Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
Common Stock, par value $0.001 per share |
| The Nasdaq Capital Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrantRegistrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒☒
Indicate by check mark if the registrantRegistrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒☒
Indicate by check mark whether the registrant:Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrantRegistrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ NO ☐☒ No ☐
Indicate by check mark whether the registrantRegistrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrantRegistrant was required to submit such files). Yes ☒ No ☐☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ | Accelerated filer | ☐ | |||
Non-accelerated filer | ☒ | Smaller reporting company | ☒ | |||
Emerging growth company | ☐ | |||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrantRegistrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐☐No No ☒☒
BasedThe aggregate market value of the voting and non-voting common equity held by non-affiliates of the Registrant as of June 30, 2023 was $10.3 million, based on the closing price of the registrant’s common stock on the last business day of the registrant’s most recently completed second fiscal quarter, which was June 30, 2021, the aggregate market value of its common stock (based on a closing price of $11.65 per share on June 30, 2021 as reported on the Nasdaq Capital Market) held by non-affiliates was approximately $110,386,092.
As of March 17, 2022, the registrant had 10,829,749shares of common stock $0.001 par value per share, issued and outstanding.on The Nasdaq Capital Market on June 30, 2023.
The number of shares of Registrant’s Common Stock outstanding as of March 18, 2024 was 10,962,460.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s Proxy Statement for its 20222024 Annual Meeting of Stockholders, which the registrant intends to file with the Securities and Exchange Commission not later than 120 days after the registrant’s fiscal year ended December 31, 2021,2023, are incorporated by reference into Part III of this Annual Report on Form 10-K.
Yumanity Therapeutics, Inc.
Table of Contents
Index
Page | ||
1 | ||
Item 1. |
| |
Item 1A. |
| |
Item 1B. |
| |
Item 1C. | 92 | |
Item 2. |
| |
Item 3. |
| |
Item 4. |
| |
PART II | ||
Item 5. |
| |
Item 6. |
| |
Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
Item 7A. |
| |
Item 8. |
| |
Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
|
Item 9A. |
| |
Item 9B. |
| |
Item 9C. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections |
|
PART III | ||
Item 10. |
| |
Item 11. |
| |
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
|
Item 13. | Certain Relationships and Related Transactions, and Director Independence |
|
Item 14. |
| |
PART IV | ||
Item 15. |
| |
Item 16. |
| |
| ||
i
EXPLANATORY NOTE
On December 22, 2020, Proteostasis16, 2022, Yumanity Therapeutics, Inc. (“Proteostasis”Yumanity”) completed its previously announced merger transaction with Yumanity,Kineta Operating, Inc. (formerly Yumanity Therapeutics,Kineta, Inc.) (“Private Kineta”) in accordance with the terms of the Agreement and Plan of Merger, and Reorganization, dated as of August 22, 2020,June 5, 2022, as amended on November 6, 2020December 5, 2022 (the “Merger Agreement”), by and among PangolinYumanity, Private Kineta and Yacht Merger Sub, Inc., a wholly-owned subsidiary of ProteostasisYumanity (“Merger Sub”), Yumanity Holdings, LLC (“Holdings”) and Yumanity, Inc., pursuant to which Merger Sub merged with and into Yumanity, Inc.,Private Kineta, with Yumanity, Inc.Private Kineta surviving such merger as a wholly ownedwholly-owned subsidiary of ProteostasisYumanity (the “Merger”). Immediately prior to the effective time ofThe surviving corporation from the Merger Holdingssubsequently merged with and into Yumanity, Inc. and Yumanity, Inc. continued to exist asKineta Operating, LLC, with Kineta Operating, LLC being the surviving corporation. On December 22, 2020,16, 2022, in connection with, and prior to the completion of, the Merger, ProteostasisYumanity effected a 1-for-201-for-7 reverse stock split of its common stock (the “Reverse Stock Split”). Immediately following the Merger, ProteostasisYumanity changed its name to “Yumanity Therapeutics,“Kineta, Inc.”
Unless the context otherwise requires, references to the “Company,” “Yumanity,“Kineta,” the “combined organization,” “we,” “our” or “us” in this reportAnnual Report on Form 10-K refer to HoldingsPrivate Kineta and its subsidiarysubsidiaries prior to completion of the Merger and to Yumanity Therapeutics,Kineta, Inc. and its subsidiarysubsidiaries after completion of the Merger. In addition, references to “Proteostasis” or “PTI”“Yumanity” refer to the registrant prior to the completion of
the Merger.
For accounting purposes, the
The Merger was treatedhas been accounted for as an “asset acquisition” undera reverse merger and asset acquisition in accordance with U.S. generally accepted accounting principles in(“U.S. GAAP”). Under this method of accounting, Private Kineta was deemed to be the United States (“GAAP”) and Yumanity was considered the acquirer. Accordingly, Yumanity’s historical results of operations will replace the Proteostasis historical results of operationsaccounting acquirer for all periods prior to the Merger and, for all periods following the Merger, the results of operations of the combined organization will be included in the Company’s financial statements.reporting purposes. Following the Merger, the business conducted by YumanityPrivate Kineta became ourthe Company’s primary business.
Except as otherwise noted, references to “common stock” in this report refer to common stock, $0.001 par value per share, of the Company.
iiCAUTIONARY STATEMENT
SummaryIn February 2024, the Company initiated a process to explore a range of strategic alternatives to maximize shareholder value. Potential strategic alternatives that may be evaluated include sale of assets of the Material Risks Associated with Our Business
We are subject to various risks associated with our businesses and industries. These risks include the following:
The summary risk factors described above should be read together with the text of the full risk factors below, in the section entitled “Risk Factors” in Part I, Item 1A. and the other information set forth in this Annual Report on Form 10-K, including our consolidated financial statements and the related notes, as well as in other documents that we file with the U.S. Securities and Exchange Commission. The risks summarized above or described in full below are not the only risks that we face. Additional risks and uncertainties not precisely known to us, or that we currently deem to be immaterial may also materially adversely affect our business, financial condition, results of operations and future growth prospects.US Bankruptcy Code.
iii
CAUTIONARYKineta, Inc. cautions that trading in the Company’s securities is highly speculative and poses substantial risks. Trading prices for the Company’s securities may bear little or no relationship to the actual value realized, if any, by holders of the Company’s securities. Accordingly, the Company urges extreme caution with respect to existing and future investments in its securities.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-lookingcertain statements that constitute “forward-looking statements” within the meaning of Section 27A of the U.S. Private Securities Litigation Reform Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and the “safe harbor” provisions of the United States Private Securities Litigation Reform Act of 1995. In some cases, you can identify these forward-looking statements by the use of terms such as “expect,” “will,” “continue,” “believe,” “estimate,” “aim,” “project,” “intend,” “should,” “is to be,” or similar expressions, and variations or negatives of these words, but the absence of these words does not mean that involve substantial risks and uncertainties.a statement is not forward-looking. All statements other than statements of historical fact containedare statements that could be deemed forward-looking statements. These forward-looking statements are subject to known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance, or achievements to differ materially from results expressed or implied in this Annual Report including statements regarding our strategy, future operations, future financial position, future revenue, projected costs, prospects, plans and objectives of management, areon Form 10-K. The following factors, among others, could cause actual results to differ materially from those described in these forward-looking statements. The words “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “might,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would,” or the negative of these words or other similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words.
The forward-looking statements in this Annual Report include, among other things, statements about:statements:
1
iv
We may not actually achieve the plans, intentions or expectations disclosed in our
The forward-looking statements and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. We have included important factors in the cautionary statements includedcontained in this Annual Report particularly inon Form 10-K and the “Risk Factors” section,documents incorporated herein by reference are based on our current expectations and beliefs concerning future developments and their potential effects on our business. There can be no assurance that future developments affecting our business will be those that we believe couldhave anticipated. These forward-looking statements involve a number of risks, uncertainties (some of which are beyond our control) or other assumptions that may cause actual results or eventsperformance to differbe materially different from those expressed or implied by these forward-looking statements. These risks and uncertainties include, but are not limited to, those factors described under the forward-looking statementscaption “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K and under similar headings in the documents that we make. are incorporated by reference herein.
2
Moreover, we operate in a very competitive and rapidly changing environment. New risk factorsrisks and uncertainties may emerge from time to time and it is not possible for managementus to predict all such risk factors, and uncertainties. Ournor can we assess the effect of all such risk factors on our business or the extent to which any factor or combination of factors may cause actual results to differ materially from those contained in any forward-looking statements do not reflectstatements. Should one or more of these risks or uncertainties materialize, or should any of the potential impact of any future acquisitions, mergers, dispositions, collaborations, joint ventures, or investments weassumptions prove incorrect, actual results may make or enter into.vary in material respects from those projected in these forward-looking statements.
You should read this Annual Report and the documents that we file with the Securities and Exchange Commission with the understanding that our actual future results may be materially different from what we expect. The forward-looking statements containedmade by us in this Annual Report are madeon Form 10-K and the documents incorporated herein by reference speak only as of the date of this Annual Report,such statement. Except to the extent required under the federal securities laws and rules and regulations of the U.S. Securities and Exchange Commission (the “SEC”), we do not assumedisclaim any obligation to update any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events. In light of these risks and uncertainties, there is no assurance that the events or results suggested by the forward-looking statements will in fact occur, and you should not place undue reliance on these forward-looking statements.
Although we undertake no obligation to revise or update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by applicable law.law, you are advised to consult any additional disclosures we make in the documents that we file with the SEC.
v
3
PART I
All brand names or trademarks appearing in this report are the property of their respective owners. Except where the context otherwise requires or where otherwise indicated, the terms “we,” “us,” “our,” “our company,” “the company,” and “our business” refer to Yumanity Therapeutics, Inc. and its consolidated subsidiary.
ITEMItem 1. BUSINESSBusiness.
Overview
On February 29, 2024, Kineta announced that it had completed a review of its business, including the status of its programs, resources and
capabilities. Following this review, Kineta determined that it would implement a significant corporate restructuring to substantially reduce expenses and preserve cash. The restructuring includes a reduction in its workforce by approximately 64% and the termination of enrollment of new patients in its ongoing VISTA-101 Phase 1/2 clinical trial evaluating KVA12123 in patients with advanced solid tumors. Patients currently enrolled in the trial will be permitted to continue to participate. The Company made this decision, in part, because certain investors have indicated they will not be able to fulfill their contractual obligation to consummate the Private Placement (as defined below). Kineta has initiated a process to explore a range of strategic alternatives to maximize shareholder value. Potential strategic alternatives that may be evaluated include sale of assets of the Company, a sale of the Company, licensing of assets, a merger, liquidation or other strategic action. There is no set timetable for this process, and there can be no assurance that this strategic review process will result in the pursuit of any transaction or that any transaction, if pursued, will be completed. Additionally, there can be no assurances that any particular course of action, business arrangement or transaction, or series of transactions, will be pursued, successfully consummated, or lead to increased stockholder value. If the strategic process is unsuccessful, the Company's Board may decide to pursue a liquidation or obtain relief under the US Bankruptcy Code. In the event of such liquidation, bankruptcy case, or other wind-down event, holders of the Company's securities will likely suffer a total loss of their investment.
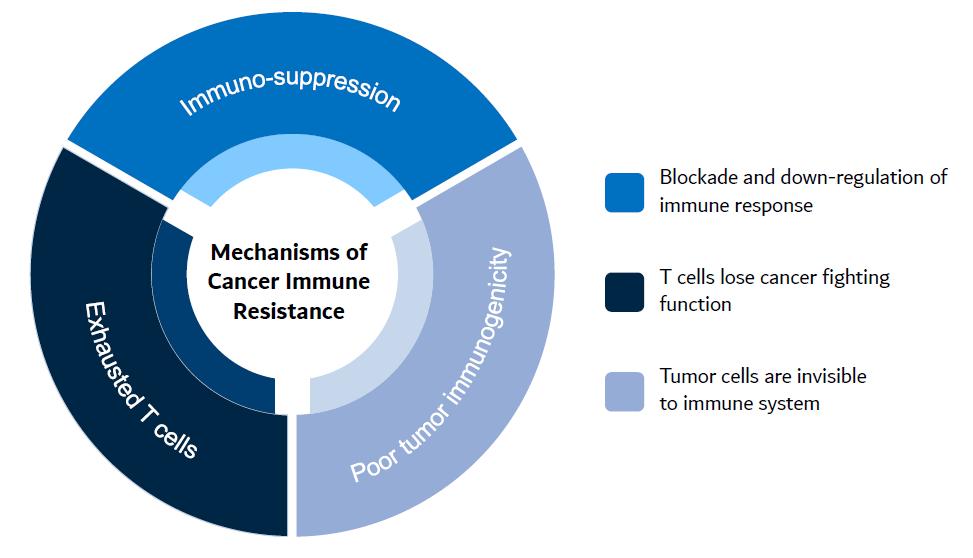
Kineta is a clinical-stage biotechnology company with a mission to develop next-generation immunotherapies that transform patients’ lives. Kineta has leveraged its expertise in innate immunity and is focused on discovering and developing potentially differentiated immunotherapies that address the mechanisms of cancer immune resistance:
Kineta’s pipeline of potentially next-generation immunotherapies includes (i) KVA12123, a monoclonal antibody (“mAb”), immunotherapy targeting VISTA (V-domain Ig suppressor of T cell activation) and (ii) an anti-CD27 agonist mAb immunotherapy. These novel immunotherapies have the potential to address disease areas with unmet medical needs and significant commercial potential.
KVA12123 is a VISTA blocking immunotherapy in development as an intravenous infusion dosed every two weeks. Kineta dosed the first patient in a Phase 1/2 clinical trial of KVA12123 in the United States in April 2023. The ongoing Phase 1/2 clinical study is designed to evaluate KVA12123 as a monotherapy and in combination with the immune checkpoint inhibitor pembrolizumab in patients with advanced solid tumors. Initial monotherapy safety, pharmacokinetic and biomarker data were presented at the Society for Immunotherapy of Cancer’s (SITC) annual meeting in November 2023. KVA12123 was designed to be a differentiated VISTA blocking immunotherapy to address the problem of immunosuppression in the tumor microenvironment (“TME”). It is a fully human engineered IgG1 monoclonal antibody that binds to VISTA through a unique epitope and across neutral and acidic pHs. KVA12123 may be an effective immunotherapy for many types of cancer, including non-small cell lung cancer (“NSCLC”), colorectal cancer (“CRC”), ovarian cancer (“OC”), renal cell carcinoma (“RCC”) and head and neck squamous cell carcinoma (“HNSCC”). These indications represent a significant unmet medical need with a large worldwide commercial opportunity for KVA12123.
Kineta is also developing an anti-CD27 agonist mAb immunotherapy to address the problem of exhausted T cells. The nominated lead candidate is a fully human mAb that demonstrates nanomolar (“nM”) binding affinity to CD27 in humans. In preclinical studies,Kineta’s lead anti-CD27 candidate demonstrated antitumor efficacy as a single agent and in combination with other immunotherapies in multiple solid and hematological preclinical tumor models. CD27 is a clinically validated target that may be an effective immunotherapy for advanced solid tumors including RCC, CRC and OC. Kineta continues to conduct preclinical studies to optimize its lead anti-CD27 agonist mAb clinical candidate and to evaluate it in combination with other checkpoint inhibitors.
According to Market Data Forecast, the immuno-oncology market generated sales of approximately $111 billion in 2023 and is forecast to reach $201 billion in 2028. If Kineta successfully completes the clinical trial program for KVA12123 and if Kineta subsequently obtains regulatory approval for KVA12123, Kineta will focus on initial target indications in NSCLC, CRC and OC. Initially the clinical development of KVA12123 will be as a second-line therapy in these indications. These three cancer therapy segments represent a forecasted $48 billion market opportunity in 2027 according to GlobalData.
Kineta is a leader in the field of innate immunity and is focused on developing potentially differentiated immunotherapies. With KVA12123 in clinical development and the lead anti-CD27 agonist mAb in preclinical development, Kineta believes it is positioned to achieve multiple value-driving catalysts. Kineta has assembled an experienced management team, a seasoned research and clinical team, an immuno-oncology focused scientific advisory board, and a leading intellectual property position to advance its pipeline of potential novel immunotherapies for cancer patients.
Kineta’s Strategy
Kineta’s immediate strategy is to continue its process to explore strategic alternatives.
4
Kineta has initiated a process to explore a range of strategic alternatives to maximize shareholder value. Potential strategic alternatives that may be evaluated include a sale or merger of the Company, the sale of all or a portion of the Company’s assets and/or intellectual property, or securing additional financing or partnerships that would enable further development of our programs. There can be no assurance that this strategic review process will result in the Company pursing any transaction or that any transaction, if pursed, will be completed. If the strategic process is unsuccessful, the Company's Board may decide to pursue a liquidation or obtain relief under the US Bankruptcy Code.
Overview
We are a biopharmaceutical company focused on the discovery and development of innovative, disease-modifying therapies for neurodegenerative diseases. Neurodegenerative diseases cause a progressive loss of structure and function in the brain, leaving patients with devastating damageKineta’s mission is to their nervous system and widespread functional impairment. Although treatments may help relieve some of the physical or mental symptoms associated with neurodegenerative diseases, few of the currently available therapies slow or stop the continued loss of neurons, resulting in a critical unmet need. We are specificallydevelop next-generation immunotherapies that transform patients’ lives. Kineta is focused on developing fully human antibodies that address the mechanisms of cancer immune resistance. Kineta is a leader in developing fully human antibody drugs directed against novel therapies to treat devastating conditions, either with large or orphan disease markets, such as Parkinson’s disease, dementia with Lewy bodies, multiple system atrophy, or MSA, amyotrophic lateral sclerosis, or ALS (also known as Lou Gehrig’s disease),innate immune targets. Kineta’s focus on innate immunity differentiates it from other immuno-oncology companies that are primarily focused on adaptive immunity and frontotemporal lobar dementia, or FTLD.T cell focused therapies.
Neurodegenerative diseases exertThe key element of Kineta’s strategy to achieve this mission is to advance the clinical development of Kineta’s lead product candidates. Kineta’s most advanced drug candidate, KVA12123, is a heavy societal burden worldwide and represent one of the largest global healthcare challenges of our time. With an increasingly aging population, diseases affecting the brain and central nervous system are rising in prevalence, with overwhelming personal and economic consequences that exact a toll on patients, caregivers and treatment providers. The rising prevalence of neurodegenerative disease and a lack of disease-modifying treatments has resultedpotentially differentiated VISTA blocking immunotherapy currently being tested in a significant and growing unmet medical need. It is estimated that more than 60 million people worldwide suffer from neurodegenerative diseases, which is expected to almost double every 20 years. Global costsPhase 1/2 clinical trial. Kineta’s IND application for treating these diseases are greater than $1 trillion annually.
In February 2022, we announced that we are exploring strategic alternatives to enhance shareholder value and engaged H.C. Wainwright as our exclusive financial advisor to assist in this process. No timetable has been established for the completion of this process, and we do not expect to disclose developments unless and until the Board of Directors has concluded that disclosure is appropriate or required.
In February we also began implementation of a strategic restructuring with the objective of preserving capital. As part of the restructuring, we are eliminating approximately 60% of its workforce and took other actions, including reducing our office and laboratory space, to reduce expenditures.
Our lead program, YTX-7739, is in development for the potential treatment and disease modification of Parkinson’s disease. YTX-7739 targets an enzyme known as stearoyl-CoA desaturase, or SCD. Inhibition of SCD in multiple cellular systems, including patient-derived neurons, as well as in a novel mouse model of Parkinson’s disease, has been demonstrated to reverse the toxicity of misfolded alpha-synuclein, or α-synuclein, a protein strongly associated with Parkinson’s disease.
In January 2022,KVA12123 was accepted by the U.S. Food and Drug Administration (FDA), placed a partial clinical hold on multidose clinical trials of YTX-7739. The partial clinical hold suspends initiation of multiple dose clinical trials(the “FDA”) in the U.S. until the FDA’s concerns have been addressed. We anticipate working closely with the FDA to adequately address their concerns. The FDA’s action has not impacted our on-going multidose clinical trial in Europe and is permitting our planned single dose formulation clinical trial to proceed.
On November 10, 2021, we announced the top-line results of2022. Kineta initiated a Phase 1b clinical trial of YTX-77391 dose escalation study with KVA12123 as a monotherapy and in combination with pembrolizumab in patients with mild-to-moderate Parkinson’s disease, which assessedadvanced solid tumors in the safety, tolerability and pharmacokinetics and pharmacodynamicsfourth quarter of YTX-7739. The Phase 1b2022. Initial data from this clinical trial was a randomized, placebo-controlled, double-blind multi dose study to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics of YTX-7739. Data were reported from 20 patients with mild-to-moderate Parkinson’s disease. Patients received once-daily oral doses of YTX-7739 (20 mg or placebo) for 28 days. YTX-7739 was shown to inhibit its primary target, SCD, an enzyme whose inhibition has been closely linked to neuronal survival and improved motor function in a Parkinson’s disease model.
After 28 days of treatment, the 20 mg dose given once-daily reduced the fatty acid desaturation index (FA-DI), a biomarker of SCD inhibition, by approximately 20%-40%, the range expected to be clinically relevant based on preclinical studies. Target engagementreleased in the cerebrospinal fluid suggested that YTX-7739 crossed the blood-brain barrier. Additionally, the PK/PD profilefourth quarter of YTX-7739 was consistent with previous2023. Kineta is also conducting preclinical studies and we believe informs dose selection for future studies.its lead anti-CD27 agonist mAb immunotherapy.
YTX-7739 was generally well toleratedUnmet medical needs for cancer patients
With improvements in screening and early diagnosis, cancer patient survival has increased considerably, since tumors that are detected and treated early with surgery, conventional chemotherapy or radiation therapy can often be cured. However, for patients who are diagnosed with more advanced or difficult to treat tumors, conventional therapies are often ineffective, and the chance of long-term survival is seriously reduced.
The discovery of novel immune checkpoint inhibitors (“CPIs”) targeting the B7/CD28 family of proteins, including programmed cell-death protein 1 (“PD1”), programmed death-ligand 1 (“PD-L1”) and cytotoxic T lymphocyte associated protein 4 (“CTLA4”) has completely revolutionized cancer treatment. These new immunotherapies provide hope for patients with advanced tumors to achieve long-term remission after treatment.
However promising the existing CPIs are in treating certain clinical indications, several key deficiencies of this approach have become apparent during clinical development and post-marketing use:
Addressing the major challenges with increased Parkinson’s symptoms, 2 patients with lower back pain, 1 patient with headache, 1 patient with myalgia, 1 patient with insomnia, 1 patient withcurrent cancer therapy
1There remains a significant unmet need to improve overall and long-term survival for cancer patients, especially those diagnosed with later stage cancers. New innovations and enhancements to the currently available therapies are urgently needed to address the treatment gaps.
Kineta is developing next-generation immunotherapies to address the major challenges with current cancer treatments. Kineta aims to improve outcomes for cancer patients by solving the major problems of cancer immune resistance.
Kineta’s development approach involves first exploring the main mechanisms of cancer resistance to existing therapies, including CPIs. Kineta focuses on the importance of the innate immune response to achieve a complete adaptive immune response. Kineta has identified that colder, less inflamed and more difficult to treat tumors have three characteristics that Kineta believes can be addressed by its pipeline. Figure 1 below represents the three major mechanisms of cancer immune resistance to therapies that Kineta’s pipeline is designed to address.
ligament strain,5
Figure 1. The major challenges with current cancer therapies
Kineta’s Product Candidate Pipeline
Kineta is devoted to the discovery and development of fully human monoclonal antibodies that target novel innate immune regulators. Kineta is developing two novel innate immune-targeted therapies that may address advanced solid tumors:
Figure 2. Kineta’s pipeline
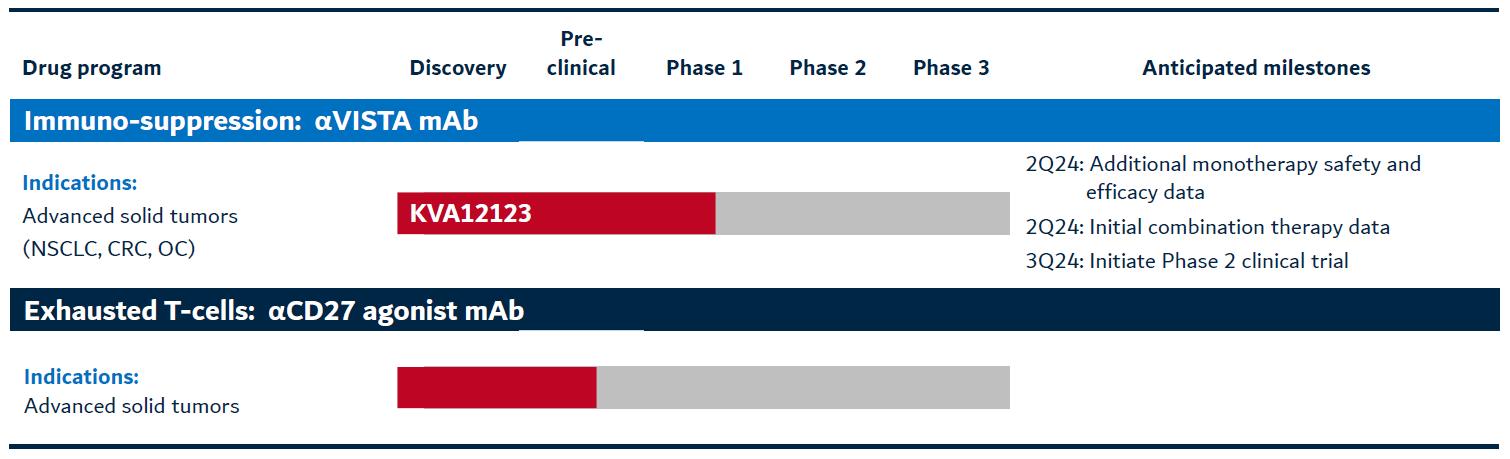
KVA12123: VISTA blocking immunotherapy
KVA12123 is designed to be a differentiated VISTA blocking immunotherapy to address the problem of immunosuppression in the TME. KVA12123 is a VISTA blocking immunotherapy in development as an infusion dosed every two weeks. The drug is being evaluated in an ongoing Phase 1/2 clinical trial as a monotherapy and in combination with pembrolizumab in patients with advanced solid tumors. Through the combination of unique epitope binding and an optimized IgG1 Fc region, KVA12123 demonstrates strong monotherapy tumor growth inhibition in preclinical models without evidence of cytokine release syndrome (“CRS”) in clinical trial participants. KVA12123 also exhibits an excellent safety profile in initial clinical trial cohorts. KVA12123 has been shown to de-risk the VISTA target and provides a novel approach to address the problem of immunosuppression in the TME with a mechanism of action that is differentiated and complementary to T cell focused therapies. KVA12123 may be an effective immunotherapy for many types of cancer including NSCLC, CRC, OC, RCC and HNSCC.
VISTA (V-domain Ig suppressor of T cell activation) is a negative immune checkpoint that suppresses T cell function in a variety of solid tumors. High VISTA expression in tumor correlates with poor survival in cancer patients and has been associated with a lack of response to other immune checkpoint inhibitors. Blocking VISTA induces an efficient polyfunctional immune response to address immunosuppression and drives anti-tumor responses.
There is a strong clinical rationale for targeting VISTA with an antibody immunotherapy. The innate immune target VISTA is highly expressed in
6
NSCLC, OC, colon cancer, melanoma, pancreatic cancer and gastric cancer and correlates with poor outcomes in cancer patients. VISTA is also up-regulated after CPI therapy (e.g., Keytruda®) and is associated with treatment failure (Figure 3).
Figure 3. VISTA expression is associated with poor overall survival and treatment failure with CPI
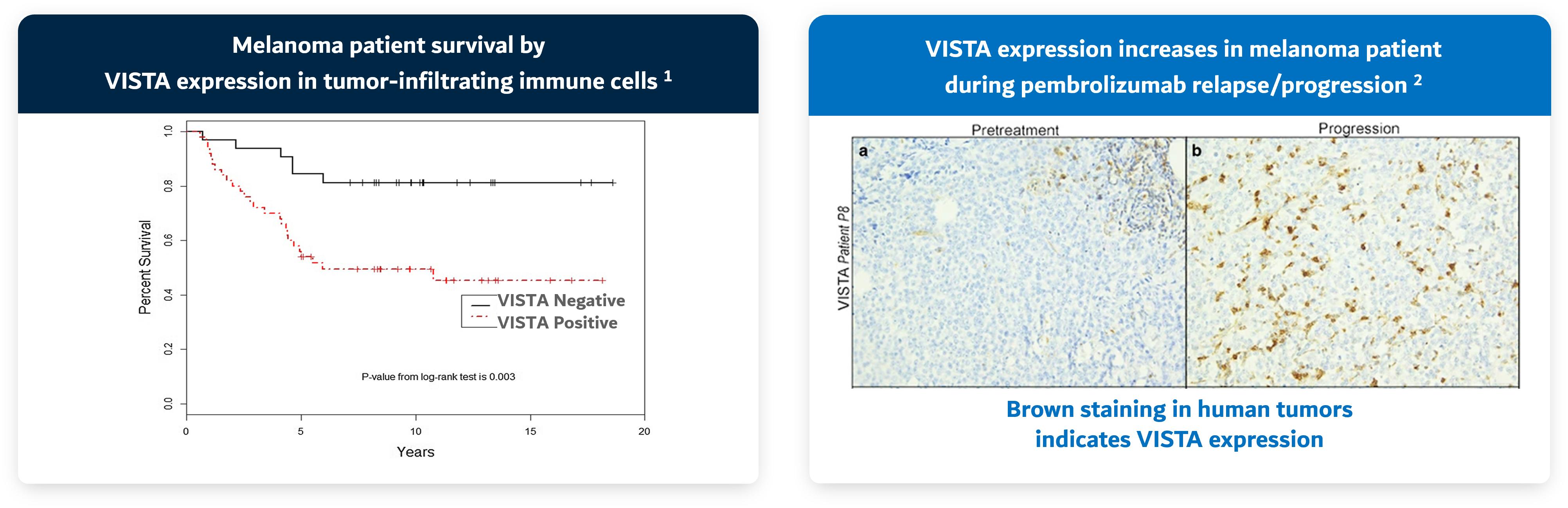
Sources: 1. Kuklinski et al. 2018; 2. Kakavand et al. 2017
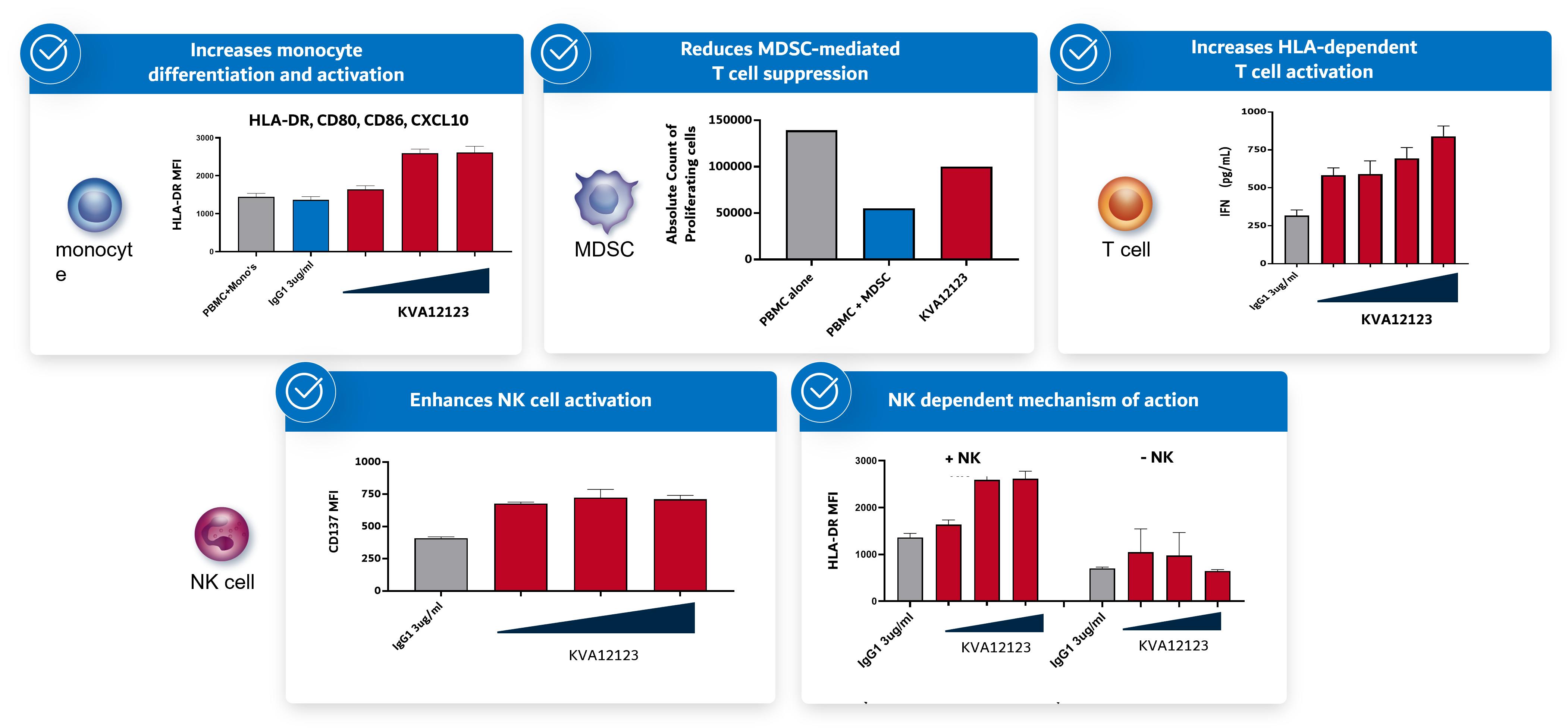
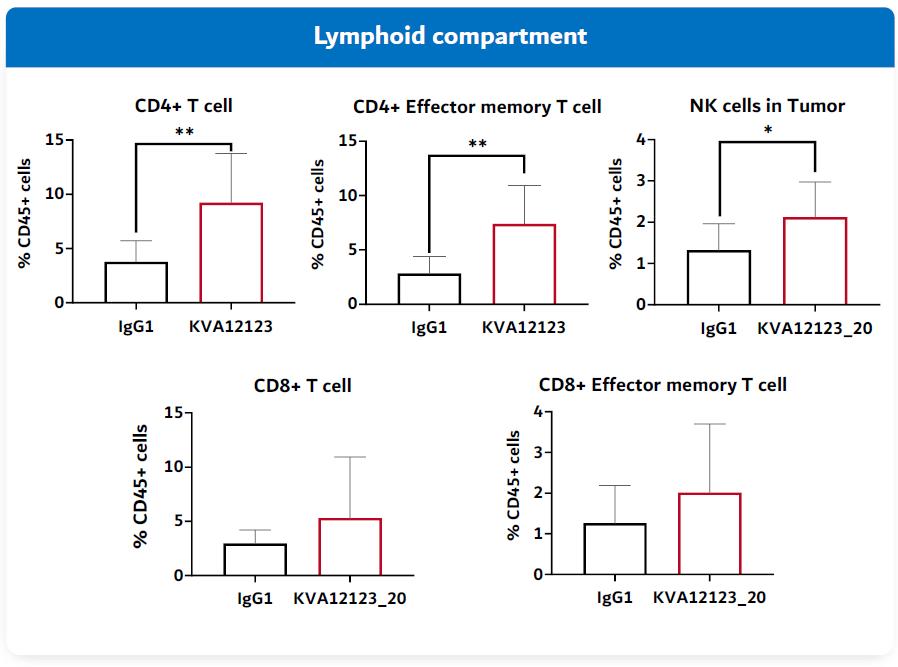
Blocking VISTA drives an efficient polyfunctional immune response to turn cold tumors hot. VISTA is a novel immuno-oncology target due to its unique expression and activity. First, high VISTA expression on immunosuppressive myeloid cells (tumor associated macrophages and myeloid-derived suppressor cells (MDSCs)) is consistent across tumor types, making it a relevant target across multiple types of cancer. Re-programmed myeloid cells can drive tumor inflammation. VISTA-blockade decreases immune suppression and provides single agent tumor growth inhibition and also improves efficacy of T cell focused therapies like anti-PD(L)1 and anti-CTLA4.
Second, blocking VISTA induces activation of dendritic cells and natural killer (“NK”) cells and ultimately proliferation and infiltration of T cells into the tumor. The combination of myeloid, NK and T cell responses can reverse immunosuppression and drive anti-tumor activity. While many immuno-oncology targets address either T cell or myeloid functions, VISTA has the potential to regulate both.
KVA12123 has demonstrated activity on important innate and adaptive immune cells (Figure 4) present in the TME in preclinical assays (in vitro).
Figure 4. Blocking VISTA with KVA12123 activates both innate and adaptive immune cells
Additionally, KVA12123 drives an integrated innate and adaptive anti-tumor immune response in tumor models like MB49 bladder tumor (ex vivo).
Figure 5. Blocking VISTA with KVA12123 drives anti-tumor responses in MB49 model
7
In preclinical models, KVA12123 has been observed to show strong single agent tumor growth inhibition in poorly immunogenic “cold tumors” models and complementary tumor growth inhibition when dosed in combination with CPIs like PD-1 or CTLA-4 as shown in Figure 6 below. Studies in preclinical tumor models demonstrate the tumor growth inhibition of Kineta’s anti-VISTA antibody as a single agent in bladder cancer, T cell lymphoma and colon cancer models. In combination studies, Kineta’s anti-VISTA antibody acts synergistically in combination with anti-PD-1 therapy to inhibit tumor growth in preclinical colon cancer and bladder cancer models.
8
Figure 6. KVA12123 demonstrates single agent tumor growth inhibition and in combination with PD-1 in preclinical models
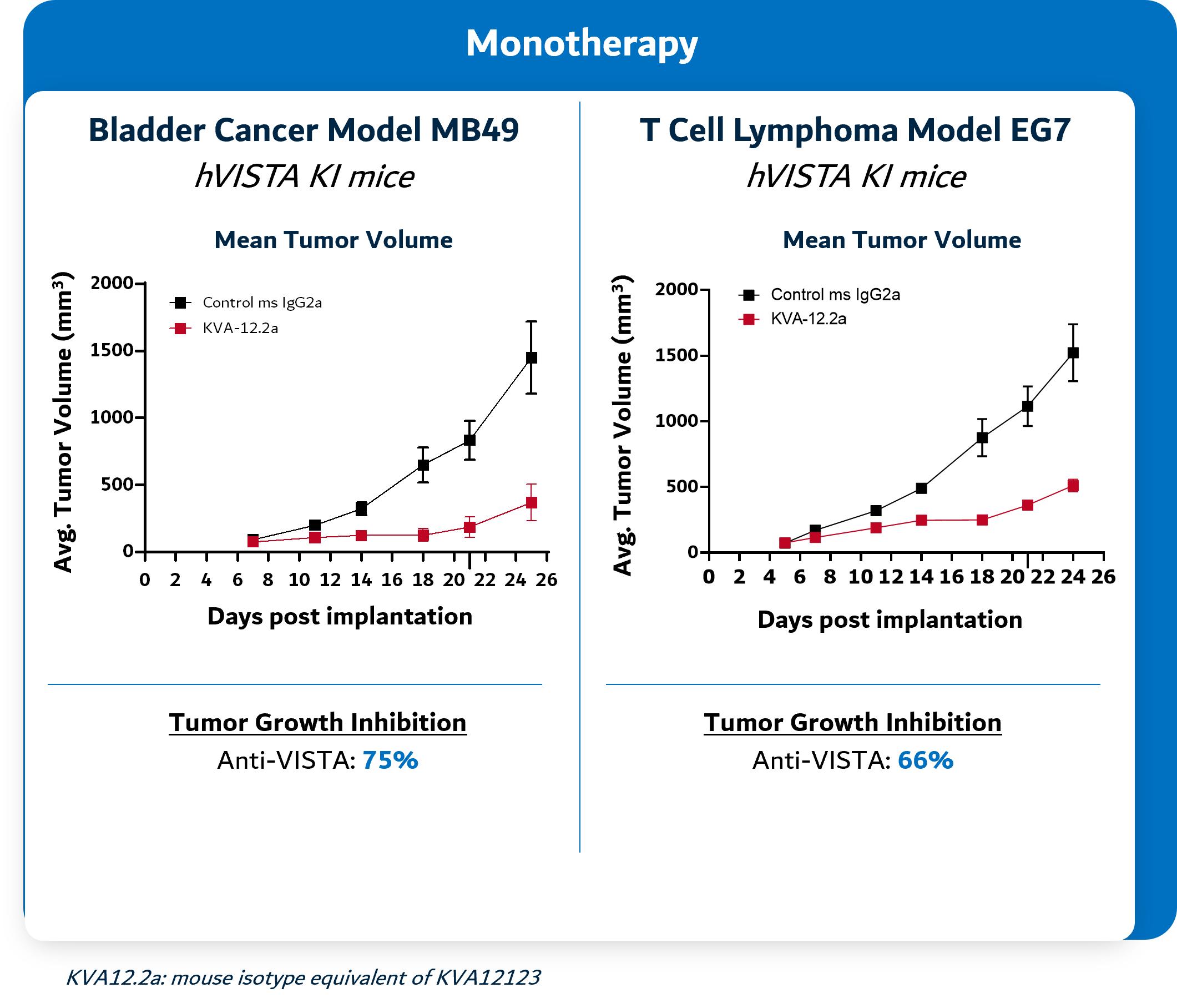
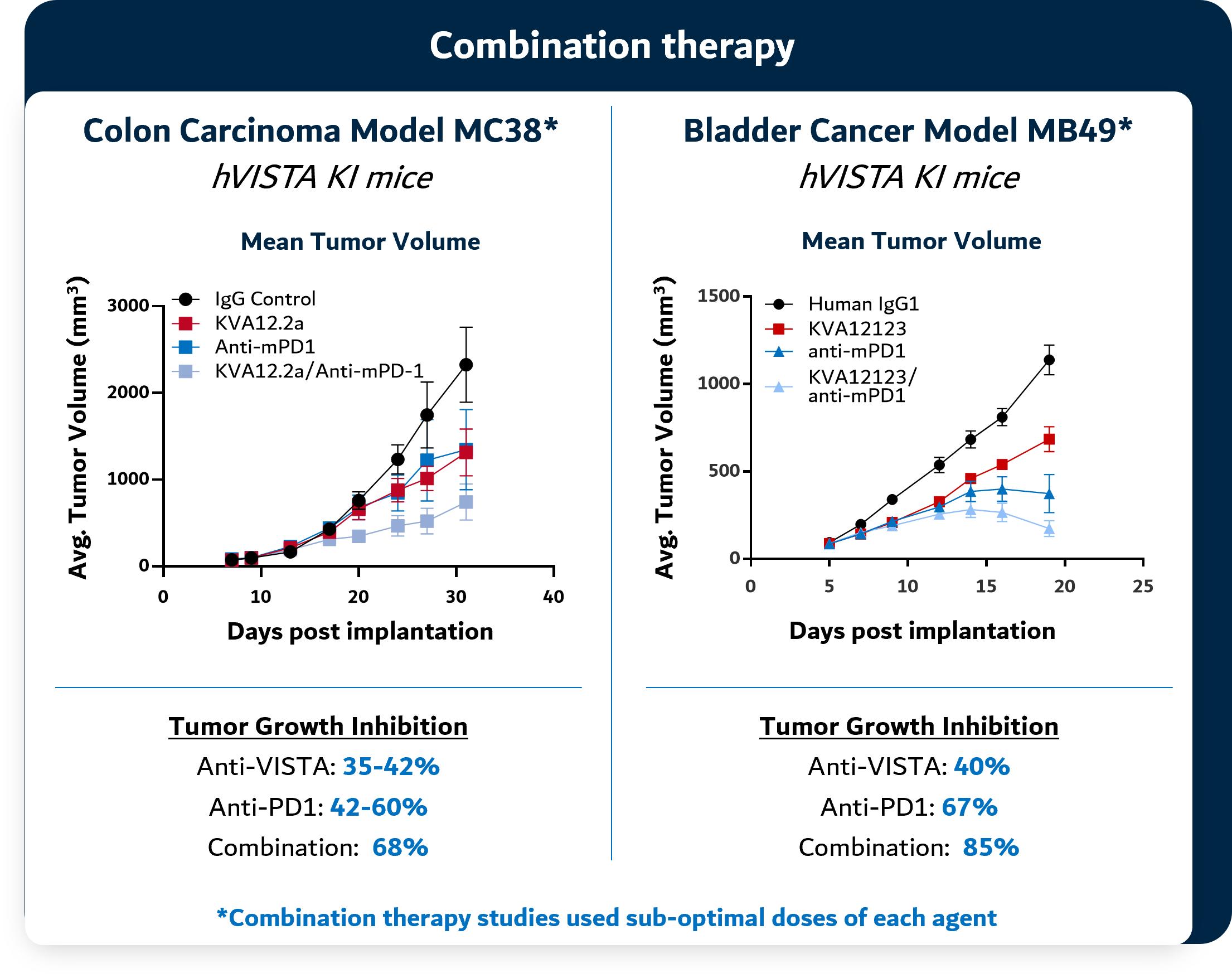
Source: Kineta data
Kineta has completed multiple, single and repeat-dose toxicology studies in non-human primates (“NHP”) with doses of KVA12123 up to 100 mg/kg (>100-fold safety margin over target human exposure). KVA12123 was observed to be well-tolerated in NHP toxicology studies with no mortality, no overt clinical signs or weight loss, no treatment-related findings and no change in CRS-associated cytokine levels (IL6 or TNFα). IL6 and TNFα are indicators of CRS.
9
KVA12123 Competitive Differentiation
The competitive landscape for VISTA blocking immunotherapies includes six primary companies (Kineta, Inc., Curis, Inc., Pierre Fabre Laboratories, Hummingbird Bioscience Pte. Ltd., PharmAbcine, Inc. and Sensei Biotherapeutics, Inc.) with assets in Phase 1 clinical development. Other discovery stage assets have been announced by Apexigen, Inc. and Five Prime Therapeutics (acquired by Amgen Inc.)/Bristol Myers Squibb Company (“BMS”). See Figure 7 below for more information on competitive products in development.
Kineta is developing a VISTA blocking immunotherapy that is designed to be differentiated from competitive products by the following:
Figure 7. KVA12123: Differentiated VISTA blocking immunotherapy
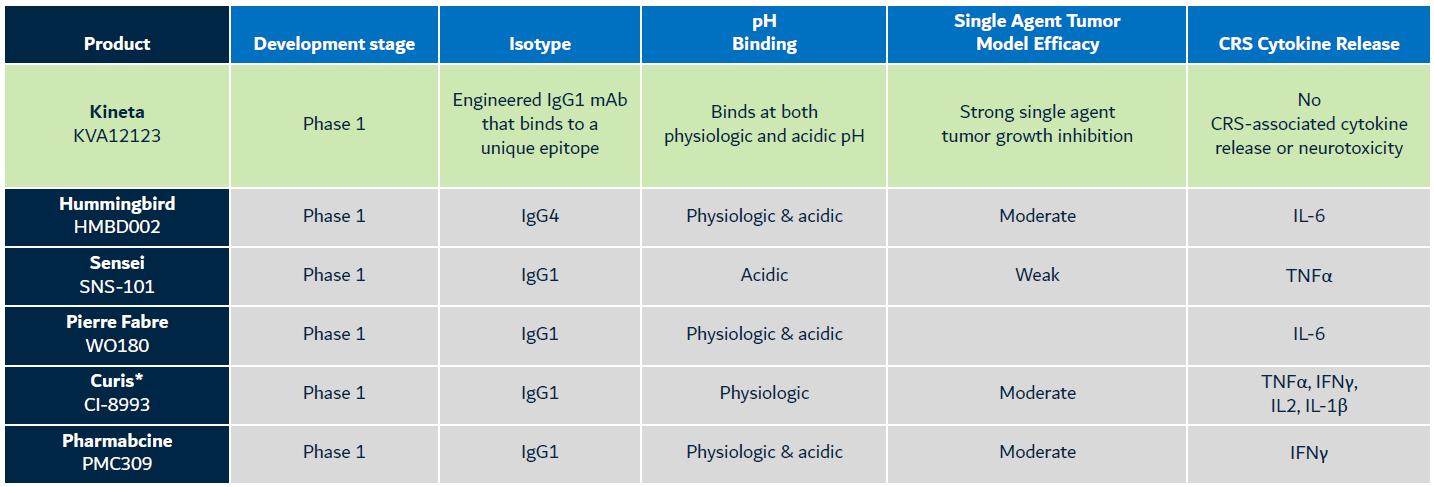
Other discovery stage programs: Apexigen and Five Prime Therapeutics/BMS. Empty cells indicate no public data available
* Curis announced 11/9/2022: “Concentrating its resources to focus on and accelerate emavusertib”, the company’s lead asset and “deprioritization of other programs” (CI-8993)
Kineta believes that KVA12123 may be differentiated as the only antibody in its class with strong single-agent tumor growth inhibition in the absence of cytokine-mediate toxicity.
Figure 8. KVA12123 binds at physiologic and acidic pH
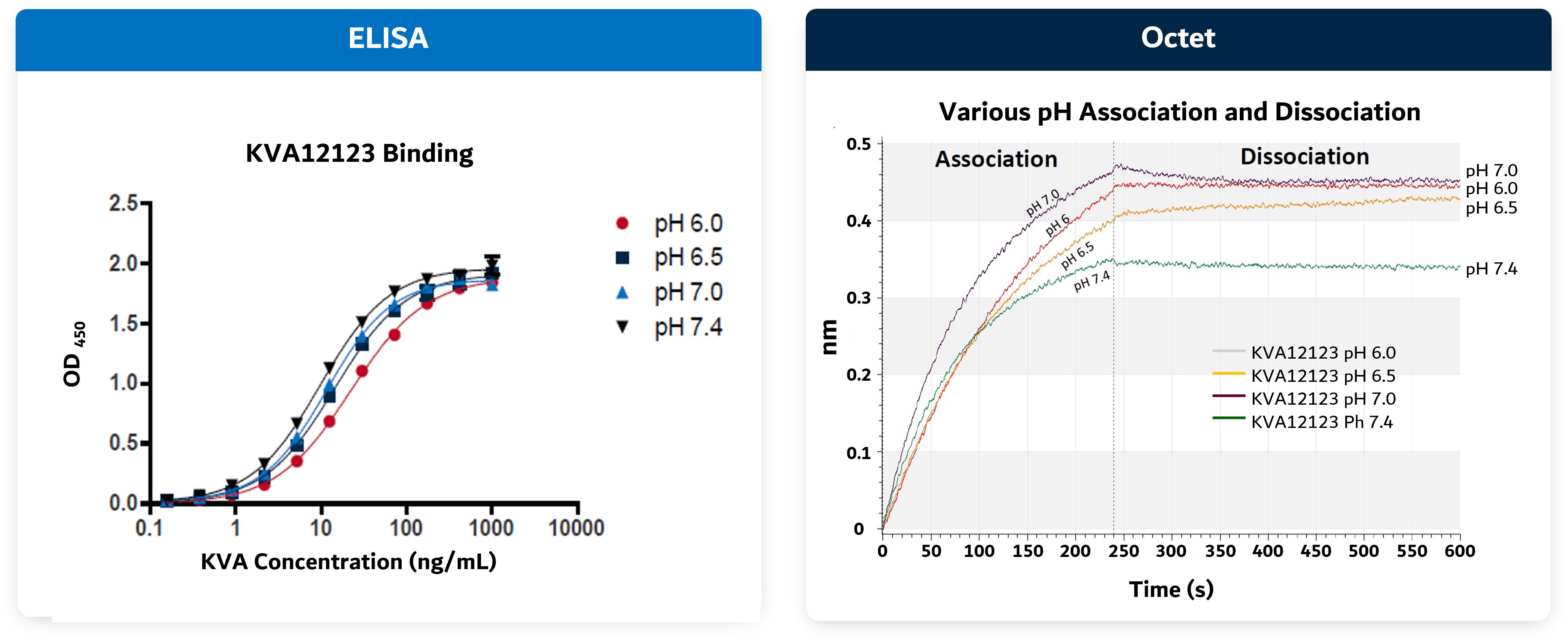
Source: Kineta data
10
Figure 9. KVA12123: No CRS-associated cytokine release seen in preclinical models in NHP toxicology and in human whole blood studies
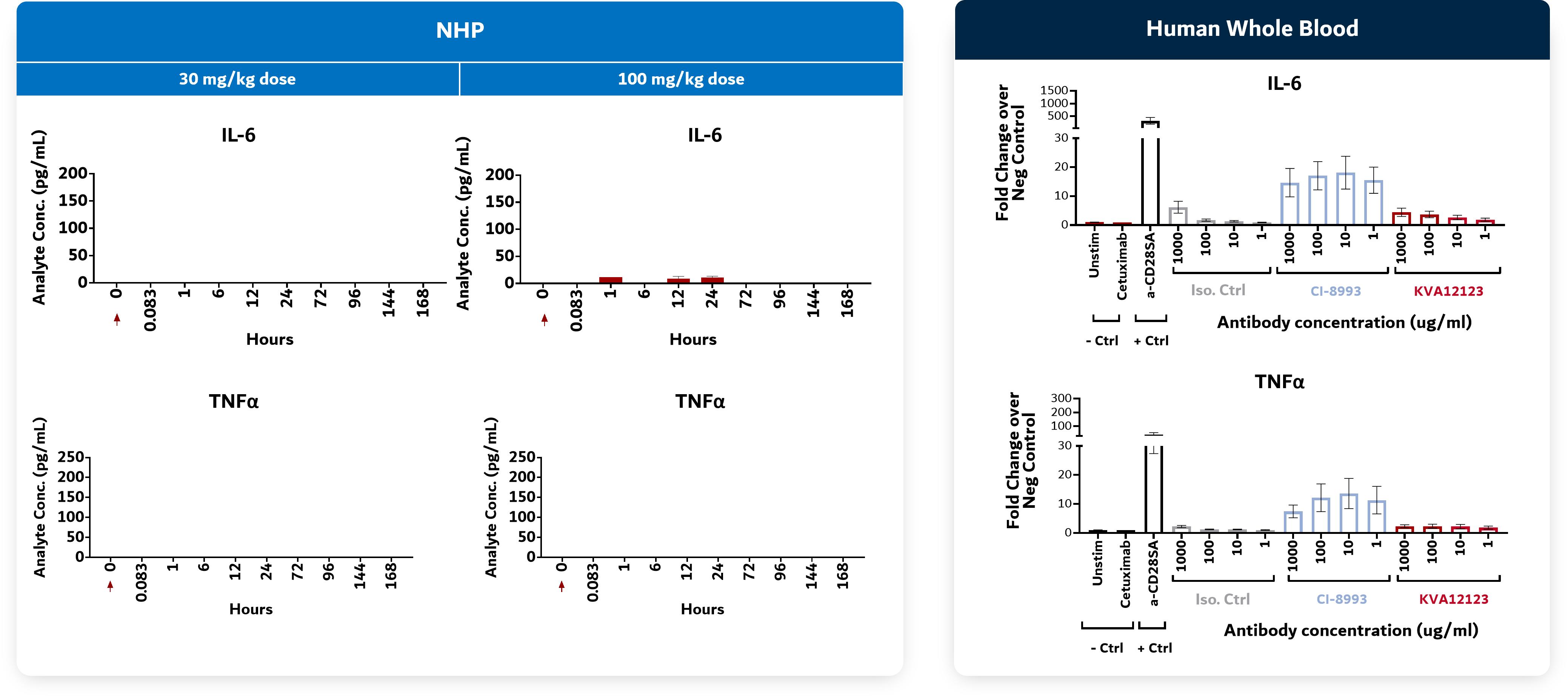
Source: Kineta data
Clinical rationale for KVA12123
Kineta is developing KVA12123 in large clinical and commercial indications where existing CPIs perform poorly, there is a high unmet medical need and VISTA expression in the TME is high. Clinical applications for KVA12123 are primarily focused on solid tumors with high levels of VISTA expression. KVA12123 may be an effective immunotherapy for many types of cancer, including NSCLC, CRC, OC, RCC and HNSCC and other “cold” difficult-to-treat solid tumors. The lead commercial and clinical indications for KVA12123 are NSCLC, CRC and OC based on the following clinical rationale.
Non-small cell lung cancer (NSCLC)
NSCLC is the leading cause of cancer-related mortality in the United States with more than 200,000 newly diagnosed cases each year. NSCLC accounts for about 85% of all diagnosed cases, and about 70% of newly diagnosed NSCLC is already locally advanced or metastatic. For NSCLC that has spread regionally, five-year relative survival rates are 35%. For NSCLC that has spread to distant locations in the body at the time of diagnosis, five-year survival rates are only 7%. More than half of all newly diagnosed NSCLC patients die within one year.
Current treatment options for advanced NSCLC include chemotherapy with cytotoxic combinations (cisplatin and carboplatin plus paclitaxel, gemcitabine, docetaxel, vinorelbine, irinotecan, protein-bound paclitaxel or pemetrexed), EGFR (epidermal growth factor receptor) tyrosine kinase inhibitors, monoclonal antibodies, and anaplastic lymphoma kinase (“ALK”) inhibitors for ALK-rearranged tumors. Targeted therapies overall show modest increases in progression-free survival (“PFS”) and overall survival (“OS”) relative to chemotherapy alone. Only 1 to 2% of lung adenocarcinomas are BRAF V600E positive, 1% of NSCLC have a ROS1 rearrangement, less than 0.5% have an nRTK (non-receptor tyrosine kinase) fusion and less than 2% have an RET fusion, making most of these additional approved targeted therapies of no benefit to most patients.
Keytruda®, Tecentriq®, Imfinzi® and Libtayo®, all targeting PD-(L)1, have been approved for first-line treatment of advanced NSCLC in combination with chemotherapy. The combination of Opdivo® and Yervoy® has also been approved in first line advanced indications. However, CR rates in this setting are low (less than 5%) and median PFS is increased by only two to seven months over conventional chemotherapy alone. In advanced NSCLC that has progressed following initial treatment, PFS and objective responses are even lower. Imfinzi® is also approved as consolidation therapy following chemoradiation therapy, Tecentriq® and Opdivo® are approved in the adjuvant setting, and Opdivo® is approved in the neoadjuvant setting.
Taken together, the above analysis shows that there is a large population of NSCLC patients globally with advanced, refractory disease that could benefit from novel immunotherapy.
The microenvironment in NSCLC is dominated by immunosuppressive innate immune cells, especially neutrophils and macrophages, making this colder tumor a candidate for treatment with KVA12123. Kineta has conducted immuno-histochemical analysis of VISTA expression on immune cell populations in NSCLC and found high levels in several NSCLC histologies (Figure 10).
11
Figure 10. VISTA expression in NSCLC. (A) Normal lung tissue and (B) NSCLC lung cancer tissue stained for VISTA expression (brown)
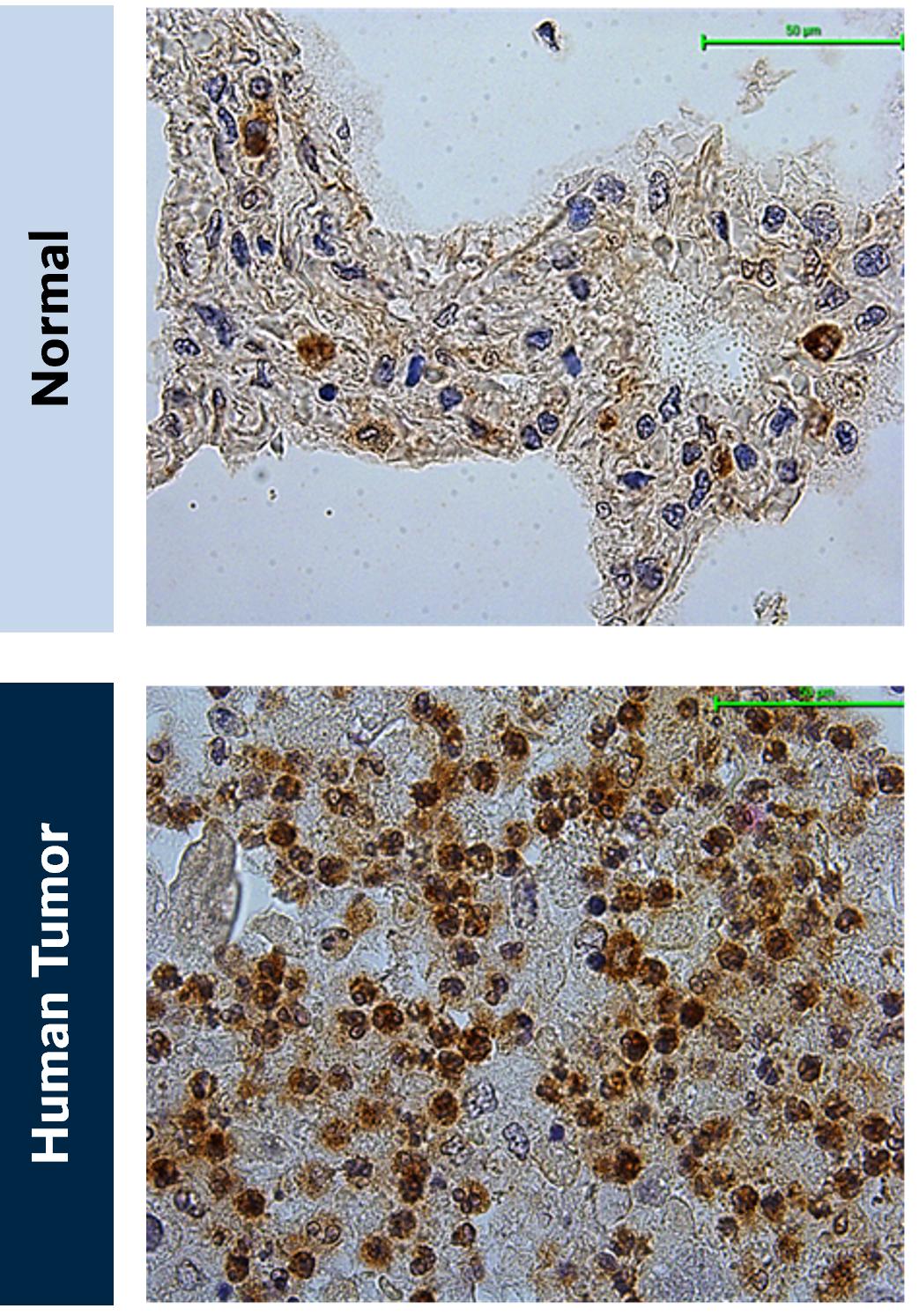
Source: Kineta data
Colorectal cancer (CRC)
More than 150,000 patients in the U.S. each year are diagnosed with CRC, and more than 50,000 deaths are attributed to the disease. In advanced and metastatic CRC, five-year survival rates are only 14%. The mainstay of treatment for CRC that is detected early is surgical resection. However, patients diagnosed with locally or regionally advanced disease can benefit from adjuvant chemotherapy, in addition to surgical resection. About 22% of patients are initially diagnosed with advanced or metastatic disease. For these patients, and for patients with recurrent disease, chemotherapy and targeted therapy result in only very slight increases in PFS and OS. Radiation therapy has no proven benefit in CRC. Keytruda®, Yervoy® and Opdivo® are approved for the treatment of mismatch repair deficient or microsatellite unstable/microsatellite instability-high tumors, but this accounts for only 4% of CRC patients.
Like NSCLC, CRC is characterized by many VISTA positive innate immune cells and presents an excellent clinical indication for KVA12123 (Figure 11).
12
Figure 11. VISTA expression in CRC. (A) Normal colon tissue and (B) colorectal cancer tissue stained for VISTA expression (brown)
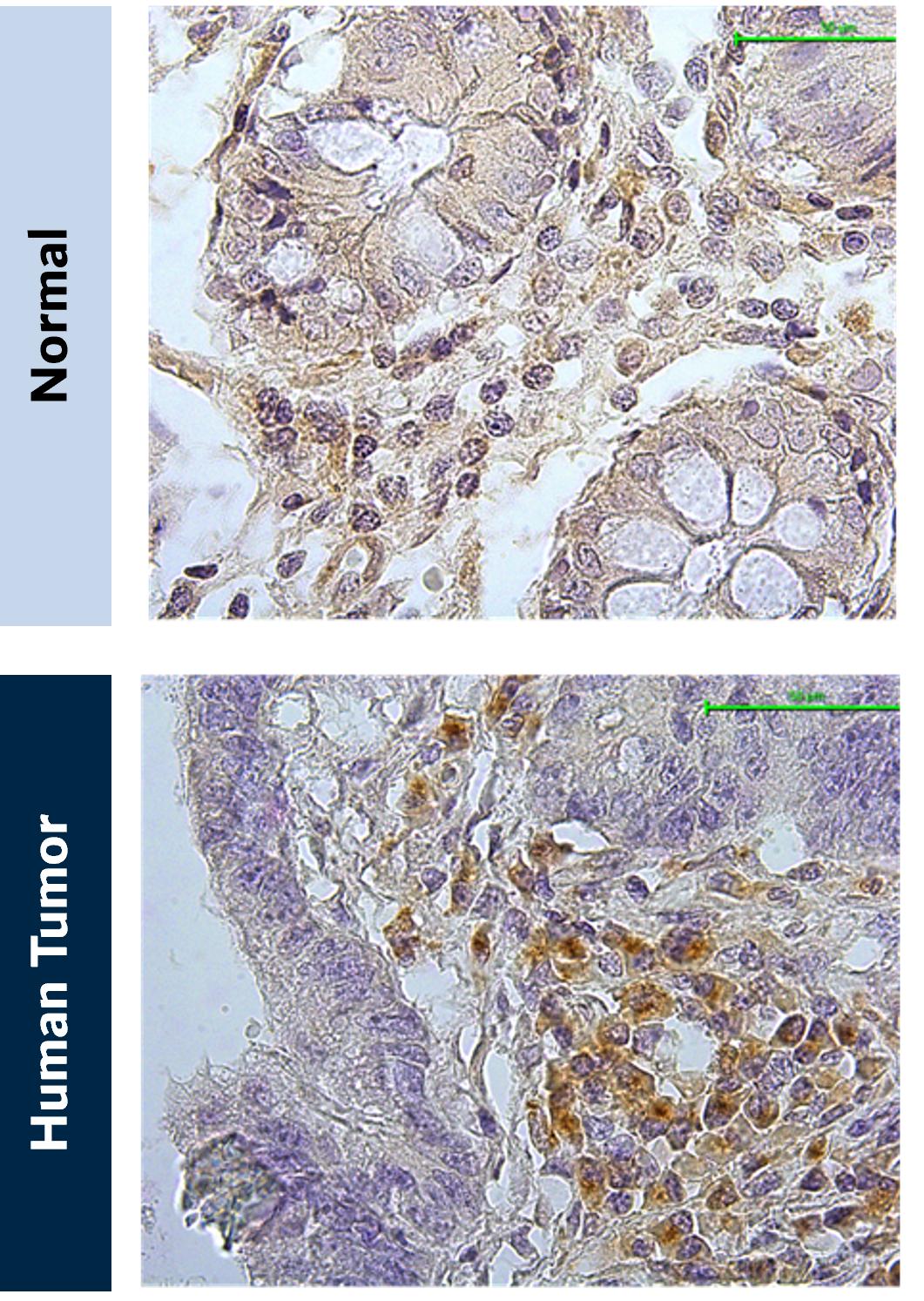
Source: Kineta data
Ovarian cancer (OC)
A small number of mostly gynecological cancers express VISTA on tumor cells and on infiltrating immune cells. One example is OC, where tumor cells express high levels of VISTA (Figure 12). More than 60% of OC cases are diagnosed at an advanced stage of disease, and five-year survival rates for these patients are less than 50%. Platinum/taxane combination chemotherapy is widely used in this indication, with modest improvements in PFS and OS. OC represents a third potential clinical indication for KVA12123.
13
Figure 12. VISTA expression in ovarian cancer. (A) Normal ovarian tissue and (B) ovarian cancer tissue stained for VISTA expression (brown)
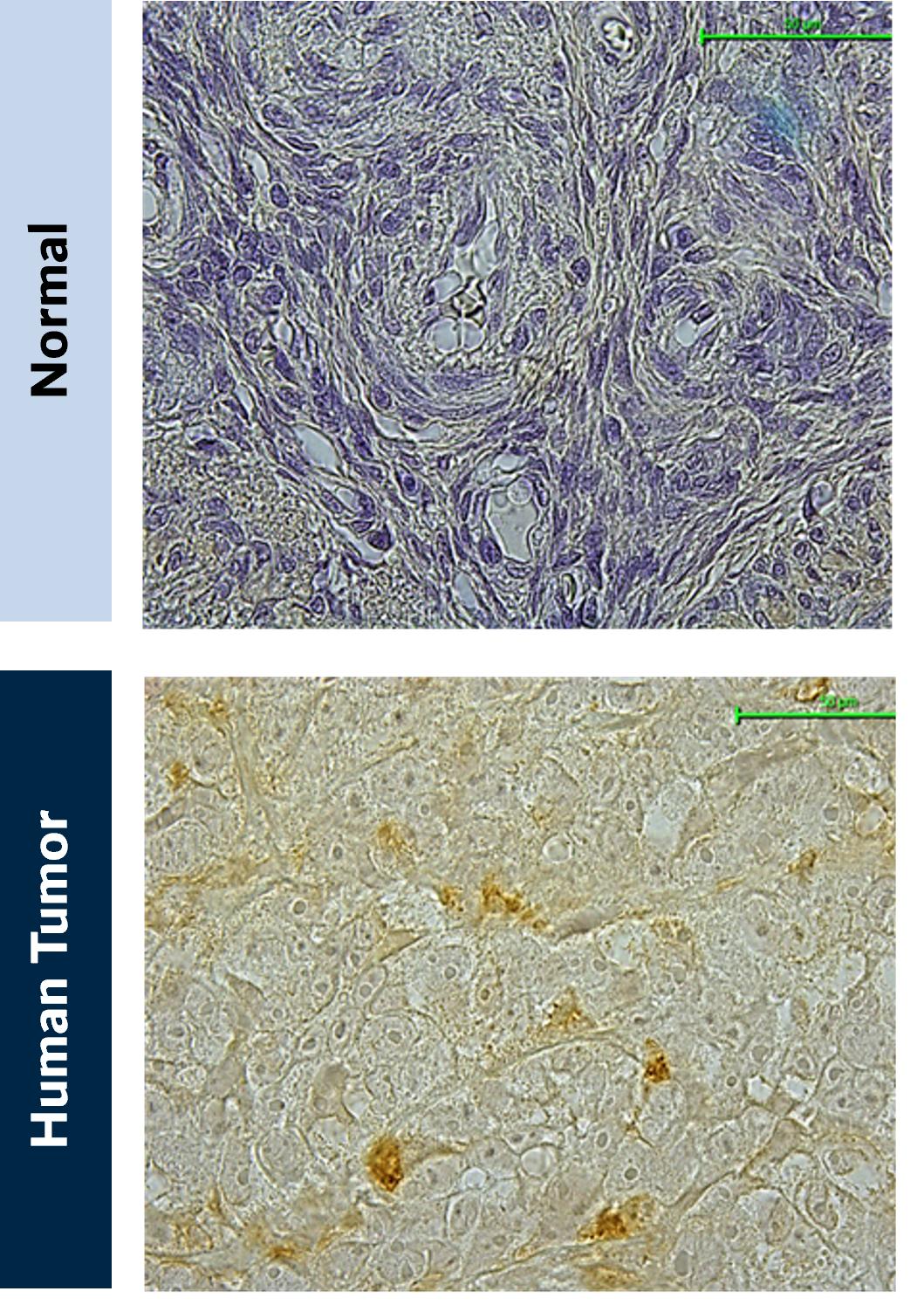
Source: Kineta data
VISTA-101 Clinical Trial (VISTA-101)
Kineta announced dosing of the first patient with vaccination complication. OneKVA12123 as a monotherapy in the VISTA-101 trial in April 2023. The ongoing Phase 1/2 clinical study of KVA12123 has continued to enroll monotherapy cohorts and dosed the first patient in combination with pembrolizumab in October 2023.
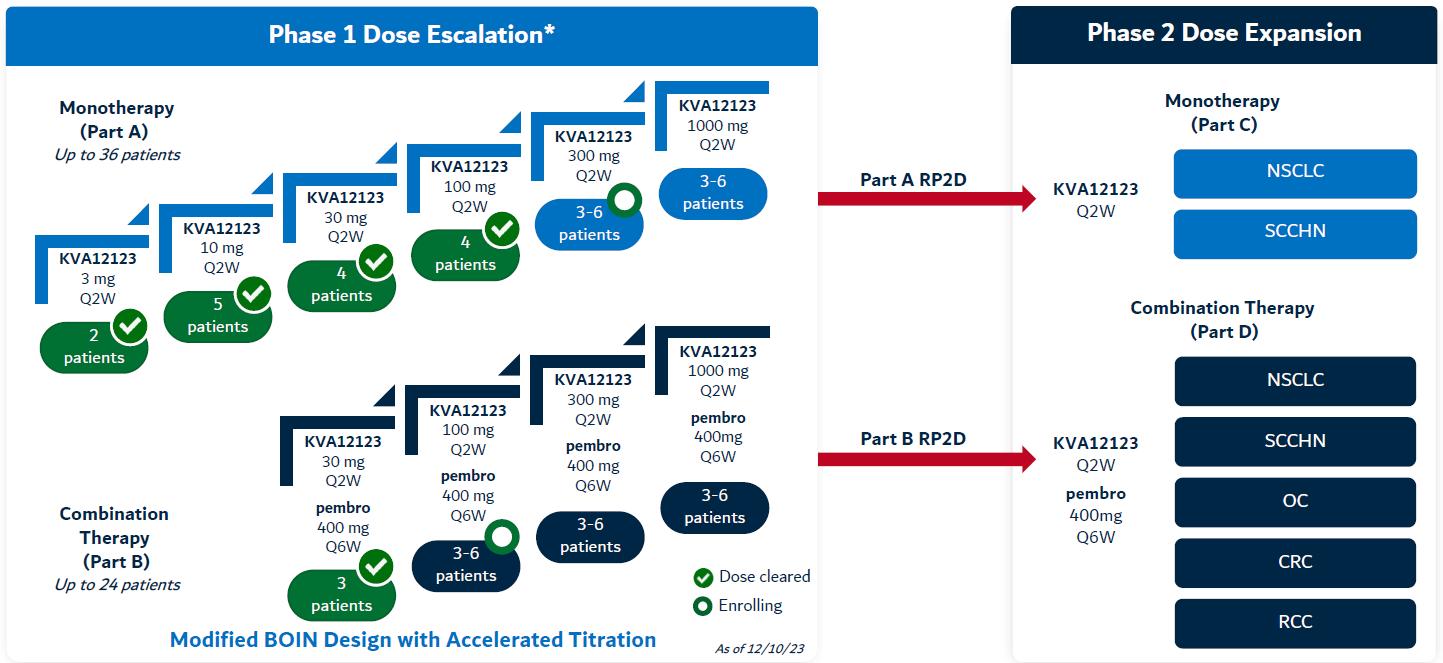
VISTA-101 is a first-in-human (FIH), Phase 1/2, open-label, multicenter, dose escalation, and dose expansion study designed to evaluate the safety, tolerability, pharmacokinetics (“PK”), immunogenicity, and tumor response of the investigational drug KVA12123 as a monotherapy and in combination with pembrolizumab in adults with relapsed or refractory advanced solid tumors.
The study is being conducted in 4 parts: Parts A, B, C and D. Parts A and B focus on placebo had moderate worseningdose escalation. Parts A (single-agent KVA12123) and B (KVA12123 + pembrolizumab) comprise up to 6 and 4 dose escalation cohorts, respectively, each treating 1-6 participants, to characterize the safety, tolerability, pharmacodynamics (“PD”), PK and preliminary tumor responses of tremorsstudy interventions.
Parts C and Parkinsonism,D will focus on dose expansion. Parts C (single-agent KVA12123) and D (KVA12123 + pembrolizumab) will comprise up to 7 disease-specific dose expansion cohorts (2 for Part C and 5 for Part D), which ledwill commence at the recommended Phase 2 dose (“RP2D”) to discontinuation. AEs occurringfurther characterize the safety, tolerability, PD, PK, and preliminary tumor response of KVA12123 as a monotherapy and in combination with pembrolizumab. Part C and Part D will enroll patients with specific tumor types including NSCLC, SCCHN, OC, CRC and RCC as determined in Parts A and B.
14
Figure 13. KVA12123 Phase 1/Phase 2 dose escalation study design
VISTA-101 study objectives
VISTA-101 study objectives are outlined below:
Primary objectives
Secondary objectives
Exploratory Objectives
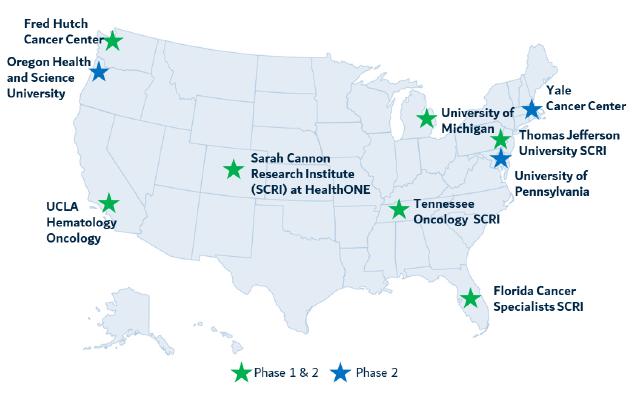
Clinical sites
Kineta has engaged seven well-known research sites to conduct the Phase 1 arm of VISTA-101 across the United States only (Figure 14). Three additional sites will be added as the study advances to the Phase 2 dose expansion cohorts.
15
Figure 14. VISTA-101 clinical trial sites
Clinical collaboration with Merck
Kineta has entered into a clinical trial collaboration and supply agreement with Merck (known as MSD outside the U.S. and Canada). Under this collaboration, Kineta will evaluate the safety, tolerability, PK and anti-tumor activity of KVA12123, its novel anti-VISTA monoclonal antibody, alone and in combination with KEYTRUDA® (pembrolizumab), Merck’s anti-PD-1 therapy, in patients with advanced solid tumors.
Kineta is conducting a Phase 1/2 clinical study evaluating KVA12123 as a single agent and in combination with KEYTRUDA® in patients with advanced solid tumors. The objectives of the study are to evaluate the safety, tolerability, PK and anti-tumor responses of KVA12123 as a monotherapy and in combination with KEYTRUDA® with initial clinical data released in the fourth quarter of 2023. Kineta is responsible for conducting this study.
Initial VISTA-101 Clinical Data
Kineta presented initial KVA12123 monotherapy clinical data from VISTA-101 at a higher percentagethe Society for Immunotherapy of Cancer’s (SITC) 38th Annual Meeting in 2 or more patients administered YTX-7739 compared to placebo were procedural pain, myalgia, dry eye, hyperbilirubinemia, hypoesthesia, lower back pain, and constipation. AEs occurring at a higher percentage with placebo included orthostatic hypotension, headache, tremor, fatigue and dizziness.November 2023.
As expected, after only 28 days of dosing, there were no statistically significant differences in clinical assessments (Unified Parkinson's Disease Rating Scale Part III (UPDRS III), Montreal Cognitive Assessment (MoCA)) or most exploratory biomarkers. Quantitative electroencephalogram (qEEG) assessments ofJanuary 31, 2024, the effect of YTX-7739 on brain activity were completed in a subset of 8Phase 1/2 VISTA-101 trial enrolled 15 patients and demonstrated a statistically significant change compared to baseline, suggestive of a potential improvement in synaptic function.
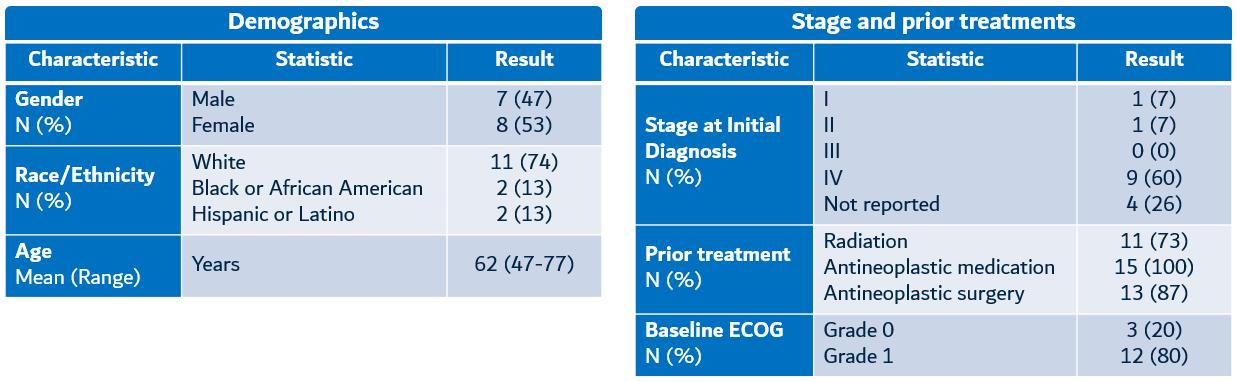
At the center of our scientific foundation is our drug discovery engine, which is based on technology licensed from the Whitehead Institute, an affiliate of the Massachusetts Institute of Technology. This core technology, combined with investments and advancements by us, is designed to enable rapid screening to identify product candidates with the potential to modify disease by overcoming toxicity in disease-causing gene networks. Toxicity in many neurodegenerative diseases results from an aberrant accumulation of misfolded proteinsadvanced solid tumors in the brain. We leverage our proprietary discovery engine to identifyfirst four monotherapy dose-escalation cohorts, where subjects received either 3, 10, 30 or 100mg of KVA12123 by intravenous infusion every two weeks, and screen novel drug targets3 patients in the first combination therapy cohort, where subjects received 30 mg of KVA12123 Q2W and drug molecules for their ability to protect nerve cells from toxicity arising from misfolded proteins. To date, we have identified over twenty targets, most400 mg of which have not previously been linked to neurodegenerative diseases.pembrolizumab Q6W. Patients enrolled in VISTA-101 monotherapy arm ranged in gender, ethnicity and age (Figure 15). Patients were heavily pretreated with multiple prior lines of therapy including chemotherapy, radiation and immunotherapy.
Figure 15. VISTA-101 baseline patient characteristics
16
Safety
Evaluating the safety and tolerability of KVA12123 is one of the primary objectives of VISTA-101. As of January 31, 2024, 15 patients were dosed in the first four monotherapy cohorts. KVA12123 was well tolerated at all doses and no dose limiting toxicities (“DLT”) were observed. All KVA12123 treatment emergent adverse events were grades 1-2.
Figure 16: VISTA-101 KVA12123 was well tolerated in 3, 10, 30 and 100 mg monotherapy cohorts
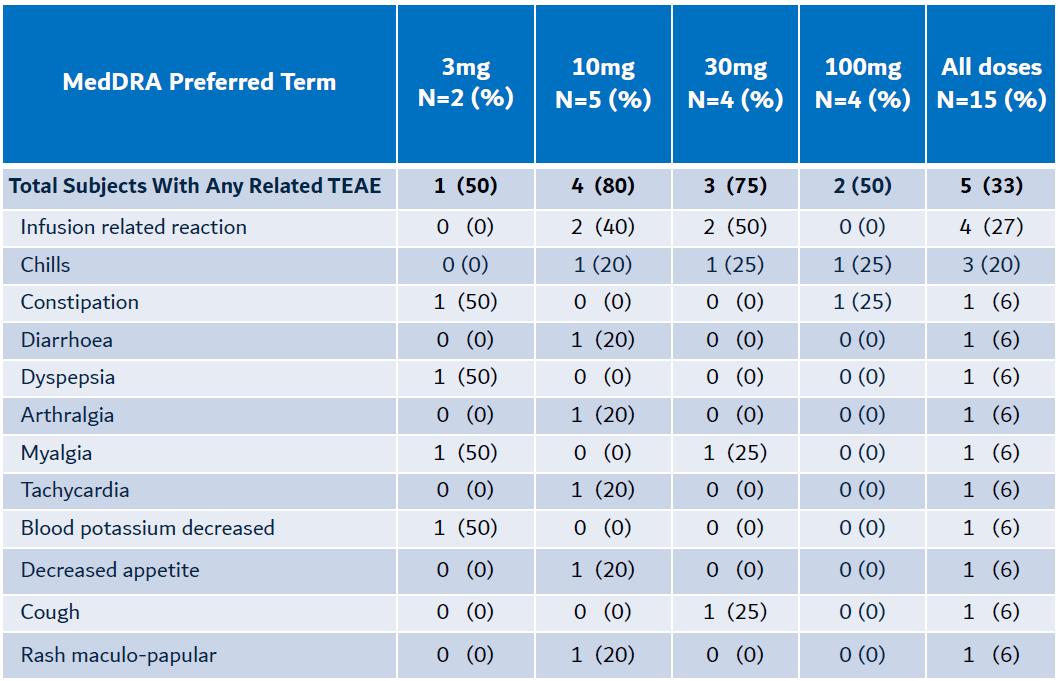
Furthermore, no evidence of CRS or associated cytokines including IL-6, TNFα & IL-10 were detected at any of the dose levels.
17
Figure 17. VISTA-101 No CRS-related cytokine induction observed
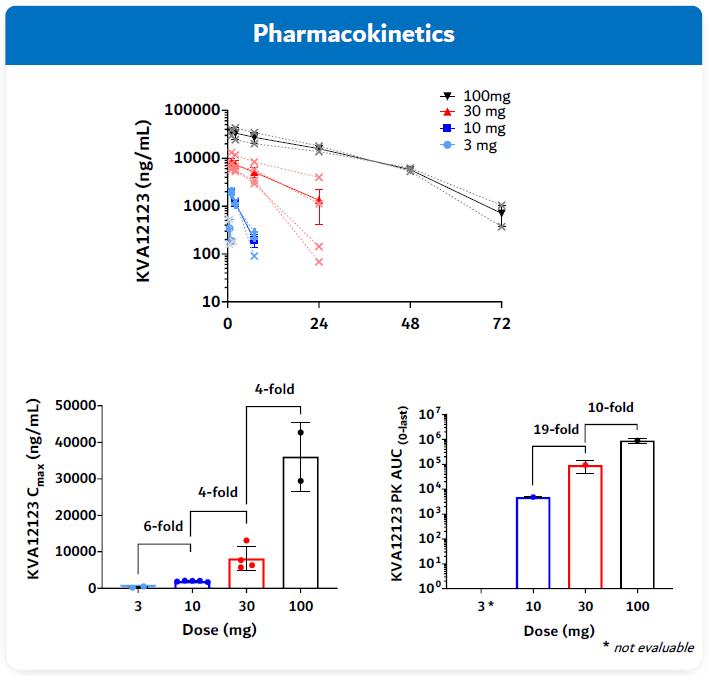
Pharmacokinetics (PK) and Receptor Occupancy (RO)
PK is the study of how the body interacts with KVA12123 for the entire duration of exposure after administration. KVA12123 exhibited a greater than dose-proportional pharmacokinetic profile in drug exposure across all evaluated doses, consistent with target-mediated drug disposition at lower doses.
Figure 18. VISTA-101 KVA12123 Pharmacokinetics
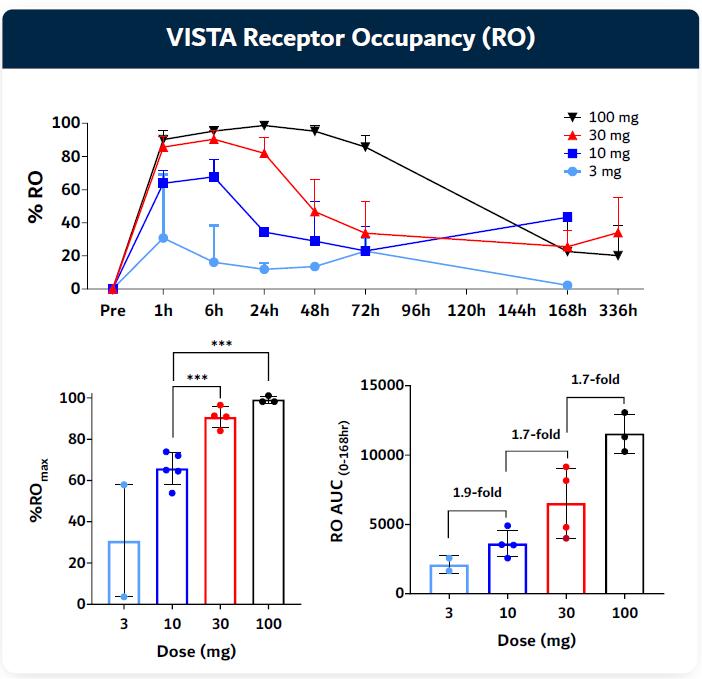
To guide the recommended phase 2 dose decision, Kineta developed a proprietary assay to evaluate VISTA receptor occupancy (“RO”) on immune cells from patients treated with KVA12123. This is an important metric for evaluating how well KVA12123 is blocking the VISTA target. KVA12123 achieved a greater than 90% VISTA RO at the 30 mg dose indicating that KVA12123 may be approaching an optimal clinical dose.
18
Figure 19. VISTA-101 KVA12123 VISTA receptor occupancy
Biomarkers
In drug development and clinical trials, biomarkers may be useful to identify patient populations for a study, monitor therapeutic response and identify side effects. KVA12123 demonstrated dose-proportional on-target biomarker immune responses involved in anti-tumor activity. KVA12123 demonstrated significant efficacy-related, dose-dependent cytokine induction of CXCL10, CCL2 (MCP1), CCL3 (MIP1α) and CCL4 (MIP1β), which are involved in immune cell activation and recruitment to the TME (Figure 19). Additionally, increases in anti-tumor immune cell subpopulations including nonclassical monocytes, NK cells, CD4+ T cells and CD8+ T cells were observed during treatment (Figure 20).
19
Figure 20. VISTA-101 KVA12123 pro-inflammatory biomarkers
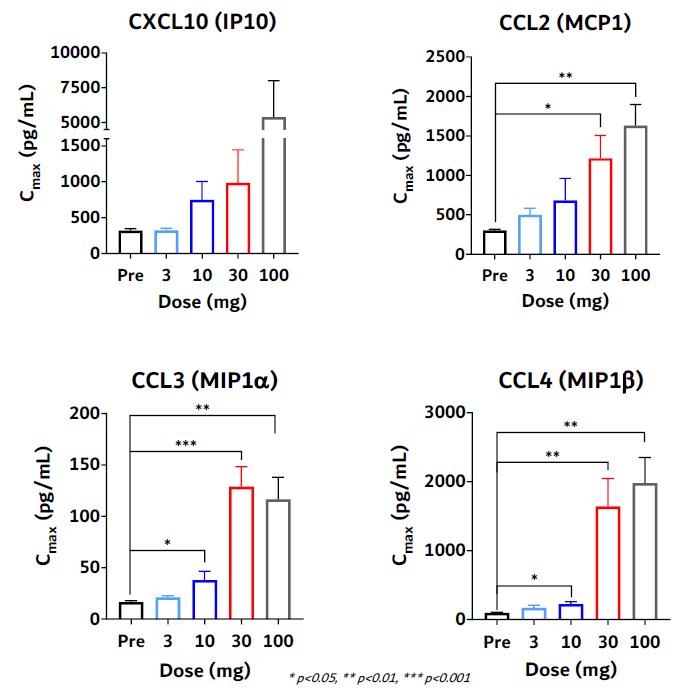
Figure 21. VISTA-101 KVA12123 immune cell responses
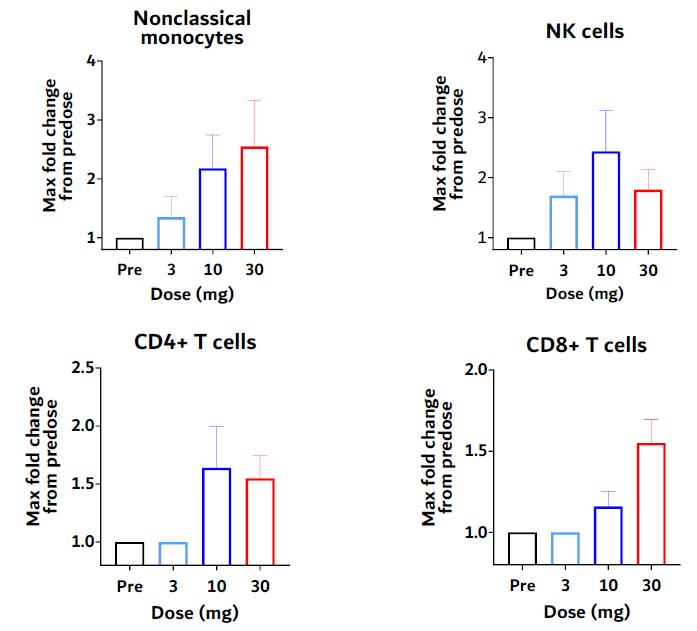
KVA12123 demonstrated induction of pro-inflammatory myeloid derived cytokines/chemokines involved in immune cell activation and recruitment in the TME. Changes in these key biomarkers and immune cell populations are indicative of the anti-tumor effects of blocking VISTA that is
20
consistent with data from preclinical models (NHP and KO mice). These data validate their use as potential biomarker of VISTA target engagement with KVA12123.
VISTA-101 Initial Monotherapy Data Summary
Safety
Pharmacokinetics (PK) and Receptor Occupancy (RO)
Biomarkers
Development timeline
The ongoing VISTA-101 clinical trial is currently enrolling patients in both monotherapy and combination therapy arms of the study. Initial monotherapy data was presented at SITC in November 2023. Additional monotherapy safety and efficacy data as well as initial combination therapy data are anticipated in the second quarter of 2020, we secured2024. The Company anticipates a strategic research and development collaboration in ALS and FTLD with Merck Sharp & Dohme Corp., or Merck, with upfront and potential milestone payments of up to $530 million plus royalties.
Neurodegenerative Disease Market and Challenges
Many factors, including too few disease-relevant biological hypotheses, the inherent complexity of the brain, and high patient heterogeneity, have led to a graveyard of failed approaches over the last several decades and have produced few approved disease-modifying therapies for neurodegenerative diseases to date.
We believe this is about to change. Recent scientific and technological advances include the improved understanding of the disease-relevant biology, innovative target discovery technologies, potentially better predictive animal models, new imaging approaches and identification of new biomarkers. These advances have ignited a renewed focus and commitment to neurodegenerative disease research and development, and we are one of the companies at the forefront of this emerging revolution.
Clinical study of neurodegenerative disease and evaluation of potential therapeutics has faced several hurdles. A primary consideration is the genetic heterogeneity of neurodegenerative diseases and subsequent variations in the disease biology in patients with similar clinical diagnoses. We believe this degree of heterogeneity is far greater than previously appreciated and is likely due to a unique combination of genetic and environmental factors which have important implications for development of therapies and their appropriate use by individual patients. We believe there is a need for a larger and more accurate set of biomarkers to aid in diagnosis as well as monitoring disease progression and treatment response in trials. Damage to brain cells early in the disease course and prior to the onset of symptoms presents further challenges for the design of clinical trials, as patients enrolling in clinical trials are typically selected based on expression of disease symptoms when significant damage to brain cells has already occurred. For example, increasingly sophisticated imaging studies have demonstrated that patients have lost at least 40% to 60% of dopaminergic neuronal integrity before qualifying for a diagnosis of Parkinson’s disease, indicating damage to brain cells begins long, often decades, before clinical symptoms manifest. As a result, many previous clinical trials in neurodegenerative disease included patients at a stage of the disease beyond which progression could no longer be modified. Thus, clinical trials would optimally be performed in patient populations that are at an early enough stage where potential disease-modifying therapies have an opportunity to preserve existing brain cells and function. Approaches to early diagnosis remain a focus in the neurodegenerative clinical research field.
In Parkinson’s disease, the cornerstone of pharmacological therapy for the past several decades has focused on either temporarily replenishing dopamine or mimicking the action of dopamine such asmeeting with the dopamine precursor levodopa. Levodopa, which is very helpful to patients in managing some of the motor symptoms of the disease, does not alter disease progression. Certain disease-modifying molecules are currently being investigated in early clinical trials for the potential of removing or reducing levels of α-synuclein. These programs, however, are predominantly focused on the development of antibodies. Therapeutic antibodies are large molecules that are administered systemically, and as such have significant challenges crossing the blood brain barrier and penetrating into the brain, which is the target tissue for neurodegenerative diseases. Even if some limited amount of antibody can penetrate into the brain, antibodies face a further challenge. α-Synuclein functions within cells to facilitate vesicle trafficking, however when α-synuclein misfolds it is thought to have an increased propensity to aggregate and disrupt multiple critical processes inside the cell. The ultimate expression of this pathology is the formation of Lewy bodies within neurons, which are a hallmark of dystrophic and degenerating cells. Therapeutics that target pathological processes within cells would be expected to prevent this toxic progression. α-Synuclein can also be secreted by neurons, and although the function is unclear, this results in α-synuclein outside of cells. It is this
2
population that would be a target for antibody therapeutics which are generally believed to interact with protein extracellularly, or outside the cell. We believe antibodies therefore have less access to α-synuclein, and recently, two antibody drug candidates that target α-synuclein failed to meet primary clinical endpoints in Phase 2 trials. By contrast, we are developing small molecules that our yeast platform pre-selects by design to cross the blood brain barrier and diffuse into the cell where α-synuclein causes cellular toxicity and damage. YTX-7739 targets the enzyme SCD, the inhibition of which has been shown to help overcome the toxicity of α-synuclein and promote protection of neurons.
Our Approach
Our approach is to unlock the path to new therapies by addressing the fundamental and persistent barriers in neurodegeneration research: the poor understanding of disease mechanisms and lack of new biological targets. We believe that programs focused on novel drug targets, which are grounded in an improved understanding of disease biology, will enable a higher likelihood of success in developing disease-modifying therapies.
Our discovery engine is built upon core enabling technology that we exclusively license from the Whitehead Institute, an affiliate of the Massachusetts Institute of Technology. The core discovery technologies were created in the laboratory of our co-founder, Dr. Susan Lindquist. Dr. Lindquist and senior scientists from her team integrated multiple technology platforms to create a drug discovery engine designed to reliably generate new insights into fundamental mechanisms of neurodegenerative disease, and also reveal new potential drug targets and drug molecules that address neurodegeneration in a range of different ways, many of which were previously unknown.
The discovery engine is centered on the key insight that protein misfolding, a phenomenon at the root of virtually all neurodegenerative diseases, can be modeled in yeast cells. These yeast models are then screened against large chemical libraries using high throughput technology, selecting for chemical hits that protect cells from the toxicities created by the misfolded human disease-relevant proteins. The biological targets and pathways for these protective molecules are then uncovered using a series of chemical genetic techniques. Our technology also allows for screening yeast collections that have individual genes deleted, such that, when rescue is observed, it can be inferred that the gene that was deleted in that yeast strain is involved in ameliorating the toxicity of the misfolded human disease-relevant protein. Since the only modification to the original yeast system was the introduction of the culprit misfolding proteins, any molecule or gene deletion that can protect cells from the resultant toxicity is of interest. The discovery of protective molecules and biological targets, especially when previously unknown, can reveal new or untapped areas for study. We believe that the complement and overlap between the small molecule and genetic rescue screens have the potential to create a powerful network of interlinked biological processes that can further identify previously unknown therapeutic targets. We explore these cell-protective discoveries from the yeast system for translation to human disease-relevant cells using informatics and cutting-edge stem cell and iPSC experimental techniques. The discovery engine is designed to ultimately output novel programs: molecules with novel biological targets that can then be progressed through the standard preclinical drug development processes.
We believe our proprietary discovery engine has the potential to dramatically expand the knowledge around the complex biology of neurodegeneration, and further allows initiation of discovery programs outside of the traditional, limited set of hypotheses that exist today. Screening for hits in a living yeast system can save time by providing a biological-relevant readout sooner than some of the more typical practice of starting in non-live, test tube systems. Additionally, shared features between yeast cell membranes and the blood brain barrier, such as comparable membrane permeability, polarity, and drug pumps for removal of non-native compounds, mean that molecules that can permeate a yeast cell to effect intracellular rescue may also be likely to penetrate the blood brain barrier. Furthermore, our molecules have been tested in diseased human cells ex-vivo at the beginning rather thanFDA at the end of preclinical development. We believe that successPhase 1 and initiating the Phase 2 arms of VISTA-101 in this setting confers increased confidence in programs compared to the more traditional paradigmthird quarter of multiple rounds of animal studies before any actual testing in human tissues.2024.
3Potentially Large Commercial Opportunity for KVA12123
Based on the strong clinical rationale and commercial opportunity, Kineta has identified NSCLC, SCCHN, OC, CRC and RCC as potential initial indications for KVA12123. Data from the Phase 1 / 2 clinical trial will more fully inform the indications to initially pursue for regulatory approval.
Key DifferentiatorsThe projected new annual patients worldwide for each of Our Discovery Enginethese initial indications in 2027 totals 984,000 for NSCLC, 243,000 for SCCHN, 142,000 for OC, 1.2 million for CRC and 372,000 for RCC, based on reports from GlobalData. In total, these five initial indications represent an estimated 2.9 million annual new patient opportunity (Figure 21). Improving survival for CPI non-responders remains a critical unmet need that affects ~70% of cancer patients representing 2.0 million patients annually who could be an ideal candidate for treatment with KVA12123.
If Kineta successfully completes the clinical trial program for KVA12123 and if Kineta subsequently obtains regulatory approval for KVA12123, Kineta will focus on initial target indications in NSCLC, CRC and OC as potential commercial opportunities with significant unmet needs. Clinical development of KVA12123 will be initially as a second-line therapy in these indications. The projected therapeutic market size in 2027 for each of these initial indications totals $31.8 billion for NSCLC, $10.3 billion for CRC and $5.9 billion for OC, based on reports from GlobalData. In total, these three initial cancer indications represent an estimated $48 billion market opportunity for KVA12123.
We
21
Figure 22. Large commercial opportunity in initial indications in solid tumors for KVA12123

Source: GlobalData: Global Drug Forecast and Market Analysis to 2028 (1. NSCLC, 2. SCCHN, 3. OC 4. CRC and 5. RCC)
Anti-CD27 agonist mAb immunotherapy
Kineta is developing an anti-CD27 agonist mAb immunotherapy to address the problem of exhausted T cells in the TME. It has been recently demonstrated that it is very difficult to reverse T cell exhaustion. As an alternative approach, Kineta is developing an agonist antibody to a receptor (CD27) present on naïve T cells circulating outside the tumor. Anti-CD27 monoclonal antibodies activate and induce the maturation and migration of naïve T cells. CD27 activation also drives the diversification of the T cell repertoire, lowering the activation threshold of T cells against low affinity tumor antigens. Recent data also suggests that an agonist anti-CD27 antibody can activate important innate immune cell populations like NK cells and inflammatory myeloid cells. These cells contribute to an effective anti-tumor response, especially in CPI-resistant patients.
Recent publications have leveragedalso demonstrated that anti-CD27 agonist antibodies can drive tumor growth inhibition as a monotherapy and in combination with CPIs.
Figure 23. Activating CD27 demonstrates tumor growth inhibition as a monotherapy and in combination with CPIs
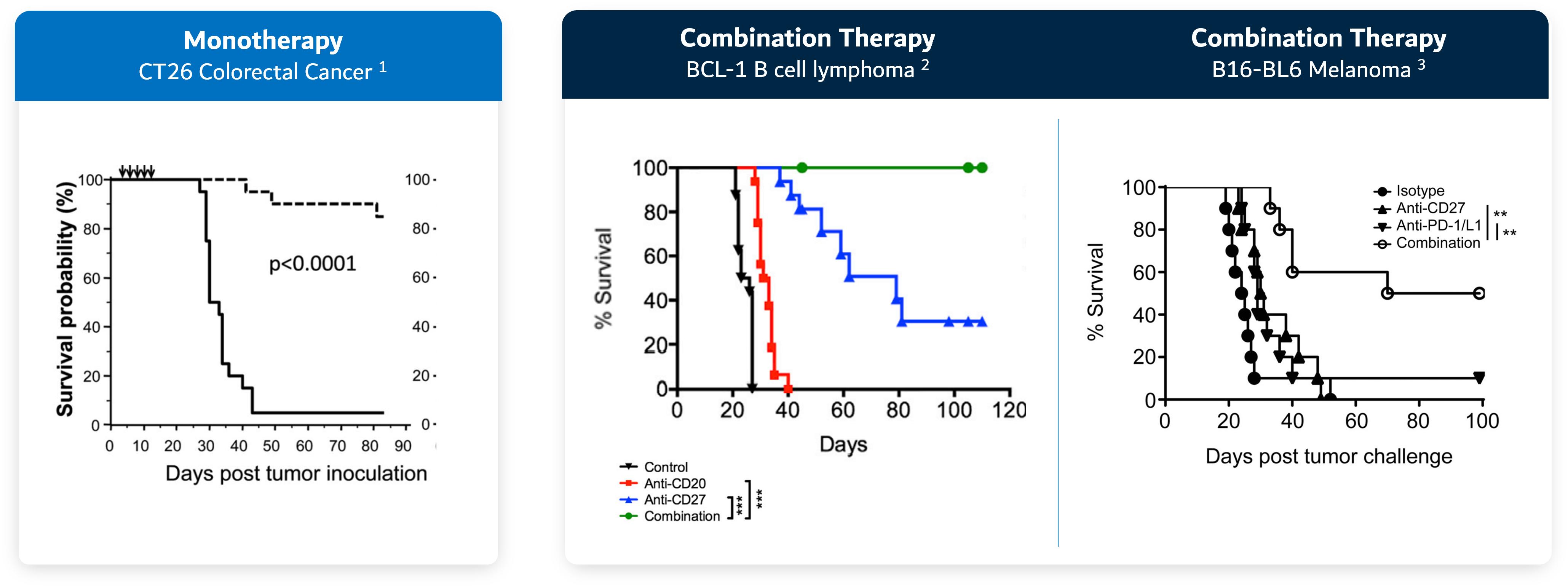
Source: 1. He et al. J. Immunol 2013 2. Turaj et al. Cancer Cell 20173. Buchan et al. Clin. Cancer Research 2018
Kineta has identified a lead candidate out of a diverse set of anti-CD27 agonist antibody sequences. The lead candidate is a fully human monoclonal antibody that has been observed to show nM binding affinity to CD27 in humans. Kineta plans to develop the powerdrug as an intravenous infusion.
