UNITED STATESSECURITIES AND EXCHANGE COMMISSIONWashington, D.C. 20549
FORM 10-K
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||
TRANSMISSION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||
$1,677,874,611. psychiatry. Our extensive expertise in product development has been built over the past 25 years: initially as a Our net product IQVIA care physicians. therapies. Our key proprietary technology platforms include: Microtrol, Solutrol and EnSoTrol. These technologies have been utilized to create approved products; invest in sales and marketing resources for existing and new products; enter into agreements to purchase products or other companies; and invest in support of our business, technology, regulatory and intellectual property portfolio. Epilepsy Overview headaches. this program, because initial studies with immediate release formulations of non-synthetic huperzine A have shown dose-limiting serious side effects. research. We currently employ internal resources to manage our manufacturing contractors. current sales force of over 200 sales representatives is effectively targeting healthcare providers, primarily neurologists, to support and grow our epilepsy and migraine product franchise. Simultaneously promoting two neurology products allows us to leverage our commercial infrastructure and gain efficiencies in operations. amitriptyline. ADHD Overview
(State or other jurisdiction of
incorporation or organization) 20-2590184
(I.R.S. Employer
Identification Number)1550 East Gude Drive, Rockville MD (301)
including area code) 20850(zip code)TITLE OF EACH CLASS: Outstanding at February 13, 2020 Trading Symbol Common Stock, $0.001 Par Value 52,533,973 SUPN The NASDAQ Stock Market LLC o☒ No ý☐o☐Noý☒ý☒ No o☐and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yesý☒ No o☐o☐or a non-accelerated filer, or a smaller reporting company, or an emerging growth company. See the definitions of "large accelerated filer",filer," "accelerated filer"filer," "smaller reporting company," and "smaller reporting"emerging growth company" in Rule 12b-2 of the Exchange Act. (Check one):Large accelerated filer ýAccelerated filer ☐ AcceleratedNon-accelerated filer o☐ Smaller reporting company ☐ Non-accelerated filero(Do not check if asmaller reporting company) Smaller reportingEmerging growth companyo☐ o☐ No ý☒2016,2019, the aggregate market value of the common stock held by non-affiliates of the registrant based on the closing price of the common stock on The NASDAQ Global Market was $966,994,822. The number of shares of the registrant's common stock outstanding as of March 9, 2017 was 50,162,496.20172020 Annual Meeting of Stockholders, which will be filed with the Securities and Exchange Commission not later than 120 days after the end of the registrant's 20162019 fiscal year end, are incorporated by reference into Part III of this Annual Report on Form 10-K.2016
2019applications(™applications(TM), including the following marks referred to in this Annual Report on Form 10-K pursuant to applicable U.S. intellectual property laws: "Supernus®," "Oxtellar XR®," "Trokendi XR®," "Microtrol®," "Solutrol®," and the registered Supernus Pharmaceuticals logo.prospectusAnnual Report are the property of their respective owners. Solely for convenience, the trademarks and trade names in this Annual Report on Form 10-K are referred to without the ® andTM™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. specialty pharmaceutical company focused on developing and commercializing products for the treatment of central nervous system (CNS) diseases. In 2013, we launched Oxtellar XR (extended-release oxcarbazepine)diseases in neurology and Trokendi XR (extended-release topiramate), our two novel treatments for patients with epilepsy. In addition, we are developing multiple product candidates in psychiatry to address significant unmet medical needs and market opportunities for the treatment of impulsive aggression (IA) and for the treatment of attention deficit hyperactivity disorder (ADHD). We are initially developing SPN-810 (molindone hydrochloride) to treat IA in patients who have ADHD. We subsequently plan to develop SPN-810 for the treatment of IA in other CNS diseases, such as autism, post traumatic stress disorder (PTSD), bipolar disorder, schizophrenia, and some forms of dementia. There are currently no approved products indicated for the treatment of IA. We are developing SPN-812 (viloxazine hydrochloride) as a candidate to treat patients who have ADHD.standaloneprivately-held stand-alone development organization,organization; then, as a U.S.United States (U.S.) subsidiary of Shire plcPlc (Shire, a subsidiary of Takeda Pharmaceutical Company Ltd.); and upon our acquisition of substantially all of the assets of Shire Laboratories Inc. in late 2005, as Supernus Pharmaceuticals.
* Prophylaxis of migraine headache in adults and adolescents. ** Prescription Drug User Fee Act (PDUFA) ourtwo products, in the United States through our own specialty sales force and have and will continue to seek strategic collaborations with other pharmaceutical companies to license our products outside the United States.Our neurology portfolio consists of Oxtellar XR and Trokendi XR whichin the U.S. Oxtellar XR and Trokendi XR are the first once-daily extended release oxcarbazepine and topiramate products respectively, indicated for the treatment of epilepsy in the U.S. market. TheseIn April 2017, we launched Trokendi XR for the prophylaxis of migraine headache in adults and adolescents. In January 2019, we launched Oxtellar XR for monotherapy treatment of partial onset epilepsy seizures in adults and in children 6 to 17 years of age. We market our products are differentiated, comparedthrough our own sales force in the U.S. and seek strategic collaborations with other pharmaceutical companies to their immediate release counterpartcommercialize our products by offering convenient once-daily dosing and unique pharmacokinetic profiles. We believe that a once-daily dosing regimen improves compliance which in turn reducesoutside of the frequency of seizures. We also believe that the unique smooth and steady pharmacokinetic profiles of once-daily dosing mitigate the blood level fluctuations typically associated with immediate release products, which can result in adverse events (AEs) or decreased efficacy.revenuessales of $210.1$383.4 million in 20162019 were driven by strongcontinued growth in prescriptions for Oxtellar XR and Trokendi XR. Total prescriptions as reported by Intercontinental Marketing Services (IMS) have shown a steady increase year over yearXR, as shown in the following graph.graph: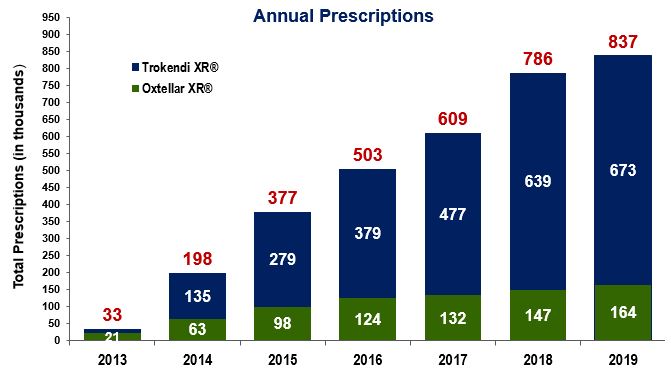
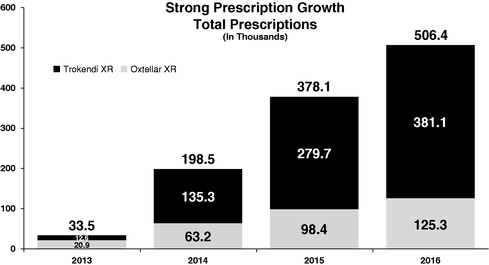
IMS Monthly Prescriptions2016, our products2019, Trokendi XR represented approximately 5% of the topiramate market, and Oxtellar XR represented approximately 3% of the large and growing base ofoxcarbazepine market. Total annual prescriptions for the topiramate and oxcarbazepine (total annual prescriptions for topiramate market and oxcarbazepine market is 14.3markets are approximately 13.4 million and 4.54.7 million, respectively). respectively.expectare also developing multiple proprietary CNS product candidates to continueaddress significant unmet medical needs and market opportunities. We are developing SPN-812 (viloxazine hydrochloride) as a novel, non-stimulant product candidate to grow our revenues for Oxtellar XR and Trokendi XR for the foreseeable future by continuing to drive penetration in these markets. We believe these products have the potential to achieve combined peak net sales in excess of $500 million annually.Oxtellar XR is indicated for add-on, adjunctive or concomitant therapy of partial seizures in adults and intreat children 6 years to 17 years of age. Trokendi XR is indicated for initial monotherapy in patients 6 years of age and older with partial onset or primary generalized tonic-clonic seizures, and as add-on therapy in patients 6 years of age and older with partial onset or primary generalized tonic-clonic seizures or with seizures associated with Lennox-Gastaut syndrome.In August 2016, we received tentative approval to expand the label for Trokendi XR to include the indication of prophylaxis of migraine headaches in adults.who have ADHD. We continue to prepare and will be readyexpect to launch SPN-812, assuming FDA approval, in the migraine indication soon after receiving fullfourth quarter of 2020. Additionally, we initiated a Phase III ADHD program to study SPN-812 in adults during the third quarter of 2019.fromfor the treatment of bipolar disorder with oxcarbazepine in the U.S. FoodDrug Administration (FDA).development expenses related to the continued development of each of our product candidates through FDA approval or until the program terminates. We incurred total research and development expenses of $69.1 million, $89.2 million and $49.6 million for the years ended December 31, 2019, 2018 and 2017, respectively.receiving this approval duringcreating a sales force to market our products to the second quarterrelevant population of 2017.Regarding SPN-810, we initiated two Phase III clinical trials in 2015 (P301psychiatrists and P302) that will continue to enroll patients through 2017. Our Phase III clinical trial (P301) is being conducted under a Special Protocol Agreement (SPA). SPN-810 has been granted fast-track designation by the FDA.We completed a Phase IIb dose ranging trial for SPN-812 and announced topline results in 2016. The trial met the primary endpoint, demonstrating that SPN-812 at daily doses of 400 mg, 300 mg, and 200 mg achieved a statistically significant improvement in the symptoms of ADHD when compared to placebo. All SPN-812 doses tested in the trial were well tolerated. Of the patients treated with SPN-812, only 6.7% discontinued due to an AE. In addition, 87% of patients who completed the trial elected to enroll in the ongoing open-label extension. Based on these positive results, we plan to have an end-of-Phase II meeting with the FDA after which we will initiate Phase III clinical testing during the second half of 2017.therapies and expand the treatment to new indications.nineten marketed products, includingincluding: Trokendi XR and Oxtellar XR,XR; Adderall XR (developed for Shire),; Intuniv (developed for Shire),; Mydayis (developed for Shire); and Orenitram (developed for United Therapeutics Corporation); as well as our key product candidates SPN-810 andcandidate SPN-812.Products and Product CandidatesThe table below summarizes our current portfolio of novel products and product candidates.ProductIndicationStatusOxtellar XREpilepsyLaunched in 2013Trokendi XREpilepsyLaunched in 2013Adult Migraine ProphylaxisTentative ApprovalSPN-810IA*Phase IIISPN-812ADHDPhase IIbSPN-809DepressionPhase II ready*Initial program is in patients with ADHD, with plansWe continue to follow on in other indications, such as IA in patients with autism, PTSD, bipolar disorder, schizophrenia, and some forms of dementia.We are continuing to expandbuild our intellectual property portfolio to provide additional protection for our technologies, products and product candidates.currently have seven U.S. patents issued covering Oxtellar XRexpect to incur significant expenses as we: invest in research and eight U.S. patents issued covering Trokendi XR, providing patent protection expiring no earlier than 2027development related to the continued development of each of our product candidates through FDA approval or until the program terminates; expand product indications for each product.bebecome a leading specialty pharmaceutical company, developing and commercializing new medicines for treatment of CNS diseases in neurology and psychiatry. Key elements of our strategy to achieve this vision are to:•Drive growth and profitability. We will continue to drive the prescription growth of Trokendi XR and Oxtellar XR by continuing to dedicate sales and marketing resources in the United States.•Advance our pipeline toward commercialization. We initiated the Phase III clinical trials for SPN-810, a novel treatment for IA in patients who have ADHD, during the third quarter of 2015. We completed a Phase IIb dose ranging study for SPN-812 during 2016 and expect to initiate Phase III clinical testing during the second half of 2017.•Target strategic business development opportunities. We are actively exploring a broad range of strategic opportunities that fit well with our strong presence in CNS. These include: in-licensing products and entering into co-promotion partnerships which are synergistic with our sales force call point for our marketed products and product candidates; co-development partnerships for our pipeline products; and growth opportunities through value-creating and transformative merger and acquisition transactions, including both commercial stage and development stage products.•Continue to grow our pipeline. We plan to continue to evaluate and develop additional CNS product candidates that we believe have significant commercial potential through our internal research and development efforts.• • • • and Trokendi XR are the firsta once-daily extended release oxcarbazepine product that was initially approved for adjunctive treatment of partial onset epilepsy seizures. During January 2019, we launched Oxtellar XR for the monotherapy treatment of partial onset epilepsy seizures in adults and topiramate products indicated for patientsin children 6 to 17 years of age; andepilepsypharmacological activities in the U.S. market. These products differ from theCNS conditions such as epilepsy.immediate release products by offering once-daily dosing and unique pharmacokinetic profiles which we believe can have very positive clinical effects for many patients. We believe a once-daily dosing regimen improves adherence, making it more probable that patients maintain sufficient levels of medication in their bloodstreams to protect against seizures. In addition, we believe that the unique smooth and steady pharmacokinetic profiles of our once-daily formulations reduce the peak to trough blood level fluctuations that are typically associated with immediate release products and may result in increased AEs, more side effects and decreased efficacy.Improved tolerabilityExtended release products may help patients improve adherence, have fewer breakthrough seizuresmigraines and, correspondingly,consequently, help patients enjoy a better quality of life.the firstindicated for: initial monotherapy in patients 6 years of age and older with partial onset or primary generalized tonic-clonic (PGTC) seizures; as add-on therapy in patients 6 years of age and older with partial onset or PGTC seizures or with seizures associated with Lennox-Gastaut syndrome; and for prophylaxis of migraine headache in adults and adolescents 12 years of age and older. Trokendi XR's once-daily extended release topiramate product indicated for patients with epilepsy in the U.S. market, anddosing is designed to improve patient adherence over the current immediate release products, which must be taken multiple times per day. We believe a once-daily dosing regimen improves compliance, making it more probable that patients take their medication and maintain sufficient levels of medication in their bloodstreams. Trokendi XR's unique smooth pharmacokinetic profile results in lower peak plasma concentrations, higher trough plasma concentrations, and slower plasma uptake rates. This results in smoother and more consistent plasma concentrations than immediate release topiramate formulations can deliver.formulations. We believe that such a profile mitigates blood level fluctuations that are frequently associated with many side effects, as well as mitigatingthereby reducing the likelihood of breakthrough seizures or migraine headaches that patients can suffer when taking immediate release products. Side effects associated with immediate release products may lead patients to skip doses, which could place them at higher risk for breakthrough seizures.In August 2016, we received tentative approval to expand the label for Trokendi XR to include the indication of prophylaxis ofseizures or migraine headache in adults. We continue to prepare and will be ready to launch the adult migraine indication soon after receiving full FDA approval, which we anticipate will occur during the second quarter of 2017.the only once-daily extended release oxcarbazepine product indicated for adjunctive treatmentas therapy of patients with epilepsypartial onset seizures in the U.S.adults and in children 6 years to 17 years of age. With its novel pharmacokinetic profile showing lower peak plasma concentrations, a slower rate of plasma input, and smoother and more consistent blood levels as compared to immediate release products, we believe Oxtellar XR improves the tolerability of oxcarbazepine and thereby reduces side effects. In addition, Oxtellar XR once-per-day dosing is designed to improve patient adherencecompliance compared to the current immediate release products that must be taken multiple times per day.Sales and MarketingWeestablished a commercial organizationnew chemical entity status (NCE) in the U.S. market. We expect to support currenthave significant intellectual property (IP) protecting this product candidate through our own research and future sales of Oxtellar XR and Trokendi XR. We believe our current sales force of over 150 sales representatives is effectively targeting healthcare providers, primarily neurologists, to support and grow our epilepsy franchise. Simultaneously promoting two epilepsy products allows us to leverage our commercial infrastructure with these prescribers. Assuming we receive FDA approvaldevelopment efforts, as well as through in-licensed IP. SPN-817 represents a novel MOA for the prophylaxis of migraine in adults, we may expand the sales force dependingan anticonvulsant. Development will initially focus on the prescription uptake post launch.Assuming we obtain FDA approvaldrug's anticonvulsant activity, which has been shown in preclinical models for treatment of partial seizures and Dravet Syndrome. SPN-817 is in clinical development, and has received an Orphan Drug designation for Dravet Syndrome from the product candidatesFDA.our pipeline, we anticipate adding sales representatives to market our productssevere pediatric epilepsy disorders. A Phase I proof-of-concept trial is currently underway in adult patients with refractory complex partial seizures, studying the safety and pharmacokinetic profile of a new extended release formulation of non-synthetic huperzine A. The Company initiated an Investigational New Drug (IND) application, enabling preclinical activities in the U.S.relevant populationsuccess of physicians, primarily psychiatrists.productproducts for commercial use, as well as productsome products for preclinical research and clinical trials.Inc.Inc; and Catalent Pharma Solutions, for the manufacture and packaging of the final commercial products Oxtellar XR and Trokendi XR.XR, as well as for our pipeline candidate, SPN-812. These CMOs offer a comprehensive range of contract manufacturing and packaging services. Commercial products as well as our product candidates are single sourced from single third-party suppliers.for Phase III clinical materials or commercial products in the foreseeable future.currently employ internal resourceshave a commercial sales and marketing organization in the U.S. to managesupport sales of Oxtellar XR and Trokendi XR. We believe our manufacturing contractors.anti-epileptic products. Oxtellar XR competes with all immediate release oxcarbazepine products including Trileptalused for the prevention of migraine headaches. Most notably, this includes a new class of products introduced in 2018, anti-CGRPs (calcitonin gene related peptide); Botox; beta-blockers; valproic acid; and its related generic products as well as other anti-epileptic products.disorders. Thedisorders:SPN-810, has fast track statusis a novel non-stimulant product being developed for the treatment of ADHD. In January 2020, the FDA accepted the review of the NDA for SPN-812 for the treatment of children and is expected to be the first product approved for IA. SPN-812adolescents with ADHD and assigned a PDUFA target action date of November 8, 2020;employwhich employs the same active ingredient as in SPN-812, is Phase II ready and areis in development for the treatment of depression; andADHD and depression, respectively. SPN-812 recently completed athe treatment of bipolar disorder. A Phase IIbIII clinical trial and SPN-809 is Phase II ready.IA OverviewOur market research shows that, for adolescents and children, child psychiatrists, psychiatrists, child neurologists, and high prescribing pediatricians write approximately 40%was initiated during the fourth quarter of their ADHD prescriptions, representing approximately 13 million prescriptions. By 2020, we project that this group of physicians will collectively write approximately 16 million prescriptions for ADHD medication. Of these 16 million ADHD prescriptions, roughly one-third will be written for patients with IA or with IA and other comorbidities.IA is not limited to individuals with ADHD. We believe IA occurs in patients with other CNS disorders, including autism, Alzheimer's, bipolar disorder, PTSD, oppositional defiant disorder, conduct disorder, and intermittent explosive disorder. Market research we have conducted indicates that the prevalence of IA in autistic children and adolescents is approximately 45%, and the prevalence of IA in children and adolescents with bipolar disorder is approximately 60%.
Current Treatments
Currently, there prescriptions. The ADHD market is projected to grow approximately 4% annually, from approximately 75 million prescriptions in 2019 to approximately 78 million prescriptions by 2020. According to data from IQVIA, the U.S. market for ADHD prescription drugs was $8.5 billion for the year ended December 31, 2019.
In response, doctors have also tried to treat this group with off-label use of prescription medicines, such as mood stabilizers, stimulants and anti-psychotic drugs. Results have varied, but anti-psychotic drugs appear to have the best therapeutic potential. Unfortunately, many of these agents are associated with adverse effects including obesity, dyskinesia, lipid abnormalities, marked increases in prolactin, and increase in diabetes, which is of particular concern when treating pediatric populations.
SPN-810 (molindone hydrochloride)
We are developing SPN-810 (molindone hydrochloride) as a novel treatment for IA in patients who have ADHD and who are being treated with standard ADHD medication. During 2014, the FDA
U.S.
- (1)Dopheide, J.A.,Attention-Deficit- Hyperactivity Disorder: An Update, published June 2009 inPharmacotherapy.(2)Floet, A.M.W.,Attention- Deficit/Hyperactivity Disorder, published February 2010 inPediatrics in Review.
(3)The MTA Cooperative Group,A 14-month randomized clinical trial of treatment strategies for attention- deficit/hyperactivity disorder, published December 1999 inArchives of General Psychiatry(4) Harvard Medical School, 2007. National Comorbidity Survey (NSC). (2017, August 21).TableThe FDA accepted the review ofContentsgranted fast track designationthe NDA forSPN-810SPN-812 for the treatment ofIA inchildren and adolescents with ADHD inpatients being treated with standard ADHD medication. The fast track designation allows for more frequent interactions with theJanuary 2020 and assigned a PDUFA target action date of November 8, 2020. We plan to launch it, pending FDAfor the early submission of some sections of the marketing application, and carries the potential for an expedited review category for the New Drug Application (NDA). Currently, we and the FDA have a SPA for the conduct of our Phase III program for SPN-810, using an agreed upon novel scale to measure IA that was developed by us. We initiated two Phase III clinical trials in 2015 (P301 and P302) that continue to enroll patients in 2017.Molindone hydrochloride was previously marketedapproval, in theUnited States as an anti-psychoticfourth quarter of 2020. We expect SPN-812, if approved, totreat schizophrenia under the trade name Moban, albeit at much higher dosages (50 to 225mg/day) than we are using in our development program (18 and 36 mg/day). Moban has not been commercially available since 2010 and the FDA has confirmed that the withdrawal from thehave five-year marketwas notexclusivity due toissues with safety or efficacy. Molindone hydrochloride is differentiated from other anti-psychoticsits NCE status inthat it is less likely to be associated with weight gain and, in preclinical models, has not caused increases in prolactin levels as seen with other anti-psychotic drugs.In addition, we believethelower doses tested for the proposed indication of IA in ADHD should be better tolerated than the higher doses approved to treat schizophrenia. The Phase IIb trial with SPN-810, which included 121 patients, showed that there was no difference in weight gain between patients treated with SPN-810 and those treated with placebo. Although initiallyU.S. Furthermore, we are developingSPN-810 as a novel treatment for IA in patients who have ADHD, if we are successful in demonstrating the effectiveness of SPN-810 in ADHD, we may then develop the product as a candidate for treating other indications; e.g., patients with IA in autism, PTSD, bipolar disorder, schizophrenia, and some forms of dementia. In the aggregate, we believe the addressable market for SPN-810 is greater than $6.3 billion, including $3.2 billion in ADHD, $0.8 billion in autism and $2.3 billion in PTSD.We are developing an intellectual property position aroundIP covering the novel synthesis process for the active ingredient in SPN-812, its novel use inIA,ADHD and its novelformulations. Patents, if issued, could expireextended release delivery system.SPN-812 Development ProgramThe Phase III pivotal program consisted of four three-arm, placebo-controlled trials: P301 and P303 trials in patients 6 to 11 years old; and P302 and P304 trials in patients 12 to 17 years old. We announced positive topline results from2029 to 2033.the pediatric trials (P301 and P303) and the first adolescent trial (P302) in December 2018. Results of the second adolescent Phase III trial (P304) were released in March of 2019.Wehave one patent issued eachinitiated a Phase III program in adults in theU.S., Mexico, Australia and Japan, covering modified release formulationsthird quarter ofmolindone hydrochloride. In another patent family, covering2019.Refer to thenovel process of synthesisCompany's Annual Report on Form 10-K for the year ended December 31, 2017 for the results of theactive ingredient, we have two patents issuedpreviously completed Phase IIb trials.Results of P301 and P303 Phase III trialsBoth studies were randomized, double‑blind, placebo controlled, multicenter, parallel group clinical trials in children 6 to 11 years of age who are diagnosed with ADHD. After titration, each treatment was administered orally once a day over five weeks in study P301, and over seven weeks in study P302. A total of 477 patients were randomized in theU.S. InP301 study, across placebo and two doses (100mg; 200mg), and athird patent family, covering usetotal ofmolindone hydrochloride313 patients were randomized intreating IA, we have one patent issuedthe P303 study, across placebo and two doses (200mg; 400mg). The primary objective of both studies was to assess the efficacy of SPN‑812 inJapan. We own allreducing the symptoms of ADHD in children. The primary outcome measure was the change, from baseline to the end of thepending applications.SPN-810 Development Programstudy, in the ADHD Rating Scale (RS‑5) total score. Safety and tolerability were assessed by monitoring: adverse events (AEs); clinical laboratory tests; vital signs; electrocardiograms (ECGs); suicidality; and physical examinations. Patients who completed the study were offered the opportunity to continue into an open‑label phase, that is currently on‑going.
On December 6, 2018, we announced positive topline results from the P301 and P303 Phase III studies of SPN‑812, having successfully met the primary endpoint. At daily doses of 100 mg and 200 mg in study P301, and at daily doses of 200mg and 400mg in study P303, statistically significant improvement in the symptoms of ADHD, from baseline to end of study, as measured by the ADHD‑RS‑5, was achieved. Patients receiving SPN‑812 100 mg and 200 mg had a −16.6 point change (p=0.0004) and a−17.7 point change (p<0.0001) from baseline, respectively, in the primary endpoint, vs. a −10.9 point change for placebo at week 6. This primary result, based on Mixed Model Repeated Measures (MMRM) analysis in the Intent‑To‑Treat (ITT) population, was confirmed by sensitivity analyses using Analysis of Covariance (ANCOVA) (100 mg, p=0.0008; 200 mg, p<0.0001). All SPN‑812 doses tested in the trials were well tolerated.The study demonstrated fast onset of action, reaching statistical significance for 100 mg and 200 mg doses as early as week 1, with p‑ values of 0.0004 and 0.0244, respectively. Statistical significance was maintained on a weekly basis through the end of the trial at week 6. In2012,addition, at the end of the study, SPN‑812 100 mg and 200 mg reached statistical significance compared to placebo on the hyperactivity/impulsivity and inattention subscales of the ADHD‑RS‑5, scale with p‑ values ranging from <0.0001 to 0.0026. Finally, SPN‑812 100 mg and 200 mg met all secondary endpoints, including the important analysis of the Clinical Global Impression Improvement (CGI‑I) secondary endpoint, with p‑ values of 0.002 and <0.0001, respectively, compared to placebo.At the end of the P303 Study, SPN‑812 200 mg and 400 mg doses reached statistical significance, as compared to placebo, in the primary endpoint. Patients receiving 200 mg and 400 mg had a −17.6 point change (p=0.0038) and a −17.5 point change (p=0.0063) from baseline to end of study, respectively, in the primary endpoint vs. a −11.7 point change for placebo at week 8. This primary result, based on MMRM analysis in the ITT population, was confirmed by sensitivity analyses using ANCOVA (200 mg, p=0.0058; 400 mg, p<0.0121).Onset of action for SPN‑812 showed clear differences compared to placebo starting by week 1, reaching statistical significance at week 5, which was sustained through the rest of the trial.As with the P301 study, at the end of the P303 study, SPN‑812 200 mg and 400 mg reached statistical significance compared to placebo on the hyperactivity/impulsivity and inattention subscales of the ADHD‑RS‑5, scale with p‑ values ranging from 0.0020 to 0.0248. In addition, 200 mg and 400 mg met the CGI‑I secondary endpoint, with p‑ values of 0.0028 and 0.0099, respectively, compared to placebo.Overall, both trials exhibited favorable tolerability and safety profiles, with low incidence of AEs across all doses. AEs were mild, leading to low discontinuation rates due to AEs, ranging from 2.2% to 4.8%. Treatment related AEs that reported at more than or equal to 5% included somnolence, headache, decreased appetite, fatigue and upper abdominal pain.Results of P302 Phase III trialOn December 20, 2018, wecompletedannounced positive topline results from the P302 Phase III study of SPN‑812 in patients 12 to 17 years old for the treatment of ADHD. The trial was successful in meeting the primary endpoint, demonstrating that SPN‑812 at daily doses of 200 mg and 400 mg achieved statistically significant improvement in the symptoms of ADHD, from baseline to end of study, as measured by the ADHD‑RS‑5. Each of the SPN‑812 doses tested in the trials was well tolerated.The study was aPhase IIbrandomized, double‑blind, placebo controlled, multicenter,randomized, double-blind, placebo-controlledparallel group clinical trial, inthe United States in pediatric subjects 6adolescents 12 to1217 years of age diagnosed withADHDADHD. Each treatment was administered orally once a day over six weeks, including the titration phase of the 400 mg dose group.A total of 310 patients were randomized across placebo andwith IA that is not controlled by optimal stimulant and behavioral therapy.two doses of SPN-812 (200 mg/400 mg). The primary objectiveof the studywas to assess the effect ofSPN-810SPN‑812 in reducingIA as measured bytheRetrospective-Modified Overt Aggression Scale (R-MOAS) after at least three weekssymptoms oftreatment. Secondary endpoints includedADHD in adolescents 12 to 17 years old. The primary outcome measure was therate of remission of IA and measurementchange, from baseline to the end of theeffectiveness of SPN-810 onstudy, in theClinical Global Impression (CGI) and ADHD scales as well as evaluation of the safetyADHD‑RS‑5 total score. Safety and tolerability of SPN‑812 were assessed by thedrug.monitoring of: AEs; clinical laboratory tests; vital signs; ECGs; suicidality; and physical examinations. Patients who completed the study were offered the opportunity to continue into an open‑label phase, currently on‑going.At the end of the P302 Study, 200 mg and 400 mg doses reached statistical significance, as compared to placebo, for the primary endpoint. Patients receiving 200 mg and 400 mg had a −16.0 point change (p=0.0232) and a −16.5 point change (p=0.0091) from baseline, respectively, in the primary endpoint, vs. a −11.4 point change for placebo, at week 6. This primary result, based on MMRM analysis in the ITT population, was confirmed by sensitivity analyses using ANCOVA (200 mg, p=0.0163; 400 mg, p=0.0055).The study demonstrated fast onset of action, reaching statistical significance for the 400 mg dose as early as week 1, with a p‑value of 0.0085, and maintaining statistical significance on a weekly basis through the end of the trial at week 6. Onset of action for the 200 mg dose showed clear difference compared to placebo starting by week 1, reaching statistical significance at week 3. This difference was sustained through the rest of the trial.As with the P301 and P303 studies, at the end of the P302 study, 200 mg and 400 mg doses reached statistical significance compared to placebo on the hyperactivity/impulsivity and inattention subscales of the ADHD‑RS‑5 scale, with p‑values ranging from 0.0005 to 0.0424. In addition, 200 mg and 400 mg doses met the CGI‑I secondary endpoint, with p‑values of 0.0042 and 0.0003, respectively, compared to placebo.Overall, the trial exhibited favorable tolerability and safety profiles, with low incidence of AEs across all doses. AEs were mild, leading to low discontinuation rates due to AEs, ranging from 1.9% to 4.1%. Treatment related AEs that reported at more than or equal to 5% for SPN‑812 included somnolence, fatigue, decreased appetite, headache and nausea.Results of P304 Phase III trialOn March 28, 2019, we announced topline results from the P304 Phase III study of SPN-812, in patients 12 to 17 years old for the treatment of ADHD.The study is a randomized, double-blind, placebo controlled, multicenter, parallel group clinical trial in adolescents 12 to 17 years of age, diagnosed with ADHD. Each treatment was administered orally once a day over seven weeks, including one week of titration for 400 mg and two weeks of titration for 600 mg.A total of 297 patients were randomized across placebo and two doses of SPN-812 (400 mg/600 mg). The primary objective was to assess the efficacy of SPN-812 in reducing the symptoms of ADHD, in adolescents 12 to 17 years old. The primary outcome measure was the change, from baseline to the end of the study, in the ADHD-RS-5 total score tested on the 600 mg followed by the 400 mg in the statistical plan. Safety and tolerability of SPN-812 were assessed by the monitoring of: AEs; clinical laboratory tests; vital signs; ECGs; suicidality; and physical examinations. Patients who completed the study were offered the opportunity to continue into an open-label phase,of six months duration.Analysis of treatment was performed using both parametric and non-parametric statistical methods. The parametric method assumes that data are normally distributed. Under this method, mean results of each treatment group at the end of three weeks of treatment were compared to the baseline R-MOAS score for each of the four dose groups (high, medium, low and placebo) using the t-test. The non-parametric method does not assume that data are normally distributed. Under this method, the median results of the change in R-MOAS score from baseline at the end of three weeks of treatment were computed for each of the four dose groups (high, medium, low and placebo). These were compared using the Wilcoxon Rank-sum test. Statistical analyses were performed to compare the median of each of the treatment groups: high, medium, and low versus placebo at the end of three weeks of treatment. The change in score from baseline to visit 10 was used as the outcome variable. There was a statistically significant difference between the low dose and placebo (p=0.031) and also between the medium dose and placebo (p=0.024) at thea=0.05 level. There was no statistically significant difference between the high dose and placebo. Both the medium dose and low dose were superior to placebo. These results convinced us that both low and medium doses were effective. This range of doses is being further evaluated in Phase III clinical trials.A secondary efficacy variable was the proportion of children whose impulsive aggressive behavior remitted, with remission defined as R-MOAS£10 at the end of the study. Low and medium doses of SPN-810 showed statistically significant results versus placebo, with percent of patients who experienced remission of impulsive aggressive behavior of 51.9% (p=0.009) and 40.0% (p=0.043), respectively.The CGI results (Severity and Improvement) are consistent with the findings on the R-MOAS scale, in that notable improvement (reduction in severity) occurred primarily in the low dose and medium dose groups. Scores on SNAP-IV Hyperactivity and Impulsivity items did not exhibit statistically significant differences across treatment groups, indicating that efficacy against IA was specific, rather than being efficacious against the underlying ADHD. Numerical trends in SNAP-IV Oppositional Defiant Disorder scores, while not always significant, consistently favored the low dose and medium dose groups over placebo.SPN-810 was well tolerated throughout the study across all doses. Sedation was the most frequently reported adverse reaction, with two subjects (7%) reporting this event in each of the four treatment groups, including the placebo group. The next most frequently reported adverse reaction was increased appetite with two subjects (7%) reporting this event in each of the three active treatment groups and one subject (3%) in the placebo group.The two serious AEs that occurred were not drug-related. One patient in the low dose arm and two patients in the medium dose arm had severe AEs that were considered either possibly or definitely related to the drug. Six patients in total discontinued the study because of AEs in the active treatment arms: one in low dose; two in medium dose; and three in high dose. AEs requiring dose reduction were infrequent.The frequency of AEs associated with extra-pyramidal symptoms was also low and the events were reversible. The data are too sparse to evaluate dose-related aspects of these reports; thus, no clear dose-response relationship can be assessed. SPN-810 exhibited a very good safety and tolerability profile, with low incidence of AEs, and no unexpected, life threatening, or dose-limiting safety issues.SPN-812 (viloxazine hydrochloride)
on-going.ADHD affects 6% to 9% of all school-age children and 3% to 5% of all adults. Current non-stimulant treatments for ADHD account for about 8% of the total ADHD prescriptions in the U.S. As a novel non-stimulant, SPN-812 has the potential to address a $2.5 billion market opportunity for the treatment of ADHD with non-stimulants. SPN-812, a norepinepherine reuptake inhibitor, would provide an additional option to the few non-stimulant therapiescurrentlyavailable. We believe that SPN-812 could be more effective than other non-stimulant therapies due to its different pharmacological profile.We expect SPN-812, if approved, to have five year market exclusivity, given its new chemical entity (NCE) status in the U.S. We are developing an intellectual property position around the novel synthesis process for the active ingredient, its novel use in ADHD and its novel extended release delivery.Our SPN-812 product candidate has three families of pending U.S. non-provisional and foreign counterpart patent applications. Patents, if issued, could expire from 2029 to 2033. We have one patent issued in Europe and one in Canada in one of these families, covering a method of treating ADHD using viloxazine hydrochloride. In another family, covering the novel process of active ingredient synthesis, we have two patents issued in the U.S. and one patent issued each in Europe, Mexico, and Australia. We have one patent issued in the U.S. covering modified release formulations of viloxazine. We own all of the pending applications.SPN-812 Development ProgramWe are developing SPN-812 as a novel non-stimulant treatment for ADHD. During 2016, we completed a Phase IIb dose ranging trial and announced topline results. The trial met the primary endpoint, demonstrating that SPN-812 at daily doses of 400 mg, 300 mg, and 200 mg achieved a statistically significant improvement in the symptoms of ADHD when compared to placebo. All SPN-812 doses tested in the trial were well tolerated. Of the patients treated with SPN-812, only 6.7% discontinued due to an AE. In addition, 87% of patients who completed the trial elected to enroll in the ongoing open-label extension.At the end of the study (EOS), SPN-812
study,400 mg300 mg and 200 mg doses were statistically significantreached statistical significance as compared to placebo,in meetingfor the primary endpoint.With respect to the primary endpoint, patientsPatients receivingSPN-812400 mg300 mg and 200 mghada –19.0 pointan -18.3 Least Squares (LS) Mean change(p=0.021), –18.6 point change (p=0.027) and a –18.4 point change (p=0.031)from baselinerespectively, as compared to –10.5(p=0.0082) vs. LS Mean change of-13.2 from baseline forplacebo.The treatment groupsplacebo at week 7. SPN-812400600 mg300 mg and 200 mg showed a standardized mean effect sizedid not reach statistical significance with an LS Mean change of0.63, 0.60 and 0.55 compared to placebo, respectively. Patients receiving SPN-812 100 mg had 16.7average mean change-16.7 (p=0.0712) from baseline in the primary endpoint at week 7. The result, based on MMRM analysis in the ITT population, was consistent with the results from sensitivity analyses using ANCOVA (400 mg, p=0.0191; 600 mg, p=0.1002) at week 7 (EOS), with placebo based imputation for missing data.
In addition,AEs across all doses. AEs were mild leading to low discontinuation rates, ranging from 4.0% to 5.1%. Treatment related AEs that reported at more than or equal to 5% for SPN-812 400were somnolence, fatigue, decreased appetite, headache and nausea.
Based on these positive results in children with ADHD and the positive Phase IIa results in adults with ADHD, Supernus plans to have an end-of-Phase II meeting with the FDA after which it plansin November 2019, and received acceptance of the filing in January 2020. The FDA has assigned a PDUFA target action date of November 8, 2020. We expect to initiate Phase III clinical testing duringlaunch SPN-812, assuming FDA approval, in the second halffourth quarter of 2017.
2020.
for this indication.
Treatment options for ADHD in the U.S. market can be broadly classified as either stimulant or as non-stimulant products. Shire plc isPlc, one of the leaders in the U.S. ADHD market, with threehas four marketed products: Vyvanse, a stimulant prodrugdrug product launched in 2007; Intuniv, a non-stimulant treatmentproduct launched in November 2009; and Adderall XR, an extended release stimulant treatment designed to provideproduct providing once-daily dosing.dosing, launched in October 2001; and Mydayis, a stimulant product launched in August 2017. Other marketed stimulant products for the treatment of ADHD in the U.S. market include the following once-daily formulations: Concerta,Concerta; Metadate CD,CD; Ritalin LA,LA; Focalin XR; Daytrana; Adzenys XR-ODT; Cotempla XR Daytrana,ODT; and Adzenys XR-ODT.Aptensio XR. Other marketed non-stimulants arein the U.S. include Strattera and Kapvay.
second line therapy.
Microtrol was also utilized to develop Mydayis, which was launched by Shire in 2017.
We cannot be sure that any patents, if granted, will sustain legal challenge.
patents; our ability to preserve the confidentiality of our trade secretssecrets; and to operate our business without infringing the patents and proprietary rights of third parties.
We have established and continue to build proprietary positions for
Patents for both Oxtellar XR andCompany entered into settlement agreements that allow third parties to enter the market by selling a generic version of Trokendi XR have received numerous Paragraph IV Notice Letters and we have filed claims for infringement of our patents against the third-parties.by January 1, 2023, or earlier under certain circumstances. For more information, please see Part I, Item 3—
In addition to the We have two issued patents for extended release oxcarbazepine in both Europe and patent applications relating to Oxtellar XR, we currently have eight U.S. patents that cover Trokendi XR. We haveAustralia, and one patent issued in each in Mexico, Australia, Japanof the following countries: Canada; Japan; China and Canada forMexico. In addition, we have a pending U.S. patent application that covers various extended release topiramate. We have two patents issued in Europe for extended release topiramate. The eight issued U.S. patents covering Trokendi XR will expire no earlier than 2027. We own all of the issued patents and pending applications.
formulations containing oxcarbazepine.
term may be lengthened byvia a patent term adjustment (PTA), which compensates a patentee for administrative delays by the USPTO in granting a patent. In viewBecause of a recent court decision, the USPTO is under greater scrutiny regarding its calculations becausein which the USPTO erred in calculating the PTA which resulted inby denying the patentee a portion of the patent term to which it was entitled. entitled, the USPTO is under greater scrutiny regarding its calculations of PTAs.
risk that one or more of our patents will be held invalid (in whole or in part,part; on a claim-by-claim basis) or held unenforceable. Such an adverse court ruling could allow third parties to commercialize products or use technologies that are similar to ours, and then compete directly with us, without paymentcompensation to us. See "Risk Factors—IfPart I, Item 1A—
Afecta Pharmaceuticals, Inc.
