•our ability to
satisfy the required conditionsobtain andotherwise completemaintain regulatory approvals for ourplanned merger, or the Merger, with Oncternal Therapeutics, Inc., or Oncternal, pursuant to the Agreement and Plan of Merger and Reorganization, dated March 6, 2019, or the Merger Agreement, by and among GTx, Grizzly Merger Sub, Inc., a wholly-owned subsidiary of GTx, and Oncternal, on a timely basis or at all;•- product candidates;
the expected
benefitstiming for achieving key milestones, including completing andpotential value created byannouncing results of clinical trials of cirmtuzumab and TK216 and announcing theproposed Mergerfirst-in-human dosing of our Receptor tyrosine kinase-like Orphan Receptor 1, or ROR-1, chimeric antigen receptor T cell, CAR-T, product candidate currently in preclinical development;the timing or likelihood of regulatory filings and approvals;
the estimated size of the patient population and anticipated market potential for our
stockholders, includingproduct candidates;the
ownership percentageimpact of products that compete with ourstockholders inproduct candidates that are or may become available;the
combined organization immediately following the consummationsize and growth potential of theproposed Mergermarkets for our product candidates, andthe potential value of the contingent value rights to be received by our stockholders in connection with the proposed Merger if it is completed;•the implementation of our business strategies, includingour ability topreserve or realize any significant value from our selective androgen receptor degrader, or SARD, program and our selective androgen receptor modulators, or SARMs;•our expectations regarding the near-term development of our SARD program, includingserve those markets;our ability to
advance a SARD compound into a first-in-human clinical trial;•- obtain and maintain favorable regulatory designations for our product candidates and preclinical programs;
the
therapeutic and commercial potentialscope ofour SARD program;•our abilityprotection we are able to establish and maintainpotential new collaborative, partnering or other strategic arrangementsforthe development ofintellectual property rights covering ourSARD program;•- product candidates and our ability to
establish and maintain potential new collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets;•our ability to raise additional capital, whether through potential new collaborative, partnering or other strategic arrangements or otherwise;•our ability to protect our intellectual property andoperate our business without infringing upon the intellectual property rights of others;•our projected operatingthe plans and
financial performance;objectives of management for future operations and•- future results of anticipated products; and
our estimates regarding the sufficiency of our cash resources and our expenses,
including those related to the consummation of the proposed Merger,capital requirements andneedsneed for additional financing, and our ability to obtain additionalfinancing and to continue as a going concern if the Merger is not completed.
financing.In some cases, you can identifyThese forward-lookingstatements by terms such as "anticipates," "believes," "could," "estimates," "expects," "intends," "may," "plans," "potential," "predicts," "projects," "should," "will," "would," and similar expressions intended to identify forward-looking statements. Forward-lookingstatements reflect ourcurrentmanagement’s beliefs and views with respect to future events and are based on estimates and assumptions as of the date of this Annual Report and are subject to risksuncertaintiesandother important factors.uncertainties. We discuss many of these risks inthis Annual Report on Form 10-K ingreater detail under “Risk Factors.” Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess thesection entitled "Risk Factors" under Part I, Item 1A below.impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. Given theserisks,uncertainties,and other important factors,you should not place undue reliance on these forward-looking statements.Also,We qualify all of the forward-looking statements
represent our estimates and assumptions only as of the date ofin this Annual Reporton Form 10-K. You should read this Annual Report on Form 10-K and the documents that we incorporatebyreference in and have filed as exhibits to this Annual Report on Form 10-K, completely and with the understanding that our actual future results may be materially different from what we expect.these cautionary statements. Except as required by law, we
assumeundertake no obligation to publicly update any forward-looking statements,publicly, or to update the reasons actual results could differ materially from those anticipated in any forward-looking statements, even ifwhether as a result of new information,becomes availablefuture events or otherwise.Overview
Oncternal Therapeutics, Inc. is a clinical-stage biopharmaceutical company focused on the development of novel oncology therapies for cancers with critical unmet medical need. Our development efforts are focused on promising, yet untapped, biological pathways implicated in cancer generation or progression. Our pipeline includes cirmtuzumab, an investigational monoclonal antibody that is designed to inhibit Receptor tyrosine kinase-like Orphan Receptor 1, or ROR1, a growth factor receptor that is widely expressed on many tumors and that activates pathways leading to increased tumor proliferation, invasiveness and drug resistance. Cirmtuzumab is being evaluated in a Phase 1/2 clinical trial in combination with ibrutinib (Imbruvica®) for the treatment of patients with B-cell lymphoid malignancies, including mantle cell lymphoma, or MCL, and chronic lymphocytic leukemia, or CLL, and in an investigator-sponsored, Phase 1b clinical trial in combination with paclitaxel for the treatment of women with HER2-negative metastatic or locally advanced, unresectable breast cancer. We are also developing TK216, an investigational small molecule that is designed to inhibit the ETS, or E26 Transformation Specific, family of oncoproteins, which have been shown in preclinical studies to alter gene transcription and RNA processing and lead to increased cell proliferation and invasion. TK216 is being evaluated in a Phase 1 clinical trial as a single agent and in combination with vincristine in patients with relapsed or refractory Ewing sarcoma, a rare pediatric cancer. In addition, we are developing a chimeric antigen receptor T cell, or CAR-T, therapy candidate that targets ROR1, which is currently in preclinical development as a potential treatment for hematologic cancers and solid tumors.
Cirmtuzumab is an investigational, humanized, potentially first-in-class, anti-ROR1 monoclonal antibody that is designed to bind to a specific functionally important epitope of ROR1, which is a protein expressed on many tumors. ROR1 is a potentially attractive target for cancer therapy because it is an onco-embryonic antigen, which is a protein typically expressed only during embryogenesis that may confer a survival and fitness advantage when reactivated and expressed by tumor cells. ROR1 overexpression in various tumor types, including MCL, CLL and breast cancer, has been associated with more aggressive disease, resistance to therapy and shorter progression-free and overall survival. In preclinical models, inhibition of ROR1 has shown anti-tumor activity, and we believe may have additive or synergistic effects when combined with either targeted therapy or chemotherapy. Preclinical data indicated that when cirmtuzumab bound to ROR1, it blocked growth factor Wnt5a signaling, inhibited tumor cell proliferation, migration and survival, and induced differentiation of CLL tumor cells. Cirmtuzumab was developed in the
future.laboratory of one of our scientific advisors, Professor Thomas Kipps, M.D., Ph.D., Professor of Medicine and Evelyn and Edwin Tasch Chair in Cancer Research at the University of California San Diego, or UC San Diego, with support from the California Institute for Regenerative Medicine, or CIRM. We have an exclusive license to cirmtuzumab for therapeutic uses from UC San Diego.We have completed a Phase 1 study of single-agent cirmtuzumab in patients with CLL and have completed enrollment of dose-finding and expansion cohorts of a Phase 1/2 clinical study of cirmtuzumab in combination with ibrutinib in patients with CLL, as well as a dose-finding cohort of cirmtuzumab in combination with ibrutinib in patients with MCL. We are currently enrolling a Phase 1b clinical trial of cirmtuzumab in combination with ibrutinib in patients with MCL and a randomized Phase 2 clinical trial in patients with CLL. Cirmtuzumab is also being evaluated in an investigator-sponsored, Phase 1b clinical trial in combination with paclitaxel in patients with HER2 negative breast cancer. In addition, based on the high levels of ROR1 expression in multiple tumors and the importance of ROR1 for tumor proliferation and metastases, we believe that cirmtuzumab has potential as a therapeutic agent in other solid tumors and hematologic malignancies with high unmet medical need.
TK216 is an investigational, potentially first-in-class, targeted small molecule that is designed to specifically inhibit the biological activity of the ETS family of oncoproteins. Tumorigenic gene fusions involving ETS factors are frequently found in tumors such as Ewing sarcoma and prostate cancer, and ETS factors are often overexpressed in other tumors, such as acute myeloid leukemia, or AML, and diffuse large B cell lymphoma, or DLBCL. Researchers in the laboratory of Professor Jeffrey Toretsky, M.D., at Georgetown Lombardi Comprehensive Cancer Center, identified the precursor to TK216 using a novel chemical screening assay they developed based on a deep understanding of the underlying biological mechanism of ETS factors. Following this early work, TK216, which is designed to be a specific inhibitor of ETS factors, was created by us through the rational design and screening of novel small molecule inhibitors of a critical protein-protein interaction. In preclinical models, TK216 inhibited the interaction between ETS family members and RNA helicase A, or RHA, and by doing so, shut down excessive cell proliferation. We own intellectual property related to TK216 and have an exclusive license to product candidates targeting ETS oncoproteins for therapeutic, diagnostic or research tool purposes from Georgetown University.
We are evaluating TK216, as a
biopharmaceutical company dedicatedsingle agent and in combination with vincristine, in a Phase 1 clinical trial in patients with relapsed or refractory Ewing sarcoma. The dose-finding portion of the study was completed in 2019, and we are enrolling patients in an expansion cohort to evaluate thediscovery, development and commercializationclinical response ofmedicinestreatment with TK216 in combination with vincristine using the recommended Phase 2 dosing regimen. Ewing sarcoma is a rare pediatric cancer that has historically been very challenging to treatserious and/effectively, particularly for recurrent and metastatic disease. ETS fusion proteins have been shown to be present in over 90% of Ewing sarcoma cases. TK216 has received an Orphan Drug Designation and Fast Track Designation from the U.S. Food and Drug Administration, or FDA, for the treatment of patients with relapsed or refractory Ewing sarcoma.We are also developing a ROR1-targeted CAR-T therapy based on the binding domain of cirmtuzumab as a single-chain fragment variable, or scFv, as a potential treatment for patients with aggressive hematological malignancies or solid tumors. Because the epitope of ROR1 recognized by cirmtuzumab has appeared to be restricted to tumor cells in preclinical studies, we believe that a cirmtuzumab-based CAR-T may be selective in distinguishing cancer from normal tissues. Our ROR1-targeted CAR-T therapy candidate is in preclinical development in collaboration with UC San Diego, supported by funding from CIRM, and with Shanghai Pharmaceuticals Holding Co., Ltd., or SPH, in China.
We have assembled a management team, board of directors and scientific founders who have significant experience in successfully developing and commercializing therapeutics in oncology and orphan diseases, having worked or served on the Board of companies such as Amgen, Inc., Bavarian Nordic, Inc. (lead cancer asset acquired by Bristol Meyers Squibb Company), Cadence Pharmaceuticals, Inc.(acquired by Mallinckrodt plc), Dynavax Technologies Corporation, Elan Corporation (acquired by Perrigo), Eli Lilly and Co., Genzyme Corporation (acquired by Sanofi S.A.), Gilead Sciences, Inc., Halozyme Therapeutics, Inc., Ignyta, Inc. (acquired by Roche Holding AG), Immune Design Corporation (acquired by Merck Co., Inc.), Janssen Pharmaceuticals, Inc., Merck & Co., Inc., Micromet, Inc. (acquired by Amgen, Inc.), Pfizer, Inc., Roche Holding AG, Sorrento Therapeutics, Inc., and Zavante Therapeutics, Inc. (acquired by Nabriva Therapeutics plc).
Our strategy
Our mission is to build a leading oncology company that creates novel and transformative treatments for a wide range of oncology indications for which there are significant unmet medical
conditions. Underneeds. We believe our investigational agents target novel cancer pathways and have unique mechanisms of action. Our current pipeline is derived from our ability to identify therapeutic candidates that have generated promising, late-stage preclinical results or early clinical data, and in-license them for further development. We are particularly focused on therapeutic approaches for which there is a genetic or protein biomarker that can be used to identify populations of patients most likely to respond. We prioritize targets that we believe have the potential to transform the treatment paradigm of difficult-to-treat cancers with either single agent or combination therapy. As is the case for many oncology products, we believe that potential efficacy in one indication suggests the potential for application in other indications that carry the same target.Key elements of our strategy are as follows:
Generateclinicalproof-of-conceptdatawithTK216 inEwingsarcoma, an orphan pediatric cancer indication;
Advancecirmtuzumabthroughclinicaldevelopment,initiallyinMCL,CLLandbreast cancer;
Advance our ROR1-targeting CAR-Ttherapy to clinical testing, initially in hematological cancers and then in solid tumors;
EvaluatecirmtuzumabinadditionalROR1-positive solid tumorssuchaslung,ovarian andprostatecancers, as well as in additional hematological malignancies; and
EvaluateTK216inadditionaltumors with ETS fusion proteins or overexpression, suchasprostatecancer, lymphoma and AML.
The following figure summarizes our current programs:

Cirmtuzumab - monoclonal antibody targeting ROR1
Cirmtuzumab is an investigational, humanized, potentially first-in-class, anti-ROR1 monoclonal antibody that was designed to bind to a specific functionally important epitope of ROR1, which is a protein expressed on many tumors. ROR1 is a potentially attractive target for cancer therapy because it is an onco-embryonic antigen, which is a protein typically expressed only during embryogenesis that may confer a survival and fitness advantage when reactivated and expressed by tumor cells. ROR1 is over-expressed in many different cancers, including MCL, CLL and breast cancer, and has been reported to be associated with more aggressive disease, resistance to therapy and shorter progression-free and overall survival. In preclinical models, inhibition of ROR1 has shown anti-tumor activity and we believe may have additive or synergistic effects when combined with other agents.Preclinical data demonstrated that when cirmtuzumab bound to ROR1, it blocked Wnt5a signaling, inhibited tumor cell proliferation, migration and survival, and induced differentiation of CLL tumor cells. In preclinical models, cirmtuzumab showed synergy with both targeted therapy (e.g., ibrutinib and venetoclax (Venclexta®) in CLL models) and chemotherapy (e.g., paclitaxel in breast cancer models).
Cirmtuzumab was developed in the laboratory of one of our scientific advisors, Thomas Kipps, M.D., Ph.D., Professor of Medicine and Evelyn and Edwin Tasch Chair in Cancer Research at UC San Diego with support from CIRM. We have an exclusive, worldwide license to cirmtuzumab for therapeutic uses from UC San Diego.
A Phase 1 study of single agent cirmtuzumab in patients with CLL was completed at UCSD, and a Phase 1/2 clinical study of cirmtuzumab in combination with ibrutinib in patients with MCL and CLL is ongoing. The combination therapy study was designed to evaluate the safety of cirmtuzumab plus ibrutinib, determine a recommended dosing regimen, and evaluate efficacy in a randomized comparison study. Based on completed cohorts of the study, a recommended dosing regimen was chosen and the randomized portion of the study, comparing ibrutinib alone to cirmtuzumab plus ibrutinib in patients with CLL, is now enrolling. Similarly, a recommended dose regimen has been chosen for patients with MCL and enrollment in an expansion arm of the study is ongoing. Cirmtuzumab is also being evaluated in an investigator-sponsored, Phase 1b clinical trial in combination with paclitaxel in patients with HER2 negative metastatic or locally advanced, unresectable breast cancer. In addition, high levels of ROR1 expression have been reported in multiple additional cancers, and this expression appears to be a driver for tumor proliferation and metastases. As a result, we believe that cirmtuzumab has potential as a therapeutic agent in other solid tumors and hematologic malignancies with high unmet medical need.
Cirmtuzumab scientific background: inhibition of ROR1 as a therapeutic strategy in cancer
ROR1 is an onco-embryonic protein essential for normal fetal development that is suppressed in adults unless reactivated as a survival factor by many different cancers. The switching-on of ROR1 is consistent with a typical pattern in cancer, in which normal cells lose their highly differentiated functions and return to a more primal state, where they exhibit a greatly increased capacity for invasion, metastasis and resistance to treatment. This de-differentiation activates a number of genes normally restricted to fetal development, one of which is ROR1. Cancer cells with the highest potential for self-renewal are sometimes referred to as tumor-initiating cells or cancer stem cells and are capable of invading other tissues or metastasizing to disseminate tumors to distant sites in the body. These tumor-initiating cells are also the cells that have been found to be the most resistant to current therapies including chemotherapy and radiation therapy. Expression of ROR1 in ovarian cancer, for example, appears highest in a subpopulation of tumor cells that also have other markers of cancer stem cells. Cells that overexpress ROR1 show increased survival, migration, and resistance to chemotherapy.
Over-expression of ROR1 has been reported in multiple hematological and solid tumor types. Histological staining of over 350 human tumor samples identified that a majority expressed ROR1, including 90% or more of uterine cancers, lymphomas, and prostate cancers.
Cancer type
ROR1
Expressed (%)
Cancer type
ROR1 Expressed (%)
Uterus
96%
Adrenal
83%
MCL
>95%
Lung
77%
CLL
95%
Breast
75%
Lymphoma
90%
Testicular
73%
Prostate
90%
Colon
57%
Skin
89%
Ovarian
54%
Pancreas
83%
Bladder
43%
High ROR1 expression on patients’ tumor cells in a variety of cancers is associated with early relapse after therapy or the development of metastases. ROR1 expression levels on patients’ tumor cells is substantially higher in cancers that are more advanced and that contain poorly differentiated cells. Whereas Grade 1 or 2 ovarian tumors have been found to be 21% positive for ROR1, Grade 3 or 4 tumors have been found to be 62% positive for ROR1. Similar patterns in the percentage of ROR1-positive tumors were seen in pancreatic cancers, with 54% of Grade 1 or 2 tumors and 100% of Grade 3 or 4 tumors testing positive for ROR1 by immunohistochemistry. High expression of ROR1 has been associated with more aggressive disease and shorter patient survival in multiple tumor types, including CLL, breast cancer and ovarian cancer. High expression of ROR1 has also been associated with resistance to targeted therapy and chemotherapy.
Inhibition of ROR1 signaling or silencing ROR1 in multiple preclinical cancer models, including breast cancer, ovarian cancer, and glioblastoma, and was associated with suppressing the expression of genes characteristic of tumor-initiating cells, and with repression of cancer migration and metastasis. Preclinical models have also demonstrated that inhibition of ROR1, and blocking of Wnt5a signaling, inhibited tumor cell proliferation, migration and survival, and induced differentiation of the tumor cells – resulting in fewer metastases and improved survival.
Inhibition of ROR1 has been demonstrated in preclinical models to be additive to, or synergistic with, chemotherapy agents such as paclitaxel, and with targeted therapy agents such as ibrutinib and venetoclax. In addition, inhibition of ROR1 has been shown to enhance sensitivity of cancer cells to targeted therapy with agents such as erlotinib and may increase apoptosis and decrease proliferation.
In summary, we believe that ROR1 is an attractive therapeutic target in oncology for several reasons:
ROR1 is widely expressed on many tumors, including hematological malignancies and solid tumors;
Expression ofhighlevelsofROR1 on patients’ tumors is associated withmorerapid disease progression, resistance to therapy andshorter patient survival;
Blocking of ROR1 in preclinical models inhibited tumor cell proliferation, migration and survival, and induced differentiation of the tumor cells; and
Inhibition of ROR1 has been observed in preclinical models to be synergistic with chemotherapy and targeted therapy.
Cirmtuzumab development in Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia
MCL disease overview
MCL is an aggressive form of non-Hodgkin’s lymphoma. There are approximately 4,200 new cases of MCL each year in the United States, with the average age at diagnosis in the mid-60s. MCL is an aggressive lymphoma and carries a poor prognosis, with a median survival of about two to five years. The 10-year survival rate is only approximately 5%-10%.
While there are several therapeutic options available to treat patients with relapsed or refractory MCL, none of these options offers long-term benefit, with most patients relapsing in less than 20 months. In an open-label Phase 2 clinical trial, ibrutinib (Imbruvica®), a BTK inhibitor that is approved by the FDA for the treatment of patients with relapsed MCL, demonstrated an overall response rate, or ORR, of 67% and a complete response, or CR, rate of 23%, with a median duration of response, or DoR, of 17.5 months, median progression-free survival, or PFS, of 13 months and median overall survival, or OS, of 22.5 months. In an open-label Phase 2 clinical trial, acalabrutinib (Calquence®), another BTK inhibitor approved by the FDA for the treatment of patients with relapsed MCL, demonstrated an ORR of 80% and CR rate of 40%. Another BTK inhibitor approved in 2019, zanubrutinib (Brukinsa®), demonstrated an ORR of 84% and CR rate of 59%, with a median DoR of 19.5 months in an open-label Phase 2 clinical trial. These therapies are given continuously for prolonged periods of time, and their use can be associated with significant toxicity. The majority of patients with MCL are older, and remissions are not durable for most patients treated with continuous ibrutinib therapy. As a result, we believe that more effective and better tolerated therapies with shorter treatment periods represent a significant unmet need.
CLL disease overview
CLL is the most common form of leukemia in adults, accounting for 25-30% of all leukemias in the United States. An estimated 20,720 new cases of CLL were expected to occur in the United States in 2019, and in 2016 the prevalence of CLL in the United States was estimated to be 178,000 patients. CLL is primarily a disease of older adults. The median age at diagnosis is 71 years of age. Most patients are diagnosed as a result of routine blood work when elevated levels of lymphocytes are detected.
Significant progress has been made in the treatment of CLL since the advent of targeted therapies and FDA approval of ibrutinib for CLL in 2014. A treatment paradigm shift has taken place, from chemotherapies to targeted therapies. Three classes of targeted therapies have been approved for the treatment of patients with CLL: inhibitors of Bruton’s tyrosine kinase, or BTK, a key component of cell signaling in B-cells, such as ibrutinib, which is marketed as Imbruvica® by AbbVie, Inc., and Johnson & Johnson, and acalabrutinib, which is marketed as Calquence® by AstraZeneca PLC; inhibitors of the protein B-cell lymphoma-2, or Bcl-2, such as venetoclax, which is marketed as Venclexta® and Venclyxto® by AbbVie, Inc., and Roche/Genentech; and inhibitors of Phosphoinositide 3-kinase, or PI3K, which include idelalisib, which is marketed as Zydelig® by Gilead Sciences, Inc., and duvelisib, which is marketed as Copiktra® by Verastem, Inc.. These targeted therapies are now the core of the recommended treatment regimens for patients with both first-line and relapsed or refractory CLL, and have achieved objective response rates of 85-90%, two-year PFS of 65-90%, and two-year overall survival of 75-95%. The outcomes are worse for patients with certain prognostic factors, such as 17p or 11q chromosome deletions; for such patients with relapsed or refractory CLL treated with ibrutinib, the reported PFS is 50-75%. While CLL is treatable, it generally remains incurable, and patients with CLL will generally experience a recurrence of their cancer.
While these new targeted therapies improve outcomes for patients with CLL, only a limited number of patients achieve a CR. The proportion of patients with treatment-naïve or relapsed or refractory CLL who achieve a CR when treated with single-agent ibrutinib after twelve months of therapy is below 10%. The trade-off of these new and more effective targeted therapies is the paradigm of continuous treatment required for BTK inhibitors, resulting in accumulating costs and toxicities. Adverse events have been shown in a real-world analysis to limit ibrutinib treatment duration for almost half of all patients.
We believe that the next goals for CLL therapies will be to achieve deeper anti-tumor responses and more complete remissions, and to progress towards achieving the ultimate goal of developing a cure, while maintaining an acceptable safety profile. An acceptable safety profile may be particularly important for patients with CLL who are older (the median age at diagnosis is 71) and have multiple co-morbidities.
The market for CLL therapies in the United States, France, Germany, Italy, Spain, the UK, and Canada is estimated to be approximately $8 billion, largely driven by recently approved therapies, including ibrutinib, venetoclax, and idelalisib. We believe that CLL represents an attractive clinical and commercial opportunity for cirmtuzumab.
Cirmtuzumab preclinical data in CLL and MCL
Cirmtuzumab is an investigational, humanized monoclonal antibody that was designed to bind to a specific epitope of ROR1. The ligand for ROR1 in hematologic malignancies is Wnt5a, a secreted glycoprotein that has a critical role in embryonic and fetal development. Researchers at the UC San Diego School of Medicine discovered that targeting a critical epitope on ROR1 was key to inhibiting Wnt5a activation, specifically targeting ROR1 expressing tumors. This led to the development of cirmtuzumab, which binds this critical epitope of ROR1. Unlike antibodies that bind to other epitopes of ROR1, cirmtuzumab was not observed to bind to normal adult tissues in a GLP tissue cross-reactivity study.
Wnt5a controls the ability of cancer stem cells to self-renew and regulates cell migration and adhesion. Cancer patients whose tumors have high levels of Wnt5a have a lower probability of long-term survival than patients with low Wnt5a levels, analogous to the situation for patients whose tumors express more ROR1. In tumor models derived from primary human tumors, such as glioblastoma, overexpression of Wnt5a has been associated with tumors with more rapid growth that have increased invasiveness into other tissues. Similarly, cells from human melanoma that were engineered to overexpress Wnt5a have shown increased motility and invasiveness.
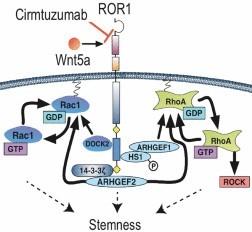
Figure 1. Cirmtuzumab blocked activation of ROR1 by Wnt5a in CLL cells, preventing a cascade of intracellular signaling events that leads to expression of genes associated with dedifferentiated stem cells.
Studies in mice have shown that ROR1 expression on the tumor cells accelerated the development and progression of leukemia in models of CLL and that Wnt5a enhanced CLL cell viability, migration and proliferation in a ROR1-dependent manner.
Patients with high levels of ROR1 on their CLL cells have more aggressive disease and have a significant reduction in survival: patients with CLL having high ROR1 expression have an approximately 50% survival rate at twenty years compared to an 80% survival rate for those with low ROR1 expression.
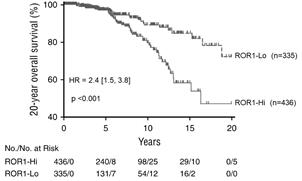
Figure 2. Patients with CLL having high levels of ROR1 expression had lower overall survival than those with low levels of ROR1.
In vivo studies conducted in mouse CLL models have shown that ibrutinib and cirmtuzumab exerted antitumor activities through independent pathways; that is, inhibition of BTK by ibrutinib did not alter ROR1 signaling nor did it impair the rate at which cirmtuzumab blocked ROR1 signaling. The combination of both drugs reduced the size of the spleen, the primary site of leukemic disease in these mice, as well as the number of CLL cells in these spleens. Further preclinical studies suggested that cirmtuzumab was synergistic with venetoclax in vitro.
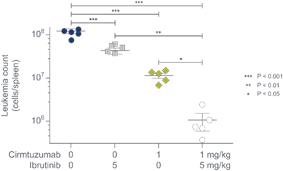
Figure 3. Combined administration of cirmtuzumab and ibrutinib reduced leukemic cell count in the spleen in a mouse model of CLL.
An analysis of patient samples indicated that ROR1 is highly and uniformly expressed on MCL cells but not on cells from multiple myeloma or follicular lymphoma patients.
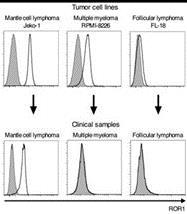
Figure 4: Analysis of cell lines and clinical samples from MCL, multiple myeloma and follicular lymphoma patients indicate that ROR1 is uniformly expressed on MCL patient cells.
The presence of ROR1 on MCL patient cells was confirmed by another analysis evaluating the expression of ROR1 using clinical samples from 21 patients with MCL. These data indicated that ROR1 was expressed by a very high percentage of the analyzed patient cells.
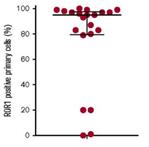
Figure 5. ROR1 is expressed on most MCL cells in most MCL patient samples.
Furthermore, an analysis of MCL and CLL patient samples has shown that ROR1 surface expression, as well as secreted Wnt5a levels, were comparable between patients with MCL and CLL.
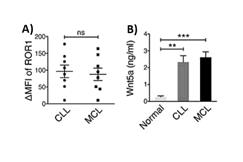
Figure 6: Analyses of samples from patients with CLL and MCL indicated that surface ROR1 expression levels as well as serum Wnt5a concentrations were similar.
In preclinical studies with cirmtuzumab, it has been demonstrated that the treatment of MCL patient cells with a combination of cirmtuzumab and ibrutinib led to reduced MCL cell proliferation.
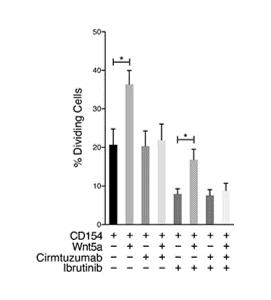
Figure 7. Cirmtuzumab inhibited Wnt5a-enhanced proliferation in ibrutinib-treated MCL Cells.
Lastly, in a published study using a research reagent anti-ROR1 antibody it was shown that pretreatment with an anti-ROR1 mAb and subsequent combination with a BCL2 inhibitor (venetoclax or navitoclax) led to an increase in anti-cancer activity against MCL cell lines as well as in several MCL patient samples tested with the combination therapy compared to the single agents.
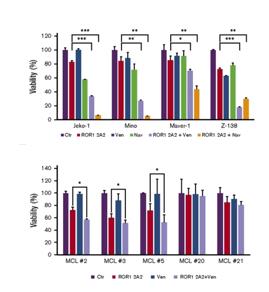
Figure 8. Percentage of cell viability (compared with controls) of untreated or treated cells with anti-ROR1 for 24 hours followed by addition of indicated drugs for 24 hours.
Cirmtuzumab clinical development in MCL and CLL
Cirmtuzumab Phase 1 clinical trial in patients with CLL
A Phase 1 dose escalation clinical trial of cirmtuzumab, which was funded jointly by us, CIRM, and others, was conducted in 26 patients with actively progressing CLL who had relapsed or refractory disease. Patients received four doses of cirmtuzumab administered every two weeks in cohorts of three, with each patient receiving escalating doses from 0.15 to 20 mg/kg/dose. Cirmtuzumab infusions were generally well tolerated. There were no dose-limiting toxicities, no serious adverse events, and no discontinuations related to adverse events. The main laboratory findings included anemia, thrombocytopenia and neutropenia, which were primarily attributed to the underlying CLL. Pharmacokinetic data showed a plasma half-life of approximately 32 days following four doses of cirmtuzumab at 16 mg/kg. In this clinical trial, 22 patients were evaluable for response assessment; four patients who discontinued cirmtuzumab early without meeting criteria for progressive disease were not considered evaluable. No patients met criteria for complete or partial remission following this brief treatment. Seventeen of 22 evaluable patients had stable disease. Five patients had progressive disease. Most patients experienced reductions in their leukemic lymphocyte counts and were able to delay initiation of further treatments for an average of 262 days, at which point plasma levels of cirmtuzumab were undetectable. Although cirmtuzumab therapy was limited to four doses, one patient who received cirmtuzumab at 20 mg/kg had a greater than 50% reduction in lymphadenopathy. Analysis of blood samples from these patients showed significantly higher plasma levels of Wnt5a compared to healthy matched controls. Patients also had high levels of expression of ROR1 on their CLL cells. Patients receiving doses of cirmtuzumab of 2 mg/kg or greater had a 33% reduction in ROR1 expression on their CLL cells relative to their baseline. In addition, when compared to baseline, cells from cirmtuzumab treated patients showed a reversal in the enrichment for genes that were identified as being the most highly correlated with stem cells and oncogenic de-differentiation. These results were consistent with other preclinical observations that cirmtuzumab induced ROR1 inhibition drove cells away from a stem-cell-like profile.
Cirmtuzumab Phase 1/2 clinical trial in combination with ibrutinib in patients with MCL and CLL (CIRLL)
We and UC San Diego, with major funding from CIRM and a donation of ibrutinib product from Pharmacyclics LLC, are conducting a Phase 1/2 trial of cirmtuzumab in combination with ibrutinib in patients with treatment-naïve or relapsed or refractory CLL and previously treated patients with MCL (Cirmtuzumab and Ibrutinib targeting ROR1 for Leukemia and Lymphoma, or CIRLL). Despite its efficacy in extending progression-free survival, ibrutinib does not provide complete tumor responses for the majority of patients even after prolonged dosing. Therefore, we believe there is potential to improve efficacy and overall outcomes by combining ibrutinib with cirmtuzumab. This clinical trial was designed to evaluate the safety, pharmacokinetics, pharmacodynamics, immunogenicity, and antitumor activity of cirmtuzumab in combination with ibrutinib in adult subjects with adequate performance status and organ function. The study has three parts:
Part 1
is a Phase 1b, open-label, sequential allocation, dose-finding evaluation of the administration of cirmtuzumab monotherapy followed by cirmtuzumab/ibrutinib combination therapy in subjects with CLL or relapsed/refractory MCL;
Part 2
is an open-label dose-confirming or expansion evaluation of the concurrent administration of cirmtuzumab and ibrutinib in CLL or MCL using the recommended cirmtuzumab dose regimen derived from Part 1; and
Part 3
is a Phase 2 open-label, randomized, controlled evaluation of the clinical activity and safety of cirmtuzumab plus ibrutinib versus ibrutinib alone in patients with CLL.
We have completed enrollment of patients with CLL in Parts 1 and 2, and those patients are completing therapy or are in long-term follow-up. Following an evaluation of safety and PK data from Part 1, the recommended dose regimen, or RDR, of cirmtuzumab for Part 2 was determined to be 600 mg of cirmtuzumab administered intravenously every two weeks for three doses, followed by dosing every four weeks until disease progression or intolerance develop. This cirmtuzumab regimen was designed and chosen to be used in combination with 420 mg of ibrutinib administered once daily for patients with CLL, or 560 mg of ibrutinib once daily for patients with MCL, which are the FDA-approved doses of ibrutinib in these indications. In August 2019, we opened enrollment in Part 3, which is a randomized Phase 2 study in patients with CLL, and in October 2019, we opened enrollment in the Part 2 expansion cohort of this clinical trial for patients with MCL. In mid-2020, we expect to announce additional data from this study of patients with CLL, including 12-month follow-up data for 34 patients enrolled in Parts 1 and 2 of the study.
Cirmtuzumab CIRLL clinical trial Part 1 interim data in MCL
Twelve patients with relapsed or refractory MCL were enrolled in the dose-finding cohort of this clinical trial. Patients had received a median of two prior therapies before participating in this study. As of the data cut-off on March 6, 2020, at a median follow-up of 6.4 months, the complete response rate was 50% (6 of 12), with an additional 33% achieving a PR (4 of 12) and 17% with stable disease (2 of 12), for a best ORR of 83% and clinical benefit rate of 100%. Of the six patients who achieved CRs, most did so within three to four months on the combination of cirmtuzumab and ibrutinib. All six patients who achieved CRs were heavily pre-treated. These patients had received, prior to participating in this clinical trial, chemotherapy and additional therapies including an autologous stem cell transplant (SCT); autologous SCT and CAR-T therapy; autologous SCT and allogeneic SCT; and ibrutinib with rituximab. As of March 6, 2020, all six CRs were ongoing and one patient had remained in CR at over 21 months on study.
��
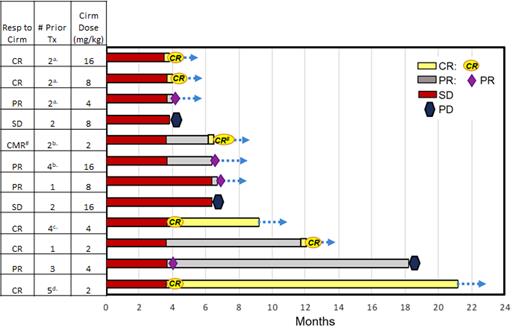
Figure 9. Best tumor response over time in the MCL cohort of 9 patients, based on investigator assessments, in Phase 1/2 clinical trial of cirmtuzumab in combination with ibrutinib as of March 6, 2020.
a. Prior ibrutinib/rituximab (7-10 months), R-HyperCVAD. b. Prior chemo, auto-stem cell transplant (SCT). c. Prior chemo, auto-SCT, CAR-T. d. Prior chemo, auto-SCT, allo-SCT.
Cirmtuzumab CIRLL clinical trial Part 1 and 2 interim data in CLL
Thirty-four patients with CLL who had never been treated with a BTK inhibitor were enrolled in the dose-finding and dose-expansion cohorts of this clinical trial, including 12 treatment-naïve and 22 relapsed/refractory patients. Patients with relapsed/refractory CLL had received a median of two prior therapies before participating in this clinical trial. As of the data cut-off on January 29, 2020, at a median follow-up of 9.9 months, 30 of the 34 patients achieved a response, for a best ORR of 88%. One patient achieved a CR and remained in remission six months after completion of the trial, without receiving any further anti-CLL therapy. In addition, three patients met radiographic and hematologic response criteria for Clinical CR, pending completion and evaluation of bone marrow biopsies. An additional four patients had stable disease, for a total clinical benefit rate of 100%. One patient was discontinued from the study after missing treatment and later passed away due to unrelated medical issues. None of the patients progressed or died while participating in the study, for a progression-free survival, or PFS, of 100%. As of an early November 2019 data cut-off, the rise in leukemic cell counts that is typically seen in the first six months with ibrutinib monotherapy was blunted with the cirmtuzumab plus ibrutinib combination, and leukemic cell counts returned toward baseline and normal levels rapidly.
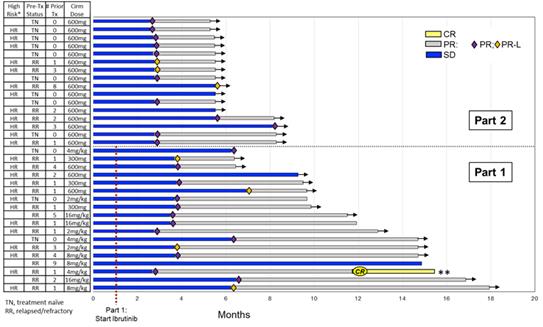
Figure 10. Best tumor response over time in the CLL cohort of 34 patients, based on investigator assessments, in Phase 1/2 clinical trial of cirmtuzumab in combination with ibrutinib, as of January 29, 2020.
* HR = known high risk factors: unmutated IgVH, del 17p/ TP53, and/or deletion 11q; ** Sustained CR for 6+ months on no CLL therapy
Genetic analysis of CLL cells from three patients showed pre-treatment transcriptome profiles associated with a stemness signature and NF-kB-driven inflammation. Both genetic signatures were reversed in these patients following cirmtuzumab treatment.
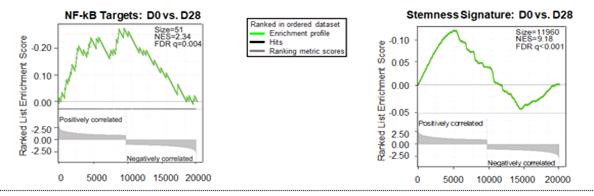
Figure 11. Reversal of NF-kB target genes and cancer-cell-stemness transcriptome signature observed in three patients with CLL in Phase 1/2 clinical trial of cirmtuzumab in combination with ibrutinib, as of November 2019.
In Part 3 of this trial, which is currently enrolling patients, up to 90 additional patients with CLL will be randomized 2:1 to receive cirmtuzumab plus ibrutinib or ibrutinib alone. The primary endpoint of Part 3 is to determine the CR rate, with a secondary endpoint of PFS. The data emerging from this trial will be used to determine our regulatory strategy, including whether we will plan to seek regulatory approval through standard review or an accelerated approval pathway. This trial is co-sponsored by UC San Diego with support from CIRM.
Cirmtuzumab as a single agent has been well tolerated in the CIRLL study. The combination of cirmtuzumab plus ibrutinib has also been well tolerated, with adverse events consistent with those reported for ibrutinib alone. As of the January 29, 2020, cut-off date, there were no dose limiting toxicities, no discontinuations and no serious adverse events attributed to cirmtuzumab alone. The rate of neutropenia, which is a common hematological side effect reported for ibrutinib in patients with CLL or MCL, was only 13% for ibrutinib plus cirmtuzumab (Grade 3-4 neutropenia of 8.7%). In mid-2020, we expect to announce additional data from this study of patients with MCL, including follow-up data for 12 patients enrolled in Part 1 of the study.
Cirmtuzumab development in breast cancer
Breast cancer disease overview
Breast cancer is the most common type of invasive cancer among women and the second leading cause of cancer deaths among women. There are approximately 266,000 new diagnoses and 41,000 breast cancer deaths in the United States each year, and 12.4% of women will develop breast cancer in their lifetime. The Centers for Disease Control and Prevention, or CDC, estimates that there are approximately one million women in the United States living with breast cancer that has been diagnosed within the past five years.
Breast cancers can be segregated into subtypes based upon the presence of three protein receptors: estrogen receptor (“ER”), progesterone receptor (“PR”) and humanepidermal growthfactorreceptor2 (“HER2”). Therapies have been developed that target tumors containing one or more of these receptors. Approximately 10% to 15% of breast cancers, however, do not express any of these three receptors and are referred to as triple-negative breast cancers, or TNBC. These tumors have a more aggressive phenotype and a poorer prognosis due to the high propensity for metastatic progression and absence of specific targeted treatments. The five-year survival rate for patients with breast cancer other than TNBC has been reported to be 80.8%, but only 62.1% for patients with TNBC. One hypothesis for the high rate of metastasis and poor response to chemotherapy for patients with TNBC is that these tumors contain a high number of tumor-initiating cells, or cancer stem cells, that are highly migratory and insensitive to standard chemotherapy. Treatment options for TNBC are limited and include chemotherapy, targeted therapy (such as PARP inhibitors), surgery, radiation, and immunotherapy. Additional targeted and immuno-therapeutic approaches are in clinical development.
ROR1 Expression and Historical Clinical Outcomes in Patients with Breast Cancer
Approximately 75% of breast tumors have been shown to express ROR1. In a retrospective analysis, patients with TNBC with high levels of ROR1 were found to have a significantly reduced disease-free survival (p< 0.00015) as well as overall survival (p<0.026) compared to patients with low ROR1 levels.
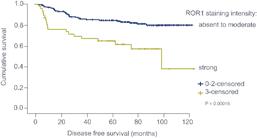
Figure 12. TNBC patients with high levels of ROR1 expression had lower disease-free survival.
Another retrospective, long-term analysis that included all breast cancer types showed that patients with tumors expressing high levels of ROR1 were at a statistically significantly higher risk of developing metastases within the first several years. Over 60% of patients with high ROR1 developed metastases, compared to only 35% of patients with the lowest levels of ROR1.
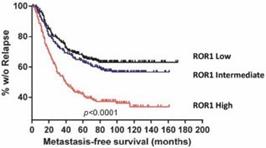
Figure 13. High levels of ROR1 in breast cancer was associated with shorter metastasis-free survival.
Preclinical experiments have shown that treatment of breast tumors with paclitaxel increased the percentage of cells with high levels of ROR1. In these experiments, immunodeficient mice were implanted with patient-derived xenografts, or PDX, then were treated with paclitaxel. While paclitaxel either slowed tumor growth or reduced the size of tumors in these mice, the surviving cells were enriched for expression of ROR1. This increased expression of ROR1 was also associated with a shift in the properties of cells from these tumors towards a more metastatic and more tumorigenic phenotype. Cells from tumors that had been treated with paclitaxel were more likely to form spheroids in tissue culture and were enriched for cells with the ability to form new tumors when transplanted, both properties that are correlated with tumor aggressiveness.
Together, these clinical and preclinical data are consistent with a model of the natural disease progression in breast cancer centered on the critical role played by tumor-initiating cells or stem-like cancer cells that express high levels of ROR1:
TNBCisinitiallyresponsivetochemotherapysuchaspaclitaxel,becausechemotherapykillsthemajorityofcancercells,leavingcells with stem-like properties that expressROR1;
TNBCreturnsmoreoftenthanothertypesofbreastcancerinpartbecausetheinitialchemotherapyenrichesforcellswithahigher propensity to formtumors;
Thesiteofrecurrenceisoftenatanotherplaceinthebodythantheoriginaltumorbecausecellswithstemcell-likepropertiesareableto metastasize; and
Therecurringtumormay beresistanttotherapybecauseitcontainsahighpercentageofcellswith stemcell-likeproperties.
Preclinical experiments in an MDA-MD-231 TNBC model in mice provided evidence that reduction in ROR1 expression levels can limit metastases and improve overall survival. In that model, ROR1 levels were selectively reduced using a genetic construct that delivers a short-hairpin RNA, or shRNA, that is designed to prevent ROR1 protein from being produced. Inhibition of ROR1 production resulted in significantly fewer cancer cells that have metastasized to the lungs.
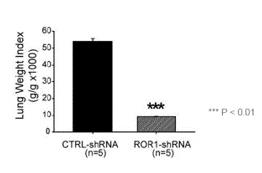
Figure 14. Suppression of ROR1 led to fewer metastases to the lungs in an MDA-MD-231 model TNBC model.
Inhibition of ROR1 production in these mice also improved overall survival to a mean of approximately 43 days compared to 30 days for mice containing control shRNA.
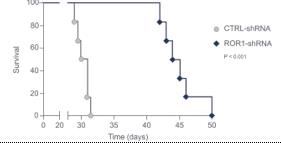
Figure 15. Inhibition of ROR1 expression led to improved survival in an MDA-MD-231 TNBC model.
In a preclinical model, cirmtuzumab reduced the growth rate of primary human breast cancers in immunodeficient mice and led to complete suppression of tumor growth for twenty days when used in combination with paclitaxel. Even after tumors did eventually grow, they lacked the ability to form new tumors. All tumor samples isolated from control mice, most of the tumor samples from paclitaxel-treated mice, and some of the cirmtuzumab-treated mice were able to establish new tumors when transplanted into other mice. No tumors, however, were formed when equal numbers of tumor cells from mice treated with the combination of cirmtuzumab and paclitaxel were introduced into other mice.
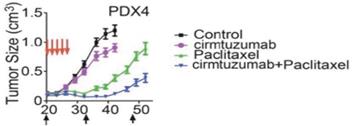
Figure 16. Combination of cirmtuzumab and paclitaxel suppressed growth of primary human breast tumors in a mouse model.
Cirmtuzumab clinical development in breast cancer
An investigator-sponsored single-arm, open-label, Phase 1b trial of cirmtuzumab in combination with paclitaxel in patients with locally-advanced, unresectable or metastatic HER2-negative breast cancer has been initiated at UC San Diego. The objectives of the trial include the evaluation of safety, tolerability, pharmacokinetics, and clinical activity. The treatment regimen is cirmtuzumab at a dose of 600 mg on days 1 and 15 of cycle 1, and then on day 1 of each subsequent 28-day cycle, and paclitaxel weekly at a dose of 80 mg/m. In December 2019, an interim clinical data update for this clinical trial was presented by the investigator at the 2019 San Antonio Breast Cancer Symposium. As of the data cut-off date of November 27, 2019, a total of eight patients with HER2-negative, metastatic or locally-advanced unresectable breast cancer were enrolled in the study. Seven of the eight patients were evaluable for safety and efficacy. Four of the patients had TNBC at study enrollment. Four of the seven evaluable patients achieved a partial response, for an ORR of 57%, including one patient who had a partial response that continued on cirmtuzumab alone for 30 weeks after discontinuing paclitaxel. It was reported that the combination of cirmtuzumab and paclitaxel was well tolerated in this trial, with no study discontinuations for toxicity and no dose-limiting toxicities observed as of the cutoff date. Adverse events were consistent with the known safety profile of paclitaxel alone. Pharmacokinetic analysis of serial plasma samples for free unbound antibody from two patients provided results similar to those observed in previous studies of patients with CLL, consistent with a projected half-life of 30 days. No abrupt decline in antibody concentration over time was observed, consistent with the absence of anti-drug or neutralizing antibodies. In the second half of 2020, we expect to announce additional data from this study of patients with HER2-negative, metastatic or locally-advanced unresectable breast cancer.
Potential additional clinical opportunities for cirmtuzumab in solid tumors
Lung cancer. ROR1 is expressed by approximately 77% to 93% of lung cancers. In adenocarcinoma of the lung, higher levels of ROR1 expression were correlated with advanced stages of disease and with positive lymph node metastases. In addition, Kaplan-Meier survival analysis indicated an association of high ROR1 expression with worse overall survival in lung adenocarcinoma patients. ROR1 expression has been shown to be correlated with the presence of other negative prognostic factors such as phosphorylated AKT, or p-AKT, or phosphorylated CREB, or p-CREB. Inhibition of ROR1 in lung cancer cell lines induced apoptosis and cell cycle arrest and led to a reduction in levels of p-CREB and p-AKT. Notably, a recent preclinical study has shown that downregulating ROR1 expression re-sensitizes erlotinib-resistant lung cancer cells to an EGFR inhibitor drug.
Ovarian cancer. ROR1 is expressed by approximately 54% of ovarian cancers, which is the most lethal gynecologic malignancy among women worldwide. Analysis of ROR1 expression on ovarian cancer patient samples revealed that disease-free survival and overall survival rate in patients with high ROR1 expression were significantly lower than in patients with low or no ROR1 expression. In a preclinical study, it was shown that a ROR1 antibody inhibited growth of ovarian cancer cell lines in vitro and slowed tumor growth in a mouse model.
Prostate cancer. ROR1 is expressed by approximately 90% of prostate cancers, and the Wnt5a signaling pathway is activated in patients with advanced prostate cancer that is progressing while on treatment with an androgen receptor, or AR, inhibitor. Treatment of prostate cancer cell lines with an AR inhibitor was found to increase the expression of Wnt5a, and the addition of Wnt5a attenuated the antiproliferative effect of AR inhibition. The expression of Wnt5a in patients with metastatic castrate resistant prostate cancer, or mCRPC, has been associated with poor overall survival. We are collaborating with academic investigators to investigate the potential effects of cirmtuzumab on this disease.
Pancreatic cancer. ROR1 is expressed by approximately 83% of pancreatic cancers. A recent preclinical study has shown that blocking ROR1 led to apoptotic cell death, which was further enhanced in combination with chemotherapeutic drugs such as erlotinib and ibrutinib, when tested against a panel of pancreatic cancer cell lines.
In mid-2020, we expect to announce data from IND-enabling preclinical studies of cirmtuzumab for additional indications.
TK216 - ETS oncoprotein inhibitor
TK216 is an investigational, potentially first-in-class, targeted small molecule that was designed to specifically inhibit the biological activity of the ETS family of oncoproteins. Tumorigenic gene fusions involving ETS factors are frequently found in tumors such as Ewing sarcoma and prostate cancer, and ETS factors are often overexpressed in other tumors, such as AML and DLBCL. Researchers in the laboratory of Professor Jeffrey Toretsky, M.D. at Georgetown Lombardi Comprehensive Cancer Center identified the precursor to TK216 by using a novel chemical screening assay that they developed based on a deep understanding of the underlying biological mechanism of ETS factors. Following this early work, TK216, which is designed to be a specific, high-affinity inhibitor of ETS factors, was created by us through the rational design and screening of novel small molecule inhibitors of a critical protein-protein interaction. In preclinical models, TK216 has inhibited the interaction between ETS family members and RNA helicase A, or RHA, and by doing so, shut down excessive cell proliferation.
We are evaluating TK216 as a single agent and in combination with vincristine in a Phase 1 clinical trial in patients with relapsed or refractory Ewing sarcoma. The dose-finding portion of the study was completed in 2019, and we are currently enrolling patients in an expansion cohort to evaluate the clinical response of treatment with TK216 in combination with vincristine using the recommended Phase 2 dosing regimen. Ewing sarcoma is a rare pediatric cancer that has historically been very challenging to treat effectively, particularly for recurrent and metastatic disease. ETS fusion proteins have been shown to be present in over 90% of Ewing sarcoma cases. TK216 has received an Orphan Drug Designation and Fast Track Designation from the FDA for the treatment of patients with relapsed or refractory Ewing sarcoma.
TK216 scientific background: ETS transcription factors and oncogenesis
TK216 targets the ETS family of oncoproteins known to be associated with both solid tumors and hematological malignancies. In normal development and physiology, ETS transcription factors govern processes such as cell cycle control, differentiation, proliferation, apoptosis, tissue remodeling and angiogenesis. However, when alterations in the functions of ETS factors develop, through overexpression, gene fusion or modulation, they have been shown to lead to tumor initiation, progression, and metastasis. Fusion proteins are a well-known category of targets for small molecule cancer therapy that have been cited in the scientific literature as providing a number of diagnostic and therapeutic advantages because of their tumor-specific expression. ETS overexpression or fusion proteins incorporating an ETS factor have been observed in multiple tumor types:
Ewing sarcoma*
98%
Prostate cancer*
55%
Diffuse Large B Cell Lymphoma
52%
Head & Neck cancer
33%
Acute Myeloid Leukemia*
30%
Breast cancer*
25%
Melanoma
25%
Ovarian cancer
23%
Lung cancer
21%
Glioblastoma multiforme
15%
* Fusion identified
Fusion proteins involving ETS factors have been implicated in various solid tumors, including Ewing sarcoma and prostate cancer. For example, approximately 85% of Ewing sarcomas contain a genomic rearrangement between chromosomes 11 and 22. DNA is exchanged between these chromosomes in a pathological manner, and this exchange results in a fusion of two genes: the FLI1 gene, an ETS family member, and the EWSR1 gene, an unrelated transcription factor. This gene fusion, known as EWS/FLI1, functions as a transcription activator that is no longer controlled by the relevant regulatory machinery in the cell. In addition to escaping regulation, the dysregulated function of the EWS/FLI1 fusion causes a series of abnormalities in RNA processing including aberrant mRNA expression and splicing, where it leads to defects in the synthesis of proteins such as BRCA1, a DNA repair protein. EWS/FLI1 fusions also cause the formation of abnormal and potentially deleterious DNA and RNA structures known as R-loops that are associated with replication and transcriptional blocks as well as being prone to increased DNA damage.
Multiple other tumors contain gene fusions of other ETS factors. For example, over 50% of metastatic prostate cancers carry a TMPRSS2-ETS gene fusion. Other tumors have genetic changes that result in overexpression of ETS factors.
ETS Fusions
ETS Overexpression
•
Ewing sarcoma
•
AML
•
EWS-FLI1
•
FLI1, ERG, ETV5, ETS2
•
Prostate cancer
•
DLBCL
•
TMPRSS2-ERG
•
ETV1, FLI1, ETV4, SPIB
•
AML
•
Prostate cancer
•
ETV6-various (20+)
•
ERG, ETV1, ETV4, ETV6
•
ALL
•
Lung cancer
•
ETV6-RUNX1
•
ETV5, ETV1, FLI1, ETS1
•
Secretory breast cancer
•
Breast cancer
•
ETV6-NTRK3
•
ETV6, ETV4, SPIB, ETV5
Despite the genetic associations between ETS factors and tumorigenesis and the reported correlation between high levels of ETS factor expression and survival, there are currently no approved therapeutics available that target these factors. It had been widely considered that transcription factors are difficult to target due to their non-enzymatic mechanism of action. Researchers in the laboratory of Jeffrey Toretsky, M.D., Professor at Georgetown University, identified the precursor to TK216 by using a chemical screening assay that they developed based on a deep understanding of the underlying biological mechanism of ETS factors. TK216 has been observed to inhibit the interaction between ETS family members and RNA helicase A, or RHA, a critical component of the human transcriptional complex, and by doing so, shuts down excessive cell proliferation in preclinical tumor models. We believe that our approach of inhibiting protein-protein interactions is novel and that our product candidate TK216 targeting ETS factors could fill an important gap in the treatment landscape for both solid tumors and hematological malignancies.
TK216 development in Ewing sarcoma
Ewing sarcoma disease overview
Ewing sarcoma is the second most common bone tumor of children that occurs most often in adolescents and accounts for approximately 2% of all childhood cancer diagnoses. The incidence of Ewing sarcoma for all ages is approximately 1.3 cases per 1 million people in the United States, corresponding to approximately 430 new patients diagnosed per year in the United States. The median age at diagnosis of patients with Ewing sarcoma is 15.
Nearly all Ewing sarcoma cases are driven by translocations of ETS family oncogenes, including 85-90% of cases driven by the EWS-FLI1 fusion, and approximately 10% by EWS-ERG.
Ewing sarcoma typically develops in the pelvis, femur, and bones of the head and trunk, but its diagnosis often takes months as other causes for non-specific symptoms such as localized pain, fever, fatigue, weight loss, or anemia are ruled out. The five-year survival of patients who are diagnosed with non-metastatic disease is between 50% and 70%. Patients diagnosed with metastatic disease have five-year survival between 18% and 30%. The prognosis for patients with recurrent Ewing sarcoma is particularly poor, and five-year survival after recurrence is approximately 10 to 15%.
Ewing sarcoma is usually treated systemically due to the fact that local treatments, even in patients without overt metastases, have an 80% to 90% relapse rate. The current standard therapy for patients with localized Ewing sarcoma in the United States is a combination of chemotherapy agents, including vincristine, doxorubicin and cyclophosphamide, with alternating cycles of ifosfamide and etoposide – a therapy known as VDC/IE. Patients that respond to this therapy may be candidates for tumor resection and continued treatment for a total of 14 to 17 cycles. This therapeutic regimen, however, is associated with significant toxicities. Patients with metastatic disease are often treated with VDC/IE or variations of this therapy with higher or more compressed dosing. This may also be supplemented by local radiation therapy or systemic radiation followed by autologous hematopoietic stem cell transplant. We believe that more effective therapies are needed for this rare and severe pediatric disease.
TK216 preclinical data in Ewing sarcoma
TK216 was the product of a novel approach based on developing small molecule inhibitors of a critical protein-protein interaction linked to the ETS family of transcription factors. Researchers at Georgetown University identified YK-4-279, the precursor to TK216, by using a novel chemical screening assay. Following this early work, TK216, a specific inhibitor of ETS factors, was then created by Oncternal through the rational design and screening of novel small molecule inhibitors of a critical protein-protein interaction linked to the ETS family of transcription factors. TK216 is a structural analog of YK-4-279 that has shown increased potency in biochemical, cellular and xenograft tumor models.
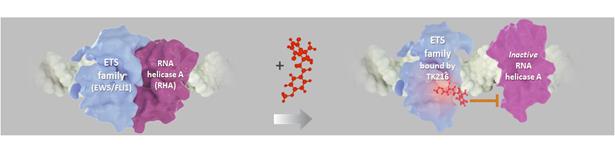
Figure 17. TK216 inhibits interaction of ETS fusion protein EWS/FLI1 with RNA helicase A (RHA).
In Ewing sarcoma, a key heterodimer between EWS/FLI1 and RHA forms the core of a transcriptome complex causing activated oncogenes, inhibited tumor suppressors, abnormal RNA transcription and abnormal RNA splicing. TK216 was developed to disrupt that heterodimer, thereby potentially preventing transcription and leading to inhibition of the oncogenic activity of EWS/FLI1, by decreasing oncogene expression, increasing tumor suppressor function, and apoptotic cell death. In preclinical models, TK216 inhibited the interaction between ETS family members and RNA helicase A (“RHA”), and by doing so, shut down excessive cell proliferation and cause apoptotic cell death.
Treatment in vitro with TK216 led to dose-dependent inhibition of transcription from a luciferase reporter assay in COS7 cells. TK216 also inhibited proliferation of Ewing sarcoma cell line A4573.
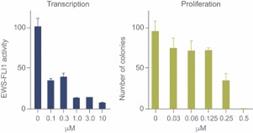
Figure 18. TK216 inhibited transcription of a reporter gene dependent on EWS/FLI (left). TK216 inhibited proliferation of a Ewing sarcoma cell line A4573 (right).
TK216 has inhibited proliferation of multiple cell lines containing EWS/FLI1 fusions, as well as other cell lines containing other ETS translocations or overexpressing ETS factors. These results suggest that TK216 binds to a site that is commonly used by multiple ETS family members to interact with other factors such as RHA. Therefore, we believe that TK216 has potential beyond targeting the EWS/FLI1 fusion that is commonly found in patients with Ewing sarcoma.
1
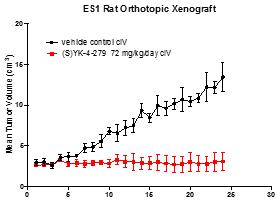
Figure 19. YK-4-279 (analog of TK216) showed activity in a Ewing sarcoma rat xenograft model.
Treatment of aggressive tumors such as Ewing sarcoma typically requires a combination of agents. A systematic analysis combining approved agents tested in combination with YK-4-279, a precursor of TK216, was conducted using Ewing sarcoma cell lines. YK-4-279 led to synergistic cytotoxicity with 28% of the agents tested including antimetabolites, nucleic acid synthesis inhibitors, immunosuppressive or immunomodulating agents and microtubule inhibitors. In vivo activity in an A4573 xenograft model of Ewing sarcoma showed tumor shrinkage and increased survival when YK-4-279 was combined with vincristine.
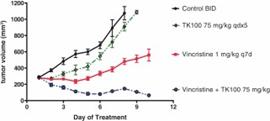
Figure 20. Combination of YK-4-279 (analog of TK216) and vincristine resulted in rapid tumor shrinkage in an A4573 model of Ewing sarcoma.
TK216 clinical development in Ewing sarcoma
We are conducting an open-label, first-in-human, multicenter Phase 1 clinical trial of TK216 as a single agent and in combination with vincristine, in patients with relapsed or refractory Ewing sarcoma. In November 2019, we reported an interim clinical data update for this clinical trial at the Connective Tissue Oncology Society (CTOS) 2019 Annual Meeting. As of the data cut-off date in November 2019, 32 patients had been treated in the dose-finding part of the trial. Patients entering the trial had previously been treated with a median of four, and up to nine, prior lines of systemic therapy. TK216 was reported to be generally well tolerated in this trial, with common side effects including myelosuppression, fatigue, nausea and alopecia. No unexpected off-target toxicities were observed. Based on the dose-finding part of the trial, the recommended Phase 2 dose (RP2D) was selected to be 200 mg/m2/day for 14 days. Clinical pharmacology data suggest that this dosing regimen may result in drug levels that meet or exceed those that killed tumor cells in vitro and inhibited tumor growth in animal models.
Subsequent to the CTOS presentation, additional patients were enrolled in the cohort treated at RP2D. Of the three evaluable patients treated at RP2D in the dose-finding part of the trial, one patient achieved a deep and sustained PR on single-agent TK216, followed by a surgical CR; another patient had stable disease; and a third had progressive disease. One additional patient had been enrolled but developed rapidly progressive disease. This patient exited the study before completing the DLT observation period and, per protocol, was replaced.
One patient who achieved a deep and sustained clinical response at RP2D had relapsed/refractory Ewing sarcoma with lung metastases and had received and failed multiple lines of therapy prior to participating in this clinical trial, including radiation, chemotherapies and targeted therapies. Multiple lung nodules in this patient regressed following two cycles of TK216 as a single agent and, after six months of treatment that included concomitant vincristine starting in the third cycle, a single remaining 7 mm lung nodule was resected, resulting in a surgical complete remission. The patient had no evidence of disease after more than ten months of treatment. TK216, with or without vincristine, was well tolerated by this patient, with minimal myelosuppression.
In December 2019, we announced that we had opened for enrollment an expansion cohort of our clinical trial in patients with relapsed or refractory Ewing sarcoma. The expansion cohort will further evaluate the recommended Phase 2 dose regimen of TK216 (200 mg/m2/day for 14 days) in combination with vincristine and is anticipated to enroll approximately 18 patients. By mid-2020, we expect to enroll seven to 12 additional patients in the expansion cohort of this study and we anticipate announcing data from these patients in the second half of 2020.
Potential additional clinical opportunities for TK216
Acute myeloid leukemia (AML). AML is a hematologic malignancy characterized by dysregulated maturation of myeloid or blood stem cells and failure of the bone marrow to properly function, leaving patients with anemia and immune deficiency, and at high risk of infections and bleeding. AML is the most common type of acute leukemia in adults. Approximately 21,450 new AML cases and 10,920 AML associated deaths occur annually in the United States. The average age of an AML patient is 68 years. The National Cancer Institute estimated in 2018 that the five-year survival rate for adult patients with AML was approximately 27%. We believe that there is a need for more effective and less toxic therapies for AML.
ETS overexpression or fusion proteins incorporating ETS family member have been observed in about 30% of AML cases. The ETS family member ERG is overexpressed in many cancers, such as AML. In a retrospective analysis of patients with AML, the quartile of patients with the highest levels of ERG expression had a significantly higher rate of relapse and poorer overall survival than patients with lower levels of ERG expression. Those with the highest levels of ERG had a five-year survival rate of 20%, while those with lower levels of ERG had a survival rate of approximately 50%. ERG overexpression was an independent negative prognostic factor. Similarly, AML patients with high levels of ETS2, another ETS family member, had a significantly lower five-year survival rate of approximately 15% compared to 40% for patients with lower levels of ETS2. ETS2 overexpression was an independent negative prognostic factor.

Figure 21.SurvivalofthequartileofAMLpatientswiththehighestERG(left)orETS2(right)expressionwassignificantlylowerthanthosewith lowerexpression.
Multiple AML cell lines have been shown to be sensitive to being killed by TK216, with sensitivity proportional to ETS expression. TK216 may provide a novel therapeutic strategy for the treatment of patients with relapsed and refractory AML, a patient population known to express, in certain cases, fusion proteins involving ETV6, and to have overexpression of ETS family members including FLI1, ERG, ETS2, and ETV5.
Prostate cancer. Approximately 174,650 new cases of prostate cancer are diagnosed annually in the United States. Incidence of metastatic prostate cancer is increasing, causing an estimated 31,620 deaths per year in the United States. New therapeutic options are needed after failure of androgen antagonism and prior to chemotherapy. Approximately 55% of men with advanced prostate cancer carry the ETS family fusion gene TMPRSS2-ERG that is related to androgen resistance.
We believe TK216 may provide a novel therapeutic strategy for the treatment of patients with advanced prostate cancer, in particular those who carry the ETS family fusion gene TMPRSS2-ERG. In a preclinical in vivo study, YK-4-279, which is an analog of TK216, showed anti-tumor activity against a prostate cancer cell line harboring the ETS-family translocation, while growth of a prostate cancer cell without the translocation was not inhibited.
In the second half of 2020, we expect to announce data from IND-enabling preclinical studies of TK216 in additional ETS-driven tumors.

Figure 22.Prostate cancer sensitivity was associated with an ETS-family fusion protein in human prostate cancer xenograft models.
ROR1 CAR-T Program
We are developing a CAR-T therapy based on the ROR1 binding domain of cirmtuzumab to treat patients with aggressive hematological malignancies or solid tumors. We believe that the selective expression of ROR1 on tumor cells and its absence on normal cells make it an ideal target for a CAR-T approach. In addition, we believe that the survival benefit imparted on cancer cells by expressing ROR1 may limit the development of ROR1-negative resistant tumors, and that tumors that generate mutations that escape an ROR1 CAR-T therapeutic by inactivating or suppressing ROR1 may lose their stem cell-like properties, limiting their ability to metastasize or establish new tumors. Our ROR1 targeted CAR-T therapy is in preclinical development at UC San Diego, with funding from CIRM.
We also plan to collaborate with our strategic partner SPH for our CAR-T program. Through its United States subsidiary Shanghai Pharmaceutical (USA) Inc., or SPH USA. SPH USA has entered into the SPH USA License Agreement with us to develop ROR1 targeted CAR-T products in greater China. Our plans to collaborate with SPH USA include a collaboration to develop processes to produce and manufacture lentiviruses carrying the ROR1 construct. We believe that this represents a potential advantage for our CAR-T program, because viral manufacturing capacity is constrained in the United States and European Union. We and SPH USA also intend to collaborate by conducting one or more initial clinical trials of our potential CAR-T product candidate at hospitals in China that have experience with processing cellular immunotherapy materials and conducting CAR-T clinical trials. Initial clinical trials of our CAR-T program may occur both in the United States, for example at UC San Diego and at sites in China. In the fourth quarter of 2020, we expect to announce first-in-human dosing of our ROR-1 CART therapy in China.
Scientific background: CAR-T therapy overview
Immuno-oncology approaches to treating cancer involve redirecting one of the pillars of the immune system, the adaptive immune system, so that it specifically and efficaciously recognizes cancerous cells that might previously have escaped immune recognition. A key element in the adaptive immune response is the T cell. T cells are white blood cells that can recognize and kill infected and abnormal cells. T cells also act to signal other immune cells to respond to threats. T cells recognize their targets because they are created in a way that allows them to specifically recognize foreign antigens on the surface of other cells.
T cells are ideally suited for immuno-oncology applications based on several characteristics. They are created to be exquisitely specific and avid killers. One T cell can eliminate numerous target cells. T cells are extremely specific, able to recognize a cancer cell and kill it, while ignoring an almost identical healthy cell. T cells are thought to be active all the time, eliminating cancer cells from the body before they can form tumors. However, tumor cells sometimes evolve to escape killing by T cells by activating a number of pathways that suppress T cell function. Adoptive T cell therapies, and specifically CAR-T, were developed to provide a method to generate large quantities of T cells capable of specifically recognizing and killing tumor cells despite tumor suppressive mechanisms.
CAR-T therapeutics are created by isolating T cells from patients and modifying them to recognize specific antigens on tumors. T cells have potent cell killing activity that is directed to target cells that are recognized by specific T cell receptors, or TCRs, that are expressed on the surface of these T cells. While some T cells have TCRs that can recognize cancer cells leading to their killing, potent T cells do not develop to all targets. In some cases, the potential cancer cell target is also a protein that has an essential role in other tissues or at other stages of development, and TCRs that recognize these targets are eliminated during normal T cell development.
CAR-T therapy has emerged as a way to engineer T cells to recognize specific targets, such as those that are selectively expressed on cancer cells. A gene encoding a chimeric protein is constructed that contains a single antigen-binding domain of an antibody that recognizes the target, which is coupled to a T cell costimulatory domain and a portion of the T cell receptor.
CAR-T therapies are typically produced from a patient’s own T cells which are isolated by leukapheresis. These cells are then genetically modified with the chimeric antigen gene construct which can be delivered by various mechanisms such as lentiviral gene delivery vectors. Transduced cells are then expanded and undergo quality testing before being reintroduced into the same patient.
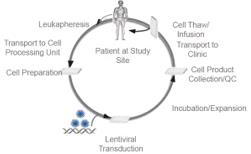
Figure 23. CAR-T production and patient treatment.
Two CAR-T cell therapies, Yescarta®, developed by Kite Pharma, Inc., a wholly-owned subsidiary of Gilead Sciences, Inc., and Kymriah®, developed by Novartis Pharmaceuticals Corporation, have been approved by the FDA. Both of these therapies target the CD19 protein, a protein expressed on the surface of the majority of B cells, including B cell tumorigenic cells. Yescarta has been approved for the treatment of relapsed or refractory large B-cell lymphoma and Kymriah for the treatment of relapsed or refractory B-cell precursor acute lymphoblastic leukemia. These therapies have shown high response rates with prolonged treatment effects for a subset of patients. No CAR-T therapies have been approved for use in patients with solid tumors. Despite the high response rates and prolonged treatment effects observed for a subset of patients, we believe that novel CAR-T approaches have the potential to improve efficacy, duration of response as well as safety.
Prior to the Merger as defined below, GTx had been developing selective androgen receptor modulators, or SARMs, under an exclusive, worldwide license agreement with the University of Tennessee Research Foundation, or
UTRF, we are developing UTRF's proprietary selective androgen receptor degrader, or SARD, technology, which we believe has the potential to provide compounds that can degrade or antagonize multiple forms of androgen receptor, or AR, thereby potentially inhibiting tumor growth in patients with progressive castration-resistant prostate cancer, or CRPC, including those patients who do not respond to or are resistant to current androgen targeted therapies. We are in the process of completing ongoing mechanistic preclinical studies in order to select the most appropriate SARD compounds to move forward into the additional preclinical studies required to submit an investigational new drug application, or IND, and potentially advance one of our SARD compounds into a first-in-human clinical trial.We had been developing selective androgen receptor modulators, or SARMs. OurUTRF. GTx’s SARM product candidate, enobosarm (GTx-024), was most recently evaluated in post-menopausal women with stress urinary incontinence, or SUI. During the third quarter of 2018,weGTx announced thatour randomized, placebo-controlled Phase 2 clinical trial, orthe ASTRID trial, evaluating the change in the mean number of daily SUI episodes following 12 weeks of enobosarm treatment failed to achieve statistical significance on the primary endpoint of the proportion of patients with a greater than 50% reduction in incontinence episodes per day compared to placebo.We haveGTx completed the ASTRID trial, includingourits review of the full data sets from the clinical trial, andhavedetermined that thereiswas not a sufficient path forward to warrant additional clinical development of enobosarm to treat SUI.We have thereforeAs a result, GTx discontinued further development of enobosarm to treat SUI, including discontinuing the related durability and open-label safety extension studiesweinitiated by GTx beforeweit received topline data from the ASTRIDtrial. We have alsotrial, and discontinued any further development ofourits SARM program generally.Following the announcement of the ASTRID trial results, our board of directors commenced a process of evaluating strategic alternatives to maximize stockholder value. To assist with this process, our board of directors engaged a financial advisory firm to help explore our available strategic alternatives, including possible mergers and business combinations, a sale of part or allOn December 31, 2019, we notified UTRF of ourassets, and collaboration and licensing arrangements. On March 6, 2019, we and Oncternal announcedintent to terminate thesigning of the Merger Agreement. Upon the terms and subject to the satisfaction of the conditions described in the MergerSARM License Agreement,including approval of the transaction by our stockholders and Oncternal's stockholders, a wholly-owned subsidiary of GTxwhich termination will bemerged with and into Oncternal, with Oncternal surviving the Merger as a wholly-owned subsidiary of GTx.The proposed Merger is structured as a stock-for-stock transaction whereby all of Oncternal's outstanding shares of common stock and securities convertible into or exercisable for Oncternal's common stock will be converted into the right to receive GTx common stock and securities convertible into or exercisable for GTx common stock. Under the exchange ratio formula in the Merger Agreement, the former Oncternal stockholders immediately before the Merger are expected to own approximately 75% of the outstanding capital stock of GTx, and the stockholders of GTx immediately before the Merger are expected to own approximately 25% of the outstanding capital stock of GTx, subject to certain assumptions. The exchange ratio formula excludes Oncternal's outstanding stock options and warrants and GTx's outstanding stock options and warrants. To the extent Oncternal's outstanding stock options or warrants are exercised in the future, it will result in further dilution toGTx's stockholders. Under certain circumstances as set forth in the Merger Agreement, the ownership percentages may be adjusted upward or downward based on cash levels of the respective companies at the closing of the Merger. We anticipate that the Merger will close in the second quarter of 2019. Following the closing of the Merger, James Breitmeyer will serve as GTx's Chief Executive Officer, Richard Vincent will serve as GTx's Chief Financial Officer, and Lauren Otsuki will serve as GTx's Chief Operating Officer. Additionally, following the closing of the Merger, our board of directors will consist of nine directors, including two current GTx board members. This transaction, which has been approved by our board of directors and the board of directors of Oncternal, is subject to the satisfaction or waiver of certain conditions, including the required approvals by the parties' stockholders (including stockholder approval from one of Oncternal's significant stockholders, Shanghai Pharmaceutical (USA) Inc., which holds all of the outstanding shares of one series of Oncternal's preferred stock that must approve the transactions contemplated by the Merger Agreement) and other customary closing conditions. Certain affiliates of ours who hold approximately 45% of our common stockeffective as ofdateMarch 31, 2020.Also under an exclusive worldwide license agreement with UTRF, GTx had been developing UTRF’s proprietary selective androgen receptor degrader, or SARD, technology, to provide compounds to degrade or antagonize multiple forms of
the Merger Agreement have agreedandrogen receptor, thereby potentially inhibiting tumor growth in patients with progressive castration-resistant prostate cancer, including those patients who do not respond tovote in favor of the Merger and certain affiliates of Oncternal, excluding Shanghai Pharmaceutical (USA) Inc., who hold approximately 42% of the outstanding capital stock of Oncternal as of date of the Merger Agreement have agreedor are resistant tovote in favor of the Merger. However, Oncternal currently expects that it will receive stockholder approval from Shanghai Pharmaceutical (USA) Inc. approximately two months after the date of the Merger Agreement based on the internal approval process required for such approval at Shanghai Pharmaceutical (USA) Inc.In addition, at the effective time of the Merger, GTx and certain other parties will enter into a Contingent Value Rights Agreement, or the CVR Agreement. Pursuant to the CVR Agreement, for each share of GTx common stock held, GTx stockholders of record as of immediately prior to the effective time of the Merger will receive one contingent value right, or CVR, entitling such holders to receive in the aggregate 50% of any net proceeds received during the 15-year period after the closing of the Merger from the grant, sale or transfer of rights to our SARD or SARM technology that occurs during the 10-year period after the closing of the Merger (or in the 11thyear if based on a term sheet approved during the initial 10-year period) and, if applicable, to receive royalties on the sale of any SARD products by the combined company during the 15-year period after the closing of the Merger. Under the CVR Agreement, Oncternal (as successor in interest to GTx) agreed to use commercially reasonable efforts to develop SARD products and to divest our SARM technology, subject to certain limitations. The CVRs will not be transferable, except in certain limited circumstances, will not be certificated or evidenced by any instrument and will not be registered with the SEC or listed for trading on any exchange.Although we have entered into the Merger Agreement and intend to consummate the proposed Merger, there is no assurance that we will be able to successfully consummate the proposed Merger on a timely basis, or at all. If, for any reason, the proposed Merger is not completed, we will reconsider our strategic alternatives and could pursue one or more of the following courses of action:•Continue development of our SARD program.As set forth above, wecurrent androgen targeted therapies. We arein the process of completing ongoingperforming additional mechanistic preclinical studies in order toselect the most appropriatedetermine if one or more of these SARD compoundsto move forwardshould be advanced into the additional preclinical studies required to submit anINDinvestigational new drug application, andpotentiallywhether we should advance one ofourthe SARD compounds into a first-in-human clinical trial.Accordingly, if, for any reason, the proposed Merger is not consummated, we may determine to move forward with our planned IND-enabling studies of our SARD compounds. However, while we believe that our existing capital resources will be adequate to enable us to conductCompetition
The biotechnology and
complete planned IND-enabling preclinical studies of our SARD compounds, we will require significant additional financial resources in order to initiatepharmaceutical industries are intensely competitive andcomplete initial human clinical trials of a SARD compoundcharacterized by rapid technology evolution. Our potential competitors include large pharmaceutical, specialty pharmaceutical andto otherwise further the development of our SARD program.