22
In in vitro studies, Kineta’s lead candidate antibodies demonstrate robust agonist activation of our discovery engineT cells and NK cells demonstrating the ability to generatepotentiate new anti-tumor responses (Figure 23).
Figure 24. CD27 T cell and NK cell activation
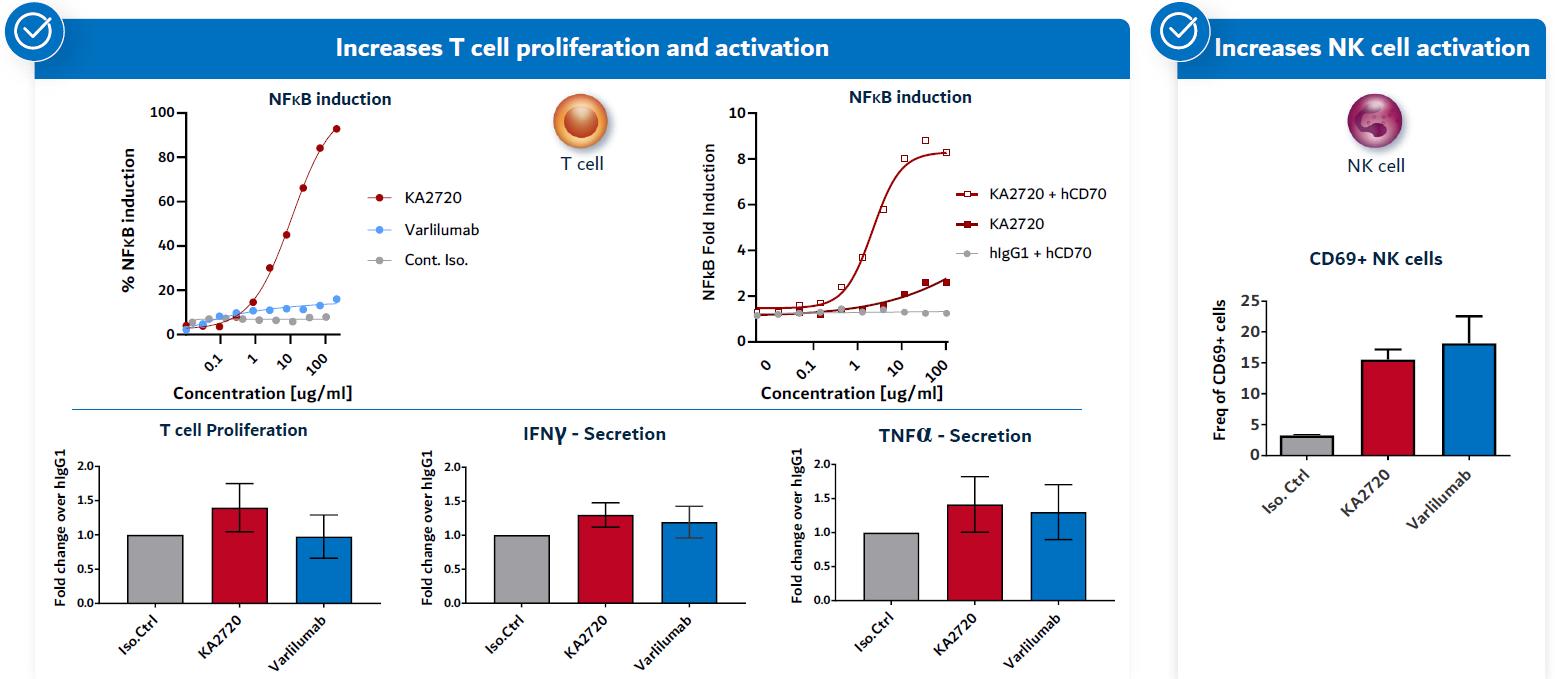
Source: Kineta data
In preclinical tumor models, Kineta’s anti-CD27 agonist lead mAb has shown strong tumor growth inhibition as a robust portfolio of promising novel drug targetsmonotherapy and molecules. We then prioritize the most promising targets to accelerate drug discovery programs and advance compounds intoin combination with other checkpoint inhibitors in preclinical and ultimately clinical development. To date, this approach has already uncovered over twenty novel targets, pathways, mechanisms, and molecules that we believe have the potential to ameliorate the fundamental cellular toxicities associated with neurodegenerative diseases.tumor models.
Discovered targets may mature into programs as they advance through the discovery process. YTX-7739, our lead program, targets the enzyme SCD, one of the early targets identified and validated in our discovery engine and is being studied for the treatment of Parkinson’s disease. We are also developing lead compounds and validating two targets that are advancing through a research collaboration with Merck. Other targets for multiple potential indications are at varying stages of the discovery process.
Our PipelineFigure 25. Lead anti-CD27 agonist mAb demonstrates monotherapy and combination therapy growth inhibition in MC38 preclinical model
All of our therapeutic candidates are small molecules
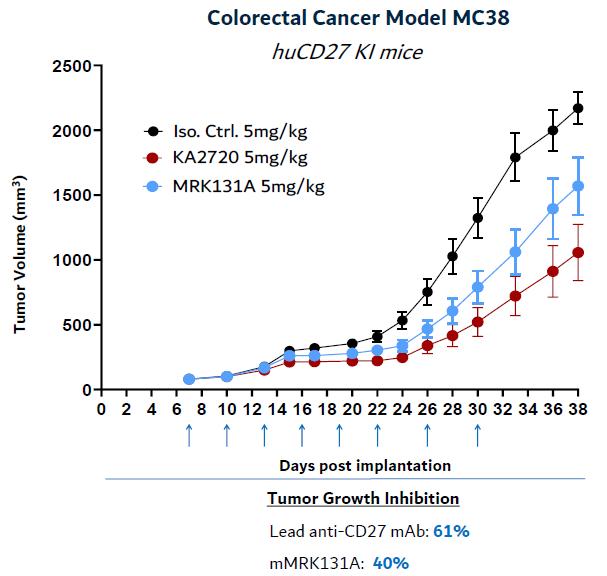
Source: Kineta data
Kineta is developing a novel anti-CD27 agonist mAb immunotherapy for advanced solid tumors including RCC, OC and are optimizedCRC.
23
Strategic Partnerships
KVA12123
Kineta has entered into a clinical trial collaboration and formulated for oral delivery. We own both development and commercialization rights to YTX-7739 and all other targets except for our research collaborationsupply agreement with Merck who has licensed these potential programs(known as MSD outside the U.S. and Canada). Under this collaboration, Kineta will conduct IND-enabling toxicologyevaluate the safety, tolerability, PK and safety pharmacology, clinical development,anti-tumor activity of KVA12123, its novel anti-VISTA monoclonal antibody, as a monotherapy and commercialization.
YTX-7739 is a novel small molecule for the potential treatment of Parkinson’s disease and related disorders of α-synuclein. The program that resulted in this lead compound was the first prioritized output program of our discovery engine. YTX-7739 is designed to ameliorate the consequences of α-synuclein toxicity in human cells that results in cellular dysfunction, specifically disruptionscombination with the directed movement, or trafficking, of proteins or lipid-bound vesicles within cells. YTX-7739 targets the enzyme SCD, that catalyzes a reaction in the lipid metabolism pathway.
α-Synuclein is a protein that is a prominent constituent of Lewy bodies, the abnormal protein aggregates that are the pathological hallmarks of Parkinson’s disease, dementia with Lewy bodies, MSA and other neurological disorders known collectively as “synucleinopathies”. Current treatments for Parkinson’s disease manage the early motor symptoms of the disease. The goal of our differentiated and potentially disease-modifying approach with YTX-7739 is to block the intracellular toxicity associated with α-synuclein misfolding and aggregation to allow the cell to continue to function normally, and to slow or possibly even halt the progressive degenerative consequences of the disease.
On November 10, 2021, we announced the top-line results from our Phase 1b clinical trial of YTX-7739KEYTRUDA® (pembrolizumab), Merck’s anti-PD-1 therapy, in patients with mild-to-moderate Parkinson’s disease.advanced solid tumors.
Effective October 14, 2022, Kineta entered into a Clinical Trial Collaboration and Supply Agreement (the “CTCSA”) with MSD International Business GmbH (“Merck”) to evaluate KVA12123 as a monotherapy and in combination KEYTRUDA® (pembrolizumab), Merck’s anti-PD-1 therapy, in patients with advanced solid tumors. Pursuant to the terms of the CTCSA, each party retains its intellectual property rights, but all joint clinical data and joint inventions shall be jointly owned by the parties. Each party shall bear its own costs related to manufacturing and supply of its compound, as well as be responsible for its own internal costs and expenses to support the clinical trial. During the term of the CTCSA and for a specified period thereafter, either party shall have the option to propose an amendment to the CTCSA or to negotiate a new agreement to conduct a subsequent study. The parties shall negotiate the terms of such amendment or new agreement in good faith.
Unless terminated earlier by either party, the CTCSA will continue in full force and effect until Kineta delivers Merck final versions of the study results memorandum and final report. Either party may terminate the CTCSA upon an uncured material breach by the other party, for reasons related to patient safety, in the event of certain regulatory actions or if development of such party’s compound is discontinued for certain reasons. If the CTCSA is terminated, Kineta is obligated to return or destroy the unused supply of pembrolizumab to Merck.
Kineta is conducting a Phase 1b1/2 clinical trial wasstudy evaluating KVA12123 as a randomized, placebo-controlled, double-blind multi dosesingle agent and in combination with KEYTRUDA® in patients with advanced solid tumors. The objectives of the study are to investigateevaluate the safety, tolerability, pharmacokinetics and pharmacodynamicsanti-tumor responses of YTX-7739. Data were reportedKVA12123 as a monotherapy and in combination with KEYTRUDA® with initial clinical data presented in the fourth quarter of 2023. Kineta is responsible for conducting this study, which was initiated in the fourth quarter of 2022.
In-License Agreements
License Agreement with GigaGen, Inc.-VISTA
In August 2020, Kineta entered into an Option and License Agreement with GigaGen, Inc. (“GigaGen”), which was amended in November 2020 and May 2023 (such agreement, as amended, the “VISTA Agreement”) to in-license certain intellectual property and antibodies for the VISTA/KVA12123 drug program. Pursuant to the terms of the VISTA Agreement, GigaGen granted Kineta an exclusive (even as to GigaGen) world-wide license, with the right to grant sublicenses to research, develop, make, have made, use, have used, offer for sale, sell, have sold, distribute, import, have imported, export and have exported and otherwise exploit the licensed antibodies and licensed products. Upon Kineta’s exercise of the option, Kineta made an upfront payment of cash to GigaGen and issued Kineta equity to GigaGen. In December 2020, Kineta exercised its exclusive option to GigaGen’s intellectual property rights to develop, manufacture and commercialize six antibodies and derivatives identified by GigaGen that target VISTA and subsequently made a cash payment of $400,000 and issued 7,818 shares of common stock to GigaGen per the terms of the VISTA Agreement. In May 2023, Kineta achieved initiation of the Phase 1 clinical trial milestone and incurred $500,000 in fees to Gigagen per the terms of the VISTA Agreement. From inception of the VISTA Agreement through December 31, 2023, Kineta has incurred $1.3 million of expense to GigaGen for the license to certain antibodies and development antibodies.
Under the VISTA Agreement, GigaGen is eligible to receive approximately $20.4 million in development and regulatory milestone payments and up to $8.0 million in sales milestone payments. In addition, GigaGen is eligible to receive low single-digit royalty percentages based on net sales. Kineta is responsible (with input from 20 patients with mild-to-moderate Parkinson’s disease. Patients received once-daily oral dosesGigaGen) for the preparation, filing, prosecution and maintenance of YTX-7739 (20 mg or placebo)all patents and patent applications, and all associated costs.
The VISTA Agreement shall remain in effect on a licensed product-by-licensed product and country-by-country basis, until the expiration of the royalty term for 28 days. YTX-7739 was shown to inhibit its primary target, SCD, an enzyme whose inhibition has been closely linked to neuronal survival and improved motor functiona licensed product in a Parkinson’s disease model.country, which, based on the expiration of the last-to-expire valid claim of the two current patent applications (without any patent term adjustment or extensions) would be February 2042 and March 2044, respectively. Kineta may terminate the VISTA Agreement with 30 days’ written notice to GigaGen. Either party has the right to terminate the VISTA Agreement upon a material breach of the other party that is not cured within 90 days after the breaching party receives written notice of such breach from the non-breaching party.
After 28 days of treatment, the 20 mg dose given once-daily reduced the fatty acid desaturation index (FA-DI), a biomarker of SCD inhibition, by approximately 20%-40%, the range expected to be clinically relevant based on preclinical studies. Target engagement inLicense Agreement with GigaGen, Inc.-CD27
4
In June 2021, Kineta entered into an Option and License Agreement with GigaGen, as amended in July 2022, December 2022, May 2023 and December 2023 (such agreement, as amended, the cerebrospinal fluid suggested“CD27 Agreement”) to in-license certain intellectual property rights and antibodies for the CD27 drug program. Pursuant to the terms of the CD27 Agreement, GigaGen granted Kineta an exclusive (even as to GigaGen) world-wide license, with the right to grant sublicenses to research, develop, make, have made, use, have used, offer for sale, sell, have sold, distribute, import, have imported, export and have exported and otherwise exploit the licensed antibodies and licensed products. Upon Kineta’s exercise of the option, Kineta made an upfront payment of cash to GigaGen and issued shares of common stock to GigaGen. From inception of the CD27 Agreement through December 31, 2023, Kineta has incurred $80,000 of expense to GigaGen to maintain its rights to exercise its license to certain antibodies and development antibodies. In January 2024, Kineta exercised its exclusive option to GigaGen’s intellectual property rights to develop, manufacture and commercialize antibodies and derivatives identified by GigaGen that YTX-7739 crossedtarget CD27 and subsequently incurred license fees of $100,000 and issued 91,240 shares of common stock to GigaGen per the blood-brain barrier. Additionally,terms of the PK/PD profile of YTX-7739 was consistent with previous studies and we believe informs dose selection for future studies.CD27 Agreement.
YTX-7739 was generally well tolerated with
24
Under the CD27 Agreement, GigaGen is eligible to receive approximately $20.4 million in development and regulatory milestone payments and up to $11.0 million in sales milestone payments. In addition, GigaGen is eligible to receive low single-digit royalty percentages based on net sales. Kineta is responsible (with input from GigaGen) for the preparation, filing, prosecution and maintenance of all treatment emergent adverse events being mild to moderate in severity. There were no serious adverse events. Moderate AEs in the active treatment group consisted of 2 patients with increased Parkinson’s symptoms, 2 patients with lower back pain, 1 patient with headache, 1 patient with myalgia, 1 patient with insomnia, 1 patient with ligament strain,patents and 1 patient with vaccination complication. One patient on placebo had moderate worsening of tremorspatent applications, and Parkinsonism, which led to discontinuation. AEs occurring at a higher percentage in 2 or more patients administered YTX-7739 compared to placebo were procedural pain, myalgia, dry eye, hyperbilirubinemia, hypesthesia, lower back pain, and constipation. AEs occurring at a higher percentage with placebo included orthostatic hypotension, headache, tremor, fatigue and dizziness.all associated costs.
As expected, after only 28 days of dosing, there were no statistically significant differencesThe CD27 Agreement shall remain in clinical assessments (UPDRS III, MoCA) or most exploratory biomarkers. Additionally, qEEG assessmentseffect on a licensed product-by-licensed product and country-by-country basis, until the expiration of the effect of YTX-7739 on brain activity were completedroyalty term for a licensed product in a subsetcountry. Kineta may terminate the CD27 Agreement with 30 days’ written notice to GigaGen. Either party has the right to terminate the CD27 Agreement upon a material breach of 8 patients and demonstrated a statistically significant change compared to baseline, suggestivethe other party that is not cured within 90 days after the breaching party receives written notice of a potential improvement in synaptic function. We plan to further validatesuch breach from the role of this diagnostic marker in future clinical studies.non-breaching party.
Out-License Agreements
Merck & Co., Inc.
In January 2022, the U.S Food and Drug Administration (FDA) placed a partial clinical hold on multidose clinical trials of YTX-7739. The partial clinical hold suspends initiation of multiple dose clinical trials in the U.S. until the FDA’s questions have been addressed. The FDA has not halted all clinical programming and is permitting our planned single dose clinical trial to proceed. We anticipates working closelyconnection with the FDAMerger, Kineta became the successor in interest to adequately address their concerns. While we work to address the FDA’s concerns, we have paused our planned window-of-opportunity clinical study of YTX-7739 in glioblastoma multiforme patients and exploration of additional indications.
Two other programs are novel targets for the treatment of ALS and FTLD. Activities for these potential programs are currently being conducted through a research collaboration with Merck, with up to $530 million in potential milestones for us plus royalties. If these potential programs are successful and achieve full target validation with small molecule agents, they will advance to IND-enabling safety pharmacology and toxicology studies to be conducted by Merck, who will also be responsible for any subsequent clinical development and commercialization.
Beyond these programs, we have additional targets that constitute a rich discovery pipeline.
Leadership Team and Scientific Team
We are led by a team of seasoned executives with prior experience in both public and private companies as well as small and large biopharmaceutical companies. The management team has deep expertise in neurology, neuroscience, rare diseases, and oncology.
Our Chief Executive Officer is Richard Peters, M.D., Ph.D., the former President, Chief Executive Officer and director at Merrimack Pharmaceuticals, and former Senior Vice President and Head of Global Rare Diseases at Sanofi Genzyme. Mike Wyzga is our Chief Financial Officer, and he was a former Vice President on the Healthcare Investment Banking Team at Needham & Company and also worked within the Healthcare Corporate and Investment Banking group at Citigroup. Paulash Mohsen is our Chief Business Officer, and he was the former Country Manager (Canada) at Cubist (acquired by Merck) and Vice President of Strategy and Business Operations at Optimer Pharmaceuticals (acquired by Cubist), and also a Vice President of Strategy and Multi-Channel Management at Pfizer.
Our co-founder and chairman of our board of directors is N. Anthony Coles, M.D., previously the Chief Executive Officer of Onyx Pharmaceuticals until its sale to Amgen in 2013, and current Chief Executive Officer of Cerevel Therapeutics. Our other co-founder is the late Susan Lindquist, Ph.D., a renowned cell biologist, National Medal of Science recipient, Howard Hughes Medical Institute investigator and former director of the Whitehead Institute.
Our Discovery Engine Platform
Overview – Protein Misfolding and Toxicity Cascades
DNA is the foundational code for all proteins. The information held in DNA, in our genes, is transcribed first into RNA and then translated into linear strands of amino acids, the building blocks of all proteins found within cells. The linear strands of amino acids then fold in very precise, highly complex ways to form proteins, each having defined shapes and structures that enable them to carry out their normal biological function. When protein folding goes awry, critical functions of proteins may be lost, or new, abnormal functions may be gained.
5
Protein misfolding plays a key role in the initiation and progression of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, dementia with Lewy bodies, MSA, ALS and FTLD. In each of these diseases, as culprit proteins misfold, they form aggregates that may combine into plaques, which form sticky deposits in the brain cells or in brain tissue. These aggregates and plaques can interfere with normal cellular function in a number of ways. For example, they may interfere with the directed movement, or trafficking, of proteins or lipid-bound vesicles within cells, or they may trigger inflammatory reactions. They may also impede chemical and enzymatic processes. Ultimately, these aggregates and plaques result in nerve cell damage and cell death.
A revolution in genetics over the past 15 years has led to the identification of genetic risk factors for neurodegenerative diseases and a number of genes that can be causally tied to protein misfolding processes. Patients who inherit mutations in a single, specific gene generally present with early-onset and aggressive forms of disease. Genetic data have enabled the development of animal and cellular pathology models based on overexpression of disease-causing genes. While undoubtedly an important advance, these models often do not replicate the full features of disease pathology. As a result, there is no conclusive demonstration to date that simply reducing the levels of misfolded proteins reduces the neurodegenerative pathology or presents an efficacious therapeutic approach to the treatment of neurodegenerative disease in humans.
Protein aggregate formation occurs in different places. In Alzheimer’s disease, protein aggregates are found in extracellular plaques, but in diseases like Parkinson’s disease and ALS, misfolded proteins tend to aggregate and create toxic effects inside of brain cells. These intracellular protein aggregates are protected by a cell membrane and therefore lie beyond the typical reach of protein-based drugs, such as antibodies, that do not effectively cross cell membranes. Ongoing trials seeking to use antibody therapy to bind to the misfolded proteins can only do so while proteins are outside cell membranes, which is a relative minority of their life cycle.
The toxic consequences of protein misfolding and aggregation ultimately result in cell death, and the accumulation of dead cells within specific brain regions marks the progression of disease symptoms and severity. It is our goal to keep cells alive by protecting them from the consequences of these misfolded proteins, thereby slowing disease progression. Our discovery engine is designed to better understand the processes through which protein misfolding and aggregation trigger cellular toxicity, and to do so in a manner that allows us to identify a network of new targets and biological processes which, when modulated using a therapeutic drug, we believe will ameliorate the toxicities initiated by protein misfolding, allow the cells to continue to function normally, and halt the progression of disease. We believe the identification of these new targets and close-in biology networks, and the molecules that modulate them, enables us to provide a new treatment approach to neurodegenerative drug discovery.
Our Programs
We have leveraged our discovery engine to identify more than twenty diverse biological targets not previously linked to neurodegenerative disease that we believe are suitable for a future disease-modifying drug discovery program. Our lead program, YTX-7739 for the potential treatment and disease-modification of Parkinson’s disease, is currently in Phase 1 clinical trials. In collaboration with Merck, we are also currently in the lead optimization process for another potential program for the potential treatment of ALS and FTLD.
Our Lead Program – YTX-7739
We have been developing YTX-7739 for the treatment of Parkinson’s disease. YTX-7739 is the first prioritized drug candidate identified by our discovery engine and is designed to reduce α-synuclein toxicity by inhibiting SCD, an enzyme that metabolizes saturated fatty acids to their monounsaturated form.
Parkinson’s Disease Overview
Parkinson’s disease is a chronic, progressive neurological disorder of the central nervous system and the second most prevalent neurodegenerative disorder in the United States after Alzheimer’s disease, with an estimated 500,000 to one million prevalent cases. More than 10 million people worldwide are believed to have Parkinson’s disease. In patients with Parkinson’s disease, the premature death of neurons in the brain reduces levels of the neurotransmitter dopamine, causing motor dysfunction including tremor, slow movement, muscle rigidity, and difficulty with balance, falling, swallowing, speech, and writing. Other changes in the brain associated with the disease may result in cognitive and sensory symptoms. Additional features of the disease include disruptions in nerves connecting the brain to other systems such as cardiovascular, gastrointestinal, and urogenital systems, as well as sleep disturbances, constipation, and loss of sense of smell. Later-stage Parkinson’s disease is severely debilitating, and its symptoms make sufferers more likely to experience life-threatening medical issues. As a result, while Parkinson’s does not directly cause death, it is nevertheless the fourteenth leading cause of death in the United States.
Approximately 60,000 people are diagnosed with Parkinson’s disease in the United States each year, most often after the age of 50. The National Institutes of Health estimates the annual cost of treating Parkinson’s disease in the United States to be $14 billion, with indirect costs such as lost productivity adding at least another $6 billion. As with many other neurodegenerative diseases, the greatest
6
risk factor for Parkinson’s disease is increasing age. The growing population of older adults and longer average lifespans are therefore likely to increase the number of Parkinson’s patients and the need for effective treatments. Currently, it is estimated that the number people diagnosed with Parkinson’s disease in the United States will double by the year 2040.
Although certain Parkinson’s disease cases have been associated with rare gene mutations, both hereditary and environmental factors are likely to contribute to the occurrence of Parkinson’s disease in the majority of cases. Additionally, while the biological cause of most cases of Parkinson’s disease is not clear, the core pathology of Parkinson’s disease is degeneration of the dopaminergic neurons in the midbrain. In certain cases, cell loss occurs in association with the formation of intraneuronal Lewy inclusion bodies. Abnormally aggregated α-synuclein is the principal component of Lewy bodies, which are the pathological hallmark of Parkinson’s disease. The presence of Lewy bodies and other associated fibrils is correlated with neuron loss and death, decline in motor function and cognitive dysfunction.
Limitations of Current Therapies
There is currently no known cure for Parkinson’s disease. Pharmacological therapies for Parkinson’s disease are aimed at either temporarily replenishing dopamine or mimicking the action of dopamine. They generally help reduce muscle rigidity, improve speed and coordination of movement and lessen tremor.
The dopamine precursor levodopa is the most commonly prescribed pharmacotherapy. While extremely helpful overall, some symptoms do not respond as well to levodopa, like difficulty with balance, falling, difficulty with speech and swallowing, and memory issues. It can also be challenging to titrate and find the optimal dose and patients may experience “on” and “off” periods when the drug concentration falls to below their individual needs. Unfortunately, the long-term use of levodopa is frequently associated with the development of additional motor complications, for example dyskinesias, or uncontrolled, involuntary movements. Additional therapies attempt to slow the degradation of dopamine using monoamine oxidase-B inhibitors. Catechol-o-methyltransferase inhibitors and carbidopa may be used to reduce levodopa degradation. Amantadine, an N-methyl-D-aspartate, or NMDA, receptor antagonist, is also used and may act through more than one mechanism. Drug treatment for Parkinson’s disease is commonly individualized by patient and disease characteristics, and patients may receive multiple drug therapies throughout their course of disease, including other medications for comorbid conditions and symptomatic management such as antidepressants, anxiolytics, and anti-psychotics. While these therapies address the symptoms of Parkinson’s disease, they are unable to halt disease progression over the longer-term. As a result, these symptomatic therapies lose their efficacy over time, leaving patients with few treatment options.
Outside of drug therapy, electrical deep brain stimulation is also be used to control motor symptoms of Parkinson’s disease, typically in the advanced stages of disease. In addition to pharmacotherapy, a holistic approach to treatment is encouraged and patients may gain benefit from regular exercise, psychological, physical, occupational and speech therapy, nutrition consultation, education, support groups, and the use of assistive devices and caregiver relief.
There are currently a small number of early clinical trials investigating the potential of directly reducing α-synuclein to change the course of the disease. These programs, however, are predominantly based on antibody therapy which, due to their large size, do not readily enter the brain or brain cells and are believed to interact only with extracellular α-synuclein that has been secreted or released from cells. Formation of pathological α-synuclein aggregates, known as Lewy bodies, occurs inside of cells, and the ability of therapeutic antibodies to impact consequent toxic α-synuclein cascades is unclear.
Our Solution
YTX-7739 is a small molecule with potential to slow or halt disease progression in patients suffering with Parkinson’s disease. Using our discovery engine and Parkinson’s disease patient cell lines, we identified SCD as a biological target enzyme for diseases caused by α-synuclein-mediated toxicity. YTX-7739 is designed to inhibit SCD to reduce α-synuclein-mediated toxicity within cells.
The α-Synuclein Toxicity Cascade
Aggregated α-synuclein protein is the primary constituent of the pathological Lewy bodies formed in the brains of patients with Parkinson’s disease. Although its precise molecular function is poorly understood, α-synuclein is known to be a membrane-associated lipid binding protein and has been implicated in vesicle trafficking, a process by which cells transport materials between different cellular destinations, as well as in membrane curvature and fusion.
7
In yeast, the overexpression of α-synuclein causes severe cellular toxicity by disrupting multiple cellular mechanisms, including vesicle trafficking. These disruptions occur in both yeast models and mammalian systems, where mutations or overexpression of α-synuclein increases the amount of membrane-associated α-synuclein, which in turn impairs vesicle trafficking and increases cellular stress and toxicity. Importantly, human genetic studies indicate mutations and overexpression of α-synuclein lead to severe and rapidly progressing forms of Parkinson’s disease, and this relationship to disease severity almost certainly involves toxic effects of α-synuclein aggregates on the functions of cell membranes.
YTX-7739’s Target
We discovered SCD as a result of our unbiased phenotypic screening efforts, which identified a series of compounds that potently protected cells against α-synuclein-mediated toxicity. Using our discovery engine’s target identification capability, we were able to identify that the specific biological target implicated in this protection was inhibition of Ole1, the single yeast fatty-acid desaturase enzyme and direct counterpart of SCD in humans.
Ole1 in yeast and SCD in humans are enzymes that metabolize saturated lipids and break them down into their unsaturated lipid components. Unsaturated lipids are important components of cell membranes because of the processes they regulate, including membrane fluidity, curvature, and fusion. Paradoxically, the greater the level of unsaturated lipid in membranes, the greater the vesicle trafficking impairment and toxicity caused by α-synuclein. Inhibiting SCD enzymatic activity reduces the levels of unsaturated lipids, which ameliorates the detrimental vesicle trafficking defects associated with increased α-synuclein expression. Our hypothesis is that reducing SCD activity will reduce abnormal vesicle trafficking within cells caused by α-synuclein, thereby reducing the accumulation of neurotoxicity and slowing the progression of neurologic impairment in patients with Parkinson’s disease and related disorders.
The chart below demonstrates the impact of inhibiting the SCD enzyme in a diseased human cell line prepared from a patient with Parkinson’s disease. This patient had a single amino acid mutation in the α-synuclein protein sequence. The red line shows the risk of cell death in cells containing the α-synuclein mutation. The black line, which shows lower risk of death, is a control cell line – genetically identical to the Parkinson’s disease patient cell line but with a correction of the α-synuclein mutation generated using CRISPR technology. The increasing shades of blue lines represent the survival of the mutation-containing cells upon exposure to increasing concentrations of a potent SCD inhibitor. As the chart shows, inhibition of SCD reduces cell death in the mutation-containing cell line down to the levels of the mutation-corrected control. The improved survival effect is dependent upon the concentration of SCD inhibitor.
Cell death in Parkinson’s patient cell line corrected by inhibition of SCD
In addition to demonstrating that inhibition of SCD can protect human neurons grown in a dish, we have also explored the effects of SCD inhibition in a new mouse model of Parkinson’s disease. This mouse was engineered to express a mutant version of human α-synuclein, which leads to progressive motor deficits and pathological neuron cell loss in the brain that are similar to the progression seen in Parkinson’s disease. Dopamine replacement therapy, the standard of care in Parkinson’s therapy, can partially reverse the motor deficits in these engineered mice, demonstrating the disease-relevance of the model. When these mice were administered YTX-7739 for 4 months, the expected motor deficits never developed, whereas similar mice in the same study that received placebo evidenced the expected altered motor behaviors. Interestingly, when the SCD1 gene was removed in these engineered α-synuclein
8
mice, known as a gene knockout, which would mimic the effects of an SCD inhibitor, these mice also had significantly reduced motor deficits and pathological neuron loss. These studies demonstrate the consistent effect of reducing SCD activity and validate SCD as a target for reducing the toxic effects of α-synuclein in disease-relevant models.
Motor Behavior at Six Months of Age
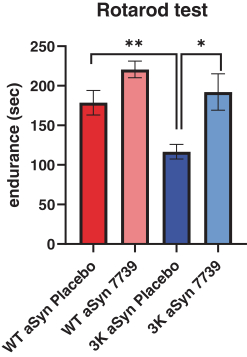
While we have established that SCD inhibition protects cells from α-synuclein toxicity, the precise mechanism of protection has not been defined. We believe there are at least three possible mechanisms of action: (1) SCD inhibition reverses a toxic increase in fatty acid desaturation triggered by α-synuclein aggregation, (2) SCD inhibition directly antagonizes toxic effects of α-synuclein on membrane properties and/or trafficking, or (3) reduced fatty acid desaturation ameliorates a direct toxic interaction of α-synuclein with cell membranes.
The knowledge of SCD enzyme biology also allows us to define a biomarker that can be used to measure target inhibition. Specifically, because the substrates for SCD are sixteen-carbon, or C16, or eighteen-carbon, or C18, saturated fatty acids, and the products are C16 and C18 monounsaturated fatty acids, we can therefore monitor drug effects on SCD by measuring the amount, or ratio, of the C16 and C18 precursors and their monounsaturated products. The result of this analysis gives us the fatty-acid desaturation index, or FA-DI, expressed as a ratio of the amount of monounsaturated C16 or C18 substrates divided by the amount of corresponding saturated fatty acid. The FA-DI gives us a biomarker that allows it to measure the effects of its compounds on SCD in vitro and in vivo.
Clinical Trials
YTX-7739 is currently in Phase 1 clinical development consisting of a single ascending dose (SAD) study in healthy volunteers, and a multiple ascending dose (MAD) study in healthy volunteers, with a Phase 1b part in patients with Parkinson’s disease. The SAD study was a randomized, double-blind, placebo-controlled single ascending dose study to investigate the safety, tolerability, and pharmacokinetics of YTX-7739 in healthy volunteers, and has completed. The SAD study was conducted in three parts at one site in the Netherlands. The first part was to investigate the safety, tolerability and pharmacokinetics of increasing doses of YTX-7739 in healthy subjects. The second and third parts were to study the effect of food on the pharmacokinetics of YTX-7739 after administration in a fed state in healthy subjects.
Fifty-six healthy volunteers (aged 19-39 years of age; 22 males; 34 females) were administered single oral dose of YTX-7739, from 5 mg to 400 mg in the SAD study. Forty subjects participated in the placebo controlled, randomized, double blind part of the study which included seven cohorts of eight subjects each, randomized to YTX-7739 or placebo in a 6:2 ratio. Sixteen of these subjects also participated in two cohorts where YTX-7739 was administered with food. In addition, two cohorts of eight subjects each (16 subjects
9
in total) participated in an open label fashion to further inform dose selection for the MAD study. Safety assessments included, but were not limited to, adverse events, serious adverse events, safety laboratory tests, vital signs and electrocardiograms (ECG). In addition, plasma drug levels were collected to assess pharmacokinetic variables and a pharmacodynamic biomarker was also included to explore the potential of YTX-7739 to change plasma levels of fatty acids. There were no safety concerns identified and YTX-7739 was found to be generally well tolerated with most adverse events being mild or moderate in severity. The half-life of YTX-7739 combined with a favorable dose-proportional pharmacokinetic (PK) profile, in the fed state, supports that low daily doses administered with food will sustain the target range of exposure.
There were dose-linear increases in the maximum serum concentration achieved and the 24-hour area under the concentration-time curve observed at 5 mg, 10 mg, 30 mg and 250 mg dose levels in the fed state, suggesting dose-proportionality of YTX-7739 exposure in the 5 – 250 mg dose range, when administered in a fed state. Drug plasma concentrations in the trial exceeded levels of exposure estimated to be sufficient for target engagement based on pharmacodynamic modeling. Consistent with preclinical data, YTX-7739 also demonstrated clinically relevant drug concentrations in the cerebral spinal fluid (CSF). The results of this SAD trial supported progression to the MAD trial.
On November 10, 2021, we announced top-line data from our Phase 1b clinical trial of YTX-7739 in patients with mild-to-moderate Parkinson’s disease. The Phase 1b clinical trial was a randomized, placebo-controlled, double-blind multi dose study to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics of YTX-7739. Data were reported from 20 patients with mild-to-moderate Parkinson’s disease. Patients received once-daily oral doses of YTX-7739 (20 mg or placebo) for 28 days.
YTX-7739 was shown to inhibit its primary target, SCD, an enzyme whose inhibition has been closely linked to neuronal survival and improved motor function in a Parkinson’s disease model. After 28 days of treatment, the 20 mg dose given once-daily reduced the fatty acid desaturation index (FA-DI), a biomarker of SCD inhibition, by approximately 20%-40%, the range expected to be clinically relevant based on preclinical studies. Target engagement in the cerebrospinal fluid suggested that YTX-7739 crossed the blood-brain barrier. Additionally, the PK/PD profile of YTX-7739 was consistent with previous studies and we believe informs dose selection for future studies.
YTX-7739 was generally well tolerated with all treatment emergent adverse events being mild to moderate in severity. There were no serious adverse events. Moderate adverse events (AEs) in the active treatment group consisted of 2 patients with increased Parkinson’s symptoms, 2 patients with lower back pain, 1 patient with headache, 1 patient with myalgia, 1 patient with insomnia, 1 patient with ligament strain, and 1 patient with vaccination complication. One patient on placebo had moderate worsening of tremors and Parkinsonism, which led to discontinuation. AEs occurring at a higher percentage in 2 or more patients administered YTX-7739 compared to placebo were procedural pain, myalgia, dry eye, hyperbilirubinemia, hypesthesia, lower back pain, and constipation. AEs occurring at a higher percentage with placebo included orthostatic hypotension, headache, tremor, fatigue and dizziness.
As expected, after only 28 days of dosing, there were no statistically significant differences in clinical assessments (Unified Parkinson's Disease Rating Scale Part III (UPDRS III), Montreal Cognitive Assessment (MoCA)) or most exploratory biomarkers. Quantitative electroencephalogram (qEEG) assessments of the effect of YTX-7739 on brain activity were completed in a subset of 8 patients and demonstrated a statistically significant change compared to baseline, suggestive of a potential improvement in synaptic function.
Our Potential Additional Programs
We currently have two preclinical programs that are being progressed through a research collaboration with Merck. On December 17, 2021, Merck notified us that the first data package that we submitted for one program met the requirements to progress to the next stage of the research collaboration. Achievement of this milestone triggered a $5.0 million milestone payment from Merck, which was received in February 2022. We cannot provide assurance as to the timing of future milestones or royalty payments or that we will receive any of the future payments at all. Merck will be responsible for IND-enabling toxicology and safety pharmacology studies, as well as subsequent clinical development and commercialization.
ALS and FTLD Disease Overview
ALS, also referred to as Lou Gehrig’s disease, is a neurodegenerative disease that affects nerve cells in the brain and the spinal cord. In healthy individuals, upper motor neurons in the brain send signals to lower motor neurons in the spinal cord and brainstem, which send signals to muscles, thereby generating body movement. In ALS, both the upper motor neurons and the lower motor neurons degenerate or die, resulting in loss of muscle function with progression to severe impairment of mobility, speech, and communication.
Early symptoms of ALS usually include muscle weakness or stiffness, and over time, all muscles under voluntary control are affected. As ALS progresses, individuals lose their strength and the ability to speak, eat and move. Many sufferers lose the muscular ability to maintain breathing, requiring permanent ventilatory support. Individuals with ALS usually retain their ability to perform higher mental
10
processes such as reasoning, remembering, understanding, and problem solving, so they are entirely aware of their progressive loss of muscle function. Continued deterioration of muscle control leads to respiratory failure and death, with an average survival time of three years. About 20 percent of people with ALS live five years, 10 percent will survive 10 years and 5 percent will live 20 years or longer.
According to the ALS Association, in 2016, between 14,000 and 15,000 Americans had ALS. ALS is most commonly diagnosed between the ages of 55 and 75, and only ten percent of people with ALS will survive for ten years or more. Medical and non-medical costs of ALS, including lost income, range between $256 million and $433 million each year in the United States, with annual costs from ALS exceeding $60,000 per patient.
FTLD is an umbrella term for a group of related syndromes and processes, often also called frontotemporal lobar dementia, frontotemporal degeneration or dementia or Pick’s disease, that impacts the frontal and temporal lobes of the brain. Formally, the process of frontotemporal degeneration results in the condition of frontotemporal dementia. Disease processes cause the degeneration of neurons and shrinking of the frontal and temporal brain regions, which causes progressive alterations in personality, behavior, and language. There are different types of FTLD, which manifest as a frontal or behavioral variant affecting behavior and personality, or as a primary progressive aphasia variant, which results in difficulty communicating due to loss of speech and inability to use and understand language. FTLD patients often exhibit aggressive and compulsive behaviors and have various changes in sexual behaviors.
It is believed that the prevalence of FTLD in the United States is around 60,000 cases. There is a wide range of onset, from 21 to 80 years of age, with most cases occurring between 45 and 64 years of age. Given the younger age of onset as compared to Alzheimer’s disease, FTLD has been cited as the most common form of dementia in people under 60. Annual medical and nonmedical costs of FTLD are estimated at approximately $120,000 per patient, indicating a societal disease impact in the United States alone of over $7 billion annually.
These conditions are generally believed to exist as a spectrum disorder, with “pure” ALS as a neuromuscular disorder at one end of the spectrum, and “pure” FTLD as a dementia-related disorder at the other. Many patients exhibit varying degrees of both types of symptoms on this spectrum.
Limitations of Current Therapies
There is no known cure for ALS. The cause of ALS in 90% or more of cases is unknown. Known as sporadic ALS, genetic and environmental factors may play a role in these cases. In contrast, up to 10% of all ALS cases are of an inherited familial form. A number of genetic mutations have been implicated in familial ALS, most frequently C9orf72 and SOD1. In all cases of ALS, upper and lower motor neurons are lost, causing muscle dysfunction and atrophy. Two drugs have been approved in the United States for the treatment of ALS, riluzole and edaravone. Riluzole has demonstrated a survival benefit and edaravone delayed decline in an assessment of daily functioning.
There are also no known cures for FTLD, nor are there any approved medications for this disease. Patients are sometimes proscribed Riluzole, although its effectiveness in this indication is uncertain. To manage quality of life, antidepressants are prescribed to help with anxiety and obsessive-compulsive behaviors, and anti-psychotics can sometimes help control irrational and risky behavior. Sleep aids are also prescribed to help with insomnia and sleep disturbances.
Competition
The biotechnology and pharmaceutical industries, including in the neurodegenerative disease field, are highly competitive and subject to rapid and significant technological change. While we believe that its discovery engine platform and its employees and consultants, scientific knowledge and development experience provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical, and biotechnology companies, academic institutions, governmental agencies, and public and private research institutions. Several of these entities have commercial products, robust drug pipelines, readily available capital, and established research and development organizations. Many of our competitors may have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical, biotechnology, and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Small or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. The key competitive factors affecting the success of our product candidates, if approved, are likely to be their efficacy, safety, convenience, price, the level of branded and generic competition, and the availability of reimbursement from government and other third-party payors.
11
Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future, including but not limited to:
Collaboration Agreement with Merck
In June 2020, we entered into an exclusive license and research collaboration agreement orwith Merck & Co., Inc. to support research, development and commercialization of products for treatment of amyotrophic lateral sclerosis and frontotemporal lobar dementia (the “Merck Neuromuscular License Agreement”).
On June 29, 2023, Kineta achieved a development milestone which triggered a $5.0 million payment. Merck will continue to advance the research program for the ALS pipeline, one of the two pipeline programs licensed under the Merck Collaboration Agreement,Neuromuscular License Agreement. As a result, the Company is eligible to receive up to an additional $255.0 million in development milestones and royalties on net sales. Following this milestone, Merck assumed sole responsibility for all future development and commercialization for the ALS program.
Genentech, Inc.
In connection with Merckthe Merger, Kineta became the successor in interest to an exclusive technology transfer and license agreement with Genentech, Inc. to support research, development and commercialization of a small molecule product with an undisclosed target (the “Genentech Small Molecule License Agreement”).
FAIR Therapeutics, B.V.
In connection with the Merger, Kineta became the successor in interest to an exclusive license agreement with FAIR Therapeutics, B.V. to support research, development and commercialization of products for the treatment of ALS and FTLD.cystic fibrosis (the “FAIR License Agreement”) .
Pursuant to the Merck Collaboration Agreement, we granted Merck an exclusive, worldwide license with the right to grant and authorize sublicenses, under certain intellectual property rights related to two certain undisclosed targets in connection with our ALS and FTLD programs to make, have made, use, import, offer to sell and sell compounds and products covered by such intellectual property rights. In the event that the exploitation of such compound or product would infringe during the term of the Merck Collaboration Agreement a claim of an issued patent controlled by us, we also granted Merck a non-exclusive, sublicensable, royalty-free license under such issued patent to exploit such compound and product.
Under the terms of the Merck, Collaboration Agreement, weGenentech and Merck are each responsible to perform certain research activities in accordance with a mutually agreed upon research plan. Upon the completion of certain stages of the research plan, Merck will elect to either advance or terminate the applicable research program. Following completion of the research program, Merck is responsibleFair Therapeutics assumed sole responsibility for theall future development and commercialization of the compounds developed pursuantlicensed products. These license agreements offer the potential for Kineta to receive up to ~$1.3 billion in potential milestone payments plus royalties on net sales for any products that make it to market. Kineta does not count potential revenues from these license agreements in the research program and any product containing such compounds.Company’s financial forecasts, but they do offer the potential for significant non-dilutive capital.
As consideration
Clinical Trial Services Agreement
In January 2023, Kineta entered into a Master Services Agreement with PPD Development L.P. (“PPD”) to provide services and support to Kineta in connection with the development and execution of a Phase 1/Phase 2 clinical trial in immuno-oncology (the “PPD Agreement”). Under the PPD Agreement, PPD will assist Kineta with, among other things, identifying clinical sites to participate in the Phase 1/Phase 2 trial of KVA12123 in treating advanced solid tumor cancer patients, identifying potential clinical sites, initiating and opening clinical sites and monitoring and validating research at each site involved in the trial. In addition, PPD will also provide support in preparing and developing interim safety data reports for review and analysis by the licenses grantedindependent safety monitoring committee. Pursuant to Merck under the Merck Collaboration Agreement, Merck paid us a one-time upfront payment and also purchased Class C preferred units of Holdings. Under the terms of the Merck CollaborationPPD Agreement, we are eligible to receive up to $280 million if Merck elects to advanceKineta will pay PPD on periodic basis and will pay the research program and upon achievement of specified research and development milestones, and up to $250 million upon achievement of specified sales-based milestones as well as a tiered, mid-single digit royalty on net sales of licensed products, subject to customary reductions. Merck’s royalty obligations for each licensed product continue on a country-by-country basis untilpass-through costs associated with the later of (i) the last-to-expire valid claimconduct of the patent rights in such country or (ii) the tenth anniversary of the first commercial sale of such product in such country.Phase 1 clinical trial.
On December 17, 2021, Merck notified us that Merck has accepted the first data package from our research collaboration relating to amyotrophic lateral sclerosis, or ALS (also known as Lou Gehrig’s disease) and frontotemporal lobar degeneration, or FTLD, and that
12
Merck has elected to continueThe PPD Agreement expires five years from the research collaboration. Achievementeffective date of this milestone triggers a $5.0 million milestone payment from Merck, which was received in February 2022.
Unless terminated earlier, the Merck CollaborationPPD Agreement will continue in full force and effect until one or more products has received marketing authorization and, thereafter, until expirationunless extended by mutual consent of all royalty obligations under the Merck Collaboration Agreement. We or Merckparties. Either party may terminate the Merck CollaborationPPD Agreement or a project addendum upon an uncured30 days’ prior written notice (and 120 days’ prior written notice for medical information contact center services) and may terminate immediately in the case of insolvency.
Drug Manufacturing Organizations Agreements
Master Development Services Agreement with Samsung Biologics Co., Ltd.
In July 2021, Kineta entered into a Master Development Services Agreement (the “Samsung Agreement”) with Samsung Biologics Co., Ltd. (“Samsung”) to perform biologics development and manufacturing services for the VISTA program. Under the Samsung Agreement, Samsung will provide services pursuant to product-specific agreements, which specify the services to be provided, deliverables, payments due and timelines, in accordance with cGMP, where applicable. The services will be performed at Samsung’s facility and Samsung will maintain manufacturing documentation for the manufacturing process. Kineta will provide adequate materials for Samsung to carry out the services and will pay Samsung pre-negotiated fees for product-specific services related to VISTA.
25
The Samsung Agreement gives Kineta a worldwide, non-exclusive sublicensable, royalty-free license to any Samsung intellectual property or invention that is incorporated into the service deliverables to further develop, manufacture, make, use, sell, offer to sell, export and import certain clinical products. Pursuant to the terms of the Samsung Agreement, Kineta and Samsung will each continue to own their respective background intellectual property and any inventions derived from their respective intellectual property and confidential information.
The Samsung Agreement expires five years from the effective date of the Samsung Agreement and will automatically renew for successive two-year terms unless either party gives the other party written notice of termination at least six months prior to the end of the then-current Samsung Agreement term. Either party may terminate the Samsung Agreement or a product-specific agreement in the event of a material breach by the other party that is not cured within 30 days’ written notice or insolvencyin the event of the other party. Merck may also terminate the Merck Collaboration Agreement for any reason upon certain notice to us.
License Agreement with Whitehead Institute
In February 2016, we entered into a tangible property and exclusive patent license agreement or the Whitehead License Agreement, with the Whitehead Institute which was subsequently amended in April 2016, August 2016 and July 2018. Pursuant to the Whitehead License Agreement, the Whitehead Institute granted us a worldwide license under certain patent rights to develop, commercialize and sell products and to develop and perform processes covered by such patents for the treatment of any disease in humans other than certain specified treatments for infectious diseases and cancer. The Whitehead Institute also granted us a non-exclusive, worldwide license to use certain know-how and to use and make certain biological materials.
The patent rights licensed to us under the Whitehead License Agreement relate to our discovery engine and are directed to compositions and methods for identifying novel disease-modifying therapies for neurodegenerative diseases. Such patent rights include patent rights developed or co-developed by Dr. Lindquist as an employee of Howard Hughes Medical Institute, as well as patents owned or jointly owned by the University of Chicago, the University of Washington, the Massachusetts Institute of Technology, the Curators of the University of Missouri and Pfizer, Inc. Our license under such patent rights is exclusive, subject to certain retained rights and certain patent rights previously licensed by the Whitehead Institute. Additionally, our exclusivity with respect to patent rights jointly owned by Pfizer, Inc. only applies to the Whitehead Institute’s right as a joint owner of such patents.
Under the terms of the Whitehead License Agreement, we must use commercially reasonable efforts to develop licensed products or processes and to introduce such licensed products or processes into the commercial market. Thereafter, we are required to use commercially reasonable efforts to make such licensed products or processes reasonably available to the public. In addition, in any given year until the first regulatory approval of a licensed product, we have diligence requirements to achieve a certain development milestone with respect to a licensed product or expend a minimum amount of money for platform development and/or development of licensed products or processes.
As consideration for the licenses granted to us under the Whitehead License Agreement, we paid an initial license fee and reimbursed the Whitehead Institute for certain expenses incurred in connection with the patent rights. Holdings also issued 3,000 common units (which, following a 100:1 unit split, represented 300,000 units) to the Whitehead Institute and certain persons and entities as directed by the Whitehead Institute in satisfaction of the Whitehead Institute’s policy on equity sharing. Under the terms of the Whitehead License Agreement, we are also required to pay an annual license maintenance fee which is creditable against royalties on net sales earned during the same calendar year. In addition, we are obligated to make payments to the Whitehead Institute upon achievement of certain milestones, the amount of which depends on the licensed product and indication, with up to an aggregate of $1.9 million for each of the first two licensed products for the first indication and less for subsequent licensed products and additional indications. We are also required to pay the Whitehead Institute a low single digit royalty percentage of net sales of licensed products and a low single digit royalty on net sales of products determined to have biological activity or utility by the use of a licensed product or process, or Identified Products. Additionally, we are required to pay a mid single to low double digit percentage of certain income received from sublicensees and certain partners. Our royalty obligation continues on a licensed product-by-licensed product and country-by-country basis for so long as the manufacture, use or sale of such licensed product in such country infringes a valid claim of the patent rights or, with respect to each Identified Product, for ten years after the first sale for consumption by an end user patient of such Identified Product.
Unless terminated earlier, the Whitehead License Agreement will expire upon the expiration or abandonment of all issued patents and filed patent applications within the licensed patent rights, which is currently projected to occur in 2035. The Whitehead Institute may terminate the Whitehead License Agreement upon notice to us if we cease to carry on our business related to the Whitehead License Agreement, if we fail to make required payments within a certain period time or if we commit a material breach and fail to cure such breach within a certain period of time. The Whitehead Institute may also terminate the Whitehead License Agreement and/or the licenses granted to us if we bring or assist others in bringing a patent challenge against the Whitehead Institute. We may terminate the Whitehead License Agreement for any reason upon certain notice to the Whitehead Institute.insolvency.
Intellectual Property
The proprietary natureKineta has established a broad intellectual property portfolio, including patent applications covering the composition of or protection for, ourKineta’s product candidates and methodsrelated technology, and other inventions that are important to Kineta’s business. Kineta works with its outside patent counsel to employ various life-cycle management patent strategies, such as managing public disclosures prior to patent application filing, timing of manufacturefiling the patent application, drafting clear claims language and clinical use are an important part of our strategy to develop and commercialize novel therapies. We have filed numerousfiling follow-on patent applications pertainingfor patents on new drug formulations and new indications (such as pediatrics or rare diseases), all of which optimize the value of the patent portfolio and can extend the product life cycle, giving Kineta an advantage for extended patent term and a broader scope of protection for novel technologies. Kineta seeks to our product
13
candidatesmaximizes patent term restoration and clinical use. We strivepatent term adjustment opportunities. When appropriate, Kineta also takes advantage of the Patent Prosecution Highway (“PPH”), which is a framework that reduces duplication of effort of multiple patent offices. The PPH allows the patent office in a country of a second filing to protect and enhancetake advantage of the proprietary technology, inventions, and improvements that are commercially importantwork of the patent office in the country of first filing by allowing the country of a second filing to use the search results related to the development of its business by seeking, maintaining and defending our intellectual property, whether developed internally or licensed from third parties. We also rely on trade secrets, know-how, continuing technological innovation, and potential in-licensing opportunities to develop, strengthen, and maintain our proprietary positionallowed claims in the fieldfirst country, accelerating the examination process, increasing the allowance rate of neurodegenerative diseasesclaims and protein misfolding. Additionally, we intend to rely on regulatory protection afforded through data exclusivity and market exclusivity, as well as patent term extensions, where available.reducing the number of office actions issued for an application.
As of March 15, 2022, ourJanuary 31, 2024, Kineta’s patent portfolio as it pertains to certain of its key product candidates included:
In addition, we have in-licensed an estate of patents and patent applications relating to our discovery engine from the Whitehead Institute which is directed to compositions and methods for identifying novel disease-modifying therapies for neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and ALS. For example, this estate includes granted and pending claims to a number of yeast models of protein misfolding and methods of use thereof. As of March 15, 2022, this estate includes twenty-four granted U.S. patents, projected to expire between 2022-2035; one pending U.S. patent applications, which if granted in the U.S., would be expected to expire in 2038, without taking a potential patent term adjustment or extension into account; thirty-one granted foreign patents, projected to expire between 2025-2035; and eight pending foreign patent applications, which if granted, would be expected to expire between 2034-2038. We do not control the prosecution and maintenance of all of our in-licensed patents and patent applications, and our rights to enforce the patents are limited in certain ways. For additional detail regarding the risks associated with our license agreements, see “Risk Factors—Risks Related to Our Intellectual Property.”September 29, 2044, respectively.
The term of individual patents may vary based on the countries in which they are obtained. Generally, patents issued for applications filed in the United States are effective for 20 years from the earliest effective non-provisional filing date in the absence, for example, of a terminal disclaimer shortening the term of the patent or patent term adjustment increasing the term of the patent. In addition, in certain instances, a patent term can be extended to recapture a portion of the term effectively lost as a result of FDA regulatory review periods. The restoration period cannot be longer than five years and the total patent term, including the restoration period, must not exceed 14 years following FDA approval. The duration of patents outside of the United States varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective non-provisional filing date.
14
In addition to patents, and patent applications that we own and license, weKineta may rely, in some circumstances, on trade secrets and know-how to develop and maintainprotect its competitive position. However, trade secrets can be difficult to protect. Wetechnology. Kineta seeks to protect ourits proprietary technology and processes, and obtain and maintain ownership of certain technologies, in part, throughby confidentiality agreements and invention assignment agreements with ourits employees, consultants, scientific advisors contractors, and commercial partners.contractors. Kineta also seeks to preserve the integrity and confidentiality of its data and trade secrets by maintaining physical security of its premises and physical and electronic security of its information technology systems.
Kineta’s patent strategy focuses on securing market exclusivity through a portfolio of patents and claim sets to ensure broad based protection for Kineta’s innovative technologies. Geographically, Kineta files patents in those countries that account for 90% of the revenue of the global pharmaceutical market as well as several additional markets due to their strategic importance, including the U.S., EU, Japan, Korea, China, India, Singapore, Switzerland, Russia, Canada and Mexico.
Kineta’s patent strategy includes filing for multiple claim sets that include both specific patent claims as well as broader based claims. This approach helps to protect the innovative science at Kineta and to protect its intellectual property. Kineta’s filing strategy includes filing for patent claims for (i) composition of matter, (ii) picture claims and sequences, (iii) product uses and indications, (iv) manufacturing and (v) pharmaceutical properties and characteristics.
The table below summarizes the high-level filing strategy of Kineta’s existing patent portfolio:
VISTA | VISTA | Anti-CD27 | |||||
Patent Family | KVA-001 | KVA-002 | KA27-001 | ||||
Composition of matter | Y | Y | Y | ||||
Methods of Manufacturing | Y | Y | Y | ||||
Sequences/Structure | Y | Y | Y | ||||
Indications | Y | Y | Y | ||||
Specification on use (mono or combo) | Y | Y | Y | ||||
Binding characteristics | Y | Y | Y | ||||
Immune cell regulation | Y | Y | Y | ||||
Physiologic properties | Y | Y | Y | ||||
Discovery Candidates | To be added on a rolling basis | To be added on a rolling basis | To be added on a rolling basis |
26
Kineta strives to protect the proprietary technologies that it believes are important to its business, including by seeking, maintaining and defending patent rights, whether developed internally or in conjunction with or in-licensed from third parties. Kineta also relies on trade secrets relating to its monoclonal antibodies, know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen and maintain its proprietary position in the field of innate immunity and fully human antibodies.
As more fully described above, as of January 31, 2024, Kineta’s patent portfolio included 13 U.S. and foreign applications, which entered national phase in 2023. The portfolio also included a provisional application in the KVA-002 family related to VISTA, scheduled for PCT filing early in 2024, and a provisional application in the KA27-001 family related to anti-CD27 antibodies, scheduled for PCT filing later in 2024.
Our future commercialKineta also relies on trade secrets and careful monitoring of its proprietary information to protect aspects of its business that are not amenable to, or that Kineta does not consider appropriate for, patent protection.
Kineta’s success depends, in part,will depend significantly on ourits ability to obtainto:
Although Kineta takes steps to stopprotect its proprietary information and trade secrets, including through contractual means with its employees and consultants, third parties from making, using, selling, offeringmay independently develop substantially equivalent proprietary information and techniques or otherwise gain access to sell,Kineta’s trade secrets or importingdisclose its technology. Thus, Kineta may not be able to meaningfully protect its trade secrets.
In addition, a third party may hold intellectual property, including patent rights that are important or necessary to the development of Kineta’s products. It may be necessary for Kineta to use the patented or proprietary technology of third parties to commercialize its products, may dependin which case Kineta would be required to obtain a license from these third parties on the extent to which we have rights under valid and enforceable patentscommercially reasonable terms, or trade secrets that cover these activities. With respect to our intellectual property, we cannotKineta’s business could be sure that patents will issue from anyharmed, possibly materially. For example, certain of the pending patent applicationsmethods for Kineta’s fully human antibodies are covered by patents held by third parties. Although Kineta has obtained exclusive licenses to which we own or that we may file in the future, nor can we be sure that anythese patents that may be issued in the future to us will befrom these third parties on what Kineta believes are commercially useful in protecting our product candidates and methods of using or manufacturing the same. Moreover, we may be unablereasonable terms, if Kineta were not able to obtain a license on similar technology, or were not able to obtain a license on commercially reasonable terms, its business could be harmed, possibly materially.
The patent protection for certain aspectspositions of biopharmaceutical companies like Kineta are generally uncertain and involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Consequently, Kineta does not know whether any of its product candidates generally,will be protectable or remain protected by enforceable patents.
Kineta cannot predict whether the patent applications it is currently pursuing will issue as wellpatents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient proprietary protection from competitors. Any patents that Kineta holds may be challenged, circumvented or invalidated by third parties.
Because patent applications in the United States and certain other jurisdictions are maintained in secrecy for 18 months, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, Kineta cannot be certain of the priority of inventions covered by pending patent applications. Moreover, Kineta may have to participate in interference proceedings declared by the United States Patent and Trademark Office (“USPTO”) or a foreign patent office to determine priority of invention or in post-grant challenge proceedings, such as with respectoppositions, that challenge priority of invention or other features of patentability. Such proceedings could result in substantial cost, even if the eventual outcome is favorable to Kineta.
The term for individual patents depends upon the legal term for patents in the countries in which they are granted. In most countries, including the United States, the patent term is 20 years from the earliest claimed filing date of a non-provisional patent application in that country or the international filing date. In the United States, a patent’s term may, in certain indications. Seecases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the section titled “Risk Factors—Risks RelatedUSPTO in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over a commonly owned patent or a patent naming a common inventor and having an earlier expiration date.
The Drug Price Competition and Patent Term Restoration Act of 1984 permits a patent term extension of up to Our Intellectual Property”five years beyond the expiration date of a U.S. patent as partial compensation for the length of time the drug is under regulatory review while the patent is in force. A patent term extension cannot extend the remaining term of a more comprehensive descriptionpatent beyond a total of risks related14 years from the date of product approval. Only one patent applicable to our intellectual property.each regulatory review period may be extended and only those claims covering the approved drug, a method for using it or a method for manufacturing it
27
may be extended. Similar provisions are available in the EU and certain other foreign jurisdictions to extend the term of a patent that covers an approved drug.
In the future, to the extent Kineta’s product candidates, including KVA12123, receive approval by the FDA or foreign regulatory authorities, Kineta expects to apply for patent term extensions on issued patents covering those products, depending upon the length of the clinical trials for each drug and other factors.
Manufacturing
We doKineta does not have anymaintain manufacturing facilities or personnel. WeKineta currently rely,relies, and expectexpects to continue to rely, on third parties for the manufacturingmanufacture of ourits product candidates for preclinical andtesting, clinical testing, as well asstudy evaluation and for commercial manufacturingmanufacture if ourits product candidates receive marketingregulatory approval.
Currently, all of ourKineta established a manufacturing agreement with Samsung in July 2021 to provide end-to-end contract development and manufacturing services, including cell line development, manufacturing process development, clinical drug substance and drug product candidates are small moleculesmanufacturing and are manufactured in reliable and reproducible synthetic processes from readily available starting materials. The chemistry does not require unusual equipment in the manufacturing process. We expectIND filing support for KVA12123. Samsung has no commercial rights to continue to develop product candidates that can be produced at contract manufacturing facilities.KVA12123 or any other Kineta assets.
Commercialization
Kineta has not yet established a sales, marketing or product distribution infrastructure for its product candidates, which are still in preclinical or early clinical development. Kineta believes that it will be possible to access the United States oncology market through a focused, specialized sales force. Kineta has not yet developed a commercial strategy outside of the United States and will likely seek a strategic partner for these markets.
We intendSubject to developreceiving marketing approvals, Kineta expects to commence commercialization activities by building a focused sales and ifmarketing organization in the United States to sell its products. Kineta believes that such an organization will be able to address the community of oncologists who are the key specialists in treating cancer patients for which its product candidates are being developed.
Competition
Some of Kineta’s proposed products will face competition from approved therapeutics. Competition for Kineta’s pipeline products comes primarily from large, well-established pharmaceutical companies, who have greater financial resources and expertise in research and development, manufacturing, conducting clinical trials and marketing approved products. Mergers and acquisitions within the pharmaceutical and biotechnology industries may further concentrate competitors’ resources. Kineta is not only competing with these companies in terms of technology, but also in recruiting and retaining qualified scientists and management personnel, in establishing partnerships with clinical trial sites and in registering patients into clinical trials.
In addition to current standard of care for patients, clinical trials are being pursued by a number of parties in the field of immuno-oncology and in Kineta’s lead indications. These products in development may provide efficacy, safety, convenience and other benefits that are not provided by currently marketed therapies. As a result, they may provide significant competition for any of Kineta’s product candidates for which it obtains marketing approval. Based on publicly available information, the following are some of the products being developed by competitors in indications overlapping with those of Kineta’s programs.
Oncology landscape
For the last 150 years, cancer treatment was dominated by surgery, chemotherapy, radiation therapy and hormonal therapy. Before 1997, all available chemotherapy drugs for cancer were generic in their mechanism of action, designed to either kill rapidly dividing cells or deprive them of essential growth factors. Since 1997 the field has witnessed an emergence of many targeted agents for cancer, including in 2011, the first CPI for cancer, ipilimumab or Yervoy®.
Immunotherapies are unique in cancer treatment in that they do not kill cancer cells directly, but rather enhance the endogenous immune response to tumors. By enhancing the immune response, it is now possible to obtain dramatic and long-lasting tumor regressions, even in patients with advanced or otherwise incurable cancers. There exist today four broad categories of marketed immunotherapies:
Immune checkpoint inhibitors (CPIs)
28
The most widely prescribed and effective group of treatments are the CPIs. Since 2011, nine CPIs have been approved in the United States, primarily for the treatment of advanced or metastatic solid tumors. CPIs that have been approved by the FDA to commercialize our product candidates aloneonly have a few different mechanisms of action. They either block the interaction of PD1 with its ligands (PD-L1 or in collaboration-L2), or they block the interaction of CTLA4 with others. We may workits ligands (CD80 or CD86). Since both PD1 and CTLA4 serve as breaks on the T-cell-driven immune response, antibodies that block these interactions enhance the activation of effector T cells. Additionally, the first LAG-3 inhibitor was FDA approved in combination with onea PD1 inhibitor in March 2022.
Because there is such a large population of advanced cancer patients for whom there are few available treatments, the CPIs have become widely used, and this is reflected in the commercial success of the group. However, despite more than a decade of development, existing CPIs still address only two distinct mechanisms of action and are effective in only a fraction of treated patients.
Several key CPI deficiencies have become apparent from the clinical data:
Because the key to successful cancer treatment often involves the use of complex combination therapies, the immuno-oncology field urgently needs additional immunotherapies that do not increase the burden of drug related toxicity. Kineta is developing novel immunotherapies that address the mechanisms of cancer resistance where specialist capabilitiescurrent therapies fail.
KVA12123 (VISTA) Competition
There are needed. We may enter into distribution or licensing arrangementscurrently no approved VISTA blocking immunotherapies on the market. The competitive landscape includes six primary companies with assets in Phase 1 clinical development (Figure 25). Other discovery stage assets have been announced by Apexigen, Inc. and Five Prime Therapeutics (acquired by Amgen Inc.)/BMS.
Figure 26. VISTA competitive landscape
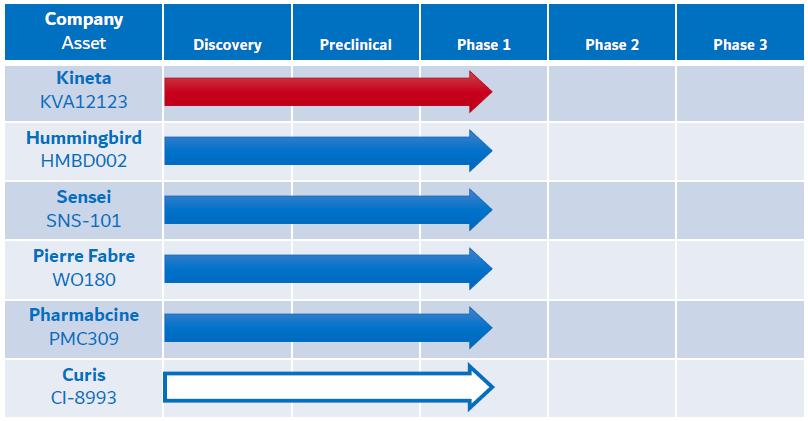
Other discovery stage programs: Apexigen and Five Prime Therapeutics/BMS.
Anti-CD27 Agonist mAb Immunotherapy Competition
The competitive landscape for commercialization rightsanti-CD27 agonist immunotherapies is led by Merck & Co., Inc. and Celldex Therapeutics, Inc. Merck is developing an anti-CD27 agonist immunotherapy that is in Phase 2 clinical trials. Celldex Therapeutics, Inc. was developing a bi-specific antibody with PD-L1 for other regions outside the United States.patients with OC that is in Phase 1 clinical trials, but was recently discontinued. Other discovery stage assets have been announced by Apogenix AG, Ligand Pharmaceuticals Incorporated, Shanghai Henlius Biotech, Avacta Life Sciences and Boston Immune Technologies and Therapeutics, Inc.
29
Government Regulation
Government authorities in the United StatesU.S., at the federal, state and local levellevels, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug products. Generally, before aproducts such as those we are developing. A new drug can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific to each regulatory authority, submitted for review and approved by the regulatory authority.FDA through the new drug application (“NDA”) process before it may be legally marketed in the U.S.
U.S. Drug Development Process
In the United States,U.S., the FDA regulates drugs under the Federalfederal Food, Drug, and Cosmetic Act or FDCA,(the “FDCA”), and its implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Additionally, a manufacturer may need to recall a product from the market. Any agency or judicial enforcement action could have a material adverse effect on us.Kineta.
Our product candidates must be approved by the FDA through the new drug application, or NDA, process before they may be legally marketed in the United States. The process required by the FDA before a drug may be marketed in the United StatesU.S. generally involves the following:
15
Preclinical Studies and Clinical Trials
The nonclinical and clinicalOnce a pharmaceutical candidate is identified for development, it enters the preclinical testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
The data required to support an NDA are generated in two distinct development stages: nonclinical and clinical. For new chemical entities, the nonclinical development stage generally involves synthesizing the active component, developing the formulation and determining the manufacturing process, as well as carrying out non-human toxicology, pharmacology and drug metabolism studies in the laboratory, which support subsequent clinical testing. These nonclinicalstage. Preclinical tests include laboratory evaluationevaluations of product chemistry, formulation, stabilitytoxicity and toxicity,formulation, as well as animal studies to assess the characteristics and potential safety and efficacy of the product. The conduct of the nonclinical tests must comply with federal and state regulations and requirements, including GLPs for safety and toxicology studies. TheAn IND sponsor must submit the results of the nonclinicalpreclinical tests, together with manufacturing information and analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND application.IND. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The sponsor will also include a protocol detailing, among other things, the objectives of the first phase of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated, if the first phase lends itself to an efficacy evaluation. Some nonclinicalpreclinical testing may continue even after the IND is submitted, but an IND application must become effective before human clinical trials may begin. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human trials.submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, raises concerns or questions regardingwithin the proposed clinical trials, including concerns that human research subjects will be exposed to unreasonable health risks, and30-day time period, places the INDclinical trial on a clinical hold within that 30-day time period.hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. TheClinical holds also may be imposed by the FDA may also impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns about on-going or non-compliance. Accordingly, we cannot be sureproposed clinical trials or non-compliance with specific FDA requirements, and the trials may not begin or continue until the FDA notifies the sponsor that submissionthe hold has been lifted. Submission of an IND willtherefore may or may not result in the FDA allowingauthorization to begin a clinical trial.
All clinical trials to begin, or that, once begun, issues will not arise that could cause the trial tomust be suspended or terminated. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development of a product candidate.
The clinical stage of development involves the administration of the product candidate to healthy volunteers or patients under the supervision of one or more qualified investigators generally physicians not employed by or under the trial sponsor’s control, in accordance with GCPs,GCP regulations, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials areThey must be conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameterssafety and effectiveness criteria to be used to monitor subject safety and assess efficacy.evaluated. Each protocol, and any subsequent amendments to the protocol must be submitted to the FDA as part of the IND. Further, each clinical trialIND as well as any subsequent protocol amendments, and timely safety reports must be reviewedsubmitted to the FDA and approved by anthe investigators for serious and unexpected adverse events. An IRB at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimizedtrial must review and are reasonable in relation to anticipated benefits. The IRBapprove each protocol before a clinical trial commences at that institution and must also approvesapprove the informedinformation regarding the trial and the consent form that must be provided to each clinical trial subject or his or her legal representative, and must monitor the clinical trial until completion. There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries. In the United States, information about applicable clinical trials, including clinical trials results, must be submitted within specific timeframes for publication on the www.clinicaltrials.gov website.
16
As part of30
monitor the 21st Century Cures Act, or the Cures Act, which was signed into law on December 13, 2016, upon request, the FDA is to establish a process for the qualification of drug development tools. A drug development tool includes a biomarker including a surrogate endpoint, a clinical outcome assessment including a patient-reported outcome,study until completed and any other method, material or measure that the FDA determines aids drug development and regulatory review. A drug development tool is qualified if the FDA has determined that the tool and its proposed context of use can be relied upon to have a specific interpretation and application in drug development and regulatory review. A qualified drug development tool may be used to support the investigational use of a drug or support or obtain NDA approval.otherwise comply with IRB regulations.
A sponsor who wishes to conduct aHuman clinical trial outside the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of an NDA so long as the clinical trial is conducted in compliance with GCP and the FDA is able to validate the data through an onsite inspection if the agency deems it necessary.
Clinical trials are generallytypically conducted in three sequential phases that may overlap known as Phase 1, Phase 2 and Phase 3 clinical trials.or be combined:
Post-approval studies,trials, sometimes referred to as Phase 4 clinical trials,studies, may be conducted after initial marketing approval. These studiestrials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA. Written IND safety reports must be submitted to the FDA and the investigators within 15 calendar days for serious and unexpected suspected adverse events, finding from other studies or animal or in vitro testing that suggests a significant risk for human subjects, and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. Additionally, a sponsor must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally,In addition, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. ThisDepending on its charter, this group provides authorization formay determine whether or not a studytrial may move forward at designated check points based on access to certain data from the study and may recommend that the clinical trial be stopped if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy.trial.
ADuring the development of a new drug, being studied insponsors are given opportunities to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2, and before an NDA or BLA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach agreement on the next phase of development. Sponsors typically use the meetings at the end of the Phase 2 trial to discuss Phase 2 clinical results and present plans for the pivotal Phase 3 clinical trials may be made available to individual patients in certain circumstances. Pursuant to the Cures Act, the manufacturer of an investigational drug for a serious disease or condition is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for individual patient access to such investigational drug. This requirement applies on the earlierthat they believe will support approval of the first initiation of a Phase 2 or Phase 3 trial of the investigational drug, or as applicable, 15 days after the drug receives a designation as a breakthrough therapy, fast track product, or regenerative advanced therapy. Further, the Right to Try Act, among other things, provides a federal framework for certain patients to request access to certain investigational new drug products that have completed a Phase 1 clinical trial and that are undergoing investigation for FDA approval. There is no obligation for a pharmaceutical manufacturer to make its drug products available to eligible patients as a result of the Right to Try Act.drug.
17
ConcurrentlyConcurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug as well asand finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsormanufacturer must develop methods for testing the identity, strength, quality and purity of the final drug product. Additionally,drug. In addition, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
While the IND is active and before approval, progress reports summarizing the results of the clinical trials and nonclinical studies performed since the last progress report must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the same or similar drugs, findings from animal or in vitro testing suggesting a significant risk to humans, and any clinically important increased incidence of a serious suspected adverse reaction compared to that listed in the protocol or investigator brochure.
There are also requirements governing the reporting of ongoing clinical trials and completed trial results to public registries. Sponsors of certain clinical trials of FDA-regulated products are required to register and disclose specified clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, trial sites and investigators and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion.
Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved.
Kineta may be required to develop and implement additional clinical trial policies and procedures designed to help protect subjects from any future pandemic or public health crisis. For example, during the COVID-19 pandemic, the FDA issued a number of COVID-19 related guidance documents for manufacturers and clinical trial sponsors, many of which have expired or were withdrawn with the termination of the COVID-19 public health emergency declaration in May 2023, although some COVID-19 related guidance documents continue in effect. Depending on the severity and duration of any resurgence of COVID-19 and its variants, the FDA may issue additional guidance and policies that may materially impact our
31
business and clinical development timelines. The ultimate impact of the COVID-19 pandemic on our business operations and clinical development plans will depend on future developments, including the severity and duration of any resurgence of COVID-19 and its variants. If new guidance and policies are promulgated by the FDA that require changes in our clinical protocol or clinical development plans, our anticipated timelines and regulatory approval may be delayed or materially impacted.
NDA Review and FDA ReviewApproval Process
The results of the nonclinicalproduct development, preclinical and other non-clinical studies and clinical trials, togetheralong with other detailed information, including extensivedescriptions of the manufacturing information and informationprocess, analytical tests conducted on the compositionchemistry of the drug, and proposed labeling and other relevant information are submitted to the FDA in the formas part of an NDA or BLA requesting approval to market the drug for oneproduct. The submission of an NDA or more specified indications. Data may come from company-sponsored clinical trials intended to evaluate the safety and efficacy of a product candidate or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational drugBLA is subject to the satisfactionpayment of the FDA.substantial user fees; a waiver of such fees may be obtained under certain limited circumstances. The FDA reviews an NDA or BLA to determine, among other things, whether a drugproduct is safe and effective for its intended use and whether the productits manufacturing is being manufactured in accordance with cGMPcGMP-compliant to assure and preserve the product’s identity, strength, quality and purity. FDA approval of an NDA must be obtained before a drug may be offered for sale in the United States.
Under the Prescription Drug User Fee Act (“PDUFA”) guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA or PDUFA, as amended, eachBLA for a new molecular entity to review and act on the submission. This review typically takes 12 months from the date the NDA must be accompanied byor BLA is submitted to the FDA because the FDA has approximately two months to make a user fee.“filing” decision after the application is submitted. The FDA adjusts the PDUFA user fees on an annual basis. According to the FDA’s fee schedule, effective from October 1, 2021 through September 30, 2022, the user fee for an application requiring clinical data, such as an NDA, is $3,117,218. PDUFA also imposes an annual prescription drug product program fee for human drugs ($369,413). Fee waiversconducts a preliminary review of all NDAs or reductions are available in certain circumstances, including a waiver of the application fee forBLAs within the first application filed by a small business. Additionally, no user fees are assessed on NDAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA reviews all NDAs submitted60 days after submission, before it acceptsaccepting them for filing, andto determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than acceptingaccept an NDA or BLA for filing. In this event, the NDA or BLA must be resubmitted with the additional information. The FDA must make a decision on accepting an NDA for filing within 60 days of receipt. Once the submissionresubmitted application also is accepted for filing,subject to review before the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under PDUFA,accepts it for drugs that contain a new chemical entity, or NCE, the FDA has 10 months from the filing date in which to complete its initial review of a standard NDA and respond to the applicant, and six months from the filing date for a priority NDA. For drugs that do not contain an NCE, these 10 and six month review timeframes are from the receipt date of an NDA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs, and the review process is often significantly extended by FDA requests for additional information or clarification.filing.
After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. Before approving an NDA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMPs. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. In addition, before approving an NDA, the FDA may also audit data from clinical trials to ensure compliance with GCP requirements. Additionally, the FDA may refer applicationsan application for a novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee. An advisory committee typicallyis a panel that includesof independent experts, including clinicians and other scientific experts, for review, evaluationthat reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. Before approving an NDA or BLA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will likely re-analyzenot approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA may inspect one or more clinical trial data, which could result in extensive discussions between the FDA and the applicant during the review process. The review and evaluation of an NDA by the FDA is extensive and time consuming and may take longer than originally plannedsites to complete, and we may not receive a timely approval, if at all.assure compliance with GCP requirements.
After the FDA evaluates an NDA or BLA, it maywill issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application iswill not ready for approval.be approved in its present form. A Complete Response Letter usually describes all of the specific deficiencies in the NDA or BLA identified by the FDA. The Complete Response LetterFDA and may require additional clinical data, and/orsuch as an additional pivotal Phase 3 clinical trial(s), and/trial or other significant and time-consuming requirements related to clinical trials, nonclinical studies or manufacturing. If a Complete Response Letter is issued, the applicant maysponsor must resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application, or request an opportunity for a hearing.application. Even if such data and information isare submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data.
18
There is no assurance that the FDA will ultimately approve a drug product for marketing in the United States and we may encounter significant difficulties or costs during the review process. If a product receives marketingregulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further,In addition, the FDA may require that certain contraindications, warnings, or precautions be included in the product labeling or may condition the approval of the NDA on other changesa sponsor to the proposed labeling, development of adequate controls and specifications, or a commitment to conduct post-marketing testing or clinical trials and surveillance to monitor the effects of the approved product. For example, the FDA may require Phase 4 testing, which involves clinical trials designed to further assess a drug’s safety and efficacyeffectiveness after NDA or BLA approval and may require testing and surveillance programs to monitor the safety of approved products thatwhich have been commercialized. The FDA may also may place other conditions on approvalsapproval including the requirement for a risk evaluation and mitigation strategy, or REMS to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA or BLA must submit a proposed REMS. The FDA will not approve the NDA or BLA without an approved REMS, if required. A REMS could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvalsMarketing approval may be withdrawn for non-compliance with regulatory requirements or if problems occur following initial marketing.
Orphan Drug Designation The Pediatric Research Equity Act (“PREA”) requires a sponsor to conduct pediatric clinical trials for most drugs, for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration. Under PREA, original NDAs or BLAs and Exclusivity
Undersupplements must contain a pediatric assessment unless the Orphan Drug Act,sponsor has received a deferral or waiver. The required assessment must evaluate the FDA may grant orphan designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United Statessafety and for which there is no reasonable expectation that the costeffectiveness of developing and making the product available in the United States for this type of disease or condition will be recovered from sales of the product.
Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or conditionclaimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which it has such designation, the product is entitled to orphan drug exclusivity, which meanssafe and effective. The sponsor or FDA may request a deferral of pediatric clinical trials for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the FDA may not approve any other applications to market the same drug is ready for the same indicationapproval for seven years from the date of such approval, exceptuse in limited circumstances, such as a showing ofadults before pediatric clinical superiority to the product with orphan exclusivity by means of greater effectiveness, greatertrials are complete or that additional safety or providingeffectiveness data needs to be collected before the pediatric clinical trials begin. The FDA must send a major contributionnon-compliance letter to patient care,any sponsor that fails to submit the required assessment, keep a deferral current or in instances of drug supply issues. Competitors, however, may receivefails to submit a request for approval of either a different product for the same indication or the same product for a different indication but that could be used off-label in the orphan indication. Orphan drug exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval before we do for the same product, as defined by the FDA, for the same indication we are seeking approval, or if our product is determined to be contained within the scope of the competitor’s product for the same indication or disease. If one of our products designated as an orphan drug receives marketing approval for an indication broader than that which is designated, it may not be entitled to orphan drug exclusivity. Orphan drug status in the European Union has similar, but not identical, requirements and benefits.pediatric formulation.
Expedited Development and Review Programs
Kineta plans to seek to accelerate regulatory approval in all major markets. The pathways outlined in Figure 26 below provide an overview of accelerated review and approval pathways with the FDA.
Kineta also plans to pursue “fast track” and “accelerated approval” for the KVA12123 and anti-CD27 mAb immunotherapy programs.
32
Figure 27. Accelerated Regulatory Approval by FDA
Fast track: A sponsor may seek approval of its product candidate under programs designed to accelerate the FDA’s review and approval of new drugs and biological products that meet certain criteria. The FDA has a fast trackfast-track designation program that is intended to expedite or facilitate the process for reviewing new drugsdrug products that meet certain criteria. Specifically, new drugs are eligible for fast trackfast-track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast track designation appliesUnique to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug may request the FDA to designate the drug as a fast track product, at any time during the clinical development of the product. The sponsor of a fast track product may request that the FDA may consider for review sections of the marketing applicationNDA or BLA on a rolling basis before the complete NDAapplication is submitted, if the sponsor provides a schedule for the submission of the sections of the application,NDA or BLA, the FDA agrees to accept sections of the applicationNDA or BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.NDA or BLA.
Any product submitted to theBreakthrough therapy: A sponsor may seek FDA for marketing, including under the fast track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review. A product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or offers a significant improvement in the treatment, diagnosis or preventiondesignation of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug designated for priority review in an effort to facilitate the review.
Additionally, a drug may be eligible for designationproduct candidate as a breakthrough therapy“breakthrough therapy” if the drugproduct is intended, alone or in combination with one or more other drugs,products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drugproduct may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as
19
substantial treatment effects observed early in clinical development. If the FDA designates a breakthrough therapy, it may take actions appropriate to expedite the development and review of the application, which may include holding meetings with the sponsor and the review team throughout the development of the therapy; providing timely advice to, and interactive communication with, the sponsor regarding the development of the drug to ensure that the development program to gather the nonclinical and clinical data necessary for approval is as efficient as practicable; involving senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review; assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor; and considering alternative clinical trial designs when scientifically appropriate, which may result in smaller trials or more efficient trials that require less time to complete and may minimize the number of patients exposed to a potentially less efficacious treatment. The benefitsdesignation includes all of the fast-track program features, which means that the sponsor may file sections of the NDA or BLA for review on a rolling basis if certain conditions are satisfied, including an agreement with the FDA on the proposed schedule for submission of portions of the application and the payment of applicable user fees before the FDA may initiate a review. The breakthrough therapy designation includeis a distinct status from both accelerated approval and priority review, which can also be granted to the same benefitsdrug if relevant criteria are met. If a product is designated as fast track designation, plus intensive guidance frombreakthrough therapy, the FDA will work to ensure an efficient drug development program. Fast Track designation, priority review, and breakthrough designation do not change the standards for approval but may expedite the development or approval process.and review of such drug.
Additionally,Accelerated approval: In addition, a drugproduct may be eligible for accelerated approval. Drugs studied for their safety and effectiveness in treatingDrug products intended to treat serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatmentsdiseases or conditions may receivebe eligible for accelerated approval which means that they may be approved on the basis of adequate and well-controlled clinical trials establishingupon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. In addition, the FDA currently requires unless otherwise informed by the agency,as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. The FDA may withdraw approval of a drug or indication approved under accelerated approval if, for products being consideredexample, the confirmatory trial fails to verify the predicted clinical benefit of the product.
33
Priority review: Any product submitted to the FDA for approval, including a product with a fast-track designation, may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. A product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the safety or effectiveness of the treatment, diagnosis or prevention of a serious disease or condition. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug designated for priority review in an effort to facilitate the review. The FDA endeavors to review applications with priority review designations within six months of the filing date as compared to 10 months for review of new molecular entity NDAs or BLAs under its current PDUFA review goals. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Fast track designation, priority review and breakthrough therapy designation and accelerated approval do not change the standards for approval but may expedite the development or approval process. Moreover, evenEven if a product candidate qualifies for one or more of these programs, the FDA may later decide that the product candidate no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. Kineta may explore some of these opportunities for its product candidates as appropriate. Depending on other factors that impact clinical trial timelines and development, such as Kineta’s ability to identify and onboard clinical sites and rates of study participant enrollment and drop-out, Kineta may not realize all the benefits of these expedited or accelerated review programs.
Pediatric TrialsPost-Approval Requirements
The FDCA requires thatOnce an approval is granted, the FDA may withdraw the approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a sponsor who is planning to submit a marketing application for a drug that includes a new active ingredient, new indication, new dosage form, new dosing regimenproduct may result in restrictions on the product or new route of administration submit an initial Pediatric Study Plan, or PSP, within sixty days of an end-of-Phase 2 meeting or as may be agreed between the sponsor and the FDA. The initial PSP must include an outlineeven complete withdrawal of the pediatric study or studies thatproduct from the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferralmarket. After approval, some types of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from nonclinical studies, early phase clinical trials and/or other clinical development programs.
Post-Marketing Requirements
Under the Pediatric Research Equity Act, or PREA,approved product, such as amended, an NDA or supplement to an NDA must contain data to assess the safetyadding new indications, certain manufacturing changes and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of pediatric data or full or partial waivers. Following approval of a new product, a pharmaceutical company and the approved productadditional labeling claims, are subject to continuing regulation by thefurther FDA including, among other things, monitoringreview and recordkeeping activities, reporting to the FDA of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements and complying with promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling (known as “off-label use”), limitations on industry-sponsored scientific and educational activities and requirements for promotional activities involving the internet. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new NDA or NDA supplement, which may require the applicant to develop additional data or conduct additional nonclinical studies and clinical trials. As with new NDAs, the review process is often significantly extended by FDA’s requests for additional information or clarification. Any distribution of prescription drug products and pharmaceutical samples must comply with the U.S. Prescription Drug Marketing Act, or the PDMA, a part of the FDCA.
In the United States, once a product is approved, its manufacture is subject to comprehensive and continuing regulation by the FDA. The FDA regulations require that products be manufactured in specific facilities and in accordance with cGMP. NDA holders using contract manufacturers, laboratories or packagers are responsible for the selection and monitoring of qualified firms, and, in certain circumstances, qualified suppliers to these firms. These manufacturers must comply with cGMP regulations that require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP.approval. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies and are
20
subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP regulations and other laws. laws and regulations. In addition, the FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
ManufacturersAny drug products manufactured or distributed by Kineta or its partners pursuant to FDA approvals will be subject to pervasive and continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the drug, providing the FDA with updated safety and efficacy information, drug sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other parties involved in the drug supply chain for prescription drug products must also comply with product tracking and tracing requirements and for notifying the FDAtypes of counterfeit, diverted, stolen and intentionally adulterated products orinformation on products that are otherwise unfitplaced on the market and imposes requirements and restrictions on drug manufacturers, such as those related to direct-to-consumer advertising, the prohibition on promoting products for distributionuses or in patient populations that are not described in the United States. Accordingly, manufacturers must continue to expend time, moneyproduct’s approved labeling (known as “off-label use”), industry-sponsored scientific and effort ineducational activities and promotional activities involving the areainternet.
Discovery of production and quality control to maintain cGMP compliance. The discovery of violative conditions, includingpreviously unknown problems or the failure to conform to cGMP, could result in enforcement actions that interruptcomply with the operation of any such facilities or the ability to distribute products manufactured, processed or tested by them. Discovery of problems with a product after approvalapplicable regulatory requirements may result in restrictions on the marketing of a product manufacturer, or holder of an approved NDA, including, among other things, recall or withdrawal of the product from the market.
Discovery of previously unknown problems with a productmarket as well as possible civil or the failurecriminal sanctions. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant or manufacturer to administrative or judicial civil or criminal sanctions and adverse publicity. FDA requirements can have negative consequences, including judicialsanctions could include refusal to approve pending applications, withdrawal of an approval, clinical holds on post-approval clinical trials, warning or administrative enforcement, warninguntitled letters, from the FDA,product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, mandated corrective advertising or communications with doctors, anddebarment, restitution, disgorgement of profits, or civil or criminal penalties, among others. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development. Changes in statutes, regulations, or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.penalties.
Orange Book ListingNDA and BLA Marketing Exclusivity
Section 505 of the FDCA describes three types of marketing applications that may be submitted to the FDA to request marketing authorization for a new drug. A Section 505(b)(1) NDA is an application that contains full reports of investigations of safety and efficacy. A Section 505(b)(2) NDA is an application in which the applicant, in part, relies on investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. Section 505(j) establishes an abbreviated approval process for a generic version of approved drug products through the submission of an Abbreviated New Drug Application, or ANDA. An ANDA provides for marketing of a generic drug product that has the same active ingredients, dosage form, strength, route of administration, labeling, performance characteristics and intended use, among other things, to a previously approved product. Limited changes must be preapproved by the FDA via a suitability petition. ANDAs are termed “abbreviated” because they are generally not required to include nonclinical and clinical data to establish safety and efficacy. Instead, generic applicants must scientifically demonstrate that their product is bioequivalent to, or performs in the same manner as, the innovator drug through in vitro, in vivo, or other testing. The generic version must deliver the same amount of active ingredients into a subject’s bloodstream in the same amount of time as the innovator drug and can often be substituted by pharmacists under prescriptions written for the reference listed drug.
In seeking approval for a drug through an NDA, including a 505(b)(2) NDA, applicants are required to list with the FDA certain patents having claims that cover the applicant’s product and method of use. Upon approval of an NDA, each of the patents listed in the application for the drug is then published in Approved Drug Products with Therapeutic Equivalence Evaluations, also known as the Orange Book. These products may be cited by potential competitors in support of approval of an ANDA or 505(b)(2) NDA.
Any applicant who files an ANDA seeking approval of a generic equivalent version of a drug listed in the Orange Book or a 505(b)(2) NDA referencing a drug listed in the Orange Book must make patent certifications to the FDA that (1) no patent information on the drug or method of use that is the subject of the application has been submitted to the FDA; (2) the patent has expired; (3) the date on which the patent has expired and approval will not be sought until after the patent expiration; or (4) the patent is invalid or will not be infringed upon by the manufacture, use, or sale of the drug product for which the application is submitted. The last certification is known as a paragraph IV certification. Generally, the ANDA or 505(b)(2) NDA cannot be approved until all listed patents have expired, except where the ANDA or 505(b)(2) NDA applicant challenges a listed patent through a paragraph IV certification or if the applicant is not seeking approval of a patented method of use. If the applicant does not challenge the listed patents or does not indicate that it is not seeking approval of a patented method of use, the ANDA or 505(b)(2) NDA application will not be approved until all of the listed patents claiming the referenced product have expired.
If the competitor has provided a paragraph IV certification to the FDA, the competitor must also send notice of the paragraph IV certification to the holder of the NDA for the reference listed drug and the patent owner within 20 days after the application has been accepted for filing by the FDA. The NDA holder or patent owner may then initiate a patent infringement lawsuit in response to the notice of the paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a paragraph IV certification notice prevents the FDA from approving the ANDA or 505(b)(2) application until the earlier of 30 months from the date of the lawsuit, expiration of the patent, settlement of the lawsuit, a decision in the infringement case that is favorable to the applicant or such shorter or longer period as may be ordered by a court. This prohibition is generally referred to as the 30-month stay.
21
In instances where an ANDA or 505(b)(2) NDA applicant files a paragraph IV certification, the NDA holder or patent owners regularly take action to trigger the 30-month stay, recognizing that the related patent litigation may take many months or years to resolve. Thus, approval of an ANDA or 505(b)(2) NDA could be delayed for a significant period of time depending on the patent certification the applicant makes and the reference drug sponsor’s decision to initiate patent litigation.
U.S. Marketing Exclusivity
MarketingMarket exclusivity provisions under the FDCA can also delay the submission or the approval of certain marketing applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United StatesU.S. to the first applicant to obtain approval of an NDA for an NCE.a new chemical entity. A drug is an NCEa new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not approve or even accept for review an abbreviated new drug application, or ANDA, or aan NDA submitted under Section 505(b)(2), or 505(b)(2) NDA, submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovatorinnovative drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder.
The FDCA alsoalternatively provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for drugs containing the active agent for the original indication or condition of use. Three-yearFive-year and five-yearthree-year exclusivity will not delay the submission
34
or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the nonclinicalpreclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and efficacy. effectiveness.
Under the FDCA, market exclusivity for biologics agents provides a 12-year period of market exclusivity within the U.S. for the first FDA approved compound.
Pediatric exclusivity is another type of regulatory marketmarketing exclusivity available in the United States.U.S. Pediatric exclusivity if granted, addsprovides for an additional six months of marketing exclusivity attached to existinganother period of exclusivity periods and patent terms. This six-month exclusivity, which runsif a sponsor conducts clinical trials in children in response to a written request from the end of other exclusivity protection or patent term, may be granted based on the voluntary completionFDA. The issuance of a pediatric trialwritten request does not require the sponsor to undertake the described clinical trials. In addition, orphan drug exclusivity may offer a seven-year period of marketing exclusivity, except in accordance with an FDA-issued “Written Request” for such a trial.certain circumstances.
U.S. Patent-Term ExtensionCoverage and Reimbursement
Depending uponSignificant uncertainty exists as to the timing, durationcoverage and specificsreimbursement status of FDA approvalany product candidate for which Kineta may seek regulatory approval. Sales in the U.S. will depend, in part, on the availability of sufficient coverage and adequate reimbursement from third-party payors, which include government health programs such as Medicare, Medicaid, TRICARE and the Veterans Administration, as well as managed care organizations and private health insurers. Prices at which Kineta or its customers seek reimbursement for our current product candidates can be subject to challenge, reduction or any futuredenial by third-party payors.
The process for determining whether a third-party payor will provide coverage for a product candidate, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch Waxman Act. The Hatch Waxman Act permits extension of the patent term of up to five years as compensation for patent term lost during FDA regulatory review process. Patent term extension, however, cannot extend the remaining term of a patent beyond a total of 14 yearsis typically separate from the product’s approval date. The patent term extension period is generally one halfprocess for setting the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application, exceptreimbursement rate that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligiblepayor will pay for the extension (andproduct. A third-party payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be available. Additionally, in the U.S. there is no uniform policy among payors for coverage or reimbursement. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own coverage and reimbursement policies, but also have their own methods and approval processes. Therefore, coverage and reimbursement for products can differ significantly from payor to payor. If coverage and adequate reimbursement are not available, or are available only those patent claims coveringat limited levels, successful commercialization of, and obtaining a satisfactory financial return on, any product Kineta develops may not be possible.
Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. In order to obtain coverage and reimbursement for any product that might be approved drug, a method for using it or a method for manufacturing itmarketing, Kineta may need to conduct expensive studies in order to demonstrate the medical necessity and cost-effectiveness of any products, which would be extended), and the application for the extension must be submitted priorin addition to the expiration ofcosts expended to obtain regulatory approvals. Third-party payors may not consider our product candidates to be medically necessary or cost-effective compared to other available therapies, or the patent. A patent that covers multiple products for which approval is sought can only be extendedrebate percentages required to secure favorable coverage may not yield an adequate margin over cost or may not enable Kineta to maintain price levels sufficient to realize an appropriate return on its investment in connection with one of the approvals. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension. In the future, we may apply for extension of a patent term for our currently owned patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA. However, there can be no assurance that the USPTO will grant us any requested patent term extension, either for the length we request or at all.drug development.
U.S. Healthcare Reform
In the United States and some foreign jurisdictions,U.S., there havehas been, and continuecontinues to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of product candidates, restrict or regulate post-approval activities and affect the ability to profitably sellprofitable sale of product candidates for which marketing approval is obtained. candidates.
Among policy makers and payors in the United States and elsewhere,U.S., there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States,U.S., the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
For example, In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act or collectively, ACA, enacted in(the “ACA”) was passed, which substantially changed the United States in March 2010, has already had,way healthcare is financed by both the government and is expected to continue to have, a significant impact onprivate insurers, and significantly impacts the healthcareU.S. pharmaceutical industry. The ACA, expanded coverage foramong other things: (1) increased the uninsured while atminimum Medicaid rebates owed by manufacturers under the same time containing overall healthcare costs. AmongMedicaid Drug Rebate Program and extended the provisions of the ACA of importancerebate program to our product candidates are the following:
22
35
Since its enactment, there have been executive, judicial Congressional and executiveCongressional challenges to certain aspects of the ACA. OnFor example, in June 17, 2021 the U.S. Supreme Court dismissed the most recent judicialheld that Texas and other challengers had no legal standing to challenge to the ACA, brought by several statesdismissing the case on procedural grounds without specifically ruling on the constitutionality of the ACA. PriorThus, the ACA will remain in effect in its current form. Further, prior to the U.S. Supreme Court’s decision,Court ruling, on January 28, 2021, President Biden issued an executive order to initiatethat initiated a special enrollment period from February 15, 2021 through August 15,in 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. Themarketplace, which began on February 15, 2021, and remained open through August 15, 2021. This executive order also instructedinstructs certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA.ACA, among others. It is possible that the ACA will be subject to judicial or Congressional challenges in the future. It is unclear how otherany such challenges and healthcare reform measures ofpromulgated by the Biden administration or other efforts, if any, to challenge, repeal or replacewill impact the ACA, will impactour business, financial condition and results of operations. Complying with any new legislation or reversing changes implemented under the ACA could be time-intensive and expensive, resulting in a material adverse effect on our business.
In addition, otherOther legislative changes have been proposed and adopted in the United States since the ACA was enacted. For example:
23
Recently,Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. Such scrutinyproducts, which has resulted in several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drugproduct pricing, review the relationship between pricing and manufacturer patient programs reduce the cost of drugs under Medicare and reform government program reimbursement methodologies for drug products. At the federal level, President Biden signed an Executive Order on July 9, 2021 affirming the administration’sTrump administration used several means to propose or implement drug pricing reform, including through federal budget proposals, executive orders and policy to (i) support legislative reforms that would lower the prices of prescription drug and biologics, including by allowing Medicare to negotiate drug prices, by imposing inflation caps, and, by supporting the development and market entry of lower-cost generic drugs and biosimilars; and (ii) support the enactment of a public health insurance option. Among other things, the Executive Order also directsinitiatives. For example, in 2020, the U.S. Department of Health and Human Services or HHS, to provide a report on actions to combat excessive pricing of prescription drugs, enhance the domestic drug supply chain, reduce the price that the Federal government pays for drugs, and address price gouging in the industry; and directs the FDA to work with states and Indian Tribes that propose to develop section 804 Importation Programs in accordance with the Medicare Prescription Drug, Improvement, and Modernization Act of 2003,(“HHS”) and the FDA’s implementing regulations. FDA released such implementing regulations on September 24, 2020, which went into effect on November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada. On September 25, 2020, CMS stated drugs imported by states under this rule will not be eligible for federal rebates under Section 1927 of the Social Security Act and manufacturers would not report these drugs for “best price” or Average Manufacturer Price purposes. Since these drugs are not considered covered outpatient drugs, CMS further stated it will not publish a National Average Drug Acquisition Cost for these drugs. If implemented, importation of drugs from Canada may materially and adversely affect the price we receive for any of our product candidates. Further, on November 20, 2020 CMS issued an Interim Final Rule implementing the Most Favored Nation, or MFN, Model under which Medicare Part B reimbursement rates would have been be calculated for certain drugs and biologicals based on the lowest price drug manufacturers receive in Organization for Economic Cooperation and Development countries with a similar gross domestic product per capita. However, on December 29, 2021 CMS rescinded the Most Favored Nations rule. Additionally, on November 30, 2020, HHS published a regulation removing safe harbor protection forvarious rules that are expected to impact, among others, price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between pharmacy benefit managers and manufacturers. Pursuantmanufacturers, manufacturer price reporting requirements under the Medicaid Drug Rebate Program, including regulations that affect manufacturer-sponsored patient assistance programs subject to court order,pharmacy benefit manager accumulator programs and Best Price reporting related to certain value-based purchasing arrangements. Multiple lawsuits have been brought against the removal and additionHHS challenging various aspects of the aforementioned safe harbors were delayed and recent legislation imposed a moratorium on implementationrules. Under the American Rescue Plan Act of the rule until2021, effective January 1, 2026. Although a number2024, the statutory cap on Medicaid Drug Rebate Program rebates that manufacturers pay to state Medicaid programs will be eliminated. Elimination of these and other proposed measuresthis cap may require authorization through additional legislationpharmaceutical manufacturers to become effective, andpay more in rebates than it receives on the sale of products, which could have a material impact on our business. Further, based on a recent executive order, the Biden administration expressed its intent to pursue certain policy initiatives to reduce drug prices. Any reduction in reimbursement from Medicare or other government programs may reverse or otherwise change these measures, bothresult in a reduction in payments from private payors. The impact of legislative, executive and administrative actions of the Biden administration on us and Congress have indicated that they will continue to seek new legislative measures to control drug costs.the pharmaceutical industry as a whole is unclear.
We expect that additional U.S. federal healthcare reform measures will be adopted inAt the future, any of which could limit the amounts that the U.S. Federal Government will pay for healthcare drugs and services, which could result in reduced demand for our drug candidates or additional pricing pressures.
Individual states in the United Statesstate level, legislatures have also become increasingly active in passingpassed legislation and implementingimplemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain drugproduct access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payorsKineta is unable to predict the future course of federal or other restrictions could harm our business, financial condition, resultsstate healthcare legislation in the U.S. directed at broadening the availability of operationshealthcare and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which supplierscontaining or lowering the cost of healthcare. Further, it is possible that additional governmental action will be includedtaken in their prescription drugresponse to the COVID-19 pandemic. If Kineta or any third parties it may engage are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if Kineta or such third parties are not able to maintain regulatory compliance, Kineta’s products candidates may lose regulatory approval that may have been obtained and other healthcare programs. This could reduce the ultimate demand for our drugsKineta may not achieve or put pressure on our drug pricing, which could negatively affect our business, financial condition, results of operations and prospects.sustain profitability.
Other U.S. Healthcare Fraud and Abuse Laws and Compliance Requirements
Manufacturing, sales, promotion,Federal and other activities following product approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including,state healthcare laws and regulations restrict business practices in the United States, CMS, other divisions of HHS including the Office of the Inspector General, the U.S. Department of Justice, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agencypharmaceutical industry. These laws include anti-kickback and statefalse claims laws and local regulatory authorities. For example, sales, marketingregulations, data privacy and scientific/educational grant programs may have to comply with statesecurity and federal fraudtransparency laws and abuse laws, the privacy and
24
security provisions of the Health Insurance Portability and Accountability Act of 1996, or HIPAA, and similar state laws, each as amended.regulations.