We have two license agreements with Afecta Pharmaceuticals, Inc. (Afecta) pursuant to which we obtained exclusive worldwide rights to selected product candidates, including an exclusive license to SPN-810. We may pay up to $300,000 upon the achievement of certain milestones. If a product candidate is successfully developed and commercialized, we will be obligated to pay royalties to Afecta based on worldwide net product sales at a rate in the low-single digits.
Product Approval
Government authorities in the United States at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, recordkeeping, promotion, advertising, distribution, marketing, export and import of products such as those we are developing. Our product candidates must receive final approval from the FDA before they may be marketed legally in the U.S.
In the U.S., the FDA regulates drugs under the Federal Food, Drug,
Table identification of Contents
FDA's refusal to approve pending applications, to withdraw an approval, to institute or issue a clinical hold, warning letters, product recalls, product seizures, product detention, total or partial suspension of production or distribution, to impose injunctions, fines, refusal of government contracts, restitution, disgorgement or civil or criminal penalties.
molecule that has the desired effect against a given disease. The process required by the FDA before a drug may be marketed in the U.S. generally involves the following:
•Completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices regulations;•Submission to the FDArequires submission of an IND, which must become effective before human clinicaltrialstrial testing maybegin;•Performancecommence. The results of pre-clinical testing, along with other information, including information about product chemistry, product manufacturing and controls, and a proposed clinical trial protocol are submitted to the FDA as part of the IND. Until the IND is approved, or becomes effective following a waiting period, we may not start the clinical trials. This is typically followed by additional preclinical laboratory and animal testing, as well as adequate and well-controlled human clinical trialsaccording to Good Clinical Practices (GCP)to establish the safety and efficacy of the proposed drug for its intendeduse;•Submissionuse. Satisfaction of FDA approval requirements typically takes many years. The actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
The testing and approval process requires substantial time, effort and financial resources and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all. Our total research and development expense was approximately $42.8 million and $29.1 million for each of 2016 and 2015, respectively. In order to continue the progress of our product candidates, significant increases in these expenditures will be required.
Once a suitable product candidate is successfully created, a preliminary development strategy is determined. Usually, an IND is opened with adequate preclinical and clinical trial material to permit initiation of the first proposed clinical trial. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. safety.
All clinical trials must be conducted underin compliance with applicable regulations and consistent with good clinical practices, as well as protocols detailing the supervisionobjectives of onethe trial, the parameters to be used in monitoring safety, and the parameters to determine effectiveness. Each protocol involving testing on patients, and subsequent protocol amendments, must be submitted to the FDA as part of the IND. The FDA may order the temporary halt or more qualified investigatorspermanent discontinuation of a clinical trial at any time, or to impose other sanctions if they believe that the clinical trial is not being conducted in accordance with GCP regulations. These regulations include the requirement that all research subjects provideapplicable requirements, or if continuing the trial presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent. Further,consent information for patients in clinical trials must also be submitted to an institutional review board (IRB) must review and approve the planor ethics committee, for any clinical trial before it commences at any institution. An IRB considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits.approval. The IRBIRB/ethics committee may also approves the protocol for conductingrequire the clinical trial andat the consent form that mustsite to be providedhalted, either temporarily or permanently, for failure to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
Once an IND is in effect, each new clinical protocol and any amendments to the protocol must be submittedcomply with the IND for FDA review, and to the IRBs for approval. The protocol details, amongIRB/ethics committee requirements, or they may impose other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety.
Human clinical trials for product candidates are typically conducted in three sequential phases that may overlap or be combined:
•Phase I.The product is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing may be conducted in patients.•Phase II.Phase II trials involve investigations in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage and schedule.•Phase III.In Phase III, clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for regulatory approval and product labeling.
Concurrent with clinical trials, companies usually complete additional animal studies, and must also develop additional information about the chemistry and physical characteristics of the product candidate andcandidate. They must finalize a process for manufacturing the product in commercial quantities, in accordance with cGMPcurrent good manufacturing practice (cGMP) requirements. Moreover, product used in late stage clinical trials must be manufactured under the proposed commercial process, and at the same scale as will be used commercially. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, thecandidate. The manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stabilitytested. Stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
The different indication. product. illegitimate products. USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. not reference to other clinical trials or data. 2019. Business If any of our major products were to become subject to problems, such as changes in prescription growth rates, unexpected side effects, loss of intellectual property protection, supply chain or product supply shortages, regulatory proceedings, changes in labeling, publicity adversely affecting doctor or patient confidence in our product, material product liability litigation, pressure from new or existing competitive products, or adverse changes in coverage under managed care programs, the adverse impact on our revenue and profit could be significant. In addition, our revenue and profit could be significantly impacted by the timing and rate of commercial acceptance of key new products. We are subject to uncertainty relating to payment or reimbursement policies which, if not favorable for our products or product candidates, could hinder or prevent our commercial success. made by payors. Reduced or partial payment, or reduced reimbursement coverage, could make our products or product candidates, including Oxtellar XR and Trokendi XR, less attractive to patients and prescribing physicians. We also may be required to sell our products or product candidates at a significant discount, which would adversely affect our ability to realize an appropriate return on our investment in our products or product candidates or to maintain profitability. If we fail to produce our products and product candidates in the volumes that we require on a timely basis, or fail to comply with stringent regulations applicable to pharmaceutical drug manufacturers, we may face delays in the development and commercialization of our products and product candidates, or be required to withdraw our products from the market. Accordingly, as it relates to SPN-812, for example, we may encounter additional manufacturing and supply-chain risks due to the regulatory and political structure of Switzerland, or as a result of the international relationship between Switzerland and the U.S. marketing exclusivity may be granted for the approval of new and sNDAs, including Section 505(b)(2) applications, for, among other things, new indications, dosage forms, routes of administration, strengths, or for a new use of an existing drug. If the clinical investigations that were conducted or sponsored by the applicant are determined by the FDA to be essential to the approval of the application, the FDA may grant exclusivity for the product, sometimes referred to as clinical investigation exclusivity. This prevents the FDA from approving an application under Section 505(b)(2) for the same conditions of use for new clinical investigations prior to the expiration of three years from the date of approval. Such exclusivity, however, would not prevent the approval of another application if the applicant submits a full NDA, and has conducted its own adequate, well-controlled clinical trials, demonstrating safety and efficacy. It would not prevent approval of a generic product or Section 505(b)(2) product that did not incorporate the exclusivity-protected changes of the approved drug product. financially significant to us. those subject areas. collaboration partners may fail to develop or effectively commercialize results of operations. differ from that required to obtain FDA approval. The regulatory approval process in other jurisdictions may include all of the risks detailed above regarding FDA approval in the U.S., as well as other risks. For example, legislation analogous to Section 505(b)(2) of the FDCA in the U.S., which relates to the ability of an NDA applicant to use published data not developed by such applicant, may not exist in other countries. In territories where data Depending on what changes the FDA may make to its orphan drug regulations and policies, our business could be adversely impacted. costs to assure compliance with post-approval regulatory requirements, and could result in potential restrictions on the sale and/or distribution of any approved product. employees, with the requisite skills and experience. may not be able to make improvements to our management information and control systems in an efficient or timely manner, and may discover deficiencies in existing systems and controls. In addition, our growth will cause us to comply with an increasing number of regulations and statutory requirements. If our management is unable to effectively manage our expected growth, our expenses may increase more than results of operations. Risks Related to Our Finances and Capital Requirements We may not be able to maintain or increase profitability. performance. If our quarterly operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Furthermore, any quarterly fluctuations in our operating results may, in turn, cause the price of our stock to fluctuate substantially. arrangement. Complying with increased financial reporting and securities laws reporting requirements has increased our costs and requires additional management resources. We may fail to meet these obligations. operations. any of the risks described in these "Risk Factors" could have a dramatic, material and adverse impact on the market price of our common stock. In addition, class action litigation has often been instituted against companies whose securities have experienced periods of volatility. Any such litigation brought against us could result in substantial costs and a diversion of management attention, which could hurt our business, operating results and financial condition. acquirers to obtain control of our board of directors or initiate actions that are opposed by the then-current board of directors. 2016 First Quarter Second Quarter Third Quarter Fourth Quarter 2015 First Quarter Second Quarter Third Quarter Fourth Quarter On December 31, May 1, 2012 December 31, 2012 December 31, 2013 December 31, 2014 December 31, 2015 December 31, 2016 Revenue Net product sales Royalty revenue Licensing revenue Total revenue Costs and expenses Cost of product sales Research and development Selling, general and administrative Total costs and expenses Operating income (loss) Other income (expense) Interest income Interest expense Interest expense-nonrecourse liability related to sale of future royalties Changes in fair value of derivative liabilities Loss on extinguishment of debt Other (loss) income Total other expense Earnings (loss) before income tax Income tax (benefit) expense Net income (loss) Cumulative dividends on Series A convertible preferred stock Net income (loss) attributable to common stockholders Income (loss) per common share: Basic Diluted Weighted-average number of common shares outstanding: Basic Diluted Consolidated Balance Sheet Data: Cash and cash equivalents and marketable securities Long term marketable securities Working capital Total assets Convertible notes, net of discount Nonrecourse liability related to sale of future royalties Secured notes payable, including current portion Accumulated deficit Total stockholders' equity candidates. ready. our intellectual property position. We Consolidated Financial Statements. and Related Accrued Research and Development Expenses whether partially or fully completed. Revenues: Net product sales Royalty revenue Licensing revenue Total revenues Costs and expenses Cost of product sales Research and development Selling, general and administrative Total costs and expenses Operating income Other income (expense) Interest income and other income, net Interest expense Interest expense-nonrecourse liability related to sale of future royalties Changes in fair value of derivative liabilities Loss on extinguishment of debt Total other expenses Earnings before income taxes Income tax (benefit) expense Net income products sold to our customers. commercial sales if the NDA is approved. securities holdings. The increase in securities holdings primarily resulted from the net proceeds of the March 2018 0.625% 2023 Convertible Senior Note issuance (2023 Notes), with a principal amount of $402.5 million. issued in March 2018. Revenues: Net product sales Royalty revenue Licensing revenue Total revenues Costs and expenses Cost of product sales Research and development Selling, general and administrative Total costs and expenses Operating income (loss) Other income (expense) Interest income and other income, net Interest expense Interest expense-nonrecourse liability related to sale of future royalties Changes in fair value of derivative liabilities Loss on extinguishment of debt Total other expenses Earnings (loss) before income taxes Income tax expense Net income (loss) the periods presented below are as follows (dollars in thousands): Net cash provided by (used in): Operating activities Investing activities Financing activities Net increase (decrease) in cash and cash equivalents Cash provided by operating income Cash provided by working capital Net cash provided by operating activities The reflects the timing impacts of cash collections on receivables and settlement of payables, as described below. (Increase) in accounts receivable (Increase) decrease in inventory Decrease (increase) in prepaid expenses and other assets Increase in accounts payable, accrued sales deduction, and accrued expenses Other 2018.Net cash used in investing activities decreased by $255.6 million, from $413.5 million in 2018 to $157.9 million in 2019, for the year ended December 31, warrants. Convertible Senior Secured Notes Interest on Convertible Notes Operating leases(1) Purchase obligations(2) Total(3) We have also entered into a purchase and sale agreement with Rune HealthCare Limited (Rune), where we obtained the exclusive worldwide rights to a product concept from Rune. There are no future milestone payments owing to Rune under this agreement. If we receive approval to market and sell any products based on the Rune product concept for SPN-809, we will be obligated to pay royalties product sales. convertible note hedges. In our opinion, the consolidated financial statements We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB. Consolidated Balance Sheets Assets Current assets: Cash and cash equivalents Marketable securities Accounts receivable, net Inventories, net Prepaid expenses and other current assets Total current assets Long term marketable securities Property and equipment, net Deferred legal fees Intangible assets, net Other non-current assets Deferred income taxes Total assets Liabilities and stockholders' equity Current liabilities: Accounts payable Accrued sales deductions Accrued expenses Non-recourse liability related to sale of future royalties, current portion Deferred licensing revenue Total current liabilities Deferred licensing revenue, net of current portion Convertible notes, net Non-recourse liability related to sale of future royalties, long term Other non-current liabilities Derivative liabilities Total liabilities Stockholders' equity: Common stock, $0.001 par value, 130,000,000 shares authorized at December 31, 2016 and 2015; 49,971,267 and 49,004,674 shares issued and outstanding at December 31, 2016 and 2015, respectively Additional paid-in capital Accumulated other comprehensive loss, net of tax Accumulated deficit Total stockholders' equity Total liabilities and stockholders' equity Earnings Revenue Net product sales Royalty revenue Licensing revenue Total revenue Costs and expenses Cost of product sales Research and development Selling, general and administrative Total costs and expenses Operating income (loss) Other income (expense) Interest income Interest expense Interest expense-nonrecourse liability related to sale of future royalties Changes in fair value of derivative liabilities Loss on extinguishment of debt Other (loss) income Total other expense Earnings (loss) before income taxes Income tax (benefit) expense Net income (loss) Basic Diluted Weighted-average number of common shares outstanding: Basic Diluted Earnings Net income (loss) Other comprehensive income (loss): Unrealized net gain (loss) on marketable securities, net of tax Other comprehensive income (loss): Comprehensive income (loss) Balance, December 31, 2013 Share-based compensation Issuance of employee stock purchase plan shares Exercise of stock options Equity issued on conversion of convertible notes Net loss Other comprehensive loss Balance, December 31, 2014 Share-based compensation Issuance of employee stock purchase plan shares Exercise of stock options Equity issued on conversion of convertible notes Exercise of warrants Net income Other comprehensive loss Balance, December 31, 2015 Share-based compensation Issuance of employee stock purchase plan shares Exercise of stock options Equity issued on conversion of convertible notes Net income Unrealized net gain (loss) on marketable securities, net of tax Other Balance, December 31, 2016 Cash flows from operating activities Net income (loss) Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: Loss on extinguishment of debt Change in fair value of derivative liability Unrealized loss on marketable securities Depreciation and amortization Amortization of deferred financing costs and debt discount Noncash interest expense on nonrecourse liability related to sale of future royalties Non-cash royalty revenue Share-based compensation expense Deferred income tax benefit Changes in operating assets and liabilities: Accounts receivable Inventories Prepaid expenses and other assets Accounts payable Accrued sales deductions Accrued expenses Deferred product revenue, net Deferred licensing revenue Other non-current liabilities Net cash provided by (used in) operating activities Cash flows from investing activities Purchases of marketable securities Sales and maturities of marketable securities Purchases of property and equipment Deferred legal fees Net cash used in investing activities Cash flows from financing activities Proceeds from issuance of common stock Cash settlement of debt to equity conversion Proceeds from sale of future royalties Net cash provided by financing activities Net change in cash and cash equivalents Cash and cash equivalents at beginning of year Cash and cash equivalents at end of year Supplemental cash flow information: Cash paid for interest Noncash financial activity: Conversion of convertible notes and interest make-whole Exercise of warrants Deferred legal fees included in accounts payable and accrued expenses April 2017. The Company January 2019. The Company evaluates the methodologies employed in making its estimates on an ongoing basis. investment grade. The Company's investments are furthermore classified as comprehensive earnings. Realized gains and losses are included in interest income and earnings. Net Payment terms for receivables are based on customary commercial terms and are predominantly less than one year. Customer A Customer B Customer C Concentration of Credit Risk share-based compensation; contract research and development services provided by third parties; costs for preclinical and clinical studies; cost of acquiring or manufacturing clinical trial material; regulatory costs; facilities costs; depreciation expense and other allocated expenses; and license fees and milestone payments related to in-licensed products and technologies. Assets acquired that are used for research and development and that have no future alternative use are expensed as in-process research and development. other current liabilities speaker programs. The costs of the Company's advertising efforts are expensed as incurred. earnings. expense in the relevant period. January 1, 2021. (Continued) Assets: Cash and cash equivalents Marketable securities Long term marketable securities Marketable securities—restricted (SERP) Total assets at fair value Liabilities: Derivative liabilities Assets: Cash and cash equivalents Marketable securities Long term marketable securities Marketable securities—restricted (SERP) Total assets at fair value Liabilities: Derivative liabilities Inputs are quoted prices for similar assets and liabilities in active markets; quoted prices for identical or similar assets and liabilities in markets that are not active; inputs other than quoted prices, that are observable for the asset or liability (e.g., interest rates; yield curves); and inputs that are derived principally from or corroborated by observable market data by correlation or by other means (i.e., market corroborated inputs). Balance at December 31, 2014 Changes in fair value of derivative liabilities included in earnings Reduction due to conversion of debt to equity Cashless exercise of common stock warrants Balance at December 31, 2015 Changes in fair value of derivative liabilities included in earnings Reduction due to conversion of debt to equity Balance at December 31, 2016 2019 or 2018. Corporate debt securities Corporate debt securities Less Than 1 Year 1 year to 2 years 3 years to 4 years Greater Than 4 Years Total The Company has not experienced any other-than-temporary losses on its marketable estimated based on actual trade information, as well as quoted prices provided by bond traders. Raw materials Work in process Finished goods Computer equipment Software Lab equipment and furniture Leasehold improvements Construction in progress Less accumulated depreciation and amortization 2019, there were no identified indicators of impairment. respective patents. Capitalized patent defense costs Less accumulated amortization January 1, 2023, or earlier under certain circumstances. impairment. and Other Current Liabilities Accrued compensation Accrued professional fees Accrued clinical trial and clinical supply costs Accrued product costs Accrued sales and marketing expenses Accrued interest expense Other accrued expenses Due 2023 2023 Notes. The Company also granted the Initial Purchasers an over-allotment option to purchase, within a 30-day period, up to an additional $52.5 million principal amount of 2023 Notes, on the same terms and conditions which the Initial Purchasers exercised in full on March 15, 2018. The total principal amount of 2023 Notes was $402.5 million. election, based on the applicable conversion rate. The initial conversion rate Indenture occurs, then noteholders may require the Company to repurchase their 2023 Notes at a cash repurchase price equal to the principal amount of the 2023 Notes to be repurchased, plus accrued and unpaid interest, if any. Gross proceeds Initial value of interest make-whole derivative reported as debt discount Conversion option reported as debt discount and APIC Conversion of debt to equity—principal Conversion of debt to equity—accretion of debt discount and deferred financing costs Accretion of debt discount and deferred financing costs December 31, 2015 carrying value Conversion of debt to equity—principal Conversion of debt to equity—accretion of debt discount and deferred financing costs Accretion of debt discount and deferred financing costs December 31, 2016 carrying value held. Plan Research and development Selling, general and administrative Total The fair value of each option award is estimated on the date of the grant, using the Black-Scholes option-pricing model and the assumptions in the following table: Fair value of common stock Expected volatility Dividend Yield Expected term Risk-free interest rate Expected forfeiture rate Outstanding, December 31, 2014 Granted Exercised Forfeited Outstanding, December 31, 2015 Granted Exercised Forfeited Outstanding, December 31, 2016 As of December 31, 2016: Vested and expected to vest Exercisable The weighted-average 2023 Notes, the Company entered into Convertible Note Hedge and Warrant Transactions as described further in Note 9, Shares underlying Convertible Senior Secured Notes Warrants to purchase common stock Stock options, stock appreciation rights, and ESPP awards Numerator, in thousands: Net income (loss) used for calculation of basic EPS Interest expense on convertible debt Changes in fair value of derivative liabilities Loss on extinguishment of debt Loss on extinguishment of outstanding debt, as if converted Total adjustments Net income used for calculation of diluted EPS Denominator: Weighted average shares outstanding, basic Effect of dilutive potential common shares: Shares underlying Convertible Senior Secured Notes Shares issuable to settle interest make-whole derivatives Stock options and stock appreciation rights Total potential dilutive common shares Weighted average shares outstanding, diluted Net income (loss) per share, basic Net income (loss) per share, diluted Current Federal State Deferred Federal State Total Income tax expense/(benefit) computed at U.S. Federal statutory tax rate Permanent items State income taxes Change in valuation allowance Uncertain income tax position Research and development credits Other Deferred rate change Income tax (benefit)/expense Deferred tax assets: Net operating loss carryforward Deferred rent credit Accrued compensation and non-qualified stock options Deferred financing costs Depreciation and amortization Research and development credits Capitalized overhead into inventory (UNICAP §263A) Nonrecourse liability related to sale of future royalties Other AMT credit Valuation allowance Net deferred tax asset Deferred tax liability: Debt discount on convertible notes Infringement legal cost Depreciation Net deferred taxes In assessing the realizability of deferred income tax assets, management considers whether it is expense in the future. may be subject to tax audit adjustment. Balance as of January 1 Gross (decreases) increases related to prior-year tax positions Gross increases related to current-year tax positions Gross decreases related to prior-year tax positions Gross decreases related to current-year tax positions Change in tax rates Balance as of December 31 expiring statutes of limitation. Year ending December 31: 2017 2018 2019 Thereafter 2016 Revenue Total costs and expenses Operating income Net income Net income per share, basic Net income per share, diluted 2015 Revenue Total costs and expenses Operating income Net income Net income per share, basic Net income per share, diluted 2019. All internal control systems, no matter how well designed, have inherent limitations. Because of Annual Report on Form 10-K. 2019. Equity compensation plans approved by security holders Equity compensation plans not approved by security holders Total Data product development,all preclinical, studiesclinical and clinical trials,other testing, along with descriptionsa description of the manufacturing process, validation of the manufacturing process, analytical tests conducted on the drug, proposed labeling and other relevant information are submitted to the FDA as part of an NDA for a new drug.information. The NDA requests approval to market the product.NDAs are Each NDA is subject to a substantial user fee at the time of submission, unless a waiver is granted by the FDA. A holder of an approved NDA may also be subject to annual product and establishment user fees. These fees typically increase annually.orNDA (Full NDA) and the Section 505(b)(2) applications. ForNDA.standard 505(b)(1) application,Full NDA, must contain all pertinent information must be partand full reports of investigations conducted by the applicant to demonstrate the safety and effectiveness of the regulatory submission under that NDA number.drug, as well as complete preclinical, clinical and manufacturing information.interprets Section 505(b)(2) of the FDCA to permitpermits the applicant to rely upon the FDA's previous findings of safety and effectiveness for an approved product. The FDA requires submission of information needed to support any changes to a previously approved drug, such as published data or new studies conducted by the applicant, including bioavailability or bioequivalence studies, or clinical trials demonstrating safety and effectiveness. The FDA may then approve the new product candidate for all or some of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.submission of an NDASection 505(b)(2) regulatory approval process is subjectdesigned to the payment ofallow for potentially expedited, lower cost and lower risk regulatory approval, based on previously established safety, efficacy and manufacturing information on a substantial user fee, although a waiver of such fee may be obtained under certain limited circumstances.In addition, under the Pediatric Research Equity Act of 2003,drug which was reauthorized under the Food and Drug Administration Safety and Innovation Act of 2012, an NDA must contain,a priori, or propose clinical work that supports the product's use in all relevant pediatric subpopulations. The FDA may grant deferrals for submission of data or full or partial waivers of the data requirements. Pursuant to the FDA's approval of Oxtellar XR, we committed to the conduct of four pediatric post-marketingstudies; however,has been already approved by the FDA granted a waiver for the pediatric study requirements for ages birth to one month andsame or a deferral for submission of post-marketing assessments for children one month to six years of age. Pursuant to the FDA's approval of Trokendi XR, the FDA granted a deferral for submission of post-marketing pediatric studies in the following categories: (1) adjunctive therapy in partial onset seizures (POS) for children one month to less than six years of age, (2) initial monotherapy in POS and primary generalized tonic-clonic (PGTC) for children two years to less than ten years of age, and (3) adjunctive therapy in PGTC and adjunctive therapy in Lennox-Gastaut Syndrome from two years to less than six years of age.Since our product approvals, we have gained more knowledge about our abilities to create formulations, and programs that would enable us to meet our deferred pediatric commitments, Supernus has identified a need to renegotiate the commitments made at the time of NDA approvals for both Oxtellar XR and Trokendi XR. Supernus is actively interfacing with the FDA on these programs and these commitments.Section 505(b)(2) New Drug Applicationsathe Section 505(b)(2) NDA reliesapplicant is relying on clinical trialsstudies conducted for a previously approved drug product or the FDA's prior findings of safety and effectiveness for aon previously approved drug product, the Section 505(b)(2) applicant must submit patent certifications in its Section 505(b)(2) application with respect to any patents for the approved product on which the application relies that are listed in the FDA's publication, Approved Drug Products with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book. Specifically, the applicant must certify for each listed patent that either: (1) the required patent information has not been filed; or (2) the listed patent has expired; or (3) the listed patent has not expired, but will expire on a particular date, and approval is not sought until after patent expiration; or (4) the listed patent is invalid, unenforceable or will not be infringed by the proposed new product. A certification that the new product will not infringe the previously approved product's listed patent, or that such patent is invalid or unenforceable, is known as a Paragraph IV certification. also not approve, as applicable, a Section 505(b)(2) NDA application until any non-patent exclusivity has expired, such as for example,example: five-year exclusivity period for obtaining approval of an NCE,NCE; or three year exclusivity period for an approval based on new clinical trials,trials; or pediatric exclusivity, listed in the Orange Book for the referenced product, has expired.NDA submissions formarket sooner than our product candidates,candidate. Further, the time and financial resources required to obtain FDA approval for SPN-812 could substantially and materially increase. After we intend to followgain approval for SPN-812 for one indication, additional indications may be submitted using the Section 505(b)(2) development pathway when appropriate.FDA Review of New Drug ApplicationsThe FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing.regulatory pathway. The FDA may request additional information rather than accept annot approve our filing under Section 505(b)(2) for SPN-812 for other indication(s), and therefore would require a full NDA for filing. In such case, the time and financial resources required to obtain approval could also significantly increase.event,policy include showing clinical superiority to the product with the orphan drug exclusivity, or if the license holder cannot supply sufficient quantities of the product. Orphan drug exclusivity in the U.S., which is seven years, does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition, provided the sponsor has conducted appropriate clinical trials required for approval. Among the other benefits of orphan drug designation are tax credits for certain research expenses and waiver of the NDA must be re-submitted withapplication user fee for the additional information. The re-submitted application alsoorphan indication.subjecteligible for priority review, or review within six months from filing, for a new molecular entity (NME). In addition, a six month review period may pertain to review beforea non-NME, if the FDA accepts itdrug candidate provides a significant improvement as compared to marketed drugs in the treatment, diagnosis, or prevention of a disease. A fast track designated drug candidate would ordinarily meet the FDA’s criteria for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantivepriority review. The FDA reviewsmakes its determination of priority or standard review during the 60-day filing period post the initial NDA submission.determine,continuing regulation by the FDA, including, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant to assure and preservethings: record-keeping requirements; reporting of AE’s with the product's identity, strength, quality and purity. Before approving an NDA,product; providing the FDA will inspectwith updated safety and efficacy information; product sampling and distribution requirements; complying with certain electronic records and signature requirements; and complying with FDA promotion and advertising requirements.facilityapproved indication and in accordance with the provisions of the approved label. Changes to some of the conditions established in an approved application, including changes in indications, labelling, or manufacturing processes or facilities, wheremay require submission to further review, and approval by the productFDA before the change can be implemented.manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications.following marketing approval. The FDA may referalso require post-marketing testing, known as Phase IV testing, REMS, and surveillance to monitor the effects of an approved product, or to place conditions on an approval that could restrict the distribution and use of the product.an advisory committee for review, evaluationinterface with the FDA on these programs and recommendationthese commitments.whetherconform to cGMPs after approval. Drug manufacturers and other entities involved in the application should bemanufacturing and distribution of approved and under what conditions. An advisory committee is a panel of independent experts who provide advice and recommendations when requesteddrugs are subject to periodic unannounced inspections by the FDA on mattersand by certain state agencies for compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of importance that come beforeproduction and quality control to maintain compliance with cGMPs. Regulatory agencies may withdraw product approval or request product recalls if a company fails to comply with regulatory standards, or if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered. In addition, prescription drug manufacturers in the agency. The FDA is not bound by the recommendation of an advisory committee.The approval process is lengthy and difficult and the FDA may refuse to approve an NDA if theU.S. must comply with applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA in its present form. The complete response letter usually describes allprovisions of the specific deficiencies that the FDA identifiedDrug Supply Chain Security Act, and: provide and receive product tracing information; maintain appropriate licenses; ensure they only work with other properly licensed entities; and have procedures in the NDA. The deficiencies identified may be minor; for example, requiring labeling changes, or major; for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might takeplace to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, withdraw the application, or then request an opportunity for a hearing.If a product receives regulatory approval, the approval may be significantly limited to specific diseasesidentify and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase IV testing which involves clinical trials designed to further assess a drug's safetyproperly handle suspect and effectiveness after NDA approval and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.patent term extensionPTE under the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent term restoration of up to five years as compensation for patent term lost during product development and during the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product's approval date. The patent term restoration period is generally one-half50% the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent and within sixty days of approval of the drug. TheFDCAFederal Food, Drug, and Cosmetic Act (FDCA) provides a five-year period of non-patent marketing exclusivity within the United StatesU.S., to the first applicant to gain approval of an NDA for an NCE. A drug is an NCE if the FDA has not previously approved any other new drug containing the same active pharmaceutical ingredient (API) or active moiety, which is the molecule or ion responsible for the therapeutic action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application (ANDA) or a Section 505(b)(2) NDA submitted by another company for another version of such drug, where the applicant does not own or have a legal right of reference to all the data required for approval. As an alternative to submission via 505(b)(2) approval, an applicant may choose to submit a full Section 505(b)(1) NDA, but such an NDAwherein the applicant would be required to conduct its own preclinical and adequate, well-controlled clinical trials to demonstrate safety and effectiveness. Further, a Section 505(b)(2) applicationThey may be submitted after four years if it contains a Paragraph IV certification.supportsupport: new indications, dosages,indications; dosages; routes of administrationadministration; or strengths of an existing drug, ordrug. Alternatively, these trials may be for a new use, if the new clinical investigations that were conducted or sponsored by the applicant are determined by the FDA to be essential to the approval of the application. This exclusivity, sometimes referred to as clinical investigation exclusivity, prevents the FDA from approving an application under Section 505(b)(2) for the same conditions of use associated with the new clinical investigations before the expiration of three years from the date of approval. Such three-year exclusivity, however, would not prevent the approval of another application if the applicant submits a Section 505(b)(1) NDA and has conducted its own adequate, well-controlled clinical trials demonstrating safety and efficacy, nor would it prevent approval of a generic product or Section 505(b)(2) product that did not incorporate the exclusivity-protected changes of the approved drug product. The FDCA, FDA regulations and other applicable regulations and policies provide incentives to manufacturers to create modified, non-infringing versions of a drug, to facilitate the approval of an ANDA or other application for generic substitutes.current pediatric exclusivity provision was reauthorizedU.S. has enacted a number of legislative and regulatory proposals to change the healthcare system in September 2007.Post-Approval RequirementsAny drugs for which we receive FDA approval are subjectways that could affect our ability to continuing regulationsell our products profitably. In the U.S., the Patient Protection and Affordable Care Act of 2010, as amended by the FDA, including, among other things, record-keeping requirements, reporting of AEs with the product, providing the FDA with updated safetyHealth Care and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. In September 2007, the Food and Drug Administration AmendmentsEducation Reconciliation Act of 2007 was enacted, giving2010 (as amended), is a sweeping measure intended to improve quality of care, constrain healthcare spending, and expand healthcare coverage within the U.S. This is accomplished primarily through imposition of health insurance mandates on employers, and individuals and expansion of the Medicaid program.enhanced post-marketing authority, including the authority to require post-marketing studies and clinical trials, labeling changes basedrestrictions on new safety information, and compliance with risk evaluations and mitigation strategies approved by the FDA. The FDA strictly regulates labeling, advertising, promotion andmarketing of pharmaceutical products, several other types of information on products that are placed onstate and federal laws have been applied to restrict certain business and marketing practices in the market. Drugs may be promoted only for the approved indicationspharmaceutical industry in recent years. These laws include: anti-kickback; false claims; patient data privacy; and in accordancesecurity and transparency statutes and regulations.with the provisions of the approved label. Further, manufacturers of drugs must continueThe U.S. Foreign Corrupt Practices Act (FCPA), to comply with cGMP requirements, which are extensive and require considerable time, resources and ongoing investment to ensure compliance. In addition, certain changes to the manufacturing process generally require prior FDA approval before being implemented. Other types of changes to the approved product, such as adding new indications and additional labeling claims,we are also subject, prohibits corporations and individuals from engaging in certain activities to further FDA review and approval.Drug manufacturersobtain or retain business, or to influence a person working in an official capacity. Under FCPA, it is illegal to pay, offer to pay or authorize the payment of anything of value to any foreign government official, government staff member, political party or political candidate, in an attempt to obtain or retain business, or to otherwise influence a person working in an official capacity. Historically, pharmaceutical companies have been the target of FCPA and other entities involved in the manufacturinganti-corruption investigations and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the drug. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards, and test each product batch or lot prior to its release. We rely, and expect to continue to rely on, third parties for the production of clinical quantities of our product candidates. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution or may require substantial resources to correct.The FDA may withdraw a product approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, warning letters, holds on clinical trials, product recalls or seizures, product detention or refusal to permit the import or export of products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, injunctions or civil or criminal penalties.From time to time, legislation is drafted, introduced and passed by the United States Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. For example, in July 2012, the Food and Drug Administration Safety and Innovation Act was enacted, expanding drug supply chain requirements and strengthening FDA's response to drug shortages, among other things. In addition to new legislation, the FDA regulations and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.Foreign RegulationUnited States,U.S., we are subject to a variety of foreign regulations governing clinical trials, and commercial sales, andas well as distribution of our product candidates, to the extent we choose to clinically evaluate or sell any products outside of the U.S. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparableappropriate regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The requirements, approval process and time frame varies from country to country and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.each jurisdiction. As in the United States,U.S., post-approval regulatory requirements, such as those regarding product manufacture, marketing, or distribution would apply to any product that is approved outside the U.S.Table We generally market our products outside of ContentsThird-Party Payor Coverage and ReimbursementIn both the U.S. and foreign markets, our abilitythrough licensing arrangements.commercializePart 1, Item 1A—Risk Factors, for discussion of risks associated with government regulations.product candidates successfully, and to attract commercialization partners for our product and product candidates, dependsdistributors who, in significant part on the availability of adequate financial coverage and reimbursement from third party payors, including, in the U.S., governmental payors such as the Medicare and Medicaid programs, managed care organizations, and private health insurers. Medicare is a federally funded program managed by the Centers for Medicare and Medicaid Services (CMS), through local fiscal intermediaries and carriers that administer coverage and reimbursement for certain healthcare items and services furnished to the elderly and disabled. Medicaid is an insurance program for certain categories of patients whose income and assets fall below state defined levels and who are otherwise uninsured. It is both federally and state funded and managed by each state. The federal government sets general guidelines for Medicaid while each state creates specific regulations that govern its individual program. Each payor has its own process and standards for determining whether it will cover and reimburse a procedure or particular product. Private payors often rely on the lead of the governmental payors in rendering coverage and reimbursement determinations. Therefore, achieving favorable CMS coverage and reimbursement is usually a significant gating issue for successful introduction of a new product. The competitive position of some of our products will depend, in part, upon the extent of coverage and adequacy of reimbursement for such products and for the indications in which such products are used. Prices at which we or our customers seek reimbursement for our product candidates can be subject to challenge, reduction or denial by the government and other payors.The United States Congress and state legislatures may, from time to time, propose and adopt initiatives aimed at cost containment, which could impact our ability toturn, sell our products profitably. For example, in March 2010, President Obama signed into law the Patient Protectionto pharmacies, hospitals and Affordable Care Act as amended by the HealthCare and Education Reconciliation Act of 2010, which we refer to collectively as the HealthCare Reform Law. This is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the healthcare industry, and impose additional healthcare policy reforms. Effective October 1, 2010, the HealthCare Reform Law revises the definition of "average manufacturer price" for reporting purposes, which could increase the amount of Medicaid drug rebates paid by drug companies to states once the provision is effective. Further, since 2011, the HealthCare Reform Law imposes a significant annual fee on companies that manufacture or import branded prescription drug products. Substantial new provisions affecting compliance have also been enacted, which may require us to modify our business practices with healthcare practitioners. We will not know the full effects of the HealthCare Reform Law until applicableother customers, including federal and state agencies issue regulations or guidance under the new law. Although it is too early to determine the effectentities. Each of the HealthCare Reform Law, the new law appears likely to continue to put pressure on pharmaceutical pricing, especially under the Medicare program,three customers, AmerisourceBergen Drug Corporation, Cardinal Health, Inc. and may also increase our regulatory burden and operating costs. Moreover, in the coming years, additional changes are likely to be made to governmental healthcare programs that could significantly impact the successMcKesson Corporation, accounted for more than 30% of our total product candidates.The cost of pharmaceuticals continues to generate substantial governmentalrevenue in 2019, and third party payor interest. We expect that the pharmaceutical industry will experience pricing pressures due to the trend toward managed healthcare, the increasing influence of managed care organizations and additional legislative proposals. Our results of operations could be adversely affected by current and future healthcare reforms. Some third party payors also require pre-approval of coveragecollectively accounted for new or innovative devices or drug therapies before they will reimburse healthcare providers that use such therapies.While we cannot predict whether any proposed cost-containment measures will be adopted or otherwise implemented in the future, the announcement or adoption of these proposals could have a materialadverse effect on our ability to obtain adequate prices for our product candidates and operate profitably.Other HealthCare Laws and Compliance RequirementsIn the United States, our activities are potentially subject to statutes and regulation by various federal, state and local authorities in addition to the FDA, including CMS, other divisions of the United States Department of Health and Human Services (e.g., the Office of Inspector General), the United States Department of Justice and individual United States Attorney offices within the Department of Justice, and state and local governments. These statutes and regulations include:•The federal healthcare program Anti-Kickback Statute, which prohibits, among other things, persons from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. The term remuneration has been interpreted broadly to include anything of value, meaning that arrangements with healthcare professionals must be scrutinized carefully for compliance with the Statute;•The Federal False Claims Act ("FCA"), which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other government reimbursement programs that are false or fraudulent. We may be subject to FCA claims for, among other things, promoting our products for off-label indications or making false and misleading statements about the products' safety and efficacy;•The federal Health Insurance Portability and Accountability Act of 1996, which prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and which also imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information and places restrictions on the use of such information for marketing communications;•The federal Physician Payments Sunshine Act within the Patient Protection and Affordable Care Act, (PPACA), and its implementing regulations, and similar state law provisions require manufacturers of drugs, devices, biologics, and medical supplies to report to the Department of Health and Human Services information related to physician payments and other transfers of value and physician ownership and investment interests;•The FDCA, which among other things, strictly regulates the distribution of drug samples and drug product marketing and prohibits manufacturers (i) from marketing drugs for off-label use; and (ii) from making false and misleading statements about a products' safety or efficacy;•State law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts;•State laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state;•Laws and ordinances in various state and local jurisdictions that require pharmaceutical manufacturers to establish and fund drug "take-back" programs that allow consumers to return unused prescription and OTC medications for safe disposal;•Various additional laws and regulations which result from our status as a supplier of pharmaceutical products to various agencies of the US federal government. Participation in certain government programs requires that discounted prices be offered for purchases by the government via a rebate system. Participation in these programs can require submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations;•The Federal Drug Supply Chain Security Act (DSCSA), which requires development of an electronic pedigree to track and trace each prescription drug at the salable unit level through the distribution system, which will be effective incrementally over a 10-year period. Compliance with DSCSA and any additional federal or state electronic pedigree requirements may increase our operational expenses and impose significant administrative burdens; and•Allmore than 90% of our activities potentially subject to federal and state consumer protection and unfair competition laws.Depending on the circumstances, failure to comply with these laws and regulations can resulttotal product revenue in penalties, including criminal, civil, and/or administrative criminal penalties, damages, fines, disgorgement, exclusion of products from reimbursement under government programs, "qui tam" actions brought by individual whistleblowers in the name of the government, refusal to allow us to enter into supply contracts, including government contracts, reputational harm and diminished profits and future earnings, any of which could adversely affect our business.2016,2019, we employed 363464 full-time employees; 85 employees are engaged in research and development activities and 278 employees are engaged in selling, general and administrative activities.employees. We consider relations with our employees to be good. None of our employees is represented by a labor union.BusinessIndustry and Industry•and intellectual property and products from competition, including generics;••inventory of our products to meet demand;•revenue growth;and grow revenue;•••••intellectual propertyIP rights alleging that our products infringe their rights; and•In addition, weWe will need to continue investing substantial financial and management resources to maintain our commercial sales and marketing infrastructure and to recruit and train qualified marketing, sales and other personnel.Increasesor Trokendi XR may slow for a variety of reasons, including competing products or safety issues. If we are notin the U.S. Our business and growth prospects could be materially impaired.broadening the current commercial acceptanceobtaining approval to manufacture and market its own version of either Oxtellar XRextended release oxcarbazepine or Trokendi XR, our business would be harmed.Any increase in sales of Oxtellar XR and Trokendi XR will be dependent on several factors, including our ability to educate physicians and to increase physician awareness and acceptance of the benefits and cost-effectiveness of our products relative to competing products. Our ability to increase market acceptance of any of our products or gain market acceptance of approved product candidates among physicians, patients, health care payors and the medical community will depend on a number of factors, including:•Acceptable evidence of safety and efficacy;•Relative convenience and ease of administration;•The prevalence, nature, and severity of any adverse side effects;•Availability of alternative treatments; and•Pricing and cost effectiveness.In addition, Oxtellar XR and Trokendi XR will be subject to continual review by the FDA. We cannot assure that newly discovered or reported safety issues will not arise. With the use of any newly marketed drug by a wider patient population, serious AEs may occur from time to time that initially do not appear to relate to the drug itself. Any safety issues could cause us to suspend or cease marketing of our approved products, cause us to modify how we market our approved products, subject us to substantial liabilities and adversely affect our revenues and financial condition. In the event of awithdrawal of either Oxtellar XR or Trokendi XR from the market, our revenues would decline significantly and our business would be seriously harmed and could fail.We are involved in lawsuits to protect or enforce our patents, which could be expensive, time consuming and unsuccessful.Competitors may infringe our patents. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming. For example, we are involved in several matters related to Paragraph IV Certification Notice Letters that we have received in connection with our products and our collaborators' products. In connection with an ANDA, a Paragraph IV Certification Notice Letter notifies the FDA that one or more patents listedtopiramate in the FDA's Orange Book is alleged to be invalid, unenforceable or will not be infringed by the ANDA product. These matters include claims related to Oxtellar XR and Trokendi XR, and are discussed in Part I, Item 3—Legal Proceedings.In any infringement proceeding, including the foregoing, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patients at risk of being invalidated or interpreted narrowly and could put our patent application at risk of not issuing.Interference proceedings brought by the USPTO may be necessary to determine the priority of inventions with respect to our patents and patent applications or those of our collaborators. An unfavorable outcome could require us to cease using the technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if a prevailing party does not offer us a license on terms that are acceptable to us or at all. Litigation or interference proceedings may fail. Even if successful, litigation may result in substantial costs and distraction of our management and other employees. We may not be able to prevent, alone or with our collaborators, misappropriation of our proprietary rights, particularly in countries where the laws may not protect those rights as fully as in the United States.Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.In addition, there could be public announcements of the results of hearings, motions or other interim proceeding or developments. If securities analysts or investors perceive these results to be negative, or perceive that the presence or continuation of these cases creates a level of uncertainty regarding our ability to increase or sustain products sales, it could have a substantial adverse effect on the price of our common stock. There can be no assurance that our product candidates will not be subject to the same risks.We are dependent on obtaining regulatory approval of our product candidates and for additional indications for existing products.Our ability to successfully commercialize any of our product candidates and to obtain additional indications for existing products will depend on, among other things, our ability to:•Successfully complete our clinical trials;•Receive marketing approvals from the FDA;•Produce, through a validated process, sufficiently large quantities of our product candidates to permit successful clinical development and commercialization;•Establish commercial manufacturing arrangements with third-party manufacturers;•Build and maintain strong sales, distribution and marketing capabilities sufficient to commercially launch our product candidates;•Secure acceptance of our product candidates from physicians, health care payors, pharmacies, wholesalers, patients and the medical community; and•Manage our spending as costs and expenses increase due to undertaking clinical trials and commercially launching product candidates.There are no guarantees that we will be successful in completing these tasks. If we are unable to successfully complete these tasks,U.S., we may not be able to commercialize any of our other product candidates in a timely manner, or at all, in which case we may be unable to maximize our revenues. In addition, if we experience unanticipated delays or problems, development costs could substantially increase and our business, financial condition and results of operations would likely be adversely affected.We may not be able to effectively market and sell our product candidates, if approved, in the U.S.We plan on building our sales and marketing capabilities in the U.S. to commercialize our product candidates, if approved. We will build such capabilities by investing significant amounts of financial and management resources. Furthermore, the cost of establishing and maintaining marketing and sales capabilities may not be justifiable in light of the revenues generated by any of our product candidates.If we are unable to establish and maintain adequate sales and marketing capabilities for our product candidates or are unable to do so in a timely manner, we may not be able to generate sufficient productprospectively realize revenues from these product candidates to be profitable.Final marketing approval of any of our product candidatesOxtellar XR or additional indications for existing products by the FDA or other regulatory authorities may be delayed, limited, or denied, any of which would adversely affect our ability to generate operating revenues.Our business depends on the successful development and commercialization of our product candidates. We are not permitted to market any of our product candidates in the U.S. until we receive approval of an NDA from the FDA, or, in any foreign jurisdiction, from the requisite authority. Satisfaction of regulatory requirements typically takes many years, is dependent upon the type, complexity and novelty of the product and requires the expenditure of substantial resources. We cannot predict whether or when we will obtain regulatory approval to commercialize our product candidates and we cannot, therefore, predict the timing of any future revenues from these product candidates. In addition, we have received tentative approval from the FDA for Trokendi XR as a treatment for prophylaxis of migraines in adults; however, we cannot predict if, or when, we will obtain full regulatory approval for this indication. Therefore, we cannot predict the timing of any future revenues, if any, from the sale of Trokendi XR for this indication.The FDA has substantial discretion in the drug approval process, including the ability to delay, limit or deny approval of a product candidate or a prior approval supplement for many reasons. For example, the FDA:
XR.•Could reject or delay the marketing application for an NCE;•Could determine that we cannot rely on Section 505(b)(2) for any of our product candidates;•Could determine that the information provided by us was inadequate, contained clinical deficiencies or otherwise failed to demonstrate the safety and effectiveness of any of our product candidates for any indication;•May not find the data from bioequivalence studies and/or clinical trials sufficient to support the submission of an NDA or to obtain marketing approval in the U.S., including any findings that the clinical and other benefits of our product candidates outweigh their safety risks;•May disagree with our trial design or our interpretation of data from preclinical studies, bioequivalence studies and/or clinical trials, or may change the requirements for approval even after it has reviewed and commented on the design for our trials, the outcome and measurement scale used in the trials, and the clinical protocols whether with or without a special protocol assessment process;•May determine that we have identified the wrong reference listed drug or drugs or that approval of our Section 505(b)(2) application of our product candidate is blocked by patent or non-patent exclusivity of the reference listed drug or drugs;•May identify deficiencies in the manufacturing processes or facilities of third-party manufacturers with which we enter into agreements for the supply of raw materials, including the API or manufactured product candidates used in our product candidates, wherein those deficiencies may result in interruption in the ability to supply product;•May approve our product candidates for fewer or more limited indications than we request, or may grant approval contingent on the performance of costly post-approval clinical trials;•May change its approval policies or adopt new regulations; or•May not approve the labeling claims that we believe are necessary or desirable for the successful commercialization of our product candidates, or the addition of new indications to the label of our existing products.Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(1) and 505(b)(2), over the last few years some pharmaceutical companies and others have objected to the FDA's interpretation of Section 505(b)(2). If the FDA changes its interpretation of Section 505(b)(2), or if the FDA's interpretation is successfully challenged in court, this could delay or even prevent the FDA from approving any Section 505(b)(2) application that we submit. Any failure to obtain regulatory approval of our product candidates would significantly limit our ability to generate revenues, and any failure to obtain such approval for all of the indications and labeling claims we deem desirable could reduce our potential revenues.and Trokendi XR, and our product candidates, including SPN-812, will depend in part on the coverage and reimbursement levels set by governmental authorities, private health insurers, managed care organizations and other third-party payors. As a threshold for coverage and reimbursement, third-party payors generally require that drug products be approved for marketing by the FDA. Third-party payors also are increasingly challenging the effectiveness of and prices charged for medical products and services. Government authorities and these third-party payors have attempted to control costs, in some instances, by limiting coverage, andby limiting the amount of reimbursement for particular medications, or by encouraging the use of lower-cost generic products. We cannot be sure that reimbursement will be available for any of the products that we develop and, if reimbursement is available, the level of reimbursement. Moreover, that level of reimbursement may change over time, as a result of decisionsatto what level.extent they will provide reimbursement. Moreover, they will consider the efficacy and cost effectiveness of comparable or competitive products, including generic products, in making reimbursement decisions for our products. Because each third-party payor individually approves payment or reimbursement, obtaining these approvals can be a time consuming and expensive process, that could requirerequiring us to provide scientific or clinical support for the use of each of our products or product candidates separately to each third-party payor. In some cases, it could take several months or years before a particular private insurer or managed care organization reviews a particular product. We may ultimately be unsuccessful in obtaining coverage. OurIn addition, our competitors may have larger organizations, as well asmore extensive existing business relationships with third-party payors relating to theirthat could have an impact on the coverage for our products.six6 to 12 months or longer after the receipt of regulatory approval and product launch. To obtain favorable reimbursement for the indications sought or to obtain pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our products or product candidates, if approved, to other available therapies. If reimbursement for our products or product candidates is unavailable in any country in which reimbursement is sought, limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be materially harmed, and could be unprofitable.and establishthrough the use of formularies, which establish pricing and reimbursement levels. Exclusion of a product from a formulary can lead to its sharply reduced usage in the managed care organization's patient population. If our products or product candidates are not included within an adequate number of managed care formularies or reimbursed at adequate payment or reimbursement levels, or if those policies increasingly favor generic products, our market share and gross margins could be negatively affected, whichaffected. This would have a material adverse effect on our overall business and financial condition.experience pricingcontinue and potentially to intensify in 2020 and following years, as political pressures duemount, and healthcare payors, including government-controlled health authorities, insurance companies and managed care organizations, step up initiatives to potentialreduce the overall cost of healthcare, reforms discussed elsewhere in this Annual Reportrestrict access to higher-priced new medicines, increase the use of generic products and to impose overall price cuts. Such pressures could have a material adverse impact on Form 10-K,our business, financial condition, or results of operations, as well as due to cost control measures instituted by health maintenance organizations.Our failure to successfully developon our reputation.market our product candidates would impair our ability to grow.As partdistributors for retail distribution of our growth strategy,Oxtellar XR and Trokendi XR. If we intend to develop and market additional product candidates. We may spend several years completing our development of a particular current or future internal product candidate, during which process we can experience failure at any stage. The product candidates to which we allocate our resources may not be commercially successful. In addition, because our internalresearch capabilities are limited, we may be dependent upon pharmaceutical companies, academic scientists and other researchers to sell or license products or technology to us. The success of this strategy depends partly upon our ability to identify, select, discover and acquire promising pharmaceutical product candidates and approved products.The process of proposing, negotiating and implementing a license or acquiring a product candidate or approved product is lengthy and complex. Other companies, including some with substantially greater financial, marketing and sales resources, may compete with us for the license or the product candidate or approved product. We have limited resources, including financial resources, to identify and execute the acquisition or in-licensing of third-party products, businesses and technologies and to integrate them into our current infrastructure. Moreover, we may devote resources to potential acquisitions or in-licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts. We may not be able to acquire the rights to additional product candidates on terms that we find acceptable, or at all.In addition, future acquisitions may entail numerous operational and financial risks, including:•Exposure to unknown liabilities;•Disruption of our business and diversion of our management's time and attention to develop acquired products or technologies;•Incurring substantial debt or dilutive issuances of securities or depletion of cash to pay for acquisitions;•Incurring higher than expected acquisition, integration, and operating costs;•Difficulty in combining the operations and personnel of any acquired businesses with our operations and personnel;•Impairing relationships with key suppliers or customers of any acquired businesses due to changes in management and ownership; and•An inability to retain and/or motivate key employees of any acquired businesses.Our clinical trials for our product candidates may fail to demonstrate acceptable levels of safety, efficacy or any other requirements, which could prevent or significantly delay regulatory approval.We may be unable to sufficiently demonstrate the safety and efficacy of our product candidates to obtain regulatory approval. We must demonstrate, with substantial evidence gathered in well-controlled studies, to the satisfaction of the relevant regulatory authorities that each product candidate is safe and effective for use in the target indication. We may be required to conduct or perform additional studies or trials to adequately demonstrate safety and efficacy, which could prevent or significantly delay our receipt of regulatory approval, increase clinical costs, and, ultimately delay the commercialization of that product candidate.Any product candidate that we acquire may require additional development prior to commercial sale, including extensive clinical testing and approval by the FDA and applicable foreign regulatory authorities. All product candidates are prone to risks of failure typical of pharmaceutical product development, including the possibility that a product candidate will not be shown to be sufficiently safe and effective for approval by regulatory authorities.In addition, the results from the trials that we have completed for our product candidates may not be replicated in future trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced development, even after promising results in earlier trials. If our product candidates are not shown to be safe and effective, our clinical development programs might be terminated.We rely on and will continue to rely on outsourcing arrangements for certain of our activities, including clinical research of our product candidates, manufacturing of our compounds and product candidates beyond Phase II clinical trials and the manufacturing of our commercial products.We rely on outsourcing arrangements for some of our activities, including manufacturing, preclinical and clinical research, data collection and analysis, and electronic submission of regulatory filings. We may have limited control over these third parties and we cannot guarantee that they will perform their obligations in an effective, competent and timely manner. Our reliance on third parties, including third-party clinical research organizations (CROs) and CMOs, entails risks including, but not limited to:•Non-compliance by third parties with regulatory and quality control standards;•Sanctions imposed by regulatory authorities if compounds supplied or manufactured by a third party supplier or manufacturer fail to comply with applicable regulatory standards;•Possible breach of the agreements by the CROs or CMOs because of factors beyond our control or the insolvency or other financial difficulties of any of these third parties, labor unrest, natural disasters or other factors adversely affecting their ability to conduct their business; and•termination or non-renewal of an agreement by a third party, at a time that is inconvenient for us, for reasons not entirely under our control.We do not own or operate manufacturing facilities for the production oflose any of our productssignificant wholesalers or product candidates beyond Phase II clinical trials, nor do we have plans in the foreseeable future to developdistributors, our own manufacturing operations for Phase III clinical materials or commercial products. We currently depend on third-party CMOs for allbusiness could be harmed.required raw materials and drug substance for our preclinical research and clinical trials. Forsales of Oxtellar XR and Trokendi XR we currently rely on single source suppliersare made to wholesalers and distributors who, in turn, sell our products to pharmacies, hospitals and other customers. For the year ended December 31, 2019, three wholesale pharmaceutical distributors, AmerisourceBergen Drug Corporation, Cardinal Health, Inc. and McKesson Corporation, each individually accounted for raw materials, including API, and rely on third-party suppliers and manufacturers for the final commercial products. If any of these vendors are unable to perform their obligations to us, including due to violations of the FDA's requirements, our ability to meet regulatory requirements, projected timelines, necessary quality standards for successful manufacturemore than 30% of our developmenttotal revenue in 2019, and commercialization product would be adversely affected. Further, if we were required to change vendors, it could resultcollectively accounted for more than 90% of our total revenue in delays in our regulatory approval efforts and significantly increase our costs. Accordingly, the2019. The loss of any of our currentthese wholesale pharmaceutical distributors' accounts, or future third-party manufacturers or suppliersa material reduction in their purchases, could have a material adverse effect on our business, results of operations, financial condition, and prospects.have entered into supply agreements for bothcannot assure you that we can manage these pricing pressures, or that wholesaler purchases will not fluctuate unexpectedly from period to period.with leading CMOs headquartered in North America forcan be greatly affected by the manufactureinventory levels our respective wholesalers and distributors carry. We monitor wholesaler and distributor inventory of the final commercial products. However, there is a risk that the counterparties to these agreements will not perform their respective obligations or will terminate these agreements. We could also become embroiled in disputes with third party manufacturers for Oxtellar XR and Trokendi XR regarding the termsusing a combination of methods. Pursuant to distribution service agreements with our agreements, the performance of a CMO or intellectual property rights, any of which could disrupt the sales of our products and adversely affect our reputation andthree largest wholesale customers, we receive product revenue. In addition,inventory reports. For other wholesalers where we do not have contractual relationshipsreceive inventory reports, our estimates of wholesaler inventories may differ significantly from actual inventory levels. Significant differences between actual and estimated inventory levels may result in excessive production, resulting in our holding substantial quantities of unsold inventory, or alternatively inadequate supplies of product in distribution channels, resulting in inability to support sales at the retail level. These changes may cause our revenues to fluctuate significantly from quarter to quarter, and in some cases may cause our operating results for a particular quarter to be below our expectations, the manufactureexpectations of commercial suppliessecurities analysts and/or investors.alla particular period, the market price of our common stock may drop significantly. The number of third-party manufacturers with the expertise, required regulatory approvalsfacilitiesmaintain adequate sales and marketing capabilities for our product candidates; if we are unable to manufacture drug substance and final drug product ondo so in a commercial scale is limited. Therefore,timely manner, we may not be able to enter into such arrangements with third-party manufacturers in a timely manner,generate sufficient product revenues from our product candidates to be profitable.acceptable terms, or at all. Failure to secure such contractual arrangements would harmobtaining regulatory approval of our product candidates and approval for additional indications for existing products. Our business depends on the commercial prospects forsuccessful clinical development; i.e., successful completion of clinical trials and commercialization of our product candidates. Our costsWe are not permitted to market any of our product candidates in the U.S. until we receive approval of an NDA from the FDA, or in any foreign jurisdiction, from the requisite authority. Satisfaction of regulatory requirements typically takes many years, is dependent upon the type, complexity and novelty of the product, and requires the expenditure of substantial resources. We cannot predict whether or when we will obtain regulatory approval to commercialize our product candidates. We cannot, therefore, predict the timing of any future revenues from these product candidates.increaselimit the acceptance of our product candidates and their commercial success; orcould be delayed.Delays or failures in the completion of clinical development of our product candidates would increase our costs and delay or limit our ability to generate revenues.Delays or failures in the completion of clinical trials for our product candidates could significantly raise our product development costs. We do not know whether current or planned trials will be completed on schedule, if at all. The commencement and completion of clinical development can be delayed or halted for a number of reasons, including:•Difficulties obtaining regulatory approval to commence a clinical trial or complying with conditions imposed by a regulatory authority regarding the scope or term of a clinical trial;•Delays in reaching orthat candidate. Any failure to reach agreement on acceptable terms with prospective CRO trial sites and investigators, the terms of which can be subject to extensive negotiation and may vary significantly;•Insufficient or inadequate supply or quantity of a product candidateobtain such approval for use in trials;•Difficulties obtaining Investigational Research Board (IRB) or ethics committee approval to conduct a trial at a prospective site;•Challenges recruiting and enrolling patients to participate in clinical trials for a variety of reasons, including competition from other programs for the treatment of similar conditions;•Severe or unexpected drug-related side effects experienced by patients in a clinical trial;•Difficulty retaining patients who have enrolled in a clinical trial but may be prone to withdraw due to side effects from the therapy, lack of efficacy or personal issues;•Clinical holds; or•Clinical trials may be delayed as a result of ambiguous or negative interim results.Clinical trials may be suspended or terminated by us, at a trial site by a Data Safety Monitoring Board (DSMB) or ethics committee overseeing the clinical trial, the FDA, or other regulatory authorities due to a number of factors, including:•Failure to conduct the clinical trial in accordance with regulatory requirements or the trial protocols;•Observations during inspectionall of the clinical trial operations or trial sites by the FDA or other regulatory authorities that ultimately result in the imposition of a clinical hold;•Unforeseen safety issues; or•Lack of adequate funding to continue the trial.Failure to conduct the clinical trial in accordance with regulatory requirements or the trial protocols may result in the inability to use the trial data to support product approval. Changes in regulatory requirementsindications and guidance may occur, andlabeling claims we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmitdeem desirable could reduce our clinical trial protocols to IRBs or ethics committees for reexamination, which may adversely impact the costs, timing or successful completion of a clinical trial.In addition, many of the factors that cause or lead to a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates. If we experience delays in completion of, or if we terminate any of our clinical trials, our ability to obtain regulatory approval for our product candidates may be materially harmed, and our commercial prospects and ability to generate product revenues diminished.If other versions of extended or controlled release oxcarbazepine or topiramate are approved and successfully commercialized, our business could be materially harmed.Third parties have and may receive approval to manufacture and market their own versions of extended release oxcarbazepine or topiramate anti-epileptic drugs in the U.S. For example, Upsher-Smith launched Qudexy XR (extended release topiramate) and its own authorized generic, both of which compete with Trokendi XR. Since Trokendi XR was not granted marketing exclusivity by the FDA, we may not be able to prevent the submission or approval of another full NDA for any competitor's extended or controlled release topiramate product candidate. However, we do have the right to defend our products against third parties who may infringe or are infringing our patents.In addition, we are aware of companies who are marketing modified-release oxcarbazepine products outside of the U.S., such as Apydan, which is developed by Desitin Arzneimittel GmbH and requires twice-daily administration. If companies with modified-release oxcarbazepine products outside of the U.S. pursue or obtain approval of their products within the U.S., such competing products may limit the potential success of Oxtellar XR in the U.S., and our business and growth prospects would be materially impaired. Accordingly, if any third party is successful in obtaining approval to manufacture and market its own version of extended release oxcarbazepine or topiramate in the U.S., we may not be able to recover expenses incurred in connection with the development of or prospectively realize revenues from Oxtellar XR or Trokendi XR.If we do not obtain marketing exclusivity for our product candidates, our business may suffer.Under the Hatch-Waxman Amendments, three years of marketing exclusivity may be granted for the approval of new and supplemental NDAs, including Section 505(b)(2) applications, for, among other things, new indications, dosage forms, routes of administration, strengths of an existing drug or for a new use, if the new clinical investigations that were conducted or sponsored by the applicant are determined by the FDA to be essential to the approval of the application. This exclusivity, which is sometimes referred to as clinical investigation exclusivity, prevents the FDA from approving an application under Section 505(b)(2) for the same conditions of use associated with the new clinical investigations before the expiration of three years from the date of approval. Such exclusivity, however, would not prevent the approval of another application if the applicant submits a Section 505(b)(1) NDA and has conducted its own adequate, well-controlled clinical trials demonstrating safety and efficacy, nor would it prevent approval of a generic product or Section 505(b)(2) product that did not incorporate the exclusivity-protected changes of the approved drug product.Under the Hatch-Waxman Amendments, newly-approved drugs and indications may also benefit from a statutory period of non-patent marketing exclusivity. The Hatch-Waxman Amendments provide five-year marketing exclusivity to the first applicant to gain approval of an NDA for an NCE, meaning that the FDA has not previously approved any other drug containing the same API, or active moiety, which is the molecule responsible for the action of the drug substance. Although protection under the Hatch-Waxman Amendments will not prevent the submission or approval of another full Section 505(b)(1) NDA, such an NDA applicant would be required to conduct its own preclinical and adequate, well-controlled clinical trials to demonstrate safety and effectiveness.While the FDA granted a three year marketing exclusivity period for Oxtellar XR, it did not grant a similar marketing exclusivity period for Trokendi XR. If we are unable to obtain marketing exclusivity for our subsequent product candidates, then our competitors may obtain approval of competing products more easily than if we had such marketing exclusivity, and our future revenues could be reduced, possibly materially.Our products and product candidates may cause undesirable side effects or have other characteristics that limit their commercial potential or delay or prevent their regulatory approval.Undesirable side effects caused by any of our product candidates could cause us or regulatory authorities to interrupt, delay or halt development and could result in the denial of regulatory approval by the FDA or other regulatory authorities, and result in potential product liability claims. Undesirable side effects caused by any of our products could cause regulatory authorities to temporarily or permanently halt product sales, which could have a material adverse effect on our business as a whole.Immediate release oxcarbazepine and topiramate products, which use the same active pharmaceutical ingredients as Oxtellar XR and Trokendi XR, are known to cause various side effects, including but not limited to dizziness, paresthesia, headaches, cognitive deficiencies such as memory loss and speech impediment, digestive problems, somnolence, double vision, gingival enlargement, nausea, weight gain, oral malformation birth defects, visual field defects, infant small for gestational age and fatigue. The use of Oxtellar XR and Trokendi XR may cause similar side effects as compared to their reference products, or may cause additional or different side effects.If our products cause side effects or if any of our product candidates receive marketing approval, and we or others later identify undesirable side effects caused by our products or product candidates, a number of potentially significant negative consequences could result, including:•Regulatory authorities may withdraw approval of the product candidate or otherwise require us to take the approved product off the market;•Regulatory authorities may require additional warnings, or a narrowing of the indication, on the product label;•We may be required to create a medication guide outlining the proper use of the medication and risks of side effects, for distribution to patients;•We may be required to modify the product in some way;•Regulatory authorities may require us to conduct additional clinical trials or costly post-marketing testing and surveillance to monitor the safety or efficacy of the product;•Sales of approved products may decrease significantly;•We could be sued and held liable for harm caused to patients; or•Our reputation may suffer.Any of these events could prevent us from achieving or maintaining the commercial success of our products and product candidates and could substantially increase commercialization costs.We may not be able to manage our business effectively if we are unable to attract, motivate and retain key members of our management team.We may not be able to attract or motivate qualified management and scientific and clinical personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses. Our industry has experienced a high rate of turnover of management personnel in recent years. If we are not able to attract and motivate necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our objectives.We are highly dependent on the development, regulatory, commercial and financial expertise of our management, particularly Jack A. Khattar, our President and Chief Executive Officer. Mr. Khattar has an employment agreement and other members of the senior management team have executive retention agreements. If we lose key members of our management team, we may not be able to findsuitable replacements in a timely fashion, if at all. We cannot be certain that future management transitions will not disrupt our operations or generate concern among employees and those with whom we do business.In addition to competition for personnel, our corporate offices are located in the greater Washington D.C. metropolitan area, an area that is characterized by a high cost of living. As such, we could have difficulty attracting experienced personnel to our Company and may be required to expend significant financial resources in our employee recruitment efforts.If our competitors develop or market alternatives for treatments of our target indications, our commercial opportunities will be reduced or eliminated.The pharmaceutical industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary therapeutics. We face competition from a number of sources, some of which may target the same indications as our products and product candidates, including large pharmaceutical companies, smaller pharmaceutical companies, biotechnology companies, academic institutions, government agencies and private and public research institutions. The availability of new products or approval for new indications for existing products may limit the demand for and the price we are able to charge for any of our products. We may be unable to differentiate our products from competitive offerings. In addition to competition with our currently marketed products, we anticipate that we will face intense competition when our pipeline product candidates are approved by regulatory authorities and we begin the commercialization process for these products.There are currently no marketed products and no known products in development for the treatment of IA in patients with ADHD, autism, or PTSD. However, the off-label use of risperidone (Risperdal) and aripiprazole (Abilify) is common. These products are approved for irritability in autism which, as a result, may influence use of products to treat IA in patients with ADHD.In addition, we are aware of several companies have various product candidates under development for ADHD which may compete with our SPN-812 product candidate. Such companies include Alcobra, Sunovion, Neos Therapeutics, and Neurovance.Further new developments, including the development of other drug technologies, may render our products or product candidates obsolete or noncompetitive. As a result, our products and product candidates may become obsolete before we recover expenses incurred in connection with their development or realize revenues from commercialization. Further, many competitors have substantially greater:•Capital resources;•Research and development resources and experience, including personnel and technology;•Drug development, clinical trial and regulatory resources and experience;•Sales and marketing resources and experience;•Manufacturing and distribution resources and experience;•Name recognition; and•Resources, experience and expertise in prosecution and enforcement of intellectual property rights.As a result of these factors, our competitors may obtain regulatory approval of their products more rapidly than we are able to or may obtain patent protection or other intellectual property rights that limit or block us from developing or commercializing our product candidates. Our competitors may also develop drugs that are more effective, more useful, better tolerated, subject to fewer or less severe sideeffects, more widely prescribed or accepted or less costly than ours and may also be more successful than us in manufacturing and marketing their products. If we are unable to compete effectively with the products of our competitors or if such competitors are successful in developing products that compete with any of our product candidates that are approved, our business, results of operations, financial condition and prospects may be materially and adversely affected. Mergers and acquisitions in the pharmaceutical industry may result in even more resources being concentrated at competitors. Competition may increase further as a result of advances made in the commercial applicability of technologies and greater availability of capital for investment.Our products and our product candidates may be subject to restrictions or withdrawal from the market. We may be subject to penalties if we fail to comply with regulatory requirements.Even though U.S. regulatory approval has been obtained for Trokendi XR and Oxtellar XR, the FDA may still impose significant restrictions on their indicated uses or marketing or impose ongoing requirements for costly post-approval studies. Our product candidates would also be, and our approved product and our collaborators' approved products are, subject to ongoing FDA requirements governing the labeling, packaging, storage, advertising, promotion, recordkeeping and submission of safety and other information. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations. If we, our collaborators or a regulatory authority discovers previously unknown problems with a product, including side effects that are unanticipated in severity or frequency, or problems with the facility where the product is manufactured, a regulatory authority may impose restrictions on that product or the manufacturer, including requiring withdrawal of the product from the market or suspension of manufacturing. If we or our collaborators, or our or our collaborators' approved products or product candidates, or the manufacturing facilities for our or our collaborators' approved products or product candidates fail to comply with applicable regulatory requirements, a regulatory authority may:•Issue warning letters or untitled letters;•Impose civil or criminal penalties;•Suspend regulatory approval;•Suspend any ongoing bioequivalence and/or clinical trials;•Refuse to approve pending applications or supplements to applications filed by us;•Impose restrictions on operations, including costly new manufacturing requirements, or suspension of production for a sustained period of time; or•Seize or detain products or require us to initiate a product recall.In addition, our product labeling, advertising and promotion of our approved products, are subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product's approved labeling. Notwithstanding, physicians may nevertheless prescribe our products to their patients in a manner that is inconsistent with the approved label, which is known as "off label use". The FDA and other authorities actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have promoted off-label uses may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. If we are found to have promoted off-label uses, we may be enjoined from such off-label promotion and becomesubject to significant liability, which would have an adverse effect on our reputation, business and revenues, if any.forat commercial productsscale in the foreseeable future. We currently depend on third-party contract manufacturersCMOs in various countries for the supply of the APIsAPI for our products and product candidates, including drug substance for our preclinical research and clinical trials. For Oxtellar XR and Trokendi XR, we currently rely on single source suppliers for raw materials, including API. We rely onAPI, as well as single manufacturerssource suppliers to produce and package final dosage forms. With respect to product candidates, we currently rely on Bachem Americas, Inc. and Bachem AG (collectively Bachem), a company based in Switzerland, as the sole supplier and manufacturer for viloxazine hydrochloride raw material, the API in SPN-812.include manufacturing difficulties relating tocan adversely affect production costs and yields, quality control, stability of the product and quality assurance testing, shortages of qualified personnel, as well as compliance with federal, state and foreign regulations. If we are unable to demonstrate stability in accordance with commercial requirements, or if our manufacturers were to encounter difficulties or otherwise fail to comply with their obligations to us, our ability to maintainobtain or obtainmaintain FDA approval and to market our products and product candidates, respectively, would be jeopardized. In addition, any delay or interruption in producing clinical trial supplies could delay or prohibit the completion of our clinical trials, increase the costs associated with conducting our clinical trials and, depending upon the period of delay, require us to commence new trials, at significant additional expense, or to terminate a trial.A failureFailure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. If the safety of any of our products or product candidates is compromised due to failure to adhere to applicable laws, or for other reasons, we may not be able to obtain regulatory approval for such product candidate, or to successfully commercialize such products, and weproducts. We may be held liable for any injuries sustained as a result. Any of these factors could cause a delay in clinical development, regulatory submissions, approvals or commercialization of our product candidates, entail higher costs, or result in our being unable to effectively commercialize our product candidates. Furthermore, if we fail to obtain the required commercial quantities on a timely basis from our suppliers and at commercially reasonable prices, we may be unable to meet demand for our approved products, and consequently lose potential revenues.or may not be able to sell our products profitably.TableContents The FDCA, FDA regulations and other applicable regulations and policies provide incentives to manufacturers to create modified, non-infringing versions of a drug to facilitate the approval of an ANDA or other application for generic substitutes. These manufacturers might only be required to conduct a relatively inexpensive study to show that their product has the same active ingredient(s), dosage form, strength, route of administration, and conditions of use or labeling, as our product or product candidate and that the generic product is bioequivalent to our product. Bioequivalence implies that a product is absorbed in the body at the same rate and to the same extent as our product or product candidate. These generic equivalents, which must meet the same quality standards as branded pharmaceuticals, would be significantly less costly than ours to bring to market. Companies that produce generic equivalents are generally able to offer their products at significantly lower prices. Thus, regardless of the regulatory approval pathway, after the introduction of a generic competitor, a significant percentage of the sales of any branded product are typically lost to the generic product, through both price and volume erosion. Accordingly, competition from generic equivalents would adversely, materially, permanently and adverselypermanently impact our revenues, profitability and cash flows andfrom those products. In this eventuality, it would substantially limit our ability to obtain a return on the investments we have made in our products and product candidates.discloseda result of advances made in the commercial applicability of technologies, and as a result of greater availability of capital for investment.3—Legal Proceedings1—Post-approval Regulatory Requirements for more information. To date, the only consequence of this Annual Reportour failure to meet our PREA commitment deadlines has been a notation on Form 10-K, we received Paragraph IV Notice Letters against our Oxtellar XR andFDA websites, making the status of PREA publicly known.Orange Book patentsfor the treatment of prophylaxis of migraine. If we do not meet our post-marketing commitments, and are unable to show good cause for our inability to adhere to the timetables laid out in the approval letters, the FDA could take enforcement action against us, including withdrawal of approval. While we believe that we can show good cause for our inability to meet the timelines for our post-approval study requirements, the FDA may disagree. Refer to Part I, Item 1—Post-approval Regulatory Requirements for more information.several generic drug makers. Wethe market or suspension of manufacturing.lawsuit against eachsustained period of these drug makers alleging infringementtime; orOxtellar XRapproved products are subject to regulatory requirements and Trokendi XR patents.continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about prescription products. In October 2015,particular, a product may not be promoted for uses that are not approved by the FDA, as reflected in the product's approved labeling. Notwithstanding, physicians may nevertheless prescribe products to their patients in a manner that is inconsistent with the approved label, which is known as "off label use". The FDA and other authorities actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have promoted off-label use may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion, and has enjoined companies from engaging in off-label promotion. If we reached a settlement agreement with oneare found to have promoted off-label use, we may be enjoined from such off-label promotion and become subject to significant liability. This could have an adverse effect on our reputation, business and revenues. these generic drug makers, Par Pharmaceutical Companies, Inc., concerning our Trokendi XR patents. In 2016, the U.S. District Court and Federal Court of Appeals ruled in our favor against Actavis concerning Oxtellar XR patents. In March 2017, we signed settlement agreements with two other generic drug makers, Actavis and Zydus, concerning our Trokendi XR patents. While we intend to vigorously defend our product rights against TWi concerning Oxtellar XR patents,candidates. If we are slow or unable to adapt to changes in the event thatexisting requirements, or to adopt new requirements or policies, or if we are not successfulable to maintain regulatory compliance, we may lose any marketing approval that we have obtained, adversely affecting our business, prospects and ability to achieve or sustain profitability.lawsuit,treatment of pulmonary arterial hypertension, and other indications. United Therapeutics Corporation launched Orenitram (treprostinil) in 2014, which triggered payment of a milestone payment to us of $2.0 million. In the third quarter of 2014, we received a cash payment of $30.0 million from HealthCare Royalty Partners III, L.P.'s (HC Royalty), for the purchase of certain of our rights under our license agreement with United Therapeutics Corporation related to the commercialization of Orenitram. Ownership of the royalty rights will return to us if/when a certain cumulative threshold payment to HC Royalty is reached. We are entitled to receive milestones and royalties for use of this formulation in indications other than arterial hypertension. If we materially breach any of our obligations under the license agreement, we could lose the right to receive any future sales of Oxtellar XR willroyalty payments thereunder, which could be significantly, adversely and permanently affected by competition from this generic drug., who may fail to effectively commercialize our products and product candidates.Outside We utilize strategic partners outside the U.S., we utilize strategic partners where appropriate, to assist in the commercialization of our products and product candidates. We currently possess limited resources, and may not be successful in establishing collaborations or licensing arrangements on acceptable terms, if at all. We also face competition in our search for collaborators and licensing partners. By entering into strategic collaborations or similar arrangements, we will rely on third parties for financial resources andto financially support their local operations, including support required for development, commercialization, sales, and marketing and regulatory expertise. Our collaborators may fail to develop or effectively commercialize our products or product candidates because they cannot obtain the necessary regulatory approvals, they lack adequate financial or other resources or they decide to focus on other initiatives. Any failure of our third-party collaborators to successfully market and commercialize our products or product candidates outside the U.S. would diminish our revenues and harm our results of operations.Limitations on our patent rights relating to our products and product candidates may limit our ability to prevent third parties from competing against us.To a significant degree, our success will depend on our ability to obtain and maintain patent protection for our proprietary technologies and our products and product candidates, preserve our trade secrets,prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others. To that end, we seek patent protection in the U.S. and internationally for our products and product candidates. Our policy is to actively seek to protect our proprietary position by, among other things, filing patent applications in the U.S. and abroad (including Europe, Canada and certain other countries when appropriate) relating to proprietary technologies that are important to the development of our business.The strength of patents in the pharmaceutical industry involves complex legal and scientific questions and can have uncertain results. Patent applications in the U.S. and most other countries are confidential for a period of time until they are published, and publication of discoveries in scientific or patent literature typically lags actual discoveries by several months or more. As a result, we cannot be certain that we were the first to conceive inventions covered by our patents and pending patent applications or that we were the first to file patent applications for such inventions. In addition, we cannot be certain that our patent applications will be granted, that any issued patents will adequately protect our intellectual property or that such patents will not be challenged, narrowed, invalidated or circumvented.We also rely upon unpatented trade secrets, unpatented know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, by confidentiality agreements with our employees and our collaborators and consultants. We also have agreements with our employees and selected consultants that obligate them to assign their inventions to us. It is possible that technology relevant to our business will be independently developed by a person that is not a party to such an agreement. Furthermore, if the employees and consultants that are parties to these agreements breach or violate the terms of these agreements, we may not have adequate remedies, and we could lose our trade secrets through such breaches or violations. Further, our trade secrets could otherwise become known or be independently discovered by our competitors. Any failure to adequately prevent disclosure of our trade secrets and other proprietary information could have a material adverse impact on our business.In addition, the laws of certain foreign countries do not protect proprietary rights to the same extent or in the same manner as the U.S., and therefore we may encounter problems in protecting and defending our intellectual property in certain foreign jurisdictions.If we are sued for infringing intellectual property rights of third parties, it could be costly and time consuming to defend such a suit. An unfavorable outcome in that litigation could have a material adverse effect on our business.Our commercial success depends upon our ability and the ability of our collaborators to develop, manufacture, market and sell our approved products and our product candidates and use our proprietary technologies without infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we and our collaborators are developing product candidates. As the pharmaceutical industry expands and more patents are issued, the risk increases that our collaborators' approved products or our product candidates may give rise to claims of infringement of the patent rights of others. There may be issued patents of third parties of which we are currently unaware that may be infringed by our collaborators' approved products or Oxtellar XR or Trokendi XR, which could prevent us from being able to maximize revenue generated by our products or our product candidates. Because patent applications can take many years to issue, there may be currently pending applications which may later result in issued patents that our collaborators' approved products or our product candidates may infringe.We may be exposed to, or threatened with, future litigation by third parties alleging that our collaborators' approved products or our products or product candidates infringe their intellectualproperty rights. If one of our collaborators' approved products or our products or product candidates is found to infringe the intellectual property rights of a third party, we or our collaborators could be enjoined by a court and required to pay damages. We could be unable to commercialize the applicable approved products and product candidates unless we obtain a license to the patent. A license may not be available to us on acceptable terms, if at all. In addition, during litigation, the patent holder could obtain a preliminary injunction or other equitable relief which could prohibit us from making, using or selling our approved products prior to a trial. Such a trial may not occur for several years.There is a substantial amount of litigation involving patent and other intellectual property rights in the pharmaceutical industry generally. If a third party claims that we or our collaborators infringe its intellectual property rights, we may face a number of issues, including, but not limited to:•Infringement and other intellectual property claims which, regardless of merit, may be expensive and time-consuming to litigate and may divert our management's attention from our core business;•Substantial damages for infringement, which we may have to pay if a court decides that the product at issue infringes on or violates the third party's rights. If the court finds that the infringement was willful, we could be ordered to pay treble damages and the patent owner's attorneys' fees;•Court rulings prohibiting us from selling our products or product candidate unless the third party licenses its rights to us, which it is not required to do;•If a license is available from a third party, we may have to pay substantial royalties, fees or grant cross-licenses to our intellectual property rights; and•Redesigning our products or product candidates so they do not infringe, which may not be possible or may require substantial monetary expenditures and time.We depend on collaborators to work with us to develop, manufacture and commercialize their and our products and product candidates.We have a license agreement with United Therapeutics Corporation to use one of our proprietary technologies for an oral formulation of treprostinil diethanolamine, or treprostinil, for the treatment of pulmonary arterial hypertension,activities, as well as for other indications. United Therapeutics Corporation launched Orenitram (treprostinil)expertise in 2014, which triggered a milestone payment to useach of $2.0 million. In the third quarter of 2014, we received a cash payment of $30.0 million as a result of HealthCare Royalty Partners III, L.P.'s (HC Royalty) purchase of certain of our rights under our license agreement with United Therapeutics Corporation related to the commercialization of Orenitram. We will retain full ownership of the royalty rights if a certain cumulative threshold payment to HC Royalty is reached. We are entitled to receive milestones and royalties for use of this formulation in other indications. If we materially breach any of our obligations under the license agreement, we could lose the right to receive any future royalty payments thereunder, which could be financially significant to us.have the effect of limitinglimit the areas of research and development that we may pursue, either alone or in collaboration with third parties. Much of the potential revenues from these future collaborations may consist of contingent payments, such as payments for achieving certain development milestones, and royalties payable on sales of developed products.product sales. The milestonemilestones and royalty revenues that we may receive under these collaborations will depend upon our collaborators' ability to successfully develop, introduce, market and sell new products. Future products using our products, product candidates or technologies because they, among other things, may:•decrease or fail to increaseapply sufficient development or commercialization efforts related to those product candidates;•or limited cash resources, or in the belief that other internal drug development programs may have a higher likelihood of obtaining marketing approval, or may potentially generate a greater return on investment;••necessary to carrydevelop the product candidate through clinical development, marketing approval and commercialization;••Be•collaboration.collaboration, if at all. Further, even if we are able to replace the collaboration partner, we may not be able to do so on commercially favorable terms. As a result, the development and commercialization of the affected product or product candidate could be delayed, curtailedimpaired, or terminated because we may not have sufficient financial resources or capabilities to continue development and commercialization of the product candidate on our own.We have in-licensed or acquired a portion Failure of our intellectual property necessarythird-party collaborators to develop certain ofsuccessfully market and commercialize our psychiatry product candidates. If we fail to comply with our obligations under any of these arrangements, we could lose such licensesproducts or intellectual property rights.We are a party to and rely on several arrangements with third parties, such as those with Afecta and Rune, which give us rights to intellectual property that is necessary for the development of certain of our product candidates including SPN-810within and SPN-809, respectively. In addition, we may enter into similar arrangements inoutside the future for other product candidates. Our current arrangements impose various development, financialU.S. could materially diminish our revenues and other obligations on us. If we materially breach these obligations or if Afecta or Rune fail to adequately perform their respective obligations, these exclusive arrangements could be terminated, which would result inharm our inability to develop, manufacture and sell products that are covered by such intellectual property.nevernot receive approval to commercialize our product candidates outside of the U.S.productsproduct outside of the U.S., we must establish and comply with numerous and varying regulatory requirements of other regulatory jurisdictions regarding safety and efficacy. Approval procedures vary among jurisdictions, and can involve product testing and administrative review periods different from, and greater than, those in the U.S. The time required to obtain approval in other jurisdictions mightisare not freely available, we may not have the ability to commercialize our products without first negotiating rights fromwith third parties to obtain their permission to refer to their clinical data in our regulatory applications, whichapplications. This process could require the expenditure of significant additional funds and time.another, but aanother. A failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory processes in others. Failure to obtain regulatory approvalsapproval in other jurisdictions, or any delay or setback in obtaining such approvals, could have the same adverse effects as detailed above regarding FDA approval. As described above, such effects include the risks that any of our product candidates may not be approved for all requested indications, requested, which could limit the uses of our product candidates, and could have an adverse effect on their commercial potential or could require costly post-marketing studies.Guidelinesrecommendations publishedrely on several arrangements with third parties. These arrangements give us rights to IP that are necessary for the development of certain of our product candidates. In addition, we may enter into similar arrangements in the future for other product candidates. Our current arrangements impose various development, financial and other obligations on us. If we materially breach these obligations, or if third parties fail to adequately perform their respective obligations, these exclusive arrangements could be terminated, which could result in our inability to develop, manufacture, market and sell products that are covered by such IP.organizations can reduceside effects, including but not limited to dizziness, paresthesia, headaches, cognitive deficiencies such as memory loss and speech impediment, digestive problems, somnolence, double vision, gingival enlargement, nausea, weight gain, oral malformation birth defects, visual field defects, infants small for gestational age, and fatigue. The use of Oxtellar XR and Trokendi XR may cause similar side effects as compared to their reference products, or may cause additional or different side effects.candidates.Government agencies promulgate regulationscandidates, and guidelines directly applicablecould substantially increase commercialization costs.usdrugs intended to treat a rare disease or condition that affects fewer than 200,000 individuals annually in the U.S. Orphan drug designation entitles a party to financial incentives, such as opportunities for grant funding towards clinical trial costs, tax credits for certain research, and to our products and product candidates.user fee waivers under certain circumstances. In addition, professional societies, practice management groups, private health and science foundations and organizations involvedif a drug receives its first FDA approval in various diseases from time to time may also publish guidelines or recommendations to the health care and patient communities. Recommendations of government agencies or these other groups or organizations may relate to such matters as usage, dosage, route of administration and use of concomitant therapies. Recommendations or guidelines suggesting the reduced use of our products or the use of competitive or alternative products that are followed by patients and health care providers could result in decreased use of our products.We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liabilities.The use of our product candidate in clinical trials and the sale of any of our products expose us to the risk of product liability claims. Product liability claims might be brought against us by consumers, healthcare providers or others selling or otherwise coming into contact with our products and product candidates. If we cannot successfully defend ourselves against product liability claims, we could incur substantial liabilities. In addition, product liability claims may result in:•Decreased demand for any product that has received approval and is being commercialized;•Impairment of our business reputation and exposure to adverse publicity;•Withdrawal of bioequivalence and/or clinical trial participants;•Initiation of investigations by regulators;•Costs of related litigation;•Distraction of management's attention from our primary business;•Substantial monetary awards to patients or other claimants;•Loss of revenues; and•Our inability to commercialize productsan indication for which we obtain marketing approval.Tableit has orphan drug designation, that drug is entitled to seven years of ContentsOur product liability insurance coverage for our clinical trials is limited to $15 million per claim and $15 million inmarket exclusivity. This implies that the aggregate, and covers bodily injury and property damage arising from our clinical trials, subject to industry-standard terms, conditions and exclusions. Our insurance coverageFDA may not be sufficientapprove any other firm's application for the same drug for that same indication for a period of seven years. Exceptions are limited, such as showing clinical superiority over the drug with orphan drug exclusivity.reimburse usexpand our designation for all expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future,alternative uses where applicable, we may not be ablereceive the benefits associated with orphan drug designation. This may result from a failure to maintain insurance coverage atorphan drug status, or it may result from a reasonable costcompeting product reaching the market with an orphan designation for the same disease indication. Under U.S. rules for orphan drugs, if such a competing product reaches the market before ours does, the competing product could potentially obtain a scope of market exclusivity that limits or precludes our product from being sold in sufficient amountsthe U.S. for seven years. Even if we obtain exclusivity, the FDA could subsequently approve an alternative drug for the same condition, if the FDA concludes that the second to protect us against losses. On occasion, large judgments have been awardedreach the market is clinically superior in class action lawsuits based on drugs that had unanticipated side effects. A successfulit is safer, more effective or makes a major contribution to patient care. In addition, a competitor may receive approval of different products for the same indication for which our orphan product liability claimhas exclusivity, or seriesmay obtain approval for the same product but for a different indication for which the orphan product has exclusivity.claims brought against us could cause our stock price2017 (FDARA) was enacted. FDARA, among other things, codified the FDA's pre-existing regulatory interpretation to decline. If judgments exceed our insurance coverage, our cash balance could decreaserequire that a drug sponsor demonstrate clinical superiority of an orphan drug that is otherwise the same as a previously approved drug for the same rare disease in order to receive orphan drug exclusivity. The new legislation reverses prior precedent holding that the Orphan Drug Act unambiguously requires that the FDA recognize the orphan exclusivity period, regardless of showing clinical superiority.adverselypolicies. We do not know if, when, or how the FDA may change the orphan drug regulations and policies in the future. It is uncertain how any changes might affect our business. government (federal, certain states, and certain states) and other non-U.S.foreign governments have shown significant, and increased interest in pursuing healthcare reform.reform and changes to the healthcare delivery system. Government-adopted reform measures could adversely impact the pricing of healthcare products and services in the U.S. or internationally, and adversely impactimpacting the amountlevel of reimbursement available from governmental agencies and/or commercial third-party payors. The continuing efforts of thethird-party payors, including U.S. federal and state agencies, foreign governments, insurance companies, managed care organizations, employers, and other payors of healthcare services to contain or reduce health carehealthcare costs may adversely affect our ability to set prices for any approved productat launch or to increase priceprices once launched. These initiatives could adversely impact our ability to generate revenues, andto achieve profitability, or to and maintain profitability.In both the U. S. (federal and certain states) and some foreign jurisdictions, there There have been a number of legislative and regulatory proposals and initiatives to change the health carehealthcare system in ways that could adversely affect our ability to profitably sell any approved product profitably.product. Some of these proposed and implemented reforms couldwould result in reduced reimbursement rates for our products, which would adversely affect our business strategy, operations and financial results. For example, inHealthCareHealth Care and Education Reconciliation Act of 2010. These laws and their regulations, which we refer to collectively as the HealthCare Reform Law, may have far reaching consequences for biopharmaceuticalpharmaceutical companies like us. As a resultPossible revisions to the HealthCare Reform Law are the subject of ongoing legislative debates and litigation.substantial changes could be madeof importance to the current system for paying for healthcare in the U.S., including changes made in order to extend benefits to those who currently lack insurance coverage or to change coverage parameters. Extending coverage to a large population could substantially change the structure of the health insurance system and the methodology for reimbursing medical services and drugs. These structural changes could entail modifications to the existing system of private payors and government programs, such as Medicare and Medicaid, create of a new government-sponsored healthcare insurance source, or some combination of both, as well as other changes. Restructuring the healthcare delivery system in the U.S., could impact the reimbursement for prescribed drugs, including our products and product candidates. If reimbursement for our approvedcandidates are the following:is substantially less than we expectto certain federal healthcare programs;future,statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program;obligations associatedliability to individuals enrolled in Medicaid managed care organizations;themfunding for such research.substantially increased,often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. It is impossible to predict whether additional legislative changes will be enacted or whether FDA regulations, guidance or interpretations will be changed, and what the impact of such changes, if any, may be. Future regulatory changes could be materiallymake it more difficult for us to maintain or attain approval to develop and adversely impacted.In 2007, the Foodcommercialize our products and Drug Administration Amendments Act of 2007 was enacted, giving thetechnologies.andor to require compliance with risk evaluationsevaluation and mitigation strategies approved bystrategies. Further, the FDA. In 2012 the Food and Drug Administration Safety and Innovation Act was enacted, expandingexpanded drug supply chain reporting requirements and strengtheningstrengthened the FDA's response to drug shortages, as well as other changes.shortages. The FDA's exercise of thisits authority could result in delays, or increasedcould increase costs during product development, clinical trials and regulatory review,review. It could also result in increased The DQSA creates the requirement for companies to trace, verify and identify all products across all changes of ownershipthrough the entire supply chain, from manufacturer to dispenser.federalhealthcare reforms in the U.S. and state proposals and health care reforms in other countries could limit the prices that can be charged for our productproducts and product candidates, that we develop andor may furtherotherwise limit our commercial opportunities. Our results of operations could be materially and adversely affected by the HealthCare Reform Law by reducing the amounts that private insurers will pay and by other health care reforms that may be enacted or adopted in the future.the HealthCare Reform Lawany change in healthcare laws could cause us to incur significant compliance expenses or could subject us to substantial penalties and fines if our business is found to violate these requirements.HealthCare Reform Law is multi-faceted and is being implemented in phases. Theassessment of the financial impact of the HealthCare Reform Law on our business is on-going, and thereon-going. There can be no assurance that our business will not be materially harmed by future implementation of or changes to the HealthCare Reform Law. In addition, ifIf we are not in full compliance with the HealthCare Reform Law, we could face enforcement action, fines and other penalties and wepenalties. We could receive adverse publicity.enforcement, such as increasedenforcement. These include increasing funding for enforcement efforts, and lowering the intent requirement of the federal anti-kickback statute and criminal health carehealthcare fraud statute, such that a person or entity no longer needs to have actual knowledge or specific intent to violatesviolate the statute.includingboth civil and criminal, penalties, damages, fines, exclusion from federal health carehealthcare programs and/or the curtailment or restructuring of our operations.of them have not been fully interpreted by the regulatory authorities or the courts, and theircourts. Their provisions are subject to a variety of interpretations. Any action against us for violation of these laws, even if we successfully defend against them,these assertions, could cause us to incur significant legal expenses, and divert our management's attention from the operation of our business.penalties and ourpenalties. Our business, operations and financial condition could be adversely affected.health carehealthcare laws and regulations pertaining to patients' rights to privacy, fraud and abuse protection, are and will be applicable to our business. We could be subject to allegations of healthcare fraud and abuse, and patient privacy regulationviolations by both the federal government and the states in which we conduct our business. The regulationsRegulations include the:•anti-kickback law,Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual for an item or service, or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs;•Falsecivil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things,things: individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent, and whichfraudulent; knowingly making a false statement material to an obligation to pay or transmit money to the federal government; or knowingly concealing or improperly avoiding or decreasing an obligation to pay money to the federal government. This may apply to entities like us, which provide coding and billing advice to customers;•ThisSimilar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge or specific intent to violate the statute in order to have committed a violation;•HealthCare Reform Law requiresAffordable Care Act, which require manufacturers of drugs, devices, biologics, and medical supplies to report to the Department of Health and Human Services information related to physician payments, and to report other transfers of value, and physician ownership and investment interests;••Sunshine Act,laws, physician payment and drug pricing transparency laws, and false claims laws which may apply to our business practices, including, but not limited to: research, distribution, sales and marketing arrangements; claims for items or services reimbursed by any third-party payor, including commercial insurers,insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry's voluntary compliance guidelines, and the applicable compliance guidance promulgated by the federal government; otherwise restrict payments that may be made to healthcare providers; and state laws governing the privacy and security of health information in certain circumstances, anycircumstances. Many of whichthese state laws differ from each otherone another in significant ways, and often are not preempted by federal laws, thus complicating compliance efforts.part,significant degree, on our ability to effectively manage our recent and any future growth. In 2016,2019, we increased employee headcount from 344448 employees to 363 employees and increased revenues464 employees. Revenues in 2019 were $392.8 million, compared to $215.0 million from $147.5$408.9 million in 2015.2018. Our need to effectively execute our growth strategy requires that we:• our regulatory approvals and clinical trials effectively;••••employees.personnel, whichpersonnel. This may result in weaknesses in our infrastructure,infrastructure; give rise to operational mistakes,mistakes; loss of business opportunities,opportunities; loss of employeesemployees; and reduced productivity.expected,expected; our ability to generate or increase our revenues could be impaired,impaired; and we may not be able to implement our business strategy.Moreover, acquiringAcquiring such additional resources could increase our legal and financial compliance costs, divert managementmanagement's attention from other matters, and/or make somecertain activities more time consuming.bodies.bodies, and as the market gains familiarity with these requirements. This could result in continuing uncertainty regarding compliance matters, and on-going financial reporting requirements.regulations, whichregulations. This can be expensive and restrict how we do business.manufacturers'manufacturers and suppliers' activitiessuppliers involve the controlled storage, use and disposal of hazardous materials owned by us.materials. We and our manufacturers and suppliers are subject to federal, state, city and local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. Although we believe that the safety procedures we use for handling and disposing of these materials comply with the standards prescribed by theseapplicable laws and regulations, we cannot eliminate the risk of accidental contamination or injury from these materials. In the event of an accident, local, city, state or federal authorities may curtail the use of these materials, and may interrupt our business operations, including our commercialization, and research and development efforts. Although we believe that the safety procedures utilized by our third-party manufacturers for handling and disposing of these materials generally comply with the standards prescribed by theseapplicable laws and regulations, we have no direct control over our third-party manufacturers, and therefore cannot guarantee that this is the case orcase. We can eliminate the risk of accidental contamination, or that such safety procedures will prevent injury from these materials. In such an event, we may be held liable for any resulting damages and suchdamages. Such liability could exceed our resources.Obtaining While we have implemented processes and maintaining our patent protection depends on complianceprocedures to ensure that the suppliers we use are complying with various procedural, document submission, fee paymentall applicable regulations, there can be no assurances that such suppliers in all instances will comply with such processes and other requirements imposed by governmental patent agencies, and our patent protection could be reducedprocedures, or eliminated for non-complianceotherwise comply with these requirements.The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.We employ individuals who were previously employed at other pharmaceutical companies, including our competitors or potential competitors and, as such, we may be subject to claims that we or these employees have used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against such claims, litigationapplicable regulations. Noncompliance could result in substantial costsour marketing and be a distraction to management.Security breaches and other disruptionsdistribution of contaminated, defective or dangerous products, which could compromise our information and exposesubject us to liability, which would cause our business and reputation to suffer.In the ordinary course of our business, we collect and store sensitive data in our data centers and on our networks, including: intellectual property; our proprietary business information; proprietary information of our customers, suppliers and business partners; and personally identifiable information of our employees and patients in our clinical trials. The secure processing, maintenance and transmission of this information is critical to our operations and business strategy. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of informationliabilities. This could result in legal claimsthe imposition by governmental authorities of procedures or proceedings, liability under laws that protect the privacy of personal information, and regulatory penalties that could disruptrestrict or eliminate our operations and damage our reputation, whichability to sell products. Any or all of these effects could adversely affect our business, revenuesfinancial condition and competitive position.(amphetamine,(i.e., amphetamine, carbamazepine, guanfacine, lanthanum and mesalamine), and any derivative thereof. Although these various restrictions and covenants on us do not currently impact our products, product candidates or business, they could in the future limit or delay our ability to take advantage of business opportunities that may relate to such compounds.•••Due 2019 (the(2019 Notes) in May 2013; and•Orenitram.WeOrenitram; andinception. Asinception through 2014, substantially as a consequence of December 31, 2016, we had an accumulated deficit of approximately $84.3 million. Substantially all of our operating losses resulted from costs incurred in connection with our development programs, expenses associated with launching our products, and from selling, general and administrative costs associated with our operations. We expect our research and development costs to continue to be substantial and to increase with respect to our product candidates, as we advance those product candidates through preclinical studies, clinical trials, manufacturing scale-up and other pre-approval activities. We expect our selling, general and administrative costs to continue to increase as we continue to support the ongoing commercialization of our products.Our prior losses have had an adverse effect onproducts, and to further increase in anticipation of launching our stockholders' equity and cash position. product candidates.20172020 and beyond, we cannot be certain that we will do so. Any potential future losses, if and when they occur, could have an adverse impact on our stockholders' equity and working capital. Furthermore, sincecompletionamount and timing of development milestones, and product revenue received under our collaboration license agreements.initial public offeringproduct development and clinical trial activities through all phases of clinical development;May 2012,regulatory review and approval of product candidates in clinical development;have incurred additional costs associated withmay become involved;as a public company.commercializationbusiness development efforts.seek to raise additional funds. We have no committed external sources of funds.•our•the•the additional product candidates or acquiring other complementary companies;•the•theThe actions of our competitors and their success in selling competitive product offerings, including generics; and•thearrangements.whenin the amount we need itrequire or may not be available on terms that are favorable to us, or at all. We may seek additional capital due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our current or future operating plans. If adequate funds are not available to us on a timely basis, or at all, we may be required to delay, reduce the scope of, or eliminate one or more of our development programs, our commercialization efforts or strategic initiatives.We may not be able to maintain or increase profitability.remain profitable depends uponuse our net operating loss carryforwards and other tax attributes may be limited, or may expire prior to utilization.generate increasing levels of revenues from sales ofuse our products, Oxtellar XRpre-change net operating loss carryforwards and Trokendi XR, while simultaneously funding the requisite research expenditurestax credit carryforwards to gain FDA approval for our product candidates. Since 2013, the first year inreduce U.S. federal and state income tax may be subject to limitations, which we generated revenue from our first commercial products, we have demonstrated the ability to become and remain profitable. Future revenues will depend highly on our ability to grow demand for our products and defend against potential generic competition, and successfully developing and commercializing our product candidates.Our operating results may fluctuate significantly.We expect that any revenues we generate will fluctuate from quarter to quarter and year to year as a result of revenue from approved products, our license agreements, the amount of and timing for development milestones and product revenues received under our collaboration license agreements.Our net income and other operating results will be affected by numerous factors, including:
