Use these links to rapidly review the documentTABLE OF CONTENTSTable of ContentsINDEX TO FINANCIAL STATEMENTSItem 8. Financial Statements and Supplementary Data
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the year ended December 31, | ||
OR | ||
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the transition period from to | ||
Commission File Number 001-36464
Agile Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) | 23-2936302 (I.R.S. Employer Identification No.) |
101 Poor Farm Road
Princeton, New Jersey 08540
(Address including zip code of principal executive offices)
(609) 683-1880
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of exchange on which registered: | ||
|---|---|---|---|---|
| Common stock, par value $0.0001 per share | AGRX | The |
Securities registered pursuant to Section 12(g) of the Act:None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company, or emerging growth company. See definition of "large accelerated filer," "accelerated filer"filer," "smaller reporting company," and "smaller reporting"emerging growth company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filer o | Accelerated filer | Non-accelerated filer | Smaller reporting company ý Emerging growth company o |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by checkmark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of the voting stock held by non-affiliates of the registrant as of June 30, 201628, 2019 was approximately $152.8$52.1 million.
As of March 10, 2017February 18, 2020, there were 28,776,39869,810,305 shares of the registrant's common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant's definitive proxy statement for its 20172020 Annual Meeting of Stockholders (the "Proxy Statement"), to be filed within 120 days of the registrant's fiscal year ended December 31, 2016,2019, are incorporated by reference in Part II and Part III of this Annual Report on Form 10-K. Except with respect to information specifically incorporated by reference in this Annual Report on Form 10-K, the Proxy Statement is not deemed to be filed as part of this Annual Report on Form 10-K10-K.
Agile Therapeutics, Inc.
Annual Report on Form 10-K
For Thethe Year Ended December 31, 20162019
Table of Contents
| | | Page | |||||||
|---|---|---|---|---|---|---|---|---|---|
PART I | |||||||||
Item 1. | Business | 3 | |||||||
Item 1A. | Risk Factors | ||||||||
Item 1B. | Unresolved Staff Comments | ||||||||
Item 2. | Properties | ||||||||
Item 3. | Legal Proceedings | ||||||||
Item 4. | Mine Safety Disclosures | ||||||||
| |||||||||
Item 5. | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | ||||||||
Item 6. | Selected Financial Data | ||||||||
Item 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations | ||||||||
Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | ||||||||
Item 8. | Financial Statements and Supplementary Data | ||||||||
Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | ||||||||
Item 9A. | Controls and Procedures | ||||||||
Item 9B. | Other Information | ||||||||
| |||||||||
Item 10. | Directors, Executive Officers and Corporate Governance | ||||||||
Item 11. | Executive Compensation | ||||||||
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | ||||||||
Item 13. | Certain Relationships and Related Transactions and Director Independence | ||||||||
Item 14. | Principal Accounting Fees and Services | ||||||||
| |||||||||
Item 15. | Exhibits, | ||||||||
Item 16. | Form 10-K Summary | 133 | |||||||
i
SPECIAL CAUTIONARY NOTICE REGARDING FORWARD-LOOKING STATEMENTS
This annual reportAnnual Report on Form 10-K includes statements that are, or may be deemed, "forward-looking statements." In some cases, these forward-looking statements can be identified by the use of forward-looking terminology, including the terms "believes," "estimates," "anticipates," "expects," "plans," "intends," "may," "designed," "could," "might," "will," "should," "approximately" or, in each case, their negative or other variations thereon or comparable terminology, although not all forward-looking statements contain these words. They appear in a number of places throughout this Annual Report on Form 10-K and include statements regarding our current intentions, beliefs, projections, outlook, analyses or current expectations concerning, among other things, our ongoing and planned manufacturing and commercialization of Twirla®, the potential market uptake of Twirla® and the development of Twirla and our otherpotential product candidates, the strength and breadth of our intellectual property, our ongoing and planned clinical trials, the timing of and our ability to make regulatory filings and obtain and maintain regulatory approvals for our potential product candidates, the legal and regulatory landscape impacting our business, the degree of clinical utility of our products, particularly in specific patient populations, expectations regarding clinical trial data, our development and validation of manufacturing capabilities, our results of operations, financial condition, liquidity, prospects, growth and strategies, the length of time that we will be able to continue to fund our operating expenses and capital expenditures, our expected financing needs and sources of financing, the industry in which we operate and the trends that may affect the industry or us.
By their nature, forward-looking statements involve risks and uncertainties because they relate to events, competitive dynamics, and healthcare, regulatory and scientific developments and depend on the economic circumstances that may or may not occur in the future or may occur on longer or shorter timelines than anticipated. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Annual Report on Form 10-K, we caution you that forward-looking statements are not guarantees of future performance and that our actual results of operations, financial condition and liquidity, and the development of the industry in which we operate may differ materially from the forward-looking statements contained in this Annual Report on Form 10-K. In addition, even if our results of operations, financial condition and liquidity, and the development of the industry in which we operate are consistent with the forward-looking statements contained in this Annual Report on Form 10-K, they may not be predictive of results or developments in future periods.
Some of the factors that we believe could cause actual results to differ from those anticipated or predicted include:
Any forward-looking statements that we make in this Annual Report on Form 10-K speak only as of the date of such statement, and we undertake no obligation to update such statements to reflect events or circumstances after the date of this Annual Report on Form 10-K. You should also read carefully the factors described in the "Risk Factors" section of this Annual Report on Form 10-K to better understand the risks and uncertainties inherent in our business and underlying any forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report on Form 10-K will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard any of these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified timeframe, or at all.
This Annual Report on Form 10-K includes statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties. Industry publications and third partythird-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe these industry publications and third partythird-party research, surveys and studies are reliable, we have not independently verified such data.
We qualify all of our forward-looking statements by these cautionary statements. In addition, with respect to all of our forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995.
Overview
We are a forward-thinking women's healthcare company dedicated to fulfilling the unmet health needs of today's women. Our currentTwirla® and our potential product candidates are designed to provide women with contraceptive options that offer greater convenience and facilitate compliance. Our leadTwirla, our first and only approved product, candidate, Twirla®, also known as AG200-15, is a once-weekly prescription combination hormonal contraceptive patch that is at the end of Phase 3 clinical development. We completed the third of three Phase 3 clinical trials for Twirla in December 2016 and expect to resubmit our new drug application, or NDA, in the first half of 2017. Our short-term goal is to establish a market-leading franchise in the U.S. hormonal contraceptive market, which had total market sales of approximately $5.5 billion in 2016. Over half of those sales were generated by branded products. Currently, there is only one other contraceptive patch available in the United States and we believe it has limitations due to its dose and physical characteristics.patch. Twirla is designed to address these limitations. We have developed ausing our proprietary transdermal patch technology, called Skinfusion®, which is designed to provide advantages over the currently available patch and is intendedwith properties to optimize patch adhesion and patient wearability.wearability, which may help support compliance while, for the first time in a contraceptive patch, delivering a dose of estrogen consistent with commonly prescribed combined hormonal contraceptives, or CHCs. We believe there is an unmet market need for a low-dose contraceptive patch that is designed to address the limitationsdeliver approximately 30 mcg of the existing patch, while increasing patient convenienceestrogen and 120 mcg of progestin in a convenient dosage form that may support compliance in a non-invasive fashion.
Twirla was approved for sale in the United States on February 14, 2020 as a method of contraception for use in women of reproductive potential with a body mass index (BMI) < 30 kg/m2 for whom a combined hormonal contraceptive is appropriate. Based on the observed relationship between efficacy and BMI in a Phase 3 clinical trial, Twirla's limitation of use instructs healthcare providers to consider Twirla's reduced effectiveness in women with a BMI³ 25 to <30 kg/m2 before prescribing. Twirla is contraindicated in women with a BMI³ 30 kg/m2 because compared to women with a lower BMI, women in this group had reduced effectiveness and may have a higher risk for VTEs.
As part of Twirla's approval, the FDA is requiring us to conduct a long-term prospective, observational post-marketing study comparing the risks for VTE and ATE in new users of Twirla to new users of other CHCs. The FDA's requirement for Twirla is similar to another post-marketing study requirement for a recently approved CHC. The final study report for the Twirla post-marketing study is scheduled to be submitted to the FDA in November 2032, with interim safety data reporting to the FDA due in November 2026. We have also agreed to a small post-marketing commitment PMC study to assess the residual drug content and strength of Twirla. The PMC study is similar to residual drug studies requested of patch developers in the FDA's November 2019 draft guidance entitled Transdermal and Topical Delivery Systems—Product Development and Quality Considerations. We are evaluating the design and cost of these post-marketing studies. With the approval of Twirla we now plan to focus on our transition from a clinical development stage company to a commercial company. During 2020, we plan to begin the implementation of our commercialization plan for Twirla and to manage the growth of our company. Our near-term plan for the commercialization of Twirla includes:
| Activity | Expected Timing | |
|---|---|---|
| Initiate coverage and reimbursement activities in the United States from third-party payors | First Quarter 2020 | |
Initiate hiring of contract sales force | Second Quarter 2020 | |
Complete pre-validation and validation of the commercial manufacturing process consistent with our approved marketing application | Second Half 2020 with first shipment of product anticipated in the Fourth Quarter 2020 |
Our Strategy
Our short-term goal is to establish an initial franchise in the multi-billion-dollar U.S. hormonal contraceptive market built on approval of Twirla in the U.S. Our resources are currently focused on the commercialization of Twirla. To that end, our goal is to begin the pre-validation and validation of the
commercial manufacturing process in the first half of 2020, manufacture three validation batches of Twirla and complete the validation process in the second half of 2020. At the same time, we will prepare for the availability of commercial product supply. In the first quarter of 2020, we plan to initiate work with managed care and patient payors to gain market access for Twirla. In the second quarter of 2020, we plan to begin hiring and training an initial sales team, which we estimate to be in the range of 70 to 100 persons. We intend to ship product to wholesalers in the fourth quarter of 2020. During 2020, we also expect to begin planning the buildout of our existing pipeline and explore other opportunities to add additional products to our business.
Our current priorities are as follows:
Twirla
Twirla is our first and only approved product, indicated as a method of contraception for use in women of reproductive potential with a BMI < 30 kg/m2 for whom a combined hormonal contraceptive is appropriate. Based on the reduced efficacy seen with increasing BMI in a Phase 3 clinical trial, Twirla's limitation of use instructs healthcare providers to consider Twirla's reduced effectiveness in women with a BMI³ 25 to <30 kg/m2 before prescribing. Twirla is contraindicated in women with a BMI³ 30 kg/m2 because compared to women with a lower BMI, women in this group had reduced effectiveness and may have a higher risk for VTEs.
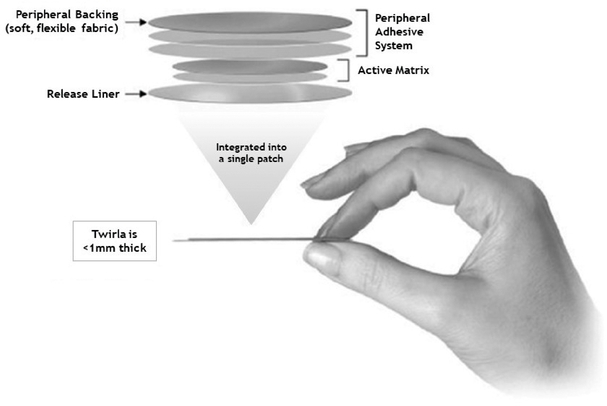
Twirla is a prescription combined hormonal contraceptive, or CHC, patch that contains the active ingredients ethinyl estradiol, or EE, which is a synthetic estrogen, and levonorgestrel, or LNG, which is a type of progestin, a synthetic steroid hormone, both of which have an established history of efficacy and safety in currently marketed combination low-dose, oral contraceptives. Twirla delivers approximately 30 micrograms of EE per day, a dose of EE consistent with the dose delivered by many commonly prescribed oral contraceptives. Our Skinfusion technology allows Twirla to be the first approved patch capable of delivering a contraceptive dose of LNG across the skin. The patch is applied once weekly for three weeks, followed by a week without a patch. Twirla is packaged with three individually wrapped patches per carton to provide for one 28-day cycle of therapy.
Twirla is designed using our proprietary Skinfusion technology to consistently deliver both hormones overfor convenient application by patients as a seven-day period at levels comparable to currently marketed low-dose oral contraceptives.method of contraception. By delivering these active ingredients over seven days, in a comfortable, convenient and easy-to-use weekly patch, Twirla is designed to promotearound principals of ease of use, andwhich may support enhanced patient compliance. It is also designed with properties to optimize patch adhesion and patient wearability with low levels of skin irritation. The patch is applied once weekly for three weeks, followed byround and made of a week withoutsoft, flexible fabric, designed to flex with the movement of a patch. If approved,woman's body. Twirla will be packaged with three patches per carton to provide for one 28-day cycleis a matrix patch consisting of therapy.
We have conducted a comprehensive clinical program, with completed Phase 1, Phase 2, and Phase 3 trials enrolling over 4,100 women, over 3,500several layers of whom received Twirla. Most recently, in December 2016, we completed a Phase 3 trial,material that contain the SECURE trial, in which we enrolled over 2,000 women for up to one year of treatment. In the Phase 1 and Phase 2 clinical trials, we demonstrated that Twirla delivers levels of bothactive ingredients EE and LNG, as well as the inactive ingredients Dimethylsulfoxide, Ethyl
Lactate, Capric Acid and Lauryl Lactate, which are ingredients to assist in the transport of EE and LNG across the skin, and adhesives that enable adherence to the blood stream that are consistent with current low-dose oral contraceptives. Priorskin. The final top layer is the one seen when placed on the skin, and consists of a thin, cloth-like material consisting only of adhesive. There is a barrier formed between the inner portion of the patch, which contains the active ingredients, and the outer portion of the patch, which only contains the adhesive. This barrier is intended to prevent the active and inactive ingredients from migrating to the SECURE trial, we completed twoperipheral portion of the patch and breaking down the adhesive there. Twirla is also designed to help prevent seepage of the adhesives from around the edge of the patch where it could collect dirt and leave a sticky black ring on the skin. The five layers of the patch are integrated to create a patch that has a slim profile and is unobtrusive when applied.
Twirla Marketing Authorization
Twirla received FDA approval on February14, 2020 as a method of contraception for use in women of reproductive potential with a BMI < 30 kg/m2 for whom a combined hormonal contraceptive is appropriate. Based on the reduced efficacy seen with increasing BMI, Twirla's limitation of use instructs healthcare providers to consider Twirla's reduced effectiveness in women with a BMI³ 25 to <30 kg/m2 before prescribing. Twirla is contraindicated in women with a BMI³ 30 kg/m2 because compared to women with a lower BMI, women in this group had reduced effectiveness and may have a higher risk for VTEs.
Pregnancy Rates (Estimated*) in Twirla-Treated Patients as BMI Increases for Women£35 Years of Age in Study ATI-CL23
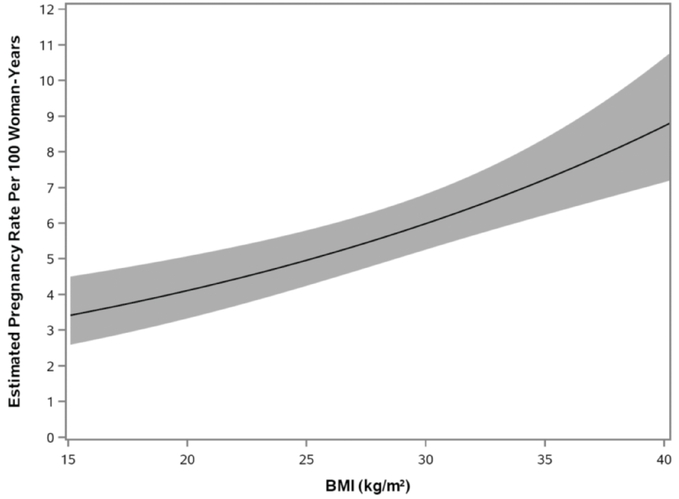
Twirla's approval is primarily based on safety and efficacy data from the Phase 3 clinical trials that enrolled over 1,900 women inSECURE trial. Because the aggregate for up to 12 months, and we demonstrated that Twirla generally had comparable efficacy and tolerability to an approved low-dose oral contraceptive. In the SECURE trial, we observed positive evidence of efficacy for Twirla based on use for up to one year. In our completed Phase 3 trials to date, over 1000 women have received Twirla for 12 months. Across all completed clinical trials, TwirlaNDA was generally well tolerated and had a favorable safety profile.
We have filed asubmitted under Section 505(b)(2) NDA, for approval of Twirla by the U.S.Federal Food, Drug and Drug Administration,Cosmetic Act, or FDA, which is required before marketing a new drug in the United States. Our 505(b)(2) NDA relies in part on clinical trials thatFDCA, and we conducted andrelied, in part, on the FDA's findings of safety and efficacy from investigations for approved products containing the active ingredientsEE and LNG and published scientific literature for which we have not obtained a right of reference.reference, we were not required to conduct preclinical studies.
The FDA has indicatedSECURE trial was a multicenter, single-arm, open-label, 13-cycle trial that evaluated the safety, efficacy and tolerability of Twirla in a Complete Response Letter, or CRL, that our NDA2,032 healthy women, aged 18 and over, at 102 experienced investigative sites across the United States. The trial was not sufficient for approval as originally submitted. After multiple communicationsdesigned in consultation with the FDA, we have received significant guidance asand incorporated a number of stringent trial design elements, including exclusion of treatment cycles not only for use of back-up contraception but also for lack of sexual activity. SECURE had broad entry criteria, placed no limitations on body mass index, or BMI, or other demographic factors during enrollment, and enrolled a large and diverse population from the United States in order to what additional clinical development and other activities needallow for efficacy to be completed priorassessed across different groups. These entry criteria resulted in the inclusion of a substantial number of women with high BMI, who have frequently been under-represented in past contraceptive studies. The efficacy measure for SECURE was the Pearl Index in an intent-to-treat population of subjects 35 years of age and under. The FDA also requested inclusion of pre-specified efficacy analyses related to approval. In accordance with the FDA's adviceBMI and comments, we conducted an additional Phase 3body weight.
clinical trial, As part of Twirla's approval, the SECURE trial, which was initiatedFDA is requiring us to conduct a long-term prospective, observational post-marketing study comparing the risks for VTE and ATE in 2014 and completed in December 2016. We announced the top-line resultsnew users of Twirla to new users of other CHCs. The FDA's requirement for Twirla is similar to another post-marketing study requirement for a recently approved CHC. The final study report for the SECURE trial in January 2017. Based on the guidance that we received fromTwirla post-marketing study is scheduled to be submitted to the FDA in connectionNovember 2032, with our discussions on clinical trial design, we believe thatinterim safety data reporting to the results fromFDA due in November 2026. We have also agreed to a post-marketing commitment, or PMC, study to assess the SECURE trial will address allresidual drug content and strength of the clinical issues raisedTwirla in a minimum of 25 women. The PMC study is similar to residual drug studies requested of patch developers in the CRL.FDA's November 2019 draft guidance entitledTransdermal and Topical Delivery Systems—Product Development and Quality Considerations. We expect to respond toare evaluating the CRLdesign and supplement our NDA with the resultscost of the trial in the first half of 2017, along with additional information relating to the manufacture of Twirla.
We intend to commercialize Twirla in the United States, if approved, through a direct sales force. Obstetricians and gynecologists, or ObGyns, contribute 43% of the U.S. contraception prescription volume, and Nurse Practitioners and Physician Assistants, or NP/PAs, who are often affiliated with an ObGyn practice, contribute an additional 29% of the U.S. prescriptions. We anticipate that a targeted sales force focused initially on ObGyns, NPs, PAs and primary care providers who comprise the top prescribers of contraceptives will be highly effective. We believe that we can address this market with a specialty sales force of approximately 70 to 100 representatives. We also intend to augment our sales force through digital marketing and other techniques to market directly to patients. We will require additional capital for the commercial launch of Twirla, if approved.
Our Skinfusion technology makes Twirla the first patch capable of delivering a contraceptive dose of LNG across the skin, allowing weekly application using a patch that is soft and flexible and is designed to adhere well with low levels of skin irritation. We, along with Corium International, Inc., or Corium, our manufacturing partner, have made a significant investment in a proprietary process to manufacture Twirla. We believe we have developed a robust process to reliably manufacture Twirla on a commercial scale. The materials produced for our clinical trials were manufactured using the same process that we expect will be used for our commercial-scale manufacturing, and we have made a significant investment in equipment for commercial-scale manufacturing if Twirla is approved. We believe that the technical challenges and know-how involved in manufacturing, including proprietary chemistry, production to scale and use of custom equipment and reproducibility, present significant barriers to entry for other pharmaceutical companies who might potentially want to replicate our Skinfusion technology.
Our intellectual property represents an additional barrier to potential competitors. We have thirteen issued U.S. patents, eight of which cover Twirla and that we intend to list in the Orange Book, the last of which expires in 2028, and five that provide additional coverage for other product candidates in our pipeline. The Orange Book lists drug products, including related patent and exclusivity information, approved by the FDA under the Federal Food, Drug, and Cosmetic Act. If a patent is listed in the Orange Book, potential competitors seeking approval of drug products under an Abbreviated New Drug Application, which provides for the marketing of a generic drug product that has the same active ingredients, dosage form, strength, route of administration, labeling, performance characteristics and intended use, among other things, of a previously approved product, or a 505(b)(2) application, for which the listed drug is a reference product, must provide a patent certification in their application stating either that (1) no patent information on the drug product has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. In addition, we continue to prosecute additional patent applications relating to Twirla, as well as our other product candidates, both in the United States and internationally. The intellectual property behind all of our product candidates in the pipeline and our Skinfusion technology consists of patent families developed and wholly-owned by us. There are no royalties or payments owed to third parties on our Skinfusion technology or any of our product candidates.
In addition to Twirla, we plan to develop a pipeline of other new transdermal contraceptive products, including AG200-ER, which is a regimen designed to allow a woman to extend the length of her cycle, AG200-SP, which is a regimen designed to provide shorter lighter periods, AG200-ER (SmP), which is a regimen designed to allow a woman to extend the length of her cycle and experience shorter,
lighter periods, and AG890, which is a progestin-only contraceptive patch intended for use by women who are unable or unwilling to take estrogen. Substantially all of our resources are currently dedicated to developing and seeking regulatory approval for Twirla. We will require additional capital to advance the development of our other product candidates.
Backgroundthese post-marketing studies.
Hormonal Contraception Overview
A woman is biologically capable of pregnancy from the time of her first menstrual cycle, at the average age of 12.6 years, to natural menopause, at the average age of 51.3 years. This is nearly half of a typical woman's lifespanContraceptive Landscape and for the typical woman, the majority of this time frame is spent trying to avoid pregnancy or is characterized by no desire to become pregnant. Nearly half of the pregnancies that occur each year in the United States are unplanned. The United States was the first country to approve a hormonal contraceptive, with the approval of the first contraceptive pill in 1960. The latest data from 2011 to 2013 from the Centers for Disease Control, or CDC, indicate that approximately 28% of women aged 15 to 44 use some form of hormonal contraception, which amounts to approximately 17 million U.S. women.
Hormonal contraceptives are composed of synthetic estrogens and progestins. Contraceptives containing both estrogen and a progestin are referred to as CHCs, and contraceptives containing only progestin are referred to as P-only. There are three synthetic estrogens approved in the United States for use in contraceptive products: EE, mestranol and estradiol valerate. EE has been available for over 40 years and is the estrogen component in nearly all CHCs today. There are 10 different progestins that have been used in contraceptives sold in the United States. The progestin component provides most of the contraceptive effect, while the estrogen component primarily provides cycle control, for example, minimizing bleeding or spotting between cycles. The progestin exerts its contraceptive effect by inhibiting ovulation, or release of an egg from the ovary, and by thickening cervical mucus. Thickening cervical mucus helps to prevent sperm entry into the upper genital tract. The estrogen component, in addition to providing cycle control, makes a small contribution to contraception by decreasing the maturation of the egg in the ovary.
Hormonal contraceptives are generally well-tolerated and are generally safer than pregnancy. A risk associated with hormonal contraceptives is a rare but serious adverse event called venous thromboembolism, or VTE, which involves the formation of a blood clot in a vein. VTEs can be life-threatening, and typically present as either deep vein thrombosis or pulmonary embolism. Evidence supports that the increased risk of VTE in CHC users is dependent upon the estrogen dose and duration of use. Estrogen increases formation of clotting factors in the liver and decreases production of elements that promote breakdown of blood clots. Most experts believe that progestins on their own have minimal to no impact on the clotting system, but some progestins, when combined with estrogen, can increase estrogen's effect on the clotting system.
The likelihood of a woman spontaneously developing a VTE is extremely low and the use of combination oral contraceptives, or COCs, increases the incidence only slightly, and less than
Table of ContentsMarket Opportunity
pregnancy. Epidemiologic studies evaluated by the FDA have demonstrated the incidence of VTE in women based on pregnancy or use of COCs as follows:
| ||
| ||
| ||
| ||
The available progestins are commonly categorized into generations, based on their history of introduction in the United States. The first and second generation progestins, including LNG, have been available in contraceptive formulations in the United States for over 25 years. The third and fourth generation progestins, for example desogestrel and drospirenone, respectively, were introduced to reduce androgenic side effects, such as oily skin and acne. Epidemiologic data suggest that CHCs containing third and fourth generation progestins are associated with an increased risk of VTE as compared to those containing the second generation progestin, LNG.
Effectiveness of Hormonal Contraceptives
For the purpose of FDA approval, contraceptive effectiveness is measured by a calculation called the Pearl Index, or PI and its associated 95% confidence interval (CI). The PI is a measure of the rate of pregnancies over a specific period of time in a clinical trial, and is expressed as the number of pregnancies per 100 woman years, or WY, of use. Each cycle lasts 28 days, so there are approximately 13 cycles in one year. According to recent FDA guidance, the PI calculation typically includes all pregnancies for which conception is estimated to have occurred while the subject was using the drug (i.e., on-treatment pregnancies), but only includes cycles where the woman indicates that she engaged in sexual activity and did not use backup contraception, such as a condom, and where she has completed a study diary. The PI values from clinical trials are affected by several factors, including differences in study design, increased sensitivity of early pregnancy tests, weight and body mass index, or BMI, of the study population, user experience and inconsistent or incorrect use of the contraceptive method. In addition, there has been an observable trend in PIs for approved combined hormonal contraceptives demonstrating an increase in the PIs over time, believed to be related to changes in study design and study populations. The FDA has not established any regulatory guidance on specific parameters for an acceptable PI or CI to support approval.
The contraceptive failure rates observed in clinical trials are generally lower than those seen once a CHC is approved and in use by a broad population, referred to as typical use, without the close monitoring of a clinical trial setting. There is a large difference in pregnancy rates under conditions of perfect use, where the method is used following the directions exactly, and typical use. For example, for CHCs, including oral contraceptives, the vaginal ring and the transdermal patch, the percent of women experiencing an unintended pregnancy during the first year of use is 0.3% for perfect use and 9.0% for typical use.
U.S. Hormonal Contraceptive Market Background
Contraceptive methods, other than sterilization, can be divided into non-hormonal and hormonal alternatives. Examples of non-hormonal products available in the United States include the diaphragm,
male condom, female condom, and female condom.non-hormonal intrauterine device, or IUD. Hormonal contraceptives containing both estrogen and a progestin are referred to as CHCs, and contraceptives containing only progestin are referred to as P-only. There are several categories of hormonal contraception products available in the United States, including:
The U.S. hormonal contraceptive market recorded annual sales in 2016is a multi-billion-dollar market. Data from 2011 to 2013 from the Centers for Disease Control, or CDC, indicate that approximately 28% of women aged 15 to 44 use some form of hormonal contraception, which amounts to approximately $5.5 billion, according to IMS Health.17 million U.S. women. The CHC portion of the market, consisting ofwhich includes pills, atwo transdermal patchpatches, including Twirla, and atwo vaginal ring,rings, generates significantly greater prescription volume and sales compared to the P-only portion of the market, consisting of IUDs, injectables, implants, and P-only pills. In 2016, IMS Health reported total U.S. sales of $3.9 billion for the CHC market and $1.6 billion for the P-only market. Twirla is a CHC and, if approved, we believe it will compete primarily with products in the CHC market.
The U.S. hormonal contraceptive market is a mature market, with many branded and generic products available. InSince the past 10 years, themid-2000s, CHC market growth was flat to declining as measured by prescription volume (TRx) has been flat to declining, with the exception of a 4.8% increase in 2013 compared to 2012. In the past three years (2017-2019), while CHC TRx growth has appeared to decline more aggressively (by 6%-12% per year), we believe this is largely due to an increase in the average TRx size (i.e. number of contraceptive cycles dispensed per TRx), which has increased from 1.4 cycles/TRx in 2016 to 1.7 cycles/TRx in 2019. The average annualtotal cycles dispensed in 2018 and 2019 has remained relatively stable reflecting the continued flat growth rate in dollarof this mature market. While gross sales were relatively flat between 2014 and 2018 due to a lack of new product entries and increased generic competition for the five years ended December 31, 2016 was 1.0% forboth the total hormonal contraceptive market and –0.7% for the CHC market. Market growthmarket, CHC market sales grew by 5.9% in gross sales is primarily2019 vs. 2018 to a total of $4.1 billion, largely due to price increases amongst branded products.increases.
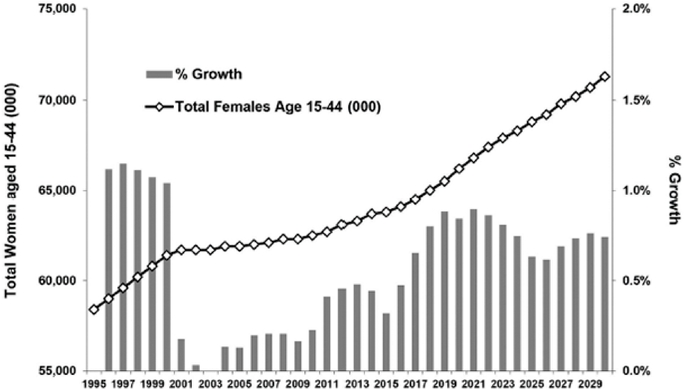
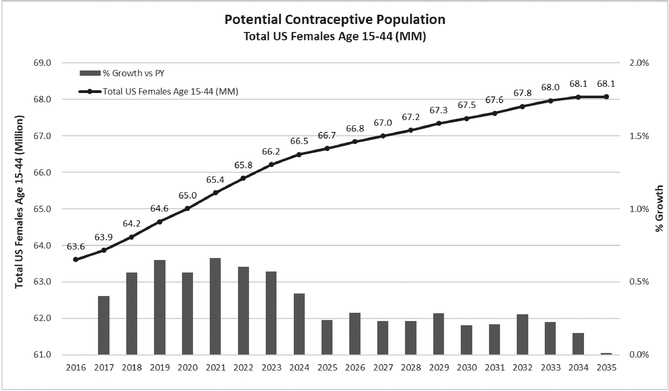
We believe there are twoseveral possible factors primarily affecting prescription volume growth in the contraceptive market. First, accordingAccording to U.S. Census Bureau data and projections, the population of women
aged 15 to 44 years has been growing at a rate of approximately 0.4%0.3% to 0.5%0.9% per year since 2011, increasing this population by 250,000approximately 200,000 to 300,000580,000 women per year.
Contraceptive Population(Total women aged 15-44 yrs)
Source:
Second,Additionally, in 2010, the Patient Protection and Affordable Care Act, as amended by the Healthcare and Education Reconciliation Act, or collectively, the ACA, was signed into law, which, among other things, requires all health plans, with limited exceptions, to cover certain preventive services for women with no cost-sharing, which means no deductible, no co-insurance and no co-payments by the patient, effective August 1, 2012. These services include those set forth in the Guidelines for Women's Preventive Services, or HRSA Guidelines, and adopted by the U.S. Department of Health and Human Services Health Resources and Services Administration. Contraceptive methods and counseling, including all FDA approved contraceptive methods as prescribed, are included in the HRSA Guidelines. Since these new ACA provisions went into effect in August 2012, quarterly prescription volume growth for the CHC market rose from negative growth year-on-year to positive growth between 4.0% and 5.0% for each of the six quarters following implementation. However, this appears to be a one-time phenomenon, as the market volume growth fell to 0.8%has been relatively flat since 2014, with the exception of a one-time drop in 2014 and –0.9%TRx cycles dispensed in 2015.2017.
Effect of ACA on Market Growth
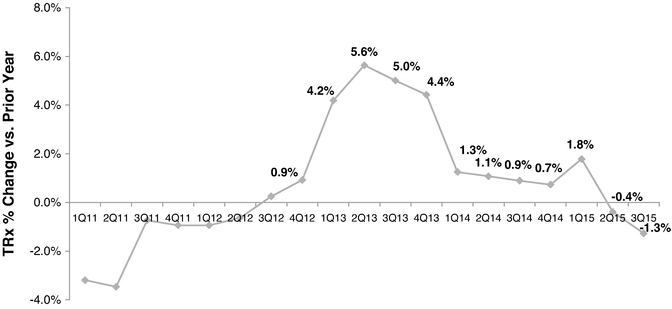
During the period following enactment of the ACA, generic oral contraceptives havecontraceptive volume has shown the greatest growth, primarily at the expense of branded oral contraceptives. This is likely due to the policies that were implemented by many managed care plans, which generally only provided generic oral contraceptives with no cost-sharing to the patient. The effect on non-oral products is less clear, but prescription volume for the vaginal ring showed a 5.1%6.8% decline from 20132015 to 2015,2019, while the prescription volume for the patch increased by 15.0%31% over the same time period. In May 2015, several government agencies, such asincluding the U.S. Department of Health and Human Services, or HHS, the Department of Labor, or DOL, and the U.S. Department of Treasury, or Treasury, jointly issued a clarification in the form of an FAQ which clarified the requirements for coverage of contraceptives under the ACA. The FAQ states
that plans and issuers must cover without cost-sharing at least one form of contraception in each of the 18 current methods that the FDA has identified for women in its current Birth Control Guide. The patch is identified as a specific method in the FDA Birth Control Guide, and therefore insurers must cover at least one patch product with no cost-sharing to the patient. Because this clarifying guidance is applied for plan years (or in the individual market, policy years) beginning on or after 60 days from the date of publication of the FAQs, patients did not have the benefit of this clarification until their new plan year, which generally started in January 2016.
In March 2017, the U.S. Congress proposed legislation, which, if signed into law by the new administration, would repeal certain aspects of the ACA. Further, on January 20, 2017, the new administration signed an Executive Order directing federal agencies with authorities and responsibilities under the ACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the ACA that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices, among others. Congress also could consider subsequent legislation to repeal and replace elements of the ACA that are repealed. Therefore, it is difficult to determine the full effect of the ACA or any other healthcare reform efforts on our business. We will continue to monitor the healthcare reform efforts. We believe the CHC market will maintain a long-term neutral to slightly positive annual growth rate in line with contraceptive population growth.
In spite ofDespite the availability of generic contraceptives for over 25 years, branded products have maintained a significant, though declining, share of the CHC market, with 55% of dollar sales and 17% of prescriptions for 2016.sales. Branded contraceptives in the CHC market have driven significant increases in the value of branded total prescriptions, or TRx. In the five years ended December 2016,2019, the average annual price increase among the top branded products was 10.6%10.5%. The average price per cycle, referred to as the wholesale acquisition cost, or WAC, for a single 28-day cycle of the top branded products was $41.53 in 2006 and rose to $131.40$160.88 by December 2016.2019. As of October 2014, the branded CHC transdermal patch (Ortho Evra) has been discontinued, and the generic CHC transdermal patch (Xulane) is currently priced at $105.92$122.15 per cycle. The other non-oral form of CHC, the vaginal ring, is currently priced at $128.21$162.63 per cycle. We cannot predict whether the manufacturers of branded products will continue to increase prices going forward,forward. We have not yet set a WAC price for Twirla, but we believe we will be able to set a WAC price for Twirla, if approved,one that is comparable to other branded and branded generic CHC products at the time of launch. Based on IMS Health data, we estimate that each percentage point of market share of CHC total prescriptions in the United States currently represents approximately $166 million of annual gross sales potential for Twirla, if approved.
Contraceptive Pills
Based on 2014 data from the CDC, of women who choose to use a hormonal contraceptive, approximately 64% use thea contraceptive pill, vaginal ring or patch, the majority of whichwhom use the contraceptive pill. The remaining 36% of women using hormonal contraception are split between using injectables, implants, or IUDs. Based on this information, we believe that contraceptive pills are the most popular choice because:
However, compliance remains a significant draw-back with pills. Published studies have shown that the average woman who uses oral contraceptives misses approximately two to four pills per month, which increases the potential for unintended pregnancies. We believe that a patch can offer greater convenience than a pill, as it does not require daily administration and, for certain women, could lead to greater compliance and ease of use.
Contraceptive Patch Market Experience
The Ortho Evra® contraceptive patch, or Evra, was introduced in early 2002 and was the first FDA-approved contraceptive patch. The initial approved labeling for Evra indicated that it delivered a daily EE dose of 20 micrograms. Evra had rapid uptake in the contraceptive market and achieved a 10% share of the CHC market by September 2003. Following FDA approval of Evra, users of Evra began to report thrombotic and thromboembolic events to the FDA. Johnson & Johnson, the manufacturer of Evra, revised the Evra labeling in November 2005 to include information that EE exposure with Evra is 60% higher than that of an oral contraceptive containing EE of 35 micrograms, based on area under the curve, a commonly-used metric for measuring EE exposure in contraceptives. This information was ultimately included in a black boxan addition to the boxed warning and bolded warningsthat was unique to the Evra label. The Evra market share declined rapidly following the labeling changes, from a peak share of 11% in 2005, to 4% by the end of 2006, to 1.4% by the end of 2013, where it stabilized, with a 1.5% share of the market based on combined prescriptions for Evra and its generic equivalent (Xulane®) in 2014. In the past two years, the patch share of the CHC market grew slightly, with a 1.6% TRx share in 2015 and 1.7% TRx share in 2016.
2014. In more recent years, the Xulane share of the CHC market TRx has grown, with a 1.8% share in 2017, 2.4% share in 2018, and 2.4% share in 2019.
In April 2014, Mylan Inc. announced the launch of Xulane™,Xulane®. In recent years, the Xulane share of the CHC market has grown slightly, with a generic version of Evra.1.7% TRx share in 2016 and 1.8% TRx share in 2017. Generic pharmaceutical products are the chemical and pharmaceutical equivalents of the brand or a reference listed drug, or RLD. Generic drugs are bioequivalent to their reference brand name counterparts. Bioequivalence studies compare the bioavailability of the proposed drug product with that of the RLD product containing the same active ingredients. Bioavailability is a measure of the rate and extent to which the active ingredient is absorbed from a drug product and becomes available at the site of action. Under pharmacy dispensing rules governed by state law, depending on the state, if an automatic generic substitute is introduced, the pharmacist may dispense either the prescribed product, or they may replace it with an equivalent generic without being required to inform the patient or healthcare professional. In addition, the FDA offers a 180-day exclusivity period for generic products in specific cases. The first generic applicants to submit a substantially complete Abbreviated New Drug Application containing a paragraph IV certification to a listed patent are protected from competition from other generic versions of the same drug for the 180 days. As of December 2016, no other generic equivalents to Evra have been introduced.
The FDA has maintained, in spite of the wording in the labeling for Evra, which has been discontinued, and its approved branded generic, that none of the epidemiologic studies to date provides a definitive answer regarding the relative risk of VTE with Evra compared to combined oral contraceptive use or whether the increased risk that some studies demonstrated is directly attributable to Evra. An advisory committee for the FDA stated that the benefits of Evra outweigh the risks. In its denial of a Citizen's Petition calling for the withdrawal of Evra, the FDA followed the committee's recommendations stating that the increased VTE risk does not warrant removal from the market, and that the labeling revisions to the Evra label provide a sufficient update and guidance on the interpretation of the epidemiologic data about the risk of VTE with Evra. In spite of the labeling changes, and Johnson & Johnson ceasing promotion of Evra in 2007, Evra and its generic equivalent generated $211 million in gross sales in 2016.continue to generate significant sales.
With its approval on February 14, 2020, Twirla is now the only other transdermal contraceptive patch approved by the FDA. We believe that the rapid uptake and acceptance of Evra upon its introduction demonstrates that there is an unmetand its (and Xulane's) continued sales over the past several years demonstrate a market needopportunity for amultiple choices in transdermal patch as a contraceptive option. Also, the epidemiologic data on VTE risk suggest that there is a need for a contraceptive patch that delivers both a low dose of EE similar to oral contraceptives and a first or second generation progestin.patches.
Our Product CandidatesTwirla Potential Market Share
EachThree of our market research studies have included an allocation exercise to estimate the potential uptake of Twirla and peak market share. In all of these studies, ObGyns and nurse practioners, or NPs, indicated their allocation of contraceptive prescriptions before and after reviewing a product candidates utilizesprofile like Twirla that reflects the safety and efficacy results from our proprietary Skinfusion technology,SECURE clinical trial. In the 2010 study, which is designed to provide advantages over the currently available patch. Skinfusion is designed to deliver contraceptive levels of hormoneswas conducted prior to the blood stream throughimplementation of the skin overACA, ObGyns estimated use of a seven-day period. It is also designed to optimize patch adhesion and patient wearability. Our lead product candidate islike Twirla a prescriptionin 17% of their CHC patch which contains both EE and LNG and is designed to deliver a low dose of EE and LNG comparablepatients. A proprietary calibration model developed by the research firm was applied to the total dose delivered with low-dose oral contraceptives.peak share estimate, to adjust for physician overstatement, resulting in an estimated peak market share of 9% of the CHC market. In addition to Twirla, we plan to develop a pipeline of other new transdermal contraceptive products, including AG200-SP, which is a regimen designed to provide shorter, lighter periods; AG200-ER, which is a regimen designed to allow a woman to extend the length of her cycle; AG200-ER (SmP), which is a regimen designed to allow a woman to extend the length of her cyclestudy completed in December 2016, ObGyns, NPs, and experience shorter, lighter periods; and AG890, which is a progestin-only contraceptive patch intended for use by women who are unablephysicians assistants, or unwilling to take estrogen. AG200-SP, AG200-ER, and AG200-ER (SmP) are intended to be Twirla line extensions that would expand thePAs, estimated use of Twirla beyond its initial, approved use. In July 2016, we began preparationsin 22% of their CHC patients, which was also calibrated to adjust for overstatement, resulting in an initial Phase 2 clinical trial examiningestimated peak market share of 14% of the CHC market. This estimate was confirmed in our most recent study completed in September of 2019, in which ObGyns and NPs/PAs estimated use of AG200-SP along with a smaller lower-dose combination ethinyl estradiol/levonorgestrel patch (SmP)Twirla in the fourth week20% of their CHC patients, calibrated to 14% of the woman's cycle. The Phase 2 clinical trial is aimed at identifying the optimal dose of the SmP, and will evaluate bleeding profiles, pharmacokinetic parameters, ovulation inhibition and safety over three cycles of treatment with AG200-SP (SmP).CHC market.
We have decided to postpone the trial and will continue to evaluate the timing for
initiating dosing of subjects for this Phase 2 clinical trial, which is dependent on financial and other capital resources.
The National Institutes of Health, through a clinical trial agreement with us, conducted a Phase 1/2 trial with AG890. The Phase1/2 study was a multicenter study to evaluate the pharmacokinetics, safety and mechanisms of potential contraceptive efficacy of AG890. The trial is complete and we continue to evaluate the findings. After we complete our evaluation, we may need to perform additional patch development work to determine the optimal formulation and dose to advance to Phase 3. Based upon a number of factors, including, but not limited to, our available capital resources and feedback from the FDA, we continue to review the clinical path and budgetary requirements for each of AG200-SP, AG200-ER and AG890.
Our current product candidate pipeline is summarized in the graphic below:

Substantially all of our resources are currently dedicated to developing and seeking regulatory approvalcommercial opportunity for Twirla. We will require additional capital to advance the development of our other product candidates.
Twirla Product Overview
Twirla is a CHC patch which contains both EE and LNG. Twirla is designed to address an unmet medical need for increased compliance and improved ease of use as compared to oral contraceptives. A single Twirla patch delivers the active ingredients LNG and EE over a seven-day dosing interval, and thereby eliminates the need to take a daily pill as is necessary with an oral contraceptive. Twirla uses a traditional 28-day contraceptive regimen, where one patch is applied weekly for three consecutive weeks and then there is a fourth, patch-free week in each 28-day time period. Twirla may be applied to the buttock, abdomen or upper torso, but not the breast. In clinical trials reported to date, women most frequently chose the buttock and abdomen for patch placement. The exact patch location needs to be rotated with each patch change. Twirla has demonstrated a therapeutically equivalent pharmacokinetic profile when worn on the buttock, abdomen or upper torso. A drug's pharmacokinetic
profile refers to the specific way in which a given drug is handled by the body over time, reflecting the particular patterns of absorption, distribution and elimination of the drug in the body.
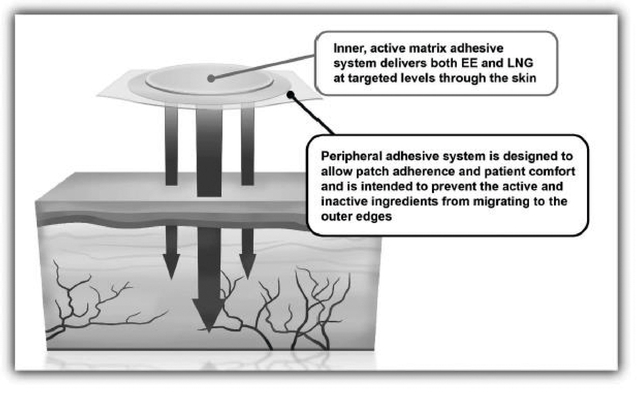
Twirla is designed to be highly appealing to patients as a method of contraception. The patch is round and made of a soft, flexible, silky fabric, designed to flex with the movement of a woman's body. Twirla is a matrix patch consisting of several layers of material that contain the active ingredients EE and LNG, as well as the inactive ingredients Dimethylsulfoxide, Ethyl Lactate, Capric Acid and Lauryl Lactate, which are ingredients to assist in the transport of EE and LNG across the skin, and adhesives that enable adherence to the skin. The final top layer is the one seen on the skin, and consists of a thin, silky material consisting of only adhesive. There is a barrier formed between the inner portion of the patch, which contains the active ingredients, and the outer portion of the patch, which only contains the adhesive. This barrier is intended to prevent the active and inactive ingredients from migrating to the peripheral portion of the patch, and from breaking down the adhesive in that portion of the patch. Twirla is also designed to help prevent seepage of the adhesives from around the edge of the patch where it could collect dirt and leave a sticky black ring on the skin. The six layers of the patch are integrated to create a patch which has a slim profile, and is unobtrusive when applied. The results of multiple clinical trials suggest that Twirla delivers the active ingredients needed for contraception over a
seven-day period and that it remains adhered to the skin of most subjects for the full seven-day period, even under conditions of heat, humidity, showering, exposure to water and vigorous exercise.
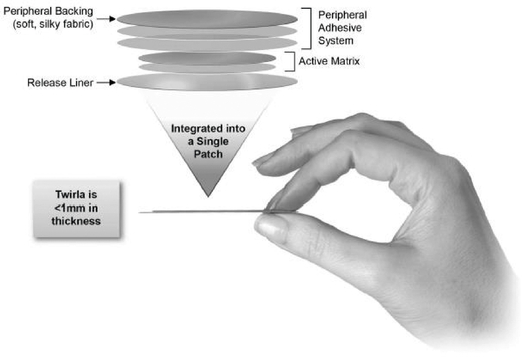
Twirla Patch Profile
The following table compares Twirla with the Evra product and its generic equivalent, Xulane, as stated in their labels, based upon publicly-available information regarding the products and the characteristics of Twirla and other Twirla attributes observed in our completed Phase 3 clinical trials. We have not performed a head-to-head comparison of Twirla to Evra.
| ||||
| ||||
| ||||
| ||||
| ||||
|
 |  |
Twirla employs our Skinfusion patch technology, resulting in a unique appearance and feel of the patch. Evra/Xulane does not utilize our Skinfusion technology; its active ingredients and adhesives are dispersed to its edges. One frequent complaint about patches that do not utilize Skinfusion is that they collect dirt and lint and may leave a sticky black ring of residue on the skin which can be difficult to remove. We do not have any direct comparison of the appearance of the patch on the skin at the end of seven days between Twirla and Evra/Xulane, but we believe, based on anecdotal feedback from our clinical trial investigators, as well as based upon the differences in the design of the patches, that Twirla may have an advantage in this regard.
We have not performed a head-to-head comparison of Twirla to Evra/Xulane, however, a pharmacokinetic study that we conducted with Twirla was similar in design to the pharmacokinetic study conducted with Evra that provided the information regarding the daily amount of EE delivered that is currently in the Evra/Xulane package insert. The figure below combines the results for average EE concentrations from these two studies, and suggests a comparison of the observed blood concentration of EE for Twirla versus Evra versus observed and estimated data for the pill. The lower amount of EE delivered from Twirla as compared to Evra can be observed. If Twirla is approved by the FDA, we will not be able to make direct comparative claims regarding the safety, efficacy or pharmacokinetics of Twirla and Evra/Xulane, since none of our completed clinical trials studied Twirla in a head-to-head comparison with Evra/Xulane.
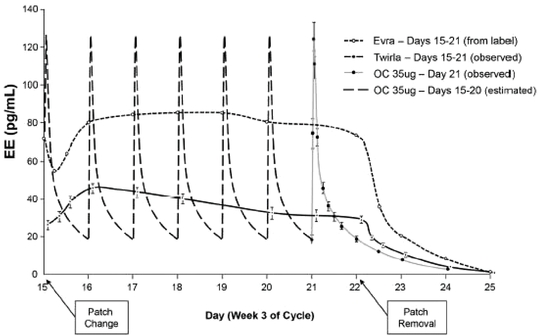
The Evra curve presented in the graphic above was estimated based on the graph provided in the Evra label. In the legend to the figure above, "OC" refers to an oral contraceptive containing 35 micrograms of EE. The OC data prior to Day 21 are estimated steady-state data based on Day 21 EE concentrations observed during our pharmacokinetic study.
Twirla contains LNG, which is the progestin used as the reference standard when comparing risk of VTE between progestins. Evra/Xulane contains the progestin norelgestromin, which is a prodrug of norgestimate, a second generation progestin that has not demonstrated an increased risk of VTE independent of EE. We do not expect any meaningful clinical differences between Twirla and Evra/Xulane based on the progestin component, but our market research with ObGyns has demonstrated that they perceive LNG to be one of the safest progestins available.
Twirla Product Profile
Assuming approval of our marketing application by the FDA based on the results of the SECURE trial, we believe the clinical trial data from the SECURE trial for Twirla will support our future marketing of Twirla as follows:
Twirla Clinical Development Program
Clinical Trials Completed prior to SECURE
Our clinical program includes three Phase 1 studies, one Phase 2 study, and three Phase 3 studies, as well as other supporting studies. In December 2016, we completed our third Phase 3 clinical trial, SECURE, in response to FDA comments and guidance. In Phase 1 and Phase 2 clinical trials, we demonstrated that Twirla delivers levels of both EE and LNG to the blood stream that are consistent with currently marketed low-dose oral contraceptives. In our Phase 3 clinical trials completed prior to SECURE, we demonstrated that Twirla was comparable to an approved low-dose oral contraceptive in two randomized studies, one that enrolled over 1,500 women over 12 months and the other that enrolled over 400 women over six months. Across all completed clinical trials, Twirla was generally well-tolerated and had a favorable safety profile. Because we relied, in part, on the FDA's findings of safety and efficacy from investigations for approved products containing EE and LNG and published scientific literature for which we have not obtained a right of reference, we were not required to conduct preclinical studies. In the pharmacokinetic study comparing Twirla to an approved low-dose oral contraceptive, results demonstrated that Twirla delivers a daily dose of EE that results in estrogen exposure similar to low-dose oral contraceptives containing approximately 30 micrograms.
Our two Phase 3 trials completed prior to SECURE enrolled over 1,900 subjects to evaluate the safety and efficacy of Twirla. Each of these studies included an active comparator arm with an approved low-dose oral contraceptive. The results of these studies demonstrated that Twirla was generally well-tolerated, with levels of adverse events generally comparable to those of low-dose oral contraceptives. In these studies, subjects had a higher rate of self-reported compliance when using the patch as compared with the group using oral contraceptives. However, as discussed further below, the FDA issued a CRL in response to our marketing application for Twirla and requested an additional Phase 3 study and additional chemistry manufacturing and control, or CMC, information. The results of our prior clinical trials demonstrated that approximately only 3% of patches became completely detached from the skin of subjects during the seven-day period, and that the patch generally remained adhered to the skin even when exposed to normal daily activities and conditions such as showering, swimming and other forms of exercise, heat and humidity.
More specifically, our safety population included subjects who received at least one dose of Twirla or COC. In the combined safety population of our two Phase 3 trials completed prior to SECURE, there were a total of 22 serious adverse events, or SAEs, of which 16 were from the Twirla cohort, which had approximately 2.3 times as many subjects as the oral contraceptive comparator cohort. Three of these SAEs (0.2% of the overall Twirla safety population) were considered to be possibly related to the study drug and included one drug overdose with Benadryl®, one case of uncontrollable nausea and vomiting and one instance of upper extremity deep vein thrombosis. In addition to the SAEs described above, some subjects taking Twirla experienced non-serious adverse events, such as nausea, headache, application site irritation and breast tenderness. Subjects receiving the oral contraceptive comparator also generally experienced similar non-serious adverse events such as nausea, headache, and breast tenderness, though at different rates. We believe that Twirla will have a label consistent with all
marketed low-dose CHC products, which include class labeling that warns of risks of certain serious conditions, including venous and arterial blood clots, such as heart attacks, thromboembolism and stroke, as well as liver tumors, gallbladder disease and hypertension, and a black box warning regarding risks of smoking and CHC use, particularly in women over 35 years old who smoke.
In our Phase 3 trials, the primary measure of efficacy is the Pearl Index, or PI, which is calculated based on the number of observed on-treatment pregnancies and total number of on-treatment cycles during the study. Specifically, the PI is expressed as the number of pregnancies per 100 WY of use. The pooled PI value in the previously completed Phase 3 trials for the Twirla patch was 5.76 and for the combined oral contraceptive control arms was 6.72, which were higher than the range of 1.34 to 3.19 in pivotal studies conducted on products approved by the FDA in the previous ten years. In addition, the upper bound of the associated confidence intervals were higher than those seen in clinical trials used for registration of other approved hormonal contraceptives.
We believe that the results for bothpotential new CHC users who are within Twirla's approved indication represent a significant population of women. Based on the patchCompany's market research, analysis of the current and oralexpected future U.S. contraceptive control armsmarket, and review of other product launches in the two Phase 3 trials completed priorcategory, the Company estimates that Twirla can potentially achieve a peak market share of 5-8%. As we prepare for the commercialization of Twirla, we will continue to SECURE were affected primarily by issues with study conduct at several study sites, including rapid enrollment which led to inability to manageanalyze the study population, poor subject compliance,contraceptive market and high rates of loss to follow-up. In the larger ofupdate our Phase 3 trials completed prior to SECURE, 96 sites enrolled subjects, 60 of which had no on-treatment pregnancies. Nineteen percent of the on-treatment pregnancies reported during this trial came from one site. This site represented approximately 8% of the randomized subject population. Thirty six percent of on-treatment pregnancies were reported at four of the 96 sites. These four sites represented approximately 15% of the randomized subject population.
Experts agree that the characteristic most likely to impact contraceptive failure and pregnancy rates is the subject's likelihood of using a method inconsistently or incorrectly. Consistent with expert opinions, our analyses have suggested that the resultsmarket research for both the patch and oral contraceptive control arms in the two Phase 3 trials completed prior to SECURE were also affected in part by the study population, which comprised a disproportionately high number of new users and minority subjects, known to be at higher risk of noncompliance and pregnancy, as compared to the majority of other recent CHC clinical trials which have gained approval in the United States.
Individuals who immediately switch from one hormonal contraception method to another, referred to as current users, or who have recently used another method of hormonal contraception, are less likely to experience contraceptive failure than a new user because they are less likely to have inconsistent or incorrect use. These experienced subjects are often selected for trial participation because their inclusion will lower failure rates. Indeed, many contraceptive trials have enrolled a high proportion of these subjects. Direct comparisons across multiple trials are limited by differences in study design and population, as well as differences in definitions of user status; however, as shown in the table below, some comparisons are possible. For example, when compared against trials that captured current hormonal contraceptive use, in the larger of our two Phase 3 trials completed prior to SECURE, we had a lower proportion of subjects randomized to receive Twirla that were current users, only 17.8%, reflecting a population with less experience using hormonal contraception, compared to two recently approved hormonal contraceptives. When compared against trials that categorized subject experience more broadly by their use of hormonal contraception within the 6 months prior to enrollment, our trial also had a lower proportion of experienced subjects, only 44%. In both the COC and Twirla groups, new users had approximately three times the rate of noncompliance compared to experienced users, as verified through blood tests revealing non-detectable blood levels of EE and LNG. Similarly, the pooled PI values from our two Phase 3 trials completed prior to SECURE were more than twice as high among new users compared to experienced users, and in the primary efficacy analysis population there were no pregnancies observed in current users of other hormonal contraception who immediately switched to the patch upon entry into the trial.
