•a license to Janssen Pharmaceuticals, Inc., or Janssen Pharmaceuticals, an affiliate of Janssen, for the research, development and commercialization of products based on specialized oligonucleotide backbone chemistry and novel amidates for disorders, excluding cancers originating from the blood or bone marrow. In connection with this license, we also granted to Janssen Pharmaceuticals a non‑exclusive worldwide license under our patent rights covering the synthesis of monomers, which are the building blocks of oligonucleotides. These non‑exclusively licensed patent rights are licensed exclusively to Janssen under the Collaboration Agreement for the imetelstat program, and our license with Janssen Pharmaceuticals expressly excludes products whose predominant or primary mechanism of action is telomerase inhibition and is subject to the rights and licenses granted to Janssen under the Collaboration Agreement;
licenses to several biotechnology and pharmaceutical companies to use or commercialize telomerase immortalized cells in drug discovery research;
•licenses to several companies to commercialize telomerase immortalized cells for drug discovery applications;•licenses to several companies to sell antibodies specific to telomerase for research purposes;
•licenses to several companies to develop and commercialize reagent kits, or to provide services, for the measurement of telomere length or telomerase activity for research purposes;
•a license to a company to develop and commercialize a particular
telomerasetelomerase‑based technology for cancer detection; and•a license to a company for the development of cancer immunotherapies for veterinary applications.
See Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Revenues” for a further discussion of revenues from our license agreements. We expect revenues under our license agreements related to our telomerase technology to decline significantly in the coming years, and to be eliminated by the end of 2019, due to upcoming patent expirations on such technology.
Concentration of Revenues
Our revenues were $1.1 million, $6.2 million and $36.4 million for the years ended December 31, 2017, 2016 and 2015, respectively. We operate in one operating segment and have operations solely in the United States. All of our long‑lived assets are maintained in the United States. See Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Revenues” for additional detail regarding the composition of our revenues.
Research and Development
Our research and development costs were $11.0 million, $18.0 million and $17.8 million for the years ended December 31, 2017, 2016 and 2015, respectively. See Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Research and Development Expenses” for additional detail regarding our research and development activities.
A typical sequence of steps in the manufacture of imetelstat drug product includes the following key components:
starting materials, which are well‑defined raw materials that are used to make bulk drug substance;
bulk drug substance, which is the active pharmaceutical ingredient in a drug product that provides pharmacological activity or other direct effect in the treatment of disease; and
final drug product, which is the finished dosage form that contains the drug substance that is shipped to the clinic for patient treatment.
In accordance with the Collaboration Agreement, Janssen is now responsible for the manufacture and management of the supply of imetelstat on a global basis for clinical trials and, after any regulatory approval, all commercial activities. Consequently, we are, and expect to remain, dependent on Janssen to appropriately supply imetelstat and other clinical trial materials. Currently, third‑party contractors perform certain process development and other technical and scientific work with respect to imetelstat, as well as supply starting materials and manufacturing drug substance and drug product. Janssen does not have direct control over third‑party personnel or operations. These third‑party contractors, and/or any other contractors that Janssen may rely upon for the manufacture and/or supply of imetelstat, typically complete their services on a proposal by proposal basis under master supply agreements and may need to make substantial investments to enable sufficient capacity increases and cost reductions, and to implement those regulatory and compliance standards necessary for successful Phase 3 clinical trials and commercial production. These third‑party contractors, and/or any other contractors that Janssen may rely upon for the manufacture and/or supply of imetelstat, may not be able to achieve such capacity increases, cost reductions, or regulatory and compliance standards, and even if they do, such achievements may not be at a commercially reasonable cost. Janssen is responsible for establishing any long‑term commitments or commercial supply agreements with any of the third‑party contractors for imetelstat. The information provided in this section should be reviewed in the context of the section entitled “Risks Related to Manufacturing” under Item 1A, “Risk Factors”.
Consultants
We have consulting agreements with drug development professionals, clinicians and regulatory experts with experience in numerous fields, including oncology and drug regulations. We retain each consultant according to the terms of a consulting agreement. Under such agreements, we pay them a consulting fee and reimburse them for out‑of‑pocket expenses incurred in performing their services for us. In addition, some consultants hold options to purchase our common stock, subject to the vesting requirements contained in the consulting agreements. Our consultants may be employed by other entities and therefore may have commitments to their employer, or may have other consulting or advisory agreements that may limit their availability to us.
The pharmaceutical and biotechnology industries are intensely competitive. Other pharmaceutical and biotechnology companies and research organizations currently engage in or have in the past engaged in efforts related to the biological mechanisms related to imetelstat,
includingthe study of telomeres, telomerase,andor our proprietary oligonucleotide chemistry, and the research and development of therapies for the treatment of hematologic myeloid malignancies. In addition, other products and therapies that could directly competedirectlywith imetelstat currently exist or are being developed by pharmaceutical and biopharmaceutical companies and by academic institutions, government agencies and other public and private research organizations.Our sole product candidate, imetelstat,We expect Janssen’s decisions regarding continued development and/or commercialization, ifapproved for marketing, will face significant competition from approved drugs, drugs currently under development and any other drugs that may be subsequently approved. Imetelstat would have to compete successfully based on efficacy, safety, convenience, price, cost-effectiveness and other relevant factors. In addition, imetelstat would have to compete against other drugs with a variety of decision makers, including physicians, patients, government and private third-party payors, technology assessment groups and patient advocacy organizations. We cannot guarantee that we, in collaboration with Janssen, will be able to compete successfully onany, ofthese factors. If weimetelstat, including completing IMerge and/orJanssen cannot compete successfully on anyIMbark, its Continuation Decision or the termination ofthe factors described previously, Janssen may terminatethe Collaboration Agreement,and our business may fail.Imetelstat is likelyto be informed inhighly competitive markets. We are awarepart by what Janssen believes is the estimated commercial potential ofproducts in researchimetelstat for the treatment of hematologic malignancies, such as MF ordevelopment by our competitors that address the diseases we are targeting, and any of these products may compete with imetelstat. Our competitors may succeed in developing their products before we do, obtaining approvals from the FDA or other regulatory agencies for their products more rapidly than we do, or developing products that are more effective than imetelstat. These products or technologies might render our technology obsolete or noncompetitive. There may also be product candidates of which we are not aware at an earlier stage of development that may compete with imetelstat. In addition, imetelstat may need to compete or combine with existing therapies, many with long histories of use.MDS.Many companies are developing alternative therapies to treat hematologic myeloid
malignancies and, in this regard, are competitors of ours and Janssen.malignancies. For example, if approved for commercial sale for the treatment of MF, imetelstat would compete against IncyteCorporation'sCorporation’s ruxolitinib, orJakafi®Jakafi®, which is orally administered. In clinical trials,Jakafi®Jakafi® reduced spleen size, abdominal discomfort, early satiety, bone pain, night sweats and itching in MF patients. Recently, there have also been reports of overall survival benefit as well as improvement in bone marrow fibrosis fromJakafi®Jakafi® treatment. Other treatment modalities for MFinclude:•- include hydroxyurea for the management of splenomegaly, leukocytosis, thrombocytosis and
constitutional symptoms;
•- splenectomy and splenic irradiation for the management of splenomegaly and
co-existingco‑existing cytopenias, or low blood cell counts;and •- chemotherapy and pegylated interferon.
Drugs for the treatment of
MF-associatedMF‑associated anemia includeerythropoiesis-stimulatingerythropoiesis stimulating agents, androgens, danazol, corticosteroids, thalidomide and lenalidomide. There are other investigational treatments for MF furtheradvancedalong in development than imetelstat, such asmomelotinib by Gilead Sciences, Inc. andpacritinib byCell Therapeutics,CTI Biopharma Corporation, or CTI Biopharma and fedratinib by Impact Biomedicines, Inc., acquired by Celgene Corporation, or Celgene, whichare currently inhave reported results from Phase 3 clinicaltrials, and othertrials. Other investigational treatments for MF include inhibitors of theJAK-STATJAK‑STAT pathway,as well as several investigational treatments in early phase testingsuch as NS‑018 by NS Pharma, Inc.; histone deacetylaseinhibitors,inhibitors; interleukin‑3 receptor targeted agents; inhibitors of heat shock protein90,90; hypomethylatingagents,agents; PI3 Kinase and mTORinhibitors,inhibitors; anti‑fibrosis antibodies such as PRM-151 from Promedior, Inc.; hedgehoginhibitors, anti-LOX2 inhibitors,and SMO inhibitors; PIM kinase inhibitors; IAP inhibitors; anti‑LOX2 inhibitors; recombinant pentraxin 2protein, KIP-1 activators, TGF-betaprotein; KIP‑1 activators; TGF‑beta superfamily inhibitors, such as sotatercept and luspatercept by Acceleron Pharma, Inc., or Acceleron, in collaboration with Celgene; FLTinhibitors,inhibitors; and other tyrosine kinase inhibitors.If approved for commercial sale for the treatment of lower risk MDS, imetelstat would compete against a number of treatment options, including erythropoiesis stimulating agents and other hematopoietic growth factors; immunomodulators, such as lenalidomide by Celgene; hypomethylating agents, such as azacitidine by Celgene and decitabine by Janssen; in addition to investigational treatments in MDS that may be further along in development than imetelstat, such as oral versions of azacitidine; histone deacetylase inhibitors; TGF‑beta superfamily inhibitors, such as luspatercept by Acceleron, in collaboration with Celgene; PI3 Kinase inhibitors; aminopeptidase inhibitors, such as tosedostat by CTI Biopharma; TLR2‑specific antibodies; anti‑CD33 antibodies; anti-CD38 antibodies, such as daratumumab by Genmab A/S in collaboration with Janssen; anti-CD123 antibodies, such as talacotuzumab by Janssen; retinoic acid receptor alpha agonists, such as SY-1425 by Syros Pharmaceuticals; hypoxia-inducible factor prolyl hydroxylase inhibitors, such as roxadustat by FibroGen, Inc.; Fas ligand inhibitors; and JAK‑STAT pathway inhibitors.
Independently, Janssen is developing therapies for hematologic malignancies, including AML, MDS,
MMmultiple myeloma andABC-subtypeABC‑subtype diffuse largeB-cellB‑cell lymphoma. Molecular and cellular pathways of interest include:•cell surface targets for
immune-directedimmune‑directed therapy;•immune checkpoint inhibition;
•leukemia stem cells;
•pathway addiction (genetic alterations,
cell-typecell‑type specific pathways);•conditional sensitivity (stress,
protein-producingprotein‑producing tumors);•targeting of
T-cellsT‑cells and natural killer"NK"“NK” cells to tumors;•identification of novel
tumor-specifictumor‑specific antigens; and•Success by Janssen in any of these approaches may compete with imetelstat or render imetelstat obsolete or
noncompetitive. Anoncompetitive, which could lead to a decision by Janssen to discontinue the imetelstat program and terminate the Collaboration Agreement, which would materially and adversely affect our business and businessprospects.prospects and might cause us to cease operations.Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. We anticipate increased competition in the future as new companies explore treatments for hematologic myeloid malignancies, which may significantly impact the commercial viability of imetelstat. Academic institutions, government agencies and other public and private research organizations may also conduct research, seek patent protection and establish collaborative arrangements for research, clinical development and marketing of products similar to
ours.imetelstat. These companies and institutions compete with us in recruiting and retaining qualifiedscientificdevelopment and management personnel as well as in acquiring technologies complementary toourthe imetelstat program.In addition to the above factors,
we and Janssen expect toimetelstat will face competitionin the following areas:based on:•product efficacy and safety;
•convenience of product administration;
•cost of manufacturing;
•the timing and scope of regulatory consents;
•status of
reimbursement coverage;•- coverage and reimbursement;
price; and
•patent position, including potentially dominant patent positions of others.
As a result of the foregoing, competitors may develop more commercially desirable or affordable products than imetelstat, or achieve earlier patent protection or product commercialization than us or Janssen. Competitors have developed, or are in the process of developing, technologies that are, or in the future may be, competitive to imetelstat. Some of these products may have an entirely different approach or means of accomplishing therapeutic effects similar or superior to those that may be demonstrated by imetelstat. Competitors may develop products that are safer, more effective, or less costly than imetelstat, or more convenient to administer to patients and, therefore, present a serious competitive threat to imetelstat. In addition, competitors may price their products below what Janssen may determine to be an acceptable price for imetelstat, may receive better
third-partythird‑party payor coverage and/or reimbursement, or may be morecostcost‑effective than imetelstat. Such competitive products or activities by competitors may render imetelstat obsolete, which may cause Janssen to terminate the Collaboration Agreement,andwhich would severely and adversely affect our financial results, businessprospects.and business prospects, and the future of imetelstat, and might cause us to cease operations.Government Regulation
Regulation by governmental authorities in the United States and other countries is a significant factor in the development, manufacture and marketing of
our sole product candidate,imetelstat, which is being developed in collaboration with Janssen.We anticipate that imetelstatImetelstat will require regulatory approval by governmental agencies prior to commercialization. In particular, potential human therapeutic products, such as imetelstat, are subject to rigorous preclinical and clinical testing and other approval procedures of the FDA and similar regulatory authorities in European and other countries. Various governmental statutes and regulations also govern or influence testing, manufacturing, safety, labeling, storage, import, export, distribution and recordkeeping related to such products and their marketing. In collaboration with Janssen, the process of obtaining these approvals and the subsequent compliance with appropriate statutes and regulations require the expenditure of substantial time and money, and there can be no guarantee that approvals will be granted.TableThe information provided in this section should be reviewed in the context ofContentsthe section entitled “Risks Related to Clinical Development, Regulatory Approval and Commercialization of Imetelstat” under Item 1A, “Risk Factors”.United States Food and Drug Administration Regulatory Approval Process
Prior to commencement of clinical trials involving humans, preclinical testing of new pharmaceutical products is generally conducted on animals in the laboratory to evaluate the potential efficacy and safety of a product candidate. The results of these trials are submitted to the FDA as part of an Investigational New Drug, or IND, application, which must
be cleared by the FDAbecome effective before clinical testing in humans can begin. The FDA can place an IND on clinical hold at any time, which prevents the conduct of clinical trials under the IND until safety concerns are addressed by the IND sponsor to the FDA’s satisfaction. Typically, clinical evaluation involves a time consuming and costly three phase trial process. In Phase 1, clinical trials are conducted with a small number of healthy volunteers or patients afflicted with a specific disease to assess safety and to evaluate the pattern of drug distribution and metabolism within the body. In Phase 2, clinical trials are conducted with groups of patients afflicted with a specific disease in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. The Phase 2 trials can be conducted comparing the investigational treatment to a comparator arm, or not. If used, a comparator usually includes standard of care therapy. Safety and efficacy data from Phase 2 clinical trials, even if favorable, may not provide sufficient rationale for proceeding to a Phase 3 clinical trial. In Phase 3, large scale,multimulti‑center, comparative trials are conducted with patientsafflicted with a target disease to provide sufficient data to demonstrate the efficacy and safety required by the FDA. The FDA closely monitors the progress of each of the three phases of clinical testing and may, at its discretion,
re-evaluate,re‑evaluate, alter, suspend, or terminate the trials. Human clinical trials must be conducted in compliance with Good Clinical Practice regulations and applicable laws, with the oversight of Institutional Review Boards for the protection of human subjects. The manufacture of drug product candidates is subject to requirements that drugs be manufactured, packaged and labeled in conformity with current Good Manufacturing Practices and applicable laws.The results of the preclinical and clinical testing of
small moleculesdrugs andmany biologic drugscomplete manufacturing information are submitted to the FDA in the form of a New Drug Application, or NDA, for review and for approval prior to commencement of commercial sales.InSubmission of an NDA requires thecasepayment ofblood products, vaccines, or gene and cell therapies, the results of clinical trials are submitteda substantial user fee to the FDA,as a Biologics License Application, or BLA.which may be waived in certain cases. In responding to anNDA/BLANDA submission, the FDA maygrant a marketing authorization,approve the drug for commercialization, impose limitations on its indications for use and labeling, including in the form of Risk Evaluation and Mitigation Strategies or may issue amarketing authorization, request additional information, deny the applicationcomplete response letter. Even ifit determines that the application does not provideanadequate basis for approval, or refuseNDA is approved, its sponsor is subject toreview an application that has been submitted if it determines that the application does not provide an adequate basis for filingongoing andreview.pervasive regulatory compliance requirements.European and Other Regulatory Approval Process
Prior to initiating clinical trials in a region outside of the United States, a clinical trial application must be submitted and reviewed by the appropriate regulatory authority regulating the country in which the trial will be conducted. Whether or not FDA clearance or approval has been obtained, approval of a product by comparable regulatory authorities in Europe and other countries is necessary prior to commencement of marketing the product in such countries. The regulatory authorities in each country may impose their own requirements and may refuse to grant an approval, or may require additional data before granting it, even though the relevant product has been cleared or approved by the FDA or another authority. As with the FDA, the regulatory authorities in the European Union, or EU, and other developed countries have lengthy approval processes for pharmaceutical products. The process for gaining approval in particular countries varies, but generally follows a similar sequence to that described for FDA approval. In Europe, the European Medicine Agency, or EMA, and the European Committee for Proprietary Medicinal Products, or CPMP, provide a mechanism for EU member states to exchange information on all aspects of product licensing. The EU has established the EMA for the evaluation of medical products, with both a centralized procedure with which the marketing authorization is recognized in all EU member states and a decentralized procedure, the latter being based on the principle of licensing within one member country followed by mutual recognition by the other member countries.
Table of ContentsOrphan Drug DesignationOther RegulationsWe are also subjectFor a drug tovariousqualify for orphan drug designation by the FDA, both the drug andoften changing federal, state, local and international laws, rules, regulations, guidelines and recommendations relating to safe working conditions and manufacturing practices.ManufacturingA typical sequence of stepsthe disease or condition must meet certain criteria specified in themanufactureOrphan Drug Act, or ODA, and FDA’s implementing regulations. Orphan drug designation is granted by the FDA’s Office ofimetelstatOrphan Drug Products in order to support development of medicines for underserved or rare diseases and patient populations that affect fewer than 200,000 people in the United States or, if the disease or condition affects more than 200,000 individuals annually in the United States, if there is no reasonable expectation that the cost of developing and making the drugproduct includeswould be recovered from sales in thefollowing key components:•starting materials, which are well-defined raw materials that are used to make bulkUnited States. Orphan drugsubstance;•bulkdesignation qualifies the sponsor of the drugsubstance, whichfor various development incentives of the ODA, including, if regulatory approval is received, theactive pharmaceutical ingredient inpotential for seven years of market exclusivity with certain limited exceptions and certain tax credits for qualified clinical testing. A marketing application for a prescription drug product that has received orphan drug designation is not subject to a prescription drug user fee unless the application includes an indication for a disease or condition other than the rare disease or condition for which the drug was granted orphan drug designation. The granting of orphan drug designation does not alter the standard regulatory requirements and process for obtaining marketing approval. The safety and effectiveness of a drug must be established through adequate and well‑controlled studies. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition.On June 11, 2015 and December 23, 2015, the FDA granted orphan drug designation to imetelstat for the treatment of MF and MDS, respectively.
Orphan drug designation by the European Commission provides
pharmacological activityregulatory and financial incentives for companies to develop and market therapies that treat a life‑threatening orotherchronically debilitating condition affecting no more than five in 10,000 persons in the EU, and where no satisfactory treatment is available. In the EU, orphan drug designation also entitles a party to financial incentives such as reduction of fees or fee waivers, as well as protocol assistance from the EMA during the product development phase, and directeffectaccess to the centralized authorization procedure. In addition, ten years of market exclusivity is granted following drug product approval, meaning that another application for marketing authorization of a later similar medicinal product for the same therapeutic indication will generally not be approved in the EU. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable to not justify maintenance of market exclusivity.On December 14, 2015, the EMA granted orphan drug designation to imetelstat for the treatment of MF.
Fast Track Designation
Fast Track designation provides opportunities for frequent interactions with FDA review staff, as well as eligibility for priority review, if relevant criteria are met, and rolling review. Fast Track designation is intended to facilitate and expedite development and review of a New Drug Application to address unmet medical needs in the treatment of
disease;serious or life-threatening conditions. However, Fast Track designation does not accelerate conduct of clinical trials or mean that the regulatory requirements are less stringent, nor does it ensure that imetelstat will receive marketing approval or that approval will be granted within any particular timeframe. In addition, the FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data emerging from the imetelstat clinical development program.In October 2017, the FDA granted Fast Track designation to imetelstat for the treatment of adult patients with transfusion-dependent anemia due to Low or Intermediate-1 risk MDS who are non-del(5q) and
•finalwho are refractory or resistant to treatment with an ESA. Janssen sponsored the application for Fast Track designation using preliminary data from Part 1 of IMerge.Fraud and Abuse, Data Privacy and Security, and Transparency Laws and Regulations
We may also be subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which we conduct our business. These additional healthcare regulations could affect our current and future arrangements with healthcare professionals, principal investigators, consultants, customers and third‑party payors. Such laws include, without limitation, state and federal anti‑kickback, fraud and abuse, false claims, privacy and security, and physician payment sunshine laws.
The federal Anti‑Kickback Statute makes it illegal for any person or entity, including a prescription drug
product, which is the finished dosage form that contains the drug substancemanufacturer (or a party acting on its behalf) to knowingly and willfully, directly or indirectly, solicit, receive, offer, or pay any remuneration that isshippedintended to induce the referral of business, including the purchase, order, or lease of any good, facility, item or service for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. The term “remuneration” has been broadly interpreted to include anything of value. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the Anti‑Kickback Statute has been violated. The Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act, collectively the Affordable Care Act or ACA, among other things, amended the intent requirement of the federal Anti‑Kickback Statute such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate, in order to commit a violation.Federal civil and criminal false claims and false statement laws, including the federal civil False Claims Act and whistleblower or qui tam actions, prohibit, among other things, any person or entity from knowingly presenting, or causing to be presented, for payment to, or approval by, federal programs, including Medicare and Medicaid, claims for items or services, including drugs, that are false or fraudulent or not provided as claimed. Entities can be held liable under these laws if they are deemed to “cause” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers, promoting a product off‑label, or for providing medically unnecessary services or items. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Criminal prosecution is also possible for making or presenting a false, fictitious or fraudulent claim to the
clinicfederal government.The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created additional federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third‑party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for
patient treatment.
UnderHIPAA, as amended by theCollaboration Agreement, afterHealth Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, imposes certain requirements relating to the privacy, security, electronic exchange and breach reporting of individually identifiable health information upon entities subject to the law, such as health plans, healthcare clearinghouses and healthcare providers and their respective business associates that perform services for them that involve individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in U.S. federal courts to enforce the federal HIPAA laws and seek attorneys' fees and costs associated with pursuing federal civil actions.The federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or CMS, information related to payments or other transfers of value made to physicians and teaching hospitals, and applicable manufacturers and applicable group purchasing organizations to report annually to CMS ownership and investment interests held by physicians and their immediate family members.
Analogous state and foreign laws and regulations, such as state anti‑kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non‑governmental third party payors, including private insurers. Additionally, we may be subject to state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government. Further, we may be subject to state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians, other healthcare providers and healthcare entities, or marketing expenditures, as well as state and foreign laws governing the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
If our operations are found to be in violation of any of these federal, state or foreign laws or regulations, we may be subject to penalties, including without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, individual imprisonment, exclusion from participation in government healthcare programs, reputational harm, diminished profits and future earnings, additional reporting requirements and oversight if we become subject to a
transition period, Janssen will be responsible forcorporate integrity agreement or similar agreement to resolve allegations of non‑compliance with these laws or themanufacture and/curtailment orsupplyrestructuring of our operations.Reimbursement and Healthcare Reform
Significant uncertainty exists as to the coverage and reimbursement status of any product candidate that receives regulatory approval. In the United States and markets in other countries, sales of imetelstat, if approved for commercial sale, will depend, in part, on
a global basisthe extent to which third‑party payors provide coverage and establish adequate reimbursement levels forclinical trialsimetelstat.In the United States, third‑party payors include federal and
after any regulatory approval, all commercial activities. Consequently, we will be, and expect to remain, dependent on Janssen to appropriately supply imetelstat. Currently, third-party contractors perform certain process developmentstate healthcare programs, government authorities, private managed care providers, private health insurers and othertechnicalorganizations. There has been increasing legislative andscientific workenforcement interest in the United States with respect to specialty drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed and enacted legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the cost of drugs under Medicare, and reform government program reimbursement methodologies for drugs. At the federal level, Congress and the Trump Administration have each indicated that they will continue to seek new legislative and/or administrative measures to control drug costs. At the state level, legislatures haveincreasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, to encourage importation from other countries and bulk purchasing. Further, third‑party payors are increasingly challenging the price, examining the medical necessity and reviewing the cost‑effectiveness of medical drug products and medical services, in addition to questioning their safety and efficacy. Such payors may limit coverage to specific drug products on an approved list, also known as a formulary, which might not include all of the FDA‑approved drugs for a particular indication. Janssen may need to conduct expensive pharmaco‑economic studies in order to demonstrate the medical necessity and cost‑effectiveness of imetelstat, in addition to
supplying starting materials and manufacturing drug substance and drug product. We or Janssen do not have direct control over their personnel or operations. These third-party contractors, and/or any other contractors that Janssen may rely upon forthemanufacture and/or supply of imetelstat, typically complete their services on a proposal by proposal basis under master supply agreements and may needcosts required tomake substantial investments to enable sufficient capacity increases and cost reductions, and to implement those regulatory and compliance standards necessary for successful Phase 3 clinical trials and commercial production. These third-party contractors, and/or any other contractors that Janssen may rely upon forobtain themanufacture and/or supply ofFDA approvals. Nonetheless, imetelstat may not beable to achieve such capacity increases, cost reductions,considered medically necessary orregulatory and compliance standards, and even if they do, such achievements may not be atcost‑effective.Moreover, the process for determining whether a
commercially reasonable cost. Neither we nor Janssen have any long-term commitments or commercial supply agreements with any of the third-party contractorsthird‑party payor will provide coverage forimetelstat.We currently haveamaster service agreement with two third-party contractors for labeling and packaging of imetelstat finaldrug productandmay be separate from the process fordistributionsetting the price ofimetelstat to clinical sites in North America. In addition, we have agreements with two third-party contractors for release and distribution of imetelstata drug product or for establishing the reimbursement rate that such a payor will pay for the drug product. A payor’s decision toclinical sites in Europe. These third-party contractorsprovideservices oncoverage for aproposal by proposal basis. If requested by Janssen, we may transfer or assign these agreements to Janssen to the extent permissible by the terms of the agreements or agreed with such third-party contractors.We have also entered into quality agreements with our imetelstat bulk drug substance and finaldrug productmanufacturers, and our labeling, packaging and distribution service providers. The master and quality agreements are designeddoes not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination toensureprovide coverage for a drug productquality, compliance with current Good Manufacturing Practices, or cGMP, and oversight of third parties for all critical aspects of imetelstat production, testing, release, labeling and packaging, storage and distribution. If requested by Janssen, we may transfer or assign these agreements to Janssen to the extent permissible by the terms of the agreements or agreed with such third-party contractors.Concentration of RevenuesIn 2014 and 2013, the majority of our revenues were from license fees and royalties under licenses granted to several biotechnology and pharmaceutical companies to use telomerase immortalized cells in drug discovery research and for drug discovery applications. Two customers accounted for approximately 31%, 42% and 59% of our 2014, 2013 and 2012 revenues, respectively. In 2012, the majority of our revenues were from license fees and royalties related to our license and collaboration agreement with GE Healthcare UK, Limited, or GE Healthcare,does not assure that other payors will also provide coverage for thedevelopmentdrug product, as there is no uniform coverage andcommercialization of cellular assay products and our license agreement with Asia Biotech Corporation related to our telomerase activation technology. Upon the closing of the stem cell divestiture under the Contribution Agreement on October 1, 2013, the license agreement with GE Healthcare, including any future revenue payments thereunder, was transferred to Asterias Biotherapeutics, Inc. In December 2012, we assigned our telomerase activation technology to Telomerase Activation Sciences, Inc. and terminated our license agreement with Asia Biotech Corporation. Future royalty obligations by Asia Biotech Corporation under the license agreement have been terminated. We operate in one operating segment and have operations solelyreimbursement policy among third-party payors in the United States.AllAdequate third‑party reimbursement may not be available to enable Janssen to maintain price levels sufficient to realize an appropriate return on Janssen’s and our investment in imetelstat.The United States and some foreign jurisdictions are considering or have enacted legislative and regulatory proposals to contain healthcare costs, as well as to improve quality and expand access. For example, in March 2010, the ACA, was signed into law that included a number of
our long-lived assets are maintainedprovisions of importance to the biopharmaceutical industry. Since January 2017, President Trump has signed two executive orders and other directives designed to delay, circumvent, or loosen certain requirements mandated by the ACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the ACA. Additionally, President Trump signed The Tax Cuts and Jobs Act of 2017 on December 22, 2017, which includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year, that is commonly referred to as the “individual mandate.” Additionally, on January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain ACA-mandated fees. Congress may also consider other legislation to repeal or replace other elements of the ACA.We expect that other healthcare reform measures that may be adopted in the future may result in more rigorous coverage criteria and lower reimbursement, and additional downward pressure on the price that may be charged for imetelstat.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. For example, in August 2011, the Budget Control Act of 2011 was enacted, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for fiscal years 2012 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect beginning on April 1, 2013 and, due to subsequent legislative amendments to the statute, including the Bipartisan Budget Act of 2018, will stay in effect through 2027 unless additional Congressional action is taken. Additionally, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers, including hospitals and imaging centers. More recently, there has been heightened governmental scrutiny in the United
States. Information regarding total revenues, net lossStates to control the rising cost of healthcare. For example, such scrutiny has resulted in several recent congressional inquiries andtotal assets is set forth in our financial statementsproposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the price of drugs under Medicare, and reform government program reimbursement methodologies for drugs, some of which are included inItem 8 of this annual report on Form 10-K.Stem Cell Divestiture; Asterias Series A DistributionBackgroundOn October 1, 2013, we closedthetransaction to divest our human embryonic stem cell assets and our autologous cellular immunotherapy program pursuant to the terms of the previously disclosed asset contribution agreement, or the Contribution Agreement, that we entered into in January 2013 with BioTime, Inc., or BioTime, and BioTime's wholly owned subsidiary, Asterias Biotherapeutics, Inc., or Asterias (formerly known as BioTime Acquisition Corporation) and received 6,537,779 shares of Asterias Series A common stock. In accordance with our contractual obligations under the Contribution Agreement, we distributed all of the shares of Asterias Series A common stock to our stockholders on a pro rata basis, or distributed cash in lieu thereof, which we refer to as the Series A Distribution. We completed the Series A Distribution to eligible stockholders on August 15, 2014 and have no remaining obligationsTrump administration’s budget proposal forthe Series A Distribution. See further discussion in Note 7 on Divestiture of Stem Cell Assets in Notes to Consolidated Financial Statements of this annual report on Form 10-K.In connection with the Contribution Agreement, BioTime made certain contributions to Asterias, including five-year warrants to purchase 8,000,000 shares of BioTime common stock at an exercise price of $5.00 per share, or the BioTime Warrants. Upon the completion of the Series A Distribution, Asterias distributed the BioTime Warrants on a pro rata basis to the holders of Asterias Series A common stock. The BioTime Warrant distribution was completed on October 1, 2014.ConsultantsWe have consulting agreements with a number of leading academic scientists, clinicians and regulatory experts. These individuals serve as key consultants, expert witnesses, or as members of clinical advisory panels with respect to our imetelstat program or in legal proceedings. They also serve as important contacts for us throughout the broader scientific and clinical communities. They are distinguished individuals with expertise in numerous fields, including telomere and telomerase biology, cellular biology, molecular biology, oncology and drug regulations.We retain each consultant according to the terms of a consulting agreement. Under such agreements, we pay them a consulting fee and reimburse them for out-of-pocket expenses incurred in performing their services for us. In addition, some consultants hold options to purchase our common stock and restricted stock awards, subject to the vesting requirements contained in the consultingfiscal year 2019.agreements. Our consultants may be employed by other entities and therefore may have commitments to their employer, or may have other consulting or advisory agreements that may limit their availability to us.Executive Officers of the Company
The following table sets forth certain information with respect to our executive officers as of January 31,
2015:2018:Name
Age
Position
NameAgePositionJohn A. Scarlett, M.D.
6366
President and Chief Executive Officer
Olivia K. Bloom
4649
Executive Vice President, Finance, Chief Financial Officer
and Treasurer
Melissa A. Kelly Behrs
5154
Executive Vice President, Business Development and Portfolio
and& Alliance Management
Andrew J. Grethlein, Ph.D.
5053
Executive Vice President, Development and Technical Operations
Stephen N. Rosenfield, J.D.
6568
Executive Vice President, General Counsel and Corporate Secretary
John A. Scarlett, M.D., has served as our Chief Executive Officer and a director since September 2011 and President since January 2012. Dr. Scarlett has served as a director for Chiasma, Inc., a biopharmaceutical company focused on transforming injectable drugs into oral medications, since February 2015 and CytomX Therapeutics, Inc., a biopharmaceutical company focused on developing antibody therapeutics for the treatment of cancer, since June 2016. Prior to joining Geron, Dr. Scarlett served as President, Chief Executive Officer and a member of the board of directors of Proteolix, Inc., a privately held,
oncology-orientedoncology‑oriented biopharmaceutical company, from February 2009 until its acquisition by Onyx Pharmaceuticals, Inc., anoncology-orientedoncology‑oriented biopharmaceutical company, in November 2009. From February 2002 until its acquisition by Ipsen, S.A. in October 2008, Dr. Scarlett served as the Chief Executive Officer and a member of the board of directors of Tercica, Inc., anendocrinology-orientedendocrinology‑oriented biopharmaceutical company, and also as its President from February 2002 through February 2007. From March 1993 to May 2001, Dr. Scarlett served as President and Chief Executive Officer of Sensus Drug Development Corporation. In 1995, heco-foundedco‑founded Covance Biotechnology Services, Inc., a contract biopharmaceutical manufacturing operation, and served as a member of its board of directors from inception to 2000. From 1991 to 1993, Dr. Scarlett headed the North American Clinical Development Center and served as Senior Vice President of Medical and Scientific Affairs at Novo Nordisk Pharmaceuticals, Inc., a wholly owned subsidiary of Novo Nordisk A/S. Dr. Scarlett received his B.A. degree in chemistry from Earlham College and his M.D. from the University of Chicago, Pritzker School of Medicine.Olivia K. Bloom has served as our Executive Vice President, Finance since February 2014, Chief Financial Officer since December 2012 and Treasurer since February 2011. Ms. Bloom previously served as our Senior Vice President, Finance from December 2012 to February 2014, Chief Accounting Officer from September 2010 to December 2012 and Vice President, Finance from January 2007 to December 2012. Ms. Bloom joined the Company in 1994 as a Senior Financial Analyst and from 1996 to 2011 served as our Controller. Prior to joining Geron, Ms. Bloom started her career in public accounting at KPMG Peat Marwick and became a Certified Public Accountant in 1994. Ms. Bloom graduated Phi Beta Kappa with a B.S. in Business Administration from the University of California at Berkeley.
Melissa A. Kelly Behrs has served as our Executive Vice President, Business Development and Portfolio & Alliance Management, since July 2014.
Prior to thatPreviously, she was our Executive Vice President, Portfolio and Alliance Managementsincestarting in February 2014, andshe wasour Senior Vice President, Portfolio and Alliance Management from September 2012 to February 2014. Ms. Behrs joined Geron in November 1998 as Director of Corporate Development. Since then, she has served in various managerial positions, including General Manager, R&D Technologies; Vice President, CorporateDevelopment; Senior Vice President, Therapeutic Development, Oncology; and Senior Vice President, Strategic Portfolio Management. From 1990 to 1998, Ms. Behrs worked at Genetics Institute, Inc., a biotechnology research and development company, serving initially as Assistant Treasurer and then as Associate Director of Preclinical Operations where she was responsible for all business development, regulatory, and project management activities for the Preclinical Development function. Ms. Behrs received a B.S. from Boston College and an M.B.A. from Babson College.
Andrew J. Grethlein, Ph.D., has served as our Executive Vice President, Development and Technical Operations, since July 2014.
Prior to that he servedHe joined Geron in September 2012 as our Executive Vice President, TechnicalOperations, since September 2012.Operations. Prior to joining Geron, Dr. Grethlein was Executive Vice President and Chief Operating Officer for Inspiration Biopharmaceuticals, a biopharmaceutical company, from January 2010 to September 2012. From October 2008 until January 2010, Dr. Grethlein was Senior Vice President of Biotechnology and Portfolio Management Team Leader for Hematology at Ipsen S.A., a global specialty pharmaceutical company. His responsibilities at Ipsenincluded planning and execution of worldwide strategy for product and portfolio development in the hematologic therapeutic area. From 2003 to 2008, Dr. Grethlein served as Senior Vice President of Pharmaceutical Operations at Tercica, Inc., an
endocrinology-orientedendocrinology‑oriented biopharmaceuticalcompany. In this role,company, where he was a member of the senior executive team that governed corporate strategy, business planning and company operations, and had responsibility for all manufacturing and quality functions. Before joining Tercica, Dr. Grethlein served in various positions at Elan Corporation, a biotechnology company, from 1997 to 2003, including as Senior Director, South San Francisco PharmaceuticalOperations, where he had responsibility as site head for commercial manufacturing operations.Operations. From 1995 to 1997, Dr. Grethlein served as Manager, Biologics Development and Manufacturing, for Athena Neurosciences, Inc., a pharmaceutical company. Prior to this, he served in various engineering positions for the Michigan Biotechnology Institute, a nonprofit technology research and business development corporation. Dr. Grethlein received his A.A. degree in liberal arts fromSimon'sSimon’s Rock Early College, his B.S. in biology from Bates College, and his M.S. and Ph.D. in chemical engineering from Michigan State University.Stephen N. Rosenfield, J.D., has served as our Executive Vice President, General Counsel and Corporate Secretary since February 2012, General Counsel and Secretary since January 2012 and Secretary since October 2011. From July 2009 to February 2012, Mr. Rosenfield
has beenserved as a consultant to private companies. FromOctober 2008June 2004 until June 2009, Mr. Rosenfieldwas theheld several positions at Tercica, Inc., an endocrinology‑oriented biopharmaceutical company, and through its acquisition by Ipsen, S.A. in October 2008, including General Counsel andSecretary of Tercica, Inc., a U.S. subsidiary of Ipsen, S.A., a global specialty pharmaceutical company. From June 2004 until October 2008, Mr. Rosenfield was the General Counsel and Secretary of Tercica, Inc., an endocrinology-oriented biopharmaceutical company, from January 2006 until October 2008, he was also the Executive Vice President of Legal Affairs, and from June 2004 until January 2006, Mr. Rosenfield was the Senior Vice President of Legal Affairs.Secretary. Prior to joining Tercica, Mr. Rosenfield served as the Executive Vice President of Legal Affairs, General Counsel and Secretary of InterMune, Inc., a biotechnology company that focusedinon pulmonology and fibrotic diseases. Prior to joining InterMune, Mr. Rosenfield was an attorney at Cooley LLP, an international law firm, where he served as outside counsel for biotechnology and technology clients. Mr. Rosenfield received a B.S. from Hofstra University and a J.D. from Northeastern University School of Law.Employees
As of December 31,
2014,2017, we had4215 full‑time and three part‑time employees. Two of our employeesof whom 7hold Ph.D. degrees and13seven hold other advanced degrees. Of this current total workforce,22two employees were engaged in, or directly supported, our research and development activities, and2016 employees were engaged in business development, legal, finance and administration. None of our employees are covered by a collective bargaining agreement; nor have we experienced work stoppages. We consider relations with our employees to be good.On March 3, 2015, we announced an organizational resizing to reduce our workforce from 39 to 21 positions, representing a reduction of approximately 46% of our workforce. For a further discussion, see Note 15 on Subsequent Event in Notes to Consolidated Financial Statements of this annual report on Form 10-K.Corporate Information
Geron Corporation was incorporated in the State of Delaware on November 28, 1990.
Available Information
Our internet address is www.geron.com. Information included on our website is not part of this annual report on Form
10-K.10‑K. We make available free of charge on our website our annual reports on Form10-K,10‑K, quarterly reports on Form10-Q,10‑Q, current reports on Form8-K,8‑K, and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with or furnished to the United States Securities and Exchange Commission, or the SEC. In addition, copies of our annual reports are available free of charge upon written request. The SEC also maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that site is www.sec.gov.Our business is subject to various risks and uncertainties that may have a material adverse effect on our business, financial condition or results of operations. You should carefully consider the risks and uncertainties described below, together with all of the other information included in this annual report on Form
10-K.10‑K. Our business faces significant risks and uncertainties, and those described below may not be the only risks and uncertainties we face. Additional risks and uncertainties not presently known to us or that we currently believe are immaterial may also significantly impair our business, financial condition or results of operations. If any of these risks or uncertainties occur, our business, financial condition or results of operationsor financial conditioncould suffer, the market price of our common stock could decline and you could lose all or part of your investment in our common stock.
RISKS RELATED TO OURBUSINESS
COLLABORATION WITH JANSSENWe
are dependent upon our collaborative relationship with Janssen to further develop, manufacture and commercialize imetelstat,have outlicensed our sole productcandidate.candidate, imetelstat, to Janssen. If Janssenfails to perform as expected,discontinues thepotential for us to generate future revenues from milestone paymentsimetelstat program and/or terminates the Collaboration Agreement, our business androyalties from imetelstatbusiness prospects would besignificantly reduced,severely harmed, and we might cease operations, the development and/or commercialization of imetelstatmaywould be terminated or substantially delayed, and the market price of our common stock would be adversely affected.Janssen may terminate the Collaboration Agreement at any time at its sole discretion. If imetelstat fails to meet criteria determined by Janssen to support an affirmative Continuation Decision, or for any other reason, Janssen may discontinue the imetelstat program and terminate the Collaboration Agreement. In this regard, we believe that without an adequate improvement in survival in relapsed or refractory MF in IMbark, with the determination of adequacy to be assessed by Janssen in its sole discretion, Janssen would decide to discontinue the imetelstat program and terminate the Collaboration Agreement.
In addition, Janssen could discontinue the imetelstat program and terminate the Collaboration Agreement at any time and for any reason, irrespective of whether there is data from IMbark suggesting an adequate improvement in survival in relapsed or refractory MF or whether there is data from IMerge to support the benefit-risk profile of imetelstat in lower risk MDS. Any discontinuation of the imetelstat program or termination of the Collaboration Agreement by Janssen at any time would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations, and the market price of our common stock would be adversely affected.
If Janssen terminates the Collaboration Agreement:
we would no longer have the right to receive any milestone payments or royalties under the Collaboration Agreement;
further development of imetelstat, if any, would be significantly delayed or terminated;
we would bear all risks and costs related to any further clinical development, manufacturing, regulatory approval and commercialization of imetelstat, if any;
we might determine that the commercial potential of imetelstat does not warrant further development of imetelstat by us, in which case the development of imetelstat would cease, which might cause us to cease operations;
we would need to raise substantial additional capital if we were to choose to pursue imetelstat development on our own, or we would need to establish alternative collaborations with third parties, which might not be possible in a timely manner, or at all, or might not be possible on terms acceptable to us, in which case it would likely be necessary for us to limit the size or scope of the imetelstat development program;
if we were to choose to pursue imetelstat development independently, we would need to hire additional qualified employees and secure multiple third-party vendors and service providers to support the development and commercialization of imetelstat, which may take significant amounts of time, may not be feasible, and which would increase our need for additional funding; and
If Janssen does not provide an affirmative Continuation Decision in a timely manner, or at all, our business and business prospects would be severely
harmed.harmed, and we might cease operations.Under the terms of the Collaboration Agreement, Janssen is not obligated to make any additional payments to us until it makes an affirmative Continuation Decision following the protocol-specified primary analysis of IMbark, or, if IMbark is terminated early, or placed on clinical hold or suspended by a regulatory authority for an extended period of time, within approximately 24 months after the initiation of IMerge. In March 2018, the JSC agreed that the protocol-specified primary analysis for IMbark will begin by the end of the second quarter of 2018. As such, we expect the Continuation Decision to occur by the end of the third quarter of 2018. If Janssen terminates IMbark early based on preliminary or ongoing data assessments, safety concerns or for any other reason, or the trial is placed on clinical hold or suspended by a regulatory authority, the protocol-specified primary analysis for IMbark may not take place at all, which would further delay the timing of any Continuation Decision or result in a negative Continuation Decision. Delays in the timing of the Continuation Decision or a negative Continuation Decision from Janssen could increase our development costs and would impair our ability to earn revenues from milestone payments or royalties under the Collaboration Agreement, any of which would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.
If there are further delays in IMerge or IMbark, Janssen may decide to cease the development of imetelstat and terminate the Collaboration Agreement, and our business and business prospects would be severely harmed.
The expansion of enrollment in Part 1 of IMerge has prolonged the development of imetelstat in lower risk MDS, and Janssen
have createdcould decide not to proceed with Part 2 of IMerge. Janssen has made no commitment to commence Part 2 of IMerge. Even if Janssen obtains additional data from the refined target patient population, such data may not support the development of imetelstat in the refined target patient population for Part 2. In any event, we believe Janssen will initiate Part 2 only following an affirmative Continuation Decision, if any. Janssen could discontinue the imetelstat program and terminate the Collaboration Agreement at any time, such as, before the start of the IMbark primary analysis, and for any reason, irrespective of whether there is data from IMbark suggesting an adequate improvement in survival in relapsed or refractory MF or whether there is data from IMerge to support the benefit-risk profile of imetelstat in lower risk MDS. In this regard, we believe that without an adequate improvement in survival in relapsed or refractory MF, with the determination of adequacy to be assessed by Janssen in its sole discretion, Janssen would decide to discontinue the imetelstat program and terminate the Collaboration Agreement. In addition, delays, including in the commencement of Part 2 of IMerge, could cause Janssen to discontinue the imetelstat development program and terminate the Collaboration Agreement.For IMbark, although we expect the protocol-specified primary analysis to begin by the end of the second quarter of 2018, the primary analysis may not occur on the timing that we expect, or at all, if any of the following occurs:
Janssen terminates IMbark based on preliminary or ongoing data assessments, safety concerns, feedback or requirements from the FDA or other regulatory authorities or terminates the Collaboration Agreement;
the FDA or any other regulatory authority places a
joint governance structure, including jointclinical hold on or suspends the trial for any reason;Janssen decides that further changes to the trial are necessary;
additional time is needed to obtain longer-term efficacy and safety data; or
sufficient efficacy and safety data are not available to assess overall survival.
Even if Janssen obtains longer-term efficacy and safety data for IMbark, Janssen or the FDA or any other regulatory authorities may determine that such data do not show an adequate improvement in survival to support further development and
steering committeespotential regulatory approval for imetelstat in relapsed or refractory MF patients, which we expect would result in a decision by Janssen to discontinue IMbark andworking groups,the imetelstat program and terminate the Collaboration Agreement. In addition, the time needed tooverseeobtain longer-term efficacy andmanagesafety data for IMbark, including sufficient data to assess overall survival, could significantly further delay the development of imetelstat in MF and in lower risk MDS, and the timing of a Continuation Decision, if any.If our collaboration with Janssen is not successful, our business and business prospects would be severely harmed.
Our collaboration with Janssen may be unsuccessful due to many factors, including the following:
Janssen may believe that any preliminary or final results of IMbark and/or IMerge, including any future internal data reviews of these trials, are negative under the criteria set forth in the respective protocols or otherwise, are inconclusive, or do not otherwise demonstrate adequate efficacy or clinical benefit to warrant further development or commercialization of imetelstat by Janssen, including if Janssen believes that there is not an adequate improvement in survival in relapsed or refractory MF in IMbark, which would likely result in a termination of the Collaboration Agreement by Janssen at any time, or a negative Continuation Decision;
Janssen may choose to terminate the Collaboration Agreement for any reason;
Janssen may provide a negative Continuation Decision and halt its development of imetelstat, in which case we would receive no further payments from Janssen under the Collaboration Agreement;
Janssen may observe additional or new safety issues in either IMbark and/or IMerge, or any potential future clinical trials of imetelstat, which may result in a termination of the Collaboration Agreement by Janssen at any time, or a negative Continuation Decision;
Janssen may conclude that the commercial potential of imetelstat does not meet Janssen’s internal thresholds or yield a timely return on its investment in imetelstat, either of which would cause Janssen to reconsider continued development of imetelstat, and could result in a renegotiation or a termination of the Collaboration Agreement by Janssen, and if we were to agree to renegotiated terms and Janssen were to continue development of imetelstat, the potential milestone payments and royalties we may receive under such renegotiated agreement would likely be less than the potential milestone payments and royalties under the current Collaboration Agreement;
Janssen may choose not to develop and commercialize imetelstat in certain, or any, markets or for one or more indications, if at all;
in the event of a dispute between us and Janssen regarding Janssen’s performance under the Collaboration Agreement, it may be difficult or impossible for us to prove that Janssen breached its obligations under the Collaboration Agreement, including the obligation to use “commercially reasonable efforts” with regard to the development, regulatory approval, manufacture and commercialization of imetelstat under the Collaboration Agreement;
Janssen may not dedicate the resources necessary to carry imetelstat through clinical development, and this would delay or preclude the achievement of development, regulatory or sales milestones under the Collaboration Agreement;
Janssen may be unable to obtain regulatory clearances or approvals to continue clinical development or commercialize imetelstat for sale in the United States and other countries, in a timely manner, or at all, or such regulatory clearances or approvals may be revoked or put on hold by governmental or regulatory authorities in any jurisdiction;
Janssen may not comply with all applicable regulatory requirements or may fail to report safety data from clinical trials of imetelstat in accordance with all applicable regulatory requirements, which could delay, suspend or stop clinical activities of imetelstat being performed by Janssen or by us;
subject to our election of the U.S. Co-Promotion Option, Janssen will be responsible for all aspects of the commercialization of imetelstat worldwide, including pricing decisions which would affect the royalties on worldwide net sales we could receive;
Janssen may fail to manufacture or supply sufficient quantities of imetelstat or other clinical trial materials for use in current and/or planned clinical trials, which could delay, suspend or stop any imetelstat clinical activities;
Janssen may fail to develop a commercially viable formulation or manufacturing process for imetelstat, and may fail to manufacture or supply sufficient quantities of imetelstat for commercial use, if approved, which would result in lost sales revenue for Janssen and reduced royalties for us;
the loss or impairment of our intellectual property rights related to imetelstat might delay or halt ongoing or potential future clinical trials of imetelstat by Janssen and any applications for regulatory approval by Janssen, and therefore delay or halt the payment of any potential milestone payments to us;
Janssen’s ability to develop, manufacture and commercialize imetelstat may be delayed or substantially impacted if we are unable to provide to Janssen in a timely manner, or at all, data or results from studies of imetelstat conducted by us and others prior to the Collaboration Agreement, or other information, related to imetelstat that may be requested by Janssen; and
if Janssen is acquired by a third party during the term of our collaboration with Janssen, the acquirer may have different strategic priorities that could cause it to terminate the Collaboration Agreement or reduce its commitment to our collaboration.
If our collaboration with Janssen is unsuccessful as a result of any of the above factors, or any other factors, then Janssen may terminate the Collaboration Agreement or cease its efforts to develop, manufacture or commercialize imetelstat, and we would not be eligible for any further payments from Janssen under the Collaboration Agreement, which would adversely impact our financial results, business and business prospects, and the future of imetelstat, and could cause us to cease operations.
If Janssen does not perform in the manner we expect or fulfill its responsibilities under the Collaboration Agreement in a timely manner, or at all, the clinical development, manufacturing,
andregulatory approval and/or commercializationactivities for imetelstat; however, Janssen is solely responsible for the operational implementationofthose activities. Accordingly, theimetelstat could be further delayed or terminated.The timely and successful completion by Janssen of
thosethe development, manufacturing, regulatory and commercialization activities for imetelstat will significantly affect the timing and amount of any revenues from milestone payments and royalties we may receive under the Collaboration Agreement, and these activities will be influenced by, among other things, the efforts and allocation of resources by Janssen, none of which we control.If Janssen does not perform in the manner we expect or fulfill its responsibilities in a timely manner, or at all, the clinical development, manufacturing, regulatory approval and/or commercialization efforts related to imetelstat could be delayed or terminated, and it could become necessary for us to assume the responsibilities for the clinical development, manufacturing, regulatory approval and/or commercialization of imetelstat at our own expense.Accordingly, there can be no assurance that any of the development, regulatory or sales milestones under the Collaboration Agreement will be achieved or that we will receive any future milestone or royalty payments under the Collaboration Agreement.In addition, because Janssen is solely responsible for the operational
implementationexecution of worldwide regulatory, development, manufacturing and commercialization activities related to imetelstat, we are solely dependent onJanssen to provide us with timely and accurate information concerning these
activities.activities as well as information about the costs incurred under the Collaboration Agreement. If we do not receive accurate information from Janssen in a timely manner, or at all, regarding these activities, including, for example, plans for, and enrollment of, and efficacy and safety results from, clinical trials of imetelstat, and commercialization assumptions or criteria set by Janssen for the continued development and commercialization of imetelstat, then the timeliness and accuracy of our public disclosures, as well as our governance-related decision-making regarding these activities, may be adversely affected.Our collaboration with Janssen may be unsuccessful due to other factors, including the following:•Janssen may choose to terminate the Collaboration Agreement for convenience;•Janssen may provide a negative Continuation Decision and halt itsAny developmentof imetelstat;•the results of the Initial Phase 2 MF Study and/or the Initial Phase 2 MDS Study may be negative or inconclusive, or Janssen may observe safety issues in either of these studies, which may result in a negative Continuation Decision by Janssen, in which case we would receive no further payments from Janssen under the Collaboration Agreement;•Janssen may choose not to develop and commercialize imetelstat in certain markets or for one or more indications, if at all;•Janssen may take considerably more time advancing imetelstat through the clinical and regulatory process than we currently anticipate, which could materially delay the achievement of milestones and, consequently the receipt of milestone payments from Janssen, and ultimately, any royalties on worldwide net sales;•in the event of a dispute between us and Janssen regarding Janssen's performance under the Collaboration Agreement, it may be difficult for us to prove that Janssen breached its obligation to use "commercially reasonable efforts" with regard to the development, regulatory approval, manufacture and commercialization of imetelstat under the Collaboration Agreement;•Janssen may not dedicate the resources necessary to carry imetelstat through clinical development or may not obtain the necessary regulatory approvals, and this would delay the achievement of development, regulatory or sales milestones;•Janssen's ability to achieve development and manufacturing objectives or milestones may be delayed or substantially impacted if we fail to transfer technology and information related to imetelstat to Janssen in a timely manner or at all;•subject to our election of the U.S. Co-Promotion Option, Janssen will be responsible for all aspects of the commercialization of imetelstat worldwide, including pricing decisions which would affect the royalties on worldwide net sales we could receive;•Janssen may change the focus of its commercialization efforts or prioritize other programs more highly and, accordingly, reduce the efforts and resources allocated to imetelstat, which would have the direct effect of reducing our royalties or share of potential co-promotionactivitiessince the extent of our U.S. Co-Promotion Option is limited to a percentage of overall promotion activities under the Collaboration Agreement;•after assuming manufacturing responsibilities for imetelstat, Janssen may fail to manufacture or supply sufficient quantities of imetelstat for use in planned clinical trials, which could delay, suspend or stop any imetelstat clinical activities;•Janssen may fail to develop a commercially viable formulation or manufacturing process for imetelstat, and may fail to manufacture or supply sufficient quantities of imetelstat for commercial use, if approved, which would result in lost sales revenue and reduced royalties for us;
•Janssen may not comply with all applicable regulatory requirements or may fail to report safety data in accordance with all applicable regulatory requirements, which could delay, suspend or stop clinical activities being performed by Janssen or by us; and•if Janssen is acquired during the term of our collaboration, the acquirer may have different strategic priorities that could cause it to terminate the Collaboration Agreement or reduce its commitment to our collaboration.
If our collaboration with Janssen is unsuccessful as a result of any of the above factors, or any other factor, then Janssen may terminate the Collaboration Agreement, and we would not be eligible for any further payments from Janssen under the Collaboration Agreement, which would severely and adversely affect our business and business prospects.Delays in the initiation of, or the inability to initiate, subsequent clinical trials of imetelstat, such as the Initial Phase 2 MF Study or the Initial Phase 2 MDS Study planned to be conducted under the Collaboration Agreement, could result in increased development costs and would delay our ability to earn revenues from milestone payments or royalties from the Collaboration Agreement with Janssen.To date, we have not initiated any clinical trials evaluating imetelstat in any hematologic myeloid malignancies (other than essential thrombocythemia), including myelofibrosis, or MF. Advancing clinical development of imetelstat will be influenced by results from existing clinical trials, such as the MF Pilot Study, and potential future clinical trials of imetelstat in hematologic myeloid malignancies, such as the Initial Phase 2 MF Study or the Initial Phase 2 MDS Study planned to beconducted by Janssenunder the Collaboration Agreement. The commencement of potential future clinical trials of imetelstat could be delayed or abandoned for a variety of reasons, including as a result of failures or delays by Janssen in:•obtaining regulatory clearance to commence subsequent clinical trials of imetelstat in a timely manner, or at all, in the United States or other countries;•properly designing, commencing, enrolling, conducting or completing potential future clinical trials, including the Initial Phase 2 MF Study or the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, and promptly or adequately reporting data from such trials;•demonstrating sufficient safety and efficacy in future Phase 2 clinical trials, including the Initial Phase 2 MF Study or the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, to obtain regulatory clearance to commence subsequent clinical trials in a timely manner, or at all, in the United States or other countries;•properly conducting and/or completing the MF Pilot Study;•manufacturing sufficient quantities of imetelstat and in a manner that meets the quality standards of the FDA and other regulatory agencies;•ensuring the ability to manufacture imetelstat at acceptable costs for Phase 3 clinical trials and commercialization;•obtaining clearance or approval of proposed trial designs or manufacturing specifications from the FDA and other regulatory authorities;•reaching agreement on acceptable terms and on a timely basis, if at all, with collaborators and vendors located in the United States or foreign jurisdictions, including contract research organizations, laboratory service providers and trial sites, on all aspects of clinical trials;•obtaining institutional review board or ethics committee approval to conduct clinical trials at prospective clinical trial sites; and
•identifying and successfully screening and enrolling appropriate subjects for participation in clinical trials and retaining those subjects in the clinical trials.