us.•the level of market acceptance for any approved product candidate, underlying demand for that product and wholesalers' buying patterns;•variations in the level of expenses related to our development programs;•the success of our bioequivalence and clinical trials through all phases of clinical development;•our execution of any collaborative, licensing or similar arrangements, and the timing of payments we may make or receive under these arrangements;•any delays in regulatory review and approval of product candidates in clinical development;•the timing of any regulatory approvals, if received, of additional indications for our existing products;•potential side effects of our products and our future products that could delay or prevent commercialization, cause an approved drug to be taken off the market, orpotentially result in litigation;•any intellectual property infringement lawsuit in which we may become involved;•our abilityincreased future cash tax liability to maintain an effective sales and marketing infrastructure;•our dependency on third-party manufacturers to supply or manufacture our products and product candidates;•competition from existing products, new products, or potential generics to our products that may emerge;•regulatory developments affecting our products and product candidates; and•changes in reimbursement environment and regulatory changes.Due to the various factors mentioned above, and others, the results of any prior quarterly period should not be relied upon as an indication of our future operating performance. If our quarterly operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Furthermore, any quarterly fluctuations in our operating results may, in turn, cause the price of our stock to fluctuate substantially.the Sarbanes-Oxley ActSOX relating to internal controls over financial reporting. We have and expect to continue to incur significant expense and to devote substantial management effort toward ensuring compliance with Section 404. We currently do not have an internal audit group, and we may need to hiregroup. We have hired additional accounting and financial staff with appropriate public company experience and technical accounting knowledge. We expect that we will have to compete in the market place for qualified accounting and financial staff and we may have difficulties identifying and attracting qualified persons.assuregive assurance that our internal controls over financial reporting will prove to be effective. have identified material weakness in our internal control over financial reporting and may identify material weaknesses in the futureour internal controls over financial reporting or otherwise fail to maintain an effective system of internal controls, which might cause stockholders to lose confidence in our financial and other public reporting, which in turn would harm our business and the trading price of our common stock.reports andreports. These are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could cause us to fail to meet our reporting obligations. In addition, any testing conducted by us conducted in connection with Section 404(a) of the Sarbanes-Oxley Act,SOX, or the subsequent testing by our independent registered public accounting firm in connection with Section 404(b) of the Sarbanes-Oxley Act,SOX, may reveal deficiencies in our internal controlcontrols over financial reporting that are deemed to be material weaknesses. These may require prospective or retroactive changes to our consolidated financial statements or may identify other areas for further attention or improvement.Our management has identified a Any system of internal controls, however well designed and operated, is based in part on certain assumptions, and can provide only reasonable, not absolute, assurances that the objectives of the system are met. Any material weakness in our internal control over financial reporting as of December 31, 2016 as described in Item 9A. Controls and Procedures below. As a result, our management, under the supervision and with the participation of our CEO and our CFO, hasconcluded that our disclosure controls and procedures were not effective as of December 31, 2016. Although our management and the audit committee of our board of directors has formulated and is implementing a plan to remediate this material weakness, we expect implementation to continue to be time consuming, and the remedial actions we take may prove to be ineffective or inadequate. Any deficiencies or material weaknessweaknesses in our internal controls could cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.TheannualThe annual independent assessment of the effectiveness of our internal controls is very expensive, and could continue to detect problems that our management's assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.use our net operating loss carryforwards and other tax attributes may be limited.Our ability to utilize our U.S. Federal and state net operating losses or U.S. Federal tax credits is currently limited, and may be limited further, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended. The limitations apply if an ownership change, as defined by Section 382, occurs. Generally, an ownership change occurs when certain shareholders change their aggregate ownership by more than 50 percentage points over their lowest ownership percentage in a testing period, which is typically three years or since the last ownership change. We are already subject to Section 382 limitations due to cumulative ownership changes that, as of November 15, 2013, totaled more than 50%. As of December 31, 2016, we had U.S. federal net operating loss carryforwards of $87.3 million and research and development tax credit carryforwards of $7.1 million available. Future changes in stock ownership may also trigger an additional ownership change and, consequently, another Section 382 limitation. Any limitation may result in expiration of a portion of the net operating loss or tax credit carryforwards before utilization which would reduce our gross deferred income tax assets. As a result, if we earn net taxable income,complete future acquisitions will depend on our ability to use our pre-change net operating loss carryforwards and tax credit carryforwards to reduce U.S. Federal and state income tax may be subject to limitations, which could potentially result in increased future cash tax liability to us.Risks Related to Our IndebtednessThe Indenture governing the Notes contains restrictions that will limit our operating flexibility.The Indenture governing the Notes contains covenants that, among other things, restrict our and our existing and future subsidiaries' ability to take specific actions, even ifidentify suitable acquisition candidates. If suitable candidates are identified, we believe them to be in our best interest. These covenants include restrictions on our ability to:•incur additional indebtedness and issue certain types of preferred stock; and•enter into mergers, consolidations or sales or leases of all or substantially all of our assets.These covenants may limit our operational flexibility and could prevent us from taking advantage of business opportunities as they arise, growing our business or competing effectively.We may not be permitted, by the agreements governingable to negotiate commercially acceptable terms for their acquisition or, if necessary, to finance those acquisitions. We anticipate competition for attractive candidates from other parties, some of whom have substantially greater financial and other resources than we have. Whether or not any particular acquisition is successfully completed, each of these activities is expensive and time consuming and would likely require our existing or future indebtedness,management to pay any interest make-whole payment upon conversion in cash, requiring usspend considerable time and effort to issue shares for such amounts,complete, which would detract from our management's ability to run our current business. Although we may spend considerable funds and efforts to pursue acquisitions, we may not be able to complete them.significant dilutionthe occurrence of one or more of the following events:stockholders.If a holder elects to convert some or allother business activities; andtheir Notes, if, for at least 20 trading days (whether or not consecutive) duringdebt and liabilities of the 30 consecutive trading day period ending within five trading days prior to a conversion date, the last reported sale price of our common stock exceeds the applicable conversion price on each such trading day,target companypaybe able to complete acquisitions that we believe are necessary to complement our growth strategy on acceptable terms, or at all. Further, if we do successfully integrate the operations of any companies that we have acquired or subsequently acquire, we may not achieve the potential benefits of such holderacquisitions. Even if we are able to consummate an interest make-whole paymentacquisition, the transaction would present many risks, including, among others: failing to achieve anticipated revenues, profits, benefits or cost savings; difficulty incorporating and integrating the acquired technologies, services or products; difficulty in cashcoordinating, establishing orTable expanding sales, distribution and marketing functions, as necessary; diversion of Contentscommon stock formanagement's attention from other business concerns; being exposed to unanticipated or contingent liabilities from the Notes being converted. We haveacquired company, or incurring the option to issue our common stock to any converting holder in lieuimpairment of makinggoodwill; the interest make-whole payment in cash.loss of key employees or distribution partners; and difficulties implementing and maintaining sufficient controls, policies and procedures over the systems, products and processes of the acquired company. If we electdo not achieve the anticipated benefits of an acquisition as rapidly or to issue our common stock for such payment, then the stock will be valued at 95%extent anticipated by management, or if others do not perceive the same benefits of the simple averageacquisition as we do, there could be a material, adverse effect on our business, cash flows, financial condition or results of the daily volume-weighted average price (VWAP) of our common stock for the 10 trading days ending on and including the trading day immediately preceding the conversion date. Agreements governing our existing or future indebtedness may prohibit us from making cash payments in respect of the interest make-whole amount upon a conversion. Notwithstanding the foregoing, in no event will the shares we deliver in connection with a conversion, including those delivered in connection with the interest make-whole amount and repayment of principal, exceed 221.7294 shares per $1,000 principal amount of Notes, subject to adjustment or, in aggregate, 19.96 million shares. If, pursuant to our election to deliver common stock in connection with the payment of the interest make-whole amount, we would be required to deliver a number of shares of common stock in excess of such threshold, we will deliver cash in lieu of any shares otherwise deliverable upon conversions in excess thereof (based on the simple average of the daily VWAP for the 10 trading days ending on and including the trading day immediately preceding the conversion date).including in connection with the conversion of our Notes, and thereby materially and adversely affect the market price of our common stock.2016,2019, we had outstanding 49,971,26752,533,348 shares of common stock, of which approximately 1,799,3561,959,294 shares are restricted securities that may be sold in accordance with the resale restrictions under Rule 144 of the Securities Act of 1933, as amended (Securities Act), or pursuant to a resale registration statement. Also, as of December 31, 2016,2019, we had outstanding options to purchase 3,644,0884,606,559 shares of common stock that, if exercised, would result in these additional shares becoming available for sale. Approximately 7.2%6% of these shares and options are held by senior management of the Company. We have also registered all common stock subject to options outstanding or reserved for issuance under our 2005 Stock Plan, 2012 Equity Incentive Plan and 2012 Employee Stock Purchase Plan. An aggregate of 4,387,4911,972,307 and 297,34054,081 shares of our common stock are reserved for future issuance under the 2012 Equity Incentive Plan and the 2012 Employee Stock Purchase Plan, respectively. In addition, as of December 31, 2016, 863,403 shares of our common stock are presently reserved for future issuance upon conversion of the Notes. These shares will be eligible for resale in the public market upon issuance.stock, and becausestock. Because we do not anticipate paying any cash dividends in the foreseeable future, capital appreciation, if any, of our common stock will be your sole source of gain on an investment in our common stock. any cash dividends on our common stock in the foreseeable future. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future. There is no guarantee that shares of our common stock will appreciate in value or even maintain the price at which our stockholders have purchased their shares.TableContents We currently have very limited research coverage by securities and industry analysts. If securities or industry analysts presently covering our business do not continue such coverage, or if additional securities or industry analysts do not commence coverage of our Company, the trading price for our stock could be negatively impacted. If one or more of the analysts who covers us downgrades our stock, our stock price would likely decline. If one or more of these analysts ceases to cover us or fails to regularly publish reports on us, interest in our stock could decrease, which could cause our stock price or trading volume to decline.•meeting.•directors.•us.•Company.•meeting.•meeting.•or repeal or to adopt any provision inconsistent with certain provisions of our certificate of incorporation and to amend our by-laws, which make it more difficult to change the provisions described above.become.be. If an active public market is not sustained, it may be difficult to sell shares of common stock at a price that is attractive to the investor, or at all. Further, an inactive market may also impair our ability to raise capital by selling shares of our common stock, andor may impair our ability to enter into strategic partnerships or acquire companies or products, product candidates or technologies by using our shares of common stock as consideration.2016,2019, we had issued options to purchase 3,644,0884,606,559 shares of common stock outstanding, with exercise prices ranging from $0.40$2.56 to $22.80$58.15 per share and a weighted average exercise price of $10.25$23.05 per share. Upon the vesting of each of these options, the holder may exercise his or her options, which would result in dilution to investors.fluctuate substantially.The market priceconduct future offerings of our common stock, preferred stock or other securities that are convertible into or exercisable for our common stock to finance our operations or fund acquisitions, or for other purposes. In addition, as of December 31, 2019, 4,606,559 shares of our common stock were reserved for future issuance upon the exercise of outstanding options, 1,972,307 shares were reserved for future issuance under our 2012 Equity Incentive Plan and 54,081 shares were reserved for future issuance under our 2012 Employee Stock Purchase Plan.be volatile. In addition,do so during any observation period (as defined in the indenture) for the 2023 Notes, by purchasing and/or selling shares of our common stock and/or other securities of ours, including the 2023 Notes, in privately negotiated transactions and/or open-market transactions, or by entering into and/or unwinding various over-the-counter derivative transactions with respect to our common stock.may fluctuate significantly in response toand the trading price of the 2023 Notes will depend on a numbervariety of factors, including:•the commercial performance of Oxtellar XR, Trokendi XR, or any of our product candidates that receive marketing approval;•substitution of our products in favor of generic versions;•status of our ongoing patent infringement law suits;•the filing of ANDAs by generic companies seeking approval to market generic versions of our products;•plans for, progress in and results from clinical trials of our product candidates generally;•FDA or international regulatory actions, including actions on regulatory applications for any of our product candidates;•announcements of new products, services or technologies, commercial relationships, acquisitions or other events by us or our competitors;•inand cannot be ascertained at this time. Any of these activities could, however, adversely affect the pharmaceutical and biotechnology sectors;•fluctuations in stock market prices and trading volumes of similar companies;•fluctuations in stock market prices for the U.S. stock market;•variations in our quarterly operating results;•changes in accounting principles;•litigation or public concern about the safety of our products and/or potential products;•actual and anticipated fluctuations in our quarterly operating results;•deviations in our operating results from the estimates of securities analysts;•additions or departures of key personnel;•sales of large blocksprice of our common stock including sales byand/or the trading price of the 2023 Notes and, consequently, adversely affect noteholders' ability to convert the 2023 Notes and/or affect the value of the consideration that you receive upon conversion of the 2023 Notes. In addition, the hedge counterparties and/or their affiliates may choose to engage in, or to discontinue engaging in, any of these transactions with or without notice at any time, and their decisions will be in their sole discretion and not within our executive officers, directorscontrol.significant stockholders;•changes in third-party coverage and reimbursement policies for our products and/or product candidates; and•discussion by us or our stock pricewe will be subject to the risk that they might default in the fulfillment of their obligations under the convertible note hedge transactions. Our exposure to the credit risk of the hedge counterparties will not be secured by any collateral.scientific press or online investor communities.The realizationviability of any hedge counterparty.risks described2023 Notes or exercise of the warrants evidenced by the warrant transactions may dilute the ownership interest of existing stockholders, including noteholders who have previously converted their 2023 Notes.these "Risk Factors"shares of our common stock. Furthermore, the warrants evidenced by the warrant transactions are expected to be settled on a net-share basis. As a result, the conversion of some or all of the 2023 Notes, or the exercise of some or all of such warrants may dilute the ownership interests of existing stockholders. Any sales in the public market of the common stock issuable upon such conversion of the 2023 Notes, or such exercise of the warrants, could have a dramatic, material and adverse impact on theadversely affect prevailing market price of our common stock. In addition, class action litigation has often been instituted against companies whose securities have experienced periodsthe existence of volatility. Any such litigation brought against usthe 2023 Notes may encourage short selling by market participants because the conversion of the 2023 Notes could result in substantial costs and a diversiondepress the price of management attention, which could hurt our business, operating results and financial condition.common stock.1550 East Gude Drive,9715 and 9717 Key West Avenue, Rockville, Maryland, 20850, where we occupy approximately 44,500136,016 square feet of laboratory and office space. OurThe term of this lease term expires incommenced on February 1, 2019 and shall continue until April 30, 2020, with an option for a five-year extension. We also lease approximately 20,530 square feet of office space in an adjacent building to our existing office space located at 1500 East Gude Drive, Rockville, MD 20850 with a co-terminus lease term date of April 30, 2020.2034. We believe that these facilities are sufficient for our present and contemplated operations.aremay be subject to various claims, charges and litigation. We may be required to file infringement claims against third parties for the infringement of our patents. We have filed such claims for infringementAs of the Orange Book patents listed for our products Oxtellar XR and Trokendi XR.Supernus Pharmaceuticals, Inc. v. Actavis, Inc., et al., C.A. Nos. 13-4740; 14-1981 (RMB)(JS) (D.N.J.)Supernus Pharmaceuticals, Inc. v. Actavis, Inc., et al., Appeal No. 2016-1619 (Fed. Cir.)We received a Paragraph IV Notice Letter against two of our Oxtellar XR Orange Book patents (United States Patent Nos. 7,722,898 and 7,910,131) from generic drug maker Watson Laboratories, Inc.—Florida (WLF) n/k/a Actavis Laboratories FL, Inc. (Actavis Labs FL) on June 26, 2013. On August 7, 2013, we filed a lawsuit against Actavis, Inc., Actavis Labs FL, Actavis Pharma, Inc., Watson Laboratories, Inc., and ANDA, Inc. (collectively Actavis) alleging infringement of United States Patent Nos. 7,722,898 and 7,910,131. We received a second Paragraph IV Notice Letter against a later-issued Oxtellar XR Orange Book Patent (United States Patent No. 8,617,600) on February 20, 2014. On March 28, 2014, we filed a second lawsuit against Actavis alleging infringement of United States Patent No. 8,617,600. We have since listed four additional Orange Book patents: United States Patent Nos. 8,821,930, 9,119,791, 9,351,975, and 9,370,525. Our United States Patent Nos. 7,722,898, 7,910,131, 8,617,600, 8,821,930, 9,119,791, 9,351,975, and 9,370,525 generally cover once-a-day oxcarbazepine formulations and methods of treating seizures using those formulations. The FDA Orange Book lists all seven of our Oxtellar XR patents as expiring on April 13, 2027.Both Complaints—filed in the U.S. District Court for the District of New Jersey—alleged, inter alia, that Actavis infringed our Oxtellar XR patents by submitting to the FDA an Abbreviated New DrugApplication (ANDA) seeking to market a generic version of Oxtellar XR prior to the expiration of our patents. The two cases were consolidated for all purposes on October 8, 2015.A seven-day bench trial for the consolidated action involving United States Patent Nos. 7,722,898, 7,910,131, and 8,617,600 was held between November 18 and December 4, 2015. On February 5, 2016, the Court issued an opinion and order finding that: (i) Actavis's ANDA products infringe United States Patent Nos. 7,722,898 and 7,910,131; (ii) Actavis's ANDA products do not infringe U.S. Patent No. 8,617,600; and (iii) United States Patent Nos. 7,722,898, 7,910,131, and 8,617,600 are not invalid. The Court entered a final judgment on February 18, 2016: (i) enjoining the FDA from approving Actavis's ANDA before the expiration date of United States Patent Nos. 7,722,898 and 7,910,131; and (ii) enjoining Actavis from commercially manufacturing, using, offering to sell, or selling within the United States, or importing into the United States, Actavis's ANDA products until the expiration of United States Patent Nos. 7,722,898 and 7,910,131. On February 19, 2016, Actavis filed a Notice of Appeal to the United States Court of Appeals for the Federal Circuit. The parties executed a Partial Settlement Agreement in May 2016 that provided for the dismissal of all appeals, cross-appeals, claims, and counterclaims concerning U.S. Patent Nos. 8,617,600, 8,821,930, and 9,119,791. The appeal with respect to United States Patent Nos. 7,722,898 and 7,910,131 (docketed on February 24, 2016) was argued on December 8, 2016. On December 12, 2016, the United States Court of Appeals for the Federal Circuit affirmed the District Court's February 18, 2016 Final Judgment.Supernus Pharmaceuticals, Inc. v. Actavis, Inc., et al., C.A. No. 15-2499 (RMB)(JS) (D.N.J.)We received a Paragraph IV Notice Letter against United States Patent No. 8,821,930 from Actavis Labs FL on February 21, 2015. On April 7, 2015, we filed a third lawsuit against Actavis alleging infringement of United States Patent No. 8,821,930.The Complaint—filed in the U.S. District Court for the District of New Jersey—alleged, inter alia, that Actavis infringed United States Patent No. 8,821,930 by submitting to the FDA an ANDA seeking to market a generic version of Oxtellar XR prior to the expiration of United States Patent No. 8,821,930.The parties executed a Partial Settlement Agreement in May 2016 that provided for the dismissal of both parties' claims and counterclaims concerning U.S. Patent No. 8,821,930.Supernus Pharmaceuticals, Inc. v. TWi Pharmaceuticals, Inc., et al., C.A. No. 15-369 (RMB)(JS) (D.N.J.)We received a Paragraph IV Notice Letter against United States Patent Nos. 7,722,898, 7,910,131, 8,617,600, and 8,821,930 from generic drug maker TWi Pharmaceuticals, Inc. on December 9, 2014. On January 16, 2015, we filed a lawsuit against TWi Pharmaceuticals, Inc. and TWi International LLC (d/b/a TWi Pharmaceuticals USA) (collectively TWi) alleging infringement of United States Patent Nos. 7,722,898, 7,910,131, 8,617,600, and 8,821,930.The Complaint—filed in the U.S. District Court for the District of New Jersey—alleged, inter alia, that TWi infringed our Oxtellar XR patents by submitting to the FDA an ANDA seeking to market a generic version of Oxtellar XR prior to the expiration of our patents. Filing the Complaint within 45 days of receiving TWi's Paragraph IV certification notice entitles Supernus to an automatic stay preventing the FDA from approving TWi's ANDA for 30 months from the date of our receipt of the first Paragraph IV certification notice. On February 13, 2015, TWi answered the Complaint and denied the substantive allegations of the Complaint. TWi also asserted Counterclaims seeking declaratory judgments of non-infringement and invalidity of United States Patent Nos. 7,722,898 and 7,910,131. On March 20, 2015, we filed our Reply, denying the substantive allegations of those Counterclaims.The parties have completed fact and expert discovery, and are preparing final joint pretrial submissions. Trial is scheduled to begin on April 3, 2017.We received a second Paragraph IV Notice Letter against United States Patent Nos. 7,722,898, 7,910,131, 8,617,600, 8,821,930, 9,119,791, 9,351,975, and 9,370,525 from generic drug maker TWi Pharmaceuticals, Inc. on February 16, 2017. We are currently evaluating this Notice Letter and determining how to proceed.Supernus Pharmaceuticals, Inc. v. Actavis, Inc., et al., C.A. No. 15-8342 (RMB)(JS) (D.N.J.)We received a Paragraph IV Notice Letter against United States Patent No. 9,119,791 from Actavis Labs FL on October 15, 2015. On November 25, 2015, we filed a fourth lawsuit against Actavis alleging infringement of United States Patent No. 9,119,791.The Complaint—filed in the U.S. District Court for the District of New Jersey—alleged, inter alia, that Actavis infringed United States Patent No. 9,119,791 by submitting to the FDA an ANDA seeking to market a generic version of Oxtellar XR prior to the expiration of United States Patent No. 9,119,791. On January 29, 2016, Actavis answered the Complaint, denying the substantive allegations of that Complaint. Actavis Labs FL also asserted Counterclaims seeking declaratory judgments of non-infringement and invalidity of United States Patent No. 9,119,791. On March 4, 2016, we filed our Reply, denying the substantive allegations of those Counterclaims.The parties executed a Partial Settlement Agreement in May 2016 that provided for the dismissal of both parties' claims and counterclaims concerning U.S. Patent No. 9,119,791.Supernus Pharmaceuticals, Inc. v. Actavis, Inc., C.A. No. 14-6102 (SDW)(LDW) (D.N.J.)We received three Paragraph IV Notice Letters against six Trokendi XR Orange Book patents, namely United States Patent Nos. 8,298,576, 8,298,580, 8,663,683, 8,877,248, 8,889,191, and 8,992,989 from generic drug maker Actavis Laboratories FL, Inc. These patents cover once-a-day topiramate formulations and methods of treating seizures using those formulations. On October 1, 2014, we initiated a lawsuit against Actavis; the lawsuit alleges infringement of the Trokendi XR Orange Book patents. The FDA Orange Book currently lists United States Patent No. 8,298,576 as expiring on April 4, 2028 and United States Patent Nos. 8,298,580, 8,663,683, 8,877,248, 8,889,191, and 8,992,989 as expiring on November 16, 2027.This action for patent infringement—filed in the U.S. District Court for the District of New Jersey—alleges that Actavis infringed the Trokendi XR patents by, inter alia, submitting to the FDA an ANDA seeking to market a generic version of Trokendi XR prior to the expiration of these patents. Actavis answered these allegations with affirmative defenses and counterclaims of noninfringement and invalidity of the patents in suit. Filing its October 1, 2014 Complaint within 45 days of receiving the first of three Actavis Laboratories FL, Inc. Paragraph IV Notice Letters entitles Supernus to an automatic stay preventing the FDA from approving Actavis's ANDA for 30 months from the date of our receipt of such Notice Letter.The Company announced on March 7, 2017 that it has entered into a binding term sheet with Actavis regarding the settlement of this case. The binding term sheet permits Actavis to begin selling a generic version of Trokendi XR on January 1, 2023, or earlier under certain circumstances. On March 13, 2017,31, 2019, the Company entered into a settlement agreement with Actavis. The agreements will be submitted to the applicable governmental agencies.Supernus Pharmaceuticals, Inc. v. Zydus Pharmaceuticals (USA) Inc., C.A. No. 14-7272 (SDW)(LDW) (D.N.J.)We received three Paragraph IV Notice Letters against six Trokendi XR Orange Book patents, namely United States Patent Nos. 8,298,576, 8,298,580, 8,663,683, 8,877,248, 8,889,191, and 8,992,989 from generic drug maker Zydus Pharmaceuticals (USA) Inc. These patents cover once-a-day topiramateformulations and methods of treating seizures using those formulations. On November 21, 2014, we initiated a lawsuit against Zydus Pharmaceuticals (USA) Inc. and Cadila Healthcare Limited (collectively Zydus); the lawsuit alleges infringement of the Trokendi XR Orange Book patents. The FDA Orange Book currently lists United States Patent No. 8,298,576 as expiring on April 4, 2028 and United States Patent Nos. 8,298,580, 8,663,683, 8,877,248, 8,889,191 and 8,992,989 as expiring on November 16, 2027.This action for patent infringement—filed in the U.S. District Court for the District of New Jersey—alleges that Zydus infringed the Trokendi XR patents by, inter alia, submitting to the FDA an ANDA seeking to market a generic version of Trokendi XR prior to the expiration of these patents. Zydus answered these allegations with affirmative defenses and counterclaims of noninfringement and invalidity of the patents in suit. Filing its November 21, 2014 Complaint within 45 days of receiving the first of three Paragraph IV Notice Letters from Zydus Pharmaceuticals (USA) Inc. entitles Supernus to an automatic stay preventing the FDA from approving Zydus's ANDA for 30 months from the date of our receipt of such Notice Letter.The Company announced on March 6, 2017 that it has entered into a settlement agreement with Zydus regarding this case. The settlement permits Zydus to begin selling a generic version of Trokendi XR on January 1, 2023, or earlier under certain circumstances. A stipulation and order of dismissal without prejudice was entered by the U.S. District Court for the District of New Jersey. The agreement will be submitted to the applicable governmental agencies.Supernus Pharmaceuticals, Inc. v. Par Pharmaceutical Companies, Inc., C.A. No. 15-326 (SDW)(LDW) (D.N.J.)We received three Paragraph IV Notice Letters against six Trokendi XR Orange Book patents, namely United States Patent Nos. 8,298,576, 8,298,580, 8,663,683, 8,877,248, 8,889,191, and 8,992,989 from generic drug maker Par Pharmaceutical, Inc. These patents cover once-a-day topiramate formulations and methods of treating seizures using those formulations. On January 16, 2015, we initiated a lawsuit against Par; the lawsuit alleges infringement of the Trokendi XR Orange Book patents. The FDA Orange Book currently lists United States Patent No. 8,298,576 as expiring on April 4, 2028 and United States Patent Nos. 8,298,580, 8,663,683, 8,877,248, 8,889,191, and 8,992,989 as expiring on November 16, 2027.This action for patent infringement—filed in the U.S. District Court for the District of New Jersey—alleges that Par infringed the Trokendi XR patents by, inter alia, submitting to the FDA an ANDA seeking to market a generic version of Trokendi XR prior to the expiration of these patents. Par answered these allegations with affirmative defenses and counterclaims of noninfringement and invalidity of the patents in suit. Filing its January 16, 2015 Complaint within 45 days of receiving the first of three Paragraph IV Notice Letters from Par Pharmaceutical, Inc. entitles Supernus to an automatic stay preventing the FDA from approving Par's ANDA for 30 months from the date of our receipt of such Notice Letter.The Company announced on October 15, 2015 that it has entered into a settlement agreement with Par regarding this case. The settlement permits Par to begin selling a generic version of Trokendi XR on April 1, 2025, or earlier under certain circumstances. The agreement is subject to a consent judgment that was entered by the U.S. District Court for the District of New Jersey. In the consent judgment, Par acknowledges that the Orange Book-listed patents for Trokendi XR owned by Supernus, namely United States Patent Nos. 8,298,576, 8,298,580, 8,663,683, 8,877,248, 8,889,191, and 8,992,989, are valid and enforceable with respect to Par's ANDA product, and would be infringed by Par's ANDA product. The agreement has been submitted to the applicable governmental agencies. The following table sets forth for the periods indicated the high and low intra-day sales prices per share of our common stock as reported on the Nasdaq Global Market. High Low $ 15.99 $ 9.51 $ 20.38 $ 14.14 $ 26.84 $ 20.19 $ 27.10 $ 17.25
$ 12.38 $ 7.97 $ 18.55 $ 11.11 $ 23.30 $ 13.32 $ 20.39 $ 12.54 2016,2019, the closing price of our common stock on The NASDAQ Global Market was $25.25$23.72 per share. As of December 31, 2016,2019, we had 1519 holders of record of our common stock. The actual number of common stockholders is greater than the number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.2016,2019, the Company granted options to employees to purchase an aggregate of 22,25013,100 shares of common stock at an exercise price of $22.80$22.99 per share. The options are exercisable for a period of ten years from the grant date. These issuances were exempt from registration in reliance on Section 4(a)(2) of the Securities Act as transactions not involving any public offering.May 1, 2012December 31, 2014 and ending December 31, 2016. 2019.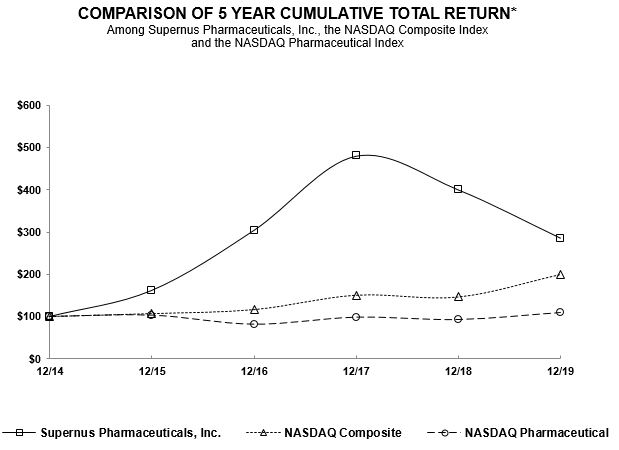
COMPARISON OF 44 MONTH CUMULATIVE TOTAL RETURN*Among Supernus Pharmaceuticals, Inc., the NASDAQ Composite Indexand the NASDAQ Pharmaceutical Index
*$100 invested on 5/1/12 in stock or 4/30/12 in each index, including reinvestment of dividends. Fiscal year ending December 31.* $100 invested on 12/31/2014 in stock or index, including reinvestment of dividends. Fiscal year ending December 31. Supernus
Pharmaceuticals, Inc. NASDAQ
Composite Index NASDAQ
Pharmaceuticals
Index $ 100.00 $ 100.00 $ 100.00 133.52 99.81 115.72 140.41 141.87 195.46 NASDAQ Composite
Index NASDAQ
Pharmaceuticals
Index 154.56 161.78 252.03 $ 100.00 $ 100.00 $ 100.00 250.28 171.75 261.96 161.93 106.96 103.06 470.20 185.66 207.12 304.22 116.45 81.93 December 31, 2017 480.12 150.96 98.23 December 31, 2018 400.24 146.67 92.83 December 31, 2019 285.78 200.49 109.06 2016, 20152019, 2018 and 20142017 and balance sheet data as of December 31, 20162019 and 20152018 set forth below have been derived from our audited consolidated financial statements included elsewhere in this Annual Report on Form 10-K. The selected statement of operationsearnings data for the years ended December 31, 20132016 and 20122015 and the balance sheet data as of December 31, 2014, 20132017, 2016 and 20122015 set forth below hashave been derived from the audited consolidated financial statements for such year not included in this Annual Report on Form 10-K. The historical periods presented here are not necessarily indicative of future results.Supernus Pharmaceuticals, Inc.Consolidated Statements of Operations Data(in thousands, except share and per share data) Year Ended December 31, 2016 2015 2014 2013 2012 $ 210,078 $ 143,526 $ 89,571 $ 11,552 $ — 4,686 3,038 633 — — 239 901 2,474 467 1,480 215,003 147,465 92,678 12,019 1,480 11,986 8,423 5,758 1,104 — 42,791 29,135 19,586 17,245 23,517 106,010 89,063 72,612 55,590 20,132 160,787 126,621 97,956 73,939 43,649 54,216 20,844 (5,278 ) (61,920 ) (42,169 ) 1,482 643 348 299 120 (543 ) (1,229 ) (4,963 ) (7,849 ) (3,575 ) (4,548 ) (3,541 ) (658 ) — — 448 193 2,809 (13,354 ) (710 ) (671 ) (2,338 ) (2,592 ) (9,550 ) — (15 ) 38 39 101 50 (3,847 ) (6,234 ) (5,017 ) (30,353 ) (4,115 ) 50,369 14,610 (10,295 ) (92,273 ) (46,284 ) (40,852 ) 666 630 — — 91,221 13,944 (10,925 ) (92,273 ) (46,284 ) — — — — (1,143 ) $ 91,221 $ 13,944 $ (10,925 ) $ (92,273 ) $ (47,427 ) $ 1.84 $ 0.29 $ (0.26 ) $ (2.90 ) $ (2.72 ) $ 1.76 $ 0.28 $ (0.26 ) $ (2.90 ) $ (2.72 ) 49,472,434 47,485,258 42,260,896 31,848,299 17,440,910 51,708,983 51,160,380 42,260,896 31,848,299 17,440,910 Years Ended December 31, 2019 2018 2017 2016 2015 Revenues $ 392,755 $ 408,897 $ 302,238 $ 215,003 $ 147,465 Net earnings 113,056 110,993 57,284 91,221 13,944 Earnings per share Basic $ 2.16 $ 2.13 $ 1.13 $ 1.84 $ 0.29 Diluted 2.10 2.05 1.08 1.76 0.28 Weighted-average shares outstanding Basic 52,412,181 51,989,824 50,756,603 49,472,434 47,485,258 Diluted 53,816,754 54,098,872 53,301,150 51,708,983 51,160,380 Balance Sheet and Other Data: Cash and cash equivalents and marketable securities $ 347,073 356,018 140,040 90,121 62,190 Long term marketable securities 591,773 418,798 133,638 75,410 55,009 Working capital 312,057 332,134 105,451 70,662 49,012 Total assets 1,160,282 977,811 424,464 309,568 188,626 Convertible notes, net 345,170 329,462 — 4,165 7,085 22,492 24,758 26,541 30,390 30,528 Retained earnings (accumulated deficit) 199,548 86,492 (26,823 ) (84,288 ) (175,509 ) Total stockholders' equity 595,428 453,023 267,480 191,755 88,007 Year Ended December 31, 2016 2015 2014 2013 2012 (in thousands) $ 90,121 $ 62,190 $ 74,336 $ 82,191 $ 88,508 75,410 55,009 19,816 8,756 — 70,662 49,012 80,603 70,761 68,479 309,568 188,626 136,784 110,995 93,989 4,165 7,085 26,223 34,393 — 30,390 30,528 30,025 — — — — — — 22,897 (84,288 ) (175,509 ) (189,453 ) (178,528 ) (86,255 ) 191,755 88,007 40,699 33,464 57,570 involvesinvolving risk and uncertainties. For example, statements regarding our expectations as to our plans and strategy for our business, future financial performance, expense levels and liquidity sources are forward-looking statements. Our actual results and the timing of those events could differ materially from those discussed in our forward-looking statements as a resultbecause of many factors, including those set forth under the "Risk Factors" section and elsewhere in this report. specialty pharmaceutical company focused on developing and commercializing products for the treatment of central nervous system (CNS) diseases. In 2013, we launched Oxtellar XR (extended-release oxcarbazepine)We have a portfolio of commercial products and Trokendi XR (extended-release topiramate), our two novel treatments for epilepsy. Since that time, we have significantly grown our net product sales.indicated for patients with epilepsy launched in the U.S.United States (U.S.) market. Net product sales from these products reached $210.1 million in 2016 representing significant growth compared to the $143.5 million in net product sales in 2015.We are continuing to expand our intellectual property portfolio to provide additional protection for our technologies, products, and product candidates. We currently have seven issued U.S. patents covering and eight issued U.S. patents covering is indicated for the treatment of epilepsy.withis indicated for the patents expiring no earlier than 2027treatment of epilepsy and for each product.Data from Intercontinental Marketing Services (IMS) shows 136,145the prophylaxis of migraine headache.were filled for both drugsas reported by IQVIA, during the three months ended December 31, 2016, representingperiods indicated below: Years Ended December 31, Change 2019 2018 Volume Percent Prescriptions Trokendi XR 672,485 638,923 33,562 5% Oxtellar XR 163,914 147,488 16,426 11% Total prescriptions 836,399 786,411 49,988 6% 22.0% increase over the 111,627novel non-stimulant product prescriptionscandidate for the fourth quartertreatment of 2015. Product prescriptions for Trokendi XR and Oxtellar XR totaled 506,542 for the year ended 2016, a 33.9% increase over the 378,173 product prescriptions for the year ended 2015. We expect the number of prescriptions filled for Oxtellar XR and Trokendi XR to continue to increase in the future.Net product sales for the year ended December 31, 2016 totaled $210.1 million, an increase of 46.4% over 2015. Net product sales for the fourth quarter of 2016 were $61.1 million, compared to net product sales of $42.6 million for the same quarter last year, an increase of 43.4%.Operating income for the year ended December 31, 2016 totaled $54.2 million compared to an operating income of $20.8 million in 2015, an increase of $33.4 million or 160.6%.We received several Paragraph IV Notice Letters concerning Oxtellar XR and Trokendi XR from various third-parties, asserting that our patents are invalid, or that our patents are not infringed by their formulations, or both. In response to these Paragraph IV notice letters, we initiated litigation against these third parties alleging infringement of our intellectual property rights. In October 2015, we reached a settlement agreement with one of these generic drug makers, Par Pharmaceutical Companies, Inc., concerning our Trokendi XR patents. In 2016, the U.S. District Court and Federal Court of Appeals ruled in our favor against Actavis concerning Oxtellar XR patents. In March 2017, we signed settlement agreements with two other generic drug makers, Actavis and Zydus, concerning our Trokendi XR patents. We intend to vigorously defend our intellectual property rights against TWi concerning our Oxtellar XR patents. We anticipate continuing to incur substantial amounts of legal feesand related expenses for these cases as they progress. (See Part I, Item 3—Legal Proceedings for additional information.)We are developing multiple product candidates in psychiatry to address large unmet medical needs and market opportunities. We are developing SPN-810 (molindone hydrochloride) to treat impulsive aggression (IA) in patients who have attention deficit hyperactivity disorder (ADHD). There are currently no approved products indicatedOn January 2020, we received the acceptance from the U.S. Food and Drug Administration (FDA) for the review of the New Drug Application (NDA) for SPN-812 for the treatment of IA.ADHD in pediatric patients. We arehave also developinginitiated a novel non-stimulant product candidate SPN-812 (viloxazine hydrochloride) to treat patients who have ADHD.We initiated two Phase III clinical trialstrial for SPN-810 duringthe treatment of adult patients with ADHD in the third quarter of 2015 and2019.IIb clinical trialIII study for SPN-812the treatment of bipolar disorder in the fourth quarter of 2015. We expect to continue recruiting2019. If approved, SPN-604 would represent the first approval for the treatment of bipolar disorder with oxcarbazepine in the two Phase III clinical trials for SPN-810 during 2017. ResultsU.S.Phase IIb clinical trialtreatment of severe epilepsy. We initiated an Investigational New Drug (IND) application enabling preclinical activities in the U.S. and have received an Orphan Drug designation for SPN-812 were announced in 2016. Subsequent to holding an endDravet Syndrome from the FDA.meeting with the FDA, we plan to initiate Phase III clinical trials for SPN-812 during the second half of 2017.with a total cost of approximately $85 million to $90 million for each of the two programs, from 20172020 through FDA approval.On January 19, 2017, Shire announced thatapproval or until the FDA acknowledged receiptprogram terminates. See Part I, Item I—the Class 2 resubmission of a New Drug Application (NDA) for SHP465, for the treatment of ADHD. The FDA is expectedour product and product candidates and development programs.decision on or around June 20, 2017. If approved by the FDA, SHP465 is expected to be launched by Shire in the second halfcomplete description of 2017. SHP465 was originally developed by Shire Laboratories, the former division of Shire which subsequently became Supernus Pharmaceuticals. Based on the agreement between Supernus and Shire, Shire will pay to Supernus a single digit percentage royalty on net sales of the product."SummarySummary of Significant Accounting Policies."Policies of the Notes to the Consolidated Financial Statements. The preparation of our consolidated financial statements in accordance with U.S. generally accepted accounting principles (GAAP) requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, and expenses, and to disclose contingent assets and liabilities. Actual results could differ materially from those estimates.from Product SalesRevenueRecognitionrecognized when: persuasive evidence of an arrangement exists; delivery has occurredtransferred to our customers, who are primarily pharmaceutical wholesalers and title to the product and associated risk of loss has passed to the customer; the price is fixed or determinable; collection from the customer has been reasonably assured; all performance obligations have been met; and returns and allowances can be reasonably estimated.distributors. Product sales are recorded net of various forms of variable consideration, including: estimated rebates, chargebacks, discounts, allowances, copay assistancerebates; sales discounts; and other deductions as well asan estimated liability for future product returns (collectively, "sales deductions"“sales deductions”).baseadjust our estimated sales deductions on an analysisestimates at the earlier of historical levelswhen the most likely amount of deductions specificconsideration we expect to each product. In addition, we also considerreceive changes, or when the impact of actual or anticipated changes in product price, sales trends and changes in managed care coverage and copay assistance programs.consideration becomes fixed. For a complete description of Trokendi XR and Oxtellar XR gross revenues and gross to net adjustments,our revenue recognition policy, see Part II, Item 8 Financial Statements and Supplemental Data,- Note 2, Sales.Deferred Legal FeesDeferred legal fees are comprised of costs incurred in connection with the defense of patents for Oxtellar XR and Trokendi XR (see Part I, Item 3—Legal Proceedings).Deferred legal fees have been incurred in connection with legal proceedings related to the defense of patents for Oxtellar XR and Trokendi XR (see Part II, Item 8—Financial Statements and Supplementary Data, Note 6). AmortizationSalesdeferred legal fees will begin upon successful outcome of the on-going litigation. Deferred legal fees will be chargedNotes to expense in the event of an unsuccessful outcome of the on-going litigation.ResearchWe estimate preclinical and development costs primarily consist of employee-relatedclinical trial expenses including salaries and benefits; share-based compensation expense; expenses incurred under agreementsbased on services performed pursuant to contracts with research institutions, clinical investigators, clinical research organizations (CROs), fees paid to investigators who are participating in our clinical trials, consultants and other vendorsservice providers that conduct activities on the Company's clinical trials;Company’s behalf. If the costactual timing of acquiringthe performance of services or the level of effort varies from our estimate, we adjust our accrued expenses or our deferred advance payments accordingly. For a complete description of our research and manufacturingdevelopment expense and preclinical and clinical trial materials;accrual policies, see Part II, Item 8 - Note 2, Summary of Significant Accounting Policies—Research and Development Expense and Related Accrued Research and Development Expenses, in the cost of manufacturing materials used in process validation,Notes to the extent that those materials are manufactured prior to receiving regulatory approval for those productsConsolidated Financial Statements.are not expected to be sold commercially; facilities costs that do not have an alternative future use; related depreciation and other allocated expenses; license fees for and milestone payments related to in-licensed products and technologies; and costs associated with animal testing activities and regulatory approvals.Accrued Clinical ExpensesClinicalclinical trials are inherently complex and often involve multiple service providers, and can include payments made to investigator physicians at study sites.providers. Because billing for services often lags delivery of service by a substantial amount of time,month or several months, we are often are required to estimate, and therefore accrue, a significant portion of our accrued clinicalthe incurred expenses. This process involves reviewing open contracts and communicating with our subject matter expert personnel, andas well as with the appropriate service provider personnel to identify services that have been performed on our behalf.behalf but for which no invoice has been received. This includes services provided by CROs, as well as services provided by clinical investigators and other service providers. We accrue for the estimated butcost for unbilled services performed, and the associated costs incurred.services providedservice or based on achievement of performance driven milestones. We work with each service provider to obtain an estimate for services provided but as yet unbilled as of the end of the calendar quarter, including estimates for payments to site investigators. When accruing clinical trial expenses, we estimate the time period over which services will be performed during the life of the entire clinical program, the total cost of the program, and the level of effort to be expended in each intervening period. To the maximum extent possible, we work with each service provider to provide an estimate for incurred but unbilled services as of the end of the calendar quarter. This includes estimates for payments to site investigators.companyCompany generated calculations.calculations by relying primarily on estimates provided by our vendors. If we and/or the service provider underestimates or overestimates the costcosts associated with a trial or service at any given point in time, adjustments to research and development expenses may be necessary in futurethe following periods. Historically, our estimated accrued clinical expenses have closely approximated the actual expenses incurred.incurred, with minimal adjustments to expense in the subsequent periods.Comparison2016 and2018 as compared to the year ended December 31, 2015 Year Ended
December 31, Increase/
(decrease) 2016 2015 (in thousands) $ 210,078 $ 143,526 66,552 4,686 3,038 1,648 239 901 (662 ) 215,003 147,465 11,986 8,423 3,563 42,791 29,135 13,656 106,010 89,063 16,947 160,787 126,621 54,216 20,844 1,467 681 786 (543 ) (1,229 ) 686 (4,548 ) (3,541 ) (1,007 ) 448 193 255 (671 ) (2,338 ) 1,667 (3,847 ) (6,234 ) 50,369 14,610 (40,852 ) 666 (41,518 ) $ 91,221 $ 13,944 Net Product Sales. Net product sales are based2017, please refer to Part II, Item 7—gross revenue from shipments to distributors, less estimates for discounts, rebates, allowances, other sales deductions and returns. Our net product sales of $210.1 millionForm 10-K for the year ended December 31, 20162018, which discussion is comprised of $51.7 million of revenue from Oxtellar XRincorporated by reference herein.$158.4 million of revenue from Trokendi XR. The increase in net product sales from 2016 to 2015 is primarily driven by increased prescriptions.Our net product sales of $143.5 millionexpenses, other (expense) income and income tax expense for the year ended December 31, 2015 were comprised of $33.2 million of revenue from Oxtellar XR and $110.3 million of revenue from Trokendi XR.Royalty Revenue. Non-cash royalty revenue of $4.7 million and $3.0 million was generated during the years ended December 31, 20162019, 2018 and December 31, 2015, respectively, pursuant2017 (dollars in thousands): 2019 vs 2018 Change 2018 vs 2017 Change 2019 2018 2017 Dollar Percent Dollar Percent Net product sales $ 383,400 $ 399,871 $ 294,097 $ (16,471 ) (4)% $ 105,774 36% Royalty revenue 9,355 8,276 6,367 1,079 13% 1,909 30% Cost of goods sold 16,660 15,356 15,215 1,304 9% 141 1% Research and development 69,099 89,209 49,577 (20,110 ) (23)% 39,632 80% Selling, general and administrative 158,425 159,888 137,905 (1,463 ) (1)% 21,983 16% Other (expense) income (1,084 ) (4,268 ) 1,077 3,184 (75)% (5,345 ) (496)% Income tax expense 34,431 29,183 43,334 5,248 18% (14,151 ) (33)% Net earnings 113,056 110,993 57,284 2,063 2% 53,709 94% 2019 vs 2018 Change 2018 vs 2017 Change 2019 2018 2017 Dollar Percent Dollar Percent Trokendi XR $ 295,214 $ 315,295 $ 226,518 $ (20,081 ) (6)% $ 88,777 39% Oxtellar XR 88,186 84,576 67,579 3,610 4% 16,997 25% Total $ 383,400 $ 399,871 $ 294,097 $ (16,471 ) (4)% $ 105,774 36% agreement8% price increase in 2019. Specifically, as regards sales deductions, patient reimbursement challenges and increased contracting pressure from managed care providers resulted in both increased per patient costs for our co-pay programs, higher per patient rebate payments to managed care providers, and higher Medicaid reimbursement payments. As a result, net product sales decreased by $16.5 million year over year.HC Royalty.Licensing Revenue. Total licensing revenuethe impact of an 8% price increase were offset by higher levels of net sales deductions. Increased sales deductions were driven primarily by increased per patient costs for our co-pay programs, higher per patient rebate payments to managed care providers, and higher Medicaid reimbursement payments. In addition, the majority of the impact of the $10 million channel inventory reduction, as described above, was reflected in lower net product sales for Trokendi XR in 2019.yearyears ended December 31, 2016 was $0.22019, 2018 and 2017 (dollars in thousands): Accrued Product Returns and Rebates Product
Rebates Product
Returns Allowance for
