related to the exchange of American Depositary Shares and common shares of Nabriva Austria for ordinary shares of Nabriva Ireland, which resulted in Nabriva Ireland, a new Irish holding company, becoming the ultimate holding company of Nabriva Austria (the predecessor registrant and former ultimate holding company) and its subsidiaries, which we refer to as the Redomiciliation Transaction. On October 19, 2017, Nabriva Austria was converted into a limited liability company under Austrian law and renamed Nabriva Therapeutics GmbH.
Unless the context requires otherwise, all references in this Annual Report to "Nabriva," "the Nabriva Group," "the Company," "we," "ours," "us," or similar terms on or prior to June 23, 2017 (the effective date of the Redomiciliation Transaction), refer to our predecessor, Nabriva Therapeutics AG, together with its subsidiaries.
Overview
Overview
We are a clinical stage biopharmaceutical company engaged in the research and development of novel anti-infective agents to treat serious infections, with a focus oninfections. We have two product candidates that have been submitted to the pleuromutilin class of antibiotics. We are developing our lead product candidate,U.S. Food and Drug Administration, or the FDA, for approval: lefamulin, to bepotentially the first pleuromutilin antibiotic available for systemicoral and intravenous (or IV) administration in humans. We are developing both intravenous, or IV, and oral formulations of lefamulinhumans, for the treatment of community-acquired bacterial pneumonia, or CABP and intend toCONTEPO, a potentially first-in-class epoxide antibiotic for IV use in the United States for complicated urinary tract infections, or cUTI. We may potentially develop lefamulin and CONTEPO for additional indicationsindications. Both lefamulin formulations and CONTEPO were granted Qualified Infectious Disease Product, or QIDP, and Fast Track designation by the FDA, enabling priority review of the New Drug Applications, or the NDAs by the FDA.
Lefamulin
Lefamulin is a semi-synthetic pleuromutilin antibiotic discovered and developed by our team with the potential to be first-in-class for IV and oral administration in humans. It inhibits the synthesis of bacterial protein, which is required for bacteria to grow by binding with high affinity and specificity at molecular targets that are different than other than pneumonia. We have completed a Phase 2 clinical trial of lefamulin for acute bacterial skin and skin structure infections, or ABSSSI.antibiotic classes. Based on the clinical results of lefamulin for ABSSSI, as well as its rapid tissue distribution, including substantial penetration into lung fluids and lung immune cells, we have initiatedfrom two pivotal, internationalglobal, Phase 3 clinical trials, ofwe believe lefamulin is well-positioned for use as first-line monotherapy for the treatment of moderateCABP due to severe CABP.
its novel mechanism of action, targeted spectrum of activity, resistance profile, achievement of substantial drug concentrations in lung tissue and fluid, availability of oral and IV formulations and a generally well-tolerated profile. We initiated the first of our Phase 3 trials, which we refer to as LEAP 1, in September 2015 and initiated the second trial, which we refer to as LEAP 2, in April 2016. These are the first clinical trials we have conducted withbelieve lefamulin represents a potentially important new treatment option for the treatment of CABP. Both trials are designedfive to follow draft guidance published bysix million adults in the United States diagnosed with CABP each year.
We submitted two NDAs to the FDA for the developmentoral and IV formulations of drugs for CABP and guidance from the European Medicines Agency, or EMA, for the development of antibacterial agents. Based on our estimates regarding patient enrollment, we expect to have top-line data from LEAP 1 in the third quarter of 2017. With respect to LEAP 2, based on current projections, we expect to complete patient enrollment in the fourth quarter of 2017, and we anticipate receiving top-line data for LEAP 2 in the first quarter of 2018. If the results of these trials are favorable, including achievement of the primary efficacy endpoints of the trials, we expect to submit applications for marketing approval for lefamulin for the treatment of CABP in December 2018. The FDA has granted us a Prescription Drug User Fee Act, or PDUFA, target action date of August 19, 2019 for lefamulin. We plan to submit a marketing authorization application for lefamulin in Europe in the first half of 2019. The two NDAs for lefamulin are supported by two pivotal, Phase 3 clinical trials (known as LEAP 1 and LEAP 2) that evaluated the safety and efficacy of IV and oral lefamulin compared to moxifloxacin in the treatment of adults with CABP, including the option to switch from IV to oral administration and a short course oral treatment with lefamulin. In both LEAP 1 and LEAP 2, lefamulin was demonstrated to be non-inferior to moxifloxacin, and met both the United StatesFDA and Europe in 2018. We believe that lefamulin is well suited for use as a first-line empiric monotherapyEuropean Medicines Agency, or EMA primary and secondary efficacy endpoints for the treatment of CABP because of its novel mechanism of action, spectrum of activity, including against multi-drug resistant pathogens, achievement of substantial drug concentrations in lung fluidsCABP. Lefamulin was also shown to be generally well-tolerated when administered both orally and lung immune cells, availability as both an IV and oral formulation and favorable safety and tolerability profile.
The U.S. Food and Drug Administration, or FDA, has designated each of the IV and oral formulations of lefamulin as a qualified infectious disease product, or QIDP, which provides for the extension of statutory exclusivity periods in the United States for an additional five years upon FDA approval of the product for the treatment of CABP and granted fast track designation to these formulations of lefamulin. Fast track designation is granted by the FDA to facilitate the development and expedite the review of drugs that treat serious conditions and fill an unmet medical need. The fast track designation for the IV and oral formulations of lefamulin will allow for more frequent interactions with the FDA, the opportunity for a rolling review of any new drug application, or NDA, we submit and eligibility for priority review and a shortening of the FDA’s goal for taking action on a marketing application from ten months to six months.
intravenously.
We believe that pleuromutilin antibiotics can help address the major public health threat posed by bacterial resistance, which the World Health Organization, or WHO, characterized in 20102017 as one of the three greatestbiggest threats to human health. Increasing resistance to antibiotics used to treat CABP is a growing concern and has become an issue in selecting the appropriate initial antibiotic treatment prior to determining the specific microbiological cause of the infection, referred to as empiric treatment. For example, the U.S. Centers for Disease Control and Prevention, or CDC, has classifiedStreptococcus pneumoniae, the most common respiratory pathogen, as a serious threat to human health as a result of increasing resistance to currently available antibiotics. In addition, the CDC recently reported on the growing evidence of widespread resistance to macrolides, widely used antibiotics that disrupt bacterial protein synthesis, inMycoplasma pneumoniae, a common cause of CABP that is associated with significant morbidity and mortality. Furthermore,Staphylococcus aureus, including methicillin-resistantS. aureus, or MRSA, which has also been designated as a serious threat to human health by the CDC,
has emerged as a more common cause of CABP in some regions of the world, and a possible pathogen to be covered with empiric therapy.
In recognition of the growing need for the development of new antibiotics, recent regulatory changes, including priority review and regulatory guidance enabling smaller clinical trials, have led to renewed interest from the pharmaceutical industry in anti-infective development. For example, the Food and Drug Administration Safety and Innovation Act became law in 2012 and included the Generating Antibiotic
Incentives Now Act, or the GAIN Act, which provides incentives, including access to expedited FDA review for approval, fast track designation and five years of potential data exclusivity extension for the development of new QIDPs.
As a result of increasing resistance to antibiotics and the wide array of potential pathogens that cause CABP, the current standard of care for hospitalized patients with CABP whose treatment is initiated in the hospital usually involves first-line empiric treatment with a combination of antibiotics (cephalosporins and macrolides) to address all likely bacterial pathogens or monotherapy with a fluoroquinolone antibiotic.respiratory fluoroquinolone. Combination therapy presents the logistical challenge of administering multiple drugs with different dosing regimens, with some drugs available only as IV, and increases the risk of drug-drug interactions and the potential for serious side effects. Fluoroquinolones are associated with safety and tolerability concerns, including a relatively high risk for developingClostridium difficile infections. In addition, infection and increasing rates of resistance for uropathogens. We believe these concerns have resulted in May 2016, thea decreasing use of fluoroquinolones and restriction of their use within a growing number of hospitals.
The FDA announcedhas communicated safety information about fluoroquinolones, advising that an FDA safety review has shown that fluoroquinolones, when used systemically, in the form of tablets, capsules and injectable, fluoroquinolones are associated with disabling and potentially permanent serious side effects thateffects. In December 2018, the FDA warned prescribers of an increase in the occurrence of rare but serious events of ruptures or tears in the main artery of the body, called the aorta. These tears, called aortic dissections, or ruptures of an aortic aneurysm can occur together. Theselead to dangerous bleeding or even death. Prior communications pertaining to the safety of fluoroquinolones occurred in July 2018 (significant decreases in blood sugar and certain mental health side effects), July 2016 (disabling side effects can involveof the tendons, muscles, joints, nerves, and central nervous system.system), May 2016 (restricting use for certain uncomplicated infections), August 2013 (peripheral neuropathy), and July 2008 (tendinitis and tendon rupture). The European Medicines Agency has also reviewed this class and have modified prescribing information restricting use, as well as outlining some of the safety risks.
Fluoroquinolones are typically administered in combination with other antibiotics, if community-acquired MRSA is suspected. In addition, many currently available antibiotic therapies are only available for IV administration and are prescribed for seven to 14fourteen days, meaning continued treatment requires prolonged hospitalization or a switch to a different antibiotic administered orally, with the attendant risk that the patient might respond differently.
Effective January 1, 2017, the Joint Commission & Center for Medicare and Medicaid Services, or CMS, began requiring all U.S. hospitals to have Antibiotic Management guidelines, also known as “Stewardship”"Stewardship" Committees, in place to identify antibiotics most appropriate and targeted to each individual patient’spatient's infection. Past efforts to “cast"cast the widest net possible”possible" with broad-spectrum antibiotics that affect many types of bacteria have caused problems, such asC. difficileinfections, by killing good bacteria or increased antibiotic resistance in other bacteria in different areas of the body. Additionally, in 2016, the Infectious DisesaseDiseases Society of America and the Society for Healthcare Epidemiology of America, or IDSA/SHEA, updated their Antibiotic Stewardship guidelines for antibiotic use. We believe that three key goals from these guidelines are applicable to the treatment of CABP:
- •
·Reduce the risk of antibiotics associated with a high risk ofC. difficile infections;·- •
- Increase use of oral antibiotics as a strategy to improve outcomes or decrease costs; and
· - •
- Reduce antibiotic therapy to the shortest effective duration.
Pleuromutilins are semi-synthetic compounds derived fromConsistent with the Antimicrobial Stewardship principles, we believe that lefamulin could be well suited as either a naturally occurring antibiotic and inhibit bacterial growth by binding to a specific site onfirst-line or second-line empiric monotherapy for the bacterial ribosome that is responsible for bacterial protein synthesis. We have developed an understandingtreatment of how to optimize characteristicsCABP patients in the hospital setting, outpatient-transition of care or in the pleuromutilin class, such as antimicrobial spectrum, potency, absorption following oral administration and tolerability, which in turn led to our selection and developmentcommunity setting, because of lefamulin, our lead product candidate. We have completed a Phase 2 clinical trial for ABSSSI in which IV lefamulin achieved a high cure rate against multi-drug resistant Gram-positive bacteria, including MRSA. In addition, in preclinical studies, lefamulin showed potent antibacterial activity against a variety of Gram-positive bacteria, Gram-negative bacteria and atypical bacteria, including multi-drug resistant strains.
The preclinical studies and clinical trials we have conducted to date suggest that lefamulin’sits novel mechanism of action, is responsiblecomplete spectrum of activity for the lackCABP pathogens, including against multidrug
resistant strains, achievement of bacterial resistance to lefamulin. As a result of the favorable safety and tolerability profile we have observedsubstantial drug concentrations in our clinical trials to date, we believe lefamulin has the potential to present fewer complications relative to the use of current therapies. Based on our research, we also believe that the availability of both IV and oral formulations of lefamulin, and an option to switch to oral treatment, could reduce the length of a patient’s hospital stay and the overall cost of care.
We have evaluated lefamulin in more than 440 patients and subjects in seventeen completed Phase 1 clinical trials and a Phase 2 clinical trial in ABSSSI. In our Phase 1 clinical trials, we have characterized the clinical pharmacology of the IV formulation of lefamulin and shown oral bioavailability of a tablet formulation of lefamulin with rapid tissue distribution, including substantial penetration into lung fluids and lung immune cells. In our Phase 2 clinical trial evaluatingcells, and flexibility as step down oral agent with both the safety and efficacy of two different doses of the IV formulation
of lefamulin administered over five to 14 days compared to the antibiotic vancomycin in patients with ABSSSI, the clinical success rate at test of cure, or TOC, for lefamulin was similar to that of vancomycin. Lefamulin has been well tolerated in all our clinical trials to date when administered by IV and oral routes. The frequencyformulations and favorable safety and tolerability profile.
CONTEPO
On July 24, 2018, we completed the acquisition of adverse events that we observed in our Phase 2 clinical trial in ABSSSI was similarZavante Therapeutics, Inc., or Zavante, a privately-held late clinical-stage biopharmaceutical company focused on developing novel therapies to improve the outcomes of hospitalized patients. Zavante's lead product candidate is CONTEPO™ (fosfomycin for patients treated with IV lefamulininjection, previously referred to as ZTI-01 and patients treated with vancomycin.ZOLYD).
Based on the clinical resultsWe submitted an NDA for marketing approval of lefamulinCONTEPO for the treatment of ABSSSI, as well as its rapid tissue distribution,cUTI including substantial penetration into the lung, we are evaluating lefamulin for the treatment of moderate to severe CABPAP in two international Phase 3 clinical trials. We are initially pursuing the development of lefamulin for CABP because of the limited development of new antibiotic classes for this indication over the past 15 years, our belief that there exists a significant unmet medical need for a first-line empiric monotherapy that addresses the growing development and spread of bacterial resistance, as well as recently clarified FDA guidance regarding the approval pathway. We initiated the first of these trials in September 2015 and the second trial in April 2016. We are also further characterizing the clinical pharmacology of lefamulin in several additional Phase 1 clinical trials.
We plan to pursue a number of additional opportunities for lefamulin, including beginning a development program for use in pediatric patients and potentially for the treatment of ABSSSI. In addition, as an antibiotic with potent activity against a wide variety of multi-drug resistant pathogens, including MRSA, we may explore development of lefamulin in other indications, including ventilator-associated bacterial pneumonia, or VABP, hospital-acquired bacterial pneumonia, or HABP, sexually transmitted infections, or STIs, osteomyelitis and prosthetic joint infections. Through our research and development efforts, we have also identified a topical pleuromutilin product candidate, BC-7013, which has completed a Phase 1 clinical trial.
We own exclusive, worldwide rights to lefamulin. Lefamulin is protected by issued patentsadults in the United States, Europeto the FDA in October 2018. The NDA submission is utilizing the 505(b)(2) regulatory pathway and Japan covering compositionis supported by a robust data package, including a pivotal Phase 2/3 clinical trial (known as ZEUS™), which met its primary endpoint of matter, whichstatistical non-inferiority to piperacillin/tazobactam in patients with cUTI, including AP. The FDA has granted us a Prescription Drug User Fee Act, or PDUFA target action date of April 30, 2019 for CONTEPO.
The prevalence of antibiotic-resistant bacteria is increasing and is considered a significant threat to global health. In particular, the CDC and the WHO consider antibiotic resistance to be an urgent and critical threat to human health. The prevalence of lactamase enzymes among Gram-negative pathogens threatens the usefulness of many beta -lactam antibiotics and has resulted in greater reliance on last line antibiotics, including carbapenems. Complicated urinary tract infections, or cUTIs, including acute pyelonephritis, or AP, are scheduledamong the most common infections due to expire no earlier than 2028. We also havemulti-drug resistant, or MDR bacteria, including carbapenem-resistant Enterobacteriaceae, or CRE, and are often healthcare-associated. Global mortality attributable to CRE infections has been granted patentsestimated in some studies to be over 20% and reflects the need for lefamulin relating to process and pharmaceutical crystalline salt formssafe, alternative, carbapenem-sparing options.
CONTEPO is a novel, potentially first-in-class investigational IV antibiotic in the United States whichwith a broad spectrum of Gram-negative and Gram-positive activity, including activity against most MDR strains such as extended-spectrum beta-lactamase-, or ESBL-producing Enterobacteriaceae. Intravenous fosfomycin has been approved for a number of indications and utilized for over 45 years in Europe to treat a variety of serious bacterial infections, including cUTIs. CONTEPO utilizes a new dosing regimen that optimizes its pharmacokinetics and pharmacodynamics. We believe these attributes, the extensive clinical experience worldwide and the positive efficacy and safety results from the Phase 2/3 clinical trial support CONTEPO as a first-line treatment for cUTIs, including AP, suspected to be caused by MDR pathogens. At least 20% of cUTIs are scheduled to expire no earlier than 2031.caused by MDR bacteria and limited treatment options are available in the U.S. In addition, we own a family of pending patent applications directednon-clinical data have shown that CONTEPO acts in combination with certain other antibiotics to pharmaceutical compositions of lefamulin, which if issued would be scheduled to expire no earlier than 2036.improve bacterial killing.
Our Strategy
Our goal is to become a fully integrated biopharmaceutical company focused on the research, development and commercialization of novel anti-infective products. The key elements of our strategy to achieve this goal are:
- •
·Complete Phase 3 clinical development of lefamulin for CABP. We are devoting a significant portion of our financial resources and business efforts to completing the clinical development of lefamulin for the treatment of CABP. We initiated two international Phase 3 clinical trials of lefamulin for the treatment of moderate to severe CABP. We initiated the first of these trials in September 2015 and the second trial in the April 2016. Based on our estimates regarding patient enrollment, we expect to have top-line data from LEAP 1 in the third quarter of 2017. With respect to LEAP 2, based on current projections, we expect to complete patient enrollment in the fourth quarter of 2017, and we anticipate receiving top-line data for LEAP 2 in the first quarter of 2018. If the results of these trials are favorable, including achievement of the primary efficacy endpoints of the trials, we expect to submit applications for marketing approval for lefamulin for the treatment of CABP in both the United States and Europe in 2018.·Maximize the commercial potential of CONTEPO for cUTI's and lefamulin forCABPCABP..We own exclusive, worldwide rights tolefamulin.lefamulin and US rights to CONTEPO and we have out licensed the rights to lefamulin in China. We expect that our initial target patient population for lefamulin will consist of patients with moderate to severeCABP.CABP and the initial target population for CONTEPO will be hospitalized patients with complicated urinary tract infections. If either or both CONTEPO or lefamulin receives marketing approval from the FDA,for the treatment of CABP,we plan to commercializeitthem in the United States with our own targetedhospitalsales and marketing organization that we plan toestablish.expand beyond its current levels. We believe both CONTEPO and lefamulin have innovative profiles which, if approved, would support their adoption in the
- •
·Pursue the continued development of lefamulin in additionalindicationsindications..Weplan to pursueare evaluating the cost and benefits of the continued development of lefamulin for indications in addition to CABP.For example, we are conductingPediatric oral formulation developmentactivities for lefamulin for useis ongoing, and we initiated a Phase 1 clinical trial in pediatric patientsand potentially for the treatment of ABSSSI. In addition, we are evaluating whether to pursue studies of lefamulininpatients with VABP or HABP.mid-2018. We believethat lefamulin’s product profile also provides the opportunitylefamulin has potential toexpand to other indications beyond pneumonia. For example, investigation of the tolerability of higher single doses of lefamulin could also support use of lefamulin for the treatment of STIs.treat acute bacterial skin and skin structure infection (ABSSSI), ventilator-associated bacterial pneumonia (VABP) or hospital-acquired bacterial pneumonia (HABP) and sexually transmitted infections (STIs). In addition, we may explore longer duration of treatment with lefamulin to support development of a treatment for osteomyelitis and prosthetic joint infections. We believe that lefamulin would be differentiated from other treatment options foreach ofthese potentialusesindications because oflefamulin’sits novel mechanism of action, spectrum of activity, including activity against multi-drug resistant pathogens, achievement of substantial concentrations in relevant tissues, availability as both an IV and oral formulation and favorable safety and tolerability profile.·- •
AdvancePursue the development ofother pleuromutilin product candidates and possibly compounds in other classesCONTEPO for a pediatric indication..We arecurrently focused on developing additional pleuromutilin product candidates through our deep understandingevaluating the continued development ofthis class of antibiotics. Our product candidate BC-7013 has completedCONTEPO for use in pediatric patients with cUTIs. In June 2018, we initiated a Phase 1,clinical trial. We believe that this pleuromutilin compound is well suited fornon-comparative, open-label study of thetopical treatmentpharmacokinetics and safety of avarietysingle dose ofGram-positive infections, including uncomplicated skin and skin structure infections, or uSSSIs. Furthermore, we own diverse librariesCONTEPO in pediatric subjects less than 12 years ofcompounds in other antibacterial classes, such as ß-lactams and acremonic acids, which are a potential basis for the discovery and development of novel antibacterial agents.age.
·- •
- Evaluate business development opportunities and potential
collaborationscollaborations..We may expand our product pipeline through opportunistically in-licensing or acquiring the rights to complementary products, product candidates and technologies for the treatment of a range of infectious diseases or other hospital-based products that could utilize our planned commercial infrastructure. We plan to evaluate the merits of entering into collaboration agreements with other pharmaceutical or biotechnology companies that may contribute to our ability to efficiently advance our product candidates, build our product pipeline,andconcurrently advance a range of research and developmentprograms.programs and leverage our commercial infrastructure. Potential collaborations may provide us with funding and access to the scientific, development, regulatory and commercial capabilities of the collaborators. We also plan to encourage local and international government entities and non-government organizations to provide additional funding and support for our development programs.We may expand our product pipeline through opportunistically in-licensing or acquiring the rights to complementary products, product candidates and technologies for the treatment of a range of infectious diseases.
United States for adult cUTI patients treaded in the hospital and CABP patients, respectively, treated as in-patients in a hospital setting. Lefamulin also has the opportunity to be adopted as outpatient transition of care from the hospital, which we believe represents a significant commercial opportunity. We believe that we will be able to effectively communicate lefamulin’sCONTEPO's and lefamulin's differentiating characteristics and key attributes to clinicians, and hospital pharmacies and payors with the goal of establishing favorable reimbursement as well as a favorable formulary status for lefamulin. If lefamulin receives marketing approval outsidein targeted hospitals. Outside the United States for the treatment of CABP, we expect to utilize a variety of types of collaboration, distribution and other marketing arrangements
with one or more third parties to commercialize lefamulin in such markets. We also are conducting pediatric formulationcurrently have a team of regional business directors and medical science liaisons in the field performing educational and market development activities, respectively. We initially plan to support clinical trials ofuse a targeted hospital-based sales force to promote both lefamulin for pediatric use for CABP.formulations and CONTEPO, if approved.
Our Product Development PipelineBackground
The following table summarizes the indications for which we are developing our product candidates and the status of development.
DEVELOPMENT STAGE

* We have initiated two international Phase 3 clinical trials of lefamulin for the treatment of moderate to severe CABP. However, we have not previously conducted any clinical trials of lefamulin specifically for CABP. Our completed Phase 2 clinical trial evaluated lefamulin in patients with ABSSSI. We have obtained input from the FDA and select European authorities, including reaching agreement with the FDA on a Special Protocol Assessment, or SPA, regarding the study design of our first Phase 3 clinical trial, in anticipation of submitting applications for marketing approval for lefamulin for the treatment of CABP in both the United States and Europe in 2018.
Background
Anti-Bacterial Market and Scientific Overview
Bacteria are broadly classified as Gram-positive or Gram-negative. Gram-positive bacteria possess a single membrane and a thick cell wall and turn dark-blue or violet when subjected to a laboratory staining method known as Gram’sGram's method. Gram-negative bacteria have a thin cell wall layered between an inner cytoplasmic cell membrane and a bacterial outer membrane and, as a result, do not retain the violet stain used in Gram’sGram's method. Antibiotics that are active against both Gram-positive
and Gram-negative bacteria are referred to as broad spectrum, while those that are active only against a select subset of Gram-positive or Gram-negative bacteria are referred to as narrow spectrum. Bacteria that cause infections are often referred to as bacterial pathogens. Because it often takes from 24 to 48 hours to definitively diagnose the particular bacterial pathogen causing an infection, the causative pathogen often remains unidentified and narrow spectrum antibiotics are not generally used as empiric monotherapy for first-line treatment of hospitalized patients with serious infections.
Since the introduction of antibiotics in the 1940s, numerous new antibiotic classes have been discovered and developed for therapeutic use. The development of new antibiotic classes and new antibiotics within a class is important because of the ability of bacteria to develop resistance to existing mechanisms of action of currently approved antibiotics. However, the pace of discovery and development of new antibiotic classes slowed considerably in the past few decades. The CDC estimates that the pathogens responsible for more than 70% of U.S. hospital infections are resistant to at least one of the antibiotics most commonly used to treat them. The CDC also estimated in 2013, based on data collected from evaluations performed between 2006 and 2011, that annually in the United States at least two million people become infected with bacteria that are resistant to antibiotics and at least 23,000 people die as a direct result of these infections.
Antibiotic resistance is primarily caused by genetic mutations in bacteria selected by exposure to antibiotics that do not kill all of the bacteria. In addition to mutated bacteria being resistant to the drug used for treatment, many bacterial strains can also become cross-resistant, meaning that they become resistant to multiple classes of antibiotics. As a result, the effectiveness of many antibiotics has declined, limiting physicians’physicians' options to treat serious infections and exacerbating a global health issue. For example, the WHO estimated in 2014 that people with infections caused by MRSA, a highly resistant form of bacteria, are 64% more likely to die than people with a non-resistant form of the infection. Resistance can increase the cost of healthcare because of the potential for lengthier hospital stays and more intensive care. Growing antibiotic resistance globally, together with the low level of investment in research and development, is considered one of the biggest global health threats. In 2010, the WHO stated that antibiotic resistance is one of the three greatest threats to human health. Partially in response to this threat, the U.S. Congress passed the GAIN Act in 2012, which provides incentives, including access to expedited FDA review for approval, fast track designation and five years of potential data exclusivity extension for the development of new QIDPs. Additional legislation is also being considered in the United States, including the Antibiotic Development to Advance Patient Treatment Act of 2013, which is intended to accelerate the development of anti-infective products, and the Developing an Innovative Strategy for Antimicrobial Resistant Microorganisms Act of 2014, which is intended to establish a new reimbursement framework to enable premium pricing of anti-infective products.
In 2009,2017, sales of antibiotics totaled approximately $42 billion globally. Although judicious use of antibiotics is important to reduce the rate of antibiotic resistance, this approach alone cannot fully address the threat from increasing antibiotic resistance. New antibiotics, and particularly new antibiotic classes, are needed to ensure the availability of effective antibiotic therapy in the future.
Community-Acquired Bacterial Pneumonia (CABP)
Market Overview
The WHO estimated in 2002 that there were approximately 450 million pneumonia cases reported per year worldwide, causing approximately 4.0 million deaths in 2002. According to an article published in 2011 in the peer-reviewed medical journal Therapeutic Advances in Respiratory Disease, the annual incidence of community-acquired pneumonia is between five and 11 cases per 1,000 people, with the incidence rate rising in elderly patients. In a study published in 2004 in the peer-reviewed medical journal Clinical Infectious Diseases in which more than 46,000 people in the state of Washington were monitored over three years, the incidence of CABP among those 65 to 69 years of age was 18.2 cases per 1,000 people per year and increased to 52.3 cases per 1,000 people per year in those over 85 years of age.