•Pursue potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets.We have discontinued further development of our SARM program, including enobosarm, and do not currently have any plans to resume development of our SARM program. We continue our efforts to seek potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets.•Pursue another strategic transaction like the proposed Merger.Our board of directors may elect to pursue an alternative strategy, one of which may be a strategic transaction similar to the proposed Merger.•Dissolve and liquidate our assets.If, for any reason, the proposed Merger is not consummated and we are unable to identify and complete an alternative strategic transaction like the Merger or potential collaborative, partnering or other strategic arrangements for our SARM assets, or to continue to operate our business due to our inability to raise additional funding for the development of our SARD program or otherwise, we may be required to dissolve and liquidate our assets. In such case, we would be required to pay all of our debts and contractual obligations, and to set aside certain reserves for potential future claims, and there can be no assurances as to the amount or timing of available cash left to distribute to our stockholders after paying our debts and other obligations and setting aside funds for reserves.
As a result, we may also resume our efforts to seek additional funds through potential collaborative, partnering or other strategic arrangements to provide us with the necessary resources for the development of our SARD program.Our SARD ProgramSARDs for the Potential Treatment of Castration Resistant Prostate CancerScientific Overview.SARDs are a novel class of drugs. The AR is a major driver of prostate tumor cell proliferation, and blocking its activity is a therapeutic target. Despite the use of therapies designed to inhibit the AR pathway in men with advanced prostate cancer, a significant number of men have tumors that do not respond to such therapeutic approaches and/or become resistant to them. This lack of response may be due to the presence of forms of the AR (splice variants and mutated) for which these therapies are not effective. SARDs are designed to not only bind to androgen receptors, but also induce androgen receptor degradation and ultimately inhibit tumor cell growth. Selective AR degradation which targets the N-terminus may be an effective therapeutic strategy where a variant or mutated AR can be degraded by the SARD. This ability to circumvent common drug resistance in prostate cancer patients may provide an important tool for effective new treatments.We believe SARDs have the potential to treat prostate cancer,biotechnology companies, as well as government, academic and otherdiseases such as benign prostatic hyperplasiaresearch institutions. Many of our competitors have significantly greater financial resources andKennedy's disease. We envision initially developing SARDs as a potentially novel treatment for menexpertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements withCRPC, including those who do not respondlarge and established companies. Our commercial opportunities may be reduced or eliminated if our competitors develop and commercialize similar products that are safer, more effective, have fewer side effects or areresistant toless expensive than any products that we or our collaborators may develop.In particular, we compete with other companies that are developing and commercializing treatments for patients with cancer. Competing therapies include chemotherapies, targeted therapies and immunotherapies and may represent various therapeutic modalities including small molecules, antibodies, cell therapies, gene therapies, and cancer vaccines. These companies may compete with us for clinical trial sites and eligible patient populations, scientific and management talent, outsourced manufacturing capacity and healthcare budgets for commercial-stage products.
While there are currently no approved
therapies. Although current therapies have improved overall survivalproducts targeting the ROR1 receptor, we are aware of therapeutics inmen with CRPC, approximately one-thirdclinical development that target ROR1, including an antibody-drug conjugate being developed by VelosBio, Inc., and a ROR1 CAR-T therapy being developed by Juno Therapeutics, Inc., a subsidiary of theCRPC patients do not respond to these therapies, due in part to the presence of splice variants, including AR-V7, as well as mutations in the androgen receptor. Splice variants of the androgen receptor have been identified in which the ligand binding domain, the binding site for androgens and necessaryBristol-Myers Squibb Company.There are numerous companies developing or marketing treatments for the
action of many of the current therapies, is lost. In addition, most patients who initially respond to available treatments eventually progress due to the emergence of resistance to these therapies. It is believedsame oncology indications thatCRPC growth remains highly dependent on androgen receptor activity, although the mechanisms which underlie this resistancewe arenot fully understood. Webelieve a therapeutic agent that would safely degrade multiple forms of the androgen receptor, including those without the ligand binding domain, would be uniquely positioned to address this patient population.Potential Market.In the United States alone, we believe there are approximately 80,000 men who have developed resistance to luteinizing hormone-releasing hormone,targeting with our cirmtuzumab program. Therapies approved orLHRH, therapies and therefore have CRPC but who have not received chemotherapy. We believe there are approximately 36,000 men diagnosed each year with metastatic hormone sensitive prostate cancer. Zytiga® and XTANDI® are currently the only drugs approvedin clinical development for the treatment ofmetastatic CRPC inpatientswho have not yet received chemotherapy, although several otherwith treatment-naïve or relapsed/refractory CLL and relapsed/refractory MCL include BTK inhibitors, Bcl-2 inhibitors, PI3K inhibitors, anti-CD20 antibodies, and cell therapies that are being marketed or developed by companies including AbbVie, Inc., ArQule, Inc., AstraZeneca PLC, BeiGene, Ltd., Eli Lilly and Company, Gilead Sciences, Inc., Johnson & Johnson, Merck KGaA, Novartis Pharmaceuticals Corporation, Roche Holding AG’s Genentech subsidiary, TG Therapeutics, Inc., and Verastem, Inc.While there are currently no approved drugs designed to target ETS oncoproteins, there are numerous companies developing or marketing treatments for the same oncology indications that we are targeting with our TK216 program. Investigational therapies in clinical development for
this indication. We believe new hormonal therapies in development, if approved, will be used prior to chemotherapy as physicians and patients look forthe treatmentoptions capable of delaying cancer progression and possibly prolonging survival prior to chemotherapy.Preclinical Development.We are in the process of completing ongoing mechanistic preclinical studies in order to select the most appropriate SARD compounds to move forward into the additional preclinical studies required to submit an IND and potentially advance one of our SARD compounds into a first-in-human clinical trial. However, while we believe that our existing capital resources will be adequate to enable us to conduct and complete planned IND-enabling preclinical studies of our SARD compounds, we will require significant additional financial resources in order to initiate and complete initial human clinical trials of a SARD compound and to otherwise further the development of our SARD program. Accordingly, if, for any reason, the proposed Merger is not consummated, we may resume our efforts to seek additional funds through potential collaborative, partnering or other strategic arrangements to provide us with the necessary resources for the development of our SARD program.Evaluation of Enobosarm for the Treatment of Postmenopausal Women with SUI.In the third quarter of 2017, we initiated the ASTRID trial at over 60 clinical trial centers in the United States to evaluate the change in the mean number of daily SUI episodes following 12 weeks of enobosarm treatment. The ASTRID trial evaluated the safety and efficacy of enobosarm (1 mg and 3 mg) compared with placebo in post-menopausal women who have demonstrated SUI symptoms for more than six months, with an average of 3 to 15 reported SUI episodes per day over a three-day period, and a positive bladder stress test. The primary endpoint for the ASTRID trial was the percentageof patients withat least a 50 percent reduction in mean leaks per day at week 12, compared to baseline. During the third quarter of 2018, we announcedrelapsed/refractory Ewing sarcoma include kinase inhibitors, LSD1 inhibitor and other targeted therapies, therapeutic antibodies and cell therapies thatthe ASTRID trial failed to achieve statistical significance on the primary endpoint of the proportion of patients with a greater than 50% reduction in incontinence episodes per day compared to placebo. The percentage of patients with a greater than 50% reduction after 12 weeks of enobosarm treatment was 58.9% for 3 mg, 57.7% for 1 mgare being developed by companies including Bayer AG, Bristol-Meyers Squibb Company, Eisai Co., Ltd., Epizyme, Inc., Gradalis, Inc., Eli Lilly and52.7% for placebo. Enobosarm was generally safeCompany, Johnson & Johnson, Exilixis, Inc., NantCell, Inc., Pharmamar S.A., Pfizer, Inc., Salarius Pharmaceuticals, Inc., Takeda Pharmaceutical Company Limited, andwell tolerated, and reported adverse events were minimal and similar across all treatment groups. We have completed the ASTRID trial, including our review of the full data sets from the clinical trial, and have determined that there is not a sufficient path forward to warrant additional clinical development of enobosarm to treat SUI. We have therefore discontinued further development of enobosarm to treat SUI, including discontinuing the related durability and open-label safety extension studies we initiated before we received topline data from the ASTRID trial.others.Evaluation of Enobosarm for the Treatment of Breast Cancer.We have previously evaluated enobosarm in a Phase 2 clinical trial designed to evaluate the efficacy and safety of a 9 mg and 18 mg dose of enobosarm in patients whose advanced breast cancer is both estrogen receptor, or ER, positive and AR positive. We announced in November 2016 that enobosarm achieved the pre-specified primary efficacy endpoint in the 9 mg dose cohort with 9 patients achieving a clinical benefit response, or CBR,defined as a complete response, partial response, or stable disease, among the first 22 evaluable patients in that cohort. In November 2017, we announced that in the 9 mg cohort, a total of 14 patients achieved a CBR following 24 weeks of treatment. We also announced in November of 2017 that the 18 mg cohort achieved the pre-specified primary efficacy endpoint as 12 patients achieved a CBR at 24 weeks. Although both the 9 mg and 18 mg cohorts met the primary efficacy endpoint in the Phase 2 clinical trial, after evaluating the breast cancer environment where the treatment paradigms are shifting to immunotherapies and/or combination therapies, we decided in the third quarter of 2017 that the time and cost of conducting the necessary clinical trials for potential approval in this indication does not warrant further development of enobosarm in this indication. In 2015, we also commenced enrollment in a Phase 2 proof-of-concept clinical trial designed to evaluate the efficacy and safety of an 18 mg dose of enobosarm in patients with advanced AR positive triple-negative breast cancer, or TNBC. This clinical trial was conducted utilizing a Simon's two-stage trial design whereby if at least 2 of the first 21 patients achieved clinical benefit, the trial was designed to enroll the second stage, which would result in enrolling 41 evaluable patients in the clinical trial. During the third quarter of 2017, we completed our review of the data from the first stage of the clinical trial. While our review of the data did not raise any safety concerns, it did confirm that there were insufficient patients achieving clinical benefit from enobosarm treatment to continue this clinical trial and we closed the clinical trial down.Discontinuation of SARM Development Efforts.Following our review of the full data sets from the ASTRID trial, we discontinued further development of enobosarm to treat SUI and otherwise discontinued any further development of our SARM program. We continue our efforts to seek potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets. If the Merger is completed, any net proceeds derived from the disposition or licensing of our SARM assets following completion of the Merger will be made available to our stockholders in accordance with the CVR Agreement. We have for many years actively pursued, but have been unable to successfully enter into, potential collaborative, partnering or other strategic arrangements for our SARM assets. If we are unable to ultimately enter into any such arrangements for our SARM assets, we will not receive any return on our investment in enobosarm and our other SARMs.
Licenses and Collaborative RelationshipsWe have in the past established and, if the proposed Merger is not completed, we may continue to pursue, in-licenses and partnering, and collaborative or other strategic relationships with academic institutions and with other pharmaceutical and biotechnology companies.UC San DiegoIn March
2015,2016, weand UTRFentered into a license agreement with the Regents of the University of California (the “Regents”), represented by UC San Diego, which was amended and restated in August 2018, and amended on March 25, 2019 and May 15, 2019 (the “UC San Diego License Agreement”), for the development, manufacturing and distribution rights to naked antibodies, including cirmtuzumab and genetically engineered cellular therapy products, including CAR-T products that are covered by licensed patents for all human therapeutic, diagnostic and preventive applications in all indications. Under the UC San Diego License Agreement, we paid an upfront license fee of $0.5 million and issued 107,108 shares of common stock. Commencing in 2017, we also pay UC San Diego an annual license maintenance fee and reimburses to UC San Diego its annual patent costs for the licensed patents. The UC San Diego License Agreement also requires the payment of certain development and regulatory milestones, aggregating from $10.0 million to $12.5 million, on a per product basis, certain worldwide sales milestones based on achievement of tiered revenue levels aggregating $75.0 million, low single-digit royalties including potential future minimum annual royalties on net sales of each product, reimbursement of certain annual patent costs, and requires certain minimum diligence efforts to advance the licensed assets, including spending at least $1.0 million in development annually through 2021. Unless terminated earlier, the UC San Diego License Agreement will expire upon the later of the expiration date of the longest-lived patent rights or theSARD15th anniversary of the first commercial sale of a licensed product. UC San Diego may terminate the UC San Diego License Agreement if a material breach by us is not cured within a reasonable time, we file a claim asserting the licensed patent rights are invalid or unenforceable, or we file for bankruptcy. We may terminate the agreement at any time upon at least 90 days’ written notice. In July 2016, we entered into a research agreement with UC San Diego (the “UC San Diego Research Agreement”), for further research on the ROR1 therapeutic development program. Under this five-year agreement, UC San Diego will have an aggregate budget of $3.6 million, with $125,000 payable quarterly. The costs paid to UC San Diego under the UC San Diego Research Agreement are included as part of our annual diligence obligations under the UC San Diego License Agreement.CIRM
In August 2017, CIRM awarded an $18.3 million grant to researchers at UC San Diego to advance our Phase 1/2 clinical trial evaluating cirmtuzumab in combination with ibrutinib for the treatment of patients with B-cell lymphoid malignancies, including MCL and CLL. We are conducting the trial in collaboration with UC San Diego, and we are responsible for study conduct and data management. We estimate we will receive approximately $14.0 million in development milestones under research subaward agreements throughout the award project period, estimated to be from October 1, 2017 to March 31, 2022. We are required to provide UC San Diego progress and financial update reports throughout the award period. The subaward does not bear a royalty payment commitment, nor is the subaward otherwise refundable. As of December 31, 2019, we believe we have met our obligations under the CIRM award and UC San Diego subawards.
In October 2017, CIRM awarded a $5.8 million grant to the researchers at UC San Diego to develop a novel anti-cancer stem cell targeted therapy for patients with advanced solid and hematological malignancies. In connection with such CIRM award, we agreed to provide up to $1.0 million in contingency funds if required during the grant period.
CIRM may suspend or permanently cease disbursements of funds under the research subaward agreements, or pursue other remedies as allowed by law, if CIRM determines that UC San Diego has not complied with the terms and conditions of the award, or if there are unexpected, substantial manufacturing failure leading to delayed enrollment in the clinical trial, failure to enroll the trial, or if FDA issues a clinical hold order with respect to the clinical trial.
Georgetown University
In March 2014, we entered into an exclusive license agreement (the “Georgetown License Agreement”), with Georgetown University, or Georgetown, pursuant to which we licensed the exclusive worldwide right to patents and technologies for the development and commercialization of certain product candidates targeting EWS-FLI1 as an anti-tumor therapy for therapeutic, diagnostics, or research tool purposes. Under the Georgetown License Agreement, we are solely responsible for all development and commercialization activities and costs in our respective territories, and are also responsible for all costs related to the filing, prosecution and maintenance of the licensed patent rights. Commencing in 2015, we are obligated to pay Georgetown an annual license maintenance fee until the first commercial sale occurs, make up to $0.2 million in aggregate milestone payments upon the achievement of certain regulatory milestones, and will be required to pay low single digit royalties based on annual net product sales. The term of the Georgetown License Agreement continues until the expiration of the last valid claim within the patent rights covering the product, but may be terminated by either party upon material breach, or by us as to one or more countries with 90 days written notice of termination. Additionally, Georgetown may terminate the agreement in the event we fail to pay any amount and fails to cure such failure within 30 days after receipt of notice, defaults in our obligation to obtain and maintain insurance and fails to remedy such breach within 60 days after receipt of notice, or declares insolvency or bankruptcy. We may terminate the agreement at any time upon at least 60 days’ written notice.
Shanghai Pharmaceutical (USA) Inc. (“SPH USA”)
In November 2018, we entered into the SPH USA License Agreement, with SPH USA under which we granted exclusive rights to SPH USA to manufacture, develop, market, distribute and sell in the People’s Republic of China, Hong Kong, Macau, and Taiwan (the “SPH USA Territory”), our product candidates under the Georgetown License Agreement and the UC San Diego License Agreement. Under the SPH USA License Agreement, SPH USA is solely responsible for all pre-clinical and clinical development activities specific to obtaining regulatory approval for such product candidates in the SPH USA Territory, any third-party license milestone or royalty payments owed under the Georgetown License Agreement and the UC San Diego License Agreement, and paying us a low single digit royalty on net sales of licensed products in the SPH USA Territory. The SPH USA License Agreement will expire on a licensed product-by-licensed product and country/region-by-country/region basis on the later of ten years from the date of first commercial sale or when there is no longer a valid patent claim covering such licensed product in such country/region. The SPH USA License Agreement may be terminated by SPH USA, on a country/region-by-country/region or product-by-product basis with 180 days written notice following the first anniversary of the effective date of the agreement or at any time on a product-by-product basis for a safety concern with respect to such product. Either party may terminate the SPH USA License Agreement in its entirety or on a licensed product-by-licensed product basis upon material breach that is not cured within 90 days, or in its entirety the event the other party becomes insolvent or enters into bankruptcy proceedings. We may terminate the agreement with 60 days written notice if SPH USA or its affiliates or sublicensees commence an action challenging the validity or enforceability of any licensed patent, or with 10 days written notice if SPH USA fails to own at least 20% of the voting securities of any assignee of the SPH USA License Agreement. Upon termination of the agreement for any reason all rights and licenses granted to SPH USA under the agreement will terminate, and in the event of termination for reasons other than our material breach, SPH USA would grant us non- exclusive, royalty-free, worldwide license to any intellectual property rights controlled by SPH USA or its affiliates to exploit the terminated program in the SPH USA Territory.
In May 2014, ROAR Therapeutics, Inc., our predecessor company, entered into a commercial license agreement (the “Selexis License Agreement”), with Selexis, S.A., a Swiss company, pursuant to which we obtained a world-wide, non-exclusive license under certain of Selexis’ patents and technology rights to use a recombinant cell line produced using the Selexis technology to produce cirmtuzumab. Under the terms of the Selexis License Agreement, we will pay Selexis milestone payments totaling, in the aggregate, CHF 1,235,000, and a royalty in the low single digits on net sales of cirmtuzumab to third parties. The Selexis License Agreement remains in effect until the last to expire of the licensed Selexis patents, but may be terminated by either party if the other party materially breaches the agreement and fails to cure the breach within sixty days after receipt of a notice of default from the other party, or in the event the other party becomes insolvent or enters into bankruptcy proceedings. Additionally, we may terminate the Selexis License Agreement and the license granted therein at any time upon sixty days prior written notice to Selexis. In May 2015, Selexis’ rights to receive future milestone payments and royalties under the Selexis License Agreement were assigned to Ligand Pharmaceuticals, Incorporated. In February 2020, the Selexis License Agreement was amended to authorize us to grant a sublicense to VelosBio, Inc.
University of Tennessee Research Foundation (“UTRF”)
In March 2015, we entered into a license agreement with UTRF (the “SARD License Agreement”), pursuant to which we were granted exclusive worldwide rights in all
existingproprietary selective androgen receptor degrader, or SARD, technologies owned or controlled by UTRF, including all improvements thereto. Under the SARD License Agreement, we are obligated to employ active, diligent efforts to conduct preclinical research and development activities for the SARD program to advance one or more lead compounds into clinical development. We are also obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and additional royalties on sublicense revenues, depending on the state of development of a clinical product candidate at the time it is sublicensed. Unless terminated earlier, the term of the SARD License Agreement will continue, on a country-by-country basis, until the expiration of the last valid claim of any licensed patent in the particular country in which a licensed patent is granted. UTRF may terminate the SARD License Agreement for our uncured breach or uponourits bankruptcy. On January 30, 2020, the SARD License Agreement was amended to extend certain diligence milestones.In July 2007, we
and UTRF also previouslyentered into a consolidated, amended and restated license agreementor the SARMwith UTRF (the “SARM LicenseAgreement,Agreement”), to consolidate and replaceourtwo previouslyexisting SARM license agreements with UTRF and to modify and expand certain rights and obligations of each of the parties under both license agreements. Pursuant to the SARM License Agreement, we were granted exclusive worldwide rights in all existing selective androgen receptor modulator, or SARM, technologies owned or controlled by UTRF, including enobosarm, and certain improvements thereto, and exclusive rights to certain future SARM technology that may be developed by certain scientists at the University of Tennessee or subsequently licensed to UTRF under certain existing inter-institutional agreements with The Ohio State University.
Unless terminated earlier, the termIn 2018, we disclosed that we had ceased its development of the SARMLicense Agreement will continue, ontechnology following the failure of acountry-by-country basis, forPhase 2 clinical study of enobosarm to achieve statistical significance with respect to thelonger of 20 years or until the expirationprimary endpoint of thelast valid claimstudy. On December 31, 2019, we notified UTRF ofany licensed patent in the particular country in which a licensed product is being sold. UTRF mayour intent to terminate the SARM License Agreement,for our uncured breach or upon our bankruptcy.Underwhich termination will be effective as of March 31, 2020. Following termination, we will no longer have the obligation to make further payments under the SARM License Agreement,we paid UTRF a one-time, upfront feeincluding payments for patent prosecution and maintenance, and will no longer have any rights to develop or sublicense the SARM Technology. We will not incur any early termination penalties due to the termination of$290,000 as consideration for entering intothe SARM License Agreement.Manufacturing
We
are also obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net saleshave adopted a manufacturing strategy ofproducts and mid-single-digit royalties on sublicense revenues. We also agreed to pay all expenses to file, prosecute and maintain the patents relating to the licensed SARM technologies, and are obligated to use commercially reasonable efforts to develop and commercialize products based on the licensed SARM technologies. While we currently have ceased development efforts for SARMs, we continue to seek potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets. In December 2008, we and UTRF amended the SARM License Agreement, or the SARM License Amendment, to, among other things, clarify the treatment of certain payments that we may receive from our current and future sublicensees for purposes of determining sublicense fees payable to UTRF, including the treatment of payments made to uscontracting with third parties inexchangeaccordance with cGMP for thesalemanufacture of drug substance and product, and additional manufacturers are used to label, package and distribute investigational drug products. This strategy allows us to maintain a more flexible infrastructure while focusing oursecurities in connection with sublicensing arrangements. In consideration for the execution of the SARM License Amendment, we paid UTRF $494,000.expertise on developing our products.We
do not currently own or operate manufacturing facilities, and we rely, andexpect to continue to rely on third parties for the production of clinical and commercial quantities of any product candidates.There are no unusually complicated
chemistriesbiochemistries or unusual equipment required in the manufacturing process for eitherSARMscirmtuzumab orSARDs. We rely and expect to continue to rely on third-party vendors for drug substance and drug product manufacturing, including drug substance for SARDs used in our current and potential future preclinical studies.The biotechnology and biopharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We face competition from many different sources, including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies and private and public research institutions.Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Our commercial opportunities will be reduced or eliminated if our competitorsTableWe have established a quality control and quality assurance program, which includes a set ofContentsstandard operating procedures and specifications designed to ensure that our products are manufactured in accordance with cGMPs, and other applicable domestic and foreign regulations.developIntellectual PropertyWe strive to protect and
commercialize similar productsenhance the proprietary technology, inventions, and improvements that aresafer, more effective, have fewer side effectscommercially important to our business, including seeking, maintaining, and defending patent rights, whether developed internally orare less expensive than any products that we and/acquired orour collaborators may develop.SARDs for the Potential Treatment of CRPCWe have entered into an exclusive worldwide license agreement with UTRFlicensed from third parties. Our policy is todevelop its proprietary SARD technology which we believe has the potential to provide compounds that can degrade or antagonize multiple forms of the AR thereby inhibiting tumor growth in patients with CRPC, including those patients who do not respond or are resistant to current therapies. Drugs in development having potentially similar approaches to removing the AR by degradation include Arvinas Inc.'s ARV-110, which is a chimera with an AR binding moiety on one end and an E3 ligase recruiting element on the other that has recently entered Phase 1 development for the treatment of advanced prostate cancer, and Androscience Corporation's androgen receptor degrader enhancer, ASC-J9, which is currently in development for acne and alopecia with the potential for development as a treatment for prostate cancer. Additionally, Essa Pharma Inc. recently completed a Phase 1 study with EPI-506, an AR antagonist that targets the N-terminal domain of the AR, and has plans to develop a second generation agent. C4 Therapeutics, Inc. is developing degronimids as means to degrade the AR through the ligand binding domain associated degradation. CellCentric is developing therapies that target the histone methyltransferase enzyme to lower AR levels, and recently initiated a clinical trial with CCS1477 in prostate cancer. Oric Pharmaceuticals is targeting the glucocorticoid receptor as a means to impact men that have CRPC, and has a lead candidate ORIC-101 in preclinical testing. In addition to this specific potential mechanistic competition, there are various products approved or under clinical development in the broader space of treating men with advanced prostate cancer who have metastatic CRPC which may compete with our proposed initial clinical objective for our SARD compounds. Pfizer and Astellas Pharma market XTANDI® (enzalutamide), an oral androgen receptor antagonist, for the treatment of metastatic CRPC in men previously treated with docetaxel as well as those that have not yet received chemotherapy. XTANDI® received FDA approval in July 2018 for the treatment of men with non-metastatic CRPC. Zytiga®, sold by Johnson & Johnson, has been approved for the treatment of metastatic CRPC and metastatic high-risk castration-sensitive prostate cancer. Johnson & Johnson also received FDA approval for a second generation anti-androgen ERLEADA (apalutamide) for the treatment of men with non-metastatic castrate-resistant prostate cancer. Bayer HealthCare and Orion Corporation recently announced that the primary endpoint of increased metastatic free survival was met in a Phase 3 study of darolutamide (ODM-201) in men with CRPC without metastases and with a rising PSA. Another target in prostate cancer that is being pursued by several companies is bromodomain inhibition. Zenith Epigenetics, Gilead Sciences Inc., CellCentric, Incyte Corporation and GlaxoSmithKline are among the companies that are evaluating BET inhibitors in Phase 1-2 trials.With respect to SARMs, there are other SARM product candidates in development that may compete with enobosarm and any future SARM product candidates, if approved for commercial sale. For example, Viking Therapeutic's VK5211 recently reported positive results from a Phase 2 study for patients recovering from non-elective hip fracture surgery. Radius Health Inc.'s RAD140 is currently being evaluated in a Phase 1 study in postmenopausal women with hormone-receptor positive locally advanced or metastatic breast cancer. GlaxoSmithKline is conducting a Phase 1 study to assess the effect of GSK2881078 on physical strength and function after 13 weeks of treatment in patients with chronic obstructive pulmonary disease, or COPD, and muscle weakness. OPKO Health's OPK88004 is enrolling in a dose ranging study to improve symptoms of benign prostatic hyperplasia (BPH) byreducing prostate size and, on the basis of data from a previous trial in 350 men, increase muscle mass and bone strength and decrease body fat.We will be ableseek to protect ourtechnology from unauthorized useproprietary position by,third parties only to the extent it is covered by valid and enforceable patents or is effectively maintained as trade secrets. Patents andamong otherproprietary rights are an essential element of our business.For our SARD compounds andmethods,of use thereof, we have filed certainfiling patent applications in the United StatesCanada, Mexico, Australia, Japan, China,andother countriesinAsia and before the European Patent Office and are the exclusive licensee of worldwide rights for the SARD technology under a license agreement with UTRF executed in 2015. Thus far we have six issued patents and one is allowed, all in the United States. The patents and patent applications (if are issued) will expire between 2036 and 2039.For enobosarm and our other SARM compounds, we have an exclusive license from UTRF under its issued patents and pending patent applications in the United States, Canada, Australia, Japan, China and other countries in Asia, before the European Patent Office designating Germany, Great Britain, Spain, France, Italy, and other European Union countries, as well as in certain other countries outside those regions, covering the composition of matter of the active pharmaceutical ingredient for pharmaceutical products, pharmaceutical compositions and methods of synthesizing the active pharmaceutical ingredients. We have also exclusively licensed from UTRF issued and pending patent applications in the United States, Canada, Australia, Japan, China and other countries in Asia, before the European Patent Office designating Germany, Great Britain, Spain, France, Italy and other European Union countries, as well as in certain other countries outside those regions, related to methods for treating muscle wasting disorders, including Duchenne Muscular Dystrophy, or DMD, and cancer cachexia, and for treating conditions such as SUI and fecal incontinence, as well as sarcopenia, and increasing muscle performance, muscle size and muscle strength and increasing the strength of or mass of a bone and for treating bone related disorders, including bone frailty and osteoporosis. Issued patents for enobosarm composition of matter that we licensed from UTRF and issued in the United States expire in 2024. Issued patents for composition of matter for our other SARM compounds in the United States will expire from 2021-2029, depending on the specific SARM compound. The issued patentsjurisdictions outside of the United States related to our proprietary technology, inventions, and improvements that are important to the development and implementation of our business. We also rely on trade secrets and know-how relating to our proprietary technology, continuing innovation, and acquisition and in-licensing opportunities to develop, strengthen, and maintain our proprietary position in the field of cancer therapeutics.Our commercial success may depend in part on our ability to (i) obtain and maintain patent and other proprietary protection for
enobosarm expire in 2025,our technology, inventions, andwith respect toimprovements; (ii) preserve the confidentiality of our trade secrets; (iii) defend and enforce our proprietary rights, including our patents; and (iv) operate without infringing the valid and enforceable patents and otherSARM compounds, expire in 2023 and 2027, depending on the specific SARM compound.proprietary rights of third parties.We have developed, licensed and acquired numerous patents and patent applications and possess substantial know-how and trade secrets relating to the development and commercialization of healthcare products and services. As of February 19, 2020, our owned and in-licensed patent portfolio consisted of approximately 28 issued U.S. patents and 21 pending U.S. patent applications related to certain of our proprietary technology, inventions, and improvements, and 30 issued patents and 120 pending patent applications in jurisdictions outside of the United States.
ROR1 Program
We have an exclusive, commercial, worldwide, transferrable license to a portfolioofpatentsandpatentapplicationsdirected to ROR1 antibodiesand CAR-T therapies for alltherapeuticindications.Thisportfolio is licensed from theRegents of the University of California. We have know-how and trade secrets related to compositions of matter for treating cancers, methods for treating cancer, and methods of screening for additionalcompositionsof matter used for treating cancer, as well as to additionalantibodiesandmoleculesthatmodulate ROR1 signaling, and we have filed one pending provisional application in the U.S. related to cirmtuzumab.
As of February 19, 2020, our licensed patent portfolio included patents related to our clinical candidate currently in phase 1/2 clinical trials, cirmtuzumab. Cirmtuzumab is a humanizedmonoclonalantibodythatspecificallybinds to the ROR1 receptor. We have two issued U.S. patents directed to the cirmtuzumab compositionof matter: U.S. Pat. No. 9,217,040, with a patent term not due to expire before 2032;and U.S. Patent No. 9,758,591, with a patent term not due to expire before March 2033. We have one patent issued in the U.S. directed to methods of using cirmtuzumab to treat cancer, U.S. Pat, No. 10,344,096. We have one patent application pending in the U.S. related to single chain variable region fragments that bind to cirmtuzumab which, if issued, would have a patent term not due to expire before 2033. We also have patents issued in Australia, China, Europe, Israel, Japan, Korea, Macao and Mexico directed to the cirmtuzumab compositions of matter. We have approximately 14 pending applications in foreign jurisdictions related to the cirmtuzumab composition of matter and methods of use in treating cancer, including Australia, Canada, China, Europe, Japan, Korea, Malaysia, Mexico, Philippines, and Thailand. Patents, if issued from these pending foreign applications, would not be due to expire before 2033.
As of February 19, 2020, we have patent applications pending in the U.S. and in 17 jurisdictions outside the U.S. related to methods of treating cancer using a combination of cirmtuzumab and small-molecule chemotherapeutics. Patents, if issued from these pending non-provisional applications, would not be due to expire before dates ranging from 2037 to 2039.
As of February 19, 2020, we have patents and patent applications related to additional ROR1 binding antibodies and chimeric antigen receptor T cells specific for
our other SARM compounds that,ROR1. We have four issued U.S. patents directed to non-cirmtuzumab antibodies: U.S. Pat. No. 8,212,009, with a patent term not due to expire before November, 2026; U.S. Patent No. 9,242,014, with a patent term not due to expire before June 2031; U.S. Patent No. 9,938,350, with a patent term not due to expire before June 2031; and U.S. Patent No. 9,217,040, with a patent term not due to expire before January 2032. We have two patent applications pending in the U.S. related to additional non-cirmtuzumab ROR1 binding antibodies and non-cirmtuzumab chimeric antigen receptor T cells specific for ROR1, which, if issued, would have a patent term not due to expire before dates ranging from 2031 to 2032. We also have patents issued in Europe and Canada directed to additional ROR1 binding antibodies. We have patent applications pending in Europe and Canada related to additional ROR1 binding antibodies and single-chain variable region fragments specific for ROR1. Patents, if issued from these pending foreign applications, would not be due to expire before 2032.As of February 19, 2020, we have intellectual property related to methods of screening for antibodies that specifically bind to ROR1. We have two issued U.S. patents, U.S. Pat. Nos. 9,523,695, and 9,933,434, with patent terms not due to expire before January 2032, directed to methods of screening for antibodies that specifically bind to ROR1. We additionally have patent applications pending directed to methods of screening for modulators of ROR1 signaling in jurisdictions including the U.S., Australia, Canada, China, Hong Kong, Japan, and Europe.
TK216 Program
We have exclusive worldwide rights to a portfolio of patents and patent applications related to small molecules, including TK216, targeting EWS-FLI1 for use in therapeutics and companion diagnostics. We hold a portfolio of patents and patent applications, the Oncternal Portfolio, related to TK216, analogs thereof, and uses thereof, as well as the Georgetown Licensed Portfolio, which is licensed from Georgetown University.
As of February 19, 2020, the Oncternal Portfolio consisted of approximately six U.S. issued patents and three pending applications in the U.S., as well as approximately three patents and approximately 36 pending patent applications in jurisdictions outside of the U.S. As of February 19, 2020, we had two U.S. patents directed to TK216: U.S. Pat. No. 9,604,927, with a patent term not due to expire before October 2035, and U.S. Pat. No. 9,987,251, with a patent term not due to expire before October 2035. We also had a patent with claims directed to methods of inhibiting proliferation of a cell that overexpresses an ETS gene or comprises an ETS fusion gene, or inhibiting growth of or killing neoplastic cells: U.S. Pat. No. 9,895,352, with a patent term not due to expire before October 2035. We had approximately one pending U.S. application and approximately 18 patents or pending applications in jurisdictions outside the U.S., including Australia, Argentina, Canada, China, Eurasia, Europe, Hong Kong, India, Israel, Japan, Korea, Mexico, New Zealand, Pakistan, and Taiwan. These patents have a patent term not due to expire before October 2035, and patents, if issued from these applications, would not be due to expire before October 2035. We also had a patent with claims covering compositions of TK216 in combination with venetoclax and associated methods of inducing apoptosis in cells in AML and DLBCL: U.S. Pat. No. 10,159,660, with a patent term not due to expire before July 2037. We had approximately one pending U.S. application and approximately 13 pending applications filed in jurisdictions outside the U.S., including Argentina, Canada, China, Europe, Hong Kong, Indonesia, Japan, Korea, Mexico, Malaysia, Philippines, Singapore, and Taiwan. Patents, if issued from these applications, would not be due to expire before July 2037. The Oncternal Portfolio further contained additional patents and pending applications related to indoline derivative compounds, which are analogs of TK216. We had two issued U.S. patents directed to compounds and methods of inhibiting proliferation of a cell expressing an ETS gene or comprising an ETS fusion gene: U.S. Pat. No. 9,822,122, with a patent term not due to expire before March 2037, and U.S. Pat. No. 10,351,569, with a patent term not due to expire before March 2037. There was one pending U.S. application and approximately eight applications pending outside the U.S. in Argentina, Pakistan, Taiwan, China, Europe, Japan, Korea, and Malaysia. Patents, if issued from these applications, would not be due to expire before March 2037.
As of February 19, 2020, the Georgetown Licensed Portfolio contained patents directed to other EWS-FLI1 inhibitor compounds. We had three U.S. patents directed to compounds and methods for treating Ewing sarcoma or pancreatic cancer: U.S. Pat. No. 8,232,310, with a patent term not due to expire before November 2028, U.S. Pat. No. 9,045,415, with a patent term not due to expire before August 2028, and U.S. Pat. No. 9,758,481, with a patent term not due to expire before December 2027. We had four issued patents in jurisdictions outside the U.S., including Australia, Canada, Europe, and Hong Kong. These patents are not due to expire before December 2027. We had two issued U.S. patents directed to compounds and methods for treating pancreatic cancer or Ewing sarcoma: U.S. Pat. No. 9,290,449, with a patent term not due to expire before April 2033, and U.S. Pat. No. 9,714,222, with a patent term not due to expire before April 2033. There are approximately 16 patents or pending applications outside the U.S. in Australia, Canada, China, Europe, Hong Kong, Israel, India, Japan, Korea, Mexico, and New Zealand. These patents have a patent term not due to expire before April 2033, and patents, if issued from these applications, would not be due to expire before April 2033. The Georgetown Licensed Portfolio contained additional patents and pending applications related to methods of treating cancers. We had one issued U.S. patent directed to methods of treating lung cancer or glioblastoma multiforme: U.S. Pat. No. 9,511,050, with a patent term not due to expire before October 2034. There were approximately two patents issued outside the U.S. in China and Japan. These patents have a patent term not due to expire before October 2034.
SARD Program
We have exclusive worldwide rights to a portfolio of patents and patent applications related to Selective Androgen Receptor Degrader (SARD) compounds for use in therapeutics. We hold a portfolio of patents and patent applications related to SARDs and jointly owned with University of Tennessee Research Foundation, including seven issued U.S. patents directed to SARD ligands and methods of use thereof: U.S. Pat. No. 9,814,698, U.S. Pat. No. 10,017,471, U.S. Pat. No. 10,035,763, U.S. Pat. No. 10,441,570, U.S. Pat. No. 9,815,776, U.S. Pat. No. 9,834,507, and U.S. Pat. No. 10,093,613, with a patent term not due to expire before April 2036. We also had a portfolio of patents and patent applications licensed from University of Tennessee Research Foundation including one issued U.S. patent directed to SARD ligands and methods of use thereof: U.S. Pat. No. 10,314,797 with a patent term not due to expire before June 2037.
Individual patents extend for varying periods of time, depending upon the date of filing of the patent application, the date of patent issuance, and the legal term of patents in the countries in which they are obtained. Generally, patents issued for applications filed in the United States are effective for 20 years from the earliest effective and non-provisional filing date. The patent term may be adjusted to compensate for delayed patent issuance, when such delays are caused by the patent office or successful appeals against patent office actions. There is no limit on this patent term adjustment. In addition, in
countries outsidecertain instances, a patent term can be extended to recapture a portion of theUnited States in 2027. We have issued patents in the United States, and issued patents and pending applications in countries outside the United States for enobosarm and certain other SARM compoundsterm effectively lost as afeed composition for animals.result of the FDA regulatory review period. Thepatents inextended restoration period cannot be longer than five years and theUnited States will expire in 2025. Issuedtotal patent term, including the restoration period, must not exceed 14 years following the date of FDA approval of the applicable drug product. The duration of patentsoutside the United States, and patent applications, if issued, which are pending outside the United States, will expire in 2027 or 2031 depending on the country. Patent applications which are pending in the United States and outside the United States using SARMs for SUI and pelvic floor disorders will expire in 2035, if the patents are issued. Our issued patent in the United States using enobosarm for DMD will expire in 2021. Our issued patent in the United States using other SARMs for DMD will expire in 2024. Patent applications, if issued, which are pending in the United States, using other SARMs for DMD will expire in 2024 or 2027 depending on the SARM.We have our own issued patents and pending patent applications in the United States, Canada, Australia, Europe, Japan, China and other countries in Asia, as well as in certain other countries outside those regions, related to solid forms of enobosarm. Issued patents covering solid forms ofenobosarm in the United States will expire in 2029. Issued patents and pending patent applications, if issued, in countriesoutside of the United Stateswillvaries in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective non-provisional filing date. Our issued patents are due to expirein 2028. We haveon dates ranging from 2026-2037. If patents are issued on ourownpending patent applications, the resulting patents would be due to expire on dates ranging from 2026-2040. However, the actual protection afforded by a patent varies on a product-by-product basis, from country-to-country, andissueddepends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country, and the validity and enforceability of the patent. Most countries require a patent owner to pay maintenance fees or annuities in order to extend the patent to the full length of its term. If these fees and annuities are not paid timely, our patents will expire prior to the expiration dates set forth herein.Government authorities in the United States, at the federal, state and
in Europe, Canada, Australia, Japan, Chinalocal level, and other countriesin Asia related to methodsextensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record‑keeping, promotion, advertising, distribution, marketing and export and import oftreating breast cancer using our SARM compounds. Such patentsproducts such as those we are developing. A new drug must be approved by the FDA through the NDA process andpatent applications, if issued, would expire in 2033a new biologic must be approved by the FDA through the BLA process before it may be legally marketed in the United States.United States
and outside of the United States. We have issued patents inDrug Development ProcessIn the United States,
directed to androgen receptor positive breast cancerthe FDA regulates drugs under the federal Food, Drug, and Cosmetic Act, or FDCA, and ingeneral, various categoriesthe case ofestrogen receptorbiologics, also under the Public Health Service Act, or “PHSA”, andandrogen receptor positive breast cancer, as well as triple negative breast cancer.We cannot be certain that anytheir implementing regulations. The process ofour pending patent applications, or those of UTRF, will result in issued patents. In addition, because the patent positions of biopharmaceutical companies are highly uncertain and involve complex legal and factual questions, the patents we own and license, or any further patents we may own or license, may not prevent other companies from developing similar or therapeutically equivalent products. Patents also will not protect our product candidates if competitors devise ways of making or using these product candidates without legally infringing our patents. In recent years, several companies have been extremely aggressive in challenging patents covering pharmaceutical products,obtaining regulatory approvals and thechallenges have often been successful. We cannot be assured that our patents will not be challenged by third parties or that we will be successful in any defense we undertake.subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure tosuccessfully defendcomply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, apatent challenge could materially and adversely affect our business.In addition, changes in patent laws, rulesclinical hold, warning letters, product recalls, product seizures, total orregulationspartial suspension of production orin their interpretations in the United States and other countries by the courts may materially diminish the valuedistribution, injunctions, fines, refusals ofour intellectual propertygovernment contracts, restitution, disgorgement ornarrow the scope of our patent protection, whichcivil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect onour business and financial condition.us.We also rely on trade secrets, technical know-how and continuing innovation to develop and maintain our competitive position. We seek to protect our proprietary informationThe process required byrequiring our employees, consultants, contractors, outside scientific collaborators and other advisors to execute non-disclosure and confidentiality agreements and our employees to execute assignment of invention agreements to us on commencement of their employment. Agreements with our employees also prevent them from bringing any proprietary rights of third parties to us. We also require confidentialitythe FDA before a drug ormaterial transfer agreements from third parties that receive our confidential data or materials.New Drug Development and Approval ProcessNumerous governmental authoritiesbiologic may be marketed in the United States generally involves the following:completion of preclinical laboratory tests, animal studies and formulation studies in accordance with GLP requirements and other
countries extensively regulate the testing, clinical development, manufacturing and marketing of pharmaceutical products and ongoing research and development activities. In the United States, the FDA rigorously reviews pharmaceutical products under the Federal Food, Drug, and Cosmetic Act andapplicableregulations. Non-compliance with FDA regulations can result in administrative and judicial sanctions, including warning or untitled letters, clinical holds, fines, recall or seizure of products, injunctions, total or partial suspension of production, refusal of the government to approve marketing applications or allow entry into supply contracts, refusal to permit import or export of products, civil penalties, criminal prosecution and other actions affecting a company and its products. The FDA also has the authority to revoke previously granted marketing authorizations.regulations;To secure FDA approval, an applicant must submit extensive preclinical and clinical data, as well as information about product manufacturing processes and facilities and other supporting informationsubmission to the FDA
for each indicationof an Investigational New Drug Application, or IND, which must become effective before human clinical trials may begin;performance of adequate and well‑controlled human clinical trials in accordance with Good Clinical Practice, or GCP, requirements to establish
a product candidate's safety and efficacy. The development and approval process takes many years, requires the expenditure of substantial resources and may be subject to delays or limitations of approval or rejection of an applicant's new drug application, or NDA. Even if the FDA approves a product, the approval is subject to post-marketing surveillance, adverse drug experience and other recordkeeping and reporting obligations, and may involve ongoing requirements for post-marketing studies. The FDA also has authority to place conditions on any approvals that could restrict the commercial applications, advertising, promotion or distribution of these products. Product approvals may be withdrawn if compliance with regulatory standards is not maintained or if problems occur following initial marketing.Preclinical and Clinical TestingPreclinical studies involve laboratory evaluation of product characteristics and animal studies to assess the biological activity and safety of the product. In some cases, long-term preclinical studies are conducted while clinical studies are ongoing. The FDA, under its Good Laboratory Practices regulations, regulates preclinical studies. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring these studies to be replicated. When the preclinical testing is considered adequate by the sponsor to demonstratethe safety andscientific rationaleefficacy of the proposed drug forinitial human studies,its intended use;submission to the FDA of an NDA or BLA;
satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with cGMP requirements to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and
FDA review and approval of the NDA or BLA.
The clinical study sponsor must submit the results of the preclinical tests, together with manufacturing information,
andanalytical data,are submittedany available clinical data or literature and a proposed clinical protocol, to the FDA as part ofanthe IND. Some preclinical testing may continue even after the INDsubmission.is submitted. The IND automatically becomes effectiveif not rejected30 days after receipt by the FDA,within 30 days afterunless the FDAreceivesplaces theIND.clinical study on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical study can begin. The FDA mayeither during the 30-day period after filing of an IND oralso impose clinical holds on a biological product candidate at anyfuturetimeimpose a clinical hold on proposedbefore orongoingduring clinical trialson various grounds, including that the study subjects aredue to safety concerns orwould be exposed to an unreasonable and significant health risk.non-compliance. If the FDA imposes a clinical hold,clinicaltrialscannot commence ormay not recommence without FDA authorization and then only under terms authorized by the FDA.ClinicalIn addition to the submission of an IND to the FDA, supervision of certain human gene transfer trials may also require evaluation and assessment by an institutional biosafety committee, or IBC, a local institutional committee that reviews and oversees research utilizing recombinant or synthetic nucleic acid molecules at that institution. The IBC assesses the safety of the research and identifies any potential risk to the public health or the environment, and such assessment may result in some delay before initiation of a clinical trial.’Clinical trials involve the administration of
the investigationala productcandidatescandidate tohumanshealthy volunteers or patients under the supervision ofaqualifiedprincipal investigator.investigators, generally physicians not employed by or under the study sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical study, dosing procedures, patient selection and exclusion criteria, and the parameters to be used to monitor patient safety, including stopping rules that assure a clinical study will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must beconducted in accordance with Good Clinical Practices under protocolssubmitted to the FDA as part of the IND.In addition,Clinical trials must be conducted and monitored in accordance with the FDA’s regulations including GCP requirements, including the requirement that all research patients provide informed consent. Further, each clinicaltrialstudy must be reviewed and approvedand conducted under the auspices ofby anInvestigational Review Board,independent institutional review board, or IRB,and with patient informed consent. The IRB typically considers, among other things, ethical factors andat or servicing each institution at which thesafety of human subjects.Clinicalclinical study will be conducted. Human clinical trials are typically conducted in three sequential phasesbutthat may overlap or be combined:Phase 1: The product candidate is initially administered to healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the
phasescase of some products for severe or life‑threatening diseases, such as cancer, especially when the product mayoverlap.be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.Phase
12: This phase involves clinical trialsusually involve healthy human subjects. The goal ofin aPhase I clinical trial islimited patient population toestablish initial data aboutidentify possible adverse effects and safety risks, to preliminarily evaluate thesafety, tolerability and pharmacokinetic propertiesefficacy of the productcandidates in humans. In Phase 2 clinical trials, controlled studies are conducted on an expanded population of patients with thefor specific targeteddisease. The primary purpose of these tests is to evaluate the initial effectiveness of the drug candidate on the intended target and to determine if there are any side effects or other risks associated with the drugdiseases and to determine theoptimal dose of the drug from theappropriate dosage for further clinical trials.Phase 3: Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety
and efficacy profile developed from thein an expanded patient population at geographically dispersed clinicalstudy. Phase 3 trials involve even larger patient populations, often with several hundred or even several thousand patients, depending on the use for which the drug is being studied. Phase 3study sites. These clinical trials are intended to establish the safety and efficacy of the product and the overallrisk-benefitrisk‑benefit ratio of thedrugproduct candidate and provide, if appropriate, an adequate basis for productlabeling. During all clinical trials, physicians monitor the patients to determine effectivenesslabeling andto observe and report any reactions or other safety risks that may result fromcommercial use of the product.Post‑approval trials, sometimes referred to as Phase 4 studies, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA or BLA.
The FDA or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug
candidate.Tablehas been associated with unexpected serious harm to patients. In addition, some clinical trials are overseen by an independent group ofContentsqualified experts organized by the sponsor, known as a data safety monitoring board or committee. Depending on its charter, this group may determine whether a trial may move forward at designated check points based on access to certain data from the trial.Product FormulationDuring the development of a new drug or biologic, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2, andManufacture
before an NDA or BLA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach agreement on the next phase of development. Sponsors typically use the meetings at the end of the Phase 2 trial to discuss Phase 2 clinical results and present plans for the pivotal Phase 3 clinical trial that they believe will support approval of the new drug or biologic.Concurrent with clinical trials, companies usually complete additional animal studies and
preclinical studies, companiesmust also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing theproduct. In addition, manufacturers, including contract manufacturers, are required to complyproduct in commercial quantities in accordance withcurrent applicable FDA Good Manufacturing Practice, orcGMPregulations. The cGMP regulations include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation.requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, qualitypurityandpotencypurity of the finaldrugs. Additionally,drug. In addition, appropriate packaging must be selected and tested, andchemistrystability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over itsshelf-life.Compliance with cGMP regulations alsoshelf life. While the IND is active and before approval, progress reports summarizing the results of the clinical trials and nonclinical studies performed since the last progress report must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and investigators for serious and unexpected suspected adverse events, findings from other studies suggesting aconditionsignificant risk to humans exposed to the same or similar drugs, findings from animal or in vitro testing suggesting a significant risk to humans, and any clinically important increased incidence ofnew drug application approval. The FDA must approve manufacturing facilities before they can be useda serious suspected adverse reaction compared to that listed in thecommercial manufactureprotocol or investigator brochure.There are also requirements governing the reporting of
drug products. In addition, manufacturing establishmentsongoing clinical trials and completed trial results to public registries. Sponsors of certain clinical trials of FDA‑regulated products aresubjectrequired topre-approval inspectionsregister andunannounced periodic inspections.Afterdisclose specified clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to thecompletionproduct, patient population, phase of investigation, trial sites and investigators and other aspects of the clinical trialphasesis then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved.United States Review and Approval Process
The results of product development,
ifpreclinical and other non‑clinical studies and clinical trials, along with descriptions of thesponsor concludes that there is substantial evidence thatmanufacturing process, analytical tests conducted on the chemistry of the drug,candidate is safeproposed labeling andeffective for its intended use, the sponsor may submit a NDAother relevant information are submitted to theFDA.FDA as part of an NDA or BLA requesting approval to market the product. Theapplication must contain all of the information on the drug candidate gathered to that date, including data from the clinical trials, and be accompanied by a user fee.Under the Prescription Drug User Fee Act, or PDUFA,submission ofaan NDAwith clinical data requiresor BLA is subject to the payment of user fees; afee, with some exceptions. In return, the FDA assigns a goalwaiver ofsix or ten months from filing of the application to return of a first "complete response," in which the FDAsuch fees mayapprove the product or request additional information. There canbeno assurance that an application will be approved within the performance goal timeframe establishedobtained underPDUFA.certain limited circumstances.The FDA
initially determines whether a NDA asreviews all NDAs and BLAs submittedis acceptableto ensure that they are sufficiently complete for substantive review before it accepts them for filing. The FDA mayrefuserequest additional information rather than accept an NDA or BLA for filing. In this event, the NDA or BLA must be resubmitted with the additional information. The resubmitted application also is subject tofile an application, in which casereview before the FDAretains one-half of the user fees. Ifaccepts it for filing.Once the submission is accepted for filing, the FDA begins an
in-depth review of the application. As part of this review, thein‑depth substantive review. The FDA may refer theapplicationNDA or BLA to anappropriateadvisory committeetypically a panel of clinicians,for review, evaluation anda recommendation.recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisorycommittee.Ifcommittee, but it generally follows such recommendations. The approval process is lengthy and often difficult, and the FDAevaluations ofmay refuse to approve an NDA or BLA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data and information. Even if such data and information are submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP‑compliant to assure and preserve the product’s identity, strength, quality and purity. The FDA reviews a BLA to determine, among other things whether the product is safe, pure and potent and themanufacturing facilities are favorable,facility in which it is manufactured, processed, packed or held meets standards designed to assure the product’s continued safety, purity and potency. Before approving an NDA or BLA, the FDAmaywill inspect the facility or facilities where the product is manufactured.After the FDA evaluates an NDA or BLA, it will issue an approval letter
authorizingor a Complete Response Letter. An approval letter authorizes commercial marketing of the drugcandidatewith prescribing information forspecifiedspecific indications.The FDA could also issue a "complete response" letter at the end of the review period.A"complete response" letter will be issued to let a company knowComplete Response Letter indicates that the reviewperiod for a drugcycle of the application is complete andthatthe applicationiswill notyet ready for approval. The letter will describebe approved in its present form. A Complete Response Letter usually describes the specific deficienciesand, when possible, will outline recommended actionsin theapplicant might take to get the application ready for approval, including calling for additional clinical trial data.Marketing Approval and Post-Marketing ObligationsIf the FDA approves an application, the drug becomes available for physicians to prescribe. Periodic reports must be submitted to the FDA, including descriptions of any adverse reactions reported. The FDA may require post-marketing studies, also known as Phase IV studies, as a condition of approval. In addition to studies required by the FDA after approval, trials and studies are often conducted to explore new indications for the drug. The purpose of these trials and studies and related publications is to develop data to support additional indications for the drug, which must be approvedNDA or BLA identified by the FDA and may require additional clinical data, such as an additional pivotal Phase 3 trial or other significant and time‑consuming requirements related toincrease its acceptanceclinical trials, nonclinical studies or manufacturing. If a Complete Response Letter is issued, the sponsor must resubmit the NDA or BLA, addressing all of the deficiencies identified in themedical community.letter, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the NDA or BLA does not satisfy the criteria for approval. If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. In addition,some post-marketing studies are done at the request ofthe FDA may require a sponsor todevelop additional information regarding theconduct Phase 4 testing, which involves clinical trials designed to further assess a drug’s safetyof a product.The FDAand effectiveness after NDA or BLA approval, and mayimpose risk evaluation mitigation strategies, or REMS, on a product if the FDA believes there is a reasonrequire testing and surveillance programs to monitor the safety of approved products which have been commercialized. The FDA may also place other conditions on approval including the requirement for a risk evaluation and mitigation strategy, or “REMS” to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS. The FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Marketing approval may be withdrawn for non‑compliance with regulatory requirements or if problems occur following initial marketing.The Food and Drug Administration Safety and Innovation Act, or FDASIA, made permanent the Pediatric Research Equity Act, or PREA, which requires a sponsor to conduct pediatric clinical trials for most drugs and biologics, for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration. Under PREA, original NDAs, BLAs and supplements thereto must contain a pediatric assessment unless the sponsor has received a deferral or waiver. The required assessment must evaluate the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The sponsor or FDA may request a deferral of pediatric clinical trials for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the drug or biologic is ready for approval for use in adults before pediatric clinical trials are complete or that additional safety or effectiveness data needs to be collected before the pediatric clinical trials begin. The FDA must send a non‑compliance letter to any sponsor that fails to submit the required assessment, keep a deferral current or fails to submit a request for approval of a pediatric formulation.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the
marketplace. REMSUnited States or, if it affects more than 200,000 individuals in the United States, there is no reasonable expectation that the cost of developing and making a drug or biologic product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan designation must be requested before submitting an NDA or BLA. After the FDA grants orphan designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug or biological product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity or inability to manufacture the product in sufficient quantities. The designation of such drug or biologic also entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user‑fee waivers. However, competitors, may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity. Orphan exclusivity also could
add training requirementsblock the approval of one of our product candidates forhealthcare professionals, safety communications efforts,seven years if a competitor obtains approval of the same drug or biologic as defined by the FDA or if our product candidate is determined to be contained within the competitor’s product for the same indication or disease. If an orphan designated product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan exclusivity. Orphan drug status in the European Union has similar but not identical benefits in that jurisdiction.Expedited Development and
limitsReview ProgramsThe FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drug products that meet certain criteria. Specifically, new drugs are eligible for Fast Track designation if they are intended to treat a serious or life‑threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. Unique to a Fast Track product, the FDA may consider for review sections of the NDA or BLA on
channelsa rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission ofdistribution, amongthe sections of the NDA or BLA, the FDA agrees to accept sections of the NDA or BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA or BLA.Any product submitted to the FDA for approval, including a product with a Fast Track designation, may also be eligible for other
things.types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. A product is eligible for priority review if it has the potential to provide a significant improvement in the treatment, diagnosis or prevention of a serious disease or condition compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug designated for priority review in an effort to facilitate the review. The FDA endeavors to review applications with priority review designations within six months of the filing date as compared to ten months for review of original BLAs and new molecular entity NDAs under its standard review goals.In addition, a product may be eligible for accelerated approval. Drug and biologic products intended to treat serious or life‑threatening diseases or conditions may be eligible for accelerated approval upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well‑controlled post‑marketing clinical trials. In addition, the FDA currently requires as a condition for accelerated approval pre‑approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
FDASIA established a category of drugs and biologics referred to as “breakthrough therapies” that may be eligible to receive Breakthrough Therapy Designation. A sponsor may seek FDA designation of a drug or biologic candidate as a “breakthrough therapy” if the product is intended, alone or in combination with one or more other products, to treat a serious or life‑threatening disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The designation includes all of the Fast Track program features, as well as more intensive FDA interaction and guidance. The Breakthrough Therapy Designation is a distinct status from both accelerated approval and priority review, which can also be granted to the same drug if relevant criteria are met. If a product is designated as breakthrough therapy, the FDA will expedite the development and review of such drug. All requests for breakthrough therapy designation will be reviewed within 60 days of receipt, and the FDA will either grant or deny the request.
Rare Pediatric Disease Priority Review Voucher Program
In 2012, Congress authorized the FDA to award priority review vouchers to sponsors of certain rare pediatric disease product applications. This program is designed to encourage development of new drug and biological products for prevention and treatment of certain rare pediatric diseases. Specifically, under this program, a sponsor who receives an approval for a drug or biologic for a “rare pediatric disease” may qualify for a voucher that can be redeemed to receive a priority review of a subsequent marketing application for a different product. The sponsor
wouldof a rare pediatric disease drug product receiving a priority review voucher may transfer (including by sale) the voucher to another sponsor. The voucher may be further transferred any number of times before the voucher is used, as long as the sponsor making the transfer has not yet submitted the application. The FDA may also revoke any priority review voucher if the rare pediatric disease drug for which the voucher was awarded is not marketed in the U.S. within one year following the date of approval.For purposes of this program, a “rare pediatric disease” is a (a) serious or life-threatening disease in which the serious or life-threatening manifestations primarily affect individuals aged from birth to 18 years, including age groups often called neonates, infants, children, and adolescents; and (b) rare diseases or conditions within the meaning of the Orphan Drug Act. Congress has only authorized the Rare Pediatric Disease Priority Review Voucher program until September 30, 2020. Consequently, sponsors of marketing applications approved after that date will not receive the voucher unless Congress reauthorizes the Rare Pediatric Disease Priority Review Voucher program, for which legislation has been proposed in the current Congress. However, even if the program is not reauthorized, if a drug candidate receives Rare Pediatric Disease Designation before October 1, 2020, the sponsor of the marketing application for such drug will be eligible to receive a voucher if the application for the designated drug is approved by the FDA before October 1, 2022.
Post‑approval requirements
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. After approval, some types of changes to the approved product, such as adding new indications, certain manufacturing changes and additional labeling claims, are subject to further FDA review and approval. Drug and biologics manufacturers and other entities involved in the manufacture and distribution of approved drugs and biologics are required to
evaluateregister their establishments with the FDA andmonitorcertain state agencies, and are subject to periodic unannounced inspections by thevarious REMS activitiesFDA andadjust them if need be. Whether a REMS would be imposedcertain state agencies for compliance with cGMP regulations and other laws and regulations. Changes to the manufacturing process are strictly regulated, and, depending ona productthe significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon us and anyresulting financial impact is uncertain at this time.third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.Any drug products
manufacturedwe ordistributedour partners manufacture or distribute pursuant to FDA approvalsarewill be subject to continuing regulation by the FDA, including,recordamong other things, record‑keeping requirements, reporting of adverse experiences with the drug, providing the FDA with updated safety and efficacy information, drug sampling and distribution requirements,notifying the FDA and gaining its approval of certain manufacturing or labeling changes,complying with certain electronic records and signature requirements, and complying with FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market and imposes requirements and restrictions on drug and biologics manufacturers, such as those related to direct‑to‑consumer advertising, the prohibition on promoting products for uses or in patient populations that are not described in the product’s approved labeling (known as “off‑label use”), industry‑sponsored scientific and educational activities, and promotional activities involving the internet. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and consistent with the provisions of the approved label. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off-label use of their products.Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant or manufacturer to administrative or judicial civil or criminal sanctions and adverse publicity. FDA sanctions could include refusal to approve pending applications or supplements to approved applications, withdrawal of an approval, clinical hold, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, mandated corrective advertising or communications with doctors, debarment, restitution, disgorgement of profits, or civil or criminal penalties.
Biosimilars and Exclusivity
The Affordable Care Act includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or “BPCIA”, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA‑licensed reference biological product. The FDA has issued several guidance documents outlining an approach to review and approval of biosimilars.
Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. However, complexities associated with the larger, and often more complex, structures of biological products, as well as the processes by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being addressed by the FDA.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12‑year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing the sponsor’s own preclinical data and data from adequate and well‑controlled clinical trials to demonstrate the safety, purity and potency of their product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products. At this juncture, it is unclear whether products deemed “interchangeable” by the FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy law.
A biological product can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
The BPCIA is complex and continues to be interpreted and implemented by the FDA. In addition, recent government proposals have sought to reduce the 12‑year reference product exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. As a result, the ultimate impact, implementation and meaning of the BPCIA is subject to significant uncertainty.
FDA Regulation of Companion Diagnostics
Our product candidates may require use of an in vitro diagnostic to identify appropriate patient populations. These diagnostics, often referred to as companion diagnostics, are regulated as medical devices. In the United States, the FDCA and its implementing regulations, and other federal and state statutes and regulations govern, among other things, medical device design and development, pre-clinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance. Unless an exemption applies, companion diagnostic tests require marketing clearance or approval from the FDA prior to commercial distribution. The two primary types of FDA marketing authorization applicable to a medical device are premarket notification, also called 510(k) clearance, and premarket approval, or PMA, approval.
If use of companion diagnostic is essential to safe and effective use of a drug or biologic product, then the FDA generally will require approval or clearance of the diagnostic contemporaneously with the approval of the therapeutic product. On August 6, 2014, the FDA issued a final guidance document addressing the development and approval process for “In Vitro Companion Diagnostic Devices.” According to the guidance, for novel candidates such as our product candidates, a companion diagnostic device and its corresponding drug or biologic candidate should be approved or cleared contemporaneously by FDA for the use indicated in the therapeutic product labeling. The guidance also explains that a companion diagnostic device used to make treatment decisions in clinical trials of a drug generally will be considered an investigational device, unless it is employed for an intended use for which the device is already approved or cleared. If used to make critical treatment decisions, such as patient selection, the diagnostic device generally will be considered a significant risk device under the FDA’s Investigational Device Exemption, or IDE, regulations. Thus, the sponsor of the diagnostic device will be required to comply with the IDE regulations. According to the guidance, if a diagnostic device and a drug are to be studied together to support their respective approvals, both products can be studied in the same investigational study, if the study meets both the requirements of the IDE regulations and the IND regulations. The guidance provides that depending on the details of the study plan and subjects, a sponsor may seek to submit an IND alone, or both an IND and an IDE. In July 2016, the FDA issued a draft guidance document intended to further assist sponsors of therapeutic products and sponsors of in vitro companion diagnostic devices on issues related to co-development of these products.
The FDA generally requires companion diagnostics intended to select the patients who will respond to cancer treatment to obtain approval of a PMA for that diagnostic contemporaneously with approval of the therapeutic. The review of these in vitro companion diagnostics in conjunction with the review of therapeutic candidates such as those we are developing involves coordination of review by the FDA’s Center for Drug Evaluation and Research and by the FDA’s Center for Devices and Radiological Health. The PMA process, including the gathering of clinical and pre-clinical data and the submission to and review by the FDA, can take several years or longer. It involves a rigorous premarket review during which the applicant must prepare and provide the FDA with reasonable assurance of the device’s safety and effectiveness and information about the device and its components regarding, among other things, device design, manufacturing and labeling. PMA applications are also subject to an application fee. In addition, PMAs for certain devices must generally include the results from extensive pre-clinical and adequate and well-controlled clinical trials to establish the safety and effectiveness of the device for each indication for which FDA approval is sought. In particular, for a diagnostic, the applicant must demonstrate that the diagnostic produces reproducible results when the same sample is tested multiple times by multiple users at multiple laboratories. In addition, as part of the PMA review, the FDA will typically inspect the manufacturer’s facilities for compliance with the Quality System Regulation, or QSR, which imposes elaborate testing, control, documentation and other quality assurance requirements.
If the FDA evaluations of both the PMA application and the manufacturing facilities are favorable, the FDA will either issue an approval letter or an approvable letter, which usually contains a number of conditions that must be met in order to secure the final approval of the PMA, such as changes in labeling, or specific additional information, such as submission of final labeling, in order to secure final approval of the PMA. If the FDA concludes that the applicable criteria have been met, the FDA will issue a PMA for the approved indications, which can be more limited than those originally sought by the applicant. The PMA can include post-approval conditions that the FDA believes necessary to ensure the safety and effectiveness of the device, including, among other things, restrictions on labeling, promotion, sale and distribution.
If the FDA’s evaluation of the PMA or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. A not approvable letter will outline the deficiencies in the application and, where practical, will identify what is necessary to make the PMA approvable. The FDA may also determine that additional clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and then the data submitted in an amendment to the PMA. Once granted, PMA approval may be withdrawn by the FDA if compliance with post approval requirements, conditions of approval or other regulatory standards is not maintained or problems are identified following initial marketing. PMA approval is not guaranteed, and the FDA may ultimately respond to a PMA submission with a not approvable determination based on deficiencies in the application and require additional clinical trial or other data that may be expensive and time-consuming to generate and that can substantially delay approval.
After a device is placed on the market, it remains subject to significant regulatory requirements. Medical devices may be marketed only for the uses and indications for which they are cleared or approved. Device manufacturers must also establish registration and
their subcontractorsdevice listings with the FDA. A medical device manufacturer’s manufacturing processes and those of its suppliers are required toregister their establishmentscomply with the applicable portions of the QSR, which cover the methods and documentation of the design, testing, production, processes, controls, quality assurance, labeling, packaging and shipping of medical devices. Domestic facility records and manufacturing processes are subject to periodicunannouncedunscheduled inspectionsfor compliance with cGMP requirements. Also, newly discovered or developed safety or effectiveness databy the FDA. The FDA also mayrequire changesinspect foreign facilities that export products toa product's approved labeling, includingtheaddition of new warnings and contraindications, or even in some instances revocation or withdrawal of the product's approval.United States.Approval Process Outside of the United States
In
orderaddition tomarket any product outside ofregulations in the United States, wemust comply with numerous and varying regulatory requirementswill be subject to a variety of regulations in othercountries regarding safety and efficacy andjurisdictions governing, among other things, clinicaltrialsstudies and any commercial sales and distribution of ourproducts, which broadly reflectproduct candidates.Whether or not we obtain FDA approval for a product candidate, we must obtain the
issues addressed byrequisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical studies or marketing of the product candidates in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical study application much like the IND prior to the commencement of human clinical studies. In the European Union, for example, a clinical trial authorization, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, much like the FDAabove. Approval procedures vary among countriesandcan involve additionalthe IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical study development may proceed.To obtain regulatory approval of an investigational biological product
testing and additional administrative review periods.under European Union regulatory systems, we must submit a marketing authorization application. Thetime requiredapplication used toobtain approval in other countries might differ from and be longer than that required to obtain FDA approval. Marketing approval in one country does not ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one country may negatively impactfile theregulatory process in other countries.AsBLA in the United States is similar to that required in the European Union, with the exception of, among other things, country‑specific document requirements. The European Union also provides opportunities for market exclusivity. For example, in the European Union, upon receiving marketing authorization, new chemical entities generally receive eight years of data exclusivity and an additional two years of market exclusivity. If granted, data exclusivity prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic application. During the additional two‑year period of market exclusivity, a generic marketing authorization can be submitted, and the innovator’s data may be referenced, but no generic product can be marketed until the expiration of the market exclusivity. However, there is no guarantee that a product will be considered by the European Union’s regulatory authorities to be a new chemical entity, and products may not qualify for data exclusivity. Products receiving orphan designation in the European Union can receive ten years of market exclusivity, during which time no similar medicinal product for the same indication may be placed on the market. An orphan product can also obtain an additional two years of market exclusivity in the European Union for pediatric studies. No extension to any supplementary protection certificate can be granted on the basis of pediatric studies for orphan indications.The criteria for designating an “orphan medicinal product” in the European Union are similar in principle to those in the United States. Under Article 3 of Regulation (EC) 141/2000, a medicinal product may be designated as orphan if (1) it is intended for the diagnosis, prevention or treatment of a life‑threatening or chronically debilitating condition; (2) either (a) such condition affects no more than five in 10,000 persons in the European Union when the application is made, or (b) the product, without the benefits derived from orphan status, would not generate sufficient return in the European Union to justify investment; and (3) there exists no satisfactory method of diagnosis, prevention or treatment of such condition authorized for marketing in the European Union, or if such a method exists, the product will be of significant benefit to those affected by the condition, as defined in Regulation (EC) 847/2000. Orphan medicinal products are eligible for financial incentives such as reduction of fees or fee waivers and are, upon grant of a marketing authorization, entitled to ten years of market exclusivity for the approved therapeutic indication. The application for orphan drug designation must be submitted before the application for marketing authorization. The applicant will receive a fee reduction for the marketing authorization application if the orphan drug designation has been granted, but not if the designation is still pending at the time the marketing authorization is submitted. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval
process in Europe and inprocess.The 10‑year market exclusivity may be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan designation, for example, if the product is sufficiently profitable not to justify maintenance of market exclusivity. In addition, marketing authorization may be granted to a similar product for the same indication at any time if:
the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior;
the applicant consents to a second orphan medicinal product application; or
the applicant cannot supply enough orphan medicinal product.
