivea sufficient amount ofsubstantial revenue from our products,drug sales, if ever, we expect to finance futureour cash needs through publica combination of equity and debt financings, strategic alliances, and license and development agreements in connection with any collaborations. We do not have any committed external source of funds. To the extent that we issue additional equity securities, our stockholders may experience substantial dilution, and the terms of these securities may include liquidation or privateother preferences that adversely affect your rights as a stockholder. In addition, we may issue equity or debt offerings. securities as consideration for obtaining rights to additional compounds.“S-3“Current S-3 Registration Statement”), which includes a prospectus covering the issuance and sale of up to $75.0 million of common stock pursuant to an at-the-market (“ATM”) offering program. As of December 31, 2021,2023, we had $250.0 million available under our Current S-3 Registration Statement, including $75.0 million available pursuant to our ATM program. Financing activitiesDebt and equity financings, if available, may have an adverse impact oninvolve agreements that include covenants limiting or restricting our stockholders’ rights as well as on our operations, and such additional funding may not be available on reasonable terms, if at all. If we raise additional funds through the issuance of additional debt or equity securities, it may result in dilutionability to our existing stockholders and/or increased fixed payment obligations. Furthermore, these securities may have rights senior to those of our common stock and could contain covenants that would restrict our operations and potentially impair our competitiveness,take specific actions, such as redeeming our shares, making investments, issuing additional equity, limitations on our ability to incurincurring additional debt, making capital expenditures, declaring dividends or placing limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adverselynegatively impact our ability to conduct our48Additionally, ifIf we seek fundsraise additional capital through future collaborations, strategic alliances or third-party licensing arrangements, with collaborative partners, these arrangementswe may require ushave to relinquish valuable rights to some of our technologiesintellectual property, future revenue streams, research programs or drug candidates, or otherwise agree togrant licenses on terms unfavorablethat may not be favorable to us. Any of these events could significantly harm our business, financial condition and prospects.2021,2023, we had outstanding options to purchase an aggregate of 10,776,75815,124,546 shares of our common stock at a weighted average exercise price of $4.97$3.87 per share and 1,250,000 shares of common stock issuable upon conversion of outstanding Series A convertible preferred stock for no additional consideration. Such Series A convertible preferred stock is convertible any time at the option of the holder thereof subject to the beneficial ownership limitations described in Note 67 to the consolidated financial statements contained in this Annual Report on Form 10-K. The exercise of such options and conversion of the Series A convertible preferred stock for shares of our common stock will result in further dilution of your investment and could negatively affect the market price of our common stock. In addition, you may experience further dilution if we issue common stock, or securities convertible into common stock, in the future. As a result of this dilution, you may receive significantly less than the full purchase price you paid for the shares in the event of liquidation.2021,2023, our executive officers, directors and stockholders who owned more than 5% of our outstanding common stock, in the aggregate, beneficially own shares representing approximately 44.4%52.4% of our outstanding common stock.Takeda License and TerminationRLT Agreement.disagree with.49Takeda License and TerminationRLT Agreement may delay or prevent a merger, acquisition or other change in control of us that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares.50If we engage in future acquisitions or strategic partnerships, this may increase our capital requirements, dilute our stockholders, cause us to incur debt or assume contingent liabilities and subject us to other risks.Our business plan is to continue to evaluate various acquisitions and strategic partnerships, including licensing or acquiring complementary drugs, intellectual property rights, technologies, or businesses. Any potential acquisition or strategic partnership may entail numerous risks, including:•increased operating expenses and cash requirements;•the assumption of additional indebtedness or contingent liabilities;•assimilation of operations, intellectual property and drugs of an acquired company, including difficulties associated with integrating new personnel;•the diversion of our management’s attention from our existing drug programs and initiatives in pursuing such a strategic partnership, merger or acquisition;•retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships;•risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing drugs or drug candidates and regulatory approvals;•our inability to generate revenue from acquired technology and/or drugs sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs;•challenges related to integrating acquired businesses or entering into or realizing the benefits of strategic transactions generally; and•risks associated with potential international acquisition transactions, including in countries where we do not currently have a material presence.In addition, if we engage in future acquisitions or strategic partnerships, we may issue dilutive securities, assume or incur debt obligations, incur large one-time expenses and acquire intangible assets that could result in significant future amortization expense. Moreover, we may not be able to locate suitable acquisition opportunities and this inability could impair our ability to grow or obtain access to technology or drugs that may be important to the development of our business.None.Significant disruptions of our information technology systems or data security incidents could result in significant financial, legal, regulatory, business and reputational harm to us1460 Broadway, New York, New York, on a monthly basis. We have also entered a 10-year lease for office space at 441 Ninth Avenue, New York, New York, and anticipate moving into this space mid-yearunder a ten-year lease agreement which commenced in March 2022. We believe our facilities are adequate to meet current needs.5152May 5, 2017 (the date our common stock commenced trading on the Nasdaq Global Select Market)December 31, 2018 through December 31, 2021,2023, of the cumulative total return for our common stock, the Nasdaq Composite Index and the Nasdaq Biotechnology Index.
May 5, 2017.December 31, 2018. The comparisons in the graph are not intended to forecast or be indicative of possible future performance of our common stock.10, 2022,5, 2024, we had approximately 1114 holders of record of our common stock. Certain shares are held in “street” name“street name” and accordingly, the number of beneficial owners of such shares is not known or included in the foregoing number. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.53None.54focused on drug discovery and development fordedicated to meaningfully improving the lives of people affected by certain epilepsies and rare CNS disorders in a manner thatbrain conditions with seizure symptoms. Our approach to achieve this goal is scientifically driven, patient focused, and is coupled with an integrated and discipleddisciplined approach to research, clinical development and business development. Our team has significant experience with and understanding of rare epilepsyepilepsies and neurological conditions, and we continue to buildgain insight into the wayways the different molecular mechanisms and pathways underlying these disorders impact the symptoms patients suffer. Ovid hasWe have set out to be a leader in the field, and hashave developed a differentiated pipeline containing threefour novel mechanisms of action to target different causes of certain epilepsies and seizures. Our knowledge of epilepsy disease biology and pathology, which was acquired through our small molecule development programs, now contributes to our pursuit of additional relevant genetic targets and molecular pathways that are the cause of seizures. Over time, webrain conditions with seizure symptoms. We have built a scalable scientific platform andwith efficient development capabilities in epilepsies and conditions with seizure symptoms that focusfocuses on clear, clinical endpoints. Three of our programs are in clinical trials in humans, and the fourth is in preclinical development and anticipated to advance into human safety studies in 2024. We are initially pursuing therapeutic assets for rare disorders as they can leverage accelerated development programs. If successfully developed and marketed in rare conditions, we intend to explore these assets for broader neurologic indications. Our cohesive focus in certain epilepsies and seizuresbrain conditions with seizure symptoms reinforces our belief that we can develop and produce multiple novel medicines, scale our infrastructure, and thereby succeed in our mission.yearyears ended December 31, 2021,2023 and 2022, we generated $208.4$0.4 million and $1.5 million of licenseroyalty and otherlicensing revenue, primarily through the license and collaboration agreement with Takeda, andrespectively. We have otherwise primarily funded our business primarily through the sale of our capital stock.stock and through the closing of the RLT Agreement with Takeda, which