The U.S. National Center for Health Statistics estimated that between 1988 and 1994 there were approximately 5.6 million cases of pneumonia per year in the United States. More recently, based on our combined analysis of the CDC’sCDC's 2007 National Ambulatory Medical Care Survey, the National Hospital Ambulatory Medical Care Survey and 2013 data from the Healthcare Cost and Utilization Project we estimate that over 5.0 million adults are treated annually for CABP in the United States and that the majority of these adult CABP patients have their treatment initiated in a hospital, including
emergency departments. According to the Healthcare Cost and Utilization Project, or HCUP, in 2013, approximately 3.1 million adults sought treatment in a U.S. hospital for CABP. In addition, in 2013, approximately 2.4 million adults were admitted to U.S. hospitals for in-patient care with a diagnosis of CABP and approximately 700,000 adults were seen in an emergency department at U.S. hospitals for treatment of CABP and then released.
Additionally, in 2014, based on CDC data approximately 50,000 patients died from CABP in the United States. Based on data collected from July 1, 2012 through June 30, 2015, on the Medicare.gov Hospital Compare website, the current national rate of readmissions for Medicare pneumonia patients is 17.1%, which is the percentage of patients who have had a recent hospital stay that must return to a hospital for unplanned care within 30 days of being discharged. The national average death rate for Medicare pneumonia patients, excluding Medicare Advantage plan data, is 16.3%, which is the percentage of patients who die, for any reason, within 30 days of admission to a hospital.
Based on data from Arlington Medical Resources, or AMR,LexisNexis® Risk Solutions, a leading provider of medicalhealthcare data and analytics solutions, as well as analysis of data from US hospitals and other healthcare facilities, who reportedwe determined that the number of adult CABP patients who were treated with antibiotic treatment courses for CABP adult patientstherapy in hospitals in the United States exceeded 6.83.8 million for full-year 2015, we estimate2016. Our analysis of the LexisNexis data also indicates that approximately 5.32.4 million of these adult CABP coursespatients were fortreated as inpatients with IV/injectable antibiotics, forand we find that the majority of CABP patients enter the hospital inpatient setting following the initiation of antibiotic therapy during an Emergency Department (ED) visit. Additionally, our analyses show that approximately 1.4 million adult CABP patients
while approximately 1.5 million CABP oral were treated with antibiotic courses were prescribed for(IV or oral) in the ED or as hospital outpatients and subsequently released without hospital admission.
Additionally, approximately 1.4 million adult CABP patients were treated with antibiotic courses (IV or oral) in the ED or as hospital setting. Additionally, for the twelve months ending September 30, 2016, Source Health Solutions estimatesoutpatients and subsequently released without hospital admission. Furthermore, as a result of our market research in 2017-18, we believe that once adult CABP patients are released from ED stays or are discharged home from U.S. hospitals, approximately 4.2 million60-70%, receive antibiotic oral outpatient prescriptions are written annually foras continuation of their outpatient antibiotic treatment. RelativeAs hospitals look to minimize the total cost of care and duration of hospital stay for CABP patients toward improved outcomes, efficient transition of adult CABP inpatients to an oral antibiotic treatment as outpatient therapy can significantly reduce days of hospitalization and overall treatment cost.
IQIVA estimated that in 2017 approximately 6.62 million adult CABP outpatient oral antibiotic prescriptions that Health Source Solutions estimates are written over the same time-period, approximately 6 out of every 10 oral antibiotic prescriptions for adult CABP results as a transition of care from hospital-initiated treatment to outpatient therapy. The remaining CABP prescriptions originatepatients were actively treated with antibiotics from prescribers in community clinics, e.g. primary care offices and at other non-hospital based sites of urgent care. As a result, we believe that approximately 6 million CABP patients are treated with antibiotics in the United States on an annual basis and 6 out of every 10 adult CABP patients have treatment initiated in a hospital setting versus. the community setting.
Causes of CABP
Pneumonia can be caused by a variety of micro-organisms, with bacteria being the most common identifiable cause. CABP refers to bacterial pneumonia that is acquired outside of a hospital setting. Signs and symptoms of CABP include cough, fever, sputum production and chest pain. A number of different types of bacteria can cause CABP, including both Gram-positive and Gram-negative bacteria. Pneumonia that is caused by atypical bacterial pathogens often has different symptoms and responds to different antibiotics than pneumonia caused by pathogens referred to as typical bacteria. However, atypical bacteria are not uncommon. The most common bacterial pathogens noted in current treatment guidelines from the Infectious Diseases Society of America, or IDSA, for hospitalized CABP patients who are not in the intensive care unit areStreptococcus pneumoniae, Mycoplasma pneumoniae, Haemophilus influenzae, Chlamydophila pneumoniae, andLegionellaspecies. In addition, IDSA notes the emergence of resistance to commonly utilized antibiotics for CABP, specifically drug-resistant
S. pneumoniae and community-acquired MRSA, or CA-MRSA, as a major consideration in choosing empiric therapy. However, a majority of patients do not have a pathogen identified using routine diagnostic tests available to physicians.
Currently Available Treatment Options
In 2007, based on the most likely bacteria to cause CABP, IDSA and the American Thoracic Society, or ATS, recommend empiric treatment of hospitalized patients with CABP who do not require treatment in an intensive care unit with either:
- •
·a combination of a cephalosporin, an antibiotic that disrupts the cell wall of bacteria, plus a macrolide, an antibiotic that disrupts bacterial protein synthesis; or·- •
- monotherapy with a respiratory fluoroquinolone, an antibiotic that disrupts bacterial protein synthesis.
In the event CA-MRSA is suspected, these guidelines recommend that vancomycin, an antibiotic that disrupts the cell wall of bacteria, or linezolid, an antibiotic that disrupts bacterial protein synthesis, be used or added to the current regimen.
In addition, physicians need to be aware of the local susceptibility profiles of the common bacterial pathogens associated with CABP because of increasing resistance to first-line antibiotics. For example, rates of pneumococcal resistance to recommended first-line macrolides exceed 40% in some areas, while resistance inM. pneumoniae associated with severe disease has been recently reported by the CDC in the United States.
Limitations of Currently Available Treatment Options
When confronted with a new patient suffering from a serious infection caused by an unknown pathogen, a physician may be required to quickly initiate first-line empiric antibiotic treatment, often with a combination of antibiotics, to stabilize the patient prior to definitively diagnosing the particular bacterial infection. However, currently available antibiotic therapies for first-line empiric treatment of CABP suffer from significant limitations.
Bacterial Resistance and Spectrum of Activity
As a result of bacterial resistance, the effectiveness of many antibiotics has declined. For example, the CDC estimates that in 30% of severeS. pneumoniae cases, the bacterial pathogen is fully resistant to one or more clinically relevant antibiotics, with 44% of strains resistant to a macrolide in the United States. In addition, fluoroquinolone resistance inS. pneumoniae has increased from less than 0.5% to more than 3% of cases in some regions of North America, which parallels increased total fluoroquinolone prescriptions. Antibiotic resistance has a significant impact on mortality and contributes heavily to healthcare system costs worldwide. According to the CDC, cases of resistant pneumococcal pneumonia result in 32,000 additional doctor visits, approximately 19,000 additional hospitalizations and 7,000 deaths each year. These cases are associated with $96 million in excess medical cost per year in the United States. IDSA/ATS guidelines recommend empiric treatment that provides broad spectrum antimicrobial coverage. None of the currently available treatment options provides a spectrum of antibacterial coverage as a monotherapy that sufficiently covers all of the most common bacterial causes of CABP, including multi-drug resistant strains.
Difficult, Inconvenient and Costly Regimens
Currently available antibiotics used to treat CABP and other serious infections can be difficult, inconvenient and costly to administer. Physicians typically prefer IV administration for patients hospitalized with more serious illness to ensure adequate delivery of the drug rapidly. Many IV antibiotics are prescribed for seven to 14 days or more and patients can be hospitalized for much or all of this period or require in-home IV therapy. The diagnosis related group, or DRG, reimbursement
system often used in the U.S. hospital setting pays a fixed fee for an episode of CABP that may not fully compensate hospitals for the duration of hospitalized care. Prolonged IV treatment that extends the period of hospitalization may cause hospital costs to increase in excess of the fixed reimbursement fee, resulting in significant negative impact on healthcare institutions. In addition, to address all likely bacterial pathogens in a patient with a more serious illness, IDSA guidelines recommend using a combination of antibiotics. Combination therapy presents the logistical challenge of administering multiple drugs with different dosing regimens and increases the risk of drug-drug interactions. While IV treatment delivers the drug more rapidly than is possible orally, once a patient is stabilized, oral treatment with the same drug would allow for more convenient and cost-effective out-patient treatment. Because many commonly used antibiotics are only available in IV form, a switch to an oral therapy requires changing to a different antibiotic, which may be less effective for the patient.
Adverse Effects
Currently available antibiotic therapies can have serious side effects. These side effects may include severe allergic reaction, decreased blood pressure, nausea and vomiting, suppression of platelets, pain and inflammation at the site of injection, muscle, renal and oto-toxicities,oto-toxicity, optic and peripheral neuropathies and headaches. At times, these side effects may be significant and require discontinuation of therapy. As a result, some treatments require clinicians to closely monitor patients’patients' blood levels and other parameters, increasing the expense and inconvenience of treatment. This risk may be increased with combination therapy, which exposes patients to potential adverse effects from each of the antibiotics used in treatment. For example, fluoroquinolones are associated with tendon rupture and peripheral neuropathy. In addition, fluoroquinolones have been associated with an increased frequency ofC. difficile colitis, an overgrowth of a bacteria in the colon that produces a toxin that results in inflammation of the colon and repeated bouts of watery diarrhea. This has resulted in limitations on the use of fluoroquinolones in several countries. In November 2015, the FDA convened an Advisory Committee meeting to review the benefits and risks of fluoroquinolones in less severe indications, such as uncomplicated UTI, acute bacterial sinusitis and acute bacterial exacerbations of chronic bronchitis. Based on the committee’scommittee's recommendation, in July 2016, the FDA approved changes to the labels of fluoroquinolones to indicate that fluoroquinolones should be reserved for use in patients who have no other treatment options for the indications mentioned above, because the risk of these serious side effects generally outweighs the benefits in these patients. These changes included a requirement that a separate patient Medication Guide be given with each prescription that describes the safety issues associated with this class of drugs. In December 2018, an FDA review found that fluoroquinolone antibiotics can increase the occurrence of rare but serious events of ruptures or tears in the main artery of the body, called the aorta. These tears, called aortic dissections, or ruptures of an aortic aneurysm can lead to dangerous bleeding or even death. They can occur with fluoroquinolones for systemic use given by mouth or through an injection.
cUTIs
LefamulinMarket Overview
OverviewInfections due to a bacterial pathogen that is resistant to three or more antibiotic classes have become increasingly common and present a risk to our fight against infectious diseases and the management of complications in vulnerable patients. According to the CDC, more than two million hospital infections caused by bacteria resistant to one or more antibiotics occur every year in the United States, and over 23,000 patients with an antibiotic-resistant pathogen die each year.
We are developing lefamulinThe prevalence of antibiotic-resistant bacteria is increasing and is considered a significant threat to global health. In particular, the CDC and the WHO consider antibiotic resistance to be an urgent and critical threat to human health. The prevalence of Beta-lactamase enzymes among Gram-negative
pathogens threatens the first pleuromutilinusefulness of many Beta-lactam antibiotics and has resulted in greater reliance on last line antibiotics, including carbapenems.
CUTIs, including AP, are among the most common infections due to MDR bacteria, including CRE, and are often healthcare-associated. Global mortality attributable to CRE infections has been estimated in some studies to be over 20% and reflects the need for safe, alternative, carbapenem-sparing
Surveillance and epidemiological studies suggest that some traditional, first-line antibiotics may no longer be acceptable choices for early therapy. In one large-scale surveillance study, approximately one out of three patients hospitalized in the United States with cUTI, a complicated intra-abdominal infection, hospital-associated pneumonia, or a bloodstream infection did not receive timely effective antibiotic therapy, and this delay was associated with increased morbidity and mortality. The rate of antibiotic resistance appears to be two to four times higher in patients who were admitted to the hospital from a nursing home or were recently hospitalized. Antibiotic therapy within the past six months has also been identified as a risk factor for antibiotic resistance.
New classes of antibiotics that are effective against drug-resistant pathogens are needed for early, appropriate treatment of serious infections in hospitalized patients and to treat patients who have failed to respond to standard, first-line antibiotics due to acquired drug resistance.
For over 45 years, oral and IV formulations of fosfomycin have been used in the European Union, Africa, Asia, and South America, and an oral formulation of fosfomycin has been used in the United States and Canada for the treatment of a variety of indications. Oral fosfomycin is available in the United States as single-dose therapy for cystitis and is noted as an appropriate treatment option for cystitis in treatment guidelines by the Infectious Diseases Society of America and the CDC. However, oral administration of fosfomycin provides inadequate concentrations required for treatment of more serious infections due to its limited bioavailability and dose-limiting gastrointestinal tolerability.
Outside of the United States, IV fosfomycin is approved for patients with a variety of infections, often severe, including cUTI, bacteremia, osteomyelitis, nosocomial lower respiratory tract infections, surgical site infections, bone and joint infections, endocarditis, skin infections and bacterial meningitis. The efficacy and safety profile of IV fosfomycin has been established by more than 45 years of clinical use outside of the United States and has been evaluated in more than 60 clinical studies. Fosfomycin has retained highin vitro activity with a low and stable resistance profile, and continues to be suitable for use as a monotherapy for cUTI despite long term use.
Causes of cUTIs
cUTI is defined as a clinical syndrome characterized by pyuria and a documented microbial pathogen on culture of urine or blood, accompanied by local and systemic administrationsigns and symptoms, including fever, chills, malaise, flank pain, back pain or costo-vertebral angle pain or tenderness that occur in humans.the presence of a functional or anatomical abnormality of the urinary tract, or in the presence of catheterization. Indwelling urethral catheters account for 70% to 80% of cUTIs, or 1 million cases per year in the United States. Catheter-associated UTI is the most common cause of secondary bloodstream infections and is linked to increased morbidity and mortality. Patients with pyelonephritis, regardless of underlying abnormalities of the urinary tract, are considered a subset of patients with cUTI.
cUTI are usually caused by a greater variety of pathogens, with a greater likelihood of associated antimicrobial resistance, than uncomplicated UTIs, or uUTIs. Escherichia coli, or E. coli, is isolated in approximately 75% to 95% of uUTIs and approximately 50% of cUTIs and is the most common etiologic agent of cUTIs. Additional commonly-identified Gram-negative uropathogens include other Enterobacteriaceae (such as Klebsiella spp., Proteus spp., Enterobacter cloacae) and non-fermenting
Gram-negative bacilli (such as Pseudomonas aeruginosa, or P. aeruginosa, and Acinetobacter spp.). Gram-positive organisms, such as Enterococci and coagulase-negative Staphylococci, may also be contributing pathogens.
Limitations of Currently Available Treatment Options
We believe bacterial resistance against antimicrobials has created the need for more antibiotic treatment options, particularly among MDR, Gram-negative bacilli (including CRE, ESBL, producers, and MDR P. aeruginosa). Gram-negative antimicrobial resistance is particularly common among urinary tract pathogens. Enterobacteriaceae, including E. coli and Klebsiella pneumoniae, or K. pneumoniae, may acquire plasmids that encode ESBLs and confer resistance to third-generation cephalosporins and other broad-spectrum antibiotics. Third-generation cephalosporins and Beta-lactamase inhibitors, or BLIs, are also commonly ineffective against Enterobacteriaceae that generate AmpC enzymes.
The recent spread into hospitals of Enterobacteriaceae expressing emergent Beta-lactamases, including members of the serine carbapenemases and metallo-Beta-lactamases, endanger antibiotic options. The lack of available and effective antibiotic classes for these organisms has created an unmet medical need. For example, infections with CRE are difficult to treat, as there are limited treatment choices available. Mortality rates as high as 40% to 50% have been associated with CRE infections, making them a serious threat to public health. The available treatment choices are associated with serious potential toxicity, in the case of colistin and aminoglycosides, or concerns of allergy or hypersensitivity, in the case of Beta-lactams or penicillin derivatives.
Our Solution: CONTEPO for the Treatment of cUTI
- •
- CONTEPO is an IV formulation of fosfomycin and the sole member of the epoxide antibiotic class.
- •
- CONTEPO has a different mechanism of action than other IV antibiotics in the United States.
- •
- CONTEPO has a broad spectrum ofin vitro activity against a variety of clinically important MDR Gram-negative pathogens, including ESBL-producing Enterobacteriaceae, CRE, and Gram-positive pathogens, including methicillin-resistantStaphylococcus aureus, or MRSA, and vancomycin-resistant enterococci.
- •
- CONTEPO has demonstrated inin vitro studies additivity or synergy when used in combination with other classes of antibiotic agents in pre-clinical trials.
- •
- CONTEPO has a small molecular size, which may enable high levels of tissue penetration and facilitates renal elimination, both of which are important for treatment of cUTIs.
- •
- CONTEPO is supported by a long history of IV fosfomycin use outside the United States in a variety of indications, including cUTI.
- •
- CONTEPO has completed the ZEUS Study, a pivotal registrational Phase2/3 clinical trial in cUTI, achieving non-inferiority to an active comparator.
CONTEPO is a potentially first-in-class epoxide IV antibiotic in the United States with a broad spectrum of bactericidal Gram-negative and Gram-positive activity, including activity against many contemporary MDR strains that threaten hospitalized patients. IV fosfomycin has an extensive commercial history in markets outside the United States, where it has been used broadly for over 45 years to treat a variety of indications, including complicated urinary tract infections, bacteremia, pneumonia and skin infections.
CONTEPO works differently than other IV antibiotics approved in the United States. CONTEPO inhibits an early step in bacterial cell wall synthesis, so the cell wall lacks integrity and the bacteria die quickly. We believe that because of its different mechanism of action, we have not observed any cross resistance to date between CONTEPO and any of the existing classes of intravenous antibiotics. In addition, CONTEPO has demonstrated inin vitro studies an additive or synergistic antibacterial effect with other classes of antibiotics when used in combination therapy, and has been shown to restore susceptibility in resistant strains.
Our Product Development Pipeline
Lefamulin
Overview
Lefamulin is a semi-synthetic derivative of the naturally occurring antibiotic, pleuromutilin, which was originally identified from a fungus calledPleurotus mutilismutilius. Lefamulin inhibits the synthesis of bacterial protein, which is required for bacteria to grow. Lefamulin acts by binding to the peptidyl transferase center, or PTC, on the bacterial ribosome in such a way that it interferes with the interaction of protein production at two key sites known as the “A”"A" site and the “P”"P" site, resulting in the inhibition of bacterial proteins and the cessation of bacterial growth. Lefamulin’sLefamulin's binding occurs with high affinity, high specificity and at molecular sites that are different than other antibiotic classes. We believe that lefamulin’slefamulin's novel mechanism of action is responsible for the lack of cross-resistance with other antibiotic classes that we have observed in our preclinical studies and clinical trials and a low propensity for development of bacterial resistance to lefamulin. The binding of lefamulin to the PTC on the bacterial ribosome is depicted in the graphic below.
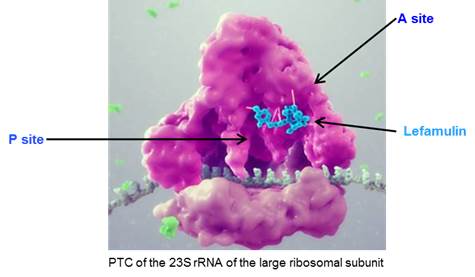
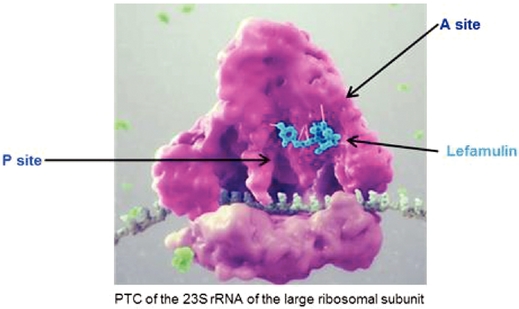
We are developing both IV and oral formulations of lefamulin. We believe that lefamulin is well suited to be used empirically as monotherapy for the treatment of respiratory tract infections, such as CABP, because of its spectrum of antibacterial activity against both the typical and atypical pathogens causing CABP, including multi-drug resistant pathogens such as MRSA. In addition, in preclinical studies, lefamulin showed potent antibacterial activity against a variety of Gram-positive bacteria, Gram-negative bacteria and atypical bacteria, including multi-drug resistant strains. In preclinical studies and in Phase 1 clinical trials, lefamulin achieved substantial concentrations in the epithelial lining fluid, or ELF, of the lung, the site infected during pneumonia. Lefamulin also provides the ability to switch from IV to oral therapy and maintain therapy with the same active ingredient.antibacterial treatment. The efficacy of lefamulin in humans has been shown in a
We have completedproof-of-concept clinical Phase 3 trial with 207 patients with ABSSSI (acute bacterial skin and skin structure infections) comparing two lefamulin doses (100 mg and 150 mg i.v. q12 h) with vancomycin (³ 1 000 mg) over 5-14 days. This trial enrolled patients with severe skin and skin structure infections, excluding any patients with minor and uncomplicated infections. In total, 90.8 % of patients in the Modified Intent to Treat, or mITT population hadS. aureus infection; 69.1 % of patients had MRSA. There were consistent high response rates seen across all three treatment arms, across a wide range of clinical and microbiological outcomes. Furthermore, no development of resistance was observed for lefamulin or for vancomycin during the trial. Lefamulin treatment administered intravenously over 5 to 14 days was well tolerated. The types of treatment-emergent adverse events were consistent with a patient population under treatment for ABSSSI. Lefamulin prolonged the cardiac de- and repolarization duration but otherwise had a similar cardiac safety profile as vancomycin as assessed with standard 12-lead ECGs. The results of the clinical Phase 2 clinical trial in ABSSSI provided the first proof of lefamulinconcept for ABSSSI. Based on the clinical resultssystemic use of lefamulin for ABSSSI, as well as its rapid tissue distribution, including substantial penetration into lung fliuds and lung immune cells, we initiated two international, pivotal Phase 3 clinical trials of lefamulina pleuromutilin antibiotic for the treatment of moderate to severe CABP. These areserious bacterial infections in humans. Thereafter, the first clinical trials we have conducted with lefamulin for the treatment of CABP. We initiated the first of these trials in September 2015 and the second trial in April 2016. We designed these trials to follow draft guidance published by the FDA for the development of drugs for CABP and guidelines from the EMA for the development of antibacterial agents, as well as our SPA with the FDA regarding the study design of our first Phase 3 clinical trial. According to the draft FDA guidance and FDA feedback, either a Phase 3 clinical trial for CABP, supported by evidence of antibacterial activity accrued during a clinical development program for another indication, such as ABSSSI, orlefamulin progressed with completion of two Phase 3 clinical trials forin CABP may provide sufficient evidence(LEAP 1, LEAP 2). These trials demonstrated that lefamulin treatment, administered as IV only, IV to oral, and oral only regimens, was non-inferior to the standard of efficacy in CABP.
Based on our estimates regarding patient enrollment, we expect to have top-line data from LEAP 1 in the third quarter of 2017. With respect to LEAP 2, based on current projections, we expect to complete patient enrollment in the fourth quarter of 2017, and we anticipate receiving top-line data for LEAP 2 in the first quarter of 2018. If the results of these trials are favorable, including achievement of the primary efficacy endpoints of the trials, we expect to submit applications for marketing approval for lefamulincare moxifloxacin for the treatment of CABPadults with CABP. Each trial provided independent evidence of the treatment effect and safety in both the United States and Europe in 2018. We submitted to the FDA an investigational new drug application, or IND, for the IV formulation of lefamulin in September 2009 and an IND for the oral formulation of lefamulin in January 2015.this population with unmet medical need.
The FDA has designated each of the IV and oral formulations of lefamulin as a QIDP which provides for the extension of statutory exclusivity periods in the United States for an additional five years upon FDA approval of the product for the treatment of CABP and also granted fast track designationsdesignation to each of these formulations of lefamulin. Fast track designation is granted by the FDA to facilitate the development and expedite the review of drugs that treat serious conditions and fill an unmet medical need. The fast track designation for the IV and oral formulations of lefamulin will allow for more frequent interactions with the FDA, the opportunity for a rolling review of any NDAs, we submit and eligibility for priority review and a shortening of the FDA's goal for taking action on a marketing application from ten months to six months. Two NDAs for IV and oral formulations of lefamulin for treatment of CABP were submitted to the FDA December 19, 2018. We plan to submit a marketing authorization application for lefamulin in Europe in the first half of 2019.
We own exclusive, worldwide rights to lefamulin, other than our rights in China which were licensed to Sinovant. Lefamulin is protected by issued patents in the United States, Europe and Japan covering composition of matter, which are scheduled to expire no earlier than 2028. We also have been granted patents for lefamulin relating to process and pharmaceutical crystalline salt forms in the United States, which are scheduled to expire no earlier than 2031. In addition, we own a family of pending patent applications directed to pharmaceutical compositions of lefamulin, which if issued would be scheduled to expire no earlier than 2036.
Key Attributes of Lefamulin
We believe that the combination of the following key attributes of lefamulin, observed in clinical trials and preclinical studies, differentiates lefamulin from currently available antibiotics and make lefamulin well suited for use as a first-line or second-line empiric monotherapy for the treatment of CABP.
The preclinical studies and clinical trials we have conducted to date suggest that lefamulin's novel mechanism of action is responsible for the low risk of cross resistance observed with other antibiotic classes and a low propensity for development of bacterial resistance to lefamulin. As a result of the favorable safety and tolerability profile we have observed in our clinical trials to date, we believe lefamulin has the potential to present fewer complications relative to the use of current therapies. Based on our research, we also believe that the availability of both IV and oral formulations of
lefamulin, and an option to switch to oral treatment, could reduce the length of a patient's hospital stay and the overall cost of care.
BroadTargeted Spectrum of Activity for CABP Pathogens and Low Propensity for the Development of Bacterial Resistance
We expect lefamulin’slefamulin's spectrum of antibacterial activity against typical and atypical pathogens could eliminate the need to use a combination of antibiotics for the treatment of CABP. In our completed Phase 2 clinical trial, IV lefamulin achieved a high cure rate against multi-drug resistant Gram-positive bacteria, including MRSA. In addition, in preclinical studies, lefamulin showed activity against a variety of Gram-positive bacteria, includingS. pneumoniae andS. aureus, that are resistant to other classes of antibiotics, Gram-negative bacteria, includingH. influenzae andM. catarrhalis, and atypical bacteria, includingC. pneumoniae,M. pneumoniae andL. pneumophila. Included in lefamulin’slefamulin's spectrum of activity are all bacterial pathogens identified by IDSA as the most common causes of CABP for hospitalized patients who are not in the intensive care unit, as well as strains of the above listed bacteria that are resistant to other classes of antibiotics, including penicillins, cephalosporins, fluoroquinolones and macrolides.
