
t
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K10-K
(Mark One)
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 20212023
or
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 001-36510
LARIMAR THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) |
| 20-3857670 (I.R.S. Employer Identification No.) |
|
|
|
Three Bala Plaza East, Suite 506 Bala Cynwyd, Pennsylvania (Address of principal executive offices) |
| 19004 (Zip Code) |
(844) 511-9056
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
Common Stock, par value $0.001 per share | LRMR | The Nasdaq Global Market |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐ Yes ☒ No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ☐ Yes ☒ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ |
| Accelerated filer | ☐ |
Non-accelerated filer | ☒ |
| Smaller reporting company | ☒ |
|
| Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
On the last business day of the most recently completed second fiscal quarter, the aggregate market value (based on the closing sale price of its common stock on that date) of the voting and non-voting stock held by non-affiliates of the registrant was $94.480,218,629 million..
As of March 23, 2022,12, 2024, the registrant had 17,710,45063,800,017 shares of the registrant’s Common Stock, $0.001 par value per share, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Part III of this Annual Report on Form 10-K incorporates certain information by reference from the registrant’s proxy statement for the 20222024 annual meeting of shareholders to be filed no later than 120 days after the end of the registrant’s fiscal year ended December 31, 2021.2023.
|
|
|
|
|
|
TABLE OF CONTENTS
Item No. |
| Page No. |
| ||
| ||
| ||
88 | ||
| ||
| ||
| ||
|
|
|
| ||
| ||
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
| |
| ||
| ||
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
| |
| ||
| ||
DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS |
| |
|
|
|
| ||
| ||
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
| |
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
| |
| ||
|
|
|
| ||
| ||
|
|
|
| ||
| ||
As previously disclosed, on May 28, 2020, Zafgen, Inc., a Delaware corporation (“Zafgen”), completed a Merger with Chondrial Therapeutics, Inc., a Delaware corporation (“Chondrial”), in accordance with the terms of the Agreement and Plan of Merger Reorganization (the “Merger Agreement”) entered into on December 17, 2019. Pursuant to the Merger Agreement, (i) a subsidiary of Zafgen merged with and into Chondrial, with Chondrial continuing as a wholly-owned subsidiary of Zafgen and the surviving corporation of the merger and (ii) Zafgen was renamed as “Larimar Therapeutics, Inc.” (the “Merger”).
For accounting purposes, the Merger is treated as a “reverse asset acquisition” under generally accepted accounting principles in the United States (“GAAP”) and Chondrial is considered the acquirer. Accordingly, Chondrial’s historical results of operations will replace the Company’s (as defined below) historical results of operations for all periods prior to the Merger and, for all periods following the Merger, the results of operations of the combined company will be included in the Company’s financial statements.
Unless the context otherwise requires, references to the “Company,” the “combined company,” “we,” “our” or “us” in this report refer to Larimar Therapeutics, Inc. and its subsidiaries, references to “Larimar” refer to the Company following the completion of the Merger and references to “Zafgen” refer to the Company prior to the completion of the Merger.
Except as otherwise noted, references to “common stock” in this report refer to common stock, $0.001 par value per share, of the Company.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Statements made in this Annual Report on Form 10-K that are not statements of historical or current facts are “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements discuss our business, operations and financial performance and conditions, as well as our plans, objectives and expectations for our business operations and financial performance and condition. In some cases, you can identify forward-looking statements by terminology such as “aim,” “anticipate,” “assume,” “believe,” “contemplate,” “continue,” “could,” “design,” “due,” “estimate,” “expect,” “goal,” “intend,” “may,” “objective,” “plan,” “predict,” “positioned,” “potential,” “seek,” “should,” “target,” “will,” “would” and other similar expressions that are predictions of or indicate future events and future trends, or the negative of these terms or other comparable terminology. In addition, statements that “we believe” or similar statements reflect our beliefs and opinions on the relevant subject only. These forward-looking statements, which are subject to risks, uncertainties and assumptions about us, may include projections of our future financial performance, our anticipated growth strategies and anticipated trends in our business.
You should understand that the following important factors could affect our future results and could cause those results or other outcomes to differ materially from those expressed or implied in our forward-looking statements:
2
These forward-looking statements are based on management’s current expectations, estimates, forecasts and projections about our business and the industry in which we operate, and management’s beliefs and assumptions are not guarantees of future performance or development and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. In light of the significant uncertainties in these forward-looking statements, you should not rely upon forward-looking statements as predictions of future events. Although we believe the expectations reflected in the forward-looking statements are reasonable, the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements may not be achieved or occur at all. Except as required by law, we undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise after the date of this Annual Report on Form 10-K or to reflect the occurrence of any unanticipated events. Comparisons of results for current and any prior periods are not intended to express any future trends or indications of future performance, unless expressed as such, and should only be viewed as historical data.
3
SUMMARY RISK FACTORS
The risk factors summarized and detailed below could materially harm our business, operating results and/or financial condition, and may impair our future prospects and/or cause the price of our common stock to decline. These are not all of the risks we face, and other factors not presently known to us or that we currently believe are immaterial may also affect our business if they occur. Material risks that may affect our business, operating results and financial condition include, but are not necessarily limited to, those relating to:
Risks Related to Our Financial Position and Need for Capital
Risks Related to Our Product Development and Regulatory Approvals
4
4
Risks Related to Our Business
Risks Related to Our Reliance on Third Parties
Risks Related to Our Intellectual Property Rights
5
Risks Related to Our Common Stock
65
PART I
ITEM 1. BUSINESS
Overview
We are a clinical-stage biotechnology company focused on developing treatments for patients suffering from complex rare diseases using our novel cell penetrating peptide ("CPP") technology platform. Our lead product candidate, CTI-1601,nomlabofusp (nomlabofusp is the International Nonproprietary Name ("INN") and the United States Adopted Name ("USAN") for CTI-1601), is a subcutaneously administered, recombinant fusion protein intended to deliver humantissue frataxin ("FXN"), an essential protein, to the mitochondria of patients with Friedreich’s ataxia. Friedreich’sFriedreich's ataxia (“FA”). FA is a rare, progressive, and fatal disease in which patients are unable to produce sufficient FXN due to a genetic abnormality. There is currentlyCurrently, there are no approvedtreatment options that address the core deficit of FA, low levels of FXN. Nomlabofusp represents the first potential therapy for Friedreich’s ataxia. We have completed two Phase 1 clinical trialsdesigned to increase FXN levels in patients with Friedreich’s ataxia but the clinical development of CTI-1601 is currently subject to a clinical hold issued by the U.S. Food and Drug Administration ("FDA") in May 2021. We submitted a response to the clinical hold in January 2022 but were informed by the FDA in February 2022 that the clinical hold would remain in place and that additional data is needed to resolve the clinical hold. We are evaluating how best to respond to their request. We do not know when, or if, the clinical hold will be lifted. We have received orphan drug status, fast track designation and rare pediatric disease designation, from the FDA for CTI-1601. In addition, we received orphan drug and Priority Medicines ("PRIME") designations for CTI-1601 from the European Commission. The receipt of such designations or positive opinions may not result in a faster development process, review or approval compared to products considered for approval under conventional FDA or European Medicines Agency ("EMA") procedures and does not assure ultimate approval by the FDA or EMA.FA.
We believe that our cell penetrating peptide ("CPP") technologyCPP platform, which enables a therapeutic molecule to cross a cell membrane in order to reach intracellular targets, has the potential to enable the treatment of other rare and orphan diseases. We intend to use our proprietary platform to target additional orphan indications characterized by deficiencies in or alterations of intracellular content or activity.
Since our inception, we have devoted substantially all of our resources to developing nomlabofusp, building our intellectual property portfolio, developing third-party manufacturing capabilities, business planning, raising capital, and providing general and administrative support for such operations.
As of December 31, 2023, we had cash, cash equivalents and marketable securities of $86.8 million. In February 2024,we raised net proceeds of $161.6 million in an underwritten public offering of common stock, We anticipate the $161.6 million in net proceeds, together with $86.8 million of cash, cash equivalents and marketable securities on hand will fund operations into 2026.
Nomlabofusp Program Update
Clinical Trials
We have assembled an experienced management team, eachcompleted two Phase 1 clinical trials and a Phase 2 dose exploration trial, and recently initiated a Phase 2 OLE trial in patients with FA.
In May 2021, we reported positive top-line data from our Phase 1 FA program after completing dosing of whom has over 20 yearsthe single ascending dose ("SAD") trial in December 2020 and of pharmaceutical industry experience. Our management teamthe multiple ascending dose ("MAD") trial in March 2021. Data from these trials demonstrated proof-of-concept by showing that daily subcutaneous injections of nomlabofusp for up to 13 days resulted in dose-dependent increases in FXN levels from baseline compared to placebo in all evaluated tissues (buccal cells, skin, and consultants have significant expertiseplatelets). There were no serious adverse events associated with either the MAD or SAD trials.
In May 2023, we reported preliminary unblinded top-line data from the 25 mg cohort of our Phase 2 four-week, placebo-controlled, dose exploration trial of nomlabofusp in discovery, non-clinicalFA patients. Data from the cohort indicated nomlabofusp was generally well tolerated and showed increases in FXN levels from baseline compared to placebo in all evaluated tissues (skin and buccal cells) at day 14.
In July 2023, following the FDA’s review of our complete response to the partial clinical development, regulatory affairshold, the FDA cleared initiation of a second cohort (50 mg) of our four-week, placebo-controlled, Phase 2 dose exploration trial of nomlabofusp in patients and the developmentinitiation of manufacturing processes utilizing good manufacturing practices (GMPs") for biologicsour OLE trial with daily dosing of 25 mg.
In February 2024, we reported positive top-line data and small molecules. We believe thatsuccessful completion of our management team’s diverse mixfour-week, placebo-controlled Phase 2 dose exploration study of skills providesnomlabofusp in participants with FA. Nomlabofusp was generally well tolerated and demonstrated dose dependent increases in FXN levels in all evaluated tissues (skin and buccal cells) after daily dosing of 14 days followed by every other day dosing until day 28 in the 25 mg and 50 mg cohorts. Participants in the 25 mg (n=13) and 50 mg (n=15) cohorts were randomized 2:1 to receive subcutaneous injections of nomlabofusp or placebo. Participants treated with 50 mg had individual frataxin levels in skin increase from less than 17% relative to healthy volunteers at baseline, to 33% to 59% of healthy volunteers after 14 days of treatment. Patients treated with placebo showed no increase in their frataxin levels during this period. Additionally, the majority of treated patients in both cohorts with quantifiable levels of frataxin at baseline and day 14 achieve at least
6
a 100% increase in tissue frataxin in skin cells, and at least a 30% increase in tissue frataxin levels in buccal cells. With both skin and buccal cells, when dosing is switched to every other day the magnitude of the increase declines.
In January 2024, we initiated the OLE trial evaluating daily subcutaneous injections of 25 mg of nomlabofusp self-administered or administered by a caregiver, with the first patient dosed in March 2024. Participants who completed treatment in the Phase 2 dose exploration trial, or who previously completed a prior clinical trial of nomlabofusp are potentially eligible to screen for the implementationOLE. The OLE will evaluate safety, tolerability, pharmacokinetics ("PK") and measures of effective approachesfrataxin levels in peripheral tissues as well as other exploratory PD (lipid profiles and gene expression data) following long-term subcutaneous administration of nomlabofusp. Clinical measures collected during the trial will be compared to drugdata from a matched control arm derived from participants in the Friedreich’s Ataxia Clinical Outcome Measures Study (FACOMS) database. Dose escalation in the OLE trial will be considered based on safety, tolerability, PK, and biologic development.tissue FXN levels from the Phase 2 trial’s 50 mg cohort as well as available data from the 25 mg dose of nomlabofusp in the OLE trial, and is contingent on FDA review as part of the partial clinical hold. Initial data from the OLE trial is expected in the fourth quarter of 2024.
Our Strategy
Our strategy is to become a leader in the treatment of rare diseases by leveraging our technology platform and applying our management team’s know how and expertise to the development of CTI-1601nomlabofusp and other future pipeline programs. Key elements of our strategy include:
7
7
Platform Technology for Treatment of Rare Genetic Diseases
There are estimated to be between 5,000 and 7,000over 9,500 rare genetic diseases, which, collectively affect hundreds of millions of people worldwide. Of the hundreds of millions of individuals suffering from these rare genetic diseases, only approximately 5% have therapeutic options available to manage their disease. Many of these diseases result from a deficiency in the amount or the function of a particular target molecule, often a protein. Particularly challenging to treat are those diseases that result from the deficiency of a molecule that is active within a cell or within a cell-based organelle. The challenge to providing treatment of these diseases is the need to improve the amount or function of the therapeutic target by transporting a therapeutic element across the cell membrane and potentially the membrane of the organelle where the target is active in the diseased patients.
The ability to transport therapeutic proteins across biological membranes has, to date, not been reproducibly achieved. The collective population of people with rare diseases stands to benefit from the emergence of a scalable treatment platform that can transport therapeutic proteins across cell membranes to deliver them to the intracellular site of activity. In addition, traditionally, medical treatment for each rare genetic disorder has been approached on a disease-by-disease basis. This approach is inefficient, as there are thousands of diseases, each with a distinct patient population, that cannot be addressed by traditional therapeutic approaches and are in need of treatment options. Our understanding of our therapeutics derived from proprietary gene expression data across several disease models supports the concept that product candidates based on our platform technology could significantly impact common pathological mechanisms in various diseases with comparable etiologies. We are utilizing this approach to identify therapeutic opportunities where our molecules and technology are more likely to be impactful.
8
Lead Product Candidate—CTI-1601
Nomlabofusp For the Treatment of Friedreich’s ataxia
Friedreich’s ataxia
Friedreich’s ataxia is a rare genetic disease that is the most commonly inherited ataxia in humans, with approximately 20,000 individuals living with Friedreich’s ataxia globally, and of these individuals, approximately 5,000 are in the United States and the majority of the remaining individuals are primarily in the European Union.Europe. Friedreich’s ataxia results from a deficiency of the mitochondrial protein, FXN. FXN is an essential and phylogenetically conserved protein that is found in cells throughout the body, with the highest levels found in the heart, spinal cord, liver, pancreas, and skeletal muscle. FXN is encoded in the nucleus of the cell, expressed in the cytoplasm and transported into the mitochondria, where it is processed to the mature form. As part of this process the mitochondrial targeting sequence is cleaved off in the mitochondria by a naturally occurring enzyme.
Friedreich’s ataxia is a progressive multi-symptom disease typically presenting in mid-childhood that affects the functioning of multiple organs and systems. It is a debilitating neurodegenerative disease that results in poor coordination of legs and arms, progressive loss of the ability to walk, generalized weakness, loss of sensation, scoliosis, diabetes and cardiomyopathy as well as impaired vision, hearing and speech. Patients suffer from progressive neurologic and cardiac dysfunction. Key among these is a primary neurodegeneration of the dorsal root ganglia and the dentate nucleus of the cerebellum, which leads to the hallmark clinical findings of progressive limb ataxia and dysarthria. A hypertrophic cardiomyopathy is common and associated with early mortality, typically between 30 and 50 years of age. As of February 2022, there are no medical treatment optionsOmaveloxolone, the first drug approved for the treatment of Friedreich’s ataxia.FA, was approved by the FDA and the European Commission in February 2023 and February 2024, respectively.
CTI-1601Nomlabofusp
CTI-1601, aNomlabofusp, an investigational biologic fusion protein that is administered subcutaneously, consists of a CPP genetically fused to human FXN, and includes a mitochondrial targeting sequence. Using our proprietary peptide delivery technology, CTI-1601 carriesnomlabofusp is designed to carry the molecule from the intravascular space across the cell membrane and into the mitochondria where the CPP and the mitochondrial targeting sequence are cleaved off to yield mature FXN. See Figure 1.
Figure 1.
We have completed two Phase 1 CTI-1601nomlabofusp clinical trials. Clinical developmenttrials, and a Phase 2 dose exploration trial and have initiated an OLE study in the U.S with patients to be dosed 25 mg of CTI-1601 is currently subject tonomlabofusp daily. The nomlabofusp program remains under a partial clinical hold issuedimposed by the FDA, in May 2021.and expansion of the OLE study above the 25 mg dose level as well as further clinical development of nomlabofusp is contingent upon FDA agreement based on its review of data from the 50 mg cohort of the Phase 2 dose exploration study and review of the available data from the OLE study at the current 25 mg dose level. Based on the results of our non-clinical development program as well as results from our Phase 12 clinical trials, we believe that administering CTI-1601nomlabofusp may increase FXN levels in the mitochondria of patients with Friedreich’s ataxia and patients could potentially experience:
9
In our Phase 1 clinical trials, CTI-1601nomlabofusp appeared to increase FXN levels in the peripheral tissues that were tested (buccal cells, skin biopsies and platelets). and data from our Phase 2 dose exploration study demonstrated dose dependent increases in FXN levels in all evaluated tissues (skin and buccal cells) after daily dosing of 14 days followed by every other day dosing until day 28 in the 25 mg and 50 mg cohorts. Based on this, we believe that our technology may allow us to address other rare genetic diseases that either require the replacement of molecules that need to target specific intracellular organelles, or that share similar clinical symptoms that overlap with Friedreich’s ataxia. Finally, the use of CTI-1601nomlabofusp to improve mitochondrial function in other rare diseases that demonstrate evidence of mitochondrial dysfunction is also being explored.
Development of CTI-1601Nomlabofusp
Non-clinical Development
Knock-Out Mice and Other Non-clinical Studies
CTI-1601Nomlabofusp has been demonstrated to prolong the life of knock-out mice or KO("KO") mice whose heart and skeletal muscles were deficient in FXN. These mice, when untreated, develop a severe hypertrophic cardiomyopathy similar to patients with Friedreich’s ataxia and, like many Friedreich’s ataxia patients, die early in life. In non-clinical studies of CTI-1601,nomlabofusp, the median survival in animals treated with vehicle of 98 days was extended to a median survival of 166 days in animals treated with CTI-1601nomlabofusp subcutaneously three times per week (p=0.0001). Furthermore, 87.5% of mice treated with CTI-1601nomlabofusp survived beyond the mean age of death in the vehicle treated group (107.5 days) whereas only 33% of vehicle treated animals survived. Results are reflected in Figure 2.
Figure 2.

10
In a separate study conducted at an independent laboratory, a similar mouse model was studied. In this study doses of 2 mg/kg, 10 mg/kg, 30 mg/kg, 60 mg/kg and 100 mg/kg administered subcutaneously every other day were compared to vehicle. After 2 weeks of dosing, mitochondrial extracts from cardiac tissue were analyzed for the presence of human FXN. In addition, activity of succinate dehydrogenase ("SDH") an enzyme whose activity is dependent on the presence of FXN, was also analyzed. Human FXN was found in the mitochondria of the cardiomyocytes and increased with increasing dose. SDH activity which was suppressed to near zero in vehicle treated animals was also suppressed to near zero in the 2 mg/kg dose group. In the 10 mg/kg dose group activity was increased and in the 30 mg/kg dose group the activity was returned to that of wild type animals. There was no further increase in activity when the animals were dosed with 60 mg/kg or 100 mg/kg but the effect was maintained at levels equivalent to that of wild type animals. See Figures 3 and 4.
10
Figure 3.

Figure 4.

11
Another study, also performed at an independent laboratory, demonstrated the maintenance of cardiac function when the same KO mouse model was studied. These mice were treated with CTI-1601nomlabofusp at doses of 10 mg/kg every other day for 6 weeks. Echocardiograms were performed prior to initiating dosing and after 4 weeks of dosing. When compared to vehicle, mice treated with CTI-1601nomlabofusp maintained their left ventricular volume and ejection fraction while vehicle treated mice deteriorated over the same 4-week period. See Figures 5 and 6.
Figure 5.

11
Figure 6.

Using a neurologic mouse model, treatment with CTI-1601nomlabofusp prevented the development of ataxia in mice whose nervous system was deficient in FXN compared to those treated with placebo.
In multiple non-clinical studies in rodents and non-human primates ("NHPs"), human FXN was found to be distributed into all tissues tested following CTI-1601nomlabofusp dosing. See Figure 7.
Figure 7.
|
|
|
|
|
|
|
|
|
|
|
Observed hFXN across all tissue and cell types tested: | ||||||||||
|
|
|
|
|
| |||||
✓ |
| Brain |
| ✓ |
| Spinal Cord |
| ✓ |
| Skin |
|
|
|
|
|
| |||||
✓ |
| Heart |
| ✓ |
| Cardiac Mitochondria |
| ✓ |
| Buccal Cells |
|
|
|
|
|
| |||||
✓ |
| Liver |
| ✓ |
| CSF (Cerebrospinal Fluid) |
| ✓ |
| Platelets |
|
|
|
|
|
| |||||
✓ |
| Dorsal Root Ganglia |
| ✓ |
| Skeletal Muscle |
|
|
|
|
12
| ||||
|
|
| ||
|
|
| ||
|
|
| ||
|
|
| ||
|
|
| ||
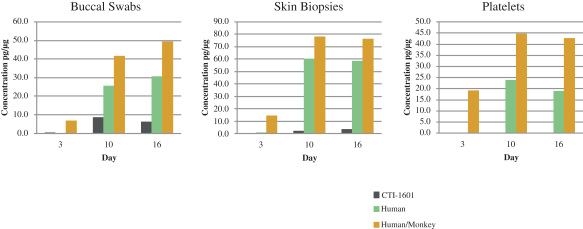
Since CTI-1601 is intended to increase FXN levels in patients with Friedreich’s ataxia who are deficient in FXN, it is important to be able to measure changes in human FXN in the clinic in easily accessible tissues. To accomplish this, we have developed a proprietary assay that can measure and quantify human FXN in buccal swabs, skin biopsies and platelets. This process is designed to allow repeated analysis of changes in human FXN in patients over time as they are dosed with CTI-1601. The effectiveness of this assay was first demonstrated by its ability to detect the presence of human FXN in those tissues in cynomolgus monkeys. In an exploratory study, NHPs were dosed for 14 days with 15 mg/kg twice a day of CTI-1601. Buccal swabs skin biopsies and platelets were obtained on Day 3 prior to administration of any CTI-1601 but after two days of dosing with vehicle. No human FXN was found. Since these were healthy NHPs there were levels of endogenous monkey FXN present. This is demonstrated by the yellow bars in the graph in Figure 8 below on Day 3. After 7 and 14 days of dosing with CTI-1601 human FXN was seen in significant amounts in all tissues analyzed. This is demonstrated by the appearance of the green bars on Day 10 and 16 in the figure below. This study demonstrates that CTI-1601 delivers human FXN to NHPs when administered subcutaneously and that we should be able to use our proprietary assay to evaluate changes in FXN levels in patients with Friedreich’s ataxia as CTI-1601 enters early-stage clinical trials.
Figure 8.
Non-Human Primate and Rat Toxicology Studies
We previously conducted 28-day and 13-week GLP toxicology studies of CTI-1601nomlabofusp in two species, rat and NHP. In the rat studies, some injection sites showed edema and erythema with associated histologic changes localized to the injection site. The rat studies showed no significant clinical observations and no significant systemic histopathological findings. In the NHP studies, some injection sites were raised and firm with dose dependent histologic changes localized to the injection sites. The NHP 28-day study showed no systemic toxicity. The NHP 13-week study showed no systemic toxicity in the low and mid-dose groups, and minimal to mild histopathological findings in the high dose group. There were also several episodes of occasional transient muscle rigidity in some animals observed immediately after dosing in the two highest dose groups at very high exposures. These clinical observations resolved with no intervention and all of the NHPs who experienced these clinical observations received all doses and completed the in-life portion of the study.
13
To support extended dosing of patients with CTI-1601,nomlabofusp, we conducted a 26-week NHP toxicology study in 2021. In May 2021, we notified the FDA of certain mortalities which occurred at the two highest dose levels in the then-ongoing study. On May 25, 2021, the FDA placed a clinical hold on the CTI-1601nomlabofusp clinical program. In the clinical hold letter, the FDA stated that it needed to review a full study report from the then-ongoing NHP study and that we could not initiate additional interventional clinical trials until we submitted such report and received notification from the FDA that additional clinical trials could commence. At the time of the FDA clinical hold, we had no interventional clinical trials with patients enrolled or enrolling.
In July 2021, we completed dosing in the 26-week NHP toxicology study. The study included four dose groups in addition to vehicle. Data from the study were collected throughout the second half of 2021 and included in the complete response to the clinical hold submitted to the FDA in January 2022.
In February 2022, we received feedback from the FDA following our submission ofin response to the complete response which included a comprehensive study report fromto the 26-week NHP toxicology study. Theclinical hold we submitted to the FDA, the FDA stated that it was maintaining itsthe clinical hold and that additional data were needed to resolve the clinical hold. We
12
subsequently submitted a request to the FDA for a Type C meeting, which was granted and held in July 2022. We submitted a complete response to clinical hold incorporating additional information requested by the FDA at this timethe meeting as well as information on the proposed study in August 2022.
In September 2022, following the Type C meeting and requested additional information. We are evaluating how best to respond to their request and also are reassessing the timingsubmission of the planned open label extensionCompany's complete response to clinical hold, the FDA allowed the 25 mg cohort of a Phase 2, four-week, placebo-controlled, dose exploration trial of nomlabofusp in FA patients discussed above to proceed. In connection with this decision, the FDA lifted its full clinical hold on the nomlabofusp clinical development program and imposed a partial clinical hold.
In June 2023, we met with the pediatric MAD studies.FDA. Following that meeting, we submitted a complete response to the FDA’s partial clinical hold that included unblinded safety, PK and frataxin data from the Phase 2 trial’s completed 25 mg cohort.
In July 2023, following the FDA’s review of our complete response to the partial clinical hold, the FDA cleared initiation of a second cohort at 50 mg of our four-week, placebo-controlled, Phase 2 dose exploration trial and initiation of our OLE trial with daily dosing of 25 mg.
Clinical Development
Non-Interventional Studies
We have conducted non-interventional studies to examine the range of tissue FXN concentrations in platelets, buccal cells and skin cells in individuals who are homozygous for the normal FXN gene. Tissue samples in these studies were collected using the same sampling techniques and proprietary assay used in the interventional studies. In a 2020 study we obtained data from 8 healthy adults, but the study was then terminated early by the study site due to competing priorities during the COVID-19 pandemic. In 2021 we initiated a second study that enrolled approximately 60 healthy adults. Enrollment in this study was completed in early 2022.2022 and data has been used for comparison purposes in the Phase 2 clinical study described below. See Figure 8.
14Figure 8.

FXN concentrations were measured in skin and buccal cells from 60 homozygous healthy volunteers utilizing the same sampling technique and assay as clinical trials of nomlabofusp; FXN levels measured via detection of peptide derived from mature FXN; FXN concentrations normalized to total cellular protein content in each sample. 1. E.C. Deutsch et al. Molecular Genetics and Metabolism 101 (2010) 238–245. 2. Friedreich’s Ataxia Research Alliance
Clinical Trials
Completed Initial Phase 1 Clinical Studies in Adults with Friedreich’s ataxiaAtaxia
We submitted our INDinvestigational New Drug application ("IND") application for CTI-1601nomlabofusp and began Phase 1 clinical trials in 2019 in patients with Friedreich’s ataxia. FA. We completed two clinical studies of CTI-1601nomlabofusp in adult subjects with Friedreich’s ataxia,FA, a single ascending dose ("SAD")SAD study (completed in 2020) and a multiple ascending dose ("MAD")MAD study (completed in 2021). The primary objective
13
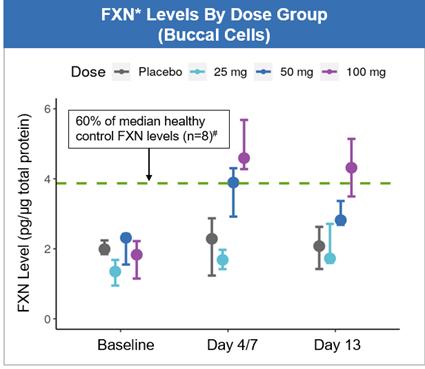
for the SAD and MAD studies was to assess the tolerability of CTI-1601nomlabofusp at doses ranging from 25 mg to 100 mg administered via subcutaneous injection ("SC") injection.. The secondary objectives were to establish the pharmacokinetics ("PK")PK of CTI-1601nomlabofusp administration in humans and to explore the pharmacodynamics ("PD")PD of CTI-1601nomlabofusp administration by measuring tissue FXN concentrations in accessible tissues, namely buccal and skin cells, and in platelets. Administration of CTI-1601nomlabofusp via SC injection appeared to be well tolerated up to doses of 100 mg administered daily for up to 13 days. No serious adverse events ("SAEs")SAEs were reported in either study. The most common treatment emergent adverse events ("TEAEs") were injection site reactions ("ISRs") that were generally mild, self-limited, and usually resolved within approximately 1 hour. There was rapid uptake of CTI-1601 into the circulation following SC injection, and exposure appears to be proportional to dose. Data from the MAD study demonstrated that after 7 days of daily dosing of 50 mg and 100 mg of CTI-1601 in subjects with Friedreich’s ataxia, mean tissue FXN concentrations increased in the tissues studied by approximately 2- to 3-fold to levels that are similar to or exceed levels we would expect to see in asymptomatic heterozygous carriers (See Figure 9). Patients in the 25 mg cohort were dosed once a day for the first 4 days followed by 1 dose every third day through Dayday 13 for a total of 7 doses, while the patients in the 50 mg cohort were dosed once a day for the first 7 days followed by 1 dose every other day through Dayday 13 for a total of 10 doses, and patients in the 100 mg cohort were dosed once a day for 13 days consecutively for a total of 13 doses. (See Figuredoses (see Figures 9 and 10). The study was designed in this fashion to ensure that the pharmacokineticsPK of CTI-1601nomlabofusp in patients with Friedreich’s ataxiaFA were characterized at the lower doses before the dose was escalated. This is typical of first in human studies such as multiple-ascending dose trials that evaluate a study drug’s clinical safety profile. We believe
There was rapid uptake of nomlabofusp into the data provide preliminary clinical evidence of the potential of CTI-1601 as a treatment for patients with Friedreich’s ataxiacirculation following subcutaneous injection, and warrant continued evaluation of the potential benefits of sustained treatment.exposure appears to be proportional to dose.
14
Figure 9.

15
Figure 10.
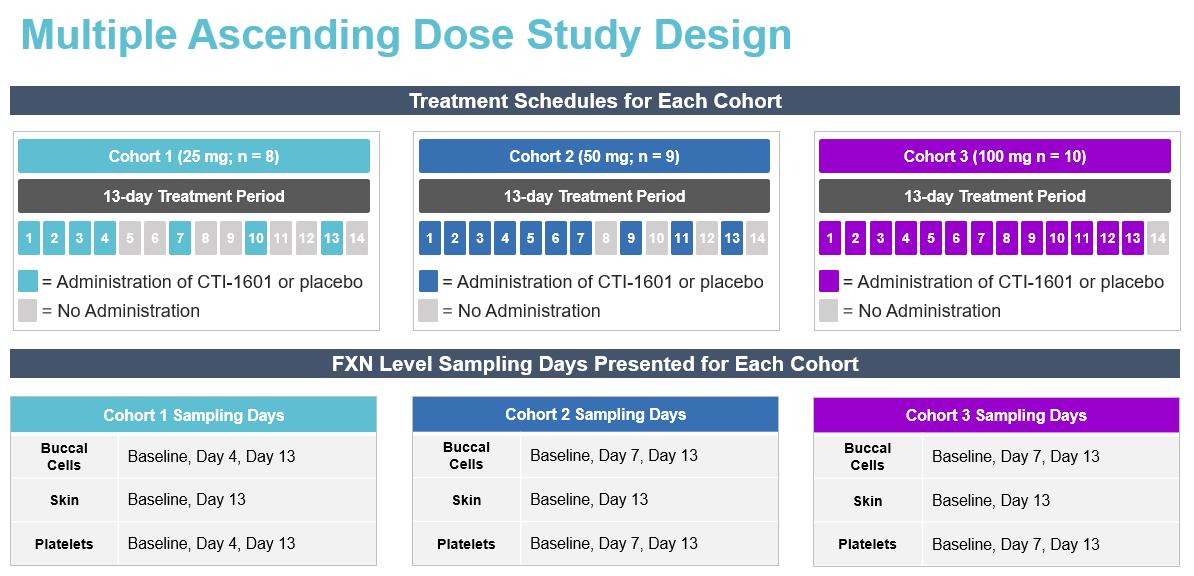

CTI-1601 Development Plan*FXN levels measured via detection of peptide derived from mature FXN; FXN concentrations are normalized to total cellular protein content in each sample; Data represent median and 25th and 75th percentiles; FXN levels from day 4, & day 13 measurements are shown for data derived from the 25 mg cohort; FXN levels from day 7 & day 13 measurements are shown for data derived from the 50 & 100 mg cohorts.
We are in
Completed Phase 2 Dose Exploration Study
The Phase 2 randomized, double-blind, placebo controlled dose exploration study evaluated the process of evaluating the timeline of the open label extension ("OLE") study which we refer to as the Jive study and the pediatric MAD placebo-controlled study. The Jive study is a clinical trial that will enroll patients who participated in the SAD or MAD study if they are eligible and is designed to gather long-term data to assess safety, and tolerability, dose, PK and PD of two doses of nomlabofusp. Cohort 1 evaluated 25 mg and potential efficacyCohort 2 evaluated 50 mg. Participants in either the 25 mg or 50 mg cohorts were randomized 2:1 to receive daily subcutaneous injections of SCeither nomlabofusp or placebo for 14 days, and then every other day for an additional 14 days for a total 28-day dosing period.
15
In May 2023, we reported preliminary unblinded top-line data from the 25 mg cohort of the Phase 2 trial. Data from the cohort indicated nomlabofusp was generally well tolerated and showed increases in FXN levels from baseline compared to placebo in all evaluated tissues (skin and buccal cells) at day 14.
In June 2023, we met with the FDA. Following that meeting, we submitted a complete response to the FDA’s partial clinical hold that included unblinded safety, PK and frataxin data from the Phase 2 trial’s completed 25 mg cohort.
In July 2023, following the FDA’s review of our complete response to the partial clinical hold, the FDA cleared initiation of the second cohort (50 mg) of our four-week, placebo-controlled, Phase 2 dose exploration trial and the initiation of our OLE trial with daily dosing of 25 mg.
In February 2024, we reported positive top-line data and successful completion of our four-week, placebo-controlled Phase 2 dose exploration study of nomlabofusp in participants with FA. Nomlabofusp was generally well tolerated and demonstrated dose response in FXN levels in all evaluated tissues (skin and buccal cells) after daily dosing of 14 days followed by every other day dosing until day 28 in the 25 mg and 50 mg cohorts.
With regard to safety in the Phase 2 dose exploration trial, there were a total of 28 participants (19 received nomlabofusp and 9 received placebo). There were no serious adverse events reported. There was one allergic reaction which was considered to be a severe adverse event in the 25 mg cohort that resolved with standard treatment. This participant withdrew from the trial. The most common adverse events reported were mild and moderate injection site reactions which resolved without any intervention and there were no study discontinuations due to injection site reactions. Consistent with findings in our Phase 1 study, nomlabofusp demonstrated quick absorption after subcutaneous administration and had dose proportional increases in exposure.
Participants treated with 50 mg for 14 days daily had frataxin levels in skin cells increase from less than 17% at baseline relative to healthy volunteers, to 33% to 59% of healthy volunteers after 14 days of treatment. Patients treated with placebo showed no increase in their frataxin levels during this period. Additionally, the majority of treated patients in both cohorts with quantifiable levels of frataxin at baseline and day 14 achieve at least a 100% increase in tissue frataxin in skin cells, and at least a 30% increase in tissue frataxin levels in buccal cells. Figures 11 and 14 show a clear dose response and increase from baseline in skin tissue and buccal tissue frataxin levels respectively.
The graph on the left in both Figures 12 and 15 show a dose dependent increase in frataxin levels after 14 days of treatment. With both skin and buccal cells, when dosing is switched to every other day the magnitude of the increase in frataxin levels declines.
The waterfall plots on the right in both Figures 12 and 15 depict the individual changes in frataxin levels from baseline in skin cells and buccal cells at day 14 respectively. Each bar represents an individual patient, with placebo treated participants in grey, the 25 mg cohort in aqua, and the 50 mg cohort in blue. The majority of patients treated with nomlabofusp had significant increases in frataxin levels while placebo patients were basically unchanged. Importantly, there also appears to be a dose response as frataxin levels are notably increased in most of the 50 mg cohort over the 25 mg cohort.
16
Figure 11.

Only participants with quantifiable levels at baseline and day 14 and day 28 are included in the tables.
Figure 12.

*FXN levels measured via detection of peptide derived from mature FXN; FXN concentrations are normalized to total cellular protein content in each sample. Data represent median and 25th and 75th percentiles. Only participants with quantifiable levels at both baseline and day 14 are included in the figures.
**Median baseline FXN levels in patients were 3.5 pg/µg for the placebo, 3.7 pg/µg for the 25 mg cohort and 2.1 pg/µg for the 50 mg cohort.
Figures 13 and 16 depict how many people had a shift in frataxin levels in skin cells and buccal cells as a percentage of average healthy volunteers after 14 days of treatment. On the left is the 25 mg cohort and on the right is the 50 mg cohort. On the far left of each figure are categories of the percentage of average healthy volunteers. Figures in black to the left of the black vertical line represent patients at baseline. Figures to the right of the black
17
vertical line represent day 14 frataxin levels as a percentage of average healthy volunteers. In blue are individuals that increase by 25% to 50% and in green are individuals that exceeded 50% of average healthy volunteers.
Figure 13.

Only participants with quantifiable levels at baseline and day 14 are included in the figures.
*% of healthy volunteer FXN level is calculated by dividing each participant's FXN level by the average FXN level (16.34 pg/µg) from the noninterventional healthy volunteer study (N=60).
Figure 14.

Only participants with quantifiable levels at baseline and day 14 and day 28 are included in the tables.
18
Figure 15.

*FXN levels measured via detection of peptide derived from mature FXN; FXN concentrations are normalized to total cellular protein content in each sample. Data represent median and 25th and 75th percentiles. Only participants with quantifiable levels at both baseline and Day 14 are included in the figures.
**Median baseline FXN level in patients were 2.1 pg/µg for the placebo, 1.8 pg/µg for the 25 mg cohort and 1.6 pg/µg for the 50 mg cohort.
Figure 16.

Only participants with quantifiable levels at baseline and day 14 are included in the figures.
*% of healthy volunteer FXN level is calculated by dividing each participant's FXN level by the average FXN level (8.24 pg/µg) from the noninterventional healthy volunteer study (N=60).
Open Label Extension
In January 2024, we initiated the OLE trial evaluating daily subcutaneous injections of 25 mg of nomlabofusp self-administered or administered by a caregiver, with the first patient dosed in March 2024. Participants who completed treatment in the Phase 2 dose exploration trial, or who previously completed a prior clinical trial of
19
nomlabofusp are potentially eligible to screen for the OLE. The OLE will evaluate the safety, tolerability, PK, and measures of frataxin levels in peripheral tissues as well as other exploratory PD (lipid profiles and gene expression data) following long-term subcutaneous administration of CTI-1601 in subjects with Friedreich’s ataxia. The pediatric MAD placebo-controlled studynomlabofusp. Clinical measures collected during the trial will be in patientscompared to data from two to seventeen years of age. These patients may also be eligible to enrolla matched control arm derived from participants in the Jive study after completing participationFriedreich’s Ataxia Clinical Outcome Measures Study (FACOMS) database. Further dose expansion in the MAD study. We expect to initiate a global registration study once sufficientOLE trial will be considered based on safety, tolerability, PK, FXN levels from the 25 mg dose of nomlabofusp in the OLE trial, as well as data from the Phase 2 trial's 50 mg cohort and is available to inform decisions regarding dosing and trial design. The design and protocolcontingent on FDA review as part of the study will be based onpartial clinical hold. Initial data from the resultsOLE trial is expected in the fourth quarter of 2024. We also are planning to include in our clinical studies, individuals between the earlier studiesages of 2 and will incorporate feedback from regulatory authorities17 and are currently in various countries. The progressiondiscussions with FDA as to what additional clinical data in adults would inform inclusion of the Jive study and the global registration study are pending lifting of the clinical hold.those pediatric individuals in our studies.
Intellectual Property
Our success depends in large part upon our ability to obtain and maintain proprietary protection for our current and future products and technologies, and to operate without infringing the proprietary rights of others. Our policy is to protect our proprietary position by, among other methods, filing for patent applications on inventions that are important to the development and conduct of our business with the U.S. Patent and Trademark Office and its foreign counterparts. We also intend to rely on regulatory exclusivity (also called data package exclusivity), which is separate and distinct from the protection afforded by patents, to protect our products. We further protect our proprietary information by requiring our employees, consultants, contractors and other advisors to execute nondisclosure and assignment of invention agreements upon commencement of their respective employment or engagement. Agreements with our employees also prevent them from bringing the proprietary rights of third parties to us. In addition, we require confidentiality or service agreements from third parties that receive our confidential information or materials.
As of February 1, 2022,23, 2024, our intellectual property portfolio was composed of numerous international Patent Cooperation Treaty ("PCT"), foreign, and United States non-provisional patent applications, a United States patent and United States provisional patent applications that we own or co-own, and fivesix issued United States patents and additional non-provisional patent applications in the United States and in certain foreign jurisdictions that we license from academic institutions. The issued patents in the United States licensed by us, which include issued patents generically covering the composition of matter for CTI-1601nomlabofusp and methods for treating Friedreich’s ataxia,FA, have
16
expiration dates between 2024 and 2034.2040 without taking potential patent term extension into consideration. The international and the United States non-provisional patent applications licensed by us relate to composition of matter and methods of use for CTI-1601.nomlabofusp.
The additional patent applications we own or co-own which include internationalPCT, foreign and United States non-provisional patent applications a United States patent, and United States provisional patent applications are related to the development of CTI-1601nomlabofusp, including methods of use of nomlabofusp, and to our peptide-delivery platform technology. If the non-provisional patent applications owned or co-owned by us result in issued patents, such patents are expected to expire between 2040 and 2041, without taking potential patent term adjustment or patent term extension into consideration.
A provisional patent application allows for an effective filing date to be established with regard to an invention, but once a provisional patent application is filed, either a corresponding non-provisional patent application or a petition to convert the provisional patent application into a non-provisional patent application must be filed within 12 months or such effective filing date will be lost. If we or our licensor timely files non-provisional patent applications in the United States and in countries outside of the United States with regard to our provisional patent applications and these non-provisional patent applications result in issued patents, such patents are expected to expire in 2041 and 2042,2044 or 2025, without taking potential patent term adjustment or patent term extension into consideration.
CTI-1601Nomlabofusp is covered by licensed issued patents (composition of matter and methods of use) in the United States which, if properly maintained, will expire in 2024, 2025 and 2025 (respectively),2040, excluding any patent term extensions that might be available following the grant of marketing authorizations. We also possess an exclusive license to non-provisional applications in the United States and certain countries outside the United States for CTI-1601nomlabofusp (composition of matter and methods of use). If thethese patent applications in the United States and other countries result in issued patents, those patents would be expected to expire in the United States and in countries outside of the United States in 2040,2040. This estimated expiration excludes any patent term adjustment that might be available following the grant of the patent and any patent term extensions that might be available following the grant of marketing authorizations. We cannot predict whether the patent applications we and our licensors are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient protection from competitors or other third parties.
20
Patents extend for varying periods according to the date of patent filing or grant and the legal term of patents in various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depends on the type of patent, the scope of its coverage and the availability of legal remedies in the country.
We also use other forms of protection besides patent protection and regulatory exclusivity, such as trademark, copyright, and trade secret protection, to enhance our intellectual property, particularly where we do not believe patent protection is appropriate or obtainable. We aim to take advantage of all of the intellectual property rights that are available to us and believe that this comprehensive approach will provide us with proprietary exclusive positions for our product candidates, such as CTI-1601,nomlabofusp, where available.
In-License Agreements
We are party to a License Agreement ("WFUHS License") dated November 30, 2016 with Wake Forest University Health Sciences ("WFUHS") and a License Agreement ("IU License") dated November 30, 2016, as amended, with Indiana University ("IU"). Such agreements provide for a transferable, worldwide license to certain patent rights regarding technology used by us with respect to the development of CTI-1601.nomlabofusp.
In partial consideration for the right and license granted under these agreements, we will pay each of WFUHS and IU a royalty of a low single digit percentage of net sales of licensed products depending on whether there is a valid patent covering such products. As additional consideration for these agreements, we are obligated to pay each of WFUHS and IU certain milestone payments of up to $2.6 million in the aggregate upon the achievement of certain developmental milestones, commencing on the enrollment of the first patient in a Phase 1 clinical trial. We will also pay each of WFUHS and IU sublicensing fees ranging up to a low double-digit percentage of sublicense consideration depending on our achievement of certain regulatory milestones as of the time of receipt of the sublicense consideration. We are also obligated to reimburse WFUHS and IU for patent-related expenses. In the event that we dispute the validity of any of the licensed patents, the royalty rate would be tripled during such
17
dispute. We are also obligated to pay to IU a minimum annual royalty of less than $0.1 million per annum starting in the 2020 calendar year for the term of the agreement.
In the event that we are required to pay IU consideration, then we may deduct 20% of such IU consideration on a dollar-for-dollar basis from the consideration due to WFUHS. In the event that we are required to pay WFUHS consideration, then we may deduct 60% of such WFUHS consideration on a dollar-for-dollar basis from the consideration due to IU.
DuringCompetition
The biopharmaceutical industry is characterized by intense and dynamic competition to develop new technologies and proprietary therapies. Any product candidates that we successfully develop into products and commercialize may compete with existing therapies and new therapies that may become available in the twelve months ended December 31, 2021future. While we believe that our platform technology, product candidates and 2020, no milestones were achieved,scientific expertise in the field of rare diseases provide competitive advantages, we face competition from various sources, including larger and no expensebetter-funded pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies, as well as from academic institutions, governmental agencies and public and private research institutions. We expect to compete with Biogen’s SKYCLARYSTM (omaveloxolone), which was recognized. Both agreements continue fromapproved for the treatment of FA in adults and adolescents aged 16 and older by the FDA and the European Commission in February 2023 and February 2024, respectively. Other competitors currently developing therapeutics to treat FA include, but are not limited to Design Therapeutics, Lexeo Therapeutics, Neurocrine Biosciences/Voyager Therapeutics and PTC Therapeutics. Many of our competitors have significantly greater financial, technical and human resources than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated amongst a smaller number of our competitors. Our commercial opportunity could be reduced or eliminated if our competitors develop or market products or other novel therapies that are more effective, safer or less costly than our product candidates or obtain regulatory approval for their effective date through the last to expire of the licensed patents unless earlier terminated by either party.products more rapidly than we may obtain approval for our product candidates.
Manufacturing and Supply
CTI-1601Nomlabofusp is a biologic fusion protein that is produced in E. coli. We have worked with contract manufacturers to develop, in accordance with current good manufacturing practice ("cGMP")cGMPs a manufacturing process and analytical methods for drug substance to support clinical trials. The drug product manufacturing process was also developed and packaged into single use vials for the clinical program. We also use third party manufacturers for the clinical packaging, storage and
21
distribution of CTI-1601.nomlabofusp. We rely on third parties to store the CTI-1601nomlabofusp master cell bank and working cell bank, each stored at a different location. We continue to advance the manufacturing of CTI-1601,nomlabofusp, obtain stability data, and produce drug product for toxicology studies and future clinical trials. We are continually trying to optimize our manufacturing process to increase yields, decrease costs and increase reliability in supply chains. The final process will need to be successfully scaled up to support commercial manufacturing.
The drug substance which is in frozen liquid form for CTI-1601nomlabofusp is currently manufactured for us by KBI Biopharma, Inc. ("KBI"). We are party to a Master Services Agreement, as amended, with KBI, pursuant to which KBI provides biological development and clinical manufacturing services with respect to CTI-1601.nomlabofusp. We currently produce a frozen liquid form of drug product at another manufacturer. We are undertakinghave also begun a program with a third-party manufacturer to begin to produce a lyophilized version of the drug product from the same KBI drug substance, that, once available, we intend to use in certain of our future planned clinical trials.trials and ultimately, commercial use, assuming nomlabofusp receives required regulatory approvals.
Human Capital Resources
Employees and Compensation
In order to achieve the goals and expectations of our Company, it is crucial that we continue to attract and retain top talent. To facilitate talent attraction and retention, we strive to make our company a safe and rewarding workplace, with opportunities for our employees to grow and develop in their careers, supported by strong compensation, benefits, paid time-off and health and wellness programs, including programs that build connections between our employees. In response to the COVID-19 pandemic, we implemented significant changes that we determined were in the best interest of our employees, and which comply with local and federal government regulations. This includes having some of our non-laboratory employees work from home, while implementing additional safety measures for employees continuing critical on-site work.
As of February 1, 2022,December 31, 2023, we employed 3142 full-time employees in the United States.States, of which 32 are directly engaged in research and development with the rest providing administrative, business and operations support. None of our employees are represented by a labor organization or under any collective-bargaining arrangements. We consider our employee relations to be good.
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and new employees, advisors and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and reward personnel through the granting of stock-based and cash-based compensation awards, in order to increase stockholder value and the success of our company by motivating such individuals to perform to the best of their abilities and achieve our objectives.
18
Diversity and Inclusion
We value the diversity of our employees and take pride in our commitment to diversity and inclusion across all levels of our organizational structure and with respect to our board of directors. We value diversity and inclusion across our entire workforce. We are committed to developing strategies for building diverse teams and promoting the advancement of employees from diverse backgrounds.