In addition, our two Phase 3 trials completed prior to SECURE also included a higher proportion of black and Hispanic subjects than most recent hormonal contraceptive trials. Although the underlying reasons are not well-understood, several articles in medical journals, such asContraception and theAmerican Journal of Obstetrics & Gynecology, and in at least one report by HHS, state that contraceptive failure rates are highest in black and Hispanic subjects. In our two Phase 3 trials completed prior to SECURE, rates of laboratory-verified noncompliance were substantially higher in blacks and Hispanics compared to non-Hispanic white subjects in the larger of our Phase 3 trials, and as shown in the table below, there were substantially higher PI values in the black and Hispanic subpopulations than in non-Hispanic white subjects. Additionally, as shown in the table the observed PI values were more dramatically increased for new users who were also black or Hispanic.
Study Population Demographics in Selected Contraception Trials
| | | Contraceptive Product (Year of Approval) % of subjects in category* | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Parameter | | Twirla | Seasonique (2006) | Yaz (2006) | Lo- Seasonique (2008) | Natazia (2010) | Quartette (2013) | ||||||||||||||
Hormonal contraception use | |||||||||||||||||||||
Current Users | 18 | (a) | — | 60 | (b) | — | 59 | (c) | — | ||||||||||||
Within 6m of enrollment | Yes(d) | 44 | 68 | — | 61 | — | 44 | ||||||||||||||
| No(e) | 56 | 32 | — | 39 | 56 | ||||||||||||||||
Race/ethnicity | Hispanic | 15 | 5 | 5 | 10 | 13 | 11 | ||||||||||||||
| Black | 22 | 11 | 4 | 12 | 7 | 18 | |||||||||||||||
Current user definitions (extrapolated for approved products):
Use within 6 months of enrollment definitions:
Twirla Pearl Indices Stratified By New Users and Minority Subjects
| ||||||
| ||||||
| ||||||
CRL and FDA InteractionsCommercialization Strategy
In February 2013,January 2018, following our receipt of the complete response letter, or CRL, we received a CRL from the FDA indicating that the results from our two completed Phase 3 trials would not be sufficient for approval, and the FDA proposed that we conduct an additional Phase 3 trial. Among the comments expressed in the letter were some regarding the PI values seen in the studies. Specifically, the FDA indicated that the PI values and the upper bound of the associated confidence intervals in the studies, in both the subjects using the Twirla patch and the control arm using oral contraceptives, were higher than seen in clinical trials used for registration of other approved hormonal contraceptives. The confidence interval is a range around a measurement that conveys how precise a measurement is. The FDA recommended that we conduct an additional Phase 3 trial with a simplified clinical trial design and improved study conduct, including site monitoring and data collection procedures. The FDA also requested that we study Twirla in a representative sample of U.S. women who are seeking hormonal contraception, without enrollment restrictions based on demographic characteristics such as contraceptive user status, age, race, ethnicity, and body mass index, or BMI. The FDA also required additional information relating to the laser etching of label information on each patch and required that the patch used in the new trial utilize the same etching as will be used for the commercial product, in order to demonstrate that it does not adversely affect the performance of the patch. Furthermore, the FDA also requested in the CRL additional information on controls and release specifications related to the patch, and manufacturing and control information related to the Drug Master File of one of the raw materials in Twirla.
In October 2013, we met with the FDA and received further guidance on requirements for our planned Phase 3 trial. In addition, we had a follow-up written interaction with the FDA in February 2014 and have received substantial written comments from the FDA in subsequent interactions. We enrolled the first subject in the SECURE clinical trial in the third quarter of 2014, and completed the clinical trial in December 2016. The patches studied in the SECURE trial were laser etched using the same process as2017, or 2017 CRL, we anticipatesignificantly scaled back our preparations for commercialization of Twirla, if approved. Weincluding commercial pre-launch and manufacturing validation activities. With the approval of Twirla, we have continuedbegun to accelerate our commercial activities. In the first quarter of 2020, we plan to
interactinitiate work with managed care and patient payors to gain market access for Twirla. In the FDA on its CMC questionssecond quarter of 2020, we plan to begin hiring and continued additional supportive testing in ordertraining an initial sales team, which we estimate will consist of 70 to respond100 persons. At the same time, we are currently preparing to initiate the FDA's CMC questions.
The SECURE trial,validation of our third Phase 3 Clinical Trial
SECURE, our third Phase 3 clinical trial, was a multicenter, single-arm, open-label, 13-cycle trial that evaluatedcommercial manufacturing process and expect to complete the safety, efficacyvalidation process and tolerability of Twirla in 2032 healthy women, aged 18 and over, at 102 experienced investigative sites across the United States. The design and execution of SECURE was intendedcommence distributing product to address a number of issues identified in the CRL, including but not limited to, improved clinical trial conduct and demonstration of efficacy as measured by an acceptable Pearl Index and related 95% confidence interval in a representative sample of U.S. women who are seeking hormonal contraception, without enrollment restrictions based on demographic characteristics, such as contraceptive user status, age, race, ethnicity, and BMI. The trial was designed in consultation with the FDA, and comprised a number of stringent trial design elements, including exclusion of treatment cycles not only for use of back-up contraception but also for lack of sexual activity. SECURE had broad entry criteria, placed no limitations on BMI or other demographic factors during enrollment, and enrolled a large and diverse population from the United States in order to allow for efficacy to be assessed across different groups, as requested by the FDA. These entry criteria resulted in the inclusion of a substantial number of women with high BMI, who have frequently been under-represented in past contraceptive studies. The efficacy measure for SECURE was the Pearl Index in an intent-to-treat population of subjects 35 years of age and under. The FDA also requested inclusion of pre-specified efficacy analyses related to BMI and body weight.
We began enrollment for SECUREwholesalers in the fourth quarter of 20142020. We will need to raise additional funds to complete these activities, and completedour ability to complete such activities according to our current planned timelines will depend on our ability to successfully raise the clinical trialnecessary capital.
Twirla Promotion Strategy
We have a limited number of sales and marketing employees. We plan to expand our commercial team, but will primarily rely on third-party agencies with experience in December 2016. In January 2017, we announcedcommercializing pharmaceutical products to advance the following highlightscommercialization of the SECURE clinical trial top-line results:
BMI Category | BMI (kg/m2) | % of Trial Population | Pearl Index | Upper Bound of 95% CI | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
Normal | < 25 | 39 | % | 3.03 | 4.62 | |||||||
Overweight | 25 - < 30 | 25 | % | 5.36 | 7.98 | |||||||
Obese* | ³ 30 | 35 | % | 6.42 | 8.88 | |||||||
Non-Obese* | < 30 | 65 | % | 3.94 | 5.35 | |||||||
Obese* | ³ 30 | 35 | % | 6.42 | 8.88 | |||||||
The Pearl Index for subjects by minority and ethnicity status was as follows:
Race/Ethnicity | % of Trial Population | Pearl Index | Upper Bound of 95% CI | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
White (not Hispanic) | 66.9 | % | 4.63 | 6.23 | ||||||
African-American | 24.3 | % | 4.05 | 6.69 | ||||||
Hispanic | 19.7 | % | 2.70 | 5.06 | ||||||
Adverse Event | SECURE Trial | Prior Agile Phase 3 Trial* | Ortho Evra Trials** | Quartette Trial** | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Total in Safety Population | 2032 | 1043 | 3322 | 3597 | |||||||||
Headache | 4.3 | % | 3.7 | % | 21.0 | % | 12.2 | % | |||||
Nausea | 4.1 | % | 4.3 | % | 16.6 | % | 6.7 | % | |||||
Breast tenderness/pain/discomfort | 2.0 | % | 1.8 | % | 22.4 | % | 2.2 | % | |||||
Mood swings/changes/depression | 2.7 | % | 2.8 | % | 6.3 | % | 2.9 | % | |||||
Heavy/irregular vaginal bleeding*** | 1.8 | % | 2.1 | % | 6.4 | % | 9.7 | % | |||||
commercial managed care plans. We believe that we can deploy a focused sales force effort targeting the efficacy results observed in SECURE were a reflectionObGyn, NP and PA prescribers who are responsible for approximately 70% of branded CHC prescriptions. In areas of the study populationcountry where it is not efficient to deploy a sales representative, remote promotion can be used to reach these prescribers.
We plan to use both branded and unbranded campaigns to create awareness of Twirla and available contraceptive options among consumers. We believe there are cost-effective means to reach our target demographic of females aged 18 to 34 years, who tend to engage in online activities to a high degree and are more likely to seek health information online and through social networks. Traditional mass-market direct-to-consumer advertising on television may not be required to reach these consumers. Marketing tactics aimed at today's female consumer need to be optimized for mobile technology because smartphones and text messaging are the clinical trial design. As recommended by the FDA, we had broad entry criteriapreferred means of communication. We believe that a focused consumer promotion plan that uses digital media, potential social media advertising, and other mass-market advertising vehicles will generate consumer awareness and demand for the trialTwirla.
Twirla Coverage and placed no limitations on BMI or other demographic factors during our enrollment. These entry criteria resultedReimbursement Strategy
We initiated research with managed care and patient payors in the inclusionfourth quarter of 2019 to ensure we have a substantial numberthorough understanding of womenthe management of the contraceptive category. In the first quarter of 2020 we will continue to monitor competitive activity, assess the most probable formulary positions with overweightidentified target accounts and obese BMI, who have frequently been under-representedprepare for commercialization, including making limited company and portfolio presentations to payors within FDA guidelines. Also, in past contraceptive studies. As noted above,the first quarter of 2020, we observedwill begin to meet with formulary decision makers as appropriate to rapidly secure positions for Twirla that BMI had an effect on the efficacy resultsminimize access barriers for Twirla.prescribers and patients. We believe these observations require further analysisthat it is important in this category for women to have equal access to all methods, dosing regimens and hormonal options so that they and their provider can select the choice that is the most appropriate to meet their lifestyle and family planning goals.
Topline Summary of whether there are other important factors at work here, such as race/ethnicity, user profile and compliance rates, which we believe may have impacted the results of our prior Phase 3 studies.
Several scientists at the FDA published a paper in 2015,"Effect of Obesity on the Effectiveness of Hormonal Contraceptives: an Individual Participant Data Meta-Analysis,"Our Managed Care Market Research: which focused on the issue of obesity and effectiveness of hormonal contraceptives (HC) by showing that obesity may increase the risk of unintended pregnancy in women using HC. The FDA's Individual Participant Data meta-analysis of pivotal Phase 3 clinical trials of combination hormonal contraceptives suggested a 44% higher pregnancy rate during use of combined oral contraceptives for obese women after adjusting for age and race. The authors of the paper highlighted the limitations of currently available prospective data due to historical exclusion of obese women from contraceptive studies, calling for more data and additional analyses on obese women from Phase 3 clinical trials to allow further assessment of the effect of weight on the probability of unintended pregnancy in women using HC. We believe our results from the SECURE clinical trial are consistent with the conclusions from the paper by the FDA scientists.
Additionally, the observed PI values were not only impacted by the number of pregnancies that occurred in the study, but also by the number of cycles included in the analysis, which affects the denominator of the PI calculation. Cycles in which a subject was not sexually active, or in which a subject used a back-up method of contraception were not counted toward the number of cycles included in the calculation of the PI. Many contraceptive clinical trials have not historically included these stringent requirements, in particular the exclusion of cycles for lack of sexual activity, in the clinical trial design. As a result, we believe that the SECURE results reflect evidence of efficacy in a real-world population.
The highest PI for a hormonal contraceptive product approved by the FDA is 3.19managed care research summarized below was conducted with medical and the highest upper-bound of the 95% confidence interval of 5.03. As with all products, ultimate approvability of a hormonal contraceptive is based on a risk/benefit assessment of the overall safety and efficacy profile of a product, not only a specific Pearl Index. For hormonal contraceptive trials, the FDA generally evaluates safety and efficacy results of each individual studypharmacy directors in the unique contextfourth quarter of the study2019. In regard to forward-looking questions, subjects were asked to
populationassume that the ACA and trial design. PIsContraceptive Mandate would still be in effect. Our recent managed care research found the following:
In SECURE, we employed several measures to improve study conduct and, in particular, improve upon the loss to follow up rate. These measures included selecting a highly experienced contract resource organization, or CRO, selecting experienced sites, increasing and improving monitoring and training, and the use of electronic diaries for subjects. We engaged Parexel International Corporation, or Parexel, a CRO with substantial experience in contraception studies and excellent site monitoring capabilities, as the CRO for the SECURE trial. We actively participated in site selection and in monitoring subject recruitment, and actively participated in site monitoring and oversight of Parexel's activities throughout the length of the trial.
Regarding site selection, the SECURE trial was conducted at 102 experienced sitesalthough there were many options in the United States. Sites were evaluated extensively throughcategory, there was a data-driven approach assessing performance on previous clinical trials, staffing with experienced study coordinators, demographics of potential study subjects,need for improvement around safety, tolerability, and audit history. We also focused on ongoing training of study coordinators at the investigator meeting and study initiation visits, at coordinator's meetings, and through ongoing communication. In addition, study sites that showed early trends toward higher rates of loss to follow-up or overall poor study management were re-trained. We also focused closely on subject recruitmentimprovement in order to achieve our goal of a population intended to provide reliable and generalizable data in the SECURE trial. We trained our sites to provide individualized attention to recruitment of subjects who were most likely to adhere to the study protocol with respect to compliance, including correct patch application, timing of patch removal and replacement, electronic diary, or e-diary, completion and study visits. Subjects were also advised through the informed consent process that noncompliance with study procedures may lead to discontinuation from the trial. Enrollment of the SECURE trial was completed in October 2015.
A number of measures were also put in place in order to facilitate compliance with study procedures. To ensure subjects were adequately educated regarding their responsibilities during the trial, a detailed subject teaching plan, along with materials, was developed and implemented, and subject education regarding the importance of compliance, including videos, brochures and one-to-one education with study coordinators, was provided at repeated intervals throughout the study. A number of measures were put in place to support and monitor compliance through the study. One key measure was the use of text message technology, which provided reminders to subjects for patch application, diary completion and study visits. Phone contact with subjects between visits was also part of the study protocol, which increased the frequency of contact with subjects throughout the study. Subjects with consistent poor compliance despite re-education by site personnel were discontinued from the trial.
An independent Pregnancy Review Committee composed of experts in early ultrasound was selected to review all pregnancies and determine on or off-treatment status, which affects the numerator of the PI calculation. Accurate and timely pregnancy adjudication is critically important in order to reduce the likelihood that pregnancies which occur off treatment will be included by the FDA during the review process. In order to avoid pre- or post-treatment pregnancies being included, every pregnancy was assessed via ultrasound as soon as possible and full data was collected regarding the relationship of the pregnancy to the subject's use of Twirla. We did not have an independent Pregnancy Review Committee for our previous clinical trials.
We plan to submit a complete response to our CRL that includes the additional clinical trial results and additional information relating to the manufacture of Twirla in a resubmission of our NDA for Twirla to the FDA in the first half of 2017.
Our Pipeline: Twirla Line Extensions and OtherPotential Product Candidates
In additionTwirla is our first and only approved product, and substantially all of our resources are committed to the manufacturing validation and commercialization of Twirla. We have halted all further work on our pipeline. We will require additional capital to conduct required post-market studies of Twirla and, should we choose, to advance the development of Twirla line extensions and our potential product candidates.
Our potential product pipeline consists of two classes of product candidates: Twirla line extensions and other transdermal contraceptive product candidates. These potential product candidates are designed to address market needs and offer additional non-daily contraceptive options. Based on the results of our market research on lineonline extension regimen concepts conducted in December 2016, we believe that our potential line extension product candidates aremay be commercially viable and could garner a share of the contraceptive market.
The current status of our potential product candidate pipeline is summarized in the graphic below:
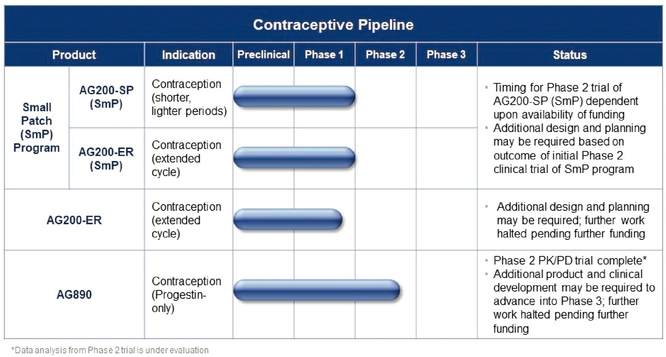
The hormonal contraceptive market has a long history of manufacturers successfully using line extensions to extend the lifecycle of a brand, often by gaining additional exclusivity periods for the product extension under the provisions of the Hatch-Waxman Act and/or with additional patents. Our lifecycle strategy with Twirla is to introduce line extensions that will have exclusivity for some time period, either due to our intellectual property estate, or due to Hatch-Waxman exclusivity. The line extensions in our pipeline include using our Skinfusion technology to allow a 28-day regimen where women will experience shorter, lighter withdrawal bleeding, as well as extending the cycle beyond the typical 28-day regimen to allow women to experience fewer withdrawal bleeds each year. In addition, the potential line extension product candidates in our pipeline will utilize a unique aspect in the regimen, where a smaller patch, or SmP, that delivers a lower dose of both EE and LNG will be worn during the final seven days of each cycle, rather than having a patch-free week, to allow for withdrawal bleedbleeding while minimizing hormonal fluctuations and potentially the side effects that accompany changes in hormone levels. These regimens are protected by patents issued to us in 2015. A study to examine the pharmacokinetics and pharmacodynamics of the SmP will be required prior to advancing the potential line extension product candidates through clinical development. In July 2016, we began preparations for an initial Phase 2 clinical trial examining the use of AG200-SP along with a smaller lower-dose combination ethinyl estradiol/levonorgestrel patch (SmP) in the fourth week of the woman's cycle. We have decided to postpone the trial and will continue to evaluate the timing for initiating dosing of subjects for this Phase 2 clinical trial, which is dependent on financial and other capital resources.
Our Twirla line extensions include the following:
Our other potential product candidate is a P-only contraceptive patch described below:
P-only market consists of pills and several non-oral options, including IUDs, implants, and injections. AG890 is intended to fulfill an unmet medical need for a non-daily, easily reversible form of contraception in the P-only market. We have conducted a Phase 1 clinical trial with AG890. In addition, the National Institutes of Health, through a clinical trial agreement with us, conducted a Phase 1/1/2 trial with AG890. The Phase 1/1/2 study was a multicenter study to evaluate the pharmacokinetics, safety, and mechanisms of potential contraceptive efficacy of AG890. The trial is complete, and we continue to evaluate the findings. Once we have completed our analysis of the data, it is possible that additional patch development work for dose selection may be required, including additional Phase 1 and Phase 2 studies to determine the optimal formulation and dose to advance to Phase 3.
We do not expect to be required to conduct preclinical studies for any of these potential product candidates. Based upon a number of factors, including, but not limited to, our available capital resources and feedback from the FDA, we continue to review the clinical path and the budgetary requirements for each of these three product candidates. Substantially all of our resources are currently dedicated to developing and seeking regulatory approval for Twirla. We will require additional capital to advance the development of our otherpotential product candidates.
Sales and Marketing
Twirla Commercialization Strategy
We expect to build a sales and marketing infrastructure in the United States to support the launch of Twirla for contraception, if approved. We anticipate that a targeted sales force focused initially on ObGyns, NPs, PAs and primary care providers who comprise the top prescribers of contraceptives will be highly effective. Outside the United States, in the future we may decide to commercialize Twirla, if approved, by entering into third-party collaboration agreements with pharmaceutical partners. We will require additional capital for the commercial launch of Twirla.
Twirla Promotion Strategy
We have employed several key strategies during the development of Twirla to prepare us for the launch of Twirla. These include:
Prescribing in the CHC category is primarily driven by ObGyns, who write 43% of the total prescriptions. In addition, NPs and PAs, who are often affiliated with an ObGyn practice but can also be in a primary care setting, write 29% of all CHC prescriptions. The ObGyns, NPs and PAs combine to write nearly 72% of total CHC prescriptions. In addition, 34% of all prescriptions written by ObGyns are for contraceptives. We plan to focus the promotion of Twirla on these key prescribers and other key customer groups, including consumers and commercial managed care plans. We believe that we can deploy a focused sales force effort targeting the approximately 22,000 prescribers responsible for 80% of branded CHC prescriptions. We believe that this universe of branded prescribers can be covered adequately by a specialty sales force of between 70 and 100 total representatives. In areas of the country where it is not efficient to deploy a sales representative, remote promotion can be used to reach these prescribers.
We plan to deploy patient promotion at the launch of Twirla, both in the physician's office, and through targeted media campaigns. We plan to use both branded and unbranded campaigns to create awareness of Twirla among consumers. We believe there are cost-effective means to reach our target demographic of females aged 18 to 34 years, the so-called Millennials, who are more likely to seek health information online and through social networks. Traditional mass-market direct-to-consumer advertising on television may not be required to reach these consumers. Marketing tactics aimed at today's female consumer need to be optimized for mobile technology, because smartphones and text messaging are the preferred means of communication. Millennials also engage in online activities to a high degree. For example, approximately 80% use a social network and approximately 40% read blogs. We believe that a focused consumer promotion plan that uses digital media and other mass-market advertising vehicles will generate consumer awareness and demand for Twirla if approved.
Managed care plans have traditionally used differential co-pays to attempt to drive patients to use either generic products or products for which they have a contract with the manufacturer. Many plans encourage patients to obtain their branded contraceptives through mail-order, incentivizing them with a 90-day co-pay that is often less on a per-month basis than that for a 30-day supply. Most manufacturers of contraceptive brands offer a coupon to patients covered by non-governmental payors to offset the difference in co-pay between a generic and Tier 2 or Tier 3 for their promoted brands. These co-pay coupons are a useful tactic to overcome barriers to initiating therapy in such patients. When used in conjunction with product samples given out by the physician, a co-pay coupon often allows the patient to then fill their first prescription for free or at a steep discount, and limits the out of pocket expenditure for the patient for several months. This co-pay assistance creates brand loyalty, particularly for a brand where there is no generic alternative. We believe that we will be able to use free product samples and co-pay coupons or vouchers at the time of Twirla's launch to gain use of the product by patients covered by non-governmental payors while we are negotiating contracts with select commercial health plans and awaiting formulary review. In addition, we believe the enactment of the ACA, and specifically the requirements for contraceptive coverage required by the ACA, provides a favorable managed care environment for Twirla. The ACA requires all insurers to provide at least one product in each of the 18 methods referenced in the FDA Birth Control Guide with no cost-sharing to the patient, including no co-pays, coinsurance, or deductibles. The FDA Birth Control Guide lists "Patch" as a unique method, therefore insurers must provide access to at least one contraceptive patch product with
no cost-sharing to the patient. Currently, there is only one other patch product available on the market, Xulane (the generic version of Ortho Evra), and we believe the safety and tolerability profile of Twirla, if approved, will be superior to that of Xulane. Therefore, we believe Twirla will be well-positioned to be the no-cost patch option on formulary, either based on its clinical profile, or based upon negotiated rebates and discounts. In addition, we expect to be able to provide co-pay assistance in the form of a coupon for patients on plans where Twirla requires a co-pay.
In March 2017, the U.S. Congress proposed legislation, which, if signed into law by the new administration, would repeal certain aspects of the ACA. Further, on January 20, 2017, the new administration signed an Executive Order directing federal agencies with authorities and responsibilities under the ACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the ACA that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices among others. Congress also could consider subsequent legislation to repeal and replace elements of the ACA that are repealed. We will continue to monitor the healthcare reform efforts. While there is uncertainty about the specific effects of healthcare reform, we expect to be able to compete in either a managed care environment that maintains elements of the ACA that require contraceptive coverage or an environment that requires negotiated rebates and discounts.
Market Research
We have conducted market research with healthcare professionals (HCPs), consumers and managed care decision-makers to determine market drivers, unmet needs and the reaction to the Twirla product profile. A total of over 800 healthcare professionals and over 3,300 consumers have participated in our market research on Twirla and the contraceptive market. The main findings of the market research conducted in December 2016 are discussed below.
Topline Summary of Our ObGyn/NP Market Research:
Two of our more recent market research studies have included an allocation exercise to estimate the potential uptake of Twirla and peak market share. In both of these studies, ObGyns and NPs indicated their allocation of contraceptive prescriptions before and after reviewing a product profile like Twirla. In the 2010 study, which was conducted prior to the implementation of the ACA, ObGyns
estimated use of a product like Twirla in 17% of their CHC patients. A proprietary calibration model developed by the research firm was applied to the peak share estimate, to adjust for physician overstatement, resulting in an estimated peak market share of 9% of the CHC market. In the most recent study completed in December 2016, ObGyns and NP/PAs estimated use of Twirla in 22% of their CHC patients, which was also calibrated to adjust for overstatement, resulting in an estimated peak market share of 14% of the CHC market.
Even with the evolving healthcare landscape, we continue to believe a peak CHC market share of 9% can be achieved with Twirla within seven years of launch, allowing us time to establish a presence in the CHC market and to overcome any perceptions or barriers among prescribers due to the past history of Evra and to account for potential changes in the ACA and overall healthcare landscape.
Topline Summary of Our Consumer Market Research:
Topline Summary of Our Managed Care Market Research:
The managed care research summarized below was conducted with medical and pharmacy directors in September 2016. In regard to forward-looking questions, subjects were asked to assume that the ACA and Contraceptive Mandate would still be in effect.
Competition
The industry for contraceptive products is characterized by intense competition and strong promotion of proprietary products. While we believe that our Skinfusion technology provides us with a competitive advantage, we face potential competition from many different sources, including large pharmaceutical companies, specialty pharmaceutical and generic drug companies, and medical device companies. Any product candidates that we successfully develop and commercialize will compete with existing products and new products that may become available in the future.
We face competition from a variety of non-permanent birth control products. There are non-hormonal barrier methods, such as the contraceptive sponge, diaphragm, cervical cap or shield and condoms. Then, there are hormonal methods, which is the category for Twirla and our potential product candidates, such as oral contraceptives, injections, implants, hormonal IUDs and vaginal ring and transdermal contraceptive products.
The following table is the 2016 FDA Birth Control Chart, which outlines the 18 unique forms of birth control and compares the effectiveness of each method.
Although there are over 200250 CHC products currently available, including brands and generics, available on the market today, 50.5%just twelve branded products make up approximately half of the total market sales, or $1.97 billion in 2016, consisted of sales of just seven branded products.sales. Our potential competitors include large, well-established pharmaceutical companies, and specialty pharmaceutical sales and marketing companies. The productbranded products with the highest dollar sales in the CHCestablished market for the 12 months ending December 2016 waspresence include, Nuvaring®, marketed by Merck, the only contraceptive vaginal ring available on the market, with over $786 million in sales for 2016. Thethe Loestrin® franchise, marketed by Allergan (formerly known as Actavis), consisting of twothree oral contraceptives, Minastrin® 24, LoLoestrin® and LoLoestrin®Taytulla®, totaled approximately $898 million in sales in 2016. Other competing products include: Gianvi® and Quartette®Beyaz®, marketed by Teva, Beyaz® and Yaz®, which totaled over $178 million in sales in 2016,Yasmin® and Natazia® marketed by Bayer. Although not a branded product, Xulane, the branded generic to Ortho Evra and the only other patch currently available on the market, generated $211$297 million in sales for Mylan in 2016 for Mylan.2019. Additionally, several generics manufacturers currently market and continue to introduce new generic contraceptives, including Sandoz, Glenmark, Lupin, Amneal and Mylan. Based on the market experience of other non-oral CHC dosage forms, including Evra and Nuvaring, we believe there is a continuing demand for an innovative transdermal contraceptive patch that can provide convenience in a low-dose transdermal format.
There are other hormonal contraceptive products, recently approved or in development that may compete with Twirla and our other potential product candidates. Kyleena®Annovera™, a Bayer product,vaginal ring developed by the Population Council, Inc. and licensed for commercialization by TherapeuticsMD, was approved on August 10, 2018 and began distribution August 15, 2019. Slynd®, a progestin-only pill containing drosperinone, was introduced by Exeltis in September 2016, isAugust of 2019. The Population Council also has a hormonal IUD that releases a small amount of hormone to prevent pregnancy for up to 5 years. Also recently approved was Taytulla® from Allegran, which is the only oraltransdermal gel contraceptive in a capsule. CompaniesPhase 2, developed in collaboration with Antares Pharma, Inc. Two generics to Nuvaring were introduced in December 2019, EluRyng™ by Amneal and EVE-112 by Prasco Labs. Other companies that have new hormonal contraceptive products in various stages of development include Bayer, with a contraceptive patch an oral contraceptive and a P-only vaginal ring, eachboth in Phase 3 development. Allergan has a vaginal ring in development, which is a generic equivalent to Nuvaring, a P-only ring for which they received a CRL from the FDA, and an additional vaginal ring. This ring is inFDA. Mithra Pharmaceuticals SA announced Phase 3 development which isdata for a 12-month vaginal ring that was developed by the Population Councilcombination oral contraceptive in January 2019, and licensed this product to Mayne Pharma for usedistribution in the developing world.U.S. In the past few years, some of the large pharmaceutical companies such as Johnson & Johnson, Pfizer, and PfizerTeva have dissolved their women's health specialty marketing and sales teams, and Bayer has shifted their focus away from their CHC products to their IUD franchise.franchise, although they recently signed a license agreement with Dare Bioscience for U.S. commercial rights to Ovaprene, a hormone-free monthly contraceptive vaginal ring.
We are aware of only one other CHC transdermal patch in development. This patch is being developed by Bayer, and contains the active ingredients EE and gestodene, a third generationthird-generation progestin. Bayer has stated that their gestodene patch is small, round, and transparent, and delivers a daily EE dose comparable to a 20 microgram EE oral contraceptive. Phase 3 studies of the Bayer gestodene patch began in 2004, and they completed a Phase 3 efficacy trial in the United States in December 2010. Bayer also completed Phase 3 efficacy trials in the European Union, or E.U., and Latin America in September 2011, submitted a marketing application to the E.U. in September 2012, and received approval to market the gestodene patch in the E.U. in February 2014. At the time of the E.U. submission, Bayer reported that they were in talks with the FDA regarding a U.S. submission, but there has been no further public information regarding a U.S. submission or approval, and the most recent Bayer pipeline information does not list the gestodene patch.
To date, there are no contraceptives containing gestodene available in the United States. We are aware that Wyeth was developing oral contraceptives containing gestodene in the late 1980s, with ana new drug application, or NDA, filed for an oral contraceptive containing gestodene and EE in 1988, and Wyeth planned on filing an NDA for a second oral contraceptive containing gestodene in 1991. These products were never approved, and in a Wyeth pipeline report from 1996, there was no mention of any gestodene-containing product candidates among its contraceptives in development. Although not available in the United States, gestodene has been widely used outside the United States for a number of years. As with other third generation progestins, epidemiologic studies have reported a two-fold increase in risk of VTE with contraceptives containing gestodene compared to those containing LNG.
We believe that if Bayer were to obtain FDA approval for the gestodene patch, the approved labeling may contain the same language that products containing third generation progestins have, which states that these contraceptives have a two-fold increase in risk of VTE as compared with contraceptives containing second generation progestins.
Manufacturing
We do not own any manufacturing facilities.facilities and rely on Corium for all aspects of the manufacturing of Twirla. We, currently rely,along with Corium, have made a significant investment in a proprietary process to manufacture Twirla. We believe we have developed a robust process to reliably manufacture Twirla on a commercial scale. We believe that the technical challenges and expectknow-how involved in manufacturing, including proprietary chemistry, production to scale and use of custom equipment and reproducibility, present significant barriers to entry for other pharmaceutical companies who might potentially want to replicate our Skinfusion technology.
We will need to continue to rely, on a third partyinvest in the manufacturing process for Twirla, and incur significant expenses, in order to complete the equipment qualification and validation related to Corium's manufacturing capabilities in order to be capable of supplying projected commercial quantities of Twirla. In September 2019, we re-started manufacturing development at Corium. We are currently working with Corium to complete manufacturing development and process improvements and plan to commence pre-validation work when that work is complete. Our goal is to manufacture three validation batches of Twirla and complete the pre-validation and validation of the commercial manufacturing process consistent with our product candidates for clinical trials, as well as for commercial manufacture if anyapproved marketing application in the second half of our product candidates receive marketing approval.2020.
In 2006, we entered into an exclusive agreement with Corium International, Inc., or Corium to develop Twirla using our Skinfusion technology, and also for AG890, which is a P-onlyprogestin-only contraceptive patch in Phase 1/2 of clinical development. Our Corium agreement is an exclusive arrangement until Corium has commercially produced a significant, agreed-upon quantity of patches, currently projected to occur no earlier than five years following the commercial launch of Twirla. Pursuant to the terms of our agreement, Corium is required to use commercially reasonable efforts to maintain sufficient manufacturing capabilities to supply the quantities of Twirla required for its initial commercial launch and commercial sales thereafter. Corium needs to conductcomplete the equipmentvalidation of the commercial manufacturing process for Twirla and facility validation and expansion ofpotentially further expand its manufacturing capabilities in order to be capable of supplying projected commercial quantities of Twirla. In 2018 Corium was acquired by Gurnet Holding Company, or GHC. Following completion of the transaction, Corium became a private company, wholly owned by GHC. Corium has announced that it plans to continue its operations in Grand Rapids, Michigan, where Twirla if approved. Based on our interactions with the FDA on the CMC issues raised in the CRL and our plan with Corium to validate the commercial scale equipment to manufacture Twirla, we expect towill be able to address these issues in the resubmission of our NDA. We expect the validation and expansion to be completed in coordination with our planned commercialization activities. Corium is responsible for all aspects of Twirla manufacturing.commercially manufactured.
Strategic Agreements
Agreement with Corium
Pursuant to our manufacturing agreement, Corium's exclusive right to manufacture Twirla and AG890 extends until Corium has commercially produced a significant, agreed-upon quantity of patches, currently projected to occur no earlier than five years following the commercial launch of Twirla, at which point the agreement will expire. Under the terms of our agreement, we will pay Corium a defined price per finished patch, whether used for samples or commercial sale. We will owe no royalties to Corium in connection with the production of finished patches. The contract may be terminated by either party for the other party's uncured material breach. Following the end of the exclusivity period, if we were to seek a second source of supply, we would be required to obtain FDA approval through an NDA supplement for an additional manufacturing sites.site(s). The process of acquiring a second source of supply and obtaining FDA approval generally takes two years or more and would require us to make substantial investments in new facilities and equipment.
Under our agreement, Corium has performed process development and manufacturing of Twirla for each of our clinical trials. For the development work performed, we paid Corium for time and materials related to the achievement of certain development goals. To date, we have made approximately $1.7 million of milestone payments to Corium, all of which were paid between the years 2006 and 2009. Corium is not eligible for any milestone payments in the future. During 2012, we paid Corium an aggregate of $3.5 million towards leasehold improvements incurred by Corium to its facilities to provide for adequate manufacturing space for our product candidates.
In order to accommodate our anticipated commercial launch of Twirla, if approved, Corium has completed a substantial build-out of its facilities in Grand Rapids, Michigan, and it has installed over $10.0 million of equipment we purchased. This additional equipment and these facilities may require FDA pre-notification, pre-approval or inspection; however, we believe we can accomplish this expansion through an Annual Report filingWe plan to complete the validation of the commercial manufacturing process for Twirla NDA.in the second half of 2020.
Reimbursement
Managed care plans have traditionally used differential co-pays to attempt to drive patients to use either generic products or products for which they have a contract with the manufacturer. Typically, a
managed care plan's formulary is organized into between three and six tiers. Each tier is then associated with a set range of co-pay amounts or a percent of the drug costs with products in the lower tiers having a lower co-pay. ManyThe Patient Protection and Affordable Care Act (ACA) was signed into law on March 23, 2010. As of August 1, 2011, female contraception was added to a list of preventive services covered by the ACA that would be provided without patient co-payment. The federal mandate applied to all new health insurance plans encourage patients to obtain their branded contraceptives through mail-order, incentivizing them with a 90-day co-pay that may be less on a per-month basis than that for a 30-day supply. Contraceptive brands are generally placed on Tier 2 only if there is a contract with the plan, although there are a few plans that place several branded products on Tier 2.in all states from August 1, 2012.
Prior to May 2015, managed care plans have individually interpreted the requirement for coverage of contraceptives under the ACA. Some plans have designated that all contraceptives containing the same progestin are equivalent, and therefore only cover a select few products containing each progestin, usually the least expensive generics, with no co-pay. Other plans have defined contraceptive methods into categories such as "hormonal", "emergency contraception", and "barrier methods", and they cover just one product for each method with no co-pay. In May 2015, a clarification in the form of an FAQ was jointly issued by the applicable government agencies (HHS, DOL, and Treasury) which clarified the requirements for coverage of contraceptives under the ACA. The FAQ states that plans and issuers must cover without cost-sharing at least one form of contraception in each of the 18 methods the FDA has identified for women in its current Birth Control Guide. The patch is identified as a specific method in the FDA Birth Control Guide, and therefore insurers must cover at least one patch product with no cost-sharing to the patient. Because this clarifying guidance is applied for plan years (or in the individual market, policy years) beginning on or after 60 days from the date of publication of the FAQs, patients did not have had the benefit of this clarification until their new plan year, which generally started in January 2016.
In March 2017, the U.S. Congress proposed legislation, which, if signed into law by the new administration, would repeal certain aspects of the ACA. Further, onOn January 20, 2017, the new administration signed an Executive Order directing federal agencies with authorities and responsibilities under the ACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the ACA that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices, among others. Congress also could consider subsequent legislation to repeal and replace elements of the ACAACA. Additionally, in October 2017, the Department of Health and Human Services, jointly with the Department of Labor and the Treasury, issued two interim rules outlining exemption processes for employers not wanting to offer contraceptive coverage based on their religious beliefs or sincerely held moral convictions. While there is an injunction against the administration prohibiting it from implementing these rules, the ultimate outcome of that are repealed.litigation, which is currently in front of the Supreme Court, cannot be predicted. Therefore, it is difficult to determine the full effect of the ACA or any other healthcare reform efforts on our business.
Before the ACA was passed, many states had enacted contraceptive equity laws that required plans to treat contraceptives in the same way they covered other services. In addition, since the ACA was
passed, a number of states have enacted laws that basically codify in state legislation the ACA benefit rules (requiring all plans to cover, without cost-sharing, each of the 18 FDA-approved contraceptive methods). Federal law applies to all plans while state law applies to only individual plans and fully-insured group plans. Currently, 30 states and the District of Columbia require insurance plans to cover contraceptives, with a wide range of coverage and cost-sharing requirements, and exemptions among these mandates. If new federal regulations become effective the scope of contraceptive benefits for women would depend on the coverage policies and exemptions established by state laws. We will continue to monitor the healthcare reform efforts.efforts and agency implementation.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, import and export of pharmaceutical products such as those we are developing. The processes for obtaining regulatory approvals in the United States and in foreign countries, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.
FDA Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA's refusal to approve pending NDAs, withdrawal of an approval, imposition of a clinical hold or termination, issuance of Warning, Untitled, or Cyber Letters, requests for product recalls, product
seizures or detention, total or partial suspension or restriction of production, marketing or distribution, injunctions, fines, debarment, refusal to allow the import or export of product, adverse publicity, modification of promotional materials or labeling, refusals of government contracts, exclusion from participation in federal and state healthcare programs, restitution, disgorgement, imprisonment, consent decrees and corporate integrity agreements, or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
Preclinical Studies and IND Submission
Preclinical studies include laboratory evaluation of drug substance chemistry, pharmacology, toxicity and drug product formulation, as well as animal studies to assess potential safety and efficacy. An IND sponsor must submit the results of the preclinical tests and preclinical literature, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND, unless the sponsor is relying on prior FDA findings of safety or efficacy of the drug product, in which case, some of the above information may be omitted. Some preclinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials
Clinical trials involve the administration of an investigational new drug to human subjects under the supervision of qualified investigators in accordance with cGCP requirements, which includes the requirements that all research subjects provide their informed consent in writing for their participation in any clinical trial, and the review and approval of the study by an IRB. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the trial procedures, the
parameters to be used in monitoring safety and the efficacy criteria to be evaluated and a statistical analysis plan. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB for each clinical trial site participating in the clinical trial must review and approve the plan for any clinical trial before it commences, and the IRB must continue to oversee the clinical trial while it is being conducted, including any changes. Information about certain clinical trials, including a description of the study and study results, must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their ClinicalTrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined. In Phase 1, the drug is initially introduced into healthy human subjects or subjects with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an initial indication of its effectiveness. In Phase 2, the drug typically is administered through controlled studies to a limited subject population with the target disease or condition to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the drug for specific targeted diseases or conditions and to determine dosage tolerance and optimal dosage. In Phase 3, the drug is administered to an expanded subject population, generally at geographically dispersed clinical trial sites, in two adequate and well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product candidate for approval, to establish the overall risk-benefit profile of the product candidate and to provide adequate information for the labeling of the product candidate. In the case of a 505(b)(2) NDA, which is a marketing application in which sponsors may rely on investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted, some of the above-described studies and preclinical studies may not be required or may be abbreviated. Bridging studies may be needed, however, to demonstrate the applicability of the studies that were previously conducted by other sponsors to the
drug that is the subject of the marketing application. In addition to the above traditional kinds of data required for the approval of an NDA, the recently passed 21st Century Cures Act provides for FDA acceptance of newadditional kinds of data such as such as patient experience data, real world evidence for already approved products, and, for appropriate indications sought through supplemental marketing applications, data summaries.
In addition, under the Pediatric Research Equity Act, or PREA, an NDA or supplement to an NDA for a new active ingredient, indication, dosage form, dosage regimen or route of administration must contain data that are adequate to assess the safety and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. We have obtained a waiver from the conduct of a PREA study.
The manufacture of investigational drugs for the conduct of human clinical trials is subject to cGMPFDA product manufacturing requirements. Investigational drugs and active pharmaceutical ingredients imported into the United States are also subject to regulation by the FDA relating to their labeling and distribution. Further, the export of investigational drug products outside of the United States is subject to regulatory requirements of the receiving country as well as U.S. export requirements under the FDCA.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and the IRB and more frequently if serious adverse events occur. Information about certain clinical trials, including a description of the study and study results, must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their ClinicalTrials.gov website. Failure to submit the required information to ClinicalTrials.gov can result in monetary penalties. Marketing application applicants must also report certain investigator financial interests to the FDA.
Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB's requirements or if the drug has been associated with unexpected serious harm to subjects. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group regularly reviews accumulated data and advises the study sponsor regarding the continuing safety of trial subjects, potential trial subjects,
and the continuing validity and scientific merit of the clinical trial. We may also suspend or terminate a clinical trial based on evolving business objectives or competitive climate.
U.S. Marketing Approval
Assuming successful completion of the required clinical testing, the results of the preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product's chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. These user fees must be filed at the time of the first submission of the application, even if the application is being submitted on a rolling basis. A user fee for the Twirla contraceptive patch was submitted with the original NDA. Application resubmissions by the same applicant do not require a new application fee. Under the Prescription Drug User Fee Act, or PDUFA guidelines that are currently in
effect, the FDA has agreed to certain performance goals regarding the timing of its review of an application. The FDA's standard review goal is to act on 90% of all Non-New Molecular Entity applications within ten months of FDA receipt of the application. The FDA's review goal for an NDA resubmission, such as Twirla, is to act on 90% of such applications within six months of FDA receipt. ThisThese time periodperiods may be extended by the FDA should an applicant submit new information to the agency during the course of the FDA's review of the marketing application. The time period is also only a goal and may not be met by FDA. We expect that our products, if and when approved, will be subject to a standard review goal.
In addition, under the Pediatric Research Equity Act, or PREA, an NDA or supplement to an NDA for a new active ingredient, indication, dosage form, dosage regimen or route of administration must contain data that are adequate to assess the safety and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. We believe that we may be able to obtain a waiver from the conduct of a PREA study as, historically, waivers have been granted for other contraceptive applicants.FDA.
The FDA conducts a preliminary review of all original NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmittedsubmitted again with the additional information. The resubmitted applicationinformation and is also subject to review before the FDA accepts it for filing.
Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDAreview to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held, as well as the manufacturing processes and controls, meet standards designed to ensure the product's continued safety, quality and purity.
The FDA may refer a marketing application to an external advisory committee for questions pertaining to issues such as clinical trial design, safety and efficacy, and public health questions. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it typically follows such recommendations and considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured, referred to as a Pre-Approval Inspection. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP
the FDA's requirements for product manufacturing and adequate to assure consistent production of the product within required specifications by the manufacturer and all of its subcontractors and contract manufacturers. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical trial sites to assure compliance with cGCP. Also, as part of its regulatory review, the FDA verifies the data contained in the NDA.
The testing and approval process for an NDAa drug product requires substantial time, effort and financial resources, and may take several years to complete. Data obtained from preclinical and clinical testing are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval of an NDAa marketing application on a timely basis, or at all.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a CRL. A CRL indicates that the review cycle of the application is complete, and the application is not ready for approval. A CRL generally contains a statement of specific conditions that must be met in order to secure final approval of the NDAdrug product and may require additional clinical or preclinical testing, or other information in order for the FDA to reconsider the application. We received
If an application receives a CRL, for Twirla, have conducted the additional required clinical trial and other analyses, and intend toapplicant may resubmit the NDAapplication, addressing all of the FDA-cited deficiencies, withdraw the application, or request the opportunity for Twirlaa hearing. If the applicant resubmits the application, the application is subject to an initial FDA review. Within 30 days of receipt, the FDA will review a resubmission to determine whether it constitutes a complete response that addresses all deficiencies identified in a complete response letter. The agency then issues a letter to the applicant, stating whether the agency agrees that the resubmission is a complete response. If the FDA with this updated information. We expectdoes not agree that the FDA'sresubmission is a complete response, the review timeline for our Twirlaclock will not start until a complete response is received. If the agency agrees that the resubmission is a complete response, the FDA will classify the resubmission as either Class 1 or 2. The FDA aims to be approximatelyreview Class 1 resubmissions within two months of receipt or Class 2 resubmissions within six months after submission.of receipt. Class 1 resubmissions are resubmissions of an NDA following a complete response letter that include minor updates or data reanalysis. Class 2 resubmissions include more complex or extensive updates to the NDA. As with the PDUFA timelines for original submissions, these are also subject to extension if the sponsor submits new information. Resubmitted applications may also be subject to FDA inspection of clinical and manufacturing sites, as well as review by FDA advisory committees. Following its review of a resubmitted NDA, the FDA may issue an approval letter or another CRL.
Even if an applicant resubmits with submission of thisthe required additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA's satisfaction, the FDA may issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product candidate, it may limit the approved indications for use of the product candidate and require that contraindications, warnings or precautions be included in the product labeling, including a black box warning. The FDA also may not approve the inclusion of labeling claims necessary for successful marketing. Moreover, the FDA may require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess certain aspects of a drug's safety and efficacy after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms. For example, the FDA may require a risk evaluation and mitigation strategy, or REMS, as a condition of approval or following approval to mitigate any identified or suspected serious risks and ensure safe use of the drug. The REMS plan could include medication guides, physician communication plans, assessment plans, and elements to assure safe use, such as restricted distribution methods, patient registries or other risk minimization tools. A REMS could materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements, submission of a supplemental application, and FDA review and approval. Further, should new safety information arise, additional testing, product labeling or FDA notification may be required.
Hatch-Waxman Act
Section 505 of the FDCA describes three types of marketing applications that may be submitted to the FDA to request marketing authorization for a new drug. A Section 505(b)(1) NDA is an application that contains full reports of investigations of safety and efficacy. A 505(b)(2) NDA is an application that contains full reports of investigations of safety and efficacy but where at least some of the information required for approval comes from investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. This regulatory pathway enables the applicant to rely,
in part, on the FDA's prior findings of safety and efficacy for an existing product, or published
literature, in support of its application. Section 505(j) establishes an abbreviated approval process for a generic version of an approved drug productsproduct through the submission of an Abbreviated New Drug Application, or ANDA. An ANDA provides for marketing of a generic drug product that has the same active ingredients, dosage form, strength, route of administration, labeling, performance characteristics and intended use, among other things, to a previously approved product. ANDAs are termed "abbreviated" because they are generally not required to include preclinical (animal) and clinical (human) data to establish safety and efficacy. Instead, generic applicants must scientifically demonstrate that their product is bioequivalent to, or performs in the same manner as, the innovator drug throughin vitro,in vivo, or other testing. The generic version must deliver the same amount of active ingredients into a subject's bloodstream in the same amount of time as the innovator drug and can often be substituted by pharmacists under prescriptions written for the reference listed drug. In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant's drug or a method of using the drug. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA's Approved Drug Products with Therapeutic Equivalence Evaluations publication, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an ANDA or 505(b)(2) NDA.
Upon submission of an ANDA or a 505(b)(2) NDA, an applicant must certify to the FDA thatthat: (1) no patent information on the drug product that is the subject of the application has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. Generally, the ANDA or 505(b)(2) NDA cannot be approved until all listed patents have expired, except where the ANDA or 505(b)(2) NDA applicant challenges a listed patent through the last type of certification, also known as a paragraphParagraph IV certification. If the applicant does not challenge the listed patents or indicatesindicate that it is not seeking approval of a patented method of use, the ANDA or 505(b)(2) NDA application will not be approved until all of the listed patents claiming the referenced product have expired.
If the ANDA or 505(b)(2) NDA applicant has provided a Paragraph IV certification to the FDA, the applicant must send notice of the Paragraph IV certification to the NDA and patent holders once the application has been accepted for filing by the FDA.within a specified timeframe. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the paragraphParagraph IV certification. If the paragraphParagraph IV certification is challenged by an NDA holder or the patent owner(s) asserts a patent challenge to the paragraphParagraph IV certification, the FDA may not make an approval effective until the earlier of 30 months from the receipt of the notice of the paragraphParagraph IV certification, the expiration of the patent, when the infringement case concerning each such patent was favorably decided in the applicant's favor or settled, or such shorter or longer period as may be ordered by a court. This prohibition is generally referred to as the 30-month stay. In instances where an ANDA or 505(b)(2) NDA applicant files a paragraphParagraph IV certification, the NDA holder or patent owner(s) regularly take action to trigger the 30-month stay, recognizing that the related patent litigation may take many months or years to resolve. Thus, approval of an ANDA or 505(b)(2) NDA could be delayed for a significant period of time depending on the patent certification the applicant makes and the reference drug sponsor's decision to initiate patent litigation.
The Hatch-Waxman Act establishes periods of regulatory exclusivity for certain approved drug products, during which the FDA cannot approve (or in some cases accept) an ANDA or 505(b)(2) application that relies on the branded reference drug. For example, the holder of an NDA, including a 505(b)(2) NDA, may obtain five years of exclusivity upon approval of a new drug containing new chemical entities, or NCEs, that have not been previously approved by the FDA. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active
moiety, which is the molecule or ion responsible for the therapeutic activity of the drug substance. During the exclusivity period, the FDA may not accept for review an ANDA or a 505(b)(2) NDA
submitted by another company that contains the previously approved active moiety. However, an ANDA or 505(b)(2) NDA may be submitted after four years if it contains a certification of patent invalidity or non-infringement.
The Hatch-Waxman Act also provides three years of marketing exclusivity to the holder of an NDA (including a 505(b)(2) NDA) for a particular condition of approval, or change to a marketed product, such as a new formulation for a previously approved product, if one or more new clinical studies (other than bioavailability or bioequivalence studies) was essential to the approval of the application and was conducted/sponsored by the applicant. This three-year exclusivity period protects against the FDA approval of ANDAsmaking an ANDA and 505(b)(2) NDAsNDA approval effective for the condition of the new drug's approval. As a general matter, the three yearthree-year exclusivity does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for generic versions of the original, unmodified drug product. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and efficacy.
Our NDA for Twirla was submitted under Section 505(b)(2), and we expect that some of our other drug candidates will utilize the Section 505(b)(2) regulatory pathway. Even though several of our drug products utilize active drug ingredients that are commercially marketed in the United States in other dosage forms, we need to establish the safety and efficacy of those active ingredients in the formulation and dosage forms that we are developing. All approved products, both innovator and generic, are listed in the FDA's Orange Book.
Recently, Congress, the Administration, and administrative agencies have taken certain measures to increase drug competition and thus, decrease drug prices. By example, in 2019 FDA introduced a proposed rule and draft guidance to facilitate drug importation. Congress also passed a bill requiring sponsors of NDA approved products to provide sufficient quantities of drug product on commercially reasonable market based terms to entities developing generic and similar drug products. This bill also included provisions on shared and individual REMS for generic drug products.
Combination Drug/Device Regulation
Twirla and our potential product candidates are considered to be drug-device combination products by the FDA. While our potential product candidates, as a whole, are subject to the NDA approval process, drug-device combination products require compliance with additional FDA regulations. For instance, drug-device combination products must comply with the drug cGMPs, as well as some of the device Quality System Regulations, or QSRs. These dual requirements will require additional effort, FDA reporting, and monetary expenditure to ensure that Twirla and our potential product candidates comply with all applicable regulatory requirements.
U.S. Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to manufacturing recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion, reporting of adverse experiences with the product and drug shortages, and compliance with any post-approval requirements imposed as a condition of approval, such as Phase 4 clinical trials, REMS and surveillance to assess safety and efficacy after commercialization. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There are also are continuing, annual prescription drug program user fee requirements for any approved products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data other than bioavailability or bioequivalence studies.products. In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with
the FDA and state agencies, list drugs manufactured at their facilities with the FDA, and are subject to periodic announced and unannounced inspections by the FDA and these state agencies for compliance with cGMPFDA and state requirements for product manufacturing and other requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented, or FDA notification. FDA regulations also require investigation and correction of any deviations from cGMPFDA requirements for product manufacturing and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMPFDA requirements for product manufacturing compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market.
Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Although physicians, in the practice of medicine, may prescribe approved drugs for unapproved indications, pharmaceutical companies are prohibited from marketing or promoting their drug products for uses outside the approved label, a practice known as off-label promotion. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including criminal and civil penalties under the FDCA and False Claims Act, exclusion from participation in federal healthcare programs, mandatory compliance programs under corporate integrity agreements, debarment and refusal of government contracts.
In addition, the distribution of prescription pharmaceutical products, including samples, is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level.level and reporting regarding drug samples. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and impose requirements to ensure accountability in distribution.
Moreover, the Drug Quality and Security Act imposes obligations on manufacturers of pharmaceutical products related to product tracking and tracing. Among the requirements of this legislation, manufacturers are required to provide certain information regarding the drug product to individuals and entities to which product ownership is transferred, will beare required to label drug product with a product identifier toward the end of 2017 and are required to keep certain records regarding the drug product. The transfer of information to subsequent product owners by manufacturers will beis also required to be done electronically toward the end of 2017.electronically. Manufacturers must also verify that purchasers of the manufacturers' products are appropriately licensed. Further, under this legislation, manufactures have drug product investigation, quarantine, disposition, and FDA and trading partner notification responsibilities related to counterfeit, diverted, stolen and intentionally adulterated products, as well as products that are the subject of fraudulent transactions or which are otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death.
Table of Contents Other persons and entities within the drug supply chain are also subject to Drug Quality and Security Act requirements.
U.S. Fraud and Abuse, Data Privacy and Security and Transparency Laws and Regulations
In addition to FDA restrictions on marketing of pharmaceutical products, federal and state fraud and abuse laws restrict business practices in the biopharmaceutical industry. These laws include, among other things, anti-kickback, physician payment transparency and false claims laws and regulations as well as data privacy and security laws and regulations.
The federal Anti-Kickback Statute prohibits, among other things, any person or entity from knowingly and willfully offering, paying, soliciting or receiving any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or in return for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any item or service reimbursable under Medicare, Medicaid or other federal healthcare programs. The term "remuneration" has been interpreted broadly to include anything of value. Additionally, the intent standard under the Anti-Kickback Statute and criminal healthcare fraud statutes was also amended by the ACA to a stricter standard such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the ACA provided that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases, or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances.
The federal civil False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to, or approval by, the federal government or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes "any request or demand" for money or property presented to the U.S. government. The civil False
Claims Act has been used to assert liability on the basis of kickbacks and other improper referrals, improperly reported government pricing metrics such as Best Price or Average Manufacturer Price, improper promotion of off-label uses not expressly approved by the FDA in a drug's label, and allegations as to misrepresentations with respect to the services rendered. Additionally, the civil monetary penalties statute, which, among other things, imposes fines against any person who is determined to have presented, or caused to be presented, claims to a federal healthcare program that the person knows, or should know, is for an item or service that was not provided as claimed or is false or fraudulent. The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, also created federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, including private third party payors and knowingly and willfully falsifying, concealing or covering up by trick, scheme or device a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services relating to healthcare matters. Also, many states have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, that apply regardless of the payor.
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their respective implementing regulations, including the final omnibus rule published on January 25, 2013, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes security standards and certain privacy standards directly applicable to business associates. HITECH also created four new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys' fees and costs associated with pursuing federal civil actions. In addition, state laws may govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. For instance, the recently enacted California Consumer Privacy Act may govern the privacy and security of health and other information in certain circumstances, many of which differ from each other in significant ways and may not be preempted by HIPAA, thus complicating compliance efforts.