Sales Discounts Total Balance at December 31, 2017 $ 49,460 $ 18,883 $ 8,892 $ 77,235 Provision Provision for sales in current year 240,368 10,767 59,245 310,380 Adjustments relating to prior year sales (1,744 ) (75 ) (3 ) (1,822 ) Total provision 238,624 10,692 59,242 308,558 Less: Actual payments/credits (203,081 ) (7,515 ) (56,586 ) (267,182 ) Balance at December 31, 2018 $ 85,003 $ 22,060 $ 11,548 $ 118,611 Balance at December 31, 2018 $ 85,003 $ 22,060 $ 11,548 $ 118,611 Provision Provision for sales in current year 307,430 10,199 61,123 378,752 Adjustments relating to prior year sales (888 ) 549 (43 ) (382 ) Total provision 306,542 10,748 61,080 378,370 Less: Actual payments/credits (302,734 ) (13,990 ) (61,615 ) (378,339 ) Balance at December 31, 2019 $ 88,811 $ 18,818 $ 11,013 $ 118,642 and $0.9from $308.6 million in 2015. There was $0.82018 to $378.4 million in revenue generated from achievement2019. Virtually all of milestonesthis increase was attributable to the year over year increase in the year ended December 31, 2015.Cost of Product Sales. Cost ofprovision for product sales during the year ended December 31, 2016 was $12.0rebates, from $238.6 million an increase of $3.6in 2018 to $306.5 million in 2019, or 42.9%, as compared to $8.4 million for the year ended December 31, 2015.$67.9 million. The year over year increase isin the provision for product rebates of $67.9 million was primarily attributable to greater utilization of our patient co-pay programs. In addition, patient reimbursement challenges and increased contracting pressure from managed care providers resulted in both increased per patient costs for our co-pay programs, higher per patient rebate payments to managed care providers, and higher Medicaid reimbursement payments. Growth in prescriptions and the impact of the 8% price increase taken in January contributed, to a lesser extent, to the increase in product rebates. 2019 2018 2017 $ 2,428 $ 2,243 $ 1,034 6,927 6,033 5,283 Total $ 9,355 $ 8,276 $ 6,317 2019 vs 2018 Change 2018 vs 2017 Change 2019 2018 2017 Dollar Percent Dollar Percent Cost of goods sold $ 16,660 $ 15,356 $ 15,215 $ 1,304 9% $ 141 1% increased net product sales.Tablehigher volume of ContentsExpense. ResearchExpensesduringfor the year ended December 31, 2016 were $42.8years indicated (dollars in thousands): 2019 vs 2018 Change 2018 vs 2017 Change 2019 2018 2017 Dollar Percent Dollar Percent Research and development expense $ 69,099 $ 89,209 $ 49,577 $ (20,110 ) (23)% $ 39,632 80% $29.12018, primarily driven by the completion of the four Phase III clinical trials for SPN-812 in late 2018/early 2019, and the one-time $14 million for the year ended December 31, 2015, an increase of $13.7 million or 46.9%. This increase isexpense incurred in 2018 due to the conductacquisition of late stage clinical trials for both of our product candidates, SPN-810 and SPN-812. During 2016, we continuedBiscayne Neurotherapeutics Inc. These reductions were partially offset by the cost to recruit patients for our two Phase III trials for SPN-810 as well as recruiting patients for our Phase IIb trial for SPN-812. The Phase IIb trialmanufacture registration/validation materials for SPN-812 was completed in 2016. We expect R&D costs to increase significantly in 2017support the NDA filing for SPN-812 and beyond, as we continue to advance both of these programs.Expenses. OurExpensewere $106.0for the years indicated (dollars in thousands): 2019 vs 2018 Change 2018 vs 2017 Change 2019 2018 2017 Dollar Percent Dollar Percent Selling and marketing expense $ 113,609 $ 121,645 $ 104,072 $ (8,036 ) (7)% $ 17,573 17% General and administrative expense 44,816 38,243 33,833 6,573 17% 4,410 13% Total $ 158,425 $ 159,888 $ 137,905 $ (1,463 ) (1)% $ 21,983 16% during the year ended December 31, 2016in 2019 as compared to $89.12018, primarily as a result of decreased professional and consulting expenses of $4.3 million for the year ended December 31, 2015,and decreased sample expense of $3.1 million to support our existing commercial products.$16.9$1.6 million or 19.0%in professional and consulting fees. Change 2019 2018 2017 2019 vs 2018 2018 vs 2017 Interest income $ 21,623 $ 13,843 $ 2,864 $ 7,780 $ 10,979 Interest expense (18,207 ) (13,840 ) (134 ) (4,367 ) (13,706 ) Interest expense on nonrecourse liability related to sale of future royalties (4,500 ) (4,271 ) (1,434 ) (229 ) (2,837 ) Changes in fair value of derivative liabilities — — 76 — (76 ) Loss on extinguishment of debt — — (295 ) — 295 Total $ (1,084 ) $ (4,268 ) $ 1,077 $ 3,184 $ (5,345 ) SG&A expenses isinterest income, $7.8 million, was primarily due to support of our commercial products, and development of promotional materials and programsan increase in preparation for the launch of the migraine indication for Trokendi XR in 2017.Interest Income and Other Income, net. During the years ended December 31, 2016 and 2015, we recognized $1.5 million and $0.7 million, respectively, of interest income earned on our cash, cash equivalents and marketable securities.Expense.Expensewas $0.5in 2019 increased by $4.4 million, during the year ended December 31, 2016 as compared to $1.2 million for the2018, because of full year ended December 31, 2015. The decrease of $0.7 million was primarily due to a decrease in the principal amount of our outstanding 7.5% Convertible Senior Secured Notes dueinterest expense recognized in 2019 (the Notes) from $8.5 million at December 31, 2015 to $4.6 million at December 31, 2016. Duringon the year ended December 31, 2016, a total of $3.9 million of2023 Notes and related accrued interest converted into 0.8 million shares of common stock.Expense—Expense on Non-recourse Liability Related to Sale of Future Royalties. Non-cashRoyaltieswas $4.5 millionremained generally unchanged, from 2018 to 2019.year ended December 31, 2016 as comparedperiods indicated (dollar in thousands): Change 2019 2018 2017 2019 vs 2018 2018 vs 2017 Income tax expense $ 34,431 $ 29,183 $ 43,334 $ 5,248 $ (14,151 ) Effective tax rate 23.3% 20.8% 43.1% $3.5 million for the year ended December 31, 2015.2018. The increase of $1.0 million for this non-cashin income tax expense item was primarily due to a low effective tax rate in 2018 as the effective tax rate was favorably impacted by employee stock option exercises. The 2019 effective tax rate is favorably impacted by a decrease in our uncertain tax position reserve due to expiring statute of limitations and partially offset by an increase in our state effective tax rates due to an increase in the number of states in which we owe taxes.projection of future royalties relatedincome tax expense during the periods indicated (dollar in thousands): 2019 vs 2018 Change 2018 vs 2017 Change 2019 2018 2017 Dollar Percent Dollar Percent Net earnings $ 113,056 $ 110,993 $ 57,284 $ 2,063 2% $ 53,709 94% Orenitram.Changes2018Fair Value of Derivative Liability. During the year ended December 31, 2016, we recognized a non-cash gain of $0.4 million related to a change in the estimated fair value of the interest make-whole derivative liability related to our Notes. This gain is attributable to the passage of time and because our stock price remains above the $5.30 conversion price. During the year ended December 31, 2015, we recognized a non-cash gain of $0.2 million related to a change in estimated fair value of the interest make-whole derivative liability related to our Notes. This gainnet earnings was primarily due to the passage of time.Loss on Extinguishment of Debt. During the year ended December 31, 2016, we recognized a non-cash loss on extinguishment of debt of $0.7 million related to the conversion of $3.9 million of our Notes. During the year ended December 31, 2015, we recognized a non-cash loss on extinguishment of debt of $2.3 million related to the conversion of $27.5 million of our Notes.Income Tax. During the year ended December 31, 2016, we recorded $40.9 million of current tax benefit related primarily to releasing all of our valuation allowance on deferred tax assets. During the year ended December 31, 2015, we recorded $0.7 million of current tax expense related to an increase in our reserve for an uncertain tax position related to the Alternative Minimum Tax.Net Income. We realized net income of $91.2 million during the year ended December 31, 2016, compared to net income of $13.9 million during the year ended December 31, 2015, an increase of $77.3 million. This change was primarily due to the revenue generated from the sale of our two commercial products,products. Trokendi XR and Oxtellar XR, and Trokendi XR, increase inpartially offset by decreased R&D and SG&A spending and the impact of the elimination of the valuation allowance onincreased income tax expense.deferred tax asset.Comparison of the year ended December 31, 2015 and December 31, 2014 Year Ended
December 31, Increase/
(decrease) 2015 2014 (in thousands) $ 143,526 $ 89,571 53,955 3,038 633 2,405 901 2,474 (1,573 ) 147,465 92,678 8,423 5,758 2,665 29,135 19,586 9,549 89,063 72,612 16,451 126,621 97,956 20,844 (5,278 ) 681 387 294 (1,229 ) (4,963 ) 3,734 (3,541 ) (658 ) (2,883 ) 193 2,809 (2,616 ) (2,338 ) (2,592 ) 254 (6,234 ) (5,017 ) 14,610 (10,295 ) 666 630 36 $ 13,944 $ (10,925 ) Net Product Sales. Netoperations primarily with cash generated from product sales, are basedsupplemented by revenues from royalty and licensing arrangements as well as proceeds from the sale of equity and debt securities. Continued cash generation is highly dependent on gross revenue from shipments to distributors, less estimates for discounts, rebates, allowances, other sales deductions and returns. Our net product sales of $143.5 million for the year ended December 31, 2015 is comprised of $33.2 million of revenue from Oxtellar XR and $110.3 million of revenue from Trokendi XR. The increase in net product sales from 2014 to 2015 is primarily driven by increased prescriptions.Our net product sales of $89.6 million for the year ended December 31, 2014 are comprised of $24.7 million of revenue from Oxtellar XR and $64.9 million of revenue from Trokendi XR.Royalty Revenue. Non-cash revenues of $3.0 million and $0.6 million were generated during the years ended December 31, 2015 and 2014, respectively, pursuant to an agreement with HC Royalty.Licensing Revenue. Total licensing revenue for the year ended December 31, 2015 was $0.9 million. There was $0.8 million in revenue generated from achievement of milestones in the year ended December 31, 2015. The Company recognized $2.5 million in licensing revenue in 2014. This consisted primarily of the United Therapeutics Corporation milestone payment of $2.0 million under the license agreement with the Company.Cost of Product Sales. Cost of product sales during the year ended December 31, 2015 was $8.4 million as compared to $5.8 million for the year ended December 31, 2014, an increase of $2.6 million or 44.8%. This increase was primarily due to sales increases.Research and Development Expense. R&D expenses during the year ended December 31, 2015 were $29.1 million as compared to $19.6 million for the year ended December 31, 2014, an increase of $9.5 million, or 48.5%. This increase was due to preclinical and clinical trials and manufacturing scale up activities for bothcommercial success of our product candidates, SPN-810 and SPN-812. During 2015, we initiated two Phase III trials for SPN-810 and a Phase IIb trial for SPN-812. We expect R&D costs to increase significantly in 2017 and beyond, as we continue to advance these trials and the related development activities for both of these programs.Selling, General and Administrative Expenses. Our SG&A expenses were $89.1 million during the year ended December 31, 2015 as compared to $72.6 million for the year ended December 31, 2014, an increase of $16.5 million, or 22.7%. The increase in SG&A expenses is primarily due to the continued expansion of our sales and marketing efforts for bothcommercial products, Trokendi XR and Oxtellar XR, including promotional material and grants. In addition, we expended effort in 2015 to prepare for the launch of the migraine indication for Trokendi XR in 2016.Interest Income and Other Income, net. During the years ended December 31, 2015 and 2014, we recognized $0.7 million and $0.4 million, respectively, of interest income earned on our cash, cash equivalents, and marketable securities.Interest Expense. Interest expense was $1.2 million during the year ended December 31, 2015 as compared to $5.0 million for the year ended December 31, 2014. The decrease of $3.8 million was primarily due to a decrease in the principal amount of our outstanding 7.5% Convertible Senior Secured Notes due in 2019 (the Notes) from $36.1 million at December 31, 2014 to $8.5 million at December 31, 2015. During the year ended December 31, 2015, $27.5 million of the Notes and related accrued interest converted into 5.7 million shares of common stock.Interest Expense—Non-recourse Liability Related to Sale of Future Royalties. Non-cash interest expense related to our royalty liability was $3.5 million during the year ended December 31, 2015 as compared to $0.7 million for the year ended December 31, 2014. The increase of $2.8 million for this non-cash expense item was primarily due to an increase in the expected royalties forecast related to Orenitram and the annualization impact as the agreement was entered into in July 2014.Changes in Fair Value of Derivative Liability. During the year ended December 31, 2015, we recognized a non-cash gain of $0.2 million related to a change in estimated fair value of the interest make-whole derivative liability related to our Notes. This gain is attributable to the passage of time and because our stock price remains above the $5.30 conversion price. During the year ended December 31, 2014, we recognized a non-cash gain of $2.8 million related to a change in estimated fair value of the interest make-whole derivative liability related to our Notes. This gain is primarily due to the passage of time.Loss on Extinguishment of Debt. During the year ended December 31, 2015, we recognized a non-cash loss on extinguishment of debt of $2.3 million related to the conversion of $27.5 million of our Notes. During the year ended December 31, 2014, we recognized a non-cash loss on extinguishment of debt of $2.6 million related to the conversion of $13.4 million of our Notes.Income Tax. During the year ended December 31, 2015, we recorded $0.7 million of current tax expense related primarily to an increase in our reserve for an uncertain tax position for the Alternative Minimum Tax. During the year ended December 31, 2014, we recorded $0.6 million of current tax expense related primarily to the establishment of a reserve for an uncertain tax position for the Alternative Minimum Tax.Net Income (Loss).XR.realized net income of $14.0 million during the year ended December 31, 2015, compared to a net loss of $10.9 million during the year ended December 31, 2014, an increase of $24.9 million. This change was primarily due to the revenue generated from our two commercial products, Oxtellar XR and Trokendi XR, offset by increased expenses incurred in preparing for the latestage studies for two product candidates and an increase in marketing expenditures associated with ongoing support of Oxtellar XR and Trokendi XR.Liquidity and Capital ResourcesWe believe our increasing levels of net product sales will be sufficient to finance our operations in 2017 and subsequent years, including the increased R&D expenses for our clinical trials. We expect to incur significantly increased R&D expenses in 2017 and in subsequent years to support the development of SPN-810 and SPN-812, including the Phase III trials for SPN-810 and for SPN-812.Our working capital at December 31, 2016 was $70.7 million, an increase of $21.7 million compared to our working capital of $49.0 million at December 31, 2015. In addition, our long term marketable securities at December 31, 2016 were $75.4 million, an increase of $20.4 million compared to our long term marketable securities of $55.0 million at December 31, 2015.Our stockholders' equity increased by $103.7 million during the year ended December 31, 2016, primarily as a result of net income, the issuance of shares related to the conversion of our Notes and share-based compensation.In July 2014, we entered into a Royalty Interest Acquisition Agreement (the Agreement) with HC Royalty. Pursuant to the Agreement, HC Royalty paid us $30.0 million in consideration for acquiring certain royalty and milestone rights related to the commercialization of Orenitram (treprostinil) Extended-Release Tablets by United Therapeutics Corporation. Full ownership of the royalty rights will revert back to us if and when a certain threshold is reached per the terms of the Agreement.In addition to income from operations, we historically financed our business through the sale of our debt and equity securities. Our two most recent financings occurred on May 3, 2013, when we issued $90.0 million aggregate principal amount of Notes to qualified institutional buyers, the initial purchasers of the Notes (Initial Purchasers), and on July 2014 when we raised $30.0 million through a non-recourse liability related to the sale of future royalties.As of December 31, 2016, holders of the Notes have converted a total of approximately $85.4 million of the Notes. Cumulatively, through December 31, 2016, we issued a total of approximately 16.1 million shares of common stock in conversion of the principal amount of the Notes and issued an additional 2.2 million shares of common stock and paid approximately $1.7 million cash in settlement of the interest make-whole provision related to the converted Notes.Subsequent to December 31, 2016, holders of the Notes converted approximately $1.0 million of the Notes. We issued a total of approximately 0.2 million shares of common stock in conversion of the principal amount of the Notes and accrued interest thereon.We believe our current working capital and long term marketable securities, along with increased revenues from increasing product sales, will be sufficient to finance the Company. We achieved positive cash flow positive and profitabilityprofitable from operations in each quarter of 2015 and 2016.2019. While we expect continued profitability in 2017 as we continue to increase sales, while also increasing spending to advance our clinical product candidates,for future years, we anticipate there may be significant variability from quarteryear to quarteryear in our levelprofitability, and particularly as we move forward with the anticipated commercial launch of profitability.TableContents Change 2019 2018 2017 2019 vs 2018 2018 vs 2017 Cash and cash equivalents $ 181,381 $ 192,248 $ 100,304 $ (10,867 ) $ 91,944 Marketable securities 165,692 163,770 39,736 1,922 124,034 Long term marketable securities 591,773 418,798 133,638 172,975 285,160 Total $ 938,846 $ 774,816 $ 273,678 $ 164,030 $ 501,138 Working capital $ 312,057 $ 332,134 $ 105,451 $ (20,077 ) $ 226,683 Convertible notes, net (2023 Notes) $ 345,170 $ 329,462 $ — $ 15,708 $ 329,462 Total stockholder's equity $ 595,428 $ 453,023 $ 267,480 $ 142,405 $ 185,543 sets forthsummarizes the major sources and uses of cash for the periods set forth below summarized,(dollars in thousands: Year Ended
December 31, Increase/
(decrease) 2016 2015 $ 66,812 $ 34,524 32,288 (35,964 ) (39,289 ) 3,325 2,052 1,867 185 $ 32,900 $ (2,898 ) December 31, 2019 2018 2017 Net cash provided by (used in): Operating activities Operating earnings $ 142,516 $ 133,720 $ 90,930 Working capital 613 (4,734 ) 23,710 Total operating activities 143,129 128,986 114,640 Investing activities (157,924 ) (413,480 ) (86,415 ) Financing activities 3,928 376,438 5,681 Net change in cash and cash equivalents $ (10,867 ) $ 91,944 $ 33,906 by/used inby operating activities is comprised of two components;components: cash provided by operating income/lossearnings; and cash provided by/used inby (used in) changes in working capital.Results for the years ended December 31, 2016 and December 31, 2015 are summarized below, in thousands: Year Ended
December 31, Increase/
(decrease) 2016 2015 $ 58,364 $ 22,351 36,013 8,448 12,173 (3,725 ) $ 66,812 $ 34,524 increase in net cash provided by operating activities, is$143.1 million, was primarily driven by increased revenue generated from the sale of Trokendi XR and Oxtellar XR. The decrease inoperating earnings, reduced by incremental cash providedabsorbed by changesincreased working capital.is primarily driven by increased net sales deductions associated with our increased revenue.thousands: Year Ended December 31, 2016 2015 Explanation of Change $ (15,619 ) $ (8,638 ) Increased sales. (4,214 ) 854 Change in product inventory. 2,306 (1,582 ) Progress of clinical trials and other receivables. 26,165 20,901 Increased expenses, primarily for clinical trial accruals and accrued net sales deductions. (190 ) 638 $ 8,448 $ 12,173 Years Ended December 31, 2019 2018 2017 Explanation of Change (Increase) Decrease in: 2019 Compared to 2018 Accounts receivable $ 15,751 $ (35,856 ) (24,059 ) Receivables decreased in 2019 because of channel inventory reduction in first quarter 2019. Inventories (969 ) (9,355 ) 497 Increased inventory to support increased product demand. Prepaid expenses, other current assets and other non-assets (2,864 ) (2,367 ) (3,566 ) Timing differences related to deposits for equipment purchases and prepaid expenses for new clinical trials costs. Increase (Decrease) in: Accounts payable and accrued other noncurrent liabilities 3,151 6,854 2,268 Timing of vendor payments. Accrued product returns and rebates 566 38,720 26,400 Timing of product rebate payments; impact of channel inventory reduction in first quarter 2019; increased provision due to greater utilization of patient co-pay program and higher per patient Medicaid rebates, managed care rebates, and patient co-pay payments. Income taxes payable (9,934 ) (3,561 ) 15,931 Increased current tax provision due to increased state taxes and higher taxable income. Other (5,088 ) 831 6,239 Decreased employee-related costs. Total $ 613 $ (4,734 ) 23,710 We invest excess cash in accordance with our investment policy. Marketable securities consist of investments which mature in four years or less, including U.S. Treasury and various government agency debt securities, as well as investment grade securities in industrial and financial institutions.Fluctuations in investing activities between periods relate exclusively2019 Compared to the timing of marketable security purchases and the related maturities of these securities.2016 of $36.0 million related to2019. This year over year change was driven by changes in the net purchase of marketable securities. In 2018, proceeds from the issuance of the 2023 Notes in March 2018 were used to purchase marketable securities of $15.6 million, deferred legal fees of $18.8 million and property and equipment purchases of $1.6 million. long term marketable securities.used in investingprovided by financing activities decreased to $3.9 million for the year ended December 31, 20152019 versus $376.4 million provided in the same period in 2018. This year over year decrease is primarily attributable to the issuance of $39.3 million consisted of deferred legal fees of $10.9 millionthe 2023 Notes in March 2018, coupled with the related convertible note hedges and property and equipment purchases of $2.1 million, and net purchase of marketable securities of $26.3 million.Financing ActivitiesNet cash provided by financing activities for the year ended December 31, 2016 was $2.1 million, resulting from proceeds received from stock option exercises. Net cash provided by financing activities for the year ended December 31, 2015 was $1.9 million, resulting from proceeds received from stock option exercises.2016 (except2019, except as noted below),below (dollars in thousands: Less than
1 Year 1 - 3
Years 3 - 5
Years Greater than
5 Years Total $ — $ 4,575 $ — $ — $ 4,575 343 486 — — 829 1,321 2,655 454 — 4,430 46,060 799 — — 46,859 $ 47,724 $ 8,515 $ 454 $ — $ 56,693 (1)Our commitments for operating leases relate to our lease of office equipment, fleet vehicles and office and laboratory space as of December 31, 2016.(2)Relates primarily to agreements and purchase orders with contractors.(3)This table does not include (a) any milestone payments which may become payable to third parties under license agreements as the timing and likelihood of such payments are not known, (b) any royalty payments to third parties as the amounts, timing and likelihood of such payments are not known and (c) contracts that are entered into in the ordinary course of business which are not material in the aggregate in any period presented above.Contractual Obligations FY2020 FY2021 - FY2022 FY2023 - FY2024 Thereafter Total 2023 Convertible Notes $ — $ — $ 402,500 $ — $ 402,500 2,516 5,031 629 — 8,176 4,212 7,727 5,124 26,784 43,847 215,240 4,524 25 84 219,873 $ 221,968 $ 17,282 $ 408,278 $ 26,868 $ 674,396 Relates to the 2023 Notes (see Note 9 in the Notes to consolidated Financial Statements in Part II, Item 8 of this report.) Our commitments for operating leases relate to our leases of office equipment, fleet vehicles and the lease of the current headquarters office and laboratory space, as of December 31, 2019. Relates primarily to agreements and purchase orders with contractors and vendors. This table does not include (i) any milestone payments which may become payable to third parties under license agreements or contractual agreements regarding our clinical trials or those which may become payable upon achieving sales and developmental milestones per contractual agreements, as the timing and likelihood of such payments are not known, (ii) any royalty payments to third parties as the amounts, timing and likelihood of such payments are not known, and (iii) contracts that are entered into in the ordinary course of business which are not material in the aggregate in any period presented above. As of December 31, 2019, we had liabilities related to uncertain tax positions. Due to uncertainties in the timing of potential tax audits, the timing and the amounts associated with the resolution of these positions is uncertain. As such, we are unable to make a reasonably reliable estimate regarding the timing of payments beyond 12 months. Liabilities related to uncertain tax positions are not included in the above table. table, we are contractually obligated to pay to HC Royalty all royalty payments earned by us under a licensing agreement with United Therapeutics Corporation.for Orenitram. Although we have recorded a liability of $30.4$22.5 million atas of December 31, 20162019 related to this obligation, it is a non-recourse liability for which we have no obligation to make any cash payments to HC Royalty.Royalty, under any circumstances. Accordingly, this obligation will havehas no impact on our liquidity at any time and therefore thetime. The non-recourse liability has not been included in the table above.We have obtained exclusive licenses from third parties for proprietary rights to support the product candidates in our psychiatry portfolio. We have two license agreements with Afecta Pharmaceuticals, Inc. (Afecta) pursuant to which we obtained exclusive worldwide rights to selected product candidates, including an exclusive license to SPN-810. We may pay up to $300,000 upon the achievement of certain milestones. If a product candidate is successfully developed and commercialized, we will be obligated to pay royalties to Afecta based on worldwide net product sales at a rate in the low-single digits.to Rune basedat a low single digit percentage rate on worldwide net sales in the low single digits.notesNotes to the consolidated financial statementsConsolidated Financial Statements in Part II, Item 8 of this report.operations.operations and to facilitate business development activities. We also seek to maximize income from our investments without assuming significant risk.interest rate risk, liquidity risk or risk of default by investing in investment grade securities, with maturities of four years or less. Our exposure to market risk is confined to ourinvestments in cash, cash equivalents, marketable securities and long term marketable securities. As of December 31, 2016,2019, we had unrestricted cash, cash equivalents, marketable securities and long term marketable securities of $165.5$938.8 million.do not engage in any hedging activities against changes in interest rates.generally hold these securities to maturities of one to four years. Because of the short-term maturities ofrelatively short period that we hold our cash, cash equivalents, marketable securities and long term marketable securitiesinvestments and because we generally hold these securities to maturity, we do not believe that an increase in marketinterest rates would have any significant impact on the realizable value of our investments. We do not have any currency or other derivative financial instruments other than outstanding warrants to purchase common stock and the interest make-whole payment associated with our Notes.do not have on-going trialsonly one ongoing trial, for SPN-817, outside of the U.S. We do not hedge our foreign currency exchange rate risk. A hypothetical 10% appreciationTransactions denominated in Euro exchange rates againstcurrencies other than the U.S. dollar from prevailing marketare recorded based on exchange rates would have decreased our net income by approximately $4,000 forat the year ended time such transactions arise. As of December 31, 2016. Conversely, a hypothetical 10% depreciation2019 and December 31, 2018, substantially all of our liabilities were denominated in Euro exchange rates against the U.S. dollar from prevailing market rates would have increaseddollar.net incomecost of labor and the cost of services provided by approximately $4,000 for the year ended December 31, 2016.our vendors. We do not believe that inflation and changing prices over the years ended December 31, 20162019 and 20152018 had a significant impact on our consolidated results of operations.Years ended December 31, 2016, 2015 and 2014The and Stockholderssubsidiarysubsidiaries (the Company) as of December 31, 20162019 and 2015, and2018, the related consolidated statements of operations,earnings, comprehensive income (loss),earnings, changes in stockholders'stockholders’ equity, and cash flows for each of the years in the two-yearthree‑year period ended December 31, 2016. These2019, and the related notes (collectively, the consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States)statements). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion. referred to above present fairly, in all material respects, the financial position of Supernus Pharmaceuticals, Inc. and subsidiarythe Company as of December 31, 20162019 and 2015,2018, and the results of theirits operations and theirits cash flows for each of the years in the two-yearthree‑year period ended December 31, 2016,2019, in conformity with U.S. generally accepted accounting principles.Supernus Pharmaceuticals, Inc.'sthe Company’s internal control over financial reporting as of December 31, 2016,2019, based on criteria established inInternal Control—Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated March 16, 2017February 28, 2020 expressed an adverseunqualified opinion on the effectiveness of the Company'sCompany’s internal control over financial reporting.March 16, 2017The and Stockholders's and subsidiaries (the "Company")Company) internal control over financial reporting as of December 31, 2016,2019, based on criteria established in theInternal Control—Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO 2013 Framework"). Commission. In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.Company'sCompany’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanyingManagement Report on Internal Control over Financial Reporting.Reporting. Our responsibility is to express an opinion on the Company'sCompany’s internal control over financial reporting based on our audit.Public Company Accounting Oversight Board (United States).PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.company'scompany’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company'scompany’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company'scompany’s assets that could have a material effect on the financial statements.A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company's annual or interim financial statements will not be prevented or detected on a timely basis. Material weaknesses related to inadequately trained resources with assigned responsibility and accountability over the design and operation of internal controls; an ineffective risk assessment process that assessed necessary changes in financial reporting and internal controls impacted by changes in information technology systems; ineffective operation of controls over the completeness and accuracy of key assumptions and data analyzed by a third party consultant and used to determine the returns portion of accrued sales deductions; and ineffective general information technology controls over the Microsoft Dynamics AX information technology system and the employee expense reimbursement system, that resulted in ineffective process-level automated and manual controls related to these IT systems, have been identified and included in management's assessment.We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Supernus Pharmaceuticals Inc. and subsidiary as of December 31, 2016 and 2015, and the related consolidated statements of operations, comprehensive income (loss), changes in stockholders' equity, and cash flows for each of the years in the two-year period ended December 31, 2016. These material weaknesses were considered in determining the nature, timing, and extent of audit tests applied in our audit of the 2016 consolidated financial statements, and this report does not affect our report dated March 16, 2017, which expressed an unqualified opinion on those consolidated financial statements.In our opinion, because of the effect of the aforementioned material weaknesses on the achievement of the objectives of the control criteria, the Company has not maintained effective internal control over financial reporting as of December 31, 2016, based on criteria established in the COSO 2013 Framework.March 16, 2017Report of Independent Registered Public Accounting FirmThe Board of Directors and ShareholdersWe have audited the accompanying consolidated balance sheets of Supernus Pharmaceuticals, Inc. as of December 31, 2014, and the related consolidated statements of operations, comprehensive income (loss), changes in stockholders' equity and cash flows for the period ended December 31, 2014. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company's internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Supernus Pharmaceuticals, Inc. at December 31, 2014, and the consolidated results of their operations and their cash flows for the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles./s/ Ernst & Young LLPMcLean, VirginiaMarch 12, 2015, except for Note 2, as to which the date is January 20, 2017Supernus Pharmaceuticals, Inc. December 31, 2016 2015 $ 66,398 $ 33,498 23,723 28,692 41,527 25,908 16,801 12,587 2,955 5,261 151,404 105,946 75,410 55,009 4,344 3,874 19,860 22,503 16,490 976 331 318 41,729 — $ 309,568 $ 188,626 $ 8,055 $ 4,314 41,943 26,794 27,434 25,153 3,101 497 209 176 80,742 56,934 1,501 1,390 4,165 7,085 27,289 30,031 4,002 4,325 114 854 117,813 100,619
50 49 276,127 263,955 (134 ) (488 ) (84,288 ) (175,509 ) 191,755 88,007 $ 309,568 $ 188,626 December 31,
2019 December 31,
2018Assets Current assets Cash and cash equivalents $ 181,381 $ 192,248 Marketable securities 165,692 163,770 Accounts receivable, net 87,332 102,922 Inventories, net 26,628 25,659 Prepaid expenses and other current assets 11,611 8,888 Total current assets 472,644 493,487 Long term marketable securities 591,773 418,798 Property and equipment, net 17,068 4,095 Intangible assets, net 24,840 31,368 Lease assets 21,279 — Deferred income taxes 32,063 29,683 Other assets 615 380 Total assets 1,160,282 977,811 Liabilities and stockholders’ equity Current liabilities Accounts payable 10,141 3,195 Accrued product returns and rebates 107,629 107,063 Accrued expenses and other current liabilities 37,130 36,535 Income taxes payable 2,443 12,377 Nonrecourse liability related to sale of future royalties; current portion 3,244 2,183 Total current liabilities 160,587 161,353 Convertible notes, net 345,170 329,462 Nonrecourse liability related to sale of future royalties; long term 19,248 22,575 Lease liabilities, long term 30,440 — Other liabilities 9,409 11,398 Total liabilities 564,854 524,788 Stockholders’ equity Common stock, $0.001 par value; 130,000,000 shares authorized; 52,533,348 and 52,316,583 shares issued and outstanding as of December 31, 2019 and December 31, 2018, respectively 53 52 Additional paid-in capital 388,410 369,637 Accumulated other comprehensive earnings (loss), net of tax 7,417 (3,158 ) Retained earnings 199,548 86,492 Total stockholders’ equity 595,428 453,023 Total liabilities and stockholders’ equity 1,160,282 977,811 Operations Year Ended December 31, 2016 2015 2014 $ 210,078 $ 143,526 $ 89,571 4,686 3,038 633 239 901 2,474 215,003 147,465 92,678 11,986 8,423 5,758 42,791 29,135 19,586 106,010 89,063 72,612 160,787 126,621 97,956 54,216 20,844 (5,278 ) 1,482 643 348 (543 ) (1,229 ) (4,963 ) (4,548 ) (3,541 ) (658 ) 448 193 2,809 (671 ) (2,338 ) (2,592 ) (15 ) 38 39 (3,847 ) (6,234 ) (5,017 ) 50,369 14,610 (10,295 ) (40,852 ) 666 630 $ 91,221 $ 13,944 $ (10,925 ) $ 1.84 $ 0.29 $ (0.26 ) $ 1.76 $ 0.28 $ (0.26 ) 49,472,434 47,485,258 42,260,896 51,708,983 51,160,380 42,260,896 Years Ended December 31, 2019 2018 2017 Revenue Net product sales $ 383,400 $ 399,871 $ 294,097 Royalty revenue 9,355 8,276 6,367 Licensing revenue — 750 1,774 Total revenues 392,755 408,897 302,238 Costs and expenses Cost of goods sold 16,660 15,356 15,215 Research and development 69,099 89,209 49,577 Selling, general and administrative 158,425 159,888 137,905 Total costs and expenses 244,184 264,453 202,697 Operating earnings 148,571 144,444 99,541 Other (expense) income Interest expense (22,707 ) (18,111 ) (1,568 ) Interest income, net 21,623 13,843 2,645 Total other (expense) income (1,084 ) (4,268 ) 1,077 Earnings before income taxes 147,487 140,176 100,618 Income tax expense 34,431 29,183 43,334 Net earnings $ 113,056 $ 110,993 $ 57,284 Earnings per share Basic $ 2.16 $ 2.13 $ 1.13 Diluted $ 2.10 $ 2.05 $ 1.08 Weighted-average shares outstanding Basic 52,412,181 51,989,824 50,756,603 Diluted 53,816,754 54,098,872 53,301,150 Income (Loss) Year Ended December 31, 2016 2015 2014 $ 91,221 $ 13,944 $ (10,925 ) 354 (334 ) (154 ) 354 (334 ) (154 ) $ 91,575 $ 13,610 $ (11,079 ) Years Ended December 31, 2019 2018 2017 Net earnings $ 113,056 $ 110,993 57,284 Other comprehensive earnings (loss) Unrealized gain (loss) on marketable securities, net of tax 10,575 (2,411 ) (613 ) Other comprehensive earnings (loss) 10,575 (2,411 ) (613 ) Comprehensive earnings $ 123,631 $ 108,582 $ 56,671 Common Stock Accumulated
Other
Comprehensive
Income (Loss) Additional
Paid-in
Capital Accumulated
Deficit Total
Stockholders'
Equity Shares Amount 39,983,437 $ 40 $ 211,952 $ — $ (178,528 ) $ 33,464 — — 2,857 — — 2,857 76,333 — 516 — — 516 17,627 — 54 — — 54 2,897,066 3 14,884 — — 14,887 — — — — (10,925 ) (10,925 ) — — — (154 ) — (154 ) 42,974,463 43 230,263 (154 ) (189,453 ) 40,699 — — 4,090 — — 4,090 98,986 — 930 — — 930 205,640 — 937 — — 937 5,693,062 6 27,083 — — 27,089 32,523 — 652 652 — — — — 13,944 13,944 — — — (334 ) — (334 ) 49,004,674 49 263,955 (488 ) (175,509 ) 88,007 — — 5,926 — — 5,926 109,244 — 1,494 — — 1,494 85,694 — 557 — — 557 771,655 1 4,161 — — 4,162 — — — — 91,221 91,221 — — — 354 — 354 — — 34 — — 34 49,971,267 $ 50 $ 276,127 $ (134 ) $ (84,288 ) $ 191,755 Common Stock Shares Amount Balance, December 31, 2016 49,971,267 $ 50 $ 276,127 $ (134 ) $ (84,288 ) $ 191,755 Cumulative-effect of adoption of ASU 2016-09 — — 211 — 181 392 Balance, January 1, 2017 49,971,267 50 276,338 (134 ) (84,107 ) 192,147 Share-based compensation — — 8,433 — — 8,433 Issuance of employee stock purchase plan shares 71,256 — 1,888 — — 1,888 Exercise of stock options 407,477 — 3,793 — — 3,793 Equity issued on conversion of convertible notes 864,850 1 4,547 — — 4,548 Net earnings — — — — 57,284 57,284 Unrealized loss on marketable securities, net of tax — — — (613 ) — (613 ) Balance, December 31, 2017 51,314,850 51 294,999 (747 ) (26,823 ) 267,480 Cumulative-effect of adoption of ASC 606 — — — — 2,322 2,322 Balance, January 1, 2018 51,314,850 51 294,999 (747 ) (24,501 ) 269,802 Share-based compensation — — 11,291 — — 11,291 Issuance of employee stock purchase plan shares 71,250 — 2,209 — — 2,209 Exercise of stock options 930,483 1 9,372 — — 9,373 Equity component of convertible notes, net of tax — — 56,215 — — 56,215 Purchase of convertible note hedges, net of tax — — (70,137 ) — — (70,137 ) Issuance of warrants — — 65,688 — — 65,688 Net earnings — — — — 110,993 110,993 Unrealized loss on marketable securities, net of tax — — — (2,411 ) — (2,411 ) Balance, December 31, 2018 52,316,583 52 369,637 (3,158 ) 86,492 453,023 Share-based compensation — — 14,846 — — 14,846 Issuance of employee stock purchase plan shares 102,012 1 2,447 — — 2,448 Exercise of stock options 114,753 — 1,480 — — 1,480 Net earnings — — — — 113,056 113,056 Unrealized gain on marketable securities, net of tax — — — 10,575 — 10,575 Balance, December 31, 2019 52,533,348 $ 53 $ 388,410 $ 7,417 $ 199,548 $ 595,428 Year Ended December 31, 2016 2015 2014 $ 91,221 $ 13,944 $ (10,925 ) 671 2,338 2,592 (448 ) (193 ) (2,809 ) — — (154 ) 2,399 921 928 520 748 2,090 4,548 3,541 658 (4,686 ) (3,038 ) (633 ) 5,926 4,090 2,857 (41,787 ) — — (15,619 ) (8,638 ) (12,216 ) (4,214 ) 854 (6,289 ) 2,306 (1,582 ) (1,144 ) 3,470 2,061 (2,054 ) 15,149 18,333 7,461 7,546 507 2,031 — — (7,882 ) 144 149 (204 ) (334 ) 489 1,198 66,812 34,524 (24,495 ) (47,364 ) (63,859 ) (53,262 ) 31,824 37,581 53,473 (1,603 ) (2,104 ) (593 ) (18,821 ) (10,907 ) (2,277 ) (35,964 ) (39,289 ) (2,659 ) 2,052 1,867 571 — — (1 ) — — 30,000 2,052 1,867 30,570 32,900 (2,898 ) 3,416 33,498 36,396 32,980 $ 66,398 $ 33,498 $ 36,396 $ 493 $ 825 $ 2,854 $ 4,162 $ 27,089 $ 14,887 $ — $ 652 $ — $ 5,122 $ 9,789 $ 2,228 Years Ended December 31, 2019 2018 2017 Cash flows from operating activities Net earnings $ 113,056 $ 110,993 $ 57,284 Adjustments to reconcile net earnings to net cash provided by operating activities: Loss on extinguishment of debt — — 295 Change in fair value of derivative liability — — (76 ) Depreciation and amortization 6,659 7,063 8,132 Noncash operating lease cost 3,566 — — Amortization of deferred financing costs and debt discount 15,708 11,848 50 Amortization of premium/discount on marketable securities (4,335 ) (1,665 ) (563 ) Noncash interest expense 5,775 4,271 1,434 Noncash royalty revenue (6,927 ) (5,914 ) (5,283 ) Share-based compensation expense 14,846 11,291 8,433 Deferred income tax (benefit) provision (5,832 ) (4,167 ) 21,224 Changes in operating assets and liabilities: Accounts receivable 15,751 (35,856 ) (24,059 ) Inventories (969 ) (9,355 ) 497 Prepaid expenses and other current assets (2,723 ) (2,367 ) (3,566 ) Other noncurrent assets (141 ) — — Accounts payable 6,962 (3,578 ) (620 ) Accrued product returns and rebates 566 38,720 26,400 Accrued expenses and other current liabilities (3,811 ) 10,432 2,888 Income taxes payable (9,934 ) (3,561 ) 15,931 Deferred licensing revenue — — (274 ) Other liabilities (5,088 ) 831 6,513 Net cash provided by operating activities 143,129 128,986 114,640 Cash flows from investing activities Purchases of marketable securities (409,707 ) (491,654 ) (101,889 ) Sales and maturities of marketable securities 253,170 79,827 28,657 Purchases of property and equipment (2,736 ) (844 ) (2,029 ) Deferred legal fees 1,349 (809 ) (11,154 ) Net cash used in investing activities (157,924 ) (413,480 ) (86,415 ) Cash flows from financing activities Proceeds from issuance of convertible notes — 402,500 — Convertible notes issuance financing costs — (10,435 ) — Proceeds from issuance of warrants — 65,688 — Purchases of convertible note hedges — (92,897 ) — Proceeds from issuance of common stock 3,928 11,582 5,681 Net cash provided by financing activities 3,928 376,438 5,681 Net change in cash and cash equivalents (10,867 ) 91,944 33,906 Cash and cash equivalents at beginning of year 192,248 100,304 66,398 Cash and cash equivalents at end of period $ 181,381 $ 192,248 $ 100,304 Supplemental cash flow information: Cash paid for interest on convertible notes $ 2,516 $ 1,342 $ 134 Cash paid for Biscayne acquisition $ — $ 15,000 $ — Income taxes paid $ 51,540 $ 34,772 $ 1,588 Noncash investing and financing activity: Conversion of convertible notes and interest make-whole $ — $ — $ 4,548 Deferred legal fees and fixed assets included in accounts payable and accrued expenses $ 1,832 $ 250 $ 521 Unsettled purchase of marketable securities included in accrued expenses $ — $ — $ 1,004 Property and equipment additions from utilization of tenant improvement allowance $ 10,151 $ — $ — Years ended December 31, 2016, 2015 and 2014on March 30, 2005, and commenced operations on December 22,in 2005. The Company is a specialty pharmaceutical company focused on developing and commercializing products for the treatment of central nervous system (CNS) diseases, including neurological and psychiatric disorders.diseases. The Company markets two2 products: Oxtellar XR for the treatment of epilepsy products,and Trokendi XR for the prophylaxis of migraine headache and the treatment of epilepsy. The Company is also developing multiple proprietary CNS product candidates to address significant unmet medical needs and market opportunities.has several proprietary product candidatesadults in clinical development that address the psychiatry market.commenced the commercialization oflaunched Oxtellar XR and Trokendi XRwith an expanded indication to include monotherapy for partial seizures in 2013.The Company's consolidated financial statements include the accounts of Supernus Pharmaceuticals, Inc. and Supernus Europe Ltd., collectively referred to herein as "Supernus" or "the Company." All significant intercompany transactions and balances have been eliminated in consolidation. U.S.United States (U.S. GAAP).U.S.United States (U.S.), operates in one1 operating segment.preparation of the financial statements in accordance with U.S. GAAP requires the Company to make estimates and judgments in certain circumstances that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. In preparing these consolidated financial statements, management has made its best estimates and judgments of certain amounts included in the financial statements, giving due consideration to materiality. On an ongoing basis, the Company evaluates its estimates, including those related to revenue recognition, future royalty revenue related to Orenitram net product sales, accrued sales deductions, fair value of financial assets and liabilities, derivative liabilities, common stock options, income taxes, preclinical study and clinical trial accruals, and other contingencies. Management bases its estimates onon: historical experience or onexperience; various forecasts, includingforecasts; information received from its service providers which itand from other sources; and other assumptions that the Company believes to beare reasonable under the circumstances. Actual results could differ materially from thesethe Company’s estimates.inin: U.S. Treasuries, certificateTreasury bills and notes; certificates of deposit,deposit; various U.S. governmental agency debt securities,securities; corporate bondsand municipal bonds; and other fixed income securities. The Company places all investments with government,governmental, industrial or financial institutions whose debt is rated asSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 20142. Summary of Significant Accounting Policies (Continued)available-for-sale. Such securitiesavailable-for-sale and are carried at estimated fair value.reported, as accumulateda component of other comprehensive loss, which is a separate componentearnings (loss) in the consolidated statement of stockholders' equity.declinesare determined using the specific identification method for determining the cost of securities sold.resultsstatement of operations.earnings. A decline in the market value of any available for saleavailable-for-sale security below cost that is deemed to be other-than-temporary results in a reduction in fair value, which iswith that reduction charged to earnings in that period, and aperiod. A new cost basis for the security is established. then established at the time the reduction is recognized.The cost ofPremiums and discounts on marketable securities sold is calculated using the specific identification method.The Company established the Supernus Supplemental Executive Retirement Plan (SERP) for the sole purpose of receiving funds for executives from a previous SERPare amortized and providing a continuing deferral program under the Supernus SERP. As of December 31, 2016accreted, respectively, to maturity and 2015, the estimated fair value of the mutual fund investment securities within the SERP was approximately $275,000 and $263,000, respectively. The fair value of these assets is included within other non-current assets on the consolidated balance sheets. A corresponding noncurrent liability is also includedin interest income in the consolidated balance sheets to reflect the Company's obligation for the SERP. The Company has not made, and has no plans to make, contributions to the SERP. The securities are restricted in nature and can only be used for purposesstatement of paying benefits under the SERP.netdiscounts.sales allowances. The Company extends credit without requiring collateral.includingincluding: the financial condition and payment history of customers,customers; an overall review of collections experience on other accounts,accounts; and economic factors or events expected to affect future collections experience.an allowance for bad debts of approximately $42,000 as of December 31, 2016. No accounts were written off in 2015. The Company recorded an allowance of approximately $5.60, $0.1 million and $3.8 million0 for expected sales discounts as of December 31, 2016 and December 31, 2015, respectively. The following table includes those customers, who are wholesalers and distributors,Supernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Yearsdoubtful accounts for the years ended December 31, 2016, 20152019, 2018 and 20142. Summary of Significant Accounting Policies (Continued)that represent more than 10% of total net product sales2017, respectively. NaN receivable was written-off for 2016 and more than 10% of the accounts receivable balance on the consolidated balance sheet as ofyears ended December 31, 2016: Percent of Net
Product Sales Percent of Accounts