The federal Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) toprohibits, among other things, individuals or entities from knowingly and willfully solicit, receive, offer,offering, paying, soliciting or pay anyreceiving remuneration, (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind to induce or in return for the referral of an individual,purchasing, leasing, ordering or arranging for or recommending the purchase, lease order, or recommendationorder of any good, facility, item or service (including the purchase, order,reimbursable under Medicare, Medicaid or prescription of a particular drug), for which payment may be made, in whole or in part, under aother federal healthcare program, such as Medicare or Medicaid. “Remuneration” has been interpreted broadly to include anything of value. Violations of this law are punishable by up to five years in prison, criminal fines, administrative civil money penalties and exclusion from participation in federal healthcare programs. In addition, the ACA, among other things, amended the intent requirement of the federal Anti-Kickback Statute. A person or entity no longer needsdoes not need to have actual knowledge of thethis statute or specific intent to violate it. Further, courtsit in order to have found that if “one purpose” of renumeration is to induce referrals, the federal Anti-Kickback statute is violated. Moreover, the ACA provides thatcommitted a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. There are a number of statutory exceptionsAct and regulatory safe harbors protecting some common activities.the Civil Monetary Penalties Statute.
Although we would not submit claims directly to payors, drug manufacturers can be held liable under theThe federal civil and criminal false claims laws and civil monetary penaltypenalties laws, including the civil False Claims Act, which prohibit, among other things, anyoneany individual or entity from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or approval by federal programs (including Medicare and Medicaid) claims for itemsknowingly making, using or services, including drugs, that arecausing to be made or used a false record or statement material to a false or fraudulent claims for items or services not provided as claimed, or claims for medically unnecessary items or services, knowingly concealing or knowingly and improperly avoiding or decreasing or concealing an obligation to pay moneyclaim to the federal government. The government may deem manufacturers to have “caused” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. In addition, our activities relating to the reporting of wholesaler or estimated retail prices for our products, the reporting of prices used to calculate Medicaid rebate information and other information affecting federal, state and third-party reimbursement for our products, and the sale and marketing of our products, are subject to scrutiny under this law. Penalties for a False Claims Act violation include three times the actual damages sustained by the government, plus mandatory civil penalties for each separate false claim, the potential for exclusion from participation in federal healthcare programs, and, although the federal False Claims Act is a civil statute, conduct that results in a False Claims Act violation may also implicate various federal criminal statutes. If the government were to allege that we were, or convict us of, violating these false claims laws, we could be subject to a substantial fine and may suffer a decline in its stock price. In addition, private individuals have the ability to bring actions under the federal False Claims Act and certain states have enacted laws modeled after the federal False Claims Act.
HIPAAThe federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) created additional federal civil and criminal statutes that prohibit, among other actions,things, knowingly and willfully executing or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private), willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the federal Anti-Kickback Statute a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.program. In addition, HIPAA, as
Many states have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs. Additionally, to the extent that any of our product candidates, if approved, are sold in a foreign country, we may be subject to similar foreign laws.
HIPAA, as 36
amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, imposeimposes certain requirements relating to the privacy, security and transmission of individually identifiableprotected health information on HIPAA covered entities, which include certain healthcare providers, health plans and healthcare clearinghouses, and their business associates who conduct certain activities for or on their behalf involving protected health plans, known as covered entities,information on their behalf as well as independent contractors, or agents oftheir covered entities that create, receive or obtain individually identifiable health information in connection with providing a service on behalf of a covered entity, known as a business associates. Among other things, HITECH makes HIPAA’s security standards directly applicable to business associates, defined as independent contractors or agents of covered entities that create, receive or obtain protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities and business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, certain state laws govern the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and criminal penalties.
25
subcontractors.
The U.S. federal transparency requirements under the ACA, including the provision commonly referred to as the Physician Payments Sunshine Act requires applicable group purchasing organizations and its implementing regulations, require applicable manufacturers of covered drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to CMS information related to certain payments or other transfers of value made to covered recipients, including physicians licensed to practice in the U.S. (defined to include doctors of medicine and osteopathy, dentists, podiatrists, optometrists podiatrists and licensed chiropractors), and teaching hospitals, in the previous year, including ownership and investment interests held by covered physicians and their immediate family members. Effective January 1, 2021, for data collected in 2021 and submitted to CMS in 2022, such reporting obligations with respect to covered recipients have been extended to include new provider types: physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists and teaching hospitals, as well as ownershipanesthesiologist assistants and investment interests held by the physicians described above and their immediate family members.certified nurse-midwives.
Additionally, we are subject toSimilar state and foreign equivalents of each of the healthcarelocal laws and regulations described above, among others, some ofmay also restrict business practices in the pharmaceutical industry, such as state anti-kickback and false claims laws, which may be broader in scope and may apply regardless of the payor. Many U.S. states have adopted laws similar to the federal Anti-Kickback Statute and False Claims Act, and may apply to our business practices, including but not limited to, research, distribution, sales orand marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers. In addition, some states have passedinsurers, or by patients themselves; state laws that require pharmaceutical companies to comply with the April 2003 Officepharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments or transfers of Inspector General Compliance Program Guidance for Pharmaceutical Manufacturers and/value that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information or the Pharmaceutical Researchwhich require tracking gifts and Manufacturersother remuneration and items of America’s Code on Interactions with Healthcare Professionals. Several states also imposevalue provided to physicians, other marketing restrictions or require pharmaceutical companies to make marketing or price disclosures to thehealthcare providers and entities; state and local laws that require the registration of pharmaceutical sales representatives. Staterepresentatives; and foreignstate and local laws including for example the European Union General Data Protection Regulation, which became effective May 2018 also governsgoverning the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. There are ambiguities as
Efforts to what is required to complyensure compliance with these state requirementsapplicable healthcare laws and if we fail to comply with an applicable state law requirement we could be subject to penalties. Finally, there are state and foreignregulations can involve substantial costs. Violations of healthcare laws governing the privacy and security of health information, many of which differ from each othercan result in significant wayspenalties, including the imposition of significant civil, criminal and often are not preempted by HIPAA, thus complicating compliance efforts.
Other regulations may affect other aspects of our business. For example, pricing and rebate programs must comply with theadministrative penalties, damages, monetary fines, disgorgement, individual imprisonment, possible exclusion from participation in Medicare, Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990 and more recent requirements in ACA. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair competition laws. There has also been a recent trend of increased federal and state regulation of payments made to physicians. Certain states mandate implementation of complianceU.S. healthcare programs, impose restrictions on drug manufacturers’ marketing practices and/or require the trackingintegrity oversight and reporting of gifts, compensation and other remuneration to physicians.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement,obligations, contractual damages, reputational harm, diminished profits and future earnings, theand curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and individual imprisonment, any of which could adversely affect our ability to operate its business and its financial results. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of its business.operations.
Coverage and ReimbursementForeign Regulation
Sales of our drugs will depend, in part, on the extentIn order to which our drugs will be covered by third-party payors, such as government health programs, commercial insurance, and managed healthcare organizations. Significant uncertainty exists as to the coverage and reimbursement status ofmarket any product candidatesoutside of the U.S., Kineta would need to comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of Kineta’s products. Whether or not Kineta obtains FDA approval for which we maya product, Kineta would need to obtain regulatory approval. Coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid in the United States, and commercial payors are critical to new product acceptance. Third-party payors decide which drugs they will pay for and establish reimbursement levels. In the United States, the principal decisions about reimbursement for new medicines are typically made by CMS. CMS decides whether and to what extent our products will be covered and reimbursed under Medicare and private payors tend to follow CMS to a substantial degree. Coverage and reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a therapeutic is:
26
We cannot be sure that reimbursement will be available for any product that we commercialize and, if coverage and reimbursement are available, we cannot be sure that the level of reimbursement will be adequate. Coverage may also be more limited than the purposes for which the product is approvednecessary approvals by the FDA or comparable foreign regulatory authorities. Limited coverageauthorities before Kineta can commence clinical trials or marketing of the product in foreign countries and less than adequate reimbursement may reduce the demand for, or the price of, any product for which we obtain regulatory approval.jurisdictions.
These third-party payors are increasingly reducing reimbursements for medical drugsAlthough many of the issues discussed above with respect to the U.S. apply similarly in the context of the EU, the approval process varies between countries and services. Additionally,jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries or jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the containment of healthcare costs has become a priority of federal and state governments, and the prices of drugs have been a focusregulatory process in this effort. The U.S. government, state legislatures, and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic drugs. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our product candidates, if approved, or a decision by a third-party payor to not cover our product candidates could reduce physician usage of such drugs and have a material adverse effect on our sales, results of operations and financial condition.others.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, established the Medicare Part D program to provideTo market a voluntary prescription drug benefit to Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for drugs for which we obtain marketing approval. However, any negotiated prices for our drugs covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal drugs for which their national health insurance systems provide reimbursement and to control the prices of medicinal drugs for human use. A member state may approve a specific price for the medicinal drug or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal drug on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical drugs will allow favorable reimbursement and pricing arrangements for any of our drugs. Historically, drugs launchedproduct in the European Union do not follow price structuresEconomic Area (“EEA”) (which is comprised of the United27 Member States and generally tend to be significantly lower.
European Drug Development
In the European Union, our product candidates may also be subject to extensive regulatory requirements. As in the United States, medicinal products can only be marketed if a marketing authorization from the competent regulatory agencies has been obtained.
Similar to the United States, the various phases of nonclinical and clinical research in the European Union are subject to significant regulatory controls.
In April 2014, the European Union adopted the new Clinical Trials Regulation (EU) No 536/2014, which replaced the EU Clinical Trials Directive 2001/20/EC on January 31, 2022. The transitory provisions of the new Regulation offer sponsors the possibility to choose between the requirements of the previous Directiveplus Norway, Iceland and the new Regulation if the request for authorization ofLiechtenstein), Kineta must obtain a clinical trial is submitted in the year after the new Regulation became applicable. If the sponsor chooses to submit under the previous Directive, the clinical trial continues to be governed by the Directive until three years after the new Regulation became applicable. If a clinical trial continues for more than three years after the Regulation became applicable, the new Regulation will at that time begin to apply to the clinical trial. The new Regulation overhauls the current system of approvals for clinical trials in the European Union. Specifically, the new Regulation, which is directly applicable in all Member States (and so does not require national implementing legislation in each Member State), aims at simplifying and streamlining the approval of clinical trials in the European Union. The main characteristics of the new Regulation include: a streamlined application procedure via a single-entry point through the Clinical Trials Information System, or CTIS; a single set of documents to be prepared and submitted for the application as well as simplified reporting procedures
27
for clinical trial sponsors; and a harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts (Part I contains scientific and medicinal product documentation and Part II contains the national and patient-level documentation)Marketing Authorization (“MA”). Part I is assessed by a coordinated review by the competent authorities of all European Union Member States in which an application for authorization of a clinical trial has been submitted (Concerned Member States) of a draft report prepared by a Reference Member State. Part II is assessed separately by each Concerned Member State. Strict deadlines have also been established for the assessment of clinical trial applications.
European Drug Review and Approval
In the European Union, medicinal products can only be commercialized after obtaining a marketing authorization, or MA. There are two main types of marketing authorizations:
37
Under the above describedabove-described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the European UnionEEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
Now that the UK (which comprises Great Britain and Northern Ireland) has left the EU, Great Britain will no longer be covered by centralized MAs (under the Northern Ireland Protocol, centralized MAs will continue to be recognized in Northern Ireland). All medicinal products with a current centralized MA were automatically converted to Great Britain MAs on January 1, 2021. For a period of two years from January 1, 2021, the Medicines and Healthcare products Regulatory Agency, or MHRA, the UK medicines regulator, may rely on a decision taken by the European Commission on the approval of a new marketing authorization in the centralized procedure, in order to more quickly grant a new Great Britain MA. A separate application will, however, still be required.
28
European Data and MarketMarketing Exclusivity
In the European Union, innovative medicinalEEA, new products approved on the basis of a complete independent data package,authorized for marketing, or reference products, qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity.exclusivity upon marketing authorization. The data exclusivity if granted,period prevents generic or biosimilar applicants from referencingrelying on the innovator’s preclinical and clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization in the European Union, forEU during a period of eight years from the date on which the reference product was first authorized in the European Union. During the additional two-year period ofEU. The market exclusivity period prevents a successful generic or biosimilar MA application can be submitted, and the innovator’s data may be referenced, but no generic or biosimilarapplicant from commercializing its product can be marketed in the European UnionEU until 10 years have elapsed from the expirationinitial authorization of the reference product in the EU. The 10-year market exclusivity period. The overall ten year period willcan be extended to a maximum of eleven11 years if, during the first eight years of those ten10 years, the marketing authorization holder obtains an MAauthorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are determinedheld to bring a significant clinical benefit in comparison with currently approvedexisting therapies. There is no guarantee that a product will be considered by the EMA to be an innovative medicinal product, and products may not qualify for data exclusivity. Even if a product is considered to be an innovative medicinal product so that the innovator gains the prescribed period of data exclusivity, another company nevertheless could also market another version of the product if such company obtained an MA based on an MA application with a complete independent data package of pharmaceutical tests, preclinical tests and clinical trials.
European Orphan Designation and ExclusivityPediatric Investigation Plan
In the European Union,EEA, marketing authorization applications for new medicinal products not previously authorized must include the results of studies conducted in the pediatric population, in compliance with a pediatric investigation plan (“PIP”) agreed with the EMA’s Pediatric Committee (“PDCO”). The PIP sets out the timing and measures proposed to generate data to support a pediatric indication of the drug for which marketing authorization is being sought. The PDCO can grant a deferral of the obligation to implement some or all of the measures of the PIP until there are sufficient data to demonstrate the efficacy and safety of the product in adults. Further, the obligation to provide pediatric clinical trial data can be waived by the PDCO when these data are not needed or appropriate because the product is likely to be ineffective or unsafe in children, the disease or condition for which the product is intended occurs only in adult populations, or when the product does not represent a significant therapeutic benefit over existing treatments for pediatric patients. Once the marketing authorization is obtained in all Member States of the EU and study results are included in the product information, even when negative, the product is eligible for six months’ supplementary protection certificate extension.
Clinical Trials
Clinical trials of medicinal products in the EU must be conducted in accordance with EU and national regulations and the International Conference on Harmonization guidelines on GCPs. Additional GCP guidelines from the European Commission, basedfocusing in particular on traceability, apply to clinical trials of advanced therapy medicinal products. If the recommendationsponsor of the EMA’s Committee for Orphan Medicinal Products, grants orphan designationclinical trial is not established within the EU, it must appoint an entity within the EU to promoteact as its legal representative. The sponsor must take out a clinical trial insurance policy, and in most EU countries, the development of products that: (1) are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions; (2) either (i) such condition affects no more than 5 in 10,000 personssponsor is liable to provide ‘no fault’ compensation to any study subject injured in the European Union when the application is made, or (ii) it is unlikely that the product, without the benefits derived from orphan status, would generate sufficient return in the European Union to justify the necessary investment in its development; and (3) there exists no satisfactory method of diagnosis, prevention or treatment of such condition authorized for marketing in the European Union or, if such a method exists, the product would be of a significant benefit to those affected by the condition.clinical trial.
InPrior to commencing a clinical trial, the European Union, orphan designation entitlessponsor must obtain a party to financial incentives such as reduction of fees or fee waiversclinical trial authorization from the competent authority, and ten years of market exclusivity is granted following marketing approvala positive opinion from an independent ethics committee. The application for the orphan medicinal product. This period is extended by two years for compliance with an agreed upon pediatric investigation plan granted at the time of reviewa clinical trial authorization must include, among other things, a copy of the orphan designation. This period may be reduced to six years if, attrial protocol and an investigational medicinal product dossier containing information about the endmanufacture and quality of the fifth year, it is established that the orphan designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. During the period of market exclusivity, an MA may only be granted to a “similar medicinal product” for the same therapeutic indication if (i) the holder of the MA for the original orphan medicinal product consents to a second orphan medicinal product application, (ii) the holder of the MA for the original orphan medicinal product cannot supply sufficient quantities of the orphan medicinal product, or (iii) the second applicant can establish that the second medicinal product, although similar, is safer, more effective or otherwise clinically superior to the authorized orphan medicinal product. A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. Orphan designation must be requested before submitting an application for marketing approval. Orphan designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.under investigation.
Regulatory Requirements After a Marketing Authorization has been Obtained
In case an MA for a medicinal productClinical trials in the EU is obtained,are regulated under European Council Directive 2001/20/EC (“Clinical Trials Directive”) on the holderimplementation of GCP in the MA is required to comply with a rangeconduct of requirements applicable to the manufacturing, marketing, promotion and saleclinical trials of medicinal products. These include:
The aforementioned European Union rules are generally applicable in the EEA.
29
Brexit and the Regulatory Framework in the United Kingdom
In June 2016, the electorate in the United Kingdom voted in favor of leaving the European Union (commonly referred to as “Brexit”), and the United Kingdom officially withdrew from the European Union on January 31, 2020. Pursuant to the formal withdrawal arrangements agreed between the United Kingdom and the European Union, the United Kingdom was subject to a transition period until December 31, 2020, or Transition Period, during which European Union rules continued to apply. However, the European Union and the United Kingdom have concluded a trade and cooperation agreement, or TCA, which was provisionally applicable since January 1, 2021 and has been formally applicable since May 1, 2021. The TCA includes specific provisions concerning pharmaceuticals, which include the mutual recognition of GMP, inspections of manufacturing facilities for medicinal products and GMP documents issued, but does not foresee wholesale mutual recognition of UK and European Union pharmaceutical regulations. At present, Great Britain has implemented European Union legislation on the marketing, promotion and sale of medicinal products through the Human Medicines Regulations 2012 (as amended) (under the Northern Ireland Protocol, the European Union regulatory framework will continue to apply in Northern Ireland). The regulatory regime in Great Britain therefore largely aligns with current European Union regulations, however it is possible that these regimes will diverge in future now that Great Britain’s regulatory system is independent from the European Union and the TCA does not provide for mutual recognition of United Kingdom and European Union pharmaceutical legislation. For example, the new Clinical Trials Regulation which became effective inis intended to simplify the EU on January 31, 2022 and provides for a streamlined clinical trial application and assessment procedure covering multiple EU Member States has not been implemented into UK law, and a separate application will need to be submittedrules for clinical trial authorization and standards of performance. The implementation of the Clinical Trials Regulation depends on confirmation of full functionality of the Clinical Trials Information System through an independent audit, which commenced in September 2020. The system went live in January 2022. The new clinical trial portal and database will be maintained by the EMA in collaboration with the European Commission and the EU Member States. The Clinical Trials Directive requires the sponsor of an investigational medicinal product to obtain a clinical trial authorization (“CTA”), much like an IND in the UK.U.S., from the national competent authority of an EU Member State in which the clinical trial is to be conducted. The CTA application must be accompanied by an investigational medicinal product dossier with supporting information prescribed by the Council Directive and corresponding national laws of the Member States and further detailed in applicable guidance, including the European Commission Communication 2010/C 82/01. A clinical trial may only be commenced after an ethics committee has given its approval. Any substantial changes to the trial protocol or other information submitted with the clinical trial applications must be notified to or approved by the relevant competent authorities and ethics committees. Medicines used in clinical trials must be manufactured in accordance with cGMP. Other national and EU-wide regulatory requirements also apply.
European UnionPrivacy and Data CollectionProtection Laws
TheKineta is also subject to laws and regulations in non-U.S. countries covering data privacy and the protection of health-related and other personal information. EU Member States and other jurisdictions have adopted data protection laws and regulations, which impose significant compliance obligations. Laws and regulations in these jurisdictions apply broadly to the collection, use, storage, disclosure, processing and usesecurity of personal information that identifies or may be used to identify an individual, such as names, contact information and sensitive personal data such as health data indata. These laws and regulations are subject to frequent revisions and differing interpretations and have generally become more stringent over time.
38
As of May 25, 2018, Regulation 2016/676, known as the EEA is governed by the EU General Data Protection Regulation 2016/679, or GDPR, which went into effect on May 25, 2018 and superseded(“GDPR”) replaced the Data Protection Directive. The GDPR appliesDirective with respect to any company established in the EEA and to companies established outside the EEA that provides goods or services to residents in the EEA. This would include companies that process personal data in connection with the offering of goods or services to data subjects in the EEA or the monitoring of the behavior of data subjects in the EEA. The GDPR enhances data protection obligations for data controllersprocessing of personal data (including stringent requirements relating to the consent of data subjects, expanded disclosures about how personal data is used, requirements to conduct privacy impact assessments for “high risk” processing, limitations on retention of personal data, mandatory data breach notification and “privacy by design” requirements), creates direct obligations on service providers acting as data processors, and imposes special protections for “sensitive information,” which includes health and genetic information of data subjects residing in the EU. The GDPR grantsimposes many requirements for controllers and processors of personal data, including, for example, higher standards for obtaining consent from individuals the opportunity to objectprocess their personal data, more robust disclosures to individuals and a strengthened individual data rights regime, shortened timelines for data breach notifications, limitations on retention and secondary use of information, increased requirements pertaining to health data and pseudonymized (i.e., key-coded) data and additional obligations when we contract third-party processors in connection with the processing of theirthe personal information,data. The GDPR allows EU Member States to make additional laws and allows them to request deletionregulations further limiting the processing of personal information in certain circumstances. Additionally, the GDPR also imposes strict rules on the transfer of personal data outside of the EEA to countries that do not ensure an adequate level of protection, like the U.S.genetic, biometric or health data. Failure to comply with the requirements of the GDPR and the relatedapplicable national data protection laws of the EEA Member States may resultEU member states could subject Kineta to regulatory sanctions, delays in clinical trials, criminal prosecution and/or civil fines of upor penalties. Changes to 20 million Euros or 4% of a company’s global annual revenues for the preceding financial year, whichever is higher. Moreover, the GDPR grantsand applicable national data subjects the rightprivacy laws, including with respect to claim material and non-material damages resulting from infringement of the GDPR. Given the breadth and depth of changes in data protection obligations, maintaining compliance with the GDPR will require significant time, resources and expense, and we mayhow these laws should be required to put in place additional mechanisms ensuring compliance with the new data protection rules. This may be onerous and adversely affect our business, financial condition, results of operations and prospects. In addition, further to the UK’s exit from the EU on January 31, 2020, the GDPR ceased to applyapplied in the UK at the end of the transition period on December 31, 2020. However, as of January 1, 2021, the UK’s European Union (Withdrawal) Act 2018 incorporated the GDPR (as it existed on December 31, 2020 but subject to certain UK specific amendments) into UK law (referred to as the ‘UK GDPR’). The UK GDPR and the UK Data Protection Act 2018 set out the UK’s data protection regime, which is independent from but aligned to the European Union’s data protection regime. Non-compliance with the UK GDPR may result in monetary penalties of up to £17.5 million or 4% of worldwide revenue, whichever is higher. Although the United Kingdom is regarded as a third country under the EU GDPR, the European Commission has now issued a decision recognizing the United Kingdom as providing adequate protection under the EU GDPR and, therefore, transfers of personal data originating in the EEA to the United Kingdom remain unrestricted. Like the EU GDPR, the UK GDPR restricts personal data transfers outside the United Kingdom to countries not regarded by the United Kingdom as providing adequate protection. The United Kingdom government has confirmed that personal data transfers from the United Kingdom to the EEA remain free flowing.
Rest of the World Regulation
For other countries outside of the European Union and the United States, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conductcontext of clinical trials product licensing, pricingor other transactions from which Kineta may gain access to personal data, could increase our compliance costs and reimbursement vary from countryexposure to country. In all cases the clinical trials must be conducted in accordance with GCP requirements and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
30
potential liability.
Employees and Human Capital Resources
As of December 31, 2021, we employed 402023, Kineta had 11 full-time employees, including 27three employees with Ph.D. degrees and one with an M.D. degree. Of these full-time employees, three are engaged in research 1 in clinicaland development activities and 12eight are engaged in general and administrative and 1 part-time employee, and seventeenactivities. None of our full-timeKineta’s employees held M.D. or Ph.D. degrees. In February we also began implementation of a strategic restructuring with the objective of preserving capital. As part of the restructuring, we are eliminating approximately 60% of our workforce and took other actions.
We are highly dependent on our management and scientific and medical personnel, and it is crucial that we continue to retain valuable employees. To facilitate retention, we strive to make us a diverse, inclusive and safe workplace, with opportunities for our employees to grow and develop in their careers, supported by strong compensation and benefits programs. We have never had a work stoppage, and none of our employees is represented by a labor organizationunion or under any collective-bargaining arrangements. We consider ourcovered by a collective bargaining agreement. Kineta considers its relationship with ourits employees to be good. In addition, Kineta also had two part-time research and development consultants engaged as a clinician and a medical monitor.
Our In February 2024, and in connection with the Company’s decision to explore strategic alternatives, the Company implemented a workforce reduction of the Company’s workforce by seven full-time employees, or approximately 64% of the Company’s then-current employee base. The workforce reduction included Kineta’s Chief Executive Officer, Shawn Iadonato, Ph.D., who will continue to serve on the Company’s Board of Directors, and Kineta’s General Counsel and Secretary, Pauline Kenny.
Kineta’s human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating Kineta’s existing and additional employees. Kineta is committed to diversity, equity and inclusion across all aspects of its organization, including in Kineta’s recruitment, advancement and development practices. Each year, Kineta reviews employee demographic information to evaluate its diversity efforts across all functions and levels of the Company. Kineta conducts annual performance and development reviews for each of its employees to discuss the individual’s strengths and development opportunities, career development goals and performance goals. Kineta also regularly surveys employees to assess employee engagement and satisfaction. The principal purposes of Kineta’s equity incentive plans are to attract, retain and motivate selected employees, consultants and directors through the granting of equity awards. Kineta values its employees and regularly benchmarks total rewards Kineta provides, such as short and long-term compensation, 401(k) contributions, health, welfare and quality of life benefits, paid time off and personal leave, against Kineta’s industry peers to ensure Kineta remains competitive and attractive to potential new hires.
Properties and Facilities
Kineta occupies approximately 14,870 square feet of office and laboratory space (1,850 square feet of which is subleased to another biotech company) in Seattle, Washington under a lease that expires in July 2024. Kineta has an option to renew for two additional five-year terms. Kineta believes that its current facilities are adequate for its current needs and that suitable additional or substitute space at commercially reasonable terms will be available as needed to accommodate any future expansion of Kineta’s operations.
Legal Proceedings
On March 20, 2024, Kineta filed a complaint in the Court of Chancery of the State of Delaware against Growth & Value Development Inc. (“GVDI”), alleging breach of contract in connection with GVDI’s recent repudiation of its obligation to provide a substantial tranche of funding for Kineta as required under the Securities Purchase Agreement. The complaint provides that Kineta will seek specific performance of GVDI’s obligations under the Securities Purchase Agreement and damages equal to the amount of the unpaid funding and any damages resulting from GVDI’s breach.
Except as disclosed in the preceding paragraph, Kineta is currently not a party to any other material legal proceedings. From time to time, however, Kineta may be a party to litigation or subject to claims incident to the ordinary course of business. Although the results of litigation and claims cannot be predicted with certainty, Kineta currently believes that the final outcome of these ordinary course matters will not have a material adverse effect on Kineta’s business. Regardless of the outcome, litigation can have an adverse impact on Kineta because of defense and settlement costs, diversion of management resources and other factors.
Corporate Information
Proteostasis was
We were incorporated in Delaware on December 13, 2006 under the name Proteoguard, Inc., and subsequently changed itsour name to Proteostasis Therapeutics, Inc., on September 17, 2007. On December 22, 2020, Proteostasiswe effected a reverse merger, pursuant to which its wholly owneda wholly-owned subsidiary of ours merged with and into Yumanity, Inc. (formerly Yumanity Therapeutics, Inc.), (“Yumanity”) with Yumanity Inc. surviving as a wholly ownedwholly-owned subsidiary of Proteostasis.ours. On December 22, 2020, Proteostasiswe changed itsour name from Proteostasis“Proteostasis Therapeutics, Inc.” to Yumanity“Yumanity Therapeutics, Inc.” On December 16, 2022, we effected a reverse merger, pursuant to which a wholly-owned subsidiary of ours merged with and into Private Kineta with
See Part II—Item 7—Management’s Discussion and Analysis of Financial Condition and Results of Operations and Note 1 to the consolidated financial statements included in Part II—Item 8 for more information about the above-mentioned transactions.
We are39
Private Kineta surviving as a “smaller reporting company” as defined inwholly-owned subsidiary of ours. Private Kineta subsequently merged with and into Kineta Operating, LLC, with Kineta Operating, LLC being the Securities and Exchange Act of 1934, as amended, or the Exchange Act. We may continuesurviving corporation. On December 16, 2022, we changed our name from “Yumanity Therapeutics, Inc.” to take advantage of certain of the scaled disclosures available to smaller reporting companies if either our voting and non-voting common stock held by non-affiliates is less than $250 million measured on the last business day of our second fiscal quarter, or our annual revenues are less than $100 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is less than $700 million measured on the last business day of our second fiscal quarter.
“Kineta, Inc.” Our principal executive offices are located at 40 Guest7683 SE 27th Street, Suite 4410, Boston, Massachusetts and our481, Mercer Island, Washington 98040. Our telephone number is (617) 409-5300.(206) 378-0400. Our website address is www.yumanity.com. The information contained on, or accessible through, our website does not constitute part of this Annual Report on Form 10-K. We have included our website address in this Annual Report on Form 10-K solely as an inactive textual reference.https://kinetabio.com.
Available Information
Our Internet address is www.yumanity.com. Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, including exhibits, proxy and information statements and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Exchange Act, are available through the “Investors” portion of our website at https://kinetabio.com free of charge as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Information on our website is not part of, or incorporated by reference into, this Annual Report on Form 10-K or any of our other securities filings unless specifically incorporated herein by reference.report we file with, or furnish to, the SEC. In addition, our filings with the SEC may be accessed through the SEC’s Electronic Data Gathering, Analysis and Retrieval system at http://www.sec.gov. All statements made in any of our securities filings, including all forward-looking statements or information, are made as of the date of the document in which the statement is included, and we do not assume or undertake any obligation to update any of those statements or documents unless we are required to do so by law.www.sec.gov.
31
40
Item 1A. Risk Factors.
ITEM 1A. RISK FACTORS
Careful considerationInvesting in the securities of Kineta, Inc. (“Kineta”) involves significant risks and uncertainties. Before making an investment decision, you should be given tocarefully consider the following risk factors,risks and uncertainties described below, together with any subsequent updates described in addition to the other information set forth in thisKineta’s Annual ReportReports on Form 10-K, Quarterly Reports on Form 10-Q and in other documents that we file with the Securities and Exchange Commission, or SEC, in evaluating the Company and our business. Investing in our common stock involves a high degree of risk. If any of the following risks and uncertainties actually occurs, our business, prospects, financial condition and results of operationsCurrent Reports on Form 8-K.
Kineta could be materially and adversely affected. Theaffected by any or all of these risks described below are not intended to be exhaustive and are not the only risks facing the Company. Additionalor by additional risks and uncertainties not presently known to usKineta or that weKineta currently deemdeems immaterial alsothat may impact ouradversely affect Kineta.
Summary of the Material Risks Associated with Kineta’s Business
Risks relating to its Strategic Alternative Process, Potential Strategic Transaction, and Financial Position include:
Kineta is also subject to various risks associated with its businesses and its industries. These risks include, but are not limited to, the following:
41
Risks Related to Our Business,Strategic Alternative Process, Potential Strategic Transaction, and Financial Position and Need for Additional Capital
We are exploring strategic alternatives for the Company that could significantly impact our future operations and financial position.
In February 2022, we announcedKineta has decided to seek a strategic transaction, and there is no guarantee that we arethis strategic path will be successful.
Kineta is exploring strategic alternatives. The alternatives withto be considered may include, but are not limited to,sale of assets of the goalCompany, a sale of enhancing shareholder valuethe Company, licensing of assets, a merger, liquidation or other strategic action. Kineta has and have engaged H.C. Wainwright as our exclusive financial advisorexpects to assistcontinue to devote substantial time and resources to exploring such strategic alternatives; however, there is no set time frame and there can be no assurance that such activities will result in this process. Potentialany agreements or transactions that will enhance stockholder value. Evaluating a strategic alternative may divert the attention of the Company’s management from ordinary operating matters. The identification, negotiation, and completion of any such transaction may also require more time and cash resources than Kineta anticipates. In addition, potential strategic alternatives that require stockholder approval may not be considered as part of this process include an acquisition, merger, reverse merger, other business combination, sales of assets, licensing or other strategic transactions involving the Company. approved by Kineta’s stockholders.
42
There can be no assurance that Kineta’s process to identify and evaluate potential strategic alternatives will result in any definitive offer to consummate a strategic transaction, or if made that the explorationterms thereof will be acceptable to Kineta. If any definitive offer to consummate a strategic transaction is received, there can be no assurance that a definitive agreement will be executed or that, if a definitive agreement is executed, the transaction will be consummated. In addition, there can be no assurance that any transaction, involving the Company and/or its assets, that is consummated would enhance stockholder value.
If Kineta is successful in completing a strategic transaction, it may be exposed to other operational and financial risks, including increased near-term or long-term expenditures, exposure to unknown liabilities, incurrence of substantial debt, higher-than-expected acquisition and integration costs, write-downs of assets or impairment charges, increased amortization expenses, difficulty and cost in combining the operations and personnel of any merged businesses with Kineta’s operations and personnel, impairment of relationships with key suppliers or customers of any acquired businesses due to changes in management and ownership, and inability to retain key employees of the Company or any acquired businesses. If a strategic transaction is not completed, Kineta may be subject to a number of material risks or suffer a number of consequences that may adversely affect its business, financial results, and operations.
If Kineta is unable to successfully complete a strategic transaction, Kineta may be forced to cease operations altogether or file for bankruptcy protection.
There can be no assurance that any of Kineta’s plans will be successful or that additional capital will be available to Kineta on reasonable terms, or at all, when needed or that Kineta will successfully complete a strategic transaction. If Kineta does not obtain governmental approval for its products, or if, subject to receiving marketing approvals, it is unsuccessful in its commercial efforts to sell its products, Kineta’s business would experience significant harm. If Kineta is unable to obtain sufficient additional capital or to conclude a successful strategic transaction, Kineta may be forced to defer, reduce or eliminate significant planned expenditures, dispose of technology or assets including intangible assets, conclude a strategic transaction that is unfavorable to stockholders, enter into arrangements that may require Kineta to relinquish rights to certain of its products or product candidates, technologies or potential markets, delay or stop ongoing clinical trials, cease operations altogether or file for bankruptcy protection.
The Board remains dedicated to diligently deliberating upon and making informed decisions that the directors believe are in the best interests of the Company and its stockholders. There can be no assurance, however, that the Company’s current strategic direction, or the Board’s evaluation of strategic alternatives, will result in any initiatives, agreements, or transactions or plans that if completed,will further enhance stockholder value.
In addition, given the current restructuring of Kineta’s operations and recent reduction in force, it may be difficult to evaluate the Company’s current business and future prospects on the basis of historical operating performance.
Kineta may not realize any agreements or transactions will be successful or on attractive terms. No timetable has been established for the completion of this process, and we do not expect to disclose developments unless and until the Board of Directors has concluded that disclosure is appropriate or required. If we determine to change our business strategy or to seek to engageadditional value in a strategic transaction.
Potential counterparties in a strategic transaction our future business, prospects, financial positioninvolving Kineta may place minimal or no value on the Company’s assets. Further, the development and operating results could be significantly different than thoseany potential commercialization of Kineta’s product candidates will require substantial additional cash to fund the costs associated with conducting the necessary preclinical and clinical testing and obtaining regulatory approval. Consequently, any potential counterparty in historical periods or projected by our management. Becausea strategic transaction involving the Company may choose not to spend additional resources for the development of the significant uncertainty regarding our future plans, we are not able to accurately predict the impact of a potential change in our business strategy and future funding requirements.
Until the review process is concluded, perceived uncertainties related to our future may result in the loss of potential business opportunities and volatility in the market price of our common stockKineta’s product candidates and may make it more difficult for usattribute little or no value, in such a transaction, to attract and retain qualified personnel and business partners.those product candidates.
Our historicalIf Kineta is successful in completing a strategic transaction, it may be exposed to other operational and financial risks.
Although there can be no assurance that a strategic transaction will result from the process Kineta has undertaken to identify and evaluate strategic alternatives, the negotiation and consummation of any such transaction will require significant time on the part of the Company’s management, and the diversion of management’s attention may disrupt the Company’s business. The negotiation and consummation of any such transaction may also require more time or greater cash resources than Kineta anticipates and expose the Company to other operational and financial risks, including:
Any of the foregoing risks could have a material adverse effect on Kineta’s business, financial condition and prospects.
43
Kineta may not fully realize the expected cost savings and/or operating efficiencies from its restructuring activities and its ability to consummate a strategic transaction depends on its ability to retain its employees required to consummate such transaction.
On February 29, 2024, Kineta announced that it had completed a review of its business, including the status of its programs, resources and capabilities. Following this review, Kineta determined that it would implement a significant corporate restructuring to substantially reduce expenses and preserve cash. The restructuring includes a reduction in its workforce by approximately 64% and the termination of enrollment of new patients in its ongoing VISTA-101 Phase 1/2 clinical trial evaluating KVA12123 in patients with advanced solid tumors. Patients currently enrolled in the trial will be permitted to continue to participate. The Company made this decision, in part, because certain investors have indicated they will not be able to fulfill their contractual obligation to consummate the Private Placement (as defined below).
The Company believes these changes were needed to streamline its organization and reallocate its resources to better align with its current strategic goals, including its current focus on pursuing strategic alternatives. However, these expense reduction measures have and may continue to yield unintended consequences and costs, such as the loss of institutional knowledge and expertise, attrition beyond the Company’s intended reductions in workforce, a reduction in morale among its remaining employees, and the risk that the Company may not achieve the anticipated benefits, all of which may have an adverse effect on the Company’s results indicateof operations or financial condition.
Kineta’s ability to consummate a strategic transaction depends upon its ability to retain its employees required to consummate such a transaction, the loss of whose services may adversely impact the ability to consummate such transaction. There is no set timetable for the process Kineta has initiated to explore strategic alternatives and there can be no assurance that this process will result in the Company pursuing a transaction or that any transaction, if pursued, will be completed on attractive terms. If the Company is unable to complete a transaction, it may be required to seek a reorganization, liquidation or other restructuring. In addition, Kineta’s cash conservation activities, as well as the announcement that the Company is seeking strategic alternatives, may yield unintended consequences, such as attrition beyond its planned reductions in workforce that took place during the first quarter of 2024 and reduced employee morale, which may cause remaining employees to seek alternative employment. Kineta’s ability to successfully complete a strategic transaction depends in large part on its ability to retain certain of its remaining personnel. If the Company is unable to successfully retain its remaining personnel, it is at risk of a disruption to its exploration and consummation of a strategic alternative as well as business operations.
Kineta may become involved in securities litigation that could divert management’s attention and harm the Company’s business, and insurance coverage may not be sufficient to cover all costs and damages.
In the past, securities litigation has often followed certain significant business transactions, such as the sale of a company or announcement of any other strategic transaction. The Company may be exposed to such litigation even if no wrongdoing occurred. Litigation is usually expensive and diverts management’s attention and resources, which could adversely affect Kineta’s business and cash resources and Kineta’s ability to consummate a potential strategic transaction or the ultimate value its stockholders receive in any such transaction.
Kineta identified conditions and events that raise substantial doubt exists relatedabout its ability to ourcontinue as a going concern, Kineta needs substantial additional funding, and if Kineta is unable to raise capital when needed or on favorable terms, its business, financial condition, and results of operation could be materially and adversely affected.
Kineta may be forced to wind-down its operations if it is unable to consummate a strategic transaction and/or obtain sufficient funding.
As of December 31, 2023, Kineta had $5.8 million in cash, and there is substantial doubt about its ability to continue as a going concern. OurBased on Kineta’s current operating plans, Kineta does not have sufficient cash and cash equivalents to fund its operating expenses and capital expenditures for at least the next 12 months from the filing date of this Annual Report on Form 10-K.
Kineta is exploring strategic alternatives that may include, but are not limited to, sale of assets of the Company, a sale of the Company, licensing of assets, a merger, liquidation or other strategic action.
Kineta may seek additional funds through equity or debt financings or through collaborations, licensing transactions or other sources that may be identified through the Company’s strategic process. However, there can be no assurance that Kineta will be able to complete any such transactions on acceptable terms or otherwise. The failure to obtain sufficient funds on commercially acceptable terms when needed would have a material adverse effect on Kineta’s business, results of operations, and financial statements have been prepared assuming that we willcondition. These factors raise substantial doubt about Kineta’s ability to continue as a going concern.
WeKineta does not currently have incurred net losses and used significant cash in operating activities since inception, and we expect to continue to generate operating lossesany commitments for future funding or additional capital other than the foreseeable future. As of December 31, 2021, wePrivate Placement. However, as noted above, certain investors have an accumulated deficit of $187.3 million and cash, cash equivalents and marketable securities of $36.5 million. These factors raise substantial doubt about our ability to continue as a going concern and to satisfy our estimated liquidity needs for twelve months from the issuance of the consolidated financial statements.
If we continue to experience operating losses, and we areindicated they will not be able to generate additional liquidity through a capital raisefulfill their contractual obligation to consummate the Private Placement. As such, Kineta has paused or other cash infusion, we might need to secure additional sources of funds, which may or may not be available to us. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to further scalesignificantly scaled back or discontinue the development or commercialization of ourits future product candidates or other research and development initiatives or initiate stepsinitiatives. If Kineta is unable to cease operations.
If we do not successfully consummatecomplete a strategic transaction our board of directors may decideor raise additional capital in sufficient amounts, Kineta will not be able to pursue a dissolutioncontinue its business and liquidation of our company. In such an event, the amount of cash available for distribution to our stockholders will depend heavily on the timing of such liquidation as well as the amount of cash that will need to be reserved for commitments and contingent liabilities.
There can be no assurance that the process to identify a strategic transaction will result in a successfully consummated transaction. If no transaction is completed, our board of directors may decide to pursue a dissolution and liquidation of our company. In such an event, the amount of cash available for distribution to our stockholders will depend heavily on the timing of such decision and, ultimately, such liquidation, since the amount of cash available for distribution continues to decrease as we fund our operations while we evaluate our strategic alternatives. In addition, if our board of directors were to approve and recommend, and our stockholders were to approve, a dissolution and liquidation of our company, we would be required under Delaware corporate law to pay our outstanding obligations, as well as to make reasonable provision for contingent and unknown obligations, prior to making any distributions in liquidation to our stockholders. Our commitments and contingent liabilities may include (i) obligations under our employment and related agreements with certain employees that provide for severance and other payments following a termination of employment occurring for various reasons, including a change in control of our company; (ii) potential litigation against us, and other various claims and legal actions arising in the ordinary course of business; and (iii) non-cancelable contractual obligations. As a result of this requirement, a portion of our assetsCompany may need to be reserved pending the resolution of such obligations. In addition, we may be subject to litigation or other claims related to a dissolution and liquidation of our company. If a dissolution and liquidation werefile for bankruptcy protection.
32
Risks Related to Kineta’s Limited Operating History, Financial Position and Capital Requirements
pursued, our board of directors, in consultation with its advisors, would need to evaluate these matters and makeKineta has a determination about a reasonable amount to reserve. Accordingly, holders of our common stock could lose all or a significant portion of their investment in the event of a liquidation, dissolution or winding up of our company.
We havelimited operating history, has incurred significant operatingnet losses since ourits inception, and anticipate weanticipates that it will continue to incur continuedsignificant losses for the foreseeable future. Kineta may never generate any revenue or become profitable or, if Kineta achieves profitability, may not be able to sustain it.
We have funded our44
Kineta is a clinical-stage biotechnology company with a limited operating history that may make it difficult to evaluate the success of Kineta’s business to date and to assess its future viability. Kineta’s operations to date through proceedshave been limited to organizing and staffing its company, business planning, raising capital, developing and optimizing its technology platform, identifying potential product candidates, undertaking research, preclinical studies and clinical trials for its product candidates, establishing and enhancing its intellectual property portfolio, and providing general and administrative support for these operations. Kineta’s KVA12123, KCP506 and LHF535 programs are in early clinical development, and Kineta’s CD27 program is in preclinical development. None of Kineta’s product candidates have been approved for commercial sale. Kineta has never generated any revenue from collaborationsproduct sales and sales of preferred units. From our inception through December 31, 2021, we have received gross proceeds of $125.5 million from such transactions. As of December 31, 2021, our cash, cash equivalents and marketable securities were $36.5 million. We havehas incurred net losses in each year since our inception, and we have an accumulated deficit of $187.3Kineta commenced operations. Kineta’s net losses were $14.1 million as offor the year ended December 31, 2021.
In February 2022, we announced2023 and $63.5 million for the year ended December 31, 2022. Kineta expects that our Boardit will be several years, if ever, before it has a product candidate ready for regulatory approval and commercialization. Kineta expects to incur increasing levels of Directors approved a strategic restructuring with the objective of preserving capital. As part of the restructuring, we eliminated approximately 60% of our workforce among other actions to reduce cash burn while we explore strategic alternatives. There can be no assurance of success in reducing our cash burn or having sufficient cash to execute on a transaction that may result from our exploration of strategic alternatives.
Substantially all of our operating losses have resulted from costs incurred in connection with generalover the next several years and administrative costs associated with our operations, and our research and development programs, including for our preclinical and clinicalthe foreseeable future as Kineta advances its product candidates and our discovery engine platform. We expect that our existing cash, cash equivalents and marketable securities will not be sufficient to fund our operating expenses for a period of twelve months from the issuance of the consolidated financial statements. Ourthrough clinical development. Kineta’s prior losses, combined with expected future losses, have had and will continue to have an adverse effect on ourKineta’s stockholders’ deficitequity and working capital. In any particular quarter or quarters, our operating results could be below the expectations of securities analysts or investors, which could cause our stock price to decline.
AsTo become and remain profitable, Kineta must develop and eventually commercialize a public company, we expectproduct or products with significant market potential. This will require Kineta to continuebe successful in a range of challenging activities, including completing preclinical studies and clinical trials of its product candidates, obtaining marketing approval for these product candidates, manufacturing, marketing and selling those products for which Kineta may obtain marketing approval and satisfying any post-marketing requirements. Kineta may never succeed in these activities and, even if Kineta succeeds in commercializing one or more of its product candidates, Kineta may never generate revenue that is significant or large enough to incur significantachieve profitability. In addition, as a young business, Kineta may encounter unforeseen expenses, difficulties, complications, delays and increasing operating losses for the foreseeable future. Because of the numerous risksother known and uncertainties associated with developing pharmaceutical products, we are also unable to predict the extent of any future losses or when we will become profitable, if at all. Even if we do become profitable, weunknown challenges. If Kineta does achieve profitability, it may not be able to sustain or increase profitability on a quarterly or annual basis and Kineta will continue to incur substantial research and development and other expenditures to develop and market additional product candidates. Kineta’s failure to become and remain profitable would decrease the value of its company and could impair its ability to raise capital, maintain its research and development efforts, expand its business or continue its operations. A decline in the value of the Company could also cause Kineta’s stockholders to lose all or part of their investment.
Kineta has incurred recurring net losses and negative cash flows from operations since inception and, as of December 31, 2023, had an accumulated deficit of $165.8 million. The net loss attributable to Kineta was $14.1 million for the year ended December 31, 2023. As of December 31, 2023, Kineta had unrestricted cash of $5.8 million, andthere is substantial doubt about its ability to continue as a going concern. For more information, see the risk factor above entitled, “Kineta identified conditions and events that raise substantial doubt about its ability to continue as a going concern, Kineta needs substantial additional funding, and if Kineta is unable to raise capital when needed or on favorable terms, its business, financial condition, and results of operation could be materially and adversely affected.”
Kineta’s limited operating history may make it difficult for you to evaluate the success of its business to date and to assess Kineta’s future viability.
Kineta has a limited operating history, and its operations to date have been limited to organizing and staffing the Company, business planning, raising capital, conducting discovery and research activities, engaging third parties for initiating manufacturing of drug product and preparing for preclinical toxicology studies, conducting clinical trials, filing patent applications and identifying and obtaining rights to potential product candidates. Kineta has not yet demonstrated an ability to successfully obtain marketing licenses, manufacture a commercial scale product directly or through a third party or conduct sales, marketing and distribution activities necessary for successful product commercialization. Consequently, any predictions you make about Kineta’s future success or viability may not be as accurate as they could be if Kineta had a longer operating history or if Kineta had already successfully completed some or all of these types of activities.
In addition, as a clinical-stage biotechnology company, Kineta may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown challenges. Kineta will need to transition at some point from a company with a research and development focus to a company capable of supporting commercial activities and it may not be successful in making that transition.
Kineta expects its financial condition and operating results to continue to fluctuate significantly from quarter to quarter and year to year due to a variety of factors, many of which are beyond Kineta’s control. Accordingly, you should not rely upon the results of any quarterly or annual periods as indications of future operating performance.
Kineta’s ability to generate revenue and achieve profitability depends significantly on its ability to achieve its objectives relating to the discovery, development and commercialization of Kineta’s product candidates.
Kineta relies on its team’s expertise in drug discovery, translational research and patient-driven precision medicine to develop its product candidates. Kineta’s business depends significantly on the success of this engine and the development and commercialization of the product candidates that Kineta discovers with this engine. Kineta has no products approved for commercial sale and does not anticipate generating any revenue from product sales in the near term, if ever. Kineta’s ability to generate revenue and achieve profitability depends significantly on its ability to achieve several objectives, including:
45
Kineta may never be successful in achieving its objectives and, even if it does, may never generate revenue that is significant or large enough to achieve profitability. If Kineta does achieve profitability, it may not be able to sustain or increase profitability on a quarterly or annual basis. Kineta’s failure to become and remain profitable would decrease the value of the Company and could impair its ability to maintain or further its research and development efforts, raise additional necessary capital, grow its business and continue its operations.
We are a clinical stage biopharmaceutical company with a very limited operating history and no products approved for commercial sale, which may make it difficult to evaluate our current business and predict our future success and viability.The second closing of the Private Placement will not close on the expected timeline.
We are a clinical stage biopharmaceutical company with a limited operating history, focused on developing therapeutics for neurodegenerative diseases. We were initially formed as a limited liability company in 2014 and converted
Kineta entered into a corporationfinancing agreement, dated as of June 5, 2022, as amended on December 5, 2022, March 29, 2023, May 1, 2023, July 21, 2023 and October 13, 2023 (such financing agreement, as amended, the “Securities Purchase Agreement”) with certain investors to sell shares of its common stock in 2015, we have no products approveda private placement (the “Private Placement”). The first closing of the Private Placement occurred on December 16, 2022 and Kineta issued 649,346 shares of its common stock and received net proceeds of $7.4 million. The second closing of the Private Placement for commercial sale, and we havean aggregate purchase price of $22.5 million is scheduled to occur on April 15, 2024. Kineta may further amend the Private Placement to close at a later date based on its funding needs. In February 2024, Kineta was informed by certain investors that they would not generated any revenue from product salesbe able to date. We began human clinical trials for YTX-7739 atconsummate the endsecond closing of 2019 and have not initiated clinical trials for any of our other current product candidates. Our operationsthe Private Placement, resulting in Kineta’s decision to date have been limited primarily to organizing and staffing, raising capital, and conducting research and development activities for our product candidates.seek strategic alternatives as discussed elsewhere in this Annual Report on Form 10-K.
To date, we have not initiatedKineta will need to obtain substantial additional funding to complete the development and commercialization of its product candidates. If Kineta is unable to raise this capital when needed, Kineta may be forced to delay, reduce or completed a pivotaleliminate its product development programs or other operations.
Since its inception, Kineta has used substantial amounts of cash to fund its operations and expects its expenses to increase substantially during the next few years. The development of biopharmaceutical product candidates, especially immuno-oncology product candidates, is capital intensive. As Kineta’s product candidates enter and advance through preclinical studies and clinical trial, obtainedtrials, Kineta will need substantial additional funds to expand its clinical, regulatory, quality and manufacturing capabilities. In addition, if Kineta obtains marketing approval for any of its product candidates, manufacturedKineta expects to incur significant commercialization expenses related to marketing, sales, manufacturing and distribution. Furthermore, Kineta expects to incur additional costs associated with operating as a commercial scalepublic company.
As of December 31, 2023, Kineta had $5.8 million in cash, and there is substantial doubt about its ability to continue as a going concern. For more information, see the risk factor above entitled, “Kineta identified conditions and events that raise substantial doubt about its ability to continue as a going concern, Kineta needs substantial additional funding, and if Kineta is unable to raise capital when needed or on favorable terms, its business, financial condition, and results of operation could be materially and adversely affected.”
Kineta has based these estimates on assumptions that may prove to be incorrect or require adjustment as a result of business decisions, and Kineta
46
could utilize its available capital resources sooner than it currently expects. Kineta’s future capital requirements will depend on many factors, some of which are outside of its control, including:
Kineta will require additional capital to achieve its business objectives. Additional funds may not be available on a timely basis, on favorable terms, or at all, and viability subjectsuch funds, if raised, may not be sufficient to significant uncertainty. We will encounter risks and difficulties frequently experienced by early-stage biopharmaceutical companies in rapidly evolving fields, and we have not yet demonstrated anenable Kineta to continue to implement its long-term business strategy. Further, Kineta’s ability to successfully overcomeraise additional capital may be adversely impacted by potential worsening global economic conditions and the disruptions to and volatility in the credit and financial markets in the United States and worldwide resulting from inflation, changes in interest rates, geopolitical instability and pandemics or other public health crises. If Kineta is unable to raise sufficient additional capital, Kineta could be forced to curtail its planned operations and the pursuit of its growth strategy.
Raising additional capital may cause dilution to Kineta’s stockholders, restrict its operations or require Kineta to relinquish rights to its technologies or product candidates.
Until such riskstime, if ever, as Kineta can generate substantial product revenue, Kineta expects to finance its operations through equity offerings, debt financings or other capital sources, including potentially grants, collaborations, licenses or other similar arrangements. To the extent that Kineta raises additional capital through the sale of equity or convertible debt securities, Kineta’s stockholders’ ownership interest will be diluted, and difficulties. the terms of these securities may include liquidation or other preferences that adversely affect the rights of holders of Kineta’s common stock. Additional debt financing, if available, may involve agreements that include covenants further limiting or restricting Kineta’s ability to take specific actions,
47
such as further limitations on Kineta’s ability to incur additional debt, make capital expenditures or declare dividends.
If we doKineta raises funds through collaborations or licensing arrangements with third parties, Kineta may have to relinquish valuable rights to its technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not address these risksbe favorable to Kineta. If Kineta is unable to raise additional funds when needed, Kineta may be required to delay, limit, reduce or terminate its product development or future commercialization efforts or grant rights to develop and difficulties successfully, our business will suffer.market product candidates that Kineta would otherwise prefer to develop and market itself.
SEC regulations limit the amount of funds wethat Kineta can raise during any 12-month period pursuant to ourits shelf registration statement on Form S-3.
SEC regulations limit the amount that companies with a public float of less than $75 million may raise during any 12-month period pursuant to a shelf registration statement on Form S-3, referred to as the baby shelf rules.S-3. As of the filing of this Annual Report on Form 10-K, we areKineta is subject to suchGeneral Instruction I.B.6 to Form S-3, referred to as the baby shelf rules. Under these regulations, the amount of funds weKineta can raise through primary public offerings of securities in any 12-month period using ourits registration statement on Form S-3 including the registration statement under which our Open Market Sale AgreementSM with Jefferies LLC, is operated is limited to one-third of the aggregate market value of the shares of ourits common stock held by non-affiliates of the Company. Therefore, weKineta will be limited in the amount of proceeds we areit is able to raise by selling shares of ourits common stock using ourits Form S-3 until such time as ourits public float exceeds $75 million. Furthermore, if we areKineta is required to file a new registration statement on another form, weit may incur additional costs and be subject to delays due to review by the SEC staff.
33
Kineta has identified material weaknesses in its internal control over financial reporting. If Kineta is unable to remedy its material weaknesses in the future, or if Kineta fails to establish and maintain effective internal controls, Kineta may be unable to produce timely and accurate financial statements. Kineta has concluded that its internal control over financial reporting is ineffective as of December 31, 2023, which could adversely impact Kineta’s investors’ confidence and Kineta’s stock price.
Drug developmentPrior to completion of the Merger, Kineta was a private company and had limited accounting and financial reporting personnel and other resources with which to address its internal controls and related procedures. In connection with the audit of Kineta’s financial statements for the years ended December 31, 2023 and 2022, Kineta and its independent registered public accounting firm identified material weaknesses in Kineta’s internal control over financial reporting. A material weakness is a highly uncertain undertakingdeficiency, or a combination of deficiencies, in internal control over financial reporting as defined under the Exchange Act and involvesby the Public Company Accounting Oversight Board (United States), such that there is a substantial degreereasonable possibility that a material misstatement of risk. We have never generated any revenue from product sales,Kineta’s annual or interim financial statements will not be prevented or detected on a timely basis. The material weaknesses for the year ended December 31, 2023 relates to accounting for complex financial instruments related to the derivative asset and we may never generate revenue or be profitable.for accounting for allocated facilities costs. The material weakness for the year ended December 31, 2022 relates to accounting for complex financial instruments related to warrants issued to certain existing stockholders.
Our abilityKineta is in the process of implementing measures designed to become profitable depends uponimprove its internal control over financial reporting to remediate these material weaknesses. For example, Kineta began to address the abilitymaterial weaknesses by implementing certain Sarbanes-Oxley controls during the first half of our product candidates2022. In October 2022, Kineta hired a Chief Financial Officer to generate revenue. Toenhance internal controls and address the material weaknesses and other control deficiencies identified during the audit of the financial statements. Kineta has designed and implemented improved processes and internal controls, including ongoing senior management review and audit committee oversight. Additionally, Kineta has implemented and upgraded accounting and reporting systems to improve accounting and financial reporting processes and has enhanced, developed and implemented formal policies, processes and documentation procedures relating to financial reporting, including the oversight of third-party service providers. The actions that Kineta has taken are subject to ongoing executive management review and will also be subject to audit committee oversight. Kineta expects to incur additional costs to remediate these material weaknesses. Kineta cannot assure you that the measures it has taken to date, we have not generatedtogether with any revenue from our product candidates and we do not know when,measures it may take in the future, will be sufficient to remediate the control deficiency that led to the material weaknesses in Kineta’s internal control over financial reporting or to avoid potential future material weaknesses. If Kineta is unable to successfully remediate its existing or any future material weakness in Kineta’s internal control over financial reporting, or if we will do so. We do not anticipate generatingKineta identifies any revenue from product sales until after we have successfully completed clinical developmentadditional material weakness, the accuracy and received regulatory approval for the commercial saletiming of a product candidate, if ever. Our ability to generate revenue depends on a number of factors, including, but not limited to:
Because of the numerous risks and uncertainties associated with drug development, we arebe unable to predict the timing or amountmaintain compliance with securities law requirements regarding timely filing of our expenses, or when we will be able to generate any meaningful revenue or achieve or maintain profitability, if ever. In addition, our expenses could increase beyond our current expectations if we are required by the U.S. Food and Drug Administration (the “FDA”), or foreign regulatory agencies, to perform studiesperiodic reports in addition to those that we currently anticipate, or if there are any delaysapplicable stock exchange listing requirements, investors may lose confidence in any of our current or future collaborators’ clinical trials orKineta’s financial reporting, and Kineta’s stock price may decline as a result. Kineta also could become subject to investigations by The Nasdaq Stock Market, LLC (“Nasdaq”), the development of any of our product candidates. Even if one or more of our product candidates is approved for commercial sale, absent our entering into a collaboration or partnership agreement, we anticipate incurring significant costs associated with commercializing any approved product candidate and ongoing compliance efforts.
Even if we are able to generate revenue from the sale of any approved products, we may not become profitable and may need to obtain additional funding to continue operations. Revenue from the sale of any product candidate for which regulatory approval is obtained will be dependent, in part, upon the size of the markets in the territories for which we gain regulatory approval, the accepted price for the product, the ability to get reimbursement at any price, and whether we own the commercial rights for that territory. The precise number of people with Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis (“ALS”) is unknown. Our projections of both the number of people who have these diseases, as well as the subset of people with these diseases who have the potential to benefit from treatment with our product candidates, are based on estimates. If the number of addressable patients is not as significant as we anticipate, the indication approved by regulatory authorities is narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice, or treatment guidelines, we may not generate significant revenue from sales of our product candidates, even if approved. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
34
Our failure to become and remain profitable would decrease our value and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our pipeline of product candidates, or continue our operations and cause a decline in the value of our common stock, all or any of which may adversely affect our viability.
Due to the significant resources required for the development of our programs, and depending on our ability to access capital, we must prioritize development of certain product candidates. Moreover, we may fail to expend our limited resources on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
We are prioritizing our lead product candidate, YTX-7739, which is in Phase 1 clinical development for the treatment of Parkinson's disease. We seek to maintain a process of prioritization and resource allocation to maintain an optimal balance between aggressively advancing product candidates, such as YTX-7739, and ensuring replenishment of our portfolio.
Due to the significant resources required for the development of our product candidates, we must focus on specific diseases and disease pathways and decide which product candidates to pursue and advance and the amount of resources to allocate to each. For example, we plan to evaluate YTX-7739 in additional preclinical studies instead of advancing our other product candidate, YTX-9184, as we believe this approach will provide an opportunity to assess results in patients sooner, given the studies and clinical trials done to date of YTX-7739. Our decisions concerning the allocation of research, development, collaboration, management, and financial resources toward particular product candidates or therapeutic areas may not lead to the development of any viable commercial product and may divert resources away from better opportunities. If we make incorrect determinations regarding the viability or market potential of any of our product candidates, including our decision to focus on YTX-7739, or misread trends in the biopharmaceutical industry, in particular for neurodegenerative diseases, our business, financial condition, and results of operations could be materially adversely affected. As a result, we may fail to capitalize on viable commercial products or profitable market opportunities, be required to forego or delay pursuit of opportunities with other product candidatesSEC, or other diseases and disease pathways that may later prove to have greater commercial potential than those we choose to pursue, or relinquish valuable rights to such product candidates through collaboration, licensing, or other royalty arrangements in cases in which it would have been advantageous for us to invest additional resources to retain sole development and commercialization rights.
We will need additional funding to advance YTX-7739 through clinical development, which funding may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit, or terminate our product development efforts or other operations.
As of December 31, 2021, our cash, cash equivalents and marketable securities were $36.5 million. We will require additional funding to advance clinical trials of YTX-7739 and other planned early development of other programs generated by our discovery engine platform. Our ability to secure this additional funding may be adversely impacted by negative or ambiguous results in future clinical trials for YTX-7739. Developing small-molecule products is expensive, and we expect our discovery, research, and development expenses to increase substantially in connection with our ongoing activities, particularly as we advance our product candidates in clinical trials. We may also need additional funds sooner if we choose to pursue additional indications and/or geographies for our product candidates or otherwise expand more rapidly than we presently anticipate.
In addition, we have identified conditions and events that raise substantial doubt about our ability to continue as a going concern.
Our operating plan may also change as a result of many factors currently unknown, and we are currently exploring strategic alternatives such as an acquisition, merger, reverse merger, other business combination, sales of assets, licensing or other strategic transactions. In any event, we will require additional capital to obtain regulatory approval for, and, if approved, to commercialize our product candidates. Raising funds in the current economic environment may present additional challenges. Even if we believe that we have sufficient funds for our current or future operating plans, we may seek additional capital if market conditions are favorable or if we have specific strategic considerations.
Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and, if approved, commercialize our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to it, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our stockholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our shares to decline. The sale of additional equity or convertible securities would dilute all of our stockholders. The incurrence of indebtedness would result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights, and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborative partners or otherwise at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results, and prospects.
35
Based on our public float, as of the date of the filing of this Annual Report on Form 10-K, we are only permitted to utilize a “shelf” registration statement, including the registration statement under which our Open Market Sale AgreementSMwith Jefferies LLC, is operated, subject to Instruction I.B.6 to Form S-3, which is referred to as the “baby shelf” rule. For so long as our public float is less than $75 million, we may not sell more than the equivalent of one-third of our public float during any 12 consecutive months pursuant to the baby shelf rules. Although alternative public and private transaction structures may be available, these may require additional time and cost, may impose operational restrictions on us, and may not be available on attractive terms.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay, or discontinue one or more of our research or development programs or the commercialization of any approved product candidate or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition, and results of operations.authorities.
Risks Related to Our Productthe Discovery, Development and CommercializationRegulatory Approval of Kineta’s Product Candidates
It may take considerable timeKineta’s development efforts are in the early stages. All of Kineta’s product candidates are in clinical development or in preclinical development. If Kineta is unable to advance its product candidates through clinical development, obtain regulatory approval and expense to respond to the partial clinical hold that has been placed on our IND by the FDA andultimately commercialize its product candidates, or experience significant delays in doing so, Kineta’s business will be materially harmed.
There is no assurance can be given that the FDA will remove the partial clinical hold in which case our business and prospects will likely suffer material adverse consequences.
In January 2022, the U.S. Food and Drug Administration (FDA) placed a partial clinical hold on multidose clinical trials of YTX-7739. The partial clinical hold suspends initiation of multiple dose clinical trials in the U.S. until the FDA’s concerns have been addressed. The FDA has not halted all clinical programming and is permitting our planned single dose formulation clinical trial to proceed. It may take a considerable period of time, the length of which is not certain at this time, and expense for us to fully address FDA’s concerns. Even if we are able to fully respond to the FDA’s questions, they may subsequently make additional requests that we would need to fulfill prior to the lifting of the partial clinical hold. It is possible that we will be unable to fully address the FDA’s questions and as a result the partial clinical hold may never be lifted and we may never be able to begin multidoseKineta’s product candidates, or any other future clinical trials of YTX-7739 in the U.S.
Research and development of biopharmaceutical products is inherently risky.
We are at an early stage of development of the product candidates currently in our pipeline and are continuing to discover additional potential product candidates leveraging our discovery engine platform. To date, we have devoted substantially all of our efforts and financial resources to identify, secure intellectual property for, and develop our discovery engine platform and our product candidates, including conducting multiple preclinical studies, and providing general and administrative support for these operations. Our business depends heavily on the successful clinical development, regulatory approval, and commercialization of our lead product candidate, YTX-7739, which is in clinical development. None of our product candidates have advanced into late-stage development or a pivotal clinical study and it may be years before any such study is initiated, if at all. YTX-7739 will require substantial additional clinical development, testing, and regulatory approval before we are permitted to commence commercialization. Further, we cannot be certain that any of ourKineta’s product candidates, will be successful or will generate positive clinical data and Kineta may not receive marketing approval from the FDA, European Commission, or other regulatory authorities for any of its product candidates. Kineta has limited experience submitting Investigational New Drug Applications (the “INDs”) to the FDA. KVA12123, KCP506 and LHF535 are in clinical trials or obtain regulatory approval.
Ourdevelopment. There can be no assurance that the FDA will permit any of Kineta’s future success is dependent on our ability to successfully develop, obtain regulatory approval for, and then successfully commercialize our product candidates, and we may fail to do so for many reasons,INDs, including the following:
36
If any of these events occur, we may be forced to abandon our development efforts for a product candidate or candidates, which would have a material adverse effect on our business and could potentially cause us to cease operations. For instance, if we observe harmful side effects or other characteristics that indicate one product candidate is unlikely to be effective or otherwise does not meet applicable regulatory criteria, these findings may implicate the discovery engine platform as a whole.
We may not be successful in our efforts to further develop our discovery engine platform technology and current product candidates. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates. Each of our product candidates are in the early stages of development and will require significant additionalconduct clinical development, management of preclinical, clinical, and manufacturing activities, regulatory approval, adequate manufacturing supply, a commercial organization, and significant marketing efforts before we could generate any revenue from product sales, if at all.
The preclinical and clinical product candidates and current clinical trials are, and the future clinical trials and the manufacturing and marketing of our product candidates will be, subject to extensive and rigorous review and regulation by numerous government authorities in the United States and in other countries where we intend to test and, if approved, market anyof such product candidate. Before obtaining regulatory approvals for the commercial sale of any product candidate, we must, among other requirements, demonstrate through preclinical studies and clinical trials that the product candidate is safe and effective for use in each target indication. Drug
Biopharmaceutical development is a difficult, long, time-consuming, expensive and uncertain process, and delay or failure can occur at any stage of any of ourKineta’s clinical trials. This process can take many yearsFailure to obtain regulatory approval for Kineta’s product candidates will prevent it from commercializing and may include post-marketing studies and surveillance, which will require the expenditure of substantial resources. Of the large number of drugs in development
48
marketing its product candidates. The success in the United States, only a small percentagedevelopment of Kineta’s product candidates will successfullydepend on many factors, including:
If any of our product candidates successfully complete clinical trials, we generally plan to seek regulatory approval to market our product candidates in the United States, the European Union (“EU”),in additional foreign countries where we believe there is a viable commercial opportunitymaintaining patent, trademark and significant patient need. We have never commenced, compiled, or submitted an application seeking regulatory approval to market any product candidate. We may never receive regulatory approval to market any product candidates even if such product candidates successfully complete clinical trials, which would adversely affect our viability. To obtain regulatory approval in countries outside the United States, we must comply with numeroustrade secret protection and varying regulatory requirements of such other countries regarding safety, efficacy, chemistry, manufacturing and controls, clinical trials, commercial sales, pricing, and distribution of our product candidates. We may also rely on collaborators or partners to conduct the required activities to support an applicationregulator exclusivity for regulatory approval, and to seek approval, for one or more of our product candidates. We cannot be sure that any collaborators or partners will conduct these activities or do so within the timeframe we desire. Even if we (or any collaborators or partners) are successful in obtaining approval in one jurisdiction, we cannot ensure that we (or any collaborators or partners) will obtain approval in any other jurisdictions. If we are unable to obtain approval for our product candidates in multiple jurisdictions, our revenue and results of operations could be negatively affected.Even if we receive regulatory approval to market any of our product candidates, we cannot assure you that any such product candidate will be successfully commercialized, widely accepted in the marketplace or more effective than other commercially available alternatives.Investment in biopharmaceutical product development involves significant risk that any product candidate will fail to demonstrate adequate efficacy or an acceptable safety profile, gain regulatory approval, and become commercially viable. We cannot provide any assurance that we will be able to successfully advance any of our product candidates through the development process or, if approved, successfully commercialize any of our product candidates.We may not be successful in our efforts to continue to create a pipeline of product candidates or to develop commercially successful products. If we fail to successfully identify and develop additional product candidates, our commercial opportunity may be limited.One of our strategies is to identify and pursue clinical development of additional product candidates. Our portfolio currently consists of four programs, one of which is in clinical development and the rest of which are in research, discovery and preclinical stages of development. Identifying, developing, obtaining regulatory approval, and commercializing additional product candidates for the treatment of neurodegenerative diseases will require substantial additional funding and is prone to the risks of failure inherent in drug development. We cannot provide you any assurance that we will be able to successfully identify or acquire additional product candidates, advance any of these additional product candidates through the development process, successfully commercialize any such37additional product candidates, if approved, or assemble sufficient resources to identify, acquire, develop or, if approved, commercialize additional product candidates. If we are unable to successfully identify, acquire, develop, and commercialize additional product candidates, our commercial opportunity may be limited.We may not be able to conduct, or contract others to conduct, animal testing in the future, which could harm our research and development activities.Certain laws and regulations relating to drug development require us to test our product candidates on animals before initiating clinical trials involving humans. Animal testing activities have been the subject of controversy and adverse publicity. Animal rights groups and other organizations and individuals have attempted to stop animal testing activities by pressing for legislation and regulation in these areas and by disrupting these activities through protests and other means. To the extent the activities of these groups are successful, our research and development activities may be interrupted or delayed.We have concentrated our research and development efforts on the treatment of neurodegenerative diseases, a field that has seen limited success in drug development. Further, our product candidates are based on new approaches and novel technology, which makes it difficult to predict the time and cost of product candidate development and subsequently obtaining regulatory approval.We have focused our research and development efforts on addressing neurodegenerative diseases, including Parkinson’s disease, ALS and Alzheimer’s disease. Efforts by biopharmaceutical companies in the field of neurodegenerative diseases have seen limited successes in drug development. There are few effective therapeutic options available for patients with Parkinson’s disease, ALS or Alzheimer’s disease. Our future success is highly dependent on the successful development of our discovery engine platform technology and our product candidates for treating neurodegenerative diseases. Developing and, if approved, commercializing our product candidates for treatment of neurodegenerative diseases subjects us to a number of challenges, including engineeringKineta’s product candidates and obtaining regulatory approval from the FDAotherwise protecting its rights in its intellectual property portfolio;
Our approach is centered on the key insight that human protein misfolding, a phenomenon at the root of virtually all neurodegenerative diseases, can be modeled effectively in yeast cells. Discoveries from the yeast system are then translated to diseased human cell lines created by adult stem cells using induced pluripotent stem cell technology (“iPSC”). This strategy may not prove to be successful. We cannot be sure that our approach will yield satisfactory therapeutic products that are safe and effective, scalable, or profitable.
Moreover, public perception of drug safety issues, including adoption of new therapeutics or novel approaches to treatment, may adversely influence the willingness of subjects to participate inproceed with Kineta’s planned clinical trials or if approved, of physicians to prescribe our products.
We may encounter difficulties in enrolling subjects in ourother future clinical trials, thereby delaying or preventing development of our product candidates.
There is no precise method of establishing the actual number of people with neurodegenerative diseases in any geography over any time period. It is estimated that more than 60 million people worldwide suffer from neurodegenerative diseases. If the actual number of people with neurodegenerative diseases is lower than we believe, we may experience difficulty in enrolling subjects in our clinical trials, thereby delaying development of our product candidates. Furthermore, we may experience difficulties in subject enrollment in our clinical trials for a variety of other reasons, including:
38
Our clinical trials may fail to demonstrate adequate safety and efficacy of our product candidates, which would prevent, delay, or limit the scope of regulatory approval and commercialization.
Before obtaining regulatory approvals for the commercial sale of any of our product candidates, we must, among other requirements, demonstrate through lengthy, complex, and expensivepositive results from Kineta’s preclinical studies and clinical trials that oursupport a demonstration of efficacy, safety, and durability of effect for its product candidates;
Many of these factors are both safebeyond Kineta’s control, including the time needed to adequately complete clinical testing, the regulatory submission process and effective for usepotential threats to Kineta’s intellectual property rights. It is possible that none of Kineta’s product candidates will ever obtain regulatory approval, even if Kineta expends substantial time and resources seeking such approval. If Kineta does not achieve one or more of these factors in each target indication. Eacha timely manner or at all, or any other factors impacting the successful development of biopharmaceutical products, Kineta could experience significant delays or an inability to successfully develop its product candidate must demonstrate an adequate risk versus benefit profile in our intended patient population and for our intended use.candidates, which would materially harm Kineta’s business.
Clinical testing is expensive and can take many years to complete, and our outcome is inherently uncertain. Failure can occur at any time during the clinical study process. The results of preclinical studies and early clinical trials are not always predictive of ourfuture results. Any product candidate that Kineta advances in clinical trials may not achieve favorable results in later clinical trials, if any, or receive marketing approval.
The research and development of drugs and biological products is extremely risky. Only a small percentage of product candidates may not be predictivethat enter the development process ever receive marketing approval. Before obtaining marketing approval from regulatory authorities for the sale of Kineta’s product candidates, Kineta must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidates in humans. The outcome of clinical testing is uncertain. Kineta may face unforeseen challenges in its product candidate development strategy, and Kineta can provide no assurances that it will ultimately be successful in its current and future clinical trials or that Kineta’s product candidates will be able to receive regulatory approval. The results of early-stage or later-stage clinical trials,preclinical studies and results of early-stageearly clinical trials of ourKineta’s product candidates may not be predictive of the results of later-stage clinical trials. The results of clinical trials in one set of subjects or disease indications mayFor example, it is not be predictive of those obtained in another. In some instances, there can be significant variability inuncommon for product candidates to exhibit unforeseen safety or efficacy issues when tested in humans despite promising results between differentin preclinical animal models. Future results of preclinical and clinical testing of Kineta’s product candidates are also less certain due to the novel and relatively untested nature of the approach of Kineta’s development platform. In general, clinical trial failure may result from a multitude of factors including flaws in study design, dose selection, patient enrollment criteria and failure to demonstrate favorable safety or efficacy traits. As such, failure in clinical trials can occur at any stage of the same product candidate due to numerous factors, including changes in study procedures set forth in protocols, differences in the size and type of the patient populations, changes in and lack of adherence to the dosing regimen or the delivery formulation of YTX-7739 or any of our other product candidates and other clinical study protocols, and the rate of dropout among clinical study participants. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy profile despite having progressed through preclinical studies and initial clinical trials.testing. A number of companies in the biopharmaceutical industry, including immuno-oncology companies, have suffered significant setbacks in later-stagethe advancement of clinical trials due to lack of efficacy or adverse safety issues,profiles, notwithstanding promising results in early-stage studies. This is particularly true in neurodegenerative diseases, where failure rates historically have been higher than in other disease areas. Most product candidates that begin clinical trials are never approved by regulatory authorities for commercialization.
We have limited experience in designing clinical trials and may be unable to design and execute a clinical study to support marketing approval. We cannot be certain that our current clinical trials or any other future clinical trials will be successful. Additionally, any safety concerns observed in any one of our clinical trials in our targeted indications could limit the prospects for regulatory approval of our product candidates in those, and other indications, which could have a material adverse effect on our business, financial condition, and results of operations.
In addition, even if such clinical trials are successfully completed, we cannot guarantee that the FDA or foreign regulatory authorities will interpret the results as we do, and more studies could be required before we submit our product candidates for approval. To the extent that the results of the studies are not satisfactory to the FDA or foreign regulatory authorities for support of a marketing application, we may be required to expend significant resources, which may not be available to us, to conduct additional studies in support of potential approval of our product candidates. Even if regulatory approval is secured for any of our product candidates, the terms of such approval may limit the scope and use of our product candidates, which may also limit their commercial potential.
We may not be able to file IND applications or related amendments or similar applications and amendments outside the United States to commence additional clinical trials on the timelines expected, and even if we are able to, regulatory authorities may not permit us to proceed.
We may not be able to file future IND applications or similar applications outside the Unites States for our product candidates on the timelines we expect. For example, we may experience manufacturing delays or other delays with preclinical studies. Moreover, we cannot be sure that submission of an IND or similar application outside the United States will result in the FDA or respective regulatory authority allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate clinicalearlier trials. Additionally, even if such regulatory authorities agree with the design and implementation of the clinical trials set forth in IND or similar application, we cannot guarantee that such regulatory authorities will not change their requirements in the future. These considerations also apply to new clinical trials we may submit as amendments to existing IND or similar applications or to a new application. Any failure to file IND or similar applications on the timelines we expect or to obtain regulatory authorizations for our trials may prevent us from completing our clinical trials or commercializing our products on a timely basis, if at all.
39
Interim, “topline,” and preliminary data from ourKineta’s clinical trials that are announced or published from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, weKineta may publicly disclose preliminary or topline data from ourits clinical trials, which are based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular trial. WeKineta also makemakes assumptions, estimations, calculations and conclusions as part of its analyses of data, and weKineta may not have received or had the opportunity to fully evaluate all data. As a result, the topline or preliminary results that we reportKineta reports may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data havehas been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data weKineta previously published. As a result, topline data should be viewed with caution until the final data are available. From time to time, weKineta may also disclose interim data from ourits clinical trials. Interim data from clinical trials that weKineta may complete areis subject to the risk that
49
one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available or as patients from ourKineta’s clinical trials continue other treatments for their disease. Adverse differences between preliminary or interim data and final data could significantly harm ourKineta’s business prospects.
In addition, the information we chooseKineta chooses to publicly disclose regarding a particular clinical trial is based on what is typically extensive information, and you or others may not agree with what we determineKineta determines is material or otherwise appropriate information to include in ourits disclosure.
If the interim, topline, or preliminary data that we reportKineta reports differs from actual or final results, or if others, including regulatory authorities, disagree with the conclusions reached, ourKineta’s ability to obtain approval for, and commercialize, ourits product candidates may be harmed, which could harm ourKineta’s business, operating results, prospects or financial condition.
Kineta’s immuno-oncology product candidates are based on novel technologies that target the TME, which makes it difficult to predict the results, timing and cost of product candidate development and likelihood of obtaining regulatory approval.
OurKineta has concentrated its research and development efforts on immuno-oncology product candidates using its development platform, and Kineta’s future success depends on the successful development of this approach. Kineta’s product candidates target the TME which is highly immunosuppressive. Kineta has not yet succeeded and may not succeed in demonstrating efficacy and safety for any product candidates based on its platform technologies in clinical trials or in obtaining marketing approval thereafter, and use of Kineta’s platform technologies may not ever result in marketable products. Kineta may also experience delays in developing a sustainable, reproducible and scalable manufacturing process or transferring that process to commercial partners or establishing its own commercial manufacturing capabilities, which may prevent Kineta from completing its clinical trials or commercializing any products on a timely or profitable basis, if at all.
In addition, the clinical trial requirements of the FDA and other regulatory authorities and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty and intended use and market of the potential products. The regulatory approval process for novel product candidates such as Kineta’s can be less predictable, more expensive and longer than for other, better known or extensively studied pharmaceutical or other product candidates.
There is no assurance that the approaches offered by Kineta’s products will gain broad acceptance among doctors or patients or that governmental agencies or third-party medical insurers will be willing to provide reimbursement coverage for proposed product candidates. Since Kineta’s current product candidates and any future product candidates will represent novel approaches to treating various conditions, it may be difficult, in any event, to accurately estimate the potential revenues from these product candidates. Accordingly, Kineta may spend significant capital trying to obtain approval for product candidates that have an uncertain commercial market. The market for any products that Kineta successfully develops will also depend on the cost of the product. Kineta does not yet have sufficient information to reliably estimate what it will cost to commercially manufacture its current product candidates, and the actual cost to manufacture these products could materially and adversely affect the commercial viability of these products. If Kineta does not successfully develop and commercialize products based upon its approach or find suitable and economical sources for materials used in the production of its products, Kineta will not become profitable, which would materially and adversely affect the value of Kineta’s common stock.
The immuno-oncology industry is also rapidly developing, and Kineta’s competitors may introduce new technologies improving the immune response to cancer that render Kineta’s technologies obsolete or less attractive. New technology could emerge at any point in the development cycle of Kineta’s product candidates.
Kineta has initiated or plans to initiate clinical trials with its immuno-oncology products, KVA12123 and CD27. If these product candidates do not show any functionality in the TME, Kineta’s development plans, financial position, results of operations and prospects may be materially adversely affected.
While Kineta plans to develop product candidates for use in solid tumors, including KVA12123 and CD27, Kineta’s immuno-oncology product candidates may not show any functionality in the TME. The cellular environment in which solid tumor cells thrive is generally hostile to T cells due to factors such as the presence of immunosuppressive cells, humoral factors and limited access to nutrients. Kineta’s product candidates may not be able to access the solid tumor, and even if they do, they may not be able to exert anti-tumor effects in a hostile TME. In addition, the safety profile of Kineta’s product candidates may differ in a solid tumor setting. As a result, Kineta’s product candidates may not demonstrate efficacy in solid tumors. If Kineta is unable to make its immuno-oncology product candidates function in tumors, Kineta’s development plans, financial position, results of operations and prospects may be materially adversely affected.
Kineta has initiated clinical trials with KVA12123, KCP506 and LHF535. If these product candidates do not show any functionality in cancer, chronic pain or anti-viral applications, Kineta’s development plans, financial position, results of operations and prospects may be materially adversely affected. If Kineta’s drugs fail to demonstrate clinically relevant activity in patients, Kineta’s development plans, financial position, results of operations and prospects may be materially adversely affected.
KVA12123 is in early stage clinical trials and may not show any functionality in cancer. If KVA121123 does not show any such functionality, Kineta’s development plans, financial position, results of operations and prospects may be materially adversely affected. LHF535, a drug candidate held by Kineta’s subsidiary, KVHF, is used in the treatment of Arenaviruses, which are very rare in developed markets. Adequate investment may not be available to advance this program, and if the drug does obtain regulatory approval, it may be difficult to find payors willing to pay for this
50
drug. On December 27, 2022, KCP received written notice from Genentech, Inc. of its termination of the Exclusive Option and License Agreement (“Genentech KCP506 License Agreement”) for KCP506. As a result of the termination of the Genentech KCP506 License Agreement, KCP may not have adequate funding to continue the development of this program.
Kineta may experience delays or difficulties in the enrollment and/or retention of patients in clinical trials, which could delay or prevent Kineta’s receipt of necessary regulatory approvals.
Successful and timely completion of clinical trials will require that Kineta enrolls a sufficient number of patients. Patient enrollment, which is an important factor in the timing of clinical trials, is affected by many factors, including the size and nature of the patient population and competition for patients eligible for Kineta’s clinical trials with competitors which may have ongoing clinical trials for product candidates that are under development to treat the same indications as one or more of Kineta’s product candidates, or approved products for the conditions for which Kineta is developing its product candidates.
Trials may be subject to delays as a result of patient enrollment taking longer than anticipated or patient withdrawal. Kineta may not be able to initiate or continue clinical trials for its product candidates if Kineta is unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA, EMA or comparable foreign regulatory authorities. Kineta cannot predict how successful it will be at enrolling subjects in future clinical trials. Subject enrollment is affected by other factors including:
Kineta’s inability to enroll a sufficient number of patients for clinical trials would result in significant delays and could require Kineta to abandon one or more clinical trials altogether. Enrollment delays in these clinical trials may result in increased development costs for Kineta’s product candidates, which would cause the value of the Company to decline and limit Kineta’s ability to obtain additional financing. Furthermore, Kineta expects to rely on Contract Research Organizations (CROs) and clinical trial sites to ensure the proper and timely conduct of its clinical trials and Kineta will have limited influence over their performance.
Kineta may not be able to submit INDs to commence additional clinical trials on the timelines Kineta expects and, even if Kineta is able to, the FDA may not permit Kineta to proceed.
Kineta plans to submit an IND for CD27 in the second half of 2024, but Kineta may not be able to submit this planned IND on the timeline it expects. For example, Kineta may experience manufacturing delays or other delays with IND-enabling studies. Moreover, Kineta cannot be sure that submission of an IND will result in the FDA allowing it to commence clinical trials or that, once begun, issues will not arise that lead to the suspension or termination of Kineta’s clinical trials. Additionally, even if the applicable regulatory authorities agree with the design and implementation of the clinical trials set forth in Kineta’s INDs, Kineta cannot guarantee that those regulatory authorities will not change their requirements in the future, or that circumstances will not arise under which FDA or other regulatory authorities may place Kineta’s clinical trials on partial or full clinical hold. These considerations apply to the INDs described above and also to new clinical trials Kineta may submit as amendments to existing INDs or as part of new INDs in the future. Any failure to submit INDs on the timelines Kineta expects or to obtain authorization to proceed with its trials may prevent Kineta from completing its clinical trials or commercializing its products on a timely basis, if at all.
The regulatory approval processes of the FDA, European Commission (based on recommendation from the EMA), and comparable foreign authorities are lengthy, time consuming and inherently unpredictable. If Kineta is not able to obtain required regulatory approval for its product candidates, Kineta’s business will be substantially harmed.
51
The time required to obtain approval or other marketing authorizations by the FDA, European Commission (based on recommendation from the EMA) and comparable foreign regulatory authorities is unpredictable, and it typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations and the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. Kineta has not obtained regulatory approval for any product candidate, and it is possible that Kineta may never obtain regulatory approval for any product candidates it may seek to develop in the future. Neither Kineta nor any current or future collaborator is permitted to market any drug product candidates in the United States until Kineta receives regulatory approval of a biologics license application (“BLA”) or an NDA from the FDA, and Kineta cannot market it in the EU until Kineta receives a marketing authorization approval from the European Commission (based on recommendation from the EMA), or in the UK until Kineta receives regulatory approval from the Medicines and Healthcare products Regulatory Agency (MHRA) or other required regulatory approval in other countries. To date, Kineta has had only limited discussions with the FDA and European Commission (based on recommendation from the EMA) regarding clinical development programs or regulatory approval for any product candidate within the United States and EU, respectively. In addition, Kineta has had no discussions with other comparable foreign authorities regarding clinical development programs or regulatory approval for any product candidate outside of those jurisdictions.
Prior to obtaining approval to commercialize any drug product candidate in the United States or abroad, Kineta must demonstrate with evidence from well-controlled clinical trials, and to the satisfaction of the FDA, European Commission (based on recommendation from the EMA) or other foreign regulatory authorities, that such product candidates are safe and effective for their intended uses. Results from preclinical studies and clinical trials can be interpreted in different ways. Even if Kineta believes the preclinical or clinical data for its product candidates are promising, such data may not be sufficient to support approval by the FDA, the European Commission (based on recommendation of the EMA) and other comparable foreign regulatory authorities. The FDA or European Commission (based on recommendation from the EMA) may also require Kineta to conduct additional preclinical studies or clinical trials for its product candidates either prior to or after approval, or it may object to elements of Kineta’s clinical development programs.
Kineta’s product candidates could fail to receive regulatory approval for many reasons, including the following:
Of the large number of products in development, only a small percentage successfully complete the FDA, European Commission (based on recommendation from the EMA) or comparable foreign regulatory approval processes and are commercialized. The lengthy approval and marketing authorization process as well as the unpredictability of future clinical trial results may result in Kineta’s failure to obtain regulatory approval and marketing authorization to market its product candidates, which would significantly harm Kineta’s business, financial condition, results of operations and prospects.
Kineta has invested a significant portion of its time and financial resources in the development of its clinical and preclinical product candidates. Kineta’s business is dependent on its ability (or its partners’ or licensees’ ability) to successfully complete preclinical and clinical development of, obtain regulatory approval for, and, if approved, successfully commercialize KCP506, LHF535, KVA12123, CD27 and any future product candidates in a timely manner.
Even if Kineta (or its partners or licensees) eventually complete clinical testing and receive approval of an NDA or a BLA or other comparable foreign marketing application for KCP506, LHF535, KVA12123, CD27 or any future product candidates, the FDA, European Commission (based on recommendation from the EMA) or other comparable foreign regulatory authorities may grant approval or other marketing authorization contingent on the performance of costly additional clinical trials, including post-marketing clinical trials. The FDA, European Commission (based on recommendation from the EMA) or other comparable foreign regulatory authorities may also approve or authorize for marketing a product candidate for a more limited indication or patient population than Kineta originally requests, and the FDA, European Commission (based on recommendation from the EMA) or other comparable foreign regulatory authorities may not approve or authorize the labeling that Kineta believes is necessary or desirable for the successful commercialization of a product candidate. Any delay in obtaining, or inability to obtain, applicable regulatory approval or other marketing authorization would delay or prevent commercialization of that product candidate and would materially adversely impact Kineta’s business and prospects.
In addition, the FDA, European Commission (based on recommendation from the EMA) or other comparable foreign regulatory authorities and regulatory review committees described above may change their policies, issue additional regulations or revise existing regulations, or take other
52
actions, which may prevent or delay approval of Kineta’s future products under development on a timely basis. Such policy or regulatory changes could impose additional requirements upon Kineta that could delay its ability to obtain approvals, increase the costs of compliance or restrict Kineta’s ability to maintain any marketing authorizations it may have obtained.
Failure to obtain marketing approval in foreign jurisdictions would prevent Kineta’s product candidates from being marketed abroad. Any approval Kineta may be granted for its product candidates in the United States would not assure approval of its product candidates in foreign jurisdictions and any of its product candidates that may be approved for marketing in a foreign jurisdiction will be subject to risks associated with foreign operations.
In order to market and sell its products in the EU and other foreign jurisdictions, Kineta must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain FDA approval. The marketing approval process outside the United States generally includes all of the risks associated with obtaining FDA approval. Kineta may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. Kineta may file for marketing approvals but not receive necessary approvals to commercialize its products in any market.
In many countries outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that country. In some cases, the price that Kineta intends to charge for its products, if approved, is also subject to approval. Obtaining non-U.S. regulatory approvals and compliance with non-U.S. regulatory requirements could result in significant delays, difficulties and costs for Kineta and could delay or prevent the introduction of its product candidates in certain countries. In addition, if Kineta fails to obtain the non-U.S. approvals required to market its product candidates outside the United States or if Kineta fails to comply with applicable non-U.S. regulatory requirements, Kineta’s target markets will be reduced and its ability to realize the full market potential of its product candidates will be harmed and its business, financial condition, results of operations and prospects may be adversely affected.
Additionally, Kineta could face heightened risks with respect to seeking marketing approval in the United Kingdom as a result of the withdrawal of the United Kingdom from the EU, commonly referred to as Brexit. As of January 1, 2021, the MHRA became responsible for supervising medicines and medical devices in Great Britain, comprising England, Scotland and Wales under domestic law, whereas Northern Ireland will continue to be subject to EU rules under the Northern Ireland Protocol. The MHRA will rely on the Human Medicines Regulations 2012 (SI 2012/1916) (as amended) (HMR) as the basis for regulating medicines. The HMR has incorporated into the domestic law of the body of EU law instruments governing medicinal products that pre-existed prior to the UK’s withdrawal from the EU.
Any delay in obtaining, or an inability to obtain, any marketing approvals, as a result of Brexit or otherwise, would prevent Kineta from or delay Kineta commercializing its product candidates in the United Kingdom and/or the EEA and restrict Kineta’s ability to generate revenue and achieve and sustain profitability. Kineta also expects that it will be subject to additional risks in commercializing any of its product candidates that receive marketing approval outside the United States, including tariffs, trade barriers and regulatory requirements; economic weakness, including inflation, or political instability in particular foreign economies and markets; compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; and workforce uncertainty in countries where labor unrest is more common than in the United States.
Kineta’s preclinical studies and clinical trials may fail to demonstrate the safety and efficacy of its product candidates, or serious adverse events or other undesirableunacceptable side effects may be identified during the development of Kineta’s product candidates, which could prevent, delay or limit the scope of regulatory approval of its product candidates, limit their commercialization, increase Kineta’s costs or necessitate the abandonment or limitation of the development of some of Kineta’s product candidates.
To obtain the requisite regulatory approvals for the commercial sale of Kineta’s product candidates, Kineta must demonstrate through lengthy, complex and expensive preclinical testing and clinical trials that its product candidates are safe and effective for use in each target indication. These trials are expensive and time consuming, and their outcomes are inherently uncertain. Failures can occur at any time during the development process. Preclinical studies and clinical trials often fail to demonstrate safety or efficacy of the product candidate studied for the target indication, and most product candidates that begin clinical trials are never approved.
Kineta may fail to demonstrate with evidence from adequate and well-controlled trials, and to the satisfaction of the FDA, European Commission (based on recommendation from the EMA) or comparable foreign regulatory authorities, that its product candidates are safe and effective for their intended uses.
Possible adverse reactions and adverse side effects that could occur with immuno-oncology treatments can be severe, for example, cytokine CRS. Depending on an evaluation of the available data, Kineta may decide or be required to perform additional preclinical studies or to halt or delay further clinical development of its product candidates or preventto limit their regulatory approval,development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe, or more acceptable from a risk-benefit perspective, which may limit the commercial profileexpectations for the product candidate, if approved.
Kineta’s clinical trials could also be suspended or terminated and the FDA, EMA or comparable foreign regulatory authorities could order Kineta to cease further development of, or deny approval of, its product candidates for any or all targeted indications. Even if this does not occur, reports of serious reactions could affect patient recruitment or the ability of enrolled patients to complete the trial. Moreover, if Kineta elects, or is required, to
53
not initiate, delay, suspend or terminate any future clinical trial of any of Kineta’s product candidates, the commercial prospects of such product candidates may be harmed, and Kineta’s ability to generate product revenues from any of these product candidates may be delayed or eliminated. Any of these occurrences may harm Kineta’s ability to develop other product candidates, and may harm Kineta’s business, financial condition and prospects significantly.
If Kineta’s product candidates are associated with side effects in clinical trials or have characteristics that are unexpected, Kineta may need to abandon their development or limit development to more narrow uses in which the side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. The FDA, EMA, an approved label,institutional review board (“IRB”) or ethics committee (“EC”), which are local institutional boards or committees, as applicable, that review, approve and oversee basic and clinical research conducted as the institution participating in the clinical trial, or comparable foreign regulatory authorities, may also require that Kineta suspend, discontinue or limit its clinical trials based on safety information, or that Kineta conducts additional animal or human studies regarding the safety and efficacy of its product candidates which Kineta has not planned or anticipated. Such findings could further result in significant negative consequences followingregulatory authorities failing to provide marketing approval,authorization for Kineta’s product candidates or limiting the scope of the approved indication, if any.approved. Many product candidates that initially showed promise in early-stage testing have later been found to cause side effects that prevented further development of the product candidate.
Preclinical development is uncertain. Kineta’s preclinical programs may experience delays or may never advance to clinical trials, which would adversely affect Kineta’s ability to obtain regulatory approvals or commercialize these programs on a timely basis or at all, which would have an adverse effect on Kineta’s business.
In the third quarter of 2020, the requisite authorities in the Netherlands authorized Kineta to initiate an initial clinical trial in healthy volunteers to demonstrate safety and tolerability of KCP506. Kineta completed this Phase 1 trial in healthy volunteers in 2022.
Serious adverse events or other undesirable side effects causedThe FDA has requested additional pre-clinical data related to Kineta’s IND for LHF535. Kineta completed the Phase 1 trial in healthy volunteers in Australia in June 2020.
In the fourth quarter of 2022, Kineta was authorized by ourthe FDA to begin a clinical trial of KVA12123 in the United States. In April 2023, the first patient was dosed in the Phase 1/2 clinical study evaluating KVA12123 alone and in combination with the immune checkpoint inhibitor pembrolizumab in patients with advanced solid tumors, and in October 2023, the first patient was dosed in combination with KEYTRUDA® (pembrolizumab) in the clinical trial. All of Kineta’s product candidates, could cause usincluding KVA12123, KCP506 and LHF535, are still in the preclinical or regulatory authorities to interrupt, delay, or haltearly clinical stage, and their risk of failure is high. Before Kineta can commence clinical trials for a product candidate, it must complete extensive preclinical testing and could resultstudies that support Kineta’s planned INDs in a more restrictive labelthe United States, or similar applications in other jurisdictions. Kineta cannot be certain of the delaytimely completion or denialoutcome of regulatory approval byits preclinical testing and studies and cannot predict if the FDA or other regulatory authorities.authorities will accept Kineta’s proposed clinical programs or if the outcome of Kineta’s preclinical or clinical testing and studies will ultimately support the further development of its programs. As a result, Kineta cannot be sure that it will be able to submit INDs or similar applications for its preclinical programs beyond KVA12123, KCP506 and LHF535 on the timelines Kineta expects, if at all, and Kineta cannot be sure that submission of INDs or similar applications will result in the FDA or other regulatory authorities allowing clinical trials to begin.
Further, clinical trials by their nature utilize a sample of the potential patient population for a limited duration of exposure. Rare and severe side effects of a product candidateKineta may only be uncovered with a significantly larger number of patients exposed to the product candidate. If our product candidates receive marketing approval and we or others identify undesirable side effects caused by such product candidates (or any other similar products) after such approval, a number of potentially significant negative consequences could result, including:
40
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidates, could substantially increase the costs of commercializing our product candidates, and could significantly impact our ability to successfully commercialize our product candidates and generate revenues.
Failures orencounter substantial delays in the commencement or completion, or termination or suspension, of or ambiguous or negative results from, ourits clinical trials, of our product candidateswhich could result in increased costs to usKineta and could delay prevent, or limit ourits ability to generate revenue and continue our business.adversely affect Kineta’s commercial prospects.
We do not know whetherBefore obtaining marketing approval from regulatory authorities for the sale of its product candidates, Kineta must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidate for its intended indications. Kineta cannot guarantee that any of our clinical trials will beginbe conducted as planned or be completed on schedule, if at all,all. Kineta may experience numerous unforeseen events during or as the commencement and completiona result of clinical trials can be delayedthat could delay or prevented for a number of reasons, including, among others:prevent its ability to receive marketing approval or commercialize Kineta’s product candidates, including:
54
If Kineta is required to conduct additional clinical trials or other testing of its product candidates beyond the clinical trials and testing that Kineta contemplates, if Kineta is unable to successfully complete clinical trials or other testing of its product candidates, if the results of these clinical trials or tests are unfavorable or are only modestly favorable, or if there are safety concerns associated with any of product candidates, Kineta may:
Conducting clinical trials in foreign countries, as Kineta may do for its product candidates, presents additional risks that may delay completion of Kineta’s clinical trials. These risks include the failure of enrolled patients in foreign countries to adhere to clinical protocols as a result of differences in healthcare services or cultural customs, managing additional administrative burdens associated with foreign regulatory schemes, as well as political and economic risks relevant to such foreign countries.
Moreover, principal investigators for Kineta’s clinical study buttrials may serve as scientific advisors or consultants to Kineta from time to time and receive compensation in connection with such services. Under certain circumstances, Kineta may be pronerequired to withdraw duereport some of these relationships to rigorsthe FDA or comparable foreign regulatory authorities. The FDA or comparable foreign regulatory authority may conclude that a financial relationship between Kineta and a principal investigator has created a conflict of interest or otherwise affected interpretation of the trial. The FDA or comparable foreign regulatory authority may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial lack of efficacy, side effects, personal issues, or loss of interest.
Clinical trials may also be delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical studyitself may be suspendedjeopardized. This could result in a delay in approval, or terminated by us, the FDA, the IRBs or ethics committees at the sites in which such clinical studies are being conducted, a data and safety monitoring board (“DSMB”) overseeing the clinical study at issue or other regulatory authorities due to a numberrejection, of factors, including, among others:
•
41
55
•
We may in the future seek orphan drug designation or exclusivity for certain of our product candidates. If our competitors are able to obtain orphan drug exclusivity for products that constitute the same drugconsuming and treat the same indications as our product candidates, we may not be able to have competing products approved by the applicable regulatory authority for a significant period of time.successful at all.
WeKineta’s failure to successfully initiate and complete clinical trials of its product candidates and to demonstrate the efficacy and safety necessary to obtain regulatory approval to market any of its product candidates would significantly harm Kineta’s business. Kineta cannot guarantee that its clinical trials will begin as planned or be completed on schedule, if at all, or that Kineta will not need to restructure its clinical trials. Significant preclinical study or clinical trial delays could also shorten any periods during which Kineta may have the exclusive right to commercialize its product candidates or allow its competitors to bring products to market before Kineta does and impair Kineta’s ability to successfully commercialize its product candidates, which may harm Kineta’s business and results of operations. In addition, many of the factors that cause, or lead to, delays of clinical trials may ultimately lead to the denial of regulatory approval of Kineta’s product candidates.
As an organization, Kineta has never conducted pivotal clinical trials, and Kineta may be unable to do so for any product candidates it may develop.
Kineta will need to successfully complete clinical trials meeting requirements for approval of the FDA or comparable foreign regulatory authorities, known as pivotal trials, to market its drugs, or any future product candidate. Carrying out pivotal clinical trials is a complicated process. As an organization, Kineta has not previously conducted any later-stage or pivotal clinical trials. In order to do so, Kineta will need to expand its clinical development and regulatory capabilities, and it may be unable to recruit and train qualified personnel. Kineta also expects to continue to rely on third parties to conduct its pivotal clinical trials. Consequently, Kineta may be unable to successfully and efficiently execute and complete necessary clinical trials in thea way that leads to NDA or BLA submission and approval of Kineta’s drugs, or future seek orphan drug designation or exclusivity for certain of our product candidates. Regulatory authoritiesKineta may require more time and incur greater costs than its competitors and may not succeed in some jurisdictions, including the United States and the European Union, may designate drugs and biologics intendedobtaining regulatory approvals of product candidates that Kineta develops. Failure to treat relatively small patient populations as orphan drugs. Under the Orphan Drug Act of 1983, FDA may designate acommence or complete, or delays in, Kineta’s planned clinical trials could prevent Kineta from or delay Kineta in commercializing its product candidate as an orphan drug if it is intended to treat a rare disease or condition, which is defined as a disease or condition having a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the European Union, the European Commission, after recommendation from the EMA’s Committee for Orphan Medicinal Products, grants orphan designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 persons in the European Union and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized for marketing in the European Union (or, if a method exists, the product would be of significant benefit to those affected by the condition). Additionally, orphan designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the product in the European Union would generate sufficient return to justify the necessary investment in developing the product.candidates.
If we request orphan drug designationSome data for our product candidates there can be no assurances that FDA or the European Commission will grant any of our product candidates such designation. Additionally, orphan drug designation does not guarantee that any regulatory authority will accelerate regulatory review of, or ultimately approve, the product candidate, nor does it limit the ability of any regulatory authority to grant orphan drug designation to product candidates of other companies that treat the same indications.
Generally, if a product candidate with an orphan drug designation receives the first marketing approval for the indication for which it has such designation, the product is entitled to a period of marketing exclusivity, which precludes FDA or the European Commissioncomes from approving another marketing application for a product that constitutes the same drug treating the same indication for that marketing exclusivity period, except in limited circumstances. If another sponsor receives such approval before we do (regardless of our orphan drug designation), we will be precluded from receiving marketing approval for our product for the applicable exclusivity period. The applicable period is seven years in the United States and 10 years in the European Union. The exclusivity period in the European Union may be reduced to six years if, at the end of the fifth year, it is established that a product no longer meets the criteria for orphan designation or if the product is sufficiently profitable so that market exclusivity is no longer justified. Orphan exclusivity may be revoked if any regulatory authority determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the product to meet the needs of patients with the rare disease or condition.
Even if we obtain orphan drug exclusivity for a product candidate, that exclusivity may not effectively protect the product candidate from competition because different drugs can be approved for the same condition. In the United States, even after an orphan drug is approved, FDA may subsequently approve another drug for the same condition if FDA concludes that the latter drug is not the same drug or is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care.