For other countries
is a lengthy, challengingoutside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical studies, product licensing, pricing andinherently uncertain process.reimbursement vary from country to country. In all cases, again, the clinical studies are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of
marketingregulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.GenerallyOther Healthcare Laws and Compliance Requirements
Pharmaceutical companies are subject to additional healthcare regulation and enforcement by the
developmentU.S. federal andapproval procedures are harmonized throughout the European Union: however, there is limited harmonization in relation to national pricingstate governments andreimbursement practices.Under European Union regulatory systems, a company may not market a medicinal product without marketing authorization. There are three procedures for submitting a MAAby authorities in theEU: (1)foreign jurisdictions in which they conduct their business. At theTablefederal level, such laws include, without limitation: the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, to induce, or in return for, either the referral ofContentsan individual, or the purchase or recommendation of an item or service for which payment may be made under any federal healthcare program; federal civil and criminal false claims laws, including the civil False Claims Act, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment to the federal government, including federal healthcare programs, that are false or fraudulent and civil monetary penalty laws; the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits, among other things, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; and the federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to annually report to the federal government, information related to payments or other transfers of value made to physicians, certain other health care professionals beginning in 2022, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members.mutual recognition procedure (MRP); (2) the decentralized procedure (DCP)Pharmaceutical companies are also subject to U.S. state and(3) the centralized procedure (CP). The submission strategy for a given product will depend on the natureforeign law equivalents of each of theproduct, the target indication(s), the historyabove federal laws, such as anti-kickback and false claims laws which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, or that apply regardless ofthe product, and the marketing plan. The centralized procedure is compulsory for medicinal productspayor; laws whichare produced by biotechnology processes, advanced therapy medicinal products and orphan drugs. Besides the products falling under the mandatory scope, the centralized procedure is also open for other innovative products that are new active substances or other medicinal products that constitute a significant therapeutic, scientific or technical innovation.The centralized procedure leadsrequire pharmaceutical companies toapproval of the product in all 27 EU member states and in Norway, Iceland and Liechtenstein. Submission of one MAA thus leads to one assessment process and one authorization that allows access to all applicable markets within the entire EU. The process of the centralized procedure is triggered when the applicant sends the letter announcing the intent to submit a MAA (letter of intent). The letter of intent also initiates the assignment of the Rapporteur and Co-Rapporteur, who are the two appointed members of the Committee for Human Medicinal Products, or CHMP, representing two EU member states. However, in light of the United Kingdom's vote in 2016 to leave the European Union, the so-called Brexit vote, there may be changes forthcoming in the scope of the centralized approval procedure as the terms of that exit are negotiated between the UK and the European Union.When using the MRP or DCP, the applicant must select which and how many EU member states in which to seek approval. In the case of an MRP, the applicant must initially receive national approval in one EU member state. This will be the so-called reference member state (RMS) for the MRP. Then, the applicant seeks approval for the product in other EU member states, the so-called concerned member states (CMS) in a second step: the mutual recognition process. For the DCP, the applicant will approach all chosen member states at the same time. To do so, the applicant will identify the RMS that will assess the submitted MAA and provide the other selected member statescomply with theconclusions and results of the assessment.When the application for marketing authorization is made, the competent authority responsible for granting a marketing authorization must verify whether the application complies with the relevant requirements, includingpharmaceutical industry’s voluntary compliancewith the agreed pediatric investigational plan, or PIP. Assuming it does, the marketing authorization may be grantedguidelines and the relevantresults are included in the summary of product characteristics (SmPC) for the product, along with a statement indicatingcompliancewith the agreed PIP. It is not necessary for the product actually to be indicated for use in the pediatric population (for example, if the results show that that would not be appropriate).Drug Price Competition and Patent Term Restoration Act of 1984Under the Drug Price Competition and Patent Term Restoration Act of 1984, known as the Hatch-Waxman Act, a portion of a product's patent term that was lost during clinical development and application reviewguidance promulgated by theFDAU.S. federal government, laws which require pharmaceutical companies to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures and information related to drug pricing, and laws requiring the registration of pharmaceutical sales and medical representatives. Violation of these laws or other governmental regulations maybe restored. The Hatch-Waxman Act also provides for a statutory protection, knownresult in penalties, including, without limitation, significant civil, criminal and administrative penalties, damages, fines, exclusion from government-funded healthcare programs, such asexclusivity, against the FDA's acceptanceMedicare and Medicaid orapprovalsimilar programs in other countries or jurisdictions, integrity oversight and reporting obligations to resolve allegations ofcertain competitor applications. The Hatch-Waxman Act also provides the legal basis for the approval of abbreviated new drug applications, or ANDAs.Patent term extension can compensate for time lost during product developmentnon-compliance, disgorgement, imprisonment, contractual damages, reputational harm, diminished profits and theregulatory review process by returning up to five yearscurtailment or restructuring ofpatent life for a patent that covers a new product or its use. This period is generally one-half the time between the effective date of an INDoperations.Coverage and
the submission date of a NDA, plus the time between the submission date of a NDA and the approval of that application. Patent term extensions, however, are subject to a maximum extension of five years, and the patent term extension cannot extend the remaining term of a patent beyond a total of 14 years.Table of ContentsReimbursementThe application for patent term extension is subject to approval by the United States Patent and Trademark Office in conjunction with the FDA. It generally takes at least six months to obtain approval of the application for patent term extension.The Hatch-Waxman Act also provides for a period of statutory protection for new drugs that receive NDA approval from the FDA. If a new drug receives NDA approvalSignificant uncertainty exists asa new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active entity, then the Hatch-Waxman Act prohibits an ANDA or a NDA submitted pursuant to section 505(b)(2) of the Federal Food, Drug, and Cosmetics Act, where the applicant does not own or have a legal right of reference to all of the data required for approval to be submitted by another company for a generic version of such drug (505(b)(2) NDA), with some exceptions, for a period of five years from the date of approval of the NDA. The statutory protection provided pursuantto theHatch-Waxman Act will not prevent the filing or approval of a full NDA, as opposed to an ANDA or 505(b)(2) NDA, for any drug, including, for example, a drug with the same active ingredient, dosage form, route of administration, strengthcoverage andconditions of use. In order to obtain a NDA, however, a competitor would be required to conduct its own clinical trials, and any use of the drug for which marketing approval is sought could not violate another NDA holder's patent claims.If NDA approval is received for a new drug containing an active ingredient that was previously approved by the FDA but the NDA is for a drug that includes an innovation over the previously approved drug, for example, a NDA approval for a new indication or formulation of the drug with the same active ingredient, and if such NDA approval was dependent upon the submission to the FDA of new clinical investigations, other than bioavailability studies, then the Hatch-Waxman Act prohibits the FDA from making effective the approval of an ANDA or 505(b)(2) NDA for a generic version of such drug for a period of three years from the date of the NDA approval. This three year exclusivity, however, only covers the innovation associated with the NDA to which it attaches. Thus, the three year exclusivity does not prohibit the FDA, with limited exceptions, from approving ANDAs or 505(b)(2) NDAs for drugs containing the same active ingredient but without the new innovation.While the Hatch-Waxman Act provides certain patent restoration and exclusivity protections to innovator drug manufacturers, it also permits the FDA to approve ANDAs for generic versions of their drugs assuming the approval would not violate another NDA holder's patent claims. The ANDA process permits competitor companies to obtain marketing approval for a drug with the same active ingredient for the same uses but does not require the conduct and submission of clinical studies demonstrating safety and effectiveness for that product. Instead of safety and effectiveness data, an ANDA applicant needs only to submit data demonstrating that its product is bioequivalent to the innovator product as well as relevant chemistry, manufacturing and product data. The Hatch-Waxman Act also instituted a third type of drug application that requires the same information as a NDA, including full reports of clinical and preclinical studies, except that some of the information from the reports required for marketing approval comes from studies which the applicant does not own or have a legal right of reference. This type of application, a 505(b)(2) NDA, permits a manufacturer to obtain marketing approval for a drug without needing to conduct or obtain a right of reference for all of the required studies.If a competitor submits an ANDA or 505(b)(2) NDA for a compound or usereimbursement status of anycompound covered by another NDA holder's patent claims, the Hatch-Waxman Act requires, in some circumstances, the applicant to notify the patent owner and the holder of the approved NDA of the factual and legal basis of the applicant's opinion that the patent is not validpharmaceutical orwill not be infringed. Upon receipt of this notice, the patent owner and the NDA holder have 45 days to bring a patent infringement suit in federal district court and obtain a 30-month stay against the company seeking to reference the NDA. The NDA holder could still file a patent suit after the 45 days, but if they miss the45-day deadline, they would not have the benefit of the 30-month stay. Alternatively, after this 45-day period, the applicant may file a declaratory judgment action, seeking a determination that the patent is invalid or will not be infringed. Depending on the circumstances, however, the applicant may not be able to demonstrate a controversy sufficient to confer jurisdiction on the court. The discovery, trial and appeals process in such suits can take several years. If such a suit is commenced, the Hatch-Waxman Act provides a 30-month stay on the approval of the competitor's ANDA or 505(b)(2) NDA. If the litigation is resolved in favor of the competitor or the challenged patent expires during the 30-month period, unless otherwise extended by court order, the stay is lifted and the FDA may approve the application. Under regulations issued by the FDA, and essentially codified under the Medicare prescription drug legislation, the patent owner and the NDA holder have the opportunity to trigger only a single 30-month stay per ANDA or 505(b)(2) NDA. Once the applicant of the ANDA or 505(b)(2) NDA has notified the patent owner and the NDA holder of the infringement, the applicant cannot be subjected to another 30-month stay, even if the applicant becomes aware of additional patents that may be infringed by its product.Pharmaceutical Pricing and ReimbursementWe currently have no marketed products. In both domestic and foreign markets, sales of any productsbiological product for which wereceiveobtain regulatoryapproval for commercial sale willapproval. Sales of any product depend, in part, on theavailabilityextent to which such product will be covered by third-party payors, such as federal, state, and foreign government healthcare programs, commercial insurance and managed healthcare organizations, and the level of reimbursementfromfor such product by third-party payors. Decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. Further, no uniform policy for coverage and reimbursement exists in the United States, and coverage and reimbursement can differ significantly from payor to payor. Third-party payorsincludeoften rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates, but also have their own methods and approval process apart from Medicare determinations. As a result, the coverage determination process is often a time-consuming and costly process that may require companies to provide scientific and clinical support for the use of a product to each payor separately. For products administered under the supervision of a physician, obtaining coverage and adequate reimbursement may be particularly difficult because of the higher prices often associated with such drugs. Additionally, separate reimbursement for the product itself or the treatment or procedure in which the product is used may not be available, which may impact physician utilization. Lastly, companion diagnostic tests require coverage and reimbursement separate and apart from the coverage and reimbursement for their companion pharmaceutical or biological products. Similar challenges to obtaining coverage and reimbursement, applicable to pharmaceutical or biological products, will apply to companion diagnostics.In addition, the U.S. government,
authorities orstate legislatures and foreign governments have continued implementing cost-containment programs,managed care providers, private health insurersincluding price controls, restrictions on coverage andother organizations. These third-partyreimbursement and requirements for substitution of generic products. Third-party payors are increasingly challenging theprice and examining the cost-effectiveness ofprices charged for medical products andservices. Inservices, examining the medical necessity and reviewing the cost effectiveness of pharmaceutical or biological products, medical devices and medical services, in additionsignificant uncertainty exists astothe reimbursement statusquestioning safety and efficacy. Adoption ofnewly approved healthcare products. We may need to conduct expensive pharmacoeconomic studiesprice controls and cost-containment measures, and adoption of more restrictive policies inorder to demonstrate the cost-effectivenessjurisdictions with existing controls and measures, could further limit sales ofour products. Our product candidates may not be considered cost-effective. Adequateany product. Decreases in third-party reimbursementmayfor any product or a decision by a third-party payor notbe availabletoenable uscover a product could reduce physician usage and patient demand for the product.Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of reform proposals to
maintain price levels sufficient to realize an appropriate return on our investmentchange the healthcare system. There is significant interest inproduct development. Third-party payors may also control access to,promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality ormanage utilization of, our products with various utilization management techniques, such as requiring prior authorization for coverage of our products.Withinexpanding access. In the United States,if we obtain appropriate approval inthefuture to market anypharmaceutical industry has been a particular focus ofour oral drug product candidates, those products could potentially be coveredthese efforts and has been significantly affected byvarious government health benefit programs as well as purchased by government agencies. The participation in such programs or the sale of products to such agencies is subject to regulation. The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement.Medicaid is a jointfederal and stateprogram that is administered bylegislative initiatives, including those designed to limit thestates for low income and disabled beneficiaries. Under the Medicaid Drug Rebate Program, participating manufacturers are required to pay a rebate for each unit of product reimbursed by the state Medicaid programs. The amount of the rebate for each product is set by law and may be subject to an additional discount if certainpricing,increases more than inflation.Medicare is a federal program that is administered by the federal government that covers individuals age 65 and over as well as those with certain disabilities. Oral drugs may be covered under Medicare Part D. Medicare Part D provides coverage to enrolled Medicare patients for self-administered drugs (i.e., drugs that do not need to be injected or otherwise administered by a physician). Medicare Part D is administered by private prescription drug plans approved by the U.S. government and each drug plan establishes its own Medicare Part D formulary for prescription drugcoverage, andpricing,reimbursement of pharmaceutical and biopharmaceutical products, especially under government-funded health care programs, and increased governmental control of drug pricing.In March 2010, the ACA was signed into law, which substantially changed the
drug plan may modify from time-to-time.way healthcare is financed by both governmental and private insurers in the United States, and significantly affected the pharmaceutical industry. Theprescription drug plans negotiate pricing withACA contains a number of provisions of particular import to the pharmaceutical and biotechnology industries, including, but not limited to, those governing enrollment in federal healthcare programs, a new methodology by which rebates owed by manufacturersand may condition formulary placement on the availability ofmanufacturer discounts. Since 2011, manufacturers with marketed brand name drugs have been required to provide a 50% discount the negotiated price for on brand name prescription drugs utilized by Medicare Part D beneficiaries when those beneficiaries reach the coverage gap in their drug benefits, and, beginning in 2019, that discount increased to 70%.Drug products are subject to discounted pricing when purchased by federal agencies via the Federal Supply Schedule (FSS). FSS participation is required for a drug product to be covered and reimbursed by certain federal agencies and for coverage under Medicaid, Medicare Part B and the Public Health Service (PHS) pharmaceutical pricing program. FSS pricing is negotiated periodically with the Department of Veterans Affairs. FSS pricing is intended not to exceed the price that a manufacturer charges its most-favored non-federal customer for its product. In addition, prices for drugs purchased by the Veterans Administration, Department of Defense (including drugs purchased by military personnel and dependents through the TRICARE retail pharmacy program), Coast Guard, and PHS are subject to a cap on pricing (known as the "federal ceiling price") and may be subject to an additional discount if pricing increases more than the rate of inflation.To maintain coverage of drugsunder the Medicaid Drug Rebate Programmanufacturersarerequiredcalculated for drugs that are inhaled, infused, instilled, implanted or injected, and annual fees based on pharmaceutical companies’ share of sales toextend discountsfederal health care programs. Since its enactment, there have been judicial, Congressional, and executive branch challenges to certainpurchasers underaspects of thePHS pharmaceutical pricing program. Purchasers eligibleACA. For example, the Tax Act, was enacted on December 22, 2017, which includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health insurance fordiscounts include hospitalsall or part of a year thatserveis commonly referred to as the “individual mandate”. On December 14, 2018, adisproportionate shareU.S. District Court Judge in the Northern District offinancially needy patients, community health clinicsTexas, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the Tax Act, the remaining provisions of the ACA are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court’s decision that the individual mandate was unconstitutional but remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. It is unclear how these decisions, subsequent appeals, if any, and otherentities that receive health services grants fromefforts to challenge, repeal or replace thePHS.ACA will impact the law.The United StatesOther legislative changes have been proposed and adopted since the ACA was enacted, including aggregate reductions of Medicare payments to providers of 2% per fiscal year and reduced payments to several types of Medicare providers. Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and stategovernments continue to propose and passlegislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reformdelivery of, or paymentgovernment program reimbursement methodologies forhealth care, which include initiatives to reducedrug products. At thecost of healthcare. For example, in March 2010, the United States Congress enacted the Patient Protection and Affordable Care Act and the Health Care and Education Reconciliation Act ("Healthcare Reform Act") which includes changes to the coverage and reimbursement of drug products under government health care programs. Underfederal level, the Trumpadministration, there have been ongoing effortsadministration’s budget proposal for fiscal year 2019 contains further drug price control measures that could be enacted during the 2020 budget process or in other future legislation, including, for example, measures tomodify or repeal all or certain provisions of the Healthcare Reform Act. For example, tax reform legislation was enacted at the end of 2017 that eliminates the tax penalty for individuals who do not maintain sufficient health insurance coverage beginning in 2019 (the so-called "individual mandate"). In a May 2018 report, the Congressional Budget Office estimated that, compared to 2018, the number of uninsured will increase by 3 million in 2019 and 6 million in 2028, in part due to the elimination of the individual mandate. The Healthcare Reform Act has also been subject to judicial challenge. In December 2018, a federal district court judge, in a challenge brought by a number of state attorneys general, found the Healthcare Reform Act unconstitutional in its entirety because, once Congress repealed the individual mandate provision, there was no longer a basis to rely on Congressional taxing authority to support enactment of the law. Pending appeals, which could take some time, the Healthcare Reform Act is still operational in all respects.There have also been other reform initiatives under the Trump Administration, including initiatives focused on drug pricing. For example, in May of 2018, President Trump and the Secretary of the Department of Health and Human Services released a "blueprint" to lower prescription drug prices and out-of-pocket costs. Certain proposals in the blueprint, and related drug pricing measures proposed since the blueprint, could cause significant operational and reimbursement changes for the pharmaceutical industry. As another example, in November of 2018, CMS issued an advance notice of proposed rulemaking that proposed revisions to Medicare Part D to support health plans' negotiation of lower drug prices with manufacturers and reduce health plan members' out-of-pocket costs. The HHS Office of Inspector General also issued a proposed rule in February of 2019 that would revise the federal anti-kickback statute to limit protection for discounts offered by pharmaceutical manufacturers to pharmacy benefit managers ("PBMs"),permit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid, andMedicaid managed care plansto eliminate cost sharing for generic drugs for low-income patients. Additionally, the Trump administration released a “Blueprint” to lower drug prices and reduce the out of pocket costs of prescription drugs that contains additional proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. Although some of these, and other, proposals will require authorization through additional legislation to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.Table of ContentsData Privacy and Security Lawsthat are not reflected in the price charged to the patient at the pharmacy counter and to provide protection only for certain types of service fees paid by pharmaceutical manufacturers to PBMs.Recently, there has been considerable public and government scrutiny in the U.S. of pharmaceutical pricing and proposals to address the perceived high cost of pharmaceuticals. There have also been several recent state legislative efforts to address drug costs, which generally have focused on increasing transparency around drug costs or limiting drug prices or price increases. Adoption of new legislation at the federal or state level could affect demand for, or pricing of, our product candidates if approved for sale.We cannot predict the ultimate content, timing or effect of any changes to the Healthcare Reform Act or other federal and state reform efforts. There is no assurance that federal or state health care reform will not adversely affect our future business and financial results.Although we currently have no products approved for commercial sale, we marketed FARESTON® through September 30, 2012 and the product was covered under various government health benefit programs as well as purchased by federal agencies. We could be subject to liability under federal laws regulating our participation in such programs or the sale of our product to such agencies if we failed to comply with applicable requirements, including reporting prices for our products or offering products for sale at certain prices.Regulations Pertaining to Sales and MarketingAlthough we currently have no products approved for commercial sale, wePharmaceutical companies may be subject tovariousU.S. federal and state health information privacy, security and data breach notification laws, which may govern the collection, use, disclosure and protection of health-related and other personal information. HIPAA imposes privacy, security and breach reporting obligations with respect to individually identifiable health information upon “covered entities” (health plans, health care clearinghouses and certain health care providers), and their respective business associates, individuals or entities that create, received, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HIPAA mandates the reporting of certain breaches of health information to HHS, affected individuals and if the breach is large enough, the media. Entities that are found to be in violation of HIPAA as the result of a breach of unsecured PHI, a complaint about privacy practices or an audit by HHS, may be subject to significant civil, criminal and administrative fines and penalties and/or additional reporting and oversight obligations if required to enter into a resolution agreement and corrective action plan with HHS to settle allegations of HIPAA non-compliance. Even when HIPAA does not apply, according to the Federal Trade Commission or the FTC, failing to take appropriate steps to keep consumers’ personal information secure constitutes unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act, or the FTCA, 15 U.S.C § 45(a). The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards. The FTC’s guidance for appropriately securing consumers’ personal information is similar to what is required by the HIPAA Security Rule.In addition, certain state laws govern the privacy and security of health information in certain circumstances, some of which may be more stringent, broader in scope or offer greater individual rights with respect to protected health information, or PHI, than HIPAA, and many of which differ from each other, thus complicating compliance efforts. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties and private litigation. For example, California enacted the California Consumer Privacy Act, or the CCPA, on June 28, 2018, which took effect on January 1, 2020. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing, and receive detailed information about how their personal information is used. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA may increase our compliance costs and potential liability.
In the European Union, as of May 25, 2018, Regulation 2016/676, known as the General Data Protection Regulation, or GDPR, replaced the Data Protection Directive with respect to the processing of personal data in the European Union. The GDPR imposes many requirements for controllers and processors of personal data, including, for example, higher standards for obtaining consent from individuals to process their personal data, more robust disclosures to individuals and a strengthened individual data rights regime, shortened timelines for data breach notifications, limitations on retention and secondary use of information, increased requirements pertaining to health
care "frauddata andabuse," including anti-kickback lawspseudonymized (i.e., key-coded) data andfalse claims laws for activities related to our previous salesadditional obligations when we contract third-party processors in connection with the processing ofFARESTON®, which we sold to a third party in 2012, or to future sales of any of our product candidates that may inthefuture receive regulatory and marketing approval. Anti-kickback laws generally prohibit a prescription drug manufacturer from soliciting, offering, receiving, or paying any remuneration to generate business, including the purchase or prescription of a particular drug. Although the specific provisions of these laws vary, their scope is generally broad and there may not be regulations, guidance or court decisions that apply the laws to particular industry practices. There is therefore a possibility that our practices might be challenged under such anti-kickback laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented, any claims for payment for reimbursed drugs or services to third party payors (including Medicare and Medicaid) that are false or fraudulent. Violations of fraud and abuse laws may be punishable by criminal or civil sanctions, including fines and civil monetary penalties, and/or exclusion from federal health care programs (including Medicare and Medicaid).Laws and regulations have been enacted by the federal government and variouspersonal data. The GDPR allows EU member states toregulate the sales and marketing practices of pharmaceutical manufacturers with marketed products. Themake additional laws and regulationsgenerally limitfurther limiting the processing of genetic, biometric or health data. Failure to comply with the requirements of GDPR and the applicable national data protection laws of the EU member states may result in fines of up to €20 million or up to 4% of the total worldwide annual turnover of the preceding financialinteractions between manufacturersyear, whichever is higher, andhealth care providers and/or require disclosureother administrative penalties.On March 6, 2019, we, then operating as GTx, Inc. (“GTx”), entered into an Agreement and Plan of Merger and Reorganization, as amended (the “Merger Agreement”), with privately-held Oncternal Therapeutics, Inc. (“Private Oncternal”) and Grizzly Merger Sub, Inc., our wholly-owned subsidiary (“Merger Sub”). Under the Merger Agreement, Merger Sub merged with and into Private Oncternal, with Private Oncternal surviving as our wholly-owned subsidiary (the “Merger”). On June 7, 2019, the Merger was completed. GTx changed its name to Oncternal Therapeutics, Inc., and Private Oncternal, which remains as our wholly-owned subsidiary, changed its name to Oncternal Oncology, Inc. On June 10, 2019, the combined company’s common stock began trading on The Nasdaq Capital Market under the ticker symbol “ONCT.”
Pursuant to the
government and publicterms ofsuch interactions. Manythe Merger Agreement, each outstanding share ofthese laws and regulations contain ambiguous requirements or require administrative guidance for implementation. Given the lack of clarity in laws and their implementation, ourPrivate Oncternal common stock outstanding immediately prioractivities (when we marketed FARESTON®) or any future activities (if we obtain approval and/or reimbursement from federal healthcare programs for our product candidates) could be subjectto thepenalty provisionsclosing of thepertinent lawsMerger was converted into approximately 0.073386 shares of our common stock (the “Exchange Ratio”), after taking into account a one-for-seven reverse stock split of our then-outstanding common stock (the “Reverse Stock Split”). Immediately prior to the closing of the Merger, all shares of Private Oncternal preferred stock then outstanding were exchanged into shares of common stock of Private Oncternal. In addition, all outstanding options exercisable for common stock of Private Oncternal andregulations.Tablewarrants exercisable for convertible preferred stock ofContentsPrivate Oncternal became options and warrants exercisable for the same number of shares of common stock of the Company multiplied by the Exchange Ratio. Immediately following the Merger, stockholders of Private Oncternal owned approximately 77.5% of the outstanding common stock of the combined company. The par value and the authorized shares of our common stock were not adjusted as a result of the Reverse Stock Split.The transaction was accounted for as a reverse asset acquisition in accordance with generally accepted accounting principles in the United States of America (“GAAP”). Under this method of accounting, Private Oncternal was deemed to be the accounting acquirer for financial reporting purposes. This determination was primarily based on the facts that, immediately following the Merger: (i) Private Oncternal’s stockholders owned a substantial majority of the voting rights in the combined company, (ii) Private Oncternal designated a majority of the members of the initial board of directors of the combined company, and (iii) Private Oncternal’s senior management holds all key positions in the senior management of the combined company. As a result, as of the closing date of the Merger, our net assets were recorded at their acquisition-date relative fair values in our condensed consolidated financial statements and the reported operating results prior to the Merger are those of Private Oncternal.
Employees
As of
December 31, 2018,March 6, 2020, we had21eleven full-time employees,6three part-time employees, and a number of consultants, most of whomwere M.D.s, Pharm.D.s and/or Ph.D.s.are engaged in research and development activities. None of our employees aresubject to arepresented by labor unions or covered by collective bargainingagreement.agreements. Webelieve that we have good relationsconsider our relationship with ouremployees.employees to be good.Our corporate headquarters are located in San Diego, California, where we currently sublease 4,677 square feet of office space used primarily for corporate, research, development, clinical, regulatory, manufacturing and quality functions. Our sublease for this facility expires in March 2021.
Corporate Information
We were
originallyincorporated under the name Genotherapeutics, Inc. in Tennessee in September 1997. We changed our name to GTx, Inc. in 2001 andwereincorporated in Delaware in 2003. On March 6, 2019, we, then operating as GTx, Inc., entered into the Merger Agreement with Private Oncternal and Merger Sub. Under the Merger Agreement, Merger Sub merged with and into Private Oncternal, with Private Oncternal surviving as our wholly-owned subsidiary. On June 7, 2019, the Merger was completed and GTx, Inc. changed its name to Oncternal Therapeutics, Inc. Private Oncternal, which remains as our wholly-owned subsidiary, changed its name to Oncternal Oncology, Inc. On June 10, 2019, the combined company’s common stock began trading on The Nasdaq Capital Market under the ticker symbol “ONCT.”Our principal executive
office isoffices are located at175 Toyota Plaza, 7th Floor, Memphis, TN 38103,12230 El Camino Real, Suite 300, San Diego, CA 92130, and our telephone number is(901) 523-9700.(858) 434-1113. Our website address is www.oncternal.com.We file electronically with the
U.S.Securities and Exchange Commission, or SEC, our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-Kand amendments to those reports filed or furnishedpursuant to Section 13(a) or 15(d) of the Securities Exchange Act of1934.1934, as amended. We make available on ourWeb sitewebsite atwww.gtxinc.com,www.oncternal.com, free of charge, copies of these reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. The SEC maintains a website that contains reports, proxy and informationprovided onstatements and other information regarding issuers that file electronically with the SEC. The address of that website is www.sec.gov. The information in or accessible through the SEC and ourWeb site iswebsite are not incorporated into, and are not considered part of, thisreport, and is therefore not incorporated by reference unless such information is otherwise specifically referenced elsewhere in this report.Executive Officers offiling. Further, our references to theRegistrant
URLs for these websites are intended to be inactive textual references only.The following table sets forth information about our executive officers as of March 12, 2019:NameAgePosition(s)Executive OfficersMarc S. Hanover56Chief Executive OfficerRobert J. Wills, Ph.D65Executive ChairmanHenry P. Doggrell70Vice President, Chief Legal Officer and SecretaryJason T. Shackelford43Vice President, Finance and Accounting, and Principal Financial and Accounting OfficerMarc S. Hanover,a co-founder of GTx, served as our President and Chief Operating Officer from our inception in September 1997 until his appointment as our permanent Chief Executive Officer in February 2015, and served as our acting Principal Financial Officer from December 31, 2013 until his appointment as our interim Chief Executive Officer on April 3, 2014. He also previously served as a member of our Board of Directors from September 1997 to August 2011. Prior to joining GTx, Mr. Hanover was a founder of Equity Partners International, Inc., a private equity firm in Memphis, Tennessee, and participated as a founder and investor in three healthcare companies. From 1985 to 1997, Mr. Hanover was a Senior Vice President and a member of the Executive Management Committee of National Bank of Commerce in Memphis, Tennessee. Mr. Hanover holds a B.S. in Biology from the University of Memphis and an MBA in Finance from the University of Memphis.Robert J. Wills, Ph.D., joined GTx as Executive Chairman of the Board of Directors and as the Chairman of the Board's Scientific and Development Committee on March 2, 2015. Prior to joining GTx, Dr. Wills served as Vice President, Alliance Manager for Johnson & Johnson (J&J) and was responsible for managing strategic alliances for J&J's Pharmaceutical Group worldwide since 2002. Prior to this, Dr. Wills spent 22 years in pharmaceutical drug development, 12 of which were at J&J and 10 of which were at Hoffmann-La Roche Inc. Before assuming his alliance management role atJ&J, Dr. Wills served as Senior Vice President Global Development at J&J where he was responsible for its late stage development pipeline and was a member of several internal commercial and research and development operating boards. Since 2015, Dr. Wills has served as the chairman of the board of Cymabay Therapeutics Inc. (Nasdaq: CBAY). Dr. Wills holds a B.S. in Biochemistry and a M.S. in Pharmaceutics from the University of Wisconsin and a Ph.D. in Pharmaceutics from the University of Texas.Henry P. Doggrellcurrently serves as our Vice President, Chief Legal Officer and Secretary, after joining GTx in October 2001 as General Counsel and Secretary. From April 1998 to August 2001, Mr. Doggrell was Senior Vice President, Corporate Affairs at Buckeye Technologies, Inc., a specialty cellulose company, where he was responsible for matters including corporate finance, investor relations, mergers and acquisitions, intellectual property and licensing and strategic development. From 1996 to 1998, Mr. Doggrell served as General Counsel and Secretary of Buckeye Technologies. Prior to joining Buckeye Technologies, Mr. Doggrell was a partner of the Baker, Donelson, Bearman, Caldwell and Berkowitz law firm from 1988 to 1996, where he served as a member of the law firm management committee and Chair of the firm's Corporate Securities department. Mr. Doggrell holds a B.S. in Commerce from the University of Virginia and a JD from Vanderbilt University.Jason T. Shackelfordcurrently serves as our Vice President, Finance and Accounting, after joining GTx in July 2007 as Director, Accounting and Corporate Controller, and has served as our principal accounting officer since December 31, 2013 and as our principal financial and accounting officer since April 3, 2014. Prior to joining GTx, Mr. Shackelford was a Senior Audit Manager at KPMG LLP. Mr. Shackelford is a Certified Public Accountant and holds a Bachelor of Business Administration and Master of Accountancy from the University of Mississippi.Item 1A.
We have identifiedYou should consider carefully the followingadditional risksrisk factors, together with the other information contained in this annual report on Form 10-K, including our financial statements anduncertaintiesthe related notes and “Management Discussion and Analysis of Financial Condition and Results of Operations,” before making a decision to purchase or sell shares of our common stock. We cannot assure you thatmay have a material adverse effect onany of the events discussed in the risk factors below will not occur. If any of the following events actually occur, our business, operating results, prospects or financial condition could be materially and adversely affected. This could cause the trading price of our common stock to decline and you may lose all orresultspart ofoperations. Investors should carefully consider theyour investment. The risks described belowbefore making an investment decision. Our business faces significant risks and the risks described below mayare notbethe onlyrisksones that we face. Additional risks not presently known to us or that we currentlybelieve aredeem immaterial may alsosignificantlyaffect our business operations or financial condition.Risks Related to Our Limited Operating History, Financial Position and Capital Requirements
We have a limited operating history, have incurred significant operating losses since our inception and expect to incur significant losses for the foreseeable future. We may never generate any revenue or become profitable or, if we achieve profitability, we may not be able to sustain it.
Biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. We are a clinical-stage biopharmaceutical company with a limited operating history upon which you can evaluate our business and prospects. To date, we have focused primarily on organizing and staffing our company, business planning, raising capital, identifying, acquiring and in-licensing our product candidates and conducting preclinical studies and early-stage clinical trials. Cirmtuzumab and TK216 are in clinical development, while our ROR1 CAR-T program is in the preclinical stage. We have not yet demonstrated an ability to successfully obtain regulatory approvals, manufacture a commercial scale product, or arrange for a third-party to do so on our behalf, or embark on sales and marketing activities necessary for successful post regulatory approval product commercialization, and have not developed any companion diagnostic test for our product candidates. Consequently, any predictions made about our future success or viability may not be as accurate as they could be if we had a history of successfully developing and commercializing biopharmaceutical products.
We have incurred significant operating losses since our inception. If our product candidates are not successfully developed and approved, we may never generate any revenue. Our net losses were $34.2 million ($18.1 million related to nonrecurring Merger costs) and $6.6 million for the years ended December 31, 2019 and 2018, respectively. As of December 31, 2019, we had an accumulated deficit of $65.6 million. Substantially all of our losses have resulted from expenses incurred in connection with our research and development programs and from general and administrative costs associated with our operations. All of our product candidates will require substantial additional development time and resources before we would be able to apply for or receive regulatory approvals and begin generating revenue from product sales. We expect to continue to incur losses for the foreseeable future, and anticipate these losses will increase substantially as we continue to develop, seek regulatory approval for and potentially commercialize any of our product candidates, and seek to identify, assess, acquire, in-license or develop additional product candidates.
To become and remain profitable, we must succeed in developing and eventually commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing clinical trials and preclinical studies of our product candidates, obtaining regulatory approval for these product candidates and manufacturing, marketing and selling any products for which we may obtain regulatory approval. We are only in the preliminary stages of most of these activities. We may never succeed in these activities and, even if we do, may never generate revenues that are significant enough to achieve profitability. In addition, we have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical industry. Because of the numerous risks and uncertainties associated with biopharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress our value and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product candidates or even continue our operations. A decline in our company’s value could also cause stockholders to lose all or part of their investment.
We will require substantial additional financing to achieve our goals, and a failure to obtain this necessary capital when needed and on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our product development programs, commercialization efforts or other operations.
The development of biopharmaceutical product candidates is capital-intensive. We expect our expenses to increase in connection with our ongoing activities, particularly as we conduct our ongoing and planned clinical trials of cirmtuzumab and TK216, continue research and development and initiate clinical trials of our other development programs and seek regulatory approval for our current product candidates and any future product candidates we may develop. In addition, as our product candidates progress through development and toward commercialization, we will need to make milestone payments to the licensors and other third parties from whom we have in-licensed or acquired our product candidates, including cirmtuzumab, TK216 and ROR1 CAR-T. If we obtain regulatory approval for any of our product candidates, we also expect to incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution. Because the outcome of any clinical trial or preclinical study is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of our product candidates. Furthermore, following the completion of the Merger, we have incurred additional costs associated with operating as a public company. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts.
We have based our estimates on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect. Our operating plans and other demands on our cash resources may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through a combination of equity financings, debt financings, government funding or other capital sources, including potentially collaborations, licenses and other similar arrangements. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. Attempting to secure additional financing may divert our management from our day-to-day activities, which may adversely affect our ability to develop our product candidates.
Our future capital requirements will depend on many factors, including:
the type, number, scope, progress, expansions, results, costs and timing of, our clinical trials of cirmtuzumab and TK216, and preclinical studies or clinical trials of other product candidates that we may choose to pursue in the future;
the costs and timing of manufacturing for our product candidates, including commercial manufacturing if any product candidate is approved;
the costs of obtaining ibrutinib, for which we currently obtain supply at no cost under our clinical supply agreement with Pharmacyclics LLC, and vincristine to conduct our clinical trials of cirmtuzumab and TK216, respectively;
the costs of obtaining, maintaining and enforcing our patents and other intellectual property rights;
our efforts to evaluate, develop or partner the SARD assets; our efforts to enhance operational systems and hire additional personnel to satisfy our obligations as a public company, including enhanced internal controls over financial reporting;
the costs associated with hiring additional personnel and consultants as our clinical and other development activities increase;
the timing and amount of the milestone or other payments we must make to the licensors and other third parties from whom we have in-licensed or acquired our product candidates or technology;
the costs and timing of establishing or securing sales and marketing capabilities if any of our product candidates are approved;
our ability to achieve sufficient market acceptance, coverage and adequate reimbursement from third-party payors and adequate market share and revenue for any approved products;
the terms and timing of establishing and maintaining collaborations, licenses and other similar arrangements; and
costs associated with any products or technologies that we may in-license or acquire.
Conducting clinical trials and preclinical studies is a time consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain regulatory approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success. Our commercial revenues, if any, will be derived from sales of products that we do not expect to be commercially available for many years, if at all.
Accordingly, we will need to continue to rely on additional financing to achieve our business objectives. Adequate additional financing may not be available to us on acceptable terms, or at all. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our current or future operating plans.
For example, we entered into an At-the-Market Equity OfferingSM Sales Agreement, or the ATM Sales Agreement, with Stifel, Nicolaus & Company, Incorporated, or Stifel, under which we may, from time to time, sell shares of the our common stock, having an aggregate offering price of up to $50.0 million, of which $25.0 remains available at December 31, 2019. However, there can be no assurance that Stifel will be successful in consummating future sales based on prevailing market conditions or in the quantities or at prices that we deem appropriate. Furthermore, under current SEC regulations, at any time during which the aggregate market value of our common stock held by non-affiliates, or public float, is less than $75.0 million, the amount we can raise through primary public offerings of securities in any twelve-month period using shelf registration statements, including sales under the ATM Sales Agreement, is limited to an aggregate of one-third of our public float. As of February 28, 2020, our calculated public float was approximately $51.8 million, which would have yielded a capacity to issue up to $17.3 million of shares of common stock pursuant to the ATM Sales Agreement. If our public float decreases in the future, the amount of securities we may sell under our Form S-3 shelf registration statement will also decrease. In addition, the ATM Sales Agreement may be terminated by us or Stifel at any time upon notice to the other party.
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through equity offerings, debt financings, government funding or other capital sources, including potentially collaborations, licenses and other similar arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, existing stockholders’ ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect stockholders’ rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
If we raise funds through future collaborations, licenses and other similar arrangements, we may have to relinquish valuable rights to our future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us and/or that may reduce the value of our common stock.
Risks Related to the Discovery, Development and Regulatory Approval of Our Product Candidates
We depend heavily on the success of cirmtuzumab and TK216, which are in Phase 1 or Phase 2 clinical trials, as well as our ROR1 CAR-T program, which is in preclinical development. If we are unable to advance our product candidates in clinical development, obtain regulatory approval and ultimately commercialize our product candidates, or experience significant delays in doing so, our business will be materially harmed.
Our two clinical-stage product candidates, cirmtuzumab and TK216, are in Phase 1 or Phase 2 clinical development. Cirmtuzumab is being evaluated in a Phase 1/2 clinical trial in combination with ibrutinib (Imbruvica®) for the treatment of patients with B-cell lymphoid malignancies, including MCL and CLL and in an investigator-sponsored, Phase 1b clinical trial in combination with paclitaxel for the treatment of women with HER2-negative metastatic or locally advanced, unresectable breast cancer. We are also developing TK216, an investigational small molecule that is designed to inhibit the ETS, or E26 Transformation Specific, family of oncoproteins, which have been shown in preclinical studies to alter gene transcription and RNA processing and lead to increased cell proliferation and invasion. TK216 is being evaluated in a Phase 1 clinical trial as a single agent and in combination with vincristine in patients with relapsed or refractory Ewing sarcoma, a rare pediatric cancer. In addition, we are developing a chimeric antigen receptor T cell, or CAR-T, therapy candidate that targets ROR1, which is currently in preclinical development as a potential treatment for hematologic cancers and solid tumors. None of our product candidates have advanced into a pivotal or registrational study for the indications for which we are studying them. Our ability to generate product revenues, which we do not expect will occur for many years, if ever, will depend heavily on the successful development and eventual commercialization of our product candidates. The success of our product candidates will depend on various factors, including the following:
successful completion of preclinical and clinical studies with favorable results;
acceptance of investigation new drug applications, or INDs, by the U.S. Food and Drug Administration, or FDA, or similar regulatory filing by comparable foreign regulatory authorities for the conduct of clinical trials of our product candidates and our proposed designs for future clinical trials;
demonstrating safety and efficacy of our product candidates to the satisfaction of applicable regulatory authorities;
receiving marketing approvals from applicable regulatory authorities, including Biologics License Applications, or BLAs, or new drug applications, or NDAs, from the U.S. Food and Drug Administration, or the FDA, and maintaining such approvals;
making arrangements with our third-party manufacturers for commercial manufacturing capabilities for our product candidates;
establishing sales, marketing and distribution capabilities and launching commercial sales of our product candidates, if and when approved, whether alone or in collaboration with others;
•
establishing and maintaining patent and trade secret protection or regulatory exclusivity for our product candidates;
the demonstration of an acceptable safety profile of our products following approval, if any;
developing, in-licensing or acquiring companion diagnostics to our product candidates; and
maintaining and growing an organization for people who can develop our product candidates and technology.
The success of our business, including our ability to finance the company and generate any revenue in the future, will primarily depend on the successful development, regulatory approval and commercialization of our product candidates, which may never occur. We have not yet succeeded and may not succeed in demonstrating efficacy and safety for any of our product candidates in clinical trials or in obtaining marketing approval thereafter. Given our early stage of development, it may be several years, if at all, before we have demonstrated the safety and efficacy of a product candidate sufficient to warrant approval for commercialization. If we are unable to develop, or obtain regulatory approval for, or, if approved, successfully commercialize our product candidates, we may not be able to generate sufficient revenue to continue our business.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome, and the results of preclinical studies and early clinical trials are not necessarily predictive of future results. Our product candidates may not have favorable results in clinical trials or receive regulatory approval on a timely basis, if at all.
Clinical drug development is expensive and can take many years to complete, and its outcome is inherently uncertain. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all, and failure can occur at any time during the preclinical study or clinical trial process. Despite promising preclinical or clinical results, any product candidate can unexpectedly fail at any stage of preclinical or clinical development. The historical failure rate for product candidates in our industry is high.
The results from preclinical studies or clinical trials of a product candidate may not predict the results of later clinical trials of the product candidate, and interim results of a clinical trial are not necessarily indicative of final results. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy characteristics despite having progressed through preclinical studies and initial clinical trials. In particular, while cirmtuzumab was well tolerated and showed favorable results in the Phase 1 portion of our ongoing Phase 1/2 clinical trial as well as the inhibition of ROR1 signaling in patients with CLL in early clinical trials, we do not know how cirmtuzumab will perform in the Phase 2 portion of the clinical trial and one or more of the reported clinical outcomes may materially change as patient enrollment continues in such trial, and such results may not be replicated in any other future clinical trials, including as a result of any differences in the target population, drug interactions or other differences in our trial design. It is not uncommon to observe results in clinical trials that are unexpected based on preclinical studies and early clinical trials, and many product candidates fail in clinical trials despite very promising early results. Moreover, this and any future preclinical and clinical data may be susceptible to varying interpretations and analyses. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical development even after achieving promising results in earlier studies. Furthermore, we cannot assure you that we will be able to successfully progress our preclinical programs from candidate identification to Phase 1 clinical development.
For the foregoing reasons, we cannot be certain that our ongoing and planned clinical trials and preclinical studies will be successful. Any safety concerns observed in any one of our clinical trials in our targeted indications could limit the prospects for regulatory approval of our product candidates in those and other indications, which could have a material adverse effect on our business, financial condition and results of operations.
Any difficulties or delays in the commencement or completion, or termination or suspension, of our current or planned clinical trials could result in increased costs to us, delay or limit our ability to generate revenue, and adversely affect our commercial prospects.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical studies to demonstrate the safety and efficacy of the product candidates in humans. We are conducting a Phase 1/2 clinical trial of cirmtuzumab in combination with ibrutinib in patients with treatment-naïve or relapsed or refractory CLL and previously treated patients with MCL. Additionally, we are evaluating TK216 as a single agent and in combination with vincristine in a Phase 1 clinical trial in patients with relapsed or refractory Ewing sarcoma. We will have to follow the same procedure for our other preclinical product candidates that we plan to advance to clinical development, and would also be required to submit regulatory filings to foreign regulatory authorities if we decide to initiate clinical trials outside of the United States.
We do not know whether our planned trials will begin on time or be completed on schedule, if at all. The commencement and completion of clinical trials can be delayed for a number of reasons, including delays related to:
the FDA or comparable foreign regulatory authorities disagreeing as to the design or implementation of our clinical studies;
difficulties in obtaining regulatory authorizations to commence a trial or reaching a consensus with regulatory authorities on trial design;
difficulties in recruiting clinical trial investigators with the appropriate competencies and experience;
failure or delay in reaching an agreement with CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
delays in obtaining approval from one or more institutional review boards, or IRBs;
IRBs refusing to approve, suspending or terminating the trial at an investigational site, precluding enrollment of additional subjects, or withdrawing their approval of the trial;
changes to clinical trial protocols;
clinical sites deviating from trial protocols or dropping out of a trial;
challenges in manufacturing sufficient quantities of product candidates or obtaining sufficient quantities of combination therapies for use in clinical trials;
subjects failing to enroll or remain in our trial at the rate we expect, or failing to return for post-treatment follow-up;
subjects choosing an alternative treatment for the indication for which we are developing our product candidates, or participating in competing clinical trials;
lack of adequate funding to continue clinical trials;
subjects experiencing severe or unexpected drug-related adverse effects;
occurrence of serious adverse events in clinical trials of the same class of agents conducted by other companies;
selection of clinical endpoints that require prolonged periods of clinical observation or analysis of the resulting data;
a facility manufacturing our product candidates or any of their components being ordered by the FDA or comparable foreign regulatory authorities to temporarily or permanently shut down due to violations of current Good Manufacturing Practices, or cGMP, regulations or other applicable requirements, or infections or cross-contaminations of product candidates in the manufacturing process;
third-party clinical investigators losing the licenses or permits necessary to perform our clinical trials, not performing our clinical trials in a timely manner or consistent with applicable clinical trial protocols, good clinical practices, or GCP, or other regulatory requirements; third-party contractors not performing data collection or analysis in a timely or accurate manner; or
third-party contractors becoming debarred or suspended or otherwise penalized by the FDA or other government or regulatory authorities for violations of regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or all of the data produced by such contractors in support of our marketing applications.
We could also encounter delays if our clinical trials are suspended or terminated by us, by the IRBs of the institutions in which such trials are being conducted, by a Data Safety Monitoring Board for such trial, or by the FDA or comparable foreign regulatory authorities. Regulatory authorities may suspend or terminate clinical trials due to a number of factors, including failure to conduct clinical trials in accordance with regulatory requirements or the applicable clinical protocols, inspection of the clinical trial operations or trial site by the FDA or comparable foreign regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. In addition, changes in regulatory requirements and policies may occur, and we may need to amend clinical trial protocols to comply with these changes. Amendments may require us to resubmit our clinical trial protocols to IRBs for reexamination, which may impact the costs, timing or successful completion of a clinical trial.
Further, if we decide to conduct clinical trials of our product candidates in foreign countries additional risks may arise that may delay completion of those clinical trials. These risks include the failure of enrolled patients in other countries to adhere to clinical protocol as a result of differences in healthcare practices or cultural customs, managing additional administrative burdens associated with the regulatory schemes of other countries, as well as political and economic risks relevant to other countries. Under our license and development agreement with SPH USA, SPH USA has the right to manufacture, develop, market, distribute and sell our cirmtuzumab, ROR1 CAR-T, and TK216 product candidates in the People’s Republic of China, Hong Kong, Macau and Taiwan, or Greater China, and the obligation to perform all preclinical and clinical development activities required to obtain regulatory approvals for such product candidates in Greater China. In the event that SPH USA’s preclinical studies or clinical trials of our product candidates raise new safety or efficacy concerns, the prospects for obtaining regulatory approval of our product candidates in the United States and other countries, and our business, could be adversely impacted.
Moreover, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, we may be required to report some of these relationships to the FDA or comparable foreign regulatory authorities. The FDA or comparable foreign regulatory authority may conclude that a financial relationship between us and a principal investigator has created a conflict of interest or otherwise affected interpretation of the study. The FDA or comparable foreign regulatory authority may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA or comparable foreign regulatory authority, as the case may be, and may ultimately lead to the denial of marketing approval of one or more of our product candidates.
If we experience delays in the completion of, or termination of, clinical trials of our product candidates, the commercial prospects of such product candidates may be harmed, and our ability to generate product revenues from such product candidates may be delayed. Moreover, delays in completing our clinical trials may increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues.
In addition, many of the factors that cause, or lead to, the termination, suspension or delay in the commencement or completion of, clinical trials may also ultimately lead to the denial of regulatory approval of a product candidate. If we make formulation or manufacturing changes to our product candidates or revise the route of administration or dosing regimen for our product candidates, we may be required to conduct additional preclinical or clinical studies to bridge our modified product candidates to earlier versions or to bridge the new dosing regimens to dosing regimens used in our clinical trials. The need to conduct additional preclinical or clinical studies could result in delays in the approval or commercialization of our product candidates, which could shorten any period during which we may have the exclusive right to commercialize our product candidates and enable our competitors to bring products to market before we do. In such an event, the commercial viability of our product candidates could be significantly reduced. Any of these occurrences may harm our business, financial condition and prospects significantly.
We may find it difficult to enroll patients in our clinical trials. If we encounter difficulties enrolling subjects in our clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
We may not be able to initiate or continue clinical trials for our product candidates if we are unable to identify and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory authorities outside the United States. Subject enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility and exclusion criteria for the trial, the design of the clinical trial, the availability of competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages and risks of the product candidate being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating as well as any drugs under development. We will be required to identify and enroll a sufficient number of subjects for each of our clinical trials. Potential subjects for any planned clinical trials may not be adequately diagnosed or identified with the diseases which we are targeting or may not meet the entry criteria for such trials. For example, a limited number of patients are affected by CLL, MCL and particularly Ewing sarcoma, which are our initial target indications for cirmtuzumab and TK216. We also may encounter difficulties in identifying and enrolling subjects with a stage of disease appropriate for our planned clinical trials. We may not be able to initiate or continue clinical trials if we are unable to locate a sufficient number of eligible subjects to participate in the clinical trials required by the FDA or comparable foreign regulatory authorities. In addition, the process of finding and diagnosing subjects may prove costly.
The timing of our clinical trials depends, in part, on the speed at which we can recruit patients to participate in our trials, as well as completion of required follow-up periods. For certain of our product candidates, including cirmtuzumab and TK216, the conditions which we currently plan to evaluate are orphan or rare diseases with limited patient pools from which to draw for clinical trials. The eligibility criteria of our clinical trials will further limit the pool of available trial participants. If patients are unwilling to participate in our clinical trials for any reason, including the existence of concurrent clinical trials for similar patient populations or the availability of approved therapies, or if we otherwise have difficulty enrolling a sufficient number of patients, the timeline for recruiting subjects, conducting studies and obtaining regulatory approval of our product candidates may be delayed. Our inability to enroll a sufficient number of subjects for any of our clinical trials would result in significant delays or may require us to abandon one or more clinical trials altogether. In addition, we expect to rely on CROs and clinical trial sites to ensure proper and timely conduct of our future clinical trials and, while we intend to enter into agreements governing their services, we will have limited influence over their actual performance.
We cannot assure stockholders that our assumptions used in determining expected clinical trial timelines are correct or that we will not experience delays in enrollment, which would result in the delay of completion of such trials beyond our expected timelines.
Use of our product candidates could be associated with side effects, adverse events or other properties or safety risks, which could delay or preclude approval, cause us to suspend or discontinue clinical trials, abandon a product candidate, limit the commercial profile of the label for an approved product candidate, or result in other significant negative consequences that could severely harm our business, prospects, operating results and financial condition.
As is the case with oncology drugs generally, it is likely that there may be side effects and adverse events associated with the use of our product candidates. Results of our clinical trials could reveal a high and unacceptable severity and prevalence, or unexpected characteristics of side effects. For example, our ongoing clinical trials of cirmtuzumab in combination with ibrutinib, and TK216 in combination with vincristine, and the ongoing investigator-initiated clinical trial of cirmtuzumab in combination with paclitaxel, may reveal adverse events based on the combination therapy under evaluation. Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials, result in a more restrictive label for the product candidate, or delay or cause the denial of regulatory approval of the product candidate by the FDA or comparable foreign regulatory authorities. The drug-related side effects could also affect patient recruitment for our clinical trials, or the ability of enrolled patients to complete the trials, or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Moreover, if our product candidates are associated with undesirable side effects in clinical trials or have characteristics that are unexpected, we may elect to abandon their development or limit their development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective, which may limit the commercial prospects for the product candidate if approved. We may also be required to modify our plans for future studies based on findings in our ongoing clinical trials. Many compounds that initially showed promise in early-stage testing have later been found to cause side effects that prevented further development of the compound. In addition, regulatory authorities may draw different conclusions or require additional testing to confirm these determinations.
It is possible that as we test our product candidates in larger, longer and more extensive clinical trials, or as the use of our product candidates becomes more widespread if they receive regulatory approval, illnesses, injuries, discomforts and other adverse events that were observed in earlier trials, as well as conditions that did not occur or went undetected in previous trials, will be reported by subjects. If such side effects become known later in development or upon approval, if any, such findings may harm our business, financial condition and prospects significantly.
In addition, if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
regulatory authorities may withdraw, suspend or limit approvals of such product;
we may be required to recall a product or change the way such product is administered to patients;
regulatory authorities may require additional warnings on the label, such as a “black box” warning or a contraindication;
we may be required to implement a Risk Evaluation and Mitigation Strategy, or REMS, or create a medication guide outlining the risks of such side effects for distribution to patients;
we may be required to change the way a product is distributed or administered, conduct additional clinical trials or change the labeling of a product or be required to conduct additional post-marketing studies or surveillance;
we could be sued and held liable for harm caused to patients;
sales of the product may decrease significantly or the product could become less competitive; and
our reputation could suffer.
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
The regulatory landscape that will apply to development of gene therapy or cell-based therapeutic product candidates by us or by our collaborators is rigorous, complex, uncertain and subject to change, which could result in delays or termination of development of such product candidates or unexpected costs in obtaining regulatory approvals.
Regulatory requirements governing products involving gene therapy treatment have changed frequently and will likely continue to change in the future. Approvals by one regulatory agency may not be indicative of what any other regulatory agency may require for approval, and there is substantial, and sometimes uncoordinated, overlap in those responsible for regulation of gene therapy products, cell therapy products and other products created with genome editing technology. For example, in addition to the submission of an IND to the FDA, before initiation of a clinical trial in the United States, certain human clinical trials for cell therapy products and gene therapy are subject to the National Institutes of Health Guidelines for Research Involving Recombinant DNA Molecules, or NIH Guidelines. The NIH Guidelines call for the supervision of human gene transfer trials including an evaluation and assessment by an institutional biosafety committee, or IBC, a local institutional committee that reviews and oversees research utilizing recombinant or synthetic nucleic acid molecules at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment, and such review may result in some delay before initiation of a clinical trial. While the NIH Guidelines are not mandatory unless the research in question is being conducted at or sponsored by institutions receiving NIH funding of recombinant or synthetic nucleic acid molecule research, many companies and other institutions not otherwise subject to the NIH Guidelines voluntarily follow them. We will therefore be subject to significant regulatory oversight by the FDA, and in addition to the government regulators, the applicable IBC and IRB of each institution at which we or our collaborators conduct clinical trials of our product candidates, or a central IRB if appropriate, would need to review and approve the proposed clinical trial.
Similar requirements apply in the European Union. The European Medicines Agency, or the EMA, has a Committee for Advanced Therapies, or CAT, that is responsible for assessing the quality, safety and efficacy of advanced therapy medicinal products. Advanced-therapy medical products include gene therapy medicine, somatic-cell therapy medicines and tissue-engineered medicines. The role of the CAT is to prepare a draft opinion on an application for marketing authorization for a gene therapy medicinal candidate that is submitted to the EMA. In the European Union, the development and evaluation of a gene therapy medicinal product must be considered in the context of the relevant European Union guidelines. The EMA may issue new guidelines concerning the development and marketing authorization for gene therapy medicinal products and require that we comply with these new guidelines. Similarly complex regulatory environments exist in other jurisdictions in which we might consider seeking regulatory approvals for our product candidates, further complicating the regulatory landscape. As a result, the procedures and standards applied to gene therapy products and cell therapy products may be applied to any of our gene therapy product candidates such as CAR-T, but that remains uncertain at this point.
The clinical trial requirements of the FDA, the EMA and other regulatory authorities and the criteria these regulators use to evaluate the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty and intended use and market of the potential products. The regulatory approval process for product candidates involving gene therapy can be more lengthy, rigorous and expensive than the process for other better known or more extensively studied product candidates and technologies. Since we are developing novel treatments for diseases in which there is little clinical experience with new endpoints and methodologies, there is heightened risk that the FDA, the EMA or comparable regulatory bodies may not consider the clinical trial endpoints to provide clinically meaningful results, and the resulting clinical data and results may be more difficult to analyze. This may be a particularly significant risk for many of the genetically defined diseases for which we may develop product candidates alone or with collaborators due to small patient populations for those diseases, and designing and executing a rigorous clinical trial with appropriate statistical power is more difficult than with diseases that have larger patient populations. Regulatory agencies administering existing or future regulations or legislation may not allow production and marketing of products utilizing gene therapy in a timely manner or under technically or commercially feasible conditions. Even if our product candidates obtain required regulatory approvals, such approvals may later be withdrawn as a result of changes in regulations or the interpretation of regulations by applicable regulatory agencies.
Changes in applicable regulatory guidelines may lengthen the regulatory review process for our product candidates, require additional studies or trials, increase development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of such product candidates, or lead to significant post-approval limitations or restrictions. Additionally, adverse developments in clinical trials of gene therapy products conducted by others may cause the FDA, the EMA and other regulatory bodies to revise the requirements for approval of any product candidates we may develop or limit the use of products utilizing gene therapy, either of which could materially harm our business. Furthermore, regulatory action or private litigation could result in increased expenses, delays or other impediments to our research programs or the development or commercialization of current or future product candidates.
As we advance our product candidates alone or with collaborators, we will be required to consult with these regulatory and advisory groups and comply with all applicable guidelines, rules and regulations. If we fail to do so, we or our collaborators may be required to delay or terminate development of such product candidates. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a product candidate to market could decrease our ability to generate sufficient product revenue to maintain our business.
As an organization, we have limited experience in the process of enrolling patients in our clinical trials, have never conducted later-stage clinical trials or submitted a BLA or an NDA, and may be unable to do so for any of our product candidates.
We are early in our development efforts for our product candidates, and will need to successfully complete later-stage and pivotal clinical trials in order to obtain FDA or comparable foreign regulatory approval to market cirmtuzumab, TK216, ROR1 CAR-T, or any future product candidates. Carrying out later-stage clinical trials and submitting a successful BLA or NDA is a complicated process. As an organization, we are in the process of conducting a Phase 1/2 clinical trial for cirmtuzumab in combination with ibrutinib and a Phase 1 clinical trial for TK216, alone and in combination with vincristine. We have not yet conducted any clinical trials for our other product candidates. We have not previously conducted any later stage or pivotal clinical trials, have limited experience as a company in preparing, submitting and prosecuting regulatory filings and have not previously submitted a BLA, an NDA or other comparable foreign regulatory submission for any product candidate. In addition, we have had limited interactions with the FDA and cannot be certain how many additional clinical trials of cirmtuzumab, TK216 or any other product candidates will be required or how such trials should be designed. We may be unable to successfully and efficiently execute and complete necessary clinical trials in a way that leads to regulatory submission and approval of any of our product candidates. We may require more time and incur greater costs than our competitors and may not succeed in obtaining regulatory approvals of product candidates that we develop. Failure to commence or complete, or delays in our planned clinical trials could delay or prevent us from submitting BLAs or NDAs for, and commercializing, our product candidates.
Our product candidates are subject to extensive regulation and compliance, which is costly and time consuming, and such regulation may cause unanticipated delays or prevent the receipt of the required approvals to commercialize our product candidates.
The clinical development, manufacturing, labeling, storage, record-keeping, advertising, promotion, import, export, marketing and distribution of our product candidates are subject to extensive regulation by the FDA in the United States and by comparable foreign regulatory authorities in foreign markets. In the United States, we are not permitted to market our product candidates until we receive regulatory approval from the FDA. The process of obtaining regulatory approval is expensive, often takes many years following the commencement of clinical trials and can vary substantially based upon the type, complexity and novelty of the product candidates involved, as well as the target indications and patient population. Approval policies or regulations may change, and the FDA has substantial discretion in the drug approval process, including the ability to delay, limit or deny approval of a product candidate for many reasons. Despite the time and expense invested in clinical development of product candidates, regulatory approval is never guaranteed. We are not permitted to market any of our product candidates in the United States until we receive approval of a BLA or an NDA from the FDA.
Prior to obtaining approval to commercialize a product candidate in the United States or abroad, we must demonstrate with substantial evidence from adequate and well-controlled clinical trials, and to the satisfaction of the FDA or comparable foreign regulatory authorities, that such product candidates are safe and effective for their intended uses, and in the case of biological products, that such product candidates are safe, pure and potent. Results from nonclinical studies and clinical trials can be interpreted in different ways. Even if we believe the nonclinical or clinical data for our product candidates are promising, such data may not be sufficient to support approval by the FDA and comparable foreign regulatory authorities. The FDA or comparable foreign regulatory authorities, as the case may be, may also require us to conduct additional preclinical studies or clinical trials for our product candidates either prior to or post-approval, or may object to elements of our clinical development program.
The FDA or comparable foreign regulatory authorities can delay, limit or deny approval of a product candidate for many reasons, including:
such authorities may disagree with the design or execution of our clinical trials;
negative or ambiguous results from our clinical trials or results may not meet the level of statistical significance required by the FDA or comparable foreign regulatory agencies for approval;
serious and unexpected drug-related side effects may be experienced by participants in our clinical trials or by individuals using drugs similar to our product candidates;
the population studied in the clinical trial may not be sufficiently broad or representative to assure safety in the full population for which we seek approval;
such authorities may not accept clinical data from trials that are conducted at clinical facilities or in countries where the standard of care is potentially different from that of their own country;
we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks;
such authorities may disagree with our interpretation of data from preclinical studies or clinical trials;
such authorities may not agree that the data collected from clinical trials of our product candidates are acceptable or sufficient to support the submission of a BLA, NDA or other submission or to obtain regulatory approval in the United States or elsewhere, and such authorities may impose requirements for additional preclinical studies or clinical trials;
such authorities may disagree with us regarding the formulation, labeling and/or the product specifications of our product candidates;
approval may be granted only for indications that are significantly more limited than those sought by us, and/or may include significant restrictions on distribution and use;
such authorities may find deficiencies in the manufacturing processes or facilities of the third-party manufacturers with which we contract for clinical and commercial supplies; or
such authorities may not accept a submission due to, among other reasons, the content or formatting of the submission.
With respect to foreign markets, approval procedures vary among countries and, in addition to the foregoing risks, may involve additional product testing, administrative review periods and agreements with pricing authorities. In addition, events raising questions about the safety of certain marketed pharmaceuticals may result in increased cautiousness by the FDA and comparable foreign regulatory authorities in reviewing new drugs based on safety, efficacy or other regulatory considerations and may result in significant delays in obtaining regulatory approvals. Any delay in obtaining, or inability to obtain, applicable regulatory approvals would prevent us or any of our potential future collaborators from commercializing our product candidates.
Of the large number of drugs in development, only a small percentage successfully complete the FDA or foreign regulatory approval processes and are commercialized. The lengthy approval process as well as the unpredictability of future clinical trial results may result in our failure to obtain regulatory approval to market our product candidates, which would significantly harm our business, financial condition, results of operations and prospects.
Even if we eventually complete clinical trials and receive approval of a BLA, NDA or comparable foreign marketing application for our product candidates, the FDA or comparable foreign regulatory authority may grant approval contingent on the performance of costly additional clinical trials, including Phase 4 clinical trials, and/or the implementation of a REMS, which may be required because the FDA believes it is necessary to ensure safe use of the drug after approval. The FDA or the comparable foreign regulatory authority also may approve a product candidate for a more limited indication or patient population than we originally requested, and the FDA or comparable foreign regulatory authority may not approve the labeling that we believe is necessary or desirable for the successful commercialization of a product. Any delay in obtaining, or inability to obtain, applicable regulatory approval would delay or prevent commercialization of that product candidate and would materially adversely impact our business and prospects.
We may expend our limited resources to pursue a particular product candidate and fail to capitalize on product candidates or indications that may be more profitable or for which there are a greater likelihood of success.
Because we have limited financial and managerial resources, we are focused on specific product candidates, indications and development programs. As a result, we may forgo or delay the pursuit of opportunities with other indications or other product candidates that could have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential for a particular product candidate, we could relinquish valuable rights to that product candidate through collaborations, licenses and other similar arrangements, when it might be more advantageous for us to retain sole development and commercialization rights to such product candidate.
Fast Track designation by the FDA for TK216 or our other product candidates may not actually lead to a faster development or regulatory review or approval process.
We have been granted a Fast Track designation for TK216 in the United States for the treatment of Ewing sarcoma and may seek Fast Track designation for cirmtuzumab or our other product candidates. The Fast Track program is intended to expedite or facilitate the process for reviewing new product candidates that meet certain criteria. Specifically, new drugs are eligible for Fast Track designation if they are intended, alone or in combination with one or more drugs, to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast Track designation applies to the combination of the product candidate and the specific indication for which it is being studied. With a Fast Track product candidate, the FDA may consider for review sections of the NDA or BLA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA or BLA, the FDA agrees to accept sections of the NDA or BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA or BLA.
Obtaining a Fast Track designation does not change the standards for product approval, but may expedite the development or approval process. Even though the FDA has granted such designation for TK216, it may not actually result in faster clinical development or regulatory review or approval. Furthermore, such a designation does not increase the likelihood that TK216 or any other product candidate that may be granted Fast Track designation will receive marketing approval in the United States.
We may not be able to obtain or maintain orphan drug designations for certain of our product candidates, and may be unable to maintain the benefits associated with orphan drug designation, including the potential for market exclusivity.
Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as orphan drugs. Under the Orphan Drug Act of 1983, the FDA may designate a product as an orphan product if it is intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals in the United States, or a patient population of greater than 200,000 individuals in the United States, but for which there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the European Union, the EMA’s, Committee for Orphan Medicinal Products grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the European Union. We have received orphan drug designation in the United States for TK216 for patients with Ewing sarcoma and we may seek orphan drug designation in the European Union for TK216 for patients with Ewing sarcoma, as well as seek orphan drug designation for certain of our other product candidates. There can be no assurance that the FDA or the EMA’s Committee for Orphan Medicinal Products will grant orphan designation for any indication for which we apply, or that we will be able to maintain such designation.
In the United States, orphan designation entitles a party to financial incentives such as opportunities for grant funding for clinical trial costs, tax advantages and user-fee waivers. In addition, if a product candidate that has orphan designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including an NDA or BLA, to market the same drug for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or where the manufacturer is unable to assure sufficient product quantity. The applicable exclusivity period is ten years in Europe, but such exclusivity period can be reduced to six years if a product no longer meets the criteria for orphan designation or if the product is sufficiently profitable so that market exclusivity is no longer justified.
Even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. Even after an orphan drug is approved, the FDA or comparable foreign regulatory authority can subsequently approve the same drug for the same condition if such regulatory authority concludes that the later drug is clinically superior if it is shown to be safer, more effective or makes a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process.
We may conduct certain of or portions of our clinical trials for our product candidates outside of the United States and the FDA may not accept data from such trials, in which case our development plans will be delayed, which could materially harm our business.
We may in the future choose to conduct one or more of our clinical trials or a portion of our clinical trials for our product candidates outside the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of this data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted and performed by qualified investigators in accordance with GCP requirements, and, and FDA must be able to validate the data from the study through an onsite inspection, if required. In general, the patient population for any clinical studies conducted outside of the United States must be representative of the population for whom we intend to label the product in the United States. In addition, while these clinical trials are subject to the applicable local laws, FDA acceptance of the data will be dependent upon its determination that the studies also complied with all applicable U.S. laws and regulations. There can be no assurance the FDA will accept data from trial conducted outside of the United States. If the FDA does not accept the data from our clinical trials of our product candidates, it would likely result in the need for additional trials, which would be costly and time consuming and delay or permanently halt our development of our product candidates.
Interim, topline and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary or topline data from our clinical studies, which are based on preliminary analyses of then-available data. Such preliminary results and related findings and conclusions are subject to change following more comprehensive reviews of the data related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results once additional data have been received and fully evaluated. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution until the final data are available.
From time to time, we may also disclose interim data from our clinical studies, such as the positive interim data we announced from our Phase 1/2 clinical trial of cirmtuzumab in combination with ibrutinib in December 2019. Interim data from this clinical trial and future clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues, following more comprehensive reviews of the data, and as more patient data become available. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses of data from preclinical studies or clinical trials of its product candidates, or may interpret or weigh the importance of data differently, which could impact the value of the particular product candidate, the approvability or prospects for commercialization of the product candidate, or our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and stockholders and others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure. Information that we decide not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular product, product candidate or our business. If the interim, topline or preliminary data that we disclose differ from actual results, or if others, including regulatory authorities, disagree with the conclusions we reach based on our analyses of such data, our ability to obtain approval for, and commercialize our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
Any breakthrough therapy designation that we may receive from the FDA for our product candidates may not lead to a faster development or regulatory review or approval process, and it does not increase the likelihood that our product candidates will receive marketing approval.
We may seek breakthrough therapy designation for some of our product candidates, including cirmtuzumab and TK216. A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. For drugs that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Drugs designated as breakthrough therapies by the FDA may also be eligible for accelerated approval. Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe one of our product candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a breakthrough therapy designation for a product candidate may not result in a faster development process, review or approval compared to drugs considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify as breakthrough therapies, the FDA may later decide that such product candidates no longer meet the conditions for qualification and rescind the designation.
We may seek a rare pediatric disease designation for TK216, however, there is no guarantee that we will obtain such designation, and even if we do, there is no guarantee that FDA approval of TK216 will result in a priority review voucher.
In 2012, Congress authorized the FDA to award priority review vouchers to sponsors of certain rare pediatric disease product applications. This program is designed to encourage development of new drug and biological products for prevention and treatment of certain rare pediatric diseases. Specifically, under this program, a sponsor who receives an approval for a drug or biologic for a “rare pediatric disease” that meets certain criteria may qualify for a voucher that can be redeemed to receive a priority review of a subsequent marketing application for a different product. The sponsor of a rare pediatric disease drug product receiving a priority review voucher may transfer (including by sale) the voucher to another sponsor. The voucher may be further transferred any number of times before the voucher is used, as long as the sponsor making the transfer has not yet submitted the application. The FDA may also revoke any priority review voucher if the rare pediatric disease drug for which the voucher was awarded is not marketed in the U.S. within one year following the date of approval.
We may seek a rare pediatric disease designation for TK216 for the treatment of Ewing’s sarcoma, however, we may not be able to obtain such designation. If we are able to obtain rare pediatric disease designation, there is no guarantee that we will be able to obtain a priority review voucher, even if TK216 is approved by the FDA. Moreover, Congress included a sunset provision in the statute authorizing the rare pediatric disease priority review voucher program. Specifically, FDA may not award the voucher to sponsors of marketing applications approved after September 30, 2020 unless either (i) the drug has received rare pediatric disease designation as of September 30, 2020, and is then approved by the FDA no later than September 30, 2022; or (ii) Congress reauthorizes the program, for which legislation has been proposed in the current Congress. If Congress does not reauthorize the rare pediatric disease priority review program in its current form, and if we do not receive rare pediatric disease designation for TK216 before September 30, 2020, we will not be issued a voucher upon any approval of TK216 that we receive. Moreover, even if we receive rare pediatric disease designation for TK216 by the current statutory deadline of September 30, 2020, we may not receive the voucher if we do not obtain approval by September 30, 2022.
Risks Related to Our Reliance on Third Parties
We rely on third parties to conduct many of our preclinical studies and clinical trials. Any failure by a third-party to conduct the clinical trials according to good laboratory practices, GCPs and other requirements and in a timely manner may delay or prevent our ability to seek or obtain regulatory approval for or commercialize our product candidates.
We are dependent on third parties to conduct our clinical trials and preclinical studies, including our ongoing clinical trials for cirmtuzumab and TK216 and preclinical studies for ROR1 CAR-T and our other development programs. Specifically, we have used and relied on, and intend to continue to use and rely on, medical institutions, clinical investigators, CROs and consultants to conduct our clinical trials in accordance with our clinical protocols and applicable regulatory requirements. These CROs, investigators and other third parties play a significant role in the conduct and timing of these trials and subsequent collection and analysis of data. While we have agreements governing the activities of our third-party contractors, we have limited influence over their actual performance. Nevertheless, we are responsible for ensuring that each of its clinical trials is conducted in accordance with the applicable protocol and legal, regulatory and scientific standards, and our reliance on the CROs and other third parties does not relieve us of our regulatory responsibilities. We and our CROs are required to comply with GCP requirements, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for all of our product candidates in clinical development. Regulatory authorities enforce these GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs or trial sites fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable, and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. In addition, our clinical trials must be conducted with product produced under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
There is no guarantee that any such CROs, investigators or other third parties will devote adequate time and resources to such trials or perform as contractually required. If any of these third parties fail to meet expected deadlines, adhere to our clinical protocols or meet regulatory requirements, or otherwise performs in a substandard manner, our clinical trials may be extended, delayed or terminated. In addition, many of the third parties with whom we contract may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other drug development activities that could harm our competitive position. In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and may receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, or the FDA concludes that the financial relationship may have affected the interpretation of the study, the integrity of the data generated at the applicable clinical trial site may be questioned and the utility of the clinical trial itself may be jeopardized, which could result in the delay or rejection of any BLA or NDA we submit to the FDA. Any such delay or rejection could prevent us from commercializing our product candidates.
If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative third parties or do so on commercially reasonable terms. Switching or adding additional CROs, investigators and other third parties involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays may occur, which can materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs, investigators and other third parties, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
We rely on third parties for the manufacture of our product candidates for clinical and preclinical development and expect to continue to do so for the foreseeable future. This reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or products or such quantities at an acceptable cost, which could delay, prevent or impair our development or commercialization efforts.
We do not own or operate manufacturing facilities and have no plans to build our own clinical or commercial scale manufacturing capabilities. We rely, and expect to continue to rely, on third parties for the manufacture of our product candidates and related raw materials for clinical and preclinical development, as well as for commercial manufacture if any of our product candidates receive marketing approval. The facilities used by third-party manufacturers to manufacture our product candidates must be approved by the FDA or other regulatory agencies pursuant to inspections that will be conducted after we submit a BLA or an NDA to the FDA or their equivalent to other regulatory agencies. We do not control the manufacturing process of, and are completely dependent on, third-party manufacturers for compliance with cGMP requirements for manufacture of our drug products. If these third-party manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, including requirements related to the manufacturing of high potency and pure compounds or other products, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. In addition, we have no control over the ability of third-party manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our product candidates, or if regulatory authorities withdraw any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our products.