Scientific Advisors
We have established a scientific advisory board and we regularly seek advice and input from these experienced thought leaders on matters related to our research and development programs. The members of our scientific advisory board consist of distinguished research scientists, professors and industry experts recognized as key opinion leaders in the fields of rare disease, pediatrics and mitochondrial disease. Their scientific perspectives will be invaluable to determine our strategic scientific pathway and support the development of other potential treatments for complex rare diseases to help fill unmet medical needs in this space. We intend to continue to leverage the broad expertise of our advisors by seeking their counsel on important topics relating to our product development and clinical development programs. Our scientific advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us. In addition, our scientific advisors may have arrangements with other companies to assist those companies in developing products or technologies that may compete with ours. All of our scientific advisors are affiliated with other entities and devote only a small portion of their time to us.
22
Our scientific advisors are set forth in the table below:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Name | Title | ||
|
| ||
Marni Joy Falk, MD |
| Executive Director, Mitochondrial Medicine Frontier Program at Children’s Hospital of Philadelphia; Professor, Department of Human Genetics and Department of Pediatrics, University of Pennsylvania Perelman School of Medicine |
|
Giovanni Manfredi, MD, PhD |
| Finbar and Marianne Kenny Professor of Clinical and Research Neurology, Weill Cornell Medicine; Professor of Neuroscience, Weil Cornell Medicine |
|
Jill Ostrem, MD | Medical director and division chief of University of California San Francisco (UCSF) Movement Disorders and Neuromodulation Center. Carlin and Ellen Wiegner Endowed Professor of Neurology. | ||
Mark Payne, MD |
| Professor of Pediatrics, Indiana University School of Medicine |
|
Facilities
We lease office and laboratory space, which consists of approximately 5,0008,000 square feet and 1,750 square feet located in Bala Cynwyd, PA and Philadelphia, PA, respectively. Our officeWe are currently on a month-to-month lease expires in August 2023on this Philadelphia lab space. We, as well as our landlord, can terminate the lease with an option to extend for three years, and our laboratory lease expires in December 2022. We believe that we will need to increase both its office and laboratory facilities in the near term. We believe that both appropriate office and laboratory space will be readily available on commercially reasonable terms.four months notice.
AsWe have entered into a lease for 3,927 square feet of new laboratory space in King of Prussia, PA to replace the Philadelphia lab space, but this lease has not yet commenced with expected occupancy in the second quarter of 2024.As part of the Merger (as defined below) with Zafgen, Inc.Inc ("Zafgen"), we acquired a non-cancellable operating lease for approximately 17,705 square feet of office space at 3 Center Plaza, Boston, Massachusetts ("the Boston(the "Boston Lease"). The Boston Lease expires on October 30, 2029. On October 27, 2020, we entered into a sublease agreement whereby we subleased all 17,705 square feet of office space leased under the Boston Lease until October 30, 2029.
19
Legal Proceedings
From time to time, we may become involved in legal proceedings arising in the ordinary course of our business. We are not presently a party to any legal proceedings that, if determined adversely to us, would individually or taken together have a material adverse effect on us.
Corporate Information
We were founded in 2005 as a Delaware corporation under the name Zafgen, Inc. ("Zafgen"). On May 28, 2020, Zafgen completed a reverse merger with Chondrial Therapeutics, Inc. ("Chondrial") in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated December 17, 2019, by and among Zafgen, Zordich Merger Sub, Inc., Chondrial and Chondrial Therapeutics Holdings, LLC, pursuant to which Zordich Merger Sub, Inc. merged with and into Chondrial, with Chondrial surviving as our wholly owned subsidiary. Following completion of the Merger,merger, Zafgen, Inc. changed its name to Larimar Therapeutics, Inc.
Our principal executive offices are located at Three Bala Plaza East, Suite 506, Bala Cynwyd, PA 19004, and our telephone number is (844) 511-9056. Our website address is www.larimartx.com. The information on, or that can be accessed through, our website is not part of this Annual Report on Form 10-K and is not incorporated by reference herein. We have included our website address as an inactive textual reference only.
Available Information
We file annual, quarterly, and current reports, proxy statements, and other documents with the Securities and Exchange Commission ("the SEC") under the Securities Exchange Act of 1934, as amended ("the Exchange Act"). The SEC maintains an internet website, www.sec.gov, that contains reports, proxy and information statements, and other information regarding issuers, including us, that file electronically with the SEC.
Copies of each of our filings with the SEC on Form 10-K, Form 10-Q and Form 8-K and all amendments to those reports, can be viewed and downloaded free of charge at our website, www.larimartx.com as soon as reasonably practicable after the reports and amendments are electronically filed with or furnished to the SEC.
Our code of ethics, other corporate policies and procedures, and the charters of our Audit Committee, Compensation Committee and Nominating and Corporate Governance Committee are available through our website
23
at www.larimartx.com. We intend to post information regarding any amendments to, or waivers from, our code of ethics on our website.
Government Regulation
In the United States, drug and biologic products are licensedsubject to extensive regulation by the FDA for marketing under the Public Health Service Act, referred to as the PHS Act, and regulated under theFDA. The Federal Food, Drug, and Cosmetic Act ("the FDCA"(“FDCA”). Both the FDCA and the PHS Actother federal and their correspondingstate statutes and regulations, govern, among other things, the research, development, testing, manufacturing, safety, purity, potency, efficacy,manufacture, storage, recordkeeping, approval, labeling, packaging, storage, record keeping,promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Biological products used for the prevention, treatment, or cure of a disease or condition of a human being are subject to regulation under the FDCA, with the exception that the section of the FDCA that governs the approval of drugs via new drug applications (“NDAs”), does not apply to the approval of biologics. In contrast, biologics are approved for marketing sales, import, export, reporting, advertisingunder provisions of the Public Health Service Act (“PHS Act”), via a Biologics License Application (“BLA”). However, the application process and other promotional practices involving drug and biological products.requirements for approval of BLAs are very similar to those for NDAs. FDA clearance must be obtained before clinical testing of drug and biologic products. FDAapproval or licensure also must be obtained before the marketing of drug and biological products. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
20
U.S. Development Process
The process required by the FDA before a drug or biologic product may be marketed in the United States generally involves the following:
Before testing any drug or biological product candidate in humans, the product candidate enters the non-clinical testing stage. Non-clinical tests include laboratory evaluations of product chemistry, pharmacology, toxicity
24
and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of the non-clinical tests must comply with federal regulations and requirements including GLPs.
The clinical study sponsor must submit the results of the non-clinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some non-clinical testing typically continues after the IND is submitted. An IND is an exemption from the FDCA that allows an unapproved product to be shipped in interstate commerce for use in an investigational clinical trial and a request for FDA authorization to administer an investigational product to humans. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA requests certain changes to a protocol before
21
the trial can begin, or the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor must resolve any deficiencies to the satisfaction of the FDA, and the FDA must resolve any outstanding concerns and lift the clinical hold before the clinical trial can begin. The FDA may also impose clinical holds on a drug or biological product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA.
Clinical trials may involve the administration of the drug or biological product candidate to healthy volunteers or subjects under the supervision of qualified investigators, generally physicians not employed by or under the study sponsor’s control. Clinical trials involving some products for certain diseases, including some rare diseases may begin with testing in patients with the disease. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND application. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research subjects or his or her legal representative provide informed consent. Further, each clinical trial must be reviewed and approved by an independent IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of study participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Additionally, some trials are overseen by an independent group of qualified experts organized by the trial sponsor, known as a data monitoring committee. The data monitoring committee may review safety data and/or efficacy data.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
An Open Label ExpansionOLE study may also be conducted. An OLE study typically enrolls participants from previous clinical trials and is designed to gather the long-term safety and tolerability data on a potential new medicine beyond the time period of the original studies.
25
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication in real-world scenarios. They provide additional information regarding risks, benefits and best use, including longer term safety data. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA or BLA.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in
22
the rate of a serious suspected adverse reactions over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. Phase 1, Phase 2, and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug or biological product has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the physical characteristics of the drug or biological product as well as finalize a process for manufacturing the product in commercial quantities in accordance with GMPcGMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHS Act emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the drug or biological product candidate does not undergo unacceptable deterioration over its shelf life.
There are also various laws and regulations regarding laboratory practices, the experimental use of animals, and the use and disposal of hazardous or potentially hazardous substances in connection with the research. In each of these areas, the FDA and other regulatory authorities have broad regulatory and enforcement powers, including the ability to levy fines and civil penalties, suspend or delay issuance of approvals, seize or recall products, and withdraw approvals.
Information about certain clinical trials must be submitted within specific timeframes to the NIH for public dissemination on its clinicaltrials.gov website. Sponsors or distributors of investigational products for the diagnosis, monitoring, or treatment of one or more serious diseases or conditions must also have a publicly available policy on evaluating and responding to requests for expanded access requests.
U.S. Review and Approval Processes
After the completion of clinical trials of a drug or biological product, FDA approval of an NDA, or BLA, must be obtained before commercial marketing of the product. A BLA submission is planned for CTI-1601. The NDA or BLA must include results of product development, laboratory and animal studies, human studies, information on the manufacture and composition of the product, proposed labeling and other relevant information. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the NDA or BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, as amended, ("PDUFA") each NDA or BLA may be accompanied by a significant user fee. Under federal law, the submission of most applications is subject to an application user fee. The sponsor ofapplicant for an approved application is also subject to an annual program fee. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs or BLAs for product candidates designated as orphan drugs, unless the product candidate also includes a non-orphan indication.
Within 60 days following submission of the application, the FDA reviews an NDA or BLA submitted to determine if it is substantially complete before the agency accepts it for filing.files it. The FDA may refuse to file any NDA or BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the NDA or BLA must be resubmitted with the additional information. The resubmitted application is
26
also subject to review before the FDA accepts it for filing. The application also needs to be published and submitted in an electronic format that can be processed through the FDA’s electronic systems. If the electronic submission is not compatible with FDA’s systems, the NDA or BLA can be refused for filing.files it. Once the submission is accepted for filing,filed, the FDA begins an in-depth substantive review of the NDA or BLA. The FDA reviews the
23
NDA or BLA to determine, among other things, whether the proposed product is safe and effective (for drugs) or safe, potent, and effective (for biologics), for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with GMPscGMPs to assure and preserve the product’s identity, safety, strength, quality, potency and purity. One of the performance goals agreed to by the FDA under the PDUFA is to review standard NDAs or BLAs in ten months from filing and priority NDAs or BLAs in six months from filing, whereupon a review decision is to be made. The review process may be extended by three months if the FDA classifies a response to an FDA request for additional information or clarification as a major amendment.
The FDA may refer applications for novel products or products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the drug or biological product approval process,review, the FDA will also determine whether a Risk Evaluation and Mitigation Strategy ("REMS")REMS is necessary to assure the safe use of the biological product. If the FDA concludes a REMS is needed, the sponsor of the NDA or BLA must submit a proposed REMS; the FDA will not approve the NDA or BLA without a REMS, if required.
Before approving an NDA or BLA, the FDA may inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with GMPcGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA will typically inspect one or more clinical trial sites to assure that the clinical trials were conducted in compliance with IND study requirements and GCP requirements. To assure GMPcGMP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the NDA or BLA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than the sponsor interprets the same data. If the agency decides not to approve the NDA or BLA in its present form, the FDA will issue a complete response letter that usually describes all of the specific deficiencies in the NDA or BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA or BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. In addition, the FDA may require post marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to further assess a drug or biological product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized. As a condition for approval, the FDA may also require additional non-clinical testing as a Phase 4 commitment.
One of the performance goals agreed to by the FDA under the PDUFA is to review standard NDAs or BLAs in 10 months from filing and priority NDAs or BLAs in six months from filing, whereupon a review decision is to be made. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs or BLAs and its review goals are subject to change from time to time. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the NDA or BLA sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
Post-Approval Requirements
Maintaining substantial compliance with applicable federal, state, and local statutes and regulations requires the expenditure of substantial time and financial resources. Rigorous and extensive FDA regulation of drug and biological products continues after approval, particularly with respect to GMP.cGMP. We will rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of any products that we may commercialize. Manufacturers of our products are required to comply with applicable requirements in the GMPcGMP regulations, including quality control and quality assurance and maintenance of records and documentation.
24
Following approval, the manufacturing facilities are subject to biennialperiodic inspections by the FDA’sFDA and such inspections may result in an issuance of FDA Form 483 deficiency observations, untitled letter, or a warning letter, which can lead to plant shutdown and other more serious penalties and fines. Prior to the institution of any manufacturing changes, a determination needs to be made whether FDA approval is required in advance. If not done
27
in accordance with FDA expectations, the FDA may restrict supply and may take further action. Annual product reports are required to be submitted annually. Other post-approval requirements applicable to drug and biological products, include reporting of GMPcGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse events, reporting updated safety and efficacy information, and complying with electronic record and signature requirements.
After an NDA or BLA is approved, the product may also be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. The FDA may also perform certain confirmatory tests on lots of some products, before releasing the lots for distribution by the manufacturer. In addition, the FDA may conduct laboratory research related to the regulatory standards on the safety, purity, potency, and effectiveness of drug and biological products. Systems need to be put in place to record and evaluate adverse events reported by health care providers and patients and to assess product complaints. An increase in severity or new adverse events can result in labeling changes or product recall. Defects in manufacturing of commercial products can result in product recalls.
WeManufacturers must also comply with the FDA’s advertising and promotion requirements, such as those related to direct-to-consumer advertising, the prohibition on promoting products for uses or inpatientin patient populations that are not described in the product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the internet. Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant or manufacturer to administrative or judicial civil or criminal sanctions and adverse publicity. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval or license revocation, clinical hold, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, mandated corrective advertising or communications with doctors, debarment, restitution, disgorgement of profits, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect.
Drug and biologic product manufacturers and other entities involved in the manufacture and distribution of approved drug and biological products are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with GMPscGMPs and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality control to maintain GMPcGMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved NDA or BLA, including withdrawal of the product from the market. In addition, changes to the manufacturing process or facility generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant Orphan Drug Designation ("ODD") to a drug or biologic product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug or biologic product available in the United States for this type of disease or condition will be recovered from sales of the product. ODD must be requested before submitting an NDA or BLA. After the FDA grants ODD, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. ODD does not convey any advantage in or shorten the duration of the regulatory review and approval process.
25
If a product that has ODD receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug or biological product for the same indication for seven years, except in limited circumstances, such as not being able to supply the product for patients or showing clinical superiority to the product with orphan exclusivity.
exclusivity by means of greater effectiveness, greater safety or providing a major contribution to patient care or in instances of drug supply issues. Competitors, however, may receive approval of either a different products for the indication for which the orphan product has exclusivity or obtain approval for the same indication or the same
28
product but for a different indication for whichbut that could be used off-label in the orphan product has exclusivity.indication. Orphan productdrug exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval ofbefore we do for the same drug or biologic product as defined by the FDA for the same indication we are seeking approval, or if our product candidate is determined to be contained within the competitor’s scope of product for the same indication or disease. If a drug or biological product designated as an orphan product receiveswe pursue marketing approval for an indication broader than what is designated, itthe orphan drug designation we have received, we may not be entitled to orphan productdrug exclusivity.
Expedited Review and Approval Programs
The FDA has various programs, including Fast Trackfast track designation, priority review, accelerated approval, and breakthrough therapy designation, that are intended to expedite or simplify the process for the development and FDA review of drug and biological products that are intended for the treatment of serious or life-threatening diseases or conditions and demonstrate the potential to address unmet medical needs. The purpose of these programs is to provide important new drug and biological products to patients earlier than under standard FDA review procedures. To be eligible for a Fast Trackfast track designation, the FDA must determine, based on the request of a sponsor, that a drug or biological product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need. The FDA will determine that a product will fill an unmet medical need if it will provide a therapy where none exists or provide a therapy that may be potentially superior to existing therapy based on efficacy or safety factors. In addition to other benefits, such as the ability to have greater interactions with the FDA, the FDA may initiate review of sections of a Fast Trackfast track NDA or BLA before the application is complete, a process known as rolling review.
The FDA may give a priority review designation, such as a rare pediatric disease designation to drug or biological products that treat a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. A priority review means that the goal for the FDA to review an application is six months, rather than the standard review of ten months under current PDUFA guidelines. Most products that are eligible for Fast Trackfast track designation may also be considered appropriate to receive a priority review.
In addition, drug and biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug or biological product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require a sponsor of a drug or biological product receiving accelerated approval to perform post-marketing studies with due diligence to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical endpoint, and the drug or biological product may be subject to accelerated withdrawal procedures.
Moreover, under theThe Food and Drug Administration SafetyOmnibus Reform Act (“FDORA”) was recently enacted, which included provisions related to the accelerated approval pathway. Pursuant to FDORA, the FDA is authorized to require a post-approval study to be underway prior to approval or within a specified time period following approval. FDORA also requires the FDA to specify conditions of any required post-approval study, which may include milestones such as a target date of study completion and Innovation Act enacted in 2012,requires sponsors to submit progress reports for required post-approval studies and any conditions required by the FDA not later than 180 days following approval and not less frequently than every 180 days thereafter until completion or termination of the study. FDORA also provides the FDA increased authority for expedited procedures to withdraw approval of a drug or biologic or indication approved under accelerated approval if, for example, the confirmatory trial fails to verify the predicted clinical benefit of the product. FDORA also enables the FDA to initiate enforcement actions for the failure to conduct with due diligence a required post-approval study, including a failure to meet any required conditions specified by the FDA or to submit timely reports. In addition, the FDA generally requires, unless otherwise informed by the agency, pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product.
Moreover, a sponsor can request designation of a product candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug or biologic product that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug or biologic product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.
29
The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decidesdecide that the time period for FDA review or approval will not be
26
shortened. Furthermore, fast-track designation, priority review, accelerated approval and breakthrough therapy designation, do not change the standards for approval and may not ultimately expedite the development or approval process.
Biologics Price CompetitionRare Pediatric Disease Vouchers
The Rare Pediatric Disease Voucher Program is intended to encourage development of new drug and Innovationbiological products for prevention and treatment of certain rare pediatric diseases. Although there are existing incentive programs to encourage the development and study of drugs and biologics for rare diseases, pediatric populations, and unmet medical needs, this program provides an additional incentive for the development of drugs and biologics for rare pediatric diseases, which may be used alone or in combination with other incentive programs. A rare pediatric disease is defined as a disease that is a serious or life-threatening disease in which the serious or life-threatening manifestations primarily affect individuals aged from birth to 18 years, including age groups often called neonates, infants, children, and adolescents; and is a rare disease or condition as defined in the FDCA, which includes diseases and conditions that affect fewer than 200,000 persons in the United States and diseases and conditions that affect a larger number of persons and for which there is no reasonable expectation that the costs of developing and making available the product in the United States can be recovered from sales of the product in the United States.
The sponsor of an application for a drug product that obtains rare pediatric disease designation may be eligible for a voucher that can be used or sold to obtain a priority review for a subsequent application submitted under section 505(b)(1) of the FDCA or section 351 of the PHS Act. A rare pediatric disease drug product must meet certain eligibility requirements for a priority voucher at the time the sponsor seeks approval. The rare pediatric disease priority review voucher program was most recently re-authorized by Congress through September 30, 2024, with the potential for priority review vouchers to be granted through September 30, 2026.
Pediatric Information and Pediatric Exclusivity
Under the Pediatric Research Equity Act (“PREA”), certain NDAs and BLAs and certain supplements to an NDA or BLA must contain data to assess the safety and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of pediatric data or full or partial waivers. A sponsor who is planning to submit a marketing application for a drug that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration must submit an initial Pediatric Study Plan (“PSP”) within 60 days of an end-of-Phase 2 meeting or, if there is no such meeting, as early as practicable before the initiation of the Phase 3 or Phase 2/3 study. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach an agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from preclinical studies, early phase clinical trials and/or other clinical development programs.
Under the Best Pharmaceuticals for Children Act (“BPCA”), a drug or biologic product can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods for all formulations, dosage forms, and indications of the active moiety and patent terms for drugs and six months to exclusivity periods for all formulations, dosage forms, and indications for biologic products. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study, provided that at the time pediatric exclusivity is granted there is not less than nine months of term remaining.
30
Biosimilars
The Biologics Price Competition and Innovation Act of 2009 ("BPCIA") which was enacted as part of the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010 ("PPACA") created an abbreviated approval pathway for biological products that are demonstrated to be "biosimilar" or "interchangeable" with an FDA-licensed reference biological product via an approved BLA. Biosimilarity to an approved reference product requires that there be no differences in conditions of use, route of administration, dosage form, and strength, and no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency. Biosimilarity is demonstrated in steps beginning with rigorous analytical studies or "fingerprinting", in vitro studies, in vivo animal studies, and generally at least one clinical study, absent a waiver from the Secretary of the U.S. Department of Health and Human Services.Services ("HHS"). The biosimilarity exercise tests the hypothesis that the investigational product and the reference product are the same. If at any point in the stepwise biosimilarity process a significant difference is observed, then the products are not biosimilar, and the development of a stand-alone NDA or BLA is necessary. In order to meet the higher hurdle of interchangeability, a sponsor must demonstrate that the biosimilar product can be expected to produce the same clinical result as the reference product, and for a product that is administered more than once, that the risk of switching between the reference product and biosimilar product is not greater than the risk of maintaining the patient on the reference product. Complexities associated with the larger, and often more complex, structures of biological products, as well as the process by which such products are manufactured, pose significant hurdles to implementation that are still being evaluated by the FDA. Under the BPCIA, a reference biologic is granted 12 years of exclusivity from the time of first licensure or BLA approval, of the reference product, and no application for a biosimilar can be submitted for four years from the date of licensure of the reference product. The first biologic product submitted under the biosimilar abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against a finding of interchangeability for other biologics for the same condition of use for the lesser of (i) one year after first commercial marketing of the first interchangeable biosimilar, (ii) 18 months after the first interchangeable biosimilar is approved if there is no patent challenge, (iii) 18 months after resolution of a lawsuit over the patents of the reference biologic in favor of the first interchangeable biosimilar applicant, or (iv) 42 months after the first interchangeable biosimilar is approved if a patent lawsuit is ongoing within the 42-month period.
Regulation Outside of the United States
In addition to regulations in the United States, we are subject to a variety of regulations in other jurisdictions governing clinical studies, commercial sales, and distribution of our products. Most countries outside of the United States require that clinical trial applications be submitted to and approved by the local regulatory authority for each clinical study. In the European Union, for example, an application must be submitted to the national competent authority and an independent ethics committee in each country in which we intend to conduct clinical trials, much like the FDA and IRB, respectively. Under the Clinical Trials Regulation (EU) No 536/2014, which replaced the Clinical Trials Directive 2001/20/EC on January 31, 2022, a single application is now made through the Clinical Trials Information System (“CTIS”), for clinical trial authorization in up to 30 European Union/European Economic Area ("EU/EEA") countries at the same time and with a single set of documentation.
The assessment of applications for clinical trials is divided into two parts (Part I contains scientific and medicinal product documentation and Part II contains the national and patient-level documentation). Part I is assessed by a coordinated review by the competent authorities of all European Union Member States in which an application for authorization of a clinical trial has been submitted (Member States concerned) of a draft report prepared by a Reference Member State. Part II is assessed separately by each Member State concerned. The role of the relevant ethics committees in the assessment procedure will continue to be governed by the national law of the Member State concerned, however overall related timelines are defined by the Clinical Trials Regulation. The new Clinical Trials Regulation also provides for simplified reporting procedures for clinical trial sponsors.
In addition, whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of countries outside the U.S. before we can commence clinical studies or marketing of the product in those countries. The approval process and requirements vary from country to country, so the number and type of non-clinical, clinical, and manufacturing studies needed may differ, and the time may be longer or shorter than that required for FDA approval.
To obtain regulatory approval of an orphan druga product under the European Union's regulatory system, we are mandated to submit a Marketing Authorization Application ("MAAs"MAA"), to be assessed in the Centralized Procedure.centralized procedure. The centralized procedure which came into operation in 1995, allows applicants to obtain a marketing authorization ("MA") that is valid throughout the European Union.Union and the additional Member States of the European Area (Iceland, Liechtenstein and Norway) ("EEA"). It is
31
compulsory for medicinal products manufactured using biotechnological processes, for orphan medicinal products, for advance therapy medicinal products (gene-therapy, somatic cell-therapy or tissue-engineered medicines) and for human products containing a new active substance which wasare not authorized in the Community before 20 May 2004 (date of entry into force of Regulation (EC) No 726/2004)European Union and which are intended for the treatment of HIV, AIDS, cancer, neurodegenerative disorderdisorders, auto-immune and other immune dysfunctions, viral diseases or diabetes. The centralized procedure is optional for any other products containing new active substances not authorized in the Community before 20 May 2004European Union or for products which constitute a significant therapeutic, scientific or technical innovation or for which a Communitycentralized authorization is in the interests of patients at Community level.in the European Union. When a company wishes to place on the market a medicinal product that is eligible for the centralized procedure, it sends an application directly to the European Medicines Agency ("EMA"),EMA, to be assessed by the Committee for Medicinal Products for Human Use ("CHMP"). The CHMP is responsible for conducting the assessment of whether a medicine meets the required quality, safety and efficacy requirements, and whether the product has a positive risk/benefit/risk profile. The procedure results in a Commission decision, which is valid in all European Union Member States. Centrally authorized products may be marketed in all Member States. Centralized procedure: Full copies of the MA applicationMAA are sent to a rapporteur and a co-rapporteur designated by the competent EMA scientific committee. They coordinate the EMA's scientific assessment of the medicinal product and prepare draft reports. Once the draft reports are prepared (other experts might be called upon for this purpose),
27
they are sent to the CHMP, whose comments or objections are communicated to the applicant. The rapporteur is therefore the privileged interlocutor of the applicant and continues to play this role, even after the MA has been granted.
The rapporteur and co-rapporteur then assess the applicant's replies, submit them for discussion to the CHMP and, taking into account the conclusions of this debate, prepare a final assessment report. Once the evaluation is completed, the CHMP gives a favorable or unfavorable opinion as to whether to grant the authorization. When the opinion is favorable, it shall include the draft summary of the product's characteristics, the package leaflet and the texts proposed for the various packaging materials. The time limit for the evaluation procedure is 210 days.days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP). The EMA then has fifteen days to forward its opinion to the Commission. This isEuropean Commission which will make a binding decision on the startgrant of an MA within 67 days of the second phasereceipt of the procedure: the decision-making process. CHMP.
The Agency sends to the Commission its opinion and assessment report, together with annexes containing: the SmPC, or Annex 1; the particulars of the MAH responsible for batch release, the particulars of the manufacturer of the active substance and the conditions of the marketing authorization, or Annex 2; and the labelling and the package leaflet, or Annex 3. The annexes are translated into the 22 other official languages of the European Union During the decision-making process, the Commission services verify that the marketing authorization complies with Union law. The Commission has fifteen days to prepare a draft decision. The medicinal product is assigned a Communityan EU registration number, which will be placed on its packaging if the marketing authorizationMA is granted. During this period, various European Commission directorates-general are consulted on the draft marketing authorizationMA decision.
The draft decision is then sent to There are two other procedures in the Standing Committee on Medicinal Products for Human Use, (Member States have one representative each in both of these committees) for their opinions. The Centralized Procedure providesEuropean Union for the grant of a single marketing authorization that is valid for allan MAA in multiple European Union member states.Member States. The Decentralized Proceduredecentralized procedure provides for approval by one or more other, or concerned, member statesMember States of an assessment of an application performed by one member state, known as the reference member state.Reference Member State. Under this procedure, an applicant submits an application, or dossier, and related materials including a draft summary of product characteristics, and draft labeling and package leaflet, to the reference member stateReference Member State and concerned member states.Concerned Member States. The reference member stateReference Member State prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state's assessment report, each concerned member stateConcerned Member State must decide whether to approve the assessment report and related materials. If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to the public health, the disputed points may eventually be referred to the European Commission, whose decision is binding on all member states. Where a product has already received an MA in one Member State, another Member State can recognize that MA under the mutual recognition procedure.
Applications from persons or companies seeking "orphanRegulation (EC) No. 141/2000, as amended, provides that a product may be designated as an orphan medicinal product designation" for products they intend to developif its sponsor can establish that it is intended for the diagnosis, prevention, or treatment of life-threatening or very seriouschronically debilitating conditions that affect not more than 5 in 10,000 persons in the European Union are reviewed bywhen the Committee for Orphan Medicinal Products, or COMP.application is made. In addition, orphan drug designation can be granted if the drugproduct is intended for a life threatening, seriously debilitating, or serious and chronic condition in the European Union and thatwhere without incentives itderived from orphan designation.it is unlikely that sales of the drugproduct in the European Union would be sufficient to justify developing the drug.necessary investment in its development. Orphan drug designation is only available if there is no other satisfactory method approved in the European Union of diagnosing, preventing, or treating the condition, or if such a method exists, the proposed orphan drugproduct will be of significant benefit to patients.patients over the existing options. We have obtained orphan drug designationin the European Union for CTI-1601.nomlabofusp.
Orphan drug designation provides opportunities for fee reductions, protocol assistance and access to the centralized procedure before and duringin the first year after marketing approval.European Union. Fee reductions are not limited to the first year after marketing approvalan MA for small and medium enterprises. In addition, if a product which has an orphan drug designation subsequently receives EMA marketing approvala centralized MA for the indication for which it has such designation, the product is entitled to orphan market exclusivity, which means the EMAregulatory authorities in the European Union may not approve any other application to market a "similar medicinal product" similar drug for the same indication for a period of 10 years. A “similar medicinal product” is defined as a
32
medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. The exclusivity period may be reduced to six years if the designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. CompetitorsAdditionally, an MA may receive marketing approval of different drugs or biologicsbe granted to a similar medicinal product for the indications for which same indication if:
28
The EMA offers a scheme thatto facilitate development of product candidates in indications, often rare, for which few or no therapies currently exist. The PRIority MEdicines, or PRIME, scheme is intended to encourage drug development in areas of unmet medical need and is intended to reinforce early dialogue with, and regulatory support from, EMA in order to stimulate innovation, optimize development and enable accelerated assessment of PRIority MEdicines ("PRIME").such product candidates. It is intended to build upon the scientific advice scheme and accelerated assessment procedure offered by the EMA. The scheme is voluntary and eligibility criteria must be met for a medicine to qualify for PRIME.
The PRIME scheme is open to medicines under development and for which the applicant intends to apply for an initial marketing authorization applicationMA through the centralized procedure. Eligible products must target conditions for which wherethere is an unmet medical need (there is no satisfactory method of diagnosis, prevention or treatment in the European Union or, if there is, the new medicine will bring a major therapeutic advantage) and they must demonstrate the potential to address the unmet medical need by introducing new methods or therapy or improving existing ones. Applicants will typically be at the exploratory clinical trial phase of development and will have preliminary clinical evidence in patients to demonstrate the promising activity of the medicine and its potential to address to a significant extent an unmet medical need. In exceptional cases, applicants from the academic sector or SMEs (small and medium sized enterprises) may submit an eligibility request at an earlier stage of development if compelling non-clinical data in a relevant model provide early evidence of promising activity to establish proof of principle, and first in man studies indicate adequate exposure for the desired pharmacotherapeutic effects and tolerability.
If a medicine is selected for the PRIME scheme, EMA:
For SMEs who enter the scheme based on data showing proof of principle, the appointment of the rapporteur occurs once they have generated data confirming eligibility at proof of concept stage. They are required to submit relevant data and justification as the product development reaches this stage.
Medicines that are selected for the PRIME scheme are also expected to benefit from EMA’s accelerated assessment procedure at the time of application for marketing authorization. Where, during the course of development, a medicine no longer meets the eligibility criteria, support under the PRIME scheme may be withdrawn.
A Pediatric Investigation Plan ("PIP") in the European Union is aimed at ensuring that the necessary data are obtained to support the authorization of a medicine for children, through studies in children. All applications for marketing authorization for new medicines have to include the results of studies as described in an agreed PIP, unless the study results are deferred, or the medicine is exempt because of a waiver. This requirement also applies when a marketing-authorization holder wants to add a new indication, pharmaceutical form, or route of
33
administration for a medicine that is already authorized and covered by intellectual property rights. Several rewards and incentives for the development of pediatric medicines for children are available in the E.U. Medicines authorized across the E.U. with the results of studies from a PIP included in the product information are eligible for an extension of their supplementary protection certificate by six months. This is the case even when the studies’ results are negative. For orphan medicines, the incentive is an additional two years of market exclusivity. Scientific advice and protocol assistance at the Agency are free of charge for questions relating to the development of pediatric medicines.
The U.K.aforementioned European Union rules are generally applicable in the EEA.
The UK left the EUEuropean Union on January 31, 2020 followingand the UK and the European Union concluded a trade and cooperation agreement (“TCA”), which existing EUwas provisionally applicable since January 1, 2021 and has been formally applicable since May 1, 2021. The TCA includes specific provisions concerning pharmaceuticals, which include the mutual recognition of cGMP, inspections of manufacturing facilities for medicinal productproducts and cGMP documents issued, but does not provide for wholesale mutual recognition of UK and European Union pharmaceutical regulations. At present, Great Britain has implemented European Union legislation continuedon the marketing, promotion and sale of medicinal products through the Human Medicines Regulations 2012 (as amended) (under the Northern Ireland Protocol, the European Union regulatory framework continues to apply in Northern Ireland). Except in respect of the U.K. duringnew European Union Clinical Trials Regulation, the transition periodregulatory regime in Great Britain therefore largely aligns with current European Union medicines regulations, however it is possible that these regimes will diverge more significantly in future now that Great Britain’s regulatory system is independent from the European Union and the TCA does not provide for mutual recognition of UK and European Union pharmaceutical legislation. However, notwithstanding that there is no wholesale recognition of European Union pharmaceutical legislation under the terms ofTCA, under the EU-UK Withdrawal Agreement. A transition period,new framework mentioned below which ended on December 31, 2020, maintained access to the EU single market and to the global trade deals
29
negotiatedwill be put in place by the EU on behalf of its members. The transition period provided time for the U.K. and EU to negotiate a framework for partnership for the future, which was then crystallized in the Trade and Cooperation Agreement ("TCA") and became effective on the January 1, 2021.
From January 1, 2021, the Medicines and Healthcare products Regulatory Agency ("MHRA"(“MHRA”), the UK’s medicines regulator, from January 1, 2024, the MHRA has stated that it will betake into account decisions on the U.K.’s standalone medicinesapproval of MAs from the EMA (and certain other regulators) when considering an application for a Great Britain MA. On February 27, 2023, the UK government and medical devices regulator. Asthe European Commission announced a result ofpolitical agreement in principle to replace the Northern Ireland protocol, different rules will apply inProtocol with a new set of arrangements, known as the “Windsor Framework”. This new framework fundamentally changes the existing system under the Northern Ireland than in England, Wales and Scotland (together Great Britain, or GB); broadly, Northern Ireland will continueProtocol, including with respect to follow the EU regulatory regime, but its national competent authority will remain the MHRA. The MHRA has published a draft guidance on how various aspectsregulation of the U.K. regulatory regime for medicines will operate in GB and in Northern Ireland following the expiry of the Brexit transition period on December 31, 2020. The guidance includes clinical trials, marketing authorizations, importing, exporting and pharmacovigilance and is relevant to any business involvedmedicinal products in the research, development or commercialization of medicines inUK. In particular, the U.K. The new guidanceMHRA will be given effect viaresponsible for approving all medicinal products destined for the Human Medicines Regulations (Amendment etc.) (EU Exit) Regulations 2019, orUK market (Great Britain and Northern Ireland), and the Exit Regulations. The U.K. regulatory regime largely mirrors that ofEMA will no longer have any role in approving medicinal products destined for Northern Ireland. Once the EU.
Windsor Framework is approved by the EU-UK Joint Committee, the UK Government and the European Union will enact legislative measures to enact it into law. The MHRA has introduced changes to national licensing procedures, including procedures to prioritize access to new medicines that will benefit patients, an accelerated assessment procedure and new routes of evaluation for novel products and biotechnological products. All existing E.U. MAs for centrally authorized products willwere automatically be converted (grand fathered) into U.K.UK MAs free-of-charge on January 1, 2021. For a period of three years from January 1, 2021, the MHRA may rely on a decision taken by the European Commission on the approval of a new MA in the centralized procedure, in order to more quickly grant a new Great Britain MA. A separate application will, however, still be required. On January 24, 2023, the MHRA announced that a new international recognition framework will be put in place from January 1, 2024, which will have regard to decisions on the approval of MAs made by the EMA and certain other regulators when determining an application for a new Great Britain MA.
There will beis now no pre-marketing authorization orphan designation.designation in Great Britain. Instead, the MHRA will review applications for orphan designation in parallel to the corresponding MA application. The criteria are essentially the same, but have been tailored for the GB market, i.e., the prevalence of the condition in GB (rather than the EU) must not be more than 5 in 10,000. Should an orphan designation be granted, the period or market exclusivity will be set from the date of first approval of the product in GB or EU/European Economic Area, wherever is earliest.
We may also apply for access to the UK’s Innovative Licensing and Access Pathway (“ILAP”) which aims to accelerate the time to market and facilitate patient access to certain types of medicinal products in development which target a life-threatening or seriously debilitating condition, or where there is a significant patient or public health need in the UK. To access the ILAP, an applicant applies for an Innovation Passport designation. Once an Innovation Passport designation is granted, the MHRA and its partner agencies (including The All Wales Therapeutics and Toxicology Centre, National Institute for Health and Care Excellence and the Scottish Medicines Consortium) will work with the Innovation Passport designee to define a Target Development Profile, (“TDP”). The
34
TDP sets out a unique product-specific roadmap towards patient access in the UK and provides access to a toolkit to support all stages of the design, development and approvals process, including continuous benefit-risk assessment, increased support for novel development approaches and enhanced patient engagement. However, although the goal of the ILAP is to reduce the time to market and enable earlier patient access, access does not accelerate conduct of clinical trials or mean that the regulatory requirements are less stringent, nor does it ensure that a marketing authorization application will be approved or that any approval will be granted within a particular timeframe or at all.
Healthcare Laws and Regulations
Coverage and Reimbursement
Our ability to successfully commercialize our product candidates will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels.
There is also significant uncertainty related to the insurance coverage and reimbursement of newly approved products and coverage may be more limited than the purposes for which the medicine is approved by the FDA or comparable foreign regulatory authorities. In the United States, the principal decisions about reimbursement for new medicines are typically made by the Centers for Medicare & Medicaid Services (“CMS”), an agency within the U.S. Department of Health and Human Services. CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare and private payors tend to follow CMS to a substantial degree. Factors payors consider in determining reimbursement are based on whether the product is a covered benefit under its health plan; safe, effective and medically necessary; appropriate for the specific patient; cost-effective; and neither experimental nor investigational. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products.
Outside of the United States, the pricing of pharmaceutical products is subject to governmental control in many countries. For example, in the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular therapy to currently available therapies or so-called health technology assessments, in order to obtain reimbursement or pricing approval. Other countries may allow companies to fix their own prices for products, but monitor and control product volumes and issue guidance to physicians to limit prescriptions. We cannot be sure that reimbursement will be available for any of our product candidates that we commercialize and, if reimbursement is available, the level of reimbursement.
Other Healthcare Laws
Sales of our product candidate, if approved, or any other future product candidate will be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments in which we might conduct our business. The healthcare laws and regulations that may affect our ability to operate include the following:
35
30
Many states have similar laws and regulations, such as anti-kickback and false claims laws that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs. Additionally, we may be subject to state laws that require pharmaceutical companies to comply with the federal government’s and/or pharmaceutical industry’s voluntary compliance guidelines, state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures, as well asexpenditures. Certain states and local jurisdictions also require the registration of pharmaceutical sales representatives. Further, certain states require the posting of information relating to clinical studies and their outcomes. A growing number of states require the reporting of certain drug pricing information, including information pertaining to and justifying price increases and the price set for newly launched drugs, or to prohibit prescription drug price gouging. We may also be subject to state and foreign laws governing the privacy and security of health information, many of which differ from each other in significant ways and often are not preempted by HIPAA. In the United States, there are numerous federal and state privacy and data security laws and regulations governing the collection, use, disclosure and protection of personal information, including federal and state health information privacy laws, federal and state security breach notification laws, and federal and state consumer protection laws. Each of these laws is subject to varying interpretations and new laws continue to be proposed. At the state level, numerous states have or are in the process of enacting or considering comprehensive
36
state-level data privacy and security laws, rules and regulations while other states have focused on more narrow aspects of privacy. In the state of Washington, for example, the My Health My Data Act will require regulated entities to obtain consent to collect health information, grant consumers certain rights, including to request deletion of their information, and provide for robust enforcement mechanisms, including enforcement by the Washington state attorney-general and a private right of action for consumer claims. Other states have also enacted, proposed, or are considering proposing, data privacy laws, which could further complicate compliance efforts, increase our potential liability and adversely affect our business. Additionally, to the extent that our product is sold in a foreign country, we may be subject to similar foreign laws.
Healthcare Reform
The United States and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system. The United States government, state legislatures and foreign governments also have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. In recent years, Congress has considered reductionsThird-party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost effectiveness of pharmaceutical or biological products, medical devices, and medical services, in Medicare reimbursement levels for drugs administered by physicians.addition to questioning safety and efficacy. CMS the agency that administers the Medicare and Medicaid programs, also has authority to revise reimbursement rates and to implement coverage restrictions for some drugs. Cost reduction initiatives and changes in coverage implemented through legislation or regulation could decrease utilization of and reimbursement for any approved products. While Medicare regulations apply only to drug benefits for Medicare beneficiaries, private payerspayors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, although private payors have their own methods and approval process apart from Medicare determinations, any reduction in reimbursement that results from federal legislation or regulation may result in a similar reduction in payments from private payers.payors.
31
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the Affordable Care Act substantially changed the way healthcare is financed by both governmental and private insurers, and significantly impactsincluded provisions that affected the pharmaceutical industry. The Affordable Care Act is intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers, and impose additional health policy reforms. Among other things, the Affordable Care Act expanded manufacturers’ rebate liability under the Medicaid Drug Rebate Program, by increasing the minimum Medicaid rebate for both branded and generic drugs, expanded the 340B program, and revised the definition of average manufacturer price ("AMP"), which could increaseincreased the amount of Medicaid drug rebates manufacturers are required to pay to states. The legislation also extended Medicaid drug rebates, previously due only on fee-for-service Medicaid utilization, to include the utilization of Medicaid managed care organizations and created an alternative rebate formula for certain new formulations of certain existing products that is intended to increase the amount of rebates due on those drugs. On February 1, 2016, CMS issued final regulations to implement the changes to the Medicaid Drug Rebate program under the Affordable Care Act. These regulations became effective on April 1, 2016. Since that time, there have been significant ongoing efforts to modify or eliminateseveral healthcare reform proposals recently culminated in the Affordable Care Act. On January 20, 2017, President Trump signed an executive order directing federal agencies to exercise existing authorities to reduce burdens associated with the Affordable Care Act pending further action by Congress. In October 2017, he signed an Executive Order which directed federal agencies to modify how the Affordable Care Act is implemented. The Tax Act, enacted on December 22, 2017, repealed the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000Aenactment of the Internal Revenue Code of 1986, as amended, or the Code, commonly referred to as the individual mandate,
Other legislative changes have been proposed and adopted since the passage of the Affordable Care Act. The Budget ControlInflation Reduction Act of 2011, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals(“IRA”) in spending reductions to Congress. The Joint Select Committee did not achieve its targeted deficit reduction of an amount greater than $1.2 trillion for the fiscal years 2012 through 2021, triggering the legislation’s automatic reductions to several government programs. These reductions included aggregate reductions to Medicare payments to healthcare providers of up to 2.0% per fiscal year, which went into effect in April 2013. Subsequent legislation extended the 2% reduction, on average, to 2030 unless additional Congressional action is taken. However, pursuant to the Coronavirus Aid, Relief and Economic Security Act ("CARES Act") the 2% Medicare sequester reductions were suspended from May 1, 2020 through December 31, 2021 due to the COVID-19 pandemic. The sequester will remain in place through 2030. On January 2, 2013, the American Taxpayer Relief Act was signed into law,August 2022, which, among other things, reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
The Affordable Care Act also has been subject to challenges in court. On December 14, 2018, a Texas U.S. District Court Judge ruled that the Affordable Care Act is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. On December 18, 2019, the Fifth Circuit U.S. Court of Appeals held that the individual mandate is unconstitutional and remanded the case to the Texas District Court to reconsider its earlier invalidation of the entire Affordable Care Act. An appeal was taken to the U.S. Supreme Court. On June 17, 2021, the Supreme Court ruled that the plaintiffs lacked standing to challenge the law as they had not alleged personal injury traceable to the allegedly unlawful conduct. As a result, the Supreme Court did not rule on the constitutionality of the Affordable Care Act.
Further changes to and under the Affordable Care Act remain possible but it is unknown what form any such changes or any law proposed to replace or revise the Affordable Care Act would take, and how or whether it may affect our business in the future. We expect that changes to the Affordable Care Act, the Medicare and Medicaid programs, changes allowing the federal governmentallows HHS to directly negotiate the selling price of a statutorily specified number of drugs and biologics each year that CMS reimburses under Medicare Part B and Part D. Only high-expenditure single-source biologics that have been approved for at least 11 years (7 years for drugs) can be selected by CMS for negotiation, with the negotiated price taking effect two years after the selection year. Negotiations for Medicare Part D products take place in 2024 with the negotiated price taking effect in 2026, and negotiations for Medicare Part B products will begin in 2026 with the negotiated price taking effect in 2028. In August 2023, HHS announced the ten Medicare Part D drugs and biologics that it selected for negotiations. HHS will announce the negotiated maximum fair prices by September 1, 2024, and this price cap, which cannot exceed a statutory ceiling price, will go into effect on January 1, 2026. A drug or biological product that has an orphan drug designation for only one rare disease or condition will be excluded from the IRA’s price negotiation requirements, but will lose that exclusion if it receives designations for more than one rare disease or condition, or if is approved for an indication that is not within that single designated rare disease or condition, unless such additional designation or such disqualifying approvals are withdrawn by the time CMS evaluates the drug for selection for negotiation. The IRA also imposes rebates on Medicare Part B and Part D drugs whose prices have increased at a rate greater than the rate of inflation. The IRA also extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. The IRA permits the Secretary of HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years. Manufacturers that fail to comply with the IRA may be subject to various penalties, including significant civil monetary penalties. These provisions have been and changes stemming from other healthcare reform measures, especially with regardmay continue to healthcare access, financing or other legislationbe subject to legal challenges. For example, the provisions related to the negotiation of selling prices of high-expenditure single-source drugs and biologics have been challenged in individual states, couldmultiple lawsuits brought by pharmaceutical manufacturers. Thus, while it is unclear how the IRA will be implemented, it will likely have a material adverse effectsignificant impact on the healthcare industry.biopharmaceutical industry and the pricing of prescription drug products.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. For example, the FDA released a final rule in
3237
September 2020 providing guidance for states to build and submit plans for importing drugs from Canada, and FDA authorized the first such plan in Florida in January 2024. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs.
We expect that additional federal, state and foreign healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in limited coverage and reimbursement and reduced demand for our products, once approved, or additional pricing pressures.
3338
ITEM 1A. RISK FACTORS
You should consider carefully the following risks and uncertainties when reading this Annual Report on Form 10-K, as well as the other information contained herein, including our audited consolidated financial statements and the related notes and the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operation.” If any of the following risks occur, our business, financial condition and results of operations could be materially and adversely affected. Although we believe that we have identified and discussed below the key risk factors affecting our business, there may be additional risks and uncertainties that are not presently known or that are not currently believed to be significant that may adversely affect our performance or financial condition. In that event, the trading price of our common stock could decline.
Risks Related to Our Financial Position and Need for Capital
We have incurred significant losses since our inception and anticipate that we will incur continued losses for the foreseeable future.
Since our inception we have devoted substantially all of our resources to the development of CTI-1601.nomlabofusp. We have incurred significant losses in each year of operation since our inception in 2016. For the yearyears ended December 31, 20212023 and 2022, we had net losses of $50.6$36.9 million and $35.4 million, respectively, and, as of December 31, 2021,2023, we had an accumulated deficit of $116.3$188.6 million and we expect to continue to incur significant expenses and net operating losses ("NOLs") for the foreseeable future.
We have devoted substantially all of our financial resources and efforts to research and development, including non-clinical studies, our clinical development program, the development of manufacturing processes as well as the manufacture of initial lots of clinical trial material. We expect to incur significant losses for the foreseeable future to further develop and commercialize our lead drug candidate, CTI-1601.nomlabofusp.
We expect that our expenses will increase substantially if and as we:
We currently have no sales, marketing or medical affairs infrastructure and have no experience in the sales, marketing, or distribution of pharmaceutical products. Assuming nomlabofusp ultimately is approved for marketing in the US and elsewhere, we will need to establish commercial capabilities as well as customer service and support, logistics, and other related functions, or make arrangements with third parties to perform these services.
Net losses and negative cash flows have had, and will continue to have, an adverse effect on our liquidity and potentially the ability for us to raise capital due to our unfavorable operating results.
We have no commercial revenue and may never become profitable.
To date, we have not generated any commercial revenue. Our ability to generate revenue and become profitable depends upon our ability to obtain regulatory approval for, and to successfully commercialize, CTI-1601nomlabofusp or other product candidates that we may develop, in-license or acquire in the future.