Additionally, federal physician payment transparency laws, including the federal Physician Payment Sunshine Act created under Section 6002 of the ACA and its implementing regulations, require that manufacturers of drugs for which payment is available under Medicare, Medicaid or the Children's Health Insurance Program, with certain exceptions, report annually to the government information related to payments or other "transfers of value" made or distributed to physicians, which is defined to include doctors of medicine, dentists, optometrists, podiatrists and chiropractors, generally, with some exceptions, and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, physicians and teaching hospitals. Additionally, applicable manufacturers and group purchasing organizations are required to report annually to the government certain ownership and investment interests held by physicians and their immediate family members. , Manufacturers must submit reports by the 90th day of each calendar year. Disclosure of such information is made on a publicly available website.
There are also an increasing number of analogous state laws that regulate price increases, require manufacturers to file reports with states on pricing and marketing information, and to track and report gifts, compensation, other remuneration and items of value provided to healthcare professionals and healthcare entities. Many of these laws contain ambiguities as to what is required in order to comply
with such laws. For example, several states have enacted legislation requiring pharmaceutical companies to, among other things, establish and implement commercial compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities, or register their sales representatives. Certain state laws also regulate manufacturers' use of prescriber-identifiable data. These laws may affect our future sales, marketing and other promotional activities by imposing administrative and compliance burdens. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions once we commercialize could be subject to the penalty provisions of the pertinent state and federal authorities.
If our operations are found to be in violation of any of the laws or regulations described above or any other laws that apply to us, we may be subject to a variety of penalties, depending upon the law found to have been violated, potentially including criminal and significant civil monetary penalties, damages, fines, imprisonment, exclusion from participation in government healthcare programs, corporate integrity agreements, refusal of government contracts, contract debarment and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. To the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
Coverage and Reimbursement Generally
The commercial success of Twirla and our other potential product candidates and our ability to commercialize any approved product candidates successfully will depend in part on the extent to which governmental payor programs at the federal and state levels, including Medicare and Medicaid, private health insurers and other third-party payors provide coverage for and establish adequate coverage of and reimbursement levels for our potential product candidates. Government authorities, private health insurers and other organizations generally decide which drugs they will pay for and establish reimbursement levels for healthcare. In particular, in the United States, private health insurers and other third-party payors often provide reimbursement for products and services based on the level at which the government provides reimbursement through the Medicare or Medicaid programs for such products and services. In the United States, the European UnionE.U. and other potentially significant markets for our potential product candidates, government authorities and third-party payors are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies, which often has resulted in average selling prices lower than they would otherwise be. Further, the increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the European UnionE.U. will put additional pressure on product pricing, reimbursement and utilization, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical coverage and reimbursement policies and pricing in general. Patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Sales of our potential product candidates will therefore depend substantially, both domestically and abroad, on the extent to which the costs of our products will be paid by health maintenance organizations, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, such as Medicare and Medicaid, private health insurers and other third-party payors.
Third-party payors are increasingly imposing additional requirements and restrictions on coverage and limiting reimbursement levels for medical products, including pharmaceuticals. For example,
federal and state governments reimburse covered prescription drugs at varying rates generally below average wholesale price. These restrictions and limitations influence the purchase of healthcare services and products. Third-party payors are developing increasingly sophisticated methods of controlling healthcare costs. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drug products for a particular indication. Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain FDA approvals. Our potential product candidates may not be considered medically necessary or cost-effective. Moreover, a payor's decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in drug development for a product candidate. Legislative proposals to reform healthcare or reduce costs under government insurance programs may result in lower reimbursement for our potential product candidates or exclusion of our potential product candidates from coverage. The cost containment measures that healthcare payors and providers are instituting and any healthcare reform could significantly reduce our revenues from the sale of any approved product candidates. We cannot provide any assurances that we will be able to obtain and maintain third-party coverage or adequate reimbursement for our potential product candidates in whole or in part.
Healthcare Reform
Legislative proposals to reform healthcare or reduce costs under government healthcare programs may result in lower reimbursement for our potential product candidates or exclusion of our potential product candidates from coverage. There have been a number of legislative and regulatory changes to the healthcare system that could affect our ability to profitably sell our potential product candidates, if approved. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
In March 2010, the ACA was enacted, which included provisions on comparative clinical effectiveness research extended the initiatives of the American Recovery and Reinvestment Act of 2009, also known as the stimulus package, which provided $1.1 billion in funding to study the comparative effectiveness of healthcare treatments. This funding was designated for, among other things, conducting, supporting or synthesizing research that compares and evaluates the risks and benefits, clinical outcomes, effectiveness and appropriateness of products. The ACA also appropriated additional funding to comparative clinical effectiveness research. Although Congress has indicated that this funding is intended to improve the quality of healthcare, it remains unclear how the research will impact current Medicare coverage and reimbursement or how new information will influence other third-party payor policies.
It is possible that comparative effectiveness research demonstrating benefits in a competitor's product could adversely affect the sales of our potential product candidates. If third-party payors do not consider our potential product candidates to be cost-effective compared to other available therapies, they may not cover our potential product candidates, once approved, as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our potential product candidates on a profitable basis.
In addition, in August 2011, President Obama signed into law the Budget Control Act of 2011, as amended, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee on Deficit Reduction did not achieve its targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation's automatic reductions to several government programs. These reductions include aggregate reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and will stay in effect through 2024 unless additional Congressional action is taken. In November 2015, the Bipartisan Budget Act was enacted into law, which, among other things, extended sequestration through 2025. These and other healthcare reform initiatives may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our financial operations. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could further limit the prices we
are able to charge, or the amounts of reimbursement available, for our potential product candidates if they are approved.
In March 2017, the U.S. Congress proposed legislation, which, if signed into law by the new administration, would repeal certain aspects of the ACA. Further, onOn January 20, 2017, the newthen-new administration signed an Executive Order directing federal agencies with authorities and responsibilities under the ACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the ACA that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices among others. Additionally, in October 2017, the Department of Health and Human Services, jointly with the Department of Labor and the Treasury, issued two interim rules outlining exemption processes for employers not wanting to offer contraceptive coverage based on their religious beliefs or sincerely held moral convictions. While there is an injunction against the administration prohibiting it from implementing these rules, the ultimate outcome of that litigation, which is currently in front of the Supreme Court, cannot be predicted. Congress also could consider subsequent legislation to repeal and replace elements of the ACA that are repealed.
Table Therefore, it is difficult to determine the full effect of Contentsthe ACA or any other healthcare reform efforts on our business.
The Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act, or FCPA, prohibits any U.S. individual or business from paying, offering, or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. Activities that violate the FCPA, even if they occur wholly outside the United States, can result in criminal and civil fines, imprisonment, disgorgement, oversight and debarment from government contracts.
Foreign Regulation
We currently have no plans to seek approval for Twirla outside of the United States. In order to market any product outside of the United States, we would need to comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of our products. For example, in the European Union, we must obtain authorization of a clinical trial application, or CTA, in each member state in which we intend to conduct a clinical trial. Whether or not we obtain FDA approval for a product, we would need to obtain the necessary approvals by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others.
Research and Development
Conducting research and development is central to our business model. We have invested and expect to continue to invest significant time and capital in our research and development operations. Our research and development expenses were $20.9$9.9 million, $25.6$9.8 million, and $13.4$14.4 million for the years ended December 31, 2016, 2015,2019, 2018, and 2014,2017, respectively. In 2017, we expect the expenses associated with the SECURE clinical trial to decrease as we complete the close-out activities associated with the trial, and no additional clinical trials are planned at this time. During 2017,2020, we expect to increase activities relatedcontinue to equipment qualificationincur research and validation ofdevelopment expenses as we refine our commercial manufacturing process as we continue to prepare for the commercializationprocess.
Intellectual Property
We strive to protect the proprietary technologies that we believe are important to our business, including seeking and maintaining patent protection intended to cover our SkinfusionSkinfusion® technology, its methods of use, related technologies and other inventions that are important to our business. As more fully described below, our patents and patent applications are directed to our Skinfusion technology or aspects thereof including certain transdermal delivery systems having an active adhesive matrix and methods of using such transdermal delivery systems for controlling fertility. We also rely on manufacturing trade secrets and careful monitoring of our proprietary information to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
Our success will depend significantly on our ability to obtain new patents and maintain existing patents and other proprietary protection for commercially important technology, inventions and
know-how related to our business, defend and enforce our patents, preserve the confidentiality of our trade secrets and operate without infringing valid and enforceable patents and other proprietary rights of third parties.
A third party may hold intellectual property, including patent rights, which are important or necessary to the development of our potential product candidates. It may be necessary for us to use the patented or proprietary technology of third parties to commercialize our potential product candidates, in which case we would be required to obtain a license from these third parties on commercially reasonable terms. If we were not able to obtain a license on commercially reasonable terms, our business could be harmed, possibly materially.
We plan to continue to expand our intellectual property estate by filing patent applications directed to novel and nonobvious transdermal contraceptive products. The active pharmaceutical ingredients, or API, in our potential product candidates are generic and therefore our patents do not include claims directed solely to the API. We anticipate seeking additional patent protection in the United States and internationally for additional transdermal delivery systems and their methods of use.
The patent positions of pharmaceutical companies like us are generally uncertain and involve complex legal, scientific and factual questions. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and the patent's scope can be modified after issuance. Consequently, we do not know whether any of our potential product candidates will remain protected by enforceable and valid patents. We cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient proprietary protection from competitors. Any patents that we hold may be challenged, circumvented or invalidated by third parties.
Because patent applications in the United States and certain other jurisdictions generally are maintained in secrecy for 18 months, and since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain of our entitlement to patent rights in the inventions covered in our issued patents and pending patent applications. Moreover, we may have to participate in interference proceedings declared by the U.S. Patent and Trademark Office, USPTO, to determine priority of invention, or in post-grant challenge proceedings in the USPTO or foreign patent offices such as oppositions, reexamination, inter-partes review, post grant review, or a derivation proceeding, that challenge our entitlement to an invention or the patentability of one or more claims in our patent applications or issued patents. Such proceedings could result in substantial cost, even if the eventual outcome is favorable to us.
More specifically, TwirlaTwirla® is a transdermal contraceptive hormone delivery system. The system is a patch for application to the skin and contains two API, the hormones levonorgestrel, or LNG, which is a synthetic progestin, and ethinyl estradiol,EE, a synthetic estrogen. The API are formulated with a combination of skin penetration enhancers, which promote penetration through the dermis and into the bloodstream, such
that effective blood levels of the active agents are achieved to suppress ovulation and thereby prevent pregnancy. One of our other potential product candidates, AG890, is similar to Twirla, except that it contains only a single API, LNG.
In both our Twirla product candidate line and in AG890, the active adhesive system consists of the active ingredients in a polyacrylate adhesive polymer matrix comprising the permeation enhancers dimethylsulfoxide, ethyl lactate, capric acid and lauryl lactate. The active blend is coated onto a release liner, and a backing layer is added on top of the active blend. The peripheral adhesive system, also called the overlay, comprising three layers is added onto the backing layer. The overlay comprises a polyisobutylene adhesive layer, an acrylic adhesive layer, and an overlay covering. The overlay covering is a commercially available silk-like polyester fabric. The adhesive components of the overlay, in addition to their adhesive function, create anin situ seal with the disposable release liner, trapping evaporable solvents in the active blend, thereby extending the usable shelf life of the product candidate
and contributing to the comfort and effectiveness of the transdermal system during use. Prior to use of any of our potential product candidates, the release liner is removed by the user and discarded. The patch is then applied to the skin.
Eight U.S. patents, issuing from threetwo patent families, have been or are being submitted to the FDA for listing in the Orange Book upon approval of Twirla. These patents include claims directed to transdermal delivery systems having an active adhesive matrix and claims directed to methods of controlling fertility by applying such transdermal delivery systems, and in all cases including a skin permeation enhancer. One of our eight issued U.S. patents will expire November 22, 2020. Four will expire March 14, 2021. Two will expire July 10, 2028. The eighth will expire August 26, 2028.
U.S. Patent No. 7,045,145 is directed to the adhesive matrix of the transdermal delivery system used in Twirla and expires in March 2021; product-by-process claims cover patches manufactured by drying wet formulations of the active adhesive matrix. U.S. Patent No. 7,384,650, U.S. Patent No. 8,221,784, and U.S. Patent No. 8,221,785 are all directed to the dry final product formulation of the transdermal delivery system used in Twirla and expire in March 2021. U.S. Patent No. 8,221,784 covers both Twirla and AG890. Foreign counterparts to these patents have been granted in Australia, Brazil, Canada, China, Europe, France, Germany, Great Britain, Ireland, Italy,Hong Kong, India, Israel, Japan, Korea, Mexico, Netherlands, New Zealand, Norway, Spain, Switzerland, and South Africa.Mexico. U.S. Patent No. 8,883,196 is directed to a method of controlling fertility by applying Twirla or AG890 once each week for three weeks followed by a one weekone-week rest interval, or in an extended regimen without a rest interval for a selected number of weeks and expires November 22, 2020.
U.S. Patent Nos. 8,246,978, 8,747,888, and 9,050,348 are directed to structural features of the transdermal delivery system used in Twirla and AG890 patch design for transdermal delivery of hormones or of other drugs. As such, these patents protect a platform technology for delivery of LNG, EE, other hormones, and other drugs. These patents expire in July and August 2028. Foreign counterparts arehave been granted in Australia, Canada, China, Spain, France, Netherlands, Italy, UK, Ireland, Germany, Switzerland, Japan, Russia and New Zealand and areone counterpart remains pending elsewhere.in India
U.S. Patent Nos. 9,198,876, 9,192,614, 9,198,919 and 9,198,920 and related patents and patent applications are directed to various novel dosing regimens, each of which employs transdermal delivery of contraceptive doses of ethinyl estradiolEE and levonorgestrelLNG during a "treatment interval" and transdermal delivery of low dose ethinyl estradiolEE and low dose levonorgestrelLNG during a "withdrawal interval". Foreign counterparts are pending or granted in Europe and Canada. We expect these patents will be relevant to two of the products in our pipeline, AG200-SP and AG200-ER, as well as other new potential regimens.
U.S. Patent No. 9,364,487 is directed to a composition and device for transdermal delivery of levonorgestrelLNG for P-only therapy. The composition contains an anti-oxidant to protect the progestin against oxidative degradation caused by other components of the composition. Foreign counterparts are pending or
granted in Canada, Europe, India, Japan and Mexico. We expect this patent to be relevant to at least one product in our pipeline, AG890.
We own a total of about 45 granted patents in jurisdictions other than the United States, including patents in New Zealand, Australia, Canada, Austria, Germany, Ireland, Italy, The Netherlands, Spain, Switzerland, Israel, India, Japan, South Korea, Mexico, Norway, the Philippines, Taiwan and South Africa. These issued foreign patents include claims directed to transdermal delivery systems having an active adhesive matrix and claims directed to methods of controlling fertility by applying such transdermal delivery systems, and in all cases including a skin permeation enhancer. In addition, we have about 37 pending patent applications pending in the United States and certain foreign jurisdictions directed to novel formulations and methods designed to improve efficacy and modulate side effects of administration, as well as to provide personalized dosing based on body weight or BMI. We also have a pending United States patent application directed to packaging for Twirla and AG890, and for unique patch dosage regimens intended to align with future label expansions and line extensions, such as AG200ER and AG200SP, including an antioxidant formulation and a desogestrel patch.
Table of Contentstransdermal systems containing certain skin permeation enhancers.
Regulatory Exclusivity
Our NDA for Twirla was submitted under Section 505(b)(2) of the Food, Drug, and Cosmetic Act, or FDCA. Even though Twirla utilizes API that were previously approved in the United States, Twirla utilizes LNG in a new dosage form, specifically a transdermal patch, and we provided new clinical data essential to approval in our NDA to establish the safety and efficacy of Twirla. Therefore, if approved by the FDA, we expect to receivereceived three years of U.S. marketing exclusivity for Twirla.Twirla under the Hatch Waxman Act. The exclusivity will prohibitprohibits the FDA from approving ANDAs and 505(b)(2) NDAs for the conditions of the Twirla approval. We will consider whether we are going to pursue patent term restoration, however, we do not expect to receive patent term restoration because, as explained above, Twirla willis not be the first approval of the API.
Employees
As of December 31, 2016,2019, we had 1915 full time employees, including elevensix in research and development and eightnine in general and administrative roles. None of our employees are represented by a labor union or subject to a collective bargaining agreement. We have not experienced a work stoppage and consider our relations with our employees to be good.
Corporate Information
We were incorporated in Delaware in December 1997. Our offices are located at 101 Poor Farm Road, Princeton, New Jersey 08540, and our telephone number is (609) 683-1880.
Available Information
Our corporate website address is www.agiletherapeutics.com. Information contained on or accessible through our website areis not a part of this annual reportAnnual Report on Form 10-K, and the inclusion of our website address in this annual report is an inactive textual reference only. We make our annual reportAnnual Report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and all amendments to those reports available free of charge on our website as soon as reasonably practicable after we file such reports with, or furnish such reports to, the Securities and Exchange Commission, or SEC.
We are an "emerging growtha "smaller reporting company," as defined in the Jumpstart Our Business Startups Act of 2012. We will remain an emerging growth company until the earlier of (1) the last dayRule 12b-2 of the fiscal year (a) followingExchange Act. Since the fifth anniversary of the completion of our initial public offering, (b) in which we have total annual gross revenue of at least $1.0 billion, or (c) in which we are deemed to be a large accelerated filer, which means theaggregate market value of our commonvoting stock that is held by non-affiliates exceeded $700was less than $75 million ason June 28, 2019, we are a non-accelerated filer under the rules of the prior March 31st,SEC, and (2)an auditor attestation report over Internal Controls over Financial Reporting does not need to be included in the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.2019 Form 10-K.
Investing in our common stock involves a high degree of risk. You should carefully consider the risk factors set forth below as well as the other information contained in this Annual Report on Form 10-K and in our other public filings in evaluating our business. Any of the following risks could materially and adversely affect our business, financial condition or results of operations. The risks described below are not the only risks facing us. Additional risks and uncertainties not currently known to us or that we currently view to be immaterial may also materially adversely affect our business, financial condition or results of operations. In these circumstances, the market price of our common stock would likely decline.
Risks Related to the Clinical Trial Process and Regulatory Approval for Our Product Candidates
We have not obtained regulatory approval for any of our product candidates in the United States or any other country.
We currently do not have any product candidates that have gained regulatory approval for sale in the United States or any other country, and we cannot guarantee that we will ever have marketable products. Our business is substantially dependent on our ability to complete the development of, obtain regulatory approval for and successfully commercialize product candidates in a timely manner. We cannot commercialize product candidates in the United States without first obtaining regulatory approval to market each product candidate from the U.S. Food and Drug Administration, or FDA; similarly, we cannot commercialize product candidates outside of the United States without obtaining regulatory approval from comparable foreign regulatory authorities. We are not currently pursuing any regulatory approvals for Twirla or any other product candidate outside the United States.
We have previously conducted two Phase 3 clinical trials for Twirla, and we filed a new drug application, or NDA, with the FDA for Twirla in April 2012. The FDA issued a Complete Response Letter, or CRL, in February 2013, identifying certain issues, including a request for additional clinical data, quality information and chemistry, manufacturing and controls information, which must be addressed before approval can be granted. We have continued to interact with the FDA on its CMC and other questions and continued additional supportive testing in order to respond to the FDA's CMC questions. In addition, we are gathering the requested information and conducted an additional Phase 3 clinical trial for Twirla®, which we refer to as the SECURE clinical trial. The SECURE clinical trial commenced enrollment during the third quarter of 2014 and completed in December 2016. In January 2017, we announced top-line results. Based on the results of the SECURE clinical trial and additional information relating to the manufacture of Twirla, we plan to resubmit our NDA in the first half of 2017. Although we met with the FDA in October 2013 to discuss our new Phase 3 clinical trial and have received substantial written comments from the FDA in subsequent interactions, we have not sought and have not obtained agreement with the FDA on a special protocol assessment regarding the new Phase 3 trial. We cannot predict whether regulators will agree with our conclusions regarding the results of the SECURE clinical trial or any clinical trials we have conducted to date, including whether our data are reliable and generalizable. For example, based on the SECURE top-line data, the Pearl Index for the overall intent to treat population of subjects 35 years of age and under was 4.80 with an upper-bound of the 95% confidence interval of 6.06, but in the obese subpopulation of subjects 35 years of age and under, the Pearl Index was 6.42 with an upper-bound of the 95% confidence interval of 8.88. If we were to exclude the top-line data on the obese subpopulation, our Pearl Index for non-obese patients was 3.94 with an upper-bound of the 95% confidence interval of 5.35. The highest Pearl Index for a hormonal contraceptive product approved by the FDA to date was 3.19 and the highest upper-bound of the 95% confidence interval was 5.03. In the combined safety database for our three Agile Phase 3 trials (n>3,000), there were 5 subjects with potentially study drug related DVTs or PEs, 4 of whom were obese (BMI³30kg/m2). Although ultimate approvability of a hormonal contraceptive is based on a risk/benefit assessment of the overall safety and efficacy profile of a product, not only a specific Pearl Index, the FDA could conclude that the Pearl Index is too high to
demonstrate efficacy and an adequate risk/benefit profile for either the overall study population or a subgroup of the study population. Accordingly, FDA may not approve our Twirla NDA. Alternatively, FDA may determine that for a specific subgroup of patients, Twirla has lower efficacy and presents a higher risk, necessitating labeling restrictions. For instance, FDA may require labeling restrictions on the use of Twirla for patients in certain BMI categories. As such, we may not obtain approval of Twirla based on these data or any other basis, or if approved, may only receive approval with significant labeling restrictions. In addition, the FDA may re-inspect our manufacturing partner's facilities as well as SECURE clinical trial sites during its review of our resubmission before approval can be granted.
Before obtaining regulatory approvals for the commercial sale of any product candidate for a target indication, we must demonstrate in preclinical studies and well-controlled clinical trials and, with respect to approval in the United States, to the satisfaction of the FDA, that the product candidate is safe and effective for use for that target indication and that the manufacturing facilities, processes and controls are adequate. In the United States, it is necessary to submit an NDA to obtain FDA approval. An NDA must include extensive preclinical and clinical data and supporting information to establish the product candidate's safety and efficacy for each desired indication, although we may partially rely on published scientific literature or the FDA's prior approval of similar products. The NDA must also include significant information regarding the chemistry, manufacturing and controls for the product. The FDA may further inspect our manufacturing facilities to ensure that the facilities can manufacture our product candidates and our products, if and when approved, in compliance with the applicable regulatory requirements, as well as inspect our clinical trial sites to ensure that our studies are properly conducted. Obtaining approval of an NDA is a lengthy, expensive and uncertain process, and approval may not be obtained. Upon submission, or resubmission, of an NDA, the FDA must make an initial determination that the application is sufficiently complete to accept the submission for filing. We cannot be certain that any submissions will be accepted for filing and review by the FDA, or ultimately be approved. If the application is not accepted for review or approved, the FDA may require that we conduct additional clinical or preclinical trials, or take other actions before it will reconsider our application. If the FDA requires additional studies or data, we would incur increased costs and delays in the marketing approval process, which may require us to expend more resources than we have available. In addition, the FDA may not consider any additional information to be complete or sufficient to support approval.
Regulatory authorities outside of the United States, such as in Europe and Japan and in emerging markets, also have requirements for approval of drugs for commercial sale with which we must comply prior to marketing in those areas. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our product candidates. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and obtaining regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. However, the failure to obtain regulatory approval in one jurisdiction could have a negative impact on our ability to obtain approval in a different jurisdiction. Approval processes vary among countries and can involve additional product candidate testing and validation and additional administrative review periods. Seeking foreign regulatory approval could require additional non-clinical studies or clinical trials, which could be costly and time consuming. Foreign regulatory approval may include all of the risks associated with obtaining FDA approval. For all of these reasons, if we seek foreign regulatory approval for Twirla or any of our other product candidates, we may not obtain such approvals on a timely basis, if at all.
The process to develop, obtain regulatory approval for and commercialize product candidates is long, complex and costly both inside and outside of the United States, and approval is never guaranteed. Even if our product candidates were to successfully obtain approval from regulatory authorities, any such approval might significantly limit the approved indications for use, including more limited patient populations, require that precautions, contraindications or warnings be included on the
product labeling, including black box warnings, require expensive and time-consuming post-approval clinical studies, risk evaluation and mitigation strategies, or REMS, or surveillance as conditions of approval, or, through the product label, the approval may limit the claims that we may make, which may impede the successful commercialization of our product candidates. For example, we believe that Twirla, if approved, will have labeling consistent with all other marketed hormonal contraceptive products, which include class labeling that warns of risks of certain serious conditions, including venous and arterial blood clots, such as heart attacks, thromboembolism and stroke, as well as liver tumors, gallbladder disease, and hypertension, and a boxed warning regarding risks of smoking and CHC use, particularly in women over 35 years old who smoke. However, regulatory authorities may require the inclusion of additional statements about adverse events in the labeling, including additional black box warnings or contraindications. Following any approval for commercial sale of our product candidates, certain changes to the product, such as changes in manufacturing processes and additional labeling claims, as well as new safety information, will be subject to additional FDA notification, or review and approval. Also, regulatory approval for any of our product candidates may be withdrawn. If we are unable to obtain regulatory approval for our product candidates in one or more jurisdictions, or any approval contains significant limitations, our ability to market to our full target market will be reduced and our ability to realize the full market potential of our product candidates will be harmed. Furthermore, we may not be able to obtain sufficient funding or generate sufficient revenue and cash flows to continue or complete the development of any of our current or future product candidates.
The reported results of the SECURE clinical trial are based on top-line data and may ultimately differ from actual results once additional data are received and fully evaluated.
The reported results of the SECURE clinical trial that we have publicly disclosed, and that are discussed herein, consist of top-line data. Top-line data are based on a preliminary analysis of currently available efficacy and safety data, and therefore the reported results, findings and conclusions related to the SECURE clinical trial are subject to change following a comprehensive review of the more extensive data that we expect to receive related to the SECURE clinical trial. Top-line data are based on important assumptions, estimations, calculations and information currently available to us, and we have not received or had an opportunity to fully and carefully evaluate all of the data related to the SECURE clinical trial. As a result, the top-line results of the SECURE clinical trial that we have reported may differ from future results, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. In addition, third parties, including regulatory agencies, may not accept or agree with our assumptions, estimations, calculations or analyses or may interpret or weigh the importance of data differently, which could impact the potential for approval of Twirla, or if approved, the labeling and commercial value of Twirla and our business in general. If the top-line data that we have reported related to the SECURE clinical trial differ from actual results, our ability to obtain approval for, and commercialize, our products may be harmed, which could harm our business, financial condition, operating results or prospects.
The FDA may disagree with our interpretation of clinical results obtained from the SECURE clinical trial, our results do not guarantee support for a resubmission of our NDA or for regulatory approval, and, even if the SECURE clinical trial data are deemed to be positive by the FDA, the FDA may disagree with other aspects of the SECURE clinical trial and decline to approve Twirla for the proposed indication.
We have reported positive top-line data from the SECURE clinical trial. However, even if we believe that the data from the SECURE clinical trial are positive, the FDA could determine that the data from the SECURE clinical trial were negative or inconclusive or could reach a different conclusion than we did on that same data. Negative or inconclusive results of a clinical trial or difference of opinion could cause the FDA to decline to approve our application or require us to repeat the trial or conduct additional clinical trials prior to obtaining approval for commercialization, and there is no guarantee that additional trials would achieve positive results to the satisfaction of the
FDA or that the FDA will agree with our interpretation of the results. Any such determination by the FDA would delay the timing of our commercialization plan for Twirla or prevent its further development, or the further development of our other product candidates, and adversely affect our business operations. Additionally, the FDA may provide review commentary at any time during the resubmission and review process which could delay the review timeline, adversely affect the review process, or even prevent the approval of Twirla, any of which would adversely affect our business. We may not be able to appropriately remedy issues that the FDA may raise in its review of our NDA resubmission, and we may not have sufficient time or financial resources to conduct future activities to remediate issues raised by the FDA.
There is no guarantee that the data obtained from the SECURE clinical trial will be supportive of, or guarantee, an NDA resubmission, or result in our successfully obtaining FDA approval of Twirla in a timely fashion and for a commercially viable indication, if at all. For example, the FDA could determine that the trial did not meet its objectives or the FDA could still have concerns regarding the conduct of the SECURE clinical trial, including regarding discontinuance of subjects from the trial. At any future point in time, the FDA could require us to complete further clinical or preclinical trials, or take other actions which could delay or preclude any NDA resubmission or approval of the NDA and would require us to obtain significant additional funding. There is no guarantee such funding would be available to us on favorable terms, if at all, nor is there any guarantee that FDA would consider any additional information complete or sufficient to support approval. If the Twirla NDA is resubmitted, the FDA may hold an advisory committee meeting to obtain committee input on the safety and efficacy of Twirla. Typically, advisory committees will provide responses to specific questions asked by the FDA, including the committee's view on the approvability of the product candidate under review. Advisory committee decisions are not binding but an adverse decision at the advisory committee may have a negative impact on the regulatory review of Twirla. Additionally, we may choose to engage in the dispute resolution process with the FDA if we do not receive approval, which could extend the timeline for any potential approval.
Further, if we are able to resubmit an NDA for Twirla with the clinical data from the SECURE clinical trial, there is no guarantee that such data will be deemed sufficient by the FDA. While we designed the protocols for the SECURE clinical trial to address the issues raised in the CRL, there is no guarantee that the FDA will deem such protocols or results from the study sufficient to address those issues when they are formally reviewed as a part of an NDA resubmission or to demonstrate safety and efficacy to the satisfaction of the FDA. The FDA has significant discretion in the review process, and we cannot predict whether the FDA will agree with our conclusions regarding the results of the SECURE clinical trial, including whether our data are reliable and generalizable. For example, the FDA may disagree with our calculations relating to the number of pregnancies occurring on study, or may view the SECURE data as insufficient to demonstrate a favorable benefit/risk profile for approval for the proposed indication. In addition, based on top-line data, the Pearl Index for the overall intent to treat population of subjects 35 years of age and under was 4.80 with an upper-bound of the 95% confidence interval of 6.06, but in the obese subpopulation of subjects 35 years of age and under, the Pearl Index was 6.42 with an upper-bound of the 95% confidence interval of 8.88. If we were to exclude the top-line data on the obese subpopulation, our Pearl Index for non-obese patients was 3.94 with an upper-bound of the 95% confidence interval of 5.35. The highest Pearl Index for a hormonal contraceptive product approved by the FDA to date was 3.19 and the highest upper-bound of the 95% confidence interval was 5.03. In the combined safety database for our three Agile Phase 3 trials (n>3,000), there were 5 subjects with potentially study drug related DVTs or PEs, 4 of whom were obese (BMI³30kg/m2). Although ultimate approvability of a hormonal contraceptive is based on a risk/benefit assessment of the overall safety and efficacy profile of a product, not only a specific Pearl Index, the FDA could conclude that our Pearl Index for either the overall study population or a subgroup of the study population or only the non-obese study population is too high to demonstrate efficacy and an adequate risk/benefit profile, and as such, the FDA could decline to approve Twirla on
this or any other basis. Further, the FDA may not agree with our analysis of the relationship between BMI and efficacy for Twirla and the FDA may interpret our overall data differently than we do and may decline to approve Twirla on this or any other basis.
Moreover, even if we obtain approval of Twirla, any such approval might significantly limit the approved indications for use, including by limiting the approved label for use by more limited patient populations than we propose, require that precautions, contraindications or warnings be included on the product labeling, including black box warnings, require expensive and time-consuming post-approval clinical studies, risk evaluation and mitigation strategies, or REMS, or surveillance as conditions of approval, or, through the product label, the approval may limit the claims that we may make, which may impede the successful commercialization of Twirla. For example, the FDA may deem the higher Pearl Index in the obese subpopulation when combined with safety findings for this subpopulation to warrant a labeling limitation or warning for such subpopulation, which could limit the commercial potential of the product, if approved. Moreover, because we did not conduct any head-to-head studies of Twirla against Ortho Evra, we will not be able to make direct comparative claims regarding the safety, efficacy or pharmacokinetics of Twirla and Ortho Evra or its generic version, Xulane®.
Failure can occur at any stage of clinical development. If the clinical trials for Twirla or any of our current or future product candidates are unsuccessful, we could be required to abandon development.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. A failure of one or more clinical trials can occur at any stage of testing for a variety of reasons. The outcome of preclinical testing and early clinical trials may not be predictive of the outcome of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. In some instances, there can be significant variability in safety or efficacy results between different trials of the same product candidate due to numerous factors, including changes in or adherence to trial protocols, differences in size and type of the subject populations and the rates of dropout among clinical trial subjects. Our future clinical trial results therefore may not demonstrate safety and efficacy sufficient to obtain regulatory approval for our product candidates. For example, we received a CRL from the FDA with respect to an NDA previously filed for Twirla, in which the FDA requested, among other items, additional Phase 3 clinical data to support the application. The SECURE Phase 3 clinical trial was designed in consultation with the FDA and is different than the design of our previous clinical trials of Twirla and it is possible that there could be significant variability in the safety and efficacy results of these trials. Additionally, while our SECURE Phase 3 clinical trial was designed and implemented in a manner to address the FDA's comments and guidance, it is possible that the trial may not be successful or the FDA could conclude the data are not reliable or generalizable. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. Our future clinical trials may not be successful.
Flaws in the design of a clinical trial may not become apparent until the clinical trial is well-advanced. We have limited experience in designing contraceptive clinical trials and may be unable to design and execute clinical trials to support regulatory approval of our product candidates. In addition, clinical trials often reveal that it is not practical or feasible to continue development efforts for a product candidate.
We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to subjects. Furthermore, regulatory agencies, Institutional Review Boards, or IRBs, or data safety monitoring boards, if utilized in our clinical trials, may at any time order the temporary or permanent discontinuation of our clinical trials or request that we cease using certain investigators in the clinical trials if such regulatory agencies or boards believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an
unacceptable safety risk to subjects. Since our inception, we have not voluntarily or involuntarily suspended or terminated a clinical trial due to unacceptable safety risks to subjects.
If the results of the clinical trials for our current product candidates or clinical trials for any future product candidates do not achieve the primary efficacy endpoints or demonstrate unexpected safety issues, the prospects for approval of our product candidates will be materially adversely affected. For example, in the CRL that we received from the FDA in connection with the NDA previously filed for Twirla, one of the FDA's comments was that acceptable evidence of efficacy was not demonstrated, as measured by Pearl Index, or PI. Specifically, in our completed Phase 3 trials, the PI was higher than that seen in registration trials for previously approved hormonal contraceptives. Experts seem to agree that inconsistent or incorrect use is a major contributor to the increased PI seen in more recent contraceptive trials. The PI values from clinical trials are also affected by additional factors, including differences in study design, increased sensitivity of early pregnancy tests, weight and body mass index, or BMI, of the study population and user experience. For example, consistent with other recent hormonal contraceptive clinical trials, including Ortho Evra® and Quartette®, and the 2015 meta-analysis conducted by FDA authors on the effect of obesity on the effectiveness of hormonal contraceptives, a relationship between obesity and efficacy was observed among subjects 35 years of age and under in our SECURE clinical trial. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have failed to achieve similar results in later clinical trials, including longer-term trials, or have failed to obtain regulatory approval of their product candidates. Many compounds that initially showed promise in clinical trials or earlier preclinical studies have later been found to cause undesirable or unexpected adverse effects that have prevented further development of the compound. Our SECURE Phase 3 clinical trial for our primary product candidate, Twirla, may not produce successful results and the FDA may interpret the data from the SECURE trial differently than we do and may decline to approve Twirla on this or any other basis.
In addition to the circumstances noted above, we may experience numerous unforeseen events that could cause our clinical trials to be delayed, suspended or terminated, or which could delay or prevent our ability to receive regulatory approval for or commercialize our product candidates, including:
If we elect or are required to suspend or terminate a clinical trial for any of our product candidates, or our product candidate development is otherwise delayed, our development costs may increase, our commercial prospects will be adversely impacted, any periods during which we may have the exclusive right to commercialize our product candidates may be shortened and our ability to generate product revenues may be delayed or eliminated.
In December 2016, we completed our SECURE Phase 3 clinical trial for Twirla and, as we have previously announced, we expect to conduct additional clinical trials in the future for our other product candidates subject to available funding. Subject enrollment for our future clinical trials, which is a significant factor in the timing of clinical trials, is affected by a variety of factors, including the following:
Furthermore, we plan to rely on a CRO and clinical trial sites to ensure the proper and timely conduct of our clinical trials, and while we may have agreements governing their committed activities, we have limited influence over their actual performance. Additionally, the CRO and clinical trial sites may have business, regulatory, personnel or other issues that keep us from satisfactorily completing our clinical trials. Any delays or unanticipated problems during clinical trials, such as additional monitoring of clinical trial sites, slower than anticipated enrollment in our clinical trials or subjects dropping out of or being excluded from participation in our clinical trials at a higher rate than we anticipate, could increase our costs, slow down our product development and approval process and harm our business. For example, we experienced a slower than expected rate of enrollment for our SECURE Phase 3 clinical trial of Twirla, which we began enrolling in the fourth quarter of 2014, and, as a result, we completed the clinical trial in December 2016.
Regulatory approval may be substantially delayed or may not be obtained for one or all of our product candidates if regulatory authorities require additional time or studies to assess the safety and efficacy of our product candidates.
We may be unable to initiate or complete development of our product candidates on schedule, if at all. The timing for the completion of the studies for our product candidates other than Twirla will require funding beyond our existing cash and cash equivalents. In addition, if regulatory authorities require additional time or studies to assess the safety or efficacy of Twirla, we may not have or be able to obtain adequate funding to complete the necessary steps for approval for any or all of our product candidates. Additional delays may result if the FDA, an FDA Advisory Committee or other regulatory authority recommends non-approval or restrictions on approval. Studies required to demonstrate the safety and efficacy of our product candidates are time consuming, expensive and together take several years or more to complete. In addition, approval policies, regulations or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate's clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval. Delays in regulatory approvals or rejections of applications for regulatory approval in the United States, Europe, Japan or other markets may result from many factors, including:
In December 2016, we completed our Phase 3 SECURE clinical trial and announced top line data in early January 2017. We plan to resubmit our NDA for Twirla in the first half of 2017. The FDA's review of our NDA is subject to all the risks described above in addition to, among other things, the FDA's assessment of our specific response to the 2013 CRL and the efficacy and safety of Twirla as demonstrated in the final SECURE clinical trial results. The lengthy and unpredictable approval process, as well as the unpredictability of future clinical trial results, may result in our failure to obtain regulatory approval to market Twirla or any of our other product candidates, which would significantly harm our business, results of operations and prospects.
Changes in regulatory requirements and guidance may also occur and we may need to amend clinical trial protocols submitted to applicable regulatory authorities or conduct additional studies to reflect these changes. Amendments and additional studies may require us to resubmit clinical trial protocols to Institutional Review Boards and regulatory authorities for re-examination, which may impact the costs, timing or successful completion of a clinical trial.
If we are required to conduct additional clinical trials or other studies with respect to any of our product candidates beyond those that we contemplated, if we are unable to successfully complete our clinical trials or other studies or if the results of these studies are not positive or are only modestly positive, we may be delayed in obtaining regulatory approval for our product candidates, we may not be able to obtain regulatory approval at all or we may obtain approval for indications that are not as
broad as intended. For example, the FDA issued a CRL in response to our NDA for Twirla requesting, among other items, an additional Phase 3 clinical study, which has delayed our ability to obtain regulatory approval for that product candidate. We may also experience delays due to changes in regulatory requirements and guidance, which may require protocol amendments or the conduct of additional studies. These amendments and additional studies may require regulatory or IRB approval. The approval and conduct of these studies may delay, limit or preclude regulatory approval for our product candidates. Our product development costs will also increase if we experience delays in testing or approvals and we may not have sufficient funding to complete the testing and approval process for any of our product candidates. Significant clinical trial delays could allow our competitors to bring products to market before we do and impair our ability to commercialize our products if and when approved. If any of this occurs, our business will be materially harmed.
Our product candidates may have undesirable adverse effects, which may delay or prevent regulatory approval or, if approval is received, require our products to be taken off the market, require them to include safety warnings or otherwise limit their sales.
Unforeseen adverse effects from any of our product candidates could arise either during clinical development or, if approved, after the approved product has been marketed. In the combined safety population of our previously completed Phase 3 trials, there were a total of 22 serious adverse events, or SAEs, of which 16 occurred in the Twirla cohort, which had approximately 2.3 times as many subjects as the oral contraceptive comparator cohort. Three of the 16 SAEs in the Twirla cohort (0.2% of the overall Twirla safety population) were considered to be possibly related to Twirla, and included one drug overdose with Benadryl, one case of uncontrollable nausea and vomiting and one instance of deep vein thrombosis, or DVT. In addition to the SAEs described above, some subjects taking Twirla experienced non-serious adverse events, such as nausea, headache, application site irritation and breast tenderness. Subjects receiving the oral contraceptive comparator also experienced non-serious adverse events such as nausea, headache and breast tenderness, though at different rates. In the SECURE clinical trial, SAEs were observed in 2.0% of the SECURE trial population, and 0.6% of subjects had SAEs that were considered potentially study drug related, including DVT, pulmonary embolism, or PE, gallbladder disease, ectopic pregnancy, and depression. In the combined safety database for the three Agile Phase 3 trials (n >3,000), there were 5 subjects with potentially study drug related DVTs or PEs, 4 of whom were obese (BMI >30kg/m2).
Any undesirable adverse effects that may be caused by our product candidates could interrupt, delay or halt clinical trials and could result in more restrictive labeling or the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications, and in turn prevent us from commercializing our product candidates and generating revenues from their sale. For instance, FDA may determine that for specific subgroups of patients. Twirla has lower efficacy and presents a higher risk. Accordingly, FDA may not approve our Twirla NDA or may require labeling restrictions. By example, FDA may require labeling restrictions on the use of Twirla for patients in certain BMI categories. Adverse effects could also impact subject recruitment or the ability or willingness of enrolled subjects to complete the trial, or result in product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
In addition, if any of our product candidates receive regulatory approval and we or others later identify undesirable adverse effects caused by the product, we could face one or more of the following consequences:
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing such product, which in turn could delay or prevent us from generating significant revenues from its sale.
Our development and commercialization strategy for Twirla depends, in part, on published scientific literature and the FDA's prior findings regarding the safety and efficacy of approved products containing Ethinyl Estradiol and Levonorgestrel based on data not developed by us, but upon which the FDA may rely in reviewing our NDA.
The Hatch-Waxman Act added Section 505(b)(2) to the Federal Food, Drug and Cosmetic Act, or FDCA, Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. The FDA interprets Section 505(b)(2) of the FDCA, for purposes of approving an NDA, to permit the applicant to rely, in part, upon published literature or the FDA's previous findings of safety and efficacy for an approved product. The FDA may also require companies to perform additional clinical trials or measurements to support any deviation from the previously approved product. The FDA may then approve the new product candidate for all or some of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant. The label, however, may require all or some of the limitations, contraindications, warnings or precautions included in the reference product's label, including a black box warning, or may require additional limitations, contraindications, warnings or precautions. We have submitted an NDA for Twirla under Section 505(b)(2) and as such the NDA relied, in part, on the FDA's previous findings of safety and efficacy from investigations for approved products containing ethinyl estradiol, or EE, and levonorgestrel, or LNG and published scientific literature for which we have not received a right of reference. We received a CRL in response to our Section 505(b)(2) NDA for Twirla, in which the FDA requested, among other things, that we conduct an additional Phase 3 clinical trial. Even though we may be able to take advantage of Section 505(b)(2) to support potential U.S. approval for Twirla, the FDA may require us to perform additional clinical trials or measurements to support approval over and above the clinical trials that we have already
completed. In addition, notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years some pharmaceutical companies and others have objected to the FDA's interpretation of Section 505(b)(2). If the FDA changes its interpretation of Section 505(b)(2), or if the FDA's interpretation is successfully challenged in court, this could delay or even prevent the FDA from approving any Section 505(b)(2) NDAs that we submit. Such a result could require us to conduct additional testing and costly clinical trials, which could substantially delay or prevent the approval and launch of our product candidates, including Twirla.
Risks Related to Our Financial Position and Need for Capital
We have never been profitable. Currently, we have no products approved for commercial sale, no source of revenue and we may never become profitable.
We have never been profitable and do not expect to be profitable in the foreseeable future. We have no products approved for commercial sale and to date have not generated any revenue from product sales. Our ability to generate revenue and become profitable depends upon our ability to successfully complete the development of and obtain the necessary regulatory approvals for our product candidates. We have been engaged in developing Twirla and our Skinfusion® technology since our inception. To date, we have not generated any revenue from Twirla, and we may never be able to obtain regulatory approval for the marketing of Twirla. Further, even if we are able to gain approval for and commercialize Twirla or any other product candidate, there can be no assurance that we will generate significant revenues or ever achieve profitability. Our ability to generate product revenue depends on a number of factors, including our ability to:
In addition, because of the numerous risks and uncertainties associated with product candidate development, we are unable to predict the timing or amount of increased expenses, or when, or if, we will be able to achieve or maintain profitability. In addition, our expenses could increase beyond our current expectations if we are required by the FDA or other regulatory authorities to perform studies in addition to those that we currently anticipate. Even if our product candidates are approved for commercial sale, we anticipate incurring significant costs associated with the commercial launch of these products.
Our ability to become and remain profitable depends on our ability to generate revenue. Even if we are able to generate revenues from the sale of our products, if approved, we may not become profitable and may need to obtain additional funding to continue operations. If we fail to become profitable or obtain additional funding, or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce our operations. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would decrease the value of our company and could impair our ability to raise capital, expand our business or continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
We have incurred operating losses in each year since our inception and expect to continue to incur substantial losses for the foreseeable future.
We have incurred losses in each year since our inception in December 1997. Our net loss was $ 28.7 million, $30.3 million and $16.1 million for the years ended December 31, 2016, 2015 and 2014, respectively. As of December 31, 2016, we had an accumulated deficit of $193.5 million.
Specialty pharmaceutical product development is a speculative undertaking, involves a substantial degree of risk and is a capital-intensive business. We expect to incur expenses without corresponding revenues until we are able to obtain regulatory approval and subsequently sell Twirla in significant quantities, which may not happen. We have devoted most of our financial resources to research and development, including our non-clinical development activities and clinical trials. We expect to incur increased expenses as we complete the development of Twirla, respond to the CRL and supplement our NDA with the results of the SECURE trial, complete the qualification and validation of our commercial manufacturing process, initiate pre-launch commercial activities, commercially launch Twirla, advance our other product candidates and expand our research and development programs. Substantially all of our resources are currently dedicated to developing and seeking regulatory approval for Twirla. We will require additional capital for the commercial launch of Twirla, if approved, as well as advancing the development of our other product candidates. To date, we have financed our operations primarily through sales of common stock, convertible preferred stock and convertible promissory notes and to a lesser extent, through term loans and government grants. Our product candidates will require the completion of regulatory review, significant marketing efforts and substantial investment before they can provide us with any revenue.
Assuming we obtain FDA approval, we expect that our expenses will increase as we prepare for the commercial launch of Twirla. As a result, we expect to continue to incur substantial losses for the foreseeable future, and these losses may increase. We are uncertain when or if we will be able to achieve or sustain profitability. If we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Failure to become and remain profitable would impair our ability to sustain operations and adversely affect the price of our common stock and our ability to raise capital.
If we fail to obtain the capital necessary to fund our operations, we may be unable to obtain regulatory approval of or commercialize Twirla in the United States and we could be forced to share our rights to commercialize Twirla with third parties on terms that may not be favorable to us.
We need large amounts of capital to support our development and commercialization efforts for Twirla. If we are unable to secure sufficient capital to fund our operations, we will not be able to continue these efforts and we might have to enter into strategic collaborations that could require us to share commercial rights to Twirla with third parties in ways that we currently do not intend or on terms that may not be favorable to us. Our cash and cash equivalents were $48.8 million as of December 31, 2016. Based on our current business plan, we believe that our cash and cash equivalents as of December 31, 2016 will be sufficient to meet our operating requirements into the second quarter of 2018. Our current business plan assumes resubmission of the NDA for Twirla in the first half of 2017, a six month FDA review of our resubmission, initiation of pre-commercial activities and initiation of validation of our commercial manufacturing process in coordination with the commercialization of Twirla. In the event of unforeseen changes to our planned timelines, we have the ability to postpone certain commercial and validation spending in order to continue the funding of our operations into the second quarter of 2018. We anticipate requiring additional capital to fund operating needs thereafter, including among other items, the commercial launch for Twirla and advancing the development of our other product candidates. We may also need to raise additional funds sooner if we choose to expand more rapidly than we presently anticipate or we encounter any unforeseen events that affect our current business plan. Adequate additional funding may not be available to us on acceptable terms, or at all. If we are unable to raise capital when needed or on attractive terms and not enter into strategic
collaborations, we would be forced to delay, reduce or eliminate our research and development programs or future commercialization efforts.
Our operating activities may be restricted as a result of covenants related to the outstanding indebtedness under our loan agreement and we may be required to repay the outstanding indebtedness in an event of default, which could have a materially adverse effect on our business.
In February 2015, we entered into a loan and security agreement, referred to herein as the Hercules Loan Agreement, with Hercules Capital, Inc., or Hercules, for a term loan of up to $25.0 million. The Hercules Loan Agreement was amended effective August 25, 2016. A first tranche of $16.5 million was funded upon execution of the Hercules Loan Agreement, approximately $15.5 million of which was used to repay our term loan with Oxford. Under terms of the Hercules Loan Agreement, we may, but are not obligated to, draw an additional tranche of up to $8.5 million through March 31, 2017, subject to the achievement of certain clinical milestones. We are currently in discussions with Hercules to extend the period during which the additional tranche of $8.5 million may be drawn. We can make no assurances that our discussions will ultimately be successful and, if such discussions result in an extension of the period in which we may draw the additional tranche of $8.5 million, we could incur additional fees payable to Hercules.
The Hercules Loan Agreement subjects us to various customary covenants, including requirements as to financial reporting and insurance, and restrictions on our ability to dispose of our business or property, change our line of business, liquidate or dissolve, enter into any change in control transaction, merge or consolidate with any other entity or acquire all or substantially all the capital stock or property of another entity, incur additional indebtedness, incur certain types of liens on our property, including our intellectual property, pay any dividends or other distributions on our capital stock other than dividends payable solely in capital stock or redeem our capital stock. Our business may be adversely affected by these restrictions on our ability to operate our business.
The Hercules Loan Agreement is secured by substantially all of our property other than our intellectual property. As a result of the amendment to the Hercules Loan Agreement, we are currently required to make interest-only payments through January 2017. On February 1, 2017, we began making principal payments with respect to the Hercules Loan. The Hercules Loan Agreement currently bears interest at rate of 9.0% per annum and matures on December 1, 2018.
Additionally, we may be required to repay the outstanding indebtedness under the term loan if an event of default occurs under the Hercules Loan Agreement. Under the Hercules Loan Agreement, an event of default will occur if, among other things, we fail to make payments under the Hercules Loan Agreement we breach any of our covenants under the Hercules Loan Agreement, subject to specified cure periods with respect to certain breaches; Hercules determines in good faith that we are unable to satisfy our obligations under the Hercules Loan Agreement as they become due and that our principal investors do not intend to fund amounts necessary to satisfy such obligations; we or our assets become subject to certain legal proceedings, such as bankruptcy proceedings; we are unable to pay our debts as they become due; or we default on contracts with third parties which would permit Hercules to accelerate the maturity of such indebtedness or that could have a material adverse effect on us. We may not have enough available cash or be able to raise additional funds through equity or debt financings to repay such indebtedness at the time any such event of default occurs. In that case, we may be required to delay, limit, reduce or terminate our product candidate development or commercialization efforts or grant to others rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves. Hercules could also exercise its rights as collateral agent to take possession and dispose of the collateral securing the loan for its benefit, which collateral includes all of our property other than our intellectual property. Our business, financial condition and results of operations could be materially adversely affected as a result of any of these events.
We will need to obtain additional financing to fund our operations and, if we are unable to obtain such financing, we may be unable to complete the development and commercialization of our product candidates.
Our operations have consumed substantial amounts of cash since inception. From our inception to December 31, 2016, we have cumulative net cash flows used by operating activities of $170.1 million. We will need to obtain additional financing to fund our future operations, including completing the development and commercialization of our product candidates. We will need to obtain additional financing to conduct additional trials for the approval of our product candidates if requested by regulatory authorities, and to complete the development of any additional product candidates we might acquire. Moreover, our fixed expenses such as rent, interest expense and other contractual commitments are substantial and are expected to increase in the future.
Our future funding requirements will depend on many factors, including, but not limited to:
Until we can generate a sufficient amount of revenue, we may finance future cash needs through public or private equity offerings, license agreements, debt financings, collaborations, strategic alliances and marketing or distribution arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available, we may be required to delay or reduce the scope of or eliminate one or more of our research or development programs or our commercialization efforts. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time. In addition, if we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or product candidates or to grant licenses on terms that may not be favorable to us.
Based on our current business plan, we believe that our cash and cash equivalents as of December 31, 2016 will be sufficient to meet our operating requirements into the second quarter of 2018. Our current business plan assumes resubmission of the NDA for Twirla in the first half of 2017, a six month FDA review of our resubmission and successful completion of validation of its commercial manufacturing process in coordination with the commercialization of Twirla. We expect that these funds will not be sufficient to enable us to complete all necessary development of our product candidates other than Twirla, or commercially launch Twirla or our other current product candidates. Accordingly, we will be required to obtain further funding through other public or private offerings, debt financing, collaboration or licensing arrangements or other sources. Adequate additional funding may not be available to us on acceptable terms, or at all. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our research and development programs or future commercialization efforts. Our forecast of the period of time through which our financial resources will be adequate to support our operating requirements is a forward- looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed elsewhere in this "Risk Factors" section. We have based this estimate on a number of assumptions that may prove to be wrong, and changing circumstances beyond our control may cause us to consume capital more rapidly than we currently anticipate. Our inability to obtain additional funding when we need it could seriously harm our business.
Raising additional capital may cause dilution to our existing stockholders or restrict our operations.
We may seek additional capital through a combination of private and public equity offerings, debt financings and strategic collaborations. The sale of additional equity or convertible debt securities could result in the issuance of additional shares of our capital stock and could result in dilution to our stockholders. The incurrence of indebtedness would result in increased fixed payment obligations and could also result in certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. We cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we will be prevented from pursuing research and development efforts. This could harm our business, operating results and financial condition and cause the price of our common stock to fall.
We are a development stage company which may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
We are a development stage company. We were incorporated and commenced active operations in 1997. Our operations to date have been limited to organizing and staffing our company, business planning, raising capital and developing our product candidates. We have not yet demonstrated our ability to successfully complete a Phase 3 registration trial for, obtain regulatory approval of, or manufacture on a commercial scale any of our product candidates, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, any predictions about our future success or viability may not be as accurate as they could be if we had a longer operating history.
In addition, as a development stage company, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors. We will need to transition from a company with a focus on product candidate development to a company capable of supporting commercial activities. We may not be successful in such a transition.
Risks Relating to the Commercialization of Our Product CandidatesTwirla
We are substantiallysignificantly dependent on the commercial success of Twirla.Twirla, our only approved product. If we are unable to successfully commercialize Twirla, our business, financial condition, results of operations, and prospects and value of our common stock will be materially adversely affected.
Assuming FDA approval, Twirla will be the first product that we commercialize. The rest of our pipeline of productspotential product candidates are in earlier stages of clinical development and will require additional clinical studies and product development and funding in order to advance towards commercialization, which could take considerable time. If Twirla is not approved, our ability to advance our pipeline would be significantly adversely affected. In addition, we will require additional capital for the validation of our commercial manufacturing process and commercial launch of Twirla. We will not be able to commercially launch Twirla until validation is complete and we may not be successful in completing validation within expected timelines or at all. We will also require capital to develop and initiate post-marketing studies of Twirla. Our ability to generate revenues and become profitable will depend in large part on the commercial success of Twirla. Potential prescribers of Twirla include physicians, nurse practitioners, (NPs),or NPs, physician's assistants, (PAs)or PAs, and pharmacists. Registered Pharmacists (RPh) are authorized to prescribe contraceptives in some states, currently, and othersother states have pending legislation that would allow pharmacists to prescribe contraceptives. If Twirla or any other product that we commercialize in the future does not gain an adequate level of acceptance among prescribers, patients and third parties, we may not generate significant product revenues or become profitable. Market acceptance of Twirla and any other product that we commercialize, by prescribers, patients and third partythird-party payors will depend on a number of factors, some of which are beyond our control, including:
thromboembolic events. Twirla's label also contains a limitation of use restrictions imposed by the FDA or to which we agree as partconsider Twirla's reduced effectiveness in women with a BMI of a mandatory REMS or voluntary risk management plan.³25 to£30 kg/m2 before prescribing.
For example, if Twirla is approved by the FDA, prescribers and patients may not be immediately receptive to a transdermal contraceptive system, as opposed to a pill or any other method, and may be slow to adopt it as an accepted treatment for the prevention of pregnancy. In addition, even though we believe Twirla has significant advantages over other treatment options, because no adequate head-to-head trials comparing the safety and efficacy of Twirla to the competing approved patch productor other contraceptive products have been conducted, the prescribing information approved by the FDA may not containwe cannot make claims that Twirla is safer or more effective than the currently approved patch product, or other claims that may be necessary for successful marketing of Twirla. Accordingly,contraceptive products, without conducting a supportive head-to-head postmarketing study. Moreover, we will not be permittedable to promotemake any other Twirla if approved, for any comparative advantagesmarketing or promotional claims to the currently marketed contraceptive patch.extent that they are inconsistent with the Twirla FDA-approved label or are not otherwise supported. The availability of numerous inexpensive generic forms of contraceptive products may also limit acceptance of Twirla among prescribers, patients and third partythird-party payors. If Twirla does not achieve an adequate level of acceptance among prescribers, patients and third partythird-party payors, we may not generate significant product revenues or become profitable.profitable, and the value of our common stock may suffer.