Failures or delays with respect to any of these events could adversely affect the ability to initiate, maintain or successfully complete any future clinical trials of imetelstat, which could increase development costs, impair our ability to earn revenues from milestone payments or royalties from the Collaboration Agreement with Janssen or cause Janssen to terminate the Collaboration Agreement, any of which could adversely impact our financial results and would have severe adverse effects on our business and business prospects.If there are any safety or efficacy results that cause the benefit-risk profile of imetelstat to become unacceptable, the clinical development of imetelstat would be delayed or halted, and Janssen may terminate the Collaboration Agreement, which would severely and adversely affect our business prospects, and may cause us to cease operations.Imetelstat may prove to have undesirable or unintended side effects or other characteristics adversely affecting its safety, efficacy, cost-effectiveness or marketability that could prevent or limit its approval for marketing and successful commercial use, or that could delay or prevent the commencement and/or completion of clinical trials for imetelstat. For example, although the FDA removed the full clinical hold on our IND for imetelstat, if patients in current or future clinical trials experience similar or more severe hepatotoxicity, including elevated LFTs or severe hepatic adverse events, such IND for imetelstat may again be placed on clinical hold, and we, in collaboration with Janssen, may be precluded from further developing imetelstat. In addition, if regulatory submissions requesting approval to market imetelstat are submitted, after reviewing the data in such submissions, the FDA and regulatory agencies in other countries may conclude that the overall benefit-risk profile of imetelstat treatment, including hepatotoxicity or severe hepatic adverse events, may preclude approval of imetelstat for marketing or further development for any indications, including hematologic malignancies. Any of these events would severely harm our business and prospects, and would likely cause us to cease operations.Further, in our Phase 1 clinical trials of imetelstat, we observed dose-limiting toxicities, including thrombocytopenia when the drug was used as a single agent, and neutropenia when the drug was used in combination with paclitaxel, as well as a low incidence of severe infusion reactions. In our Phase 2 clinical trials of imetelstat in ET, MM, and solid tumors, we have observed hematologic toxicities as well as gastrointestinal events, infections, muscular and joint pain, fatigue and infusion reactions. In addition, in our Phase 2 clinical trials of imetelstat, we have observed LFT abnormalities, the clinical significance and long-term consequences of which are currently undetermined. In our Phase 2 trial in ET, one patient died of bleeding esophageal varices, a complication of chronic liver disease, for which imetelstat could not be excluded as a causative agent. In the MF Pilot Study, myelosuppression has been the primary dose-limiting toxicity reported to date, consistent with our observations in previous Geron-sponsored imetelstat studies. However, during the MF Pilot Study, more persistent and profound myelosuppression, particularly thrombocytopenia, was observed with imetelstat administered on a weekly basis. This included one case of febrile neutropenia after prolonged myelosuppression with intracranial hemorrhage resulting in patient death, which was assessed as possibly related to imetelstat by the investigator. Since the MF Pilot Study is an ongoing study with additional data being generated, the benefit-risk profile of imetelstat in MF will continue to be assessed, including the risk of hepatotoxicity and severe cytopenias that may be associated with life-threatening clinical outcomes.Clinical trials by their nature examine the effect of a potential therapy in a sample of the potential future patient population. As such, clinical trials conducted with imetelstat, to date and in the future, may not uncover all possible adverse events that patients treated with imetelstat may experience. In collaboration with Janssen, we may observe or report dose-limiting toxicities or other safety issues in potential future clinical trials of imetelstat, including the Initial Phase 2 MF Study and the InitialPhase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement. Likewise, because previously enrolled patients continue to receive imetelstat in the MF Pilot Study, additional or more severe toxicities or safety issues in the MF Pilot Study, including additional serious adverse events and clinically significant LFT abnormalities, may be observed or reported as patient treatment continues and more data become available. If such toxicities or other safety issues in any clinical trial of imetelstat result in an unacceptable benefit-risk profile, then:•the commencement and/or completion of any current or future clinical trials, including the MF Pilot Study and the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, would likely be delayed or prevented;•the MF Pilot Study or any potential future clinical trials, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, may be placed on clinical hold or halted by regulatory authorities, such as the previous clinical holds placed by the FDA on our IND for imetelstat and the IND for the MF Pilot Study; or•additional, unforeseen trials or preclinical studies may be required to be conducted.
The occurrence of any of these events would likely cause Janssen to abandon their development of imetelstat entirely and terminate the Collaboration Agreement. Any termination of the Collaboration Agreement by Janssen would have a material adverse effect on our results of operations, financial condition, business prospects and the future of imetelstat, any of which may cause us to cease operations.If Janssen does not elect to continue the development of imetelstat in a timely manner, or at all, our business and business prospects would be severely harmed.Under the terms of the Collaboration Agreement, Janssen is not obligated to make any additional payments to us until it makes an affirmative Continuation Decision following the results of the Initial Phase 2 MF Study, or, if the Initial Phase 2 MF Study is terminated early or suspended for an extended period of time, within a certain time period thereafter as set forth in the Collaboration Agreement. The timing of Janssen's Continuation Decision also affects the timing and availability of our decision regarding U.S. Opt-In Rights, as well as our election of the U.S. Co-Promotion Option. If the Initial Phase 2 MF Study is terminated early, suspended for an extended period of time, or is otherwise unsuccessful, Janssen may provide a negative Continuation Decision, in which case, the Collaboration Agreement would terminate, we would not be eligible for any further payments from Janssen under that agreement and our business and business prospects would be severely and adversely affected.In addition, Janssen may terminate the Collaboration Agreement at any time for convenience. If Janssen terminates the Collaboration Agreement, then, depending on the timing of such event:•we would no longer have the right to receive any milestone payments or royalties under the Collaboration Agreement;•the development of imetelstat would likely be terminated or significantly delayed;•we would bear all of the risks and costs related to the further clinical development, manufacturing, regulatory approval and commercialization of imetelstat;•we would need to raise additional capital if we were to choose to pursue imetelstat development on our own, or we would need to establish alternative collaborations with third-party collaboration partners, which may not be possible in a timely manner or at all, or may not be possible on terms acceptable to us, in which case it would likely be necessary for us to limit the
•we would need to hire additional employees to support the development and commercialization of imetelstat, which would increase our need for additional funding.
size or scope of the imetelstat development program or seek additional funding by other means to accommodate the increased expenditures; andAny termination of the Collaboration Agreement by Janssen at any time would have a material adverse effect on our results of operations, financial condition, business prospects and the future of imetelstat, any of which would have severe adverse effects on our business and business prospects, and may cause us to cease operations.Our decision to exercise our U.S. Opt-In Rights under the Collaboration Agreement with Janssen for imetelstat must be made within a limited time after Janssen makes an affirmative Continuation Decision and, as a result, we may be required to make a substantial capital investment based on limited clinical data.We must elect to exercise our U.S. Opt-In Rights within a short timeframe following Janssen's Continuation Decision. Although we expect to receive information from Janssen regarding data from the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study, proposed future clinical development plans and costs, estimates in timing for commercializing imetelstat and related promotional activities, and calculation of our share of development costs incurred to date by Janssen that we will be required to reimburse if we exercise our U.S. Opt-In Rights, we will be required to rapidly decide whether to make a substantial capital investment in imetelstat prior to the conclusion of any Phase 3 registration-enabling clinical trial. Accordingly, if imetelstat were to become unsuccessful in any Phase 3 registration-enabling clinical trial or fails to receive regulatory approval, we would not receive any financial return on this substantial capital investment. Such an occurrence would negatively impact our financial condition and results of operations.Our Collaboration Agreement with Janssen prohibits us from developing or commercializing any product that operates through the same mechanism of action as imetelstat, and our U.S. co-promotion rights may be terminated if we market or promote any such products for any oncology indication. As a result of this, or for any other reason, we may not be able to successfully acquire or in-license promising product opportunities for development, which would limit our growth and revenue potential.We plan to seek to diversify our sole product candidate development risk by identifying promising product opportunities for development, which we may seek to acquire or in-license. However, the Collaboration Agreement with Janssen prohibits us from commercializing, under the intellectual property we have licensed exclusively to Janssen, any substance whose identified or known mechanism of action is telomerase inhibition. Further, if we exercise our U.S. Co-Promotion Option under the Collaboration Agreement, we will be required to certify at the time of exercising our U.S. Co-Promotion Option that we are not marketing or promoting, and have no right to market or promote, any such products for any oncology indication. Our right to co-promote in the U.S. may be terminated by Janssen if we develop or commercialize a product for treating an oncology indication that acts through the same mechanism of action as imetelstat or that is substitutable for imetelstat. Accordingly, our Collaboration Agreement with Janssen could adversely affect our ability to acquire or in-license, or to research, develop or market, promising product candidates.In addition, we may not be able to identify promising product candidates. The competition to acquire or in-license rights to promising product candidates is fierce, and many of our competitors are large, multinational pharmaceutical and biotechnology companies with considerably more financial, development and commercialization resources and experience than we have. Thus, even if we succeed in identifying promising product candidates, we may not be able to acquire rights to them on acceptable terms, or at all. In any event, any growth through acquisition or in-licensing will depend upon our identifying and obtaining promising product candidates, our ability to develop those productcandidates and the availability of funding to complete the development of, obtain regulatory approval for and commercialize these product candidates. If we are unable to identify and acquire promising product candidates, we will be unable to diversify our sole product candidate development risk, and our growth and revenue potential could be limited.We may not be able to successfully retain key personnel to support our collaboration with Janssen or to manage any future growth.Under the terms of the Collaboration Agreement, we and Janssen have created a joint governance structure, including committees and working groups, to manage worldwide regulatory, development, manufacturing and commercialization activities for imetelstat, and we will have ongoing responsibilities to oversee and participate in the collaboration with Janssen. In addition, we will remain responsible for prosecuting, at Janssen's direction, the patents we licensed to Janssen, and have sole responsibility for those patents that were not licensed to Janssen. If we are unable to successfully retain, motivate and incentivize our personnel, our ability to support the Collaboration Agreement with Janssen could be impaired, and our business and the price of our common stock would be adversely impacted.In addition, our future growth and success depend to a significant extent on the skills, experience and efforts of our executive officers and key members of our clinical and scientific staff. We face intense competition for qualified individuals from numerous pharmaceutical, biopharmaceutical and biotechnology companies, as well as academic and other research institutions. The previous restructurings we implemented and the recently announced organizational resizing, as well as our collaboration with Janssen and uncertainties regarding our ability to diversify our sole product candidate development risk, could have an adverse impact on our ability to retain and recruit qualified personnel or we may incur unanticipated inefficiencies caused by our reduced personnel resources. We may be unable to retain our current personnel or attract or assimilate other highly qualified management and development personnel in the future on acceptable terms. The loss of any or all of these individuals could harm our business and could impair our ability to support our collaboration with Janssen or to support future growth.We and certain of our officers have been named as defendants in three purported securities lawsuits, two of which are securities class action lawsuits, and certain of our officers and directors have been named as defendants in a derivative lawsuit. These, and potential similar or related lawsuits, could result in substantial damages, divert management's time and attention from our business, and have a material adverse effect on our results of operations. These lawsuits and any other lawsuits will be costly to defend or pursue and are uncertain in their outcome.Securities-related class action lawsuits and derivative litigation has often been brought against companies, including many biotechnology companies, which experience volatility in the market price of their securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies often experience significant stock price volatility in connection with their product development programs.On March 14, 2014, a purported securities class action lawsuit was commenced in the United States District Court for the Northern District of California, or the California District Court, naming as defendants us and certain of our officers. The lawsuit alleges violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements made by us related to our Phase 2 trial of imetelstat in patients with ET or PV. The plaintiff alleges, among other things, that we failed to disclose facts related to the occurrence of persistent low-grade LFT abnormalities observed in our Phase 2 trial of imetelstat in ET or PV patients and the potential risk of chronic liver injury following long-term exposure to imetelstat. The plaintiff seeks damages and an award of reasonable costs and expenses, including attorneys' fees.On March 28, 2014, a second purported securities class action lawsuit was commenced in the California District Court, naming as defendants us and certain of our officers. This lawsuit, which is based on the same factual background as the purported securities class action lawsuit that commenced on March 14, 2014, also alleges violations of the Securities Exchange Act of 1934 and seeks damages and an award of reasonable costs and expenses, including attorneys' fees.On June 30, 2014, both of the foregoing lawsuits, or the Class Action Lawsuits, were consolidated for all purposes, and a lead plaintiff and lead counsel were appointed by the California District Court. On July 21, 2014, the California District Court ordered the lead plaintiff to file its consolidated amended complaint in the Class Action Lawsuits, which was filed on September 19, 2014. We filed our motion to dismiss the consolidated amended complaint on November 18, 2014. The plaintiff's opposition to our motion to dismiss was filed on January 20, 2015, and we filed our reply on February 25, 2015. The court hearing for the motion to dismiss has been scheduled for April 10, 2015.On June 6, 2014, a purported securities lawsuit, not styled as a class action, was commenced in the United States District Court for the Southern District of Mississippi, or the Mississippi District Court, naming as defendants us and certain of our officers. This lawsuit, which is based on the same factual background as the Class Action Lawsuits, also alleges violations of the Securities Exchange Act of 1934 and seeks damages and an award of reasonable costs and expenses, including attorneys' fees. On August 11, 2014, we filed a motion to transfer the purported securities lawsuit filed in the Mississippi District Court to the California District Court so it could be consolidated with the purported Class Action Lawsuits. On November 4, 2014, the Mississippi District Court granted our motion and transferred the case to the California District Court, and the transferred case has been consolidated by the California District Court with the purported Class Action Lawsuits filed in the California District Court.On April 21, 2014, a stockholder purporting to act on our behalf filed a derivative lawsuit in the Superior Court of California for the County of San Mateo against certain of our officers and directors. The lawsuit alleges breaches of fiduciary duties by the defendants and other violations of law. In general, the lawsuit alleges that the defendants caused or allowed the dissemination of allegedly false and misleading statements related to our Phase 2 trial of imetelstat in patients with ET or PV. The plaintiff is seeking unspecified monetary damages and other relief, including reforms and improvements to our corporate governance and internal procedures.It is possible that additional suits will be filed, or allegations received from stockholders, with respect to these same or other matters and also naming us and/or our officers and directors as defendants. These lawsuits and any other related lawsuits are subject to inherent uncertainties, and the actual defense and disposition costs will depend upon many unknown factors. The outcome of these lawsuits is necessarily uncertain. We could be forced to expend significant resources in the defense against these lawsuits and we may not prevail. In addition, we may incur substantial legal fees and costs in connection with these lawsuits. We currently are not able to estimate the possible cost to us from these matters, as these lawsuits are currently at an early stage, and we cannot be certain how long it may take to resolve these matters or the possible amount of any damages that we may be required to pay. We have not established any reserve for any potential liability relating to these lawsuits. It is possible that we could, in the future, incur judgments or enter into settlements of claims for monetary damages. A decision adverse to our interests on these actions could result in the payment of substantial damages, or possibly fines, and could have a material adverse effect on our cash flow, results of operations and financial position.We may also be subject to litigation arising from our proposed or completed strategic transactions or if the results of our business and collaboration activities are not successful.On October 1, 2013, we closed the transaction to divest our human embryonic stem cell assets and our autologous cellular immunotherapy program pursuant to the terms of the Contribution Agreement that we entered into in January 2013 with BioTime and Asterias. On November 13, 2014, we announced that we had entered into the Collaboration Agreement with Janssen to develop and commercialize imetelstat worldwide. We may face litigation arising from or related to the value received by our stockholders, if any, from our distribution of the Asterias Series A common stock and/or the BioTime Warrants distributed by Asterias under the Contribution Agreement, or our role as a named underwriter with respect to our distribution of the Asterias Series A common stock, including the delays we experienced with respect to completing our distribution of the Asterias Series A common stock, or we may face litigation based on other matters related to the Contribution Agreement and the Collaboration Agreement or the transactions contemplated thereby, including if we are unable to generate substantial value under the Collaboration Agreement with Janssen or such collaboration is not otherwise successful.As a result of these and other factors, we may be exposed to a number of litigation risks related to the transactions contemplated by the Contribution Agreement and the Collaboration Agreement, including declines or fluctuations in our stock price, additional advisor and legal fees, distractions to our management caused by activities undertaken in connection with resolving any disputes related to the transactions, or the loss of important contractual rights. As another example, some of our investors purchased shares of our common stock because they were interested in the opportunities presented by our human embryonic stem cell programs. Thus, certain stockholders may have attributed substantial financial value to our stem cell assets and may believe that the Asterias Series A common stock, BioTime Warrants and/or cash received in the distributions pursuant to the Contribution Agreement were inadequate consideration for such assets.Similarly, the announcement and/or completion of these strategic transactions could result in litigation arising out of any claims that our stockholders suffered financial losses due to the transactions, the approval of our stockholders was required under applicable law or otherwise should have been obtained prior to the completion of either or both of these transactions, or that our officers and directors breached their fiduciary duties in connection with the approval and completion of these transactions. Although we believe that stockholder approval was not required under applicable law in order to complete either or both of these transactions and therefore we neither sought nor intend to seek such stockholder approval, it is possible that persons who were stockholders at the time of the applicable transaction may claim that their approval was required, in which case litigation could follow, which could result in substantial damages to us and/or could negatively affect our rights and obligations under either of these agreements or, in the case of the Collaboration Agreement, could result in the termination of that agreement.Likewise, our stockholders may believe that the financial and other terms of the Collaboration Agreement are not favorable to either us or our stockholders, including any belief that the potential payments we may receive under the Collaboration Agreement are inadequate. Litigation brought by our stockholders challenging the validity of, or financial losses resulting from, these transactions could also result in claims against us by Asterias and/or Janssen, and each of the Contribution Agreement and the Collaboration Agreement provide for indemnification by us of BioTime and Janssen, respectively, against all losses and expenses relating to breaches of our representations, warranties and covenants in the applicable agreement, which could expose us to further financial obligations and damages. The occurrence of any one or more of the above could have a significant adverse impact on our business and financial condition.In addition, if the results of our business and collaboration activities are not successful, including without limitation, if:•we or Janssen are otherwise unable to continue development of imetelstat due to actions by regulatory authorities, such as the previous full clinical hold that was placed by the FDA on our IND for imetelstat in March 2014;•we, Janssen or any investigators ascertain that the use of imetelstat results in significant systemic or organ toxicities, including hepatotoxicity, or other safety issues resulting in an unacceptable benefit-risk profile;•the conduct of previous clinical trials, such as the MF Pilot Study, and future clinical trials, such as the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, results in patient injury or death, or any failure to meet regulatory and compliance requirements;•the final or any preliminary results from the MF Pilot Study, or any subsequent clinical trial of imetelstat, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, are not deemed to be successful;•Janssen discontinues the further development of imetelstat and terminates the Collaboration Agreement; or•Asterias is unable to develop our stem cell assets, and we are not able to receive any royalties from the sale of any potential stem cell products by Asterias,
our stock price would likely decline, and future litigation may result. A decision adverse to our interests in any such lawsuits could result in the payment of substantial damages by us, and could have a material adverse effect on our cash flow, results of operations and financial position or could otherwise severely harm our business.Our business may also bring us into conflict with our licensees, licensors, or others with whom we have contractual or other business relationships, or with our competitors or others whose interests differ from ours. If we are unable to resolve those conflicts on terms that are satisfactory to all parties, we may become involved in litigation brought by or against us. For example, we are subject to the risk of possible disagreements with Janssen, including those regarding the development and/or commercialization of imetelstat, interpretation of the Collaboration Agreement and ownership of proprietary rights. In addition, in certain circumstances we may believe that we have achieved a particular milestone and Janssen may disagree with our belief. In that case, receipt of that milestone payment may be delayed or may never be received, which would adversely affect our financial condition and may require us to adjust our operating plans. While the Collaboration Agreement provides for a joint governance structure to oversee and manage worldwide regulatory, development, manufacturing and commercialization activities for imetelstat, Janssen generally will, subject to limited exceptions, have the deciding vote in the event of any disagreement. In any event, the joint governance structure contemplated by the Collaboration Agreement will be dissolved in the event that we do not exercise our U.S. Opt-In Rights, which would preclude our ability to participate in any further decision-making for imetelstat. Reliance on a joint governance structure also subjects us to the risk that changes in key management personnel that are members of the various joint committees may materially and adversely affect the functioning of these committees, which could significantly delay or preclude imetelstat development and/or commercialization. As a result of possible disagreements with Janssen, we also may become involved in litigation or arbitration, which would be time-consuming and expensive.Monitoring, initiating and defending against legal actions, including our currently-pending securities-related lawsuits and derivative litigation, are time-consuming for our management, likely to be expensive and may detract from our ability to fully focus our internal resources on our businessactivities. The outcome of litigation is always uncertain, and in some cases could include judgments against us that require us to pay damages, enjoin us from certain activities, or otherwise negatively affect our legal or contractual rights, which could have a significant adverse effect on our business. In addition, the inherent uncertainty of such litigation, including our currently-pending securities-related lawsuits and derivative litigation, could lead to increased volatility in our stock price and a decrease in the value of our stockholders' investment in our common stock.RISKS RELATED TO CLINICAL AND COMMERCIALIZATION ACTIVITIESThe research and development of imetelstat is subject to numerous risks and uncertainties.The science and technology of telomere biology, telomerase and our proprietary oligonucleotide chemistry are relatively new. There is no precedent for the successful commercialization of a therapeutic product candidate based on these technologies. In collaboration with Janssen, we must undertake significant research and development activities to develop imetelstat, our sole product candidate, based on these technologies, which may take years to accomplish, if at all.Because of the significant scientific, regulatory and commercial challenges that must be overcome for the research, development and commercialization of imetelstat to be successful, the development of imetelstat in hematologic myeloid malignancies, including MF and MDS, or any other indications, may be delayed or abandoned, even after significant resources have been expended on it. Our decisions to discontinue our Phase 2 clinical trial of imetelstat in metastatic breast cancer in September 2012, and to discontinue our development of imetelstat in solid tumors with short telomeres in April 2013, are examples of this. Any further delay or abandonment of the development of imetelstat in hematologic myeloid malignancies would have a material adverse effect on our collaboration with Janssen which could result in the termination of the Collaboration Agreement. Any of these events would have severe adverse effects on our business and business prospects and likely result in the failure of our business.Success in early clinical trials may not be indicative of results in subsequent clinical trials. Likewise, preliminary data reported by investigators from time-to-time are subject to review or verification procedures that could result in material differences to final data and may change as more patient data become available.A number of new drugs and biologics have shown promising results in initial clinical trials, but subsequently failed to establish sufficient safety and efficacy data to obtain necessary regulatory approvals. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval.Data from our preclinical studies and Phase 1 and Phase 2 clinical trials of imetelstat, as well as preliminary, additional or updated data from the MF Pilot Study, should not be relied upon as evidence that subsequent or larger-scale clinical trials of imetelstat will succeed. The positive efficacy results we have obtained from the Phase 2 clinical trial of imetelstat in ET may not predict the future therapeutic benefit of imetelstat, if any, in other hematologic myeloid malignancies, including MF. For example, the known LFT abnormalities and dose-limiting toxicities associated with imetelstat, such as profound thrombocytopenia and neutropenia and other safety issues, including death, that have been observed in both Geron-sponsored and investigator-sponsored trials, including the MF Pilot Study, could cause complexities in treating patients with MF and could result in the discontinuation of the MF Pilot Study and any future clinical trials of imetelstat, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement. Also, the criteria used to assess efficacy in the MF Pilot Study have not been validated for clinical use and may not be considered by the FDA or other regulatory agencies to be accurate predictors of efficacy for different endpoints that may be required by the FDA or other regulatory agencies for Phase 3 clinical trials.The preliminary results of the MF Pilot Study presented by the investigator at the ASH annual meeting in December 2013 and the updated preliminary results presented at ASH in December 2014 will need to be confirmed in one or more larger Phase 2 and Phase 3 trials in MF at multiple treating centers. The results reported by us, Janssen or by the investigator in the MF Pilot Study may not be reproduced in any subsequent imetelstat trials conducted in the future, including the Initial Phase 2 MF Study or the Initial Phase 2 MDS Study planned to be conducted under the Collaboration Agreement, or by any other investigator or group of investigators, or in any trial enrolling a larger number of patients or conducted at multiple treating centers, and thus should not be relied upon as indicative of future clinical results of imetelstat in MF or any other hematologic myeloid malignancy.In addition, from time-to-time, we or Janssen may report or announce preliminary data from current or potential future clinical trials, such as the Initial Phase 2 MF Study and Initial Phase 2 MDS Study planned to be conducted under the Collaboration Agreement. For example, the investigator for the MF Pilot Study reported preliminary data from the trial in December 2013 and updated preliminary data in December 2014. Since those data were preliminary, the final data from the MF Pilot Study may be materially different than the data reported in December 2013 or December 2014. Since patients previously enrolled in the MF Pilot Study continue to receive imetelstat, safety and efficacy data continue to be generated, and such additional and updated data may materially change the overall conclusions from the preliminary data reported in December 2013 or December 2014. Therefore, such preliminary data should be considered carefully and with caution. Additional and updated data from the MF Pilot Study are also subject to any review or verification procedures that Janssen may conduct as the trial sponsor for the MF Pilot Study after it assumes responsibility for the conduct of the MF Pilot Study, and since this could result in material differences from the data reported by the investigator or us, additional or updated data that may be reported from the MF Pilot Study should be considered carefully and with caution. Analyses performed by Janssen may result in conclusions that are materially different from the investigator's analyses or ours, and therefore such preliminary data should be considered carefully and with caution.Material adverse changes in final data from the MF Pilot Study could jeopardize our Collaboration Agreement with Janssen and if Janssen were to terminate the Collaboration Agreement, our business prospects would be severely and adversely affected. Even if final safety and efficacy data from the MF Pilot Study are positive, significant additional clinical testing will be necessary for the future development of imetelstat in MF. Any such final safety and efficacy data from the MF Pilot Study may not be reproducible in future clinical trials, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement.Before Janssen can seek to obtain regulatory approval for the commercial sale of imetelstat, multiple clinical trials, including larger-scale Phase 3 clinical trials, will need to be conducted to demonstrate that imetelstat is safe and effective for use in a diverse population. There is typically an extremely high rate of attrition from the failure of drug candidates proceeding through clinical trials. If imetelstat cannot be developed in future clinical trials, including Phase 3 clinical trials, our Collaboration Agreement with Janssen will be negatively impacted and could be terminated altogether, which would have severe adverse effects on our business and business prospects, and likely result in the failure of our business.Conducting and completing potential future clinical trials of imetelstat, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, on a timely basis is subject to risks and uncertainties.Delays or terminations of potential future clinical trials, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, could be caused by matters such as:•not receiving timely regulatory clearances or approvals in any jurisdiction, whether within or outside of the United States, including, if we, Janssen or future investigators do not obtain and maintain regulatory clearance to commence studies of imetelstat in MF, MDS or any additional hematologic myeloid malignancies in a timely manner or at all, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement;•the inability by Janssen to maintain the INDs for imetelstat that we expect to transfer to Janssen, including the IND for the MF Pilot Study, without such INDs being placed on full or partial clinical hold by the FDA;•the inability to properly design, conduct and/or complete current and potential future clinical trials of imetelstat including the MF Pilot Study and the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study;•data showing lack of effectiveness of imetelstat during clinical trials, or results that do not demonstrate statistically significant efficacy;•safety issues, side effects or dose-limiting toxicities, including any additional or more severe safety issues related to imetelstat in addition to those which have been observed to date in Geron-sponsored or investigator-sponsored trials, whether or not in the same indications or therapeutic areas;•disruptions due to drug supply or quality issues;•not receiving acceptance of new manufacturing specifications or procedures or clinical trial protocol amendments by regulatory authorities;•failure by investigators conducting future clinical trials of imetelstat, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, to timely commence, enroll, complete or report data from such clinical trials;•not receiving timely institutional review board or ethics committee approval of clinical trial protocols or protocol amendments;•delays in patient enrollment due to size or nature of patient population, nature of protocols, proximity of patients to clinical sites, availability of effective treatments for the relevant disease and eligibility criteria for the trial;•inability to retain patients to complete clinical trials or to return for post-treatment follow-up;•difficulty in obtaining or accessing necessary clinical data, including additional and future data from the MF Pilot Study, which may result in incomplete data sets;•unavailability of any study-related treatment (including comparator therapy);•issues or disputes with key vendors of clinical services, such as contract research organizations, clinical trial sites and laboratory service providers; or
•governmental or regulatory delays in any jurisdiction, whether within or outside of the United States, information requests, clinical holds, such as the previous clinical holds placed by the FDA on our IND for imetelstat and the IND for the MF Pilot Study, and changes in regulatory requirements, policies and guidelines.
Advancing clinical development of imetelstat in the United States is dependent on obtaining positive results from existing and potential future clinical trials of imetelstat in hematologic myeloid malignancies, including the MF Pilot Study and the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement. Obtaining additional and future data from the MF Pilot Study may provide additional insights into the further development of imetelstat for MF, MDS or AML, including with respect to Janssen's ability to initiate the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement. Accordingly, a delay in the timely completion of the MF Pilot Study, including any delay caused by any future clinical hold placed by the FDA on the INDs for imetelstat, including the IND for the MF Pilot Study, that we plan to transfer to Janssen prior to initiation of the Initial Phase 2 MF Study, could have a material adverse effect on advancing the development of imetelstat to subsequent clinical trials, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement. Also, adverse safety results from clinical trials of imetelstat, including those results that have been reported and those that may in the future be reported from the MF Pilot Study or the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, could delay or prevent the initiation or continuation of further clinical development of imetelstat, whether under the Collaboration Agreement or otherwise. Occurrence of any of these events would delay the timing of any Continuation Decision Janssen could provide to us or could cause Janssen to terminate the Collaboration Agreement which would severely and adversely affect the future of imetelstat and our business prospects.In addition, enrollment goals for potential future clinical trials of imetelstat, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, may not be met. The inability to retain or treat patients who have enrolled in a clinical trial but may be prone to withdraw due to side effects from imetelstat, lack of efficacy or personal issues, or who are lost to further follow-up, could result in clinical trial delays, the inability to complete clinical trials, or incomplete data sets. Further, any future clinical trials may be overseen by a safety monitoring committee, which may determine to significantly modify, delay or suspend one or more of these trials due to safety or futility findings based on emerging data occurring during a clinical trial. Data that we have received or that we or Janssen may in the future receive from investigators may be flawed or incomplete if the investigators fail to follow appropriate clinical or quality practices. Delays in timely initiation or completion of clinical testing of imetelstat could increase research and development costs and could prevent or would delay obtaining regulatory approval for imetelstat, either of which would delay the timing of any Continuation Decision Janssen could provide to us or could cause Janssen to terminate the Collaboration Agreement which would severely and adversely affect the future of imetelstat and our business prospects.Obtaining regulatory clearances and approvals to develop and market imetelstat in the United States and other countries is a costly and lengthy process, and we cannot predict whether or when regulatory authorities will permit additional imetelstat development or approve imetelstat for commercial sale.Federal, state and local governments in the United States and governments in other countries have significant regulations in place that govern drug research and development and may prevent us, in collaboration with Janssen, from successfully conducting development efforts or from commercializing imetelstat. Imetelstat must receive all relevant regulatory approvals before it may be marketed in the United States or other countries. Obtaining regulatory approval is a lengthy, expensive and uncertainprocess. Because imetelstat involves the application of new technologies and a new therapeutic approach, it may be subject to substantial additional review by various government regulatory authorities, and, as a result, the process of obtaining regulatory approvals for imetelstat may proceed more slowly than for product candidates based upon more conventional technologies, and any approval that may be received could limit the use of imetelstat.Prior to initiating future clinical trials of imetelstat, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement, the clinical trial protocols must be submitted to the FDA or regulatory authorities in other countries. Questions or comments from these agencies that must be addressed would likely delay further clinical development of imetelstat and the timing of any Continuation Decision by Janssen or could cause Janssen to terminate the Collaboration Agreement, which would severely and adversely affect the future of imetelstat and our business prospects.Prior to submission of any regulatory application seeking approval to commence commercial sales of imetelstat, extensive preclinical and clinical testing will be required to be conducted. If the interpretation of safety and efficacy data obtained from these preclinical and clinical studies varies from interpretations by the FDA or regulatory authorities in other countries, this would likely delay, limit or prevent further development and approval of imetelstat which may cause Janssen to terminate the Collaboration Agreement. For example, the FDA and regulatory authorities in other countries may require more or different data than what has been generated from our preclinical studies and our prior Geron-sponsored Phase 2 clinical trials or the MF Pilot Study or that may be generated by potential future clinical trials, including the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement. In addition, delays or rejections of regulatory approvals, or limitations in marketing authorizations, may be encountered as a result of changes in regulatory environment or regulatory agency policy during the period of product development and/or the period of review of any application for regulatory agency approval for imetelstat. We do not expect imetelstat to be approved for commercial sale for many years, if at all.Delays in obtaining regulatory agency clearances and approvals or limitations in the scope of such clearances or approvals could:•significantly harm the commercial potential of imetelstat;•impose costly procedures upon future development activities;•diminish any competitive advantages that may have been available; or•adversely limit the amount of, or affect our ability to receive, any milestone payments or royalties under the Collaboration Agreement with Janssen.
Even if the necessary time and resources are committed by us and Janssen, the required regulatory agency clearances and approvals may not be obtained for imetelstat. Even if regulatory agency clearances and approvals are obtained to commence commercial sales of imetelstat, they may entail limitations on the indicated uses or other aspects of the product label for which imetelstat can be marketed, which could limit the potential commercial use of imetelstat, or an approval might be contingent on the performance of costly additional clinical trials that would be required after approval. The occurrence of any of these events could delay any applications for regulatory approval and therefore delay the payment of potential milestone payments to us, or, if approved for commercial sale, could reduce the market demand for imetelstat and therefore result in decreased sales and reduced royalties for us under the Collaboration Agreement. Occurrence of any of these events could negatively impact our collaboration with Janssen or cause Janssen to terminate the Collaboration Agreement, which would severely and adversely affect our business and business prospects.Failure to achieve continued compliance with government regulation could delay or halt commercialization of imetelstat, our sole product candidate.Approved products and their manufacturers are subject to continual review, and discovery of previously unknown problems with a product or its manufacturer may result in restrictions on the product or manufacturer, including import restrictions, seizure and withdrawal of the product from the market. If approved for commercial sale, future sales of imetelstat will be subject to government regulation related to numerous matters, including the processes of:•manufacturing;•advertising and promoting;•selling and marketing;•labeling; and•distribution.
If, and to the extent that, we are or Janssen is unable to comply with these regulations, our ability to earn potential milestone payments and royalties from worldwide net sales of imetelstat would be materially and adversely impacted.Failure to comply with regulatory requirements can result in severe civil and criminal penalties, including but not limited to:•recall or seizure of products;•injunctions against the import, manufacture, distribution, sales and/or marketing of products; and•criminal prosecution.
The imposition of any of these penalties or other commercial limitations could negatively impact our collaboration with Janssen or cause Janssen to terminate the Collaboration Agreement, either of which would severely and adversely affect our business and business prospects.Janssen's development activities conductedunder a Janssen Independent Development Plan, or IDP, may create significant reimbursement obligations for us, which could result in reduced cash inflow from future milestone payments and royalties until we have fully paid ourobligations.reimbursement obligations under the Collaboration Agreement.Under the Collaboration Agreement, Janssen may conduct certain development activities for imetelstat under a Janssen IDP, if we and Janssen agree that such activities should be performed outside of the mutually agreed global clinical development plan. Although Janssen would bear all of the costs for such Janssen IDP, if we exercised our U.S. Opt-In Rights and if any data from a Janssen IDP supports approval by a regulatory
agencyauthority in the United States or other countries, then we would be required to reimburse Janssen for our share of the costs of that Janssen IDP plus a premium pursuant to the terms of the Collaboration Agreement. This cost reimbursement is payable as a lump sum up to a certain threshold upon receipt of regulatory approval for the Janssen IDP. Any remaining amounts in excess of the threshold are payable in installments by offsetting milestone payments or royalties received by us over a certain period of time, at which time any remaining reimbursement amount would be payable in a lump sum. This payment mechanism could result in reduced cash inflow from future milestone payments and royalties, which would adversely affect our results of operations and financial condition.Under the Collaboration Agreement,
with Janssen,if we develop imetelstat independently under our own IDP, the success of that IDPmay dependdepends onprovidingour ability to provide adequate financial and technical resources.Failure to successfully conduct or fund our own IDP activities may adversely affect our business.Under the Collaboration Agreement,
with Janssen,we may conduct certain development activities for imetelstat under a Geron IDP if we and Janssen agree that such activities should be performed outside of the mutually agreed global clinical development plan. In the event we conduct any clinical activities under a Geron IDP, we will be responsible for paying all of the development costs for the Geron IDP. Because the outcome of any clinical activities and/or regulatory approval process is highly uncertain, we cannot reasonably estimate whether any Geron IDP activities we may undertake will succeed. Since we are only eligible for reimbursement from Janssen for their share of the Geron IDP costs plus a premium if any data from a Geron IDP supports approval by a regulatoryagencyauthority in the United States or other countries, we may not recoup our investment in any Geron IDP, which could adversely affect our financial condition. In addition, we may need additional capital to support any Geron IDP activities and we cannot assure you that our existing capital resources, future interest income, potential milestone payments and royalties under the Collaboration Agreement and potential future sales of our common stock will be sufficient to fund these future activities. If sufficient capital is not available, we may be unable to pursue activities under a Geron IDP, which could adversely affect our business.To execute activities under a Geron IDP, we likely would be required to collaborate with contract research organizations, investigators, academic institutions, vendors, clinical trial sites, scientific consultants and others. We would be dependent upon the ability of these parties to perform their responsibilities reliably. In addition, we would have limited control over the activities of these organizations, investigators, scientific consultants and vendors. Except as otherwise required by our agreements with them, we could expect only limited amounts of their time to be dedicated to our activities. If any of these third parties were unable or
refuserefused to contribute to projects on which we needed their help, our ability to conduct activities under a Geron IDP could be significantly harmed. Also, if the performance of these services is not of the highest quality, does not achieve necessary regulatory compliance standards, or if such organization or vendor stops or delays its performance for any reason, it would impair and delay our ability to report data from clinical activities under a Geron IDP which would, in turn, hinder our ability to make the necessary representations or provide the necessary information to regulatory authorities, if at all. As a result, we may not obtain regulatory approval and receive any reimbursement from Janssen for their share of the costs for the Geron IDP, which could adversely affect our business and financial condition.WeIf Janssen makes an affirmative Continuation Decision under the Collaboration Agreement, our decision to exercise our U.S. Opt-In Rights must thereafter be made within a short timeframe and, as a result, we may be required to invest substantial capital based on limited clinical data and information.If Janssen makes an affirmative Continuation Decision under the Collaboration Agreement, we must decide whether to elect to exercise our U.S. Opt-In Rights within a short timeframe following such a decision, and although we expect to receive information from Janssen regarding data from IMbark and IMerge, proposed future clinical development plans and costs for imetelstat, estimates in timing for commercializing imetelstat and related promotional activities, and a calculation of our share of development costs incurred to date by Janssen that we will be
dependentrequired to reimburse if we exercise our U.S. Opt-In Rights, we will be required to rapidly decide whether to make a substantial capital investment in imetelstat prior to the conclusion of any Phase 3 registration-enabling clinical trial. Accordingly, if we exercise our U.S. Opt-In Rights and imetelstat were to become unsuccessful in any Phase 3 registration-enabling clinical trial or were to fail to receive regulatory approval, we would not receive any financial return on this substantial capital investment. Such an occurrence would negatively impact our financial condition and results of operations, and might cause us to cease operations.RISKS RELATED TO CLINICAL DEVELOPMENT, REGULATORY APPROVAL AND COMMERCIALIZATION OF IMETELSTAT
The research and development of imetelstat is subject to numerous risks and uncertainties.
The science and technology of telomere biology, telomerase and our proprietary oligonucleotide chemistry are relatively new. There is no precedent for the successful commercialization of a therapeutic product candidate based on these technologies. Significant research and development activities will be necessary to further develop imetelstat, which is our sole product candidate that we have exclusively outlicensed to Janssen, which may take years to accomplish, if at all.
Because of the significant scientific, regulatory and commercial challenges that must be overcome to successfully research, develop and commercialize imetelstat, the development of imetelstat in hematologic myeloid malignancies, including MF and MDS, or any other indications, may be further delayed or abandoned, even after significant resources have been expended on it. Examples of such decisions include:
the discontinuation of our Phase 2 clinical trial of imetelstat in metastatic breast cancer in September 2012;
the discontinuation of our development of imetelstat in solid tumors with short telomeres in April 2013;
Janssen’s decisions in the third quarter of 2016 to close the 4.7 mg/kg dosing arm in IMbark to new patient enrollment and to suspend enrollment in the 9.4 mg/kg dosing arm in IMbark because an insufficient number of patients in the 9.4 mg/kg dosing arm met the protocol defined interim efficacy criteria at 12 weeks; and
Janssen’s decision in the third quarter of 2017 to expand enrollment in Part 1 of IMerge to include approximately 20 additional lower risk MDS patients in a refined target population.
Any further delay, suspension or abandonment of the development of imetelstat in hematologic myeloid malignancies, including delays resulting from potential future protocol amendments for IMerge, IMbark or potential future clinical trials of imetelstat, would have a material adverse effect on our collaboration with Janssen, which could result in the termination of the Collaboration Agreement. Any of these events would have severe adverse effects on the future of imetelstat and our business prospects and likely result in the failure of our business.
Imetelstat may cause, or have attributed to it, undesirable or unintended side effects or other adverse events that delay or prevent the commencement and/or completion of clinical trials for imetelstat, delay or prevent its regulatory approval, or limit its commercial potential.
Imetelstat may cause, or have attributed to it, undesirable or unintended side effects or other adverse events affecting its safety or efficacy that could cause Janssen to interrupt, delay or halt current or potential future clinical trials of imetelstat. For example, adverse events and dose limiting toxicities observed in previous clinical trials of imetelstat include:
hematologic toxicities, such as profound and/or prolonged thrombocytopenia or neutropenia, including one case of febrile neutropenia after prolonged myelosuppression with intracranial hemorrhage resulting in patient death, which the investigator assessed as possibly related to imetelstat;
bleeding events, with or without thrombocytopenia;
liver function test, or LFT, abnormalities, the clinical significance and long-term consequences of which are currently undetermined;
gastrointestinal events;
infections;
muscular and joint pain;
fatigue; and
infusion reactions.
Such adverse events and other safety issues, including deaths, have also been observed by Janssen in IMbark and IMerge. If patients in current or potential future clinical trials of imetelstat experience similar or more severe adverse events, or new or unusual adverse events, or if the FDA or other regulatory authorities determine that efficacy and safety data in current or potential future clinical trials of imetelstat, do not support an adequate benefit-risk profile to justify continued treatment of patients, then the FDA or other regulatory authorities may again place the INDs for imetelstat on clinical hold, as occurred in March 2014.
Further, clinical trials by their nature examine the effect of a potential therapy in a sample of the potential future patient population. As such, clinical trials conducted with imetelstat, to date and in the future, may not uncover all possible adverse events that patients treated with imetelstat may experience. Because remaining patients in the treatment phase continue to receive imetelstat in the Pilot Study, IMbark and IMerge, including the expanded Part 1, additional or more severe toxicities or safety issues, including additional serious adverse events and dose limiting toxicities, may be observed as patient treatment continues and more data become available. In addition, since IMbark, IMerge and the Pilot Study are ongoing studies in which additional data are being generated, the benefit-risk profile of imetelstat will continue to be assessed, including the risk of hepatotoxicity, severe cytopenias, fatal bleeding with or without any associated thrombocytopenia, patient injury or death, and any other severe adverse effects that may be associated with life-threatening clinical outcomes. If such toxicities or other safety issues in any clinical trial of imetelstat are determined by Janssen, the FDA or any other regulatory authority to result in an unacceptable benefit-risk profile, then:
additional information supporting the benefit-risk profile of imetelstat may be requested by the FDA or other regulatory authorities and if any such information supplied by Janssen is not deemed acceptable, current clinical trials of imetelstat could be suspended, terminated, or placed on clinical hold by the FDA or other regulatory authorities;
patient recruitment and the ability to retain enrolled patients in current clinical trials may be negatively affected, resulting in incomplete data sets and the inability to adequately assess the benefit-risk profile of imetelstat in a specific patient population, such as the inability to assess overall survival in IMbark or the benefit-risk profile of imetelstat in the refined target patient population in IMerge; or
additional, unexpected clinical trials or preclinical studies may be required to be conducted.
The occurrence of any of these events could cause Janssen to further delay its Continuation Decision, or abandon the development of imetelstat entirely and terminate the Collaboration Agreement. Any termination of the
Collaboration Agreement by Janssen would have a severe adverse effect on our results of operations, financial condition, business prospects and the future of imetelstat, any of which might cause us to cease operations.
Success in early clinical trials may not be predictive or indicative of results in current clinical trials or potential future clinical trials. Likewise, preliminary data from clinical trials should be considered carefully and with caution since the final data may be materially different from the preliminary data, particularly as more patient data become available.
A number of new drugs and biologics have shown promising results in preclinical studies and initial clinical trials, but subsequently have failed to establish sufficient safety and efficacy data to obtain necessary regulatory approvals to initiate commercial sale. There is typically an extremely high rate of attrition from the failure of product candidates proceeding through clinical trials. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent regulatory approval. Product candidates in later stages of clinical trials may fail to show the desired benefit-risk profile despite having progressed through preclinical studies and initial clinical trials.
Data from our preclinical studies and Phase 1 and Phase 2 clinical trials of imetelstat, including the Pilot Study, as well as the results of the past or future internal data reviews conducted by Janssen for IMbark and IMerge, should not be relied upon as predictive or indicative of future clinical results, including any final results in the Pilot Study, IMbark or IMerge or the results in potential subsequent or larger-scale clinical trials of imetelstat. The results we obtained from the ET Trial and the Pilot Study and the results that have been obtained by Janssen in the internal data reviews conducted for IMbark and IMerge, as well as any future results that may be obtained by Janssen from IMbark and IMerge, may not predict the future therapeutic effect of imetelstat, if any, in hematologic myeloid malignancies, including MF and MDS. For example, the potential disease-modifying activity observed through molecular responses in the ET trial and partial or complete remissions observed in the Pilot Study may not be seen in current or future clinical trials of imetelstat. Since remaining patients in the treatment phase for the Pilot Study, IMbark and IMerge, including the expanded Part 1, continue to receive imetelstat, efficacy and safety data continue to be generated. Such additional and updated data may materially change the overall conclusions from the preliminary data reported for the Pilot Study, IMbark or IMerge. In addition, such additional and updated efficacy and safety data may not support an adequate benefit-risk profile to justify continued treatment of patients enrolled in current clinical trials of imetelstat. Also, the criteria used to assess efficacy in the Pilot Study have not been validated for clinical use and may not be considered by the FDA or other regulatory authorities to be accurate predictors of efficacy for different endpoints that may be required by the FDA or other regulatory authorities for Phase 3 clinical trials.
From time-to-time, preliminary or interim data from current clinical trials, such as the Pilot Study, IMbark and IMerge, or potential future clinical trials, may be reported or announced by Janssen, its investigators, or us. For example, preliminary results of the Pilot Study were presented by the investigator at the American Society of Hematology, or ASH, annual meeting in December 2013, and updated by the investigator at ASH in December 2014, and preliminary data were reported by the investigator from a cohort of MDS patients in the Pilot Study in December 2015. In addition, preliminary data from Part 1 of IMerge was announced in November 2017 and presented at ASH in December 2017. Since such data are preliminary, the final data from any final analysis which may be conducted, or any future analyses of the Pilot Study, IMbark or IMerge, or potential future clinical trials of imetelstat, may be materially different. In addition, changes in study design, including changes in patient enrollment criteria and target patient population, such as the decision to expand enrollment in Part 1 of IMerge for patients in a refined target population, may cause data from later stage clinical trials to differ significantly from data obtained in earlier clinical trials. Preliminary or interim results from the Pilot Study, IMbark or IMerge reported by us, Janssen or by investigators in those trials may not be reproduced in any potential future clinical trials of imetelstat, and thus should not be relied upon as indicative of future clinical results of imetelstat in MF, MDS or in any other hematologic myeloid malignancy. Preliminary or interim data should be considered carefully and with caution.
Material adverse differences in final data, compared to preliminary or interim data, from the Pilot Study, IMbark or IMerge, or potential future clinical trials of imetelstat, could result in a decision by Janssen to discontinue the imetelstat program, further delay its Continuation Decision, or terminate the Collaboration Agreement, any of which would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations. Even if final safety and/or efficacy data from the Pilot Study, IMbark or IMerge or potential future clinical trials of imetelstat are positive, significant additional clinical testing will be
necessary to advance the future development of imetelstat in hematologic myeloid malignancies, including MF or MDS.
Clinical development involves a lengthy and expensive process with uncertain outcomes. Current clinical trials of imetelstat being conducted by Janssen, including IMbark, IMerge and the Pilot Study, and potential future clinical trials of imetelstat may fail to demonstrate sufficient safety and efficacy of imetelstat to warrant further development of the drug, which could prevent or further delay regulatory approval and commercialization of imetelstat.
Before regulatory approvals for the commercial sale of imetelstat can be obtained, clinical testing must be conducted to show that imetelstat is both safe and effective for use in each target indication. Such clinical testing is expensive, can take many years to complete and is inherently uncertain. Failure can occur at any time during clinical testing. Most product candidates that commence clinical trials are never approved as commercial products.
The clinical development of imetelstat will be influenced by results from current clinical trials being conducted by Janssen and
third partiespotential future clinical trials of imetelstat. The advancement of current clinical trials of imetelstat and commencement of potential future clinical trials of imetelstat could be further delayed or abandoned for a variety of reasons, including as a result of failures or delays by Janssen in:demonstrating an adequate improvement in survival in relapsed or refractory MF in IMbark;
otherwise demonstrating sufficient safety and efficacy of imetelstat in IMbark, IMerge and potential future clinical trials without safety issues, side effects or dose-limiting toxicities, including any additional or more severe safety issues in addition to those that have been observed to date in previous or ongoing clinical trials related to imetelstat, whether or not in the same indications or therapeutic areas;
obtaining or maintaining regulatory clearances in the United States or other countries to conduct clinical trials, such as obtaining or maintaining regulatory clearances to commence, conduct or modify current or potential future clinical trials of imetelstat, in a timely manner, or at all;
maintaining the INDs for imetelstat without such INDs being placed on full or partial clinical hold, suspended or subject to other requirements by the FDA or other regulatory authorities;
properly designing, enrolling, conducting or completing: (i) IMerge, including collecting sufficient efficacy and safety data from the expanded Part 1 to assess the benefit-risk profile of imetelstat in the refined target patient population; and (ii) IMbark, by collecting longer-term efficacy and safety data to enable an assessment of overall survival, and promptly or adequately reporting data from such trials;
properly conducting and/or completing the Pilot Study and promptly or adequately reporting data from such trial;
obtaining or accessing necessary clinical data in accordance with appropriate clinical or quality practices to ensure complete data sets;
responding to safety or futility findings by the data review committees of current clinical trials, including IMbark, IMerge and the Pilot Study, and potential future clinical trials of imetelstat, based on emerging data occurring during such clinical trials, such as significant systemic or organ toxicities, including severe cytopenias, hepatotoxicity, fatal bleeding with or without any associated thrombocytopenia, patient injury or death, or other safety issues, resulting in an unacceptable benefit-risk profile;
manufacturing sufficient quantities of imetelstat or other clinical trial materials in a manner that meets the quality standards of the FDA and other regulatory authorities, and responding to any disruptions to drug supply, clinical trial materials or quality issues that may arise;
ensuring the ability to manufacture imetelstat at acceptable costs for potential Phase 3 clinical trials and commercialization;
obtaining sufficient quantities of any study-related treatments, materials (including comparator products, placebo or combination therapies) or ancillary supplies;
•
obtaining acceptance by regulatory authorities of manufacturing changes, as well as successfully implementing any such manufacturing changes;
complying with current and future regulatory requirements, policies or guidelines, including domestic and international laws and regulations pertaining to fraud and abuse, transparency, and the privacy and security of health information;
reaching agreement on acceptable terms and on a timely basis, if at all, with collaborators and vendors located in the United States or foreign jurisdictions, including contract research organizations, laboratory service providers and clinical trial sites, on all aspects of clinical development;
obtaining timely review and clearances by regulatory authorities of future protocol amendments which may be sought for IMbark, IMerge and potential future clinical trials of imetelstat; and
obtaining institutional review board or ethics committee approval of clinical trial protocols or protocol amendments, including any future refinements to the trial design sought for Part 1 and Part 2, if any, of IMerge, or any potential protocol amendments for IMbark.
Failures or delays with respect to any of these events could adversely affect Janssen’s ability to continue or successfully complete any current clinical trials of imetelstat or to initiate potential future clinical trials of imetelstat, which could increase development costs, further delay the timing of the Continuation Decision from Janssen, impair our ability to earn revenues from milestone payments or royalties under the Collaboration Agreement or cause Janssen to terminate the Collaboration Agreement, any of which could severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.
If Janssen encounters difficulties enrolling or retaining patients in current or potential future clinical trials of imetelstat, including the expanded Part 1 of IMerge, clinical development and commercialization activities could be further delayed or otherwise adversely affected, which would cause our business and business prospects to be severely harmed.
The timely completion of a clinical trial in accordance with its protocol depends, among other things, on the ability to enroll a sufficient number of patients who remain in the trial until its conclusion. Janssen may experience difficulties in patient enrollment or retention in IMbark and IMerge, including the expanded Part 1, or potential future clinical trials of imetelstat, for a variety of reasons. The enrollment and retention of patients depends on many factors, including:
the patient eligibility criteria in the protocol;
the size of the patient population required for analysis of the trial’s primary endpoint;
the proximity of patients to trial sites;
the design of the trial;
Janssen’s ability to recruit and retain clinical trial investigators with the appropriate competencies and experience;
clinicians’ and patients’ perceptions of the potential advantages of imetelstat, both in relation to other available therapies, including any new drugs that may be approved for the indications being investigated, and as a result of any preliminary data from current clinical trials;
the ability to obtain and maintain patient consents; and
the risk that patients enrolled in any imetelstat clinical trial will drop out of the trial before completion due to lack of efficacy, adverse side effects, investigator decision, slow progress to later stage clinical trials or personal issues.
In addition, IMbark and IMerge compete, and potential future clinical trials of imetelstat will compete, with other clinical trials for product candidates that are in the same therapeutic areas with imetelstat, and this competition will reduce the number and type of patients available to enroll or remain in the imetelstat clinical trials. Since the number of qualified clinical investigators is limited, IMbark and IMerge are being conducted, and potential future
clinical trials of imetelstat are expected to be conducted, at the same clinical trial sites that competitors use, which will reduce the number of patients who are available for the imetelstat clinical trials at such clinical trial sites. Moreover, because imetelstat represents a departure from more commonly used methods for cancer treatment, potential patients and their doctors may be inclined to use conventional therapies, rather than enroll patients into imetelstat clinical trials.
Delays in patient enrollment or the inability to retain or treat patients could result in increased costs, lead to incomplete data sets, or adversely affect the timing or outcome of current or potential future clinical trials of imetelstat, which could prevent completion of these trials and adversely affect the clinical development and commercialization of imetelstat. For example, delays in or the inability to collect sufficient safety and efficacy data from the expanded Part 1 of IMerge will delay or potentially preclude an assessment of clinical benefit, if any, in the refined target patient population in Part 1. For IMbark, if the data necessary for the protocol-specified primary analysis are not available as a result of patient withdrawals from the trial, insufficient follow-up time, and/or inability to collect follow-up data on such patients, then Janssen will be unable to assess overall survival in IMbark. If Janssen is unable to assess overall survival in IMbark or believes there is not an adequate improvement in survival, we believe Janssen would decide to discontinue the imetelstat program and terminate the Collaboration Agreement. However, Janssen could discontinue the imetelstat program and terminate the Collaboration Agreement at any time and for any reason, irrespective of whether there is data from IMbark suggesting an adequate improvement in survival in relapsed or refractory MF or whether there is data from IMerge to support the benefit-risk profile of imetelstat in lower risk MDS. Such occurrences would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.
Obtaining regulatory clearances and approvals to continue clinical development of, and in the future, to potentially market imetelstat in the United States and other countries, is a costly and lengthy process, and we cannot predict whether or when regulatory authorities will permit additional imetelstat development or approve imetelstat for commercial sale.