resulted in a one-time up-front payment of $196.0 million in 2021. Through December 31, 2021,2023, we have raised net proceeds of $275.4 million from the sale of our convertible preferred stock and our common stock. As of December 31, 2021,2023, we had $187.8$105.8 million in cash, cash equivalents and cash equivalents.marketable securities. We recorded net incomeloss of $122.8$52.3 million and net loss $81.0$54.2 million for the years ended December 31, 20212023 and 2020,2022, respectively. As of December 31, 2021,2023, we had an accumulated deficit of $171.4$277.9 million.55Recent DevelopmentsLicense AgreementAstraZeneca ABOn December 30, 2021, we entered into an exclusive license agreement with AstraZeneca AB, forpublic health crises and global geopolitical tensions, like the ongoing war between Russia and Ukraine and the war in Israel, may have a librarymaterial adverse effect on our business, financial condition, results of early-stage small molecules targetingoperations and growth prospects. The resulting high inflation rates may materially affect our business and corresponding financial position and cash flows. Inflationary factors, such as increases in the KCC2 transporter, including lead candidate OV350. Upon execution of the agreement, we owed an upfront cash payment of $5.0 million and issued sharescost of our common stockclinical trial materials and supplies, interest rates and overhead costs may adversely affect our operating results. High interest rates also present a recent challenge impacting the U.S. economy and could make it more difficult for us to obtain traditional financing on acceptable terms, if at all, in an amountthe future. Furthermore, economic conditions have produced downward pressure on share prices. Although we do not believe that equaled $7.3 million basedinflation has had a material impact on our financial position or results of operations to date, we may experience increases in the volume-weighted average price of shares ofnear future (especially if inflation rates remain high or begin to rise again) on our common stock for the 30 business days immediately preceding the execution date of the transaction. The cash payment of $5.0 million was paid in January 2022.Pursuantoperating costs, including our labor costs and research and development costs, due to the AstraZeneca license agreement, we agreed to potential milestone payments of up to $203.0 million upon the achievement of certain developmental, regulatory and sales milestones. The first payment of $3.0 million is due upon the successful completion of the first Phase 2 clinical study ofsupply chain constraints, global geopolitical tensions as a licensed product following a positive biomarker readout in a Phase 1 clinical study. In addition, if a licensed product achieves regulatory approval, AstraZeneca is entitled to tiered royalty payments ranging from the single digits up to 10 percent on net sales subject to standard reductions in certain circumstances.COVID-19 UpdateWe have implemented business continuity plans designed to address and mitigate the impactresult of the ongoing COVID-19 pandemicwar between Russia and Ukraine and the war in Israel, worsening global macroeconomic conditions and employee availability and wage increases, which may result in additional stress on our employeesworking capital resources.business. We continueability to operate normallyexecute on our strategy, as well as risks and uncertainties common to companies in the pharmaceutical industry with development and commercial operations, including, without limitation, risks and uncertainties associated with: identifying, acquiring or in-licensing products or product candidates; obtaining regulatory approval of product candidates; pharmaceutical product development and the exceptioninherent uncertainty of enabling allclinical success; and the challenges of protecting and enhancing our employees to work productively at home and abiding by travel restrictions issued by federal, state and local governments. Our current plans to return to the office remain fluid as federal, state and local guidelines, rules and regulations continue to evolve.Takeda LicenseRLT Agreement, as well as nominal amounts from other licensing agreements and Termination Agreement and under the Angelini License Agreement.royalties. We have not generated any revenue from commercial drug sales and we do not expect to generate any further revenue from commercial drug sales unless or until we obtain regulatory approval and commercialize one or more of our current or future drug candidates.candidates, or if we become entitled to revenue from our licensing agreements. In the future, we may also seek to generate revenue from a combination of research and development payments, license fees and other upfront or milestone payments.56described below, travel expenses, conferences, professional fees for auditing, tax and legal services and facility-related costs.(expense)Income (Expense), netnetOther (expense) income,, net, consists primarily of interest income earned on our cash and cash equivalents maintained in money market funds and accretion of discount on short-term investments and unrealized gains/losses on long-term equity investments.5720212023 and 2020Year Ended December 31, 2023 Year Ended December 31, 2022 Change $ (in thousands) Revenue: License and other revenue $ 392 $ 1,503 $ (1,111) Total revenue 392 1,503 (1,111) Operating expenses: Research and development 28,588 24,618 3,970 General and administrative 31,085 32,433 (1,348) Total operating expenses 59,673 57,051 2,622 Loss from operations (59,281) (55,548) (3,733) Other income (expense), net 6,943 1,379 5,563 Loss before provision for income taxes (52,339) (54,169) 1,830 Provision for income taxes — — — Net loss $ (52,339) $ (54,169) $ 1,830 Total revenue$208.4recognized for the year ended December 31, 2023 related to royalties. Revenue was $1.5 million for the year ended December 31, 2021 as a result of revenue recorded in connection with the Takeda License2022 related to licensing and Termination Agreementother agreements.the Angelini License Agreement. Revenue was $12.6Development ExpensesYear Ended December 31, 2023 Year Ended December 31, 2022 Change $ (in thousands) Preclinical and development expenses $ 14,605 $ 9,715 $ 4,883 Payroll and payroll-related expenses 10,541 11,498 (957) Other expenses 3,442 3,405 37 Total research and development $ 28,588 $ 24,618 $ 3,963 2020.Research and Development ExpensesResearch and development expenses were $46.92023 compared to $24.6 million for the year ended December 31, 2021 compared2022. The increase of $4.9 million in preclinical and development expenses was due to $63.4additional activities related to our ongoing development programs, primarily relating to OV888 (GV101) and OV329. The decrease of $1.0 million in payroll and payroll-related expenses was primarily due to the impact of a reorganization in early 2022 which resulted in approximately $1.0 million in severance costs during the period.Year Ended December 31, 2023 Year Ended December 31, 2022 Change $ (in thousands) Payroll and payroll-related expenses $ 17,131 $ 16,071 $ 1,060 Legal and professional fees 7,610 9,253 (1,643) General office expenses 6,345 7,108 (763) Total general and administrative $ 31,085 $ 32,432 $ (1,347) 2020. The decrease of $16.5 million was primarily due2023 compared to a decrease in activities related to our ongoing development programs, including as a result of the termination of the development of OV101 and the transfer of the development of OV935, which was taken over by Takeda. During the year ended December 31, 2021, research and development expenses consisted of $30.4 million in preclinical and development expenses, $13.5 million in payroll and payroll-related expenses, of which $1.7 million related to stock-based compensation, and $3.1 million in other expenses. During the year ended December 31, 2020, total research and development expenses consisted of $43.6 million in preclinical and development expenses, $15.4 million in payroll and payroll-related expenses, of which $2.8 million related to stock-based compensation, and $4.4 million in other expenses.General and Administrative Expenses58General and administrative expenses were $37.2$32.4 million for the year ended December 31, 2021 compared2022. The decrease of $1.3 million was primarily due to $30.6reduced legal and professional fees and general office expenses, partially offset by an increase in non-cash compensation expenses.2020. The increase2023, comprised of $6.6$4.9 million primarily consistedin interest and accretion income on investments in U.S. treasuries and $2.0 million of increases in business development costs related tounrealized gain on long-term equity investments. For the Takeda transaction, compliance and pre-commercialization expenses and professional fees.Income TaxesThe provision recorded for income taxes for the yearsyear ended December 31, 20212022, other income (expense), net of $1.4 million was comprised of $1.8 million in interest and 2020 is $1.3accretion income on investments in U.S. treasuries and $0.4 million and zero, respectively, with an effective rate of 1.07% and 0%, respectively. There was an increase in the provision for income taxes due to the significant one-time, upfront payment from Takeda pursuant to the Takeda License and Termination Agreement, as well as disallowed use of net operating losses. The Company has historically incurred operating losses and maintains a full valuation allowance against net deferred tax assets. The valuation allowance was approximately $63.3 million and $89.8 million at December 31, 2021 and 2020, respectively.2021,2023 and 2022, we had total cash, and cash equivalents and marketable securities of $187.8$105.8 million as compared to $72.0and $129.0 million, as of