Based on observations from our preclinical studies and clinical trials of lefamulin, as well as industry experience with pleuromutilins used in veterinarianveterinary medicine over the last 3039 years, we believe that lefamulin’slefamulin's novel mechanism of action is responsible for the lacklow risk of cross-resistance observed with other antibiotic classes and a low propensity for development of bacterial resistance to lefamulin.
Convenient Dosing Regimen; Potential for Switching from IV to Oral Treatment
We have developed both an IV and oral formulation of lefamulin, which we are utilizingutilized in our Phase 3 clinical trials of lefamulin for the treatment of CABP. The administration of lefamulin as a monotherapy avoids the need for the complicated dosing regimens typical of multi-drug cocktails. We believe the availability of both IV and oral administration, and an option to switch to oral treatment, would be more convenient for patients and could reduce the length of a patient’spatient's hospital stay and the overall cost of care. The potential reduction in the overall cost of care could be particularly meaningful to healthcare institutions, as the DRG reimbursement system pays a fixed fee for the treatment of CABP regardless of the length of hospital stay. We believe that our Phase 3 trial design will permit us to submit for approval of both IV
The efficacy and oral formulations of lefamulin, subject to obtaining favorable results, including achievement of the primary efficacy endpoints of the trials.
Favorable Safety and Tolerability Profile
We have evaluated lefamulin in over 440 subjects and patients in our completed Phase 1 and Phase 2 clinical trials. In these trials, lefamulin has exhibited a favorable safety and tolerability profile. In our Phase 2 clinical trial of lefamulin, no patient suffered any serious adverse events that were determined to be related to lefamulin, and safety and tolerability were comparable to vancomycin, the control therapy in the trial. In addition, no clinically significant change in electrocardiogram, or ECG, was measured, and no drug-related cardiovascular adverse events were reported. Furthermore, we believe the use of lefamulin as a monotherapy would present fewer potential complications relative to the use of multiple antibiotics as combination therapy. We are also continuing to evaluate the safety and tolerability of lefamulin in our Phase 3 clinical trials.
Phase 3 Clinical Trials
We are conducting aadult patients with CABP was shown in two pivotal clinical trial program of lefamulin for the treatment of CABP consisting of two international Phase 3 clinical trials. We initiated the first of these trials in September 2015 and the second
trial in April 2016. We designed these trials to comply with the guidelines of The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, which are currently used as guidance by the FDA, and good clinical practices. We are conducting these trials at centers in the United States, Europe, Asia and selected countries in the southern hemisphere. We are currently enrolling patients in each of these clinical trials in several countries and are continuing with the regulatory steps necessary to initiate and conduct these trials, including submission of the trial protocol and relevant information about lefamulin to local regulatory authorities and ethics review committees in other countries.
First Phase 3 Clinical Trial
We designed our Phase 3 clinical trials (LEAP 1 and LEAP 2). The two trials were designed in accordance with US and EU regulatory guidelines and conducted in parallel from 2016 to follow2018. Design elements of the draft guidance published byPhase 3 clinical trials were broadly comparable. Both were global, multicenter, randomized, double-blind, active-controlled, non-inferiority studies to establish the efficacy and safety of lefamulin against the standard-of-care moxifloxacin in the treatment of adult subjects with CABP. In LEAP 1, subjects were treated with IV study drug and could be switched to oral study drug at the discretion of the Investigator after 3 full days (6 doses) of IV treatment if, in the opinion of the Investigator, pre-defined criteria were met. In LEAP 2, subjects were treated with lefamulin for five days (ten doses) compared to seven days of moxifloxacin (seven doses).
LEAP 1 (IV to Oral) Phase 3 Clinical Trial
In LEAP 1, a total of 551 subjects with Pneumonia Outcomes Research Team (PORT) Risk Class III to V who required IV antibiotic therapy as initial treatment for the current episode of CABP were randomized 1:1 to treatment with lefamulin 150 mg IV every 12 hours (n=276) or moxifloxacin 400 mg IV every 24 hours (n=275). Subjects could be switched from IV to oral study drug (lefamulin 600 mg orally every 12 hours or moxifloxacin 400 mg orally every 24 hours) at the discretion of the Investigator after three full days (six doses) of IV treatment if pre-defined criteria were met. If the
investigator determined that MRSA was a probable pathogen at Screening, adjunctive linezolid 600 mg IV every 12 hours was to be added to the moxifloxacin group and linezolid placebo was to be added to the lefamulin group.
The protocol defined different primary endpoints for the FDA for the development of drugs for CABP and guidance from the EMA to address regional differences in regulatory requirements for the development of antibacterial agents with the goal of positioning lefamulin as a first-line empiric monotherapy for the treatment of CABP. We reached agreement with the FDA in September 2015 on a SPA regarding the study design for our first Phase 3 clinical trial and obtained input from select European authorities in anticipation of submitting a new drug application with the FDA and a marketing authorization application, or MAA, with the EMA, in each case, for the treatment of CABP. In April 2016, we reached agreement with the FDA regarding an amendment to the SPA. We also plan to conduct a number of studies to support FDA approval of lefamulin, including studies in patients with hepatic insufficiency and renal impairment. If we complete the two Phase 3 clinical trials of lefamulin when we anticipate and obtain favorable results, we expect to submit an NDA to the FDA and an MAA to the EMA in 2018.
Our first Phase 3 clinical trial of lefamulin for the treatment of CABP is a multi-center, randomized, controlled, double-blind study comparing lefamulin to moxifloxacin, a fluoroquinolone antibiotic. Linezolid (or matching placebo for the lefamulin arm), can be added to treatment if an investigator suspects that a patient is infected with MRSA prior to randomization, as moxifloxacin is not approveddrugs to treat MRSA. This trial is designed to assessCABP. The FDA primary endpoint (EMA secondary endpoint) was the non-inferioritypercentage of lefamulin compared to moxifloxacin,subjects with an Early Clinical Response, or without linezolid. We expect the study population will include male and female patientsECR, of responder at least 18 years of age. Our study design targets the enrollment of approximately 550 patients, of which we expect a small proportion will require linezolid to be added.
Lefamulin will be dosed at 150 mg IV every 12 hours. The comparator drugs will be dosed according to their approved labeling, with moxifloxacin dosed at 400 mg IV daily and linezolid at 600 mg IV every 12 hours. Based on pre-defined criteria, investigators will have the option to switch patients to oral therapy96 ± 24-hours after three days (at least six doses) of IV study medication. Lefamulin will be administered orally as one 600 mg tablet every 12 hours, moxifloxacin at 400 mg daily and linezolid at 600 mg every 12 hours. Based on the pharmacokinetic profile of lefamulin, we expect oral dosing of one 600 mg tablet every 12 hours to have a similar therapeutic benefit as IV dosing of 150 mg every 12 hours.
All patients enrolled in this trial will be classified as Pneumonia Outcomes Research Team, or PORT, severity of at least 3 on a scale of 1 to 5, which corresponds to moderate to severe clinical disease. Patients who have previously taken no more than one dose of a short acting, potentially effective antibiotic for the treatment of the current CABP episode within 24 hours of receiving the first dose of study medication will be allowed to participate in the trial but will comprise only up to 25% of the total intent to treat, or ITT, population. Patients with confirmed S. aureus bacteremia will be discontinued from the trial. Investigators will obtain baseline Gram’s stain and culture of suitable specimens from the site of infection. Patients will be treated for a minimum seven days and a maximum of ten days.
We will assess patients between 72 and 120 hours from the start of treatment, at the end of treatment, or EOT, within 48 hours of administration of the final dose of study medication, at a TOC visit between five and ten days after administration of the final dose of study medication and at a telephone follow-up 30 days after administration of the first dose of study medication.
We will evaluate the following patient subsets:
· an ITT population consisting of all randomized patients regardless of whether they have received study medication;
· a modified intent to treat, or MITT, population consisting of all randomized subjects who receive any amount of study drug;
· a microbiological intent to treat, or microITT, population consisting of all subjects in the ITT population who have at least one baseline bacterial pathogen known to cause CABP, Legionella pneumophila from an appropriate microbiological specimen, or CABP caused by Mycoplasma pneumoniae or Chlamydophila pneumoniae;
· a clinically evaluable, or CE, population which is a subset of the ITT population that will include subjects who meet the criteria for CABP and who have received at least the pre-specified minimal amount of the intended dose of study drug and duration of treatment, do not have an indeterminate response based on the investigator’s assessment of clinical response at EOT for the CE-EOT population and at TOC for the CE-TOC population, did not receive concomitant antibacterial therapy, other than adjunctive linezolid, that is potentially effective against CABP pathogens (except in the case of clinical failure) from the first dose of study drug through the EOT visit for the CE-EOT population and through the TOC visit for the CE-TOC population, and for whom there are no other confounding factors that interfere with the assessment of the outcome; and
· a microbiologically evaluable, or ME, population consisting of all subjects who meet the criteria for inclusion in both the microITT, CE-EOT and ME-EOT populations or the CE-TOC and ME-TOC populations.
The primary efficacy endpoint for the trial for the FDA is the proportion of patients in the Intent-to-treat, or ITT, population for eachAnalysis Set. The EMA co-primary endpoints (FDA secondary endpoints) were the percentages of the lefamulin treatment group and the moxifloxacin treatment group who are alive, have improvement in at least two of the four cardinal symptoms of CABP as outlined in the current FDA guidance, have no worsening in any of the four cardinal symptoms of CABP and have not received a concomitant antibiotic (other than linezolid) for the treatment of CABP up through 120 hours after the first dose of lefamulin. This endpoint is also referred to as early clinical response. The four cardinal symptoms of CABP, as outlined in the current FDA guidance, are difficulty breathing, cough, production of purulent sputum and chest pain.
The primary efficacy endpoint for the EMA is the clinical success rate at the TOC visit for lefamulin in both the CE and MITT populations compared to moxifloxacin. Clinical success is based on the investigator’s assessment that a patient has clinically responded to lefamulin, which means that the patient has complete resolution or significant improvement of all local and systemic signs and symptoms of infection such that no additional antibiotic treatment is administered for the treatment of the current episode of CABP.
Key secondary efficacy and exploratory endpoints for our first Phase 3 clinical trial include the following:
· assessment of response for the primary efficacy outcome of early clinical response (the FDA primary endpoint) in the microITT population;
· assessment of response in each treatment groupsubjects with an investigator assessmentInvestigator's Assessment of clinical responseClinical Response (IACR) of success at TOC (the EMA primary endpoint) in the microITT and ME-TOC populations;
· assessmentTest of the microbiological response by pathogen for the microITT and ME-TOC populations at TOC; and
· assessment of all-cause mortality through day 28 in the ITT population.
In April 2016, we reached an agreement with the FDA under the Special Protocol Assessment procedureCure (TOC) Visit (5 to amend the design of our first Phase 3 clinical trial of lefamulin for the treatment of CABP. The amendment to the SPA followed discussions with the FDA regarding the design of our second Phase 3 clinical trial of lefamulin as well as discussions regarding the overall Phase 3 development program for lefamulin for the treatment of CABP. We believe the design of the study remains within the parameters set forth in the FDA Guidance for Industry Community-Acquired Bacterial Pneumonia: Developing Drugs for Treatment. Key amendments to the study include:
· modify the non-inferiority margin of the primary FDA endpoint of early clinical response from 10% to 12.5%;
· reduce the total number of patients to be enrolled in the study from 740 to as low as 550 as a result of a revision of the non-inferiority margin;
· simplify the treatment regimens; and
· increase the duration of therapy from a minimum of five days to a minimum of seven days.
In December 2016, we reached an enrollment target of 60% in our first Phase 3 clinical trial, and in February 2017, we received a recommendation from an independent interim analysis committee that we continue the trial with no adjustment to its sample size.
Second Phase 3 Clinical Trial
Our second Phase 3 clinical trial of lefamulin for the treatment of CABP is a multi-center, randomized, controlled, double-blind study comparing oral lefamulin to moxifloxacin, a fluoroquinolone antibiotic. This trial is designed to assess the non-inferiority of oral lefamulin compared to moxifloxacin. We expect the study population will include male and female patients of at least 18 years of age. We are targeting the enrollment of approximately 738 patients in this trial.
Lefamulin will be dosed orally at 600mg every 12 hours. The comparator drug moxifloxacin will be dosed according to approved labeling at 400 mg daily. All medications will be administered according to a double-blind and double-dummy design.
All patients enrolled in this trial will be classified as PORT severity of at least 2 and no greater than 4 on a scale of 1 to 5 which corresponds to moderate disease. Patients who have previously taken no more than one dose of a short acting, potentially effective antibiotic for the treatment of the current CABP episode within 24 hours of receiving the first dose of study medication will be allowed to participate in the trial but will comprise only up to 25% of the total intent to treat, or ITT, population. Investigators will obtain baseline Gram’s stain and culture of suitable specimens from the site of infection. Patients will be treated for five days with lefamulin or for seven days with moxifloxacin. We will assess patients between 72 and 120 hours from the start of treatment, at the end of treatment, or EOT, within 48 hours of administration of the final dose of study medication, at a TOC visit between five and ten days after administration of the final dose of study medication and at a telephone follow-up 30 days after administration of the first dose of study medication.
We will evaluate the following patient subsets:
· an ITT population consisting of all randomized patients regardless of whether they have received study medication;
· a MITT population consisting of all randomized subjects who receive any amount of study drug;
· a microITT, population consisting of all subjects in the ITT population who have at least one baseline bacterial pathogen known to cause CABP, Legionella pneumophila from an appropriate microbiological specimen, or CABP caused by Mycoplasma pneumoniae or Chlamydophila pneumoniae;
· a clinically evaluable, or CE, population which is a subset of the ITT population that will include subjects who meet the criteria for CABP and who have received at least the pre-specified minimal amount of the intended dose of study drug and duration of treatment, do not have an indeterminate response based on the investigator’s assessment of clinical response at EOT for the CE-EOT population and at TOC for the CE-TOC population, did not receive concomitant antibacterial therapy, other than adjunctive linezolid, that is potentially effective against CABP pathogens (except in the case of clinical failure) from the first dose of study drug through the EOT visit for the CE-EOT population and through the TOC visit for the CE-TOC population, and for whom there are no other confounding factors that interfere with the assessment of the outcome; and
· a microbiologically evaluable, or ME, population consisting of all subjects who meet the criteria for inclusion in both the microITT, CE-EOT and ME-EOT populations or the CE-TOC and ME-TOC populations.
The primary efficacy endpoint for the trial for the FDA is the proportion of patients in the ITT population for each lefamulin treatment group and the moxifloxacin treatment group who are alive, have improvement in at least two of the four cardinal symptoms of CABP as outlined in the current FDA guidance, have no worsening in any of the four cardinal symptoms of CABP and have not received a concomitant antibiotic for the treatment of CABP up through 120 hours after the first dose of lefamulin. This endpoint is also referred to as early clinical response.
The primary efficacy endpoint for the EMA is the clinical success rate at the TOC visit for lefamulin in both the CE and MITT populations compared to moxifloxacin. Clinical success is based on the investigator’s assessment that a patient has clinically responded to lefamulin, which means that the patient has complete resolution or significant improvement of all local and systemic signs and symptoms of infection such that no additional antibiotic treatment is administered for the treatment of the current episode of CABP.
Key secondary efficacy and exploratory endpoints for our second Phase 3 clinical trial include the following:
· assessment of response for the primary efficacy outcome of early clinical response (the FDA primary endpoint) in the microITT population;
· assessment of response in each treatment group with an investigator assessment of clinical response at TOC (the EMA primary endpoint) in the microITT and ME-TOC populations;
· assessment of the microbiological response by pathogen for the microITT and ME-TOC populations at TOC; and
· assessment of all-cause mortality through day 28 in the ITT population.
Completed Phase 2 Clinical Trial in ABSSSI
In 2011, we completed a multi-center, randomized, double-blind Phase 2 clinical trial in the United States evaluating the efficacy, safety and pharmacokinetics of the IV formulation of lefamulin compared to vancomycin in patients with ABSSSI. We selected ABSSSI as the indication for the trial to ensure that there would be a significant population of patients with multi-drug resistant Gram-positive bacteria. Gram-positive bacteria are the prevalent pathogens in ABSSSI. We selected vancomycin as the comparison therapy because vancomcyin is one of the antibiotics recommended by IDSA guidelines for the treatment of ABSSSI.
Trial Design
We enrolled 210 hospitalized patients with ABSSSI in the trial. The study population included male and female patients of at least 18 years of age and documented ABSSSI known or suspected to have been caused by a Gram-positive pathogen. Patients must have exhibited two signs of systemic inflammation or evidence of a significant underlying systemic or local medical condition at the time of enrollment and have required IV antibiotic therapy for the treatment of the ABSSSI.
We randomized patients on a 1:1:1 basis to three treatment groups to receive:
· 100 mg of IV lefamulin every 12 hours;
· 150 mg of IV lefamulin every 12 hours; or
· 1,000 mg of IV vancomycin every 12 hours or otherwise dosed as local practice dictated based upon a patient’s kidney function.
Investigators obtained baseline Gram’s stain and culture of suitable specimens from the site of infection. We treated patients for a minimum of five days and a maximum of 14 days. We assessed patients on day 3, at the end of treatment within 24 hours of administration of the final dose of study medication, at a TOC visit between seven and 14 days after administration of the final dose of study medication and at telephone follow-up at 3010 days after the last dose of study medication was administered.
The trial protocol specifieddrug) in the following four patient subsets for evaluation:
mITT and Clinically Evaluable at Test of Cure, or CE-TOC Analysis Sets.
· an ITT population consisting of all Of the 551 subjects randomized, patients who546 received at least one doseany amount of study medication;drug (Safety Analysis Set: 273 lefamulin, 273 moxifloxacin). The mean total duration of study drug treatment (IV and oral combined) was approximately seven days in each treatment group. The two treatment groups were generally well balanced with respect to demographics and baseline characteristics. Overall, 59.9% of subjects were male. The overall mean age was 60.3 years; 43.6% were³65 years and 18.1% were³75 years. Overall, 72.1% of subjects were classified as PORT Risk Class III, 26.5% were PORT Risk Class IV, and 1.3% were PORT Risk Class V.
· an MITT population consistingLEAP 1 met its primary objective and demonstrated that lefamulin is non-inferior to moxifloxacin with or without adjunctive linezolid for the treatment of all patientsadult subjects with CABP based on the FDA and EMA primary endpoints. The ECR responder rate (FDA primary endpoint) was 87.3% in the ITT population who had a documented Gram-positive pathogen culture at baseline;
· a CE population consisting of patients who had a confirmed diagnosis of ABSSSI, received study medication as randomized, received at least 80% of expected study medication, did not receive any potentially concomitant antibiotics, were not unblindedlefamulin group and had a response assessment at the TOC visit; and
· an ME population consisting of patients90.2% in the CE population who had a documented Gram-positive pathogen culture at baseline.
moxifloxacin group (treatment difference –2.9%; 95% CI: –8.5, 2.8). The primary efficacy endpointlower limit of the trial was the clinical success rate at the TOC visit95% CI for the 100 mgdifference in ECR responder rates was greater than the non-inferiority margin of –12.5%. Success rates for IACR at TOC (EMA co-primary endpoints) were 81.7% in the lefamulin group and 150 mg dosage forms84.2% in the moxifloxacin group (treatment difference –2.6%; 95% CI: –8.9, 3.9) in the mITT group, and 86.9% in the lefamulin group and 89.4% in the moxifloxacin group (treatment difference –2.5%; 95% CI: –8.4, 3.4) in the CE-TOC group. The lower limit of IVthe 95% CI for the difference in IACR success rates was greater than the non-inferiority margin of –10% for both groups.
Early Clinical Response rates for the most frequently identified baseline pathogens in the Microbiological Intent-to-treat, or microITT, group were:S. pneumoniae (88.2% lefamulin vs 93.8% moxifloxacin),H. influenzae (92.2% lefamulin vs 94.7% moxifloxacin), M. catarrhalis (92.0% lefamulin vs 100.0% moxifloxacin),M. pneumoniae (84.2% lefamulin vs 90.0% moxifloxacin),L. pneumophila (88.9% lefamulin vs 85.7% moxifloxacin), andC. pneumoniae (90.9% lefamulin vs 94.7% moxifloxacin). ECR responder rates forS. aureus were 100.0% in both the CE and MITT populations compared to vancomycin. Clinical success was defined as complete resolution or significant improvement of all local and systemic signs and symptoms of infection with no further systemic antibiotic treatment required.
Key secondary efficacy and exploratory endpoints of the trial included the following:
· assessment of clinical responsegroups. Responder rates among resistant pathogens were high in the ITTlefamulin group (eg, 100.0% for penicillin-intermediateS. pneumoniae [PISP], penicillin-resistantS. pneumoniae [PRSP], multi-drug resistantS. pneumoniae, or MDRSP, and ME populations;macrolide-resistantS. pneumoniae), although the number of resistant pathogens was low.
· comparison of clinical response by pathogenBoth lefamulin and microbiological response by pathogen;
· change in lesion size and resolution of fever; and
· clinical response at day 3.
Evaluation of pharmacokinetic parametersmoxifloxacin were well tolerated in the trial included analysis of plasma concentrations of lefamulin in blood samples after the first dose on day 1, on day 5 and on the finalIV ± oral treatment day.
Three of the 210 patients enrolledregimens administered in the trial did not receive study medication, resulting in 207 patientsstudy. A similar rate of treatment-emergent adverse events, or TEAEs, was observed (38.1% vs 37.7% in the ITT population. Oflefamulin and moxifloxacin groups, respectively). Gastrointestinal events were the patientsmost frequently reported TEAEs in the ITT population, 105 patients had cellulitis (50.7%), 64 patients had abscess with cellulitis (30.9%), 37 patients had wound infections (17.9%) and one patient had burns (0.5%). At least one pathogen responsible for ABSSSI was identified in 155 patients. Of these patients, 152 patients (97.4%) had at least one Gram-positive pathogen, comprising the MITT population. The most frequent Gram-positive pathogen was S. aureusboth treatment groups (6.6% lefamulin, 13.6% moxifloxacin), with the majority, 69.1%difference between groups driven by an imbalance in TEAEs of patientsdiarrhea (0.7% lefamulin, 7.7% moxifloxacin). No gastrointestinal TEAEs led to discontinuation of study drug in the MITT population, being methicillin-resistant strains.either treatment group.
Administration site reactions of any type occurred more frequently for lefamulin (7.7%) than moxifloxacin (3.7%). The CE population included 165 patients. The ME population included 129 patients.most common individual TEAE was infusion site pain, affecting eight (2.9%)
Patient demographics were similar across all three treatment groups, except for the presence of diabetes at baseline. The 150 mg lefamulin dose group included a slightly greater proportion of patients with diabetes than the other treatment groups.
Efficacy
In the trial, the patientssubjects in the lefamulin treatment groups experienced a similar clinical success rate at the TOC visit as patientsgroup, and no subjects in the vancomycinmoxifloxacin group. One subject in each treatment group in eachhad an infusion site reaction that led to discontinuation of the ITT, MITT, CE and ME patient subsets. These results are summarized in the table below. In addition, the clinical success rate in the trial was high for important subgroups of patients based on factors such as primary infection type and diabetes mellitus status. The table below also shows the 95% confidence interval, a statistical determination that demonstrates the range of possible differences in the point estimate of success that will arise 95% of the time that the endpoint is measured. However, this trial was not statistically powered to determine differences between treatment groups. The sample size chosen was to provide clinically meaningful information on efficacy, safety and tolerability. In this table and other tables appearing below, the abbreviation “N” refers to the number of patients or subjects in each group or subgroup.study drug.
Clinical Success Rate atThe incidence of TEAEs leading to discontinuation of study drug was 2.9% and 4.4% for the TOC Visit (ITT, MITT, CElefamulin and ME Populations)moxifloxacin groups, respectively. The only TEAE preferred terms leading to discontinuation for more than 1 subject per treatment group were electrocardiogram (ECG) QT prolonged (one lefamulin-treated subject and three moxifloxacin-treated subjects) and infectious pleural effusion (one lefamulin-treated subject and two moxifloxacin-treated subjects).
|
|
|
|
| ||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
| |||||
|
|
|
|
In the trial, the patientsSerious TEAEs occurred in 7.0% of subjects in the lefamulin treatment groups also experienced a similar clinical response at the day 3 visit as patientsgroup and 4.8% of subjects in the vancomycin treatmentmoxifloxacin group in each of the ITT, MITT, CE and ME patient subsets. The clinical response results for the ITT patient subset are presentedwere most frequently reported in the table below. Importantly, the assessment at day 3 included evaluation of a new primary endpoint recommended by the FDA of at least a 20% reduction in area of erythema, Infections and Infestations System Organ Class,or redness.SOC (2.9% lefamulin, 1.5% moxifloxacin).
Clinical Response at Day 3 (ITT Population)
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
|
A list of all pathogens identified at baseline along with the corresponding eradication rate by treatment group in the MITT patient subset is presented in the table below. Microbiological eradication rate was defined as the proportion of patients with a microbiological outcome of eradication or presumed eradication based on cultures from both the primary infection site and blood cultures. Patients with indeterminate or missing clinical
responses were considered non-eradication. Overall, in the MITT population, microbiological success was achieved in 40 of 50 patients (80.0%)Nine deaths (five in the lefamulin 100 mg group 43 of 51 patients (84.3%)and, four in the lefamulin 150 mg group,moxifloxacin group) occurred by Day 28. Two additional deaths were reported after Day 28 (i.e., after the intended Late Follow-up [LFU] Visit): one lefamulin-treated subject on Day 32 and 42 of 51 patients (82.4%) in the vancomycin group. We did not observe development of decreased susceptibility to lefamulin or vancomycin during the trial. In this table, the abbreviation “n” refers to the number of patients who had a microbiological outcome of eradication or presumed eradication for each specified pathogen.
Sponsor-Assessed Microbiological Eradication Rate at the TOC Visit by Baseline Target Pathogen (MITT Population)
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
|
We evaluated the clinical success of lefamulin against S. aureus, which is the most commonly identified cause of ABSSSI. The clinical success rate against a variety of subsets of S. aureus basedone moxifloxacin-treated subject on in vitro antibiotic susceptibility (methicillin-resistance), as well as the presence or absenceDay 48. None of the virulence factors PVL-positivitydeaths was assessed as related to study drug by the Investigators.
There were no clinically meaningful trends or USA300, are clinically important, as limited therapeutic options exist to treat such infection. A summarypattern of the clinical success rate against S. aureus is presentedchanges identified in the table below. The clinical success rates for lefamulin against PVL-positive MRSA and USA300 MRSA strains were similar to,hematology or numerically higher than, the corresponding clinical success rates for vancomycin. In this table, the abbreviation “n” refers to the number of patients with clinical success for each specified pathogen.chemistry laboratory parameters. No subjects met Hy's Law criteria.