34
This will require success in a range of challenging activities, including completing numerous clinical trials of CTI-1601nomlabofusp or any future product candidates, obtaining marketing approval for CTI-1601nomlabofusp and any future product candidates, manufacturing, marketing and selling those products for which we, or any future collaborators
39
or partners, may obtain marketing approval, satisfying any post-marketing requirements and obtaining reimbursement for our products from private insurance and/or government payors. Even if we are able to successfully achieve the above, we do not know what the reimbursement status of CTI-1601nomlabofusp or any other future product candidates will be or when any of these products will generate revenue for us, if at all. We have not generated, and do not expect to generate, any product revenue for the foreseeable future, and expect to continue to incur significant operating losses for the foreseeable future due to the cost of research and development, non-clinical studies and clinical trials and the regulatory approval process for CTI-1601nomlabofusp and any future product candidates.
Our ability to generate revenue from CTI-1601nomlabofusp or any future product candidates also depends on a number of additional factors, including our ability to:
Because of the uncertainties and risks associated with these activities, we are unable to accurately predict the timing and amount of increased expenses, and if or when we might achieve or maintain profitability. We and any future collaborators may never succeed in these activities and, even if we do, or any future collaborators do, we may never generate revenues that are large enough for us to achieve profitability. Even if we are able to complete the processes described above, we anticipate incurring significant costs associated with commercializing CTI-1601nomlabofusp or any of our future product candidates. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our failure to become and remain profitable could decrease the value of our business and could impair our ability to raise capital, maintain our discovery and clinical development efforts, expand our business or continue our operations and may require us to raise additional capital that may dilute the ownership interest of shareholders. A decline in the value of our business could also cause shareholders to lose all or part of their investment.
We may need to raise additional funding in order to continue our planned operations.complete the development and commercialization of nomlabofusp. This funding may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed would force us to delay, limit or terminate our product development efforts or other operations.
As of December 31, 2023, our existing cash, cash equivalents and marketable securities were $86.8 million, excluding restricted cash of $1.3 million. In February 2024, we completed an underwritten public offering of 19,736,842 shares of our common stock at an offering price of $8.74 per share with net proceeds, after deducting underwriting commissions and offering costs, of approximately $161.6 million. Together with our existing cash, cash equivalents and marketable securities, we have adequate resources to fund our operations into 2026.
We expect to continue to spend substantial and increasing amounts to conduct clinical trials of CTI-1601nomlabofusp and further research and development activities for CTI-1601,nomlabofusp, and for any additional product candidates that we may develop, in-license or acquire in the future. In addition, raising funds in the current economic environment may present substantial challenges, and our expenses will increase as we expand, through development, in-license or acquisition,
35
our pipeline of product candidates. If we obtain marketing approval for any of our product candidates, we will likely incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution to the extent that such sales, marketing, manufacturing and distribution are not the responsibility of a future collaborator. Accordingly, we will need to obtain additional funding in connection with our continuing operations.
As of December 31, 2021, our existing Our current cash, and cash equivalents were $70.1 million, excluding restricted cash of $1.3 million. This amount willand marketable securities may not be sufficient to fund all of the
40
efforts that we plan to undertake or to fund the completion of the development and commercialization of CTI-1601.nomlabofusp. Accordingly, we willmay be required to obtain further funding through public or private equity offerings, debt financings, collaborations and licensing arrangements or other sources. The incurrence of indebtedness would result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates.
ThereIf additional capital is needed to complete the development and commercialization of nomlabofusp, there can be no assurance that we will be able to raise sufficient additional capital on acceptable terms or at all. If such additional financing is not available on satisfactory terms, or is not available in sufficient amounts, or we do not have sufficient authorized shares, we may be required to delay, limit, or eliminate the development of business opportunities and our ability to achieve our business objectives, our competitiveness, and our business, financial condition, and results of operations will be materially adversely affected. We could also be required to seek funds through arrangements with collaborative partners or otherwise at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results, financial condition and prospects.prospects. In addition, geopolitical unresttension including, for example, the potentialbroader impact of the Russian invasion ofongoing conflict between Russia and Ukraine and the possibility that thecurrent conflict could expand beyond eastern Europe,in Israel and Gaza (including any escalation or expansion), and the impact of thea future pandemic, epidemic or outbreak of an infectious disease, such as COVID-19, pandemic or other health crisisrecent liquidity constraints, failures and instability in U.S. and international financial banking systems on thethe global financial markets may reduce our ability to access capital, which could negatively affect our liquidity.
If additional capital is needed to complete the development and commercialization of nomlabofusp, and if we are unable to obtain funding when needed and/or on acceptable terms, we may be required to significantly curtail, delay or discontinue one or more of itsour research and development programs, the manufacture of clinical and commercial supplies, product portfolio expansion or pre commercialization efforts, which could adversely affect our business prospects, or we may be unable to continue operations.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies, CTI-1601nomlabofusp or other product candidates that we may develop, in-license or acquire in the future.
We may seek additional capital through a combination of private or public equity offerings, debt financings, collaborations and licensing arrangements or other sources. To the extent we raise additional capital through the sale of equity or convertible debt securities, existing ownership interests will be diluted and the terms of such financings may include liquidation or other preferences that adversely affect the rights of existing stockholders. Debt or equity financings may be coupled with an additional equity component, such as warrants to purchase shares, which could also result in dilution of our existing stockholder’s ownership.
If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights, including future revenue streams, to CTI-1601nomlabofusp or other product candidates that we may develop, in-license or acquire in the future, or grant licenses on terms that are not favorable to us.
Our ability to use our NOLs and certain other tax attributes may be limited.
As of December 31, 2023 we had NOL carryforwards that expire for U.S. federal income tax purposes of $179.0 million, a portion of which begin to expire in 2026. Our NOLs could expire unused and be unavailable to offset future income tax liabilities because of their limited duration or because of restrictions under U.S. tax law. NOLs generated in taxable years beginning before January 1, 2018 are permitted to be carried forward for 20 taxable years under applicable U.S. federal income tax law. Under current U.S. federal income tax law, NOLs arising in tax years beginning after December 31, 2020 may not be carried back. Moreover, NOLs generated in taxable years beginning after December 31, 2017 may be carried forward indefinitely. As of December 31, 2023, the Company had federal net operating loss carryforwards that were generated after December 31, 2017 of $140.3 million that do not expire, however these carryforwards are limited to 80% of the taxable income in any one tax period.
In general, under Section 382 of the Internal Revenue Code ("the Code"(the "Code") if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change (by value) in its equity ownership over a
41
three-year period, the corporation’s ability to use its pre-change Net Operating Losses ("NOLs")NOLs and other pre-change tax attributes (such as capitalized research and development costs and research tax credits) to offset its post-change income may be limited. We believe that as a result of our merger with Zafgen, Inc., our ability to utilize NOLs acquired in this
36
the transaction and our other NOLs is expected to be severely limited by Section 382 of the Code. Additionally, our July 2021, September 2022 and February 2024 equity transactions could also limit our ability to utilize NOLs in the future. We may also experience ownership changes in the future as a result of subsequent shifts in our stock ownership. As a result, if we earn net taxable income, our ability to use our pre-change NOLs to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. The Tax Cuts and Jobs Act of 2017 ("Tax Act") which significantly reformed the Code, also reduced the corporate income tax rate to 21%, from a prior rate of 35%. This may cause a reduction in the economic benefit of our NOLs and other deferred tax assets available to us. In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed and would adversely affect our business, financial condition and results of operations.
Changes in tax laws and regulations may have a material adverse effect on our business, financial condition and results of operations.
New income, sales, use or other tax laws, statutes, rules, regulations or ordinances could be enacted at any time, which could affect the tax treatment of any of our future domestic and foreign earnings. Any new taxes could adversely affect our domestic and international business operations, and our business and financial performance. Further, existing tax laws, statutes, rules, regulations or ordinances could be interpreted, changed, modified or applied adversely to us. For example, under Section 174 of the U.S. government enacted significant tax reformscode, in the past, and certain provisions of any new laws may adversely affect us. Changes in recent years include, but are not limited to, a federal corporate tax rate decrease to 21% for taxtaxable years beginning after December 31, 2017, a reduction to2021, expenses that are incurred for research and development in the maximum deduction allowed for net operating losses generated in tax years after December 31, 2017, eliminating carrybacks of net operating losses,U.S. are capitalized and providing for indefinite carryforwards for losses generated in tax years after December 31, 2017. The legislation is unclear in many respects and could be subject to potential amendments and technical corrections, and will be subject to interpretations and implementing regulations by the Treasury and Internal Revenue Service, any ofamortized, which could mitigate or increase certainmay have an adverse effects of the legislation.effect on our cash flow. In addition, it is unclear how these U.S. federal income tax changes will affect state and local taxation. Generally, future changes in applicable U.S. tax laws and regulations, or their interpretation and application could have an adverse effect on our business, financial conditions and results of operations.
Risks Related to Our Product Development and Regulatory Approvals
The FDA has placed a partial clinical hold on CTI-1601.
Our business may be adversely affectednomlabofusp and there is uncertainty as to when, or if, the FDA does notwill lift the partial clinical hold in a timely manneron our nomlabofusp program, or if additional non-clinical orit will ever allow further clinical studies are required. This may cause significant delays or expensedevelopment of nomlabofusp beyond our currently ongoing OLE study at the 25 mg dose level.
To support extended dosing of patients with nomlabofusp, we conducted a 26-week NHP toxicology study. In May 2021, we notified the FDA of certain mortalities which occurred at the two highest dose levels in developing CTI-1601.the then ongoing study. On May 25, 2021, the FDA placed a clinical hold on the CTI-1601 investigational new drug application (IND)nomlabofusp clinical program following our notification to the FDA of mortalities which occurred at the highest dose levels in an ongoing 26-week non-human primate (NHP) toxicology study, which is designed to support extended dosing of patients with CTI-1601.program. In the clinical hold letter, the FDA stated that it needed to review a full study report from the then ongoingthen-ongoing NHP study and that we maycould not initiate additional interventional clinical trials until we have submitted thesuch report and received notification from the FDA that additional clinical trials maycould commence. At the time of the FDA clinical hold, we had no interventional clinical trials with patients enrolling or enrolled.
In July 2021, we completed dosing in the 26-week NHP toxicology study. The study included four dose groups in addition to vehicle. Data from the study were collected throughout the second half of 2021 and included in the complete response to the clinical hold submitted to the FDA in January 2022. The study included four dose groups in addition to vehicle and the mortalities were seen in the two highest dose groups.
In February 2022, in response to the complete response to clinical hold, we received a response fromsubmitted to the FDA, following our submission of a complete response including a comprehensive study report from the 26-week NHP toxicology study. The FDA stated that it was maintaining its clinical hold at this time and requested additional information. We are evaluating how best to respond to their request. We are reassessing the timing of the planned open label extension and the pediatric MAD studies.
We cannot be certain whether or when the FDA will lift the clinical hold and allow usthat additional data were needed to pursue furtherresolve the clinical hold. We subsequently submitted a request to the FDA for a Type C meeting, which was granted and held in July 2022. We submitted a complete response to clinical hold incorporating additional information requested by the FDA at the meeting as well as information on the proposed study in August 2022.
In September 2022, following the Type C meeting and the submission of the complete response to clinical hold, the FDA allowed the 25 mg cohort of a Phase 2, four-week, placebo-controlled, dose exploration trial of nomlabofusp in FA patients discussed above to proceed. In connection with this decision, the FDA lifted its full clinical hold on the nomlabofusp clinical development program and imposed a partial clinical hold.
In June 2023, we met with the FDA. Following that meeting, we submitted a complete response to the FDA’s partial clinical hold that included unblinded safety, PK and frataxin data from the Phase 2 trial’s completed 25 mg cohort.
42
In July 2023, following the FDA’s review of CTI-1601our complete response to the partial clinical hold, the FDA cleared initiation of a second cohort (50 mg) in our four-week, placebo-controlled, Phase 2 dose exploration trial and the initiation of our OLE trial with daily dosing of 25 mg.
In January 2024, we initiated the OLE trial evaluating daily subcutaneous injections of 25 mg of nomlabofusp self-administered or administered by a caregiver, with the first patient dosed in March 2024. Dose escalation in the United States. IfOLE trial will be considered based on safety, tolerability, PK, and tissue FXN levels from the Phase 2 trial’s 50 mg cohort as well as available data from the 25 mg dose of nomlabofusp in the OLE trial, and is contingent on FDA does not liftreview as part of the partial clinical holdhold. Initial data from the OLE trial is expected in a timely manner, or atthe fourth quarter of 2024.
In February 2024, we reported positive top-line data and successful completion of both the 25 mg cohort and the 50 mg cohort of our four-week, placebo-controlled Phase 2 dose exploration study of nomlabofusp in participants with FA. Nomlabofusp was generally well tolerated and demonstrated dose dependent increases in FXN levels in all our development timelinesevaluated tissues (skin and ourbuccal cells) after daily dosing of 14 days followed by every other day dosing until day 28 in the 25 mg and 50 mg cohorts.
Our business may be adversely affected and our stock price may decline. Further, even if the FDA lifts thepartial clinical hold ordoes not enable the development program to proceed as planned, if the FDA places a full clinical hold on the development of nomlabofusp, or, other regulatory agencies continue to express safety concerns after the hold is lifted, future CTI-1601if additional non-clinical or clinical studies are required. This may be required. In such instances, our progresscause significant delays or expense in developing nomlabofusp.
37
the development of CTI-1601 may be significantly slowed and the associated costs may be significantly increased, adversely affecting our business, which could impair our ability to ultimately obtain FDA approval for CTI-1601
Our success is currently dependent upon the success of our solelead product candidate, CTI-1601,for which we have completed two Phase 1 clinical trials in patients with Friedreich’s ataxia. Clinical development of CTI-1601 is currently under clinical hold.nomlabofusp. We cannot be certain that data from both cohorts of our Phase 2 dose exploration study augmented by data from the OLE trial will provide the FDA with adequate data to remove the partial clinical hold on nomlabofusp, allow the nomlabofusp development program to proceed as planned in part or in full, or that we will ultimately be successful with our clinical development or that we will be ever able to obtain regulatory approval for CTI-1601.nomlabofusp.
We currently have no drug products for sale and our business is currently wholly dependent on our successful clinical development, regulatory approval and commercialization of CTI-1601,nomlabofusp, our lead product candidate and our only product candidate in clinical development, for which we have completed two Phase 1 clinical trialsstudies and a four-week, placebo-controlled Phase 2 dose exploration study in patients with Friedreich’s ataxia. ClinicalFA. In January 2024, as permitted by the FDA, we initiated the OLE trial evaluating daily subcutaneous injections of 25 mg of nomlabofusp self-administered or administered by a caregiver, with the first patient dosed in March 2024. Participants who completed treatment in the Phase 2 dose exploration trial, or who previously completed a prior clinical trial of nomlabofusp are potentially eligible for the OLE. Dose escalation in the OLE trial will be considered based on safety, PK, and tissue FXN levels from the 25 mg dose of nomlabofusp in the OLE trial, as well as other data from the Phase 2 trial’s 50 mg cohort, and is contingent on FDA review as part of the partial clinical hold. We cannot provide any assurance that the data from our Phase 1 and Phase 2 trials augmented by data from the OLE trial will prove the FDA with adequate data to remove the partial clinical hold, allow us to continue our development of CTI-1601 is currently undernomlabofusp, or that we will ultimately be successful in our clinical hold.development.
In addition to the regulatory and manufacturing hurdles faced by our product candidate, the administration of a protein such as nomlabofusp, may cause an immune response, resulting in the creation of antibodies directed against the protein. These anti-drug antibodies can have no effect or can neutralize the effectiveness of the protein or require that higher doses be used to obtain a therapeutic effect. Neutralizing antibodies may be detected at a later date or upon longer exposure periods and there can be no assurance that neutralizing antibodies will not be detected in the future.
If our efforts to develop and commercialize CTI-1601nomlabofusp for the treatment of Friedreich’s ataxiaFA are unsuccessful, or we experience significant delays in doing so, our business could also be substantially harmed. The success of CTI-1601nomlabofusp will depend on several factors, including the following:
43
38
Many of these factors are outside of our control, including the clinical development and regulatory approval processes, results of non-clinical and toxicology studies and clinical trials, potential threats to our intellectual property rights and the manufacturing, marketing and sales efforts, respectively. The process of obtaining regulatory approval is expensive and time consuming. The FDA and foreign regulatory authorities may never approve CTI-1601nomlabofusp for sale and marketing, and even if CTI-1601nomlabofusp is ultimately approved, regulatory approval may be delayed or limited in the United States or in other jurisdictions. Even if we are authorized to sell and market CTI-1601nomlabofusp in one or more markets, there is no assurance that we will be able to successfully market CTI-1601nomlabofusp or that CTI-1601nomlabofusp will achieve market acceptance sufficient to generate profits. If we are unable to successfully develop and commercialize CTI-1601nomlabofusp due to failure to obtain regulatory approval for CTI-1601,nomlabofusp, to successfully market CTI-1601,nomlabofusp, to generate profits from the sale of CTI-1601,nomlabofusp, or due to other risk factors outlined in this report, it would have material adverse effects on our business, financial condition, and results of operations as CTI-1601nomlabofusp is currently our sole product candidate.
Clinical development is a lengthy and expensive process with an uncertain outcome, and the results of non-clinical studies, toxicology studies or clinical trials may not be predictive of future non-clinical studies, toxicology studies or clinical trial results.
Clinical development is expensive and can take many years to complete, and its outcome is inherently uncertain. We cannot guarantee that any non-clinical studies, toxicology studies or clinical trials will be conducted as planned or completed on schedule, if at all, and failure can occur at any time during the non-clinical study, toxicology study or clinical trial process. Despite promising non-clinical, toxicology or clinical results, any product candidate can unexpectedly fail at any stage of non-clinical, toxicology or clinical development. The historical failure rate for product candidates in our industry is high, especially for products in early stages of development.
The results from non-clinical studies, toxicology studies or clinical trials of a product candidate may not predict the results of later non-clinical or clinical trials of the product candidate, or in clinical trials with different patient populations such as children and adolescents and interim results of a clinical trial are not necessarily
44
indicative of final results. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy characteristics despite having progressed through non-clinical studies and initial clinical trials. It is not uncommon to observe results in clinical trials that are unexpected based on non-clinical studies and early clinical trials, and many product candidates fail in clinical trials despite very promising early results. Favorable safety and efficacy outcomes in adult clinical trials may not be seen in pediatric clinical trials.
Moreover, current and future non-clinical and clinical data may be susceptible to varying interpretations and analyses. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical development even after achieving promising results in earlier studies. Furthermore, we cannot provide assurance that we will be able to successfully progress any future non-clinical programs from candidate identification to Phase 1 clinical development. As is typical in candidate development, we have a program of ongoing toxicology studies in animals for CTI-1601nomlabofusp and cannot provide assurance that the findings from such studies or any ongoing or future clinical trials will not adversely affect the clinical development of CTI-1601.nomlabofusp. For the foregoing reasons, we cannot be certain that our ongoing and planned non-clinical studies and clinical trials will be successful. If non-clinical studies or clinical trials for CT1-1601nomlabofusp or any future product candidates or indications fail to demonstrate safety or efficacy to the satisfaction of the FDA or the equivalent regulatory authorities in other countries, the FDA or equivalent regulatory authority will not approve our product candidates in those and other indications, which could have a material adverse effect on our business, financial condition results of operations and prospects.
We do not know whether any ongoing or future clinical trials for CTI-1601nomlabofusp will be completed on schedule, if at all, as the commencement and completion of clinical trials can be delayed, prevented or terminated for a number of reasons, including as a result of safety concerns, or ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated by us, the FDA, other regulatory authorities, the institutional review boards
39
("IRBs")IRBs or ethics committees, aan independent data monitoring committee, or safety review committee, overseeing the clinical trial at issue or other regulatory authorities due to a number of factors, including, among others:
45
Failures or delays in the completion of our clinical trials could result in increased costs and could delay, prevent or limit our ability to generate revenue and continue our business.
For our lead product candidate CTI-1601,nomlabofusp, we have completed two Phase 1 clinical trials in patients with Friedreich’s ataxia butFA and our four-week, placebo-controlled Phase 2 dose exploration study of nomlabofusp in FA patients. The FDA has agreed that we may commence our OLE study in patients who previously participated in one of our previous nomlabofusp clinical development is currently subjecttrials at the 25 mg level. We cannot be certain that data from both cohorts of our Phase 2 dose exploration study augmented by data from the OLE study will provide the FDA with adequate data to aremove the current partial clinical hold issued by the FDA. To receive approval of CTI-1601 in other countries we will also have to satisfy their regulatory requirements. Satisfaction of these requirements will entail substantial time, effort and financial resources. We may never satisfy these requirements. on nomlabofusp.
Clinical trials may also be delayed or terminated as a result of safety issues in non-clinical or clinical trials, ambiguous or negative interim results or events outside of our control. Because Friedreich’s ataxia is a rare disease, there are a limited number of patients in close proximity to clinical trial sites and clinical trial patients must travel from throughout the United States to the clinical trial sites to participate if clinical development is allowed to continue. The travel advisories and risk of infection related to COVID-19 may present increased risks to patients traveling to our clinical trial sites for dosing, if clinical trials are allowed to continue. The evolution of COVID-19 variants could be resistant to current vaccines which, if the clinical hold is lifted, may preclude us from conducting
40
clinical studies unless or until an effective new or modified vaccine is available. If future clinical trials of CTI-1601nomlabofusp fail or further delays occur in the United States and or other countries, we may not be able to develop and commercialize CTI-1601nomlabofusp and could fail to realize the potential advantages of doing so, and it could materially adversely affect our business, financial condition and results of operations.
In addition, disruptions caused by future pandemic, epidemic or outbreak of an infectious disease, such as the COVID-19 pandemic may increase the likelihood that we encounter such difficulties or delays in initiating, enrolling, conducting or completing our planned and ongoing clinical trials. Further, FA is a rare disease, there are a limited number of patients in close proximity to clinical trial sites and clinical trial patients may need to travel from other countries to the clinical trial sites in order to participate. In addition, given the limited number of FA patients, the recent approval of a competing therapy for the treatment of FA may make patients less likely to enroll in our clinical trials or less likely to be eligible for our clinical trials. Any inability to successfully initiate or complete clinical trials could result in additional costs to us or impair our ability to generate revenue from product sales. In addition, if we make manufacturing or formulation changes to our product candidates, we may be required to or we may elect to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical trial delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may seriously harm our business.
We may not be successful in our efforts to identify, discover or acquire additional product candidates.
We currently only have one product candidate CTI-1601.nomlabofusp in clinical development, although we have other product candidates in pre-clinical development. Therefore, the success of our business largely depends upon our ability to identify, develop, in-license or acquire and commercialize products targeting rare diseases. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful. In addition, our research methodology may be unsuccessful in identifying potential product candidates or our potential product candidates may be shown to have harmful side effects or may have other characteristics that may make the products unmarketable or unlikely to receive marketing approval.
Research programs to identify new product candidates require substantial technical, financial and human resources. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful. If any of these events occur, we may be forced to abandon our development efforts for a program or programs, which would have a material adverse effect on our business, financial condition and results of operations.
We have no marketed proprietary products and have not yet advanced a product candidate beyond Phase 12 clinical trials, which makes it difficult to assess our ability to develop CTI-1601nomlabofusp or any future product candidates and commercialize any resulting products independently.
We have nolittle experience in later stage clinical development, and related regulatory requirements or the commercialization of products. As a result, we have not yet demonstrated our ability to independently and repeatedly conduct clinical development after Phase 1, which has not yet been successfully completed, successfully conduct an international multi-center clinical trial, conduct a pivotal clinical trial, obtain regulatory approval, manufacture drug product on a commercial scale or
46
arrange for a third party to do so on our behalf, and commercialize therapeutic products. We will need to develop such abilities if we are to execute on our business strategy to develop and independently commercialize product candidates for orphan and niche indications. To execute on our business plan for the development of independent programs, we will need to successfully:
41
If we are unsuccessful in accomplishing these objectives, we will not be able to develop and commercialize any product candidates independently and could fail to realize the potential advantages of doing so, and it would materially adversely affect our business, financial condition and results of operations.
We cannot be certain that we will be able to successfully complete clinical trials for CTI-1601nomlabofusp or any other product candidates.
We have advanced only one product candidate into clinical development, CTI-1601.nomlabofusp. Our business currently depends primarily on CTI-1601’snomlabofusp’s successful clinical development, regulatory approval and commercialization. We submitted our IND and it was accepted, permitting the conduct of clinical trials. We completed two Phase 1 clinical trials but, asand our four-week, placebo-controlled Phase 2 dose exploration study. As a result of certain mortalities in a non-clinical study of non-human primates,NHPs, the FDA issued a clinical hold. We respondedhold in May 2021.
In September 2022, following a Type C meeting and the submission of our complete response to the partial clinical hold, the FDA allowed the 25 mg cohort of a Phase 2, four-week, placebo-controlled, dose exploration trial of nomlabofusp in January 2022.FA patients to proceed. In connection with this decision, the FDA lifted its full clinical hold on the nomlabofusp clinical development program and imposed a partial hold.
In June 2023, we met with the FDA. Following that meeting, we submitted a complete response to the FDA’s partial clinical hold that included unblinded safety, PK and frataxin data from the Phase 2 trial’s completed 25 mg cohort.
In July 2023, following the FDA’s review of our complete response to the partial clinical hold, the FDA maintainedcleared initiation of a second cohort at 50 mg of our four-week, placebo-controlled, Phase 2 dose exploration trial and to initiate our OLE trial with daily dosing of 25 mg.
In February 2024, we reported positive top-line data and successful completion of our four-week, placebo-controlled Phase 2 dose exploration study of nomlabofusp in participants with FA. Nomlabofusp was generally well tolerated and demonstrated dose dependent increases in FXN levels in all evaluated tissues (skin and buccal cells) after daily dosing of 14 days followed by every other day dosing until day 28 in the 25 mg and 50 mg cohorts. Participants in the 25 mg (n=13) and 50 mg (n=15) cohorts were randomized 2:1 to receive subcutaneous injections of nomlabofusp or placebo. The initiation of additional U.S. clinical trials evaluating nomlabofusp are contingent on FDA review of data under the partial clinical hold.
The FDA has agreed that we may commence our OLE study in patients who previously participated in one of our previous nomlabofusp clinical trials at the 25 mg level dosed daily. We are workingcannot be certain that data from both cohorts of our Phase 2 dose exploration study augmented by data from the OLE study will provide the FDA with adequate data to addressremove the issues raised by the FDA. Clinical development of CTI-1601 cannot continue until thecurrent partial clinical hold is lifted. on nomlabofusp, allow the nomlabofusp development program to proceed as planned in part or in full, or that we will ultimately be successful with our clinical development or that we will be ever able to obtain regulatory approval for.
In clinical development of any product candidate, the outcome of toxicology studies and early clinical trials may not be positive and may not be predictive of the success of later non-clinical studies or clinical trials. Adverse toxicology or safety results could lead to clinical holds or other developmental delays. Interim results of clinical
47
trials do not necessarily predict success in those or future clinical trials. Success in adult clinical trials may not predict the outcome of pediatric clinical trialstrials.
Published clinical data or case reports from third parties or early clinical trial data of CTI-1601nomlabofusp or any future product candidates may not be predictive of the results of later-stage clinical trials. Interpretation of results from early, usually smaller, studies that suggest a clinically meaningful response in some patients, requires caution. Results from later stages of clinical trials enrolling more patients, or different patient populations, such as pediatric patients, may fail to show the desired safety or efficacy results or otherwise fail to be consistent with the results of earlier trials of the same product candidate. Later clinical trial results may not replicate earlier clinical trials for a variety of reasons, including differences in trial design, different trial endpoints (or lack of trial endpoints in exploratory studies), different patient population, number of patients, patient selection criteria, trial duration, drug dosage and formulation and lack of statistical power. These uncertainties are enhanced where the diseases under study lack established clinical endpoints, validated measures of efficacy, as is often the case with orphan diseases for which no drugs have been developed previously and where the product candidates target novel mechanisms. For example, to our knowledge, CTI-1601nomlabofusp is the only protein replacement therapy being developed for the treatment of Friedreich’s ataxiaFA and therefore non-clinical studies may not be adequate to predict efficacy in a clinical trial due to our novel protein replacement therapy platform.
In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, such as pediatric patients, variability of the disease being studied, changes in and adherence to the dosing regimen and other clinical trial protocols and the rate of dropout among clinical trial participants. If we fail to ultimately receive positive results in clinical trials of CTI-1601,nomlabofusp, the development timeline and regulatory approval and commercialization prospects for CTI-1601,nomlabofusp, and, correspondingly, our business, financial prospects and results of operation would be negatively impacted.
Further, CTI-1601nomlabofusp or any future product candidates may not be approved even if they achieve their primary endpoint in clinical trials. The FDA, EMA or foreign regulatory authorities may disagree with our trial design and our interpretation of data from non-clinical studies and clinical trials. In addition, anyFDA, EMA or foreign regulatory authorities may disagree with the extent of population exposure to assess clinical safety. Any of these regulatory authorities may change its requirements for the approval of a product candidate even after reviewing and providing comments or advice on a protocol for a pivotal clinical trial that, if successful, would potentially form the basis for an application for approval by the FDA, EMA or another regulatory authority. Furthermore, any of these regulatory authorities may also approve CTI-1601nomlabofusp or any future product candidates for a narrower indication than we may request or may grant approval contingent on the performance of costly post-marketing clinical trials. Any of the above could materially adversely affect our business, financial condition and results of operations.
42
We may experience difficulties identifying and enrolling patients in our clinical trials given the limited number of patients who have the disease for which CTI-1601nomlabofusp is being studied or for any other product candidate we may study in the future. Difficulty in enrolling patients could delay or prevent clinical trials of CTI-1601nomlabofusp or any future product candidate. There are also competing FA therapeutics, other competing studies and potentially other FA therapeutics that could be approvedthat may also limit the availability of prospective participants in CTI-1601nomlabofusp clinical trials.
Identifying and qualifying patients to participate in clinical trials of CTI-1601nomlabofusp is critical to our success. The timing of our clinical trials depends in part on the speed at which we can recruit patients to participate in testing CTI-1601,nomlabofusp, and we may experience delays in our clinical trials if we encounter difficulties in enrollment, such as difficulties with enrollment in pediatric clinical trials.
The conditions for which we are planning to evaluate CTI-1601nomlabofusp and any product candidates we may evaluate in the future, are rare genetic diseases. Accordingly, there are limited patient pools from which to draw for clinical trials. We are investigating our product candidate in Friedreich’s ataxia, a rare disease. Arranging for investigative sites and recruiting patients for clinical trials in this disease may be very difficult. In addition, ifThe recent FDA approval of a product for the treatment of FA may impact our ability to enroll patients in our clinical trials as patients using other FA treatments may be excluded from participation in our nomlabofusp studies or may be less likely to participate in our nomlabofusp clinical trials due to the availability of another FA therapy. If other companies are studying their investigational products in Friedreich’s ataxia and/or if other companies have their products approved for the treatment of FA, it may be more difficult to enroll eligible patients into our clinical trials. Competing priorities at sites and participation of subjects in other studies may limit our ability to execute clinical trials in a timely fashion, if at all.
48
In addition to the rarity of Friedreich’s ataxiaFA and other diseases that we are studying, the eligibility criteria of our clinical trials will further limit the pool of available study participants as it will require patients to have specific characteristics that we can measure to assure their disease is either severe enough or not too advanced to include them in a clinical trial. The process of finding and diagnosing patients may prove costly, especially since the diseases we are studying are rare. We also may not be able to identify, recruit, and enroll a sufficient number of appropriate patients to complete our clinical trials because of demographic criteria for prospective patients, the perceived risks and benefits of the product candidate under study, the proximity and availability of clinical trial sites for prospective patients, and the patient referral practices of physicians. The availability and efficacy of approved and competing therapies and clinical trials can also adversely impact enrollment. Furthermore, our inability to enroll a sufficient number of patients for our clinical trials, including pediatric clinical trials, could result in significant delays or may require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for CTI-1601nomlabofusp or any future product candidates, and jeopardize our ability to achieve our clinical development timeline and goals, including the dates by which we will commence, complete and receive results from clinical trials. If patients are unwilling to participate in our trials for any reason, the timeline for recruiting patients, conducting trials, and obtaining regulatory approval of potential products may be delayed. Enrollment delays in our clinical trials may also jeopardize our ability to commence sales of and generate revenues from CTI-1601,nomlabofusp, which could cause the value of our company to decline and limit our ability to obtain additional financing, if needed. Any of these occurrences may harm our business, financial condition, and prospects significantly.
Friedreich’s ataxiaFA has no FDA-approved treatments,therapies that address frataxin deficiency, which is the underlying cause of the disease, and clinical endpoints required to obtain approval are not well defined.
There are currently no therapies approvedFDA-approved products to treat Friedreich’s ataxia; however, Reata Pharmaceuticals Inc. announcedFA that it had initiated a rolling submission of an NDAare designed to the FDA for omaveloxolone for the treatment of patients with Friedreich’s ataxia.increase frataxin levels. We have concentrated our research and development efforts on developing a novel, therapeutic forFA therapy designed to address frataxin deficiency, which is the treatmentunderlying cause of Friedreich’s ataxia,the disease, and our future success depends on the success of this therapeutic approach. The clinical trial requirements of the FDA and other comparable regulatory agencies and the criteria these regulators use to determine the safety and efficacy of any product candidate vary substantially according to the type, complexity, novelty and intended use and market of the potential product. Given the nature of Friedreich’s ataxia, we may have to devise novel clinical endpoints to be tested in our clinical trials, which can lead to some subjectivity in interpreting trial results and could result in regulatory agencies not agreeing with the validity of our endpoints, or our interpretation of the clinical data, and therefore denying approval, which would materially adversely affect our business, financial condition and results of operations. As a result, the design and conduct of clinical trials for a therapeutic product candidate such as CTI-1601 that is intended to deliver human frataxin ("FXN") through a subcutaneously administered, recombinant fusion protein in Friedreich’s ataxia patients are subject to unknown
43
risks, andRecently, we may experience setbackshad discussions with our planned clinical trialsthe FDA regarding the use of CTI-1601 in Friedreich’s ataxia because oftissue FXN levels as a novel surrogate endpoint. The FDA has acknowledged that frataxin deficiency appears to be critical to the limited clinical experience with ourpathogenic mechanism of actionFA, and that there continues to be an unmet need for treatments for FA patients that address the underlying disease pathophysiology. We intend to pursue an accelerated approval using FXN levels, supportive PD and clinical information, and safety data from the OLE study, along with additional non-clinical pharmacology information needed to support the novel surrogate endpoint approach. We are beginning to plan for a confirmatory study and are targeting a BLA submission in these patients.the second half of 2025. The FDA or other regulatory authorities may not agree with this approach.
In particular, regulatoryRegulatory authorities in the United States, the United Kingdom and the European Union have not issued definitive guidance as to how to measure and achieve efficacy in treatments for Friedreich’s ataxia.FA. As a result, the design and conduct of clinical trials of CTI-1601nomlabofusp may take longer, be more costly or be less effective as part of the novelty of development in Friedreich’s ataxia. WeFA. The FDA may not accept that the supportive PD and clinical information, and safety data from the OLE study, along with additional non-clinical pharmacology data we ultimately submit in our BLA, adequately supports the use new orof FXN levels as a novel endpoints or methodologies, andsurrogate endpoint. Even if the FDA or other regulatory authoritiessupports the use of FXN levels as a novel surrogate endpoint, the FDA may not consideragree with the endpointsadequacy of ourthe design of clinical trials intended to provide clinically meaningful results. assess this novel surrogate endpoint.
Even if applicable regulatory authorities do not object to our proposed endpoints in an earlier stage clinical trial, such regulatory authorities may require evaluation of additional or different clinical endpoints in later-stage clinical trials.
CTI-1601Nomlabofusp, including the effects of long-term daily patient dosing, may cause adverse events or undesirable side effects in non-clinical or clinical trials that could delay or prevent its regulatory approval, limit the commercial profile of an approved label,labeling, or result in significant negative consequences following marketingregulatory approval, if any.
We completed two Phase 1 clinical trials. As a result of certain mortalities in a non-clinical study of non-human primates, the FDA issued a clinical hold in May 2021. We responded to the clinical hold in January 2022. Following review of our response, the FDA declined to lift the clinical hold. We are continuing our efforts to address the issues raised by the FDA. Clinical development of CTI-1601 cannot continue until the clinical hold is lifted by the FDA. Any adverse events or undesirable side effects caused by, or other unexpected properties of, CTI-1601nomlabofusp in non-clinical or clinical studies could cause us, any future collaborators, an IRB or ethics committee or regulatory authorities to interrupt, delay or halt clinical trials of our product candidate and could result in a more restrictive
49
label or the delay or denial of regulatory approval by the FDA or other regulatory authorities. IfShould the FDA permit us to continue with the clinical hold is lifted,development of nomlabofusp, it is possible that as we progress CTI-1601nomlabofusp through clinical trials and toxicology studies, or as the use of CTI-1601nomlabofusp becomes more widespread if it receives regulatory approval, illnesses, injuries, discomforts and other adverse events that were not observed in earlier trials, as well as conditions that did not occur or went undetected in previous trials, may be reported by patients. If such side effects become known later in development or after approval, such findings may harm our business, financial condition and prospects significantly. Further, if a serious safety issue is identified in connection with the use of CTI-1601nomlabofusp commercially or in third-party clinical trials elsewhere, such issues may adversely affect the development potential of CTI-1601nomlabofusp elsewhere or result in regulatory authorities restricting our ability to develop or commercialize CTI-1601,nomlabofusp, if approved.
Further, if CTI-1601nomlabofusp were to receive marketing approval and we or others identify undesirable side effects caused by the product (or any other product) after the approval, a number of potentially significant negative consequences could result, including:
44
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidate and could substantially increase the costs of commercializing our product candidates and significantly impact our ability to successfully commercialize our product candidates and generate revenues, all of which would materially adversely affect our business, financial condition and results of operations. In addition, the patient populations under investigation with nomlabofusp have many co-morbidities that may cause severe illness or death unrelated to our product candidate, which may be attributed to nomlabofusp in a manner that negatively affects the safety profile of our product candidate.
Interim, “top-line,” and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available or as additional analyses are conducted, and as the data are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publish interim, preliminary or “top-line” data from our clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Preliminary or “top-line” data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are available. Material adverse changes between interim, preliminary or “top-line” data and final data could significantly harm our business, financial condition, results of operations and prospects.
Our approach to discover and develop fusion proteins for delivering proteins is novelnovel and may never lead to marketable products.
50
We have concentrated our efforts and research and development activities on delivering proteins (FXN or other) to intracellular targets. Our future success depends on the successful development and manufacturing of such therapeutics and the effectiveness of our platform. The scientific discoveries that form the basis for our research are relatively new.
CTI-1601Nomlabofusp uses a novel and unproven approach and mechanism to treat Friedreich’s ataxiaFA and therefore its efficacy and safety are difficult to predict, and there is no guarantee that CTI-1601nomlabofusp will be approved by the FDA, the EMA, or any other regulatory authorities.
If CTI-1601nomlabofusp proves to be ineffective, unsafe or commercially unviable, it is possible that our platform and pipeline would have little, if any, value, which would substantially harm our business, financial condition, results of operations and prospects. In addition, our approach may expose us to additional financial risks and make it more difficult to raise additional capital than other, more advanced proven technologies, which would materially adversely affect our business, financial condition and results of operations.
Protein replacement therapies are novel, complex and difficult to manufacture. We could experience manufacturing problems that result in delays in the development or commercialization of our protein replacement therapy platform or product candidates or otherwise harm our business.
The manufacture of fusion proteins, such as CTI-1601nomlabofusp and any fusion protein candidates, is technically complex and necessitates substantial expertise and capital investment. Production difficulties caused by unforeseen events may delay the availability of material for clinical trials and commercial products for CTI-1601nomlabofusp or any fusion protein product that may receive regulatory approval in the future. Additionally, because biologic products are complex, the manufacture of such products and product candidates is more difficult and costly. We may not be able to have such products reliably manufactured in accordance with the applicable regulatory requirements in sufficient quantities to support our development programs and, if ultimately approved, commercial supply. We currently contract with third parties for the manufacturing of program materials for CTI-1601.
There are a limited number of contract manufacturers who specialize in the manufacture of biologic products and those that do may still be developing appropriate processes, controls and facilities for large-scale production. While we believe that there will be sufficient sources of supply that can satisfy our clinical and commercial requirements, we cannot be certain that we will be able to identify and establish additional relationships with such sources, if necessary, in a timely manner or at all, and what the terms and costs of such new arrangements would be, or that such suppliers would be able to supply our potential commercial needs. Furthermore, in the event our primary manufacturer cannot meet our needs, any switch to an alternative manufacturer, if available, would result in a significant delay, would require FDA approval, and cause material additional costs.
As further described in these risk factors, the manufacturers of biologic products must comply with strictly enforced cGMP requirements, state and federal regulations, as well as foreign requirements when applicable. Any failure by us or our contract manufacturing organizations to adhere to or document compliance to such regulatory requirements could lead to a delay or interruption in the availability of our program materials for clinical trials or commercial use, among other consequences. If we or our manufacturers fail to comply with the FDA, EMA, or other regulatory authorities, it could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, clinical holds or termination of clinical trials, Form 483s, warning or untitled letters, regulatory communications warning the public about safety issues with a product, import or export refusals, license revocation, seizures, detentions, or recalls of product candidates or product, operating restrictions, criminal
45
prosecutions or debarment, suits under the civil False Claims Act, corporate integrity agreements, or consent decrees any of which could significantly and adversely affect supplies of our product candidates and our business, financial conditions and results of operations could be materially adversely affected.
Our current dependence upon others for the manufacture of our product candidates may also adversely affect our business, results of operations, financial condition, and our ability to commercialize any product candidates that receive regulatory approval on a timely and competitive basis.
Fast Tracktrack designation by the FDA or any future expedited program designations may not lead to a faster development, regulatory review or approval process and it does not increase the likelihood that any of our product candidates will receive marketing approval.
We have received Fast Trackfast track therapy designation for CTI-1601nomlabofusp for the treatment of Friedreich’s ataxia.FA. We may, in the future, apply for other accelerated programsexpedited program designations from the FDA (such as breakthrough therapy or accelerated approval)therapy) for CTI-1601nomlabofusp or future product candidates. Designation for these programs is within the discretion of the FDA. Accordingly, even if we believe CTI-1601nomlabofusp or a future product candidate meets the criteria for designation, the
51
FDA may disagree. In any event, theThe receipt of a designation may not result in a faster development process, review or approval compared to products considered for approval under conventional FDA procedureswithout these expedited program designations and, in any event, does not assure ultimate approval by the FDA. In addition, even though CTI-1601nomlabofusp has been designated as Fast Track,obtained fast track designation, the FDA may later decide that it no longer meets the criteria for designation and revoke it. IfApproval of other therapies for the treatment Friedreich’s ataxia could negatively impact our continued fast track therapy designation for nomlabofusp for the treatment of FA. In addition, if we apply for designation to additional accelerated programs from the FDA for CTI-1601other designations for nomlabofusp or future product candidates, the FDA might not grant the designation.such designations. If we apply for any similar programs in foreign countries for CTI-1601nomlabofusp or future product candidates, those designations also might not be granted by the regulatory authorities of those countries. Any of the above could adversely affect our business, financial condition and results of operations.
We intend to pursue accelerated approval from FDA for nomlabofusp for the treatment of Friedreich’s ataxia, however this may not lead to a faster development or regulatory review or approval process and does not increase the likelihood that we will receive marketing approval. If we are unable to obtain approval under an accelerated pathway, we may be required to conduct additional clinical trials beyond those that we contemplate, which could increase the expense of obtaining, reduce the likelihood of obtaining, and/or delay the timing of obtaining, necessary marketing approvals.
We intend to pursue an accelerated approval from FDA for nomlabofusp for the treatment of FA using FXN levels, supportive pharmacodynamic and clinical information and safety data from the OLE study, along with additional non-clinical pharmacology information needed to support our surrogate biomarker approach. Under the FDA’s accelerated approval program, the FDA may approve a drug or biologic for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Although we have initiated discussions with FDA concerning our surrogate biomarker approach, there can be no assurance that data we intend to generate will be successful in establishing that increases in FXN levels are reasonably likely to predict clinical benefit. In addition, future nonclinical pharmacology and clinical studies may not provide adequate information or may fail to support the predictive value of increases in FXN and, therefore, its ability to serve as a surrogate endpoint. Even if these evidentiary requirements are met, we may not be able to accrue adequate exposures to assess clinical safety or expedite the scale up of manufacturing or meet other CMC requirements in a timeframe commensurate with the expedited assessment of clinical efficacy.
Approval of other therapies for the treatment of FA, including the approval of omaveloxolone for the treatment of FA, could negatively impact our ability to utilize the accelerated approval pathway, and/or get approval for nomlabofusp. For drugs or biologics granted accelerated approval, post-marketing confirmatory trials are required to describe the anticipated effect on irreversible morbidity or mortality or other clinical benefit. These confirmatory trials must be completed with due diligence and, in some cases, the FDA may require that the trial be designed, initiated and/or fully enrolled prior to approval.
Moreover, the FDA may withdraw approval of any product candidate approved under the accelerated approval pathway if, for example:
In addition, the FDA may terminate the accelerated approval program or change the standards under which accelerated approvals are considered and granted in response to public pressure or other concerns regarding the accelerated approval program. Changes to or termination of the accelerated approval program could prevent or limit our ability to obtain accelerated approval of any of our clinical development programs. Recently, the accelerated
52
approval pathway has come under scrutiny within the FDA and by Congress. The FDA has put increased focus on ensuring that confirmatory studies are conducted with diligence and, ultimately, that such studies confirm the benefit. In addition, the Food and Drug Omnibus Reform Act (“FDORA”), included provisions related to the accelerated approval pathway. Pursuant to FDORA, the FDA is authorized to require, as appropriate, that a post-approval study to be underway prior to approval or within a specified time period following approval. FDORA also requires the FDA to specify conditions of any required post-approval study and requires sponsors to submit progress reports for required post-approval studies and any conditions required by the FDA. FDORA also gives the FDA increased authority to withdraw approval of a drug or biologic granted accelerated approval on an expedited basis if the sponsor fails to verify the drug or biologic’s predicted clinical benefit. FDORA enables the FDA to initiate enforcement action for a sponsor's failure to conduct with due diligence a required post-approval study, including a failure to meet any required conditions specified by the FDA or to submit timely reports.
If we fail to maintain orphan drug designation or other regulatory exclusivity for CTI-1601nomlabofusp or obtain such exclusivity for any of our other product candidates in the future, our competitive position would be harmed.
We received orphan drug designation from the FDA for CTI-1601nomlabofusp for the treatment of FA in July 2017. In the United States, orphan drug designation entitles a party to financial incentives such as tax advantages and user-fee waivers. In addition, if a product candidate that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including an NDA or BLA, to market the same drug for the same indication for seven years, except in limited circumstances, including if the FDA concludes that the later drug is clinically superior to or safer than, the approved drug. A drug is clinically superior if it is safer, more effective, or makes a major contribution to patient care. In the case of a biological product, whether a drug is the same drug is based on the principal molecular structural features of the product. Orphan drug designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process. We could lose orphan drug designation in this scenario.
Additionally, we may lose orphan drug exclusivity if we are unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition. Moreover, orphan drug exclusivity may not effectively protect our product candidates from competition because different drugs can be approved for the same condition. For example, in January 2022, Reata Pharmaceuticals,Inc. announced that it had initiated a rolling submission of an NDA to the FDA for omaveloxolone for the treatment of patients with Friedreich’s ataxia. Our orphan drug designation for CTI-1601 will not prohibit the FDA from potentially approving a competitor’s NDA for omaveloxolone. Further, even after an orphan drug is approved, the FDA or comparable foreign regulatory authority can subsequently approve another drug for the same condition if such regulatory authority concludes that the later drug is clinically superior, if it is shown to be safer, more effective or makes a major contribution to patient care.
We have also received orphan drug designation for CTI-1601nomlabofusp in the European Union. In the European Union, the European Medicines Agency, or EMA’s Committee for Orphan Medicinal Products grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the European Union. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the drug. In the European Union, orphan drug designation also entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity following drug approval. This period may be reduced to
46
six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable so that market exclusivity is no longer justified. Loss of orphan drug designation for CTI-1601nomlabofusp or the failure to obtain such designation in other countries or for any future product candidates could adversely affect our business, financial condition and results of operation.
If another product has received approval in the indications for which we have received orphan drug designation, we may still receive approval in that indication if we can demonstrate that our product candidate is clinically superior to the existing orphan product. This demonstration of clinical superiority may be done at the time of initial approval or in post-approval studies, depending on the type of marketing authorization granted. Having to comply with these additional requirements to obtain orphan drug exclusivity could adversely affect our business, financial condition and results of operation.
Although we have obtained rare pediatric disease designation for CTI-1601,nomlabofusp, we may not be eligible to receive a priority review voucher in the event the FDA determines we no longer meet the criteria for designation, revokes the designation or that FDA approval does not occur prior to September 30, 2026.