We are currently, and may in the future, conduct clinical trials for our product candidatesconducted outside the United States, EU and the UK, and the FDA, and similarEMA or comparable foreign regulatory authorities may not accept data from such trials.
We are currently, and may in the future, conduct additional clinical trials outside the United States, including in Europe or other foreign jurisdictions. The acceptance of trial data from clinical trials conducted outside the United States or another jurisdiction by the FDA may be subject to certain conditions. The FDA will generallyconditions or may not considerbe accepted at all. Similarly, the EMA and other equivalent foreign regulatory authorities may not accept data from a foreign clinical trial nottrials conducted under an IND unless (i) the trial was well-designed and well-conducted in accordance with GCP requirements, including requirements for the design, conduct, performance, monitoring, auditing, recording, analysis, and reporting of clinical trials in a way that provides assurance that the data and reported results are credible and accurate and that the rights, safety, and well-being of trial subjects are protected, and (ii) the FDA is able to validate the data from the trial through an onsite inspection, if necessary.outside their jurisdiction. In cases where data from foreign clinical trials conducted outside the United States are intended to serve as the sole basis for marketing approval in the United States, the FDA will generally not approve the application on the basis of foreign data alone unless (i) the data are applicable to the United StatesU.S. population and United StatesU.S. medical practice; and (ii) the trials were performed by clinical investigators of recognized competence;competence and (iii)pursuant to good clinical practice (“GCP”) regulations. In general, the datapatient population for any clinical trials conducted outside the United States must be considered valid withoutrepresentative of the needpopulation for an on-site inspection bywhom Kineta intends to label the FDA or, ifproduct candidate in the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means.United States. Additionally, the FDA’s
42
clinical trial requirements, including sufficient size of patient populations and statistical powering, must be met. Many foreign regulatory bodiesauthorities have similar approval requirements.requirements for clinical trials. In addition, such foreign trials would be subject to the applicable local laws of the foreign jurisdictions where the trials are conducted. There can be no assurance that the FDA, EMA or any similarcomparable foreign regulatory authority will accept data from trials conducted outside of the United States or the applicable jurisdiction. If the FDA, EMA or any similarcomparable foreign regulatory authority does not accept such data, it would result in the need for additional trials, which wouldcould be costly and time-consuming, and delay aspects of our business plan, and which may result in our product candidates that Kineta may develop not receiving approval for commercialization in the applicable jurisdiction.
ChangesBreakthrough therapy designation by the FDA for any product candidate may not lead to a faster development or regulatory review or approval process, and it does not increase the likelihood that the product candidate will receive marketing approval.
Kineta may seek breakthrough therapy designation for its product candidates or programs. A breakthrough therapy is defined as a product candidate that is intended, as a monotherapy or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the product candidate may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For product candidates that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Product candidates designated as breakthrough therapies by the FDA are also eligible for priority review if supported by clinical data at the time of the submission of the NDA or BLA.
Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if Kineta believes that one of its product candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a breakthrough therapy designation for a product candidate may not result in a faster development process, review or approval compared to product candidates considered for approval under conventional FDA procedures and it would not assure ultimate approval by the FDA. In addition, even after a product candidate qualifies as a breakthrough therapy, the FDA may later decide that the product candidate no longer meets the conditions for qualification or it may decide that the time period for FDA review or approval will not be shortened.
A Fast Track designation by the FDA, even if granted for other current or future product candidates, may not lead to a faster development or regulatory review or licensure process and does not increase the likelihood that Kineta’s product candidates will receive marketing licensure.
Kineta may seek Fast Track designation for one or more of its future product candidates. If a drug or biological product is intended for the treatment of a serious or life-threatening disease or condition and it demonstrates the potential to address unmet medical needs for such a disease or condition,
56
the drug sponsor may apply for FDA Fast Track designation for a particular indication. Kineta may seek Fast Track designation for its product candidates, but there is no assurance that the FDA will grant this designation to any of Kineta’s proposed product candidates. Marketing applications submitted by sponsors of products in Fast Track development may qualify for priority review under the policies and procedures offered by the FDA, but the Fast Track designation does not assure any such qualification or ultimate marketing licensure by the FDA. The FDA has broad discretion whether or not to grant Fast Track designation, so even if Kineta believes a particular product candidate is eligible for this designation, there can be no assurance that the FDA would decide to grant it. Even if Kineta does receive Fast Track designation, Kineta may not experience a faster development process, review or licensure compared to conventional FDA procedures or pathways, and receiving a Fast Track designation does not provide assurance of ultimate FDA licensure. In addition, the FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data from Kineta’s clinical development program. In addition, the FDA may withdraw any Fast Track designation at any time.
Accelerated approval by the FDA, even if granted for Kineta’s current or any other future product candidates, may not lead to a faster development or regulatory review or approval process and it does not increase the likelihood that Kineta’s product candidates will receive regulatory approval.
Kineta may seek accelerated approval of its current or future product candidates using the FDA’s accelerated approval pathway. A product may be eligible for accelerated approval if it treats a serious or life-threatening condition and generally provides a meaningful advantage over available therapies. In addition, it must demonstrate an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality (“IMM”) that is reasonably likely to predict an effect on IMM or other clinical benefit. As a condition of approval, the FDA requires that a sponsor of a drug or biologic receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. These confirmatory trials must be completed with due diligence. In addition, the FDA currently requires, unless otherwise informed by the agency, pre-approval of promotional materials for products receiving accelerated approval, which could adversely impact the timing of the commercial launch of the product. Even if Kineta does receive accelerated approval, Kineta may not experience a faster development or regulatory review or approval process, and receiving accelerated approval does not provide assurance of ultimate FDA approval.
Even if Kineta receives regulatory approval for any of its product candidates, Kineta will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense. Additionally, Kineta’s product candidates, if approved, could be subject to post-market study requirements, marketing and labeling restrictions, and even recall or market withdrawal if unanticipated safety issues are discovered following approval. In addition, Kineta may be subject to penalties or other enforcement action if Kineta fails to comply with regulatory requirements.
If the FDA, the European Commission (based on recommendation from the EMA) or a comparable foreign regulatory authority approves any of Kineta’s product candidates, the manufacturing processes, labeling, packaging, distribution, storage, advertising, promotion, import, export, recordkeeping, monitoring and reporting for Kineta’s product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, establishment registration and listing, as well as continued compliance with current Good Manufacturing Practice requirements (“cGMPs”) and GCP requirements for any clinical trials that Kineta conducts post-approval. Any regulatory approvals that Kineta receives for its product candidates may also be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing studies, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product.
The FDA may require a REMS in order to approve Kineta’s product candidates, which could entail requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with Kineta’s third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
In the EU, the European Commission (based on recommendation from the EMA) may require an equivalent risk management plan. The FDA’s, European Commission’s, EMA’s and other comparable foreign regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of Kineta’s product candidates. If Kineta is slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if Kineta is not able to maintain regulatory compliance, Kineta may lose any marketing approval that it may have obtained, which would adversely affect Kineta’s business, prospects and ability to achieve or sustain
57
profitability.
Kineta anticipates that some of its current product candidates and any future product candidates may be used in combination with third-party drugs or biologics, some of which are still in development, and Kineta has limited or no control over the supply, regulatory status or regulatory approval of such drugs or biologics.
Kineta’s immuno-oncology drugs target both immune suppressive host and tumor cells in the TME initiating a dynamic process of activating the host immune system, which response can be further exploited by concurrent or subsequent therapies including checkpoint inhibitors such as the dominant PD-1 monoclonal antibodies, pembrolizumab and nivolumab. Accordingly, it is expected that Kineta’s product candidates, if approved, would be used in combination with third-party drugs or biologics. Kineta’s ability to develop and ultimately commercialize its current product candidates and any future product candidates used in combination with other therapies, including for example, pembrolizumab, nivolumab, or any other checkpoint inhibitor immunotherapies, will depend on Kineta’s ability to access such drugs or biologics on commercially reasonable terms for the clinical trials and their availability for use with the commercialized product, if approved. Kineta cannot be certain that current or potential future commercial relationships will provide it with a steady supply of such drugs or biologics on commercially reasonable terms or at all.
Any failure to maintain or enter into new successful commercial relationships, or the expense of purchasing checkpoint inhibitor immunotherapies or other comparator therapies in the market, may delay Kineta’s development timelines, increase Kineta’s costs and jeopardize Kineta’s ability to develop its current product candidates and any future product candidates as commercially viable therapies. If any of these occur, Kineta’s business, financial condition, results of operations, stock price and prospects may be materially harmed.
Moreover, the development of product candidates for use in combination with another product or product candidate may present challenges that are not faced for single agent product candidates. Kineta is currently developing immuno-oncology drugs for use in monotherapy and in combination with checkpoint inhibitors, targeted therapies and chemotherapeutics. The FDA, EMA or comparable foreign regulatory authorities may require Kineta to use more complex clinical trial designs in order to evaluate the contribution of each product and product candidate to any observed effects. It is possible that the results of such trials could show that any positive previous trial results are attributable to the combination therapy and not Kineta’s current product candidates and any future product candidates. It is also possible that trial results for Kineta’s product candidates may differ significantly if Kineta’s product candidates are investigated with different combination therapies in different trials. Moreover, following product approval, the FDA, EMA or comparable foreign regulatory authorities may require that products used in conjunction with each other be cross labeled for combined use. To the extent that Kineta does not have rights to the other product, this may require Kineta to work with a third party to satisfy such a requirement. Moreover, developments related to the other product may impact Kineta’s clinical trials for the combination as well as Kineta’s commercial prospects should Kineta receive marketing approval. Such developments may include changes to the other product’s safety or efficacy profile, changes to the availability of the approved product, quality, manufacturing and supply issues and changes to the standard of care.
In the event that any of Kineta’s collaborators or suppliers cannot continue to supply their products on commercially reasonable terms, Kineta would need to identify alternatives for accessing such checkpoint inhibitor immunotherapies. Additionally, should the supply of products from any collaborator or supplier be interrupted, delayed or otherwise be unavailable to Kineta, Kineta’s clinical trials may be delayed. In the event Kineta is unable to source an alternative supply, or is unable to do so on commercially reasonable terms, Kineta’s business, financial condition, results of operations, stock price and prospects may be materially harmed.
Kineta may expend its limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because Kineta has limited financial and managerial resources, Kineta must prioritize its research programs and will need to focus its discovery and development on select product candidates and indications. Correctly prioritizing Kineta’s research and development activities is particularly important for Kineta due to the breadth of potential product candidates and indications that it believes could be pursued using Kineta’s platform technologies. As a result, Kineta may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Kineta’s resource allocation decisions may cause it to fail to capitalize on viable commercial products or profitable market opportunities. Kineta’s spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable products. If Kineta does not accurately evaluate the commercial potential or target market for a particular product candidate, Kineta may also relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for Kineta to retain sole development and commercialization rights to such product candidate.
If Kineta does not achieve its projected development goals in the timeframes it announces and expects, the commercialization of its products may be delayed and, as a result, Kineta’s stock price may decline.
From time to time, Kineta estimates the timing of the anticipated accomplishment of various scientific, clinical, regulatory and other product development goals, which Kineta sometimes refers to as milestones. These milestones may include the commencement or completion of preclinical studies and clinical trials and the submission of ourregulatory filings and may be associated with payments from collaborators. From time to time, Kineta may publicly announce the expected timing of some of these milestones. All of these milestones are and will be based on numerous assumptions. The actual timing of these milestones may vary dramatically compared to Kineta’s estimates, in some cases for reasons beyond Kineta’s control. If Kineta does not meet these milestones as publicly announced, or at all, its revenue may be lower than expected, the commercialization of its products may be delayed or never achieved and, as a result, Kineta’s stock price may decline.
58
If Kineta decides to seek Orphan Drug Designation for any of its current or future product candidates, Kineta may occur,be unsuccessful or may be unable to maintain the benefits associated with Orphan Drug Designation, including the potential for supplemental market exclusivity.
Kineta may seek Orphan Drug Designation for one or more of its current or future product candidates. Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs or biological products for relatively small patient populations as orphan drugs. Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, defined as a disease or condition with a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States when there is no reasonable expectation that the cost of developing and making available the drug in the United States will be recovered from sales in the United States for that drug or biological product. In the United States, Orphan Drug Designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. After the FDA grants Orphan Drug Designation, the identity of the drug or biological product and its potential orphan use are disclosed publicly by the FDA. Orphan Drug Designation does not convey any advantage in, or shorten the duration of, the regulatory review and licensure process.
If a product that has Orphan Drug Designation subsequently receives the first FDA approval or licensure for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including an NDA or BLA, to market the same drug or biological product for the same indication for seven years, except in limited circumstances such as a showing of clinical superiority to the product with orphan drug exclusivity or if the FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the biological product was designated. As a result, even if one of Kineta’s product candidates receives orphan exclusivity, the FDA can still approve or license other drugs or biological products that have a different active ingredient for use in changestreating the same indication or disease. Further, the FDA can waive orphan exclusivity if Kineta is unable to preclinicalmanufacture sufficient supply of its product.
Kineta may seek Orphan Drug Designation for its product candidates in additional orphan indications in which there is a medically plausible basis for the use of these product candidates. Even when Kineta obtains Orphan Drug Designation, exclusive marketing rights in the United States may be limited if Kineta seeks licensure for an indication broader than the orphan designated indication and may be lost if the FDA later determines that the request for designation was materially defective or if Kineta, through its manufacturer, is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. In addition, although Kineta intends to seek Orphan Drug Designation for other product candidates, Kineta may never receive these designations.
If Kineta fails to develop additional product candidates, its commercial opportunity could be limited.
Kineta expects initially to focus on the development of KVA12123, its lead immuno-oncology drug candidate. A key part of Kineta’s strategy, however, is to continue to pursue clinical study protocolsdevelopment of additional product candidates utilizing its development platform or in-licensed from third parties. Developing, obtaining marketing approval for, and commercializing any future product candidates will require substantial additional preclinicalfunding and will be subject to the risks of failure inherent in drug product development. Kineta cannot assure you that it will be able to successfully advance any future product candidates through the development process.
Even if Kineta obtains approval from the FDA, European Commission (based on recommendation from the EMA) or clinical study requirements, which could resultcomparable foreign regulatory authorities to market any future product candidates for the treatment of tumors, Kineta cannot assure that any such product candidates will be successfully commercialized, widely accepted in increased coststhe marketplace, or more effective than other commercially available alternatives. If Kineta is unable to ussuccessfully develop and commercialize additional product candidates, its commercial opportunity may be limited and Kineta’s business, financial condition, results of operations, stock price and prospects may be materially harmed.
Difficulty in enrolling patients could delay our development timeline.or prevent clinical trials of Kineta’s current product candidates and any future product candidates. Kineta may find it difficult to enroll patients in its ongoing clinical trials or any subsequent trials it may conduct and Kineta’s receipt of necessary regulatory approvals could be delayed or prevented.
ChangesIdentifying and qualifying patients to participate in clinical studies of Kineta’s current product candidates and any future product candidates is critical to Kineta’s success. The timing of completion of Kineta’s clinical trials depends in part on the speed at which Kineta can recruit patients to participate in testing its current product candidates and any future product candidates, and Kineta may experience delays in its clinical trials if it encounters difficulties in enrollment or patient retention due to other unforeseen factors. Kineta may not be able to initiate or continue clinical trials for its current product candidates and any future product candidates if Kineta is unable to locate and enroll and retain a sufficient number of eligible patients to participate in these trials as required by the FDA, EMA or comparable foreign regulatory requirements, FDA guidance,authorities outside the United States. For example, the COVID-19 pandemic in the past has impacted, and in the future may impact, Kineta’s ability to initiate clinical sites and recruit, enroll and retain patients or unanticipated events during our preclinicalmay divert healthcare resources away from clinical trials. In addition, some of Kineta’s competitors have ongoing clinical trials for product candidates that treat the same indications as Kineta’s current product candidates, and patients who would otherwise be eligible for Kineta’s clinical trials may instead enroll in clinical trials of Kineta’s competitors’ product candidates or future product candidates.
In addition to the competitive trial environment, the eligibility criteria of Kineta’s planned clinical trials will further limit the pool of available study participants as Kineta will require that patients have specific characteristics that it can measure to assure their cancer is either severe enough or not too advanced to include them in a study. Additionally, the process of finding patients may prove costly. Kineta also may not be able to identify, recruit and enroll a sufficient number of patients to complete Kineta’s clinical studies because of the perceived risks and benefits of the product candidates under study, the availability and efficacy of competing therapies and clinical trials, may force usthe proximity and availability of clinical trial sites for prospective patients, and the patient referral practices of physicians. If patients are unwilling to amend preclinicalparticipate in Kineta’s studies for any reason, the timeline for recruiting patients, conducting studies and obtaining regulatory approval of potential products may be delayed.
59
The enrollment of patients further depends on many factors, including:
In addition, Kineta’s clinical trials will compete with other clinical trials for reviewproduct candidates that are in the same therapeutic areas as Kineta’s current product candidates and approval,any future product candidates, and this competition will reduce the number and types of patients available to Kineta because some patients who might have opted to enroll in Kineta’s clinical trials may instead opt to enroll in a clinical trial being conducted by one of Kineta’s competitors. Since the number of qualified clinical investigators is limited, Kineta expects to conduct some of its clinical trials at the same clinical trial sites that some of Kineta’s competitors use, which will reduce the number of patients who are available for Kineta’s clinical trials at such clinical trial sites. Furthermore, even if Kineta is able to enroll a sufficient number of patients for its clinical trials, Kineta may adversely impacthave difficulty maintaining enrollment of such patients in its clinical trials.
If Kineta experiences delays in the cost, timing, or successful completion of, or termination of, any clinical trials. Similarly, amendments to our preclinical studies may adversely impact the cost, timing, or successful completiontrial of those preclinical studies. If we experiences delays completing, or if we terminate,its current product candidates and any of our preclinical studies or clinical trials, or if we are required to conduct additional preclinical studies or clinical trials,future product candidates, the commercial prospects for ourof Kineta’s current product candidates mayand any future product candidates will be harmed, and ourKineta’s ability to generate product revenue from such product candidates could be delayed or prevented.
Kineta’s future growth depends, in part, on its ability to penetrate multiple markets in which Kineta would be subject to additional regulatory burdens and other risks and uncertainties.
Kineta’s future profitability will depend, in part, on its ability to commercialize its product candidates, if approved, in markets in the United States, Europe, the UK and other countries where Kineta maintains commercialization rights. As Kineta begins to commercialize its product candidates, if approved, in multiple markets, Kineta is subject to additional risks and uncertainties, including:
These and other risks associated with international operations may adversely affect Kineta’s ability to attain or maintain profitable operations. Future sales of Kineta’s products or its product candidates, if they are approved, will be delayed.dependent on purchasing decisions of and reimbursement from government health administration authorities, distributors and other organizations. As a result of adverse conditions affecting the global economy and credit and financial markets, including disruptions due to political instability or otherwise, these organizations may defer purchases, may be unable to
60
satisfy their purchasing or reimbursement obligations, or may affect milestone payments or royalties for Kineta’s products or any of its product candidates that are approved for commercialization in the future. Should any of these risks materialize, this could have a material adverse effect on Kineta’s business, prospects, financial condition and results of operations.
Risks Related to Manufacturing and Commercialization
The manufacture of Kineta’s product candidates is complex and Kineta may encounter difficulties in production, particularly with respect to process development or scaling-out of Kineta’s manufacturing capabilities. If Kineta encounters such difficulties, Kineta’s ability to provide supply of its product candidates for clinical trials or its products for patients, if approved, could be delayed or stopped.
Kineta has not yet manufactured or processed its product candidates on a commercial scale and may not be able to do so for any of its product candidates. Kineta may encounter difficulties in production, particularly in scaling up or out, validating the production process, and assuring high reliability of the manufacturing process. These problems include delays or break-downs in logistics and shipping, difficulties with production costs and yields, failure to maintain adequate quality control, product testing issues, operator error and lack of availability of qualified personnel, as well as failure to comply with strictly enforced federal, state and foreign regulations.
Furthermore, if contaminations are discovered in Kineta’s supply of product candidates or in the manufacturing facilities, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination. Kineta cannot assure you that any of these or other issues relating to the manufacture of its product candidates will not occur in the future. Any delay or interruption in the supply of clinical trial supplies could delay the completion of clinical trials, increase the costs associated with maintaining clinical trial programs and, depending upon the period of delay, require Kineta to begin new clinical trials at additional expense or terminate clinical trials completely.
Manufacturing facilities also require commissioning and validation activities to demonstrate that they operate as designed, and are subject to government inspections by the FDA, EU Member States, coordinated by the EMA, and other comparable foreign regulatory authorities. If Kineta is unable to reliably produce products to specifications acceptable to the regulatory authorities, Kineta may not obtain or maintain the approvals it needs to manufacture its products. Further, manufacturing facilities may fail to pass government inspections prior to or after the commercial launch of Kineta’s product candidates, which would cause significant delays and additional costs required to remediate any deficiencies identified by the regulatory authorities. Any of these challenges could delay completion of clinical trials, require bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of Kineta’s product candidate, impair commercialization efforts, increase Kineta’s cost of goods and have an adverse effect on Kineta’s business, financial condition, results of operations and growth prospects.
Changes in product candidate manufacturing or formulation may result in additional costs or delay.
As product candidates are developed through preclinical studies to later-stage clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an effort to optimize processes and results. Any of these changes could cause Kineta’s current product candidates or any future product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the altered materials. Such changes may also require additional testing, or notification to, or approval by the FDA, European Commission, EMA or a comparable foreign regulatory authority. This could delay completion of clinical trials, require the conduct of bridging clinical trials or studies, require the repetition of one or more clinical trials, increase clinical trial costs, delay approval of Kineta’s current product candidates and any future product candidates and/or jeopardize Kineta’s ability to commence product sales and generate revenue.
Even if any of Kineta’s product candidates receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
Even if Kineta obtains marketing approvals from the FDA, the European Commission (based on recommendation from the EMA) or other comparable foreign regulatory agencies and is able to initiate commercialization of Kineta’s clinical-stage product candidates or any other product candidates Kineta develops, the product candidate may not achieve market acceptance among physicians, patients, hospitals, including pharmacy directors, and third-party payors and, ultimately, may not be commercially successful. The degree of market acceptance of Kineta’s product candidates, if approved for commercial sale, will depend on a number of factors, including:
61
Kineta’s efforts to educate physicians, patients, third-party payors and others in the medical community on the benefits of Kineta’s product candidates, if approved, may require significant resources and may never be successful.
Such efforts may require more resources than are typically required due to the complexity and uniqueness of Kineta’s product candidates. Because Kineta expects sales of its product candidates, if approved, to generate substantially all of Kineta’s product revenue for the foreseeable future, the failure of Kineta’s product candidates to find market acceptance would harm Kineta’s business and could require Kineta to seek additional financing. Even if Kineta’s product candidates, if approved, achieve market acceptance, Kineta may not be able to maintain that market acceptance over time if new products or technologies are introduced that are more favorably received than Kineta’s products, are more cost effective or render Kineta’s products obsolete.
Kineta may not be able to successfully commercialize its product candidates, if approved, due to unfavorable pricing regulations or third-party coverage and reimbursement policies, which could make it difficult for Kineta to sell its product candidates profitably.
Obtaining coverage and reimbursement approval for a product from a government or other third-party payor is a time-consuming and costly process, with uncertain results, that could require Kineta to provide supporting scientific, clinical and cost effectiveness data for the use of Kineta products to the payor. There may be significant delays in obtaining such coverage and reimbursement for newly approved products, and coverage may not be available, or may be more limited than the purposes for which the product is approved by the FDA or other comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a product will be paid for in all cases or at a rate that covers Kineta’s costs, including research, development, intellectual property, manufacture, sale and distribution expenses. Interim reimbursement levels for new products, if applicable, may also not be sufficient to cover Kineta’s costs and may not be made permanent. Reimbursement rates may vary according to the use of the product and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost products and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors, by any future laws limiting drug prices and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the United States.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, there is no uniform policy among third-party payors for coverage and reimbursement. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting reimbursement policies, but also have their own methods and approval process apart from Medicare coverage and reimbursement determinations. Therefore, one third-party payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage for the product.
Coverage and reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a product is:
Kineta cannot be sure that reimbursement will be available for any product that it commercializes and, if coverage and reimbursement are available, what the level of reimbursement will be. Kineta’s inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products that Kineta develops could have a material adverse effect on Kineta’s operating results, Kineta’s ability to raise capital needed to commercialize products and Kineta’s overall financial condition.
Reimbursement may impact the demand for, and the price of, any product for which Kineta obtains marketing approval. Even if Kineta obtains coverage for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. Patients who are prescribed medications for the treatment of their conditions, and their prescribing physicians, generally rely on third-party payors to reimburse all or part of the costs associated with those medications. Patients are unlikely to use Kineta’s products unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the cost of Kineta’s products. Therefore, coverage and adequate reimbursement are critical to a new product’s acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new products when more established or lower cost therapeutic alternatives are already available or subsequently become
62
available.
For products administered under the supervision of a physician, obtaining coverage and adequate reimbursement may be particularly difficult because of the higher prices often associated with such drugs. Additionally, separate reimbursement for the product itself may or may not be available. Instead, the hospital or administering physician may be reimbursed only for providing the treatment or procedure in which Kineta’s product is used. Further, from time to time, the Centers for Medicare & Medicaid Services (“CMS”), the federal agency responsible for administering the Medicare program, revises the reimbursement amounts paid to health care providers, including the Medicare Physician Fee Schedule and Hospital Outpatient Prospective Payment System, which may result in reduced Medicare payments.
Kineta expects to experience pricing pressures in connection with the sale of any of its product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription medicines, medical devices and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the successful commercialization of new products. Further, the adoption and implementation of any future governmental cost containment or other health reform initiative may result in additional downward pressure on the price that Kineta may receive for any approved product.
Outside of the United States, many countries require approval of the sale price of a product before it can be marketed, and the pricing review period only begins after marketing or product licensing approval is granted. To obtain reimbursement or pricing approval in some of these countries, Kineta may be required to conduct a clinical trial that compares the cost-effectiveness of Kineta’s product candidate to other available therapies. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, Kineta might obtain marketing approval for a product candidate in a particular country, but then be subject to price regulations that delay its commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenue, if any, Kineta is able to generate from the sale of the product in that country. Adverse pricing limitations may hinder Kineta’s ability to recoup its investment in one or more product candidates, even if such product candidates obtain marketing approval.
Reimbursement and healthcare payment systems vary significantly by country outside the U.S., and many countries have instituted price ceilings on specific products and therapies. In the EU and the UK, similar political, economic and regulatory developments may affect Kineta’s ability to profitably commercialize its product candidates, if approved. In addition to continuing pressure on prices and cost containment measures, legislative developments at the EU, UK or at an EU Member State level may result in significant additional requirements or obstacles that may increase Kineta’s operating costs. The delivery of healthcare in the EU and the UK, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than EU, law and policy. National governments and health service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most EU Member States and the UK have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory burdens on those wishing to develop and market products in these countries, this could prevent or delay marketing approval of Kineta’s product candidates, restrict or regulate post-approval activities and affect Kineta’s ability to commercialize its product candidates, if approved.
Kineta cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action in the U.S., the EU, UK or any other jurisdiction. If Kineta, or any third parties Kineta may engage, are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if Kineta or such third parties are not able to maintain regulatory compliance, Kineta’s product candidates may lose any regulatory approval that may have been obtained and Kineta may not achieve or sustain profitability.
If the regulatory authorities in such jurisdictions set prices or make reimbursement criteria that are not commercially attractive for Kineta or its collaborators, Kineta’s revenues and the potential profitability of Kineta’s products in those countries would be negatively affected.
If approved, Kineta’s product candidates that are licensed and regulated as biological products, or biologics, may face competition from biosimilars approved through an abbreviated regulatory pathway.
The Biologics Price Competition and Innovation Act of 2009 (the “BPCIA”) was enacted as part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the “ACA”), to establish an abbreviated pathway for the approval of biosimilar and interchangeable with an FDA-licensed reference biologic product. The regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an approved biologic. Under the BPCIA, reference biological product is granted 12 years of non-patent data exclusivity from the time of first licensure of the product, and the FDA will not accept an application for a biosimilar or interchangeable product based on the reference biological product until four years after the date of first licensure of the reference product. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still develop and receive approval of a competing biologic, so long as their BLA does not rely on the reference product or sponsor’s data or submit the application as a biosimilar application. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation and meaning are subject to uncertainty, and any new policies or processes adopted by the FDA could have a material adverse effect on the future commercial prospects for Kineta’s biological products.
Kineta believes that any of the product candidates it develops that is approved in the future, weUnited States as a biological product under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to congressional action or otherwise, or that the FDA will not consider the subject product candidate to be a reference product for competing products, potentially creating the
63
opportunity for biosimilar competition sooner than anticipated. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of the reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. The approval of a biosimilar of Kineta’s product candidates could have a material adverse impact on Kineta’s business due to increased competition and pricing pressure.
If competitors are able to obtain regulatory approval for biosimilars referencing Kineta’s product candidates, Kineta’s product candidates may become subject to competition from such biosimilars, with the attendant competitive pressure and consequences.
If the market opportunities for any of Kineta’s product candidates are smaller than it believes they are, Kineta’s revenue may be adversely affected, and Kineta’s business may suffer.
Kineta is focused on the development of treatments for cancer. Kineta’s projections of addressable patient populations that have the potential to benefit from treatment with Kineta’s product candidates are based on estimates, including estimated incidence rates of specific forms of cancer. If any of Kineta’s estimates are inaccurate, the market opportunities for any of Kineta’s product candidates could be significantly diminished and have an adverse material impact on Kineta’s business.
If any of Kineta’s product candidates are approved for marketing and commercialization and Kineta is unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market anyits product candidates, we may develop, we may notKineta will be successful in commercializing thoseunable to successfully commercialize its product candidates if and when they are approved.
We do not currently have an infrastructure for theKineta has no sales, marketing or distribution capabilities or experience. To achieve commercial success for any approved product for which Kineta retains sales and distributionmarketing responsibilities, Kineta must either develop a sales and marketing organization, which would be expensive and time consuming, or outsource these functions to other third parties. In the future, Kineta may choose to build a focused sales and marketing infrastructure to sell, or participate in sales activities with its collaborators for, some of pharmaceutical products. In order to market ourKineta’s product candidates if approved by the FDA or any other regulatory body, we must build our sales, marketing, managerial, and other non-technical capabilities, or make arrangements with third parties to perform these services. when they are approved.
There are risks involved with both establishing ourKineta’s own commercialsales and marketing capabilities and entering into arrangements with third parties to perform these services. For example, recruiting and training a sales force or reimbursement specialists is expensive and time-consumingtime consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruitKineta recruits a sales force and establishestablishes marketing and other commercialization capabilities is delayed or does not occur for any reason, weKineta would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and ourKineta’s investment would be lost if weit cannot retain or reposition our commercializationits sales and marketing personnel. Factors that may inhibit Kineta’s efforts to commercialize future products on its own include:
If we enterKineta enters into arrangements with third parties to perform sales, marketing commercial support, and distribution services, ourKineta’s product revenue or the profitability of these product revenue mayto Kineta are likely to be lower than if weKineta were to market and sell any products we may develop ourselves.that it develops itself. In addition, weKineta may not be successful in entering into arrangements with third parties to commercialize oursell and market its product candidates or may be unable to do so on terms that are favorable to us. We may have little control overKineta. In entering into third-party marketing or distribution arrangements, any revenue Kineta receives will depend upon the efforts of the third parties and Kineta cannot assure you that such third parties will establish adequate sales and any of them may fail todistribution capabilities or devote the necessary resources and attention to sell and market ourany future products effectively. If weKineta does not establish commercializationsales and marketing capabilities successfully, either on ourits own or in collaboration with third parties, weKineta will not be successful in commercializing ourits product candidates if approved.candidates.
If we are unable to establish adequate sales, marketing, and distribution capabilities, whether independently or with third parties, or if we are unable to do so on commercially reasonable terms, our business, results of operations, financial condition, and prospects will be materially adversely affected.
Even if we receive marketing approval for our product candidates, our product candidates may not achieve broad market acceptance by physicians, patients, healthcare payors, or others in the medical community, which would limit the revenue that we generate from their sales.
The commercial success of our product candidates, if approved by the FDA or other applicable regulatory authorities, will depend upon the awareness and acceptance of our product candidates among the medical community, including physicians, patients, and healthcare payors. If any of our product candidates are approved but do not achieve an adequate level of acceptance by physicians, patients, healthcare payors, and others in the medical community, we may not generate sufficient revenue to become or remain profitable. Market acceptance of our product candidates, if approved, will depend on a number of factors, including, among others:
43
If our product candidates are approved but do not achieve an adequate level of acceptance by patients, physicians, and payors, we may not generate sufficient revenue from our approved product candidates to become or remain profitable. Before granting reimbursement approval, healthcare payors may require us to demonstrate that our product candidates, in addition to treating these target indications, also provide incremental health benefits to patients. Our efforts to educate the medical community and third-party payors about the benefits of our product candidates may require significant resources and may never be successful.
We face significant competition in an environment of rapid technological and scientific change, and there is a possibility that our competitors may achieve regulatory approval before we do or develop therapies that are safer, more advanced, or more effective, which may negatively impact our ability to successfully market or commercialize any product candidates we may develop and ultimately harm our financial condition.
The development and commercialization of new drug products is highly competitive. Moreover, the neurodegenerative field is characterized by strong and increasing competition, with a strong emphasis on intellectual property. We may face competition with respect to any product candidates that we seek to develop or commercialize in the future from major pharmaceutical companies, specialty pharmaceutical companies, and biotechnology companies worldwide. Potential competitors also include academic institutions, government agencies, and other public and private research organizations that conduct research, seek patent protection, and establish collaborative arrangements for research, development, manufacturing, and commercialization.
There are a number of large pharmaceutical and biotechnology companies that are currently pursuing the development of products for the treatment of the neurodegenerative disease indications for which we have research programs, including Parkinson’s disease, ALS and Alzheimer’s disease. Companies that we are aware of are developing therapeutics in the neurodegenerative disease area include large companies with significant financial resources, such as AbbVie, AstraZeneca, Biogen, Bristol-Myers Squibb, Lilly, GlaxoSmithKline, Johnson & Johnson, Novartis, Roche, Sanofi, and Takeda. In addition to competition from other companies targeting neurodegenerative indications, any products we may develop may also face competition from other types of therapies, such as gene-editing therapies.
Many of our current or potential competitors, either alone or with their strategic partners, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical study sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for,
44
our programs. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient, or are less expensive than any products that we may develop. Furthermore, currently approved products could be discovered to have application for treatment of neurodegenerative disease indications, which could give such products significant regulatory and market timing advantages over any of our product candidates. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our product candidates and may obtain orphan product exclusivity from the FDA for indications our product candidates are targeting, which could result in our competitors establishing a strong market position before we are able to enter the market. Additionally, products or technologies developed by our competitors may render our potential product candidates uneconomical or obsolete, and we may not be successful in marketing any product candidates we may develop against competitors.
In addition, we could face litigation or other proceedings with respect to the scope, ownership, validity and/or enforceability of our patents relating to our competitors’ products and our competitors may allege that our products infringe, misappropriate, or otherwise violate their intellectual property. The availability of our competitors’ products could limit the demand, and the price we are able to charge, for any products that we may develop and commercialize. See “Risks Related to Our Intellectual Property Rights.”
The ongoing pandemic of COVID-19 and the future outbreak of other highly infectious or contagious diseases has and may continue to impact our research, development and potential future commercialization efforts, increase our costs and expenses and have a material adverse effect on our business, financial condition and results of operations.
Broad-based business or economic disruptions could adversely affect our ongoing or planned research and development activities. For example, in December 2019, an outbreak of a novel strain of coronavirus originated in Wuhan, China, and has since spread to a number of other countries, including the United States. To date, the COVID-19 pandemic has caused significant disruptions to the U.S. and global economy and has contributed to significant volatility and negative pressure in financial markets. The global impact of the outbreak is continually evolving and, as additional cases and new variants of the virus are identified, many countries, including the U.S., have reacted by instituting quarantines, restrictions on travel and mandatory closures of businesses. Certain states and cities, including where we or the third parties with whom we engage operate, have also reacted by instituting quarantines, restrictions on travel, “shelter in place” rules, restrictions on types of business that may continue to operate, and/or restrictions on the types of construction projects that may continue.
The extent to which COVID-19 may impact our preclinical studies or clinical trial operations will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration of the outbreak, the identification of new variants of the virus, the severity of COVID-19, or the effectiveness of actions to contain and treat COVID-19. The continued spread of COVID-19 globally has and may continue to adversely impact our preclinical studies or clinical trial operations, including our ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 if an outbreak occurs in their geography. COVID-19 may also affect employees of third-party CROs located in affected geographies that we rely upon to carry out our clinical trials. Any negative impact COVID-19 has to patient enrollment or treatment or the execution of our current product candidates and any future product candidates could cause costly delays to clinical trial activities, which could adversely affect our ability to obtain regulatory approval for and to commercialize our current product candidate and any future product candidates, increase our operating expenses, and have a material adverse effect on our financial results.
Further, the COVID-19 outbreak caused delays in our Phase 1 single ascending dose trial of YTX-7739, and certain of the sites in the YTX-7739 Phase 1b clinical trial incurred delays due to COVID-19, resulting in a delay in the expected timing of early results from that trial. COVID-19 may cause delays in our other clinical trials, including delays in enrollment, due to diversion or prioritization of trial site resources away from the conduct of clinical trials and toward the COVID-19 pandemic. Key clinical trial activities, such as site monitoring, may be interrupted due to restrictions in travel, and some patients may be unwilling to enroll in our trials or be unable to comply with clinical trial protocols if quarantines or travel restrictions impede patient movement or interrupt healthcare services, which would delay our ability to conduct clinical trials or release clinical trial results. The spread of COVID-19, or another infectious disease, could also negatively affect the operations at our third-party manufacturers and suppliers, which could result in delays or disruptions in the supply of our current product candidates and any future product candidates. In addition, we may take temporary precautionary measures intended to help minimize the risk of the virus to our employees, including temporarily requiring all employees to work remotely, suspending all non-essential travel worldwide for our employees, and discouraging employee attendance at industry events and in-person work-related meetings, which could negatively affect our business.
We cannot presently predict the scope and severity of any potential business shutdowns or disruptions. If we or any of the third parties with whom we engage, however, were to experience shutdowns or other business disruptions, our ability to conduct our business in the manner and on the timelines presently planned could be materially and negatively affected, which could have a material adverse impact on our business and our results of operation and financial condition.
45
Risks Related to Our Regulatory Approval and Other Legal Compliance Matters
The regulatory approval processes of the FDA and comparable foreign regulatory authorities are lengthy, time-consuming, and inherently unpredictable. If we are ultimately unable to obtain regulatory approval for our product candidates, we will be unable to generate product revenue and our business will be substantially harmed.
The time required to obtain approval by the FDA, andEuropean Commission (based on recommendation from the EMA) or comparable foreign regulatory authorities is unpredictable, typically takes many years following the commencement of clinical trials,limited to those specific indications and depends upon numerous factors, including the type, complexity,conditions for which approval has been granted, and novelty of the product candidates involved. In addition, approval policies, regulations, or the type and amount of clinical data necessaryKineta may be subject to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions, which may cause delays in the approval or the decision not to approve an application. Regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinicalfines, criminal penalties, injunctions or other studies. We have not submittedenforcement actions if Kineta is determined to be promoting the use of its products for unapproved or obtained regulatory approval for any product candidate, and it is possible that none of our existing product candidates“off-label” uses or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
Applications for our product candidates could be delayed in receiving or fail to receive regulatory approval for many reasons, including but not limited to the following:
This lengthy approval process, as well asinconsistent with the unpredictability of the results of clinical trials, may resultapproved labeling, resulting in our failingdamage to obtain regulatory approval to market any of our product candidates, which would significantly harm our business, results of operations,Kineta’s reputation and prospects.business.
Even if we obtain regulatory approval for our product candidates, our products will remain subject to extensive regulatory scrutiny.
Even if we receive marketing approval for our product candidates, regulatory authorities may still impose significant restrictions on our product candidates, indicated uses or marketing, or impose ongoing requirements for potentially costly post-approval studies. If any of our product candidates are approved, they will be subject to ongoing regulatory requirements, including for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing studies, and submission of safety, efficacy, and other post-marketing information, including both federal and state requirements in the United States and requirements of comparable foreign regulatory authorities.
Manufacturers and manufacturers’ facilities are required to comply with extensive requirements imposed by the FDA and comparable foreign regulatory authorities, including, for example, ensuring that quality control and manufacturing procedures conform to current Good Manufacturing Practice (“cGMP”) regulations and applicable product tracking and tracing requirements. As such, we and our contract manufacturers will be subject to continual review and inspections to assess compliance with cGMP and adherence to commitments made in any new drug application (“NDA”) or comparable marketing approval. Accordingly, we and others with whom
46
we workKineta must continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, and quality control.
The FDA has significant post-marketing authority, including, for example, the authority to require labeling changes based on new safety information and to require post-marketing studies or clinical trials to evaluate serious safety risks related to the use of a drug. The FDA also has the authority to require, as part of an NDA or post-approval, the submission of a REMS. Any REMS required by the FDA may lead to increased costs to assure compliance with new post-approval regulatory requirements and potential requirements or restrictions on the sale of approved products, all of which could lead to lower sales volume and revenue.
Any regulatory approvals that we receive for our product candidates will be subject to limitations on the approved indicated uses for which the product may be marketed and promoted or to the conditions of approval (including the requirement to implement a REMS), or contain requirements for potentially costly post-marketing testing. We will be required to report certain adverse reactions and production problems, if any, to the FDA and comparable foreign regulatory authorities. Any new legislation addressing drug safety issues could result in delays in product development or commercialization, or increased costs to assure compliance. The FDA and other agencies, including the U.S. Department of Justice, closely regulate and monitor the post-approval marketing and promotion of products to ensure that they are manufactured, marketed, and distributed only for the approved indications and in accordance with the provisions of the approved labeling. We will have to comply with requirements concerning advertising and promotion for our products.any product candidates for which Kineta obtains marketing approval. Promotional communications with respect to prescription drugstherapeutics are subject to a variety of legal and regulatory restrictions and continuing review by the FDA or comparable foreign regulatory and governmental authorities, Department of Justice, Department of Health and Human Services’ (“HHS”) Office of Inspector General, state attorneys general, members of Congress and the public. When the FDA or comparable foreign regulatory authorities issue regulatory approval for a product candidate, the regulatory approval is limited to those specific uses and indications for which a product is approved. If Kineta is not able to obtain FDA or comparable foreign regulatory authority approval for desired uses or indications for its current product candidates and any future product candidates, Kineta may not market or promote them for those indications and uses, referred to as off-label uses, and Kineta’s business, financial condition, results of operations, stock price and prospects will be materially harmed. Kineta also must be consistent withsufficiently substantiate any claims that it makes for its products, including claims comparing Kineta’s products to other companies’ products, and must abide by the informationFDA or a comparable foreign regulatory or governmental authority’s strict requirements regarding the content of promotion and
64
advertising.
While physicians may choose to prescribe products for uses that are not described in the product’s labeling and for uses that differ from those tested in clinical trials and approved label. As such, we may not promote ourby the regulatory authorities, Kineta and any third parties engaged on its behalf are prohibited from marketing and promoting the products for indications orand uses for which they dothat are not have approval. The holder of anspecifically approved NDAby the FDA or comparable marketing approval must submit new or supplemental applications and obtain approval for certain changes to the approved product, product labeling, or manufacturing process. We could also be asked to conduct post-marketing studies or clinical trials to verify the safety and efficacy of our products in general or in specific patient subsets.
If aforeign regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or disagrees with the promotion, marketing or labeling of a product, such regulatory agency may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If we fail to comply with applicable regulatory requirements, a regulatory agency or enforcement authority may, among other things:
Any government investigation of alleged violations of law could require us to expend significant time and resources in response, and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize and generate revenue from our products, if approved. If regulatory sanctions are applied or if regulatory approval is withdrawn, the value of the company and our operating results will be adversely affected.
We are subject to healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, and diminished profits and future earnings.
Although we do not currently have any products on the market, if we obtain FDA approval for any of our product candidates and begins commercializing our products, we may be subject to additional healthcare statutory and regulatory requirements and enforcement by the federal government and the states and foreign governments in which we conduct our business. Healthcare providers, physicians, third-party payors, and others play a primary role in the recommendation and prescription of our product candidates, if approved. Our future arrangements with third-party payors will expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we
47
market, sell, and distribute our product candidates, if we obtain marketing approval. See section entitled “Business – Government Regulation – Other U.S. Healthcare Laws and Compliance Requirements.”
In the event we decide to conduct clinical trials or continue to enroll subjects in our ongoing or future clinical trials, we may be subject to additional privacy requirements. The collection, use, storage, disclosure, transfer, or other processing of personal data regarding individuals in the EEA, including personal health data, for example, is subject to the EU GDPR. The GDPR is wide-ranging in scope and imposes numerous requirements on companies that process personal data, including requirements relating to processing health and other sensitive data, obtaining consent of the individuals to whom the personal data relates, providing information to individuals regarding data processing activities, implementing safeguards to protect the security and confidentiality of personal data, providing notification of data breaches, and taking certain measures when engaging third-party processors. The GDPR also imposes strict rules on the transfer of personal data to countries outside the EEA, including the United States, and permits data protectionauthorities. Regulatory authorities to impose large penalties for violations of the GDPR, including potential fines of up to €20 million or 4% of annual global revenues, whichever is greater. The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the GDPR. The GDPR may increase our responsibility and liability in relation to personal data that we process where such processing is subject to the GDPR, and we may be required to put in place additional mechanisms to ensure compliance with the GDPR, including as implemented by individual countries. Compliance with the GDPR is a rigorous and time-intensive process that may increase our cost of doing business or require us to change our business practices, and despite those efforts, there is a risk that we may be subject to fines and penalties, litigation, and reputational harm in connection with our European activities. Further to the United Kingdom’s exit from the EU, the UK GDPR and the UK Data Protection Act 2018 set out the United Kingdom’s data protection regime, which is independent from but aligned to the European Union’s data protection regime. Non-compliance with the UK GDPR may result in monetary penalties of up to £17.5 million or 4% of worldwide revenue, whichever is higher. Although the United Kingdom is regarded as a third country under the EU’s GDPR, the European Commission has now issued a decision recognizing the United Kingdom as providing adequate protection under the EU GDPR and, therefore, transfers of personal data originating in the EEA to the United Kingdom remain unrestricted. Like the EU GDPR, the UK GDPR restricts personal data transfers outside the United Kingdom to countries not regarded by the United Kingdom as providing adequate protection. The United Kingdom government has confirmed that personal data transfers from the United Kingdom to the EEA remain free flowing.
Additionally, other countries outside of Europe have enacted or are considering enacting similar privacy and data protection laws, which could increase the cost and complexity of delivering our services and operating our business. For example, Brazil enacted the General Data Protection Law, New Zealand enacted the New Zealand Privacy Act, China released a second draft of the Personal Information Protection Law, and Canada introduced the Digital Charter Implementation Act.
California recently enacted the California Consumer Privacy Act, or CCPA, which creates new individual privacy rights for California consumers (as defined in the law) and places increased privacy and security obligations on entities handling personal data of consumers or households. The CCPA requires covered businesses to provide certain disclosures to consumers about their data collection, use and sharing practices, and to provide affected California residents with ways to opt-out of certain sales or transfers of personal information, as well as the right to request, modify, and delete personal information. The CCPA went into effect on January 1, 2020, and the California State Attorney General submitted final regulations for review on June 2, 2020, which were finalized and are now effective. The California State Attorney General has commenced enforcement actions against violators as of July 1, 2020. Further, a new California privacy law, the California Privacy Rights Act, or CPRA, was passed by California voters on November 3, 2020. The CPRA will create additional obligations with respect to processing and storing personal information that are scheduled to take effect on January 1, 2023 (with certain provisions having retroactive effect to January 1, 2022). Other U.S. states also are considering omnibus privacy legislation and industry organizations regularly adopt and advocate for new standards in these areas. For example, Virginia recently enacted the Consumer Data Protection Act that will become effective on January 1, 2023, and is similar to the CCPA and CPRA, and Colorado recently enacted the Colorado Privacy Act, which will become effective on July 1, 2023, and which is similar to the CCPA and Virginia law. While the CCPA and CPRA provide exceptions for certain activities involving data collected in the context of clinical trials, health data governed by California’s Confidentiality of Medical Information Act, and PHI governed by HIPAA, and other state laws may contain similar exceptions, we cannot yet determine the impact the CCPA, CPRA, or other such existing or future laws, regulations and standards may have on our business.
Pharmaceutical and other healthcare companies have been prosecuted under these laws for a variety of promotional and marketing activities, such as: providing free trips, free goods, sham consulting fees and grants and other monetary benefits to prescribers; reporting to pricing services inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in off-label promotion; and submitting inflated best price information to the Medicaid Rebate Program to reduce liability for Medicaid rebates.
Ensuring that our internal operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, or case law involving applicable fraud and abuse or other healthcare laws and
48
regulations. If our operations, including anticipated activities to be conducted by our sales team, were found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal, and administrative penalties, damages, fines, and exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations, any of which could substantially disrupt our operations. If any of the physicians or other providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to criminal, civil, or administrative sanctions, including exclusions from government funded healthcare programs.
If the physicians or other providers or entities with whom we expect to do business are found not to be in compliance with applicable laws, they may be subject to significant criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. Even if resolved in our favor, litigation or other legal proceedings relating to healthcare laws and regulations may cause us to incur significant expenses and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development, manufacturing, sales, marketing or distribution activities. Uncertainties resulting from the initiation and continuation of litigation or other proceedings relating to applicable healthcare laws and regulations could have an adverse effect on our ability to compete in the marketplace.
If any of our product candidates obtain regulatory approval, additional competitors could enter the market with generic or other versions of such drugs, which may result in a material decline in sales of affected products.
Under the Drug Price Competition and Patent Term Restoration Act of 1984 (the “Hatch-Waxman Act”) a pharmaceutical manufacturer may file an abbreviated new drug application (“ANDA”) seeking approval of a generic copy of an approved, small-molecule innovator product. Under the Hatch-Waxman Act, a manufacturer may also submit an NDA under Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act that references the FDA’s prior approval of the small-molecule innovator product. A 505(b)(2) NDA product may be for a new or improved version of the original innovator product. The Hatch-Waxman Act also provides for certain periods of regulatory exclusivity, which preclude FDA approval (or in some circumstances, FDA filing and reviewing) of an ANDA or 505(b)(2) NDA. For example, a drug that is granted regulatory approval may be eligible for five years of marketing exclusivity in the United States following regulatory approval if that drug is classified as a new chemical entity (“NCE”). A drug can be classified as a NCE ifgenerally do not restrict or regulate the FDA has not previously approved any other drug containingbehavior of physicians in their choice of treatment within the same active moiety.practice of medicine. Regulatory authorities do, however, restrict communications by biopharmaceutical companies concerning off-label use.
In additionIf Kineta is found to the benefits of regulatory exclusivity, an innovator NDA holder may have patents claiming the active ingredient, product formulation or an approved use of the drug, which would be listed with the product in the FDA publication, “Approved Drug Products with Therapeutic Equivalence Evaluations,” known as the “Orange Book.” If there are patents listed in the Orange Book, a generic or 505(b)(2) applicant that seeks to market our product before expiration of the patents must include in the ANDA or 505(b)(2) NDA a “Paragraph IV certification,” challenging the validity or enforceability of, or claiming non-infringement of, the listed patent or patents. Appropriate notice of the certification must be given to the innovator, too, and if within 45 days of receiving such notice the innovator sues to protect our patents, approval of the ANDA or 505(b)(2) is stayed for 30 months, or as lengthened or shortened by the court.
Accordingly, ifimpermissibly promoted any of ourits current product candidates are approved, competitors could file ANDAs for generic versions of our small-molecule drug products or 505(b)(2) NDAs that reference our small-molecule drug products, respectively. If there are patents listed for our small-molecule drug products in the Orange Book, those ANDAs and 505(b)(2) NDAs would be requiredany future product candidates, Kineta may become subject to include a certification as to each listed patent indicating whether the ANDA or 505(b)(2) NDA applicant does or does not intend to challenge the patent. We cannot predict which, if any, patents in our current portfolio or patents we may obtain in the future will be eligible for listing in the Orange Book, how any generic competitor would address such patents, whether we would sue on any such patents, or the outcome of any such suit.
We may not be successful in securing or maintaining proprietary patent protection for productssignificant liability and technologies we develop or license. Moreover, if any of our owned or in-licensed patents that are listed in the Orange Book are successfully challenged by way of a Paragraph IV certification and subsequent litigation, the affected product could immediately face generic competition and our sales would likely decline rapidly and materially. See “Risks Related to Our Intellectual Property Rights.”
government fines. The FDA and other regulatory agencies actively enforce the laws and regulations regarding product promotion, particularly those prohibiting the promotion of off-label uses. If we areuses, and a company that is found to have improperly promoted off-label uses, wea product may becomebe subject to significant liability.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA or such other regulatory agencies as reflected in the product’s approved labeling. For example, if we receive marketing approval for YTX-7739 as a treatment for Parkinson’s disease,
49
physicians may nevertheless prescribe YTX-7739 to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability.sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. If we cannot successfully manage the promotion of our product candidates, if approved, we could become subject to significant liability, which would materially adversely affect our business and financial condition.
Even if approved, reimbursement policies could limit our ability to sell our product candidates.
In the United States, engaging in the impermissible promotion of Kineta’s products, following approval, for off-label uses can also subject Kineta to false claims and marketsother litigation under federal and state statutes. These include fraud and abuse and consumer protection laws, which can lead to civil and criminal penalties and fines and agreements with governmental authorities that materially restrict the manner in which Kineta promotes or distributes therapeutic products and conducts its business. These restrictions could include corporate integrity agreements, suspension or exclusion from participation in federal and state healthcare programs, and suspension and debarment from government contracts and refusal of orders under existing government contracts. These False Claims Act lawsuits against manufacturers of drugs and biologics have increased significantly in volume and breadth, leading to several substantial civil and criminal settlements pertaining to certain sales practices and promoting off-label uses. In addition, False Claims Act lawsuits may expose manufacturers to follow-on claims by private payors based on fraudulent marketing practices. This growth in litigation has increased the risk that a biopharmaceutical company will have to defend a false claim action, pay settlement fines or restitution, as well as criminal and civil penalties, agree to comply with burdensome reporting and compliance obligations and be excluded from Medicare, Medicaid or other federal and state healthcare programs. If Kineta does not lawfully promote its approved products, if any, Kineta may become subject to such litigation and, if Kineta does not successfully defend against such actions, those actions may have a material adverse effect on Kineta’s business, financial condition, results of operations, stock price and prospects.
In the United States, the promotion of biopharmaceutical products is subject to additional FDA requirements and restrictions on promotional statements. If after one or more of Kineta’s current or future product candidates obtains marketing approval the FDA determines that Kineta’s promotional activities violate its regulations and policies pertaining to product promotion, it could request that Kineta modify its promotional materials or subject Kineta to regulatory or other enforcement actions, including issuance of warning letters or untitled letters, suspension or withdrawal of an approved product from the market, requests for recalls, payment of civil fines, disgorgement of money, imposition of operating restrictions, injunctions or criminal prosecution and other enforcement actions. Similarly, industry codes in foreign jurisdictions may prohibit companies from engaging in certain promotional activities and regulatory agencies in various countries may enforce violations of such codes with civil penalties. If Kineta becomes subject to regulatory and enforcement actions, Kineta’s business, financial condition, results of operations, stock price and prospects will be materially harmed.
Furthermore, the use of Kineta’s products for indications other than those approved by the FDA or comparable foreign regulatory authorities may not effectively treat such conditions. Any such off-label use of Kineta’s product candidates could harm Kineta’s reputation in the marketplace among physicians and patients. There may also be increased risk of injury to patients if physicians attempt to use Kineta’s products for these uses for which they are not approved, which could lead to product liability suits that might require significant financial and management resources and that could harm Kineta’s reputation.
Even if Kineta obtains FDA or European Commission (based on recommendation of the EMA) approval any of its product candidates in the United States or EU, Kineta may never obtain approval for or commercialize any of them in any other jurisdiction, which would limit Kineta’s ability to realize their full market potential.
In order to market any products in any particular jurisdiction, Kineta must establish and comply with numerous and varying regulatory requirements on a country-by-country basis regarding safety and efficacy.
Approval by the FDA in the United States or the European Commission (based on recommendation of the EMA) in the EU does not ensure approval by regulatory authorities in other countries or jurisdictions. However, the failure to obtain approval in one jurisdiction may negatively impact Kineta’s ability to obtain approval elsewhere. In addition, clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not guarantee regulatory approval in any other country.
Approval processes vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approval could result in difficulties and increased costs for Kineta and require additional preclinical studies or clinical trials which could be costly and time consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of Kineta’s products in those countries. Kineta does not have any product candidates approved for sale in any jurisdiction, including in international markets, and Kineta does not have experience in obtaining regulatory approval in international markets. If Kineta fails to comply with regulatory requirements in international markets or to obtain and maintain required approvals, or if regulatory approvals in international markets are delayed, Kineta’s target market will be reduced and its ability to realize the full market potential of any product it develops will be unrealized.
65
Risks Related to Kineta’s Reliance on Third Parties
Some of Kineta’s product candidates may be studied in clinical trials sponsored by organizations or agencies other than Kineta, or in investigator-initiated clinical trials, which means Kineta will have minimal or no control over the conduct of such trials.
Kineta has and may continue to supply and otherwise support third party research, including investigator-initiated clinical trials. Investigator-initiated clinical trials pose similar risks as those set forth elsewhere in this “Risk Factor” section relating to Kineta’s internally-sponsored clinical trials, but because Kineta may not be the sponsors of these trials, Kineta has less control over the protocols, administration or conduct of these trials, including follow-up with patients generally relyand ongoing collection of data after treatment. The conduct or findings of these trials may have a negative impact on third-party payorsKineta’s development programs notwithstanding that Kineta has little involvement or control over these trials. As a result, Kineta is subject to reimburse all or part ofadditional risks associated with the way investigator-initiated trials are conducted. In particular, Kineta may be named in lawsuits that would lead to increased costs associated with legal defense. Additional risks include difficulties or delays in communicating with investigators or administrators, procedural delays and other timing issues and difficulties or differences in interpreting data. Third-party investigators may design clinical trials with clinical endpoints that are more difficult to achieve, or in other ways that increase the risk of negative clinical trial results compared to clinical trials that Kineta may design on its own. Negative results in investigator-initiated clinical trials could have a material adverse effect on Kineta’s efforts to obtain regulatory approval for Kineta’s product candidates and the public perception of Kineta’s product candidates. As a result, Kineta’s lack of control over the conduct and timing of and communications with the FDA and other regulatory authorities regarding investigator-sponsored trials may expose Kineta to additional risks and uncertainties, many of which are outside Kineta’s control, and the occurrence of which could adversely affect the commercial prospects for Kineta’s product candidates.
Kineta relies on third parties to conduct, supervise and monitor its clinical trials and perform some of its research and preclinical studies. If these third parties do not satisfactorily carry out their treatment. Salescontractual duties or fail to meet expected deadlines, Kineta’s development programs may be delayed or subject to increased costs, each of ourwhich may have an adverse effect on Kineta’s business and prospects.
Kineta does not have the ability to conduct all aspects of its preclinical testing or clinical trials itself. As a result, Kineta is and expects to remain dependent on third parties to conduct its current and future preclinical studies and clinical trials. CROs that manage Kineta’s preclinical studies and clinical trials as well as clinical investigators, including in investigator-initiated clinical trials, and consultants play a significant role in the conduct of Kineta’s preclinical studies and clinical trials and the subsequent collection and analysis of data. The timing of the initiation and completion of these studies and trials will therefore be partially controlled by such third parties and may result in delays to Kineta’s development programs. Nevertheless, Kineta is responsible for ensuring that each of its preclinical studies and clinical trials is conducted in accordance with the applicable protocol, legal requirements and scientific standards, and Kineta’s reliance on the CROs and other third parties does not relieve Kineta of its regulatory responsibilities. Kineta and its CROs are required to comply with Good Laboratory Practice (“GLP”) and GCP requirements, which are regulations and guidelines enforced by the FDA, the EMA and comparable foreign regulatory authorities for all of Kineta’s product candidates in clinical development. Regulatory authorities enforce these GLP and GCP requirements through periodic inspections of preclinical study sites, trial sponsors, clinical trial investigators and clinical trial sites. If Kineta or any of its CROs or clinical trial sites, including clinical trial sites in investigator-initiated clinical trials, fail to comply with applicable GLP or GCP requirements, the data generated in Kineta’s preclinical studies and clinical trials may be deemed unreliable, and the FDA, EMA or comparable foreign regulatory authorities may require Kineta to perform additional preclinical or clinical trials before approving Kineta’s marketing applications. In addition, Kineta’s clinical trials must be conducted with products produced under cGMP regulations. Kineta’s failure to comply with these regulations may require Kineta to stop and/or repeat clinical trials, which would delay the marketing approval process.
There is no guarantee that any such CROs, clinical trial investigators or other third parties on which Kineta relies will devote adequate time and resources to Kineta’s development activities or perform as contractually required. If any of these third parties fail to meet expected deadlines, adhere to Kineta’s clinical protocols or meet regulatory requirements, otherwise performs in a substandard manner, or terminates its engagement with Kineta, the timelines for Kineta’s development programs may be extended or delayed or Kineta’s development activities may be suspended or terminated. If any of Kineta’s clinical trial sites terminates for any reason, Kineta may experience the loss of follow-up information on subjects enrolled in such clinical trials unless Kineta is able to transfer those subjects to another qualified clinical trial site, which may be difficult or impossible. In addition, clinical trial investigators for Kineta’s clinical trials or investigator-initiated clinical trials may serve as scientific advisors or consultants to Kineta from time to time and may receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, or the FDA or any comparable foreign regulatory authority concludes that the financial relationship may have affected the interpretation of the trial, the integrity of the data generated at the applicable clinical trial site may be questioned and the utility of the clinical trial itself may be jeopardized, which could result in the delay or rejection of any marketing application Kineta submits by the FDA or any comparable foreign regulatory authority. Any such delay or rejection could prevent Kineta from commercializing its product candidates.
Furthermore, these third parties may also have relationships with other entities, some of which may be Kineta’s competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct Kineta’s clinical trials in accordance with regulatory requirements or Kineta’s stated protocols, Kineta will not be able to obtain, or may be delayed in obtaining, marketing approvals for its product candidates and will not be able to, or may be delayed in its efforts to, successfully commercialize its products.
Kineta relies on third parties to manufacture its product candidates, and Kineta expects to continue to rely on third parties for the clinical as well as any future commercial supply of its product candidates and other future product candidates. The development of Kineta’s current and future product candidates, and the commercialization of any approved products, could be stopped, delayed or made less profitable if any such third party fails to provide Kineta with sufficient clinical or commercial quantities of such product candidates or products, fails to do so at acceptable quality levels or prices or fails to achieve or maintain satisfactory regulatory compliance.
66
Kineta does not currently have, and Kineta does not plan to build, the infrastructure or capability internally to manufacture current product candidates or any future product candidates for use in the conduct of its clinical trials or, if approved, for commercial supply. Kineta relies on, and expects to continue to rely on, contract manufacturing organizations (“CMOs”). Reliance on third-party providers may expose Kineta to more risk than if it were to manufacture its product candidates itself. Kineta does not control the manufacturing processes of the CMOs it contracts with and is dependent on those third parties for the production of its product candidates in accordance with relevant applicable regulations such as cGMP, which includes, among other things, quality control, quality assurance and the maintenance of records and documentation.
In complying with the manufacturing regulations of the FDA and other comparable foreign regulatory authorities, Kineta and its third-party suppliers must spend significant time, money and effort in the areas of design and development, testing, production, record-keeping and quality control to assure that the products meet applicable specifications and other regulatory requirements. The failure to comply with these requirements could result in an enforcement action against Kineta, including the seizure of products and shutting down of production. Kineta and any of these third-party suppliers may also be subject to inspections by the FDA, EU Member States, coordinated by the EMA, or comparable foreign regulatory authorities. If any of Kineta’s third-party suppliers fail to comply with cGMP or other applicable manufacturing regulations, Kineta’s ability to develop and commercialize its product candidates could suffer significant interruptions.
Kineta’s failure, or the failure of Kineta’s third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on Kineta, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or drugs, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of Kineta’s products.
Any disruption, such as a fire, natural hazards or vandalism at Kineta’s CMOs, or any impacts on Kineta’s CMOs due to pandemics or other public health crises, could significantly interrupt Kineta’s manufacturing capability. Kineta currently does not have alternative production plans in place or disaster-recovery facilities available. In case of a disruption, Kineta will depend,have to establish alternative manufacturing sources. This would require substantial capital on Kineta’s part, which it may not be able to obtain on commercially acceptable terms or at all. Additionally, Kineta would likely experience months of manufacturing delays as Kineta builds facilities or locates alternative suppliers and seeks and obtains necessary regulatory approvals. If this occurs, Kineta will be unable to satisfy manufacturing needs on a timely basis, if at all. If changes to CMOs occur, then there also may be changes to manufacturing processes inherent in the setup of new operations for Kineta’s product candidates and any products that may obtain approval in the future. Any such changes could require the conduct of bridging studies before Kineta can use any materials produced at new facilities or under new processes in clinical trials or, for any products reaching approval, in Kineta’s commercial supply. Further, business interruption insurance may not adequately compensate Kineta for any losses that may occur and Kineta would have to bear the additional cost of any disruption. For these reasons, a significant disruptive event of any CMOs could have drastic consequences, including placing Kineta’s financial stability at risk.
Kineta’s product candidates and any drugs that Kineta may develop may compete with other product candidates and drugs for access to manufacturing facilities. There are no assurances Kineta would be able to enter into similar commercial arrangements with other manufacturers that operate under cGMP regulations and that might be capable of manufacturing for Kineta. Any performance failure on the part onof Kineta’s existing or future manufacturers could delay clinical development or marketing approval.
If Kineta were to experience an unexpected loss of supply of or if any supplier were unable to meet Kineta’s clinical or commercial demand for any of Kineta’s product candidates, Kineta could experience delays in its planned clinical studies or commercialization. For example, the extent to which our drugsthe COVID-19 pandemic may impact Kineta’s ability to procure sufficient supplies for the development of Kineta’s current and future product candidates, and the extent of such impacts will depend on future developments, including the severity and duration of any resurgence of COVID-19 and its variants. Kineta could be unable to find alternative suppliers of acceptable quality and experience that can produce and supply appropriate volumes at an acceptable cost or on favorable terms. Moreover, Kineta’s suppliers are often subject to strict manufacturing requirements and rigorous testing requirements, which could limit or delay production. The long transition periods necessary to switch manufacturers and suppliers, if necessary, would significantly delay Kineta’s clinical trials and, for any product candidates that reach approval, the commercialization of Kineta’s products, which would materially adversely affect Kineta’s business, financial condition and results of operation.
Kineta depends on third-party suppliers for materials that are necessary for the conduct of preclinical studies and manufacture of Kineta’s product candidates for clinical trials, and the loss of these third-party suppliers or their inability to supply Kineta with sufficient quantities of adequate materials, or to do so at acceptable quality levels and on a timely basis, could harm Kineta’s business.
Manufacturing Kineta’s product candidates requires many reagents, which are substances used in Kineta’s manufacturing processes to bring about chemical or biological reactions, and other specialty materials and equipment, some of which are manufactured or supplied by small companies with limited resources and experience to support commercial biologics production. Kineta currently depends on a limited number of vendors for certain materials and equipment used in the manufacture of its product candidates. For example, Kineta currently uses facilities and equipment at external CMOs, as well as supply sources internal to the collaboration for vector supply. Kineta’s use of CMOs increases the risk of delays in production or insufficient supplies as Kineta transfers its manufacturing technology to these CMOs and as they gain experience with Kineta’s supply requirements. Some of these suppliers may not have the capacity to support clinical trials and commercial products manufactured under cGMP by biopharmaceutical firms or may otherwise be ill-equipped to support Kineta’s needs. Kineta also does not have supply contracts with many of these suppliers and may not be able to obtain supply contracts with them on acceptable terms or at all. Accordingly, Kineta may experience delays in receiving key materials and equipment to support clinical or commercial manufacturing.
For some of these reagents, equipment and materials, Kineta relies and may in the future rely on sole source vendors or a limited number of vendors. The supply of the reagents and other specialty materials and equipment that are necessary to produce Kineta’s product candidates could be reduced or interrupted at any time. In such case, identifying and engaging an alternative supplier or manufacturer could result in delay, and Kineta may not be
67
able to find other acceptable suppliers or manufacturers on acceptable terms, or at all. Switching suppliers or manufacturers may involve substantial costs and is likely to result in a delay in Kineta’s desired clinical and commercial timelines. If Kineta changes suppliers or manufacturers for commercial production, applicable regulatory agencies may require Kineta to conduct additional studies or trials. If key suppliers or manufacturers are lost, or if the supply of the materials is diminished or discontinued, Kineta may not be able to develop, manufacture and market its product candidates in a timely and competitive manner, or at all. An inability to continue to source product from any of these suppliers, which could be due to a number of issues, including regulatory actions or requirements affecting the supplier, adverse financial or other strategic developments experienced by a supplier, labor disputes or shortages, unexpected demands or quality issues, could adversely affect Kineta’s ability to satisfy demand for its product candidates, which could adversely and materially affect Kineta’s product sales and operating results or Kineta’s ability to conduct clinical trials, either of which could significantly harm Kineta’s business.
As Kineta continues to develop and scale its manufacturing process, Kineta expects that it will need to obtain rights to and supplies of certain materials and equipment to be used as part of that process. Kineta may not be able to obtain rights to such materials on commercially reasonable terms, or at all, and if Kineta is unable to alter its process in a commercially viable manner to avoid the use of such materials or find a suitable substitute, it would have a material adverse effect on Kineta’s business. Even if Kineta is able to alter its process so as to use other materials or equipment, such a change may lead to a delay in Kineta’s clinical development and/or commercialization plans. If such a change occurs for a product candidate that is already in clinical testing, the change may require Kineta to perform both ex vivo comparability studies and to collect additional data from patients prior to undertaking more advanced clinical trials. These factors could cause the delay of studies or trials, regulatory submissions, required approvals or commercialization of product candidates that Kineta develops, cause Kineta to incur higher costs and prevent Kineta from commercializing its product candidates successfully.
If Kineta is unable to obtain sufficient raw and intermediate materials on a timely basis or if Kineta experiences other manufacturing or supply difficulties, Kineta’s business may be adversely affected.
The manufacture of certain of Kineta’s product candidates requires the timely delivery of sufficient amounts of raw and intermediate materials. Kineta works closely with its suppliers to ensure the continuity of supply but cannot guarantee these efforts will always be successful. Further, while efforts are made to diversify Kineta’s sources of raw and intermediate materials, in certain instances Kineta acquires raw and intermediate materials from a sole supplier. While Kineta believes that alternative sources of supply exist where it relies on sole supplier relationships, there can be no assurance that Kineta will be coveredable to quickly establish additional or replacement sources for some materials. A reduction or interruption in supply, and an inability to develop alternative sources for such supply, could adversely affect Kineta’s ability to manufacture its product candidates in a timely or cost-effective manner.
Kineta’s reliance on third parties requires Kineta to share its trade secrets, which increases the possibility that a competitor will discover them or that Kineta’s trade secrets will be misappropriated or disclosed.
Because Kineta relies on third parties to research and develop and to manufacture Kineta’s product candidates, Kineta must share trade secrets with them. Kineta seeks to protect its proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, consulting agreements or other similar agreements with Kineta’s advisors, employees, third-party payors,contractors and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose Kineta’s confidential information, including Kineta’s trade secrets. Despite the contractual provisions employed when working with third parties, the need to share trade secrets and other confidential information increases the risk that such as government healthtrade secrets become known by Kineta’s competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that Kineta’s proprietary position is based, in part, on Kineta’s know-how and trade secrets, a competitor’s independent discovery of Kineta’s trade secrets or other unauthorized use or disclosure would impair Kineta’s competitive position and may have a material adverse effect on Kineta’s business.
In addition, these agreements typically restrict the ability of Kineta’s advisors, employees, third-party contractors and consultants to publish data potentially relating to Kineta’s trade secrets, although Kineta’s agreements may contain certain limited publication rights. For example, any academic institution that Kineta may collaborate with will likely expect to be granted rights to publish data arising out of such collaboration and any joint research and development programs commercial insurance,may require Kineta to share trade secrets under the terms of its research and managed healthcare organizations. Significant uncertainty exists asdevelopment or similar agreements. Despite Kineta’s efforts to protect its trade secrets, Kineta’s competitors may discover Kineta’s trade secrets, either through breach of Kineta’s agreements with third parties, independent development or publication of information by any of Kineta’s third-party collaborators. A competitor’s discovery of Kineta’s trade secrets would impair Kineta’s competitive position and have an adverse impact on Kineta’s business.
Kineta has already entered into collaborations with third parties for the research, development and commercialization of certain of the product candidates Kineta may develop. Kineta may form or seek additional collaborations or strategic alliances or enter into additional licensing arrangements in the future. If any of these collaborations, strategic alliances or additional licensing arrangements are not successful, Kineta may not be able to capitalize on the market potential of those product candidates.
Kineta has already entered into licenses and collaborations with third parties and may seek other third-party collaborators for the research, development and commercialization of Kineta’s current or future product candidates. The collaboration with drug discovery vendors and any other collaboration agreements Kineta enters into will likely limit Kineta’s control over the amount and timing of resources that its collaborators dedicate to the coverage and reimbursement statusdevelopment or commercialization of any product candidates for which weKineta may obtain regulatory approval. See section entitled “Business – Government Regulation – Coverage and Reimbursement.”
Market acceptance and sales of our product candidatesseek to develop with them. Kineta’s ability to generate revenues from these arrangements will depend on reimbursementKineta’s collaborators’ abilities to successfully perform the functions assigned to them in these arrangements. Kineta cannot predict the success of any collaboration in which Kineta has entered or may enter.
68
Kineta may in the future form or seek strategic alliances, create joint ventures or collaborations or enter into additional licensing arrangements with third parties that Kineta believes will complement or augment its development and commercialization efforts with respect to Kineta’s product candidates and any future product candidates that Kineta may develop. Any of these relationships may require Kineta to incur non-recurring and other charges, increase Kineta’s near and long-term expenditures, issue securities that dilute Kineta’s existing stockholders or disrupt Kineta’s management and business.
In addition, Kineta faces significant competition in seeking appropriate strategic partners and the negotiation process for these sorts of transactions is time-consuming, complex and expensive. Moreover, Kineta may not be successful in its efforts to establish a strategic partnership or other alternative arrangements for Kineta’s product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view Kineta’s product candidates as having the requisite potential to demonstrate safety, potency, purity and efficacy and obtain marketing approval. Additionally, Kineta’s existing partners may decide to acquire or partner with other companies developing oncology therapeutics, which may have an adverse impact on Kineta’s business prospects, financial condition and results of operations.
As a result, if Kineta enters into additional collaboration agreements and strategic partnerships or licenses its product candidates, Kineta may not be able to realize the benefit of those transactions if Kineta is unable to successfully integrate them with Kineta’s existing operations and company culture, which could delay Kineta’s timelines or otherwise adversely affect Kineta’s business prospects, financial condition and results of operations. Kineta also cannot be certain that, following a strategic transaction or license, it will achieve the revenue or specific net income that justifies the entry into the transaction in the first place. Any delays in entering into new collaborations or strategic partnership agreements related to Kineta’s product candidates could delay the development and commercialization of Kineta’s product candidates in certain geographies for certain indications, which would harm Kineta’s business prospects, financial condition and results of operations.
Risks Related to Kineta’s Industry and Business Operations
Public health crises, including the COVID-19 pandemic, could have a material adverse impact on Kineta’s business, financial condition and results of operations, including through disruption to Kineta’s planned clinical trials, supply chains, business operations and commercialization efforts, or through delay in the FDA’s approval of Kineta’s product candidates.
The COVID-19 pandemic and government measures taken in response to the pandemic in the past have had a significant impact, both direct and indirect, on businesses and commerce, as worker shortages have occurred, supply chains have been disrupted, facilities and production have been suspended, and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen. The extent to which COVID-19 impacts Kineta’s business and operating results will depend on future developments, including the severity and duration of any resurgence of COVID-19 and its variants. For example, ineffective or uncoordinated vaccine deployment in the future or other responses to COVID-19, the emergence of more virulent or infectious variants of the virus or limitations on vaccine availability could risk increasing the duration and severity of the pandemic, which could have various negative impacts on Kineta’s business, the extent of which Kineta cannot fully predict.
If there is a resurgence of COVID-19 or its variants, site initiation, participant recruitment and enrollment, participant dosing, distribution of clinical trial materials, study monitoring and data analysis for Kineta’s planned clinical trials may be delayed due to changes in hospital or university policies, federal, state or local regulations, prioritization of hospital resources toward pandemic efforts, or other reasons related to the pandemic. Additionally, some participants and clinical investigators may not be able to comply with clinical trial protocols. For example, quarantines or other travel limitations (whether voluntary or required) may impede participant movement, affect sponsor access to study sites, or interrupt healthcare services, and Kineta may be unable to conduct its planned clinical trials. Any delays to Kineta’s planned clinical trials for its current product candidates and any future clinical trials as a result of a resurgence of COVID-19 or its variants could impact the use and sufficiency of Kineta’s existing cash reserves, and Kineta may be required to raise additional capital earlier than it had previously planned. Kineta may be unable to raise additional capital if and when needed, which may result in further delays or suspension of Kineta’s development plans.
Further, as a result of a resurgence of COVID-19 or its variants, Kineta may be required in the future to develop and implement additional clinical trial policies and procedures based on new guidance and regulatory requirements promulgated by the FDA or other regulatory authorities.
Kineta currently utilizes third parties to, among other things, manufacture raw materials and Kineta’s product candidates, components, parts and consumables, and to perform quality control and testing. If either Kineta or any third-party in the supply chain for materials used in the production of Kineta’s product candidates are adversely impacted by restrictions resulting from a resurgence of COVID-19 or its variants, Kineta’s supply chain may be disrupted, limiting Kineta’s ability to manufacture product candidates for its clinical trials.
The ultimate impact of the COVID-19 pandemic, or any other health epidemic, will depend on future developments that cannot be predicted with confidence. Kineta cannot be certain what the overall impact of a resurgence of COVID-19 or its variants or future pandemics or other health crises will be on its business.
Disruptions at the FDA, EMA, SEC and other government agencies and regulatory authorities caused by funding shortages or global health concerns could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal governmental functions on which the operation of Kineta’s business may rely, which could negatively impact Kineta’s business.
The ability of the FDA, EMA and other comparable foreign regulatory authorities to review and approve new products can be affected by a variety of factors, including government budget and funding levels, the ability to hire and retain key personnel and accept the payment of user fees, and
69
statutory, regulatory and policy changes. Average review times at regulatory authorities and government agencies have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which Kineta’s operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies such as the EMA following its post-Brexit relocation and resulting staff changes may also slow the time necessary for new products to be reviewed and/or approved by necessary government agencies, which would adversely affect Kineta’s business. For example, in recent years, including in 2018 and 2019, the U.S. government shut down several times and certain regulatory agencies, such as the FDA and the SEC, had to furlough critical employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to review and process Kineta’s regulatory submissions, which could have a material adverse effect on Kineta’s business. Further, in Kineta’s operations as a public company, future government shutdowns could impact Kineta’s ability to access the public markets and obtain necessary capital in order to properly capitalize and continue Kineta’s operations.
Separately, in response to the COVID-19 pandemic, the FDA postponed most inspections of domestic and foreign manufacturing facilities at various points. Even though the FDA has resumed standard inspection operations of domestic facilities where feasible, the FDA has continued to monitor and implement changes to its inspectional activities to ensure the safety of its employees and those of the firms it regulates as it adapts to the evolving COVID-19 pandemic and any resurgence of the virus or emergence of new variants may lead to further inspectional delays. Regulatory authorities outside the United States have adopted similar restrictions or other policy measures in response to the COVID-19 pandemic. If a prolonged government shutdown occurs, or if global health concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process Kineta’s regulatory submissions, which could have a material adverse effect on Kineta’s business.
Kineta may be exposed to significant foreign exchange risk.
Kineta conducts research and business activities in foreign countries and it incurs portions of its expenses, and may in the future derive revenues, in a variety of currencies. As a result, Kineta is exposed to foreign currency exchange risk as its results of operations and cash flows are subject to fluctuations in foreign currency exchange rates. Fluctuations in currency exchange rates have had, and will continue to have, an impact on Kineta’s results as expressed in U.S. dollars. Kineta currently does not engage in hedging transactions to protect against uncertainty in future exchange rates between particular foreign currencies and the U.S. dollar. Kineta cannot predict the impact of foreign currency fluctuations, and foreign currency fluctuations in the future may adversely affect Kineta’s financial condition, results of operations and cash flows.
Kineta’s employees, principal investigators, consultants and commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
Kineta is exposed to the risk of fraud or other misconduct by its employees, principal investigators, consultants and commercial partners. Misconduct by these parties could include intentional failures to comply with the regulations of the FDA and other comparable foreign regulatory authorities, provide accurate information to the FDA and other comparable foreign regulatory authorities, comply with healthcare reform measures. Coveragefraud and adequate reimbursementabuse laws and regulations in the United States, EU, UK and in other jurisdictions, report financial information or data accurately or disclose unauthorized activities to Kineta. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and cause serious harm to Kineta’s reputation. It is not always possible to identify and deter employee misconduct, and the precautions Kineta takes to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting Kineta from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against Kineta, those actions could have a significant impact on Kineta’s business, including the imposition of significant civil, criminal and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, diminished profits and future earnings, additional reporting obligations and oversight if subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of Kineta’s operations.
Kineta faces potential product liability, and, if successful claims are brought against it, Kineta may incur substantial liability and costs. If the use of Kineta’s product candidates harms patients or is perceived to harm patients even when such harm is unrelated to Kineta’s product candidates, Kineta’s regulatory approvals could be revoked or otherwise negatively impacted and Kineta could be subject to costly and damaging product liability claims.
The use of Kineta’s product candidates in clinical trials and the sale of any products for which Kineta obtains marketing approval exposes Kineta to the risk of product liability claims. Product liability claims might be brought against Kineta by consumers, healthcare providers, pharmaceutical companies or others selling or otherwise coming into contact with Kineta’s products. There is a risk that Kineta’s product candidates may induce adverse events. If Kineta cannot successfully defend against product liability claims, Kineta could incur substantial liability and costs. In addition, regardless of merit or eventual outcome, product liability claims may result in:
70
Kineta believes its product liability insurance coverage is sufficient in light of its current clinical programs; however, Kineta may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect Kineta against losses due to liability. If and when Kineta obtains marketing approval for product candidates, Kineta intends to expand its insurance coverage to include the sale of commercial products; however, Kineta may be unable to obtain product liability insurance on commercially reasonable terms or in adequate amounts. On occasion, large judgments have been awarded in class action lawsuits based on drugs or medical treatments that had unanticipated adverse effects. A successful product liability claim, or series of claims brought against Kineta, could cause Kineta’s stock price to decline and, if judgments exceed Kineta’s insurance coverage, could adversely affect Kineta’s results of operations and business.
Patients with cancer and other diseases targeted by Kineta’s product candidates are often already in severe and advanced stages of disease and have both known and unknown significant pre-existing and potentially life-threatening health risks. During the course of treatment, patients may suffer adverse events, including death, for reasons that may be related to Kineta’s product candidates. Such events could subject Kineta to costly litigation, require Kineta to pay substantial amounts of money to injured patients, delay, negatively impact or end Kineta’s opportunity to receive or maintain regulatory approval to market Kineta’s products, or require Kineta to suspend or abandon its commercialization efforts. Even in a circumstance in which Kineta does not believe that an adverse event is related to its products, the investigation into the circumstance may be time-consuming or inconclusive. These investigations may divide the attention of Kineta’s management team, interrupt Kineta’s sales efforts, delay Kineta’s regulatory approval process in other countries or impact and limit the type of regulatory approvals Kineta’s product candidates receive or maintain. As a result of these factors, a product liability claim, even if successfully defended, could have a material adverse effect on Kineta’s business, financial condition or results of operations.
Kineta’s future success depends on its ability to retain key members of senior management and to attract, retain and motivate qualified personnel.
Kineta’s ability to compete in the highly competitive biopharmaceutical industry depends upon its ability to attract and retain highly qualified management, research and development, clinical, financial and business development personnel. Kineta is highly dependent on its management, scientific and medical personnel, including Shawn Iadonato, Ph.D., Kineta’s Chief Executive Officer, Craig Philips, Kineta’s President, Keith Baker, Kineta’s Chief Financial Officer, Pauline Kenny, Kineta’s General Counsel, Thierry Guillaudeux, Ph.D., Kineta’s Chief Scientific Officer and Jacques Bouchy, Kineta’s EVP Investor Relations & Business Development. Kineta’s senior management may terminate their employment with Kineta at any time. Kineta does not maintain “key person” insurance for any of its employees.
Recruiting and retaining qualified scientific and clinical personnel and, if Kineta progresses the development of any of its product candidates, commercialization, manufacturing and sales and marketing personnel, will be critical to Kineta’s success. The loss of the services of members of Kineta’s senior management or other key employees could impede the achievement of Kineta’s research, development and commercialization objectives and seriously harm Kineta’s ability to successfully implement its business strategy. Furthermore, replacing members of Kineta’s senior management and key employees may be difficult and may take an extended period of time because of the limited number of individuals in Kineta’s industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize Kineta’s product candidates. Kineta’s success also depends on its ability to continue to attract, retain and motivate highly skilled junior, mid-level and senior managers, as well as junior, mid-level and senior scientific and medical personnel. Competition to hire from this limited candidate pool is intense, and Kineta may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. Kineta also experiences competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, Kineta relies on consultants and advisors, including scientific and clinical advisors, to assist Kineta in formulating its research and development and commercialization strategy. Kineta’s consultants and advisors may have commitments under consulting or advisory contracts with other entities that may limit their availability to Kineta. If Kineta is unable to continue to attract and retain high-quality personnel, Kineta’s ability to pursue its growth strategy will be limited.
Kineta expects to expand its clinical development and regulatory capabilities and potentially implement sales, marketing and distribution capabilities, and as a result, Kineta may encounter difficulties in managing its growth, which could disrupt Kineta’s operations.
As of December 31, 2023, Kineta had 11 full-time employees. As Kineta’s development progresses, Kineta expects to experience growth in the number of its employees and the scope of its operations, particularly in the areas of clinical product development, regulatory affairs, manufacturing and, if any of Kineta’s product candidates receives marketing approval, sales, marketing and distribution. To manage Kineta’s anticipated future growth, Kineta must continue to implement and improve its managerial, operational and financial systems, expand its facilities and continue to recruit and train additional qualified personnel. Due to Kineta’s limited financial resources and the limited experience of Kineta’s management team in managing a company with such anticipated growth, Kineta may not be able to effectively manage the expansion of its operations or recruit and train additional qualified personnel. The expansion of Kineta’s operations may lead to significant costs and may divert its management and business development resources. Any inability to manage growth could delay the execution of Kineta’s business plans or disrupt Kineta’s operations.
Kineta faces substantial competition, which may result in others discovering, developing or commercializing products more quickly or marketing them more successfully than Kineta.
71
The development and commercialization of new products is highly competitive. Kineta expects to compete in the segments of the pharmaceutical, biotechnology and other related markets that pursue immuno-oncology treatments. Kineta’s commercial opportunity could be reduced or eliminated if its competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient, or are less expensive than any products that Kineta may develop. Kineta’s competitors also may obtain regulatory approval from the FDA or other comparable foreign regulatory authorities for their products more rapidly than Kineta may obtain approval for its products, if ever, which could result in Kineta’s competitors establishing a strong market position before Kineta is able to enter the market or make Kineta’s development more complicated. Moreover, with the proliferation of new drugs and therapies into oncology, Kineta expects to face increasingly intense competition as new technologies become available. If Kineta fails to stay at the forefront of technological change, it may be unable to compete effectively. Any product candidates that Kineta successfully develops and commercializes will compete with existing therapies and new therapies that may become available in the future. The highly competitive nature of and rapid technological changes in the biotechnology and pharmaceutical industries could render Kineta’s product candidates or its technology obsolete, less competitive or uneconomical.
Other products in a similar class as some of Kineta’s product candidates have already been approved and other products in the same class are further along in development. As more product candidates within a particular class of biopharmaceutical products proceed through clinical development to regulatory review and approval, the amount and type of clinical data that may be required by regulatory authorities may increase or change. Consequently, the results of Kineta’s clinical trials for product candidates in those classes will likely need to show a risk benefit profile that is competitive with or more favorable than those products and product candidates in order to obtain marketing approval or, if approved, a product label that is favorable for commercialization. If the risk benefit profile is not competitive with those products or product candidates, Kineta may have developed a product that is not commercially viable, that Kineta is not able to sell profitably or that is unable to achieve favorable pricing or reimbursement. In such circumstances, Kineta’s future product revenue and financial condition would be materially and adversely affected.
Specifically, there are many companies that have commercialized or are developing immuno-oncology treatments for cancer including large pharmaceutical and biotechnology companies such as Amgen Inc., AstraZeneca plc and its subsidiary, MedImmune, LLC, Bristol-Myers Squibb Company (“BMS”), Merck, Novartis AG, Pfizer Inc., Curis, Inc., Hummingbird Bioscience, Pte. Ltd., and Roche, and its subsidiary Genentech. Kineta is also aware of several companies testing their compounds in combination with nivolumab or pembrolizumab. Select programs in late-stage development include lymphocyte activation gene-3 (“LAG-3”) assets from BMS (relatlimab) and modified interleukin-2 (“IL-2”) assets from Nektar Therapeutics bempegaldesleukin). In earlier stage development there are also BioNTech SE with NEO-PV-01 and Karyopharm Therapeutics, Inc. with selinexor.
In addition, there are large pharmaceutical and biotech companies developing therapeutics for the treatment of chronic pain and viral diseases.
Many of Kineta’s competitors, either alone or with their collaboration partners, have significantly greater financial resources and expertise in research and development, preclinical testing, clinical trials, manufacturing and marketing than Kineta does. Future collaborations and mergers and acquisitions may result in further resource concentration among a smaller number of competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors will also compete with Kineta in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and subject registration for clinical trials, as well as in acquiring technologies complementary to, or that maybe necessary for, Kineta’s programs.
The key competitive factors affecting the success of all of Kineta’s programs are likely to be efficacy, safety, convenience, price, the level of generic competition and the availability of reimbursement from government and other third-party payors. If Kineta is not successful in developing, commercializing and achieving higher levels of reimbursement than its competitors, Kineta will not be able to compete against them and Kineta’s business would be materially harmed.
Kineta has net operating losses (“NOL”) to be carried forward, which may become devalued if Kineta does not generate sufficient future taxable income, applicable corporate tax rates are reduced or if Kineta experiences an ownership change.
Kineta’s total gross deferred tax assets as of December 31, 2023 were $174.8 million. Utilization of most deferred tax assets is dependent on generating sufficient future taxable income in the appropriate jurisdiction and/or entity. Kineta has provided a valuation allowance of $174.6 million on its net deferred tax assets as of December 31, 2023. Based on all available evidence, it is considered more likely than not that all the recorded deferred tax assets will not be realized in a future period. Accordingly, in the event of a reduction of any such corporate income tax rates, the carrying value of certain of Kineta’s deferred tax assets would decrease. Moreover, Kineta’s ability to use its NOL and other deferred tax assets to offset future taxable income may be limited if Kineta experiences an ownership change. Kineta may experience ownership changes in the future as a result of subsequent shifts in its stock ownership, some of which are outside Kineta’s control.
For U.S. federal income tax purposes, an ownership change will generally occur when the percentage of Kineta’s stock (by value) owned by one or more “5% shareholders” (as defined in the U.S. Internal Revenue Code of 1986, as amended (the “Code”)) has increased by more than 50% over the lowest percentage owned by such shareholders at any time during the prior three years (calculated on a rolling basis). Kineta anticipates that it will incur losses in the United States in the foreseeable future related to Kineta’s research and development activities. Due to potential ownership changes under Section 382 of the Code, Kineta may be limited in its ability to realize a tax benefit from the use of such losses, whether or not Kineta attains profitability in future years.
In addition, Kineta’s ability to utilize any future NOL may be limited by Pub. L. 115-97, enacted in 2017 and commonly known as the Tax Cuts and Jobs Act of 2017 (the “TCJA”). Under the TCJA, the amount of Kineta’s NOL that Kineta is permitted to deduct in any taxable year is limited to 80% of its taxable income in such year, where taxable income is determined without regard to the NOL deduction itself, while allowing unused NOL
72
to be carried forward indefinitely.
For these reasons, a material devaluation in Kineta’s deferred tax assets due to insufficient taxable income, lower corporate income tax rates or ownership change would have an adverse effect on Kineta’s results of operations and financial condition.
Foreign subsidiaries may directly become subject to U.S. federal income tax and be subject to a branch profits tax in the United States, which could reduce Kineta’s after-tax returns and the value of Kineta’s shares.
Kineta currently intends to conduct substantially all of its businesses and operations in a manner such that any foreign subsidiaries, if applicable, will not be treated as engaged in a trade or business in the United States and commercial payors are criticalwill not be subject to new product acceptance. Third-party payors decide which drugs they will pay for and establish reimbursement levels. Inadditional U.S. income tax or branch profits tax. However, it is not entirely clear when a foreign subsidiary is treated as being engaged in a trade or business in the United States for U.S. federal income tax purposes. Accordingly, Kineta cannot assure you that the principal decisions about reimbursementInternal Revenue Service (the “IRS”) will not contend, perhaps successfully, that Kineta’s foreign subsidiaries were engaged in a trade or business in the United States or are subject to more U.S. income tax than they currently incur. A foreign corporation deemed to be so engaged would be subject to U.S. federal income tax on its income that is treated as effectively connected with the conduct of that trade or business, as well as to branch profits tax on its “dividend equivalent amount,” unless the corporation is entitled to relief under an applicable tax treaty, which is determined on an annual basis.
Kineta’s business operations and current and future relationships with investigators, health care professionals, consultants, third-party payors and customers are subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, transparency laws and other healthcare laws and regulations. If Kineta is unable to comply, or has not fully complied, with such laws, Kineta could face substantial penalties.
Healthcare providers and others play a primary role in the recommendation and prescription of any products for new medicines are typically madewhich Kineta obtains marketing approval. Although Kineta does not currently have any products on the market, Kineta’s operations and current and future arrangements with investigators, healthcare professionals, customers and third-party payors may be subject to various U.S. federal and state healthcare laws and regulations, including, without limitation, the U.S. federal Anti-Kickback Statute, the U.S. federal civil and criminal false claims laws and the Physician Payments Sunshine Act and regulations. These laws may impact, among other things, Kineta’s current business operations, including its clinical research activities and proposed sales, marketing and education programs and constrain the business of financial arrangements and relationships with healthcare providers and other parties through which Kineta may market, sell and distribute its products for which Kineta obtains marketing approval. In addition, Kineta may be subject to additional healthcare, statutory and regulatory requirements and enforcement by CMS. CMS decides whether andforeign regulatory authorities in jurisdictions in which Kineta conducts its business. The laws that may affect Kineta’s ability to what extent our products will be covered and reimbursed under Medicare and private payors tend to follow CMS to a substantial degree. Coverage and reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a therapeutic is:operate include:
73
GovernmentEnsuring that Kineta’s internal operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that Kineta’s business practices, including certain arrangements with physicians who receive stock, warrants or stock options as compensation for services provided to Kineta, do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other third-party payors,healthcare laws and regulations. If Kineta’s operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to Kineta, Kineta may be subject to significant penalties, including civil, criminal and administrative penalties, damages, fines, exclusion from U.S. government funded healthcare programs, such as private health insurersMedicare and health maintenance organizations, decide which medications they will pay forMedicaid, or similar programs in other countries or jurisdictions, disgorgement, imprisonment, contractual damages, reputational harm, diminished profits, additional reporting requirements and establish reimbursement levels for those medications. Cost containment isoversight if Kineta becomes subject to a primary concern in the U.S. healthcare industry and elsewhere. Government authorities andcorporate integrity agreement or similar agreement to resolve allegations of non-compliance with these third-party payors have attempted to control costs by limiting coveragelaws and the amountdelay, reduction, termination or restructuring of reimbursement for particular medications. We cannotKineta’s operations. Further, defending against any such actions can be surecostly and time-consuming, and may require significant financial and personnel resources. Therefore, even if Kineta is successful in defending against any such actions that reimbursement willmay be available for our product candidates and, if reimbursementbrought against it, Kineta’s business may be impaired. If any of the physicians or other providers or entities with whom Kineta expects to do business is available, the level of such reimbursement. Reimbursement may impact the demand for, or the price of, our product candidates. If reimbursement is not available or is available only at limited levels, we mayfound to not be ablein compliance with applicable laws, they may be subject to successfully commercialize our product candidates. Limited coveragesignificant criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs and less than adequate reimbursement may reduceimprisonment. If any of the demand for, or the priceabove occur, it could adversely affect Kineta’s ability to operate its business and its results of any product for which we obtain regulatory approval.operations.
In some foreign countries, particularly in CanadaHealthcare legislative reform measures may have a material adverse effect on Kineta’s business and European countries, the pricingresults of prescription pharmaceuticals is subject to strict governmental control. In these countries, pricing negotiations with governmental authorities can take six to 12 months or longer after the receipt of regulatory approval and product launch. To obtain favorable reimbursement for the indications sought or pricing approval in some countries, we may be required to conduct a clinical study that compares the cost-effectiveness of our product candidates with other available therapies. If reimbursement for our product candidates is unavailable in any country in which we seek reimbursement, if it is limited in scope or amount, if it is conditioned upon our completion of additional clinical trials, or if pricing is set at unsatisfactory levels, our operating results could be materially adversely affected.operations.
Recently enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain.
In theThe United States and somemany foreign jurisdictions there have been a number ofenacted or proposed legislative and regulatory changes and proposed changes regardingaffecting the healthcare system that could prevent or delay regulatorymarketing approval of ourKineta’s current product candidates and any future product candidates, restrict or regulate post-approval activities and affect ourKineta’s ability to profitably sell anya product candidates for which we obtainit obtains marketing approval. See section entitled “Business – Government Regulation – Healthcare Reform.”Changes in regulations, statutes or the interpretation of existing regulations could impact Kineta’s business in the future by requiring, for example: (i) changes to Kineta’s manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of Kineta’s products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of Kineta’s business.
Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality, and/or expanding access. In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs. For example, in March 2010, the ACA was passed, which substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impacted the U.S. pharmaceutical industry has beenindustry. The ACA, among other things, subjected biological products to potential competition by lower-cost biosimilars, addressed a particular focusnew methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs and biologics that are inhaled, infused, instilled, implanted or injected, increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, established annual fees and taxes on manufacturers of these effortscertain branded prescription drugs and has been significantly affected by major legislative initiatives.biologics, and created a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% (increased from 50% pursuant to the Bipartisan Budget Act of 2018) point-of-sale discounts off negotiated prices of applicable brand drugs and biologics to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs or biologics to be covered under Medicare Part D.
50
In March 2010, President Obama signed into lawSince its enactment, there have been judicial, executive and Congressional challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Prior to the Supreme Court’s decision, President Biden issued an executive order to initiate a sweeping law intendedspecial enrollment period from February 15, 2021 through August 15, 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. The executive order also instructed certain governmental agencies to broadenreview and reconsider their existing policies and rules that limit access to healthcare, including among others, re-examining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance reducecoverage through Medicaid or constrain the growthACA. It is unclear how other healthcare reform measures of healthcare spending, enhance remedies against fraudthe Biden administration or other efforts, if any, to challenge, repeal or replace the ACA will impact the ACA or Kineta’s business.
74
In addition, other legislative changes have been proposed and abuse, add new transparency requirementsadopted since the ACA was enacted. On August 2, 2011, the Budget Control Act of 2011 was signed into law, which, among other things, resulted in reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2030, with the exception of a temporary suspension from May 1, 2020 through December 31, 2021, unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the healthcare and health insurance industries, impose new taxes and fees on the health industry, and impose additional health policy reforms.government to recover overpayments to providers from three to five years.
ThereFurther, there has been heightened governmental scrutiny recently overin the mannerUnited States of pharmaceutical pricing practices in which drug manufacturers set prices for their marketed products, whichlight of the rising cost of prescription drugs. Such scrutiny has resulted in several Congressionalrecent congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. However, noneAlthough a number of these executive orders make immediate policy changes. For policies contained withinand other measures may require additional authorization to become effective, Congress and the executive orderscurrent U.S. administration have each indicated that it will continue to have any effect, agencies would needseek new legislative and/or administrative measures to take additional administrative action. How these executive orders will be implementedcontrol drug costs. Any reduction in reimbursement from Medicare and their impact on the industry remain uncertain. Depending on the details of further administrative actions, some of these proposals could have significant impacts for drug manufacturers and providers.other government programs may result in a similar reduction in payments from private payors.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. We cannot be sure whether additional legislative changesLegally mandated price controls on payment amounts by third-party payors or other restrictions could harm Kineta’s business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be enacted, or whetherincluded in their prescription drug and other healthcare programs. This could reduce the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of ourultimate demand for Kineta’s product candidates, if any, may be. In addition, increased scrutiny byapproved, or put pressure on Kineta’s product pricing, which could negatively affect Kineta’s business, results of operations, financial condition and prospects.
Kineta expects that the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent labeling and post-marketing testingACA, these new laws, and other requirements. It is likely that federal and state legislatures within the United States and foreign governments will continue to consider changes to existing healthcare legislation. We cannot predict the reform initiativesmeasures that may be adopted in the future may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, new payment methodologies and additional downward pressure on the price that Kineta receives for any approved product. The implementation of cost containment measures or other healthcare reforms may prevent Kineta from being able to generate revenue, attain profitability or commercialize its product candidates, if approved.
Current and future legislative efforts may limit the prices for Kineta’s products, if and when they are licensed for marketing, and that could materially impact Kineta’s ability to generate revenues.
The prices of prescription pharmaceuticals have also been the subject of considerable discussion in the United States. There have been several recent U.S. congressional inquiries, as well as proposed and enacted state and federal legislation designed to, among other things, bring more transparency to pharmaceutical pricing, review the relationship between pricing and manufacturer patient programs and reduce the costs of pharmaceuticals under Medicare and Medicaid. In 2020, President Trump issued several executive orders intended to lower the costs of prescription products and certain provisions in these orders have been incorporated into regulations. These regulations include an interim final rule implementing a most favored nation model for prices that would tie Medicare Part B payments for certain physician-administered pharmaceuticals to the lowest price paid in other economically advanced countries, effective January 1, 2021. That rule, however, has been subject to a nationwide preliminary injunction and, on December 29, 2021, CMS issued a final rule to rescind it. With issuance of this rule, CMS stated that it will explore all options to incorporate value into payments for Medicare Part B pharmaceuticals and improve beneficiaries’ access to evidence-based care.
In addition, in October 2020, HHS and the FDA published a final rule allowing states and other entities to develop a Section 804 Importation Program (“SIP”) to import certain prescription drugs from Canada into the United States. The final rule is currently the subject of ongoing litigation, but at least six states (Vermont, Colorado, Florida, Maine, New Mexico and New Hampshire) have passed laws allowing for the importation of drugs from Canada with the intent of developing SIPs for review and approval by the FDA. Further, on November 20, 2020, HHS finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The final rule would eliminate the current safe harbor for Medicare drug rebates and create new safe harbors for beneficiary point-of-sale discounts and pharmacy benefit manager service fees. It originally was set to go into effect on January 1, 2023, but with passage of the Inflation Reduction Act has been delayed by Congress to January 1, 2032.
On July 9, 2021, President Biden signed Executive Order 14063, which focuses on, among other things, the price of pharmaceuticals. The Order directs HHS to create a plan within 45 days to combat “excessive pricing of prescription pharmaceuticals and enhance domestic pharmaceutical supply chains, to reduce the prices paid by the federal government for such pharmaceuticals, and to address the recurrent problem of price gouging.” On September 9, 2021, HHS released its plan to reduce pharmaceutical prices. The key features of that plan are to: (a) make pharmaceutical prices more affordable and equitable for all consumers and throughout the health care system by supporting pharmaceutical price negotiations with manufacturers; (b) improve and promote competition throughout the prescription pharmaceutical industry by supporting market changes that strengthen supply chains, promote biosimilars and generic drugs, and increase transparency; and (c) foster scientific innovation to promote better healthcare and improve health by supporting public and private research and making sure that market incentives promote discovery of valuable and accessible new treatments.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional healthcare organizations and individual hospitals are increasingly using bidding procedures to determine what
75
pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. These measures could reduce the ultimate demand for Kineta’s products, once approved, or put pressure on Kineta’s product pricing. Kineta expects that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for Kineta’s product candidates or additional pricing pressures.
Finally, outside the United States, in some nations, including those of the EU, the pricing of prescription pharmaceuticals is subject to governmental control and access. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, Kineta or its collaborators may be required to conduct a clinical trial that compares the cost-effectiveness of Kineta’s product to other available therapies. If reimbursement of Kineta’s products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, Kineta’s business could be materially harmed.
Kineta is subject to a variety of privacy and data security laws, and Kineta’s failure to comply with them could harm Kineta’s business.