LEAP 2 (Oral Only) Phase 3 Clinical Trial
In LEAP 2, a total of 738 subjects with PORT Risk Class II to IV who were appropriate candidates for oral antibiotic therapy for the current episode of CABP were randomized 1:1 to treatment with lefamulin (n=370) or moxifloxacin (n=368). Subjects received either lefamulin 600 mg orally every 12 hours for 5 days (10 doses) or moxifloxacin 400 mg orally every 24 hours for seven days (seven doses). The primary and secondary objectives were identical to those in LEAP 1.
Clinical Success Rate at Of the TOC Visit by Baseline Target Pathogens (S. aureus) (MITT Population)
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
|
738 subjects randomized, 736 received any amount of study drug (Safety Analysis Set: 368 lefamulin, 368 moxifloxacin). The mean duration of exposure to active lefamulin was 5.0 days, compared with 6.7 days of active moxifloxacin, which reflects the intended duration of active treatment for each drug as per the study medicationdesign. The two treatment groups were generally well balanced with respect to demographics and baseline characteristics. Overall, 52.4% of subjects were male. The overall mean age was approximately seven days for all groups,57.5 years; 37.5% were³65 years and almost 70%16.3% were³75 years. Overall, 50.4% of patients completed therapy within that time.subjects were classified as PORT Risk Class II, 37.7% were PORT Risk Class III, and 11.1% were PORT Risk Class IV.
SafetyLEAP 2 met its primary objective and tolerability
Lefamulin was well tolerated in this trial. No patient in the trial suffered any serious adverse eventsdemonstrated that were found to be related to lefamulin, and no patient in the trial died. The percentage of patients in the trial arms that experienced any treatment emergent adverse event were similar across treatment groups: 71.4% in the lefamulin 100 mg group, 69.0% in the lefamulin 150 mg group and 74.2% in the vancomycin group. Most of the treatment emergent adverse events were mild to moderate in severity. The table below shows the adverse events experienced by patients in the trial that were assessed by the investigator as possibly or probably related to study medication.
Drug-Related Treatment-Emergent Adverse Events by Preferred Term Reported by More Than 2% of Patients in the ITT Population
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
| |||
|
|
|
|
The incidences of pain, tenderness, itching, erythema, swelling and thrombosis at the infusion site were higher for the lefamulin 100 mg group and the lefamulin 150 mg group than for the vancomycin group. The majority of these local tolerability symptoms were mild in severity. No patient had a severe local tolerability issue of erythema or swelling. No patient had a local tolerability issue of necrosis. When summarized on an infusion basis, the proportions of infusions with local tolerability events were similar for the treatment groups.
Four patients discontinued study medication following a drug-related adverse event: one patient (1.4%) in the lefamulin 100 mg group (events of hyperhidrosis, vomiting and headache), two patients (2.8%) in the lefamulin 150 mg group (infusion site pain in one patient and dyspnea in the other), and one patient (1.5%) in the vancomycin group (drug eruption).
Because the potential for mild effect on ECG measurements was observed in preclinical studies, we have continued to assess this potential in all human clinical trials we have conducted. In the Phase 2 clinical trial, no change in ECG measurements was considered to be of clinical significance, and no drug-related cardiovascular adverse event was reported. Both lefamulin and vancomycin treatment were associated with a small increase in the QT interval. The QT interval is a measure of the heart’s electrical cycle, with a lengthened QT interval representing a marker for potential ventricular arrhythmia. We plan to continue to evaluate the effect of lefamulin on the QT interval in our Phase 3 clinical trials of lefamulin for CABP.
Pharmacokinetics
The table below summarizes selected pharmacokinetic parameters we obtained from pharmacokinetic sampling in the trial. Cmax refers to the maximum observed peak blood plasma concentration of study medication. AUC refers to the area under the curve in a plot of concentration of study medication in blood plasma over time, representing total drug exposure over time. In this table, the abbreviation “SD” refers to the standard deviation of the results. Standard deviation is a statistical measure used to quantify the amount of variation within a set of data values. A standard deviation close to zero indicates that the data points do not vary greatly from the mean, while a high standard deviation indicates that the data points are spread over a wider range of values.
Summary Statistics for PK Exposure and Secondary PK Parameters
|
| |||
|
|
| ||
|
|
| ||
|
|
| ||
|
|
| ||
|
|
| ||
|
|
| ||
|
|
| ||
|
|
| ||
|
|
| ||
|
|
|
Efficacy for the pleuromutilin class of antibiotics is related to the ratio of total drug exposure over time, measured by the AUC, to minimum inhibitory concentration, or MIC. MIC is the minimum concentration of an antibiotic needed to inhibit growth of an organism. The plasma concentration data obtained from the Phase 2 clinical trial are the first data that describe how lefamulin is absorbed, distributed around the body, metabolized and eliminated in patients suffering from an infection.
Phase 1 Clinical Development
We conducted seventeen Phase 1 clinical trials of lefamulin in Austria, Germany, the United States and the United Kingdom between 2009 and 2015. In these trials, we exposed 321 healthy subjects, including elderly subjects 65 years of age or older,non-inferior to single or multiple doses of IV or oral lefamulin. The objectives of our Phase 1 clinical trial program have been to understand the absorption and distribution of lefamulin in the blood and target tissues, evaluate the metabolism and elimination route of lefamulin and obtain safety and tolerability data to help predict safe and effective doses of lefamulinmoxifloxacin for the treatment of patients. We have additional Phase 1adult subjects with CABP based on the FDA and EMA primary endpoints. The ECR responder rate (FDA primary endpoint) was 90.8% in the lefamulin group and 90.8% in the moxifloxacin group (treatment difference 0.1%; 95% CI: –4.4, 4.5). The lower limit of the 95% CI for the difference in ECR responder rates was greater than the non-inferiority margin of –10%. Success rates for IACR at TOC (EMA co-primary endpoints) were 87.5% in the lefamulin group and 89.1% in the moxifloxacin group (treatment difference –1.6%; 95% CI: –6.3, 3.1) in the mITT group, and 89.7% in the lefamulin group and 93.6% in the moxifloxacin group (treatment difference –3.9%; 95% CI: –8.2, 0.5) in the CE-TOC group. The lower limit of the 95% CI for the difference in IACR success rates was greater than the non-inferiority margin of –10% for both groups.
Early Clinical Response rates for the most frequently identified baseline pathogens in the microITT group were:S. pneumoniae (89.4% lefamulin vs 91.3% moxifloxacin),H. influenzae (89.3% lefamulin vs 91.7% moxifloxacin),M. pneumoniae (100.0% in both groups), M. catarrhalis (85.7% lefamulin vs 100.0% moxifloxacin),L. pneumophila (81.3% lefamulin vs 94.1% moxifloxacin), andC. pneumoniae (93.8% lefamulin vs 100.0% moxifloxacin). ECR responder rates forS. aureus were 100.0% in both treatment groups. Responder rates among resistant pathogens were high in the
lefamulin group (eg, 100.0% for PISP, PRSP, MDRSP, and MRSA), although the number of resistant pathogens was low.
Both lefamulin and moxifloxacin were well tolerated in the oral treatment regimens administered in the study. The overall incidence of TEAEs was higher in the lefamulin group (32.6%) than in the moxifloxacin group (25.0%), which was driven by a difference in the incidence of mild/moderate Gastrointestinal Disorders. For lefamulin and moxifloxacin, respectively, the most frequently reported individual TEAEs in this category (and for the study overall) were diarrhea (12.2% vs 1.1%), nausea (5.2% vs 1.9%), and vomiting (3.3% vs 0.8%). Among the lefamulin-treated subjects reporting each of these TEAEs, approximately 75% had mild events and the remainder had moderate events. The only severe gastrointestinal adverse event, which was also serious, was a case of inguinal hernia strangulated in a moxifloxacin-treated subject. There were no severe or serious gastrointestinal adverse events among lefamulin-treated subjects. Furthermore, gastrointestinal events led to study drug discontinuation for 3 lefamulin-treated subjects (due to vomiting or abdominal pain) and one moxifloxacin-treated subject (due to vomiting). One patient who had a positive clinical trials ongoing, includingresponse to lefamulin was later diagnosed with aC. difficile infection during an extended hospital stay.
The incidence of TEAEs leading to discontinuation of study drug interaction studies as well as studieswas 3.3% for the lefamulin group and 2.4% for the moxifloxacin group.
Serious TEAEs occurred in special populations (hepatic4.6% of lefamulin-treated subjects and renally impaired subjects)4.9% of moxifloxacin-treated subjects, most frequently in the Infections and Infestations category (2.4% and 1.4%, respectively).
Pharmacokinetic Overview
Our key observations from our Phase 1 clinical trials include the following:
· lefamulin is rapidly absorbed and distributed throughout the bodyIn each treatment group, three (0.8%) subjects died by Day 28. Deaths of two additional lefamulin-treated subjects were reported after either IV or oral administration;
· lefamulin achieves therapeutic concentrations in a variety of target tissues, including the lung, skin and soft tissue;
· lefamulin has a half-life between 8.6 and 11.8 hours, which enables a twice daily regimen, and is eliminated primarily through non-renal pathways;
· lefamulin is a weak inhibitor of some liver enzymes, but we do not expect to need to adjust the dose of lefamulin when it is co-administered with other drugs;
· no statistically significant effects of age, gender, body weight or height, body mass index or other demographics on the pharmacokinetic parameters of lefamulin;
· the absolute oral bioavailability of a 600 mg immediate release, or IR, tablet formulation of lefamulin were 25.8 % in the fasted and 21% in the fed condition in healthy subjects; and
· in the assessment of relative bioavailability, bioequivalence was demonstrated for the fasted IR tablet and the IV dose, exposure was slightly lower for the fed IR tablet than the IV dose.
Absorption
Lefamulin is absorbed rapidly after oral dosing with or without food. In our Phase 1 clinical trials, steady state blood levels were achieved after two days of dosing every 12 hours, irrespective of the route of administration, and the variability after oral dosing was similar to the variability after IV infusion. Because the ability of pleuromutilin antibiotics to kill bacteria is dependent on the AUC, or total lefamulin exposure over time, to MIC ratio, and IV doses of 150 mg every 12 hours and oral doses of 600 mg every 12 hours achieve similar AUCs, we believe that both regimens are capable of providing a similar therapeutic benefit.
Distribution
Following IV infusion, lefamulin is rapidly distributed throughout the body over approximately 30 minutes. We have observed rapid distribution of lefamulin into tissues, including the skin and ELF of the lung. In CABP, the lung is the target organ where pathogens replicate and cause inflammation that results in mucous production, cough and shortness of breath. Therefore, in 2010, we conducted a Phase 1 clinical trial to assess the pharmacokinetics of lefamulin in 12 healthy subjects. After a single IV administration of 150 mg of lefamulin over 60 minutes, we performed a bronchoalveolar lavage, or BAL, a medical procedure to collect fluid from the lung. We performed BAL analyses in groups of three subjects at 1, 2, 4 and 8 hoursDay 28 (i.e., after the start of the lefamulin infusionintended LFU Visit): one subject on Day 57 and measured lefamulin concentrations in the ELF, the muscle tissue, soft tissue and blood plasma. In this trial, the exposure of free lefamulin, or the amount of lefamulin not bound to proteins and therefore available to inhibit bacterial growth, achieved in the ELF was approximately six times greater than free lefamulin exposure observed in blood plasma.
Metabolism
The average half-life, or the time it takes the body to eliminate one-half of the concentration of lefamulin present, is 9 to 12 hours. The major route lefamulin is eliminated from the body is the gastrointestinal tract, with limited metabolism of lefamulin occurring mainly through a liver and gut wall enzyme called CYP3A4, which is responsible for the metabolism of a wide variety of medication. We have identified only one metabolite, called BC-8041, as exceeding 10% of lefamulin concentrations in the plasma and only when lefamulin was given orally.subject on Day 271. None of the metabolitesdeaths was assessed as related to study drug by the Investigators.
No clinically meaningful trends or pattern of lefamulin have any antibacterial properties.changes were identified in hematology or chemistry laboratory parameters. Two unique lefamulin-treated subjects had either an ALT or an aspartate aminotransferase (AST) value >10 × the upper limit of normal, or ULN; in both cases the transaminase increases were transient with no associated increase in serum bilirubin. No subjects met Hy's Law criteria.
Drug Interaction Potential
We continue to perform studies recommended by regulatory authorities to assessElectrocardiogram analyses demonstrated increases from baseline in the drug-drug interaction potential of new drug products, includingQTcF interval in both treatment groups, but the assessmentmagnitude of the impact of potent P-glycoprotein and CYP3A4 inducers onchange in the PK oflefamulin treatment group was smaller than that caused by moxifloxacin. In this study the mean change from baseline in QTcF interval at the steady state assessment was 9.5 msec for lefamulin and the impact11.6 msec for moxifloxacin. Post-baseline QTcF increases of lefamulin on drugs metabolized via CYP3A4.
Safety>60 msec occurred in 1.1% of lefamulin-treated subjects and Tolerability
Lefamulin has been well tolerated1.9% of moxifloxacin-treated subjects. No associated cardiac arrhythmias of concern were observed. No adverse trends in all Phase 1 trials completed to date. We did not observe any systemic adverse events of clinical concern or any drug-related serious adverse events in these trials. In addition, we did not observe any changes of clinical concern in laboratory safety parameters or vital signs in any subject in any of the trials. The most commonly observed adverse effects with oral administration of lefamulineither treatment group were related to the gastrointestinal tract, including nausea and abdominal discomfort, while the most commonly observed adverse effects related to IV administration were related to irritation at the infusion site. In addition, lefamulin produced a transient, predictable and reproducible prolongation of the QT interval based on the maximum concentration of the drug in the blood plasma. At therapeutic doses, we expect that the drug will not produce large effects on the QT interval that would be of clinical relevance. We did not observe any drug-related cardiacobserved.
adverse events, such as increase in ectopic ventricular activity or other cardiac arrhythmia, or clinically relevant ECG findings during the conduct of any of our Phase 1 or Phase 2 clinical trials.
Intravenous FormulationPediatric Clinical Trial
We have administered IV lefamulin as single and repeat doses every 12 hours for up to 14 days. The most frequently reported adverse events in our Phase 1 clinical trials were pain or erythema at the site of the IV infusion. To further assess local tolerability, we conducted a Phase 1 clinical trial in 2013 to evaluate the local tolerability of two different IV formulations of lefamulin dosed every twelve hours for 7.5 days. In this trial, we compared lefamulin infusions given in normal saline solution, a sterile sodium chloride solution commonly used to administer IV medications, with lefamulin infusions given in a sterile saline solution buffered by a citrate salt that adjusted the pH, or level of acidity, of the solution. We enrolled 60 healthy subjects in the trial, of which 25 received the normal saline solution, 25 received the citrate buffered solution and ten received a matching placebo solution. Although we did not observe any difference between treatment arms over the first three days of study infusions, over the entire treatment period, the incidence of local pain or redness of at least moderate severity was statistically higher with lefamulin in the saline solution (84%), as compared to the citrate buffered infusions (36%) or placebo (10%). There was no statistical difference between citrate buffered infusions and placebo at any time period during the trial. As a result, we will administer lefamulin IV infusions in a citrate buffered saline solution in our Phase 3 clinical trials for CABP.
Oral Formulation
Initially, we administered lefamulin orally in capsules as single and repeat doses for up to five days. Oral administration of lefamulin was generally well tolerated with infrequent reports of mild gastrointestinal findings, such as nausea, abdominal pain and diarrhea. We subsequently developed 600 mg tablets that we have investigated in single and repeat dose studies. These tablets have been well tolerated and shown favorable pharmacokinetics. We will utilize the 600 mg tablets in our Phase 3 clinical trials.
Electrocardiogram Measurements
In our Phase 1 clinical trials, lefamulin was associated with a Cmax-dependent, transient, predictable, reversible and reproducible prolongation of the QT interval. We have closely monitored ECG measurements in all our trials. We did not observe any drug-related cardiac adverse events, such as increase in ectopic ventricular activity or other cardiac arrhythmia, or clinically relevant ECG findings during the conduct of any of our Phase 1 clinical trials. None of the ECG stopping criteria defined in the trial protocols was reached in any clinical trial. We plan to continue to assess the effects of lefamulin on the QT interval in our planned clinical trials.
Preclinical Development
In our preclinical studies, administration of lefamulin was well tolerated in a variety of animal models. Lefamulin was active against a broad range of bacteria, suggesting possible use as monotherapy for CABP with a low propensity for development of bacterial resistance.
Nonclinical Safety
In several preclinical safety and toxicity studies, including repeat dose toxicity, local tolerance, genotoxicity, development and reproductive toxicity, and safety pharmacology testing in both rodent and non-rodent species, lefamulin was safe and well tolerated. When we treated rats or monkeys for up to four weeks with either oral or IV lefamulin, we did not identify any specific target organ toxicity. Lefamulin was not associated with genetic damage, effects on fertility or birth defects. We are also conducting longer term toxicity studies in a rodent and a non-rodent species.
Antimicrobial Spectrum of Lefamulin
We have extensively studied the antimicrobial in vitro activity of lefamulin against a variety of respiratory, or aerobic, and non-respiratory, or anaerobic, bacterial pathogens representing more than 17,700 clinical isolates collected from patients worldwide. A summary of our observations is presented in the table
below. MIC90 indicates the concentration of drug that inhibits 90% of the pathogens in vitro, while MIC50 indicates the concentration of drug that inhibits 50% of the pathogens in vitro.
Antimicrobial Activity of Lefamulin Against Gram-Positive, Gram-Negative and Atypical Bacteria
|
|
|
| MIC [µg/mL] |
| ||
Organism |
| N |
| 50% |
| 90% |
|
Aerobic and facultative anaerobic Gram-positive microorganisms |
|
|
|
|
|
|
|
S. aureus |
| 7984 |
| 0.12 |
| 0.12 |
|
S. aureus, MSSA |
| 4365 |
| 0.06 |
| 0.12 |
|
S. aureus, MRSA |
| 3619 |
| 0.12 |
| 0.25 |
|
CA-MRSA (USA 300/400) |
| 50 |
| 0.12 |
| 0.12 |
|
VRSA, VISA, hVISA |
| 30 |
| 0.06 |
| 0.25 |
|
Coagulase-negative Staphylococcus species |
| 1133 |
| 0.06 |
| 0.12 |
|
S. epidermidis |
| 474 |
| 0.06 |
| 0.25 |
|
S. pneumoniae |
| 3570 |
| 0.12 |
| 0.25 |
|
S. pyogenes (Group A Streptococcus species) |
| 472 |
| 0.03 |
| 0.03 |
|
S. agalactiae (Group B Streptococcus species) |
| 503 |
| 0.03 |
| 0.06 |
|
Group C Streptococcus species |
| 116 |
| 0.03 |
| 0.06 |
|
Group G Streptococcus species |
| 160 |
| 0.03 |
| 0.06 |
|
Viridans Group Streptococcus species |
| 445 |
| 0.12 |
| 0.5 |
|
E. faecalis |
| 50 |
| >32 |
| >32 |
|
E. faecium |
| 850 |
| 0.12 |
| 8 |
|
E. faecium, VSE |
| 361 |
| 0.12 |
| >32 |
|
E. faecium, VRE |
| 389 |
| 0.06 |
| 0.5 |
|
Aerobic and facultative anaerobic Gram-negative microorganisms |
|
|
|
|
|
|
|
H. influenzae |
| 1078 |
| 0.5 |
| 1 |
|
L. pneumophila |
| 30 |
| 0.12 |
| 0.5 |
|
M. catarrhalis |
| 855 |
| 0.12 |
| 0.25 |
|
N. gonorrhoeae, resistant isolates |
| 93 |
| 0.12 |
| 0.5 |
|
E. coli |
| 40 |
| 16 |
| 32 |
|
Anaerobic microorganisms |
|
|
|
|
|
|
|
C. difficile |
| 43 |
| 4 |
| 8 |
|
Clostridium species |
| 10 |
| 1 |
| >16 |
|
Peptostreptococcus species |
| 10 |
| 0.06 |
| 1 |
|
Porphyromonas species |
| 10 |
| 0.03 |
| 0.03 |
|
B. fragilis and B. fragilis group |
| 22 |
| >32 |
| >32 |
|
Other microorganisms |
|
|
|
|
|
|
|
C. pneumoniae |
| 50 |
| 0.02 |
| 0.04 |
|
C. trachomatis |
| 15 |
| 0.02 |
| 0.04 |
|
M. pneumoniae |
| 60 |
| <0.001 |
| 0.002 |
|
The tables below compare the in vitro activity of lefamulin and various antibiotics for CABP and ABSSSI pathogens against various strains of bacteria, including those resistant to current antibiotics. Unlike other CABP antibiotics, such as ß-lactam/ß-lactamase inhibitor combinations, glycopeptides and oxazolidinones, lefamulin was active against the vast majority of potential respiratory pathogens collected in 2015. When an alternative antibiotic from the same drug class was utilized, it is footnoted within the table and below.
Lefamulin in vitro Activity Against CABP Bacteria
Organism |
| Lefamulin |
| Levofloxacin |
| Azithromycin |
| Tigecycline |
| Ceftriaxone |
Streptococcus pneumoniae (1835) |
| 0.12 |
| <0.5 |
| >4 |
| 0.06 |
| 1 |
Haemophilus influenzae (536) |
| 1 |
| <0.015 |
| 1 |
| 0.25 |
| <0.015 |
Moraxella catarrhalis (446) |
| 0.12 |
| 0.03 |
| <0.03 |
| 0.06 |
| 0.5 |
Staphylococcus aureus incl. MRSA (1273) |
| 0.12 |
| >4 |
| >4 |
| 0.12 |
| * |
Legionella pneumophila (30) |
| 0.5 |
| 0.015 |
| 0.015 |
| * |
| * |
Mycoplasma pneumoniae (60) |
| 0.002 |
| 0.25 (a) |
| <0.0003 |
| 0.25 (b) |
| * |
Chlamydophila pneumoniae (50) (c) |
| 0.04 |
| 0.32-1.28 |
| 0.08-0.16 |
| 0.04 |
| * |
(a) Moxifloxacin used instead of levofloxacin against M. pneumoniae.
(b) Doxycycline used instead of tigecycline
(c) Only two strains tested for comparators.
* Not determined.
Lefamulin displayed potent antibacterial activity against bacterial pathogens predominantly causing skin and blood stream infections, such as S. aureus, coagulase-negative staphylococci, ß-hemolytic and viridans group streptococci, as well as E. faecium, including vancomycin-resistant strains, or VRE.
Lefamulin in vitro Activity Against ABSSSI Bacteria and Pathogens Causing Bacteremia
Organism |
| Lefamulin |
| Erythromycin |
| Doxycycline |
| Vancomycin |
|
S. aureus (5527) |
| 0.12 |
| >4 |
| 0.25 |
| 1 |
|
MSSA (3157) |
| 0.12 |
| >4 |
| 0.25 |
| 1 |
|
MRSA (2370) |
| 0.25 |
| >4 |
| 1 |
| 1 |
|
CoNS (878) |
| 0.12 |
| >4 |
| 2 |
| 2 |
|
E. faecium (536) |
| 4 |
| >4 |
| >8 |
| >16 |
|
VRE (304) |
| 0.25 |
| >4 |
| >8 |
| >16 |
|
ß-hemolytic Streptococcus species (763) |
| 0.03 |
| >4 |
| 8 |
| 0.5 |
|
S. pyogenes (267) |
| 0.03 |
| <0.25 |
| 8 |
| 0.5 |
|
S. agalactiae (334) |
| 0.03 |
| >4 |
| 8 |
| 0.5 |
|
Viridans group Streptococcus species (245) |
| 0.5 |
| >4 |
| >8 |
| 0.5 |
|
Activity Against Resistant Strains and Low Propensity for Development of Bacterial Resistance
When tested against bacterial organisms resistant to macrolides, tetracyclines, quinolones, trimethoprim/sulfamethoxazole, vancomycin, mupirocin or ß-lactams, we did not observe any cross-resistance with lefamulin. Lefamulin displayed activity in vitro against drug-resistant N. gonorrhoeae, VRE, MRSA, multi-drug resistant S. pneumoniae, VISA/hVISA, erythromycin-resistant group A Streptococcus species and clindamycin-resistant group B Streptococcus species, all of which are listed as urgent, serious or concerning threats by the CDC. We utilized the interpretative criteria of the Clinical and Laboratory Standards Institute, or CLSI, to categorize the in vitro activity of each comparator against the organisms listed in the table below as sensitive (%S), intermediate (%I) or resistant (%R). Bold and underlined data indicate resistance according to CLSI criteria.