We may not be able to successfully commercialize Twirla, and the revenue that we generate from its sales, if any, may be limited.
The commercial success of Twirla will depend upon the contraceptive market landscape as well as acceptance and uptake of Twirla by prescribers, patients and third-party payors.
Risks related to the contraceptive market landscape include:
The degree of acceptance and uptake of Twirla by prescribers, patients and third-party payors will depend upon a number of factors, including:
In addition, we may face additional generic or other drug product competition sooner than we anticipate for Twirla or our potential product candidates, which would potentially limit their commercial success. We believe that Twirla is eligible for three years of FDA marketing exclusivity for Twirla. The FDCA provides a period of three years of marketing exclusivity for an NDA, Section 505(b)(2) NDA or supplement to an existing NDA for a drug product that contains a previously approved active moiety, if new clinical investigations, other than bioavailability or bioequivalence studies, were conducted or sponsored by the applicant and are determined by the FDA to be essential to the approval of the application. This three-year marketing exclusivity, however, does not protect drug products from all competition. For instance, it does not protect against the approval of a full NDA. It also would only protect against the approval of a product that contains the same conditions of approval as Twirla. We, however, may not receive the three-year exclusivity for them, and, even if we do, it may not adequately protect us from competition. Competition that Twirla and our potential product candidates may face from generic or similar versions of the same or similar products could materially and adversely impact our future revenue, profitability and cash flows and substantially limit our ability to obtain a return on the investments we have made in Twirla or our potential product candidates.
If Twirla does not achieve an adequate level of acceptance by prescribers, third-party payors and patients, we may not generate sufficient revenue and we may not be able to achieve or sustain profitability. Our efforts to educate prescribers, patients and third-party payors on the benefits of Twirla may require significant resources and may never be successful. Even if we are able to demonstrate and maintain a competitive advantage over our competitors and become profitable, if the market for hormonal contraceptives fails to achieve expected future growth or decreases, we may not be able to generate sufficient revenue or sustain profitability. Our ability to generate sufficient revenue from Twirla will also be dependent on our ability to support the commercial demand for Twirla and we cannot assure you that we along with our manufacturing partner Corium will be able to complete validation of our commercial manufacturing successfully and in a timely manner, and, ultimately, to commercially market Twirla. We also cannot assure that we and Corium will be able to manufacture sufficient quantities of Twirla in order to meet commercial demand.
It will be difficult for us to profitably sell Twirla if approved, or any other product that we obtain marketing approval for in the future if coverage and reimbursement for such product is limited.
Market acceptance and sales of Twirla if approved, or any other product that we obtain marketing approval for in the future, will depend on coverage and reimbursement policies and may be affected by future healthcare reform measures. Government authorities and third partythird-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels for approved medications. A primary trend in the U.S. healthcare industry is cost containment. Government authorities and these third partythird-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. We cannot be sure that coverage or reimbursement will be available for Twirla if approved, or any other product that we obtain marketing approval for in the future and, if coverage is available, we cannot be sure of the level of reimbursement. Even when a payor determines that a product is eligible for reimbursement, the payor may set a reimbursement rate that is too low to support a profitable sales price for the product. Subsequent approvals of competitive products could result in a detrimental change to the reimbursement of our products. Reimbursement may impact the demand for, or the price of, Twirla, if approved, and any other products that we obtain marketing approval for and commercialize.Twirla. Numerous generic products may be available at lower prices than branded therapy products, such as Twirla, which may also reduce the likelihood and level of reimbursement for Twirla or other products.Twirla. If coverage and reimbursement are not available or are available only at limited levels, we may not be able to successfully commercialize Twirla, if approved, or any other product for which we obtain marketing approval.would adversely impact our business, financial condition, results of operations and prospects and the value of our common stock.
If we are unable to establish effective marketing and sales capabilities for Twirla if approved, or enter into agreements with third parties to market and sell Twirla, we may be unable to generate product revenues.
We are seeking approval for Twirla from the FDA for a contraception indication. Following our original submission of the NDA, we received a CRL from the FDA requesting, among other things, additional Phase 3 data. Our ability to commercialize Twirla, and the timing of Twirla commercialization, is dependent on FDA's review of our data from the SECURE trial and our NDA for Twirla, and other items such as timely and successful completion of validation of equipment for commercial manufacturing, ultimate FDA approval, and additional capital. In our current business plan, we have assumed resubmission of our NDA for Twirla to the FDA in the first half of 2017, a six-month FDA review of our resubmission and completion of validation of our commercial manufacturing process in coordination with our commercialization of Twirla. We cannot assure you that the FDA will approve Twirla or that the FDA's timeline for review will be within six months.
At present, we have no sales personnel and a limited number of marketing personnel. Depending on our available capital resources,Initially, we do not intendplan to begin to hire additional marketing personnel until shortly prior to the final submission to our NDA or establish our own sales force orforce. Rather, we plan to engage a contract sales organization in the United States until shortly priorand, depending on our available capital resources, we plan to FDA approvalalso hire a limited number of Twirla.additional marketing personnel in the second and third quarters of 2020. At the time of our anticipated commercial launch of Twirla, assuming regulatory approval by the FDA, our sales and marketing team will have worked together for only a limited period of time. If our regulatory review period by the FDA is extended beyond six months, we may need to postpone initiating certain commercial activities in order to preserve cash, in which case our ability to launch Twirla would be compromised. We cannot guarantee that we will be successful in marketing Twirla in the United States.
We may not be able to establish our own sales forcemarketing capabilities or a contract sales force in a cost-effective manner or realize a positive return on this investment. In addition, we will have to compete with other pharmaceutical and biotechnology companies to recruit, hire, train and retain sales and marketing personnel. Factors that may inhibit our efforts to commercialize Twirla if approved, in the United States without strategic partners or licensees include:
If we are not successful in recruiting sales and marketing personnel or in building a sales and marketing infrastructure, or if we do not successfully enter into appropriate collaboration arrangements, we will have difficulty commercializing Twirla, which would adversely affect our business, operating results and financial condition.
If we intend to commercialize Twirla outside the United States, we will likely enter into collaboration agreements with pharmaceutical partners, and we may have limited or no control over the sales, marketing and distribution activities of these third parties. Our future revenues may depend on the success of the efforts of these third parties.
To the extent that we rely on, or partner with, third parties to commercialize Twirla, if approved, or any other product candidate for which we obtain marketing approval in the future, we may receive less revenue than if we commercialized these products ourselves. In addition, we would have less control over the sales efforts of any other third parties involved in our commercialization efforts. We, however, will remain responsible for the conduct of any contract sales force, which could expose us to legal and regulatory enforcement actions and liability. In the event that we are unable to partner with a third partythird-party marketing and sales organization, our ability to generate product revenues may be limited in the United States, internationally or both.
A varietyIf estimates of risks associated withthe size of the potential internationalmarket for Twirla are overstated or data we have used to identify physicians is inaccurate, our ability to earn revenue to support our business relationships could be materially adversely affect our business.affected.
We may enter into agreements with third partieshave relied on a number of external sources, as well as market research funded by us and internal analyses and calculations, to estimate the potential market opportunity for Twirla in the United States. We have not independently verified the externally sourced information used to develop the estimates for the development and commercialization ofpotential market for Twirla, and possibly othertheir accuracy and completeness cannot be assured. Similarly, our internal analyses and calculations are based upon analysis of the current and expected future U.S. contraceptive market and management's understanding and assessment of numerous inputs and market conditions, including, but not limited to, the addressable market segment for combined hormonal contraceptives and the reimbursement status of contraceptives under the federal Affordable Care Act and similar state laws. These understandings and assessments necessarily require assumptions subject to significant judgment and may prove to be inaccurate. As a result, our estimates of the size of the potential market for Twirla could prove to be overstated, perhaps materially.
In addition, we are relying on third party data to identify the healthcare providers who prescribe contraception in the U.S. and to determine how to deploy resources to market to those healthcare providers; however, we may not be marketing to the appropriate physicians and may therefore be limiting our market opportunity.
We may develop estimates with respect to market opportunities for potential product candidates in international markets. If we do so, wethe future, and such estimates would be subject to additional risks related to entering into international business relationships, including:
These and other risks may materially adversely affect our ability to develop and commercialize products in international markets and may harm our business.
similar risks. Even if we receive regulatory approval for Twirla, we still may not be able to successfully commercialize it and the revenue that we generate from its sales, if any, may be limited.
The commercial success of Twirla in any indication for which we obtain marketing approval from the FDA or other regulatory authorities will depend upon the contraceptive market landscape as well as acceptance and uptake of Twirla by prescribers, patients and third-party payors.
Risks related to the contraceptive market landscape include:
The degree of acceptance and uptake of Twirla, if approved, by prescribers, patients and third-party payors will depend upon a number of factors, including:
In addition, even if we obtain regulatory approval the timing of an approval may reduce our ability to commercialize Twirla successfully. For example, if the approval process takes too long, we may miss market opportunities, give other companies the ability to develop competing products, and require us to raise additional capital, which could delay our commercial launch. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render Twirla not commercially viable. For example, regulatory authorities may grant approval contingent on the performance of costly post- marketing clinical trials or other post-marketing commitments, including REMS, or may approve Twirla with a label that contains fewer,for one or more limited, indications than requested, warnings, precautions or contraindications, including black box warnings, and the label may not include the claims necessary or desirable for the successful commercialization of Twirla. Any of the foregoing scenarios could materially harm the commercial prospects for Twirla.
Moreover, we may face additional generic or other drugpotential product competition sooner than we anticipate for Twirla or our other product candidates, which would potentially limit their commercial success. We believe that we may be eligible for three yearsunable to commercialize the product on a scale sufficient to generate significant revenue, which could have a material adverse effect on our business, financial condition, results of FDA marketing exclusivity for Twirlaoperations and our other product candidates. The FDCA provides a period of three years of marketing exclusivity for an NDA, Section 505(b)(2) NDA or supplement to an existing NDA for a drug product that contains a previously approved active moiety, if new clinical investigations, other than bioavailability or bioequivalence studies, were conducted or sponsored byprospects and the applicant and are determined by the FDA to be essential to the approval of the application. This three year marketing exclusivity, however, does not protect drug products from all competition. For instance, it does not protect against the approval of a full NDA. It also would only protect against the approval of a product that contains the same conditions of approval as our product candidates. We may not receive the three year exclusivity for anyvalue of our product candidates, and, even if we do, it may not adequately protect us from competition. Competition that our product candidates may face from generic or similar versions of our product candidates could materially and adversely impact our future revenue, profitability and cash flows and substantially limit our ability to obtain a return on the investments we have made in those product candidates.
If Twirla is approved, but does not achieve an adequate level of acceptance by prescribers, third-party payors and patients, we may not generate sufficient revenue and we may not be able to achieve or sustain profitability. Our efforts to educate prescribers, patients and third party payors on the benefits of Twirla may require significant resources and may never be successful. Even if we are able to demonstrate and maintain a competitive advantage over our competitors and become profitable, if the
market for hormonal contraceptives fails to achieve expected future growth or decreases, we may not generate sufficient revenue or sustain profitability.common stock.
The proportion of the contraceptive market that is made up of generic products continuescould continue to increase, making introduction of a branded contraceptive difficult and expensive.
The proportion of the U.S. market that is made up of generic products has been increasing over time. InFor example, in 2005, generic contraceptive products held 47%49% of prescription volume and 34%36% of sales and, by 2011,2019, those values had risen to 68%88% and 44%43%, respectively. ForRecently, Congress and the year ended December 31, 2016, approximately 83% of the prescription volume and approximately 43% of sales of combined hormonal contraceptives, or CHCs,FDA have taken steps to increase generic competition in the U.S. were generated by generic products.market. If this trend continues, it may be more difficult to introduce Twirla if approved, as a branded contraceptive at a price that will maximize our revenue and profits. Also, there may be additional marketing costs to introduce Twirla in order to overcome the
trend towards generics and to gain access to reimbursement by payors. If we are unable to introduce Twirla at a price that is commensurate with that of current branded contraceptive products, or we are unable to gain reimbursement from payors for Twirla, or if patients are unwilling to pay any price differential between Twirla and a generic contraceptive, our revenues will be limited. For example, in light of the introduction of the branded generic version of the Ortho Evra product by Mylan Inc. in April 2014, and the subsequent discontinuation of distribution of Ortho Evra in October 2014 by Janssen we may have to take additional measures in order to be competitive and gain market share,share. For example, we may increase the rebates available to commercial payors or we may provide incentives to consumers covered by non-governmental payors, such as coupons or rebates, in order to make up for the difference in the co-payment for Twirla and the generic patch product.
Prescribers, patients and payors may not adopt a new contraceptive patch due to concerns based upon the prior experience with or perception of the previously marketed contraceptive patch and its currently marketed contraceptive patch.generic equivalent product.
The Ortho Evra® contraceptive patch, or Evra, was introduced in early 2002 and was the first FDA-approved contraceptive patch. The following is a brief history of the Evra market experience:
vote, concluded that the benefits of Evra outweighed the risks, but that the current package insert did not adequately reflect the risk/benefit profile.
We have conducted pharmacokinetic studies of Twirla to demonstrate that it delivers a daily EE dose of approximately 30 micrograms, comparablewhich is less than that delivered by Xulane, according to a low-dose oral contraceptive.its FDA approved label. However, because none of our completed or plannedPhase 3 clinical trials studied or expect to study Twirla in a head-to-head comparison with Evra, if Twirla is approved by the FDA,Xulane, we will not be able to make direct comparative claims regarding the EE exposure, safety, andor efficacy of Twirla as compared to Evra. While we expect Twirla, if approved, to have the same black box warning currently required for all CHCs, we cannot predict whether the FDA will require that we include information in the Twirla labeling or black box warning regarding the additional risks associated with the Evra patch. Assuming approval, if we are not able to convince prescribers, patients and payors that Twirla deliversXulane without conducting a low daily dose of EE, this may limitsupportive head-to-head post-marketing study., As a result, uptake and usage of Twirla and our revenuerelated revenues could ultimately be limited.
Twirla could develop unexpected safety, efficacy or quality concerns, which would likely have a material adverse effect on us.
Twirla was approved in the U.S. based on the SECURE clinical trial, in which patients were enrolled for 13 cycles of treatment. Twirla will now be limited.used by larger numbers of patients, potentially for longer periods of time, and we and others (including regulatory agencies and private payors) will endeavor to collect extensive information on the efficacy and safety of Twirla by monitoring its use in the marketplace. In addition, we will endeavor to conduct a long-term post marketing safety study required by the FDA to compare the risks for venous thromboembolism (VTE) and arterial thromboembolism (ATE) in new users of Twirla to new users of oral combined hormonal contraceptives (CHCs) and new users of Xulane in U.S. women of reproductive age. Further, we may conduct additional trials in connection with lifecycle management programs for Twirla. New safety or efficacy data from both market surveillance and our post-marketing clinical trials may result in negative consequences including:
Furthermore, the discovery of significant problems with a product similar to Twirla that implicate (or are perceived to implicate) the entire class of products could have an adverse impact on our ability to commercialize Twirla. Any of these circumstances could reduce Twirla's market acceptance and would inhibit or delay our ability to commercialize Twirla or gain and/or sustain market share, any of which would adversely affect sales of Twirla.
We face competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive. We would have significant competition with contraceptive products already in the marketplace, many of which have substantially greater name recognition, commercial infrastructures and financial, technical and personnel resources than we have. Any new product that competes with a previously approved product may need to demonstrate compelling advantages in efficacy, convenience, tolerability or safety to be commercially successful. In addition, new products developed by others could emerge as competitors to Twirla, if approved.Twirla. If we are not able to compete effectively against our current and future competitors, our business will not grow, and our financial condition and operations will suffer.
Our potential competitors include, but are not limited to, large, well-established pharmaceutical companies, and specialty pharmaceutical sales and marketing companies. These companies include Merck & Co., Inc., or Merck, which markets Nuvaring®, TherapeuticsMD, Inc., or TherapeuticsMD, which has licensed and will market Annovera®, a recently approved contraceptive ring, Allergan, Inc., or Allergan, which markets several branded and generic contraceptives including Loestrin® 24, Minastrin® 24, and LoLoestrin®, Teva Pharmaceutical Industries Ltd., or Teva, which markets several branded and generic contraceptives including Gianvi® and Quartette®Taytulla®, Bayer AG, or Bayer, which markets Beyaz®, Yaz®, Yasmin®, and Mirena®Natazia®, Johnson & Johnson, which markets Ortho-Tri-Cyclen® Lo, Pfizer Inc.and Mylan N.V., which markets Alesse® and Mylan Inc. which markets Xulane™Xulane®, a generic version of Ortho Evra. Additionally, several generic manufacturers currently market and continue to introduce new generic contraceptives, including Sandoz International GmbH, Glenmark Pharmaceuticals Ltd., Lupin Pharmaceuticals, Inc., and Amneal Pharmaceuticals, LLC.Inc.
There are other contraceptive product candidates in development that, if approved, would potentially compete with Twirla. Specifically, Bayer has a contraceptive patch approved in the European Union, or E.U. Bayer entered into a license and distribution agreement for the sale of this contraceptive patch in Europe with Gedeon Richter Ltd. Other companies that have new hormonal contraceptive product candidates in various stages of development include Teva (oral contraceptiveAllergan (progestin-only vaginal ring for which they received a CRL from the FDA), The Population Council in Phase 3), Merck (vaginal ring and oral contraceptive in Phase 3), Allergan (vaginal ring in Phase 3) andcollaboration with Antares Pharma, Inc. (transdermal gel contraceptive in Phase 2), Mithra Pharmaceuticals SA (combination oral contraceptive in Phase 3), and Panterhei Bioscience (combination oral contraceptive in Phase 2).
Sales of our products, if approved,Twirla may be adversely affected by the consolidation among wholesale drug distributors and the growth of large retail drug store chains.
The network through which we will sell Twirla and our products,potential product candidates, if and when approved, has undergone significant consolidation marked by mergers and acquisitions among wholesale distributors and the growth of large retail drugstore chains. As a result, a small number of large distributors control a significant share of the market. In 2012,2018, three companies generated about 85%95% of all revenues from drug distribution in the United States, and in 2010, four2019, the top five chain pharmacy companies owned about 30%35% of all retail pharmacy outlets. Consolidation of drug wholesalers and retailers, as well as any increased pricing pressure that those entities face from their customers, including the U.S. government, may increase pricing pressure and place other competitive pressures on drug manufacturers, including us.
Recently enactedExisting and future legislation may increase the difficulty and cost for us to obtain marketing approval of and to commercialize Twirla and our other product candidates and may affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for Twirla, restrict or regulate post-approval activities and affect our ability to profitably sell Twirla.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We do not know whether additional legislative changes will be enacted, or whether the FDA's regulations, guidance or interpretations will change, or what the impact of such changes on the potential marketing approvalour ability to market of Twirla if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA's approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
In March 2010, President Obama signed into law the ACA, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the healthcare industry and impose additional healthcare policy reforms. The ACA, among other things, increased the Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program for both branded and generic drugs, extended the rebate program to certain individuals enrolled in Medicaid managed care organizations, addressed new methodologies by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are line extension products and expanded the 340B drug discount program (excluding orphan drugs) to other entities. Further, the ACA imposed a significant annual tax on companies that manufacture or import branded prescription drug products. Substantial new provisions affecting compliance have also been enacted, which may require us to modify our business practices with regard to healthcare practitioners.
Of particular relevance to our business is the ACA requirement that all health plans, with limited exceptions, cover certain preventive services for women with no cost-sharing, which means no deductible, no co-insurance and no co-payments by the patient. Contraceptive methods and counseling, including all FDA-approved contraceptive methods as prescribed, are included in the ACA mandate, and this has come to be known as the "contraceptive mandate." Under the ACA, payors are only required to cover one favored product within each contraceptive "method" without imposing any cost-sharing obligations on the patient. For example, the introduction of a generic contraceptive patch product with a price that will likely be lower than the price of Twirla makes it less clear that Twirla would have a preferred position, such as coverage without a co-insurance payment, under the ACA contraceptive mandate. Other products within the same method may also be covered, but payors are allowed to use reasonable medical management techniques, such as the application of cost-sharing
obligations. An amendment was issued that provided an exemption to the contraceptive mandate for group health plans established or maintained by religious employers. However, the contraceptive mandate has remained controversial, with several legal challenges filed around the country. In June 2014, the U.S. Supreme Court ruled that owners of certain private companies can object to the contraceptive mandate on religious grounds and in November 2015, the Court agreed to hear arguments from non-profit organizations requesting similar treatment. AlthoughIn October 2017, the U.S. Department of Health and Human Services announced it is too earlywill seek to determineissue regulations that will allow all companies to qualify for the full effect ofexemption from the contraceptive mandate on the basis of religious and other provisionsmoral grounds. While there is an injunction against the administration prohibiting it from implementing these rules, the ultimate outcome of that litigation, which is currently in front of the Supreme Court, cannot be predicted. The ACA on our business, the law appears likely to continue theto apply pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs. In March 2017, the U.S. Congress proposed legislation, which, if signed into law by the new administration, would repeal certain aspects of the ACA. Further, on January 20, 2017, the newcurrent administration signed an Executive Order directing federal agencies with authorities and responsibilities under the ACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the ACA that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices, among others. Congress also could consider subsequent legislation to repeal and replace elements of the ACA that are repealed. There are several proposals to reform the federal healthcare laws being advocated and it is still unclear whether such reform efforts will succeed and if so, which proposals will ultimately be successful. Therefore, it is difficult to determine the full effect of the ACA or any other healthcare reform efforts on our business.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation's automatic reduction in funding to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and will stay in effect through 2024 unless additional Congressional action is taken. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or ATRA, which among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. We expect that additional federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, and in turn could significantly reduce the projected value of Twirla and our potential product candidates and reduce our profitability.
Moreover, the Drug Quality and Security Act imposes obligations on manufacturers of pharmaceutical products related to product tracking and tracing. Among the requirements of this legislation, manufacturers are required to provide certain information regarding the drug product to individuals and entities to which product ownership is transferred, will beare required to label the drug product with a product identifier toward the end of 2017 and are required to keep certain records regarding the drug product. The transfer of information to subsequent product owners by manufacturers will beis required to be done electronically toward the end of 2017.electronically. Manufacturers must also verify that purchasers of the manufacturers' products are appropriately licensed. Further, under this legislation, manufacturesmanufacturers have drug product investigation, quarantine, disposition, and FDA and trading partner notification responsibilities related to counterfeit, diverted, stolen and intentionally adulterated products, as well as products that are the subject of fraudulent transactions or which are otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death.
Table Other measures have also been taken by Congress, the Administration, and administrative agencies to increase drug competition and thus, decrease drug prices. By example, in 2019 FDA introduced a proposed rule and draft guidance to facilitate drug importation. Congress also passed a bill requiring sponsors of ContentsNDA approved products to provide sufficient quantities of drug product on commercially reasonable market based terms to entities developing generic and similar drug products. New legislative and regulatory efforts could ultimately have an adverse impact on our business and results of operation.
Third partyThird-party coverage and reimbursement and healthcare cost containment initiatives and treatment guidelines may constrain our future revenues.
Our ability to successfully market Twirla and other product candidates, if approved, will depend in part on the level of coverage and reimbursement that government authorities, private health insurers and other organizations provide for Twirla or our other product candidates and contraceptives in general. Countries in which Twirla or our other product candidates areis sold through reimbursement schemes under national health insurance programs frequently require that manufacturers and sellers of pharmaceutical products obtain governmental approval of initial prices and any subsequent price increases. In certain countries, including the United States, government-funded and private medical care plans can exert significant indirect pressure on prices. We may not be able to sell Twirla or our other product candidates profitably if adequate prices are not approved or coverage and reimbursement are unavailable or limited in scope. Increasingly, third party payors attempt to contain healthcare costs in ways that are likely to impact our development of products including:
We may never seek or receive marketing approval for or commercialize Twirla outside the United States.
In order to market Twirla outside the United States, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy and governing, among other things, clinical trials and commercial sales, pricing and distribution of Twirla. The time required to obtain approval in other countries might differ from and be longer than the time required to obtain FDA approval. The marketing approval process in other countries may include all of the risks associated with obtaining FDA approval in the United States, as well as other risks. For example, legislation analogous to Section 505(b)(2) of the FDCA in the United States may not exist in other countries. In territories where data is not freely available, we may not have the ability to commercialize Twirla, or any products approved in the future, without negotiating the rights from third parties to refer to their clinical data in our regulatory applications, which could require the expenditure of significant additional funds. Further, we may be unable to obtain rights to the necessary clinical data and may be required to develop our own proprietary safety and efficacy dossiers. In addition, in many countries outside the United States, it is required that a product receive pricing and reimbursement approval before the product can be commercialized. This can result in substantial delays in the advancement of our products in such countries. Further, product labeling requirements outside the United States may be different and inconsistent with the U.S. labeling, potentially negatively affecting our ability to market in countries outside the United States.
Marketing approval in one country does not ensure marketing approval in another, but failure or delay in obtaining marketing approval in one country may have a negative effect on the regulatory process in others. In addition, we may be subject to fines, suspension or withdrawal of marketing approvals, product recalls, seizure of products, operating restrictions, and criminal prosecution if we fail to comply with applicable foreign regulatory requirements. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, our ability to market to our full target market will be reduced and our ability to realize the full marketing potential of our products will be harmed.
Risks Related to ManufacturingOur Financial Position and Our Reliance on Third PartiesNeed for Capital
We have no manufacturing capacityincurred operating losses in each year since our inception and anticipate continued reliance on Corium, our third party manufacturer,expect to continue to incur substantial losses for the development and commercialization offoreseeable future. Management has concluded that these factors raise substantial doubt about our product candidates in accordance with manufacturing regulations.ability to continue as a going concern.
We rely on Corium International, Inc.,have incurred losses in each year since our inception in December 1997. Our net loss was $18.6 million, $19.8 million and $28.3 million for the years ended December 31, 2019, 2018 and 2017, respectively. As of December 31, 2019, we had an accumulated deficit of approximately $260.4 million. In February 2020, we entered into a Credit Agreement and Guaranty with Perceptive Credit Holdings III, LP, or Corium,Perceptive, for a senior secured term loan facility of up to $35 million, which we refer to as the Perceptive Credit Agreement. We believe that our third party manufacturer,cash and cash equivalents as of December 31, 2019, along with the proceeds of the Perceptive Credit Agreement we have received to produce clinical supplies of Twirla and our other product candidates, and we plan to continue relying on them for commercial supplies and samples of our product candidates, if approved. We do not own or operate, and have no plans to establish, any manufacturing facilities for our product candidates. We lack the resources and the capabilities to manufacture Twirla or any of our product candidates on a clinical or commercial scale. The facilities used by Corium to manufacture our product candidates must be approved by the FDA pursuant to inspections thatdate, will be conducted after submissionsufficient to meet our projected operating requirements through the end of an NDA to the FDA. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with the regulatory requirements, known as Current Good Manufacturing Practices, or cGMPs, for manufacture of our product candidates and our products, if and when approved. If Corium or other contract manufacturers that we may use cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, they2020. They will not be ablesufficient to secure or maintain regulatory approval for their manufacturing facilities. In addition, we have no control overfund our current and planned operations through the ability of our contract manufacturer to maintain adequate quality control, quality assurance and qualified personnel. If12 months following the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities that would also require FDA approval and which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved. Moreover, if our contract manufacturer cannot successfully manufacture materials that conform to our specifications and the strict regulatory requirements of the FDA or others, we may be subject to other regulatory enforcement action such as adverse inspectional findings, Warning Letters, Untitled Letters, recall requests, withdrawal of product or investigational approvals, clinical holds ordate
termination, disgorgement, restitution, exclusion from federal healthcare programs product seizures and detention, consent decrees, corporate integrity agreements, criminal and civil penalties, including imprisonment, refusal to permit import or export of the product and injunction against or restriction of manufacture or distribution. If our contract manufacturer experiences issues in its manufacturing process oron which this Annual Report on Form 10-K is unable to produce clinical supplies in adequate quantity and quality, our clinical trial could be delayed orfiled, which raises substantial doubt about our ability to receive regulatory approvalcontinue as a going concern. Substantial doubt about our ability to continue as a going concern may create negative reactions to the price of our common stock and we may have a more difficult time obtaining financing in the future.
Specialty pharmaceutical product development is a speculative undertaking, involves a substantial degree of risk and is a capital-intensive business. We expect to incur expenses without corresponding revenues until we are able to sell Twirla in significant quantities, which may not happen. We have devoted most of our financial resources to research and development, including our non-clinical development activities and clinical trials. We expect we will need to incur additional expenses as we complete the qualification and validation of our commercial manufacturing process, initiate pre-launch commercial activities, commercially launch Twirla, advance our other potential product candidates could be negatively affected. Additionally, if there are changesand expand our research and development programs. We will require additional capital to fund our operating needs beyond 2020, including among other items, the completion of our commercial plan for Twirla, which primarily includes validation of our commercial manufacturing process, as well as the commercial launch of Twirla and advancing the development of our other potential product candidates. We may not be able to obtain sufficient additional funding to continue our operations at planned levels and be forced to reduce, or even terminate, our operations. To date, we have financed our operations primarily through sales of common stock, convertible preferred stock and convertible promissory notes and to a lesser extent, through term loans and government grants. Our potential product candidates will also require the completion of regulatory review, significant marketing efforts and substantial investment before they can provide us with any revenue.
We expect that our expenses will increase as we prepare for Twirlathe commercial launch of Twirla. As a result, we expect to continue to incur substantial losses for the foreseeable future, and these losses may increase. We are uncertain when or if we will be able to our formulation for Twirla that require a changeachieve or sustain profitability. If we achieve profitability in the manufacturing process,future, we could experience significant additional costmay not be able to sustain profitability in subsequent periods. Any failure to become and remain profitable would impair our ability to sustain operations and adversely affect the price of our common stock and our ability to receive regulatory approval could be delayed.
The machinery to produceraise additional capital. We are significantly dependent on the success of Twirla, and if we do not achieve the commercial supplysuccess of Twirla must be qualified and validated, which is time-consuming and expensive, and this machinery is located within one manufacturing site and is customized to the particular manufacturing specifications of Twirla. If Corium isand/or are unable to qualifyobtain additional funding, we will need to reassess our operating capital needs and validate this equipmentmay be unable to continue our operations at planned levels and be forced to reduce, or even terminate, our operations.
We have never been profitable. Currently, we have no products available for commercial sale, no source of revenue pending the commercial launch of Twirla, and we may never become profitable.
We have never been profitable and do not expect to be profitable in the foreseeable future. We have no products currently available for commercial sale, and will not have any products available for sale until the commercial launch of Twirla, which we currently anticipate will commence in the fourth quarter of 2020. To date, we have not generated any revenue from product sales. Even if we are able to commercialize Twirla or any other potential product candidate, there can be no assurance that we will generate significant revenues or ever achieve profitability. Our ability to generate product revenue depends on a timely manner and successfully produce validation batches,number of factors, including our ability to launch and commercialize Twirla will be compromised and we could requireto:
Although we have manufacturing agreements with Corium for the clinical and commercial supply of Twirla, Corium and several of its suppliers of raw materials will be single source providers to us for a significant period of time. In particular, Corium manufactures Twirla using EE and LNG and components that it purchases from third parties, most of which are single source suppliers of the applicable material. We do not have any control over the process or timing of the acquisition of these raw materials by Corium. Although we generally do not begin a clinical trial unless we believe we have a sufficient supply of a product candidate to complete the clinical trial, any significant delay in the supply of a product candidate, or the raw material components thereof, for an ongoing clinical trial due to the need to replace a third party manufacturer could considerably delay completion of our clinical trials, product testing and potential regulatory approval of our product candidates.
Because we outsource all of our manufacturing processes, there is no guarantee that there will be sufficient supplies to fulfill our requirements or that we may obtain such supplies on acceptable terms. Although Corium intends to enter into agreements with critical manufacturers, component fabricators and secondary service providers to secure commercial supply of Twirla, not all of such suppliers and service providers will be under contract. Any delays in obtaining adequate supplies of our product candidates could limit our ability to meet commercial demand for Twirla.
In addition, in the event Twirla is approved and achieves significant market share, Corium may not possess adequate manufacturing capabilities to meet market demand for Twirla. If it becomes necessary to engage an additional third party manufacturer to produce Twirla, we may need to license certain manufacturing know-how from Corium, or our commercial supply will be limited while the new third party manufacturer develops the necessary know-how to manufacture Twirla and while we obtain regulatory approval for the addition of a new manufacturer.
Reliance on a third party manufacturer subjects us to risks that would not affect us if we manufactured the product candidates ourselves, including:
In addition, because of the numerous risks and uncertainties associated with product commercialization and product candidate development, we are unable to predict the timing or amount of increased expenses, or when, or if, we will be able to achieve or maintain profitability. In addition, our expenses could increase beyond our current expectations and resources if we are required to provide increased rebates to managed care payors, experience set-backs in the validation of our commercial manufacturing process, we need to increase our manufacturing capacity sooner than planned, experience disruptions in our manufacturing capabilities, or need to alter our marketing strategy. We anticipate incurring significant costs associated with the commercial launch of Twirla and our other potential product candidates, if approved.
Our ability to become and remain profitable depends on our ability to generate revenue in excess of our increasing costs. Even if we are able to generate revenues from the sale of Twirla and our other potential product candidates, if approved, we may not become profitable and may need to obtain additional funding to continue operations. If we fail to become profitable or obtain additional funding or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce our operations. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would decrease the value of our company and could impair our ability to raise additional capital, expand our business or continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
Our operating activities may be restricted as a result of covenants related to the outstanding indebtedness under our loan agreement and we may be required to repay the outstanding indebtedness in an event of default, which could have a materially adverse effect on our business.
In February 2020, we entered into the Perceptive Credit Agreement for a senior secured term loan facility of up to $35.0 million. A first tranche of $5 million was funded on execution of the Perceptive Credit Agreement. A second tranche of $15 million was funded as a result of the approval of Twirla by the FDA. Another $15 million tranche will be available upon the achievement of certain revenue milestones. The facility will be interest only until the third anniversary of the closing date.
The Perceptive Credit Agreement subjects us to various customary affirmative and negative covenants, including requirements as to financial reporting and insurance, and restrictions on our ability to dispose of our business or property, change our line of business, liquidate or dissolve, enter into any change in control transaction, merge or consolidate with any other entity or acquire all or substantially all the capital stock or property of another entity, incur additional indebtedness, incur certain types of liens on our property, including our intellectual property, pay any dividends or other distributions on our capital stock other than dividends payable solely in capital stock or redeem our capital stock. Our business may be adversely affected by these restrictions on our ability to operate our business. The Perceptive Credit Agreement also subjects us to financial covenants in respect of minimum liquidity and minimum product revenue.
The loans provided under the Perceptive Credit Agreement are secured by substantially all of our property. We are currently required to make interest-only payments through February 2023. Loans under the Perceptive Credit Agreement currently bear interest at rate of 10.25% per annum plus one-month LIBOR, and mature on February 10, 2024.
The Credit Agreement contains certain customary Events of Default, which include, among others, non-payment of principal, violation of covenants, inaccuracy of representations and warranties,
bankruptcy and insolvency events, material judgments, certain regulatory-related events and events constituting a Change of Control (as defined in the Credit Agreement). We may not have enough available cash or be able to raise additional funds through equity or debt financings to repay such indebtedness at the time any such event of default occurs. In that case, we may be required to delay, limit, reduce or terminate our potential product candidate development or commercialization efforts or grant to others rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves. Perceptive could also exercise its rights as collateral agent to take possession and dispose of the collateral securing the loan for its benefit, which collateral includes substantially all of our property. Our business, financial condition and results of operations could be materially adversely affected as a result of any of these events.
We will need to obtain additional financing to fund our operations and, if we are unable to obtain such financing, we may be unable to commercialize Twirla or to complete the development and commercialization of our other potential product candidates.
Our operations have consumed substantial amounts of cash since inception. From our inception to December 31, 2019, we have cumulative net cash flows used by operating activities of $227.3 million. We will need to obtain additional capital to fund our future operations, including the commercialization of Twirla. We will need to obtain additional financing to develop and commercialize our other potential product candidates and to complete the development of any additional product candidates we might acquire. Moreover, our fixed expenses such as rent, interest expense and other contractual commitments are substantial and are expected to increase in the future.
Our future funding requirements will depend on many factors, including, but not limited to:
Our product candidatesUntil we can generate a sufficient amount of revenue, we may compete with other productsfinance future cash needs through public or private equity offerings, license agreements, debt financings, collaborations, strategic alliances and product candidates for access to manufacturing resources and facilities. There are a limited number of manufacturers that operate under cGMP requirements andmarketing or distribution arrangements. Additional funds may not be available when we need them on terms that are both capable of manufacturing foracceptable to us, and willing to do so.or at all. If our existing third party manufacturer, or the third parties that we may engage in the future to manufacture a product for commercial sale or for our clinical trials, should cease to continue to manufacture our product candidates for any reason, we likely would experience delays in obtaining sufficient quantities of our product candidates for us to meet commercial demand or to advance our clinical trials while we identify and qualify replacement suppliers. If for any reason weadequate funds are unable to obtain adequate supplies of our product candidates or the drug substances used to manufacture them, it will be more difficult for us to develop our product candidates and compete effectively.
Our third party manufacturer is subject to regulatory requirements, covering manufacturing, testing, quality control and record keeping relating to our product candidates, and subject to ongoing inspections by the regulatory agencies. In addition to the above-described regulatory actions, failures by our third party manufacturer to comply with applicable regulations may result in long delays and interruptions to our manufacturing capacity while we seek to secure another third party manufacturer that meets all regulatory requirements.
We are dependent on numerous third parties in Corium's supply chain for the supply of our product candidates, and if Corium fails to maintain supply relationships with these third parties, develop new relationships with other third parties or suffers disruptions in supply,not available, we may be unablerequired to continue to develop our product candidates,delay or assuming FDA approval, commercialize Twirla.
We, through our manufacturing partner Corium, rely on a number of third parties forreduce the supply of active ingredients, other raw materials and laboratory services for the supplyscope of our product candidates and, assuming FDA approval, commercialization efforts or one or more of Twirla. Our abilityour research or development programs. We may seek to develop our product candidates depends, in part, on Corium's ability to successfully obtainaccess the active pharmaceutical ingredients used in our product candidates, in accordancepublic or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time. In addition, if we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with regulatory requirements and in sufficient quantities for clinical testing and later commercialization. If Corium fails to develop and maintain supply relationships with these third parties, we may be unablehave to continuerelinquish valuable rights to develop our technologies, future revenue streams or potential product candidates or to grant licenses on terms that may not be favorable to us.
Our planned timeline for the commercial launch of Twirla and our ability to fund our operations through the period of time necessary to successfully commercialize any approved productsTwirla could be adversely affected based on our inability to timely and successfully complete the validation of our commercial manufacturing process, the failure of Twirla to gain acceptance in the future.
marketplace, our inability to successfully compete with other contraceptive products and the need to provide higher rebates in order to gain a competitive formulary status. We through Corium, also rely on certain third parties as the current sole source of the materials they supply. Although many of these materials are produced in more than one locationmay not be able to obtain sufficient additional funding to continue our operations at planned levels and be forced to reduce, or are available from another supplier, if any of these materials becomes unavailable to us for any reason, we likely would incur added costs and delays in identifying or qualifying replacement materials and there can be no assurance that replacements wouldeven terminate, our operations. Adequate additional funding may not be available to us on acceptable terms, or at all. In certain casesIf we are unable to raise additional capital when needed or on attractive terms, or we are unable to enter into strategic collaborations, we then may be unable to complete the commercialization of Twirla and may also be required to further cut operating costs, delay, reduce or eliminate our research and development programs or future commercialization efforts or even terminate our operations, which may involve seeking bankruptcy protection. Our forecast of the period of time through which our financial resources will be adequate to support our operating requirements is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed elsewhere in this "Risk Factors" section. We have based this estimate on a number of assumptions that may prove to be wrong and changing circumstances beyond our control may cause us to consume capital more rapidly than we currently anticipate. If we choose to accelerate elements of our commercial plan or we encounter any unforeseen events that affect our business plan, we may choose to raise additional funds to provide us with additional working capital. Our inability to obtain additional funding when we need it could seriously harm our business and we may be requiredunable to get regulatory approvalcontinue our operations at planned levels and be forced to use alternative suppliers,reduce, or even terminate, our operations.
Raising additional capital may cause dilution to our existing stockholders or restrict our operations.
We may seek additional capital through a combination of private and this processpublic equity offerings, debt financings and strategic collaborations. The sale of approvaladditional equity or convertible debt securities could delay developmentresult in the issuance of additional shares of our product candidatescapital stock and assuming FDA approval, commercial productioncould result in dilution to our stockholders. The incurrence of Twirla, indefinitely. For example, the sole manufacturer of one of the components of the packaging ofindebtedness would result in increased fixed payment obligations and could also result in certain restrictive covenants, such as limitations on our Twirla patch notified usability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that it would be discontinuing manufacture of the component in 2016, although it has now extended the period during which it will continuecould adversely impact our ability to manufacture the component. In conjunction with Corium, we were able to secure an amount of inventory of the packaging componentconduct our business. We cannot guarantee that we believe will last until 2019. We are currently evaluating sources for a replacement for this discontinued component and, assuming FDA approval of this replacement
material,future financing will be available in sufficient amounts or on terms acceptable to us, if at all. If we plan to use the replacement material in connection with the commercial production of Twirla.
If Corium's third party suppliers fail to deliver the required quantities of sub-components and starting materials, in accordance with all regulatory requirements, and on a timely basis and at commercially reasonable prices, and we and Corium are unable to find oneraise additional capital in sufficient amounts or more replacement suppliers capable of production at a substantially equivalent cost in substantially equivalent volumeson terms acceptable to us, we will be prevented from pursuing research and quality, and on a timely basis, the continued development of our product candidates, and assuming FDA approval, commercialization of Twirla, would be impeded, delayed, limited or prevented, whichefforts. This could harm our business, operating results of operations,and financial condition and prospects.
Ifcause the manufacturing facilities of Corium are not maintained in a manner that is compliant with cGMP requirements, we may need to find alternative manufacturers and suppliers, which could result in supply interruptions of Twirla and our other product candidates, additional costs and lost revenues.
Corium's facilities used for the manufactureprice of our product candidates must be maintained in a manner compliant with cGMP requirements, including obtaining favorable inspection reports. We do not control the manufacturing processcommon stock to fall.
Risks Relating to Maintaining Regulatory Compliance and are dependent on Corium for compliance with the FDA's requirements for manufactureApproval of Twirla and our other product candidates. If Corium cannot successfully manufacture material components and finished products that conform to our specifications and the FDA's strict regulatory requirements, they and we may be subject to regulatory action, including adverse inspectional findings, Warning Letters, Untitled Letters, product recall requests, withdrawal of product or investigational approvals, clinical holds or termination, disgorgement, restitution, exclusion from federal healthcare programs, detentions or seizures, refusal to allow the import or export of a product, injunction against or restriction of manufacture or distribution, consent decrees, corporate integrity agreements, criminal and civil penalties, including imprisonment, and Corium may not be able to maintain FDA approval for its manufacturing facilities or acceptance of its manufacturing data in regulatory filings. If Corium's facilities cannot maintain compliance with FDA requirements, we may need to find and successfully qualify alternative manufacturing facilities, which could result in supply interruptions of Twirla and our other product candidates and substantial additional costs as a result of such delays, including costs with respect to finding alternative manufacturing facilities, and lost revenues.
We rely on third partiesremain subject to conduct aspects of our clinical trials. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or comply with applicablesubstantial ongoing regulatory requirements, we may be delayed in obtaining or ultimately not be able to obtain marketing approval for our product candidates.
We currently rely on CROs for most aspects of our clinical trials, including trial conduct, data management, statistical analysis and electronic compilation of our NDA. We may enter into agreements with CROs to obtain additional resources and expertise in an attempt to accelerate our progress with regard to new or ongoing clinical and preclinical programs. Entering into relationships with CROs involves substantial cost and requires extensive management time and focus. In addition, typically there is a transition period between engagement of a CRO and the time the CRO commences work. As a result, delays may occur, which may materially impact our ability to meet our desired clinical development timelines and ultimately have a material adverse impact on our operating results, financial condition or future prospects.
As CROs are not our employees, we cannot control whether or not they devote sufficient time and resources to our clinical trials for which they are engaged to perform, and whether they comply with the applicable regulatory requirements, known as Current Good Clinical Practices, or cGCPs, which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area, or EEA, and comparable foreign regulatory authorities for all of our
product candidates in clinical development, which include requirements related to the conduct of the study, subject informed consent,Twirla, and IRB approval. Regulatory authorities enforce these cGCPs through periodic inspections of trial sponsors, principal investigators and trial sites. Although we may rely on third parties for the execution of our trials, we are nevertheless responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on CROs does not relieve us of our regulatory responsibilities. If we or any of our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, European Medicines Agency or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications, in addition to SECURE. We cannot assure you that, upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials complies with cGCP regulations. In addition, our clinical trials must be conducted with product candidate materials produced under cGMP regulations. Our failure to comply with these regulations may require us to discontinue or repeat clinical trials, which would delay the regulatory approval process. If the CROs we engage do not successfully carry out their contractual duties or obligations, conduct the clinical trials in accordance with all regulatory requirements or meet expected deadlines, or if they need to be replaced, or the quality or accuracy of the data they provide is compromised due to the failure to adhere to regulatory requirements or for other reasons, then our development programs may be extended, delayed or terminated, or we may not be able to obtain marketing approval for or successfully commercialize our product candidates. Failure to comply with clinical trial regulatory requirements may further subject us to regulatory action, including Warning Letters, Untitled Letters, adverse inspectional findings, clinical holds or termination, criminal and civil penalties, including imprisonment, injunction against manufacture or distribution and debarment. As a result, our financial results and the commercial prospects for our product candidates would be harmed and our costs would increase.
Any collaboration arrangements that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our product candidates.
We may seek partnerships, collaborations and other strategic transactions to maximize the commercial potential of Twirla, our other product candidates and our proprietary technologies in the United States and territories throughout the world. We may enter into such arrangements on a selective basis depending on the merits of retaining commercialization rights for ourselves as compared to entering into selective collaboration arrangements with leading pharmaceutical or biotechnology companies for Twirla and each of our other product candidates and technologies, both in the United States and internationally. We face competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements should we choose to enter into such arrangements. The terms of any collaborations or other arrangements that we may establish may not be favorable to us.
Any future collaborations that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations.
Disagreements between parties to a collaboration arrangement regarding clinical development and commercialization matters could lead to delays in the development process or commercialization of our product candidates and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision making authority.
Collaborations with pharmaceutical or biotechnology companies and other third parties often are terminated or allowed to expire by the other party. Any such termination or expiration could adversely affect us financially and could harm our business reputation.
If we fail to establish an effective distribution process our business may be adversely affected.
We do not currently have the infrastructure necessary for distributing pharmaceutical products. We intend to contract with third party logistics wholesalers to warehouse these products and distribute them to pharmacies. This distribution network will require significant coordination with our sales and marketing and finance organizations. Failure to secure contracts with wholesalers could negatively impact the distribution of our products, if and when approved, and failure to coordinate financial systems could negatively impact our ability to accurately report product revenue. If we are unable to effectively establish and manage the distribution process, the commercial launch and sales of our products, if and when approved, will be delayed or severely compromised and our results of operations may be harmed. Distribution practices will also need to comply with the applicable regulatory requirements. If our distributors do not comply with the applicable regulatory requirements, we could be exposed to potential enforcement actions.
Risks Related to Regulatory Matters Following Approval
Even if we obtain marketing approval for Twirla or other product candidates, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. Additionally, Twirla or other product candidates could be subject to labeling and other restrictions,penalties, including withdrawal from the market, and we may be subject to penalties if we fail to comply with regulatory requirementssuspension, or if we experience unanticipated problems.withdrawal of product approval.
Even if we obtain U.S. regulatory approval of Twirla or other product candidates, the FDA may still impose significant restrictions on their indicated uses, including more limited patient populations, require that precautions, contraindications, or warnings be included on the product labeling, including black box warnings, or impose ongoing requirements for potentially costly and time-consuming post-approval studies, including Phase 4 clinical trials, and post-market surveillance to monitor safety and efficacy. Claims that we may make may also be restricted through our approved labeling. For example, based on the SECURE top-line data, the Pearl Index for the overall intent to treat population of subjects 35 years of age and under was 4.80 with an upper-bound of the 95% confidence interval of 6.06, but in the obese subpopulation of subjects 35 years of age and under, the Pearl Index was 6.42 with an upper-bound of the 95% confidence interval of 8.88. The highest Pearl Index for a hormonal contraceptive product approved by the FDA to date was 3.19 and the highest upper-bound of the 95% confidence interval was 5.03. In the combined safety database for our three Phase 3 trials (n>3,000), there were 5 subjects with potentially study drug related DVTs or PEs, 4 of whom were obese (BMI³30kg/m2). Although ultimate approvability of a hormonal contraceptive is based on a risk/benefit assessment of the overall safety and efficacy profile of a product, not only a specific Pearl Index, the FDA could conclude that the Pearl Index in the overall population or a subpopulation is too high to demonstrate efficacy and an adequate risk/benefit profile. As such, we may not obtain approval of Twirla based on these data or any other basis. Even if we receive approval of Twirla, FDA may determine that for a specific subgroup of patients, Twirla has lower efficacy and presents a higher risk, necessitating labeling restrictions. For instance, FDA may require labeling restrictions on the use of Twirla for patients in certain BMI categories or otherwise require labeling limitations or warnings for such subpopulation, which could limit the commercial potential of the product, if approved. FDA may further require us to include other information and/or data in the label for Twirla that may make it more difficult for us to successfully commercialize the product, if approved. For instance, FDA may require us to include the Pearl Index results from the previously conducted Phase 3 trials, which were higher than the SECURE trial's overall and certain sub-group Pearl Index results. We will discuss specific labeling requirements with FDA in the future.
If approved, Twirla and our other product candidates will also be subject to ongoing regulatory requirements governing the manufacturing, labeling, packaging, storage, distribution, import, export, safety surveillance, advertising, marketing promotion, recordkeeping, reporting of adverse events and other post-market information, and further development. Thesedevelopment, including ongoing requirements for costly post-marketing studies, including Phase 4 clinical trials or post-market surveillance. For example, as part of the FDA's approval of Twirla, the FDA has required a long-term post-marketing safety study to assess and describe the risks of Twirla, including the risk of VTE and ATE compared to oral combined hormonal contraceptives and Xulane. This study is similar to one recently required by FDA for another contraceptive product. We will also conduct a second small post-marketing study, required by FDA, to assess Twirla's residual drug content, strength, and adhesion. The results generated in these post-approval clinical trials could result in loss of marketing approval, changes in product labeling, or new or increased concerns about side effects or efficacy of a product. Failure to comply with post-market study requirements can also result in enforcement action or FDA removal of the product from the market.
Other post-approval requirements include registration with the FDA, listing of our drug products, payment of annual fees, as well as continued compliance with cGCPs for any clinical trials that we conduct post-approval. Application holders must notify the FDA, and depending on the nature of the change, obtain FDA pre-approval for product manufacturing changes. In addition, manufacturers of drug products and their facilities are subject to continual review and periodicroutine inspections by the FDA and other regulatory authorities for compliance with cGMPthe FDA's manufacturing requirements relating to quality control, quality assurance and corresponding maintenance of records and documents. If we are found to be noncompliant with applicable requirements, the FDA and other government authorities may issue a Warning Letter or Untitled Letter, or take other regulatory action such as a product seizure and detention, withdrawal of product approval, requestrequests for a recall, refusal to allow the import or export of the product, criminal or civil penalties, injunction against or restriction of product manufacture or distribution, consent decrees, disgorgement, restitution, clinical holds or terminations of clinical trials, FDA debarment, debarment from government contracts or refusal of orders under existing contracts, exclusion from federal healthcare programs, corporate integrity agreements, or imprisonment.
The FDA has the authority to require a REMS as part of an NDA or after approval, which may impose further requirements or restrictions on the information that patients must be provided, distribution or use of an approved drug, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria or requiring treated patients to enroll in a registry.
With respect to sales and marketing activities by us or any future collaborative partner, advertising and promotional materials must comply with the FDA's rules in addition to other applicable federal and local laws in the United States and similar legal requirements in other countries. In the United States, the distribution of product samples to physicians must comply with the requirements of the U.S. Prescription Drug Marketing Act.Act and is subject to certain requirements. We may also be subject, directly or indirectly through our customers and partners, to various fraud and abuse laws, including,
without limitation, the U.S. Anti-Kickback Statute, U.S. False Claims Act and similar state laws, which impact, among other things, our proposed sales, marketing and scientific/educational grant programs.efforts. If we participate in the U.S. Medicaid Drug Rebate Program, the Federal Supply Schedule of the U.S. Department of Veterans Affairs, or other government drug programs, we will be subject to complex laws and regulations regarding reporting and payment obligations. All of these activities are also potentially subject to U.S. federal and state consumer protection and unfair competition laws. Similar requirements exist in many of these areas in other countries.
In addition, if Twirla and our other product candidates are approved, our product labeling, advertising and promotional materials wouldfor Twirla will be subject to regulatory requirements and continuing review by the FDA, Department of Justice, Department of Health and Human Services' Office of Inspector General, state attorneys general, members of Congress and the public. The FDA strictly regulates the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product's approved labeling, a practice known as off-label promotion. If we receive marketing approval for Twirla or our other product candidates, physiciansPhysicians may nevertheless prescribe the products to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability and government fines. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees of permanent injunctions under which
specified promotional conduct is changed or curtailed. For example, we believe that Twirla, if approved, will have a label consistent with all other marketed hormonal contraceptive products, which include class labeling that warns of risks of certain serious conditions, including venous and arterial blood clots, such as heart attacks, thromboembolism and stroke, as well as liver tumors, gallbladder disease, and hypertension, and a black box warning regarding risks of smoking and CHC use, particularly in women over 35 years old that smoke. However, regulatory authorities may require the inclusion of additional statements about adverse events in the label, including additional black box warnings or contraindications.
In the United States, engaging in the impermissible promotion of our products following approval, for off-label uses can also subject us to false claims litigation under federal and state statutes, which can lead to civil and criminal penalties and fines, agreements with governmental authorities that materially restrict the manner in which we promote or distribute drug products through, for example, corporate integrity agreements, and debarment, suspension or exclusion from participation in federal and state healthcare programs. These false claims statutes include the federal civil False Claims Act, which allows any individual to bring a lawsuit against a pharmaceutical company on behalf of the federal government alleging submission of false or fraudulent claims or causing others to present such false or fraudulent claims, for payment by a federal program such as Medicare or Medicaid. If the government decides to intervene and prevails in the lawsuit, the individual will share in the proceeds from any fines or settlement funds. If the government declines to intervene, the individual may pursue the case alone. Since 2004, these False Claims Act lawsuits against pharmaceutical companies have increased significantly in volume and breadth, leading to several substantial civil and criminal settlements regarding certain sales practices promoting off-label drug uses involving fines that are as much as $3.0 billion. This growth in litigation has increased the risk that a pharmaceutical company will have to defend a false claim action, pay settlement fines or restitution, as well as criminal and civil penalties, agree to comply with burdensome reporting and compliance obligations, and be excluded from Medicare, Medicaid and other federal and state healthcare programs. If we do not lawfully promote our approved products, if any, we may become subject to such litigation and, if we do not successfully defend against such actions, those actions may have a material adverse effect on our business, financial condition, results of operations and prospects.
If we or a regulatory agency discover previously unknown problems with Twirla or with a potential product candidate, once approved, such as adverse events of unanticipated severity or frequency, data integrity issues with regulatory filings, problems with the facility where the product is manufactured or we or our manufacturers or others working on our behalf fail to comply with applicable regulatory
requirements before or after marketing approval, we may be subject to reporting obligations as well as the following administrative or judicial sanctions:
Tabletermination of Contentsclinical trials;
The occurrence of any event or penalty described above may inhibit our ability to commercialize Twirla or our otherpotential product candidates, if approved, and generate revenue. Adverse regulatory action, whether pre- or post-approval, can also potentially lead to product liability claims and increase our product liability exposure.
Moreover, the FDA's policies may change, and additional government regulations may be enacted that could prevent, limit or delay the sale and promotion of Twirla or marketing approval and the sale and promotion of our potential product candidates.candidates, if approved. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any obtained marketing approval, that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
Even if Twirla receives marketing approval by the FDA in the United States, we may never receive marketing approval for or commercialize Twirla or any other product candidates outside the United States.