Federal, state and local governments in the United States and governments in other countries have significant regulations in place that govern drug research and development and may prevent us, in collaboration with Janssen, from successfully conducting development efforts or from commercializing imetelstat. Delays in obtaining regulatory clearances and approvals or limitations in the scope of such clearances or approvals could:
cause Janssen to terminate the Collaboration Agreement;
impede or halt clinical development activities and plans;
significantly harm the commercial potential of imetelstat;
impose additional development costs;
diminish any competitive advantages that may have been available; or
adversely limit the amount of, or affect our ability to receive, any milestone payments or royalties under the Collaboration Agreement with Janssen.
Prior to initiating potential future clinical trials of imetelstat, clinical trial protocols must be submitted to the FDA or regulatory authorities in other countries. Questions or comments from these agencies regarding any protocol amendments of current clinical trials of imetelstat, including IMbark or IMerge, or protocols for potential future clinical trials of imetelstat, must be addressed in a timely and adequate manner. The inability to timely or adequately address any questions, comments or requests for information from regulatory authorities could impede further clinical development of imetelstat, which could cause Janssen to further delay its Continuation Decision or discontinue the imetelstat program entirely and terminate the Collaboration Agreement, which would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.
Before Janssen can seek to obtain regulatory approval for the commercial sale of imetelstat, multiple clinical trials, including larger-scale Phase 3 clinical trials, will need to be conducted to demonstrate if imetelstat is safe and effective for use in a diverse population. If imetelstat cannot be developed in potential future clinical trials, including Phase 3 clinical trials, our Collaboration Agreement with Janssen will be negatively impacted and likely be terminated
altogether, which would have severe adverse effects on our business and business prospects, and might result in the failure of our business.
If the interpretation by Janssen or us of safety and efficacy data obtained from preclinical and clinical studies varies from interpretations by the FDA or regulatory authorities in other countries, this would likely delay, limit or prevent further development and approval of imetelstat which may cause Janssen to terminate the Collaboration Agreement. For example, the FDA and regulatory authorities in other countries may require more or different data than what has been generated from our preclinical studies and previous or ongoing clinical trials, such as IMbark, IMerge or the Pilot Study. In addition, delays or rejections of regulatory approvals, or limitations in marketing authorizations, may be encountered as a result of changes in the regulatory environment or regulatory policy during the period of product development and/or the period of review of any application for regulatory approval for imetelstat.
The benefit-risk profile of imetelstat will also affect the assessment by the FDA and regulatory authorities in other countries of the drug’s cost-effectiveness and/or marketability, which assessment could prevent or limit its approval for marketing and successful commercial use. If regulatory submissions requesting approval to market imetelstat are submitted, the FDA and regulatory authorities in other countries may conclude that the overall benefit-risk profile of imetelstat treatment does not merit approval of imetelstat for marketing or further development for any indication. Any of these events could cause Janssen to terminate the Collaboration Agreement, which would severely harm our business and business prospects, and might cause us to cease operations.
Imetelstat must receive all relevant regulatory approvals before it may be marketed in the United States or other countries. Obtaining regulatory approval is a lengthy, expensive and uncertain process. For example in June 2016, the electorate in the United Kingdom voted in favor of exiting the European Union, and in March 2017, the Government of the United Kingdom initiated the formal procedure of withdrawal from the European Union. Although the impact of the withdrawal of the United Kingdom from the European Union will not be known for some time, this could lead to a period of considerable uncertainty in relation to the regulatory process in Europe, which could result in a delay in the review of regulatory submissions made in Europe by biotechnology and pharmaceutical companies, and could also lead to less efficient, more expensive, and potentially lengthier regulatory review processes for companies, including Janssen and us, who may seek to obtain regulatory approval for drug products in the European Union or the United Kingdom. Likewise, the Trump Administration has appointed and employed and will appoint and employ many new secretaries, directors and the like into positions of authority in the U.S. federal government dealing with the pharmaceutical and healthcare industries that may potentially have a negative impact on the prices and the regulatory pathways for pharmaceuticals. Such changes could adversely affect and/or delay the ability of Janssen to obtain approval of, and market and sell, imetelstat in the United States. In addition, because imetelstat involves the application of new technologies and a new therapeutic approach, it may be subject to substantial additional review by various government regulatory authorities, and, as a result, the process of obtaining regulatory approvals for imetelstat may proceed more slowly than for product candidates based upon more conventional technologies, and any approval that may be received could limit the use of imetelstat. We do not expect imetelstat to be approved for commercial sale for many years, if at all.
Even if the necessary time and resources are committed by Janssen, the required regulatory clearances and approvals may not be obtained for imetelstat. Further, if regulatory clearances and approvals are obtained to commence commercial sales of imetelstat, they may impose significant limitations on the indicated uses or other aspects of the product label for which imetelstat can be marketed. An approval might also be contingent on the performance of costly additional post-marketing clinical trials. The occurrence of any of these events could limit the potential commercial use of imetelstat, and therefore delay the payment of potential milestone payments to us, or, if approved for commercial sale, could reduce the market demand for imetelstat and therefore result in decreased sales for Janssen and reduced royalties for us under the Collaboration Agreement. Occurrence of any of these events could negatively impact our collaboration with Janssen or cause Janssen to terminate the Collaboration Agreement, which would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.
Although orphan drug designation has been granted to imetelstat for the treatment of MF and MDS, these designations may not be maintained, which would eliminate the benefits associated with orphan drug designation, including the potential for market exclusivity, which would likely result in the reduction of potential imetelstat sales revenue for Janssen, if any, and would likely harm our business and business prospects.
Although the FDA granted orphan drug designation to imetelstat in June 2015 for the treatment of MF and for the treatment of MDS in December 2015, and the European Medicines Agency, or EMA, granted it in December 2015 for the treatment of MF, Janssen may not be the first to obtain marketing approval of a product candidate for the orphan-designated indication due to the uncertainties associated with developing pharmaceutical products. In addition, exclusive marketing rights in the United States or the European Union, if granted, may be limited if Janssen seeks approval for an indication broader than the orphan-designated indication or such marketing exclusivity may be lost if the FDA or the EMA later determines that the request for orphan drug designation was materially defective, or if Janssen is unable to ensure and provide sufficient quantities of imetelstat to meet the needs of patients with the rare disease or condition. Further, even if Janssen obtains orphan drug exclusivity for imetelstat, that exclusivity may not effectively protect imetelstat from competition because different drugs with different active moieties can be approved for the same condition. Even after an orphan drug product is approved, the FDA or EMA can subsequently approve a different drug with the same active moiety for the same condition if the FDA or EMA concludes that the later drug is safer, more effective, or makes a major contribution to patient care. The occurrence of any of these events could result in decreased sales for Janssen and reduced royalties for us, and may harm our business and business prospects. In addition, orphan drug designation will neither shorten the development time nor regulatory review time for imetelstat, and does not give imetelstat any advantage in the regulatory review or approval process.
A fast track designation by the FDA, such as the Fast Track designation received for imetelstat, does not guarantee approval and may not lead to a faster development, regulatory review or approval process.
In October 2017, following submission by Janssen of an application to the FDA requesting fast track designation for the imetelstat clinical development program for the treatment of adult patients with transfusion-dependent anemia due to Low or Intermediate-1 risk MDS who are non-del(5q) and who are refractory or resistant to treatment with an ESA, the FDA notified Janssen that imetelstat has been granted such fast track designation. Fast track designation provides opportunities for frequent interactions with FDA review staff, as well as eligibility for priority review, if relevant criteria are met, and rolling review. Fast track designation is intended to facilitate and expedite development and review of a New Drug Application to address unmet medical needs in the treatment of serious or life-threatening conditions. However, fast track designation does not accelerate conduct of clinical trials or mean that the regulatory requirements are less stringent, nor does it ensure that imetelstat will receive marketing approval or that approval will be granted within any particular timeframe. In addition, the FDA may withdraw fast track designation if it believes that the designation is no longer supported by data emerging from the imetelstat clinical development program.
Failure to achieve continued compliance with government regulations could delay or halt commercialization of imetelstat, which we have exclusively outlicensed to Janssen.
Approved products and their manufacturers are subject to continual review, and discovery of previously unknown problems with a product or its manufacturer may result in restrictions on the product or manufacturer, including import restrictions, seizure and withdrawal of the product from the market. If approved for commercial sale, future sales of imetelstat will be subject to government regulation related to numerous matters, including the processes of:
manufacturing;
advertising and promoting;
selling and marketing;
labeling; and
distribution.
If, and to the extent that, we are or Janssen is unable to comply with these regulations, our ability to earn potential milestone payments and royalties from worldwide net sales of imetelstat would be materially and adversely impacted.
Failure to comply with regulatory requirements can result in severe civil and criminal penalties, including but not limited to:
recall or seizure of products;
injunctions against the import, manufacture, distribution, sales and/or marketing of products; and
criminal prosecution.
The imposition of any of these penalties or other commercial limitations could negatively impact our collaboration with Janssen or cause Janssen to terminate the Collaboration Agreement, either of which would severely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.
RISKS RELATED TO MANUFACTURING IMETELSTAT
Failure by Janssen to manufacture or provide adequate clinical and commercial quantities of imetelstat
which couldon a timely basis, or at all, would result in a delay of clinical trials or regulatoryapprovalapprovals, or lostsales.sales, and our business and business prospects could be severely harmed.UnderIn accordance with the Collaboration Agreement,after a transition period,Janssenwill beis responsible for the manufactureand/or manageand management of the supply of imetelstat on a global basis for all clinical trials andallcommercial activities. Consequently, wewill be,are, and expect to remain, dependent on Janssen to appropriately supplyimetelstat. Janssen may encounter difficulties inimetelstat and other clinical trial materials. The process of manufacturing imetelstat is complex and subject to several risks, including:the ability to scale-up and attain sufficient production
scale-up, including problems involving productionyields with appropriate quality control and qualityassurance,assurance;reliance on third-party manufacturers and suppliers;
supply chain issues, including the timely availability and shelf life requirements of raw materials and other supplies;
shortage of qualified
personnel.personnel; andcompliance with regulatory requirements, which are less well-defined for oligonucleotide products than for small molecule drugs and vary in each country where imetelstat might be sold or used.
As a result of these risks, Janssen may not perform as agreed or may default in its obligations to supply imetelstat or other clinical trial materials for clinical trials and/or commercial activities. Janssen also may fail to deliver the required quantities of imetelstat or other clinical trial materials on a timely
basis.basis, or at required or applicable quality standards. If Janssen were to terminate the Collaboration Agreement, and we chose to pursue imetelstat development independently, we would be reliant upon Janssen for the manufacture and supply of adequate clinical quantities of imetelstat or other clinical trial materials, until such time as we could establish our own independent third-party manufacturers or suppliers, which might not be feasible for a significant period of time, and could significantly delay our ability to further develop imetelstat independently. Any such failure by Janssen to supply imetelstat or other clinical trial materials for clinical trials and/or commercial activities, including to us in the event that the Collaboration Agreement was terminated, could delay current and/or potential future clinical trials and any applications for regulatory approval and therefore delay the payment of potential milestone payments to us, or, if approved for commercial sale, could impairJanssen'sJanssen’s ability to meet the market demand for imetelstat and therefore result in decreased sales for Janssen and reduced royalties forus.us which would severely and adversely affect our financial results, business and business prospects, and might cause us to cease operations.If third parties that manufacture imetelstat fail to perform as needed, then the clinical and commercial supply of imetelstat will be limited.
Currently, third-party contractors perform certain process development or other technical and scientific work with respect to imetelstat, as well as supply starting materials and manufacture drug
substance and drug product.
We do not have direct control over their personnel or operations. We relyJanssen, which is responsible for the manufacture and management of the supply of imetelstat on a global basis for clinical trials and, after any regulatory approval, all commercial activities, currently relies on these third-party contractors to produce and deliver sufficient quantities of imetelstat and other clinical trial materials to support clinical trials on a timely basis and to comply with applicable regulatory requirements.If requested byJanssenwedoes not have direct control over these third-party personnel or operations. Reliance on these third-party manufacturers is subject to numerous risks, including:being unable to identify suitable third-party manufacturers, because the number of potential manufacturers is limited and regulatory authorities may
transfer certainrequire significant activities to validate and qualify any replacement manufacturer, which could involve new testing and compliance inspections;being unable to contract with third-party manufacturers on acceptable terms, or at all;
the inability of third-party manufacturers to timely formulate and manufacture imetelstat or to produce imetelstat in the quantities or of the
agreements with these third partiesquality required toJanssen, if permittedmeet clinical and commercial needs;decisions by third-party manufacturers to exit the
terms and conditions ofcontract manufacturing business during therespective agreements or otherwise allowed by the third parties. If these companies do not perform the work which they are contractedtime required toperform, fail to comply with applicable cGMP regulations, do not complete the work within the expected timelines, fail to produce materials which are suitable for use insupply clinical trials orchoosetoexitsuccessfully produce, store and distribute products;compliance by third-party manufacturers with current Good Manufacturing Practice, or cGMP, standards mandated by the
business, the abilityFDA and state agencies and other government regulations corresponding todevelopforeign regulatory authorities;breach or
manufacturetermination of manufacturing contracts;capacity limitation and scheduling imetelstat
could be significantly harmed. For example, changes to one or more suppliers due toas a priority in contracted facilities; andnatural disasters that affect contracted facilities.
Each of these
or other reasonsrisks could lead to delays or shortages in drugsupply.supply, or the inability to manufacture drug supply necessary for preclinical and clinical activities, and commercialization. In addition, any decision by Janssen to self-manufacture imetelstat, change third-party manufacturers or make changes to manufacturing processes, product vial size or packaging, or formulations for imetelstat, could result in manufacturing delays. Manufacturing delays could adversely impact theinitiation orcompletion offuturecurrent clinical trials, such asthe Initial Phase 2 MF StudyIMbark and IMerge, or theInitial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement,initiation of potential future clinical trials, which may cause Janssen to terminate the Collaboration Agreement or further delay the timing of any Continuation Decision that Janssen could provide to us, either of which would severely and adversely affect our financial results, businessprospects.and business prospects, and the future of imetelstat, and might cause us to cease operations.In addition, current third-party contractors and/or any other contractors utilized by Janssen may need to make substantial investments to enable sufficient capacity increases and cost reductions, and to implement those regulatory and compliance standards necessary for successful Phase 3 clinical trials and commercial
production.production of imetelstat. These third-party contractors may not be able to achieve such capacity increases, cost reductions, or regulatory and compliance standards, and even if they do, such achievements may not be at commercially reasonable costs.Neither we norJanssen currently does not have any long-term commitments or commercial supply agreements with any of the third-party contractors for imetelstat, and changing manufacturers may be prolonged and difficult due to inherent technical complexities and because the number of potential manufacturers is limited. It may be difficult or impossible for Janssen to find a replacement manufacturer on acceptable terms, or at all.There are other risks and uncertainties with respect to manufacturing that could adversely impact the initiation or completion of future clinical trials. For example, certain commonly used reagents and solvents may experience market shortages and, if these shortages occur, such shortages may adversely impact the ability to manufacture imetelstat. If a significant issue arises regarding manufacturing, this may cause Janssen to terminate the Collaboration Agreement or delay the timing of any Continuation Decision that Janssen could provide to us, either of which would severely and adversely affect our business prospects.ImetelstatIt may not beablepossible tobe manufacturedmanufacture imetelstat at costs or scales necessary to conduct clinical trials or potential future commercialization activities.Oligonucleotides are relatively large molecules produced using complex chemistry, and the cost of manufacturing an oligonucleotide like imetelstat is greater than the cost of making typical small-molecule drugs. Therefore, imetelstat for clinical use is more expensive to manufacture than most other treatments currently available today or that may be available in the future. Similarly, the cost of manufacturing imetelstat for commercial use will need to be significantly lower than
ourcurrent costs in order for imetelstat to become a commercially successful product.However,Janssen may not be able to achieve sufficient scale increases or cost reductions necessary for successful commercial production ofimetelstat, whichimetelstat. Failure to achieve necessary cost reductions could result in decreased sales for Janssen and reduced royalties forus.us, could negatively impact our collaboration with Janssen or could cause Janssen to terminate the Collaboration Agreement, any of which would materially and adversely affect our financial results, business and business prospects, and the future of imetelstat, and might cause us to cease operations.Manufacturing imetelstat is subjectRISKS RELATED TO MANAGING OUR GROWTH AND OTHER BUSINESS OPERATIONSWe may not be able to
processsuccessfully identify andtechnical challengesacquire and/or in-license other oncology products, product candidates, programs or companies to grow andregulatory risks.diversify our business, and, even if we are able to do so, we may not be able to successfully manage the risks associated with integrating any such products, product candidates, programs or companies into our business or we may otherwise fail to realize the anticipated benefits of these licenses or acquisitions.We have
facedexclusively outlicensed imetelstat, which was our sole product candidate, to Janssen. Accordingly, we are relying exclusively upon our collaborative relationship with Janssen to further develop, manufacture andJanssencommercialize imetelstat. To grow and diversify our business, we plan to continue our business development efforts to identify and seek to acquire and/or in-license other oncology products, product candidates, programs or companies. Such efforts have not yet resulted in any transaction, and may never result in a transaction. Future growth through acquisition or in-licensing willcontinuedepend upon the availability of suitable products, product candidates, programs or companies for acquisition or in-licensing on acceptable prices, terms and conditions. Even if appropriate opportunities are available, we may not be able toface numerousacquire rights to them on acceptable terms, or at all. The competition to acquire or in-license rights to promising products, product candidates, programs and companies is fierce, and many of our competitors are large, multinational pharmaceutical and biotechnology companies with considerably more financial, development and commercialization resources, personnel, and experience than we have. In order to compete successfully in the current business climate, we may have to pay higher prices for assets than may have been paid historically, which may make it more difficult for us to realize an adequate return on any acquisition. In addition, even if we succeed in identifying promising products, product candidates, programs or companies, we may not have the ability to develop, obtain regulatory approval for and commercialize such opportunities, or the financial resources necessary to pursue them.Even if we are able to successfully identify and acquire or in-license new products, product candidates, programs or companies, we may not be able to successfully manage the risks associated with integrating such products, product candidates, programs or companies into our business or the risks arising from anticipated and unanticipated problems in connection with an acquisition or in-licensing, including risks related to intellectual property, research, manufacturing, regulatory approval and/or commercialization. Further, while we seek to mitigate risks and liabilities of potential acquisitions through, among other things, due diligence, there may be risks and liabilities that such due diligence efforts fail to discover, that are not disclosed to us, or that we inadequately assess. Any failure in identifying and managing these risks and uncertainties
with regardeffectively would have a material adverse effect on our business. In any event, we may not be able tomanufacturing imetelstat. Regulatory requirementsrealize the anticipated benefits of any acquisition or in-licensing foroligonucleotide products are less well-defined than for small-molecule drugs, and there is no guaranteea variety of reasons, including the possibility thatJanssen will achieve sufficienta productquality standards required for Phase 3candidate fails to advance to clinical development, proves not to be safe or effective in clinical trials, orforfails to reach its forecasted commercialapprovalpotential or that the integration of a product, product candidate, program or company gives rise to unforeseen difficulties andmanufacturing ofexpenditures. Any failure in identifying and managing these risks and uncertainties would have a material adverse effect on our business.TableIn addition, acquisitions create other uncertainties and risks, particularly when the acquisition takes the form ofContentsa merger or other business consolidation. We may encounter unexpected difficulties, or incur unexpected costs, in connection with transition activities and integration efforts, which include:imetelstat. Changeshigh acquisition costs;the need to incur substantial debt or engage in dilutive issuances of equity securities to pay for acquisitions;
the potential disruption of our historical business and our activities under the Collaboration Agreement;
the strain on, and need to expand, our existing operational, technical, financial and administrative infrastructure;
our lack of experience in late-stage product development and commercialization;
the difficulties in assimilating employees and corporate cultures;
the difficulties in hiring qualified personnel and establishing necessary development and/or commercialization capabilities;
the failure to retain key management and other personnel;
the challenges in controlling additional costs and expenses in connection with and as a result of the acquisition;
the need to write down assets or recognize impairment charges;
the diversion of our management’s attention to integration of operations and corporate and administrative infrastructures; and
any unanticipated liabilities for activities of or related to the acquired business or its operations, products or product candidates.
If we fail to integrate or otherwise manage an acquired business successfully and in a timely manner, resulting operating inefficiencies could increase our costs more than we planned, could negatively impact the market price of our common stock and could otherwise distract us from execution of our strategy. Failure to maintain effective financial controls and reporting systems and procedures could also impact our ability to produce timely and accurate financial statements.
In addition, the Collaboration Agreement with Janssen prohibits us from commercializing, under the intellectual property we have licensed exclusively to Janssen, any substance whose identified or known mechanism of action is telomerase inhibition. Further, if we exercise our U.S. Co-Promotion Option under the Collaboration Agreement, we will be required to certify to Janssen at the time of exercising our U.S. Co-Promotion Option that we are not marketing or promoting, and have no right to market or promote, any such products for any oncology indication. Our right to co-promote in the
manufacturing processes or formulations for imetelstat thatUnited States may bemade during later stagesterminated by Janssen if we develop or commercialize a product for treating an oncology indication that acts through the same mechanism ofclinical development, including during Phase 3 clinical trials, may result in regulatory delays, the needaction as imetelstat or that is substitutable forfurther clinical trials, rejection of a marketing application, or limitation on marketing authorization by regulatory authorities, and therefore delay the payment of potential milestone payments to us, or, if approved for commercial sale,imetelstat. Accordingly, our Collaboration Agreement with Janssen couldimpair Janssen'sadversely affect our ability tomeetacquire or in-license, or to research, develop or market, promising products, product candidates or programs.We may be unable to successfully retain or recruit key personnel to support our collaboration with Janssen or to manage any future growth.
Our future growth and success depend to a significant extent on the
market demandskills, experience and efforts of our executive officers and key members of our staff. We face intense competition for qualified individuals from numerous pharmaceutical, biopharmaceutical and biotechnology companies, as well as academic and other research institutions. The previous restructurings we implemented, as well as the fact that we exclusively outlicensed imetelstat, which was our sole product candidate, to Janssen, and the uncertainties regarding our ability to diversify our business or related to the continued development of imetelstat by Janssen, could have an adverse impact on our ability to retain and recruit qualified personnel or we may incur unanticipated inefficiencies caused by our reduced personnel resources. In addition, if we acquire or in-license new products, product candidates, programs or companies as a result of ourbusiness development efforts, we may not be able to successfully retain or recruit any executive officers or key staff members knowledgeable about such new products, product candidates, programs or companies. Under the terms of the Collaboration Agreement, we and Janssen have created a joint governance structure, including joint committees and working groups, to manage worldwide regulatory, development, manufacturing and commercialization activities for imetelstat, and
therefore resultwe have ongoing responsibilities to oversee and participate indecreased salesthe collaboration with Janssen. In addition, we remain responsible for prosecuting, at Janssen’s direction, the patents we exclusively licensed to Janssen, andreduced royaltieshave sole responsibility forus.those patents that were non-exclusively licensed to Janssen. If we are unable to successfully retain, motivate and incentivize our personnel or attract or assimilate other highly qualified management and development personnel in the future on acceptable terms, our ability to support the Collaboration Agreement with Janssen and any future growth could be impaired, and our business and the price of our common stock would be adversely impacted.We have not yet negotiated our agreement with Janssen specifying all of the terms for our co-promotion of imetelstat should we exercise our U.S. Co-Promotion Option. In addition, we do not have a sales force and may not develop
one.an effective one, if at all.Pursuant to the Collaboration Agreement with Janssen, we have a U.S. Co-Promotion Option if we exercise our U.S. Opt-In Rights. Assuming we exercise the U.S. Co-Promotion Option, we can elect to provide 20% of the U.S. imetelstat selling effort with Geron sales force personnel, in lieu of funding 20% of U.S. promotion costs upon regulatory approval and commercial launch of imetelstat in the United States. While the Collaboration Agreement includes the material terms of our U.S. Co-Promotion Option, we and Janssen mutually agreed to negotiate a separate agreement specifying
thedetailed activities and responsibilities with respect to the marketing and co-promotion of imetelstat following our election to exercise our U.S. Co-Promotion Option.WeIf Janssen makes an affirmative Continuation Decision, and we subsequently exercise our U.S. Opt-In Rights and U.S. Co-Promotion Option, we will need to negotiate this separate agreement with Janssen and, as a result, Janssen mayplaceimpose restrictions or additional obligations on us, including financial obligations. Any restrictions or additional obligations may restrict our co-promotion activities or involve more significant financial or other obligations than we currently anticipate. In addition, we have no sales experience as a company, and there are risks involved with establishing our own sales forcecapabilities. Developing an internalcapabilities, including:incurring substantial expenditures to develop a sales force and
function will require substantial expenditures and will be time-consuming, may expose usfunction;exposure to unforeseen costs and
expenses,expenses; andwe may not be ablebeing unable to effectively recruit, train or retain sales personnel.
Accordingly, we may be unable to establish our own sales force, which
could effectivelywould delay or preclude us from participating in co-promoting imetelstat in the United States. In addition, because of our current lack of expertise in sales operations, any sales force we establish may not be effective, or may be less effective than any sales force that Janssen utilizes to promote imetelstat. In such event, the commercialization of imetelstat may be adversely affected,whichsince we would be wholly reliant on Janssen’s sales efforts, and this could materially and adversely affect any sales milestone or royalties we may receive under the Collaboration Agreement.The Collaboration Agreement limits our ability to transfer our U.S. Co-Promotion Option to a potential acquirer.
Although the Collaboration Agreement permits us to be acquired by any company, our right to transfer our U.S. Co-Promotion Option
toin the case of an acquisition, merger, consolidation, share exchange, business combination, recapitalization, sale of apotential acquirermajority of assets or similar transaction is limited, and subject toJanssen'sJanssen’s sole discretion under certain circumstances. If we are acquiredunderoutside of such limited circumstances, then we may not be able to transfer the U.S. Co-Promotion Option to such acquirer as part of the acquisition. This limiting provision may discourage potential acquisition bids for us or lower our value, thus preventing holders of our common stock from benefiting from what they may believe are the positive aspects of an acquisition, including the potential realization of a higher rate of return on their investment from this type of transaction.We may not be able to obtain or maintain sufficient insurance on commercially reasonable terms or with adequate coverage against potential liabilities in order to protect ourselves against product liability claims.
Our business exposes us to potential product liability risks that are inherent in the testing, manufacturing and marketing of human therapeutic and diagnostic products. We may become subject to product liability claims if the use of imetelstat is alleged to have injured patients, including any injuries alleged to arise from any hepatotoxicity or hemorrhagic event associated with the use of imetelstat. We currently have limited clinical trial liability insurance, and we may not be able to maintain this type of insurance for any clinical trials, including clinical trials that we may conduct under a Geron IDP or in collaboration with Janssen under the Collaboration Agreement. In addition, product liability insurance is becoming increasingly expensive. Being unable to obtain or maintain product liability insurance in the future on acceptable terms or with adequate coverage against potential liabilities could have a material adverse effect on our business.
We have been, and may in the future be, involved in securities-related legal actions that are expensive and time consuming. Any securities-related legal actions, if resolved adversely, could harm our business, financial condition, or results of operations.
Securities-related class action lawsuits and/or derivative lawsuits have often been brought against companies which experience volatility in the market price of their securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies often experience significant stock price volatility in connection with their product development programs.
We and certain of our officers were named as defendants in two purported class action securities lawsuits filed in the United States District Court for the Northern District of California, or the California District Court, as well as a third securities lawsuit, not styled as a class action, which was transferred to the California District Court. These three cases, or the Class Action Lawsuits, were consolidated for all purposes and settled in July 2017. In connection with the settlement, in April 2017, we paid $250,000 and our insurance providers paid $6.0 million to a settlement escrow account to be paid to members of the settlement class, less payment of attorneys’ fees and costs to plaintiff’s counsel.
It is possible that additional suits will be filed, or allegations received from stockholders naming us and/or our officers and directors as defendants with respect to these same or other matters. Monitoring, initiating and defending against legal actions is time-consuming for our management, is likely to be expensive and may detract from our ability to fully focus our internal resources on our business activities. We could be forced to expend significant resources in the settlement or defense of any additional lawsuits, and we may not prevail in such lawsuits. We have not established any reserve for any potential liability relating to any additional lawsuits. It is possible that we could, in the future, incur judgments or enter into settlements of claims for monetary damages. A decision adverse to our interests in any such lawsuit, or in similar or related litigation, could result in the payment of substantial damages, or possibly fines, and could have a material adverse effect on our business, cash flow, results of operations and financial condition.
We may be subject to litigation, including securities-related litigation, if the results of our business and collaboration activities are not successful, and such litigation would be costly to defend or pursue and uncertain in its outcome.
Our business may bring us into conflict with our licensees, licensors, or others with whom we have contractual or other business relationships, or with our competitors or others whose interests differ from ours. If we are unable to resolve those conflicts on terms that are satisfactory to all parties, we may become involved in litigation brought by or against us.
On November 13, 2014, we announced that we had entered into the Collaboration Agreement with Janssen. We may face litigation arising from or related to the Collaboration Agreement or the transactions contemplated thereby, including if we are unable to generate substantial value under the Collaboration Agreement with Janssen or such collaboration is otherwise unsuccessful. For example, as a result of possible disagreements with Janssen, we may become involved in litigation or arbitration, which would be time-consuming and expensive. Possible disagreements with Janssen could include disagreements regarding the development and/or commercialization of imetelstat, interpretation of the Collaboration Agreement and ownership of proprietary rights. In addition, in certain circumstances we may believe that a particular milestone under the Collaboration Agreement has been achieved, and
Janssen may disagree with our belief. In that case, receipt of that milestone payment may be delayed or may never be received, which would adversely affect our financial condition and may require us to adjust our operating plans. While the Collaboration Agreement provides for a joint governance structure to oversee and manage worldwide regulatory, development, manufacturing and commercialization activities for imetelstat, Janssen generally will, subject to limited exceptions, have the deciding vote in the event of any disagreement. In any event, the joint governance structure contemplated by the Collaboration Agreement will be dissolved in the event that Janssen makes an affirmative Continuation Decision and we do not exercise our U.S. Opt-In Rights, which would preclude our ability to participate in any further decision-making for imetelstat. Reliance on a joint governance structure also subjects us to the risk that changes in key Janssen management personnel that are members of the various joint committees may materially and adversely affect the functioning of these committees, which could significantly delay or preclude imetelstat development and/or commercialization.
The Collaboration Agreement could also result in litigation arising out of any claims that our stockholders suffered financial losses due to the transaction, the approval of our stockholders was required under applicable law or otherwise should have been obtained prior to the completion of the transaction, or that our officers and directors breached their fiduciary duties in connection with the approval and completion of the transaction. Although we believe that stockholder approval was not required under applicable law in order to complete our transaction with Janssen, and therefore we neither sought nor intend to seek such stockholder approval, it is possible that persons who were stockholders at the time of the transaction may claim that their approval was required, in which case litigation could follow, which could result in substantial damages to us and/or could negatively affect our rights and obligations, or result in the termination of, the Collaboration Agreement.
Likewise, our stockholders may believe that the financial and other terms of the Collaboration Agreement are not favorable to either us or our stockholders, including any belief that the potential payments we may receive under the Collaboration Agreement are inadequate. Litigation brought by our stockholders challenging the validity of, or financial losses resulting from the Collaboration Agreement could also result in claims against us by Janssen, and the Collaboration Agreement provides for indemnification by us of Janssen against all losses and expenses relating to breaches of our representations, warranties and covenants in the Collaboration Agreement, which could expose us to further financial obligations and damages. The occurrence of any one or more of the above could have a significant adverse impact on our business and financial condition.
In addition, if the results of our business and collaboration activities are not successful, including without limitation, for example, if:
•
we receive a negative Continuation Decision from Janssen or Janssen otherwise terminates the Collaboration Agreement;
•
serious adverse events are encountered in current and potential future clinical trials of imetelstat; or
our stock price would decline significantly, and future litigation may result. In addition, allegations against us may be made related to the duration and nature of follow-up of patients, or any other activities conducted by Janssen or us in current and potential future clinical trials of imetelstat, and the nature and timing of our disclosures related to efficacy or safety data observed in current and potential future clinical trials of imetelstat may be alleged to have been inadequate or incomplete.
Monitoring, initiating and defending against legal actions is time-consuming for our management, likely to be expensive and may detract from our ability to fully focus our internal resources on our business activities. In addition,
despite the availability of insurance, we may incur substantial legal fees and costs in connection with litigation. Lawsuits are subject to inherent uncertainties, and defense and disposition costs depend upon many unknown factors. Lawsuits could result in judgments against us that require us to pay damages, enjoin us from certain activities, or otherwise negatively affect our legal or contractual rights, which could have a significant adverse effect on our business. In addition, the inherent uncertainty of such litigation could lead to increased volatility in our stock price and a decrease in the value of our stockholders’ investment in our common stock.
RISKS RELATED TO PROTECTING OUR INTELLECTUAL PROPERTY
We remain responsible for prosecuting, at
Janssen'sJanssen’s direction, the patents we have exclusively licensed to Janssen. The success of our collaboration with Janssen will depend on our ability to protect our technologies and imetelstat through patents and other intellectual property rights.Protection of our proprietary technology is critically important to our business, especially with respect to our collaboration with Janssen. Our success will depend in part on our ability to obtain, maintain, enforce and extend our patents and maintain trade secrets, both in the United States and in other
countries.
If we are unsuccessful in any of these regards, the value of our technologies and imetelstat will be adversely affected, and we and/or Janssen may be unable to continue development of imetelstat. By way of example, we do not yet have issued compound patent coverage for imetelstat in Europe after 2020. Further, ourOur patents may be challenged, invalidated or circumvented, and our patent rights may not provide proprietary protection or competitive advantages to us or Janssen. In the event that weor our licensorsare unsuccessful in obtaining, maintaining, and enforcing our patents and other intellectual property rights, the value of our technologies and imetelstat will be adversely affected, and we or Janssen may not be able or willing to further develop or commercialize imetelstat. Loss or impairment of our intellectual property related to imetelstatany of which couldmight delay or halt ongoing or potential future clinical trials of imetelstat and any applications for regulatory approval, and therefore delay or halt the payment of potential milestone payments to usor,under the Collaboration Agreement. Further, if imetelstat is approved for commercial sale, such events could impairJanssen'sJanssen’s ability to sell imetelstat and therefore result in decreased sales for Janssen and reduced royalties for us. Occurrence of any of these events could negatively impact our collaboration with Janssen or cause Janssen to terminate the Collaboration Agreement, which would materially and adversely affect our financial results, business andwe may be unablebusiness prospects, and the future of imetelstat, and might cause us tocontinuecease operations.Changes in U.S. or foreign patent law or interpretations of such patent laws could diminish the value of our
operations.patents in general, thereby impairing our ability to protect our technologies and imetelstat.The patent positions of pharmaceutical and biopharmaceutical companies, including ours, are highly uncertain and involve complex legal and technical questions. In particular, legal principles for biotechnology and pharmaceutical patents in the United States and in other countries are evolving, and the extent to which we will be able to obtain patent coverage to protect our technologies and imetelstat, or enforce or defend issued patents, is uncertain.
If we or Janssen infringe the patents of others, we or Janssen may be blocked from continuing development work or be required to obtain licenses on terms that may impact the value of imetelstat or cause it to be commercially impracticable.A number of significant changes to U.S. patent law occurred when the Leahy-Smith America Invents Act, or the AIA, was signed into law on September 16, 2011. These include provisions that affect the way patent applications will be prosecuted and may affect patent litigation. The United States Patent and Trademark Office, or the Patent Office, has developed new and untested regulations and procedures to govern the full implementation of the AIA. Many of the substantive changes to patent law associated with the AIA, and in particular, the first to file provisions, became effective on March 16, 2013. For example, under the AIA, patent rights are awarded to the first inventor to file a patent application with respect to a particular invention.Since the publication of discoveries in scientific or patent literature tends to lag behind actual discoveries by at least several months and sometimes several years, the persons or entities that weor our licensorsname as inventors in our patents and patent applications may not have been the first to invent the inventions disclosed in the patent applications or patents, or the first to file patent applications for these inventions. As a result, we may not be able to obtain patents for discoveries that we otherwise would consider patentable and that we consider to be extremely significant to the future success of imetelstat. Thus, our ability to protect our patentable intellectual property depends, in part, on our ability to be the first to file patent applications with respect to our inventions or any joint inventions that we may develop with Janssen. Delay in the filing of a patent application for any purpose, including further development or refinement of an invention, may result in the risk of loss of patent rights.A number of significant changes to U.S. patent law occurred when the Leahy-Smith America Invents Act, or the AIA, was signed into law on September 16, 2011. These include provisions that affect the way patent applications are examined and may affect patent litigation. Many of the substantive changes to patent law associated with the AIA, and in particular, the first to file provisions, became effective on March 16, 2013. For example, the AIA limits where a patentee may file a patent infringement suit. The AIA and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
Significant impairmentU.S. court rulings have narrowed the scope of
our imetelstatpatent protection available in certain circumstances and weakened the rightswould have a material adverse effectof patent owners in certain situations. For example, onour business and could cause Janssen to terminateJune 13, 2013, theCollaboration Agreement which would severely and adversely affect our business prospects.TheU.S. Supreme Court, or the Court,has alsoissueddecisions affecting patents. On June 13, 2013,a decision inAssociation for Molecular Pathology v. Myriad Genetics, Inc.the Court heldholding that claims to isolated genomic DNA were not patentable subject matter, but claims to complementary DNA, or cDNA, molecules werepatentable subject matter.
The effect of the decision on patents for other isolated natural products is uncertain.On March 20, 2012, inMayo Collaborative Services, DBA Mayo Medical Laboratories, et al. v. Prometheus Laboratories, Inc., the Court held that several claims drawn to measuring drug metabolite levels from patient samples and correlating them to drug doses were notpatentable subject matter.
The decision has created uncertainty aroundIn addition, court rulings in cases such as BRCA1- & BRCA2-Based Hereditary Cancer Test Patent Litig. and Promega Corp. v. Life Technologies Corp. have also narrowed theabilityscope of patent protection available in certain circumstances. In addition topatent certain biomarker-related method patents. These decisions have increased theincreasing uncertainty with regard to our ability to obtain patents in the future,as well asthis combination of events may have created uncertainty with respect to the value ofcurrentcertain patents we have previously obtained or in-licensed.In addition, in June 2016, the electorate of the United Kingdom voted to exit the European Union, and
futurein March 2017 the Government of the United Kingdom initiated the formal procedure of withdrawal from the European Union. While the exit of the United Kingdom from the European Union is to be completed in 2019, the exact timing of the withdrawal and the resulting effect of withdrawal will not be known for some time, which could lead to a period of considerable uncertainty relating to our ability to obtain and maintain Supplementary Protection Certificates (SPCs) of our products based on our United Kingdom patentsonce obtained.and our ability to establish and maintain European trademarks in the United Kingdom.Depending on decisions by the U.S. federal courts,
andthe Patent Office and similar authorities in foreign jurisdictions, the interpretation of laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents. Occurrence of these eventscould significantly impairand/or significant impairment of our imetelstat patent rightswhichwouldhave a material adverse effect onseverely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and could cause Janssen to terminate the CollaborationAgreement.Agreement, which might cause us to cease operations.Challenges to our patent rights
canwould result in costly and time-consuming legal proceedings thatmaycould prevent or limit development of imetelstat.Our patents may be challenged through administrative or judicial
proceedings. Suchproceedings,are typically lengthy and complex, and an adverse decision canwhich could result in the loss of important patent rights. For example, where more than one party seeks U.S. patent protection for the same technology, the Patent Office may declare an interference proceeding in order to ascertain the party to which the patent should be issued. Patent interferences are typically complex, highly contested legal proceedings, subject to appeal. They are usually expensive and prolonged, and can cause significant delay in the issuance of patents. Our pending patent applications, or our issued patents, may be drawn into interference proceedings or be challenged through post-grant review procedures or litigation, any of whichmaycould delay or prevent the issuance of patents, or result in the loss of issued patent rights.Under the AIA, interference proceedings
have been eliminated forbetween patent applications filed on or after March 16, 2013andhave been replaced with other types of proceedings, including derivation proceedings. The AIA also includes post-grant review procedures subjecting U.S. patents to post-grant review procedures similar to Europeanoppositions.oppositions, such as inter partes review, or IPR, covered business method post-grant reviews and other post-grant reviews. This applies to all of our U.S. patents, even those issued before March 16, 2013. Because of a lower evidentiary standard necessary to invalidate a patent claim in Patent Office proceedings compared to the evidentiary standard in U.S. federal court, a third party could potentially provide evidence in a Patent Office proceeding sufficient for the Patent Office to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party could attempt to use the Patent Office procedures to invalidate patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. U.S. patents owned or licensed by us may therefore be subject to post-grant review procedures, as well as other forms of review and re-examination.A decision in such proceedings adverseIn addition, the IPR process under the AIA permits any person, whether they are accused of infringing the patent at issue or not, to challenge the validity of certain patents. As a result, entities associated with hedge funds have challenged valuable pharmaceutical patents through the IPR process. Significant impairment of ourinterests could result in the loss of valuableimetelstat patent rightswhichwouldhave a material adverse effect onseverely and adversely affect our financial results, business and business prospects, and the future of imetelstat, and could cause Janssen to terminate the Collaboration Agreement, whichwould severely and adversely affect our business prospects.might cause us to cease operations.Certain jurisdictions, such as Europe, New Zealand and Australia, permit oppositions to be filed against granted patents or patents proposed to be granted. Under the Collaboration Agreement, Janssen could commercialize imetelstat internationally if approved by regulatory authorities for commercial sale. Therefore, securing both
proprietary protection and freedom to operate outside of the United States is important to the Collaboration Agreement with Janssen and our business.
We have been involved in both opposing the grant of patents to others through such oppositionOpposition proceedingsand in defending our patent applications against oppositions filed by others. These opposition proceedings requiredrequire significant time and costs,to protect our intellectual property rights. Ifand if we are unsuccessful or are unable to commit these types of resourcesforto protect our imetelstat patent rights, we could lose our patent rights and we and/or Janssen could be prevented or limited in the development and commercialization of imetelstat.Occurrence of any of these events could negatively impact our collaboration with Janssen or cause Janssen to terminate the Collaboration Agreement which would severely and adversely affect our business prospects.As more groups become engaged in scientific research and product development in the areas of telomerase biology, the risk of our patents, or patents that we have in-licensed, being challenged through patent interferences, derivation proceedings, IPRs, post-grant proceedings, oppositions, re-examinations, litigation or other means will likely increase. For example, litigation may arise as a result of our decision to enforce our patent rights against third parties. Challenges to our patents through these procedures
canwould be extremely expensive and time-consuming, even if the outcomeiswas favorable to us. An adverse outcome in a patent dispute could severely harm our collaboration with Janssen or cause Janssen to terminate the Collaboration Agreement, or could otherwise have a material adverse effect on our business, and might cause us to cease operations, by:•causing us to lose patent rights in the relevant jurisdiction(s);
•subjecting us to litigation, or otherwise preventing Janssen or us from commercializing imetelstat in the relevant jurisdiction(s);
•requiring Janssen or us to obtain licenses to the disputed patents;
•forcing Janssen or us to cease using the disputed technology; or
•requiring Janssen or us to develop or obtain alternative technologies.
We or Janssen may be subject to infringement claims that are costly to defend, and as to which we may be obligated to indemnify Janssen or obtain unblocking licenses, and such claims may limit our or
Janssen'sJanssen’s ability to use disputed technologies and prevent us or Janssen from pursuing research,anddevelopment, manufacturing or commercialization of imetelstat.The commercial success of imetelstat will depend upon our and
Janssen'sJanssen’s ability to research, develop, manufacture, market and sell imetelstat without infringing or otherwise violating the intellectual property and other proprietary rights of third parties. There is considerable intellectual property litigation in the biotechnology and pharmaceutical industries, and many pharmaceutical companies, including potential competitors, have substantial patent portfolios. In the event our technologies infringe the rights of others or require the use of discoveries and technologies controlled by third parties, we or Janssen may be prevented from pursuing research, development, manufacturing or commercialization of imetelstat, or may be required to obtain licenses to those patents or other proprietary rights or develop or obtain alternative technologies. For example, we are aware that certain third parties have or may be prosecuting patents and patent estates that may relate to imetelstat, and while we believe these patents will expire before imetelstat is commercialized and/or that these patents are invalid and/or would not be infringed by the manufacture, use or sale of imetelstat, it is possible that the owner(s) of these patents will assert claims against us and/or Janssen in the future. Under the Collaboration Agreement, we are obligated under certain circumstances to indemnify Janssen from any claim of infringement of the patent rights of third parties inJanssen'sJanssen’s development, manufacture or commercialization of imetelstat, or to obtain unblocking licenses from such third parties, at our cost.Since we cannot be aware of all intellectual property rights potentially relating to imetelstat and its uses, we do not know with certainty that imetelstat, or the intended commercialization thereof, does not and will not infringe or otherwise violate any third
party'sparty’s intellectual property. Any infringement claims against us or Janssen would likely be expensive to resolve, and the cost of any indemnification of Janssen or unblocking license that we could be required to obtain under the Collaboration Agreement is unpredictable and could be significant. If we or Janssen are unable to resolve an infringement claim successfully, we or Janssen could be subject to an injunction which would prevent us or Janssen from commercializing imetelstat, and could also require us or Janssen to pay substantial damages. In addition to infringement claims, in the future we or Janssen may also be subject to other claims relating to intellectual property, such as claims that we or Janssen have misappropriated the trade secrets of third parties.We expectProvided thatas imetelstatJanssen continues to progressinthe development of imetelstat, wewillexpect to see more efforts by others to obtain patents that are positioned to cover imetelstat. Our success thereforedepends significantly on our and
Janssen'sJanssen’s ability to operate without infringing patents and the proprietary rights of others.We or Janssen may become aware of discoveries and technologies controlled by third parties that are advantageous to developing or manufacturing imetelstat. Under such circumstances, we or Janssen may initiate negotiations for licenses to other technologies as the need or opportunity arises. We or Janssen may not be able to obtain a license to a technology required for the research, development,
manufacturingmanufacture or commercialization of imetelstat on commercially favorable terms, or at all, oroursuch licenses may be terminated on certain grounds, including as a result of our or Janssen’s failure to comply withourthe obligations under such licenses. If we or Janssen do not obtain a necessary license or if such a license is terminated, we or Janssen may need to redesignoursuch technologies or obtain rights toalternatealternative technologies, which may not be possible, and even if possible, could cause delays in the development efforts for imetelstat and could increase the development and/or production costs of imetelstat. In cases where we or Janssen are unable to license necessary technologies, we and/or Janssen could be subject to litigation and prevented from researching, developing, manufacturing or commercializing imetelstat, and in certain circumstances we may be required to indemnify Janssen for infringement claims arising fromJanssen'sJanssen’s research, development, manufacture or commercialization of imetelstat, which could materially and adversely impact our business. Failure by us or Janssen to obtain rights to alternative technologies or a license to any technology that may be required to research, develop, manufacture or commercialize imetelstat would delay potential future clinical trials of imetelstat and any applications for regulatory approval and therefore delay the payment of potential milestone payments to us, or, if imetelstat is approved for commercial sale, could impairJanssen'sJanssen’s ability to sell imetelstat and therefore result in decreased sales for Janssen and reduced royalties for us. Occurrence of any of these events could negatively impact our collaboration with Janssen or cause Janssen to terminate the Collaboration Agreement, which would materially and adversely affect our business, andwe may be unablemight cause us tocontinue ourcease operations.We may become involved in disputes with Janssen or any past or future collaborator(s) over intellectual property inventorship or ownership, and publications by
ourus or Janssen, or by investigators, scientific consultants,andresearch collaborators or others could impair our ability to obtain patent protection or protect our proprietary information, which, in either case, could have a significant impact on our business.Inventions discovered under research, material transfer or other such collaborative agreements, including our Collaboration Agreement with Janssen, may become jointly owned by us and the other party to such agreements in some cases and the exclusive property of either party in other cases. Under some circumstances, it may be difficult to determine who invents and owns a particular invention, or whether it is jointly owned, and disputes
couldcan arise regarding inventorship and ownership of those inventions. These disputes could be costly andtime consumingtime-consuming and an unfavorable outcome could have a significant adverse effect on our business if we were not able to protect or license rights to these inventions. In addition, clinical trial investigators, scientific consultants and research collaborators generally have contractual rights to publish data and other proprietary information, subject to review by us and/or Janssen. Publications by us or Janssen, or by investigators, scientific consultants,andprevious employees, research collaboratorscontaining such information,or others, either with permission or in contravention of the terms of their agreements or otherwise, may impair the ability to obtain patent protection or protect proprietary information which would have a material adverse effect on our business and could cause Janssen to terminate the CollaborationAgreement.Agreement, which might cause us to cease operations.Much of the information and know-how that is critical to our business is not patentable, and we may not be able to prevent others from obtaining this information and establishing competitive enterprises.
We sometimes rely on trade secrets to protect our proprietary technology, especially in circumstances in which we believe patent protection is not appropriate or available. We attempt to protect our proprietary technology in part by confidentiality agreements with our employees, consultants, collaborators and contractors. We cannot provide assurance that these agreements will not
be breached, that we would have adequate remedies for any breach, or that our trade secrets will not otherwise become known or be independently discovered by competitors, any of which would harm our business significantly.
In May 2016, the Defend Trade Secrets Act of 2016, or the DTSA, was enacted, providing a federal cause of action for misappropriation of trade secrets. Under the DTSA, an employer may not collect enhanced damages or attorney fees from an employee or contractor in a trade secret dispute brought under the DTSA, unless certain advanced provisions are observed. We cannot provide assurance that our existing agreements with employees and contractors contain notice provisions that would enable us to seek enhanced damages or attorneys’ fees in the event of any dispute for misappropriation of trade secrets brought under the DTSA.
Significant disruptions of information technology systems, including cloud-based systems, or breaches of data security could adversely affect our business.Our business is increasingly dependent on critical, complex and interdependent information technology systems, including cloud-based systems, to support business processes as well as internal and external communications. Our computer systems are potentially vulnerable to breakdown, malicious intrusion and computer viruses that may result in the impairment of key business processes. Such disruptions and breaches of security could have a material adverse effect on our business, financial condition and results of operations.
In addition, our data security and information technology systems are potentially vulnerable to data security breaches, whether by employees or others, that may expose sensitive data to unauthorized persons. Such data security breaches could lead to the loss of trade secrets or other intellectual property, public disclosure of sensitive clinical or commercial data, and the exposure of personally identifiable information (including sensitive personal information) of our employees, collaborators, clinical trial patients and others. A data security breach or privacy violation that leads to disclosure or modification of or prevents access to patient information, including personally identifiable information or protected health information, could result in increased costs or loss of revenue as a result of:
harm to our reputation;
additional compliance obligations under federal and/or state breach notification laws;
requirements for mandatory corrective action to be taken by us; and
requirements to verify the correctness of database contents and otherwise subject us to liability under laws and regulations that protect personal data.
If we are unable to prevent such data security breaches or privacy violations or implement satisfactory remedial measures, our operations could be disrupted, and we may suffer loss of reputation, financial loss and other regulatory penalties because of lost or misappropriated information, including sensitive patient data. In addition, these breaches and other inappropriate access can be difficult to detect, and any delay in identifying them may lead to increased harm of the type described above. Moreover, the prevalent use of mobile devices that access confidential information increases the risk of data security breaches, which could lead to the loss of confidential information, trade secrets or other intellectual property. While we have implemented security measures to protect our data security and information technology systems, such measures may not prevent such events.
RISKS RELATED TO OUR FINANCIAL POSITION AND NEED FOR ADDITIONAL FINANCING
WeAlthough we reported a small profit for the year ending December 31, 2015, we have a history of losses and anticipate continued future losses, and our continued losses could impair our ability to sustain operations.We haveUntil 2015, we had never been profitable and we had incurred operating losses every year since our operations began in 1990. While we were profitable in 2015 due to the recognition of revenue in connection with the upfront payment from Janssen under the Collaboration Agreement, we expect to incur additional operating losses and, as clinical development activities for imetelstat continue under our Collaboration Agreement with Janssen, our operating losses may increase in size. As of December 31,2014,2017, our accumulated deficit was approximately$928.4$985.8 million. Losses have resulted principally from costs incurred in connection with our research and development activities and from general and administrative costs associated with our operations.We expect to incur additional operating losses and, as clinical development activities continue under our Collaboration Agreement with Janssen, our operating losses may increase in size.Substantially all of our revenues to date have been
research supportpayments under collaborative agreements and milestones, royalties and other revenues from our licensing arrangements. Any revenues generated from our licensing arrangements or ongoing collaborative agreements,and revenues from our licensing arrangements,including the Collaboration Agreement with Janssen, may not be sufficient alone to sustain our operations. For example, we expect revenues under our license agreements related to our telomerase technology to decline significantly in the coming years, and to be eliminated by the end of 2019, due to upcoming patent expirations on such technology. In addition, there can be no assurance that we will receive any milestone payments or royalties from Janssen in the future. We may be unsuccessful in entering into any new corporate collaboration, partnership or license agreements that result in revenues, or existing collaborative agreements or license arrangements, such as the Collaboration Agreement with Janssen, may be terminated or expire.We also expect to experience negative cash flow for the foreseeable future as we fund our
operating lossesoperations and capital expenditures. This will result in decreases in our working capital, total assets andstockholders'stockholders’ equity, which may not be offset by milestone payments or royalties from Janssen or by future financings. We will need to generate significant revenues to achieve consistent future profitability. We may not be able to generate these revenuesfromunder the Collaboration Agreement with Janssen through milestone payments or royalties, and we may never achieve consistent future profitability. Even if we do become profitable in the future, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to achieve consistent future profitability could negatively impact the market price of our common stock and our ability to sustain operations.Even if we do become profitable, we may not be able to sustain or increase profitability on a quarterly or annual basis.We may require additional capital to support development and commercialization of imetelstat in collaboration with Janssen and to otherwise grow our business, and our ability to obtain the necessary funding is uncertain.
We may need additional capital resources in order to support development and commercialization of imetelstat, especially if we elect to exercise our U.S. Opt-In Rights and U.S. Co-Promotion Option under the Collaboration Agreement and potentially independently pursue imetelstat development under our own IDP, and to otherwise support the future growth of our business through the potential acquisition and/or in-licensing of other oncology products, product candidates, programs or companies. We cannot assure you that our existing capital resources, future interest income, potential milestone payments and royalties under the Collaboration Agreement with Janssen and potential future sales of our common stock, including pursuant to our
At-The-Market Issuance2015 Sales Agreementor sales agreement,withMLV & Co. LLC, orMLV, will be sufficient to fund future planned activities. The timing and degree of any future capital requirements will depend on many factors, including:•the accuracy of the assumptions underlying our estimates for our capital needs;
•whether Janssen discontinues development of imetelstat and/or terminates the Collaboration Agreement, and we
elect U.S. Opt-In Rightschoose toshare future U.S.develop imetelstat ourselves;further changes or delays in Janssen’s development
and promotion costsplans for imetelstat, including changes to or further expansion of or delays in ongoing clinical trials decided upon by Janssen or required by regulatory authorities, such as clinical holds or other requirements, or any other factors;the achievement of development, regulatory and sales milestones resulting in payments to us from Janssen under the Collaboration Agreement
with Janssen;Tableand the timing ofContentsreceipt of such payments, if any;•
to the extent permitted under the Collaboration Agreement, whether we independently pursue imetelstat development under our own IDP;
•our potential reimbursement obligations to Janssen if any data from a Janssen IDP support approval by
aregulatoryagencyauthorities in the United States or other countries;•the achievement of development, regulatory and commercial milestones resultingin the
paymentevent that Janssen provides an affirmative Continuation Decision to us,from Janssenwhether we then elect our U.S. Opt-In Rights to share further U.S. development and promotion costs for imetelstat beyond IMbark or IMerge under the Collaboration Agreement,and the timingincluding our share ofreceipt of such payments, if any;•changes or delays in our and Janssen'sdevelopmentplans for imetelstat, including changes which may result from any future clinical holds on the INDs for imetelstat, including the IND for the MF Pilot Study,costs incurred to date by Janssen that weexpectwill be required totransfer to Janssen;•Janssen'sreimburse if we exercise our U.S. Opt-In Rights;Janssen’s ability to meaningfully reduce manufacturing costs of imetelstat;
•the progress, timing, magnitude, scope and costs of clinical development, manufacturing and commercialization
forof imetelstat, including the number of indications being pursued, subject topermission fromclearances and approvals by theFDA;•- FDA and other regulatory authorities;
the time and costs involved in obtaining regulatory clearances and approvals in the United States and in other countries;
•Janssen'sJanssen’s ability to successfully market and sell imetelstat, upon regulatory approval or clearance, in the United States and other countries;
•if we exercise our U.S. Opt-In Rights, our decision to also exercise our U.S. Co-Promotion Option, including the costs and timing of building a U.S. sales force;
the sales price for imetelstat;
•the timing, receipt and amount of royalties under the Collaboration Agreement on worldwide net sales of imetelstat, upon regulatory approval or clearance, if any;
•the timing, receipt and amount of royalties on sales of any stem cell products by Asterias, upon development, regulatory approval or clearance, if any;•the sales price and availability of adequate third-party reimbursement for imetelstat;•the cost of acquiring and/or
licensing any newin-licensing other oncology products, product candidates, programs or companies, if any;•the progress, timing, magnitude, scope and costs of clinical development, manufacturing and commercialization of any acquired or in-licensed oncology products, product candidates, programs, or companies, including the number of indications being pursued, subject to clearances and approvals by the FDA and other regulatory authorities;
expenses associated with
the pending andpotentialadditional related purported securities lawsuits and derivative lawsuits, as well as any otherfuture litigation; and•the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims.