Lefamulin in vitro Activity Against Resistant Bacterial Pathogens Listed as Urgent, Serious or Concerning Threats According to CDC
|
|
|
| MIC [µg/mL] |
| CLSI |
| ||
Organism |
| N |
| 50% |
| 90% |
| %S /%I /%/R |
|
|
|
|
|
|
|
|
|
|
|
Urgent Threats |
|
|
|
|
|
|
|
|
|
Drug-resistant Neisseria gonorrhoeae |
|
|
|
|
|
|
|
|
|
Lefamulin |
| 93 |
| 0.12 |
| 0.5 |
| — / — / — |
|
Azithromycin |
| 58 |
| 0.12 |
| 1 |
| 81.0 /6.9 / 12.1(b) |
|
Tetracycline |
| 58 |
| 0.5 |
| 2 |
| 19.0 / 56.9 / 24.1(c) |
|
Ciprofloxacin |
| 58 |
| 0.25 |
| 16 |
| 37.9 / 20.7 /41.4(c) |
|
Ceftriaxone |
| 54 |
| 0.015 |
| 0.06 |
| 100.0 / 0.0 /0.0(c) |
|
|
|
|
|
|
|
|
|
|
|
Serious Threats |
|
|
|
|
|
|
|
|
|
Methicillin-resistant Staphylococcus aureus (MRSA) |
|
|
|
|
|
|
|
|
|
Lefamulin |
| 3,570 |
| 0.12 |
| 0.25 |
| — / — / — |
|
Clindamycin |
| 2,370 |
| <0.25 |
| >2 |
| 63.5 / — / 36.4 |
|
Doxycycline |
| 2,370 |
| 0.12 |
| 1 |
| 96.2 / — / 0.8 |
|
Erythromycin |
| 2,370 |
| >4 |
| >4 |
| 15.0 / — / 84.1 |
|
Levofloxacin |
| 2,370 |
| >4 |
| >4 |
| 26.8 / — / 71.2 |
|
Linezolid |
| 2,370 |
| 1 |
| 1 |
| 100.0 / — / 0.0 |
|
Oxacillin |
| 2,370 |
| >2 |
| >2 |
| 0.0 / — / 100.0 |
|
Trimethoprim/sulfamethoxazole |
| 2,370 |
| <0.5 |
| <0.5 |
| 95.8 / — / 4.2 |
|
Vancomycin |
| 2,370 |
| 1 |
| 1 |
| 100.0 / — / 0.0 |
|
Drug-resistant Streptococcus pneumoniae |
|
|
|
|
|
|
|
|
|
Lefamulin |
| 633 |
| 0.06 |
| 0.12 |
| — / — / — |
|
Azithromycin |
| 633 |
| >4 |
| >4 |
| 0.3 / 0.9 / 98.7 |
|
Ceftriaxone |
| 633 |
| 0.25 |
| 1 |
| 90.2 / 7.3 / 2.5 |
|
Erythromycin |
| 633 |
| >2 |
| >2 |
| 0 / 0 / 100.0 |
|
Levofloxacin |
| 633 |
| 1 |
| 1 |
| 97.9 / — / 2.1 |
|
Penicillin (oral penicillin V) |
| 633 |
| 0.5 |
| 4 |
| 45.8 / 26.7 / 27.5 |
|
Tetracycline |
| 633 |
| >4 |
| >4 |
| 37.9 / 0.8 / 61.3 |
|
Trimethoprim/sulfamethoxazole |
| 633 |
| <0.5 |
| >4 |
| 50.1 / 16.9 / 33.0 |
|
Vancomycin |
| 633 |
| 0.25 |
| 0.25 |
| 100.0 / — / 0.0 |
|
|
|
|
|
|
|
|
|
|
|
Vancomycin-resistant Enterococcus faecium |
|
|
|
|
|
|
|
|
|
Lefamulin |
| 304 |
| 0.06 |
| 0.25 |
| — / — / — |
|
Ampicillin |
| 304 |
| >8 |
| >8 |
| 0.3 / — / 99.7 |
|
Daptomycin |
| 304 |
| 2 |
| 2 |
| 100.0 / — / — |
|
Linezolid |
| 304 |
| 1 |
| 1 |
| 98.7 / — / 0.7 |
|
Vancomycin |
| 304 |
| >16 |
| >16 |
| 0.0 / 99.3 |
|
|
|
|
| MIC [µg/mL] |
| CLSI |
| ||
Organism |
| N |
| 50% |
| 90% |
| %S /%I /%/R |
|
|
|
|
|
|
|
|
|
|
|
Concerning Threats |
|
|
|
|
|
|
|
|
|
Vancomycin-resistant Staphylococcus aureus |
|
|
|
|
|
|
|
|
|
Lefamulin |
| 10 |
| 0.06 |
| 0.12 |
| — / — / — |
|
Ceftaroline |
| 10 |
| 1 |
| 1 |
| 100.0 / — / — |
|
Daptomycin |
| 10 |
| 0.5 |
| 0.5 |
| 100.0 / — / — |
|
Linezolid |
| 10 |
| 1 |
| 1 |
| 100.0 / — / — |
|
Oxacillin |
| 10 |
| >4 |
| >4 |
| — / — / 100.0 |
|
Quinupristin/dalfopristin |
| 10 |
| 0.25 |
| 0.5 |
| 100.0 /— / — |
|
Tigecycline |
| 10 |
| 0.06 |
| 0.12 |
| 100.0 / — / — |
|
Vancomycin |
| 10 |
| >32 |
| >32 |
| — / — /100.0 |
|
|
|
|
|
|
|
|
|
|
|
Erythromycin-resistant Group A Streptococcus species |
|
|
|
|
|
|
|
|
|
Lefamulin |
| 25 |
| 0.015 |
| 0.03 |
| — /— /— |
|
Clindamycin |
| 25 |
| <0.25 |
| >2 |
| 56.0 / — / 44.0 |
|
Doxycycline |
| 25 |
| <0.05 |
| >8 |
| — / — /— |
|
Erythromycin |
| 25 |
| >4 |
| >4 |
| — / — /100.0 |
|
Levofloxacin |
| 25 |
| <0.5 |
| 1 |
| 96.0 / — / 4.0 |
|
Penicillin |
| 25 |
| <0.03 |
| <0.03 |
| 100.0 /— / — |
|
Vancomycin |
| 25 |
| 0.25 |
| 0.5 |
| 100.0 / — / — |
|
|
|
|
|
|
|
|
|
|
|
Clindamycin-resistant Group B Streptococcus species |
|
|
|
|
|
|
|
|
|
Lefamulin |
| 69 |
| 0.03 |
| 0.03 |
| — / — /— |
|
Clindamycin |
| 69 |
| >2 |
| >2 |
| — / — /100.0 |
|
Doxycycline |
| 69 |
| 8 |
| >8 |
| 8.7 / 2.9 / 88.4(e) |
|
Erythromycin |
| 69 |
| >4 |
| >4 |
| 1.4 / 0.0 / 98.6 |
|
Levofloxacin |
| 69 |
| <0.5 |
| 1 |
| 100.0 / — / — |
|
Penicillin |
| 69 |
| <0.03 |
| 0.06 |
| 100.0 / — / — |
|
Vancomycin |
| 69 |
| 0.5 |
| 0.5 |
| 100.0 /— / — |
|
(a) Criteria as published by CLSI (2011).
(b) No breakpoints by CLSI available; criteria as published by the European Committee on Antimicrobial Susceptibility Testing (2014).
(c) Criteria as published by CLSI (2014).
(d) No breakpoints by CLSI available; criteria as published by the European Committee on Antimicrobial Susceptibility Testing (2011).
(e) Breakpoints of tetracycline applied.
Lefamulin has shown low potential for resistance development in vitro, which we believe is the result of the specific interaction with a binding site on the ribosome. Repeated exposure to low levels of lefamulin in laboratory tests resulted in a slow and step-wise development of resistance in S. aureus, Streptococcus species, and E. faecium. We believe that lefamulin’s low potential for resistance is further supported by the fact that we observed isolates with consistently low MICs during our Phase 2 clinical trial in ABSSSI and that, despite the use of pleuromutilins in veterinary medicine for decades, the incidence of pleuromutilin-resistant isolates remains relatively low. Cross-resistance between lefamulin and other classes of antibiotics has also been rarely observed in our completed studies to date. Based on global surveillance studies in more than 13,600 clinical isolates, fewer than 0.02% of isolates contain mutations responsible for methylating, or chemically modifying, the interaction between lefamulin and other protein synthesis inhibiting antibiotics. One example of this mutation is called cfr mutation, which when present has resulted in observed elevations in the MIC90 to lefamulin as well as other antibiotics, such as chloramphenicol and linezolid.
Activity in Animals
We evaluated the activity of lefamulin in a number of murine, or mouse, infection models, including pneumonia, septicemia, and thigh infection models. In these models, lefamulin was efficacious against S.
pneumoniae (penicillin- and macrolide-resistant) and S. aureus (methicillin-susceptible, or MSSA, and MRSA). Lefamulin was active against serious lung infections caused by clinically relevant strains of S. pneumoniae or S. aureus. Investigations of the exposure levels in the ELF in the lungs of mice showed rapid distribution of lefamulin into the lung compartment with penetration rates into the ELF of 12-fold compared to free plasma concentration measured in the same mice. We confirmed this result by whole-body imaging using radioactive labeled lefamulin.
Lefamulin also showed high intracellular activity and rapid accumulation in macrophages, or immune cells that are responsible for assisting in clearing bacterial infections from the lung and other body sites, (30- to 50-fold) dependent on the time of incubation and lefamulin concentration. Azithromycin showed a 15- to 20-fold accumulation in the same experiments. Furthermore, the activity of lefamulin was unaffected by lung surfactant, a naturally occurring substance found in the lung that has the ability to inactivate some antibiotics.
Concentrations of Lefamulin Predicted to be Effective in Treating Lung Infections
We believe that results from preclinical analyses of concentrations of lefamulin in lung tissue indicate the potential for favorable outcomes in CABP patients treated with lefamulin. We have provided and discussed with regulatory authorities, including the FDA, these preclinical results and the safety and efficacy of lefamulin observed in subjects and patients with ABSSSI. Based on these discussions, we intend to evaluate the efficacy and safety of lefamulin in our Phase 3 clinical trials for CABP. We used results obtained from pharmacokinetic analyses of lefamulin concentrations over time and pharmacodynamic analyses of the relationship of concentrations of lefamulin and effect, also called PK-PD, to determine the predicted likelihood of achieving lefamulin concentrations in the lung that would be effective at inhibiting the growth of common bacterial causes of CABP. This assessment utilized a population pharmacokinetic model that describes the behavior of lefamulin in blood plasma in subjects and patients with infection, the ELF concentrations achieved in healthy volunteers, in vitro MIC targets for the most common causative pathogens associated with CABP accrued from a robust, global in vitro surveillance library, and mathematical simulations replicating thousands of scenarios that could represent the many possible combinations of lefamulin concentrations achieved and the MIC required to inhibit bacterial growth. Based on these assessments, we believe that a lefamulin regimen of either 150 mg administered by IV every 12 hours or 600 mg administered orally every 12 hours has a probability of 96% or more to achieve concentrations in the ELF that would inhibit the growth of both S. pneumoniae and S. aureus.
Based on all of the available evidence, including lefamulin’s in vitro activity, clinical pharmacokinetics and tissue penetration, and safety and efficacy observed in our Phase 2 clinical trial for ABSSSI, we are using a dose of 150 mg IV or 600 mg orally every 12 hours in our Phase 3 clinical trials of lefamulin for CABP.
Earlier Stage Product Pipeline
Additional Indications for Lefamulin
ABSSSI
Acute bacterial skin and skin structure infections are common and are characterized by a wide range of disease presentations. Gram-positive bacteria, in particular S. aureus, S. pyogenes and S. agalactiae, are the most common pathogens in ABSSSI. The rising frequency of ABSSSI caused by MRSA and the significant increase in the occurrence of CA-MRSA infections over the past 15 years is an increasing concern. According to IDSA Skin and Skin Structure Infection guidelines 2014, in most U.S. cities, CA-MRSA is now the most common pathogen cultured from patients with ABSSSI in emergency departments. While the current standard of care for MRSA infections is vancomycin, the efficacy of this treatment is being compromised because of decreased susceptibility, or even resistance, of S. aureus to vancomycin. In addition, although linezolid is approved for ABSSSI due to MRSA, its use has been limited because of potential adverse events and drug-drug interactions with commonly prescribed concomitant medications such as antidepressants.
The emerging incidence of resistance to multiple antibiotics in pathogens makes ABSSSI increasingly difficult to treat and results in a need for alternate therapies. Based on our preclinical studies and clinical trials, we believe that lefamulin has potential to treat ABSSSI. In preclinical studies, lefamulin has shown in vitro antibacterial activity against the most relevant pathogens responsible for ABSSSI including S. aureus (MSSA, MRSA, and CA-MRSA), S. pyogenes, and S. agalactiae. In our Phase 2 clinical trial evaluating the safety and efficacy of two different doses of the IV formulation of lefamulin administered over five to 14 days compared to vancomycin in patients with ABSSSI, the clinical success rate at test of cure for lefamulin was similar to that of
vancomycin. We have discussed the design of a proposed Phase 3 clinical trial to evaluate the efficacy and safety of lefamulin for the treatment of ABSSSI with the FDA and several E.U. regulatory authorities.
Pediatric Indications
Not unlike treatment of infectious diseases in adults, the management of pediatric infections has become more difficult due to the continuing rise in resistance in bacteria. Further complicating antimicrobial selection in the pediatric population is the need for agents to be very well tolerated and available in a final dosage form that can be easily administered to children. Based upon thein vitro antimicrobial spectrum of activity, along with the safety profile observed to date, we believe lefamulin is appropriate for evaluation for the treatment of a variety of pediatric infections, including those affecting the respiratory tract and skin and skin structure. We have begun pediatrican agreed Pediatric Investigation Plan, or PiP, and Pediatric Study Plan, or PSP, with the EMA and FDA, respectively. Pediatric oral formulation development activities to support clinical trials in the pediatric population.
HABP/VABP
One of the major causative organisms of hospital-acquired bacterial pneumoniais ongoing and ventilator-associated bacterial pneumonia is S. aureus, including MRSA. We are evaluating whether to investigate the utility of lefamulin in the treatment of HABP and VABP. We have deferred commencement ofwe initiated a previously planned Phase 1 clinical trial in pediatric patients in mid-2018.
STIsAdditional Potential Indications for Lefamulin
STIs
Urethritis and cervicitis caused byN. gonorrhoeae,C. trachomatis orM. genitalium are frequently occurring sexually transmitted infections in the United States and Europe. Left untreated, these infections can cause serious health problems, particularly in women, including chronic pelvic pain, life-threatening ectopic pregnancy and infertility. Resistance in these organisms to the most commonly prescribed antibacterial treatments poses a serious public health threat. For example, the CDC estimates that 30% of the clinical isolates ofN. gonorrhoeae are resistant to at least one currently available antibiotic.
In preclinical studies, lefamulin has shown high potency againstN. gonorrhoeae,C. trachomatis andM. genitalium, including strains resistant to currently available antibacterial agents. As a result, we are actively assessing a non-clinical and clinical development plan to support the development of lefamulin as a first-line treatment for urethritis, cervicitis and pelvic inflammatory disease.
Osteomyelitis
The incidence of osteomyelitis, which is an infection of the bone, is increasing. The most common causative organism isS. aureus. In the United States, the prevalence of MRSA in these cases ranges from 33% to 55%. Up to 90% of cases of hematogenous osteomyelitis, most frequently in children, are caused byS. aureus. We believe that lefamulin has the potential to be an effective treatment option for osteomyelitis. Lefamulin has shown substantial tissue penetration and activity against the most common causative organism in all forms of osteomyelitis. We believe that based on the safety profile observed to date, lefamulin will be well tolerated for the long term use necessary for the treatment of both adult and pediatric patients with osteomyelitis. The current standard of care for these infections is treatment with vancomycin. We believe the ability to administer lefamulin by either the IV or oral route would provide a significant advantage over agents, such as vancomycin, that can only be administered by IV.
Prosthetic Joint Infections
Infection occurs in approximately 1% of joint replacement surgeries. Although the incidence of infection has been decreasing, the total number of replacement operations has been rising, such that, overall, there is increasing morbidity. The majority of these infections are caused by three organisms: coagulase negative staphylococci,S. aureus(including (including MRSA) and streptococci, all organisms that are sensitivesusceptible to lefamulin. The preferred treatment for joint infections with MRSA is vancomycin, with daptomycin and linezolid as alternatives. Vancomycin and daptomycin are administered only by IV for this indication, and linezolid has side effects that affect long term use. We believe that lefamulin could provide an alternative for both IV and oral therapy for these infections cases.
BC-7013 (Topical)
BC-7013 is a semi-synthetic compound derived from pleuromutilin with the potentialAlthough we have no current plans to be developeddevelop lefamulin for the topical treatment of Gram-postivie infections, including uSSSIs.
BC-7013 is highly active against key bacterial pathogens causing skin and ocular infections. The MIC90 values for BC-7013 against MRSA are up to 20-fold lowerindications other than for mupirocin and 8-fold lower than for retapamulin, an FDA-approved topical pleuromutilin. Furthermore, BC-7013 has demonstrated potent activity against Chlamydia trachomatis, the leading cause of blindnessCABP, we may advance these programs in the world, and Propionibacterium acnes, the causative agent of acne.
We observed activity in a superficial skin infection model in mice infected with MRSA. BC-7013 was well tolerated following intranasal administration of an ointment formulation in a Phase 1 clinical trial.
Pleuromutilin Molecule Platform
Our pleuromutilin research program isclinic based on our large and diverse proprietary compound library. We believe that our expertiseavailable funding.
CONTEPO Clinical Development Program
Overview
CONTEPO is under development in the areas of medicinal chemistry, pharmacology and toxicology have enabled targeted discovery of novel pleuromutilins through modification of side chains and core positions in the mutilin moiety. These modifications have resulted in alterations in microbial activity, ADME and toxicity of the semi-synthetic molecules.
We are actively pursuing an in-house discovery program to sustain our pipeline with future product candidates. The aim of this program is the development of novel pleuromutilins with enhanced affinity for the bacterial ribosome directed at increasing the antimicrobial potency and broadening the spectrum of activity to include rare strains with known mechanisms of resistance to the pleuromutilin class (e.g. cfr or Vga mutants). We believe next generation pleuromutlins have the potential to exhibit improved antibacterial activity and a pharmacokinetic profile that may make them well suitedUnited States for the treatment of respiratory tractcUTI, including AP. The clinical development plan for CONTEPO utilized a modernized dosing regimen to optimize coverage of the predominant pathogens in hospital infections, acute/complicatedincluding strains recognized by the CDC as an urgent or serious antibiotic resistant threat to public health in the United States. We submitted an NDA for marketing approval of CONTEPO for the treatment of cUTI in adults in the United States, to the FDA in October 2018. The NDA submission is utilizing the 505(b)(2) regulatory pathway and is supported by a robust data package, including a pivotal Phase 2/3 clinical trial (known as
ZEUS™), which met its primary endpoint of statistical non-inferiority to piperacillin/tazobactam in patients with cUTI, including acute pyelonephritis. The FDA has granted us a PDUFA target action date of April 30, 2019 for CONTEPO.
Phase 2/3 Clinical Trial
The ZEUS Study was a multicenter, randomized, parallel-group, double-blind, pivotal Phase 2/3 clinical trial designed to evaluate safety, tolerability, efficacy and pharmacokinetics of CONTEPO compared to PIP-TAZ in the treatment of hospitalized adults with cUTI or AP. PIP-TAZ is a combination antibiotic consisting of a broad-spectrum antibiotic, piperacillin, plus a Beta-lactamase inhibitor, tazobactam, which extends the antibiotic spectrum of piperacillin to include many Beta-lactamase-producing bacteria that have acquired resistance to piperacillin alone. PIP-TAZ is widely used to treat serious Gram-negative infections. The primary objective of the ZEUS Study was to demonstrate that CONTEPO was non-inferior to PIP-TAZ in overall success based on clinical cure and microbiologic eradication in the microbiologic modified intent-to-treat, or m-MITT, population at the test-of-cure visit, or TOC, which occurred on the 19th to 21st day after completion of seven days of treatment with the study drug, or after up to 14 days of treatment for patients with concurrent bacteremia. The m-MITT population consisted of 362 patients, each of whom met the study's inclusion criteria, was randomized, received any amount of study drug, and had one or more baseline Gram-negative pathogens growing at greater than or equal to 10(5) CFU/mL from an appropriately collected, pre-treatment baseline urine or blood sample. The primary endpoint was a composite of the investigator's determination of clinical cure, meaning complete resolution or significant improvement of signs and symptoms that were present at baseline and no new symptoms, such that no further antimicrobial therapy is warranted, plus microbiologic eradication, meaning that the baseline bacterial skin infections, sexually transmitted infectionspathogen was reduced to less than 10(4) CFU/mL on urine culture and, biothreat organisms.if applicable, negative on repeat blood culture, both in the m-MITT population at TOC. Any missing or presumed eradications were classified as indeterminates, and conservatively counted as failures in the overall success analysis.
Compounds in Other Antibiotic Classes
In addition toAll pathogens isolated from patients who had a baseline and TOC pathogen underwent blinded, post-hoc, pulsed-field gel electrophoresis, or PFGE, typing analysis. Microbiologic outcome was also defined utilizing the pleuromutilin research program, we own a ß-lactam library encompassing approximately 2,000 novel broad spectrum cephalosporinsPFGE results, whereby microbiologic persistence required the same genus and approximately 150 novel ß-lactamase inhibitor molecules. We own all rightsspecies of baseline and hold one active patent application on file covering ß-lactamase inhibitors.
We also own a library of approximately 200 acremonic acid derivatives which inhibit bacterial protein translation and have an antibacterial profile that covers primarily Gram-positive bacteria, such as S. aureus, MRSA and mupirocin-resistant strains,post-baseline pathogens, as well as ß-hemolytic streptococci (StreptococciPFGE-confirmed genetic identity.
Patients eligible for the trial were required to be 18 years of age or older and have cUTI or AP that are not S. pneumoniaewas considered by the clinical investigator to be serious enough to require hospitalization and IV antibiotic therapy. The diagnosis was based on pyuria, or membersthe presence of pus or white blood cells in the urine, and cUTI or AP with at least two additional symptoms such as chills, rigors, or warmth associated with fever, nausea or vomiting, painful, difficult or frequent urination, lower abdominal or pelvic pain, or acute flank pain. Patients with cUTI were also required to have at least one risk factor, such as use of intermittent or indwelling bladder catheterization; functional or anatomical abnormality of the Viridans family)urogenital tract; complete or partial hindrance of normal urine flow; blood urea nitrogen greater than 20 mg/dL, blood urea greater than 42.8 mg/dL, or serum creatinine greater than 1.4 mg/dL, due to known prior renal disease; or, in male patients, chronic urinary retention. A baseline urine culture specimen was obtained within 48 hours prior to randomization, and any indwelling bladder catheters were required to be removed or replaced, unless such removal was considered unsafe or contraindicated, before or within 24 hours after randomization.
Eligible patients were randomly assigned to receive either CONTEPO (6 grams IV fosfomycin) or PIP-TAZ (4 grams piperacillin/0.5 grams tazobactam) as one-hour infusions three times daily for seven days, except patients with concurrent bacteremia, who could have received treatment for up to 14 days at the clinical investigator's discretion. Oral step down therapy was prohibited. Throughout the study, all patients were monitored for signs and symptoms of cUTI or AP and the occurrence of adverse events. Laboratory data, including chemistry panels, complete blood counts, electrocardiograms, and
samples for urine and blood cultures were collected from all patients at specified times throughout the study.
Of the total of 465 patients randomized across 92 sites in 16 countries, with studies conducted at 74 sites in 15 countries, 464 (99.8%) received at least one dose of the study drug. Of the 464 patients who received at least one dose of study drug, 233 patients were in the CONTEPO treatment group, and 231 patients were in the PIP-TAZ treatment group. The incidence of premature discontinuation from study drug was low and similar between treatment groups (6.0% in the CONTEPO treatment group compared to 3.9% in the PIP-TAZ treatment group), and the incidence of not completing the study through the last follow-up visit, or LFU, which occurred on the 24th through 28th day after completion of seven days of treatment with the study drug, or after up to 14 days of treatment for patients with concurrent bacteremia, was 5.2% in the CONTEPO group compared to 0.9% in the PIP-TAZ group.
In the ZEUS Study, CONTEPO was non-inferior to PIP-TAZ for the primary efficacy outcome of overall success, which was defined as clinical cure and microbiologic eradication at TOC. Overall success occurred in 64.7% of CONTEPO patients and 54.5% of PIP-TAZ patients. The treatment difference between the CONTEPO and PIP-TAZ groups was 10.2%, with a 95% confidence interval (–0.4, 20.8). Additionally, the lower bound of the 95% confidence interval met the pre-specified non-inferiority margin of –15%, demonstrating that CONTEPO was non-inferior to PIP-TAZ in the study. In a post-hoc primary efficacy analysis using results of blinded PFGE molecular typing of urinary tract pathogens, this difference was even greater (69.0% CONTEPO patients compared to 57.3% PIP-TAZ patients, with a treatment difference of 11.7%, with a 95% confidence interval (1.3, 22.1). Overall success rates were driven by microbiologic eradication rates, as clinical cure rates were greater than 90% and treatment differences were small at TOC. Using the PFGE molecular typing, the microbiologic eradication rates in the m-MITT population at the TOC were 70.7% for patients receiving CONTEPO compared to 60.1% for patients receiving PIP-TAZ. These rates were consistent with those observed in some contemporary cUTI studies, and most patients with microbiologic persistence at TOC had identifiable reasons or risk factors for persistence, such as functional or anatomical abnormalities of the urogenital tract, recent or indwelling urinary tract catheterization, elevated minimum inhibitory concentration, or MIC, to the study drug received, abbreviated study drug therapy, or other underlying co-morbidities. Of note, a majority of patients with microbiologic persistence at TOC were clinical cures at TOC, did not require rescue antimicrobial therapy, and remained sustained cures at LFU.
The identity and frequency of pathogens recovered at baseline from patients in the ZEUS Study were similar in both the CONTEPO and PIP-TAZ treatment groups. The most common pathogens identified were Enterobacteriaceae, identified in 96.2% of the CONTEPO patients and 94.9% of the PIP-TAZ patients, including E. coli, identified in 72.3% of the CONTEPO patients and 74.7% of the PIP-TAZ patients; K. pneumoniae, identified in 14.7% of the CONTEPO patients and 14.0% of the PIP-TAZ patients; Enterobacter cloacae species complex, identified in 4.9% of the CONTEPO patients and 1.7% of the PIP-TAZ patients; and Proteus mirabilis, identified in 4.9% of the CONTEPO patients and 2.8% of the PIP-TAZ patients. Gram-negative aerobes other than Enterobacteriaceae included Pseudomonas aeruginosa, which was identified in 4.3% of the CONTEPO patients and 5.1% of the PIP-TAZ patients, and Acinetobacter baumannii-calcoaceticus species complex, identified in 1.1% of the CONTEPO patients and no PIP-TAZ patients. These pathogens are representative of the pathogens that have been recovered in other studies of patients with cUTI or AP. For the predominant pathogens E. coli and K. pneumoniae, the clinical cure rates at TOC for CONTEPO were greater than 90% for both pathogens, and microbiologic eradication rates were 68.4%, or 72.9% with PFGE analysis, for E. coli, and 66.7% for K. pneumoniae on both a non-PFGE analysis and PFGE analysis-basis.
A total of 42.1% of CONTEPO patients and 32.0% of PIP-TAZ patients experienced at least one TEAE. Most TEAEs were mild or moderate in severity, and severe TEAEs were uncommon (2.1% of
CONTEPO patients and 1.7% of PIP-TAZ patients). The first moleculesmost common TEAEs in both treatment groups were transient, asymptomatic laboratory abnormalities and gastrointestinal events. Treatment-emergent serious adverse events, or SAEs, were uncommon in both treatment groups (2.1% of CONTEPO patients and 2.6% of PIP-TAZ patients). There were no deaths in the study and one SAE in each treatment group was deemed related to the study drug (hypokalemia in a CONTEPO patient and renal impairment in a PIP-TAZ patient), leading to study drug discontinuation in the PIP-TAZ patient. Study drug discontinuations due to TEAEs were infrequent and similar between treatment groups (3.0% of CONTEPO patients and 2.6% of PIP-TAZ patients).
The most common laboratory abnormality TEAEs were increases in the levels of alanine aminotransferase (8.6% of CONTEPO patients and 2.6% of PIP-TAZ patients) and aspartate transaminase (7.3% of CONTEPO patients and 2.6% of PIP-TAZ patients). None of the aminotransferase elevations were symptomatic or treatment-limiting, and none of the patients met the criteria for Hy's Law. Outside of the United States, elevated liver aminotransferases are listed among undesirable effects in labeling for IV fosfomycin.