Our or a third-party’s failure to execute on our manufacturing requirements, to do so on commercially reasonable terms, or to comply with cGMP could adversely affect our business in a number of ways, including:
an inability to initiate or continue clinical trials of cirmtuzumab, TK216 or any future product candidates under development;
delay in submitting regulatory applications, or receiving marketing approvals, for our product candidates;
subjecting third-party manufacturing facilities to additional inspections by regulatory authorities;
requirements to cease development or to recall batches of our product candidates; and
in the event of approval to market and commercialize our product candidates, an inability to meet commercial demands for our product candidates.
In addition, we may be unable to establish any agreements with third-party manufacturers or to do so on acceptable terms. Even if we are able to establish agreements with third-party manufacturers, reliance on third-party manufacturers entails additional risks, including:
failure of third-party manufacturers to comply with regulatory requirements and maintain quality assurance;
breach of the manufacturing agreement by the third-party;
failure to manufacture our product according to our specifications;
failure to manufacture our product according to our schedule, or at all;
misappropriation of our proprietary information, including our trade secrets and know-how; and
•
termination or nonrenewal of the agreement by the third-party at a time that is costly or inconvenient for us.
Our product candidates and any products that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us.
Any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval, and any related remedial measures may be costly or time consuming to implement. We do not currently have arrangements in place for redundant supply or a second source for all required raw materials used in the manufacture of our product candidates. If our current third-party manufacturers cannot perform as agreed, we may be required to replace such manufacturers and we may be unable to replace them on a timely basis or at all.
Our current and anticipated future dependence upon others for the manufacture of our product candidates or products may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that our trade secrets will be misappropriated or disclosed.
Because we currently rely on third parties to manufacture our product candidates and to perform quality testing, we must, at times, share our proprietary technology and confidential information, including trade secrets, with them. We seek to protect our proprietary technology, in part, by entering into confidentiality agreements, consulting agreements or other similar agreements with our advisors, employees, consultants and contractors prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information. Despite the contractual provisions employed when working with third parties, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors, are intentionally or inadvertently incorporated into the technology of others or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets and despite our efforts to protect our trade secrets, a competitor’s discovery of our proprietary technology and confidential information or other unauthorized use or disclosure would impair our competitive position and may have a material adverse effect on our business, financial condition, results of operations and prospects.
We have entered into and may seek to enter into additional collaborations, licenses and other similar arrangements, and we may not be successful in doing so, and we may not realize the benefits of such relationships.
We may seek to enter into collaborations, joint ventures, licenses and other similar arrangements for the development or commercialization of our product candidates, due to capital costs required to develop or commercialize the product candidate or manufacturing constraints, in addition to our collaboration with Shanghai Pharmaceutical Holding Co., Ltd. and SPH USA. We may not be successful in our efforts to establish such collaborations for our product candidates because our research and development pipeline may be insufficient, our product candidates may be deemed to be at too early of a stage of development for collaborative effort or third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy or significant commercial opportunity. In addition, we face significant competition in seeking appropriate strategic partners, and the negotiation process can be time consuming and complex. Further, any future collaboration agreements may restrict us from entering into additional agreements with potential collaborators. We cannot be certain that, following a strategic transaction or license, we will achieve an economic benefit that justifies such transaction.
Even if we are successful in our efforts to establish such collaborations, the terms that we agree upon may not be favorable to us, and we may not be able to maintain such collaborations if, for example, development or approval of a product candidate is delayed, the safety of a product candidate is questioned or sales of an approved product candidate are unsatisfactory.
In April 2018, we entered into a clinical trial and supply agreement with Pharmacyclics, LLC, an AbbVie Company, in support of our clinical trial to evaluate the combination of cirmtuzumab with ibrutinib. Ibrutinib is an inhibitor of Bruton’s tyrosine kinase, a key component of cell signaling in B-cells, and is marketed by Pharmacyclics for treatment in patients with CLL and MCL. We initiated a Phase 1/2 clinical trial in May 2018 to assess cirmtuzumab in combination with ibrutinib in patients with CLL and MCL. Pursuant to the agreement, Pharmacyclics has supplied ibrutinib up to a maximum aggregate amount at no cost to us for part 1 (a dose-finding arm) and part 2 (dose expansion arm) of the ongoing Phase 1/2 clinical trial evaluating cirmtuzumab in combination with ibrutinib. Under the clinical trial and supply agreement with Pharmacyclics, we are required to provide periodic reports to Pharmacyclics, including safety data reports, and to collaborate with Pharmacyclics in relation to any interactions with regulatory authorities regarding ibrutinib. The agreement includes no upfront costs, milestone or royalty payment commitments. In August 2019, Pharmacyclics agreed to provide additional quantities of ibrutinib at no cost to us for part 3 of the clinical trial, and so that patients who participated in parts 1 and 2 of the study may continue to receive ibrutinib in combination with cirmtuzumab for as long as their disease is responding. In the event the clinical supply agreement is terminated, we would likely incur substantial additional costs in order to obtain and purchase ibrutinib from a source other than Pharmacyclics and the Phase 2 part 3 of the Phase 1/2 clinical trial may be delayed.
In addition, any potential future collaborations may be terminable by our strategic partners, and we may not be able to adequately protect our rights under these agreements. Furthermore, strategic partners may negotiate for certain rights to control decisions regarding the development and commercialization of our product candidates, if approved, and may not conduct those activities in the same manner as we would. Any termination of collaborations we enter into in the future, or any delay in entering into collaborations related to our product candidates, could delay the development and commercialization of our product candidates and reduce their competitiveness if they reach the market, which could have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Commercialization of Our Product Candidates
Even if we receive regulatory approval for any product candidate, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense. Additionally, our product candidates, if approved, could be subject to labeling and other restrictions on marketing or withdrawal from the market, and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our product candidates, when and if any of them are approved.
Following potential approval of any of our product candidates, the FDA may impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly and time-consuming post-approval studies, post-market surveillance or clinical trials to monitor the safety and efficacy of the product. The FDA may also require a REMS as a condition of approval of our product candidates, which could include requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. In addition, if the FDA or a comparable foreign regulatory authority approves our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for our products will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMPs and GCP requirements for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with our products, including adverse events of unanticipated type, severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
restrictions on the marketing or manufacturing of our products, withdrawal of the product from the market or voluntary or mandatory product recalls;
restrictions on product distribution or use, or requirements to conduct post-marketing studies or clinical trials;
fines, restitutions, disgorgement of profits or revenues, warning letters, untitled letters or holds on clinical trials;
refusal by the FDA to approve pending applications or supplements to approved applications we filed or suspension or revocation of approvals;
product seizure or detention, or refusal to permit the import or export of our products; and
The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and generate revenue and could require us to expend significant time and resources in response and could generate negative publicity.
In addition, if any of our product candidates are approved, our product labeling, advertising and promotion will be subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about drug products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. If we receive marketing approval for a product candidate, physicians may nevertheless prescribe it to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed.
The FDA and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad. For example, certain policies of the current U.S. administration may impact our business and industry. Namely, the current U.S. administration has taken several executive actions, including the issuance of a number of Executive Orders, that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine regulatory and oversight activities such as implementing statutes through rulemaking, issuance of guidance, and review and approval of marketing applications. It is difficult to predict how these executive actions, including any Executive Orders, will be implemented, and the extent to which they will impact the FDA’s ability to exercise its regulatory authority. If these executive actions impose constraints on the FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would adversely affect our business, prospects, financial condition and results of operations.
Changes in funding for the FDA and other government agencies could hinder their ability to hire and retain key leadership and other personnel, or otherwise prevent new products and services from being developed or commercialized in a timely manner, which could negatively impact our business.
The ability of the FDA and other regulatory agencies to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs and biologics to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including for 35 days beginning on December 22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
The commercial success of our product candidates will depend upon the degree of market acceptance of such product candidates by physicians, patients, healthcare payors and others in the medical community.
Our product candidates may not be commercially successful. Even if any of our product candidates receive regulatory approval, they may not gain market acceptance among physicians, patients, healthcare payors or the medical community. The commercial success of any of our current or future product candidates will depend significantly on the broad adoption and use of the resulting product by physicians and patients for approved indications. The degree of market acceptance of our products will depend on a number of factors, including:
demonstration of clinical efficacy and safety compared to other more-established products;
the indications for which our product candidates are approved;
the limitation of our targeted patient population and other limitations or warnings contained in any FDA-approved labeling;
acceptance of a new drug for the relevant indication by healthcare providers and their patients;
the pricing and cost-effectiveness of our products, as well as the cost of treatment with our products in relation to alternative treatments and therapies;
our ability to obtain and maintain sufficient third-party coverage and adequate reimbursement from government healthcare programs, including Medicare and Medicaid, private health insurers and other third-party payors;
the willingness of patients to pay all, or a portion of, out-of-pocket costs associated with our products in the absence of sufficient third-party coverage and adequate reimbursement;
any restrictions on the use of our products, and the prevalence and severity of any adverse effects;
potential product liability claims;
the timing of market introduction of our products as well as competitive drugs;
the effectiveness of our or any of our potential future collaborators’ sales and marketing strategies; and
unfavorable publicity relating to the product.
If any product candidate is approved but does not achieve an adequate level of acceptance by physicians, hospitals, healthcare payors or patients, we may not generate sufficient revenue from that product and may not become or remain profitable. Our efforts to educate the medical community and third-party payors regarding the benefits of our products may require significant resources and may never be successful.
The market opportunities for our product candidates may be limited to patients who are ineligible for or have failed prior treatments and may be small or different from our estimates.
Cancer therapies are sometimes characterized as first line, second line or third line, and the FDA often approves new therapies initially only for third line use. When cancer is detected early enough, first line therapy is sometimes adequate to cure the cancer or prolong life without a cure. Whenever first line therapy, including targeted therapy, immunotherapy, chemotherapy, hormone therapy, surgery or a combination of these, proves unsuccessful, second line therapy may be administered. Second line therapies often consist of more chemotherapy, radiation, antibody drugs, tumor targeted small molecules or a combination of these. Third line therapies can include bone marrow transplantation, antibody and small molecule targeted therapies, more invasive forms of surgery and new technologies. In markets with approved therapies, there is no guarantee that our product candidates, even if approved, would be approved for second line or first line therapy. This could limit our potential market opportunity. In addition, we may have to conduct additional clinical trials prior to gaining approval for second line or first line therapy.
Our projections of both the number of people who have the cancers we are targeting, as well as the subset of people with these cancers in a position to receive later stage therapy and who have the potential to benefit from treatment with our product candidates, are based on our beliefs and estimates. These estimates have been derived from a variety of sources, including scientific literature, surveys of clinics, patient foundations or market research and may prove to be incorrect. Further, new studies may change the estimated incidence or prevalence of these cancers. The number of patients may turn out to be lower than expected. In addition, the potentially addressable patient population for our product candidates may be limited or may not be amenable to treatment with our product candidates. Even if we obtain significant market share for our product candidates, we may never achieve profitability without obtaining regulatory approval for additional indications, including use as a first- or second-line therapy.
Any product candidates for which we intend to seek approval as biologic products may face competition sooner than anticipated.
The Affordable Care Act includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of its product. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation and meaning are subject to uncertainty, and any processes adopted by the FDA to implement the BPCIA could have a material adverse effect on the future commercial prospects for our biological products.
We believe that any of our future product candidates approved as a biological product under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to Congressional action or otherwise, or that the FDA will not consider our product candidates to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. Moreover, the extent to which a biosimilar, once approved, could be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products will depend on a number of marketplace and regulatory factors that are still developing.
The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. If we are found or alleged to have improperly promoted off-label uses, we may become subject to significant liability.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products, as our product candidates would be, if approved. In particular, a product may not be promoted for uses that are not approved by the FDA or such other regulatory agencies as reflected in the product’s approved labeling. If we are found to have promoted such off-label uses, we may become subject to significant liability. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. If we cannot successfully manage the promotion and avoid off-label promotion of our product candidates, if approved, we could become subject to significant liability, which would materially adversely affect our business and financial condition.
The successful commercialization of our product candidates, if approved, will depend in part on the extent to which governmental authorities and health insurers establish coverage, adequate reimbursement levels and favorable pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our products could limit our ability to market those products and decrease our ability to generate revenue.
The availability of coverage and the adequacy of reimbursement by governmental healthcare programs such as Medicare and Medicaid, private health insurers and other third-party payors are essential for most patients to be able to afford prescription medications such as our product candidates, if approved. Our ability to achieve coverage and acceptable levels of reimbursement for our products by third-party payors will have an effect on our ability to successfully commercialize those products. Even if we obtain coverage for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. We cannot be sure that coverage and reimbursement in the United States, the European Union or elsewhere will be available for any product that we may develop, and any reimbursement that may become available may be decreased or eliminated in the future.
Third-party payors increasingly are challenging prices charged for pharmaceutical products and services, and many third-party payors may refuse to provide coverage and reimbursement for particular drugs when an equivalent generic drug or a less expensive therapy is available. It is possible that a third-party payor may consider our products as substitutable and only offer to reimburse patients for the less expensive product. Even if we are successful in demonstrating improved efficacy or improved convenience of administration with our products, pricing of existing drugs may limit the amount we will be able to charge for our products. These payors may deny or revoke the reimbursement status of a given product or establish prices for new or existing marketed products at levels that are too low to enable us to realize an appropriate return on our investment in product development. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our products and may not be able to obtain a satisfactory financial return on products that we may develop.
There is significant uncertainty related to third-party payor coverage and reimbursement of newly approved products. In the United States, third-party payors, including private and governmental payors, such as the Medicare and Medicaid programs, play an important role in determining the extent to which new drugs will be covered. Some third-party payors may require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse healthcare providers who use such therapies. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our products.
Obtaining and maintaining reimbursement status is time consuming, costly and uncertain. The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs. However, no uniform policy for coverage and reimbursement for products exists among third-party payors in the United States. Therefore, coverage and reimbursement for products can differ significantly from payor to payor. As a result, the coverage determination process is often a time consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Furthermore, rules and regulations regarding reimbursement change frequently, in some cases at short notice, and we believe that changes in these rules and regulations are likely.
Additionally, we or our collaborators may develop companion diagnostic tests for use with our product candidates as we are targeting certain defined populations for our treatments. We, or our collaborators, will be required to obtain coverage and reimbursement for these tests separate and apart from the coverage and reimbursement sought for our product candidates, once approved. While we, or our collaborators, have not yet developed any companion diagnostic test for our product candidates, if we do, there is significant uncertainty regarding our ability to obtain approval, coverage and adequate reimbursement for the same reasons applicable to our product candidates.
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe and other countries has and will continue to put pressure on the pricing and usage of our products. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for medical products but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our products. Accordingly, in markets outside the United States, the reimbursement for our products may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profits.
Moreover, increasing efforts by governmental and third-party payors in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our products. We expect to experience pricing pressures in connection with the sale of any of our products due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
We face significant competition, and if our competitors develop technologies or product candidates more rapidly than we do, or their technologies are more effective, our ability to develop and successfully commercialize products may be adversely affected.
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary and novel products and product candidates. Our competitors have developed, are developing or may develop products, product candidates and processes competitive with our product candidates. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. We believe that a significant number of products are currently under development, and may become commercially available in the future, for the treatment of conditions for which we may attempt to develop product candidates. In particular, there is intense competition in the fields of immunology, inflammation and oncology. Our competitors include larger and better funded pharmaceutical, biopharmaceutical, biotechnological and therapeutics companies. Moreover, we may also compete with universities and other research institutions who may be active in the indications we are targeting and could be in direct competition with us. We also compete with these organizations to recruit management, scientists and clinical development personnel, which could negatively affect our level of expertise and our ability to execute our business plan. We will also face competition in establishing clinical trial sites, enrolling subjects for clinical trials and in identifying and in-licensing new product candidates. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
If any of our product candidates are approved in oncology indications such as CLL or MCL, they will compete with small molecule therapies, biologics, cell-based therapies and vaccines, either approved or under development, that are intended to treat the same cancers that we are targeting, including through approaches that may prove to be more effective, have fewer side effects, be less costly to manufacture, be more convenient to administer or have other advantages over any product candidates we develop. In addition to competing with other therapies targeting similar indications, there are numerous other companies and academic institutions focused on similar targets as our product candidates and/or different scientific approaches to treating the same indications. We face competition from such companies in seeking any future potential collaborations to partner our product candidates, as well as potentially competing commercially for any approved products.
Significant progress has been made in the treatment of CLL since the advent of targeted therapies and FDA approval of ibrutinib for CLL in 2014. Three classes of targeted therapies have now been approved for the treatment of patients with CLL: inhibitors of Bruton’s tyrosine kinase, or BTK, a key component of cell signaling in B-cells, such as ibrutinib, which is marketed as Imbruvica® by AbbVie, Inc., and Johnson & Johnson, and acalabrutinib, which is marketed as Calquence® by AstraZeneca PLC; inhibitors of the protein B-cell lymphoma-2, or Bcl-2, such as venetoclax, which is marketed as Venclexta® and Venclyxto® by AbbVie, Inc., and Roche/Genentech; and inhibitors of Phosphoinositide 3-kinase, or PI3K, which include idelalisib, which is marketed as Zydelig® by Gilead Sciences, Inc., and duvelisib, which is marketed as Copiktra® by Verastem, Inc. These targeted therapies are now the core of the recommended treatment regimens for patients with both first-line and relapsed or refractory CLL, and have achieved objective response rates of 85-90%, two-year PFS of 65-90%, and two-year overall survival of 75-95%. The outcomes are worse for patients with certain prognostic factors, such as 17p or 11q chromosome deletions; for such patients with relapsed or refractory CLL treated with ibrutinib, the reported PFS is 50-75%. While CLL is treatable, it generally remains incurable, and patients with CLL will generally experience a recurrence of their cancer. Additionally, clinicians are investigating their potential in earlier stage disease in multiple clinical trials.
There are several therapeutic options available to treat MCL. Newly diagnosed patients are typically treated with rituximab combined with a chemotherapy regimen known as CHOP, comprised of cyclophosphamide, doxorubicin, vincristine, and prednisone. Alternative chemotherapy regimens include bortezomib or bendamustine. Patients with clinical responses to chemotherapy may become candidates for another therapeutic approach, autologous stem cell transplantation, a procedure in which radiation and/or chemotherapy is used to eliminate the patient’s immune cells, including residual MCL cells. Recently, ibrutinib was granted accelerated approval by the FDA for the treatment of relapsed MCL. Additionally, two other BTK inhibitors, acalabrutinib (Calquence®) and zanubrutinib (Brukinsa®) have been approved by the FDA for the treatment of patients with relapsed MCL. These therapies are given continuously for prolonged periods of time, and their use can be associated with significant toxicity.
The current standard therapy for patients with localized Ewing sarcoma in the U.S. is a combination of chemotherapy agents, including vincristine, doxorubicin and cyclophosphamide, with alternating cycles of ifosfamide and etoposide – a therapy known as VDC/IE. Patients that respond to this therapy may be candidates for tumor resection and continued treatment for a total of 14 to 17 cycles. This therapeutic regimen, however, is associated with significant toxicities. Patients with metastatic disease are often treated with VDC/IE or variations of this therapy with higher or more compressed dosing. This may also be supplemented by local radiation therapy or systemic radiation followed by autologous hematopoietic stem cell transplant.
Many of our competitors have significantly greater financial, technical, manufacturing, marketing, sales and supply resources or experience than we do. If we successfully obtain approval for any product candidate, we will face competition based on many different factors, including the safety and effectiveness of our products, the ease with which our products can be administered and the extent to which patients accept relatively new routes of administration, the timing and scope of regulatory approvals for these products, the availability and cost of manufacturing, marketing and sales capabilities, price, reimbursement coverage and patent position. Competing products could present superior treatment alternatives, including by being more effective, safer, more convenient, less expensive or marketed and sold more effectively than any products we may develop. Competitive products may make any products we develop obsolete or noncompetitive before we recover the expense of developing and commercializing our product candidates. If we are unable to compete effectively, our opportunity to generate revenue from the sale of products we may develop, if approved, could be adversely affected.
If the market opportunities for our products are smaller than we believe they are, our revenue may be adversely affected, and our business may suffer.
The precise incidence and prevalence for all the conditions we aim to address with our product candidates are unknown. Our projections of both the number of people who have these diseases, the number who have the specific indicated stage or treatment history we believe will be the approved indication, as well as the subset of people with these diseases who have the potential to benefit from treatment with our product candidates, are based on our beliefs and estimates. These estimates have been derived from a variety of sources, including the scientific literature, surveys of clinics, patient foundations or market research, and may prove to be incorrect. Further, new trials may change the estimated incidence or prevalence of these diseases. The total addressable market across all of our product candidates will ultimately depend upon, among other things, the indication approved by regulatory agencies and the diagnostic criteria included in the final label for each of our product candidates approved for sale for these indications, the availability of alternative treatments and the safety, convenience, cost and efficacy of our product candidates relative to such alternative treatments, acceptance by the medical community and patient access, drug pricing and reimbursement. The number of patients in the United States and other major markets and elsewhere may turn out to be lower than expected, patients may not be otherwise amenable to treatment with our products or new patients may become increasingly difficult to identify or gain access to, all of which would adversely affect our results of operations and our business. Further, even if we obtain significant market share for our product candidates, because some of our potential target populations are very small, we may never achieve profitability despite obtaining such significant market share.
We currently have no marketing and sales organization and have no experience as a company in commercializing products, and we may have to invest significant resources to develop these capabilities. If we are unable to establish marketing and sales capabilities or enter into agreements with third parties to market and sell our products, we may not be able to generate product revenue.
We have no internal sales, marketing or distribution capabilities, nor have we commercialized a product. If any of our product candidates ultimately receives regulatory approval, we must build a marketing and sales organization with technical expertise and supporting distribution capabilities to commercialize each such product in major markets, which will be expensive and time consuming, or collaborate with third parties that have sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. We have no prior experience as a company in the marketing, sale and distribution of biopharmaceutical products and there are significant risks involved in building and managing a sales organization, including our ability to hire, retain and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of these products. We may not be able to enter into collaborations or hire consultants or external service providers to assist us in sales, marketing and distribution functions on acceptable financial terms, or at all. In addition, our product revenues and our profitability, if any, may be lower if we rely on third parties for these functions than if we were to market, sell and distribute any products that we develop. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we are not successful in commercializing our products, either on our own or through arrangements with one or more third parties, we may not be able to generate any future product revenue and we would incur significant additional losses.
Our future growth may depend, in part, on our ability to operate in foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future growth may depend, in part, on our ability to develop and commercialize our product candidates in foreign markets. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from applicable regulatory authorities in foreign markets, and we may never receive such regulatory approvals for any of our product candidates. To obtain separate regulatory approval in most other countries, we must comply with numerous and varying regulatory requirements regarding safety and efficacy and governing, among other things, clinical trials, commercial sales, manufacturing, pricing and distribution of our product candidates. If we receive regulatory approval of our product candidates and ultimately commercialize our products in foreign markets, we would be subject to additional risks and uncertainties, including:
different regulatory requirements for approval of drugs in foreign countries;
reduced protection for intellectual property rights;
the existence of additional third-party patent rights of potential relevance to our business;
unexpected changes in tariffs, trade barriers and regulatory requirements;
economic weakness, including inflation, public health emergencies, or political instability in particular foreign economies and markets;
compliance with tax, employment, immigration and labor laws for employees living or traveling abroad;
foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country;
foreign reimbursement, pricing and insurance regimes;
workforce uncertainty in countries where labor unrest is common;
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
Risks Related to Our Business Operations and Industry
Our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations or any guidance we may provide.
Our quarterly and annual operating results may fluctuate significantly, which makes it difficult for us to predict our future operating results. These fluctuations may occur due to a variety of factors, many of which are outside of our control, including, but not limited to:
the timing and cost of, and level of investment in, research, development, regulatory approval and commercialization activities relating to our product candidates, which may change from time to time;
coverage and reimbursement policies with respect to our product candidates, if approved, and potential future drugs that compete with our products;
the cost of manufacturing our product candidates, which may vary depending on the quantity of production and any manufacturing issues or challenges requiring additional manufacturing activities, and the terms of our agreements with third-party manufacturers;
business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters such as earthquakes, typhoons, floods and fires or public health emergencies or pandemics such as the recent coronavirus (COVID-19) outbreak;
the timing and amount of any milestone or other payments we must make to the licensors and other third parties from whom we have in-licensed or acquired our product candidates;
expenditures that we may incur to acquire, develop or commercialize additional product candidates and technologies;
the level of demand for any approved products, which may vary significantly;
future accounting pronouncements or changes in our accounting policies; and
the timing and success or failure of preclinical studies or clinical trials for our product candidates or competing product candidates, or any other change in the competitive landscape of our industry, including consolidation among our competitors or partners.
The cumulative effects of these factors could result in large fluctuations and unpredictability in our quarterly and annual operating results. As a result, comparing our operating results on a period-to-period basis may not be meaningful. Investors should not rely on our past results as an indication of our future performance.
This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any forecasts we may provide to the market, or if the forecasts we provide to the market are below the expectations of analysts or investors, the price of our common stock could decline substantially. Such a stock price decline could occur even when we have met any previously publicly stated revenue or earnings guidance we may provide.
We are dependent on the services of our management and if we are not able to retain these individuals or recruit additional management or other key personnel, our business will suffer.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel. We are highly dependent upon our senior management, particularly our Chief Executive Officer, as well as other members of our senior management team. The loss of services of any of these individuals could delay or prevent the successful development of our product pipeline, initiation or completion of our planned operations, planned clinical trials or the commercialization of our product candidates. Although we have executed employment agreements or offer letters with each member of our senior management team, these agreements are terminable at will with or without notice and, therefore, we may not be able to retain their services as expected. We do not currently maintain “key person” life insurance on the lives of any of our employees. This lack of insurance means that we may not have adequate compensation for the loss of the services of these individuals.
We will need to expand and effectively manage our managerial, operational, financial and other resources in order to successfully pursue our clinical development and commercialization efforts. We may not be successful in maintaining our unique company culture and continuing to attract or retain qualified management and scientific and clinical personnel in the future due to the intense competition for qualified personnel among pharmaceutical, biotechnology and other businesses, particularly in the San Diego area. Our industry has experienced a high rate of turnover of management personnel in recent years. If we are not able to attract, integrate, retain and motivate necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
We may encounter difficulties in managing our growth and expanding our operations successfully.
As of March 6, 2020, we had eleven full-time employees and three part-time employees. As we continue research and development activities and pursue the potential commercialization of our product candidates, as well as function as a public company, we will need to expand our financial, research, development, regulatory, manufacturing, marketing and sales capabilities or contract with third parties to provide these capabilities for the company. As our operations expand, we expect that we will need to manage additional relationships with various strategic partners, suppliers and other third parties. Our future financial performance and our ability to develop and commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively.
We are subject to various foreign, federal, and state healthcare and privacy laws and regulations, and our failure to comply with these laws and regulations could harm our results of operations and financial condition.
Our business operations and current and future arrangements with investigators, healthcare professionals, consultants, third-party payors and customers expose us to broadly applicable foreign, federal and state fraud and abuse and other healthcare and privacy laws and regulations. These laws may constrain the business or financial arrangements and relationships through which we conduct our operations, including how we research, market, sell and distribute any products for which we obtain marketing approval. Such laws include:
the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving or providing any remuneration (including any kickback, bribe or certain rebates), directly or indirectly, overtly or covertly, in cash or in kind, in return for, either the referral of an individual or the purchase, lease, or order, or arranging for or recommending the purchase, lease, or order of any good, facility, item or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the federal Anti- Kickback Statute or specific intent to violate it in order to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act;
the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for, healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and their implementing regulations, also impose obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information without appropriate authorization by covered entities subject to the rule, such as health plans, healthcare clearinghouses and certain healthcare providers as well as their business associates that perform certain services for or on their behalf involving the use or disclosure of individually identifiable health information;
the federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the Centers for Medicare and Medicaid Services, CMS, information related to payments and other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by the physicians described above and their immediate family members;
federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and
analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to our business practices, including but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by non- governmental third-party payors, including private insurers, or by the patients themselves; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information or which require tracking gifts and other remuneration and items of value provided to physicians, other healthcare providers and entities; state and local laws that require the registration of pharmaceutical sales representatives; state and foreign laws governing the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA; state and foreign governments that have enacted or proposed requirements regarding the collection, distribution, use, security, and storage of personally identifiable information and other data relating to individuals (including the European Union General Data Protection Regulation 2016/679, or GDPR, and the California Consumer Protection Act), and federal and state consumer protection laws are being applied to enforce regulations related to the online collection, use, and dissemination of data, thus complicating compliance efforts.
As of May 25, 2018, the GDPR replaced the Data Protection Directive with respect to the processing of personal data in the European Union. The GDPR imposes many requirements for controllers and processors of personal data, including, for example, higher standards for obtaining consent from individuals to process their personal data, more robust disclosures to individuals and a strengthened individual data rights regime, shortened timelines for data breach notifications, limitations on retention and secondary use of information, increased requirements pertaining to health data and pseudonymised (i.e., key-coded) data and additional obligations when we contract third-party processors in connection with the processing of the personal data. The GDPR allows European Union member states to make additional laws and regulations further limiting the processing of genetic, biometric or health data. Failure to comply with the requirements of GDPR and the applicable national data protection laws of the European Union member states may result in fines of up to €20 million or up to 4% of the total worldwide annual turnover of the preceding financial year, whichever is higher, and other administrative penalties.
Ensuring that our internal operations and business arrangements with third parties comply with applicable healthcare laws and regulations could involve substantial costs. It is possible that governmental authorities will conclude that our business practices, including our consulting arrangements with physicians and other healthcare providers, some of whom received stock options as compensation for services provided, do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to significant penalties, including civil, criminal and administrative penalties, damages, fines, exclusion from U.S. government funded healthcare programs, such as Medicare and Medicaid, or similar programs in other countries or jurisdictions, disgorgement, individual imprisonment, contractual damages, reputational harm, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, diminished profits and the curtailment or restructuring of our operations. Further, defending against any such actions can be costly, time consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired. If any of the physicians or other providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusion from government funded healthcare programs and imprisonment. If any of the above occur, it could adversely affect our ability to operate our business and our results of operations.
Recently enacted legislation, future legislation and healthcare reform measures may increase the difficulty and cost for us to obtain marketing approval for and commercialize our product candidates and may affect the prices we may set.
In the United States and some foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system, including cost-containment measures that may reduce or limit coverage and reimbursement for newly approved drugs and affect our ability to profitably sell any product candidates for which we obtain marketing approval. In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs and improve the quality of healthcare.
For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, referred to collectively as the Affordable Care Act, was enacted in the United States. Among the provisions of the Affordable Care Act of importance to our potential product candidates, the Affordable Care Act: establishes an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents; extends manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; expands eligibility criteria for Medicaid programs; expands the entities eligible for discounts under the Public Health program; increases the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; creates a new Medicare Part D coverage gap discount program; establishes a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical effectiveness research, along with funding for such research; and establishes a Center for Medicare Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending.
At this time, we are unsure of the full impact that the Affordable Care Act will have on our business. There have been judicial and political challenges to certain aspects of the Affordable Care Act. For example, since January 2017, President Trump has signed two executive orders and other directives designed to delay, circumvent, or loosen certain requirements of the Affordable Care Act. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the Affordable Care Act. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the Affordable Care Act have been signed into law. The Tax Cuts and Jobs Act, or the Tax Act, includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” On January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain Affordable Care Act-mandated fees, including the so-called “Cadillac” tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share. The Bipartisan Budget Act of 2018, or the BBA, among other things, amends the Affordable Care Act, effective January 1, 2019, to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole,” by increasing from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D. In July 2018, CMS published a final rule permitting further collections and payments to and from certain Affordable Care Act qualified health plans and health insurance issuers under the Affordable Care Act risk adjustment program in response to the outcome of federal district court litigation regarding the method CMS uses to determine this risk adjustment. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, or Texas District Court Judge, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the Tax Act, the remaining provisions of the ACA are invalid as well. While the Texas District Court Judge, as well as the Trump Administration and CMS, have stated that the ruling will have no immediate effect, it is unclear how this decision, subsequent appeals, and other efforts to repeal and replace the ACA will impact the ACA and our business.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. On August 2, 2011, the Budget Control Act of 2011 was signed into law, which, among other things, resulted in reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, including the BBA, will remain in effect through 2027 unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Further, there has been heightened governmental scrutiny in the United States of pharmaceutical pricing practices in light of the rising cost of prescription drugs. Such scrutiny has resulted in several recent congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products.
At the federal level, the Trump administration’s budget proposal for fiscal year 2019 contains further drug price control measures that could be enacted during the 2019 budget process or in other future legislation, including, for example, measures to permit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid and to eliminate cost sharing for generic drugs for low-income patients. Additionally, the Trump administration released a “Blueprint” to lower drug prices through proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. The U.S. Department of Health and Human Services has begun the process of soliciting feedback on some of these measures and, at the same time, is implementing others under our existing authority. Although some of these, and other, proposals will require authorization through additional legislation to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our product candidates, if approved, or put pressure on our product pricing, which could negatively affect our business, results of operations, financial condition and prospects.
Additionally, on May 30, 2018, the Trickett Wendler, Frank Mongiello, Jordan McLinn, and Matthew Bellina Right to Try Act of 2017, or the Right to Try Act, was signed into law. The law, among other things, provides a federal framework for certain patients with life-threatening diseases or conditions to access certain investigational new drug products that have completed a Phase 1 clinical trial. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA approval under the FDA expanded access program. There is no obligation for a drug manufacturer to make our drug products available to eligible patients as a result of the Right to Try Act.
We expect that the Affordable Care Act, these new laws and other healthcare reform measures that may be adopted in the future may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, new payment methodologies and additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our product candidates, if approved.
We and any of our third-party manufacturers or suppliers may use potent chemical agents and hazardous materials, and any claims relating to improper handling, storage or disposal of these materials could be time consuming or costly.
We and any of our third-party manufacturers or suppliers will use biological materials, potent chemical agents and may use hazardous materials, including chemicals and biological agents and compounds that could be dangerous to human health and safety of the environment. Our historical operations and the operations of our third-party manufacturers and suppliers also produce hazardous waste products. Federal, state and local laws and regulations govern the use, generation, manufacture, storage, handling and disposal of these materials and wastes. Compliance with applicable environmental laws and regulations may be expensive, and current or future environmental laws and regulations may impair our product development efforts. In addition, we cannot eliminate the risk of accidental injury or contamination from these materials or wastes. We do not carry specific biological or hazardous waste insurance coverage, and our property, casualty and general liability insurance policies specifically exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination. In the event of contamination or injury, we could be held liable for damages or be penalized with fines in an amount exceeding our resources, and our clinical trials or regulatory approvals could be suspended.
Although we maintain workers’ compensation insurance for certain costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for toxic tort claims that may be asserted against us in connection with our storage or disposal of biologic, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations, which have tended to become more stringent over time. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions or liabilities, which could materially adversely affect our business, financial condition, results of operations and prospects.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our products.
We face an inherent risk of product liability as a result of the clinical trials of our product candidates and will face an even greater risk if we commercialize our product candidates. For example, we may be sued if our product candidates allegedly cause injury or are found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product candidate, negligence, strict liability and a breach of warranties. Claims may be brought against us by clinical trial participants, patients or others using, administering or selling products that may be approved in the future. Claims could also be asserted under state consumer protection acts.
If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit or cease the commercialization of our products. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
decreased demand for our products;
injury to our reputation and significant negative media attention;
withdrawal of clinical trial participants;
costs to defend the related litigation;
a diversion of management’s time and our resources;
substantial monetary awards to trial participants or patients;
product recalls, withdrawals or labeling, marketing or promotional restrictions;
significant negative financial impact;
the inability to commercialize our product candidates; and
a decline in our stock price.
We currently hold $10.0 million in product liability insurance coverage in the aggregate. We may need to increase our insurance coverage as we expand our clinical trials or if we commence commercialization of our product candidates. Insurance coverage is increasingly expensive. Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of our product candidates. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies will also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
We and any of our potential future collaborators will be required to report to regulatory authorities if any of our approved products cause or contribute to adverse medical events, and any failure to do so would result in sanctions that would materially harm our business.
If we and any of our potential future collaborators are successful in commercializing our products, the FDA and foreign regulatory authorities would require that we and any of our potential future collaborators report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We and any of our potential future collaborators or CROs may fail to report adverse events within the prescribed timeframe. If we or any of our potential future collaborators or CROs fail to comply with such reporting obligations, the FDA or a foreign regulatory authority could take action, including criminal prosecution, the imposition of civil monetary penalties, seizure of our products or delay in approval or clearance of future products.
Our internal computer systems, or those of any of our CROs, manufacturers, other contractors or consultants or potential future collaborators, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
The United States federal and various state and foreign governments have adopted or proposed requirements regarding the collection, distribution, use, security, and storage of personally identifiable information and other data relating to individuals, and federal and state consumer protection laws are being applied to enforce regulations related to the online collection, use, and dissemination of data. Despite the implementation of security measures, our internal computer systems and those of our current and any future CROs and other contractors, consultants and collaborators are vulnerable to damage from computer viruses, cybersecurity threats, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations or result in the unauthorized disclosure of or access to personally identifiable information or individually identifiable health information (violating certain privacy laws such as GDPR or the California Consumer Privacy Act, which became effective January 1, 2020), it could result in a material disruption of our development programs and our business operations, whether due to a loss of our trade secrets or other similar disruptions. Some of the federal, state and foreign government requirements include obligations of companies to notify individuals of security breaches involving particular personally identifiable information, which could result from breaches experienced by us or by our vendors, contractors, or organizations with which we have formed strategic relationships. Even though we may have contractual protections with such vendors, contractors, or other organizations, notifications and follow-up actions related to a security breach could impact our reputation, cause us to incur significant costs, including legal expenses, harm customer confidence, hurt our expansion into new markets, cause us to incur remediation costs, or cause us to lose existing customers. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. We also rely on third parties to manufacture our product candidates, and similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability, the further development and commercialization of our product candidates could be delayed, and we could be subject to significant fines, penalties or liabilities for any noncompliance to certain privacy and security laws.
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or manmade disasters or business interruptions, for which we are predominantly self-insured. We rely on third- party manufacturers to produce our product candidates. Our ability to obtain clinical supplies of our product candidates could be disrupted if the operations of these suppliers were affected by a man-made or natural disaster or other business interruption. In addition, our corporate headquarters is located in San Diego, California near major earthquake faults and fire zones, and the ultimate impact on us of being located near major earthquake faults and fire zones and being consolidated in a certain geographical area is unknown. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
The COVID-19 novel coronavirus outbreak may adversely impact our business.
The public health emergency caused by the COVID-19 novel coronavirus outbreak first identified in Wuhan, Hubei Providence, China, may adversely impact our business. The COVID-19 outbreak and other public health emergencies may negatively affect clinical trial site activation, as well as patient enrollment and retention, at sites in a region or a city whose health care system becomes overwhelmed due to the illness. For example, our expectations that we will announce the first-in-human dosing of our ROR-1 CAR-T therapy in China may be delayed to clinical site activation or other delays caused by the COVID-19 outbreak. Additionally, our collaborator and an affiliate of our largest stockholder is SPH, which is based in China may delay work on our collaboration or focus on other investments during a continued outbreak of COVID-19. Two of our directors are affiliates of SPH, including Cho Man, the Chief Executive Officer of SPH, who may be focused on other responsibilities during the COVID-19 outbreak which may impair the ability of our board to act by written consent. Further, if our third-party manufacturers, particularly those located in China, experience manufacturing difficulties due to resource constraints or as a result of natural disasters, labor disputes, unstable political environments, or public health emergencies such as the COVID-19 outbreak, our ability to provide our product candidates to patients in clinical trials, or to provide product for treatment of patients if approved, would be jeopardized. If the current outbreak of the strain of COVID-19 continues and results in a prolonged period of travel and other similar logistics restrictions, this may reduce our capabilities to travel, domestically and internationally, which may impact our ability to raise funds, develop and renew contracts, or could otherwise disrupt portions of our business. It is not currently possible to ascertain the overall impact of the COVID-19 outbreak, if any, on our business. The extent to which COVID-19 could have a material impact on our business, financial condition and results of operations will depend on future developments as to the geographic presence of COVID-19 and government and healthcare responses to such spread, which are presently highly uncertain.
Our employees and independent contractors, including principal investigators, CROs, consultants and vendors, may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees and independent contractors, including principal investigators, CROs, consultants and vendors may engage in misconduct or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violate: (1) the laws and regulations of the FDA and other similar regulatory requirements, including those laws that require the reporting of true, complete and accurate information to such authorities, (2) manufacturing standards, including cGMP requirements, (3) federal and state data privacy, security, fraud and abuse and other healthcare laws and regulations in the United States and abroad or (4) laws that require the true, complete and accurate reporting of financial information or data. Activities subject to these laws also involve the improper use or misrepresentation of information obtained in the course of clinical trials, the creation of fraudulent data in our preclinical studies or clinical trials, or illegal misappropriation of drug product, which could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. In addition, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and financial results, including, without limitation, the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgements, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, individual imprisonment, contractual damages, reputational harm, diminished profits and future earnings, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
We are subject to U.S. and certain foreign export and import controls, sanctions, embargoes, anti-corruption laws and anti-money laundering laws and regulations. Compliance with these legal standards could impair our ability to compete in domestic and international markets. We could face criminal liability and other serious consequences for violations, which could harm our business.
We are subject to export control and import laws and regulations, including the U.S. Export Administration Regulations, U.S. Customs regulations, and various economic and trade sanctions regulations administered by the U.S. Treasury Department’s Office of Foreign Assets Controls, and anti-corruption and anti-money laundering laws and regulations, including the U.S. Foreign Corrupt Practices Act of 1977, as amended, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, and other state and national anti-bribery and anti-money laundering laws in the countries in which we conduct activities. Anti-corruption laws are interpreted broadly and prohibit companies and their employees, agents, clinical research organizations, contractors and other collaborators and partners from authorizing, promising, offering, providing, soliciting or receiving, directly or indirectly, improper payments or anything else of value to recipients in the public or private sector. We may engage third parties for clinical trials outside of the United States, to sell our products abroad once we enter a commercialization phase, and/or to obtain necessary permits, licenses, patent registrations and other regulatory approvals. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities and other organizations. We can be held liable for the corrupt or other illegal activities of our employees, agents, clinical research organizations, contractors and other collaborators and partners, even if we do not explicitly authorize or have actual knowledge of such activities. Any violations of the laws and regulations described above may result in substantial civil and criminal fines and penalties, imprisonment, the loss of export or import privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm and other consequences.
We may engage in strategic transactions that could impact our liquidity, increase our expenses and present significant distractions to our management.
From time to time, we may consider strategic transactions, such as acquisitions of companies, asset purchases and out-licensing or in-licensing of intellectual property, products or technologies, similar to our approach in in-licensing and acquiring our current product candidates. Any future transactions could increase our near and long-term expenditures, result in potentially dilutive issuances of our equity securities, including our common stock, or the incurrence of debt, contingent liabilities, amortization expenses or acquired in-process research and development expenses, any of which could affect our financial condition, liquidity and results of operations. Additional potential transactions that we may consider in the future include a variety of business arrangements, including spin-offs, strategic partnerships, joint ventures, restructurings, divestitures, business combinations and investments. Future acquisitions may also require us to obtain additional financing, which may not be available on favorable terms or at all. These transactions may never be successful and may require significant time and attention of management. In addition, the integration of any business that we may acquire in the future may disrupt our existing business and may be a complex, risky and costly endeavor for which we may never realize the full benefits of the acquisition. Accordingly, although there can be no assurance that we will undertake or successfully complete any additional transactions of the nature described above, any additional transactions that we do complete could have a material adverse effect on our business, results of operations, financial condition and prospects.
Risks Related to Our Intellectual Property
Our success depends on our ability to protect our intellectual property and our proprietary technologies.
Our commercial success depends in part on our ability to obtain and maintain patent protection and trade secret protection for our product candidates, proprietary technologies and their uses as well as our ability to operate without infringing upon the proprietary rights of others. If we are unable to protect our intellectual property rights or if our intellectual property rights are inadequate for our technology or our product candidates, our competitive position could be harmed. We generally seek to protect our proprietary position by licensing or filing patent applications in the United States and abroad related to our product candidates, proprietary technologies and their uses that are important to our business. Our or our licensor’s patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless, and until, patents issue from such applications, and then only to the extent the issued claims cover the technology. There can be no assurance that our or our licensor’s patent applications will result in patents being issued or that issued patents will afford sufficient protection against competitors with similar technology, nor can there be any assurance that the patents if issued will not be infringed, designed around or invalidated by third parties. Even issued patents may later be found invalid or unenforceable or may be modified or revoked in proceedings instituted by third parties before various patent offices or in courts. The degree of future protection for our proprietary rights is uncertain. Only limited protection may be available and may not adequately protect our rights or permit us to gain or keep any competitive advantage. These uncertainties and/or limitations in our ability to properly protect the intellectual property rights relating to our product candidates could have a material adverse effect on our financial condition and results of operations.
Although we own and license issued patents in the United States and foreign countries, we cannot be certain that the claims in our or our licensor’s other U.S. pending patent applications, corresponding international patent applications and patent applications in certain foreign countries will be considered patentable by the United States Patent and Trademark Office, or USPTO, courts in the United States or by the patent offices and courts in foreign countries, nor can we be certain that the claims in our or our licensor’s issued patents will not be found invalid or unenforceable if challenged.
The patent application process is subject to numerous risks and uncertainties, and there can be no assurance that we, our licensors or any of our potential future collaborators will be successful in protecting our product candidates by obtaining and defending patents. These risks and uncertainties include the following:
the USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process, the noncompliance with which can result in abandonment or lapse of a patent or patent application, and partial or complete loss of patent rights in the relevant jurisdiction;
patent applications may not result in any patents being issued;
patents may be challenged, invalidated, modified, revoked, circumvented, found to be unenforceable or otherwise may not provide any competitive advantage;
our competitors, many of whom have substantially greater resources than we do and many of whom have made significant investments in competing technologies, may seek or may have already obtained patents that will limit, interfere with or block our ability to make, use and sell our product candidates;
there may be significant pressure on the U.S. government and international governmental bodies to limit the scope of patent protection both inside and outside the United States for disease treatments that prove successful, as a matter of public policy regarding worldwide health concerns; and
countries other than the United States may have patent laws less favorable to patentees than those upheld by U.S. courts, allowing foreign competitors a better opportunity to create, develop and market competing products.
The patent prosecution process is also expensive and time consuming, and we and our licensors may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner or in all jurisdictions where protection may be commercially advantageous. It is also possible that we or our licensors will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. Moreover, in some circumstances, we do not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, directed to technology that we license from third parties. We may also require the cooperation of our licensor in order to enforce the licensed patent rights, and such cooperation may not be provided. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. We cannot be certain that patent prosecution and maintenance activities by our licensors have been or will be conducted in compliance with applicable laws and regulations, which may affect the validity and enforceability of such patents or any patents that may issue from such applications. If they fail to do so, this could cause us to lose rights in any applicable intellectual property that we in-license, and as a result our ability to develop and commercialize products or product candidates may be adversely affected and we may be unable to prevent competitors from making, using and selling competing products.
In addition, although we enter into non-disclosure and confidentiality agreements with parties who have access to patentable aspects of our research and development output, such as our employees, outside scientific collaborators, CROs, third-party manufacturers, consultants, advisors, licensees, collaboration partners, and other third parties, any of these parties may breach such agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties, including with respect to cirmtuzumab and TK216, or otherwise experiences disruptions in our business relationships with our licensors, we could lose license rights that are important to our business.
We are a party to several license agreements under which we are granted rights to intellectual property that are important to our business and we may enter into additional license agreements in the future. For example, in March 2014, we entered into an exclusive license agreement with Georgetown University, or Georgetown, to obtain an exclusive license to certain intellectual property rights to develop and commercialize compounds targeting EWS-FLI1. In March 2016, we entered into an exclusive license agreement with the Regents of the University of California to obtain an exclusive license to certain intellectual property rights to develop and commercialize cirmtuzumab and other ROR1 related naked antibodies.
These license agreements impose, and we expect that any future license agreements where we in-license intellectual property, will impose on us, various development, regulatory and/or commercial diligence obligations, payment of milestones and/or royalties and other obligations. If we fail to comply with our obligations under these agreements, or we are subject to bankruptcy-related proceedings, the licensor may have the right to terminate the license, in which event we would not be able to market products covered by the license.
We may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we cannot provide any assurances that third-party patents do not exist which might be enforced against our product candidates in the absence of such a license. We may fail to obtain any of these licenses on commercially reasonable terms, if at all. Even if we are able to obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. In that event, we may be required to expend significant time and resources to develop or license replacement technology. If we are unable to do so, we may be unable to develop or commercialize the affected product candidates, which could materially harm our business and the third parties owning such intellectual property rights could seek either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation. Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues. Disputes may arise between us and our licensors regarding intellectual property subject to a license agreement, including:
the scope of rights granted under the license agreement and other interpretation-related issues;
whether and the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement;
our right to sublicense patents and other rights to third parties;
our diligence obligations with respect to the use of the licensed technology in relation to our development and commercialization of our product candidates, and what activities satisfy those diligence obligations;
our right to transfer or assign the license; and
the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners.
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may not be able to successfully develop and commercialize the affected product candidates, which would have a material adverse effect on our business.
If the scope of any patent protection we obtain is not sufficiently broad, or if we lose any of our patent protection, our ability to prevent our competitors from commercializing similar or identical product candidates would be adversely affected.
The patent position of biopharmaceutical companies generally is highly uncertain, involves complex legal and factual questions, and has been the subject of much litigation in recent years. As a result, the issuance, scope, validity, enforceability and commercial value of our and our licensor’s patent rights are highly uncertain. Our and our licensor’s pending and future patent applications may not result in patents being issued which protect our product candidates or which effectively prevent others from commercializing competitive product candidates.
Moreover, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Even if patent applications we own or license currently or in the future issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us, or otherwise provide us with any competitive advantage. Any patents that we own or license may be challenged or circumvented by third parties or may be narrowed or invalidated as a result of challenges by third parties. Consequently, we do not know whether our product candidates will be protectable or remain protected by valid and enforceable patents. Our competitors or other third parties may be able to circumvent our or our licensor’s patents by developing similar or alternative technologies or products in a non-infringing manner which could materially adversely affect our business, financial condition, results of operations and prospects.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our and our licensor’s patents may not cover our product candidates or may be challenged in the courts or patent offices in the United States and abroad. Our and our licensor’s patents may be subject to a third-party pre-issuance submission of prior art to the USPTO, or become involved in opposition, derivation, revocation, reexamination, post-grant review, or PGR, and inter partes review, or IPR, or other similar proceedings in the USPTO or foreign patent offices challenging our or our licensor’s patent rights. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we or our predecessors or our licensor and the patent examiner were unaware during prosecution. There is no assurance that all potentially relevant prior art relating to our patents and patent applications or those of our licensors has been found. There is also no assurance that there is not prior art of which we, our predecessors or licensors are aware, but which we do not believe affects the validity or enforceability of a claim in our patents and patent applications or those of our licensors, which may, nonetheless, ultimately be found to affect the validity or enforceability of a claim. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate or render unenforceable, our or our licensor’s patent rights, allow third parties to commercialize our product candidates and compete directly with us, without payment to us. Such loss of patent rights, loss of exclusivity or in patent claims being narrowed, invalidated or held unenforceable could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our product candidates. Such proceedings also may result in substantial cost and require significant time from our scientists and management, even if the eventual outcome is favorable to us. In addition, if the breadth or strength of protection provided by our or our licensor’s patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.
The patent protection and patent prosecution for some of our product candidates may be dependent on third parties.
We or our licensors may fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them. Therefore, we or our licensors may miss potential opportunities to strengthen our patent position. It is possible that defects of form in the preparation or filing of our or our licensor’s patents or patent applications may exist, or may arise in the future, for example with respect to proper priority claims, inventorship, claim scope, or requests for patent term adjustments. If there are material defects in the form, preparation, prosecution, or enforcement of our or our licensor’s patents or patent applications, such patents may be invalid and/or unenforceable, and such applications may never result in valid, enforceable patents. If we or our licensors, whether current or future, fail to establish, maintain or protect our patents and other intellectual property rights, such rights may be reduced or eliminated. If our licensors are not fully cooperative or disagree with us as to the prosecution, maintenance or enforcement of any patent rights, such patent rights could be compromised. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business.
As a licensee of third parties, we rely on third parties to file and prosecute patent applications and maintain patents and otherwise protect the licensed intellectual property under some of our license agreements. We have not had and do not have primary control over these activities for certain of our patents or patent applications and other intellectual property rights. We cannot be certain that such activities by third parties have been or will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents or other intellectual property rights. Pursuant to the terms of the license agreements with some of our licensors, the licensors may have the right to control enforcement of our licensed patents or defense of any claims asserting the invalidity of these patents and even if we are permitted to pursue such enforcement or defense, we will require the cooperation of our licensors. We cannot be certain that our licensors will allocate sufficient resources or prioritize their or our enforcement of such patents or defense of such claims to protect our interests in the licensed patents. Even if we are not a party to these legal actions, an adverse outcome could harm our business because it might prevent us from continuing to license intellectual property that we may need to operate our business. If any of our licensors or any of our future licensors or future collaborators fail to appropriately prosecute and maintain patent protection for patents covering any of our product candidates, our ability to develop and commercialize those product candidates may be adversely affected and we may not be able to prevent competitors from making, using and selling competing products.
In addition, even where we have the right to control patent prosecution of patents and patent applications we have acquired or licensed from third parties, we may still be adversely affected or prejudiced by actions or inactions of our predecessors or licensors and their counsel that took place prior to our assuming control over patent prosecution.
Our technology acquired or licensed from various third parties may be subject to retained rights. Our predecessors or licensors often retain certain rights under their agreements with us, including the right to use the underlying technology for noncommercial academic and research use, to publish general scientific findings from research related to the technology, and to make customary scientific and scholarly disclosures of information relating to the technology. It is difficult to monitor whether our predecessors or licensors limit their use of the technology to these uses, and we could incur substantial expenses to enforce our rights to our licensed technology in the event of misuse.
If we are limited in our ability to utilize acquired or licensed technologies, or if we lose our rights to critical in-licensed technology, we may be unable to successfully develop, out-license, market and sell our products, which could prevent or delay new product introductions. Our business strategy depends on the successful development of licensed and acquired technologies into commercial products. Therefore, any limitations on our ability to utilize these technologies may impair our ability to develop, out-license or market and sell our product candidates.
Some of our intellectual property has been discovered through government funded programs and thus may be subject to federal regulations such as “march-in” rights, certain reporting requirements and a preference for U.S.-based companies. Compliance with such regulations may limit our exclusive rights, and limit our ability to contract with non-U.S. manufacturers.
Some of the intellectual property rights we have acquired or licensed or may acquire or license in the future may have been generated through the use of U.S. government funding and may therefore be subject to certain federal regulations. For example, some of the research and development work on cirmtuzumab and TK216 was funded by government research grants. As a result, the U.S. government may have certain rights to intellectual property embodied in our product candidates pursuant to the Bayh-Dole Act of 1980, or Bayh-Dole Act. These U.S. government rights include a non-exclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right, under certain limited circumstances, to require us to grant exclusive, partially exclusive, or non-exclusive licenses to any of these inventions to a third-party if it determines that: (i) adequate steps have not been taken to commercialize the invention; (ii) government action is necessary to meet public health or safety needs; or (iii) government action is necessary to meet requirements for public use under federal regulations (also referred to as “march-in rights”). The U.S. government also has the right to take title to these inventions if the grant recipient fails to disclose the invention to the government or fails to file an application to register the intellectual property within specified time limits. Intellectual property generated under a government funded program is also subject to certain reporting requirements, compliance with which may require us to expend substantial resources. In addition, the U.S. government requires that any products embodying any of these inventions or produced through the use of any of these inventions be manufactured substantially in the United States. This preference for U.S. industry may be waived by the federal agency that provided the funding if the owner or assignee of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the United States or that under the circumstances domestic manufacture is not commercially feasible. This preference for U.S. industry may limit our ability to contract with non-U.S. product manufacturers for products covered by such intellectual property. To the extent any of our future intellectual property is also generated through the use of U.S. government funding, the provisions of the Bayh-Dole Act may similarly apply.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
others may be able to develop products that are similar to our product candidates but that are not covered by the claims of the patents that we own or license;
•
we or our licensors or predecessors might not have been the first to make the inventions covered by the issued patents or patent applications that we own or license;
we or our licensors or predecessors might not have been the first to file patent applications covering certain of our inventions;
others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights;
it is possible that our or our licensor’s pending patent applications will not lead to issued patents;
issued patents that we own or license may be held invalid or unenforceable, as a result of legal challenges by our competitors;
our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets;
we may not develop additional proprietary technologies that are patentable; and
the patents of others may have an adverse effect on our business.
Should any of these events occur, it could significantly harm our business, results of operations and prospects.
We rely on licensee relationships, and any disputes or litigation with our partners or termination or breach of any of the related agreements could reduce the financial resources available to us, including milestone payments and future royalty revenues.
Our existing collaborations may not continue or be successful, and we may be unable to enter into future collaborative arrangements to develop and commercialize our unpartnered assets. If any of our collaborative partners breach or terminate their agreements with us or otherwise fail to conduct their collaborative activities successfully, our product development under these agreements will be delayed or terminated. Disputes or litigation may also arise with our collaborators (with us and/or with one or more third parties), including those over ownership rights to intellectual property, know-how or technologies developed with our collaborators. Such disputes or litigation could adversely affect our rights to one or more of our product candidates and could delay, interrupt or terminate the collaborative research, development and commercialization of certain potential products, create uncertainty as to ownership rights of intellectual property, or could result in litigation or arbitration. In addition, a significant downturn or deterioration in the business or financial condition of our collaborators or partners could
suffer,result in a loss of expected revenue and our expected returns on investment. The occurrence of any of these problems could be time-consuming and expensive and could adversely affect our business.Our commercial success depends significantly on our ability to operate without infringing the patents and other proprietary rights of third parties. Claims by third parties that we infringe their proprietary rights may result in liability for damages or prevent or delay our developmental and commercialization efforts.
Our commercial success depends in part on avoiding infringement of the patents and proprietary rights of third parties. However, our or our licensee’s research, development and commercialization activities may be subject to claims that we or our licensee infringes or otherwise violates patents or other intellectual property rights owned or controlled by third parties. Other entities may have or obtain patents or proprietary rights that could limit our or our licensee’s ability to make, use, sell, offer for sale or import our product candidates and products that may be approved in the future, or impair our competitive position. There is a substantial amount of litigation, both within and outside the United States, involving patent and other intellectual property rights in the biopharmaceutical industry, including patent infringement lawsuits, oppositions, reexaminations, IPR proceedings and PGR proceedings before the USPTO and/or foreign patent offices. Numerous third-party U.S. and foreign issued patents and pending patent applications exist in the fields in which we are developing product candidates. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates.
As the biopharmaceutical industry expands and more patents are issued, the risk increases that our product candidates may be subject to claims of infringement of the patent rights of third parties. Because patent applications are maintained as confidential for a certain period of time, until the relevant application is published we may be unaware of third-party patents that may be infringed by commercialization of any of our product candidates, and we cannot be certain that we were the first to file a patent application related to a product candidate or technology. Moreover, because patent applications can take many years to issue, there may be currently-pending patent applications that may later result in issued patents that our product candidates may infringe. In addition, identification of third-party patent rights that may be relevant to our technology is difficult because patent searching is imperfect due to differences in terminology among patents, incomplete databases and the difficulty in assessing the meaning of patent claims. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Any claims of patent infringement asserted by third parties would be time consuming and could:
result in costly litigation that may cause negative publicity;
divert the time and attention of our technical personnel and management;
cause development delays;
subject us to an injunction preventing us from making, using, selling, offering for sale, or importing our products;
prevent us from commercializing any of our product candidates until the asserted patent expires or is held finally invalid or not infringed in a court of law;
require us to develop non-infringing technology, which may not be possible on a cost-effective basis;
subject us to significant liability to third parties; or
require us to enter into royalty or licensing agreements, which may not be available on commercially reasonable terms, or at all, or which might be non-exclusive, which could result in our competitors gaining access to the same technology.
Although no third-party has asserted a claim of patent infringement against us as of the date of this prospectus, others may hold proprietary rights that could prevent our product candidates from being marketed. Any patent-related legal action against us claiming damages and seeking to enjoin activities relating to our product candidates or processes could subject us to potential liability for damages, including treble damages if we were determined to willfully infringe, and require us to obtain a license to manufacture or develop our product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. We cannot predict whether we would prevail in any such actions or that any license required under any of these patents would be made available on commercially reasonable terms, if at all. Moreover, even if we or our future strategic partners were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. In addition, we cannot be certain that we could redesign our product candidates or processes to avoid infringement, if necessary. Accordingly, an adverse determination in a judicial or administrative proceeding, or the failure to obtain necessary licenses, could prevent us from developing and commercializing our product candidates, which could harm our business, financial condition and operating results.
Parties making claims against us may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise additional funds or otherwise have a material adverse effect on our business, results of operations, financial condition and prospects.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful. Further, our issued patents could be found invalid or unenforceable if challenged in court.
Competitors may infringe our intellectual property rights or those of our licensors. To prevent infringement or unauthorized use, we and/or our licensors may be required to file infringement claims, which can be expensive and time consuming. In addition, in a patent infringement proceeding, a court may decide that a patent we own or license is not valid, is unenforceable and/or is not infringed. If we or any of our licensors or potential future collaborators were to initiate legal proceedings against a third-party to enforce a patent directed at one of our product candidates, the defendant could counterclaim that our or our licensor’s patent is invalid and/or unenforceable in whole or in part. In patent litigation, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, lack of written description or non-enablement. Grounds for an unenforceability assertion could include an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO or made a misleading statement during prosecution.
If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on such product candidate. In addition, if the breadth or strength of protection provided by our patents and patent applications or those of our licensors is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates. Such a loss of patent protection would have a material adverse impact on our business.
Even if resolved in our favor, litigation or other legal proceedings relating to our or our licensor’s intellectual property rights may cause us to incur significant expenses and could distract our technical and management personnel from their normal responsibilities. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We or our licensor may not have sufficient financial or other resources to conduct or participate in such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we or our licensor can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could compromise our ability to compete in the marketplace.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or other legal proceedings relating to our intellectual property rights, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation or other proceedings.
Intellectual property litigation may lead to unfavorable publicity that harms our reputation and causes the market price of our common shares to decline.
During the course of any intellectual property litigation, there could be public announcements of the initiation of the litigation as well as results of hearings, rulings on motions, and other interim proceedings in the litigation. If securities analysts or investors regard these announcements as negative, the perceived value of our existing products, programs or intellectual property could be diminished. Accordingly, the market price of shares of our common stock may decline. Such announcements could also harm our reputation or the market for our future products, which could have a material adverse effect on our business.
Derivation or interference proceedings may be necessary to determine priority of inventions, and an unfavorable outcome may require us to cease using the related technology or to attempt to license rights from the prevailing party.
Derivation or interference proceedings provoked by third parties or brought by us or our licensors or declared by the USPTO or similar proceedings in foreign patent offices may be necessary to determine the priority of inventions with respect to our or our licensor’s patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our or our licensor’s defense of such proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. In addition, the uncertainties associated with such proceedings could have a material adverse effect on our ability to raise the funds necessary to continue our clinical trials, continue our research programs, license necessary technology from third parties or enter into development or manufacturing partnerships that would help us bring our product candidates to market.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. In particular, under the Leahy-Smith Act, the United States transitioned in March 2013 to a “first inventor to file” system in which, assuming that other requirements of patentability are met, the first inventor to file a patent application will be entitled to the patent regardless of whether a third-party was first to invent the claimed invention. A third-party that files a patent application in the USPTO after March 2013 but before we could therefore be awarded a patent covering an invention of our even if we had made the invention before it was made by such third-party. This will require us to be cognizant going forward of the time from invention to filing of a patent application. Furthermore, our ability to obtain and maintain valid and enforceable patents depends on whether the differences between our technology and the prior art allow our technology to be patentable over the prior art. Since patent applications in the United States and most other countries are confidential for a period of time after filing or until issuance, we cannot be certain that we or our licensor was the first to either (1) file any patent application related to our product candidates or (2) invent any of the inventions claimed in our or our licensor’s patents or patent applications.
The Leahy-Smith Act also includes a number of significant changes that affect the way patent applications will be prosecuted and also may affect patent litigation. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including PGR, IPR, and derivation proceedings. An adverse determination in any such submission or proceeding could reduce the scope or enforceability of, or invalidate, our patent rights, which could adversely affect our competitive position.
Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal courts necessary to invalidate a patent claim, a third-party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third-party may attempt to use the USPTO procedures to invalidate our or our licensor’s patent claims that would not have been invalidated if first challenged by the third-party as a defendant in a district court action. Thus, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our or our licensor’s patent applications and the enforcement or defense of our or our licensor’s issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
Changes in U.S. patent law, or laws in other countries, could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves a high degree of technological and legal complexity. Therefore, obtaining and enforcing biopharmaceutical patents is costly, time consuming and inherently uncertain. Changes in either the patent laws or in the interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property rights and may increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. We cannot predict the breadth of claims that may be allowed or enforced in our or our licensor’s patents or in third-party patents. In addition, Congress or other foreign legislative bodies may pass patent reform legislation that is unfavorable to us.
For example, the U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our and our licensor’s ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the U.S. federal courts, the USPTO, or similar authorities in foreign jurisdictions, the laws and regulations governing patents could change in unpredictable ways that would weaken our or our licensor’s ability to obtain new patents or to enforce our existing patents and patents we might obtain in the future.
We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We may also be subject to claims that former employees or other third parties have an ownership interest in our patents or other intellectual property. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and distraction to management and other employees.
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired, we may be open to competition from competitive products. Given the amount of time required for the development, testing and regulatory review of product candidates, patents protecting our product candidates might expire before or shortly after such candidates are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to our product candidates.
If we do not obtain patent term extension for our product candidates, our business may be harmed.
Depending upon the timing, duration and specifics of FDA marketing approval of our product candidates, one or more of our or our licensor’s U.S. patents may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. A maximum of one patent may be extended per FDA approved product as compensation for the patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only those claims covering such approved drug product, a method for using it or a method for manufacturing it may be extended. Patent term extension may also be available in certain foreign countries upon regulatory approval of our product candidates. However, we may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or restoration or the term of any such extension is less than we request, our competitors may obtain approval of competing products following our or our licensor’s patent expiration, and our revenue could be reduced, possibly materially. Further, if this occurs, our competitors may take advantage of our investment in development and trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
We may not be able to protect our intellectual property rights throughout the world.
Although we and our licensors have issued patents and pending patent applications in the United States and certain other countries, filing, prosecuting and defending patents in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we or our licensor has not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection but enforcement is not as strong as that in the United States. These products may compete with our product candidates, and our and our licensor’s patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of many foreign countries do not favor the enforcement of patents and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We or our licensor may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our or our licensor’s efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or our licensor is forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired, and our business, financial condition, results of operations and prospects may be adversely affected.
Obtaining and maintaining our patent protection depends on compliance with various procedural, documentary, fee payment and other requirements imposed by regulations and governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to the USPTO and various foreign patent offices at various points over the lifetime of our and our licensors’ patents and/or applications. We have systems in place to remind us to pay these fees, and we rely on third parties to pay these fees when due. Additionally, the USPTO and various foreign patent office’s require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with rules applicable to the particular jurisdiction. However, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If such an event were to occur, it could have a material adverse effect on our business.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition, we rely on the protection of our trade secrets, including unpatented know-how, technology and other proprietary information to maintain our competitive position. Although we have taken steps to protect our trade secrets and unpatented know-how, including entering into confidentiality agreements with third parties, and confidential information and inventions agreements with employees, consultants and advisors, we cannot provide any assurances that all such agreements have been duly executed, and any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets.
Moreover, third parties may still obtain this information or may come upon this or similar information independently, and we would have no right to prevent them from using that technology or information to compete with us. If any of these events occurs or if we otherwise lose protection for our trade secrets, the value of this information may be greatly reduced and our competitive position would be harmed. If we do not apply for patent protection prior to such publication or if we cannot otherwise maintain the confidentiality of our proprietary technology and other confidential information, then our ability to obtain patent protection or to protect our trade secret information may be jeopardized.
We may be subject to claims that we have wrongfully hired an employee from a competitor or that we or our employees have wrongfully used or disclosed alleged confidential information or trade secrets of their former employers.
As is common in the biopharmaceutical industry, in addition to our employees, we engage the services of consultants to assist us in the development of our product candidates. Many of these consultants, and many of our employees, were previously employed at, or may have previously provided or may be currently providing consulting services to, other biopharmaceutical companies including our competitors or potential competitors. We may become subject to claims that we, our employees or a consultant inadvertently or otherwise used or disclosed trade secrets or other information proprietary to their former employers or their former or current clients. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely affect our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to our management team and other employees.
Risks Related to Our Common Stock
An active, liquid and orderly market for our common stock may not be maintained.
Although our common stock is listed on the Nasdaq Capital Market, or Nasdaq, an active trading market for our common stock may never develop or, if it develops, many not be sustained. The lack of an active market may impair your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. An inactive market may also impair our ability to raise capital by selling shares and may impair our ability to acquire other businesses or technologies using our shares as consideration, which, in turn, could materially adversely affect our business.
The trading price of the shares of our common stock may be highly volatile, and purchasers of our common stock may incur substantial losses.
Our stock price is likely to be volatile. The stock market in general and the market for stock of biopharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may not be able to sell their common stock at or above their purchase price. The market price for our common stock may be influenced by those factors discussed in this “Risk Factors” section and many others, including:
our ability to enroll subjects in our ongoing and planned clinical trials;
results of our clinical trials and preclinical studies, and the results of the trials of our competitors or those of other companies in our market sector;
regulatory approval of our product candidates, or limitations to specific label indications or patient populations for its use, or changes or delays in the regulatory review process;
regulatory developments in the United States and foreign countries;
changes in the structure of healthcare payment systems, especially in light of current reforms to the U.S. healthcare system;
the success or failure of our efforts to acquire, license or develop additional product candidates;
innovations or new products developed by us or our competitors;
announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments;
manufacturing, supply or distribution delays or shortages;
any changes to our relationship with any manufacturers, suppliers, licensors, future collaborators or other strategic partners;
achievement of expected product sales and profitability;
market conditions in the biopharmaceutical sector and issuance of securities analysts’ reports or recommendations;
trading volume of our common stock;
an inability to obtain additional funding;
sales of our stock by insiders and stockholders;
general economic, industry and market conditions or other events or factors, many of which are beyond our control;
additions or departures of key personnel; and
intellectual property, product liability or other litigation against us.
In addition, in the past, stockholders have initiated class action lawsuits against biopharmaceutical companies following periods of volatility in the market prices of these companies’ stock. Such litigation, if instituted against us, could cause us to incur substantial costs and divert management’s attention and resources, which could have a material adverse effect on our business, financial condition and results of operations.
Our failure to meet the continued listing requirements of Nasdaq could result in a delisting of our common stock.
If we fail to satisfy the continued listing requirements of Nasdaq, such as the corporate governance requirements or the minimum closing bid price requirement, Nasdaq may take steps to delist our common stock. Such a delisting would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. In the event of a delisting, we can provide no assurance that any action we take to restore compliance with listing requirements would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below the Nasdaq minimum bid price requirement or prevent future non-compliance with Nasdaq’s listing requirements.
Our executive officers, directors and principal stockholders, if they choose to act together, will continue to control or significantly influence all matters submitted to stockholders for approval. Furthermore, two of our directors have been appointed by one of our principal stockholders.
As of December 31, 2019, our executive officers, directors and greater than 5% stockholders, in the aggregate, owned approximately 29.0% of our outstanding common stock. Furthermore, two of our directors have been appointed by our largest stockholder, SPH USA. As a result, such persons or their appointees to our board of directors, acting together, will have the ability to control or significantly influence all matters submitted to our board of directors or stockholders for approval, including the appointment of our management, the election and removal of directors and approval of any significant transaction, as well as our management and business affairs. This concentration of ownership may have the effect of delaying, deferring or preventing a change in control, impeding a merger, consolidation, takeover or other business combination involving us, or discouraging a potential acquiror from making a tender offer or otherwise attempting to obtain control of our business, even if such a transaction would benefit other stockholders.
We do not currently intend to pay dividends on our common stock, and, consequently, your ability to achieve a return on your investment will depend on appreciation, if any, in the price of our common stock.
We have never declared or paid any cash dividend on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. In addition, the terms of any future debt agreements may preclude us from paying dividends. Any return to stockholders will therefore be limited to the appreciation of their stock. There is no guarantee that shares of our common stock will appreciate in value or even maintain the price at which stockholders have purchased their shares.
Sales of a substantial number of shares of our common stock by our stockholders in the public market could cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market or the perception that these sales might occur could significantly reduce the market price of our common stock and impair our ability to raise adequate capital through the sale of additional equity securities. As of December 31, 2019, 9,583,867 shares of our outstanding common stock are freely tradable, without restriction, in the public market, unless they are purchased by one of our affiliates.
Holders of approximately 58.3% of our outstanding securities, including our directors and executive officers, entered into lock-up agreements in connection with the Merger pursuant to which they could not, with limited exceptions, for a period of 180 days from the date of the effective time of the Merger, offer, sell or otherwise transfer or dispose of any of our securities, without our prior written consent, subject to certain exceptions. Sales of these shares, or perceptions that they will be sold, could cause the trading price of our common stock to decline. The lock-up agreements expired in December 2019 and, as a result, up to an additional 8,967,006 shares of common stock became eligible for sale in the public market.
In addition, as of December 31, 2019, up to 3,293,674 shares of common stock that are either subject to outstanding warrants, options or reserved for future issuance under our equity incentive plans will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules and Rule 144 and Rule 701 under the Securities Act of 1933, as amended, or the Securities Act. If these additional shares of common stock are sold, or if it is perceived that they will be sold, in the public market, the trading price of our common stock could decline.
We incur significant costs as a result of operating as a public company, and our management is required to devote substantial time to compliance initiatives.
As a public company, we incur significant legal, accounting and other expenses that we did not incur as a private company. We are subject to the reporting requirements of the Exchange Act, which will require, among other things, that we file with the Securities and Exchange Commission, or the SEC, annual, quarterly and current reports with respect to our business and financial condition. In addition, Sarbanes-Oxley, as well as rules subsequently adopted by the SEC and Nasdaq to implement provisions of Sarbanes-Oxley, impose significant requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. Further, pursuant to the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, the SEC has adopted additional rules and regulations in these areas, such as mandatory “say on pay” voting requirements that apply to us. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business in ways we cannot currently anticipate.
We expect the rules and regulations applicable to public companies to substantially increase our legal and financial compliance costs and to make some activities more time consuming and costly. If these requirements divert the attention of our management and personnel from other business concerns, they could have a material adverse effect on our business, financial condition and results of operations. The increased costs will increase our net loss and may require us to reduce costs in other areas of our business or increase the prices of our products or services. For example, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to incur substantial costs to maintain the same or similar coverage. We cannot predict or estimate the amount or timing of additional costs we may incur to respond to these requirements. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as executive officers.