In addition, changes in our business may occur that would consume available capital resources sooner than we expect. If our existing capital resources, future interest income, and potential milestone payments and royalties under the Collaboration Agreement with Janssen are insufficient to meet future capital requirements, we will need to raise additional capital to fund our
operations.operations, including pursuant to our 2015 Sales Agreement with MLV.Further, if the Collaboration Agreement is terminated, including as a result of
Janssen'sJanssen’s failure to provide an affirmative Continuation Decision to us, or for any other reason, we would not receive any milestone payments or royalties under the Collaboration Agreement, and then, depending on the timing of such event, we would be required to fund all clinical development, manufacturing and commercial activities for imetelstat should we elect to continue the development of imetelstat ourselves, which would require us to raise substantial additional capital or establish alternative collaborations with third-party collaboration partners, which may not be possible. If the Collaboration Agreement is terminated and we are unable to raise additional capital or establish alternative collaborations with third-party collaboration partners for imetelstat, the development of imetelstat would be discontinued, which might cause us to cease operations. Additional financing through public or private equity financings, including pursuant to oursales agreement2015 Sales Agreement with MLV,prior to its expiration in October 2015,capital lease transactions or other financing sources may not be available on acceptable terms, or at all. We may raise equity capital at a stock price or on other terms that could result in substantial dilution ofownership for our stockholders. The receptivity of the public and private equity markets to proposed financings is substantially affected by the general economic, market and political climate and by other factors which are unpredictable and over which we have no control. In this regard, continued volatility and instability in the global financial markets and political climate could adversely affect our ability to raise additional funds through financings and the terms upon which we may raise those funds.
Our ability to raise additional funds will be severely impaired in the event of:
•any future clinical holds on any INDfurther changes or delays in Janssen’s development plans for imetelstat;
•a failure or inability to show adequate safety or efficacy of imetelstat in
existingcurrent or potential future clinicaltrials;trials, which may result in a decision by Janssen to delay or•- discontinue further development of imetelstat; or
a termination of the Collaboration Agreement or if our collaboration with Janssen is otherwise unsuccessful.
If sufficient capital is not available, we may be unable to fulfill our funding obligations under the Collaboration Agreement with Janssen, resulting in our breach of the Collaboration Agreement, which could lead to Janssen paying lower milestone payments and lower royalties to us under a reduced royalty
tier which couldtier. This would have a material adverse effect on our results of operations and financial condition.Moreover, in order to grow and diversify our business, we plan to
diversifycontinue oursole product candidatebusiness developmentrisk by identifyingefforts to identify andseekingseek to acquire and/or in-licensenewother oncology products, productopportunities for development.candidates, programs or companies. Acquisition or in-licensing opportunities that we may pursue could materially affect our liquidity and capital resources and may require us to incur indebtedness, seek equity capital orboth.both, including pursuant to our 2015 Sales Agreement with MLV. In addition, there can be no assurance that sufficient additional capital would be available to us in order to pursue any of these opportunities.The recently passed comprehensive tax reform bill could adversely affect our business and financial condition.
On December 22, 2017, President Trump signed into law new legislation that significantly revises the Internal Revenue Code of 1986, as amended. The newly enacted federal income tax law, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, limitation of the tax deduction for interest expense to 30% of adjusted earnings (except for certain small businesses), limitation of the deduction for net operating losses to 80% of current year taxable income and elimination of net operating loss carrybacks, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits (including reducing the business tax credit for certain clinical testing expenses incurred in the testing of certain drugs for rare diseases or conditions). Notwithstanding the reduction in the corporate income tax rate, the overall impact of the new federal tax law is uncertain and our business and financial condition could be adversely affected. In addition, it is uncertain if and to what extent various states will conform to the newly enacted federal tax law. The impact of this tax reform on holders of our common stock is also uncertain and could be adverse. We urge our stockholders to consult with their legal and tax advisors with respect to this legislation and the potential tax consequences of investing in or holding our common stock.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Our net operating loss carryforwards could expire unused and be unavailable to offset future income tax liabilities. Under the newly enacted federal income tax law, federal net operating losses incurred in 2018 and in future years may be carried forward indefinitely, but the deductibility of such federal net operating losses is limited. It is uncertain if and to what extent various states will conform to the newly enacted federal tax law. In addition, under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, and corresponding provisions of state law, if a corporation undergoes an
"ownership“ownership change,"” generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period, thecorporation'scorporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes (such as research tax credits) to offset its post-change taxable income or taxes may be limited. Changes in our stock ownership, some of which are outside of our control, may have resulted or could in the future result in an ownership change. If a limitation were to apply, utilization of a portion of our domestic net operating loss and tax credit carryforwards could be limited in future periods. In addition, a portion of the carryforwards may expire before being available to reduce future income taxliabilities.liabilities which could adversely impact our financial position.
RISKS RELATED TO OUR COMMON STOCK AND FINANCIAL REPORTINGHistorically, our stock price has been extremely volatile.
Historically, our stock price has been extremely volatile. Between January 1,
20052008 and December 31,2014,2017, our stock has traded as high as$12.18$9.24 per share and as low as $0.91 per share. Between January 1,20122015 and December 31,2014,2017, the price has ranged between a high of$7.79$5.30 per share and a low of$0.91$1.74 per share. The significant market price fluctuations of our common stock have been due to and may in the future be influenced by a variety of factors, including:•announcements regarding the research and development of imetelstat, including results of, further delays in, discontinuation of, or further modifications or refinements to any clinical trials of imetelstat as a result of any internal data reviews or decisions by joint governance committees, and investor perceptions thereof;
not receiving timely regulatory clearances or approvals in any jurisdiction, whether within or outside of the United States, including, if we, Janssen or future investigators do not obtain regulatory clearance to commence,
studiesconduct or continue clinical trials of imetelstat in MF, MDS or any additional hematologic myeloid malignancies in a timely manner or at all,includingor to amend any clinical trial protocol with respect to theInitial Phase 2 MF Study and the Initial Phase 2 MDS Study planned to be conducted by Janssen under the Collaboration Agreement;Tableconduct ofContentsIMerge, IMbark or any future clinical trial of imetelstat;•
developments in our collaboration with Janssen, including the termination, modification or
modificationamendment of the Collaboration Agreement, or disputes regarding the collaboration;•announcements regarding the research and development of imetelstat, including results of or delays in any clinical trials of imetelstat, and investor perceptions thereof;•announcements regarding regulatory developments concerning imetelstat, including announcements similar to our March 2014
announcementsannouncement that the FDA had placed a full clinical hold on our IND forimetelstat and a partial clinical hold onimetelstat;the
investigator's IND for the MF Pilot Study due to safety concerns;•announcements regarding our plans to discontinue certain programs or clinical trials, such as our prior announcements regarding the discontinuationexperimental nature ofour stem cell programs and certain clinical trials;•- imetelstat;
perception by our stockholders about the adequacy of
the consideration received for the divestiture of our stem cell assets to Asterias orpotential payments we may receive under the Collaboration Agreement;•the demand in the market for our common stock;
•the experimental nature of imetelstat;•fluctuations in our operating results;•our declining cash balance as a result of operating losses;•general market conditions or market conditions relating to the biopharmaceutical and pharmaceutical industries;•announcements of technological innovations, new commercial products, or clinical progress or lack thereof by us, our collaborators, licensees, partners or our competitors;
•fluctuations in our operating results;
our declining cash balance as a result of operating losses;
general market conditions or market conditions relating to the biopharmaceutical and pharmaceutical industries;
announcements concerning
regulatory developments andimetelstat proprietary rights;•comments by securities analysts;
•large stockholders exiting their position in our common stock;
•announcements of or developments concerning
pending and/orpotential future litigation;•the issuance of common stock to partners, vendors or investors to raise additional
capital;capital or to acquire other oncology products, product candidates, programs or companies; and•the occurrence of any other risks and uncertainties discussed under the heading
"Risk“Risk Factors."”Stock prices and trading volumes for many biopharmaceutical companies fluctuate widely for a number of reasons, including factors which may be unrelated to their businesses or results of operations, such as media coverage, statements made on message boards and social media forums, legislative and regulatory measures and the activities of various interest groups or organizations. In addition to
otherthe risk factors described in this section, overall market volatility, as well as general domestic or international economic, market and political conditions, could materially and adversely affect the market price of our common stock and the return on your investment.If we fail to continue to meet
continuedthe listing standards of NASDAQ, our common stock may be delisted, which could have a material adverse effect on the liquidity of our common stock.Our common stock is currently traded on the Nasdaq Global Select Market. The NASDAQ Stock Market LLC has requirements that a company must meet in order to remain listed on NASDAQ. In particular, NASDAQ rules require us to maintain a minimum bid price of $1.00 per share of our common stock. If the closing bid price of our common stock were to fall below $1.00 per share for 30 consecutive trading days or we do not meet other listing requirements, we would fail to be in
compliance with
NASDAQ'sNASDAQ’s listing standards. There can be no assurance that we will continue to meet the minimum bid price requirement, or any other requirement in the future. If we fail to meet the minimum bid price requirement, The NASDAQ Stock Market LLC may initiate the delisting process with a notification letter. If we were to receive such a notification, we would be afforded a grace period of 180 calendar days to regain compliance with the minimum bid price requirement. In order to regain compliance, shares of our common stock would need to maintain a minimum closing bid price of at least $1.00 per share for a minimum of 10 consecutive trading days. In addition, we may be unable to meet other applicable NASDAQ listing requirements, including maintaining minimum levels ofstockholders'stockholders’ equity or market values of our common stock, in which case our common stock could be delisted. If our common stock were to be delisted, the liquidity of our common stock would be adversely affected and the market price of our common stock could decrease.The sale of a substantial number of shares may adversely affect the market price of our common stock.
The sale of a substantial number of shares of our common stock in the public market, or the perception that such sales could occur, could significantly and negatively affect the market price of our common stock.As of December 31,2014,2017, we had 300,000,000 shares of common stock authorized for issuance and157,429,871159,877,239 shares of common stock outstanding. In addition, we had reserved32,327,34530,044,457 shares of our common stock for future issuance pursuant to our option and equity incentive plans and outstanding warrants as of December 31,2014. Issuing additional shares could negatively affect2017. In addition, under themarket priceuniversal shelf registration statement filed by us in August 2015 and declared effective by the SEC in September 2015, we may sell any combination ofourcommon stock, preferred stock, debt securities andthe return on your investment.warrants in one or more offerings, up to a cumulative value of $250 million.Future sales of our common stock or the perception that such sales could occur, including pursuant to our
sales agreement2015 Sales Agreement with MLV,prior to its expiration in October 2015,or the issuance of common stock to satisfy our current or future cash payment obligations or to acquire technology, property, or other businesses, could cause immediate dilution and adversely affect the market price of our common stock.In addition, under the universal shelf registration statement filed by us in July 2012 and declared effective by the SEC in October 2012, we may sell any combination of common stock, preferred stock, debt securities and warrants in one or more offerings, up to a cumulative value of $96.5 million.The sale or issuance of our securities, as well as the existence of outstanding options and shares of common stock reserved for issuance under our option and equity incentive plans and outstanding warrants, also may adversely affect the terms upon which we are able to obtain additional capital through the sale of equity securities, which could negatively affect the market price of our common stock and the return on your investment.Our undesignated preferred stock may inhibit potential acquisition bids; this may adversely affect the market price of our common stock and the voting rights of holders of our common stock.
Our certificate of incorporation provides our board of directors with the authority to issue up to 3,000,000 shares of undesignated preferred stock and to determine or alter the rights, preferences, privileges and restrictions granted to or imported upon these shares without further vote or action by our stockholders. The issuance of shares of preferred stock may delay or prevent a change in control transaction without further action by our stockholders. As a result, the market price of our common stock may be adversely affected.
In addition, if in the future, we issue preferred stock that has preference over our common stock with respect to the payment of dividends or upon our liquidation, dissolution or winding up, or if we issue preferred stock with voting rights that dilute the voting power of our common stock, the rights of holders of our common stock or the market price of our common stock could be adversely affected.
Provisions in our charter, bylaws and Delaware law may inhibit potential acquisition bids for us, which may prevent holders of our common stock from benefiting from what they believe may be the positive aspects of acquisitions and takeovers.
Provisions of our charter documents and bylaws may make it substantially more difficult for a third party to acquire control of us and may prevent changes in our management, including provisions that:
•prevent stockholders from taking actions by written consent;
•divide the board of directors into separate classes with terms of office that are structured to prevent all of the directors from being elected in any one year; and
•set forth procedures for nominating directors and submitting proposals for consideration at
stockholders'stockholders’ meetings.Provisions of Delaware law may also inhibit potential acquisition bids for us or prevent us from engaging in business combinations. In addition, we have severance agreements with several employees and a company-wide severance plan, either of which could require a potential acquirer to pay a higher price. Either collectively or individually, these provisions may prevent holders of our common stock from benefiting from what they may believe are the positive aspects of acquisitions and takeovers, including the potential realization of a higher rate of return on their investment from these types of transactions.
We do not intend to pay cash dividends on our common stock in the foreseeable future.
We do not anticipate paying cash dividends on our common stock in the foreseeable future. Any payment of cash dividends will depend upon our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors.
Failure to achieve and maintain effective internal controls in accordance with Section 404 of the Sarbanes-Oxley Act of 2002 could have a material adverse effect on our business and stock price.
Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, requires that we establish and maintain an adequate internal control structure and procedures for financial reporting. Our annual reports on Form 10-K must contain an annual assessment by management of the effectiveness of our internal control over financial reporting and must include disclosure of any material weaknesses in internal control over financial reporting that we have identified. In addition, our independent registered public accounting firm must
annuallyprovide an opinion annually on the effectiveness of our internal control over financial reporting.The requirements of Section 404 are ongoing and also apply to future years. We expect that our internal control over financial reporting will continue to evolve as our business develops. Although we are committed to continue to improve our internal control processes and we will continue to diligently and vigorously review our internal control over financial reporting in order to ensure compliance with Section 404 requirements, any control system, regardless of how well designed, operated and evaluated, can provide only reasonable, not absolute, assurance that its objectives will be met. Therefore, we cannot be certain that
in the futurematerial weaknesses or significant deficiencies will not exist or otherwise bediscovered.discovered in the future. If material weaknesses or other significant deficiencies occur,thesesuch weaknesses or deficiencies could result in misstatements of our results of operations, restatements of our financial statements, a decline in our stock price, or other material adverse effects on our business, reputation, results of operations, financial condition or liquidity.
RISKS RELATED TO COMPETITIVE FACTORSCompetitors may develop technologies that are superior to or more cost-effective than ours, which may significantly impact the commercial viability of imetelstat, which could cause Janssen to terminate the Collaboration Agreement and damage our ability to sustain operations.
The pharmaceutical and biotechnology industries are intensely competitive. Other pharmaceutical and biotechnology companies and research organizations currently engage in or have in the past engaged in efforts related to the biological mechanisms related to imetelstat,
includingthe study of telomeres, telomerase,andor our proprietary oligonucleotide chemistry, and the research and development of therapies for the treatment of hematologic myeloid malignancies. In addition, other products and therapies that could directly compete with imetelstat currently exist or are being developed by pharmaceutical and biopharmaceutical companies and by academic institutions, government agencies and other public and private research organizations. We expect Janssen’s decisions regarding continued development and/or commercialization, if any, of imetelstat, including completing IMerge and/or IMbark, its Continuation Decision or the termination of the Collaboration Agreement, to be informed in part by what Janssen believes is the estimated commercial potential of imetelstat for the treatment of hematologic malignancies, such as MF or MDS.Many companies are developing alternative therapies to treat hematologic myeloid
malignancies and, in this regard, are competitors of ours and Janssen.malignancies. For example, if approved for commercial sale for the treatment of MF, imetelstat would compete against IncyteCorporation'sCorporation’s ruxolitinib, or Jakafi®, which is orally administered. In clinical trials, Jakafi® reduced spleen size, abdominal discomfort, early satiety, bone pain, night sweats and itching in MF patients. Recently, there have also been reports of overall survival benefit as well as improvement in bone marrow fibrosis from Jakafi® treatment. Other treatment modalities for MF include hydroxyurea for the management of splenomegaly, leukocytosis, thrombocytosis and constitutional symptoms; splenectomy and splenic irradiation for the management of splenomegaly and co-existing cytopenias, or low blood cell counts; chemotherapy and pegylated interferon. Drugs for the treatment of MF-associated anemia include erythropoiesis-stimulating agents, androgens, danazol, corticosteroids, thalidomide and lenalidomide. There are other investigational treatments for MF further along in development than imetelstat, such asmomelotinib by Gilead Sciences, Inc. andpacritinib byCell Therapeutics,CTI Biopharma Corporation, or CTI Biopharma, and fedratinib by Impact Biomedicines, Inc., acquired by Celgene Corporation, or Celgene, whichare currently inhave reported results from Phase 3 clinicaltrials, and othertrials. Other investigational treatments for MF include inhibitors of the JAK-STAT pathway,as well as several investigational treatments in early phase testingsuch as NS-018 by NS Pharma, Inc.; histone deacetylaseinhibitors,inhibitors; interleukin-3 receptor targeted agents; inhibitors of heat shock protein90,90; hypomethylatingagents,agents; PI3 Kinase and mTORinhibitors,inhibitors; anti-fibrosis antibodies, such as PRM-151 from Promedior, Inc.; hedgehoginhibitors,and SMO inhibitors; PIM kinase inhibitors; IAP inhibitors; anti-LOX2inhibitors,inhibitors; recombinant pentraxin 2protein,protein; KIP-1activators,activators; TGF-beta superfamily inhibitors, such as sotatercept and luspatercept by Acceleron Pharma, Inc., or Acceleron, in collaboration with Celgene; FLTinhibitors,inhibitors; and other tyrosine kinase inhibitors.If approved for commercial sale for the treatment of lower risk MDS, imetelstat would compete against a number of treatment options, including erythropoiesis stimulating agents and other hematopoietic growth factors; immunomodulators, such as lenalidomide by Celgene; hypomethylating agents, such as azacitidine by Celgene and decitabine by Janssen; in addition to investigational treatments that may be further along in development than imetelstat, such as oral versions of azacitidine; histone deacetylase inhibitors; TGF-beta superfamily inhibitors, such as luspatercept by Acceleron, in collaboration with Celgene; PI3 Kinase inhibitors; aminopeptidase inhibitors, such as tosedostat by CTI Biopharma; TLR2-specific antibodies; anti-CD33 antibodies; anti-CD38 antibodies, such as daratumumab by Genmab A/S in collaboration with Janssen; anti-CD123 antibodies, such as talacotuzumab by Janssen; retinoic acid receptor alpha agonists, such as SY-1425 by Syros Pharmaceuticals; hypoxia-inducible factor prolyl hydroxylase inhibitors, such as roxadustat by FibroGen, Inc.; Fas ligand inhibitors; and JAK-STAT pathway inhibitors.
Independently, Janssen is developing therapies for hematologic malignancies, including AML, MDS,
MMmultiple myeloma and ABC-subtype diffuse large B-cell lymphoma. Molecular and cellular pathways of interest include:•cell surface targets for immune-directed therapy;
•immune checkpoint inhibition;
•leukemia stem cells;
•pathway addiction (genetic alterations, cell-type specific pathways);
•conditional sensitivity (stress, protein-producing tumors);
•targeting of T-cells and natural killer
"NK"“NK” cells to tumors;•identification of novel tumor-specific antigens; and
•progression from early MDS to AML and cancer interception.
Success by Janssen in any of these approaches may compete with imetelstat or render imetelstat obsolete or noncompetitive, which could lead to a decision by Janssen to discontinue the imetelstat program and terminate the Collaboration Agreement, which would materially and adversely affect our business and business
prospects.Table of Contentsprospects and might cause us to cease operations.Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. We anticipate increased competition in the future as new companies explore treatments for hematologic myeloid malignancies, which may significantly impact the commercial viability of imetelstat. Academic institutions, government agencies and other public and private research organizations may also conduct research, seek patent protection and establish collaborative arrangements for research, clinical development and marketing of products similar to
ours.imetelstat. These companies and institutions compete with us in recruiting and retaining qualifiedscientificdevelopment and management personnel as well as in acquiring technologies complementary toourthe imetelstat program.In addition to the above factors,
we and Janssen expect toimetelstat will face competitionin the following areas:based on:•product efficacy and safety;
•convenience of product administration;
•cost of manufacturing;
•the timing and scope of regulatory consents;
•status of
reimbursement coverage;•- coverage and level of reimbursement;
price; and
•patent position, including potentially dominant patent positions of others.
As a result of the foregoing, competitors may develop more commercially desirable or affordable products than imetelstat, or achieve earlier patent protection or product commercialization than us or Janssen. Competitors have developed, or are in the process of developing, technologies that are, or in the future may be, competitive to imetelstat. Some of these products may have an entirely different approach or means of accomplishing therapeutic effects similar or superior to those that may be demonstrated by imetelstat. Competitors may develop products that are safer, more effective, or less costly than imetelstat, or more convenient to administer to patients and, therefore, present a serious competitive threat to imetelstat. In addition, competitors may price their products below what Janssen may determine to be an acceptable price for imetelstat, may receive better third-party payor coverage and/or reimbursement, or may be more
cost effectivecost-effective than imetelstat. Such competitive products or activities by competitors may render imetelstat obsolete, which may cause Janssen to terminate the Collaboration Agreement, which would severely and adversely affect our financial results, businessprospects.and business prospects, and the future of imetelstat, and might cause us to cease operations.To be successful, imetelstat must be accepted by the health care community, which can be very slow to adopt or unreceptive to new technologies and products.
If approved for marketing, imetelstat may not achieve market acceptance since hospitals, physicians, patients or the medical community in general may decide not to accept and utilize imetelstat. If approved for commercial sale, imetelstat will compete with a number of conventional and widely accepted drugs and therapies manufactured and marketed by major pharmaceutical companies. The degree of market acceptance of imetelstat will depend on a number of factors, including:
•the clinical indications for which imetelstat is approved;
the country and/or regions within which imetelstat is approved;
the establishment and demonstration to the medical community of the clinical efficacy and safety of imetelstat;
•the ability to demonstrate that imetelstat is superior to alternatives
currentlyon themarket;•- market at the time;
the ability to establish in the medical community the potential
advantageadvantages of imetelstat over alternative treatmentmethods;•- methods, including with respect to efficacy, safety, cost or route of administration;
the label and promotional claims allowed by the FDA or other regulatory
agenciesauthorities for imetelstat, if any;
the effectiveness of sales, marketing and distribution support for imetelstat;
the availability of coverage, adequate reimbursement and
•reimbursement policies ofpricing by government and third-partypayors.the willingness of patients to pay out-of-pocket in the absence of coverage by third-party payors, including governmental authorities.
The established use of conventional products competitive with imetelstat may limit or preclude the potential for imetelstat to receive market acceptance upon any commercialization.
We orJanssen may be unable to demonstrate any pharmacoeconomic advantage for imetelstat compared to established or standard-of-care therapies, or newly developed therapies, for hematologic myeloid malignancies. Third-party payors may decide that any potential improvement that imetelstat may provide to clinical outcomes in hematologic myeloid malignancies is not adequate to justify the costs of treatment with imetelstat. Ifthird-party payors do not view imetelstat as offering a better balance between clinical benefit and treatment cost compared to standard-of-care therapies or other treatment modalities currently in development, imetelstat may not be commercially viable. Ifthe health care community does not accept imetelstat for any of the foregoing reasons, or for any other reason, our ability to earn potential milestone payments and royalties under the Collaboration Agreement with Janssen would be negatively impacted and our business and business prospects would be severely and adversely affected.If acceptable prices or adequate reimbursement for imetelstat is not obtained, the use of imetelstat could be severely limited.
The ability to successfully commercialize imetelstat will depend significantly on obtaining acceptable prices and the availability of coverage and adequate reimbursement to the patient from third-party payors. Government payors, such as the Medicare and Medicaid programs, and other third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels. Assuming Janssen obtains coverage for imetelstat by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. If approved for commercial sale, patients are unlikely to use imetelstat unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the cost of imetelstat. Therefore, coverage and adequate reimbursement is critical to new product acceptance.
Government authorities and other third-party payors are developing increasingly sophisticated methods of controlling healthcare costs, such as by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices as a condition of coverage, are using restrictive formularies and preferred drug lists to leverage greater discounts in competitive classes, and are challenging the prices charged for medical products. Further, no uniform policy requirement for coverage and reimbursement for drug products exists among third-party payors in the United States. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require Janssen to provide scientific and clinical support for the use of imetelstat to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
We cannot be sure that coverage and reimbursement will be available for imetelstat, if approved for commercial sale, and, if reimbursement is available, what the level of reimbursement will be. There may also be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which marketing approval is obtained. If coverage and reimbursement are not available or reimbursement is available only to limited levels, Janssen may not successfully commercialize imetelstat, even if marketing approval is obtained.
The adoption of health policy changes and health care reform in the United States may adversely affect our business and financial results.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, collectively known as the Affordable Care Act, or ACA, became law and substantially changed the way healthcare
will be financedis funded by both governmental and private insurers, and significantly impacted the pharmaceutical industry. TheAffordable Care ActACA contains a number of provisionsincluding those governing enrollment in federal healthcare programs, reimbursement changes and fraud and abuse, which willthat may have a significant impactexisting government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program. Additionally, the Affordable Care Act:•mandates a further shift in the burden of Medicaid payments to the states;•increases the minimum level of Medicaid rebates payable by manufacturers of brand-name drugs from 15.1% to 23.1%;•requires collection of rebates for drugs paid by Medicaid managed care organizations;•requires manufacturers to participate in a coverage gap discount program, under which they must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer's outpatient drugs to be covered under Medicare Part D, beginning January 2011; and•imposes a non-deductible annual feeonpharmaceutical manufacturers or importers who sell "branded prescription drugs" to specified federal government programs.
While the
U.S.Supreme Court upheld the constitutionality of most elements of theAffordable Care ActACA in June 2012 and upheld the ACA against challenges to nationwide tax subsidies in July 2015, otherlegaljudicial and Congressional challenges against the ACA have been brought, and arestill pending final adjudicationlikely to be brought inseveral jurisdictions. In addition,theUnited Statesfuture. Since January 2017, President Trump has signed two executive orders and other directives designed to delay, circumvent, or loosen certain requirements mandated by the ACA. Concurrently, Congress hasalso proposed a number of legislative initiatives, including possibleconsidered legislation that would repeal or repeal and replace all or part of theAffordable Care Act. At this time, it remains unclear whether there will be any changes madeACA. While Congress has not passed repeal legislation, the Tax Cuts and Jobs Act of 2017 includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as theAffordable Care Act, whether“individual mandate”. Additionally, on January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain ACA-mandated fees. Congress may consider additional legislation tocertain provisionsrepeal orits entirety. Werepeal other elements of the ACA. Therefore, we cannotassureassume that theAffordable Care Act,ACA, as currently enacted or as amended in the future, or any legislation that may replace, or repeal other elements of the ACA, will not adversely affect our business and financial results and we cannot predict how future federal or state legislative or administrative changes relating to healthcare reform will affect our business.In addition, other legislative changes have been proposed and adopted since the
Affordable Care ActACA was enacted. For example, in August 2011, the Budget Control Act of 2011 was enacted, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for fiscal years 2012 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect beginning on April 1, 2013 and, due to subsequent legislative amendments to the statute, including the Bipartisan Budget Act of 2018, will stay in effect through 2027 unless additional Congressional action is taken. Further, the American Taxpayer Relief Act of 2012, signed into law in January 2013, among other things, also reduced Medicare payments toseveralcertain providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.On March 1, 2013,In the
President signed an executive order implementing sequestration,future, we anticipate additional proposals relating to the reform of the U.S. healthcare system, some of which could further limit the prices, or the amounts of reimbursement available for imetelstat. There has been increasing legislative and enforcement interest in the United States with respect to specialty drug pricing practices in light of the rising cost of prescription drugs and biologics. Specifically, there have been several recent U.S. Congressional inquiries and proposed and enacted legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the price of drugs under Medicare, and reform government program reimbursement methodologies for drugs, some of which are included in the Trump administration’s budget proposal for fiscal year 2019. At the federal level, Congress and the Trump Administration have each indicated that they will continue to seek new legislative and/or administrative measures to control drug costs. If future legislation were to impose direct governmental price controls and access restrictions, it could have a significant adverse impact onApril 1, 2013, Medicare payment reductionsour business and financial results. Managed care organizations, as well as Medicaid and other government agencies, continue to seek price discounts. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biologic product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, to encourage importation from other countries and bulk purchasing. Due to the volatility in the current economic and market dynamics, we are unable to predict the impact of2% went into effect and will remain in effect through 2024 unless additional Congressional action is taken.While the Affordable Care Act has likely increased the number of patients who have insurance coverage for imetelstat, it is uncertain whether itsany unforeseen or unknown legislative, regulatory, payor or policy actions, which may include cost containmentmeasures will adversely affect reimbursement for imetelstat.and healthcare reform measures. Such policy actions could have a material adverse impact on the potential royalties under the Collaboration Agreement with Janssen on worldwide net sales of imetelstat, if approved.Cost control initiatives also could decrease the price that Janssen may receive for imetelstat in the future. If imetelstat is not considered cost-effective or adequate third-party reimbursement for the users of imetelstat cannot be obtained, then Janssen may be unable to maintain price levels sufficient to realize an appropriate return on the investment in imetelstat, which
could impairwould have a material adverse effect on our ability to earn potential milestone payments and royalties under the Collaboration Agreement,with Janssen and our financial condition, operating results and business prospects would be severely and adversely affected.RISKS RELATED TO ENVIRONMENTAL AND PRODUCT LIABILITYActivities conducted by usorJanssen involve hazardous materials, and improper handling of these materials by employees, contractors, or agentscouldexpose us or Janssen to significant legal and financial penalties.We, Janssen, or contractors or agents may be required to incur significant costs to comply with current or future environmental laws and regulations and may be adversely affected by the cost of compliance with these laws and regulations. Additionally, an accident could damage the manufacturing facilities and operations of any third-party contracted by us or Janssen to perform services with respect to imetelstat. If we, Janssen or contractors or agents are unable to comply with federal, state and county environmental and safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials, chemicals and various radioactive compounds, or an accident occurs, considerable additional costs, fines, penalties or liabilities could be assessed which could negatively impact our collaboration with Janssen orcause Janssen to terminate the CollaborationAgreement, eitherAgreement. Any ofwhichthese events would severely and adversely affect our financial results, businessprospects.and business prospects, and might cause us to cease operations.If we fail to comply with federal and state healthcare laws, including fraud and abuse, transparency, and health information privacy and security laws, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected.
WeOur business operations and current and future arrangements with investigators, healthcare professionals, consultants, third-party payors and customers, maynotexpose us to broadly applicable fraud and abuse and other healthcare laws and regulations. These laws may constrain the business or financial arrangements and relationships through which we conduct our operations, including how we research, market, sell and distribute any product of ours for which marketing approval is obtained. Such laws include:the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities, including prescription drug manufactures (or a party acting on its behalf), from knowingly and willfully, directly or indirectly, soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order, lease or recommendation of, any good, facility, item or service for which payment may be
ablemade under a federal healthcare program such as Medicare and Medicaid. The term “remuneration” has been broadly interpreted toobtaininclude anything of value. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the federal Anti‑Kickback Statute has been violated. The ACA, among other things, amended the intent requirement of the federal Anti‑Kickback Statute such that a person ormaintain sufficient insurance on commercially reasonable termsentity no longer needs to have actual knowledge of the statute orwith adequate coverage against potential liabilitiesspecific intent to violate, in order toprotect ourselvescommit a violation;the federal civil and criminal false claims and civil monetary penalties laws, including the civil False Claims Act and its qui tam or whistleblower provisions, which impose criminal and civil penalties against
product liability claims.Our business exposes usindividuals or entities for, among other things, knowingly presenting, or causing topotential product liability risksbe presented, to the federal government, claims for payment that areinherentfalse or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. Entities can be held liable under these laws if they are deemed to “cause” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers, promoting a product off‑label, or for providing medically unnecessary services or items. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act;the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false or fraudulent statements in connection with the delivery of or payment for healthcare benefits, items or services;
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, which imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security, transmission and breach reporting of individually identifiable health information, upon entities subject to the law, such as health plans, healthcare clearinghouses and healthcare providers and their respective business associates that perform services for them that involve individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in U.S. federal courts to enforce the federal HIPAA laws and seek attorneys' fees and costs associated with pursuing federal civil actions;
analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians, other healthcare providers, and healthcare entities, or marketing expenditures; and state and foreign laws governing the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Efforts to ensure that our current and future business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable healthcare laws, in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the
testing, manufacturinghealthcare industry. If our operations are found to be in violation of any of these or any other health regulatory laws that may apply to us, we may be subject to significant penalties, including the imposition of significant civil, criminal andmarketing of human therapeuticadministrative penalties, damages, monetary fines, disgorgement, individual imprisonment, possible exclusion from participation in Medicare, Medicaid anddiagnostic products. We mayother federal healthcare programs, reputational harm, diminished profits and future earnings, additional reporting requirements and oversight if we become subject toproduct liability claims if the usea corporate integrity agreement or similar agreement to resolve allegations ofimetelstat is alleged to have injured patients, including any injuries alleged to arise from any hepatotoxicity from imetelstat. We currently have limited clinical trial liability insurance,non-compliance with these laws, andwe may not be able to maintain this typecurtailment ofinsurance forour operations, any ofour clinical trials or clinical trials that we may conduct in collaboration with Janssen under the Collaboration Agreement. In addition, product liability insurance is becoming increasingly expensive. Being unable to obtain or maintain product liability insurance in the future on acceptable terms or with adequate coverage against potential liabilities could have a material adverse effect on our business.Significant disruptions of information technology systems or breaches of data securitywhich could adversely affectour business.Our business is increasingly dependent on critical, complex and interdependent information technology systems, including Internet-based systems, to support business processes as well as internaland external communications. The size and complexity of our computer systems make them potentially vulnerable to breakdown, malicious intrusion and computer viruses that may result in the impairment of key business processes.In addition, our systems are potentially vulnerable to data security breaches—whether by employees or others—that may expose sensitive data to unauthorized persons. Such data security breaches could lead to the loss of trade secrets or other intellectual property, or could lead to the public exposure of personal information (including sensitive personal information) of our employees, collaborators, clinical trial patients, customers and others.Such disruptions and breaches of security could have a material adverse effect on our business, financial condition and results of operations.Our headquarters are located near known earthquake fault zones, and the occurrence of an earthquake or other catastrophic disaster could cause damage to our offices and equipment, which could cause delays or even require us to cease or curtail operations.Our headquarters are located in the San Francisco Bay Area near known earthquake fault zones and are vulnerable to significant damage from earthquakes. We do not carry earthquake insurance. We are also vulnerable to damage from other types of disasters, including fires, floods, power loss, communications failures, terrorism and similar events. If any disaster were to occur,our ability to operate our businessatand ouroffices wouldresults of operations. Defending against any such actions can beseriously, or potentially completely,costly, time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired.The insurance we maintain may not be adequate to cover our losses from such disasters or other business interruptions.ITEM 1B. UNRESOLVED STAFF COMMENTSNone.
In
February 2014,September 2017, we amended the lease agreement for our premises at 149 Commonwealth Drive, Menlo Park, California, to extend the lease term from February 2018 through January2016 and reduce2020. During thespace leased by usterm of the amended lease, we will continue to occupy approximately24,00014,500 square feet of officespace effective July 2014.space. Our amended lease at 149 Commonwealth Drive includes an option to extend the lease for one additional period of18 months.two years. We believe that our facilities are adequate to meet our requirements for the near term.On March 14, 2014,
athe first of two substantially similar purportedsecuritiesclass actionlawsuitsecurities lawsuits wascommencedfiled in the United States District Court for the Northern District of California, or the California District Court, naming as defendants us and certain of our officers. The second such lawsuitalleges violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements made by us related to our Phase 2 trial of imetelstat in patients with ET or PV. The plaintiff alleges, among other things, that we failed to disclose facts related to the occurrence of persistent low-grade LFT abnormalities observed in our Phase 2 trial of imetelstat in ET or PV patients and the potential risk of chronic liver injury following long-term exposure to imetelstat. The plaintiff seeks damages and an award of reasonable costs and expenses, including attorneys' fees. Onwas filed on March 28,2014, a second purported securities class action lawsuit was commenced in the California District Court, and on2014. On June 6, 2014, athird purportedsecurities lawsuit, not styled as a class action, wascommencedfiled in the United States District Court for the Southern District of Mississippi, or the Mississippi District Court, naming as defendants us and certain of our officers.These lawsuits, which areThis lawsuit was based on the same factual background as thepurported securitiesclass actionlawsuit that commenced on March 14, 2014, also allege violations ofsecurities lawsuits. These three cases, or theSecurities Exchange Act of 1934 and seek damages and an award of reasonable costs and expenses,Table of ContentsClass Action Lawsuits, were consolidated for all purposes into a single case.including attorneys' fees.OnJune 30, 2014,July 21, 2017, the California District Courtconsolidated bothentered an order and final judgment that dismissed with prejudice and released the claims asserted in the Class Action Lawsuits against all named defendants in connection with the Class Action Lawsuits, including us, and any claims that could have been asserted that arise or relate to the facts alleged in the Class Action Lawsuits, such that every member of thepurportedsettlement classactions filedwill be barred from asserting such claims in theCalifornia District Courtfuture. In connection with the settlement of the Class Action Lawsuits, in April 2017, we paid $250,000 andappointedour insurance providers paid $6.0 million to alead plaintiffsettlement escrow account, to be paid to members of the settlement class, less payment of attorneys’ fees andlead counselcosts torepresentplaintiff’s counsel. The settlement does not constitute any admission of fault or wrongdoing by us or any of thepurported class. On July 21, 2014,individual defendants.We do not expect to make any additional payments for and do not expect, and are not aware of, any additional claims arising from or related to the
California District Court ordered the lead plaintiff to file its consolidated amended complaint, which was filed on September 19, 2014. On August 11, 2014, we filed a motion to transfer the purported securities lawsuit filedfacts alleged in theMississippi District Court toClass Action Lawsuits and asserted by stockholders who have opted out of theCalifornia District Court. On November 4, 2014,settlement class in theMississippi District Court granted our motion and transferred the case to the California District Court, which was thereafter consolidated with the class actions. We filed our motion to dismiss the consolidated amended complaint on November 18, 2014. The plaintiff's opposition was filed on January 20, 2015, and we filed our reply on February 25, 2015. The court hearing for the motion to dismiss has been scheduled for April 10, 2015. ItClass Action Lawsuits. However, it is possible that additionalsuits willlawsuits may be filed, or allegations may be made by stockholders, with respect to these same or other matters and also naming us and/or our officers and directors as defendants.We believe that we have meritorious defensesMonitoring, initiating andintenddefending against legal actions is time-consuming for our management, is likely todefend against these lawsuits vigorously.On April 21, 2014, a stockholder purportingbe expensive and may detract from our ability toactfully focus our internal resources on ourbehalf filed a derivative lawsuit inbusiness activities. In addition, despite theSuperior Courtavailability ofCalifornia for the County of San Mateo against certain of our officers and directors. The lawsuit alleges breaches of fiduciary duties by the defendants and other violations of law. In general, the lawsuit alleges that the defendants caused or allowed the dissemination of allegedly false and misleading statements related to our Phase 2 trial of imetelstat in patients with ET or PV. The plaintiff is seeking unspecified monetary damages and other relief, including reforms and improvements to our corporate governance and internal procedures. It is possible that additional derivative lawsuits will be filed with respect to these same or other matters and also naming our officers and directors as defendants. We have not yet responded to the derivative lawsuit, but intend to vigorously defend against the claims alleged and to seek dismissal of the lawsuit.These lawsuits and any other related lawsuits are subject to inherent uncertainties, and the actual defense and disposition costs will depend upon many unknown factors. The outcome of these lawsuits is necessarily uncertain. We could be forced to expend significant resources in the defense against these and any other related lawsuits andinsurance, we maynot prevail. We currently are not able to estimate the possible cost to us from these lawsuits, as they are currently at an early stage,incur substantial legal fees and costs in connection with any additional litigation, and such amounts could be material to our financialstatements even ifstatements. We may expend significant resources in the settlement or defense of any additional lawsuits, and we may not prevail inthe defense against thesesuch lawsuits. Wecannot behave not established any reserve for any potential liability relating to any additional lawsuits. Lawsuits could result in judgments against us that require us to pay damages, enjoin us from certainhow long it may takeactivities, or otherwise negatively affect our legal or contractual rights, which could have a significant adverse effect on our business. In addition, the inherent uncertainty of such litigation could lead toresolve these lawsuits orincreased volatility in our stock price and a decrease in thepossible amountvalue ofany damages that we may be required to pay.our stockholders’ investment in our common stock.ITEM 4. MINE SAFETY DISCLOSURESNot applicable.
ITEM 5.
MARKET FOR THE REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
ITEM 5. MARKET FOR THE REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIESMarket Information
Our common stock is quoted on the Nasdaq Global Select Market under the symbol GERN. The high and low intraday sales prices as reported by the Nasdaq Global Select Market of our common stock for each of the quarters in the years ended December 31,
20142017 and20132016 were as follows:High
Low
High Low Year ended December 31, 2014
Year Ended December 31, 2017:
First quarter
$ 5.92 $ 1.39 $
2.45
$
1.87
Second quarter
$ 3.47 $ 1.69 $
3.15
$
2.05
Third quarter
$ 3.30 $ 1.98 $
3.01
$
1.95
Fourth quarter
$ 3.96 $ 1.76 $
2.36
$
1.74
Year ended December 31, 2013
Year Ended December 31, 2016:
First quarter
$ 1.78 $ 1.05 $
4.77
$
2.30
Second quarter
$ 1.55 $ 0.98 $
3.35
$
2.42
Third quarter
$ 3.95 $ 1.27 $
3.13
$
1.84
Fourth quarter
$ 7.79 $ 2.65 $
2.45
$
1.81
As of March
6, 2015,7, 2018, there were approximately654574 stockholders of record of our common stock. This number does not include"street name"“street name” or beneficial holders, whose shares are held of record by banks, brokers and other financial institutions. We are engaged in a highly dynamic industry, which often results in significant volatility of our common stock price. On March6, 2015,7, 2018, the closing sales price for our common stock was$3.50$2.68 per share.Dividend Policy
We have never paid cash dividends on our capital stock and do not anticipate paying cash dividends in the foreseeable future, but intend to retain our capital resources for reinvestment in our business. Any future determination to pay cash dividends will be at the discretion of the board of directors and will be dependent upon our financial condition, results of operations, capital requirements and other factors our board of directors deems relevant.
Performance Measurement Comparison(1)
The following graph compares total stockholder returns of Geron Corporation for the last five fiscal years beginning December 31,
20092012 to two indices: the Nasdaq CRSP Total Return Index for the Nasdaq StockMarket-U.S.Market‑U.S. Companies, or theNasdaq-US,Nasdaq‑US, and the NasdaqPharmaceuticalBiotech Index, or theNasdaq-Pharmaceutical.Nasdaq‑Biotech. The total return for our stock and for each index assumes the reinvestment of dividends, although we have never declared cash dividends on Geron stock, and is based on the returns of the component companies weighted according to their capitalizations as of the end of each quarterly period. TheNasdaq-USNasdaq‑US tracks the aggregate price performance of equity securities of U.S. companies traded on the Nasdaq Global Select Market, or NGSM. TheNasdaq-Pharmaceutical,Nasdaq‑Biotech, which is calculated and supplied by Nasdaq, representspharmaceutical companies, includingbiotechnology companies trading on Nasdaq under the Standard Industrial Classification (SIC) Code No. 283 Drugs main category (2833—Medicinals & Botanicals, 2834—Pharmaceutical Preparations, 2835—Diagnostic Substances, 2836—Biological Products). Geron common stock trades on the NGSM and is a component of both theNasdaq-USNasdaq‑US and theNasdaq-Pharmaceutical.Nasdaq‑Biotech. The stockholder return shown in the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
Comparison of Five Year Cumulative Total Return on Investment AmongGeron Corporation, the
Nasdaq-USNasdaq‑US Index and theNasdaq-PharmaceuticalNasdaq‑Biotech Index(2)

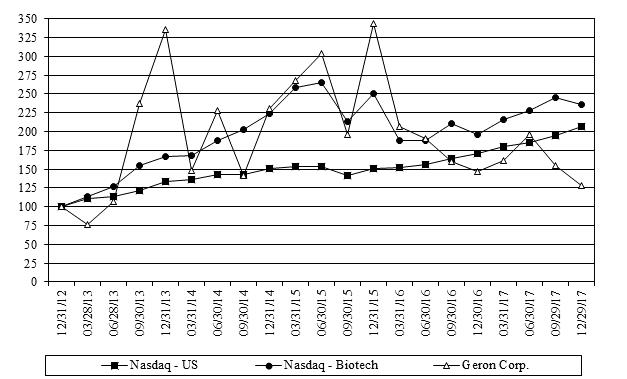
(1)This Section is not "soliciting material," is not deemed "filed" with the SEC and is not to be incorporated by reference in any filing of Geron Corporation under the Securities Act, or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.(2)Shows the cumulative total return on investment assuming an investment of $100 in each of Geron, the Nasdaq-US and the Nasdaq-Pharmaceutical on December 31, 2009. The cumulative total return on Geron stock has been computed based on a price of $5.55 per share, the price at which Geron common stock closed on December 31, 2009.
(1)
This Section is not “soliciting material,” is not deemed “filed” with the SEC and is not to be incorporated by reference in any filing of Geron Corporation under the Securities Act of 1933, as amended, or the Exchange Act of 1934, as amended, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
(2)
Shows the cumulative total return on investment assuming an investment of $100 in each of Geron, the Nasdaq‑US and the Nasdaq‑Biotech on December 31, 2012. The cumulative total return on Geron stock has been computed based on a price of $1.41 per share, the price at which Geron common stock closed on December 31, 2012.
Recent Sales of Unregistered Securities
InDuring the year ended December2014, we issued an aggregate31, 2017, there were no unregistered sales of168,039 shares of our common stock pursuant to the net, or cashless, exercise of warrants that were originally issued in connection with a loan agreement with the California Institute for Regenerative Medicine, or CIRM, in November 2011. These warrants were exercisable for an aggregate of 461,382 shares of our common stock and had an exercise price of $2.32 per share.In issuing the above-mentioned shares, we relied on the exemptions providedequity securities bySection 3(a)(9) of the Securities Act of 1933, as amended.us.ITEM 6. SELECTED FINANCIAL DATAThe following selected
consolidatedfinancial data should be read together with ourconsolidatedaudited financial statements and accompanying notes and"Management's“Management’s Discussion and Analysis of Financial Condition and Results ofOperations"Operations” appearing elsewhere in this annual report on Form10-K.10‑K. The selectedconsolidatedfinancial data in this section is not intended to replace ourconsolidatedfinancial statements and the accompanying notes. Our historical results are not necessarily indicative of our future results.Year Ended December 31, 2014 2013 2012 2011 2010 (In thousands, except share and per share data) Consolidated Statements of Operations Data:
Revenues from collaborative agreements
$ — $ — $ — $ 300 $ 925 License fees and royalties
1,153 1,283 2,709 2,138 2,638 Total revenues
1,153 1,283 2,709 2,438 3,563 Operating expenses:
Research and development
20,707 23,155 51,368 69,316 61,687 Acquired in-process research and development(1)
— — — — 35,000 Restructuring charges(2)
— 1,462 2,702 5,449 — General and administrative
16,758 15,624 20,397 23,789 18,043 Total operating expenses
37,465 40,241 74,467 98,554 114,730 Loss from operations
(36,312 ) (38,958 ) (71,758 ) (96,116 ) (111,167 ) Unrealized gain (loss) on derivatives
351 (316 ) 13 643 190 Interest and other income
373 951 3,097 1,024 2,045 Losses recognized under equity method investment
— — — (503 ) (2,347 ) Losses recognized from debt extinguishment(3)
— — — (1,664 ) — Interest and other expense
(82 ) (56 ) (233 ) (237 ) (98 ) Net loss
$ (35,670 ) $ (38,379 ) $ (68,881 ) $ (96,853 ) $ (111,377 ) Basic and diluted net loss per share:
Net loss per share
$ (0.23 ) $ (0.30 ) $ (0.54 ) $ (0.78 ) $ (1.14 ) Shares used in computing net loss per share
153,540,341 128,380,800 126,941,024 124,506,763 97,601,520 Year Ended December 31,
2017
2016
2015
2014
2013
(In thousands, except share and per share data)
Statements of Operations Data:
Revenues:
Collaboration revenue(1)
$
—
$
—
$
35,000
$
—
$
—
License fees and royalties(2)
1,065
6,162
1,371
1,153
1,283
Total revenues
1,065
6,162
36,371
1,153
1,283
Operating expenses:
Research and development
11,033
18,047
17,831
20,707
23,155
Restructuring charges(3)
—
—
1,306
—
1,462
General and administrative
19,287
18,761
17,793
16,758
15,624
Total operating expenses
30,320
36,808
36,930
37,465
40,241
Loss from operations
(29,255
)
(30,646
)
(559
)
(36,312
)
(38,958
)
Unrealized gain (loss) on derivatives
—
—
16
351
(316
)
Interest and other income
1,416
1,192
677
373
951
Interest and other expense
(77
)
(83
)
(88
)
(82
)
(56
)
Net (loss) income
$
(27,916
)
$
(29,537
)
$
46
$
(35,670
)
$
(38,379
)
Net (loss) income per share:
Basic
$
(0.18
)
$
(0.19
)
$
0.00
$
(0.23
)
$
(0.30
)
Diluted
$
(0.18
)
$
(0.19
)
$
0.00
$
(0.23
)
$
(0.30
)
Shares used in computing net (loss)
income per share:
Basic
159,224,986
159,045,644
158,036,162
153,540,341
128,380,800
Diluted
159,224,986
159,045,644
162,663,894
153,540,341
128,380,800
(1)
In November 2014, we entered into a collaboration and license agreement, or the Collaboration Agreement, pursuant to which we granted to Janssen Biotech Inc., or Janssen, the exclusive rights to develop and commercialize imetelstat worldwide for all indications in oncology, including hematologic myeloid malignancies, and all other human therapeutic uses. The Collaboration Agreement became effective in December 2014 and we received $35 million from Janssen as an upfront payment, which was classified as deferred revenue on our balance sheet as of December 31, 2014. Upon delivery of the imetelstat license rights and completion of our performance of the technology transfer‑related activities to Janssen as outlined under the Collaboration Agreement, we fully recognized the $35 million upfront payment as collaboration revenue in the third quarter of 2015.
(1)In December 2010, we and Angiochem, Inc., or Angiochem, entered into an exclusive license agreement that provided us with a worldwide exclusive license, with the right to grant sublicenses, to Angiochem's proprietary peptide technology that facilitates the transfer of anti-cancer compounds across the blood-brain barrier to be used with tubulin disassembly inhibitors to enable the treatment of primary brain cancers and cancers that have metastasized to the brain. As consideration for the license rights, we paid Angiochem an upfront payment of $7.5 million in cash and issued to Angiochem 5,261,144 shares of common stock on January 5, 2011 as payment of our obligation to issue $27.5 million in stock to Angiochem. Because further clinical and process development of GRN1005 was required before any viable commercial application could be identified or utilized, we concluded that the technology had no alternative future use, and
(2)
In September 2016, we entered into a license agreement, or License Agreement, with Janssen Pharmaceuticals, Inc., or Janssen Pharmaceuticals, pursuant to which we granted to Janssen Pharmaceuticals an exclusive worldwide license under our proprietary patents for the research, development and commercialization of products based on specialized oligonucleotide backbone chemistry and novel amidates for ribonucleic acid interference, or RNAi, for the prevention, treatment and/or diagnosis of any and all human disorders, excluding cancers originating from the blood or bone marrow, and products whose predominant or primary mechanism of action is telomerase inhibition. In accordance with the terms of the License Agreement, we received $5 million from Janssen Pharmaceuticals as an upfront payment, which we recognized as license fee revenue in the third quarter of 2016 upon our delivery of the license rights and transfer of know‑how to Janssen Pharmaceuticals under the License Agreement.
accordingly, expensed the total upfront payment of $35 million as acquired in-process research and development at the time of acquisition in 2010.On December 3, 2012, we provided to Angiochem notice of termination of the exclusive license agreement. We returned the asset to Angiochem in May 2013 and the license agreement terminated effective June 1, 2013.(2)- In April 2013, we
announced the decision to discontinuediscontinued our discovery research programs and companion diagnostics program based on telomere length andcloseclosed our research laboratory facility located at 200 Constitution Drive, Menlo Park, California.With this decision, a total of 20 positions were eliminated, representing approximately 31% of our workforce at that time.In connection with this restructuring, we incurred aggregate restructuring charges of approximately $1.4 million in 2013. All actions associated with this restructuring were completed in2013, and we do not anticipate incurring any further charges in connection with this restructuring. In December 2012, we announced the decision to discontinue development of GRN1005. With this decision, a total of 43 positions were eliminated, representing a reduction of approximately 40% of our workforce at that time. In connection with the restructuring, we incurred aggregate restructuring charges of approximately $2.8 million, of which $2.7 million was recorded in 2012 and $92,000 was recorded in2013.All actions associated with this restructuring were completed in 2013, and we do not anticipate incurring any further charges in connection with this restructuring.In November 2011, we discontinued further development of our stem cell programs. With this decision, a total of 66 positions were eliminated, representing a reduction of approximately 38% of our workforce at that time. In connection with the restructuring, we recorded aggregate restructuring charges of approximately $5.4 million in 2011. All actions associated with this restructuring were completed in 2012, and we do not anticipate incurring any further charges in connection with this restructuring. See Note 6 on Restructurings in Notes to Consolidated Financial Statements of this annual report on Form 10-K.(3)In November 2011, we repaid the outstanding principal balance, including accrued interest, or Loan Balance, to CIRM representing our entire Loan Balance under our loan agreement with CIRM. In addition, we relinquished our right to future disbursements from CIRM under the loan agreement and gave notice of termination. With the repayment of the entire outstanding Loan Balance, we have no further amounts owed to CIRM. In connection with the early termination of the loan agreement with CIRM, we recognized a debt extinguishment charge of $1.7 million for the unamortized debt discount associated with the loan.
December 31,
(In thousands)
2017
2016
2015
2014
2013
Balance Sheets Data:
Cash, restricted cash, cash equivalents and
marketable securities
$
109,195
$
129,067
$
146,700
$
170,639
$
66,019
Working capital
89,454
108,243
109,258
111,607
59,470
Total assets
110,313
130,249
148,760
172,511
67,344
Accumulated deficit
(985,840
)
(957,924
)
(928,387
)
(928,433
)
(892,763
)
Total stockholders' equity
103,797
122,380
142,126
130,712
59,757
December 31, (In thousands) 2014 2013 2012 2011 2010 Consolidated Balance Sheets Data:
Cash, restricted cash, cash equivalents and marketable securities
$ 170,639 $ 66,019 $ 96,329 $ 154,239 $ 221,274 Working capital
111,607 59,470 84,269 112,181 154,168 Total assets
172,511 67,344 99,801 160,047 233,584 Accumulated deficit
(928,433 ) (892,763 ) (854,384 ) (785,503 ) (688,650 ) Total stockholders' equity
130,712 59,757 85,653 146,603 192,735 ITEM 7.