December 31, 2020.respectively. We believe that our cash, and cash equivalents and marketable securities as of December 31, 20212023 will fund our projected operating expenses and capital expenditure requirements for at least 12 months from the next 12 months.Our net revenue in the year ended December 31, 2021 was primarily due to a significant one-time, upfront payment from Takeda pursuant to the Takeda License and Termination Agreement. Otherwise, weWe have incurred losses and experienced negative operating cash flows in most years since our inception and anticipate that we will continue to incur losses for at least the next several years. We incurred net incomelosses of approximately $122.8$52.3 million and net losses of approximately $81.0$54.2 million for the years ended December 31, 20212023 and 2020,2022, respectively. As of December 31, 2021,2023, we had an accumulated deficit of $171.4$277.9 million and working capital of $175.7$98.1 million.and cash equivalents and marketable securities are sufficient to fund existing and planned cash requirements.requirements into the first half of 2026. Our primary uses of capital are, and we expect will continue to be, compensation and related expenses, third-party clinical research and development services, clinical costs, legal and other regulatory expenses and general overhead costs. We have based our estimates on assumptions that may prove to be incorrect, and we could use our capital resources sooner than we currently expect. Additionally, the process of testing drug candidates in clinical trials is costly, and the timing of progress in these trials is uncertain. We cannot estimate the actual amounts necessary to successfully complete the development and commercialization of our product candidates or whether, or when, we may achieve profitability.2021,2023, we had no long-term debt and no material non-cancelable purchase commitments with service providers, as we have generally contracted on a cancelable, purchase order basis. We cannot estimate whether we will receive or the timing of any potential contingent payments upon the achievement by us of clinical, regulatory and commercial events, as applicable, or royalty payments that we may be required to make under license agreements we have entered into with various entities pursuant to which we have in-licensed certain intellectual property as contractual obligations or commitments, including agreements with AstraZeneca, AB, H. Lundbeck A/S,Gensaic and Northwestern. Pursuant to these license agreements, we have agreed to make milestone payments up to an aggregate of $279.3$660.3 million upon the achievement of certain development, regulatory and sales milestones. We excluded these contingent payments from the consolidated financial statements given that the timing, probability, and amount, if any, of such payments cannot be reasonably estimated at this time. product revenues from product sales to date. Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of equity offerings, debt financings and additional funding from license and collaboration arrangements. Except for any obligations of our collaborators to reimburse us for research and development expenses or to make milestone or royalty payments under our agreements with them, we will not have any committed external source of liquidity. To the extent that we raise additional capital through future equity offerings or debt financings, ownership interests may be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt and equity financings, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. There can be no assurance that such financings will be obtained on terms acceptable to us, if at all. The ongoing COVID-19 pandemic continuesAdditionally, while the long-term economic impact of geopolitical tensions, including the war between Russia and Ukraine and war in Israel, is difficult to rapidly evolve andassess or predict, each of these events has already resulted in acaused significant disruption ofdisruptions to the global financial markets.markets and contributed to a general global economic slowdown. Furthermore, inflation rates have increased recently to levels not seen in decades. In addition, the U.S. Federal Reserve has raised interest rates in response to concerns about inflation. High interest rates, especially if coupled with reduced government spending and volatility in financial markets, may further increase economic uncertainty and heighten these risks. If the disruption persistsdisruptions and deepens,slowdown deepen or persist, we could experience an inabilitymay not be able to access additional capital on favorable terms, or at all, which could in the future59operations.ability to pursue our business strategy. If we raise additional funds through collaborations, strategic alliances or licensing agreements with third parties for one or more of our current or future drug candidates, we may be required to relinquish valuable rights to our technologies, future revenue streams, research programs or drug candidates or to grant licenses on terms that may not be favorable to us. Our failure to raise capital as and when needed would have a material adverse effect on our financial condition and our ability to pursue our business strategy.“S-3“Current S-3 Registration Statement”), which includes a prospectus covering the issuance and sale of up to $75.0 million of common stock pursuant to an at-the-market (“ATM”) offering program, including the unsold securities under the Prior Registration Statement. During the years ended December 31, 2023 and 2022, we did not sell any shares under our ATM program. As of December 31, 2021,2023, we had $250.0 million available under our Current S-3 Registration Statement, including $75.0 million available pursuant to our ATM program.Year Ended December 31, 2023 Year Ended December 31, 2022 (in thousands) Net cash (used in) provided by: Operating activities $ (45,781) $ (55,170) Investing activities (2,581) (87,940) Financing activities 30,535 181 Net decrease in cash, cash equivalents, and restricted cash $ (17,827) $ (142,930) Provided by (Used in)Used in Operating Activitiesprovided byused in operating activities was $118.6$45.8 million for the year ended December 31, 2021,2023, which consisted of net incomeloss of $122.5$52.3 million offset by a net of $6.6 million of various non-cash charges and indirectoperating cash changes, primarily related to $12.3most significantly $7.3 million related to an asset acquisition via stock issuance and cash,in stock-based compensation expense of $5.0 million, and $12.4 million of deferred revenue reversal.compensation. Net cash used in operating activities was $51.6$55.2 million for the year ended December 31, 2020,2022, which consisted of a net loss of $81.0$54.2 million and $8.3 million decrease in accounts payable and accrued expenses, partially offset by a net of $29.5 million ofvarious non-cash charges and indirect cash changes, primarily related to $7.2 million of stock-based compensation expense and $12.4 million in deferred revenue.(Used in) Provided byUsed in Investing Activities$1.8$2.6 million for the year ended December 31, 2021,2023, which was primarily comprisedrelated to our purchases and sales/maturities of $1.6 millioninvestments in U.S. treasury funds and the purchase of a long-term equity investment. For the year ended December 31, 2020, $34.62022, $87.9 million was provided byused in investing activities, which was primarily due to net proceeds from short-termcomprised of purchases and sales/maturities of investments during the year.of $0.9was $30.5 million for the year ended December 31, 20212023, which was primarily due to the $30.0 million received in connection with the Ligand Agreement. For the same period in 2022, cash provided by financing activities was $0.2 million, related to proceeds from the exercise of options. Foroptions and purchases made under the year ended December 31, 2020, net cash provided by financing activities of $47.0 million was primarily due to the net proceeds from the August 2020 Offering.managementmanagement’s discussion and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements, as well as the revenue and expenses incurred during the reported periods. On an ongoing basis, we evaluate our estimates and judgments, including those related to revenue recognition and accrued clinical expenses.judgments. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not apparent from other sources. Changes in estimates are reflected in reported results for the period in which they become known.60Angelini License Agreement and the Takeda License and TerminationRLT Agreement. The terms of the agreements within this scope may contain multiple performance obligations, including but not limited to licenses and research and development activities. ASC 606 requires that we evaluate these agreements to determine the distinct performance obligations. Non-refundable, up-frontupfront fees that are not contingent on any future performance and require no consequential continuing involvement by us, are recognized as revenue when the license term commences and the licensed data, technology or product is delivered. We defer recognition of non-refundable upfront fees if the performance obligations are not satisfied.ForTakeda License and Termination Agreement that closed on March 29, 2021, we received an upfront payment of $196.0 million. We determined that the transaction price was equal to the upfront fee of $196.0 million and was associated with several material conditions that were satisfied at the closing date. We recognized the full upfront payment of $196.0 million as revenue at the closing date upon the satisfaction of the material conditions outlined in the termination agreement.On March 29, 2021, we received a notice of termination of the Angelini License Agreement. Subsequently, we and Angelini mutually agreed to waive the six month termination notice provisions and the Angelini License Agreement terminated effective Marchyear ended December 31, 2021. We have been released from our performance obligations and will not be entitled to any future milestone payments under the Angelini License Agreement. As a result of being released from the performance obligations,2023, we recognized $12.4 millionrevenue of revenue at the termination date, consisting of $5.4 million of license revenue related to ongoing trials and $7.0approximately $0.4 million related to the potential 35% funding of the cost for Angelini's future trials.Accrued Clinical Expensesclinicalresearch and development expenses. This process involves reviewing open contracts and communicating with our personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost. Payments under certain contracts we have with third parties depend on factors, such as the successful enrollment of certain numbers of patients, site initiation and the completion of clinical trial milestones.clinicalresearch and development expenses, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If possible, we obtain information regarding unbilled services directly from our service providers. However, we may be required to estimate the cost of these services based only on information available to us. If we underestimate or overestimate the cost associated with a trial or service at a given point in time, adjustments to research and development expenses may be necessary in future periods. Historically, our estimated accrued clinicalresearch and development expenses have approximated actual expense incurred.Emerging Growth Company Status and an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and may remain an emerging growth company until December 31, 2022. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include:•reduced disclosure about our executive compensation arrangements;•no non-binding stockholder advisory votes on executive compensation or golden parachute arrangements; and•exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting.We have taken advantage of reduced reporting requirements in this Annual Report on Form 10-K and may continue to do so until such time that we are no longer an emerging growth company. We will remain an “emerging growth company” until the earliest of (a) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more, (b) December 31, 2022, the last day of the fiscal year following the fifth anniversary of the completion of the our IPO, (c) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years or (d) the date on which we are deemed to be a large accelerated filer61under the rules of the SEC. Section 107 of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period for complying with new or revised accounting standards. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies.In addition, we are also a smaller reporting company as defined in the Exchange Act. We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies and will be able to take advantage of these scaled disclosures for so long as (i) our voting and non-voting common stock held by non-affiliates is less than $250.0 million measured on the last business day of our second fiscal quarter or (ii) our annual revenue is less than $100.0 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is less than $700.0 million measured on the last business day of our second fiscal quarter.62Risk.Risk2021,2023, we had cash, and cash equivalents and marketable securities of $187.8 million that were held in an interest-bearing money market account.$105.8 million. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates. Due to the short-term maturities of our cash equivalents and marketable securities and the low risk profile of our investments, an immediate 100 basis point change in interest rates would not have a material effect on the fair market value of our cash equivalents.equivalents and marketable securities. To minimize the risk in the future, we intend to maintain our portfolio of cash equivalents and marketable securities in institutional market funds that are comprised of U.S. Treasury and U.S. Treasury-backed repurchase agreements.agreements as well as treasury notes and high quality short-term corporate bonds.2021,2023, our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act). Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Our principal executive officer and principal financial officer have concluded based upon the evaluation described above that, as of December 31, 20212023 our disclosure controls and procedures were effective at the reasonable assurance level.20212023 based on the framework in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework). Based on the results of its evaluation, management concluded that our internal control over financial reporting was effective as of December 31, 2021.ofon our internal control over financial reporting from our registered public accounting firm due to an exemption established byas a smaller reporting company and as a non-accelerated filer for the JOBS Act for “emerging growth companies.”our internal controlcontrols over financial reporting during our most recent quarter ended December 31, 20212023, that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.None.6320222024 Proxy Statement.20222024 Proxy Statement.ManagementManagement” and “Equity Compensation Plan Information” in our 20222024 Proxy Statement.F-1367 – STOCKHOLDERS’ EQUITY AND PREFERRED STOCKPreferred Stock.preferred stock. Pursuant to the Company’s amended and restated certificate of incorporation, as amended, the Company is authorized to issue up to 125,000,000 shares of common stock and 10,000,000 shares of Preferred Stock.preferred stock. The Company has designated 10,000 of the 10,000,000 authorized shares of Preferred Stockpreferred stock as non-voting Series A Convertible Preferred Stock (“Series A Preferred Stock”).held.held. The holders of common stock have no preemptive or other subscription rights, and there are no redemption or sinking fund provisions with respect to such shares. Subject to preferences that may apply to any outstanding series of Preferred Stock,preferred stock, holders of the common stock are entitled to receive ratably any dividends declared on a non-cumulative basis. Shares of Series A Preferred Stock will be entitled to receive dividends at a rate equal to (on an as-if-converted-to-common stock basis), and in the same form and manner as, dividends actually paid on shares of common stock. The common stock is subordinate to all series of Preferred Stock with respect to rights upon liquidation, winding up and dissolution of the Company. The holders of common stock are entitled to liquidation proceeds after all liquidation preferences for the Preferred Stockpreferred stock are satisfied.In November 2020, the Company entered into a sales agreement (the “2020 ATM agreement”) with Cowen and Company, LLC ("Cowen") under which the Company may offer and sell in “at the market offerings,” from time to time at its sole discretion, shares of its common stock having an aggregate offering price of up to $75.0 million through Cowen acting as sales agent. As of December 31, 2021, the Company has not sold any shares of its common stock under the 2020 ATM agreement. and 3,250 shares of Series A Preferred Stock outstanding as of December 31, 20212023 and December 31, 2020, respectively.2022. Each share of Series A Preferred Stock is convertible into 1,000 shares of common stock at any time at the holder’s option. However, the holder will be prohibited, subject to certain exceptions, from converting shares of Series A Preferred Stock into shares of common stock if, as a result of such conversion, the holder, together with its affiliates, would own more than, at the written election of the holder, either 9.99%9.99% or 14.99%14.99% of the total number of shares of common stock then issued and outstanding, which percentage may be changed at the holder’s election to any other number less than or equal to 19.99% upon 61 days’ notice to the Company; provided, however, that effective 61 days after delivery of such notice, such beneficial ownership limitations shall not be applicable to any holder that beneficially owns either 10.0% or 15.0%, as applicable based on the holder’s initial written election noted above, of the total number of shares of common stock issued and outstanding immediately prior to delivery of such notice. In the event of a liquidation, dissolution, or winding up of the$0.001$0.001 per share of Series A Preferred Stock before any proceeds are distributed to the holders of common stock.In March 2021, certain of the Company's stockholders elected to convert an aggregate of 2,000 shares of Series A Preferred Stock owned by such holders into an aggregate of 2,000,000 shares of the Company's common stock.In2020, entities affiliated with Biotechnology Value Fund, L.P. elected to convert an aggregate of 2,256 shares of Series A Preferred Stock owned by such holders into an aggregate of 2,256,000 shares of the Company’s common stock.In August 2020,31, 2023, the Company sold 6,250,000 shares of its common stock at a public offering price of $8.00 per share, for net proceeds of $46.7 million after deducting underwriting discounts and commissions and other offering expenses payable by the Company, (the “August 2020 Offering”).In May 2020, entities affiliated with Biotechnology Value Fund, L.P. elected to convert an aggregate of 2,256 shares of Series A Preferred Stock owned by such holders into an aggregate of 2,256,000 shares of the Company’s common stock.DividendsPreferred Stockpreferred stock have been declared and paid. Through December 31, 2021, the Company has 0t declared any dividends.78 – STOCK-BASED