If securities or industry analysts do not publish research or reports or publish unfavorable research or reports about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us, our business, our market or our competitors. We do not currently have and may never obtain research coverage by securities and industry analysts. If no securities or industry analysts commence coverage of our company, the trading price for our common stock would be negatively impacted. In the event we obtain securities or industry analyst coverage, if one or more of the analysts who covers us downgrades our stock, our stock price would likely decline. If one or more of these analysts ceases to cover us or fails to regularly publish reports on us, interest in our stock could decrease, which could cause our stock price or trading volume to decline.
If we fail to maintain proper and effective internal control over financial reporting, our ability to produce accurate and timely financial statements could be impaired, investors may lose confidence in our financial reporting and the trading price of our common stock may decline.
Pursuant to Section 404 of Sarbanes-Oxley, we are required to report upon the effectiveness of our internal control over financial reporting. Additionally, our independent registered public accounting firm is required to attest to the effectiveness of our internal control over financial reporting. The rules governing the standards that must be met for management to assess our internal control over financial reporting are complex and require significant documentation, testing and possible remediation. To comply with the requirements of being a reporting company under the Exchange Act, we have been required to upgrade our information technology systems; implement additional financial and management controls, reporting systems and procedures; and hire additional accounting and finance staff. If we or, if required, our auditors are unable to conclude that our internal control over financial reporting is effective, investors may lose confidence in our financial reporting and the trading price of our common stock may decline.
We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and
youwe couldlose allbe subject to sanctions orpart of your investmentinvestigations by Nasdaq, the SEC or other regulatory authorities. Failure to remedy any material weakness in ourcommon stock.Risks Relatedinternal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to theProposedcapital markets.Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable and may lead to entrenchment of management.
Our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could significantly reduce the value of our shares to a potential acquiror or delay or prevent changes in control or changes in our management without the consent of our board of directors. The provisions in our charter documents include the following:
a classified board of directors with three-year staggered terms, which may delay the ability of stockholders to change the membership of a majority of our board of directors;
no cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates;
the exclusive right of our board of directors, unless the board of directors’ grants such right to the stockholders, to elect a director to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors;
the prohibition on removal of directors without cause due to the classified board of directors;
the ability of our board of directors to alter our amended and restated bylaws without obtaining stockholder approval;
the required approval of at least 66-2/3% of the shares entitled to vote to adopt, amend or repeal our amended and restated bylaws or repeal certain provisions of our amended and restated certificate of incorporation;
a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders;
an exclusive forum provision providing that the Court of Chancery of the State of Delaware will be the exclusive forum for certain actions and proceedings;
the requirement that a special meeting of stockholders may be called only by the chairman of the board of directors, the chief executive officer or the board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; and
advance notice procedures that stockholders must comply with in order to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquiror from conducting a solicitation of proxies to elect the acquiror’s own slate of directors or otherwise attempting to obtain control of us.
We are also subject to the anti-takeover provisions contained in Section 203 of the Delaware General Corporation Law. Under Section 203, a corporation may not, in general, engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other exceptions, the board of directors has approved the transaction.
Our amended and restated bylaws provide that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated bylaws provide that the Court of Chancery of the State of Delaware is the exclusive forum for any derivative action or proceeding brought on our behalf, any action asserting a breach of fiduciary duty, any action asserting a claim against us arising pursuant to the Delaware General Corporation Law, our amended and restated certificate of incorporation or our amended and restated bylaws, or any action asserting a claim against us that is governed by the internal affairs doctrine. These choice of forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. By agreeing to this provision, however, stockholders will not be deemed to have waived our compliance with the federal securities laws and the rules and regulations thereunder. Furthermore, the enforceability of similar choice of forum provisions in other companies’ certificates of incorporation has been challenged in legal proceedings, and it is possible that a court could find these types of provisions to be inapplicable or unenforceable. If a court were to find the choice of forum provisions in our amended and restated bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could adversely affect our business and financial condition.
If the Merger
does not qualify as a “reorganization” for U.S. federal income tax purposes, U.S. Holders of our common stock will be required to recognize gain or loss for U.S. federal income tax purposes upon the exchange of their Private Oncternal common stock for our common stock in the Merger.The
exchange ratio set forthU.S. federal income tax consequences of the Merger to U.S. Holders will depend on whether the merger qualifies as a “reorganization” for U.S. federal income tax purposes. Our and Private Oncternal’s obligations to effect the Merger were subject to the satisfaction, or waiver, at or prior to the effective time of the Merger, of the condition that each company receive an opinion of counsel, dated as of the closing date of the Merger, to the effect that the Merger will qualify as a “reorganization” within the meaning of Section 368(a) of the Code. If, contrary to the opinions from counsel, the Merger fails to qualify as a reorganization within the meaning of Section 368(a) of the Code, a U.S. Holder of Private Oncternal common stock would recognize gain or loss for U.S. federal income tax purposes on each share of Private Oncternal common stock surrendered in the MergerAgreementfor our common stock and any cash received in lieu of a fractional share. For purposes of this discussion, a U.S. Holder is a beneficial owner of Oncternal common stock that, for U.S. federal income tax purposes, is or is treated as: an individual who is a citizen or resident of the United States; a corporation created or organized under the laws of the United States, any state thereof, or the District of Columbia; an estate, the income of which is subject to U.S. federal income tax regardless of its source; or a trust that (i) is subject to the primary supervision of a U.S. court and the control of one or more “United States persons” (within the meaning of Section 7701(a)(30) of the Code) over all of its substantial decisions or (ii) has a valid election in effect to be treated as a United States person for U.S. federal income tax purposes.Our ability to use net operating loss (“NOL”) carryforwards and other tax attributes may be limited.
We have incurred substantial losses during our history and do not
adjustable basedexpect to become profitable in the near future, and we may never achieve profitability. To the extent that we continue to generate taxable losses, unused losses will carry forward to offset future taxable income, if any, until such unused losses expire (if at all). As of December 31, 2019, we had federal and state NOL carryforwards of approximately $70.0 million and $48.7 million, respectively. Approximately $44.2 million of NOLs do not expire and the remaining federal and state NOL carryforward will begin to expire in 2033, unless previously utilized. At December 31, 2019, we had federal and state research and development credit carryforwards of approximately $0.9 million and $0.7 million, respectively. The federal research and development credit carryforwards will begin expiring in 2034, unless previously utilized. The state research and development credits do not expire.Under the Tax Act, federal NOLs generated in taxable years ending after December 31, 2017, may be carried forward indefinitely but federal NOLs generated in taxable years beginning after December 31, 2017 may only be used to offset 80% of our taxable income annually. Under Sections 382 and 383 of the Code, our NOL and research and development tax credit carryforwards may become subject to an annual limitation in the event of certain cumulative changes in the ownership interest of significant stockholders over a three-year period in excess of 50 percentage points. Our ability to utilize our NOL carryforwards and other tax attributes to offset future taxable income or tax liabilities may be limited as a result of ownership changes, including changes resulting from our Merger. We have not yet determined the amount of the cumulative change in our ownership resulting from the Merger or other transactions, or any resulting limitations on our ability to utilize our NOL carryforwards and other tax attributes. If we earn taxable income, such limitations could result in increased future tax liability to us and our future cash flows could be adversely affected. We have recorded a full valuation allowance related to our NOLs and other deferred tax assets due to the uncertainty of the ultimate realization of the future benefits of those assets.
U.S. tax legislation may materially adversely affect our financial condition, results of operations and cash flows.
The Tax Act has significantly changed the U.S. federal income taxation of U.S. corporations, including by reducing the U.S. corporate income tax rate and revising the rules governing NOLs. Many of these changes became effective beginning in 2018, without any transition periods or grandfathering for existing transactions. The legislation is unclear in many respects and could be subject to potential amendments and technical corrections, as well as interpretations and implementing regulations by the U.S. Treasury Department and the IRS, any of which could lessen or increase certain adverse impacts of the legislation. In addition, it is unclear how these U.S. federal income tax changes will affect state and local taxation, which often uses federal taxable income as a starting point for computing state and local tax liabilities. There may be other material adverse effects resulting from the legislation that we have not yet identified. While some of the changes made by the tax legislation may adversely affect us in one or more reporting periods and prospectively, other changes may be beneficial on a going forward basis. We continue to work with our tax advisors to determine the full impact that the recent tax legislation as a whole will have on us. We urge our investors to consult with their legal and tax advisors with respect to such legislation.
We are currently involved, and may become involved in the future, in securities class action litigation that could divert management’s attention, adversely affect our business and subject us to significant liabilities.
In the past, securities class action litigation has often been brought against a company following volatility in the market price of its securities. This risk is especially relevant for us, because biotechnology and pharmaceutical companies have experienced significant stock price volatility in recent years. Between April 10, 2019, and May 1, 2019, three putative class action lawsuits and one individual lawsuit were filed in the U.S. District Court for the District of Delaware, and two putative class actions were filed in the U.S. District Court for the Southern District of New York, naming us and our board of directors as defendants and alleging that the defendants violated Sections 14(a) and 20(a) of the Exchange Act, as well as Rule 14a-9 promulgated thereunder, in connection with our filing of the Registration Statement in connection with the Merger. The Delaware actions have now been voluntarily dismissed with prejudice.
Additionally, on October 15, 2019, a lawsuit was filed against us in the U.S. District Court for the District of Delaware alleging that GTx management engaged in illegal insider trading and false, manipulative and deceptive practices in violation of Sections 10(b) (and Rule 10b-5 promulgated thereunder), with respect to the timing of the disclosure of failed clinical trial results of GTx’s enobosarm product candidate in September 2018.
These lawsuits and any future lawsuits to which we may become a party are subject to inherent uncertainties and will likely be expensive and time-consuming to investigate, defend and resolve, and will divert our management’s attention and financial and other resources. The outcome of litigation is necessarily uncertain, and we could be forced to expend significant resources in the defense of this and other suits, and we may not prevail. Any litigation to which we are a party may result in an onerous or unfavorable judgment that may not be reversed upon appeal or in payments of substantial monetary damages or fines, or we may decide to settle this or other lawsuits on similarly unfavorable terms, which could adversely affect our business, financial condition, results of operations or stock price. See Part II, Item 1 “Legal Proceedings” in this Quarterly Report on Form 10-Q for more information about the lawsuits that have been filed. In addition, stock markets have experienced significant price and volume fluctuations that have affected the market prices for the common stock of pharmaceutical companies. These broad market fluctuations as well a broad range of other factors, including the realization of any of the risks described in these “Risk Factors,” may cause the market price of our common stock
to decline.so the merger consideration at the closing of the Merger may have a greater or lesser value than at the time the Merger Agreement was signed.The Merger Agreement has set the exchange ratio for the Oncternal capital stock, and the exchange ratio is based on the outstanding capital stock of Oncternal and the outstanding common stock of GTx, in each case immediatelyOur stockholders prior to the
closing of the Merger. Applying the exchange ratio formula in theMergerAgreement, the former Oncternal stockholders immediately before the Merger are expected to own approximately 75% of the outstanding capital stock of GTx immediately following the Merger, and the stockholders of GTx immediately before the Merger are expected to own approximately 25% of the outstanding capital stock of GTx immediately following the Merger, subject to certain assumptions. Under certain circumstances further described in the Merger Agreement, however, these ownership percentages may be adjusted upward or downward based on cash levels ofthe respective companies at the closing of the Merger, and as a result, either our stockholders or the Oncternal stockholders could own less of the combined company than expected.Any changes in the market price of our common stock before the completion of the Merger will not affect the number of shares of our common stock issuable to Oncternal's stockholders pursuant to the Merger Agreement. Therefore, if before the completion of the Merger the market price of our common stock declines from the market price on the date of the Merger Agreement, then Oncternal's stockholders could receive merger consideration with substantially lower value than the value of such merger consideration on the date of the Merger Agreement. Similarly, if before the completion of the Merger the market price of our common stock increases from the market price of our common stock on the date of the Merger Agreement, then Oncternal's stockholders could receive merger consideration with substantially greater value than the value of such merger consideration on the date of the Merger Agreement. The Merger Agreement does not include a price-based termination right. Because the exchange ratio does not adjust as a result of changes in the market price of our common stock, for each one percentage point change in the market price of our common stock, there is a corresponding one percentage point rise or decline, respectively, in the value of the total merger consideration payable to Oncternal's stockholders pursuant to the Merger Agreement.Failure to complete the proposed Merger may result in GTx and Oncternal paying a termination fee to the other party and could significantly harm the market price of our common stock and negatively affect the future business and operations of each company.If the proposed Merger is not completed and the Merger Agreement is terminated under certain circumstances, we or Oncternal may be required to pay the other party a termination fee of up to $2.0 million. Even if a termination fee is not payable in connection with a termination of the Merger Agreement, each of GTx and Oncternal will have incurred significant fees and expenses, which must be paid whether or not the Merger is completed. Further, if the proposed Merger is not completed, it could significantly harm the market price of our common stock.In addition, if the Merger Agreement is terminated and the board of directors of GTx or Oncternal determines to seek another business combination, there can be no assurance that either we or Oncternal will be able to find a partner and close an alternative transaction on terms that are as favorable or more favorable than the terms set forth in the Merger Agreement.The proposed Merger is subject to approval of the Merger Agreement by our stockholders and the Oncternal stockholders. Failure to obtain these approvals would prevent the closing of the Merger.Before the proposed Merger can be completed, the stockholders of each of GTx and Oncternal must approve the Merger Agreement. Additionally, the Merger Agreement must be approved by multiple classes of Oncternal preferred stockholders, one class of which is held by a sole stockholder, Shanghai Pharmaceutical (USA) Inc., which has not executed a voting agreement and has not otherwise agreed to vote in favor of the Merger Agreement. Although Oncternal expects to receive stockholder approval from Shanghai Pharmaceutical (USA) Inc. approximately two months after the date of the Merger Agreement, there can be no assurance that all of the necessary stockholder approvals will be obtained. Failure to obtain the required stockholder approvals, including as a result of Shanghai Pharmaceutical (USA) Inc. refusing to approve the transactions contemplated by the Merger Agreement, may result in a material delay in, or the abandonment of, the Merger. Any delay in completing the proposed Merger may materially adversely affect the timing and benefits that are expected to be achieved from the proposed Merger.The Merger may be completed even though certain events occur prior to the closing that materially and adversely affect GTx or Oncternal.The Merger Agreement provides that either GTx or Oncternal can refuse to complete the proposed Merger if there is a material adverse change affecting the other party between March 6, 2019, the date of the Merger Agreement, and the closing of the Merger. However, certain types of changes do not permit either party to refuse to complete the proposed Merger, even if such change could be said to have a material adverse effect on GTx or Oncternal, including:•general business, economic or political conditions or conditions generally affecting the industries in which Oncternal or GTx, as applicable, operates;•any natural disaster or any acts of war, armed hostilities or terrorism;•any changes in financial, banking or securities markets;•with respect to GTx, any change in the stock price or trading volume of GTx excluding any underlying effect that may have caused such change;•with respect to GTx, failure to meet internal or analysts' expectations or projects or the results of operations;•any clinical trial programs or studies, including any adverse data, event or outcome arising out of or related to any such programs or studies;•any change in accounting requirements or principles or any change in applicable laws, rules, or regulations or the interpretation thereof;•any effect resulting from the announcement or pendency of the proposed Merger or any related transactions; and•the taking of any action, or the failure to take any action, by either GTx or Oncternal required to comply with the terms of the Merger Agreement.
If adverse changes occur and GTx and Oncternal still complete the Merger, the market price of the combined organization's common stock may suffer. This in turn may reduce the value of the Merger to the stockholders of GTx, Oncternal or both.Some GTx and Oncternal officers and directors have interests in the proposed Merger that are different from the respective stockholders of GTx and Oncternal and that may influence them to support or approve the Merger without regard to the interests of the respective stockholders of GTx and Oncternal.Certain officers and directors of GTx and Oncternal participate in arrangements that provide them with interests in the proposed Merger that are different from the interests of the respective stockholders of GTx and Oncternal, including, among others, the continued service as an officer or director of the combined organization, severance benefits, the acceleration of stock option vesting, continued indemnification and the potential ability to sell an increased number of shares of common stock of the combined organization in accordance with Rule 144 under the Securities Act of 1933, as amended.For example, we have entered into certain employment and severance benefits agreements with certain of our executive officers that may result in the receipt by such executive officers of cash severance payments and other benefits in the event of a covered termination of employment of each executive officer's employment. The closing of the Merger will also result in the acceleration of vesting of options to purchase shares of our common stock held by our executive officers and directors, whether or not there is a covered termination of such officer's employment. In addition, and for example, certain of Oncternal's directors and executive officers have options, subject to vesting, to purchase shares of Oncternal's common stock which, at the closing of the Merger, shall be converted into and become options to purchase shares of our common stock, certain of Oncternal's directors and executive officers are expected to become directors and executive officers of GTx upon the closing of the Merger, and all of Oncternal's directors and executive officers are entitled to certain indemnification and liability insurance coverage pursuant to the terms of the Merger Agreement. These interests, among others, may influence the officers and directors of GTx and Oncternal to support or approve the proposed Merger.The market price of our common stock following the Merger may decline as a result of the Merger.The market price of our common stock may decline as a result of the Merger for a number of reasons including if:•investors react negatively to the prospects of the combined organization's product candidates, business and financial condition following the Merger;•the effect of the Merger on the combined organization's business and prospects is not consistent with the expectations of financial or industry analysts; or•the combined organization does not achieve the perceived benefits of the Merger as rapidly or to the extent anticipated by financial or industry analysts.
GTx and Oncternal securityholders will have a reduced ownership and voting interest in, and will exercise less influence over the management of, the combined organization following the closing of the Merger as compared to their current ownership and voting interest in the respective companies.After the completion of the Merger, the current securityholders of GTx and Oncternal will own a smaller percentage of the combined organization than their ownership in their respective companies prior to the Merger. Immediately after the Merger, it is currently estimated that Oncternal securityholders will own approximately 75% of the common stock of the combined organization, and GTx securityholders, whose shares of GTx common stock will remain outstanding after the Merger, will own approximately 25% of the common stock of the combined organization. These estimates are based on the anticipated exchange ratio and are subject to adjustment as provided in the Merger Agreement. See also the risk factor above titled, "The exchange ratio is not adjustable based on the market price of GTx common stock, so the merger consideration at the closing may have a greater or lesser value than at the time the Merger Agreement was signed."In addition, the nine member board of directors of the company will initially include seven individuals with prior affiliations with Oncternal and two individuals with prior affiliations with GTx. Consequently, securityholders of GTx and Oncternal will be able to exercise less influence over the management and policies of the combined organization following the closing of the Merger than they currently exercise over the management and policies of their respective companies.GTx and Oncternal stockholders may not realize a benefit from the Merger commensurate with the ownership dilution they will experience in connection with the Merger.If the combined organization is unable to realize the strategic and financial benefits currently anticipated from the proposed Merger, GTx's and Oncternal's stockholders will have experienced substantial dilution of their ownership interests in their respective companies without receiving the expected commensurate benefit, or only receiving part of the commensurate benefit to the extent the combined organization is able to realize only part of the expected strategic and financial benefits currently anticipated from the proposed Merger.The combined company will need to raise additional capital by issuing securities or debt or through licensing or other strategic arrangements, which may cause dilution to the combined company's stockholders or restrict the combined company's operations or impact its proprietary rights.The combined company may be required to raise additional funds sooner than currently planned. In this regard, while the exchange ratio may be impacted by cash levels of the respective companies at the closing of the Merger, the Merger Agreement does not condition the completion of the Merger upon either company holding a minimum amount of cash at the effective time of the Merger. If either or both of GTx or Oncternalwho holdless cash at the time of the closing Merger than the parties currently expect, the combined company will need to raise additional capital sooner than expected. Additional financing may not be available to the combined company when it needs it or may not be available on favorable terms. To the extent that the combined company raises additional capital by issuing equity securities, such an issuance may cause significant dilution to the combined company's stockholders' ownership and the terms of any new equity securities may have preferences over the combined company's common stock. Any debt financing the combined company enters into may involve covenants that restrict its operations. These restrictive covenants may include limitations on additional borrowing and specific restrictions on the use of the combined company's assets, as well as prohibitions on its ability to create liens, pay dividends, redeem its stock or make investments. In addition, if the combined company raises additional funds through licensing, partnering or other strategic arrangements, it may be necessary to relinquish rights to some of the combined company's technologies or product candidates and proprietary rights, or grant licenses on terms that are not favorable to the combined company.During the pendency of the proposed Merger, GTx and Oncternal may not be able to enter into a business combination with another party at a favorable price because of restrictions in the Merger Agreement, which could adversely affect their respective businesses.Covenants in the Merger Agreement impede the ability of GTx and Oncternal to make acquisitions, subject to certain exceptions relating to fiduciary duties, as set forth below, or to complete other transactions that are not in the ordinary course of business pending completion of the proposed Merger. As a result, if the Merger is not completed, the parties may be at a disadvantage to their competitors during such period. In addition, while the Merger Agreement is in effect, each party is generally prohibited from soliciting, initiating, encouraging or entering into certain extraordinary transactions, such as a merger, sale of assets, or other business combination outside the ordinary course of business with any third party, subject to certain exceptions relating to fiduciary duties. Any such transactions could be favorable to such party's stockholders.Certain provisions of the Merger Agreement may discourage third parties from submitting alternative takeover proposals, including proposals that may be superior to the arrangements contemplated by the Merger Agreement.The terms of the Merger Agreement prohibit each of GTx and Oncternal from soliciting alternative takeover proposals or cooperating with persons making unsolicited takeover proposals,except in limited circumstances when such party's board of directors determines in good faith that an unsolicited alternative takeover proposal is or is reasonably likely to lead to a superior takeover proposal and that failure to cooperate with the proponent of the proposal would be reasonably likely to be inconsistent with the applicable board's fiduciary duties.Because the lack of a public market for Oncternal's capital stock makes it difficult to evaluate the value of Oncternal's capital stock, the stockholders of Oncternal may receive shares of our common stock in the Merger that have a value that is less than, or greater than, the fair market value of Oncternal's capital stock.The outstanding capital stock of Oncternal is privately held and is not traded in any public market. The lack of a public market makes it extremely difficult to determine the fair market value of Oncternal. Because the percentage of our common stock to be issued to Oncternal's stockholders was determined based on negotiations between the parties, it is possible that the value of our common stock to be received by Oncternal's stockholders will be less than the fair market value of Oncternal, or GTx may pay more than the aggregate fair market value for Oncternal.If the conditions to the Merger are not met, the Merger will not occur.Even if the Merger is approved by the stockholders of GTx and Oncternal, specified conditions must be satisfied or waived to complete the Merger. We cannot assure you that all of the conditions will be satisfied or waived. If the conditions are not satisfied or waived, the Merger will not occur or will be delayed, and GTx and Oncternal each may lose some or all of the intended benefits of the proposed Merger.Litigation relating to the proposed Merger could require GTx or Oncternal to incur significant costs and suffer management distraction, and could delay or enjoin the proposed Merger.GTx and Oncternal could be subject to demands or litigation related to the proposed Merger, whether or not the Merger is consummated. Such actions may create uncertainty relating to the Merger, or delay or enjoin the Merger, and responding to such demands.Risks Related to Our Financial Condition and Our Need for Additional Financing, and Additional Risks Related to the MergerThere is no assurance that the proposed Merger will be completed in a timely manner or at all. If the proposed Merger is not consummated, our business could suffer materially and our stock price could decline.The closing of the proposed Merger is subject to the satisfaction or waiver of a number of closing conditions, as described above, including the required approvals by GTx and Oncternal stockholders (including stockholder approval from one of Oncternal's significant stockholders, Shanghai Pharmaceutical (USA) Inc., which holds all of the outstanding shares of one series of Oncternal's preferred stock that must approve the transactions contemplated by the Merger Agreement) and other customary closing conditions. See the risk factors above titled, "The proposed Merger is subject to approval of the Merger Agreement by our stockholders and the Oncternal stockholders. Failure to obtain these approvals would prevent the closing of the Merger" and "If the conditions to the Merger are not met, the Merger will not occur." If the conditions are not satisfied or waived, including as a result of Shanghai Pharmaceutical (USA) Inc. refusing to approve the transactions contemplated by the Merger Agreement, the proposed Merger may be materially delayed or abandoned. If the proposed Merger is not consummated, our ongoing business may be adversely affected and, without realizing any of thebenefits of having consummated the proposed Merger, we will be subject to a number of risks, including the following:•we have incurred and expect to continue to incur significant expenses related to the proposed Merger even if the Merger is not consummated;•we could be obligated to pay Oncternal a termination fee of up to $2.0 million under certain circumstances set forth in the Merger Agreement;•the market price of our common stock may decline to the extent that the current market price reflects a market assumption that the proposed Merger will be completed; and•matters relating to the proposed Merger have required and will continue to require substantial commitments of time and resources by our remaining management and employees, which could otherwise have been devoted to other opportunities that may have been beneficial to us.
We also could be subject to litigation related to any failure to consummate the proposed Merger or to perform our obligations under the Merger Agreement. If the proposed Merger is not consummated, these risks may materialize and may adversely affect our business, financial condition and the market price of our common stock.If the proposed Merger is not completed, we may be unsuccessful in completing an alternative transaction on terms that are as favorable as the terms of the proposed Merger with Oncternal, or at all, and we may otherwise be unable to continue to operate our business. Our board of directors may decide to pursue a dissolution and liquidation of GTx. In such an event, the amount of cash available for distribution to our stockholders will depend heavily on the timing of such liquidation as well as the amount of cash that will need to be reserved for commitments and contingent liabilities.Our assets currently consist primarily of cash, cash equivalents and short-term investments, our SARD and SARM assets, the remaining value, if any, of our deferred tax assets, our listing on The Nasdaq Capital Market and the Merger Agreement with Oncternal. While we have entered into the Merger Agreement with Oncternal, the closing of the proposed Merger may be delayed or may not occur at all and there can be no assurance that the proposed Merger will deliver the anticipated benefits we expect or enhance stockholder value. If we are unable to consummate the proposed Merger, our board of directors may elect to pursue an alternative strategy, one of which may be a strategic transaction similar to the proposed Merger. Attempting to complete an alternative transaction like the proposed Merger will be costly and time consuming, and we can make no assurances that such an alternative transaction would occur at all. Alternatively, our board of directors may elect to continue our operations to advance appropriate SARD compounds into the additional preclinical studies required to submit an IND and potentially advance one of our SARD compounds into a first-in-human clinical trial, which would require that we obtain additional funding, and to resume our efforts to seek potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets, or our board of directors could instead decide to pursue a dissolution and liquidation of our company. In such an event, the amount of cash available for distribution to our stockholders will depend heavily on the timing of such decision, as with the passage of time the amount of cash available for distribution will be reduced as we continue to fund our operations. In addition, if our board of directors were to approve and recommend, and our stockholders were to approve, a dissolution and liquidation of our company, we would be required under Delaware corporate law to pay our outstanding obligations, as well as to make reasonable provision for contingent and unknown obligations, prior to making any distributions in liquidation to our stockholders. Our commitments and contingent liabilities may include severance obligations,regulatory and preclinical obligations, and fees and expenses related to the proposed Merger. As a result of this requirement, a portion of our assets may need to be reserved pending the resolution of such obligations. In addition, we may be subject to litigation or other claims related to a dissolution and liquidation. If a dissolution and liquidation were pursued, our board of directors, in consultation with its advisors, would need to evaluate these matters and make a determination about a reasonable amount to reserve. Accordingly, holders of our common stock could lose all or a significant portion of their investment in the event of a liquidation, dissolution or winding up of the company.The issuance of shares of our common stock to Oncternal stockholders in the proposed Merger will substantially dilute the voting power of our current stockholders.If the proposed Merger is completed, each outstanding share of Oncternal common stock will be converted into the right to receive a number of shares of our common stock equal to the exchange ratio determined pursuant to the Merger Agreement. Immediately following the Merger, the former Oncternal stockholders immediately before the Merger are expected to own approximately 75% of our outstanding capital stock, and our stockholders immediately before the Merger are expected to own approximately 25% of our outstanding capital stock, subject to certain assumptions. Accordingly, the issuance of shares of our common stock to Oncternal stockholders in the Merger will reduce significantly the relative voting power of each share of GTx common stock held by our current stockholders. Consequently, our stockholders as a group will have significantly less influence over the management and policies of the combined company after the Merger than prior to the Merger. These estimates are based on the anticipated exchange ratio and are subject to adjustment as provided in the Merger Agreement. See also the risk factor above titled, "The exchange ratio is not adjustable based on the market price of GTx common stock, so the merger consideration at the closing may have a greater or lesser value than at the time the Merger Agreement was signed."GTx stockholdersCVRs may not receive any payment on the CVRs and the CVRs may otherwise expire valueless.If the proposed Merger is completed, weWe and certain other partieswill enterhave entered into the CVR Agreement pursuant to which, for each share of GTx common stock held,GTxstockholders of record as of immediately prior to theeffective time of theMergerwill receivereceived one CVR entitling such holders to receive in the aggregate50%75% of any net proceeds received during the 15-year period after the closing of the Merger from the grant, sale or transfer of rights to our SARD or SARM technology that occurs during the 10-year period after the closing of the Merger (or in the 11th year if based on a term sheet approved during the initial 10-year period) and, if applicable, to receive royalties on the sale of any SARD products or SARM products bythe combined companyus during the 15-year period after the closing of the Merger.In 2018, we announced that we had ceased development of the SARM technology following the failure of a Phase 2 clinical study of enobosarm to achieve statistical significance with respect to the primary endpoint of the study. On December 31, 2019, we disclosed that we had provided notice of termination of the amended and restated license agreement with the University of Tennessee Research Foundation, or UTRF, for the development and production of the SARM technology, which termination will be effective on March 31, 2020. Following termination, we will no longer have the obligation to make further payments to UTRF under the SARM license agreement, including payments for patent prosecution and maintenance, and will no longer have any rights to develop or sublicense the SARM technology.
The CVRs
willare notbetransferable, will not have any voting or dividend rights, and interest will not accrue on any amounts potentially payable on the CVRs. Accordingly, the right of anyGTxstockholder of record as of immediately prior to the Merger to receive any future payment on or derive any valueformfrom the CVRs will be contingent solely upon the achievement of the foregoing events within the time periods specified in the CVR Agreement and if these events are not achieved for any reason within the time periods specified in the CVR Agreement, no payments will be made under the CVRs, and the CVRs will expire valueless. In addition,Oncternalwe (as successor in interest to GTx)hashave agreed only to use commercially reasonable efforts to develop SARD products,and to divest our SARM technology,subject to certain limitations, which allows for the consideration of a variety of factors in determining the efforts thatthe combined company iswe are required to use to develop SARD products, andto divest our SARM technology, and it doeswe are notrequire the combined companyrequired to take all possible actions to continue efforts to develop SARDproducts and to divest our SARM technology.products. Accordingly, under certain circumstancesthe combined companywe may not be required to continue efforts to develop SARD products,and to divest our SARM technology,or may allocate resources to other projects, which would have an adverse effect on the value, if any, of the CVRs. Furthermore, the CVRs will be unsecured obligations ofthecombinedour company and all payments under the CVRs, all other obligations under the CVR Agreement and the CVRs and any rights or claims relating thereto will be subordinated in right of payment to the prior payment in full of all of our current or future seniorobligations of the combined company.obligations. Finally, the U.S. federal income tax treatment of the CVRs is unclear. There is no legal authority directly addressing the U.S. federal income tax treatment of the receipt of, and payments on, the CVRs, and there can be no assurance that theInternal Revenue Service,IRS, would not assert, or that a court would not sustain, a position that could result in adverse U.S. federal income tax consequences to holders of the CVRs.We have incurred losses since inception, and we anticipate that we will incur continued losses for the foreseeable future.As of December 31, 2018, we had an accumulated deficit of $600.1 million. Our net loss for the year ended December 31, 2018 was $38.4 million and we expect to incur significant operating losses for the foreseeable future depending on the extent of our preclinical and any clinical development activities and, if any such development activities are successful, potentially seeking regulatory approval of any potential future product candidates. These losses, among other things, have had and will continue to have an adverse effect on our stockholders' equity and working capital.A substantial portion of our recent efforts and expenditures have been devoted to, and our prospects were substantially dependent upon, the development of enobosarm for the treatment of postmenopausal women with SUI. However, in September 2018, we announced that our placebo-controlled Phase 2 clinical trial of enobosarm to evaluate the change in frequency of daily SUI episodes following 12 weeks of treatment, or the ASTRID trial, failed to achieve statistical significance on the primary endpoint of the proportion of patients with a greater than 50% reduction in incontinence episodes per day compared to placebo. The failure of the ASTRID trial to achieve its primary endpoint has significantly depressed our stock price and has severely harmed our ability to raise additional capital and to secure potential collaborative, partnering or other strategic arrangements for our SARM assets, and consequently, our prospects to continue as a going concern have been severely diminished. Following our review of the full data sets from the ASTRID trial, we determined to discontinue further development of enobosarm to treat SUI and to otherwise discontinue any further development of our SARM program generally. We continue our efforts to seek potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets. We have for many years actively pursued, but have been unable to successfully enter into, potential collaborative, partnering or other strategic arrangements for our SARM assets. If we are unable to ultimately enter into any such arrangements for our SARM assets, we will not receive any return on our investment in enobosarm and our other SARMs.As a result of our decision to discontinue our SARM development efforts, our development activities are focused solely on completing ongoing mechanistic preclinical studies in order to select the most appropriate SARD compounds to move forward into the additional preclinical studies required to submit an IND and potentially advance one of our SARD compounds into a first-in-human clinical trial. However, while we believe that our existing capital resources will be adequate to enable us to conduct and complete planned IND-enabling preclinical studies of our SARD compounds, we will require significant additional financial resources in order to initiate and complete initial human clinical trials of a SARD compound and to otherwise further the development of our SARD program. Accordingly, if, for any reason, the proposed Merger is not consummated, we may resume our efforts to seek additional funds through potential collaborative, partnering or other strategic arrangements to provide us with the necessary resources for the development of our SARD program. In addition, our preclinical evaluation of our SARD technology is at very early stage and is subject to the substantial risk and probability of failure inherent in the development of early-stage programs.Because of the numerous risks and uncertainties associated with developing and commercializing small molecule drugs, we are unable to predict the extent of any future losses or when we will become profitable, if at all. We have funded our operations primarily through public offerings and private placements of our securities, as well as payments from our former collaborators. We also previously recognized product revenue from the sale of FARESTON®, the rights to which we sold to a third party in the third quarter of 2012. Currently, we have no ongoing collaborations for the development and commercialization of our product candidates, and as a result of the sale of our rights and certain assets related to FARESTON®, we also currently have no sources of revenue.If the proposed Merger is not completed and we are unable to raise sufficient additional funds for the development of our SARD program, whether through potential collaborative, partnering or other strategic arrangements or otherwise, or if we otherwise determine to discontinue the development of our SARD program, we will likely determine to cease operations. Even if we are able to raise additional funds to permit the continued development of our SARD program, if we and/or any potential collaborators are unable to develop and commercialize our SARDs or SARM technology, if development is further delayed or is eliminated, or if sales revenue from any SARD or partnered SARM products upon receiving marketing approval, if ever, is insufficient, we may never become profitable and we will not be successful.If we do not successfully complete the proposed Merger, we will need to raise substantial additional capital and may be unable to raise the capital necessary to permit the continued development of our SARD program, which would force us to delay, reduce or eliminate our SARD program and would likely cause us to cease operations.At December 31, 2018, we had cash, cash equivalents and short-term investments of $28.5 million. If the proposed Merger is not completed, based on our current business plan and spending assumptions as a standalone company, we estimate that our current cash, cash equivalents and short-term investments, together with interest thereon, will be sufficient to meet our projected operating requirements for at least the next 12 months. We have based our cash sufficiency estimates on our current business plan and our assumptions that may prove to be wrong. We could utilize our available capital resources sooner than we currently expect, and we could need additional funding sooner than currently anticipated.While we believe that our existing capital resources will be adequate to enable us to conduct and complete planned IND-enabling preclinical studies of SARD compounds, we will require significant additional financial resources in order to initiate and complete initial human clinical trials of a SARD compound and to otherwise further the development of our SARD program. If we are unable to raise sufficient additional funds for the development of our SARD program, whether through potential collaborative, partnering or other strategic arrangements or otherwise, or if we otherwise determine to discontinue the development of our SARD program, we will likely determine to cease operations.Our future funding requirements will depend on many factors, including:•our ability to successfully complete the Merger;•the scope, rate of progress and cost of our preclinical and potential future clinical development programs;•the terms and timing of any potential collaborative, partnering and other strategic arrangements that we may establish;
•the amount and timing of any licensing fees, milestone payments and royalty payments from potential collaborators, if any;•potential future clinical trial results;•the cost and timing of regulatory filings and/or approvals to commercialize any potential future product candidates and any related restrictions, limitations, and/or warnings in the label of an approved product candidate;•the effect of competing technological and market developments; and•the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights, and the cost of defending any other litigation claims.
While we have been able to fund our operations to date, we have no ongoing collaborations for the development and commercialization of any product candidates and no source of revenue, nor do we expect to generate product revenue for the foreseeable future. We do not have any commitments for future external funding. In addition, although we have entered into an At-the-Market Equity OfferingSMSales Agreement with Stifel, Nicolaus & Company, Incorporated, or the ATM Sales Agreement, under which approximately $25.0 million of shares of our common stock remained available for sale at December 31, 2018, it is unlikely we could raise sufficient funds under the ATM Sales Agreement to permit us to initiate and complete initial human clinical trials of a SARD compound, and given our currently-depressed stock price, the ATM Sales Agreement is not otherwise expected to be a practical source of liquidity for us at this time. Further, given our currently-depressed stock price, we are significantly limited in our ability to sell shares of common stock under the ATM Sales Agreement since the issuance and sale of our common stock under the ATM Sales Agreement, if it occurs, would be effected under a registration statement on Form S-3 that we filed with the Securities and Exchange Commission, and in accordance with the rules governing those registration statements, we generally can only sell shares of our common stock under that registration statement in an amount not to exceed one-third of our public float, which limitation for all practical purposes precludes our ability to obtain any meaningful funding through the ATM Sales Agreement at this time.Until we can generate a sufficient amount of product revenue, which we may never do, we will need to finance future cash needs through potential collaborative, partnering or other strategic arrangements, as well as through public or private equity offerings or debt financings or a combination of the foregoing. If we are unable to raise additional funds, we will need to continue to reduce our expenditures in order to preserve our cash. Further cost-cutting measures that we may take may not be sufficient to enable us to meet our cash requirements, and they may negatively affect our business and our ability to derive any value from our SARD program. In any event, in order to further the development of our SARD program, we will need to raise substantial additional capital. Our failure to do so would likely result in our determining to cease operations.To the extent that we raise additional funds through potential collaborations, partnering or other strategic arrangements, it may be necessary to relinquish rights to some of our technologies or product candidates and intellectual property rights thereof, or grant licenses on terms that are not favorable to us, any of which could result in our stockholders having little or no continuing interest in our SARD program and/or SARM assets as stockholders or otherwise. To the extent we raise additional funds by issuing equity securities, our stockholders may experience significant dilution, particularly given our currently-depressed stock price, and debt financing, if available, may involve restrictive covenants. For example, we completed substantially dilutive private placements of our common stock and warrants in March 2014, November 2014 and September 2017, in addition to a registered direct offering of ourcommon stock that we completed in October 2016 and the sale of our common stock pursuant to the ATM Sales Agreement. Our stockholders will experience additional, perhaps substantial, dilution should we again raise additional funds by issuing equity securities. Any additional debt or equity financing that we raise may contain terms that are not favorable to us or our stockholders. Our ability to raise additional funds and the terms upon which we are able to raise such funds have been severely harmed by the failure of the ASTRID trial to meet its primary endpoint and the resulting significant uncertainty regarding our prospects to continue as a going concern. If we are unable to complete the proposed Merger, our ability to raise additional funds and the terms upon which we are able to raise such funds may also be adversely affected by the uncertainties regarding our financial condition, uncertainties with respect to the prospects for our early-stage SARD program, the sufficiency of our capital resources, potential future management turnover, and volatility and instability in the global financial markets. As a result of these and other factors, there is no guarantee that sufficient additional funding will be available to us on acceptable terms, or at all.We are substantially dependent on our remaining employees to facilitate the consummation of the proposed Merger.We have substantially reduced our workforce since November 2018 and as of March 12, 2018, we had only 13 full-time employees. Our ability to successfully complete the proposed Merger depends in large part on our ability to retain our remaining personnel. Despite our efforts to retain these employees, one or more may terminate their employment with us on short notice. The loss of the services of any of these employees could potentially harm our ability to consummate the proposed Merger, to run our day-to-day business operations, as well as to fulfill our reporting obligations as a public company.The pendency of the proposed Merger could have an adverse effect on the trading price of our common stock and our business, financial condition and prospects.While there have been no significant adverse effects to date, the pendency of the proposed Merger could disrupt our business in many ways, including:•the attention of our remaining management and employees may be directed toward the completion of the proposed Merger and related matters and may be diverted from our day-to-day business operations; and•third parties may seek to terminate or renegotiate their relationships with us as a result of the proposed Merger, whether pursuant to the terms of their existing agreements with us otherwise.
Should they occur, any of these matters could adversely affect the trading price of our common stock or harm our business, financial condition and prospects.Risks Related to Our Development ActivitiesWe were substantially dependent on the success of enobosarm, and the recent failure of the ASTRID trial to meet its primary endpoint has severely diminished enobosarm's prospects and our prospects to continue as a going concern. As we are now focused solely on our SARD program, our failure to obtain funding for and to advance the development of our SARD program would likely require us to cease operations.A substantial portion of our recent efforts and expenditures have been devoted to, and our prospects were substantially dependent upon, the development of enobosarm for the treatment of postmenopausal women with SUI. However, in September 2018, we announced that the ASTRID trial failed to achieve statistical significance on the primary endpoint of the proportion of patients with agreater than 50% reduction in incontinence episodes per day compared to placebo. The failure of the ASTRID trial to achieve its primary endpoint has significantly depressed our stock price and has severely harmed our ability to raise additional capital and to secure potential collaborative, partnering or other strategic arrangements for our SARM assets, and consequently, our prospects to continue as a going concern have been severely diminished. Following our review of the full data sets from the ASTRID trial, we determined to discontinue further development of enobosarm to treat SUI and to otherwise discontinue any further development of our SARM program generally. We continue our efforts to seek potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets. We have for many years actively pursued, but have been unable to successfully enter into, potential collaborative, partnering or other strategic arrangements for our SARM assets. If we are unable to ultimately enter into any such arrangements for our SARM assets, we will not receive any return on our investment in enobosarm and our other SARMs.As a result of our decision to discontinue our SARM development efforts, our development activities are focused solely on completing ongoing mechanistic preclinical studies in order to select the most appropriate SARD compounds to move forward into the additional preclinical studies required to submit an IND and potentially advance one of our SARD compounds into a first-in-human clinical trial. However, while we believe that our existing capital resources will be adequate to enable us to conduct and complete planned IND-enabling preclinical studies of our SARD compounds, we will require significant additional financial resources in order to initiate and complete initial human clinical trials of a SARD compound and to otherwise further the development of our SARD program. In addition, our preclinical evaluation of our SARD technology is at very early stage and is subject to the substantial risk and probability of failure inherent in the development of early-stage programs.In any event, if the proposed Merger is not completed and we are unable to raise sufficient additional funds for the development of our SARD program, whether through potential collaborative, partnering or other strategic arrangements or otherwise, or if we otherwise determine to discontinue the development of our SARD program, we will likely determine to cease operations.We and any potential collaborators will not be able to commercialize any SARD product candidates if our preclinical studies do not produce successful results or if our or their SARD or SARM clinical trials do not adequately demonstrate safety and efficacy in humans.Significant additional clinical development, financial resources and personnel would be required to obtain necessary regulatory approvals for any potential future product candidates and to develop them into commercially viable products. Preclinical and clinical testing is expensive, can take many years to complete and has an uncertain outcome. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and top-line or interim results of a clinical trial do not necessarily predict final results. In this regard, from time to time, we have and may in the future publish or report top-line, interim or other preliminary data from our clinical trials, which data is based on a preliminary analysis of then-available efficacy and safety data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data from the applicable trial. As a result, the top-line results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Similarly, interim or other preliminary data from clinical trials that we may conduct may not be indicative of the final results of the trial and are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Top-line, interim and otherpreliminary data also remain subject to audit and verification procedures that may result in the final data being materially different from such top-line, interim or other preliminary data we previously published. As a result, top-line, interim or preliminary data should be viewed with caution until the final data are available.Typically, the failure rate for development candidates is high. If a product candidate fails at any stage of development, we will not have the anticipated revenues from that product candidate to fund our operations, and we will not receive any return on our investment in that product candidate. For example, in September 2018, we announced that the ASTRID trial failed to achieve statistical significance on the primary endpoint of the proportion of patients with a greater than 50% reduction in incontinence episodes per day compared to placebo. The failure of the ASTRID trial to achieve its primary endpoint has significantly depressed our stock price and has severely harmed our ability to raise additional capital and to secure potential collaborative, partnering or other strategic arrangements for our SARM assets, and consequently, our prospects to continue as a going concern have been severely diminished. Likewise, during the third quarter of 2017, we determined that there were insufficient patients achieving clinical benefit from enobosarm treatment to continue our Phase 2 proof-of-concept clinical trial evaluating enobosarm in patients with advanced AR positive triple-negative breast cancer, or TNBC. Additionally, in the third quarter of 2017, we decided not to pursue additional clinical development of enobosarm to treat women with ER positive, AR positive advanced breast cancer after evaluating the breast cancer environment where the treatment paradigms are shifting to immunotherapies and/or combination therapies, along with the time and cost of conducting the necessary clinical trials for potential approval, even though we announced that our Phase 2 clinical trial of enobosarm in this indication achieved its primary endpoint in both the 9 mg and 18 mg cohorts of the clinical trial. Following our review of the full data sets from the ASTRID trial, we determined to discontinue further development of enobosarm to treat SUI and to otherwise discontinue any further development of our SARM program generally. We continue our efforts to seek potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets. We have for many years actively pursued, but have been unable to successfully enter into, potential collaborative, partnering or other strategic arrangements for our SARM assets. If we are unable to ultimately enter into any such arrangements for our SARM assets, we will not receive any return on our investment in enobosarm and our other SARMs.In the first quarter of 2015, we entered into an exclusive worldwide license agreement with UTRF to develop its proprietary SARD technology and we are currently focused solely on the further development of our SARD program. Our preclinical evaluation of our SARD technology is at an early stage and is subject to the substantial risk and probability of failure inherent in the development of early-stage programs. While we believe that our existing capital resources will be adequate to enable us to conduct and complete planned IND-enabling preclinical studies of our SARD compounds, we will require significant additional financial resources in order to initiate and complete initial human clinical trials of a SARD compound and to otherwise further the development of our SARD program. If our research and preclinical development of our SARD program is unsuccessful, is discontinued and/or we are not able to obtain sufficient funding to advance the development of our SARD program, we will likely cease operations.Significant delays in preclinical and clinical testing could materially impact our product development costs. We do not know whether our planned preclinical and potential future clinical trials will need to be modified or will be completed on schedule, if at all. We or any potential collaborators may experience numerous unforeseen and/or adverse events during, or as a result of, preclinical testingand the clinical trial process that could delay or prevent our or our potential collaborators' ability to commercialize any product candidates, including:•regulators or institutional review boards may not authorize us or any potential collaborators to commence a clinical trial or conduct a clinical trial at a prospective trial site, or we or any potential collaborators may experience substantial delays in obtaining these authorizations;•we or any potential collaborators may be delayed in reaching, or may fail to reach, agreement on acceptable terms with prospective clinical research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;•preclinical or clinical trials may produce negative or inconclusive results, which may require us or any potential collaborators to conduct additional preclinical or clinical testing or to abandon projects that we expect to be promising;•even if preclinical or clinical trial results are positive, the FDA or foreign regulatory authorities could nonetheless require us to conduct unanticipated additional preclinical development or clinical trials;•patient registration or enrollment in clinical trials may be slower than we anticipate resulting in significant delays, additional costs and/or study terminations;•we or any potential collaborators may suspend or terminate clinical trials if the participating patients are being exposed to unacceptable health risks;•regulators or institutional review boards may suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements;•our product candidates may not have the desired effects or may include undesirable side effects; and•changes in regulatory requirements, policies and guidelines.
If any of these events were to occur in the future and, as a result, we or any potential collaborators have significant delays in or termination of potential future clinical trials, our costs could increase and our ability to generate revenue could be impaired, which would materially and adversely impact our business, financial condition and growth prospects.If we or any potential collaborators observe serious or other adverse events during the time any potential future product candidates are in development or after our products are approved and on the market, we or any potential collaborators may be required to perform lengthy additional clinical trials, may be required to cease further development of such product candidates, may be denied regulatory approval of such products, may be forced to change the labeling of such products or may be required to withdraw any such products from the market, any of which would hinder or preclude our ability to generate revenues.In our Phase 2 clinical trials for enobosarm for the treatment of muscle wasting in patients with cancer and healthy older males and postmenopausal females, we observed mild elevations of hepatic enzymes, which in certain circumstances may lead to liver failure, in a few patients in both the placebo and enobosarm treated groups. Reductions in high-density lipoproteins, or HDL, have also been observed in subjects treated with enobosarm. Lower levels of HDL could lead to increased risk ofadverse cardiovascular events. Mild transient elevations in liver enzymes that were within normal limits were observed in our Phase 2 proof-of-concept clinical trial of enobosarm to treat postmenopausal women with SUI, except for one patient with levels greater than 1.5 times the upper limit of normal which returned to normal following her 12-week treatment period. Reductions in total cholesterol, low-density lipoproteins, or LDL, HDL and triglycerides were also observed. Results of the placebo-controlled ASTRID study in postmenopausal women with SUI indicated that enobosarm was generally safe and well tolerated, and reported adverse events were generally mild to moderate in intensity and similar across all treatment groups. Mild transient elevations in hepatic enzymes and changes in lipid profile were dose dependent, and consistent with results seen in previous trials. In addition, in our Phase 2 proof-of-concept clinical trial evaluating enobosarm in a 9 mg daily dose for the treatment of patients with ER positive and AR positive metastatic breast cancer, bone pain of the chest cage, a serious adverse event, or SAE, was assessed as possibly related to enobosarm. Although doses up to 30 mg have been evaluated in short duration studies, the 3 mg dose that was the subject of the ASTRID trial and higher enobosarm doses that may potentially be tested by potential future collaborators in later stage longer duration trials, if any, may increase the risk or incidence of known potential side effects of SARMs, including elevations in hepatic enzymes and further reductions in HDL, in addition to the emergence of side effects that have not been seen to date.If the incidence of serious or other adverse events related to enobosarm or any other SARD or SARM product candidates increases in number or severity, if a regulatory authority believes that these or other events constitute an adverse effect caused by the drug, or if other effects are identified during clinical trials that we or any potential collaborators may conduct in the future or after any potential future product candidates are approved and marketed:•we or any potential collaborators may be required to conduct additional preclinical or clinical trials, make changes in the labeling of any such approved products, reformulate any such products, or implement changes to or obtain new approvals of our contractors' manufacturing facilities;•regulatory authorities may be unwilling to approve our product candidates or may withdraw approval of our products;•we may experience a significant drop in the sales of the affected products;•our reputation in the marketplace may suffer; and•we may become the target of lawsuits, including class action suits.
Any of these events could prevent approval or harm adoption and sales of the affected product candidates or products, or could substantially increase the costs and expenses of commercializing and marketing any such products.Risks Related to Our Dependence on Third PartiesIf the proposed Merger is not completed and we do not establish collaborative, partnering or other strategic arrangements for our SARD program and SARM assets or otherwise raise substantial additional capital, we will likely determine to cease operations.Our current strategy is dependent on our ability to secure potential collaborative, partnering or other strategic arrangements with other pharmaceutical and biotechnology companies to assist us in furthering development and potential commercialization of any SARD and SARM product candidates,and to otherwise obtain funding for such activities. For example, we are currently focused solely on the further development of our SARD program and while we believe that our existing capital resources will be adequate to enable us to conduct and complete planned IND-enabling preclinical studies of SARD compounds, we will require significant additional financial resources in order to initiate and complete initial human clinical trials and to otherwise further the development of our SARD program. Accordingly, if, for any reason, the proposed Merger is not consummated, we may resume our efforts to seek additional funds through potential collaborative, partnering or other strategic arrangements to provide us with the necessary resources for the development of our SARD program. We face significant competition in seeking such arrangements, and such arrangements are complex and time consuming to negotiate and document. In any event, we may not be successful in entering into new collaborative, partnering or other strategic arrangements with third parties for the further development of our SARD program (or our SARD assets) on acceptable terms, or at all. In this regard, we have for many years actively pursued, but have been unable to successfully enter into, potential collaborative, partnering or other strategic arrangements for our SARM assets and we likewise have not been successful to date in entering into potential collaborative, partnering or other strategic arrangements for our SARD program. In addition, we are unable to predict when, if ever, we will enter into any potential collaborative, partnering or other such strategic arrangements because of the numerous risks and uncertainties associated with establishing such arrangements, and we have otherwise been unsuccessful, for many years, in our efforts to establish such arrangements. In any event, if the proposed Merger is not completed and we are unable to raise sufficient additional funds for the development of our SARD program, whether through potential collaborative, partnering or other strategic arrangements or otherwise, or if we otherwise determine to discontinue the development of our SARD program, we will likely determine to cease operations. In addition, because we have discontinued our SARM development efforts, if we are unable to ultimately enter into any potential collaborative, partnering or other such strategic arrangements for our SARM assets, we will not receive any return on our investment in enobosarm and our other SARMs.Any collaborative arrangements that we establish in the future may not be successful or we may otherwise not realize the anticipated benefits from these collaborations. In addition, any future collaborative arrangements may place the development and commercialization of our product candidates outside our control, may require us to relinquish important rights or may otherwise be on terms unfavorable to us.We have in the past established, and, if the proposed Merger is not completed, we intend to continue to seek to establish, partnering, collaborative and similar strategic arrangements with third parties to develop and commercialize any potential future product candidates, and these collaborations may not be successful or we may otherwise not realize the anticipated benefits from these collaborations. For example, in March 2011, we and Ipsen Biopharm Limited, or Ipsen, mutually agreed to terminate our collaboration for the development and commercialization of our toremifene-based product candidate. As of the date of this report, we have no ongoing collaborations for the development and commercialization of any product candidate. We may not be able to locate third-party collaborators to develop and market any product candidates, and we lack the necessary financial resources to develop any product candidates alone.Dependence on collaborative arrangements subjects us to a number of risks, including:•we may not be able to control the amount and timing of resources that our potential collaborators may devote to our product candidates;•potential collaborations may experience financial difficulties or changes in business focus;•we may be required to relinquish important rights such as marketing and distribution rights;
•should a collaborator fail to develop or commercialize one of our compounds or product candidates, we may not receive any future milestone payments and will not receive any royalties for the compound or product candidate;•business combinations or significant changes in a collaborator's business strategy may also adversely affect a collaborator's willingness or ability to complete its obligations under any arrangement;•under certain circumstances, a collaborator could move forward with a competing product candidate developed either independently or in collaboration with others, including our competitors; and•collaborative arrangements are often terminated or allowed to expire, which could delay the development and may increase the cost of developing our product candidates.
If third parties do not manufacture our clinical and commercial drug supplies in sufficient quantities, in the required timeframe, at an acceptable cost, and with appropriate quality control, clinical development and commercialization of any potential future product candidates would be delayed.We do not currently own or operate manufacturing facilities, and we rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of any product candidates. Our current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our future profit margins, if any, and our ability to develop product candidates and commercialize any product candidates on a timely and competitive basis.We rely and expect to continue to rely on third-party vendors for drug substance and drug product manufacturing, including drug substance for SARDs used in our current and potential future preclinical studies. If the contract manufacturers that we are currently utilizing to meet our supply needs for SARD compounds or any potential future SARD product candidates prove incapable or unwilling to continue to meet our supply needs, we could experience a delay in conducting any additional preclinical or clinical trials of SARD compounds or any potential future SARD product candidates. We may not be able to maintain or renew our existing or any other third-party manufacturing arrangements on acceptable terms, if at all. If our suppliers fail to meet our requirements for our product candidates for any reason, we would be required to obtain alternate suppliers. Any inability to obtain alternate suppliers, including an inability to obtain approval from the FDA of an alternate supplier, would delay or prevent the clinical development and commercialization of any potential future product candidates.Use of third-party manufacturers may increase the risk that we will not have adequate drug supplies for preclinical, clinical and commercial use.Reliance on third-party manufacturers entails risks, to which we would not be subject if we manufactured our product candidates ourselves, including:•reliance on the third party for regulatory compliance and quality assurance;•the possible breach of the manufacturing agreement by the third party because of factors beyond our control;•the possible termination or non-renewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us; and•drug product supplies not meeting the requisite requirements for clinical trial use.
If we are not able to obtain adequate drug supplies, including SARD compounds, it will be more difficult for us to develop any product candidates and compete effectively. Our potential future product candidates and any products that we and/or our potential collaborators may develop may compete with other product candidates and products for access to manufacturing facilities.Our present or future manufacturing partners may not be able to comply with FDA-mandated current Good Manufacturing Practice regulations, other FDA regulatory requirements or similar regulatory requirements outside the United States. Failure of our third-party manufacturers or us to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our product candidates.If third parties on whom we rely do not perform as contractually required or expected, we may not be able to obtain regulatory approval for or successfully commercialize any potential future product candidates.We do not have the ability to independently conduct clinical trials for our product candidates, and we must rely on third parties, such as CROs, medical institutions, clinical investigators and contract laboratories to conduct our clinical trials. In addition, we rely on third parties to assist with our preclinical development of product candidates. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if the third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize any potential future product candidates.Risks Related to Our Intellectual PropertyIf we lose our licenses from UTRF, we may be unable to continue our business.We have licensed intellectual property rights and technology from UTRF used in substantially all of our business. Our license agreements with UTRF, under which we were granted rights to enobosarm and other SARM compounds, and to SARD compounds and, for both, to methods of use thereof, may be terminated by UTRF if we are in breach of our obligations under, or fail to perform any terms of, the relevant agreement and fail to cure that breach. If one or both of these agreements are terminated, then we may lose our rights to utilize enobosarm and other SARM compounds and/or SARD compounds and the intellectual property covered by those agreements to market, distribute and sell licensed products, which may prevent us from continuing our business and would likely cause us to cease operations altogether.If some or all of our or our licensor's patents expire or are invalidated or are found to be unenforceable, or if some or all of our patent applications do not result in issued patents or result in patents with narrow, overbroad, or unenforceable claims, or claims that are not supported in regard to written description or enablement by the specification, or if we are prevented from asserting that the claims of an issued patent cover a product of a third party, we may be subject to competition from third parties with products in the same class of products as our product candidates or products with the same active pharmaceutical ingredients as our product candidates, including in those jurisdictions in which we have no patent protection.Our commercial success, if any, will depend in part on obtaining and maintaining patent and trade secret protection for any product candidates that we may develop, as well as the methods for treatingpatients in the product indications using these product candidates. We will be able to protect any potential future product candidates and the methods for treating patients in the product indications using these product candidates from unauthorized use by third parties only to the extent that we or our exclusive licensor owns or controls such valid and enforceable patents or trade secrets.Even if any potential future product candidates and/or the methods for treating patients for prescribed indications using these product candidates are covered by valid and enforceable patents and have claims with sufficient scope, disclosure and support in the specification, the patents will provide protection only for a limited amount of time. Our and our licensor's ability to obtain patents can be highly uncertain and involve complex and in some cases unsettled legal issues and factual questions. Furthermore, different countries have different procedures for obtaining patents, and patents issued in different countries provide different degrees of protection against the use of a patented invention by others. Therefore, if the issuance to us or our licensor, in a given country, of a patent covering an invention is not followed by the issuance, in other countries, of patents covering the same invention, or if any judicial interpretation of the validity, enforceability, or scope of the claims in, or the written description or enablement in, a patent issued in one country is not similar to the interpretation given to the corresponding patent issued in another country, our ability to protect our intellectual property in those countries may be limited. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may materially diminish the value of our intellectual property or narrow the scope of our patent protection.We may be subject to competition from third parties with products in the same class of products as our product candidates or products with the same active pharmaceutical ingredients as our product candidates in those jurisdictions in which we have no patent protection. Even if patents are issued to us or our licensor regarding our product candidates or methods of using them, those patents can be challenged by our competitors who can argue such patents are invalid or unenforceable, lack of utility, lack sufficient written description or enablement, or that the claims of the issued patents should be limited or narrowly construed. Patents also will not protect our product candidates if competitors devise ways of making or using these product candidates without legally infringing our patents. The Federal Food, Drug, and Cosmetic Act and FDA regulations and policies create a regulatory environment that encourages companies to challenge branded drug patents or to create non-infringing versions of a patented product in order to facilitate the approval of abbreviated new drug applications for generic substitutes. These same types of incentives encourage competitors to submit new drug applications that rely on literature and clinical data not prepared for or by the drug sponsor, providing another less burdensome pathway to approval.We also rely on trade secrets to protect our technology, especially where we do not believe that patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our confidential information to competitors, and confidentiality agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Enforcing a claim that a third party illegally obtained and is using our trade secrets is expensive and time-consuming, and the outcome is unpredictable. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how. Failure to obtain or maintain trade secret protection could adversely affect our competitive business position.If we infringe intellectual property rights of third parties, it may increase our costs or prevent us from being able to commercialize our product candidates.There is a risk that we are infringing the proprietary rights of third parties because numerous United States and foreign issued patents and pending patent applications, which are owned by thirdparties, exist in the fields that are the focus of our development and manufacturing efforts. Others might have been the first to make the inventions covered by each of our or our licensor's pending patent applications and issued patents and/or might have been the first to file patent applications for these inventions. In addition, because patent applications take many months to publish and patent applications can take many years to issue, there may be currently pending applications, unknown to us or our licensor, which may later result in issued patents that cover the production, manufacture, synthesis, commercialization, formulation or use of our product candidates. In addition, the production, manufacture, synthesis, commercialization, formulation or use of our product candidates may infringe existing patents of which we are not aware. Defending ourselves against third-party claims, including litigation in particular, would be costly and time consuming and would divert management's attention from our business, which could lead to delays in our development or commercialization efforts. If third parties are successful in their claims, we might have to pay substantial damages or take other actions that are adverse to our business.As a result of intellectual property infringement claims, or to avoid potential claims, we might:•be prohibited from selling or licensing any product that we and/or any potential collaborators may develop unless the patent holder licenses the patent to us, which the patent holder is not required to do;•be required to pay substantial royalties or other amounts, or grant a cross license to our patents to another patent holder; or•be required to redesign the formulation of a product candidate so that it does not infringe, which may not be possible or could require substantial funds and time.
Risks Related to Regulatory ApprovalIf we or any potential collaborators are not able to obtain required regulatory approvals, we or such collaborators will not be able to commercialize our product candidates, and our ability to generate revenue will be materially impaired.The activities associated with the development and commercialization of drug candidates are subject to comprehensive regulation by the FDA, other regulatory agencies in the United States and by comparable authorities in other countries, including the European Medicines Agency, or EMA. Failure to obtain regulatory approval for a product candidate will prevent us or any potential collaborator from commercializing the product candidate. We have not received regulatory approval to market any product candidate in any jurisdiction, and we do not expect to obtain FDA, EMA or any other regulatory approvals to market any potential future product candidates for the foreseeable future, if at all. The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon the type, complexity and novelty of the product candidates involved.Changes in the regulatory approval policy during the development period, changes in or the enactment of additional regulations or statutes, or changes in regulatory review for each submitted product application may cause delays in the approval or rejection of an application. Even if the FDA or the EMA approves a product candidate, the approval may impose significant restrictions on the indicated uses, conditions for use, labeling, advertising, promotion, marketing and/or production of such product, and may impose ongoing requirements for post-approval studies, including additional research and development and clinical trials. Any FDA approval may also impose Risk Evaluation Mitigation Strategy, or REMS, on a product if the FDA believes there is a reason to monitor the safety of thedrug in the market place. REMS may include requirements for additional training for health care professionals, safety communication efforts and limits on channels of distribution, among other things. The sponsor would be required to evaluate and monitor the various REMS activities and adjust them if need be. The FDA and EMA also may impose various civil or criminal sanctions for failure to comply with regulatory requirements, including withdrawal of product approval.Furthermore, the approval procedure and the time required to obtain approval varies among countries and can involve additional testing beyond that required by the FDA. Approval by one regulatory authority does not ensure approval by regulatory authorities in other jurisdictions. Failure to obtain approval in one jurisdiction may negatively impact our ability to obtain approval elsewhere.The FDA, the EMA and other foreign regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional preclinical, clinical or other studies, including Phase 4 clinical studies. For example, in October 2009, we received a Complete Response Letter from the FDA regarding our new drug application, or NDA, for toremifene 80 mg to reduce fractures in men with prostate cancer on androgen deprivation therapy notifying us that the FDA would not approve our NDA as a result of certain clinical deficiencies identified in the Complete Response Letter. We have since discontinued our toremifene 80 mg development program, as well as other toremifene-based products. Although we evaluated the potential submission of a marketing authorization application, or MAA, to the EMA seeking marketing approval of enobosarm 3 mg in the European Union, or EU, for the prevention and treatment of muscle wasting in patients with advanced NSCLC, based on input from the Medicines and Healthcare Products Regulatory Agency, or MHRA, we determined that the data from the POWER trials was not sufficient to support the filing and approval of a MAA without confirmatory data from another Phase 3 clinical trial of enobosarm 3 mg. As a result of this input, we elected not to submit a MAA in the absence of such confirmatory data. In addition, since data from the two POWER trials failed to meet the primary statistical criterion pre-specified for the co-primary endpoints of lean body mass and physical function, we were unable to file with the FDA a NDA for enobosarm 3 mg for the prevention and treatment of muscle wasting in patients with advanced NSCLC.In addition, varying interpretations of the data obtained from preclinical and clinical testing could delay, limit, or prevent regulatory approval of a product candidate. Even if we submit an application to the FDA, the EMA and other foreign regulatory authorities for marketing approval of a product candidate, it may not result in any marketing approvals.We do not expect to receive regulatory approval for the commercial sale of any product candidates for the foreseeable future, if at all. The inability to obtain approval from the FDA, the EMA and other foreign regulatory authorities for our product candidates would prevent us or any potential collaborators from commercializing these product candidates in the United States, the EU, or other countries. See the section entitled "Business — Government Regulation" under Part 1, Item 1 of this Annual Report on Form 10-K for additional information regarding risks associated with marketing approval, as well as risks related to potential post-approval requirements.Risks Related to CommercializationThe commercial success of any products that we and/or any potential collaborators may develop and for which we may obtain regulatory approval will depend upon the market and the degree of market acceptance among physicians, patients, health care payors and the medical community.Any products that we and/or any potential collaborators may develop may not gain market acceptance for its stated indication among physicians, patients, health care payors and the medicalcommunity despite regulatory approval. If these products do not achieve an adequate level of acceptance, we may not generate material product revenues or receive royalties to the extent we currently anticipate, and we may not become profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, including:•efficacy and safety results in clinical trials;•the prevalence and severity of any side effects;•potential advantages over alternative treatments;•whether the products we commercialize become and/or remain a preferred course of treatment;•the ability to offer our product candidates for sale at competitive prices;•relative convenience and ease of administration compared to alternative treatment;•the strength of marketing and distribution support; and•sufficient third-party coverage or reimbursement.
If we are unable to establish sales and marketing capabilities or establish and maintain agreements with third parties to market and sell our product candidates, we may be unable to generate product revenue from such candidates.We have limited experience as a company in the sales, marketing and distribution of pharmaceutical products. In the event one of our potential future product candidates is approved, we will need to establish sales and marketing capabilities or establish and maintain agreements with third parties to market and sell any such product candidates. Either of these options would be expensive and time-consuming. We may be unable to build our own sales and marketing capabilities, and there are risks involved with entering into arrangements with third parties to perform these services, which could delay the commercialization of any of our product candidates if approved for commercial sale. In addition, to the extent that we enter into arrangements with third parties to perform sales, marketing and distribution services, our product revenues are likely to be lower than if we market and sell any products that we develop ourselves.If we and/or any potential collaborators are unable to obtain reimbursement or experience a reduction in reimbursement from third-party payors for products we sell, our revenues and prospects for profitability will suffer.Sales of products developed by us and/or any potential collaborators are dependent on the availability and extent of reimbursement from third-party payors, both governmental and private. Changes in the coverage and/or reimbursement policies of these third-party payors that reduce reimbursements for any products that we and/or any potential collaborators may develop and sell could negatively impact our future operating and financial results.Medicare coverage and reimbursement of prescription drugs exists under Medicare Part D for oral drug products capable of self-administration by patients. Our oral drug product candidates would likely be covered by Medicare Part D (if covered by Medicare at all). In March 2010, the United States Congress enacted the Healthcare Reform Act, which, among other initiatives, implemented cost containment and other measures that could adversely affect revenues from sales of product candidates, including an increase in the drug rebates that manufacturers must pay under Medicaid for brand nameprescription drugs and extension of these rebates to Medicaid managed care and a requirement that manufacturers provide a 50% discount on the negotiated price of Medicare Part D brand name drugs utilized by Medicare Part D beneficiaries during the coverage gap (the so-called "donut hole")(which discount has subsequently been increased to 70% in 2019).The provisions of the Healthcare Reform Act have been subject to judicial and Congressional challenges, as well as efforts by the Trump administration to modify certain requirements of the Healthcare Reform Act by executive branch order. For example, on January 20, 2017, President Trump signed an Executive Order directing federal agencies with authorities and responsibilities under the Healthcare Reform Act to waive, defer, grant exemptions from, or delay the implementation of any provision of the Healthcare Reform Act that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. On October 12, 2017, President Trump signed another Executive Order directing certain federal agencies to propose regulations or guidelines to provide small businesses with greater opportunities to form association health plans, expand the availability of short-term, limited duration insurance, and allow employees to make use of certain employer-paid health benefits, called health reimbursement arrangements, to pay for health insurance that does not meet all Healthcare Reform Act requirements. In addition, citing legal guidance from the U.S. Department of Justice, the U.S. Department of Health and Human Services, or HHS, concluded that cost-sharing reduction, or CSR, payments to insurance companies required under the Healthcare Reform Act had not received necessary appropriations from Congress. President Trump subsequently discontinued these payments. The loss of the CSR payments is expected to increase premiums on certain policies issued by qualified health plans under the Healthcare Reform Act. Certain administrative actions have been subject to judicial challenge. In Congress, there have been a number of legislative initiatives to modify, repeal and/or replace portions of the Healthcare Reform Act. Tax reform legislation enacted at the end of 2017 eliminated the tax penalty for individuals who do not maintain sufficient health insurance coverage beginning in 2019. The Bipartisan Budget Act of 2018 contained various provisions that affect coverage and reimbursement of drugs, including an increase in the discount that manufacturers of Medicare Part D brand name drugs must provide to Medicare Part D beneficiaries during the coverage gap from 50% to 70% starting in 2019. Congress may consider other legislation to modify, repeal and/or replace certain elements of the Healthcare Reform Act. In December 2018, a federal district court judge, in a challenge brought by a number of state attorneys general, found the Healthcare Reform Act unconstitutional in its entirety because, once Congress repealed the individual mandate provision, there was no longer a basis to rely on Congressional taxing authority to support enactment of the law. Pending appeals, which could take some time, the Healthcare Reform Act is still operational in all respects. We continue to evaluate the effect that the Healthcare Reform Act and its possible repeal, replacement or modification may have on our business. Such legislation and other healthcare reform measures that may be adopted in the future could have a material adverse effect on our industry generally and on our ability to successfully commercialize our product candidates, if approved.Economic pressure on state budgets may result in states increasingly seeking to achieve budget savings through mechanisms that limit coverage or payment for drugs. State Medicaid programs are increasingly requesting manufacturers to pay supplemental rebates and requiring prior authorization for use of drugs where supplemental rebates are not provided. Private health insurers and managed care plans are likely to continue challenging the prices charged for medical products and services, and many of these third-party payors may limit reimbursement for newly-approved health care products. In particular, third-party payors may limit the indications for which they will reimburse patients who use any products that we and/or any potential collaborators may develop or sell. These cost-control initiatives could decrease the price we might establish for products that we or any potential collaborators may develop or sell, which would result in lower product revenues or royalties payable to us.Similar cost containment initiatives exist in countries outside of the United States, particularly in the countries of the EU, where the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can extend well beyond the receipt of regulatory marketing approval for a product and may require us or any potential collaborators to conduct a clinical trial that compares the cost effectiveness of our product candidates or products to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in our or a potential collaborators' commercialization efforts. Third-party payors are challenging the prices charged for medical products and services, and many third-party payors limit reimbursement for newly-approved health care products. Recently budgetary pressures in many EU countries are also causing governments to consider or implement various cost-containment measures, such as price freezes, increased price cuts and rebates. If budget pressures continue, governments may implement additional cost containment measures. Cost-control initiatives could decrease the price we might establish for products that we or any potential collaborators may develop or sell, which would result in lower product revenues or royalties payable to us.Another development that could affect the pricing of drugs would be if the Secretary of HHS allowed drug reimportation into the United States. The Medicare Prescription Drug, Improvement and Modernization Act of 2003 gives discretion to the Secretary of Health and Human Services to allow drug reimportation into the United States under some circumstances from foreign countries, including from countries where the drugs are sold at a lower price than in the United States. If the circumstances were met and the Secretary exercised the discretion to allow for the direct reimportation of drugs, it could decrease the price we or any potential collaborators receive for any products that we and/or any potential collaborators may develop, negatively affecting our revenues and prospects for profitability.Health care reform measures could hinder or prevent our product candidates' commercial success.Among policy makers and payors in the United States and elsewhere, there is significant interest in health care reform, as evidenced by the initial enactment of, as well as the efforts to repeal, replace and/or modify the Healthcare Reform Act in the United States. Federal and state legislatures within the United States and foreign governments will likely continue to consider other changes to existing health care legislation. These changes adopted by governments may adversely impact our business by lowering the price of health care products in the United States and elsewhere. For example, there has been increasing administrative, legislative and enforcement interest in the United States with respect to drug pricing practices. There have been several U.S. Congressional inquiries and legislative and administrative initiatives at the federal and state levels intended to, among other things, bring more transparency to drug pricing and modify government program reimbursement for drugs. We cannot predict what health care reform initiatives may be adopted in the future. Further federal, state and foreign legislative and regulatory developments are likely, and we expect ongoing initiatives to increase pressure on drug pricing, which could decrease the price we might establish for products that we or any potential collaborators may develop or sell, which would result in lower product revenues or royalties payable to us.We operate in a highly regulated industry and new laws, regulations or judicial decisions, or new interpretations of existing laws, regulations or decisions, related to health care availability, method of delivery or payment for health care products and services, or sales, marketing and pricing practices could negatively impact our business, operations and financial condition.If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of any products that we may develop.We face an inherent risk of product liability exposure related to our prior commercial sales of FARESTON® and the testing of our product candidates in human clinical trials, and we will face aneven greater risk if we commercially sell any product that we may develop. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:•decreased demand for any product candidates or products;•injury to our reputation;•withdrawal of clinical trial participants;•costs to defend the related litigation;•substantial monetary awards to trial participants or patients;•loss of revenue; and•the inability to commercialize any products for which we obtain or hold marketing approvals.