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONSOverviewThe following discussion should be read in conjunction with the section entitled “Business” in Part I, Item 1 and the audited
consolidatedfinancial statements and notes thereto included in Part II, Item 8 of this annual report on Form 10‑K. The information provided should be reviewed in the context of the sections entitled “Risks Related to Our Collaboration with Janssen” and “Risks Related to Clinical Development, Regulatory Approval and Commercialization of Imetelstat” in Part I, Item 1A entitled “Risk Factors” and elsewhere in this annual report on Form 10-K.Business Overview
We are a
clinical stagebiopharmaceutical companyfocused onthat currently supports the clinical stage development of a telomerase inhibitor, imetelstat, in hematologic myeloidmalignancies. The discovery and early development of imetelstat, our sole product candidate, was based on our core expertise in telomerase and telomere biology. Telomerase is an enzyme that enables cancer cells, including malignant progenitor cells, to maintain telomere length, which provides them with the capacity for limitless, uncontrolled proliferation. Using our proprietary nucleic acid chemistry, we designed imetelstat to be an oligonucleotide that targets and binds with high affinity to the active site of telomerase, thereby directly inhibiting telomerase activity and impeding malignant cell proliferation. Molecular responsesmalignancies, by Janssen Biotech, Inc., or Janssen. Early clinical data in essential thrombocythemia, or ET,and remission responses, including reversal of bone marrow fibrosis, inmyelofibrosis, or MF, and myelodysplastic syndromes, or MDS, suggest imetelstathas disease-modifyingmay have disease‑modifying activity by inhibiting the progenitor cells of the malignantcloneclones for the underlyingdisease in a relatively selective manner.diseases.On November 13, 2014, we entered into
an exclusivea collaboration and license agreement, or the Collaboration Agreement,withpursuant to which we granted JanssenBiotech, Inc., or Janssen,the exclusive rights to develop and commercialize imetelstat worldwide for all indications in oncology, including hematologic myeloid malignancies, and all other human therapeutic uses. The Collaboration Agreement became effective on December 15, 2014, and we received $35 million from Janssen as an upfrontpayment, which has been recorded as deferred revenue on our consolidated balance sheet as of December 31, 2014.payment. Additional consideration under the Collaboration Agreement includes potential payments of up toa potentialan aggregate maximum total of $900 million for the achievement of development, regulatory and commercial milestones, as well as royalties on worldwide netsales.sales of imetelstat. The Collaboration Agreement also provides for a joint governance structure that includes a Joint Steering Committee, or JSC, with equal membership from both companies. See “Licensing—Collaboration and License Agreement with Janssen” under Item 1, “Business” for more information about the Collaboration Agreement, including economic terms and termination provisions of the Collaboration Agreement. The information provided should be reviewed in the context of the sections entitled “Risks Related to Our Collaboration with Janssen” and “Risks Related to Clinical Development, Regulatory Approval and Commercialization of Imetelstat” under Item 1A, “Risk Factors”.Under the Collaboration Agreement, Janssen is wholly responsible for
the development,developing, manufacturing,and commercialization of, andseeking regulatory approval for, and commercialization of, imetelstat worldwide.DevelopmentJanssen is currently conducting two clinical trials of imetelstat: IMbark, a Phase 2 trial in MF, in which the first patient was dosed in September 2015 and the last patient was enrolled in October 2016; and IMerge, a Phase 2/3 trial in MDS, in which the first patient was dosed in January 2016. We contribute 50% of the development costs for these trials, which Janssen is solely conducting.For IMbark, Janssen completed internal data reviews in September 2016, April 2017 and March 2018. In these data reviews, activity within multiple outcome measures was observed with imetelstat treatment that suggest potential
clinical benefit in patients with MF who are relapsed after or refractory to prior treatment with a janus kinase, or JAK, inhibitor. However, new patient enrollment in IMbark was suspended in October 2016 because an insufficient number of patients met the protocol defined interim efficacy criteria to continue enrollment. In March 2018, Janssen will officially close the trial to new patient enrollment. The JSC expects that the over 100 patients enrolled in IMbark to date will be adequate to assess overall survival. Patients who remain in the treatment phase of IMbark may continue to receive imetelstat, and until the protocol-specified primary analysis, all safety and efficacy assessments are being conducted as planned in the protocol, including following patients, to the extent possible, until death, to enable an assessment of overall survival. In March 2018, based on the rate of deaths occurring in the trial, the JSC determined that the protocol-specified primary analysis of IMbark, which includes an assessment of overall survival, will begin by the end of the second quarter of 2018. Upon the protocol-specified primary analysis, the main trial will be completed. The IMbark protocol is being amended to establish an extension phase of the trial to enable patients remaining in the treatment phase to continue to receive imetelstat treatment, per investigator discretion. Following completion of the primary analysis, Janssen must notify us of its decision, or the Continuation Decision, whether to: (i) maintain the license rights granted under the Collaboration Agreement and continue the development of imetelstat
will proceed under a mutually agreed clinicalor (ii) discontinue the developmentplan, which includes two Phase 2 studies to be pursued initially, one in myelofibrosis, referred to asof imetelstat and terminate theInitial Phase 2 MF Study, and one in myelodysplastic syndrome, referred to as the Initial Phase 2 MDS Study.Collaboration Agreement. We expect Janssen toinitiate the Initial Phase 2 MF Study in mid-2015, followedinform us of its decision bythe Initial Phase 2 MDS Study to be initiated atthe end of2015.the third quarter of 2018.For IMerge, Janssen completed internal data reviews in September 2016 and April 2017. In addition, preliminary data from Part 1 of IMerge were presented at the
clinicalAmerican Society of Hematology Annual Meeting, or ASH, in December 2017. These data showed that among the 32 red blood cell transfusion-dependent MDS patients enrolled in Part 1of the trial, a subset of 13 patients who had not received prior treatment with either a hypomethylating agent or lenalidomide and did not have a deletion 5q chromosomal abnormality, or non-del(5q), exhibited an increased rate and durability of transfusion independence compared to the overall trial population. Based on the preliminary data from this 13-patient subset, Janssen has expanded new patient enrollment in Part 1 of IMerge to enroll approximately 20 additional patients to increase the experience and confirm the benefit-risk profile of imetelstat in this refined target patient population. In November 2017, the first patient was dosed in the expanded Part 1 and enrollment was completed in February 2018. Using the preliminary data from Part 1, Janssen sponsored an application to the United States Food and Drug Administration, or the FDA, for Fast Track designation for the potential treatment of adult patients with transfusion-dependent anemia due to Low or Intermediate-1 risk MDS who are non-del(5q) and who are refractory or resistant to treatment with an erythropoiesis stimulating agent. The FDA granted Fast Track designation to imetelstat in October 2017.Financial Overview
We had approximately $109.2 million in cash and investments as of December 31, 2017. To grow and diversify our business, we plan to continue our business development
planefforts to identify, and seek to acquire and/or in-license other oncology products, product candidates, programs or companies. Acquisition or in-licensing opportunities that we mayalso include additional, possible registration studiespursue could materially affect our liquidity and capital resources and may require us to incur indebtedness or seek equity capital, or both. While we reported a small profit for the year ended December 31, 2015 due to our recognition of revenue inMF and myelodysplastic syndrome, or MDS, and possible exploratory Phase 2 and potential follow-on Phase 3 studies in acute myelogenous leukemia, or AML. For a further discussion regarding the Collaboration Agreement, see Note 10 on License Agreements in Notes to Consolidated Financial Statements of this annual report on Form 10-K.We believe our current operational and financial resources, includingconnection with the upfront paymentreceivedfrom Janssen under the Collaboration Agreement,may enable us to acquire other oncology products, programs or companies to diversify our business.until 2015 we had never been profitable. We have incurred
operatingsignificant net lossesevery yearsince ouroperations beganinception in1990. Losses have resulted1990, resulting principally from costs incurred in connection with our research and development activities and from general and administrative costs associated with our operations.Substantially allAs ofour revenues to date have been research support payments under collaborative agreements, and milestones, royalties and other revenues from our licensing arrangements. We currently have no sourceDecember 31, 2017, we had an accumulated deficit ofproduct revenue. Our revenues for 2015 are expected to primarily consist of revenue from the upfront payment from Janssen upon the completion of technology transfer-related activities in 2015, and future revenues are substantially dependent on Janssen successfully developing and commercializing imetelstat inaccordance with the Collaboration Agreement.$985.8 million. Since our inception, we primarily haveprimarilyfinanced our operations through the sale of equity securities, interest income on our marketable securities and payments we received under our collaborative and licensing arrangements.For the years ended December 31, 2014, 2013Substantially all of our revenues to date have been payments under collaborative agreements, and2012, we incurred net lossesmilestones, royalties and other revenues from our licensing arrangements. We currently have no source of$35.7 million, or $0.23 per share, $38.4 million, or $0.30 per share, and $68.9 million, or $0.54 per share, respectively. As of December 31, 2014, we had an accumulated deficit of $928.4 million.product revenue. The significance of future losses, future revenues and any potential future profitability will depend primarily on whether Janssen continues to develop and advance imetelstat and the clinical and commercial success of imetelstat, which would result in potential future revenues to us in the form of milestone payments and royalties under the Collaboration Agreement,as described above,and whether we in-license or acquire other oncology products, product candidates, programs or companies in order to grow and diversify our business. There can be no assurance that we will receive any milestone payments or royalties from Janssen in the future, or at all.Imetelstat, which isIn addition, if Janssen does not perform in the manner we expect or fulfill its responsibilities in a timely manner, or at all, including with respect to obtaining sufficient efficacyand safety data from the additional patients enrolled in Part 1 of IMerge and/or obtaining longer-term efficacy and safety data from IMbark to enable an assessment of overall survival, the clinical development, manufacturing, regulatory approval and/or commercialization of imetelstat could be delayed or terminated, and it could become necessary for us to assume responsibility for the clinical development, manufacturing, regulatory approval and/or commercialization of imetelstat at our
sole product candidate,own expense. In any event, imetelstat will require significant additional clinical testing prior to possible regulatory approval in the United States and other countries, and we do not expect imetelstat to be commercially available for many years, if at all.As of December 31, 2014, we had cash, restricted cash, cash equivalents and marketable securities of $170.6 million compared to $66.0 million at December 31, 2013 and $96.3 million at December 31, 2012. The increase in cash, restricted cash, cash equivalents and marketable securities in 2014 was primarily the result of the receipt of net cash proceeds of approximately $96.8 million, after deducting the underwriting discount and offering expenses payable by us, from an underwritten public offering of 25,875,000 shares of our common stock at a public offering price of $4.00 per share that we completed in February 2014 and the receipt of $35.0 million from Janssen as an upfront payment in December 2014 upon the effectiveness of the Collaboration Agreement. We estimate that our existing capital resources and future interest income will be sufficient to fund our current level of operations through at least the next 12 months. However, we may use our available capital resources sooner than we anticipate.Critical Accounting Policies and Estimates
Our
consolidatedfinancial statements have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of theseconsolidatedfinancial statements requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses. Note 1 of Notes toConsolidatedFinancial Statements describes the significant accounting policies used in the preparation ofthe consolidatedour financial statements. Certain of these significant accounting policies are considered to be critical accounting policies, as defined below.A critical accounting policy is defined as one that is both material to the presentation of our
consolidatedfinancial statements and requires management to make difficult, subjective or complex judgments that could have a material effect on our financial condition and results of operations. Specifically, critical accounting estimates have the following attributes: (i) we are required to make assumptions about matters that are highly uncertain at the time of the estimate; and (ii) different estimates we could reasonably have used, or changes in the estimate that are reasonably likely to occur, would have a material effect on our financial condition or results of operations.Estimates and assumptions about future events and their effects cannot be determined with certainty. We base our estimates on historical experience and on various other assumptions believed to be applicable and reasonable under the circumstances. These estimates may change as new events occur, as additional information is obtained and as our operating environment changes. These changes historically have
historicallybeen minor and have been included in theconsolidatedfinancial statements as soon as they became known. Based on a critical assessment of our accounting policies and the underlying judgments and uncertainties affecting the application of those policies, management believes that ourconsolidatedfinancial statements are stated fairlystatedin accordance with accounting principles generally accepted in the United States, and meaningfully present our financial condition and results of operations.We believe the following critical accounting policies reflect our more significant estimates and assumptions used in the preparation of our
consolidatedfinancial statements:Fair Value of Financial Instruments
We categorize financial instruments recorded at fair value on our
consolidatedbalancesheetsheets based upon the level of judgment associated with inputs used to measure their fair value. The categories are as follows:Level 1—Inputs are unadjusted, quoted prices in active markets for identical assets or liabilities at the measurement date. An active market for the asset or liability is a market in which transactions for the asset or liability occur with sufficient frequency and volume to provide pricing information on an ongoing basis.
Level 2—Inputs (other than quoted market prices included in Level 1) are either directly or indirectly observable for the asset or liability through correlation with market data at the measurement date and for the duration of the
instrument'sinstrument’s anticipated life.Level 3—Inputs reflect
management'smanagement’s best estimate of what market participants would use in pricing the asset or liability at the measurement date. Consideration is given to the risk inherent in the valuation technique and the risk inherent in the inputs to the model.A financial
instrument'sinstrument’s categorization within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Following is a description of the valuation methodologies used for financial instruments measured at fair value on ourconsolidatedbalancesheet,sheets, including the category for such financial instruments.InstrumentsFinancial instruments classified as Level 1 include money market funds and certificates of deposit, representing24%approximately 10% of our total financial instruments classified as assets measured at fair valueclassified as assetsas of December 31,2014. Instruments2017. Financial instruments classified as Level 2 include U.S.government-sponsoredgovernment‑sponsored enterprise securities, commercial paper and corporate notes, representing76%approximately 90% of our total financial instruments classified as assets measured at fair valueclassified as assetsas of December 31,2014.2017. The price for each security at the measurement date is derived from various sources. Periodically, we assess the reasonableness of these sourced prices by comparing them to the prices provided by our portfolio managers from broker quotes as well as reviewing the pricing methodologies used by our portfolio managers. Historically, we have not experienced significant deviation between the sourced prices and our portfoliomanager'smanagers’ prices.Warrants to purchase common stock and non-employee options are normally traded less actively, have trade activity that is one way, and/or are traded in less-developed markets and are therefore valued based upon models with significant unobservable market parameters, resulting in Level 3 categorization. Instruments classified as Level 3 include derivative liabilities from non-employee options, representing all of our financial instruments measured at fair value classified as liabilities as of December 31, 2014. The fair value for these instruments is calculated using the Black Scholes option-pricing model. The model's inputs reflect assumptions that market participants would use in pricing the instrument in a current period transaction. Use of this model requires us to make assumptions regarding stock volatility, dividend yields, expected term of the non-employee options and risk-free interest rates. Changes to the model's inputs are not changes to valuation methodologies, but instead reflect direct or indirect impacts from changes in market conditions. Accordingly, results from the valuation model in one period may not be indicative of future period measurements. Expected volatilities are based on historical volatilities of our stock. The expected term of non-employee options represents the remaining contractual term of the instruments. The risk-free interest rate is based on theU.S. Zero Coupon Treasury Strip Yields for the remaining term of the instrument. If factors change and we employ different assumptions in future periods, the fair value of these non-employee options reflected as of each balance sheet date and the resulting change in fair value that we record may differ significantly from what we have recorded in previous periods. As of December 31, 2014, we have not revised the method in which we derive assumptions in order to estimate fair values of non-employee options classified as liabilities, and we do not expect revisions in the future.For a further discussion regarding fair value measurements, see Note 2 on Fair Value Measurements in Notes to
ConsolidatedFinancial Statements of this annual report on Form10-K.10‑K.Revenue Recognition
In general, weWe recognize revenue for each unit of accounting when all of the following criteria have been met: (a) persuasive evidence of an arrangement exists, (b) delivery has occurred or services have been rendered, (c) theseller'sseller’s price to the buyer is fixed or determinable, and (d) collectability is reasonably assured. Amounts received prior to satisfying these revenue recognition criteria are recorded as deferred revenue. Amounts expected to be recognized as revenue within the 12 months following the balance sheet date are classified as current deferred revenue. Amounts not expected to be recognized as revenue within the 12 months following the balance sheet date are classified as noncurrent deferred revenue.Since our inception, substantially all of our revenues have been generated from license and collaboration
agreements and licensing arrangements.agreements. Economic terms in these agreements may includenon-refundablenon‑refundable upfront license payments in cash or equity securities, option payments in cash or equity securities, cost reimbursements,cost-sharingcost‑sharing arrangements, milestone payments, royalties on future sales of products, or any combination of these items. In applying the appropriate revenue recognition guidance related to these agreements, we first assess whether the arrangement contains multiple elements. In this evaluation, we consider:(1)(i) the deliverables included in the arrangement and(2)(ii) whether the individual deliverables represent separate units of accounting or whether they must be accounted for as a combined unit of accounting. This evaluation involves subjective determinations and requires us to make judgments about the individual deliverables and whether such deliverables are separable from the other aspects of the contractual relationship. Deliverables are considered separate units of accounting provided that: (i) the delivered item(s) has value to the customer on a standalone basis, and (ii) if(ii)the arrangement includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control. In assessing whether an item has standalone value, we consider factors such as the research, manufacturing and commercialization capabilities of the collaboration partner or licensee and the availability of the associated expertise in the general marketplace. In addition, we consider whether the collaboration partner or licensee can use the other deliverable(s) for their intended purpose without the receipt of the remaining element(s), whether the value of the deliverable is dependent on the undelivered item(s) and whether there are other vendors that can provide the undelivered element(s).Arrangement consideration that is fixed or determinable is allocated among the separate units of accounting using the relative selling price method. We then apply the applicable revenue recognition criteria noted above to each of the separate units of accounting in determining the appropriate period and pattern of recognition. We determine how to allocate arrangement consideration to identified units of accounting based on the selling price hierarchy provided under relevant accounting guidance. The estimated fair value of deliverables under the arrangement may be derived using a best estimate of selling price if
vendor-specific-objectivevendor‑specific‑objective evidence andthird-partythird‑party evidence are not available.Upfront
non-refundablenon‑refundable signing, license ornon-exclusivenon‑exclusive option fees are recognized as revenue: (i) when rights to use the intellectual propertyrelated to ahave been delivered, if the licensethathas standalone value from the other deliverables to be provided under the agreement,have been deliveredor (ii) over the term of the agreement if we have continuing performance obligations, as the arrangement would be accounted for as a single unit of accounting. When payments are received in equity securities, we do not recognize any revenue unless such securities are determined to be realizable in cash.At the inception of an arrangement that includes milestone payments, we assess whether each milestone is substantive and at risk
to both partieson the basis of the contingent nature of the milestone. This evaluation includes an assessment of whether: (i) the consideration is commensurate with eitherthe(1) our performance to achieve the milestone or (2) the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting fromtheour performance to achieve the milestone, (ii) the consideration relates solely to past performance and (iii) the consideration is reasonable relative to all of the deliverables and payment terms (including other potential milestone consideration) within the arrangement. We consider various factors, such as the scientific, clinical, regulatory, commercial and other risks that must be overcome to achieve the respective milestone and the level of effort and investment required to achieve the respective milestone, in making this assessment. There is considerable judgment involved in determining whether a milestone satisfies all of the criteria required to conclude that a milestone is substantive. Milestone payments for milestones that are considered substantive would be recognized as revenue in their entirety upon successful accomplishment of the milestone, assuming all other revenue recognition criteria are met. Milestone payments for milestones that are not considered substantive would be recognized as revenue over the remaining period of performance, assuming all other revenue recognition criteria are met.Our license and collaboration agreements with certain partners also provide for contingent payments to us based solely upon the performance of the respective partner. For such contingent amounts, we recognize the payments as revenue when earned under the applicable contract, which is generally upon completion of performance by the respective partner, provided that collection is reasonably assured.
Royalties are recognized as earned in accordance with contract terms when royalties from licensees can be reasonably estimated and
collectabilitycollection is reasonably assured. If royalties cannot be reasonably estimated orcollectabilitycollection of a royalty amount is not reasonably assured, royalties are recognized as revenue when the cash is received. Revenue from commercial milestone paymentswill beis accounted for as royalties and recorded as revenue upon achievement of the milestone, assuming all other revenue recognition criteria aremet.met and we have no performance obligations related to the milestone.Cost-sharingCost‑sharing expenses are recorded as earned or owed based on the performance requirements by both parties under the respective contracts. For arrangements in which we and our collaboration partner in the agreement are exposed to significant risks and rewards depending on the commercial success of the activity, we recognize payments between the parties on a net basis and record such amounts as a reduction or addition to research and development expense. For arrangements in which we have agreed to perform certain research and development services for our collaboration partner and are not exposed to significant risks and rewardsdependingthat depend on the commercial success of the activity, we recognize the respective cost reimbursements as revenue under the collaborative agreement as the related research and development services are rendered.Revenue recognition for licenses and collaboration agreements requires significant judgment.
We estimate the projected future term of license agreements over which we recognize revenue.We evaluate the deliverables under an arrangement and estimate the fair value of those deliverables. We also assess the substantive nature of milestones. Our assessments and estimates are based on contractual terms, historical experience and general industry practice. Revisions in these values or estimations have the effect of increasing or decreasing license fee or collaboration revenue in the period of revision. As of December 31,2014,2017, we have not made any revisions to revenue recognitionestimates and we do not expect revisions to currently active agreements in the future.estimates.Clinical Trial Accruals
Substantial portions of our preclinical studies and all of our clinical trials have been performed by third-party contract research organizations, or CROs, and other vendors. We accrue expenses forpreclinical studies performed by our vendors based on certain estimates over the term of the service period and adjust our estimates as required. We accrue expenses for clinical trial activities performed by CROs based upon the estimated amount of work completed on each study.Forclinical trial expenses, the significant factors used in estimating accruals include the number of patients enrolled, the number of active clinical sites, and the duration for which the patients will be enrolled in the study. Pass through costs from CROs include, but are not limited to, regulatory expenses, investigator fees, lab fees, travel costs and other miscellaneous costs, including shipping and printing fees. We accrue pass through costs based on estimates of the amount of work completed forthe clinicaltrial. Wedevelopment activities being conducted by Janssen under the Collaboration Agreement, we monitor patient enrollment levels and related activities to the extent possible throughinternal reviews, review of contractual termsdiscussions with Janssen personnel andcorrespondence with CROs. Webase our estimates on the best information available at the time. However, additional information may become available to us whichwillwould allow us to make a more accurate estimate in future periods. In this event, we may be required to record adjustments to research and development expenses in future periods when the actual level of activity becomes more certain.Valuation of
Stock-BasedStock‑Based CompensationWe measure and recognize compensation expense for all
stock-basedshare‑based payment awards to our employees and directors, including stock options, restricted stock awards and employee stock purchases related to our Employee Stock Purchase Plan, or ESPP, based on estimated grant‑date fairvalues. We use the Black Scholes option-pricing model to estimate the grant-datevalues for these instruments. The grant‑date fair value ofour stock options and employee stock purchases. Option-pricing valuation model assumptions such as expected volatility, risk-free interest rate and expected term impact the fair value estimate.Further, the estimated forfeiture rate impacts the amount of aggregate stock-based compensation expense recognized during the period. The fair value of stock options and employee stock purchasesshare‑based payment awards is amortized over the vesting period of the awards using astraight-line method.straight‑line method and reduced for estimated forfeitures. We use the Black Scholes option‑pricing model to estimate the grant‑date fair value of our stock options and employee stock purchases. The grant-date fair value for service-based restricted stock awards is determined using the fair value of our common stock on the date of grant.Option‑pricing model assumptions, such as expected volatility, expected term and risk‑free interest rate, impact the fair value estimate. Expected volatilities are based on historical volatilities of our stock since traded options on Geron common stock do not correspond to option terms and trading volume of options is limited. The expected term of options represents the period of time that options granted are expected to be outstanding. In deriving this assumption, we
reviewedreview actual historical exercise and post‑vesting cancellation data and the remaining outstanding options not yet exercised or cancelled. The expected term ofemployees'employees’ purchase rights under our ESPP is equal to the purchase period. Therisk-freerisk‑free interest rate is based on the U.S. Zero Coupon Treasury Strip Yields for the expected term in effect on the date of grant. Forfeiture rates are estimated based on historical data and are adjusted, if necessary, over the requisite service period based on the extent to which actual forfeitures differ, or are expected to differ, from their estimate.We have granted restricted stock awards to employees and directors with three types of vesting schedules: (i) service-based, (ii) performance-based or (iii) market-based. Service-based restricted stock awards generally vest annually over four years. Performance-based restricted stock awards vest upon achievement of discrete strategic corporate goals within a specified performance period, generally three years. Market-based restricted stock awards vest upon achievement of certain market price thresholds of our common stock within a specified performance period, generally three years.The fair value for service-based restricted stock awards is determined using the fair value of our common stock on the date of grant. The fair value is amortized as stock-based compensation expense over the requisite service period of the award, which is generally the vesting period, on a straight-line basis and is reduced for estimated forfeitures, as applicable.The fair value for performance-based restricted stock awards is determined using the fair value of our common stock on the date of grant. Stock-based compensation expense for awards with vesting based on performance conditions is recognized over the period from the date the performancecondition is determined to be probable of occurring through the date the applicable condition is expected to be met and is reduced for estimated forfeitures, as applicable. If the performance condition is not considered probable of being achieved, no stock-based compensation expense is recognized until such time as the performance condition is considered probable of being met, if at all. If that assessment of the probability of the performance condition being met changes, the impact of the change in estimate would be recognized in the period of the change. If the requisite service period has been met prior to the change in estimate, the effect of the change in estimate would be immediately recognized. We evaluate whether performance conditions are probable of occurring, as well as the expected performance period, on a quarterly basis.The fair value for market-based restricted stock awards is determined using a lattice valuation model with a Monte Carlo simulation. The model takes into consideration the historical volatility of our stock and the risk-free interest rate at the date of grant. In addition, the model is used to estimate the derived service period for the awards. The derived service period is the estimated period of time that would be required to satisfy the market condition, assuming the market condition will be satisfied. Stock-based compensation expense is recognized over the derived service period for the awards using the straight-line method and is reduced for estimated forfeitures, as applicable, but is accelerated if the market condition is achieved earlier than estimated. If a market-based restricted stock award is forfeited or expires after completion of the derived service period, any previously recognized stock-based compensation expense is not reversed.We evaluate the assumptions used in estimating grant‑date fair values of our
stock-basedshare‑based payment awards by reviewing current trends in comparison to historical data on an annual basis. We have not revised the methods by which we derive assumptions in order to estimate grant‑date fair values of ourstock-basedshare‑based payment awards. If factors change and we employ different assumptions in future periods, thestock-basedstock‑based compensation expense that we record for share‑based payment awards to employees and directors may differ significantly from what we have recorded in the current period.Compensation expense recognized for stock-based awards to employees and directors was $7.7 million, $4.4 million and $5.3 million for the years ended December 31, 2014, 2013 and 2012, respectively. As of December 31, 2014, total compensation cost related to unvested stock-based awards not yet recognized, net of estimated forfeitures, was $15.0 million, which is expected to be recognized over the next 27 months on a weighted-average basis.For our
non-employee stock-basednon‑employee stock‑based awards, the measurement date on which the fair value of thestock-basedstock‑based award is calculated is equal to the earlierofof: (i) the date at which a commitment for performance by the counterparty to earn the equity instrument is reached or (ii) the date at which thecounterparty'scounterparty’s performance is complete.We recognized stock-based compensation expense of $94,000, $92,000 and $135,000 for the fair value of the vested portion of non-employee options and restricted stock awards in our consolidated statements of operations for the years ended December 31, 2014, 2013 and 2012, respectively.Results of Operations
Our results of operations have fluctuated from period to period and may continue to fluctuate in the future, based primarily upon the progress of research and development efforts in collaboration with Janssen and whether we are able to acquire and/or in-license other oncology products, product candidates, programs or companies in order to grow and diversify our business. Results of operations for any period may be unrelated to results of operations for any other period.
In addition,Thus, historical results should not be viewed as indicative of future operating results. For example, in 2015 we reported net income for the first time due to recognition of revenue in connection with the upfront payment from Janssen under the Collaboration Agreement. However, we expect to incur operating losses in the future as clinical development activities for imetelstat continue under our Collaboration Agreement with Janssen, and our operating losses may increase in size. We are subject to risks common to companies in our industry and at our stage of development, including, but not limited to, risks inherent in research and development efforts, our dependence on Janssen for the development, manufacture, regulatory approvalmanufacturefor and commercialization of,our sole product candidate,imetelstat, uncertainty of preclinical and clinical trial results or regulatory approvals or clearances,needTablethe future development ofContentsimetelstat, including any future efficacy or safety results that may cause the benefit-risk profile of imetelstat to become unacceptable, the possibility that Janssen could discontinue the imetelstat program and terminate the Collaboration Agreement at any time and for any reason, irrespective of whether there is data from IMbark suggesting an adequate improvement in survival in relapsed or refractory MF, with the determination of adequacy to be assessed by Janssen in its sole discretion, or whether there is data from IMerge to support the benefit-risk profile of imetelstat in lower risk MDS; our need for future capital, enforcement of our patent and proprietary rights, reliance upon our collaborators, licensees, investigators and other third parties, and potential competition. In order for imetelstat to be commercialized,
basedwe are wholly dependent onour research, we andJanssenmustto conduct preclinical tests and clinical trials to demonstrate the safety and efficacy of imetelstat, obtain regulatory approvals or clearances and enter into manufacturing, distribution and marketing arrangements, as well as obtain market acceptance. We do not expect to receive royalties based on sales of imetelstat for many years, if at all.Collaboration Revenue
Upon the effectiveness of the Collaboration Agreement with Janssen in December 2014, we received $35 million as an upfront payment, which we classified as deferred revenue upon receipt. We determined delivery of the imetelstat license rights granted by us to Janssen, together with our performance of the technology transfer‑related activities outlined in the Collaboration Agreement, represented the sole non‑contingent deliverable associated with the upfront payment. Therefore, we accounted for our delivery of the imetelstat license rights and our performance of the technology transfer‑related activities as a single unit of accounting. During the third quarter of 2015, we completed performance of the technology transfer‑related activities to Janssen as outlined under the Collaboration Agreement. Combining this performance with the delivery of the imetelstat license rights, we fully recognized the $35 million upfront payment from Janssen as collaboration revenue on our statements of operations during the third quarter of 2015. No further payments have been made by Janssen to us under the Collaboration Agreement. Any future collaboration revenue is substantially dependent on Janssen’s ability to successfully develop and commercialize imetelstat in accordance with the Collaboration Agreement. See further discussion of revenue recognition for the Collaboration Agreement in Note 4 on License Agreements in Notes to Financial Statements of this annual report on Form 10‑K.
License Fees and Royalties
In addition to the Collaboration Agreement with Janssen for imetelstat, we have entered into several license or collaboration agreements with companies involved with oncology, diagnostics, research tools and biologics
production. In each of these agreements,production, whereby we have granted certain rights to our non‑imetelstat related technologies. In connection withthethese agreements, we are eligible to receive license fees, option fees, milestone payments and royalties on future sales of products, or any combination thereof. We recognized license fee revenues of$738,000, $933,000$667,000, $5.6 million and$1.6 million$722,000 in2014, 20132017, 2016 and2012,2015, respectively, related to our various agreements.We have not recognized any revenue related to the Collaboration Agreement in 2014 since we have determined that the sole non-contingent deliverable under the Collaboration Agreement is our delivery of the license rights to Janssen and our complete performance of the technology transfer-related activities. We currently expect completion of the technology transfer-related activities to occur by September 30, 2015, at which point we expect to fully recognize the $35.0 million upfront payment from Janssen as license fee revenue.The decrease in license fee revenues in20142017 compared to20132016 and the increase in license fee revenues in 2016 compared to 2015 primarily reflects the full recognition of an upfront payment of $5 million from Janssen Pharmaceuticals, Inc., or Janssen Pharmaceuticals, under anon-refundable up-frontlicense agreement that was executed in September 2016, or the License Agreement, related to license rights to commercialize products based on specialized oligonucleotide backbone chemistry and novel amidates for RNAi for the prevention, treatment and/or diagnosis of any and all human disorders, excluding cancers originating from the blood or bone marrow, and products whose predominant or primary mechanism of action is telomerase inhibition. See further discussion of revenue recognition under and description of the terms of the License Agreement in Note 4 on License Agreements in Notes to Financial Statements of this annual report on Form 10 K.We determined delivery of the license rights granted by us to Janssen Pharmaceuticals under the License Agreement, together with the transfer of certain know‑how necessary for the research, development and commercialization of products under the License Agreement, represented the sole non‑contingent deliverable under the License Agreement. Accordingly, we accounted for our delivery of the license rights and transfer of know‑how under the License Agreement as a single unit of accounting. Since we completed the delivery of the license rights and transfer of know‑how to Janssen Pharmaceuticals under the License Agreement during the third quarter of 2016, we fully recognized the upfront payment
in 2013 for an exclusive commercial license using our telomerase promoter technology for oncology-related in vitro assays. The decrease infrom Janssen Pharmaceuticals as license feerevenuesrevenue on our statements of operations in2013 comparedthe third quarter of 2016. Since the research and development of the technology we licensed to2012 primarily reflectsJanssen Pharmaceuticals under thefull recognition ofLicense Agreement is at alicense payment from GE Healthcare in 2012 uponvery early stage, we do not expect significant milestone payments under theexercise of an option to expandLicense Agreement unless and until a product that utilizes or incorporates thescope of their original license agreement with us to obtain exclusive global rights to intellectual property and know-how for thelicensed technology enters clinical development andsalereceives marketing approval, which may take many years, if ever. As such, future revenues under the License Agreement involve a substantial degree ofcellular assays derived from induced pluripotent stem cells. In connection with the closing of the divestiture of our human embryonic stem cell assets in October 2013, our license agreement with GE Healthcare, including any future revenue payments thereunder, was transferred to Asterias.uncertainty and risk that they may never be realized.We recognized royalty revenues of
$415,000, $350,000$398,000, $537,000 and$1.1 million$649,000 in2014, 20132017, 2016 and2012,2015, respectively, on product sales of telomerase detection and telomere measurement kits to theresearch-use-onlyresearch‑use‑only market,cell-basedcell‑based research products and nutritional products. Theincrease in royalty revenues in 2014 compared to 2013 primarily reflects the receipt of a milestone fee in 2014 in connection with the achievement of a net sales milestone by a licensee of our hTERT technology. Thedecrease in royalty revenues in20132017 compared to20122016 and 2016 compared to 2015 primarily reflects lower product sales by our licensees.In 2017, the
assignmentmajority of our revenues were from license fees and royalties under licenses granted to several biotechnology and pharmaceutical companies to use telomeraseactivation technology to Telomerase Activation Sciences, Inc., or TA Sciences,immortalized cells inDecember 2012drug discovery research andterminationfor drug discovery applications. Two customers accounted for approximately 39% of ourlicense agreement with Asia Biotech Corporation. See further discussion of the agreement with TA Sciences under the sub-section entitled "Interest and Other Income".We expect our revenues for 2015 to primarily consist of license fee revenue from the2017 revenues. The upfront payment from Janssenunder the Collaboration Agreement uponPharmaceuticals represented approximately 81% of ourcompletion2016 revenues. The upfront payment from Janssen represented approximately 96% ofthe technology transfer-related activities inour 2015and future license fee and royalty revenues are substantially dependent on Janssen successfully developing and commercializing imetelstat in accordance with the Collaboration Agreement. See further discussion of revenue recognition for the Janssen collaboration in Note 10 on License Agreements in Notes to Consolidated Financial Statements of this annual report on Form 10-K.revenues.Future license fee and royalty revenues are
alsodependent on additional agreements being signed,andif any, current agreements beingmaintained.maintained and the underlying patent rights for the licenses remaining active. We expect revenues under our license agreements related to our human telomerase reverse transcriptase technology to decline significantly in the coming years, and to be eliminated by the end of 2019, due to upcoming patent expirations on such technology. Current revenues may not be predictive of future revenues.Research and Development Expenses
For each of ourDuring the years ended December 31, 2017, 2016 and 2015, imetelstat was the sole research and developmentprograms,program we supported. For the imetelstat research and development program, we incur direct external, personnel related and other research and development costs.DirectFor the years ended December 31, 2017, 2016 and 2015, direct external expenses primarilyconsistconsisted of our proportionate share of research and development coststo outside parties to perform laboratory studies, develop manufacturing processes and manufacture raw materials and clinical trial drug materials, conduct and manage clinical trials, including investigator-sponsored clinical trials, and provide advice and consultation for scientific and clinical strategies.incurred by Janssen under the Collaboration Agreement. Personnel related expenses primarily consist of salaries and wages,stock-basedstock‑based compensation, payroll taxes and benefits forthose individualsGeron employees involved with ongoing research and development efforts. Other research and development expenses primarily consist oflaboratory supplies, research-relatedresearch related overhead associated withleasing, operatingallocated expenses for rent andmaintaining ourmaintenance of facilities andequipment depreciation and maintenance. All of these costs apply to our current and historical clinical programs and our historical preclinical programs and discovery research efforts. A product candidate is designated a clinical candidate once an investigational new drug application has been filed with the FDA, or a similar filing with regulatory agencies outside the United States, for the purpose of commencing clinical trials in humans. Preclinical programs include product candidates undergoing toxicology, pharmacology, metabolism and efficacy studies and manufacturing process development required before testing in humans can commence.other supplies.Research and development expenses were
$20.7$11.0 million,$23.2$18.0 million and$51.4$17.8 million for the years ended December 31,2014, 20132017, 2016 and2012,2015, respectively.As shown in the table below, the decrease in research and development expenses in 2014 compared to 2013 and 2013 compared to 2012 primarily reflects the net result of lower direct external costs due to the wind-down of our GRN1005 trials in patients with brain metastases and imetelstat trials in solid tumors, reduced personnel related costs resulting from previous restructurings and lower costs for scientific supplies and services and other research-related overhead costs due to the discontinuation of our discovery research programs in April 2013.The decrease in research and development expenses in20142017 compared to2013 was2016 primarily reflects lower direct external costs for our proportionate share of clinical development expenses under the collaboration with Janssen and reduced personnel related expenses. The increase in research and development expenses in 2016 compared to 2015 primarily reflects the net result of higher direct external costs for our proportionate share of clinical development expenses under the collaboration with Janssen, partially offset byan increase inreduced personnel related expenses and research related overhead as a result of the March 2015 restructuring and lower direct externalcostsexpenses for the manufacturing of imetelstat drug product.Overall, in 2015 we expect direct external research and development expenses to increase as we collaborate with Janssen on the development of imetelstat in hematologic myeloid malignancies and personnel related research and development expenses to decrease as a result of the recently announced organizational resizing.Research and development expenses for the years ended December 31,
2014, 20132017, 2016 and20122015 were as follows:Year Ended December 31, (In thousands) 2014 2013 2012 Direct external research and development expenses:
Clinical program: Imetelstat
$ 8,901 $ 7,665 $ 12,907 Clinical program: GRN1005(1)
— 1,039 10,723 Clinical program: GRNOPC1(2)
— 202 393 Preclinical programs(3)
— 228 1,155 Personnel related expenses
9,674 10,753 19,008 All other research and development expenses
2,132 3,268 7,182 Total
$ 20,707 $ 23,155 $ 51,368 (1)In December 2012, we discontinued the GRN1005 program and returned the asset to Angiochem in May 2013.(2)In October 2013, we closed the transaction to divest our human embryonic stem cell assets and our autologous cellular immunotherapy program to Asterias. Asterias assumed all post-closing liabilities with respect to all of the assets contributed by us, including any liabilities related to the
GRNOPC1 and autologous cellular immunotherapy clinical trials. See Note 7 on Divestiture of Stem Cell Assets in Notes to Consolidated Financial Statements of this annual report on Form 10-K for further discussion.(3)In April 2013, we announced the decision to discontinue our discovery research programs and companion diagnostics program based on telomere length.
Year Ended December 31,
(In thousands)
2017
2016
2015
Direct external research and development expenses:
Clinical program: Imetelstat
$
8,437
$
14,695
$
9,574
Personnel related expenses
2,063
2,729
6,478
All other research and development expenses
533
623
1,779
Total
$
11,033
$
18,047
$
17,831
At this time, we cannot provide reliable estimates of how much time or investment will be necessary to commercialize imetelstat. For a more complete discussion of the risks and uncertainties associated with the development of imetelstat in collaboration with Janssen, see the
sub-sectionssub‑sections entitled"Risks“Risks Related to OurBusiness"Collaboration with Janssen” and"Risks“Risks Related to Clinical Development, Regulatory Approval and CommercializationActivities"of Imetelstat” in Part I, Item 1A entitled"Risk Factors"“Risk Factors” and elsewhere in this annual report on Form10-K.10‑K.In
April 2013, we announced the decision to discontinue our discovery research programs and companion diagnostics program based on telomere length and close our research laboratory facility located at 200 Constitution Drive, Menlo Park, California. With this decision, a total of 20 positions were eliminated. InMarch 2015, in connection withthis restructuring,projected reduced operational demands as a result of the Collaboration Agreement with Janssen, weincurredimplemented an organizational resizing which resulted in aggregate restructuring charges of approximately$1.4$1.3 million in2013, of which $624,0002015 related toone-timeone‑time termination benefits, including$28,000$307,000 ofnon-cash stock-basednon‑cash stock‑based compensation expense relating to the extension of thepost-terminationpost‑termination exercise periodthrough the end of December 2013for certain stock options previously granted toterminatedemployees$200,000 related to non-cash charges for write-downs of excess equipment and leasehold improvements and $546,000 related to costs associated withaffected by theclosure of our research laboratory facility. In connection with the decision to close our research laboratory facility, we entered into an amendment to the lease agreement for the 200 Constitution Drive facility under which the lease terminated effective December 31, 2013. As consideration for the early termination of the lease, we paid the landlord the remaining rents due under the original term of the lease as well as certain facility maintenance costs, all of which have been included in restructuring charges. In 2013, we received proceeds of approximately $1.1 million from the sale of excess laboratory equipment in connection with the closure of our research laboratory facility.restructuring. All actions associated with this restructuring were completed in2013, and we do not anticipate incurring any further charges in connection with this restructuring.In December 2012, we announced the decision to discontinue development of GRN1005. With this decision, a total of 43 positions were eliminated. In connection with this restructuring, we incurred aggregate restructuring charges of approximately $2.8 million, of which $2.7 million was recorded in 2012 and $92,000 was recorded in 2013. The aggregate restructuring charges consisted of $2.5 million related to one-time termination benefits, including $107,000 of non-cash stock-based compensation expense relating to the extension of the post-termination exercise period through the end of December 2013 for certain stock options previously granted to terminated employees, and $271,000 related to write-downs of GRN1005 manufacturing equipment. All actions associated with this restructuring were completed in 2013, and we do not anticipate incurring any further charges in connection with this restructuring.2015. See Note 6 onRestructuringsRestructuring in Notes toConsolidatedFinancial Statements of this annual report on Form10-K10‑K for further discussion of the restructuring charges.General and Administrative Expenses
General and administrative expenses were
$16.8$19.3 million,$15.6$18.8 million and$20.4$17.8 million for the years ended December 31,2014, 20132017, 2016 and2012,2015, respectively. The increase in general and administrative expenses in20142017 compared to20132016 primarily reflects higher non‑cash stock‑based compensation expense, an increased allocation of facilities and other overhead costs to general and administrative activities and higher consulting costs, partially offset by lower legal costs. The increase in general and administrative expenses in 2016 compared to 2015 primarily reflects the net result of highernon-cash stock-basednon‑cash stock‑based compensation expense and an increasedlegal fees associated with the purported securities lawsuitsallocation of facilities andthe derivative lawsuit filed against us and/or certain of our officersother overhead costs to general anddirectors and transactioncosts associated with the Collaboration Agreement we entered into with Janssen in November 2014,administrative activities, partially offset byreduced patent costslower consulting andtransaction fees associated with the stem cell divestiture which closed in October 2013. The decrease in general and administrative expenses in 2013 compared to 2012 primarily reflects lower personnel related expenses, primarily resulting from previous restructurings, and reducedlegaland consulting fees associated with our intellectual property portfolio and our stem cell divestiture efforts.costs. We expect general and administrative expenses in 2018 toincrease in 2015 as a result of higher anticipated legal fees as we intend to vigorously defend against the lawsuits filed against us.remain consistent with 2017.Unrealized Gain
(Loss)on DerivativesUnrealized gain (loss) on derivatives reflects a non-cash adjustment for changes in fair value ofNon‑employee optionsheld by non-employees that areclassified ascurrent liabilities. Derivatives classified asderivative liabilities are marked to fair value at each financial reporting date with any resulting changes in fair value being recorded as unrealized gain (loss)recordedin theconsolidatedstatements of operations.The derivatives continue to be reported as a liability until such time as the instruments are exercised or expire or are otherwise modified to remove the provisions which require them to be recorded as liabilities, at which time these instruments are marked to fair value and reclassified from liabilities to stockholders' equity.We incurred an unrealizedgainsgain on derivatives of$351,000$16,000 for the year ended December 31, 2015, which reflected a decline in the fair value of non‑employee options classified as derivative liabilities. No comparable amounts were incurred in 2017 and$13,0002016 as all non‑employee options classified as derivative liabilities expired unexercised during the first quarter of 2015.Interest and Other Income
Interest income was $1.4 million, $1.2 million and $677,000 for the years ended December 31,
20142017, 2016 and2012, respectively, compared to unrealized losses on derivatives of $316,000 for the year ended December 31, 2013. The unrealized gains and losses on derivatives primarily reflect the change in fair values of derivative liabilities as a result of fluctuations in the market value of our stock and changes in other inputs factored into the estimate of their fair value such as the volatility of our stock. See Note 2 on Fair Value Measurements in Notes to Consolidated Financial Statements of this annual report on Form 10-K for further discussion of the fair value of derivatives.Interest and Other IncomeInterest income was $373,000, $219,000 and $597,000 for the years ended December 31, 2014, 2013 and 2012,2015, respectively. The increase in interest income in20142017 compared to20132016 and 2016 compared to 2015 primarily reflectsan increase inhigher yields on ourcash and investment balances in connection with the receipt of the net cash proceeds from the underwritten public offering of shares of our common stock that we completed in February 2014. The decrease in interest income in 2013 compared to 2012 primarily reflects lower cash and investment balances resulting from the use of cash for operations.marketable securities portfolio. Interest earned in future periods will depend on the size of our marketable securities portfolio and prevailing interest rates.Other income
was $732,000 and $2.5 millionof $16,000 for theyearsyear ended December 31,2013 and 2012, respectively.2016 reflects gains on sales of excess equipment. No other income was recognized for theyearyears ended December 31,2014. Other income recognized in 2013 reflects a net gain on the sale of excess laboratory equipment in connection with the closure of our research laboratory facility. Other income recognized in 2012 reflects the receipt a non-refundable upfront payment of $2.5 million for the assignment of our telomerase activation technology to TA Sciences, pursuant to the Termination2017 andAssignment Agreement that we entered into with Asia Biotech Corporation, or Asia Biotech, and TA Sciences in December 2012. See Note 10 on License Agreements in Notes to Consolidated Financial Statements of this annual report on Form 10-K for further discussion of the Termination and Assignment Agreement with Asia Biotech and TA Sciences.2015.Interest and Other Expense
Interest and other expense was
$82,000, $56,000$77,000, $83,000 and$233,000$88,000 for the years ended December 31,2014, 20132017, 2016 and2012,2015, respectively.The increase in interestInterest and other expensein 2014 compared to 2013primarily reflectshigherbank charges related tothe increase inour cash operating accounts andinvestment balances in connection with the receipt of the net cash proceeds from the underwritten public offering of shares of our common stock that we completed in February 2014. The decrease in interest and other expense in 2013 compared to 2012 primarily reflects the recognition of accumulated foreign currency translation adjustments in connection with the dissolution of Geron Bio-Med Ltd. in August 2012 and reduced bank charges as a result of lower cash and investment balances in 2013.marketable securities portfolio.Net LossNet loss was $35.7 million, $38.4 million and $68.9 million for the years ended December 31, 2014, 2013 and 2012, respectively. The decrease in net loss in 2014 compared to 2013 and 2013 compared to 2012 primarily reflects lower clinical trial costs as a result of the wind-down of our GRN1005 trials in patients with brain metastases and imetelstat trials in solid tumors, decreased personnel related costs resulting from previous restructurings, reduced costs for scientific supplies and services and other research-related overhead costs with the discontinuation of our discovery research programs in April 2013 and lower patent costs and transaction fees associated with the stem cell divestiture which closed in October 2013. The decrease in net loss in 2014 compared to 2013 was partially offset by increased costs for the manufacturing of imetelstat drug product, higher non-cash stock-based compensation expense and increased legal fees associated with the purported securities lawsuits and transaction costs for the Collaboration Agreement we entered into with Janssen in November 2014.Liquidity and Capital Resources
Cash,As of December 31, 2017, we had cash, restricted cash, cash equivalents and marketable securitiesat December 31, 2014 were $170.6of $109.2 million, compared to$66.0$129.1 million at December 31,20132016 and$96.3$146.7 million at December 31,2012.2015. The decrease in cash, restricted cash, cash equivalents and marketable securities in 2017 and 2016 was the result of cash being used for operations. We expect to experience negative cash flow and to incur significant and increasing operating expenses for the foreseeable future as the development of imetelstat continues in collaboration with Janssen. We estimate that our existing capital resources and future interest income will be sufficient to fund our current level of operations through at least the next 12 months. However, we may use our available capital resources sooner than we anticipate. For example, in order to grow and diversify our business, we plan to continue our business development efforts to identify and seek to acquire and/or in‑license other oncology products, product candidates, programs or companies. Acquisition or in‑licensing opportunities that we may pursue could materially affect our liquidity and capital resources and may require us to incur indebtedness, seek equity capital or both. In addition, there can be no assurance that sufficient additional capital would be available to us in order to pursue any of these opportunities.We have an investment policy to invest
these fundsour cash in liquid, investment grade securities, such asinterest-bearinginterest‑bearing money market funds, certificates of deposit, municipal securities, U.S. government and agency securities, corporate notes and commercial paper. Our investment portfolio does not contain securities with exposure tosub-primesub‑prime mortgages, collateralized debt obligations,asset-backedasset‑backed securities or auction rate securities and, to date, we have not recognized anyother-than-temporaryother‑than‑temporary impairment charges on our marketable securities or any significant changes in aggregate fair value that would impact our cash resources or liquidity. To date, we have not experienced lack of access to our invested cash and cash equivalents; however, access to our invested cash and cash equivalents may be impacted by adverse conditions in the financial and credit markets.The increase in cash, restricted cash, cash equivalents and marketable securities in 2014 was primarily the result of the receipt of net cash proceeds of approximately $96.8 million, after deducting the underwriting discount and offering expenses payable by us, from an underwritten public offering of 25,875,000 shares of our common stock at a public offering price of $4.00 per share that we completed in February 2014 and the receipt of $35.0 million from Janssen as an upfront payment in December 2014 upon the effectiveness of the Collaboration Agreement.In
October 2012,August 2015, we entered into anAt-The-MarketAt Market Issuance Sales Agreement, orsales agreement,2015 Sales Agreement, with MLV & Co. LLC, or MLV, which provides that, upon the terms and subject to the conditions and limitations set forth in thesales agreement,2015 Sales Agreement, we may elect to issue and sell shares of our common stock having an aggregate offering price of up to$50.0$50 million from time to time through MLV as our sales agent. We are not obligated to make any sales of common stock under thesales agreement. To date, we have not sold any2015 Sales Agreement. Prior to 2017, no shares of our common stockpursuant towere sold under the 2015 Sales Agreement. In December 2017 and January 2018, 614,230 and 776,788 shares, respectively, of our common stock were sold under the 2015 Sales Agreement for aggregate net cash proceeds of approximately $2.6 million, after deducting salesagreement.commissions and offering expenses payable by us. As of March 16, 2018, approximately $47.2 million of our common stock remained available for issuance under the 2015 Sales Agreement. Thesales agreement2015 Sales Agreement will expire inOctober 2015August 2018 unless extended by the parties.We may need additional capital resources in order to support development and commercialization of imetelstat, especially if we elect to exercise
our U.S. Opt-In Rights and U.S. Co-Promotion Optioncertain options under the Collaboration Agreement and potentially independently pursue imetelstat development under our own independent development plan, or IDP, under the Collaboration Agreement, and to otherwise support the future growth of our business through the potential acquisition and/or in‑licensing of other oncology products, product candidates, programs or companies. We cannot assure you that our existing capital resources, future interest income, potential milestone payments and royalties under the Collaboration Agreement with Janssen, and potential future sales of our common stock, including pursuant to oursales agreement2015 Sales Agreement with MLV, will be sufficient to fund future planned activities. The timing and degree of any future capital requirements will depend on many factors, including:•the accuracy of the assumptions underlying our estimates for our capital needs;
•whether
we elect U.S. Opt-In Rights to share future U.S.Janssen discontinues developmentand promotion costs forof imetelstatunderand/or terminates the Collaboration Agreement,with Janssen;•- and we choose to
the extent permitted under the Collaboration Agreement, whether we independently pursuedevelop imetelstat ourselves;further changes or delays in Janssen’s development
under our own IDP;•our potential reimbursement obligationsplans for imetelstat, including changes to or further expansion of or delays in ongoing clinical trials decided upon by Janssenif any data from a Janssen IDP support approvalor required byaregulatoryagency in the United Statesauthorities, such as clinical holds or othercountries;•- requirements, or any other factors;
the achievement of development, regulatory and
commercialsales milestones resulting inthe paymentpayments to us from Janssen under the Collaboration Agreement and the timing of receipt of such payments, if any;•changes•
to the extent permitted under the Collaboration Agreement, whether we independently pursue imetelstat development under our own IDP;
our potential reimbursement obligations to Janssen if any data from a Janssen IDP support approval by regulatory authorities in the United States or
delaysother countries;in the event that Janssen provides an affirmative Continuation Decision to us, whether we then elect our option, or the U.S. Opt-In Rights, to share further U.S. development and
Janssen's development planspromotion costs for imetelstat beyond IMbark or IMerge under the Collaboration Agreement, includingchanges which may result from any future clinical holds on the INDs for imetelstat, including the IND for the MF Pilot Study,our share of development costs incurred to date by Janssen that weexpectwill be required totransfer to Janssen;•Janssen'sreimburse if we exercise our U.S. Opt-In Rights;Janssen’s ability to meaningfully reduce manufacturing costs of imetelstat;
•the progress, timing, magnitude, scope and costs of clinical development, manufacturing and commercialization
forof imetelstat, including the number of indications being pursued, subject topermission fromclearances and approvals by theFDA;•- FDA and other regulatory authorities;
the time and costs involved in obtaining regulatory clearances and approvals in the United States and in other countries;
•Janssen'sJanssen’s ability to successfully market and sell imetelstat, upon regulatory approval or clearance, in the United States and other countries;
•if we exercise our U.S. Opt-In Rights, our decision to also exercise our co-promotion option under the Collaboration Agreement with Janssen, or the U.S. Co-Promotion Option, including the costs and timing of building a U.S. sales force;
•the sales price for imetelstat;
the availability of coverage and adequate third-party reimbursement for imetelstat;
the timing, receipt and amount of royalties under the Collaboration Agreement on worldwide net sales of imetelstat, upon regulatory approval or clearance, if any;
•the timing, receipt and amount of royalties on sales of any stem cell products by Asterias, upon development, regulatory approval or clearance, if any;•the sales price and availability of adequate third-party reimbursement for imetelstat;•the cost of acquiring and/or
licensing any newin-licensing other oncology products, product candidates, programs or companies, if any;•the progress, timing, magnitude, scope and costs of clinical development, manufacturing and commercialization of any acquired or in-licensed oncology products, product candidates, programs, or companies, including the number of indications being pursued, subject to clearances and approvals by the FDA and other regulatory authorities;
expenses associated with
the pending andpotentialadditional related purported securities lawsuits and derivative lawsuits, as well as any otherfuture litigation; and•the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims.
In addition, changes in our business may occur that would consume available capital resources sooner than we expect. If our existing capital resources, future interest income, and potential milestone payments and royalties under the Collaboration Agreement with Janssen are insufficient to meet future capital requirements, we will need to raise additional capital to fund our
operations.operations, including pursuant to our 2015 Sales Agreement with MLV. Further, if the Collaboration Agreement is terminated, including as a result ofJanssen'sJanssen’s failure to provide an affirmative Continuation Decision to us, or for any other reason, we would not receive any milestone payments or royalties under the Collaboration Agreement, and then, depending on the timing of such event, we would be required to fund all clinical development,manufacturing and commercial activities for imetelstat should we elect to continue the development of imetelstat ourselves, which would require us to raise substantial additional capital or establish alternative collaborations with third‑party collaboration partners, which may not be possible. If the Collaboration Agreement is terminated and we are unable to raise additional capital or establish alternative collaborations with
third-partythird‑party collaboration partners for imetelstat, the development of imetelstat would be discontinued, whichmay not be possible.might cause us to cease operations. Additional financing through public or private equity financings, including pursuant to oursales agreement2015 Sales Agreement with MLV, capital lease transactions or other financing sources may not be available on acceptable terms, or at all. We may raise equity capital at a stock price or on other terms that could result in substantial dilution of ownership for our stockholders. The receptivity of the public and private equity markets to proposedfinancings is substantially affected by the general economic, market and political climate and by other factors which are unpredictable and over which we have no control. In this regard, continued volatility and instability in the global financial markets and political climate could adversely affect our ability to raise additional funds through financings and the terms upon which we may raise those funds.
Our ability to raise additional funds will be severely impaired in the event of:
•any future clinical holds on any INDfurther changes or delays in Janssen’s development plans for imetelstat;
•a failure or inability to show adequate safety or efficacy of imetelstat in
existingcurrent or potential future clinicaltrials;trials, which may result in a decision by Janssen to delay or•- discontinue further development of imetelstat; or
a termination of the Collaboration Agreement or if our collaboration with Janssen is otherwise unsuccessful.