Receivable, net 29 % 43 % 30 % 26 % 37 % 28 % 96 % 97 % receivable and marketable securities.receivable. The counterparties are various corporations, governmental institutions, and financial institutions of high credit standing.with well known,in U.S. government agencies,agency debt and debt of well-known, investment grade corporations. Deposits held with banks may exceed the amount of governmental insurance provided on such deposits. Generally, these deposits may be redeemed upon demand and, therefore, management believes theythese bear minimal default risk.Inventory Percentage to Net Product Sales Percentage to Accounts Receivable, net 2019 2018 2017 2019 2018 Customer A 32 % 33 % 30 % 45 % 46 % Customer B 32 % 33 % 30 % 21 % 24 % Customer C 34 % 32 % 37 % 30 % 27 % 98 % 98 % 97 % 96 % 97 % market,net realizable value, include materials, labor, and other direct costs and indirect costs and are valued using the first-in, first-out method. The Company writes down inventory that has become obsolete or has a cost basis in excess of its expected net realizable value. Expired inventory is disposed of and the related costs are recognized as Cost of goods sold in the consolidated statement of earnings.commercialproduct launches sufficient to support estimated initial market demand when future commercialization of a product is probable and future economic benefit is expected to be realized. The determination to capitalize is based on the particular facts and circumstances relating to the product. Capitalization of such inventory begins when the Company determines that (i) positive results have been obtained for the clinical trials that are necessary to support regulatory approval; (ii) uncertainties regarding regulatory approval have been significantly reduced; and (iii) it becomesis probable that these capitalized costs will provide some future economic benefit in excess of capitalized costs.candidates will receive regulatorysafety or efficacy concerns, potential labeling restrictions and other impediments: historical experience with manufacturing and commercializing similar products and the relevant product candidate; and the Company’s manufacturing environment including its supply chain in determining logistical constraints that could hamper approval or commercialization. In assessing the economic benefit that the Company is likely to realize, the Company considers the shelf life of the product in relation to the expected timeline for approval and patent related or contract issues that may prevent or delay commercialization and product stability data of all pre-approval production to date to determine whether there is adequate expected shelf life; viability of commercialization taking into account trends in the related costs will be recoverable through the commercial sale ofmarketplace and market acceptance; and anticipated future sales and anticipated reimbursement strategies that may prevail with respect to the product.PropertyEquipmentPropertysales deductions.equipment2018, there was 0 pre-launch inventory recognized on the consolidated balance sheets.stated at cost. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts and any resulting gain or loss is credited or charged to operations. Repairs and maintenance costs are expensed as incurred. Depreciation and amortization are computed using the straight-line method over the following average useful lives:Computer equipment3 yearsSoftware3 yearsLab equipment and furniture5 - 10 yearsLeasehold improvementsShorter of lease term or useful lifeDeferred Legal FeesLegaldeferred legal fees that have been incurred in connection with legal proceedings related to the defense of patents for Oxtellar XR and Trokendi XR (see Notes 6 and 17). Amortization of the deferred legal fees willSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 20142. Summary of Significant Accounting Policies (Continued)begin upon successful outcome of the on-going litigation. Deferred legal fees will beXR. Patent defense costs are charged to expense in the event of an unsuccessful outcome of the on-going litigation.Intangible AssetsIntangible assets consist primarily of purchased patents and deferred legal fees related to patents. Patentsstraight-linestraight line basis over the estimated useful lives of the patents. Amortization commences in the quarter after the costs are incurred. The carrying valueamortization period is based initially upon the remaining patent life and is adjusted, if necessary, for any subsequent settlements or other changes to the expected useful life of the patents and deferred legal fees arepatent. Carrying value is assessed for impairment annually during the fourth quarter of each year, or more frequently if impairment indicators exist. There were no indicators of impairment identified at December 31, 2016 or 2015.purchased patents, deferred legal fees, and property and equipment.equipment and patent defense costs. The Company assesses the recoverability of its long-lived assets whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. If indications of impairment exist, projected future undiscounted cash flows associated with the asset are compared to the carrying amountvalue of the asset to determine whether the asset's value is recoverable. Evaluating for impairment requires judgment, including the estimation ofestimating future cash flows, future growth rates and profitability, and the expected life over which cash flows will occur. Changes in the Company's business strategy or adverse changes in market conditions could impactadversely affect impairment analyses, and require therequiring recognition of an impairment charge equal to the excess of the carrying value of the long-lived assetsasset over its estimated fair value.Forvalue at the years ended December 31, 2016, 2015 and 2014, the Company determinedtime at which that there was no impairmentdetermination is made.consist of financing costswere incurred by the Company in connection with the closingissuance of the Company's 7.50%$402.5 million of 0.625% Convertible Senior Secured Notes and Secured Notes Payable offeringdue 2023 (2023 Notes), (see Note 8)9). The Company amortizes deferred financing costs over the term of the related debt, using the effective interest method.• • extinguishing debt,the Company adjusts its estimates for product returns, the adjustment affects the estimated liability, product sales and earnings in the period of adjustment. Those adjustments may be material to our financial results.• deferred financingevent, such as regulatory approval, occurs. Milestones that are not within the control of the Company, such as approval from regulatory authorities, or where attainment of the specified event is dependent on the success of a third-party, are not considered probable until the specified event occurs.written off.Preclinical Studyexpensed as incurred. These expenses include: salaries, benefits and Clinical Trial Accruals study and clinical trial expenses based on the services performed pursuant to contracts with research institutions, clinical investigators, and clinical research organizations (CROs) and other service providers that conduct these activitiesperform services on ourthe Company’s behalf. In recording service fees, the Company estimates the timecost of those services which have been performed on behalf of the Company during the current period, over which the related services will be performed and compares the level of effort expended through the end of each period tothose costs with the cumulative expenses recorded and payments made for such services. As appropriate, itthe Company accrues additional service fees, or defers any non-refundablenonrefundable advance payments, until the related services are performed. If the actual timing of the performance of services or the level of effort varies from the estimate, the Company will adjustadjusts its accrualaccrued expenses, or its deferred advanceSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 20142. Summary of Significant Accounting Policies (Continued)payment payments, accordingly. If the Company latersubsequently determines that it no longer expects the services associated with a nonrefundable advance payment to be rendered, the remaining portion of that advance payment will beis charged to expense in the period thatin which such determination is made.Revenue from Product SalesRevenue from product salesrecognized when persuasive evidencemeasured based on estimated fair value as of an arrangement exists; deliverythe grant date, using the Black-Scholes option-pricing model in calculating the fair value of option grants as of the grant date. The Company uses the following assumptions for estimating fair value of option grants:occurred and titlehistorically fluctuated or is expected to fluctuate (i.e., expected volatility) during a period. Beginning in the first quarter of 2019, the Company began using the historical volatility of its common stock to measure expected volatility. Prior to the productfirst quarter of 2019, volatility was estimated using the observed volatility of the common stock of several public entities of similar size, complexity, and associated riskstage of loss has passed to the customer; the price is fixed or determinable; collection from the customer has been reasonably assured; all performance obligations have been met; and returns and allowances can be reasonably estimated. Product sales are recorded net of estimated rebates, chargebacks, allowances, discounts, co-pay assistance and other deductionsdevelopment, as well as estimated product returns (collectively, "sales deductions").Our products are distributed through wholesalerstaking into consideration the Company’s actual volatility since the Company’s IPO in 2012.pharmaceutical distributors. Each of these wholesalers and distributors will take title and ownershiphas no plans to do so in the product upon physical receipt of the product and then distribute our products to pharmacies.During the year ended December 31, 2015, the Company recorded a $2.9 million reduction to net revenue related to a change in estimate associated with its accrued sales deductions of $26.8 million at December 31, 2015. The change in estimate reflects returns experience associated with our initial launch shipments, which have now passed their expiry dating.Sales DeductionsAllowances for estimated sales deductions are provided for the following:•Rebates. Rebates include mandated discounts under the Medicaid Drug Rebate Program, the Medicare coverage gap program, as well as negotiated discounts with commercial healthcare providers. Rebates are amounts owed after the final dispensing of products to a benefit plan participant and are based upon contractual agreements or legal requirements with the public sector (e.g. Medicaid) and with private sector benefit providers. The allowance for rebatesforeseeable future. based on statutory and contractual discount rates and expected claimed rebates paid based on a plan provider's utilization. Rebates are generally invoiced and paid quarterly in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter's activity, plus an accrual balance for known or estimated prior quarters' unpaid rebates. If actual future rebates vary from estimates, we may need to adjust prior period accruals, which would affect revenue in the period of adjustment.•Co-pay assistance. Patients who paytime during which options are expected to remain unexercised. Options have a maximum contractual term of ten years. Beginning in cash or have commercial insurancethe first quarter of 2019, the Company began estimating the average expected life of stock options using its historical experience. Prior to the first quarter of 2019, the Company determined the average expected life of stock options according to the “simplified method”, as described in Staff Accounting Bulletin 110, which is the mid-point between the vesting date and meet certain eligibility requirements may receive co-pay assistance from the Company. The intentend of this programthe contractual term.to reduce the patient's outobserved U.S. Treasury Note rate as of pocket costs. Liabilitiesthe week each option grant is issued, with a term that most closely resembles the expected term of the option.co-pay assistance are based on actual program participation and estimates of program redemption using data provided by third-party administrators.•Distributor/Wholesaler deductions and discounts. U.S. specialty distributors and wholesalers are offered various forms of consideration including allowances, service fees and prompt payment discounts as consideration for distributing our products. Distributor allowances and service feesYears ended December 31, 2016, 2015 and 2014arise from contractual agreements with distributors and are generally a percentagethe purchase price paid by the distributors and wholesalers. Wholesale customers are offered a prompt pay discount for payment within a specified period.•Returns. Sales of our products are not subject to a general right of return; however,January 1, 2019, the Company will accept product thatself-insures its employee medical insurance liability. The self-insurance liability is damaged or defective when shipped directly from our warehouseundiscounted and expired product six months prior to, and up to twelve months subsequent to, its expiry date. Product that has been used to fill patient prescriptions is no longer subject to any right of return.•Chargebacks. Chargebacks are discounts that occur when contracted customers purchase directly from an intermediary distributor or wholesaler. Contracted customers, which currently consist primarily of Public Health Service institutions and federal government entities purchasing via the Federal Supply Schedule, generally purchase the product at a discounted price. The distributor or wholesaler, in turn, charges back the difference between the price initially paid by the distributor or wholesaler and the discounted price paid to the distributor or wholesaler by the customer. The allowance for distributor/wholesaler chargebacksdetermined actuarially, is based on known salesclaims filed, historical and industry claims experience, and an estimate of claims incurred but not yet paid. The Company has established stop-loss amounts that limit the Company’s further exposure after any individual claim reaches the designated stop-loss threshold, which effectively transfers any additional liability to contracted customers.Revenue Recognitiona third party. The stop-loss limit for self-insured employee medical claims is $150,000 per employee per year.License RevenueLicenseapproximately $600,000 as of December 31, 2019 in Collaboration Agreementsentered into collaboration agreements to have both Oxtellar XRnot been adjusted and Trokendi XR commercialized outside of the U.S. These agreements generally include an up-front license fee and ongoing milestone payments upon the achievement of specific events. We believe that when milestones meet all of the necessary criteriacontinue to be considered substantive, these should be recognized as revenue when achieved. For up-front license fees, we have estimatedreported in accordance with our historical accounting under Topic 840.service periodpackage of practical expedients permitted under the transition guidance, which, among other things, allowed the Company to carryforward the historical lease classification, the assessment on whether a contract was or contains a lease, and are recognizing this payment as revenuethe initial direct costs for any leases that existed prior to January 1, 2019. The Company also elected to combine the lease and non-lease components, and to keep leases with an initial term of 12 months or less off the balance sheet. We recognize the associated lease payments in the consolidated statements of earnings on a straight-line basis over the respective service period.Milestone PaymentsMilestone payments on licensing agreementslease term. Additionally, for certain equipment leases, we apply a portfolio approach to effectively account for the operating lease right-of-use (ROU) assets and lease liabilities.as revenue whenat commencement date based on the collaborative partner acknowledges completionpresent value of remaining lease payments over the lease term. For this purpose, the Company considers only payments that are fixed and determinable at the time of commencement. The Company calculates the present value of future payments by using an estimated incremental borrowing rate which approximates the rate at which the Company would borrow, on a secured basis and over a similar term. This rate is estimated based on information available at commencement date of the milestonelease, and substantive effort was necessarymay differ for individual leases or for portfolios of leased assets.achieveterminate prior to the milestone. Management may recognize revenue contingent uponend of the achievement of a milestone in its entiretylease term, or to extend the lease for one or more years. These options are included in the period in whichlease term when it is reasonably certain that the milestone is achieved only ifoption will be exercised. When determining the milestone meets allprobability of exercising such options, the criteria to be considered substantive. Substantive milestone paymentsCompany considers contract-based, asset-based, entity-based, and market-based factors.recognized upon achievement of the milestone only if all of the following conditions are met:•the milestone payments are non-refundable;•achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement;•substantive effortexpensed as incurred on the partner's partconsolidated statements of earnings. The Company’s lease agreements generally do not contain any material residual value guarantees or material restrictive covenants.involved in achieving the milestone; and•the amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with achievement of the milestone.Years ended December 31, 2016, 2015 and 2014Determinationto whether a payment meets the aforementioned conditions involves management's judgment. If any of these conditions are not met, the resulting payment would not be considered a substantive milestone,marketing materials, marketing programs and therefore the resulting payment would be considered part of the consideration for the single unit of accounting and amortized over the appropriate period.There was no milestone revenue during the year ended December 31, 2016. The Company recorded $0.8 million and $2.0 million, during the years ended December 31, 2015 and 2014, respectively.Royalty RevenueWe recognize non-cash royalty revenue for royalty amounts earned pursuant to a royalty agreement with United Therapeutics. In 2014, the Company sold certain of these royalty rights to HC Royalty (see Note 15). Accordingly, the Company records non-cash royalty revenue when payments are made from United Therapeutics to HC Royalty in connection with these agreements.Cost of Product SalesThe cost of product sales consist primarily of materials, third-party manufacturing costs, freight and distribution costs, allocation of labor, quality control and assurance, and other manufacturing overhead costs.Research and Development CostsResearch and development costs are expensed as incurred. Research and development costs primarily consist of employee-related expenses, including salaries and benefits; share-based compensation expense; expenses incurred under agreements with CROs, payments to investigators, and consultants that conduct the Company's clinical trials; the cost of acquiring and manufacturing clinical trial materials; the cost of manufacturing materials used in process validation, to the extent that those materials are manufactured prior to receiving regulatory approval for those products and are not expected to be sold commercially; facilities costs that do not have an alternative future use; related depreciation and other allocated expenses; license fees for, and milestone payments related to, in-licensed products and technologies; and costs associated with animal testing activities and regulatory approvals.Advertising Expense$21.9$40.8 million, $19.3$43.3 million and $14.8$33.8 million in advertising costs for the years ended December 31, 2016, 2015,2019, 2018 and 2014, respectively, which2017, respectively. These expenses are recorded in the selling,Selling, general and administrative expense line of the Statement of Operations.Share-Based CompensationEmployee share-based compensation is measured based on the estimated fair value on the grant date. The grant date fair value is calculated using the Black-Scholes option-pricing model, which requires the use of subjective assumptions including volatility, expected term, risk-free rate, and the fair value of theSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 20142. Summary of Significant Accounting Policies (Continued)underlying common stock. The Company recognizes expense using the straight-line method less estimated forfeitures.The Company records the expense for stock option grants to non-employees based on the estimated fair value of the stock option using the Black-Scholes option-pricing model. The fair value of non-employee awards is re-measured at each reporting period. As a result, stock compensation expense for non-employee awards with vesting is affected by subsequent changesexpensesfair valueconsolidated statement of the Company's common stock.measuredestimated as the largest amount of tax benefit that has a greater than 50% likelihood of being realized upon ultimate settlement with the tax authority,authorities, assuming full knowledge of the position and relevant facts. The Company's policy is to recognize any interest and penalties related to income taxes inas income tax expense.In August 2016,Financial Accounting Standards Board (FASB) issuedCompany adopted Accounting Standards Update (ASU) No. 2016-15, "Classification2014-9, "Revenue from Contracts with Customers," and has subsequently issued a number of Certain Cash Receiptsamendments to ASU 2014-9. ASU 2014-9 and Cash Payments."all the related amendments are codified in ASC 606, "Revenue from Contracts with Customers" (the New Revenue Standard). The standard eliminates diversityNew Revenue Standard provides a comprehensive model to be used in practice in how certain cash receiptsthe accounting for revenue arising from contracts with customers and cash paymentssupersedes current revenue recognition guidance, including industry-specific guidance.classifiedcontinue to be reported under the accounting standards in effect for the statementprior periods. The Company recognized the cumulative effect of initially applying the New Revenue Standard as an adjustment to the opening balance of retained earnings. Adjustments January 1, 2018 Accounts receivable, net $ 65,586 $ 1,620 $ 67,206 Deferred licensing revenue 287 (287 ) — Deferred licensing revenue, net of current portion 1,149 (1,149 ) — Deferred income taxes (asset) 20,843 (734 ) 20,109 Accumulated deficit 26,823 (2,322 ) 24,501 under Topic 230, Statement of Cash Flows, and other Topics. ASU 2016-15 is effective for annual reporting periods, and interim periods therein, beginning after December 15, 2017.arising from leasing arrangements. The Company doesadopted the new guidance using the modified retrospective transition approach by applying the new standard to all leases existing at the date of initial application and not expectrestating comparative periods. The most significant impact was the recognition of ROU assets and lease liabilities for operating leases. For information regarding the impact of the New Lease Standard adoption, see Significant Accounting Policies - Leases above and Note 14 - Leases.of this guidance towill not have a material impact on its Consolidated Financial Statements.In March 2016,consolidated financial statements.FASB issuedrequirements for capitalizing implementation costs incurred in a hosting arrangement that is a service contract with the requirements for capitalizing implementation costs incurred to develop or to obtain internal-use software. This includes hosting arrangements that include an internal-use software license. This ASU No. 2016-09, "Compensation-Stock Compensation (Topic 718): Improvementsalso requires the implementation costs of a hosting arrangement that is a service contract to Employee Share-Based Payment Accounting." The standard is intended to simplify several areasbe expensed over the term of accounting for share-based compensation arrangements, including the income tax impact, classification on the statement of cash flows and forfeitures. ASU 2016-09 is effective for fiscal years, and interim periods within those years, beginning after December 15, 2016, and early adoption is permitted.hosting arrangement, which includes reasonably certain renewals. The Company does not expect thatwill adopt the new standard effective January 1, 2020, and expects the adoption of this ASU will not have a material impact on Consolidated Financial Statementsits consolidated financial statements.related disclosures.In February 2016,Topic 606FASB issued ASU No. 2016-02, "Leases (Topic 842)."scope of the FASB’s revenue standard, Topic 606. The Company will adopt the new standard requires a lessee to recognize assetseffective January 1, 2020, and liabilities onexpects the balance sheet for leases with lease terms greater than 12 months. ASU 2016-02 is effective for fiscal years, and interim periods within those years, beginning after December 15, 2018, and early adoption is permitted. We expect the ASU to have a materialSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 20142. Summary of Significant Accounting Policies (Continued)impact on our assets and liabilities due to the addition of previously classified operating leases, but we dowill not expect it to have a material impact on our cash flows or resultsits consolidated financial statements.operations.In November 2015, the FASB issued ASU No. 2015-17, "Income Taxes (Topic 740): Balance Sheet Classification of Deferred Taxes."disclosure requirements for recurring and nonrecurring fair value measurements. The standard requires that deferred tax assetsremoves, modifies, and liabilities be classified as noncurrent onadds certain disclosure requirements. The Company will adopt the balance sheet rather than being separated into currentnew standard effective January 1, 2020, and noncurrent. ASU 2015-17 is effective for fiscal years, and interim periods within those years, beginning after December 15, 2016. Early adoption is permitted and the standard may be applied either retrospectively or on a prospective basis to all deferred tax assets and liabilities. We early adopted ASU 2015-17 during the fourth quarter of fiscal year 2015 on a prospective basis. As of December 31, 2015, the impact ofexpects the adoption of this standard was immaterial.In July 2015, the FASB issued ASU No. 2015-11, "Inventory (Topic 330): Simplifying the Measurement of Inventory." Under this new guidance, entities that measure inventory using any method other than last-in, first-out or the retail inventory method will be required to measure inventory at the lower of cost and net realizable value. The amendments in this ASU, which should be applied prospectively, are effective for annual and interim periods beginning after December 15, 2016. Early adoption is permitted. The Company does not expect that the adoption of this ASU will have a material impact on Consolidated Financial Statements and related disclosures.In April 2015,its consolidated financial statements.FASBAccounting for Income Taxes - The new standard, issued ASU No. 2015-03, "Simplifyingin December 2019, simplifies the Presentation of Debt Issuance Costs." This ASU more closely aligns the treatment of debt issuance costs with debt discounts and premiums and requires debt issuance costs to be presented as a direct deduction from the carrying amount of the related debt. The amendments in this ASU are effectiveaccounting for financial statements issued for fiscal years beginning after December 15, 2015, and interim periods within those fiscal years.income taxes. This guidance has been appliedwill be effective on January 1, 2021 on a retrospective basis. As of December 31, 2015,prospective basis, with early adoption permitted. The Company is currently evaluating the impact of the adoption of thisnew guidance on its consolidated financial statements and will adopt the new standard was immaterial.In May 2014, the FASB issued ASU No. 2014-09, "Revenue from Contracts with Customers." ASU 2014-09 will eliminate transaction-and industry-specific revenue recognition guidance under current U.S. GAAP and replace it with a principles-based approach for determining revenue recognition. ASU 2014-09 will require that companies recognize revenue based on the value of transferred goods or services as they occur in the contract. The ASU also will require additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. ASU 2014-09 is effective for annual reporting periods beginning after December 15, 2017, with early adoption being permitted for periods ending after December 15, 2016. Entities can transition to the standard either retrospectively or as a cumulative effect adjustment as of the date of adoption. We are in the process of evaluating the potential revenue implications of the standard change, which may result in changes to our revenue recognition practices around license and collaboration agreements.of the consolidated financialsfinancial statements were issued in this Annual Report on Form 10-K, and believes that no other ASU will have a material impact on the Company's consolidated financial statements.Years ended December 31, 2016, 2015 and 2014should representrepresents the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. Such transactions to sell an asset or transfer a liability are assumed to occur in the principal or most advantageous market for the asset or liability. Accordingly, fair value is determined based on a hypothetical transaction at the measurement date, considered from the perspective of a market participant rather than from a reporting entity's perspective. that are measured at fair value using a three level fair value hierarchy that prioritizes the inputs used to measure fair value. This hierarchy maximizes the use of observable inputs and minimizes the use of unobservable inputs. The three levels of inputs used to measure fair value are as follows:•assets that theassets. The Company has the ability to access atthese prices as of the measurement date.•Inputs are quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (interest rates, yield curves, etc.) and inputs that are derived principally from or corroborated by observable market data by correlation or other means (market corroborated inputs).•Level 3—Unobservable inputs that reflect the Company's own assumptions, based on the best information available, including the Company's own data.In accordance with the fair value hierarchy described above, the following tables show the fair value of the Company's financial assets and liabilities that are required to be measured at fair value, in thousands: Fair Value Measurements at December 31, 2016 Total Carrying
Value at
December 31,
2015 Quoted Prices
in Active
Markets
(Level 1) Significant
Other
Observable
Inputs
(Level 2) Significant
Unobservable
Inputs
(Level 3) $ 66,398 $ 66,398 $ — $ — 23,723 656 23,067 — 75,410 — 75,410 — 275 — 275 — $ 165,806 $ 67,054 $ 98,752 $ — $ 114 $ — $ — $ 114 Supernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 20143. Fair Value of Financial Instruments (Continued) Fair Value Measurements at December 31, 2015 Total Carrying
Value at
December 31,
2015 Quoted Prices
in Active
Markets
(Level 1) Significant
Other
Observable
Inputs
(Level 2) Significant
Unobservable
Inputs
(Level 3) $ 33,498 $ 33,498 $ — $ — 28,692 654 28,038 — 55,009 — 55,009 — 263 — 263 — $ 117,462 $ 34,152 $ 83,310 $ — $ 854 $ — $ — $ 854 The fair value of the restricted marketable securities is included within other non-current assets in the consolidated balance sheets.The Company's Level 1 assets include cash held with banks, certificate of deposits, and money market funds.Level 2 assets include the SERP (Supplemental Executive Retirement Plan) assets, commercial paper and investment grade corporate bonds and other fixed income securities. Level 2 securities are valued using third-party pricing sources that apply applicable inputs and other relevant data intoin their models to estimate fair value.3 liabilities include3—Unobservable inputs that reflect the estimatedCompany’s own assumptions. These are based on the best information available, including the Company’s own data.of the interest make-whole liability associated with the Company's 7.50% Convertible Senior Secured Notes due 2019 (the Notes), which is recordedon a recurring basis are as a derivative liability.The fair value of the interest make-whole liability of the Notes was calculated using a binomial-lattice model with the following key assumptions as of December 31 2016:Volatility45%Stock Price as of December 31, 2016$25.25 per shareCredit Spread900 bpsTerm4 monthsDividend Yield0.0% Fair Value Measurements as of December 31,
2019 Total Fair
value at
December 31, 2019 Quoted Prices
in Active Markets
for Identical Assets
(Level 1) Significant
Other
Observable
Inputs
(Level 2)Assets: Cash and cash equivalents Cash $ 78,912 $ 78,912 $ — Money market funds 102,469 102,469 — Marketable securities Corporate debt securities 165,527 — 165,527 Municipal debt securities 165 — 165 Long term marketable securities Corporate debt securities 571,828 254 571,574 U.S. government agency debt securities 19,945 — 19,945 Other noncurrent assets Marketable securities - restricted (SERP) 418 3 415 Total assets at fair value $ 939,264 $ 181,638 $ 757,626 Changes in the fair value of the warrants and the interest make-whole liability are recognized as a component of Other Income (Expense) in the Consolidated Statements of Operations. The following table presents information about the Company's Level 3 liabilities as of December 31, 2015 andYears ended December 31, 2016, 2015 and 2014December 31, 2016 that are included in the Non-Current Liabilities section Fair Value Measurements as of December 31,
2018 Total Fair Value at
December 31, 2018 Quoted Prices
in Active Markets
for Identical Assets
(Level 1) Significant
Other
Observable
Inputs
(Level 2)Assets: Cash and cash equivalents Cash $ 106,918 $ 106,918 $ — Money market funds 85,330 85,330 — Marketable securities Corporate debt securities 163,770 245 163,525 Long term marketable securities Corporate debt securities 415,650 445 415,205 U.S. government agency debt securities 2,983 — 2,983 Municipal debt securities 165 — 165 Other noncurrent assets Marketable securities - restricted (SERP) 326 1 325 Total assets at fair value $ 775,142 $ 192,939 $ 582,203 the Consolidated Balance Sheets, in thousands: Year Ended
December 31,
2015 and 2016 $ 6,564 (193 ) (4,865 ) (652 ) 854 (448 ) (292 ) $ 114 The carrying value, face valueLevel 2 assets include: commercial paper; investment grade corporate, U.S. government agency, state and estimatedmunicipal debt securities; other fixed income securities and SERP (Supplemental Executive Retirement Plan) assets. The fair value of the Notes was approximately $4.2 million, $4.6 million and $21.8 million, respectively,restricted marketable securities is recorded in Other assets on the consolidated balance sheets.2016. The fair value was estimated based on actual trade information as well as quoted prices provided by bond traders, which would be characterized within Level 2 of the fair value hierarchy. This fair value amount gives recognition to the value of the interest make-whole liability and the value of the conversion option. These items have been accounted for as derivative liabilities and additional paid-in-capital, respectively.wereare as follows (dollars in thousands:At December 31, 2016: December 31,
2019 December 31, 2018 Corporate and U.S. government agency and municipal debt securities Amortized cost $ 747,598 $ 586,726 Gross unrealized gains 10,031 55 Gross unrealized losses (164 ) (4,213 ) Total fair value $ 757,465 $ 582,568 Amortized
Cost Gross
Unrealized
Gains Gross
Unrealized
Losses Fair Value $ 99,487 86 (440 ) $ 99,133 At December 31, 2015: Amortized
Cost Gross
Unrealized
Gains Gross
Unrealized
Losses Fair Value $ 84,189 5 (493 ) $ 83,701 Years ended December 31, 2016, 2015 and 2014available for saleavailable-for-sale marketable securities held by the Company wereare as follows (dollars in thousands: December 31,
2019Less Than 1 Year $ 165,692 1 year to 2 years 188,378 2 year to 3 years 201,491 3 years to 4 years 201,904 Greater Than 4 Years — Total $ 757,465 December 31,
2016 $ 23,723 24,318 51,092 — $ 99,133 securities and restricted marketable securities.cost of securities soldfollowing table sets forth the Company’s financial liabilities that are not carried at fair value (dollars in thousands): December 31, 2019 December 31, 2018 Carrying Value Fair Value (Level 2) Carrying Value Fair Value (Level 2) 2023 Notes $ 345,170 $ 366,023 $ 329,462 $ 375,834 calculated using the specific identification method.thousands: December 31,
2019 December 31,
2018Raw materials $ 4,582 $ 5,742 Work in process 11,428 7,275 Finished goods 10,618 12,642 $ 26,628 $ 25,659 December 31,
2016 December 31,
2015 $ 2,091 $ 2,887 8,874 3,946 5,836 5,754 $ 16,801 $ 12,587 thousands: December 31,
2019 December 31,
2018Lab equipment and furniture $ 11,053 $ 8,995 Leasehold improvements 14,217 2,731 Software 2,225 2,181 Computer equipment 1,839 1,313 Construction-in-progress 433 94 29,767 15,314 Less accumulated depreciation and amortization (12,699 ) (11,219 ) Total $ 17,068 $ 4,095 December 31,
2016 December 31,
2015 $ 1,206 $ 1,112 1,807 307 6,758 5,667 2,642 2,642 28 1,114 12,441 10,842 (8,097 ) (6,968 ) $ 4,344 $ 3,874 $1.1$1.5 million, $0.7$1.9 million, and $0.7$1.2 million for the years ended December 31, 2016, 20152019, 2018 and 2014,2017, respectively.TableThe Company annually performs an impairment assessment of ContentsSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years endedits property and equipment in the fourth quarter of each year, or earlier if impairment indicators exist. As of December 31, 2016, 2015 and 2014 Deferred Legal Fees and Intangible AssetsDeferredhave been incurred in connectionconjunction with patent litigationdefending patents for Oxtellar XR and Trokendi XR. AsThe Company amortizes these costs over the useful life of December 31, 2016 and 2015, the Company had deferred legal fees of $19.9 million and $22.5 million, respectively.thesethe intangible assets (dollars in thousands: December 31,
2019 December 31,
2018Capitalized patent defense costs 3.01 - 7.25 years $ 43,375 $ 44,724 Less accumulated amortization (18,535 ) (13,356 ) Total $ 24,840 $ 31,368 Weighted-Average
Life December 31,
2016 December 31,
2015 9.5 - 11 years $ 17,773 $ 994 (1,283 ) (18 ) $ 16,490 $ 976 The Company prevailed in a lawsuit related toin 2016, at which timeand Trokendi XR will expire no earlier than 2027. As regards Trokendi XR, the Company reduced deferred legal fees,entered into settlement agreements that allow third parties to enter the market by $16.6 million, and transferred these amounts to intangible assets. The Company subsequently began amortizing the costs associated with that litigation.The net book value of intangible assets was $16.5 million as of December 31, 2016 and was $1.0 million as of December 31, 2015. The increase in intangible assets reflects the successful outcome of the lawsuit related to Oxtellar XR in February 2016. There is an offsetting reduction in the amount carried as deferred legal fees, as described above.$1.3$5.2 million, $0.2$5.2 million and $0.2$6.9 million for the years ended December 31, 2016, 20152019, 2018 and 2014,2017, respectively. Amortization expense in 2015 and 2014 includedassociated with purchased patents that were fully amortized asfor intangible assets from 2020 to 2022 is estimated at $5.0 million per year. Anticipated annual amortization expense for intangible assets in 2023 and 2024 is estimated at $2.3 million per year.2015 and are therefore not included in the above table.There2019, there were no identified indicators of impairment identified at December 31, 2016 or December 31, 2015.are comprisedand other current liabilities consist of the following (dollars in thousands: December 31,
2019 December 31,
2018 $ 13,285 $ 14,034 Accrued compensation 11,223 13,546 Accrued professional fees 3,936 3,706 Lease liabilities, current 2,825 — Other accrued expenses 5,861 5,249 Total $ 37,130 $ 36,535 December 31,
2016 December 31,
2015 $ 9,145 $ 7,519 5,919 10,057 4,350 3,677 1,035 113 528 434 61 295 6,396 3,058 $ 27,434 $ 25,153 Years ended December 31, 2016, 201520148.Rebates December 31,
2019 December 31,
2018Accrued rebates $ 88,811 $ 85,003 Accrued product returns 18,818 22,060 Total $ 107,629 $ 107,063 Secured NotesMay 3, 2013,March 14, 2018, the Company issued $90.0entered into a Purchase Agreement (the Purchase Agreement) with Jefferies LLC, J.P. Morgan Securities LLC and Cowen and Company, LLC, as the initial purchasers (collectively, the Initial Purchasers), in connection with the offering and sale of $350 million aggregate principal amount of Notes in a private placement offering.the Notes underpursuant to an Indenture, dated May 3, 2013as of March 19, 2018 (the Indenture), between the Company and U.S. BankWilmington Trust, National Association, as Trusteetrustee. The Indenture includes customary terms and Collateral Agent.covenants, including certain events of default upon which the 2023 Notes may be due and payable immediately. The Indenture governing the 2023 Notes provide for 7.50% interest per annumdoes not contain any financial or operating covenants or any restrictions on the principal amountpayment of dividends, the issuance of other indebtedness, or the issuance or repurchase of securities by the Company.MayApril 1 and NovemberOctober 1 of each year.year, beginning on October 1, 2018. The 2023 Notes will mature on MayApril 1, 2019,2023, unless earlier converted redeemed or repurchased by the Company. Theare convertible intoat their option only in the following circumstances: (1) during any calendar quarter, if the last reported sale price per share of the Company's common stock (Common Stock) as described below.The Notes are the Company's senior secured obligations and (i) rank senior in right of payment to any of the indebtedness that is expressly subordinated in right of payment to the Notes; (ii) rank effectively senior to any of the unsecured indebtedness to the extent of the value of the collateral securing the Notes; (iii) rank equal in right of payment with all of the Company's indebtedness that is not subordinated to the Notes; and (iv) are structurally subordinated to all indebtedness and liabilities, including trade payables, of the Company's existing and future subsidiaries.The Notes are secured by a first-priority lien, other than customary permitted liens, on substantially all of the Company's and its domestic subsidiaries' assets, whether now owned or hereafter acquired, including license agreements, general intangibles, accounts, instruments, investment property, intellectual property and any proceeds of the foregoing pursuant to that certain Security and Pledge Agreement, dated May 3, 2013 (the Security Agreement), between the Company and U.S. Bank National Association, as Collateral Agent. The Indenture restricts the ability of the Company and its existing and future subsidiaries to make investments, including transfers of the Company's assets that constitute collateral securing the Notes, in its existing and future foreign subsidiaries.Prior to November 1, 2018, a holder of Notes may convert all or a portion of its Notes, in principal amounts equal to $1,000 or an integral multiple thereof, only if one or more of the following conditions has been satisfied: (1) if, for at least 20 trading days (whether or not consecutive) during the 30 consecutive trading day perioddays ending within five trading days prior to a conversion date,on, and including the last reported sale pricetrading day of the Company's Common Stockimmediately preceding calendar quarter, exceeds 130% of the conversion price, or a price of approximately $77.13 per share on each such trading day; (2) during the five5 consecutive business day perioddays immediately followingafter any five10 consecutive trading day period (the Measurement Period),(such 10 consecutive trading day period, the "measurement period") in which for each trading day of that Measurement Period, the trading price (as defined in the Indenture) per $1,000 principal amount of Notes for sucheach trading day of the measurement period was less than 98% of the product of the last reported sale price per share of the Company's Common Stockcommon stock on such trading day and the applicable conversion rate on such trading day; (3) upon the occurrence of certain corporate events or distributions on the Company's common stock, as specified corporate transactions; orin the Indenture; and (4) if the Company calls the Notes for redemption, at any time prior tofrom and including October 1, 2022, until the close of business on the businesssecond scheduled trading day immediately preceding the redemption date. On and after November 1, 2018, a holder of Notes may convert all or a portion of its Notes, in principal amounts equal to $1,000 or an integral multiple thereof, at any time prior to the close of business on the business day immediately precedingbefore the maturity date of the Notes, regardless of the foregoing circumstances.date. The Company will settle conversion of the Notes through paymentconversions by paying or delivery,delivering, as the case may be ofapplicable, cash, shares of Common Stockthe Company's common stock, or a combination thereof,of cash and shares of the Company's common stock, at its election.for the Notes is equal to 188.705916.8545 shares of Common Stock per $1,000 principal amount of notes (which is equivalent tothe 2023 Notes, which represents an initial conversion price of approximately $5.30$59.33 per share, ofSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 20148. Convertible Senior Secured Notes (Continued)Common Stock). The conversion rate is subject to adjustment uponas specified in the occurrence of certain specified events but will not be adjusted for accrued and unpaid interest. In addition, upon the occurrence ofIndenture. (as, as defined in the Indenture), the Company will, in certain circumstances, increase the conversion rate by a number of additional shares for a holder that elects to convert its notes in connection with such make-whole fundamental change as described in the Indenture.Effective November 1, 2013, if, for at least 20 trading days (whether or not consecutive) during the 30 consecutive trading day period ending within five trading days prior to a conversion date, the last reported sale price of the Company's common stock exceeds the conversion price on each such trading day, the Company became required, in certain circumstances, to make an interest make-whole payment to converting holders equal to the sum of the present value of the remaining scheduled payments of interest that would have been made on the Notes to be converted had such notes remained outstanding until May 1, 2017 computed using a discount rate equal to 2%. The Company may pay an interest make-whole payment either in cash or in Common Stock, at its election. If the Company elects to pay an interest make-whole payment in Common Stock,Indenture occurs, then the stock will be valued at 95% of the simple average of the daily volume- weighted average price (VWAP) per share for the 10 trading days ending on and including the trading day immediately preceding the conversion date. Notwithstanding the foregoing, the number of shares the Company may deliver in connection with an interest make-whole payment and repayment of principal will not exceed 221.7294 shares per $1,000 principal amount of Notes, subject to adjustment. If, pursuant to its election to deliver Common Stock in connection with the payment of the interest make-whole amount, the Company would be required to deliver a number of shares of Common Stock in excess of such threshold, the Company would deliver cash in lieu of shares otherwise deliverable upon conversions in excess thereof (based on the simple average of the daily VWAP for the 10 trading days ending on and including the trading day immediately preceding the conversion date).Upon (i) the occurrence of a fundamental change (as defined in the Indenture) or (ii) if the Company calls the Notes for redemption as described below (either event, a "make-whole fundamental change") and a holder elects to convert its Notes in connection with such make-whole fundamental change, the Company will, in certain circumstances, increase the conversion rate by a number of additional shares (the "Additional Shares") as described below. The Company will notify holders within one business day after the first public announcement by it or a third party of an event or transaction that the Company reasonably determines would, if consummated, constitute a make-whole fundamental change. Upon receiving notice or otherwise becoming aware of a potential make-whole fundamental change described, the Company will use commercially reasonable efforts to announce or cause the announcement of such potential make-whole fundamental change in time to deliver such notice at least 50 scheduled trading days prior to the anticipated effective date for such transaction. The Company will notify the Trustee and holders of the effective date of any make-whole fundamental change no later than one business day after such effective date.The number of additional shares by which the Company will increase the conversion rate will be determined based on the date on which the make-whole fundamental change occurs or becomes effective (the Effective Date) and the price (the Stock Price) paid (or deemed paid) per share of the Company's Common Stock in the fundamental change. If the holders of the Company's common stockSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 20148. Convertible Senior Secured Notes (Continued)receive only cash in a make-whole fundamental change (i) the Stock Price shall be the cash amount paid per share and (ii) the Company will satisfy its conversion obligation to a holder that converts its Notes any time after such make-whole fundamental change by delivering to such holder, on the third business day immediately following the relevant conversion date, an amount of cash, for each $1,000 principal amount of Notes converted, equal to the product of (x) the conversion rate in effect on the relevant conversion date (as increased by the Additional Shares, if any) and (y) the Stock Price. Otherwise, (i) the Stock Price will equal the average of the last reported sale prices of the Company's Common Stock over the five trading day period ending on, and including, the trading day immediately preceding the Effective Date of the make-whole fundamental change and (ii) the Company will satisfy its conversion obligation to a holder that converts its Notes in connection with such make-whole fundamental change based on the conversion rate as increased by the number of Additional Shares. In connection with a make-whole fundamental change triggered by redemption of the Notes, the Effective Date of such make-whole fundamental change will be the date on which the Company delivers notice of the redemption. Notwithstanding the foregoing, in no event will the conversion rate exceed the maximum conversion rate, which is 221.7294 shares per $1,000 principal amount of Notes, which amount is inclusive of repayment of the principal of the Notes.If a fundamental change occurs at any time, holders will have the right, at their option, to require the Company to purchase for cash any or all of the Notes, or any portion of the principal amount thereof, that is equal to $1,000 or an integral multiple of $1,000 in excess thereof, on a date of the Company's choosing that is not less than 20 calendar days nor more than 35 calendar days after the date on which it delivers a fundamental change notice. The price the Company is required to pay for a Note is equal to 100% of the principal amount of such Note plus accrued and unpaid interest, if any, to, but excluding, the fundamental change purchase date. Any Notes purchased by the Company will be paid for in cash.The Company may not redeem the Notes prior to May 1, 2017. On or after May 1, 2017, the Company may redeem for cash all, but not less than all, of the Notes if the last reported sale price of the Company's Common Stock equals or exceeds 140% of the applicable conversion price, or $7.42 per share, for at least 20 trading days during the 30 consecutive trading day period ending on the trading day immediately prior to the date the Company delivers written notice of the redemption. The redemption price will be equal to 100% of the principal amount of the Notes to be redeemed, plus accrued and unpaid interest to, but excluding, the redemption date. If the Company calls the Notes for redemption, a make-whole fundamental change will be deemed to occur and the Company will in certain circumstances increase the conversion rate for holders who convert their notes in connections with such make-whole fundamental changea specified period of time. If a "fundamental change", as describeddefined in the Indenture.incurred approximately $3.5 millionmay not redeem the 2023 Notes at its option before maturity.financing costs (includingconversion, if converted in cash, holders would forgo all future interest payments, any unpaid accrued interest and the underwriters' fee) in connection withpossibility of further stock price appreciation. Upon the issuancereceipt of conversion requests, the settlement of the Notes. Approximately $0.9 million of this amount was allocated2023 Notes will be paid pursuant to additional paid-in capital and the remaining $2.6 million is recorded as a deferred cost being amortized over the termterms of the Notes. AsIndenture. In the event that all of December 31, 2016, approximately $30,000 remained unamortized.the 2023 Notes are converted, the Company would be required to repay the $402.5 million in principal value and any conversion premium in cash, shares, or any combination of cash and shares of its common stock (at the Company's option).Years ended December 31, 2016, 201520148. Convertible Senior Securedwill be equal in right of payment with the Company's future senior, unsecured indebtedness. The 2023 Notes (Continued)The table below summarizes activity relatedare senior in right of payment to the Company's future indebtedness that is expressly subordinated to the 2023 Notes. The 2023 Notes from issuance on May 3, 2013 through December 31, 2016, in thousands: $ 90,000 (9,270 ) (22,336 ) (81,463 ) 25,003 5,151 7,085