We have product liability insurance that covers our clinical trials and any commercial products up to a $25 million annual aggregate limit. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost, and we may not be able to obtain insurance coverage that will be adequate to satisfy any liability that may arise.If our competitors are better able to develop and market products than any products that we and/or any potential collaborators may develop, our commercial opportunity will be reduced or eliminated.We face competition from commercial pharmaceutical and biotechnology enterprises, as well as from academic institutions, government agencies and private and public research institutions. Our commercial opportunities will be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than any products that we and/or any potential collaborators may develop. Competition could result in reduced sales and pricing pressure on our product candidates, if approved, which in turn would reduce our ability to generate meaningful revenue and have a negative impact on our results of operations. In addition, significant delays in the development of our product candidates could allow our competitors to bring products to market before us and impair any ability to commercialize any potential future product candidates.Various products are currently marketed or used off-label for some of the diseases and conditions that we are targeting in our pipeline, and a number of companies are or may be developing new treatments. These product uses, as well as promotional efforts by competitors and/or clinical trial results of competitive products, could significantly diminish any ability to market and sell any products that we and/or any potential collaborators may develop.We believe SARDs have the potential to provide compounds that can degrade or antagonize multiple forms of the AR thereby inhibiting tumor growth in patients with CRPC, including those patients who do not respond or are resistant to current therapies. Drugs in development having potentially similar approaches to removing the AR by degradation include Arvinas Inc.'s ARV-110, which is a chimera with an AR binding moiety on one end and an E3 ligase recruiting element on the other that has recently entered Phase 1 development for the treatment of advanced prostate cancer, and Androscience Corporation's androgen receptor degrader enhancer, ASC-J9, which is currently in development for acne and alopecia with the potential for development as a treatment for prostatecancer. Additionally, Essa Pharma Inc. recently completed a Phase 1 study with EPI-506, an AR antagonist that targets the N-terminal domain of the AR, and has plans to develop a second generation agent. C4 Therapeutics, Inc. is developing degronimids as means to degrade the AR through the ligand binding domain associated degradation. CellCentric is developing therapies that target the histone methyltransferase enzyme to lower AR levels, and recently initiated a clinical trial with CCS1477 in prostate cancer. Oric Pharmaceuticals is targeting the glucocorticoid receptor as a means to impact men that have CRPC, and has a lead candidate ORIC-101 in preclinical testing. In addition to this specific potential mechanistic competition, there are various products approved or under clinical development in the broader space of treating men with advanced prostate cancer who have metastatic CRPC which may compete with our proposed initial clinical objective for our SARD compounds. Pfizer and Astellas Pharma market XTANDI® (enzalutamide), an oral androgen receptor antagonist, for the treatment of metastatic CRPC in men previously treated with docetaxel as well as those that have not yet received chemotherapy. XTANDI® received FDA approval in July 2018 for the treatment of men with non-metastatic CRPC. Zytiga®, sold by Johnson & Johnson, has been approved for the treatment of metastatic CRPC and metastatic high-risk castration-sensitive prostate cancer. Johnson & Johnson also received FDA approval for a second generation anti-androgen ERLEADA (apalutamide) for the treatment of men with non-metastatic castrate-resistant prostate cancer. Bayer HealthCare and Orion Corporation recently announced that the primary endpoint of increased metastatic free survival was met in a Phase 3 study of darolutamide (ODM-201) in men with CRPC without metastases and with a rising PSA. Another target in prostate cancer that is being pursued by several companies is bromodomain inhibition. Zenith Epigenetics, Gilead Sciences Inc., CellCentric, Incyte Corporation and GlaxoSmithKline are among the companies that are evaluating BET inhibitors in Phase 1-2 trials.With respect to SARMs, there are other SARM product candidates in development that may compete with enobosarm and any future SARM product candidates, if approved for commercial sale. For example, Viking Therapeutic's VK5211 recently reported positive results from a Phase 2 study for patients recovering from non-elective hip fracture surgery. Radius Health Inc.'s RAD140 is currently being evaluated in a Phase 1 study in postmenopausal women with hormone-receptor positive locally advanced or metastatic breast cancer. GlaxoSmithKline is conducting a Phase 1 study to assess the effect of GSK2881078 on physical strength and function after 13 weeks of treatment in patients with chronic obstructive pulmonary disease, or COPD, and muscle weakness. OPKO Health's OPK88004 is enrolling in a dose ranging study to improve symptoms of benign prostatic hyperplasia (BPH) by reducing prostate size and, on the basis of data from a previous trial in 350 men, increase muscle mass and bone strength and decrease body fat.Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business.Risks Related to Employees, Growth and Other Aspects of Our OperationsOur internal computer and information technology systems, or those of our CROs or other contractors or consultants, may fail or suffer security breaches, or could otherwise face serious disruptions, which could result in a material disruption of our product development efforts and could result in significant financial, legal, regulatory, business and reputational harm to us.Despite the implementation of security measures, our internal computer and information technology systems and those of our CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, and telecommunication and electrical failures. Such events could cause interruptions of our operations. For instance, the loss of preclinical data or data from potential future clinical trials involving our product candidates, if any, could result in delays in our development and regulatory filing efforts and significantly increase our costs. In addition, while all information technology operations are inherently vulnerable to inadvertent or intentional security breaches, incidents, attacks and exposures, the size, complexity, accessibility and distributed nature of our information technology systems, and the large amounts of sensitive information stored on those systems, make such systems potentially vulnerable to unintentional or malicious, internal and external attacks on our technology environment. Potential vulnerabilities can be exploited from inadvertent or intentional actions of our employees, third-party vendors, business partners, or by malicious third parties. Attacks of this nature are increasing in their frequency, levels of persistence, sophistication and intensity, and are being conducted by sophisticated and organized groups and individuals with a wide range of motives (including, but not limited to, industrial espionage) and expertise, including organized criminal groups, "hacktivists," nation states and others. To the extent that any disruption or security breach or incident were to result in a loss of, or damage to, our data, or inappropriate disclosure of confidential, proprietary or protected health information, we could be subject to significant legal, financial and regulatory exposure and suffer reputational harm, and the development of our product candidates could be delayed. In addition, security breaches and other inappropriate access events can be difficult to detect, and any delay in identifying them may lead to increased harm of the type described above. Moreover, the prevalent use of mobile devices to access confidential information increases the risk of security breaches. While we have implemented security measures to protect our information technology systems and infrastructure, there can be no assurance that such measures will prevent service interruptions or security breaches that could adversely affect our business. In addition, our information technology and other internal infrastructure systems, including corporate firewalls, servers, leased lines and connection to the Internet, face the risk of systemic failure that could disrupt our operations. A significant disruption in the availability of our information technology and other internal infrastructure systems could cause delays in our research and development work and could otherwise adversely affect our business. In addition, failure to maintain effective internal accounting controls related to security breaches and cybersecurity in general could impact our ability to produce timely and accurate financial statements and subject us to regulatory scrutiny.If we fail to attract and keep senior management and key scientific personnel, we may be unable to continue our business operations.Our success depends on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel and on our ability to develop and maintain important relationships with leading academic institutions, clinicians and scientists. Significant competition exists for qualified personnel in the biotechnology field, particularly clinical development personnel. We may incur greater costs than anticipated, or may not be successful, in attracting new scientists or management or in retaining or motivating our existing personnel. If we are not able to attract and keep senior management and key scientific personnel, our ability to progress the development of any product candidates and any future growth could be impaired, and our business and the value of your investmentwould be adversely impacted. All of our employees are at-will employees and can terminate their employment at any time.To conserve our cash resources, we have substantially reduced our workforce since November 2018 and have ceased our SARM development activities and all other operations except for day-to-day business operations, completing ongoing mechanistic SARD preclinical studies and those activities necessary to complete the proposed Merger. As of March 12, 2018, we had only 13 full-time employees. Accordingly, we have been and are continuing operating with a shortage of resources and may not be able to effectively conduct our operations with this limited number of employees. In addition, our ability to successfully complete the proposed Merger depends in large part on our ability to retain our remaining personnel. Despite our efforts to retain these employees, one or more may terminate their employment with us on short notice. The loss of the services of any of these employees could potentially harm our ability to consummate the Merger, to run our day-to-day business operations, as well as to fulfill our reporting obligations as a public company.If the proposed Merger is not completed and we are able to raise sufficient additional funds necessary to pursue the continued development of our SARD program, we will need to hire a substantial number of additional employees. Any inability to manage future growth could harm our ability to develop and commercialize any potential future product candidates, increase our costs and adversely impact our ability to compete effectively.As of March 12, 2018, we had only 13 full-time employees. If the proposed Merger is not completed and we are able to raise sufficient additional funds necessary to pursue the continued development of our SARD program, we will need to hire experienced personnel to continue to develop our SARD program and to develop and commercialize any potential future product candidates, and we will need to expand the number of our managerial, operational, financial and other employees to support that growth. Significant competition exists for qualified personnel in the biotechnology field, particularly clinical development personnel.Future growth, if any, will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain and integrate additional employees. Our future financial performance and our ability to develop and commercialize any potential future product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively.Management transition creates uncertainties and could harm our business.We have in the past, and may again in the future, experience significant changes in executive leadership. Changes to company strategy, which can often times occur with the appointment of new executives, can create uncertainty, may negatively impact our ability to execute quickly and effectively, and may ultimately be unsuccessful. In addition, executive leadership transition periods are often difficult as the new executives gain detailed knowledge of our operations, and friction can result from changes in strategy and management style. Management transition inherently causes some loss of institutional knowledge, which can negatively affect strategy and execution. Until we integrate new personnel, and unless they are able to succeed in their positions, we may be unable to successfully manage and grow our business, and our results of operations and financial condition could suffer as a result. In any event, changes in our organization as a result of executive management transition may have a disruptive impact on our ability to implement our strategy and could have a material adverse effect on our business, financial condition and results of operations.Risks Related to Our Common StockThe market price of our common stock has been volatile and may continue to be volatile in the future. This volatility may cause our stock price and the value of your investment to decline.The market prices for securities of biotechnology companies, including ours, have been highly volatile and may continue to be so in the future. In this regard, the market price for our common stock has varied between a high of $25.60 on September 13, 2018, and a low of $0.74 on December 24, 2018, in the twelve-month period ended December 31, 2018. The market price of our common stock is likely to continue to be volatile and subject to significant price and volume fluctuations. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock:•our ability to consummate the transactions contemplated by the Merger Agreement, including the proposed Merger;•our ability to execute on our SARD development program, including our ability to conduct and complete IND-enabling studies and potentially advance one of our SARD compounds into a first-in-human clinical trial;•our ability to raise sufficient additional funds necessary for the continued development of our SARD program, whether through potential collaborative, partnering or other strategic arrangements or otherwise;•our ability to realize any value from our SARM assets, particularly in light of our decision to discontinue the development of enobosarm and our SARM program generally;•the terms and timing of any future collaborative, licensing or other strategic arrangements that we may establish;•uncertainties created by our potential future management turnover;•our inability to comply with the minimum listing requirements of The Nasdaq Stock Market LLC;•the timing of achievement of, or failure to achieve, our and any potential collaborators' clinical, regulatory and other milestones, such as the commencement of clinical development, the completion of a clinical trial or the receipt of regulatory approval;•reports of unacceptable incidences of adverse events observed in any future clinical trials of any product candidates that we and/or any potential collaborators may develop;•announcement of FDA approval or non-approval of any potential future product candidates or delays in or adverse events during the FDA review process;•actions taken by regulatory agencies with respect to any potential future product candidates or our potential future clinical trials, if any, including regulatory actions requiring or leading to a delay or stoppage of any clinical trials;•introductions or announcements of technological innovations or new products by us, our potential collaborators, or our competitors, and the timing of these introductions or announcements;
•the commercial success of any product approved by the FDA or its foreign counterparts;•market conditions for equity investments in general, or the biotechnology or pharmaceutical industries in particular;•regulatory developments in the United States and foreign countries;•changes in the structure or reimbursement policies of health care payment systems;•if our patents covering our products candidates expire or are invalidated or are found to be unenforceable, or if some or all of our patent applications do not result in issued patents or result in patents with narrow, overbroad, or unenforceable claims;•competition from third parties with products in the same class of products as any potential future product candidates or products with the same active pharmaceutical ingredients as those product candidates;•any intellectual property infringement lawsuit involving us;•actual or anticipated fluctuations in our results of operations;•changes in financial estimates or recommendations by securities analysts;•hedging or arbitrage trading activity that may develop regarding our common stock;•sales of our common stock and other securities by us;•sales of our common stock by our executive officers, directors and significant stockholders;•the low trading volume of our common stock;•changes in accounting principles; and•additional losses of any of our key scientific or management personnel.
In addition, the stock markets in general, and the markets for biotechnology and pharmaceutical stocks in particular, have experienced significant volatility that has often been unrelated to the operating performance of particular companies. For example, negative publicity regarding drug pricing and price increases by pharmaceutical companies has negatively impacted, and may continue to negatively impact, the markets for biotechnology and pharmaceutical stocks. Likewise, as a result of significant changes in U.S. social, political, regulatory and economic conditions or in laws and policies governing foreign trade and health care spending and delivery, including the possible repeal and/or replacement of all or portions of the Healthcare Reform Act or changes in tariffs and other restrictions on free trade stemming from the Trump Administration and foreign government policies, the financial markets could experience significant volatility that could also negatively impact the markets for biotechnology and pharmaceutical stocks. These broad market fluctuations may adversely affect the trading price of our common stock.In the past, class action litigation has often been instituted against companies whose securities have experienced periods of volatility in market price. Any such litigation brought against us could result in substantial costs, which would hurt our financial condition and results of operations and divertmanagement's attention and resources, which could result in delays of our clinical trials or commercialization efforts.If we fail to meet continued listing standards of The Nasdaq Stock Market LLC, our common stock may be delisted. Delisting could adversely affect the liquidity of our common stock and the market price of our common stock could decrease, and our ability to obtain sufficient additional capital to fund our operations would be substantially impaired.Our common stock is currently listed on The Nasdaq Capital Market. The Nasdaq Stock Market LLC, or Nasdaq, has minimum requirements that a company must meet in order to remain listed on The Nasdaq Capital Market. These requirements include maintaining a minimum closing bid price of $1.00 per share, or the Bid Price Requirement, and the closing bid price of our common stock has in the past been well below $1.00 per share. In this regard, on December 5, 2016, we effected one-for-ten reverse stock split of our outstanding common stock, or the Reverse Stock Split, the primary purpose of which was to enable us to regain compliance with the Bid Price Requirement, which compliance was regained on December 20, 2016. However, the closing bid price of our common stock has recently been well below $1.00 per share, and there can be no assurance that we will meet the Bid Price Requirement, or any other Nasdaq continued listing requirement, in the future. If we fail to meet these requirements, including the Bid Price Requirement and requirements to maintain minimum levels of stockholders' equity or market values of our common stock, Nasdaq may notify us that we have failed to meet the minimum listing requirements and initiate the delisting process.In addition, we are required pursuant to the terms of the Merger Agreement to submit to our stockholders a proposal to approve an amendment to our restated certification of incorporate to authorize our board of directors to effect a reverse stock split of all outstanding shares of our common stock. The approval of the reverse stock split by the stockholders is a condition to closing, pursuant to the Merger Agreement. If this reverse stock split proposal is not approved by our stockholders, and if the parties waive this closing condition, the combined company resulting from the proposed Merger will likely not be able to obtain compliance with the minimum bid price requirement for an initial listing on The Nasdaq Capital Market and, as a consequence, Nasdaq will immediately provide the combined company with written notification that our common stock will be delisted.If our common stock is delisted, we would expect our common stock to be traded in the over-the-counter market, which could adversely affect the liquidity of our common stock. Additionally, we could face significant material adverse consequences, including:•a limited availability of market quotations for our common stock;•a reduced amount of news and analyst coverage for us;•a decreased ability to issue additional securities and a concomitant substantial impairment in our ability to obtain sufficient additional capital to fund our operations and to continue as a going concern;•reduced liquidity for our stockholders;•potential loss of confidence by employees and potential future partners or collaborators; and•loss of institutional investor interest and fewer business development opportunities.
Our executive officers, directors and largest stockholders have the ability to control all matters submitted to stockholders for approval.Based solely on the most recent Schedules 13G and 13D filed with the SEC and reports filed with the SEC under Section 16 of the Exchange Act, our executive officers, directors and holders of 5% or more of our outstanding common stock, including their affiliated or associated entities, held approximately 53.5% of our outstanding common stock, and our executive officers and directors alone, including their affiliated or associated entities, held approximately 30.0% of our outstanding common stock as well as warrants to purchase up to an additional 3.2 million shares of common stock. As a result, these stockholders, acting together, have the ability to control all matters requiring approval by our stockholders, including the election of directors, the approval of the issuance of shares of our common stock pursuant to the Merger Agreement, and the approval of potential alternative mergers or other business combination transactions. The interests of this group of stockholders may not always coincide with our interests or the interests of other stockholders.Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.We have a significant amount of federal and state net operating loss carryforwards. In this regard, as of December 31, 2018, we had net federal operating loss carryforwards of approximately $472.1 million. The federal operating loss carryforwards originating prior to 2018 will expire from 2019 to 2037 if not utilized, and state operating loss carryforwards of approximately $411.4 million, which expire from 2019 to 2038 if not utilized. Our ability to use our federal and state net operating loss carryforwards to offset potential future taxable income and related income taxes that would otherwise be due is dependent upon our generation of future taxable income before the expiration dates of the net operating loss carryforwards, and we cannot predict with certainty when, or whether, we will generate sufficient taxable income to use all of our net operating loss carryforwards. On December 22, 2017, President Trump signed into law new tax legislation, or the Tax Reform Act. Under the Tax Reform Act, federal net operating losses incurred in 2018 and in future years may be carried forward indefinitely, but the deductibility of such federal net operating losses is limited. It is uncertain if and to what extent various states will conform to the Tax Reform Act. In addition, under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an "ownership change," generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period, the corporation's ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes (such as research tax credits) to offset its post-change taxable income or taxes may be limited. We completed a study through December 31, 2016 to determine whether any Section 382 limitations exist and, as a result of this study and our analysis of subsequent ownership changes, we do not believe that any Section 382 limitations exist through December 31, 2018, though we have not yet conducted an in-depth analysis since the last study. Section 382 of the Internal Revenue Code is an extremely complex provision with respect to which there are many uncertainties and we have not established whether the IRS agrees with our determination. In any event, our 2016 and 2017 equity offerings, our past and potential future issuances of common stock pursuant to the ATM Sales Agreement, other future equity offerings and/or changes in our stock ownership, some of which are outside of our control, could in the future result in an ownership change and an accompanying Section 382 limitation. If a limitation were to apply, utilization of a portion of our domestic net operating loss and tax credit carryforwards could be limited in future periods and a portion of the carryforwards could expire before being available to reduce future income tax liabilities. In this regard, the proposed Merger, if consummated, will constitute an ownership change (within the meaning Section 382 of the Internal Revenue Code) which would eliminate or otherwise substantially limit our federal and state net operating loss carryforwards.Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.Provisions in our certificate of incorporation and our bylaws may delay or prevent an acquisition of us or a change in our management. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our Board of Directors. Because our Board of Directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. These provisions include:•a classified Board of Directors;•a prohibition on actions by our stockholders by written consent;•the ability of our Board of Directors to issue preferred stock without stockholder approval, which could be used to institute a "poison pill" that would work to dilute the stock ownership of a potential hostile acquirer, effectively preventing acquisitions that have not been approved by our Board of Directors; and•limitations on the removal of directors.
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns 15% or more of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired 15% or more of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner. Finally, these provisions establish advance notice requirements for nominations for election to our Board of Directors or for proposing matters that can be acted upon at stockholder meetings. These provisions would apply even if the offer may be considered beneficial by some stockholders.Our amended and restated bylaws provide that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders' ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.Our amended and restated bylaws provide that the Court of Chancery of the State of Delaware is the exclusive forum for any derivative action or proceeding brought on behalf of GTx, for any action asserting a claim of breach of a fiduciary duty owed by any current or former director, officer, other employee or stockholder of GTx to GTx or to our stockholders, for any action asserting a claim arising pursuant to any provision of the General Corporation Law of the State of Delaware, or the DGCL, our restated certificate of incorporation or our amended and restated bylaws or as to which the DGCL confers jurisdiction on the Court of Chancery of the State of Delaware, or for any action asserting a claim governed by the internal affairs doctrine. The choice of forum provision may limit a stockholder's ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. If a court were to find the choice of forum provision contained in our amended and restated bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could harm our financial condition.If there are substantial sales of our common stock, the market price of our common stock could drop substantially, even if our business is doing well.For the 12-month period ended December 31, 2018, the average daily trading volume of our common stock on The Nasdaq Capital Market was only 705,027 shares. As a result, future sales of a substantial number of shares of our common stock in the public market, or the perception that such sales may occur, could adversely affect the then-prevailing market price of our common stock. As of December 31, 2018, we had 24,051,844 shares of common stock outstanding. In addition, as a result of the low trading volume of our common stock, which was exacerbated by the one-for-ten reverse stock split of our outstanding common stock effected on December 5, 2016, or the Reverse Stock Split, the trading of relatively small quantities of shares by our stockholders may disproportionately influence the market price of our common stock in either direction. The price for our shares could, for example, decline significantly in the event that a large number of our common shares are sold on the market without commensurate demand, as compared to an issuer with a higher trading volume that could better absorb those sales without an adverse impact on its stock price. In addition, due to the limitations of our market, the volatility in the market price of our common stock and our currently-depressed stock price, stockholders may face difficulties in selling shares at attractive prices when they want to sell.In September 2017, we completed a private placement of 5.5 million shares of our common stock and warrants to purchase 3.3 million shares of our common stock. In November 2014, we completed a private placement of 6.4 million shares of our common stock and warrants to purchase 6.4 million shares of our common stock (as adjusted to give effect to the Reverse Stock Split). Similarly, in March 2014 we completed a private placement of 1.2 million shares of our common stock and warrants to purchase 1.0 million shares of our common stock (as adjusted to give effect to the Reverse Stock Split). Pursuant to the terms of the registration rights or securities purchase agreements we entered into in connection with these private placements, we have filed registration statements under the Securities Act registering the resale of an aggregate of approximately 23.8 million shares of common stock that we issued to, or are issuable upon the exercise of warrants that we issued to, the investors in these private placements, which investors include our largest stockholders. Moreover, J.R. Hyde, III and certain of his affiliates, have rights under a separate registration rights agreement with us to require us to file resale registration statements covering an additional 785,000 shares of common stock held in the aggregate or to include these shares in registration statements that we may file for ourselves or other stockholders. If Mr. Hyde or his affiliates or any of our other significant stockholders, including the other investors in our private placements, were to sell large blocks of shares in a short period of time, the market price of our common stock could drop substantially.The comprehensive U.S. tax reform bill passed in 2017 could adversely affect our business and financial condition.On December 22, 2017, President Trump signed the Tax Reform Act into law, which significantly revises the Internal Revenue Code of 1986, as amended. The Tax Reform Act, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, limitation of the tax deduction for interest expense to 30% of adjusted earnings (except for certain small businesses), limitation of the deduction for net operating losses to 80% of current year taxable income and elimination of net operating loss carrybacks, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits (including reducing the business tax credit for certain clinical testing expenses incurred in the testing of certain drugs for rare diseases or conditions). Notwithstanding the reduction in the corporate income tax rate, the overall impact of the new Tax Reform Act is uncertain and our business and financial conditioncould be adversely affected. In addition, it is uncertain if and to what extent various states will conform to the Tax Reform Act. The impact of the Tax Reform Act on holders of our common stock is also uncertain and could be adverse. We urge our stockholders to consult with their legal and tax advisors with respect to this legislation and the potential tax consequences of investing in or holding our common stock.Item 1B.
Not applicable.
Our principal executive offices are
ITEM 1B. UNRESOLVED STAFF COMMENTSNone.Welocated in San Diego, California, where we currently subleaseapproximately 26,0004,677 square feet of office spacelocated at 175 Toyota Plaza, Memphis, Tennessee, underused primarily for corporate, research, development, clinical, regulatory, manufacturing and quality functions. Our sublease for this facility expires in March 2021.Litigation Related to the Merger
Between April 10 and May 1, 2019, three putative class action lawsuits and one individual lawsuit were filed in the U.S. District Court for the District of Delaware: Wheby v. GTx, Inc. et al., Miller v. GTx, Inc. et al., Tabb v. GTx, Inc. et al., and Living Seas LLC v. GTx, Inc. et al. (collectively, the “Delaware Actions”). On April 11 and 23, 2019, two putative class actions were filed in the U.S. District Court for the Southern District of New York: Kopanic v. GTx, Inc. et al. and Cooper v. GTx, Inc. et al. (collectively, the “New York Actions” and, together with the Delaware Actions, the “Actions”). The Actions name as defendants us and our former board of directors, and, in the case of the Wheby and Miller actions, Private Oncternal and Merger Sub. The Actions allege that defendants violated Sections 14(a) and 20(a) of the Exchange Act, as well as Rule 14a-9 promulgated thereunder, in connection with our filing of the Registration Statement in connection with the Merger. The Delaware Actions have now been voluntarily dismissed with prejudice: the Wheby action on June 12, 2019; the Miller action on July 15, 2019; the Living Seas action on June 26, 2019; and the Tabb action on October 21, 2019. On September 16, 2019, Plaintiffs in the New York Actions filed an
operating lease which expiresamended complaint, alleging violations of Sections 14(a) and 20(a) of the Exchange Act related to the value GTx’s stockholders received in the Merger. The complaint seeks damages and other unspecified relief. On January 10, 2020, the defendants filed their motion to dismiss the amended complaint, onApril 30, 2019.January 31, 2020, the plaintiffs filed their opposition to defendants’ motion to dismiss, and on February 14, 2020, the defendants filed a reply in support of their motion to dismiss. The defendants’ motion to dismiss is pending. We believe that the New York Actions are without merit and intend to vigorously defend these actions. We cannot predict the outcome of or estimate the possible loss or range of loss from any of these matters.Zappia vs. GTx Incorporated
On October 15, 2019, Joseph Zappia and Karen Zappia filed a lawsuit against us in the U.S. District Court for the District of Delaware. The complaint alleges that our
facilities are currently adequateformer management (prior tomeet our needs.the Merger) engaged in illegal insider trading and false, manipulative and deceptive practices in violation of Sections 10(b) (and Rule 10b-5 promulgated thereunder), with respect to the timing of the disclosure of failed clinical trial results of GTx’s enobosarm product candidate in September 2018. The plaintiffs seek damages, interest, costs, attorneys' fees. We
are not currently involved in any material legal proceedings.believe that this lawsuit is without merit and intend to vigorously defend this matter. We cannot predict the outcome of or estimate the possible loss or range of loss from this matter.ITEM 4. MINE SAFETY DISCLOSURESNot applicable.
Item 5.
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market InformationMarket for Registrant's Common EquityOur common stock
tradesis listed on The Nasdaq Capital Market under thetradingticker symbol"GTXI." On“ONCT”. As of March12, 2019, the closing price of our common stock as reported on The Nasdaq Capital Market was $1.42 per share and6, 2020, there were approximately69156 holders of record of our common stock.This number was derived from our stockholder records and does not include beneficial owners of our common stock whose shares are held in the name of various dealers, clearing agencies, banks, brokers and other fiduciaries.We have never declared or paid any
cashdividends on ourcapitalcommon stock. We currently intend to retain all available funds and any future earnings, if any, to fund the development and expansion of our business andthereforewe do not anticipate paying any cash dividendson our common stockin the foreseeable future.Any future determination to pay dividends will be at the discretion of our Board of Directors.
Recent Sales of UnregisteredEquity Securities
Securities.Except as previously reported in our Quarterly Reports on Form 10-Q and Current Reports on Form 8-K filed with the Securities and Exchange Commission during the year ended December 31, 2018, there were no unregistered sales of equity securities by us during the year ended December 31, 2018.None.Item 6.
ITEM 6. SELECTED FINANCIAL DATA
Not applicable.You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and the related notes and other financial information included elsewhere in this Annual Report. Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report, including information with respect to our plans and strategy for our business and future financial performance, includes forward-looking statements that are based upon current beliefs, plans and expectations and involve risks, uncertainties and assumptions. You should review the “Risk Factors” section of this Annual Report for a discussion of important factors that could cause our actual results and the timing of selected events to differ materially from those described in or implied by the forward-looking statements contained in the following discussion and analysis. Please also see the section within Part I of this Annual Report entitled “Forward-Looking Statements.”
Unless otherwise indicated or as the context otherwise requires, the historical financial
data belowinformation included inconjunction with "Management'sthis Management’s Discussion and Analysis of Financial Condition and Results ofOperations"Operations is that of Private Oncternal prior to the Merger because Private Oncternal was deemed to be the accounting acquirer in the Merger for financial reporting purposes.Overview
We are a clinical-stage biopharmaceutical company focused on the development of novel oncology therapies for cancers with critical unmet medical need. Our development efforts are focused on promising, yet untapped, biological pathways implicated in cancer generation or progression. Our pipeline includes cirmtuzumab, an investigational monoclonal antibody that is designed to inhibit ROR1, a growth factor receptor that is widely expressed on many tumors and that activates pathways leading to increased tumor proliferation, invasiveness and drug resistance. Cirmtuzumab is being evaluated in a Phase 1/2 clinical trial in combination with ibrutinib (Imbruvica®) for the treatment of patients with B-cell lymphoid malignancies, including MCL and CLL and in an investigator-sponsored, Phase 1 clinical trial in combination with paclitaxel for the treatment of women with HER2-negative metastatic or locally advanced, unresectable breast cancer. We are also developing TK216, an investigational small molecule that is designed to inhibit the ETS, or E26 Transformation Specific, family of oncoproteins, which have been shown in preclinical studies to alter gene transcription and RNA processing and lead to increased cell proliferation and invasion. TK216 is being evaluated in a Phase 1 clinical trial as a single agent and in combination with vincristine in patients with relapsed or refractory Ewing sarcoma, a rare pediatric cancer. In addition, we are developing a chimeric antigen receptor T cell, or CAR-T, therapy candidate that targets ROR1, which is currently in preclinical development as a potential treatment for hematologic cancers and solid tumors.
Since Private Oncternal’s inception in 2013, we have devoted most of our resources to organizing and staffing, business planning, raising capital, acquiring product candidates and securing related intellectual property rights and advancing our cirmtuzumab and TK216 clinical development programs. Under research subaward agreements between us and the
auditedUniversity of California San Diego (“UC San Diego”), we are eligible to receive approximately $14.0 million in development milestones throughout the award project period, estimated to be from October 1, 2017 to March 31, 2022. Through December 31, 2019, we have funded our operations primarily through: (i) gross proceeds of $49.0 million from the issuance of convertible preferred stock, (ii) receipt of $10.3 million in subaward grant payments received from UC San Diego, and (iii) cash proceeds of $18.3 million received in connection with the closing of the Merger described below. As of December 31, 2019, we had cash and cash equivalents of $20.1 million.We have incurred net losses in each year since inception. Our ability to generate product revenue sufficient to achieve profitability will depend heavily on the successful development and eventual commercialization of one or more of our current or future product candidates. Our net losses were $34.2 million (including $18.1 million related to nonrecurring merger costs) and $6.6 million for the years ended December 31, 2019 and 2018, respectively. As of December 31, 2019, we had an accumulated deficit of $65.6 million. Substantially all of our net losses have resulted from costs incurred in connection with: (i) advancing our research and development programs, (ii) general and administrative costs associated with our operations, including the costs associated with operating as a public company, and (iii) in-process research and development costs associated with the Merger. We expect to continue to incur significant and increasing operating losses for at least the next several years. We expect that our expenses and capital funding requirements will increase substantially in connection with our ongoing activities, particularly if and as we:
generateclinical proof-of-concept data with TK216 in Ewing sarcoma, an orphan pediatric cancer indication;
advance cirmtuzumab through clinical development, initially in MCL, CLL and breast cancer;
advance our ROR1-targeting CAR-T therapy candidate to clinical testing, initially in hematological cancers and then in solid tumors;
Evaluate cirmtuzumab in additional ROR1-positive solid tumors such as lung, ovarian, liver and prostate cancers, as well as in additional hematological malignancies;
Evaluate TK216 in additional tumors with ETS fusion proteins or overexpression, such as prostate cancer, lymphoma and AML;
continue to develop additional product candidates;
acquire or in‑license other product candidates and technologies;
maintain, expand and protect our intellectual property portfolio;
establish a commercial manufacturing source and secure supply chain capacity sufficient to provide commercial quantities of any product candidates for which we may obtain regulatory approval;
seek regulatory approvals for any product candidates that successfully complete clinical trials;
establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain regulatory approval; and
add operational, financial and management information systems and personnel, including personnel to support our planned product development and future commercialization efforts, as well as to support our transition to a public reporting company.
We will not generate product sales revenue unless and until we successfully complete clinical development and obtain regulatory approval for our product candidates. If we obtain regulatory approval for any of our product candidates and do not enter into a commercialization partnership, we expect to incur significant expenses related to developing our internal commercialization capability to support product sales, marketing and distribution. In addition, we expect to incur additional costs associated with operating as a public company.
As a result, we believe we will need substantial additional funding to support our continuing operations and pursue our business strategy. Until such time as we can generate significant product sales revenue, if ever, we expect to finance our operations through a combination of equity offerings, debt financings, government funding, or other sources, including potentially collaborations, licenses and other similar arrangements. We may not be able to raise additional funds or enter into such other agreements or arrangements when needed on favorable terms, or at all. If we fail to raise capital or enter into such agreements as and when needed, we may have to significantly delay, reduce or eliminate the development and commercialization of one or more of our product candidates or delay our pursuit of potential in‑licenses or acquisitions.
Because of the numerous risks and uncertainties associated with product development, we are unable to predict the timing or amount of increased expenses or when or if we will be able to achieve or maintain profitability. Even if we are able to generate product sales, we may not become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.
We expect that our existing cash and cash equivalents will be sufficient to fund our operating expenses and capital expenditure requirements into the third quarter of 2020. We have based this estimate on assumptions that may prove to be wrong, and we could exhaust our available capital resources sooner than we expect. See “Liquidity and Going Concern.” Beyond that point, we will need to raise additional capital to finance our operations, which cannot be assured. We have concluded that this circumstance raises substantial doubt about our ability to continue as a going concern within one year after March 16, 2020, the issuance date of our annual consolidated financial statements for the year ended December 31, 2019. See Note 1 of our consolidated financial statements and the related notes
theretoand other financial information included elsewhere in this Annual Report.Similarly, in our report on
Form 10-K. The following selectedour financialdata have been derived from our audited historical financialstatementscertain of which are included elsewhere in the Annual Report on Form 10-K. Historical results are not indicative of the results to be expected in the future.Years Ended December 31, 2018 2017 2016 2015 2014 (in thousands, except per share data) Statement of Operations Data:
Expenses:
Research and development expenses
$ 29,669 $ 21,467 $ 17,228 $ 13,607 $ 20,870 General and administrative expenses
9,390 9,188 8,705 8,234 9,478 Total expenses
39,059 30,655 25,933 21,841 30,348 Loss from operations
(39,059 ) (30,655 ) (25,933 ) (21,841 ) (30,348 ) Other income (expense), net
641 216 46 57 (259 ) Gain (loss) on change in fair value of warrant liability (a)
— — 8,163 3,081 (8,804 ) Loss from operations before income taxes
(38,418 ) (30,439 ) (17,724 ) (18,703 ) (39,411 ) Income tax benefit
— — — — — Net loss
$ (38,418 ) $ (30,439 ) $ (17,724 ) $ (18,703 ) $ (39,411 ) Net loss per share — basic and diluted: (b)
Net loss per share — basic
$ (1.65 ) $ (1.75 ) $ (1.22 ) $ (1.33 ) $ (4.82 ) Net loss per share — diluted
$ (1.65 ) $ (1.75 ) $ (1.22 ) $ (1.47 ) $ (4.82 ) As of December 31, 2018 2017 2016 2015 2014 (in thousands) Balance Sheet Data: Cash, cash equivalents and short-term investments (c) $ 28,458 $ 43,899 $ 21,869 $ 29,256 $ 49,295 Working capital 25,998 38,102 19,687 1,717 17,359 Total assets 31,321 46,236 24,502 32,031 50,651 Accumulated deficit (600,055 ) (561,637 ) (531,198 ) (513,474 ) (494,771 ) Total stockholders' equity 26,111 38,261 19,891 1,859 17,829 (a)The gain (loss) on the change in fair value of warrant liability is related to the private placement of warrants completed in November 2014. See Note 6,Stockholders' Equity, for further information.(b)Net loss per share — basic and diluted disclosures have been adjusted to give effect to the one-for-ten reverse stock split of our outstanding common stock effected on December 5, 2016.(c)Cash, cash equivalents and short-term investmentsfor the year ended December 31,2018 includes2019, our independent registered public accounting firm included an explanatory paragraph stating that our recurring losses from operations and required additional funding to finance our operations raise substantial doubt about our ability to continue as a going concern.Merger with GTx
On March 6, 2019, we, then operating as GTx, Inc. (“GTx”), entered into an Agreement and Plan of Merger and Reorganization, as amended (the “Merger Agreement”), with privately-held Oncternal Therapeutics, Inc. (“Private Oncternal”) and Grizzly Merger Sub, Inc., our wholly-owned subsidiary (“Merger Sub”). Under the
net proceedsMerger Agreement, Merger Sub merged with and into Private Oncternal, with Private Oncternal surviving as our wholly-owned subsidiary (the “Merger”). On June 7, 2019, the Merger was completed. GTx changed its name to Oncternal Therapeutics, Inc., and Private Oncternal, which remains as our wholly-owned subsidiary, changed its name to Oncternal Oncology, Inc. On June 10, 2019, the combined company’s common stock began trading on The Nasdaq Capital Market under the ticker symbol “ONCT.”Pursuant to the terms of
$24.5 million received fromthesaleMerger Agreement, each outstanding share of Private Oncternal common stock outstanding immediately prior to the closing of the Merger was converted into approximately 0.073386 shares of our common stock (the “Exchange Ratio”), after taking into account a one-for-seven reverse stock split of our then-outstanding common stock (the “Reverse Stock Split”). Immediately prior to the closing of the Merger, all shares of Private Oncternal preferred stock then outstanding were exchanged into shares of common stockunder our At-the-Market Equity OfferingSMSales Agreement with Stifel, Nicolaus & Company, Incorporated, in May 2017. Cash, cash equivalentsof Private Oncternal. In addition, all outstanding options exercisable for common stock of Private Oncternal andshort-term investmentswarrants exercisable for convertible preferred stock of Private Oncternal became options and warrants exercisable for theyear ended December 31, 2017 includes the net proceedssame number of$45.6 million received from the private placementshares of common stockand warrants completed in September 2017. Cash, cash equivalents and short-term investments forof theyear ended December 31, 2016 includesCompany multiplied by thenet proceedsExchange Ratio. Immediately following the Merger, stockholders of$13.7 million received fromPrivate Oncternal owned approximately 77.5% of theregistered direct offering ofoutstanding common stockcompleted in October 2016. Cash, cash equivalentsof the combined company. The par value andshort-term investments fortheyear ended December 31, 2014 includes the net proceedsauthorized shares of$21.1 million and $42.8 million received from the private placements ofour common stockand warrants completed in March and November 2014, respectively. See Note 6,Stockholders' Equity, for further information.
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONSThe following discussion and analysis should be read in conjunction with our financial statements and related notes included elsewhere in this Annual Report on Form 10-K. This discussion contains forward-looking statements based upon current expectations that involve risks and uncertainties. Our actual results and the timing of selected events could differ materially from those anticipated in these forward-looking statementswere not adjusted as a result ofseveral factors, including those set forth under Part I, Item 1A "Risk Factors"the Reverse Stock Split.The transaction was accounted for as a reverse asset acquisition in accordance with generally accepted accounting principles in the United States of America (“GAAP”). Under this method of accounting, Private Oncternal was deemed to be the accounting acquirer for financial reporting purposes. This determination was primarily based on the facts that, immediately following the Merger: (i) Private Oncternal’s stockholders owned a substantial majority of the voting rights in the combined company, (ii) Private Oncternal designated a majority of the members of the initial board of directors of the combined company, and
elsewhere(iii) Private Oncternal’s senior management holds all key positions inthis Annual Report on Form 10-K. See "Special Note Regarding Forward-Looking Statements"the senior management of the combined company. As a result, as of the closing date of the Merger, (i) the merger is being treated as the equivalent of Private Oncternal issuing stock to acquire the net assets of GTx, (ii) the net assets of GTx are recorded based upon the fair values inthis Annual Report on Form 10-K.Business Overviewthe financial statements at the time of closing andHighlightsWe are a biopharmaceutical(iii) the reported historical operating results of the combined companydedicatedprior to thediscovery, development and commercializationmerger will be those ofmedicinesPrivate Oncternal.Prior to
treat serious and/or significant unmet medical conditions. Under an exclusive worldwide license agreement withtheUniversity of Tennessee Research Foundation, or UTRF,Merger, weare developing UTRF's proprietaryhad been evaluating enobosarm, a selective androgen receptordegrader, or SARD, technology, which we believe hasmodulator (“SARM”), for thepotential to provide compounds that can degrade or antagonize multiple formstreatment ofandrogen receptor, or AR, thereby potentially inhibiting tumor growth in patients with progressive castration-resistant prostate cancer, or CRPC, including those patients who do not respond to or are resistant to current androgen targeted therapies. We are in the process of completing ongoing mechanistic preclinical studies in order to select the most appropriate SARD compounds to move forward into the additional preclinical studies required to submit an investigational new drug application, or IND, and potentially advance one of our SARD compounds into a first-in-human clinical trial.We had been developing selective androgen receptor modulators, or SARMs. Our SARM product candidate, enobosarm (GTx-024), was most recently evaluated inpost-menopausal women with stress urinary incontinenceor SUI. During(“SUI”). However, based on thethird quarterresults of2018, we announced that our randomized, placebo-controlled Phase 2 clinical trial, orthe ASTRID trialevaluating the changeinthe mean number of daily SUI episodes following 12 weeks of enobosarm treatment failed to achieve statistical significance on the primary endpoint of the proportion of patients with a greater than 50% reduction in incontinence episodes per day compared to placebo. We have completed the ASTRID trial, including our review of the full data sets from the clinical trial, and have2018, we determined that thereiswas not a sufficient path forward to warrant additional clinical development of enobosarm to treatSUI. We have thereforeSUI, and discontinued further development of enobosarm to treat SUI, as well as our SARM technology generally. On December 31, 2019, we provided notice of termination of the amended and restated license agreement (the “SARM License Agreement”), dated July 24, 2007, by and between us and the University of Tennessee Research Foundation related to the development of the SARM Technology, which termination will be effective three months following such notice. We have the right to terminate the SARM License Agreement at any time, effective upon three months’ notice. Following termination, we will no longer have the obligation to make further payments under the SARM License Agreement, includingdiscontinuingpayments for patent prosecution and maintenance, and we will no longer have any rights to develop or sublicense therelated durabilitySARM Technology. We will not incur any early termination penalties due to the termination of the SARM License Agreement.Components of Results of Operations
Grant Revenue
We have not and
open-label safety extension studiesdo not expect to generate any product sales revenue in the foreseeable future. If our development efforts for our product candidates are successful and result in regulatory approval, weinitiated beforemay generate product sales revenue in the future. We cannot predict if, when, or to what extent wereceived topline datawill generate revenue from theASTRID trial.commercialization and sale of our product candidates. We may never succeed in obtaining regulatory approval for any of our product candidates. Our total revenue to date has been derived from a California Institute for Regenerative Medicine (“CIRM”) grant subaward with UC San Diego.In August 2017, CIRM awarded an $18.3 million grant to researchers at UC San Diego to advance our Phase 1/2 clinical trial evaluating cirmtuzumab in combination with ibrutinib for the treatment of patients with B-cell lymphoid malignancies, including MCL and CLL. Oncternal is conducting this study in collaboration with UC San Diego and estimates it will receive approximately $14.0 million in development milestones under research subaward agreements throughout the award project period, estimated to be from October 1, 2017 to March 31, 2022. In addition, we are committed to certain co-funding requirements and are required to provide UC San Diego progress and financial update reports throughout the award project period. We received subaward payments of $6.2 million and $0.5 million in the years ended December 31, 2019 and 2018, respectively. As of December 31, 2019, we believe we have
also discontinued any furthermet our obligations under the CIRM award and UC San Diego subawards.Operating Expenses
Research and Development
Research and development expenses consist primarily of costs incurred for the preclinical and clinical development of our
SARM program generally.product candidates, cirmtuzumab, TK216 and our ROR1-targeting CAR-T therapy candidate, which include:expenses under agreements with third-party contract organizations, investigative clinical trial sites that conduct research and development activities on our behalf, and consultants;
costs related to develop and manufacture preclinical study and clinical trial material;
salaries and employee-related costs, including stock-based compensation;
costs incurred under our collaboration and third-party licensing agreements; and
laboratory and vendor expenses related to the execution of preclinical and clinical trials.
FollowingWe accrue all research and development costs in theannouncementperiod they are incurred. Costs for certain development activities are recognized based on an evaluation of theASTRIDprogress towards completion of specific tasks using information and data provided to us by our vendors, collaborators and third-party service providers. Advance payments for goods or services to be received in future periods for use in research and development activities are deferred and then expensed as the related goods are delivered and as services are performed. Any unearned advances would be refunded when known.We expect our research and development expenses to increase substantially for the foreseeable future as we: (i) invest in additional operational personnel to support our planned product development efforts, and (ii) continue to invest in developing our product candidates preclinically, advance them into later stages of clinical development, and as we begin to conduct larger clinical trials. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials.
Our direct research and development expenses are tracked by product candidate and consist primarily of external costs, such as fees paid under third-party license agreements and to outside consultants, contract research organizations (“CROs”), contract manufacturing organizations and research laboratories in connection with our preclinical development, process development, manufacturing and clinical development activities. We do not allocate employee costs and costs associated with our discovery efforts, laboratory supplies and facilities, including other indirect costs, to specific product candidates because these costs are deployed across multiple programs and, as such, are not separately classified. We use internal resources primarily to conduct our research as well as for managing our preclinical development, process development, manufacturing and clinical development activities. These employees work across multiple programs and, therefore, we do not track our costs by product candidate unless such costs are includable as subaward costs.
We cannot determine with certainty the timing of initiation, the duration or the completion costs of current or future preclinical studies and clinical trials of our product candidates due to the inherently unpredictable nature of preclinical and clinical development, including any potential expanded dosing beyond the original protocols based in part on ongoing clinical success. Clinical and preclinical development timelines, the probability of success and development costs can differ materially from expectations. We anticipate that we will make determinations as to which product candidates to pursue and how much funding to direct to each product candidate on an ongoing basis in response to the results of ongoing and future preclinical studies and clinical trials, regulatory developments and our ongoing assessments of each product candidate’s commercial potential. We will need to raise substantial additional capital in the future. In addition, we cannot forecast which product candidates may be subject to future collaborations, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
General and Administrative
General and administrative expenses consist primarily of personnel-related costs, insurance costs, facility costs and professional fees for legal, patent, consulting, investor and public relations, accounting and audit services. Personnel-related costs consist of salaries, benefits and stock-based compensation. We expect our general and administrative expenses will increase substantially as we: (i) incur additional costs associated with being a public company, including audit, legal, regulatory, and tax-related services associated with maintaining compliance with exchange listing and SEC requirements, director and officer insurance premiums, and investor relations costs, (ii) hire additional personnel, and (iii) protect our intellectual property.
Other Income (Expense)
Change in Fair Value of Preferred Stock Warrant Liability
In connection with Private Oncternal’s Series B-2 preferred stock financing in 2017, Private Oncternal issued warrants to purchase shares of its Series B-2 preferred stock. We classified these warrants as a liability on our consolidated balance sheets and remeasured them to fair value at each reporting date, and we recognized changes in the fair value of the warrant liability as a component of other income (expense), net in our consolidated statements of operations.
Upon the closing of the Merger, all outstanding warrants to purchase Private Oncternal Series B-2 preferred stock were converted into warrants to purchase our common stock. As a result, such warrants no longer require liability accounting and the fair value of the warrant liability has been reclassified to stockholders’ equity.
Interest income consists of interest earned on our cash equivalents, which consist of money market funds. Our interest income has not been significant due to low interest earned on invested balances.
Results of Operations
Comparison of the Years Ended December 31, 2019 and 2018
The following table summarizes our results of operations for the years ended December 31, 2019 and 2018 (in thousands):
Years Ended
December 31,
2019
2018
Change
Grant revenues
$
2,425
$
2,521
$
(96
)
Operating expenses:
Research and development
10,159
8,287
1,872
In-process research and development
18,088
—
18,088
General and administrative
7,286
1,820
5,466
Total operating expenses
35,533
10,107
25,426
Loss from operations
(33,108
)
(7,586
)
(25,522
)
Other income (expense):
Change in fair value of warrant liability
(1,268
)
713
(1,981
)
Other income
—
216
(216
)
Interest income
188
79
109
Interest expense
—
(1
)
1
Total other income (expense)
(1,080
)
1,007
(2,087
)
Net loss
$
(34,188
)
$
(6,579
)
$
(27,609
)
Grant Revenue
Grant revenue for the year ended December 31, 2019 was $2.4 million, consistent with the $2.5 million for the year ended December 31, 2018.
Research and Development Expenses
The following table summarizes our research and development expenses for the periods indicated (in thousands):
Years Ended
December 31,
Increase/
2019
2018
(Decrease)
Cirmtuzumab
$
6,156
$
5,561
$
595
TK216
1,101
1,465
(364
)
Other
408
—
408
Unallocated research and development expenses
2,494
1,261
1,233
Total research and development expenses
$
10,159
$
8,287
$
1,872
Research and development expenses for the years ended December 31, 2019 and 2018 were $10.2 million and $8.3 million, respectively, an increase of $1.9 million. The increase was primarily due to: (i) a $0.7 million net increase in direct product candidate costs, and (ii) a $1.2 million increase in unallocated research and development expenses.
Direct expenses for cirmtuzumab increased $0.6 million for the year ended December 31, 2019, compared to the year ended December 31, 2018, primarily due to the following partially offsetting factors: (i) a $1.9 million increase in clinical trial
results,activities related to ourboardongoing Phase 1/2 clinical trial ofdirectorscirmtuzumab in combination with ibrutinib for the treatment of patients with B-cell lymphoid malignancies, including MCL and CLL, that commenced in the latter part of 2017, (ii) aprocess$0.3 million increase in regulatory and quality activities, and (iii) a $1.6 million decrease in manufacturing clinical trial material costs.Direct expenses for TK216 decreased $0.4 million for the year ended December 31, 2019, compared to the year ended December 31, 2018, primarily due to a corresponding decrease in clinical trial activities related to our continuing Phase 1 clinical trial of
evaluating strategic alternativesTK216 in refractory Ewing sarcoma.Direct expenses other increased $0.4 million for the year ended December 31, 2019, compared to
maximize stockholder value. To assist withthe year ended December 31, 2018, primarily due to a $0.4 million increase in research activities for other program initiatives.Unallocated expenses increased $1.2 million for the year ended December 31, 2019, compared to the year ended December 31, 2018, primarily due to higher personnel costs.
In-Process Research and Development Expenses
In-process research and development expenses increased $18.1 million for year ended December 31, 2019, compared to the year ended December 31, 2018, due solely to the Merger.
General and Administrative Expenses
General and administrative expenses for the years ended December 31, 2019 and 2018 were $7.3 million and $1.8 million, respectively, an increase of $5.5 million. The increase is primarily due to higher: (i) legal fees of $1.8 million incurred to expand our intellectual property portfolios on our platforms and product candidates in 2019 and for additional services incurred as a public company, (ii) personnel-related costs and professional fees of $2.3 million related primarily to additional headcount and other activities to operate as a publicly-traded company, (iii) director and officer liability insurance costs of $0.6 million, and (iv) other operating expenses of $0.8 million.
Other Income (Expense)
Other expense was ($1.1) million for the year ended December 31, 2019, compared to other income of $1.0 million for the year ended December 31, 2018, a change of ($2.1) million in additional expense. The change was primarily due to a $2.0 million increase in the fair value of the preferred stock warrant liability prior to the Merger closing.
Liquidity and Going Concern
From our inception through December 31, 2019, we have devoted substantially all of our efforts to organizational activities including raising capital, building infrastructure, acquiring assets, developing intellectual property, and conducting preclinical studies, clinical trials and product development activities. We have a limited operating history and the sales and income potential of our business and market are unproven. We have experienced recurring net losses and negative cash flows from operating activities. At December 31, 2019, we had an accumulated deficit of $65.6 million and had cash and cash equivalents of $20.1 million. We will need to continue to raise a substantial amount of funds until we are able to generate revenues to fund our development and operating activities.
We expect to continue to incur net losses into the foreseeable future. Successful transition to attaining profitable operations is dependent upon achieving a level of revenues adequate to support our cost structure. We have incurred net losses since inception and have relied on our ability to fund our operations through debt and equity financings and grant funding. These conditions raise substantial doubt about our ability to continue as a going concern. The accompanying consolidated financial statements have been prepared assuming that we will continue as a going concern and do not include any adjustments that might result from the outcome of this
process, our boarduncertainty. This basis ofdirectors engaged a financial advisory firm to help explore our available strategic alternatives, including possible mergers and business combinations, a sale of part or allaccounting contemplates the recovery of our assets andcollaboration and licensing arrangements. On March 6, 2019, we and Oncternal Therapeutics Inc., or Oncternal, announced the signing of an Agreement and Plan of Merger and Reorganization, or the Merger Agreement. Upon the terms and subject tothe satisfaction ofthe conditions describedliabilities in theMerger Agreement, including approvalnormal course ofthe transaction by our stockholders and Oncternal's stockholders, a wholly-owned subsidiary of GTx will be merged with and into Oncternal, or the Merger, with Oncternal surviving the Merger as a wholly-owned subsidiary of GTx. For more information on the terms of the Merger Agreement and the transactions contemplated thereby, see the section entitled "Business — Overview" under Part 1, Item 1 of this Annual Report on Form 10-K and Note 2,Significant Accounting Policies — Subsequent Events, in the accompanying Notes to Financial Statements.business.Although we have entered into the Merger Agreementequity offerings, debt financings, government funding, or other sources, including potentially collaborations, licenses andintend to consummate the proposed Merger, there isother similar arrangements. There can be no assurance that we will be able tosuccessfully consummate the Mergerobtain any sources of financing ona timely basis,acceptable terms, or at all.If, for any reason,To theMerger is not completed,extent that wewill reconsider our strategic alternatives and could pursue one or more of the following courses of action:•Continue development of our SARD program.As set forth above, we are in the process of completing ongoing mechanistic preclinical studies in order to select the most appropriate SARD compounds to move forward into the additional preclinical studies required to submit an IND and potentially advance one of our SARD compounds into a first-in-human clinical trial. Accordingly, if, for any reason, the Merger is not consummated, we may determine to move forward with our planned IND-enabling studies of our SARD compounds. However, while we believe that our existing capital resources will be adequate to enable us to conduct and complete planned IND-enabling preclinical studies of our SARD compounds, we will require significant additional financial resources in order to initiate and complete initial human clinical trials of a SARD compound and to otherwise further the development of our SARD program. As a result, we may also resume our efforts to seekcan raise additional fundsthrough potential collaborative, partnering or other strategic arrangements to provide us with the necessary resources for the development of our SARD program.•Pursue potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets.We have discontinued further development of our SARM program, including enobosarm, and do not currently have any plans to resume development of our SARM program. We continue our efforts to seek potential collaborative, partnering or other strategic arrangements for our SARM assets, including a sale or other divestiture of our SARM assets.•Pursue another strategic transaction like the proposed Merger.Our board of directors may elect to pursue an alternative strategy, one of which may be a strategic transaction similar to the proposed Merger.•Dissolve and liquidate our assets.If, for any reason, the Merger is not consummated and we are unable to identify and complete an alternative strategic transaction like the Merger or potential collaborative, partnering or other strategic arrangements for our SARM assets, or to continue to operate our business due to our inability to raise additional funding for the development of our SARM program or otherwise, we may be required to dissolve and liquidate our assets. In such case, we would be required to pay all of our debts and contractual obligations, and to set aside certain reserves for potential future claims, and there can be no assurances as to the amount or timing of available cash left to distribute toby issuing equity securities, our stockholdersafter paying our debts and other obligations and setting aside funds for reserves.
Financial HighlightsOur net loss for the year ended December 31, 2018 was $38.4 million. We expect to incurmay experience significantoperating losses for the foreseeable future depending on the extent of our preclinical and any clinical development activities and,dilution. Any debt financing, ifany such development activities are successful, potentially seeking regulatory approval of any potential future product candidates. We have funded our operations primarily through the sale of equity securities, collaboration and license agreements, and prior to September 2012, product revenue from sales of FARESTON®, the rights to which we sold to a third party in the third quarter of 2012. We do not expect to receive regulatory approval for the commercial sale of any product candidates for the foreseeable future, if at all.At December 31, 2018, we had cash, cash equivalents and short-term investments of $28.5 million compared to $43.9 million at December 31, 2017. In May 2018, we sold 1.5 million shares of common stock under our At-the-Market Equity OfferingSMSales Agreement, or the ATM Sales Agreement, with Stifel, Nicolaus & Company, Incorporated, or Stifel, and raised net proceeds of $24.5 million.To conserve our cash resources, we have substantially reduced our workforce since November 2018 and have ceased our SARM development activities and all other operations except for day-to-day business operations, completing ongoing mechanistic SARD preclinical studies and those activities necessary to complete the proposed Merger. In the first quarter of 2019, due to the entry into the Merger Agreement with Oncternal, our board of directors committed to reducing our workforce down to a total of eleven full-time employees, who will remain with us until the closing of the transaction to assist with our day-to-day business operations, including continuing our ongoing mechanistic SARD preclinical studies, and those activities necessary to complete the proposed Merger. All employees affected by the workforce reduction will be eligible to receive, among other things, specified severance payments based on the applicable employee's level and years of service with us and the continuation of group health insurance coverage. In addition, the affected employees will also be eligible for full vesting acceleration of their outstanding stock options as well as an extension of the post-termination exercise period for their outstanding stock options. As a result of the workforce reduction and prior termination of three employees earlier in the first quarter of 2019, we estimateavailable, may involve restrictive covenants thatwe will incur total severance-related charges for these employees of approximately $1.0 million in the first quarter of 2019 and up to an additional $500,000 contingent upon the closing of the Merger. We do not expect to record a non-cash charge related to the modification of outstanding stock options in connection with the workforce reduction.If the proposed Merger is not completed, based on our current business plan and spending assumptions as a standalone company, we estimate that our current cash, cash equivalents and short-term investments, together with interest thereon, will be sufficient to meet our projected operating requirements for at least the next 12 months. We have based our cash sufficiency estimates on our current business plan and our assumptions that may prove to be wrong. We could utilize our available capital resources sooner than we currently expect, and we could need additional funding sooner than currently anticipated.While we believe that our existing capital resources will be adequate to enable us to conduct and complete planned IND-enabling preclinical studies of SARD compounds, we will require significant additional financial resources in order to initiate and complete initial human clinical trials of a SARD compound and to otherwise further the development of our SARD program. If we are unable to raise sufficient additional funds for the development of our SARD program, whether through potential collaborative, partnering or other strategic arrangements or otherwise, or if we otherwise determine to discontinue the development of our SARD program, we will likely determine to cease operations.While we have been able to fund our operations to date, we have no ongoing collaborations for the development and commercialization of any product candidates and no source of revenue, nor do we expect to generate product revenue for the foreseeable future. We do not have any commitments for future external funding. In addition, although we have entered into an At-the-Market Equity OfferingSMSales Agreement with Stifel, Nicolaus & Company, Incorporated, or the ATM Sales Agreement, under which approximately $25.0 million of shares of our common stock remained available for sale at December 31, 2018, it is unlikely we could raise sufficient funds under the ATM Sales Agreement to permit us to initiate and complete initial human clinical trials of a SARD compound, and given our currently-depressed stock price, the ATM Sales Agreement is not otherwise expected to be a practical source of liquidity for us at this time. Further, given our currently-depressed stock price, we are significantly limited inimpact our ability tosell shares of common stock under the ATM Sales Agreement since the issuance and sale ofconduct ourcommon stock under the ATM Sales Agreement, if it occurs, would be effected under a registration statement on Form S-3 that we filed with the Securities and Exchange Commission, and in accordance with the rules governing those registration statements, we generally can only sell shares of our common stock under that registration statement in an amount not to exceed one-third of our public float, which limitation for all practical purposes precludes our ability to obtain any meaningful funding through the ATM Sales Agreement at this time.business.Until we can generate a sufficient amount of product revenue, which we may never do, we will need to finance future cash needs through potential collaborative, partnering or other strategic arrangements, as well asthrough public or private equity offerings or debt financings or a combination of the foregoing. If we are unable to raise additional funds, we will need to continue to reduce our expenditures in order to preserve our cash. Further cost-cutting measures that we may take may not be sufficient to enable us to meet our cash requirements, and they may negatively affect our business and our ability to derive any value from our SARD program. In any event, in order to further the development of our SARD program, we will need to raise substantial additional capital. Our failure to do so would likely result in our determining to cease operations.Since our inception in 1997, we have been focused on drug discovery and development programs. Research and development expenses include, but are not limited to, our expenses for personnel and supplies associated with our research activities, screening and identification of product candidates, formulation and synthesis activities, manufacturing, preclinical studies, toxicology studies, clinical trials, regulatory and medical affairs activities, quality assurance activities and license fees. We expect that our research and development expenses for fiscal year 2019 to be significantly less than fiscal year 2018 primarily due to the completion of the ASTRID trial and termination of the related extension studies and due to the reductions in headcount during the fourth quarter of 2018 and the first quarter of 2019.There is a substantial risk that any development program may not produce revenue. Moreover, because of uncertainties inherent in drug development, including those factors described in Part I, Item 1A "Risk Factors" of this Annual Report on Form 10-K, we and/or potential future collaborators may not be able to successfully develop and commercialize any of our product candidates.The successful development and commercialization of our product candidates is highly uncertain. We cannot reasonably estimate or know the nature, timing and estimated costs of the efforts necessary to complete the development and commercialization of, or the period in which material net cash inflows are expected to commence from, any of our product candidates due to the numerous risks and uncertainties associated with developing and commercializing drugs, including the uncertainty of:•the scope, rate of progress and cost of our preclinical and potential future clinical development programs;•the terms and timing of any potential collaborative, partnering and other strategic arrangements that we may establish;•the amount and timing of any licensing fees, milestone payments and royalty payments from potential collaborators, if any;•potential future clinical trial results;•the cost and timing of regulatory filings and/or approvals to commercialize any potential future product candidates and any related restrictions, limitations, and/or warnings in the label of an approved product candidate;•the effect of competing technological and market developments; and•the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights, and the cost of defending any other litigation claims.
Any failure to complete the development of any potential future product candidates in a timely manner could have a material adverse effect on our operations, financial position and liquidity. A discussion of the risksand uncertainties associated with completing our development efforts on schedule, or at all, and some consequences of failing to do so, are set forth under Part I, Item 1A "Risk Factors" of this Annual Report on Form 10-K.General and Administrative ExpensesOur general and administrative expenses consist primarily of salaries and other related costs for personnel serving executive, finance, legal, human resources, information technology, and investor relations functions. General and administrative expenses also include facility costs, insurance costs, and professional fees for legal, accounting, and public relations services. We expect our general and administrative expenses for fiscal year 2019 to decrease in comparison to fiscal year 2018 due to the reductions in headcount during the fourth quarter of 2018 and the first quarter of 2019.Critical Accounting Policies and Significant Judgments and EstimatesOur management's discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments related to revenue recognition, income taxes, intangible assets, long-term service contracts, share-based compensation, and other contingencies. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.While our significant accounting policies are more fully described in Note 2 to our financial statements appearing at the end of this Annual Report on Form 10-K, we believe that the following accounting policies are most critical to aid you in fully understanding and evaluating our reported financial results.Research and Development ExpensesResearch and development expenses include, but are not limited to, our expenses for personnel and supplies associated with our research activities, screening and identification of product candidates, formulation and synthesis activities, manufacturing, preclinical studies, toxicology studies, clinical trials, regulatory and medical affairs activities, quality assurance activities and license fees. We expense these costs in the period in which they are incurred. We estimate our liabilities for research and development expenses in order to match the recognition of expenses to the period in which the actual services are received. As such, accrued liabilities related to third party research and development activities are recognized based upon our estimate of services received and degree of completion of the services in accordance with the specific third party contract.Share-Based CompensationWe have stock option and equity incentive plans that provide for the purchase or acquisition of our common stock by certain of our employees and non-employees. We measure compensation expense for our share-based payments based on the fair value of the awards on the grant date and recognize the expense over the period during which an employee or non-employee director is required to provide service in exchange for the award.The determination of the fair value of stock options on the date of grant include the expected life of the award, the expected stock price volatility over the expected life of the awards, and risk-free interest rate. We estimate the expected life of options by calculating the average of the vesting term and contractual term of the options. We estimate the expected stock price volatility based on the historical volatility of our common stock. The risk-free interest rate is determined using U.S. Treasury rates where the term is consistent with the expected life of the stock options. Expected dividend yield is not considered as we have not made any dividend payments and have no plans of doing so in the foreseeable future. The fair value of each stock option is amortized into compensation expense on a straight-line basis between the grant date for the award and each vesting date. During the first quarter of 2017, we adopted the Financial Accounting Standards Board Accounting Standards Update 2016-09,Improvements to Employee Share Based Payment Accounting. This guidance addresses the income tax effects of stock-based payments and eliminates the windfall pool concept, as all of the tax effects related to stock-based payments are now being recorded at settlement (or expiration) through the income statement. The new guidance also permits entities to make an accounting policy election for the impact of forfeitures on the recognition of expense for stock-based payment awards, allowing for forfeitures to be estimated or recognized when they occur. We elected to prospectively adopt the policy that forfeitures be recorded when they occur. The adoption of this guidance did not have a material impact on our financial position or results of operations.The following table summarizes share-based compensation expense included within the statements of operations for the years ended December 31, 2018, 2017 and 2016:Years ended December 31, 2018 2017 2016 (in thousands) Research and development expenses
$ 807 $ 1,171 $ 1,260 General and administrative expenses
1,556 2,146 1,829 Total share-based compensation
$ 2,363 $ 3,317 $ 3,089 Share-based compensation expense recorded in the statement of operations as general and administrative expense for the years ended December 31, 2018, 2017 and 2016 included share-based compensation expense related to deferred compensation arrangements for our non-employee directors of $166,000, $166,000 and $132,000, respectively. At December 31, 2018, the total compensation cost related to non-vested stock options not yet recognized was approximately $7.7 million with a weighted average expense recognition period of 2.95 years.Income TaxesWe account for deferred taxes by recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. Accordingly, at December 31, 2018 and 2017, net of the valuation allowance, the net deferred tax assets were reduced to zero.Research and Development ExpensesThe following table identifies the research and development expenses for our SARD program and our discontinued SARM program, as well as research and development expenses pertaining to our other research and development efforts, for each of the periods presented. Research and development spending for past periods is not indicative of spending in future periods.Years Ended December 31, Proposed Candidate / Proposed Indication Program 2018 2017 2016 (in thousands) Enobosarm Treatment of postmenopausal women with SUI (1 mg and 3 mg) SARM $ 25,576 $ 11,279 $ 1,286
Enobosarm
Treatment of women with ER positive and AR positive advanced breast cancer (9 mg and 18 mg) SARM 1,957 5,541 7,316
SARDs
Treatment of castration resistant prostate cancer SARD 1,052 1,772 2,157
Enobosarm
Treatment of women with advanced AR positive TNBC (18 mg) SARM 801 2,348 4,853
Other research and development
283
527
1,616
Total research and development expenses
$
29,669
$
21,467
$
17,228
Research and development expenses increased 38% to $29.7 million for the year ended December 31, 2018 from $21.5 million for the year ended December 31, 2017. Research and development expenses increased 25% to $21.5 million for the year ended December 31, 2017 from $17.2 million for the year ended December 31, 2016.Research and development expenses for enobosarm for the treatment of postmenopausal women with SUI substantially increased from the years ended December 31, 2017 and 2016 due to the initiation of a placebo-controlled Phase 2 clinical trial of enobosarm to treat postmenopausal women with SUI, which opened for enrollment in the third quarter of 2017 and completed enrollment in the second quarter of 2018, and due to the related durability and open-label safety extension studies, which were initiated in the second quarter of 2018. During the third quarter of 2018, we announced that the ASTRID trial failed to achieve statistical significance on the primary endpoint of the proportion of patients with a greater than 50% reduction in incontinence episodes per day compared to placebo. The years ended December 31, 2016 and 2017 also included expenses related to the Phase 2 open-label, non-placebo controlled, proof-of-concept clinical trial of enobosarm to treat postmenopausal women with SUI that initiated enrollment in the first quarter of 2016.Research and development expenses for enobosarm for the treatment of women with ER positive and AR positive advanced breast cancer decreased from the years ended December 31, 2017 and 2016 due primarily to the timing and nature of activities related to conducting the Phase 2 clinical trial evaluating enobosarm 9 mg and enobosarm 18 mg in this indication. The clinical trial commenced enrollment during the third quarter of 2015 and completed enrollment in the first quarter of 2017.Research and development expenses for the SARD program for the year ended December 31, 2018 decreased from the prior years due to fewer drug formulation and preclinical research expenses being incurred during 2018 than in the comparable periods. If the proposed Merger is not completed, we expect increased research and development expenses for the SARD program in 2019 as we plan to complete ongoing mechanistic preclinical studies, and to select the most appropriate SARD compounds to move forward with IND-enabling preclinical studies.Research and development expenses for enobosarm for the treatment of women with AR positive TNBC decreased from the years ended December 31, 2017 and 2016 due to the timing and nature of activities related to conducting the first stage of the Phase 2 clinical trial, which commenced enrollment during the fourth quarter of 2015. During the third quarter of 2017, we determined that there were insufficient patients achieving clinical benefit from enobosarm treatment to continue this clinical trial.General and Administrative ExpensesGeneral and administrative expenses for the year ended December 31, 2018 of $9.4 million remained relatively consistent with the year ended December 31, 2017 of $9.2 million. General and administrative expenses increased 6% to $9.2 million for the year ended December 31, 2017 from $8.7 million for the year ended December 31, 2016. The increase during the year ended December 31, 2017 from the prior year was due primarily to an increase in share-based compensation expense.Other Income (Expense), NetOther income, net for the years ended December 31, 2018, 2017, and 2016 was $641,000, $216,000 and $46,000, respectively, and consisted of interest earned on our cash, cash equivalents and short-term investments, foreign currency transaction gains and losses, and other non-operating income or expense. The increase in other income, net for each year over year was primarily due to interest earned on the net proceeds received from issuances of common stock by the Company.Liquidity and Capital ResourcesWe have financed our operations to date primarily through public offerings and private placements of our securities, as well as payments from our former collaborators. We have incurred significant losses since our inception in 1997 as we have devoted substantially all of our resources to research and development, including our clinical trials. As of December 31, 2018, we had an accumulated deficit of $600.1 million, which resulted primarily from:•our research and development activities associated with:•the preclinical development of our SARD program;•the preclinical and clinical development of our SARM compounds, including enobosarm;•the preclinical and clinical development of our discontinued GTx-758 product candidate for the treatment of advanced prostate cancer;
•the development of our discontinued toremifene 80 mg product candidate to reduce fractures and treat other estrogen deficiency side effects of androgen deprivation therapy in men with prostate cancer, including two Phase 2 clinical trials, a Phase 3 clinical trial, and the preparation and submission of a NDA to the FDA;•the development of our discontinued toremifene 20 mg product candidate for the prevention of prostate cancer in high risk men with high grade prostatic intraepithelial neoplasia, including a Phase 2b clinical trial and a Phase 3 clinical trial; and•the preclinical development of other product candidates; and•general and administrative expenses.