If sufficient capital is not available, we may be unable to fulfill our funding obligations under the Collaboration Agreement with Janssen, resulting in our breach of the Collaboration Agreement, which could lead to Janssen paying lower milestone payments and lower royalties to us under a reduced royalty
tier which couldtier. This would have a material adverse effect on our results of operations and financial condition.Moreover, in order to grow and diversify our business, we plan to
diversifycontinue oursole product candidatebusiness developmentrisk by identifyingefforts to identify andseekingseek to acquire and/orin-license newin‑license other oncology products, productopportunities for development.candidates, programs or companies. Acquisition orin-licensingin‑licensing opportunities that we may pursue could materially affect our liquidity and capital resources and may require us to incur indebtedness, seek equity capital orboth.both, including pursuant to our 2015 Sales Agreement with MLV. In addition, there can be no assurance that sufficient additional capital would be available to us in order to pursue any of these opportunities.Cash Flows from Operating Activities
Net cash provided by operations was $9.4 million in 2014.Net cash used in operations was$36.7$20.6 million, $18.4 million and$55.1$24.2 million in20132017, 2016 and2012,2015, respectively. The increase in net cash used in operations in 2017 compared to 2016 primarily reflects the net result of the receipt of the $5 million upfront payment from Janssen Pharmaceuticals under the License Agreement in 2016, partially offset by lower payments to Janssen in 2017 under the cost-sharing arrangement for imetelstat clinical development. The decrease in net cash used in operations in20142016 compared to20132015 primarily reflects the net result of the receipt of$35.0the $5 million upfront payment from Janssen Pharmaceuticals under the License Agreement, reduced costs for the manufacturing of imetelstat drug product and lower personnel related expenses asan upfront payment in December 2014 upon the effectivenessa result of theCollaboration Agreement. Additionally,restructuring announced in March 2015, partially offset by higher payments to Janssen in 2016 under thedecrease in net cash used in operations in 2014 compared to 2013 and in 2013 compared to 2012 reflects reduced operating expenses due to previous restructurings and the wind-down of our GRN1005 trials in patients with brain metastases andcost‑sharing arrangement for imetelstattrials in solid tumors.clinical development.Cash Flows from Investing Activities
Net cash used in investing activities was $77.9 million in 2014.Net cash provided by investing activities was$21.1$23.0 million, $8.8 million and$61.0 million$73,000 in20132017, 2016 and2012,2015, respectively. Thedecreaseincrease in net cash provided by investing activities in20142017 compared to20132016 and 2016 compared to 2015 primarily reflects a higher rate of maturities than purchases of marketable securitieswith the net cash proceeds received from an underwritten public offering of shares of our common stock that we completedinFebruary 2014. The decrease in net cash provided by investing activities in 2013 compared to 2012 was primarily the result of lower proceeds from maturities of marketable securities relative to purchases of marketable securities.2017 and 2016, respectively.For the three years ended December 31,
2014,2017, wehavepurchased approximately$1.0 million$147,000 in property and equipment, none of which was financed through equipment financing arrangements.In2014, we closed the certificate of deposit for our unused equipment line of credit upon maturity and transferred the cash proceeds to our cash operating accounts. This action also cancelled the availability of the equipment line of credit.Cash Flows from Financing Activities
Net cash provided by financing activities in
2014, 20132017, 2016 and20122015 was$98.4$1.1 million,$6.6$1.2 million and$150,000,$2.6 million, respectively.NetThe decrease in net cash provided by financing activities in20142017 compared to 2016 primarily reflects the net result of the receipt of higher cash proceeds in 2016 from the exercise of outstanding stock options under our equity plans, partially offset by the receipt of net cash proceeds of approximately$96.8$1.1 million, after deductingthe underwriting discountsales commissions and offering expenses payable by us, from theunderwritten public offeringsale of25,875,000an aggregate of 614,230 shares of our common stock pursuant to the 2015 Sales Agreement with MLV. The decrease in net cash provided by financing activities in 2016 compared to 2015 primarily reflects the receipt of higher cash proceeds in 2015 from the exercise of outstanding stock options under our equity plans and the receipt of cash proceeds of approximately $881,000 in March 2015 from the exercise of warrants to purchase 235,000 shares of our common stock ata public offeringan exercise price of$4.00$3.75 pershare that we completed in February 2014. Net cash provided by financing activities in 2013 and 2012 reflects proceeds from the issuance of common stock under our employee equity plans.share.Significant Cash and Contractual Obligations
As of December 31,
2014,2017, our contractual obligations for the next five years and thereafter were as follows:Payments Due by Period Contractual Obligations(1)Total Less Than
1 Year1 - 3 Years 4 - 5 Years After
5 Years(In thousands) Equipment lease
$ 20 $ 10 $ 10 $ — $ — Operating leases(2)
959 885 74 — — Research funding and license fees(3)
313 88 90 90 45 Total contractual cash obligations
$ 1,292 $ 983 $ 174 $ 90 $ 45 Payments Due by Period
Less Than
After
Contractual Obligations (1)
Total
1 Year
1-3 Years
4 - 5 Years
5 Years
(In thousands)
Equipment lease
$
33
$
11
$
22
$
—
$
—
Operating lease(2)
1,435
678
757
—
—
License fees(3)
240
60
75
30
75
Total contractual cash obligations
$
1,708
$
749
$
854
$
30
$
75
(1)
This table does not include payments under our severance plan if there were a change in control of Geron or severance payments to employees in the event of an involuntary termination. In addition, this table does not include any royalty obligations under our license agreements as the timing and likelihood of such payments are not known.
(1)This table does not include payments under our severance plan if there were a change in control of Geron or severance payments to employees in the event of an involuntary termination. In addition, this table does not include any royalty obligations under our license agreements as the timing and likelihood of such payments are not known.(2)In February 2014, we amended the lease agreement for our premises at 149 Commonwealth Drive to extend the lease term through January 2016. Our amended lease at 149 Commonwealth Drive includes an option to extend the lease for one additional period of 18 months. Operating lease obligations in the table above do not include payments due under the amended lease agreement for the extended lease term or assume the exercise by us of any right of termination.(3)Research funding is comprised of sponsored research commitments at various laboratories around the world. License fees are comprised of minimum annual license payments under our existing license agreements with several universities and companies for the right to use intellectual property related to technologies that we have in-licensed.
(2)
In September 2017, we amended the lease agreement for our premises at 149 Commonwealth Drive, Menlo Park, California, to extend the lease term from February 2018 through January 2020. Our amended lease at 149 Commonwealth Drive includes an option to extend the lease for one additional period of two years. Operating lease obligations in the table above do not assume the exercise by us of the option to extend the lease or any right of termination.
(3)
License fees are comprised of minimum annual license payments under our existing license agreements with several universities and companies for the right to use intellectual property related to technologies that we have in‑licensed.
Off-BalanceOff‑Balance Sheet ArrangementsWe have not engaged in any
off-balanceoff‑balance sheet arrangements, including the use of structured finance, special purpose entities or variable interest entities.ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISKThe following discussion about our market risk disclosures contains
forward-lookingforward‑looking statements. Actual results could differ materially from those projected in theforward-lookingforward‑looking statements. We are exposed to credit risk and interest rate risk. We do not use derivative financial instruments for speculative or trading purposes.Credit Risk. We currently place our cash, restricted cash, cash equivalents and marketable securities with four financial institutions in the United States. Deposits with banks may exceed the amount of insurance provided on such deposits. While we monitor the cash balances in our operating accounts and adjust the cash balances as appropriate, these cash balances could be impacted if the underlying financial institutions fail or could be subject to other adverse conditions in the financial markets. Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash equivalents and marketable securities. Cash equivalents and marketable securities currently consist of money market funds, U.S.
government-sponsoredgovernment‑sponsored enterprise securities, commercial paper and corporate notes. Our investment policy, approved by the audit committee of our board of directors, limits the amount we may invest in any one type of investment issuer, thereby reducing credit risk concentrations. We limit our credit and liquidity risks through our investment policy and through regular reviews of our portfolio against our policy. To date, we have not experienced any loss or lack of access to cash in our operating accounts or to our cash equivalents and marketable securities in our investment portfolio. The effect of a hypothetical decrease of 10% in the average yield earned on our cash equivalents and marketable securities would have resulted in an immaterial decrease in our interest income for the year ended December 31,2014.2017.Interest Rate Risk. The primary objective of our investment activities is to manage our marketable securities portfolio to preserve principal and liquidity while maximizing the return on the investment portfolio through the full investment of available funds without significantly increasing risk. To achieve this objective, we primarily invest in widely diversified investments with fixed interest rates, which carry a degree of interest rate risk. Fixed rate securities may have their fair value adversely impacted due to a rise in interest rates. Due in part to these factors, our future interest income may fall short of expectations due to changes in market conditions and in interest rates or we may suffer losses in principal if forced to sell securities which may have declined in fair value due to changes in interest rates.
The fair value of our cash equivalents and marketable securities at December 31,
20142017 was$167.9$107.6 million. These investments include$40.4$15.0 million of cash equivalents whichare due inhave an original maturity of three months or less,than 90 days, $108.6$78.4 million ofshort-termshort‑term investments which are due in less than one year and$18.9$14.2 million oflong-termlong‑term investments which are due in one to two years.WeIn accordance with our investment policy, which is approved by our audit committee, we primarily investourin marketablesecurities portfolio insecurities with at least an investment grade rating to minimize interest rate and credit risk as well as to provide for an immediate source of funds. Although changes in interest rates may affect the fair value of the marketable securities portfolio and cause unrealized gains or losses, such gains or losses would not be realized unless the investments are sold. Due to the nature of our investments, which are primarily money market funds, U.S.government-sponsoredgovernment‑sponsored enterprise securities, commercial paper and corporate notes, we have concluded that there is no material interest rate risk exposure and a hypothetical movement of 1%movementin market interest rates would not have a significant impact on the total realized value of our portfolio.ITEM 8.
CONSOLIDATEDFINANCIALSTATEMENTSSTATEMENTS AND SUPPLEMENTARY DATAThe following
consolidatedfinancial statements and the related notes thereto, of Geron Corporation and the Report of Independent Registered Public Accounting Firm, Ernst & Young LLP, are filed as a part of this annual report on Form10-K.10‑K.Page
PageReport of Independent Registered Public Accounting Firm
8179
ConsolidatedBalance Sheets8280
ConsolidatedStatements of Operations8381
ConsolidatedStatements of Comprehensive Loss8482
ConsolidatedStatements ofStockholders'Stockholders’ Equity8583
ConsolidatedStatements of Cash Flows8684
Notes to
ConsolidatedFinancial Statements8785
Supplemental Data: Selected Quarterly Financial Information (Unaudited)
120108
REREPORTPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRMTheTo the Stockholders and the Board of Directorsand Stockholdersof Geron CorporationOpinion on the Financial Statements
We have audited the accompanying
consolidatedbalance sheets of Geron Corporation (the Company) as of December 31,20142017 and2013, and2016, the relatedconsolidatedstatements of operations, comprehensive loss,stockholders'stockholders’ equity and cash flows for each of the three years in the period ended December 31,2014. These2017, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statementsarepresent fairly, in all material respects, theresponsibilityfinancial position of theCompany's management. Our responsibility is to express an opinion on these financial statements based on our audits.Company at December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.We
conducted our auditsalso have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States).(PCAOB), the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated March 16, 2018 expressed an unqualified opinion thereon.Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material
misstatement. An audit includesmisstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidencesupportingregarding the amounts and disclosures in the financial statements.An auditOur audits alsoincludes assessingincluded evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financialstatement presentation.statements. We believe that our audits provide a reasonable basis for our opinion.In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Geron Corporation at December 31, 2014 and 2013, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Geron Corporation's internal control over financial reporting as of December 31, 2014, based on criteria established inInternal Control—Integrated Frameworkissued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 11, 2015 expressed an unqualified opinion thereon./s/ Ernst & Young LLP
We have served as the Company’s auditor since 1992.
Redwood City, California
March
11, 201516, 2018GERON CORPORATIONCONSOLIDATEDBALANCE SHEETSDecember 31, December 31,
December 31,
2014 2013 2017
2016
(In thousands, except share and per share data) (In thousands, except share and per share data)
ASSETS
Current assets:
Cash and cash equivalents
$ 42,796 $ 12,990 $
16,335
$
12,810
Restricted cash
266 795 268
268
Marketable securities
108,645 52,234 78,351
102,035
Interest and other receivables
963 564 436
475
Prepaid assets
736 474 580
524
Total current assets
153,406 67,057 95,970
116,112
Noncurrent marketable securities
18,932 — 14,241
13,954
Property and equipment, net
173 92 102
183
Deposits and other assets
— 195 $ 172,511 $ 67,344 $
110,313
$
130,249
LIABILITIES AND STOCKHOLDERS' EQUITY
Current liabilities:
Accounts payable
$ 1,033 $ 1,397 $
503
$
225
Accrued compensation and benefits
4,213 3,946 3,385
2,843
Accrued restructuring charges
— 94 Accrued collaboration charges
1,702
3,367
Accrued liabilities
1,537 1,783 926
1,434
Deferred revenue
35,000 — Fair value of derivatives
16 367 Total current liabilities
41,799 7,587 6,516
7,869
Commitments and contingencies
Stockholders' equity:
Preferred stock, $0.001 par value; 3,000,000 shares authorized; no shares issued and outstanding at December 31, 2014 and 2013
— — Common stock, $0.001 par value; 300,000,000 shares authorized; 157,429,871 and 130,677,949 shares issued and outstanding at December 31, 2014 and 2013, respectively
157 131 Preferred stock, $0.001 par value; 3,000,000 shares authorized; no
shares issued and outstanding at December 31, 2017 and 2016
—
—
Common stock, $0.001 par value; 300,000,000 shares authorized;
159,877,239 and 159,158,636 shares issued and outstanding
at December 31, 2017 and 2016, respectively
160
159
Additional paid-in capital
1,059,072 952,403 1,089,684
1,080,198
Accumulated deficit
(928,433 ) (892,763 ) (985,840
)
(957,924
)
Accumulated other comprehensive loss
(84 ) (14 ) (207
)
(53
)
Total stockholders' equity
130,712 59,757 103,797
122,380
$ 172,511 $ 67,344 $
110,313
$
130,249
See accompanying notes.
GERON CORPORATIONCONSOLIDATEDSTATEMENTS OF OPERATIONSYear Ended December 31, 2014 2013 2012 (In thousands, except share and per share data) Revenues:
License fees and royalties
$ 1,153 $ 1,283 $ 2,709 Operating expenses:
Research and development
20,707 23,155 51,368 Restructuring charges
— 1,462 2,702 General and administrative
16,758 15,624 20,397 Total operating expenses
37,465 40,241 74,467 Loss from operations
(36,312 ) (38,958 ) (71,758 ) Unrealized gain (loss) on derivatives
351 (316 ) 13 Interest and other income
373 951 3,097 Interest and other expense
(82 ) (56 ) (233 ) Net loss
$ (35,670 ) $ (38,379 ) $ (68,881 ) Basic and diluted net loss per share
$ (0.23 ) $ (0.30 ) $ (0.54 ) Shares used in computing basic and diluted net loss per share
153,540,341 128,380,800 126,941,024 Year Ended December 31,
2017
2016
2015
(In thousands, except share and per share data)
Revenues:
Collaboration revenue
$
—
$
—
$
35,000
License fees and royalties
1,065
6,162
1,371
Total revenues
1,065
6,162
36,371
Operating expenses:
Research and development
11,033
18,047
17,831
Restructuring charges
—
—
1,306
General and administrative
19,287
18,761
17,793
Total operating expenses
30,320
36,808
36,930
Loss from operations
(29,255
)
(30,646
)
(559
)
Unrealized gain on derivatives
—
—
16
Interest and other income
1,416
1,192
677
Interest and other expense
(77
)
(83
)
(88
)
Net (loss) income
$
(27,916
)
$
(29,537
)
$
46
Net (loss) income per share:
Basic
$
(0.18
)
$
(0.19
)
$
0.00
Diluted
$
(0.18
)
$
(0.19
)
$
0.00
Shares used in computing net (loss) income per share:
Basic
159,224,986
159,045,644
158,036,162
Diluted
159,224,986
159,045,644
162,663,894
See accompanying notes.
GERON CORPORATIONCONSOLIDATEDSTATEMENTS OF COMPREHENSIVE LOSSYear Ended December 31, 2014 2013 2012 (In thousands) Net loss
$ (35,670 ) $ (38,379 ) $ (68,881 ) Other comprehensive income (loss):
Net unrealized loss on marketable securities
(70 ) (54 ) (38 ) Foreign currency translation adjustments
— — 16 Reclassification of accumulated foreign currency translation adjustments
— — 153 Other comprehensive (loss) income
(70 ) (54 ) 131 Comprehensive loss
$ (35,740 ) $ (38,433 ) $ (68,750 ) Year Ended December 31,
2017
2016
2015
(In thousands)
Net (loss) income
$
(27,916
)
$
(29,537
)
$
46
Net unrealized (loss) gain on marketable securities
(154
)
160
(129
)
Comprehensive loss
$
(28,070
)
$
(29,377
)
$
(83
)
See accompanying notes.
GERON CORPORATIONCONSOLIDATEDSTATEMENTS OFSTOCKHOLDERS'STOCKHOLDERS’ EQUITYCommon Stock Accumulated
Other
Comprehensive
Income (Loss)Additional
Paid-In
CapitalAccumulated
DeficitTotal
Stockholders'
EquityShares Amount (In thousands, except share data) Balances at December 31, 2011
131,443,148 $ 131 $ 932,066 $ (785,503 ) $ (91 ) $ 146,603 Net loss
— — — (68,881 ) — (68,881 ) Other comprehensive income
— — — — 131 131 Stock-based compensation related to issuance of common stock and options in exchange for services
170,298 — 135 — — 135 Cancellations of non-vested restricted stock under equity plans, net of issuances of common stock
(2,592,375 ) (2 ) 269 — — 267 Stock-based compensation for equity-based awards to employees and directors
— — 5,311 — — 5,311 401(k) contribution
1,221,624 1 2,086 — — 2,087 Balances at December 31, 2012
130,242,695 130 939,867 (854,384 ) 40 85,653 Net loss
— — — (38,379 ) — (38,379 ) Other comprehensive loss
— — — — (54 ) (54 ) Stock-based compensation related to issuance of common stock and options in exchange for services
66,853 — 252 — — 252 Cancellations of non-vested restricted stock under equity plans, net of issuances of common stock
(388,056 ) — 6,553 — — 6,553 Stock-based compensation for equity-based awards to employees and directors
— — 4,435 — — 4,435 401(k) contribution
756,457 1 1,296 — — 1,297 Balances at December 31, 2013
130,677,949 131 952,403 (892,763 ) (14 ) 59,757 Net loss
— — — (35,670 ) — (35,670 ) Other comprehensive loss
— — — — (70 ) (70 ) Issuance of common stock in connection with public offering, net of issuance costs of $6,695
25,875,000 26 96,779 — — 96,805 Stock-based compensation related to issuance of common stock and options in exchange for services
71,239 — 253 — — 253 Issuance of common stock upon net exercise of warrants
168,039 — — — — — Issuances of common stock under equity plans, net of cancellations of non-vested restricted stock
564,950 — 1,555 — — 1,555 Stock-based compensation for equity-based awards to employees and directors
— — 7,658 — — 7,658 401(k) contribution
72,694 — 424 — — 424 Balances at December 31, 2014
157,429,871 $ 157 $ 1,059,072 $ (928,433 ) $ (84 ) $ 130,712 Accumulated
Additional
Other
Total
Common Stock
Paid-In
Accumulated
Comprehensive
Stockholders'
Shares
Amount
Capital
Deficit
Loss
Equity
(In thousands, except share data)
Balances at December 31, 2014
157,429,871
$
157
$
1,059,072
$
(928,433
)
$
(84
)
$
130,712
Net income
—
—
—
46
—
46
Other comprehensive loss
—
—
—
—
(129
)
(129
)
Stock-based compensation related to
issuance of common stock and
options in exchange for services
18,077
—
364
—
—
364
Issuance of common stock upon exercise
of warrants
235,000
1
880
—
—
881
Issuances of common stock under equity
plans, net of cancellations of
non-vested restricted stock
1,098,411
1
1,693
—
—
1,694
Stock-based compensation for equity-
based awards to employees and
directors
—
—
8,397
—
—
8,397
401(k) contribution
—
—
161
—
—
161
Balances at December 31, 2015
158,781,359
159
1,070,567
(928,387
)
(213
)
142,126
Net loss
—
—
—
(29,537
)
—
(29,537
)
Other comprehensive income
—
—
—
—
160
160
Stock-based compensation related to
issuance of common stock and
options in exchange for services
21,541
—
156
—
—
156
Issuances of common stock under equity
plans
355,736
—
1,169
—
—
1,169
Stock-based compensation for equity-
based awards to employees and
directors
—
—
8,245
—
—
8,245
401(k) contribution
—
—
61
—
—
61
Balances at December 31, 2016
159,158,636
159
1,080,198
(957,924
)
(53
)
122,380
Net loss
—
—
—
(27,916
)
—
(27,916
)
Other comprehensive loss
—
—
—
—
(154
)
(154
)
Issuance of common stock in connection
with at market offering, net of
issuance costs of $114
614,230
1
1,059
—
—
1,060
Stock-based compensation related to
issuance of common stock and
options in exchange for services
72,066
—
200
—
—
200
Issuances of common stock under
equity plans
32,307
—
51
—
—
51
Stock-based compensation for equity-
based awards to employees and
directors
—
—
8,144
—
—
8,144
401(k) contribution
—
—
32
—
—
32
Balances at December 31, 2017
159,877,239
$
160
$
1,089,684
$
(985,840
)
$
(207
)
$
103,797
See accompanying notes.
GERON CORPORATIONCONSOLIDATEDSTATEMENTS OF CASH FLOWSYear Ended December 31, Year Ended December 31,
2014 2013 2012 2017
2016
2015
(In thousands) (In thousands)
Cash flows from operating activities:
Net loss
$ (35,670 ) $ (38,379 ) $ (68,881 ) Adjustments to reconcile net loss to net cash provided by (used in) operating activities:
Net (loss) income
$
(27,916
)
$
(29,537
)
$
46
Adjustments to reconcile net (loss) income to net cash used in
operating activities:
Depreciation and amortization
47 320 830 76
81
56
Loss (gain) on retirement/sales of property and equipment
5
(16
)
—
Accretion and amortization on investments, net
2,889 1,322 2,184 273
552
2,098
Loss (gain) on retirement/sales of property and equipment, net
3 (831 ) (142 ) Loss on write-downs of property and equipment
— 200 271 Loss on sales of marketable securities
—
—
1
Stock-based compensation for services by non-employees
253 252 183 200
156
364
Stock-based compensation for employees and directors
7,658 4,435 5,311 8,144
8,245
8,397
Amortization related to 401(k) contributions
111 458 726 32
61
161
Unrealized (gain) loss on derivatives
(351 ) 316 (13 ) Unrealized gain on derivatives
—
—
(16
)
Changes in assets and liabilities:
Interest and other receivables
(399 ) 188 646 39
731
(243
)
Prepaid assets
(72 ) 1,081 1,311 (56
)
123
89
Deposits and other assets
5 (4 ) 112 Accounts payable
(364 ) (2,032 ) 449 278
65
(873
)
Accrued compensation and benefits
580 (431 ) 3,548 542
(183
)
(1,187
)
Accrued restructuring charges
(94 ) (1,878 ) (1,758 ) Accrued collaboration charges
(1,665
)
1,039
2,328
Accrued liabilities
(246 ) (1,697 ) (92 ) (508
)
314
(417
)
Deferred revenue
35,000 — — —
—
(35,000
)
Translation adjustment
— — 169 Net cash provided by (used in) operating activities
9,350 (36,680 ) (55,146 ) Net cash used in operating activities
(20,556
)
(18,369
)
(24,196
)
Cash flows from investing activities:
Restricted cash transfer
529 (1 ) (1 ) —
(1
)
(1
)
Purchases of property and equipment
(131 ) (3 ) (862 ) —
(57
)
(90
)
Proceeds from sales of property and equipment
— 1,196 170 —
16
—
Purchases of marketable securities
(190,263 ) (88,977 ) (79,369 ) (100,006
)
(129,250
)
(206,459
)
Proceeds from sales/calls of marketable securities
10,549 — — —
—
4,242
Proceeds from maturities of marketable securities
101,412 108,839 141,016 122,976
138,054
202,381
Net cash (used in) provided by investing activities
(77,904 ) 21,054 60,954 Net cash provided by investing activities
22,970
8,762
73
Cash flows from financing activities:
Proceeds from issuance of common stock, net of issuance costs
98,360 6,553 150 Proceeds from issuances of common stock
1,111
1,169
2,575
Net cash provided by financing activities
98,360 6,553 150 1,111
1,169
2,575
Net increase (decrease) in cash and cash equivalents
29,806 (9,073 ) 5,958 3,525
(8,438
)
(21,548
)
Cash and cash equivalents, at beginning of year
12,990 22,063 16,105 Cash and cash equivalents, at end of year
$ 42,796 $ 12,990 $ 22,063 Cash and cash equivalents at the beginning of the period
12,810
21,248
42,796
Cash and cash equivalents at the end of the period
$
16,335
$
12,810
$
21,248
See accompanying notes.
84
GERON CORPORATION
NOTES TOCONSOLIDATEDFINANCIAL STATEMENTS1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Organization
Geron Corporation, or we or Geron, was incorporated in the State of Delaware on November 28, 1990. We are a
clinical stagebiopharmaceutical companyfocused onthat currently supports the clinical stage development of a telomerase inhibitor, imetelstat, in hematologic myeloidmalignancies.malignancies, by Janssen Biotech, Inc., or Janssen. Principal activities to date have included obtaining financing, securing operating facilities and conducting research and development. In November 2014, we entered into an exclusive collaboration and license agreement, or the Collaboration Agreement, with JanssenBiotech, Inc., or Janssen,to develop and commercialize imetelstat worldwide for all indications in oncology, including hematologic myeloid malignancies, and all other human therapeutic uses.The significance of future losses will depend on whetherUnder the Collaboration Agreement, Janssencontinues to developis wholly responsible for the worldwide development, manufacturing, seeking regulatory approval for andadvance imetelstat and the clinical and commercial successcommercialization of, imetelstat, whichwould result in future revenues to us in the form of milestone payments and royalties under the Collaboration Agreement, as described below, and whether we acquire other oncology products, programs or companies to diversify our business. There can be no assurance that we will receive any milestone payments or royalties from Janssen in the future. Imetelstat, which iswas our sole productcandidate, will require significant additional clinical testing prior to possible regulatory approval in the United States and other countries, and we do not expect imetelstat to be commercially available for many years, if at all.candidate.Principles of ConsolidationThe consolidated financial statements include the accounts of Geron and our former wholly-owned subsidiary, Geron Bio-Med Ltd. (Geron Bio-Med), a United Kingdom company. In March 2012, the board of directors and shareholders of Geron Bio-Med approved actions to commence a voluntary winding up of the company. The full wind up of Geron Bio-Med was completed in August 2012. Prior to 2013, we eliminated intercompany accounts and transactions and prepared the financial statements of Geron Bio-Med using the local currency as the functional currency. We translated the assets and liabilities of Geron Bio-Med at rates of exchange at the balance sheet date and translated income and expense items at average monthly rates of exchange. The resultant translation adjustments were included in accumulated other comprehensive income (loss), a separate component of stockholders' equity.Net
LossIncome (Loss) Per ShareBasic
earningsnet income (loss) per share is calculatedbased onby dividing net income (loss) by theweightedweighted‑average number of shares of common stock outstanding during theperiod.periods presented, without consideration for potential common shares. Dilutedearnings (loss)net income per share is calculatedbased onby adjusting theweightedweighted‑average number of shares of common stockandoutstanding for the dilutive effect of potentialdilutive securitiescommon shares outstandingduringfor theperiod.periods presented, as determined using the treasury‑stock method. Potential dilutive securities primarily consist of outstanding stock options, restricted stock awards and warrants to purchase our commonstock and arestock. For periods in which we have incurred a net loss, potential common shares outstanding for the periods presented, as determined using thetreasurytreasury‑stock method,at an average market price duringare excluded, as their effect would be anti‑dilutive, resulting in theperiod.same number of shares being used for the calculation of basic and diluted net loss per share. For all periods presented in the accompanying statements of operations, the net income (loss) applicable to common stockholders is equal to the reported net income (loss).Year Ended December 31,
(In thousands, except share and per share data)
2017
2016
2015
Net (loss) income
$
(27,916
)
$
(29,537
)
$
46
Weighted-average shares:
Basic
159,224,986
159,045,644
158,036,162
Effect of dilutive securities:
Stock options and restricted stock awards
—
—
4,627,732
Diluted
159,224,986
159,045,644
162,663,894
Net (loss) income per share:
Basic
$
(0.18
)
$
(0.19
)
$
0.00
Diluted
$
(0.18
)
$
(0.19
)
$
0.00
Because we
arewere in a net loss positiondiluted loss per share excludes the effects offor 2017 and 2016, 2,389,310 and 3,023,520 potentialdilutive securities. Had we been in a net income position, diluted earnings per share would have included thecommon shares,used in the computation of basic net loss per share as well as an additional 3,072,340, 532,120 and 11,497 shares for 2014, 2013 and 2012,respectively, related to outstanding stock options and restricted stock awardsand warrants(as determined using thetreasurytreasury‑stock method at the estimated average market value).Tablewere excluded from the diluted net loss per share calculation as their effect would have been anti‑dilutive. In addition for 2017, 2016 and 2015, 14,475,616, 11,352,766 and 9,375,851 potentially dilutive securities, respectively, were excluded from the treasury‑stock method and calculation ofContentsdiluted net income (loss) per share as their effect would have been anti‑dilutive.
85GERON CORPORATION
NOTES TO
CONSOLIDATEDFINANCIAL STATEMENTS (Continued)1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Use of Estimates
The accompanying financial statements have been prepared in accordance with U.S. generally accepted accounting principles, or GAAP. The preparation of financial statements in conformity with
accounting principles generally accepted in the United StatesGAAP requires us to make estimates and assumptions that affect the reported amountsreported inof assets and liabilities and disclosure of contingent assets and liabilities as of the date of the financial statements andaccompanying notes.the reported amounts of revenues and expenses during the reporting period. Ona regularan ongoing basis,management evaluates thesewe evaluate our estimates, including those related to accrued liabilities, revenue recognition, fair value of marketable securities, income taxes, andassumptions.stock-based compensation. We base our estimates on historical experience and on various other market specific and relevant assumptions that we believe to be reasonable under the circumstances. Actual results could differ from those estimates.Fair Value of Financial Instruments
Cash Equivalents and Marketable Securities
We consider all highly liquid investments with an original maturity of three months or less to be cash equivalents. We are subject to credit risk related to our cash equivalents and marketable securities. We place our cash and cash equivalents in money market funds, commercial paper, corporate notes and cash operating accounts. Our marketable securities include U.S.
government-sponsoredgovernment‑sponsored enterprise securities, commercial paper and corporatenotes with original maturities ranging from four to 19 months.notes.We classify our marketable securities as
available-for-sale.available‑for‑sale. We recordavailable-for-saleavailable‑for‑sale securities at fair value with unrealized gains and losses reported in accumulated other comprehensive income (loss) instockholders'stockholders’ equity. Realized gains and losses are included in interest and other income and are derived using the specific identification method for determining the cost of securities sold and have been insignificant to date. Dividend and interest income are recognized when earned and included in interest and other income in ourconsolidatedstatements of operations. We recognize a charge when the declines in the fair values below the amortized cost basis of ouravailable-for-saleavailable‑for‑sale securities are judged to beother-than-temporary.other‑than‑temporary. We consider various factors in determining whether to recognize another-than-temporaryother‑than‑temporary charge, including whether we intend to sell the security or whether it is more likely than not that we would be required to sell the security before recovery of the amortized cost basis. Declines in market valueassociated with credit lossesjudged asother-than-temporaryother‑than‑temporary result in a charge to interest and other income.Other-than-temporary charges not related to credit losses are included in accumulated other comprehensive income (loss) in stockholders' equity.We have not recorded anyother-than-temporaryother‑than‑temporary impairment chargesforon ouravailable-for-saleavailable‑for‑sale securities for the years ended December 31,2014, 20132017, 2016 and2012.2015. See Note 2 on Fair Value Measurements.Non-Marketable EquityCost Method InvestmentsNon-marketableWe use the cost method of accounting for non-marketable equityinvestments in companies in which we ownsecurities where our ownership represents less than 20% ofthe outstanding voting stocksuch entity, anddo not otherwise have the ability towe cannot exert significant influence overthe investeesits operations. These securities are carried at costasand adjusted forother-than-temporaryimpairments.We apply the equity method of accounting for investments in non-marketable nonpublic companies in which we own more than 20% of the outstanding voting stock or otherwise have the ability to exert significant influence over the investees. Under this method, we increase (decrease) the carrying value of our investment by a proportionate share of the investee's earnings (losses). If losses exceed the carrying value of the investment, losses are then applied against any advances to the investee, including any commitment to provide financial support, until those amounts are reduced to zero. Commitments to provide financial support include formal guarantees, implicit arrangements, reputational expectations, intercompany relationships or a consistent past history of providing financial support. The equity method is then suspended until the investee has earnings. Any proportionate share of investee earnings is first applied to the share of accumulated losses not recognized during the period the equity method was suspended.GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)We recognize previously suspended losses to the extent additional investment is determined to represent the funding of prior losses. See Note 7 on Divestiture of Stem Cell Assets.Fair Value of Derivatives
For non-employeeNon‑employee options classified as derivative liabilitiesthe fair value of these instruments is recorded on the consolidated balance sheet at inception and adjustedare marked to fair value at each financial reportingdate. The changedate with any resulting changes in fair valueof the non-employee options isbeing recorded in theconsolidatedstatements of operations as unrealized gain (loss) on derivatives.Fair value of non-employee options is estimated using the Black Scholes option-pricing model.Thenon-employeenon‑employee options continue to be reported as a derivative liability until such time as the instruments are exercised or expire,or are otherwise modified to remove the provisions which require this treatment,at which time these instruments are marked to fair value and reclassified from liabilities tostockholders'stockholders’ equity.For non-employeeAs of March 31, 2015, all non‑employee options classified aspermanent equity, the fair value of the non-employee options is recorded in stockholders' equity as of their respective vesting dates and no further adjustments are made. See Note 2 on Fair Value Measurements.derivative liabilities expired unexercised.86
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
Nonmonetary Transactions1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)We account for nonmonetary transactions based on the fair values of the assets (or services) involved. The cost of a nonmonetary asset acquired in exchange for another nonmonetary asset is the fair value of the asset surrendered to obtain it with a gain or loss recognized on the exchange. We use the fair value of the asset received to measure the cost if it is more clearly evident than the fair value of the asset surrendered. If the fair value of neither the assets received nor the assets relinquished is determinable within reasonable limits, we use the recorded amount (or carrying value) of the nonmonetary assets relinquished to account for the exchange. Similarly, we use carrying value for an exchange of controlled assets that do not meet the definition of a business for a non-controlling non-marketable equity interest in a company with no gain or loss recognized on the exchange. See Note 7 on Divestiture of Stem Cell Assets.Revenue Recognition
In general, weWe recognize revenue for each unit of accounting when all of the following criteria have been met: (a) persuasive evidence of an arrangement exists, (b) delivery has occurred or services have been rendered, (c) theseller'sseller’s price to the buyer is fixed or determinable, and (d) collectability is reasonably assured. Amounts received prior to satisfying these revenue recognition criteria are recorded as deferred revenue. Amounts expected to be recognized as revenue within the 12 months following the balance sheet date are classified as current deferred revenue. Amounts not expected to be recognized as revenue within the 12 months following the balance sheet date are classified as noncurrent deferred revenue.License and/or Collaboration Agreements
In addition to the Collaboration Agreement
with Janssen,(which is more fully described in Note 4 on License Agreements), we have entered into several license or collaboration agreements with various oncology, diagnostics, research tools and biologics production companies. Economic terms in these agreements may includenon-refundablenon‑refundable upfront license payments in cash or equity securities, option payments in cash or equity securities, cost reimbursements,cost-sharingGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)cost‑sharing arrangements, milestone payments, royalties on future sales of products, or any combination of these items. In applying the appropriate revenue recognition guidance related to these agreements, we first assess whether the arrangement contains multiple elements. In this evaluation, we consider:
(1)(i) the deliverables included in the arrangement and(2)(ii) whether the individual deliverables represent separate units of accounting or whether they must be accounted for as a combined unit of accounting. This evaluation involves subjective determinations and requires us to make judgments about the individual deliverables and whether such deliverables are separable from the other aspects of the contractual relationship. Deliverables are considered separate units of accounting provided that: (i) the delivered item(s) has value to the customer on a standalone basis, and (ii) if(ii)the arrangement includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control. In assessing whether an item has standalone value, we consider factors such as the research, manufacturing and commercialization capabilities of the collaboration partner or licensee and the availability of the associated expertise in the general marketplace. In addition, we consider whether the collaboration partner or licensee can use the other deliverable(s) for their intended purpose without the receipt of the remaining element(s), whether the value of the deliverable is dependent on the undelivered item(s) and whether there are other vendors that can provide the undelivered element(s).Arrangement consideration that is fixed or determinable is allocated among the separate units of accounting using the relative selling price method. We then apply the applicable revenue recognition criteria noted above to each of the separate units of accounting in determining the appropriate period and pattern of recognition. We determine how to allocate arrangement consideration to identified units of accounting based on the selling price hierarchy provided under relevant accounting guidance. The estimated fair value of deliverables under the arrangement may be derived using a best estimate of selling price if
vendor-specific-objectivevendor‑specific‑objective evidence andthird-partythird‑party evidence are not available.Upfront
non-refundablenon‑refundable signing, license ornon-exclusivenon‑exclusive option fees are recognized as revenue: (i) when rights to use the intellectual propertyrelated to ahave been delivered, if the licensethathas standalone value from the other deliverables to be provided under the agreement,have been deliveredor (ii) over the term of the agreement if we have continuing performance obligations, as the arrangement would be accounted for as a single unit of accounting. When payments are received in equity securities, we do not recognize any revenue unless such securities are determined to be realizable in cash.87
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
At the inception of an arrangement that includes milestone payments, we assess whether each milestone is substantive and at risk
to both partieson the basis of the contingent nature of the milestone. This evaluation includes an assessment of whether: (i) the consideration is commensurate with eitherthe(1) our performance to achieve the milestone or (2) the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting fromtheour performance to achieve the milestone, (ii) the consideration relates solely to past performance and (iii) the consideration is reasonable relative to all of the deliverables and payment terms (including other potential milestone consideration) within the arrangement. We consider various factors, such as the scientific, clinical, regulatory, commercial and other risks that must be overcome to achieve the respective milestone and the level of effort and investment required to achieve the respective milestone, in making this assessment. There is considerable judgment involved in determining whether a milestone satisfies all of the criteria required to conclude that a milestone is substantive. Milestone payments for milestones that are considered substantive would be recognized as revenue in their entirety upon successful accomplishment of the milestone, assuming all other revenue recognition criteria are met.GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)Milestone payments for milestones that are not considered substantive would be recognized as revenue over the remaining period of performance, assuming all other revenue recognition criteria are met.
Our license and collaboration agreements with certain partners also provide for contingent payments to us based solely upon the performance of the respective partner. For such contingent amounts, we recognize the payments as revenue when earned under the applicable contract, which is generally upon completion of performance by the respective partner, provided that collection is reasonably assured.
Royalties are recognized as earned in accordance with contract terms when royalties from licensees can be reasonably estimated and
collectabilitycollection is reasonably assured. If royalties cannot be reasonably estimated orcollectabilitycollection of a royalty amount is not reasonably assured, royalties are recognized as revenue when the cash is received. Revenue from commercial milestone paymentswill beis accounted for as royalties and recorded as revenue upon achievement of the milestone, assuming all other revenue recognition criteria aremet.met and we have no performance obligations related to the milestone.Cost-sharingCost‑sharing expenses are recorded as earned or owed based on the performance requirements by both parties under the respective contracts. For arrangements in which we and our collaboration partner in the agreement are exposed to significant risks and rewardsdependingthat depend on the commercial success of the activity, we recognize payments between the parties on a net basis and record such amounts as a reduction or addition to research and development expense. For arrangements in which we have agreed to perform certain research and development services for our collaboration partner and are not exposed to significant risks and rewardsdependingthat depend on the commercial success of the activity, we recognize the respective cost reimbursements as revenue under the collaborative agreement as the related research and development services are rendered.Restricted Cash
Restricted cash consists of funds maintained in a separate certificate of deposit
accountsaccount forspecified purposes. The components of restricted cash were as follows:December 31, (In thousands) 2014 2013 Certificate of deposit for unused equipment line of credit
$ — $ 530 Certificate of deposit for credit card purchases
266 265 $ 266 $ 795 In 2014, we closed the certificate of deposit for our unused equipment line ofcreditupon maturity and transferred the cash proceeds to our cash operating accounts. This action also cancelled the availability of the equipment line of credit.card purchases.Research and Development Expenses
Research and development expenses consist of expenses incurred in identifying, developing and testing product candidates resulting from our independent efforts as well as efforts associated with collaborations. These expenses include, but are not limited to,
in-processin‑process research and development acquired in an asset acquisition and deemed to have no alternative future use, payroll and personnel expense, lab supplies, preclinical studies, clinical trials, including support forinvestigator-sponsoredinvestigator‑sponsored clinical trials, raw materials to manufacture clinical trial drugs, manufacturing costs for research and clinical trial materials, sponsored research at other labs, consulting, costs to maintain technology licenses, our proportionate share of research andresearch-relateddevelopment costs under cost‑sharing arrangements with collaboration partners and research‑related overhead. Research and development costs are expensed as incurred, includingpayments madecosts incurred under our collaboration and/or license agreements.
GERON CORPORATIONNOTES TO
CONSOLIDATEDFINANCIAL STATEMENTS (Continued)1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Clinical Trial CostsA significant component of our research and development expenses has historically been clinical trial costs. Substantial portions of our preclinical studies and all of our clinical trials have been performed by third-party contract research organizations, or CROs, and other vendors. We accrue expenses for preclinical studies performed by our vendors based on certain estimates over the term of the service period and adjust our estimates as required. We accrue expenses for clinical trial activities performed by CROs based upon the estimated amount of work completed on each study.Forclinical trial expenses, the significant factors used in estimating accruals include the number of patients enrolled, the number of active clinical sites and the duration for which the patients will be enrolled in the study. Pass through costs from CROs include, but are not limited to, regulatory expenses, investigator fees, lab fees, travel costs and other miscellaneous costs, including shipping and printing fees. We accrue pass through costs based on estimates of the amount of work completed forthe clinicaltrial. Wedevelopment activities being conducted by Janssen under the Collaboration Agreement, we monitor patient enrollment levels and related activities to the extent possible throughinternal reviews, review of contractual termsdiscussions with Janssen personnel andcorrespondence with CROs. Webase our estimates on the best information available at the time. However, additional information may become available to us which would allow us to make a more accurate estimate in future periods. In this event, we may be required to record adjustments to research and development expenses in future periods when the actual level of activity becomes more certain.Depreciation and Amortization
We record property and equipment at cost and calculate depreciation using the
straight-linestraight‑line method over the estimated useful lives of the assets, generally four years. Leasehold improvements are amortized over the shorter of the estimated useful life or remaining term of the lease.Stock-BasedStock‑Based CompensationWe maintain various stock incentive plans under which stock options and restricted stock awards are granted to employees, directors and consultants. We also have an employee stock purchase plan for all eligible employees. We recognize
stock-basedstock‑based compensation expense based on the grant-date fair values of these instruments on astraight-linestraight‑line basis over the requisite service period, which is generally the vesting period.For additional information, see Note 9 on Stockholders' Equity.Stock Options and Employee Stock Purchase PlanWe grant service-based stock options under our equity plans to employees, directors and consultants.Thevesting period for employee options is generally four years. We use the Black Scholes option-pricing model to estimate thedetermination of grant-date fairvalue ofvalues for our stock options and employee stockplan purchases. The determination of fair value for these stock-based awards on the date of grantpurchases using the Black Scholesoption-pricingoption‑pricing model is affected by our stock price as well as assumptions regarding a number of complex and subjective variables.For additional information, see Note 9 on Stockholders' Equity.Restricted Stock AwardsWe have granted restricted stock awards to employees and directors with three types of vesting schedules: (i) service-based, (ii) performance-based or (iii) market-based. Service-based restricted stockGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)awards generally vest annually over four years. Performance-based restricted stock awards vest upon achievement of discrete strategic corporate goals within a specified performance period, generally three years. Market-based restricted stock awards vest only upon achievement of certain market price thresholds of our common stock within a specified performance period, generally three years.The grant-date fair value for service-based restricted stock awards is determined using the fair value of our common stock on the date of grant.
The fair value is amortized as stock-based compensation expense over the requisite service period of the award, which is generally the vesting period, on a straight-line basis and is reduced for estimated forfeitures, as applicable.The fair value for performance-based restricted stock awards is determined using the fair value of our common stock on the date of grant. Stock-based compensation expense for awards with vesting based on performance conditions is recognized over the period from the date the performance condition is determined to be probable of occurring through the date the applicable condition is expected to be met and is reduced for estimated forfeitures, as applicable. If the performance condition is not considered probable of being achieved, no stock-based compensation expense is recognized until such time as the performance condition is considered probable of being met, if at all. If that assessment of the probability of the performance condition being met changes, the impact of the change in estimate would be recognized in the period of the change. If the requisite service period has been met prior to the change in estimate, the effect of the change in estimate would be recognized immediately. All previously granted performance-based restricted stock awards have been cancelled unvested as the performance conditions were not achieved within the respective performance periods.The fair value for market-based restricted stock awards is determined using a lattice valuation model with a Monte Carlo simulation. The model takes into consideration the historical volatility of our stock and the risk-free interest rate at the date of grant. In addition, the model is used to estimate the derived service period for the awards. The derived service period is the estimated period of time that would be required to satisfy the market condition, assuming the market condition will be satisfied. Stock-based compensation expense is recognized over the derived service period for the awards using the straight-line method and is reduced for estimated forfeitures, as applicable, but is accelerated if the market condition is achieved earlier than estimated. If a market-based restricted stock award is forfeited or expires after completion of the derived service period, any previously recognized stock-based compensation expense is not reversed. All previously granted market-based restricted stock awards have been cancelled unvested as the market conditions were not achieved within the specified performance period.Non-Employee Stock-Based AwardsFor our
non-employee stock-basednon‑employee stock‑based awards, the measurement date on which the fair value of thestock-basedstock‑based award is calculated is equal to the earlier of: (i) the date at which a commitment for performance by the counterparty to earn the equity instrument is reached or (ii) the date at which thecounterparty'scounterparty’s performance is complete. We recognizestock-basedstock‑based compensation expense for the fair value of the vested portion ofnon-employeenon‑employee stock‑based awards in ourconsolidatedstatements of operations.Table of ContentsFor additional information, see Note 8 on Stockholders’ Equity.GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)Accumulated Other Comprehensive Loss
Accumulated other comprehensive loss includes certain changes in
stockholders'stockholders’ equity which are excluded from netloss. The components of accumulatedincome (loss). Accumulated other comprehensive losswereon our balance sheets asfollows:of December 31, 2017 and 2016 is solely comprised of net unrealized losses on marketable securities.December 31, (In thousands) 2014 2013 Unrealized loss on marketable securities
$ (84 ) $ (14 ) Income Taxes
We maintain deferred tax assets and liabilities that reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes and are subject to tests of recoverability. Our deferred tax assets include net operating loss carryforwards, research credits and capitalized research and development. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Our net deferred tax asset has been fully offset by a valuation allowance because of our history of losses. Any potential accrued interest and penalties related to unrecognized tax benefits
within operationswould be recorded as income tax expense.89
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Concentrations of Customers and Suppliers
The majority of our revenues was earned in the United States. Two customers accounted for approximately
31%, 42% and 59%39% of our2014, 20132017 revenues. Approximately 81% of our 2016 revenues represented an upfront payment from Janssen Pharmaceuticals, Inc., or Janssen Pharmaceuticals, in connection with a license agreement signed in September 2016, or the License Agreement. Approximately 96% of our 2015 revenues represented an upfront payment from Janssen under the imetelstat Collaboration Agreement.In accordance with the Collaboration Agreement, Janssen is now responsible for the manufacture and
2012 revenues, respectively.We contract third-partymanagement of the supply of imetelstat on a global basis for clinical trials and, after any regulatory approval, all commercial activities. Janssen contracts third‑party manufacturers to produceGMP-gradeGMP‑grade drugs for preclinical and clinical studies.WeJanssen alsocontractcontracts for starting materials to supply those manufacturers andus.for its own use. Certain development and clinical activities may be delayed ifwe orJanssenareis unable to obtain sufficient quantities of starting materials orGMP-gradeGMP‑grade drugs from currentthird-partythird‑party suppliers or otherthird-partythird‑party sources.Segment Information
Our executive management team represents our chief decision maker. We view our operations as a single segment, the development of therapeutic products for oncology. As a result, the financial information disclosed herein materially represents all of the financial information related to our principal operating segment.
Recent Accounting Pronouncements Not Yet Effective
In
AprilMay 2014, the Financial Accounting Standards Board, or FASB, issued AccountingStandardStandards Update No.2014-08, Presentation of Financial Statements and Property, Plant, and Equipment: Reporting Discontinued Operations and Disclosures of Disposals of Components of an Entity,2014-09, or ASU2014-08. ASU 2014-08 raised2014-09, which created Accounting Standards Codification Topic 606, Revenue from Contracts with Customers, or Topic 606, and superseded thethresholdrevenue recognition requirements in Accounting Standards Codification Topic 605, Revenue Recognition, including most industry-specific revenue recognition guidance throughout the Industry Topics of the Codification. In summary, the core principle of Topic 606 is to recognize revenue when promised goods or services are transferred to customers in an amount that reflects the consideration that is expected to be received for those goods or services. Companies are allowed to select between two transition methods: (1) adisposalfull retrospective transition method with the application ofassets to qualify as a discontinued operation and requires new disclosures for both discontinued operations and disposals of individually significant components of a business that do not qualify as discontinued operations. Underthe new guidanceonly disposals of assets representingto each prior reporting period presented, or (2) astrategic shift in operationsmodified retrospective transition method thathas a majorrecognizes the cumulative effect on prior periods at theentity's operationsdate of adoption together with additional footnote disclosures. The amendments in ASU 2014-09 are effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period. In March, April, May andfinancial results should be presented as discontinued operations. IfDecember 2016, thedisposal does qualify as a discontinued operation, the entity will be requiredFASB issued Accounting Standards Update No. 2016-08 (Topic 606), Revenue From Contracts With Customers: Principal vs. Agent Considerations, or ASU 2016-08, Accounting Standards Update No. 2016-10 (Topic 606), Revenue From Contracts with Customers: Identifying Performance Obligations and Licensing, or ASU 2016-10, Accounting Standards Update No. 2016-12 (Topic 606), Revenue From Contracts with Customers: Narrow-Scope Improvements and Practical Expedients, or ASU 2016-12, and Accounting Standards Update No. 2016-20 (Topic 606), Revenue from Contracts with Customers: Technical Corrections and Improvements to Topic 606, or ASU 2016-20, respectively, to provideexpanded disclosures,supplemental adoption guidance and clarification to ASU 2014-09. We will adopt ASU 2014-09 and its related supplemental guidance on January 1, 2018 using the modified retrospective transition method.The new revenue standard is principles-based and the interpretation of those principles may vary from company to company based on their unique circumstances. It is possible that interpretations, industry practice, and guidance may evolve as
well as disclosure ofcompanies and thepretax income attributableaccounting profession work to implement this new standard. We have assessed the differences in accounting for our existing contracts under the new guidance compared to current revenue accounting standards. We have not identified any material differences in the accounting treatment under ASU 2014-09 compared to thedisposal of acurrent accounting treatment for the Collaboration Agreement with Janssen and the License Agreement with Janssen Pharmaceuticals, which are our most significantpart of an entity that does not qualify as a discontinued operation. ASU 2014-08 is effective for us beginning January 1, 2015 and subsequent interim periods. We do not expectlicense agreements. With the adoption of ASU2014-082014-09, we expect royalty revenues from product sales by licensees of our human telomerase reverse transcriptase, or hTERT, technology and cell-based research products tohavebe recognized earlier than under our current accounting policy for revenue recognition. Accordingly, we expect to record amaterial effect on our consolidated financial statements.Table of Contentsone-time cumulative catch-up adjustment in 2018 to reflect
90GERON CORPORATION
NOTES TO
CONSOLIDATEDFINANCIAL STATEMENTS (Continued)1. ORGANIZATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
In May 2014, the FASB issued Accounting Standard Update No. 2014-09, Revenue from Contracts with Customers, or ASU 2014-09. ASU 2014-09 provides a single comprehensive modelestimated royalty revenues on product sales earned in 2017 forentities to use in accounting for revenue arising from contracts with customers and supersedes most current revenue recognition guidance, including industry-specific guidance. ASU 2014-09 will require an entity to recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration towhichthe entity expects to be entitled in exchange for those goods or services. This update creates a five-step model that requires entities to exercise judgment when considering the terms of the contract(s). The five-step model includes: (i) identifying the contract(s) with the customer, (ii) identifying the separate performance obligations in the contract, (iii) determining the transaction price, (iv) allocating the transaction price to the separate performance obligations, and (v) recognizing revenue when each performance obligation is satisfied. ASU 2014-09 also requires additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. ASU 2014-09 will be effective for us beginning January 1, 2017 and subsequent interim periods.payments have not yet been received. Wehave the option to apply the provisions of ASU 2014-09 either retrospectively to each prior reporting period presented or retrospectively with the cumulative effect of applying this accounting standard recognized at the date of initial application. Early adoption isdo notpermitted. We are currently evaluating the transition method and the impactexpect that the adoption of ASU 2014-09 will have a material impact on ourconsolidatedfinancial statements and related disclosures.In
August 2014,January 2016, the FASB issued AccountingStandardStandards Update No.2014-15, Presentation2016-01, Financial Instruments - Overall: Recognition and Measurement of FinancialStatements—Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern,Assets and Financial Liabilities, or ASU2014-15. ASU 2014-15 is intended to define management's responsibility to evaluate whether there is substantial doubt about an organization's ability to continue as a going concern and to provide related footnote disclosures. Substantial doubt about an entity's ability to continue as a going concern exists when relevant conditions and events, considered in the aggregate, indicate that it is probable that the entity will be unable to meet its obligations as they become due within one year after the date that the financial statements are issued (or available2016-01, which requires equity investments to beissued).measured at fair value with changes in fair value recognized in net income. However, equity investments without readily determinable fair values can either be measured at fair value or use a measurement alternative which adjusts cost for changes in observable prices minus impairment. ASU2014-15 provides guidance to an organization's management, with principles2016-01 requires separate presentation of financial assets anddefinitions that are intended to reduce diversity in the timingliabilities by category andcontent of disclosures that are commonly provided by organizations in financial statement footnotes.form. ASU2014-15 will be2016-01 is effective forusannual periods beginning after December31, 201615, 2017, andsubsequentinterim periods within those annual periods. To further clarify ASU 2016-01, the FASB issued Accounting Standards Update No. 2018-03, Technical Corrections and Improvements to Financial Instruments - Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities, or ASU 2018-03, in February 2018. ASU 2018-03 requires application of a prospective transition approach only for those equity investments for which the new measurement alternative is being applied. Additionally, if an entity voluntarily discontinues using the measurement alternative, the investment and all identical or similar investments of the same issuer must be measured at fair value. ASU 2018-03 is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years beginning after June 15, 2018. Early adoption is permitted. We currently hold equity investments without readily determinable fair values that arecurrently inaccounted for under theprocess ofcost method and are evaluating theimpactmethod and timing of adoption of ASU2014-152016-01 and ASU 2018-03. The adoption of ASU 2016-01 and ASU 2018-03 may have a material impact on ourconsolidatedfinancial statements and related disclosures.In February 2016, the FASB issued Accounting Standards Update No. 2016-02, Leases (Topic 842)
Table, or ASU 2016-02. ASU 2016-02 requires an entity to recognize a right-of-use asset and lease liability for all leases with terms ofContentsmore than 12 months. Recognition, measurement and presentation of expenses will depend on classification as a finance or operating lease. Certain quantitative and qualitative disclosures about leasing arrangements also are required. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. Early adoption is permitted. The updated guidance requires a modified retrospective adoption. We are currently evaluating the impact that the adoption of ASU 2016-02 will have on our financial statements and related disclosures and have not made any decision regarding the timing of adoption.In August 2016, the FASB issued Accounting Standards Update No. 2016-15,
Classification of Certain Cash Receipts and Cash Payments, or ASU 2016-15, to clarify how certain cash receipts and cash payments are presented and classified in the statement of cash flows. ASU 2016-15 is effective for annual periods beginning after December 15, 2017, and interim periods within those annual periods. ASU 2016-15 must be applied retrospectively to each period presented. We will adopt ASU 2016-15 on January 1, 2018. We do not expect that the adoption of ASU 2016-15 will have a material impact on our financial statements and related disclosures.In November 2016, the FASB issued Accounting Standards Update No. 2016-18, Statement of Cash Flows (Topic 230) Restricted Cash, or ASU 2016-18, to address the diversity in practice in the classification and presentation of changes in restricted cash on the statement of cash flows. ASU 2016-18 is effective for annual periods beginning after December 15, 2017, and interim periods within those annual periods. ASU 2016-18 must be applied using a retrospective transition method to each period presented. We will adopt ASU 2016-18 on January 1, 2018. We do not expect that the adoption of ASU 2016-18 will have a material impact on our financial statements and related disclosures.
In May 2017, the FASB issued Accounting Standards Update No. 2017-09, Compensation — Stock Compensation: Scope of Modification Accounting, or ASU 2017-09. ASU 2017-09 clarifies when to account for a change to the terms or conditions of a share-based payment award as a modification. Under the new guidance, modification accounting is required only if the fair value, the vesting conditions, or the classification of the award (as equity or liability) changes as a result of the change in terms or conditions. ASU 2017-09 is effective for annual periods beginning after December 15, 2017, and interim periods within those annual periods. The amendments in ASU 2017-09 should be applied prospectively to an award modified on or after the adoption date. We will adopt ASU 2017-09 on January 1, 2018. We do not expect that the adoption of ASU 2017-09 will have a material impact on our financial statements and related disclosures.