COMPENSATION(the “2014(“2014 Plan”), which authorized the Company to grant shares of common stock in the form of incentive stock options, nonstatutory stock options, stock appreciation rights, restricted stock and restricted stock units. The types of stock-based awards, including share purchase rights amount, terms, and exercisability provisions offor exercising grants were determined by the Company’sBoard. of Directors.The Company's Board of Directors adopted, and the Company's stockholders approved, the 2017 equity incentive plan (“2017 Plan”), which became effective on May 4, 2017. The initial reserve of shares of common stock under the 2017 Plan was 3,052,059 shares. The 2017 Plan provides for the grant of incentive stock options, non-statutory stock options, restricted stock awards, restricted stock unit awards, stock appreciation rights, performance-based stock awards, and other forms of stock-based awards. Additionally, the 2017 PlanF-14cash awards. The Company's employees, officers, directors, and consultants and advisors are eligible to receive awards under the 2017 Plan. Upon the adoption of the 2017 Plan, no further awards will bewere granted under the 2014 Plan. Pursuant to the terms of the 2017 Plan, on each January 1st, the plan limit shall be increased by the lesser of (x) 5%5% of the number of shares of common stock outstanding as of the immediately preceding December 31 and (y) such lesser number as the Board of Directors may determine in its discretion. On January 1, 2021,2022, an additional 3,287,1581,000,000 shares were reserved for issuance under the 2017 Plan. On January 1, 2023, an additional 3,523,344 shares were reserved for issuance under the 2017 Plan. As of December 31, 2021,2023, there were 4,397,0674,400,759 shares of the Company’s common stock reserved for issuance under the 2017 Plan.Company's Board of Directors adopted, and the Company's stockholders approved the 2017 employee stock purchase plan (the “2017 (“ESPP”), which became effective on May 4, 2017. The initial reserve of shares of common stock that may be issued under the 2017 ESPP was 279,069 shares. The ESPP allows employees to purchase common stock of the Company at a 15%15% discount to the market price on designated purchase dates. During the years ended December 31, 20212023 and 2020, 60,4902022, 63,761 and 105,46476,455 shares were purchased under the ESPP and the Company recorded expense of $83,787$57,148 and $152,973,$85,319, respectively. The number of shares of common stock reserved for issuance under the 2017 ESPP will automatically increase on January 1 of each year, beginning on January 1, 2018 and continuing through and including January 1, 2027, by the lesser of (i) 1%1% of the total number of shares of the Company’s capital stock outstanding on December 31 of the preceding calendar year, (ii) 550,000 shares or (iii) such lesser number of shares determined by ourthe Board. The Board acted prior to January 1, 20212024 to provide that there be 0no increase in the number of shares reserved for issuance under the 2017 ESPP on either such date.ESPP. As of December 31, 2021,2023 and 2022, there were 493,062352,846 and 416,607 shares of the Company’s common stock reserved for issuance under the 2017 ESPP.generally have a ten-year term and a four-year graded vesting period. The vesting requirement is generally conditioned upon the grantee’s continued service with the Company during the vesting period. Once vested, all awardsoptions granted are exercisable from the date of grant until they expire. The option grants are non-transferable. Vested options generally remain exercisable for 90 days under the 2017 Plan and 30 days under the 2014 Plan subsequent to the termination of the option holder’s service with the Company. In the event of the option holder’s death or disability while employed by or providing service to the Company, the exercisable period extends to 18 months or 12 months.towith the specific terms of the agreement. At December 31, 2021,2023 and 2022, there were 150,000zero and 100,000 performance-based options outstanding and unvested, respectively, that include options to vest upon the achievement of certain research and development milestones.20212023 and 20202022 was estimated using the Black-Scholes option valuation model. The inputs for the Black-Scholes option valuation model require significant assumptions made by management and are detailed in the table below. The risk-free interest rates were based on the rate for U.S. Treasury securities at the date of grant with maturity dates approximately equal to the expected life at the grantvolatility information of peer companies that is publicly available.170,00045,000 and 10,000zero stock options to nonemployee consultants for services rendered during the years ended December 31, 20212023 and 2020,2022, respectively. There were 181,25098,542 and 32,500127,459 unvested nonemployee options outstanding as of December 31, 20212023 and 2020,2022, respectively. Total expense recognized related to the nonemployee stock options for the years ended December 31, 20212023 and 20202022 was $584,092$202,114 and $257,431,$575,995, respectively. Total unrecognized compensation expenses related to the nonemployee stock options was $67,471$133,769 and $626,977 as of December 31, 2021.2023 and 2022, respectively. During the years ended December 31, 2021,2023 and 2020, the Company recognized 0 and $115,240, respectively, in2022, there were no expenses for nonemployee performance-based option awards.1,875,9132,993,000 and 3,573,1604,575,641 stock options to employees during the years ended December 31, 20212023 and 2020,2022, respectively. There were 4,407,3085,376,910 and 4,975,2626,090,889 unvested employee options outstanding as of December 31, 20212023 and 2020,2022, respectively. Total expense recognized related to the employee stock options for the years ended December 31, 20212023 and 20202022 was $4.4$7.5 million and $7.1$5.9 million, respectively. Total unrecognized compensation expense related to employee stock options was $10.3F-152021.2023 and 2022, respectively. During the years ended December 31, 20212023 and 2020,2022, the Company recognized 0zero and $2.3$0.1 million, respectively, in expenses for employee performance-based option awards.For the Year Ended December 31, 2023 2022 Research and development $ 1,936,746 $ 1,770,599 General and administrative 5,348,446 4,786,087 Total $ 7,285,192 $ 6,556,686 For the Year Ended December 31, 2023 2022 Stock options $ 7,228,044 $ 6,471,367 Employee stock purchase plan 57,148 85,319 Total $ 7,285,192 $ 6,556,686 20212023 and 2020,2022, respectively, was estimated by utilizing the following assumptions:For the Year Ended December 31, 2023 2022 Weighted
AverageWeighted
AverageVolatility 84.52 % 87.16 % Expected term in years 6.07 6.07 Dividend rate 0.00 % 0.00 % Risk-free interest rate 3.97 % 2.21 % Fair value of option on grant date $ 1.92 $ 2.12
Average
Average20212023 and 2020,2022, respectively, was estimated by utilizing the following assumptions:
Average
AverageF-16For the Year Ended December 31, 2023 2022 Weighted
AverageWeighted
AverageVolatility 83.73 % — % Expected term in years 5.32 0.00 Dividend rate 0.00 % 0.00 % Risk-free interest rate 3.86 % — % Fair value of option on grant date $ 2.21 $ — Number of
SharesWeighted
Average
Exercise
PriceWeighted
Average
Remaining
Contractual
Life in YearsAggregate
Intrinsic
ValueOptions outstanding at December 31, 2021 10,776,758 $ 4.97 6.07 $ 2,389,890 Vested and exercisable at December 31, 2021 6,188,200 $ 5.98 4.63 $ 1,531,907 Granted 4,575,641 2.89 9.26 Exercised (25,518) 1.91 Forfeited or expired (2,365,643) 5.56 Options outstanding December 31, 2022 12,961,238 $ 4.13 7.42 $ 62,158 Vested and exercisable at December 31, 2022 6,742,890 $ 5.05 6.20 $ 61,214 Granted 3,038,000 2.63 9.20 Exercised (146,346) 2.76 Forfeited or expired (728,346) 3.44 Options outstanding December 31, 2023 15,124,546 $ 3.87 6.90 $ 5,212,586 Vested and exercisable at December 31, 2023 9,649,094 $ 4.47 5.97 $ 2,464,620 2021,2023, there was approximately $10.4$9.4 million of unamortized share–basedstock-based compensation expense, which is expected to be recognized over a remaining average vesting period of 2.61 2.23 years. At December 31, 2022, there was $12.1 million of unamortized stock-based compensation expense, which is expected to be recognized over a remaining average vesting period of 2.26 years.89 – INCOME TAXES2021,2023, the Company has available approximately $124.8$150.2 million and $171.3$190.2 million of unused net operating loss ("NOL"(“NOL”) carryforwards for federal and state tax purposes, respectively, that may be applied against future taxable income. The Company also has approximately $163.9$163.9 million of unused NOL carryforwards for New York City purposes. These NOL carryforwards reflect the result of the Section 382 limitation. The NOL carryforwards will begin to expire in the year 20352037 if not utilized prior to that date. (generally defined as a greater than 50% change in its equity ownership over a three-year period), the corporation’s ability to use its pre-change NOL carryforwards and other pre-change tax attributes to offset its post-change income may be limited. On each of August 10, 2015 and February 22, 2019 the Company experienced an ownership change. A formal Section 382 analysis has yet to be completed. The Company anticipates a significant portion of its pre-change NOLs to be limited.Aslimited, however has not yet completed a result of theformal Section 382 limitation,analysis subsequent to the Company was subject to U.S. Federal, state and local income taxes for 2021 of approximately $1.3 million.decreasedincreased by approximately $26.5$18.9 million and $10.4 million during the year 2021, respectively, whichyears 2023 and 2022, respectively. The increase in