Kineta maintains a large quantity of sensitive information, including confidential business and personal information in connection with the conduct of Kineta’s clinical trials and related to Kineta’s employees, and Kineta is subject to laws and regulations governing the privacy and security of such information. In the United States, there are numerous federal and state privacy and data security laws and regulations governing the collection, use, disclosure and protection of personal information, including federal and state health information privacy laws, federal and state security breach notification laws, and federal and state consumer protection laws. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues, including with respect to regulatory enforcement and private litigation, which may affect Kineta’s business and is expected to increase its compliance costs and exposure to liability. In the United States, numerous federal and state laws and regulations could apply to Kineta’s operations or the operations of Kineta’s partners, including state data breach notification laws, state health information privacy laws and federal and state consumer protection laws and regulations (e.g., Section 5 of the FTC Act), that govern the collection, use, disclosure and protection of health-related and other personal information. In addition, Kineta may obtain health information from third parties (including research institutions from which Kineta obtains clinical trial data) that are subject to privacy and security requirements under HIPAA, as amended by HITECH and regulations promulgated thereunder. Depending on the facts and circumstances, Kineta could be subject to significant penalties if Kineta obtains, uses or discloses, or is subject to an actual or alleged data breach regarding, individually identifiable health information in a manner that is not authorized or permitted by HIPAA.
In the EEA, Kineta is subject to the EU General Data Protection Regulation (the “EU GDPR”), which took effect in May 2018. The EU GDPR governs the collection, use, disclosure, transfer or other processing of personal data (i.e., data which identifies an individual or from which an individual is identifiable), including clinical trial data, and grants individuals various data protection rights (e.g., the right to erasure of personal data). The EU GDPR imposes a number of obligations on companies, including inter alia: (i) accountability and transparency requirements, and enhanced requirements for obtaining valid consent; (ii) obligations to consider data protection as any new products or services are developed and to limit the amount of personal data processed; and (iii) obligations to implement appropriate technical and organizational measures to safeguard personal data and to report certain personal data breaches to the supervisory authority without undue delay (and no later than 72 hours where feasible). In addition, the EU GDPR prohibits the transfer of personal data from the EEA to the United States and other jurisdictions that the European Commission does not recognize as having “adequate” data protection laws unless a data transfer mechanism has been put in place. In July 2020, the Court of Justice of the EU (the “CJEU”) limited how organizations could lawfully transfer personal data from the EEA to the United States by invalidating the EU-US Privacy Shield for purposes of international transfers and imposing further restrictions on use of the standard contractual clauses (the “SCCs”), including a requirement for companies to carry out a transfer privacy impact assessment, which, among other things, assesses laws governing access to personal data in the recipient country and considers whether initiativessupplementary measures that provide privacy protections additional to those provided under SCCs will need to be implemented to ensure an essentially equivalent level of data protection to that afforded in the EEA. The European Commission subsequently issued new SCCs in June 2021 to account for the decision of the CJEU and recommendations made by the European Data Protection Board and which are in turn relatively more onerous. The EU GDPR imposes substantial fines for breaches and violations (up to the greater of €20 million or 4% of consolidated annual worldwide gross revenue), and confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies and obtain compensation for damages resulting from violations of the EU GDPR. Relatedly, following the United Kingdom’s withdrawal from the EU (i.e., Brexit), and the expiry of the Brexit transition period, which ended on December 31, 2020, the EU GDPR has been implemented in the United Kingdom (as the “UK GDPR”). The UK GDPR sits alongside the UK Data Protection Act 2018 which implements certain derogations in the EU GDPR into UK law. Under the UK GDPR, companies not established in the UK but who process personal data in relation to the offering of goods or services to individuals in the UK, or to monitor their behavior will be subject to the UK GDPR, the requirements of which are (at this time) largely aligned with those under the EU GDPR and as such, may lead to similar compliance and operational costs with potential fines of up to the greater of £17.5 million or 4% of global turnover.
Compliance with these and any other applicable privacy and data security laws and regulations is a rigorous and time-intensive process, and Kineta may be required to put in place additional mechanisms ensuring compliance with the new data protection rules. Furthermore, the laws are not consistent, and compliance in the event of a widespread data breach is costly. In addition, states are constantly adopting new laws or amending existing laws, requiring attention to frequently changing regulatory requirements. For example, California enacted the California Consumer Privacy Act (the “CCPA”), which took effect on January 1, 2020, became enforceable by the California Attorney General on July 1, 2020, and has been dubbed the first “GDPR-like” law in the United States. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing and receive detailed information about how their personal information is used by requiring covered companies to provide new disclosures to California consumers (as that term is broadly defined) and provide such consumers new ways to opt-out of certain sales of personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. Further, the California Privacy Rights Act (the “CPRA”) recently passed in California. The CPRA will impose additional data protection obligations on companies doing business in California, including additional consumer rights processes, limitations on data uses, new audit requirements for higher risk data and opt outs for certain uses of sensitive data. It will also create a new California data protection agency authorized to issue substantive regulations and could result in increased privacy and information security
76
enforcement. The majority of the provisions will go into effect on January 1, 2023, and additional compliance investment and potential business process changes may be required. Although the CCPA currently exempts certain health-related information, including clinical trial data, the CCPA and the CPRA may increase Kineta’s compliance costs and potential liability. Similar laws have been adopted will be repealedin other states (for example Nevada, Virginia and Colorado) or modified. The continuing effortsproposed in other states and at the federal level, and if passed, such laws may have potentially conflicting requirements that would make compliance challenging.
Any actual or perceived failure by Kineta to comply with applicable privacy and data security laws and regulations could result in regulatory investigations, reputational damage, orders to cease/change Kineta’s processing of the government, insurance companies, managed care organizations,its data, enforcement notices and/or assessment notices (for a compulsory audit). Kineta may also face civil claims including representative actions and other healthcare payorsclass action type litigation (where individuals have suffered harm), potentially amounting to significant compensation or damages liabilities, as well as associated costs, diversion of to contain or reduce costs of healthcare may adversely affect the demand for any product candidates for which we may obtain regulatory approval, our ability to set a price that we believe is fair for our products, our ability to obtain coverageinternal resources and reimbursement approval for a product, our ability to generate revenue and achieve or maintain profitability; and the level of taxes that we are required to pay.reputational harm.
OurAny future growthacquisitions, in-licensing or strategic partnerships may depend, in part, on our abilityincrease Kineta’s capital requirements, dilute Kineta’s stockholders, divert Kineta’s management’s attention, cause Kineta to commercialize our product candidates in foreign markets, where we would beincur debt or assume contingent liabilities and subject Kineta to additional regulatory burdens and other risks and uncertainties.risks.
OurKineta may engage in various acquisitions and strategic partnerships in the future, profitabilityincluding licensing or acquiring complementary products, intellectual property rights, technologies or businesses. Any acquisition or strategic partnership may depend, in part, on our ability to commercialize our product candidates in foreign markets for which we may rely on collaboration with third parties. If we commercialize our product candidates in foreign markets, we would be subject to additionalentail numerous risks, and uncertainties, including:
In addition, if Kineta undertakes such a transaction, Kineta may incur large one-time expenses and acquire intangible assets that could result in significant future amortization expense.
Risks Related to Intellectual Property
If Kineta is unable to obtain and maintain sufficient intellectual property protection for its platform technologies and product candidates, or if the scope of the intellectual property protection is not sufficiently broad, Kineta’s competitors could develop and commercialize products similar or identical to Kineta’s, and Kineta’s ability to successfully commercialize its products may be adversely affected.
Kineta relies upon a combination of patents, know-how and confidentiality agreements to protect the intellectual property related to Kineta’s products and technologies and to prevent third parties from copying and surpassing Kineta’s achievements, thus eroding Kineta’s competitive position in Kineta’s market.
Kineta’s success depends in large part on its ability to obtain and maintain patent protection, know-how and trade secrets for its development platform, product candidates and their uses, as well as Kineta’s ability to operate without infringing the proprietary rights of others. Kineta seeks to protect its proprietary position by filing patent applications in the United States and abroad related to Kineta’s novel discoveries and technologies that are important to Kineta’s business. Kineta cannot guarantee that its pending and future patent applications will result in patents being issued or that issued patents will afford sufficient protection of Kineta’s product candidates or their intended uses against competitors, nor can there be any assurance that the patents issued will not be infringed, designed around, invalidated by third parties, or will effectively prevent others from commercializing competitive technologies, products or product candidates.
Obtaining and enforcing patents is expensive and time-consuming, and Kineta may not be able to file and prosecute all necessary or desirable patent applications or maintain and/or enforce patents that may issue based on Kineta’s patent applications, at a reasonable cost or in a timely manner, including delays as a result of pandemics or other health crises impacting Kineta’s or its licensors’ operations. It is also possible that Kineta will fail to identify patentable aspects of Kineta’s research and development results before it is too late to obtain patent protection. Although Kineta enters into non-disclosure and confidentiality agreements with parties who have access to patentable aspects of Kineta’s research and development output, such as Kineta’s employees, corporate collaborators, outside scientific collaborators, contract research organizations, contract manufacturers, consultants, advisors and other third parties, any of these parties may breach these agreements and disclose such results before a patent application is
77
filed, thereby jeopardizing Kineta’s ability to seek patent protection.
Composition of matter patents for biological and pharmaceutical product candidates often provides a strong form of intellectual property protection for those types of products, as such patents provide protection without regard to any method of use. However, Kineta cannot be certain that the claims in its pending patent applications directed to composition of matter of Kineta’s product candidates will be considered patentable by the United States Patent and Trademark Office (the “USPTO”) or by patent offices in foreign countries, or that the claims in any of Kineta’s issued patents will be considered valid and enforceable by courts in the United States or foreign countries. Method of use patents protect the use of a product for the specified method. This type of patent does not prevent a competitor from making and marketing a product that is identical to Kineta’s product for an indication that is outside the scope of the patented method. Moreover, even if competitors do not actively promote their product for Kineta’s targeted indications, physicians may prescribe these products “off-label.” Although off-label prescriptions may infringe or contribute to the infringement of method of use patents, the practice is common and such infringement is difficult to prevent or prosecute.
The patent position of biotechnology and biopharmaceutical companies generally is highly uncertain, involves complex legal, scientific and factual questions and has in recent years been the subject of much litigation, resulting in court decisions, including Supreme Court decisions, which have increased uncertainties as to the ability to enforce patent rights in the future. The standards that the USPTO and its foreign counterparts use to grant patents are not always applied predictably or uniformly. In addition, the laws of foreign countries may not protect Kineta’s rights to the same extent as the laws of the United States, or vice versa.
The patent application process is subject to numerous risks and uncertainties, and there can be no assurance that Kineta or any of its potential future collaborators will be successful in protecting Kineta’s product candidates by obtaining and defending patents. For example, Kineta may not be aware of all third-party intellectual property rights potentially relevantrelating to Kineta’s product candidates or their intended uses, and as a result the impact of such third-party intellectual property rights upon the patentability of Kineta’s own patents and patent applications, as well as the impact of such third-party intellectual property upon Kineta’s freedom to operate, is highly uncertain. Patent applications in the United States and other jurisdictions are typically not published until 18 months after filing or, in some cases, not at all. Therefore, Kineta cannot know with certainty whether Kineta was the first to make the inventions claimed in its patents or pending patent applications, or that Kineta was the first to file for patent protection of such inventions. As a result, the issuance, inventorship, scope, validity, enforceability and commercial value of Kineta’s patent rights are highly uncertain. Kineta’s pending patent applications may be challenged in patent offices in the United States and abroad. The issuance of a patent is not conclusive as to its inventorship, ownership, scope, validity or enforceability. Even issued patents may later be found invalid or unenforceable or may be modified or revoked in proceedings instituted by third parties before various patent offices or in courts. For example, Kineta’s pending patent applications may be subject to third-party pre-issuance submissions of prior art to the USPTO or Kineta’s issued patents may be subject to post-grant review proceedings, oppositions, derivations, reexaminations, interference or inter partes review proceedings, in the United States or elsewhere, challenging Kineta’s patent rights or the patent rights of others. An adverse determination in any such challenges may result in loss of exclusivity or in patent claims being narrowed, invalidated, or held unenforceable, in whole or in part, which could limit Kineta’s ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of Kineta’s technology and products. In addition, given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. The degree of future protection for Kineta’s proprietary rights is uncertain. Only limited protection may be available and may not adequately protect Kineta’s rights or permit Kineta to gain or keep any competitive advantage. Any failure to obtain or maintain patent protection with respect to Kineta’s product candidates or their uses could have a material adverse effect on Kineta’s business, financial condition, results of operations and prospects.
In addition to the protection afforded by patents, Kineta relies on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of Kineta’s discovery and development processes that involve proprietary know-how, information or technology that is not covered by patents. Kineta may also rely on trade secret protection as temporary protection for concepts that may be included in a future patent filing. However, trade secret protection will not protect Kineta from innovations that a competitor develops independently of Kineta’s proprietary know how. If a competitor independently develops a technology that Kineta protects as a trade secret and files a patent application on that technology, then Kineta may not be able to patent that technology in the future, may require a license from the competitor to use Kineta’s own know-how, and if the license is not available on commercially viable terms, then Kineta may not be able to launch its product. Although Kineta requires all of its employees to assign their inventions to Kineta, and requires all of its employees, consultants, advisors and any third parties who have access to Kineta’s proprietary know-how, information or technology to enter into confidentiality agreements, Kineta cannot be certain that its trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to Kineta’s trade secrets or independently develop substantially equivalent information and techniques. Furthermore, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, Kineta may encounter significant problems in protecting and defending its intellectual property both in the United States and abroad. If Kineta is unable to prevent unauthorized material disclosure of its intellectual property to third parties, Kineta will not be able to establish or maintain a competitive advantage in Kineta’s market, and this scenario could materially adversely affect Kineta’s business, financial condition and results of operations.
Intellectual property rights do not necessarily address all potential threats to Kineta’s competitive advantage.
The degree of future protection afforded by Kineta’s intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect Kineta’s business or permit Kineta to maintain its competitive advantage. For example:
78
Should any of these or similar events occur, they could significantly harm Kineta’s business, results of operations and prospects.
If Kineta fails to comply with its obligations imposed by any intellectual property licenses with third parties that Kineta may need in the 79 agreement, which would affect Kineta’s patent rights worldwide. Termination of Kineta’s current or any future license agreements would reduce or eliminate Kineta’s rights under these agreements and may result in Kineta having to negotiate new or reinstated agreements with less favorable terms or cause Kineta to lose its rights under these agreements, including Kineta’s rights to important intellectual property or technology. Any of the foregoing could prevent Kineta from commercializing its other product candidates, which could have a material adverse effect on Kineta’s operating results and overall financial condition. In addition, intellectual property rights that Kineta may in-license in the future may be sublicenses under intellectual property owned by third parties, in some cases through multiple tiers. The actions of Kineta’s licensors may therefore affect Kineta’s rights to use its sublicensed intellectual property, even if Kineta is in compliance with all of the obligations under its license agreements. Should Kineta’s licensors or any of the upstream licensors fail to comply with their obligations under the agreements pursuant to which they obtain the rights that are sublicensed to Kineta, or should such agreements be terminated or amended, Kineta’s ability to develop and commercialize its product candidates may be materially harmed. Licensing of intellectual property is of critical importance to Kineta’s business and involves complex legal, business and scientific issues. If Kineta breaches its in-license agreements or any of the other agreements under which Kineta acquired, or will acquire, intellectual property rights covering Kineta’s product candidates, Kineta could lose the ability to continue the development and commercialization of the related product. The licensing of intellectual property is of critical importance to Kineta’s business and to Kineta’s current and future product candidates, and Kineta expects to enter into additional such agreements in the future. In particular, certain rights to the intellectual property covering Kineta’s product candidates are in-licensed from third parties. Kineta may acquire the rights to the intellectual property covering future product candidates from other third-party licensors. If Kineta fails to meet its obligations under any of its in-license agreements, then the licensor may terminate the license agreement. If one of Kineta’s material in-license agreements is terminated, Kineta will lose the right to continue to develop and commercialize the product candidate(s) covered by such in-license agreement. While Kineta would expect to exercise all rights and remedies available to it, including seeking to cure any breach by Kineta, and otherwise seek to preserve Kineta’s rights under its in-license agreements, Kineta may not be able to do so in a timely manner, at an acceptable cost or at all. In the future, Kineta may need to obtain additional licenses of third-party technology that may not be available to it or are available only on commercially unreasonable terms, and which may cause Kineta to operate its business in a more costly or otherwise adverse manner that was not anticipated. Kineta currently owns or has the exclusive or non-exclusive rights to intellectual property directed to Kineta’s product candidates and other proprietary technologies, including Kineta’s development platform. Other pharmaceutical companies and academic institutions may also have filed or are planning to file patent applications potentially relevant to Kineta’s business. From time to time, in order to avoid infringing these third-party patents, Kineta may be required to license technology from additional third parties to further develop or commercialize Kineta’s product candidates. Should Kineta be required to obtain licenses to any third-party technology, including any such patents required to manufacture, use or sell Kineta’s product candidates, such licenses may not be available to Kineta on commercially reasonable terms, or at all. The inability to obtain any third-party license required to develop or commercialize any of Kineta’s product candidates could cause Kineta to abandon any related efforts, which could seriously harm Kineta’s business and operations. The licensing or acquisition of third-party intellectual property rights is a competitive area, and several more established companies may pursue strategies to license or acquire third-party intellectual property rights Kineta may consider attractive or necessary. These established companies may have a competitive advantage over Kineta due to their size, capital resources and greater clinical development and commercialization capabilities. In addition, companies that perceive Kineta to be a competitor may be unwilling to assign or license rights to Kineta. Even if Kineta is able to obtain a license under such intellectual property rights, any such license may be non-exclusive, which may allow Kineta’s competitors to access the same technologies licensed to Kineta. Moreover, some of Kineta’s owned and in-licensed patents or patent applications or future patents may be co-owned with third parties. If Kineta is unable to obtain an exclusive license to any such third-party co-owners’ interest in such patents or patent applications, such co-owners may be able to license their rights to other third parties, including Kineta’s competitors, and Kineta’s competitors could market competing products and technology. In addition, Kineta may need the cooperation of any such co-owners of Kineta’s patents in order to enforce such patents against third parties, and such cooperation may not be provided to Kineta. Furthermore, Kineta’s owned and in-licensed patents may be subject to a reservation of rights by one or more third parties. Any of the foregoing could have a material adverse effect on Kineta’s competitive position, business, financial conditions, results of operations and prospects. If Kineta is sued for infringing intellectual property rights of third parties, such litigation could be costly and time consuming and could prevent or delay Kineta from developing or commercializing its product candidates. 80 unpredictable and generally expensive and time consuming and, even if resolved in Kineta’s favor, is likely to divert significant resources from Kineta’s core business, including distracting Kineta’s technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of Kineta’s common stock. Such litigation or proceedings could substantially increase Kineta’s operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. Kineta may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of Kineta’s competitors may be able to sustain the costs of such litigation or proceedings more effectively than Kineta can because of their greater financial resources and more mature and developed intellectual property portfolios. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on Kineta’s ability to compete in the marketplace. In addition, patent holding companies that focus solely on extracting royalties and settlements by enforcing patent rights may target Kineta. There is a substantial amount of intellectual property litigation in the biotechnology and pharmaceutical industries, and Kineta may become party to, or threatened with, litigation or other adversarial proceedings regarding intellectual property rights with respect to Kineta’s product candidates. Kineta cannot be certain that its product candidates and other proprietary technologies it may develop will not infringe existing or future patents owned by third parties. Third parties may assert infringement claims against Kineta based on existing or future intellectual property rights. If Kineta is found to infringe a third party’s intellectual property rights, Kineta could be forced, including by court order, to cease developing, manufacturing or commercializing the infringing candidate product or product. Alternatively, Kineta may be required to obtain a license from such third party in order to use the infringing technology and continue developing, manufacturing or marketing the infringing candidate product or product. However, Kineta may not be able to obtain any required license on commercially reasonable terms or at all. Even if Kineta were able to obtain a license, it could be non-exclusive, thereby giving Kineta’s competitors access to the same technologies licensed to Kineta. In addition, Kineta could be found liable for monetary damages, including treble damages and attorneys’ fees if Kineta is found to have willfully infringed a patent. A finding of infringement could prevent Kineta from commercializing its investigational products or force Kineta to cease some of its business operations, which could materially harm Kineta’s business. Claims that Kineta has misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on Kineta’s business. Kineta cannot guarantee that any of its or its licensors’ patent searches or analyses, including but not limited to the identification of relevant patents, analysis of the scope of relevant patent claims or determination of the expiration of relevant patents, are complete or thorough. Kineta may not be aware of patents that have already been issued and that a third party, for example, a competitor in the fields in which Kineta is developing its product candidates, might assert are infringed by Kineta’s current or future product candidates, including claims to compositions, formulations, methods of manufacture or methods of use or treatment that cover Kineta’s product candidates. It is also possible that patents owned by third parties of which Kineta is aware, but which Kineta does not believe are relevant to Kineta’s product candidates and other proprietary technologies Kineta may develop, could be found to be infringed by Kineta’s product candidate. In addition, because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that Kineta’s product candidates may infringe. Additionally, pending patent applications that have been published can, subject to certain limitations, be later amended in a manner that could cover Kineta’s product candidates or the use of Kineta’s product candidates. Kineta’s determination of the expiration date of any patent in the United States, Europe or elsewhere that Kineta considers relevant may be incorrect, which may negatively impact Kineta’s ability to develop and market its product candidates. Kineta’s competitors in both the United States and abroad, many of which have substantially greater resources and have made substantial investments in patent portfolios and competing technologies, may have applied for or obtained or may in the future apply for and obtain, patents that will prevent, limit or otherwise interfere with Kineta’s ability to make, use and sell Kineta’s product candidates. The pharmaceutical and biotechnology industries have produced a considerable number of patents, and it may not always be clear to industry participants, including Kineta, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If Kineta were sued for patent infringement, it would need to demonstrate that its product candidates, products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid or unenforceable, and Kineta may not be able to do this. Proving invalidity may be difficult. For example, in the United States, proving invalidity in court requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents, and there is no assurance that a court of competent jurisdiction would invalidate the claims of any such U.S. patent. Even if Kineta were successful in these proceedings, it may incur substantial costs and the time and attention of its management and scientific personnel could be diverted in pursuing these proceedings, which could have a material adverse effect on Kineta’s business and operations. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of Kineta’s confidential information could be compromised by disclosure during litigation. In addition, Kineta may not have sufficient resources to bring these actions to a successful conclusion. Kineta may choose to challenge the enforceability or validity of claims in a third party’s U.S. patent by requesting that the USPTO review the patent claims in an ex-parte re-exam, inter partes review or post-grant review proceedings. These proceedings are expensive and may consume Kineta’s time or other resources. Kineta may choose to challenge a third party’s patent in patent opposition proceedings in the European Patent Office (the “EPO”), or other foreign patent office. The costs of these opposition proceedings could be substantial, and may consume Kineta’s time or other resources. If Kineta fails to obtain a favorable result at the USPTO, the EPO or other patent office then Kineta may be exposed to litigation by a third party alleging that the patent may be infringed by Kineta’s product candidates or proprietary technologies. Kineta may become involved in lawsuits to protect or enforce its patents or other intellectual property, which could be expensive, time consuming and unsuccessful. Competitors or other third parties may infringe Kineta’s patents, trademarks or other intellectual property. To counter infringement or unauthorized use, Kineta may be required to file infringement claims, which can be expensive and time consuming and divert the time and attention of Kineta’s 81 management and scientific personnel. Kineta’s pending patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications. Any claims Kineta asserts against perceived infringers could provoke these parties to assert counterclaims against Kineta alleging that Kineta infringes their patents, in addition to counterclaims asserting that Kineta’s patents are invalid or unenforceable, or both. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, non-enablement or insufficient written description. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO or made a misleading statement during prosecution. The outcome following legal assertions of invalidity and unenforceability is unpredictable. In any patent infringement proceeding, there is a risk that a court will decide that a patent of Kineta’s is invalid or unenforceable, in whole or in part, and that Kineta does not have the right to stop the other party from using the invention at issue. There is also a risk that, even if the validity of such patents is upheld, the court will construe the patent’s claims narrowly or decide that Kineta does not have the right to stop the other party from using the invention at issue on the grounds that Kineta’s patent claims do not cover the invention, or decide that the other party’s use of Kineta’s patented technology falls under the safe harbor to patent infringement under 35 U.S.C. §271(e)(1). An adverse outcome in a litigation or proceeding involving Kineta’s patents could limit Kineta’s ability to assert its patents against those parties or other competitors and may curtail or preclude Kineta’s ability to exclude third parties from making and selling similar or competitive products. Any of these occurrences could adversely affect Kineta’s competitive business position, business prospects and financial condition. Similarly, if Kineta asserts trademark infringement claims, a court may determine that the marks Kineta has asserted are invalid or unenforceable, or that the party against whom Kineta has asserted trademark infringement has superior rights to the marks in question. In this case, Kineta could ultimately be forced to cease use of such trademarks. Even if Kineta establishes infringement, the court may decide not to grant an injunction against further infringing activity and instead award only monetary damages, which may or may not be an adequate remedy. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of Kineta’s confidential information could be compromised by disclosure during litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of shares of Kineta’s common stock. Moreover, Kineta cannot assure you that it will have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are concluded. Even if Kineta ultimately prevails in such claims, the monetary cost of such litigation and the diversion of the attention of Kineta’s management and scientific personnel could outweigh any benefit Kineta receives as a result of the proceedings. Because of the expense and uncertainty of litigation, Kineta may not be in a position to enforce its intellectual property rights against third parties. Because of the expense and uncertainty of litigation, Kineta may conclude that even if a third party is infringing Kineta’s issued patent, any patents that may be issued as a result of Kineta’s pending or future patent applications or other intellectual property rights, the risk-adjusted cost of bringing and enforcing such a claim or action may be too high or not in the best interest of Kineta or its stockholders. In such cases, Kineta may decide that the more prudent course of action is to simply monitor the situation or initiate or seek some other non-litigious action or solution. Kineta may be subject to claims that its employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties. Kineta employs and may employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including Kineta’s competitors or potential competitors. Although Kineta tries to ensure that its employees, consultants Changes in patent law in the United States and other jurisdictions could diminish the value of patents in general, thereby impairing Kineta’s ability to protect its product candidates. As is the After March 2013, under the Leahy-Smith Act, the United States transitioned to a first inventor to file system in which, assuming that the other statutory requirements are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third-party was the first to invent the claimed invention. A third party that files a patent application in the In addition, the patent positions of companies in the development and commercialization of biologics and pharmaceuticals are particularly uncertain. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Depending on future actions by the U.S. Congress, the U.S. courts, the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken Kineta’s ability to obtain new patents or to enforce Kineta’s existing patents and patents that Kineta might obtain in the future. For example, in the 2013 case Assoc. for Molecular Pathology v. Myriad Genetics, Inc., the U.S. Supreme Court held that certain claims to DNA molecules are not patentable. While Kineta does not believe that any of the patents owned or licensed by it will be found invalid based on this decision, Kineta cannot predict how future decisions by the courts, the U.S. Congress or the USPTO may Obtaining and Periodic maintenance fees, renewal fees, annuities fees and various other governmental fees on patents and/or patent applications are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent and/or patent application. The USPTO and various foreign governmental patent agencies also require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse, including due to the effect of a pandemic or other public health crises on Kineta or its patent maintenance vendors, can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If Kineta fails to maintain the patents and patent applications covering its product candidates, Kineta’s competitive position would be adversely affected. Patent terms may be inadequate to protect Kineta’s competitive position on its product candidates for an adequate amount of time. The term of any individual patent depends on applicable law in the country where the patent is granted. In the United States, provided all maintenance fees are timely paid, a patent generally has a term of 20 years from its application filing date or earliest claimed non-provisional filing date. Extensions may be available under certain circumstances, but the life of a patent and, correspondingly, the protection it affords is limited. Even 83 if Kineta or its licensors obtain patents covering Kineta’s product candidates, when the terms of all patents covering a product expire, Kineta’s business If Kineta does not obtain patent term extension in the United States under the Drug Price Competition and Patent Term Restoration Act of 1984 (the “Hatch-Waxman Act”) and in foreign countries under similar legislation, thereby potentially extending the term of marketing exclusivity for its product candidates, Kineta’s business may be harmed. In the United States, a patent that covers an FDA-approved drug or biologic may be eligible for a term extension designed to restore the period of the patent term that is lost during the premarket regulatory review process conducted by the FDA. Depending upon the timing, duration and conditions of FDA marketing approval of Kineta’s product candidates, one or more of Kineta’s U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Act, which permits a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only claims covering such approved drug product, a method for using it or a method for manufacturing it may be extended. In Europe, Kineta’s product candidates may be eligible for term extensions based on similar legislation. In either jurisdiction, however, Kineta may not receive an extension if it fails to apply within applicable deadlines, fails to apply prior to expiration of relevant patents or otherwise fails to satisfy applicable requirements. Even if Kineta is granted such extension, the duration of such extension may be less than Kineta’s request. If Kineta is unable to obtain a patent term extension, or if the term of any such extension is less than Kineta’s request, the period during which Kineta can enforce its patent rights for that product will be in effect shortened and Kineta’s competitors may obtain approval to market competing products sooner. The resulting reduction of years of revenue from applicable products could be substantial. Kineta enjoys only limited geographical protection with respect to certain patents and Kineta may not be able to protect its intellectual property rights throughout the world. Filing, prosecuting and defending patents covering Kineta’s product candidates in all countries throughout the world would be prohibitively expensive, and even in countries where Kineta has sought protection for its intellectual property, such protection can be less extensive than those in the United States. The requirements for patentability may differ in certain countries, particularly developing countries, and the breadth of patent claims allowed can be inconsistent. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. In-licensing patents covering Kineta’s product candidates in all countries throughout the world may similarly be prohibitively expensive, if such opportunities are available at all. And in-licensing or filing, prosecuting and defending patents even in only those jurisdictions in which Kineta develops or commercializes its product candidates may be prohibitively expensive or impractical. Competitors may use Kineta’s and its licensors’ technologies in jurisdictions where Kineta has not obtained patent protection or licensed patents to develop their own products and, further, may export otherwise infringing products to territories where Kineta and its licensors have patent protection, but where enforcement is not as strong as that in the United States or Europe. These products may compete with Kineta’s product candidates, and Kineta or its licensors’ patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws or regulations in the United States and Europe, and many companies have encountered significant difficulties in protecting and defending proprietary rights in such jurisdictions. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets or other forms of intellectual property, particularly those relating to biotechnology products, which could make it difficult for Kineta to prevent competitors in some jurisdictions from marketing competing products in violation of Kineta’s proprietary rights generally. Proceedings to enforce Kineta’s patent rights in foreign jurisdictions, whether or not successful, are likely to result in substantial costs and divert Kineta’s efforts and attention from other aspects of its business, and additionally could put at risk Kineta’s or its licensors’ patents of being invalidated or interpreted narrowly, could increase the risk of Kineta’s or its licensors’ patent applications not issuing, or could provoke third parties to assert claims against Kineta. Kineta may not prevail in any lawsuits that it initiates, while damages or other remedies may be awarded to the adverse party, which may be commercially significant. If Kineta prevails, damages or other remedies awarded to Kineta, if any, may not be commercially meaningful. Accordingly, Kineta’s efforts to enforce its intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that Kineta develops or licenses. Furthermore, while Kineta intends to protect its intellectual property rights in its expected significant markets, Kineta cannot ensure that it will be able to initiate or maintain similar efforts in all jurisdictions in which Kineta may wish to market its product candidates. Accordingly, Kineta’s efforts to protect its intellectual property rights in such countries may be inadequate, which may have an adverse effect on Kineta’s ability to successfully commercialize its product candidates in all of its expected significant foreign markets. If Kineta or its licensors encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for Kineta’s business in such jurisdictions, the value of these rights may be diminished and Kineta may face additional competition in those jurisdictions. In some jurisdictions including European countries, compulsory licensing laws compel patent owners to grant licenses to third parties. In addition, some countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If Kineta or any of its licensors are forced to grant a license to third parties under patents relevant to Kineta’s business, or if Kineta or its licensors are prevented from enforcing patent rights against third parties, Kineta’s competitive position may be substantially impaired in such jurisdictions. 84 Kineta may rely on trade secret and proprietary know-how which can be difficult to trace and enforce and, if Kineta is unable to protect the confidentiality of its trade secrets, its business and competitive position would be harmed. In addition to seeking patents for some of its technology and current product candidates or any future product candidates, Kineta may also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain its competitive position. Elements of Kineta’s current product candidates or any future product candidates, including processes for their preparation and manufacture, as well as Kineta’s development platform, may involve proprietary know-how, information or technology that is not covered by patents, and thus for these aspects Kineta may consider trade secrets and know-how to be its primary intellectual property. Any disclosure, either intentional or unintentional, by Kineta’s employees, the Trade secrets and know-how can be difficult to protect. Kineta requires its employees to enter into written employment agreements containing provisions of confidentiality and obligations to assign to Kineta any inventions generated in the course of Kineta’s proprietary information, including Kineta’s trade secrets, and Kineta may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of Kineta’s trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, Kineta would have no right to prevent them from using that technology or information to compete with Kineta. If any of Kineta’s trade secrets were to be disclosed to or independently developed by a competitor or other third party, Kineta’s competitive position would be harmed. Kineta may become subject to claims challenging the inventorship or ownership of its patents and other intellectual property. Kineta may be subject to claims that former employees, collaborators or other third parties have an interest in Kineta’s patents or other intellectual property as an inventor or co-inventor. The failure to name the proper inventors on a patent application can result in the patents issuing thereon being unenforceable. Inventorship disputes may arise from conflicting views regarding the contributions of different individuals named as inventors, the effects of foreign laws where foreign nationals are involved in the development of the subject matter of the patent, conflicting obligations of third parties involved in developing Kineta’s product candidates or as a result of questions regarding co-ownership of potential joint inventions. Litigation may be necessary to resolve these and other claims challenging inventorship and/or ownership. Alternatively, or additionally, Kineta may enter into agreements to clarify the scope of its rights in such intellectual property. If Kineta fails in defending any such claims, in addition to paying monetary damages, Kineta may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on Kineta’s business. Even if Kineta is successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Kineta’s licensors may have relied on third-party consultants or collaborators or on funds from third parties, such as the U.S. government, such that Kineta’s licensors are not the sole and exclusive owners of the patents Kineta may in-license in the future. If other third parties have ownership rights or other rights to Kineta’s in-licensed patents, they may be able to license such patents to Kineta’s competitors, and Kineta’s competitors could market competing products and technology. This could have a material adverse effect on Kineta’s competitive position, business, financial conditions, results of operations and prospects. In addition, while it is Kineta’s policy to require its employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to Kineta, Kineta may be unsuccessful in executing such an agreement with each party who, in fact, conceives or develops intellectual property that Kineta regards as its own. The assignment of intellectual property rights may not be self-executing, or the assignment agreements may be breached, and Kineta may be forced to bring claims against third parties, or defend claims that they may bring against Kineta, to determine the ownership of what Kineta regards as its intellectual property. Such claims could have a material adverse effect on Kineta’s business, financial condition, results of operations and prospects. If Kineta’s trademarks and trade names are not adequately protected, then Kineta may not be able to build name recognition in Kineta’s markets of interest and its business may be adversely affected. Kineta’s current or future trademarks or trade names may be challenged, infringed, circumvented or declared generic or descriptive or determined to be infringing on other marks. Kineta may not be able to protect its rights to these trademarks and trade names or may be forced to stop using these names, which Kineta needs for name recognition by potential partners or customers in Kineta’s markets of interest. During trademark registration proceedings, Kineta may receive rejections of its applications by the USPTO or in other foreign jurisdictions. Although Kineta would be given an opportunity to respond to those rejections, Kineta may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications 85 and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against Kineta’s trademarks, and Kineta’s trademarks may not survive such proceedings. If Kineta is unable to establish name recognition based on its trademarks and trade names, Kineta may not be able to compete effectively and Kineta’s business may be adversely affected. Kineta may license its trademarks and trade names to third parties, such as distributors. Although these license agreements may provide guidelines for how Kineta’s trademarks and trade names may be used, a breach of these agreements or misuse of Kineta’s trademarks and trade names by Kineta’s licensees may jeopardize Kineta’s rights in or diminish the goodwill associated with Kineta’s trademarks and trade names. Moreover, any name Kineta has proposed to use with its product candidate in the United States must be approved by the FDA, regardless of whether Kineta has registered it, or applied to register it, as a trademark. Similar requirements exist in Europe. The FDA typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA (or an equivalent administrative body in a foreign jurisdiction) objects to any of Kineta’s proposed proprietary product names, Kineta may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA. Furthermore, in many countries, owning and maintaining a trademark registration may not provide an adequate defense against a subsequent infringement claim asserted by the owner of a senior trademark. At times, competitors or other third parties may adopt trade names or trademarks similar to Kineta’s, thereby impeding Kineta’s ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other registered trademarks or trademarks that incorporate variations of Kineta’s registered or unregistered trademarks or trade names. If Kineta asserts trademark infringement claims, a court may determine that the marks Kineta has asserted are invalid or unenforceable, or that the party against whom Kineta has asserted trademark infringement has superior rights to the marks in question. In this case, Kineta could ultimately be forced to cease use of such trademarks. Numerous factors may limit any potential competitive advantage provided by Kineta’s intellectual property rights. The degree of future protection afforded by Kineta’s intellectual property rights, whether owned or in-licensed, is uncertain because intellectual property rights have limitations and may not adequately protect Kineta’s business, provide a barrier to entry against Kineta’s competitors or potential competitors or permit Kineta to maintain its competitive advantage. Moreover, if a third party has intellectual property rights that cover the practice of Kineta’s technology, Kineta may not be able to fully exercise or extract value from Kineta’s intellectual property rights. The factors that may limit any potential competitive advantage provided by Kineta’s intellectual property rights include: Should any of these events occur, they could significantly harm Kineta’s business and results of operation. General Risk Factors Related to Kineta Kineta will incur significantly increased costs as a result of operating as a public company, and its management will be required to devote substantial time to new compliance initiatives. Kineta began operating as a public company as a result of the Merger. As a public company, Kineta will incur significant legal, accounting, compliance and other expenses that it did not incur as a private company. In addition, the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), as well as rules subsequently implemented by the SEC, and Nasdaq have imposed various requirements on public companies. In July 2010, the Dodd-Frank Wall Street Reform and Consumer Protection Act (the “Dodd-Frank Act”) was enacted. There are significant corporate governance and executive compensation related provisions in the Dodd-Frank Act that require the SEC to adopt additional rules and regulations in these areas such as “say on pay” and proxy access. Stockholder activism, the current political environment and the current high level of government intervention and 86 regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which Kineta operate its business in ways Kineta cannot currently anticipate. Kineta’s management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase Kineta’s legal and financial compliance costs and will make some activities more time-consuming and costlier. For example, Kineta expects these rules and regulations to make it more difficult and more expensive for Kineta to obtain director and officer liability insurance and Kineta may be required to incur substantial costs to maintain its current levels of such coverage. Failure to build Kineta’s finance infrastructure and improve its accounting systems and controls could impair Kineta’s ability to comply with the financial reporting and internal controls requirements for publicly traded companies. As a public company, Kineta operates in an increasingly demanding regulatory environment, which requires Kineta to comply with the Sarbanes-Oxley Act, the regulations of Nasdaq, the rules and regulations of the SEC, expanded disclosure requirements, accelerated reporting requirements and more complex accounting rules. Company responsibilities required by the Sarbanes-Oxley Act include establishing corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for Kineta to produce reliable financial reports and are important to help prevent financial fraud. Kineta performed testing of its internal controls over financial reporting for the year ended December 31, 2023, as required by Section 404 of the Sarbanes-Oxley Act. Management determined that as of December 31, 2023, Kineta’s internal controls over financial reporting were effective. However, due to the identification of a material weakness during quarterly review for the six months ended June 30, 3023 and for the audit for the year ended December 31, 2023, Kineta is not able to conclude that its internal controls over financial reporting was effective during the full year ended December 31, 2023. Further, in connection with the audit of Kineta’s financial statements for the years ended December 31, 2022 and 2021, Kineta and its independent registered public accounting firm identified material weaknesses in Kineta’s internal control over financial reporting. Kineta anticipates that the process of remediating the before mentioned material weaknesses in its internal control over financial reporting and building its accounting and financial functions and infrastructure will require significant additional professional fees, internal costs and management efforts. Kineta expects that it will need to implement a new internal system to combine and streamline the management of its financial, accounting, human resources and other functions. However, such a system would likely require Kineta to complete many processes and procedures for the effective use of the system or to run its business using the system, which may result in substantial costs. Any disruptions or difficulties in implementing or using such a system could adversely affect Kineta’s controls and harm Kineta’s business. Moreover, such disruption or difficulties could result in unanticipated costs and diversion of management attention. In addition, Kineta may discover weaknesses in its system of internal financial and accounting controls and procedures that could result in a material misstatement of Kineta’s financial statements. Kineta’s internal control over financial reporting will not prevent or detect all errors and all fraud. If Kineta is not able to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, or if Kineta is unable to maintain proper and effective internal controls, Kineta may not be able to produce timely and accurate financial statements. If Kineta cannot provide reliable financial reports or prevent fraud, its business and results of operations could be harmed, investors could lose confidence in Kineta’s reported financial information and Kineta could be subject to sanctions or investigations by Nasdaq, the SEC or other regulatory authorities. Kineta’s disclosure controls and procedures may not prevent or detect all errors or acts of fraud. Upon the completion of the Merger, Kineta became subject to the periodic reporting requirements of the Exchange Act. Kineta designed its disclosure controls and procedures to reasonably assure that information Kineta must disclose in reports it files or submits under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. Kineta believes that any disclosure controls and procedures or internal controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. For example, Kineta’s directors or executive officers could inadvertently fail to disclose a new relationship or arrangement causing Kineta to fail to make any related party transaction disclosures. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in Kineta’s control system, misstatements due to error or fraud may occur and not be detected. Future changes in financial accounting standards or practices may cause adverse and unexpected revenue fluctuations and adversely affect Kineta’s reported results of operations. Future changes in financial accounting standards may cause adverse, unexpected revenue fluctuations and affect Kineta’s reported financial position or results of operations. Financial accounting standards in the United States are constantly under review and new pronouncements and varying interpretations of pronouncements have occurred with frequency in the past and are expected to occur again in the future. As a result, Kineta may be required to make changes in its accounting policies. Those changes could affect Kineta’s financial condition and results of operations or the way in which such financial condition and results of operations are reported. Kineta intends to invest resources to comply with evolving standards, and this investment may result in increased general and administrative expenses and a diversion of management time and attention from business activities to compliance activities. 87 Changes in tax laws or regulations that are applied adversely to Kineta or its customers may have a material adverse effect on Kineta’s business, cash flow, financial condition or results of operations. New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could adversely affect Kineta’s business operations and financial performance. For instance, the recently enacted Inflation Reduction Act imposes, among other rules, a 15% minimum tax on the book income of certain large corporations for tax years beginning after December 31, 2022 and a 1% excise tax on certain corporate stock repurchases made after December 31, 2022. Further, existing tax laws, statutes, rules, regulations, or ordinances could be interpreted, changed, modified, or applied adversely to Kineta. For example, the TCJA enacted many significant changes to the U.S. tax laws. Future guidance from the IRS and other tax authorities with respect to the TCJA may affect Kineta, and certain aspects of the TCJA could be repealed or modified in future legislation. For example, the Coronavirus Aid, Relief, and Economic Security Act modified certain provisions of the TCJA. In addition, it is uncertain if and to what extent various states will conform to the TCJA or any newly enacted federal tax legislation. Changes in corporate tax rates, the realization of net deferred tax assets relating to Kineta’s operations, the taxation of foreign earnings, and the deductibility of expenses under the TCJA or future reform legislation could have a material impact on the value of Kineta’s deferred tax assets, could result in significant one-time charges, and could increase Kineta’s future U.S. tax expense. In addition, the presidential and congressional elections in the United States could also result in significant changes in, and uncertainty with respect to, tax legislation, regulation and government policy directly affecting Kineta and its business. For example, the United States government may enact significant changes to the taxation of business entities including, among others, a permanent increase in the corporate income tax rate, an increase in the tax rate applicable to the global intangible low-taxed income and elimination of certain exemptions, and the imposition of minimum taxes or surtaxes on certain types of income. The likelihood of these changes being enacted or implemented is unclear. Unstable market and economic conditions may have serious adverse consequences on Kineta’s business, financial condition and stock price. As a result of the COVID-19 pandemic and actions taken to slow its spread, the global credit and financial markets have experienced extreme volatility and disruptions, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates and uncertainty about economic stability. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not occur. Kineta’s general business strategy may be adversely affected by any such economic downturn, volatile business environment or continued unpredictable and unstable market conditions. If the current equity and credit markets deteriorate, it may make any necessary debt or equity financing more difficult, more costly and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on Kineta’s growth strategy, financial performance and stock price and could require Kineta to delay or abandon clinical Geopolitical developments, such as the Russian invasion of Ukraine, the conflict in Israel and the Gaza Strip or deterioration in the bilateral relationship between the United States and China, may impact government spending, international trade and market stability, and cause weaker macro-economic conditions. The impact of these developments, including any resulting sanctions, export controls or other restrictive actions that may be imposed against governmental or other entities in, for example, Russia, have in the past contributed and may in the future contribute to disruption, instability and volatility in the global markets, which in turn could adversely impact Kineta’s operations and weaken its financial results. Certain political developments may also lead to uncertainty to regulations and rules that may materially affect Kineta’s business. Kineta’s internal information technology systems, or those of Kineta’s third-party CROs or other contractors or consultants, may fail or suffer security breaches, loss or leakage of data and other disruptions, which could result in Kineta is increasingly dependent upon information technology systems, infrastructure and Despite the Hackers and data thieves are increasingly sophisticated and operate large-scale and complex automated attacks which may remain undetected until 88 after they occur. Kineta cannot assure you that its data protection efforts and its investment in The costs of mitigating cybersecurity risks are significant and are likely to increase in the future. These costs include, but are not limited to, retaining the services of cybersecurity providers; compliance costs arising out of existing and future cybersecurity, data protection and privacy laws and regulations; and costs related to maintaining redundant networks, data backups and other damage-mitigation measures. Kineta also cannot be certain that its existing insurance coverage will continue to be available on acceptable terms or in amounts sufficient to cover the potentially significant losses that may result from a security incident or breach or that the insurer will not deny coverage of any future claim. Kineta’s operations as a global company subject it to various risks, and Kineta’s failure to manage these risks could adversely affect its business, results of operations, cash flows, financial condition and/or prospects. Kineta faces significant operational risks as a result of doing business globally, such as: If one or more of these risks are realized, it could have a material adverse effect on Kineta’s business, results of operations, cash flows, financial condition and/or prospects. Kineta or the third parties upon whom it depends may be adversely affected by earthquakes, fires or other If Kineta is subject to certain U.S. and foreign anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations. Kineta can face serious consequences for violations. U.S. and foreign anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations (collectively, “Trade Laws”) prohibit, among other things, companies and their employees, agents, CROs, legal counsel, accountants, consultants, contractors and other partners 89 from authorizing, promising, offering, providing, soliciting, or receiving directly or indirectly, corrupt or improper payments or anything else of value to or from recipients in the public or private sector. Violations of Trade Laws can result in substantial criminal fines and civil penalties, imprisonment, the loss of trade privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm and other consequences. Kineta has direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities and other organizations. Kineta also expects to continue its non-U.S. activities, which may increase over time. Kineta expects to rely on third parties for research, preclinical studies and clinical trials and/or to obtain necessary permits, licenses, patent registrations and other marketing approvals. Kineta can be held liable for the corrupt or other illegal activities of its personnel, agents, or partners, even if Kineta does not explicitly authorize or have prior knowledge of such activities. If Kineta or any Risks Related to The The 90 In addition, The Delaware Forum Provision and the Federal Forum Provision may impose additional litigation costs on stockholders in pursuing the claims identified above, particularly if the stockholders do not reside in or near the State of Delaware. Additionally, the Delaware Forum Provision and the Federal Forum Provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with Kineta may issue a substantial number of additional shares of common stock under an employee incentive plan. Any such issuances would dilute the interest of Kineta’s stockholders and likely present other risks. Kineta may issue additional shares of common stock under an employee incentive plan. The issuance of additional common stock: An active trading market for Kineta’s common stock may not be sustained. Although Kineta’s common stock is listed on The Nasdaq Capital Market, an active trading market for Kineta’s shares may never be sustained. If an active market for Kineta’s common stock is not sustained, it may be difficult for you to sell shares you purchased without depressing the market price for the shares, or at all. An inactive trading market may also impair Kineta’s ability to raise capital to continue to fund operations by selling additional shares and may impair Kineta’s ability to acquire other companies or technologies by using its shares as consideration. Kineta’s failure to meet the continued listing requirements of Nasdaq could result in a delisting of On June 27, 2023, Kineta received written notice from the Listing Qualifications Department of Nasdaq stating that Kineta was not in compliance with Nasdaq Listing Rule 5550(b)(2) because Kineta did not maintain a minimum Market Value of Listed Securities of at least $35 million for the last 30 consecutive business days. On August 15, 2023, Kineta received written notice from the Listing Qualifications Department of Nasdaq notifying Kineta that, based on its Quarterly Report on Form 10-Q for the quarter ended June 30, 2023, Kineta had regained compliance with the alternative standard under Nasdaq Listing Rule 5550(b)(1). If compliance with listing requirements would allow In the past, securities class action litigation has often been brought against a company following a decline in the Item 1B. Unresolved Staff Comments. None. Item 1C. Cybersecurity. Cybersecurity Risk Management and Strategy We recognize the importance of assessing, identifying, and managing material risks associated with cybersecurity threats, as such term is defined in Item 106(a) of Regulation S-K. These risks include, among other things, operational risks; intellectual property theft; fraud; extortion; harm to 92 employees or customers; violation of privacy or security laws and other litigation and legal risk; and reputational risks. We have implemented several cybersecurity processes, technologies, and controls to aid in our efforts to assess, identify, and manage such material risks. Our process for identifying and assessing material risks from cybersecurity threats operates alongside our broader overall risk assessment process, covering all company risks. As part of this process, appropriate disclosure personnel will collaborate with subject matter specialists, as necessary, to gather insights for identifying and assessing material cybersecurity threat risks, their severity, and potential mitigations. We also have a cybersecurity specific risk assessment process, which helps identify our cybersecurity threat risks. As part of this process, and our processes to provide for the availability of critical data and systems, maintain regulatory compliance, identify and manage our risks from cybersecurity threats, and to protect against, detect, and respond to cybersecurity incidents, as such term is defined in Item 106(a) of Regulation S-K, we undertake the below listed activities, among others: In the event of a cybersecurity incident, we will coordinate with our third-party cybersecurity advisor to prepare for, detect, respond to and recover from cybersecurity incidents, which include processes to triage, assess severity for, escalate, contain, investigate, and remediate the incident, as well as to comply with potentially applicable legal obligations and mitigate brand and reputational damage. As part of the above processes, we regularly engage with assessors, consultants, auditors, and other third parties, including by regularly having a third party review our cybersecurity program to help identify areas for continued focus, improvement and/or compliance. Our internal processes also address cybersecurity threat risks associated with our use of third-party service providers, including those in our supply chain or who have access to our customer and employee data or our systems. We determine an overall risk assessment of potential cybersecurity issues with our third-party suppliers. In addition, cybersecurity considerations affect the selection and oversight of our third-party service providers. Additionally, if appropriate, we require those third parties that could introduce significant cybersecurity risk to us to agree by contract to manage their cybersecurity risks in specified ways, and to agree to be subject to cybersecurity audits, which we conduct as appropriate. Cybersecurity Governance Cybersecurity is an important part of our risk management processes and an area of increasing focus for our Board and management. Our audit committee is responsible for the oversight of risks from cybersecurity threats. At least annually, the audit committee receives an overview from management of our cybersecurity threat risk management and strategy processes covering topics such as data security posture, results from third-party assessments, progress towards pre-determined risk-mitigation-related goals, our incident response plan, and material cybersecurity threat risks or incidents and developments, as well as the steps management has taken to respond to such risks. In such sessions, the audit committee generally receives materials including a cybersecurity scorecard and other materials indicating current and emerging material cybersecurity threat risks, and describing the Company’s ability to mitigate those risks, and discusses such matters with certain members of management, which include our CFO and President. Members of the audit committee are also encouraged to regularly engage in ad hoc conversations with management on cybersecurity-related news events and discuss any updates to our cybersecurity risk management and strategy programs. Material cybersecurity threat risks are also considered during separate Board meeting discussions of important matters like risk management, operational budgeting, business continuity planning, mergers and acquisitions, brand management, and other relevant matters. Our cybersecurity risk management and strategy processes, which are discussed in greater detail above, are led by our President. Such individual completed a certificate program with Stanford University on Foundations of Information Security in the first quarter of 2024. In addition, he has over 20 years of executive responsibility for managing public company IT operations, including managing information security, developing cybersecurity 93 strategy, implementing effective information and cybersecurity programs and deployment of internal training and company technology standards. This member of management is informed about and monitors the prevention, mitigation, detection, and remediation of cybersecurity incidents through his management of, and participation in, the cybersecurity risk management and strategy processes described above, including the operation of our incident response plan. As discussed above, this member of management reports to the audit committee about cybersecurity threat risks, among other cybersecurity related matters. On March 20, 2024, Kineta filed a complaint in the Court of Chancery of the State of Delaware against Growth & Value Development Inc. (“GVDI”), alleging breach of contract in connection with GVDI’s recent repudiation of its obligation to provide a substantial tranche of funding for Kineta as required under the Securities Purchase Agreement. The complaint provides that Kineta will seek specific performance of GVDI’s obligations under the Securities Purchase Agreement and damages equal to the amount of the unpaid funding and any damages resulting from GVDI’s breach. Except as disclosed in the preceding paragraph, Kineta is currently not a party to any other material legal proceedings. From time to time, Not applicable. 94 PART II Certain Information Regarding the Trading of Our Common Stock Our common stock Holders of Our Common Stock As of March Dividends Securities Authorized for Issuance Under Equity Compensation Plans The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report. Recent Sales of Unregistered Securities 95 The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and related notes appearing at the end of this Annual Report on Form 10-K. Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report on Form 10-K, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the “Risk Factors” section of this Annual Report on Form 10-K, our actual results could differ materially from the results described in, or implied by, the forward-looking statements contained in the following discussion and analysis. capabilities. Following this review, we determined that we would implement a significant corporate restructuring to substantially reduce expenses and preserve cash. The restructuring included a reduction in The Company cautions that trading in the Company’s securities is highly speculative and poses substantial risks. Trading prices for the Company’s securities may bear little or no relationship to the actual value realized, if any, by holders of the Company’s securities. In the event of liquidation, bankruptcy or other wind-down event, holders of our securities will likely suffer a total loss of their investment. Accordingly, the Company urges extreme caution with respect to existing and future investments in its securities. We are a clinical-stage biotechnology company with a mission to develop next-generation immunotherapies that transform patients’ lives. We have leveraged our expertise in innate immunity and are focused on discovering and developing potentially differentiated immunotherapies that address the mechanisms of cancer immune resistance: Our pipeline of next-generation immunotherapies includes (i) KVA12123, a monoclonal antibody (“mAb”) immunotherapy targeting VISTA (V-domain Ig suppressor of T cell activation) and (ii) an anti-CD27 agonist mAb immunotherapy. These novel immunotherapies have the potential to address disease areas with unmet medical needs and significant commercial potential. KVA12123 is a VISTA blocking immunotherapy in development as an intravenous infusion dosed every two weeks. We dosed the first patient in a Phase 1/2 clinical trial of KVA12123 in the United States in April 2023. The ongoing Phase 1/2 clinical study is designed to evaluate KVA12123 as a monotherapy and in combination with the immune checkpoint inhibitor pembrolizumab in patients with advanced solid tumors. Initial monotherapy safety, pharmacokinetic and biomarker data were presented at the Society for Immunotherapy of Cancer’s (SITC) annual meeting in November 2023. KVA12123 was designed to be a differentiated VISTA blocking immunotherapy to address the problem of immunosuppression in the TME. It is a fully human engineered IgG1 monoclonal antibody that binds to VISTA through a unique epitope and across neutral and acidic pHs. KVA12123 may be an effective immunotherapy for many types of cancer, including non-small cell lung cancer (“NSCLC”), colorectal cancer (“CRC”), ovarian cancer (“OC”), renal cell carcinoma (“RCC”) and head and neck squamous cell carcinoma (“HNSCC”). These indications represent a significant unmet medical need with a large worldwide commercial opportunity for KVA12123. We are also developing an anti-CD27 agonist mAb immunotherapy to address the problem of exhausted T cells. The nominated lead candidate is a fully human mAb that demonstrates nanomolar (“nM”) binding affinity to CD27 in humans. In preclinical studies, our lead anti-CD27 candidate demonstrated antitumor efficacy as a single agent and in combination with other immunotherapies in multiple solid and hematological preclinical tumor models. CD27 is a clinically validated target that may be an effective immunotherapy for advanced solid tumors including RCC, CRC and OC. We continue to conduct preclinical studies to optimize its lead anti-CD27 agonist mAb clinical candidate and to evaluate it in combination with other checkpoint inhibitors. According to Market Data Forecast, the immuno-oncology market generated sales of approximately $111 billion in 2023 and is forecast to reach $201 billion in 2028. If we successfully complete the clinical trial program for KVA12123 and if we subsequently obtain regulatory approval for KVA12123, we will focus on initial target indications in NSCLC, CRC and OC. Initially the clinical development of KVA12123 will be as a second-line therapy in these indications. These three cancer therapy segments represent a forecasted $48 billion market opportunity in 2027 according to GlobalData. 96 We are a leader in the field of innate immunity and are focused on developing potentially differentiated immunotherapies. With KVA12123 in clinical development and the lead anti-CD27 agonist mAb in preclinical development, we believe we are positioned to achieve multiple value-driving catalysts. We have assembled an experienced management team, a seasoned research and clinical team, an immuno-oncology focused scientific advisory board, and a leading intellectual property position to advance our pipeline of potential novel immunotherapies for cancer patients. Since our inception in 2007, we have devoted substantially all of our resources to raising capital, licensing certain technology and intellectual property rights, identifying and developing potential product candidates, conducting research and development activities, including preclinical studies and clinical trials, organizing and staffing operations and providing general and administrative support for these operations. We have no products approved for commercial sale and have not generated any revenue from product sales. To date, revenue has been generated from the out-licensing of certain rights to third parties, providing research services under licensing and collaboration agreements as well as revenue from government grants. We have never been profitable and have incurred We expect borrowings under notes payable. As of December 31, Private Placement In connection and concurrently with the execution of the Merger Agreement, on June 5, 2022, we entered into a financing agreement (such financing agreement, as amended, the “Securities Purchase Agreement”), with certain investors to The first closing of the Private Placement occurred on December 16, 2022 and we issued 649,346 shares of our common stock and received net proceeds of $7.4 million. The second closing of the Private Placement for an aggregate purchase price of $22.5 million was expected to occur on April 15, 2024. However, in February 2024, certain investors indicated they will not be able to consummate the second closing of the Private Placement. The Company has the ability to unilaterally terminate the Securities Purchase Agreement until the date of the second closing. On December 27, 2022, we, through our subsidiary KCP, received written notice from Genentech, Inc. (“Genentech”) of its termination of the Exclusive Option and License Agreement entered into by and between KCP and Genentech dated April 11, 2018, as amended on November 27, 2019 and October 1, 2020 COVID-19 While we continue to monitor the impact of the COVID-19 pandemic 97 Geopolitical Developments Geopolitical developments, such as the At-the-Market Offering Program In On April 19, 2023, we delivered written notice to Jefferies that we were suspending and terminating the prospectus supplement (the “ATM Prospectus Supplement”) related to our common stock issuable pursuant to the New Sales Agreement. We will not make any sales of our securities pursuant to the New Sales Agreement, unless and until a new prospectus supplement is filed. Other than the termination and suspension of the ATM Prospectus Supplement, the New Sales Agreement remains in full force and effect. We During the year ended December 31, Registered Direct Offerings April 2023 On April 20, 2023, we Each April 2023 Pre-Funded Warrant represents the right to purchase one share of common stock at an exercise price of $0.001 per share. The April 2023 Pre-Funded Warrants are exercisable immediately and may be exercised at any time until the April 2023 Pre-Funded Warrants are exercised in full. In a concurrent private placement (the “April 2023 Private Placement” and, together with the April 2023 Registered Offering, the “April 2023 Offering”), we issued to the April 2023 Investor warrants to purchase up to 1,425,179 shares of common stock (the “April 2023 Common Warrants”) at an exercise price of $4.08 per share. The April 2023 Common Warrants are exercisable immediately and will expire five and one-half years from the initial exercise date. In connection with the April 2023 Offering, we entered into an engagement letter with H.C. Wainwright & Co., LLC (“Wainwright”), pursuant to which Wainwright agreed to serve as the exclusive placement agent for the issuance and sale of securities of the Company pursuant to the April 2023 Purchase Agreement. As compensation for such placement agent services, we paid Wainwright an aggregate cash fee equal to $420,000, a non-accountable expense of $35,000 and $50,000 for legal and other expenses as actually incurred. The total offering-related fees were approximately $520,000, which resulted in net proceeds to the Company of $5.5 million. On April 24, 2023, we also issued to Wainwright or its designees warrants to purchase 71,259 shares of common stock (the “April 2023 Wainwright Warrants”). The April 2023 Wainwright Warrants have a term of five years from the commencement of sales in the April 2023 Offering, and have an exercise price of $5.2625 per share. October 2023 On October 3, 2023,we entered into a Securities Purchase Agreement (the “October 2023 Purchase Agreement”) with an institutional investor (the “October 2023 Investor”) pursuant to which the Company issued and sold, in a registered direct offering priced at-the-market under the rules of Nasdaq (such offering, the “October 2023 Registered Offering”), (i) an aggregate of 110,000 shares of our common stock, at a purchase price of $3.37 per share, and (ii) pre-funded warrants exercisable for up to 780,208 shares of our common stock (the “October 2023 Pre-Funded Warrants”) to 98 the October 2023 Investor at a purchase price of $3.369 per October 2023 Pre-Funded Warrant, for aggregate gross proceeds from the October 2023 Registered Offering of approximately $3.0 million before deducting the placement agent fee (as described in greater detail below) and related offering expenses. Each October 2023 Pre-Funded Warrant represents the right to purchase one share of common stock at an exercise price of $0.001 per share. The October 2023 Pre-Funded Warrants are exercisable immediately and may be exercised at any time until the October 2023 Pre-Funded Warrants are exercised in full. In a concurrent private placement (the “October 2023 Private Placement” and, together with the October 2023 Registered Offering, the “October 2023 Offering”), we issued to the October 2023 Investor warrants to purchase up to 890,208 shares of common stock (the “October 2023 Common Warrants”) at an exercise price of $3.25 per share. The October 2023 Common Warrants are exercisable immediately and will expire five and one-half years from the initial exercise date. In connection with the October 2023 Offering, we entered into an engagement letter with Wainwright, pursuant to which Wainwright agreed to serve as the exclusive placement agent for the issuance and sale of securities of the Company pursuant to the October 2023 Purchase Agreement. As compensation for such placement agent services, the Company paid Wainwright an aggregate cash fee equal to $210,000, a non-accountable expense of $35,000 and $50,000 for legal and other expenses as actually incurred. The total offering-related fees were approximately $310,000, which resulted in net proceeds to the Company of $2.7 million. On October 5, 2023, the Company also issued to Wainwright or its designees warrants to purchase 44,510 shares of Common Stock (the “October 2023 Wainwright Warrants” and, together with the Pre-Funded Warrants and the Common Warrants, the “Warrants”). The October 2023 Wainwright Warrants have a term of five years from the commencement of sales in the October 2023 Offering, and have an exercise price of $4.2125 per share. Nasdaq Compliance On June 27, 2023, the Company received written notice from the Listing Qualifications Department of Nasdaq stating that the Company was not in compliance with Nasdaq Listing Rule 5550(b)(2) because the Company did not maintain a minimum Market Value of Listed Securities of at least $35 million for the last 30 consecutive business days. On August 15, 2023, the Company received written notice from the Listing Qualifications Department of Nasdaq notifying the Company that, based on its Quarterly Report on Form Financial Operations Overview To date, we have not generated any revenue from product sales and do not expect to generate any revenue from Licensing Revenues Our license agreements may include the transfer of intellectual property rights in the form of licenses, promises to provide research and development services and promises to participate on certain development committees with the collaboration party. The terms of such agreements include payment to us of one or more of the following: nonrefundable upfront fees, payment for research and development services, development, regulatory and commercial milestone payments and sales-based milestones and royalties on net Revenue associated with nonrefundable upfront license fees where the license fees and research and development activities cannot be accounted for as separate performance obligations is deferred and recognized as revenue over the We expect that any revenue generated from our current collaboration, research and license agreements and any future collaboration partners will Collaboration Revenues In connection with the Merger, we became the successor in interest to an exclusive license and research 99 Grant Revenues Under our grant arrangements with government-sponsored and charitable organizations, we receive payment for providing research and development services. Revenue associated with grant arrangements is based on a cost-based reimbursement model that recognizes revenue over time as we perform work under the grants and incur qualifying research and development costs. We Operating Expenses Research and Development Expenses Research and development expenses The largest component of our As we are working on multiple research and development programs at any one time, we track our external expenses by The General and Administrative Expenses General and administrative expenses consist primarily of In-Process Research and Development Assets Acquired We acquired in-process research and development assets in connection with the Merger. As the acquired in-process research and development assets were deemed to have no current or alternative future use, the entire amount was recognized as expense in the consolidated statements of operations for the year ended December 31, Other (Expense) Income 100 Interest Income Interest income consists of interest earned on short-term money market accounts. Interest Expense Interest expense consists of interest charged on outstanding invoices and outstanding borrowings associated with our Change in fair value of Change in Fair Value Measurement of Notes Payable Change in fair value of notes payable relates to the remeasurement of the notes payable that we Gain on Extinguishments of Debt, Net Gain on extinguishments of debt, net Other (Expense) Income, Net Net (Loss) Income Attributable to Noncontrolling Interest Net (loss) income Results of Operations Comparison of the Years Ended December 31, The following table summarizes our results of operations for the Year Ended December 31, 2021 2020 Change (in thousands) Collaboration revenue $ 8,044 $ 6,896 $ 1,148 Operating expenses: Research and development 26,410 22,310 4,100 General and administrative 20,379 11,881 8,498 In-process research and development assets acquired — 28,336 (28,336 ) Total operating expenses 46,789 62,527 (15,738 ) Loss from operations $ (38,745 ) (55,631 ) 16,886 Other income (expense): Change in fair value of preferred unit warrant liability — 72 (72 ) Interest expense (1,817 ) (1,900 ) 83 Interest income and other income (expense), net (75 ) (28 ) (47 ) Gain on debt extinguishment 1,134 — 1,134 Total other expense, net (758 ) (1,856 ) 1,098 Net loss $ (39,503 ) $ (57,487 ) $ 17,984 Years Ended December 31, 2023 2022 Change (in thousands) Revenues: Licensing revenues $ 5,000 $ 1,041 $ 3,959 Collaboration revenues 442 — 442 Grant revenues — 912 (912 ) Total revenues 5,442 1,953 3,489 Operating expenses: Research and development 9,023 15,928 (6,905 ) General and administrative 12,142 8,696 3,446 In-process research and development — 18,860 (18,860 ) Total operating expenses 21,165 43,484 (22,319 ) Loss from operations (15,723 ) (41,531 ) 25,808 Other (expense) income: Interest income 325 9 316 Interest expense (337 ) (3,737 ) 3,400 Change in fair value of rights from Private Placement 1,582 — 1,582 Change in fair value of measurement of notes payable (22 ) (15,280 ) 15,258 Warrant expense — (3,309 ) 3,309 Gain on extinguishments of debt expense — 341 (341 ) Other income (expense), net 99 54 45 Total other (expense) income, net 1,647 (21,922 ) 23,569 Net loss (14,076 ) (63,453 ) 49,377 Net income (loss) attributable to noncontrolling interest 23 (45 ) 68 Net loss attributable to Kineta, Inc. $ (14,099 ) $ (63,408 ) $ 49,309 Licensing Revenues Licensing revenues were $5.0 million for the year ended December 31, Collaboration Revenues Collaboration revenues were $442,000 for the year ended December 31, 2023 and zero for the year ended December 31, 2022 as a result of research services provided under the Merck Neuromuscular License Agreement pursuant to which the Company became a successor in interest in connection with Grant Revenues Grant revenues were zero for the year ended December 31, 102 Research and Development Expenses The following table summarizes our research and development expenses by program and category for the periods presented: Year Ended December 31, 2021 2020 Change (in thousands) Direct research and development expenses by program: YTX-7739 $ 8,230 $ 5,449 2,781 YTX-9184 1,873 1,826 47 Platform, research and discovery, and unallocated expenses: Platform and other early stage research external costs 3,045 2,478 567 Personnel related (including stock/equity-based compensation) 7,825 7,293 532 Facility related and other 5,437 5,264 173 Total research and development expenses $ 26,410 $ 22,310 $ 4,100 Years Ended December 31, 2023 2022 Change (in thousands) Direct external program expenses: KVA12123 program $ 6,157 $ 7,567 $ (1,410 ) ALS target program 307 — 307 CD27 program 279 583 (304 ) KCP-506 program 39 344 (305 ) Other programs — 402 (402 ) Internal and unallocated expenses: Personnel-related costs 1,636 5,907 (4,271 ) Facilities and related costs 428 851 (423 ) Other costs 177 274 (97 ) Total research and development expenses $ 9,023 $ 15,928 $ (6,905 ) Research and development expenses were General and Administrative Expenses Year Ended December 31, 2021 2020 Change (in thousands) Personnel related (including equity-based compensation) $ 8,765 $ 5,837 $ 2,928 Professional and consultant fees 6,307 5,090 1,217 Facility related and other 5,307 954 4,353 Total general and administrative expenses $ 20,379 $ 11,881 $ 8,498 General and administrative expenses were In-process research and development expenses Interest Income Interest income was $325,000 for the year ended December 31, Interest Expense Interest expense was $337,000 for the year ended December 31, 2023 and $3.7 million for the year ended December 31, 2022. Interest expense decreased due to a significantly lower balance of notes in 2023 as the majority of notes were converted to equity in December 2022. Change in Fair Value Measurement of Private Placement Change in fair value of Private Placement was a gain of $1.6 million for the year ended December 31, 2023 and relates to the remeasurement of the rights from the Private Placement that we determined was a derivative, which requires the asset to be accounted for at fair value. We determined the rights from Private Placement to be a derivative asset when we added a minimum purchase price of $3.18 upon an amendment in 2023. There was no change in fair value of the rights from Private Placement during 2022. Change in Fair Value Measurement of Notes Payable 103 Change in fair value of notes payable was a loss of $22,000 for the year ended December 31, 2023 and a loss of $15.3 million for the year ended December 31, 2022. The decrease in 2023 resulted as the majority of notes were converted to equity in December 2022. Warrant Expense Warrant expense was zero for the year ended December 31, 2023 and $3.3 million for the year ended December 31, 2022. Warrant expense in 2022 represents the fair value of warrants issued to current debt holders that converted their debt to equity in 2022. Gain on Extinguishments of Debt Gain on extinguishment of debt was zero for the year ended December 31, 2023 and $341,000 for the year ended December 31, 2022. There have been no settlement of notes payable during year ended December 31, 2023 as the majority of notes were converted to equity in December 2022. In 2022, the gains resulted primarily from the settlements of notes payable accounted for under the fair value election. Going Concern and Capital Resources Exploring Strategic Alternatives We require substantial additional capital to sustain our operations and pursue our growth strategy, including the development of our product candidates. We are exploring strategic alternatives that may include, but are not limited to, sale of assets of the Company, a sale of the Company, licensing of assets, a merger, liquidation or other strategic action. If a strategic process is unsuccessful, the Board may decide to pursue a liquidation or obtain relief under the US Bankruptcy Code. These factors raise substantial doubt about our ability to continue as a going concern. Sources of Liquidity Since our inception In December During the year ended December 31, 2023, we sold 126,503 shares of our common stock to individual investors under the New Sales Agreement and received net proceeds of $600,000 in connection with the ATM equity offering program. During the year ended December 31, 2023, we sold 1,058,000 shares of our common stock and issued pre-funded warrants exercisable for up to 1,257,387 shares of our common stock in the April 2023 Registered Offering and October 2023 Registered Offering and received net proceeds of Future Funding Requirements Our revenues to date have been primarily derived from our collaboration, research and license agreements as well as grants awarded by government agencies. We, Our future funding requirements will depend on many factors, including: 104 As of December 31, 2023, we had cash of $5.8 million,and there is substantial doubt about our ability to continue as a going concern. Based on our current operating plans, we do not have sufficient cash and cash equivalents We are exploring strategic alternatives that may include, but are not limited to, sale of assets of the Company, a sale of the Company, licensing of assets, a merger, liquidation or other strategic action. We may seek additional funds through equity or debt financings or through collaborations, licensing transactions or other sources that may be identified through our strategic process. However, there can be no assurance that we will be able to complete any such transactions on acceptable terms or otherwise. The failure to obtain sufficient funds on commercially acceptable terms when needed would have a material adverse effect on our business, results of operations, and financial condition. These We do not currently have any commitments for future funding or additional capital other than the Private Placement. However, as noted above, certain investors have indicated they will not be able to fulfill their contractual obligation to consummate the Private Placement. As such, we have paused or significantly scaled back the development or commercialization of its future product candidates or other research and development initiatives. If we are unable to complete a strategic transaction or raise additional capital in sufficient amounts, we will not be able to continue our business and we may need to file for bankruptcy protection. Cash Flows The following table summarizes our Year Ended December 31, 2021 2020 (in thousands) Cash used in operating activities $ (48,915 ) $ (17,938 ) Cash provided by investing activities 3,093 31,041 Cash (used in) provided by financing activities (1,033 ) 55,536 Net (decrease) increase in cash, cash equivalents, and restricted cash $ (46,855 ) $ 68,639 Years Ended December 31, 2023 2022 (in thousands) Net cash provided by (used in): Operating activities $ (16,209 ) $ (19,029 ) Investing activities 331 9,270 Financing activities 8,468 11,808 Net change in cash and cash equivalents $ (7,410 ) $ 2,049 Cash used in operating activities Investing Activities Cash provided by investing activities for the year ended December 31, 2022 was $9.3 million, consisting primarily of Cash provided by financing activities for the year ended December 31, Debt Obligations Notes Payable As of December 31, 2023, we had outstanding notes payable in an aggregate principal amount of $779,000 at interest rates that range from 3.75% to 6%, of which $629,000 matures within the next 12 months. The principal amount of each note payable is due at a specified periodic repayment date and/or at maturity, with such dates ranging from June 2024 to on or after September 2050. See Note 6 to our consolidated financial Other Contractual Obligations and Commitments Our cash requirement greater than 12 months are related to We have entered into a number of strategic license agreements pursuant to which we have acquired rights to specific assets, technology and intellectual property. In accordance with these agreements, we are obligated to pay, among other items, future contingent payments that are dependent upon future events such as our We lease office and laboratory space for our We have entered into an In addition, we enter into agreements in the normal course of business with various third parties for preclinical research studies, clinical trials, testing and other Critical Accounting Estimates Our management’s discussion and analysis of financial condition and results of operations Revenue Recognition License Revenues We 106 these arrangements typically include payment to us of one or more of the following: nonrefundable upfront fees, payment for research and development services provided by us under approved work plans, development, regulatory and commercial milestone payments, and sales-based milestones and royalties on net sales of licensed products. Each of these payments results in license revenues, except for revenues from royalties, which are classified as other revenues. In determining the appropriate amount of revenue to be recognized as we fulfill our We recognize collaboration revenue using the cost-to-cost method, which we believe best depicts the transfer of control to the customer. Under the cost-to-cost method, the extent of progress towards completion is measured based on the ratio of actual costs incurred to the total estimated costs expected upon satisfying the identified performance obligation. Under this method, revenue is Accrued Research and Development Expenses We record accrued expenses for estimated costs of our research and development activities conducted by third-party service providers, such as contract research organizations, contract manufacturing and other vendors, which include the conduct of preclinical studies, clinical trials and contract manufacturing activities. We record the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced and include these costs in accrued liabilities in the consolidated balance sheets and within research and development expenses in the consolidated statements of operations. These costs are a significant component of our research and development expenses. We record accrued expenses for these costs based on the We make significant judgments and estimates in determining the Change in Fair Value of Rights from Private Placement We Notes Payables accounted for under the Fair Value Option 107 We have elected the fair value option to account for certain of our notes payable and record these notes payable at fair value with changes in fair value recorded as a component of other income (expense) in the consolidated statements of operations. As a result of applying the fair value option, direct costs and fees related to the notes payable are expensed as incurred. For our convertible notes payable, the probability-adjusted model used in valuing the fair value of such convertible notes payable is based on significant unobservable inputs, including but not limited to, the timing and probability of a qualified financing event, discount rates and the fair value of the underlying common stock. For our notes payable that are not convertible, the discounted cash-flow model used in valuing the fair value of such notes payable is based on significant unobservable inputs, including but not limited to, discount rates and expected payment dates. See Note 6 to our consolidated financial statements for more details on the assumptions used. Increases or decreases in the fair value of the notes payable can result from updates to assumptions such as the expected timing or probability of a qualified financing event, or changes in discount rates. Judgment is used in determining these assumptions as of the initial valuation date and at each subsequent reporting period. Updates to assumptions could have a significant impact on our results of operations in any given period. Stock-Based Compensation We recognize noncash stock-based compensation related to stock-based awards to employees, non-employees, and directors, including stock options, based on the fair value on the grant date In determining the fair value of Fair Value of Common Stock – The fair value of the shares of common stock underlying stock options has historically been determined by our board of directors. In order to determine the fair value of Expected Term – Our expected term represents the period that the stock-based awards are expected to be outstanding and is determined using Expected Volatility – As we have only been a publicly-traded company for one year, the expected volatility is estimated based on the average volatility for a group of comparable publicly traded biotechnology companies over a period equal to Risk-Free Interest Rate – The risk-free interest rate is based on the U.S. Treasury zero coupon issues in effect at the time of grant for Expected Dividend – Other than the Distribution, we have never paid dividends on our common stock and have no plans to pay dividends on our common stock. Therefore, we use an expected dividend yield of zero. Stock-based compensation was $3.9 million for the year ended December 31, 2023 and $5.2 million for the year ended December 31, 2022. As of December 31, 2023, we had $2.9 million of total unrecognized stock-based compensation related to stock options and We are a smaller reporting company, as defined in Rule 12b-2 under the Item 8. Financial Statements and Supplementary Data. INDEX TO CONSOLIDATED FINANCIAL STATEMENTS 110 Report of Independent Registered Public Accounting Firm To the Board of Directors and Stockholders of Opinion on the Financial Statements We have audited the accompanying consolidated balance sheets of Explanatory Paragraph – Going Concern The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As Basis for Opinion These We conducted our audits Our audits included performing procedures to assess the risks of material misstatement of the Critical Audit Critical audit March We have served as the Company’s auditor since KINETA, INC. (in thousands) December 31, 2023 2022 Assets Current assets: Cash $ 5,783 $ 13,143 Restricted cash 75 — Prepaid expenses and other current assets 119 457 Total current assets 5,977 13,600 Property and equipment, net — 249 Operating right-of-use asset 472 1,211 Rights from Private Placement 3,832 2,250 Restricted cash — 125 Total assets $ 10,281 $ 17,435 Liabilities and Stockholders’ Equity Current liabilities: Accounts payable $ 3,694 $ 6,635 Accrued expenses and other current liabilities 2,211 3,527 Deferred revenue — 442 Notes payable, current portion 620 — Operating lease liability, current portion 547 843 Finance lease liabilities, current portion — 40 Total current liabilities 7,072 11,487 Notes payable, net of current portion 150 748 Operating lease liability, net of current portion — 547 Finance lease liabilities, net of current portion — 83 Total liabilities 7,222 12,865 Commitments and contingencies (Note 6) Stockholders’ equity: Common stock, $0.001 par value; 125,000 shares authorized as of December 31, 2023 and December 31, 2022; 10,397 and 8,318 shares issued and outstanding as of December 31, 2023 and December 31, 2022, respectively 10 8 Additional paid-in capital 168,669 156,106 Accumulated deficit (165,789 ) (151,690 ) Total stockholders’ equity attributable to Kineta, Inc. 2,890 4,424 Noncontrolling interest 169 146 Total stockholders’ equity 3,059 4,570 Total liabilities and stockholders’ equity $ 10,281 $ 17,435 See the accompanying notes to the consolidated financial statements. 112 KINETA, INC. Consolidated Statements of Operations (in thousands, except per share amounts) Years Ended December 31, 2023 2022 Revenues: Licensing revenues $ 5,000 $ 1,041 Collaboration revenues 442 — Grant revenues — 912 Total revenues 5,442 1,953 Operating expenses: Research and development 9,023 15,928 General and administrative 12,142 8,696 In-process research and development — 18,860 Total operating expenses 21,165 43,484 Loss from operations (15,723 ) (41,531 ) Other (expense) income: Interest income 325 9 Interest expense (with related parties $0 for the year ended December 31, 2023 and $1,659 for the year ended December 31, 2022) (337 ) (3,737 ) Change in fair value of rights from Private Placement 1,582 — Change in fair value measurement of notes payable (22 ) (15,280 ) Warrant expense — (3,309 ) Gain on extinguishments of debt, net — 341 Other income, net 99 54 Total other (expense) income, net 1,647 (21,922 ) Net loss $ (14,076 ) $ (63,453 ) Net (loss) income attributable to noncontrolling interest 23 (45 ) Net loss attributable to Kineta, Inc. $ (14,099 ) $ (63,408 ) Net loss per share, basic and diluted $ (1.28 ) $ (12.87 ) Weighted-average shares outstanding, basic and diluted 11,054 4,926 See the accompanying notes to the consolidated financial statements. 113 KINETA, INC. (in thousands) Common Stock Additional Paid-In Capital Accumulated Total Stockholders’ Equity (Deficit) Attributable Noncontrolling Total Stockholders’ Shares Amount Amount Deficit to Kineta Interest Equity (Deficit) Balance as of December 31, 2021 4,656 $ 5 $ 76,137 $ (88,282 ) $ (12,140 ) $ 191 $ (11,949 ) Issuance of common stock upon 1,338 1 37,518 — 37,519 — 37,519 Issuance of common stock in connection with the Merger 1,553 1 20,550 — 20,551 — 20,551 Issuance of common stock in 649 1 7,406 — 7,407 — 7,407 Issuance of warrants to existing stockholders — — 3,309 — 3,309 — 3,309 Rights from Private Placement — — 2,250 — 2,250 — 2,250 Issuance of warrants in connection with convertible debt amendments — — 1,639 — 1,639 — 1,639 Issuance of common stock 58 — 1,581 — 1,581 — 1,581 Note conversion discount — — 414 — 414 — 414 Issuance of common stock upon 53 — 71 — 71 — 71 Issuance of warrants for services — — 62 — 62 — 62 Issuance of common stock upon 11 — — — — — — Stock-based compensation — — 5,169 — 5,169 — 5,169 Net loss — — — (63,408 ) (63,408 ) (45 ) (63,453 ) Balance as of December 31, 2022 8,318 $ 8 $ 156,106 $ (151,690 ) $ 4,424 $ 146 $ 4,570 Issuance of common stock and pre-funded warrants 1,185 2 8,559 — 8,561 — 8,561 Issuance of common stock for services 63 — 189 — 189 — 189 Issuance of common stock upon 696 — 30 — 30 — 30 Issuance of common stock upon vesting of RSUs 135 — (69 ) — (69 ) — (69 ) Stock-based compensation — — 3,854 — 3,854 — 3,854 Net loss — — — (14,099 ) (14,099 ) 23 (14,076 ) Balance as of December 31, 2023 10,397 $ 10 $ 168,669 $ (165,789 ) $ 2,890 $ 169 $ 3,059 See the accompanying notes to the consolidated financial statements. 114 KINETA, INC. Consolidated Statements of Cash Flows ( December 31, 2021 2020 Assets Current assets: Cash and cash equivalents $ 35,102 $ 80,819 Marketable securities 1,399 4,498 Accounts receivable 5,000 — Prepaid expenses and other current assets 1,207 2,264 Total current assets 42,708 87,581 Property and equipment, net 387 874 Operating lease right-of-use assets 18,543 23,678 Deposits 366 386 Restricted cash 928 2,066 Assets held-for-sale 0 250 Total assets $ 62,932 $ 114,835 Liabilities and Stockholders’ Equity Current liabilities: Accounts payable $ 1,839 $ 7,384 Accrued expenses and other current liabilities 4,846 7,851 Current portion of long-term debt 5,805 2,891 Operating lease liabilities 5,064 4,468 Current portion of finance lease obligation 48 166 Deferred revenue 5,061 8,104 Total current liabilities 22,663 30,864 Long-term debt, net of discount and current portion 7,357 13,237 Operating lease liabilities, net of current portion 9,415 14,479 Finance lease obligation, net of current portion 0 48 Total liabilities 39,435 58,628 Commitments and contingencies (Note 12) Stockholders’ equity: Preferred stock, $0.001 par value; 5,000,000 shares authorized; 0 shares issued and outstanding as of December 31, 2021 and 2020, respectively 0 0 Common stock, $0.001 par value; 125,000,000 shares authorized; 10,644,714 shares and 10,193,831 shares issued and outstanding as of December 31, 2021 and 2020, respectively 11 10 Additional paid-in capital 210,799 204,007 Accumulated deficit (187,313 ) (147,810 ) Total stockholders’ equity 23,497 56,207 Total liabilities and stockholders’ equity $ 62,932 $ 114,835 Years Ended December 31, 2023 2022 Operating activities: Net loss $ (14,076 ) $ (63,453 ) Adjustments to reconcile net loss to net cash used in operating activities: Acquired in process research and development — 18,860 Change in fair value of rights from Private Placement (1,582 ) — Change in fair value of notes payable 22 15,280 Non-cash stock-based compensation 3,854 5,169 Warrant expense — 3,309 Issuance of warrants in connection with convertible debt amendments — 1,639 Non-cash operating lease expense 739 661 Depreciation and amortization 9 73 Common stock issued for services 189 62 Gain on extinguishments of debt, net — (341 ) Gain on disposal of asset (91 ) (62 ) Changes in operating assets and liabilities: Prepaid expenses and other current assets 338 (108 ) Accounts payable (2,941 ) (34 ) Accrued expenses and other current liabilities (1,385 ) 1,694 Operating lease liability (843 ) (737 ) Deferred revenue (442 ) (1,041 ) Net cash used in operating activities (16,209 ) (19,029 ) Investing activities: Cash acquired in connection with reverse merger — 9,276 Purchases of property and equipment — (71 ) Proceeds from sale of property and equipment 331 65 Net cash provided by investing activities 331 9,270 Financing activities: Proceeds from private placement — 7,407 Proceeds from notes payable — 6,746 Proceeds from issuance of common stock and pre-funded warrants 8,561 1,581 Proceeds from exercise of warrants 30 71 Repayments of notes payable — (4,000 ) Repayments of finance lease liabilities (123 ) 3 Net cash provided by financing activities 8,468 11,808 Net change in cash and restricted cash (7,410 ) 2,049 Cash and restricted cash at beginning of year 13,268 11,219 Cash and restricted cash at end of year $ 5,858 $ 13,268 Components of cash and restricted cash: Cash $ 5,783 $ 13,143 Restricted cash 75 125 Total cash and restricted cash $ 5,858 $ 13,268 Supplemental disclosure of cash flow information: Cash paid for interest $ 48 $ 2,371 Supplemental disclosure of noncash investing and financing activities: Issuance of common stock as non cash consideration for asset acquisition $ — $ 20,551 Issuance of common stock upon extinguishment of notes payable and accrued interest $ — $ 22,239 Net liabilities assumed in connection with asset acquisition $ — $ 1,944 Withholding to cover taxes from RSU vesting $ 69 $ — Rights from Private Placement $ — $ 2,250 Finance lease liabilities arising from obtaining new right-of-use assets $ — $ 40 Year Ended December 31, 2021 2020 Collaboration revenue $ 8,044 $ 6,896 Operating expenses: Research and development 26,410 22,310 General and administrative 20,379 11,881 In-process research and development assets acquired 0 28,336 Total operating expenses 46,789 62,527 Loss from operations (38,745 ) (55,631 ) Other income (expense): Change in fair value of preferred unit warrant liability 0 72 Interest expense (1,817 ) (1,900 ) Interest income and other income (expense), net (75 ) (28 ) Gain on debt extinguishment 1,134 0 Total other income (expense), net (758 ) (1,856 ) Net loss $ (39,503 ) $ (57,487 ) Gain on extinguishment of Class B preferred units 0 6,697 Net loss applicable to common shareholders (39,503 ) (50,790 ) Net loss per share, basic and diluted $ (3.84 ) $ (21.57 ) Weighted average common shares outstanding, basic and diluted 10,283,172 2,354,143 Year Ended December 31, 2021 2020 Net loss $ (39,503 ) $ (57,487 ) Other comprehensive loss: Unrealized gains on marketable securities, net of tax of $0 0 0 Comprehensive loss $ (39,503 ) $ (57,487 ) Preferred Units Common Units Defaulting Common Stock Additional Accumulated Accumulated Total Units Amount Units Amount Units Amount Shares Amount Capital Gain (Loss) Deficit Equity/(Deficit) Balances at December 31, 2019 12,391,101 $ 89,699 2,163,099 $ 5,120 — $ — — $ — $ — $ — $ (97,020 ) $ (91,900 ) Issuance of Class C preferred 5,404,588 21,235 — — — — — — — — — — Exchange of Class B preferred (836,319 ) (288 ) — — 836,319 288 — — — — — 288 Gain on extinguishment of — (6,697 ) — — — — — — — — 6,697 6,697 Forfeiture of unvested incentive — — (790 ) — — — — — — — — — Stock/equity-based compensation — — — 2,266 — — — — — — — 2,266 Exchange of preferred units of (16,959,370 ) (103,949 ) — — (836,319 ) (288 ) 3,745,983 4 104,233 — — 103,949 Exchange of common units of — — (2,162,309 ) (7,386 ) — — 2,278,450 2 7,384 — — — Exchange of common stock in — — — — — — 2,708,537 3 60,127 — — 60,130 Fair value of replacement equity — — — — — — — — 471 — — 471 Reclassification of warrant — — - — — — — — 189 — — 189 Private placement of common — — — — — — 1,460,861 1 31,603 — — 31,604 Net loss — — — — — — — — — — (57,487 ) (57,487 ) Balances at December 31, 2020 — $ — — $ — — — 10,193,831 $ 10 $ 204,007 — $ (147,810 ) $ 56,207 Issuance of common stock from — — — — — — 112,833 — 1,419 1,419 Exercises of common stock — — — — — — 9,241 — 84 84 Vesting of restricted stock units — — — — — — 23,146 — — — Issuance of restricted stock — — — — — — 305,663 1 (1 ) — Stock/equity-based compensation — — — — — — — — 5,290 5,290 Net loss — — — — — — — — — (39,503 ) (39,503 ) Balances at December 31, 2021 — $ — — $ — — $ — 10,644,714 $ 11 $ 210,799 $ — $ (187,313 ) $ 23,497 December 31, 2021 2020 Cash flows from operating activities: Net loss $ (39,503 ) $ (57,487 ) Adjustments to reconcile net loss to net cash used in operating activities: Non-cash expense for in-process research and development acquired 0 28,336 Depreciation and amortization expense 631 770 Non-cash lease expense 5,135 2,501 Stock/equity-based compensation expense 5,290 2,266 Other non-cash expense 58 0 Accretion of discounts on marketable securities (9 ) (6 ) Non-cash interest expense 527 535 Gain on debt extinguishment (1,134 ) 0 Change in fair value of preferred unit warrant liability 0 (72 ) Loss on assets held-for-sale 63 — (Gain) on sale of property and equipment 0 (2 ) Changes in operating assets and liabilities, excluding the effect of acquisition: Accounts receivable (5,000 ) — Prepaid expenses and other current assets 1,057 (1,497 ) Deposits 20 (346 ) Operating lease liabilities (4,468 ) (1,688 ) Accounts payable (5,545 ) 2,802 Accrued expenses and other current liabilities (2,994 ) (2,154 ) Deferred revenue (3,043 ) 8,104 Net cash used in operating activities (48,915 ) (17,938 ) Cash flows from investing activities: Purchases of marketable securities (11,267 ) (4,495 ) Proceeds from sales and maturities of marketable securities 14,375 1,350 Purchases of property and equipment (138 ) (246 ) Proceeds from assets held-for-sale 123 — Proceeds from sale of property and equipment — 13 Cash, cash equivalents, and restricted cash acquired in connection with the Merger — 35,939 Merger transaction costs — (1,520 ) Net cash provided by investing activities 3,093 31,041 Cash flows from financing activities: Proceeds from issuance of Class C preferred units, net of offering costs paid — 21,235 Proceeds from private placement of common stock, net of issuance costs — 33,597 Proceeds from Paycheck Protection Program loan — 1,123 Proceeds from at the market offering, net of issuance costs 1,419 — Proceeds from exercise of stock options 84 — Payments of principal portion of long-term debt (2,267 ) — Payments of debt issuance costs related to long-term debt (103 ) (72 ) Payments of finance lease obligations (166 ) (347 ) Net cash (used in) provided by financing activities (1,033 ) 55,536 Net (decrease) increase in cash, cash equivalents and restricted cash (46,855 ) 68,639 Cash, cash equivalents and restricted cash at beginning of period 82,885 14,246 Cash, cash equivalents and restricted cash at end of period $ 36,030 $ 82,885 Supplemental cash flow information: Cash paid for interest $ 1,298 $ 1,287 Supplemental disclosure of noncash investing and financing activities: Additions to property and equipment under finance lease $ — $ 102 Merger transaction costs included in accounts payable and accrued expenses $ — $ 1,169 Offering costs included in accounts payable $ — $ 1,993 Operating lease liabilities arising from obtaining right-of-use assets $ 0 $ 10,219 Fair value of net assets acquired in the Merger, excluding cash, cash equivalents $ — $ 24,662 Conversion of preferred units to common stock $ — $ 104,237 Conversion of preferred unit warrants into common stock warrants $ — $ 189 Kineta, Inc. (formerly Yumanity Therapeutics, Inc.) (together with its The Company is As of December 31, 2022, the Company owned a majority interest of the outstanding issued equity of KCP and all of the outstanding issued equity of KVHF. On November 30, 2023, the Company dissolved KVHF and assumed all of the outstanding issued equity. As of December 31, 2023, the Company owns a majority interest of the outstanding issued equity of KCP. On February 29, 2024, Kineta announced that it had completed a review of its business, including the status of its programs, resources and capabilities. Following this review, Kineta determined that it would implement a significant corporate restructuring to substantially reduce expenses and preserve cash. The restructuring includes a reduction in its workforce by approximately 64% and the termination of enrollment of new patients in its ongoing VISTA-101 Phase 1/2 clinical trial evaluating KVA12123 in patients with advanced solid tumors. Patients currently enrolled in the Reverse Merger and Private Placement On December At the effective time of the Merger, Unless otherwise noted herein, references to the Company’s common share and per-share amounts give retroactive effect to the Reverse Stock Split and Exchange Ratio. The Going The Company has 116 KINETA, INC. Notes to Consolidated Financial Statements Kineta is exploring strategic alternatives that may include, but are not limited to, sale of assets of the action. Kineta may seek additional funds through equity or debt financings or through collaborations, licensing transactions or other sources that may be identified through the Company’s strategic process. However, there can be no assurance that Kineta will be able to complete any such transactions on acceptable terms or otherwise. The failure to obtain sufficient funds on commercially acceptable terms when needed would have a material adverse effect on Kineta’s business, results of operations, and financial condition. These factors raise substantial doubt about Kineta does not currently have any commitments for future funding or additional capital other than the Private Placement. However, as noted above, certain investors have indicated they will not be able to fulfill their contractual obligation to consummate the Private Placement. As such, Kineta has paused or significantly scaled back the development or commercialization of its future product candidates or other research and development initiatives. If Kineta is unable to complete a strategic transaction or raise additional capital in sufficient amounts, Kineta will not be able to continue its business and the Company may need to file for bankruptcy protection. COVID-19 While the Company continues to monitor the impact of the COVID-19 pandemic variants. Clinical trial sites in many countries, including those in which the Company operates, in the past have incurred delays due to COVID-19. Certain of the sites in the The pandemic in the past has caused significant disruptions in the financial markets, and in the future may cause such disruptions, which could impact the Company’s To date, the Company has not incurred impairment losses in the carrying values of its assets as a result of the pandemic and it is not aware of any specific related event or circumstance that would require it to revise its estimates reflected in these consolidated financial statements. Geopolitical Developments Geopolitical developments, such as the current conflict in Ukraine and the conflict in Israel and the Gaza Strip or deterioration in the bilateral relationship between the United States and China, may impact government spending, international trade and market stability, and cause weaker macro-economic conditions. The Basis of Presentation and Consolidation The accompanying consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”) and applicable SEC rules regarding annual financial reporting. The consolidated financial statements include all accounts of the Company, its majority owned subsidiary KCP, and its wholly owned subsidiary, KVHF. On November 30, 2023, the Company dissolved KVHF and assumed all of the outstanding issued equity. All intercompany transactions and balances have been eliminated upon consolidation. Noncontrolling interest in the accompanying consolidated financial statements represents the proportionate share of equity which is not held by the Company. Net income (loss) of the non wholly-owned consolidated subsidiary is allocated to the Company and the holder(s) of the noncontrolling interests in proportion to their percentage ownership considering any preferences specific to the form of equity of the subsidiaries. Use of The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts in the consolidated financial statements and accompanying notes. These estimates form the basis for judgments the Company makes about the carrying values of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of 117 KINETA, INC. Notes to Consolidated Financial Statements on management’s knowledge about current events and expectations about actions the Company may undertake in the future. These judgments, estimates and assumptions Foreign Currencies The Company’s subsidiaries are all located in the U.S. with the U.S. dollar as the functional currency. Certain insignificant transactions during the years ended December 31, 2023 and 2022 were denominated in currencies other than the U.S. dollar. Gains and losses from foreign currency transactions, translated using the average exchange rates prevailing during the respective periods, were not material for all periods presented and are reflected in the consolidated statements of operations as a component of other (expense) income, net. Segment Reporting Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker (the “CODM”), or The Company Cash and Restricted Cash Cash includes cash deposited at several financial institutions in operating accounts. Restricted cash relates to a certificate of deposit with a financial institution to secure a letter of credit obtained for the Company’s leased premises. Restricted cash unavailable for a period longer than one year from the consolidated balance sheet date is classified as a Concentrations of Financial instruments that potentially expose the Company to significant concentrations of credit risk consist primarily of cash Fair To the extent that the valuation is based on models 118 KINETA, INC. Notes to Consolidated Financial Statements Rights from Private Placement The Company determined that the rights from Private Placement is a derivative asset, which requires the asset to be accounted for and reported at fair value on the balance sheet. The fair value is determined using a Monte Carlo simulation based on the contractual funding date at the measurement date, minimum contractual purchase price and historical stock prices. The significant unobservable inputs used in the fair value measurement are volatility, risk-free interest rates and funding probability. Property and Equipment, Net Property and equipment, net is stated at cost less accumulated depreciation and amortization. Depreciation of property and equipment is computed using the straight-line method over the estimated useful lives of the assets, which is five to seven years. Costs of major additions and betterments are capitalized and depreciated on a straight-line basis over their Impairment of Long-Lived Assets The Company reviews the carrying amount of its long-lived assets, including property and equipment and right-of-use assets, for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. If indicators of impairment exist, an impairment loss is recognized when the estimated undiscounted future cash flows expected to result from the The Company has elected the fair value option to account for certain of its notes payable (see Note 6). The Company concluded that it was appropriate to apply the fair value option to these certain notes payable because no component of the notes payable were required to be recognized as a component of stockholders’ equity. The Company recorded these notes payable at their estimated fair value with changes in estimated fair value recorded as a component of other (expense) income in the consolidated statement of operations. Under the fair value option, any direct costs and fees related to the notes payable are expensed as incurred. Leases The Company determines at the inception of a contract if such arrangement is or contains a lease by assessing whether it conveys the right to control the use of an identified asset for a period of time in exchange for consideration. If a lease is identified, classification is determined at lease commencement as an operating lease or finance lease. The Company recognizes a right-of-use (“ROU”) asset and a lease liability in the consolidated balance sheets for all leases with an initial term of greater than 12 months. Leases with an initial term of 12 months or less are not recognized in the consolidated balance sheets, with payments recognized as expense on a straight-line basis over the lease term. Lease liabilities are recognized at the present value of the future lease payments at the lease commencement date. The present value of future lease payments is determined by using the implicit interest rate in the lease, if readily determinable, otherwise, the Company estimates its incremental borrowing rate at the inception of the lease to discount lease payments. The incremental borrowing rate reflects the estimated interest rate that the Company would have to pay to borrow on a collateralized basis an amount equal to the lease payments in a similar economic environment over a similar term. ROU assets are determined based on the corresponding lease liability adjusted for any lease payments made at or before commencement, initial direct costs, and lease incentives. The ROU asset also includes impairment charges if the Company determines the ROU asset is impaired. Lease expenses are recognized, and the ROU assets are amortized on a straight-line basis over the lease term. The Company has elected to not separate lease and non-lease components for its leased assets and accounts for all lease and non-lease components of its agreements as a single lease component. Variable costs are not included in the measurement of ROU assets and lease liabilities, which are expensed as incurred. The Company considers a lease term to be the noncancelable period that it has the right to use the underlying asset, including any periods where it is reasonably assured the Company will exercise the option to extend the contract. Warrants to Purchase Common Stock 119 KINETA, INC. Notes to Consolidated Financial Statements The Company has issued warrants to purchase the Company’s Asset Acquisitions Acquisitions of assets or a group of assets that do not meet the Revenue Collaboration Revenues The Company License Revenues The Company enters into license agreements under which it licenses certain intellectual property rights to its product candidates to third parties. The terms of these In determining the appropriate amount of revenue For arrangements KINETA, INC. Notes to Consolidated Financial Statements or less. Grant Revenues Grants received, including cost reimbursement agreements, are assessed to determine if the Research and Development Expenses Research and development expenses represent costs Accrued Research The Company The Company makes significant judgments and estimates General and Administrative Expenses General and administrative expenses consist primarily of employee-related expenses, including salaries, benefits and noncash stock-based compensation for personnel in executive, finance and accounting, and other administrative functions, as well as fees paid for legal, accounting and tax services, consulting fees and facilities costs The Company measures stock-based compensation related to stock-based awards The Black-Scholes option pricing model requires the Company to make assumptions and judgments about the inputs used in the calculations as follows: Expected Term – The Company’s expected term represents the period that the stock-based awards are expected to be outstanding and is determined using the simplified method (based on the mid-point between the vesting date and the end of the contractual term) for employee options. Exercise Price – The Company Expected Volatility – As the Company has only been a publicly-traded company for 121 KINETA, INC. Notes to Consolidated Financial Statements option grants. The comparable companies were chosen based on their similar industry, size, or stage in the product development life cycle and financial leverage. Risk-Free Interest Rate – The risk-free interest rate is based on the U.S. Treasury zero coupon issues in effect at the time of grant for periods corresponding with the expected term of option. Expected Dividend – Other than the Distribution, the Company has never paid dividends on its common stock and has no plans to pay dividends on its common stock. Therefore, it uses an expected dividend yield of zero. Other (Expense) Income Interest Income Interest income consists of interest earned on short term money market accounts. Interest Expense Interest expense consists of interest charged on outstanding invoices and outstanding borrowings associated with the Company’s debt arrangements primarily consisting of borrowings under several notes payable agreements. Interest is expensed when incurred. Change in Fair Value of Rights from Private Placement The Company Change in Change in fair value of notes payable relates to the Warrant expense Warrant expense relates to warrants issued to current debt holders that converted their debt to equity in 2022. The expense was determined as the fair value of the warrants provided upon issuance. Income The Company recognizes the tax benefit from Basic net Comprehensive loss represents the change in the Company’s stockholders’ equity from all sources other than investments by or distributions to stockholders. The Company has no items of other comprehensive loss, and as such, net loss is the same as comprehensive loss. Accounting Pronouncements 122 KINETA, INC. Notes to Consolidated Financial Statements In On December Number of shares owned by Proteostasis stockholders (1) 2,708,537 Multiplied by fair value per share of Proteostasis common stock (2) $ 22.20 Fair value of shares of combined organization owned by Proteostasis Stockholders $ 60,130 Fair value of Proteostasis stock options assumed in Merger (3) 471 Transaction costs 2,689 Total purchase price $ 63,290 (in thousands) Number of shares owned by Yumanity shareholders (1) 1,553 Multiplied by fair value per share of Yumanity common stock (2) $ 13.23 Fair value of shares of combined organization owned by Yumanity shareholders 20,551 Transaction costs (3) 5,641 Total purchase price $ 26,192 The purchase price for the Merger was allocated to the (in thousands) Assets: Cash and cash equivalents $ 35,111 $ 9,226 Accounts receivable 100 Prepaid expenses and other current assets 703 176 Assets held-for-sale 250 Property and equipment, net 290 65 Restricted cash 50 In-process research and development 28,336 18,860 Operating lease right-of-use assets 15,166 Restricted cash 828 Current liabilities (7,171 ) Operating lease liabilities (10,223 ) Liabilities: Accounts payable (296 ) Accrued expenses and other current liabilities (1,547 ) Deferred revenue (442 ) Total purchase price $ 63,290 $ 26,192 The acquired in-process research and development The carrying amounts of the Company’s financial instruments, including cash, restricted cash, and accounts payable, approximate fair value due to the short-term nature of those instruments. Rights from Private Placement In connection and concurrently with the execution of the Merger Agreement, on June 5, 2022, the Company entered into a financing agreement, as amended on October 24, 2022, December 5, 2022 and March 29, 2023 May 1, 2023, July 21, 2023 and October 13, 2023 (such financing agreement, 123 KINETA, INC. Notes to Consolidated Financial Statements as amended, the “Securities Purchase Agreement”), to sell shares of the Company’s common stock in a private placement (the “Private Placement”). The first closing of the Private Placement closed on December 16, 2022, and the Company issued 649,346 shares of its common stock and received net proceeds of $7.4 million. The second closing of the Private Placement for an aggregate purchase price of $22.5 million was expected to occur on April 15, 2024. However, in February 2024, certain investors indicated they will not be able to consummate the second closing of the Private Placement. With respect to the second closing, the Company is obligated to sell and issue a number of shares of its common stock and the investors are obligated to buy such shares by the specified date and price equal to the volume-weighted average price of Company common stock for the five trading days prior to closing on April 15, 2024 (“VWAP”) plus 10% of the VWAP; provided, however, that the share purchase price shall be at least equal to the closing price of the Company’s common stock on March 29, 2023. The Company determined that the rights from Private Placement is a derivative asset, which requires the asset to be accounted for at fair value. The fair value was determined using a Monte Carlo simulation based on the contractual funding date at the measurement date, minimum contractual purchase price of $3.18 and The following Years Ended December 31, 2023 2022 (in thousands) Balance at beginning of period $ 2,250 $ — Increase from Private Placement — 2,250 Change in fair value of rights from Private Placement 1,582 — Balance at end of period $ 3,832 $ 2,250 2022 & 2020 Notes Payable The Company elected the fair value option to account for certain convertible notes payable and notes payable, referred to as the 2022 convertible notes, 2020 convertible notes and 2020 notes (see Note 6), respectively, and collectively the 2022 & 2020 notes payable. The 2020 convertible notes and 2020 notes are referred to as the 2020 notes payable. Upon the closing of the Merger in December 2022, the 2022 convertible notes and 2020 convertible notes were settled with shares of the Company’s common stock (see Note 6). 