Hypokalemia occurred in 71 of 232 (30.6%) CONTEPO patients and 29 of 230 (12.6%) PIP-TAZ patients. Most decreases in potassium levels were mild to moderate in severity. Shifts in potassium levels from normal at baseline to hypokalemia, as determined by worst post-baseline hypokalemia values, were more frequent in the CONTEPO group than the PIP-TAZ group for mild (17.7% compared to 11.3%), moderate (11.2% compared to 0.9%), and severe (1.7% compared to 0.4%) categories of hypokalemia. Hypokalemia was deemed a TEAE in 6.4% of patients receiving CONTEPO and 1.3% of patients receiving PIP-TAZ, and all cases were transient and asymptomatic. While no significant cardiac adverse events were observed in the ZEUS Study, post-baseline QT intervals calculated using Fridericia's formula, or QTcF, of greater than 450 to less than or equal to 480 msec (baseline QTcF of less than or equal to 450 msec) occurred at a higher frequency in CONTEPO patients (7.3%) compared to PIP-TAZ patients (2.5%). In the CONTEPO arm, these results appear to be associated with the hypokalemia associated with the salt load of the IV formulation.
Phase 1 Pediatric Clinical Trial
In June 2018, we initiated a Phase 1, non-comparative, open-label study of the pharmacokinetics and safety of a single dose of CONTEPO in pediatric subjects less than 12 years of age receiving standard-of-care antibiotic therapy for proven or suspected infection or peri-operative prophylaxis. A total of 24 patients are expected to be enrolled at up to ten clinical sites in the United States. We anticipate completing enrollment in this series also displayed improved activity against isolates showing resistance against fusidic acidtrial in late 2020.
Potential Additional Indications for CONTEPO
Fosfomycin has a long history of use outside the United States in a variety of indications beyond cUTI. The FDA has granted both Fast Track and showedQIDP designations for the investigation of CONTEPO for the following indications in addition to cUTI:
- •
- Complicated intra-abdominal infections (cIAI)
- •
- Hospital-acquired bacterial pneumonia (HABP)
- •
- Ventilator-associated bacterial pneumonia (VABP)
- •
- Acute bacterial skin and skin structure infections (ABSSSI)
Although we have no cross-resistancecurrent plans to develop CONTEPO for indications other than cUTI, including AP, these designations make CONTEPO eligible for Fast Track and GAIN incentives. We may advance these programs in the clinic based on available non-dilutive funding.
In August 2017, Zavante entered into an agreement with other classesthe United States National Institute of Allergy and Infectious Diseases, or NIAID, under which NIAID will conduct a clinical trial to assess CONTEPO's intrapulmonary penetration and pharmacokinetics in support of the product candidate's potential future development as a treatment for HABP and VABP. This bronchoalveolar lavage study, or the BAL study, will measure CONTEPO's pulmonary penetration by assessing drug concentrations in the lining of study subjects' bronchial pathways. Diffusion and saturation of antibiotics tested.in patients' airways are considered important factors in assessing a drug's ability to effectively treat lower-respiratory tract infections. Prior preclinical and clinical investigations of IV fosfomycin have demonstrated that the product candidate penetrates rapidly into tissues and achieves clinically relevant concentrations in urine, soft tissues, lungs and other organs, supporting CONTEPO's potential versatility as an antibiotic treatment option. The protocol for the BAL study is currently under development in conjunction with NIAID, and we anticipate that the study will be initiated in mid-2019.
License Agreement with Sinovant Sciences
The existing compound librariesIn March 2018, we entered into a license agreement, or the License Agreement, with Sinovant Sciences, Ltd. or Sinovant, an affiliate of ß-lactam/ß-lactamase inhibitorsRoivant Sciences, Ltd., to develop and acremonic acid derivatives represent an unrecognized portion of our pipeline. The current allocation of our funds and staff are dedicated to advancingcommercialize lefamulin in the pleuromutilin compounds. Assessmentgreater China region. As part of the ß-lactam/ß-lactamase inhibitorsLicense Agreement, Nabriva Therapeutics Ireland DAC and acremonic acid derivatives compound librariesNabriva Therapeutics GmbH, our wholly owned subsidiaries, granted Sinovant an exclusive license to develop and commercialize, and a non-exclusive license to manufacture, certain products containing lefamulin, or the Licensed Products, in the People's Republic of China, Hong Kong, Macau, and Taiwan (together the "Territory"). We retain development and commercialization rights in the rest of the world.
Under the License Agreement, Sinovant and our subsidiaries established a joint development committee, or the JDC, to review and oversee development and commercialization plans in the Territory. We received a $5.0 million upfront payment pursuant to the terms of the License Agreement and are eligible for up to an additional $91.5 million in milestone payments upon the achievement of certain regulatory and commercial milestone events related to lefamulin for CABP, plus an additional $4.0 million in milestone payments if any Licensed Product receives a second or any subsequent regulatory approval in the People's Republic of China. The first milestone was a $1.5 million payment for the submission of a clinical trial application by Sinovant to the Chinese Food and Drug Administration that was received in February 2019. The remaining milestone payments are tied to additional regulatory approvals and annual sales targets. In addition, we will be dependenteligible to receive low double-digit royalties on sales, if any, of Licensed Products in the Territory.
Sinovant will be solely responsible for all costs related to developing, obtaining regulatory approval of and commercializing Licensed Products in the Territory and is obligated to use commercially reasonable efforts to develop, obtain regulatory approval for, and commercialize Licensed Product in the Territory. We are obligated to use commercially reasonable efforts to supply, pursuant to supply agreements to be negotiated by the parties, to Sinovant sufficient supply of lefamulin for Sinovant to manufacture finished drug products for development and commercialization of the Licensed Products in the Territory.
Unless earlier terminated, the License Agreement will expire upon additional funding.the expiration of the last royalty term for the last Licensed Product in the Territory, which we expect will occur in 2033. Following the expiration of the last royalty term, the license granted to Sinovant will become non-exclusive, fully-paid, royalty-free and irrevocable. The License Agreement may be terminated in its entirety by Sinovant upon 180 days' prior written notice at any time. Either party may, subject to specified cure periods, terminate the License Agreement in the event of the other party's uncured material breach. Either party may also terminate the License Agreement under specified circumstances relating to the other party's insolvency. We have the right to terminate the License Agreement immediately if Sinovant does not reach certain development milestones by certain specified dates
(subject to specified cure periods). The License Agreement contemplates that the we will enter into ancillary agreements with Sinovant, including clinical and commercial supply agreements and a pharmacovigilance agreement.
Commercialization Strategy
WeOther than in greater China where we have licensed development and commercialization rights to lefamulin, we own exclusive, worldwide rights to lefamulin.lefamulin and U.S. rights to CONTEPO. We expect that our initial target population for lefamulin will consist of patients with moderate to severe CABP whose antibiotic treatment is hospital-initiated. If lefamulin receives marketing approval from the FDA for the treatment of CABP, we plan to pursue commercialization strategies that maximize the value of lefamulin in the United States with our own targeted hospital sales and marketing organization that we plan to establish.expand beyond its current levels. Based on our market research, we believe lefamulin has an innovative profile supporting adoption in the United States for adult hospital- initiated CABP patients, treated both as in-patients as well as outpatient-transition of care from the hospital to the community, which we believe represents a significant commercial opportunity. We believe that we will be able to effectively communicate lefamulin’slefamulin's differentiating characteristics positioning and key attributes to clinicians, hospital pharmacistspharmacies and managed care organizations,payors with the goal of establishing favorable reimbursement for outpatients as well as a favorable formulary status
for lefamulin. Based on our market research, we believe lefamulin has a novel position supporting adoption in the United States for adult CABP hospital-initated treatments, which we believe represents a significant commercial opportunity.targeted hospitals. Additionally, we believe that our plans for aan initial targeted hospital-focused sales force should allow us to address address—on our own own—the hospital-initiated treatment market for CABP in the United States.States in an efficient and effective way. There is also a significant opportunity for lefamulin to be used in the community for CABP patients, given its versatile profile, and this may represent a future expanded use. We plan to continue our pre-commercialization activities to prepare for a potential commercial launch of lefamulin, subject to receiving marketing approval in the United States. If lefamulin receives marketing approval outsideOutside of the United States, for the treatment of CABP, we expect to utilize a variety of types of collaboration, distribution and other marketing arrangements with one or more third parties to commercialize lefamulin in such markets.lefamulin.
Along with additional market research, we believe that medical education will be a key component of our commercialization efforts and, following potential commercial launch, plan to invest in these activities to maximizeoptimize the commercial potential of lefamulin. With a targeted initial prescribing base predominantly in the hospital setting, we expect that a targeted hospital sales and marketing organization would be relatively smaller than competitors who are focused on both the hospital and community prescribing base. We believe that lefamulin’slefamulin's novel mechanism of action, status as the only member of a new class of systemically administered pleuromutilins and anticipated clinical profile will support its potential favorable reimbursement, its potential inclusion on formularies and in local and national treatment guidelines, subject to and following marketing approval.
We plan to evaluate the merits of entering into collaboration agreements with other pharmaceutical or biotechnology companies that may contribute to our ability to efficiently advance our product candidates, build our product pipeline and concurrently advance a range of research and development programs for a variety of indications outside the United States.
We own exclusive U.S. rights to CONTEPO. Our strategic intention, supported by CONTEPO's differentiated profile, is to establish CONTEPO as the standard of care in the United States for hospitalized patients with serious infections caused by suspected or confirmed MDR bacteria. We plan to use a targeted sales force to promote CONTEPO to hospital-based healthcare professionals in key locations within the United States where MDR infections, including CRE, are concentrated. These include roughly 900 hospitals in high resistance locations such as New York City, Los Angeles and Chicago, and other major population centers.
We currently have a team of regional business directors and medical science liaisons in the field performing educational and market development activities, respectively, for lefamulin and CONTEPO.
We initially plan to use a targeted hospital-based sales force to promote both lefamulin formulations and CONTEPO, if approved.
Manufacturing
We do not own or operate, and currently have no plans to establish, manufacturing facilities for the production of clinical or commercial quantities of lefamulin, CONTEPO, or any of the other compounds that we are evaluating in our discovery program. We currently rely, and expect to continue to rely, on third parties for the manufacture of lefamulin, CONTEPO and any further products that we may develop. We have significant in-house knowledge and experience in (i) the relevant chemistry associated with our drug candidateproduct candidates; and use(ii) the relevant manufacturing and supply chain processes associated with the commercial supply of our product candidates. In addition to these internal resources, alongsidewe engage third-party consultants, to manageassist in the management of our manufacturing contractors.third-party manufacturers.
We have engaged a limited number of third-party manufacturers to provide all of our rawstarting materials, drug substance and finished product for use in clinical trials. The active pharmaceutical ingredients, or API, and drug products have been produced under master service contracts and specific work orders from these manufacturers pursuant to agreements that include specific supply timelines and volume and quality expectations. We choose the third-party manufacturers of the drug substance based on the volume required and the regulatory requirements at the relevant stage of development. All lots of drug substance and drug products used in clinical trials are manufactured under current good manufacturing practices. Separate third-party manufacturers have been responsible for fill and finish services, and for labeling and shipment of the final drug product to the clinical trial sites.
Lefamulin
We have entered into a long-term commercial supply agreement with SEL Biochem Xinjiang Co., Ltd, or SEL, and Fountain International Development Holding Limited for the supply of pleuromutilin, which is the key intermediate for lefamulin API production. Under this agreement, SEL is required to manufacture and supply and we are required to purchase from SEL a specified percentage of our commercial requirements of pleuromutilin. The agreement expires on August 28, 2022, subject to automatic renewal for successive three-year periods. Either party may elect not to renew the agreement by providing two-year prior written notice before the end of the initial term or the then-current renewal term. Either party may terminate the agreement for the other party's uncured material breach or upon the occurrence of specified bankruptcy events.
The agreement includes customary supply terms, including product specifications, price, payment terms, requirements forecasting, delivery mechanics and quality assurance. Under the agreement, we have also negotiated a quality technical agreement pursuant to which SEL will conduct all quality control and release testing for the pleuromutilin produced under the agreement.
In November 2018 we entered into a long-term commercial supply agreement with Arran Chemical Company Limited, or Arran, for the supply of the chiral acid starting material required in the synthesis of lefamulin API. Under this agreement Arran is required to manufacture and supply, and we are required to purchase from Arran the amount forecast for the first six months of a twelve-month rolling forecast provided monthly by us. The agreement term expires on November 12, 2023 and continues thereafter unless terminated by either party with not less than twelve months written notice. Either party may terminate the agreement for the other party's uncured material breach or upon the occurrence of insolvency or bankruptcy events. The agreement includes customary supply terms including material specifications, price, payment terms, demand forecasting, delivery mechanics, and quality assurance.
We have entered into a long-term commercial supply agreement with Hovione Limited, or Hovione, for the supply of lefamulin API. Under this agreement, Hovione is required to manufacture and supply and we are required to purchase from Hovione a specified percentage of our commercial requirements of lefamulin API. The agreement expires on November 22, 2025, subject to automatic renewal for successive two-year periods. Either party may elect not to renew the agreement by providing two-year prior written notice before the end of the initial term or the then-current renewal term. Either party may terminate the agreement for the other party's uncured material breach or upon the occurrence of specified bankruptcy events.
The agreement includes customary supply terms, including product specifications, price, payment terms, requirements forecasting, delivery mechanics and quality assurance. We are required to purchase a minimum of 750 kg to 1,500 kg per year of lefamulin API dependent on potential regulatory approval timing. Under the agreement, we have also negotiated a quality technical agreement pursuant to which Hovione will conduct all quality control and release testing for the pleuromutilin produced under the agreement.
We have also entered into a long-term commercial supply agreement with Patheon UK Limited, or Patheon, for the supply of IV vials of lefamulin. Under this agreement, Patheon is required to supply and we are required to purchase a specified percentage of our commercial requirements of IV vials of lefamulin. The agreement expires on December 31, 2023, subject to automatic renewal for successive two-year periods. Either party may elect not to renew the agreement by providing two-year prior written notice before the end of the initial term, and after the initial term, either party may terminate the agreement with 24-months prior written notice at any time. Either party may also terminate the agreement for the other party's uncured material breach or upon the occurrence of specified bankruptcy events. We may also terminate the agreement if a governmental authority takes action that prevents us from importing, exporting, purchasing or selling the IV vials of lefamulin. Finally, Patheon may terminate the agreement if we assign any of our rights under the agreement to an assignee that it does not consider to be a creditworthy substitute or is a competitor of Patheon.
The agreement includes customary supply terms, including product specifications, batch size requirements, price, payment terms, requirements forecasting, delivery mechanics and quality assurance. Under the agreement, we have also negotiated a quality agreement pursuant to which Patheon will conduct all quality control testing for the IV vials of lefamulin.
In addition, we have entered into a long-term commercial supply agreement with Almac Pharma Services Limited, or Almac, for the commercial supply of lefamulin tablets. Under this agreement, Almac is required to supply and we are required to purchase services relating to the manufactured tablets equaling a specified minimum annual spend. The initial term of the agreement expires on August 7, 2022, but it will remain in force until it is terminated by either party with 24-months prior written notice, expiring on or at any time after the expiry of the initial term. Either party may also terminate the agreement for the other party's uncured material breach or upon the occurrence of specified bankruptcy events. The agreement includes customary supply terms, including payment terms, requirements forecasting, delivery mechanics and quality assurance. Under the agreement, we have also negotiated a quality technical agreement pursuant to which Almac will conduct all quality control testing for the tablets.
In August 2018, we entered into a commercial packaging and supply agreement, or the Packaging Agreement with Sharp Corporation, or Sharp, for the commercial packaging of lefamulin acetate for oral and intravenous administration. Under the Packaging Agreement, Sharp has agreed to provide certain packaging services to us, including labeling, serialization and final packaging of the packaged products.
The Packaging Agreement has an initial five-year term ending December 31, 2023 and will automatically renew after the initial term for additional one-year terms unless either party gives notice
of its intention to terminate the Packaging Agreement at least 90 days prior to the end of the then-current term. In addition, either party may terminate the Packaging Agreement for the other party's uncured material breach, in addition to other specified events, including with respect to bankruptcy proceedings and governmental actions, in each case subject to notice, cure periods and other conditions set forth in the Packaging Agreement.
The Packaging Agreement includes customary supply terms, including product specifications, batch size requirements, price, payment terms, requirements forecasting, delivery mechanics and quality assurance. Under the agreement, we have also negotiated a quality agreement pursuant to which Sharp will conduct quality control testing for the packaged products.
These six commercial supply agreements relating to lefamulin are filed as exhibits to this Form 10-K. Other than these six agreements, we do not currently have long-term agreements with any of theseother third parties. We also do not have any current contractual relationshipsparties for the manufacture of commercial supplies of lefamulin, if it receives marketing approval. Webut we intend to enter into additional agreements with suitable third-party contract manufacturers for theadditional commercial productionsupplies of this productlefamulin pending potential regulatory approval.
Our product candidateLefamulin is a semi-synthetic organic compound of low molecular weight. The pleuromutilin core of the molecule is produced by fermentation and is manufactured on a significant scale by various manufacturers. The second part of the molecule is established from a readily accessible chiral starting material. We have selected the compoundlefamulin based on its efficacy and safety profile, although it is also associated with reasonable cost of goods, ready availability of starting materials and ease of synthesis. The development stage production of lefamulin has beenwas carried out at a significant scale and we believe, if required, the synthetic route to lefamulin is amenable to further scale-up. The synthetic route does not require unusual, or specialized, equipment in the manufacturing process. Therefore, if any of theour current or future drug substance manufacturers were to become unavailable for any reason, we believe there are a number of potential replacements, although delays may be incurred in identifying and qualifying such replacements.
CONTEPO
Effective July 28, 2016, Zavante, Laboratorios ERN, S.A. ("ERN") and Ercros, S.A. ("Ercros") entered into an amended and restated three-way agreement (the "Three-Way Agreement"), which established the basis for related supply agreements with ERN and Ercros in anticipation of FDA approval of fosfomycin disodium and succinic acid injection for intravenous use filled, finished and packaged into containers for use by end users ("Product") in the United States. Pursuant to the Three-Way Agreement, Zavante has the direct responsibility for the manufacture and supply of the commercial Product in the United States.
Under the Three-Way Agreement, (i) ERN has agreed to provide Zavante with certain technical documentation (Technical Documentation") and data required for submission of an NDA or Abbreviated New Drug Application("ANDA"), as applicable, for the Product , and other assistance in connection with the submission of an NDA or ANDA, pursuant to the ERN Supply Agreement (as defined below); (ii) Ercros has agreed to provide Zavante with certain Technical Documentation and the manufacture and supply of a blend of fosfomycin disodium and succinic acid (the "API Mixture") for the manufacture of the Product, pursuant to the terms of the Ercros Supply Agreement (as defined below); and (iii) Zavante has agreed to obtain the commercial supply of the Product, under one or more separate agreements with third party manufacturers. The rights and obligations of each of the parties are set forth in each of the ERN Supply Agreement and the Ercros Supply Agreement.
In addition, pursuant to the Three-Way Agreement, Zavante is required to (i) contract with one or more third party manufacturers to provide quantities of the Product required by Zavante for commercial sale in the United States, perform validation activities as required by the FDA, and obtain FDA approval of such third party manufacturer's facilities and quality systems; (ii) use commercially
reasonable efforts to file an NDA within one year of its receipt of all Technical Documentation for the NDA from ERN and Ercros; (iii) obtain and own all trademarks to be used for the Product in the United States and (iv) bear the cost and manage all clinical trials necessary for obtaining FDA approval of the Product and keep ERN and Ercros updated regarding the progress of such clinical trials.
The Three-Way-Agreement will continue in force and effect until the Ercros Supply Agreement and the ERN Supply Agreement have both been terminated or expired in accordance with the respective terms therein, or if the Three-Way Agreement is terminated upon mutual written agreement of all of the parties. The Three-Way Agreement contains, among other provisions, customary provisions relating to legal compliance and publicity.
Effective July 28, 2016, Zavante and Ercros entered into a manufacturing and supply agreement (the "Ercros Supply Agreement") pursuant to the Three-Way Agreement. Under the Ercros Supply Agreement, Ercros has agreed, pursuant to purchase orders entered into under the Ercros Supply Agreement, to manufacture (i) the exclusive supply of the API Mixture for Zavante in support of filing an NDA or an ANDA, as applicable, and (ii) the commercial supply of fosfomycin disodium and succinic acid injection for intravenous use in the United States. In addition, Ercros has agreed to provide access to certain technical documentation as may be requested by Zavante in connection with the filing of an NDA.
The Ercros Supply Agreement has an initial ten-year term ending July 28, 2026 and will automatically renew after the initial term for additional two-year terms unless either party gives notice of its intention to terminate the Ercros Supply Agreement at least 18 months prior to the end of the then-current term. Either party may terminate the Ercros Supply Agreement for the other party's uncured material breach, in addition to other specified events, including with respect to bankruptcy proceedings, governmental actions and legal proceedings, in each case subject to notice, cure periods and other conditions set forth in the Ercros Supply Agreement.
The Ercros Supply Agreement contains customary supply terms, including requirements forecasting, purchase orders, product specifications, price, payment terms, delivery mechanics and quality insurance. In addition, the Ercros Supply Agreement contains, among other provisions, customary representations and warranties by the parties, a grant by Ercros to Zavante of certain limited license rights to Ercros' intellectual property in connection with Zavante's performance under the Ercros Supply Agreement, certain indemnification rights in favor of both parties and customary confidentiality provisions.
Under the Ercros Supply Agreement, Zavante and Ercros have also entered into a quality agreement, pursuant to which Ercros will conduct all quality control and release testing for the API Mixture produced under the Ercros Supply Agreement.
Effective July 28, 2016, Zavante and ERN entered into an amended and restated pharmaceutical manufacturing and exclusive supply agreement, as amended on December 1, 2016, March 1, 2017, May 1, 2017 and December 20, 2017, pursuant to the Three-Way Agreement (the ERN Supply Agreement). Under the ERN Supply Agreement, each party is required to use commercially reasonable efforts to complete certain development activities required for submission of an NDA or an ANDA for fosfomycin sodium and succinic acid (the bulk formulation of CONTEPO). In addition, ERN has agreed to provide to Zavante (i) certain technical documentation and data as required by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use's guidelines and the FDA for submission of an NDA or an ANDA for the bulk formulation of CONTEPO, and (ii) certain regulatory support in connection with the bulk formulation of CONTEPO sold or intended for commercial sale and human use.
Upon the first commercial sale of the bulk formulation of CONTEPO, Zavante is obligated to make a one-time cash payment to ERN and subsequent quarterly payments thereafter based on the number of vials of the bulk formulation of CONTEPO sold during each calendar quarter.
The ERN Supply Agreement has an initial ten-year term ending July 28, 2026 and will automatically renew after the initial term for additional two-year terms unless either party gives notice of its intention to terminate the ERN Supply Agreement at least 18 months prior to the end of the then-current term. Either party may terminate the ERN Supply Agreement by mutual written agreement and for the other party's uncured material breach, in addition to other specified events, including with respect to bankruptcy proceedings and governmental actions, in each case subject to notice, cure periods and other conditions set forth in the ERN Supply Agreement.
The ERN Supply Agreement contains, among other provisions, customary representations and warranties by the parties, a grant to each party by the other party of certain limited license rights to such other party's intellectual property in connection with the parties' performance of the services under the ERN Supply Agreement, certain indemnification rights in favor of both parties and customary confidentiality provisions.
On April 25, 2017, Zavante and Fisiopharma, S.r.l. ("Fisiopharma") entered into a manufacturing and supply agreement, as amended on May 8, 2017 (the "Fisiopharma Supply Agreement"). Under the Fisiopharma Supply Agreement, Fisiopharma has agreed, pursuant to purchase orders entered into under the Fisiopharma Supply Agreement, to manufacture and supply fosfomycin disodium for intravenous injection in bulk drug vials (the "Bulk Drug Vials") to Zavante in support of filing an NDA or an ANDA, as applicable, and a specified percentage of Zavante's commercial requirements of Bulk Drug Vials for the United States.
The Fisiopharma Supply Agreement has an initial ten-year term ending April 25, 2027 and will automatically renew after the initial term for additional one-year terms unless Zavante gives notice of its intention to terminate the Fisiopharma Supply Agreement at least six months prior to the end of the then-current term. Either party may terminate the Fisiopharma Supply Agreement for the other party's uncured material breach or upon the occurrence of specified bankruptcy events, and Zavante may terminate the Fisiopharma Supply Agreement upon the occurrence of other specified events, including with respect to governmental actions and legal proceedings instituted against Fisiopharma, in each case subject to notice, cure periods and other conditions set forth in the Fisiopharma Supply Agreement.
The Fisiopharma Supply Agreement contains customary supply terms, including requirements forecasting, purchase orders, product specifications, price, payment terms, delivery mechanics and quality insurance. In addition, it contains, among other provisions, customary representations and warranties by the parties, a grant to Fisiopharma of certain limited license rights of Zavante's intellectual property in connection with Fisiopharma's performance of services under the Fisiopharma Supply Agreement, certain indemnification rights in favor of both parties, limitations of liability and customary confidentiality provisions.
Under the Fisiopharma Supply Agreement, Zavante and Fisiopharma have also entered into a quality control agreement, pursuant to which Fisiopharma will conduct all quality control and release testing for the bulk drug vials produced under the Fisiopharma Supply Agreement. Any default under the quality control agreement constitutes a default under the Ercros Supply Agreement.
On December 26, 2017, Zavante entered into a commercial packaging agreement (the "PCI Packaging Agreement") with AndersonBrecon Inc., doing business as PCI of Illinois ("PCI") for the commercial packaging of fosfomycin disodium for intravenous injection in bulk drug vials (the "Packaged Product"). Under the PCI Packaging Agreement, PCI has agreed to provide certain packaging services to Zavante, including labeling, serialization and final packaging of the PCI Packaged Product.
The PCI Packaging Agreement has an initial three-year term ending December 26, 2020 and will automatically renew after the initial term for additional one-year terms unless either party gives notice of its intention to terminate the PCI Packaging Agreement at least 120 days prior to the end of the then-current term. In addition, either party may terminate the PCI Packaging Agreement for the other party's uncured material breach, in addition to other specified events, including with respect to bankruptcy proceedings and governmental actions, in each case subject to notice, cure periods and other conditions set forth in the PCI Packaging Agreement.
The PCI Packaging Agreement contains customary supply terms, including product specifications, price, payment terms, delivery mechanics and quality insurance. In addition, it contains, among other provisions, customary representations and warranties by the parties, a grant to PCI of certain limited license rights of Zavante's intellectual property in connection with PCI's performance of services under the PCI Packaging Agreement certain indemnification rights in favor of both parties, limitations of liability and customary confidentiality provisions.
Under the PCI Packaging Agreement, Zavante and PCI have also entered into a contract services quality agreement, which governs the responsibilities of each party regarding the quality aspects of packaging and release of PCI Packaged Product.
These five commercial supply agreements relating to CONTEPO are filed as exhibits to this Form 10-K. Other than these five agreements, we do not have long-term agreements with any other third parties for the manufacture of commercial supplies of CONTEPO, but we intend to enter into additional agreements with third-party contract manufacturers for additional commercial supplies of CONTEPO pending potential regulatory approval.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. While we believe that our technologies, knowledge, experience and scientific resources provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions, government agencies and private and public research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
Many of our competitors may have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain marketing approvals for their products more rapidly than we obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, our ability to compete may be affected because in some cases insurers or other third-party payors seek to encourage the use of generic products. This may have the effect of making branded products less attractive, from a cost perspective, to buyers. We expect that if lefamulin is
approved for CABP, it will be priced at a significant premium over competitive generic products. This may make it difficult for us to replace existing therapies with lefamulin.