valuation allowance in 2023 is primarily due to the anticipated utilizationincreases in NOL carryforwards and write-off of NOLs that will expire due to the limitations under Section 382.capitalized research and experimental costs.F-17December 31, 2023 2022 Deferred tax assets/liabilities: Net operating loss carryovers $ 55,653,286 $ 55,472,573 Intangible assets 7,522,067 6,332,176 Capitalized research and experimental costs 10,909,248 4,246,071 Stock-based compensation 7,248,199 4,384,751 Royalty monetization liability 8,427,970 — Lease liability 4,495,402 3,544,624 Research and development tax credits 2,877,649 3,046,253 Accrued compensation — — Charitable contributions — — Depreciation (166,075) (241,096) Right-of-use asset (3,903,379) (3,198,857) Other (434,957) 97,494 Total gross deferred tax assets/liabilities 92,629,410 73,683,989 Valuation allowance (92,629,410) (73,683,989) Net deferred tax assets (liabilities) $ — $ — December 31, 2023 2022 Federal income tax benefit at statutory rate 21.00 21.00 State income tax, net of federal benefit 9.12 (0.27) Permanent items (1.32) (1.26) Change in valuation allowance (29.23) (18.50) Research and development tax credits 1.05 1.28 Other (0.62) (2.25) Effective income tax expense rate 0.00 % 0.00 % 20212023 and 2020,2022, the Company had 0no unrecognized tax benefits or related interest and penalties accrued. The Company has not yet conducted a study of research and development credit carryforwards. This study may result in an adjustment to the Company’s research and development credit carryforwards; however, until a study is completed and any adjustment is known, 0 amounts are being presented as an uncertain tax position. A full valuation allowance has been provided against the Company’s research and development credits and, if an adjustment is required, this adjustment would be offset by an adjustment to the valuation allowance. Thus, there would be no impact to the balance sheet or statement of operations if an adjustment were required. The Company would recognize both accrued interest and penalties related to unrecognized benefits in provision for income tax expense.taxes. The Company’s uncertain tax positions yet to be determined would be related to years that remain subject to examination by relevant tax authorities. Since the Company is in a loss carryforward position, the Company is generally subject to examination by the U.S. federal, state and local income tax authorities for all tax years in which a loss carryforward is available.910 – COMMITMENTS AND CONTINGENCIESOn March 26, 2015, the Company entered into an exclusive agreement with H. Lundbeck A/S (“Lundbeck”) for a worldwide perpetual licensing right related to the research, development and commercialization of OV101. On May 10, 2019, the parties amended the license agreement.Pursuant to the Lundbeck license agreement, as of the first amendment, the Company agreed to make milestone payments totaling up to $189.0 million upon the achievement of certain developmental, regulatory and sales milestones. The first payment of $1.0 million is due upon the successful completion of the first Phase 3 trial for a product in which OV101 is an active ingredient. addition, the agreement calls for the Company to pay royalties for an initial term based on a low double-digit percentage of sales and provides for the reduction of royalties in certain limited circumstances.F-18Thereafter, we closed our OV101 (gaboxadol) program in Angelman syndrome in early 2021. On February 1, 2022, we entered into Amendment No. 3 to the Lundbeck agreement, or Amendment No. 3, to permit our performance under the Healx License and Option Agreement. Under the terms of Amendment No. 3, if Healx exercises its option, we will owe Lundbeck a share of all milestone and royalty payments received from Healx if we choose not to exercise the Ovid Opt-In Right. If we choose to exercise the Ovid Opt-In Right and to co-develop and co-commercialize the program with Healx, we will owe a share of the net profit share to LundbeckIn December 2016,, the Company entered into a license agreement with(“Northwestern Agreement”) ///8with Northwestern University or Northwestern,(“Northwestern”), pursuant to which Northwestern granted usthe Company an exclusive, worldwide license to patent rights inof certain inventions or the (“Northwestern Patent Rights,Rights”) which relate to a specific compound andKnow-Howknow-how related to the practice of the inventions claimed in the Northwestern Patents.agreement,Agreement, the Company was granted exclusive rights to research, develop, manufacture and commercialize products utilizing the Northwestern Patent Rights for all uses. The Company has agreed that it will not use the Northwestern Patent Rights to develop any products for the treatment of cancer, but Northwestern may not grant rights in the technology to others for use in cancer. The Company also has an option, exercisable during the term of the agreement to an exclusive license under certain intellectual property rights covering novel compounds with the same or similar mechanism of action as the primary compound that is the subject of the license agreement. Northwestern has retained the right, on behalf of itself and other non-profit institutions, to use the Northwestern Patent Rights and practice the inventions claimed therein for educational and research purposes and to publish information about the inventions covered by the Northwestern Patent Rights.agreement,Agreement, the Company paid an upfront non-creditable one-time license issuance fee of $75,000,$75,000 and is required to pay an annual license maintenance fee of $20,000,$20,000, which will be creditable against any royalties payable to Northwestern following first commercial sale of licensed products under the agreement. The Company is responsible for all ongoing costs of filing, prosecuting and maintaining the Northwestern Patents,Patent Rights, but also has the right to control such activities using its own patent counsel. In consideration for the rights granted to the Company under the Northwestern agreement, the Company is required to pay to Northwestern up to an aggregate of $5.3$5.3 million upon the achievement of certain development and regulatory milestones for the first product covered by the Northwestern Patents,Patent Rights, and upon commercialization of any such products, will be required to pay to Northwestern a tiered royalty on net sales of such products by the Company, its affiliates or sublicensees, at percentages in the low to mid single-digits,mid-single-digits, subject to standard reductions and offsets. The Company’s royalty obligations continue on a product-by-product and country-by-country basis until the later of the expiration of the last-to-expire valid claim in a licensed patent covering the applicable product in such country and 10 years following the first commercial sale of such product in such country. If the Company sublicenses a Northwestern Patent Right, it will be obligated to pay to Northwestern a specified percentage of sublicense revenue received by the Company, ranging from the high single digitssingle-digits to the low-teens.1one product that is covered by the Northwestern Patent Rights.On 30, 2021, the Company entered into an exclusive license agreement with AstraZeneca AB (“AstraZeneca”), for a library of early-stage small molecules targeting the KCC2 transporter, including lead candidate OV350. Upon execution of the agreement, the Company was obligated to pay an upfront cash payment of $5.0$5.0 million and issued shares of the Company's common stock in an amount that equaled $7.3$7.3 million based on the volume-weighted average price of shares of the Company's common stock for the 30 business days immediately preceding the execution date of the transaction. Since the intangibles acquired in the AstraZeneca license agreement do not have an alternative future use, all costs incurred were treated as research and development expense. The Company recorded a total of $12.3$12.3 million as research and development expense related to this agreement induring December 2021.$203.0$203.0 million upon the achievement of certain developmental, regulatory and sales milestones. The first payment of $3.0$3.0 million is due upon the successful completion of the first Phase 2 clinical study of a licensed product following a positive biomarker readout in a Phase 1 clinical study.2021,2023, none of thethese contingent payments related to these licensing agreements were considered probable.F-19ourthe Company's named executive officers is eligible to receive severance payments and benefits upon a termination without “cause” or due to “permanent disability,” or upon “resignation for good reason,” contingent upon the named executive officer’s delivery to the Company of a satisfactory release of claims, and subject to the named executive officer’s compliance with non-competition and non-solicitation restrictive covenants for two years following the termination date.1011 – COLLABORATION AGREEMENTAngelini CollaborationOn July 9, 2020, the Company entered into the Angelini License Agreement, pursuant to which the Company granted to Angelini exclusive rights to develop and commercialize OV101, a selective agonist of the GABAA receptor, for the treatment of Angelman syndrome in the European Economic Area as well as Switzerland, the United Kingdom, Russia and Turkey (the “European Territory”).On March 29, 2021, the Company received a notice of termination of the Angelini License Agreement. Subsequently, Angelini and the Company mutually agreed to waive the six month termination notice provisions and the Angelini License Agreement terminated effective March 31, 2021. The Company has been released from its performance obligations and will not be entitled to any future milestone payments under the