91
GERON CORPORATION
NOTES TO
CONSOLIDATEDFINANCIAL STATEMENTS (Continued)We categorize financial instruments recorded at fair value on our consolidated balance sheets based upon the level of judgment associated with inputs used to measure their fair value. The categories are as follows:Level 1—Inputs are unadjusted, quoted prices in active markets for identical assets or liabilities at the measurement date. An active market for the asset or liability is a market in which transactions for the asset or liability occur with sufficient frequency and volume to provide pricing information on an ongoing basis.Level 2—Inputs (other than quoted market prices included in Level 1) are either directly or indirectly observable for the asset or liability through correlation with market data at the measurement date and for the duration of the instrument's anticipated life.Level 3—Inputs reflect management's best estimate of what market participants would use in pricing the asset or liability at the measurement date. Consideration is given to the risk inherent in the valuation technique and the risk inherent in the inputs to the model.A financial instrument's categorization within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Below is a description of the valuation methodologies used for financial instruments measured at fair value on our consolidated balance sheets, including the category for such financial instruments.Cash Equivalents and Marketable Securities
Available-for-SaleCertificates of deposit and money market funds are categorized as Level 1 within the fair value hierarchy as their fair values are based on quoted prices available in active markets. U.S. government-sponsored enterprise securities, commercial paper and corporate notes are categorized as Level 2 within the fair value hierarchy as their fair values are estimated by using pricing models, quoted prices of securities with similar characteristics or discounted cash flows.GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)2. FAIR VALUE MEASUREMENTS (Continued)Cash equivalents, restricted cash and marketable securities by security type at December 31,
20142017 were as follows:(In thousands) Amortized Cost Gross Unrealized Gains Gross Unrealized Losses Estimated Fair Value Included in cash and cash equivalents:
Money market funds
$ 40,342 $ — $ — $ 40,342 Restricted cash:
Certificate of deposit
$ 266 $ — $ — $ 266 Marketable securities:
Government-sponsored enterprise securities (due in less than 1 year)
$ 401 $ — $ (1 ) $ 400 Government-sponsored enterprise securities (due in 1 to 2 years)
6,556 — (7 ) 6,549 Commercial paper (due in less than 1 year)
10,985 14 — 10,999 Corporate notes (due in less than 1 year)
97,307 2 (63 ) 97,246 Corporate notes (due in 1 to 2 years)
12,412 — (29 ) 12,383 $ 127,661 $ 16 $ (100 ) $ 127,577 Gross
Gross
Amortized
Unrealized
Unrealized
Estimated
(In thousands)
Cost
Gains
Losses
Fair Value
Included in cash and cash equivalents:
Money market funds
$
11,030
$
—
$
—
$
11,030
Commercial paper
2,242
—
—
2,242
Corporate notes
1,750
—
(1
)
1,749
$
15,022
$
—
$
(1
)
$
15,021
Restricted cash:
Certificate of deposit
$
268
$
—
$
—
$
268
Marketable securities:
Government-sponsored enterprise securities
(due in less than one year)
$
12,500
$
—
$
(40
)
$
12,460
Commercial paper (due in less than one year)
10,928
4
(1
)
10,931
Corporate notes (due in less than one year)
55,067
—
(107
)
54,960
Corporate notes (due in one to two years)
14,303
—
(62
)
14,241
$
92,798
$
4
$
(210
)
$
92,592
Cash equivalents, restricted cash and marketable securities by security type at December 31,
20132016 were as follows:Gross
Gross
Amortized
Unrealized
Unrealized
Estimated
(In thousands)
Cost
Gains
Losses
Fair Value
Included in cash and cash equivalents:
Money market funds
$
11,193
$
—
$
—
$
11,193
Restricted cash:
Certificate of deposit
$
268
$
—
$
—
$
268
Marketable securities:
Government-sponsored enterprise securities
(due in less than one year)
$
5,000
$
—
$
(3
)
$
4,997
Government-sponsored enterprise securities
(due in one to two years)
12,500
—
(42
)
12,458
Commercial paper (due in less than one year)
31,024
50
(5
)
31,069
Corporate notes (due in less than one year)
66,012
4
(47
)
65,969
Corporate notes (due in one to two years)
1,506
—
(10
)
1,496
$
116,042
$
54
$
(107
)
$
115,989
(In thousands) Amortized Cost Gross Unrealized Gains Gross Unrealized Losses Estimated Fair Value Included in cash and cash equivalents:
Money market funds
$ 8,079 $ — $ — $ 8,079 Corporate notes
2,206 — — 2,206 $ 10,285 $ — $ — $ 10,285 Restricted cash:
Certificates of deposit
$ 795 $ — $ — $ 795 Marketable securities:
Government-sponsored enterprise securities (due in less than 1 year)
$ 7,369 $ 1 $ (1 ) $ 7,369 Commercial paper (due in less than 1 year)
5,496 3 — 5,499 Corporate notes (due in less than 1 year)
39,383 1 (18 ) 39,366 $ 52,248 $ 5 $ (19 ) $ 52,234
92GERON CORPORATION
NOTES TO
CONSOLIDATEDFINANCIAL STATEMENTS (Continued)2. FAIR VALUE MEASUREMENTS (Continued)
MarketableCash equivalents and marketable securities with unrealized losses at December 31,20142017 and20132016 were as follows:Less Than 12 Months 12 Months or Greater Total (In thousands) Estimated Fair Value Gross Unrealized Losses Estimated Fair Value Gross Unrealized Losses Estimated Fair Value Gross Unrealized Losses As of December 31, 2014:
Government-sponsored enterprise securities (due in less than 1 year)
$ 400 $ (1 ) $ — $ — $ 400 $ (1 ) Government-sponsored enterprise securities (due in 1 to 2 years)
5,549 (7 ) — — 5,549 (7 ) Corporate notes (due in less than 1 year)
92,989 (63 ) — — 92,989 (63 ) Corporate notes (due in 1 to 2 years)
12,383 (29 ) — — 12,383 (29 ) $ 111,321 $ (100 ) $ — $ — $ 111,321 $ (100 ) As of December 31, 2013:
Government-sponsored enterprise securities (due in less than 1 year)
$ 3,947 $ (1 ) $ — $ — $ 3,947 $ (1 ) Corporate notes (due in less than 1 year)
37,060 (18 ) — — 37,060 (18 ) $ 41,007 $ (19 ) $ — $ — $ 41,007 $ (19 ) Less Than 12 Months
12 Months or Greater
Total
Gross
Gross
Gross
Estimated
Unrealized
Estimated
Unrealized
Estimated
Unrealized
(In thousands)
Fair Value
Losses
Fair Value
Losses
Fair Value
Losses
As of December 31, 2017:
Government-sponsored enterprise
securities (due in less than one year)
$
—
$
—
$
12,460
$
(40
)
$
12,460
$
(40
)
Commercial paper (due in less than one
year)
7,717
(1
)
—
—
7,717
(1
)
Corporate notes (due in less than one year)
55,210
(106
)
1,499
(2
)
56,709
(108
)
Corporate notes (due in one to two years)
14,241
(62
)
—
—
14,241
(62
)
$
77,168
$
(169
)
$
13,959
$
(42
)
$
91,127
$
(211
)
As of December 31, 2016:
Government-sponsored enterprise
securities (due in less than one year)
$
4,997
$
(3
)
$
—
$
—
$
4,997
$
(3
)
Government-sponsored enterprise
securities (due in one to two years)
12,458
(42
)
—
—
12,458
(42
)
Commercial paper (due in less than one
year)
8,365
(5
)
—
—
8,365
(5
)
Corporate notes (due in less than one year)
39,218
(37
)
6,944
(10
)
46,162
(47
)
Corporate notes (due in one to two years)
1,496
(10
)
—
—
1,496
(10
)
$
66,534
$
(97
)
$
6,944
$
(10
)
$
73,478
$
(107
)
The gross unrealized losses related to
government-sponsoredgovernment‑sponsored enterprise securities, commercial paper and corporate notes as of December 31,20142017 and20132016 were due to changes in interest rates. We determined that the gross unrealized losses on our cash equivalents and marketable securities as of December 31,20142017 and20132016 were temporary in nature. We review our investments quarterly to identify and evaluate whether any investments have indications of possible impairment. Factors considered in determining whether a loss is temporary include the length of time and extent to which the fair value has been less than the cost basis and whether we intend to sell the security or whether it is more likely than not that we would be required to sell the security before recovery of the amortized cost basis. We currently do not intend to sell these securities before recovery of their amortized costbasis.bases.DerivativesNon-employee options are normally traded less actively, have trade activity that is one way, and/or are traded in less-developed markets and are therefore valued based upon models with significant unobservable market parameters, resulting in Level 3 categorization.Options held by non-employees whose performance obligations are complete are classified as derivative liabilities on our consolidated balance sheets. These options are marked to fair value at each reporting period, and upon the exercise of these options, the instruments are marked to fair value and reclassified from derivative liabilities to stockholders' equity. We have not recorded any reclassifications from current liabilities to stockholders' equity for non-employee option exercises in 2014 and 2013.GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)2. FAIR VALUE MEASUREMENTS (Continued)As of December 31, 2014 and 2013, the following non-employee options to purchase common stock were considered derivatives and classified as current liabilities:Number of Shares at December 31, Fair Value at December 31, Exercise
PriceExercisable
DateExpiration
DateIssuance Date2014 2013 2014 2013 (In thousands) March 2005
$ 6.39 284,600 284,600 January 2007 March 2015 $ 16 $ 367 The fair value of derivatives has been calculated at each reporting date using the Black Scholes option-pricing model with the following assumptions:December 31, 2014 2013 Dividend yield
0% 0% Expected volatility
0.895 0.844 Risk-free interest rate
0.04% 0.13% Expected term
0.25 yr 1 yr Dividend yield is based on historical cash dividend payments. The expected volatility is based on historical volatilities of our stock since traded options on Geron common stock do not correspond to derivatives' terms and trading volume of Geron options is limited. The risk-free interest rate is based on the U.S. Zero Coupon Treasury Strip Yields for the expected term in effect on the reporting date. The expected term of derivatives is equal to the remaining contractual term of the instruments.Fair Value on a Recurring Basis
We categorize financial instruments recorded at fair value on our balance sheets based upon the level of judgment associated with inputs used to measure their fair value. The categories are as follows:
Level 1
—
Inputs are unadjusted, quoted prices in active markets for identical assets or liabilities at the measurement date. An active market for the asset or liability is a market in which transactions for the asset or liability occur with sufficient frequency and volume to provide pricing information on an ongoing basis.
Level 2
—
Inputs (other than quoted market prices included in Level 1) are either directly or indirectly observable for the asset or liability through correlation with market data at the measurement date and for the duration of the instrument’s anticipated life.
Level 3
—
Inputs reflect management’s best estimate of what market participants would use in pricing the asset or liability at the measurement date. Consideration is given to the risk inherent in the valuation technique and the risk inherent in the inputs to the model.
93
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
2. FAIR VALUE MEASUREMENTS (Continued)
A financial instrument’s categorization within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Below is a description of the valuation methodologies used for financial instruments measured at fair value on our balance sheets, including the category for such financial instruments.
Money market funds are categorized as Level 1 within the fair value hierarchy as their fair values are based on quoted prices available in active markets. U.S. government‑sponsored enterprise securities, commercial paper and corporate notes are categorized as Level 2 within the fair value hierarchy as their fair values are estimated by using pricing models, quoted prices of securities with similar characteristics or discounted cash flows.
The following table presents information about our financial instruments that are measured at fair value on a recurring basis as of December 31,
20142017 and 2016 and indicates the fair value category assigned.Fair Value Measurements at Reporting Date Using
Quoted Prices in
Significant
Active Markets for
Significant Other
Unobservable
Identical Assets
Observable Inputs
Inputs
(In thousands)
Level 1
Level 2
Level 3
Total
As of December 31, 2017:
Money market funds(1)
$
11,030
$
—
$
—
$
11,030
Government-sponsored enterprise securities(2)
—
12,460
—
12,460
Commercial paper(1)(2)
—
13,173
—
13,173
Corporate notes(1)(2)(3)
—
70,950
—
70,950
Total
$
11,030
$
96,583
$
—
$
107,613
As of December 31, 2016:
Money market funds(1)
$
11,193
$
—
$
—
$
11,193
Government-sponsored enterprise securities(2)(3)
—
17,455
—
17,455
Commercial paper(2)
—
31,069
—
31,069
Corporate notes(2)(3)
—
67,465
—
67,465
Total
$
11,193
$
115,989
$
—
$
127,182
(1)
Included in cash and cash equivalents on our balance sheets.
Fair Value Measurements at Reporting Date Using Quoted Prices in
Active Markets for
Identical Assets /
LiabilitiesSignificant
Other
Observable
InputsSignificant
Unobservable
Inputs(In thousands) Level 1 Level 2 Level 3 Total Assets
Money market funds(1)
$ 40,342 $ — $ — $ 40,342 Government-sponsored enterprise securities(2)(3)
— 6,949 — 6,949 Commercial paper(2)
— 10,999 — 10,999 Corporate notes(2)(3)
— 109,629 — 109,629 Total
$ 40,342 $ 127,577 $ — $ 167,919 Liabilities
Derivatives(4)
$ — $ — $ 16 $ 16 (2)
Included in current portion of marketable securities on our balance sheets.
(3)
Included in noncurrent portion of marketable securities on our balance sheets.
In December 2007, we granted a license to our hTERT technology for use in human diagnostics to Sienna Cancer Diagnostics Limited, or Sienna, which was a privately held company in Australia. In connection with the license, we received 13,842,625 ordinary shares in Sienna which we recorded at a zero cost basis under the cost method of
Contentsaccounting. On August 3, 2017, Sienna became a publicly traded company on the Australian Securities Exchange Limited, or ASX, under the ticker symbol SDX. Since our shares are subject to a 24-month trading restriction from the effective date of Sienna’s listing on the ASX, we account for our investment in Sienna under the cost method of accounting since there is no readily determinable fair value for our shares, and such shares do not meet the definition of a marketable security. With the adoption of ASU 2016-01 and ASU 2018-03 in 2018, the method in which we account for our shares of Sienna will change. For additional information on ASU 2016-01 and ASU 2018-03, see the section entitled “Recent Accounting Pronouncements Not Yet Effective” in Note 1 on Organization and Summary of Significant Accounting Policies.
94GERON CORPORATION
NOTES TO
CONSOLIDATEDFINANCIAL STATEMENTS (Continued)2. FAIR VALUE MEASUREMENTS (Continued)
The following table presents information about our financial instruments that are measured at fair value on a recurring basis as of December 31, 2013 and indicates the fair value category assigned.Fair Value Measurements at Reporting Date Using Quoted Prices in
Active Markets for
Identical Assets /
LiabilitiesSignificant
Other
Observable
InputsSignificant
Unobservable
Inputs(In thousands) Level 1 Level 2 Level 3 Total Assets
Money market funds(1)
$ 8,079 $ — $ — $ 8,079 Government-sponsored enterprise securities(2)
— 7,369 — 7,369 Commercial paper(2)
— 5,499 — 5,499 Corporate notes(1)(2)
— 41,572 — 41,572 Total
$ 8,079 $ 54,440 $ — $ 62,519 Liabilities
Derivatives(4)
$ — $ — $ 367 $ 367 (1)Included in cash and cash equivalents on our consolidated balance sheets.(2)Included in current portion of marketable securities on our consolidated balance sheets.(3)Included in noncurrent portion of marketable securities on our consolidated balance sheets.(4)Included in fair value of derivatives on our consolidated balance sheets.
Changes in Level 3 Recurring Fair Value MeasurementsThe table below includes a rollforward of the balance sheet amounts for the year ended December 31, 2014, including the change in fair value, for financial instruments in the Level 3 category. When a determination is made to classify a financial instrument within Level 3, the determination is based upon the significance of the unobservable parameters to the overall fair value measurement. However, Level 3 financial instruments typically include, in addition to the unobservable components, observable components (that is, components that are actively quoted and can be validated to externalGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)2. FAIR VALUE MEASUREMENTS (Continued)sources). Accordingly, the gain in the table below includes changes in fair value due in part to observable factors that are part of the methodology.Fair Value Measurements Using Significant Unobservable Inputs (Level 3)
Year Ended December 31, 2014(In thousands) Fair Value at
December 31,
2013Total
Unrealized
Gain
Included in
Earnings(1)Purchases,
Sales,
Issuances,
SettlementsTransfers
In and/or
Out of
Level 3Fair Value at
December 31,
2014Change in
Unrealized Gain
Related to
Financial
Instruments
Held at
December 31,
2014(1)Derivative liabilities
$ 367 $ (351 ) $ — $ — $ 16 $ (351 ) (1)Reported as unrealized gain on derivatives in our consolidated statements of operations.
Credit Risk
We currently place our cash, restricted cash, cash equivalents and marketable securities with four financial institutions in the United States. Generally, these deposits may be redeemed upon demand and therefore, bear minimal risk. Deposits with banks may exceed the amount of insurance provided on such deposits. Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash equivalents and marketable securities. Cash equivalents and marketable securities currently consist of money market funds, U.S.
government-sponsoredgovernment‑sponsored enterprise securities, commercial paper and corporate notes. Our investment policy, approved by the audit committee of our board of directors, limits the amount we may invest in any one type of investment issuer, thereby reducing credit risk concentrations.3. PROPERTY AND EQUIPMENT
Property and equipment, stated at cost, is comprised of the following:
December 31, December 31,
(In thousands) 2014 2013 2017
2016
Furniture and computer equipment
$ 1,158 $ 1,092 $
711
$
1,268
Lab equipment
130 118 —
12
Leasehold improvements
74 74 111
111
1,362 1,284 822
1,391
Less accumulated depreciation and amortization
(1,189 ) (1,192 ) (720
)
(1,208
)
$ 173 $ 92 $
102
$
183
4.
EQUIPMENT LINELICENSE AGREEMENTSIn 2009, we renewed our equipment financing facilityJanssen Biotech, Inc. Collaboration andhad approximately $500,000 available for borrowing. This facility was secured by a certificate of deposit. InLicense AgreementOn November 13, 2014, we
closedand Janssen entered into thecertificate of depositCollaboration Agreement under which we granted to Janssen exclusive worldwide rights to develop and commercialize imetelstat forour unused equipment line of credit upon maturity and transferredall human therapeutic uses, including hematologic myeloid malignancies. Upon thecash proceeds to our cash operating accounts. This action also cancelled the availabilityeffectiveness of theequipment lineCollaboration Agreement, we received $35,000,000 from Janssen as an upfront payment, which we classified as deferred revenue upon receipt.Under the Collaboration Agreement, Janssen is wholly responsible for the development, manufacturing, seeking regulatory approval for and commercialization of,
credit. We had no amounts dueimetelstat worldwide. Janssen is currently conducting two clinical trials of imetelstat: a Phase 2 trial in myelofibrosis, referred to as IMbark, and a Phase 2/3 trial in myelodysplastic syndromes, referred to as IMerge. Development costs for IMbark and IMerge are being shared between us and Janssen on a 50/50 basis. Additionally, underthis facility asthe terms of the Collaboration Agreement, we remain responsible for prosecuting, at Janssen’s direction, the patents licensed to Janssen at the time we entered into the Collaboration Agreement, with costs shared between us and Janssen on a 50/50 basis. The cost‑sharing arrangement with Janssen began in January 2015. As of December 31,2013.Table2017, accrued collaboration charges ofContents$1,702,000 on our balance sheet represent the net amount owed to Janssen for our proportionate share of development costs incurred by Janssen under the Collaboration Agreement for the three months ended December 31, 2017.
Following completion of the protocol-specified primary analysis of IMbark by Janssen, if completed, we expect Janssen to notify us of their decision, or a Continuation Decision, as to whether they elect to maintain the license rights granted to them under the Collaboration Agreement and continue to advance the development of imetelstat in any indication. In the event that IMbark is terminated early, or placed on clinical hold or suspended by a regulatory authority for an extended period of time, then Janssen must instead notify us of their Continuation Decision by the date that is approximately 24 months after the initiation of IMerge.95
GERON CORPORATION
NOTES TO
CONSOLIDATEDFINANCIAL STATEMENTS (Continued)4. LICENSE AGREEMENTS (Continued)
In the event that Janssen provides an affirmative Continuation Decision, we then would have an option, or the U.S. Opt‑In Rights, to share further U.S. development and promotion costs, including our share of development costs incurred to date by Janssen beyond IMbark or IMerge, in exchange for higher tiered royalty rates and higher future development and regulatory milestone payments if imetelstat is successfully developed and approved. If we exercise the U.S. Opt‑In Rights, then we and Janssen would share U.S. development and promotion costs beyond IMbark and IMerge on a 20/80 basis (Geron 20%, Janssen 80%), we would receive a $65,000,000 milestone payment, or the Continuation Fee, at the time of an affirmative Continuation Decision, and would be eligible to receive additional potential payments of up to $470,000,000 for the achievement of certain development and regulatory milestones, up to $350,000,000 for the achievement of certain sales milestones, and tiered royalties ranging from a mid‑teens up to low twenties percentage rate on worldwide net sales of imetelstat in any countries where regulatory exclusivity exists or there are valid claims under the patent rights exclusively licensed to Janssen. In addition, if we exercise the U.S. Opt‑In Rights, we then would also have a separate option, or the U.S. Co‑Promotion Option, to provide 20% of the U.S. selling effort with our sales force personnel, in lieu of funding 20% of U.S. promotion costs, upon regulatory approval and commercial launch of imetelstat in the United States. Such co‑promotion would be conducted under a Janssen prepared promotion plan, and in accordance with a co‑promotion agreement to be agreed by the parties at the time of our exercise of the U.S. Co‑Promotion Option. We would be responsible for all costs associated with establishing and maintaining our sales force in any conduct of such co‑promotion. All product sales would be booked by Janssen. If we do not exercise the U.S. Opt‑In Rights, then all further development and promotion costs beyond IMbark and IMerge would be borne by Janssen, we would receive the $65,000,000 Continuation Fee at the time of an affirmative Continuation Decision plus a $70,000,000 payment, or the Full U.S. Rights Fee, for Janssen’s retention of full U.S. rights to imetelstat, and would be eligible to receive additional potential payments of up to $415,000,000 for the achievement of certain development and regulatory milestones, up to $350,000,000 for the achievement of certain sales milestones, and tiered royalties ranging from a double‑digit up to mid‑teens percentage rate on worldwide net sales of imetelstat in any countries where regulatory exclusivity exists or there are valid claims under the patent rights exclusively licensed to Janssen.
Under the terms of the Collaboration Agreement, we and Janssen have created a joint governance structure, including joint development and steering committees and working groups, to oversee and manage worldwide regulatory, development and manufacturing work under the joint clinical development plan and promotional activities (assuming we exercise the U.S. Opt‑In Rights) for imetelstat, with Janssen responsible for the operational execution of those activities. In addition, both we and Janssen may propose to the joint development committee imetelstat development for any new indications not then provided for in the joint clinical development plan and if we and Janssen agree such development should be conducted outside of the joint clinical development plan, both we and Janssen would be entitled to independently undertake such development at the developing party’s own cost, subject to the other party’s obligation to provide reimbursement for its specified portion of the development costs plus a premium following marketing approval of imetelstat in such newly proposed indication as a result of such independent development. In the event that we do not exercise the U.S. Opt‑In Rights following an affirmative Continuation Decision by Janssen, if any, the joint governance structure under the Collaboration Agreement would be dissolved, a joint oversight committee would monitor the progress of the collaboration, and we would have no further rights to conduct any independent imetelstat development.
After an affirmative Continuation Decision by Janssen, the Collaboration Agreement would remain in effect until the expiration of the last‑to‑expire patent or the royalty obligations on sales of imetelstat cease, unless terminated earlier. If Janssen does not effect an affirmative Continuation Decision, then the Collaboration Agreement would terminate and all rights to the imetelstat program would revert to us. Janssen may terminate the Collaboration Agreement at any time for convenience or due to a safety‑related concern. If a notice of termination from Janssen occurs, we would be entitled to certain continued operational support and cost sharing under various circumstances and all rights to the imetelstat program would revert to us.
96
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
4. LICENSE AGREEMENTS (Continued)
The terms of the Collaboration Agreement contain multiple deliverables, which included at inception: (i) exclusive worldwide rights to develop and commercialize imetelstat for all indications, (ii) transfer of know‑how and intellectual property, including our obligation to procure supply for manufacturing imetelstat for up to nine months after the effective date of the Collaboration Agreement, (iii) participation on the joint committees and working groups and (iv) potential participation in promoting imetelstat in the United States, if approved for commercial sale. We concluded the license for exclusive worldwide rights to develop and commercialize imetelstat has standalone value to Janssen based on the technical and financial resources of Janssen, including Janssen’s drug development experience, sizeable employee base with specific experience in hematologic malignancies, and sufficient capital to independently develop imetelstat on a global basis. Since Janssen has final decision‑making authority in the event a unanimous decision cannot be reached by the joint committees, we determined our participation on the joint committees does not represent a non‑contingent deliverable under the Collaboration Agreement. In addition, we determined our potential participation in promoting imetelstat in the United States does not represent a non‑contingent deliverable because such participation is uncertain and dependent on imetelstat being approved for commercial sale, which is not within our control. Accordingly, we determined delivery of the license rights granted by us to Janssen, together with our performance of certain technology transfer‑related activities under the Collaboration Agreement, represents the sole non‑contingent deliverable under the Collaboration Agreement associated with the upfront payment. Therefore, we accounted for our delivery of the imetelstat license rights and our performance of the technology transfer‑related activities as a single unit of accounting. During the third quarter of 2015, we completed performance of the technology transfer‑related activities to Janssen as outlined under the Collaboration Agreement. Combining this performance with the delivery of the imetelstat license rights, we fully recognized the $35,000,000 upfront payment from Janssen as collaboration revenue on our statements of operations in the third quarter of 2015.
We have determined that each of the additional potential milestone payments to us under the Collaboration Agreement, including: (i) the Continuation Fee at the time of an affirmative Continuation Decision, (ii) the Full U.S. Rights Fee, if we do not exercise the U.S. Opt‑In Rights and (iii) payments based on the achievement of certain development, regulatory or sales milestones, represent contingent payments. Consequently, we will recognize revenue for these payments in their entirety upon successful accomplishment of the respective milestone. Royalties on future product sales of imetelstat, if successfully commercialized under the Collaboration Agreement, will be recognized as revenue when earned.
Janssen Pharmaceuticals, Inc. License Agreement
On September 15, 2016, we entered into the License Agreement with Janssen Pharmaceuticals whereby we granted to Janssen Pharmaceuticals an exclusive worldwide license, or the Exclusive License, under our proprietary patents for the research, development and commercialization of products based on specialized oligonucleotide backbone chemistry and novel amidates for ribonucleic acid interference, or RNAi, for the prevention, treatment and/or diagnosis of any and all human disorders, excluding cancers originating from the blood or bone marrow, and products whose predominant or primary mechanism of action is telomerase inhibition.
In addition to the Exclusive License, we granted to Janssen Pharmaceuticals a non‑exclusive worldwide license, or the Non‑Exclusive License, under our patents covering the synthesis of monomers, which are the building blocks of oligonucleotides, and certain know‑how necessary for the research, development and commercialization of products under the Exclusive License. The patent rights under the Non‑Exclusive License are also licensed exclusively to Janssen under the Collaboration Agreement, as described in the section above titled “Janssen Biotech, Inc. Collaboration and License Agreement”, for the development and commercialization of imetelstat, and the License Agreement with Janssen Pharmaceuticals expressly excludes, and is subject to, the rights and licenses granted to Janssen under the Collaboration Agreement.
Under the terms of the License Agreement, Janssen Pharmaceuticals, at its sole expense, is required to use reasonable efforts to perform research, development and commercialization activities to obtain at least one licensed product to be researched, developed and commercialized under the License Agreement. We remain responsible for prosecuting the patent rights under the Exclusive License, with reasonable input provided by Janssen Pharmaceuticals, and the costs for such prosecution will be shared between us and Janssen Pharmaceuticals on a 50/50 basis. In addition, we remain responsible for prosecuting the patent rights under the Non‑Exclusive License, as set forth under the terms of the Collaboration Agreement with Janssen.
97
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
4. LICENSE AGREEMENTS (Continued)
Under the terms of the License Agreement, we received $5,000,000 from Janssen Pharmaceuticals as a non‑refundable upfront payment. We are also eligible to receive additional potential payments of up to an aggregate maximum total of $75,000,000 for the achievement of certain development and regulatory milestones and tiered royalties in the low single digit percentage range on worldwide net sales of each licensed product commercialized under the License Agreement in any countries where there are valid claims under the patent rights licensed to Janssen Pharmaceuticals.
The License Agreement will remain in effect until the expiration of the last‑to‑expire patent, unless terminated earlier. Janssen Pharmaceuticals may also terminate the License Agreement at will upon prior written notice to us. In the event of an early termination of the License Agreement, all licenses to Janssen Pharmaceuticals would terminate.
The terms of the License Agreement contain multiple deliverables, which included at inception the transfer of: (i) license rights under the Exclusive License and (ii) license rights and certain know‑how under the Non‑Exclusive License. We concluded the License Agreement has standalone value to Janssen Pharmaceuticals based on Janssen Pharmaceuticals’ technical and financial resources, including drug development experience, sizeable employee base with specific knowledge of oligonucleotide chemistry, and sufficient capital to independently research, develop and commercialize products under the License Agreement on a global basis. Accordingly, we have determined delivery of the license rights granted by us to Janssen Pharmaceuticals under the Exclusive License and Non‑Exclusive License, together with the transfer of certain know‑how under the Non‑Exclusive License, represents the sole non‑contingent deliverable under the License Agreement associated with the upfront payment. Therefore, we accounted for our delivery of the license rights and transfer of know‑how under the License Agreement as a single unit of accounting. During the third quarter of 2016, we completed the delivery of the license rights and transfer of know‑how to Janssen Pharmaceuticals under the License Agreement. Accordingly, we fully recognized the $5,000,000 upfront payment from Janssen Pharmaceuticals as license fee revenue on our statements of operations in the third quarter of 2016.
We have determined that each of the additional potential development and regulatory milestone payments to us under the License Agreement represent contingent payments. Consequently, we will recognize revenue for these payments in their entirety upon successful accomplishment of the respective milestone. Royalties on potential future product sales under the License Agreement will be recognized as revenue when earned.
5. ACCRUED LIABILITIES
Accrued liabilities consisted of the following:
December 31, (In thousands) 2014 2013 Service provider obligations
$ 408 $ 840 Clinical trial costs
513 326 Other
616 617 $ 1,537 $ 1,783 December 31,
(In thousands)
2017
2016
Professional legal and accounting fees
$
272
$
350
Clinical trial costs
516
723
Other
138
361
$
926
$
1,434
6.
RESTRUCTURINGSRESTRUCTURINGApril 2013 RestructuringOn April 25, 2013,With projected reduced operational demands as a result of the Collaboration Agreement with Janssen, on March 3, 2015, we announcedthe decisionan organizational resizing todiscontinuereduce ourdiscovery research programs and companion diagnostics program based on telomere length and close our research laboratory facility located at 200 Constitution Drive, Menlo Park, California. With this decision, a total of 20 positions were eliminated. In connection with this restructuring, we incurred aggregate restructuring charges of $1,370,000 forworkforce. For the year ended December 31,2013,2015, we recorded restructuring charges ofwhich $624,000approximately $1,306,000, net of non‑cash adjustments, related toone-timeone‑time terminationbenefits, including $28,000benefits. These charges included $307,000 ofnon-cash stock-basednon‑cash stock‑based compensation expense relating to the extension of thepost-terminationpost‑termination exercise periodthrough the end of December 2013for certain stock options previously granted toterminatedemployees$200,000 related to non-cash charges for write-downs of excess equipment and leasehold improvements and $546,000 related to costs associated withaffected by theclosure of our research laboratory facility. In connection with the decision to close our research laboratory facility, we entered into an amendment to the lease agreement for the 200 Constitution Drive facility under which the lease terminated effective December 31, 2013. As consideration for the early termination of the lease, we paid the landlord the remaining rents due under the original term of the lease as well as certain facility maintenance costs, all of which have been included in restructuring charges.restructuring. The restructuring resulted in aggregate cash expenditures of$1,085,000approximately $988,000 after adjustments andnon-cash credits. In 2013, we received proceeds of $1,080,000 from the sale of excess laboratory equipment in connection with the closure of our research laboratory facility. All actions associated with this restructuring were completed in 2013, and we do not anticipate incurring any further charges in connection with this restructuring.The components of the accrued restructuring charges included on our consolidated balance sheet relating to the April 2013 restructuring are summarized in the following table. As of December 31, 2014, we had no remaining obligations related to the April 2013 restructuring:(In thousands) Employee
Severance and
Other BenefitsFacility
Related
ChargesTotal Beginning accrual balance as of December 31, 2013
$ 21 $ 73 $ 94 Cash payments
(19 ) (73 ) (92 ) Adjustments or non-cash credits
(2 ) — (2 ) Ending accrual balance as of December 31, 2014
$ — $ — $ — GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)6. RESTRUCTURINGS (Continued)December 2012 RestructuringOn December 3, 2012, we announced the decision to discontinue development of GRN1005. With this decision, a total of 43 positions were eliminated. In connection with this restructuring, we incurred aggregate restructuring charges of $2,794,000, of which $2,702,000 was recognized for the year ended December 31, 2012 and $92,000 was recognized for the year ended December 31, 2013. The aggregate restructuring charges consisted of $2,523,000 related to one-time termination benefits, including $107,000 of non-cash stock-based compensation expense relating to the extension of the post-termination exercise period through the end of December 2013 for certain stock options previously granted to terminated employees, and $271,000 related to non-cash charges for write-downs of GRN1005 manufacturing equipment. The restructuring resulted in aggregatenon‑cashexpenditures of $2,271,000 after adjustments and non-cashcredits. All actions associated with this restructuring were completed in2013, and we do not anticipate incurring any further charges in connection with this restructuring.See Note 15 on Subsequent Event for discussion of an organizational resizing announced in March2015.7. DIVESTITURE OF STEM CELL ASSETSOn October 1, 2013, we closed the transaction to divest our human embryonic stem cell assets and our autologous cellular immunotherapy program pursuant to the terms of the previously disclosed asset contribution agreement, or the Contribution Agreement, that we entered into in January 2013 with BioTime, Inc., or BioTime, and BioTime's subsidiary, Asterias Biotherapeutics, Inc., or Asterias (formerly known as BioTime Acquisition Corporation).In accordance with the terms of the Contribution Agreement, on October 1, 2013 we received 6,537,779 shares of Asterias Series A common stock representing 21.4% of Asterias' outstanding common stock as a class as of that date. We are also entitled to receive royalties from Asterias on the sale of products that are commercialized, if any, in reliance upon the patents we contributed to Asterias. In accordance with our contractual obligations under the Contribution Agreement, we distributed all of the shares of Asterias Series A common stock we received from Asterias to our stockholders on a pro rata basis, other than with respect to fractional shares and shares that would otherwise have been distributed to Geron stockholders residing in certain excluded jurisdictions, which shares, as required by the Contribution Agreement, were sold with the net cash proceeds therefrom distributed ratably to the stockholders who would otherwise have been entitled to receive such shares. We refer to the distribution by us of the Asterias Series A common stock, or cash in lieu thereof, as the Series A Distribution. We completed the Series A Distribution to eligible stockholders on August 15, 2014.As of December 31,2014,2017 and 2016, we had no remaining obligationswith respectrelated to theSeries A Distribution.restructuring.We accounted for the divestiture of the stem cell assets as a nonmonetary transaction since we transferred intangible assets in exchange for a non-controlling interest in Asterias. The stem cell assets we contributed consisted primarily of intellectual property and know-how and did not meet the definition of a business for accounting purposes. A business consists of three elements: (i) inputs, (ii) processes and (iii) outputs. To be considered a business, only inputs and processes are required, which together form an integrated set of activities used to create outputs. Since we did not contribute any processes, such as operational processes or an organized workforce with the skills and experience98
GERON CORPORATIONNOTES TO
CONSOLIDATEDFINANCIAL STATEMENTS (Continued)7. DIVESTITURE OF STEM CELL ASSETS (Continued)to provide the necessary processes capable of being applied to inputs to create outputs, we determined the stem cell assets only represented inputs and therefore were not considered an integrated set of activities. Due to the significant research and development necessary to realize the commercial potential of the stem cell assets, we expensed all research and development costs associated with the stem cell assets as incurred and therefore, there was no recorded amount, or carrying value, for the stem cell assets on our consolidated balance sheets. Since the divestiture of the stem cell assets represented the transfer of nonfinancial assets that do not meet the definition of a business in exchange for a non-controlling equity interest in Asterias, we accounted for the transaction using the carrying amount, or book value, of the assets surrendered with no gain or loss recognized on the exchange, consistent with our accounting policy for such transactions. Because the stem cell assets had a carrying amount of zero, we applied a carrying amount of zero to the Asterias Series A common stock received in the divestiture.We applied the equity method of accounting to our investment in Asterias Series A common stock during the period of ownership from October 1, 2013 through August 15, 2014. Since our investment in Asterias had an initial carrying amount of zero upon the closing of the transactions contemplated by the Contribution Agreement on October 1, 2013 and we had no commitments to provide financial support or obligations to perform services or other activities for Asterias, we suspended the equity method of accounting on October 1, 2013. In addition, since Asterias incurred net losses during our period of ownership, no additional value has been recognized for Asterias Series A common stock. Accordingly, the completion of the Series A Distribution had no impact on our consolidated financial statements.8.7. COMMITMENTS AND CONTINGENCIESPurportedSecurities LawsuitsWe and
Derivative LawsuitsOn March 14, 2014, acertain of our officers were named as defendants in two purportedsecuritiesclass actionlawsuit was commencedsecurities lawsuits filed in the United States District Court for the Northern District of California, or the California District Court,namingasdefendants us and certain of our officers. The lawsuit alleges violations of the Securities Exchange Act of 1934 in connection with allegedly false and misleading statements made by us related to our Phase 2 trial of imetelstat in patients with ET or PV. The plaintiff alleges, among other things, that we failed to disclose facts related to the occurrence of persistent low-grade LFT abnormalities observed in our Phase 2 trial of imetelstat in ET or PV patients and the potential risk of chronic liver injury following long-term exposure to imetelstat. The plaintiff seeks damages and an award of reasonable costs and expenses, including attorneys' fees. On March 28, 2014, a second purported securities class action lawsuit was commenced in the California District Court, and on June 6, 2014,well as a thirdpurportedsecurities lawsuit, not styled as a class action, which wascommencedoriginally filed in the United States District Court for the Southern District of Mississippi, but subsequently transferred to the California District Court. These three cases, or theMississippi District Court, naming as defendants us and certain of our officers. These lawsuits,Class Action Lawsuits, whicharewere based on the same factual background,as the purported securities class action lawsuit that commenced on March 14, 2014, also allege violations of the Securities Exchange Act of 1934 and seek damages and an award of reasonable costs and expenses, including attorneys' fees.were consolidated for all purposes.On
June 30, 2014,July 21, 2017, the California District Courtconsolidated bothentered an order and final judgment that dismissed with prejudice and released the claims asserted in the Class Action Lawsuits against all named defendants in connection with the Class Action Lawsuits, including us, and any claims that could have been asserted that arise or relate to the facts alleged in the Class Action Lawsuits, such that every member of thepurportedsettlement classactions filedwill be barred from asserting such claims in theCalifornia District Courtfuture. In connection with the settlement of the Class Action Lawsuits, in April 2017, we paid $250,000 andappointedour insurance providers paid $6,000,000 to alead plaintiffsettlement escrow account to be paid to members of the settlement class, less payment of attorneys’ fees andlead counselcosts torepresentplaintiff’s counsel. The settlement does not constitute any admission of fault or wrongdoing by us or any of thepurported class. On July 21, 2014,individual defendants.We do not expect to make any additional payments for and do not expect, and are not aware of, any additional claims arising from or related to the
California District Court ordered the lead plaintiff to file its consolidated amended complaint, which was filed on September 19, 2014. On August 11, 2014, we filed a motion to transfer the purported securities lawsuit filedfacts alleged in theMississippiTableClass Action Lawsuits and asserted by stockholders who have opted out ofContentsGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)8. COMMITMENTS AND CONTINGENCIES (Continued)District Court totheCalifornia District Court. On November 4, 2014, the Mississippi District Court granted our motion and transferred the case to the California District Court, which was thereafter consolidated with thesettlement classactions. On November 18, 2014, we filed a motion with the California District Court to dismiss the consolidated amended complaint. The plaintiff's opposition to our motion to dismiss was filed on January 20, 2015, and we filed our reply on February 25, 2015. The court hearing for the motion to dismiss has been scheduled for April 10, 2015. We believe that we have meritorious defenses and intend to defend against these lawsuits vigorously.On April 21, 2014, a stockholder purporting to act on our behalf filed a derivative lawsuitin theSuperior Court of California for the County of San Mateo against certain of our officers and directors. The lawsuit alleges breaches of fiduciary duties by the defendants and other violations of law. In general, the lawsuit alleges that the defendants caused or allowed the dissemination of allegedly false and misleading statements related to our Phase 2 trial of imetelstat in patients with ET or PV. The plaintiff is seeking unspecified monetary damages and other relief, including reforms and improvements to our corporate governance and internal procedures. ItClass Action Lawsuits. However, it is possible that additionalderivativelawsuitswillmay be filed, or allegations may be made by stockholders, with respect to these same or other matters and also naming us and/or our officers and directors as defendants.We have not yet respondedMonitoring, initiating and defending against legal actions is time-consuming for our management, is likely to be expensive and may detract from our ability to fully focus our internal resources on our business activities. In addition, despite thederivative lawsuit, but intend to vigorously defend against the claims alleged and to seek dismissalavailability ofthe lawsuit.For a further discussion of these ongoing lawsuits, refer to the section entitled "Legal Proceedings" in Part I, Item 3 of this annual report Form 10-K. These lawsuits and any other related lawsuits are subject to inherent uncertainties, and the actual defense and disposition costs will depend upon many unknown factors. The outcome of these lawsuits is necessarily uncertain. We could be forced to expend significant resources in the defense against these and any other related lawsuits andinsurance, we maynot prevail. We currently are not able to estimate the possible cost to us from these lawsuits, as they are currently at an early stage,incur substantial legal fees andwe cannot be certain how long it may take to resolve these lawsuits or the possible amount ofcosts in connection with anydamages that we may be required to pay. Suchadditional litigation and such amounts could be material to ourconsolidatedfinancialstatements even ifstatements. We may expend significant resources in the settlement or defense of any additional lawsuits, and we may not prevail inthe defense against thesesuch lawsuits. We have not established anyreservesreserve for any potential liability relating totheseany additional lawsuits.It is possible that we could, in the future, incur judgments or enter into settlements of claims for monetary damages.Indemnifications to Officers and Directors
Our corporate bylaws require that we indemnify our officers and directors, as well as those who act as directors and officers of other entities at our request, against expenses, judgments, fines, settlements and other amounts actually and reasonably incurred in connection with any proceedings arising out of their services to Geron. In addition, we have entered into separate indemnification agreements with each of our directors and officers which provide for indemnification of these directors and officers under similar circumstances and under additional circumstances. The indemnification obligations are more fully described in our bylaws and the indemnification agreements. We purchase standard insurance to cover claims or a portion of the claims made against our directors and officers. Since a maximum obligation is not explicitly stated in our bylaws or in our indemnification agreements and will depend on the facts and circumstances that arise out of any future claims, the overall maximum amount of the obligations cannot be reasonably estimated.
We have no such obligations on our consolidated balance sheets as of December 31, 2014 and 2013.GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)8. COMMITMENTS AND CONTINGENCIES (Continued)Operating Lease Commitment
In February 2014,On September 21, 2017, we amended the lease agreement for our premises at 149 Commonwealth Drive, Menlo Park, California, to extend the lease term from February 2018 through January2016.2020. As of December 31,2014,2017, operating lease obligations under the amended lease agreement include aggregate future minimum paymentsunder our operating lease for our premises at 149 Commonwealth Drive wereof approximately$959,000.$1,435,000, of which payments of approximately $678,000, $699,000 and $58,000 are due in 2018, 2019 and 2020, respectively. Rent expense under our operating leases was approximately$936,000, $1,422,000$691,000, $708,000 and$1,474,000$878,000 for the years ended December 31,2014, 20132017, 2016 and2012,2015, respectively.99
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
7. COMMITMENTS AND CONTINGENCIES (Continued)
Severance Plan
We have an Amended and Restated Severance Plan, or Severance Plan, that applies to all employees that are not subject to performance improvement plans, and
most significantlyprovides for, among other benefits: (i) a severance payment upon a Change of Control Triggering Event and Separation from Service(as defined in the Severance Plan)and (ii) a severance payment for eachnon-executivenon‑executive employee upon aNon-ChangeNon‑Change of Control Triggering Event and Separation from Service. As defined in the Severance Plan, a Change of Control Triggering Event and Separation from Service(as defined in the Severance Plan). A Change of Control Triggering Event is defined as an eventrequires a “double trigger” where: (i) an employee is terminated by us without cause in connection with a change of control or within 12 months following a change of control provided, however, that if an employee is terminated by us in connection with a change of control but immediately accepts employment with our successor or acquirer, the employee will not be eligible for the benefits outlined in the Severance Plan, (ii) an employee resigns because in connection with a change of control, the offered terms of employment (new or continuing) by us or our successor or acquirer within 30 days after the change of control results in a material change in the terms of employment, or (iii) after accepting (or continuing) employment with us after a change of control, an employee resigns within 12 months following a change of control due to a material change in the terms of employment.A Non-ChangeUnder the Severance Plan, a Non‑Change of Control Triggering Event and Separation from Service is defined as an event where anon-executivenon‑executive employee is terminated by us without cause. Severance payments range from two to 18 months of base salary, depending on theemployee'semployee’s position with us, payable in a lump sum payment. The Severance Plan also provides that the provisions of employment agreements entered into between us and executive ornon-executivenon‑executive employees supersede the provisions of the Severance Plan. As of December 31,2014,2017, all our executive officers have employment agreements with provisions that may provide greater severance benefits than those in the Severance Plan.9. STOCKHOLDERS'8. STOCKHOLDERS’ EQUITYPublic OfferingSales AgreementOn
February 4, 2014,August 28, 2015, wecompletedentered into anunderwritten public offering of 25,875,000At Market Issuance Sales Agreement, or the 2015 Sales Agreement, with MLV & Co. LLC, or MLV, pursuant to which we may elect to issue and sell shares of our common stockat a publichaving an aggregate offering price of$4.00up to $50,000,000 from time to time into the open market at prevailing prices through MLV as our sales agent. We will pay MLV an aggregate commission rate equal to up to 3.0% of the gross proceeds of the sales price per share for common stock sold through MLV under the 2015 Sales Agreement. Pursuant to the 2015 Sales Agreement, sales of common stock will be made in such quantities and on such minimum price terms as we may set from time to time. We are not obligated to make any sales of common stock under the 2015 Sales Agreement. In December 2017, we sold an aggregate of 614,230 shares of our common stock pursuant to the 2015 Sales Agreement, resulting in net cash proceeds of approximately$96,805,000$1,060,000 after deductingthe underwriting discountsales commissions and offering expenses payable by us.Table of ContentsThe 2015 Sales Agreement will expire in August 2018 unless extended by the parties.GERON CORPORATIONWarrantsNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)9. STOCKHOLDERS' EQUITY (Continued)WarrantsAs of December 31, 2014, the following warrants to purchase our common stock were outstanding and classified as equity:Issuance DateExercise
PriceNumber of
SharesExercisable
DateExpiration
DateAugust 2011(1)
$ 3.98 537,893 August 2011 August 2021 April 2005
$ 3.75 470,000 April 2005 April 2015 1,007,893 stock was exercised at an exercise price of $3.75 per share. We received cash proceeds of approximately $881,000 from the exercise of this warrant.(1)- In connection with each disbursement under
thea previous loan agreement with the California Institute for Regenerative Medicine, or CIRM, we were obligated to issue to CIRM a warrant to purchase Geron common stock. Such warrants and the underlying common stock were unregistered. We have no further obligations to issue any additional warrants to CIRM.InAs of December2014, CIRM exercised31, 2017, a warrant to purchase461,382537,893 shares of our common stockutilizing the netremained outstanding. The warrant was issued to CIRM in August 2011 at an exerciseprovisionprice of $3.98 per share and expires intheAugust 2021.On March 31, 2015, a warrant
resulting in the issuance of 168,039to purchase 235,000 shares of our commonstock.
100
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
8. STOCKHOLDERS’ EQUITY (Continued)
Equity Plans
2002 Equity Incentive Plan
The 2002 Equity Incentive Plan, or 2002 Plan, expired in May 2012. Upon the adoption of the 2011 Incentive Award Plan in May 2011 (see below), no further grants of options or stock purchase rights were made from the 2002 Plan. Options granted under the 2002 Plan expire no later than ten years from the date of grant. Option exercise prices were equal to 100% of the fair market value of the underlying common stock on the date of grant.
Service-basedService‑based stock options under the 2002 Plan generally vested over a period of four years from the date of the option grant. Other stock awards (restricted stock awards and restricted stock units) had variable vesting schedules which were determined by our board of directors on the date of grant. All outstanding awards granted under the 2002 Plan remain subject to the terms of the 2002 Plan and the individual award agreements thereunder.2011 Incentive Award Plan
In May 2011, our stockholders approved the adoption of the 2011 Incentive Award Plan, or 2011 Plan. Our board of directors administers the 2011 Plan. The 2011 Plan provides for grants
to employees (including officers and employee directors)of either incentive stock options or nonstatutory stock options and stock purchase rights to employees (including officers and employee directors) and consultants (includingnon-employeenon‑employee directors). As of December 31,2014,2017, an aggregate of13,384,8836,202,727 shares of our common stock were available for future grants of equity awards under the 2011 Plan. Pursuant to the terms of the 2011 Plan, any shares subject to outstanding stock optionsoriginally granted under the 2002 Plan or 1996 Directors' Stock Option Plan,or outstanding unvested restricted stock awards originally granted under the 2002 Plan that expire or terminate for any reason prior to exercise or settlement or are forfeited because of the failure to meet a contingency or condition required to vest such shares shall become available for issuance under the 2011 Plan. Options granted under the 2011 Plan expire no later than ten years from the date of grant. Option exercise prices shall be equal to100% ofthe fair market value of the underlying common stock on the date of grant. If, at the time weGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)9. STOCKHOLDERS' EQUITY (Continued)grant an option, the optionee directly or by attribution owns stock possessing more than 10% of the total combined voting power of all classes of our stock, the option exercise price shall be at least 110% of the fair market value of the underlying common stock and shall not be exercisable more than five years after the date of grant.
We grant
service-basedservice‑based stock options to employees under our 2011 Plan that generally vest over a period of four years from the date of the option grant. Other stock awards (restricted stock awards and restricted stock units) have variable vesting schedules as determined by our board of directors on the date of grant.Under certain circumstances, options may be exercised prior to vesting, subject to our right to repurchase shares subject to such option at the exercise price paid per share. Our repurchase rights would generally terminate on a vesting schedule identical to the vesting schedule of the exercised option. During
2014,2017, we have not repurchased any shares under the 2011 Plan. As of December 31,2014,2017, we have no shares outstanding subject to repurchase.Our Non-EmployeeAs of December 31, 2017, our Non‑Employee Director Compensation Policy adopted by our board of directors in March 2014 and amended by our board of directors in February 2015, May 2015 and February 2016 provides for the automatic grant to non‑employee directors of the following types of equity awards under the 2011 Plan:First Director Option. Each person who becomes a
non-employeenon‑employee director, whether by election by our stockholders or by appointment by our board of directors to fill a vacancy, will automatically be granted an option to purchase70,000100,000 shares of common stock, or First Director Option, on the date such person first becomes anon-employee director, or First Director Option.non‑employee director. The First Director Optionshall vestvests annually over three years upon each anniversary date of appointment to our board of directors.Subsequent Director Option. Each
non-employeenon‑employee director (other than any director receiving a First Director Option on the date of the annual meeting) will automatically be granted a subsequent option to purchase35,00050,000 shares of common stock, a Subsequent Director Option, on the date of the annual meeting of stockholders in each year during suchdirector'sdirector’s service on our board of directors. The Subsequent Director Option vestsone year fromin full on the earlier of: (i) the date of the next annual meeting of our stockholders or (ii) the first anniversary of the date of grant.101
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
1996 Directors'8. STOCKHOLDERS’ EQUITY (Continued)2006 Directors’ Stock Option Plan
The 1996 Directors' Stock Option Plan, or 1996 Directors Plan, expired in July 2006 upon which no further option grants were made from the 1996 Directors Plan. The options granted under the 1996 Directors Plan were nonstatutory stock options and expire no later than ten years from the date of grant. The option exercise price was equal to the fair market value of the underlying common stock on the date of grant. Options to purchase shares of common stock generally were 100% vested upon grant, except for options granted upon first appointment to the board of directors. These initial options vested annually over three years upon each anniversary date of appointment to the board of directors.GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)9. STOCKHOLDERS' EQUITY (Continued)2006 Directors' Stock Option PlanThe 2006
Directors'Directors’ Stock Option Plan, or 2006 Directors Plan, was terminated by our board of directors and replaced by the 2011 Plan in March 2014. No further grants of options were made from the 2006 Directors Plan upon the 2006 DirectorsPlan'sPlan’s termination. All outstanding awards granted under the 2006 Directors Plan remain subject to the terms of the 2006 Directors Plan and the individualgrantsaward agreements made thereunder.The options granted to non‑employee directors under the 2006 Directors Plan were nonstatutory stock options, and they expire no later than ten years from the date of grant. The option exercise price was equal to the fair market value of the underlying common stock on the date of grant. The First Director Option granted to
non-employee members of the board ofnon‑employee directors under the 2006 Directors Plan vested annually over three years upon each anniversary date of appointment to the board of directors. The Subsequent Director Option granted tonon-employee members of the board ofnon‑employee directors on the date of the annual meeting of stockholders in each year during suchdirector'sdirector’s service on our board of directors under the 2006 Directors Plan vested one year from the date of grant.Aggregate option and award activity for the 2002 Plan, 2011 Plan
1996 Directors Planand 2006 Directors Plan is as follows:Outstanding Options Shares
Available
For GrantNumber of
SharesWeighted Average
Exercise Price
Per ShareWeighted Average
Remaining
Contractual Life
(In years)Aggregate
Intrinsic
Value
(In thousands)Balance at December 31, 2013
16,207,250 15,576,216 $ 3.04 $ 33,798 Options granted
(5,658,931 ) 5,658,931 $ 4.85 Awards granted
(59,330 ) — $ — Options exercised
— (662,626 ) $ 1.94 Options cancelled/forfeited
3,613,577 (3,613,577 ) $ 5.48 Awards cancelled/forfeited
142,375 — $ — 2006 Directors Plan termination
(860,058 ) — $ — Balance at December 31, 2014
13,384,883 16,958,944 $ 3.16 7.38 $ 16,038 Options exercisable at December 31, 2014
9,129,576 $ 3.12 6.47 $ 9,231 Options fully vested and expected to vest at December 31, 2014
16,225,022 $ 3.14 7.32 $ 15,546 Outstanding Options
Weighted Average
Aggregate
Shares
Weighted Average
Remaining
Intrinsic
Available
Number of
Exercise Price
Contractual Life
Value
For Grant
Shares
Per Share
(In years)
(In thousands)
Balance at December 31, 2016
9,570,535
19,125,287
$
3.15
Options granted
(3,484,000
)
3,484,000
$
2.19
Awards granted
(72,066
)
—
$
—
Options exercised
—
(12,500
)
$
1.41
Options cancelled/forfeited
188,258
(188,258
)
$
7.99
Balance at December 31, 2017
6,202,727
22,408,529
$
2.96
6.24
$
2,034
Options exercisable at December 31, 2017
17,249,032
$
3.03
5.57
$
2,034
Options fully vested and expected to vest at
December 31, 2017
22,171,142
$
2.96
6.21
$
2,034
The aggregate intrinsic value in the preceding table represents the total intrinsic value, based on
Geron'sGeron’s closing stock price of$3.25$1.80 per share as of December31, 2014,29, 2017, which would have been received by the option holders had all the option holders exercised their options as of that date.There were noWe have not granted any optionsgrantedwith an exercise price below or greater than the fair market value of our common stock on the date of grant in2014, 20132017, 2016 or2012.2015. As of December 31,2014, 20132017, 2016 and2012,2015, there were9,129,576, 8,144,04017,249,032, 14,074,457 and10,410,19411,356,232 exercisable options outstanding at weighted average exercise prices per share of$3.12, $4.26$3.03, $2.99 and$5.49,$2.98, respectively.GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)9. STOCKHOLDERS' EQUITY (Continued)The total pretax intrinsic value of stock options exercised during
2014, 20132017, 2016 and20122015 was$989,000, $2,787,000$15,000, $595,000 and$100,$2,398,000, respectively. Cash received from the exercise of options in2014, 20132017, 2016 and20122015 totaled approximately$1,286,000, $6,567,000$18,000, $493,000 and$1,000,$2,205,000, respectively.No income tax benefit was realized from stock options exercised in 2014 since we reported an operating loss.102
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
8. STOCKHOLDERS’ EQUITY (Continued)
Information about stock options outstanding as of December 31,
20142017 is as follows:Options Outstanding Exercise Price RangeNumber of
SharesWeighted Average
Exercise Price
Per ShareWeighted Average
Remaining
Contractual Life
(In years)$1.10 - $1.50
4,284,937 $ 1.42 7.41 $1.51 - $2.14
4,278,382 $ 1.64 7.84 $2.16 - $5.01
5,376,639 $ 4.29 7.91 $5.05 - $9.32
3,018,986 $ 5.80 5.72 $1.10 - $9.32
16,958,944 $ 3.16 7.38 Options Outstanding
Weighted Average
Weighted Average
Remaining
Number of
Exercise Price
Contractual Life
Exercise Price Range
Shares
Per Share
(In years)
$1.10 - $1.51
5,655,662
$
1.45
4.89
$1.55 - $2.16
5,631,031
$
2.08
7.04
$2.22 - $4.34
6,000,783
$
3.37
7.60
$4.42 - $7.31
5,121,053
$
5.10
5.27
$1.10 - $7.31
22,408,529
$
2.96
6.24
Aggregate restricted stock activity for the
2002 Plan,2011Plan and 2006 DirectorsPlan is as follows:Number of
SharesWeighted
Average
Grant Date
Fair Value
Per ShareWeighted Average
Remaining
Contractual Term
(In years)Non-vested restricted stock at December 31, 2013
409,437 $ 4.84 0.82 Granted
59,330 $ 2.67 Vested
(265,422 ) $ 4.47 Cancelled/forfeited
(142,375 ) $ 4.66 Non-vested restricted stock at December 31, 2014
60,970 $ 4.73 0.37 Weighted Average
Weighted Average
Grant Date
Remaining
Number of
Fair Value
Contractual Term
Shares
Per Share
(In years)
Non-vested restricted stock at December 31, 2016
—
$
—
—
Granted
72,066
$
2.20
Vested
(72,066
)
$
2.20
Non-vested restricted stock at December 31, 2017
—
$
—
—
The weighted average grant date fair value of restricted stock granted during the years ended December 31, 2017, 2016 and 2015 was $2.20, $2.44 and $3.75 per share, respectively. The total fair value of restricted stock that vested during
2014, 20132017, 2016 and20122015 was$782,000, $252,000$159,000, $54,000 and$936,000,$275,000, respectively.Employee Stock Purchase Plan
In March 2014, our board of directors adopted the 2014 Employee Stock Purchase Plan, or 2014 Purchase Plan. The 2014 Purchase Plan was approved by our stockholders in May 2014. The 2014 Purchase Plan replaced the 1996 Employee Stock Purchase Plan, or 1996 Purchase Plan, which was terminated effective as of the date the 2014 Purchase Plan was approved by our stockholders.