We expect to incur significant operating losses for the foreseeable future depending on the extent of our preclinical and any clinical development activities and, if any such development activities are successful, potentially seeking regulatory approval of any potential future product candidates. These losses, among other things, have had and will continue to have an adverse effect on our stockholders' equity and working capital. We do not expect to receive regulatory approval for the commercial sale of any product candidates for the foreseeable future, if at all.At December 31, 2018, we had cash, cash equivalents and short-term investments of $28.5 million, compared to $43.9 million at December 31, 2017 and $21.9 million at December 31, 2016.In February 2018, we entered into the ATM Sales Agreement, pursuant to which we may offer and sell, from time to time, through Stifel, shares of our common stock having an aggregate offering price of up to
$50 million.$50.0 million, of which approximately $25.0 was available for sale at December 31, 2019. We are not obligated to sell any shares under the ATM Sales Agreement. Subject to the terms and conditions of the sales agreement, Stifel will use commercially reasonable efforts, consistent with its normal trading and sales practices, applicable state and federal law, rules and regulations and the rules of theNASDAQNasdaq Capital Market, to sell shares from time to time based upon our instructions, including any price, time or size limits specified by us. Under the ATM Sales Agreement, Stifel may sell shares by any method deemed to be an "at-the-market" offering as defined in Rule 415 under the Securities Act of 1933, as amended, or any other method permitted by law, including in privately negotiated transactions. We will pay Stifel a commission of up to 3.0% of the aggregate gross proceeds from each sale of shares.In May 2018,Under current SEC regulations, if at any time our public float is less than $75.0 million, and for so long as our public float remains less than $75 million, the amount we
sold 1.5 million sharescan raise through primary public offerings ofcommon stock under the ATM Sales Agreement for net proceeds of $24.5 million. As of December 31, 2018, we had approximately $25.0 million of common stock remaining available to be sold under the ATM Sales Agreement. However, it is unlikely we could raise sufficient funds under the ATM Sales Agreement to permit us to initiate and complete initial human clinical trials of a SARD compound, and given our currently-depressed stock price, the ATM Sales Agreement is not otherwise expected to be a practical source of liquidity for us at this time. Further, given our currently-depressed stock price, we are significantly limitedsecurities inour ability to sell shares of common stock under the ATM Sales Agreement since the issuance and sale of our common stock under the ATM Sales Agreement, if it occurs, would be effected under a registration statement on Form S-3 that we filed with the Securities and Exchange Commission, and in accordance with the rules governing thoseany twelve-month period using shelf registration statementswe generally can only sell sharesis limited to an aggregate ofour common stock under that registration statement in an amount not to exceedone-third of our public float, whichlimitation for all practical purposes precludesis referred to as the baby shelf rules. As of February 28, 2020, ourabilitypublic float calculated pursuant toobtainthe instructions set forth in Form S-3 was approximately $51.8 million, based on 11,800,518 shares of outstanding common stock held by non-affiliates at a price of $4.39 per share, which is the last reported sale price of our common stock on the Nasdaq Capital Market on February 20, 2020. So long as our public float is below $75.0 million, we will be limited by the baby shelf rules until such time as our public float exceeds $75.0 million, which means we only have the capacity to sell shares up to one-third of our public float under shelf registration statements in anymeaningful funding throughtwelve-month period. As of February 28, 2020, we calculated our future capacity to issue up to approximately $17.3 million of additional shares of common stock pursuant to the ATM Sales Agreement. If our public float decreases, the amount of securities we may sell under our Form S-3 shelf registration statement will also decrease. Future sales under the ATM Sales Agreementat this time.On September 29, 2017, we completedwill depend on aprivate placementvariety ofunits consisting of an aggregate of 5.5 million shares of common stock and warrantsfactors including, but not limited to,purchase an aggregate of 3.3 million sharesprevailing market conditions, the trading price of our common stockfor net proceeds to us of approximately $45.6 million. The purchasersand our capital needs. There can be no assurance that Stifel will be successful in consummating future sales based on prevailing market conditions or in theregisteredTable of Contentsquantities or at the prices that we deem appropriate.direct offering consisted solely of accredited investors that included certain institutional and existing stockholders, including a member of our board of directors.Cash FlowsOn October 14, 2016, we completed a registered direct offering of our common stock consisting of 1.7 million shares of our common stock for net proceeds of approximately $13.7 million. The purchasers in the registered direct offering consisted of certain existing GTx stockholders and certain members of the GTx management team and board of directors.The following table
shows a summarysummarizes our sources and uses ofourcashflowsfor each of the periodsindicated:presented (in thousands):Years Ending December 31, 2018 2017 2016 (in thousands) Net cash used in operating activities
$ (39,346 ) $ (23,460 ) $ (20,778 ) Net cash provided by (used in) investing activities
27,883 (15,126 ) 2,151 Net cash provided by financing activities
23,905 45,492 13,481 Net increase (decrease) in cash and cash equivalents
$ 12,442 $ 6,906 $ (5,146 ) Years Ended
December 31,
2019
2018
Net cash provided by (used in):
Operating activities
$
(16,746
)
$
(7,417
)
Investing activities
16,137
—
Financing activities
15
17,874
Increase in cash and cash equivalents
$
(594
)
$
10,457
NetOperating Activities
During the year ended December 31, 2019, operating activities used $16.7 million of cash,
usedresulting from our net loss of $34.2 million, which included non-cash charges of: (i) $18.1 million related to the acquisition of in-process research and development, (ii) $1.3 million related to the change in fair value of warrant liability, and (iii) $0.5 million related to stock-based compensation charges; partially offset by a $2.4 million change in operating assets and liabilities. The $2.4 million change in operating assets and liabilities primarily consisted of a $6.0 million decrease in accounts payable and accrued liabilities and a $3.6 million increase in deferred revenue.During the year ended December 31, 2018, operating activities
in all periods resultedused $7.4 million of cash, resulting primarily fromfundingouroperations.net loss of $6.6 million, non-cash change in fair value of warrant liability of $0.7 million and non-cash other income of $0.2 million, a $0.1 million change in operating assets and liabilities, partially offset by stock-based compensation charges of $0.2 million. The $0.1 million change in operating assets and liabilities primarily consisted of a $0.4 million increase in prepaid expenses and other assets, a $1.9 million decrease in deferred revenue, offset by a $2.2 million increase in accounts payable and accrued liabilities.Investing Activities
Net cash provided by investing activities was
$27.9$16.1 million for the year ended December 31, 2019, primarily resulting from cash received in connection with the Merger. Net cash provided by investing activities was none for the year ended December 31, 2018.Financing Activities
Net cash provided by financing activities was $15,000 for the year ended December 31, 2019 consisting of option and warrant exercises.
Net cash provided by financing activities was $17.9 million for the year ended December 31, 2018
and resulted primarily from maturitiesconsisting ofshort-term investmentsnet proceeds of$72.0$16.8 millionoffset by the purchase of short-term investments of $44.2 million. Net cash used in investing activities for the year ended December 31, 2017 primarily resulted from the purchase of short-term investments of $39.3 million offset by the maturities of short-term investments of $24.2 million. Net cash provided by investing activities for the year ended December 31, 2016 primarily resulted from the maturities of short-term investments of $37.6 million offset by the purchase of short-term investments of $35.4 million.Net cash provided by financing activities for the year ended December 31, 2018 of $23.9 million resultedfrom the sale ofcommonour Series C convertible preferred stockunder the ATM Sales Agreement with Stifel and proceeds from the exercise of stock options of $103,000, offset slightly by $672,000 of tax payments related to shares withheld for vested restricted stock units. Net cash provided by financing activities for the year ended December 31, 2017 reflected net proceeds of $45.6 million from the issuance of common stock and warrants related to the September 2017 private placement, partially offset by $156,000 of employee withholding tax payments related to vested RSUs. Net cash provided by financing activities for the year ended December 31, 2016 reflected net proceeds of $13.7 million from the issuance of common stock related to the October 2016 registered direct offering, partially offset by $208,000 of employee withholding tax payments related to vested RSUs.To conserve our cash resources, we have substantially reduced our workforce sincein November 2018 andhave ceased our SARM development activities and all other operations except for day-to-day business operations, completing ongoing mechanistic SARD preclinical studies and those activities necessary to completetheproposed Merger. If the proposed Merger is not completed, based on our current business plan and spending assumptions as a standalone company, we estimatecollection of $1.1 million of Series B-2 convertible preferred stock subscriptions receivable.We expect that our
currentexisting cash and cash equivalentsand short-term investments, together with interest thereon,will be sufficient tomeetfund ourprojected operating requirements for at leastoperations into thenext 12 months. We have basedthird quarter of 2020. However, ourcash sufficiency estimates on our current business plan and our assumptions that may prove to be wrong. We could utilize our available capital resources sooner than we currently expect, and we could need additional funding sooner than currently anticipated.While we believe that our existing capital resources will be adequate to enable us to conduct and complete planned IND-enabling preclinical studies of SARD compounds, we will require significant additional financial resources in order to initiate and complete initial human clinical trials of a SARD compound and to otherwise further the development of our SARD program. If we are unable to raise sufficient additional funds for the development of our SARD program, whether through potential collaborative, partnering or other strategic arrangements or otherwise, or if we otherwise determine to discontinue the development of our SARD program, we will likely determine to cease operations.Our estimateforecast of the period of timeor eventsthrough which our financial resources will be adequate to support ourprojected operating requirementsoperations is a forward-looking statementandthat involves risks and uncertainties, and actual results could varyas a resultmaterially. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we expect. Additionally, the process ofa number of factors, including the factors discussed under Part II, Item 1A "Risk Factors" section of this Annual Report on Form 10-K. Because of the numerous risks and uncertainties associated with the development and potential commercialization of ourtesting product candidates in clinical trials is costly, andother research and development activities, including risks and uncertainties that could impacttheratetiming of progress,ofpotential dose expansions beyond ourdevelopment activities, we are unable to estimate with certainty the amounts of increased capital outlaysplanned study protocols based in part on our clinical progress, andoperating expenditures associated with the future development of potential future product candidates, if any.expenses in these trials is uncertain.Our future
fundingcapital requirements will depend on many factors, including:•our ability to successfully completethe
Merger;•thetype, number, scope,rate ofprogress, expansions, results, costs andcosttiming of, our preclinical studies andpotential futureclinicaldevelopment programs;trials of our product candidates which we are pursuing or may choose to pursue in the future;the costs and timing of manufacturing for our product candidates, including commercial manufacturing if any product candidate is approved;
the costs of obtaining ibrutinib, for which we currently obtain supply at no cost under our clinical supply agreement with Pharmacyclics LLC, and vincristine to conduct our clinical trials of cirmtuzumab and TK216, respectively;
the costs, timing and outcome of regulatory review of our product candidates;
the costs of obtaining, maintaining and enforcing our patents and other intellectual property rights;
our efforts to enhance operational systems and hire additional personnel to satisfy our obligations as a public company, including enhanced internal controls over financial reporting;
the costs associated with hiring additional personnel and consultants as our preclinical and clinical activities increase;
the costs and timing of establishing or securing sales and marketing capabilities if any product candidate is approved;
••
our ability to achieve sufficient market acceptance, adequate coverage and reimbursement from third-party payors and adequate market share and revenue for any approved products;
the terms and timing of
any potential collaborative, partneringestablishing and maintaining collaborations, licenses and otherstrategic arrangementssimilar arrangements; andcosts associated with any products or technologies that we may
establish;•the amount and timing of any licensing fees, milestone payments and royalty payments from potential collaborators,in-license or acquire.Until such time, if
any;•potential future clinical trial results;•theever, as we can generate substantial product revenues to support our costand timing of regulatory filings and/or approvals to commercialize any potential future product candidates and any related restrictions, limitations, and/or warnings in the label of an approved product candidate;•the effect of competing technological and market developments; and•the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights, and the cost of defending any other litigation claims.
While we have been able to fund our operations to date, we have no ongoing collaborations for the development and commercialization of any product candidates and no source of revenue, nor dostructure, we expect togenerate product revenue for the foreseeable future. We do not have any commitments for future external funding.Until we can generate a sufficient amount of product revenue, which we may never do, we will need tofinancefuture cashour losses from operations and capital funding needs throughpotential collaborative, partnering or other strategic arrangements, as well as through public or private equity offerings or debt financings ora combination of equity offerings, debt financings, government funding and other sources, including potentially collaborations, licenses and other similar arrangements. To theforegoing.extent we raise additional capital through the sale of debt or equity securities, the ownership interest of our stockholders will be or could be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise funds through collaborations, licenses and other similar arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us and/or may reduce the value of our common stock. If we are unable to raise additional fundswe will need to continue to reduce our expenditures in order to preserve our cash. Further cost-cutting measures that we may take may not besufficient to enable us to meet our cash requirements, and they may negatively affect our business and our ability to derive any value from our SARD program. In any event, in order to further the development of our SARD program, we will need to raise substantial additional capital. Our failure to do so would likely result in our determining to cease operations.To the extent that we raise additional fundsthroughpotential collaborations, partnering or other strategic arrangements, it may be necessary to relinquish rights to some of our technologies or product candidates and intellectual property rights thereof, or grant licenses on terms that are not favorable to us, any of which could result in our stockholders having little or no continuing interest in our SARD program and/or SARM assets as stockholders or otherwise. To the extent we raise additional funds by issuing equity securities, our stockholders may experience significant dilution, particularly given our currently-depressed stock price, and debt financing, if available, may involve restrictive covenants. For example, we completed substantially dilutive private placements of our common stock and warrants in March 2014, November 2014 and September 2017, in addition to a registered direct offering of our common stock that we completed in October 2016 and the sale of our common stock pursuant to the ATM Sales Agreement. Our stockholders will experience additional, perhaps substantial, dilution should we again raise additional funds by issuing equity securities. Any additionaldebt or equityfinancingfinancings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market our product candidates even if we would otherwise prefer to develop and market such product candidates by ourselves. There can be no assurance that weraise may contain terms that are not favorable to us or our stockholders. Our ability to raise additional funds and the terms upon which we arewill be able toraise such funds have been severely harmed by the failureobtain any sources ofthe ASTRID trial to meet its primary endpoint and the resulting significant uncertainty regarding our prospects to continue as a going concern. If we are unable to complete the proposed Merger, our ability to raise additional funds and the terms upon which we are able to raise such funds may also be adversely affected by the uncertainties regarding our financial condition, uncertainties with respect to the prospects for our early-stage SARD program, the sufficiency of our capital resources, potential future management turnover, and volatility and instability in the global financial markets. As a result of these and other factors, there is no guarantee that sufficient additional funding will be available to usfinancing on acceptable terms, or at all.Contractual Obligations and Commitments
AtThe following table summarizes our contractual obligations at December 31,2018, we had contractual obligations as follows:2019 (in thousands)::Payments Due by Period
Less than
1-3
3-5
More than
Total
1 Year
Years
Years
5 Years
Operating lease obligations (1)
$
207
$
166
$
41
$
—
$
—
Total
$
207
$
166
$
41
$
—
$
—
(1)
Our operating lease obligations relate to our corporate headquarters in San Diego, California. We lease 4,677 square feet of office space under an operating lease that expires in March 2021.
Payment Due by Period
(in thousands)Contractual Obligations(1) Total Less than
1 year1-3 years 4-5 years More than
5 yearsOperating lease obligations(2)
$ 162 $ 162 $ — $ — $ — the related qualifying costs are incurred and services are rendered, provided that the applicable conditions under the subaward agreement have been met.(1)This table does not include any royalty obligations under our SARM and SARDWe are party to a number of license agreements, pursuant to which we have payment obligations that are contingent upon future events such as our achievement of specified development, regulatory and commercial milestones and are required to make royalty payments in connection with
UTRF asthe sale of products developed under those agreements. As of December 31, 2019, we were unable to estimate the timingandor likelihood ofsuchachieving the milestones or making future product sales and, therefore, any related payments arenot known.excluded from the table above. See Note 4 to our consolidated financial statements included elsewhere in this Annual Report for a description of these agreements.We enter into contracts in the normal course of business with clinical trial sites and clinical supply manufacturers and with vendors for preclinical studies, research supplies and other services and products for operating purposes. These contracts generally provide for termination after a notice period, and, therefore, are cancelable contracts and are excluded from the table above.
Georgetown University (“Georgetown”)
In
additionMarch 2014, we entered into an Exclusive License Agreement (the “Georgetown License Agreement”) with Georgetown, pursuant to which we: (i) licensed the exclusive worldwide right to patents and technologies for the development and commercialization of certain product candidates targeting EWS-FLI1 as an anti-tumor therapy for therapeutic, diagnostics, or research tool purposes, (ii) are solely responsible for all development and commercialization activities and costs in our respective territories, and (iii) are also responsible for all costs related to theminimumfiling, prosecution and maintenance of the licensed patent rights.Under the terms of the Georgetown License Agreement, commencing in 2015, we: (i) shall pay and have paid an annual license maintenance fee of $10,000 until the first commercial sale occurs, (ii) are required to make up to $200,000 in aggregate milestone payments
due under our SARMupon the achievement of certain regulatory milestones, andSARD license agreements, we may(iii) will be required to pay low single digit royalties based on annual net product sales. We accounted for the licensed technology as an asset acquisition because it did not meet the definition of a business. All milestone payments under the Georgetown License Agreement will be recognized as research and development expense upon completion of the required events, as the triggering events are not considered to be probable until they are achieved. As of December 31, 2019, we had not triggered or made any milestone payments under the Georgetown License Agreement.The Georgetown License Agreement may be terminated by either party upon material breach or may be terminated by us as to one or more countries with 90 days written notice of termination. The term of the Georgetown License Agreement will continue until the expiration of the last valid claim within the patent rights covering the product. Georgetown may terminate the agreement in the event: (i) we fail to pay any amount and fail to cure such failure within 30 days after receipt of notice, (ii) we default in our obligation to obtain and maintain insurance and fail to remedy such breach within 60 days after receipt of notice, or (iii) we declare insolvency or bankruptcy. We may terminate the agreement at any time upon at least 60 days’ written notice.
In 2017, we entered into a research agreement with Georgetown for up to $150,000. For the years ended December 31, 2019 and 2018, we recorded research and development expenses of $27,000 and $53,000, respectively.
The University of Texas MD Anderson Cancer Center (“MD Anderson”)
In December 2014, we entered into a collaboration agreement (the “MD Anderson Collaboration”) with MD Anderson, which, as amended, provides for the conduct of preclinical and clinical research on TK216 in exchange for certain program payments. If MD Anderson successfully completes all the requirements of the MD Anderson Collaboration in full and the program is successfully commercialized, we will be required to pay aggregate milestone payments of $1.0 million based on net product sales. For the years ended December 31, 2019 and 2018, the recorded research and development expenses were not significant.
Agreements with the Regents of the University of California (the “Regents”)
In March 2016, we entered into a license agreement with the Regents, which was amended and restated in August 2018, for the development, manufacturing and distribution rights to naked antibodies, including cirmtuzumab and genetically engineered cellular therapy products, including CAR-T products that are covered by licensed patents for all human therapeutic, diagnostic and preventive applications in all indications. The Regents license agreement was amended on March 25, 2019 and May 15, 2019, to update the patents covered under the agreement.
The Regents License Agreement provides for the following: (i) in May 2016, an upfront license fee of $0.5 million was paid and 107,108 shares of our common stock were issued, (ii) $25,000 in annual license maintenance fees commencing in 2017, (iii) reimbursement of certain patent costs; (iv) certain development and regulatory milestones aggregating from $10.0 million to $12.5 million, on a per product basis, (v) certain worldwide sales milestones based on achievement of tiered revenue levels aggregating $75.0 million, (vi) low single-digit royalties, including potential future minimum annual royalties, on
anynet sales ofproduct ifeach target, and (vii) minimum diligence to advance licensed assets consisting of at least $1.0 million in development spend annually through 2021. Under the Regents License Agreement in 2019 and 2018, wereceive regulatory approvalrecorded $25,000 in annual license maintenance fees as research and development expense. As of December 31, 2019, we believe we have met our obligations under the Regents License Agreement.In July 2016, and as modified by the amended and restated Regents License Agreement in August 2018, we entered into a Research Agreement (the “Research Agreement”) with the Regents for further research on a ROR1 therapeutic development program. Under this five-year agreement, the Regents will have an aggregate budget of $3.6 million, with $125,000 payable quarterly. For the years ended December 31, 2019 and 2018, we recorded $0.5 million in research and development costs under this Research Agreement. Such costs are includable as part of our annual diligence obligations under the Regents License Agreement.
The Regents License Agreement will expire upon the later of the expiration date of the longest-lived patent rights or the 15th anniversary of the first commercial sale of a licensed product. The Regents may terminate the Regents License Agreement if: (i) a material breach by us is not cured within a reasonable time, (ii) we file a claim asserting the Regents licensed patent rights are invalid or unenforceable, and (iii) we file for bankruptcy. We may terminate the agreement at any time upon at least 60 days’ written notice.
University of Tennessee Research Foundation (“UTRF”)
In July 2007, we and UTRF entered into a consolidated, amended and restated license agreement (the “SARM License Agreement”), pursuant to which we were granted exclusive worldwide rights in all existing selective androgen receptor modulator (“SARM”) technologies owned or controlled by UTRF, including all improvements thereto, and exclusive rights to future SARM technology that may be developed by certain scientists at the University of Tennessee or subsequently licensed to UTRF under certain existing inter-institutional agreements with The Ohio State University. We recorded research and development expense under this agreement of $0.1 million and none for the years ended December 31, 2019 and 2018, respectively. On December 31, 2019, we provided UTRF notice of termination of the SARM License Agreement by and between us and UTRF, which termination will be effective three months following such notice. Following termination, we will no longer have the obligation to make further payments under the SARM License Agreement, and will no longer have any rights to develop or sublicense the SARM Technology.
In March 2015, we and UTRF also entered into a license agreement (the “SARD License Agreement”) pursuant to which we were granted exclusive worldwide rights in all existing selective androgen receptor degrader (“SARD”) technologies owned or controlled by UTRF, including
enobosarm, orall improvements thereto. Under the SARDproduct candidate and successfully market the product. Additionally, ifLicense Agreement, wesublicense rights under our SARM or SARD license agreements, we alsoare obligated to employ active, diligent efforts to conduct preclinical research and development activities for the SARD program to advance one or more lead compounds into clinical development. We are also obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and additional royalties on sublicense revenues, depending on the state of development of a clinical product candidate at the time it is sublicensed. We recorded research and development expense under this agreement of $0.4 million and none for the years ended December 31, 2019 and 2018, respectively.As of December 31, 2019, we believe we have met our obligations under each of the UTRF agreements.
Velos Biopharma Holdings, LLC (“VBH”) and VelosBio, Inc. (“VelosBio”) Spin-off Transactions
In November and December 2017, we formed VBH and made an in-kind tax-free distribution of 100% of our interest in VBH to our stockholders, option holders and warrant holders of record. On February 6, 2018, we licensed and assigned our rights to two preclinical product candidates, previously under the Regents License Agreement, to VBH. In consideration for the license, we: (i) received a promissory note receivable from VBH of $0.1 million, with an annual interest rate of 2.64% and a due date of 10 years, and (ii) made a partial assignment of our March 2016 Regents License Agreement. Pursuant to the partial assignment, VBH assumed certain obligations related to the licensed products under the Regents License Agreement as follows: (i) reimbursement of certain historical and future patent costs related to the licensed products, (ii) certain development and sales milestones for advancing licensed products targets, (iii) low single-digit royalties, including potential future minimum annual royalties, on net sales of each licensed product target are to be allocated between us and VBH, (iv) certain third-party agreements and related obligations specifically related to the licensed products, (v) minimum diligence obligations to advance licensed assets consisting of a minimum of $0.5 million in development spend annually through 2021, and (vi) obligations under the Research Agreement equal to $0.5 million annually commencing January 1, 2018. Due to the high uncertainty of the success of VBH ever repaying the note and associated interest, we provided a full valuation allowance for these amounts as of December 31, 2019 and 2018.
In December 2017, VelosBio was incorporated with VBH being its sole stockholder with VelosBio common shares only. On February 6, 2018, VBH sublicensed and assigned its intellectual property rights to its two preclinical product candidates to VelosBio. In consideration for the license, VelosBio agreed to use commercially reasonable efforts to develop the licensed products as well as the following payment obligations: (i) the assumption of each of the VBH assumed obligations under the partial assignment between us and VBH as outlined above, and (ii) certain tiered development milestone and royalty payments to VBH. In August 2018, we entered into the amended and restated Regents License Agreement and VelosBio entered into their own license agreement directly with the Regents. In February 2018, VelosBio secured substantially independent preferred stock financings for its programs and there is no common control overlap between the companies.
Also on February 6, 2018, we and VelosBio entered into: (i) an asset purchase agreement whereby VelosBio purchased our right, title and interest in our nominal assets related to the two preclinical product candidates and assumed our $0.2 million convertible note payable and related $16,000 of accrued interest which has been recorded as other income in our consolidated financial statements, and (ii) a transition services agreement whereby we agreed to provide VelosBio with certain transition services, which expired as of December 31, 2018, as follows: (i) access to certain common laboratory equipment at our lab facility, (ii) certain named employees were to devote up to 80% of their time supporting VelosBio related activities, (iii) cirmtuzumab manufacturing, process optimization and ancillary activities until VelosBio was able to establish their own, and (iv) agreement to cost share the purchase of certain antibody materials with VelosBio. Such services were to be provided at cost or cost plus. During 2018, we incurred $3.0 million of costs on behalf of VelosBio that were substantially reimbursed and recorded on a net basis within operating expenses. As of December 31, 2018, there are no ongoing rights or commitments under the asset purchase or transition services agreements. In February 2020, we entered into a sublicense agreement with VelosBio pursuant to which we granted certain sublicenses to VelosBio under the Selexis License Agreement.
SPH USA License Agreement
In November 2018, we entered into the SPH USA License Agreement with SPH USA for: (i) the territory of Greater China, and (ii) rights to manufacture, develop, market, distribute and sell all of our product candidates under the Georgetown License Agreement and the Regents License Agreement (exclusive to Greater China only). Under the SPH USA License Agreement, SPH USA is solely responsible for: (a) all preclinical and clinical development activities specific to obtaining regulatory approval in Greater China for such product candidates, (b) any third-party license milestone or royalty payments owed under the License Agreement and the Regents License Agreement, and (c) paying us a low single digit royalty on net sales in the territory.
Government Contracts, Grant Agreements and Incentive Programs
The CIRM Award
In August 2017, CIRM awarded an $18.3 million grant to researchers at UC San Diego, to advance our Phase 1/2 clinical trial evaluating cirmtuzumab in combination with ibrutinib for the treatment of patients with B-cell lymphoid malignancies, including MCL and CLL. We: (i) are conducting this study in collaboration with UC San Diego, (ii) estimate we will receive approximately $14.0 million in development milestones under research subaward agreements throughout the award project period, estimated to be from October 1, 2017, to March 31, 2022, (iii) are committed to certain co-funding requirements, (iv) received subaward payments of $6.2 million and $0.5 million for years ended December 31, 2019 and 2018, respectively, and (v) are required to provide UC San Diego progress and financial update reports throughout the award project period. The subaward does not bear a royalty payment commitment, nor is the subaward otherwise refundable. For the years ended December 31, 2019 and 2018, we recorded revenue of $2.4 million and $2.5 million, respectively. Related qualifying subaward costs during the years ended December 31, 2019 and 2018 were $5.4 million and $4.6 million, respectively. As of December 31, 2019, we believe we have met our obligations under the CIRM award and UC San Diego subawards.
In October 2017, CIRM awarded a $5.8 million grant to the researchers at UC San Diego to develop a novel anti-cancer stem cell targeted therapy for patients with advanced solid and hematological malignancies. In connection with such CIRM award, we agreed to provide up to $1.0 million in contingency funds if required during the grant period. We recorded no research and development expense under such CIRM award for the years ended December 31, 2019 and 2018.
Critical Accounting Policies
Our management’s discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with United States generally accepted accounting principles (“U.S. GAAP”). The preparation of the financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported expenses incurred during the reporting periods.
Our estimates are based on our historical trends and other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are described in more detail in Note 1, “Description of Business, Basis of Presentation and Summary of Significant Accounting Policies,” in the notes to our consolidated financial statements as of December 31, 2019 and 2018 and for each of the years ended December 31, 2019 and 2018, appearing elsewhere in this annual report. However, we believe that the following accounting policies are the most critical for fully understanding and evaluating our financial condition and results of operations.
Research and Development Expenses and Accruals
Research and development expenses consist of costs incurred for our own and for sponsored and collaborative research and development activities. Research and development costs are expensed as incurred and include manufacturing process development costs, manufacturing costs, costs associated with preclinical studies and clinical trials, regulatory and medical affairs activities, quality assurance activities, salaries and benefits, including stock-based compensation, fees paid to third-party consultants, license fees and overhead.
We have entered into various research and development contracts with research institutions, clinical research organizations, clinical manufacturing organizations and other companies. Payments for these activities are based on the terms of the individual agreements, which may differ from the pattern of costs incurred, and payments made in advance of performance are reflected in the consolidated balance sheets as prepaid expenses and other assets or accrued liabilities. We record accruals for estimated costs incurred for ongoing research and development activities. When evaluating the adequacy of the accrued liabilities, we analyze progress of the services, including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates may be made in determining the prepaid or accrued balances at the end of any
licensing fee or milestone paymentsreporting period. Actual results could differ from our estimates.Valuation of Warrants to Purchase Convertible Preferred Stock
Prior to the Merger, we
may receiveclassified outstanding warrants to purchase shares of our Series B-2 convertible preferred stock as a liability on our consolidated balance sheets as these warrants were free-standing financial instruments exercisable into contingently redeemable shares. The warrants were initially recorded at fair value on the date of grant, and were subsequently remeasured to fair value at each balance sheet date while the instrument was outstanding. Changes in the fair value of these warrants were recognized as a component of other income (expense) in our consolidated statements of operations. Upon the completion of the Merger, the Series B-2 warrants were amended such that they were converted into warrants to purchase our common stock. As amended, warrant liability accounting is no longer required and the fair value of the warrant liability has been reclassified into stockholders’ equity.Revenue Recognition
We currently generate revenue from a
sublicensee.(2)Our operating lease obligations consistresearch subaward agreement with UC San Diego, which provides us with payments for certain types ofpayments relating toexpenditures in return for research and development activities over alease for office space at 175 Toyota Plaza, Memphis, Tennessee,contractually defined period. Revenue from such subaward is recognized in the period during whichexpires on April 30, 2019.
Off-Balance Sheet Arrangements
WeDuring the periods presented, we did not have,not engaged innor do we currently have, any off-balance sheet arrangementsincludingas defined under theuseapplicable rules ofstandard finance, special purpose entities or variable interest entities.the SEC.Item 7A.
We are a smaller reporting company, as defined by Rule 12b-2 of the Exchange Act, and are not required to provide the information required under this item.
The Consolidated Financial Statements and supplementary data of Oncternal Therapeutics, Inc. required by this Item are described in Item 15 of this Annual Report on Form 10-K and are presented beginning on page F-1.
Report of Independent Registered Public Accounting Firm
Shareholders and Board of Directors
Oncternal Therapeutics, Inc.
San Diego, California
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Oncternal Therapeutics, Inc. and subsidiaries (the “Company”) as of December 31, 2019 and 2018, the related consolidated statements of operations, convertible preferred stock and stockholders’ equity (deficit), and cash flows for the years then ended, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2019 and 2018, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company's internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control – Integrated Framework
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK(2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) and our report dated March 16, 2020 expressed an unqualified opinion thereon.Change in Accounting Principle
As discussed in Note 1 to the consolidated financial statements, the Company has changed its method for accounting for leases in 2019 due to the adoption of Accounting Standards Codification Topic 842, Leases.
Going Concern Uncertainty
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As described in Note 1 to the consolidated financial statements, the Company has suffered recurring losses and negative cash flows from operations that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ BDO USA, LLP
We have served as the Company's auditor since 2016.
San Diego, California
March 16, 2020
Our exposureConsolidated Balance Sheets(in thousands, except par value)
December 31,
2019
2018
Assets
Current assets:
Cash and cash equivalents
$
20,051
$
20,645
Prepaid and other assets
736
565
Total current assets
20,787
21,210
Right-of-use asset
190
—
Other assets
767
752
Total assets
$
21,744
$
21,962
Liabilities, Convertible Preferred Stock and Stockholders’ Equity
(Deficit)
Current liabilities:
Accounts payable
$
871
$
3,440
Accrued liabilities
2,731
891
Deferred grant revenue
3,640
—
Current portion of lease liability
99
—
Total current liabilities
7,341
4,331
Preferred stock warrant liability
—
674
Lease liability
91
—
Commitments and contingencies (Notes 3 and 5)
Convertible preferred stock, $0.0001 par value; authorized shares - none and
130,100 at December 31, 2019 and 2018, respectively; issued and
outstanding – none and 8,148 at December 31, 2019 and 2018, respectively;
liquidation preference of none and $48,954 at December 31, 2019 and
2018, respectively
—
46,588
Stockholders’ equity (deficit):
Preferred stock, $0.001 par value, authorized shares – 5,000 and none at
December 31, 2019 and 2018, respectively; issued and outstanding
shares – none
—
—
Common stock, $0.001 par value; authorized shares – 60,000 and 200,000
at December 31, 2019 and 2018, respectively; issued and outstanding
shares – 15,387 and 3,762 at December 31, 2019 and 2018, respectively
15
5
Additional paid-in capital
79,869
1,748
Accumulated deficit
(65,572
)
(31,384
)
Total stockholders’ equity (deficit)
14,312
(29,631
)
Total liabilities and stockholders’ equity (deficit)
$
21,744
$
21,962
See accompanying notes.
Consolidated Statements of Operations
(thousands, except per share data)
Years Ended December 31,
2019
2018
Grant revenue
$
2,425
$
2,521
Operating expenses:
Research and development
10,159
8,287
In-process research and development
18,088
—
General and administrative
7,286
1,820
Total operating expenses
35,533
10,107
Loss from operations
(33,108
)
(7,586
)
Other income (expense):
Change in fair value of warrant liability
(1,268
)
713
Other income
—
216
Interest expense
—
(1
)
Interest income
188
79
Total other income (expense)
(1,080
)
1,007
Net loss
$
(34,188
)
$
(6,579
)
Net loss per share, basic and diluted
$
(3.31
)
$
(1.83
)
Weighted-average shares outstanding, basic and diluted
10,329
3,591
See accompanying notes.
Consolidated Statements of Cash Flows
(in thousands)
Years Ended December 31,
2019
2018
Cash flows from operating activities
Net loss
$
(34,188
)
$
(6,579
)
Adjustments to reconcile net loss to net cash used in operating activities:
In-process research and development
18,088
—
Noncash other income
—
(216
)
Stock-based compensation
507
180
Noncash interest expense
—
1
Noncash compensation expense
—
10
Change in fair value of preferred stock warrants liability
1,268
(713
)
Noncash lease expense
92
—
Changes in operating assets and liabilities:
Prepaid and other assets
(44
)
(436
)
Accounts payable
(4,762
)
2,242
Accrued liabilities
(1,255
)
(4
)
Change in operation lease liability
(92
)
—
Deferred grant revenue
3,640
(1,902
)
Net cash used in operating activities
(16,746
)
(7,417
)
Cash flows from investing activities
Cash acquired in connection with the Merger
18,292
—
Acquisition related costs paid
(2,155
)
—
Net cash provided by investing activities
16,137
—
Cash flows from financing activities
Proceeds from exercise of stock options
14
1
Proceeds from exercise of common stock warrants
1
—
Proceeds from the issuance of convertible preferred stock, net
—
17,873
Net cash provided by financing activities
15
17,874
Net (decrease) increase in cash and cash equivalents
(594
)
10,457
Cash and cash equivalents at beginning of period
20,645
10,188
Cash and cash equivalents at end of period
$
20,051
$
20,645
Supplemental disclosure of non-cash investing and financing activities:
Conversion of convertible preferred stock into common stock
$
46,588
$
—
Issuance of common stock to GTx stockholders
$
29,049
$
—
Reclassification of preferred stock warrants liability to additional paid-in
capital
$
1,942
$
—
Net liabilities assumed in Merger
$
5,177
$
—
See accompanying notes.
Consolidated Statements of Convertible Preferred Stock and Stockholders' Equity (Deficit)
(in thousands)
Convertible Preferred
Stock
Common Stock
Additional
Paid-In
Accumulated
Total
Stockholders’
Shares
Amount
Shares
Amount
Capital
Deficit
Equity (Deficit)
Balance at December 31, 2017
5,653
$
28,715
3,745
$
5
$
1,522
$
(24,805
)
$
(23,278
)
Collection of stock subscription receivable
—
1,100
—
—
—
—
—
Issuance of Series C convertible preferred stock for cash,
net of issuance costs
2,495
16,773
—
—
—
—
—
Exercise of stock options for cash
—
—
2
—
1
—
1
Vesting related to repurchase liability
—
—
—
—
35
—
35
Issuance of restricted common shares
—
—
15
—
10
—
10
Stock-based compensation
—
—
—
—
180
—
180
Net loss
—
—
—
—
—
(6,579
)
(6,579
)
Balance at December 31, 2018
8,148
46,588
3,762
5
1,748
(31,384
)
(29,631
)
Exercise of stock options for cash
—
—
19
—
14
—
14
Exercise of warrants for cash
—
—
—
—
1
—
1
Vesting related to repurchase liability
—
—
—
—
30
—
30
Issuance of common stock to former stockholders of GTx
upon Merger
—
—
3,458
2
29,047
—
29,049
Conversion of convertible preferred stock into common
stock upon Merger
(8,148
)
(46,588
)
8,148
8
46,580
—
46,588
Reclassification of convertible preferred stock warrant
liability
—
—
—
—
1,942
—
1,942
Stock-based compensation
—
—
—
—
507
—
507
Net loss
—
—
—
—
—
(34,188
)
(34,188
)
Balance at December 31, 2019
—
$
—
15,387
$
15
$
79,869
$
(65,572
)
$
14,312
See accompanying notes.
Notes to Consolidated Financial Statements
1.
Description of Business, Basisof Presentation and Summary of Significant Accounting Policies
Description of Business
Oncternal Therapeutics, Inc. (the “Company,” “Oncternal,” or the “combined company”), formerly known as GTx, Inc., was incorporated in Tennessee in September 1997 and reincorporated in Delaware in 2003 and is based in San Diego, California. The Company is a clinical-stage biopharmaceutical company focused on the development of novel oncology therapies for the treatment of cancers with critical unmet medical need. The Company’s clinical pipeline consists of its lead program, cirmtuzumab, a humanized monoclonal antibody that binds to ROR1 (Receptor-tyrosine kinase-like Orphan Receptor 1), and TK216, a small molecule inhibiting the biological activity of ETS-family transcription factor oncoproteins. The Company is also developing a CAR-T (chimeric antigen receptor T-cells) product candidate that targets ROR1.
Merger
On June 7, 2019, the Company, then operating as GTx, Inc. (“GTx”), completed its Agreement and Plan of Merger and Reorganization, as amended (the “Merger Agreement”), with privately-held Oncternal Therapeutics, Inc. (“Private Oncternal”) and Grizzly Merger Sub, Inc., a wholly-owned subsidiary of the Company (“Merger Sub”), dated March 6, 2019. Under the Merger Agreement, Merger Sub merged with and into Private Oncternal, with Private Oncternal surviving as a wholly-owned subsidiary of the Company (the “Merger”). GTx changed its name to Oncternal Therapeutics, Inc., and Private Oncternal, which remains as a wholly-owned subsidiary of the Company, changed its name to Oncternal Oncology, Inc. On June 10, 2019, the combined company’s common stock began trading on The Nasdaq Capital Market under the ticker symbol “ONCT.”
Except as otherwise indicated, references herein to “Oncternal,” “the Company,” and the “combined company,” refer to Oncternal Therapeutics, Inc. on a post-Merger basis, and the term “Private Oncternal” refers to the business of privately-held Oncternal Therapeutics, Inc., prior to completion of the Merger. References to GTx refer to GTx, Inc. prior to completion of the Merger.
Pursuant to the terms of the Merger Agreement, each outstanding share of Private Oncternal common stock outstanding immediately prior to the closing of the Merger was converted into approximately 0.073386 shares of Company common stock (the “Exchange Ratio”), after taking into account the Reverse Stock Split, as defined below. Immediately prior to the closing of the Merger, all shares of Private Oncternal preferred stock then outstanding were exchanged into shares of common stock of Private Oncternal. In addition, all outstanding options exercisable for common stock of Private Oncternal and warrants exercisable for convertible preferred stock of Private Oncternal became options and warrants exercisable for the same number of shares of common stock of the Company multiplied by the Exchange Ratio. Immediately following the Merger, stockholders of Private Oncternal owned approximately 77.5% of the outstanding common stock of the combined company.
The transaction was accounted for as a reverse asset acquisition in accordance with generally accepted accounting principles in the United States of America (“GAAP”). Under this method of accounting, Private Oncternal was deemed to be the accounting acquirer for financial reporting purposes. This determination was primarily based on the facts that, immediately following the Merger: (i) Private Oncternal’s stockholders owned a substantial majority of the voting rights in the combined company, (ii) Private Oncternal designated a majority of the members of the initial board of directors of the combined company, and (iii) Private Oncternal’s senior management holds all key positions in the senior management of the combined company. As a result, as of the closing date of the Merger, the net assets of the Company were recorded at their acquisition-date relative fair values in the consolidated financial statements of the Company and the reported operating results prior to the Merger will be those of Private Oncternal.
Reverse Stock Split and Exchange Ratio
On June 7, 2019, in connection with, and prior to the completion of, the Merger, GTx effected a one-for-seven reverse stock split of its then outstanding common stock (the “Reverse Stock Split”). The par value and the authorized shares of the common stock were not adjusted as a result of the Reverse Stock Split. All of the Company’s issued and outstanding common stock have been retroactively adjusted to reflect this Reverse Stock Split for all periods presented. All issued and outstanding Private Oncternal common stock, preferred stock, options and warrants prior to the effective date of the Merger have been retroactively adjusted to reflect the Exchange Ratio for all periods presented.
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries, Oncternal Oncology, Inc. and Oncternal, Inc. All intercompany accounts and transactions have been eliminated in the preparation of the consolidated financial statements.
Liquidity and Going Concern
From its inception through December 31, 2019, the Company has devoted substantially all of its efforts to organizational activities including raising capital, building infrastructure, acquiring assets, developing intellectual property, and conducting preclinical studies, clinical trials and product development activities. The Company has a limited operating history and the sales and income potential of the Company’s business and market
risk for changes in interest rates relatesare unproven. Since inception, the Company has experienced recurring net losses and negative cash flows from operating activities and expects toourcontinue to incur losses into the foreseeable future. At December 31, 2019, the Company had an accumulated deficit of $65.6 million and had cash and cash equivalents of $20.1 million. The Company believes that its existing cash and cash equivalents will be sufficient to fund its operations into the third quarter of 2020. The Company will need to continue to raise a substantial amount of funds until it is able to generate revenues to fund its development activities and operations. The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern, which contemplates the realization of assets and settlement of liabilities in the normal course of business. However, based ondepositthe Company’s current working capital, anticipated operating expenses and net losses and the uncertainties surrounding its ability to raise additional capital as needed, as discussed below, the Company believes that there is substantial doubt about its ability to continue as a going concern for one year after the date these consolidated financial statements are issued.The Company plans to continue to fund its losses from operations and capital funding needs through a combination of equity offerings, debt financings, government funding, or other sources, including, potentially, collaborations, licenses and other similar arrangements. There can be no assurance that the Company will be able to obtain any sources of financing on acceptable terms, or at all. To the extent that the Company can raise additional funds by issuing equity securities, the Company’s stockholders may experience significant dilution. Any debt financing, if available, may involve restrictive covenants that impact the Company’s ability to conduct its business.
Use of Estimates
The Company’sconsolidatedfinancialstatements are prepared inaccordance with GAAP. The preparation oftheCompany’sconsolidatedfinancialstatementsandaccompanyingnotes requires it to make estimates and assumptions that impact the reported amounts of assets, liabilities, revenues and expenses and the disclosure of contingent assets and liabilities. Significant estimates consist of those used to determine the fair value of the Company’s preferred stock, preferred stock warrant liability and stock-based awards, and those used to determine grant revenue and accruals for research and development costs. Although these estimates are based on the Company’sknowledgeof current eventsandactions it may undertake in the future, actual results may ultimately materially differ from these estimates andassumptions.
Cash and Cash Equivalents
The Company considers all highly liquid investments with maturities of three months or less when purchased to be cash equivalents. Cash and cash equivalents include cash in readily available checking accounts and money market
fundsaccounts.Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash and cash equivalents. The Company maintains deposits in federally insured financial institutions in excess of federally insured limits. The Company has not experienced any losses in such accounts and believes it is not exposed to significant risk on its cash balances due to the financial position of the depository institution in which those deposits are held. Additionally, the Company established guidelines regarding approved investments and maturities of investments, which are designed to maintain safety and liquidity.
Patent Costs
Costs related to filing and pursuing patent applications are recorded as general and administrative expense and expensed as incurred since recoverability of such expenditures is uncertain.
Research and Development Expenses and Accruals
Research and development expenses consist of costs incurred for the Company’s own and for sponsored and collaborative research and development activities. Research and development costs are expensed as incurred and include manufacturing process development costs, manufacturing costs, costs associated with preclinical studies and clinical trials, regulatory and medical affairs activities, quality assurance activities, salaries and benefits, including stock-based compensation, fees paid to third-party consultants, license fees and overhead.
The Company has entered into various research and development contracts with research institutions, clinical research organizations, clinical manufacturing organizations and other companies. Payments for these activities are based on the terms of the individual agreements, which may differ from the pattern of costs incurred, and payments made in
Federal Deposit Insurance Corporation insured certificatesadvance ofdeposit.performance are reflected in the accompanying consolidated balance sheets as prepaid and other assets or accrued liabilities. Theprimary objectiveCompany records accruals for estimated costs incurred for ongoing research and development activities. When evaluating the adequacy ofour cash investment activities is to preserve principal whilethe accrued liabilities, the Company analyzes progress of the services, including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates may be made in determining the prepaid or accrued balances at thesame time maximizingend of any reporting period. Actual results could differ from theincome we receive from our invested cash without significantly increasing riskCompany’s estimates.Preferred Stock Warrant Liability
Prior to the Merger, Private Oncternal had outstanding freestanding warrants to purchase shares of
loss. We do not use derivative financial instruments in our investment portfolio. The effect ofits Series B-2 convertible preferred stock (the “Series B-2 warrants”). Because the underlying Series B-2 convertible preferred stock was classified as temporary equity, the Series B-2 warrants were classified as ahypothetical decrease of ten percentliability in theaverage yield earnedaccompanying consolidated balance sheets. Private Oncternal adjusted the carrying value of such Series B-2 warrants to their estimated fair value at each reporting date, with any related increases or decreases in the fair value recorded as an increase or decrease to other income (expense) in the consolidated statements of operations. Upon the completion of the Merger, the Series B-2 warrants were amended such that they were converted into warrants to purchase the Company’s common stock. As amended, warrant liability accounting is no longer required and the fair value of the warrant liability has been reclassified into stockholders’ equity.The accounting guidance defines fair value, establishes a consistent framework for measuring fair value and expands disclosure for each major asset and liability category measured at fair value on
our cash equivalents and short-term investmentseither a recurring or non-recurring basis. Fair value is defined as an exit price, representing the amount that wouldhave resultedbe received to sell an asset or paid to transfer a liability in animmaterial decreaseorderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use inour interest incomepricing an asset or liability. As a basis for considering such assumptions, the accounting guidance establishes a three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value as follows:Level 1: Observable inputs such as quoted prices in active markets.
Level 2: Inputs, other than the quoted prices in active markets that are observable either directly or indirectly.
Level 3: Unobservable inputs in which there is little or no market data, which require the reporting entity to develop its own assumptions.
The carrying amounts of the Company’s current financial assets and liabilities are considered to be representative of their respective fair values because of the short-term nature of those instruments. The Company has no financial assets or liabilities, other than the preferred stock warrant liability described below, measured at fair value on a recurring basis. No transfers between levels have occurred during the periods presented.
Liabilities measured at fair value on a recurring basis are as follows (in thousands):
Fair Value Measurements at Reporting Date Using
Quoted Prices in
Active Markets
for Identical
Assets
Significant
Other
Observable
Inputs
Significant
Unobservable
Inputs
Total
(Level 1)
(Level 2)
(Level 3)
At December 31, 2018
Preferred stock warrant liability
$
674
$
—
$
—
$
674
As of December 31, 2018, the preferred stock warrant liability was recorded at fair value utilizing the Black-Scholes option pricing model using significant unobservable inputs consistent with the inputs used for the Company’s stock-based compensation expense adjusted for the preferred stock warrants’ expected term and the fair value of the underlying preferred stock.
The Company calculated the final remeasurement of the preferred stock warrant liability on June 7, 2019, the Merger closing date, using the closing price of GTx’s common stock on that date to determine the fair value of the warrants, and recorded a $1.3 million change in the fair value of the preferred stock warrant liability for the year ended December 31,
2018.2019.The assumptions used in the Black-Scholes option pricing model to determine the fair value of the preferred stock warrant liability as of December 31, 2018 were as follows:
Fair value of underlying preferred stock
$
0.29
Risk-free interest rate
2.4% — 2.7%
Expected volatility
75.3% — 76.4%
Expected term (in years)
3.7 — 4.0
Expected dividend yield
—%
The following table provides a reconciliation of the preferred stock warrant liability measured at fair value using Level 3 significant unobservable inputs (in thousands):
Preferred
Stock
Warrant
Liability
Balance at December 31, 2018
$
674
Change in fair value
1,268
Reclassification of preferred stock warrant
liability to equity
(1,942
)
Balance at December 31, 2019
$
—
Revenue Recognition
The Company currently generates revenue from the California Institute for Regenerative Medicine pursuant to a research subaward agreement (see Note 4), which provides the Company with payments in return for certain research and development activities over a contractually defined period. Revenue from such subaward is recognized in the period during which the related qualifying services are rendered and costs are incurred, provided that the applicable conditions under the subaward agreement have been met.
The subaward agreement is on a best-effort basis and does not require scientific achievement as a performance obligation. All fees received under the agreement are non-refundable. The costs associated with the agreement are expensed as incurred and reflected as a component of research and development expense in the accompanying consolidated statements of operations.
Funds received from the subaward agreement are recorded as revenue as the Company is the principal participant in the arrangement because the activities under the subaward are part of the Company’s development programs. In
addition, wethose instances where the Company first receives consideration in advance of providing underlying services, the Company classifies such consideration as deferred revenue until (or as) the Company provides the underlying services. In those instances where the Company first provides the underlying services prior to its receipt of consideration, the Company records a grant receivable. At December 31, 2019, the Company had deferred grant revenue of $3.6 million, and at December 31, 2018, the Company had a grant receivable of $0.1 million.Stock-Based Compensation
Stock-based compensation expense represents the fair value of equity awards, on the grant date, recognized in the period using the Black- Scholes option pricing model. The Company recognizes expense for awards with graded vested schedules over the requisite service period of the awards (usually the vesting period) on a straight-line basis. For equity awards for which vesting is subject to performance-based milestones, the expense is recorded over the remaining service period after the point when the achievement of the milestone is probable or the performance condition has been achieved.
Income Taxes
The Company accounts for income taxes under the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have
exposure to fluctuationsbeen included incertain foreign currenciesthe financial statements. Under this method, deferred tax assets and liabilities are determined on the basis of the differences between the financial statements and tax basis of assets and liabilities using enacted tax rates incountrieseffect for the year in whichwe conduct clinical trials. Mostthe differences are expected to reverse. The effect ofour foreign expenses incurred were associated with initiatinga change in tax rates on deferred tax assets and liabilities is recognized in income in the period that includes the enactment date.The Company recognizes net deferred tax assets to the extent that the Company believes these assets are more likely than not to be realized. In making such a determination, management considers all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax-planning strategies, and results of recent operations. If management determines that the Company would be able to realize its deferred tax assets in the future in excess of their net recorded amount, management would make an adjustment to the deferred tax asset valuation allowance, which would reduce the provision for income taxes.
Segment Reporting
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker in making decisions regarding resource allocation and assessing performance. The Company views its operations and manages its business in one operating segment in the United States.
Net Loss Per Share
Basic net loss per share is computed by dividing the net loss by the weighted-average number of common shares outstanding for the period, without consideration for potentially dilutive securities and adjusted for the weighted-average number of common shares outstanding that are subject to repurchase. The Company has excluded weighted-average shares subject to repurchase of 56,000 shares and 164,000 shares from the weighted-average number of common shares outstanding for the years ended December 31, 2019 and 2018, respectively. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of shares of common stock and dilutive common stock equivalents outstanding for the period determined using the treasury-stock and if-converted methods. For all periods presented, there is no difference in the number of shares used to calculate basic and diluted shares outstanding as inclusion of the potentially dilutive securities would be antidilutive.
Potentially dilutive securities not included in the calculation of diluted net loss per share, because to do so would be anti-dilutive, are as follows (in common stock equivalent shares; in thousands):
December 31,
2019
2018
Redeemable convertible preferred stock
—
8,148
Warrants to purchase convertible preferred stock
—
372
Warrants to purchase common stock
841
—
Common stock options
1,958
504
Common stock subject to repurchase
35
100
2,834
9,124
Recently Issued Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (“FASB”) or
conducting clinical trials for enobosarm at clinical trial sites in Europe. Consequently, changes in exchange rates could result inother standard setting bodies that are adopted by the Company as of the specified effective date. Unless otherwise discussed, the Company believes that the impact of recently issued standards that are not yet effective will not have a materialexchange losses and could unpredictably, materially and adversely affect ourimpact on its consolidated financial position or results of operations upon adoption.In August 2018, the FASB issued Accounting Standards Update (“ASU”) 2018-13, Fair Value Measurement: Disclosure Framework – Changes to the Disclosure Requirements for Fair Value Measurement, which modifies the disclosure requirements for fair value measurements. The amendments relate to disclosures regarding unrealized gains and
cash flows. A hypothetical 10% increaselosses, the range and weighted average of significant unobservable inputs used to develop Level 3 fair value measurements, and the narrative description of measurement uncertainty and are to be applied prospectively for only the most recent interim ordecreaseannual period presented inforeignthe initial fiscal year of adoption. All other amendments should be applied retrospectively to all periods presented upon their effective date. The amendments are effective for fiscal years beginning after December 15, 2019, and interim periods within those fiscal years, and early adoption is permitted. The Company expects no impact on the consolidated financial statements from the adoption of this standard effective January 1, 2020.Recently Adopted Accounting Pronouncements
In February 2016, the FASB issued ASU 2016-02, Leases, which, for operating leases, requires a lessee to recognize a right-of-use asset and a lease liability, initially measured at the present value of the lease payments, in its balance sheet. The standard also requires a lessee to recognize a single lease cost, calculated so that the cost of the lease is allocated over the lease term, generally on a straight-line basis. The Company adopted this standard on January 1, 2019 using the modified retrospective approach. As the Company has elected the practical expedient for short-term leases, the adoption of this standard had no impact on the consolidated financial statements on the date of adoption as the Company’s only lease was on a month-to-month basis for a contract period of less than one year and expired in May 2019. Subsequent to its adoption, the Company entered into a new office lease agreement and has applied the provisions of this guidance (See Note 3).
2.
Balance Sheet Details
Accrued liabilities consist of the following (in thousands):
December 31,
2019
2018
Research and development
$
1,206
$
720
Legal fees
424
20
Unvested share liability
24
54
Compensation
825
85
Other
252
12
$
2,731
$
891
3.
Commitments, Contingencies and Related Party Transactions
Lease
Rent expense was $0.1 million and $12,000 for the years ended December 31, 2019 and 2018, respectively. On May 22, 2019, the Company entered into an office sublease agreement for 4,677 square feet in San Diego, California (“San Diego Lease”) which expires on March 31, 2021. Base rent is approximately $166,000 annually and the monthly rent expense is being recognized on a straight-line basis over the lease term.
The San Diego Lease is included in the accompanying consolidated balance sheet at the present value of the lease payments. As the San Diego Lease does not have an implicit interest rate, the present value reflects a 10.0% discount rate which is the estimated rate of interest that the Company would have to pay in order to borrow an amount equal to the lease payments on a collateralized basis over a similar term and in a similar economic environment. The Company recognized a net operating lease right-of-use asset and an aggregate lease liability of $0.2 million as of December 31, 2019, in the accompanying consolidated balance sheet. The weighted average remaining lease term was 1.25 years.
Maturities of lease liabilities due under this lease agreement as of December 31, 2019, are as follows (in thousands):
Maturity of lease liabilities
Operating
Leases
2020
$
166
2021
41
Total lease payments
207
Less imputed interest
(17
)
Total operating lease liabilities
190
Less current portion of lease liability
(99
)
Lease liability
$
91
In January 2019, the Company engaged Newfront Insurance as its primary insurance broker. The son of Richard Vincent, the Company’s Chief Financial Officer, acted as the Company’s agent at Newfront Insurance. The Company paid total related policy premiums of approximately $1.2 million during the year ending December 31, 2019, for which Mr. Vincent’s son received a commission of approximately $0.1 million.
In September 2019, the Company entered into a consulting agreement with Robert J. Wills. Ph.D., a member of the Company’s Board of Directors, whereby Dr. Wills will provide services related to the potential out-licensing or sale of the SARM and/or SARD technology. The Company recorded approximately $3,500 in related services during the year ended December 31, 2019.
Effective in September 2019, the Company and Shanghai Pharmaceutical (USA) Inc. (“SPH USA”) entered into a Materials Supply and Services Agreement (“SPH USA Services Agreement”), pursuant to which the Company and SPH USA will execute various statements of work for the transfer to SPH USA of key reagents and other materials, and for the supply of certain services by the Company to SPH USA, as contemplated under and in furtherance of the License and Development Agreement between the Company and SPH USA effective as of November 2018. During the year ended December 31, 2019, the Company recorded amounts receivable from SPH USA related to statements of work totaling $0.2 million. See Notes 4 and 6.
Litigation Related to the Merger
Between April 10 and May 1, 2019, three putative class action lawsuits and one individual lawsuit were filed in the U.S. District Court for the District of Delaware: Wheby v. GTx, Inc. et al., Miller v. GTx, Inc. et al., Tabb v. GTx, Inc. et al., and Living Seas LLC v. GTx, Inc. et al. (collectively, the “Delaware Actions”). On April 11 and 23, 2019, two putative class actions were filed in the U.S. District Court for the Southern District of New York: Kopanic v. GTx, Inc. et al. and Cooper v. GTx, Inc. et al. (collectively, the “New York Actions” and, together with the Delaware Actions, the “Actions”). The Actions name as defendants the Company and its former board of directors, and, in the case of the Wheby and Miller actions, Private Oncternal and Merger Sub. The Actions allege that defendants violated Sections 14(a) and 20(a) of the Exchange Act, as well as Rule 14a-9 promulgated thereunder, in connection with the company's filing of the Registration Statement in connection with the Merger. The Delaware Actions have now been voluntarily dismissed with prejudice: the Wheby action on June 12, 2019; the Miller action on July 15, 2019; the Living Seas action on June 26, 2019; and the Tabb action on October 21, 2019. On September 16, 2019, Plaintiffs in the New York Actions filed an amended complaint, alleging violations of Sections 14(a) and 20(a) of the Exchange Act related to the value GTx’s stockholders received in the Merger. The complaint seeks damages and other unspecified relief. On January 10, 2020, the defendants filed their motion to dismiss the amended complaint, on January 31, 2020, the plaintiffs filed their opposition to defendants’ motion to dismiss, and on February 14, 2020, the defendants filed a reply in support of their motion to dismiss. The defendants’ motion to dismiss is pending. The Company believes that the New York Actions are without merit and intends to vigorously defend these actions. The Company cannot predict the outcome of or estimate the possible loss or range of loss from any of these matters.
Zappia vs. GTx Incorporated
On October 15, 2019, Joseph Zappia and Karen Zappia filed a lawsuit against us in the U.S. District Court for the District of Delaware. The complaint alleges that our former management (prior to the Merger) engaged in illegal insider trading and false, manipulative and deceptive practices in violation of Sections 10(b) (and Rule 10b-5 promulgated thereunder), with respect to the timing of the disclosure of failed clinical trial results of GTx’s enobosarm product candidate in September 2018. The plaintiffs seek damages, interest, costs, attorneys' fees. We believe that this lawsuit is without merit and intend to vigorously defend this matter. We cannot predict the outcome of or estimate the possible loss or range of loss from this matter.
Georgetown University (“Georgetown”)
In March 2014, the Company entered into an Exclusive License Agreement (the “Georgetown License Agreement”) with Georgetown, pursuant to which the Company: (i) licensed the exclusive worldwide right to patents and technologies for the development and commercialization of certain product candidates targeting EWS-FLI1 as an anti-tumor therapy for therapeutic, diagnostics, or research tool purposes, (ii) is solely responsible for all development and commercialization activities and costs, and (iii) is responsible for all costs related to the filing, prosecution and maintenance of the licensed patent rights.
Under the terms of the Georgetown License Agreement, commencing in 2015, the Company: (i) shall pay and has paid an annual license maintenance fee of $10,000 until the first commercial sale occurs, (ii) is required to make up to $0.2 million in aggregate milestone payments upon the achievement of certain regulatory milestones, and (iii) will be required to pay low single digit royalties based on annual net product sales. The Company accounted for the licensed technology as an asset acquisition because it did not meet the definition of a business. All milestone payments under the Georgetown License Agreement will be recognized as research and development expense upon completion of the required events, as the triggering events are not considered to be probable until they are achieved. As of December 31, 2019, the Company had not triggered or made any milestone payments under the Georgetown License Agreement.
The Georgetown License Agreement may be terminated by either party upon material breach or may be terminated by the Company as to one or more countries with 90 days written notice of termination. The term of the Georgetown License Agreement will continue until the expiration of the last valid claim within the patent rights covering the product. Georgetown may terminate the agreement in the event: (i) the Company fails to pay any amount and fails to cure such failure within 30 days after receipt of notice, (ii) the Company defaults in its obligation to obtain and maintain insurance and fails to remedy such breach within 60 days after receipt of notice, or (iii) the Company declares insolvency or bankruptcy. The Company may terminate the Georgetown License Agreement at any time upon at least 60 days’ written notice.
In 2017, the Company entered into a research agreement with Georgetown for up to $150,000. The Company recorded research and development expense under this agreement of $27,000 and $53,000 for the years ended December 31, 2019 and 2018, respectively.
The University of Texas MD Anderson Cancer Center (“MD Anderson”)
In December 2014, the Company entered into a collaboration agreement (as amended, the “Collaboration”) with MD Anderson, which provides for the conduct of preclinical and clinical research for TK216 in exchange
rates would resultfor certain program payments. If MD Anderson successfully completes all the requirements of the Collaboration in full and the program is successfully commercialized, the Company will be required to pay aggregate milestone payments of $1.0 million based on net product sales. The amounts recorded as research and development expense for the years ended December 31, 2019 and 2018 were not significant.Agreements with the Regents of the University of California (the “Regents”)
In March 2016, and as amended and restated in August 2018 in connection with the spin-off transactions described below, the Company entered into a license agreement (as amended, the “Regents License Agreement”) for the development, manufacturing and distribution rights related to the development and commercialization of ROR1 related naked antibodies, antibody fragments or synthetic antibodies, and genetically engineered cellular therapy. The Regents License Agreement was amended on March 25, 2019 and May 15, 2019, to update the patents covered under the agreement. The Regents License Agreement provides for the following: (i) in May 2016, an
immaterialupfront license fee of $0.5 million was paid and 107,108 shares of common stock were issued, (ii) $25,000 in annual license maintenance fees commencing in 2017, (iii) reimbursement of certain annual patent costs, (iv) certain development and regulatory milestones aggregating from $10.0 million to $12.5 million, on a per product basis, (v) certain worldwide sales milestones based on achievement of tiered revenue levels aggregating $75.0 million, (vi) low single-digit royalties, including potential future minimum annual royalties, on net sales of each target, and (vii) minimum diligence to advance licensed assets consisting of at least $1.0 million in development spend annually through 2021. Under the Regents License Agreement, the Company recorded: (i) $25,000 in licensemaintenance fees as research anddevelopmentexpense for each of the years ended December 31, 2019 and 2018, and (ii) $0.2 millionand $0.1 million in patentcosts as general and administrative expense for the years ended December 31, 2019 and 2018, respectively. As of December 31, 2019, the Company believes it has met its obligations under the Regents License Agreement.In July 2016, and as modified by the amended and restated Regents License Agreement in August 2018, the Company entered into a Research Agreement (the “Research Agreement”) with the Regents for further research on a ROR1 therapeutic development program. Under this five-year agreement, the Regents will have an aggregate budget of $3.6 million, with $125,000 payable quarterly. The Company recorded $0.5 million in research and development expense under this agreement for each of the years ended December 31, 2019 and 2018. Such costs are includable as part of the Company’s annual diligence obligations under the Regents License Agreement. The Regents License Agreement will expire upon the later of the expiration date of the longest-lived patent rights or the fifteenth anniversary of the first commercial sale of a licensed product.
The Regents may terminate the Regents License Agreement if: (i) a material breach by the Company is not cured within a reasonable time, (ii) the Company files a claim asserting the Regents licensed patent rights are invalid or unenforceable and (iii) the Company files for bankruptcy. The Company may terminate the agreement at any time upon at least 60 days’ written notice.
University of Tennessee Research Foundation (“UTRF”)
In July 2007, the Company and UTRF entered into a consolidated, amended and restated license agreement (the “SARM License Agreement”), pursuant to which the Company was granted exclusive worldwide rights in all existing selective androgen receptor modulator (“SARM”) technologies owned or controlled by UTRF, including all improvements thereto, and exclusive rights to future SARM technology that may be developed by certain scientists at the University of Tennessee or subsequently licensed to UTRF under certain existing inter-institutional agreements with The Ohio State University. Under the SARM License Agreement, the Company is obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and mid-single-digit royalties on sublicense revenues. The Company recorded research and development expense under this agreement of $0.1 million and none for the years ended December 31, 2019 and 2018, respectively. On December 31, 2019, the Company provided UTRF notice of termination of the SARM License Agreement by and between the Company and UTRF, which termination will be effective three months following such notice. Following termination, the Company will no longer have the obligation to make further payments under the SARM License Agreement, and will no longer have any rights to develop or sublicense the SARM Technology.
In March 2015, the Company and UTRF entered into a license agreement (the “SARD License Agreement”) pursuant to which the Company was granted exclusive worldwide rights in all existing selective androgen receptor degrader (“SARD”) technologies owned or controlled by UTRF, including all improvements thereto. Under the SARD License Agreement, the Company is obligated to employ active, diligent efforts to conduct preclinical research and development activities for the SARD program to advance one or more lead compounds into clinical development. The Company is also obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and additional royalties on sublicense revenues, depending on the state of development of a clinical product candidate at the time it is sublicensed. The Company recorded research and development expense under this agreement of $0.2 million and none for the years ended December 31, 2019 and 2018, respectively.
As of December 31, 2019, the Company believes it has met its obligations under each of the UTRF agreements.
Velos Biopharma Holdings, LLC (“VBH”) and VelosBio, Inc. (“VelosBio”) Spin-off Transactions
In November 2017, the Company formed VBH and in December 2017, made an in-kind tax-free distribution of 100% of its interest in VBH to the Company’s stockholders, option holders and warrant holders of record. On February 6, 2018, the Company licensed and assigned its rights to two preclinical product candidates, previously under the Regents License Agreement, to VBH. In consideration for the license, the Company: (i) received a promissory note receivable from VBH of $0.1 million, with an annual interest rate of 2.64% and a due date of 10 years, and (ii) made a partial assignment of its March 2016 Regents License Agreement. Pursuant to the partial assignment, VBH assumed certain obligations related to the licensed products under the Regents License Agreement as follows: (i) reimbursement of certain historical and future patent costs related to the licensed products, (ii) certain development and sales milestones for advancing licensed products targets, (iii) low single-digit royalties, including potential future minimum annual royalties, on net sales of each licensed product target are to be allocated between the Company and VBH, (iv) certain third party agreements and related obligations specifically related to the licensed products, (v) minimum diligence obligations to advance licensed assets under the Research Agreement equal to $0.5 million in developmentspendannuallythrough2021,and (vi) Research Agreement obligationsequal to $0.5 million annually commencing January 1, 2018. Due to the high uncertainty of the success of VBH ever repaying the note receivable and associated interest, theCompanyprovided a full valuationallowance for theseamounts as of December 31, 2019 and 2018.
In December 2017, VelosBio was incorporated with VBH being its sole stockholder. On February 6, 2018, VBH sublicensed and assigned its intellectual property rights to its two preclinical product candidates to VelosBio. In consideration for the license, VelosBio agreed to use commercially reasonable efforts to develop the licensed products as well as the following payment obligations: (i) the assumption of each of the VBH assumed obligations under the partial assignment between the Company and VBH as outlined above, and (ii) certain tiered development milestone and royalty payments to VBH. In August 2018, the Company entered into the amended and restated Regents License Agreement and VelosBio entered into their own license agreement directly with the Regents. There is no common control overlap between the companies.
Also on February 6, 2018, the Company and VelosBio entered into: (i) an asset purchase agreement whereby VelosBio purchased the Company’s right, title and interest in the Company’s nominal assets related to the two preclinical product candidates and assumed the Company’s $0.2 million convertible note payable and related $16,000 of accrued interest which has been recorded as other income, and (ii) a transition services agreement whereby the Company agreed to provide VelosBio with certain transition services, as follows: (a) access to certain common laboratory equipment at the Company’s lab facility, (b) certain named employees were to devote up to 80% of their time supporting VelosBio related activities, (c) cirmtuzumab manufacturing, process optimization and ancillary activities until VelosBio was able to establish their own, and (d) agreement to cost share the purchase of certain antibody materials with VelosBio. Such services were to be provided at cost or cost plus. During the year ended December 31, 2018, the Company incurred $3.0 million, of costs on behalf of VelosBio that were substantially reimbursed and recorded on a net basis within operating expenses in the accompanying consolidated statements of operations. As of December 31, 2018, there were no ongoing rights or commitments under the asset purchase or transition services agreements. In February 2020, the Company entered into a sublicense agreement with VelosBio pursuant to which it granted certain sublicenses to VelosBio under the Selexis License Agreement.
The California Institute for Regenerative Medicine (“CIRM”) Award
In August 2017, CIRM awarded an $18.3 million grant to researchers at UC San Diego to advance the Company’s Phase 1/2 clinical trial evaluating cirmtuzumab in combination with ibrutinib for the treatment of patients with B-cell lymphoid malignancies, including chronic lymphocytic leukemia and mantle cell lymphoma. The Company: (i) is conducting this study in collaboration with UC San Diego, (ii) estimates it will receive approximately $14.0 million in development milestones under research subaward agreements throughout the award project period, estimated to be from October 1, 2017 to March 31, 2022, (iii) is committed to certain co-funding requirements, (iv) received subaward payments of $6.2 million and $0.5 million in the years ended December 31, 2019 and 2018, respectively, and (v) is required to provide UC San Diego progress and financial update reports throughout the award period. The subaward does not bear a royalty payment commitment, nor is the subaward otherwise refundable. For the years ended December 31, 2019 and 2018, the Company recorded revenue of $2.4 million and $2.5 million, respectively. Related qualifying subaward costs for the years ended December 31, 2019 and 2018 were $5.4 million and $4.6 million, respectively. As of December 31, 2019, the Company believes it has met its obligations under the CIRM award and UC San Diego subawards.
In October 2017, CIRM awarded a $5.8 million grant to the researchers at the University of California San Diego School of Medicine (“UC San Diego”) to develop a novel anti-cancer stem cell targeted therapy for patients with advanced solid and hematological malignancies. In connection with such CIRM award, the Company agreed to provide up to $1.0 million in contingency funds if required during the grant period. The Company recorded no research and development expense under such CIRM award for the years ended December 31, 2019 and 2018.
Clinical Trial and Supply Agreement
In April 2018, the Company entered into a Clinical Trial and Supply Agreement with Pharmacyclics, LLC, an AbbVie Company (“Pharmacyclics”) to supply ibrutinib for the Company’s Phase 1/2 clinical trial evaluating cirmtuzumab in combination with ibrutinib, which agreement was amended in August 2019. Such agreement does not bear any upfront costs, inventory purchase costs, milestone or royalty payment commitments or other financial obligations.
License and Development Agreement with SPH USA, a Related Party
In November 2018, the Company entered into a License and Development Agreement (“LDA”) with SPH USA for: (i) the territory of the People’s Republic of China, Hong Kong, Macau, and Taiwan (“Greater China”), and (ii) rights to manufacture, develop, market, distribute and sell all of the Company’s product candidates under the Georgetown License Agreement and the Regents License Agreement (exclusive to Greater China only). Under the LDA, SPH USA is solely responsible for: (a) all preclinical and clinical development activities required in order to obtain regulatory approval in Greater China for such product candidates, (b) any third-party license milestone or royalty payments owed under the Georgetown License Agreement and the Regents License Agreement, and (c) paying the Company a low single digit royalty on net sales in the territory.
The LDA will expire upon the expiration of the last royalty term for the last licensed product. The LDA may be terminated by: (i) SPH USA on a country by country or product by product basis with 180 days written notice, (ii) either party upon material breach that is not cured within 90 days, and (iii) either party in theevent theother party declares insolvencyorbankruptcy.
The Merger, which closed on June 7, 2019, was accounted for as a reverse asset acquisition pursuant to Topic 805, Business Combinations, as substantially all of the fair value of the assets acquired were concentrated in a group of similar non-financial assets, and the acquired assets did not have outputs or employees. Because the assets had not yet received regulatory approval, the fair value attributable to these assets was recorded as in-process research and development (“IPR&D”) expenses in the Company’s consolidated statement of operations in the year ended December 31, 2019.
Pursuant to the Merger Agreement on June 7, 2019, the Company, a representative of holders of the contingent value rights (“CVRs”), and Computershare, Inc. as rights agent entered into a Contingent Value Rights Agreement (the “CVR Agreement”). Pursuant to the CVR Agreement, the Company’s stockholders of record as of immediately prior to the Merger received one CVR for each share of the Company’s common stock held immediately prior to the Merger. CVR holders are entitled to receive 75% of the aggregate amount of any net proceeds received by the Company during the 15-year period after the closing of the Merger from the grant, sale or transfer of rights to the Company’s SARD or SARM technology that occurs during the 10-year period after the closing (or in the 11th year if based on a term sheet approved during the initial 10-year period) and, if applicable, to receive royalties on the sale of any SARD or SARM products by the Company during the 15-year period after the closing. The CVR Agreement will continue in effect until the payment of all amounts payable thereunder. As of the June 7, 2019 closing date and December 31, 2019, no milestones had been accrued as there were no potential milestones yet considered probable.
The total purchase price paid in the Merger has been allocated to the net assets acquired and liabilities assumed based on their fair values as of the completion of the Merger. The following summarizes the purchase price paid in the Merger (in thousands, except share and per share amounts):
Number of shares of the combined organization owned
by the Company’s pre-Merger stockholders
3,458,170
Multiplied by the fair value per share of GTx common
stock (1)
$
8.40
Fair value of consideration issued to effect the Merger
$
29,049
Transaction costs
2,154
Purchase price
$
31,203
(1)
Based on the last reported sale price of the Company’s common stock on the Nasdaq Capital Market on June 7, 2019, the closing date of the Merger, and gives effect to the Reverse Stock Split.
The allocation of the purchase price is as follows:
Cash acquired
$
18,292
Net liabilities assumed
(5,177
)
IPR&D (2)
18,088
Purchase price
$
31,203
(2)
Represents the research and development projects of GTx which were in-process, but not yet completed, and which the Company plans to advance, consisting primarily of GTx’s preclinical SARD technology. Current accounting standards require that the fair value of IPR&D projects acquired in an asset acquisition with no alternative future use be allocated a portion of the consideration transferred and charged to expense on the acquisition date. The acquired assets did not have outputs or employees.
Amended and Restated Articles of Incorporation
On June 7, 2019, the Company’s certificate of incorporation was amended and restated to authorize 60,000,000 shares of common stock and 5,000,000 shares of undesignated preferred stock, each with a par value of $0.001 per share.
Convertible Preferred Stock
In connection with the Merger, all of the outstanding shares of Private Oncternal’s convertible preferred stock were converted into 8,148,268 shares of the Company’s common stock. As of December 31, 2018, Private Oncternal’s convertible preferred stock is classified as temporary equity on the accompanying consolidated balance sheets in accordance with authoritative guidance for the classification and measurement of potentially redeemable securities whose redemption is based upon certain change in
our financial assetscontrol events outside of Private Oncternal’s control, including liquidation, sale or transfer of control of Private Oncternal. Private Oncternal did not adjust the carrying values of the convertible preferred stock to the liquidation preferences of such shares because the occurrence of any such change of control event was not deemed probable.Sales of Convertible Preferred Stock
In September, November and
liabilities denominatedDecember 2017, Private Oncternal issued an aggregate of 1,662,494 shares of Series B-2 preferred stock at a per share purchase price of $6.13, raising net cash proceeds of $8.9 million, of which $1.1 million was collected inforeign currencies. This potential changeFebruary 2018.In November 2018, contemporaneous with entering into the LDA, Private Oncternal issued 2,495,114 shares of Series C preferred stock to SPH USA, at a per share purchase price of $6.81, raising net cash proceeds of $16.8 million. Private Oncternal concluded that the shares were issued at fair value and therefore no value was ascribed to the LDA.
Common Stock Warrants
In September, November and December 2017, Private Oncternal issued warrants to purchase of 371,624 shares of Series B-2 preferred stock, which converted into rights to purchase common stock of the Company at the Merger closing, at an exercise price of $6.13 per share. The warrants expire on various dates in September, November and December 2022. As of December 31, 2019, warrants to purchase 196 shares of common stock have been exercised.