In order to market Twirla or any other product candidate outside the United States, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy and governing, among other things, clinical trials and commercial sales, pricing and distribution of our product candidates. The time required to obtain approval in other countries might differ from and be longer than that required to obtain FDA approval. The marketing approval process in other countries may include all of the risks associated with obtaining FDA approval in the United States, as well as other risks. For example, legislation analogous to Section 505(b)(2) of the FDCA in the United States, which relates to the ability of an NDA applicant to use published data not developed by such applicant, may not exist in other countries. In territories where data is not freely available, we may not have the ability to commercialize our products, when and if approved, without negotiating rights from third parties to refer to their clinical data in our regulatory applications, which could require the expenditure of significant additional funds. Further, we may be unable to obtain rights to the necessary clinical data and may be required to develop our own proprietary safety and efficacy dossiers. In addition, in many countries outside the United States, it is required that a product receive pricing and reimbursement approval before the product can be commercialized. This can result in substantial delays in such countries. Further, the product labeling requirements outside the United States may be different and inconsistent with the U.S. labeling and to the detriment to the product, and therefore negatively affect the ability to market in countries outside the United States.
Marketing approval in one country does not ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one country may have a negative effect on the regulatory
process in others. In addition, we may be subject to fines, suspension or withdrawal of marketing approvals, product recalls, seizure of products, operating restrictions and criminal prosecution if we fail to comply with applicable foreign regulatory requirements. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, our ability to market to our full target market will be reduced and our ability to realize the full market potential of our product candidates will be harmed.
We will need to obtain FDA approval of any proposed product names, and any failure or delay associated with such approval may adversely affect our business.
We have received conditional approval from the FDA for the use of Twirla as the proprietary name for our lead product candidate, AG200-15. However, this approval is conditional upon a further and final review by the FDA at the time of NDA approval. Additionally, any name we intend to use for our other product candidates will require approval from the FDA regardless of whether we have secured a formal trademark registration from the U.S. Patent and Trademark Office, or USPTO. The FDA typically conducts a review of proposed product names, including an evaluation of the potential for confusion with other product names. The FDA may also object to a product name if it believes the name inappropriately implies medical claims or contributes to an overstatement of efficacy. If the FDA objects to any of our proposed product names, we may be required to adopt alternative names for our product candidates. If we adopt alternative names, we would lose the benefit of our existing trademark applications for such product candidate and may be required to expend significant additional resources in an effort to identify a suitable product name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA. We may be unable to build a successful brand identity for a new trademark in a timely manner or at all, which would limit our ability to commercialize our product candidates.
Our relationships with physicians, customers and payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, exclusion from government healthcare programs, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and others play a primary role in the recommendation and prescription of any product candidatesproducts that we commercialize. Our arrangements with third-party payors,
including government healthcare programs, and customers will expose us to broadly-applicablebroadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute Twirla, if approved, and any other product candidates we commercialize.Twirla. Restrictions under applicable federal and state healthcare laws and regulations include the following:
TheCompliance with these and other federal and state laws applicable to the sale, marketing, and distribution of commercial drug products will require that we expend time and financial resources to maintain compliance. Additionally, the risk of our being found in violation of these laws and regulations is increased by the fact that many of them have not been fully interpreted by the relevant government or regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Moreover, recent healthcare reform legislation has strengthened these laws. For example, the ACA, among other things, amended the intent requirement of the federal anti-kickback and criminal healthcare fraud statutes; such that a person or entity no longer needs to have actual knowledge of these statutes or specific intent to violate them. In addition, the ACA providedprovides that the government
may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the false claims statutes.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations are costly. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations, including anticipated activities conducted by our sales team in the sale of Twirla or our other product candidates, if approved, are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to a variety of different consequences, depending upon which law we are found to have violated, including significant civil, criminal and administrative penalties, damages, fines, exclusion from government funded healthcare programs, such as Medicare and Medicaid, corporate integrity agreements, refusal of government contracts, contract debarment and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom we expect to do business is found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Risks Related to Manufacturing and Our Reliance on Third Parties
We have no manufacturing capacity and anticipate continued reliance on Corium, our third-party manufacturer, for the commercialization of Twirla and development of our potential product candidates. We may not have or be able to obtain sufficient quantities of Twirla or our potential product candidates to meet our required supply for commercialization or clinical trials, which would materially harm our business.
Corium is a biopharmaceutical company that focuses on the development, manufacture, and commercialization of specialty transdermal products. In addition to partnering with other companies to manufacture transdermal products, Corium also engages in the research and development of its own proprietary transdermal drug delivery products. We rely on Corium, our third-party manufacturer, to produce commercial supplies and samples of Twirla. We also plan to rely on them for clinical and commercial supplies and samples of our potential product candidates, if approved. We do not own or operate, and have no plans to establish, any manufacturing facilities for Twirla or our potential product candidates. We lack the resources and the capabilities to manufacture Twirla or any of our potential product candidates on a clinical or commercial scale.
As a third-party manufacturer, Corium's business operations are completely beyond our control, and we have no influence over whether Corium changes its management or its business operations or discontinues them entirely. For example, in 2018 Corium was acquired by Gurnet Holding Company, or GHC. Following completion of the transaction, Corium became a private company, wholly owned by GHC. Corium has announced that it plans to continue its operations in Grand Rapids, Michigan, where commercial supplies of Twirla are being manufactured.
Furthermore, we do not control the manufacturing process of Twirla, and are completely dependent on Corium for compliance with the FDA's manufacturing regulatory requirements for the manufacture of Twirla, our potential product candidates and our other future products, if and when approved. As a manufacturer, Corium or other contract manufacturers that we may use are subject to routine inspection by regulatory authorities, including FDA. If Corium or other contract manufacturers that we may use cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA, they may receive adverse inspectional findings, may need to undertake costly and time consuming corrective actions, and may not be able to maintain regulatory approval for their manufacturing facilities.
In addition, while the contracts with our manufacturers contain provisions to help ensure that quality standards and compliance with laws and regulations are maintained, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and
qualified personnel. If in the future, the FDA withdraws its approval of Corium's facilities for the manufacture of Twirla, or if Corium experiences quality or other regulatory issues, we may need to find alternative manufacturing facilities that would also require FDA approval, which would significantly impact our ability to develop and sustain our market share of Twirla and to develop, and obtain, regulatory approval for and market our potential product candidates, if approved. Moreover, if our contract manufacturer cannot successfully manufacture materials that conform to our specifications and the strict regulatory requirements of the FDA, we may be subject to regulatory enforcement action such as adverse inspectional findings, Warning Letters, Untitled Letters, recall requests, withdrawal of product or investigational approvals, clinical holds or termination of clinical trials, refusals to approve pending applications, disgorgement, restitution, exclusion from federal healthcare programs, product seizures and detention, consent decrees, corporate integrity agreements, criminal and civil penalties, including imprisonment, refusal to permit import or export of the product and injunction against or restriction of manufacture or distribution. If our contract manufacturer experiences issues in its manufacturing process or is unable to produce commercial supplies in adequate quantity and quality, our commercialization of Twirla could be delayed. An inability of our contract manufacturer to produce supplies in adequate quantity and quality of our Twirla and our potential product candidates could also delay our ability to conduct clinical trials. This may adversely impact our ability to fulfil our post-marketing study requirements for Twirla and obtain regulatory approval of our potential product candidates.
The machinery and process to produce the commercial supply of Twirla must be qualified and validated, which is time-consuming and expensive, and this machinery is located within one manufacturing site and is customized to the particular manufacturing specifications of Twirla. If Corium is unable to qualify and validate this equipment and the processes in a timely manner and successfully produce validation batches, our ability to launch and commercialize Twirla will be compromised and we could require additional capital to complete the validation process. If this customized equipment malfunctions at any time during the production process, the time it may take Corium to secure replacement parts, to undertake repairs and to revalidate the equipment and process could limit our ability to meet the commercial demand for Twirla. Similar manufacturing conditions may also apply to our potential product candidates. This may increase the risk that the third-party manufacturer may not manufacture Twirla in accordance with the applicable regulatory requirements, that we may not have sufficient quantities of Twirla or our potential product candidates or that we may not have such quantities at an acceptable cost, any of which could delay, prevent, or impair the commercialization of Twirla and the development of our potential product candidates.
Although we have manufacturing agreements with Corium for the commercial supply of Twirla, Corium and several of its suppliers of raw materials will be single source providers to us for a significant period of time. In particular, Corium manufactures Twirla using EE and LNG and components that it purchases from third parties, most of which are single source suppliers of the applicable material. We do not have any control over the process or timing of the acquisition of these raw materials by Corium. Corium's failure to timely obtain, or a disruption in the supply of, these raw materials could lead to an inability to adequately supply the commercial market with finished product of Twirla and in turn adversely affect our business.
Because we outsource all of our manufacturing processes, there is no guarantee that there will be sufficient supplies to fulfill our requirements or that we may obtain such supplies on acceptable terms. Although Corium intends to enter into agreements with critical manufacturers, component fabricators and secondary service providers to secure commercial supply of Twirla, not all of such suppliers and service providers will be under contract. Any delays in obtaining adequate supplies of our potential product candidates could limit our ability to meet clinical and commercial demand for Twirla.
In addition, in the event Twirla achieves significant market share, Corium may not possess adequate manufacturing capabilities to meet market demand for Twirla. If it becomes necessary to engage an additional third-party manufacturer to produce Twirla, we may need to license certain manufacturing know-how from Corium, or our commercial supply will be limited while the new third-party manufacturer develops the necessary know-how to manufacture Twirla and while we obtain regulatory approval for the addition of a new manufacturer and processes.
Reliance on a third-party manufacturer subjects us to risks that would not affect us if we manufactured Twirla and our potential product candidates ourselves, including:
Twirla and our potential product candidates may compete with other products and product candidates for access to manufacturing resources and facilities. There are a limited number of manufacturers that operate in compliance with the FDA's manufacturing requirements and that are both capable of manufacturing for us and willing to do so. If our existing third-party manufacturer, or the third parties that we may engage in the future to manufacture a product for commercial sale or for our clinical trials, should cease to continue to manufacture our products or potential product candidates for any reason, we likely would experience delays in obtaining sufficient quantities of our products or potential product candidates for us to meet commercial demand or to advance our clinical trials while we identify and qualify alternative suppliers. If for any reason we are unable to obtain adequate supplies of our products or potential product candidates or the components used to manufacture them, it will be more difficult for us to develop our potential product candidates and compete effectively.
Our third-party manufacturer is subject to regulatory requirements, covering manufacturing, testing, quality control and record keeping relating to Twirla and our potential product candidates, and subject to ongoing inspections by the regulatory agencies. In addition to the above-described regulatory actions, failures by our third-party manufacturer to comply with applicable regulations may result in long delays and interruptions to our manufacturing capacity while we seek to secure another third-party manufacturer that meets all regulatory requirements.
We are dependent on numerous third parties in Corium's supply chain for the supply of Twirla and our potential product candidates, and if Corium fails to maintain supply relationships with these third parties, develop new relationships with other third parties or suffers disruptions in supply, we may be unable to continue to commercialize Twirla or develop our potential product candidates.
We, through our manufacturing partner Corium, rely on a number of third parties for the supply of active ingredients, other raw materials and laboratory services for the commercialization or Twirla and supply of our potential product candidates. Our ability to commercialize Twirla and to develop our potential product candidates depends, in part, on Corium's ability to successfully obtain the components used to manufacture Twirla and our potential product candidates, in accordance with regulatory
requirements and in sufficient quantities for commercialization and clinical testing meeting the applicable quality standards. If Corium fails to develop and maintain supply relationships with these third parties, or if Corium is unable to develop new relationships to replace any existing relationships that are lost, we may be unable to commercialize Twirla or to continue to develop our potential product candidates. Moreover, these third parties will be subject to FDA inspection. If these third parties do not comply with the FDA's regulatory requirements, we may not be able to maintain approval for Twirla or receive or maintain approval for any of our potential product candidates, and we may be subject to other regulatory enforcement action such as adverse inspectional findings, Warning Letters, Untitled Letters, recall requests, withdrawal of investigational approvals, clinical holds, or termination of clinical trials, refusals to approve pending applications, disgorgement, restitution, exclusion from federal healthcare programs, product seizures and detention, consent decrees, corporate integrity agreements, criminal and civil penalties, including imprisonment, refusal to permit import or export of the product and injunction against or restriction of manufacture or distribution.
We, through Corium, also rely on certain third parties as the current sole source of the materials they supply. Although many of these materials are produced in more than one location or are available from another supplier, if any of these materials becomes unavailable to us for any reason, we likely would incur added costs and delays in identifying or qualifying replacement materials and there can be no assurance that replacements would be available to us on acceptable terms, or at all. In certain cases, we may be required to get regulatory approval to use alternative suppliers, and this process of approval could delay the commercialization of Twirla or the development of our potential product candidates, indefinitely.
If Corium's third-party suppliers fail to deliver the required quantities of sub-components and starting materials, in accordance with all regulatory requirements, and on a timely basis and at commercially reasonable prices, and we and Corium are unable to find one or more alternative suppliers capable of production at a substantially equivalent cost in substantially equivalent volumes and quality, and on a timely basis, the commercialization of Twirla and the continued development of our potential product candidates, would be impeded, delayed, limited or prevented, which could harm our business, results of operations, financial condition and prospects. We could also face regulatory enforcement actions.
If the manufacturing facilities of Corium are not maintained in a manner that is compliant with FDA's manufacturing requirements, we may need to find alternative manufacturers and suppliers, which could result in supply interruptions of Twirla and our potential product candidates, additional costs and lost revenues.
Corium's facilities used for the manufacture of Twirla and our potential product candidates must be maintained in a manner compliant with the FDA's manufacturing requirements, including obtaining favorable inspection reports. We do not control the manufacturing process and are dependent on Corium for compliance with the FDA's requirements for manufacture of Twirla and our potential product candidates. If Corium cannot successfully manufacture material components and finished products that conform to our specifications and the FDA's strict regulatory requirements, they and we may be subject to regulatory action, including adverse inspectional findings, Warning Letters, Untitled Letters, product recall requests, withdrawal of product or investigational approvals, non-approval of marketing applications, clinical holds or termination of clinical trials, disgorgement, restitution, exclusion from federal healthcare programs, detentions or seizures, refusal to allow the import or export of a product, injunction against or restriction of manufacture or distribution, consent decrees, corporate integrity agreements, criminal and civil penalties, including imprisonment, and Corium may not be able to maintain FDA approval for its manufacturing facilities or acceptance of its manufacturing data in regulatory filings. If Corium's facilities cannot maintain compliance with FDA requirements, we may need to find and successfully qualify alternative manufacturing facilities, which could result in supply interruptions of Twirla and our potential product candidates and substantial
additional costs as a result of such delays, including costs with respect to finding alternative manufacturing facilities, and lost revenues. There is further no guarantee that the FDA would approve these alternative facilities.
We rely on third parties to conduct aspects of our clinical trials and post marketing studies. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or comply with applicable regulatory requirements, we may not be able to maintain regulatory approval for Twirla or be delayed in obtaining or ultimately not be able to obtain marketing approval for our potential product candidates.
We currently rely and plan to continue to rely on CROs and clinical trial sites for most aspects of our post-marketing study and any other clinical trials of our potential product candidates, such as trial conduct, data management, statistical analysis and electronic compilation of our FDA submission. We may enter into agreements with additional CROs and clinical trial sites to obtain additional resources and expertise in an attempt to accelerate our progress with regard to new or ongoing clinical and preclinical programs. Entering into relationships with CROs and clinical trial sites involves substantial cost and requires extensive management time and focus. In addition, typically there is a transition period between engagement of a CRO and clinical trial sites and the time the CRO and sites commences work. As a result, delays may occur, which may materially impact our ability to meet our desired post-marketing and clinical development timelines and ultimately have a material adverse impact on the commercialization of Twirla, our ability to maintain our marketing authorization for Twirla, our operating results, financial condition or future prospects. For example, as part of Twirla's approval, the FDA is requiring that we conduct a post-marketing study comparing the risks for venous thromboembolism (VTE) and arterial thromboembolism (ATE) in new users of Twirla to new users of oral combined hormonal contraceptives (CHCs) and new users of Xulane in U.S. women of reproductive age. The FDA has also required a second small post-marketing study to assess Twirla's residual drug content, strength, and adhesion. We plan to engage the services of a CRO to design, enroll, and complete this study, which will likely involve thousands of subjects and hundreds of clinical trial sites and will require substantial time and resources. If the CRO cannot enroll subjects and complete the trial in a timely manner, we may be unable to complete the study required by the FDA and subsequently may lose our marketing authorization for Twirla or be subject to other enforcement actions, and be forced to suspend commercial activities regarding the product.
As CROs and clinical investigators are not our employees, we cannot control whether or not they devote sufficient time and resources to our clinical trials for which they are engaged to perform, and whether they comply with the applicable regulatory requirements, known as Current Good Clinical Practices, or cGCPs, which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area, or EEA, and comparable foreign regulatory authorities for all of our products and potential product candidates in clinical trials, which include requirements related to the conduct of the study, subject informed consent, and IRB approval. Regulatory authorities enforce these cGCPs through routine inspections of trial sponsors, principal investigators and trial sites. Although we may rely on third parties for the execution of our trials, we are nevertheless responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on CROs and clinical trial sites does not relieve us of our regulatory responsibilities. If we, any of our CROs, or clinical trial sites fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, European Medicines Agency or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications for potential product candidates in development, or to perform additional post marketing studies for approved products or determine that data from the post marketing study is not sufficient to support maintaining marketing authorization for the product at issue. We cannot assure you that, upon
inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials or post marketing studies complies with cGCP regulations.
In addition, our clinical trials must be conducted with product and potential product candidates' materials produced in compliance with the FDA's manufacturing regulations. Our failure to comply with these regulations may require us to discontinue or repeat clinical trials, which would delay the regulatory approval process for our potential product candidates or impact our ability to meet our post-market study requirements. If the CROs or clinical trial sites we engage do not successfully carry out their contractual duties or obligations, conduct the clinical trials in accordance with all regulatory requirements and the applicable protocols, or meet expected deadlines, or if they need to be replaced, or the quality or accuracy of the data they provide is compromised due to the failure to adhere to regulatory requirements or for other reasons, then our development programs may be extended, delayed or terminated, we may not be able to obtain marketing approval for or successfully commercialize our potential product candidates, or we may not be able to meet our post-market study requirements. Failure to comply with clinical trial regulatory requirements may further subject us to regulatory action, including Warning Letters, Untitled Letters, adverse inspectional findings, clinical holds or termination of clinical trials, non-approval of marketing applications, criminal and civil penalties, including imprisonment, injunction against manufacture or distribution and debarment. As a result, our financial results and the commercial prospects for Twirla or our potential product candidates would be harmed and our costs would increase.
Any collaboration arrangements that we may enter into in the future may not be successful, which could adversely affect our ability commercialize Twirla and to develop and commercialize our potential product candidates.
We may seek partnerships, collaborations and other strategic transactions to maximize the commercial potential of Twirla, our potential product candidates and our proprietary technologies in the United States and territories throughout the world. We may enter into such arrangements on a selective basis depending on the merits of retaining commercialization rights for ourselves as compared to entering into selective collaboration arrangements with leading pharmaceutical or biotechnology companies for Twirla and each of our potential product candidates and technologies, both in the United States and internationally. We face competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex, and time consuming to negotiate, document and implement. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements should we choose to enter into such arrangements. The terms of any collaborations or other arrangements that we may establish may not be favorable to us.
Any future collaborations that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations. Collaborators, also, may not comply with the applicable regulatory requirements, which may subject them or us to enforcement actions.
Disagreements between parties to a collaboration arrangement regarding clinical development and commercialization matters could lead to delays in the commercialization of Twirla or the development process or commercialization of our potential product candidates and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority.
Collaborations with pharmaceutical or biotechnology companies and other third parties often are terminated or allowed to expire by the other party. Any such termination or expiration could adversely affect us financially and could harm our business reputation.
If we fail to establish an effective distribution process our business may be adversely affected.
We do not currently have the infrastructure necessary for distributing pharmaceutical products. We intend to contract with a third-party logistics wholesaler to warehouse these products and distribute them to pharmacies. This distribution network will require significant coordination with our sales and marketing and finance organizations. Failure to secure contracts with third party logistics providers s could negatively impact the distribution of Twirla and our potential product candidates, if and when approved, and failure to coordinate financial systems could negatively impact our ability to accurately report product revenue. If we are unable to effectively establish and manage the distribution process, the commercial launch and sales of Twirla and of our potential product candidates, if and when approved, will be delayed or severely compromised and our results of operations may be harmed. Distribution practices will also need to comply with the applicable regulatory requirements and we and our distributors will be required to hold state licenses in the States to which Twirla or any of our potential product candidates, if approved, are distributed. If we or our distributors do not comply with the applicable regulatory requirements, we could be exposed to potential enforcement actions and product distribution may be interrupted or discontinued.
We may rely on third parties to perform many essential services for any products that we commercialize, including services related to government price reporting, customer service, accounts receivable management, cash collection, and pharmacovigilance and adverse event reporting. If these third parties fail to perform as expected or to comply with legal and regulatory requirements, our ability to commercialize our potential product candidates will be significantly impacted and we may be subject to regulatory sanctions.
We may retain third-party service providers to perform a variety of functions related to Twirla, key aspects of which will be out of our direct control. These service providers may provide key services related to customer service, accounts receivable management, and cash collection. If we retain a service provider, we would substantially rely on it as well as other third-party providers that perform services for us. If these third-party service providers fail to comply with applicable laws and regulations, fail to meet expected deadlines, or otherwise do not carry out their contractual duties to us, or encounter physical or natural damage at their facilities, our ability to deliver product to meet commercial demand would be significantly impaired and we may be subject to regulatory enforcement action.
In addition, we may engage third parties to perform various other services for us relating to pharmacovigilance and adverse event reporting, safety database management, fulfillment of requests for medical information regarding Twirla and related services. If the quality or accuracy of the data maintained by these service providers is insufficient, or these third parties otherwise fail to comply with regulatory requirements, we could be subject to regulatory sanctions.
We may further contract with a third party to calculate and report pricing information mandated by various government programs. If a third party fails to timely report or adjust prices as required, or errors in calculating government pricing information from transactional data in our financial records, it could impact our discount and rebate liability, and potentially subject us to regulatory sanctions or False Claims Act lawsuits.
Risks Related to Intellectual Property Rights
We may not be able to protect our proprietary technology in the marketplace.
We depend on our ability to protect our proprietary technology. We rely on trade secret, patent, copyright and trademark laws, and confidentiality, licensing and other agreements with employees and third parties, all of which offer only limited protection. Our success depends in large part on our ability and any future licensee's ability to maintain our patents and to obtain additional patent protection in the United States and other countries with respect to our proprietary technology and products. We believe we will be able to obtain, through prosecution of our pending patent applications, additional
patent protection for our proprietary technology. If we are compelled to spend significant time and money protecting or enforcing our patents, designing around patents held by others or licensing or acquiring, potentially for large fees, patents or other proprietary rights held by others, our business and financial prospects may be harmed. If we are unable to effectively protect the intellectual property that we own, other companies may be able to offer for sale the same or similar products containing the generically available active pharmaceutical ingredients in Twirla and our potential product candidates, which could materially adversely affect our competitive business position and harm our business prospects. Our patents may be challenged, narrowed, invalidated or circumvented, which could limit our ability to stop competitors from marketing the same or similar products or limit the length of the term of patent protection that we may have for our potential product candidates. Even if our patents are unchallenged, they may not adequately protect our intellectual property, provide exclusivity for our potential product candidates or prevent others from designing around our claims. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business.
The patent positions of pharmaceutical products are often complex and uncertain. The breadth of claims allowed in pharmaceutical patents in the United States and many jurisdictions outside of the United States is not consistent. For example, in many jurisdictions the support standards for pharmaceutical patents are becoming increasingly strict. Some countries prohibit method of treatment claims in patents. Changes in either the patent laws or interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or create uncertainty. In addition, publication of information related to our current product and potential product candidates and potential products may prevent us from obtaining or enforcing patents relating to thesethis product and potential product candidates and potential products, including without limitation transdermal delivery systems and methods of using such transdermal delivery systems. Our product and potential product candidates contain generically available active pharmaceutical ingredients. As a result, new chemical entity patents directed to the active pharmaceutical ingredients in our product and potential product candidates, which are generally believed to offer the strongest form of patent protection, are not available for our potential product candidates.
Patents that we own or may license in the future do not necessarily ensure the protection of our intellectual property for a number of reasons, including without limitation the following:
If we encounter delays in our development or clinical trials, the period of time during which we could market our product candidates under patent protection would be reduced.
Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner. Our competitors may seek to market generic, similar or strength modified versions of any approved products by submitting abbreviated new drug or 505(b)(2) NDA applications to the FDA in which our competitors claim that our patents are invalid, unenforceable or not infringed. Alternatively, our competitors may seek approval to market their own products that are the same as, similar to or otherwise competitive with Twirla or our potential product candidates. In these circumstances, we may need to defend or assert our patents, by means including filing lawsuits alleging patent infringement. In any of these types of proceedings, a court or government agency with jurisdiction may find our patents invalid, unenforceable or not infringed. We may also fail to identify patentable aspects of our research and development before it is too late to obtain patent protection. Even if we have valid and enforceable patents, these patents still may not provide protection against competing products or processes sufficient to achieve our business objectives.
The issuance of a patent is not conclusive as to its inventorship, scope, ownership, priority, validity or enforceability. In that regard, third parties may challenge our patents in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and potential products. In addition, given the amount of time required for the development, testing and regulatory review, and commercial launch of new
product candidates, products, patents protecting such candidatesany approved product we may have might expire or be held invalid or unenforceable before our company can realize sufficient economic value following commercialization of our product candidates.commercialization.
Our intellectual property portfolio is currently comprised of issued patents and pending patent applications. If our issued patents are found to be invalid, not enforceable or not infringed by competitor products, or pending patent applications fail to issue or fail to issue with a scope that is meaningful to Twirla or our potential product candidates, our business will be adversely affected.
There can be no assurance that our pending patent applications will result in issued patents in the United States or foreign jurisdictions in which such applications are pending. Even if patents do issue on any of these applications, there can be no assurance that a third partythird-party will not challenge their validity or enforceability, that we will obtain sufficient claim scope or term in those patents to prevent a
third party from competing successfully with Twirla or our potential product candidates, or that, even if our patents are found to be valid, enforceable, and infringed, a legal tribunal would enjoin infringing activity.
We may not be able to enforce our intellectual property rights throughout the world.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to life sciences. To the extent that we have obtained or are able to obtain patents or other intellectual property rights in any foreign jurisdictions, it may be difficult for us to stop the infringement of our patents or the misappropriation of other intellectual property rights. For example, some foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the availability of certain types of patent rights and enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit.
Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology, Twirla and potential product candidates, and the enforcement of intellectual property.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications will beare prosecuted and may also affect patent litigation. In particular, under the Leahy-Smith Act, the United States transitioned in March 2013 to a "first to file" system in which the first inventor to file a patent application will be entitled to the patent. Third parties are allowed to submit prior art before the issuance of a patent by the USPTO and may become involved in post-grant proceedings including reexamination, post-grant review, inter-partesinter partes review, or derivation or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope or enforceability of, or invalidate, our patent rights, which could adversely affect our competitive position.
The USPTO has developed regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, did not become effective until March 16, 2013. However, the full impact of the Leahy-Smith Act, andas well as the courts' reviewtreatment of any appeals to related proceedings, is in its early stages.remain unclear. Accordingly, the full impact that the Leahy-Smith Act will have on the operation of our business is not clear. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, as well as our ability to bring about timely favorable resolution of any disputes involving our patents and the patents of others.
Obtaining and maintaining our patent protection depends on compliance with various procedural, documentary, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for noncompliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in unenforceability, invalidity, abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Noncompliance events that could result in unenforceability, invalidity, abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or any future licensors fail to maintain the patents and patent applications covering Twirla or our potential product candidates, our competitive position would be adversely affected.
We may infringe the intellectual property rights of others, which may prevent or delay our commercialization and product development efforts and stop us from commercializing or increase the costs of commercializing Twirla or our products,potential product candidates, when and if approved.
Our commercial success depends significantly on our ability to operate without infringing the patents and other intellectual property rights of third parties. For example, there could be issued patents of which we are not aware that Twirla or our current or future potential product candidates infringe. There also could be patents that we believe we do not infringe, but that we may ultimately be found to infringe.
Moreover, patent applications are in some cases maintained in secrecy until patents are issued. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made and patent applications were filed. There may be currently pending applications of which we are unaware that may later result in issued patents that Twirla or our current or future potential product candidates infringe. For example, pending applications may exist that claim or can be amended to claim subject matter that Twirla or our current or future potential product candidates infringe. Competitors may file continuing patent applications claiming priority to already issued patents in the form of continuation, divisional or continuation-in-part applications, in order to maintain the pendency of a patent family and attempt to cover Twirla or our potential product candidates.
Third parties may assert that we are employing their proprietary technology without authorization and may sue us for patent or other intellectual property infringement or misappropriation. These lawsuits are costly and could adversely affect our results of operations and divert the attention of managerial and scientific personnel. If we are sued for patent infringement, we would need to demonstrate that our product, potential product candidates or methods either do not infringe the claims of the relevant patent or that the patent claims are invalid or unenforceable, and we may not be able to do this.
Proving invalidity or unenforceability is difficult. For example, in the United States, proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could have a material adverse effect on us. In addition, we may not have sufficient resources to bring these actions to a successful conclusion. If a court holds that any third-party patents are valid, enforceable and cover our product, potential product candidates, or their use, the holders of any of these patents may be able to block our ability to commercialize
Twirla or our potential product candidates unless we acquire or obtain a license under the applicable patents or until the patents expire. We may not be able to enter into licensing arrangements or make other arrangements at a reasonable cost or on reasonable terms. Any inability to secure licenses or alternative technology could result in delays in the introduction of our product or potential product candidates or lead to prohibition of the manufacture or sale of our product or potential product candidates by us. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, in any such proceeding or litigation, we could be found liable for monetary damages, including treble damages and attorneys' fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product or potential product candidates or force us to cease some of our business operations, which could materially harm our business. Any claims by third parties that we have misappropriated their confidential information, know-how or trade secrets could have a similar negative impact on our business. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
We may be subject to claims that we or our employees have misappropriated the intellectual property, including know-how or trade secrets, of a third party, or that claim ownership of what we regard as our own intellectual property.
Many of our employees, consultants and contractors were previously employed at or engaged by biotechnology companies or other pharmaceutical companies, including our competitors or potential competitors. Some of these employees, consultants and contractors, including each member of our senior management, executed proprietary rights, non-disclosure and non-competition agreements in connection with such previous employment. Although we try to ensure that our employees, consultants and contractors do not use the intellectual property and other proprietary information or know-how or trade secrets of others in their work for us, we may be subject to claims that we or these employees, consultants and contractors have used or disclosed such intellectual property, including know-how, trade secrets or other proprietary information. Litigation may be necessary to defend against these claims. We are not aware of any threatened or pending claims related to these matters or concerning agreements with our senior management, or other of our employees, consultants and contractors, but litigation may be necessary in the future to defend against such claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, or personnel or access to consultants and contractors. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
In addition, while we typically require our employees, consultants and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own, which may result in claims by or against us related to the ownership of such intellectual property. If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to our management and scientific personnel.
We may be unable to adequately prevent disclosure of trade secrets and other proprietary information.
We rely on trade secrets to protect our proprietary technological advances and know-how, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, contractors, outside scientific collaborators, sponsored researchers and other advisors,
including the third parties we rely on to manufacture Twirla and our potential product candidates, to protect our trade secrets and other proprietary information. However, any party with whom we have executed such an agreement may breach that agreement and disclose our proprietary information, including our trade secrets. Accordingly, these agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights. In addition, others may independently discover our trade secrets and proprietary information. Further, the FDA, as part of its Transparency Initiative, a proposalpreviously took steps to increase disclosure and make data more accessible to the public is currently considering whether to make additionaldisclosure of information publicly available on a routine basis,regarding FDA-regulated products, including information that we may consider to be trade secrets or other proprietary information, and itinformation. It is not clear at the present time how the FDA's disclosure policies may change in the future, if at all. Failure to obtain or maintain trade secret protection could enable competitors to use our proprietary information to develop products that compete with our products or cause additional, material adverse effects upon our competitive business position and financial results.
Any lawsuits relating to infringement of intellectual property rights brought by or against us will be costly and time consuming and may adversely impact the price of our common stock.
We may be required to initiate litigation to enforce or defend our intellectual property rights. These lawsuits can be very time consuming and costly. There is a substantial amount of litigation involving patent and other intellectual property rights in the pharmaceutical industry generally. Such litigation or proceedings could substantially increase our operating expenses and reduce the resources available for development activities or any future sales, marketing or distribution activities.
In infringement litigation, any award of monetary damages we receive may not be commercially valuable. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information and trade secrets could be compromised by disclosure during litigation. Moreover, there can be no assurance that we will have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are resolved. Further, any claims we assert against a perceived infringer could provoke these parties to assert counterclaims against us alleging that we have infringed their patents. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
In addition, our patents and patent applications in the United States and other jurisdictionjurisdictions could face other challenges, such as derivation or interference proceedings, opposition proceedings,inter partes review, reexamination proceedings, third party submissions of prior art, and other forms of post-grant challenges. In the United States, for example, post-grant review, which is similar to opposition proceedings available in many countries other than the U.S., was newly established by the Leahy-Smith Act. Any of these challenges, if successful, could result in the invalidation of, or in a narrowing of the scope or preventing the issuance of, any of our patents and patent applications subject to challenge. Any of these challenges, regardless of their success, would likely be time consuming and expensive to defend and resolve and would divert our management and scientific personnel's time and attention.
In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the market price of our common stock.
Intellectual property disputes could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings.
Risks Related to the Development of Our Additional Potential Product Candidates
If we fail to develop and commercialize Twirla and our current pipeline of additional potential product candidates, our prospects for future growth and our ability to reach or sustain profitability may be limited.limited or never achieved.
A key element of our long-term strategy is to develop, obtain regulatory approval for and commercialize our portfolio of potential product candidates in addition to Twirla. To do so, we plan to utilize our proprietary transdermal delivery technology, Skinfusion,Skinfusion®, to develop additional potential product candidates. We may not be successful in our efforts to develop our portfolio of additional potential product candidates, and any potential product candidates we do develop may not produce commercially viable products that safely and effectively treat their indicated conditions. To date, our efforts have identified three additional potential product candidates, in addition to Twirla, including AG200-ER, which is a regimen designed to allow a woman to extend the length of her cycle, AG200-SP, which is a regimen designed to provide shorter, lighter periods, and AG890, which is a progestin-only contraceptive patch intended for use by women who are unable or unwilling to take estrogen. AG200-SP and AG200-ER are intended to be Twirla line extensions that would expand the use of Twirla beyond its initial approved use. In July 2016, we began preparations for an initial Phase 2 clinical trial examining the use of AG200-SP along with a smaller lower dose combination ethinyl estradiol/levonorgestrelEE/LNG patch (SmP) in the fourth week of the woman's cycle. TheWe have decided to postpone the trial and will continue to evaluate the timing for initiating dosing of subjects for this Phase 2 clinical trial, which is designed to identify the optimal dose of the SmP,dependent on financial and will evaluate bleeding profiles, pharmacokinetic parameters, ovulation inhibition and safety over 3 cycles of treatment with AG200-SP. We expect to initiate dosing of the AG200-SP (SmP) clinical trial in the first quarter of 2017.other capital resources. Our planned Phase 2 clinical trial of AG200-SP (SmP) is only the initial clinical trial in this program and AG200-SP (SmP) maywill require additional clinical trials to establish the safety and efficacy of this potential product candidate. The other potential product candidates in our pipeline will likely require additional product development efforts to optimize patch formulations and dosing. In addition, we will need to conduct additional clinical trials to establish the safety and efficacy of these potential product candidates, which will require additional capital. Our abilitySubstantially all of our resources are currently dedicated to develop thesethe manufacturing, validation and commercialization of Twirla. We will require additional capital to complete the commercialization plan for Twirla and to advance the development of our other potential product candidates, in particular AG200-SP and AG200-ER could be significantly affected by our inability to get Twirla approved.candidates.
Our development programs may initially show promise in identifying potential product leads yet fail to produce potential product candidates for clinical development. In addition, identifying new treatment needs and potential product candidates requires substantial technical, financial and human resources on our part. If we are unable to obtain development partners or additional development program funding, or to continue to devote substantial technical and human resources to such programs, we may have to delay or abandon these programs. Any potential product candidate that we successfully identify may require substantial additional development efforts prior to commercial sale, including preclinical studies, extensive clinical
testing and approval by the FDA and applicable foreign regulatory authorities. All potential product candidates are susceptible to the risks of failure that are inherent in pharmaceutical product development.
Any clinical development activities, including clinical trials, necessary to obtain and maintain regulatory approvals may not be successfully completed.
Twirla is our first approved product. While we have one approved product, we may not be successful in conducting and managing the clinical activities, including clinical trials, necessary to obtain and maintain regulatory approvals, which might prevent us from successfully designing, implementing, or completing the clinical trials required to support regulatory approval of our potential product candidates and meet our post-marketing study requirements. The application, approval and maintenance process for the FDA and other regulatory agencies are complex and difficult and vary by regulatory agency, and we might not be able to demonstrate that our potential product candidates or Twirla meet the appropriate standards for initial and continued regulatory approval or initiate and continue to commercialize Twirla or our potential product candidates, if approved, in the U.S. or elsewhere, or we might be significantly delayed in doing so. In such circumstances, our business, financial condition, results of operations and prospects and the value of our common stock may be materially adversely affected.
If we experience any of a number of possible unforeseen events in connection with clinical trials related to our potential product candidates, or Twirla, any potential marketing authorization or commercialization of our potential product candidates could be delayed or prevented or we may not be able to meet our post-marketing study requirements for Twirla.
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to receive marketing authorization or commercialize our potential product candidates, may prevent us from meeting our Twirla post-marketing study requirements, or which may adversely impact our commercialization of Twirla including:
Our costs will increase if we experience delays in testing, completion of post-marketing studies, or, for our potential product candidate marketing authorizations. We do not know whether any clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all.
For Twirla, failure to complete post-marketing studies may also result in enforcement actions or removal of the product from the market. Adverse post-marketing study results may also result in withdrawal or limitations on marketing applications, label changes, or other restrictions or requirements, such as REMS or additional study requirements.
For our potential product candidates, we cannot commercialize any potential product candidates in the U.S. without first obtaining FDA approval. Before obtaining regulatory approvals for commercial sale, however, we must demonstrate in, or rely on data from preclinical studies and well controlled clinical trials that the potential product candidate is safe and effective for use in the target indication and that the manufacturing processes, facilities, and controls are adequate. Obtaining marketing approval in the U.S. is a lengthy, expensive and uncertain process, and approval may not be obtained or may be subject to significant restrictions. Significant clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our potential product candidates, allow our competitors to bring products to market before we do, or impair our ability to successfully commercialize our potential product candidates, and so may harm our business, results of operations and financial condition. Delays in regulatory approvals or failure to obtain regulatory approval for a potential product candidate may result from many factors, including:
Delays or failure to receive regulatory approval for any additional potential product candidates may materially impact our business.
We may be unable to license or acquire suitable additional potential product candidates or technologies from third parties for a number of reasons.
The licensing and acquisition of pharmaceutical products is competitive. A number of more established companies are also pursuing strategies to license or acquire products. These established companies may have a competitive advantage over us due to their size, cash resources or greater clinical development and commercialization capabilities. In addition, we expect competition in acquiring potential product candidates to increase, which may lead to fewer suitable acquisition opportunities for us as well as higher acquisition prices.
Other factors that may prevent us from licensing or otherwise acquiring suitable potential product candidates include the following:
Risks Related to Our Business Operations and Industry
In order to establish our sales and marketing infrastructure, we will need to grow the size of our organization, and we may experience difficulties in managing this growth.
As of December 31, 2016,2019, we had a total of 1915 full-time employees and wereflecting a resumption of hiring to advance the commercialization of Twirla. We use third-party consultants to assist with our current sales and marketing functions. As our development and commercialization plans and strategies develop,of Twirla advances, we expect to need to expand the size of our employee base for managerial, operational, commercial, sales, marketing, financialcompliance, regulatory, finance and other resources. Future growth would impose significant added responsibilities on members of management, including the need to identify, recruit, maintain, motivate and integrate additional employees. In addition, our management may have to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. Our future financial performance and our ability to commercialize Twirla if approved, and any other future potential product candidates and our ability to compete effectively will depend, in part, on our ability to effectively manage any future growth.
If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive pharmaceuticals industry depends in large part upon our ability to attract and retain highly qualified managerial, scientific and medical personnel. We are highly dependent on our management, scientific and medical personnel. In order to induce valuable employees to remain with us, we have provided these employees with stock options that vest over time. The value to employees of stock options that vest over time is significantly affected by movements in our stock price that we cannot control and may at any time be insufficient to counteract more lucrative offers from other companies.
Table of Contents Additionally, at times, we have also implemented programs that included cash retention bonuses and/or restricted stock units as incentives to retain employees.
Our management team has expertise in many different aspects of drug development and commercialization. Competition for skilled personnel in our market is intense and competition for experienced personnel may limit our ability to hire and retain highly qualified personnel on acceptable terms. Despite our efforts to retain valuable employees, members of our management, scientific and medical teams may terminate their employment with us on short notice. We have an employment agreementagreements with only one of our employees,named executive officers which includes Alfred Altomari, our PresidentChairman and Chief Executive Officer. The employment agreement providesagreements provide for at-will employment, which means that Mr. Altomari or any of our other employees could leave our employment at any time, with or without notice. The loss of the services of any of our executive officers or other key employees could potentially harm our business, operating results or financial condition. In particular, we believe that the loss of the services of Mr. Altomari or Dr. Elizabeth Garner, our Chief Medical Officer, may have a material adverse effect on our business. We do not currently carry "key person" insurance on the lives of members of executive management. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior, mid-level and senior managers as well as junior, mid-level and senior scientific and medical personnel.
Other pharmaceutical companies with which we compete for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high-quality candidates than those that we have to offer. If we are unable to continue to attract and retain high-quality personnel, the rate of and success with which we can develop and commercialize Twirla, as well as our potential product candidates, would be limited.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of Twirla or our otherpotential product candidates, if approved.
We face a potential riskrisks of product liability as a result of the clinical testing and commercial availability of Twirla and the clinical testing of our other potential product candidates and will face an even greater risk if we commercialize Twirla or our other product candidates, if approved or any other current or future product candidate.candidates. For example, we may be sued if Twirla or any potential product candidate we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization or development of the product or potential product candidate subject to such claims. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
We have obtained limited product liability insurance coverage for our productsTwirla and our clinical trials with a $10.0 million annual aggregate coverage limit. Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization Twirla or of potential product candidates we develop. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
We may acquire businesses or products, or form strategic alliances in the future, and we may not realize the benefits of such acquisitions or alliances.
We may acquire additional businesses or products, form strategic alliances or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter numerous difficulties in developing, manufacturing and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. We cannot assure you that, following any such acquisition, we will achieve the expected synergies to justify the transaction.
Our business is affected by macroeconomic conditions.
Various macroeconomic factors could adversely affect our business and the results of our operations and financial condition, including changes in inflation, interest rates and foreign currency exchange rates, and overall economic conditions and uncertainties, including those resulting from political instability and the current and future conditions in the global financial markets. For instance, if inflation or other factors were to significantly increase our business costs, it may not be feasible to pass through price increases to patients. Interest rates, the liquidity of the credit markets and the volatility of the capital markets could also affect the value of our investments and our ability to liquidate our investments in order to fund our operations, if necessary.
Interest rates and the ability to access credit markets could also adversely affect the ability of patients, payors and distributors to purchase, pay for and effectively distribute our products if and when approved. Similarly, these macroeconomic factors could affect the ability of our current or potential future contract manufacturers, sole-source or single-source suppliers, or licensees to remain in business or otherwise manufacture or supply our product candidates. Failure by any of them to remain in business could affect our ability to manufacture product candidates.
We continue to incur significant increased costs as a result of operating as a public company, and our management is required to devote substantial time to compliance initiatives.
As a public company, we continue to incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC and the NASDAQ GlobalNasdaq Capital Market, impose various requirements on public companies, including requiring the establishment and maintenance of effective disclosure controls and internal control over financial reporting and changes in corporate governance practices. Our management and other personnel devote a substantial amount of time to these compliance initiatives.
Moreover, these rules and regulations have increased our legal and financial compliance costs and have made some activities more time-consuming and costly. We estimate that we will annually incur approximately $2.0 million in expenses in response to these requirements.
Section 404(a) of the Sarbanes-Oxley Act requires annual management assessments of the effectiveness of our internal control over financial reporting, starting with the second annual report that we would expect to file with the SEC. However, for as long as we remain an "emerging growth company" as defined in the Jumpstart Our Business Startups Act of 2012, or JOBS Act, we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not "emerging growth companies" including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act. We may take advantage of these reporting exemptions until we are no longer an "emerging growth company." We will remain an "emerging growth company" until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.0 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of the completion of our initial public offering; (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
reporting. Our testing, orand the subsequent testing by
our independent registered public accounting firm, may reveal deficiencies in our internal control over financial reporting that are deemed to be material weaknesses. We will incur substantial accounting expense and expend significant management efforts to comply with internal control over financial reporting requirements. We currently do not have an internal audit group, and we may need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge. Moreover, if we are not able to comply with these requirements in a timely manner or if we or our independent registered public accounting firm identifies deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our common stock could decline, and we could be subject to sanctions or investigations by the NASDAQ GlobalNasdaq Capital Market, the SEC or other regulatory authorities, which would require additional financial and management resources.
Business interruptions could delay us in the process of developing our potential product candidates and could disrupt our sales.
Our headquarters are located in Princeton, New Jersey, and Corium, our contract manufacturer, is located in Grand Rapids, Michigan. We are vulnerable to natural disasters, such as severe storms and other events that could disrupt our or Corium's operations. We do not carry insurance for natural disasters, and we may not carry sufficient business interruption insurance to compensate us for losses that may occur. Any losses or damages we incur could have a material adverse effect on our business operations.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems, and those of our CROs and other third parties on which we rely, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures.failures, cyber-attacks or cyber-intrusions over the internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyber-attacks or cyber-intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further commercialization of Twirla and/or development of our potential product candidates could be delayed.
Our employees, independent contractors, principal investigators, CROs, manufacturers, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading, which could significantly harm our business.
We are exposed to the risk that employees, independent contractors, principal investigators, CROs, manufacturers, consultants, commercial partners and vendors may engage in fraudulent or other illegal activity, fraud or other misconduct. Misconduct by these parties could include intentional, reckless or negligent conduct or disclosure of unauthorized activities to us that violates: (i) the law and regulations of the FDA and non-U.S. regulators, including those laws that require the reporting of true, complete and accurate information to the FDA and non-U.S. regulators, (ii) healthcare fraud and abuse laws and regulations in the United States and abroad and (iii) laws that require the true, complete and accurate
reporting of financial information or data. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Misconduct in violation of these laws may also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of conduct, but it is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including regulatory enforcement actions, the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, corporate integrity agreements, contractual damages, reputational harm, diminished profits and future earnings and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Our ability to use net operating loss and tax credit carryforwards and certain built-in losses to reduce future tax payments may be limited by provisions of the Internal Revenue Code of 1986, as amended, and may be subject to further limitation as a result of our initial public offering.
Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code, contain rules that limit the ability of a company that undergoes an ownership change, which is generally any change in ownership of more than 50% of its stock over a three-year period, to utilize its net operating loss and tax credit carryforwards and certain built-in losses recognized in years after the ownership change. These rules generally operate by focusing on ownership changes involving stockholders owning, directly or indirectly, 5% or more of the stock of a company and any change in ownership arising from a new issuance of stock by the company. Generally, if an ownership change occurs, the yearly taxable income limitation on the use of net operating loss and tax credit carryforwards and certain built-in losses is equal to the product of the applicable long-term tax exempttax-exempt rate and the value of the company's stock immediately before the ownership change. We may be unable to offset future taxable income, if any, with losses, or our tax liability with credits, before such losses and credits expire and therefore would incur larger federal income tax liability. Our net operating loss carryforwards arising in taxable years ending on or prior to December 31, 2017 will expire between 2019 and 2037 if we have not used them. Net operating loss carryforwards arising in taxable years ending after December 31, 2017 are no longer subject to expiration under the Code.
In addition, it is possible that the transactions relating to our initial public offering or subsequent public offerings, either on a standalone basis or when combined with future transactions, hashave caused us to undergo one or more additional ownership changes. In that event, we generally would not be able to use our pre-change loss or credit carryovers or certain built-in losses prior to such ownership change to offset future taxable income in excess of the annual limitations imposed by Sections 382 and 383.383 of the Code. We have not completed a
study to assess whether an ownership change has occurred, or whether there have been multiple ownership changes since our inception.
Risks Related to Ownership of Our Common Stock
An active trading market for our common stock may not be sustained.
In May 2014, we closed our initial public offering. Prior to our initial public offering, there was no public market for shares of our common stock. Although we have completed our initial public offering and shares of our common stock are listed and trading on The NASDAQ Global Market, an active trading market for our shares may not be sustained. If an active market for our common stock does not continue, it may be difficult for our stockholders to sell their shares without depressing the market price for the shares or sell their shares at or above the prices at which they acquired their shares or sell their shares at the time they would like to sell. Any inactive trading market for our common stock may also impair our ability to raise capital to continue to fund our operations by selling shares.
We expect that our stock price may fluctuate significantly.
Prior to our initial public offering, you could not buy or sell our common stock publicly. The trading price of our common stock is highly volatile and is subject to wide fluctuations in response to various factors, some of which are beyond our control, including limited trading volume. In
addition to the factors discussed in this "Risk Factors" section and elsewhere in this quarterlyannual report, these factors include:
These and other market and industry factors may cause the market price and demand for our common stock to fluctuate substantially, regardless of our actual operating performance, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock. In addition, the stock market in general, and the NASDAQ GlobalNasdaq Capital Market and the stock prices of pharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. In the past, when the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert the time and attention of our management. For example, two complaints have been filed in federal court in the District of New Jersey on January 6, 2017 and January 20, 2017, titledPeng v. Agile Therapeutics, Inc., Alfred Altomari, and Elizabeth Garner, No. 17-cv-119 (D.N.J.), andLichtenthal v. Agile Therapeutics, Inc., Alfred Altomari, and Elizabeth Garner, No. 17-cv-405 (D.N.J.), respectively, on behalf of a putative class of investors who purchased stock from March 9, 2016 through January 3, 2017. The complaints allege violations of the federal securities laws based on public statements made regarding the Company's Phase 3 "SECURE" clinical trial. We deny all allegations in the complaints and we plan to vigorously defend the complaints that have been filed.
Future sales of shares of our common stock by existing stockholders could cause our stock price to decline.
If our existing stockholders sell substantial amounts of our common stock in the public market, or if the public perceives that such sales could occur, this could have an adverse impact on the market price of our common stock, even if there is no relationship between such sales and the performance of our business.
As of March 10, 2017, we had 28,776,398 shares of common stock outstanding. Of these shares, 25,150,683 shares of common stock are freely tradeable, without restriction, in the public market. Moreover, a relatively small number of our stockholders own large blocks of shares. We cannot predict the effect, if any, that public sales of these shares or the availability of these shares for sale will have on the market price of our common stock
In addition, the 3,983,387 shares subject to outstanding options under our stock option plans and the 724,030 shares reserved for future issuance under our stock option plans will become eligible for sale in the public market in the future, subject to certain legal and contractual limitations.
We may be subject to securities litigation, which is expensive and could divert management attention.
OurThe market price of our common stock may be volatile, and in the past companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation. Litigation of this type could result in
substantial costs and diversion of management's attention and resources, which could adversely impact
our business. Any adverse determination in litigation could also subject us to significant liabilities. For example, two complaints have been filed in federal court in the District of New Jersey on January 6, 2017 and January 20, 2017, titledPeng v. Agile Therapeutics, Inc., Alfred Altomari, and Elizabeth Garner, No. 17-cv-119 (D.N.J.), andLichtenthal v. Agile Therapeutics, Inc., Alfred Altomari, and Elizabeth Garner, No. 17-cv-405 (D.N.J.), respectively, on behalf of a putative class of investors who purchased stock from March 9, 2016 through January 3, 2017. The complaints allege violations of the federal securities laws based on public statements made regarding the Company's Phase 3 "SECURE" clinical trial. We deny all allegations in the complaints and we plan to vigorously defend the complaints that have been filed.
Our existing principal stockholders, executive officers and directors own a significant percentage of our common stock and will be able to exert a significant control over matters submitted to our stockholders for approval.
As of December 31, 2016,2019, our executive officers, directors, director nominees, holders of 5% or more of our capital stock and their respective affiliates together beneficially owned approximately 66.9%17.7% of our outstanding voting stock.
This significant concentration of share ownership may adversely affect the trading price for our common stock because investors often perceive disadvantages in owning stock in companies with controlling stockholders. As a result, these stockholders, if they acted together, could significantly influence all matters requiring approval by our stockholders, including the election of directors and the approval of mergers or other business combination transactions. These stockholders may be able to determine all matters requiring stockholder approval. The interests of these stockholders may not always coincide with our interests or the interests of other stockholders. This may also prevent or discourage unsolicited acquisition proposals or offers for our common stock that other stockholders may feel are in their best interest and our large stockholders may act in a manner that advances their best interests and not necessarily those of other stockholders, including seeking a premium value for their common stock, and might affect the prevailing market price for our common stock.
We will have broad discretion in how we use the net proceeds from our initial public offering, ourand private placement and our recently completed public offering.offerings. We may not use these proceeds effectively, which could affect our results of operations and cause our stock price to decline.
We will have considerable discretion in the application of the net proceeds from our initial public offering, our private placement and our recently completed public offering. We intend to use the majority of the net proceeds from our initial public offering, ourand private placement and our recently completed public offering to conduct a Phase 3 clinical trial for Twirla, obtain marketing approval and begin preparations for the U.S. commercial launch of Twirla, continue the equipment qualification and validation related to the expansion of Corium's manufacturing capabilities, develop our product pipeline, and for working capital and other general corporate purposes, which may include funding for the hiring of additional personnel, validation of capital equipment and the costs of operating as a public company.offerings. As a result, investors will be relying upon management's judgment with only limited information about our specific intentions for the use of the balance of the net proceeds from our initial public offering, our private placement and our recently completed public offering.and private offerings. We may use the net proceeds for purposes that do not yield a significant return or any return at all for our stockholders. In addition, pending their use, we may invest the net proceeds from our initial public offering, our private placement and our recently completed public offeringand private offerings in a manner that does not produce income or that loses value.
We are an "emerging growth company" and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make our common stock less attractive to investors.
We are an "emerging growth company," as defined in the JOBS Act, and we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not "emerging growth companies" including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. We may take advantage of these reporting exemptions until we are no longer an "emerging growth company." We will remain an "emerging growth company" until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.0 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date we completed our initial public offering; (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
Our status as an "emerging growth company" under the JOBS Act may make it more difficult to raise capital as and when we need it.
Because of the exemptions from various reporting requirements allowed to us as an "emerging growth company" we may be less attractive to investors and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with other companies in our industry if they believe that our financial accounting is not as transparent as other companies in our industry. If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially and adversely affected.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act, or the subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our financial statements or require us to identify other areas for further attention or improvement. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting once that firm beginconducts its Section 404 reviews, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by the NASDAQ GlobalNasdaq Capital Market, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial
reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We are subject to the periodic reporting requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements or insufficient disclosures due to error or fraud may occur and not be detected.
We have never paid dividends on our common stock and we do not anticipate paying any dividends in the foreseeable future. Consequently, any gains from an investment in our common stock will likely depend on whether the price of our common stock increases.
We have not paid dividends on our common stock to date and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future. Consequently, in the foreseeable future, you will likely only experience a gain from your investment in our common stock if the price of our common stock increases.
If equity research analysts do not publish research or reports about our business or if they issue unfavorable commentary or downgrade our common stock, the price of our common stock could decline.
The trading market for our common stock relies in part on the research and reports that equity research analysts publish about us and our business. We do not control these analysts. The price of our common stock could decline if one or more equity analysts downgrade our common stock or if analysts issue other unfavorable commentary or cease publishing reports about us or our business.
Anti-takeover provisions in our organizational documents and Delaware law may discourage or prevent a change of control, even if an acquisition would be beneficial to our stockholders, which could affect our stock price adversely and prevent attempts by our stockholders to replace or remove our current management.
Our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could delay or prevent a change of control of our company or changes in our board of directors that our stockholders might consider favorable. Some of these provisions:
In addition, we are subject to the provisions of Section 203 of the Delaware General Corporation Law, which may prohibit certain business combinations with stockholders owning 15% or more of our outstanding voting stock. These and other provisions in our amended and restated certificate of incorporation, amended and restated bylaws and Delaware law could make it more difficult for stockholders or potential acquirers to obtain control of our board of directors or initiate actions that are opposed by our then-current board of directors, including a merger, tender offer or proxy contest involving our company. This provision could have the effect of delaying or preventing a change of control, whether or not it is desired by or beneficial to our stockholders. Any delay or prevention of a change of control transaction or changes in our board of directors could cause the market price of our common stock to decline.
Item 1B. Unresolved Staff Comments
None.
Our principal offices occupy approximately 8,200 square feet of leased office space in Princeton, New Jersey pursuant to a lease agreement that expires in November 2020. We believe that our current facilities are suitable and adequate to meet our current needs. We intend to add new facilities or expand existing facilities as we add employees, and we believe that suitable additional or substitute space will be available as needed to accommodate any such expansion of our operations.