(3,962
) 764 278 $ 4,165 Duringare effectively subordinated to the year ended December 31, 2016, approximately $3.9 millionCompany's future secured indebtedness, to the extent of the value of the collateral securing that indebtedness, and will be structurally subordinated to all future indebtedness and other liabilities, including trade payables.were presentedHedge and Warrant Transactionsfor conversion. Accordingly,in an amount approximately equivalent to the value that the Company issued approximately 0.7 milliondelivers to the holders of the 2023 Notes, based on a conversion price of $59.33 per share. The total cost of the convertible note hedge transactions was $92.9 million.conversionexcess of the principal amount of converted 2023 Notes, as the Notes.case may be. The Company issued an additional 24,000 shares of common stock in settlement ofWarrant Transactions were entered into to partially offset the interest make-whole provision related to the converted Notes. As a result of the conversions, the Company incurred a loss on extinguishment of debt of approximately $0.7 million during the year ended December 31, 2016.During the year ended December 31, 2015, approximately $27.5 million of the Notes were presentedcost to the Company of the purchased Convertible Note Hedge Transactions; however, the Warrant Transactions could have a dilutive effect with respect to the Company's common stock to the extent that the market price per share of the Company's common stock, as measured under the terms of the Warrant Transactions, exceeds the strike price of the warrants.conversion. Accordingly,as derivatives. The net cost incurred in connection with the convertible note hedges and warrant transactions was recorded as a reduction to additional paid-in capital.issued approximately 5.2valued and bifurcated the conversion option associated with the 2023 Notes from the respective host debt instrument, which is referred to as debt discount. The Company initially recorded the conversion option of $76.4 million sharesin additional paid-in capital. The resulting debt discount of common stock in conversion$76.4 million on the 2023 Notes is being amortized to interest expense at an effective interest rate of 5.41% over the contractual term of the principal2023 Notes. December 31,
2019 December 31,
20182023 Notes $ 402,500 $ 402,500 Unamortized debt discount and deferred financing costs (57,330 ) (73,038 ) Total carrying value $ 345,170 $ 329,462 Years Ended December 31, 2019 2018 2017 Interest income $ 21,623 $ 13,843 $ 2,864 Interest expense (18,207 ) (13,840 ) (134 ) Interest expense on nonrecourse liability related to sale of future royalties (4,500 ) (4,271 ) (1,434 ) Changes in fair value of derivative liabilities — — 76 Loss on extinguishment of debt — — (295 ) Total $ (1,084 ) $ (4,268 ) $ 1,077 Notes. The Company issued an additional 0.5 million shares of common stock in settlement of the interest make-whole provision related to the converted Notes. As a result of the conversions, the Company incurred a loss on extinguishment of debt of approximately $2.3 million during the yearyears ended December 31, 2015.9.our Common Stockthe Company's common stock are entitled to one1 vote for each share of Common Stock held. On May 1, 2012, the Company completed its IPO, in which 10 million shares of the Company's Common Stock were sold at a price of $5 per share. Additionally, the underwriters of the Company's IPO exercised the full amount of their over-allotment option resulting in the sale of an additional 449,250 shares of the Company's Common Stock at a price of $5 per share, resulting in cash proceeds to the Company of $52.3 million. The Company realized net proceeds of $47.6 million from the IPO, after issuance costs of approximately $4.7 million.On December 5, 2012, the Company completed a follow-on offering, in which 6 million shares of the Company's Common Stock were sold at a price of $8 per share. Additionally, the underwriters of theSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 20149. Stockholders' Equity (Continued)Company's follow-on offering exercised their over-allotment options in January 2013 resulting in the sale of an additional 239,432 shares of the Company's Common Stock at a price of $8 per share, resulting in total cash proceeds to the Company of $49.9 million. The Company realized net proceeds of $46.6 million from the follow-on offering, after issuance costs of approximately $3.3 million.During the period from November 1, 2013 through December 31, 2016, the Company issued 16,120,128 shares of common stock as a result of the conversion of approximately $85.4 million of Convertible Notes and approximately 2,219,908 shares of common stock in settlement of the interest-make whole provision associated with those conversions.10. Share-Based PaymentsPlansapproved, andapproved. This plan provides for the grant of stock options and certain other equity awards, includingincluding: stock appreciation rights (SAR),(SARs); restricted and unrestricted stock; stock stock units,units; performance awards,awards; cash awardsawards; and other awards that are convertible into or otherwise based on the Company's common stock, to the Company's key employees, directors, and consultants and advisors. The 2012 Plan is administered by the Company's Board of Directors and the Company's Compensation Committee of the Board, and provides for the issuance of up to 8,000,0008 million shares of the Company's Common Stock.common stock. Option awards are granted with an exercise price equal to the estimated fair valueclosing price of the Company's Common Stock atcommon stock as of the grant date; those optiondate. Option awards granted to employees, consultants and advisors generally vest in four4 equivalent annual installments, starting on the first anniversary of the date of grant andthe grant. Awards have ten-yearten year contractual terms. Option awards granted to the directors generally vest over a one year term, and have a ten year contractual term. Share-based compensation recognized relatedgrant of employee and non-employee stock options, SAR,Supernus Pharmaceuticals, Inc. 2012 Employee Stock Purchase Plan, (ESPP) awardsas amended (the ESPP). The ESPP allows eligible employees the opportunity to acquire shares of the Company's common stock at periodic intervals through accumulated payroll deductions. These deductions are applied at the semi-annual purchase dates of June 30 and non-vestedDecember 31, to purchase shares of common stock wasat a discount. Eligible employees may purchase shares at the lower of 85% of the fair market value at either the first day of the purchase period or the fair market value at the end of the purchase period. The ESPP provides for issuance of up to 700,000 shares of the Company's common stock. The Company records compensation expense related to its ESPP.thousands: Years Ended December 31, 2019 2018 2017 Research and development $ 2,599 $ 1,943 $ 1,387 Selling, general and administrative 12,247 9,348 7,046 Total $ 14,846 $ 11,291 $ 8,433 Year Ended December 31, 2016 2015 2014 $ 1,107 $ 874 $ 728 4,819 3,216 2,129 $ 5,926 $ 4,090 $ 2,857 Years Ended December 31, 2019 2018 2017 Fair value of common stock $22.99 - $37.78 $37.20 - $58.15 $25.30 - $41.00 Expected volatility 61.36% - 63.28% 57.95% - 60.56% 53.61% - 60.60% Dividend yield 0% 0% 0% Expected term 5.53 years - 6.18 years 6.25 years 6.25 years Risk-free interest rate 1.69% - 2.55% 2.69% - 2.85% 1.90% - 2.18% Expected forfeiture rate 0% 0% 0% Year Ended December 31, 2016 2015 2014 $12.98 - $22.80 $9.13 - $21.21 $7.63 - $10.02 60.9% - 64.5% 60.9% - 64.6% 64.5% - 68.3% 0% 0% 0% 6.25 years 6.25 years 6.25 years 1.14% - 2.15% 1.54% - 1.74% 1.67% - 1.97% 5% 5% 5% Years ended December 31, 2016, 2015 and 201410. Share-Based PaymentsFair Value of Common Stock—For option grants that occurred after the Company's IPO on May 1, 2012, the fair value of the Common Stock underlying the option grants was determined based on observable market prices of the Company's Common Stock.Expected Volatility—Volatility is a measure of the amount by which a financial variable such as a share price has fluctuated (historical volatility) or is expected to fluctuate (expected volatility) during a period. The Company has identified several public entities of similar size, complexity, and stage of development. Accordingly, historical volatility has been calculated using the volatility of these companies, as well as taking into consideration the Company's actual volatility since our IPO. As our historical experience is not sufficient to calculate volatility for our option grants, the Company will continue to use guideline peer group volatility information until the historical volatility of its own Common Stock is sufficient on its own to measure expected volatility for future option grants.Dividend Yield—The Company has never declared or paid dividends and has no plans to do so in the foreseeable future.Expected Term—This is the period of time that the options granted are expected to remain unexercised. Options granted have a maximum term of ten years. The Company determines the average expected life of stock options according to the "simplified method" as described in Staff Accounting Bulletin 110, which is the mid-point between the vesting date and the end of the contractual term. Over time, management will track estimates of the expected life of the option term so that estimates will approximate actual behavior for similar options.Risk-Free Interest Rate—This is the U.S. Treasury note rate for the week of each option grant during the year, having a term that most closely resembles the expected term of the option.Expected Forfeiture Rate—The forfeiture rate is the estimated percentage of options granted that are expected to be forfeited or canceled on an annual basis before becoming fully vested.Supernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 201410. Share-Based Payments (Continued) Outstanding , December 31, 2017 4,280,670 $ 14.50 7.37 $ 108,520 Granted 762,915 $ 39.91 Exercised (930,483 ) $ 10.07 $ 36,317 Forfeited (196,139 ) $ 25.01 Outstanding, December 31, 2018 3,916,963 $ 19.98 7.10 $ 57,220 Granted 880,235 $ 36.43 Exercised (114,753 ) $ 12.90 $ 2,423 Forfeited (75,886 ) $ 34.80 Outstanding, December 31, 2019 4,606,559 $ 23.05 6.66 $ 27,716 As of December 31, 2019 Vested and expected to vest 4,606,559 $ 23.05 6.66 $ 27,716 Exercisable 2,598,112 $ 15.68 5.48 $ 25,594 Number of
Options Weighted-Average
Exercise Price Weighted-Average
Remaining
Contractual
Term (in years) 2,080,749 $ 7.93 8.04 971,500 $ 10.12 (205,640 ) $ 4.56 (147,602 ) $ 8.60 2,699,007 $ 8.94 7.92 1,058,850 $ 13.32 (85,694 ) $ 6.51 (28,075 ) $ 12.23 3,644,088 $ 10.25 7.59 3,591,528 $ 10.22 7.57 1,503,004 $ 8.62 6.49 The aggregate intrinsic value of options outstanding, vested and expected to vest, and exercisable as of December 31, 2016 is approximately $54.7 million, $54.0 million and $25.0 million, respectively. The aggregate intrinsic value of options outstanding, vested and expected to vest, and exercisable as of December 31, 2015 is approximately $12.6 million, $12.4 million and $5.0 million, respectively. The aggregate intrinsic value of options outstanding, vested and expected to vest, and exercisable as of December 31, 2014 is approximately $2.0 million, $2.0 million and $1.5 million, respectively.grant-dategrant date fair value of options granted for the years ended December 31, 2016, 20152019, 2018 and 20142017 was $7.66, $6.05$21.50, $23.43 and $5.79$14.35 per share, respectively.Common Stockcommon stock related to shares that vested during the years ended December 31, 2016, 20152019, 2018, and 20142017 was approximately $3.9$10.8 million, $2.6$8.3 million and $1.9$5.4 million, respectively.The total intrinsic value of options exercised amounted to approximately $1.1 million, $1.6 million and $0.1 million, respectively, during the years ended December 31, 2016, 2015 and 2014.As of December 31, 2016 and 2015, the total unrecognized compensation expense, net of estimated forfeitures, was approximately $9.8 million and $7.2 million, respectively, which the Company expects to recognize over a weighted-average period of 2.7 and 2.5 years, respectively.11.income (loss)earnings per share (EPS) is calculated using the weighted-average number of common shareshares outstanding. Diluted EPS is determined by dividing income (loss) attributable to common stockholders bycalculated using the weighted-average number of common shares outstanding, during the period, without consideration of common stock equivalents. Diluted income (loss) per share is computed by dividing the income (loss) attributable to common stockholders by the weighted-average number of commonSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 201411. Earnings per Share (Continued)share equivalents outstanding for the period. The treasury stock method is used to determineincluding the dilutive effect of the Company'sCompany’s: stock option grants, SAR, and potentialgrants; SARs; warrants; ESPP awards,awards; and the if-converted method is used to determine2023 Notes, as determined per the dilutive effecttreasury stock method.Company's Notes.following common stock equivalents wereexpected collective impact of the Convertible Note Hedge and Warrant Transactions is to reduce the potential dilution that may occur between the conversion price of $59.33 per share and the strike price of the warrants of $80.9063 per share.lossEPS because their inclusion would be anti-dilutive. Specifically, the denominator of the diluted EPS calculation excludes the additional shares related to the 2023 Notes and warrants, because the average price of the Company's common stock was less than the conversion price of the 2023 Notes of $59.33 per share and the strike price of the warrants of $80.9063 per share. Prior to actual conversion, the Convertible Note Hedge Transactions are not considered in calculating diluted earnings per share, as their impact would be anti-dilutive.effectinclusion would be anti-dilutive as applied to the loss from continuing operations applicable to common stockholders for the years ended December 31, 2016, 2015 and 2014: Years Ended December 31, 2019 2018 2017 Stock options 1,145,446 199,982 40,009 Year Ended December 31, 2016 2015 2014 — — 7,995,340 — 20,957 20,499 — — 306,776 incomeearnings per share for the years ended December 31, 2016, 20152019, 2018 and 2014,2017: Years Ended December 31, 2019 2018 2017 Numerator, dollars in thousands: Net earnings $ 113,056 $ 110,993 $ 57,284 Adjustments: Interest expense on Convertible Senior Secured Notes due 2019 — — 134 Changes in fair value of derivative liabilities — — (76 ) Loss on extinguishment of debt — — 295 Loss on extinguishment of outstanding debt, as if converted — — (321 ) Total adjustments — — 32 Net earnings used for calculation of diluted EPS $ 113,056 $ 110,993 $ 57,316 Denominator: Weighted average shares outstanding, basic 52,412,181 51,989,824 50,756,603 Effect of dilutive securities: Shares underlying Convertible Senior Secured Notes due 2019 — — 285,257 Shares issuable to settle interest make-whole derivatives — — 7,012 Stock options and SAR 1,404,573 2,109,048 2,252,278 Total dilutive potential common shares 1,404,573 2,109,048 2,544,547 Weighted average shares outstanding, diluted 53,816,754 54,098,872 53,301,150 Earnings per share, basic $ 2.16 $ 2.13 1.13 Earnings per share, diluted $ 2.10 $ 2.05 1.08 thousands, except share and per share amounts: Years Ended December 31, 2019 2018 2017 Current Federal $ 29,333 $ 26,772 $ 18,288 State 10,930 5,621 3,822 Deferred Federal (4,551 ) (2,450 ) 21,493 State (1,281 ) (760 ) (269 ) Total income tax expense $ 34,431 $ 29,183 $ 43,334 Year ended December 31, 2016 2015 2014 $ 91,221 $ 13,944 $ (10,925 ) 543 1,229 — (448 ) (589 ) — 671 2,338 — (1,182 ) (2,494 ) — (416 ) 484 — $ 90,805 $ 14,428 $ (10,925 ) 49,472,434 47,485,258 42,260,896 — 1,222,363 2,459,009 71,537 804,507 — 942,649 411,606 — 2,236,549 3,675,122 — 51,708,983 51,160,380 42,260,896 $ 1.84 $ 0.29 $ (0.26 ) $ 1.76 $ 0.28 $ (0.26 ) Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 201412.The components of the income tax (benefit)/ expense for the years ended December 31, 2016, 2015 and 2014 were as follow, in thousands: Year Ended December 31, 2016 2015 2014 $ 544 $ 624 $ 630 78 42 — (39,898 ) — — (1,576 ) — — $ (40,852 ) $ 666 $ 630 the expected income tax (benefit)/ expense computed usingat the U.S. Federalfederal statutory income tax rate to annual income tax expense at the Company's effective income tax rate is as follows (dollars in thousands:thousands): Years Ended December 31, 2019 2018 2017 Income tax expense computed at U.S. federal statutory income tax rate $ 30,972 $ 29,437 $ 35,217 State income taxes 7,543 3,674 2,714 Permanent items 1,332 (2,196 ) (2,311 ) Research and development credits (2,071 ) (3,199 ) (2,196 ) Uncertain income tax position (2,992 ) 716 (1,137 ) — — 9,694 Other (353 ) 751 1,353 Income tax expense $ 34,431 $ 29,183 $ 43,334 Year Ended December 31, 2016 2015 2014 $ 17,629 $ 5,114 $ (3,603 ) 715 601 610 (1,523 ) 42 (245 ) (56,019 ) (4,705 ) 4,413 143 533 (329 ) (1,902 ) (979 ) (535 ) 105 60 (125 ) — — 444 $ (40,852 ) $ 666 $ 630
(1) Due to the 2017 Tax Cuts and Job Act, which lowered the U.S. Corporate income tax rate from 35% to 21% effective January 1, 2018. As a result, existing deferred taxes were remeasured. Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 201412.wereare as follow (dollars in thousands: As of December 31, 2019 2018 Deferred tax assets: Convertible bond hedge $ 17,197 $ 21,412 Accrued product returns and rebates 15,123 13,205 Accrued compensation and stock based compensation 10,349 8,218 Non-recourse liability related to sale of future royalties 5,320 5,571 Research and development credit carryforwards 3,817 3,817 Amortization of intangibles 4,617 3,289 Net operating loss carryforwards 2,245 2,900 Operating lease liability 8,187 — Inventory 1,385 499 Alternative Minimum Tax (AMT) credit 926 978 Other 199 1,268 Total deferred tax assets 69,365 61,157 Less: valuation allowance (11 ) (9 ) Total deferred tax asset, net of valuation allowance 69,354 61,148 Deferred tax liability: Debt discount on 2023 Notes (14,109 ) (17,568 ) Patent infringement legal costs (10,613 ) (10,697 ) Operating lease assets (5,237 ) — Depreciation (2,778 ) (236 ) IRC Section 481(a) liability (2,126 ) (2,964 ) Unrealized gain on marketable securities (2,428 ) — Total deferred tax liabilities (37,291 ) (31,465 ) Net deferred tax assets $ 32,063 $ 29,683 As of December 31, 2016 2015 $ 24,926 $ 34,610 417 532 8,128 5,886 128 187 706 290 7,119 5,529 1,086 543 11,223 11,526 878 498 1,581 1,108 — (60,090 ) 56,192 619
(141 ) (509 ) (13,899 ) — (423 ) (110 ) $ 41,729 $ — more likely than notmore-likely-than-not that some or all of the deferred income tax assets will not be realized. The ultimate realization of the deferred income tax assets is dependent upon the generation of future taxable income during the periods in which the net operating loss (NOL) and tax credit carryforwards are available. Management considers projected future taxable income, the scheduled reversal of deferred income tax liabilities, and available tax planning strategies that can be implemented by the Company in making this assessment. Based upon the level of historical taxable income and projections for future taxable income over the periods in which the NOL and credit carryforwards are available to reduce income taxes payable, management had established in 2015 a full valuation allowance as the Company was not more likely than notdetermined it is more-likely-than-not to realize such net deferred tax assets.Duringthird quarterCompany's ownership, the utilization of 2016,net operating loss carryforwards and research and development credit carryforwards, that can be used to offset future taxable income, are subject to annual limits in accordance with Internal Revenue Code (IRC) provisions, as well as similar state provisions. In addition, states may also impose other future limitations through state legislation or similar measures. Despite the NOL carryforwards, the Company determined the positive evidence regarding the valuation of the deferred tax assets outweighed the negative. Accordingly, the Company eliminated the valuation allowance of $60.1 million and recorded the assessment to the deferredmay incur higher state income tax expense.2016,2019, the U.S. Federalfederal and state NOL carryforwards amounted to approximately $87.3$10.8 million ($30.5and $9.9 million, tax effected) and $28.2 million ($1.6 million tax effected), respectively, and will expire in various years beginning in 2030. 2033. For the year ended December 31, 2019, the Company utilized NOLs of $10.2 million and expects the remaining federal and state NOL carryforwards to become available in the future years.2016,2019, the Company has available research and development credit carryforwards of approximately $7.1$4.2 million, which expire, if unused,Supernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 201412. Income Taxes (Continued)starting in 2026. The use of the Company's U.S. Federal and state NOL carryforwards and research and development credits are restricted in annual use due to changes in the Company's ownership. For the year ended December 31, 2016, the Company utilized NOL's of approximately $48.1 million and expects the remaining $87.3 million of Federal NOL carryforwards to become available over the years from 2017 to 2020, in amounts ranging from $7.1 million to $20.3 million per year. In addition, the Company has available research and development credits of approximately $7.1 million, expected towill become available in 2020 and will expire, if unused, starting in 2026.2021. The Company's state NOL's will have a similar limitation to the amount noted for US Federal. Additionally, despite the NOL carryforwards, the Company may have a future tax liability due to state and localU.S. Federal income tax requirements. The Company paid no Federal income taxes inexaminations for years prior to 2016, with the years ended December 31,exception that operating loss or tax credit carryforwards generated prior to 2016 2015 or 2014.FASB ASC Topic 740,Income Taxes. The Company recognizes interest and penalties related to uncertain tax positions, if any, in income tax expense. As of December 31, 2016, the Company accrued interest of a nominal amount and penalties of $0.1 million related to uncertain tax positions. The Company's income taxes have not been subject to examination by any tax jurisdictions since its inception in 2005. Due to NOL and research and development credit carryforwards, all U.S. Federal and state income tax returns filed by the Company are subject to examination by the taxing jurisdictions. Some uncertain income tax position liabilities have been recorded toagainst the Company's deferred income tax assets to offset such tax attribute carryforwards and other positions that can'tcannot be offset by tax attributes until a liability has been booked.thousands: Years Ended December 31, 2019 2018 2017 Balance as of January 1 $ 8,848 $ 8,859 $ 9,299 Gross increases related to current year tax positions 208 1,108 1,178 Gross increases related to prior year tax positions — — 947 Gross decreases related to prior year tax positions (49 ) (484 ) — Lapse of statute of limitations (3,029 ) (635 ) — Change in tax rates — — (2,565 ) Balance as of December 31 $ 5,978 $ 8,848 $ 8,859 Year Ended December 31, 2016 2015 2014 $ 9,341 $ 8,964 $ 9,828 — (5 ) 18 662 646 710 (375 ) — — (169 ) (243 ) (1,057 ) (160 ) (21 ) (535 ) $ 9,299 $ 9,341 $ 8,964 As of December 31, 2016 and 2015, theThe Company recorded $0.5$3.0 million and $0.6 million of current tax expense on setting up anbenefit in 2019 and 2018, respectively, as a result of the expiration of statutes of limitation. The Company also recorded $0.2 million and $0.3 million for uncertain tax positionpositions related to the Alternative Minimum Tax.research and development tax credits in 2019 and 2018, respectively. The Company does not anticipate a significant increase or decreasematerial impact to the financial statements in the uncertain income tax benefits within the next 12 months.13. Commitmentsmonths as a result of uncertain tax positions and Contingenciesconcurrentoperating leases for its former headquarters office and lab space that extendat 1550 East Gude Drive in Rockville, MD and for its fleet vehicles. The Company’s existing leases for its former headquarters office and lab space run through April 2020. With respect to the fleet vehicle leases, given the volume of individual leases involved in the overall arrangement, the Company applies a portfolio approach to effectively account for the operating lease assets and liabilities.may electentered into a new lease agreement, effective January 31, 2019, with Advent Key West, LLC (Landlord), for its new headquarters in Rockville, MD (Premises). The term of the new headquarters lease commenced on February 1, 2019 (the Commencement Date) and will continue until April 30, 2034, unless earlier terminated in accordance with the terms of the lease. The lease includes options to extend the termlease for up to 10 years. Fixed rent with respect to the Premises began on the Commencement Date; however, the Landlord agreed to a rent abatement from the Commencement Date through April 30, 2020.leasesCommencement Date. Under the terms of the Lease, the Company provided a security deposit of approximately $195,000, and will be required to pay all utility charges for an additional five-year term.the Premises in addition to its pro rata share of any operating expenses and real estate taxes. The leasesTableCompany will occupy the Premises upon completion of ContentsSupernus Pharmaceuticals, Inc.Notesthe build-out of the Premises, which is anticipated to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 201413. Commitments and Contingencies (Continued)provideoccur in the first half of 2020.$2.1$10.2 million, in aggregate. During the year ended December 31, 2016, none of theAll tenant improvement allowance was utilized. During the year ended December 31, 2015, approximately $0.2 million of the allowance washave been utilized and is included in fixed assets and deferred rent. Asas of December 31, 2016, $0.5 million2019 (see Note 5). The full amount of the tenant improvement allowance was initially recorded in Prepaid expenses and other current assets on the consolidated balance sheets. December 31,
2019 Assets Lease assets $ 21,279 Liabilities Accrued expenses and other current liabilities Lease liabilities, current $ 2,825 Noncurrent Lease liabilities, long term 30,440 Total lease liabilities $ 33,265 December 31, 2019 Fixed lease cost $ 4,990 Variable lease cost 1,887 Total operating leases cost $ 6,877 availableas follows (dollars in thousands): December 31, 2019 2018 Cash paid for operating leases $ 5,337 $ 5,196 Lease assets and tenant receivables obtained for new operating leases 35,594 — tenant improvements. operating leases as of December 31, 2019, are as follows:Weighted-average remaining lease term (years) 12.48 Weighted-average discount rate 4.39 % Year ending December 31: 2020 $ 4,212 2021 4,130 2022 3,597 2023 2,537 Thereafter 29,372 Total future minimum lease payments $ 43,848 (10,583 ) Present value of lease liabilities $ 33,265 December 31, 2016, 20152018 and 20142017 was approximately $3.6 million and $2.7 million, $2.6 million and $2.3 million, respectively.non-cancelablenoncancelable operating leases as of December 31, 2016 are2018 were as follows (dollars in thousands:Year ending December 31: 2019 $ 3,400 2020 2,287 Thereafter 1,840 Total $ 7,527 1,321 1,314 1,341 454 $ 4,430 Years Ended December 31, 2019 2018 2017 Net product sales Trokendi XR $ 295,214 $ 315,295 $ 226,518 Oxtellar XR 88,186 84,576 67,579 Total net product sales 383,400 399,871 294,097 Royalty revenues 9,355 8,276 6,367 Licensing revenues — 750 1,774 Total revenues $ 392,755 $ 408,897 $ 302,238 Company'sCompany’s neurology and psychiatry portfolio. Under these license agreements, with Afecta Pharmaceuticals, Inc. (Afecta), the Company has an exclusive optionmay be required to evaluate Afecta's CNS pipeline and to obtain exclusive worldwide rights to selected product candidates, including an exclusive license to SPN-810. Thepay certain amounts upon the achievement of defined milestones. If these products are ultimately commercialized, the Company does not owe any future milestone payments for SPN-810. The Company is also obligated to pay royalties to Afecta based on worldwidethird parties, as percentage of net product sales, infor each respective product under a license agreement.low-single digits.Thethird quarter of 2014, the Company has also entered intoreceived a $30.0 million payment pursuant to a Royalty Interest Acquisition Agreement related to the purchase and saleby HC Royalty of certain of the Company’s rights under the Company’s agreement with Rune HealthCare Limited (Rune), whereUnited Therapeutics related to the commercialization of Orenitram (treprostinil) Extended-Release Tablets. Full ownership of the royalty rights will revert to the Company obtainedif and when a certain cumulative payment threshold is reached, per the exclusive worldwide rightsterms of the agreement (see Note 2, Note 10 and Note 16).a product concept from Rune. There are no future milestone payments due to Rune under this agreement. If the Company receives approval to market and sell any products based on the Rune product concept for SPN-809, the Company is obligated to pay royalties to Rune based on net sales worldwide in the low single digits.14.Consolidated Financial Statements (Continued)Employees are 100% vested in their contributions to the 401(k) Plan. approximated $1.6were approximately $2.3 million, $1.4$2.1 million, and $1.1$1.8 million for the years ended December 31, 2016, 20152019, 2018 and 2014,2017, respectively.Supernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 201415. Collaboration AgreementsRoyalty RevenueIn the third quarter of 2014, the Company received a $30.0 million payment pursuant to a Royalty Interest Acquisition Agreement related to the purchase by Healthcare Royalty Partners III, L.P. (HC Royalty), of certain of the Company's rights under the agreement with United Therapeutics Corporation related to the commercialization of Orenitram (treprostinil) Extended-Release Tablets. We will retain full ownership of the royalty rights if and when a certain threshold is reached per the terms of the Agreement. We have recorded a non-recourse liability related to this transaction and have begun to amortize this amount to recognize non-cash royalty revenue as royalties are received by HC Royalty from United Therapeutics. We also recognized non-cash interest expense related to this liability that accrues at an effective interest rate, which is determined based on projections of HC Royalty's rate of return. We recognized royalty revenue of $4.7 million and $3.0 million for the years ended December 31, 2016 and 2015, respectively. We recognized non-cash interest expense of $4.5 million and $3.5 million for the years ended December 31, 2016 and 2015, respectively.The Company has a license agreement with United Therapeutics Corporation to use one of its proprietary technologies for an oral formulation of Remodulin for the treatment of pulmonary arterial hypertension and potentially for additional indications. The revenue generated in the year ended December 31, 2014 was $2.0 million for a milestone payment.16.2016years 2019 and 20152018 are presented in the following table (dollars in thousands,thousands), except per share data, unaudited: 2019 Revenues $ 85,474 $ 104,695 $ 102,140 100,446 Total costs and expenses 60,046 62,097 62,411 59,630 Operating earnings 25,428 42,598 39,729 40,816 Net earnings 18,340 32,727 28,860 33,129 Earnings per share, basic 0.35 0.62 0.55 0.63 Earnings per share, diluted 0.34 0.61 0.54 0.62 2018 Revenues $ 90,429 $ 99,538 $ 102,996 $ 115,934 Total costs and expenses 59,035 63,818 65,521 76,079 Operating earnings 31,394 35,720 37,475 39,855 Net earnings 26,352 30,737 28,011 25,893 Earnings per share, basic 0.51 0.59 0.54 0.50 Earnings per share, diluted 0.49 0.57 0.52 0.48 1st Quarter 2nd Quarter 3rd Quarter 4th Quarter $ 44,194 $ 51,626 $ 56,810 $ 62,374 37,757 39,981 36,971 46,078 6,437 11,645 19,839 16,296 4,825 10,251 61,826 14,320 0.10 0.21 1.25 0.29 0.08 0.18 1.18 0.26
$ 28,738 $ 35,678 $ 39,362 $ 43,687 24,704 31,834 34,277 35,806 4,034 3,844 5,085 7,881 738 2,437 3,916 6,853 0.02 0.05 0.08 0.14 0.02 0.04 0.08 0.14 17. Subsequent EventsSubsequent to December 31, 2016, holders of the Notes converted approximately $1.0 million of the Notes. We issued a total of approximately 0.2 million shares of common stock in conversion of theSupernus Pharmaceuticals, Inc.Notes to Consolidated Financial Statements (Continued)Years ended December 31, 2016, 2015 and 201417. Subsequent Events (Continued)principal amount of the Notes and accrued interest thereon resulting in a remaining outstanding balance of $3.6 million.During the first quarter of 2017, the Company entered into settlement and license agreements with Zydus Pharmaceutical (USA), Inc. and Cadila Healthcare Limited (collectively, "Zydus") and with Actavis Laboratories, FL, Inc. et al. (collectively, "Actavis," now a subsidiary of Teva Pharmaceuticals Industries, Ltd.) to settle ongoing patent litigation regarding Zydus' and Actavis' respective ANDA filings seeking approval to market a generic version of the Company's Trokendi XR (extended-release topiramate) capsules. These agreements prohibit Zydus and Actavis from selling a generic version of Trokendi XR before January 1, 2023 except under certain circumstances.2016,2019, the end of the period covered by this report. Based on that evaluation, under the supervision and with the participation of our management, including our CEO and CFO, we concluded that our disclosure controls and procedures were not effective as of December 31, 2016 at the reasonable assurance level because of the material weaknesses in our internal control over financial reporting described below.Notwithstanding the identified material weaknesses, management has concluded that the consolidated financial statements included in this Annual Report on Form 10-K fairly present in all material respects our financial condition, results of operations and cash flows at and for the periods presented in accordance with U.S. GAAP.2016 was the first year that the Company was subject to compliance and testing procedures under Section 404(b) of the Sarbanes-Oxley Act relating to internal controls over financial reporting.itstheir inherent limitations, internal control over financial reporting may not prevent or detect all misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.A material weakness is defined as a deficiency, or combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of annual or interim consolidated financial statements will not be prevented or detected on a timely basis. A deficiency exists when the design or operation of a control does not allow management or employees, in the normal course of performing their assigned functions, to prevent or detect misstatements on a timely basis.20162019 based on criteria related to internal control over financial reporting described inInternal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO 2013 Framework). Based on management's assessment using these criteria, our management concluded that as of December 31, 2016, ourthe Company's internal control over financial reporting was not effective due to the following control deficiencies.The Company did not have adequately trained resources with assigned responsibility and accountability over the design and operation of internal controls. Specifically, Company personnel did not have a sufficient understanding of the COSO 2013 Framework and its application to internal controls over financial reporting, and their responsibilities for effective internal control. Also, the Company did not have an effective risk assessment process that assessed necessary changes in financial reporting and internal controls impacted by changes in information technology systems.As a consequence, the Company did not have effective control activities over the following.•The Company did not have effective operation of controls over the completeness and accuracy of key assumptions and data analyzed by a third party consultant and ultimately used by management to determine the returns portion of accrued sales deductions.•The Company did not have effective general information technology controls ("GITCs") over the Microsoft Dynamics AX information technology system and the employee expense reimbursement system. Specifically, the Company did not have IT user access controls designed to restrict privileges to IT applications and the AX database commensurate with their assigned authorities and responsibilities. Furthermore, the Company did not have adequate program change controls over the AX IT applications and the AX database, designed to actively monitor program changes so as to ensure that changes were appropriate and that any deficiencies were investigated and remediated. As a result, process-level automated and manual controls related to these IT systems were also ineffective. These IT systems affect all financial reporting processes.The control deficiencies described above resulted in no misstatements in our consolidated financial statements as of and for the fiscal year ended December 31, 2016. However, these control deficiencies create a reasonable possibility that a material misstatement to our consolidated financial statements will not be prevented or detected on a timely basis. We concluded that the deficiencies represent material weaknesses in our internal control over financial reporting and our internal control over financial reporting was not effective as of December 31, 2016.The2019.LLP has expressed an adverse report onalso audited the effectiveness of ourCompany's internal control over financial reporting as of December 31, 2016.2019. Their responsibility is to evaluate whether internal controls over financial reporting was designed and operating effectively. Their report is included herein.Tableon the effectiveness of ContentsManagement's Remediation PlanThe Company will execute the following steps in 2017 to remediate the aforementioned material weaknesses in itsCompany's internal control over financial reporting:•The Company is actively looking to recruit personnel that have requisite experience working with the implementationreporting as of financial accounting and internal controls policies and procedures.•The Company will sponsor ongoing training related to the COSO 2013 Framework best practices for personnel that are accountable for internal control over financial reporting.•The Company has taken certain actions and plans to take further action to strengthen our control procedures surrounding GITCs, IT user access review and program change controls including the logging of changes to the IT applications and the database.While the audit committee of our board of directors and senior management are closely monitoring this remediation, until the remediation efforts discussedDecember 31, 2019 included in this section, including any additional remediation efforts that our senior management identifies as necessary, are complete, tested and determined effective, we will not be able to conclude that the material weaknesses have been remediated. In addition, we may need to incur incremental costs associated with this remediation, primarily due to the hiring and training of finance and accounting personnel, and the implementation of improved training procedures.2016. Other than the material weaknesses identified and assessed during the quarter described above under "Management Report on Internal Control over Financial Reporting," there2019. There were no changes in our internal control over financial reporting identified in connection with the evaluation required by paragraph (d) of Exchange Act Rule 13a-15 that occurred during the quarter ended December 31, 2016,2019, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.We identified material weaknesses as of December 31, 2015 in internal control over financial reporting. Management's review revealed that our risk assessment process and our review controls over the accounting for significant, complex, and unusual accounting transactions were deficient, in that these controls were not designed to ensure that sufficient technical accounting expertise was applied to assess and document the appropriate accounting over such transactions. The presence of these control deficiencies created a reasonable possibility that a material misstatement to the consolidated financial statements would not be prevented or detected on a timely basis. Therefore, we concluded that the deficiencies represented a material weakness in the Company's internal control over financial reporting and that our internal control over financial reporting was not effective as of December 31, 2015. We believe the processes and control activities specific to determining and documenting the appropriate accounting for significant, complex and unusual accounting transactions were remediated as of December 31, 2016. During 2016 and particularly in the fourth quarter of 2016, we took action to strengthen our internal control procedures regarding the review of the accounting for significant, complex, and unusual transactions. Specifically, during 2016 we engaged third party accounting service providers with appropriate and relevant subject matter expertise to supplement our existing resources related to several accounting matters. This engagement included, among other actions, thorough considerations of potential alternative accounting treatment regarding significant, complex, and unusual transactions. We will continue this practice on a going forward basis.20172020 Annual Meeting to be filed with the Securities and Exchange Commission not later than 120 days after December 31, 2016.2019.20172020 Annual Meeting to be filed with the Securities and Exchange Commission not later than 120 days after December 31, 2016.2019.fromto our definitive proxy statement for our 20172020 Annual Meeting to be filed with the Securities and Exchange Commission not later than 120 days after December 31, 2016.2016: Number of securities to
be issued upon exercise
of outstanding options,
warrants and rights(1) Weighted-average exercise
price of outstanding options,
warrants and rights(1) Number of securities
remaining available for future
issuance under equity
compensation plans
(excluding securities
reflected in the first
column(2)) 3,644,088 $ 10.25 4,387,491 — — — 3,644,088 $ 10.25 4,387,491 (1)The securities that may be issued are shares of the Company's Common Stock, issuable upon conversion of outstanding stock options.(2)The securities that remain available for future issuance are issuable pursuant to the 2012 Equity Incentive Plan.Plan category Equity compensation plans approved by security holders 4,606,559 $ 23.05 1,972,307 Equity compensation plans not approved by security holders — — — Total 4,606,559 $ 23.05 1,972,307 The securities that may be issued are shares of the Company's Common Stock, issuable upon conversion of outstanding stock options. The securities that remain available for future issuance are issuable pursuant to the 2012 Equity Incentive Plan. 20172020 Annual Meeting to be filed with the Securities and Exchange Commission not later than 120 days after December 31, 2016.2019.20172020 Annual Meeting to be filed with the Securities and Exchange Commission not later than 120 days after December 31, 2016.2019.(a)(1) Index to consolidated Financial Statements(a)(1) Index to consolidated Financial Statements Consolidatedconsolidated Financial Statements are filed as part of this Annual Report on Form 10-K. See Part II, Item 8, "Financial8—Financial Statement and Supplementary Data."(a)(2) Financial Statement Schedules(a)(2) Financial Statement Schedules 20162019 and 20152018 have been omitted since they are either not required, not applicable, or the information is otherwise included in the consolidated financial statements or the notes to consolidated financial statements.(a)(3) Exhibits(a)(3) Exhibits Not applicable.Pursuant to the requirements of Securities 13 or 15(d) of the Securities and Exchange Act of 1934, as amended, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized. SUPERNUS PHARMACEUTICALS, INC.Description2.1 By:/s/ JACK A. KHATTAR Name:Jack A. Khattar3.1 Title:President and Chief Executive OfficerDate: March 15, 2017Pursuant to the requirements of the Securities Act of 1934, as amended, this report has been signed by the following persons on behalf of the registrant and in the capacities and the dates indicated below:SignatureTitleDate/s/ JACK A. KHATTARPresident and Chief Executive Officer and Director (Principal Executive Officer)March 15, 2017/s/ GREGORY S. PATRICKVice President and Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer)March 15, 2017/s/ CHARLES W. NEWHALL, III.Director and Chairman of the BoardMarch 15, 2017/s/ GEORGES GEMAYELDirectorMarch 15, 2017/s/ FREDERICK M. HUDSONDirectorMarch 15, 2017/s/ WILLIAM A. NUERGEDirectorMarch 15, 2017/s/ JOHN M. SIEBERT, PH.D.DirectorMarch 15, 2017ExhibitNumberDescription3.1* 3.2 3.2* 4.1 4.1* 4.2 4.2* 4.3*Form of 7.50% Convertible Senior Secured Note due 2019trustee (incorporated by reference to Exhibit 4.2 to the Form 8-K filed on May 9, 2013,March 20, 2018, File No. 001-35518). 4.3 4.4* Security and Pledge Agreement dated as 10.1 4.5*First Supplemental Indenture dated as of October 24, 2013 by and between the Company and U.S. Bank National Association as Trustee and Collateral Agent (incorporated by reference to Exhibit 4.1 to the Form 8-K filed on October 24, 2013, File No. 001-35518).10.1*+ 10.2 10.2*+ 10.3 10.3*+ 10.4 10.4*+ 10.5 10.5* 10.6 10.6* Description 10.8 * 10.9 *Investor Rights Agreement, dated as of December 22, 2005, by and among the Registrant and the holders of shares of Series A convertible preferred stock identified therein, as amended (incorporated by reference to Exhibit 10.9 to the Company's Registration Statement on Form S-1, File No. 333-171375, as amended on December 23, 2011).10.10†* 10.10 10.11†* 10.11 10.12†* 10.12 10.13†* 10.13 10.14†* 10.14 10.15†* 10.15 10.16* 10.16 10.17*+ 10.17 10.18*+ 10.18 ExhibitNumberDescription10.20*+ 10.19 10.21*+ 10.23*Amendment No. 2 to Investor Rights Agreement dated April 6, 2012 by and among the Registrant and the holders of shares of Series A convertible preferred stock identified therein (incorporated by reference to Exhibit 10.29 to the Company's Registration Statement on Form S-1, File No. 333-171375, as amended on April 11, 2012). Description 10.20 10.24*+ 10.21 10.25†* 10.22 10.26* 10.23 10.27†* 10.24 10.28*+ 10.25 10.29* 10.26 10.30* 10.27 10.31*+ 10.28 10.32*+ 10.29 10.33* 10.30 10.34* 10.31 10.35*+ Description 10.33 10.37*+ 10.34 10.38*+ 10.35 14†* Code of Ethics. 10.36 21†* Subsidiaries of the Registrant 10.37 23.1*†*Consent of Ernst & Young LLP 10.38 23.2* *Consent of KPMG LLP 10.39 * 31.110.40 * 10.41 * 10.42 * 10.43 * Description 10.44 * 10.45 * 10.46 * 10.47 * 10.48 * 10.49 * 10.50 *+ 10.51 *+ 10.52 * 10.53 * 10.54 * 14 * 21 ** 23.1 ** Description 31.1 ** 31.2 31.2** 32.1 32.1** 32.2 32.2** 101 ** The following financial information from the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2019, formatted in Inline XBRL: (i) Cover Page; (ii) Consolidated Statement of Earnings; (iii) Consolidated Statement of Comprehensive Earnings; (iv) Consolidated Balance Sheets; (v) Consolidated Statements of Equity; (vi) Consolidated Statements of Cash Flows; and (vii) the Notes to Consolidated Financial Statements, tagged in summary and detail. 104 ** The Cover Page of the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2019, formatted in Inline XBRL (included with the Exhibit 101 attachments). † Confidential treatment requested under 17 C.F.R. §§200.80(b)(4) and 230.406. The confidential portions of this exhibit have been omitted and are marked accordingly. The confidential portions have been filed separately with the Securities and Exchange Commission pursuant to the Confidential Treatment Request. + Indicates a management contract or compensatory plan, contract or arrangement in which directors or officers participate. * Previously filed. ** Filed herewith. SUPERNUS PHARMACEUTICALS, INC. By: /s/ JACK A. KHATTAR Name: Jack A. Khattar Title: President and Chief Executive Officer Signature Title Date /s/ JACK A. KHATTAR 101 INSPresident and Chief Executive Officer and Director (Principal Executive Officer)**XBRL Instance Document.February 28, 2020 /s/ GREGORY S. PATRICK 101 SCHSenior Vice President and Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer)**XBRL Taxonomy Extension Schema Documents.February 28, 2020 /s/ CHARLES W. NEWHALL, III. 101 CALDirector and Chairman of the Board**XBRL Taxonomy Extension Calculation Linkbase Document.February 28, 2020 /s/ CARROLEE BARLOW, M.D., PH.D. 101 DEFDirector**XBRL Taxonomy Extension Definition Linkbase Document.February 28, 2020 /s/ GEORGES GEMAYEL, PH.D. 101 LABDirector**XBRL Taxonomy Extension Label/Linkbase Document.February 28, 2020 /s/ FREDERICK M. HUDSON Director February 28, 2020 101 PRE**XBRL Taxonomy Extension Presentation Linkbase Document./s/ JOHN M. SIEBERT, PH.D. Director February 28, 2020 †Confidential treatment requested under 17 C.F.R. §§200.80(b)(4) and 230.406. The confidential portions of this exhibit have been omitted and are marked accordingly. The confidential portions have been filed separately with the Securities and Exchange Commission pursuant to theConfidential Treatment Request.+Indicates a management contract or compensatory plan, contract or arrangement in which directors or officers participate.*Previously filed.**Filed herewith.