53
The sponsor of an application for a rare pediatric disease drug product may be eligible for a voucher that can be used or sold to obtain a priority review for a subsequent application submitted under section 505(b)(1) of the FDCA or section 351 of the PHS Act. The rare pediatric disease priority review voucher program was most recently reauthorized by Congress through September 30, 2024, with the potential for priority review vouchers to be granted through September 30, 2026. We received rare pediatric disease designation from the FDA for CTI-1601nomlabofusp in 2019. We may, in the future, apply for rare pediatric disease designation from the FDA for future product candidates that may qualify for designation. Vouchers for rare pediatric disease drugs are awarded for qualifying applications when the designated drug receives approval. CTI-1601
Although nomlabofusp has received rare pediatric disease designation, nomlabofusp may not receive approval, or wea priority review voucher for a number of reasons: nomlabofusp may not receive approval; nomlabofusp may receive approval in adults, but not pediatric patients, and therefore,patients; nomlabofusp may not meet the eligibility requirements for a priority voucher at the time we seek approval for nomlabofusp; or we may not receivemeet the current deadline for receiving a voucher. In addition, even though CTI-1601 has been designated aspriority review voucher (September 30, 2026), in which case we would not be able to obtain a drug forvoucher unless Congress further reauthorizes the program. Finally, a rare pediatric disease designation does not necessarily lead to faster development or regulatory review of the FDA may later decideproduct or increase the likelihood that it no longer meets the criteria for designation, the designation may be revoked, or we could not be awarded the voucher.will receive marketing approval. If we apply for designation for future product candidates as drugs for rare pediatric diseases, the FDA may not grant the designation. The failure to maintain rare pediatric disease designation for CTI-1601nomlabofusp or if FDA approval does not occur prior to September 30, 2026 could result in the inability to receive a rare pediatric disease designationpriority review voucher which could adversely affect our business, financial condition and results of operations.
If we fail to maintain PRIME designation in the European Union for CTI-1601,nomlabofusp, our competitive position would be harmed.
The PRIME scheme is open to medicines under development and for which the applicant intends to apply for an initial marketing authorization applicationMAA through the centralized procedure. Eligible products must target conditions for which where is an unmet medical need (there is no satisfactory method of diagnosis, prevention or treatment in the European Union or, if there is, the new medicine will bring a major therapeutic advantage) and they must demonstrate the potential to address the unmet medical need by introducing new methods or therapy or improving existing ones. Applicants will typically be at the exploratory clinical trial phase of development and will have preliminary clinical evidence in patients to demonstrate the promising activity of the medicine and its potential to address to a significant extent an unmet medical need. In exceptional cases, applicants from the academic sector or SMEs (small and medium sized enterprises) may submit an eligibility request at an earlier stage of development if compelling non-clinical data in a relevant model provide early evidence of promising activity to establish proof of principle, and first in man studies indicate adequate exposure for the desired pharmacotherapeutic effects and tolerability.
If a medicine is selected for the PRIME scheme, the EMA:
For SMEs who enter the scheme based on data showing proof of principle, the appointment of the rapporteur occurs once they have generated data confirming eligibility at proof of concept stage. They are required to submit relevant data and justification as the product development reaches this stage.
47
Medicines that are selected for the PRIME scheme are also expected to benefit from EMA’s accelerated assessment procedure at the time of application for marketing authorization. Where, during the course of development, a medicine no longer meets the eligibility criteria, or if a medicine granted early access to the PRIME scheme cannot later demonstrate proof of concept, support under the PRIME scheme may be withdrawn. Approval of other therapies for the treatment Friedreich’s ataxia, including the recent approval of omaveloxolone could negatively impact our continued access to this and similar programs. Loss of PRIME designation for CTI-1601nomlabofusp or
54
the failure to obtain such designation for any future product candidates could adversely affect our business, financial condition and results of operation.
Changes in regulatory requirements, FDA guidance, guidance from other regulatory authorities or unanticipated events during our non-clinical or clinical trials of CTI-1601nomlabofusp or future product candidates may result in changes to clinical trial protocols or additional non-clinical or clinical trial requirements, which could result in increased costs to us and could delay our development timeline.
Changes in regulatory requirements, FDA guidance or guidance from EMA or unanticipated events during our non-clinical or clinical trials may force us to terminate or adjust our clinical program.programs. The FDA, or theother applicable regulatory authorities may impose additional clinical trial and/or non-clinical study requirements. Amendments to our clinical trial protocols would require resubmission to the FDA, or the applicable regulatory authorities as well as IRBs and ethics committees for review and approval, which may adversely impact the cost, timing or successful completion of a clinical trial. If we experience delays completing, or if we terminate, any of our clinical trials, or if we are required to conduct additional clinical trials, or non-clinical studies and/or post-market studies, the commercial prospects for CTI-1601nomlabofusp or any other potential product candidates may be harmed and our ability to generate product revenue will be delayed or eliminated, and it would materially adversely affect our business, financial condition and results of operations.
In order to market any product outside of the United States, we must comply with numerous and varying regulatory requirements of other countries regarding drug development and commercialization. The approval processprocesses varies from country to country and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. Even if we receive marketing approval for CTI-1601nomlabofusp in the United States, we may never receive regulatory approval to market CTI-1601nomlabofusp outside of the United States.
48
Disruptions at the FDA and other government agencies caused by funding shortages or global health concerns could hinder their ability to hire, retain or deploy key leadership and other personnel, or otherwise prevent new or modified products from being developed, approved or commercialized in a timely manner or at all, which could negatively impact our business.
The ability of the FDA to review and/or approve new products can be affected by a variety of factors, including government budget and funding levels, statutory, regulatory, and policy changes, the FDA’s ability to hire and retain key personnel, and accept the payment of user fees, and other events that may otherwise affect the FDA’s ability to perform routine functions. Average review times at the FDA have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. On March 18, 2020,Furthermore, government shutdowns could also impact the ability of regulatory authorities and government agencies to function normally and support our operations. For example, the U.S. federal government has shut down repeatedly since 1980, including for a period of 35 days beginning on December 22, 2018. During a shutdown, certain regulatory authorities and agencies, such as the FDA, announced its intentionhave had to temporarily postpone routine surveillance inspections of domestic manufacturing facilities. Regulatory authorities outside the United States may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic. furlough key personnel and stop critical activities.
If a prolonged government shutdown occurs, or if global health concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
Regulatory requirements governing biologic products have changed frequently and may continue to change in the future. Such requirements may lengthen the regulatory review process, require us to perform additional non-clinical studies or clinical trials, and increase our costs, or may force us to delay, limit or terminate certain of our programs.
Regulatory requirements governing biologic drug products are still evolving and may continue to change in the future. As a result, it is difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for CTI-1601nomlabofusp for the treatment of Friedreich’s ataxiaFA or any other future protein replacement therapy product candidates in any indication, if at all. Regulatory review agencies and the new requirements and guidelines they promulgate may lengthen the regulatory review process, require us to perform additional or larger studies, increase
55
our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our product candidates or lead to significant post-approval studies, limitations or restrictions. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
Delays, failure or unexpected costs in obtaining, the regulatory approval necessary to bring our product candidates to market could have a material adverse effect on our business, results of operations, financial condition and prospects.
In addition, the clinical trial requirements of the FDA, the EMA and other regulatory authorities and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty and intended use and market of such product candidates. The regulatory approval process for novel product candidates such as ours can be more expensive and take longer than for other, better known or more extensively studied product candidates.
49
The clinical trials of CTI-1601nomlabofusp and any future product candidates are, and the manufacturing and marketing of CTI-1601nomlabofusp and any future product candidates, will be subject to extensive and rigorous review and regulation by numerous government authorities in the United States and in other countries, such as within the European Union, where we intend to seek regulatory approval of, and market, any product candidate.candidates.
Before obtaining regulatory approvals for the commercial sale of any product candidate, we must demonstrate through non-clinical testing and clinical trials that the product candidate is safe and effective for use in each target indication. This process can take many years. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions, which may cause delays in the approval of or may result in the decision not to approve our product candidates. We have not obtained regulatory approval for CTI-1601,nomlabofusp, and it is possible that ourthis product candidate or any product candidates we may seek to develop in the future will never obtain regulatory approval. If marketing approval is obtained, it will likely include post-marketing studies, and other post-marketing requirements, and surveillance such as Risk Evaluation and Mitigation Strategies ("REMS")REMS which will require the expenditure of substantial resources beyond the proceeds we currently have on hand.
Furthermore, we are not permitted to market CTI-1601nomlabofusp in the United States, or the European Union until we receive approval of a BLA from the FDA or a MAA from the EMA, or in any other foreign countries until we receive the requisite marketing approval from such countries. The development of drugs for Friedreich’s ataxiaFA or other rare diseases may require initial non-clinical studies, early and usually smaller, clinical trials and randomized, double-blind placebo controlled long-term safety and efficacy trials in order to test the safety and efficacy of the drug.
We have completed two Phase 1 clinical trials in patients with Friedreich’s ataxia in the United States but clinical development is currently subject to clinical hold. If we are successful in our efforts to have the clinical hold lifted, CTI-1601Nomlabofusp requires substantial further clinical development before we can submit a BLA to the FDA. Development and/or regulatory programs for CTI-1601nomlabofusp in any countries other than the United States (such as a MAA to the EMA) isare only in very preliminary stages and may require substantial further development in those countries prior to regulatory submissions seeking regulatory approval for marketing.
Even after successful completion of clinical trials, there is a risk that the FDA or other regulatory agencies may request further information from us, disagree with our findings or otherwise undertake a lengthy review of our submission.
The FDA and certain European regulatory authorities may delay, limit or deny testing or approval of CTI-1601nomlabofusp for many reasons, including, among others:
56
50
Any of these factors, many of which are beyond our control, could jeopardize our ability to obtain and/or maintain regulatory approval for and successfully market CTI-1601.nomlabofusp. Any delay or failure in obtaining required approvals could have a material adverse effect on our business, financial condition and results of operations. This process can take many years and will likely require the expenditure of substantial resources. Of the large number of drugs in development in the United States, only a small percentage will successfully complete the FDA regulatory approval process and be commercialized. It is possible that the FDA or other regulatory agencies will not approve any application that we submit. It is possible that our product candidates may not obtain appropriate regulatory approvals necessary for us to commence clinical trials for our product candidates. Accordingly, even if we are able to obtain the requisite financing to continue to fund our development and clinical trials, we cannot assureensure that CTI-1601,nomlabofusp, or any other of our potential product candidates will be successfully developed or commercialized.
We are subject to healthcare laws and regulations, and health information privacy and security laws, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and others will play a primary role in the recommendation and prescription of CTI-1601nomlabofusp or any potential product candidates, if approved. Our future arrangements with third-party payors will expose us broadly to applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute CTI-1601nomlabofusp or potential product candidates, if we obtain marketing approval. In addition, we may be subject to patient privacy regulation by both the federal government and the states or other countries in which we conduct our business. Restrictions under applicable federalFor more information, see the section of this report titled “Business – Healthcare Laws and state healthcare laws and regulations include the following:Regulations – Other Healthcare Laws.”
51
Ensuring that our future business arrangements with third parties comply with applicable healthcare laws and regulations could be costly. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations, including anticipated activities to be conducted by our sales team, were found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines and exclusion from
57
government funded healthcare programs, such as Medicare and Medicaid, any of which could substantially disrupt our operations and would materially adversely affect our business, financial condition and results of operations. If any of the physicians or other providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.programs, which could have a material adverse effect on our business, results of operations, financial condition and prospects.
Healthcare legislation, including potentially unfavorable pricing regulations or other healthcare reform initiatives may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates.
The commercial potential for our approved products, if any, could be affected by changes in healthcare spending and policy in the United States and abroad. We operate in a highly regulated industry. New laws, regulations or judicial decisions or new interpretations of existing laws, regulations or decisions, related to healthcare availability, the method of delivery or payment for healthcare products and services could adversely affect our business, operations and financial condition. The United States and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system that may affect our ability to profitably sell our product and product candidates, if approved. The United States government, state legislatures and foreign governments also have shown significant interest in implementing cost-containment programs to limit the growth of government-paid
52
healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs.
The Affordable Care Actpharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. Previously, in March 2010, the ACA was enacted, which was intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcarehealth care and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. There have been significant ongoing administrative, executive and legislative efforts to modify or eliminateHealthcare reform initiatives recently culminated in the Affordable Care Act. For example, the Tax Act enacted on December 22, 2017, repealed the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000Aenactment of the Internal Revenue Code, commonly referred to as the individual mandate. Other legislative changes have been proposed and adopted since the passage of the Affordable Care Act. The Budget ControlIRA Inflation Reduction Act of 2011, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve its targeted deficit reduction of an amount greater than $1.2 trillion for the fiscal years 2012 through 2021, triggering the legislation’s automatic reductions to several government programs. These reductions included aggregate reductions to Medicare payments to healthcare providers of up to 2.0% per fiscal year, which went into effect in April 2013.Subsequent legislation extended the 2% reduction, on average, to 2030 unless additional Congressional action is taken, However, pursuant to the Coronavirus Aid, Relief and Economic Security Act ("CARES Act") the 2% Medicare sequester reductions have been suspended from May 1, 2020 through December 31, 2021 due to the COVID-19 pandemic. On January 2, 2013, the American Taxpayer Relief Act was signed into law,2022 or IRA, which, among other things, reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
The Affordable Care Act has also been subject to challenges in the courts. On December 14, 2018, a Texas U.S. District Court Judge ruled that the Affordable Care Act is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. On December 18, 2019, the Fifth Circuit U.S. Court of Appeals held that the individual mandate is unconstitutional and remanded the case to the Texas District Court to reconsider its earlier invalidation of the entire Affordable Care Act
An appeal was taken to the U.S. Supreme Court. On June 17, 2021. , the Supreme Court ruled that the plaintiffs lacked standing to challenge the law as they had not alleged personal injury traceable to the allegedly unlawful conduct. As a result, the Supreme Court did not rule on the constitutionality of the ACA or any of its provisions.
Further changes to and under the Affordable Care Act remain possible, but it remains unknown what form any such changes or any law proposed to replace or revise the Affordable Care Act would take, and how or whether it may affect our business in the future. We expect that changes to the Affordable Care Act, the Medicare and Medicaid programs, changes allowing the federal governmentallow allows HHS to directly negotiate drugthe ceiling price of a statutorily specified number of drugs and biologic each year that CMS reimburses under Medicare Part B and Part D, requires the payment of rebates on Medicare Part B and Part D drugs whose prices have increased at a rate faster than the rate of inflation, and changes stemming from other healthcare reform measures, especially with regard to healthcare access, financing or other legislation in individual states, could have a material adverse effect onredesign the healthcare industry.
AtMedicare Part D cost sharing structure, including revising manufacturer financial liability for covered products. For more information, see the state level, legislatures have increasingly passed legislationsection of this report titled “Business – Healthcare Laws and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.Regulations – Healthcare Reform.”
We expect that additional federal, state and foreign healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in limited coverage and reimbursement and reduced demand for our products, once approved, or additional pricing pressures.
Even if approved, reimbursement policies could limit our ability to sell product candidates that we elect to sell on our own.
If approved by regulatory authorities, market acceptance and sales of product candidates that we elect to sell on our own will depend on reimbursement policies and may be affected by healthcare reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels for those medications. Cost containment is a
53
primary concern in the U.S. healthcare industry and elsewhere. Government authorities and these third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. We cannot be sure that reimbursement will be available for CTI-1601,nomlabofusp, if approved, or future product candidates that we elect to sell on our own and, if reimbursement is available, the level of such reimbursement. Reimbursement may impact the demand for, or the price of, product candidates that we elect to.to sell. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize product candidates that we elect to sell. For more information, see the section of this report titled “Business – Healthcare Laws and Regulations – Coverage and Reimbursement.”
In some foreign countries, particularly in Canada and European countries, the pricing of prescription pharmaceuticals is subject to strict governmental control. In these countries, pricing negotiations with governmental authorities can take six to 12 months or longer after the receipt of regulatory approval and product launch. To obtain favorable reimbursement for the indications sought or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of product candidates that we elect to sell on our own
58
with other available therapies. If reimbursement for product candidates that we elect to sell on our own is unavailable in any country in which we seek reimbursement, if it is limited in scope or amount, if it is conditioned upon our completion of additional clinical trials, or if pricing is set at unsatisfactory levels, our business, financial conditions and results of operations could be materially adversely affected.
Even if we obtain regulatory and marketing approval for a product candidate, our product candidates will remain subject to regulatory oversight.
Even if we receive marketing and regulatory approval for CTI-1601nomlabofusp or a future product candidate, regulatory authorities may still impose significant restrictions on the indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies. CTI-1601Nomlabofusp or future product candidates will also be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping and submission of safety and other post-market information. The FDA has significant post-market authority, including, for example, the authority to require labeling changes based on new safety information and to require post-market studies or clinical trials to evaluate serious safety risks related to the use of a drug. An unsuccessful post-marketing study or failure to complete such a study could result in the withdrawal of marketing approval. Any regulatory approvals that we receive for CTI-1601nomlabofusp may also be subject to a REMS, limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including post-approval clinical trials, and surveillance to monitor the quality, safety and efficacy of the product, all of which could lead to lower sales volume and revenue. For example, the holder of an approved BLA or NDA is obligated to monitor and report adverse events and any failure of a product to meet the specifications in the BLA or NDA. The holder of an approved BLA or NDA also must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling or manufacturing process. Advertising and promotional materials must comply with FDA rules and are subject to FDA review, in addition to other potentially applicable federal and state laws.
In addition, product manufacturers and their facilities are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP requirements and adherence to commitments made in the BLA or NDA or foreign marketing application. If we, or a regulatory authority, discover(s) previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured or if a regulatory authority disagrees with the promotion, marketing or labeling of that product, a regulatory authority may impose restrictions relative to that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If we or our contractors fail to comply with applicable regulatory requirements following approval of CTI-1601,nomlabofusp, a regulatory authority may:
54
59
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize CTI-1601,nomlabofusp, if approved, and adversely affect our business, financial condition, results of operations and prospects.
In addition, the FDA’s policies, and those of equivalent foreign regulatory agencies, may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of CTI-1601.nomlabofusp. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would materially and adversely affect our business, financial condition, results of operations and prospects.
Even if we receive marketing approval for CTI-1601nomlabofusp in the United States, we may never receive regulatory approval to market CTI-1601nomlabofusp outside of the United States.
We may pursue marketing approval for CTI-1601nomlabofusp in the United States, the European Union and in other jurisdictions worldwide. In order to market any product outside of the United States, we must establish and comply with the numerous and varying safety, efficacy and other regulatory requirements of other jurisdictions, including potential additional clinical trials and/or non-clinical studies. Approval procedures vary among jurisdictions and can involve additional testing and additional administrative review periods. The time required to obtain approvals in other jurisdictions might differ from that required to obtain FDA approval. The marketing approval processes in other jurisdictions may implicate all of the risks detailed above regarding FDA approval in the United States as well as other risks. In particular, in many countries outside of the United States, products must receive pricing and reimbursement approval before the product can be commercialized. Obtaining this approval can result in substantial delays in bringing products to market in such countries. Marketing approval in one jurisdiction does not necessarily ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one country may have a negative effect on the regulatory process or commercial activities in others. Failure to obtain marketing approval in other countries or any delay or other setback in obtaining such approval would impair our ability to market a product candidate in such foreign markets. Any such impairment would reduce the size of our potential market, which could have a material adverse impact on our business, financial condition, results of operations and prospects.
55
Our future growth depends, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
The prevalence of Friedreich's ataxiaFA is estimated to be approximately three times greater in the European Union than in the United States, and, therefore, represents our largest potential market for CTI-1601.nomlabofusp. Our future profitability will depend, in part, on our ability to commercialize CTI-1601nomlabofusp and future product candidates in the European Union and other foreign markets for which we may rely on collaborations with third parties. If we commercialize a product candidate in foreign markets, we would be subject to additional risks and uncertainties, including:
60
Sales in the European Union and other foreign markets of a product candidate could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell CTI-1601,nomlabofusp, if approved, we may not be able to generate any revenue.
We do not currently have an established infrastructure for the sales, marketing and distribution of biologic or drug products in the United States or foreign countries. In order to market a product candidate, if approved by the FDA or any other regulatory authority, we must build our sales, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, or if we are unable to do so on commercially reasonable terms, our business, results of operations, financial condition and prospects will be materially adversely affected.
Even if we receive marketing approval for CTI-1601,nomlabofusp, we may not achieve broad market acceptance, which would limit the revenue that we generate from our sales.
The commercial success of CTI-1601,nomlabofusp, if developed and approved for marketing by the FDA or EMA or other applicable regulatory authorities, will depend upon the market size for, and the awareness and acceptance of
56
CTI-1601 nomlabofusp among the medical community, including physicians, patients, advocacy groups and healthcare payors. Market acceptance of CTI-1601,nomlabofusp, if approved, will depend on a number of factors, including, among others:
61
If CTI-1601nomlabofusp is approved but does not achieve an adequate level of acceptance by patients, advocacy groups, physicians and payors, we may not generate sufficient revenue from CTI-1601nomlabofusp to become or remain profitable and our business, financial condition and results of operations could be materially adversely affected. Our efforts to educate the medical community and third-party payors about the benefits of CTI-1601nomlabofusp may require significant resources and may never be successful.
57
The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. If we are found to have improperly promoted off-label uses, we may become subject to significant liability.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products, such as CTI-1601nomlabofusp or any potential product candidates, if approved. If we receive marketing approval for CTI-1601,nomlabofusp, or any potential product candidates, physicians may prescribe our product candidates to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion and required that they enter into corporate integrity agreements with the Office of Inspector General of the Department of Health and Human Services. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. If we cannot successfully manage the promotion of CTI-1601nomlabofusp or any potential product candidates, if approved, we could become subject to significant liability, which would materially adversely affect our business, financial condition and results of operations.
CompetingAdditional competing technologies could emerge, adversely affecting our opportunity to generate revenue from the sale of CTI-1601,nomlabofusp, if approved.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. AnyFor example, omaveloxolone, was approved for the treatment of FA in adults and adolescents aged 16 and older by the FDA and the European Commission in February 2023 and February 2024, respectively. We expect nomlabofusp, if approved, will compete with omaveloxolone and other new, product that competes with anfuture approved productproducts and may need to demonstrate compelling advantages in efficacy, convenience, tolerability and safety to be commercially successful. Other competitive factors, including biosimilar and gene therapy competition, could force us to lower prices or could result in reduced sales. Many of our current or potential competitors, either alone or with strategic partners, have significantly greater financial resources and expertise in research and development, manufacturing, non-clinical testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved products than we do. New products developed by others could emerge as competitors to CTI-1601nomlabofusp or any other potential product candidates, resulting in CTI-1601nomlabofusp or other product candidates being obsolete before we are able to recover expenses incurred in connection with their development or realize revenues from any commercialized product. The pricing of our current product candidate, if and when approved for marketing, will depend, in part, on the pricing strategies adopted by our competitors. If these or other companies enact pricing strategies that impact the price we can charge for our product candidate, if approved, we may reduce our prices and our revenue and results of operations could be affected. Any new product could also affect our ability to recruit and retain clinical trial patients, to obtain and maintain potentialdesignations or eligibility for expedited regulatory pathways, and to commercialize current and future product candidates (including the impact of potential barriers to entry if a competitor is able to establish a strong market position before we are able to commercialize our products). For example, in January 2022, a pharmaceutical company announced that it had initiated a rolling submission of an NDA to the FDA for omaveloxolone for the treatment of patients with Friedreich’s ataxia, which if approved, would compete directly with CTI-1601.candidates. Given that we are still in a relatively early phase of development for CTI-1601,nomlabofusp, the successfulrecent approval and commercialization of omaveloxolone and the approval of any future competing technologies could provide a competitorcompetitors with a significant competitive advantage and may create an additional barrier to market acceptance of CTI-1601, if approved.nomlabofusp. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and results of operations will be adversely affected.
Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established
62
companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient, or are less expensive than any products that we may develop. Furthermore, currently approved products could be discovered to have application for treatment of age-related degenerative diseases and disorders, which could give such products significant regulatory and market timing advantages over any of our product candidates. Our competitors also may obtain FDA, EMA or other regulatory approval for their products more rapidly than we may obtain approval for ours and may obtain orphan product exclusivity from the FDA for indications our product candidates are targeting, which could result in our competitors establishing a strong market position before we are able to enter the market. Additionally, products or technologies developed by our competitors may render our potential product candidates uneconomical or obsolete, and we may not be successful in marketing any product candidates we may develop against competitors.
58
We may face competition from biosimilars and may face increasing competition over time.
We may face competition from biosimilars in both the United States and Europe, and over time we may face increasing biosimilar competition. To the extent that governments adopt more permissive approval frameworks and competitors are able to obtain broader or expedited marketing approval for biosimilars, the rate of increased competition for our biologic drug products could accelerate. Expiration or successful challenge of applicable patent rights could trigger such competition, and we could face more litigation regarding the validity and/or scope of our patents. Our products may also experience greater competition from lower cost biosimilars or generics that come to market when branded products that compete with our products lose their own patent protection.
In the European Union, the European Commission has granted marketing authorizations for biosimilars pursuant to a set of general and product class-specific guidelines for biosimilar approvals issued in 2005. In addition, in an effort to spur biosimilar utilization and/or increase potential healthcare savings, some countries in the European Union have adopted biosimilar uptake measures such as requiring physician prescribing quotas or promoting switching or pharmacy substitution of biosimilars for the corresponding reference products, and other countries may adopt similar measures. Some countries in the European Union may impose automatic price reductions upon market entry of the second or third biosimilar competitor.
In the United States, the ACA authorized the FDA to approve biosimilars via a separate, abbreviated pathway. A growing number of companies have announced that they are in varying stages of development of biosimilar versions of existing biotechnology products. Some companies pursuing development of biosimilars may challenge our patents well in advance of the expiration of our material patents. The U.S. pathway includes the option for biosimilar products meeting certain criteria to be approved as interchangeable with their reference products. Some companies developing biosimilars may seek to register their products as interchangeable biologics, which could make it easier for prescribers or pharmacists to substitute those biosimilars for our products. In addition, critics of the 12-year exclusivity period in the biosimilar pathway law will likely continue to seek to shorten the data exclusivity period and/or to encourage the FDA to interpret narrowly the law’s provisions regarding which new products receive data exclusivity. While we are unable to predict the precise impact of biosimilars, we expect in the future for there to be greater competition in the United States as a result of biosimilars and downward pressure on product prices and sales. These biosimilars or generics may affect the tier designation by third party payors and may require prior authorization for use of CTI-1601,nomlabofusp, thereby adding barriers to access. This additional competition could have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Business
If we are unable to manage expected growth in the scale and complexity of our operations, including attracting and hiring additional qualified management, our performance may suffer.
We are an early-stage clinical biotechnology company with a small number of employees, and our management systems currently in place are not likely to be adequate to support our future growth plans. As a result, we are highly dependent on our management and scientific personnel. The loss of the services of any of our executive officers, other key employees or consultants and other scientific advisors in the foreseeable future, might impede the achievement of our research, development and commercialization objectives. Competition for qualified personnel in the biopharmaceutical field is intense due to the limited number of individuals who possess the skills and experience required by our industry. We will need to continue to hire additional personnel as we expand our clinical development and if we initiate commercial activities. We may not be able to attract and retain quality personnel on acceptable terms, or at all. In addition, to the extent we hire personnel from competitors, we may be
63
subject to allegations that they have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers own their research output.
We rely on consultants and advisors, including scientific, non-clinical, manufacturing and clinical advisors, to assist us in formulating our development and commercialization strategy. These consultants and advisors may be employed by other employers and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key
59
employees may be difficult and may take an extended period of time because of the competition for talent, particularly with the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize products. In addition, COVID-19 posesa possible future pandemic, epidemic or outbreak of an infectious disease may pose a risk to our ability to retain and rely on our executive officers and key employees, including the potential that one or more of such employees or members of their families may contract the virus, which could impact the ability of such employees to perform as expected, which in turn would adversely impact our current and planned operations.
Recruiting and retaining qualified scientific, medical clinical, manufacturing, quality assurance, regulatory, legal, public company financial, business, sales, marketing and commercial personnel and implementing and improving our operational, financial and management systems will be critical to our ability to grow and succeed. These demands also will require the hiring of additional executive or management-level personnel or the development of additional expertise by our senior management personnel. Hiring a significant number of additional employees, particularly those at the executive or management level, would increase our expenses significantly. In addition, we may not be able to attract and retain these personnel on acceptable terms given the competition among numerous pharmaceutical companies for similar personnel. The ongoing COVID-19 pandemic may also affect our ability to attract and successfully recruit these personnel, for one or more reasons. We also experience competition for the hiring of scientific personnel from universities and research institutions. Moreover, delays or failures in clinical trials may also make it more challenging to recruit and retain qualified scientific personnel. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our business strategy will be limited and our business, financial condition and results of operations would be adversely affected.
Further, if we fail to expand and enhance our operational, financial, management and managementcompliance systems in conjunction with potential future growth, such failure could have a material adverse effect on our business, financial condition and results of operations. We may be unable to successfully implement these tasks on a larger scale and, accordingly, may not achieve our research, development, business and growth goals.
Our internal computer systems, or those of any contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Despite the implementation of security measures, our internal computer systems and those of third parties with which we contract are vulnerable to damage from cyber-attacks, computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. The prevalent use of mobile devices that access confidential information also increases the risk of lost or stolen devices, security incidents and data security breaches, which could lead to the loss of confidential information or other intellectual property. As a result of the COVID-19 pandemic, we may face increased risks of a security breach or disruption due to our reliance on internet technology and the number of our employees who are working remotely, which may create additional opportunities for cybercriminals to exploit vulnerabilities. Any system failure, accident or security breach that causes interruptions in our operations could result in a material disruption of our product development programs and business operations, in addition to possibly requiring substantial expenditures of resources to remedy. For example, the loss of clinical trial data from completed clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur liabilities and the further development of our product candidates may be delayed. In addition, we may not have adequate insurance coverage to provide compensation for any losses associated with such events.
We could be subject to risks caused by misappropriation, misuse, leakage, falsification or intentional or accidental release or loss of information maintained in the information systems and networks of our company, including personal information of our employees. In addition, outside parties may attempt to penetrate our systems or those of our vendors or fraudulently induce our employees or employees of our vendors to disclose sensitive information in order to gain access to our data. Like other companies, we may experience threats to our data and systems, including malicious codes and viruses, and other cyber-attacks. The number and complexity of these threats continue to increase over time. If a material breach of our security or that of our vendors occurs, the market perception of the effectiveness of our security measures could be harmed, we could lose business and our reputation and credibility could be damaged, all of which would materially adversely affect our business, financial condition and results of operations. We could be required to expend significant amounts of money and other resources to repair or replace information systems or networks. Although we develop and maintain systems and controls designed to prevent these
60
events from occurring, the development and maintenance of these systems, controls and processes is costly and requires ongoing monitoring and updating as technologies change and efforts to overcome security measures become more sophisticated. Moreover, despite our efforts, the possibility of these events occurring cannot be eliminated entirely.
We may acquire businesses or products, or form strategic alliances, in the future, and we may not realize the benefits of such acquisitions or alliances.
We may acquire additional businesses or products, form strategic alliances or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate such businesses with our existing operations and company culture.
We may encounter numerous difficulties in developing, manufacturing and marketing any new products resulting from a strategic alliance or acquisition that delays or prevents us from realizing their expected benefits or enhancing our business. We cannot be certain that, following any such transaction, we will achieve the expected synergies to justify the transaction and it could adversely affect our business, financial condition and results of operations.
We may seek to establish collaborations and, if we are not able to establish them on commercially reasonable terms, we may have to alter our development and commercialization plans or expand our internal efforts and growth.
Our development programs and the potential commercialization of our product candidates will require substantial additional cash to fund expenses. For CTI-1601,nomlabofusp, and any future product candidates, we may decide to collaborate with pharmaceutical andand/or biotechnology companies for the development and potential commercialization of those product candidates in some or all markets.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration for CTI-1601nomlabofusp or other potential product candidates will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the applicable product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty
64
with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such collaboration could be more attractive than the one with us for our product candidate. The terms of any collaboration or other arrangement that we may establish may not be favorable to us.
We may also be restricted under existing license agreements from entering into future agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable or unwilling to do so, we may have to curtail the development potential product candidates for which we are seeking to collaborate, reduce or delay our development program or one or more of our other development programs, delay potential commercialization in some or all markets or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense, including potentially increasing our infrastructure and investment outside the United States. If we elect to increase our expenditures to fund development or commercialization activities on our own, we will need to obtain additional capital, which may not be available to us on acceptable terms orif at all. If we do not have sufficient funds, we may not be able to further develop our product candidates or bring them to market and generate product revenue. In addition, such efforts may require diversion of a disproportionate amount of our attention away from other day-to-day activities and require devotion of a substantial amount of our time to managing these activities.
61
In addition, any future collaborations that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations. Disagreements between parties to a collaboration arrangement regarding clinical development and commercialization matters can lead to delays in the development process or commercializing the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority. Collaborations with pharmaceutical or biotechnology companies and other third parties often are terminated or allowed to expire by the other party. Any such termination or expiration could adversely affect our business, financial condition, results of operations and could harm our business reputation.
We face risks related to health epidemics, including the ongoing COVID-19 pandemic, andand/or other outbreaks of communicable diseases, which could significantly disrupt our operations and may materially and adversely affect our business and financial conditions.
Our business could be adversely impacted by the effects of the ongoinga global pandemic, epidemic or outbreak of an infectious disease, such as, for example, a possible resurgence of vaccine resistant or more highly contagious or deadly variants of COVID-19 pandemic and the efforts to mitigate it. The extent to which the COVID-19 pandemic andsuch outbreaks. Such global efforts to contain its spread will impact our operations will depend on future developments, which are highly uncertain and cannot be predicted at this time, and include the duration, severity and scopeoutbreaks of the outbreak and the actions taken to contain or treat the coronavirus outbreak. The continued spread of the coronaviruscommunicable diseases globally could materially and adversely impact our operations, including without limitation, our manufacturing and supply chain for CTI-1601nomlabofusp and our planned clinical trials, which have faced, and could continue to face, enrollment difficulties as hospitals or clinical trials sites experience closures. Because Friedreich’s ataxiaFA is a rare disease, there are a limited number of patients in close proximity to clinical trial sites and clinical trial patients travel from throughout the United States to clinical trial sites to participate. TheAny travel advisories and risk ofor infection related to Covid-19risks could present increased risks to patients traveling to a clinical trial site for dosing if clinical trials are allowed to continue. Further, there is currently a global shortage of non-human primates available for drug development, due in part to an increase in demand from companies and other institutions developing vaccines and treatments for COVID-19. If the shortage continues, this could substantially increase the cost of conducting our non-clinical development and could also result in delays to our development timelines. These events could delay our clinical trials, increase the cost of completing our clinical trials and negatively impact the integrity, reliability or robustness of the data from our clinical trials. In addition, employee health and availability could be impacted, which may have a material and adverse effect on our business, financial condition and results of operations. The ongoing pandemic or futureFuture pandemics could adversely affect global economies and financial markets resulting in an economic downturn that could have a material adverse effect on our business and prospects.
Compliance with globalWe are subject to stringent and evolving U.S. and foreign laws, regulations and rules, contractual obligations, industry standards, policies and other obligations related to data privacy and data security requirements could result in additional costs and liabilities to ussecurity. Our actual or inhibit our ability to collect and process data globally, and theperceived failure to comply with such requirementsobligations could exposelead to regulatory investigations or actions; litigation; fines and penalties; disruptions of our business operations; reputational harm; loss of revenue or profits; and other adverse business consequences.
65
In the ordinary course of business, we collect, receive, store, process, generate, use, transfer, disclose, make accessible, protect, secure, dispose of, transmit, and share (collectively, process) personal data and other sensitive information, including proprietary and confidential business data, trade secrets, intellectual property, data we collect about trial participants in connection with clinical trials, sensitive third-party data, business plans, transactions, financial information and medical information collected by our patient access management team (collectively, sensitive data). Our data processing activities may subject us to criminal sanctions, civil penalties,numerous data privacy and security obligations, such as various laws, regulations, guidance, industry standards, external and internal privacy and security policies, contractual damages, reputational harm, administrative burdensrequirements, and diminished profitsother obligations relating to data privacy and future earnings.security.
The regulatory framework forIn the collection, use, safeguarding, sharing, transferUnited States, federal, state, and local governments have enacted numerous data privacy and security laws, including data breach notification laws, personal data privacy laws, consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), and other similar laws (e.g., wiretapping laws). For example, HIPAA, as amended by HITECH, imposes specific requirements relating to the privacy, security, and transmission of individually identifiable health information. Additionally, in the past few years, numerous U.S. states—including California, Virginia, Colorado, Connecticut, and Utah—have enacted comprehensive privacy laws that impose certain obligations on covered businesses, including providing specific disclosures in privacy notices and affording residents with certain rights concerning their personal data. As applicable, such rights may include the right to access, correct, or delete certain personal data, and to opt-out of certain data processing activities, such as targeted advertising, profiling, and automated decision-making. The exercise of these rights may impact our business and ability to provide our products and services. Certain states also impose stricter requirements for processing certain personal data, including sensitive information, worldwide is rapidly evolvingsuch as conducting data privacy impact assessments. These state laws allow for statutory fines for noncompliance. For example, the California Consumer Privacy Act of 2018, as amended by the California Privacy Rights Act of 2020 (CPRA) (collectively, CCPA) requires businesses to provide specific disclosures in privacy notices and is likelyhonor requests of California residents to remain uncertainexercise certain privacy rights. The CCPA provides for fines of up to $7,500 per intentional violation and allows private litigants affected by certain data breaches to recover significant statutory damages. Although some U.S. comprehensive privacy laws exempt some data processed in the foreseeable future. Globally, virtually every jurisdiction in whichcontext of clinical trials, these laws may increase compliance costs and potential liability with respect to other personal data we may operate has established its ownmaintain about California residents. Similar laws are being considered in several other states, as well as at the federal and local levels, and we expect more jurisdictions to pass similar laws in the future.
Outside the United States, an increasing number of laws, regulations, and industry standards may govern data securityprivacy and privacy frameworks with which we must comply.security. For example, the European Union’s General Data Protection Regulation 2016/679 ("GDPR") imposes(EU GDPR), United Kingdom’s GDPR (UK GDPR) (collectively, the GDPR), Brazil’s General Data Protection Law (Lei Geral de Proteção de Dados Pessoais, or LGPD) (Law No. 13,709/2018), and China’s Personal Information Protection Law (PIPL) impose strict obligationsrequirements for processing personal data. For example, under the GDPR, companies may face temporary or definitive bans on data processing and other corrective actions; fines of up to 20 million Euros under the EU GDPR / 17.5 million pounds sterling under the UK GDPR or 4% of annual global revenue, whichever is greater; or private litigation related to processing of personal data brought by classes of data subjects or consumer protection organizations authorized at law to represent their interests.
The Swiss Federal Act on Data Protection, or the FADP, also applies to the collection and processing of personal data, including personal health data,health-related information, by companies located in Switzerland, or in certain circumstances, by companies located outside of Switzerland. The FADP has been revised and adopted by the free movementSwiss Parliament. Companies must comply with the revised version of such data. The GDPR appliesthe FADP and its revised ordinances from September 1, 2023, which may result in an increase of costs of compliance, risks of noncompliance and penalties for noncompliance.
In addition, we may be unable to any company established in the European Union as well as any company outside the European Union that processestransfer personal data in connection withfrom Europe and other jurisdictions to the offering of goodsUnited States or servicesother countries due to individuals indata localization requirements or limitations on cross-border data flows. Europe and other jurisdictions have enacted laws requiring data to be localized or limiting the European Union or the monitoring of their behavior. The GDPR enhances data protection obligations for processors and controllerstransfer of personal data including, for example, obligations relating to: processing healthto other countries. In particular, the European Economic Area (EEA) and other sensitive data; obtaining consentthe UK have significantly restricted the transfer of individuals; providing notice to individuals regarding data processing activities; responding to data subject requests; taking certain measures when engaging third-party processors; notifying data subjects and regulators of data breaches; implementing safeguards to protect the security and confidentiality of personal data; and transferring personal data to the United States and other countries outsidewhose privacy laws it believes are inadequate. Other jurisdictions may adopt similarly stringent interpretations of their data localization and cross-border data transfer laws. Although there are currently various mechanisms that may be used to transfer personal data from the European Union, includingEEA and UK to the United States in compliance with law, such as the EEA standard contractual clauses, the UK’s International Data Transfer Agreement / Addendum, and the EU-U.S. Data Privacy Framework and the UK extension thereto (which allows for transfers to relevant U.S.-based organizations who self-certify compliance and
66
participate in the Framework), these mechanisms are subject to legal challenges, and there is no assurance that we can satisfy or rely on these measures to lawfully transfer personal data to the United States. The GDPR imposes additional obligationsIf there is no lawful manner for us to transfer personal data from the EEA, the UK, or other jurisdictions to the United States, or if the requirements for a legally-compliant transfer are too onerous, we could face significant adverse consequences, including by limiting our ability to conduct clinical trial activities in Europe and risks uponelsewhere, the interruption or degradation of our operations, the need to relocate part of or all of our business or data processing activities to other jurisdictions (such as Europe) at significant expense, increased exposure to regulatory actions, substantial fines and substantially increasespenalties, the penaltiesinability to whichtransfer data and work with partners, vendors and other third parties, and injunctions against our processing or transferring of personal data necessary to operate our business. Some European regulators have ordered certain companies to suspend or permanently cease certain transfers of personal data to recipients outside Europe for allegedly violating the GDPR’s cross-border data transfer limitations. Additionally, companies that transfer personal data to recipients outside of the EEA and/or UK to other jurisdictions, particularly to the United States, are subject to increased scrutiny from regulators individual litigants and activist groups.
In addition to data privacy and security laws, we couldmay be contractually subject to industry standards adopted by industry groups and may become subject to such obligations in the event of any non-compliance, including fines of upfuture. We are also bound by other contractual obligations related to €20 million or 4% of total worldwide annual revenue, whichever is higher. The GDPR also confers a private right of action on data subjectsprivacy and consumer associations to lodge complaints with supervisory authorities, seek judicial remediessecurity, and obtain compensation for damages. Given the breadth and depth of changes in data protection obligations, if we are
62
requiredour efforts to comply with the GDPR’s requirements,such obligations may not be successful. We publish privacy policies, marketing materials, and other statements, such as compliance with certain certifications or self-regulatory principles, regarding data privacy and security. If these policies, materials or statements are found to be deficient, lacking in transparency, deceptive, unfair, or misrepresentative of our practices, we willmay be subject to investigation, enforcement actions by regulators, or other adverse consequences.
Additionally, under various privacy laws and other obligations, we may be required to spendobtain certain consents to process personal data. For example, some of our data processing practices may be challenged under wiretapping laws, if we obtain consumer information from third parties through various methods, including chatbot and session replay providers, or via third-party marketing pixels. These practices may be subject to increased challenges by class action plaintiffs. Our inability or failure to obtain consent for these practices could result in adverse consequences, including class action litigation and mass arbitration demands.
Obligations related to data privacy and security (and consumers’ data privacy expectations) are quickly changing, becoming increasingly stringent, and creating uncertainty. Additionally, these obligations may be subject to differing applications and interpretations, which may be inconsistent or conflict among jurisdictions. Preparing for and complying with these obligations requires us to devote significant timeresources and resourcesmay necessitate changes to review our services, information technologies, systems, and practices as well asand to those of any third-party service providers, contractors or consultantsthird parties that process or transfer personal data collectedon our behalf.
We may at times fail (or be perceived to have failed) in the European Union. The GDPRour efforts to comply with our data privacy and other changes in lawssecurity obligations. Moreover, despite our efforts, our personnel or regulations associatedthird parties on whom we rely on may fail to comply with the enhanced protection of certain types of sensitive data, such as healthcare data or other personal information from our clinical trials,obligations, which could require us to changenegatively impact our business practicesoperations. If we or leadthe third parties on which we rely fail, or are perceived to have failed, to address or comply with applicable data privacy and security obligations, we could face significant consequences, including but not limited to: government enforcement actions private(e.g., investigations, fines, penalties, audits, inspections, and similar); litigation (including class-action claims) and mass arbitration demands; additional reporting requirements and/or significant finesoversight; bans on processing personal data; and penaltiesorders to destroy or not use personal data. In particular, plaintiffs have become increasingly more active in bringing privacy-related claims against us, reputational harmcompanies, including class claims. Some of these claims allow for the recovery of statutory damages on a per violation basis, and, if viable, carry the potential for monumental statutory damages, depending on the volume of data and the number of violations. Any of these events could have a material adverse effect on our reputation, business, or financial condition, including but not limited to loss of customers; inability to process personal data or resultsto operate in certain jurisdictions; limited ability to develop or commercialize our products; expenditure of time and resources to defend any claim or inquiry; adverse publicity; or substantial changes to our business model or operations.
Our internal computer systems, oras well as those of any of our CROs, manufacturers, otherCMOs, vendors, contractors, orand consultants, orand potential future collaborators may failexperience failures, unauthorized access, or suffer security or data privacy breaches, or other unauthorized or improper access to, use of, or destruction ofpotentially causing a material disruption in our proprietary or confidential data, employee data, or personal data, which could result in additional costs, loss of revenue, significant liabilities,product development programs, operations, harm to our brand, significant liabilities, loss of revenue, and material disruption of our operations.additional costs.
Despite the implementation ofimplementing comprehensive security measures, both our internal computer systems and those of our contracted third party service providers are susceptible to various threats, including cyber-attacks, computer viruses,
67
unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. The extensive use of mobile devices accessing confidential information further heightens the risks, leading to potential device loss, security incidents, and data breaches that could result in an effortthe loss of confidential information and intellectual property. Any system failure, accident, or security breach causing interruptions in our operations may lead to a material disruption in our product development programs and business operations, potentially requiring substantial resources for recovery. For instance, the loss of clinical trial data from completed trials could lead to delays in regulatory approval efforts and significantly increase costs for data recovery or reproduction.
We are exposed to risks stemming from misappropriation, misuse, leakage, falsification, or intentional or accidental release or loss of information maintained in our information systems and networks, including personal information of our employees. External parties may attempt to penetrate our systems or induce our employees or those of our vendors to disclose sensitive information. As with other companies, we face threats to our data and systems, including malicious codes, viruses, and cyber-attacks. The increasing number and complexity of these threats over time pose a challenge. A material breach in our security or that of our vendors, contractors, or consultants could harm the market perception of our security measures, resulting in business loss and damage to our reputation and credibility. This could significantly adversely affect our business, financial condition, and results of operations, necessitating substantial amounts of money and resources to repair or replace information systems or networks. Despite our continuous efforts, the possibility of such events occurring cannot be entirely eliminated.
In our endeavor to protect systems that store ourstoring critical information, we have implemented security measures. However, given their size and complexity and the increasing amounts of information maintained, onthese systems, including our internal information technology systems and those of our third-party CROs, otherentities like Contract Research Organizations, Contract Manufacturing Organizations, contractors, (including sites performing our clinical trials) and consultants, these systems areremain potentially vulnerable to breakdown or other damage or interruption frombreakdowns, interruptions, and security breaches. Incidents such as service interruptions, system malfunction,malfunctions, natural disasters, terrorism, war, and telecommunication, and electrical failures, as well as security breaches from inadvertent or intentional actions by our employees, contractors, consultants, business partners, and/orand other third parties, or fromincluding cyber-attacks by malicious third parties (including supply chain cyber attacks or the deployment of harmful malware, ransomware, denial-of-service attacks, social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information), whichentities, may compromise our system infrastructure orinfrastructure. This could lead to the loss, destruction, alteration, prevention of access to, disclosure, or dissemination of, or damage or unauthorized access to our data, (includingincluding trade secrets, or other confidential information, intellectual property, proprietary business information, and personal information)information, or data that is processed or maintained on our behalf, or other assets, whichbehalf. Such incidents could result in financial, legal, business, and reputational harm to us. Companies have, in general, experienced an increaseharm.
The rise in phishing and social engineering attacks from third parties in connection with the COVID-19 pandemic, and the increase in remote working further increaseselevate security threats. To the extent that anyAny disruption or security incident werecould lead to result in any loss, destruction, unavailability, alteration, disclosure, or dissemination of, or damage, or unauthorized access to our applications, any other data, processed or maintained on our behalf or other assets, or for itpotentially exposing us to be believed or reported that any of these occurred, we could incur liability, financial harmlitigation and reputational damage andgovernmental investigations, delaying the development and commercialization of our product candidates, could be delayed. We cannot assure you that our data protection efforts and our investment in information technology, or the efforts or investments of CROs, consultants or other third parties, will prevent significant breakdowns or breaches in systems or other cyber incidents that cause loss, destruction, unavailability, alteration or dissemination of, or damage or unauthorized access to, our data and other data processed or maintained on our behalf or other assets that could have a material adverse effect upon our reputation, business, operations or financial condition. For example, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs and the development of our product candidates could be delayed. In addition, the loss of clinical trial data for our product candidates could result in delays in our marketing approval efforts and significantly increase our costs to recover or reproduce the data. Further, any such event that leads to loss, damage, or unauthorized access to, or use, alteration, or disclosure or dissemination of, personal information, including personal information regarding our clinical trial subjects or employees, could harm our reputation directly, compelsubjecting us to complyfines or penalties for noncompliance with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, and otherwise subject us to liability under laws and regulations that protect the privacy and security of personal information, which could result in significant legal and financial exposure and reputational damages that could potentially have an adverse effect on our business.
laws. Notifications and follow-up actions related tofollowing a security incident could impact our reputation and cause us to incur significant costs, including legal expenses and remediation costs.remediation. Significant efforts and costs are expected for detection and prevention. Our reliance on third parties for manufacturing introduces an additional layer of risk to our business.
Insurance policies, including a specific policy related to cybersecurity losses, may be insufficient, with potential availability issues in the future, high deductibles, and limitations in coverage, posing challenges in mitigating losses from disruptions, failures, or security breaches.
If our information technology systems or data, and/or those of third parties upon which we rely, are or were compromised, we could experience adverse consequences resulting from such compromise, including but not limited to regulatory investigations or actions, interruptions to operations or clinical trials, reputational harm, litigation, fines and penalties, disruptions of our business operations, and a loss of customers or sales.