The key competitive factors affecting the success of our product candidates are likely to be their efficacy, safety, convenience, price and the availability of coverage and reimbursement from government and other third-party payors.
There are a variety of available therapies marketed for the treatment of CABP. Currently, the treatment of CABP is dominated by generic products. For hospitalized patients, combination therapy is frequently used. Many currently approved drugs are well establishedwell-established therapies and are widely usedaccepted by physicians, patients and third-party payors. We also are aware of various drugs under development for the treatment of CABP, including solithromycin (NDA filedomadacycline which was launched in February 2019 by CempraParatek Pharmaceuticals Inc. and complete response letter issued, delafloxacin which was approved by the FDA in December 2016), omadacyclineJune 2017 for ABSSSI, and is marketed by Melinta Therapeutics Inc. which also have an ongoing Phase 3 clinical trial of delafloxacin for CABP and oral nafithromycin which is in Phase 2 clinical development by Wockhardt Ltd.
If approved, we expect CONTEPO will face competition from commercially available antibiotics such as ceftazidime-avibactam, meropenem-vaborbactam, tigecycline, plazomicin, from other product candidates currently in development for the treatment of cUTI, including AP, such as imipenem-relebactam (under Phase 3 clinical development by Paratek Pharmaceuticals Inc.) and delafloxacinMerck), cefiderocol (under Phase 3 clinical development by Melinta Therapeutics Inc.).Shionogi), ETX0282-cefpodoxime proxetil (under development by Entasis Therapeutics), and LYS228 (under development by Boston Pharma), as well as generically available agents including carbapenems, aminoglycosides, and polymyxins.
Intellectual Property
Our success depends in large part on our ability to obtain and maintain proprietary protection for our product candidates, technology and know-how, to operate without infringing the proprietary rights of others and to prevent others from infringing our proprietary rights. We strive to protect the proprietary technology that we believe is important to our business by, among other methods, seeking and maintaining patents, where available, that are intended to cover our product candidates, compositions and formulations, their methods of use and processes for their manufacture and any other inventions that are commercially important to the development of our business. We also rely on trade secrets, know-how, continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary and competitive position.
As of February 10, 2017,January 31, 2019, we owned 2122 different families of patents and patent applications, including 2021 families directed to the various pleuromutilin derivatives as compositions of matter, processes for their manufacture, and their use in pharmaceutical compositions and methods of treating disease. The remaining family is directed to ß-lactamase inhibitors. Our patent portfolio includes 2223 issued U.S. patents, 2023 granted European patents and 1618 granted Japanese patents, as well as patents in other jurisdictions. We also have pending patent applications in the United States, Europe, Japan and other countries and regions, including Asia,
Australia, Eastern Europe, and South America, including notably Canada, Brazil, China, Israel, India and Taiwan among others.
All of these patents and patent applications are assigned solely to us and were either originally filed by us or originally filed by Sandoz and subsequently assigned to us.
As of February 10, 2017,January 31, 2019, our lead product candidate, lefamulin, was protected by the following sixseven patent families:
·
- •
- The first patent family includes patents and applications with claims directed to generic classes of compounds that include lefamulin and/or their use in the treatment of microbial infections. This family includes issued patents in the United States, Europe and Japan, as well as issued patents in
1011 other jurisdictions. The standard term for patents in this family expires in 2021.
- •
- The second patent family includes patents and applications with claims that specifically recite lefamulin and/or its use in the treatment of microbial infections. This family includes two issued patents in each of the United States, Europe and Japan, as well as issued patents in
1921 other jurisdictions and85 pending patent applications in other jurisdictions, including one divisional application in the United States. The standard term for patents in this family expires in 2028. A patent term adjustment of 303 days has already been obtained in the United States for one patent.· - •
- The third patent family includes patents and applications with claims directed to the processes for the manufacture of lefamulin, crystalline intermediates useful in the processes, and the resulting crystalline salts. This family includes
1116 granted patents including issued patents in the United States, Europe and Japan and 13 pending patent applications one each in Europe and16Japan and 13 in other jurisdictions. The standard term for patents in this family expires in 2031.· - •
- The fourth patent family includes patents and applications with claims directed to processes for the synthetic manufacture of crystalline intermediates useful in the manufacture of lefamulin. This family includes granted patents in Europe,
andthe United States and Japan and granted patents in other jurisdictions and further pending applications. The standard term for patents in this family expires in 2031.· - •
- The fifth patent family includes patents and applications with claims directed to pharmaceuticals and treatments forHelicobacter infection, including pleuromutilins, such as lefamulin. This family includes issued patents in the United States, Europe and one other jurisdiction. The standard term for patents in this family expires in 2023. A patent term adjustment of 921 days has already been obtained for the U.S. patent.
· - •
- A further patent family is directed to pharmaceutical compositions of lefamulin and covers 17 pending patent applications in various jurisdictions.
- •
- The seventh patent family covers methods for purification of pleuromutilins and the PCT-application was recently published.
Our second most advanced product candidate, BC-7013, is covered specifically in one patent family with patents granted in the United States, Europe and Japan, as well as eightnine other jurisdictions, and pending patent applications in other jurisdictions. The standard term for patents in this family expires in 2027.
The remaining 13 pleuromutilin patent families are directed to either molecules in the intellectual property landscape surrounding our product candidates in development or molecules which can be potentially further developed by us but have not yet been pursued. All patent applications in these families have been filed at least in the United States and Europe, and most have been filed in other countries. The majority of these patent applications have already resulted in granted patents.
Finally, we own one patent family directed to ß-lactamase inhibitor compounds. Patent applications in this family have been filed and granted in the United States and Europe. The standard term for patents in this family expires in 2030.
Zavante holds one issued United States patent (U.S. 9,345,717) directed to methods for identifying dosing regimens that decrease the potential for on-therapy drug resistance. Additionally, Zavante has filed a patent application based on results from the ZEUS Study that relates to methods for treating patients with resistant bacterial infections and, specifically, Gram-negative bacterial infections. However, this patent may not ensure exclusivity through the patent term and we may not be able to secure any additional patent protection. We plan to rely on regulatory protection afforded to CONTEPO through orphan drug designations, data exclusivity, and market exclusivity where available.
The term of individual patents depends upon the legal term for patents in the countries in which they are obtained. In most countries, including the United States, the patent term is 20 years from the filing date of a non-provisional patent application. In the United States, a patent’spatent's term may, in certain cases, be lengthened by
patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office, or the USPTO, in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier filed patent. The term of a U.S. patent that covers a drug, biological product or medical device approved pursuant to a pre-market approval, or PMA, may also be eligible for patent term extension when FDA approval is granted, provided that certain statutory and regulatory requirements are met. The length of the patent term extension is related to the length of time the drug is under regulatory review while the patent is in force. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration date set for the patent. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent applicable to each regulatory review period may be granted an extension and only those claims reading on the approved drug may be extended. Similar provisions are available in Europe and certain other foreign jurisdictions to extend the term of a patent that covers an approved drug, provided that statutory and regulatory requirements are met. Thus, in the future, if and when our product candidates receive approval by the FDA or foreign regulatory authorities, we expect to apply for patent term extensions on issued patents covering those products, depending upon the length of the clinical trials for each drug and other factors. The expiration dates of our patents and patent applications referred to above are without regard to potential patent term extension or other market exclusivity that may be available to us.
In addition to patents, we may rely, in some circumstances, on trade secrets to protect our technology and maintain our competitive position. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements with our employees, corporate and scientific collaborators, consultants, scientific advisors, contractors and other third parties. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries and jurisdictions, including the European Union, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, pricing, reimbursement, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, and import and export of pharmaceutical products. The processes for obtaining regulatory approvals in the United States and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
Review and Approval of Drugs in the United States
In the United States, the FDA reviews, approves and regulates drugs under the federal Food, Drug, and Cosmetic Act, or FDCA, and associated implementing regulations. The failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approvalpost-approval may result in delays to the conduct of study, regulatory review and subject an applicant and/or sponsor to a variety of administrative or judicial sanctions, including refusal by the FDA to approve pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters and other types of letters, product recalls, product seizures, total or partial suspension of production or distribution, adverse publicity, injunctions, fines, refusals of government
contracts, restitution, disgorgement of profits, orand civil or criminal investigations and penalties brought by the FDA and the U.S. Department of Justice, or DOJ, or other governmental entities.entities, including state agencies.
An applicant seeking approval to market and distribute a new drug product in the United States must typically undertake the following:following before a product candidate will be approved by the FDA:
- •
·completion of preclinical laboratory tests, animal studies and formulation studies in compliance with theFDA’sFDA's good laboratory practice, or GLP, regulations;·- •
- submission to the FDA of an investigational new drug application, or IND, which must take effect before human clinical trials may begin;
· - •
- approval by an independent institutional review board, or IRB, representing each clinical site before each clinical trial may be initiated;
· - •
- performance of adequate and well-controlled human clinical trials in accordance with good clinical practices, or GCP, to establish the safety and efficacy of the proposed drug product for each indication;
· - •
- preparation and submission to the FDA of a new drug application, or NDA, summarizing available data to support the proposed approval of the new drug product for the proposed use;
· - •
- review of the product application by an FDA advisory committee, where appropriate or if applicable and as may be requested by the FDA;
· - •
- satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with current Good Manufacturing Practices, or cGMP, requirements and to assure that the facilities, methods and controls are adequate to preserve the
product’sproduct's identity, strength, quality and purity;· - •
- satisfactory completion of FDA audits of clinical trial sites to assure compliance with GCPs and the integrity of the clinical data;
· - •
- payment of
userPDUFA fees(per published PDUFA guidelines for the applicable year)and securing FDA approval of the NDA; and· - •
- compliance with any post-approval requirements, including the potential requirement to implement a Risk Evaluation and Mitigation Strategy, or REMS, where applicable, and the potential to conduct post-approval studies required by the FDA.
Preclinical Studies
Before an applicant begins testing a compound with potential therapeutic value in humans, the drug candidate enters the preclinical testing stage. Preclinical studies include laboratory evaluation of the purity and stability of the manufactured drug substance or active pharmaceutical ingredient and the formulated drug or drug product, as well asin vitro and animal studies to assess the safety and activity of the drug for initial testing in humans and to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations. The results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical studies, among other things, are submitted to the FDA as part of an IND. Additional long-term preclinical testing, such as animal tests of reproductive adverse events and carcinogenicity, may continue after the IND is submitted.
Companies usually must complete some long-term preclinical testing, such as animal tests of long term exposure and reproductive adverse events, and must also develop additional information about the chemistry and physical characteristics of the investigational product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The
manufacturing process must be capable of consistently producing quality batches of the candidate product and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the candidate product does not undergo unacceptable deterioration over its shelf life.
The IND and IRB Processes
An IND is an exemption from the FDCA that allows an unapproved drug to be shipped in interstate commerce for use in an investigational clinical trial and a request for FDA authorization to administer an investigational drug to humans. Such authorization must be secured prior to interstate shipment and administration of any new drug that is not the subject of an approved NDA. In support of a request for an IND, applicants must submit a protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical trials, among other things, are submitted to the FDA as part of an IND. The FDA requires a 30-day waiting period after the filing of each IND before clinical trials may begin. This waiting period is designed to allow the FDA to review the IND to determine whether human research subjects will be exposed to unreasonable health risks. At any time during this 30-day period, the FDA may raise concerns or questions about the conduct of the trials as outlined in the IND and impose a clinical hold or partial clinical hold. In this case, the IND sponsor and the FDA must resolve any
outstanding concerns before clinical trials can begin.
Following commencement of a clinical trial under an IND, the FDA may also place a clinical hold or partial clinical hold on that trial. A clinical hold is an order issued by the FDA to the sponsor to delay a proposed clinical investigation or to suspend an ongoing investigation. A partial clinical hold is a delay or suspension of only part of the clinical work requested under the IND. For example, a specific protocol or part of a protocol is not allowed to proceed, while other protocols may do so. No more than 30 days after imposition of a clinical hold or partial clinical hold, the FDA will provide the sponsor a written explanation of the basis for the hold. Following issuance of a clinical hold or partial clinical hold, an investigation may only resume after the FDA has notified the sponsor that the investigation may proceed. The FDA will base that determination on information provided by the sponsor correcting the deficiencies previously cited or otherwise satisfying the FDA that the investigation can proceed.
A sponsor may choose, but is not required, to conduct a foreign clinical study under an IND. When a foreign clinical study is conducted under an IND, all FDA IND requirements must be met unless waived. When the foreign clinical study is not conducted under an IND, the sponsor must ensure that the study complies with FDA certain regulatory requirements in order to use the study as support for an IND or application for marketing approval. Specifically, on April 28, 2008, the FDA amended its regulations governing the acceptance of foreign clinical studies not conducted under an investigational new drug application as support for an IND or a new drug application. The final rule provides that such studies must be conducted in accordance with good clinical practice, or GCP, including review and approval by an independent ethics committee, or IEC, and informed consent from subjects. The GCP requirements in the final rule encompass both ethical and data integrity standards for clinical studies. The FDA’sFDA's regulations are intended to help ensure the protection of human subjects enrolled in non-IND foreign clinical studies, as well as the quality and integrity of the resulting data. They further help ensure that non-IND foreign studies are conducted in a manner comparable to that required for IND studies.
In addition to the foregoing IND requirements, an IRB representing each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review and reapprove the study at least annually. The
IRB must review and approve, among other things, the study protocol and informed consent information to be provided to study subjects. An IRB must operate in compliance with FDA regulations. An IRB can suspend or terminate approval of a clinical trial at its institution, or an institution it represents, if the clinical trial is not being conducted in accordance with the IRB’sIRB's requirements or if the product candidate has been associated with unexpected serious harm to patients.
Additionally, some trials are overseen by an independent group of qualified experts organized by the trial sponsor, known as a data safety monitoring board or committee.committee, or DSMB. This group provides recommendations onauthorization as to whether or not a trial shouldmay move forward at designated check points based on unblinded safety data from the study thatto which only the groupDSMB has access to..access. Suspension or termination of development during any phase of clinical trials may occur if it is determined that the participants or patients are being exposed to an unacceptable health risk. Other reasons for suspension or termination may be made by us based on evolving business objectives and/or competitive climate.
Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on its ClinicalTrials.gov website.
Human Clinical Studies in Support of an NDA
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include, among other things, the requirement that all research subjects provide their informed consent in writing before their participation in any clinical trial. Clinical trials are conducted under written study protocols detailing, among other things, the inclusion and exclusion criteria, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated.
Human clinical trials are typically conducted in the following four sequential phases, which may overlap or be combined:
- •
·Phase 1: The drug is initially introduced into healthy human subjects or, in certain indications suchas cancer, patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness and to determine optimal dosage.
·- •
- Phase 2: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
· - •
- Phase 3: The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product.
· - •
- Phase 4: Post-approval studies, which are conducted following initial approval, are typically conducted to gain additional experience and data from treatment of patients in the intended therapeutic indication.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. In addition, IND safety reports must be submitted to the FDA for any of the following: serious and unexpected suspected adverse reactions; findings from other studies or animal orin vitro testing that suggest a significant risk in humans exposed to the drug; and any clinically important increase in the case of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or
the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution, or an institution it represents, if the clinical trial is not being conducted in accordance with the IRB’sIRB's requirements or if the drug has been associated with unexpected serious harm to patients. The FDA will typically inspect one or more clinical sites to assure compliance with GCP and the integrity of the clinical data submitted.
Concurrent with clinical trials, companies often complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, must develop methods for testing the identity, strength, quality, purity, and potency of the final drug. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
Under the Pediatric Research Equity Act of 2003, an NDA or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. With enactment of the Food and Drug Safety and Innovation Act, or the FDASIA, in 2012, sponsors must also submit pediatric study plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric study or studies the applicant plans to conduct, including study objectives and design, any deferral or waiver requests, and other information required by regulation. The applicant, the FDA, and the FDA's internal review committee must then review the information submitted, consult with each other, and agree upon a final plan. The FDA or the applicant may request an amendment to the plan at any time. For drugs intended to treat a serious or life-threatening disease or condition, the FDA must, upon the request of an applicant, meet to discuss preparation of the initial pediatric study plan or to discuss deferral or waiver of pediatric assessments.
The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. Additional requirements and procedures relating to deferral requests and requests for extension of deferrals are contained in FDASIA. Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation.
Expanded Access to an Investigational Drug for Treatment Use
Expanded access, sometimes called "compassionate use," is the use of investigational new drug products outside of clinical trials to treat patients with serious or immediately life-threatening diseases or conditions when there are no comparable or satisfactory alternative treatment options. The rules and regulations related to expanded access are intended to improve access to investigational drugs for patients who may benefit from investigational therapies. FDA regulations allow access to investigational drugs under an IND by the company or the treating physician for treatment purposes on a case-by-case basis for: individual patients (single-patient IND applications for treatment in emergency settings and non-emergency settings); intermediate-size patient populations; and larger populations for use of the drug under a treatment protocol or Treatment IND Application.
When considering an IND application for expanded access to an investigational product with the purpose of treating a patient or a group of patients, the sponsor and treating physicians or investigators will determine suitability when all of the following criteria apply: patient(s) have a serious or immediately life-threatening disease or condition, and there is no comparable or satisfactory alternative
therapy to diagnose, monitor, or treat the disease or condition; the potential patient benefit justifies the potential risks of the treatment and the potential risks are not unreasonable in the context or condition to be treated; and the expanded use of the investigational drug for the requested treatment will not interfere initiation, conduct, or completion of clinical investigations that could support marketing approval of the product or otherwise compromise the potential development of the product.
On December 13, 2016, the 21st Century Cures Act established (and the 2017 Food and Drug Administration Reauthorization Act later amended) a requirement that sponsors of one or more investigational drugs for the treatment of a serious disease(s) or condition(s) make publicly available their policy for evaluating and responding to requests for expanded access for individual patients. Although these requirements were rolled out over time, they have now come into full effect. This provision requires drug and biologic companies to make publicly available their policies for expanded access for individual patient access to products intended for serious diseases. Sponsors are required to make such policies publicly available upon the earlier of initiation of a Phase 2 or Phase 3 study; or 15 days after the drug or biologic receives designation as a breakthrough therapy, fast track product, or regenerative medicine advanced therapy.
In addition, on May 30, 2018, the Right to Try Act, was signed into law. The law, among other things, provides a federal framework for certain patients to access certain investigational new drug products that have completed a Phase I clinical trial and that are undergoing investigation for FDA approval. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA permission under the FDA expanded access program. There is no obligation for a drug manufacturer to make its drug products available to eligible patients as a result of the Right to Try Act, but the manufacturer must develop an internal policy and respond to patient requests according to that policy.
Special Protocol Assessment Agreements
A Special Protocol Assessment, or SPA, agreement is an agreement between a drug manufacturer and the FDA on the design and size of studies and clinical trials that can be used for approval of a drug or biological product. The FDA’sFDA's guidance on such agreements states that an agreement may not be changed by the manufacturer or the agency unless through a written agreement of the two entities or if FDA determines a substantial scientific issue essential to determining the safety or effectiveness of the drug. The protocols that are eligible for SPA agreements are: animal carcinogenicity protocols, final product stability protocols and clinical protocols for Phase 3 trials whose data will form the primary basis for an efficacy claim.
Specifically, under the FDCA, the FDA may meet with sponsors, provided certain conditions are met, for the purpose of reaching a SPA agreement on the design and size of clinical trials intended to form the primary basis of an efficacy claim in a marketing application. If a sponsor makes a reasonable written request to meet with the FDA for the purpose of reaching agreement on the design and size of a clinical trial, then the FDA will meet with the sponsor. If an agreement is reached, the FDA will reduce the agreement to writing and make it part of the administrative record. An agreement may not be changed by the sponsor or FDA after the trial begins, except with the written agreement of the sponsor and FDA, or if the director of the FDA reviewing division determines that “a"a substantial scientific issue essential to determining the safety or effectiveness of the drug”drug" was identified after the testing began. If a sponsor and the FDA meet regarding the design and size of a clinical trial and the parties cannot agree that the trial design is adequate to meet the goals of the sponsor, the FDA will clearly state the reasons for the disagreement in a letter to the sponsor. We reached agreement with
the FDA in September 2015 on a SPA regarding the study design of our first Phase 3 clinical trial of lefamulin for the treatment of CABP.
Submission of an NDA to the FDA
Assuming successful completion of required clinical testing and other requirements, the results of the preclinical and clinical studies, together with detailed information relating to the product’sproduct's chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the drug product for one or more indications. Under federal law, the submission of most NDAs is additionally subject to an application user fee, which for federal fiscal year 20172019 is $2,038,100, and the$2,588,478 for an application requiring clinical data. The sponsor of an approved NDA is also subject to an annual product and establishment user fees,program fee, which for fiscal year 2017 are $97,750 per product and $512,000 per establishment2019 is $309,154. Exceptions or waivers for userthese fees exist for a small company (fewer than 500 employees, including employees and affiliates) satisfying certain requirements and products with orphan drug designation for a particular indication are not subject to an application usera fee provided there are no other intended uses in the NDA. We believe that we will not be subject to an application user fee.
Following submission of an NDA, the FDA conducts a preliminary review of an NDA generally within 60 calendar days of its receipt and strives to inform the sponsor by the 74th74th day after the FDA’sFDA's receipt of the submission whether the application is sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA has agreed to certain performance goals in the review process of NDAs. Standard review, representing most such applications are meant to be reviewed within ten months from the date of filing. Priority review applications are meant to be reviewed within six months of filing. The review process and the Prescription Drug User Fee Act goal date may be extended by the FDA for three additional months to consider new information or clarification provided by the applicant to address an outstanding deficiency identified by the FDA following the original submission. Under the FDA Reauthorization Act of 2017, the FDA must implement a protocol to expedite review of responses to inspection reports pertaining to certain applications, including applications for products in shortage or those for which approval is dependent on remediation of conditions identified in the inspection report.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is or will be manufactured. These pre-approval inspections may cover all facilities associated with an NDA submission, including drug component manufacturing (such as active pharmaceutical ingredients), finished drug product manufacturing, and control testing laboratories. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP.
In addition, as a condition of approval, the FDA may require an applicant to develop a REMS. REMS use risk minimization strategies beyond the professional labeling to ensure that the benefits of the product outweigh the potential risks. To determine whether a REMS is needed, the FDA will consider the size of the population likely to use the product, seriousness of the disease, expected benefit of the product, expected duration of treatment, seriousness of known or potential adverse events, and whether the product is a new molecular entity. REMS can include medication guides, physician communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU may include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The FDA may require a REMS before approval or post-approval if it becomes aware of a serious risk associated with use of the product. The requirement for a REMS can materially affect the potential market and profitability of a product.
The FDA may refer an application for a novel drug to an advisory committee or explain why such referral was not made. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions relating to approval of a new drug product.
Fast Track, Breakthrough Therapy,, Priority Review and Regenerative Advanced Therapy Designations
The FDA is authorized to designate certain products for expedited review if they are intended to address
an unmet medical need in the treatment of a serious or life-threatening disease or condition. These programs are referred to as fast track designation, breakthrough therapy designation, priority review designation and regenerative advanced therapy designation.
Specifically, the FDA may designate a product for Fast Track review if it is intended, whether alone or in combination with one or more other products, for the treatment of a serious or life-threatening disease or condition, and it demonstrates the potential to address unmet medical needs for such a disease or condition. For Fast Track products, sponsors may have greater interactions with the FDA and the FDA may initiate review of sections of a Fast Track product’sproduct's application before the application is complete. This rolling review may be available if the FDA determines, after preliminary evaluation of clinical data submitted by the sponsor, that a Fast Track product may be effective. The sponsor must also provide, and the FDA must approve, a schedule for the submission of the remaining information and the sponsor must pay applicable user fees. However, the FDA’sFDA's time period goal for reviewing a Fast Track application does not begin until the last section of the application is submitted. In addition, the Fast Track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
Second, a product may be designated as a Breakthrough Therapy if it is intended, either alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA may take certain actions with respect to Breakthrough Therapies, including holding meetings with the sponsor throughout the development process; providing timely advice to the product sponsor regarding development and approval; involving more senior staff in the review process; assigning a cross-disciplinary project lead for the review team; and taking other steps to design the clinical trials in an efficient manner.
Third, the FDA may designate a product for priority review if it is a product that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. The FDA determines, on a case- by-case basis, whether the proposed product represents a significant improvement when compared with other available therapies. Significant improvement may be illustrated by evidence of increased effectiveness in the treatment of a condition, elimination or substantial reduction of a treatment-limiting product reaction, documented enhancement of patient compliance that may lead to improvement in serious outcomes, and evidence of safety and effectiveness in a new subpopulation. A priority designation is intended to direct overall attention and resources to the evaluation of such applications, and to shorten the FDA’sFDA's goal for taking action on a marketing application from ten months to six months.
With passage of the 21st Century Cures Act, or the Cures Act, in December 2016, Congress authorized the FDA to accelerate review and approval of products designated as regenerative advanced therapies. A product is eligible for this designation if it is a regenerative medicine therapy that is intended to treat, modify, reverse or cure a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product has the potential to address unmet medical
Fast Track, Breakthrough Therapyneeds for such disease or condition. The benefits of a regenerative advanced therapy designation include early interactions with FDA to expedite development and Priority Review Designationsreview, benefits available to breakthrough therapies, potential eligibility for priority review and accelerated approval based on surrogate or intermediate endpoints.
Accelerated Approval Pathway
The FDA may grant accelerated approval to a drug for a serious or life-threatening condition that provides meaningful therapeutic advantage to patients over existing treatments based upon a determination that the drug has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a drug when the product has an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, or IMM, and that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Drugs granted accelerated approval must meet the same statutory standards for safety and effectiveness as those granted traditional approval.
For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign, or other measure that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. An intermediate clinical endpoint is a measurement of a therapeutic effect that is considered reasonably likely to predict the clinical benefit of a drug, such as an effect on IMM. The FDA has limited experience with accelerated approvals based on intermediate clinical endpoints, but has indicated that such endpoints generally may support accelerated approval where the therapeutic effect measured by the endpoint is not itself a clinical benefit and basis for traditional approval, if there is a basis for concluding that the therapeutic effect is reasonably likely to predict the ultimate clinical benefit of a drug.
The accelerated approval pathway is most often used in settings in which the course of a disease is long and an extended period of time is required to measure the intended clinical benefit of a drug, even if the effect on the surrogate or intermediate clinical endpoint occurs rapidly. Thus, accelerated approval has been used extensively in the development and approval of drugs for treatment of a variety of cancers in which the goal of therapy is generally to improve survival or decrease morbidity and the duration of the typical disease course requires lengthy and sometimes large trials to demonstrate a clinical or survival benefit. The accelerated approval pathway is usually contingent on a sponsor’ssponsor's agreement to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the drug’sdrug's clinical benefit. As a result, a drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, would allow the FDA to withdraw the drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by the FDA.