However, outstanding purchase rights granted under the 1996 Purchase Plan prior to its termination remained subject to the terms of the 1996 Purchase Plan. A total of 968,829 shares of our common stock were issued under the 1996 Purchase Plan since its adoption in July 1996 and reserves of 231,171 shares of our common stock for future issuance under the 1996 Purchase Plan were cancelled as of the date of termination and became available for future issuance for other corporate purposes.Under the 2014 Purchase Plan, we are authorized to sell to eligible employees up to an aggregate of 1,000,000 shares ofGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)9. STOCKHOLDERS' EQUITY (Continued)Geron common stock. As of December 31,
2014, 24,3752017, an aggregate of 104,692 shares of our common stock have been issued under the 2014 Purchase Plan since its adoption.Under the terms of the 2014 Purchase Plan, employees can choose to have up to 10% of their annual salary withheld to purchase our common stock. An employee may not make additional payments into such account or increase the withholding percentage during the offering period.The 2014 Purchase Plan is comprised of a series of offering periods, each with a maximum duration (not to exceed 12 months) with new offering periods commencing on January 1st and July 1st of each year. The date an employee enters the offering period will be designated as the entry date for purposes of that offering period. An employee may participate only
participatein one offering period at a time. Each offering period consists of two consecutive purchase periods of sixmonths'months’ duration, with the last day of such period designated a purchase date.Under the terms of the 2014 Purchase Plan, employees can choose to have up to 10% of their annual salary withheld to purchase our common stock. An employee may not make additional payments into such account or increase the withholding percentage during the offering period.
The purchase price per share at which common stock is purchased by the employee on each purchase date within the offering period is equal to 85% of the lower of (i) the fair market value per share of
Geron'sGeron common stock on theemployee'semployee’s entry date into that offering period or (ii) the fair market value per share ofGeron'sGeron common stock on the purchase date. If the fair market value per share ofGeron'sGeron common stock on the purchase date is less than the fair market value at the beginning of the offering period, a new 12 month offering period will automatically begin on the first business day following the purchase date with a new fair market value.103
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
Stock-Based8. STOCKHOLDERS’ EQUITY (Continued)Stock‑Based Compensation for Employees and Directors
We measure and recognize compensation expense for all
share-basedshare‑based payment awards made to employees and directors, including employee stock options, restricted stock awards and employee stock purchases, based ongrant-dategrant‑date fair values for these instruments. Wegrant service-based stock options and restricted stock awards under our equity plans to employees, directors and consultants. The vesting period for employee options is generally four years. Weuse the Black Scholesoption-pricingoption‑pricing model to estimate thegrant-dategrant‑date fair value of our stock options and employee stock purchases. The fair value forservice-basedservice‑based restricted stock awards is determined using the fair value of our common stock on the date of grant.In the past, our board of directors has awarded to our employees and directors performance-based restricted stock awards and market-based restricted stock awards. We have not recognized any stock-basedAs stock‑based compensation expensefor performance-based restricted stock awardsrecognized inour consolidatedthe statements of operations for the years ended December 31,2014, 20132017, 2016 and2012 as the achievement of the specified performance criteria was not considered probable during that time. All of these awards have been cancelled unvested as the performance conditions were not achieved within the respective performance periods. The fair value for market-based restricted stock awards was determined using a lattice valuation model with a Monte Carlo simulation. All previously granted market-based restricted stock awards have been cancelled unvested as the market conditions were not achieved within the specified performance period.As stock-based compensation expense recognized in the consolidated statements of operations for the years ended December 31, 2014, 2013 and 20122015 is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures but at a minimum, reflects thegrant-dategrant‑date fair value of those awards that actually vested in the period. Forfeitures have been estimated at the time of grant based onGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)9. STOCKHOLDERS' EQUITY (Continued)historical data and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
We recognizeWith the adoption of Accounting Standards Update No. 2016-09, Improvements to Employee Share Based Payment Accounting, or ASU 2016-09, in the first quarter of 2017, we elected to continue to estimate forfeitures expected to occur to determine the amount of stock-based compensation expense to be recognized in each period. The adoption of ASU 2016-09 did not impact our accounting for or presentation of excess tax benefits recognized on stock-based compensation expense on our financial statements since our net deferred tax assets are fully offset by astraight-linevaluation allowance due to our history of operating losses. In addition, presentation requirements for cash flows related to employee taxes paid for withheld shares had no impact to all periods presented.We recognize stock‑based compensation expense on a straight‑line basis over the requisite service period, which is generally the vesting period. The following table summarizes the
stock-basedstock‑based compensation expense related to stock options, restricted stock awards and employee stock purchases for the years ended December 31,2014, 20132017, 2016 and20122015 which was allocated as follows:Year Ended December 31, (In thousands) 2014 2013 2012 Research and development
$ 2,545 $ 1,741 $ 2,336 Restructuring charges
— 28 107 General and administrative
5,113 2,666 2,868 Stock-based compensation expense included in operating expenses
$ 7,658 $ 4,435 $ 5,311 Year Ended December 31,
(In thousands)
2017
2016
2015
Research and development
$
988
$
1,275
$
2,139
Restructuring charges
—
—
307
General and administrative
7,156
6,970
5,951
Stock-based compensation expense included
in operating expenses
$
8,144
$
8,245
$
8,397
Modifications to the post-termination exercise period of outstanding options held by certain members of our executive management team resulted in additional stock-basedStock‑based compensation expense
of $205,000 for the year ended December 31, 2013 and have been reflected in the above table. In addition, stock-based compensation expensealso has been recognized for the modification of thepost-terminationpost‑termination exercise period for certain stock options previously granted to employees affected by theApril 2013 and December 2012 restructurings,March 2015 restructuring, which has been included in restructuring charges in ourconsolidatedstatements of operations. See Note 6 onRestructuringsRestructuring for further discussion of therestructurings.restructuring.The fair value of stock options granted in
2014, 20132017, 2016 and20122015 has been estimated at the date of grant using the Black Scholesoption-pricingoption‑pricing model with the following assumptions:Year Ended December 31,
2014 2013 2012 2017
2016
2015
Dividend yield
0% 0% 0% 0%
0%
0%
Expected volatility range
0.898 to 0.922 0.742 to 0.792 0.631 to 0.740 0.884 to 0.892
0.888 to 0.890
0.874 to 0.884
Risk-free interest rate range
1.64% to 1.92% 0.80% to 1.97% 0.81% to 1.25% 1.98% to 1.99%
1.21% to 1.38%
1.68% to 1.71%
Expected term
5.5 yrs 6 yrs 6 yrs 5.5 yrs
5.5 yrs
5.5 yrs
104
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
8. STOCKHOLDERS’ EQUITY (Continued)
The fair value of employee stock purchases in
2014, 20132017, 2016 and2012 under the 2014 Purchase Plan and 1996 Purchase Plan2015 has been estimated using the Black Scholesoption-pricingoption‑pricing model with the following assumptions:Year Ended December 31,
2014 2013 2012 2017
2016
2015
Dividend yield
0% 0% 0% 0%
0%
0%
Expected volatility range
0.835 to 1.666 0.506 to 1.391 0.458 to 0.774 0.577 to 0.641
0.641 to 0.684
0.654 to 1.392
Risk-free interest rate range
0.06% to 0.15% 0.09% to 0.21% 0.06% to 0.21% 0.45% to 0.89%
0.28% to 0.45%
0.11% to 0.28%
Expected term range
6 mos to 12 mos 6 mos to 12 mos 6 mos to 12 mos 6 - 12 mos
6 - 12 mos
6 - 12 mos
Dividend yield is based on historical cash dividend payments and Geron has paid no cash dividends to date. The expected volatility range is based on historical volatilities of our stock since traded options on
GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)9. STOCKHOLDERS' EQUITY (Continued)Geron common stock do not correspond to option terms and the trading volume of options is limited. The
risk-freerisk‑free interest rate range is based on the U.S. Zero Coupon Treasury Strip Yields for the expected term in effect on the date of grant for an award. The expected term of options is derived from actual historical exercise andpost-vestingpost‑vesting cancellation data and represents the period of time that options granted are expected to be outstanding. The expected term ofemployees'employees’ purchase rights is equal to the purchase period.Based on the Black Scholes
option-pricingoption‑pricing model, the weighted average estimated fair value ofemployeestock options granted during the years ended December 31,2014, 20132017, 2016 and20122015 was$3.57, $1.03$1.58, $1.83 and$0.89$3.06 per share, respectively. The weighted average estimated fair value ofemployees'employees’ purchase rights for the years ended December 31,2014, 20132017, 2016 and20122015 was$2.10,$0.75, $1.01 and$0.59$1.64 per share, respectively. As of December 31,2014,2017, total compensation cost related to unvestedshare-basedshare‑based payment awards not yet recognized, net of estimated forfeitures, was$15,032,000,$8,008,000, which is expected to be recognized over the next2726 months on aweighted-averageweighted‑average basis.Stock-Based401(k) Plan Matching ContributionsWe sponsor a defined‑contribution savings plan under Section 401(k) of the Internal Revenue Code covering all full‑time U.S. employees, or the Geron 401K Plan. Participating employees may contribute up to the annual Internal Revenue Service contribution limit. The Geron 401K Plan also permits us to provide discretionary matching and profit sharing contributions. Prior to 2014, our board of directors approved matching contributions for the Geron 401K Plan in our common stock, which vested ratably over four years for each year of service completed by our employees, commencing from the date of hire.
Stock‑Based Compensation to Service Providers
We grant stock options and restricted stock awards to consultants from time to time in exchange for services performed for us. In general, the stock options and restricted stock awards vest over the contractual period of the consulting arrangement.
We granted stock options to purchase 75,000, 80,000 and 50,000 shares of our common stock to consultants in 2014, 2013 and 2012, respectively.The fair value of stock options and restricted stock awards held by consultants is recorded as operating expenses over the vesting term of the respective equity awards. In addition, we will record any increase in the fair value of the stock options and restricted stock awards as the respective equity award vests. We recordedstock-basedstock‑based compensation expense of$94,000, $92,000$41,000, $104,000 and$135,000$311,000 for the vested portion of the fair value of stock options and restricted stock awards held by consultants in2014, 20132017, 2016 and2012,2015, respectively.We have also issued common stock to
consultantsnon‑employee directors andvendors in exchange forconsultants. For stock issuances where serviceseither performed orare to be performed forus. For these stock issuances,us, we record a prepaid asset equal to the fair market value of the shares on the date of issuance and amortize the fair value of the shares to our operating expenses on apro-ratapro‑rata basis as services are performed. For stock issuances where services have been performedor goods are received.for us, we record the fair market value of the shares on the date of issuance to offset the amounts owed. In2014, 20132017, 2016 and2012,2015, we issued71,239, 66,85372,066, 21,541 and170,29818,077 shares of common stock, respectively, in exchange forgoods or services.services provided. In2014, 20132017, 2016 and2012,2015, we recognized approximately$158,000, $202,000$159,000, $52,000 and$1,010,000,$53,000, respectively, of expense in connection with stock grants toconsultantsnon‑employee directors andvendors. As of December 31, 2014, $7,000 related to consultant and vendor stock issuances remained as a prepaid asset which is being amortized to our operating expenses on a pro-rata basis as services are incurred or goods are received.Table of Contentsconsultants.
105GERON CORPORATION
NOTES TO
CONSOLIDATEDFINANCIAL STATEMENTS (Continued)9. STOCKHOLDERS'8. STOCKHOLDERS’ EQUITY (Continued)Common Stock Reserved for Future Issuance
Common stock reserved for future issuance as of December 31,
20142017 is as follows:Outstanding stock options
22,408,529
Outstanding stock options16,958,944Options and awards available for grant
13,384,8836,202,727
Employee stock purchase plan
975,625895,308
WarrantsWarrant outstanding1,007,893537,893
Total
32,327,34530,044,457
401(k) PlanWe sponsor a defined-contribution savings plan under Section 401(k) of the Internal Revenue Code covering all full-time U.S. employees, or the Geron 401K Plan. Participating employees may contribute up to the annual Internal Revenue Service contribution limit. The Geron 401K Plan also permits us to provide discretionary matching and profit sharing contributions. The Geron 401K Plan is intended to qualify under Section 401 of the Internal Revenue Code so that contributions by employees or by us, and income earned on the contributions, are not taxable to employees until withdrawn from the Geron 401K Plan. Our contributions, if any, will be deductible by us when made.In 2014, our board of directors approved a cash matching contribution equal to 50% of each employee's contributions, which was fully vested when paid. We provided the 2014 matching contribution in February 2015. In 2013 and 2012, our board of directors approved a matching contribution equal to 75% and 100% of each employee's contributions, respectively. Those matching contributions were made in our common stock and vest ratably over four years for each year of service completed by the employee, commencing from the date of hire, until they are fully vested when the employee has completed four years of service.For the vested portion of the 2014 match, we recorded $175,000 as research and development expense and $143,000 as general and administrative expense. For the vested portion of the 2013 match, we recorded $156,000 as research and development expense and $157,000 as general and administrative expense. For the vested portion of the 2012 match, we recorded $616,000 as research and development expense and $259,000 as general and administrative expense. Due to the number of positions eliminated in the previous restructurings, a partial plan termination was triggered in both 2013 and 2012. We accelerated the vesting of unvested prior employer matches for employees affected by the respective restructurings, which resulted in $266,000 and $370,000 of operating expenses in 2013 and 2012, respectively. As of December 31, 2014, approximately $273,000 remained unvested for the 2013, 2012 and 2011 matches which will be amortized to operating expenses as the corresponding years of service are completed by the employees.Sales AgreementOn October 8, 2012, we entered into an At-the-Market Issuance Sales Agreement, or sales agreement, with MLV & Co. LLC, or MLV, pursuant to which we may elect to issue and sell shares of our common stock having an aggregate offering price of up to $50,000,000 from time to time into the open market at prevailing prices through MLV as our sales agent. We will pay MLV an aggregate commission rate equal to up to 3.0% of the gross proceeds of the sales price per share for commonGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)9.
STOCKHOLDERS' EQUITY (Continued)stock sold through MLV under the sales agreement. Pursuant to the sales agreement, sales of common stock will be made in such quantities and on such minimum price terms as we may set from time to time. We are not obligated to make any sales of common stock under the sales agreement. As of December 31, 2014, we had not sold any common stock pursuant to the sales agreement. The sales agreement will expire in October 2015 unless extended by the parties.10. LICENSE AGREEMENTSJanssen Biotech, Inc.In November 2014, we and Janssen Biotech, Inc., or Janssen, entered into an exclusive collaboration and license agreement, or Collaboration Agreement, to develop and commercialize imetelstat worldwide for oncology, including hematologic myeloid malignancies, and all other human therapeutic uses. Upon the early termination of the waiting periods under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, the Collaboration Agreement became effective on December 15, 2014. Upon the effectiveness of the Collaboration Agreement, we received $35,000,000 from Janssen as an upfront payment.Under the Collaboration Agreement, we granted to Janssen exclusive worldwide rights to develop and commercialize imetelstat for all indications, and Janssen is responsible for the development, manufacturing and commercialization of, and seeking regulatory approval for, imetelstat worldwide. Under the Collaboration Agreement, development of imetelstat will initially proceed under a mutually agreed joint clinical development plan, or CDP, which includes two agreed upon Phase 2 studies to be pursued initially, one in myelofibrosis, or the Initial Phase 2 MF Study, and one in myelodysplastic syndrome, or the Initial Phase 2 MDS Study, as well as additional, possible registration studies in myelofibrosis, or MF, and myelodysplastic syndrome, or MDS, and possible exploratory Phase 2 and potential follow-on Phase 3 studies in acute myelogenous leukemia, or AML. Development costs for the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study will be shared between the parties on a 50/50 basis. We expect Janssen to initiate the Initial Phase 2 MF Study in mid-2015, followed later by the Initial Phase 2 MDS Study to be initiated at the end of 2015.Following the protocol-specified primary analysis of the Initial Phase 2 MF Study or after a certain time period after the initiation of the first Phase 3 MF study, Janssen must notify us whether it elects to maintain its license rights and continue to advance the development of imetelstat in any indication. In the event that the Initial Phase 2 MF Study has been terminated early or suspended, Janssen must instead notify us of its election by the date that is the later of 24 months from the initiation of the planned Initial Phase 2 MDS Study or 24 months from the termination of the planned Initial Phase 2 MF Study or commencement of the suspension period, as applicable.In the event that Janssen elects to continue to maintain its license rights and advance the development of imetelstat in any indication within the applicable timeframe set forth in the Collaboration Agreement (such election, the Continuation Election), we then would have an option, or the U.S. Opt-In Rights, to share further U.S. development and promotion costs in exchange for higher tiered royalty rates and higher future milestone payments if imetelstat is successfully developed and approved. If we exercise our U.S. Opt-In Rights, then the parties would share U.S. development and promotion costs on a 20/80 basis (Geron 20%, Janssen 80%), we would receive a $65,000,000 milestone payment, or Continuation Fee, at the time of the Continuation Election, and would be eligible to receive additional potential payments of up to $470,000,000 in development and regulatory milestones,GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)10. LICENSE AGREEMENTS (Continued)up to $350,000,000 in sales milestones, and tiered royalties ranging from a mid-teens up to low twenties percentage rate on worldwide net sales of imetelstat in any countries where regulatory exclusivity exists or there are valid claims under the patent rights exclusively licensed to Janssen. In addition, if we exercise our U.S. Opt-In Rights, we then would also have a separate option, or the Co-Promotion Option, to provide 20% of the U.S. selling effort with sales force personnel, in lieu of funding 20% of U.S. promotion costs, upon regulatory approval and commercial launch of imetelstat in the United States. Such co-promotion would be conducted under a Janssen prepared promotion plan, and in accordance with a co-promotion agreement to be agreed by the parties at the time of our exercise of our Co-Promotion Option. We would be responsible for all costs associated with establishing and maintaining a sales force in any conduct of such co-promotion. All product sales would be booked by Janssen. If we do not exercise our U.S. Opt-In Rights, then all further development and promotion costs beyond the Initial Phase 2 MF Study or Initial Phase 2 MDS Study would be borne by Janssen, we would receive the $65,000,000 Continuation Fee at the time of the Continuation Election plus a $70,000,000 payment, or Full U.S. Rights Fee, for Janssen's retention of full U.S. rights to imetelstat, and would be eligible to receive additional potential payments of up to $415,000,000 in development and regulatory milestones, up to $350,000,000 in sales milestones, and tiered royalties ranging from a double-digit up to mid-teens percentage rate on worldwide net sales of imetelstat in any countries where regulatory exclusivity exists or there are valid claims under the patent rights exclusively licensed to Janssen.Under the terms of the Collaboration Agreement, we remain responsible for prosecuting, at Janssen's direction, the patents licensed to Janssen at the time we entered into the Collaboration Agreement, with costs shared between us and Janssen on a 50/50 basis. For intellectual property developed under the Collaboration Agreement, or Development IP, the party having sole ownership interest in such Development IP would be responsible for prosecuting the patents, with Janssen bearing all of the costs for Development IP solely owned by Janssen and costs shared between the parties on a 50/50 basis for Development IP either jointly owned or solely owned by us.Under the terms of the Collaboration Agreement, we and Janssen created a joint governance structure, including joint development and steering committees and working groups, to oversee and manage worldwide regulatory, development and manufacturing work under the joint CDP and promotional activities (assuming we exercises our U.S. Opt-In Rights) for imetelstat, with Janssen responsible for the operational implementation of those activities. In addition, either of the parties may propose to the joint development committee imetelstat development for any new indications not then provided for in the joint CDP and if the parties agree such development should be conducted outside of the joint CDP, each of Geron and Janssen would be entitled to independently undertake such development at its own cost, subject to the other party's obligation to provide reimbursement for its specified portion of the costs plus a premium for such independent development following marketing approval of imetelstat in such newly proposed indication as a result of such independent development. In the event that we do not exercise our U.S. Opt-In Rights following Janssen's Continuation Election, the joint governance structure under the Collaboration Agreement would be dissolved, a joint oversight committee would monitor the progress of the collaboration, and we would have no further rights to conduct any independent imetelstat development.After a Continuation Election by Janssen, the Collaboration Agreement would remain in effect until the expiration of the last-to-expire patent or the royalty obligations on sales of imetelstat cease, unless terminated earlier. If Janssen does not effect a Continuation Election, then the CollaborationGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)10. LICENSE AGREEMENTS (Continued)Agreement would terminate and all rights would revert to us. Janssen may terminate the Collaboration Agreement at any time for convenience and due to a safety-related concern. If a notice of termination from Janssen occurs, we would be entitled to certain continued operational support and cost-sharing under various circumstances and all rights would revert to us.The terms of the Janssen Collaboration Agreement contain multiple deliverables, which include at inception: (i) exclusive worldwide rights to develop and commercialize imetelstat for all indications, (ii) transfer of know-how and intellectual property, including our obligation to procure supply for manufacturing imetelstat for up to nine months after the effective date of the Collaboration Agreement, (iii) participation on the joint steering committees and working groups and (iv) potential participation in selling imetelstat in the United States, if approved for commercial sale. We concluded the license for exclusive worldwide rights to develop and commercialize imetelstat has standalone value to Janssen based on the technical and financial resources of Janssen, including Janssen's drug development experience, sizeable employee base with specific experience in hematologic malignancies, and sufficient capital to independently develop imetelstat on a global basis. Since Janssen has final decision-making authority in the event a unanimous decision cannot be reached by the joint steering committees, we determined our participation on the joint steering committees does not represent a non-contingent deliverable under the Collaboration Agreement. In addition, we determined our potential participation in selling imetelstat in the United States does not represent a non-contingent deliverable because such participation is uncertain and dependent on the drug being approved for commercial sale, which is not within our control. Accordingly, we have determined delivery of the license rights granted by us to Janssen, together with our performance of the technology transfer-related activities, represents the sole non-contingent deliverable under the Collaboration Agreement. Therefore, we will account for our delivery of the license rights and our performance of the technology transfer-related activities as a single unit of accounting. We currently expect completion of the technology transfer-related activities to occur by September 30, 2015 at which point we expect to fully recognize the $35,000,000 upfront payment from Janssen as license fee revenue. As a result, we have not recognized any revenue related to the Collaboration Agreement in 2014. We have determined that each of the additional potential milestone payments to us under the Collaboration Agreement, including: (i) the Continuation Fee at the time of the Continuation Election, (ii) the Full U.S. Rights Fee if we do not exercise our U.S. Opt-In Rights and (iii) payments based on the achievement of certain development, regulatory or commercial milestones, represent substantive milestones. Consequently, we will recognize revenue for these payments in their entirety upon successful accomplishment of the respective milestone. Royalties on future product sales of imetelstat, if successfully commercialized under the Collaboration Agreement, will be recognized as revenue when earned.The cost-sharing arrangement with Janssen for the Initial Phase 2 MF Study and the Initial Phase 2 MDS Study began in 2015. Therefore, we have not recorded any payables to Janssen or receivables from Janssen in 2014.GE Healthcare UK LimitedIn June 2009, we entered into a worldwide exclusive license and alliance agreement with GE Healthcare UK, Limited, or GEHC, to develop and commercialize cellular assay products derived from human embryonic stem cells, or hESCs, for use in drug discovery, development and toxicity screening. In connection with the GEHC agreement, we recognized $825,000 as license fee revenue in ourGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)10. LICENSE AGREEMENTS (Continued)consolidated statements of operations for the year ended December 31, 2012 which reflects the full recognition of a license payment from GEHC related to the exercise of an option to expand the scope of their original 2009 license agreement to include exclusive global rights to our intellectual property and know-how for the development and sale of cellular assays derived from induced pluripotent stem cells. Upon the closing of the divestiture of our stem cell assets on October 1, 2013, the GEHC agreement, including any future revenue payments thereunder, was transferred to Asterias. No license fee revenue was recognized under the GEHC agreement in 2013. For a further discussion of the divestiture of our stem cell assets, see Note 7 on Divestiture of Stem Cell Assets.Telomerase Activation Sciences, Inc.In December 2012, we entered into a Termination and Assignment Agreement, or the Assignment Agreement, with Asia Biotech Corporation, or Asia Biotech, and Telomerase Activation Sciences, Inc., or TA Sciences, pursuant to which we agreed to assign to TA Sciences the intellectual property, including patents previously licensed to Asia Biotech, related to our telomerase activation technology. As consideration for the assignment and fulfillment of the obligations set forth in the Assignment Agreement, we received a non-refundable, upfront payment of $2,500,000 from TA Sciences, which we recognized in full as other income in our consolidated statements of operations for the year ended December 31, 2012, and TA Sciences does not have further payment obligations to us. In addition, Asia Biotech's future royalty obligations under the original license agreement have been terminated.11.INCOME TAXESDeferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets are as follows:
December 31, December 31,
2014 2013 2017
2016
(In thousands) (In thousands)
Net operating loss carryforwards
$ 281,300 $ 271,800 $
187,700
$
280,400
Purchased technology
— 6,300 Research credits
23,400 22,700 34,300
29,800
Capitalized research and development
2,100 6,600 2,500
300
License fees
500 700 100
200
Other—net
7,600 10,100 Other-net
8,200
11,500
Total deferred tax assets
314,900 318,200 232,800
322,200
Valuation allowance for deferred tax assets
(314,900 ) (318,200 ) (232,800
)
(322,200
)
Net deferred tax assets
$ — $ — $
—
$
—
GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)11. INCOME TAXES (Continued)We record net deferred tax assets to the extent we believe these assets will more likely than not be realized. In making such determination, we consider all available positive and negative evidence, including scheduled reversals of deferred tax liabilities, projected future taxable income, tax planning strategies and recent financial performance. Forming a conclusion that a valuation allowance is not required is difficult when there is negative evidence such as cumulative losses in recent years. Because of our history of losses, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance decreased by
$3,300,000$89,400,000 and$5,500,000 during$2,300,000 for the years ended December 31,20142017 and2013,2015, respectively, and increased by$14,200,000$9,600,000 during the year ended December 31,2012. Approximately $4,900,000 of the valuation allowance for deferred2016. No income taxassets relates to benefits ofbenefit was realized from stockoption deductions which, when recognized, will be allocated directly to contributed capital.options exercised in 2017.As of December 31,
2014,2017, we had domestic federal net operating loss carryforwards of approximately$774,000,000$793,900,000 expiring at various dates beginning in 2018 through2034,2037 and state net operating loss carryforwards of approximately$395,000,000$300,200,000 expiring at various dates beginning in20152017 through2034,2037, if not utilized. We also had federal research and development tax credit carryforwards of approximately$14,900,000$34,000,000 expiring at various dates beginning in 2018 through2034,2037, if not utilized. Our state research and development tax credit carryforwards of approximately$13,000,000$19,100,000 carry forward indefinitely.Due to the change of ownership provisions of the Tax Reform Act of 1986, utilization of a portion of our domestic net operating loss and tax credit carryforwards may be limited in future periods. Further, a portion of the carryforwards may expire before being applied to reduce future income tax liabilities.
106
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
On December 22, 2017, the Tax Cuts and Jobs Act of 2017, or 2017 Tax Act, was signed into law. Among other things, the 2017 Tax Act permanently lowers the corporate federal income tax rate to 21% from the existing maximum rate of 35%, effective for tax years including or commencing January 1, 2018. In accordance with GAAP, we remeasured the carrying value of our deferred tax assets as of December 31, 2017 using the new enacted corporate federal income tax rate of 21%. This remeasurement reduced our aggregate deferred tax assets and correspondingly reduced the valuation allowance by approximately $102,300,000. The remeasurement did not impact our financial statements.
In accordance with Staff Accounting Bulletin 118, as of December 31, 2017, we have made a reasonable estimate of the effects of the 2017 Tax Act on our existing deferred tax assets. Our preliminary estimate and the remeasurement of our deferred tax assets are subject to further analysis related to certain matters, such as developing interpretations of the provisions of the 2017 Tax Act, changes to certain estimates and the filing of our tax returns. U.S. Treasury regulations, administrative interpretations or court decisions interpreting the 2017 Tax Act may require further adjustments and changes in our estimates. The final determination of the 2017 Tax Act and the remeasurement of our deferred assets will be completed as additional information becomes available, but we expect no later than one year from the enactment of the 2017 Tax Act.
We adopted the provision of the standard for accounting for uncertainties in income taxes on January 1, 2007. Upon adoption, we recognized no material adjustment in the liability for unrecognized tax benefits. At December 31,
2014,2017, we had approximately$17,100,000$15,900,000 of unrecognized tax benefits, none of which would currently affect our effective tax rate if recognized due to our deferred tax assets being fully offset by a valuation allowance.A reconciliation of the beginning and ending amounts of unrecognized tax benefits is as follows (in thousands):
Balance as of December 31, 2013
$ 11,600 Increase related to prior year tax positions
5,100 Increase related to current year tax positions
400 Settlements
— Reductions due to lapse of applicable statute of limitations
— Balance as of December 31, 2014
$ 17,100 Balance as of December 31, 2016
$
14,700
Increase related to current year tax positions
1,200
Balance as of December 31, 2017
$
15,900
If applicable, we would classify interest and penalties related to uncertain tax positions in income tax expense. Through December 31,
2014,2017, there has been no interest expense or penalties related to unrecognized tax benefits.We do not currently expect any significant changes to unrecognized tax benefits during the fiscal year ended December 31,
2015.2018. In certain cases, our uncertain tax positions are related to tax years that remain subject to examination by the relevant tax authorities. Tax years for which we haveGERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)11. INCOME TAXES (Continued)carryforward net operating loss and credit attributes remain subject to examination by federal and most state tax authorities.
12. SEGMENT INFORMATIONOur executive management team represents our chief decision maker. We view our operations as one segment, the discovery and development of therapeutic products for oncology. As a result, the financial information disclosed herein materially represents all of the financial information related to our principal operating segment.13. CONSOLIDATED10. STATEMENTS OF CASH FLOWS DATAYear Ended December 31, 2014 2013 2012 (In thousands) Supplemental operating activities:
Issuance of common stock for services rendered to date or to be received in future periods
$ — $ — $ 69 Issuance of common stock for 401(k) matching contributions
$ 313 $ 839 $ 1,361 Reclassification between deposits and other current assets
$ 190 $ 219 $ 526 Supplemental investing activities:
Net unrealized loss on marketable securities
$ (70 ) $ (54 ) $ (38 ) Year Ended December 31,
2017
2016
2015
(In thousands)
Supplemental investing activities:
Net unrealized (loss) gain on marketable securities
$
(154
)
$
160
$
(129
)
We have not made any cash payments for taxes or interest for the years ended December 31,
2014, 20132017, 2016 and2012.2015.107
GERON CORPORATION
NOTES TO FINANCIAL STATEMENTS (Continued)
14.SELECTED QUARTERLY FINANCIAL INFORMATION (UNAUDITED)First
Second
Third
Fourth
First
QuarterSecond
QuarterThird
QuarterFourth
QuarterQuarter
Quarter
Quarter
Quarter
(In thousands, except per share amounts) (In thousands, except per share amounts)
Year Ended December 31, 2014
Year Ended December 31, 2017:
Revenues
$ 474 $ 341 $ 160 $ 178 $
537
$
174
$
163
$
191
Operating expenses
9,205 9,004 10,067 9,189 8,031
6,905
7,407
7,977
Net loss
(8,440 ) (8,734 ) (9,549 ) (8,947 ) (7,183
)
(6,405
)
(6,899
)
(7,429
)
Basic and diluted net loss per share
$ (0.06 ) $ (0.06 ) $ (0.06 ) $ (0.06 ) $
(0.05
)
$
(0.04
)
$
(0.04
)
$
(0.05
)
Year Ended December 31, 2013
Revenues
$ 765 $ 112 $ 181 $ 225 Year Ended December 31, 2016:
Revenues(1)
$
749
$
211
$
5,108
$
94
Operating expenses
(1)12,750 9,077 8,914 9,500 9,826
9,122
8,985
8,875
Net loss
(11,897 ) (8,947 ) (8,254 ) (9,281 ) (8,842
)
(8,637
)
(3,576
)
(8,482
)
Basic and diluted net loss per share
$ (0.09 ) $ (0.07 ) $ (0.06 ) $ (0.07 ) $
(0.06
)
$
(0.05
)
$
(0.02
)
$
(0.05
)
(1)
The third quarter of 2016 includes the full recognition of the $5,000,000 upfront payment from Janssen Pharmaceuticals as license fee revenue. See Note 4 on License Agreements.
(1)The fourth quarter of 2013 includes approximately $430,000 in restructuring charges in connection with the closure our research laboratory facility located at 200 Constitution Drive, Menlo Park, California. See Note 6 on Restructurings.
GERON CORPORATIONNOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)14. SELECTED QUARTERLY FINANCIAL INFORMATION (UNAUDITED) (Continued)Basic and diluted net
lossesloss per share are computed independently for each of the quarters presented. Therefore, the sum of the quarters may not be equal to the full year net loss per share amounts.15.12. SUBSEQUENT EVENT
In
lightJanuary 2018, we sold an aggregate ofprojected reduced operational demands as a result776,788 shares of our common stock pursuant to the 2015 Sales Agreement with MLV, resulting in net cash proceeds of approximately $1,553,000 after deducting sales commissions and offering expenses payable by us. For further discussion of theCollaboration2015 Sales Agreement,with Janssen,see Note 8 onMarch 3, 2015, we announced an organizational resizing to reduce our workforce from 39 to 21 positions, representing a reduction of approximately 46% of our workforce. We expect the majority of the reduction in our workforce to be completed by the end of the third quarter of 2015. In connection with the resizing, we anticipate incurring aggregate restructuring charges of approximately $1,900,000, of which approximately $1,500,000 is expected be paid in cash during 2015. The aggregate projected restructuring charges represent one-time termination benefits, comprised principally of severance, benefit continuation costs, outplacement services and non-cash stock-based compensation expense associated with the elimination of 18 positions. The majority of these charges are expected to be recognized in the first half of 2015. We may incur other charges and will record these expenses in the appropriate period as they are determined.ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTINGAND FINANCIAL DISCLOSURE
Stockholders’ Equity.108
None.
ITEM 9A. CONTROLS AND PROCEDURES(I) Evaluation of Disclosure Controls and Procedures
We have carried out an evaluation under the supervision and with the participation of management, including our
principal executive officerChief Executive Officer andprincipal financial officer,our Chief Financial Officer, of our disclosure controls and procedures (as defined in Rule13a-15(e)13a‑15(e) of the Securities Exchange Act of 1934, as amended) as of the end of the period covered by this annual report on Form10-K.10‑K. Based ontheirthat evaluation, ourprincipal executive officerChief Executive Officer andprincipal financial officerour Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of December 31,2014.2017.It should be notedIn designing and evaluating disclosure controls and procedures, our management recognizes that any system of controls, however well designed and operated, can provide only reasonable assurance, and not absolute assurance, that the desired control objectives of the system are met. In addition, the design of any control system is based in part upon certain assumptions about the likelihood of future events. Because of these and other inherent limitations of control systems, there can be no assurance that any design will succeed in achieving its stated goals in all future circumstances. Accordingly, our disclosure controls and procedures are designed to provide reasonable, not absolute, assurance that the objectives of our disclosure control system are met and, as set forth above, ourprincipal executive officerChief Executive Officer andprincipal financial officerour Chief Financial Officer have concluded, based on their evaluation as of the end of the period covered by this annual report on Form 10‑K, that our disclosure controls and procedures were effective to provide reasonable assurance that the objectives of our disclosure control system were met.(II) Changes in Internal Control over Financial Reporting
There
waswere nochangechanges in our internal control over financial reporting during the quarter ended December 31,20142017 thathashave materially affected, orisare reasonably likely to materially affect, our internal control over financial reporting.(III)
Management'sManagement’s Report on Internal Control over Financial ReportingInternal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
(1)Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;(2)Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and(3)Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
(1)
Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
(2)
Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
(3)
Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Management is responsible for establishing and maintaining an adequate internal control over financial reporting for us. Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Under the supervision and with the participation of our management, including our
principal executive officerChief Executive Officer andprincipal financial officer,our Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework set forth in"Internal“Internal Control—IntegratedFramework"Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Based on our evaluation under the framework set forth in"Internal“Internal Control—Integrated Framework,"” our management concluded that our internal control over financial reporting was effective as of December 31,2014.2017. The effectiveness of our internal control over financial reporting as of December 31,20142017 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report which is included herein.(IV) Report of Independent Registered Public Accounting Firm
TheTo the Stockholders and the Board of Directorsand Stockholdersof Geron CorporationOpinion on Internal Control over Financial Reporting
We have audited Geron
Corporation'sCorporation’s internal control over financial reporting as of December 31,2014,2017, based on criteria established inInternalControl—IntegratedControl-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, GeronCorporation'sCorporation (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the balance sheets of the Company as of December 31, 2017 and 2016, the related statements of operations, comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes and our report dated March 16, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying
Management'sManagement’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on thecompany'sCompany’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.We conducted our audit in accordance with the standards of the
Public Company Accounting Oversight Board (United States).PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A
company'scompany’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Acompany'scompany’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of thecompany'scompany’s assets that could have a material effect on the financial statements.Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Geron Corporation maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014 based on the COSO criteria.We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Geron Corporation as of December 31, 2014 and 2013, and the related consolidated statements of operations, comprehensive loss, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2014 of Geron Corporation and our report dated March 11, 2015 expressed an unqualified opinion thereon./s/ Ernst & Young LLP
Redwood City, California
March
11, 201516, 2018None.
Certain information required by Part III is omitted from this annual report on Form
10-K10‑K because we will file with the U.S. Securities and Exchange Commission a definitive proxy statement pursuant to Regulation 14A in connection with the solicitation of proxies forGeron'sGeron’s Annual Meeting of Stockholders expected to be held in May2015,2018, or the Proxy Statement, not later than 120 days after the end of the fiscal year covered by this annual report on Form10-K,10‑K, and certain information included therein is incorporated herein by reference.ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCEIdentification of Directors and Nominees for Director
The information required by this item concerning our directors and nominees for director is incorporated by reference from the section captioned
"Proposal“Proposal 1: Election ofDirectors"Directors” contained in our Proxy Statement.Identification of Executive Officers
The information required by this item concerning our executive officers is set forth in Part I, Item 1 of this annual report on Form
10-K.10‑K.Code of Ethics
We have adopted a Code of Conduct with which every person who works for Geron, including our board of directors, is expected to comply. The Code of Conduct is publicly available on our website under the Investor Relations section at www.geron.com. This website address is intended to be an inactive, textual reference only; none of the material on this website is part of this annual report on Form
10-K.10‑K. If any substantive amendments are made to the Code of Conduct or any waiver granted, including any implicit waiver, from a provision of the Code to our Chief Executive Officer, Chief Financial Officer or Corporate Controller, we will disclose the nature of such amendment or waiver on that website or in a report on Form8-K.8‑K.Copies of the Code of Conduct will be furnished without charge to any person who submits a written request directed to the attention of our Corporate Secretary, at our offices located at 149 Commonwealth Drive, Suite 2070, Menlo Park, California, 94025.
Section 16(a) Compliance
Information concerning Section 16(a) beneficial ownership reporting compliance is incorporated by reference from the section captioned
"Section“Section 16(a) Beneficial Ownership ReportingCompliance"Compliance” contained in the Proxy Statement.Certain Corporate Governance Matters
The information required by this item concerning our audit committee, audit committee financial expert and procedures by which stockholders may recommend nominees to our board of directors, may be found under the
sectionsections captioned"Corporate Governance Matters"“Board Leadership and Governance” and “Other Matters” contained in the Proxy Statement.ITEM 11. EXECUTIVE COMPENSATIONThe information required by this item is incorporated by reference from the sections captioned
"Compensation“Compensation Discussion and Analysis," "Compensation” “Compensation Committee Report," "Executive” “Executive Compensation Tables" "Compensationand Related Narrative Disclosure,” “Compensation ofDirectors"Directors” and"Compensation“Compensation Committee Interlocks and InsiderParticipation"Participation” contained in the Proxy Statement.ITEM 12.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENTAND RELATED STOCKHOLDER MATTERSThe information required by this item is incorporated by reference from the sections captioned
"Equity“Equity CompensationPlans"Plan Information” and"Security“Security Ownership of Certain Beneficial Owners andManagement"Management” contained in the Proxy Statement.ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, ANDDIRECTOR INDEPENDENCEThe information required by this item is incorporated by reference from the sections captioned
"Proposal“Proposal 1: Election ofDirectors"Directors” and"Certain Transactions"“Certain Transactions” contained in the Proxy Statement.ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICESThe information required by this item is incorporated by reference from the section captioned
"Principal“Principal Accountant Fees andServices"Services” contained in the Proxy Statement.ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
(a)(1) Consolidated Financial Statements
(a)
(1) Financial Statements
Included in Part II, Item 8 of this Report:
Page
PageReport of Independent Registered Public Accounting Firm
8179
ConsolidatedBalance Sheets—December 31,20142017 and201320168280
ConsolidatedStatements of Operations—YearsendedEnded December 31,2014, 20132017, 2016 and201220158381
ConsolidatedStatements of Comprehensive Loss—YearsendedEnded December 31,2014, 20132017, 2016 and201220158482
ConsolidatedStatements ofStockholders'Stockholders’ Equity—YearsendedEnded December 31,2014, 20132017, 2016 and201220158583
ConsolidatedStatements of Cash Flows—YearsendedEnded December 31,2014, 20132017, 2016 and201220158684
Notes to
ConsolidatedFinancial Statements8785
(2)Financial Statement Schedules
(2)
Financial Statement Schedules
Financial statement schedules are omitted because they are not required or the information is disclosed in the financial statements listed in Item 15(a)(1) above.
(3)Exhibits
(3)
Exhibits
See Exhibit Index.Incorporation by Reference
Exhibit
NumberDescription
Exhibit
NumberFiling
Filing Date
File No.
2.1
2.1
8‑K
January 8, 2013
000‑20859
3.1
3.3
8‑K
May 18, 2012
000‑20859
3.2
Certificate of Amendment of the Restated Certificate of Incorporation
3.1
8‑K
May 18, 2012
000‑20859
3.3
3.1
8‑K
March 19, 2010
000‑20859
3.4
3.4
8-K
November 22, 2017
000‑20859
4.1
4.1
10‑K
March 15, 2013
000‑20859
4.2
Attachment to 10.1
10‑Q
November 3, 2011
000‑20859
10.1
10.1
10‑K
March 7, 2012
000‑20859
10.2
4.1
S‑8
June 4, 2010
333‑167349
10.3
Form of Stock Option Agreement under 2002 Equity Incentive Plan*
10.6
10‑K
March 15, 2013
000‑20859
10.4
10.5
10‑Q
November 7, 2013
000‑20859
10.5
10.1
8‑K
May 16, 2011
000‑20859
10.6
Form of Stock Option Agreement under 2011 Incentive Award Plan*
10.11
10‑K
March 15, 2013
000‑20859
Incorporation by Reference
Exhibit
NumberDescription
Exhibit
NumberFiling
Filing Date
File No.
Form of Restricted Stock Award Agreement under 2011 Incentive Award Plan*
10.12
10‑K
March 15, 2013
000‑20859
10.8
Form of Non‑Employee Director Stock Option Agreement under 2011 Incentive Award Plan*
10.2
10‑Q
May 7, 2015
000‑20859
10.9
10.1
8‑K
May 23, 2014
000‑20859
10.10
Amended and Restated Severance Plan, effective as of May 23, 2013*
10.1
8‑K
May 24, 2013
000‑20859
10.11
10.2
10‑Q
November 3, 2011
000‑20859
10.12
10.5
8‑K
February 14, 2014
000‑20859
10.13
10.1
8-K
February 2, 2018
000‑20859
10.14
10.32
10‑K
March 7, 2012
000‑20859
10.15
10.4
8‑K
September 27, 2013
000‑20859
10.16
10.2
10‑Q
November 2, 2012
000‑20859
10.17
10.4
8‑K
February 14, 2014
000‑20859
10.18
Employment agreement between the Registrant and Olivia K. Bloom, effective as of December 7, 2012*
10.26
10‑K
March 15, 2013
000‑20859
10.19
10.2
8‑K
September 27, 2013
000‑20859
10.20
10.1
8‑K
February 14, 2014
000‑20859
10.21
10.28
10‑K
March 15, 2013
000‑20859
10.22
10.1
8‑K
September 27, 2013
000‑20859
Incorporation by Reference
Exhibit
NumberDescription
Exhibit
NumberFiling
Filing Date
File No.
10.2
8‑K
February 14, 2014
000‑20859
10.24†
California Institute for Regenerative Medicine Notice of Loan Award
10.1
10‑Q
November 3, 2011
000‑20859
10.25†
10.36
10-K/A
March 27, 2012
000‑20859
10.26
10.1
8‑K
September 18, 2015
000‑20859
10.27
10.1
8-K
September 22, 2017
000‑20859
10.28
10.1
8‑K
August 28, 2015
000‑20859
10.29†
10.36
10‑K
March 11, 2015
000‑20859
10.30
Non‑Employee Director Compensation Policy, as amended February 11, 2016*
10.30
10-K
March 10, 2016
000‑20859
10.31
Non‑Employee Director Compensation Policy, as amended January 31, 2018*
12.1
23.1
24.1
31.1
31.2
32.1
Incorporation by Reference
Exhibit
NumberDescription
Exhibit
NumberFiling
Filing Date
File No.
101
The following materials from the Registrant’s annual report on Form 10‑K for the year ended December 31, 2017, formatted in Extensible Business Reporting Language (XBRL) include: (i) Balance Sheets as of December 31, 2017 and 2016, (ii) Statements of Operations, Comprehensive Loss, Stockholders’ Equity and Cash Flows for each of the three years in the period ended December 31, 2017, and (iii) Notes to Financial Statements
†
Confidential treatment has been granted for certain portions of this exhibit. Omitted information has been filed separately with the Securities and Exchange Commission.
*
Management contract or compensation plan or arrangement.
**
The certifications attached as Exhibits 32.1 and 32.2 that accompany this annual report on Form 10‑K, are not deemed filed with the Securities and Exchange Commission and are not to be incorporated by reference into any filing of the Company under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended (whether made before or after the date of this Form 10‑K), irrespective of any general incorporation language contained in such filing.
None.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
GERON CORPORATION
GERON CORPORATIONDate: March
16, 201811, 2015By:
/s/ Olivia K. Bloom
OLIVIA K. BLOOM
OLIVIA K. BLOOMExecutive Vice President, Finance,
Chief Financial Officer and Treasurer
KNOW BY ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints, jointly and severally, John A. Scarlett, M.D., and Olivia K. Bloom, and each one of them,
attorneys-in-factattorneys‑in‑fact for the undersigned, each with the power of substitution, for the undersigned in any and all capacities, to sign any and all amendments to this annual report on Form10-K,10‑K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of saidattorneys-in-fact,attorneys‑in‑fact, or his or her substitutes, may do or cause to be done by virtue hereof.IN WITNESS WHEREOF, each of the undersigned has executed this Power of Attorney as of the date indicated opposite his/her name.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
Signature
SignatureTitle
DateDate
/s/
JOHNJohn A.SCARLETT
ScarlettJOHN A. SCARLETTPresident, Chief Executive Officer and
Director (Principal Executive Officer)
March
11, 201516, 2018JOHN A. SCARLETT
/s/
OLIVIAOlivia K.BLOOM
BloomOLIVIA K. BLOOMExecutive Vice President, Finance, Chief
Financial Officer and Treasurer (Principal
Financial and Accounting Officer)
March
16, 201811, 2015OLIVIA K. BLOOM
/s/ Daniel M. Bradbury
Director
March 16, 2018
DANIEL M. BRADBURY
DANIEL M. BRADBURYDirectorMarch 11, 2015/s/ KARIN EASTHAMKARIN EASTHAMDirectorMarch 11, 2015/s/
THOMAS HOFSTAETTER
Karin EasthamTHOMAS HOFSTAETTERDirector
DirectorMarch
16, 201811, 2015/s/ HOYOUNG HUHHOYOUNG HUHDirectorMarch 11, 2015KARIN EASTHAM
/s/ Hoyoung Huh
Director
March 16, 2018
HOYOUNG HUH
/s/ V. Bryan Lawlis
Director
March 16, 2018
V. BRYAN LAWLIS
V. BRYAN LAWLISDirectorMarch 11, 2015/s/ Susan M. Molineaux
Director
March 16, 2018
SUSAN M. MOLINEAUX
SUSAN M. MOLINEAUXDirectorMarch 11, 2015/s/ Robert J. Spiegel
Director
March 16, 2018
ROBERT J. SPIEGEL
ROBERT J. SPIEGELDirectorMarch 11, 2015Incorporation by Reference Exhibit Number Description Exhibit
NumberFiling Filing Date File No. 2.1
Asset Contribution Agreement by and among Geron Corporation, BioTime, Inc. and Asterias Biotherapeutics, Inc. (formerly known as BioTime Acquisition Corporation) 2.1 8-K January 8, 2013 000-20859 3.1
Restated Certificate of Incorporation 3.3 8-K May 18, 2012 000-20859 3.2
Certificate of Amendment of the Restated Certificate of Incorporation 3.1 8-K May 18, 2012 000-20859 3.3
Amended and Restated Bylaws of Registrant 3.1 8-K March 19, 2010 000-20859 4.1
Form of Common Stock Certificate 4.1 10-K March 15, 2013 000-20859 4.2
Form of 2005 Warrant 4.2 8-K April 25, 2005 000-20859 4.3
Form of 2011 Warrant Attachment to 10.1 10-Q November 3, 2011 000-20859 10.1
Form of Indemnification Agreement 10.1 10-K March 7, 2012 000-20859 10.2
Amended and Restated 1996 Employee Stock Purchase Plan* 10.2 10-Q July 31, 2009 000-20859 10.3
1996 Directors' Stock Option Plan, as amended* Appendix B Def 14A April 15, 2003 000-20859 10.4
Amended and Restated 2002 Equity Incentive Plan* 4.1 S-8 June 4, 2010 333-167349 10.5
Form of Stock Option Agreement under 2002 Equity Incentive Plan* 10.6 10-K March 15, 2013 000-20859 10.6
Form of Restricted Stock Award Agreement under 2002 Equity Incentive Plan* 10.7 10-K March 15, 2013 000-20859 10.7
Amended and Restated 2006 Directors' Stock Option Plan* 10.5 10-Q November 7, 2013 000-20859 10.8
2011 Incentive Award Plan* 10.1 8-K May 16, 2011 000-20859 10.9
Form of Stock Option Agreement under 2011 Incentive Award Plan* 10.11 10-K March 15, 2013 000-20859 10.10
Form of Restricted Stock Award Agreement under 2011 Incentive Award Plan* 10.12 10-K March 15, 2013 000-20859 10.11
Form of Non-Employee Director Stock Option Agreement under 2011 Incentive Award Plan* 10.37 10-K March 17, 2014 000-20859 Incorporation by Reference Exhibit Number Description Exhibit
NumberFiling Filing Date File No. 10.12
2014 Employee Stock Purchase Plan* 10.1 8-K May 23, 2014 000-20859 10.13
Stock Purchase Agreement between the Registrant and Angiochem, Inc., effective as of January 5, 2011 10.1 8-K January 7, 2011 000-20859 10.14
† California Institute for Regenerative Medicine Notice of Loan Award 10.1 10-Q November 3, 2011 000-20859 10.15
Amended and Restated Severance Plan, effective as of May 23, 2013* 10.1 8-K May 24, 2013 000-20859 10.16
Employment agreement between the Registrant and John A. Scarlett, M.D., effective as of September 29, 2011* 10.2 10-Q November 3, 2011 000-20859 10.17
First Amendment to Employment Agreement between the Registrant and John A. Scarlett, M.D., effective as of February 11, 2014* 10.5 8-K February 14, 2014 000-20859 10.18
Employment agreement between the Registrant and Stephen N. Rosenfield, effective as of February 16, 2012* 10.32 10-K March 7, 2012 000-20859 10.19
First Amendment to Employment Agreement between the Registrant and Stephen N. Rosenfield, effective as of September 24, 2013* 10.4 8-K September 27, 2013 000-20859 10.20
Employment agreement between the Registrant and Andrew J. Grethlein, effective as of September 17, 2012* 10.2 10-Q November 2, 2012 000-20859 10.21
First Amendment to Employment Agreement between the Registrant and Andrew J. Grethlein, effective as of February 11, 2014* 10.4 8-K February 14, 2014 000-20859 10.22
Employment agreement between the Registrant and Craig C. Parker, effective as of December 3, 2012* 10.25 10-K March 15, 2013 000-20859 10.23
First Amendment to Employment Agreement between the Registrant and Craig C. Parker, effective as of September 24, 2013* 10.3 8-K September 27, 2013 000-20859 10.24
Second Amendment to Employment Agreement between the Registrant and Craig C. Parker, effective as of February 11, 2014* 10.3 8-K February 14, 2014 000-20859 Incorporation by Reference Exhibit Number Description Exhibit
NumberFiling Filing Date File No. 10.25
Employment agreement between the Registrant and Olivia K. Bloom, effective as of December 7, 2012* 10.26 10-K March 15, 2013 000-20859 10.26
First Amendment to Employment Agreement between the Registrant and Olivia K. Bloom, effective as of September 24, 2013* 10.2 8-K September 27, 2013 000-20859 10.27
Second Amendment to Employment Agreement between the Registrant and Olivia K. Bloom, effective as of February 11, 2014* 10.1 8-K February 14, 2014 000-20859 10.28
Employment agreement between the Registrant and Melissa A. Kelly Behrs, effective as of January 31, 2013* 10.28 10-K March 15, 2013 000-20859 10.29
First Amendment to Employment Agreement between the Registrant and Melissa A. Kelly Behrs, effective as of September 24, 2013* 10.1 8-K September 27, 2013 000-20859 10.30
Second Amendment to Employment Agreement between the Registrant and Melissa A. Kelly Behrs, effective as of February 11, 2014* 10.2 8-K February 14, 2014 000-20859 10.31
† Office Lease Agreement by and between the Registrant and Exponent Realty, LLC, effective as of February 29, 2012 10.36 10-K/A March 27, 2012 000-20859 10.32
Third Amendment to Office Lease Agreement by and between the Registrant and Exponent Realty, LLC, effective as of February 27, 2014 10.1 8-K March 4, 2014 000-20859 10.33
At-the-Market Issuance Sales Agreement, dated October 8, 2012, by and between the Registrant and MLV & Co. LLC 10.1 8-K October 9, 2012 000-20859 10.34
Transfer Agreement by and between Registrant and Mayo Clinic, effective July 31, 2014 10.1 10-Q November 5, 2014 000-20859 10.35
Non-Employee Director Compensation Policy, as amended* 10.36
†† Collaboration and License Agreement by and between the Registrant and Janssen Biotech, Inc., dated November 13, 2014 Incorporation by ReferenceExhibit NumberDescriptionExhibitNumberFilingFiling DateFile No.12.1Computation of Ratio of Earnings to Fixed Charges23.1Consent of Independent Registered Public Accounting Firm24.1Power of Attorney (see signature page)31.1Certification of Chief Executive Officer pursuant to Form of Rule 13a-14(a), as Adopted Pursuant to Section 302(a) of the Sarbanes-Oxley Act of 2002, dated March 11, 201531.2Certification of Chief Financial Officer pursuant to Form of Rule 13a-14(a), as Adopted Pursuant to Section 302(a) of the Sarbanes-Oxley Act of 2002, dated March 11, 201532.1Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated March 11, 2015**32.2Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, dated March 11, 2015**101The following materials from the Registrant's annual report on Form 10-K for the year ended December 31, 2014, formatted in Extensible Business Reporting Language (XBRL) include: (i) Consolidated Balance Sheets as of December 31, 2014 and 2013, (ii) Consolidated Statements of Operations, Comprehensive Loss, Stockholders' Equity and Cash Flows for each of the three years in the period ended December 31, 2014, and (iii) Notes to Consolidated Financial Statements.†Confidential treatment has been granted for certain portions of this exhibit. Omitted information has been filed separately with the Securities and Exchange Commission.††Confidential treatment has been requested for certain portions of this exhibit. Omitted information has been filed separately with the Securities and Exchange Commission.*Management contract or compensation plan or arrangement.
**The certifications attached as Exhibits 32.1 and 32.2 that accompany this annual report on Form 10-K, are not deemed filed with the Securities and Exchange Commission and are not to be incorporated by reference into any filing of the Company under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended (whether made before or after the date of this Form 10-K), irrespective of any general incorporation language contained in such filing.