In the ordinary course of our business, we, or the third parties upon which we rely, process proprietary, confidential, and sensitive data, including personal data (such as health-related data), intellectual property, and trade secrets.
Cyberattacks, malicious internet-based activity, online and offline fraud and other similar activities threaten the confidentiality, integrity, and availability of our sensitive data and information technology systems, and those of the third parties upon which we rely. These threats are prevalent, continue to rise, and are becoming increasingly difficult to detect. These threats come from a variety of sources, including traditional computer “hackers,” hacktivists, threat actors, personnel misconduct or error (such as through theft or misuse), organized criminal threat actors, sophisticated nation-states, and nation-state-supported actors. Some actors now engage and are expected to continue to engage in cyber-attacks, including without limitation nation-state actors for geopolitical reasons and in
68
conjunction with military conflicts and defense activities. During times of war and other major conflicts, we and the third parties upon which we rely may be vulnerable to a heightened risk of these attacks, including retaliatory cyber-attacks, that could materially disrupt our systems and operations, supply chain, and ability to produce, sell and distribute our goods and services.
We and the third parties upon which we rely are subject to a variety of evolving threats, including but not limited to, social engineering attacks (including through deep fakes, which may be increasingly more difficult to identify as fake, and phishing attacks), malicious code (such as viruses and worms), malware (including as a result of advanced persistent threat intrusions), denial-of-service attacks, credential stuffing, credential harvesting, personnel misconduct or error, ransomware attacks, supply-chain attacks, software bugs, server malfunction, software or hardware failures, loss of data or other information technology assets, adware, attacks enhanced or facilitated by AI, telecommunications failures, earthquakes, fire, flood, and other similar threats.
Ransomware attacks, including by organized criminal threat actors, nation-states, and nation-state-supported actors, are becoming increasingly prevalent and severe and can lead to significant interruptions, delays, or outages in our operations, disruption of clinical trials or otherwise affecting our ability to provide our products or product candidates, loss of sensitive data (including data related to clinical trials) and income, significant extra expenses to restore data or systems, reputational harm and the diversion of funds. Extortion payments may alleviate the negative impact of a ransomware attack, but we may be unwilling or unable to make such payments (including, for example, if applicable laws or regulations prohibit such payments). Remote work has become more common and has increased risks to our information technology systems and data, as more of our employees work from home, utilizing network connections, computers and devices outside our premises, including at home, while in transit or in public locations. Future business transactions (such as acquisitions or integrations) could expose us to additional cybersecurity risks and vulnerabilities, as our systems could be negatively affected by vulnerabilities present in acquired or integrated entities’ systems and technologies. Furthermore, we may discover security issues that were not found during due diligence of such acquired or integrated entities, and it may be difficult to integrate companies into our information technology environment and security program.
We rely on third-party service providers and technologies to operate critical business systems to process sensitive data in a variety of contexts, including, without limitation, cloud-based infrastructure, drug suppliers, data center facilities, encryption and authentication technology, employee email, content delivery to customers, and other functions. Our ability to monitor these third parties’ information security practices and posture (including whether any unremediated vulnerabilities exist or have been exploited) is limited, and these third parties may not have adequate information security measures in place. In addition, supply-chain attacks have increased in frequency and severity, and we cannot guarantee that third parties’ infrastructure in our supply chain or our third-party partners’ supply chains have not been compromised.
While we have implemented security measures designed to protect against security incidents, there can be no assurance that these measures will be effective. We take steps designed to detect, mitigate, and remediate vulnerabilities in our information security systems (such as our hardware and/or software, including that of third parties upon which we rely). We and the third parties upon which may rely may not, however, detect and remediate all such vulnerabilities including on a timely basis. For example, we have identified certain vulnerabilities in our information systems, and we have taken steps to mitigate the lossrisks associated with known vulnerabilities. These steps include implementing compensating controls and other protective measures. Further, we and the third parties upon which we rely may experience delays in developing and deploying remedial measures and patches designed to address identified vulnerabilities. Vulnerabilities could be exploited and result in a security incident.
Any of clinical trial data from completedthe previously identified or future clinical trialssimilar threats could cause a security incident or other interruption that could result in delaysunauthorized, unlawful, or accidental acquisition, modification, destruction, loss, alteration, encryption, disclosure of, or access to our sensitive information or our information technology systems, or those of the third parties upon whom we rely. A security incident or other interruption could disrupt our ability (and that of third parties upon whom we rely) to provide our products.
We may expend significant resources or fundamentally change our business activities and practices (including our clinical trials) to try to protect against security incidents. Certain data privacy and security obligations may require us to implement and maintain specific security measures or industry-standard or reasonable security measures to protect our information technology systems and sensitive data.
69
Applicable data privacy and security obligations may require us to notify relevant stakeholders, including affected individuals, customers, regulators, and investors, of security incidents. Such disclosures are costly, and the disclosure or the failure to comply with such requirements could lead to adverse consequences. If we (or a third party upon whom we rely) experience a security incident or are perceived to have experienced a security incident, we may experience adverse consequences. These consequences may include: government enforcement actions (for example, investigations, fines, penalties, audits, and inspections); additional reporting requirements and/or oversight; restrictions on processing sensitive data (including personal data); litigation (including class claims); indemnification obligations; negative publicity; reputational harm; monetary fund diversions; diversion of management attention; interruptions in our regulatory approval effortsoperations (including availability of data); financial loss; and significantly increase our costs to recover or reproduce the lost data. We expect to incur significant costs in an effort to detect and prevent securityother similar harms. Security incidents and weattendant consequences may face increased costsprevent or cause customers to stop using our products, deter new customers from using our products, and requirementsnegatively impact our ability to expend substantial resources in the event of an actual or perceivedgrow and operate our business.
In addition to experiencing a security incident. We also rely onincident, third parties may gather, collect, or infer sensitive data about us from public sources, data brokers or other means that reveals competitively sensitive details about our organization and could be used to manufactureundermine our product candidates,competitive advantage or market position. Additionally, sensitive information of the Company could be leaked, disclosed, or revealed as a result of or in connection with the use of generative AI technologies.
Our contracts may not contain limitations of liability, and similar events relatingeven where they do, there can be no assurance that limitations of liability in our contracts are sufficient to their computer systemsprotect us from liabilities, damages, or claims related to our data privacy and security obligations.
In addition, our insurance coverage may not be adequate or sufficient in type or amount to protect us from or to mitigate liabilities arising out of our privacy and security practices. The successful assertion of one or more large claims against us that exceeds our available insurance coverage, or results in changes to our insurance policies (including premium increases or the imposition of large deductible or co-insurance requirements), could also have a materialan adverse effect on our business. To the extent that any disruption or security incident were to result in any loss, destruction, or alteration of, or damage or unauthorized access to, our data or other information that is processed or maintained on our behalf, or inappropriate disclosure of or dissemination of any such information, we could be exposed to litigation and governmental
63
investigations, the further development and commercialization of our product candidates could be delayed, and we could be subject to significant fines or penalties for any noncompliance with certain state, federal and/or international privacy and security laws.
Our insurance policies may not be adequate to compensate us for the potential losses arising from any such disruption in or, failure or security breach of our systems or third-party systems where information important to our business operations or commercial development is stored. In addition, such insurance may not be available to us in the future on economically reasonable terms, or at all. Further, our insurance may not cover all claims made against us and could have high deductibles in any event, and defending a suit, regardless of its merit, could be costly and divert management attention.
We face potential product liability exposure, and, if claims are brought against us, we may incur substantial liability.
The use of CTI-1601nomlabofusp and other potential product candidates in clinical trials, if any, and the sale of CTI-1601nomlabofusp and other potential product candidates, if developed and approved, exposes us to the risk of product liability claims. Product liability claims might be brought against us by patients, healthcare providers or others selling or otherwise coming into contact with CTI-1601nomlabofusp or other potential product candidates. For example, we may be sued if any product we develop allegedly causes injury or death or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Physicians and patients may not comply with any warnings that identify known potential adverse effects and patients who should not use our products. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to stop development or, if approved, limit commercialization of our product candidates. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, including as a result of interactions with alcohol or other drugs, negligence, strict liability and a breach of warranties. Claims could also be asserted under consumer protection acts in other jurisdictions. If we become subject to product liability claims and cannot successfully defend ourselves against them, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in, among other things:
70
64
We maintain product liability insurance coverage for our clinical trials with a $5 million aggregate coverage limit. Nevertheless, our insurance coverage may be insufficient to reimburse us for any expenses or losses we may suffer. Moreover, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses, including if insurance coverage becomes increasingly expensive. If we obtain marketing approval for CTI-1601nomlabofusp or other potential product candidates, we intend to expand our insurance coverage to include the sale of commercial products; however, we may not be able to obtain this product liability insurance on commercially reasonable terms. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. The cost of any product liability litigation or other proceedings, even if resolved in our favor, could be substantial, particularly in light of the size of our business and financial resources. A product liability claim or series of claims brought against us could cause our stock price to decline and, if we are unsuccessful in defending such a claim or claims and the resulting judgments exceed our insurance coverage, our financial condition, business, results of operations and prospects could be materially adversely affected.
Risks Related to Our Reliance on Third Parties
We have limited experience in conducting or supervising clinical trials and must outsource all clinical trials. As a result, many important aspects of our drug development programs are outside of our direct control.
We have limited experience in conducting or supervising clinical trials that must be performed to obtain data to submit in concert with applications for approval by the FDA, the EMA or other comparable foreign regulatory authorities. As a result, we expect to continue to rely on CROs, clinical trial sites, clinical data management organizations and consultants to design, conduct, supervise and monitor our non-clinical studies and clinical trials. We, and our CROs, and contractors are required to comply with various regulations, including the FDA’s regulations commonly referred to as good clinical practicesregarding current Good Clinical Practices ("GCPs"cGCPs") which are enforced by regulatory agencies, including the FDA, and comparable foreign regulatory authorities to ensure the health, safety and rights of patients are protected in clinical development and clinical trials, and that trial data integrity is assured. Regulatory authorities ensure compliance with these requirements through periodic inspections of trial sponsors, principal investigators and clinical trial sites. Our expected reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. If we or any of our CROs, contractors, or clinical trial sites fail to comply with applicable requirements, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, EMA or other comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure youensure that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with such requirements. In addition, our clinical trials must be conducted with products produced under cGMP requirements, which mandate, among other things, the methods, facilities and controls used in manufacturing, processing and packaging of a drug product to ensure its safety and identity. Failure to comply with these regulations may require us to repeat non-clinical studies and/or clinical trials, which would delay the regulatory approval process, and could also subject us to enforcement action, up to and including, civil and criminal penalties, which would materially adversely affect our business, financial condition and results of operations.
Our CROs and contractors are not our employees, and except for remedies available to us under our agreements with such CROs and contractors, we cannot control whether or not they devote sufficient time and resources to our ongoing clinical and non-clinical programs. If our CROs and contractors do not successfully carry out their contractual duties or obligations or meet expected deadlines or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other
71
reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, our operations and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenue could be delayed or reduced. In addition, operations of our CROs and contractors could be affected by earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions. If their facilities are unable to operate because of an accident or incident, even for a short period of time, some or all of our research and development programs may be harmed or delayed, and our operations and financial condition could suffer.
We have less direct control over the conduct, timing and completion of these clinical trials and the management of data developed through the clinical trials than would be the case if we were relying entirely upon our own staff. Communicating with outside parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. These factors may materially adversely affect the willingness or ability of third parties to
65
conduct our clinical trials and may subject us to unexpected cost increases that are beyond our control. Nevertheless, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the applicable protocol, legal and regulatory requirements and scientific standards, and our reliance on CROs and contractors does not relieve us of our regulatory responsibilities.
Because we rely on third parties to develop and manufacture our product candidate,In addition, we must, at times, share confidential information with such third parties. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements, or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets, intellectual property, data from clinical studies and future development plans. Despite the contractual provisions employed when working with third parties, the need to share confidential information increases the risk that such confidential information become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our confidential information, a competitor’s discovery of our confidential information or other unauthorized use or disclosure would impair our competitive position and may have an adverse effect on our business.
In addition,Moreover, because we have relied on third parties, our internal capacity to perform these functions is limited. Outsourcing these functions involves risk that third parties may not perform to our standards, may not produce results in a timely manner or may fail to perform at all. We currently have a small number of employees, which limits the internal resources we have available to engage new third-party providers, if necessary, and monitor existing third-party providers. To the extent we are unable to engage new third-party providers, if necessary, and successfully manage the performance of third-party service providers in the future, our business may be adversely affected. Though we carefully manage our relationships with CROs, and contractors there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition, results of operation and prospects.
66
We rely on third-party supply and manufacturing partners for drug supplies for our research and development, non-clinical activities, and clinical activities, and may do the same for any commercial supplies of our product candidates.
We rely on third-party supply and manufacturing partners to supply the materials and components for, and manufacture, our research and development, non-clinical and clinical study drug substance and drug product. We have not yet manufactured or formulated nomlabofusp or any other product candidate on a commercial scale and may not be able to do so for any of our product candidates. We will work to develop and optimize our manufacturing process; however, we cannot be sure that the process will result in therapies that are safe, potent or effective. Given our reliance on third parties and the risk of manufacturing commercial scale quantities, our ability to adequately commercially launch and/or supply nomlabofusp could be adversely affected.
We do not own manufacturing facilities or supply sources for such components, non-clinical and clinical study drug substance, product and materials, including devices that may be required for administration, but may develop these capabilities in the future. There can be no assurance that our supply of research and development, non-clinical and clinical development of drugs and other materials will not be limited, interrupted, restricted in certain geographic regions or will be of satisfactory quality or continue to be available at acceptable prices. In particular, replacement of any product formulation manufacturer we may engage could require significant effort and expertise
72
because there may be a limited number of qualified replacements. For example, we rely and expect to continue to rely on a small number of manufacturers to supply us with our requirements for drug substance and formulated drugsdrug product related to our CTI-1601nomlabofusp clinical program. The drug substance which is in frozen liquid form for CTI-1601nomlabofusp is currently manufactured for us by a third-party manufacturer, and the frozen liquid form of drug product is made at another manufacturer. We are undertaking a program with a third party manufacturer to begin to produce a lyophilized version of the drug product from the same drug substance, that, once available, we intend to use in certain of our future planned clinical trials. Our research and development programs could be adversely affected by a significant interruption in these manufacturing services or in the supply of drug substance and formulated drugs. In addition, because we rely on multiple manufacturers for our CTI-1601nomlabofusp clinical program, termination of our agreements with any of these manufacturers could significantly adversely impact our current and planned operations.
In the event that any of our suppliers or manufacturers fails to perform itstheir obligations to us in relation to quality, timing or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may be forced to manufacture the materials ourselves, for which we currently do not have the capabilities or resources, or enter into an agreement with another third party, which we may not be able to do on reasonable terms, if at all. In some cases, the technical skills or technology required to manufacture our product candidates may be unique or proprietary to the original manufacturer and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills or technology to another third party and a feasible alternative may not exist. These factors would increase our reliance on such manufacturer or require us to obtain a license from such manufacturer in order to have another third party manufacture our product candidates. If we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop product candidates in a timely manner or within budget.
We also rely on third parties to store master and working cell banks. We currently have one master cell bank and one working cell bank for CTI-1601nomlabofusp and believe we would have adequate backup should any cell bank be lost in a catastrophic event. However, it is possible that we could lose multiple cell banks and have our manufacturing severely impacted by the need to replace the cell banks, which could materially and adversely affect our business, financial condition and results of operations.
We may rely on third party manufacturers if we receive regulatory approval for any product candidate. To the extent that we have existing, or enter into future, manufacturing arrangements with third parties, we will depend on these third parties to perform their obligations in a timely manner consistent with contractual and regulatory requirements, including those related to quality control and assurance. If we are unable to obtain or maintain third-partythird party manufacturing for product candidates, or to do so on commercially reasonable terms, we may not be able to develop and commercialize our product candidates successfully. Our or a third party’s failure to execute on our
67
manufacturing requirements could adversely affect our business, financial condition and results of operations in a number of ways, including:
We and our contract manufacturers are subject to significant regulation with respect to manufacturing our products. The manufacturing facilities on which we rely may not continue to meet regulatory requirements and have limited capacity.
73
All entities involved in the preparation of therapeutics for clinical trials or commercial sale, including our existing contract manufacturers for CTI-1601,nomlabofusp, are subject to extensive regulation. Some components of a finished therapeutic product approved for commercial sale or used in late-stage clinical trials must be manufactured in accordance with cGMP. These regulations govern manufacturing processes and procedures (including record keeping) and the implementation and operation of quality systems to control and assure the quality of investigational products and products approved for sale. Poor control of production processes can lead to the introduction of adventitious agents or other contaminants, or to inadvertent changes in the properties or stability of our product candidates that may not be detectable in final product testing. We or our contract manufacturers must supply all necessary documentation in support of a BLA or NDA on a timely basis and where required, must adhere to the FDA’s or other regulator’s good laboratory practices ("GLPs")GLPs and cGMP regulations enforced by the FDA or other regulatorregulators through facilities inspection programs. The facilities and quality systems of some or all of our third-partythird party contractors must pass a pre-approval inspection for compliance with the applicable regulations as a condition of regulatory approval of CTI-1601nomlabofusp or any of our other potential products. In addition, the regulatory authorities may, at any time, audit or inspect a manufacturing facility involved with the preparation of CTI-1601nomlabofusp or our other potential products or the associated quality systems for compliance with the regulations applicable to the activities being conducted. If these facilities do not pass a pre-approval plant inspection, the FDA or other regulatory approval of the products will not be granted.
The regulatory authorities also may, at any time following approval of a product for sale, audit the manufacturing facilities of our third-partythird party contractors. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulations occurs independent of such an inspection or audit, we or the relevant regulatory authority may require remedial measures that may be costly and/or time-consuming for us or a third party to implement and that may include the temporary or permanent suspension of a clinical trial or commercial sales or the temporary or permanent closure of a facility. Any such remedial measures imposed upon us or third parties with whom we contract could materially harm our business.
If we or any of our third-partythird party manufacturers fail to maintain regulatory compliance, the FDA or other regulators can impose regulatory sanctions including, among other things, refusal to approve a pending application for a biologic product, or revocation of a pre-existing approval. As a result, our business, financial condition and results of operations may be materially harmed.
Additionally, if supply from one approved manufacturer is interrupted, there could be a significant disruption in commercial supply. The number of manufacturers with the necessary manufacturing capabilities is limited. In addition, an alternative manufacturer would need to be qualified through a BLA or NDA supplement or similar regulatory submission which could result in further delay. The regulatory agencies may also require additional
68
studies if a new manufacturer is relied upon for commercial production. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop product candidates in a timely manner or within budget.
These factors could also cause the delay of manufacturing development, clinical trials, regulatory submissions, required approvals or commercialization of CTI-1601nomlabofusp or any other product candidates, cause us to incur higher costs and prevent us from commercializing our products successfully. Furthermore, if our suppliers fail to meet contractual requirements, and we are unable to secure one or more replacement suppliers capable of production at a substantially equivalent cost, our clinical trials may be delayed, or we could lose potential revenues. Any of the above would materially adversely affect our business, financial condition and results of operations.
Changes in methods of product candidate manufacturing may result in additional costs and/or delays.
As product candidates progress through clinical to late-stage clinical trials to marketing approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, are altered along the way in an effort to optimize yield, manufacturing batch size, change drug product dosage form, minimize costs and achieve consistent quality and results. Such changes carry the risk that they will not achieve these intended objectives. Any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the altered materials. This could delay completion of clinical trials, require the conduct of bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidates and jeopardize our ability to commercialize our product candidates and generate revenue.
74
We enter into various contracts in the normal course of our business in which we indemnify the other party to the contract. In the event we have to perform under these indemnification provisions, it could materially increase our costs and potential liability.
In the normal course of business, we periodically enter into academic, commercial, service, collaboration, licensing, consulting and other agreements that contain indemnification provisions. With respect to our academic and other research agreements, we typically indemnify the institution and related parties from losses arising from claims relating to the products, processes or services made, used, sold or performed pursuant to the agreements for which we have secured licenses, and from claims arising from our or our sublicensees’ exercise of rights under the agreement. With respect to our collaboration and contract service agreements, we typically indemnify our collaborators from any third-partythird party product liability claims that could result from the production, use or consumption of the product, as well as for alleged infringements of any patent or other intellectual property right by a third party. With respect to consulting agreements, we typically indemnify consultants from claims arising from the good faith performance of their consulting services.
Should our obligation under an indemnification provision exceed applicable insurance coverage or should we be denied insurance coverage, our business, financial condition and results of operations could be adversely affected. Similarly, if we are relying on a collaborator to indemnify us and the collaborator is denied insurance coverage or the indemnification obligation exceeds thetheir applicable insurance coverage, and if the collaborator does not have other assets available to indemnify us, our business, financial condition and results of operations could be adversely affected.
To the extent we are able to enter into collaborative arrangements or strategic alliances, we may be exposed to risks related to those collaborations and alliances.
Biotechnology companies sometimes become dependent upon collaborative arrangements or strategic alliances to complete the development and commercialization of product candidates. In seeking collaborative arrangements and strategic partners, we face significant competition from other companies as well as public and private research institutions. There can be no assurance that we will be able to enter into or maintain strategic alliances on terms favorable to us, or at all. If we elect to enter into collaborative arrangements or strategic alliances, these arrangements may place the development of our product candidates outside our control, may require us to relinquish important rights or may otherwise be on terms unfavorable to us, which could adversely affect our business, financial condition and results of operations.
69
Dependence on collaborative arrangements or strategic alliances would subject us to a number of risks, including the risk that:
Risks Related to Our Intellectual Property Rights
If, in the United States and other countries, we are unable to adequately protect our proprietary technology or maintain issued patents which are sufficient to protect CTI-1601nomlabofusp or potential product candidates, third parties could compete against us more directly, which would have a material adverse impact on our business, results of operations, financial condition and prospects.
Our commercial success will depend in part on our success in obtaining and maintaining issued patents and other intellectual property rights in the United States and elsewhere and protecting our proprietary technology. If we do not adequately protect our intellectual property and proprietary technology, competitors may be able to use our
75
technologies and erode or negate any competitive advantage we may have, which could harm our business and ability to achieve profitability.
With respect to our patent portfolio, we in-license from Wake Forest University Health Sciences ("WFUHS")WFUHS certain issued U.S. patents that relate to CTI-1601nomlabofusp and its use for treating Friedreich’s ataxia.FA. We also in-license from Indiana University ("IU") a pending international andIU a United States patent and pending non-provisional applicationapplications in the United States and certain foreign countries that relatesrelate to the composition of CTI-1601nomlabofusp and methods of use, and certain U.S. patents relating to materials and methods of use relating to the development of CTI-1601.nomlabofusp. We also own or co-own pending international PCT, foreign and United States non-provisional applications, a United States Patent, and United States provisional applications relating to the development of nomlabofusp, including methods of use of CTI-1601,nomlabofusp, biomarkers and to our peptide delivery platform technology.
In some cases, we have only filed provisional patent applications on certain aspects of our technologies and each of these provisional patent applications is not eligible to become an issued patent until, among other things, we file a non-provisional patent application within 12 months of the filing date of the applicable provisional patent application. Any failure to file a non-provisional patent application within this timeline could cause us to lose the ability to obtain patent protection for the inventions disclosed in the associated provisional patent applications.
With respect to both in-licensed and owned intellectual property, we cannot predict whether the patent applications we and our licensors are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient protection from competitors or other third parties.
We cannot provide any assurances that any of our pending patent applications that mature into issued patents will include claims with a scope sufficient to protect CTI-1601,nomlabofusp, or other potential product candidates. Other parties have developed technologies that may be related or competitive to our approach and may have filed or may file patent applications and may have received or may receive patents that may overlap or conflict with our patent applications, either by claiming the same methods or formulations or by claiming subject matter that could dominate our patent position. The patent positions of biotechnology and pharmaceutical companies, including our patent position,
70
involve complex legal and factual questions, and, therefore, the issuance, scope, validity and enforceability of any patent claims that we may obtain cannot be predicted with certainty. Patents, if issued, may be challenged, deemed unenforceable, invalidated, or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, ex parte reexamination, or inter partes review proceedings, supplemental examination and challenges in district court. Patents may be subjected to opposition, post-grant review, or comparable proceedings lodged in various foreign, both national and regional, patent offices. These proceedings could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such proceedings may be costly. Thus, any patents that we may own or exclusively license may not provide any protection against competitors. Furthermore, an adverse decision in an interference proceeding can result in a third party receiving the patent right sought by us, which in turn could affect our ability to develop, market or otherwise commercialize CTI-1601,nomlabofusp, and other potential product candidates.
Furthermore, though an issued patent is presumed valid and enforceable, its issuance is not conclusive as to its validity or its enforceability and it may not provide us with adequate proprietary protection or competitive advantages against competitors with similar products. Competitors may also be able to design around our patents. Other parties may develop and obtain patent protection for more effective technologies, designs or methods. The laws of some foreign countries do not protect our proprietary rights to the same extent as the laws of the United States, and we may encounter significant problems in protecting our proprietary rights in these countries. If these developments were to occur, they could have a material adverse effect on our potential future sales.
Our ability to enforce our patent rights depends on our ability to detect infringement. It is difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product. Any litigation to enforce or defend our patent rights, even if we were to prevail, could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.
In addition, proceedings to enforce or defend our patents could put our patents at risk of being invalidated, held unenforceable, or interpreted narrowly. Such proceedings could also provoke third parties to assert claims against us, including that some or all of the claims in one or more of our patents are invalid or otherwise unenforceable. If any of our patents covering CTI-1601nomlabofusp are invalidated or found unenforceable, our financial
76
position and results of operations would be materially and adversely impacted. In addition, if a court found that valid, enforceable patents held by third parties covered CTI-1601,nomlabofusp, our financial position and results of operations would also be materially and adversely impacted.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
71
We rely upon unpatented trade secrets, unpatented know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, by confidentiality agreements with our employees and consultants. We also have agreements with our employees and selected consultants that obligate them to assign their inventions to us and have non-compete agreements with some, but not all, of our consultants. It is possible that technology relevant to our business will be independently developed by a person that is not a party to such an agreement. Furthermore, if the employees and consultants who are parties to these agreements breach or violate the terms of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets through such breaches or violations. Further, our trade secrets could otherwise become known or be independently discovered by our competitors. If we are unable to adequately protect our proprietary technology or maintain issued patents which are sufficient to protect CTI-1601nomlabofusp or potential future product candidates, third parties could compete against us more directly, which would have a material adverse impact on our business, results of operations, financial condition and prospects.
Certain of the patentsOur key patent, which we license relatingrelates to CTI-1601nomlabofusp will expire by 2025in 2040 and we will lose our ability to rely upon these patentsthis patent to prevent competing products from entering the market, which may impair our ability to generate revenue.
We have in-licensed certain patents relating to CTI-1601nomlabofusp from WFUHS. The U.S. patents relating to CTI-1601nomlabofusp and its use for the treatment of Friedreich’s ataxiaFA expire in 2024 and 2025, respectively. When these patents expire, we will be unable to use these patents to try to block others from marketing CTI-1601nomlabofusp in the United States We have also in-licensed an international and aissued United States patent and pending non-provisional patent applicationapplications in the United States and certain foreign countries relating to the composition of CTI-1601nomlabofusp and methods of use from IU, which,IU. This United States patent will expire in 2040 at the earliest. Pending applications if issued as a patent,patents, would also expire in 2040 at the earliest in 2040.earliest. We cannot predict whether these patent applications we and our licensors are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient protection from competitors or other third parties. When these various patents, if issued, expire, we will be unable to use the patents to try to block others from marketing CTI-1601nomlabofusp in the United States.
We own a certain United States provisional applicationsapplication and certain United States and internationalforeign non-provisional applications and a United States patent relating to our platform technology which,technology. The United States Patent will expire in 2041 at the earliest, and the pending applications, if issued as patents, would be expected to expire in 2040-2041.2041-2044. The provisional applicationsapplication may not be timely converted into a non-provisional applications,application, and we cannot predict
77
whether these provisional applications and non-provisional patent applications will issue as patents in any particular jurisdiction, or whether the claims of any issued patents will provide sufficient protection from competitors or third parties for potential product candidates. When these various patents expire, we will be unable to use the patents to try to block others from marketing products pertaining to our platform technology.
In addition, given the amount of time required for the development, testing, and regulatory review of new product candidates, patents protecting such product candidates might expire before or shortly after such product candidates are commercialized. As a result, our intellectual property may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
Once our patents expire, we will be subject to competition from third parties who will be able to use the intellectual property covered by these patents, which could impair our ability to generate revenue and could adversely affect our business, financial condition and results of operations.
72
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts and stop us from commercializing or may increase the costs of commercializing CTI-1601nomlabofusp or other potential product candidates, if approved.
Our success will depend in part on our ability to operate without infringing the intellectual property and proprietary rights of third parties. We cannot ensure that our business, products and methods do not or will not infringe the patents or other intellectual property rights of third parties.
The pharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. Third parties may allege that CTI-1601nomlabofusp or our other potential product candidates or the use of our technologies infringes patent claims or other intellectual property rights held by them or that we are employing their proprietary technology without authorization. Patent and other types of intellectual property litigation can involve complex factual and legal questions, and their outcome is uncertain. Any claim relating to intellectual property infringement that is successfully asserted against us may require us to pay substantial damages, including treble damages and attorneys’ fees if we are found to be willfully infringing another party’s patents, for past use of the asserted intellectual property and royalties and other consideration going forward if we are forced to take a license. In addition, we may also be required to indemnify certain of our licensors, vendors or suppliers from any damages they incur related to any infringement of any third-partythird party intellectual property by our product candidates.
In addition, if any such claim were successfully asserted against us and we could not obtain such a license, we may be forced to stop or delay developing, manufacturing, selling or otherwise commercializing CTI-1601.nomlabofusp.
Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court, or redesign our products. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, intellectual property litigation or claims could force us to do one or more of the following:
Any of these risks coming to fruition could have a material adverse effect on our business, results of operations, financial condition and prospects.
We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
78
We may also be subject to claims that former employees or other third parties have an ownership interest in our patents or other intellectual property. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business, financial condition and results of operations. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
73
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
We have systems in place to remind us to pay periodic maintenance fees, renewal fees, annuity fees and various other patent and application fees, and we employ an outside law firm to pay these fees. The U.S. Patent and Trademark Office and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patentapplication process. We employ an outside law firm and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If this occurs, our competitors may be able to enter the market, which would have a material adverse effect on our business.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe on our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of our or our licensors is not valid, is unenforceable and/or is not infringed, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing which could materially adversely affect our business, financial condition and results of operations.
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to us from the prevailing party. Our business, financial condition and results of operations could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States, which could adversely affect our business, financial condition and results of operations.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
Issued patents covering our product candidates could be found invalid or unenforceable if challenged in court.
If we or one of our licensing partners initiated legal proceedings against a third party to enforce a patent covering our product candidate, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for unenforceability assertions include allegations that someone connected with prosecution of the patent withheld relevant information from the U.S. PTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of
79
litigation. Such mechanisms include re-examination, post grant review and equivalent proceedings in foreign jurisdictions, e.g., opposition proceedings. Such proceedings could result in revocation or amendment of our patents in such a way that they no longer cover our product candidates or competitive products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot ensure that there is no invalidating prior act, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the
74
patent protection on our product candidates. Such a loss of patent protection would have a material adverse impact on our business, financial condition, and results of operations.
We do not seek to protect our intellectual property rights in all jurisdictions throughout the world and we may not be able to adequately enforce our intellectual property rights even in the jurisdictions where we seek protection.
Filing, prosecuting and defending patents on product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States could be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. For example, an April 2020 report from the Office of the United States Trade Representative identified a number of countries, including India and China, where challenges to the procurement and enforcement of patent rights have been reported. Several countries, including India and China, have been listed in the report every year since 1989. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license, which would materially adversely affect our business, financial condition and results of operations.
We are dependent on licensed intellectual property for CTI-1601.nomlabofusp. If we were to lose our rights to licensed intellectual property, we may not be able to continue developing or commercializing CTI-1601,nomlabofusp, if approved.
We have an exclusive license with WFUHS, pursuant to which we exclusively license certain patent rights relating to the TAT-frataxin fusion protein and its use, on a worldwide basis. We have an exclusive license with IU, pursuant to which we exclusively license certain patent rights relating to CTI-1601nomlabofusp and its use for the treatment of mitochondrial diseases, on a worldwide basis.
Our license agreements with WFUHS and IU impose, and we expect our future license agreements will impose, various development, diligence, commercialization, and other obligations on us in order to maintain the licenses. In spite of our efforts, WFUHS, IU, or a future licensor might conclude that we have materially breached our obligations under such license agreements and seek to terminate the license agreements, thereby removing or limiting our ability to develop and commercialize products and technology covered by these license agreements. If these licenses are terminated, or if the underlying patent rights licensed thereunder fail to provide the intended exclusivity, competitors or other third parties would have the freedom to seek regulatory approval of, and to market, products identical to ours and we may be required to cease our development and commercialization of certain of our product candidates or of CTI-1601.nomlabofusp. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.
75
Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including:
80
The agreements under which we currently license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations, and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize CTI-1601,nomlabofusp, which could have a material adverse effect on our business, financial conditions, results of operations, and prospects.
Some intellectual property may have been discovered through government funded programs and thus may be subject to federal regulations such as “march-in” rights, certain reporting requirements and a preference for U.S.-based companies. Compliance with such regulations may limit our exclusive rights and limit our ability to contract with non-U.S. manufacturers.
Our in-licensed patent rights from WFUHS and IU were funded in part by the U.S. government and are therefore subject to certain federal regulations. When new technologies are developed with U.S. government funding, the U.S. government generally obtains certain rights in any resulting patents, including a non-exclusive license authorizing the U.S. government to use the invention or to have others use the invention on its behalf. The U.S. government’s rights may also permit it to disclose the funded inventions and technology to third parties and to exercise march-in rights to use or allow third parties to use the technology we have licensed that was developed using U.S. government funding. The U.S. government may exercise its march-in rights if it determines that action is necessary because we fail to achieve practical application of the government-funded technology, or because action is necessary to alleviate health or safety needs, to meet requirements of federal regulations, or to give preference to U.S. industry. In addition, our rights in such inventions may be subject to certain requirements to manufacture products embodying such inventions in the United States in certain circumstances and if this requirement is not waived. Any exercise by the U.S. government of such rights or by any third party of its reserved rights could have a material adverse effect on our competitive position, business, financial condition, results of operations, and prospects.
We have not yet registered trademarks for a commercial trade name for CTI-1601nomlabofusp or other potential product candidates and failure to secure such registrations could adversely affect our business, financial condition and results of operations.
We have not yet registered trademarks for a commercial trade name for CTI-1601nomlabofusp or other potential product candidates. Any future trademark applications may be rejected during trademark registration proceedings. Although
76
we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, the U.S. PTO and comparable agencies in many foreign jurisdictions give third parties an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. Moreover, any name we propose to use with our product candidates in the United States must be approved by the FDA, and similarly in many foreign jurisdictions regardless of whether we have registered it, or applied to register it, as a trademark. The FDA typically conducts a review of proposed product names, including an
81
evaluation of potential for confusion with other product names. If the FDA or a foreign regulatory authority objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA.FDA or other regulatory authorities.
Any trademarks we have obtained or may obtain may be infringed or successfully challenged, resulting in harm to our business.
We expect to rely on trademarks as one means to distinguish any of our products that are approved for marketing from the products of our competitors. Once we select new trademarks and apply to register them, our trademark applications may not be approved. Third parties may oppose or attempt to cancel our trademark applications or trademarks, or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could be forced to rebrand our drugs, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing new brands. Our competitors may infringe our trademarks and we may not have adequate resources to enforce our trademarks.
If we do not obtain additional protection under the Hatch-Waxman Amendments and similar foreign legislation by extending the patent terms and obtaining data exclusivity for CTI-1601,nomlabofusp, our business may be materially harmed.
Depending upon the timing, duration and specifics of development and FDA marketing approval of CTI-1601nomlabofusp or our other potential product candidates, one or more of our U.S. patents may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Amendments.(the "Hatch-Waxman Amendments"). The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, we may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain a patent term extension or restoration or the term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and our ability to generate revenues, business, financial condition and results of operations could be materially adversely affected.
Our proprietary rights may not adequately protect our technologies, which may adversely affect our position in the market, business, financial condition and results of operations.
We rely on unpatented trade secrets, know-how, and technology, which are difficult to protect, especially in the pharmaceutical industry, where much of the information about a product must be made public during the regulatory approval process. We seek to protect trade secrets, in part, by entering into confidentiality agreements with employees, consultants and others. These parties may breach or terminate these agreements or may refuse to enter into such agreements with us, and we may not have adequate remedies for such breaches. Furthermore, these agreements may not provide meaningful protection for our trade secrets or other proprietary information or result in the effective assignment to us of intellectual property and may not provide an adequate remedy in the event of unauthorized use or disclosure of confidential information or other breaches of the agreements. Despite our efforts to protect our trade secrets, we or our board members, employees, consultants, contractors or scientific and other advisors may unintentionally or willfully disclose our proprietary information to competitors.
If we fail to maintain trade secret protection, our competitive position may be adversely affected. Competitors may also independently discover our trade secrets. Enforcement of claims that a third party has illegally obtained and is using trade secrets is expensive, time consuming and uncertain. If our competitors independently develop equivalent
77
knowledge, methods and know-how, we would not be able to assert our trade secrets against them and our business, financial condition and results of operations could be harmed.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is therefore costly, time consuming and inherently uncertain. Patent reform legislation could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. The Leahy Smith America Invents Act ("the Leahy Smith Act") enacted in
82
September 2011, brought significant changes to the U.S. patent law system. These include provisions that affect the way patent applications are prosecuted and may affect patent litigation. The United States Patent Office continues to develop and implement new regulations and procedures to govern administration of the Leahy Smith Act, and many of the substantive changes to patent law associated with the Leahy Smith Act became effective on March 16, 2013. The Leahy Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of our issued patents, all of which could harm our business, results of operations, financial condition and prospects.
In addition, the U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Additionally, there have been recent proposals for additional changes to the patent laws of the United States and other countries that, if adopted, could impact our ability to enforce our proprietary technology. Depending on future actions by the U.S. Congress, the U.S. courts, the U.S. PTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of our employees’ former employers.
OurMany of our employees have been previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we are not aware of any claims currently pending against us, we may be subject to claims that we or our employees inadvertently or otherwise used or disclosed the trade secrets or other proprietary information of our employees’ former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying moneymonetary claims, we may lose valuable intellectual property rights or personnel. A loss of key personnel or their work product could hamper or prevent our ability to develop and commercialize CTI-1601nomlabofusp or our other potential product candidates, which would materially adversely affect our business, financial condition and results of operations.
Risks Related to Our Common Stock
Our stock price could be highly volatile, and purchasers of our common stock could incur substantial losses.
The market price of our common stock could be subject to significant fluctuations. Market prices for securities of early-stage pharmaceutical, biotechnology, and other life sciences companies have historically been particularly volatile. Some of the factors that may cause the market price of our common stock to fluctuate include:
78
83
There is no guarantee that our common stock will appreciate or even maintain the price at which our holders have purchased it. Therefore, there is a risk that investors may lose all or part of their investment in our securities.
Moreover, the stock markets in general have experienced substantial volatility that has often been unrelated to the operating performance of individual companies or the biotechnology sector. These broad market fluctuations may also adversely affect the trading price of our common stock.
In the past, following periods of volatility in the market price of a company’s securities, stockholders have often instituted class action securities litigation against those companies. Such litigation, if instituted, could result in substantial costs and diversion of management’s attention and resources, which could significantly impact our profitability and reputation. Any lawsuit to which we are a party, with or without merit, may result in an unfavorable judgment. We also may decide to settle lawsuits on unfavorable terms. Any such negative outcome could result in payments of substantial damages or fines, damage to our reputation or adverse changes to our offerings or business practices.
79
We must maintain effective internal controls over financial reporting, and if we are unable to do so, the accuracy and timeliness of our financial reporting may be adversely affected, which could have a material adverse effect on our business and stock price.
We must maintain effective internal control over financial reporting in order to accurately and timely report our results of operations and financial condition. In addition, as a public company, the Sarbanes-Oxley Act requires, among other things, that we assess the effectiveness of our disclosure controls and procedures quarterly and the effectiveness of our internal control over financial reporting at the end of each fiscal year.
The rules governing the standards that must be met for our management to assess our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act are complex and require significant documentation, testing and possible remediation. These stringent standards require that our audit committee be advised and regularly updated on management’s review of internal control over financial reporting.
Our management may not be able to effectively and timely implement controls and procedures which respond to the increased regulatory compliance and reporting requirements that are applicable to us as a public company. If we fail to staff our accounting, finance and information technology functions adequately or maintain internal control over financial reporting adequate to meet the demands that are placed upon us as a public company, including the requirements of the Sarbanes-Oxley Act, or to otherwise prevent material weaknesses in internal control over financial reporting, or identify any additional material weaknesses, our business and reputation may be harmed and our stock price may decline. Furthermore, investor perceptions of us may be adversely affected, which could cause a decline in the market price of our common stock.
84
Ownership of our common stock is highly concentrated, and it may prevent other stockholders from influencing significant corporate decisions.
Entities affiliated with Deerfield Management Company beneficially own or control approximately 31.9%38.6% of our outstanding common stock (assuming full exercise of our outstanding pre-funded warrants and no exercise of outstanding options) as of December 31, 2021,2023, on a fully-diluted basis. Accordingly, such entities have substantial influence over the outcome of a corporate action by us requiring stockholder approval, including the election of directors, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transaction. These stockholders also may exert influence in delaying or preventing a change in control of the combined company, even if such change in control would benefit our other stockholders.
We are a smaller reporting company. We cannot be certain whether the reduced disclosure requirements applicable to smaller reporting companies will make our common shares less attractive to investors or otherwise limit our ability to raise additional funds.
We are currently a “smaller reporting company” as defined in the Securities Exchange Act of 1934 as amended, or the Exchange Act, and have elected to take advantage of certain of the scaled disclosures available to smaller reporting companies, including simplified executive compensation disclosures in our filings, exemption from the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that an independent registered accounting firm provide an attestation report on the effectiveness of internal control over financial reporting and certain other decreased disclosure obligations in our SEC filings, including, among other things, only being required to provide two years of audited financial statements in annual reports. Reduced disclosure in our SEC filings due to our status as a smaller reporting company may make it harder for investors to analyze our results of operations and financial prospects. We cannot predict whether investors will find our common stock less attractive because of our reliance on any of these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us more difficult and may prevent attempts by our stockholders to replace or remove our management.
Provisions in our certificate of incorporation and bylaws may delay or prevent an acquisition or a change in management. These provisions include a classified board of directors, a prohibition on actions by written consent of
80
our stockholders, and the ability of the board of directors to issue preferred stock without stockholder approval. In addition, because we are in Delaware, we are governed by the provisions of Section 203 of the Delaware General CorporationsCorporation Law ("DGCL") which prohibits stockholders owning in excess of 15% of our outstanding voting stock from merging or combining with us. Although we believe these provisions collectively will provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if the offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove then current management by making it more difficult for stockholders to replace members of the board of directors, which is responsible for appointing the members of management.
We do not anticipate that we will pay any cash dividends in the foreseeable future.
The current expectation is that we will retain our future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of our common stock will be stockholders’ sole source of gain, if any, for the foreseeable future.
General Risk Factors
Our failure to meet the continued listing requirements of The Nasdaq Stock Market LLC could result in a delisting of our Common Stock.
If we fail to satisfy the continued listing requirements of The Nasdaq Stock Market LLC ("Nasdaq") such as the corporate governance requirements or the minimum closing bid price requirement, Nasdaq may take steps to delist our common stock. Such a delisting would likely have a negative effect on the price of our common stock and would impair youra stockholders ability to sell or purchase shares of common stock when youthey wish to do so. In the event of a delisting, we can provide no assurance that any action taken by us to restore compliance with listing requirements would allow the common stock to become listed again, stabilize the market price or improve the liquidity of the common stock, prevent the common stock from dropping below the Nasdaq minimum bid price requirement or prevent future noncompliance with Nasdaq’s listing requirements.
General Risk Factors
85
Financial reporting obligations of being a public company in the United States are expensive and time-consuming, and our management will be required to devote substantial time to new compliance matters.
The obligations of being a public company in the United States requiresrequire significant expenditures and will place significant demands on our management and other personnel, including costs resulting from public company reporting obligations under the Securities Exchange Act of 1934, as amended, and the rules and regulations regarding corporate governance practices, including those under the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010 ("the Dodd-Frank(the "Dodd-Frank Act") and the listing requirements of Nasdaq on which our securities are listed. These rules require the establishment and maintenance of effective disclosure and financial controls and procedures, internal control over financial reporting and changes instrong corporate governance practices, among many other complex rules that are often difficult to implement, monitor and maintain compliance with. Moreover, despite recent reforms made possible by the Tax Act, the reporting requirements, rules, and regulations will make some activities more time-consuming and costly. As a result, ourOur management and other personnel will need to devote a substantial amount of time to remedy the identified material weaknesses and otherwise ensure that we comply with all of these requirements and to keep pace with new regulations, otherwise we may fall out of compliance and risk becoming subject to litigation or being delisted, among other potential problems.
Our employees may engage in misconduct or other improper activities, including violating applicable regulatory standards and requirements or engaging in insider trading, which could significantly impact our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional, reckless or negligent failures to comply with the regulations of the FDA and applicable non-U.S. regulators, provide accurate information to the FDA and applicable non-U.S. regulators, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. Employees may also unintentionally or willfully disclose our proprietary
81
and/or confidential information to competitors. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of, including trading on, information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may be ineffective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. In addition, we are subject to the risk that a person or government could allege fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Unfavorable global economic conditions could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy, geopolitical events and general conditions in the global financial markets. A severe or prolonged economic downturn due to geopolitical unresttension resulting from ongoing conflict between Russia and Ukraine and the Russian invasion of Ukraine, the possibility that thecurrent conflict could expand beyond eastern Europe as well as the impact ofin Israel and Gaza (including any potential new outbreaks related to the COVID-19 pandemicescalation or expansion) or other health crises, couldfactors could result in a variety of risks to our business, including, weakened demand for our product candidates and our ability to raise additional capital when needed on acceptable terms, if at all. A weak or declining economy could also strain our suppliers, possibly resulting in supply disruption, or cause our customers to delay making payments for our services.products. Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current economic climate and financial market conditions could adversely impact our business.
82Adverse developments affecting the financial services industry, including events or concerns involving liquidity, defaults or non-performance by financial institutions or transactional counterparties, could adversely affect our business, financial condition or results of operations.
Events involving limited liquidity, defaults, non-performance or other adverse developments that affect financial institutions, transactional counterparties or other companies in the financial services industry or the financial services industry generally, or concerns or rumors about any events of these kinds or other similar risks, have in the past and may in the future lead to market-wide liquidity problems. For example, in early 2023, several financial institutions closed and were taken into receivership by the Federal Deposit Insurance Corporation
86
(“FDIC”). Although we assess our banking and customer relationships as we believe necessary or appropriate, our access to funding sources and other credit arrangements in amounts adequate to finance or capitalize our current and projected future business operations could be significantly impaired by factors that affect us, the financial services industry or economy in general. These factors could include, among others, events such as liquidity constraints or failures, the ability to perform obligations under various types of financial, credit or liquidity agreements or arrangements, disruptions or instability in the financial services industry or financial markets, or concerns or negative expectations about the prospects for companies in the financial services industry.
In addition, investor concerns regarding the U.S. or international financial systems could result in less favorable commercial financing terms, including higher interest rates or costs and tighter financial and operating covenants, or systemic limitations on access to credit and liquidity sources, thereby making it more difficult for us to acquire financing on acceptable terms or at all. Any decline in available funding or access to our cash and liquidity resources could, among other risks, adversely impact our ability to meet our operating expenses, financial obligations or fulfill our other obligations, result in breaches of our contractual obligations or result in violations of federal or state wage and hour laws. Any of these impacts, or any other impacts resulting from the factors described above or other related or similar factors not described above, could have material adverse impacts on our liquidity and our business, financial condition or results of operations.
87
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 1C. CYBERSECURITY
Cyber Risk Management and Strategy
We have implemented and maintain various information security processes designed to manage cybersecurity risks relating to our critical computer networks, third party hosted services, communications systems, hardware and software, and our critical data, including intellectual property, confidential information that is proprietary, strategic or competitive in nature, and data related to our clinical trials and products (Information Systems and Data).
Our Director of Information Technology, assisted by our managed information technology service provider and other third party service providers, lead the Company’s cybersecurity risk management processes. This group works to identify and assess risks from cybersecurity threats by monitoring and evaluating our threat environment and the Company’s risk profile using various methods in certain contexts, including, manual tools, subscribing to reports and services that identify cybersecurity threats, analyzing threat intelligence reports and feeds, conducting scans of certain environments, and other tests.
We implement and maintain various technical, physical, and organizational measures, processes, standards and policies designed to manage risks from cybersecurity threats to our Information Systems and Data, as applicable depending on the environment, including incident detection and response, disaster recovery/business continuity policies, network security controls and data segmentation, access controls, physical security, asset management and tracking, systems monitoring, and employee training.
Our assessment and management of risks from cybersecurity threats are part of the Company’s overall risk management processes. Our Director of Information Technology works with management to prioritize our risk management processes and mitigate cybersecurity threats that are more likely to lead to a material impact to our business.
We use third-party service providers to assist us to identify, assess, and manage risks from cybersecurity threats. These third-party service providers may include threat intelligence service providers, cybersecurity consultants, cybersecurity software providers, managed cybersecurity service providers, and dark web monitoring services.