Angelini License Agreement.The Company evaluated the Angelini License Agreement to determine whether it was a collaborative arrangement for purposes of ASC 808. The Company concluded that because Angelini was not the ultimate decision maker or the legal owner of the license, Angelini was not considered an active participant and therefore the Angelini License Agreement was outside of the scope of ASC 808. The Company concluded that Angelini was a customer with regard to the combined license and research and development activities and as such the Angelini License Agreement should be evaluated under ASC 606.The Company identified the following material promises under the Angelini License Agreement: (1) licensing of intellectual property with respect to OV101; (2) completion of certain ongoing trials; (3) transfer of a specified amount of compound and related information; (4) potential for funding 35% of the cost for Angelini future trials limited to $7.0 million; and (5) completion of the manufacturing process technology transfer.The Company determined that the $7.0 million represented a potential payment to a customer and was deferred. The transfer of compound and related information was considered a contingent milestone payment that will be recognized upon acceptance by Angelini of the milestone. The Company further determined that the license and the completion of ongoing trials were distinct from each other, as each had value without the other. As such, for the purposes of ASC 606, the Company determined that these two material promises, represented distinct performance obligations.Pursuant to the Angelini License Agreement and during the year ended December 31, 2020, Angelini made an upfront payment to the Company of $20.0 million. Upon the transfer of the specified amount of compound and related information and acceptance by Angelini, Angelini paid the Company an additional $5.0 million. This performance obligation was determined to be variable consideration which was constrained and not considered part of the upfront transaction price allocation. The Company determined the transaction price was equal to the upfront fee of $20.0 million. The transaction price was allocated based on the standalone selling price of the license and the ongoing trials.During the year ended December 31, 2021 and effective upon the termination of the Angelini License Agreement, the Company recognized $12.4 million of revenue consisting of $5.4 million of license revenue related to ongoing trials and the $7.0 million related to the potential 35% funding of the cost for Angelini future trials. During the year ended December 31, 2020, the Company recognized $12.6 million of revenue consisting of $6.1 million of license revenue and $1.5 million relating to the progress of the ongoing trials as well as $5.0 million for the transfer of the specified amount of compound and related information.On 6, 2017, the Company entered into a license and collaboration agreement with Takeda under which the Company licensed from Takeda certain exclusive rights to develop and commercialize soticlestat in certain territories., the Company entered into the RLT Agreement with Takeda, License and Termination Agreement, pursuant to which Takeda secured rights to the Company’s 50%50% global share in soticlestat, and the Company granted to Takeda an exclusive worldwide license under the Company’s relevant intellectual property rights to develop and commercialize the investigational medicine soticlestat for the treatment of developmental and epileptic encephalopathies, including Dravet syndrome and Lennox-Gastaut syndrome.F-20Takeda License and TerminationRLT Agreement, all rights in soticlestat wereare owned by Takeda or exclusively licensed to Takeda by the Company. Takeda assumed all responsibility for, and costs of, both development and commercialization of soticlestat, and the Company will no longer have any financial obligation to Takeda under the original collaboration agreement, including milestone payments or any future development and commercialization costs. OnIn March 29, 2021, upon the closing of the Takeda License and TerminationRLT Agreement, the Company received an upfront payment of $196.0$196.0 million and, if soticlestat is successfully developed, will be eligible to receive up to an additional $660.0$660.0 million upon Takeda achieving developmental, regulatory and sales milestones. In addition, the Company will be entitled to receive tiered royalties beginning in the low double-digits, and up to 20%20% on sales of soticlestat if regulatory approval is achieved. Royalties will be payable on a country-by-country and product-by-product basis for any indications that soticlestat is approved for and sold during the period beginning on the date of the first commercial sale of such product in such country and ending on the later to occur of the expiration of patent rights covering the product in such country and a specified anniversary of such first commercial sale.The In 2023, the Company identifiedsold a 13% stake in the following material promisesroyalty, regulatory and commercial milestone payments that the Company is eligible to receive under the TakedaRLT Agreement to Ligand for $30.0 million.Termination Agreement: (1) no later thanOption Agreementsecond business day prior toCompany entered an exclusive license option agreement (“Healx License and Option Agreement”) with Healx, Ltd. (“Healx”). Under the closingterms of the TakedaHealx License and TerminationOption Agreement, (the “Closing Date”Healx secured a one-year option to investigate gaboxadol (“OV101”), the Company as part of a potential combination therapy for Fragile X syndrome in a Phase 1B/2A clinical trial, as well as a treatment for other indications, for an upfront payment of $0.5 million, and Takeda were requiredfees to agree on an estimatesupport prosecution and maintenance of the development expenses that accrued, or would accrue,Company's relevant intellectual property rights. At the end of the one-year option period, Healx had the option to secure rights to an exclusive license under the original collaboration agreement as of March 31, 2021; (2) on the Closing Date, the Company was required to (i) provide and transfer to Takeda the materials, information and data relating to the soticlestat program, including clinical trial data and results, as further set forth in the Takeda License and Termination Agreement, (ii) assign to Takeda certain agreements applicable to the soticlestat program, and (iii) assign to Takeda all of its right, title and interest in, to and under allCompany's relevant intellectual property rights, developedin exchange for an additional payment of $2.0 million, development and commercial milestone payments, and low to mid-tier double-digit royalties. In February 2023, the Company granted an extension of the option period for up to four months for Healx to continue to investigate gaboxadol. Royalties are payable on a country-by-country and product-by-product basis during the period beginning on the date of the first commercial sale of such product in such country and ending on the later to occur of the expiration of patent rights covering the product in such country and a specified anniversary of such first commercial sale.created pursuantin the event that Healx does not exercise its option during the option period defined in the Healx License and Option Agreement (“Option Period”), the expiration of such period, or (b) in the event that Healx does exercise its option during the Option Period, and the Company does not exercise the Ovid Opt-In Right during the period of time it has to opt-in (“Opt-In Period”) or the opt-in terms are otherwise terminated, upon the expiration of all payment obligations, or (c) in the event that Healx does exercise the Option during the Option Period, and the Company does exercise the Ovid Opt-In Right during the Opt-In Period, such time as neither Healx nor the Company is continuing to exploit gaboxadol. Further, if the Company exercises the Ovid Opt-In Right to co-develop and co-commercialize the program, it will owe an equal share of any net profits to a third party with which it previously established a licensing agreement. If the Company does not exercise the Ovid Opt-In Right, it will owe the third party an equal share of all milestone and royalty payments received.original collaboration agreementHealx License and owned jointly byOption Agreement whereby revisions were made to terms regarding the Company and Takeda astiming of the Closing Date; (3) within 45 days after March 31, 2021, the Company and Takeda were required to provide a written report to the finance officer designatedoption exercise fee payable by the other party setting forth a final total of the development expenses that accrued as of March 31, 2021 and, within 10 business days after receipt of such report, the finance officers shall agree on whether a net settlement payment is due from TakedaHealx to the Company, or fromthe clinical and regulatory milestone payment structure, and the royalty payment structure. Additionally, the parties agreed that following the exercise of the option, Healx would assume direct responsibility for patent maintenance and prosecution and that the Company would transfer to Takeda; and (4) within 75 days after the Closing Date,Healx all supply obligations with respect to the extent not provided onactive pharmaceutical ingredient and finished gaboxadol products and any related licensed technology and know-how in the Closing Date, Ovid shall provideCompany's possession that is relevant to Takeda (i) any materials, information and datathe manufacture of such licensed products.soticlestat program, including clinical trial data and results, as further set forth in the Takeda License and Termination Agreement, (ii) other documents (including all expired agreements and related data developed thereunder) to the extent relating to the soticlestat program that are necessary for the exploitation, development, commercialization and manufacture of soticlestat, as further set forth in the Takeda License and Termination Agreement and (iii) any tangible embodiments of the intellectual property rights controlled by Ovid that are reasonably necessary for, used in or held for use in Takeda’s exploitation of the soticlestat program.The Company determined the transaction price is equal to the upfront fee of $196.0 million and is associated with all four performance obligations identified above. It is noted that the incremental effort associated with performance obligations three and four is negligible and not material in the context of the Takeda License and Termination Agreement since all of the information is related to the collaboration period for which the Company already has the information readily available. Therefore, since they are not material in the context of the Takeda License and Termination Agreement, the full upfront fee was allocated to the two performance obligations satisfied at closing2021,2022, the Company recognized a creditrecorded revenue of $0.5 million associated with the Healx License and Option Agreement.researchthe territory (which consists of the United States, the European Economic Area, United Kingdom and development expensesSwitzerland) for the treatment of $2.5 million and an expenseCDKL5 deficiency disorders. Following the date of $0.1 millionregulatory approval by the FDA of the first licensed product in general and administrative expensesthe territory which was received on March 18, 2022, Marinus issued, at the Company's option, 123,255 shares of Marinus common stock, par value $0.001 per share, as payment. The Marinus License Agreement also provides for a net credit amountpayment of $2.4 million reimbursedroyalties from Marinus to the Company from Takeda. Duringin single-digits on net sales of each such licensed product sold.2020,2023, and unrealized loss of $0.5 million for the year ended December 31, 2022, which were recorded as unrealized gains (losses) on equity securities and reflected in other income (expenses), net in the consolidated statements of operations.$0.7 millionan unrealized gain on the investment due to an observable change in researchprice, and development expenses representing research and development expenses reimbursable torecorded the Company from Takeda,gain in other income (expense), net, in the consolidated statements of which $0.1 million was included in related party receivable and $2.1 million was included in related party payable as of December 31, 2020.operations.1112 – RELATED PARTY TRANSACTIONSTakeda License and TerminationRLT Agreement with Takeda. For a description of the Takeda License and TerminationRLT Agreement, see Note 10. The Company recognized a long-term liability11.2020 representing long-term prepaid expenses to be reimbursed to Takeda as part of the Company’s previous collaboration agreement with Takeda.In May 2020, entities affiliated with Biotechnology Value Fund, L.P. elected to convert an aggregate of 2,256 shares of Series A Preferred Stock owned by such holders into an aggregate of 2,256,000 shares of the Company’s common stock.In August 2020,which the Company issued and sold an aggregate of 1,250,000 sharesrecords net income, diluted net income per share is calculated based upon the weighted-average number of common shares outstanding during the period plus the dilutive impact of weighted-average common equivalent shares outstanding during the period resulting from the assumed exercise of outstanding stock options determined under the treasury stock method and the assumed conversion of preferred stock into common shares determined using the if-converted method. Diluted net loss per share is equivalent to entities affiliated with Biotechnology Value Fund, L.P.,the basic net loss per share due to the exclusion of outstanding stock options and convertible preferred stock because the inclusion of these securities would result in an existing stockholder for aggregate gross proceeds of $10.0 million.In December 2020, entities affiliated with Biotechnology Value Fund, L.P. elected to convert an aggregate of 2,256 shares of Series A Preferred Stock owned by such holders into an aggregate of 2,256,000 shares of the Company’s common stock.F-21NOTE 12 – EARNINGS (LOSS) PER SHARE following tables set forth the computation of basic and diluted earnings (loss) per share for the years ended December 31, 2021 and 2020:
computing net income (loss) per share - basic
computing net income (loss) per share - dilutedNet income (loss)net loss per common share is determinedpresented in conformity with the two-class method required for participating securities and multiple classes of shares. The Company considers its preferred stock to be participating securities.income (loss)loss attributable to common stockholders by the basic and diluted weighted-average number of common shares outstanding during the period. Participating securities are excluded from basic weighted-average common shares outstanding.Company appliesfollowing tables summarizes the two-class method to allocate earnings between common stockcalculation of basic and participating securities.Net income (loss)diluted net loss per diluted share attributable to common stockholders adjusts the basic earnings per share attributable to common stockholders and the weighted-average number of shares of common stock outstanding for the potentialshare:For the Year Ended December 31, 2023 2022 Net loss $ (52,338,959) $ (54,169,029) Net income attributable to participating securities — — Net loss attributable to common stockholders $ (52,338,959) $ (54,169,029) For the Year Ended December 31, 2023 2022 Net loss attributable to common stockholders $ (52,338,959) $ (54,169,029) Weighted average common shares outstanding used in 70,580,604 70,424,819 Weighted average common shares outstanding used in 70,580,604 70,424,819 Net loss per share, basic $ (0.74) $ (0.77) Net loss per share, diluted $ (0.74) $ (0.77) impact of stock options using the treasury-stock method.NOTE 13 – SUBSEQUENT EVENTSEquity AwardsFrom January 1, 2022 through the date of the filing of this Form 10-K, the Company has granted option awards for an aggregate of 2,351,750 shares of common stock to employees with a weighted average exercise price of $2.73.Healx License AgreementOn February 1, 2022, the Company entered into an agreement with Healx, whereby Healx has securedsecurities have been excluded from the Company, an exclusive option to license rights to develop and commercialize gaboxodol. Under the agreement, Healx plans to investigate the compoundcomputations of diluted weighted-average shares outstanding as part of a potential combination therapy for Fragile X syndrome, as well as a treatment for other indications. Should Healx exercise its option, the Company will receive milestone payments for specific clinical, regulatory, and commercial achievements associated with gaboxodol's development. Additionally, Healx will pay the Company tiered royalties on net sales associated with marketed therapies containing gaboxodol. The Company retained the option to become Healx's co-development and co-commercialization partner and will share net profits and losses in lieu of the milestones and royalty payments.Amendment to Lundbeck AgreementOn February 1, 2022, the Company amended the Lundbeck agreement with Amendment No. 3 due to certain changes required tothey would be made following the execution and allowing the effective performance of the Healx License and Option Agreement. Under the terms of Amendment No. 3, the Company will owe Lundbeck a share of all milestone and royalty payments received from Healx if the Company chooses not to opt into the program. If the Company chooses to exercise the option to co-develop and co-commercialize the program with Healx, the Company will owe a share of the net profit share to Lundbeck. If the Healx License and Option Agreement is not executed within thirty (30) days of this Amendment No. 3 or terminated, Article 1 of Amendment No.3 shall become null and void.Out-License Agreement with Marinus PharmaceuticalsF-22On March 1, 2022, we entered into an exclusive patent license agreement with Marinus Pharmaceuticals, Inc. or the Marinus License Agreement. Under the Marinus License Agreement, we granted Marinus an exclusive, non-transferable (except as expressly provided therein), royalty-bearing right and license under certain Ovid patents relating to ganaxolone to develop, make, have made, commercialize, promote, distribute, sell, offer for sale and import licensed products in the territory (which consist of the United States, the European Economic Area, United Kingdom and Switzerland) for the treatment of CDKL5 deficiency disorders in humans. Following the potential date of regulatory approval by the FDA of the first licensed product in the territory, Marinus will, at our option, (i) pay to us the sum of $1,500,000 in cash; or (ii) issue us 123,255 shares of Marinus common stock, par value $0.001 per share. The Marinus License Agreement also provides for payment of royalties from Marinus to us in single digits on net sales of each such licensed product sold..Corporate Expense Reduction InitiativeOn March 15, 2022, the Company announced it is reshaping the organization to focus resources primarily on the advancement of its epilepsy and seizure-related programs. The organizational changes will reduce the Company’s workforce by approximately 20%.For the Year Ended December 31, 2023 2022 Stock options to purchase common stock 15,124,546 12,961,238 Common stock issuable upon conversion of Series A convertible preferred stock 1,250,000 1,250,000