Two complaints have been filed in federal court in the District of New Jersey on January 6, 2017 and January 20, 2017, titledPeng v. Agile Therapeutics, Inc., Alfred Altomari, and Elizabeth Garner, No. 17-cv-119 (D.N.J.), andLichtenthal v. Agile Therapeutics, Inc., Alfred Altomari, and Elizabeth Garner, No. 17-cv-405 (D.N.J.), respectively, on behalf of a putative class of investors who purchased stock from March 9, 2016 through January 3, 2017. The complaints allege violations of the federal securities laws based on public statements made regarding our Phase 3 "SECURE" clinical trial. We deny all allegations in the complaints and we plan to vigorously defend the complaints that have been filed. None.
Item 4. Mine Safety Disclosures
Not applicable.
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information and Holders of Record
Our common stock has beenwas listed on the Nasdaq Global Market under the symbol "AGRX" sincefrom May 23, 2014. Prior to that date, there was no public trading market for our common stock. The following table sets forth for the periods indicated the high and low sales prices per share of2014 through January 2, 2019. Beginning on January 3, 2019, our common stock as reportedhas been listed on the NASDAQ Global Market:Nasdaq Capital Market under the symbol "AGRX".
| | High | Low | High | Low | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Year Ended December 31, 2016 | ||||||||||||||
Year Ended December 31, 2019 | ||||||||||||||
Fourth Quarter | $ | 7.95 | $ | 5.62 | $ | 2.97 | $ | 0.35 | ||||||
Third Quarter | $ | 8.15 | $ | 6.53 | $ | 1.64 | $ | 0.95 | ||||||
Second Quarter | $ | 8.65 | $ | 5.60 | $ | 1.56 | $ | 1.02 | ||||||
First Quarter | $ | 10.00 | $ | 5.32 | $ | 1.70 | $ | 0.66 | ||||||
Year Ended December 31, 2015 | ||||||||||||||
Year Ended December 31, 2018 | ||||||||||||||
Fourth Quarter | $ | 10.41 | $ | 6.38 | $ | 1.30 | $ | 0.33 | ||||||
Third Quarter | $ | 11.30 | $ | 6.07 | $ | 0.92 | $ | 0.23 | ||||||
Second Quarter | $ | 13.19 | $ | 8.52 | $ | 3.00 | $ | 0.49 | ||||||
First Quarter | $ | 11.18 | $ | 5.80 | $ | 3.92 | $ | 2.40 | ||||||
As of March 10, 2017,February 18, 2020, we had 3634 holders of record of our common stock. The actual number of shareholders is greater than this number of record holders and includes shareholders who are beneficial owners but whose shares are held in street name by brokers and other nominees. The number of holders of record also does not include shareholders whose shares may be held in trust by other entities. The closing price of our common stock on March 10, 2017February 18, 2020 was $2.40.$3.94.
Dividends
We have never declared or paid a cash dividend on our capital stock. We currently intend to retain any future earnings and do not expect to pay any dividends in the foreseeable future. AnyIn addition, our Credit Agreement and Guaranty among us, the gurantors from time to time party thereto, the lenders from time to time party thereto and Perceptive Credit Holdings III, LP, as a lender and as Administrative Agent for the lenders, contains, and any other loan facilities that we may enter into may contain, restrictions on our ability to pay dividends. Subject to such restrictions, any future determinations to pay cash dividends will be made at the discretion of our board of directors, subject to applicable laws, and will depend on a number of factors, including our financial condition, results of operations, capital requirements, contractual restrictions, general business conditions, and any other factors that our board may deem relevant.
Stock Performance Graph
This performance graph shall not be deemed "soliciting material" or to be "filed" with the SEC for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, (Exchange Act),or the Exchange Act, or otherwise subject to the liabilities under that Section, and shall not be deemed to be incorporated by reference into any filing of Agile Therapeutics, Inc.our filings under the Exchange Act or the Securities Act of 1933, as amended or the Exchange Act.amended.
The following graph shows a comparison from May 23,December 31, 2014 (the date our common stock commenced trading of the Nasdaq Global Market) through December 31, 20152019 of the cumulative total return for our common stock, and the NASDAQNasdaq Composite Index and The NASDAQNasdaq Biotechnology Index. The graph assumes that $100 was invested at the market close on May 23,December 31, 2014 in the common stock of Agile Therapeutics, Inc., the NASDAQNasdaq Composite Index and The NASDAQ Nasdaq
Biotechnology Index and assumes reinvestments of dividends. The stock price performance of the following graph is not necessarily indicative of future stock price performance.
Comparison of Cumulative Total Return
December 31, 20162019
| | 5/23/2014 | 6/30/2014 | 9/30/2014 | 12/31/2014 | 3/31/2015 | 6/30/2015 | 9/30/2015 | 12/31/2015 | 3/31/2016 | 6/30/2016 | 9/30/2016 | 12/31/2016 | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Agile Therapeutics, Inc. | $ | 100.00 | $ | 157.40 | $ | 131.77 | $ | 110.83 | $ | 167.33 | $ | 155.05 | $ | 121.66 | $ | 176.17 | $ | 112.09 | $ | 137.36 | $ | 125.99 | $ | 102.89 | |||||||||||||
NASDAQ Composite | $ | 100.00 | $ | 105.31 | $ | 107.35 | $ | 113.15 | $ | 117.08 | $ | 119.04 | $ | 110.38 | $ | 119.63 | $ | 116.34 | $ | 115.69 | $ | 126.91 | $ | 128.60 | |||||||||||||
NASDAQ Biotechnology | $ | 100.00 | $ | 109.63 | $ | 116.68 | $ | 129.67 | $ | 146.80 | $ | 157.71 | $ | 129.33 | $ | 144.48 | $ | 111.29 | $ | 109.92 | $ | 123.53 | $ | 113.15 | |||||||||||||
Recent Sales of Unregistered Securities and Use of Proceeds from Registered SecuritiesSecurities
None
On May 22, 2014, the Company's registration statement on Form S-1 (File No. 333-194621) for our IPO was declared effective by the Securities and Exchange Commission, or SEC. On May 29, 2014, we completed our IPO whereby we sold 9,166,667 shares of common stock, at a public offering price of $6.00 per share, before underwriting discounts and expenses. The aggregate net proceeds received by us from the offering were $49.7 million after deducting the underwriting discounts and commissions and offering expenses paid by us.None.
As of December 31, 2016, we have used all of our net proceeds from the IPO primarily to fund the Phase 3 clinical trial for Twirla and for general working capital purposes; and to a lesser extent, for activities related to the completion of the equipment qualification related to the expansion for Corium's manufacturing capabilities.
There was no material change in the planned use of proceeds from our IPO as described in our prospectus dated May 22, 2014, filed with the SEC pursuant to Rule 424(b)(4) under the Securities Act of 1933, as amended, as revised in our Quarterly Report on Form 10-Q for the period ended June 30, 2014, filed with the SEC on August 14, 2014.
None.
Item 6. Selected Financial Data
The following table sets forth our selected financial data for the periods indicated. You should read the following selected financial data in conjunction with our audited financial statements and the related notes thereto included elsewhere in this Annual Report on Form 10-K and the "Management's Discussion and Analysis of Financial Condition and Results of Operations" section of this Annual Report.Report on Form 10-K.
We have derived the statement of operations data for the years ended December 31, 2019, 2018 and 2017 and the balance sheet data as of December 31, 2019 and 2018 from our audited financial statements included elsewhere in this Annual Report on Form 10-K. The statement of operations data for the years ended December 31, 2016 2015 and 2014,2015 and the balance sheet data as of December 31, 2017, 2016 2015 and 2014,2015 are derived from our audited financial statements that are not included elsewhere in this Annual Report.
Report on Form 10-K. Our historical results are not necessarily indicative of the results that may be expected in the future.
| | Year ended December 31, | Year ended December 31, | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | 2019 | 2018 | 2017 | 2016 | 2015 | ||||||||||||||||||
| | (In thousands, except share and per share amounts) | (In thousands, except share and per share amounts) | ||||||||||||||||||||||||
Statement of Operations Data: | ||||||||||||||||||||||||||
Operating expenses: | ||||||||||||||||||||||||||
Research and development | $ | 20,929 | $ | 25,622 | $ | 13,365 | $ | 9,858 | $ | 9,777 | $ | 14,428 | $ | 20,929 | $ | 25,622 | ||||||||||
General and administrative | 8,792 | 7,467 | 5,150 | 9,000 | 8,739 | 12,383 | 8,792 | 7,467 | ||||||||||||||||||
Restructuring costs | — | 1,019 | — | — | — | |||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
Total operating expenses | 29,721 | 33,089 | 18,515 | 18,858 | 19,535 | 26,811 | 29,721 | 33,089 | ||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
Loss from operations | (29,721 | ) | (33,089 | ) | (18,515 | ) | (18,858 | ) | (19,535 | ) | (26,811 | ) | (29,721 | ) | (33,089 | ) | ||||||||||
| | | | | | | | | | | | | | | | | | ||||||||||
Other income (expense) | ||||||||||||||||||||||||||
Interest income | 252 | 366 | 282 | 117 | 5 | |||||||||||||||||||||
Interest expense | (2,446 | ) | (2,077 | ) | (1,566 | ) | — | (1,116 | ) | (1,918 | ) | (2,446 | ) | (2,077 | ) | |||||||||||
Interest income | 117 | 5 | 3 | |||||||||||||||||||||||
Change in fair value of warrants | 234 | (110 | ) | 348 | — | 29 | 143 | 234 | (110 | ) | ||||||||||||||||
Loss on extinguishment of debt | — | (1,036 | ) | — | — | — | — | — | (1,036 | ) | ||||||||||||||||
| | | | | | | | | | | | | | | | | | ||||||||||
Total other income (expense), net | 252 | (721 | ) | (1,493 | ) | (2,095 | ) | (3,218 | ) | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
Loss before benefit from income taxes | (31,816 | ) | (36,307 | ) | (19,730 | ) | (18,606 | ) | (20,256 | ) | (28,304 | ) | (31,816 | ) | (36,307 | ) | ||||||||||
Benefit from income taxes | 3,075 | 5,972 | 3,653 | — | 477 | — | 3,075 | 5,972 | ||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | $ | (28,741 | ) | $ | (30,335 | ) | $ | (16,077 | ) | $ | (18,606 | ) | $ | (19,779 | ) | $ | (28,304 | ) | $ | (28,741 | ) | $ | (30,335 | ) | ||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss per share (basic and diluted) | $ | (1.02 | ) | $ | (1.38 | ) | $ | (1.41 | ) | $ | (0.38 | ) | $ | (0.58 | ) | $ | (0.91 | ) | $ | (1.02 | ) | $ | (1.38 | ) | ||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
Weighted-average shares outstanding (basic and diluted) | 28,273,331 | 22,017,229 | 11,394,971 | |||||||||||||||||||||||
Weighted-average common shares (basic and diluted) | 49,432,487 | 34,315,931 | 30,940,831 | 28,273,331 | 22,017,229 | |||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | As of December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | |||||||
| | (In thousands) | |||||||||
Balance Sheet Data: | ||||||||||
Cash and cash equivalents | $ | 48,750 | $ | 34,395 | $ | 40,182 | ||||
Working capital | 40,840 | 30,151 | 31,993 | |||||||
Total assets | 63,866 | 50,712 | 54,826 | |||||||
Accounts payable | 2,050 | 2,387 | 2,631 | |||||||
Loan payable, long-term | 10,899 | 13,035 | 9,828 | |||||||
Convertible preferred stock | — | — | — | |||||||
Total stockholders' equity (deficit) | 42,289 | 29,743 | 36,006 | |||||||
| | As of December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2019 | 2018 | 2017 | 2016 | 2015 | |||||||||||
| | (In thousands) | |||||||||||||||
Balance Sheet Data: | ||||||||||||||||
Cash and cash equivalents | $ | 34,479 | $ | 7,851 | $ | 35,952 | $ | 48,750 | $ | 34,395 | ||||||
Working Capital | 31,524 | 6,240 | 22,442 | 40,548 | 30,151 | |||||||||||
Total Assets | 49,540 | 22,392 | 50,595 | 63,866 | 50,712 | |||||||||||
Accounts Payable | 1,819 | 875 | 2,784 | 2,050 | 2,387 | |||||||||||
Loan payable, current | — | — | 10,607 | 5,104 | — | |||||||||||
Loan payable, long-term | — | — | — | 10,607 | 13,035 | |||||||||||
Total stockholders' equity | 45,745 | 20,174 | 36,323 | 42,289 | 29,743 | |||||||||||
Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of financial condition and results of operations is provided to enhance the understanding of, and should be read in conjunction with, Part I, Item 1, "Business" and Item 8, "Financial Statements and Supplementary Data." For information on risks and uncertainties related to our business that may make past performance not indicative of future results or cause actual results to differ materially from any forward-looking statements, see "Special Note Regarding Forward-Looking Statements," and Part I, Item 1A, "Risk Factors." Dollars in tabular format are presented in thousands, except per share data, or as otherwise indicated.
Overview
We are a forward-thinking women's healthcare company dedicated to fulfilling the unmet health needs of today's women. Our currentTwirla® and our potential product candidates are designed to provide women with contraceptive options that offer greater convenience and facilitate compliance. Our leadTwirla, our first and only approved product, candidate, Twirla®, also known as AG200-15, is a once-weekly prescription combination hormonal contraceptive patch. Twirla is designed using our proprietary transdermal patch technology, called Skinfusion®, designed with properties to optimize patch adhesion and patient wearability, which may help support compliance while, for the first time, delivering a dose of estrogen consistent with commonly prescribed combined hormonal contraceptives, or CHCs. We believe there is an unmet market need for a contraceptive patch that is atdesigned to delivers approximately 30 mcg of estrogen and 120 mcg of progestin in a convenient dosage form that may support compliance in a non-invasive fashion.
Twirla was approved for sale in the endUnited States on February 14, 2020 as a method of contraception for use in women of reproductive potential with a BMI < 30 kg/m2 for whom a combined hormonal contraceptive is appropriate. Based on the observed relationship between efficacy and BMI in a Phase 3 clinical development.trial, Twirla's limitation of use instructs healthcare providers to consider Twirla's reduced effectiveness in women with a BMI³ 25 to <30 kg/m2 before prescribing. Twirla is contraindicated in women with a BMI³ 30 kg/m2 because compared to women with a lower BMI, women in this group had reduced effectiveness and may have a higher risk for VTEs.
As part of Twirla's approval, the FDA is requiring us to conduct a long-term prospective, observational post-marketing study comparing the risks for VTE and ATE in new users of Twirla to new users of other CHCs. The FDA's requirement for Twirla is similar to another post-marketing study requirement for a recently approved CHC. The final study report for the Twirla post-marketing study is scheduled to be submitted to the FDA in November 2032, with interim safety data reporting to the FDA due in November 2026. We have also agreed to a small post-marketing commitment, or PMC, study to assess the residual drug content and strength of Twirla. The PMC study is similar to residual drug studies requested of patch developers in the FDA's November 2019 draft guidance entitled Transdermal and Topical Delivery Systems—Product Development and Quality Considerations. We are evaluating the design and cost of these post-marketing studies
With the approval of Twirla we now plan to focus on our transition from a clinical development stage company to a commercial company. During 2020, we plan to begin the implementation of our
commercialization plan for Twirla and to manage the growth of our company. Our near term plan for the commercialization of Twirla includes:
| Activity | Expected Timing | |
|---|---|---|
| Initiate coverage and reimbursement activities in the United States from third-party payors. | First Quarter 2020 | |
Initiate hiring of contract sales force | Second Quarter 2020 | |
Complete pre-validation and validation of the commercial manufacturing process consistent with our approved marketing application | Second Half 2020 with first shipment of product in the Fourth Quarter 2020. |
Our short-term goal is to establish an initial franchise in the multi-billion-dollar U.S. hormonal contraceptive market built on approval of Twirla in the U.S. Our resources are currently focused on the commercialization of Twirla. To that end, our goal is to begin the pre-validation and validation of the commercial manufacturing process in the first half of 2020, manufacture three validation batches of Twirla and complete the process in the second half of 2020. At the same time, we will prepare for the availability of commercial product supply. In the first quarter of 2020, we plan to initiate work with managed care and patient payors to gain market access for Twirla. In the second quarter of 2020, we plan to begin hiring and training an initial sales team, which we estimate to be in the range of 70 to 100 persons. We intend to ship product to wholesalers in fourth quarter of 2020. We also expect to explore the advancement of our existing pipeline and its possible expansion through business development activities.
Our current priorities are as follows:
For more information about the regulatory history of Twirla, please seePart 1, Item 1, "Business"
Financial Overview
Since our inception in 1997, we have devoted substantial resources to developing and seeking regulatory approval for Twirla, building our intellectual property portfolio, business planning, raising capital and providing general and administrative support for these operations. We incurred research and development expenses of $20.9$9.9 million, $25.6$9.8 million and $13.4$14.4 million during the years ended December 31, 2016, 20152019, 2018 and 2014,2017, respectively. WeWhile we anticipate that a significant portion of our operating expenses will continue to be related to research and development as we continuecomplete the pre-validation manufacturing activities related to develop Twirla, conduct our Phase 4 study, and advanceplan the development of our pipeline, we expect our operating expenses to substantially shift towards commercialization. A
Table of product candidates. Substantially allContents
substantial amount of our resources are currently dedicated to developingcompleting manufacturing validation and seeking regulatory approval forcommercializing Twirla. We will require additional capital to advance the development of our other product candidates.
We have funded our operations primarily through sales of common stock, convertible preferred stock, convertible promissory notes and term loans. As of December 31, 20162019, and 2015,2018, respectively, we had $48.8$34.5 million and $34.4$7.9 million in cash and cash equivalents.
On May 29, 2014,In January 2019, we completed our initial public offering wherebyentered into a common stock sales agreement or the "2019 ATM Agreement," under which we sold 9,166,667were authorized to sell up to an aggregate of $10.0 million in gross proceeds through the sale of shares of common stock atfrom time to time in "at-the-market" equity offerings (as defined in Rule 415 promulgated under the Securities Act of 1933, as amended). We agreed to pay a public offering pricecommission of $6.00 per share, before underwriting discounts3% of the gross proceeds of any common stock sold under this agreement. During the year ended December 31, 2019, we issued and expenses. The aggregatesold a total of 1,801,528 shares of common stock under the 2019 ATM Agreement resulting in net proceeds received by us fromof approximately $2.5 million. We terminated the initial public offering were approximately $49.7 million.2019 ATM Agreement on July 31, 2019.
On January 19, 2015,In March 2019, we completed a private placement of approximately 3.4 million8,426,750 shares of common stock at $5.85$0.93 per share. Proceeds from the private placement, net of commissions and other offering costs, were approximately $7.8 million.
In August 2019, we completed a public offering of 14,526,315 shares of common stock at a price of $0.95 per share. Proceeds from the public offering, net of underwriting discounts, commissions and offering expenses were approximately $12.7 million.
In November 2019, we entered into a second ATM Agreement, or the "Second 2019 ATM Agreement," under which we were authorized to issue and sell shares of our common stock having aggregate sales proceeds of up to $20.0 million from time to time. We paid a commission of 3% of the gross proceeds from the sales of our common stock under the Second 2019 ATM Agreement. In the year ended December 31, 2019, we issued and sold 10,440,908 shares of common stock under the second 2019 ATM Agreement, representing all the capacity of Second ATM Agreement, resulting in net proceeds of approximately $19.3 million.
In February 2015,2020, we entered into a loanCredit Agreement and security agreementGuaranty with Hercules Technology Growth Capital, Inc.Perceptive Credit Holdings III, LP, or Hercules,Perceptive, for a senior secured term loan facility of up to $25.0 million.$35 million, which we refer to as the Perceptive Credit Agreement. A first tranche of $16.5$5 million was funded uponon execution of the loan agreement, approximately $15.5 million of which was used to repay our existing term loan. The Hercules Loan Agreement was amended in August 2016 to, among other things, extend the period during which we can draw thePerceptive Credit Agreement. A second tranche of $8.5$15 million to March 31, 2017 and extend the period during which we make interest-only payments until January 31, 2017. We are currently in discussions with Hercules to extend the period beyond March 31, 2017 during which the additional tranche of $8.5 million may be drawn. We can make no assurances that our discussions will ultimately be successful and, if such discussionswas funded as a result in an extension of the period in which we may drawapproval of Twirla by the additionalFDA. Another $15 million tranche will be available upon the achievement of $8.5 million, we could incur additional fees payable to Hercules. On February 1, 2017, we began making principal payments with respect tocertain revenue milestones. The facility will be interest only until the Hercules Loan. See further discussion in "Funding Requirements and Other Liquidity Matters" below.
In January 2016, we closed an underwritten public offering of 5,511,812 shares of common stock at a public offering price of $6.35 per share. In February 2016, the underwritersthird anniversary of the public offering of common stock exercised in full their option to purchase an additional 826,771 shares of common stock at the public offering price of $6.35 per share, less underwriting discounts and commissions. A total of 6,338,583 shares of common stock were sold in the public offering, resulting in total net proceeds of approximately $37.5 million.closing date.
We have not generated any revenue and have never been profitable for any year. Our net loss was $28.7$18.6 million, $30.3$19.8 million and $16.1$28.3 million for the years ended December 31, 2016, 20152019, 2018 and 2014,2017, respectively. We expect to incur increased expenses and increasing operating losses for the foreseeable future as we complete the development of Twirla, respond to the CRL and supplement our NDA with the results of the SECURE trial, completecommercialize Twirla. This includes completing the qualification and validation of our commercial manufacturing process, initiateinitiating pre-launch commercial activities, commercially launchlaunching Twirla, advanceadvancing our other potential product candidates and expandexpanding our research and development programs. Substantially all of our resources are currently dedicated to developing and seeking regulatory approval for Twirla. We will require additional capital for the commercial launch of Twirla, if approved, as well as advancingto fund these activities and to advance the development of our other potential product candidates.
Going Concern
As of December 31, 2019, we had cash and cash equivalents of $34.5 million. Additionally, in February 2020, we received $20.0 million in gross proceeds under the Perceptive Credit Agreement. We believe that our cash and cash equivalents as of December 31, 2019, along with the proceeds of the Perceptive Credit Agreement we have received to date, will be sufficient to meet our projected operating requirements through the end of 2020. We will require additional capital to fund our
operating needs beyond 2020, which we expect primarily will consist of commercializing Twirla, and exploring the advancement of our existing pipeline and its possible expansion through business development activities.
Our future success depends on our ability to raise additional capital and/or implement various strategic alternatives. Our ability to continue operations beyond 2020 will depend on our ability to obtain additional funding, as to which no assurances can be given. Based upon the foregoing, management has concluded that there is substantial doubt about our ability to continue as a going concern for twelve months from the date of filing of this Annual Report on Form 10-K. There can be no assurance that any financing by us can be realized, or if realized, what the terms of any such financing may be, or that any amount that we are able to raise will be adequate.
We continue to analyze strategic and financing alternatives, potential asset sales as well as mergers and acquisitions. We cannot be certain that these initiatives or raising additional capital, whether through selling additional debt or equity securities or obtaining a line of credit or other loan, will be available to us or, if available, will be on terms acceptable to us. If we issue additional securities to raise funds, whether through the issuance of equity or convertible debt securities, or any combination thereof, these securities may have rights, preferences, or privileges senior to those of our common stock, and our current stockholders will experience dilution. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances or licensing arrangements with pharmaceutical partners, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates, including Twirla, or grant licenses on terms that may not be favorable to us. If we are unable to obtain funds when needed or on acceptable terms, we then may be unable to complete the commercialization of Twirla and may also be required to further cut operating costs, forego future development and other opportunities and may need to seek bankruptcy protection.
The financial statements as of December 31, 2019 have been prepared under the assumption that we will continue as a going concern for the next 12 months. Our ability to continue as a going concern is dependent upon our uncertain ability to obtain additional capital, reduce expenditures and/or execute on our business plan and successfully launch Twirla. These financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We do not own any manufacturing facilities and rely on our third partycontract manufacturer, Corium International, Inc., or Corium, for all aspects of the manufacturing of Twirla. We will need to continue to invest in the manufacturing process for Twirla, and incur significant expenses, in order to complete the equipment qualification and validation related to the expansion of Corium's commercial manufacturing capabilities in order toline for Twirla and be capable of supplying projected commercial quantities of Twirla, if approved. Based on our interactions with the FDA on the CMC issues raised in the CRL and our planTwirla. In September 2019, we re-started manufacturing development at Corium. We are currently working with Corium to validate the commercial scale equipmentcomplete manufacturing development, process improvements, and pre-validation work. Our goal is to manufacture three validation batches of Twirla we expect to be able to address these issues inand complete the resubmissionvalidation of our NDA. We continue to plan the process of scaling up the commercial manufacturing capabilities for Twirla with Corium andprocess in the associated costs and timelines.second half of 2020. We expect the validation and expansion to be completed in coordination with our planned commercialization activities. If we obtain regulatory approval for Twirla, we expect to incur significant expenses in order to create an infrastructure to support the commercialization of Twirla, including sales, marketing, distribution, medical affairs and compliance functions, which will require additional capital.
In December 2016, we completed a Phase 3 trial, the SECURE trial, in which we enrolled over 2,000 women for up to one year of treatment. We announced top-line data in early January 2017 and expect to file our resubmission to the U.S. Food and Drug Administration, or FDA, in the first half of 2017.functions.
We have incurred and will continue to incur additional costs associated with operating as a public company. Accordingly, we will need additional financingcapital to support our continuing operations and other potential product candidates in our pipeline in addition to the commercial activities required for the pre-launch and launch of Twirla. We will seek to fund our operations through public or private equity or debt financings or other sources, which may include collaborations with third parties. Adequate additional financingcapital may not be available to us on acceptable terms, or at all. Our failure to raise additional capital as and when needed would have a negative impact on our financial condition and our
ability to pursue our business strategy. We will need to generate significant revenue to achieve profitability, and we may never do so.
Financial Operations Overview
Revenue
To date, we have not generated any revenue. In the future, we may generate revenue from product sales, license fees, milestone payments and royalties from the sale of products developed using our intellectual property. Our ability to generate revenue and become profitable depends on our ability to successfully commercialize Twirla and any product candidates that we may advance in the future. If we
fail to complete the development ofsuccessfully commercialize Twirla, or any other product candidates we advance in a timely manner or obtain regulatory approval for them, our ability to generate future revenue, and our results of operations and financial position, will be adversely affected.
Research and Development Expenses
Since our inception, we have focused our resources on our research and development activities. Research and development expenses consist primarily of costs incurred for the development of Twirla and other current and future potential product candidates, and include:
Research and development costs are expensed as incurred. Costs for certain development activities, such as clinical trials, are recognized based on an evaluation of the progress to completion of specific tasks using data such as subject enrollment, clinical site activations or information provided to us by our third partythird-party vendors.
Research and development activities are central to our business model.model and to date, our research and development expenses have been related primarily to the development of Twirla. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We do not currently utilize a formal time allocation system to capture expenses on a project-by-project basis, as the majority of our past and planned expenses have been and will be in support of Twirla. In 2017, we expect the expenses associated with the SECURE clinical trial to decrease as we complete the close-out activities associated with the trial and no additional clinical trials are planned at this time. During 2017, we expect to increase activities related to equipment qualification and validation of our commercial manufacturing process as we continue to prepare for the commercialization of Twirla.
To date, our research and development expenses have related primarily to the development of Twirla. As we complete the close-out activities associated with the SECURE clinical trial, we expect research and development expenses to begin to shift away from costs associated with our SECURE clinical trial and toward the costs associated with preparing the resubmission of our new drug application, or NDA, and completing the qualification and validation of our commercial manufacturing process. In July 2016, we began preparations for an initial Phase 2 clinical trial examining the use of AG200-SP along with a smaller lower-dose combination ethinyl estradiol/levongestrel patch (SmP) in the fourth week of the woman's cycle. We have decided to postpone the trial and will continue to evaluate the timing for initiating dosing of the subjects for this Phase 2 clinical trial, which is dependent on available capital resources. We began incurring expenses for the clinical development of AG200-SP in the second half of 2016. For the years ended December 31, 2016, 2015 and 2014, our research and
For the years ended December 31, 2019, 2018 and 2017, our research and development expenses were approximately $20.9$9.9 million, $25.6$9.8 million and $13.4$14.4 million, respectively. The following table summarizes our research and development expenses by functional area.
| | Year ended December 31, | Year ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | 2019 | 2018 | 2017 | ||||||||||||||
| | (In thousands) | (In thousands) | ||||||||||||||||||
Clinical development | $ | 13,184 | $ | 19,117 | $ | 7,916 | $ | 1,781 | $ | 1,318 | $ | 2,386 | ||||||||
Regulatory | 342 | 269 | 300 | 2,990 | 562 | 1,348 | ||||||||||||||
Personnel related | 2,669 | 2,111 | 1,942 | 1,669 | 2,162 | 2,440 | ||||||||||||||
Manufacturing—commercialization | 2,290 | 2,427 | 1,303 | 2,876 | 4,306 | 5,917 | ||||||||||||||
Manufacturing | 1,381 | 537 | 1,287 | 20 | 155 | 1,153 | ||||||||||||||
Stock-based compensation | 1,063 | 1,161 | 617 | 522 | 1,274 | 1,184 | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Total research and development expenses | $ | 20,929 | $ | 25,622 | $ | 13,365 | $ | 9,858 | $ | 9,777 | $ | 14,428 | ||||||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Although we are currently in the process of completing the close-out activities associated with our SECURE Phase 3 clinical trial for Twirla, itIt is difficult to determine with any certainty the exact duration and completion costs of any of our future clinical trials of Twirla and any ofor our other current and future potential product candidates we may advance, including AG200-SP.advance. It is also difficult to determine if, when or to what extent we will generate revenue from the commercialization and sale of Twirla our potential product candidates that obtain regulatory approval. Our current business plan contemplates resubmission of our NDA in the first half of 2017 and assumes a six month review by the FDA. We may, however, never succeed in achieving regulatory approval for Twirla or any of our product candidates.
The duration, costs and timing of clinical trials and development of our other potential product candidates in addition to conducting required post-marketing studies for Twirla will depend on a variety of factors, including the uncertainties of future clinical trials and preclinical studies, the slower than expected rate of subject enrollment, we experienced for our SECURE Phase 3 clinical trial for Twirla, obtaining additional capital, and significant and changing government regulation. In addition, the probability of success for each product candidate will depend on numerous factors, including competition, manufacturing capability and commercial viability. A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA, or another regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate will be required for the completion of clinical development of a product candidate, or if we experience significant delays in enrollment in any of our clinical trials, or experience issues with our manufacturing capabilities we could be required to expend significant additional financial resources and time with respect to the development of that product candidate. We will determine which programs to pursue and how much to fund each program in response to the scientific and clinical success of each product candidate, as well as an assessment of each product candidate's commercial potential. Substantially all of our resources are currently dedicated to developing and seeking regulatory approval forcommercializing Twirla. We will require additional capital forto fund our operating needs beyond 2020, which we expect primarily will consist of commercializing Twirla, and exploring the commercial launch of Twirla, if approved, as well as advancing the developmentadvancement of our other product candidates.existing pipeline and its possible expansion through business development activities.
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for personnel in executive, finance and administrative functions including payroll taxes and health insurance, stock-based compensation and travel expenses. Other general and administrative expenses include facility-related costs, insurance and professional fees for legal, patent review, consulting and accounting services. General and administrative expenses are expensed as incurred.
For the years ended December 31, 2016, 20152019, 2018 and 2014,2017, our general and administrative expenses totaled approximately $8.8$9.0 million, $7.5$8.7 million and $5.2$12.4 million, respectively. In January 2018, following our receipt of the 2017 CRL, we significantly scaled back our preparations for the commercialization of Twirla, including commercial pre-launch activities, pending our ability to address
the 2017 CRL and receive approval of Twirla. With the recent approval of Twirla, we intend to commercialize Twirla in the United States through a contract sales force. We anticipate that our general and administrative expenses will increase in the future with the continued research,
development and potential commercialization of Twirla, its planned line extensions, and any of our other product candidates, and as we operate as a public company.Twirla. These increases will likely include increased selling and marketing costs, including payroll and operating costs, related to the commercial launch of Twirla, legal and accounting services, stock registration and printing fees, addition of new personnel to support compliance and communication needs, increased insurance premiums, outside consultants and investor relations. Additionally, if in the future we believe regulatory approval of Twirla or any of our other product candidates appears likely, we anticipate that we would begin preparations for commercial operations, which would result in an increase in payroll and other expenses, particularly with respect to the sales and marketing of our product candidates.
Critical Accounting Policies and Significant Judgments and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles, or U.S. GAAP. The preparation of these financial statements requires us to make significant estimates and judgments that affect the reported amounts of assets, liabilities and expenses and related disclosures. On an ongoing basis, our actual results may differ significantly from our estimates.
Our significant accounting policies are described in more detail in the notes to our financial statements appearing elsewhere in this annual reportAnnual Report on Form 10-K. We believe the following accounting policies to be most critical to the judgments and estimates used in the preparation of our financial statements.
Accrued Research and Development Expenses
As part of the process of preparing our financial statements, we are required to estimate our accrued expenses, particularly for product development costs. This process involves reviewing open contracts and purchase orders, communicating with our personnel to identify services that have been performed on our behalf and estimating the level of services performed and the associated costs incurred for the services when we have not yet been invoiced or otherwise notified of the actual costs. The majority of our service providers invoice us monthly in arrears for services performed or when contractual milestones are met. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us at that time. We periodically confirm the accuracy of our estimates with service providers and make adjustments as necessary. Examples of estimated accrued research and development expenses include:
We base our expenses related to clinical studies on our estimates of the services received and efforts expended pursuant to contracts with multiple CROs that conduct and manage clinical studies on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the clinical expense. Payments under some of these contracts depend on factors such as the successful enrollment of subjects and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed, enrollment of subjects, number of sites activated and the level of effort to be expended in each period. If the actual timing of the performance of services or the
level of effort varies from our estimate, we adjust the accrued liability or prepaid expense accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, our
understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in our reporting amounts that are too high or too low in any particular period. Based on historical experience, actual results have not been materially different from our estimates. As of December 31, 2019, we did not have any ongoing clinical trials.
Warrant Liability
We account for detachablewarrants to purchase common stock in accordance with Accounting Standards Codification, or ASC, 480,Distinguishing Liabilities from Equity. ASC 480 requires that a financial instrument, other than an outstanding share, that, at inception, is indexed to an obligation to repurchase the issuer's equity shares, regardless of the timing or the probability of the redemption feature and may require the issuer to settle the obligation by transferring assets classified as a liability. We measure the fair value of our warrant liability using the Black-Scholes option-pricing model with changes in fair value recognized as increases or reductions to other income (expense) in the statement of operations.
In connection with the completion of our initial public offering in May 2014, the warrants to purchase shares of Series A-1 and Series A-2 preferred stock expired unexercised and the warrants to purchase shares of Series C preferred stock automatically converted into warrants to purchase shares of common stock. Prior to January 1, 2019, warrants with non-standard anti-dilution provisions (referred to as down round protection) were classified as liabilities and re-measured each reporting period. On January 1, 2019, we adopted the provisions of Accounting Standards Update ("ASU") 2017-11Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part 1) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Non-controlling Interests with a Scope Exception, which indicates that a down round feature no longer precludes equity classification when assessing whether an investment is indexed to an entity's own stock. We used a modified retrospective approach to adoption, which does not restate our financial statements as of the prior year end (December 31, 2018). The cumulative effect of adoption of ASU 2017-11 resulted in an adjustment to accumulated deficit as of January 1, 2019 of $0.2 million with a corresponding adjustment to additional paid-in capital. Warrants to purchase convertible preferred stock (prior to our IPO) and62,505 shares of common stock at $6.00 per share expired on December 14, 2019, and none of these warrants were outstanding as liabilities, as they are freestanding derivative financial instruments.of December 31, 2019.
The warrants issued in connection with our debt financing completed in February 2015 are recordedclassified as liabilities at fair value, estimated using a Black-Scholes option pricing model, and are subject to re-adjustment at each balance sheet date, otherwise known as marked to market, with changes in the faircomponent of stockholders' equity. The value of such warrants was determined using the Black-Scholes option-pricing model. As of December 31, 2019, there were outstanding 180,274 warrants recorded into purchase common stock at $5.89 per share related to this debt financing. These warrants expire on February 24, 2020.
As part of the February 2020 Perceptive Credit Agreement, we issued Perceptive warrants to purchase 1,400,000 shares of Agile common stock. The per share exercise price for 700,000 shares is $3.74, which is equal to the 5-day volume weighted average exercise price ("5 Day VWAP") as of the trading day immediately prior to closing. The per share exercise price for the remaining 700,000 shares of our statements of operations.common stock is $4.67, which is 1.25 times the 5 Day VWAP.
Stock-Based Compensation
We account for stock-based compensation under ASC 718, "AccountingAccounting for Stock Based Compensation." All stock-based awards granted to nonemployees are accounted for at their fair value in accordance with ASC 718, and ASC 505, "Accounting for Equity Instruments that are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services,"Compensation, under which compensation expense is generally recognized over the vesting period of the award. Determining the amount of stock-based compensation to be required requires us to develop estimates of fair values of stock options as of the grant date.
We account for stock-based compensation by measuring and recognizing expense for all stock-based payments made to employees and directors based on estimated grant date fair values. We use the straight-line method to allocate compensation cost to reporting periods over each optionee's requisite service period, which is generally the vesting period. We estimate the fair value of our stock-based awards to employees and directors using the Black-Scholes option valuation model, or Black-Scholes model. The Black-Scholes model requires the input of subjective assumptions, including the expected stock price volatility, the calculation of expected term and the fair value of the underlying common stock on the date of grant, among other inputs. The risk-free interest rate was determined with the implied yield currently available for zero-coupon U.S. government issues with a remaining term approximating the expected life of the options.
Table We also award restricted stock units ("RSUs") to employees and our board of Contentsdirectors (the "Board"). RSUs are generally subject to forfeiture if employment terminates prior to the completion of the vesting restrictions. We expense the cost of the RSUs, which is determined to be the fair market value of the shares of common stock underlying the RSUs at the date of grant, ratably over the period during which the vesting restrictions lapse. Cost associated with performance-based restricted stock units with a performance condition which affects the vesting is recognized only if the performance condition is probable of being satisfied.
Comparison of Years Ended December 31, 20162019 and 20152018
| | Year ended December 31, | | Year ended December 31, | | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | Change | 2019 | 2018 | Change | ||||||||||||||
| | (In thousands) | | (In thousands) | | ||||||||||||||||
Operating expenses: | ||||||||||||||||||||
Research and development | $ | 20,929 | $ | 25,622 | $ | (4,693 | ) | $ | 9,858 | $ | 9,777 | $ | 81 | |||||||
General and administrative | 8,792 | 7,467 | 1,325 | 9,000 | 8,739 | 261 | ||||||||||||||
Restructuring costs | — | 1,019 | (1,019 | ) | ||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Total operating expenses | 29,721 | 33,089 | (3,368 | ) | 18,858 | 19,535 | (677 | ) | ||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Other income (expenses) | ||||||||||||||||||||
Other income (expense) | ||||||||||||||||||||
Interest income | 252 | 366 | (114 | ) | ||||||||||||||||
Interest expense | (2,446 | ) | (2,077 | ) | (369 | ) | — | (1,116 | ) | 1,116 | ||||||||||
Interest income | 117 | 5 | 112 | |||||||||||||||||
Change in fair value of warrants | 234 | (110 | ) | 344 | — | 29 | (29 | ) | ||||||||||||
Loss on extinguishment of debt | — | (1,036 | ) | 1,036 | ||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Loss before income taxes | (31,816 | ) | (36,307 | ) | 4,491 | |||||||||||||||
Income tax benefit | 3,075 | 5,972 | (2,897 | ) | ||||||||||||||||
Total other income (expense), net | 252 | (721 | ) | 973 | ||||||||||||||||
Loss before benefit from income taxes | (18,606 | ) | (20,256 | ) | 1,650 | |||||||||||||||
Benefit from income taxes | — | 477 | (477 | ) | ||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Net loss | $ | (28,741 | ) | $ | (30,335 | ) | $ | 1,594 | $ | (18,606 | ) | $ | (19,779 | ) | $ | 1,173 | ||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Research and development expenses. Research and development expenses decreasedincreased by $4.7$0.1 million, or 18%0.8%, from $25.6$9.8 million for the year ended December 31, 20152018 to $20.9$9.9 million for the year ended December 31, 2016.2019. This overall decreaseincrease in research and development expenses was primarily due to the following:
costs associated with the comparative wear study of Twirla and Xulane which was initiated and completed during the first quarter of 2019;
General and administrative expenses. General and administrative expenses increased by $1.3$0.3 million, or 18%3.0%, from $7.5$8.7 million for the year ended December 31, 20152018 to $8.8$9.0 million for the year ended December 31, 2016.2019. This overall increase in general and administrative expenseexpenses was primarily due to the following:
Restructuring costs. In June 2018, we announced a reduction in our workforce, which resulted in the termination of several employees primarily from our commercial and clinical teams, representing approximately thirty percent of our employees. This workforce reduction, along with other reductions in planned operating expenses was designed to preserve cash while we pursued formal dispute resolution with the FDA for Twirla and as we determine the regulatory path forward for the resubmission of our NDA for Twirla. In addition, in June 2018, we also announced that we had adopted a retention plan to provide (i) cash retention payments to be made to all remaining employees in order to induce such employees to remain employed by us through December 31, 2018 and (ii) stock option grants to all remaining employees in order to induce such employees to remain employed by us through December 31, 2019. Restructuring costs of $1.0 million for the year ended December 31, 2018 represent $0.4 million of severance-related costs and $0.6 million of costs related to the accrual of the retention bonus.
Interest income. Interest income comprises interest income earned on cash and cash equivalents.
Interest expense. Interest expense iswas primarily attributable to our term loan with Hercules for the year ended December 31, 2016 and our term loans with Hercules and Oxford for the year ended December 31, 2015.2018. Interest expense also includes the amortization of the discount associated with allocating value to the common stock warrants issued to Hercules, and Oxford, the amortization of the deferred financing costs associated with the term loansloan and the accrual of the final payment due to Hercules.
Interest income. Interest income comprises We had no interest income earned on cash and cash equivalents.expense for the year ended December 31, 2019 as the Hercules loan was paid in full in 2018.
Change in fair value of warrants. CertainPrior to our adoption of Accounting Standards Update, or ASU 2017-1, on January 1, 2019 (See Note 2 to the financial statements), certain of our warrants to purchase shares of our convertible preferred stock (prior to our IPO) and common stock (post IPO) arewere recorded at fair value and arewere subject to re-measurement at each balance sheet date. These liabilities are re-measured at each balance sheet date with the corresponding charge to earnings recorded within change in fair value of warrant liability. The fair value of the convertible preferred stock warrants (prior to the IPO) and common stock warrants with non-standard anti-dilution provisions are determined using the Black-Scholes option pricing model which incorporates a number of assumptions and judgments to estimate the fair value of these warrants including the fair value per share of the underlying stock, the remaining contractual term of the warrants, risk-free interest rate, expected dividend yield, credit spread and expected volatility of the price of the underlying stock. During the year ended December 31, 2016, the fair value2018, we reported income of our warrant liability changed by $0.2 million compared to year ended December 31, 2015, primarily due$29 thousand related to the decrease in the fair value of the underlying common stock.
Loss on extinguishment of debt. In February 2015, we entered into the Hercules Loan Agreement with Hercules for a term loan of up to $25.0 million. A first tranche of $16.5 million was funded upon execution of the Hercules Loan Agreement, approximately $15.5 million of which was used to repay our existing loan with Oxford. As a result of the repayment of the loan with Oxford, we recorded a loss on the extinguishment of debt of approximately $1.0 million representing the difference between the amount paid to Oxford and the carrying amount of the Oxford loan. Included in the loss on extinguishment of debt is the prepayment premium, the unamortized discount and the write off of deferred financing costs.warrants.
Benefit from income taxes. Benefit from income taxes forFor the years ended December 31, 20162019 and 2015 represents the proceedsDecember 31, 2018, we received $0.0 million and $0.5 million, respectively, from the sale of New Jersey net operating losses,state Net Operating Loss Carryovers, or NOLs, as part of the Technology and Business Tax Certificate Program, sponsored byor the Program. The Program enables approved biotechnology companies to sell their unused NOLs and unused Research and Development Tax Credits for at least 80% of the value of the tax benefits to unaffiliated, profitable corporate taxpayers in the State of New Jersey. The New Jersey Economic Development Authority. UnderAuthority and the program, emerging biotechnology companies with unused state NOLs are allowed to sell these NOLs to other companies. In November 2016, we completed the sale of New Jersey state NOLs totaling approximately $28.2Department of the Treasury's Division of Taxation administer the Program. We have reached the maximum lifetime benefit of $15.0 million under the Program and research and development credits totaling approximately $0.8 million for net proceeds of approximately $3.0 million. In November 2015, we completedare no longer eligible to participate in the sale of New Jersey state NOLs totaling approximately $59.8 million and research and development credits totaling approximately $1.1 million for net proceeds of approximately $6.0 million.
Table of ContentsProgram.
Comparison of Years Ended December 31, 20152018 and 20142017
| | Year ended December 31, | | Year ended December 31, | | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2015 | 2014 | Change | 2018 | 2017 | Change | ||||||||||||||
| | (In thousands) | | (In thousands) | | ||||||||||||||||
Operating expenses: | ||||||||||||||||||||
Research and development | $ | 25,622 | $ | 13,365 | $ | 12,257 | $ | 9,777 | $ | 14,428 | $ | (4,651 | ) | |||||||
General and administrative | 7,467 | 5,150 | 2,317 | 8,739 | 12,383 | (3,644 | ) | |||||||||||||
Restructuring costs | 1,019 | — | 1,019 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Total operating expenses | 33,089 | 18,515 | 14,574 | 19,535 | 26,811 | (7,276 | ) | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Other income (expenses) | ||||||||||||||||||||
Other income (expense) | ||||||||||||||||||||
Interest income | 366 | 282 | 84 | |||||||||||||||||
Interest expense | (2,077 | ) | (1,566 | ) | (511 | ) | (1,116 | ) | (1,918 | ) | 802 | |||||||||
Interest income | 5 | 3 | 2 | |||||||||||||||||
Change in fair value of warrants | (110 | ) | 348 | (458 | ) | 29 | 143 | (114 | ) | |||||||||||
Loss on extinguishment of debt | (1,036 | ) | — | (1,036 | ) | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Loss before income taxes | (36,307 | ) | (19,730 | ) | (16,577 | ) | ||||||||||||||
Income tax benefit | 5,972 | 3,653 | 2,319 | |||||||||||||||||
Total other income (expense), net | (721 | ) | (1,493 | ) | 772 | |||||||||||||||
Loss before benefit from income taxes | (20,256 | ) | (28,304 | ) | 8,048 | |||||||||||||||
Benefit from income taxes | 477 | — | 477 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Net loss | $ | (30,335 | ) | $ | (16,077 | ) | $ | (14,258 | ) | $ | (19,779 | ) | $ | (28,304 | ) | $ | 8,525 | |||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Research and development expenses. Research and development expenses increaseddecreased by $12.3$4.6 million, or 92%32%, from $13.4$14.4 million for the year ended December 31, 20142017 to $25.6$9.8 million for the year ended December 31, 2015.2018. This overall increasedecrease in research and development expenses was primarily due to the following:
General and administrative expenses. General and administrative expenses increaseddecreased by $2.33.7 million, or 45%29%, from $5.2$12.4 million for the year ended December 31, 20142017 to $7.5$8.7 million for the year ended December 31, 2015.2018. This increasedecrease in general and administrative expense was primarily due to the following:
Restructuring costs. In June 2018, we announced a reduction in our workforce, which resulted in the termination of several employees primarily from our commercial and clinical teams, representing approximately thirty percent of our employees. This workforce reduction, along with other reductions in planned operating expenses was designed to preserve cash while we pursued formal dispute resolution with the FDA for Twirla and as we determine the regulatory path forward for the resubmission of our NDA for Twirla. In addition, in June 2018, we also announced that we had adopted a retention plan to provide (i) cash retention payments to be made to all remaining employees in order to induce such employees to remain employed by us through December 31, 2018 and (ii) stock compensation expenseoption grants to all remaining employees in order to induce such employees to remain employed by us through December 31, 2019. Restructuring costs of $1.0 million for the year ended December 31, 2015 as compared to the year ended December 31, 2014, primarily associated with stock option grants in February 2015;2018
represent $0.4 million of severance-related costs and $0.6 million of costs related to the accrual of the retention bonus.
Interest income. Interest income comprises interest income earned on cash and cash equivalents.
Interest expense. Interest expense is primarily attributable to our term loansloan with Hercules and Oxford Finance LLC, or Oxford for the yearyears ended December 31, 20152018 and our term loan with Oxford for the year ended December 31, 2014.2017. Interest expense also includes the amortization of the discount associated with allocating value to the common stock warrants issued to Hercules, and Oxford and the amortization of the deferred financing costs associated with the term loans.
loan and the accrual of the final payment due to Hercules. Interest income. Interest income comprisesexpense decreased by $0.8 million, or 42% from $1.9 million for the year ended December 31, 2017 to $1.1 million for the year ended December 31, 2018. This decrease is primarily the result of a decrease in the principal outstanding under our term loan with Hercules for the year ended December 31, 2018 as compared to the year ended December 31, 2017. The term loan with Hercules was paid off on December 1, 2018 and accordingly, we expect no interest income earned on cash and cash equivalents.expense with respect to the Hercules loan in 2019.
Change in fair value of warrants. Certain of our warrants to purchase shares of our preferred stock (prior to the IPO) and common stock are recorded at fair value and are subject to re-measurement at each balance sheet date. These liabilities are re-measured at each balance sheet date with the corresponding charge or credit to earnings recorded within change in fair value of warrant liability. The fair value of the convertible preferredcommon stock warrants (prior to the IPO) and warrants to purchase common stock with non-standard anti-dilution provisions are determined using the Black-Scholes option pricing model which incorporates a number of assumptions and judgments to estimate the fair value of these warrants including the fair value per share of the underlying stock, the remaining contractual term of the warrants, risk-free interest rate, expected dividend yield, credit spread and expected volatility of the price of the underlying stock. During the year ended December 31, 2015, the fair value2018, we reported income of our warrant liability changed by $0.1 million compared$29 thousand related to the year ended December 31, 2014, primarily due to the changedecrease in the fair value of the underlying common stock.
Loss on extinguishmentwarrants as compared to income of debt. In February 2015, we entered into a loan and security agreement with Hercules$143 thousand for a term loan of up to $25.0 million. A first tranche of $16.5 million was funded upon execution of the loan and security agreement, approximately $15.5 million of which was used to repay our existing loan with Oxford. As a result of the repayment of the loan with Oxford, we recorded a loss on the extinguishment of debt of approximately $1.0 million representing the difference between the amount paid to Oxford and the carrying amount of the Oxford loan. Included in the loss on extinguishment of debt is the prepayment premium, the unamortized discount and the write off of deferred financing costs.year ended December 31, 2017.
Benefit from income taxes. Benefit from income taxes forFor the yearsyear ended December 31, 2015 and 2014 represents the proceeds2018, we received $0.5 million from the sale of New Jersey net operating losses,state Net Operating Loss Carryovers, or NOLs, as part of the Technology and Business Tax Certificate Program, sponsored byor the Program. We did not receive any payments under the Program during the year ended December 31, 2017. The Program enables approved biotechnology companies to sell their unused NOLs and unused Research and Development Tax Credits for at least 80% of the value of the tax benefits to unaffiliated, profitable corporate taxpayers in the State of New Jersey. The New Jersey Economic Development Authority. UnderAuthority and the program, emerging biotechnology companies with unused state NOLs are allowed to sell these NOLs to other companies. In November 2015, we completed the sale of New Jersey state NOLs totaling approximately $59.8Department of the Treasury's Division of Taxation administer the Program. We have reached the maximum lifetime benefit of $15.0 million under the Program and research and development credits totaling approximately $1.1 million for net proceeds of approximately $6.0 million. In February 2014, we completedare no longer eligible to participate in the sale of New Jersey state NOLs totaling approximately $39.1 million and research and development credits totaling approximately $0.4 million for net proceeds of approximately $3.6 million.Program.
Net Operating Losses and Tax Carryforwards
As of December 31, 2016,2019, we had approximately $177.4$231.5 million of federal and $44.2$92.4 million of state net operating loss carryforwards. We also potentially have federal and state research and development tax credits which would offset future taxable income. We have not completed a study to assess whether an ownership change has occurred, or whether there have been multiple ownership changes since our inception, due to the significant costs and complexities associated with such studies. Accordingly, our ability to utilize the aforementioned carryforwards may be limited. Additionally, for federal net operating losses generated prior to 2018, U.S. tax laws limit the time during which these carryforwards may be utilized against future taxes. As a result, we may not be able to take full advantage of these carryforwards for federal and state tax purposes. As of December 31, 2016,2019, all of our net operating losses were fully offset by a valuation allowance.
On December 22, 2017, the United States Congress and the Administration approved a bill reforming the US corporate income tax code which reduced our corporate tax rate from 34% to 21%
effective January 1, 2018. The carrying value of our deferred tax assets is also determined by the enacted US corporate income tax rate. Consequently, any changes in the US corporate income tax rate will impact the carrying value of our deferred tax assets. Under the new corporate income tax rate of 21%, deferred income tax assets decreased by $26.5 million with a corresponding decrease to the valuation allowance. There was no net effect of the tax reform enactment on financial statements.
Liquidity and Capital Resources
On May 29, 2014,In January 2019, we completedentered into the 2019 ATM Agreement under which we were authorized to issue and sell shares of our initial public offering wherebycommon stock having aggregate sales proceeds of up to $10.0 million from time to time. We paid a commission of 3% of the gross proceeds from the sales of our common stock under the 2019 ATM Agreement. In the year ended December 31, 2019, we sold 9,166,6671,801,528 shares of common stock at a public offering price of $6.00 per share, before underwriting discounts and expenses. The aggregateunder the 2019 ATM Agreement, resulting in net proceeds received by us fromof approximately $2.5 million. We terminated the offering were $49.7 million.2019 ATM Agreement on July 31, 2019.
In January 2015,March 2019, we completed a private placement of approximately 3.4 million8,426,750 shares of common stock at $5.85$0.93 per share. Proceeds from our private placement, net of commissions and other offering costs were $19.3approximately $7.8 million.
In February 2015,August 2019, we completed a public offering of 14,526,315 shares of our common stock at a price of $0.95 per share. Proceeds from the public offering, net of underwriting discounts, commissions and offering expenses were approximately $12.7 million.
In November 2019, we entered into a loanthe second 2019 ATM Agreement under which we were authorized to issue and security agreement with Hercules Technology Growth Capital, Inc. or Hercules, for a term loansell shares of our common stock having aggregate sales proceeds of up to $25.0 million. A first tranche$20.0 million from time to time. We paid a commission of $16.5 million was funded upon execution3% of the loan agreement, approximately $15.5 milliongross proceeds from the sales of which was used to repay our existing term loan. The Hercules Loan Agreement was amended in August 2016 to, among other things, extend the period during which we can drawcommon stock under the second tranche of $8.5 million to March2019 ATM Agreement. In the year ended December 31, 2017 and extend the period during which2019, we make interest-only payments to January 31, 2017. We are currently in discussions with Hercules to extend the period beyond March 31, 2017 during which the additional tranche of $8.5 million may be drawn. We can make no assurances that our discussions will ultimately be successful and, if such discussions result in an extension of the period in which we may draw the additional tranche of $8.5 million, we could incur additional fees payable to Hercules. On February 1, 2017, we began making principal payments with respect to the Hercules Loan.
In January 2016, we closed an underwritten public offering of 5,511,812sold 10,440,908 shares of common stock at a public offering price of $6.35 per share. In February 2016,under the underwriters ofsecond 2019 ATM Agreement, representing all the public offering of common stock exercised in full their option to purchase an additional 826,771 shares of common stock at the public offering price of $6.35 per share, less underwriting discounts and commissions. A total of 6,338,583 shares of common stock were sold in the public offering,capacity available, resulting in total net proceeds of approximately $37.5$19.3 million.
At December 31, 2016,2019, we had cash and cash equivalents totaling $48.8$34.5 million. We invest our cash equivalents in short-term highly liquid, interest-bearing investment-grade and government securities in order to preserve principal.