We also have established a process to conduct diligence on certain third parties before engaging with those third parties, such as those that support our GxP processes, which may include a security assessment.
We have not identified any cybersecurity incidents or threats that have materially affected us or are reasonably likely to materially affect us, including our business strategy, results of operations or financial condition; however, like other companies in our industry, we and our third-party vendors may, from time to time, experience threats and security incidents relating to our and our third-party vendors’ information systems and infrastructure. For more information, please see our risk factors under Part 1 Item 1A. Risk Factors.
Governance
Our board of directors holds oversight responsibility over the Company’s cybersecurity risk management as part of its general oversight function. The audit committee of the board of directors is responsible for overseeing the Company’s cybersecurity risk management processes, including oversight of the Company’s processes for monitoring and controlling cybersecurity risks.
Our cybersecurity risk assessment and management processes are implemented and maintained by certain Company management, including our Director of Information Technology, who has over 10 years of information technology management experience.
The Director of Information Technology, along with management, is responsible for helping to integrate cybersecurity risk considerations into the Company’s overall risk management strategy and communicating key priorities to relevant personnel. Management is responsible for approving budgets, helping prepare for cybersecurity incidents, approving cybersecurity processes, and reviewing security-related reports.
We have processes designed to escalate certain cybersecurity incidents to members of management depending on the circumstances, including our Vice President Legal and Compliance, Vice President Finance and
88
Administration, Chief Financial Officer and Director of Information Technology. In addition, the Company’s incident response policy includes a process for reporting to the audit committee of the board of directors for certain cybersecurity incidents.
The audit committee receives reports from members of the Company’s management concerning the Company’s significant cybersecurity threats and risk and the processes the Company has implemented to address them, as applicable.
ITEM 2. PROPERTIES
We currently lease office and laboratory space, which consists of approximately 5,000 square feet and 1,750 square feet locatedof laboratory space in Bala Cynwyd, PA and Philadelphia, PA, respectively. Our officePA. We are currently on a month-to-month lease expires in August 2023 with an option to extendon this Philadelphia lab space. We, as well as our landlord, can terminate the lease with four months notice.
We have entered into a lease for an additional three years, and our3,927 square feet of new laboratory space in King of Prussia, PA to replace the Philadelphia lab space, but this lease expireshas not yet commenced with expected occupancy in December 2022.the second quarter of 2024.
We are party to an operating lease for approximately 17,705 square feet of office space in Boston, Massachusetts, which we refer to as the Boston Lease. The Boston Lease expires in October 2029. On October 27, 2020, we entered into a sublease agreement whereby we subleased all 17,705 square feet of office space leased under the Boston Lease until October 2029.
We believe that we will need to increase both our office and laboratory facilities lease spacein the near and intermediate term. We believe that both appropriate office and laboratory space will be readily available on commercially reasonable terms.
ITEM 3. LEGAL PROCEEDINGS
We may be subject to other legal proceedings and claims in the ordinary course of business. We cannot predict the results of any such disputes, and despite the potential outcomes, the existence thereof may have an adverse material impact on us due to diversion of management time and attention as well as the financial costs related to resolving such disputes.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
8389
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Market Information
Our common stock is publicly traded on the Nasdaq Global Market under the symbol “LRMR.”
Holders
As of March 23, 2022,14, 2024, we had approximately 17,710,45062 record holders of itsour common stock.
Dividends
We have not declared or paid any dividends since our inception nor doesdo we expect to pay dividends in the foreseeable future.
Recent Sales of Unregistered Securities
There have been no sales of unregistered securities other than as previously disclosed by us in our Current Reports on Form 8-K as filed with the SEC.
Issuer Purchases of Equity Securities
None.
ITEM 6. [Reserved.]
8490
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with the audited consolidated financial statements and the related notes included elsewhere in this Annual Report on Form 10-K. In addition to historical financial information, the following discussion contains forward-looking statements based upon our current plans, expectations and beliefs that involve risks, uncertainties and assumptions. Our actual results may differ materially from those described in or implied by these forward-looking statements as a result of many factors, including those set forth under the section titled “Risk Factors” and in other parts of this Annual Report on Form 10-K.
Overview
We are a clinical-stage biotechnology company focused on developing treatments for patients suffering from complex rare diseases using our novel cell penetrating peptide ("CPP") technology platform. Our lead product candidate, CTI-1601,nomlabofusp (nomlabofusp is the International Nonproprietary Name ( "INN") and the United States Adopted Name ("USAN") for CTI-1601), is a subcutaneously administered, recombinant fusion protein intended to deliver humantissue frataxin ("FXN"), an essential protein, to the mitochondria of patients with Friedreich’sFriedreich's ataxia ("FA"(“FA”). FA is a rare, progressive, and fatal disease in which patients are unable to produce sufficient FXN due to a genetic abnormality. There is currentlyCurrently, there are no effectivetreatment options that address the core deficit of FA, low levels of FXN. Nomlabofusp represents the first potential therapy for FA.
We have completed two Phase 1 clinical trialsdesigned to increase FXN levels in patients with FA. We have received orphan drug status, fast track designation and rare pediatric disease designation, from the U.S. Food and Drug Administration ("the FDA") for CTI‑1601. In addition, we received orphan drug and a Priority Medicines ("PRIME") designation for CTI-1601 from the European Commission and a Priority Medicines ("PRIME") designation from the European Medicines Agency ("EMA"). The receipt of such designations or positive opinions may not result in a faster development process, review or approval compared to products considered for approval under conventional FDA or EMA procedures and does not assure ultimate approval by the FDA or EMA.
We believe that our cell penetrating peptide ("CPP") technologyCPP platform, which enables a therapeutic molecule to cross a cell membrane in order to reach intracellular targets, has the potential to enable the treatment of other rare and orphan diseases. We intend to use our proprietary platform to target additional orphan indications characterized by deficiencies in or alterations of intracellular content or activity.
Since our inception, we have devoted substantially all of our resources to developing CTI-1601,nomlabofusp, building our intellectual property portfolio, developing third-party manufacturing capabilities, business planning, raising capital, and providing general and administrative support for such operations.
CTI-1601Nomlabofusp Program Update
On May 20, 2021 we announced that we had received an EMA PRIME designation for CTI-1601 in FA. Through PRIME, the EMA offers early and proactive support to medicine developers to optimize the generation of robust data on a medicine’s benefits and risks and enable accelerated assessment of medicines applications so that these medicines can reach patients earlier. The PRIME designation was based on both non-clinical data as well as tolerability data from the CTI-1601Clinical Trials
We have completed two Phase 1 programclinical trials and a Phase 2 dose exploration trial in patients with FA.
In May 2021, we reported positive toplinetop-line data from our Phase 1 FA program after completing dosing of the single ascending dose ("SAD") trial in December 2020 and of the multiple ascending dose ("MAD") trial in March 2021. Data from these trials demonstratedemonstrated proof-of-concept by showing that daily subcutaneous injections of CTI-1601nomlabofusp for up to 13 days resulted in dose-dependent increases in FXN levels from baseline compared to placebo in all evaluated tissues (buccal cells, skin, and platelets). FXN levels achieved in peripheral tissues (buccal cells) following daily 50 mg and 100 mg subcutaneous injections of CTI-1601 were at or in excess of FXN levels that would be expected in
85
phenotypically normal heterozygous carriers. There were no serious adverse events ("SAEs") associated with either the MAD or SAD trials.
To support extended dosing of patients with CTI-1601, we conducted a 26-week NHP toxicology study in 2021. In May 20212023, we notified the FDA of certain mortalities which occurred at the highest dose levels in the then-ongoing study. On May 25, 2021 the FDA placed a clinical hold on the CTI-1601 clinical program. In the clinical hold letter, the FDA stated that it needed to review a full study reportreported preliminary unblinded top-line data from the then-ongoing NHP study and that we may not initiate additional interventional clinical trials until we have submitted such report and received notification25 mg cohort of our Phase 2 four-week, placebo-controlled, dose exploration trial of nomlabofusp in FA patients. Data from the FDA that additional clinical trials may commence. At the time of the FDA clinical hold, we had no interventional clinical trials with patients enrolled or enrollingcohort indicated nomlabofusp was generally well tolerated and as of the date of this report, do not have interventional clinical trials with patients enrolled or enrolling.showed increases in FXN levels from baseline compared to placebo in all evaluated tissues (skin and buccal cells) at day 14.
We have completed two Phase 1 clinical trials in patientsIn June 2023, we met with FA but the clinical development of CTI-1601 is currently subject to a clinical hold. WeFDA. Following that meeting, we submitted a complete response to the FDA’s partial clinical hold that included unblinded safety, PK and frataxin data from the Phase 2 trial’s completed 25 mg cohort.
In July 2023, following the FDA’s review of our complete response to the partial clinical hold, the FDA cleared initiation of a second cohort (50 mg) of our four-week, placebo-controlled, Phase 2 dose exploration trial and to initiate our OLE trial with daily dosing of 25 mg.
In January 2024, we initiated the OLE trial evaluating daily subcutaneous injections of 25 mg of nomlabofusp self-administered or administered by a caregiver, with the first patient dosed in January 2022. March 2024. Participants who complete treatment in the Phase 2 dose exploration trial, or who previously completed a prior clinical trial of nomlabofusp are potentially eligible to screen for the OLE. The OLE will evaluate the safety and tolerability and PK and measures of frataxin levels in peripheral tissues as well as other exploratory PD (lipid profiles and gene expression data) following long-term subcutaneous administration of nomlabofusp. Clinical measures collected during the trial will be compared to data from a synthetic control arm derived from participants in the Friedreich’s Ataxia Clinical Outcome Measures Study ("FACOMS") database. Dose escalation in the OLE trial will be
91
considered based on safety, tolerability, PK, and tissue FXN levels from the Phase 2 trial’s 50 mg cohort as well as available data from the 25 mg dose of nomlabofusp in the OLE trial and is contingent on FDA review as part of the partial clinical hold. Initial data from the OLE trial is expected in the fourth quarter of 2024.
In February 2022,2024, we received feedbackreported positive top-line data and successful completion of the two cohorts (25 mg and 50 mg groups) of our four-week, placebo-controlled Phase 2 dose exploration study of nomlabofusp in participants with FA. Nomlabofusp was generally well tolerated and demonstrated dose dependent increases in FXN levels in all evaluated tissues (skin and buccal cells) after daily dosing of 14 days followed by every other day dosing until day 28 in the 25 mg and 50 mg cohorts. Participants in the 25 mg (n=13) and 50 mg (n=15) cohorts were randomized 2:1 to receive subcutaneous injections of nomlabofusp or placebo.
Recently, we had discussions with the FDA regarding the use of tissue FXN levels as a novel surrogate endpoint. The FDA has acknowledged that frataxin deficiency appears to be critical to the pathogenic mechanism of FA, and that there continues to be an unmet need for treatments for FA patients that address the underlying disease pathophysiology. We intend to pursue an accelerated approval using FXN levels, supportive PD and clinical information, and safety data from the FDA followingOLE study, along with additional non-clinical pharmacology information needed to support the novel surrogate endpoint approach. We are beginning to plan for a confirmatory study and are targeting a BLA submission in the second half of 2025.
Nomlabofusp has been granted Orphan Drug (U.S. and Europe), Rare Pediatric Disease (U.S.), Fast Track (U.S.), and PRIME (Europe) designations for FA. We have also begun to engage with regulators and investigators outside the U.S. as we prepare to expand our submissionclinical program to additional geographies. With approximately 75% of individuals with FA living outside the U.S., establishing global clinical trial capabilities is important for addressing the pressing unmet needs of the complete response. The FDA stated that it was maintaining its clinical hold at this time and requested additional information. We are evaluating how best to respond to their request. We do not know when, or if, the clinical hold will be lifted.FA community.
Recent Financing Activities, Including Recent Material Financings
We have funded our operating and capital requirementsoperations to date primarily with proceeds from sales of common stock, proceeds from the sale of prefunded warrants for the purchase of common stock, the acquisition in 2020 of cash, cash equivalents, marketable securities and restricted cash upon the merger with Zafgen, Inc. ("Zafgen") and, prior to the 2020 merger with Zafgen, described below (see "Merger with Zafgen"), with capital contributions from Chondrial Holdings, LLC ("Holdings").
In June 2020, we completed the Merger, as defined below, and acquired $42.9 million of cash, cash equivalents, restricted cash and marketable debt securities that were held by Zafgen immediately prior to the Merger. We also raised $75.4 million, net of offering costs, through a private offering of common stock and prefunded warrants to purchase shares of our common stock in connection with, and immediately after, the closing of the Merger in June 2020.
In August 2020, we entered into an Equity Distribution Agreement ("the ATM Agreement") with an investment bank in connection with the establishment of an “at-the-market” offering program under which we could sell up to an aggregate of $50,000,000 of shares of our common stock from time to time through this investment bank as sales agent.LLC.
In July 2021,September 2022, we sold 2,342,72025,558,750 shares pursuant to the ATM Agreementof common stock in an underwritten offering for net proceeds of $19.9$75.2 million, after issuance costs. As of March 23, 2022, $29.2 million of additional
In February 2024, we sold 19,736,842 shares of common stock remained availablein an underwritten public offering, for sale by us under the ATM Agreement.net proceeds of approximately $161.6 million.
Merger with ZafgenCritical Accounting Policies and Significant Judgments and Estimates
On December 17, 2019, we entered into an Agreement and Plan of Merger ("the Merger Agreement") with Zordich Merger Sub, Inc. ("the Merger Sub") our wholly owned subsidiary, Chondrial Therapeutics Inc. ("Chondrial") and Chondrial Holding, LLC ("Holdings") pursuant to which the Merger Sub merged with and into Chondrial, with Chondrial surviving the merger as our wholly owned subsidiary ("the Merger"). The Merger was completed on May 28, 2020 pursuant to the terms of the Merger Agreement.
Pursuant to the terms of the Merger Agreement, upon closing of the Merger, all of Chondrial’s outstanding common stock was exchanged for our common stock and all outstanding options exercisable for units of Holdings were exchanged for options to purchase our common stock. In addition, immediately prior to the closing of the Merger, we effected a 1 for 12 reverse stock split and changed our name from Zafgen, Inc. to Larimar Therapeutics, Inc. Following the Merger, the business conducted by Chondrial became our primary business.
The business combination was accounted for as a reverse acquisitionOur consolidated financial statements are prepared in accordance with U.S. generally acceptedGAAP. The preparation of our consolidated financial statements and related disclosures requires us to make estimates and assumptions that affect the reported amount of assets, liabilities, costs and expenses, and related disclosures. We believe that the estimates and assumptions involved in the accounting principles ("GAAP"). Under this method of accounting, Chondrial was deemedpolicies described below may have the greatest potential impact on our consolidated financial statements and, therefore, consider these to be theour critical accounting acquirerpolicies. We evaluate these estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions and conditions.
Research and Development Expense
Costs for financial reporting purposes. This determination was primarilycertain research and development activities, such as manufacturing, non-clinical studies and clinical trials are generally recognized based on the facts that, immediately following the merger: (i) Chondrial's stockholders owned a substantial majorityevaluation of the voting rights inprogress of completion of specific tasks using information and data provided by our vendors and collaborators, and accordingly, are considered an area of significant judgment and management’s review of manufacturing, non-clinical and clinical expenses. This process involves reviewing open contracts and purchase orders, communicating with our personnel and outside vendors to identify services that have been performed on our behalf and estimating the combined
86
company, (ii)level of service performed and the majorityassociated costs incurred for the services when we have not yet been invoiced or otherwise notified of the boardactual costs. We work with vendors and suppliers to ensure that our estimates of directors of the combined company was composed of directors designated by Chondrial under the terms of the Merger Agreementour research and (iii) existing members of Chondrial management became the management of the combined company. Accordingly, for accounting purposes, the business combination was treated as the equivalent of Chondrial issuing stockdevelopment expenses are reasonable. We expect to acquire Zafgen’s net assets.increase our investment in research and development in order to advance nomlabofusp through additional clinical trials. As a result, we expect that our research and development expenses will increase in the foreseeable future as we pursue clinical development of nomlabofusp and/or any other product candidates we develop.
92
Stock Compensation Expense
We measure all stock-based awards granted to employees and directors based on the closingfair value on the date of grant using the Merger, Zafgen’s net assets were recorded at their acquisition-date fair values,Black-Scholes option-pricing model. The Black-Scholes option-pricing model requires the use of highly subjective assumptions which were then adjusted for the difference between the purchase price anddetermine the fair value of stock-based awards. The assumptions used in our option-pricing model represent management’s best estimates. These estimates are complex, involve a number of variables, uncertainties and assumptions and the assets acquired,application of management’s judgment, and thus are inherently subjective. If factors change and different assumptions are used, our stock-based compensation expense could be materially different in the financial statementsfuture.
Prior to May 28, 2020, we were a private company and lacked company-specific historical and implied volatility information for our common stock. Prior to January 1, 2023, the Company estimated its expected common stock price volatility solely based on the historical volatility of Chondrialpublicly traded peer companies with comparable characteristics including enterprise value, risk profiles and the reported operating results prior to the business combination are those of Chondrial.As the Merger has been accounted for as an asset acquisition, goodwill has not been recordedposition within the consolidated balance sheet.
COVID-19 Update
In Marchindustry. Beginning on January 1, 2023, the Company began blending its historical data starting in June 2020 the World Health Organization ("WHO") declared the outbreak of COVID-19, a novel strain of Coronavirus, a global pandemic. The pandemic resulted(following its merger with Zafgen in 2020) with its historical peer group. We regularly evaluate our peer group to assess changes in circumstances where identified companies may no longer be similar to us, in which case, more suitable companies whose share prices are publicly available would be utilized in the temporary stoppagecalculation. We expect to continue to do so until we have full historical data regarding the volatility of our CTI-1601 Phase 1 clinical trials in patients with Friedreich’s ataxia in March 2020. In July 2020, we resumed these clinical trials, and have since completed both the SAD and MAD clinical trials. Since being discovered, new variants of COVID-19 have emerged, including the Omicron variant, the Alpha variant, the Beta variant, the Gamma variant, and the Delta variant.
Vaccines manufactured by Moderna, Pfizer and Johnson & Johnson were introduced late in the fourth quarter of 2020 and became widely available by the end of the first quarter of 2021. While the vaccines have proven effective in reducing the severity and mortality of COVID-19, including the variants that have evolved to date, the overall vaccination rate in the United States has not reached the level required for herd immunity in some states, particularly in some regions of the country. Further, additional doses of vaccines may be required, and individuals may refuse or fail to receive one or more of such doses. The incidence of variants of COVID-19 has been increasing, particularly among unvaccinated individuals, and the Omicron variant has proven to be more easily spread than earlier variants.own traded stock price.
The low vaccinationexpected term of our stock options has been determined utilizing the “simplified” method for awards that qualify as “plain-vanilla” options. The risk-free interest rate is determined by reference to the spreadU.S. Treasury yield curve in effect at the time of grant of the variantsaward for time periods approximately equal to the expected term of the award. The expected dividend yield considers the fact that we have never paid cash dividends on common stock and do not expect to pay any cash dividends in the evolutionforeseeable future.
Compensation expense of additional mutations againstthose awards is recognized over the requisite service period, which is generally the vesting period of the respective award. Typically, we issue awards with only service-based vesting conditions and record the expense for these awards using the straight-line method. We account for forfeitures as they occur.
We classify stock-based compensation expense in our consolidated statements of operations and comprehensive loss in the same manner in which the current vaccines may prove ineffective could again resultaward recipient’s payroll costs are classified or in major disruptions to businesses and markets worldwide. Furthermore,which the incidence of "breakthrough" infections even among vaccinated individuals have been increasing, resulting in the need for booster doses which could make COVID-19 a long-term infectious disease concern. Our business, results of operations, financial condition and cash flows could be materially and adversely affected. Specifically, we could experience additional delays in future clinical trial timelines as a result of additional travel and hospital restrictions related to the COVID-19 pandemic and the efforts to mitigate it which may be imposed or could experience supply shortages or manufacturer shutdowns impacting the manufacture of drug substance or drug product, which could also impact clinical trial timelines. The financial statements do not reflect any adjustments as a result of the pandemic.award recipient’s service payments are classified.
Financial Operations Overview
Revenue
To date, we have not generated any revenue from product sales, and do not expect to generate any revenue from the sale of products in the foreseeable future. If our development efforts result in clinical success and regulatory approval or collaboration agreements with third parties for our product candidates, we may generate revenue from those product candidates or collaborations.
Operating Expenses
The majority of our operating expenses since inception have consisted primarily of research and development activities, and general and administrative costs.
87
Research and Development Expenses
Research and development expenses, which consist primarily of costs associated with our product research and development efforts, are expensed as incurred. Research and development expenses consist primarily of:
93
Costs for certain activities, such as manufacturing, non-clinical studies and clinical trials are generally recognized based on the evaluation of the progress of completion of specific tasks using information and data provided by our vendors and collaborators. Research and development activities are central to our business. We expect to increase our investment in research and development in order to advance CTI-1601 through additional clinical trials. As a result, we expect that our research and development expenses will increase in the foreseeable future as we pursue clinical development of CTI-1601 and/or any other product candidates we develop.
At this time, we cannot reasonably estimate or know the nature, timing and estimated costs of the efforts that will be necessary to complete the clinical and commercial development of CTI-1601, if clinical development is allowed to continue,nomlabofusp, or any other product candidates we develop. We are also unable to predict when, if ever, material net cash inflows will commence from sales of our product candidates. The duration, costs, and timing of clinical trials and development of CTI-1601nomlabofusp or any other product candidates we develop will depend on a variety of factors, including:
88
A change in the outcome of anyone or more of these variables with respect to the development of a product candidate could significantly change the costs, timing and viability associated with the development of that product candidate. For example, if the FDA or another regulatory authority were to require us to conduct additional non-clinical or clinical trials beyond those that we currently anticipate will be required for the completion of clinical development of a product candidate, or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development.
General and Administrative Expenses
General and administrative expenses consist primarily of personnel costs, consisting of salaries, related benefits and stock-based compensation, costs related to our executive, finance, information technology, and costs related to other administrative functions. General and administrative expenses also include insurance expenses and professional fees for auditing, tax, and legal services, including legal expenses to pursue patent protection for our intellectual property. We expect that our general and administrative expenses will increase in the foreseeable future as we hire additional employees to implement, improve and scale our operational, financial, commercial and management systems.
Critical Accounting Policies and Significant Judgments and EstimatesResults of Operations
Our audited consolidated financial statements are prepared in accordance with GAAP. The preparationfollowing commentary is a discussion and analysis of our auditedfinancial condition as of December 31, 2023 and results of operations and cash flows for the year ended December 31, 2023 compared to the year ended December 31, 2022 and should be read in conjunction with the consolidated financial statements and related disclosures requires us to make estimates and assumptions that affect the reported amount of assets, liabilities, costs and expenses, and related disclosures. We believe that the estimates and assumptions involved in the accounting policies described below may have the greatest potential impact on our audited consolidated financial statements and, therefore, consider these to be our critical accounting policies. We evaluate these estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions and conditions.accompanying notes.
ResearchThe discussion and Development Expenses
As part of the process of preparing our consolidated financial statements, we are required to estimate our accrued research and development expenses and evaluate payments made to vendors in advance of actual work activities being performed. This process involves reviewing open contracts and purchase orders, communicating with our personnel and outside vendors to identify services that have been performed on our behalf and estimating the level of service performed and the associated costs incurred for the services when we have not yet been invoiced or otherwise notified of the actual costs. The majorityanalysis of our service providers invoice us in arrears for services performed, on a pre-determined schedule or when contractual milestones are met. We make estimates of our accrued expensesfinancial condition as of each balance sheet date in our consolidated financial statements based on factsDecember 31, 2022, and circumstances known to us at that time. Examples of estimated accrued research and development expenses include fees paid to:
We base our expenses related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple CROs or CMOs that conduct and manage clinical trials or manufacture clinical trial material on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. There may be instances in which payments made to our vendors
89
will exceed the level of services provided and result in a prepayment of the clinical expense, non-clinical expense or manufacturing activities. Payments under some of these contracts depend on factors such as the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed, enrollment of patients, number of sites activated and the level of effort to be expended in each period. In accruing CMO costs, we estimate the time period that manufacturing will be completed, the achievement of milestones and the percentage completion of each specific CMO agreement. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the accrual or prepaid accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in us recognizing adjustments in future periods as additional information becomes available.
Stock-Based Compensation
We measure all stock-based awards granted to employees, non-employee consultants and directors based on the fair value on the date of grant using the Black-Scholes option-pricing model. Compensation expense of those awards is recognized over the requisite service period, which is generally the vesting period of the respective award. Typically, we issue awards with only service-based vesting conditions and record the expense for these awards using the straight-line method. We account for forfeitures as they occur.
We classify stock-based compensation expense in our consolidated statementsresults of operations and comprehensive losscash flows for the year ended December 31, 2022, compared to the year ended December 31, 2021, is included in the same manner in which the award recipient’s payroll costs are classified or in which the award recipient’s service payments are classified.
Prior to May 28, 2020, we were a private companyitem 7, Management’s Discussion and lacked company-specific historicalAnalysis of Financial and implied volatility information for our common stock. Therefore, we estimate our expected common stock price volatility based on the historical volatilityResults of publicly traded peer companies and expect to continue to do so until we have adequate historical data regarding the volatilityOperations of our own traded stock price. The expected term of our stock options has been determined utilizingAnnual Report on Form 10-K for the “simplified” method for awards that qualify as “plain-vanilla” options. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield considers the fact that we have never paid cash dividends on common stock and do not expect to pay any cash dividends in the foreseeable future.year ended December 31, 2022.
Results of Operations94
Comparison of the years ended December 31, 20212023 and 20202022
The following table summarizes our results of operations for the years ended December 31, 20212023 and 2020:2022:
|
| Year Ended December 31, |
|
| Year Ended December 31, |
| ||||||||||||||||||
|
|
|
|
|
|
|
| Increase |
|
|
|
|
|
|
|
| Increase |
| ||||||
|
| 2021 |
|
| 2020 |
|
| (Decrease) |
|
| 2023 |
|
| 2022 |
|
| (Decrease) |
| ||||||
|
| (in thousands) |
|
| (in thousands) |
| ||||||||||||||||||
Statement of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Research and development |
| $ | 38,396 |
| $ | 31,407 |
|
| $ | 6,989 |
|
| $ | 27,670 |
|
| $ | 24,250 |
|
| $ | 3,420 |
| |
General and administrative |
|
| 12,069 |
|
|
| 11,397 |
|
|
| 672 |
|
|
| 14,088 |
|
|
| 12,276 |
|
|
| 1,812 |
|
Total operating expenses |
|
| 50,465 |
|
|
| 42,804 |
|
|
| 7,661 |
|
|
| 41,758 |
|
|
| 36,526 |
|
|
| 5,232 |
|
Loss from operations |
| (50,465 | ) |
| (42,804 | ) |
| (7,661 | ) |
|
| (41,758 | ) |
|
| (36,526 | ) |
|
| (5,232 | ) | |||
Other income (expense), net |
|
| (171 | ) |
|
| 322 |
|
|
| (493 | ) |
|
| 4,809 |
|
|
| 1,171 |
|
|
| 3,638 |
|
Net loss |
| $ | (50,636 | ) |
| $ | (42,482 | ) |
| $ | (8,154 | ) |
| $ | (36,949 | ) |
| $ | (35,355 | ) |
| $ | (1,594 | ) |
Research and development expenses
Research and development expenses for the yeartwelve months ended December 31, 2021 were $38.42023 increased $3.4 million compared to $31.4 million for the same period in 2020.twelve months ended December 31, 2022 due to continued and increased clinical development of nomlabofusp and related regulatory and bioanalysis. The increase in research and development expenses compared to the prior year
90
period was primarily driven by higher non-clinical and internal laboratory costs of $3.2 million, an increase of $2.0$1.9 million in personnel related costs due to increased headcount in our research and development functions,expense, an increase of $1.3 million in clinical expense, an increase of $1.1 million in regulatory and statistical consulting expenditures, an increase of $0.5 million in assay development costs, and an increase of $0.3 million in stock-based compensation expense associated with stock option grants made in the second half of 20202022 and in 2021, higher clinical supply manufacturing costs of $0.4 million, and an increase of $0.4 million in professional fees primarily related to consulting services, partly2023, partially offset by a decrease of $0.6$2.0 million in clinical trialdrug manufacturing costs.
General and administrative expenses
General and administrative expenses for the yeartwelve months ended December 31, 2021 were $12.12023 increased $1.8 million compared to $11.4 million for the same period in 2020.twelve months ended December 31, 2022. The increase in general and administrative expenses as compared to the prior year periodexpense was primarily driven by an increase of $2.0$0.7 million in stock-based compensation expense associated with stock option grants made in the second half of 20202022 and thus far in 2021,2023, an increase of $1.1$0.5 million in other operating expenses including insurance, recruiting and informationpersonnel expense, an increase of $0.5 million in operational costs primarily related to technology costs required to function as a public company, and an increase of $0.3$0.4 million in personnelprofessional fees primarily related costs due to increased headcount,legal, accounting and consulting services, partially offset by a decrease of $2.1$0.3 million in professional fees primarily associated with accounting, legal, communication and consulting fees and a decreaseinsurance expense.
Other income (expense), net
Other income (expense), net was $4.8 million of $0.6income in the twelve months ended December 31, 2023 compared to $1.2 million in facility costs associated with the sublease agreement entered into in late 2020.twelve months ended December 31, 2022. The increase primarily relates to interest income on a higher investment base and higher investment yields on that base during the current period.
Cash FlowsLiquidity and Capital Resources
Since our inception, we have not generated any revenue from any sources, including from product sales, and have incurred significant operating losses and negative cash flows from our operations. We have devoted substantially all of our resources to developing CTI-1601,nomlabofusp, building our intellectual property portfolio, developing third-party manufacturing capabilities, business planning, capital raising, and providing general and administrative support for such operations.
The following table summarizes our sources and uses of cash for each of the periods presented below:
|
| Year Ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
|
| (in thousands) |
| |||||
Net cash used in operating activities |
| $ | (33,459 | ) |
| $ | (27,569 | ) |
Net cash provided by (used in) investing activities |
|
| 33,353 |
|
|
| (90,960 | ) |
Net cash provided by financing activities |
|
| 30 |
|
|
| 75,257 |
|
Net decrease in cash, cash equivalents and restricted cash |
| $ | (76 | ) |
| $ | (43,272 | ) |
|
| Year Ended December 31, |
| |||||
|
| 2021 |
|
| 2020 |
| ||
|
| (in thousands) |
| |||||
Net cash used in operating activities |
| $ | (42,105 | ) |
| $ | (42,199 | ) |
Net cash provided by investing activities |
|
| 24,169 |
|
|
| 17,090 |
|
Net cash provided by financing activities |
|
| 19,885 |
|
|
| 93,587 |
|
Net increase in cash, cash equivalents and restricted cash |
| $ | 1,949 |
|
| $ | 68,478 |
|
95
Net cash used in operating activities
During the year ended December 31, 2021,2023, net operating activities used $42.1$33.5 million of cash, resulting from our net loss of $50.6$36.9 million, adjusted for noncash expenses of $5.8$6.0 million, and changes in our operating assets and liabilities of resulting in a sourceuse of cash of $2.7$2.5 million. Our net loss was primarily attributed to operating expenses of $50.5$41.8 million offset by other income (net) of $4.8 million. The change in operating assets and liabilities was primarily due to a decrease in prepaidaccounts payable and accrued expenses due to the recognition of expense of amounts previously paid,and an increase in accrued expenses due to the growth in our operating activities, and partially offset by a decrease in accounts payable due to the timing of invoice processing.prepaid assets.
During the year ended December 31, 2020,2022, operating activities used $42.2$27.6 million of cash, resulting from our net loss of $42.5$35.4 million, adjusted for noncash expenses of $2.3$6.1 million, and changes in our operating assets and liabilities resulting in a use of cash of $2.0$1.7 million. Our net loss was primarily attributed to operating expenses of $42.8$36.5 million. The change in operating assets and liabilities was primarily due to an increase in accrued expense and prepaid expenses due to the growth in our operating activities and partially offset by a decreasefollowing the partial release of the clinical hold in accounts payable due to the timing of invoice processing.late 2022.
91
Net cash provided by (used in) investing activities
During the year ended December 31, 2021,2023, investing activities provided $24.2$33.4 million of net cash, resulting from $32.8$134.8 million representingof maturities and sales of marketable securities, which was offset by purchases of $8.2$101.2 million in marketable securities and $0.3 million used for the purchase of equipment.securities.
During the year ended December 31, 2020,2022, investing activities provided $17.1used $91.0 million of net cash, resulting from $41.9purchases of $133.6 million of cash acquired from our merger with Zafgen and $1.0 million representing maturities and sales ofin marketable securities, which was offset by transaction costs associated with the Merger$42.8 million of $1.3 million, the purchasematurities and sales of $24.5 million in marketable securities and $0.1 million used for the purchase of equipment.securities.
Net cash provided by financing activities
During the year ended December 31, 2021,2023, financing activities provided $19.9less than $0.1 million of cash from the saleexercise of common stock under the ATM Agreement (as defined below), after issuance costs.options and warrants.
During the year ended December 31, 2020,2022, financing activities provided $93.6$75.3 million of cash that was the result of proceeds from the sale of common stock in an underwritten public offering, net of $75.6 million and capital contributions from related parties prior to the merger with Zafgen of $18 million.issuance costs.
Operating Capital Requirements
We have not yet commercialized any products and do not expect to generate revenue from the commercial sale of any products for several years, if at all.
We have to date incurred net losses. We incurred net losses of approximately $51.2$36.9 million and $42.5$35.4 million for the yeartwelve months ended December 31, 20212023 and 2020,2022, respectively. As of December 31, 2021,2023, we had an accumulated deficit of $116.8$188.6 million and a cash, and cash equivalents and marketable securities balance of $70.1$86.8 million, excluding restricted cash of $1.3 million. These losses
Losses have resulted principally from costs incurred in connection with research and development activities, and general and administrative costs associated with the development of CTI-1601nomlabofusp and our operations. We expect to incur significant expenses and operating losses for the foreseeable future as we expect to continue to incur expenses in connection with our ongoing activities, if and as we:
In February 2024, we sold 19,736,842 shares of common stock in an underwritten public offering, net of underwriting discounts and commissions and offering costs of approximately $161.6 million. We believeanticipate that based on
96
these net proceeds, together with our current operating plan our cash, and cash equivalents and marketable securities will be able fund operating expenses and capital expenditure requirements for at least the next twelve months.operations into 2026. If the timing ofwe encounter unexpected delays in our clinical assumptions were delayedtrials or if there wereare other forecasted assumptionunanticipated changes to our operating plan from our current assumptions that negatively impact our operating plan, the Company couldoperations, we may reduce expenditures in order to further extend our existing cash resources.
Until we can generate substantial revenue, if ever, we expect to seek additional funding through a combination of public or private equity offerings, debtdebt/royalty financings, collaborations, strategic alliances and licensing arrangements or other sources. The incurrence of indebtedness would result in increased fixed payment obligations and we may be required to agree to
92
certain restrictive covenants, such as limitations on our ability to incur additional debt, minimum cash balances, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates.
There can be no assurance that we will be able to raise sufficient additional capital on acceptable terms, orif at all. If such additional financing is not available on satisfactory terms, or is not available in sufficient amounts, or we do not have sufficient authorized shares, we may be required to delay, limit, or eliminate the development of business opportunities and our ability to achieve our business objectives, our competitiveness, and our business, financial condition, and results of operations will be materially adversely affected. We could also be required to seek funds through arrangements with collaborative partners, strategic alliances or otherwise at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us, any of which may have a material adverse effect on our business, operating results and prospects. In addition, geopolitical unresttensions, volatility of capital markets, and other adverse macroeconomic events, including those due to inflationary pressures, rising interest rates, bank instability and the ability of the U.S. government to manage federal debt limits, as well as the potential impact of the Russian invasion of Ukraine and the possibility that the conflict could expand beyond eastern Europe, the impact of the COVID-19 pandemic and/or other health crises onon the global financial markets may reduce our ability to access capital, which could negatively affect our liquidity and ability to continue as a going concern.
If we are unable to obtain sufficient funding when needed and/or on acceptable terms, we may be required to significantly curtail, delay or discontinue one or more of itsour research and development programs, the manufacture of clinical and commercial supplies, product portfolio expansion and/or pre commercialization efforts, which could adversely affect our business prospects, or we may be unable to continue operations.
Off-Balance Sheet Arrangements
During the periods presented we did not have, and we currently do not have, any off-balance sheet arrangements, as defined under applicable SEC rules, Certain restrictive covenants, such as relationships with unconsolidated entities or financial partnerships, which are often referred to as structured finance or special purpose entities, established for the purpose of facilitating financing transactions that are not required to be reflectedlimitations on our balance sheets.
Contractual Obligations and Commitments
The following table summarizes our contractual obligations as of December 31, 2021 and the effects that such obligations are expectedability to haveincur additional debt, limitations on our liquidityability to acquire, sell or license intellectual property rights and cash flows in future periods:
|
| Payments due by Period |
| |||||||||||||||||
|
| Total |
|
| 2022 |
|
| 2023-2024 |
|
| 2025-2026 |
|
| Thereafter |
| |||||
|
| (in thousands) |
| |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Lease Obligations |
| $ | 8,930 |
|
| $ | 1,322 |
|
| $ | 2,211 |
|
| $ | 2,184 |
|
| $ | 3,213 |
|
Total |
| $ | 8,930 |
|
| $ | 1,322 |
|
| $ | 2,211 |
|
| $ | 2,184 |
|
| $ | 3,213 |
|
other operating restrictions that could adversely impact our ability to conduct our business. Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates.
Recently Issued Accounting Pronouncements
Please read Note 2 to our audited consolidated financial statements included in Part IV, Item 15, of this Annual Report for a description of recent accounting pronouncements applicable to our business.
Other Company Information
None.
9397
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not
required to provide the information required under this item.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our audited consolidated financial statements and the report of our independent registered public accounting firm are included in this Annual Report on Form 10-K on the pages indicated in Part IV, Item 16.15.
94
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, that are designed to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to our management, including our principal executive and our principal financial officers, as appropriate, to allow timely decisions regarding required disclosure.
With respect to the year ended December 31, 2021,2023, under the supervision and with the participation of our management, we conducted an evaluation of the effectiveness of the design and operations of our disclosure controls and procedures. Based upon this evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures were effective as of December 31, 20212023 to provide reasonable assurance that the information required to be disclosed by us in this Annual Report was (a) reported within the time periods specified by SEC rules and regulations and (b) communicated to our management, including our Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding any required disclosure.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under that framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2021.2023.
Changes in Internal Control Over Financial Reporting
There have beenwere no changes in our internal control over financial reporting identified in connection with the evaluation required by Rule 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the fiscal yearquarter ended December 31, 20212023, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
98
ITEM 9B. OTHER INFORMATION
(a) None.
(b) During the fourth quarter of the fiscal year ended December 31, 2023, no director or “officer” as defined in Rule 16a-1(f) under the Exchange Act adopted or terminated any Rule 10b5-1 trading plan or arrangements or any non-Rule 10b5-1 trading plan or arrangements, in both cases as defined in Item 408(a) of Regulation S-K.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable
9599
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by Item 10 of Form 10-K is incorporated by reference to the information contained in our definitive proxy statement for the 20222024 annual meeting of stockholders.
ITEM 11. EXECUTIVE COMPENSATION
The information required by Item 11 of Form 10-K is incorporated by reference to the information contained in our definitive proxy statement for the 20222024 annual meeting of stockholders.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by Item 12 of Form 10-K is incorporated by reference to the information contained in our definitive proxy statement for the 20222024 annual meeting of stockholders.
The information required by Item 13 of Form 10-K is incorporated by reference to the information contained in our definitive proxy statement for the 20222024 annual meeting of stockholders.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
Our independent accounting firm is PricewaterhouseCoopers LLP, Philadelphia, Pennsylvania, USA, PCAOB Auditor ID: 238.
The information required by Item 14 of Form 10-K is incorporated by reference to the information contained in our definitive proxy statement for the 20222024 annual meeting of stockholders.
96100
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
(a) 1. Financial Statements
See Index to the Consolidated Financial Statements on page F-1 of this Annual Report.
2. Financial Statement Schedules
None, as all information required in these schedules is included in the Notes to the Consolidated Financial Statements.
3. Exhibits
Reference is made to the Exhibit Index below for a list of exhibits required by Item 601 of Regulation S-K to be filed as part of this Annual Report.
The following exhibits are being filed herewith:
101
EXHIBIT INDEX
* Filed Herewith
**+ Certain confidential portions (indicated by brackets and asterisks) have been omitted from this exhibit
** Furnished herewith. This certification will not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or otherwise subject to the liability of that section. Such certification will not be deemed to be incorporated by reference into any filing under the Securities Act of 1933, as amended, except to the extent specifically incorporated by reference into such filing.
Exhibit No. |
| Exhibit |
|
|
|
2.1 |
| |
2.2 |
| |
3.1 |
| |
3.2 |
| |
3.3 |
| |
3.4 |
| |
4.1 |
| |
| Description of the Company’s Securities Registered Pursuant to Section 12 of the Securities Exchange Act of 1934 (incorporated by reference to Exhibit 4.2 of the Company’s Annual Report on Form 10-K filed March 4, 2021). | |
10.1 |
|
97
10.2 | ||
| ||
| ||
|
| |
| ||
10.4* | Non-Qualified Stock Option Grant Notice and Award Agreement. | |
10.5 |
| |
|
| |
|
| |
|
| |
|
| |
|
102
10.11 | ||
10.12** |
| |
|
| |
|
| |
|
| |
|
| |
| ||
10.18 | ||
10.19 |
| |
| ||
10.21* | ||
10.22+ |
| |
|
| |
|
| |
|
| |
10.26+ | ||
21.1* |
| |
23.1* |
| Consent of PricewaterhouseCoopers LLP, independent registered public accounting firm. |
98
31.1* |
| Rule 13a-14(a)/15d-14(a) certification of Principal Executive Officer. |
31.2* |
| Rule 13a-14(a)/15d-14(a) certification of Principal Financial Officer. |
32.1** |
| |
97.1* | ||
101.INS | Inline XBRL Instance Document | |
101.SCH | Inline XBRL Taxonomy Extension Schema Document | |
101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document | |
101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document | |
101.LAB | Inline XBRL Taxonomy Extension Label Linkbase Document | |
101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document | |
104 |
| Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) |
103
ITEM 16. Form 10-K Summary
None.
99104
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1933, as amended,1934, the registrant has duly caused this registration statementreport to be signed on its behalf by the undersigned, thereunto duly authorized in the authorized.Unincorporated Community of Bala Cynwyd, Commonwealth of Pennsylvania, on the 25th
day of March 2022.
|
|
|
| LARIMAR THERAPEUTICS, INC. | |
|
|
|
| By: | /s/ Carole S. Ben-Maimon, M.D. |
|
| Name: Carole S. Ben-Maimon, M.D. |
|
| Title: President and Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1933,1934, this registration statementreport has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature |
| Title |
| Date |
|
|
|
|
|
/s/ Carole S. Ben-Maimon, M.D. |
| President, Chief Executive Officer and Director (principal executive officer) |
| March |
Carole S. Ben-Maimon, M.D. |
|
| ||
|
|
|
|
|
/s/ Michael Celano |
| Chief Financial Officer |
| March |
Michael Celano |
|
| ||
|
|
|
|
|
/s/ Joseph Truitt |
| Chairman, Board of Directors |
| March |
Joseph Truitt |
|
| ||
|
|
|
|
|
/s/ |
| Director |
| March |
| ||||
/s/ Jonathan Leff | Director | March 14, 2024 | ||
Jonathan Leff | ||||
/s/ Jeffrey W. Sherman, M.D., FACP | Director | March 14, 2024 | ||
Jeffrey W. Sherman, M.D., FACP |
|
| ||
|
|
|
|
|
/s/ Frank E. Thomas |
| Director |
| March |
Frank E. Thomas |
|
| ||
|
|
|
|
|
|
|
| ||
| ||||
|
|
| ||
| ||||
|
|
| ||
| ||||
100105
INDEX
101106
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Larimar Therapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Larimar Therapeutics, Inc. and its subsidiariessubsidiary (the “Company”) as of December 31, 20212023 and 2020,2022, and the related consolidated statements of operations and comprehensive loss, of changes in stockholders’stockholders' equity (deficit) and of cash flows for the years then ended, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 20212023 and 2020,2022, and the results of its operations and its cash flows for the years then ended in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’sCompany's management. Our responsibility is to express an opinion on the Company’sCompany's consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditaudits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that (i) relates to accounts or disclosures that are material to theconsolidated financial statements and (ii) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidatedfinancial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Accrued Research and Development Expenses
As described in Notes 2 and 76 to the consolidated financial statements, the Company recognizes external research and development costs based on an evaluation of the progress to completion of specific tasks using information provided to the Company by its key service providers. Within accrued expenses, total accrued research and development expenses amounted to $5.0$4.6 million as of December 31, 2021. As disclosed, management’s2023. Management’s process involves reviewing open contracts and purchase orders, communicating with personnel and outside vendors to identify services that have been performed, and estimating the level of service performed and the associated costs incurred for the services when invoices or other notification of actual costs have not been received.
The principal considerations for our determination that performing procedures relating to accrued research and development expenses is a critical audit matter are (i) the significant judgment by management in developing the estimates of accrued research and development expenses which in turn led toand (ii) a high degree of auditor judgment,
F-1
subjectivity and effort in performing procedures and evaluating audit evidence for these accrued research and development expenses
F-1
related to management’s estimates of the level of service performed and the associated costs incurred for the services when invoices or other notification of actual costs have not yet been received.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures also included, among others,others; (i) testing management’s process for developing the estimates ofestimating accrued research and development expenses, which included (i)expenses; (ii) evaluating the reasonablenessappropriateness of management’sthe method used by management to develop the estimate; (iii) testing the completeness and accuracy of the data used by management to develop the estimates ofrelated to the level of service performed and the associated costs incurred for the services when invoices or other notification of actual costs have not yet been received; and (ii) testing the completeness and accuracy of data used by management in accrued research and development expenses calculations by comparing amounts, on a test basis, to contracts and invoices. Evaluating(iv) evaluating the reasonableness of management’s estimate of the level of service performed and the associatedestimated costs incurred involved assessing management’s abilityfor the services which have not been invoiced or other notification of actual costs have not yet been received, on a sample basis, by tracing to reasonably estimate the levelunderlying supporting documentation, such as underlying contracts, invoices and information received from certain third party service vendors, where applicable, and; (v) testing a selection of service and associated costs by comparing such estimates to invoices received provisions inby the contracts and other evidence including communications the company received from third-party vendors.Company subsequent to December 31, 2023.
/s/ PricewaterhouseCoopers LLP
Philadelphia, Pennsylvania
March 25, 202214, 2024
We have served as the Company'sCompany’s auditor since 2020.
F-2
PART I-FINANCIAL INFORMATION
Item 1. Financial Statements
LARIMAR THERAPEUTICS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share data)
|
| December 31, |
|
| December 31, |
|
| December 31, |
|
| December 31, |
| ||||
|
| 2021 |
|
| 2020 |
|
| 2023 |
|
| 2022 |
| ||||
Assets |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Current assets: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Cash and cash equivalents |
| $ | 70,097 |
| $ | 68,148 |
|
| $ | 26,749 |
|
| $ | 26,825 |
| |
Marketable debt securities |
| — |
| 24,490 |
| |||||||||||
Marketable securities |
|
| 60,041 |
|
|
| 91,603 |
| ||||||||
Prepaid expenses and other current assets |
|
| 2,107 |
|
|
| 5,314 |
|
|
| 3,385 |
|
|
| 2,311 |
|
Total current assets |
| 72,204 |
| 97,952 |
|
|
| 90,175 |
|
|
| 120,739 |
| |||
Property and equipment, net |
| 1,049 |
| 1,040 |
|
|
| 684 |
|
|
| 831 |
| |||
Operating lease right-of-use assets |
| 3,406 |
| 3,936 |
|
|
| 3,078 |
|
|
| 2,858 |
| |||
Restricted cash |
| 1,339 |
| 1,339 |
|
|
| 1,339 |
|
|
| 1,339 |
| |||
Other assets |
|
| 669 |
|
|
| 419 |
|
|
| 659 |
|
|
| 638 |
|
Total assets |
| $ | 78,667 |
|
| $ | 104,686 |
|
| $ | 95,935 |
|
| $ | 126,405 |
|
Liabilities and Stockholders’ Equity |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Current liabilities: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Accounts payable |
| $ | 1,660 |
| $ | 2,634 |
|
| $ | 1,283 |
|
| $ | 1,686 |
| |
Accrued expenses |
| 6,592 |
| 5,843 |
|
|
| 7,386 |
|
|
| 8,408 |
| |||
Operating lease liabilities, current |
|
| 594 |
|
|
| 515 |
|
|
| 837 |
|
|
| 611 |
|
Total current liabilities |
| 8,846 |
| 8,992 |
|
|
| 9,506 |
|
|
| 10,705 |
| |||
Operating lease liabilities |
|
| 5,408 |
|
|
| 6,002 |
|
|
| 4,709 |
|
|
| 4,797 |
|
Total liabilities |
|
| 14,254 |
|
|
| 14,994 |
|
|
| 14,215 |
|
|
| 15,502 |
|
Commitments and contingencies (See Note 9) |
|
|
|
|
|
| ||||||||||
Commitments and contingencies (See Note 8) |
|
|
|
|
|
| ||||||||||
Stockholders’ equity: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Preferred stock; $0.001 par value per share; 5,000,000 shares authorized |
| — |
| — |
| |||||||||||
Common stock, $0.001 par value per share; 115,000,000 shares |
| 18 |
| 15 |
| |||||||||||
Preferred stock; $0.001 par value per share; 5,000,000 shares authorized |
|
| — |
|
|
| — |
| ||||||||
Common stock, $0.001 par value per share; 115,000,000 shares |
|
| 43 |
|
|
| 43 |
| ||||||||
Additional paid-in capital |
| 180,645 |
| 155,290 |
|
|
| 270,150 |
|
|
| 262,496 |
| |||
Accumulated deficit |
| (116,250 | ) |
| (65,614 | ) |
|
| (188,554 | ) |
|
| (151,605 | ) | ||
Accumulated other comprehensive gain |
|
| — |
|
|
| 1 |
| ||||||||
Accumulated other comprehensive gain (loss) |
|
| 81 |
|
|
| (31 | ) | ||||||||
Total stockholders’ equity |
|
| 64,413 |
|
|
| 89,692 |
|
|
| 81,720 |
|
|
| 110,903 |
|
Total liabilities and stockholders’ equity |
| $ | 78,667 |
|
| $ | 104,686 |
|
| $ | 95,935 |
|
| $ | 126,405 |
|
The accompanying notes are an integral part of these consolidated financial statements.