On September 29, 2017, the Company completed a private placement transaction that included warrants to purchase an aggregate of 469,996 shares of the Company’s common stock at an exercise price of $63.14 per share. The five-year warrants expire on September 29, 2022. As of December 31, 2019, no such warrants have been exercised.
The Company assessed whether the above warrants require accounting as derivatives after the Merger closing. The Company determined that the warrants were indexed to the Company's own stock. As such, the Company has concluded the warrants meet the scope exception for determining whether the instruments require accounting as warrant liabilities.
Common Stock and Unvested Share Liability
The Company has issued restricted common stock subject to vesting and repurchase by the Company. For employee and non-employee awards, the issuance date fair value is recognized over the requisite service period of the award (usually the vesting period) on a straight-line basis. In addition, the Company has outstanding unvested shares related to the early exercise of stock options. The Company has the right, but not the obligation, to repurchase any unvested shares at the original purchase price upon any voluntary or involuntary termination. The consideration received in exchange for unvested shares is recorded as an unvested share liability on the accompanying consolidated balance sheets and is reclassified into common stock and additional paid-in capital as the shares vest. At December 31, 2019 and 2018, the unvested share liability was $24,000 and $54,000, respectively.
A summary of the Company’s unvested shares is as follows (in thousands):
Number of
Shares
Balance at December 31, 2018
100
Vested shares
(65
)
Balance at December 31, 2019
35
Equity Incentive Plans
Contemporaneous with the Merger closing: (i) Private Oncternal’s 2015 Equity Incentive Plan, as amended (the “2015 Plan”) was assumed by the Company, and (ii) the Company adopted the 2019 Incentive Award Plan (“2019 Plan”) under which the sum of: (a) 1,678,571 shares of common stock, (b) up to 275,579 shares of common stock which were subject to outstanding awards under the GTx 2013 Equity Incentive Plan (the “2013 Plan”) as of June 7, 2019, that are subsequently cancelled will become available for issuance under the 2019 Plan, and (c) an annual increase on the first day of each calendar year beginning January 1, 2020, and ending on and including January 1, 2029, equal to the lesser of (A) 5% of the aggregate number of shares of common stock outstanding on the final day of the immediately preceding calendar year and (B) such smaller number of shares of common stock as is determined by the Board, are reserved for issuance. At December 31, 2019, 494,583 shares remain available for future issuance under the 2019 Plan (see Note 9).
Number of
Shares
Weighted-
Average
Exercise
Price
Weighted-
Average
Remaining
Contractual
Term
(in years)
Aggregate
Intrinsic
Value
December 31, 2019:
Options outstanding
1,662,253
$
4.17
9.2
$
1,460
Options vested and expected to vest
1,662,253
$
4.17
9.2
$
1,460
Options exercisable
244,170
$
1.16
7.2
$
718
As of December 31, 2019 under the 2013 Plan, there were: (i) 257,067 outstanding and fully vested options with a weighted average exercise price of $59.98 per share, and (ii) 18,512 cancelled options that were added back to the 2019 Plan as of December 31, 2019. As of December 31, 2019, the former GTx stock option plans had an aggregate of 295,414 outstanding and fully vested and exercisable options with a weighted average exercise price of $71.34 and a weighted average remaining contractual term of 0.7 years.
In July 2015, Private Oncternal adopted the 2015 Plan which provided for the issuance of up to 631,120 shares of common stock for incentive stock options, non-statutory stock options, restricted stock awards, restricted stock unit awards and other stock awards to its employees, members of its board of directors and consultants. In general, the options issued under the 2015 Plan expire ten years from the date of grant and vest over a four-year period. Certain grants vest based on the achievement of development or regulatory milestones. The 2015 Plan allowed for the early exercise of all stock option grants if authorized by the board of directors at the time of grant. The Company has the option to repurchase any unvested shares at the original purchase price upon any voluntary or involuntary termination.
No further awards will be made under the 2015 Plan, which was terminated in June 2019.
A summary of the Company’s stock option activity under the 2019 Plan and 2015 Plan is as follows:
Number of
Options
Weighted-
Average
Exercise
Price
Balance at December 31, 2018
504,019
$
0.81
Granted
1,230,000
$
5.47
Cancelled
(52,726
)
$
3.38
Exercised
(19,040
)
$
0.69
Balance at December 31, 2019
1,662,253
$
4.17
Information about the Company’s outstanding stock options under the 2019 Plan and 2015 Plan is as follows (in thousands, except share and per share data and contractual term):
The weighted average grant date fair value per share of option grants for the years ended December 31, 2019 and 2018 was $3.69 and $0.55 per share, respectively. The aggregate intrinsic value used in the above table of options at December 31, 2019 is based on the Company’s closing market price per common share on December 31, 2019 of $3.95. The intrinsic value is calculated as the difference between the fair value of the Company’s common stock at the time of the option exercise and the exercise price of that stock option. The aggregate intrinsic value of stock options exercised during the years ended December 31, 2019 and 2018 was not material.
Stock-Based Compensation Expense
The weighted-average assumptions used in the Black-Scholes option pricing model to determine the fair value of stock option grants were as follows:
Years Ended
December 31,
2019
2018
Risk-free interest rate
1.6
%
2.9
%
Expected volatility
77.6
%
64.7
%
Expected term (in years)
6.0
6.1
Expected dividend yield
—
%
—
%
Expected volatility. Prior to the Merger, Private Oncternal did not have a
sensitivity analysis performedtrading history for its common stock. Accordingly, the expected volatility assumption is based onour financial positionvolatilities of a peer group of similar companies whose share prices are publicly available. The peer group was developed based on companies in the life sciences industry. The Company will continue to apply this process until a sufficient amount of historical information regarding the volatility of its own stock price becomes available.Expected term. The expected term represents the period of time that options are expected to be outstanding. Because Private Oncternal did not have historical exercise behavior, it determined the expected life assumption using the simplified method for employees, which is an average of the contractual term of the option and its vesting period. The expected term for nonemployee options is generally the remaining contractual term.
Risk-free interest rate. The risk-free interest rate is based on the implied yield on the U.S. Treasury securities with a maturity date similar to the expected term of the associated stock option award.
Expected dividend yield. The Company bases the expected dividend yield assumption on the fact that it has never paid cash dividends and has no present intention to pay cash dividends and, therefore, used an expected dividend yield of zero.
Stock-based compensation expense recognized for all equity awards has been reported in the statements of operations as follows (in thousands):
Years Ended
December 31,
2019
2018
Research and development
$
237
$
141
General and administrative
270
39
$
507
$
180
At December 31, 2019, the total compensation cost related to nonvested awards not yet recognized was $4.2 million and the weighted-average period over which it is expected to be recognized was 3.5 years.
Common Stock Reserved for Future Issuance
Common stock reserved for future issuance is as follows (in thousands):
December 31,
2019
2018
Redeemable convertible preferred stock
—
8,148
Warrants to purchase convertible preferred stock
—
372
Common stock warrants
841
—
Common stock options issued and outstanding
1,958
504
Common stock available for issuance under equity
plans
495
80
3,294
9,104
7.
Income Taxes
A reconciliation of the Company’s effective tax rate and federal statutory tax rate is as follows (in thousands):
Years Ended
December 31,
2019
2018
Federal income taxes
$
(7,179
)
$
(1,379
)
State income taxes, net of federal benefit
(968
)
(500
)
Permanent items
873
(147
)
In-process research and development
3,354
Research and development credit carryforwards
(217
)
(468
)
Other, net
18
51
Change in valuation allowance
4,119
2,443
Provision for income taxes
$
—
$
—
Significant components of the Company’s net deferred tax assets are as follows (in thousands):
December 31,
2019
2018
Deferred tax assets:
Net operating loss carryforwards
$
18,108
$
8,321
Research and development credit carryforwards
1,446
1,231
Accrued expenses
214
24
Capitalized research and development costs
7,688
—
Other, net
143
288
Total deferred tax assets
27,599
9,864
Valuation allowance
(27,546
)
(9,864
)
53
—
Deferred tax liabilities:
Right of use asset
(53
)
—
Total deferred tax liabilities
(53
)
—
Net deferred tax assets
$
—
$
—
Based on the Company’s history of operating losses, the Company is unable to conclude that it is more likely than not that the benefit of its deferred tax assets will be realized. Accordingly, the Company has provided a full valuation allowance for its deferred tax assets as of December 31, 2019 and 2018. As a result of the Merger in 2019, the Company recorded deferred tax assets of $13.1 million which are fully offset by a valuation allowance. The $13.1 million net deferred tax assets do not include federal and state net operating loss carryforwards and federal research and development credit carryforwards that are estimated to expire under Internal Revenue Code Sections 382 and 383 as a result of the Merger.
At December 31, 2019, the Company had federal and state net operating loss (NOL) carryforwards of approximately $70.0 million and $48.7 million, respectively. Of the federal net operating losses at December 31,
2018. Actual results may differ materially. We have elected2019, $44.2 million do notto hedge our exposure to foreign currency fluctuations.ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATAOur financial statementsexpire, and thereportsremaining federal and state net operating loss carryforwards will begin expiring in 2034 and 2030, respectively, unless previously utilized. At December 31, 2019, the Company also had federal and state research and development credit carryforwards ofour independent registered public accounting firmapproximately $0.9 million and $0.7 million, respectively. The federal research and development credit carryforwards will begin expiring in 2034 unless previously utilized. The state research and development credits do not expire.Pursuant to Internal Revenue Code Sections 382 and 383, annual use of the Company’s net operating loss and research and development tax credit carryforwards may be limited in the event a cumulative change in ownership of more than 50% occurs within a three-year period. The Company has not completed a Section 382 study to assess whether an ownership change has occurred or whether there have been multiple ownership changes since the Company’s formation due to the complexity and cost associated with such a study and the fact that there may be additional such ownership changes in the future. If eliminated, the related asset would be removed from the deferred tax asset schedule with a corresponding reduction in the valuation allowance. Due to the existence of the valuation allowance, limitations created by future ownership changes, if any, will not impact the Company’s effective tax rate.
The Company recognizes a tax benefit from an uncertain tax position when it is more likely than not that the position will be sustained upon examination, including resolutions of any related appeals or litigation processes, based on the technical merits. Income tax positions must meet a more likely than not recognition at the effective date to be recognized. At December 31, 2019 and 2018, there were no unrecognized tax benefits recorded in the consolidated financial statements. The Company does not expect any material changes to unrecognized tax benefits within the next twelve months.
The Company is subject to taxation in the United States federal and state jurisdictions. The Company’s 2013 through 2019 federal income tax and state income tax returns are
includedsubject to examination by federal and state tax authorities due to the carryforward of unutilized net operating losses and research and development credits. The Company is not currently under examination by any tax authority.The Company’s policy is to recognize interest and penalties related to income tax matters in income tax expense. The Company has not recognized interest or penalties in its consolidated statements of operations since inception.
The Company adopted Accounting Standards Codification Topic 842 – Leases, on January 1, 2019. Under Topic 842, the Company is required to recognize a right-of-use asset and a lease liability, initially measured at the present value of the lease payments for operating leases on the balance sheet. Upon adoption, no change in retained earnings was recorded related to income taxes as the Company maintains a full valuation allowance. As of the implementation date, an adjustment of $0.1 million was recorded as a deferred tax liability and an adjustment of $0.1 million was recorded as a deferred tax asset.
The information required by this
AnnualItem 9 was previously reported in the Company’s Current Report on Form10-K beginning8‑K that was filed with the Securities and Exchange Commission onpage F-1. The index to these reports and our financial statements is included in Part IV, Item 15 below.June 20, 2019
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
.None.ITEM 9A. CONTROLS AND PROCEDURESDisclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended (the "Exchange Act")) that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC's rules and forms and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow for timely decisions regarding required disclosures.
We have carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of December 31, 2019, the end of the period covered by this report. Based on the evaluation of these disclosure controls and procedures, our principal executive officer and principal financial officer have concluded that our disclosure controls and procedures were effective as of December 31,
2018.Table of Contents2019.Management's Report on Internal Control Over Financial Reporting
We, as management of
GTx,Oncternal Therapeutics, Inc., are responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Securities Exchange Act Rule 13a-15(f). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with United States generally accepted accounting principles. Any system of internal control, no matter how well designed, has inherent limitations, including the possibility that a control can be circumvented or overridden and misstatements due to error or fraud may occur and not be detected. Also, because of changes in conditions, internal control effectiveness may vary over time. Accordingly, even an effective system of internal control will provide only reasonable assurance that the objectives of the internal control system are met.Under the supervision and with the participation of management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31,
20182019 using the criteria for effective internal control over financial reporting as described in "Internal Control — Integrated Framework," issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Based on this evaluation, we concluded that, as of December 31,2018,2019, our internal control over financial reporting was effective. The effectiveness of our internal control over financial reporting has been audited byErnst & YoungBDO USA LLP, independent registered public accounting firm.Attestation Report of the Independent Registered Public Accounting Firm
Ernst & YoungBDO USA LLP, an independent registered public accounting firm, has issued an audit report on our internal control over financial reporting, which report is included elsewhere herein.Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting during the fourth quarter of
20182019 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.None.Certain information required by Part III is omitted from this AnnualReporton Form 10-K and incorporated by reference to our definitive proxy statement for our 2019 Annual Meeting of Stockholders, or our 2019 Proxy Statement, to be filed pursuant to Regulation 14A of the Securities Exchange Act of 1934, as amended, or Exchange Act. If our 2019 Proxy Statement is not filed within 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, the omitted information will be included in an amendment to this Annual Report on Form 10-K filed not later than the end of such 120-day period.ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE(1) The information required by this Item concerning our directors and nominees for director, including information with respect to our audit committee and audit committee financial experts, may be found under the section entitled "Election of Directors" and "Additional Information About the Board of Directors and Certain Corporate Governance Matters" appearing in the 2019 Proxy Statement. Such information is incorporated herein by reference.(2) The information required by this Item concerning compliance with Section 16(a) of the Securities Exchange Act of 1934 may be found in the section entitled "Section 16(a) Beneficial Ownership Reporting Compliance" appearing in the 2019 Proxy Statement. Such information is incorporated herein by reference.(3) The information required by this Item concerning our executive officers is set forth in the section entitled "Executive Officers of the Registrant" in Part I, Item 1 of this Form 10-K.(4) Our Board has adopted a Code of Business Conduct and Ethics applicable to all officers, directors and employees as well as Guidelines on Governance Issues. These documents are available on our Web site (www.gtxinc.com) under "Investors" at "Corporate Governance." We will provide a copy of these documents to any person, without charge, upon request, by writing to us at GTx, Inc., Chief Legal Officer, 175 Toyota Plaza, Suite 700, Memphis, Tennessee 38103. We intend to satisfy the disclosure requirement under Item 5.05 of Form 8-K regarding an amendment to, or waiver from, a provision of the Code of Business Conduct and Ethics by posting such information on our Web site at the address and the location specified above.ITEM 11. EXECUTIVE COMPENSATIONThe information required by this Item concerning director and executive compensation is incorporated herein by reference to the information from the 2019 Proxy Statement under the sections entitled "Executive Compensation" and "Director Compensation."ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS(1) The information required by this Item with respect to security ownership of certain beneficial owners and management is incorporated herein by reference to the information from the 2019 Proxy Statement under the section entitled "Security Ownership of Certain Beneficial Owners and Management."(2) The information required by this Item with respect to securities authorized for issuance under our equity compensation plans is incorporated herein by reference to the information from the 2019 Proxy Statement under the section entitled "Equity Compensation Plan Information."ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE(1) The information required by this Item concerning related party transactions is incorporated herein by reference to the information from the 2019 Proxy Statement under the section entitled "Related Party Transactions and Indemnification."(2) The information required by this Item concerning director independence is incorporated herein by reference to the information from the 2019 Proxy Statement under the section entitled "Additional Information About the Board of Directors and Certain Corporate Governance Matters — Director Independence."ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICESThe information required by this Item is incorporated herein by reference to the information from the 2019 Proxy Statement under the section entitled "Ratification of Appointmentof Independent Registered Public AccountingFirm."FirmTableShareholders and Board ofContentsDirectorsPART IV
Oncternal Therapeutics, Inc.ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
San Diego, California(a)(1) Index toOpinion on Internal Control over FinancialStatementsPageDescriptionF-2Report of Independent Registered Public Accounting FirmF-3Balance Sheets at December 31, 2018 and 2017F-4Statements of Operations for the Years Ended December 31, 2018, 2017 and 2016F-5Statements of Stockholders' Equity for the Years Ended December 31, 2018, 2017 and 2016F-6Statements of Cash Flows for the Years Ended December 31, 2018, 2017 and 2016F-7Notes to Financial Statements(a)(2) Financial statement schedules are omitted as they are not applicable.Reporting(a)(3) See Item 15(b) below.(b) Exhibits — The following exhibits are included herein or incorporated herein by reference:Incorporation By ReferenceExhibitNumberExhibit DescriptionFormSEC File No.ExhibitFiling Date32.2+Certification of Principal Financial Officer, as required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350)(1)----101.INS+XBRL Instance Document----101.SCH+XBRL Taxonomy Extension Schema Document----101.CAL+XBRL Taxonomy Extension Calculation Linkbase Document----101.DEF+XBRL Taxonomy Extension Definition Linkbase Document----101.LAB+XBRL Taxonomy Extension Labels Linkbase Document----101.PRE+XBRL Taxonomy Extension Presentation Linkbase Document----**Schedulesandexhibits have been omitted pursuant to Item 601(b)(2) of Regulation S-K. A copy of any omitted schedule and/or exhibit will be furnished to the SEC upon request.†Confidential treatment has been granted with respect to certain portions of this exhibit. This exhibit omits the information subject to the related confidentiality order. Omitted portions have been filed separately with the SEC.††Confidential treatment has been requested with respect to certain portions of this exhibit. This exhibit omits the information subject to this confidentiality request. Omitted portions have been filed separately with the SEC.*Indicates a management contract or compensation plan or arrangement.+Filed herewith(1)This certification accompanies the Form 10-K to which it relates, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of the Registrant under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended (whether made before or after the date of the Form 10-K), irrespective of any general incorporation language contained in such filing.
None provided.Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.GTx, Inc.By/s/ Marc S. HanoverMarc S. HanoverChief Executive OfficerDate: March 18, 2019(Principal Executive Officer)KNOW ALL PERSONS BY THESE PRESENT, that each person whose signature appears below constitutes and appoints Marc S. Hanover and Jason T. Shackelford, and each of them, acting individually, as his attorney-in-fact, each with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith and about the premises, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or any of them, or their or his or her substitute or substitutes, may lawfully do or cause to be done by virtue hereof.Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated./s/ Marc S. HanoverMarc S. HanoverChief Executive Officer(Principal Executive Officer)March 18, 2019/s/ Jason T. ShackelfordJason T. ShackelfordVice President, Finance and Accounting and Principal Financial and Accounting Officer(Principal Financial and Accounting Officer)March 18, 2019/s/ Robert J. WillsRobert J. Wills, B.S., M.S., Ph.D.Executive Chairman of the Board of DirectorsMarch 18, 2019/s/ Michael G. CarterMichael G. Carter, M. D.DirectorMarch 18, 2019/s/ J. Kenneth GlassJ. Kenneth GlassDirectorMarch 18, 2019/s/ J. R. Hyde, IIIJ. R. Hyde, IIIDirectorMarch 18, 2019/s/ Garry A. NeilGarry A. Neil, M.D.DirectorMarch 18, 2019/s/ Kenneth S. RobinsonKenneth S. Robinson, M.D.DirectorMarch 18, 2019MANAGEMENT'S REPORT ONINTERNAL CONTROL OVER FINANCIAL REPORTINGWe, as management of GTx, Inc., are responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Securities Exchange Act Rule 13a-15(f). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with United States generally accepted accounting principles. Any system of internal control, no matter how well designed, has inherent limitations, including the possibility that a control can be circumvented or overridden and misstatements due to error or fraud may occur and not be detected. Also, because of changes in conditions, internal control effectiveness may vary over time. Accordingly, even an effective system of internal control will provide only reasonable assurance that the objectives of the internal control system are met.Under the supervision and with the participation of management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of oursubsidiaries’ (the “Company’s”) internal control over financial reporting as of December 31,2018 using the2019, based on criteriafor effective internal control over financial reporting as describedestablished in"InternalInternal Control—– Integrated Framework"(2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission(2013 framework). Based on this evaluation, we concluded that, as of December 31, 2018, our internal control over financial reporting was effective. The effectiveness of our internal control over financial reporting has been audited by Ernst & Young LLP, independent registered public accounting firm who also audited the Company's financial statements included in this Annual Report on Form 10-K. Ernst & Young LLP's report on the Company's internal control over financial reporting is included in this Annual Report on the 10-K./s/ Marc S. HanoverMarc S. HanoverChief Executive OfficerPrincipal Executive Officer/s/ Jason T. ShackelfordJason T. ShackelfordVice President, Finance and AccountingPrincipal Financial and Accounting OfficerMemphis, TennesseeMarch 18, 2019Report of Independent Registered Public Accounting FirmThe Board of Directors and Stockholders of GTx, Inc.Opinion on Internal Control over Financial ReportingWe have audited GTx, Inc.'s internal control over financial reporting as of December 31, 2018, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework),(theCOSO criteria)“COSO criteria”). In our opinion,GTx, Inc. (the Company)the Company maintained, in all material respects, effective internal control over financial reporting as of December 31,2018,2019, based on the COSOcriteria.criteria.We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States)
(PCAOB)(“PCAOB”), the consolidated balance sheets of the Company as of December 31,20182019 and2017,2018, the related consolidated statements of operations,stockholders'convertible preferred stock and stockholders’ equity (deficit), and cash flows foreach ofthethreeyearsin the periodthen ended,December 31, 2018,and the related notes, and our report dated March18, 201916, 2020 expressed an unqualified opinion thereon.Basis for Opinion
The
Company'sCompany’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanyingManagement'sItem 9A, Management’s Report on Internal ControlOverover Financial Reporting. Our responsibility is to express an opinion on theCompany'sCompany’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance withtheU.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.We conducted our audit of internal control over financial reporting in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed
risk, andrisk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.Definition and Limitations of Internal Control
Overover Financial ReportingA
company'scompany’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Acompany'scompany’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of thecompany'scompany’s assets that could have a material effect on the financial statements.Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/
Ernst & YoungBDO USA, LLPMemphis, Tennessee
San Diego, CaliforniaMarch
18, 201916, 2020Table of ContentsNone.
PARTReport of Independent Registered Public Accounting Firm
IIIThe Board of Directors and Stockholders of GTx, Inc.Opinion on the Financial StatementsWe have audited the accompanying balance sheets of GTx, Inc. as of December 31, 2018 and 2017, the related statements of operations, stockholders' equity and cash flows for each of the three yearsInformation required by this item will be contained inthe period ended December 31, 2018, and the related notes (collectively referred to as the "financial statements"). Inouropinion, the financial statements present fairly, in all material respects, the financial position of GTx, Inc. at December 31, 2018 and 2017, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2018, in conformity with U.S. generally accepted accounting principles.We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2018, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 18, 2019 expressed an unqualified opinion thereon.Basis for OpinionThese financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are requireddefinitive proxy statement to beindependentfiled withrespect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations ofthe Securities and Exchange Commission on Schedule 14A in connection with our 2020 Annual Meeting of Stockholders or the Proxy Statement, which is expected to be filed not later than 120 days after the end of our fiscal year ended December 31, 2019, under the headings “Executive Officers,” “Election of Directors,” “Information Regarding the Board of Directors and Corporate Governance,” and “Delinquent Section 16(a) Reports,” and is incorporated herein by reference.The information required by this item regarding executive compensation is incorporated by reference to the
PCAOB.information set forth in the sections titled “Executive Compensation” in our Proxy Statement.Item 12.
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this item regarding security ownership of certain beneficial owners and management is incorporated by reference to the information set forth in the section titled “Security Ownership of Certain Beneficial Owners and Management” in our Proxy Statement.
We conductedThe information required by Item 201(d) of Regulation S-K is incorporated by reference to the information set forth in the section titled “Executive Compensation” in ourauditsProxy Statement.The information required by this item regarding certain relationships and related transactions and director independence is incorporated by reference to the information set forth in
accordancethe sections titled “Transactions withthe standardsRelated Parties” and “Election of Directors – Independence of thePCAOB. Those standards require that we planBoard of Directors,” respectively, in our Proxy Statement.The information required by this item regarding principal accountant fees and
performservices is incorporated by reference to theaudit to obtain reasonable assurance about whetherinformation set forth in the section titled “Principal Accountant Fees and Services” in our Proxy Statement.(a) Documents filed as part of this report.
1. Financial Statements
The consolidated financial statements
are freeofmaterial misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion./s/ Ernst & Young LLPWe have served as the Company's auditor since 1998.Memphis, TennesseeMarch 18, 2019GTx, Inc.BALANCE SHEETS(in thousands, except share and per share data)December 31, 2018 2017 ASSETS
Current assets:
Cash and cash equivalents
$ 28,258 $ 15,816 Short-term investments
200 28,083 Prepaid expenses and other current assets
2,750 2,178 Total current assets
31,208 46,077 Property and equipment, net
19 51 Intangible assets, net
94 108 Total assets
$ 31,321 $ 46,236 LIABILITIES AND STOCKHOLDERS' EQUITY
Current liabilities:
Accounts payable
$ 3,279 $ 2,604 Accrued expenses and other current liabilities
1,931 5,371 Total current liabilities
5,210 7,975 Commitments and contingencies
Stockholders' equity:
Common stock, $0.001 par value: 60,000,000 shares authorized at December 31, 2018 and December 31, 2017; 24,051,844 and 21,541,909 shares issued and outstanding at December 31, 2018 and December 31, 2017, respectively
24 22 Additional paid-in capital
626,142 599,876 Accumulated deficit
(600,055 ) (561,637 ) Total stockholders' equity
26,111 38,261 Total liabilities and stockholders' equity
$ 31,321 $ 46,236 The accompanying notes are an integral part of these financial statements.GTx, Inc.STATEMENTS OF OPERATIONS(in thousands, except share and per share data)Years Ended December 31, 2018 2017 2016 Expenses:
Research and development expenses
$ 29,669 $ 21,467 $ 17,228 General and administrative expenses
9,390 9,188 8,705 Total expenses
39,059 30,655 25,933 Loss from operations
(39,059 ) (30,655 ) (25,933 ) Other income, net
641 216 46 Gain on change in fair value of warrant liability
- - 8,163 Net loss
$ (38,418 ) $ (30,439 ) $ (17,724 ) Net loss per share:
Basic and Diluted
$ (1.65 ) $ (1.75 ) $ (1.22 ) Weighted average shares outstanding:
Basic and Diluted
23,346,231 17,441,280 14,559,541 The accompanying notes are an integral part of these financial statements.GTx, Inc.STATEMENTS OF STOCKHOLDERS' EQUITYFor the Years Ended December 31, 2018, 2017 and 2016(in thousands, except share data)Stockholders' Equity Common Stock Additional
Paid-in
CapitalAccumulated
DeficitTotal
Stockholders'
EquityShares Amount Balances at January 1, 2016
14,037,411 $ 14 $ 515,319 $ (513,474 ) $ 1,859 Issuance of common stock in October 2016 registered direct offering, net of offering costs
1,728,395 2 13,690 - 13,692 Vesting of restricted stock units, net of shares withheld for tax payments
154,170 - (208 ) - (208 ) Directors' deferred compensation
- - 132 - 132 Share-based compensation
- - 2,957 - 2,957 Warrant liability reclassification
- - 19,186 - 19,186 Settlement of fractional shares upon reverse stock split
(404 ) - (3 ) - (3 ) Net loss
- - - (17,724 ) (17,724 ) Balances at December 31, 2016
15,919,572 16 551,073 (531,198 ) 19,891 Issuance of common stock and warrants in September 2017 private placement, net of offering costs
5,483,320 6 45,642 - 45,648 Vesting of restricted stock units, net of shares withheld for tax payments
139,017 - (156 ) - (156 ) Directors' deferred compensation
- - 166 - 166 Share-based compensation
- - 3,151 - 3,151 Net loss
- - - (30,439 ) (30,439 ) Balances at December 31, 2017
21,541,909 22 599,876 (561,637 ) 38,261 Issuance of common stock upon exercise of warrants
674,579 1 (1 ) - - Exercise of stock options
6,000 - 103 - 103 Issuance of shares under "At-The-Market" sales agreement, net of issuance costs
1,501,501 1 24,473 - 24,474 Vesting of restricted stock units, net of shares withheld for tax payments
327,855 - (672 ) - (672 ) Directors' deferred compensation
- - 166 - 166 Share-based compensation
- - 2,197 - 2,197 Net loss
- - - (38,418 ) (38,418 ) Balances at December 31, 2018
24,051,844 $ 24 $ 626,142 $ (600,055 ) $ 26,111 The accompanying notes are an integral part of these financial statements.GTx, Inc.STATEMENTS OF CASH FLOWS(in thousands)Years Ended December 31, 2018 2017 2016 Cash flows from operating activities:
Net loss
$ (38,418 ) $ (30,439 ) $ (17,724 ) Adjustments to reconcile net loss to net cash used in operating activities:
Gain on change in fair value of warrant liability
- - (8,163 ) Share-based compensation
2,197 3,151 2,957 Directors' deferred compensation
166 166 132 Depreciation and amortization
46 47 28 Changes in assets and liabilities:
Prepaid expenses and other assets
(572 ) 251 204 Accounts payable
675 1,384 838 Accrued expenses and other liabilities
(3,440 ) 1,980 950 Net cash used in operating activities
(39,346 ) (23,460 ) (20,778 ) Cash flows from investing activities:
Purchase of property and equipment
- (2 ) (90 ) Purchase of short-term investments, held to maturity
(44,155 ) (39,283 ) (35,404 ) Proceeds from maturities of short-term investments, held to maturity
72,038 24,159 37,645 Net cash provided by (used in) investing activities
27,883 (15,126 ) 2,151 Cash flows from financing activities:
Net proceeds from the issuance of common stock and warrants
24,474 45,648 13,692 Tax payments related to shares withheld for vested restricted stock units
(672 ) (156 ) (208 ) Proceeds from exercise of employee stock options
103 - - Settlement of fractional shares upon reverse stock split
- - (3 ) Net cash provided by financing activities
23,905 45,492 13,481 Net increase (decrease) in cash and cash equivalents
12,442 6,906 (5,146 ) Cash and cash equivalents, beginning of period
15,816 8,910 14,056 Cash and cash equivalents, end of period
$ 28,258 $ 15,816 $ 8,910 The accompanying notes are an integral part of these financial statements.GTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data)1. BusinessGTx, Inc. ("GTx" or the "Company"), a Delaware corporation incorporated on September 24, 1997 and headquartered in Memphis, Tennessee, is a biopharmaceutical company dedicated to the discovery, development and commercialization of medicines to treat serious and/or significant unmet medical conditions.In 2015, the Company entered into an exclusive license agreement with the University of Tennessee Research Foundation ("UTRF") to develop UTRF's proprietary selective androgen receptor degrader ("SARD") technology which may have the potential to provide compounds that can degrade or antagonize multiple forms of androgen receptor to treat those patients who do not respond or are resistant to current androgen targeted therapies by inhibiting tumor growth in patients with progressive castration-resistant prostate cancer ("CRPC"). The Company is in the process of completing ongoing mechanistic preclinical studies in order to select the most appropriate SARD compounds to move forward into the additional preclinical studies required to submit an investigational new drug application ("IND"), and potentially advance one of its SARD compounds into a first-in-human clinical trial.The Company had been developing selective androgen receptor modulators ("SARMs"), including enobosarm (GTx-024). Most recently, enobosarm was evaluated in post-menopausal women with stress urinary incontinence ("SUI") compared to placebo. During the third quarter of 2018, the Company announced that the Phase 2 double-blind, placebo-controlled clinical trial of orally-administered enobosarm (3 mg or 1 mg) in post-menopausal women with SUI (the "ASTRID trial") did not achieve statistical significance on the primary endpoint for the trial. The Company has completed the ASTRID trial, including its review of the full data sets from the clinical trial, and has determined that there is not a sufficient path forward to warrant additional clinical development of enobosarm to treat SUI. The Company has therefore discontinued further development of enobosarm to treat SUI, including discontinuing the related durability and open-label safety extension studies that the Company initiated before it received topline data from the ASTRID trial. The Company has also discontinued any further development of its SARM program generally.Following the announcement of the ASTRID trial results, the Company's board of directors commenced a process of evaluating strategic alternatives to maximize stockholder value. To assist with this process, the Company's board of directors engaged a financial advisory firm to help explore the Company's available strategic alternatives, including possible mergers and business combinations, a sale of part or all of the Company's assets, and collaboration and licensing arrangements. On March 6, 2019, the Company andOncternal Therapeutics, Inc.("Oncternal") announcedlisted below are set forth in Item 8 of this Annual Report for thesigning of an Agreement and Plan of Merger and Reorganization (the "Merger Agreement"). Upon the terms and subject to the satisfaction of the conditions described in the Merger Agreement, including approval of the transaction by the Company's stockholders and Oncternal's stockholders, a wholly-owned subsidiary of the Company will be merged with and into Oncternal (the "Merger"), with Oncternal surviving the Merger as a wholly-owned subsidiary of the Company. See Note 2, Significant Accounting Policies — Subsequent Events, for further discussion regarding the proposed Merger.Atyear ended December 31,2018, the Company had cash, cash equivalents and short-term investments of $28,458 compared to $43,899 at December 31, 2017. To conserve its cash resources, the Company has substantially reduced its workforce since November 2018 and has ceased its SARM developmentGTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)activities and all other operations except for day-to-day business operations, completing ongoing mechanistic SARD preclinical studies and those activities necessary to complete the proposed Merger.2. Significant Accounting PoliciesBasis of Presentation2019:The accompanying financial statements have been prepared in accordance with U.S. generally accepted accounting principles ("U.S. GAAP"). Additionally, GTx operates in one business segment.Use of EstimatesThe preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual amounts and results could differ from those estimates.Cash and Cash EquivalentsThe Company considers highly liquid investments with initial maturities of three months or less to be cash equivalents.Short-term InvestmentsAt December 31, 2018 and 2017, short-term investments consisted of Federal Deposit Insurance Corporation ("FDIC") insured certificates of deposit with original maturities of greater than three months and less than one year.Property and EquipmentProperty and equipment is stated at cost. Amortization of leasehold improvements is recognized over the shorter of the estimated useful life of the leasehold improvement or the lease term. Depreciation is computed using the straight-line method over the estimated useful lives as follows:F-1
Office equipment3 to 5 yearsF-2
Leasehold improvements3 to 7 yearsF-3
Furniture and fixtures5 yearsF-4
Computer equipment and softwareF-5
3 yearsF-6
Warrant LiabilityIn November 2014, the Company issued warrants to purchase 6,430,948 shares of its common stock. The Company classified these warrants as a liability on its balance sheet since the warrants contained certain terms that could have required the Company (or its successor) to purchase the warrants for cash in an amount equal to the value (as calculated utilizing a contractually-agreed Black-Scholes-Merton option pricing valuation model ("Black-Scholes Model")) of the unexercised portion of the warrants in connection with certain change of control transactions occurring on or prior toTable of Contents2. Financial Statement SchedulesGTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)December 31, 2016, with such cash payment capped at an amount equal to $1.25 per unexercised share underlying each warrant. As a result of the provision of the warrants requiring cash settlement upon certain change of control transactions, the Company was required to account for these warrants as a liability at fair value and the estimated warrant liability was required to be revalued at each balance sheet date until the earlier of the exercise of the warrants, the modification to remove the provision that could require cash settlement upon certain change of control transactions or the expiration of such provision on December 31, 2016. Effective March 25, 2016, each of the warrants was amended by agreement of the warrant holders to remove the provision that could require cash settlement upon certain change of control transactions.Thesewarrants were no longer accounted for as a liability as of March 31, 2016. The Company recorded a non-cash reclassification of the warrant fair value to stockholders' equity based on the warrants' fair value as of the March 25, 2016 modification date, with no further adjustments to the fair value of these warrants being required.Fair Value of Financial Instruments and Warrant LiabilityThe carrying amounts of the Company's financial instruments (which include cash, cash equivalents, short-term investments, and accounts payable) and its prior warrant liability approximate their fair values. The fair value of the warrant liability was estimated using the Black-Scholes-Merton Model. See Note 6, Stockholders' Equity, for additional disclosure on the valuation methodology and significant assumptions. The Company's financial assets and liabilities are classified within a three-level fair value hierarchy that prioritizes the inputs used to measure fair value, which is defined as follows:Level 1 — Quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement dateLevel 2 — Inputs other than quoted prices in active markets that are observable for the asset or liability, either directly or indirectlyLevel 3 — Inputs that are unobservable for the asset or liabilityAs the Company has the positive intent and ability to hold its certificates of deposit classified as short-term investments until maturity, these investmentsschedules have beenclassified as held to maturity investments and are stated at cost, which approximates fair value. The Company considers these to be Level 2 investments asomitted because thefair values of these investments are determined using third-party pricing sources, which generally utilize observable inputs, such as interest rates and maturities of similar assets.Concentration of RiskFinancial instruments that potentially subject the Company to a concentration of credit risk consist of cash and cash equivalents and short-term investments. The Company has established guidelines relating to diversification and maturities of its cash equivalents and short-term investments which are designed to manage risk. The Company's cash and cash equivalents consist of bank deposits, certificates of deposit, and money market mutual funds. Bank deposits may at times be in excess of FDIC insurance limits. The Company's short-term investments consist of FDIC insured certificates of deposit with original maturities of greater than three months and less than one year.GTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)Research and Development ExpensesResearch and development expenses include, but are not limited to, the Company's expenses for personnel, supplies, and facilities associated with research activities, screening and identification of product candidates, formulation and synthesis activities, manufacturing, preclinical studies, toxicology studies, clinical trials, regulatory and medical affairs activities, quality assurance activities and license fees. The Company expenses these costs in the period in which they are incurred. The Company estimates its liabilities for research and development expenses in order to match the recognition of expenses to the period in which the actual services are received. As such, accrued liabilities related to third party research and development activities are recognized based upon the Company's estimate of services received and degree of completion of the services in accordance with the specific third party contract.Patent CostsThe Company expenses patent costs, including legal expenses, in the period in which they are incurred. Patent expenses are included in general and administrative expenses in the Company's statements of operations.Income TaxesThe Company accounts for deferred taxes by recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have beenrequired information is included in the financial statements ortax returns. Under this method, deferred tax assets and liabilitiesnotes thereto or because they aredetermined basednot applicable or not required.3. Exhibits
A list of exhibits is set forth on the
difference betweenExhibit Index immediately preceding thefinancial statementsignature page of this annual report on Form 10-K andtax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. A valuation allowanceisprovided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. Accordingly, at December 31, 2018 and December 31, 2017, net of the valuation allowance, the net deferred tax assets were reduced to zero. See Note 8,Income Taxes, for further discussion.Share-Based CompensationThe Company has stock option and equity incentive plans that provide for the purchase or acquisition of the Company's common stockincorporated herein bycertain of the Company's employees and non-employees. The Company recognizes compensation expense for its share-based payments based on the fair value of the awards over the period during which an employee or non-employee is required to provide service in exchange for the award. See Note 3,Share-Based Compensation, for further discussion.reference.Other Income (Expense), NetNone.Other income (expense), net consists of interest earned on the Company's cash, cash equivalents and short-term investments, foreign currency transaction gains and losses, and other non-operating income or expense.Basic and Diluted Net Loss Per ShareBasic and diluted net income (loss) per share attributable to common stockholders is calculated based on the weighted average number of common shares outstanding during the period. Diluted netGTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)income (loss) per share gives effect to the dilutive potential of common stock consisting of stock options, unvested RSUs and common stock warrants.Weighted average potential shares of common stock of 11,191,431, 9,438,236, and 8,162,347 were excluded from the calculation of diluted net loss per share for the years ended December 31, 2018, 2017 and 2016, respectively, as inclusion of the potential shares would have had an anti-dilutive effect on the net loss per share for the periods. At December 31, 2018, the Company had 24,051,844 shares of common stock outstanding.Comprehensive LossFor all periods presented, there were no differences between net loss and comprehensive loss.Recent Accounting PronouncementsIn February 2016, the Financial Accounting Standards Board issued Accounting Standard Update ("ASU") 2016-02,Leases (Topic 842).This ASU requires that lessees recognize assets and liabilities on the balance sheet for the present value of the rights and obligations created by all leases with terms of more than 12 months. The ASU also will require disclosures designed to give financial statement users information on the amount, timing, and uncertainty of cash flows arising from leases. This new guidance will be effective for the Company as of January 1, 2019. The Company does not expect the adoption of the standard update to have a significant impact on its financial position or results of operations.Subsequent EventsThe Company has evaluated all events or transactions that occurred after December 31, 2018 up through the date the financial statements were issued. Other than as set forth below, there were no material recognizable or nonrecognizable subsequent events during the period evaluated.Merger Agreement with Oncternal and Related MattersMerger AgreementOn March 6, 2019, the Company entered into the Merger Agreement with Oncternal and Grizzly Merger Sub, Inc., a wholly-owned subsidiary of the Company ("Merger Sub"). Upon the terms and subject to the satisfaction of the conditions described in the Merger Agreement, including approval of the transaction by the Company's stockholders and Oncternal's stockholders, Merger Sub will be merged with and into Oncternal, with Oncternal surviving the Merger as a wholly-owned subsidiary of the Company.Subject to the terms and conditions of the Merger Agreement, at the effective time of the Merger (the "Effective Time"): (i) each share of Oncternal common stock outstanding immediately prior to the Effective Time (excluding shares held by the Company, Merger Sub or Oncternal and dissenting shares) will be converted solely into the right to receive a number of shares of the Company's common stock (the "Shares") equal to the exchange ratio described below, (ii) each outstanding Oncternal stock option will be assumed by the Company, and (iii) each outstanding Oncternal warrant will be assumed by the Company.GTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)3. ExhibitsUnder the exchange ratio formula in the Merger Agreement, the former Oncternal stockholders immediately before the Merger are expected to own approximately 75% of the outstanding capital stock of the Company, and the stockholders of the Company immediately before the Merger are expected to own approximately 25% of the outstanding capital stock of the Company, subject to certain assumptions. The exchange ratio formula excludes Oncternal's outstanding stock options and warrants and the Company's outstanding stock options and warrants.Exhibit
Number
Description
Form
File No.
Exhibit No
Filing Date
Filed/
Furnished
Herewith
2.1
8-K
000-50549
2.1
October 3, 2012
2.2*
8-K
000-50549
2.1
March 7, 2019
2.3
8-K
000-50549
2.1
April 30, 2019
2.4
8-K
000-50549
10.1
June 10, 2019
3.1
S-3
333-127175
4.1
August 4, 2005
3.2
Certificate of Amendment of Restated Certificate of Incorporation of the Registrant
8-K
000-50549
3.2
May 6, 2011
3.3
Certificate of Amendment of Restated Certificate of Incorporation of the Registrant
8-K
000-50549
3.3
May 9, 2014
3.4
Certificate of Amendment of Restated Certificate of Incorporation of the Registrant
10-Q
000-50549
3.4
May 11, 2015
3.5
Certificate of Amendment of Restated Certificate of Incorporation of the Registrant
8-K
000-50549
3.1
December 5, 2016
3.6
8-K
000-50549
3.1
June 10, 2019
3.7
Certificate of Amendment of the Restated Certificate related to the Name Change of the Registrant
8-K
000-50549
3.2
June 10, 2019
3.8
8-K
000-50549
3.3
June 10, 2019
4.1
10-Q
000-50549
4.2
August 9, 2019
4.2
S-3
333-221040
4.9
October 20, 2017
4.3
Form of Warrant to purchase shares of Series B-2 Preferred Stock of Oncternal Therapeutics, Inc.
S-4
333-230758
4.11
April 8, 2019
4.4
10-Q
000-50549
4.1
August 9, 2019
4.5
X
10.1†
S-4
333-230758
10.46
April 8, 2019
10.2†
X
10.3†
S-4
333-230758
10.47
April 8, 2019
10.4
S-4
333-230758
10.48
April 8, 2019
10.5†
S-4
333-230758
10.49
April 8, 2019
10.6†
S-4
333-230758
10.50
April 8, 2019
10.7†
S-4
333-230758
10.51
April 8, 2019
10.8†
S-4
333-230758
10.52
April 8, 2019
10.9†
S-4
333-230758
10.53
April 8, 2019
10.10†
S-4
333-230758
10.54
April 8, 2019
10.11†
S-4
333-230758
10.55
April 8, 2019
10.12†
S-4
333-230758
10.56
April 8, 2019
10.13†
X
10.14#
Oncternal Therapeutics, Inc. 2015 Equity Incentive Plan, as amended
S-4
333-230758
10.57
April 8, 2019
10.15#
S-4
333-230758
10.58
April 8, 2019
10.16#
S-4
333-230758
10.59
April 8, 2019
10.17#
S-4
333-230758
10.60
April 8, 2019
10.18#
S-4
333-230758
10.61
April 8, 2019
10.19#
S-4
333-230758
10.62
April 8, 2019
10.20#
S-4
333-230758
10.63
April 8, 2019
10.21#
Employment Agreement dated August 26, 2019 between Oncternal Therapeutics, Inc. and Frank Hsu, M.D.
10-Q
000-50549
10.1
November 8, 2019
10.22#
10-Q
000-50549
10.2
November 8, 2019
10.23#
10-Q
000-50549
10.3
November 8, 2019
10.24#
10-Q
000-50549
10.4
November 8, 2019
10.25#
10-Q
000-50549
10.5
November 8, 2019
10.26#
Employment Agreement dated September 5, 2019 between Oncternal Therapeutics, Inc. and Hazel M. Aker
10-Q
000-50549
10.6
November 8, 2019
10.27#
Registrant’s 2019 Incentive Award Plan effective June 7, 2019
8-K
000-50549
10.2
June 10, 2019
10.28
10-Q
000-50549
10.7
November 8, 2019
10.29
10-Q
000-50549
10.23
August 9, 2019
10.30
8-K
000-50549
10.1
February 9, 2018
10.31
X
10.32#
X
10.33#
10-K
000-50549
10.6
March 24, 2017
10.34#
10-K
000-50549
10.7
March 24, 2017
10.35#
GTx, Inc. 2004 Equity Incentive Plan, as originally adopted, and Form of Stock Option Agreement
S-1
333-109700
10.5
January 15, 2004
10.36#
GTx, Inc. 2004 Equity Incentive Plan, as amended effective April 30, 2008
8-K
000-50549
10.6
May 6, 2008
10.37#
10-K
000-50549
10.10
March 24, 2017
10.38#
S-1
333-109700
10-6
January 15,2004
10.39#
8-K
000-50549
10-1
April 27, 2006
10.40#
10-Q
000-50549
10.35
August 9,2006
10.41#
10-K
000-50549
10.14
Mar 24, 2017
10.42#
S-8
333-188377
99.1
May 6, 2013
10.43#
GTx, Inc. 2013 Equity Incentive Plan, as amended effective May 6, 2015
10-K
000-50549
10.16
March 24, 2017
10.44#
10-Q
000-50549
10.2
July 22, 2013
10.45#
10-Q
000-50549
10.3
November 12, 2013
10.46#
10-Q
000-50549
10.4
November 12, 2013
10.47#
10-Q
000-50549
10.5
May 11, 2015
10.48#
10-K
000-50549
10.21
March 24, 2017
10.49#
10-Q
000-50549
10.4
Jul 22, 2013
21.1
X
23.1
X
24.1
X
31.2
X
32.2*
X
31.1
X
32.1*
X
XBRL Instance Document
X
101.SCH
XBRL Taxonomy Extension Schema Linkbase Document
X
101.CAL
XBRL Taxonomy Extension Calculation Linkbase Document
X
101.DEF
XBRL Taxonomy Definition Linkbase Document
X
101.LAB
XBRL Taxonomy Extension Label Linkbase Document
X
101.PRE
XBRL Taxonomy Extension Presentation Linkbase Document
X
*
These certifications are being furnished solely to accompany this quarterly report pursuant to 18 U.S.C. Section 1350, and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934 and are not subject to the liability of that section. These certifications are not to be incorporated by reference into any filing of Oncternal Therapeutics, Inc., whether made before or after the date hereof, regardless of any general incorporation language in such filing.
#
Management compensatory plan or arrangement.
†
Pursuant to Item 601(b)(10) of Regulation S-K, certain confidential portions of this exhibit were omitted by means of marking such portions with an asterisk because the identified confidential portions (i) are not material and (ii) would be competitively harmful if publicly disclosed.
Under certain circumstances further described in the Merger Agreement, the ownership percentages may be adjusted upward or downward based on cash levels of the respective companies at the closing of the Merger (the "Closing").The Merger Agreement contains customary representations, warranties and covenants made by the Company and Oncternal, including covenants relating to obtaining the requisite approvals of the stockholders of the Company and Oncternal, indemnification of directors and officers, the Company's and Oncternal's conduct of their respective businesses between the date of signing of the Merger Agreement and the Closing. The Closing is subject to satisfaction or waiver of certain conditions included in the Merger Agreement.Following the Closing, Oncternal's Chief Executive Officer, Chief Financial Officer, and Chief Operating Officer will serve in these positions for the Company. Additionally, following the Closing, the Company's board of directors will consist of nine directors, including two current GTx board members.The Merger Agreement also includes termination provisions for both the Company and Oncternal. In connection with a termination of the Merger Agreement under specified circumstances, either party may be required to pay the other party a termination fee ranging between $500 to $2,000.Contingent Value Rights AgreementAt the Effective Time, the Company will enter into a Contingent Value Rights Agreement (the "CVR Agreement").Pursuant to theCVR Agreement, for each sharerequirements of theCompany's common stock held,Section 13 or 15(d) of theCompany's stockholdersSecurities and Exchange Act ofrecord as of immediately prior1934, the registrant has duly caused this report tothe Effective Time will receive one contingent value right ("CVR") entitling such holders to receive in the aggregate 50% of any net proceeds received during the 15-year period after closing from the grant, sale or transfer of rights to the Company's SARD or SARM technology that occurs during the 10-year period after the Closing (or in the 11thyear if basedbe signed ona term sheet approved during the initial 10-year period) and, if applicable, to receive royalties on the sale of any SARD productsits behalf by thecombined company during the 15-year period after Closing. The CVR Agreement will be effective prior to the Closing and will continue in effect until the payment of all amounts payable thereunder, unless terminated upon termination of the Merger Agreement.Workforce ReductionIn the first quarter of 2019, due to the entry into the Merger Agreement with Oncternal, the Company's board of directors committed to reducing its workforce by seven employees. All employees affected by the workforce reduction will be eligible to receive, among other things, specified severance payments based on the applicable employee's level and years of service with the Company and the continuation of group health insurance coverage. In addition, the affected employees will also beGTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)eligible for full vesting acceleration of their outstanding stock options as well as an extension of the post-termination exercise period for their outstanding stock options.undersigned, thereunto duly authorized.As a result of the workforce reduction and prior termination of three employees earlier in the first quarter of 2019, the Company estimates that it will incur total severance-related charges for these employees of approximately $1,000 in the first quarter of 2019 and up to an additional $500 contingent upon the closing of the Merger. The Company does not expect to record a non-cash charge related to the modification of outstanding stock options in connection with the workforce reduction.Termination of Directors' Deferred Compensation PlanPrior to the Effective Time (but in no event more than 30 days prior to the Effective Time), the Company's board of directors will take all actions necessary to terminate and liquidate the Company's 2018 Amended and Restated Directors' Deferred Compensation Plan (the "Directors' Deferred Compensation Plan") and all rights or other deferrals thereunder effective immediately prior to the Effective Time, subject to the consummation of the Merger. Any future board compensation under the Directors' Deferred Compensation Plan on or after January 3, 2019 shall be settled only in cash.3. Share-Based CompensationShare-based payments include stock option and RSU grants under the Company's stock option and equity incentive plans and deferred compensation arrangements for the Company's non-employee directors.The Company has granted to employees and non-employees options to purchase common stock under various plans at prices equal to the fair market value of its common stock on the dates the options are granted as determined in accordance with the terms of the applicable plan. The options have a term of ten years from the grant date and generally vest over three years from the grant date for director and non-employee options and over periods of up to five years from the grant date for employee options. Under the terms of the Company's stock option and equity incentive plans, employees generally have three months after the employment relationship ends to exercise all vested options except in the case of voluntary retirement, disability or death, where post-termination exercise periods are generally longer. The Company issues new shares of common stock upon the exercise of options. The Company estimates the fair value of stock option awards as of the date of the grant by applying the Black-Scholes Model. The application of this valuation model involves assumptions that are judgmental and highly sensitive in the determination of compensation expense.The fair value of each stock option is amortized into compensation expense on a straight-line basis between the grant date for the award and each vesting date. During 2017, the Company adopted the Financial Accounting Standards Board Accounting Standards Update 2016-09,Improvements to Employee Share Based Payment Accounting. This guidance addresses the income tax effects of stock-based payments and eliminates the windfall pool concept, as all of the tax effects related to stock-based payments are now being recorded at settlement (or expiration) through the income statement. The new guidance also permits entities to make an accounting policy election for the impact of forfeitures on the recognition of expense for stock-based payment awards, allowing for forfeitures to be estimated or recognized when they occur. The Company elected to prospectively adopt the policy that forfeitures be recorded when they occur and prior periods have not been adjusted. The adoption of this guidance did not have a material impact on the Company's financial position or results of operations.GTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)Additionally, the Company periodically grants RSUs to its employees. The Company estimates the fair value of RSUs using the closing price of its common stock on the grant date. The fair value of the RSUs is amortized on a straight-line basis over the requisite service period of the awards. All RSUs were fully vested at December 31, 2018.The following table summarizes share-based compensation expense included within the statements of operations for each of the three years in the period ended December 31, 2018:Years Ended December 31, 2018 2017 2016 Research and development expenses
$ 807 $ 1,171 $ 1,260 General and administrative expenses
1,556 2,146 1,829 Total share-based compensation
$ 2,363 $ 3,317 $ 3,089 Share-based compensation expense recorded in the statement of operations as general and administrative expense for the years ended December 31, 2018, 2017 and 2016 included share-based compensation expense related to deferred compensation arrangements for the Company's non-employee directors of $166, $166 and $132, respectively. See Note 9,Directors' Deferred Compensation Plan, for further discussion of deferred compensation arrangements for the Company's non-employee directors.For the years ended December 31, 2018, 2017 and 2016, the weighted average grant date fair value per share of stock options granted was $10.36, $3.80 and $5.45, respectively. The key assumptions used in determining the grant date fair value of options granted in 2018, 2017 and 2016, and a summary of the methodology applied to develop each assumption is as follows:Years Ended December 31, 2018 2017 2016 Expected price volatility
93.1% 88.6% 91.3% Risk-free interest rate
2.4% 2.2% 2.0% Weighted average expected life in years
6.9 years 6.9 years 6.9 years Dividend yield
0% 0% 0% Expected Price Volatility— This is a measure of the amount by which a price has fluctuated or is expected to fluctuate. The Company based its determination of expected volatility on its historical stock price volatility. An increase in the expected price volatility will increase compensation expense.Risk-Free Interest Rate— This is determined using U.S. Treasury rates where the term is consistent with the expected life of the stock options. An increase in the risk-free interest rate will increase compensation expense.Expected Life— This is the period of time over which the options granted are expected to remain outstanding and is determined by calculating the average of the vesting term and the contractual term of the options. The Company has utilized this method due to the lack of historical option exerciseGTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)information related to the Company's stock option and equity incentive plans. Options granted have a maximum term of ten years. An increase in the expected life will increase compensation expense.Dividend Yield— The Company has not made any dividend payments nor does it have plans to pay dividends in the foreseeable future. An increase in the dividend yield will decrease compensation expense.The following is a summary of stock option transactions for all of the Company's stock option and equity incentive plans for the three year period ended December 31, 2018:Number of
SharesWeighted
Average
Exercise Price
Per ShareOptions outstanding at January 1, 2016
798,309 $ 38.80 Options granted
363,500 6.94 Options forfeited or expired
(71,829 ) 54.65 Options exercised
- - Options outstanding at December 31, 2016
1,089,980 27.13 Options granted
977,350 4.97 Options forfeited or expired
(166,834 ) 48.71 Options exercised
- - Options outstanding at December 31, 2017
1,900,496 13.84 Options granted
472,000 13.32 Options forfeited or expired
(31,049 ) 168.76 Options exercised
(6,000 ) 17.23 Options outstanding and vested or expected to vest at December 31, 2018
2,335,447 $ 11.67 The following table summarizes information about stock options outstanding at December 31, 2018:Options Outstanding Options Exercisable Exercise PriceNumber
OutstandingWeighted
Average
Remaining
Contractual
Life (years)Weighted
Average
Exercise
PriceNumber
ExercisableWeighted
Average
Exercise
Price$4.29 - $4.29 56,250 8.36 $ 4.29 18,750 $ 4.29 $4.71 - $4.71 825,000 8.02 4.71 22,500 4.71 $4.77 - $12.71 826,800 7.67 9.97 134,404 8.25 $12.95 - $108.90 627,397 4.20 23.71 508,691 25.59 2,335,447 6.88 11.67 684,345 20.91 GTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)At December 31, 2018, the aggregate intrinsic value of all outstanding options was zero with a weighted average remaining contractual term of 6.88 years. Of the Company's outstanding options, 684,345 options were exercisable and had a weighted average remaining contractual term of 4.05 years and no aggregate intrinsic value. Additionally, the Company's vested and expected to vest options had a weighted average remaining contractual term of 6.88 years and no aggregate intrinsic value.Options to purchase 6,000 shares were exercised during the years ended December 31, 2018. The total intrinsic value of options exercised during the years ended December 31, 2018 was $39. At December 31, 2018, the total compensation cost related to non-vested options not yet recognized was $7,697, with a weighted average expense recognition period of 2.95 years. Shares available for future issuance under the Company's stock option and equity incentive plans were 1,167,162 at December 31, 2018. On January 1, 2019, shares available for future issuance under the 2013 equity incentive plan and the 2013 non-employee director equity incentive plan increased by an aggregate of 1,012,074 shares in accordance with the automatic increase provisions of such plans.The following is a summary of the RSU transactions for all of the Company's equity incentive plans for the three year period ended December 31, 2018:Number ofSharesNonvested RSUs outstanding at January 1, 2016820,000Oncternal Therapeutics, Inc.
RSUs granted11,000RSUs vestedDate: March 16, 2020By:
(184,001)/s/ James B. Breitmeyer, M.D., PH.D.
RSUs forfeited(62,000)Nonvested RSUs outstanding at December 31, 2016584,999RSUs granted-RSUs vestedJames B. Breitmeyer, M.D., Ph.D.(168,499)RSUs forfeitedPresident and Chief Executive Officer(36,000)Nonvested RSUs outstanding at December 31, 2017380,500RSUs granted-RSUs vested(380,500)RSUs forfeited-Nonvested RSUs outstanding at December 31, 2018-The numberKNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Dr. James B. Breitmeyer, M.D., Ph.D and Richard G. Vincent, and each of
RSUs vested during 2018, 2017,them, as his or her true and. lawful attorneys-in-fact and2016 included 52,645, 29,482,agents, each with full power of substitution and29,829 shares, respectively,resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments (including post-effective amendments) to this report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all thatwere withheldsaid attorneys-in-fact and agents, or either of them, or their or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the
Company's employees to satisfy the statutory tax withholding requirements.GTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except shareregistrant andper share data) (Continued)4. Property and Equipment, NetProperty and equipment, net consisted of the following:December 31, 2018 2017 Computer equipment and software
$ 1,225 $ 1,299 Furniture and fixtures
853 853 Leasehold improvements
355 355 Office equipment
211 211 2,644 2,718 Less: accumulated depreciation
(2,625 ) (2,667 ) $ 19 $ 51 Depreciation and amortization expense for the years ended December 31, 2018, 2017 and 2016 was $32, $32, and $14, respectively. Of these amounts, $1, $2 and $2, respectively, were included in research and development expensesin thestatements of operations.5. Accrued Expensescapacities andOther Current LiabilitiesAccrued expenses and other current liabilities consisted of the following:December 31, 2018 2017 Clinical trials
$ 1,492 $ 4,742 General and administrative
101 314 Research and development
272 312 Employee compensation
66 3 $ 1,931 $ 5,371 6. Stockholders' EquityAuthorized CapitalOn December 5, 2016, the Company filed a Certificate of Amendment to the Company's Restated Certificate of Incorporation with the Secretary of State of the State of Delaware to effect a one-for-ten reverse stock split of its outstanding common stock and to effect a reduction in the number of authorized shares of common stock from 400,000,000 to 60,000,000 shares. The Company's certificate of incorporation currently authorizes the Company to issue 60,000,000 shares of common stock, $0.001 par value per share, and 5,000,000 shares of preferred stock, $0.001 par value per share.Common StockOn February 9, 2018, the Company entered into an At-the-Market Equity Offering Sales Agreement (the "ATM Sales Agreement") with Stifel, Nicolaus & Company, Incorporated, as sales agent ("Stifel"), pursuant to which the Company may offer and sell, from time to time, through Stifel, shares of the Company's common stock, having an aggregate offering price of up to $50,000. OnGTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)May 16, 2018, the Company sold 1,501,501 shares of common stock under the ATM Sales Agreement for net proceeds of $24,474. As of December 31, 2018, the Company had approximately $25,000 of common stock remaining available to be sold under the ATM Sales Agreement.On September 29, 2017, the Company completed a private placement of units consisting of an aggregate of 5,483,320 shares of common stock and warrants to purchase an aggregate of 3,289,988 shares of its common stock for net proceeds of $45,648, after deducting placement agent fees and other offering expenses. The purchasers in the private placement consisted solely of accredited investors that included certain institutional and existing stockholders, including a member of the Company's board of directors. The warrants, which have five year terms expiring on September 29, 2022, are immediately exercisable and have a per share exercise price of $9.02. The Company assessed whether the warrants require accounting as derivatives. The Company determined that the warrants were indexed to the Company's own stock. As such, the Company has concluded the warrants meet the scope exception for determining whether the instruments require accounting as derivatives and are classified in stockholders' equity. The fair value of the warrants was estimated at $21,069 using the Black-Scholes Model with the following assumptions: expected volatility of 97%, risk free interest rate of 1.92%, expected life of five years and no dividends. The net proceeds from the private placement were allocated to the common stock and warrants based upon their relative fair values.On October 14, 2016, the Company completed a registered direct offering of its common stock. Under the terms of the offering, the Company sold 1,728,395 shares of its common stock for net proceeds of $13,692, after deducting offering expenses.On November 14, 2014, the Company completed a private placement of units consisting of an aggregate of 6,431,111 shares of common stock and warrants to purchase an aggregate of 6,430,948 shares of its common stock for net proceeds of $42,814, after deducting offering expenses. The net proceeds from the private placement were allocated to the common stock and warrants based upon the fair value method. Similarly, the offering expenses were allocated between the common stock and warrants with the portion allocated to common stock offset against the proceeds allocated to stockholders' equity, whereas the portion allocated to the warrants was expensed immediately. The warrants have a per share exercise price of $8.50, became exercisable on May 6, 2015 and will continue to be exercisable for four years thereafter. Prior to May 6, 2015, each warrant was subject to net cash settlement if, at the time of any exercise, there was then an insufficient number of authorized and reserved shares of common stock to effect a share settlement of the warrant. Under the terms of the warrants, as of May 6, 2015, the net cash settlement feature of the warrants automatically became inoperative; accordingly, the warrants are exercisable only for shares of the Company's common stock. The warrants, however, also contained certain terms that could have required the Company (or its successor) to purchase the warrants for cash in an amount equal to the value (as calculated utilizing a contractually-agreed Black-Scholes Model) of the unexercised portion of the warrants in connection with certain change of control transactions occurring on or prior to December 31, 2016, with the cash payment capped at an amount equal to $1.25 per unexercised share underlying each warrant. Due to the provision of the warrants that could have required cash settlement upon certain change of control transactions, the Company was required to account for these warrants as a liability at fair value using the Black-Scholes Model and the estimated warrant liability was required to be revalued at each balance sheet date until the earlier of the exercise of the warrants, the modification to remove the provision that could require cash settlement upon certain change of control transactions or the expiration of such provision on December 31, 2016. Effective March 25, 2016, each of the warrants wasGTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)amended by agreement of the warrant holders to remove the provision that could require cash settlement upon certain change of control transactions. These warrants were no longer accounted for as a liability at March 31, 2016. The Company recorded a non-cash reclassification of the warrant fair value to stockholders' equity basedon thewarrants' fair value as of the March 25, 2016 modification date, with no further adjustments to the fair value of these warrants being required. In March 2018, certain holders of warrants issued in November 2014 exercised warrants covering 1,111,082 shares of common stock in a cashless exercise for which the Company issued an aggregate of 674,579 shares of common stock upon exercise.dates indicatedEach of these completed offerings included certain existing GTx stockholders and/or certain members of the GTx management team and/or board of directors.7. License AgreementsUniversity of Tennessee Research Foundation License AgreementsThe Company and the University of Tennessee Research Foundation ("UTRF") are parties to a consolidated, amended and restated license agreement (the "SARM License Agreement") pursuant to which the Company has been granted exclusive worldwide rights in all existing SARM technologies owned or controlled by UTRF, including all improvements thereto, and exclusive rights to future SARM technology that may be developed by certain scientists at the University of Tennessee or subsequently licensed to UTRF under certain existing inter-institutional agreements with The Ohio State University. Under the SARM License Agreement, the Company is obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and mid single-digit royalties on sublicense revenues.In accordance with the terms of the SARM License Agreement that the Company entered into with UTRF in July 2007, the Company paid a one-time up-front fee of $290, which was recorded as an intangible asset by the Company. This intangible asset, net at December 31, 2018 and 2017 was $94 and $108, respectively.The Company and UTRF also entered into a license agreement in March 2015 pursuant to which the Company was granted exclusive worldwide rights in all existing SARD technologies owned or controlled by UTRF, including all improvements thereto (the "SARD License Agreement"). Under the SARD License Agreement, the Company is obligated to employ active, diligent efforts to conduct preclinical research and development activities for the SARD program to advance one or more lead compounds into clinical development. The Company is also obligated to pay UTRF annual license maintenance fees, low single-digit royalties on net sales of products and additional royalties on sublicense revenues, depending on the state of development of a clinical product candidate at the time it is sublicensed.8. Income TaxesThe Tax Cuts and Jobs Act ("Tax Reform Act") was enacted on December 22, 2017. The Tax Reform Act significantly revised the U.S. corporate income tax regime by, among other things, lowering the U.S. corporate tax rate from 35% to 21% effective January 1, 2018. On December 22, 2017, the SEC staff issued Staff Accounting Bulletin No. 118 ("SAB 118") to address the application of U.S. GAAP in situations when a registrant does not have the necessary information available, prepared,GTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the Tax Reform Act. The Company recognized the provisional tax impacts related to the revaluation of deferred tax assets and liabilities and included these amounts in its financial statements for the year ended December 31, 2017. During 2018 the Company completed the accounting under the Tax Reform Act as allowed under SAB 118 to revalue its deferred tax assets and liabilities which resulted in no change to the amounts previously recorded by the Company for year ended December 31, 2017.Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The principal components of the Company's net deferred income tax assets and liabilities consisted of the following:December 31, 2018 2017 Deferred income tax assets:
Net federal and state operating loss carryforwards
$ 120,555 $ 110,145 Research and development credits
16,383 14,757 Share-based compensation
3,130 3,994 Depreciation and amortization
17 21 Total deferred tax assets
140,085 128,917 Deferred income tax liabilities:
Other
461 92 Total deferred tax liabilities
461 92 Net deferred tax assets
139,624 128,825 Valuation allowance
(139,624 ) (128,825 ) $ - $ - Signature
Title
Date
/s/ James B. Breitmeyer
President, Chief Executive Officer and Member of the Board of Directors
March 16, 2020
James B. Breitmeyer, M.D., Ph.D.
(Principal Executive Officer)
/s/ Richard G. Vincent
Chief Financial Officer
March 16, 2020
Richard G. Vincent
(Principal Financial Officer)
/s/ David F. Hale
Chairman of the Board of Directors
March 16, 2020
David F. Hale
/s/ Michael G. Carter
Director
March 16, 2020
Michael G. Carter, M.D., ChB, FRCP
/s/ William R. LaRue
Director
March 16, 2020
William R. LaRue
/s/ Xin Nakanishi
Director
March 16, 2020
Xin Nakanishi, Ph.D.
/s/ Robert Wills
Director
March 16, 2020
Robert Wills, Ph.D.
/s/ Charles P. Theuer
Director
March 16, 2020
Charles P. Theuer, M.D., Ph.D.
Realization of deferred income tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, due to the Company's history of net operating losses, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $10,799 in 2018, decreased by $47,132 in 2017 and increased in 2016 by $9,347. The valuation allowance decrease in 2017 was due primarily to the passage of the Tax Reform Act and the reduction in the valuation of the Company's net deferred tax assets as a result of the lowering of the corporate tax rate from 35% to 21% effective January 1, 2018.At December 31, 2018, the Company had net federal operating loss carryforwards of approximately $472,054. The federal operating loss carryforwards originating prior to 2018 will expire from 2019 to 2037 if not utilized. The Company had state operating loss carryforwards of approximately $411,396, which expire from 2019 to 2038 if not utilized. The Company also had research and development credits at December 31, 2018 of approximately $16,383, which expire from 2020 to 2038 if not utilized.129The Company will recognize the impact of a tax position in the financial statements if that position is more likely than not of being sustained on audit based on the technical merits of the position. As ofGTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)December 31, 2018, the Company had no unrecognized tax benefits. Utilization of the Company's net operating loss carryforwards may be subject to a substantial annual limitation due to ownership change limitations provided by Section 382 of the Internal Revenue Code of 1986, as amended, and similar state provisions. The annual limitations may result in the expiration of net operating loss carryforwards before utilization. The Company completed a study of its net operating losses through December 31, 2016 to determine whether such amounts are likely to be limited by Section 382. As a result of this study and its analysis of subsequent ownership changes, the Company does not currently believe any Section 382 limitation exists through December 31, 2018 though the Company has not yet conducted an in-depth analysis since the last study. However, any future ownership changes under Section 382 may limit the Company's ability to fully utilize these tax benefits. The Company has not yet conducted an in-depth study of its research and development credits, although the Company periodically reviews assumptions used in its calculations to reflect its best estimate of expected credit. An in-depth study may result in an increase or decrease to the Company's research and development credits and until such study is conducted of the Company's research and development credits, no amounts are being presented as an uncertain tax position. The Company's net deferred income tax assets have been fully offset by a valuation allowance. Therefore, future changes to the Company's unrecognized tax benefits would be offset by an adjustment to the valuation allowance and there would be no impact on the Company's balance sheet, statement of operations, or cash flows. The Company does not expect its unrecognized tax benefits to change significantly over the next 12 months.The Company is currently open to audit under the statute of limitations by the Internal Revenue Service and the appropriate state income taxing authorities for all years due to the net loss carryforwards from those years. The Company is currently not under examination by the Internal Revenue Service or any other taxing authorities. The Company has not recorded any interest and penalties on any unrecognized tax benefits since its inception.9. Directors' Deferred Compensation PlanNon-employee directors may defer all or a portion of their fees under the Company's Directors' Deferred Compensation Plan until termination of their status as directors. Deferrals can be made into a cash account, a stock account, or a combination of both. Stock accounts will be paid out in the form of Company common stock, except that any fractional shares will be paid out in cash valued at the then current market price of the Company's common stock. Cash accounts and stock accounts under the Directors' Deferred Compensation Plan are credited with interest or the value of any cash and stock dividends, respectively. Non-employee directors are fully vested in any amounts that they elect to defer under the Directors' Deferred Compensation Plan.For the years ended December 31, 2018, 2017 and 2016, the Company incurred non-employee director fee expense of $291, $291 and $257, respectively, of which $166, $166 and $132 was deferred into stock accounts and will be paid in common stock following separation from service as a director. At December 31, 2018, 122,725 shares of the Company's common stock had been credited to individual director stock accounts under the Directors' Deferred Compensation Plan, and no amounts had been credited to individual director cash accounts under the Directors' Deferred Compensation Plan.GTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)10. 401(k) PlanThe Company sponsors a 401(k) retirement savings plan that is available to all eligible employees. The plan is intended to qualify under Section 401(k) of the Internal Revenue Code of 1986, as amended. The plan provides that each participant may contribute up to a statutory limit of their pre-tax compensation which was $18.5 for employees under age 50 and $24.5 for employees 50 and older in calendar year 2018. Employee contributions are held in the employees' name and invested by the plan's trustee. The plan also permits the Company to make matching contributions, subject to established limits. The Company elected to match a portion of employee's contributions to the plan in the amount of $185, $186 and $200 in 2018, 2017 and 2016, respectively.11. Commitments and ContingenciesOperating Lease CommitmentsIn 2015, the Company entered into a new office lease with respect to the Company's current office space. The new office lease term commenced on May 1, 2015 with a three year term ending on April 30, 2018, with an option to extend the lease for an additional three years, and was accounted for as an operating lease. In March 2018, the Company amended the lease to extend the term of the lease for an additional 12-month term expiring on April 30, 2019. Total rent expense under the operating leases was approximately $509, $506 and $495 for the years ended December 31, 2018, 2017 and 2016, respectively. As of December 31, 2018, future annual minimum payments under operating lease arrangements were $162.12. Quarterly Financial Data (Unaudited)(1)The following is a summary of the quarterly results of operations for the years ended December 31, 2018 and 2017:2018 Quarters Ended March 31 June 30 September 30 December 31 Expenses: Research and development expenses
$ 11,000 $ 7,962 $ 7,467 $ 3,240 General and administrative expenses
2,688 2,196 2,160 2,346 Total expenses 13,688 10,158 9,627 5,586 Loss from operations (13,688 ) (10,158 ) (9,627 ) (5,586 ) Other income, net 131 143 196 171 Net loss $ (13,557 ) $ (10,015 ) $ (9,431 ) $ (5,415 ) Net loss per share: Basic and Diluted $ (0.62 ) $ (0.43 ) $ (0.39 ) $ (0.23 ) Weighted average shares outstanding: Basic and Diluted
21,967,805 23,288,691 24,045,992 24,051,844 GTx, Inc.NOTES TO FINANCIAL STATEMENTS(in thousands, except share and per share data) (Continued)2017 Quarters Ended March 31 June 30 September 30 December 31 Expenses:
Research and development expenses
$ 4,193 $ 4,448 $ 5,914 $ 6,912 General and administrative expenses
2,087 1,997 2,617 2,487 Total expenses
6,280 6,445 8,531 9,399 Loss from operations
(6,280 ) (6,445 ) (8,531 ) (9,399 ) Other income, net
27 40 27 122 Net loss
$ (6,253 ) $ (6,405 ) $ (8,504 ) $ (9,277 ) Net loss per share:
Basic and Diluted
$ (0.39 ) $ (0.40 ) $ (0.53 ) $ (0.43 ) Weighted average shares outstanding:
Basic and Diluted
16,018,342 16,041,923 16,115,835 21,541,909 (1)The sum of quarterly earnings per share amounts may not equal the annual amounts as the quarterly amounts are computed independently for each quarter while the full year is based on the annual weighted average shares outstanding.