2022 Convertible Notes February 2022 and April 2022 Convertible Notes The 2022 convertible notes issued in February 2022 and April 2022 were valued using a scenario-based analysis and a discounted cash flow model. Two primary scenarios were considered: the qualified financing scenario and the automatic conversion scenario. The value of these 2022 convertible notes under each scenario was probability weighted to arrive at the estimated fair value The significant unobservable inputs used in the fair value measurement of these 2022 convertible notes during 2022 prior to settlement in December 2022 were as follows: discount rate ranging from 33.6% to 41.2%, timing of the qualified financing ranging from 0.2 years to 0.6 years, timing of the automatic conversion scenario ranging from 0.4 years to 1.0 year, probability of a qualified financing ranging from 80% to 90% and August 2022, September 2022 and October 2022 Convertible Notes The Company also issued 2022 convertible notes in August 2022, September 2022 and October 2022 that were issued and accounted for at fair value The significant unobservable inputs used in the fair value measurement of these 2022 convertible notes from inception prior to settlement in December 2022 were as follows: discount rate of 41.2%, timing of the repayment scenario based on contractual maturity date of 2.0 years and timing of the automatic conversion scenario of 0.2 years, which resulted in a fair value of these 2022 convertible notes of $0.8 million. Fair Value Measurements at December 31, 2021 Using: Level 1 Level 2 Level 3 Total Assets: Cash equivalents: Money market funds $ 34,136 $ 0 $ 0 $ 34,136 Marketable securities: Commercial paper 0 1,399 0 1,399 $ 34,136 $ 1,399 $ 0 $ 35,535 Fair Value Measurements at December 31, 2020 Using: Level 1 Level 2 Level 3 Total Assets: Cash equivalents: Money market funds $ 77,129 $ 0 $ 0 $ 77,129 Commercial paper 0 1,800 0 1,800 Marketable securities: Commercial paper 0 4,498 0 4,498 $ 77,129 $ 6,298 $ 0 $ 83,427 2020 Convertible Notes KINETA, INC. Notes to Consolidated Financial Statements The 2020 convertible notes were valued The significant unobservable inputs used in 2020 Notes The 2020 notes were valued using a discounted cash flow model based on the contractual payment dates, a discount rate and the contractual maturity date. The significant unobservable inputs used in the fair value measurement of the 2020 note for the year ended December 31, 2023 were as follows: discount rates ranging from 13.0% to 15.0% and contractual payment dates ranging from 0.6 years to 1.3 years, which resulted in a fair value range for the 2020 note of $225,000 to $241,000. The significant unobservable inputs used in the fair value measurement of the 2020 notes for the year ended December 31, 2022 were as follows: discount rate ranging from 13.0% to 41.2% and contractual payment dates ranging from 0.1 years to 1.8 years, which resulted in a fair value range for the 2020 notes of $0.2 million to $1.6 million. The following table provides a Preferred Unit Fair value at December 31, 2019 $ 261 Change in fair value (72 ) Reclassification of warrant liability to permanent equity (189 ) Fair value at December 31, 2020 $ 0 December 31, 2021 Amortized Gross Gross Fair Commercial paper $ 1,399 $ 0 $ 0 $ 1,399 $ 1,399 $ 0 $ 0 $ 1,399 �� December 31, 2020 Amortized Gross Gross Fair Commercial paper $ 4,498 $ 0 $ 0 $ 4,498 $ 4,498 $ 0 $ 0 $ 4,498 Years Ended December 31, 2023 2022 (in thousands) Balance at beginning of period $ 219 $ 17,830 Change in fair value of 2022 & 2020 notes payable 22 15,280 Issuance of 2022 convertible notes — 6,746 Change in fair value of debt extinguishment — (673 ) Partial settlement of 2020 notes payable — (4,000 ) Settlement of 2022 & 2020 notes payable — (34,964 ) Balance at end of period $ 241 $ 219 Property and equipment, net consisted of the December 31, 2021 2020 Laboratory equipment $ 1,339 $ 1,674 Office equipment, computers and software 211 209 Furniture and fixtures 170 170 $ 1,720 2,053 Less: Accumulated depreciation and amortization (1,333 ) (1,179 ) $ 387 $ 874 Assets held-for-sale $ 0 $ 250 December 31, 2023 2022 (in thousands) Laboratory equipment $ — $ 779 Computer and software — 73 Leasehold improvements — 14 Total property and equipment — 866 Less: Accumulated depreciation and amortization — 617 Total property and equipment, net $ — $ 249 Depreciation and amortization expense was $ 125 KINETA, INC. Notes to Consolidated Financial Statements Accrued Expenses and Other Current Liabilities Accrued expenses and other current liabilities consisted of the following as of the periods presented: December 31, 2023 2022 (in thousands) Compensation and benefits $ 1,312 $ 745 Accrued interest 417 132 Accrued clinical trial and preclinical costs 251 404 Professional services 97 2,176 Other 134 70 Total accrued expenses and other current liabilities $ 2,211 $ 3,527 Notes payable outstanding consisted of the following as of the periods presented: December 31, 2023 December 31, 2022 Principal Fair Value Principal Fair Value (in thousands) Notes payable: 2020 notes $ 250 $ 241 $ 250 $ 219 Other notes payable 379 379 379 379 Small Business Administration loan 150 150 150 150 Total notes payable $ 779 770 $ 779 748 Less: current portion 620 — Notes payable, net of current portion $ 150 $ 748 The Company elected the fair value option for the 2020 notes (see Note 4). The other notes payable approximate their fair value because interest rates are at prevailing market rates. Expected future minimum principal payments under the Company’s notes payables as of December 31, 2023 were as follows: Total (in thousands) Years 2024 $ 629 2025 — 2026 — 2027 2 2028 3 Thereafter 145 Total notes payable $ 779 Less: current portion 620 Notes payable, net of current portion $ 159 2022 Convertible Notes In February 2022 and April 2022, the Company raised $4.8 million in total from two investors, including one investor that was previously a related party at the time of investment, pursuant to convertible notes purchase agreements (see Note 16). These 2022 convertible notes purchase agreements provided that the notes mature upon demand of the holder at any time 24 months after the date of issuance and pay a 6% interest. Additionally, these 2022 convertible notes automatically would convert into the Company’s non-voting common stock at 85% of the then current share price on the earlier of (i) the date that is 12 months from the date of issuance, or (ii) at a public market event such as an initial public offering or merger. These 2022 convertible notes also allowed for optional conversion at any time during the 12-month period after issuance and could be repaid at any time without penalty. The use of proceeds could be used to repay other debt obligations and for general corporate use. In August 2022, September 2022 and October 2022, the Company raised $1.9 million in total from several investors, pursuant to convertible notes purchase agreements, which were issued at fair value. Three investors were also issued 5,000 warrants to purchase shares of the Company’s non-voting common stock (see Note 9) with a fair value of $146,000 upon issuance that qualified for equity classification and were accounted for as 126 KINETA, INC. Notes to Consolidated Financial Statements interest expense. These convertible notes purchase agreements provide that the 2022 convertible notes mature upon demand of the holder at any time 24 months after the date of issuance and pay a 6% interest. Additionally, these 2022 convertible notes would automatically convert into the Company’s non-voting common stock at the lesser of (a) $1.61 per share or (b) 85% of the then current share price on the earlier of (i) the date that is 12 months from the date of issuance, or (ii) a change of control event such as a merger, consolidation or other capital reorganization or business combination. These 2022 convertible notes also allowed for optional conversion at any time during the 12-month period after issuance and could be repaid at any time without penalty. The use of proceeds could be used to repay other debt obligations and for general corporate use. In October 2022 and December 2022, the 2022 convertible notes were amended to provide (i) that the conversion price would be equal to the conversion amount divided by $0.995 upon automatic conversion and (ii) for the issuance of 55,000 warrants to purchase shares of the Company’s non-voting common stock (see Note 9), with exercise contingent upon the Merger closing, including to one investor that was previously a related party (see Note 16), with a fair value of $1.5 million upon issuance. The Company determined the contingent exercise provision was indexed to the Company’s operations and the warrants qualified for equity classification. As the 2022 convertible notes were accounted for under the fair value option, all lender fees, including the cost of the warrants, were expensed as incurred. Upon the closing of the Merger, the outstanding principal and accrued interest under the 2022 convertible notes was $6.8 million, with a fair value of $13.0 million, and was settled by issuing 471,000 shares of the Company’s non-voting common stock. The 2022 convertible notes were fair valued immediately prior to settlement based on the Company’s market stock price of the shares issued on the date the Merger closed, such that there was no gain or loss recognized upon extinguishment. 2020 Convertible Notes In October 2020, the Company refinanced certain convertible notes payable, or the 2020 convertible notes, with an aggregate principal amount of $ In February 2022, the Company made a $4.0 million cash payment of principal to one of its creditors that is a related party (see Note 15) as a partial repayment for a note issued pursuant to the 2020 convertible notes and recognized a $0.7 million gain on extinguishment. In December 2022, the 2020 convertible notes were amended to provide for automatic conversion upon a merger at a conversion price equal to the conversion amount divided by $0.995. Upon the closing of the Merger, the outstanding principal and accrued interest under the 2020 convertible notes was $10.9 million, with a fair value of $21.8 million, and was settled by issuing 754,000 shares of the Company’s non-voting common stock, including with related parties (see Note 15). The 2020 convertible notes were fair valued immediately prior to settlement based on the Company’s market stock price of the shares issued on the date the Merger closed, such that there was no gain or loss recognized upon extinguishment. 2020 Notes In October 2020, the Company refinanced certain notes payable (the “2020 notes”), with an aggregate principal amount of $3.0 million with various investors, including one investor that is a related party (see Note 13). The interest rate was reduced on the 2020 notes from 16.0% to 6.0% from October 2020 until the earlier of (i) the Company raises at least $25.0 million in a single transaction or series of transactions after October 2020 and (ii) the original maturity dates (that is, various dates in the first quarter of 2022), after which the interest rate increases to 16.0%. The outstanding principal is due upon demand of the majority of the lenders with respect to (i) 50% on or after nine months after the original maturity date (or on or after various dates in the fourth quarter of 2022) and (ii) 50% on or after fifteen months after the original maturity date (or on or after various dates in the second quarter of 2023). The Company may repay the 2020 notes at any time without penalty. Upon bankruptcy the lender can accelerate all amounts due immediately. In August 2021 and September 2021, outstanding principal and accrued interest under the 2020 notes with a fair value of $0.9 million was settled by issuing 33,000 shares of the Company’s non-voting common stock at fair value (based on a recent valuation) to the holders. As the 2020 notes were valued pursuant to the fair value election, an immaterial gain was recognized upon extinguishment. In August 2022, the Company settled $1.4 million in outstanding principal and accrued interest, including with a person that was a related party at the time of conversion (see Note 15) by issuing 59,000 shares of the Company’s non-voting common stock at a 15% discount, recognizing a $0.2 million loss upon extinguishment. The Company extended the maturity date for the remaining 2020 Notes with a principal balance of $0.25 million to July 31, 2024 and reduced the interest rate to 6%, which was accounted for as a modification. 127 KINETA, INC. Notes to Consolidated Financial Statements Other Notes Payable The Company issued several other notes payable in 2019 and early 2020 at a 12.0% interest rate per annum, with the principal amounts due in full at maturity and interest due monthly or quarterly. The other notes payable were due to mature at various dates between December 2020 through early 2022. The other notes payable were amended in October 2020 to increase the interest rate to 13.0% and extend the maturity date to be on demand by a majority of the holders on or after April 7, 2022, which resulted in a modification of the other notes payable. The Company may prepay the other notes payable at any time without penalty. In June 2021 and July 2021, outstanding principal and accrued interest under the other notes payable of $1.4 million was settled by issuing 52,000 shares of the Company’s non-voting common stock at fair value (based on a recent valuation) to the holders. As the other notes payable approximated to their fair value, no gain or loss was recognized upon extinguishment. In February 2022 and April 2022, outstanding principal and accrued interest under the other notes payable of $0.3 million was settled by issuing 2,400 shares of Small Business Administration Loan In August 2020, the Company Leases Operating Lease The Company leases office and laboratory premises in Seattle, Washington pursuant to a lease agreement that commenced in April 2011 and expires in July 2024. The agreement requires monthly lease payments, is subject to annual rent escalations during the lease term, and contains two five-year options to extend the lease term.In June 2020, the Company amended the lease agreement to reduce the leased space for the premises from approximately 22,064 square feet to approximately 14,870 square feet, which was accounted for as a lease modification and partial termination of the lease. Under the lease agreement, the Company is required to pay certain operating costs, in addition to rent, such as common area maintenance, taxes, and utilities. Such additional charges are considered variable lease costs and are recognized in the period in which they are incurred. Rent expense was $866,000 for the year ended December 31, 2023 and variable costs were $597,000. Rent expense was $889,000 for the year ended December 31, 2022 and variable costs were $511,000. The Company’s operating leases include various covenants, indemnities, defaults, termination rights, security deposits and other provisions customary for lease transactions of this nature. Future undiscounted payments due under the operating lease as of December 31, 2023 were as follows: Years (in thousands) 2024 $ 561 Less: Imputed interest (14 ) Operating lease liability 547 Less: Operating lease liability, current portion (547 ) Operating lease liability, net of current portion $ — Supplemental information on the Company's operating leases was as follows: 128 KINETA, INC. Notes to Consolidated Financial Statements Years Ended December 31, 2023 2022 Cash paid for operating lease agreement (in thousands) $ 936 $ 909 Remaining lease term (in years) 0.6 1.6 Incremental borrowing rate 10 % 10 % The Company subleases portions of its premises in Seattle to third parties. Under the first sublease agreement, which commenced in December 2017, the Company subleases approximately 1,850 square feet. In October 2020 the sublease expiration date was extended from December 2020 to December 2022. In September 2022, the sublease expiration date was extended from December 2022 to December 2023. In December 2023, the sublease expiration date was extended from December 2023 to July 2024. Sublease income was $194,000 for the year ended December 31, 2023 and $188,000 for the year ended December 31, 2022 and was recorded within operating expenses. As of December 31, 2023, the total minimum rentals to be received under the remaining noncancelable sublease was $70,000. Finance Leases During the year ended December 31, 2023, the Company paid off the remaining finance leases and has no finance lease liabilities as of December 31, 2023. Supplemental information on the Company’s financing leases was as follows (cash paid for finance lease agreements was not material): December 31, 2023 2022 Weighted average remaining lease term (in years) - 3.2 Incremental borrowing rate 0.0 % 9.3 % Indemnification In the ordinary course of business, the Company enters into agreements that may include indemnification provisions. Pursuant to such agreements, the Company may indemnify, hold harmless and defend an indemnified party for losses suffered or incurred by the indemnified party. Some of the provisions will limit losses to those arising from third-party actions. In some cases, the indemnification will continue after the termination of the agreement. The maximum potential amount of future payments the Company could be required to make under these provisions is not determinable. The Company has not incurred material costs to defend lawsuits or settle claims related to these indemnification provisions. The Company has also entered into indemnification agreements with its directors and officers that may require the Company to indemnify its directors and officers against liabilities that may arise by reason of their status or service as directors or officers to the fullest extent permitted under the Delaware General Corporation Law. The Company currently has directors’ and officers’ insurance. Other Commitments The Company has various manufacturing, clinical, research and other contracts with vendors in the conduct of the normal course of its business. Such contracts are generally terminable with advanced written notice and payment for any products or services received by the Company through the effective time of termination and any noncancelable and nonrefundable obligations incurred by the vendor at the effective time of the termination. In the case of terminating a clinical trial agreement at a particular site, the Company would also be obligated to provide continued support for appropriate medical procedures at that site until completion or termination. Executive Employment Agreements Effective September 20, 2022, the Company entered into an at-will employment agreement (“Baker Employment Agreement”) with Keith Baker, its Chief Financial Officer; and effective September 28, 2022, the Company entered into at-will employment agreements (together with the Baker Employment Agreement, the “Executive Employment Agreements”) with Shawn Iadonato, its Chief Executive Officer, Craig Philips, its President and Pauline Kenny, its General Counsel. The Executive Employment Agreements provide that, if the executive’s employment is terminated without Cause (as defined in the Executive Employment Agreements) or the executive resigns for Good Reason (as defined in the Executive Employment Agreements), provided that the executive signs the Release (as defined in the Executive Employment Agreement), the executive will be entitled to (i) accrued compensation, (ii) 39 weeks of pay (52 weeks in the case of Chief Executive Officer) (currently estimated at approximately $1.3 million in the aggregate), (iii) nine (9) months of COBRA benefits (12 months in the case of Chief Executive Officer) for executive and eligible dependents, and (iv) three (3) additional months of vesting of unvested and outstanding equity awards. If executive’s employment is terminated without Cause or the executive resigns for Good Reason within the Change in Control Protection Period (as defined in the Executive Employment Agreements), then in addition to (i)-(iv) above, executive will receive current year pro-rated cash bonus. 129 KINETA, INC. Notes to Consolidated Financial Statements Anti-VISTA Antibody Program In-License Agreement In August 2020, Kineta entered into an Option and License Agreement with GigaGen, Inc. (“GigaGen”), which was amended in November 2020 and further amended in May 2023 (such agreement, as amended, the “VISTA Agreement”) to in-license certain intellectual property and antibodies for the VISTA/KVA12123 drug program. Pursuant to the terms of the VISTA Agreement, GigaGen granted Kineta an exclusive (even as to GigaGen) world-wide license, with the right to grant sublicenses to research, develop, make, have made, use, have used, offer for sale, sell, have sold, distribute, import, have imported, export and have exported and otherwise exploit the licensed antibodies and licensed products. License expenses for the VISTA Agreement were $250,000 for the year ended December 31, 2023 and $250,000 for the year ended December 31, 2022. Under the VISTA Agreement, GigaGen is eligible to receive approximately $20.4 million in development and regulatory milestone payments and up to $11.0 million in sales milestone payments. In addition, GigaGen is eligible to receive low single-digit royalty percentages based on net sales. Kineta is responsible (with input from GigaGen) for the preparation, filing, prosecution and maintenance of all patents and patent applications, and all associated costs. The VISTA Agreement shall remain in effect on a licensed product-by-licensed product and country-by-country basis, until the expiration of the royalty term for a licensed product in a country, which, based on the expiration of the last-to-expire valid claim of the two current patent applications (without any patent term adjustment or extensions) would be February 2042 and March 2044, respectively. Kineta may terminate the VISTA Agreement with 30 days’ written notice to GigaGen. Either party has the right to terminate the VISTA Agreement upon a material breach of the other party that is not cured within 90 days after the breaching party receives written notice of such breach from the non-breaching party. Anti-CD27 Agonist Antibody Program In-License Agreement In June 2021, Kineta entered into an Option and License Agreement with GigaGen, as amended in July 2022, December 2022, May 2023 and December 2023 (such agreement, as amended, the “CD27 Agreement”) to in-license certain intellectual property rights and antibodies for the CD27 drug program. Pursuant to the terms of the CD27 Agreement, GigaGen granted Kineta an exclusive (even as to GigaGen) world-wide license, with the right to grant sublicenses to research, develop, make, have made, use, have used, offer for sale, sell, have sold, distribute, import, have imported, export and have exported and otherwise exploit the licensed antibodies and licensed products. License expenses for the CD27 Agreement were zero for the year ended December 31, 2023 and zero for the year ended December 31, 2022. Under the CD27 Agreement, GigaGen is eligible to receive approximately $20.4 million in development and regulatory milestone payments and up to $11.0 million in sales milestone payments. In addition, GigaGen is eligible to receive low single-digit royalty percentages based on net sales. Kineta is responsible (with input from GigaGen) for the preparation, filing, prosecution and maintenance of all patents and patent applications, and all associated costs. The CD27 Agreement shall remain in effect on a licensed product-by-licensed product and country-by-country basis, until the expiration of the royalty term for a licensed product in a country, which, based on the expiration of the last-to-expire valid claim of the current provisional patent application (without any patent term adjustment or extensions) would be September 2044. Kineta may terminate the CD27 Agreement with 30 days’ written notice to GigaGen. Either party has the right to terminate the CD27 Agreement upon a material breach of the other party that is not cured within 90 days after the breaching party receives written notice of such breach from the non-breaching party. Merck Neuromuscular License Agreement In connection with the Merger, the Company became the successor in interest to an exclusive license and research collaboration agreement (the Genentech In connection with the Merger, Kineta became the successor in interest to an exclusive technology transfer and license agreement with Genentech, Inc. to support research, development and commercialization of a small molecule product with an undisclosed target (the “Genentech Small Molecule License Agreement”). The Company did not earn revenues under the Genentech Small Molecule License Agreement for the years ended December 31, 2023 and 2022. FAIR Therapeutics 130 KINETA, INC. Notes to Consolidated Financial Statements In connection with the Merger, Kineta became the successor in interest to an exclusive license agreement with FAIR Therapeutics, B.V. to support research, development and commercialization of products for the treatment of December 31, 2021 2020 Accrued employee compensation and benefits $ 1,763 $ 4,295 Accrued external research and development expenses 1,633 1,780 Accrued professional fees 901 987 Other 549 789 $ 4,846 $ 7,851 December 31, 2021 2020 Principal amount of long-term debt $ 12,733 $ 16,123 Less: Current portion of long-term debt (5,805 ) (2,891 ) Long-term debt, net of current portion 6,928 13,232 Debt discount, net of accretion (217 ) (348 ) Accrued end-of-term payment 646 353 Long-term debt, net of discount and current portion $ 7,357 $ 13,237 Purchase Common Stock As of December 31, Year Expiration Number Outstanding as of December 31, 2022 Issued Exercised Cancelled/Expired Number Outstanding as of December 31, 2023 Range of 2013 12 — — (12 ) — 2017 March 2025 - June 2025 131 — — (5 ) 126 $0.14 - $21.80 2019 March 2025 - April 2027 44 — — — 44 $0.14 - $21.80 2020 45 — (41 ) (4 ) — 2022 August 2025 - December 2029 301 — (178 ) — 123 $0.14 - $168.35 2023 April 2028 - October 2033 — 3,688 (477 ) — 3,211 $0.001 - $5.26 Total number of shares 533 3,688 (696 ) (21 ) 3,504 Equity Raises - Registered Direct Offering April 2023 Year Ending December 31, 2022 $ 5,805 2023 6,341 2024 586 2025 0 2026 0 $ 12,732 On Each April 2023 Pre-Funded Warrant represents the right to purchase one share of In a concurrent private placement (the “April 2023 Private Placement” and, together with the April 2023 Registered Offering, the “April 2023 Offering”), the Company issued to the April 2023 Investor warrants to purchase up to 1,425,179 shares of common stock (the “April 2023 Common Warrants”) at an exercise price of $4.08 per share. The April 2023 Common Warrants are exercisable immediately and will expire five and one-half years from the initial exercise date. In connection with the April 2023 Offering, the Company entered into an engagement letter with H.C. Wainwright & Co., LLC (“Wainwright”), pursuant to which Wainwright agreed to serve as the exclusive placement agent for the issuance and sale of October 2023 131 KINETA, INC. Notes to Consolidated Financial Statements On October 3, 2023, the Company, entered into a Securities Purchase Agreement (the “October 2023 Purchase Agreement”) with an institutional investor (the “October 2023 Investor”) pursuant to which the Company issued and sold, in a registered direct offering priced at-the-market under the rules of Each October 2023 Pre-Funded Warrant represents the In a concurrent private placement (the “October 2023 Private Placement” and, together with the October 2023 Registered Offering, the “October 2023 Offering”), the Company issued to the October 2023 Investor warrants to purchase up to 890,208 shares of common stock (the “October 2023 Common Warrants”) at an exercise price of $3.25 per share. The October 2023 Common Warrants are exercisable immediately and will expire five and one-half years from the initial exercise date. In connection with the October 2023 Offering, the Company entered into an engagement letter with Wainwright, pursuant to which Wainwright agreed to serve as the exclusive placement agent for the issuance and sale of securities of the Company pursuant to the October 2023 Purchase Agreement. As compensation for such placement agent services, the Company paid Wainwright an aggregate cash fee equal to $210,000, a non-accountable expense of $35,000 and $50,000 for legal and other expenses as actually incurred. The total offering-related fees were approximately $310,000, which resulted in net proceeds to the Company of $2.7 million. On October 5, 2023, the Company also issued to Wainwright or its designees warrants to purchase 44,510 shares of common stock (the “October 2023 Wainwright Warrants”). The October 2023 Wainwright Warrants have a term of five years from the commencement of sales in the October 2023 Offering, and have an exercise price of $4.2125 per share. Warrant Exercises During the year ended December 31, 2023, the Company issued 696,000 shares of its common stock upon exercise of warrants and received proceeds of $30,000. The exercise price of all shares exercised during the year ended December 31, 2023 ranged from $0.001 to $0.14. In August 2022, the Company issued 2,000 warrants with a fair value of $62,000 to purchase share of its common stock for professional services that was recorded as compensation within general and In September 2022 and October 2022, the Company In October 2022, the Company issued 415,000 warrants to purchase shares of the Company’s non-voting common In December 2022, the Company issued 121,000 warrants to purchase shares of its common stock to existing stockholders, each at an exercise price of $0.14, with exercise contingent upon the Merger closing. The Company determined that the contingent exercise provision was indexed to the Company’s operations and the warrants qualified for equity classification. As the warrants issued to the certain existing stockholders results in In December 2022, the Company issued 104,000 warrants Upon completion of the Merger in December 2022, the Company issued 14,000 warrants to purchase shares of its common stock to former Yumanity warrant holders. During the year ended December 31, 2022, the Company issued 53,000 shares of its common stock upon exercise of warrants and received proceeds of $71,000. The exercise price of all shares exercised Common Stock 132 KINETA, INC. Notes to Consolidated Financial Statements Upon completion of the Merger in December Common stock reserved for future issuance consisted of the December 31, (in thousands) Shares reserved for stock options and restricted stock units to purchase 1,983 Shares reserved for future issuance of equity awards 932 Shares reserved for exercise of warrants 3,504 Total 6,419 For the year ended December 31, 2023, the Company issued 1.2 million shares of its common stock and issued pre-funded warrants exercisable for up to 1,257,387 shares of our common stock to institutional and individual investors, raising net proceeds of $8.6 million. For the year ended December 31, 2022, the Company issued 58,000 shares of its common stock to individual investors, raising net proceeds of $1.6 million, excluding the Private Placement (see below). Private Placement The Private Placement (see Note 1) provides for the issuance of shares of the Company’s common stock in two closings, one of which occurred immediately following the closing of the Merger and one of which was expected to occur on April 15, 2024. However, in February 2024, certain investors indicated they will not be able to consummate the second closing of the Private Placement. The first closing of the Private Placement occurred on December 16, 2022 and the Company issued 649,346 shares of its common stock and received net proceeds of $7.4 million to investors that are related parties (see Note 16). Issuance Date Contractual Class of Number of Exercise August 14, 2015 10 Common 74,622 $ 24.05 October 9, 2015 10 Common 7,798 $ 24.05 June 14, 2018 10 Common 2,152 $ 30.13 December 20, 2019 10 Common 15,414 $ 18.98 99,986 In connection with the Private Placement in December 2022, the Company issued National Institutes of Health The Company was awarded a cost-reimbursable grant from the National Institutes of Health (the “NIH”), a federal medical research agency supporting scientific studies, to support the Company’s research studies for arenavirus hemorrhagic fever. This award was based on budgeted direct and indirect costs and may only be used for budgeted costs as allowable under certain government regulations and NIH’s policy and compliance requirements, subject to government audit. This award was $1.1 million for the The Company recognized KINETA, INC. Notes to Consolidated Financial Statements The following table shows the activity for the Company’s licensing revenue agreements and deferred revenue (in thousands): December 31, 2023 2022 (in thousands) Balance as of beginning of period $ — $ 1,041 Decrease for provision of research services — (1,041 ) Balance as of end of period $ — $ — Merck In June 2023, the Company achieved a development milestone pursuant to the Merck Neuromuscular License Agreement, which triggered a $5.0 million payment. This collaboration focused on the discovery and development of novel candidates for the treatment of ALS. Merck will continue to advance the research program for the ALS pipeline, one of the two pipeline programs licensed under the Merck Neuromuscular License Agreement. As a result, the Company is eligible to receive up to an additional $255.0 million in development milestones, sales milestones and royalties on net sales. Following this milestone, Merck will assume sole responsibility for all future development and commercialization for the ALS program. The Company recognized one-time licensing revenue of $5.0 million for the year ended December 31, 2023 and zero or the year ended December 31, 2022 under the Merck Neuromuscular License Agreement. Genentech, Inc. In April 2018, the Company entered into an exclusive option and license agreement with Genentech, as amended in November 2019 and October 2020 (such agreement, as amended, the “Genentech KCP506 License Agreement”), to develop the Company’s α9/α10 nicotinic acetylcholine receptor (“nAChR”) antagonists for the treatment of chronic pain. On December 27, 2022, the Company through its subsidiary KCP, received written notice from Genentech of its termination of the Genentech KCP506 License Agreement. The Company recognized license revenue of zero for the year ended December 31, 2023 and $1.0 million for the year ended December 31, 2022. There was no deferred revenue related to this license as of December 31, 2023 as the Genentech KCP506 License Agreement was terminated in December 2022. The following table shows the activity for the Company’s collaboration revenue agreement and deferred revenue (in thousands): December 31, 2023 2022 (in thousands) Balance as of beginning of period $ 442 $ — Increase due to acquisition — 442 Decrease for provision of research services (442 ) — Balance as of end of period $ — $ 442 Merck In connection with the Merger, the Company became the successor in interest to the Merck Neuromuscular License Agreement with Merck to support research, development and commercialization of products for treatment of neuromuscular diseases, including amyotrophic lateral sclerosis (“ALS”). As of December 31, RSUs Weighted Unvested at December 31, 2020 0 $ 0 Issued 122,469 $ 17.89 Vested (23,146 ) $ 17.89 Forfeited (13,098 ) $ 17.89 Unvested at December 31, 2021 86,225 $ 17.89 The Company’s 2008 Equity Incentive Plan (the “2008 Plan”) provided for the grant of incentive stock options, non-statutory stock options, restricted stock awards and restricted stock units to employees and non-employee service providers of the Company. Under the 2008 Plan, the exercise price of stock options granted were at 100% of the estimated fair market value of the Company’s common stock on the date of grant and the 134 KINETA, INC. Notes to Consolidated Financial Statements contractual term of stock options granted were between five and ten years. Options become vested and, if applicable, exercisable based on terms determined by the Company’s board of directors or other plan administrator on the date of grant, which is continued employment or service as defined in each option agreement. In 2018, the 2008 Plan expired and 209,000 stock options granted prior to the 2008 Plan expiration remain outstanding as of December 31, 2023. The Company’s 2010 Equity Incentive Plan (the “2010 Plan”) provided for the grant of incentive stock option, non-statutory stock options, stock appreciation rights, restricted stock awards and restricted stock unit awards to employees and non-employee service providers of the Company. Under the 2010 Plan, the exercise price of stock options granted were at 100% of the estimated fair market value of the Company’s During October 2022, three employees exercised 5,000 SARs and received cash payments of $ RSAs Weighted Unvested at December 31, 2020 0 $ 0 Issued 305,663 $ 3.83 Vested 0 $ 0 Forfeited 0 $ 0 Unvested at December 31, 2021 305,663 $ 3.83 2020 Equity Incentive Plan In December 2022, the 2020 Plan expired and 201,000 stock options granted prior to the 2020 Plan expiration remain outstanding as of December 31, 2023. 2022 Equity Incentive Plan In December 2022, the Company grant, which is Notes to Consolidated Financial Statements Stock Option Activity The following table Year Ended December 31, 2021 2020 Risk-free interest rate 1.0 % 1.1 % Expected volatility 81.3 % 70.9 % Expected dividend yield 0 0 Expected term (in years) 6.3 7.8 Outstanding Stock Options Weighted-Average Exercise Price Per Share Weighted-Average Remaining Contractual Term (years) Aggregate Intrinsic Value (in thousands, except per share amounts and years) December 31, 2022 734 $ 22.67 5.4 $ — Granted 1,383 $ 3.19 Exercised — $ — Forfeited (99 ) $ 25.82 Expired (43 ) $ 16.54 Outstanding as of December 31, 2023 1,975 $ 9.00 7.6 $ 604 Exercisable as of December 31, 2023 1,072 $ 12.80 6.5 $ 186 In March 2021, an employee exercised 56,000 vested stock options and entered into a nonrecourse promissory note in the amount of $0.4 million with the Company. The promissory note provides for a fixed interest rate of 2.0% and payment is required upon the earlier of (i) the sale of the Company, (ii) the borrower’s sale of any of the shares, (iii) five years from the date the promissory note agreement was executed, and (iv) material breach by borrower of any written agreements with the Company, including but not limited to the employment agreement and Company policies. Payment may also be triggered in other specified circumstances. The promissory note remains outstanding as of December 31, 2023. Fair Value of Stock Options The fair value of stock options granted for employee and non-employee awards was estimated at the grant date using the Black-Scholes option pricing model based on the following assumptions: Years Ended December 31, 2023 2022 Expected volatility 111.1% - 113.1% 84.2% - 86.0% Expected term (years) 5.35 - 6.08 3.0 - 7.0 Risk-free interest rate 3.4% - 4.4% 1.6% - 2.9% Expected dividend yield 0% - 0% 0% - 0% Restricted Stock The Company has granted RSUs under its equity incentive plans with both service-based and performance-based vesting conditions. As of December 31, 2023, the Company’s outstanding RSUs all related to RSUs with service-based conditions that vest over time, with a grant date fair value of $204,000. The following table summarizes the Company’s Number Weighted Weighted Aggregate (in years) (in thousands) Outstanding as of December 31, 2020 944,961 $ 20.70 8.29 $ 6,522 Granted 994,014 $ 16.63 Exercised (9,241 ) $ 8.97 Cancelled (150,560 ) $ 14.60 Outstanding as of December 31, 2021 1,779,174 $ 18.99 7.67 $ — Vested and expected to vest as of December 31, 2021 1,764,174 $ 19.00 7.66 $ — Options exercisable as of December 31, 2021(1) 1,024,379 $ 20.99 6.48 $ — Number of Restricted Stock (RSUs) Weighted-Average Grant Date Fair Value Per Share (in thousands, excepts per share amounts) Outstanding and unvested as of December 31, 2022 175 $ 26.89 Exercised/Released (159 ) $ 26.90 Cancelled/Forfeited (8 ) $ 26.48 Outstanding and unvested as of December 31, 2023 8 $ 27.14 136 KINETA, INC. Notes to Consolidated Financial Statements The Year Ended December 31, 2021 2020 Research and development expenses $ 1,352 $ 663 General and administrative expenses 3,938 1,603 $ 5,290 $ 2,266 Years Ended December 31, 2023 2022 (in thousands) Research and development $ 608 $ 2,957 General and administrative 3,246 2,231 Total stock-based compensation $ 3,854 $ 5,188 In October 2022, three employees exercised 5,000 SARs and the Company paid $19,000 in cash to the employees and recognized cash-based stock compensation expense. As of December 31, Years Ended December 31, 2023 2022 (in thousands) Deferred $ (2,000 ) $ (6,923 ) Change in valuation allowance 2,000 6,923 Total $ — $ — A reconciliation of Year Ended December 31, 2021 2020 Federal statutory income tax rate (21.0 )% (21.0 )% State taxes, net of federal benefit (10.0 ) (1.6 ) Federal and state research and development tax credits (4.1 ) (2.5 ) In-process research and development (1) 0 10.4 Other (1.8 ) 1.2 Change in deferred tax asset valuation allowance 36.9 13.5 Effective income tax rate 0.0 % 0.0 % Years Ended December 31, 2023 2022 Federal income taxes 21.0 % 21.0 % Research and development tax credits 2.7 % 0.8 % Debt fair value adjustment 2.3 % (4.9 )% Change in valuation allowance (14.2 )% (10.9 )% Stock based compensation (6.7 )% (0.1 )% Deferred reconciliation adjustments (2.4 )% 1.5 % Expiration of capital loss carryforward (2.1 )% 0.0 % Transaction costs (0.4 )% (1.9 )% In-process research and development 0.0 % (4.4 )% Other, net (0.2 )% (1.1 )% Effective income tax rate 0.0 % 0.0 % 137 KINETA, INC. Notes to Consolidated Financial Statements Deferred tax assets and liabilities reflect the net tax effects of net operating loss and tax credit carryforwards and temporary differences between the carrying amount of assets and liabilities for financial reporting and the amounts used for tax purposes. Significant components of the Company’s deferred tax assets Years Ended December 31, 2023 2022 (in thousand) Deferred tax assets: Net operating losses $ 145,232 $ 144,729 Research and development credits 22,899 22,384 Capitalized research and development expenses 4,409 3,586 Stock-based compensation 1,807 1,353 Operating lease liability 115 318 Capital loss carryforward 19 316 Accrued expenses 214 103 Intangibles 57 61 Total deferred tax assets 174,752 172,850 Less: Valuation allowance (174,637 ) (172,637 ) Total deferred tax assets less valuation allowance 115 213 Deferred tax liabilities: Partnership basis deferred (15 ) 85 Right-of-use asset (100 ) (280 ) Fixed assets — (18 ) Total deferred tax liabilities (115 ) (213 ) Net deferred tax assets $ — $ — The Company determines its valuation allowance on deferred tax assets by considering both positive and negative evidence in order to ascertain whether it is more likely than not that deferred tax assets will be realized. Realization of deferred tax assets is dependent upon the generation of future taxable income, if any, the timing and amount of which are uncertain. Due to the Company’s recent history of operating losses, the Company believes that recognition of the December 31, 2021 2020 Deferred tax assets: Net operating loss carryforwards $ 134,395 $ 122,460 Research and development tax credit carryforwards 20,246 18,654 Property and equipment 242 184 Accrued expenses 521 539 Capitalized intellectual property costs 102 89 Stock/equity-based compensation expense 1,712 1,084 Operating lease liabilities 4,534 4,670 Other 290 0 Total deferred tax assets 162,042 147,680 Deferred tax liabilities: Operating lease right-of-use assets (5,806 ) (5,836 ) Other 0 (172 ) Total deferred tax liabilities (5,806 ) (6,008 ) Valuation allowance (156,236 ) (141,672 ) Net deferred tax assets $ 0 $ 0 As of December 31, Utilization of On August 16, 2022, the Inflation Reduction Act ("IRA") was enacted into US law. Effective for tax years beginning after December 31, 2022, the IRA imposes a 15% corporate minimum tax, a 1% excise tax on share repurchases, and creates and extends certain tax-related energy incentives. Management does not expect the tax-related provisions of the IRA to have a material impact on the Company's consolidated financial statements. Unrecognized Tax Benefits 138 KINETA, INC. Notes to Consolidated Financial Statements The unrecognized tax benefits, if recognized, would Years Ended December 31, 2023 2022 (in thousands) Beginning balance of unrecognized tax benefits $ 828 $ 630 Gross increases based on tax positions related to current year 172 198 Ending balance of unrecognized tax benefits $ 1,000 $ 828 Interest and penalties related to the Company’s unrecognized tax benefits accrued as of December 31, 2023 were not material. The Company does not expect its uncertain tax positions to have material impact on its consolidated financial statements within the next twelve months. All of the unrecognized tax benefits as of December 31, 2023 are accounted for as a reduction in the Company’s deferred tax assets. The Company files federal and state income tax returns subject to Year Ended December 31, 2021 2020 Valuation allowance as of beginning of year $ 141,672 $ 26,724 Increases recorded to income tax provision 14,564 7,777 Amounts from Merger with PTI 0 107,171 Valuation allowance as of end of year $ 156,236 $ 141,672 Year Ended December 31, 2021 2020 Numerator: Net loss $ (39,503 ) $ (57,487 ) Gain on extinguishment of Class B preferred units 0 6,697 Net loss applicable to common shareholders $ (39,503 ) $ (50,790 ) Denominator: Weighted average common shares outstanding, basic and diluted 10,283,172 2,354,143 Net loss per share, basic and diluted $ (3.84 ) $ (21.57 ) Years Ended December 31, 2023 2022 (in thousands, excepts per share amounts) Numerator: Net loss attributable to Kineta, Inc. $ (14,099 ) $ (63,408 ) Denominator: Weighted-average common shares outstanding, basic and diluted(1) 11,054 4,926 Net loss per share, basic and diluted $ (1.28 ) $ (12.87 ) 1. Included in the denominator for the years ended December 31, 2023 and 2022, were 628,000 and 260,000 weighted-average shares of common stock warrants, respectively, with exercise prices of $0.001 and $0.14 issued for nominal consideration. The following outstanding potentially dilutive common stock equivalents As of December 31, 2021 2020 Options to purchase common stock 1,779,174 944,961 Warrants to purchase common stock or shares convertible into common stock 99,986 99,986 Unvested RSUs 86,225 0 1,965,385 1,044,947 Years Ended December 31, 2023 2022 (in thousands) Warrants to purchase common stock 2,560 533 Common stock options 1,975 733 Vested restricted stock subject to recall 56 56 Unvested restricted stock subject to repurchase 8 175 Total 4,599 1,497 Defined Contribution Plan The Company sponsors a 401(k) Plan whereby all employees are eligible to participate in the 401(k) Plan after meeting certain eligibility requirements. Participants may elect to have a portion of their salary deferred and contributed to the 401(k) plan, subject to certain limitations. The Company provided matching contributions of $115,000 for the year ended December 31, 2023 and $129,000 for the year ended December 31, 2022. Stock Purchases During the year ended December 31, 2023, five members of the Company’s executive management purchased 43,000 shares of the Company’s common stock on the open market and one director of the Company purchased 5,000 shares of the Company’s common stock on the open market. 139 KINETA, INC. Notes to Consolidated Financial Statements RSU Vesting During the year ended December 31, 2023, the Company Warrant Exercises During the Private Placement In December 2022, the Company issued415,000 shares of its common stock for an aggregate purchase price of $4.8 million to four related parties and issued 66,000 warrants to purchase shares of the 2022 Convertible Notes In December 2022, upon the closing of the Merger, the Company settled $4.8 million in outstanding principal and accrued interest, held by three entities affiliated with a 2020 In December 2022, upon the closing of the Merger, the Company settled $2.0 million in 2020 Notes In August 2022, the Company settled $0.5 million in outstanding principal and accrued interest with the related party by issuing 23,000 shares of the Company’s non-voting common stock at a 15% discount, recognizing a $0.1 million Year Ended December 31, 2021 2020 Operating lease cost $ 6,665 $ 3,097 Short-term lease cost $ 0 $ 0 Variable lease cost $ 844 $ 271 Finance lease cost: Amortization of lease assets $ 152 $ 361 Interest on lease liabilities 7 20 Total finance lease cost $ 159 $ 381 Year Ended December 31, 2021 2020 Cash paid for amounts included in the measurement of operating lease liabilities $ 5,998 $ 2,461 Cash paid for amounts included in the measurement of finance lease liabilities (operating $ 7 $ 20 Cash paid for amounts included in the measurement of finance lease liabilities (financing $ 166 $ 347 Operating lease liabilities arising from obtaining right-of-use assets $ 0 $ 10,219 Finance lease liabilities arising from obtaining right-of-use assets $ 0 $ 102 As of December 31, 2021 2020 Weighted-average remaining lease term (in years) used for: Operating leases 5.22 5.03 Finance leases 0.60 1.26 Weighted-average discount rate used for: Operating leases 9.10 % 9.01 % Finance leases 3.41 % 6.46 % As of December 31, The Company evaluated subsequent events through the date these consolidated financial statements were issued to determine if they must be reported. The management of the Company determined there were no reportable subsequent events other than as described below. Year Ending December 31, Operating Leases Finance Leases 2022 $ 6,173 $ 49 2023 2,977 0 2024 1,931 0 2025 1,985 0 2026 2,039 0 Thereafter 2,801 0 Total future lease payments 17,906 49 Less: Imputed interest (3,427 ) (1 ) Total lease liabilities $ 14,479 $ 48 As of December 31, Leases Consolidated Balance Sheet Classification 2021 2020 Assets: Operating lease assets Operating lease right-of- use assets $ 18,543 $ 23,678 Finance lease assets Property and equipment, net 315 199 Total leased assets $ 18,858 $ 23,877 Liabilities: Current: Operating lease liabilities Operating lease liabilities $ 5,064 $ 4,468 Finance lease liabilities Current portion of finance lease obligation 48 166 Non-current: Operating lease liabilities Operating lease liabilities, net of current portion 9,415 14,479 Finance lease liabilities Finance lease obligation, net of current portion 0 48 Total lease liabilities $ 14,527 $ 19,161 In connection with the Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure. None. Evaluation of Disclosure Controls and Procedures We are in the process of implementing measures designed to improve our internal control over financial reporting to remediate the material weaknesses. For example, we began to address the material weaknesses by implementing certain Sarbanes-Oxley controls during the first half of 2022. In October 2022, we hired a Chief Financial Officer to enhance internal controls and Notwithstanding the material weaknesses in internal control over financial reporting described above, our management has concluded that our consolidated financial statements included in this Annual Report on Form 10-K are Management’s Annual Report on Internal Control over Financial Reporting Our management is responsible for the preparation and fair presentation of the consolidated financial statements and for establishing and maintaining Management conducted an evaluation of Changes in Internal Control over Financial Reporting Not applicable. 142 PART III The information required by this Item 10 will be included in our definitive proxy statement to be filed with the SEC with respect to our The information required by this Item 11 will be included in our definitive proxy statement to be filed with the SEC with respect to our The information required by this Item 12 will be included in our definitive proxy statement to be filed with the SEC with respect to our The information required by this Item 13 will be included in our definitive proxy statement to be filed with the SEC with respect to our The information required by this Item 14 will be included in our definitive proxy statement to be filed with the SEC with respect to our 143 PART IV 1. Financial Statements For a list of the financial statements included herein, see Index to the Consolidated Financial Statements on page 2. Financial Statement Schedules Financial statement schedules have been omitted because they are either not required or not applicable or the information is included in the consolidated financial statements or the notes thereto. 3. Exhibits The exhibits required by Item 601 of Regulation S-K and Item 15(b) of this Annual Report on Form 10-K are listed in the Exhibit Index immediately preceding the signature page of this Annual Report on Form 10-K. The exhibits listed in the Exhibit Index are incorporated by reference herein. Not 144 Exhibit Index Exhibit Number Description 1.1 2.2 2.3++ 3.1 3.2 3.3 3.4 3.6 4.1 4.3 4.6 4.7 4.8 10.1 10.2 10.3 10.4 145 10.6 10.7 10.8 10.9# 10.10# 10.11# 10.12 10.13 10.14 10.15++ 10.16 10.17 10.18 10.19 10.20 10.21 10.22++ 10.23 10.24 10.25# 10.26# 10.27# 146 10.28# 10.29# 10.30# 10.31# 10.32# 10.33 10.34 10.35 10.36 10.37 10.38 10.39+ 10.40 10.41+ 10.42+ 10.43 10.44 10.45 10.46 10.47+ 10.48 10.49 10.50 147 10.51 10.52 10.53 10.54 10.55 10.56 10.57 10.58 10.59 10.60 10.61 10.62 21.1* 23.1* Consent of 31.1* 31.2* 32.1* Compensation Recovery Policy approved on September 6, 2023 and effective as of October 2, 2023. Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document. 101.SCH Inline XBRL Taxonomy Extension Schema Document 101.CAL Inline XBRL Taxonomy Extension Calculation Linkbase Document 101.DEF Inline XBRL Taxonomy Extension Definition Linkbase Document 101.LAB Inline XBRL Taxonomy Extension Label Linkbase Document 101.PRE Inline XBRL Taxonomy Extension Presentation Linkbase Document 104 Cover Page Interactive Data File (formatted as Inline XBRL * Filed herewith. # Indicates a management contract or any compensatory plan, contract or arrangement. + Portions of this Exhibit (indicated with [***]) have been omitted as the Company has determined that (i) the omitted information is not material and (ii) the omitted information is the type that the Company treats as private or confidential. ++ Certain schedules and exhibits have been omitted pursuant to Item 601(b)(2) of Regulation S-K. A copy of any omitted schedule and/or exhibit will be furnished to the SEC upon request. 148 SIGNATURES Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Kineta, Inc. Date: March 21, 2024 By: /s/ Craig Philips Craig Philips President Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the Registrant in the capacities and on the dates indicated. Signature Title Date /s/ Craig Philips President March 21, 2024 Craig Philips (Principal Executive Officer) /s/ Keith A. Baker Chief Financial Officer March 21, 2024 Keith A. Baker (Principal Financial Officer) /s/ David Arkowitz Director March 21, 2024 David Arkowitz /s/ Raymond Bartoszek Director March 21, 2024 Raymond Bartoszek /s/ Kimberlee Drapkin Director March 21, 2024 Kimberlee Drapkin March 21, 2024 Scott Dylla /s/ Director March 21, 2024 Marion R. Foote /s/ Shawn Iadonato Director March Shawn Iadonato /s/ Richard Peters Director March 21, 2024 Richard Petersevent of a contract dispute.Foreign sales of our product candidatesfuture, Kineta could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions, and changes in tariffs.lose rights that are important to its business.51Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions.In order to market any product outside of the United States, however, we must establish and comply with the numerous and varying safety, efficacy, and other regulatory requirements of other countries. Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, but a failure or delay in obtaining regulatory approval in one jurisdictionKineta may have a negative effect on the regulatory approval process in others. For example, even if the FDA or other comparable foreign regulatory authority grants marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing, and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the United States, including additional preclinical studies or clinical trials as clinical trials conducted in one jurisdictionfuture require licenses to third-party technology and materials. Such licenses may not be accepted by regulatory authorities in other jurisdictions. The marketing approval processes in other countries may implicate all of the risks detailed above regarding FDA approvalavailable in the United States, as well as other risks. In many jurisdictions outside the United States, a product candidate mustfuture or may not be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval.Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties, and costs for we and could delayavailable on commercially reasonable terms, or prevent the introduction of our products in certain countries. Failure to obtain marketing approval in other countries or any delay or other setback in obtaining such approval would impair our ability to market our product candidates in such foreign markets. Any such impairment would reduce the size of our potential market,at all, which could have a material adverse impacteffect on ourKineta’s business and financial condition. Kineta may rely on third parties from whom it licenses proprietary technology to file and prosecute patent applications and maintain patents and otherwise protect the intellectual property Kineta licenses from them. Kineta may have limited control over these activities or any other intellectual property that may be related to Kineta’s in-licensed intellectual property. For example, Kineta cannot be certain that such activities by these licensors will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents and other intellectual property rights. Kineta may have limited control over the manner in which its licensors initiate an infringement proceeding against a third-party infringer of the intellectual property rights, or defend certain of the intellectual property that may be licensed to Kineta. It is possible that the licensors’ infringement proceeding or defense activities may be less vigorous than if Kineta conducts them itself. Even if Kineta acquires the right to control the prosecution, maintenance and enforcement of the licensed and sublicensed intellectual property relating to Kineta’s product candidates, Kineta may require the cooperation of its licensors and any upstream licensor, which may not be forthcoming. Therefore, Kineta cannot be certain that the prosecution, maintenance and enforcement of these patent rights will be in a manner consistent with the best interests of Kineta’s business. If Kineta or its licensor fails to maintain such patents, or if Kineta or its licensor loses rights to those patents or patent applications, the rights Kineta has licensed may be reduced or eliminated and Kineta’s right to develop and commercialize any of its product candidates that are the subject of such licensed rights could be adversely affected. In addition to the foregoing, the risks associated with patent rights that Kineta licenses from third parties will also apply to patent rights Kineta may own in the future. Further, if Kineta fails to comply with its diligence, development and commercialization timelines, milestone payments, royalties, insurance and other obligations under its license agreements, Kineta may lose its patent rights with respect to suchOurKineta’s commercial success depends, in part, on Kineta’s ability to develop, manufacture, market and sell its product candidates without infringing the intellectual property and other proprietary rights of third parties. Third parties may allege that Kineta has infringed, misappropriated or otherwise violated their intellectual property. Litigation or other legal proceedings relating to intellectual property claims, with or without merit, iscommercial partners, and vendorsadvisors do not use the proprietary information or know-how of others in their work for Kineta, Kineta may engage in misconductbe subject to claims that it or its employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of Kineta’s employees’ former employers or other improper activities, including noncompliance with regulatory standardsthird parties. Kineta may also be subject to claims that former employers or other third parties have an ownership interest in Kineta’s future patents. Litigation may be necessary to defend against these claims. If Kineta fails in defending any such claims, in addition to paying monetary damages, Kineta may lose valuable intellectual property rights or personnel. There is no guarantee of success in defending these claims, and requirements.even if Kineta is successful, litigation could result in substantial cost and be a distraction to Kineta’s management and other employees.We are exposed82risk of fraud, misconduct,case with other biopharmaceutical companies, Kineta’s success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is therefore costly, time consuming and inherently uncertain. Changes in either the patent laws or other illegal activity by our employees, independent contractors, consultants, commercial partners, and vendors. Misconduct by these parties could include intentional, reckless, and negligent conduct that fails to: comply with the lawsinterpretation of the FDA and other comparable foreign regulatory authorities; provide true, complete and accurate information to the FDA and other comparable foreign regulatory authorities; comply with manufacturing standards we have established; comply with healthcare fraud and abusepatent laws in the United States could increase the uncertainties and similar foreign fraudulent misconduct laws;costs, and may diminish Kineta’s ability to protect its inventions, obtain, maintain and enforce its intellectual property rights and, more generally, could affect the value of its intellectual property or report financial information or data accurately or to disclose unauthorized activities to us. If we obtain FDA approvalnarrow the scope of any of our product candidatesKineta’s owned and begins commercializing those productslicensed patents. Patent reform legislation in the United States our potential exposure under such laws will increase significantly, and our costs associated with compliance with such laws are also likely to increase. In particular, sales, marketing, and other countries, including the Leahy-Smith America Invents Act (the “Leahy-Smith Act”), signed into law on September 16, 2011, could increase those uncertainties and costs surrounding the prosecution of Kineta’s patent applications and the enforcement or defense of Kineta’s issued patents. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art and provide more efficient and cost-effective avenues for competitors to challenge the validity of patents. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including post-grant review, inter partes review and derivation proceedings. Further, because of a lower evidentiary standard in these USPTO post-grant proceedings compared to the evidentiary standard in United States federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate Kineta’s patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. Thus, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of Kineta’s patent applications and the enforcement or defense of Kineta’s issued patents, all of which could have a material adverse effect on Kineta’s business, arrangementsfinancial condition, results of operations and prospects.healthcare industryUSPTO after March 2013, but before Kineta files an application covering the same invention, could therefore be awarded a patent covering an invention of Kineta’s even if Kineta had made the invention before it was made by such third party. This will require Kineta to be cognizant going forward of the time from invention to filing of a patent application, but circumstances could prevent Kineta from promptly filing patent applications on its inventions. Since patent applications in the United States and most other countries are subjectconfidential for a period of time after filing or until issuance, Kineta cannot be certain that it or its licensors were the first to extensive laws designedeither (i) file any patent application related to prevent fraud, kickbacks, self-dealing,Kineta’s product candidates and other abusive practices. Theseproprietary technologies Kineta may develop or (ii) invent any of the inventions claimed in Kineta’s or its licensor’s patents or patent applications. Even where Kineta has a valid and enforceable patent, Kineta may not be able to exclude others from practicing the claimed invention where the other party can show that they used the invention in commerce before Kineta’s filing date. Thus the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of Kineta’s patent applications and the enforcement or defense of Kineta’s issued patents, all of which could have a material adverse effect on Kineta’s business, financial condition, results of operations and prospects.restrict or prohibit a wide rangeimpact the value of pricing, discounting, marketingKineta’s patents.promotion, sales and commission, certain customer incentive programs,maintaining patent protection depends on compliance with various procedural, document submissions, fee payment and other requirements imposed by governmental patent agencies, and Kineta’s patent protection could be reduced or eliminated for non-compliance with these requirements.arrangements generally. Activitiesmay become subject to competition from competitive medications, including generic medications. Given the amount of time required for the development, testing and regulatory review and approval of new product candidates, patents protecting such candidates may expire before or shortly after such candidates are commercialized. As a result, Kineta’s owned and licensed patent portfolio may not provide Kineta with sufficient rights to exclude others from commercializing products similar or identical to Kineta’s.improper useemployees of third parties with whom Kineta shares its facilities or third party consultants and vendors that Kineta engages to perform research, clinical trials or manufacturing activities, or misappropriation by third parties (such as through a cybersecurity breach) of Kineta’s trade secrets or proprietary information obtainedcould enable competitors to duplicate or surpass Kineta’s technological achievements, thus eroding Kineta’s competitive position in its market.patient recruitmenttheir employment. Kineta enters into written agreements that include confidentiality and intellectual property obligations to protect each party’s property, potential trade secrets, proprietary know-how and information. Kineta further seeks to protect its potential trade secrets, proprietary know-how and information in part by entering into non-disclosure and confidentiality agreements with parties who are given access to them, such as Kineta’s corporate collaborators, outside scientific collaborators, CROs, CMOs, consultants, advisors and other third parties. With Kineta’s consultants, contractors, and outside scientific collaborators, these agreements typically include invention assignment obligations. Despite these efforts, any of these parties may breach the agreements and disclosetrials,development plans. In addition, there is a risk that one or more of Kineta’s current service providers, manufacturers and other partners may not survive an economic downturn, which could directly affect Kineta’s ability to attain its operating goals on schedule and on budget.regulatory sanctionsa material disruption of Kineta’s product candidates’ development programs, compromise sensitive information related to Kineta’s business or prevent Kineta from accessing critical information, potentially exposing Kineta to liability or otherwise adversely affecting Kineta’s business.cause serious harmdata to our reputation. We have adopted a codeoperate its business. In the ordinary course of business, conductKineta collects, stores and ethics,transmits confidential information (including but itnot limited to intellectual property, proprietary business information and personal information). It is not always possiblecritical that Kineta does so in a secure manner to identifymaintain the confidentiality and deter misconduct by employees andintegrity of such confidential information. Kineta has also outsourced elements of its operations to third parties, and as a result Kineta manages a number of third-party contractors who have access to Kineta’s confidential information.precautions we takeimplementation of security measures, given their size and complexity and the increasing amounts of confidential information that they maintain, Kineta’s internal information technology systems and those of its third-party CROs and other contractors and consultants are potentially vulnerable to detectbreakdown or other damage or interruption from service interruptions, system malfunction, natural disasters, terrorism, war and prevent this activitytelecommunication and electrical failures, as well as security breaches from inadvertent or intentional actions by Kineta’s employees, contractors, consultants, business partners and/or other third parties, or from cyber-attacks by malicious third parties (including the deployment of harmful malware, ransomware, extortion, account takeover attacks, degradation of service attacks, denial-of-service attacks, “phishing,” or social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information), which may compromise Kineta’s system infrastructure or lead to data leakage. Kineta has technology security initiatives and disaster recovery plans in place to mitigate its risk to these vulnerabilities, but these measures may not be effectiveadequately designed or implemented to ensure that Kineta’s operations are not disrupted or that data security breaches do not occur. To the extent that any disruption or security breach were to result in controlling unknowna loss of, or unmanaged risksdamage to, Kineta’s data or lossesapplications, or inappropriate disclosure of confidential or proprietary information, Kineta could incur liability and reputational damage.protecting us from governmental investigationsinformation technology will prevent significant breakdowns, data leakages, breaches in Kineta’s systems or other actions or lawsuits stemming from a failure to be in compliance with such laws. If any such actions are instituted against us, and we are not successful in defending itself or asserting our rights, those actionscyber incidents that could have a material adverse effect upon Kineta’s reputation, business, operations or financial condition. For example, if such an event were to occur and cause interruptions in Kineta’s operations, it could result in a material disruption of Kineta’s programs and the development of its product candidates could be delayed. In addition, the loss of clinical trial data for Kineta’s product candidates could result in delays in Kineta’s marketing approval efforts and significantly increase Kineta’s costs to recover or reproduce the data. Furthermore, significant impactdisruptions of Kineta’s internal information technology systems or security breaches could result in the loss, misappropriation and/or unauthorized access, use or disclosure of, or the prevention of access to, confidential information (including trade secrets or other intellectual property, proprietary business information and personal information), which could result in financial, legal, business and reputational harm to Kineta. Like all businesses, Kineta may be increasingly subject to ransomware or other malware that could significantly disrupt its business operations, or disable or interfere with necessary access to essential data or processes. Numerous recent attacks of this nature have also involved exfiltration and disclosure of sensitive or confidential personal or proprietary information, or intellectual property, when victim companies have not paid the cyber criminals substantial ransom payments. For example, any such event that leads to unauthorized access, use, disclosure, unavailability or compromised integrity of personal or other sensitive or essential information, including personal information regarding Kineta’s clinical trial subjects or employees, could harm Kineta’s reputation directly, compel Kineta to comply with federal and/or state breach notification laws and foreign law equivalents, subject Kineta to mandatory corrective action, increase the costs Kineta incurs to protect against such information security breaches, such as increased investment in technology, render key personnel unable to perform duties or communicate throughout the organization and otherwise subject Kineta to fines and other liability under laws and regulations that protect the privacy and security of personal information, which could result in significant legal and financial exposure and reputational damages that could potentially have an adverse effect on ourKineta’s business.impositioncomplexities of significant finesforeign value-added tax systems, tax inefficiencies related to Kineta’s corporate structure and potential restrictions on the repatriation of earnings;sanctions.natural disasters and Kineta’s business continuity and disaster recovery plans may not adequately protect Kineta from a serious disaster.weearthquakes, fires, other natural disasters, terrorism and similar unforeseen events beyond Kineta’s control prevent it from using all or a significant portion of its headquarters or other facilities, it may be difficult or, in certain cases, impossible for Kineta to continue its business for a substantial period of time. Kineta does not have a disaster recovery or business continuity plan in place and may incur substantial expenses as a result of the absence or limited nature of Kineta’s internal or third-party service provider disaster recovery and business continuity plans, which could have a material adverse effect on Kineta’s business. In addition, the long-term effects of climate change on general economic conditions and the pharmaceutical manufacturing and distribution industry in particular are unclear, and changes in the supply, demand or available sources of energy and the regulatory and other costs associated with energy production and delivery may affect the availability or cost of goods and services, including raw materials and other natural resources, necessary to run Kineta’s business. Furthermore, certain parties in Kineta’s supply chain are operating from single sites, increasing their vulnerability to natural disasters or other sudden, unforeseen and severe adverse events. If such an event were to affect Kineta’s supply chain, it could have a material adverse effect on Kineta’s ability to conduct its clinical trials, its development plans and business.contract manufacturersCMOs and suppliers we engageKineta engages fail to comply with environmental, health and safety laws and regulations, weKineta could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of ourKineta’s business.WeKineta and any contract manufacturersCMOs and suppliers we engageit engages are subject to numerous federal, state and local environmental, health and safety laws, regulations and permitting requirements, including those governing laboratory procedures; the generation, handling, use, storage, treatment and disposal of hazardous and regulated materials and wastes; the emission and discharge of hazardous materials into the ground, air and water; and employee health and safety. Kineta’s operations involve the use of hazardous and flammable materials, including chemicals and biological and radioactive materials. Kineta’s operations also produce hazardous waste. Kineta generally contracts with third parties for the disposal of these materials and wastes. Kineta cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from Kineta’s use of hazardous materials, Kineta could be held liable for any resulting damages, and any liability could exceed Kineta’s resources. Under certain environmental laws, weKineta could be held responsible for costs relating to any contamination at our current or past facilities and at third-party facilities. WeKineta could also could incur significant costs associated with civil or criminal fines and penalties.52We could be adversely affected by violations of the U.S. Foreign Corrupt Practices Act (“FCPA”) and other worldwide anti-bribery laws.Our business activities may be subject to the FCPA and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we operate, including the U.K. Bribery Act. The FCPA generally prohibits offering, promising, giving, or authorizing others to give anything of value, either directly or indirectly, to a non-U.S. government official in order to influence official action, or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise and maintain an adequate system of internal accounting controls. Our business is heavily regulated and therefore involves significant interactionCompliance with public officials, including officials of non-U.S. governments. Additionally, in many other countries, the healthcare providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are government entities; therefore, our dealings with these prescribers and purchasers are subject to regulation under the FCPA. Recently the SEC and Department of Justice have increased their FCPA enforcement activities with respect to biotechnology and pharmaceutical companies. There is no certainty that all of our employees, agents, contractors, or collaborators, or those of our affiliates, will comply with all applicable environmental laws and regulations particularly given the high level of complexity of these laws. Violations of thesemay be expensive, and current or future environmental laws and regulations could result inmay impair Kineta’s research and product development efforts. In addition, Kineta cannot entirely eliminate the risk of accidental injury or contamination from these materials or wastes. Although Kineta maintains workers’ compensation insurance to cover it for costs and expenses it may incur due to injuries to its employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. Kineta does not carry specific biological or hazardous waste insurance coverage, and Kineta’s property, casualty and general liability insurance policies specifically exclude coverage for damages and fines criminal sanctions against us, our officers,arising from biological or our employees, the closing down of our facilities, requirements to obtain export licenses, cessation of business activities in sanctioned countries, implementation of compliance programs, and prohibitions on the conduct of our business. Any such violations could include prohibitions on our ability to offer our products in onehazardous waste exposure or more countries and could materially damage our reputation, our brand, our international expansion efforts, our ability to attract and retain employees, and our business, prospects, operating results, and financial condition.Risks Related to Our Reliance on Third PartiesWe depend on our collaboration with Merck and maycontamination. Accordingly, in the future depend on other collaborationsevent of contamination or injury, Kineta could be held liable for damages or be penalized with third parties for the research, developmentfines in an amount exceeding its resources, and commercialization of certain of the product candidates we may develop. If any such collaborations are not successful, we may not be able to realize the market potential of those product candidates.We have entered into a collaboration agreement with Merck and may seek other third-party collaborators for the research, development, and commercialization of certain of the product candidates we may develop. Our likely collaborators for any other collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies, biotechnology companies and academic institutions. Under our collaboration with Merck, we have, and if we enter into any such arrangements with any other third parties, we will likely have, shared or limited control over the amount and timing of resources that our collaborators dedicate to the development or potential commercialization of any product candidates we may seek to develop with them. Our ability to generate revenue from these arrangements with commercial entities will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements. We cannot predict the success of any collaboration that we enter into.Collaborations involving our research programs, or any product candidates we may develop, pose the following risks to we:•collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations;•collaborators may not properly obtain, maintain, enforce, or defend intellectual property or proprietary rights relating to our product candidates or research programs or may use our proprietary information in such a way as to expose us to potential litigation or other intellectual property related proceedings, including proceedings challenging the scope, ownership, validity and enforceability of our intellectual property;•collaborators may own or co-own intellectual property covering our product candidates or research programs that results from our collaboration with them, and in such cases, we may not have the exclusive right to commercialize such intellectual property or such product candidates or research programs;•we may need the cooperation of our collaborators to enforce or defend any intellectual property we contribute to or that arises out of our collaborations, which may not be provided to us;•disputes may arise between the collaborators and us that result in the delay or termination of the research, development, or commercialization of our product candidates or research programs or that result in costly litigation or arbitration that diverts management attention and resources;•collaborators may decide to not pursue development and commercialization of any product candidates we develop or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborator’s strategic focus or available funding or external factors such as an acquisition that diverts resources or creates competing priorities;53•collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct newKineta’s clinical trials or require a new formulation of a product candidate for clinical testing;•collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates or research programs if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours;•collaborators with marketing and distribution rights to one or more product candidates may not commit sufficient resources to the marketing and distribution of such product candidates;•we may lose certain valuable rights under circumstances identified in our collaborations, including if we undergo a change of control;•collaborators may undergo a change of control and the new owners may decide to take the collaboration in a direction which is not in our best interest;•collaborators may become bankrupt, which may significantly delay our research or development programs, or may cause us to lose access to valuable technology, know-how or intellectual property of the collaborator relating to our products, product candidates or research programs;•key personnel at our collaborators may leave, which could negatively impact our ability to productively work with our collaborators;•collaborations may require us to incur short and long-term expenditures, issue securities that dilute our stockholders, or disrupt our management and business;•collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates or our discovery engine platform; and•collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all. If a present or future collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our development or commercialization program under such collaborationregulatory approvals could be delayed, diminished, or terminated.We may face significant competition in seeking appropriate collaborations. Recent business combinations among biotechnology and pharmaceutical companies have resulted in a reduced number of potential collaborators. In addition, the negotiation process is time-consuming and complex, and we may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of the product candidate forsuspended, which we are seeking to collaborate, reduce or delay our development program or one or more of our other development programs, delay our potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to it on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop product candidates or bring them to market and generate product revenue.Under our collaboration with Merck, and if we enter into other collaborations to develop and potentially commercialize any product candidates, we may not be able to realize the benefit of such transactions if us or our collaborator elects not to exercise the rights granted under the agreement or if us or our collaborator are unable to successfully integrate a product candidate into existing operations and company culture. In addition, if our agreement with any of our collaborators terminates, our access to technology and intellectual property licensed to us by that collaborator may be restricted or terminate entirely, which may delay our continued development of our product candidates utilizing the collaborator’s technology or intellectual property or require us to stop development of those product candidates completely. We may also find it more difficult to find a suitable replacement collaborator or attract new collaborators, and our development programs may be delayed or the perception of our in the business and financial communities could be adversely affected. Many of the risks relating to product development, regulatory approval, and commercialization described in this “Risk Factors” section also applies to the activities of our collaborators and any negative impact on our collaborators may adversely affect us.Our drug development programs and the potential commercialization of our product candidates will require substantial additional cash to fund expenses. For some of our product candidates, we may decide to collaborate with pharmaceutical and biotechnology companies for the development and potential commercialization of those product candidates, such as those that may result from our collaboration with Merck.Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical studies, the likelihood of approval by the54FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge, and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidate. The terms of any collaborations or other arrangements that we may establish may not be favorable to it.In addition, our collaboration with Merck and any future collaborations that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations. Disagreements between parties to a collaboration arrangement regarding clinical development and commercialization matters can lead to delays in the development process or commercializing the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority. Collaborations with pharmaceutical or biotechnology companies and other third parties often are terminated or allowed to expire by the other party. Termination of our collaboration with Merck or any such termination or expiration of future collaborations would adversely affect us financially and could harm our business reputation.We rely, and expect to continue to rely, on third parties to conduct any preclinical studies and clinical trials for our product candidates. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.We do not have the ability to independently conduct preclinical studies and clinical trials. We rely on medical institutions, clinical investigators, contract laboratories, and other third parties, such as CROs, to conduct preclinical studies and clinical trials on our product candidates. We enter into agreements with third-party CROs to provide monitors for and to manage data for our clinical trials. We will rely heavily on these parties for execution of clinical trials for our product candidates and control only certain aspects of their activities. As a result, we will have less direct control over the conduct, timing, and completion of these clinical trials and the management of data developed through clinical trials than would be the case if we were relying entirely upon our own staff. Communicating with outside parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. Outside parties may:•have staffing difficulties;•fail to comply with contractual obligations;•experience regulatory compliance issues;•undergo changes in priorities or become financially distressed; or•form relationships with other entities, some of which may be our competitors.These factors may materially adversely affect the willingness or ability of third parties to conduct our clinical trials and may subject us to unexpected cost increases that are beyond our control. Nevertheless, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the applicable protocol, legal, regulatory, and scientific requirements and standards, and our reliance on CROs does not relieve us of our regulatory responsibilities. We and our CROs are required to comply with regulations and guidelines, including Good Clinical Practices (“GCPs”) for conducting, monitoring, recording, and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the study patients are adequately informed of the potential risks of participating in clinical trials. These regulations are enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area and comparable foreign regulatory authorities for any product candidates in clinical development. The FDA enforces GCP regulations through periodic inspections of clinical study sponsors, principal investigators and study sites. If we or our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply with GCPs. In addition, our clinical trials must be conducted with product candidates produced under cGMP regulations and will require a large number of test patients. Our failure or the failure of our CROs to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process and could also subject it to enforcement action up to and including civil and criminal penalties.Although we design our clinical trials for our product candidates, CROs conduct all of the clinical trials. As a result, many important aspects of the clinical trials are outside of our direct control. In addition, the CROs may not perform all of their obligations under arrangements with us or in compliance with regulatory requirements, but we remain responsible and are subject to enforcement action that may include civil penalties and criminal prosecution for any violations of FDA laws and regulations during the conduct of our clinical trials. If the CROs do not perform clinical trials in a satisfactory manner, breach their obligations to us, or fail to comply with55regulatory requirements, the development and commercialization of our product candidates may be delayed or our development program materially and irreversibly harmed. We cannot control the amount and timing of resources these CROs devote to our program or our clinical products. If we are unable to rely on clinical data collected by our CROs, we could be required to repeat, extend the duration of, or increase the size of our clinical trials and this could significantly delay commercialization and require significantly greater expenditures.If any of our relationships with these third-party CROs terminate, we may not be able to enter into arrangements with alternative CROs. For example, the sponsored research agreement with Northwestern may be terminated by either party upon 60 days’ written notice to the other party. If our collaboration is delayed or terminated or our ability to continue to use the current research space is terminated as a result of conflicts of interest, we may not be able to continue our planned research projects and related clinical trials on the expected timeline and may need to spend significant time and efforts to secure alternative lab facilities and equipment. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, any clinical trials such CROs are associated with may be extended, delayed, or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, we believe that our financial results and the commercial prospects for our product candidates in the subject indication would be harmed, our costs could increase and our ability to generate revenue could be delayed.The manufacture of our product candidates, particularly those that utilize our discovery engine platform, is complex and we may encounter difficulties in production. If we or any of our third-party manufacturers encounter such difficulties, or fail to meet rigorously enforced regulatory standards, our ability to provide supply of our product candidates for preclinical studies and clinical trials or our products for patients, if approved, could be delayed or stopped, or we may be unable to maintain a commercially viable cost structure.The processes involved in manufacturing our drug product candidates, particularly those that utilize our discovery engine platform, are complex, expensive, highly-regulated, and subject to multiple risks. Further, as product candidates are developed through preclinical studies to late-stage clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, are altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives, and any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials.In addition, the manufacturing process for any products that we may develop is subject to FDA and other comparable foreign regulatory authority approval processes and continuous oversight, and we will need to contract with manufacturers who can meet all applicable FDA and foreign regulatory authority requirements, including, for example, complying with cGMPs, on an ongoing basis. If we or our third-party manufacturers are unable to reliably produce products to specifications acceptable to the FDA or other regulatory authorities, we may not obtain or maintain the approvals we need to commercialize such products. Even if we obtain regulatory approval for any of our product candidates, there is no assurance that either we or our contract manufacturers will be able to manufacture the approved product to specifications acceptable to the FDA or other regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential launch of the product, or to meet potential future demand. Any of these challenges could delay completion of clinical trials, require bridging clinical trials or the repetition of one or more clinical trials, increase clinical study costs, delay approval of our product candidate, impair commercialization efforts, increase our cost of goods, and have an adverse effect on our business, financial condition, results of operations, and growth prospects.We rely completely on third-party suppliers to manufacture our clinical drug supplies for our product candidates, and we intend to rely on third parties to produce preclinical, clinical, and commercial supplies of any future product candidates.We do not currently have, nor do we plan to acquire, the infrastructure or capability to internally manufacture our clinical drug supply of our product candidates, or any future product candidates, for use in the conduct of our preclinical studies and clinical trials, and we lack the internal resources and the capability to manufacture any product candidates on a clinical or commercial scale. The facilities used by our contract manufacturers to manufacture the active pharmaceutical ingredient and final drug product must complete a pre-approval inspection by the FDA and other comparable foreign regulatory agencies to assess compliance with applicable requirements, including cGMPs, after we submit an NDA or relevant foreign regulatory submission to the applicable regulatory agency.We do not control the manufacturing process of, and are completely dependent on, our contract manufacturers to comply with cGMPs for manufacture of both active drug substances and finished drug products. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or applicable foreign regulatory agencies, they will not be able to pass a pre-approval inspection and we would be unable to obtain regulatory approval for our product candidate. We may be required to change contract manufacturers and verify that the new contract manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturing process will produce our product candidate according to the56specifications previously submitted to the FDA or another regulatory authority. The delays associated with the verification of a new CMO could negatively affect our ability to develop product candidates or commercialize our products in a timely manner or within budget. In addition, we have no direct control over our contract manufacturers’ ability to maintain adequate quality control, quality assurance, and qualified personnel. Furthermore, all of our contract manufacturers are engaged with other companies to supply and/or manufacture materials or products for such companies, which exposes our manufacturers to regulatory risks for the production of such materials and products. As a result, failure to satisfy the regulatory requirements for the production of those materials and products may affect the regulatory clearance of our contract manufacturers’ facilities generally. If the FDA or an applicable foreign regulatory agency determines now or in the future that these facilities for the manufacture of our product candidates are noncompliant, we may need to find alternative manufacturing facilities, which would adversely impact our ability to develop, obtain regulatory approval for or market our product candidates. Our reliance on contract manufacturers also exposes us to the possibility that they, or third parties with access to their facilities, will have access to and may appropriate our trade secrets or other proprietary information.We do not have long-term supply agreements in place with our contractors, and each batch of our product candidates is individually contracted under a quality and supply agreement. If we engage new contractors, such contractors must complete an inspection by the FDA and other applicable foreign regulatory agencies. We plan to continue to rely upon contract manufacturers and, potentially, collaboration partners to manufacture commercial quantities of our product candidates, if approved. Our current scale of manufacturing is adequate to support all of our needs for preclinical studies and clinical study supplies.Risks Related to Our Intellectual Property RightsIf we are unable to adequately protect our proprietary technology, or obtain and maintain issued patents that are sufficient to protect our product candidates, others could compete against us more directly by developing and commercializing products similar or identical to ours, which would have a material adverse impacteffect on our business, results of operations, financial condition, and prospects.Our success will depend significantly on our ability to obtain and maintain patent and other proprietary protection in the United States and other countries for commercially important technology, inventions, and know-how related to our business, defend and enforce our patents, should they issue, preserve the confidentiality of our trade secrets, and operate without infringing the valid and enforceable patents and proprietary rights of third parties. We strive to protect and enhance the proprietary technologies that we believe are important to our business, including seeking patents intended to cover our products and compositions, their methods of use, and any other inventions that are important to the development of our business. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.We do not currently have any issued patents covering our clinical-stage product candidate YTX-7739 as a composition of matter. We cannot provide any assurances that any of our pending patent applications will mature into issued patents in any particular jurisdiction and, if they do, that such patents will include claims with a scope sufficient to protect our product candidates or otherwise provide any competitive advantage. The patent application and approval process is expensive, complex, and time-consuming. We may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output in time to obtain patent protection. If we are unable to obtain or maintain patent protection with respect to any of our proprietary products and technology we develop, our business, financial condition, results of operations, and prospects could be materially harmed.If the scope of any patent protection we obtain is not sufficiently broad, or if we lose any of our patent protection, our ability to prevent our competitors from commercializing similar or identical technology and product candidates would be adversely affected.The patent positions of biotechnology and pharmaceutical companies, including our patent position, involve complex legal and factual questions, which in recent years have been the subject of much litigation, and, therefore, the issuance, scope, validity, enforceability, and commercial value of any patent claims that we may obtain cannot be predicted with certainty. No consistent policy regarding the breadth of claims allowed in biotechnology and pharmaceutical patents has emerged to date in the United States or in many foreign jurisdictions. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection. The laws of some foreign countries do not protect our proprietary rights to the same extent as the laws of the United States, and we may encounter significant problems in protecting our proprietary rights in these countries.Patent applications are generally maintained in confidence until publication. In the United States, for example, patent applications are typically maintained in secrecy for up to 18 months after their filing. Similarly, publication of discoveries in scientific or patent literature often lags behind actual discoveries. Consequently, we cannot be certain that we were the first to file patent applications on our product candidates. There is also no assurance that all of the potentially relevant prior art relating to our patents and patent applications has been found, which could be used by a third party to challenge the validity of our patents, should they issue, or prevent57a patent from issuing from a pending patent application. Any of the foregoing could harm our competitive position,Kineta’s business, financial condition, results of operations and prospects.Moreover, our patents, if issued,In addition, Kineta may be challenged, deemed unenforceable, invalidated,incur substantial costs in order to comply with current or circumvented in the United Statesfuture environmental, health and abroad. U.S. patentssafety laws, regulations and patent applicationspermitting requirements. These current or future laws, regulations and permitting requirements may impair Kineta’s research, development or production efforts. Failure to comply with these laws, regulations and permitting requirements also be subject to interference, derivation, exparte reexamination, post-grant review, or inter partes review proceedings, supplemental examination and challenges in district court. Patents may also be subjected to opposition, post-grant review, or comparable proceedings lodged in various foreign, both national and regional, patent offices or courts. An adverse determination in any such proceeding could result in either loss of the patentsubstantial fines, penalties or denial of the patent application,other sanctions or loss or reduction in the scope of one or more of the claims of the patent or patent application,business disruption, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. In addition, such proceedings may be costly. Thus, any patents, should they issue, that we may own or exclusively license may not provide any protection against competitors. Furthermore, an adverse decision in an interference proceeding can result in a third party receiving the patent right sought by us, which in turn could affect our ability to develop, market, or otherwise commercialize our product candidates.Furthermore, though a patent, if it were to issue, is presumed valid and enforceable, our issuance is not conclusive as to our validity or our enforceability and it may not provide us with adequate proprietary protection or competitive advantages against competitors with similar products. Even if a patent issues and is held to be valid and enforceable, competitors may be able to design around or circumvent our patents, such as using pre-existing or newly developed technology or products in a non-infringing manner. Other parties may develop and obtain patent protection for more effective technologies, designs, or methods. If these developments were to occur, they could have a material adverse effect on ourKineta’s business, financial condition, results of operations and prospects.Our abilityAny third-party CMOs and suppliers Kineta engages will also be subject to enforce our patent rights depends on our abilitythese and other environmental, health and safety laws and regulations. Liabilities they incur pursuant to detect infringement. It is difficult to detect infringers who do not advertise the components that are usedthese laws and regulations could result in their products. Moreover, it may be difficultsignificant costs or impossible to obtain evidence of infringementan interruption in a competitor’s or potential competitor’s product. Any litigation to enforce or defend our patent rights, even if we were to prevail, could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.We will incur significant ongoing expenses in maintaining our patent portfolio. Should we lack the funds to maintain our patent portfolio or to enforce our rights against infringers, we could be adversely impacted.We may in the future co-own patent rights relating to future product candidates and our discovery engine platform with third parties. Some of our in-licensed patent rights are, and may in the future be, co-owned with third parties. In addition, our licensors may co-own the patent rights us in-licenses with other third parties with whom we do not have a direct relationship. Our exclusive rights to certain of these patent rights are dependent, in part, on inter-institutional or other operating agreements between the joint owners of such patent rights, who are not parties to our license agreements. If our licensors do not have exclusive control of the grant of licenses under any such third-party co-owners’ interest in such patent rights or we are otherwise unable to secure such exclusive rights, such co-owners may be able to license their rights to other third parties, including our competitors, and our competitors could market competing products and technology. In addition, we may need the cooperation of any such co-owners of our patent rights in order to enforce such patent rights against third parties, and such cooperation may not be provided to it. Any of the foregoingoperations, which could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.Intellectual property rights do not necessarily address all potential threats.The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example, we do not know whether:•any of our pending patent applications, if issued, will include claims having a scope sufficient to protect our product candidates or any other products or product candidates;•any of our pending patent applications will issue as patents at all;•we will be able to successfully commercialize our product candidates, if approved, before our relevant patents expire;•we will be the first to make the inventions covered by each of our patents and pending patent applications;•we will be the first to file patent applications for these inventions;•others will not develop similar or alternative technologies that do not infringe our patents;58•others will not use pre-existing technology to effectively compete against it;•any of our patents, if issued, will be found to ultimately be valid and enforceable;•any patents issued to us will provide a basis for an exclusive market for our commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties;•we will develop additional proprietary technologies or product candidates that are separately patentable; or•that our commercial activities or products will not infringe upon the patents or proprietary rights of others.Should any of these events occur, they could have a material adverse effect on ourKineta’s business, financial condition, results of operations and prospects.Intellectual property rights do not necessarily address all potential threats.The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example, we do not know whether:•any of our pending patent applications, if issued, will include claims having a scope sufficient to protect our product candidates or any other products or product candidates;•any of our pending patent applications will issue as patents at all;•we will be able to successfully commercialize our product candidates, if approved, before our relevant patents expire;•we will be the first to make the inventions covered by each of our patents and pending patent applications;•we will be the first to file patent applications for these inventions;•others will not develop similar or alternative technologies that do not infringe our patents;•others will not use pre-existing technology to effectively compete against it;•any of our patents, if issued, will be found to ultimately be valid and enforceable;•any patents issued to us will provide a basis for an exclusive market for our commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties;•we will develop additional proprietary technologies or product candidates that are separately patentable; or•that our commercial activities or products will not infringe upon the patents or proprietary rights of others.Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations and prospects.If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.We have entered into license agreements with third parties and may need to obtain additional licenses from others to advance our research or allow commercialization of product candidates we may develop or our discovery engine platform technology. It is possible that we may be unable to obtain additional licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be required to expend significant time and resources to redesign our technology, product candidates, or the methods for manufacturing them or to develop or license replacement technology, all of which may not be feasible on a technical or commercial basis. If we are unable to do so, we may be unable to develop or commercialize the affected product candidates or continue to utilize our existing discovery engine platform, which could harm our business, financial condition, results of operations, and prospects significantly. We cannot provide any assurances that third-party patents do not exist which might be enforced against our current technology, including our discovery engine platform technology, manufacturing methods, product candidates, or future methods or products resulting in either an injunction prohibiting our manufacture or future sales, or, with respect to our future sales, an obligation on our part to pay royalties and/or other forms of compensation to third parties, which could be significant.In addition, each of our license agreements, and we expect our future agreements, will impose various development, diligence, commercialization, and other obligations on it. Certain of our license agreements also require us to meet development timelines, or to exercise commercially reasonable efforts to develop and commercialize licensed products, in order to maintain the licenses. In spite of59our efforts, our licensors might conclude that we have materially breached our obligations under such license agreements and might therefore terminate the license agreements, thereby removing or limiting our ability to develop and commercialize products and technology covered by these license agreements. If these in-licenses are terminated, or if the underlying patents fail to provide the intended exclusivity, competitors or other third parties would have the freedom to seek regulatory approval of, and to market, products identical to our and we may be required to cease our development and commercialization of certain of our product candidates or of our current discovery engine platform technology. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including:•the scope of rights granted under the license agreement and other interpretation-related issues;•the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;•the sublicensing of patent and other rights under our collaborative development relationships;•our diligence obligations under the license agreement and what activities satisfy those diligence obligations;•the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and•the priority of invention of patented technology.In addition, the agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations, and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates, which could have a material adverse effect on our business, financial conditions, results of operations, and prospects.If we are unable to protect the confidentiality of our trade secrets, our business and competitive position may be harmed.We may also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. Additionally, we rely on unpatented know-how, continuing technological innovation to develop, strengthen, and maintain the proprietary and competitive position of our product candidates, which we seek to protect, in part, by confidentiality agreements with our employees and our collaborators and consultants. However, trade secrets are difficult to protect. For example, we may be required to share our trade secrets with third-party licensees, collaborators, consultants, contractors, or other advisors and we have limited control over the protection of trade secrets used by such third parties. Although we use reasonable efforts to protect our trade secrets, including by entering into confidentiality agreements, our employees, consultants, contractors, outside scientific collaborators, and other advisors may unintentionally or willfully disclose our trade secrets and proprietary information to competitors and we may not have adequate remedies for any such disclosure. Enforcing a claim that a third party illegally obtained and used, disclosed, or misappropriated any of our trade secrets is difficult, expensive, and time-consuming, and the outcome is unpredictable. Furthermore, we may not obtain these agreements in all circumstances, and the employees and consultants who are parties to these agreements may breach or violate the terms of these agreements, thus we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets through such breaches or violations. In addition, trade secret laws in the United States vary, and some U.S. courts as well as courts outside the United States are sometimes less willing or unwilling to protect trade secrets. Moreover, it is possible that technology relevant to our business will be independently developed by a person that is not a party to such an agreement. Further, our trade secrets could otherwise become known or be independently discovered by our competitors or other third parties. We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or trade secrets by consultants, vendors, former employees, and current employees. If our trade secrets or confidential or proprietary information is divulged to or acquired by third parties, including our competitors, our competitive position in the marketplace, business, financial condition, results of operations, and prospects may be materially adversely affected.We may be sued for infringing the intellectual property rights of others, which may be costly and time-consuming and may prevent or delay our product development efforts and stop it from commercializing or increase the costs of commercializing our product candidates, if approved.Our success will depend in part on our ability to operate without infringing, misappropriating, or otherwise violating the intellectual property and proprietary rights of third parties. We cannot assure you that our business, products, and methods do not or will not60infringe the patents or other intellectual property rights of third parties. We may in the future become party to, or threatened with, adversarial proceedings or litigation regarding intellectual property rights with respect to our product candidates and technologies we use in our business.The pharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. Other parties may allege that our product candidates or the use of our technologies infringe or otherwise violates patent claims or other intellectual property rights held by them or that we are employing their proprietary technology without authorization. As we continue to develop and, if approved, commercialize our current product candidates and future product candidates, competitors may claim that our technology infringes their intellectual property rights as part of business strategies designed to impede our successful commercialization. There may be third-party patents or patent applications with claims to compositions, materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. In particular, we are aware of an issued patent in each of the United States and Japan that expires in 2030 that covers one of our preclinical assets. We do not know if we will have reasonable defenses against a claim of infringement or if we will be able to obtain a license to such patent on commercially reasonable terms, if at all. As a result, we may not be able to commercialize such asset, if approved, prior to such patent’s expiration.We are also aware of U.S. and foreign patents and applications owned by a third party claiming certain compositions of matter and methods of use which we expect to expire in 2031. While we believe that we have reasonable defenses against a claim of infringement, including non-infringement and invalidity, there can be no assurance that we will prevail in any such action by the holder of these patents. In the event such patents were enforced against us and deemed to cover one or more of our products, and our defenses were unsuccessful, we would then need to obtain a license to these patents, which license may not be available on commercially reasonable terms, or at all. As a result, we may not be free to manufacture or market our products, including YTX-7739, if approved, prior to expiration of such patents.Additionally, because patent applications can take many years to issue and may be confidential for 18 months or more after filing, and because patent claims can be revised before issuance, third parties may have currently pending patent applications which may later result in issued patents that our product candidates may infringe, or which such third parties claim are infringed by our technologies. If a patent holder believes one or more of our product candidates infringe its patent rights, the patent holder may sue us even if we have received patent protection for our technology. Moreover, we may face patent infringement claims from non-practicing entities that have no relevant drug revenue and against whom our own patent portfolio may thus have no deterrent effect.The outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our product candidates, products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid or unenforceable, and we may not be able to do this. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could have a material adverse effect on our business and operating results. In addition, we may not have sufficient resources to bring these actions to a successful conclusion.Patent and other types of intellectual property litigation can involve complex factual and legal questions, and their outcome is uncertain. If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our product candidates and technology. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were to obtain a license, it could be granted on non-exclusive terms, thereby providing our competitors and other third parties access to the same technologies licensed to it. In addition, if any such claim were successfully asserted against us and we could not obtain such a license, we may be forced to stop or delay developing, manufacturing, selling or otherwise commercializing our product candidates. Any claim relating to intellectual property infringement that is successfully asserted against we may require us to pay substantial damages, including treble damages and attorney’s fees if we are found to be willfully infringing another party’s patents, for past use of the asserted intellectual property and royalties and other consideration going forward if we are forced to take a license.Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on it. There could also be public announcements of the results of the hearing, motions, or other interim proceedings or developments and if securities analysts or investors perceive those results to be negative, it could cause the price of shares of our common stock to decline. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action, or challenge the validity of the patents in court, or redesign our products. Patent litigation is costly and time-consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, intellectual property litigation or claims could force us to do one or more of the following:•cease developing, selling or otherwise commercializing our product candidates;•pay substantial damages for past use of the asserted intellectual property;61•obtain a license from the holder of the asserted intellectual property, which license may not be available on reasonable terms, if at all; and•in the case of trademark claims, redesign, or rename, some or all of our product candidates to avoid infringing the intellectual property rights of third parties, which may not be possible and, even if possible, could be costly and time-consuming.Any of these risks coming to fruition could have a material adverse effect on our business, results of operations, financial condition, and prospects.We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.We enter into confidentiality and intellectual property assignment agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors. These agreements generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. The assignment of intellectual property rights under these agreements may not be automatic upon the creation of the intellectual property or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against it, to determine the ownership of what we regard as our intellectual property. For example, even if we have a consulting agreement in place with an academic advisor pursuant to which such academic advisor is required to assign any inventions developed in connection with providing services to it, such academic advisor may not have the right to assign such inventions to us, as it may conflict with his or her obligations to assign all such intellectual property to his or her employing institution.Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.Periodic maintenance fees, renewal fees, annuity fees, and various other government fees on our owned and in-licensed patents and patent applications are or will be due to be paid to the U.S. Patent and Trademark Office (“USPTO”) in several stages and various government patent agencies outside of the United States over the lifetime of such patents and patent applications and any patent rights we may own or license in the future. We have systems in place to remind us to pay these fees, and we employ outside firms to remind us or our licensors to pay annuity fees due to foreign patent agencies on our foreign patents and pending foreign patent applications. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment, and other similar provisions over the lifetime of our owned patents and applications. In some cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors or other third parties might be able to enter the market earlier than would otherwise have been the case and this circumstance could have a material adverse effect on our business, financial condition, results of operations, and prospects.We may be involved in lawsuits or other proceedings to protect or enforce our intellectual property, which could be expensive, time-consuming, and unsuccessful.Even if our patent applications are issued, competitors and other third parties may infringe, misappropriate, or otherwise violate our patents and other intellectual property rights. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming and divert the attention of our management and key personnel from our business operations.Furthermore, many of our adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we can. Our ability to enforce our patent rights also depends on our ability to detect infringement. It is difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product.62In an infringement proceeding, a court may disagree with our allegations and refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question, or may decide that a patent of ours is invalid, unenforceable or not infringed. An adverse result in any litigation, defense or post-grant proceedings could result in one or more of our patents being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing. If any of our patents, if and when issued, covering our product candidates are invalidated or found unenforceable, our financial position and results of operations would be materially and adversely impacted. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to us from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our involvement in litigation or interference proceedings may fail and, even if successful, may result in substantial costs, and distract our management and other employees. We may not be able to prevent infringement, misappropriation of, or other violations of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions, or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.Issued patents covering our discovery engine platform and our product candidates could be found invalid or unenforceable if challenged.If we initiated legal proceedings against a third party to enforce a patent, if and when issued, covering our discovery engine platform or one of our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. The outcome of any such proceeding is generally unpredictable.In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for unenforceability assertions of a patent include allegations that someone connected with prosecution of the patent application that matured into the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution of the patent application. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, inter partes review, post grant review and equivalent proceedings in foreign jurisdictions, e.g., opposition proceedings. Such proceedings could result in revocation or amendment of our patents in such a way that they no longer cover our product candidates or competitive products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates. Such a loss of patent protection would have a material adverse impact on our business.We may not seek to protect our intellectual property rights in all jurisdictions throughout the world and we may not be able to adequately enforce our intellectual property rights even in the jurisdictions where we seek protection.Filing and prosecuting patent applications, and defending patents on our discovery engine platform and product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States could be less extensive than those in the United States, assuming that rights are obtained in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. In addition, the statutory deadlines for pursuing patent protection in individual foreign jurisdictions are based on the priority date of each of our patent applications and we may not timely file foreign patent applications. For the patent families related to YTX-7739, as well as for many of the patent families that we own, the relevant statutory deadlines have not yet expired.63Thus, for each of the patent families that we believe provides coverage for our lead product candidate, we will need to decide whether and where to pursue protection outside the United States.Competitors may use our technologies in jurisdictions where we do not pursue and obtain patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Even if we pursue and obtain issued patents in particular jurisdictions, our patent claims or other intellectual property rights may not be effective or sufficient to prevent third parties from so competing.The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to biotechnology or pharmaceuticals. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of or marketing of competing products in violation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing, and could provoke third parties to assert claims against it. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.If we do not obtain additional protection under the Hatch-Waxman Act and similar foreign legislation by extending the patent terms and obtaining data exclusivity for our product candidates, our business may be materially harmed.Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from our earliest U.S. non-provisional filing date in our chain of priority. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired for a product candidate, we may be open to competition from competitive medications, including generic medications. Given the amount of time required for the development, testing, and regulatory review of new product candidates, patents protecting such product candidates might expire before or shortly after such product candidates are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing product candidates similar or identical to ours.Depending upon the timing, duration, and specifics of FDA marketing approval of our product candidates, one or more of the U.S. patents we own may be eligible for a limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent term extension of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. However, we may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain a patent term extension or the term of any such extension is less than we request, the duration of patent protection we obtain for our product candidates may not provide us with any meaningful commercial or competitive advantage, our competitors may obtain approval of competing products earlier than they would otherwise be able to do so, and our ability to generate revenues could be materially adversely affected.Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.As is the case with other biotechnology companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biotechnology industry involve both technological and legal complexity, and is therefore costly, time-consuming, and inherently uncertain. In addition, the United States has recently enacted and is currently implementing wide-ranging patent reform legislation: the Leahy-Smith America Invents Act. The America Invents Act includes a number of64significant changes to U.S. patent law. After March 2013, under the America Invents Act, the United States transitioned to a first-inventor-to-file system in which, assuming that other requirements for patentability are met, the first-inventor-to-file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. The America Invents Act also includes provisions that affect the way patent applications will be prosecuted and that may also affect patent litigation. It is not yet clear what, if any, impact the America Invents Act will have on the operation of our business. However, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of any patents that may issue from our patent applications, all of which could have a material adverse effect on our business and financial condition.In addition, recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. The full impact of these decisions is not yet known. For example, on March 20, 2012, in Mayo Collaborative Services, DBA Mayo Medical Laboratories, et al. v. Prometheus Laboratories, Inc., the Court held that several claims drawn to measuring drug metabolite levels from patient samples and correlating them to drug doses were not patentable subject matter. The decision appears to impact diagnostics patents that merely apply a law of nature via a series of routine steps and it has created uncertainty around the ability to obtain patent protection for certain inventions. Additionally, on June 13, 2013, in Association for Molecular Pathology v. Myriad Genetics, Inc., the Court held that claims to isolated genomic DNA are not patentable, but claims to complementary DNA molecules are patent eligible because they are not a natural product. The effect of the decision on patents for other isolated natural products is uncertain. However, on March 4, 2014, the USPTO issued a memorandum to patent examiners providing guidance for examining claims that recite laws of nature, natural phenomena or natural products under the Myriad and Prometheus decisions. This guidance did not limit the application of Myriad to DNA but rather applied the decision to other natural products.In addition to increasing uncertainty with regard to our ability to obtain future patents, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on these and other decisions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce any patents that may issue in the future.We may be subject to damages resulting from claims that us or our employees, consultants, or advisors have wrongfully used or disclosed alleged trade secrets of their current or former employers.Our employees have been previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We also engage advisors and consultants who are concurrently employed at universities or who perform services for other entities.Although we try to ensure that our employees, consultants, and advisors do not use the proprietary information or know-how of others in their work for it, and although we are not aware of any claims currently pending against it, we may be subject to claims that us or our employees, advisors, or consultants have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third party. We have and may in the future also be subject to claims that an employee, advisor, or consultant performed work for us that conflicts with that person’s obligations to a third party, such as an employer, and thus, that the third party has an ownership interest in the intellectual property arising out of work performed for it. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying money claims, we may lose valuable intellectual property rights or personnel. A loss of key personnel or their work product could hamper or prevent our ability to commercialize our product candidates, which would materially adversely affect our commercial development efforts.We may not be successful in obtaining, through acquisitions, in-licenses or otherwise, necessary rights to our discovery engine platform, product candidates or other technologies.We currently have rights to intellectual property, through licenses from third parties, to identify and develop our discovery engine platform technology and product candidates. Many pharmaceutical companies, biotechnology companies, and academic institutions are competing with it in the field of neurodegeneration and discovery engine platform and may have patents and have filed and are likely filing patent applications potentially relevant to our business. In order to avoid infringing these third party patents, we may find it necessary or prudent to obtain licenses to such patents from such third party intellectual property holders. In addition, with respect to any patents we co-own with third parties, we may require licenses to such co-owners’ interest to such patents. However, we may be unable to secure such licenses or otherwise acquire or in-license any compositions, methods of use, processes, or other intellectual property rights from third parties that we identify as necessary for our current or future product candidates and our discovery engine platform technology. The licensing or acquisition of third party intellectual property rights is a competitive area, and several more established companies may pursue strategies to license or acquire third party intellectual property rights that we may consider65attractive or necessary. These established companies may have a competitive advantage over it due to their size, capital resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to it. We also may be unable to license or acquire third party intellectual property rights on terms that would allow us to make an appropriate return on our investment or at all. If we are unable to successfully obtain rights to required third party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of the relevant program or product candidate, which could have a material adverse effect on our business, financial condition, results of operations and prospects.Numerous factors may limit any potential competitive advantage provided by our intellectual property rights.The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, provide a barrier to entry against our competitors or potential competitors, or permit us to maintain our competitive advantage. Moreover, if a third party has intellectual property rights that cover the practice of our technology, we may not be able to fully exercise or extract value from our intellectual property rights. The following examples are illustrative:•others may be able to make products that are similar to our product candidates or utilize similar technology but that are not covered by the claims of the patents that we license or may own;•we, or our current or future licensors or collaborators, might not have been the first to make the inventions covered by the issued patent or pending patent application that we license or own now or in the future;•we, or our current or future licensors or collaborators, might not have been the first to file patent applications covering certain of our or their inventions;•others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our owned or licensed intellectual property rights;•it is possible that our current or future pending owned or licensed patent applications will not lead to issued patents;•issued patents that we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors or other third parties;•our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets;•we may not develop additional proprietary technologies that are patentable;•the patents of others may harm our business; and•we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property.Should any of these events occur, they could significantly harm our business and results of operations.Our Business Operations, Employee Matters and Managing GrowthOur corporate restructuring and the associated headcount reduction announced in February 2022 may not result in anticipated savings, could result in total costs and expenses and attrition that are greater than expected and could disrupt our business.On February 17, 2022, we announced an approximate 60% reduction in headcount as part of a corporate restructuring. We may not realize, in full or in part, the anticipated benefits, savings and improvements in our cost structure from our restructuring efforts due to unforeseen difficulties, delays or unexpected costs. If we are unable to realize the expected operational efficiencies and cost savings from the restructuring, our operating results and financial condition would be adversely affected. We also cannot guarantee that we will not have to undertake additional headcount reductions or restructuring activities in the future. Furthermore, our restructuring activities may be disruptive to our operations. For example, our headcount reductions could yield unanticipated consequences, such as attrition beyond planned staff reductions, or increase difficulties in our day-to-day operations. Our headcount reductions could also harm our ability to attract and retain qualified management, scientific, clinical, manufacturing and other personnel who are critical to our business. Any failure to attract or retain qualified personnel could prevent us from successfully developing and commercializing our product candidates in the future.66Our future success depends on our ability to retain our management team and to retain and motivate qualified personnel.Our ability to compete in the highly competitive biotechnology and biopharmaceuticals industries depends upon our ability to retain highly qualified managerial, scientific, and medical personnel. In order to induce valuable employees to continue their employment with us, we have provided stock options and restricted stock that vest over time. The value to employees of stock options that vest over time is significantly affected by movements in our stock price that are beyond our control, and may at any time be insufficient to counteract more lucrative offers from other companies.We are highly dependent on our management, scientific and medical personnel, including our Chief Executive Officer, Richard Peters, M.D., Ph.D. Despite our efforts to retain valuable employees, members of our management, scientific, and development teams may terminate their employment with us on short notice. The loss of the services of any of our executive officers, including Dr. Peters, other key employees and other scientific and medical advisors, and an inability to find suitable replacements could result in delays in product development and harm our business. Pursuant to their employment arrangements, each of our executive officers, and other employees may voluntarily terminate their employment at any time, with or without notice. Our success also depends on our ability to continue to attract, retain, and motivate highly skilled junior, mid-level, and senior managers as well as junior, mid-level, and senior scientific and medical personnel. While we committed to pay one-time employee retention payments to certain employees at the time we effected our February 2022 corporate restructuring, the combination of having effected such restructuring and our announced plans to explore strategic alternatives may make it increasingly difficult for us to retain our executive officers, other key employees and other scientific and medical advisors.We may not be able to attract or retain qualified management and scientific personnel in the future due to the intense competition for a limited number of qualified personnel among biopharmaceutical, biotechnology, pharmaceutical, and other businesses. Many of the other pharmaceutical companies that we compete against for qualified personnel have greater financial and other resources, different risk profiles, and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high quality candidates than what we may be able to offer. We also experience competition for the hiring of scientific personnel from universities and research institutions. If we are unable to continue to attract and retain high quality personnel, the rate and success at which we can develop and commercialize product candidates will be limited.We may need to grow the size of our organization and we may experience difficulties identifying and hiring the right employees and in managing this growth.Although we announced in February 2022 an approximate 60% reduction in our headcount, it is possible that, in the future, we may experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of research and development, product development and manufacturing, regulatory affairs and, if any product candidates are submitted for or receive marketing approval, sales, marketing and distribution. Future growth would impose significant added responsibilities on members of management, including:•identifying, recruiting, integrating, maintaining and motivating additional employees;•managing our internal development efforts effectively, including the clinical and FDA review process for our product candidates, while complying with our contractual obligations to contractors and other third parties; and•improving our operational, financial and management controls, reporting systems and procedures.Our future financial performance and our ability to commercialize our product candidates may depend, in part, on our ability to effectively manage any future growth and our management may also have to divert a disproportionate amount of its attention away from day-to-day activities in order to devote a substantial amount of time to managing these growth activities.We currently rely, and for the foreseeable future will continue to rely, in substantial part on certain independent organizations, advisors, contractors and consultants to provide certain services, including substantially all aspects of regulatory approval, clinical management and manufacturing. There can be no assurance that the services of independent organizations, advisors, contractors and consultants will continue to be available to us on a timely basis when needed or that we will be able to find qualified replacements. In addition, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by independent organizations, advisors, contractors or consultants is compromised for any reason, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval of our product candidates or otherwise advance our business. There can be no assurance that we will be able to manage our existing independent organizations, advisors, contractors or consultants or find other competent resources on economically reasonable terms, or at all.If we are not able to effectively expand our organization by hiring new employees and expanding the roster of independent organizations, advisors and consultants on whom we rely on an outsourced basis, we may not be able successfully to implement the tasks necessary to further develop and commercialize our product candidates and, accordingly, may not achieve our research, development and commercialization goals.67We face potential product liability exposure, and, if claims are brought against us, we may incur substantial liability.Kineta’s Common Stockuse of our product candidates in clinical trials and the sale of our product candidates, if approved, exposes us to the risk of product liability claims. Product liability claims might be brought against us by patients, healthcare providers, or others selling or otherwise coming into contact with our product candidates. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, including as a result of interactions with alcohol or other drugs, negligence, strict liability, and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we become subject to product liability claims and cannot successfully defend itself against them, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in, among other things:•withdrawal of subjects from our clinical trials;•substantial monetary awards to patients or other claimants;•decreased demand for our product candidates or any future product candidates following marketing approval, if obtained;•damage to our reputation and exposure to adverse publicity;•increased FDA warnings on product labels;•litigation costs;•distraction of management’s attention from our primary business;•loss of revenue; and•the inability to successfully commercialize our product candidates or any future product candidates, if approved.We maintain product liability insurance coverage for our clinical trials with a €5 million annual aggregate coverage limit. Nevertheless, our insurance coverage may be insufficient to reimburse us for any expenses or losses we may suffer. Moreover, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses, including if insurance coverage becomes increasingly expensive. If and when we obtain marketing approval for our product candidates, we intend to expand our insurance coverage to include the sale of commercial products; however, we may not be able to obtain this product liability insurance on commercially reasonable terms. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. The cost of any product liability litigation or other proceedings, even if resolved in our favor, could be substantial, particularly in light of the size of our business and financial resources. A product liability claim or series of claims brought against us could cause our stock price to decline and, if we are unsuccessful in defending such a claim or claims and the resulting judgments exceed our insurance coverage, our financial condition, business, and prospects could be materially adversely affected.Inadequate funding for the FDA, the SEC and other government agencies, including from government shut downs, or other disruptions to these agencies’ operations, could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, the ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.Disruptions at the FDA and other agencies may also slow the time necessary for new product candidates to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.68We will continue to incur increased costs as a result of operating as a public company, and our management team will be required to devote substantial time to new compliance initiatives.As a public company, we will continue to incur significant legal, accounting, and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”) and rules subsequently implemented by the SEC and The Nasdaq Stock Market have imposed various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly.Pursuant to Section 404 of the Sarbanes-Oxley Act (“Section 404”), we will be required to furnish a report by our management on our internal control over financial reporting, which may include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. While we remain a “smaller reporting company” with less than $100 million in annual revenues, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants, and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented, and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that neither we nor our independent registered public accounting firm will be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.If we fail to maintain proper and effective internal controls, our ability to produce accurate and timely financial statements could be impaired, which could result in sanctions or other penalties that would harm our business.After the completion of the Merger, we became subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act, and the rules and regulations of The Nasdaq Capital Market. The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal controls over financial reporting. Commencing with our fiscal year ending the year that the Merger was completed, we must perform system and process design evaluation and testing of the effectiveness of our internal controls over financial reporting to allow management to report on the effectiveness of our internal controls over financial reporting in our Form 10-K filing for that year, as required by Section 404 of the Sarbanes-Oxley Act. This will require that we incur substantial additional professional fees and internal costs to expand our accounting and finance functions and that we expend significant management efforts. Prior to the Merger, we had never been required to test our internal controls within a specified period and, as a result, we may experience difficulty in meeting these reporting requirements in a timely manner.We may discover weaknesses in our system of internal financial and accounting controls and procedures that could result in a material misstatement of our financial statements. Our internal control over financial reporting will not prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud will be detected.If we are not able to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, or if we are unable to maintain proper and effective internal controls over financial reporting, we may not be able to produce timely and accurate financial statements. If that were to happen, our investors could lose confidence in our reported financial information, the market price of our stock could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities.Changes in tax law could adversely affect our business and financial condition.The rules dealing with U.S. federal, state, and local and non-U.S. taxation are constantly under review by persons involved in the legislative process, the Internal Revenue Service, the U.S. Treasury Department and other taxing authorities. Changes to tax laws or tax rulings, or changes in interpretations of existing laws (which changes may have retroactive application), could adversely affect us or holders of our common stock. These changes could subject us to additional income-based taxes and non-income taxes (such as payroll, sales, use, value-added, digital tax, net worth, property, and goods and services taxes), which in turn could materially affect our financial position and results of operations. Additionally, new, changed, modified, or newly interpreted or applied tax laws could increase our customers’ and our compliance, operating and other costs, as well as the costs of our products. In recent years, many such changes have been made, and changes are likely to continue to occur in the future. As we expand the scale of our business activities, any changes in the U.S. and non-U.S. taxation of such activities may increase our effective tax rate and harm our business, financial condition, and results of operations.69Our ability to use our net operating loss carryforwards and certain tax credit carryforwards may be subject to limitation.As of December 31, 2021, we had federal and state net operating loss (“NOL”) carryforwards of $486.6 million and $497.2 million, respectively. Of the federal NOL carryforwards, $228.1 million begin to expire in 2026, and $258.5 million can be carried forward indefinitely. As of December 31, 2021, we had $0.1 million of foreign net operating loss carryforwards that do not expire. Under Section 382 of the Code changes in our ownership may limit the amount of our net operating loss carryforwards and research and development tax credit carryforwards that could be utilized annually to offset our future taxable income, if any. This limitation would generally apply in the event of a cumulative change in ownership of our company of more than 50% within a three-year period. Any such limitation may significantly reduce our ability to utilize our net operating loss carryforwards and research and development tax credit carryforwards before they expire. The completion of the Merger, together with private placements and other transactions that have occurred since our inception, may trigger such an ownership change pursuant to Section 382. Any such limitation, whether as the result of the Merger, prior private placements, sales of our common stock by our existing stockholders, or additional sales of our common stock by us after the Merger, could have a material adverse effect on our results of operations in future years. We have not yet completed a Section 382 analysis, and therefore, there can be no assurances that the NOL is already not limited.In addition, the reduction of the corporate tax rate under the TCJA may cause a reduction in the economic benefit of our NOL carryforwards and other deferred tax assets available to it. For example, while the TCJA allows for federal NOLs incurred in tax years beginning after December 31, 2017 to be carried forward indefinitely, the TCJA also imposes an 80% limitation on the use of NOLs that are generated in tax years beginning after December 31, 2017. Net operating losses generated prior to December 31, 2017, however, will still have a 20-year carryforward period, but are not subject to the 80% limitation.The Coronavirus Aid, Relief and Economic Security Act (the “CARES Act”) modifies, among other things, the rules governing NOLs. NOLs arising in tax years 2019, 2020, and 2021 are subject to a five year carryback and indefinite carryforward, while NOLs arising in tax years beginning after December 31, 2020 also are subject to indefinite carryforward but cannot be carried back. The CARES Act also suspends the 80% limitation mentioned above for NOLs generated in taxable years ending after December 31, 2017 that are used in taxable years ending on or prior to December 31, 2020. In future years, if and when a net deferred tax asset is recognized related to our NOLs, the changes in the carryforward/carryback periods as well as the new limitation on use of NOLs may significantly impact our valuation allowance assessments for NOLs generated after December 31, 2017.Furthermore, our ability to utilize NOLs is conditioned upon our maintaining profitability in the future and generating U.S. federal taxable income. Since we do not know whether or when we will generate the U.S. federal taxable income necessary to utilize our remaining NOLs, these NOL carryforwards generated prior to December 31, 2017 could expire unused. Notwithstanding the foregoing discussion of NOLs, we have recorded a full valuation allowance related to our NOLs due to the uncertainty of the ultimate realization of the future benefits of such NOLs.We may acquire businesses or products, or form strategic alliances, in the future, and we may not realize the benefits of such acquisitions.We may acquire additional businesses or products, form strategic alliances, or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter numerous difficulties in developing, manufacturing, and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. We cannot provide assurance that, following any such acquisition, we will achieve the synergies expected in order to justify the transaction.Risks Related to our Common StockThe market price of ourKineta’s common stock may be highly volatile and youor may not be able to resell your shares at or above the price at which you purchased our shares.decline regardless of its operating performance.markettrading price for ourof the common stock historically has been highlywill be volatile and could continue to be subject to wide fluctuations in response to various factors. The daily closing market price for our common stock has varied between a high pricefactors, some of $19.64 on April 1, 2021 and a low price of $0.97 on March 11, 2022 in the twelve-month period ending on March 15, 2022. The stock market in general, and the market for biopharmaceutical companies in particular, has experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, you may not be able to sell your common stock at or above the price at which you purchased your shares. The market price for our common stock may be influenced by manyare beyond Kineta’s control. These factors including:include:failure to lift the partial clinical hold on our IND for YTX-7739 by the FDA ;70•adverse resultsactual or delaysanticipated fluctuations in preclinical studies or clinical trials;•an inability to obtain additional funding;operating results; by us to successfully develop and commercialize our product candidates;•failure by us to maintain our existing strategic collaborations or enter into new collaborations;•failure by us or our licensors and strategic partners to prosecute, maintain or enforce our intellectual property rights;•changes in laws or regulations applicable to future products;•an inability to obtain adequate product supply for our product candidates or the inability to do so at acceptable prices;•adverse regulatory decisions;•the introduction of new products, services or technologies by our competitors;•failure by us to meet or exceed financial estimates and projections we may provideof the investment community or that Kineta provides to the public;failureissuance of new or updated research or reports by us to meetsecurities analysts or exceedchanged recommendations for the financial projections of the investment community;•the perception of the pharmaceutical industry by the public, legislatures, regulators and the investment community;in general;commitments by us, our strategic partners or our competitors;commitments;disputesoperating and share price performance of other companies in the industry or other developments relating to proprietary rights, including patents, litigation mattersrelated markets;our ability to obtain patent protection for our technologies;magnitude of investments in the growth of the business;scientificmanagement or managementother personnel;lawsuits, including patentstockholders or stockholder litigation;the perception that such sales could occur;market valuationsincurrence of similar companies;•sales of our common stock by us or our stockholders in the future;debt; andthe trading volume of our common stock.general economic, political and market conditions. companies trading in the stock market in general, and The Nasdaq Capital Marketthe stock prices of bio-pharmaceutical and biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of thesethose companies. Broad market and industry factors may negativelyseriously affect the market price of our common stock, regardless of our actual operating performance.If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our share price and trading volume could decline.The trading market for our common stock will likely depend in part on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. Although we have obtained research coverage from certain analysts, there can be no assurance that analysts will continue to cover us or provide favorable coverage. If one or more analysts downgrade our stock or change their opinion of our stock, our share price would likely decline. In addition, if one or more analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our sharepast, following periods of volatility in the overall market and the market price or trading volume to decline.Our principal stockholders and management ownof a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.As of December 31, 2021, our executive officers, directors, five percent or greater stockholders and their affiliates beneficially own approximately 46.3% of our outstanding voting stock. These stockholders will have the ability to influence us through their ownership positions. These stockholders may be able to determine all matters requiring stockholder approval. For example, these stockholders, acting together, may be able to control elections of directors or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may believe are in your best interest as one of our stockholders.We could be subject toparticular company’s securities, class action litigation.In the past, securities class action litigation has often been broughtinstituted against a company following a decline in the market price of its securities.these companies. This risk is especially relevant for us because pharmaceutical companies have experienced significant stock price volatility71in recent years. If we face such litigation, itif instituted, could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.resources.An active trading market for our common stock may not be sustained.Although our common stock is listed on The Nasdaq Capital Market, an active trading market for our shares may never be sustained. If an active market for our common stock is not sustained, it may be difficult for you to sell shares you purchased without depressing the market price for the shares, or at all.An inactive trading market may also impair our ability to raise capital to continue to fund operations by selling additional shares and may impair our ability to acquire other companies or technologies by using our shares as consideration.We have never paid and doKineta does not intend to pay cash dividends in the foreseeable future. As a result, capital appreciation, if any, will be your sole source of gain.We have never paid cash dividends on any of our capital stock and weKineta currently intendintends to retain any future earnings if any, to fund the development and growth of ourits business. In addition, we may enter into future debt agreements that restrict our abilityAny determination to pay dividends.dividends in the future will be at the discretion of the Board of Directors of Kineta (the “Board”) and will depend on Kineta’s financial condition, operating results, capital requirements, general business conditions and other factors that the Board may deem relevant. As a result, capital appreciation, if any, of ourKineta’s common stock will be yourthe sole source of gain for the foreseeable future.OurKineta’s amended and restated bylaws contain exclusive forum provisions, which may limit a stockholder’s ability to bring a claim in a judicial forum it finds favorable and may discourage lawsuits with respect to such claims.Our thirdKineta’s fourth amended and restated bylaws provide that, unless we consentKineta consents in writing to an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for the following types of actions or proceedings under Delaware statutory or common lawlaw: (1) any derivative action or proceeding brought on ourKineta’s behalf; (2) any action or proceeding asserting a claim of breach of a fiduciary duty owed by any of ourKineta’s current or former directors, officers or other employees to usKineta or ourits stockholders; (3) any action or proceeding asserting a claim against usKineta or any of ourits current or former directors, officers, employees arising out of or pursuant to any provision of the Delaware General Corporation Law, ourDGCL, Kineta’s amended and restated certificate of incorporation or ourKineta’s amended and restated bylaws (each as may be amended from time to time); (4) any action or proceeding to interpret, apply, enforce or determine the validity of ourKineta’s amended and restated certificate of incorporation or ourKineta’s amended and restated bylaws (including any right, obligation, or remedy thereunder); (5) any action or proceeding as to which the Delaware General Corporation LawDGCL confers jurisdiction to the Court of Chancery of the State of Delaware; and (6) any action or proceeding asserting a claim against usKineta or any director, officer or other employee, governed by the internal affairs doctrine (the(the “Delaware Forum Provision”). The Delaware Forum Provision will not apply to any causes of action arising under the Securities Act, the Exchange Act or for which the federal courts have exclusive jurisdiction.Our thirdKineta’s fourth amended and restated bylaws further provide that, unless we consentKineta consents in writing to an alternative forum, the federal district courts of the United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act (the “Federal Forum Provision”). In addition, our thirdKineta’s fourth amended and restated bylaws provide that any person or entity holding, owning or otherwise acquiring any interest in shares of ourKineta’s capital stock is deemed to have notice of and consented to the foregoing Delaware Forum Provision and the Federal Forum Provision.usKineta or ourits directors, officers or other employees, which may discourage such lawsuits against usKineta and ourits directors, officers and other employees. Alternatively, if a court were to find the Delaware Forum Provision and the Federal Forum Provision to be inapplicable or unenforceable in an action, weKineta may incur additional costs associated with resolving such action in other jurisdictions, which could adversely affect ourKineta’s business and financial condition. The Court of Chancery of the State of Delaware may also reach different judgments or results than would other courts, including courts where a stockholder considering an action may be located or would otherwise choose to bring the action, and such judgments may be more or less favorable to usKineta than ourits stockholders.Our91ourits common stock.we failin the future Kineta fails to satisfy the continued listing requirements of Nasdaq, such as the corporate governance requirements, the market value standard or the minimum closing bid price requirement, Nasdaq may take steps to delist ourits common stock. Such a delisting would likely have a negative effect on the price of ourKineta’s common stock and would impair ourKineta’s stockholders’ ability to sell or purchase ourits common stock when they wish to do so. Delisting of ourKineta’s common stock could depress ourKineta’s stock price, substantially limit liquidity of ourKineta’s common stock and materially adversely affect ourKineta’s ability to raise capital on terms acceptable to us,Kineta, or at all. Further, delisting of the common stock would likely result in the common stock becoming a “penny stock” under the Exchange Act. In the event of non-compliance with the continued listing requirements or the delisting of ourKineta’s common stock, weKineta can provide no assurance that any action taken by usKineta to restore72ourits common stock to become listed again, stabilize the market price or improve the liquidity of ourits common stock, prevent ourits common stock from dropping below the Nasdaq minimum bid price requirement or prevent future non-compliance with Nasdaq’s listing requirements.General Risk FactorsFuture sales of shares by existing stockholders and future exercise of registration rights may adversely affect the market price of Kineta’s common stock.Unfavorable global economic conditionsSales of a substantial number of shares of Kineta’s common stock in the public market, or the perception that such sales could occur, could adversely affect our business, financial condition,the market price of Kineta’s common stock and may make it more difficult for you to sell your shares of Kineta’s common stock at a time and price that you deem appropriate. Kineta is unable to predict what effect, if any, sales of its shares in the public market or resultsthe availability of operations.shares for sale will have on the market price of its common stock. Moreover, as restrictions on resale end, the market price of Kineta’s shares of common stock could decline if the holders of currently restricted shares sell them or are perceived by the market as intending to sell them.Our results of operationsKineta could be adversely affected by general conditionssubject to securities class action litigation.global economymarket price of its securities. This risk is especially relevant for Kineta because biotechnology and pharmaceutical companies have experienced significant stock price volatility in the global financial markets. The recent global financial crisis caused extreme volatility and disruptions in the capital and credit markets. A severe or prolonged economic downturn,years. If Kineta faces such as the recent global financial crisis, could result in a variety of risks to our business, including, weakened demand for our product candidates and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption, or cause our customers to delay making payments for our services. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.We, or the third parties upon whom we depend, may be adversely affected by earthquakes or other natural disasters and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.Earthquakes or other natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition, and prospects. If a natural disaster, power outage, or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers and suppliers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, particularly when taken together with our lack of earthquake insurance, could have a material adverse effect on our business.Our internal computer systems, or those of our third-party CROs or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product candidates’ development programs.Despite the implementation of security measures, our internal computer systems and those of our third-party CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. While we have not experienced any such system failure, accident, or security breach to date, if such an event were to occur and cause interruptions in our operations,litigation, it could result in substantial costs and a material disruptiondiversion of our programs. For example, the loss of clinical study data for our product candidatesmanagement’s attention and resources, which could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications or other data or applications relating to our technology or product candidates, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities and the further development of our product candidates could be delayed.harm Kineta’s business.We may be unable to adequately protect our information systems from cyberattacks, whichIf securities or industry analysts do not publish or cease publishing research or reports about Kineta, its business or its market, or if they change their recommendations regarding the common stock adversely, the price and trading volume of the common stock could result in the disclosure of confidential or proprietary information, including personal data, damage our reputation, and subject us to significant financial and legal exposure.decline.We rely on information technology systemsThe trading market for the common stock will be influenced by the research and reports that weindustry or our third-party providers operatesecurities analysts may publish about Kineta, its business, its market or its competitors. If any of the analysts who may cover Kineta change their recommendation regarding the common stock adversely, or provide more favorable relative recommendations about its competitors, the price of the common stock would likely decline. If any analyst who may cover Kineta were to process, transmit and store electronic information in our day-to-day operations. In connection with our product discovery efforts, we may collect and use a variety of personal data, such as names, mailing addresses, email addresses, phone numbers and clinical trial information. A successful cyberattack could result in the theft or destruction of intellectual property, data or other misappropriation of assets, or otherwise compromise our confidential or proprietary information and disrupt our operations. Cyberattacks are increasing incease their frequency, sophistication and intensity, and have become increasingly difficult to detect. Cyberattacks could include wrongful conduct by hostile foreign governments, industrial espionage, wire fraud and other forms of cyber fraud, the deployment of harmful malware, denial-of-service, social engineering fraud or other means to threaten data security, confidentiality, integrity and availability. A successful cyberattack could cause serious negative consequences for us, including, without limitation, the disruption of operations, the misappropriation of confidential business information, including financial information, trade secrets, financial loss and the disclosure of corporate strategic plans. Although we devote resources to protect our information systems, we realize that cyberattacks are a threat, and there can be no assurance that our efforts will prevent information security breaches that would result in business, legal, financial or reputational harm to us, or would have a material adverse effect on our results of operations and financial condition. Any failure to prevent or mitigate security breaches or improper access to, use of, or disclosure of our clinical data or patients’ personal data could result in significant liability under state (e.g., state breach notification laws), federal (e.g., HIPAA, as amended by HITECH), and international law (e.g.,73the EU General Data Protection Regulation, or GDPR) and may cause a material adverse impact to our reputation, affect our ability to use collected data, conduct new studies and potentially disrupt our business.We rely on our third-party providers to implement effective security measures and identify and correct for any such failures, deficiencies or breaches. We also rely on our employees and consultants to safeguard their security credentials and follow our policies and procedures regarding use and access of computers and other devices that may contain our sensitive information. If we or our third-party providers fail to maintain or protect our information technology systems and data integrity effectivelycoverage or fail to anticipate, plan for or manage significant disruptions to our information technology systems,regularly publish reports on Kineta, we or our third-party providers could have difficulty preventing, detecting and controlling such cyber-attacks and any such attacks could resultlose visibility in losses described above, as well as disputes with physicians, patients and our partners, regulatory sanctions or penalties, increases in operating expenses, expenses or lost revenues or other adverse consequences, any ofthe financial markets, which could have a material adverse effect on our business, resultscause the stock price or trading volume of operations, financial condition, prospects and cash flows. Any failure by such third partiesKineta securities to prevent or mitigate security breaches or improper access to or disclosure of such information could have similarly adverse consequences for us. If we are unable to prevent or mitigate the impact of such security or data privacy breaches, we could be exposed to litigation and governmental investigations, which could lead to a potential disruption to our business.decline.74ITEM 1B. UNRESOLVED STAFF COMMENTSNone.ITEMItem 2. PROPERTIESProperties.Our headquarters are located at 40 Guest Street, Suite 4410, Boston, Massachusetts, where we license research and development, laboratory and office space. Our license to use such space will terminate on March 31, 2022. Commencing April 1, 2022, we will licenseKineta occupies approximately 20%14,870 square feet of the office and laboratory space previously licensed at the same premises. Such license will expire on December 31, 2022. We also lease approximately 30,000(1,850 square feet of laboratory and office space located at 80 Guest Street, Suite 500, Boston, Massachusetts. Thiswhich is subleased to another biotech company) in Seattle, Washington under a lease that expires in April 2028. We believeJuly 2024. Kineta has an option to renew for two additional five-year terms. Kineta believes that our office and laboratory space is sufficient to meet ourits current facilities are adequate for its current needs for the foreseeable future and that suitable additional or substitute space at commercially reasonable terms will be available as and when needed.needed to accommodate any future expansion of Kineta’s operations.ITEMItem 3. LEGAL PROCEEDINGSLegal Proceedings.wehowever, Kineta may be a party to litigation or subject to legal proceedings and claims inincident to the ordinary course of business. InformationAlthough the results of litigation and claims cannot be predicted with respect to legal proceedings and this item is included in Note 16certainty, Kineta currently believes that the final outcome of these ordinary course matters will not have a material adverse effect on Kineta’s business. Regardless of the Notes to Consolidated Financial Statements contained in Part II, Item 8outcome, litigation can have an adverse impact on Kineta because of this Annual Report on Form 10-K, which is incorporated herein by reference.defense and settlement costs, diversion of management resources and other factors.ITEMItem 4. MINE SAFETY DISCLOSURESMine Safety Disclosures75ITEMItem 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIESMarket for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.commenced tradinghas traded on The Nasdaq Capital Market under the symbol “KA” since December 19, 2022. From December 23, 2020 to December 18, 2022, our common stock was listed on The Nasdaq Capital Market under the symbol “YMTX”. Prior to December 23, 2020, our common stock was listed on The Nasdaq Global Market under the symbol “PTI” on February 11, 2016. Prior to that date, there was no public market for our common stock. On December 22, 2020, we completed our previously announced merger transaction with Yumanity, Inc. (formerly Yumanity Therapeutics, Inc.) in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated as of August 22, 2020, as amended on November 6, 2020, (the “Merger Agreement”), by and among Pangolin Merger Sub, Inc., a wholly-owned subsidiary of Proteostasis (“Merger Sub”), Yumanity Holdings, LLC (“Holdings”) and Yumanity, Inc., pursuant to which Merger Sub merged with and into Yumanity, Inc., with Yumanity, Inc. surviving as a wholly owned subsidiary of Proteostasis (“the Merger”). Immediately prior to the effective time of the Merger, Holdings merged with and into Yumanity, Inc. and Yumanity, Inc. continued to exist as the surviving corporation. On December 22, 2020, in connection with, and prior to the completion of, the Merger, Proteostasis effected a 1-for-20 reverse stock split of its common stock (the “Reverse Stock Split”). On December 23, 2020, following the completion of the Merger, our common stock began trading on the Nasdaq Capital Market under the symbol “YMTX”.17, 2022,18, 2024, there were approximately 74270 holders of record of shares of our common stock. This number does not include stockholders for whom shares are held in “nominee” or “street” name.We have never declared or paidOn December 14, 2022, pursuant to the Asset Purchase Agreement (the “Asset Purchase Agreement”) by and between Yumanity and Janssen Pharmaceutica NV (“Janssen”), Yumanity sold to Janssen (such transaction, the “Asset Sale”) all of its rights, title and interest in and to clinical-stage product candidate YTX-7739 as well Yumanity’s unpartnered pre-clinical and discovery-stage product candidates and related intellectual property rights for a purchase price of $26.0 million in cash. In connection with the Asset Sale, on December 19, 2022, the Company distributed $15.5 million in remaining available cash dividendsproceeds from the Asset Sale, net of net cash requirements associated with the closing of the Merger and amounts retained for outstanding obligations, to stockholders of record as of the close of business on our capital stock. WeDecember 15, 2022, via a one-time dividend (the “Distribution”). Other than this Distribution, we anticipate that we will retain all of our future earnings to advance the clinical trials and preclinical studies for our products and to finance the operation of our business and do not anticipate declaring or paying any cash dividends on our capital stock in the foreseeable future. Any future determination to declare and pay cash dividends, if any, will be made at the discretion of our board of directors and will depend on a variety of factors, including applicable laws, our financial condition, results of operations, contractual restrictions, capital requirements, business prospects, general business or financial market conditions, and other factors our board of directors may deem relevant. In addition, our loan and security agreement contains covenants that could restrict our ability to pay cash dividends.None.In connection and concurrently with the execution of the Merger Agreement, on June 5, 2022, we entered into a financing agreement, as amended on October 24, 2022, December 5, 2022, March 29, 2023, May 1, 2023, July 21, 2023 and October 13, 2023 (such financing agreement, as amended, the “Securities Purchase Agreement”), to sell shares of our common stock in a private placement (the “Private Placement”). The first closing of the Private Placement occurred on December 16, 2022 and we issued 649,346 shares of our common and received net proceeds of $7.4 million. The second closing of the Private Placement for an aggregate purchase price of $22.5 million was expected to occur on April 15, 2024. However, in February 2024, certain investors indicated they will not be able to consummate the second closing of the Private Placement.ITEMItem 6. RESERVEDReservedNot applicable.ITEMItem 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONSManagement’s Discussion and Analysis of Financial Condition and Results of Operations.76OverviewOverviewWe are a biopharmaceutical company focused on the discovery and development of innovative, disease-modifying therapies for neurodegenerative diseases. Neurodegenerative diseases cause a progressive loss of structure and function in the brain, leaving patients with devastating damage to their nervous system and widespread functional impairment. Although treatments may help relieve some of the physical or mental symptoms associated with neurodegenerative diseases, few of the currently available therapies slow or stop the continued loss of neurons, resulting in a critical unmet need. We are specifically focused on developing novel therapies to treat devastating conditions, either with large or orphan disease markets, including Parkinson’s disease, dementia with Lewy bodies, multiple system atrophy, amyotrophic lateral sclerosis, or ALS (also known as Lou Gehrig’s disease), frontotemporal lobar degeneration, or FTLD, and Alzheimer’s disease.Exploration of Strategic Alternatives and RestructuringInOn February 2022,29, 2024, we announced that we are exploring strategic alternatives to enhance shareholder value and engaged H.C. Wainwright as our exclusive financial advisor to assist in this process. No timetable has been established for the completion of this process, and the we do not expect to disclose developments unless and until the Board of Directors has concluded that disclosure is appropriate or required.In February we also began implementation of a strategic restructuring with the objective of preserving capital. In the first quarter of 2022, our board of directors approved a restructuring plan followinghad completed a review of our operations, cost structurebusiness, including the status of our programs, resources and growth opportunities.headcountour workforce by approximately 64% and the termination of approximately 60% across the Company. We recorded a chargeenrollment of $0.4 millionnew patients in the first quarter of 2022 as a result of the restructuring, which consisted of one-time termination benefits for employee severance, benefits and related costs, all of which are expected to result in cash expenditures and substantially all of which will be paid out by the end of 2022.Clinical and Regulatory UpdateOur lead program, YTX-7739, is in development for the potential treatment and disease modification of Parkinson’s disease. YTX-7739 targets an enzyme known as stearoyl-CoA desaturase, or SCD. Inhibition of SCD in multiple cellular systems, including patient-derived neurons, as well as in a novel mouse model of Parkinson’s disease, has been demonstrated to overcome the toxicity of misfolded alpha-synuclein or α-synuclein, a protein strongly associated with Parkinson’s disease.In January 2022, the U.S Food and Drug Administration (FDA) placed a partial clinical hold on our multidose clinical trials of YTX-7739. The partial clinical hold suspends initiation of multiple dose clinical trials in the U.S. until the FDA’s questions have been addressed. The FDA has not halted all clinical programming and is permitting our planned single dose formulationongoing VISTA-101 Phase 1/2 clinical trial to proceed. We anticipate working closely with the FDA to adequately address their concerns. While we work to address the FDA’s concerns, we have paused our planned window-of-opportunity clinical study of YTX-7739 in glioblastoma multiforme patients and exploration of YTX-7739 in additional indications.On November 10, 2021, we announced the top-line results of a Phase 1b clinical trial of YTX-7739evaluating KVA12123 in patients with mild-to-moderate Parkinson’s disease. The Phase 1b clinical trial was a randomized, placebo-controlled, double-blind multi dose study to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics of YTX-7739. Data were reported from 20 patients with mild-to-moderate Parkinson’s disease.advanced solid tumors. Patients received once-daily oral doses of YTX-7739 (20 mg or placebo) for 28 days. YTX-7739 was shown to inhibit its primary target, SCD, an enzyme whose inhibition has been closely linked to neuronal survival and improved motor function in a Parkinson’s disease model.After 28 days of treatment, the 20 mg dose given once-daily reduced the fatty acid desaturation index (FA-DI), a biomarker of SCD inhibition, by approximately 20%-40%, the range expected to be clinically relevant based on preclinical studies. Target engagementcurrently enrolled in the cerebrospinal fluid suggested that YTX-7739 effectively crossedtrial will be permitted to continue to participate. We have made this decision, in part, because certain investors have indicated they will not be able to fulfill their contractual obligation to consummate the blood-brain barrier. Additionally, the pharmacokinetic/pharmacodynamic profile of YTX-7739 was consistent with previous studies and we believe informs dose selection for future studies.YTX-7739 was generally well tolerated with all treatment emergent adverse events being mild to moderate in severity. There were no serious adverse events. Moderate adverse events (AEs) in the active treatment group consisted of 2 patients with increased Parkinson’s symptoms, 2 patients with lower back pain, 1 patient with headache, 1 patient with myalgia, 1 patient with insomnia, 1 patient with ligament strain, and 1 patient with vaccination complication. One patient on placebo had moderate worsening of tremors and Parkinsonism, which led to discontinuation. AEs occurring at a higher percentage in 2 or more patients administered YTX-7739 compared to placebo were procedural pain, myalgia, dry eye, hyperbilirubinemia, hypesthesia, lower back pain, and constipation. AEs occurring at a higher percentage with placebo included orthostatic hypotension, headache, tremor, fatigue and dizziness.As expected, after only 28 days of dosing, there were no statistically significant differences in clinical assessments (Unified Parkinson's Disease Rating Scale Part III (UPDRS III), Montreal Cognitive Assessment (MoCA)) or most exploratory biomarkers.77Quantitative electroencephalogram (qEEG) assessments of the effect of YTX-7739 on brain activity were completed in a subset of 8 patients and demonstrated a statistically significant change compared to baseline, suggestive of a potential improvement in synaptic function.Research and DiscoveryAt the center of our scientific foundation is our drug discovery engine, which is based on technology licensed from the Whitehead Institute, an affiliate of the Massachusetts Institute of Technology. This core technology, combined with investments and advancements by us, is designed to enable rapid screening to identify drugs with the potential to modify disease by overcoming toxicity in disease-causing gene networks. Toxicity in many neurodegenerative diseases results from an aberrant accumulation of misfolded proteins in the brain. We leverage our proprietary discovery engine to identify and screen novel drug targets and drug molecules for their ability to protect nerve cells from toxicity arising from misfolded proteins. To date, we have identified over 20 targets, most of which have not previously been linked to neurodegenerative diseases.Private Placement (as defined below).Financial UpdateDue to the fact that we are unable to consummate the Private Placement, management and the Board has determined that it was in the best interests of the stockholders to seek a strategic alternative so that we could continue to operate. If the strategic process is unsuccessful, the Board may decide to pursue a liquidation or obtain relief under the US Bankruptcy Code.significant operating losses in each period since inception. Our ability to generate product revenue sufficient to achieve profitability will depend on the successful development and eventual commercialization of one or more of our current or future product candidates. Our net losses were $39.5$14.1 million and $57.5 million, respectively, for the yearsyear ended December 31, 20212023 and 2020.$63.5 million for the year ended December 31, 2022. As of December 31, 2021,2023, we had an accumulated deficit of $187.3$165.8 million. to continue to incur significant expenses and continued operating losses.We will not generate revenue from product sales unlesslosses for at least the next several years as we initiate and until we successfully completecontinue the clinical development of, and obtainseek regulatory approval for, our product candidates. If we obtain regulatory approval for any of our product candidates and do not enter intoadd personnel necessary to advance our pipeline of clinical-stage product candidates. In addition, operating as a commercialization partnership, we expect to incur significant expenses related to developing our internal commercialization capability to support product sales, marketing, manufacturing and distribution activities. We also expect to incur ongoingpublicly-traded company will involve the incurrence of substantial other costs associated with operating as a public company. We expect to continue to incur significantthat our operating losses for the foreseeable future.will fluctuate significantly from quarter to quarter and year to year due to timing of clinical development programs and efforts to achieve regulatory approval.As a result,From inception to December 31, 2023, we will need substantial additional funding to support our continuing operations. Potential strategic alternatives that may be considered as parthave raised cash from sales and issuances of this process include an acquisition, merger, reverse merger, other business combination, sales of assets, licensing or other strategic transactions involving the Company. We may be unable to raise additional funds or enter into other agreements or arrangements when needed on favorable terms, or at all. If we fail to raise capital or enter into such agreements as,common stock and when, needed, we could have to further delay, reduce or eliminate development of one or more of our product candidates or delay our pursuit of potential in-licenses or acquisitions.Because of the numerous risks and uncertainties associated with product development, we are unable to predict the timing or amount of increased expenses or when or if we will be able to achieve or maintain profitability. Even if we are able to generate product sales, we may not become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.Biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. Typically, it takes many years to develop one new product from the time it is discovered to when it is available for treating patients, and development may cease for a number of reasons. Because of the numerous risks and uncertainties associated with product development, including any impact from the ongoing COVID-19 pandemic, we are unable to predict the timing or amount of increased expenses or when or if we will be able to achieve or maintain profitability. Even if we are able to generate product sales, we may not become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.2021,2023, we had cash cash equivalentsof $5.8 million, and marketable securities of $36.5 million. We don't believe that our existing cash, cash equivalents and marketable securities will enable us to fund our operating expenses and capital expenditure requirements for a period of twelve months from the date of issuance of the consolidated financial statements included in this Annual Report and we and our independent registered public accounting firm have expressedthere is substantial doubt about our ability to continue as a going concern. See “— LiquidityFor more information, see the risk factor in Item 1A. of this Annual Report on Form 10-K entitled, “Kineta identified conditions and Capital Resources.” Our future viability beyondevents that point is dependent on ourraise substantial doubt about its ability to continue as a going concern, Kineta needs substantial additional funding, and if Kineta is unable to raise additional capital when needed or on favorable terms, its business, financial condition, and results of operation could be materially and adversely affected.”financesell shares of our operations.common stock to such investors in a private placement (the “Private Placement”). We and the investors entered into an amendment to the Securities Purchase Agreement on October 13, 2023 to, among other things, extend the date of the second closing from October 31, 2023 to April 15, 2024.COVID-19In MarchLicense Agreement with GenentechCOVID-19 was declared a global pandemic(as amended, the “Genentech KCP506 License Agreement”). Pursuant to the Genentech KCP506 License Agreement, KCP out-licensed certain intellectual property rights to Genentech for KCP’s KCP506 program. KCP506 is an α9α10 nicotinic acetylcholine receptor antagonist developed by KCP for the World Health Organizationtreatment of neuropathic pain and neurogenic inflammation. The termination of the Genentech KCP506 License Agreement does not affect the development of any of our core oncology products, and no revenue or expenses from the Genentech KCP506 License Agreement were expected for the years ending December 31, 2023 or 2024.We intend to date,evaluate strategic alternatives for the development of this program.continues to present a substantial public health and economic challenge around the world. The length of time and full extent to which the COVID-19 pandemic will directly or indirectly impacton our business, resultsthe extent of the impact of the pandemic on our business, operations and financial conditionclinical development timelines and plans will depend on future developments, including the severity and duration of any resurgence of COVID-19 and its variants. Clinical trial sites in many countries, including those in which we operate, in the past have incurred delays due to COVID-19. Certain of the sites in the KCP-506 Phase 1 clinical trial incurred delays due to COVID-19 that are highly uncertain, subject to change and difficult to predict. While we continue to conduct ourresulted in a delay in the results from that study.78research and development activities, theThe COVID-19 pandemic may cause disruptions that affect our ability to initiate and complete preclinical studies and clinical trials or to procure items that are essential for our research and development activities. The pandemicin the past has already caused significant disruptions in the financial markets, and in the future may continue to cause such disruptions, which could impact our ability to raise additional funds to support our operations. Moreover,pandemic has significantly impacted economies worldwideRussian invasion of Ukraine, the conflict in Israel and could result in adverse effects on our business and operations. Clinical trial sites in many countries, including those in which we operate, have incurred delays due to COVID-19. Certain of the sitesGaza Strip or deterioration in the YTX-7739 Phase 1b clinical trial incurred delays due to COVID-19, resulting in a delay of early results from that study.We plan to continue to closely monitor the ongoing impact of the COVID-19 pandemic on our employees and our business operations. In an effort to provide a safe work environment for our employees, we have, among other things, implemented measures to enable remote work whenever possible. We expect to continue to take actions as may be required or recommended by government authorities or as we determine are in the best interests of our employees and other business partners in light of the pandemic.Merger with ProteostasisOn August 22, 2020, Proteostasis Therapeutics, Inc, a Delaware corporation (“Proteostasis”), Pangolin Merger Sub, Inc. (“Merger Sub”), Yumanity, Inc. (formerly Yumanity Therapeutics, Inc.), and Yumanity Holdings, LLC (“Holdings”), entered into the Merger Agreement, as amended on November 6, 2020, pursuant to which Merger Sub merged with and into Yumanity, Inc. Immediately prior to the closing of the transaction, Holdings merged with and into Yumanity, Inc. with Yumanity, Inc. surviving the Merger (the “Yumanity Reorganization”) and, upon the closing of the Merger, Yumanity, Inc. became a wholly owned subsidiary of Proteostasis. The Merger was completed on December 22, 2020 pursuant to the terms of the Merger Agreement. In connection with the completion of the Merger, Proteostasis changed its name to Yumanity Therapeutics, Inc., and the trading symbol changed from “PTI” to “YMTX.” We refer to the historical operations of Holdings and Yumanity, Inc. as Yumanity and following the Merger, the business conducted by Yumanity became our primary business.Pursuant to the terms of the Merger Agreement, upon closing of the Merger, all of Yumanity, Inc.’s outstanding common stock was exchanged for common stock of Proteostasis and all outstanding options and warrants to purchase common stock of Yumanity, Inc. were exchanged for options and warrants to purchase common stock of Proteostasis.The transaction was accounted for as a reverse merger and as an asset acquisition in accordance with Generally Accepted Accounting Principles inbilateral relationship between the United States and China, may impact government spending, international trade and market stability, and cause weaker macro-economic conditions. The impact of these developments, including any resulting sanctions, export controls or GAAP. Under this method of accounting, Yumanity was deemed toother restrictive actions that may be the accounting acquirerimposed against governmental or other entities in, for financial reporting purposes. This determination was primarily based on the facts that, immediately following the Merger: (i) Yumanity’s equity holders owned a substantial majority of the voting rightsexample, Russia, have in the combined organization, (ii) Yumanity designated a majority of the members (7 of 9) of the initial board of directors of the combined organizationpast contributed and (iii) Yumanity’s senior management held all key positionsmay in the senior management of the combined organization. Accordingly, for accounting purposes, the transaction was treated as the equivalent of Yumanity issuing stockfuture contribute to acquire the net assets of Proteostasis. As a result, as of the closing date of the Merger, the net assets of Proteostasis were recorded at their acquisition-date fair valuesdisruption, instability and volatility in the global markets, which in turn could adversely impact our operations and weaken our financial statements of the Companyresults. Certain political developments may also lead to uncertainty to regulations and the reported operating results prior to the Merger are those of Yumanity.Private PlacementOn December 14, 2020, we entered into a subscription agreement with certain accredited investors for the sale by us in a private placement of 1,460,861 shares ofrules that may materially affect our common stock for a price of $23.00 per share. We refer to this sale herein as the Private Placement. The Private Placement closed on December 22, 2020. The aggregate gross proceeds for the issuance and sale of the common stock were $33.6 million and, after deducting certain of our expenses, the net proceeds we received in the Private Placement were $31.6 million.business.AprilDecember 2021, we entered into a sales agreement (the "Prior“Prior Sales Agreement"Agreement”) with Jefferies LLC (“Jefferies”) with respect to an at-the-market (“ATM”) offering program under which we issued and sold, from time-to-timetime to time at our sole discretion, shares of our common stock, in an aggregate offering amount of up to $60.0 million. In December 2021,February 2023, we terminated the Prior Sales Agreement and entered into a new sales agreement with Jefferies with respect to an ATM offering under which we may issue and sell, from time-to-timetime to time and at our sole discretion, shares of our common stock, in an aggregate offering amount of up to $60.0$17.5 million (the “New Sales Agreement”)., subject to the offering limits in General Instruction I.B.6 to Form S-3. Jefferies acts as our sales agent and will use commercially reasonable efforts to sell shares of common stock from time-to-time,time to time, based upon instruction from us. We will pay Jefferies 3.0% of the gross proceeds from the sales of any common stock sold pursuant to the New Sales Agreement.will pay Jefferies up to 3% of the gross proceeds from any common stock sold through the New Sales Agreement. We sold 112,83330,905 shares of common stock under the Prior Sales Agreement during the twelve months ended December 31, 2021 for gross proceeds of $1.5 million2022 for aggregate net proceeds to usthe Company of approximately $1.4 million, after deducting sales commissions. As of$0.4 million.792021, $60.0 million2023, we sold 126,503 shares of our common stock remained available for future issuanceto individual investors under the New Sales Agreement although these amounts may be limited asand received net proceeds of $600,000 in connection with the ATM equity offering program.will be subjectentered into a Securities Purchase Agreement (the “April 2023 Purchase Agreement”) with an institutional investor (the “April 2023 Investor”), pursuant to which we issued and sold, in a registered direct offering priced at-the-market under the general instructionsrules of Form S-3 known asNasdaq (such offering, the "baby shelf rules." Under these instructions, the amount“April 2023 Registered Offering”), (i) an aggregate of funds we can raise through primary public offerings of securities in any twelve-month period using our registration statement on Form S-3 is limited to one-third of the aggregate market value of the948,000 shares of our common stock, held by non-affiliatesat a purchase price of the Company. Therefore, we will be limited in the amount of proceeds it is able$4.21 per share and (ii) pre-funded warrants exercisable for up to raise by selling477,179 shares of our common stock using(the “April 2023 Pre-Funded Warrants”) to the April 2023 Investor at a purchase price of $4.209 per April 2023 Pre-Funded Warrant, for aggregate gross proceeds from the April 2023 Registered Offering (as defined below) of approximately $6.0 million before deducting the placement agent fee (as described in greater detail below) and related offering expenses.S-3 until such time as our public float exceeds $75 million.10-Q for the quarter ended June 30, 2023, the Company had regained compliance with the alternative standard under Nasdaq Listing Rule 5550(b)(1). Nasdaq considers the matter closed.RevenueRevenuesthe sale of products for the foreseeable future. If our development efforts for product candidates are successful and result in regulatory approval or licenses with third parties, we may generate revenuesales in the futurenear future. Our revenues have been primarily derived from product sales, milestone payments under our existing collaboration, agreement or payments from other license agreements that we may enter into with third parties.In June 2020, we entered into a research collaboration and license agreement (the “Collaboration Agreement”) with Merck Sharp & Dohme Corp. (“Merck”), focused on accelerating the development of new treatments for neurodegenerative diseases. Under the terms of the Collaboration Agreement, Merck will gain exclusive rights to two novel pipeline programs for the treatment of ALS and FTLD. We and Merck will collaborate to advance the two preclinical programs during the research term, after which Merck has the right to continue clinical development and commercialization. Under the Collaboration Agreement, we received an upfront payment totaling $15.0 million and are eligible to receive future milestone payments of up to $530.0 million associated with the successful research, development and sales of marketed products for pipeline programs, of which $5 million was paid to us in February, 2022 as discussed below,agreements as well as grants awarded by government agencies.sales. We will perform certainsales of licensed products.research term pursuant to the Collaboration Agreement and will participateexpected period of performance based on a Joint Steering Committee to oversee researchcost-based input method. Revenue from contingent development, regulatory and development activities.On December 17, 2021, Merck notified us thatcommercial milestones, when not deemed probable of significant reversal of cumulative revenue, is also recognized over the first data package thatperformance period based on a similar method. Where we submitted for one program has methave no remaining performance obligations, revenue from such milestones is recognized when the requirements to progress to the next stageaccomplishment of the research collaboration. Achievement of this milestone triggered a $5.0 million milestone payment from Merck, which was received in February 2022. We cannot provide assurance as to the timing of future milestones or royalty payments or that we will receive any of the future payments at all.is deemed probable.record revenue overfluctuate from year to year as a result of the timing and number of milestones and other payments.termcollaboration agreement (the “Merck Neuromuscular License Agreement”) with Merck (known as we satisfy our performance obligations underMSD outside the Collaboration Agreement. Accordingly, the upfront paymentUnited States and Canada) to support research, development and commercialization of $15.0 million as well as the $5.0 million milestone payment will be recognized asproducts for treatment of neuromuscular diseases, including amyotrophic lateral sclerosis. We recognize revenue using the cost-to-cost method, which we believe best depicts the transfer of control to the customer. Under the cost-to-cost method, the extent of progress towards completion is measured based on the ratio of actual costs incurred to the total estimated costs expected upon satisfying the identified performance obligation. Under this method, revenue is recognized as a percentage of actual cost incurred to the estimated costs to complete.recorded $8.0 million of collaborationdo not have any active grant agreements and do not expect any grant revenue for the twelve months ended December 31, 2021 related to the Collaboration Agreement.foreseeable future.consist primarily ofrepresent costs incurred in connection with the discovery, research, preclinical and clinical development, and manufacture of our product candidates, which will decrease substantiallycandidates. We recognize all research and development costs as a resultthey are incurred. Research and development expenses consist primarily of our recent restructuring, but historically have included:the following:stock/equity-basedstock-based compensation, consultantsresearch and consulting arrangements and other related costs for individuals involved in research and development activities;(“CROs”), investigative sites and other scientific development services;contract developmentcontracted research and manufacturing organizations (“CDMOs”) for developing and manufacturing materialmaterials for preclinical studies, clinical trials and clinical trials;laboratory supplies;milestones;costs;lab suppliesfacilities and other lab related expenses;allocated expenses for rent and insurance; andfacilities, depreciation and other allocated expenses which include direct and allocated expenses for rent, insurance and other operating costs.80We expenseincurred to advance research and development activities including manufacturing costs as incurredassociated with production, scale up, testing and recognize external development costs based on an evaluationoptimization of methods associated with the progress to completionproduction of specific tasks using information provided to us bymaterials.service providers. This process involves reviewing open contracts and purchase orders, communicating withoperating expenses has historically been our personnel to identify services that have been performed on our behalf, and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual costs. Nonrefundable advance payments for goods and services to be received in the future for useinvestment in research and development activities are deferred and capitalized in prepaid expenses and other current assets. The capitalized amounts are expensed as the related goods are delivered or the services are performed. Upfront payments, milestone payments and annual maintenance fees under license agreements are expensed in the period in which they are incurred.Our external directactivities. We expect our research and development expenses are tracked bywill increase in the future as we advance our product candidatecandidates into and consist primarilythrough clinical trials and pursue regulatory approvals, which will require a significant investment in costs of costs that include feesclinical trials, regulatory support and contract manufacturing. In addition, we continue to evaluate opportunities to acquire or in-license other expenses paid to outside consultants, CROs, CDMOsproduct candidates and research laboratoriestechnologies, which may result in connection with our preclinical development, process development, manufacturing and clinical development activities. Our directhigher research and development expenses due to license fee and/or milestone payments, as well as added clinical development costs.product candidate also include fees incurred under third-party license agreements. We do not allocate employeethe stage of program, clinical or preclinical. However, our internal expenses, including unallocated costs, personnel costs and infrastructure costs, associated with our platform technology, early stage discovery efforts, laboratory suppliesare not directly related to any one program and facilities, including depreciation or other indirect costs, to specific product candidates because these costs are deployed across multiple programs and our platform and, asprograms. As such, arewe do not separately classified.Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We cannot accurately project total program-specifictrack internal expenses through commercialization. There are numerous factors associated with the successful commercialization of product candidates including future trial design and various regulatory requirements, many of which cannot yet be determined with accuracy based on our stage of development. Additionally, future commercial and regulatory factors beyond our control will impact our clinical developmenta specific program and plans.basis.successful developmentprocess of conducting clinical trials necessary to obtain regulatory approval is costly and commercializationtime consuming. We may never succeed in timely developing and achieving regulatory approval for our product candidates. The probability of YTX-7739 and any othersuccess of our product candidates may be affected by numerous factors, including clinical data, competition, manufacturing capability and commercial viability. As a result, we may develop inare unable to determine the future is highly uncertain. At this time, we cannot reasonably estimate or know the nature, timingduration and completion costs of our development projects or when and to what extent we will generate revenue from the efforts that will be necessary to complete the preclinicalcommercialization and clinical developmentsale of any of our future product candidates. This is due to the numerous risks and uncertainties associated with product development and commercialization, including the following:•the partial clinical hold that has been placed on our IND for YTX-7739 by the FDA;•the timing and progress of preclinical and clinical development activities;•the number and scope of preclinical and clinical programs we decide to pursue;•the ability to maintain current research and development programs and to establish new ones;•establishing an appropriate safety profile with IND-enabling or foreign equivalent studies;•successful patient enrollment in, and the initiation and completion of, clinical trials;•the successful completion of clinical trials with safety, tolerability and efficacy profiles that are satisfactory to the FDA or any comparable foreign regulatory authority;•the receipt of regulatory approvals from applicable regulatory authorities;•the timing, receipt and terms of any marketing approvals from applicable regulatory authorities;•our ability to establish new licensing or collaboration arrangements;•the performance of our future collaborators, if any;•establishing commercial manufacturing capabilities or making arrangements with third-party manufacturers;•development and timely delivery of commercial-grade drug formulations that can be used in our planned clinical trials and for commercial launch;•obtaining, maintaining, defending and enforcing patent claims and other intellectual property rights;•launching commercial sales of product candidates, if approved, whether alone or in collaboration with others; and•maintaining a continued acceptable safety profile of the product candidates following approval.Any changes in the outcome of any of these variables with respect to the development of our product candidates in preclinical and clinical development could mean a significant change in the costs and timing associated with the development of these product candidates. For example, we could be required to expend significant additional financial resources and time to complete clinical81development of YTX-7739 due to the partial clinical hold that the FDA placed on our multidose clinical trials of YTX-7739 in the U.S. We may never obtain regulatory approval for any of our product candidates. Drug commercialization will take several years and millions of dollars in development costs.personnel-relatedemployee-related expenses, including salaries, benefits and stock/equity-basednoncash stock-based compensation expenses for personnel in executive, finance and accounting, human resources and other administrative functions. Other significant general and administrative expenses include legal fees relating to patent, intellectual property and corporate matters, andfunctions, as well as fees paid for legal, accounting audit,and tax services, consulting fees and other professional services, as well as facilities costs not otherwise included in research and other allocated expenses, whichdevelopment expenses. Legal costs include direct and allocated expenses for rent, insurance and other operating costs.We anticipate that our general and administrative expenses will increase slightly in the near future as we explore our strategic alternatives. These increases will likely includecorporate legal fees and feespatent costs. We expect to outside consultants, amongincur additional expenses as a result of operating as a public company, including expenses related to compliance with the rules and regulations of the SEC and Nasdaq, additional insurance, investor relations and other expenses.administrative expenses and professional services.2020.2022.Other Income (Expense)Change in Fair Value of Warrant LiabilitiesIn connection with our loan and security arrangements, we issued warrants to purchase preferred units. These warrants were liability classified and remeasured to fair value at each reporting date, with changes in the fair value recognized as a component of other income (expense) in our statement of operations.Immediately prior to the Merger, all of our outstanding warrants to purchase preferred units were exchanged and became warrants to purchase shares of common stock. As a result, the fair value of the warrants was reclassified to additional paid-in capital and there is no longer a warrant liability to remeasure for the year ended December 31, 2021.loan and security agreements, as well as amortizationdebt arrangements primarily consisting of debt issuance costs and accretion of a final paymentborrowings under several notes payable upon the maturity or the repayment in full of all obligations under such loans. Interest expense also consists of interest related to finance leases.agreements.Interest Income and Other Income (Expense), NetChange in Fair Value Measurement of Rights from Private PlacementInterest income consistsinterest earned on our invested cash balances. Other income (expense), net includes a gain onother asset relates to the extinguishment of debt upon forgivenessremeasurement of the PPP loan (see Paycheck Protection Loan section ofrights from the Description of Indebtedness below).Income TaxesPriorPrivate Placement that we determined was a derivative, which requires the asset to the Yumanity Reorganization, Holdings was treated as a partnershipbe accounted for federal income tax purposes and, therefore, its owners, and not Holdings, were subject to U.S. federal or state income taxation. Holdings’ directly held subsidiary was treated as a corporation for U.S. federal income tax purposes and subject to taxation in the United States. After the Yumanity Reorganization, the Company and its subsidiary are both taxpaying entities. Inat fair value. Until settlement, this other asset is remeasured at fair value at each reporting period our tax provision includedwith the effects of consolidating our subsidiary’s resultschanges in fair value recorded in the statement of operations. Since our inception,have notelected to account for under the fair value option. Until settlement, these notes payable are remeasured at fair value at each reporting period with the changes in fair value recorded any income tax benefits forthrough the statement of operations.losses we incurred in each year or for our earned research and development tax credits, as we believe, basedconsists of the gain upon the weight of available evidence, that it is more likely than not that allsettlement of our net operating loss carryforwards and tax credits will not be realized. Utilization of U.S. federal and state net operating loss carryforwards and research and development tax credit carryforwards may be subject to a substantial annual limitation under Section 382 of the Internal Revenue Code of 1986, and corresponding provisions of state law, due to ownership changes that be occurred previously or that could occur in the future. These ownership changes may limit the amount of carryforwards that can be utilized annually to offset future taxable income. The Company has not conducted a study to assess whether a change of control has occurred or whether there have been multiple changes of control since inception due to the significant complexity and cost associated with such a study. As of December 31, 2021, we had U.S. federal and state net operatingnotes payable.82loss carryforwardsOther (expense) income, net consists of $486.6 millionitems that are of a non-recurring nature and $497.2 million, respectively, which may be availableprimarily relate to offset futureitems that are immaterial.tax liabilities. Asattributable to noncontrolling interest reflects investors’ share of December 31, 2021, $228.1 million of federal net operating loss carryforwards will begin to expire(loss) income in 2026, and $258.5 million can be carried forward indefinitely. State net operating loss carryforwards will begin to expire in 2030. As of December 31, 2021, the Company had $0.1 million of foreign net operating loss carryforwards that do not expire. As of December 31, 2021, we also had U.S. federal, state, and foreign research and development tax credit carryforwards of $15.4 million, $6.2 million, and $0.1 million, respectively, which may be available to offset future tax liabilities and each begin to expire in 2027 for domestic credits, and foreign credits do not expire. We have recorded a full valuation allowance against our net deferred tax assets at each balance sheet date.majority owned subsidiary.20212023 and 20202022years ended December 31, 2021 and 2020:periods presented:Collaboration Revenue101Collaboration revenue recognized duringRevenues20212023 and $1.0 million for the year ended December 31, 2022. The licensing revenues in 2023 were due to the achievement of $8.0a development milestone pursuant to the Merck Neuromuscular License Agreement and the licensing revenues in 2022 were due to research and development services from the Genentech KCP506 License Agreement, which was terminated in December 2022. Under the Merck Neuromuscular License Agreement, we are eligible to receive up to an additional $255.0 million was relatedin development milestones, sales milestones and royalties on net sales. We do not expect to our earn any revenue from this license in 2024 or 2025.Merck. The upfront paymentthe Merger. Upon completion of $15.0 million receivedthe Merger, we had $442,000 in July 2020 was initially recorded as deferred revenue and is being recognized as revenue under the cost-to-cost method as research and development is being performed.Merck Neuromuscular License Agreement. As of December 31, 2021, substantially all of2023, we have completed the initial $15.0 million upfront payment has been recognized as revenue.project services and had zero in deferred revenue under the Merck Neuromuscular License Agreement. We do not expect to recognizeearn any revenue from this remaining deferred revenue of $.1 million and amounts deferred atlicense in 2024 or 2025.2021 totaling $5 million, as revenue2023 and $912,000 for the year ended December 31, 2022. The grant revenues in 2022 as research and developmentwere due to services are provided.provided under a grant that was concluded in December 2022. We do not expect to earn any grant revenues in 2024.$26.4$9.0 million for the year ended December 31, 2021, an increase of $4.1 million from $22.32023 and $15.9 million for the year ended December 31, 2020. Direct2022 and decreased by $6.9 million, or 43%. The decrease in direct external program expenses of our YTX-7739 program increased by $2.8$2.1 million in the year ended December 31, 2021, compared to the year ended December 31, 2020. The change was due primarily to an increase in clinical and consultant costs as YTX-7739 progressed from a SAD study in 2020 to MAD clinical studies starting in the first quarter of 2021. Direct expenses of our YTX-9184 program in 2021 increased by less than $0.1 million primarily due to preclinical and manufacturing83costs. Platformlower activities for KVA12123 as we began securing clinical trial sites in advance of enrolling the first patient in a Phase 1/2 clinical trial of KVA12123, which occurred in April 2023. We also incurred lower costs for CD27, KCP-506 and other early stageprograms as we prioritized efforts on KVA12123, our lead product candidate. These decreases in direct external program expenses were partially offset by ALS program expenses incurred to complete project services under the Merck Neuromuscular License Agreement. Subject to receiving adequate funding, we expect our direct external program expenses to increase during the next few years as we advance our clinical trials of KVA12123. The decrease in our internal and unallocated research external costs increased by $0.6and development expenses of $4.8 million was primarily due to the resumption of a more normal level of spend after the second quarter of 2020 saw decreased laboratory activitieslower personnel costs as a result of COVID-19reducing research and the movedevelopment staff in December 2022 as we transitioned to new office and laboratory space. Personnel related costs increased by $0.5 million primarily due to the impact of a $0.5 million R&D tax credit that was recorded as a reduction of personnel expense in the third quarter of 2020.clinical trials.$20.4$12.1 million for the year ended December 31, 2021, an increase of $8.5 million from $11.92023 and $8.7 million for the year ended December 31, 2020.2022 and increased by $3.4 million, or 40%. The increase of $2.9 million in personnel related costs was primarily due to an increase in personnel costs of $2.3 million in stock/equity-based compensation and $0.7 millioninsurance and other administrative expenses of $1.1 million. Personnel costs increased due to additional hiring in the generalhigher salaries and administrative function. Personnel-related costs for eachbenefits of the years ended December 31, 2021$1.2 million from increased headcount to support public company responsibilities and 2020 included stock/equity-basedhigher stock-based compensation of $3.9$1.0 million, which increased due to options granted during 2023. Insurance and $1.6 million, respectively. Professional and consultant feesother administrative expenses increased by $1.2 million primarily due to higher audit expensespublic company directors and legal and Board fees related to operating as a public company. Facility and other related costs increased by $4.4 million primarily due to incremental public companyofficers insurance premiums of $2.1 million$601,000 and $1.4 millionboard fees of lease expense in excess of sublease income.$270,000.In-Process Research and Development Assets AcquiredIn connection with the Merger, we recognized a charge of $28.3 million of acquired in-processIn-process research and development expensesfor assets with no alternative usewas $18.9 million for the year ended December 31, 2020.Other Income (Expense)Other income (expense) net increased by $1.1 million resulting primarily from a $1.1 million gain on2022 in connection with the extinguishmentMerger. As the acquired in-process research and development assets were deemed to have no current or alternative future use, the entire amount was recognized as expense in the consolidated statements of debt upon forgiveness of the PPP loan (see Paycheck Protection Loan section of the Description of Indebtedness below). This loan was obtained in April 2020, prior to entering into the Merger Agreement with Proteostasis in August 2020.Interest income and other income (expense) remained relatively flat fromoperations for the year ended December 31, 2021 to2022.2020.2023 and $9,000 for the year ended December 31, 2022 and increased by $316,000. Interest income increased due to higher balances in interest-bearing accounts in 2023 and higher interest rates.Liquiditywethrough December 31, 2023, our operations have not generated revenuebeen financed primarily by net cash proceeds from product salesthe sale and issuance of our common stock and borrowings under notes payable. We have incurred significant operating losses and negative cash flowsalso received upfront payments from our operations.license agreements. As of December 31, 2023, we had $5.8 million in cash and an accumulated deficit of $165.8 million. We have fundedexpect that our research and development and general and administrative expenses will increase, and, as a result, anticipate that we will continue to incur increasing losses for the foreseeable future. Therefore, we will need to raise additional capital to fund our operations, to date primarily with proceeds from saleswhich may be through the issuance of preferred units and an upfront payment from our collaboration agreement with Merck received in July 2020. additional equity or through borrowings.2020,2022, we completedreceived net cash of $7.8 million upon the close of the Merger with Proteostasis and acquired its $35.9net proceeds of $7.4 million from the Private Placement. The second closing of cash, cash equivalents and restricted cash. Immediately following the Merger, we also completed a private placement ofPrivate Placement for an aggregate purchase price of 1,460,861$22.5 million was expected to occur on April 15, 2024. However, in February 2024, certain investors indicated they will not be able to consummate the second closing of the Private Placement.approximately $31.6$8.0 million. We have also funded operations using borrowings under loan and security agreements.believehowever, have not generated any revenue from product sales, and do not know when, or if, we will generate any revenue from product sales. We do not expect to generate any revenue from product sales unless and until we obtain regulatory approval of and commercialize any of our product candidates. At the same time, we expect our expenses to increase in connection with our ongoing development activities, particularly as we continue the research, development and clinical trials of, and seeks regulatory approval for, our product candidates. In addition, subject to obtaining regulatory approval of any of our product candidates, we anticipate that we will need substantial additional funding in connection with our continuing operations. We plan to continue to fund our operations and capital requirements through equity and/or debt financing, but there are no assurances that we will be able to raise sufficient amounts of funding in the future on acceptable terms, or at all. and marketable securities will not enable us to fund our operating expenses and capital expenditure requirementsexpenditures for a period of twelveat least the next 12 months from the filing date of issuance of the consolidated financial statements included in this Annual Report on Form 10-K.conditions give rise to afactors raise substantial doubt overabout our ability to continue as a going concern.84sources and uses of cash flows for each of the periods presented:indicated:Net Cash Used in Operating ActivitiesDuring the year ended December 31, 2021, net cashCash used in operating activities was $48.9 million, resulting from our net loss of $39.5 million, as well as by net cash used by changes in our operating assets and liabilities of $20.0 million and partially offset by non-cash charges of $10.6 million, which includes $5.1 million of non-cash lease expense and $5.3 million of stock/equity-based compensation expense. Net cash provided by changes in our operating assets and liabilities for the year ended December 31, 2021 consisted2023 was $16.2 million, consisting of decreasesa net loss of $8.5$14.1 million, a change in other net operating assets and liabilities of $5.3 million, partially offset by noncash charges of $3.1 million. Our change in net operating assets and liabilities primarily resulted from a $4.3 million decrease in accounts payable and accrued expenses and other current liabilities, $5.0 million in accounts receivable, $4.5 millionas we paid down Merger-related costs owed as of December 31, 2022, a $843,000 decrease in operating lease liabilities,liability and $2.9 milliona $442,000 decrease in deferred revenue due to completion of research and development services provided by us related to the Merck Neuromuscular License Agreement with Merck to support research, development and commercialization of products for treatment of neuromuscular diseases, partially offset by a $1.2 million increase$338,000 decrease in prepaid expensesexpenses. The noncash charges primarily consisted of $3.9 million in stock-based compensation and other current assets.$739,000 noncash operating lease expense, partially offset by a $1.6 million gain on change in fair value of rights from the Private Placement.During the year ended December 31, 2020, net cash was $17.9 million, resulting from our net loss of $57.5 million partially, offset by net cash provided by changes in our operating assets and liabilities of $5.2 million and non-cash charges of $34.3 million, including the non-cash charge of $28.3 million for in-process research and development acquired as well as $2.5 million of non-cash lease expense and $2.3 million of stock/equity-based compensation expense. Net cash provided by changes in our operating assets and liabilities for the year ended December 31, 2020 consisted2022 was $19.0 million, consisting of an $8.1a net loss of $63.5 million, increasepartially offset by $18.9 million in-process research and development expenses acquired from the Merger, a change in deferred revenueother net operating assets and liabilities of $0.3 million and noncash charges of $25.8 million. Our change in net operating assets and liabilities primarily resulted from a $0.6$1.7 million increase in accounts payable and accrued expenses and other current liabilities mainly due to increased costs associated with our KVA12123 program and the Merger as well as the timing of payments, partially offset by a $1.7 million$108,000 increase in prepaid expenses, a $737,000 decrease in operating lease liabilitiesliability and a $1.5$1.0 million increasedecrease in prepaid expenses and other current assets.Changes in accounts payable, accrued expenses and prepaid expenses in all periods were generallydeferred revenue mainly due to growth in our businesscompletion of research and the timing of vendor invoicing and payments.Net Cash Provided by Investing ActivitiesDuring the year ended December 31, 2021, net cashdevelopment services provided by investing activities was $3.1 million, primarilyus related to $3.1the Phase 1 clinical trial under the Genentech KCP506 License Agreement. The noncash charges primarily consisted of $15.3 million in change in fair value measurement of notes payable, $5.2 million in stock-based compensation, $3.3 million in warrant expense for net purchases of marketable securities.During the year ended December 31, 2020, net cash provided by investing activities was $31.0warrants issued to current debt holders that converted their debt to equity in 2022, $1.6 million primarilyexpense related to $35.9 million of cashconvertible notes payable that were converted to equity in connection with the Merger and restricted cash acquired from our merger with Proteostasis,$661,000 noncash operating lease expense, partially offset by a $341,000 gain on debt extinguishment driven by our settlement of notes payable accounted for under the net cash used of $3.1 million for net purchases of marketable securities and $1.5 million of transaction costs paid associated with the Merger.fair value election.Net Cash Provided by Financing105Net cash used in financingCash provided by investing activities for the year ended December 31, 20212023 was $1.0$331,000 from proceeds from sales of property and equipment,payments of debt principal of $2.3 million and finance lease obligations of $0.3 million, offset by $1.4 million of net proceeds from our ATM offering.cash acquired in connection with the Merger.Net cashFinancing Activities20202023 was $55.5$8.5 million, consisting primarily ofrelated to $8.0 million in net proceeds from the sale of common stock of $33.6 million, netPrivate Placement and $600,000 in proceeds from the issuance of Class C preferred units of $21.2 million and proceeds from a government loan (Paycheck Protection Program (“PPP”) loan) of $1.1 million,our common stock, partially offset by payments$123,000 in repayments of finance lease obligations of $0.3 million.obligations.Description of IndebtednessLoan and Security AgreementWe entered into a loan and security agreement with Hercules Capital, Inc. (the “Lender”) in December 2019 (the “Term Loan”), pursuant to which had $12.7 million in outstanding principal borrowings as of December 31, 2021. Another $5.0 million became available upon the occurrence of a developmental milestone and an equity event defined in the agreement, but we elected not to draw it. An additional $10.0 million could have become available to be drawn upon lender approval. On February 25, 2022, we repaid to85Hercules a payoff amount of $12.8 million and terminated the Term Loan,Cash provided that we continue to be bound by certain indemnification obligations under Section 6.3 of the Term Loan. The payoff amount included payment of approximately $0.9 million consisting of the final and additional final payment fees outlined above, as well as an interest/non-use fee of less than $0.1 million.Borrowings under the Term Loan were repayable in monthly interest-only payments until August 1, 2021. The interest-only period was followed by monthly payments of equal principal plus interest until the loan maturity date of January 1, 2024. Outstanding borrowings bore interest at the greater of (i) 8.75% and (ii) the prime rate as reported in the Wall Street Journal plus 4.00%. A final payment fee of 5.25% of the amounts drawn under the Term Loan was due upon the earlier of the maturity date or the repayment date if paid early, whether voluntary or upon acceleration due to default. We were permitted to repay the Term Loan at any time by paying the outstanding principal balance in full, along with any unpaid accrued interest, the final payment fees of 5.25% of the amounts drawn and a prepayment fee calculated on amounts being prepaid. The prepayment fee was 3.0% if the Term Loan is repaid within the one-year anniversary of the draw date, 2.0% if paid between the first and second-year anniversary of the draw date and 1.0% if paid after the second anniversary of the draw date but before the maturity date.In April 2020, the Term Loan was amended to permit indebtedness consisting of a loan under the PPP of the Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”), provided that such loan shall be unsecured, shall not contain any terms or conditions that are adverse to the lender’s rights under the loan and that we will not prepay such loan. In June 2020, the Term Loan was amended and an additional final payment fee of $0.3 million became due upon repayment of the loan.On December 22, 2020, we entered into an Unconditional Secured Guaranty and Pledge Agreement (the “Guaranty”) with the Lender as a condition to the Lender’s consent to the Merger under the Term Loan between us as borrower and the Lender. Immediately prior to the Merger, we entered into a Fourth Amendment and Consent to Loan and Security Agreement dated as of December 22, 2020 with the Lender (the “Loan Amendment”). The Guaranty providedfinancing activities for our guaranty of our obligations under the Loan Agreement and provided the Lender a security interest in all of our assets other than intellectual property as collateral. The Loan Amendment provided for the Lender’s consent to the Merger and to the creation and funding of a Silicon Valley Bank Paycheck Protection Program escrow account to hold funds in connection with our outstanding Paycheck Protection Program loan amounts for which we have submitted a forgiveness application. The Loan Amendment also amended the definition of “Change in Control” to include the situations in which we no longer control Yumanity, Inc., our wholly-owned subsidiary. The remaining terms and conditions of the Loan Agreement generally continued in the form existing prior to the Loan Amendment.On March 29, 2021, the Term Loan was amended again to allow for the creation of a new foreign subsidiary, as well as changing certain covenants related to the financial operations of said subsidiary. The subsidiary was formed on April 23, 2021.On April 13, 2021, the Term Loan was amended to reduce the additional final payment fee from $0.3 million to $0.1 million and to extend the availability of Tranche 2 from March 31, 2021 to June 30, 2021.Borrowings under the Term Loan were collateralized by substantially all of our personal property, other than our intellectual property. There were no financial covenants associated with the Term Loan; however, we are subject to certain affirmative and negative covenants restricting our activities, including limitations on dispositions, mergers or acquisitions; encumbering our intellectual property; incurring indebtedness or liens; paying dividends; making certain investments; and engaging in certain other business transactions. The obligations under the Term Loan were subject to acceleration upon the occurrence of specified events of default, including a material adverse change to our business, operations or financial or other condition. As of December 31, 2021,the Company has assessed that the risk of subjective acceleration under the material adverse events clause is not probable and therefore have classified the outstanding principal on the consolidated balance sheet based on the contractually scheduled principal payments. Upon the occurrence of an event of default and until such event of default is no longer continuing, the annual interest rate would have been 5.0% above the otherwise applicable rate.Paycheck Protection Program LoanIn April 2020, prior to entering into the Merger Agreement with Proteostasis in August 2020, we issued a Promissory Note to Silicon Valley Bank, pursuant to which we received loan proceeds of $1.1 million (the “PPP Loan”), provided under the PPP established under the CARES Act and guaranteed by the U.S. Small Business Administration. The PPP Loan was unsecured, was scheduled to mature on April 24, 2022, and had a fixed interest rate of 1.0% per annum. Equal monthly payments of principal and interest were to begin commencing in August 2021 until the maturity date. Interest would have accrued on the unpaid principal balance from the inception date of the loan. Forgiveness of the PPP Loan was only available for principal that is used for the limited purposes that expressly qualify for forgiveness under U.S. Small Business Administration requirements. On April 3, 2021, we were notified by Silicon Valley Bank that our forgiveness application was accepted by the Small Business Association as of March 30,862021. Accordingly, we have recognized $1.1 million in income for debt extinguishment in our consolidated statement of operations during the year ended December 31, 2021.Funding requirementsWe expect2022 was $11.8 million, primarily related to continue to incur significant expenses$7.4 million in connection with our ongoing activities, particularly as we explore our strategic alternatives and operate as a public company.Our future funding requirements, both near- and long-term, will depend on many factors, including, but not limited to:•professional services and other fees associated with exploring our strategic alternatives;•initiation, progress, timing, costs and results of clinical trials for our product candidates;•the outcome, timing and cost of the regulatory approval process for our product candidates by the FDA;•the cost of filing, prosecuting, defending and enforcing our patent claims and other intellectual property rights;•the cost of defending intellectual property disputes, including patent infringement actions brought by third parties against us;•the costs of operating as a public company; and•the extent to which we in-license or acquire other products, product candidates or technologies.After our recent restructuring, we believe that the existing cash and cash equivalents will not be sufficient to fund our operations for a period of twelve monthsPrivate Placement, $6.7 million in proceeds from the issuance of notes payable and $1.6 million in proceeds from the issuance of our common stock, partially offset by $4.0 million in repayments of notes payable.statements. Substantialstatements included in this Annual Report for additional financing will be neededinformation regarding our notes payable.fundother contractual obligations and commitments related to license agreements.operations thereafterachievement of certain development, regulatory and to commercially develop any current or future product candidates. We currently do not have any commitments to obtain additional fundscommercial milestones royalties, and may be unable to obtain sufficient fundingsublicensing revenue in the future, on acceptable terms, if at all. However,as applicable. As of December 31, 2023, the timing and likelihood of achieving the milestones and generating future product sales, and therefore payments that may become payable to these third parties, are uncertain.management is currently evaluating strategic alternatives includingcorporate headquarters in Seattle, Washington under a lease agreement that expires in July 2024. As of December 31, 2023, undiscounted future minimum lease payments of $549,000 remain pursuant to the lease agreement.acquisition, merger, reverse merger,engagement letter with an investment bank whereby we may be obligated to pay certain fees for financings and investment related transaction costs. business combination, sales of assets, licensing or other strategic transactions involving the Company. There can be no assurance that these future funding efforts will be successful. If we cannot obtain the necessary funding, we will need to eliminate research and development programs; consider other various strategic alternatives, including a mergerservices. Such agreements generally provide for termination upon notice, although obligate us to reimburse vendors for any time or sale; or initiate steps to cease operations. If we cannot raise adequate financing, our business,costs incurred through the date of termination.could be materially adversely affected.Critical Accounting Policies and EstimatesOuris based on our consolidated financial statements, arewhich have been prepared in accordance with U.S. GAAP. The preparation of ourthese consolidated financial statements and related disclosures requires us to make judgmentsestimates and estimatesjudgments that affect the reported amounts of assets, liabilities costs and expenses, and the disclosure of contingent assets and liabilities in our financial statements. We base ourrelated disclosures. Our estimates are based on historical experience known trends and events andon various other factorsassumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the recording of expenses that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actualActual results may differ materially from these estimates under different assumptions or conditions.estimates.While our accounting policies are described in more detail in Note 2 to our consolidated financial statements, weWe believe that the following accounting policies requirediscussed below are critical to understanding our historical and future performance, as these policies relate to the mostmore significant areas involving management’s judgments and estimates used in the preparation of our financial statements.estimates.accountenter into license agreements under which we license certain intellectual property rights to our product candidates to third parties. The terms ofone collaboration arrangement, entered into in June 2020,obligations under Accounting Standards Codification Topic 606, Revenue From Contracts With Customers (“ASC 606”). Under ASC 606, an entity recognizes revenue when its customer obtains controleach of promised goods or services in an amount that reflects the consideration that the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements within the scope of ASC 606, the entity performsour agreements, we perform the following five steps: (i) identifyidentification of the contract(s) with a customer;customer, (ii) identifydetermination of whether the promised goods or services are performance obligations, including whether they are distinct in the contract;context of the contract, (iii) determinemeasurement of the transaction price, including the constraint on any variable consideration, if any; (iv) allocateallocation of the transaction price to the performance obligations in the contract;contract, and (v) recognizerecognition of revenue when (or as) the entity satisfies awe satisfy each performance obligation. We only apply the five-step model to contracts when it is probable that we will collect the consideration to which it is entitled in exchange for the goods or services it transfers to the customer.We assess the goods or services promised within each contract and determines those that are performance obligations. Arrangements that include rights to additional goods or services that are exercisable at a customer’s discretion are generally considered options. We assess if these options provide a material right to the customer and if so, they are considered performance87obligations. The identification of material rights requires judgments related to the determinationAs part of the value ofaccounting for these arrangements containing multiple performance obligations, we must develop assumptions that require judgment to determine the underlying license relative to the option exercisestand-alone selling price including assumptions about technical feasibility and the probability of developing a candidate that would be subject to the option rights. The exercise of a material right is accounted for as a contract modification for accounting purposes.We assess whether each promised good or service is distinct for the purpose of identifying the performance obligationsobligation identified in the contract. This assessment involves subjective determinations and requires management to make judgments about the individual promised goods or services and whether such are separable from the other aspects of the contractual relationship. Promised goods and services are considered distinct provided that: (i) the customer can benefit from the good or service either on its own or together with other resources that are readily available to the customer (that is, the good or service is capable of being distinct) and (ii) the entity’s promise to transfer the good or service to the customer is separately identifiable from other promises in the contract (that is, the promise to transfer the good or service is distinct within the context of the contract). In assessing whether a promised good or service is distinct, we consider factors such as the research, manufacturing and commercialization capabilities of the collaboration partner and the availability of the associated expertise in the general marketplace. We also consider the intended benefit of the contract in assessing whether a promised good or service is separately identifiable from other promises in the contract. If a promised good or service is not distinct, an entity is required to combine that good or service with other promised goods or services until it identifies a bundle of goods or services that is distinct.The transaction price is then determined and allocated to the identified performance obligations in proportion to their standalone selling prices (“SSP”) on a relative SSP basis. SSP is determined at contract inception and is not updated to reflect changes between contract inception and when the performance obligations are satisfied. Determining the SSP for performance obligations requires significant judgment. In developing the SSP for a performance obligation, we consider applicable market conditions and relevant entity-specific factors, including factors that were contemplated in negotiating the agreement with the customer and estimated costs. We validate the SSP for performance obligations by evaluating whether changes in theuse key assumptions used to determine the SSP will have a significant effect on the allocationstand-alone selling price, which may include forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of arrangement consideration between multiple performance obligations.If the consideration promised in a contract includes a variable amount, we estimate the amount of considerationtechnical and regulatory success. We expect to which we will be entitled in exchangerecognize revenue for transferring the promised goods or services to a customer. We determine the amount of variable consideration by using the expected value method or the most likely amount method. We include the unconstrained amount of estimated variable consideration in the transaction price. The amount included in the transaction price isbeing constrained to the amount for whichwhen it is probable that a significant reversal of cumulative revenue recognizedreversal will not occur. AtFor performance obligations satisfied over time, we estimate the end of each subsequent reporting period, we re-evaluateefforts needed to complete the estimated variable consideration included inperformance obligation and recognize revenue by measuring the transaction price and any related constraint, and if necessary, adjusts our estimateprogress towards complete satisfaction of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis in the period of adjustment.performance obligation using an input measure.If an arrangement includesFor arrangements that include development and regulatory milestone payments,milestones, we evaluate whether the milestones are considered probable of being reached and estimatesestimate the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within our control or the licensee’s control, such as regulatory approvals, are generally not considered probable of being achieved until those approvals are received.For arrangements with licenses of intellectual property that include sales-based royalties, including milestone payments based on the level of sales, and the license is deemedOur management may be required to exercise considerable judgment in estimating revenue to be the predominant item to which the royalties relate, we recognize royalty revenue and sales-based milestones at the later of (i) when the related sales occur, or (ii) when therecognized. Judgment is required in identifying performance obligation to which the royalty has been allocated has been satisfied.We record amounts as accounts receivable when the right to consideration is deemed unconditional. When consideration is received, or such consideration is unconditionally due, from a customer prior to transferring goods or services to the customer under the terms of a contract, a contract liability is recorded for deferred revenue.In determiningobligations, estimating the transaction price, we adjust consideration forestimating the effectsstandalone selling price of identified performance obligations, and estimating the time valueprogress towards satisfaction of money if the timing of payments provides us with a significant benefit of financing. We do not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the licensees and the transfer of the promised goods or services to the licensees will be one year or less. We then recognize as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) each performance obligation is satisfied, either at a point in time or over time, and if over time, recognition is based on the use of an output or input method.obligations.We assessed the promised goods and services within the Collaboration Agreement with Merck to determine if they are distinct. Based on this assessment, we determined that Merck cannot benefit from the promised goods and services separately from the others as they are highly interrelated and therefore not distinct. Accordingly, the promised goods and services related to the initial upfront88payment represent one combined performance obligation and the entire transaction price was allocated to that single combined performance obligation. That performance obligation is being satisfied over the research term as we perform the research and development activities through the first substantive option period and participate in a Joint Steering Committee to oversee research and development activities. Collaboration Revenuesrecordedrecognized as a percentage of actual cost incurred to the estimated transaction pricecosts to complete.extentestimated amount of work completed and in accordance with agreements established with these third parties, according to the progress towards completion. At contract inception,of preclinical studies, clinical trials or related activities, and discussions with applicable personnel and service providers as to the potential milestone payments that we are eligible to receive were excluded fromprogress or state of consummation of goods and services.transaction priceaccrued balance as they were fully constrained. At the end of each reporting period,period. As actual costs become known, we reevaluate the transaction price and as uncertain events are resolved or other changes in circumstances occur, and if necessary, we will adjust our estimate of the transaction price. Any additions to the transaction price would be reflected in the period as a cumulative revenue catch-up. At the inception of the arrangement, we evaluated the options held by Merck to either advance or terminate the applicable research program to determine if they provided Merck with any material rights. We concluded that the options were not issued at a significant and incremental discount, and therefore do not provide Merck with a material right. As such, these options were excluded as performance obligations and will be accounted for if and when they occur. Upon Merck electing to advance the research program into the second phase (the "Second Phase"), we assessed whether the promised goods and services for the Second Phase are distinct. Basedaccrued estimates based on the facts and circumstances we determined that the Second Phase represents a separate contract with its own performance obligation.We assessed the Collaboration Agreement to determine whether a significant financing component exists and concluded that a significant financing component does not exist.Research and DevelopmentAs part of the process of preparing our financial statements, we are required to estimate our accrued research and development expenses. This process involves reviewing open contracts and purchase orders, communicating with our applicable personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual costs. The majority of our service providers invoice us in arrears for services performed, on a pre-determined schedule or when contractual milestones are met; however, some require advance payments. We make estimates of our accrued expenses as of each balance sheet date in the financial statements based on facts and circumstances known to us at that time. We periodically confirm the accuracy of these estimates with the service providers and makes adjustments, if necessary. Examples of estimated accrued research and development expenses include fees paid to:•vendors in connection with clinical and preclinical development activities;•CROs and investigative sites in connection with clinical trials; and•CDMOs in connection with the production of preclinical and clinical trial materials.We base the expense recorded related to external research and development on our estimates of the services received and efforts expended pursuant to quotes and contracts with multiple CDMOs, CROs and other vendors that supply, conduct and manage preclinical studies and clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the expense. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from the estimate, we adjust the accrual or the amount of prepaid expenses accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed, relative toincluding the actual status and timinglevel of services performedpatient enrollment, may vary from our estimates and maycould result in us reporting amounts that are too highoverestimated or too lowunderestimated in any particular period. Our accrued research and development expenses are dependent, in part, upon the receipt of timely and accurate reporting from our third-party service providers. To date, there have been no material differences from our accrued expenses to our actual expenses.Stock/Equity-Based Compensationmeasure stock/equity-baseddetermined that the rights from Private Placement is a derivative asset, which requires the asset to be accounted for at fair value. Until settlement, the rights from Private Placement are remeasured at fair value at each reporting period with the changes in fair value recorded in other income (expense) in the Statement of Operations. The fair value was determined using a Monte Carlo simulation based on the contractual funding date at the measurement date, minimum contractual purchase price of $3.18 and historical stock prices. We make significant judgments and estimates in determining the significant unobservable inputs used in the fair value measurement for volatility, risk-free interest rates and funding probability.ofusing the grant and recognizeBlack-Scholes option pricing model. The related stock-based compensation is recognized as expense on a straight line-basis over the employee’s, non-employee’s or director’s requisite service period for employees and directors and as services are delivered for non-employees, both of which are generally(generally the vesting period of the respective award. We have issued stock/equity-basedperiod). Noncash stock-based compensation is based on awards with only service-basedultimately expected to vest and performance-based vesting conditions. We record the expenseis reduced by an estimate for awards with only service-based vesting conditions using the straight-line method. We record the expense for awards with both service-based and performance-based vesting conditions using the graded vesting method, commencing once achievement of the performance condition becomes probable. Prior to the Yumanity Reorganization, Holdings had granted restricted incentive units, which were accounted forfuture forfeitures. Forfeitures are recorded as equity-classified awards. Holdings determinedincurred.restricted unit awards usingstock options, we use the Black-Scholes option-pricing model and assumptions discussed below. Each of these inputs is subjective and generally requires significant judgment to determine.itsthe common units less any applicable purchase price.89The fair value of each option grant was estimated onstock at the datetime of grant usingof the Black-Scholes option-pricing model, which used as assumption inputs:option, the board of directors considers, among other things, valuations performed by an independent third-party. Because there has been no public market for our common stock (prior to the closing of the Merger), the board of directors exercises reasonable judgment and considers a number of objective and subjective factors to determine the best estimate of the fair value of our common stock/units, calculationstock, including important developments in our operations, sales of common stock, actual operating results and financial performance, the conditions in the life sciences industry and the economy in general, the stock price performance and volatility of comparable public companies, and the lack of liquidity of our common stock/unitsstock, among other factors.historical benchmarkingthe simplified method (based on the mid-point between the vesting date and the end of the contractual term) for employee options. peer companies, the expected term of the options,stock option grants. The comparable companies were chosen based on their similar industry, size, or stage in the product development life cycle and financial leverage.a period that approximatesperiods corresponding with the expected term of option.our expected dividend yield.RSUs, which we expect to recognize over a weighted-average period of 1.8 years.Recently Issued and Adopted Accounting Pronouncements108A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 2 to our consolidated financial statements included in this Annual Report on Form 10-K.90ITEMItem 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISKQuantitative and Qualitative Disclosures About Market Risk.Securities Exchange Act, of 1934, as amended, for this reporting period and are not required to provide the information required under this item.91ITEM109 AND SUPPLEMENTARY DATAYUMANITY THERAPEUTICS, INC.Index to Consolidated Financial Statements92Yumanity Therapeutics,Kineta, Inc.Yumanity Therapeutics,Kineta, Inc. and its subsidiaries (the “Company”) as of December 31, 20212023 and 2020, and2022, the related consolidated statements of operations, of comprehensive loss, of preferred units and stockholders’ equity (deficit) and of cash flows for each of the two years thenin the period ended includingDecember 31, 2023, and the related notes (collectively referred to as the “consolidated financial“financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 20212023 and 2020,2022, and the results of its operations and its cash flows for each of the two years thenin the period ended December 31, 2023, in conformity with accounting principles generally accepted in the United States of America.Substantial Doubt about the Company’s Ability to Continue as adiscussedmore fully described in Note 1, to the consolidated financial statements, the Company has a significant working capital deficiency, has incurred recurringsignificant losses and negative cash flows from operations since inception thatneeds to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt about itsthe Company's ability to continue as a going concern. Management’sManagement's plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty. consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB)(“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB. of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditaudits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company'sCompany’s internal control over financial reporting. Accordingly, we express no such opinion. consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.MattersMatterThe criticalmatter communicated below is a mattermatters are matters arising from the current period audit of the consolidated financial statements that waswere communicated or required to be communicated to the audit committee and that (i) relatesthat: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (ii)(2) involved our especially challenging, subjective, or complex judgments. The communication ofWe determined that there are no critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.matters.Accrued external research and development expenses/s/ Marcum LLPAs described in Notes 2 and 7 to the consolidated financial statements, the Company has entered into various research, development, and manufacturing contracts with research institutions and other companies. When billing terms under these contracts do not coincide with the timing of when the work is performed, management is required to estimate the amount of outstanding obligations to those third parties as of period end. Within accrued expenses and other current liabilities, total accrued external research and development expenses amounted to $1.6 million as of December 31, 2021. The accrual estimate is based on a number of factors, including management’s knowledge of the progress towards completion of the research, development, and manufacturing activities, invoicing to date under the contracts, communication from the research institutions and other companies of any actual costs incurred during the period that have not yet been invoiced, and the costs included in the contracts. Significant judgments and estimates may be made in determining the accrued balances at the end of any reporting period.93The principal considerations for our determination that performing procedures relating to accrued external research and development expenses is a critical audit matter are (i) the significant judgment by management in developing the estimate of actual costs incurred during the period that have not yet been invoiced and (ii) a high degree of auditor judgment, subjectivity, and effort in performing procedures to evaluate management’s estimate of the actual costs incurred during the period that have not yet been invoiced and in evaluating the audit evidence obtained for estimating the accrued external research and development expenses.Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included, among others (i) testing management’s process for estimating the accrued external research and development expenses; (ii) evaluating the appropriateness of the method used by management to develop the estimate; (iii) testing the completeness and accuracy of the data used to develop the estimate related to invoicing to date under the contracts and contractual rates for services received; and (iv) evaluating the reasonableness of the estimated actual costs incurred during the period that have not yet been invoiced. Evaluating the reasonableness of management’s estimate of the costs incurred involved performing a comparison of the estimated and actual costs incurred to cost information and contracted fees supporting documentation on a test basis./s/ PricewaterhouseCoopersMarcum LLPBoston, MassachusettsNew York, New York24, 202221, 20242018.2022.94YUMANITY THERAPEUTICS,111CONSOLIDATED BALANCE SHEETSConsolidated Balance Sheets
extinguishment of notes payable and accrued interest
connection with Private Placement, net of transaction costs
exercise of warrants
cashless exercise of stock options
exercise of warrantsIn thousands, except share amounts)in thousands)TheSee the accompanying notes are an integral part of theseto the consolidated financial statements.95YUMANITY THERAPEUTICS, INC.115CONSOLIDATED STATEMENTS OF OPERATIONS(In thousands, except share/unit and per share/unit amounts)Description of BusinessThe accompanying notes are an integral part of these consolidated financial statements.96YUMANITY THERAPEUTICS, INC.CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS(In thousands)The accompanying notes are an integral part of these consolidated financial statements.97YUMANITY THERAPEUTICS, INC.CONSOLIDATED STATEMENTS OF PREFERRED UNITS AND STOCKHOLDERS’ EQUITY (DEFICIT)(In thousands, except share/unit amounts)
Class B
Preferred Units
Paid-in
Other
Comprehensive
Stockholders’
units, net of issuance costs of
$388
units for Defaulting Class B
�� preferred units
Class B preferred units
units
expense
Yumanity Holdings, LLC for
shares of common stock of
Yumanity Therapeutics, Inc.,
adjusted to reflect
the Exchange Ratio
Yumanity Holdings, LLC for
shares of common stock of
Yumanity Therapeutics, Inc.,
adjusted to reflect the
Exchange Ratio
connection with the Merger
liability to permanent equity
stock, net of issuance costs
of $1,996
at the market offering, net of
issuance costs of $44
options
awards
expense98The accompanying notes are an integral part of these consolidated financial statements.99YUMANITY THERAPEUTICS, INC.CONSOLIDATED STATEMENTS OF CASH FLOWS(In thousands)
and restricted cash acquiredThe accompanying notes are an integral part of these consolidated financial statements.100YUMANITY THERAPEUTICS, INC.NOTES TO CONSOLIDATED FINANCIAL STATEMENTS1. Nature of Business and Basis of Presentationwholly owned subsidiaries, the “Company” or “Yumanity”) is a clinical stage biopharmaceutical company engagedheadquartered in the research and development of treatments for neurodegenerative diseases caused by protein misfolding.Seattle, Washington.subject to risks similar to those of other earlya clinical stage companiesbiotechnology company with a mission to develop next-generation immunotherapies that transform patients’ lives. Kineta has leveraged its expertise in innate immunity and is focused on discovering and developing potentially differentiated immunotherapies that address the mechanisms of cancer immune resistance. Kineta Chronic Pain, LLC (“KCP”) was formed to develop new innovative therapies for pain management. Kineta Viral Hemorrhagic Fever, LLC (“KVHF”) was formed to develop a direct acting anti-viral therapy for the treatment of emerging diseases.biopharmaceutical industry, including dependence on key individuals,trial will be permitted to continue to participate. The Company made this decision, in part, because certain investors have indicated they will not be able to fulfill their contractual obligation to consummate the needPrivate Placement (as defined below). In February 2024, the Company initiated a process to develop commercially viable products, competition from other companies, manyexplore a range of whom are larger and better capitalized, the impactstrategic alternatives to maximize shareholder value. Potential strategic alternatives that may be evaluated include sale of assets of the ongoing COVID-19 pandemicCompany, a sale of the Company, licensing of assets, a merger, liquidation or other strategic action. There is no set timetable for this process and the need to obtain adequate additional financing to fund the development of its product candidates. Therethere can be no assurance that this process will result in the Company’s research and development will be successfully completed, that adequate protection for the Company’s intellectual property will be maintained, that any product candidates developed will obtain required regulatory approvalCompany pursuing a transaction or that any approved productstransaction, if pursued, will be commercially viable. Even ifcompleted on attractive terms. Additionally, there can be no assurances that any particular course of action, business arrangement or transaction, or series of transactions, will be pursued, successfully consummated, or lead to increased stockholder value. If the Company’s development efforts are successful, itstrategic process is uncertain when, if ever,unsuccessful, our board of directors may decide to pursue a liquidation or obtain relief under the Company will generate significant revenue from the sale of its products.US Bankruptcy Code.Exploration of Strategic Alternatives and RestructuringIn February 2022, the Company announced that it is exploring strategic alternatives to enhance shareholder value and engaged H.C. Wainwright as its exclusive financial advisor to assist in this process. No timetable has been established for the completion of this process, and the Company does not expect to disclose developments unless and until the Board of Directors has concluded that disclosure is appropriate or required.In February the Company also began implementation of a strategic restructuring with the objective of preserving capital. As part of the restructuring, it is eliminating approximately 60% of its workforce and has taken other actions, including reducing its office and laboratory space, to reduce expenditures (see Note 19).Clinical and Regulatory UpdateIn January 2022, the U.S Food and Drug Administration (FDA) placed a partial clinical hold on the Company's multidose clinical trials of YTX-7739. The partial clinical hold suspends initiation of multiple dose clinical trials in the U.S. until the FDA’s questions have been addressed. The FDA has not halted all clinical programming and is permitting the Company's planned single dose formulation clinical trial to proceed. The Company anticipates working closely with the FDA to try to adequately address their concerns. While the Company works to address the FDA’s concerns, it has paused its planned clinical study of YTX-7739 in glioblastoma multiforme patients and the exploration of additional indications.Merger with Proteostasis Therapeutics, Inc.22, 2020, Proteostasis16, 2022, Yumanity Therapeutics, Inc. (“Proteostasis” or “PTI”Yumanity”) completed its previously announced merger transaction with Yumanity,Kineta Operating, Inc. (formerly Yumanity Therapeutics,Kineta, Inc.) (“Private Kineta”) in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated as of August 22, 2020,June 5, 2022, as amended on November 6, 2020December 5, 2022 (the “Merger Agreement”), by and among Pangolinpursuant to which Yacht Merger Sub, Inc., a wholly-owned subsidiary of ProteostasisYumanity (“Merger Sub”), Yumanity Holdings, LLC (“Holdings”) and Yumanity, Inc., pursuant to which Merger Sub merged with and into Yumanity, Inc.,Private Kineta, with Yumanity, Inc.Private Kineta surviving such merger as a wholly ownedwholly-owned subsidiary of ProteostasisYumanity (the “Merger”). Immediately prior to the effective time ofThe surviving corporation from the Merger Holdingssubsequently merged with and into Yumanity, Inc. and Yumanity, Inc. continued to exist asKineta Operating, LLC, with Kineta Operating, LLC being the surviving corporation. On December 22, 2020,16, 2022, in connection with, and prior to the completion of the Merger, ProteostasisYumanity effected a 1-for-201-for-7 reverse stock split of its common stock (the “Reverse Stock Split”). Immediately following the Merger, ProteostasisYumanity changed its name to “Yumanity Therapeutics,“Kineta, Inc.” and the business conducted by Private Kineta became the primary business conducted by the Company.(the “Effective Time”), each outstanding share of Yumanity Inc.’sPrivate Kineta common stock par value $0.01 (the “Yumanity Common Stock”), outstanding immediately prior to the Effective Time was converted into the right to receive shares of PTI based on an exchange ratio set forth in the Merger Agreement. At the Effective Time following the Reverse Stock Split, the exchange ratio was determined to be 0.21080.0688 shares of PTI Common Stock for each share of Yumanity Common Stock (the “Exchange Ratio”). At the closing of the Merger on December 22, 2020, PTI issued an aggregate of 6,024,433 shares of its common stock to Yumanity, based on the Exchange Ratio. In addition, all options and warrants exercisable for shares of common stock of Yumanity, Inc. became options and warrants exercisable for shares of common stock of PTI equalthe Company (after giving effect to the Exchange Ratio multiplied byReverse Stock Split). In addition, the numberCompany also assumed all of shares of Yumanity Inc.’s common stock previously represented by suchPrivate Kineta’s outstanding stock options, warrants, and warrants, as applicable, with a proportionate adjustment in exercise price. No fractional shares were issued in connection withrestricted stock at the Exchange Ratio.101transaction wasMerger has been accounted for as a reverse merger and as an asset acquisition in accordance with GAAP. Under this method of accounting, Yumanity was deemed to be the accounting acquirer for financial reporting purposes. This determination was primarily based on the fact that, immediately following the Merger: (i) Yumanity’s equityholders own a majority of the voting rights in the combined organization, (ii) Yumanity designated a majority of the members (7 of 9) of the initial board of directors of the combined organization and (iii) Yumanity’s senior management hold all key positions in the senior management of the combined organization. Accordingly, for accounting purposes, (i) the Merger was treated as the equivalent of the Yumanity issuing stock to acquire the net assets of PTI, (ii) the net assets of PTI were allocated a portion of the transaction price and recorded based upon their relative fair values in the financial statements at the time of closing, (iii) the reported historical operating results of the combined organization prior to the Merger will be those of Yumanity and (iv) for periods prior to the transaction, shareholders’ equity of the combined organization is presented based on the historical equity structure of Yumanity. As a result, as of the closing date of the Merger, the net assets of PTI were recorded at their acquisition-date fair values in the financial statements of Yumanity and the reported operating results prior to the Merger will be those of Yumanity. As used herein, the words “the Company” refer to, for periods following the Merger, Yumanity Therapeutics, Inc., together with its wholly owned subsidiaries, and for periods prior to the Merger, Holdings, and its wholly owned subsidiary, as applicable.(see Note 3).The Yumanity ReorganizationOn December 22, 2020, immediately prior toIn connection and concurrently with the closing of the Merger, pursuant to the termsexecution of the Merger Agreement, on June 5, 2022, the Company completedentered into a financing agreement, as amended on October 24, 2022, December 5, 2022, March 29, 2023, May 1, 2023, July 21, 2023 and October 13, 2023 (such financing agreement, as amended, the Yumanity Reorganization whereby Holdings, the sole stockholder and holding company parent of Yumanity, Inc.“Securities Purchase Agreement”), merged with and into Yumanity, Inc., with Yumanity, Inc. as the surviving corporation. In connection with the Yumanity Reorganization, each outstanding common unit of Holdings was exchanged forto sell shares of the Company’s common stock of Yumanity, Inc. based uponin a ratio associated with the terms of each common unit, each outstanding preferred unit of Holdings was converted into shares of common stock of Yumanity, Inc. based upon the ratio associated with each individual series of preferred units, each outstanding option to purchase shares of common units of Holdings was converted into an outstanding option to purchase shares of common stock of Yumanity, Inc. on a 1-for-1 basis, with a corresponding adjustment to the exercise price, and each outstanding warrant to purchase preferred units or common units of Holdings was converted into a warrant to purchase shares of common stock of Yumanity, Inc. based upon the ratio associated with each individual series of preferred units or on a 1-for-1 basis, respectively, with a corresponding adjustment to the exercise price, as applicable.Basis of presentationThe Company’s consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“GAAP”). Any reference in these notes to applicable guidance is meant to refer to the authoritative GAAP as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Update (“ASU”) of the Financial Accounting Standards Board (“FASB”private placement (the “Private Placement”). The accompanying consolidated financial statements include the accountsfirst closing of the Private Placement occurred on December 16, 2022, and the Company issued 649,346 shares of its common stock and its wholly owned subsidiaries. All intercompany accountsreceived net proceeds of $7.4 million. The second closing of the Private Placement for an aggregate purchase price of $22.5 million was expected to occur on April 15, 2024 (see Notes 9 and transactions have been eliminated16). However, in consolidation. Unless otherwise noted, all referencesFebruary 2024, certain investors indicated they will not be able to common stock/unit share and per share amounts have also been adjusted to reflectconsummate the Exchange Ratio.second closing of the Private Placement.concernConcern and Capital Resourcesevaluated whether there are certain conditions and events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern within one year after the issuance date of the consolidated financial statements.Since its inception, the Company has funded its operations primarily with equity and debt including proceeds from the Merger. The Company has incurred recurring net losses and negative cash flows from operations since inception including net losses of $39.5 million and, $57.5 million for the years ended December 31, 2021 and 2020, respectively. In addition, as of December 31, 2021, the Company2023, had an accumulated deficit of $187.3165.8 million. The net loss attributable to the Company expects to continue to generate operating losses for the foreseeable future.was $14.1As of the issuance date of the consolidated financial statements million for the year ended December 31, 2021,2023. As of December 31, 2023, the Company expects thathad unrestricted cash of $5.8 million, and there is substantial doubt about its ability to continue as a going concern. Based on Kineta’s current operating plans, Kineta does not have sufficient cash and cash equivalents and marketable securities will not be sufficient to fund its operating expenses and capital expenditure requirementsexpenditures for a period of twelveat least the next 12 months from the issuancefiling date of this Annual Report on Form 10-K.consolidated financial statements.The Company, is currently evaluating strategic alternatives includingan acquisition, merger, reverse merger, other business combination, salesa sale of the Company, licensing of assets, licensinga merger, liquidation or other strategic transactions involving the Company. There is no assurance that the Company will be successful in executing such transaction or obtaining sufficient funding on terms acceptable to the Company to fund continuing operations, if at all. If the Company is unable to obtain additional funding or enter into strategic alternatives, the Company will be forced to further delay, reduce or eliminate its research and development programs or initiate steps to cease operations.102the Company'sKineta’s ability to continue as a going concern. The accompanying financial statements do not include any adjustments related to the recoverability and classification of assets or the amounts and classification of liabilities or any other adjustments that might result from the outcome of this uncertainty.ImpactThe COVID-19 pandemic, which began in December 2019 and has spread worldwide, has caused many governments to implement measures to slow on its business, the spreadextent of the outbreak. The outbreak and government measures taken in response have had a significant impact both direct and indirect, on businesses and commerce, as worker shortages have occurred, supply chains have been disrupted, and facilities and production have been suspended. The future progression of the pandemic and its effects on the Company’s business and operations are uncertain. The COVID-19 pandemic is ongoing and may affect the Company’s ability to initiate and complete preclinical studies, delay its clinical trial or future clinical trials, disrupt regulatory activities, or have other adverse effects on its business, operations and operations. The pandemic has already caused significant disruptions inclinical development timelines and plans will depend on future developments, including the financial markets,severity and may continue to cause such disruptions, which could impact the Company’s ability to raise additional funds to supportduration of any resurgence of COVID-19 and its operations. Moreover, the pandemic has significantly impacted economies worldwide and could result in adverse effects on the Company’s business and operations.YTX-7739KCP-506 Phase 1b1 clinical trial incurred delays due to COVID-19 that resulted in a delay in the results from that study. There continues to be a risk of additional delays toclinical programs.ability to raise additional funds to support its operations.extentimpact of these developments, including any resulting sanctions, export controls or other restrictive actions that may be imposed against governmental or other entities in, for example, Russia, have in the past contributed and may in the future contribute to disruption, instability and volatility in the global markets, which the COVID-19 pandemic will directly or indirectlyin turn could adversely impact the Company’s business, results of operations and weaken the Company’s financial condition, including currentresults. Certain political developments may also lead to uncertainty to regulations and future clinical trials and research and development costs, will depend on future developments that are highly uncertain, including as a result of new informationrules that may emerge concerning COVID-19,materially affect the actions taken to contain or treat it, and the duration and intensity of the related effects.At-the-Market Offering ProgramIn April 2021, the Company entered into a sales agreement (the "Prior Sales Agreement") with Jefferies LLC (“Jefferies”) with respect to an at-the-market (“ATM”) offering program under which it issued and sold, from time-to-time at its sole discretion, shares of its common stock, in an aggregate offering amount of up to $60.0 million. In December 2021, the Company terminated the Prior Sales Agreement and entered into a new sales agreement with Jefferies with respect to an ATM offering under which it may issue and sell, from time-to-time and at its sole discretion, shares of its common stock, in an aggregate offering amount of up to $60.0 million (the “New Sales Agreement”). Jefferies acts as the Company's sales agent and will use commercially reasonable efforts to sell shares of common stock from time-to-time, based upon instruction by the Company.The Company will pay Jefferies up to 3% of the gross proceeds from any common stock sold through the New Sales Agreement. The Company sold 112,833 shares of common stock under the Prior Sales Agreement during the twelve months ended December 31, 2021 for gross proceeds of $1.5 million for aggregate net proceeds to the Company of approximately $1.4 million, after deducting sales commissions. As of December 31, 2021, $60.0 million of common stock remained available for future issuance under the New Sales Agreement, although these amounts may be limited as the Company will be subject to the general instructions of Form S-3 known as the "baby shelf rules." Under these instructions, the amount of funds the Company can raise through primary public offerings of securities in any 12-month period using its registration statement on Form S-3 is limited to one-third of the aggregate market value of the shares of its common stock held by non-affiliates of the Company. Therefore, the Company will be limited in the amount of proceeds it is able to raise by selling shares of its common stock using its Form S-3 until such time as its public float exceeds $75 million.Private PlacementCompany’s business.