The following table sets forth the primary sources and uses of cash for the periods indicated:
| | Year ended December 31, | Year Ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | 2019 | 2018 | 2017 | ||||||||||||||
| | (In thousands) | | (In thousands) | | ||||||||||||||||
Cash used in operating activities | $ | (23,301 | ) | $ | (25,478 | ) | $ | (14,503 | ) | |||||||||||
Cash used in investing activities | $ | (31 | ) | $ | (290 | ) | $ | (96 | ) | |||||||||||
Cash provided by financing activities | $ | 37,687 | $ | 19,981 | $ | 52,661 | ||||||||||||||
Net cash used in operating activities | $ | (15,689 | ) | $ | (16,895 | ) | $ | (24,560 | ) | |||||||||||
Net cash used in investing activities | (98 | ) | (318 | ) | (1,313 | ) | ||||||||||||||
Net cash provided by (used in) financing activities | 42,415 | (10,888 | ) | 13,075 | ||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Net increase (decrease) in cash and cash equivalents | $ | 14,355 | $ | (5,787 | ) | $ | 38,062 | $ | 26,628 | $ | (28,101 | ) | $ | (12,798 | ) | |||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Operating Activities
We have incurred significant costs in the area of research and development, including CRO fees, manufacturing, regulatory and other clinical trial costs, as our primary product candidate Twirla was being developed. Net cash used in operating activities was $23.3$15.7 million for the year ended December 31, 20162019 and consisted of a net loss of $28.7$18.6 million and an increase in prepaid expenses of $0.2 million, which was offset by non-cash stock-based compensation expense of $1.8 million and depreciation and amortization of $0.2 million as well as an increase in accounts payable, accrued expenses and other liabilities of $1.1 million which reflects increased commercial development and commercial manufacturing expenses related to the initialization of pre-commercialization activities for Twirla. Net cash used in operating activities was $16.9 million for the year ended December 31, 2018 and consisted of a net loss of $19.8 million which
was offset, in part, by non-cash stock-based compensation expense of $3.6 million and non-cash interest expense of $0.3 million as well as a decrease in accounts payable and accrued liabilities of $1.2 million which reflects higher manufacturing commercialization expenses and the accrued loan fee which were both paid in 2018. Net cash used in operating activities was $24.6 million for the year ended December 31, 2017 and consisted of a net loss of $28.3 million which was offset, in part, by non-cash compensation and non-cash interest expense of $4.4$4.3 million as well as a decrease in prepaid clinical trial costs of $0.8 million. Net cash used in operating activities was $25.5 million for the year ended December 31, 2015 and consisted of a net loss of $30.3 million which was offset, in part, by non-cash
stock compensation expense of $3.0 million and a loss on extinguishment of debt of $1.0 million. Net cash used in operating activities was $14.5 million for the year ended December 31, 2014 and consisted of a net loss of $16.1 million which was offset, in part, by non-cash stock based compensation expense of $1.4$1.8 million. Cash used in operations in both 2016 and 2015 has been2018 was offset, in part, by the proceeds received from the sale of New Jersey NOLs.
Investing Activities
Net cash used in investing activities for the years ended December 31, 2016, 20152019, 2018 and 20142017 was $31 thousand,$0.1 million, $0.3 million and $0.1$1.3 million, respectively. Cash used in investing activities for these years primarily represents the acquisition of equipment to be used in the commercialization of Twirla.
Financing Activities
Net cash provided by financing activities for the year ended December 31, 20162019 was $37.7$42.4 million which included (i)primarily represented net proceeds of $37.5$7.8 million received from the issuance of 8,426,750 shares of our common stock in a private placement, net proceeds of $12.7 million from the sale of 6,338,58314,526,315 shares of common stock through a public offering, and (ii) $0.3net proceeds of approximately $21.8 million from the exercisesale of a total of 12,242,436 shares of our common stock options.through two at-the-market, or ATM, sales programs. Net cash used in financing activities for the year ended December 31, 2018 was $10.9 million which represented principal payments under the Hercules Loan Agreement which began on February 1, 2017 and were completed on December 1, 2018. Net cash provided by financing activities for the year ended December 31, 20152017 was $20.0 million which included (i) net proceeds of $19.3 million from the private placement of approximately 3.4 million shares of our common stock, (ii) net proceeds of $16.3 million from a term loan with Hercules and (iii) the repayment of our term loan with Oxford of $15.8 million. Net cash provided by financing activities for the year ended December 31, 2014 was $52.7$13.1 million which included net proceeds of (i) $49.7$18.5 million received from our initial public offeringthe sale of 9,166,6675,333,334 shares of common stock, and (ii) $3.0offset, in part, by principal payments of $5.6 million received fromunder the issuance of convertible bridge notes.Hercules Loan Agreement, which began on February 1, 2017.
Funding Requirements and Other Liquidity Matters
We believe that our cash and cash equivalents as of December 31, 2019, along with the proceeds of the Perceptive Credit Agreement that we have received to date, will be sufficient to meet our projected operating requirements through 2020. We will require additional capital to fund our operating needs beyond 2020, including, among other items, the commercialization of Twirla, is still in clinical development.and advancing the development of our other potential product candidates.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We anticipate that our expenses will increase substantially if and as we:
Based onWe may also need to raise additional funds sooner if we choose to accelerate components of our commercial plan or we encounter any unforeseen events that affect our current business plan, or we expect our existing cash and cash equivalents as of December 31, 2016, will enablemay choose to raise additional funds to provide us to fund our operating expenses and capital expenditures requirements into the second quarter of 2018. Our current business plan assumes resubmission of the NDA for Twirla in the first half of 2017, a six month FDA review of our resubmission, initiation of pre-commercial activities and initiation of validation of our commercial manufacturing process in coordination with the commercialization of Twirla. In the event of unforeseen changes to our planned timelines, we have the ability to postpone certain commercial and validation spending in order to continue the funding of our operations into the second quarter of 2018. We will require additional capital for the commercial launch of Twirla, if approved, as well as advancing the development of ourworking capital. Adequate additional
funding may not be available to us on acceptable terms, or at all. If we are unable to raise additional capital when needed or on attractive terms or are unable to enter into strategic collaborations, we then may be unable to successfully commercialize Twirla and may also be required to further cut operating costs, forgo future development and other product candidates. We have based this estimate on assumptions thatopportunities or even terminate our operations, which may prove to be wrong, and we may use our available capital resources sooner than we currently expect.involve seeking bankruptcy protection. Because of the numerous risks and uncertainties associated with the development and commercialization of Twirla, if approved,such developments, including, among other things, manufacturing scale up, we are unable to estimate the amounts of increased capital outlays and operating expenses associated with completing the developmentcommercialization of Twirla. Our future capital requirements will depend on many factors, including:
Except for the remaining tranche under the Perceptive Credit Agreement, which is contingent upon achieving certain revenue milestones, we do not have any committed external source of funds. Until such time, if ever, as we can generate substantial cash flows from product revenues, we expect to finance our cash needs through a combination of equity offerings, debt financings, collaborations, strategic alliances and licensing arrangements.
Going Concern
As of December 31, 2019, we had cash and cash equivalents of $34.5 million. Additionally, in February 2020, we entered into the Perceptive Credit Agreement. A first tranche of $5 million was funded on execution of the agreement. A second tranche of $15 million was funded as a result of the approval of Twirla by the FDA (see Note 15). We do notbelieve that our cash and cash equivalents as of December 31, 2019 along with the proceeds of the Perceptive Credit Agreement we have any committed external sourcereceived to date, will be sufficient to meet our projected operating requirements through the end of funds. To2020. We will require additional capital to fund our operating needs beyond 2020, which we expect primarily will consist of commercializing Twirla, and exploring the extent that weadvancement of our existing pipeline and its possible expansion through business development activities.
Our future success depends on our ability to raise additional capital and/or implement various strategic alternatives. We continue to analyze strategic and financing alternatives, potential asset sales as well as mergers and acquisitions. We cannot be certain that these initiatives or raising additional capital, whether through selling additional debt or equity securities or obtaining a line of credit or other loan, will be available to us or, if available, will be on terms acceptable to us. If we issue additional securities to raise funds, whether through the saleissuance of equity or convertible debt securities, the ownership interest of our stockholders will be diluted, and the terms ofor any combination thereof, these securities may include liquidationhave rights, preferences, or other preferences that adversely affect your rights as aprivileges senior to those of our common stockholder.stock, and our current shareholders may experience dilution. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances or licensing arrangements with pharmaceutical partners, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates, including Twirla, or grant licenses on terms that may not be favorable to us. If we are unable to raise additionalobtain funds through equity or debt financings when needed or on acceptable terms, we may
be required to delay, limit, reduce or terminatecurtail our productcurrent development orprograms, cut operating costs, forego future commercialization efforts or grant rightsdevelopment and other opportunities and may need to develop and market Twirlaseek bankruptcy protection.
The financial statements as of December 31, 2019 have been prepared under the assumption that we would otherwise preferwill continue as a going concern for the next 12 months following the date this Annual Report on Form 10-K is filed. Our ability to developcontinue as a going concern is dependent upon our uncertain ability to obtain additional capital, reduce expenditures and/or execute on our business plan and market ourselves.successfully launch Twirla. The audited financial statements as of December 31, 2019 do not include any adjustments that might result from the outcome of this uncertainty.
Contractual Obligations and Commitments
The following table summarizes our contractual obligations and commitments as of December 31, 20162019 that will affect our future liquidity:
| | Total | Less than 1 year | 1 - 3 years | 3 - 5 years | More than 5 years | Total | Less than 1 year | 1 - 3 years | 3 - 5 years | More than 5 years | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | (In thousands) | | | (In thousands) | | | ||||||||||||||||||||||||||
Term loan | $ | 19,124 | $ | 6,907 | $ | 12,217 | — | — | ||||||||||||||||||||||||
Operating lease | 783 | 192 | 591 | — | 191 | 191 | — | — | — | |||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Total | $ | 19,907 | $ | 7,099 | $ | 12,808 | $ | — | $ | 191 | $ | 191 | $ | — | $ | — | $ | — | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Our operating lease commitment relates to our lease of office space in Princeton, New Jersey. In August 2015, we renewed this lease with the new term to expire in November 2020.
January 2015 Private Placement
In January 2015, we completed a private placement of approximately 3.4 million shares of common stock at $5.85 per share. Proceeds from our private placement, net of commissions and other offering costs, were $19.3 million.
February 2015 Loan and Security Agreement—Hercules Capital, Inc.
The first tranche of the Hercules Loan was funded in February 2015. In August 2016, we entered into the First Amendment to Loan and Security Agreement, or the "First Amendment" with Hercules which amends certain terms of the Hercules Loan Agreement.
The First Amendment extends our option to draw down the second tranche of $8.5 million referred to as the "Second Term Loan Advance", of the term loan facility provided under the Hercules Loan, or the Term Loan, until March 31, 2017 and makes the Second Term Loan Advance subject to the consent of Hercules, among other customary conditions. We are currently in discussions with Herculesseeking new facilities or considering expanding existing facilities as we consider adding employees, and we believe that suitable additional or substitute space will be available as needed to extend the period beyond March 31, 2017 during which the additional tranche of $8.5 million may be drawn. We can make no assurances that our discussions will ultimately be successful and, ifaccommodate any such discussions result in an extension of the period in which we may draw the additional tranche of $8.5 million, we could incur additional fees payable to Hercules. The First Amendment also extends the interest-only payments until January 31, 2017, in connection with the first tranche of $16.5 million, or the First Term Loan Advance, and together with the Second Term Loan Advance, referred to as the Term Loan Advances. The First Amendment also provides us the ability to extend further the interest-only payments for two successive periods as follows: (i) until April 30, 2017, subject to us successfully completing our SECURE clinical trial, and receiving data that supports the filing of a response to the FDA's complete response letter relating to the new drug application filed by us referred to as the First Interest Only Period Extension, and (ii) until July 31, 2017, provided that (x) we have received the First Interest Only Period Extension and (y) we have received unrestricted gross cash proceeds in an aggregate amount greater than or equal to $40.0 million from the issuance and saleexpansion of our equity securities, referred to as the Second Interest Only Period Extension.
The First Amendment provides that the Term Loan will mature on December 1, 2018; provided, however, that if the First Interest Only Period Extension occurs on or prior to January 31, 2017, the Term Loan will mature on March 1, 2019; and provided further, however, that if both (a) the First Interest Only Period Extension occurs on or prior to January 31, 2017, and (b) the Second Interest Only Period Extension occurs on or prior to April 30, 2017, the Term Loan will mature on June 1, 2019.
The First Amendment also provides that as part of the extension of the interest-only period from the First Term Loan Advance, Hercules returned to us the principal payments paid by us in July and August 2016, which such refunded payments will once again constitute Term Loan Advances under the Hercules Loan. In connection with the execution of the First Amendment, we paid Hercules a facility fee of $0.165 million.
The Hercules Loan accrues interest at a rate of the greater of 9.0% or 9.0% plus Prime minus 4.25% and is payable monthly. Principal is due in 23 equal consecutive monthly installments beginning on February 1, 2017 and ending on December 1, 2018. In addition, we are required to make a final payment of $610,500 on the maturity date of the Hercules Loan, December 1, 2018. The final payment is being accrued and recorded to interest expense over the life of the Hercules Loan. On February 1, 2017, we began making principal payments with respect to the Hercules Loan.
We may prepay all, but not less than all, of the Hercules Loan subject to a prepayment premium of 3.0% of the outstanding principal if prepaid during the first year, 2.0% of the outstanding principal if prepaid during the second year and 1.0% of the outstanding principal if prepaid after the second
year. Our obligations under the Hercules Loan are secured by a perfected first position lien on all of our assets, excluding intellectual property assets.
In connection with the Hercules Loan, we issued Hercules a warrant to purchase 180,274 shares of our common stock at an exercise price of $5.89 per share and granted Hercules the right to participate in future equity financings in an amount up to $2.0 million while the loan and warrant are outstanding.
We allocated the proceeds of $16.5 million in accordance with ASC 470 based on the relative fair values. The relative fair value of the warrants of approximately $1.2 million at the time of issuance, which was determined using the Black-Scholes option-pricing model, was recorded as additional paid-in capital and reduced the carrying value of the debt. The discount on the debt is being amortized to interest expense over the term of the debt.
In December 2012, we entered into a Loan and Security Agreement, the Oxford Loan, with Oxford Finance, LLC, or Oxford, pursuant to which we borrowed a total of $15.0 million from Oxford.
In February 2015, we terminated and repaid all amounts outstanding under the Oxford Loan. As a result of this repayment, we recorded a loss on the extinguishment of debt of approximately $1.0 million on our statement of operations for the year ended December 31, 2015, primarily representing a prepayment premium and the write off of deferred financing costs.operations.
Shelf Registration StatementStatements
On June 19, 2015,November 2, 2018, we filed a universal shelf registration statement with the SEC for the issuance of common stock, preferred stock, warrants, rights, debt securities and units up to an aggregate amount of $150.0$100.0 million, which we refer to as the 20152018 Shelf Registration Statement. On July 1, 2015,November 14, 2018, the 20152018 Shelf Registration Statement was declared effective by the SEC. We completed an offering of common stock utilizing the 2015
On January 23, 2019, we filed a prospectus supplement to our 2018 Shelf Registration Statement (see below).registering an at-the-market offering program we entered into for the sale of up to $10.0 million of shares of our common stock. In the future,year ended December 31, 2019, we may periodically offer one or moresold a total of these securities1,801,528 shares of our common stock under this ATM program resulting in amounts, prices and termsnet proceeds of approximately $2.5 million. We terminated this at-the-market offering program on July 31, 2019.
In August 2019, we filed a prospectus supplement to be announced when and if the securities are offered. At the time any of the securities covered by the 2015our 2018 Shelf Registration Statement are offered for sale,registering a prospectus supplement will be prepared and filed with the SEC containing specific information about the terms of any such offering.
2016 Public Offering of Common Stock
In January 2016, we closed an underwritten public offering of 5,511,812 shares of common stock registered under the 2015 Shelf Registration Statement at a public offering price of $6.35 per share. In February 2016, the underwriters of the public offering of common stock exercised in full, their option to purchase an additional 826,77114,526,315 shares of common stock at a price of $0.95 per share. Proceeds from the public offering, pricenet of $6.35 per share, less underwriting discounts, commissions and commissions. Aoffering expenses, were approximately $12.7 million.
On November 8, 2019, we filed a prospectus supplement to our 2018 Shelf Registration Statement registering an at-the-market offering program we entered into for the sale of up to $20.0 million of shares of our common stock. In the year ended December 31, 2019, we sold a total of 6,338,58310,440,908 shares of our common stock were sold inunder this ATM program, representing all the public offeringcapacity, resulting in total net proceeds of approximately $37.5$19.3 million. One of our stockholders, who is also affiliated with an individual that was at the time a member of our Board of Directors, purchased 393,700 shares of common stock for approximately $2.5 million in the public offering.
Recent Accounting Pronouncements
In August 2014, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2014-15,Presentation of Financial Statements—Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity's Ability See Note 2 to Continue as a Going Concern, which defines management's responsibility to assess an entity's ability to continue as a going concern, and to provide related footnote disclosures if there is substantial doubt about its ability to continue as a going concern. Theour financial statements that discusses new standard is effective for the annual period ending after December 15, 2016, and for interim periods thereafter. We adopted ASU 2014-15 in the fourth quarter of 2016, which resulted in noaccounting pronouncements.
change to our financial statements. Additionally, we will perform quarterly evaluations to identify current conditions which may raise substantial doubt about the entity's ability to continue as a going concern within one year after the date that the financial statements are issued. See Note 1 to our financial statements for additional information on our liquidity risks and management's plans.
In April 2015, the FASB issued an amendment to U.S. GAAP to simplify the balance sheet presentation of the costs for issuing debt. The changes were adopted in ASU No. 2015-03,Interest—Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issue Costs. ASU 2015-03 amends current presentation guidance by requiring that debt issuance costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts. Prior to the issuance of ASU 2015-03, debt issuance costs were required to be presented as an asset in the balance sheet. We adopted the provisions of ASU 2015-03 on January 1, 2016 and prior period balances have been reclassified to conform to the current period presentation. As of December 31, 2015, $152 thousand of debt issuance costs were reclassified in the balance sheet from "deferred financing costs, net" to "loan payable, current portion" and $139 thousand was reclassified from "deferred financing costs, net" to "loan payable, long-term". The adoption of ASU 2015-03 did not have an impact on our operations or cash flows.
In February 2016, the FASB issued ASU No. 2016-02,Leases. The new standard establishes a right-of-use (ROU) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the statement of operations. The new standard is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. A modified retrospective transition approach is required for leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements. We will be evaluating the impact of the pending adoption of the new standard on our financial statements.
In March 2016, the FASB issued ASU No. 2016-09,Improvements to Employee Share-Based Payment Accounting. This ASU requires all tax effects of share-based payment settlements to be recorded through the statement of operations. Currently, tax benefits in excess of compensation cost, or "windfalls", are recorded in equity, and tax deficiencies, or "shortfalls", are recorded to equity to the extent of previous windfalls, and then to the statement of operations. In addition, under the new guidance, companies will be permitted to make a policy election to recognize the impact of forfeitures either when they occur, or on an estimated basis. Finally, this update simplifies withholding requirements to allow companies to withhold up to an employee's maximum tax rate without resulting in liability classification for the award. ASU 2016-09 is effective for annual reporting periods beginning after December 15, 2016, and early adoption is permitted. We adopted the provisions of this standard early and the impact on our financial statements was not significant.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under SEC rules, such as relationships with unconsolidated entities or financial partnerships, which are often referred to as structured finance or special purpose entities, established for the purpose of facilitating financing transactions that are not required to be reflected on our balance sheets.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Interest Rate Risk
We are exposed to market risks in the ordinary course of our business. Market risk is the risk of change in fair value of a financial instrument due to changes in interest rates, equity prices, financing, exchange rates or other factors. These market risks are principally limited to interest rate fluctuations.
We had cash and cash equivalents of $48.8$34.5 million and $34.4$7.8 million at December 31, 20162019 and 2015,December 31, 2018, respectively consisting primarily of funds in cash and money market accounts. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes. Due to the short-term nature of our investment portfolio, we do not believe an immediate 10.0% increase in interest rates would have a material effect on the fair market value of our portfolio, and accordingly we do not expect our operating results or cash flows to be materially affected by a sudden change in market interest rates.
Our results of operations and cash flows were subject to fluctuations due to changes in interest rates, principally in connection with our loan agreement with Hercules (through November 30, 2018) and interest income on cash balances. We do not believe we are materially exposed to changes in interest rates. We do not currently use interest rate derivative instruments to manage exposure to interest rate changes. Based on average invested cash of $14.8 million for the year ended December 31, 2019, a 1% increase or decrease in interest rates would have increased or decreased interest income by $148,000 for the year ended December 31, 2019.
Inflation Risk
Inflation generally affects us by increasing our cost of labor and pricing of contracts and agreements. We do not believe that inflation had a material effect on our business, financial condition, or results of operations during the year ended December 31, 2019.
Item 8. Financial Statements and Supplementary Data
Agile Therapeutics, Inc.
Index to Financial Statements
Report of Independent Registered Public Accounting Firm | ||||
Balance Sheets | ||||
Statements of Operations | ||||
Statements of | ||||
Statements of Cash Flows | ||||
Notes to Financial Statements |
Report of Independent Registered Public Accounting Firm
The BoardTo the stockholders and the board of Directors and Stockholders
directors of Agile Therapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Agile Therapeutics, Inc. (the Company)"Company") as of December 31, 20162019 and 2015, and2018, the related statements of operations, convertible preferred stock andstatements of changes in stockholders' equity, (deficit), and cash flows for each of the three years in the period ended December 31, 2016.2019, and the related notes (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2019 and 2018, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2019, in conformity with U.S. generally accepted accounting principles.
The Company's Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations, requires additional capital to fund its commercialization activities and has stated that substantial doubt exists about the Company's ability to continue as a going concern. Management's evaluation of the events and conditions and management's plans regarding these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on thesethe Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the US federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States).PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. Wemisstatement, whether due to error or fraud. The Company is not required to have, nor were notwe engaged to perform, an audit of the Company'sits internal control over financial reporting. OurAs part of our audits included considerationwe are required to obtain an understanding of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence supportingregarding the amounts and disclosures in the financial statements, assessingstatements. Our audits also included evaluating the accounting principles used and significant estimates made by management, andas well as evaluating the overall presentation of the financial statement presentation.statements. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Agile Therapeutics, Inc. at December 31, 2016 and 2015, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2016, in conformity with U.S. generally accepted accounting principles.
/s/ Ernst & Young LLP
We have served as the Company's auditor since 2010.
Iselin, New JerseyMarch 15, 2017February 20, 2020
Agile Therapeutics, Inc.
Balance Sheets
(in thousands, except par value and share data)
| | December 31 | December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2019 | 2018 | ||||||||||
Assets | ||||||||||||||
Current assets: | ||||||||||||||
Cash and cash equivalents | $ | 48,750 | $ | 34,395 | $ | 34,479 | $ | 7,851 | ||||||
Prepaid expenses | 2,768 | 3,690 | 840 | 607 | ||||||||||
| | | | | | | | | | | | | | | |
Total current assets | 51,518 | 38,085 | 35,319 | 8,458 | ||||||||||
Property and equipment, net | 12,330 | 12,318 | 14,044 | 13,916 | ||||||||||
Other assets | 18 | 18 | ||||||||||||
Right of use and other assets | 177 | 18 | ||||||||||||
| | | | | | | | | | | | | | | |
Total assets | $ | 63,866 | $ | 50,421 | $ | 49,540 | $ | 22,392 | ||||||
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
Liabilities and stockholders' equity | ||||||||||||||
Current liabilities: | ||||||||||||||
Accounts payable | $ | 2,050 | $ | 2,387 | $ | 1,819 | $ | 875 | ||||||
Accrued expenses | 3,352 | 2,653 | 1,804 | 1,343 | ||||||||||
Loan payable, current portion | 5,104 | 2,336 | ||||||||||||
Warrant liability | 172 | 406 | ||||||||||||
Lease payable, curent portion | 172 | — | ||||||||||||
| | | | | | | | | | | | | | | |
Total current liabilities | 10,678 | 7,782 | 3,795 | 2,218 | ||||||||||
Loan payable, long-term | 10,899 | 12,896 | ||||||||||||
Total liabilities | 3,795 | 2,218 | ||||||||||||
| | | | | | | | | |||||||
Commitments and contingencies(Note 13) | ||||||||||||||
Stockholders' equity: | ||||||||||||||
Common stock, $.0001 par value, authorized 150,000,000 shares; 28,759,731 shares issued and outstanding as of December 31, 2016 and 22,315,612 shares issued and outstanding as of December 31, 2015; | 3 | 2 | ||||||||||||
Stockholders' equity | ||||||||||||||
Common stock, $.0001 par value, 150,000,000 shares authorized, 69,810,305 and 34,377,329 issued and outstanding at December 31, 2019 and 2018, respectively | 7 | 3 | ||||||||||||
Additional paid-in capital | 235,754 | 194,468 | 306,108 | 261,722 | ||||||||||
Accumulated deficit | (193,468 | ) | (164,727 | ) | (260,370 | ) | (241,551 | ) | ||||||
| | | | | | | | | | | | | | | |
Total stockholders' equity | 42,289 | 29,743 | 45,745 | 20,174 | ||||||||||
| | | | | | | | | | | | | | | |
Total liabilities and stockholders' equity | $ | 63,866 | $ | 50,421 | $ | 49,540 | $ | 22,392 | ||||||
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
See accompanying notes.
Agile Therapeutics, Inc.
Statements of Operations
(in thousands, except share and per share data)
| | Year Ended December 31 | Year ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | 2019 | 2018 | 2017 | ||||||||||||||
Operating expenses: | ||||||||||||||||||||
Research and development | $ | 20,929 | $ | 25,622 | $ | 13,365 | $ | 9,858 | $ | 9,777 | $ | 14,428 | ||||||||
General and administrative | 8,792 | 7,467 | 5,150 | 9,000 | 8,739 | 12,383 | ||||||||||||||
Restructuring costs | — | 1,019 | — | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Total operating expenses | 29,721 | 33,089 | 18,515 | 18,858 | 19,535 | 26,811 | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Loss from operations | (29,721 | ) | (33,089 | ) | (18,515 | ) | (18,858 | ) | (19,535 | ) | (26,811 | ) | ||||||||
| | | | | | | | | | | | ||||||||||
Other income (expense) | ||||||||||||||||||||
Interest income | 252 | 366 | 282 | |||||||||||||||||
Interest expense | (2,446 | ) | (2,077 | ) | (1,566 | ) | — | (1,116 | ) | (1,918 | ) | |||||||||
Interest income | 117 | 5 | 3 | |||||||||||||||||
Change in fair value of warrants | 234 | (110 | ) | 348 | — | 29 | 143 | |||||||||||||
Loss on extinguishment of debt | — | (1,036 | ) | — | ||||||||||||||||
| | | | | | | | | | | | ||||||||||
Total other income (expense), net | 252 | (721 | ) | (1,493 | ) | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Loss before benefit from income taxes | (31,816 | ) | (36,307 | ) | (19,730 | ) | (18,606 | ) | (20,256 | ) | (28,304 | ) | ||||||||
Benefit from income taxes | 3,075 | 5,972 | 3,653 | — | 477 | — | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Net loss | $ | (28,741 | ) | $ | (30,335 | ) | $ | (16,077 | ) | $ | (18,606 | ) | $ | (19,779 | ) | $ | (28,304 | ) | ||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Net loss per share (basic and diluted) | $ | (1.02 | ) | $ | (1.38 | ) | $ | (1.41 | ) | $ | (0.38 | ) | $ | (0.58 | ) | $ | (0.91 | ) | ||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Weighted-average shares outstanding (basic and diluted) | 28,273,331 | 22,017,229 | 11,394,971 | |||||||||||||||||
Weighted-average common shares (basic and diluted) | 49,432,487 | 34,315,931 | 30,940,831 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
See accompanying notes.
Statements of Convertible Preferred Stock and Changes in Stockholders' Equity (Deficit)
(in thousands, except share data)
| | Series A-1 Convertible Preferred Stock | Series A-2 Convertible Preferred Stock | Series B Convertible Preferred Stock | Series C Convertible Preferred Stock | | | | | | |||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Common Stock | | Deficit Accumulated During the Development Stage | | ||||||||||||||||||||||||||||||||||||
| | | Net Stockholders' Equity (Deficit) | ||||||||||||||||||||||||||||||||||||||
| | Number of Shares | Amount | Number of Shares | Amount | Number of Shares | Amount | Number of Shares | Amount | Number of Shares | Amount | Additional Paid-in Capital | |||||||||||||||||||||||||||||
Balance December 31, 2013 | 137,787 | 898 | 66,116 | 544 | 4,510,066 | 44,928 | 1,578,400 | 22,862 | 109,321 | — | 46,873 | (118,315 | ) | (71,442 | ) | |||||||||||||||||||||||||
Share-based compensation—stock options | — | — | — | — | — | — | — | — | — | — | 1,381 | — | 1,381 | |||||||||||||||||||||||||||
Issuance of common stock for employee bonuses | — | — | — | — | — | — | — | — | 9,983 | — | 80 | — | 80 | |||||||||||||||||||||||||||
Conversion of preferred stock to common stock | (137,787 | ) | (898 | ) | (66,116 | ) | (544 | ) | (4,510,066 | ) | (44,928 | ) | (1,578,400 | ) | (22,862 | ) | 8,803,547 | 1 | 69,232 | — | 69,233 | |||||||||||||||||||
Conversion of notes and accrued interest | — | — | — | — | — | — | — | — | 503,450 | — | 3,020 | — | 3,020 | |||||||||||||||||||||||||||
Common stock issued in IPO, net of expenses | — | — | — | — | — | — | — | — | 9,166,667 | 1 | 49,743 | — | 49,744 | |||||||||||||||||||||||||||
Issuance of common stock upon exercise of options | — | — | — | — | — | — | — | — | 41,904 | — | 67 | — | 67 | |||||||||||||||||||||||||||
Net loss for the year ended December 31, 2014 | — | — | — | — | — | — | — | — | — | — | — | (16,077 | ) | (16,077 | ) | |||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance December 31, 2014 | — | — | — | — | — | — | — | — | 18,634,872 | 2 | 170,396 | (134,392 | ) | 36,006 | ||||||||||||||||||||||||||
Share-based compensation—stock options | — | — | — | — | — | — | — | — | — | — | 2,965 | — | 2,965 | |||||||||||||||||||||||||||
Issuance of common stock in Private Placement, net of expenses | — | — | — | �� | — | — | — | — | — | 3,418,804 | — | 19,330 | — | 19,330 | ||||||||||||||||||||||||||
Fair value of common stock warrants issued with debt financing | — | — | — | — | — | — | — | — | — | — | 1,184 | — | 1,184 | |||||||||||||||||||||||||||
Issuance of common stock upon exercise of options | — | — | — | — | — | — | — | — | 261,936 | — | 593 | — | 593 | |||||||||||||||||||||||||||
Net loss for the year ended December 31, 2015 | — | — | — | — | — | — | — | — | — | — | — | (30,335 | ) | (30,335 | ) | |||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2015 | — | — | — | — | — | — | — | — | 22,315,612 | 2 | 194,468 | (164,727 | ) | 29,743 | ||||||||||||||||||||||||||
Share-based compensation—stock options and RSUs | — | — | — | — | — | — | — | — | — | — | 3,425 | — | 3,425 | |||||||||||||||||||||||||||
Vesting of RSUs | — | — | — | — | — | — | — | — | 16,666 | — | — | — | — | |||||||||||||||||||||||||||
Issuance of common stock in public offering, net of expenses | — | — | — | — | — | — | — | — | 6,338,583 | 1 | 37,527 | — | 37,528 | |||||||||||||||||||||||||||
Exercise of stock options | — | — | — | — | — | — | — | — | 88,870 | — | 334 | — | 334 | |||||||||||||||||||||||||||
Net loss for the year ended December 31, 2016 | — | — | — | — | — | — | — | — | — | — | — | (28,741 | ) | (28,741 | ) | |||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2016 | — | — | — | — | — | — | — | — | 28,759,731 | $ | 3 | $ | 235,754 | $ | (193,468 | ) | $ | 42,289 | ||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Common Stock | | | | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Number of Shares | Amount | Additional Paid-in Capital | Accumulated Deficit | Net Stockholders' Equity | |||||||||||
Balance December 31, 2016 | 28,759,731 | $ | 3 | $ | 235,754 | $ | (193,468 | ) | $ | 42,289 | ||||||
Share-based compensation—stock options and RSUs | — | — | 3,651 | — | 3,651 | |||||||||||
Vesting of RSUs | 16,667 | — | — | — | — | |||||||||||
Issuance of common stock in public offering, net of expenses | 5,333,334 | — | 18,535 | — | 18,535 | |||||||||||
Issuance of common stock upon exercise of options | 76,610 | — | 152 | — | 152 | |||||||||||
Net loss | — | — | — | (28,304 | ) | (28,304 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balance December 31, 2017 | 34,186,342 | $ | 3 | $ | 258,092 | $ | (221,772 | ) | $ | 36,323 | ||||||
Share-based compensation—stock options and RSUs | — | — | 3,630 | — | 3,630 | |||||||||||
Vesting of RSUs | 190,987 | — | — | — | — | |||||||||||
Net loss | — | — | — | (19,779 | ) | (19,779 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balance December 31, 2018 | 34,377,329 | $ | 3 | $ | 261,722 | $ | (241,551 | ) | $ | 20,174 | ||||||
Adjustment to derivitive liabilities upon adoption of ASU 2017-11 | — | — | 213 | (213 | ) | — | ||||||||||
Share-based compensation— stock options and RSUs | — | — | 1,762 | — | 1,762 | |||||||||||
Issuance of common stock in private placement, net of expenses | 8,426,750 | 1 | 7,809 | — | 7,810 | |||||||||||
Issuance of common stock pursuant to at-the-market stock sales, net of expenses | 12,242,436 | 1 | 21,753 | — | 21,754 | |||||||||||
Issuance of common stock upon exercise of stock options | 92,271 | 164 | — | 164 | ||||||||||||
Proceeds from issuance of common stock in public offering, net of expenses | 14,526,315 | 2 | 12,685 | — | 12,687 | |||||||||||
Vesting of RSUs | 145,204 | — | — | — | — | |||||||||||
Net loss | — | — | — | (18,606 | ) | (18,606 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balance December 31, 2019 | 69,810,305 | $ | 7 | $ | 306,108 | $ | (260,370 | ) | $ | 45,745 | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
See accompanying notes.
Agile Therapeutics, Inc.
Statements of Cash Flows
(in thousands)
| | Year Ended December 31 | Year Ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | 2019 | 2018 | 2017 | ||||||||||||||
Cash flows from operating activities | ||||||||||||||||||||
Cash flows from operating activities: | ||||||||||||||||||||
Net loss | $ | (28,741 | ) | $ | (30,335 | ) | $ | (16,077 | ) | $ | (18,606 | ) | $ | (19,779 | ) | $ | (28,304 | ) | ||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||||||||||
Depreciation | 19 | 18 | 12 | 18 | 23 | 23 | ||||||||||||||
Noncash stock bonus | — | — | 80 | |||||||||||||||||
Amortization | 145 | — | — | |||||||||||||||||
Noncash stock based compensation | 3,425 | 2,965 | 1,381 | 1,762 | 3,630 | 3,651 | ||||||||||||||
Loss on extinguishment of debt | — | 1,036 | — | |||||||||||||||||
Noncash interest | 946 | 590 | 185 | — | 282 | 667 | ||||||||||||||
Change in fair value of warrants | (234 | ) | 110 | (348 | ) | — | (29 | ) | (143 | ) | ||||||||||
Changes in operating assets and liabilities: | ||||||||||||||||||||
Prepaid expenses and other current assets | 922 | (1,209 | ) | (2,335 | ) | |||||||||||||||
Prepaid expenses and other assets | (233 | ) | 155 | 2,006 | ||||||||||||||||
Accounts payable and accrued expenses | 362 | 1,347 | 2,599 | 1,377 | (1,177 | ) | (2,460 | ) | ||||||||||||
Lease liability | (152 | ) | — | — | ||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Net cash used in operating activities | (23,301 | ) | (25,478 | ) | (14,503 | ) | (15,689 | ) | (16,895 | ) | (24,560 | ) | ||||||||
Cash flows from investing activities | ||||||||||||||||||||
| | | | | | | | | | | | ||||||||||
Cash flows from investing activities: | ||||||||||||||||||||
Acquisition of property and equipment | (31 | ) | (290 | ) | (96 | ) | (98 | ) | (318 | ) | (1,313 | ) | ||||||||
| | | | | | | | | | | | | | | | | | | | | |
Net cash used in investing activities | (31 | ) | (290 | ) | (96 | ) | (98 | ) | (318 | ) | (1,313 | ) | ||||||||
Cash flows from financing activities | ||||||||||||||||||||
Proceeds from convertible bridge notes | — | — | 3,000 | |||||||||||||||||
Proceeds from issuance of term loan | — | 16,265 | — | |||||||||||||||||
Repayment of term loan | — | (15,784 | ) | — | ||||||||||||||||
Cash flows from financing activities: | ||||||||||||||||||||
Proceeds from issuance of common stock, net of offering costs | 42,251 | — | 18,535 | |||||||||||||||||
Principal payments of long-term debt | (985 | ) | — | — | — | (10,888 | ) | (5,612 | ) | |||||||||||
Return of principal payments of long-term debt | 985 | — | — | |||||||||||||||||
Proceeds from issuance of common stock, in public offering, net of offering costs | 37,528 | — | 49,744 | |||||||||||||||||
Proceeds from issuance of common stock in private placement, net of offering costs | — | 19,330 | — | |||||||||||||||||
Cash paid for financing costs | (175 | ) | (423 | ) | (150 | ) | ||||||||||||||
Proceeds from exercise of stock options | 334 | 593 | 67 | 164 | — | 152 | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Net cash provided by financing activities | 37,687 | 19,981 | 52,661 | |||||||||||||||||
Net cash provided by (used in) financing activities | 42,415 | (10,888 | ) | 13,075 | ||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Net increase (decrease) in cash and cash equivalents | 14,355 | (5,787 | ) | 38,062 | 26,628 | (28,101 | ) | (12,798 | ) | |||||||||||
Cash and cash equivalents, beginning of year | 34,395 | 40,182 | 2,120 | 7,851 | 35,952 | 48,750 | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Cash and cash equivalents, end of year | $ | 48,750 | $ | 34,395 | $ | 40,182 | $ | 34,479 | $ | 7,851 | $ | 35,952 | ||||||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Supplemental disclosure of noncash financing activities | ||||||||||||||||||||
Supplemental cash flow information | ||||||||||||||||||||
Interest paid | $ | 1,500 | $ | 1,474 | $ | 1,380 | ||||||||||||||
| | | | | | | | | | | |||||||||||
| | | | | | | | ||||||||||||||
| | | | | | | | | | | |||||||||||
Income taxes paid | $ | — | $ | — | $ | — | ||||||||||||||
| | | | | | | | | | | |||||||||||
| | | | | | | | ||||||||||||||
| | | | | | | | | | | |||||||||||
Supplemental disclosure of noncash financing activities | ||||||||||||||||||||
Fair value of common stock warrants issued | $ | $ | 1,184 | $ | — | |||||||||||||||
| | | | | | | | | | | |||||||||||
| | | | | | | | ||||||||||||||
| | | | | | | | | | | |||||||||||
Conversion of preferred stock into common stock | $ | — | $ | — | $ | 69,233 | ||||||||||||||
| | | | | | | | | | | |||||||||||
| | | | | | �� | | | |||||||||||||
| | | | | | | | | | | |||||||||||
Conversion of notes payable and interest into common stock | $ | — | $ | — | $ | 3,021 | ||||||||||||||
| | | | | | | | | | | |||||||||||
| | | | | | | | ||||||||||||||
| | | | | | | | | | | |||||||||||
Interest paid during the year | $ | — | $ | 1,370 | $ | 1,295 | ||||||||||||||
Cash paid for income taxes | $ | — | $ | — | $ | — | ||||||||||||||
Non-cash transactions | ||||||||||||||||||||
Property and equipment purchases included in accounts payable | $ | 49 | $ | — | $ | 242 | ||||||||||||||
See accompanying notes.
Agile Therapeutics, Inc.
Notes to Financial Statements
December 31, 20162019
(in thousands, except share and per share data)
1. Organization and Description of Business
Nature of Operations
Agile Therapeutics, Inc. ("Agile" or the "Company") was incorporated in Delaware on December 22, 1997. Agile is a forward-thinking women's healthcare company dedicated to fulfilling the unmet health needs of today's women. The Company's activities since inception have consisted principally of raising capital and performing research and development.development, including development of the Company's lead product candidate. The Company is headquartered in Princeton, New Jersey.
The Company's leadsole approved product, candidate, Twirla®, also known as AG200-15, is a once-weekly prescription contraceptive patch that is atreceived approval from the end of Phase 3 clinical development.U.S. Food and Drug Administration, or FDA in February 2020. Substantially all of the Company's resources are currently dedicated to developing and seeking regulatory approval for Twirla.commercializing Twirla in the United States. The Company has not generated product revenue to date and is subject to a number of risks similar to those of other early stage companies, including, but not limited to, dependence on key individuals, the difficulties and uncertainties inherent in the development of commercially usable products, market acceptance of products, protection of proprietary technology, the potential need to obtain additional capital necessary to fund the development of its products, and competition from larger companies.companies and compliance with the FDA and other government regulations. If the Company does not successfully commercialize any product candidates, it will be unable to generate recurring product revenue or achieve profitability. The Company has incurred operating losses and negative cash flows from operating activities each year since inception. As of December 31, 2016,2019, the Company had an accumulated deficit of approximately $193.5$260 million. The Company expects to continue to incur net losses into the foreseeable future
The Company has financed its operations to date primarily through the issuance and sale of its common stock in both public and private offerings (see Note 9), private placements of its convertible preferred stock, venture loans, and non-dilutive grant funding. The Company expects to continue to incur net losses into the foreseeable future.
Going Concern
As of December 31, 2016,2019, the Company had cash and cash equivalents of $48.8$34.5 million. AlthoughAdditionally, in February 2020, the Company has incurred recurring losses in each year since inception,entered into a Credit Agreement and Guaranty with Perceptive Credit Holdings III, LP, or the Perceptive Credit Agreement. A first tranche of $5.0 million was funded on execution of the Perceptive Credit Agreement. A second tranche of $15.0 million was funded as a result of the approval of Twirla by the FDA (see Note 15). The Company expectsbelieves that its cash and cash equivalents as of December 31, 2019 along with the proceeds of the Perceptive Credit Agreement received by the date of this report will be sufficient to meet its projected operating requirements through the end of 2020. The Company will require additional capital to fund its operating needs beyond 2020, which primarily will include commercializing Twirla, and exploring the advancement of its existing pipeline and its possible expansion through business development activities.
The Company anticipates it will continue to incur net losses for the foreseeable future and the Company's ability to continue operations beyond 2020 will depend on its ability to obtain additional capital, as to which no assurances can be given. There can be no assurance that any financing by the Company can be realized by the Company, or if realized, what the terms of any such financing may be, or that any amount that the Company is able to raise will be adequate. Based upon the foregoing,
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
1. Organization and Description of Business (Continued)
management has concluded that there is substantial doubt about the Company's ability to continue as a going concern through the 12 months following the date on which this Annual Report on Form 10-K is filed.
The Company continues to analyze various alternatives, including strategic and refinancing alternatives, asset sales and mergers and acquisitions. The Company's future success depends on its ability to raise additional capital and/or implement the various strategic alternatives discussed above. The Company cannot be certain that these initiatives or raising additional capital, whether through selling additional debt or equity securities or obtaining a line of credit or other loan, will be available to it or, if available, will be on terms acceptable to the Company. If the Company issues additional securities to raise funds, these securities may have rights, preferences, or privileges senior to those of its common stock, and the Company's current stockholders will experience dilution. If the Company is unable to obtain funds when needed or on acceptable terms, the Company then may be unable to complete the commercialization of Twirla, and may also be required to cut operating costs, and forego future development and other opportunities.
The audited financial statements as of December 31, 2019 have been prepared under the assumption that the Company will continue as a going concern for at least the next twelve12 months. The Company's ability to continue as a going concern is dependent upon its uncertain ability to obtain additional capital, reduce expenditures and/or execute on its business plan and successfully launch Twirla. The audited financial statements as of December 31, 2019 do not include any adjustments that might result from the outcome of this uncertainty. If the Company is unable to continue as a going concern, it may have to liquidate its assets and may receive less than the value at which those assets are carried on the financial statements.
2. Summary of Significant Accounting Polices
Basis of Presentation
The accompanying financial statements have been prepared in accordance with United States ("U.S.") generally accepted accounting principles ("U.S. GAAP") and include all adjustments necessary for the fair presentation of the Company's financial position for the periods presented.
Use of Estimates
The preparation of the Company's financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. The Company bases its estimates and judgments on historical experience and various other assumptions that it believes are reasonable under the circumstances. The amounts of assets and liabilities reported in the Company's balance sheets and the amounts of expenses reported for each of the periods presented are affected by estimates and assumptions, which are used for, but not limited to, the accounting for common stock warrants, stock-based compensation, income taxes, and accounting for research and development costs. Actual results could differ from those estimates.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 20162019
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
Risks and Uncertainties
ProductWhile Twirla has been approved by the FDA, other potential product candidates developed by the Company typically will require approval from the FDA prior to commercial sales. There can be no assurance that the Company's other product candidates will receive the required approval. If the Company wasis denied approval or such approval wasis delayed, or is unable to obtain the necessary financing to complete development and approval, there willcould be a material adverse impact on the Company's financial condition and results of operations.
Stock Split
On May 5, 2014, the Company effected a 1.4-for-1 stock split of the Company's common stock. All share and per share amounts of common stock contained in the Company's financial statements have been restated for all periods to give retroactive effect to the stock split. The shares of common stock retained a par value of $0.0001 per share. Accordingly, the stockholders' deficit reflects the stock split by reclassifying from "Additional paid-in Capital" to "Common Stock" in an amount equal to the par value of the increased shares resulting from the stock split.
Cash and Cash Equivalents
The Company considers all highly-liquid investments with an original maturity of three months or less when purchased to be cash equivalents. All cash and cash equivalents are held in United States financial institutions. Cash and cash equivalents include money market funds that invest primarily in commercial paper and U.S. government and U.S. government agency obligations.
The Company maintains balances with financial institutions in excess of the FDICFederal Deposit Insurance Corporation limit.
Fair Value of Financial Instruments
In accordance with ASCAccounting Standards Codification ("ASC") 825,Financial Instruments, disclosures of fair value information about financial instruments are required, whether or not recognized in the balance sheet, for which it is practicable to estimate that value. Cash and cash equivalents are carried at fair value (see Note 3).
FinancialOther financial instruments, including accounts payable and accrued liabilities, are carried at cost, which approximates fair value given their short-term nature.
Property and Equipment
Property and equipment, consisting of manufacturing, office and computer equipment, is stated at cost, less accumulated depreciation. Depreciation is computed using the straight-line, method over the estimated useful lives of the assets.
Expenditures incurred after the fixed assets have been put into operation, such as repairs and maintenance, are charged to earnings in the period in which costs are incurred. Improvements and additions are capitalized in accordance with Company policy.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
Long-Lived Assets
In accordance with ASC 360,Property, Plant and Equipment, the Company's policy is to review long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Management does not believe that there has been any impairment of the carrying value of any long-lived assets as of December 31, 2016.2019.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
Research and Development Expense
Research and development costs are expensed as incurred. Research and development expense consists primarily of costs related to personnel, including salaries and other personnel-related expenses, expenses related to manufacturing, clinical trial expenses, consulting fees and support services used in drug development. All research and development costs are charged to operations as incurred in accordance with ASC 730,Research and Development.
In certain circumstances, the Company is required to make advance payments to vendors for goods or services that will be received in the future for use in research and development activities. In such circumstances, the advance payments are deferred and are expensed when the activity has been performed or when the goods have been received.
Deferred Financing Costs
Costs directly attributable to the Company's term loan (see Note 8) are deferred and reported as a reduction of the related term loan. These costs represent legal fees and other costs related to the term loan and are being amortized over the term of the loan. Amortization of deferred financing costs charged to interest expense was $256, $211approximately $0, $133 and $59$239 for the years ended December 31, 2016, 20152019, 2018 and 2014,2017, respectively.
Concentrations of Credit Risk
Financial instruments which potentially subject the Company to credit risk consist principally of cash and cash equivalents. All cash and cash equivalents are held in business checking and money market accounts in United States financial institutions the balances of which exceed federally insured limits. The Company has not recognized any losses from credit risks on such accounts. The Company believes it is not exposed to significant credit risks on cash and cash equivalents. The Company has no financial instruments with off-balance sheet risk of accounting loss.
Warrants
The Company accounts for its warrants to purchase redeemable convertible stock in accordance with ASC 480,Distinguishing Liabilities from Equity. ASC 480 requires that a financial instrument, other than an outstanding share, that, at inception, is indexed to an obligation to repurchase the issuer's equity shares, regardless of the timing or the probability of the redemption feature and may require the issuer to settle the obligation by transferring assets be classified as a liability. The Company measures the fair
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
value of its warrant liability using the Black-Scholes option pricingoption-pricing model with changes in fair value recognized as increases or reductions to other income (expense) in the statement of operations.
In connection with the completion of the Company's initial public offering in May 2014, the warrants to purchase shares of Series A-1 and Series A-2 preferred stock expired unexercised and the warrants to purchase shares of Series C preferred stock automatically converted into warrants to purchase shares of common stock. Warrants with non-standard anti-dilution provisions (referred to as down round protection) are classified as liabilities and re-measured each reporting period. AsOn
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016, there were outstanding2019
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
January 1, 2019, the Company adopted the provisions of Accounting Standards Update ("ASU") 2017-11Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part 1) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Non-controlling Interests with a Scope Exception, which indicates that a down round feature no longer precludes equity classification when assessing whether an investment is indexed to an entity's own stock. The Company used a modified retrospective approach to adoption, which does not restate its financial statements as of the prior year end (December 31, 2018). The cumulative effect of adoption of ASU 2017-11 resulted in an adjustment to accumulated deficit as of January 1, 2019 of $213 with a corresponding adjustment to additional paid-in capital. Warrants to purchase 62,505 warrants to purchaseshares of common stock at $6.00 per share. These warrants expireshare expired on December 14, 2019, and none of these warrants are outstanding as of December 31, 2019.
The warrants issued in connection with the Company's debt financing completed in February 2015 (see Note 8) are classified as a component of stockholders' equity. The value of such warrants was determined using the Black-Scholes option-pricing model. As of December 31, 2019, there were outstanding 180,274 warrants to purchase common stock at $5.89 per share related to this debt financing. These warrants expire on February 24, 2020.
Income Taxes
The Company accounts for deferred taxes using the asset and liability method as specified by ASC 740,Income Taxes. Deferred income tax assets and liabilities are determined based on differences between the financial statement reporting and the tax basis of assets and liabilities, operating losses and tax credit carryforwards. Deferred income taxes are measured using the enacted tax rates and laws that are anticipated to be in effect when the differences are expected to reverse. The measurement of deferred income tax assets is reduced, if necessary, by a valuation allowance for any tax benefits which are not expected to be realized. The effect on deferred income tax assets and liabilities of a change in tax rates is recognized in the period that such tax rate changes are enacted.
The Company has adopted the authoritative guidance on accounting for and disclosure of uncertainty in tax positions which prescribes a comprehensive model for the financial statement recognition, measurement, presentation and disclosure of uncertain tax positions taken or expected to be taken in income tax returns. The Company has no uncertain tax positions as of December 31, 20162019 that qualifies for either recognition or disclosure in the financial statements under this guidance.
Stock-Based Compensation
The Company accounts for stock-based compensation in accordance with ASC 718,Compensation—Stock Compensation. The Company grants stock options for a fixed number of shares to employees and non-employees with an exercise price equal to the fair value of the shares at grant date. Compensation cost is recognized for all share-based payments granted and is based on the grant-date fair value estimated using the weighted-average assumption of the Black-Scholes option pricing model based on key assumptions such as stock price, expected volatility and expected term. The Company
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
elects to account for forfeitures when they occur. The equity instrument is not considered to be issued until the instrument vests. As a result, compensation cost is recognized over the requisite service period with an offsetting credit to additional paid-in capital.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
The Company also awards restricted stock units ("RSUs") to employees.employees and its board of directors. RSUs are generally subject to forfeiture if employment terminates prior to the completion of the vesting restrictions. The Company expenses the cost of the RSUs, which is determined to be the fair market value of the shares of common stock underlying the RSUs at the date of grant, ratably over the period during which the vesting restrictions lapse.
Awards for consultants are accounted for under ASC 505-50,Equity Based Payments to Non-Employees. Any compensation expense related to consultants Cost associated with performance-based restricted stock units with a performance condition which affects the vesting is marked-to-market overrecognized only if the applicable vesting period as they vest.
Prior to May 22, 2014, the Company utilized various methodologies in accordance with the frameworkperformance condition is probable of the American Institute of Certified Public Accountants Technical Practice Aid,Valuation of Privately-Held Company Equity Securities Issued as Compensation, to estimate the fair value of its stock. The methodologies included an option pricing method and a probability-weighted expected return methodology that determined an estimated value under an initial public offering (IPO) scenario and a sale scenario based upon an assessment of the probability of occurrence of each scenario. Each valuation methodology includes estimates and assumptions that require the Company's judgment. These estimates include assumptions regarding future performance, including the successful completion of clinical trials and the time to completing an IPO or sale of the Company. As with any valuation, significant changes to the key assumptions used in the valuations could result in different fair values of common stock at each valuation date.being satisfied.
Segment Information
Operating segments are defined as components of an enterprise about which separate discrete information is available for evaluation by the chief operating decision maker, or decision makingdecision-making group, in deciding how to allocate resources and in assessing performance. The Company views its operations and manages its business in one operating and reporting segment, which is the business of developing its transdermal patch for use in contraception.
Net Loss Per Share
Basic net loss per share is calculated by dividing the net loss attributable to common stockholders by the weighted average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted net loss per share is calculated by dividing the net loss attributable to common stockholders by the weighted-average number of common shares outstanding plus the effect of dilutive potential common shares outstanding during the period determined using the treasury-stock and if-converted methods. For purposes of diluted net loss per share calculation, common stock warrants, unvested RSUs and stock options are considered to be potentially dilutive securities but are excluded from the calculation of diluted net loss per share because their effect would be anti-dilutive and therefore, basic and diluted net loss per share were the same for all periods presented.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
The following table sets forth the outstanding potentially dilutive securities that have been excluded from the calculation of diluted net loss per share for the years ended December 31, 2019, 2018 and 2017, respectively, because to do so would be anti-dilutive (in common equivalent shares):
| | Year Ended December 31, | Year Ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | 2019 | 2018 | 2017 | ||||||||||||||
Convertible preferred stock | — | — | — | |||||||||||||||||
Convertible preferred stock warrants | — | — | — | |||||||||||||||||
Common stock warrants | 242,779 | 242,779 | 62,505 | 180,274 | 242,779 | 242,779 | ||||||||||||||
Unvested restricted stock units | 33,334 | — | — | — | 147,554 | 264,361 | ||||||||||||||
Common stock options | 2,844,970 | 2,165,065 | 1,817,548 | 7,192,357 | 5,687,901 | 3,805,305 | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Total | 3,121,083 | 2,407,844 | 1,880,053 | 7,372,631 | 6,078,234 | 4,312,445 | ||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
Recent Accounting Pronouncements
In August 2014, From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2014-15,Presentation of Financial Statements—Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern, which defines management's responsibility to assess an entity's ability to continue as a going concern, and to provide related footnote disclosures if there is substantial doubt about its ability to continue as a going concern. The new("FASB") or other standard is effective for the annual period ending after December 15, 2016, and for interim periods thereafter. The Company adopted ASU 2014-15 in the fourth quarter of 2016, which resulted in no change to the Company's financial statements. Additionally,setting bodies that the Company will perform quarterly evaluations to identify current conditions which may raise substantial doubt aboutadopts as of the entity's ability to continue as a going concern within one year afterspecified effective date. Unless otherwise discussed below, the dateCompany does not believe that the adoption of recently issued standards have or may have a material impact on our consolidated financial statements are issued.
In April 2015, the FASB issued an amendment to U.S. GAAP to simplify the balance sheet presentation of the costs for issuing debt. The changes were adopted in ASU No. 2015-03,Interest—Imputation of Interest (Subtopic 835-30): Simplifying the Presentation of Debt Issue Costs. ASU 2015-03 amends current presentation guidance by requiring that debt issuance costs related to a recognized debt liability be presented in the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts. Prior to the issuance of ASU 2015-03, debt issuance costs were required to be presented as an asset in the balance sheet. The Company adopted the provisions of ASU 2015-03 on January 1, 2016 and prior period balances have been reclassified to conform to the current period presentation. As of December 31, 2015, $152 of debt issuance costs were reclassified in the balance sheet from "deferred financing costs, net" to "loan payable, current portion" and $139 was reclassified from "deferred financing costs, net" to "loan payable, long-term". The adoption of ASU 2015-03 did not have an impact on the Company's operations or cash flows.disclosures.
In February 2016, the FASB issued ASU No. 2016-02,Leases. The new standard establishes a right-of-use (ROU) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
or operating, with classification affecting the pattern of expense recognition in the statement of operations. The new standard is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. A modified retrospective transition approach is required for leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements. The Company will be evaluatingadopted ASU No. 2016-02 on January 1, 2019. The Company recorded a lease asset and lease liability of approximately $0.3 million on its balance sheet as of January 1, 2019, with no impact on its statement of operations.
In July 2017, the impactFASB issued ASU No. 2017-11,Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part 1) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the pendingIndefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception. This ASU eliminates the requirement to consider "down round" features when determining whether certain equity-linked financial instruments or embedded features are indexed to an entity's own stock. On January 1, 2019, the Company adopted the provisions of ASU No. 2017-11 using a modified retrospective approach, which does not restate its financial statements as of the prior year end (December 31, 2018). The cumulative effect of adoption of ASU 2017-11 resulted in an adjustment to accumulated deficit as of January 1, 2019 of $213 with a corresponding adjustment to additional paid-in capital. As a result of the new standardadoption of ASU 2017-11, effective January 1, 2019, the Company no longer measures these warrants at fair value.