F-3
LARIMAR THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(In thousands, except share and per share data)
|
| Year Ended December 31, |
|
| Year Ended December 31, |
| ||||||||||
|
| 2021 |
|
| 2020 |
|
| 2023 |
|
| 2022 |
| ||||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Research and development |
| $ | 38,396 |
| $ | 31,407 |
|
| $ | 27,670 |
|
| $ | 24,250 |
| |
General and administrative |
|
| 12,069 |
|
|
| 11,397 |
|
|
| 14,088 |
|
|
| 12,276 |
|
Total operating expenses |
|
| 50,465 |
|
|
| 42,804 |
|
|
| 41,758 |
|
|
| 36,526 |
|
Loss from operations |
| (50,465 | ) |
| (42,804 | ) |
|
| (41,758 | ) |
|
| (36,526 | ) | ||
Other income (expense), net |
|
| (171 | ) |
|
| 322 |
|
|
| 4,809 |
|
|
| 1,171 |
|
Net loss |
| $ | (50,636 | ) |
| $ | (42,482 | ) |
| $ | (36,949 | ) |
| $ | (35,355 | ) |
Net loss per share, basic and diluted |
| $ | (2.95 | ) |
| $ | (3.57 | ) |
| $ | (0.84 | ) |
| $ | (1.37 | ) |
Weighted average common shares outstanding, basic and diluted |
|
| 17,164,284 |
|
|
| 11,883,155 |
|
|
| 43,901,241 |
|
|
| 25,761,394 |
|
Comprehensive income (loss): |
|
|
|
|
| |||||||||||
Comprehensive loss: |
|
|
|
|
| |||||||||||
Net loss |
| $ | (50,636 | ) |
| $ | (42,482 | ) |
| $ | (36,949 | ) |
| $ | (35,355 | ) |
Other comprehensive loss: |
|
|
|
|
|
|
|
|
|
| ||||||
Unrealized gain (loss) on marketable debt securities |
|
| (1 | ) |
|
| 1 |
| ||||||||
Total other comprehensive income (loss) |
|
| (1 | ) |
|
| 1 |
| ||||||||
Unrealized gain (loss) on marketable securities |
|
| 112 |
|
|
| (31 | ) | ||||||||
Total other comprehensive gain (loss) |
|
| 112 |
|
|
| (31 | ) | ||||||||
Total comprehensive loss |
| $ | (50,637 | ) |
| $ | (42,481 | ) |
| $ | (36,837 | ) |
| $ | (35,386 | ) |
The accompanying notes are an integral part of these consolidated financial statements.
F-4
LARIMAR THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN
STOCKHOLDERS’ EQUITY (DEFICIT)
(In thousands, except share data)
|
|
|
|
|
|
|
|
|
|
|
|
|
| Accumulated |
|
|
|
| ||||||
|
|
|
|
|
|
|
| Additional |
|
|
|
|
| Other |
|
| Total |
| ||||||
|
| Common Stock |
|
| Paid-in |
|
| Accumulated |
|
| Comprehensive |
|
| Stockholders’ |
| |||||||||
|
| Shares |
|
| Par Value |
|
| Capital |
|
| Deficit |
|
| Loss |
|
| Equity |
| ||||||
Balances as of December 31, 2020 |
|
| 15,367,730 |
|
| $ | 15 |
|
| $ | 155,290 |
|
| $ | (65,614 | ) |
| $ | 1 |
|
| $ | 89,692 |
|
Issuance of Common Stock. Net |
|
| 2,342,720 |
|
|
| 3 |
|
|
| 19,882 |
|
|
| — |
|
|
| — |
|
|
| 19,885 |
|
Stock-based compensation expense |
|
| — |
|
|
| — |
|
|
| 5,473 |
|
|
| — |
|
|
| — |
|
|
| 5,473 |
|
Unrealized loss on marketable debt securities |
|
| — |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
| (1 | ) |
|
| (1 | ) |
Net loss |
|
| — |
|
|
| — |
|
|
| — |
|
|
| (50,636 | ) |
|
| — |
|
|
| (50,636 | ) |
Balances as of December 31, 2021 |
|
| 17,710,450 |
|
| $ | 18 |
|
| $ | 180,645 |
|
| $ | (116,250 | ) |
| $ | — |
|
| $ | 64,413 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Accumulated |
|
|
|
| ||||||
|
|
|
|
|
|
|
| Additional |
|
|
|
|
| Other |
|
| Total |
| ||||||
|
| Common Stock |
|
| Paid-in |
|
| Accumulated |
|
| Comprehensive |
|
| Stockholders’ |
| |||||||||
|
| Shares |
|
| Par Value |
|
| Capital |
|
| Deficit |
|
| Gain (Loss) |
|
| Equity |
| ||||||
Balances as of December 31, 2022 |
|
| 43,269,200 |
|
| $ | 43 |
|
| $ | 262,496 |
|
| $ | (151,605 | ) |
| $ | (31 | ) |
| $ | 110,903 |
|
Stock-based compensation expense |
|
| — |
|
|
| — |
|
|
| 7,615 |
|
|
| — |
|
|
| — |
|
|
| 7,615 |
|
Exercise of stock options |
|
| 11,466 |
|
|
| — |
|
|
| 33 |
|
|
| — |
|
|
| — |
|
|
| 33 |
|
Exercise of warrants |
|
| 628,403 |
|
|
| — |
|
|
| 6 |
|
|
| — |
|
|
| — |
|
|
| 6 |
|
Unrealized gain on marketable securities |
|
| — |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
| 112 |
|
|
| 112 |
|
Net loss |
|
| — |
|
|
| — |
|
|
| — |
|
|
| (36,949 | ) |
|
| — |
|
|
| (36,949 | ) |
Balances as of December 31, 2023 |
|
| 43,909,069 |
|
| $ | 43 |
|
| $ | 270,150 |
|
| $ | (188,554 | ) |
| $ | 81 |
|
| $ | 81,720 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Accumulated |
|
|
|
| ||||||
|
|
|
|
|
|
|
| Additional |
|
|
|
|
| Other |
|
| Total |
| ||||||
|
| Common Stock |
|
| Paid-in |
|
| Accumulated |
|
| Comprehensive |
|
| Stockholders’ |
| |||||||||
|
| Shares |
|
| Par Value |
|
| Capital |
|
| Deficit |
|
| Loss |
|
| Equity (Deficit) |
| ||||||
Balances as of December 31, 2019 |
|
| 6,091,250 |
|
| $ | 6 |
|
| $ | 22,432 |
|
| $ | (23,132 | ) |
| $ | — |
|
| $ | (694 | ) |
Capital contributions from related party |
|
| — |
|
|
| — |
|
|
| 17,995 |
|
|
| — |
|
|
| — |
|
|
| 17,995 |
|
Merger with Zafgen Inc. |
|
| 3,124,337 |
|
|
| 3 |
|
|
| 37,116 |
|
|
| — |
|
|
| — |
|
|
| 37,119 |
|
Private Placement, net of transaction costs |
|
| 6,105,359 |
|
|
| 6 |
|
|
| 74,844 |
|
|
| — |
|
|
| — |
|
|
| 74,850 |
|
Issuance of Common Stock to placement agent for advisory fees in lieu of cash and other issuances of common stock |
|
| 46,784 |
|
|
| — |
|
|
| 742 |
|
|
| — |
|
|
| — |
|
|
| 742 |
|
Stock-based compensation expense |
|
| — |
|
|
| — |
|
|
| 2,161 |
|
|
| — |
|
|
| — |
|
|
| 2,161 |
|
Unrealized gain on marketable debt securities |
|
| — |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
| 1 |
|
|
| 1 |
|
Net loss |
|
| — |
|
|
| — |
|
|
| — |
|
|
| (42,482 | ) |
|
| — |
|
|
| (42,482 | ) |
Balances as of December 31, 2020 |
|
| 15,367,730 |
|
| $ | 15 |
|
| $ | 155,290 |
|
| $ | (65,614 | ) |
| $ | 1 |
|
| $ | 89,692 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| Accumulated |
|
|
|
| |||||||
|
|
|
|
|
|
| Additional |
|
|
|
|
| Other |
|
| Total |
| |||||||
| Common Stock |
|
| Paid-in |
|
| Accumulated |
|
| Comprehensive |
|
| Stockholders’ |
| ||||||||||
| Shares |
|
| Par Value |
|
| Capital |
|
| Deficit |
|
| Loss |
|
| Equity |
| |||||||
Balances as of December 31, 2021 |
|
| 17,710,450 |
|
| $ | 18 |
|
| $ | 180,645 |
|
| $ | (116,250 | ) |
| $ | — |
|
| $ | 64,413 |
|
Issuance of Common Stock, net |
|
| 25,558,750 |
|
|
| 25 |
|
|
| 75,232 |
|
|
| — |
|
|
| — |
|
|
| 75,257 |
|
Stock-based compensation expense |
| — |
|
|
| — |
|
|
| 6,619 |
|
|
| — |
|
|
| — |
|
|
| 6,619 |
| |
Unrealized loss on marketable securities |
| — |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
| (31 | ) |
|
| (31 | ) | |
Net loss |
|
| — |
|
|
| — |
|
|
| — |
|
|
| (35,355 | ) |
|
| — |
|
|
| (35,355 | ) |
Balances as of December 31, 2022 |
|
| 43,269,200 |
|
| $ | 43 |
|
| $ | 262,496 |
|
| $ | (151,605 | ) |
| $ | (31 | ) |
| $ | 110,903 |
|
The accompanying notes are an integral part of these consolidated financial statements.
F-5
LARIMAR THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
|
| Year Ended December 31, |
| Year Ended December 31, |
| |||||||||||
|
| 2021 |
|
| 2020 |
| 2023 |
|
| 2022 |
| |||||
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
| ||||||
Net loss |
| $ | (50,636 | ) |
| $ | (42,482 | ) |
| $ | (36,949 | ) |
| $ | (35,355 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
| |||||||
Stock-based compensation expense |
|
| 5,473 |
|
|
| 2,161 |
|
| 7,615 |
|
|
| 6,619 |
| |
Gain on disposal of fixed asset |
|
| (1 | ) |
|
| — |
| ||||||||
Lease expense |
|
| (82 | ) |
|
| (46 | ) | ||||||||
Depreciation expense |
|
| 326 |
|
|
| 155 |
|
| 311 |
|
|
| 318 |
| |
Amortization of premium (discount) on marketable securities |
|
| (14 | ) |
|
| 10 |
| ||||||||
Amortization of discount on marketable securities |
| (1,843 | ) |
|
| (774 | ) | |||||||||
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
| ||||||
Prepaid expenses and other current assets |
|
| 3,207 |
|
|
| (1,647 | ) |
| (1,065 | ) |
|
| (204 | ) | |
Accounts payable |
|
| (974 | ) |
|
| (3,288 | ) |
|
| (403 | ) |
|
| 26 |
|
Accrued expenses |
|
| 749 |
|
|
| 3,232 |
|
|
| (1,022 | ) |
|
| 1,816 |
|
Right-of-use assets |
|
| 530 |
|
|
| 405 |
| ||||||||
Operating lease liabilities |
|
| (515 | ) |
|
| (427 | ) | ||||||||
Other assets |
|
| (250 | ) |
|
| (318 | ) |
|
| (21 | ) |
|
| 31 |
|
Net cash used in operating activities: |
|
| (42,105 | ) |
|
| (42,199 | ) |
|
| (33,459 | ) |
|
| (27,569 | ) |
Cash flows from investing activities: |
|
|
|
|
|
|
|
| ||||||||
Purchase of property and equipment |
| (333 | ) |
| (62 | ) |
|
| (164 | ) |
|
| (100 | ) | ||
Purchase of marketable debt securities |
| (8,248 | ) |
| (24,486 | ) | ||||||||||
Maturities and sales of marketable debt securities |
| 32,750 |
| 1,000 |
| |||||||||||
Cash, cash equivalents, and restricted cash acquired in connection with the Merger |
| — |
| 41,934 |
| |||||||||||
Merger transaction costs |
|
| — |
|
|
| (1,296 | ) | ||||||||
Net cash provided by investing activities |
|
| 24,169 |
|
|
| 17,090 |
| ||||||||
Purchase of marketable securities |
|
| (101,233 | ) |
|
| (133,610 | ) | ||||||||
Maturities and sales of marketable securities |
|
| 134,750 |
|
|
| 42,750 |
| ||||||||
Net cash provided by (used in) investing activities |
|
| 33,353 |
|
|
| (90,960 | ) | ||||||||
Cash flows from financing activities: |
|
|
|
|
|
|
|
| ||||||||
Capital contribution from related party |
| — |
| 17,995 |
| |||||||||||
Proceeds from sale of common stock and prefunded warrants, net of issuance costs |
|
| 19,885 |
|
|
| 75,592 |
| ||||||||
Proceeds from sale of common stock, net of issuance costs |
|
| — |
|
|
| 75,257 |
| ||||||||
Proceeds from exercise of stock options and warrants |
|
| 30 |
|
|
| — |
| ||||||||
Net cash provided by financing activities |
|
| 19,885 |
|
|
| 93,587 |
|
|
| 30 |
|
|
| 75,257 |
|
Net increase in cash, cash equivalents and restricted cash |
| 1,949 |
| 68,478 |
| |||||||||||
Net decrease in cash, cash equivalents and restricted cash |
|
| (76 | ) |
|
| (43,272 | ) | ||||||||
Cash, cash equivalents and restricted cash at beginning of period |
|
| 69,487 |
|
|
| 1,009 |
|
|
| 28,164 |
|
|
| 71,436 |
|
Cash, cash equivalents and restricted cash at end of period |
| $ | 71,436 |
|
| $ | 69,487 |
|
| $ | 28,088 |
|
| $ | 28,164 |
|
Supplemental disclosure of non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
|
| ||||||
Fair value of net assets acquired in the Merger, including $1.0 million of marketable debt securities and excluding cash acquired |
| $ | — |
|
| $ | (4,815 | ) | ||||||||
Property and equipment included in accounts payable and accrued expenses |
| $ | — |
|
| $ | 460 |
| ||||||||
Leased assets obtained in exchange for new operating lease liabilities |
| $ | — |
|
| $ | 448 |
|
| $ | 790 |
|
| $ | — |
|
Proceeds from exercise of stock options included in other current assets |
|
| 9 |
|
|
| — |
| ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
LARIMAR THERAPEUTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Larimar Therapeutics, Inc., together with its subsidiariessubsidiary (the “Company” or “Larimar”), is a clinical-stage biotechnology company focused on developing treatments for patients suffering from complex rare diseases using its novel cell penetrating peptide technology platform. Larimar's lead product candidate, CTI-1601,nomlabofusp (nomlabofusp is the International Nonproprietary Name ("INN") and the United States Adopted Name ("USAN") for CTI-1601), is a subcutaneously administered, recombinant fusion protein intended to deliver human frataxin ("FXN"), an essential protein, to the mitochondria of patients with Friedreich’s ataxia. Friedreich’s ataxia ("FA"). FA is a rare, progressive and fatal disease in which patients are unable to produce sufficient FXN due to a genetic abnormality.
The Company has completed two phase 1 studies of nomlabofusp and a Phase 2 dose exploration study in patients with FA.
In May 2021, Larimar reportedafter reporting positive toplinetop-line data from the Company’s Phase 1 FA program, the U.S. Food and Drug Administration (“FDA”) placed a clinical hold on the Company’s nomlabofusp clinical program after the Company notified the FDA of mortalities at the highest dose levels of a 26-week non-human primate toxicology study that was designed to support extended dosing of patients with nomlabofusp. In August 2022, the Company submitted a complete response to the clinical hold following a Type C Meeting with the FDA, and proposed as nomlabofusp's next clinical trial a Phase 2, four-week, dose exploration study in FA patients starting at the lower dose levels tested in the Company’s Phase 1 multiple-ascending dose clinical trial. In September 2022, the FDA lifted its full clinical hold on the nomlabofusp program and imposed a partial clinical hold.
In May 2023, the Company announced top-line data from its completed 25 mg cohort of a Phase 1 Friedreich’s ataxia,2, four-week, dose exploration trial of nomlabofusp in patients with FA and provided a complete response to the FDA in June 2023, which included unblinded safety, pharmacokinetic ("FA") program after completing dosing of the single ascending dose, ("SAD"PK"), trial in December 2020 and ofpharmacodynamic ("PD") data from the multiple ascending dose, ("MAD"), trial in March 2021.Phase 2 trial’s completed 25 mg cohort. Data from these trials demonstrate proof-of-concept by showingthe completed 25 mg cohort (n = 13) indicated that daily subcutaneous injections of CTI-1601 for up to 13 days resulted in dose-dependentnomlabofusp was generally well tolerated and showed increases in FXN levels from baseline compared to placebo in all evaluated tissues (buccal cells, skin,(skin and platelets). FXN levels achievedbuccal cells) at day 14 (the final day of daily dosing in peripheral tissues (buccal cells) following daily 50 mg and 100 mg subcutaneous injections of CTI-1601 were at or in excess of FXN levels that would be expected in phenotypically normal heterozygous carriers. There were no serious adverse events, ("SAEs"), associated with either the MAD or SAD trials.trial).
To support extended dosing of patientsIn June 2023, the Company met with CTI-1601, we conducted a 26-week NHP toxicology study in 2021. In May 2021 we notified the FDA of certain mortalities which occurred atFDA. Following that meeting, the highest dose levels in the then-ongoing study. On May 25, 2021 the FDA placed a clinical hold on the CTI-1601 clinical program. In the clinical hold letter, the FDA stated that it needed to review a full study report from the then-ongoing NHP study and that we may not initiate additional interventional clinical trials until we have submitted such report and received notification from the FDA that additional clinical trials may commence. At the time of the FDA clinical hold, we had no interventional clinical trials with patients enrolled or enrolling and as of the date of this report, do not have interventional clinical trials with patients enrolled or enrolling.
The Company submitted a complete response to the FDA’s partial clinical hold in January 2022. that included unblinded safety, PK and frataxin data from the Phase 2 trial’s completed 25 mg cohort.
In July 2023, following the FDA’s review of the Company's complete response to the partial clinical hold, the FDA cleared initiation of a second cohort at 50 mg of our four-week, placebo-controlled, Phase 2 dose exploration trial and initiation of the OLE trial with daily dosing of 25 mg.
In February 2022,2024, the Company received feedbackreported positive top-line data and successful completion of their four-week, placebo-controlled Phase 2 dose exploration study of nomlabofusp in participants with FA. Nomlabofusp was generally well tolerated and demonstrated dose dependent increases in FXN levels in all evaluated tissues (skin and buccal cells) after daily dosing of 14 days followed by every other day dosing until day 28 in the 25 mg and 50 mg cohorts. Participants in the 25 mg (n=13) and 50 mg (n=15) cohorts were randomized 2:1 to receive subcutaneous injections of nomlabofusp or placebo. The initiation of additional U.S. clinical trials evaluating nomlabofusp are contingent on FDA review of data under the partial clinical hold.
In January 2024, the Company initiated an OLE trial evaluating daily subcutaneous injections of 25 mg of nomlabofusp self-administered or administered by a caregiver, with the first patient dosed in March 2024. Participants who complete treatment in the Phase 2 dose exploration trial, or who previously completed a prior clinical trial of nomlabofusp are potentially eligible to screen for the OLE. The OLE will evaluate the safety, tolerability and PK and measures of frataxin levels in peripheral tissues as well as other exploratory PD (lipid profiles and gene expression data) following long-term subcutaneous administration of nomlabofusp. Clinical measures collected during the trial will be compared to data from a synthetic control arm derived from participants in the Friedreich’s Ataxia Clinical Outcome Measures Study (FACOMS) database. Dose escalation in the OLE trial will be considered based on safety, tolerability, PK, and tissue FXN levels from the Phase 2 trial’s 50 mg cohort as well as available data from the 25 mg dose of nomlabofusp in the OLE trial and is contingent on FDA following its submissionreview as part of the complete response. The FDA stated that it was maintaining itspartial clinical hold at this time and requested additional information. The Companyhold. Initial data from the OLE trial is evaluating how best to respond to their request. The Company does not know when, or if,expected in the clinical hold will be lifted.fourth quarter of 2024.
The Company is subject to risks and uncertainties common to pre-commercial companies in the biotechnology industry, including, but not limited to, development and commercialization by competitors of new technological innovations, dependence on key personnel, protection of proprietary technology, compliance with governmental
F-7
regulations, failure to secure regulatory approval for its drug candidates or any other product candidates and the ability to secure additional capital to fund its operations. DrugProduct candidates currently under development will require extensive non-clinical and clinical testing and regulatory approval prior to commercialization. These efforts require significant amounts of additional capital, adequate personnel, and infrastructure and extensive compliance-reporting capabilities. Even if ourthe Company's drug development efforts are successful, it is uncertain when, if ever, weit will realize significant revenue from product sales.
In March 2020, the World Health Organization ("WHO"), declared the outbreak of COVID-19, a novel strain of Coronavirus, a global pandemic. The pandemic resulted in the temporary stoppage of the Company’s CTI-1601 Phase 1 clinical trials in patients with Friedreich’s ataxia in March 2020. In July 2020, the Company resumed these clinical trials, and has since completed both of the Company's SAD and MAD clinical trials. Since being discovered, new variants of COVID-19 have emerged. In November 2021, the Omicron variant was identified in South Africa, and has been deemed a variant of concern by the WHO. In addition to the Omicron variant, the WHO has deemed four other variants to be variants of concern: the Alpha variant, the Beta variant, the Gamma variant, and the Delta variant. All of the aforementioned variants have at least one of the following characteristics resulting in the WHO deeming them variants of concern: an increase in transmissibility or detrimental change in COVID-19 epidemiology,
F-7
an increase in virulence or change in clinical disease presentation, or a decrease in effectiveness of public health and social measures or available diagnostics, vaccines, or therapeutics.
Vaccines manufactured by Moderna, Pfizer and Johnson & Johnson were introduced late in the fourth quarter of 2020 and became widely available by the end of the first quarter of 2021. While the vaccines have proven effective in reducing the severity and mortality of COVID-19, including the variants that have evolved to date, the overall vaccination rate has not reached the level required for herd immunity in some areas of the country. Further, additional doses of vaccines may be required and individuals may refuse or fail to receive one or more such doses. The incidence of variants of COVID-19 has been increasing, particularly among unvaccinated individuals, and the Omicron variant has proven to be more easily spread than earlier variants.
The low vaccination rate, the spread of the variants and the evolution of additional mutations against which the current vaccines may prove ineffective could again result in major disruptions to businesses and markets worldwide. Furthermore, the incidence of "breakthrough" infections even among vaccinated individuals have been increasing, resulting in the need for booster doses which could make COVID-19 a long-term infectious disease concern. The Company’s business, results of operations, financial condition and cash flows could be materially and adversely affected. Specifically, the Company could experience additional delays in future clinical trial timelines as a result of additional travel and hospital restrictions related to the COVID-19 pandemic and the efforts to mitigate it which may be imposed or could experience supply shortages or manufacturer shutdowns impacting the manufacture of drug substance or drug product, which could also impact clinical trial timelines. The financial statements do not reflect any adjustments as a result of the pandemic.
Merger with Zafgen
On December 17, 2019, Zafgen, Inc. (“Zafgen”), Chondrial Therapeutics Inc. (“Chondrial”), Zordich Merger Sub, Inc. (“Merger Sub”) and Chondrial Holdings, LLC (“Holdings”), the sole stockholder of Chondrial, entered into an Agreement and Plan of Merger, as amended on March 9, 2020 (the “Merger Agreement”), pursuant to which Merger Sub merged with and into Chondrial, with Chondrial surviving as a wholly owned subsidiary of the Company and the surviving corporation of the merger (the “Merger”).
The transaction was accounted for as a reverse acquisition in accordance with accounting principles generally accepted in the United States of America ("GAAP"). Under this method of accounting, Chondrial was deemed to be the accounting acquirer for financial reporting purposes. This determination was primarily based on the facts that, immediately following the Merger: (1) former shareholders of Chondrial owned a substantial majority of the voting rights of the combined company; (2) the majority of the board of directors of the combined company was composed of directors designated by Chondrial under the terms of the Merger Agreement; and (3) existing members of Chondrial management constituted the management of the combined company. Because Chondrial has been determined to be the accounting acquirer in the Merger, but not the legal acquirer, the Merger is deemed a reverse acquisition under the guidance of the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 805, Business Combinations. As a result, the historical financial statements of Chondrial are the historical financial statements of the combined company. As the Merger has been accounted for as an asset acquisition, goodwill has not been recorded within the consolidated balance sheet.
The Merger was completed on May 28, 2020 pursuant to the terms of the Merger Agreement. In addition, immediately prior to the closing of the Merger, Zafgen effected a 1-for-12 reverse stock split (the “Reverse Stock Split”) of Zafgen’s common stock, par value $0.001 per share (the “Zafgen Common Stock”). At the effective time of the Merger (the “Effective Time”), each share of Chondrial’s common stock, par value $0.001 per share (“Chondrial Common Stock”), outstanding immediately prior to the Effective Time was converted into the right to receive shares of Zafgen Common Stock based on an exchange ratio set forth in the Merger Agreement. At the Effective Time following the Reverse Stock Split, the exchange ratio was determined to be 60,912.5005 shares of Zafgen Common Stock for each share of Chondrial Common Stock (the “Exchange Ratio”). At the closing of the Merger on May 28, 2020, Zafgen issued an aggregate of 6,091,250 shares of its common stock to Holdings (the “Merger Shares”), based on the Exchange Ratio after giving effect to the Reverse Stock Split described below. Holdings subsequently distributed the Merger Shares to its members.
F-8
In addition, all outstanding options exercisable for common units of Holdings became options exercisable for the shares of common stock of Zafgen based on the conversion factor discussed within the Merger Agreement. In connection with the Merger, Zafgen changed its name to Larimar Therapeutics, Inc. Following the closing of the Merger, Chondrial Therapeutics, Inc. became a wholly owned subsidiary of the Company. In December 2020, Chondrial Therapeutics was legally merged into Larimar Therapeutics, Inc. As used herein, the words “the Company” refers to, for periods following the Merger, Larimar, together with its subsidiaries, and for periods prior to the Merger, Chondrial Therapeutics Inc., and its direct and indirect subsidiaries, as applicable.
Basis of Presentation
The consolidated financial statements include the accounts of Larimar and its wholly owned subsidiaries.subsidiary. All intercompany balances and transactions have been eliminated. The accompanying consolidated financial statements have been prepared in conformity with GAAP. Unless otherwise noted, all references to common stock share and per share amounts have also been adjusted to reflect the Exchange Ratio.
Reverse Stock Split
On May 28, 2020, immediately prior to the closing of the Merger, Zafgen effected the Reverse Stock Split. Accordingly, all shareLiquidity and per share amounts for all periods presented in the accompanying consolidated financial statements and notes thereto have been adjusted retroactively, where applicable, to reflect the Reverse Stock Split. No fractional shares were issued in connection with the Reverse Stock Split.
Going ConcernCapital Resources
The Company’s consolidated financial statements have been presented on the basis that it will continue as a going concern, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business.
Since its inception, the Company has incurred significant recurring operating losses and negative cash flows from operations. The Company has incurred net losses of $50.636.9 million and $42.535.4 million for the years ended December 31, 2021,2023, and 2020,2022, respectively. In addition, as of December 31, 2021,2023, the Company had an accumulated deficit of $116.3188.6 million. The Company expects to continue to generate operating losses for the foreseeable future. As of December 31, 2021,2023, the Company had approximately $70.186.8 million of cash, and cash equivalents and marketable securities available for use to fund its operations and capital requirements.
The Company has funded its operations to date primarily with proceeds from sales of common stock and proceeds from the sale of prefunded warrants for the purchase of common stock, the acquisition in 2020 of cash, cash equivalents and marketable securities upon the merger with Zafgen, Inc. ("Zafgen") and, prior to the 2020 merger with Zafgen, described above,capital contributions from Holdings.Chondrial Holdings, LLC.
In June 2020,February 2024, the Company completed the Merger and acquired $sold 42.919,736,842 million of cash, cash equivalents, restricted cash and marketable debt securities that were held by Zafgen immediately prior to the Merger. The Company also raised $75.4 million, net of offering costs, through a private offering of common stock and prefunded warrants to purchase shares of common stock in connection with and immediately after the closing of the Merger in June 2020.
In August 2020, the Company entered into an Equity Distribution Agreement (the “ATM Agreement”) with an investment bank in connection with the establishment of an “at-the-market” offering program under which the Company could sell up to an aggregate of $50,000,000 of shares of its common stock from time to time through this investment bank as sales agent. In July 2021, the Company sold in an underwritten public offering at a price of $2,342,7208.74 shares pursuant to the ATM Agreement for grossper share and received net proceeds of approximately $20.5161.6 million. As of March 23, 2022, $29.2 million of additional shares of common stock remained available for sale by the Company under the ATM Agreement. See Note 8 for a further discussion of the ATM Agreement.
In accordance with Accounting Standards Update (“ASU”) No. 2014-15, Disclosure"Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern,Concern", the Company has evaluated whether there are certain conditions
F-9
and events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that thethese consolidated financial statements are issued. As of the issuance date of these consolidated financial statements, the Company expects its cash, and cash equivalents and marketable securities, including its net proceeds from the February 2024 public offering, will be sufficient to fund its forecasted operating expenses and capital expenditure requirements for at least the next twelve months from the issuance of these financial statements.into 2026. If the timing of ourthe Company's clinical assumptions wereare delayed or if there wereare other forecasted assumption changes that negatively impact ourits operating plan, the Company couldwould reduce expenditures in order to further extend cash resources.
The Company has not yet commercialized any products and does not expect to generate revenue from the commercial sale of any products for several years, if at all. The Company expects that its research and development and general and administrative expenses will continue to increase and, as a result, that it will need additional capital to fund its future operating and capital requirements. Management is currently evaluatingUnless and until the Company can generate substantial revenue, management continuously evaluates different strategies to obtain the required funding for future operations. Until the Company can generate substantial revenue, if ever, the Company expects to seekThese strategies include seeking additional funding through a combination of public or private equity offerings, debt or royalty financings, collaborations and licensing arrangements, strategic partnerships with pharmaceutical and/or larger biotechnology companies, or other sources. The incurrence of indebtedness would result in increased fixed payment obligations and the Company may be required to agree to certain restrictive covenants, such as limitations on its ability to incur additional debt, limitations on its ability to acquire, sell or license intellectual property rights, minimum required cash balances and other operating restrictions that could adversely impact the Company's ability to conduct ourits business. Any additional fundraising efforts may divert the Company's management from their day-to-day activities, which may adversely affect its ability to develop and commercialize ourits product candidates.
There can be no assurance that the Company will be able to raise sufficient additional capital on acceptable terms, orif at all. If such additional financing is not available on satisfactory terms, or is not available in sufficient amounts, or if the Company does not have sufficient authorized shares, the Company may be required to delay,
F-8
limit, or eliminate the development of business opportunities and its ability to achieve its business objectives, its competitiveness, and its business, financial condition, and results of operations will be materially adversely affected. The Company could also be required to seek funds through arrangements with collaborative partners or otherwise at an earlier stage than otherwise would be desirable and it may be required to relinquish rights to some of its technologies or product candidates or otherwise agree to terms unfavorable to it, any of which may have a material adverse effect on the Company's business, operating results and prospects.prospects. In addition, geopolitical unresttensions, volatility of capital markets, and other adverse macroeconomic events, including those due to inflationary pressures, rising interest rates, bank instability and the ability of the U.S. government to manage federal debt limits as well as the potential impact of the Russian invasion of Ukraine, the possibility that the conflict could expand beyond eastern Europe, the impact of the COVID-19 pandemic and/or other health crises on the global financial markets may reduce the Company's ability to access capital, which could negatively affect its liquidity and ability to continue as a going concern.
If the Company is unable to obtain sufficient funding when needed and/or on acceptable terms, the Company may be required to significantly curtail, delay or discontinue one or more of its research and development programs, the manufacture of clinical and commercial supplies, product portfolio expansion, or pre commercialization efforts and/or commercial operations, which could adversely affect its business prospects, or the Company may be unable to continue operations.
Use of Estimates
The preparation of consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. This process involves reviewing open contracts and purchase orders, communicating with our personnel and outside vendors to identify services that have been performed on our behalf and estimating the level of service performed and the associated costs incurred for the services when we have not yet been invoiced or otherwise notified of the actual costs. Significant estimates and assumptions reflected in these consolidated financial statements include, but are not limited to, the accrual of research and development expense, the recording as prepaid expense of payments made in advance of the actual provision of goods or services, valuation of stock-based awards and valuation of leases. Due to inherent uncertainty involved in making estimates, actual results reported in future periods may be affected by changes in these estimates. On an ongoing basis, the Company evaluates its estimates and assumptions.
Concentrations of Credit Risk and Significant Suppliers
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash, cash equivalents and marketable debt securities. The Company generally maintains cashCash balances may be held in various
F-10
operating accounts at financial institutions that management believes to be of high credit quality in amounts thatwhich may exceed federally insured limits. The Company has not experienced realized losses related to its cash, cash equivalents andor marketable debt securities.
The Company is highly dependent on third-party manufacturers to supply products for research and development activities in its programs, to scale and optimize their manufacturing processes and, ultimately, to provide commercial supply. The Company relies and expects to continue to rely on a small number of manufacturers to supply it with its requirements for drug substance and formulated drugs related to these programs. The drug substance which is in frozen liquid form for CTI-1601nomlabofusp is currently manufactured for the Company by a third-party manufacturer, and the frozen liquid form of drug product is made at another manufacturer. The Company is undertaking a program with a third manufacturer to begin to produce a lyophilized version of the drug product from the same drug substance, that, once available, the Company intends to use in certain of its future planned clinical trials. The Company’s research and development programs could be adversely affected by a significant interruption in these manufacturing services or in the supply of drug substance and formulated drugs.
Cash and Cash Equivalents
The Company considers all highly liquid investments with maturities of three months or less at the date of purchase to be cash equivalents. Cash equivalents consisted of commercial paper, corporate bonds and money market funds as of December 31, 20212023 and 2020.2022.
F-9
Marketable debt securities
Marketable debt securities consist of debt investments with original maturities greater than ninety days. The Company classifies its marketable debt securities as available-for-sale. Accordingly, these investments are recorded at fair value, which is based on quoted market prices. When the fair value is below the amortized cost the amount of the expected credit loss is estimated. The credit-related impairment amount, isif any, would be recognized in net income; the remaining impairment amount and unrealized gains are reported as a component of accumulated other comprehensive income in stockholders’ equity. Credit losses, if any, are recognized through the use of an allowance for credit losses account and subsequent improvements in expected credit losses are recognized as a reversal of the allowance account. If the Company has the intent to sell the security or if it is more likely than not that the Company willwould be required to sell the security prior to recovery of its amortized cost basis, the allowance for credit loss iswould be written off and the excess of the amortized cost basis of the asset over its fair value is recorded in net income.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation. Depreciation expense is recognized using the straight-line method over a five or seven-year estimated useful life for equipment, furniture and fixtures and office equipment. Leasehold improvements are amortized over the shorter of the asset life or the term of the lease agreement. Expenditures for repairs and maintenance of assets are charged to expense as incurred. Upon retirement or sale, the cost and related accumulated depreciation of assets disposed of are removed from the accounts and any resulting gain or loss is included in loss from operations.
Impairment of Long-Lived Assets
Long-lived assets consist of property and equipment, net¸ and net operating leaseright-of-use assets. Long-lived assets to be held and used are tested for recoverability whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. Factors that the Company considers in deciding when to perform an impairment review include significant underperformance of the business in relation to expectations, significant negative industry or economic trends, and significant changes or planned changes in the use of the assets. If an impairment review is performed to evaluate a long-lived asset for recoverability, the Company compares forecasts of undiscounted cash flows expected to result from the use and eventual disposition of the long-lived asset to its carrying value. Any impairment loss, if indicated, is measured as the amount by which the carrying amount of the asset exceeds the estimated fair value of the asset.
F-11
Segment Information
The Company manages its operations as a single operating segment for the purposes of assessing performance and making operating decisions. The Company’s focus is on the research, development and commercialization of novel therapeutics for the treatment of rare diseases.
Research and Development Costs
Costs associated with internal research and development and external research and development services, including drug development, clinical studies and non-clinical studies, are expensed as incurred. Research and development expenses include costs for salaries, employee benefits, subcontractors, facility-related expenses, depreciation, stock-based compensation, third-party license fees, laboratory supplies, and external costs of outside vendors engaged to conduct discovery, non-clinical and clinical development activities and clinical trials as well as to manufacture clinical trial materials, and other costs. The Company recognizes external research and development costs based on an evaluation of the progress to completion of specific tasks using information provided to the Company by its key service providers.
Nonrefundable advance payments for goods or services to be received in the future for use in research and development activities are recorded as prepaid expenses. Such prepaid expenses are recognized as an expense when the goods have been delivered or the related services have been performed, or when it is no longer expected that the goods will be delivered, or the services rendered.
Upfront payments, milestone payments and annual maintenance fees under license agreements are currently expensed in the period in which they are incurred.
F-10
Patent Costs
All patent-related costs incurred in connection with filing and prosecuting patent applications are expensed as incurred due to the uncertainty about the recovery of the expenditure. Amounts incurred are classified as general and administrative expenses.
Stock-Based Compensation
The Company measures all stock-based awards granted to employees non-employee consultants and directors based on the fair value on the date of grant using the Black-Scholes option-pricing model. Compensation expense of those awards is recognized over the requisite service period, which is the vesting period of the respective award. Typically, the Company issues awards with only service-based and market-based vesting conditions and records the expense for these awards using the straight-line method. The Company accounts for forfeitures as they occur.
The Company classifies stock-based compensation expense in its consolidated statements of operations and comprehensive loss in the same manner in which the award recipient’s payroll costs are classified or in which the award recipient'srecipient’s service payments are classified.
Prior to May 28, 2020, the Company had been a private company and lacked company-specific historical and implied volatility information for its common stock. Therefore,Prior to January 1, 2023, the Company estimatesestimated its expected common stock price volatility solely based on the historical volatility of publicly traded peer companiescompanies. Beginning on January 1, 2023, based on the availability of sufficient historical trading data of the Company's own common stock on the Nasdaq Global Market to calculate accurately its volatility, the Company began blending its volatility starting from June 2020 (following its merger with Zafgen in 2020) to the date of each stock-based award, and expects to continue to do so until it has adequate historical data regardingweighing the volatility of its own traded stock price.peer group for the amount of time from May 31, 2020 backwards so that the blended volatility equals the expected term of the related stock-based award. The expected term of the Company’s stock options has been determined utilizing the “simplified” method for awards that qualify as “plain-vanilla” options. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. ExpectedThe expected dividend yield considers the fact that the Company has never paid cash dividends on common stock and does not expect to pay any cash dividends in the foreseeable future.
F-12
Income Taxes
The Company accounts for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the consolidated financial statements or in the Company’s tax returns. Deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. The Company assesses the likelihood that its deferred tax assets will be recovered from future taxable income and, to the extent it believes, based upon the weight of available evidence, that it is more likely than not that all or a portion of the deferred tax assets will not be realized, a valuation allowance is established through a charge to income tax expense. Potential for recovery of deferred tax assets is evaluated by estimating the future taxable profits expected and considering prudent and feasible tax planning strategies.
The Company accounts for uncertainty in income taxes recognized in the consolidated financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the consolidated financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The provision for income taxes includes the effects of any resulting tax reserves, or unrecognized tax benefits, that are considered appropriate as well as the related net interest and penalties.
Comprehensive LossGain (Loss)
Comprehensive lossgain (loss) includes net loss as well as other changes in stockholders’ equity that result from transactions and economic events other than those with stockholders. For the years ended December 31, 20212023 and 2020,2022, the Company’s only element of other comprehensive income (loss)loss was unrealized gain (loss)loss on marketable securities.
F-11
Net Loss Per Share
Basic net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted average number of common shares outstanding for the period. BasicPrior to August 11, 2023, basic shares outstanding includes the weighted average effect of the Company’s prefunded warrants issued in June 2020, the exercise of which requires little or no consideration for the delivery of shares of common stock. BasicThese prefunded warrants were exercised on August 11, 2023 and diluted weighted average sharesthe Company received cash proceeds of common stock outstanding forless than $0.1 million. Accordingly, the twelve months ended December 31, 2021 and 2020 includes the weighted average effect of 628,403 prefundedshares were issued upon the exercise of these warrants for the purchase of shares ofand are included in issued and outstanding common stock for which the remaining unfunded exercise price is $0.01 per share.stock.
Diluted net loss per share attributable to common stockholders is computed by dividing the diluted net loss attributable to common stockholders by the weighted average number of common shares, including potentially dilutive common sharesstock equivalents assuming the dilutive effect of outstanding stock options, outstanding restricted stock units, and unvested restricted common shares, as determined using the treasury stock method. For periods in which the Company has reported net losses (all periods since inception), diluted net loss per common share attributable to common stockholders is the same as basic net loss per common share attributable to common stockholders, since dilutive common sharesstock equivalents are not assumed to have been issued if their effect is antidilutive.
The Company excluded 2,523,3054,888,502 and2,008,9023,071,528 options to purchase common stock, outstanding as of December 31, 20212023 and 20202022 respectively, from the computation of diluted net loss per share for the twelve months ended December 31, 20212023 and 2020,2022, respectively, because they had an anti-dilutive impact due to the net loss incurred for the periods.
Prior to the Merger the Company did not have options to purchase common stock or unvested restricted common stock to exclude from the calculation of earnings per share as all outstanding options were for common
F-13
units of Holdings that upon the Merger converted into options exercisable for the shares of common stock of the Company.
Recently Issued and Adopted Accounting Pronouncements
In June 2016,From time to time, new accounting guidance is issued by the FASB issued ASU No. 2016-13, Financial Instruments—Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. The FASB subsequently issued amendments to ASU 2016-13. Thisor other standard requires entities to estimate an expected lifetime credit loss on financial assets ranging from short-term trade accounts receivable to long-term financings and report credit losses using an expected losses model rather than the incurred losses modelsetting bodies that was previously used, and establishes additional disclosures related to credit risks. For available-for-sale debt securities with unrealized losses, the standard now requires allowances to be recorded instead of reducing the amortized costis adopted by us as of the investment. This standard limitseffective date or, in some cases where early adoption is permitted, in advance of the amount of credit losses to be recognized for available-for-sale debt securities toeffective date. We have assessed the amount by which carrying value exceeds fair valuerecently issued guidance that is not yet effective and requiresbelieve the reversal of previously recognized credit losses if fair value increases. The Company adopted the standard on January 1, 2020. The adoption of this standard didnew guidance will not have a material impact on the Company’s consolidated results of operations, cash flows or financial statements or related disclosures.position.
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement (Topic 820): Disclosure Framework—Changes to the Disclosure Requirements for Fair Value Measurement. This standard modifies certain disclosure requirements on fair value measurements. This standard became effective and was adopted by the Company on January 1, 2020. The adoption of this standard did not have a material impact on the Company’s disclosures.
F-14
On May 28, 2020, the Company completed its merger with Zafgen. Based on the Exchange Ratio, immediately following the Merger, former Zafgen stockholders, Zafgen option holders and other persons holding securities or other rights directly or indirectly convertible, exercisable or exchangeable for Zafgen Common Stock (collectively, the “Zafgen Securityholders”) owned approximately 34% of the outstanding capital stock of the combined company, and Holdings, the former Chondrial stockholder, owned approximately 66% of the outstanding capital stock of the combined company. At the closing of the Merger, all shares of Chondrial Common Stock were exchanged for an aggregate of 6,091,250 shares of Zafgen Common Stock, after giving effect to the Reverse Stock Split.
In addition, pursuant to the terms of the Merger Agreement, the Company assumed all outstanding stock options to purchase shares of Zafgen common stock at the closing of the Merger. At the closing of the Merger, such stock options became options to purchase an aggregate of 328,770 shares of the Company’s common stock after giving effect to the Reverse Stock Split.
The total purchase price paid in the Merger was allocated to the tangible and intangible assets acquired and liabilities assumed of Zafgen based on their fair values as of the completion of the Merger.Transaction costs primarily included bank fees and professional fees associated with legal counsel, auditors and printers. The following summarizes the purchase price paid in the Merger (in thousands, except share and per share amounts):
Number of shares of the combined organization owned by Zafgen stockholders(1) |
|
| 3,124,337 |
|
Multiplied by the fair value per share of Zafgen common stock(2) |
| $ | 11.88 |
|
Fair value of consideration issued in effect of the Merger |
| $ | 37,119 |
|
Transaction costs |
| $ | 1,715 |
|
Purchase price: |
| $ | 38,834 |
|
The allocation of the purchase price for the Merger was based on estimates of the fair value of the net assets acquired, which was then adjusted for the difference between the purchase price and the fair value of the assets acquired. The following summarizes the allocation of the purchase price to the net tangible and intangible assets acquired (in thousands):
Cash and cash equivalents |
| $ | 40,595 |
|
Marketable debt securities |
|
| 1,014 |
|
Other current and noncurrent assets |
|
| 357 |
|
Property and equipment, net |
|
| 398 |
|
Restricted cash |
|
| 1,339 |
|
Right-of-use asset |
|
| 3,806 |
|
Current liabilities |
|
| (2,685 | ) |
Lease liability, net of current portion |
|
| (5,990 | ) |
Purchase price |
| $ | 38,834 |
|
F-15
Fair Value Measurements
The Company’s assets and liabilities that are measured at fair value on a recurring basis as of December 31, 20212023 and December 31, 20202022 are measured in accordance with the standards of ASC 820, Fair Value Measurements and Disclosures, which establishes a three-level valuation hierarchy for measuring fair value and expands financial statement disclosures about fair value measurements. The valuation hierarchy is based on upon the transparency of inputs to the valuation of an asset or liability as of the measurement date. The three levels are defined as follows:
Level – 1 | Inputs to the valuation methodology are quoted prices (unadjusted) for identical assets or liabilities in active markets. |
|
|
Level – 2 | Inputs to the valuation methodology include quoted prices for similar assets and liabilities in active markets, and inputs that are observable for the asset or liability, either directly or indirectly, for substantially the full term of the financial instrument. |
|
|
Level – 3 | Inputs to the valuation methodology are unobservable and significant to the fair value measurement. |
The Company’s financial instruments consist primarily of cash and cash equivalents, marketable securities, accounts payable and accrued liabilities. For accounts payable and accrued liabilities, the carrying amounts of these financial instruments as of December 31, 20212023 and 20202022 were considered representative of their fair values due to their short term to maturity.
F-12
The following tables summarize the Company’s cash equivalents and marketable debt securities as of December 31, 20212023 and 2020:2022:
|
| Total |
|
| Quoted |
|
| Significant |
|
| Significant |
| ||||
|
| (in thousands) |
| |||||||||||||
December 31, 2021 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Money market funds |
| $ | 6,137 |
|
| $ | 6,137 |
|
| $ | — |
|
| $ | — |
|
Commercial paper |
|
| 7,549 |
|
|
| 7,549 |
|
|
| — |
|
|
| — |
|
Corporate bonds |
|
| 1,219 |
|
|
| 1,219 |
|
|
| — |
|
|
| — |
|
Total cash equivalents |
|
| 14,905 |
|
|
| 14,905 |
|
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
December 31, 2020 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Money market funds |
| $ | 4,229 |
|
| $ | 4,229 |
|
| $ | — |
|
| $ | — |
|
Commercial paper |
|
| 6,499 |
|
|
| — |
|
|
| 6,499 |
|
|
| — |
|
Corporate bonds |
|
| 1,907 |
|
|
| — |
|
|
| 1,907 |
|
|
| — |
|
Total cash equivalents |
|
| 12,635 |
|
|
| 4,229 |
|
|
| 8,406 |
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Marketable securities: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
U.S Government securities |
|
| 2,005 |
|
|
| — |
|
|
| 2,005 |
|
|
| — |
|
Commercial paper |
|
| 22,485 |
|
|
| — |
|
|
| 22,485 |
|
|
| — |
|
Total marketable debt securities |
|
| 24,490 |
|
|
| — |
|
|
| 24,490 |
|
|
| — |
|
Total cash equivalents and marketable debt securities |
| $ | 37,125 |
|
| $ | 4,229 |
|
| $ | 32,896 |
|
| $ | — |
|
|
| Total |
|
| Quoted |
|
| Significant |
|
| Significant |
| ||||
|
| (in thousands) |
| |||||||||||||
December 31, 2023 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Money market funds invested in government securities |
| $ | 24,701 |
|
| $ | 24,701 |
|
| $ | — |
|
| $ | — |
|
Total cash equivalents |
|
| 24,701 |
|
|
| 24,701 |
|
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Marketable securities: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
U.S. Treasury Bills |
|
| 17,334 |
|
|
| 17,334 |
|
|
| — |
|
|
| — |
|
U.S. Government securities |
|
| 35,719 |
|
|
| — |
|
|
| 35,719 |
|
|
| — |
|
Corporate bonds |
|
| 6,988 |
|
|
| — |
|
|
| 6,988 |
|
|
| — |
|
Total marketable securities |
|
| 60,041 |
|
|
| 17,334 |
|
|
| 42,707 |
|
|
| — |
|
Total cash equivalents and marketable securities |
| $ | 84,742 |
|
| $ | 42,035 |
|
| $ | 42,707 |
|
| $ | — |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
December 31, 2022 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Money market funds invested in government securities |
| $ | 22,184 |
|
| $ | 22,184 |
|
| $ | — |
|
| $ | — |
|
Total cash equivalents |
|
| 22,184 |
|
|
| 22,184 |
|
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Marketable securities: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
U.S. Government securities |
|
| 91,603 |
|
|
| — |
|
|
| 91,603 |
|
|
| — |
|
Total marketable securities |
|
| 91,603 |
|
|
| — |
|
|
| 91,603 |
|
|
| — |
|
Total cash equivalents and marketable securities |
| $ | 113,787 |
|
| $ | 22,184 |
|
| $ | 91,603 |
|
| $ | — |
|
The accrued interest receivable related to the Company’s investments was $0.3 million and $0.1 million as of December 31, 2023 and 2022, respectively, and is included in prepaid expenses and other current assets on the consolidated balance sheet.