On December 14, 2020, the Company entered into a subscription agreement with certain accredited investors for the sale by it in a private placement of 1,460,861 shares of its common stock for a price of $23.00 per share. The Company refers to this sale herein as the Private Placement. The Private Placement closed on December 22, 2020. The aggregate gross proceeds for the issuance and sale of the common stock were $33.6 million and, after deducting certain of its expenses, the net proceeds it received in the Private Placement were $31.6 million.103estimatesEstimatesexpensesrevenue and expense during the reporting periods. SignificantThe Company bases its estimates and judgments on historical experience and on various other assumptions that the Company believes are reasonable under the circumstances. These estimates are basedreflected in these consolidated financial statements include,are used for, but are not limited to, revenue recognition, the accrual ofaccrued research and development expenses, the valuationfair value of notes payable, the fair value of the Company’s common unitsstock prior to the Merger, stock-based compensation, uncertain tax positions and the valuation of stock/unit-based awards. The Company bases its estimates on historical experience, known trends and other market-specific or other relevant factors that it believes to be reasonable under the circumstances. On an ongoing basis, management evaluates its estimates as there are changes in circumstances, facts, and experience.allowance for net deferred tax assets. Actual results may differ from those estimatesthe Company’s estimates.assumptions.decision-making group, in making decisions on how to allocate resources and assess performance. The Company’s Chief Executive Officer and President collectively serve as the CODM. The Company views its operations and manages its business in one operating segment.Segment informationRisks and Uncertaintiesmanages its operationsis subject to certain risks and uncertainties associated with companies at a similar stage of development, including, but not limited to: successfully develop, manufacture, and market any approved therapies and products, obtain regulatory approval from the U.S. Food and Drug Administration or foreign regulatory agencies prior to commercial sales, new technological innovations, dependence on key personnel, protection of intellectual property, compliance with governmental regulations, uncertainty of market acceptance of any approved therapies and products, competition from companies with greater financial and technical resources, and the need to obtain additional financing.single segment for the purposes of assessing performance and making operating decisions. All of the Company’s tangible assets are heldnoncurrent asset in the United States.consolidated balance sheets.credit risk and of significant suppliersCredit Riskand cash equivalents and marketable securities. At times the Companydeposited in accounts at several financial institutions that may maintain cash and investment balances in excess ofexceed federally insured limits. The Company does not believe thatis exposed to credit risk in the event of a default by the financial institutions holding its cash to the extent recorded in the consolidated balance sheets. The Company believes it is subjectnot exposed to unusual credit risk beyond the normal credit risk associated with commercial banking relationships.relationships and has not incurred any such losses to date.The Company relies, and expects to continue to rely, on a small number of vendors to provide services, supplies and materials related to its discovery programs. These programs could be adversely affected by a significant interruption in these services or the availability of materials.Deferred financing costsThe Company capitalizes certain legal and other third-party fees that are directly associated with obtaining access to capital under credit facilities. Deferred financing costs incurred in connection with obtaining access to capital are recorded in prepaid expenses and other current assets and are amortized over the term of the credit facility. Deferred financing costs related to a recognized debt liability are recorded as a reduction of the carrying amount of the debt liability and amortized to interest expense using the effective interest method over the repayment term.Cash equivalentsThe Company considers all highly liquid investments with original maturities of three months or less at the date of purchase to be cash equivalents.Restricted cashAmounts included in restricted cash represent amounts pledged as collateral for Company credit cards as part of the terms of the “Term Loan” (see note 8) and for its office and laboratory space lease. These amounts are classified as restricted cash (non-current) in the Company’s consolidated balance sheet. In December 2020, in connection with the Merger, the Company acquired Proteostasis’ restricted cash pledged as collateral for its office and laboratory space lease and amended its loan and security agreement to establish an escrow account in the amount of its Paycheck Protection Program loan. After forgiveness of the PPP Loan, in April 2021 the escrowed cash was released and reclassified into cash and cash equivalents. The cash pledged as collateral is classified as restricted cash (non-current) in the Company’s consolidated balance sheet as of December 31, 2021. As of December 31, 2021 and 2020, the cash and restricted cash of $36.0 million and $82.9 million, respectively, presented in the consolidated statements of cash flows included cash and cash equivalents of $35.1 million and $80.8 million, respectively, and restricted cash of $0.9 million and $2.1 million, respectively.Property and equipmentProperty and equipment are stated at cost less accumulated depreciation and amortization. Depreciation and amortization expense is recognized using the straight-line method over the estimated useful life of each asset as follows:104Estimated Useful LifeLaboratory equipment2 - 3 yearsOffice equipment, computers and software2 - 5 yearsFurniture and fixtures2 - 7 yearsLeasehold improvementsShorter of remaining term of lease or useful lifeCosts for capital assets not yet placed into service are capitalized as construction-in-progress and depreciated once placed into service. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation and amortization are removed from the accounts and any resulting gain or loss is included in loss from operations. Expenditures for repairs and maintenance are charged to expense as incurred.Assets held-for-saleThe Company classifies assets as held-for-sale when the following conditions are met: (1) management has committed to a plan to sell, (2) the assets are available for immediate sale in their present condition, (3) the Company has initiated an active program to identify a buyer, (4) it is probable that a sale will occur within one year, (5) the assets are actively marketed for sale at a reasonable price in relation to their current fair value, and (6) there is a low likelihood of significant changes to the plan or that the plan will be withdrawn. If all of the criteria are met as of the balance sheet date, the assets are presented separately in the consolidated balance sheet as held-for-sale at the lower of the carrying amount or fair value less costs to sell. The assets are then no longer depreciated or amortized while classified as held-for-sale.Impairment of long-lived assetsThe Company evaluates its long-lived assets, which consist primarily of property and equipment and right-of-use assets, for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the future undiscounted net cash flows expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the asset exceeds the fair value of the asset. The Company did 0t record any impairment losses on long-lived assets during the years ended December 31, 2021 or 2020.AcquisitionsAcquisitions of assets or a group of assets that do not meet the definition of a business are accounted for as asset acquisitions using the cost accumulation method, whereby the cost of the acquisition, including certain transaction costs, is allocated to the assets acquired on the basis of relative fair values. No goodwill is recognized in an asset acquisition. Intangible assets that are acquired in an asset acquisition for use in research and development activities which have an alternative future use are capitalized as in-process research and development (“IPR&D”). Acquired IPR&D which has no alternative future use is recognized as research and development expense at acquisition. Contingent milestone payments associated with asset acquisitions are recognized when probable and estimable. These amounts are expensed to research and development if there is no alternative future use associated with the asset, or capitalized as an intangible asset if alternative future use of the asset exists.value measurementsValue of Financial InstrumentsCertain assets and liabilities are carried at fair value under GAAP. Fair value is defined as the exchange price that would be received forto sell an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants onat the measurement date. Valuation techniques used to measureThe Company measures fair value must maximizeby maximizing the use of observable inputs, where available, and minimizeminimizing the use of unobservable inputs.inputs when measuring fair value. Financial assets and liabilities carriedrecorded at fair value in the consolidated balance sheets are to be classified and disclosedcategorized in one of the following three levels of the fair value hierarchy based upon the lowest level of whichinput that is significant to the first two are considered observable and the last is considered unobservable:fair value as follows:••1 quoted prices)1), such as quoted prices in active markets for identical or similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets orof liabilities in markets, or other inputs that are observable or can be corroborated by observable market data.•liabilities, including pricingliabilities.discounted cash flow methodologies and similar techniques.The Company’s cash equivalents and marketable securitiesor inputs that are carried atless observable or unobservable in the market, the determination of fair value determined accordingrequires more judgment. Accordingly, the degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3. A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value hierarchy described above (see Note 4). The carrying values of the Company’s accounts payable and accrued expenses approximateinstrument.105fair values dueestimated useful lives. Leasehold improvements are amortized using the straight-line method over the lesser of the estimated useful lives of the assets or the remaining term of the lease. Upon sale or retirement of the assets, the cost and related accumulated depreciation are removed from the consolidated balance sheets and the resulting gain or loss is recognized in the consolidated statements of operations. Expenditures for maintenance and repairs are expensed as incurred.short-term natureuse of these liabilities.the asset and its eventual disposition are less than its carrying amount. The impairment charge is determined based upon the excess of the carrying value of the Company’s long-term debt underasset over its loan and security agreement approximates itsestimated fair value, duewith estimated fair value determined based upon an estimate of discounted future cash flows or other appropriate measures of estimated fair value. Estimating discounted cash flows requires the Company to its variable interest rate.make significant judgments and assumptions. Actual results may vary from the Company’s estimates as of the date of impairment testing and adjustments may occur in future periods. For the years ended December 31, 2023 and 2022, there were no impairments of long-lived assets.Marketable securitiesFair Value Optionmarketable securities, which consistcommon stock in connection with the execution of certain equity and debt securities, are classified as available-for-salefinancings and are carried atother agreements. The fair value. Realized gainsvalue of warrants is determined using the Black- Scholes option-pricing model using assumptions regarding volatility of the Company’s common share price, remaining life of the warrant, and losses are reported in other income (expense), net,risk-free interest rates. The Company classifies warrants indexed to its own equity and meeting the criteria for equity classification within the consolidated statements of operations and comprehensive loss on a specific identification basis.stockholders’ equity.The Company conducts periodic reviews to identify and evaluate each investment inCompany’s portfolio that has an unrealized lossdefinition of a business are accounted for as asset acquisitions, with a cost accumulation model used to determine whether a credit loss exists. An unrealized loss exists when the currentcost of the acquisition. Common stock issued as consideration in an acquisition of assets is generally measured based on the acquisition date fair value of an individual security is less than its amortized cost basis. A credit loss is estimated by considering available information relevant to the collectabilityequity interests issued. Direct transaction costs are recognized as part of the security and information about past events, current conditions, and reasonable and supportable forecasts. Any credit loss is recorded as a charge to other income (expense), net, not to exceed the amountcost of the unrealized loss. Unrealized losses other than the credit lossan acquisition of assets. Intangible assets that are recognized in accumulated other comprehensive income (loss). When determining whether a credit loss exists, the Company considers several factors, including whether the Company has the intent to sell the security or whether it is more likely than not that the Company will be required to sell the security prior to recovery of its amortized cost basis. If the Company has an intent to sell, or if it is more likely than not that the Company will be required to sell a debt securityacquired in an unrealized loss position before recoveryasset acquisition for use in research and development activities that have an alternative future use are capitalized as in-process research and development ("IPR&D"). Acquired IPR&D that has no alternative future use is expensed immediately in the consolidated statements of its amortized cost basis, the Company will write down the security to its fair value and record the corresponding charge as a component of other income (expense), net. No declines in value were deemed to be credit losses or other than temporary during the year ended December 31, 2021.operations.recognitionRecognitionaccounts for its onerecognizes collaboration arrangement, entered into in June 2020, under ASC Topic 606, Revenue From Contracts With Customers (ASC 606). For additional informationrevenue using the cost-to-cost method, which it believes best depicts the transfer of control to the customer. Under the cost-to-cost method, the extent of progress towards completion is measured based on the Company’s collaboration agreement, see Note 6, Collaboration Agreement,ratio of actual costs incurred to the total estimated costs expected upon satisfying the identified performance obligation. Under this method, revenue is recognized as a percentage of actual cost incurred to the estimated costs to complete.consolidated financial statements. Under ASC 606, an entity recognizesarrangements typically include payment to the Company of one or more of the following: nonrefundable upfront fees, payment for research and development services provided by the Company under approved work plans, development, regulatory and commercial milestone payments, and sales-based milestones and royalties on net sales of licensed products. Each of these payments results in license revenues, except for revenues from royalties, which are classified as other revenues.whento be recognized as the Company fulfills its customer obtains controlobligations under each of promised goods or services in an amount that reflectsits agreements, the consideration that the entity expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements within the scope of ASC 606, the entityCompany performs the following five steps: (i) identifyidentification of the contract(s) with a customer;customer, (ii) identifydetermination of whether the promised goods or services are performance obligations, including whether they are distinct in the contract;context of the contract, (iii) determinemeasurement of the transaction price, including the constraint on any variable consideration, if any; (iv) allocateallocation of the transaction price to the performance obligations in the contract;contract, and (v) recognizerecognition of revenue when (or as) the entityCompany satisfies aeach performance obligation. The Company only applies the five-step model to contracts when it is probable that the entity will collect the consideration to which it is entitled in exchange for the goods or services it transfers to the customer.The Company assesses the goods or services promised within each contract and determines those that are performance obligations. Arrangements that include rights to additional goods or services that are exercisable at a customer’s discretion are generally considered options. The Company assesses if these options provide a material right to the customer and if so, they are considered performance obligations. The identification of material rights requires judgments related to the determinationAs part of the value ofaccounting for arrangements containing multiple performance obligations, the underlying license relativeCompany develops assumptions that require judgment to determine the option exercisestand-alone selling price including assumptions about technical feasibility and the probability of developing a candidate that would be subject to the option rights. The exercise of a material right is accounted for as a contract modification for accounting purposes.The Company assesses whether each promised good or service is distinct for the purpose of identifying the performance obligationsobligation identified in the contract. This assessment involves subjective determinations and requires management to make judgments about the individual promised goods or services and whether such are separable from the other aspects of the contractual relationship. Promised goods and services are considered distinct provided that: (i) the customer can benefit from the good or service either on its own or together with other resources that are readily available to the customer (that is, the good or service is capable of being distinct) and (ii) the entity’s promise to transfer the good or service to the customer is separately identifiable from other promises in the contract (that is, the promise to transfer the good or service is distinct within the context of the contract). In assessing whether a promised good or service is distinct, the Company considers factors such as the research, manufacturing and commercialization capabilities of the collaboration partner and the availability of the associated expertise in the general marketplace. The Company also considers the intended benefit of the contract in assessing whether a promised good or service is separately identifiable from other promises in the contract. If a promised good or service is not distinct, an entity is required to combine that good or service with other promised goods or services until it identifies a bundle of goods or services that is distinct.The transaction price is then determined and allocated to the identified performance obligations in proportion to their standalone selling prices (“SSP”) on a relative SSP basis. SSP is determined at contract inception and is not updated to reflect changes between contract inception and when the performance obligations are satisfied. Determining the SSP for performance obligations requires significant judgment. In developing the SSP for a performance obligation, the Company considers applicable market conditions and106relevant entity- specific factors, including factors that were contemplated in negotiating the agreement with the customer and estimated costs. The Company validates the SSP for performance obligations by evaluating whether changes in theuses key assumptions used to determine the SSP will have a significant effect on the allocationstand-alone selling price, which may include forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of arrangement consideration between multiple performance obligations.If the consideration promised in a contract includes a variable amount, the Company estimates the amount of consideration to which it will be entitled in exchange for transferring the promised goods or services to a customer.technical and regulatory success. The Company determines the amount ofexpects to recognize revenue for variable consideration by using the expected value method or the most likely amount method. The Company includes the unconstrained amount of estimated variable consideration in the transaction price. The amount included in the transaction price isbeing constrained to the amount for whichwhen it is probable that a significant reversal of cumulative revenue recognizedreversal will not occur. At the end of each subsequent reporting period,For performance obligations satisfied over time, the Company re-evaluatesestimates the estimated variable consideration included inefforts needed to complete the transaction priceperformance obligation and any related constraint, and if necessary, adjusts its estimaterecognizes revenue by measuring the progress towards complete satisfaction of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis in the period of adjustment.performance obligation using an input measure.If an arrangement includesFor arrangements that include development and regulatory milestone payments,milestones, the Company evaluates whether the milestones are considered probable of being reached and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. Milestone payments that are not within the Company’s control or the licensee’s control, such as regulatory approvals, are generally not considered probable of being achieved until those approvals are received. with licenses of intellectual property that include sales-based royalties, including commercial milestone payments based on thepre-specified level of sales, and the license is deemed to be the predominant item to which the royalties relate, the Company recognizes royalty revenue and sales-based milestones at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied. Achievement of these royalties and commercial milestones may solely depend upon the performance of the licensee.TheUpfront payments are recorded as deferred revenue upon receipt or when due and may require deferral of revenue recognition to a future period until the Company records amountsperforms its obligations under these arrangements. Amounts are recorded as accounts receivable when the Company’s right to consideration is deemed unconditional. When consideration is received, or such consideration is unconditionally due, from a customer prior to transferring goods or services to the customer under the terms of a contract, a contract liability is recorded for deferred revenue.In determining the transaction price, the Company adjusts consideration for the effects of the time value of money if the timing of payments provides the Company with a significant benefit of financing. The Company does not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the licenseescustomer and the transfer of the promised goods or services to the licenseescustomer will be one year or less. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) each performance obligation is satisfied, either at a point in time or over time, and if over time, recognition is based on the use of an output or input method.Classification and accretion of preferred unitsThe Company’s preferred units were classified outside of stockholders' equity(deficit) on120consolidated balance sheets becauseagreement should be accounted for as an exchange transaction or a contribution. An agreement is accounted for as a contribution if the holders of such units had redemption rightsresource provider does not receive commensurate value in return for the event of a deemed liquidation that, in certain situations, were not solely within the control of the Company. The occurrence of a deemed liquidation event was not determined to be probable in any period prior to the Merger, therefore the carrying values of the preferred units were not being accreted to their redemption values.assets transferred. Contributions are recognized as grant revenue when all donor-imposed conditions have been met.Costs for incurred in connection with the discovery, research, preclinical and clinical development, and manufacture of our product candidates. Research and development costs are expensed as incurred and consist of salaries, benefits, and other personnel related costs, including stock-based compensation, fees paid to other entities to conduct certain research and development activities are expensed inon the period in which they are incurred. ResearchCompany’s behalf, materials for preclinical studies, clinical studies and development expenses consist of costs incurred in performing research and development activities, including salaries and bonuses, stock/equity-based compensation, employee benefits, facilities costs, laboratory supplies, depreciationlicensing agreements and amortization, manufacturing expenses, and externalassociated costs of vendors engaged to conduct research and preclinical development activities and clinical trials as well as the cost of licensing technology.Upfront payments under license agreements are expensed upon receipt of the license,allocated facility and annual maintenance fees under license agreements are expensed in the period in which they are incurred. Milestone payments under license agreements are accrued, with a corresponding expense being recognized, in the period in which the milestone is determined to be probable of achievementallocated expenses for rent, insurance and theother related amount is reasonably estimable.Non-refundablecosts. Nonrefundable advance payments for goods or services tothat will be received in theused or rendered for future for use in research and development activities are recordeddeferred and capitalized as prepaid expenses. The prepaid amounts are expensed asexpenses until the related goods are delivered or the services are performed.107development, and manufacturing contract costs and accrualsDevelopment Expenseshas entered into variousrecords accrued expenses for estimated costs of its research and development andactivities conducted by third-party service providers, such as contract research organizations, contract manufacturing contracts with research institutions and other companies. These agreements are generally cancelable,vendors, which include the conduct of preclinical studies, clinical trials and relatedcontract manufacturing activities. The Company records the estimated costs are recorded asof research and development activities based upon the estimated amount of services provided but not yet invoiced and includes these costs in accrued expenses and other current liabilities in the consolidated balance sheets and within research and development expenses as incurred.in the consolidated statements of operations. The Company records accrualsaccrued expenses for estimated ongoing research and development costs. When billing terms under these contracts do not coincide withcosts based on the timing of when the work is performed, management is required to estimate theestimated amount of outstanding obligations to thosework completed and in accordance with agreements established with these third parties, as of period end. Any accrual estimates are based on a number of factors, including the Company’s knowledge ofaccording to the progress towards completion of preclinical studies, clinical trials or related activities, and discussions with applicable personnel and service providers as to the research, development,progress or state of consummation of goods and manufacturing activities, invoicing to date under the contracts, communication from the research institutions and other companies of any actual costs incurred during the period that have not yet been invoiced, and the costs included in the contracts. Significantservices. may be made in determining the accrued balances at the endbalance as of anyeach reporting period. Actual results could differAs actual costs become known, the Company adjusts its accrued estimates based on the facts and circumstances known at that time. The Company’s accrued research and development expenses are dependent, in part, upon the receipt of timely and accurate reporting from its third-party service providers. To date, there have been no material differences from the estimates made by the Company. The historical accrual estimates made by the Company have not been materially different from theCompany’s accrued expenses to its actual costs.expenses.Patent costsAll patent-relatedincurrednot otherwise included in connection with filingresearch and prosecutingdevelopment expenses. Legal costs include general corporate legal fees and patent applicationscosts. General and administrative expenses are expensed as incurred due to the uncertainty about the recovery of the expenditure. Amounts incurred are classified as general and administrative expenses..Stock/equity-based compensationStock-Based Compensation with service-based vesting or performance-based vesting granted to employees, non-employees and directors based on the estimated grant-date fair value of the awardawards and recognizes the related expense on the date of grant. Compensation expense for the awards is recognizeda straight-line basis over the requisite service period for employees and directors and as services are delivered for non-employees, both of which are generally(generally the vesting period of the respective award.period). The Company uses the straight-line methodBlack-Scholes option-pricing model to recordestimate the expensefair value of awardsits stock options. The fair value of restricted stock units (“RSUs”) is estimated based on the fair value of the Company’s common stock at the grant date. For RSUs with only service-based vesting conditions. The Company uses the graded-vesting method to record the expense of awards with both service-based and performance-basedperformance vesting conditions, commencing once achievementthe Company evaluates the probability of achieving the performance condition becomes probable.at each reporting date and recognizes expense for such performance awards over the requisite service period using the accelerated attribution method. Forfeitures are recorded as incurred.accountsgrants stock-based awards using the current share price of its common stock on the date of the award.forfeituresone year, the expected volatility is estimated based on the average volatility for a group of stock/equity-based awards as they occur.comparable publicly traded biotechnology companies over a period equal to the expected term of the stockclassifies stock/equity-based compensation expensedetermined that the rights from Private Placement is a derivative asset, which requires the asset to be accounted for at fair value. Until settlement, the rights from Private Placement are remeasured at fair value at each reporting period with the changes in its consolidated statements of operationsfair value recorded in other income (expense) in the same mannerStatement of Operations.whichFair Value Measurement of Notes Payableaward recipient’s payroll costsremeasurement of the notes payable that the Company elected to account for under the fair value option. Until settlement, these notes payable are classified orremeasured at fair value at each reporting period with the changes in whichfair value recorded in other income (expense) in the award recipient’s service payments are classified.Statement of Operations.taxesTaxesPrior to the Yumanity Reorganization, Holdings was organized as a Limited Liability Company and subject to the provisions of Subchapter K of the Internal Revenue Code. As such, Holdings was not viewed as a taxpaying entity in any jurisdiction and did not require a provision for income taxes. Each member was responsible for the tax liability, if any, related to its proportionate share of the member’s taxable income. The Company’s wholly owned corporate subsidiary was a taxpaying entity. After the Yumanity Reorganization, the Company and its subsidiary are both taxpaying entities.The Company accountsIncome taxes are accounted for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the consolidated financial statements or in the Company’s tax returns.method. Deferred tax assets and liabilities are determined onrecognized for the basis of thefuture tax consequences attributable to differences between the financial statements and tax basis ofstatement carrying amounts or existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using the enacted tax rates expected to apply to taxable income in effect for the yearyears in which thethose temporary differences are expected to reverse. Changes inbe recovered or settled. The effect on deferred tax assets and liabilities are recordedof a change in tax rates is recognized in income in the provision for income taxes.period of enactment. The Company assesses the likelihood that itsrecords a valuation allowance to reduce deferred tax assets willto an amount expected to be recoveredrealized.future taxable income and, to the extent it believes, based upon the weight of available evidence, thatan uncertain tax position if it is more likely than not that all or a portion of the deferred tax assets will not be realized, a valuation allowance is established through a charge to income tax expense. Potential for recovery of deferred tax assets is evaluated by estimating the future taxable profits expected and considering prudent and feasible tax planning strategies.The Company accounts for uncertainty in income taxes recognized in the consolidated financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. Iftax authorities, based on the tax positionmerits of the position. The Company’s policy is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize ininterest and penalties related to the consolidated financial statements. The amountunderpayment of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The provision for income taxes includesas a component of income tax expense or benefit. To date, there have been no interest or penalties charged in relation to the effects of any resulting tax reserves, or unrecognized tax benefits, that are considered appropriate as well as the related net interest and penalties.benefits.108Comprehensive lossNet Loss Per ShareComprehensive loss is comprised of net loss and other comprehensive loss. The Company’s only elements of other comprehensive loss are unrealized gains (losses) on marketable securities.Net loss per shareincome (loss)loss per common share is computedcalculated by dividing net loss attributable to the net income (loss)Company by the weighted averageweighted-average number of shares of common stock outstanding for the period. Diluted net income (loss) per common share is computed by dividing net income (loss) by the weighted average number of common shares outstanding forduring the period, including potential dilutive common shares assuming the dilutive effect of outstandingwithout consideration for common stock equivalents. For periods in which the Company reported aDiluted net loss diluted net loss per common share is the same as basic net loss per common share, since the effects of potentially dilutive securities are antidilutive given the net loss for each period presented. In computing basic net loss per share, nominal issuances of common sharesstock, including warrants to purchase the Company’s common stock with exercise prices of $0.001 and $0.14 per share, are not assumed to have been issuedreflected in basic net loss per share for all periods, even if their affect is anti-dilutive.antidilutive.LeasesComprehensive Lossaccordance with ASC 842,December 2023, the Company determines at the inception of a contract if such arrangement is or contains a lease. A contract is or contains a lease if the contract conveys the rightFinancial Accounting Standards Board issued Accounting Standards Update (“ASU”) No. 2023-09, Improvements to control the use of an identified asset for a period of time in exchange for consideration. The Company classifies leases at the lease commencement date as operating or finance leases and records a right-of-use asset and a lease liability on the consolidated balance sheet for all leases with an initial lease term of greater than 12 months. Leases with an initial term of 12 months or less are not recorded on the balance sheet, but payments are recognized as expense on a straight-line basis over the lease term.Income Tax Disclosures.The Company often enters into contracts that contain both lease and non-lease components. Non-lease components may include maintenance, utilities, and other operating costs. Subsequent to the Company’s adoption of ASC 842 as of January 1, 2019, the Company combines the lease and non-lease components of fixed costs in its lease arrangements as a single lease component. Variable costs, such as utilities or maintenance costs, are not included in the measurement of right-of-use assets and lease liabilities, but rather are expensed when the event determining the amount of variable consideration to be paid occurs.Lease liabilities and their corresponding right-of-use assets are recorded based on the present value of future lease payments over the expected lease term. The present value of future lease payments is determined by using the interest rate implicit in the lease if that rate is readily determinable; otherwise, the Company uses its estimated secured incremental borrowing rate for that lease term. The Company estimates its secured incremental borrowing rate for each lease based on the rate of interest that the Company would have to pay to borrow an amount equal to the lease payments on a collateralized basis over a similar term.Certain of the Company’s leases include options to extend or terminate the lease. The amounts determined for the Company’s right-of-use assets and lease liabilities generally do not assume that renewal options or early-termination provisions, if any, are exercised, unless it is reasonably certain that the Company will exercise such options.Recently adopted accounting pronouncementsIn June 2016, the FASB issued ASU No. 2016-13, Financial Instruments — Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments (“ASU 2016-13”), which requires the measurement and recognition of expected credit losses for financial assets held at amortized cost. ASU 2016-13 replaces the existing incurred loss impairment model with an expected loss model. It also eliminates the concept of other- than-temporary impairment and requires credit losses This new guidance enhances income tax disclosures related to available-for-sale debt securitieseffective tax rates and cash income taxes paid and aims to be recorded through an allowance for credit losses rather than as a reduction in the amortized cost basis of the securities. These changes may result in earlier recognition of credit losses. In November 2018, the FASB issuedenhance transparency and provide investors with better insights into income tax matters. ASU No. 2018-19, Codification Improvements to Topic 326, Financial Instruments — Credit Losses, which narrowed the scope and changed the effective date for non-public entities for ASU 2016-13. The FASB subsequently issued supplemental guidance within ASU No. 2019-05, Financial Instruments — Credit Losses (Topic 326): Targeted Transition Relief (“ASU 2019-05”). ASU 2019-05 provides an option to irrevocably elect the fair value option for certain financial assets previously measured at amortized cost basis. For public entities that are Securities and Exchange Commission filers, excluding entities eligible to be smaller reporting companies, ASU 2016-132023-09 is effective for annual periods beginning after December 15, 2019, including interim periods within those fiscal years.2024 and can be applied prospectively or retrospectively. The Company has not adopted this standard on January 1, 2020ASU 2023-09 yet and does not expect the adoption had no impact on its consolidated financial statements.In December 2019, the FASB issuedof this ASU No. 2019-12, Income Taxes – Simplifying the Accounting for Income Taxes (Topic 740). The amendments in this update simplify the accounting for income taxes by removing certain exceptions to the general principles as well as clarifying and amending existing guidance to improve consistent application. For public entities, the guidance is effective for annual reporting periods beginning after December 15, 2020 and for interim periods within those fiscal years. For nonpublic entities, the guidance is effective for annual reporting periods beginning after December 15, 2021 and to interim periods within fiscal years beginning after December 15, 2022. Early adoption is permitted for all entities. Depending on the amendment,109adoption may be applied on the retrospective, modified retrospective or prospective basis. The Company adopted this standard on January 1, 2021 and it did not have a material impact on the Company’s consolidated financial statements and related disclosures.statements.3. Merger Accounting22, 2020,16, 2022, the Company completed itsthe Merger with Private Kineta (see Note 1). The transaction was determined to be a reverse merger with PTI. Basedprimarily based on the Exchange Ratio,fact that, immediately following the Merger, former PTI stockholders, PTI option holders and other persons holding securities or other rights directly or indirectly convertible, exercisable or exchangeable for PTI Common Stock owned approximately Merger: (i) Private Kineta’s shareholders own a majority (29.780%) of the outstanding capitalcommon stock of the Company, (ii) Private Kineta designated a majority of the members of the initial board of directors of the combined organization and (iii) Private Kineta’s senior management hold all key positions in the former Yumanity stockholders, Yumanity option holders and other persons holding securities or other rights directly or indirectly convertible, exercisable or exchangeable for Yumanity Common Stock owned approximately 70.3% of the outstanding capital stocksenior management of the combined organization. At the closing of the Merger, all shares of Yumanity Common StockPrivate Kineta common stock were exchanged for an aggregate of 6,024,4336,115,000 shares the Company’s common stock. The reverse merger was accounted for as an acquisition of PTI Common Stock.assets as substantially all of the fair value was concentrated in cash and IPR&D. In connection with the Merger, the authorized shares of common stock of the Company are 125,000,000 with par value of $0.001. The total purchase price paid in the Merger, including certain transaction costs, has been allocated to the tangible and intangible assets acquired and liabilities assumed of PTI based on their relative fair values as of the completion of the Merger. Transaction costs primarily included bank fees and professional fees associated with legal counsel, auditors and printers. The following summarizes the purchase price paid in the Merger (in thousands, except share and per share amounts):2,609,4891,551,000 shares of PTIYumanity common stock outstanding as of December 22, 2020, plus 25,719 shares issued for the settlement of severance obligations16, 2022 and 21,739 shares issued as compensation for investment banking fees related to the Merger. Additionally, 51,5902,000 shares of restricted stock units were issued as compensation for two consultants hired by PTI. The number of sharesand reflects the impact of the Reverse Stock Split.last reported saleclosing price of PTIYumanity common stock on the Nasdaq GlobalCapital Market on December 22, 2020,16, 2022, the closing date of the Merger and after giving effect to the Reverse Stock Split.Represents the fair value of the PTI optionsTransaction costs primarily relate to purchase 194,550 shares of common stock outstanding at the time of the Merger.bank fees and professional fees associated with legal counsel.110net assets acquired and liabilities on the basis ofa relative fair values. The following summarizes the allocation of the purchase price to the net tangible and intangible assets acquired (in thousands):value basis as followsasset relatesassets relate to two leadthree product candidates for the treatment of cystic fibrosis.candidates. Due to the stageearly stages of development of these assets at the date of acquisition, significant risk remained and it was not yet probable that there was future economic benefit from these assets. Absent successful clinical results and regulatory approval for the assets and there was no alternative future use associated with the assets. Accordingly, the value of the assets wereacquired IPR&D was expensed in the consolidated statementsstatement of operations for the year ended December 31, 2020.2022.4. Fair Value MeasurementsMarketable Securitieshistorical stock prices. The significant unobservable inputs used in the fair value measurement for the year ended December 31, 2023 were as follows: volatility ranging from of 76.0% to 114.0%, risk-free interest rates ranging from 4.7% to 5.5% and funding probability of 75%, which resulted in a gain in fair value of $1.6 million for the year ended December 31, 2023, which is recorded in other income (expense) in the Statement of Operations. The fair value measurement as of December 31, 2023 was approximately $3.8 million. The fair value measurement as of December 31, 2022 was approximately $2.3 million and there was no change in fair value for the year ended December 31, 2022.tables presenttable provides a summary of the changes in the fair value of the rights from Private Placement measured using Level 3 inputs:hierarchy for its assetsthe notes. The qualified financing scenario considers the value impact of conversion at the stated discount to the issue price if the Company completes a qualifying financing event before the maturity date. The automatic conversion scenario estimates the timing of such conversion.liabilities,probability of automatic conversion ranging from 10% to 20%, which are measuredresulted in a fair value of these 2022 convertible notes ranging from $4.8 million to $5.3 million.on a recurring basis (in thousands):(see Note 6).Marketable securities124byusing a scenario-based analysis and a discounted cash flow model. Two primary scenarios were considered: the qualified financing scenario and the repayment scenario. The value of the 2020 convertible notes under each scenario was probability weighted to arrive at the estimated fair value for the notes. The qualified financing scenario considers the value impact of conversion at the stated discount to the issue price if the Company using quoted pricescompletes a qualifying financing event before the maturity date. The repayment scenario considers payment of principal at the contractual maturity dates. active markets for similar securities, which represent a Level 2 measurement within the fair value hierarchy.measurement of the 2020 convertible notes during 2022 prior to settlement in December 2022 were as follows: discount rates ranging from 13.0% to 41.2%, timing of the qualified financing ranging from 0.2 years to 0.75 years, timing of the repayment scenarios based on contractual maturity dates ranging from 0.25 years to 1.25 year, probability of a qualified financing ranging from 80% to 90% and probability of repayment ranging from 10% to 20%, which resulted in a fair value range for the 2020 convertible notes of $11.3 million to $16.2 million.roll-forwardsummary of the aggregate fair values of the Company’s preferred units warrant liability, for which fair value was determined by Level 3 inputs (in thousands):
Warrant LiabilityThe preferred unit warrant liability in the table above consisted of the fair value of warrants to purchase preferred units issued in 2019 (see Note 11) and was based on significant inputs not observable in the market, which represented a Level 3 measurement within111the fair value hierarchy. The Company’s valuation of the preferred unit warrants utilized the Black-Scholes option-pricing model, which incorporated assumptions and estimates to value the preferred unit warrants. The Company assessed these assumptions and estimates at the end of each reporting period. Changeschanges in the fair value of the preferred unit warrants were recognized within other income (expense) in the consolidated statements of operations. The most significant assumption in the Black-Scholes option-pricing model impacting the fair value of the preferred unit warrant liability was the fair value of the underlying preferred units as of each remeasurement date. The Company determined the fair value per unit of these preferred units by taking into consideration its most recent sales of its preferred units as well as additional factors that the Company deemed relevant. Immediately prior to the Merger, all of Holdings’ outstanding warrants were exchanged and became warrants to purchase shares of Yumanity Common Stock. As a result, the fair value of the warrants was reclassified to additional paid-in capital and there is no longer a warrant liability subject to remeasurement. There were 0 preferred unit warrants issued as of December 31, 2021.Marketable securities by security type consisted of the following (in thousands):Company’s 2022 & 2020 notes payable measured using Level 3 inputs:
Cost
Unrealized
Gains
Unrealized
Losses
Value
Cost
Unrealized
Gains
Unrealized
Losses
ValueThe Company’s marketable securities are due within one year5..1125. Property and Equipment, Netfollowing (in thousands):following:0.69,000 for the year ended December 31, 2023 and $73,000 for the year ended December 31, 2022. The Company has acquired certain laboratory equipment under agreements that are classified as finance leases. The carrying value of the equipment under finance leases included in the balance sheet as property and equipment was zero as of December 31, 2023 and $123,000 as of December 31, 2022, net of accumulated depreciation. During the year ended December 31, 2023, the Company disposed of assets with a net carrying value of $240,000 and received proceeds of $331,000. During the year ended December 31, 2022, the Company disposed of assets with a net carrying value of $3,000 and received proceeds of $65,000. The Company recorded a gain on disposal of fixed assets, which is recorded in other income (expense) in the Statement of Operations.0.813.8 million forwith various investors that are related parties (see Note 13). The interest rate was reduced on the years ended December 31, 20212020 convertible notes from 16.0% to 6.0% from October 2020 to until the earlier of (i) the Company raises at least $25.0 million in a single transaction or series of transactions after October 2020 and 2020, respectively. As(ii) the original maturity date of December 31, 2021, after which the interest rate increases 16.0%. The outstanding principal is due upon demand of the majority of the lenders with respect to (i) 50% on or after nine months after the original maturity date, September 30, 2022, and (ii) 50% on or after fifteen months after the original maturity date, March 31, 2023. The Company may prepay the 2020 convertible notes at any time without penalty. Upon default the lenders may apply a default interest rate of 20% and accelerate all amounts due upon bankruptcy. Repayment of the principal amount is required on a pro rata basis should the Company hadreceive excess proceeds from (i) commercial revenues exceeding $3.0 million in any 12-month period and (ii) the Company receives any funding proceeds from a capital financing transaction. The holders may at any time convert the 2020 convertible notes into shares of the Company’s non-voting common stock at a conversion price equal to 85% of the then-fair value of non-voting common stock but not less than $0.50 per share.gross assets under finance leases, which primarily consistedthe Company’s voting common stock and 8,500 shares of laboratory equipment,the Company’s non-voting common stock at fair value (based on a recent valuation) to the holders. As the other notes payable approximated to their fair value, no gain or loss was recognized upon extinguishment. In June 2022, the Company settled $1.0 million in outstanding principal and related accumulated amortizationaccrued interest by issuing 43,000 shares of the Company’s non-voting common stock at a 15% discount, recognizing a $0.2 million loss on extinguishment. The Company extended the maturity date for the remaining other notes payable with a principal balance of $0.30.4 million. At December 31,million to June 30, 2024 and decreased the interest rate to 6.0% interest, which was accounted for as a modification.had $0.8 million of gross assets under finance leases and related accumulated amortizationreceived a U.S. Small Business Administration (“SBA”) loan of $0.6150,000 million.at a 3.75% interest rate and maturing in August 2050. Repayments of principal are due monthly beginning in June 2027 and interest is due monthly.6. Collaboration Agreement“Collaboration“Merck Neuromuscular License Agreement”) with Merck Sharp & Dohme Corp. (“Merck”) to support research, development and commercialization of products for treatment of neuromuscular diseases, including amyotrophic lateral sclerosis. In June 2023, the Company achieved a development milestone pursuant to the Merck Neuromuscular License Agreement, which triggered a $5.0 million payment. Merck will continue to advance the research program for the ALS pipeline, one of the two pipeline programs licensed under the Merck Neuromuscular License Agreement. Following this milestone, Merck will assume sole responsibility for all future development and commercialization for the ALS program. The Company recognized licensing revenues of $5.0 million for the year ended December 31, 2023 and zero for the year ended December 31, 2022 under the Merck Neuromuscular License Agreement and has no further obligations under the Merck Neuromuscular License Agreement.amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (FTLD)cystic fibrosis (the “FAIR License Agreement”). Pursuant toThe Company did not earn revenues under the CollaborationFAIR License Agreement the Company granted Merck an exclusive, worldwide license with the right to grant and authorize sublicenses, under certain intellectual property rights related to two certain undisclosed targets in connection with the Company’s ALS and FTLD programs to make, have made, use, import, offer to sell and sell compounds and products covered by such intellectual property rights. In the event that the exploitation of such compound or product would infringe during the term of the Merck Collaboration Agreement a claim of an issued patent controlled by Yumanity, Yumanity also granted Merck a non-exclusive, sublicensable, royalty-free license under such issued patent to exploit such compound and product.Under the terms of the Collaboration Agreement, the Company and Merck are each responsible to perform certain research activities in accordance with a mutually agreed upon research plan. Upon the completion of certain stages of the research plan, Merck will elect to either advance and make certain contractual option payments or terminate the applicable research program. If Merck elects not to advance a research program, such program terminates and the rights granted to Merck in the program revert to the Company. Following completion of the research program, Merck is responsible for the development and commercialization of the compounds developed pursuant to the research program and any product containing such compounds.Under the terms of the Collaboration Agreement, the Company received an upfront payment totaling $15.0 million in July 2020 and is eligible to receive up to $280.0 million if Merck elects to advance the research program and upon achievement of specified research and development milestones, and up to $250.0 million upon achievement of specified sales- based milestones as well as a tiered, mid-single digit royalty on net sales of licensed products, subject to customary reductions.Unless terminated earlier, the Collaboration Agreement will continue in full force and effect until one or more products has received marketing authorization and, thereafter, until expiration of all royalty obligations under the Collaboration Agreement. The Company or Merck may terminate the Collaboration Agreement upon an uncured material breach by the other party or insolvency of the other party. Merck may also terminate the Merck Collaboration Agreement for any reason upon certain notice to the Company.Merck also participated in the Company’s Class C preferred units financing in June 2020 with terms consistent with those of other investors that purchased Class C preferred units in June 2020. The Class C preferred units were issued at a price of $4.0008 per unit, which was determined to be fair value based on the same price paid by other investors that purchased Class C preferred units in the financing. The equity investment was considered to be distinct from the Collaboration Agreement.113The Company assessed the promised goods and services expected to be delivered as part of the first stage of the research program (the "Initial Phase") to determine if they are distinct. Based on this assessment, the Company determined that Merck cannot benefit from the promised goods and services separately from the others as they are highly interrelated and therefore not distinct. Accordingly, the promised goods and services represent one combined performance obligation and the entire transaction price was allocated to that single combined performance obligation. The performance obligation is being satisfied over the research term as the Company performs the research and development activities through the first substantive option period, and participates in a Joint Steering Committee to oversee research and development activities. Accordingly, the upfront payment of $15.0 million was recorded as deferred revenue and is being recognized as revenue as the performance obligation is satisfied. The Company recognizes revenue using the cost-to-cost method, which it believes best depicts the transfer of control to the customer. Under the cost-to-cost method, the extent of progress towards completion is measured based on the ratio of actual costs incurred to the total estimated costs expected upon satisfying the identified performance obligation. Under this method, revenue is recorded as a percentage of the estimated transaction price based on the extent of progress towards completion. As of December 31, 2021, the aggregate amount of the transaction price related to the unsatisfied portion of the performance obligation associated with Initial Phase is $0.1 million, which is expected to be recognized as revenue within the next year. During the yearyears ended December 31, 2021, the Company recorded $8.0 million of collaboration revenue related to the Collaboration Agreement associated with providing the Initial Phase research2023 and development services.At contract inception, the potential milestone payments that the Company is eligible to receive were excluded from the transaction price as they were fully constrained. At the end of each reporting period, the Company reevaluates the transaction price and as uncertain events are resolved or other changes in circumstances occur, and if necessary, the Company will adjust its estimate of the transaction price. Any additions to the transaction price would be reflected in the period as a cumulative revenue catch-up. At the inception of the arrangement, the Company evaluated the options held by Merck to either advance or terminate the applicable research program to determine if they provided Merck with any material rights. The Company concluded that the options were not issued at a significant and incremental discount, and therefore do not provide Merck with a material right. As such, these options were excluded as performance obligations and will be accounted for if and when they occur.In November 2021, the Company delivered one of two data packages associated with the Initial Phase of the research and development services. On December 17, 2021, Merck notified Yumanity Therapeutics, Inc. (the “Company”) that Merck has accepted the first data package for one program from their research collaboration with the Company relating to amyotrophic lateral sclerosis, or ALS (also known as Lou Gehrig’s disease) and frontotemporal lobar degeneration, or FTLD, and that Merck has elected to continue the research collaboration. Achievement of this milestone triggered a $5 million milestone payment due from Merck. Upon Merck electing to advance the research program into the second phase (the "Second Phase"), the Company assessed whether the promised goods and services for the Second Phase are distinct. Based on the facts and circumstances, including but not limited to key differences between the Initial Phase research plan and the Second Phase research plans, including experiments performed and personnel utilized, the Company determined that the Second Phase represents a separate contract with its own performance obligation. The Second Phase performance obligation is being satisfied over the Second Phase research term as the Company performs the research and development activities through the second substantive option period, and participates in a Joint Steering Committee to oversee research and development activities. Accordingly, the option payment of $5.0 million was recorded to deferred revenue at December 31, 2021 and will be recognized as revenue as the performance obligation is satisfied. The Company will recognize the Second Phase revenue using the cost-to-cost method, which it believes best depicts the transfer of control to the customer.The Company assessed the Collaboration Agreement to determine whether a significant financing component exists and concluded that a significant financing component does not exist.7. Accrued ExpensesAccrued expenses consisted of the following (in thousands):1148. DebtLong-term debt consisted of the following (in thousands):2022.We have outstanding principal borrowings of $12.7 million under a loan and security agreement entered into in December 2019 (the “Term Loan”) with Hercules Capital, Inc. (the “Lender”). Another $5.0 million became available upon the occurrence of a developmental milestone and an equity event defined in the agreement, but we elected notWarrants to draw it. An additional $10.0 million may become available to be drawn upon lender approval. Borrowings under the Term Loan were repayable in monthly interest-only payments until August 1, 2021. The interest-only period is followed by monthly payments of equal principal plus interest until the loan maturity date of January 1, 2024. Outstanding borrowings bear interest at the greater of (i) 8.75% and (ii) the prime rate as reported in the Wall Street Journal plus 4.00%. A final payment fee of 5.25% of the amounts drawn under the Term Loan is due upon the earlier of the maturity date or the repayment date if paid early, whether voluntary or upon acceleration due to default. We may repay the Term Loan at any time by paying the outstanding principal balance in full, along with any unpaid accrued interest, the final payment fees of 5.25% of the amounts drawn and a prepayment fee calculated on amounts being prepaid. The prepayment fee is 3.0% if the Term Loan is repaid within the one-year anniversary of the draw date, 2.0% if paid between the first and second-year anniversary of the draw date and 1.0% if paid after the second anniversary of the draw date but before the maturity date.In April 2020, the Term Loan was amended to permit indebtedness consisting of a loan under the Paycheck Protection Program (“PPP”) of the Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) provided that such loan shall be unsecured, shall not contain any terms or conditions that are adverse to the Lender’s rights under the loan and that the Company will not prepay such loan. In June 2020, the Term Loan was amended and an additional final payment fee of $0.3 million became due upon repayment of the loan.On December 22, 2020, the Company entered into an Unconditional Secured Guaranty and Pledge Agreement (the “Guaranty”) with the Lender as a condition to the Lender’s consent to the Merger under the Term Loan between Yumanity, Inc. as borrower and the Lender. Immediately prior to the Merger, Yumanity, Inc. entered into a Fourth Amendment and Consent to Loan and Security Agreement dated as of December 22, 2020 with the Lender (the “Loan Amendment”). The Guaranty provided for the Company’s guaranty of Yumanity Inc.’s obligations under the Loan Agreement and provided the Lender a security interest in all of Company’s assets other than intellectual property as collateral. The Loan Amendment provided for the Lender’s consent to the Merger and to the creation and funding of a Silicon Valley Bank Paycheck Protection Program escrow account to hold funds in connection with Yumanity’s outstanding Paycheck Protection Program loan amounts for which Yumanity submitted a forgiveness application. The Loan Amendment also amended the definition of “Change in Control” to include the situations in which the Company no longer controls Yumanity, Inc. The remaining terms and conditions of the Loan Agreement generally continued in the form existing prior to the Loan Amendment.As of December 31, 2021 and 2020, the interest rate applicable to borrowings under the Term Loan was 8.75%. During the year ended December 31, 2021, the weighted average effective interest rate on outstanding borrowings under the Term Loan was approximately 12.52%. On March 29, 2021, the Term Loan was amended again to allow for the creation of a new foreign subsidiary, as well as changing certain covenants related to the financial operations of said subsidiary. The subsidiary was formed on April 23, 2021.On April 13, 2021, the Term Loan was amended to reduce the additional final payment fee of $0.3 million to $0.1 million upon repayment of the loan.Borrowings under the Term Loan are collateralized by substantially all of the Company’s personal property, other than its intellectual property. There were no financial covenants associated with the Term Loan; however, the Company is subject to certain affirmative and negative covenants restricting the Company’s activities, including limitations on dispositions, mergers or acquisitions; encumbering its intellectual property; incurring indebtedness or liens; paying dividends; making certain investments; and engaging in certain other business transactions. The obligations under the Term Loan are subject to acceleration upon the occurrence of specified events of default, including a material adverse change in the Company’s business, operations or financial or other condition. As of December 31, 2021, the Company has assessed that the risk of subjective acceleration under the material adverse events clause is not115probable and therefore have classified the outstanding principal on the consolidated balance sheet based on the contractually scheduled principal payments. Upon the occurrence of an event of default and until such event of default is no longer continuing, the annual interest rate will be 5.0% above the otherwise applicable rate.In April 2020, prior to entering into the Merger Agreement with PTI in August 2020, the Company issued a Promissory Note to Silicon Valley Bank, pursuant to which it received loan proceeds of $1.1 million (the “Loan”) provided under the PPP established under the CARES Act and guaranteed by the U.S. Small Business Administration. On April 3, 2021, the Company was notified by Silicon Valley Bank that the Loan forgiveness application was accepted by the Small Business Association as of March 30, 2021. Accordingly, the Company has recognized $1.1 million in income for debt extinguishment in the consolidated statement of operations as of December 31, 2021.1162021, future principal payments due are2023, the Company has issued and outstanding warrants to purchase shares of the Company’s common stock as follows, which all met the condition for equity classification (in thousands):
Issued
Date
Exercise
Price
underlying warrantsFebruary 25, 2022, Term Loan terminated upon the payment byApril 20, 2023, the Company entered into a Securities Purchase Agreement (the “April 2023 Purchase Agreement”) with an institutional investor (the “April 2023 Investor”), pursuant to Hercules of a voluntary payoff amount of $12.8 million, provided that the Company continues to be bound by certain indemnification obligations under Section 6.3 of the Term Loan. The payoff amount included payment of approximately $0.9 million consisting of the final and additional final payment fees outlined above, as well as an interest/non-use fee of less than $0.1 million.9. Preferred UnitsPrior to the Merger, the Company had issued Class A preferred units, Class B preferred units, and Class C preferred units, collectively referred to as the “Preferred Units”. In June 2020,which the Company issued and sold, in a registered direct offering priced at-the-market under the rules of The Nasdaq Stock Market LLC (“Nasdaq”) (such offering, the “April 2023 Registered Offering”), (i) an aggregate of 5,404,588948,000 Class C preferred unitsshares of its common stock, at a purchase price of $4.00084.21 per unit, resulting in cash proceedsshare and (ii) pre-funded warrants exercisable for up to 477,179 shares of its common stock (the “April 2023 Pre-Funded Warrants”) to the April 2023 Investor at a purchase price of $21.24.209 per April 2023 Pre-Funded Warrant, for aggregate gross proceeds from the April 2023 Registered Offering of approximately $6.0 million netbefore deducting the placement agent fee (as described in greater detail below) and related offering expenses.issuance costscommon stock at an exercise price of $0.40.001 million.per share. The April 2023 Pre-Funded Warrants are exercisable immediately and may be exercised at any time until the April 2023 Pre-Funded Warrants are exercised in full.Class C preferred units, a majoritysecurities of the Company’s voting preferred and common unit holders voted to amend the Company’s operating agreement such that Class B preferred unitholders who did not participate in a minimum purchase of Class C preferred units, referred to as non-participating holders, became holders of Class B preferred units referred to as “Defaulting Class B Preferred Units.” Class B preferred units other than the Defaulting Class B Preferred Units are referred to as Ordinary Class B preferred units. The terms of the Defaulting Class B Preferred Units are similar to the terms of common units with respect to distributions, except that Defaulting Class B Preferred Units are treated as one-fifth (1/5) of a common unit. The Defaulting Class B Preferred Units lose their rights associated with Preferred Units and have no voting rights. For accounting purposes, this transaction was treated as an extinguishment of the existing Class B preferred units held by the non-participating holders and the issuance of a new security. The carrying value of $7.0 million for the Class B preferred units exchanged for Defaulting Class B Preferred Units was removed from Preferred units on the balance sheet and the Defaulting Class B Preferred Units were reflected in permanent equity at their issuance date fair value of $0.3 million with the difference $6.7 million reflected as a reduction of accumulated deficit.Yumanity Reorganization and MergerImmediately prior to the Merger on December 22, 2020,Company pursuant to the Yumanity Reorganization, allApril 2023 Purchase Agreement. As compensation for such placement agent services, the Company paid Wainwright an aggregate cash fee equal to $420,000, a non-accountable expense of the Class A, Class B,$35,000 and Class C preferred units converted to shares of Yumanity, Inc. common stock. Pursuant$50,000 for legal and other expenses as actually incurred. The total offering-related fees were approximately $520,000, which resulted in net proceeds to the Merger, these sharesCompany of Yumanity Inc. common stock were then exchanged for shares of PTI common stock based upon the Exchange Ratio and the related carrying value was reclassified to common stock and additional paid-in capital. There were $05.5 preferred units outstanding after the Yumanity Reorganization.10. Common Stock/UnitsAs of December 31, 2021, the Company’s certificate of incorporation, as amended and restated, authorizedmillion. On April 24, 2023, the Company also issued to issueWainwright or its designees warrants to purchase 125,000,00071,259 shares of common stock par value $0.001 per share, and 5,000,000 shares(the “April 2023 Wainwright Warrants”). The April 2023 Wainwright Warrants have a term of preferred stock, par value $0.001 per share, all of which is undesignated. Each share of common stock entitles the holder to one vote for the election of directors and on all matters submitted to a vote of the Company’s stockholders. Common stockholders are entitled to receive dividends, as may be declared by the board of directors, if any, subject to the preferential dividend rights of the preferred stockholders. NaN cash dividends have been declared or paid to date.Prior to the Yumanity Reorganization, the Company had issued common units. Each common unit entitled the holder to one vote on all matters submitted to a vote of the Company’s members. In the event of any deemed liquidation, dissolution, or winding-up of the Company, the assets of the Company would have been distributed in accordance with the order of distributions described under the rights and preferences of the Preferred Units.Prior to the Yumanity Reorganization, the Company also had outstanding restricted incentive units, a form of common units, that generally vested over fourfive years (see Note 12).117Yumanity ReorganizationImmediately prior tofrom the Merger, pursuant tocommencement of sales in the Yumanity Reorganization, all of Holdings’ outstanding common units, including the outstanding incentive units, were exchangedApril 2023 Offering, and became shares of common stock of Yumanity, Inc.Private PlacementFollowing the Merger, on December 22, 2020, pursuant to the Subscription Agreement, dated as of December 14, 2020, by and among the Company and the purchasers named therein, the Company completed the sale of $33.6 million of the Company’s common stock, par value $0.001 per share to the purchasers in a private placement.11. Warrants for Common Stock and Preferred and Common UnitsPrior to the Merger, in December 2019, in connection with the Term Loan (see Note 8), the Company issued 34,946 Class B preferred warrants withhave an exercise price of $8.375.2625 per unit. Upon issuanceshare.Class C preferred unitsNasdaq (such offering, the “October 2023 Registered Offering”), (i) an aggregate of 110,000 shares of its common stock, at a purchase price of $3.37 per share, and (ii) pre-funded warrants exercisable for up to 780,208 shares of its common stock (the “October 2023 Pre-Funded Warrants”) to the October 2023 Investor at a purchase price of $3.369 per October 2023 Pre-Funded Warrant, for aggregate gross proceeds from the October 2023 Registered Offering of approximately $3.0 million before deducting the placement agent fee (as described in June 2020,greater detail below) and related offering expenses.warrants for Class B preferred units issued in December 2019 became warrantsright to purchase one share of 73,109 Class C preferred units withcommon stock at an exercise price of $4.00080.001 per unit (see Note 9)share. The October 2023 Pre-Funded Warrants are exercisable immediately and may be exercised at any time until the October 2023 Pre-Funded Warrants are exercised in full.Yumanity ReorganizationMergeradministrative expense.As of December 31, 2019,had outstanding warrants for the purchase of common units, Class A preferred units, and Class B preferred units (which became warrants to purchase Class C preferred units as described above). Immediately prior to the Merger, pursuant to the Yumanity Reorganization, all of Holdings’ outstanding warrants were exchanged and becameissued 5,000 warrants to purchase shares of Yumanityits common stock. Upon consummationstock in connection with the issuance of its 2022 convertible notes and in October 2022 and December 2022, the Merger, theCompany issued 55,000 warrants to purchase shares of Yumanityits common stock becamein connection with amendments to its 2022 convertible notes (see Note 6). The Company recorded non cash interest expense of $1.6 million on the statement of operations.stock. The contractual termstock to investors in the Private Placement, each at an exercise price of each warrant remained unchanged. No additional$0.14, with exercise contingent upon the Merger closing and exercisable following the first closing of the Private Placement. These warrants were subsequently cancelled in December 2022 upon amendment of the Securities Purchase Agreement.2021value being transferred, the Company recorded warrant expense of $3.3 million within other income and no(expense) on the statement of operations.wereto purchase shares of its common stock in connection with the Private Placement (see below).in 2021.during the year ended December 31, 2022 ranged from $0.14 to $26.88.As31, 2021 and 2020,2022, the Company’s outstanding warrants to purchasenumber of authorized shares of common stock of the Company was adjusted to 125,000,000 with a par value of $0.001 and all non-voting shares became voting shares. As of December 31, 2023, there were 10,396,614 shares issued and outstanding.following:following as the period presented:
2023
common stock under equity incentive plans
Term
(in Years)
Stock
Shares of
Common
Stock Issuable
Price12. Stock/Equity-Based Compensation104,000 warrants to purchase shares of the Company’s non-voting common stock to investors in the Private Placement, each at an exercise price of $0.14, with exercise contingent upon the Merger closing and exercisable following the first closing of the Private Placement. The Company determined the contingent exercise provisions were indexed to the Company’s operations and the warrants qualified for equity classification.Incentive unitsPrior to the Yumanity Reorganization, the Company’s operating agreement, as amended and restated, provided for the granting of incentive units, a type of common units, to officers, directors, employees, consultants and advisors. Holders of incentive units were entitled to receive distributions in proportion to their ownership percent interest, upon liquidation, that were in excessThe second closing of the strike pricePrivate Placement was expected to occur on April 15, 2024. However, in February 2024, certain investors indicated they will not be able to consummate the second closing of the award, (the “Participation Threshold”) set byPrivate Placement. If the boardsecond closing of directors on the datePrivate Placement were to occur, the Company would be obligated to issue a number of grant. The Participation Threshold wasshares of its common stock based on the amount that would be distributed in respectaggregate purchase price of a$22.5 million divided by the purchase price equal to (a) the volume-weighted average price of Company common unit pursuantstock for the five trading days prior to its liquidation preferences, if, upon a hypothetical liquidationApril 15, 2024 (“VWAP”), plus (b) 10% of the Company on the date of issuance of such Incentive Unit, the Company sold its assets for their fair market value, satisfied its liabilities and distributed its remaining net assets to holders of units in liquidation. The Company determinedVWAP; provided, however, that the underlying terms ofshare purchase price shall be at least equal to the incentive units and the intended purpose of the awards were more akin to an equity-based compensation award than a performance bonus or profit-sharing arrangement and, therefore, the incentive units were equity-classified awards.Restricted Stock Units (RSUs)On January 14, 2021, the Company’s compensation committee of the board approved payment to be made to Company employees through a grant of RSUs based on the February 1, 2021 closing share price of the Company’s common stock withon March 29, 2023. The Company determined that its obligation to issue additional shares of its common stock in the second closing at a premium to the VWAP was a freestanding financial instrument and a future right, which is subject to fair value. Accordingly, at inception the future right was recorded as an other asset in the Company’s consolidated balance sheet at its fair value equal to 10% of the second closing amount, or $2.22.3 million. The requisite service periodremaining proceeds from the first closing were allocated to the shares of common stock issued in the first closing and to the warrants as such instruments are equity-classified. The future right is subject to remeasurement at each reporting date, however, as the fair value will always equal 10% of the value of the future second closing until settlement, no changes in fair value are expected to be recorded in the Company’s consolidated statements of operations. The Company incurred insignificant issuance costs related to the Private Placement.awards ranges from onebudget period January 2021 to four years (the vesting period). December 2021, which was later extended to December 31, 2022.employee stock-based compensation expensegrant revenue from this grant of zero for the RSU grant on a straight-line basis overyear ended December 31, 2023, and $912,000 for the vesting period of theyear ended December 31, 2022.118awards.1332021, 122,469 RSUs were granted and 86,225 were outstanding, and2022, the Company recognizedhad $0.9442,000 million stock-based compensation expense duringin deferred revenue under the twelve monthsMerck Neuromuscular License Agreement. The Company recognized collaboration revenue of $442,000 for the year ended December 31, 2021.2023, due to completion of research and development services provided by us, and The following table summarizes the Company’s RSU activityzero for the twelve monthsyear ended December 31, 2021:2022. As of December 31, 2023, the Company had zero in deferred revenue under the Merck Neuromuscular License Agreement.
Average Grant
Date Fair ValueRestricted Stock Awards (RSAs)2008 Equity Incentive PlanOn November 22, 2021,2010 Equity Incentive Plancompensation committeecommon stock on the date of grant and the board approved payment to be made to Company employees through a grantcontractual term of RSAsstock options granted did not exceed ten years. Options become vested and, if applicable, exercisable based on terms determined by the December 1, 2021 closing shareCompany’s board of directors or other plan administrator on the date of grant, which is continued employment or service as defined in each option agreement. Stock appreciation rights (“SARs”) provide a participant with the right to receive the aggregate appreciation in stock price over the market price of the Company’s common stock with aat the date of grant, payable in cash. The rights granted have varying vesting terms, including SARs that vest immediately on the grant date and upon satisfaction of the service-based requirement, typically three to five years. The maximum fair value is limited to four times the exercise price.1.247,000. In February 2020, the 2010 Plan expired and 181,000 million. The requisite service period forstock options granted prior to the awards is one year (the vesting period). The Company recognized employee stock-based compensation expense for the RSA grant on a straight-line basis over the vesting period of the awards. Asexpiration remain outstanding as of December 31, 2021, 2023.305,663 RSAs were granted and outstanding, and the Company recognized $0.1 million stock-based compensation expense during the twelve months ended December 31, 2021.The following table summarizes the Company's RSA activity for the twelve months ended December 31, 2021:
Average Grant
Date Fair ValueSummary of plansUpon completion of the Merger, the Company assumed PTI’s 2016 Stock Option andThe Company’s 2020 Equity Incentive Plan (the “2016“2020 Plan”) authorizes the grant of equity awards for up to 206,000 shares of the Company’s voting common stock and PTI’s 2016 Employee Stock Purchase Plan (the “2016 ESPP”).206,000 of the Company’s non-voting common stock.2016 Stock Option and Incentive PlanOn February 3, 2016, PTI’s stockholders approved the 2016 Plan, which became effective on February 9, 2016. The 20162020 Plan provides for the grant of incentive stock options, nonqualifiednon-statutory stock options stock appreciation rights,and restricted stock units, restrictedto employees and non-employee service providers. Under the 2020 Plan, the contractual term of stock awardsoptions shall not exceed ten years and other stock-based awards. The numberthe exercise price of shares initially reserved for issuance under the 2016 Plan wasstock options granted shall not be less than 79,092 shares. The number of shares of common stock that may be issued under the 2016 Plan will automatically increase each January 1, beginning January 1, 2017, by the lesser of 3100% of the sharesestimated fair market value of the Company’s common stock outstanding on the immediately preceding December 31, or an amountdate of grant. However, the exercise price of incentive stock options granted to a 10% stockholder shall not be less than 110% of the fair market value of the common stock on the date of grant and the contractual term shall not exceed ten years. Options become vested and, if applicable, exercisable based on terms determined by the Company’s board of directors or other plan administrator on the compensation committeedate of grant, which is continued employment or service as defined in each option agreement. Restricted stock has vesting terms that vest immediately on the grant date or upon satisfaction of the boardservice-based requirement, typically four years or the performance-based requirement. The Company has a repurchase right exercisable upon termination of directors. Thecontinuous service with respect to restricted stock for any shares of common stock underlying any awards that are forfeited, canceled, repurchased, or are otherwise terminated byissued and unvested.underapproved the 2016 Plan and the 20082022 Equity Incentive Plan as amended (the “2008“2022 Plan”) will be added back. The 2022 Plan provides for the grant of incentive stock option, non-statutory stock options, restricted stock, restricted stock units, stock appreciation rights (“SARs”), performance units and performance shares to the shares of common stock available for issuance under the 2016 Plan. On January 1, 2020, an additional 78,175 shares were reserved for issuance under the 2016 Plan. The number of shares reserved for issuance under the 2016 Plan was increased by 303,495 shares effective as of January 1, 2021 in accordance with the provisionsemployees, directors and independent contractors of the 2016Company. Under the 2022 Plan, described above. Options granted under the 2016 Plan with service-based vesting conditions generally vest overexercise price of stock options grants shall be at four years100 and expire after ten years. As of December 31, 2021, the total number of shares% fair market value of the Company’s common stock reserved for issuance under the 2016 Plan was 379,720, of which 138,163 shares are available for future issuance under the 2016 Plan.1192016 Employee Stock Purchase PlanOn February 3, 2016, PTI’s stockholders approved the 2016 ESPP, which became effective in connection with the completion of the PTI’s initial public offering. A total of 6,938 shares of common stock were initially reserved for issuance under the 2016 ESPP. In addition, the number of shares of common stock that may be issued under the 2016 ESPP will automatically increase each January 1, beginning January 1, 2017, by the lesser of (i) 6,938 shares of common stock, (ii) 1% of the Company’s shares of common stock outstanding on the immediately preceding December 31, or (iii) an amountdate of grant and the contractual term of stock options granted shall not exceed ten years. Options become vested and, if applicable, exercisable based on terms determined by the Company’s board of directors or the compensation committee of the board of directors. The number of shares reserved for issuance under the 2016 ESPP was increased by 6,938 shares effective as of January 1, 2021 in accordance with the provisions of the 2016 ESPP described above. As of December 31, 2021, the total number of shares reserved under the 2016 ESPP was 41,626 shares.Yumanity Therapeutics, Inc. Amended and Restated 2018 Stock Option and Grant PlanOn December 4, 2018, the Company’s board of directors adopted the 2018 Unit Option and Grant Plan (the “2018 Plan”), which was approved by the Company’s membersother plan administrator on December 5, 2018. The 2018 Plan provided for the Company to grant unit options, restricted unit awards and unrestricted unit awards to employees, directors and consultants of the Company. As part of the Yumanity Reorganization and the Merger, the 2018 Plan was amended and restated as the “Yumanity Therapeutics, Inc. Amended and Restated 2018 Stock Option and Grant Plan”. Each stock option outstanding under the 2018 Plan at the Effective Time of the Merger was automatically converted into a stock option exercisable for the same number of shares of Yumanity common stock, and then assumed by the Company, based on the Exchange Ratio and the exercise price per share of such outstanding stock option, as adjusted for the Exchange Ratio. The 2018 Plan is administered by the board of directors or, at the discretion of the board of directors, by a committee of the board of directors. The exercise prices, vesting and other restrictions are determined at the discretion of the board of directors, or its committee if so delegated.Options granted under the 2018 Plan with service-based vesting conditions generally vest over four years and expire after ten years. The total number of common shares that may be issued under the 2018 Plan is 1,527,210 as of December 31, 2021. Shares that are forfeited, canceled, reacquired by the Company prior to vesting, satisfied without the issuance of shares or otherwise terminated (other than by exercise) and units that are withheld upon the exercise of an option or settlement of an award to cover exercise price or tax withholding shall be added back to units available under the 2018 Plan. As of December 31, 2021, 33,209 shares remain available for issuance under the 2018 Plan.Under each plan, the exercise price per option granted is not less than the fair market value of common stock as of the date of grant.2021 Inducement PlanOn June 2, 2021, the Board of Directors approved the adoption of the Company’s 2021 Inducement Plan (the “2021 Plan”),used exclusively for the grant of equity awards to individuals who were not previously employees of the Company (or followingcontinued employment or service as defined in each option agreement. SARs provide a bona fide period of non-employment), as an inducement material to such individual’s entering into employmentparticipant with the Company, pursuantright to Rule 416 underreceive the Securities Act of 1933. Duringaggregate appreciation in stock price over the year ended December 31, 2021, the Company issued 174,400 options from the 2021 Plan to purchase common stock. As of December 31, 2021, the total number of sharesmarket price of the Company’s common stock that may be issued underat the 2021 Plan is 400,000date of grant, payable in cash or in shares of which equivalent value.227,600 shares are available for future issuance under the 2021 Plan. Shares that are expired, forfeited, canceled or otherwise terminated without having been fully exercised will be available for future grant under the 2021 Plan.On April 13, 2021, Board of Directors approved the issuance of stock options to purchase 135104,000 shares of its common stock. The stock options were issued outside of the Company’s 2021 Plan as an inducement material to the individual’s acceptance of an offer of employment with the Company in accordance with Nasdaq Listing Rule 5635(c)(4).KINETA, INC.valuationThe fair value of option grants is estimated using the Black-Scholes option-pricing model. Prior to the Merger, the Company was a private company and lacks company-specific historical and implied volatility information. Therefore, it estimates its expected stock volatility based on the historical volatility of a publicly traded set of peer companies. The expected term of the Company’s options has been determined utilizing a midpoint convention estimate. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield is based on the fact that the Company has never paid cash dividends and does not expect to pay any cash dividends in the foreseeable future.120presents, on a weighted average basis,summarizes stock option activity under the assumptions used in the Black-Scholes option-pricing model to determine the grant-date fair value of options granted:Company’s equity incentive plans:Option activityNonrecourse Promissory Notes for Stock Options Exercisedoptionrestricted stock activity during the year ended December 31, 2021:consisting of RSUs:
of Shares/
Units
Average
Exercise
Price
Average
Remaining
Contractual
Term
Intrinsic
Value(1)Certain options were immediately exercisable for restricted common stock which vest accordingthe original vesting terms of the option grant. No options have been exercised prior to vesting.The aggregate intrinsic value of options is calculated as the difference between the exercise price of the options and the fair value of the Company’s common stock/units for those stock/unit options that had exercise prices lower than the fair value of the Company’s common stock/units.The weighted average grant-date fair value of stock/unit options granted during the years ended December 31, 2021 and 2020 was $11.56 per share and $11.39 per share/unit, respectively.Stock/equity-based compensationStock-Based CompensationCompany recorded stock/equity-basedfollowing table summarizes total stock-based compensation expense related to common stock/unit options, restricted stock units, and restricted stock awardsincluded in the following expense categories in itsCompany’s consolidated statements of operations (in thousands):operations:2021, total2023, there was $2.9 million of unrecognized stock-based compensation cost related to unvestedstock options and restricted common stock was $12.3 million,outstanding, which is expected to be recognized over a weighted averageweighted-average remaining service period of 2.651.8 years.13. 14.Prior to the Yumanity Reorganization, Holdings was treated as a partnership for federalThe Company had no income tax purposes and, therefore, its owners, and not Holdings, were subject to U.S. federal or state income taxation on the income of Holdings. Holdings’ directly held subsidiary Yumanity Therapeutics, Inc. was treated as a corporationexpense for U.S. federal income tax purposes and was subject to taxation in the United States. Subsequent to the Yumanity Reorganization, the Company is a corporation and is subject to taxation in the United States. In each reporting period, the Company’s tax provision includes the effects of consolidating the results of the operations of its subsidiary.121During the years ended December 31, 20212023 and 2020, the Company recorded 0 income tax benefits for the net operating losses incurred or for the research and development tax credits generated in each year2022 due to its uncertaintyhistory of realizing a benefit from those items.operating losses. The Company has established a foreign subsidiary in Australia in 2021 and is components of income tax expense (benefit) are as follows:0t subject to foreign taxes in the current year due to a loss generated in the current year.income tax expense at the statutoryCompany’s federal income tax rate and effective income taxes as reflected in the financial statementstax rate is as follows:(1)Represents the tax effect on the in-process research and development expense recorded on the acquisition of PTINetconsistedand liabilities are summarized as follows:following (in thousands):deferred tax assets arising from the above-mentioned future tax benefits is currently not likely to be realized and, accordingly, has provided a valuation allowance on its deferred tax assets. The valuation allowance increased by $2.0 million for the year ended December 31, 2023 and $158.5 million for the year ended December 31, 2022. During the year ended December 31, 2022, $151.6 million of the increase in valuation allowance relates to the Merger.2021,2023, the Company had U.S.has federal andnet operating loss carryforwards of approximately $549.6 million of which approximately $304.2 million begins to expire in 2027. The remaining balance can be carried forward indefinitely with utilization limited to 80% of future taxable income. The Company has general business credit carryforwards of $23.9 million as of December 31, 2023, which will begin to expire in 2027. As of December 31, 2023, the Company has state net operating loss carryforwards of $486.6489.9 million, and $497.2 million, respectively, which may be available to offset future taxable income. Federal and state net operating loss carryforwards of $228.1 million and $497.2 million, respectively,will begin to expire in 2026 and 2030, respectively. Federal net operating loss carryforwards of $.258.5million do not expire but may be limited in their usage to an annual deduction equal to 80% of annual taxable income. As of December 31, 2021, the Company had $0.1 million of foreign net operating loss carryforwards that do not expire. As of December 31, 2021, the Company also had U.S. federal, state, and foreign research and development tax credit carryforwards of $15.4 million, $6.2 million, and $0.1 million, respectively, which may be available to offset future tax liabilities and begin to expire in 2027 for domestic credits, and foreign credits do not expire. the U.S. federal and state net operating loss carryforwards and research and development tax credit carryforwards may be subject to a substantial annual limitation under SectionsSection 382 and 383 of the Internal Revenue Code of 1986 ("Section 382"), and corresponding provisions of state law, due to ownership changes that may have occurred previously or that could occur in the future, including those tax attributes acquired from PTI via the Merger.future. These ownership changes may limit the amount of carryforwards that can be utilized annually to offset future taxable income or tax liabilities. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of certain stockholders or public groups in the stock of a corporation by more than 50% over a three-year period.income. The Company has not conducted a studycontinues to assessevaluate whether a change of control has occurred or whether therefor previous mergers and other shareholder events. We have been multiple changes of control since inception due to the significant complexity and cost associated with suchrecorded a study. If the Company has experienced a change of control, as defined by Section 382,full valuation allowance against our net deferred tax assets at any time since inception, utilization of the net operatingeach balance sheet date.122loss carryforwardsThe Tax Cuts and Jobs Act contained a provision which requires the capitalization of Section 174 costs incurred in years beginning on or after January 1, 2022. Section 174 costs are expenditures which represent research and development costs that are incident to the development or improvement of a product, process, formula, invention, computer software, or technique. This provision changes the treatment of Section 174 costs such that the expenditures are no longer allowed as an immediate deduction but rather must be capitalized and amortized. The Company has included the impact of this provision, which results in a deferred tax credit carryforwardsasset of approximately $4.4 million as of December 31, 2023 and approximately $3.6 million as of December 31, 2022.benot have an impact on the Company’s effective tax rate assuming the Company continues to maintain a full valuation allowance position. As of December 31, 2023, no significant increases or decreases are expected to the Company’s uncertain tax positions within the next twelve months.an annual limitation under Section 382, which is determined by first multiplying the valuevarying statutes of the Company’s stock at the time of the ownership change by the applicable long-term tax-exempt rate, and then could be subjectlimitations. Due to additional adjustments, as required. Any limitation may result in expiration of a portion of the net operating loss carryforwards, or research and development tax credit carryforwards before utilization. Further, until a study is completed by the Company and any limitation is known, no amounts are being presented as an uncertain tax position.The Company has evaluated the positive and negative evidence bearing upon its ability to realize the deferred tax assets. Management has considered the Company’s history of cumulative net losses incurred since inception and its lack of commercialization of any products or generation of any revenue from product sales since inception and has concluded that it is more likely than not that the Company will not realize the benefits of the deferred tax assets. Accordingly, a full valuation allowance has been established against the net deferred tax assets as of December 31, 2021 and 2020. Management reevaluates the positive and negative evidence at each reporting period.Changes in the valuation allowance for deferred tax assets during the years ended December 31, 2021 and 2020 related primarily to the increase in net operating loss carryforwards and research and development tax credit carryforwards and were as follows (in thousands):As of December 31, 2021, the Company had 0t recorded any amounts for unrecognized tax benefits. The Company’s policy is to record interest and penalties related to income taxes as part of its income tax provision. As of December 31, 2021, the Company had 0 accrued interest or penalties related to uncertain tax positions and no amounts had been recognized in the Company’s consolidated statements of operations. The Company files income tax returns as prescribed by the tax laws of the jurisdictions in which it operates. In the normal course of business, the Company isgenerally remain subject to examination by federal and state jurisdictions, where applicable. There are currently no pending tax examinations. The Company is open to future tax examination under statute from 2017 to the present; however, carryforward attributes that were generated prior to December 31, 2017 may still be adjusted upon examination by federal, state or local tax authorities if they either have been or will be used in a future period.As part of Congress’ response to the COVID-19 pandemic, the Families First Coronavirus Response Act, or FFCR Act, was enacted on March 18, 2020, and the Coronavirus Aid, Relief, and Economic Security Act, or CARES Act, was enacted on March 27, 2020. COVID-19 relief provisions were also included in the Consolidated Appropriations Act, 2021, or CAA, which was enacted on December 27, 2020. The FFCR Act, the CARES Act, and the CAA contain numerous tax provisions. In particular, the CARES Act retroactively and temporarily (for taxable years beginning before January 1, 2021) suspends application of the 80%-of-income limitation on the use of net operating losses, which was enacted as part of the TCJA. It also provides that net operating losses arising in any taxable year beginning after December 31, 2017, and before January 1, 2021, are generally eligible to be carried back up to five years. The CARES Act also temporarily (for taxable years beginning in 2019 or 2020) relaxes the limitation on the tax deductibility of net interest expense by increasing the limitation from 30% to 50% of adjusted taxable income.authorities.12315.14. Net Loss Per ShareBasicThe following table summarizes the computation of basic and diluted net loss per share was calculated as follows (in thousands, except share and per share amounts):share:presented based on amounts outstanding at each period end, have beenwere excluded from the calculationcomputation of diluted net loss per share as of the periods presented because including them would have had an anti-dilutive impact:been antidilutive:15. LeasesThe Company leased office and laboratory facilities in Cambridge, Massachusetts (the “Old Premises”) from an investor inunder a noncancelable operating lease that began in April 2015issued 123,000 shares of its common stock to members of the Company’s executive management and expired in March 2020. In February 2020,10,000 shares to directors of the Company, amendedupon vesting of restricted stock units.leaseyear ended December 31, 2023, the Company issued 3,000 shares of its common stock to members of the Company’s executive management and 64,000 shares to a director of the Company, upon exercise of outstanding warrants. During the year ended December 31, 2022, the Company issued 43,000 shares of its common stock to a director of the Company, upon exercise of outstanding warrants.Old PremisesCompany’s non-voting common stock to extend the lease expiration to April 30, 2020. The amendment was accounted for assame related parties in connection with such Private Placement (see Note 9). Two of the related parties are members of the Company’s board of directors and two are members of the Company’s senior management team.lease reassessment andprevious member of the right-of-use asset and lease liability were remeasuredCompany’s board of directors, by issuing 335,000 shares of the Company’s non-voting common stock at the reassessment dateconversion price of February $0.995 (see Note 6). As of December 31, 2023 and 2022, the Company had no outstanding principal for its 2022 convertible notes with related parties.resultingConvertible Notesan increaseoutstanding principal and accrued interest, held by two members of the Company’s board of directors, by issuing 139,000 shares of the Company’s non-voting common stock at the conversion price of $0.995 (see Note 6). As of December 31, 2023 and 2022, the Company had no outstanding principal for its 2020 convertible notes with related parties.to the right-of-use asset for prepaid rent and a reduction of $0.1 million to the lease liability. In May 2020, the Company amended the lease for the Old Premises to extend the lease expiration to May 23, 2020 and recognized the final rent payment of less than $0.1 million in expense.In February 2020, the Company entered into a license agreement with a third party for the use of office and laboratory space in Boston, Massachusetts, commencing in May 2020 (the “New Premises”)loss upon extinguishment (see Note 6). The Company determined that this license agreement qualified as a lease under ASC 842, Leases (“ASC 842”). The initial term of the license agreement is three years with the option to extend for a total of three one-year periods at fair-market rent at the time of each extension. In addition to use of office and laboratory space, the license fee includes various laboratory, office, and operational support services to be provided by the licensor. The initial monthly license fee escalates 3% annually and totals approximately $12.0 million for the three-year term. Additionally, the licensee agreement for the New Premises requires the Company to pay for a non-exclusive, irrevocable license to use forty-two unreserved parking spaces adjacent to the New Premises at the prevailing monthly parking rate. On May 1, 2020, the lease commencement date was met and the Company recorded an operating lease asset of $10.6 million and a corresponding lease liability of $10.2 million.On December 22, 2020, as part of the Merger, the Company acquired a lease for approximately 30,000 square feet of office and laboratory space in Boston, Massachusetts (the "Merger Premises"). The lease commenced in January 2018 with rent payments commencing in April 2018. The initial term of the lease was ten years with the option to extend for an additional seven years at fair-market rent at the time of the extension. In addition to use of office and laboratory space, the Company is responsible for paying its allocable portion of building and laboratory operating expenses separately from rent, based on actual costs incurred. Remaining fixed lease payments at the time of the Merger were approximately $14.2 million. On December 22, 2020, the Company recorded an operating lease asset and corresponding lease liability of $10.2 million associated with this lease. The operating lease asset was increased by the value attributable to the below-market lease of $3.1 million and an allocated portion of the excess purchase price for the Merger of $1.9 million.On January 7, 2021 the Company entered into a sublease agreement (the “Sublease”) with Moma Therapeutics, Inc. (the “Subtenant”), whereby the Company subleased the entire Merger Premises to the Subtenant. The initial term of the Sublease commenced on February 3, 2021, which was the date the Company received consent to the Sublease from the landlord, and shall continue until 18 months from the commencement date.124
The Sublease provides for an initial annual base rent of $1.9 million, which increases annually up to a maximum annual base rent of $2.0 million. The Subtenant also is responsible for paying to the Company operating costs, annual tax costs and all utility costs attributable to the Premises during the term of the Sublease. Expense arising from the Merger Premises of $1.9 million for the twelve months ended December 31, 2021 and lease income from the Sublease of $1.8 million for the twelve months ended December 31, 2021 are classified in operating expense on a net basis.The Company also leases property and equipment under agreements that are accounted for as finance leases.The components of lease cost were as follows (in thousands):Supplemental disclosure of cash flow information related to leases was as follows (in thousands):
(operating cash flows)
cash flows)
cash flows)The weighted-average remaining lease term and discount rate were as follows:Because the interest rates implicit in the license agreement and lease agreement assumed from PTI were not readily determinable, the Company’s incremental borrowing rate was used to calculate the present value of each. The present value of the Company’s finance leases was calculated using the rate implicit in the lease.1252021, future annual lease payments under2023 and 2022, the Company’s real estate operating leases and equipment finance leases were as follows (in thousands):Company had no outstanding principal for its 2020 notes with related parties.126The following table presents lease assetsAs described above, the Company initiated a process to explore a range of strategic alternatives to maximize stockholder value and liabilities and their classification onhas engaged professional advisors. Management can make no assurances that any particular course of action, business arrangement or transaction, or series of transactions, will be pursued, successfully consummated or lead to increased stockholder value. If the consolidated balance sheet (in thousands):strategic process is unsuccessful, the Company’s board of directors may decide to pursue a liquidation or obtain relief under the US Bankruptcy Code.16. Commitments and ContingenciesLicense agreementThe Company has a tangible property and patent license agreement with Whitehead Institute for Biomedical Research (“Whitehead”) entered into in 2016 and subsequently amended in 2016 and 2018, under which the Company obtained a certain exclusive and non-exclusive, royalty-bearing, sublicensable, worldwide license to make, sell and distribute products under certain patents owned by Whitehead for certain know-how related to specific neurodegenerative diseases. In consideration for the rights granted by the agreement, the Company paid a one-time license fee of less than $0.1 million and issued 300,000 common units valued at $0.8 million. The Company is required to pay annual maintenance fees of up to $0.1 million through the termination of the agreement. The Company is also required to pay up to an aggregate of approximately $1.9 million upon the achievement of certain developmental and regulatory milestones for the first two licensed products under its first indication. The Company is also required to pay additional milestone amounts for subsequent licensed products under its first or subsequent indications, but at a lower rate. The Company did not meet any milestones for the years ended December 31, 2021 or 2020. The Company must also pay a royalty in the low single digits on future sales by the Company and a mid single to low double digit percentage of certain income received from sublicensees and certain partners. The license agreement remains in effect until the expiration of the last-to-expire patent licensed under the agreement. Whitehead may terminate the agreement upon the Company’s uncured material breach of the agreement, including failure to make required payments under the agreement or to achieve certain milestones, or if the Company becomes insolvent or bankrupt. The Company may terminate the license agreement at any time upon providing certain written notice to Whitehead.Contingent Value Rights AgreementMerger,Company’s decision to explore strategic alternatives, the Company entered intoimplemented a Contingent Value Rights Agreement (the “CVR Agreement”) with Shareholder Representative Services LLC as representativeworkforce reduction of the PTI stockholders. The CVR Agreement entitled each holder of Company Common Stock as of immediately prior to the effective time of the Merger (the “Effective Time”) to receive certain net proceeds, if any, derived from the grant, sale or transfer of rights of the Cystic Fibrosis Assets ( the “CF Assets”) to any one of three specified counterparties completed during the 9-month period after the Effective Time (with any potential payment obligations continuing until the 10-year anniversary of the closing of the Merger Agreement). The CVR agreement became effective at Closing of the Merger. Due to the fact that no CF Asset sale was completed by the nine-month anniversary of the Effective Time, the CVRs expired. NaN liability has been recorded at December 31, 2021 or previous periods associated with the CVRs.Indemnification agreementsIn the ordinary course of business, the Company may provide indemnification of varying scope and terms to vendors, lessors, contract research organizations, business partners and other parties with respect to certain matters including, but not limited to, losses arising out of breach of such agreements or from intellectual property infringement claims made by third parties. In addition, the Company has entered into indemnification agreements with members of its board of directors and its executive officers that will require the Company, among other things, to indemnify them against certain liabilities that may arise by reason of their status or127service as directors or officers. The maximum potential amount of future payments the Company could be required to make under these indemnification agreements is, in many cases, unlimited. The Company has not incurred any material costs as a result of such indemnifications and is not currently aware of any indemnification claims.Legal MattersBetween October 14 and December 7, 2020, following the announcement of the proposed merger among PTI, Yumanity, Inc. and Merger Sub, a wholly owned subsidiary of PTI, nine lawsuits were filed by purported stockholders of PTI challenging the Merger. The first lawsuit, brought as a putative class action, is captioned Aniello v. Proteostasis Therapeutics, Inc., et al., No. 1:20-cv-08578 (S.D.N.Y. filed Oct. 14, 2020). The remaining eight lawsuits, brought by the plaintiffs individually, are captioned Culver v. Proteostasis Therapeutics, Inc., et al., 1:20-cv-08595 (S.D.N.Y. filed Oct. 15, 2020); Donolo v. Proteostasis Therapeutics, Inc. et al., 1:20-cv-01400 (D. Del. filed Oct. 16, 2020); Straube v.Proteostasis Therapeutics, Inc., et al., 1:20-cv-08653 (S.D.N.Y. filed Oct. 16, 2020); Beck v. ProteostasisTherapeutics, Inc., et al., 1:20-cv-08783 (S.D.N.Y. filed Oct. 21, 2020); Dreyer v. Proteostasis Therapeutics, Inc., et al., 1:20-cv-05193 (E.D.N.Y. filed Oct. 28, 2020); Kopkin v. Proteostasis Therapeutics, Inc. et al., No. 1:20-cv-12103 (D. Mass. filed Nov. 23, 2020); Merritt v. Proteostasis Therapeutics, Inc., et al., No. 1:20-cv-10275 (S.D.N.Y. filed Dec. 6, 2020); and Koh v. Proteostasis Therapeutics, Inc., et al., No. 1:20-cv-10296 (S.D.N.Y. filed Dec. 7, 2020). All of the complaints named PTI and the individual members of PTI’s board of directors as defendants. The Aniello complaint also named Yumanity, Inc. as an additional defendant, and the Donolo complaint named Yumanity, Inc. and Merger Sub as additional defendants. The complaints asserted violations of Section 14(a) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and Rule 14a-9 promulgated thereunder against PTI and its directors, and violations of Section 20(a) of the Exchange Act against PTI’s directors. The Donolo complaint asserted an additional violation of Section 20(a) of the Exchange Act against Yumanity, Inc. The Aniello complaint asserted additional claims for breach of fiduciary duty against PTI’s directors and aiding and abetting against PTI and Yumanity, Inc.. The plaintiffs contended that the registration statement on Form S-4 filed by PTI with the Securities and Exchange Commission on September 23, 2020 (the “Registration Statement”) or the proxy statement/prospectus on Form 424B3 filed by PTI with the SEC on November 12, 2020 (the “Definitive Proxy”) omitted or misrepresented certain material information regarding the Merger. The complaints sought injunctive relief, rescission, or rescissory damages, dissemination certain information requested by the plaintiffs, and an award of plaintiffs’ costs, including attorneys’ fees and expenses. While PTI and Yumanity, Inc. believed that the disclosures set forth in the Registration Statement and Definitive Proxy complied fully with all applicable law and denied the allegations in the pending actions described above, in order to moot plaintiffs’ disclosure claims, avoid nuisance and possible expense and business delays, and provide additional information to its stockholders, on December 9, 2020, PTI filed a Form 8-K voluntarily to supplement certain disclosures in the Definitive Proxy related to plaintiffs’ claims with the supplemental disclosures (the “Supplemental Disclosures”). Following the filing of the Supplemental Disclosures, all of the actions discussed above were voluntarily dismissed by the respective plaintiffs. On March 18, 2021, the parties executed a confidential fee and settlement agreement, pursuant to which all claims were released by plaintiffs and their counsel and an immaterial payment of a mootness fee was paid to plaintiffs’ counsel, a portion of which was paid by the Company’s insurer.17. Defined Contribution PlanThe Company has a 401(k) defined contribution plan (the “401(k) Plan”) for its employees. Eligible employees may make pretax contributions to the 401(k) Plan up to statutory limits. To date, the Company has 0t made any contributions to the plan.18. Related PartiesThere were 0 related party transactions for the twelve months ended December 31, 2021. The Company leased certain office and laboratory space from an investor in the Company until May 2020 (see Note 15). Lease expense and amounts paid to the investor under the lease agreement during the twelve months ended December 31, 2020 was $0.4 million and $0.6 million, respectively. There were 0 amounts payable to the investor as of December 31, 2021 or 2020.19. Subsequent EventsOn February 17, 2022, the Company announced that it was reducing its workforce by seven full-time employees, or approximately 6064% of its current headcount with the objective of preserving capital. ThisCompany’s then-current employee base. The workforce reduction includes Kineta’s Chief Executive Officer, Shawn Iadonato, Ph.D., who will take place primarily duringcontinue to serve on the first quarterCompany’s Board of 2022. As a resultDirectors, and Kineta’s General Counsel and Secretary, Pauline Kenny. Each of these actions,Dr. Iadonato and Ms. Kenny will continue to support the Company expectsin a consulting capacity until December 31, 2024. In connection with this workforce reduction, the affected employees will be provided severance benefits, including cash severance payments. Each affected employee’s eligibility for these severance benefits is contingent upon such employee’s entering into an effective separation agreement, which includes a general release of claims against the Company. The reduction of workforce is expected to incur personnel-related restructuring charges ofresult in approximately $0.4246,000 million in connection with one-time employee terminationseverance costs. The incremental costs including severance and other benefits, which are expected to be incurred in the first quarter of 2022. In addition, the Company has committed to pay one-time employee retention costs to certain employees of up to $0.4 million, which are expected to be incurred through the second quarter of 2022.2024.On February 25, 2022, the Term Loan entered into with Hercules Capital, Inc. (“Hercules”) in December 2019 and most recently amended in April 2021 terminated upon the receipt by Hercules of a payoff amount of $12.8million from the Company. The128payoff amount paid by the Company included payment of $0.9 million as an end of term fee and $0.1 million as an interest/non-use fee.On February 28, 2022, the Company entered into two agreements that effectively amended the “New Premises” license agreement for laboratory space in Boston, Massachusetts (see Note 15, Leases). The first agreement terminated the existing license, due to expire in May of 2023, effective March 31, 2022. The second agreement, effective April 1, 2022, created a new license for approximately 20 percent of the space covered by the original license with an expiration date of December 31, 2022. The Company agreed to surrender to Licensor the full amount of both the security deposit and the last month’s license fee held by licensor pursuant to the agreement, totaling approximately $0.8 million, in consideration of the agreement to terminate the original license. The new license agreement decreases the monthly license fee amount from $0.4 million to $0.1 million.140129ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE141None.ITEMItem 9A. CONTROLS AND PROCEDURESControls and Procedures.Our management,Prior to completion of the Merger, we were a private company and had limited accounting and financial reporting personnel and other resources with which to address our internal controls and related procedures. In connection with the participationaudit of our Presidentfinancial statements for the years ended December 31, 2022 and Chief Executive Officerin connection with the review of our financial statements for the six months ended June 30, 2023, our management and our Chief Business Officer (our principal executive officerindependent registered public accounting firm identified material weaknesses in our internal control over financial reporting. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting as defined under the Securities Exchange Act of 1934, as amended (the “Exchange Act”) and principal financial officer, respectively)by the Public Company Accounting Oversight Board (United States), evaluated the effectivenesssuch that there is a reasonable possibility that a material misstatement of our disclosureannual or interim financial statements will not be prevented or detected on a timely basis. The material weaknesses for the year ended December 31, 2023 relate to accounting for complex financial instruments related to the derivative asset and for accounting for allocated facilities costs. The material weakness for the year ended December 31, 2022 relates to accounting for complex financial instruments related to warrants issued to certain existing stockholders. The material weaknesses are still present and have not been remediated.procedures as of December 31, 2021. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) underaddress the Exchange Act, means controlsmaterial weaknesses and other control deficiencies identified during the 2021 audit of the financial statements. We have designed and implemented improved processes and internal controls, including ongoing senior management review and audit committee oversight. We have also implemented and upgraded accounting and reporting systems to improve accounting and financial reporting processes. Additionally, we have enhanced, developed and implemented formal policies, processes and documentation procedures relating to our financial processes, including the oversight of a companythird-party service providers. Our actions are subject to ongoing executive management review and will also be subject to audit committee oversight.designed to ensure that information required to be disclosed by a companyfairly stated in all material respects in accordance with accounting principles generally accepted in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assuranceUnited States of achieving their objectives. Based on the evaluation of our disclosure controls and procedures as of December 31, 2021, our President and Chief Executive Officer and our Chief Business Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.America.adequateeffective internal control over financial reporting (as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f) under. The consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the Exchange Act).United States, and any deviations from such principles have been disclosed in the notes to the financial statements. Management is also responsible for assessing the effectiveness of internal control over financial reporting and for providing reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations,However, internal control over financial reporting may not prevent or detect misstatements. Also, projectionsall misstatements due to inherent limitations, including the possibility of anycollusion or improper management override of controls. effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2021. In making this assessment, it usedbased on the criteria establishedset forth in the Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on such assessment, ourthis evaluation, management has concludeddetermined that as of December 31, 2023, our internal control over financial reporting wasis not effective as of December 31, 2021.based on the material weaknesses identified above.NoExcept as disclosed above, there has been no change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act)that occurred during the three months ended December 31, 2021fourth quarter of 2023 that has materially affected, or is reasonably likely to materially affect, our internal control over current or future financial reporting.ITEMItem 9B. OTHER INFORMATIONOther Information.None.During the fiscal quarter ended December 31, 2023, none of our directors or officers informed us of the adoption, modification or termination of a “Rule 10b5-1 trading arrangement” or “non-Rule 10b5-1 trading arrangement,” as those terms are defined in Regulation S-K, Item 408.ITEMItem 9C. Disclosure Regarding Foreign Jurisdictions that Prevent InspectionsInspections.130ITEMItem 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCEDirectors, Executive Officers and Corporate Governance.20222024 Annual Meeting of Stockholders and is incorporated herein by reference.ITEMItem 11. EXECUTIVE COMPENSATIONExecutive Compensation.20222024 Annual Meeting of Stockholders and is incorporated herein by reference.ITEMItem 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERSSecurity Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.20222024 Annual Meeting of Stockholders and is incorporated herein by reference.ITEMItem 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCECertain Relationships and Related Transactions, and Director Independence.20222024 Annual Meeting of Stockholders and is incorporated herein by reference.ITEMItem 14. PRINCIPAL ACCOUNTING FEES AND SERVICESPrincipal Accounting Fees and Services.20222024 Annual Meeting of Stockholders and is incorporated herein by reference.131ITEMItem 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULESExhibits, Financial Statement Schedules.117110 of this Annual Report on Form 10-K, incorporated into this Item by reference.ITEMItem 16. FORMForm 10-K SUMMARYSummaryapplicable.provided.Exhibit
NumberDescription 2.1++ 2.22.1++First Amendment to 3.2 3.3 3.43.5 4.14.24.2*132 10.4# 10.5# 10.6# 10.7 10.8 10.9+ 10.10+ 10.11 10.12 10.134.4 10.144.510 10.15# 10.16# 10.17# 10.18# 10.19# 10.20#133 10.21 10.22 10.23 10.2410.5 10.25 10.26 10.27 10.29 10.30 23.1*PricewaterhouseCoopersMarcum LLP, independent registered public accounting firm. 31.1* 31.2* 32.1**134101.INS*97.1*101.SCH*101.CAL*101.INS101.DEF*101.LAB*101.PRE*104*with applicable taxonomy extension informationand contained in ExhibitsExhibit 101).in accordance with the rules of the Securities and Exchange Commission.# Indicates a management contract or any compensatory plan, contract or arrangement.** The certifications furnished in Exhibit 32.1 hereto are deemed to accompany this Annual Report on Form 10-K and will not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. Such certifications will not be deemed to be incorporated by reference into any filings under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, except to the extent that the Registrant specifically incorporates it by reference.135registrantRegistrant has duly caused this reportReport to be signed on its behalf by the undersigned, thereunto duly authorized.authorized.Yumanity Therapeutics, Inc.Date: March 24, 2022By:/s/ Richard PetersRichard PetersPresident, Chief Executive Officer andPrincipal Executive OfficerPursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.Signature/s/ Scott DyllaTitleDateDirectorRichard Peters Marion R. Foote President, Chief Executive Officer
(Principal Executive Officer)24, 202221, 2024/s/ Michael D. WyzgaSenior Vice President and Chief Financial Officer(Principal Financial Officer)March 24, 2022Michael D. Wyzga/s/ Marie EpsteinVice President, Finance(Principal Accounting Officer)March 24, 2022Marie Epstein/s/ N. Anthony ColesDirectorMarch 24, 2022N. Anthony Coles/s/ Patricia L. AllenDirectorMarch 24, 2022Patricia L. Allen/s/ David ArkowitzDirectorMarch 24, 2022David Arkowitz/s/ Kim C. DrapkinDirectorMarch 24, 2022Kim C. Drapkin/s/ Richard A. HeymanDirectorMarch 24, 2022Richard A. Heyman/s/ Jeffery W. KellyDirectorMarch 24, 2022Jeffery W. Kelly/s/ Cecil B. PickettDirectorMarch 24, 2022Cecil B. Pickett/s/ Lynne ZydowskyDirectorMarch 24, 2022Lynne Zydowsky136149