In May 2017, the FASB issued ASU 2017-09, Compensation—Stock Compensation (Topic 718): Scope of Modification Accounting to provide clarity and reduce both (1) diversity in practice and (2) cost and complexity when applying the guidance in Topic 718, Compensation—Stock Compensation, to change the terms or conditions of a share-based payment award. The amendments in this ASU provide guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. This Update is the final version of Proposed ASU 2016-360—Compensation—Stock Compensation (Topic 718)—Scope of Modification Accounting, which has been deleted. The amendments in this ASU are effective for all entities for annual periods, and interim periods within those annual periods, beginning after December 15, 2017. The adoption of this ASU did not have a material impact on the Company's financial statements.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
2. Summary of Significant Accounting Polices (Continued)
In MarchJune 2016, the FASB issued ASU No. 2016-09,Improvements2016-13, Financial Instruments—Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments ("ASU 2016-13"), which amends the impairment model by requiring entities to Employee Share-Based Payment Accounting. Thisuse a forward-looking approach based on expected losses rather than incurred losses to estimate credit losses on certain types of financial instruments, including trade receivables. ASU requires all tax effects of share-based payment settlements to be recorded through2016-13 was adopted by the statement of operations. Currently, tax benefits in excess of compensation cost, or "windfalls", are recorded in equity,Company on January 1, 2020 and tax deficiencies, or "shortfalls", are recorded to equity to the extent of previous windfalls, and then to the statement of operations. In addition, under the new guidance, companies will be permitted to make a policy election to recognize the impact of forfeitures either when they occur, or on an estimated basis. Finally, this update simplifies withholding requirements to allow companies to withhold up to an employee's maximum tax rate without resulting in liability classification for the award. ASU 2016-09 is effective for annual reporting periods beginning after December 15, 2016, and early adoption is permitted. The Company has adopted the provisions of this standard early and theno current impact on itsthe Company as we do not have any financial statements wasinstruments covered by the topic.
Management does not material.believe that any other recently issued, but not yet effective accounting pronouncements, if adopted, would have a material impact on the accompanying financial statements.
3. Fair Value Measurements
ASC 820,Fair Value Measurements and Disclosures, describes the fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value.
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants at the measurement date. Assets and liabilities that are measured at fair value are reported using a three-level fair value hierarchy that prioritizes the inputs used to measure fair value. This hierarchy maximizes the use of observable inputs and minimizes the use of unobservable inputs. The three levels of inputs used to measure fair value are as follows:
The Company is required to mark the value of its warrant liability to market and recognize the change in valuation in its statements of operations each reporting period.
The following table setstables set forth the Company's financial instruments measured at fair value by level within the fair value hierarchy as of December 31, 20162019 and 2015.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
3. Fair Value Measurements (Continued)2018:
| | Level 1 | Level 2 | Level 3 | Level 1 | Level 2 | Level 3 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
2016 | ||||||||||||||||||||
2019 | ||||||||||||||||||||
Assets: | ||||||||||||||||||||
Cash equivalents | $ | 48,659 | $ | — | $ | — | $ | 34,444 | $ | — | $ | — | ||||||||
| | | | | | | | | | | | | | | | | | | | | |
Total assets at fair value | $ | 48,659 | $ | — | $ | — | $ | 34,444 | $ | — | $ | — | ||||||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Liabilities: | ||||||||||||||||||||
Common stock warrants | — | — | 172 | |||||||||||||||||
| | | | | | | | | | | |||||||||||
Total liabilities at fair value | $ | — | $ | — | $ | 172 | ||||||||||||||
| | | | | | | | | | | |||||||||||
| | | | | | | | ||||||||||||||
| | | | | | | | | | | |||||||||||
The significant assumptions used in preparing the option pricing model for valuing the Company's warrants as of December 31, 2016 include (i) volatility (75.0%), (ii) risk free interest rate of 1.47% (estimated using treasury bonds with a 3 year life), (iii) strike price ($6.00) for the common stock warrants, (iv) fair value of common stock ($5.70) and (v) expected life (three years).
The following is a roll forward of the fair value of Level 3 warrants:
Beginning balance at December 31, 2013 | $ | 644 | ||
Expiration of Series A-1 and Series A-2 warrants | (493 | ) | ||
Change in fair value | 145 | |||
| | | | | |
Ending balance at December 31, 2014 | 296 | |||
Change in fair value | 110 | |||
| | | | | |
Ending balance at December 31, 2015 | 406 | |||
Change in fair value | (234 | ) | ||
| | | | | |
Ending balance at December 31, 2016 | $ | 172 | ||
| | | | | |
| | | | | |
| | | | | |
| | Level 1 | Level 2 | Level 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
2015 | ||||||||||
Assets: | ||||||||||
Cash equivalents | $ | 34,324 | $ | — | $ | — | ||||
| | | | | | | | | | | |
Total assets at fair value | $ | 34,324 | $ | — | $ | — | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Liabilities: | ||||||||||
Common stock warrants | — | — | 406 | |||||||
| | | | | | | | | | | |
Total liabilities at fair value | $ | — | $ | — | $ | 406 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The significant assumptions used in preparing the option pricing model for valuing the Company's warrants as of December 31, 2015 include (i) volatility (75.0%), (ii) risk free interest rate of 1.54% (estimated using treasury bonds with a 4 year life), (iii) strike price ($6.00) for the common stock warrants, (iv) fair value of common stock ($9.76) and (v) expected life (four years).
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 20162019
(in thousands, except share and per share data)
3. Fair Value Measurements (Continued)
| | Level 1 | Level 2 | Level 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
2018 | ||||||||||
Assets: | ||||||||||
Cash equivalents | $ | 7,776 | $ | — | $ | — | ||||
| | | | | | | | | | | |
Total assets at fair value | $ | 7,776 | $ | — | $ | — | ||||
| | | | | | | | | | | |
| | ��� | | | | | | | | | |
| | | | | | | | | | | |
The following is a roll forward of the fair value of Level 3 warrants:
Beginning balance at December 31, 2016 | $ | 172 | ||
Change in fair value | (143 | ) | ||
| | | | | |
Ending balance at December 31, 2017 | 29 | |||
Change in fair value | (29 | ) | ||
| | | | | |
Ending balance at December 31, 2018 | — | |||
Change in fair value | — | |||
| | | | | |
Ending balance at December 31, 2019 | $ | — | ||
| | | | | |
| | | | | |
| | | | | |
There were no transfers between Level 1, 2 or 3 during 20162019 or 2015. If the Company's estimates regarding the fair value of its warrants are inaccurate, a future adjustment to these estimated fair values may be required. Additionally, these estimated fair values could change significantly.2018.
4. Prepaid Expenses
Prepaid expenses consist of the following:
| | December 31 | December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2019 | 2018 | ||||||||||
Prepaid clinical trial expense | $ | 2,005 | $ | 2,803 | ||||||||||
Prepaid insurance | 665 | 780 | $ | 656 | $ | 484 | ||||||||
Other | 98 | 107 | 184 | 123 | ||||||||||
| | | | | | | | | | | | | | | |
Total prepaid expenses | $ | 2,768 | $ | 3,690 | $ | 840 | $ | 607 | ||||||
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
5. Property and Equipment
Property and equipment, consisting of manufacturing, office and computer equipment, is stated at cost, less accumulated depreciation. Depreciation is computed using the straight-line, method over the estimated useful lives of the assets. Property and equipment consist of the following:
| | December 31 | | December 31, | | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Estimated Life | Estimated Life | ||||||||||||||
| | 2016 | 2015 | 2019 | 2018 | ||||||||||||
Office equipment | $ | 55 | $ | 55 | 3 - 10 years | $ | 49 | $ | 49 | 3 - 10 years | ||||||
Computer equipment | 133 | 106 | 3 years | 179 | 175 | 3 years | ||||||||||
Manufacturing equipment | 12,465 | 12,461 | 5 years | 14,203 | 14,061 | 5 years | ||||||||||
| | | | | | | | | | | | | | | | | |
| 12,653 | 12,622 | 14,431 | 14,285 | |||||||||||||
Less: accumulated depreciation | (323 | ) | (304 | ) | (387 | ) | (369 | ) | ||||||||
| | | | | | | | | | | | | | | | | |
Property and equipment, net | $ | 12,330 | $ | 12,318 | ||||||||||||
Property and equipment | $ | 14,044 | $ | 13,916 | ||||||||||||
| | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
As of December 31, 20162019, and 2015,2018, manufacturing equipment includes approximately $12.4$14.0 million and $13.9 million, respectively, of equipment which is in the process of being constructed and qualified and is not currently being depreciated.
6. Accrued Liabilities
Accrued liabilities consist of the following:
| | December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2019 | 2018 | |||||
Employee bonuses | $ | 1,437 | $ | 621 | |||
Accrued retention bonus | — | 638 | |||||
Accrued professional fees and other | 367 | 84 | |||||
| | | | | | | | |
Total accrued liabilities | $ | 1,804 | $ | 1,343 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
7. Leases
In February 2016, the FASB issued ASU No. 2016-02,Leases. The new standard establishes a right-of-use (ROU) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases are classified as either finance or operating, with classification affecting the pattern of expense recognition in the statement of operations. The Company adopted ASU No. 2016-02 on January 1, 2019 for leases that existed on that date. The Company has elected to apply the provisions of ASC 842 modified retrospectively at January 1, 2019 through a cumulative-effect adjustment. Prior period results continue to be presented under ASC 840 based on the accounting standards originally in effect for such periods. The company recorded a lease asset and lease liability of approximately $0.3 million on its balance sheet as of January 1, 2019, with no impact on its statement of operations.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 20162019
(in thousands, except share and per share data)
6. Accrued Liabilities7. Leases (Continued)
Accrued liabilities consistThe Company has no finance leases and one operating lease for office space in Princeton, NJ. Operating lease expense was $193 for the year ended December 31, 2019.
Operating cash flows used for operating leases during the year ended December 31, 2019 were $152. As of December 31, 2019, the following:weighted-average remaining lease term was 0.92 years and the weighted average discount rate was 21.2%.
Future minimum lease payments under non-cancellable leases as of December 31, 2019 were as follows:
| | December 31 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | |||||||||
Employee bonuses | $ | 1,041 | $ | 938 | |||||||
Accrued clinical trial costs | 1,908 | 1,507 | |||||||||
Accrued professional fees and other | 403 | 208 | |||||||||
2020 | $ | 191 | |||||||||
| | | | | | | | | | | | |
Total accrued liabilities | $ | 3,352 | $ | 2,653 | |||||||
Total | $ | 191 | |||||||||
Less: Interest | (19 | ) | |||||||||
| | | | | | | | | | | | |
Present value of lease liability | $ | 172 | |||||||||
| | | | | | | | | | | ||
| | | | | | | | |||||
7. Convertible Note Financing
On April 28, 2014, the Company and certain of the Company's existing preferred stockholders, all of whom qualify as accredited institutional investors, entered into a Convertible Subordinated Note Purchase Agreement pursuant to which such holders agreed to loan the Company an aggregate of $3.0 million. The Company issued Convertible Promissory Notes (the "Notes") to evidence its payment obligations with respect to the $3.0 million. The Notes had an interest rate of 8%, accruing daily and compounding annually. The Notes are convertible into unregistered equity securities of the Company upon the occurrence of events stated therein. The Notes and accrued interest automatically converted into 503,450 shares of common stock at $6.00 per share which was equal to the purchase price at which shares were sold to the public in an underwritten public offering (see Note 9). The Notes were subordinate to the Company's term loan with Oxford Finance LLC.
8. Loan and Security Agreements
Oxford Finance LLC
In December 2012, the Company entered into a Loan and Security Agreement (the "Oxford Loan") with Oxford Finance LLC ("Oxford") pursuant to which the Company borrowed a total of $15.0 million from Oxford. The Oxford Loan accrued interest at a fixed annual rate equal to 9.20% (Three-month U.S. Libor rate of 0.47% plus 8.73%).
Interest on the Oxford Loan was payable monthly and principal was due in 30 equal consecutive monthly installments beginning on February 1, 2015 and ending on July 1, 2017. In addition, the Company was required to make a final payment of $675 on the maturity date of the Oxford Loan (July 1, 2017).
In connection with the Oxford Loan, the Company issued Oxford warrants to purchase 62,505 shares of common stock at $6.00 per share. These warrants expire on December 14, 2019.
In February 2015, the Company terminated and repaid all amounts outstanding under the Oxford Loan and recorded a loss on the extinguishment of the Oxford Loan (see further discussion below).
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
8. Loan and Security Agreements (Continued)
Hercules Capital, Inc.
In February 2015, the Company entered into a loan and security agreement (the "Hercules Loan"Loan Agreement") with Hercules Capital, Inc. ("Hercules") for a term loan of up to $25.0 million. In August 2016, the Company entered into the First Amendment to Loan and Security Agreement (the "First Amendment") with Hercules which amendsamended certain terms of the Hercules Loan.Loan Agreement. In May 2017, the Company entered into the Second Amendment to Loan and Security Agreement (the "Second Amendment") with Hercules which further amended certain terms of the Hercules Loan Agreement. A first tranche of $16.5 million was funded upon execution of the Hercules Loan Agreement, approximately $15.5 million of which was used to repay the Company's existingprevious term loan with Oxford.Oxford Finance LLC.
The First Amendment extendsextended the Company's option to draw down the second tranche of $8.5 million (the "Second Term Loan Advance") of the term loan facility provided under the Hercules Loan Agreement (the "Term Loan") until March 31, 2017 and makesmade the Second Term Loan Advance subject to the consent of Hercules, among other customary conditions. The Company is currently in discussions withSecond Amendment further extended the Company's option to draw the Second Term Loan Advance until January 31, 2018 and continued to make the Second Term Loan Advance subject to the consent of Hercules, to extend the period beyond March 31, 2017 during which the additional tranche of $8.5 million may be drawn.among other customary conditions. The First Amendment also extendsextended the interest-only payments until January 31, 2017, in connection with the first tranche of $16.5 million (the "First Term Loan Advance" and together with the Second Term Loan Advance, the "Term Loan Advances"). The First Amendment also providesperiod during which the Companyadditional tranche of $8.5 million may be drawn has expired and therefore the ability to extend further the interest-only payments for two successive periods as follows: (i) until April 30, 2017, subject to the Company successfully completing its SECURE clinical trial and the Company receiving data that supports the filing of a response to the U.S. Food and Drug Administration's complete response letter relating to the new drug application filed$8.5 million can no longer be drawn by the Company ("First Interest Only Period Extension") and (ii) until July 31, 2017, provided that (x) the Company has received the First Interest Only Period Extension and (y) the Company has received unrestricted gross cash proceeds in aggregate amount greater than or equal to $40.0 million from the issuance and sale by the Company of its equity securities ("Second Interest Only Period Extension").Company.
The First Amendment providesprovided the Term Loan will maturematured on December 1, 2018; provided, however, that if the First Interest Only Period Extension occurs on or prior to January 31, 2017, the Term Loan will mature on March 1, 2019; and provided further, however that if both (a) The First Interest Only Period Extension occurs on or prior to January 31, 2017, and (b) the Second Interest Only Period Extension occurs on or prior to April 30, 2017, the Term loan will mature on June 1, 2019.
2018. As a result of the First Amendment, and in connection with the extension of the interest-only period from the First Term Loan Advance, Hercules returned to the Company the principal payments paid by the Company in July and August 2016, which such returned payments will once again constitute outstanding Term Loan advances under the Hercules Loan. In connection with the execution of the First Amendment, the Company paid Hercules a facility fee of $165. The facility fee represents a debt issue cost which is being reflected as a reduction to the carrying amount of loan payable in accordance with ASU 2015-03. Such issue costs are being amortized to interest expense over the life of the loan using the effective interest method.
The Hercules Loan accrues interest at a rate of the greater of 9.0% or 9.0% plus Prime minus 4.25% and is payable monthly. Principal is due in 23 equal consecutive monthly installments beginning on February 1, 2017 and ending on December 1, 2018. In addition, the Company is required to make a
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 20162019
(in thousands, except share and per share data)
8. Loan and Security AgreementsAgreement (Continued)
and August 2016, which such returned payments once again constituted outstanding Term Loan advances under the Hercules Loan Agreement. In connection with the execution of the First Amendment, the Company paid Hercules a facility fee of $165. The facility fee represented a debt issue cost which was reflected as a reduction to the carrying amount of the loan payable in accordance with ASU 2015-03. Such issue costs were amortized to interest expense over the life of the Term Loan using the effective interest method. As of December 31, 2019, and 2018, the Company had no outstanding borrowings related to the Hercules Loan Agreement.
The Term Loan accrued interest at a rate of the greater of 9.0% or 9.0% plus Prime minus 4.25% and was payable monthly. Principal was due in 23 consecutive monthly installments beginning on February 1, 2017 and ending on December 1, 2018. In addition to the outstanding principal balance, the Company was required to make a final payment of $610.5approximately $611 on the maturity date of the Hercules Loan (December 1, 2018). The amount of the end of term final payment is beingwas accrued over the loan term as interest expense.
The Company may prepay all, but not less than all, of the Hercules Loan subject to a prepayment premium of 2.0% of the outstanding principal if prepaid during the second year (through February 24, 2017) and 1.0% of the outstanding principal if prepaid after February 24, 2017. The obligations of the Company under the Hercules Loan areAgreement were secured by a perfected first position lien on all of the assets of the Company, excluding intellectual property assets.
In connection with the Hercules Loan Agreement, the Company issued Hercules a warrant to purchase 180,274 shares of the Company's common stock at an exercise price of $5.89 per share which expires on February 24, 2020 and granted Hercules the right to participate in future equity financings in an amount up to $2.0 million while the loan andor warrant are outstanding.
The Company allocated the proceeds of $16.5 million in accordance with ASC 470 based on the relative fair values. The relative fair value of the warrants of approximately $1.2 million at the time of issuance, which was determined using the Black-Scholes option-pricing model, was recorded as additional paid-in capital and reduced the carrying value of the debt. The significant assumptions used in preparing the option pricing model for valuing the Company's warrant issued to Hercules include (i) volatility (75.0%), (ii) risk free interest rate of 1.22% (estimated using treasury bonds with a 4 year4-year life), (iii) strike price ($5.89) for the common stock warrant, (iv) fair value of common stock ($9.82) and (v) expected life (four(4 years). The discount on the debt is beingwas amortized to interest expense over the term of the debt.
As a result of the repayment of the Oxford Loan, the Company recorded a loss on the extinguishment of debt of approximately $1.0 million on the Company's statement of operations for the year ended December 31, 2015, representing a prepayment premium, the unamortized discount of the Oxford Loan and the write off of deferred financing costs.
Interest expense on the OxfordHercules Loan and the Hercules LoanAgreement including the accretion of the value of the related warrants, accrual of term loan back-end fee and amortization of the deferred financing costs was approximately $2,446, $2,077$0, $1.1 million and $1,545,$1.9 million, for the years ended December 31, 2016, 20152019, 2018 and 2014,2017, respectively.
The annual maturities of the Hercules Loan, as of December 31, 2016, are as follows:
2017 | $ | 5,612 | ||
2018 | 10,888 | |||
| | | | | |
Total | $ | 16,500 | ||
| | | | | |
| | | | | |
| | | | | |
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
9. Stockholders' Equity
Initial Public Offering and Related Transactions
On May 29, 2014, the Company completed its initial public offering selling 9,166,667 shares of common stock at $6.00 per share. Proceeds from the Company's initial public offering, net of underwriting discounts and commissions and other offering costs, were $49.7 million.
In addition, each of the following occurred in connection with the completion of the Company's IPO on May 29, 2014:
On May 7, 2014, the Company filed an amendment to its amended and restated certificate of incorporation which, among other things, revised the automatic conversion provision relating to the Series C Preferred Stock, Series B Preferred Stock, Series A-1 Preferred Stock and Series A-2 Preferred Stock. Following such amendment, the Series C, the Series B, the Series A-1 and A-2 Preferred Stock automatically converted into shares of common stock at the then effective conversion price upon:
(i) the closing of an underwritten public offering pursuant to an effective registration statement under the Securities Act of 1933, covering the offer and sale of common stock from which the Company receives gross proceeds of at least $45,000,000 or (ii) the affirmative vote of the holders of at least a majority of the voting power the Series C Preferred Stock, the Series B Preferred Stock and the Series A-1 Preferred Stock, respectively, after first giving effect, if in conjunction with a public offering which does not meet the standards set forth in clause (i) above, to any adjustment of the conversion price for each series of preferred stock to which it would otherwise be entitled by virtue of such public offering.
On May 29, 2014, the Company filed an amended and restated certificate of incorporation (the "Restated Certificate") with the Secretary of State of the State of Delaware in connection with the closing of the Company's initial public offering of shares of its common stock. The Company's boardCertificate of directors (the "Board") and stockholders previously approved the Restated Certificate effective as of and contingent upon the closing of the Company's initial public offering.
The Restated Certificate amends and restates in its entirety the Company's second amended and restated certificate of incorporation, as amended. The Restated Certificate,Incorporation, among other things: (i) authorizes 150,000,000 shares of common stock; (ii) eliminates all references to the previously existing series of preferred stock; (iii) authorizes 10,000,000 shares of undesignated preferred stock that may be issued from time to time by the Board in one or more series; (iv)(iii) provides that the Board be divided into three classes with staggered three-year terms, with one class of directors to be elected at each annual meeting of the Company's stockholders; (v)(iv) provides that directors may only be removed with cause and only upon the affirmative vote of holders of at least 75% of the voting power of all
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 20162019
(in thousands, except share and per share data)
9. Stockholders' Equity (Continued)
and only upon the affirmative vote of holders of at least 75% of the voting power of all then-outstanding shares of capital stock of the Company entitled to vote generally in the election of directors; (vi)(v) provides that only the Board, the chairman of the Board if one is appointed, or the chief executive officer may call a special meeting of stockholders; and (vii)(vi) requires that any action instituted against the Company's officers or directors in connection with their service to the Company be brought in the State of Delaware.
2015 Private Placement of Common Stock
In January 2015, the Company completed a private placement of approximately 3.4 million shares of common stock at $5.85 per share. Proceeds from the Company's private placement, net of commissions and other offering costs, were approximately $19.3 million. Two of the Company's stockholders, who are also affiliated with members of the Company's Board of Directors, purchased a total of 1,623,932 shares of common stock for approximately $9.5 million in the private placement.
Shelf Registration StatementStatements
On June 19, 2015, the Company filed a universal shelf registration statement with the SEC for the issuance of common stock, preferred stock, warrants, rights, debt securities and units up to an aggregate amount of $150.0 million (the "2015 Shelf Registration Statement"). On July 1, 2015, the 2015 Shelf Registration Statement was declared effective by the SEC. The Company completed an offering of common stock in both January 2016 and August 2017 utilizing the 2015 Shelf Registration Statement. The 2015 Shelf Registration Statement (see Note 14)expired on June 30, 2018.
On November 2, 2018, the Company filed a universal shelf registration statement with the Securities and Exchange Commission ("SEC") for the issuance of common stock, preferred stock, warrants, rights, debt securities and units up to an aggregate amount of $100.0 million (the "2018 Shelf Registration Statement"). On November 14, 2018, the 2018 Shelf Registration Statement was declared effective by the SEC. In the future, the Company may also periodically offer one or more of these securities in amounts, prices and terms to be announced when and if the securities are offered. At the time any of the securities covered by the 20152018 Shelf Registration Statement are offered for sale, a prospectus supplement will be prepared and filed with the SEC containing specific information about the terms of any such offering.
On January 23, 2019, the Company filed a prospectus supplement to its 2018 Shelf Registration Statement registering an at-the-market offering program we entered into for the sale of up to $10.0 million of shares of our common stock. In the year ended December 31, 2019, the Company sold a total of 1,801,528 shares of our common stock under this ATM program resulting in net proceeds of approximately $2.5 million. We terminated this at-the-market offering program on July 31, 2019.
In August 2019, the Company filed a prospectus supplement to its 2018 Shelf Registration Statement registering a public offering of 14,526,315 shares of common stock at a price of $0.95 per share. Proceeds from the public offering, net of underwriting discounts, commissions and offering expenses, were approximately $12.7 million.
On November 8, 2019, the Company filed a prospectus supplement to its 2018 Shelf Registration Statement registering an at-the-market offering program we entered into for the sale of up to $20.0 million of shares of our common stock. In the year ended December 31, 2019, we sold a total of 10,440,908 shares of our common stock under this ATM program resulting in net proceeds of approximately $19.3 million.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
9. Stockholders' Equity (Continued)
Private Placement
In March 2019, the Company completed a private placement of 8,426,750 shares of common stock at $0.93 per share. Proceeds from the Company's private placement, net of offering costs were approximately $7.8 million.
2016 Public Offering of Common Stock
In January 2016, the Company completed an underwritten public offering of 5,511,812 shares of its common stock at a public offering price of $6.35 per share. In February 2016, the underwriters of the public offering of common stock exercised in full their option to purchase an additional 826,771 shares of common stock at the public offering price of $6.35 per share, less underwriting discounts and commissions. A total of 6,338,583 shares of common stock were sold in the public offering resulting in total net proceeds of approximately $37.5 million. One of the Company's stockholders, who is also affiliated with a member of the Company's Board, of Directors, purchased 393,700 shares of common stock for approximately $2.5 million in the public offering.
Convertible Preferred2017 Public Offering of Common Stock (Prior to IPO)
Prior to its conversion in the IPO, the Company's convertible preferred stock was classified as temporary equity on its balance sheets instead of stockholders' (deficit) in accordance with authoritative guidance for the classification and measurement or redeemable securities. Upon certain change in
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
9. Stockholders' Equity (Continued)
control events that were outside of the Company's control, including liquidation, sale or transfer of control ofIn August 2017, the Company holderscompleted an underwritten public offering of the convertible preferred5,333,334 shares of its common stock could cause its redemption.at a public offering price of $3.75 per share. Proceeds from this offering, net of underwriting discounts, commissions and other offering costs were approximately $18.5 million.
10. Equity Incentive Plans
Stock options
The Company had granted stock options under an amended and restated 1997 Equity Incentive Plan (the "1997 Plan") and a 2008 Equity Incentive Plan (the "2008 Plan"). The plans provided for the granting of incentive and non-statutory options and stock awards to consultants, directors, officers and employees. Such options are exercisable for a period of ten years and generally vest over a four-year period. In conjunction with the adoption of the 2008 Plan in April 2008, no additional grants were made from the 1997 Plan and issued options from the 1997 Plan remain outstanding. In 2014, the Company's Board of Directors approved the 2014 Equity Incentive Plan (the "2014 Plan"). The 2014 Plan is the successor to the Company's 2008 Plan and 1997 Plan. In conjunction with the adoption of the 2014 Plan in 2014, no additional grants were made from the 2008 Plan and options from the 1997 Plan and the 2008 Plan remain outstanding. In June 2018, the 2014 Plan was amended and restated, and the Amended and Restated 2014 Incentive Compensation is now referred to as the Amended 2014 Plan. As of December 31, 2016,2019, there were 605,3902,170,175 shares available for future grant under the Amended 2014 Plan.
Through December 31, 2016,2019, the Company granted options to certain employees and nonemployees to purchase shares of common stock at exercise prices ranging from $0.71$0.58 to $285.71$10.75 per share. The Company recorded non cash stock basednoncash stock-based compensation expense of $3,425, $2,965 and $1,381 for the years ended
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016, 20152019
(in thousands, except share and 2014, respectively,per share data)
10. Equity Incentive Plans (Continued)
December 31, 2019, 2018 and 2017 based on the fair market value of the options and shares granted at the grant date. Stock-based compensation expense was as follows:
| | Year Ended December 31, | Year Ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | 2019 | 2018 | 2017 | ||||||||||||||
Employee | $ | 3,456 | $ | 2,662 | $ | 1,185 | ||||||||||||||
Non-employee | (31 | ) | 303 | 196 | ||||||||||||||||
Research and development | $ | 522 | $ | 1,274 | $ | 1,184 | ||||||||||||||
General and administrative | 1,240 | 2,356 | 2,467 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | |
Total | $ | 3,425 | $ | 2,965 | $ | 1,381 | $ | 1,762 | $ | 3,630 | $ | 3,651 | ||||||||
| | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
Stock-based compensation expense was allocated as follows:
| | Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | |||||||
Research and development | $ | 1,063 | $ | 1,161 | $ | 617 | ||||
General and administrative | 2,362 | 1,804 | 764 | |||||||
| | | | | | | | | | | |
Total stock-based compensation expense | $ | 3,425 | $ | 2,965 | $ | 1,381 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
10. Equity Incentive Plans (Continued)
The following assumptions were used to compute employee stock-based compensation under the Black-Scholes option pricing model:
| | 2016 | 2015 | 2014 | 2019 | 2018 | 2017 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Risk-free interest rate | 1.48 | % | 1.92 | % | 1.84 | % | 1.74% - 2.61% | 2.57% | 2.27% | |||||||
Expected volatility | 75.0 | % | 75.0 | % | 104.8 | % | ||||||||||
Expective volatility | 65.0% | 70.0% | 73.9% | |||||||||||||
Expected dividend yield | 0 | % | 0 | % | 0 | % | 0% | 0% | 0% | |||||||
Expected life (in years) | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | ||||||||||
Risk-free interest rate. The Company bases the risk-free interest rate assumption on observed interest rates appropriate for the expected term of the stock option grants.
Expected dividend yield. The Company bases the expected dividend yield assumption on the fact that it has never paid cash dividends and has no present intention to pay cash dividends.
Expected volatility. The expected volatility assumption is based on volatilities of a peer group of similar companies whose share prices are publicly available. The peer group was developed based on comparable companies in the biotechnology and pharmaceutical industries.
Expected term. The expected term represents the period of time that options are expected to be outstanding. Because the Company does not have historic exercise behavior, management determined the expected life assumption using the simplified method, which is an average of the contractual term of the option and its ordinary vesting period.
Forfeitures. The Company has elected to record forfeitures as they occur.
As of December 31, 2016,2019, the unrecorded deferred stock-based compensation balance related to stock options was approximately $5.9$1.9 million and will be recognized over an estimated weighted-average amortization period of 1.711.4 years. The weighted average grant date fair value of options granted during the year ended December 31, 20162019 was $4.21.$0.59.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 20162019
(in thousands, except share and per share data)
10. Equity Incentive Plans (Continued)
The following table summarizes the options outstanding, options vested and the options exercisable as of December 31, 2016, 20152019, 2018 and 2014:2017:
| | Options | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Life (Years) | Aggregate Intrinsic Value | Options | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (Years) | Aggregate Intrinsic Value | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Options outstanding at December 31, 2014 | 1,817,548 | 5.56 | 7.9 years | ||||||||||||||||||||||
Options outstanding at December 31, 2017 | 3,805,305 | 5.74 | 7.4 | ||||||||||||||||||||||
Options granted | 620,600 | 9.84 | 2,230,000 | 1.96 | |||||||||||||||||||||
Options exercised | (261,936 | ) | 2.26 | — | — | ||||||||||||||||||||
Options cancelled/forfeited | (11,147 | ) | 2.14 | (347,404 | ) | 4.19 | |||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | |||||||
Options outstanding at December 31, 2015 | 2,165,065 | 7.19 | 7.8 years | ||||||||||||||||||||||
Options outstanding at December 31, 2018 | 5,687,901 | 4.34 | 7.4 | ||||||||||||||||||||||
Options granted | 825,500 | 2,805,600 | 1.18 | ||||||||||||||||||||||
Options exercised | (88,870 | ) | (92,271 | ) | 1.78 | ||||||||||||||||||||
Options cancelled/forfeited | (56,725 | ) | (1,208,873 | ) | 2.70 | ||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | |||||||
Options outstanding at December 31, 2016 | 2,844,970 | 7.50 years | $ | 1,772 | |||||||||||||||||||||
Options outstanding at December 31, 2019 | 7,192,357 | 3.42 | 7.2 | $ | 5,256 | ||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | ||||||
| | | | | | | | | | | | | | | | | | | | |||||||
Options exercisable at December 31, 2016 | 1,482,812 | 6.45 years | $ | 1,772 | |||||||||||||||||||||
Options exercisable at December 31, 2019 | 4,396,577 | 4.60 | 6.1 | $ | 2,228 | ||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | ||||||
| | | | | | | | | | | | | | | | | | | | |||||||
Vested and expected to vest at December 31, 2016 | 2,844,970 | 7.50 years | |||||||||||||||||||||||
Vested and expected to vest at December 31, 2019 | 7,192,357 | $ | 5,256 | ||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | ||||||
| | | | | | | | | | | | | | | | | | | | |||||||
Intrinsic value in the above tabletables was calculated as the difference between the Company's estimated stock price at December 31, 2016,2019, of $5.70,$2.50 per share, and the exercise price, multiplied by the number of options. Intrinsic value for options exercised during 2016 amounts to $227.
Restricted Stock Units
During the year ended December 31, 2016, there was one RSU grantthe Company granted 50,000 RSUs to an employee of 50,000 shares of common stock (of whichthe Company, 16,666 sharesRSUs vested at issuance). Theon the grant date, fair value was $5.93 per share and there was no intrinsic value at December 31, 2016. The remaining16,667 RSUs vestvested in February 2017 (16,667 shares) and February 2018 (16,667 shares) and the remaining 16,667 RSUs vested in February 2018.
During the year ended December 31, 2017, the Company granted a total of 247,694 RSUs to executive officers and directors of the Company. These RSUs vested ratably over a two-year period for the executive officers and on the one-year anniversary of the grant date for the directors.
During the year ended December 31, 2018, the Company granted a total of 108,254 RSUs to executive officers of the Company representing payment for 2017 target bonuses. These RSUs vested on the one-year anniversary of the grant date.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
10. Equity Incentive Plans (Continued)
The following table shows the Company's restricted stock unit activity during the years ended December 31, 2019, 2018 and 2017:
| | Shares | Weighted Average Grant Date Fair Value | Aggregate Intrinsic Value | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Restricted stock units outstanding at December 31, 2017 | 264,361 | 3.16 | ||||||||
Granted | 108,254 | 3.46 | ||||||||
Vested | (225,061 | ) | 3.39 | $ | 370 | |||||
Restricted stock units outstanding at December 31, 2018 | 147,554 | 3.03 | ||||||||
Vested | (147,554 | ) | 3.03 | $ | 129 | |||||
| | | | | | | | | | | |
Restricted stock units outstanding at December 31, 2019 | — | |||||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Performance Based Restricted Stock Awards
In addition to the RSUs detailed in the table above, during 2017 the Company granted up to 260,000 shares of performance-based restricted stock units ("Performance Units") under the Company's Amended 2014 Incentive Compensation Plan, to executive officers which are primarily contingent upon achievement of performance goals during the performance period beginning on the date of grant and ending on December 31, 2018 as set forth in each officer's performance unit agreement. For awards with a performance condition which affects the vesting of the Performance Units, cost is recognized only if the performance condition is probable of being satisfied. Given the uncertainty of the achievement of the performance goals during the performance period, the Company did not record compensation expense related to be recognized is $109.these awards for the year ended December 31, 2017. These performance-based restricted stock units expired and were subsequently replaced with new awards in January 2018 (see below).
In January 2018, the Company granted up to 365,000 shares of performance-based restricted stock units ("Performance Units") under the Company's 2014 Incentive Compensation Plan primarily to executive officers, which were largely contingent upon the achievement of performance goals during the performance period beginning on the date of grant and ending on December 31, 2019 as set forth in each individual's Performance Unit agreement. Performance Units granted in January 2018 replaced Performance Units granted in April 2017 which expired. During 2018, 50,000 Performance Units were cancelled and as of December 31, 2018 315,000 Performance Units remained outstanding. The remaining 315,000 Performance Units expired in December 2019 as the performance goals were not achieved, and there are no Performance Units outstanding as of December 31, 2019.
11. Income Taxes
On December 22, 2017, the President of the United States signed into law an Act to provide for reconciliation pursuant to titles II and V of the concurrent resolution on the budget for fiscal year 2018 (commonly known as "the Tax Cuts and Jobs Act or the "TCJA""), which introduced a comprehensive set of tax reforms. The Tax Cuts and Jobs Act significantly revises U.S. tax law by, among other
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
11. Income Taxes (Continued)
provisions, lowering the Company's corporate tax rate from 34% to 21% and eliminating or reducing certain income tax deductions.
In December 2017, in accordance with the SEC Staff Accounting Bulletin ("SAB") 118—Income Tax Accounting Implications of the TCJA, the Company recorded tax effects on a provisional basis based on a reasonable estimate. The TCJA did not have a material impact on the Company's financial statements because its deferred temporary differences are fully offset by a valuation allowance and the Company does not have any offshore earnings from which to record the mandatory transition tax. During 2018, the Company completed its analysis under SAB 118 and no additional tax effects due to rate-remeasurement were required to be recorded.
As of December 31, 2016,2019, the Company had available net operating loss carryforwards ("NOL"NOLs") of approximately $177.4 million and $44.2$231.5 million for federal and $92.4 million for state income tax reporting purposes, respectively, whichpurposes. Under TCJA, the federal NOLs generated in 2019 and 2018, approximately $32.5 million, can be carried forward indefinitely, while the NOLs generated through taxable years ending December 31, 2017, approximately $198.9 million, are available to offset future federal and state taxable income, if any, through 2036.2037. The Company also has research and development tax credit carryforwards of approximately $4.9$6.3 million and $0.4$1.6 million for federal and state income tax reporting purposes, respectively, which are available to reduce federal income taxes, if any, through 2039 and state income taxes, if any, through 2036 and state income taxes, if any, through 2031.2034.
The Internal Revenue Code of 1986, as amended (the "Code") provides for a limitation on the annual use of NOLNOLs and other tax attributes (such as research and development tax credit carryforwards) following certain ownership changes, as defined by the Code that could significantly limit the Company's ability to utilize these carryforwards. At this time, the Company has not completed a study to assess whether an ownership change under Section 382 of the Code has occurred, or whether
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
11. Income Taxes (Continued)
there have been multiple ownership changes since the Company's formation, due to the costs and complexities associated with such a study. The Company is likely to have experienced various ownership changes, as defined by the Code, as a result of past financings. Accordingly, the Company's ability to utilize the aforementioned carryforwards may be limited. Additionally, U.S. tax laws limit the time during which these carryforwards may be applied against future taxes. Therefore, the Company may not be able to take full advantage of these carryforwards for federal and state income tax purposes.
The Company does not have any significant unrecognized tax benefits.
As of December 31, 2016,2019, the Company has not accrued interest or penalties related to uncertain tax positions. The Company's tax returns for the years ended December 31, 20132016 through December 31, 20152018 are still subject to examination by major tax jurisdictions. However, the Internal Revenue Service ("IRS") and state tax jurisdictions can audit the NOLs generated in prior years in the years that those NOLs are utilized.
For all years through December 31, 2016,2019, the Company generated research creditcredits but has not conducted a study to document the qualified activities. This study may result in an adjustment to the Company's research and development credit carryforwards; however, until a study is completed and any adjustment in known, no amounts are being presented as an uncertain tax position. A full valuation allowance has been provided against the Company's research and development credits and, if an
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
11. Income Taxes (Continued)
adjustment is required, this adjustment would be offset by an adjustment to the deferred tax asset established for the research and development credit carryforwards and the valuation allowance.
The tax effects of temporary differences that give rise to significant portions of the deferred tax assets are presented below:
| | December 31 | December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2019 | 2018 | ||||||||||
Deferred tax assets: | ||||||||||||||
Net operating loss carryforwards | $ | 63,068 | $ | 54,197 | $ | 55,216 | $ | 51,240 | ||||||
Research credit carryforward | 5,284 | 4,527 | 7,609 | 6,904 | ||||||||||
Stock options and other | 2,250 | 1,474 | 3,225 | 2,655 | ||||||||||
| | | | | | | | | | | | | | | |
Total gross deferred tax assets | 70,602 | 60,198 | 66,050 | 60,799 | ||||||||||
Valuation allowance for deferred tax assets | (70,602 | ) | (60,198 | ) | (66,050 | ) | (60,799 | ) | ||||||
| | | | | | | | | | | | | | | |
Net deferred tax assets | $ | — | $ | — | $ | — | $ | — | ||||||
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | |
The gross deferred tax assets and the valuation allowance shown above represent the items which reduce the income tax benefit which would result from applying the federal statutory tax rate to the pretax loss and cause no income tax expense or benefit to be recorded for the years ended December 31, 2016 and 2015.
The net change in the valuation allowance for the years ended December 31, 20162019 and 20152018 was an increase of $10.4$5.3 million and $8.9an increase of $4.8 million, respectively, related primarilyrespectively.
A reconciliation of the U.S. statutory income tax rate to net operating losses incurredthe Company's effective tax rate is as follows:
| | December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2019 | 2018 | 2017 | |||||||
Federal income tax at statutory rate | 21.0 | % | 21.0 | % | 34.0 | % | ||||
State income tax benefit, net of federal benefit | 7.0 | % | 6.0 | % | 6.0 | % | ||||
Research and development tax credits | 4.0 | % | 3.0 | % | 3.0 | % | ||||
Effect of tax rate changes | 0.0 | % | 0.0 | % | –94.0 | % | ||||
Other | –4.0 | % | –4.0 | % | –1.0 | % | ||||
Decrease (increase) to valuation allowance | –28.0 | % | –24.0 | % | 52.0 | % | ||||
| | | | | | | | | | | |
Effective income tax rate | 0.0 | % | 2.0 | % | 0.0 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Sale of New Jersey Net Operating Losses
2018
The Company has participated in the State of New Jersey's Technology Business Tax Certificate Transfer Program (the "Program") sponsored by The New Jersey Economic Development Authority. The Program enables approved biotechnology companies with unused NOLs and unused research and development credits to sell these benefits for at least 80% of the value of the tax benefits to unaffiliated, profitable corporate taxpayers in the State of New Jersey. The Program is administered by The New Jersey Economic Development Authority and the New Jersey Department of the Treasury's Division of Taxation. In January 2018, the Company which are not currently deductible.completed the sale of NOLs totaling approximately
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 20162019
(in thousands, except share and per share data)
11. Income Taxes (Continued)
A reconciliation$0.5 million. This amount is a current state tax benefit and is reflected in the statement of operations for the year ended December 31, 2018. The Company has now reached the maximum lifetime benefit of $15.0 million under the Program and will no longer be eligible to participate in the Program.
12. Restructuring Costs
In June 2018, the Company announced a reduction in its workforce, which resulted in the termination of several employees primarily from the Company's commercial and clinical teams, representing approximately thirty percent of its employees. This workforce reduction, along with other reductions in planned operating expenses is designed to preserve cash while the Company pursued formal dispute resolution with the FDA for Twirla and determines a regulatory path forward for the resubmission of the U.S. statutory income tax rateCompany's NDA for Twirla.
In June 2018, the Company also announced that it had adopted a retention plan (the "Retention Plan") to provide (i) cash retention payments to all remaining employees in order to induce such employees to remain employed by the Company through December 31, 2018 and (ii) stock option grants to all remaining employees in order to induce such employees to remain employed by the Company through December 31, 2019.
Each employee who participated in the Retention Plan and (i) remained continuously employed by the Company through December 31, 2018 or (ii) was terminated by the Company other than for cause (as defined in an applicable employment agreement, or, if no employment agreement exists, as determined by the Company in good faith) prior to December 31, 2018, was paid a lump-sum cash payment in an amount determined by the compensation committee ("Compensation Committee") of the Company's board of directors at the time of the adoption of the Retention Plan. If an eligible employee terminated service prior to December 31, 2018 for any reason other than termination of employment by the Company without cause, no such cash retention payment was made to the eligible employee. The total amount of the cash portion of the Retention Plan was approximately $0.6 million.
In addition, all remaining employees were granted a stock option to purchase the number of shares of common stock as approved by the Compensation Committee, with a per share exercise price of $0.58, representing the closing price of the Company's effective tax ratecommon stock as reported by Nasdaq on the date the Retention Plan was approved by the Compensation Committee. Each option vested in four equal 25% installments on the following dates: (i) June 20, 2018, (ii) December 31, 2018, (iii) June 30, 2019 and (iv) December 31, 2019, subject to the employee's continuing service to the Company.
A summary of accrued restructuring costs, included as a component of accrued liabilities on the Company's unaudited December 31, 2019 balance sheet is as follows:
| | December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2016 | 2015 | 2014 | |||||||
Federal income tax at statutory rate | 34.0 | % | 34.0 | % | 34.0 | % | ||||
State income tax benefit, net of federal benefit | 6.0 | % | 6.0 | % | 6.0 | % | ||||
Research and development tax credits | 2.0 | % | 2.0 | % | 3.0 | % | ||||
Other | 1.0 | % | (2.0 | )% | — | % | ||||
Increase to valuation allowance | (33.0 | )% | (24.0 | )% | (24.0 | )% | ||||
| | | | | | | | | | | |
Effective income tax rate | 10.0 | % | 16.0 | % | 19.0 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | December 31, 2018 | Charges | Payments | December 31, 2019 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Accrued retention bonus | 638 | — | (638 | ) | — | ||||||||
| | | | | | | | | | | | | | |
Total | $ | 638 | $ | — | $ | (638 | ) | $ | — | ||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Sale of New Jersey Net Operating Losses
The Company received approval to sell a portion of the Company's New Jersey net operating losses (NOLs) as part of the Technology Business Tax Certificate Program sponsored by The New Jersey Economic Development Authority. Under the program, emerging biotechnology companies with unused NOLs and unused research and development credits are allowed to sell these benefits to other companies. In December 2016, the Company completed the sale of NOLs totaling approximately $28.2 million and research and development credits totaling approximately $0.8 million for net proceeds of approximately $3.0 million. Such proceeds are reflected as a tax benefit for the year ended December 31, 2016. On November 30, 2015, the Company completed the sale of NOLs totaling approximately $59.8 million and research and development credits totaling $1.1 million for net proceeds of approximately $6.0 million. Such proceeds are reflected as a tax benefit for the year ended December 31, 2015. On February 27, 2014, the Company completed the sale of NOLs totaling approximately $39.1 million and research and development credits totaling approximately $0.4 million for net proceeds of approximately $3.6 million. Such proceeds are reflected as a tax benefit for year ended December 31, 2014.
12. Related Party Transactions
Between March 17, 2014 and July 6, 2016, one of the Managing Partners of SmartPharma LLC, or SmartPharma, an entity which provides commercial and business development consulting services to the Company, served as Chief Commercial Officer of the Company. In connection with the appointment of this individual as Chief Commercial Officer, the Company amended its consulting agreement with SmartPharma to remove this individual from the list of persons providing service under the consulting agreement. SmartPharma invoiced the Company $3, $73 and $126 of fees for the years ended December 31, 2016 (through July 6, 2016), 2015 and 2014, respectively. In connection with the resignation of our Chief Commercial Officer who was affiliated with SmartPharma on July 6, 2016, the Company appointed a new Chief Commercial Officer.
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 20162019
(in thousands, except share and per share data)
13. Commitments and Contingencies
Operating Leases2019 Retention Plan
TheIn July 2019, the Company leases approximately 8,200 square feet of office space in Princeton, NJ. The current termadopted a retention plan (the "2019 Retention Plan") for all employees (with the exception of the leaseChairman and Chief Executive Officer) in order to induce such employees to remain employed by the Company through at least the PDUFA goal date of November 16, 2019.
Each employee who participates in the 2019 Retention Plan and remains continuously employed by the Company through the Approval shall be paid a lump-sum cash payment in an amount determined for each eligible employee by the Compensation Committee at the time of the adoption of the 2019 Retention Plan. If an eligible employee terminates employment prior to the Approval for any reason, no such retention payment shall be made to the eligible employee. The total amount of the cash portion of the 2019 Retention Plan is for a five year period ending on November 30, 2020. Theapproximately $0.3 million. Given that the PDUFA goal date was extended to February 16, 2020 and the ultimate uncertainty of the approval of Twirla, the Company has the rightnot recorded compensation expense related to terminate the lease after November 30, 2018 under certain circumstances as defined in the lease.
Rent expense was $195, $163 and $159these potential cash awards for the yearsyear ended December 31, 2016, 20152019.
All employees (with the exception of the Chairman and 2014, respectively.
Future minimum annual lease commitments underChief Executive Officer) who were employed by the non-cancelable operating lease in effectCompany as of July 3, 2019 were also granted a stock option to purchase the number of shares of common stock as approved by the Compensation Committee, with a per share exercise price of $1.48, representing the closing price of the Company's common stock as reported by Nasdaq on the date of grant. Each option will vest in two equal 50% installments on the following dates (i) July 3, 2020 and (ii) December 31, 2016 are2020.
In addition, the vesting for the annual stock option grant in January 2019 were amended for all employees holding such options who were employed on July 3, 2019 as follows:
2017 | $ | 192 | ||
2018 | $ | 200 | ||
2019 | $ | 200 | ||
2020 | $ | 191 | ||
2021 | $ | — |
Legal Proceedings
Two complaints have been filed in federal court in 50% of the District of New Jerseyoption will vest on January 6, 201729, 2020, 25% on June 30, 2020 and January 20, 2017, titledPeng v. Agile Therapeutics, Inc., Alfred Altomari,the remaining 25% on December 31, 2020. The change in vesting schedule was approved by the Compensation Committee and Elizabeth Garner, No. 17-cv-119 (D.N.J.), andLichtenthal v. Agile Therapeutics, Inc., Alfred Altomari, and Elizabeth Garner, No. 17-cv-405 (D.N.J.), (collectively, the "Complaints") respectively,did not have a material impact on behalf of a putative class of investors who purchased stock from March 9, 2016 through January 3, 2017. The complaints allege violations of the federal securities laws based on public statements made regarding the Company's Phase 3 "SECURE" clinical trial. Agile denies all allegations in the complaintsstatement of operations.
14. Commitments and the Company plans to vigorously defend the complaints that have been filed.Contingencies
The Company records a provision for contingent losses when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. An unfavorable outcome to any legal matter, if material, could have an adverse effect on the Company's operations or its financial position. Based on its current knowledge,As of December 31, 2019, the Company doeshas not believe thatrecorded a provision for any contingent losses.
15. Subsequent Event
In February 2020, the amountCompany entered into a Credit Agreement and Guaranty with Perceptive Credit Holdings III, LP, a related party, or Perceptive, for a senior secured term loan credit facility of such possible loss or rangeup to $35 million, (the Perceptive Credit Agreement"). A first tranche of potential loss relating$5 million was funded on execution of the Perceptive Credit Agreement. A second tranche of $15 million was funded as a result of the approval of Twirla by the FDA. Another $15 million tranche will be available to the ComplaintsCompany based on the achievement of certain revenue milestones. The facility will be interest only until the third anniversary of the closing date. As part of the Perceptive Credit Agreement, the Company issued Perceptive warrants to purchase 1,400,000 shares of Agile common stock. The per share exercise price
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2019
(in thousands, except share and per share data)
15. Subsequent Event (Continued)
for 700,000 shares is reasonably estimable.$3.74, which is equal to the 5-day volume weighted average exercise price ("5 Day VWAP") as of the trading day immediately prior to closing. The per share exercise price for the remaining 700,000 shares of Agile common stock is $4.67, which is 1.25 times the 5 Day VWAP.
14.16. Quarterly Data (Unaudited)
The following tables summarize the quarterly results of operations for each of the quarters in 20162019 and 2015.2018. These quarterly results are unaudited, but in the opinion of management, have been prepared on the same basis as our audited financial information and include all adjustments (consisting
Agile Therapeutics, Inc.
Notes to Financial Statements (Continued)
December 31, 2016
(in thousands, except share and per share data)
14. Quarterly Data (Unaudited) (Continued)
only of normal recurring adjustments) necessary for a fair presentation of the information set forth herein.herein (in thousands, except per share amounts).
| | March 31, 2016 | June 30, 2016 | September 30, 2016 | December 31, 2016 | March 31, 2019 | June 30, 2019 | September 30, 2019 | December 31, 2019 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Total revenue | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
Operating expenses | $ | 6,980 | $ | 7,841 | $ | 7,091 | $ | (7,810 | ) | $ | 4,707 | $ | 3,547 | $ | 4,499 | $ | 6,105 | |||||||||
Net loss | $ | (7,318 | ) | $ | (8,418 | ) | $ | (7,804 | ) | $ | (5,201 | ) | $ | (4,669 | ) | $ | (3,484 | ) | $ | (4,432 | ) | $ | (6,021 | ) | ||
Basic and diluted net loss per common share | $ | (0.27 | ) | $ | (0.29 | ) | $ | (0.27 | ) | $ | (0.18 | ) | $ | (0.13 | ) | $ | (0.08 | ) | $ | (0.08 | ) | $ | (0.10 | ) | ||
| | March 31, 2015 | June 30, 2015 | September 30, 2015 | December 31, 2015 | March 31, 2018 | June 30, 2018 | September 30, 2018 | December 31, 2018 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Total revenue | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
Operating expenses | $ | 6,977 | $ | 7,982 | $ | 8,965 | $ | 9,165 | $ | 7,046 | $ | 5,147 | $ | 3,615 | $ | 3,727 | ||||||||||
Net income (loss) | $ | (8,538 | ) | $ | (8,486 | ) | $ | (9,411 | ) | $ | (3,899 | ) | ||||||||||||||
Basic and diluted net income loss per common share | $ | (0.40 | ) | $ | (0.38 | ) | $ | (0.42 | ) | $ | (0.17 | ) | ||||||||||||||
Net loss | $ | (6,833 | ) | $ | (5,344 | ) | $ | (3,792 | ) | $ | (3,810 | ) | ||||||||||||||
Basic and diluted net loss per common share | $ | (0.20 | ) | $ | (0.16 | ) | $ | (0.11 | ) | $ | (0.11 | ) | ||||||||||||||
The net loss and basic and diluted net loss per share for the quarter ended DecemberMarch 31, 2016 includes2018 include a tax benefit of $3,075$0.5 million from the sale of New Jersey state net operating losses. The net loss and basic and diluted net loss per share for the quarter ended December 31, 2015 includes a tax benefit of $5,972 from the sale of New Jersey state net operating lossesNOLs. (see Note 11).
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
Our management, with the participation of our chief executive officer and chief financial officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2016.2019. The term "disclosure controls and procedures," as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, mean controls and other procedures of a company that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms. Disclosure controls include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2016,2019, our chief executive officer and chief financial officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable level.
Management's Annual Report on Internal Controls Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act and is a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our board of directors, management and other personnel, to:
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Our management assessed the effectiveness of the Company's internal control over financial reporting as of December 31, 2016.2019. In making this assessment, the Company's management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework.
Based on its evaluation, our management has concluded that, as of December 31, 2016,2019, our internal control over financial reporting was effective.
Attestation Report of the Registered Public Accounting Firm
This annual report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Management's report was not subject to the attestation by our independent registered public accounting firm because emerging growth companiesas a non-accelerated filer, we are exempt from this requirement.
Changes in Internal Control Over Financial Reporting
No change in our internal control over financial reporting occurred during the quarter ended December 31, 20162019 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
None.
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this item will be included in an amendment to this Annual Report on Form 10-K or incorporated by reference from our definitive proxy statement to be filed pursuant to Regulation 14A.
Item 11. Executive Compensation
The information required by this item will be included in an amendment to this Annual Report on Form 10-K or incorporated by reference from our definitive proxy statement to be filed pursuant to Regulation 14A.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this item will be included in an amendment to this Annual Report on Form 10-K or incorporated by reference from our definitive proxy statement to be filed pursuant to Regulation 14A.
Item 13. Certain Relationships and Related Transactions and Director Independence
The information required by this item will be included in an amendment to this Annual Report on Form 10-K or incorporated by reference from our definitive proxy statement to be filed pursuant to Regulation 14A.
Item 14. Principal Accounting Fees and Services
The information required by this item will be included in an amendment to this Annual Report on Form 10-K or incorporated by reference from our definitive proxy statement to be filed pursuant to Regulation 14A.
Item 15. Exhibits, and Financial Statement Schedules
The following documents are filed as a part of this Annual Report on Form 10-K:
(a)
The information concerning our financial statements, and Report of Independent Registered Public Accounting Firm required by this Item is incorporated by reference herein to the section of this Annual Report on Form 10-K in Item 8, entitled "Financial Statements and Supplementary Data."
(b)
All schedules have been omitted because the required information is not present or not present in amounts sufficient to require submission of the schedules, or because the information required is included in the Financial Statements or notes thereto.
(c)
The list of exhibits filed with this report is set forth in the Exhibit Index followingimmediately preceding the signature pagespage and is incorporated herein by reference.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on March 15, 2017.
Pursuant to the requirements of the Securities Act of 1934, this report has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated.
None.
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on February 20, 2020.
| AGILE THERAPEUTICS, INC. | ||||
By | /s/ ALFRED ALTOMARI Alfred Altomari Chief Executive Officer | |||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated.
Signature | Title | Date | ||
|---|---|---|---|---|
| /s/ ALFRED ALTOMARI Alfred Altomari | Chief Executive Officer and Director (Principal Executive Officer) | February 20, 2020 | ||
/s/ DENNIS P. REILLY Dennis P. Reilly | Chief Financial Officer (Principal Financial and Accounting Officer) | February 20, 2020 | ||
/s/ SETH H.Z. FISCHER Seth H.Z. Fischer | Director | February 20, 2020 | ||
/s/ JOHN HUBBARD John Hubbard, Ph.D. | Director | February 20, 2020 | ||
/s/ ABHIJEET LELE Abhijeet Lele | Director | February 20, 2020 | ||
/s/ WILLIAM T. MCKEE William T. McKee | Director | February 20, 2020 | ||
/s/ AJIT S. SHETTY Ajit S. Shetty, Ph.D. | Director | February 20, 2020 | ||
/s/ JAMES TURSI James Tursi, M.D. | Director | February 20, 2020 |