The Company classifies its money market funds and U.S. treasury bills, which are valued based on quoted market prices in active markets with no valuation adjustment, as Level 1 assets within the fair value hierarchy.
The Company classifies its investments in U.S. government and agency securities, corporate commercial paper, and corporate bonds, if any, as Level 2 assets within the fair value hierarchy. The fair values of these investments are estimated by taking into consideration valuations obtained from third-party pricing services. The pricing services utilize industry standard valuation models, including both income- and market-based approaches, for which all significant inputs are observable, either directly or indirectly, to estimate fair value. These inputs include reported trades of and broker/dealer quotes on the same or similar securities, issuer credit spreads, benchmark securities, prepayment/default projections based on historical data and other observable inputs.
As of December 31, 2022, the unrealized loss for available-for-sale investments were non-credit related, and the Company does not intend to sell the investments that were in an unrealized loss position, nor will it be required to sell those investments before recovery of their amortized cost basis, which may be maturity. As of December 31, 2023 and 2022, no allowances for credit losses for the Company’s investments were recorded. During the twelve months ended December 31, 2023 and 2022, the Company did not recognize any impairment losses related to investments.
F-13
F-16
As of December 31, 2023, the Company's cash equivalents and marketable securities consisted of a U.S. government money market fund, U.S. Treasury Bills, U.S. government and agency securities and corporate bonds, all held in our name in a separate custody account with U.S. Bank. The U.S. government money market fund has same-day liquidity access and the U.S. government and agency securities all have maturities of 90 days or less.
Marketable Debt Securities
The following tables summarize the Company’s marketable debt securities as of December 31, 2020. There were 0 marketable debt securities as of December 31, 2021:2023 and 2022:
|
| Amortized |
|
| Gross |
|
| Gross |
|
| Fair Value |
|
| Amortized |
|
| Gross |
|
| Gross |
|
| Fair Value |
| ||||||||
|
| (in thousands) |
|
| (in thousands) |
| ||||||||||||||||||||||||||
December 31, 2020 |
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||
December 31, 2023 |
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||
Assets: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||
U.S Government securities (due within 1 year) |
| 2,005 |
| — |
| — |
| 2,005 |
| |||||||||||||||||||||||
U.S. Treasury Bills |
|
| 17,330 |
|
|
| 4 |
|
|
| — |
|
|
| 17,334 |
| ||||||||||||||||
U.S. Government securities (due within 1 year) |
|
| 35,653 |
|
|
| 66 |
|
|
| — |
|
|
| 35,719 |
| ||||||||||||||||
Commercial paper (due within 1 year) |
|
| 22,484 |
|
|
| 2 |
|
|
| (1 | ) |
|
| 22,485 |
|
|
| 6,977 |
|
|
| 11 |
|
|
| — |
|
|
| 6,988 |
|
Total marketable securities |
| $ | 59,960 |
|
| $ | 81 |
|
| $ | — |
|
| $ | 60,041 |
| ||||||||||||||||
December 31, 2022 |
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||
Assets: |
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||
U.S. Government securities (due within 1 year) |
|
| 91,634 |
|
|
| 12 |
|
|
| (43 | ) |
|
| 91,603 |
| ||||||||||||||||
|
| $ | 24,489 |
|
| $ | 2 |
|
| $ | (1 | ) |
| $ | 24,490 |
|
| $ | 91,634 |
|
| $ | 12 |
|
| $ | (43 | ) |
| $ | 91,603 |
|
Prepaid expenses and other current assets consisted of the following:
|
| December 31, |
|
| December 31, |
|
| December 31, |
|
| December 31, |
| ||||
|
| 2021 |
|
| 2020 |
|
| 2023 |
|
| 2022 |
| ||||
|
| (in thousands) |
|
| (in thousands) |
| ||||||||||
Prepaid research and development expenses |
| $ | 676 |
| $ | 4,460 |
|
| $ | 1,994 |
|
| $ | 1,394 |
| |
Prepaid insurance |
|
| 944 |
|
|
| 571 |
|
| 682 |
|
| 679 |
| ||
Payroll tax receivable |
| 208 |
| 32 |
| |||||||||||
Other prepaid expenses and other assets |
|
| 279 |
|
|
| 251 |
|
|
| 709 |
|
|
| 238 |
|
|
| $ | 2,107 |
|
| $ | 5,314 |
| ||||||||
Total prepaid expenses and other assets |
| $ | 3,385 |
|
| $ | 2,311 |
| ||||||||
Fixed assets, net consisted of the following:
|
| December 31, |
|
| December 31, |
|
| December 31, |
|
| December 31, |
| ||||||||
|
| Useful Life |
| 2021 |
|
| 2020 |
|
| Useful Life |
| 2023 |
|
| 2022 |
| ||||
|
| (in thousands) |
|
| (in thousands) |
| ||||||||||||||
Computer equipment |
| 5 years |
| $ | 66 |
| $ | 66 |
|
| 5 years |
| $ | 117 |
|
| $ | 66 |
| |
Lab equipment |
| 5 years |
| 1,092 |
| 849 |
|
| 5 years |
|
| 1,192 |
|
|
| 1,192 |
| |||
Furniture and fixtures |
| 7 years |
| 456 |
| 460 |
|
| 7 years |
|
| 555 |
|
|
| 456 |
| |||
Leasehold improvements |
| lease term |
|
| 31 |
|
|
| — |
|
| lease term |
|
| 45 |
|
|
| 31 |
|
|
|
|
| 1,645 |
| 1,375 |
| |||||||||||||
Total property, plant and equipment |
|
|
|
| 1,909 |
|
|
| 1,745 |
| ||||||||||
Less: Accumulated depreciation |
|
|
|
| (596 | ) |
|
| (335 | ) |
|
|
|
| (1,225 | ) |
|
| (914 | ) |
|
|
|
| $ | 1,049 |
|
| $ | 1,040 |
| ||||||||||
Property, plant and equipment, net |
|
|
| $ | 684 |
|
| $ | 831 |
| ||||||||||
Depreciation expense was $0.2 million for the years ended December 31, 20212023 and 2020,2022, respectively. In addition, for the years ended December 31, 2023 and 2022, there was $0.1 million of depreciation related to sublet assets recorded as other expense.
F-17F-14
Accrued expenses consisted of the following:
|
| December 31, |
|
| December 31, |
|
| December 31, |
|
| December 31, |
| ||||
|
| 2021 |
|
| 2020 |
|
| 2023 |
|
| 2022 |
| ||||
|
| (in thousands) |
|
| (in thousands) |
| ||||||||||
Accrued research and development expenses |
| $ | 5,042 |
| $ | 3,409 |
|
| $ | 4,594 |
|
| $ | 5,921 |
| |
Accrued payroll and related expenses |
| 1,098 |
| 1,350 |
|
|
| 2,365 |
|
|
| 2,046 |
| |||
Accrued professional fees |
| — |
| 924 |
| |||||||||||
Accrued other |
|
| 452 |
|
|
| 160 |
|
|
| 427 |
|
|
| 441 |
|
|
| $ | 6,592 |
|
| $ | 5,843 |
| ||||||||
Total accrued expenses and other current liabilities |
| $ | 7,386 |
|
| $ | 8,408 |
| ||||||||
Common Stock and Prefunded warrants
On May 28, 2020, the Company entered into a securities purchase agreement with certain accredited investors (the “Purchasers”) for the sale by the Company in a private placement of 6,105,359 shares of the Company’s common stock and prefunded warrants to purchase an aggregate of 628,403 shares of the Company’s common stock, for a price of $11.88 per share of the common stock and $11.87 per prefunded warrant. The prefunded warrants were exercisable at an exercise price of $0.01 and were exercisable indefinitely. In August 2023, the 628,403 shares of prefunded warrants were exercised and the Company received cash proceeds of six thousand two hundred and eighty-four dollars. The private placement closed on June 1, 2020. The aggregate gross proceeds for the issuance and sale of the common stock and prefunded warrants were $80.0 million, transaction costs totaled $4.6 million and resulted in net proceeds of $75.4 million. The Company’s Registration Statement on Form S-3, filed with the SEC on June 26, 2020, registered the resale of 6,105,359 shares of common stock sold and the 628,403 shares of common stock underlying the prefunded warrants. MTS Health Partners served as placement agent to the Company in connection with the private placement. As partial compensation for these services, the Company issued MTS Health Partners 35,260 shares of common stock.
As of December 31, 2021,2023, the Company’s Amended and Restated Certificate of Incorporation, as amended and restated, authorized the Company to issue up to 115,000,000 shares of $0.001 par value common stock, of which 17,710,45043,909,069 shares were issued and outstanding, and up to 5,000,000 shares of $0.001 par value undesignated preferred stock, of which 0no shares were issued or outstanding. The voting, dividend and liquidation rights of the holders of the Company’s common stock are subject to and qualified by the rights, powers and preferences of the holders of the preferred stock. Each share of common stock entitles the holder to one vote on all matters submitted to a vote of the Company’s stockholders. Common stockholders are entitled to receive dividends, as may be declared by the board of directors of the Company (the “Board”), if any. NaNNo cash dividends have been declared or paid to date.
On May 28, 2020,In September 2022, the Company sold 25,558,750 shares of common stock in an underwritten public offering price of $3.15 per share and received net proceeds, net of underwriting discounts and commissions and offering costs of $75.2 million.
In February 2024, the Company sold 19,736,842 shares of its common stock in an underwritten public offering price of $8.74 per share and received net proceeds of approximately $161.6 million.
2022 ATM Agreement
In November 2022, the Company entered into a securities purchasesales agreement (the "ATM Agreement") with certain accredited investors (the “Purchasers”) for the sale by the Company in a private placement of 6,105,359 shares of the Company’s common stock and prefunded warrants to purchase an aggregate of 628,403 shares of the Company’s common stock, for a price of $11.88 per share of the common stock and $11.87 per prefunded warrant. The prefunded warrants are exercisable at an exercise price of $0.01 and are exercisable indefinitely. The Purchasers may exercise the prefunded warrants on a cashless basis in the event that there is no effective registration statement covering the resale of the shares of common stock underlying the prefunded warrants on the date in which the Company is required to deliver the shares. The private placement closed on June 1, 2020. The aggregate gross proceeds for the issuance and sale of the common stock and prefunded warrants were $80.0 million; transaction costs totaled $4.6 million and resulted in net proceeds of $75.4 million. The Company’s Registration Statement on Form S-3, filed with the SEC on June 26, 2020, registered the resale of 6,105,359 shares of common stock sold and the 628,403 shares of common stock underlying the prefunded warrants.MTS Health Partners served as placement agent to the Company in connection with the private placement. As partial compensation for these services, the Company issued MTS Health Partners 35,260 shares of common stock.
Equity Distribution Agreement
On August 14, 2020, the Company entered into the ATM Agreement with an investment bank,Guggenheim Securities, LLC in connection with the establishment of an “at-the-market” offering program under which the Company maycould sell up to an aggregate of $50,000,00050.0 million of shares of common stock (the “ATM Shares”) from time to time (the “Offering”).
F-18
Under the ATM Agreement, the Company sets the parameters for the sale of ATM Shares, including the number of ATM Shares to be issued, the time period during which sales are requested to be made, limitations on the number of ATM Shares that may be sold in any one trading day and any minimum price below which sales may not be made. Sales of the ATM Shares, if any, under the ATM Agreement may be made in transactions that are deemed to be “at-the-market offerings” as defined in Rule 415 under the Securities Act. The Company pays its investment bank a commission equal to 3.0% of the gross proceeds of any ATM Shares sold through its investment bank under the ATM Agreement and reimburses investment bank for certain specified expenses. The Agreement contains customary representations, warranties and agreements by the Company, indemnification obligations of the Company and its investment bank, other customary obligations of the parties and termination provisions. The Company has no obligation to sell any of the ATM Shares and may at any time suspend offers under the ATM Agreement.time.
In July 2021,February 2024, in connection with the underwritten public offering described above, the Company sold 2,342,720 shares under the ATM Agreement for net proceeds of $19.9 million, after issuance costs.
As of December 31, 2021, 2,354,244 shares have been sold under the ATM Agreement for net proceeds of $20.1 million, after issuance costs. As of March 23, 2022, approximately $29.2 million of additional shares of common stock remained available for sale by the Company underterminated the ATM Agreement.
Summary of PlansNo
Upon completion of ATM Shares were ever sold pursuant to the Merger with Zafgen, Zafgen’s 2014 Stock Option and Incentive Plan (the “2014 Plan”) and Zafgen’s 2006 Stock Option Plan (the “2006 Plan” and together with the 2014 Plan the “Prior Plans”) were assumed by the Company. As described below, the Company adopted a new equity incentive plan in July 2020 that was approved by the stockholders in September 2020. These three plans are administered by the Board or, at the discretion of the Board, by a committee of the Board.ATM Agreement.
2020 Equity Incentive Plan
The Board adopted the 2020 Equity Incentive Plan (the "2020 Plan") on July 16, 2020 and the stockholders of the Company approved the 2020 Plan on September 29, 2020. The 2020 Plan replacesreplaced the 2014 Plan. Optionpredecessor plans (the
F-15
"Prior Plans") that the Company assumed following its merger with Zafgen in May 2020. Options outstanding under the Prior Plans will remain outstanding, unchanged, and subject to the terms of the Prior Plans and the respective award agreements, and no further awards will be made under the 2014 Plan.Prior Plans. However, if any award previously granted under the Prior Plans, expires, terminates, is canceled, or is forfeited for any reason after the approval of the 2020 Plan, the shares subject to that award will be added to the 2020 Plan share pool so that they can be utilized for new grants under the 2020 Plan.
The 2020 Plan provides for the grant of incentive stock options (“ISOs”), nonstatutory stock options (”(“NSOs”), stock appreciation rights, restricted stock awards, restricted stock unit awards, and cash or other stock-based awards. ISOs may be granted only to the Company’s employees, including the Company’s officers, and the employees of the Company’s affiliates. All other awards may be granted to the Company’s employees, including the Company’s officers, the Company’s non-employee directors and consultants, and the employees and consultants of the Company’s affiliates.
The maximum number of shares that may be issued in respect of any awards under the 2020 Plan is the sum of: (i) 1,700,000 shares plus (ii) an annual increase on January 1, 2021 and each anniversary of such date thereafter through January 1, 2030, equal to the lesser of (A) 4% of the shares issued and outstanding on the last day of the immediately preceding fiscal year, andor (B) such smaller number of shares as determined by the Board (collectively, the “Plan Limit”). The maximum aggregate number of shares that may be issued under the 2020 Plan is 8,000,000 over the ten-year term of the 2020 Plan. In 2021, options to purchase 56,966 shares issued under the Prior Plans were cancelled and became available for grant under the 2020 Plan. As of December 31, 2021, 993,419 shares of common stock were available for grant under the 2020 Plan.
As permitted by the 2020 Plan, the Company added 708,4181,756,363 and 1,730,768 shares available for grant underto the 2020 Plan on January 1, 20222024 and January 1, 2023, respectively, increasing the maximum number of shares of the Company’s common stock that may be issued under the 2020 Plan as of January 1, 2024 to 1,701,8373,042,968 shares.
F-19
2014 Stock OptionDuring the twelve months ended December 31, 2023 and Incentive Plan2022, respectively, options to purchase 224,437 and 2006 Stock Option Plan
In 2014, the Board and stockholders of Zafgen adopted the 2014 Plan. The 2014 Plan provided for the grant of stock options, stock appreciation rights, restricted stock awards, restricted stock units, unrestricted stock awards, performance-share awards, cash-based awards and dividend equivalent rights to employees, members of the Board and consultants of the Company. The number of148,623 shares initially reserved for issuanceissued under the 2014 Plan wasPrior Plans were canceled and became available for grant under the 2020 Plan. In addition, as of December 31, 2023, 180,6851,286,605 shares of common stock. Asstock were available for grant under the 2020 Plan was adopted by the Company and approved by the Company’s stockholders, no further awards will be made under the Prior Plans.
2016 Equity and Incentive Plan
Under the 2016 Equity Plan adopted by Holdings on November 30, 2016, (the “2016 Equity Incentive Plan”), the Board of Managers of Holdings (the “Board of Managers”) or a committee thereof was authorized to issue 122,133 Common Units of Holdings or combination of Common Units, Common Unit options or profit interest units. On March 23, 2018, the Board of Managers increased the number of Common Units reserved for grant and issuance pursuant to the 2016 Plan from 122,133 to 138,133 and on April 29, 2019 increased the number of Common Units reserved for grant and issuance pursuant to the 2016 Plan by an additional 101,500 to 239,633. The Company has recorded costs incurred as stock-based compensation with a corresponding capital contribution from Holdings.
From January 1, 2020 through the Merger date Holdings did not issue options to purchase Common Units to employees of the Company.
The Company assumed all of the outstanding and unexercised options to purchase units of Holdings upon consummation of the Merger. Pursuant to the terms of the Merger Agreement, options to purchase 330,818 shares of the Company’s common stock at a weighted average exercise price of $12.14 per share were substituted for the 202,392 options to purchase Common Units, with a weighted average exercise price of $10.36 per Common Unit, that were outstanding immediately prior to the Merger.
The Company treated the conversion as a modification pursuant to ASC 718, Compensation—Stock Compensation, andcalculated the pre- and post-modification value of the options. The increase in fair value of the options was calculated to be $1.2 million. As $0.7 million related to vested options the expense was recognized immediately on the Merger date, the remaining $0.5 million is recognized over the remaining vesting term with the original grant date fair value remaining of less than $0.1 million.Plan.
Stock Valuation
The following table presents, on a weighted average basis, the assumptions used in the Black-Scholes option-pricing model to determine the grant-date fair value of stock options granted to employees:
|
| 2021 |
|
| 2020 |
| ||
Risk-free interest rate |
| 0.89% |
|
| 0.37% |
| ||
Expected term (in years) |
|
| 6.19 |
|
|
| 6.08 |
|
Expected volatility |
| 91% |
|
| 91% |
| ||
Dividend yield |
| 0.00% |
|
| 0.00% |
| ||
F-20
|
| 2023 |
|
| 2022 |
| ||
Risk-free interest rate |
| 3.72% |
|
| 2.15% |
| ||
Expected term (in years) |
|
| 6.23 |
|
|
| 6.21 |
|
Expected volatility |
| 94% |
|
| 89% |
| ||
Dividend yield |
| 0.00% |
|
| 0.00% |
| ||
Stock Options
The following table summarizes the Company’s stock option activity for the twelve months ended December 31, 20212023 (amounts in millions, except for share and per share data):
|
|
|
|
| Weighted |
|
| Weighted Average |
|
| Aggregate |
| ||||
|
|
|
|
| Average |
|
| Remaining |
|
| Intrinsic |
| ||||
|
| Number of |
|
| Exercise |
|
| Contractual |
|
| Value (a) |
| ||||
|
| Shares |
|
| Price |
|
| Term (in years) |
|
| (in millions) |
| ||||
Outstanding as of December 31, 2020 |
|
| 2,008,902 |
|
| $ | 22.31 |
|
|
| 7.9 |
|
|
|
| |
Granted |
|
| 626,750 |
|
|
| 15.56 |
|
|
|
|
|
|
| ||
Forfeited/Expired |
|
| (112,347 | ) |
|
| 61.80 |
|
|
|
|
|
|
| ||
Outstanding as of December 31, 2021 |
|
| 2,523,305 |
|
| $ | 18.88 |
|
|
| 7.6 |
|
| $ | 0.2 |
|
Exercisable as of December 31, 2021 |
|
| 1,027,858 |
|
| $ | 26.52 |
|
|
| 5.9 |
|
| $ | — |
|
Vested and expected to vest as of December 31, 2021 |
|
| 2,523,305 |
|
| $ | 18.88 |
|
|
| 7.6 |
|
| $ | 0.2 |
|
|
|
|
|
| Weighted |
|
| Weighted Average |
|
| Aggregate |
| ||||
|
|
|
|
| Average |
|
| Remaining |
|
| Intrinsic |
| ||||
|
| Number of |
|
| Exercise |
|
| Contractual |
|
| Value (a) |
| ||||
|
| Shares |
|
| Price |
|
| Term (in years) |
|
| (in millions) |
| ||||
Outstanding as of December 31, 2022 |
|
| 3,071,528 |
|
| $ | 12.13 |
|
|
| 7.6 |
|
|
|
| |
Granted |
|
| 1,748,200 |
|
|
| 4.68 |
|
|
|
|
|
|
| ||
Exercised |
|
| (11,466 | ) |
|
| 2.93 |
|
|
|
|
|
|
| ||
Forfeited/Expired |
|
| (534,760 | ) |
|
| 12.52 |
|
|
|
|
|
|
| ||
Outstanding as of December 31, 2023 |
|
| 4,273,502 |
|
| $ | 9.06 |
|
|
| 7.8 |
|
| $ | 0.9 |
|
Exercisable as of December 31, 2023 |
|
| 1,930,100 |
|
| $ | 12.55 |
|
|
| 6.6 |
|
| $ | 0.2 |
|
Vested and expected to vest as of December 31, 2023 |
|
| 4,273,502 |
|
| $ | 9.06 |
|
|
| 7.8 |
|
| $ | 0.9 |
|
2021F-16
2023 Option Grants
During the yeartwelve months ended December 31, 2021,2023, the Company granted options to purchase 626,750 shares of common stock to employees and directors under the 2020 Plan. The options have an exercise price equal to the closing stock price as of the grant date. Of the 626,750 options granted in 2021, 576,950 were granted to employees and vest over four years, with 25% vesting on the first anniversary of the grant and the remainder vesting in equal monthly installments thereafter. The remaining 49,800 options were annual grants to the Company's directors and vest one year from the grant date.
January 2022 Option Grants
On January 18, 2022, the Company granted options to purchase 582,7501,388,200 shares of common stock to employees under the 2020 Plan. The options have an exercise price equal to the closing stock price as of the grant date, and vest over four years, with 25% vesting on the first anniversary of the grant and the remainder vesting in equal monthly installments thereafter. The weighted-average grant date fair value of options granted under the 2020 Plan during the twelve months ended December 31, 2023 and 2022 was $3.70 and $5.21, respectively.
As of December 31, 2023, total unrecognized compensation expense related to unvested stock options granted under the 2020 Plan was $8.8 million, which is expected to be recognized over a weighted average period of 1.91 years.
Inducement Stock Option Grant
During the twelve months ended December 31, 2023, the Company granted options to purchase 360,000 shares of common stock granted outside of the 2020 Plan. These grants were made pursuant to the Nasdaq inducement grant exception in accordance with Nasdaq listing rule 5635(c)(4). The options issued under this inducement grant have an exercise price equal to the closing stock price as of the grant date, and vest over four years, with 25% vesting on the first anniversary of the grant and the remainder vesting in equal monthly installments thereafter. The weighted-average grant date fair value of options granted under this inducement grant during the twelve months ended December 31, 2023 was $3.59. There were no inducement grants in twelve months ended December 31, 2022.
As of December 31, 2023, total unrecognized compensation expense related to unvested inducement options granted under the 2020 Plan was $1.1 million, which is expected to be recognized over a weighted average period of 3.28 years.
Restricted Stock Units
In January 2023, RSUs were granted under the 2020 Plan to the Company's employees in order to maintain retention of key employees. The value of an RSU award is based on the Company's stock price on the date of the grant. The shares underlying the RSUs are not issued until the RSUs vest.
Activity with respect to the Company's RSUs during the twelve months ended December 31, 2023 was as follows (in millions, except share, contractual term, and per share data):
|
|
|
|
| Weighted |
|
| Weighted Average |
|
| Aggregate |
| ||||
|
|
|
|
| Average |
|
| Remaining |
|
| Intrinsic |
| ||||
|
| Number of |
|
| Grant Date |
|
| Contractual |
|
| Value (a) |
| ||||
|
| Shares |
|
| Fair Value |
|
| Term (in years) |
|
| (in millions) |
| ||||
Outstanding as of December 31, 2022 |
|
| — |
|
| $ |
|
|
|
| — |
|
|
|
| |
Restricted stock units granted |
|
| 650,000 |
|
|
| 4.94 |
|
|
|
|
|
|
| ||
Restricted stock units forfeited |
|
| (35,000 | ) |
|
| 4.94 |
|
|
|
|
|
|
| ||
Outstanding as of December 31, 2023 |
|
| 615,000 |
|
| $ | 4.94 |
|
|
| 1.6 |
|
| $ | 2.8 |
|
Unvested and expected to vest as of December 31, 2023 |
|
| 615,000 |
|
| $ | 4.94 |
|
|
| 1.6 |
|
| $ | 2.8 |
|
Restricted Stock Unit Grants
The RSUs vest annually over four years and have a weighted-average grant date fair value of $4.94 per unit.
As of December 31, 2023, total unrecognized compensation expense for RSUs was $2.3 million, which is expected to be recognized over a weighted-average period of 3.09 years.
January 2024 Option and Restricted Stock Unit Grants
On January 17, 2024, the Company granted options to purchase 1,472,175 shares of common stock and 245,363 shares of restricted stock to employees under the 2020 Plan. The options have an exercise price equal to the closing stock price as of the grant date, and vest over four years, with 25% vesting on the first anniversary of the
F-17
grant and the remainder vesting in equal monthly installments thereafter. The shares of restricted stock will vest annually over four years.
Stock-Based Compensation
Stock-based compensation expense was classified in the consolidated statements of operations as follows:
|
| Year Ended December 31, |
|
|
| Year Ended December 31, |
|
| ||||||||||
|
| 2021 |
|
| 2020 |
|
|
| 2023 |
|
| 2022 |
|
| ||||
|
| (in thousands) |
|
|
| (in thousands) |
|
| ||||||||||
Research and development |
| $ | 2,104 |
| $ | 788 |
|
|
| $ | 3,077 |
|
| $ | 2,771 |
|
| |
General and administrative |
|
| 3,369 |
|
|
| 1,373 |
|
|
|
| 4,538 |
|
|
| 3,848 |
|
|
|
| $ | 5,473 |
|
| $ | 2,161 |
|
|
| $ | 7,615 |
|
| $ | 6,619 |
|
|
As of December 31, 2021, total unrecognized compensation expense related to unvested stock options and restricted stock units was $14.6 million, which is expected to be recognized over a weighted average period of 2.66 years.
F-21
Intellectual Property Licenses
The Company is party to an exclusive License Agreement (the “WFUHS License”), dated November 30, 2016, as amended, with Wake Forest University Health Sciences (“WFUHS”) and an exclusive License Agreement (the “IU License”), dated November 30, 2016, as amended, with Indiana University (“IU”). Such agreements provide for a transferable, worldwide license to certain patent rights regarding technology used by the Company with respect to the development of CTI-1601.nomlabofusp. Both agreements continue from their effective date through the last to date of expiration of the licensed patents, unless earlier terminated by either party in accordance with their terms.
In partial consideration for the right and license granted under these agreements, the Company will pay each of WFUHS and IU a royalty of a low single digit percentage of net sales of licensed products depending on whether there is a valid patent covering such products. As additional consideration for these agreements, the Company is obligated to pay each of WFUHS and IU certain milestone payments of up to $2.6 million in the aggregate upon the achievement of certain developmental milestones, commencing onwhich commenced with the enrollment of the first patient in a Phase 1 clinical trial. The Company enrolled the first patient in its SAD trial on December 11, 2019 and paid WFUHS and IU less than $0.1 million. The Company will also pay each of WFUHS and IU sublicensing fees ranging from a high singlehigh-single digit to a low double-digit percentage of sublicense consideration depending on the Company’s achievement of certain regulatory milestones as of the time of receipt of the sublicense consideration. The Company is also obligated to reimburse WFUHS and IU for patent-related expenses. In the event that the Company disputes the validity of any of the licensed patents, the royalty rate would be tripled during such dispute. The Company is also obligated to pay to IU a minimum annual royalty of less than $0.1 million per annum starting in the 2020 calendar year for the term of the agreement.annum.
In the event that the Company is required to pay IU consideration, then the Company may deduct 20% of such IU consideration on a dollar-for-dollar basis from the consideration due to WFUHS. In the event that the Company is required to pay WFUHS consideration, then the Company may deduct 60% of such WFUHS consideration on a dollar-for-dollar basis from the consideration due to IU.
DuringIn October 2022, the twelve months ended December 31, 2021Company initiated dosing of a Phase 2 study. Pursuant to the terms of both the WFUHS License and 2020, 0 milestones were achieved and 0the IU License, the company recognized milestone expense was recognized. of $0.3 million within research and development expenses.
Both agreements continue from their effective date through the last to expiredate of expiration of the licensed patents, unless earlier terminated by either party in accordance with their terms.
Leases
Bala Cynwyd Office Space
On August 8, 2019, the Company entered into an operating lease for office space in Bala Cynwyd, Pennsylvania, effective as of December 15, 2019, for a period of three years and six months with an option to extend the lease for three additional years. Due to required tenant improvements to be completed by the landlord, the Company did not take immediate possession of the leased property and the lease term commenced on February 15, 2020. In
F-18
On March 9, 2023, the quarter ended March 31, 2020,Company executed a lease extension agreement on its original 4,642 square footage of office space in Bala Cynwyd, Pennsylvania (which was set to expire in August 2023) and agreed to lease an additional 3,462 square feet of office space from the same landlord.
The lease extension on the original 4,642 square footage commenced on September 1, 2023 and the Company recorded an operating lease right-of-usea right of use asset and operating lease liability of $0.40.5 million.million as of that date.
OnThe new lease on 3,462 additional square footage commenced on October 1, 2023 and the Company recorded a right of use asset and lease liability of $0.3 million as of that date.
The right of use assets and lease liabilities with both these leases are reflected in the financial statements for the year ended December 31, 2023 as are the right of use asset and lease liability of the Company's Boston office space discussed below.
Boston Office Lease
In connection with the Company's 2020 merger with Zafgen described in footnote 1, on May 28, 2020, as part of the Merger with Zafgen, the Company acquired a non-cancellable operating lease for approximately 17,705 square feet of office space (the “Premises”). The lease expires on October 30, 2029.2029. As part of the agreement, the Company is required to maintain a letter of credit, which upon signing was $1.3 million and is classified as restricted cash within the condensed consolidated financial statements. In addition to the base rent, the Company is also responsible for its share of operating expenses, electricity and real estate taxes, which costs are not included in the determination of the leases’ right-of-use assets or lease liabilities. Starting in 2021, theThe right-of-use asset is being amortized to other income income/(expense) over the remaining lease term as a result of the sublease agreement discusseddescribed below.
On October 27, 2020, the Company entered into a sublease agreement (the “Sublease”) with Massachusetts Municipal Association, Inc. (the “Subtenant”), whereby the Company sublet the entire Premises to the Subtenant. The initial term of the Sublease commenced on December 4, 2020 and continues until October 30, 2029. In connection with the Sublease, the Company evaluated the need for impairment under ASC 360 "Impairment Testing: Long-Lived Assets Classified as Held and Used," and determined there was no impairment.
F-22
The Sublease providesprovided for an initial annual base rent of $0.8 million, which increases annually up to a maximum annual base rent of $1.0 million. The Subtenant also is responsible for paying to the Company future increases in operating costs (commencing on January 1, 2022), future increases in annual tax costs (commencing July 1, 2021) and all utility costs (commencing March 1,2021)1, 2021) attributable to the Premises during the term of the Sublease. As part of the Sublease, the subtenant deposited a letter of credit in the amount of $0.8 million to assure their performance under the sublease. If there are no uncured events of default under the sublease, the amount of this security deposit decreases over time to $0.4 million on the sixth anniversary of the Sublease. The Company records sublease income on the Subleasethis sublease on a straight-line basis as a component of other income income/(expense).
Lab Space
On November 5, 2018, the Company entered into an operating lease for office and lab space in Philadelphia, Pennsylvania, effective as of January 1, 2019, and expiring on December 31, 2020 with an option to extend the lease for each of the two additional years. On August 4, 2020, the Company executed the first option to extend the lease for an additional year, expiring on December 31, 2021. On August 9, 2021, the Company executed the remaining option to extend the lease for an additional year, expiring on December 31, 2022. In January 2023, the Company executed an extension of this lease for an additional year, expiring on December 31, 2023. The Company has determined this lease extension qualifies as a short-term lease and have applied the accounting policy election to not record the related right-of-use asset and lease liabilities.
On October 16, 2023, the Company entered into an operating lease for lab space in King of Prussia, Pennsylvania for a period of four years. Due to required tenant improvements to be completed by the landlord, the Company did not take immediate possession of the leased property and the lease term is expected to commence in the second quarter of 2024, no right of use asset or lease liability will be recorded until the lease commencement date.
Lease Expense
Expense arising from operating leases was $0.30.4 million and $0.70.3 million during the twelve months ended December 31, 20212023 and 2020,2022, respectively. For operating leases, the weighted-average remaining lease term for leases at December 31, 20212023 and 20202022 was 7.65.5 and 8.66.8 years, respectively. For operating leases, the weighted
F-19
average discount rate for leases at December 31, 20212023 and 20202022 was 11.0%. The Company has not entered into any financing leases.
Maturities of lease liabilities due under these lease agreements as of December 31, 20212023 are as follows:
Year Ending December 31, |
| Operating |
|
| Operating |
| ||
(in thousands) | (in thousands) | Leases |
| (in thousands) | Leases |
| ||
2022 |
| $ | 1,197 |
| ||||
2023 |
|
| 1,146 |
| ||||
2024 |
|
| 1,065 |
|
| $ | 1,380 |
|
2025 |
|
| 1,083 |
|
|
| 1,403 |
|
2026 |
|
| 1,101 |
|
|
| 1,328 |
|
2027 |
|
| 1,118 |
| ||||
2028 |
|
| 1,136 |
| ||||
Thereafter |
|
| 3,213 |
|
|
| 959 |
|
Total lease payments |
|
| 8,805 |
|
|
| 7,324 |
|
Less: imputed interest |
|
| (2,803 | ) |
|
| (1,778 | ) |
Present value of lease liabilities |
| $ | 6,002 |
|
| $ | 5,546 |
|
Legal Proceedings
The Company is not currently a party to any litigation, nor is management aware of any pending or threatened litigation against the Company, that it believes would materially affect the Company’s business, operating results, financial condition or cash flows.
During the years ended December 31, 2021,2023, and 2020,2022, the Company recorded 0no income tax benefits for the net operating losses incurred in each year due to its uncertainty of realizing a benefit from those items.
The domestic and foreign components of loss before income taxes are as follows.
|
| Years ended December 31, |
| |||||
|
| 2021 |
|
| 2020 |
| ||
Domestic |
| $ | (50,617 | ) |
| $ | (42,608 | ) |
Foreign |
|
| (19 | ) |
|
| 126 |
|
|
| $ | (50,636 | ) |
| $ | (42,482 | ) |
F-23
|
| Years ended December 31, |
| |||||
|
| 2023 |
|
| 2022 |
| ||
Domestic |
| $ | (36,948 | ) |
| $ | (35,339 | ) |
Foreign |
|
| (1 | ) |
|
| (16 | ) |
| $ | (36,949 | ) |
| $ | (35,355 | ) | |
A reconciliation of the U.S. federal statutory income tax rate to the Company’s effective income tax rate is as follows:
|
| Year Ended December 31, |
|
| Year Ended December 31, |
| ||||||||||
|
| 2021 |
|
| 2020 |
|
| 2023 |
|
| 2022 |
| ||||
Federal statutory income tax rate |
| 21.0 | % |
| 21.0 | % |
|
| 21.0 | % |
|
| 21.0 | % | ||
State taxes, net of federal benefit |
| 3.4 |
| 7.8 |
|
|
| 4.9 |
|
|
| 6.2 |
| |||
Change in state tax rate |
| (14.5 | ) |
| — |
|
|
| (1.7 | ) |
|
| (7.9 | ) | ||
Federal and state research and development tax credit |
| 7.9 |
| 13.5 |
|
|
| 8.2 |
|
|
| 1.1 |
| |||
Nondeductible permanent differences |
| (0.6 | ) |
| (0.2 | ) |
|
| (1.0 | ) |
|
| (1.0 | ) | ||
Stock compensation forfeitures - DTA Write Off |
|
| (1.8 | ) |
|
| — |
| ||||||||
Change in deferred tax asset valuation allowance |
|
| (17.2 | ) |
|
| (42.1 | ) |
|
| (29.6 | ) |
|
| (19.3 | ) |
Effective income tax rate |
|
| 0.0 | % |
|
| 0.0 | % |
|
| 0.0 | % |
|
| 0.0 | % |
F-20
Net deferred tax assets as of December 31, 20212023 and 20202022 consisted of the following:
|
| 2021 |
|
| 2020 |
|
| 2023 |
|
| 2022 |
| ||||
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Capitalized R&D Expenses |
| $ | 65,781 |
| $ | 73,014 |
| |||||||||
Stock Based Compensation |
| 1,458 |
| 490 |
| |||||||||||
Net operating Loss Carryforwards |
| 43,219 |
| 32,081 |
| |||||||||||
Capitalized R&D - acquired in merger with Zafgen |
| $ | 66,678 |
|
| $ | 66,941 |
| ||||||||
Capitalized R&D - Section 174 Costs post 2021 |
|
| 9,411 |
|
|
| 5,019 |
| ||||||||
Stock based compensation |
|
| 3,490 |
|
|
| 2,709 |
| ||||||||
Net operating loss carryforwards |
|
| 45,195 |
|
|
| 42,204 |
| ||||||||
Tax credit carryforwards |
| 12,912 |
| 8,904 |
|
|
| 16,319 |
|
|
| 13,284 |
| |||
Other Temporary Differences |
| 24 |
| 50 |
| |||||||||||
Fixed Assets & Intangibles |
| 44 |
| 75 |
| |||||||||||
Operating Lease Liability |
|
| 1,477 |
|
|
| 1,786 |
| ||||||||
Other temporary differences |
|
| 21 |
|
|
| 20 |
| ||||||||
Accruals & reserves |
|
| 19 |
|
|
| 19 |
| ||||||||
Fixed assets & intangibles |
|
| 107 |
|
|
| 81 |
| ||||||||
Operating lease liability |
|
| 1,298 |
|
|
| 1,355 |
| ||||||||
Total deferred tax assets |
| $ | 124,915 |
| $ | 116,400 |
|
| $ | 142,538 |
|
| $ | 131,632 |
| |
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Operating Right of Use Asset |
|
| (899 | ) |
|
| (1,081 | ) | ||||||||
Operating right of use asset |
|
| (768 | ) |
|
| (801 | ) | ||||||||
Total deferred tax liabilities |
| $ | (899 | ) |
| $ | (1,081 | ) |
| $ | (768 | ) |
| $ | (801 | ) |
Less: Valuation allowance |
| $ | (124,016 | ) |
| $ | (115,319 | ) |
| $ | (141,770 | ) |
| $ | (130,831 | ) |
Net deferred tax assets / (liabilities) |
| $ | 0 |
|
| $ | 0 |
|
| $ | — |
|
| $ | — |
|
Changes in the valuation allowance for deferred tax assets during the year ended December 31, 20212023 and 2022 related primarily to the increase in net operating loss carryforwards and tax credit carryforwards and a decrease other deferred tax assets associated with a reduction in the Company’s effective state rate. The change in the valuation allowance during the year ended December 31, 2020 related primarily to deferred tax assets acquired as a result of the Merger with Zafgen, increase in net operating loss carryforwards, and tax credit carryforwards, Changes to the valuation allowance were as follows:
|
| Years ended December 31, |
|
| Years ended December 31, |
| ||||||||||
|
| 2021 |
|
| 2020 |
|
| 2023 |
|
| 2022 |
| ||||
Valuation allowance as of the beginning of the year |
| $ | 115,319 |
| $ | 11,735 |
|
| $ | 130,831 |
|
| $ | 124,016 |
| |
Increases acquired as a result of the Merger with Zafgen |
| — |
| 85,689 |
| |||||||||||
Increases recorded to income tax provision |
|
| 8,697 |
|
|
| 17,895 |
|
|
| 10,939 |
|
|
| 6,815 |
|
Valuation Allowance at end of Year |
| $ | 124,016 |
|
| $ | 115,319 |
| ||||||||
Valuation allowance at end of year |
| $ | 141,770 |
|
| $ | 130,831 |
| ||||||||
As of December 31, 2021,2023, the Company had net operating loss carryforwards that expire for federal, foreign and state income tax purposes of $158.8179.0 million, $1.2 million and $131.3148.1 million, respectively. The federal and state operating losses begin to expire in 2026 and 2030, while the foreign net loss carryforward can be carried forward indefinitely. As of December 31, 2021,2023, the Company had federal net operating loss carryforwards that were generated after December 31, 2017 of $120.0140.3 million that do not expire, however these carryforwards are limited to 80% of the taxable income in any one tax period. As of December 31, 2021,2023, the Company also had available tax credit carryforwards for federal and state income tax purposes of $12.916.3 million which begin to expire in 2039. Utilization of the pre-Merger net operating loss carryforwards attributable to Zafgen, of approximately $33.5 million, are subject to a substantial annual limitation under Section 382 of the Internal Revenue Code of 1986 due to ownership changes occurred during the tax year associated with the Merger. In addition to the limitation of the
F-24
pre-Merger NOL’s of Zafgen, the net capitalized R&D deferred tax assets in the amount of $73.066.9 million is subject to the built-in loss rules under Section 382 and may not be realized if the underlying asset associated with the R&D is disposed within five years of the Merger, or May 28, 2025. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of certain stockholders or public groups in the stock of a corporation by more than 50% over a three-year period. The ownership changes will limit the amount of pre-merger Zafgen carryforwards that can be utilized annually to offset future taxable income with an annual limitation of approximately $35 thousand per year. The Company has reduced their NOL and R&D tax credit deferred tax assets associated with the pre-Merger Zafgen operations as a result of the 382 analysis.
F-21
The Company believes that as a result of its merger with Zafgen, its ability to utilize NOLs acquired in the transaction and our other NOLs is expected to be severely limited by Section 382 of the Code. Additionally, the Company’s July 2021, September 2022 and February 2023 equity transactions could also limit its ability to utilize NOLs in the future. The Company has not conducted a study to assess whether a change of control has occurred or whether there have been multiple changes of control since inception associated with the pre-Merger Chondrial tax attributes.inception. If the Company experienced a change of control, as defined by Section 382, at any time since inception, utilization of the pre-Merger Chondrialits net operating loss carryforwards or tax credit carryforwards would be subject to an annual limitation under Section 382, which is determined by first multiplying the value of the Company’s stock at the time of the ownership change by the applicable long-term tax-exempt rate, and then could be subject to additional adjustments, as required. Additionally, our July 2021, September 2022 and February 2024 equity transactions could also limit our ability to utilize NOLs in the future. Any limitation may result in expiration of a portion of the net operating loss carryforwards or tax credit carryforwards before utilization. Further, until a study is completed, and any limitation is known, no amounts are being presented as an uncertain tax position.
The Tax Cuts and Jobs Act ("TCJA") requires taxpayers to capitalize and amortize research and experimental expenditures under IRC Section 174 for tax years beginning after December 31, 2021. This rule became effective for the Company during the year ended December 31, 2022 and resulted in the capitalization of research and development costs in both 2023 and 2022. The Company is amortizing these costs for tax purposes over five years if the research and development was performed in the U.S. and over 15 years if the research and development was performed outside the U.S.
As of December 31, 20212023 and 2020,2022, the Company’s net deferred tax asset balance before the valuation allowance was $124.0141.8 million and $115.3130.8 million, respectively, and was comprised principally of net operating loss carryforwards, capitalized research and development expenses and tax credit carryforwards. During the years ended December 31, 20212023 and 2020,2022, gross deferred tax assets increased due to deferred tax assets acquired as a result of the Merger with Zafgen, additional net operating loss carryforwards and research and development tax credits generated.
The Company has evaluated the positive and negative evidence bearing upon its ability to realize the deferred tax assets. Management has considered the Company’s history of cumulative net losses incurred since inception and its lack of commercialization of any products or generation of any revenue from product sales since inception and has concluded that it is more likely than not that the Company will not realize the benefits of the deferred tax assets. Accordingly, a full valuation allowance has been established against the deferred tax assets as of December 31, 20212023 and 2020.2022. Management reevaluates the positive and negative evidence at each reporting period.
The Company has 0not recorded any amounts for unrecognized tax benefits as of December 31, 20212023 and 2020.2022. The Company files tax returns as prescribed by the tax laws of the jurisdictions in which it operates. In the normal course of business, the Company is subject to examination by federal and state jurisdictions, where applicable. There are currently no pending income tax examinations. The Company’s tax years are still open under statute from 20162019 to the present. Earlier years may be examined to the extent that tax credit or net operating loss carryforwards are used in future periods. The Company’s policy is to record interest and penalties related to income taxes as part of its income tax provision.
In November 2016, the Company entered into a consulting agreement with Mark Payne, M.D (the “Consulting Engagement”). Dr. Payne was a director of Chondrial at that time, a full-time employee of IU and one of the inventors of the licensed IU intellectual property, and as such is entitled to a certain share of the revenues received by IU under the IU License. Pursuant to the terms of his consulting agreement the Company agreed to pay Dr. Payne $0.1 million per year over the term of the agreement and granted Dr. Payne 123,853 restricted Common Units in Holdings. On November 30, 2016, 30% immediately vested and was associated with Chondrial Therapeutics IP, LLC (“IP LLC”) becoming a subsidiary of Holdings, which was subsequently contributed to the Company on December 31, 2018. The remaining 70% vested ratably over 48 months beginning on December 1, 2016. The consulting agreement had a four-year term, subject to earlier termination. On November 30, 2020, the Company entered into a 1-month extension of the Consulting Engagement, expiring on December 31, 2020.On January 1, 2021, the Company entered into a new consulting agreement with Mark Payne, M.D. which extended the term of the Consulting Engagement for a four-year term beginning on January 1, 2021. During each of the twelve months
F-25
ended December 31, 2021 and 2020, the Company recognized $0.1 million, related to this consulting agreement, recorded as research and development expense in the Statement of Operations.
The funding to the Company originated from Holdings’ sale of Series A Preferred Units and Series B convertible preferred units (the "Units") with Deerfield Private Design Fund IV, L.P., Deerfield Private Design Fund III, L.P. and Deerfield Health Innovations Fund, L.P. (together, the “Deerfield Funds”), and certain other purchasers, from inception through May 28, 2020 and the contribution of the proceeds received by Holdings on such sales to the Company in order to fund the Company’s operations.
Under a November 30, 2016 Series A Preferred Unit Purchase Agreement, as amended on September 8, 2017, November 15, 2017, November 14, 2018 and April 29, 2019, Holdings sold Series A Preferred Units for gross proceeds of $35.6 million. The gross proceeds of $35.6 million were contributed to the Company.
On November 21, 2019 (as amended on December 20, 2019), Holdings entered into a Second Amended and Restated LLC Agreement and entered into a Series B Bridge Unit Purchase Agreement with the Deerfield Funds and certain other purchasers to sell Series B convertible preferred units (“Series B Bridge Units”) for gross proceeds of up to $10.0 million. The gross proceeds of $10.0 million were contributed to the Company.
On January 16, 2020, Holdings entered into a Third Amended and Restated LLC Agreement and entered into a Second Series B Bridge Unit Purchase Agreement with the Deerfield Funds and certain other purchasers to sell Second Series B convertible preferred units (“Second Series B Bridge Units”) for gross proceeds of up to $15.0 million. The gross proceeds of $11.4 million were contributed to the Company.
During the twelve months ended December 31, 2020, Holdings provided the Company non-interest bearing, permanent funding from the above Series A and Series B preferred unit transactions, totaling $18.0 million which has been recorded as capital contributions with the balance of combined equity and additional paid in capital on the consolidated balance sheets and consolidated statements of changes in stockholders’ equity for each respective period. No contributions were made by Holdings subsequent to the Merger.
During 2020, the Company purchased a piece of laboratory equipment for $0.5 million from a supplier of which one the Company's directors is also a current director. During 2021, the Company purchased a piece of laboratory equipment and lab supplies for a cumulative $0.1 million from the same supplier.F-22
F-26