Limited Population Antibacterial Drug Pathway
With passage of the CURES Act, Congress authorized FDA to approve an antibacterial or antifungal drug, alone or in combination with one or more other drugs, as a “limited"limited population drug.”" To qualify for this approval pathway, the drug must be intended to treat a serious or life-threatening infection in a limited population of patients with unmet needs; the standards for approval of drugs and biologics under the FDCA and PHSA must be satisfied; and FDA must receive a written request from the sponsor to approve the drug as a limited population drug pursuant to this provision. The FDA’sFDA's determination of safety and effectiveness for such a product must reflect the benefit-risk profile of such
drug in the intended limited population, taking into account the severity, rarity, or prevalence of the infection the drug is intended to treat and the availability or lack of alternative treatment in such a limited population.
Any drug or biologic approved under this pathway must be labeled with the statement “Limited Population”"Limited Population" in a prominent manner and adjacent to the proprietary name of the drug or biological product. The prescribing information must also state that the drug is indicated for use in a limited and specific population of patients and copies of all promotional materials relating to the drug must be submitted to FDA at least 30 days prior to dissemination of the materials. If FDA subsequently approves the drug for a broader indication, the agency may remove any post-marketing conditions, including requirements with respect to labeling and review of promotional materials applicable to the product. Nothing in this pathway to approval of a limited population drug prevents sponsors of such products from seeking designation or approval under other provisions of the FDCA, such as accelerated approval.
The FDA’sFDA's Decision on an NDA
On the basis of the FDA’sFDA's evaluation of the NDA and accompanying information, including the results of the inspection of the manufacturing facilities, the FDA may issue an approval letter or a complete response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A complete response letter generally outlines the deficiencies in the submission and may require additional, sometimes substantial, testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’sFDA's satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
If the FDA approves a new product, it may limit the approved indications for use for the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess the drug’sdrug's safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, including REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs. After approval, many types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Regulation
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with or without clinical data.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with
cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
- •
·restrictions on the marketing or manufacturing of the product, suspension of the approval, or complete withdrawal of the product from the market or product recalls;·- •
- fines, warning letters or holds on post-approval clinical trials;
· - •
- refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals;
· - •
- product seizure or detention, or refusal to permit the import or export of products; or
· - •
- injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates the marketing, labeling, advertising and promotion of prescription drug products that are placed on the market. DrugsThis regulation includes, among other things, standards and regulations for direct-to-consumer advertising, communications regarding unapproved uses, industry-sponsored scientific and educational activities, and promotional activities involving the Internet and social media. Promotional claims about a drug's safety or effectiveness are prohibited before the drug is approved. After approval, a drug product generally may not be promoted only for uses that are not approved by the approved indicationsFDA, as reflected in the product's prescribing information. In the United States, health care professionals are generally permitted to prescribe drugs for such uses not described in the drug's labeling, known as off-label uses, because the FDA does not regulate the practice of medicine. However, FDA regulations impose rigorous restrictions on manufacturers' communications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibitingprohibit the promotion of off-label uses, anduses. It may be permissible, under very specific, narrow conditions, for a manufacturer to engage in non-promotional, non-misleading communication regarding off-label information, such as distributing scientific or medical journal information.
If a company that is found to have improperly promoted off-label uses, it may bebecome subject to adverse public relations and administrative and judicial enforcement by the FDA, the Department of Justice, or the Office of the Inspector General of the Department of Health and Human Services, as well as state authorities. This could subject a company to a range of penalties that could have a significant liability.commercial impact, including civil and criminal fines and agreements that materially restrict the manner in which a company promotes or distributes drug products. The federal government has levied large civil and criminal fines against companies for alleged improper promotion, and has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, and its implementing regulations, as well as the Drug Supply Chain Security Act, or DSCA, which regulate the distribution and tracing of prescription drugs and prescription drug samples at the federal level, and set minimum standards for the regulation of drug distributors by the states. The PDMA, its implementing regulations and state laws limit the distribution of prescription pharmaceutical product samples and the DSCA imposes requirements to ensure accountability in distribution and to identify and remove counterfeit and other illegitimate products from the market.
Section 505(b)(2) NDAs
NDAs for most new drug products are based on two full clinical studies which must contain substantial evidence of the safety and efficacy of the proposed new product.product for the proposed use. These applications are submitted under Section 505(b)(1) of the FDCA. The FDA is, however, authorized to approve an alternative type of NDA under Section 505(b)(2) of the FDCA. This type of application allows the applicant to rely, in part, on the FDA’sFDA's previous findings of safety and efficacy for a similar product, or published literature. Specifically, Section 505(b)(2) applies to NDAs for a drug for which the investigations made to show whether or not the drug is safe for use and effective in use and relied upon by the applicant for approval of the application “were"were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted.”"
Thus, Section 505(b)(2) authorizes the FDA to approve an NDA based on safety and effectiveness data that were not developed by the applicant. NDAs filed under Section 505(b)(2) may provide an alternate and potentially more expeditious pathway to FDA approval for new or improved formulations or new uses of previously approved products. If the Section 505(b)(2) applicant can establish that reliance on the FDA’sFDA's previous approval is scientifically appropriate, the applicant may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new drug candidate for all or some of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
Abbreviated New Drug Applications for Generic Drugs
In 1984, with passage of the Hatch-Waxman Amendments to the FDCA, Congress authorized the FDA to approve generic drugs that are the same as drugs previously approved by the FDA under the NDA provisions of the statute. To obtain approval of a generic drug, an applicant must submit an abbreviated new drug application, or ANDA, to the agency. In support of such applications, a generic manufacturer may rely on the preclinical and clinical testing previously conducted for a drug product previously approved under an NDA, known as the reference listed drug, or RLD.
Specifically, in order for an ANDA to be approved, the FDA must find that the generic version is identical to the RLD with respect to the active ingredients, the route of administration, the dosage form, and the strength of the drug. At the same time, the FDA must also determine that the generic drug is “bioequivalent”"bioequivalent" to the innovator drug. Under the statute, a generic drug is bioequivalent to a RLD if “the"the rate and extent of absorption of the drug do not show a significant difference from the rate and extent of absorption of the listed drug.”"
Upon approval of an ANDA, the FDA indicates whether the generic product is “therapeutically equivalent”"therapeutically equivalent" to the RLD in its publication “Approved"Approved Drug Products with Therapeutic Equivalence Evaluations,”" also referred to as the “Orange"Orange Book.”" Physicians and pharmacists consider a therapeutic equivalent generic drug to be fully substitutable for the RLD. In addition, by operation of certain state laws and numerous health insurance programs, the FDA’sFDA's designation of therapeutic equivalence often
results in substitution of the generic drug without the knowledge or consent of either the prescribing physician or patient.
Under the Hatch-Waxman Amendments, the FDA may not approve an ANDA until any applicable period of non-patent exclusivity for the RLD has expired. The FDCA provides a period of five years of non-patent data exclusivity for a new drug containing a new chemical entity. For the purposes of this provision, a new chemical entity, or NCE, is a drug that contains no active moiety that has previously been approved by the FDA in any other NDA. An active moiety is the molecule or ion responsible for the physiological or pharmacological action of the drug substance. In cases where such exclusivity has been granted, an ANDA may not be filed with the FDA until the expiration of five years unless the submission is accompanied by a Paragraph IV certification, in which case the applicant may submit its application four years following the original product approval. The FDCA also provides for a period of three years of exclusivity if the NDA includes reports of one or more new clinical investigations, other than bioavailability or bioequivalence studies, that were conducted by or for the applicant and are essential to the approval of the application. This three-year exclusivity period often protects changes to a previously approved drug product, such as a new dosage form, route of administration, combination or indication.
The FDCA also provides for a period of three years of exclusivity if the NDA includes reports of one or more new clinical investigations, other than bioavailability or bioequivalence studies, that were conducted by or for the applicant and are essential to the approval of the application. This three-year exclusivity period often protects changes to a previously approved drug product, such as a new dosage form, route of administration, combination or indication. Three-year exclusivity would be available for a drug product that contains a previously approved active moiety, provided the statutory requirement for a new clinical investigation is satisfied. Unlike five-year NCE exclusivity, an award of three-year exclusivity does not block the FDA from accepting ANDAs seeking approval for generic versions of the drug as of the date of approval of the original drug product. The FDA typically makes decisions about awards of data exclusivity shortly before a product is approved.
The FDA must establish a priority review track for certain generic drugs, requiring the FDA to review a drug application within eight (8) months for a drug that has three (3) or fewer approved drugs listed in the Orange Book and is no longer protected by any patent or regulatory exclusivities, or is on the FDA's drug shortage list. The new legislation also authorizes FDA to expedite review of "competitor generic therapies" or drugs with inadequate generic competition, including holding meetings with or providing advice to the drug sponsor prior to submission of the application.
Hatch-Waxman Patent Certification and the 30-Month Stay
Upon approval of an NDA or a supplement thereto, NDA sponsors are required to list with the FDA each patent with claims that cover the applicant’sapplicant's product or an approved method of using the product. Each of the patents listed by the NDA sponsor is published in the Orange Book. When an ANDA applicant files its application with the FDA, the applicant is required to certify to the FDA concerning any patents listed for the reference product in the Orange Book, except for patents covering methods of use for which the ANDA applicant is not seeking approval. To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would.
Specifically, the applicant must certify with respect to each patent that:
- •
·the required patent information has not been filed;·- •
- the listed patent has expired;
- •
- the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or
· - •
- the listed patent is invalid, unenforceable or will not be infringed by the new product.
A certification that the new product will not infringe the already approved product’sproduct's listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the applicant does not challenge the listed patents or indicates that it is not seeking approval of a patented method of use, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired (other than method of use patents involving indications for which the ANDA applicant is not seeking approval).
If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days after the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months after the receipt of the Paragraph IV notice, expiration of the patent, or a decision in the infringement case that is favorable to the ANDA applicant.
To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. As a result, approval of a Section 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
Pediatric Studies and Exclusivity
Under the Pediatric Research Equity Act of 2003, an NDA or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. With enactment of the FDASIA in 2012, sponsors must also submit pediatric study plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric study or studies the applicant plans to conduct, including study objectives and design, any deferral or waiver requests, and other information required by regulation. The applicant, the FDA, and the FDA’s internal review committee must then review the information submitted, consult with each other, and agree upon a final plan. The FDA or the applicant may request an amendment to the plan at any time.
The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. Additional requirements and procedures relating to deferral requests and requests for extension of deferrals are contained in the FDASIA. Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation.
Pediatric exclusivity is another type of non-patent marketing exclusivity in the United States and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity, including the non-patent and orphan exclusivity. This six-month exclusivity may be granted if an NDA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’sFDA's request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot approve another application.
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may designate a drug product as an “orphan drug” if it is intended to treat a rare disease or condition (generally meaning that it affects fewer than 200,000 individuals in the United States, or more in cases in which there is no reasonable expectation that the cost of developing and making a drug product available in the United States for treatment of the disease or condition will be recovered from sales of the product). A company must request orphan product designation before submitting an NDA. If the request is granted, the FDA will disclose the identity of the therapeutic agent and its potential use. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product with orphan status receives the first FDA approval for the disease or condition for which it has such designation or for a select indication or use within the rare disease or condition for which it was designated, the product generally will receive orphan product exclusivity. Orphan product exclusivity means that the FDA may not approve any other applications for the same product for the same indication for seven years, except in certain limited circumstances. Competitors may receive approval of different products for the indication for which the orphan product has exclusivity and may obtain approval for the same product but for a different indication. If a drug or drug product designated as an orphan product ultimately receives marketing approval for an indication broader than what was designated in its orphan product application, it may not be entitled to exclusivity.
GAIN Exclusivity for Antibiotics
In 2012, Congress passed legislation known as the Generating Antibiotic Incentives Now Act, or GAIN Act. This legislation is designed to encourage the development of antibacterial and antifungal drug products that treat pathogens that cause serious and life-threatening infections. To that end, the law grants an additional five years of exclusivity upon the approval of an NDA for a drug product designated by FDA as a QIDP. Thus, for a QIDP, the periods of five-year new chemical entity exclusivity, three-year new clinical investigation exclusivity, and seven-year orphan drug exclusivity, would become ten years, eight years, and twelve years, respectively.
A QIDP is defined in the GAIN Act to mean “an"an antibacterial or antifungal drug for human use intended to treat serious or life-threatening infections, including those caused by (1) an antibacterial or antifungal resistant pathogen, including novel or emerging infectious pathogens”pathogens" or (2) certain “qualifying"qualifying pathogens.”" A “qualifying pathogen”"qualifying pathogen" is a pathogen that has the potential to pose a serious threat to public health (such as resistant Gram-positive pathogens, multi-drug resistant Gram-negative bacteria, multi-drug resistant tuberculosis, andC. difficile) and that is included in a list established and maintained by FDA. A drug sponsor may request the FDA to designate its product as a QIDP any time before the submission of an NDA. The FDA must make a QIDP determination within 60 days of the designation request. A product designated as a QIDP will be granted priority review by the FDA and can qualify for “fast track”"fast track" status.
The additional five years of exclusivity under the GAIN Act for drug products designated by the FDA as QIDPs applies only to a drug that is first approved on or after July 9, 2012. Additionally, the five year
exclusivity extension does not apply to: a supplement to an application under FDCA Section 505(b) for any QIDP for which an extension is in effect or has expired; a subsequent application filed with respect to a product approved by the FDA for a change that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength; or a product that does not meet the definition of a QIDP under Section 505(g) based upon its approved uses. The FDA has designated IV fosfomycin, and each of the IV and oral formulations of lefamulin as a QIDP and also granted fast track designations to each of these formulations of lefamulin.designations.
Patent Term Restoration and Extension
A patent claiming a new drug product may be eligible for a limited patent term extension under the Hatch-Waxman Amendments,Act, which permits a patent restoration of up to five years for patent term lost during product development and the FDA regulatory review. The restoration period granted on a patent covering a product is typically one-half the time between the effective date of an INDa clinical investigation involving human beings is begun and the submission date of an NDA,application, plus the time between the submission date of an NDAapplication and the ultimate approval date. Patent term restoration cannot be used to extend the remaining term of a patent past a total of 14 years from the product’sproduct's approval date. Only one patent applicable to an approved drug product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent in question. A patent that covers multiple drugsproducts for which approval is sought can only be extended in connection with one of the approvals. The USPTOUnited States Patent and Trademark Offices reviews and approves the application for any patent term extension or restoration in consultation with the FDA.
The 21st Century Cures Act
On December 13, 2016, President Obama signed the 21st Century Cures Act, or Cures Act, into law. The Cures Act is designed to modernize and personalize healthcare, spur innovation and research, and streamline the discovery and development of new therapies through increased federal funding of particular programs. It authorizes increased funding for the FDA to spend on innovation projects. The new law also amends the Public Health Service Act to reauthorize and expand funding for the National Institutes of Health. The Act establishes the NIH Innovation Fund to pay for the cost of development and implementation of a strategic plan, early stage investigators and research. It also charges NIH with leading and coordinating expanded pediatric research. Further, the Cures Act directs the Centers for Disease Control and Prevention to expand surveillance of neurological diseases.
With amendments to the FDCA and the Public Health Service Act, or PHSA, Title III of the Cures Act seeks to accelerate the discovery, development, and delivery of new medicines and medical technologies. To that end, and among other provisions, the Cures Act reauthorizes the existing priority review voucher program for certain drugs intended to treat rare pediatric diseases until 2020; creates a new priority review voucher program for drug applications determined to be material national security threat medical countermeasure applications; revises the FDCA to streamline review of combination product applications; requires FDA to evaluate the potential use of “real world evidence” to help support approval of new indications for approved drugs; provides a new “limited population” approval pathway for antibiotic and antifungal drugs intended to treat serious or life-threatening infections; and authorizes FDA to designate a drug as a “regenerative advanced therapy,” thereby making it eligible for certain expedited review and approval designations.
Regulation Outside the United States
In order to market any product outside of the United States, a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of drug products. Whether or not it obtainswe obtain FDA approval for a product,lefamulin, the company would need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can commence clinical trials or marketing of the product in those countries or jurisdictions. The approval process ultimately varies between countries and jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others.
Regulation and Marketing Authorization in the European Union
The process governing approval of medicinal products in the European Union follows essentially the same lines as in the United States and, likewise, generally involves satisfactorily completing each of the following:
- •
·preclinical laboratory tests, animal studies and formulation studies all performed in accordance with the applicable E.U. Good Laboratory Practice regulations;·- •
- submission to the relevant national authorities of a clinical trial application, or CTA, which must be approved before human clinical trials may begin;
· - •
- performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product for each proposed indication;
· - •
- submission to the relevant competent authorities of a marketing authorization application, or MAA, which includes the data supporting safety and efficacy as well as detailed information on the manufacture and composition of the product in clinical development and proposed labelling;
· - •
- satisfactory completion of an inspection by the relevant national authorities of the manufacturing facility or facilities, including those of third parties, at which the product is produced to assess compliance with strictly enforced cGMP;
· - •
- potential audits of the non-clinical and clinical trial sites that generated the data in support of the MAA; and
· - •
- review and approval by the relevant competent authority of the MAA before any commercial marketing, sale or shipment of the product.
Preclinical Studies
Preclinical Studies
Preclinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animal studies, in order to assess the potential safety and efficacy of the product. The conduct of the preclinical tests and formulation of the compounds for testing must comply with the relevant E.U. regulations and requirements. The results of the preclinical tests, together with relevant manufacturing information and analytical data, are submitted as part of the CTA.
Clinical Trial Approval
Requirements for the conduct of clinical trials in the European Union including Good Clinical Practice, or GCP, are set forth in the Clinical Trials Directive 2001/20/EC and the GCP Directive 2005/28/EC. Pursuant to Directive 2001/20/EC and Directive 2005/28/EC, as amended, a system for the approval of clinical trials in the European Union has been implemented through national legislation of the E.U. member states. Under this system, approval must be obtained from the competent national authority of each E.U. member state in which a study is planned to be conducted. To this end, a CTA is submitted, which must be supported by an investigational medicinal product dossier, or IMPD, and further supporting information prescribed by Directive 2001/20/EC and Directive 2005/28/EC and other applicable guidance documents. Furthermore, a clinical trial may only be started after a competent ethics committee has issued a favorable opinion on the clinical trial application in that country.
In April 2014, the E.U. passed the new Clinical Trials Regulation, (EU) No 536/2014, which will replace the current Clinical Trials Directive 2001/20/EC. The Regulation was published on June 16, 2014 but is not expected to apply until later in 2019. To ensure that the rules for clinical trials are identical throughout the European Union, the new E.U. clinical trials legislation was passed as a
regulation that is directly applicable in all E.U. member states. All clinical trials performed in the European Union are required to be conducted in accordance with the Clinical Trials Directive 2001/20/EC until the new Clinical Trials Regulation (EU) No 536/2014 becomes applicable. According to the current plans of EMA, the new Clinical Trials Regulation will become applicable in October 2018. The Clinical Trials Directive 2001/20/EC will, however, still apply three years from the date of entry into application of the Clinical Trials Regulation to (i) clinical trials applications submitted before the entry into application and (ii) clinical trials applications submitted within one year after the entry into application if the sponsor opts for the old system.
The new Clinical Trials Regulation aims to simplify and streamline the approval of clinical trial in the European Union. The main characteristics of the regulation include:
- •
·a streamlined application procedure via a single entry point, the E.U. portal;·- •
- a single set of documents to be prepared and submitted for the application as well as simplified reporting procedures that will spare sponsors from submitting broadly identical information separately to various bodies and different member states;
· - •
- a harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts. Part I is assessed jointly by all member states concerned. Part II is assessed separately by each member state concerned;
· - •
- strictly defined deadlines for the assessment of clinical trial applications; and
· - •
- the involvement of the ethics committees in the assessment procedure in accordance with the national law of the member state concerned but within the overall timelines defined by the Clinical Trials Regulation.
Marketing Authorization
Marketing Authorization
Authorization to market a product in the member states of the European Union proceeds under one of four procedures: a centralized authorization procedure, a mutual recognition procedure, a decentralized procedure or a national procedure.
Centralized Authorization Procedure
The centralized procedure enables applicants to obtain a marketing authorization that is valid in all E.U. member states based on a single application. Certain medicinal products, including products developed by means of biotechnological processes must undergo the centralized authorization procedure for marketing authorization, which, if granted by the European Commission, is automatically valid in all 28 E.U. member states. The EMA and the European Commission administer this centralized authorization procedure pursuant to Regulation (EC) No 726/2004.
Pursuant to Regulation (EC) No 726/2004, this procedure is mandatory for:
- •
·medicinal products developed by means of one of the following biotechnological processes:·- •
- recombinant DNA technology;
· - •
- controlled expression of genes coding for biologically active proteins in prokaryotes and eukaryotes including transformed mammalian cells; and
· - •
- hybridoma and monoclonal antibody methods;
·
- •
- advanced therapy medicinal products as defined in Article 2 of Regulation (EC) No 1394/2007 on advanced therapy medicinal products;
- •
- medicinal products for human use containing a new active substance that, on the date of effectiveness of this regulation, was not authorized in the European Union, and for which the therapeutic indication is the treatment of any of the following diseases:
· - •
- medicinal products that are designated as orphan medicinal products pursuant to Regulation (EC) No 141/2000.
The centralized authorization procedure is optional for other medicinal products if they contain a new active substance or if the applicant shows that the medicinal product concerned constitutes a significant therapeutic, scientific or technical innovation or that the granting of authorization is in the interest of patients in the European Union.
Administrative Procedure
Under the centralized authorization procedure, the EMA’sEMA's Committee for Medicinal Products for Human Use, or CHMP serves as the scientific committee that renders opinions about the safety, efficacy and quality of medicinal products for human use on behalf of the EMA. The CHMP is composed of experts nominated by each member state’sstate's national authority for medicinal products, with one of them appointed to act as Rapporteur for the co-ordination of the evaluation with the possible assistance of a further member of the Committee acting as a Co-Rapporteur. After approval, the Rapporteur(s) continue to monitor the product throughout its life cycle. The CHMP has 210 days, to adopt an opinion as to whether a marketing authorization should be granted. The process usually takes longer in case additional information is requested, which triggers clock-stops in the procedural timelines. The process is complex and involves extensive consultation with the regulatory authorities of member states and a number of experts. When an application is submitted for a marketing authorization in respect of a drug that is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation, the applicant may pursuant to Article 14(9) Regulation (EC) No 726/2004 request an accelerated assessment procedure. If the CHMP accepts such request, the time-limit of 210 days will be reduced to 150 days but it is possible that the CHMP can revert to the standard time-limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment. Once the procedure is completed, a European Public Assessment Report, or EPAR, is produced. If the opinion is negative, information is given as to the grounds on which this conclusion was reached. After the adoption of the CHMP opinion, a decision on the MAA must be adopted by the European Commission, after consulting the E.U. member states, which in total can take more than 60 days.
Conditional Approval
In specific circumstances, E.U. legislation (Regulation (EC) No 726/2004 and Regulation (EC) No 507/2006 on Conditional Marketing AuthorisationsAuthorizations for Medicinal Products for Human Use) enables applicants to obtain a conditional marketing authorization prior to obtaining the comprehensive clinical data required for an application for a full marketing authorization. Such conditional approvals may be granted for product candidates (including medicines designated as orphan medicinal products), if (1) the risk-benefit balance of the product candidate is positive, (2) it is likely that the applicant will be
in a position to provide the required comprehensive clinical trial data, (3) the product fulfills unmet medical needs and (4) the benefit to public health of the immediate availability on the market of the medicinal product concerned outweighs the risk inherent in the fact that additional data are still required. A conditional marketing authorization may contain specific obligations to be fulfilled by the marketing authorization holder, including obligations with respect to the completion of ongoing or new studies, and with respect to the collection of pharmacovigilance data. Conditional marketing authorizations are valid for one year, and may be renewed annually, if the risk-benefit balance remains positive, and after an assessment of the need for additional or modified conditions and/or specific obligations. The timelines for the centralized procedure described above also apply with respect to the review by the CHMP of applications for a conditional marketing authorization.
Marketing Authorization Under Exceptional Circumstances
Under Regulation (EC) No 726/2004, products for which the applicant can demonstrate that comprehensive data (in line with the requirements laid down in Annex I of Directive 2001/83/EC, as amended) cannot be provided (due to specific reasons foreseen in the legislation) might be eligible for marketing authorization under exceptional circumstances. This type of authorization is reviewed annually to reassess the risk-benefit balance. The fulfillment of any specific procedures/obligations imposed as part of the marketing authorization under exceptional circumstances is aimed at the provision of information on the safe and effective use of the product and will normally not lead to the completion of a full dossier/approval.
Market Authorizations Granted by Authorities of E.U. Member States
In general, if the centralized procedure is not followed, there are three alternative procedures to obtain a marketing authorization in (one or several) E.U. member states as prescribed in Directive 2001/83/EC:
- •
·The decentralized procedure allows applicants to file identical applications to several E.U. member states and receive simultaneous national approvals based on the recognition by E.U. member states of an assessment by a reference member state.·- •
- The national procedure is only available for products intended to be authorized in a single E.U. member state.
· - •
- A mutual recognition procedure similar to the decentralized procedure is available when a marketing authorization has already been obtained in at least one E.U. member state.
A marketing authorization may be granted only to an applicant established in the European Union.
Pediatric Studies
Prior to obtaining a marketing authorization in the European Union,E.U., applicants have to demonstrate compliance with all measures included in an EMA-approved Pediatric Investigation Plan, or PIP, covering all subsets of the pediatric population, unless the EMA has granted a product-specific waiver, a class waiver, or a deferral for one or more of the measures included in the PIP. The respective requirements for all marketing authorization procedures are set forth in Regulation (EC) No 1901/2006, which is referred to as the Pediatric Regulation. This requirement also applies when a company wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorized. The Pediatric Committee of the EMA, or PDCO, may grant deferrals for some medicines, allowing a company to delay development of the medicine in children until there is enough information to demonstrate its effectiveness and safety in adults. The PDCO may also grant
waivers when development of a medicine in children is not needed or is not appropriate, such as for diseases that only affect the elderly population.
Before a marketing authorization application can be filed, or an existing marketing authorization can be amended, the EMA determines that companies actually comply with the agreed studies and measures listed in each relevant PIP.
Period of Authorization and Renewals
A marketing authorization, other than a conditional marketing authorization, is initially valid for five years and the marketing authorization may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the authorizing member state. To this end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least six months before the marketing authorization ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal. Any authorization which is not followed by the actual placing of the drug on the E.U. market (in case of centralized procedure) or on the market of the authorizing member state within three years after authorization ceases to be valid (the so-called sunset clause).
Regulatory Data Protection
European Union legislation also provides for a system of regulatory data and market exclusivity. According to Article 14(11) of Regulation (EC) No 726/2004, as amended, and Article 10(1) of Directive 2001/83/EC, as amended, upon receiving marketing authorization, new chemical entities approved on the basis of complete independent data package benefit from eight years of data exclusivity and an additional two years of market exclusivity. Data exclusivity prevents regulatory authorities in the European Union from referencing the innovator’sinnovator's data to assess a generic (abbreviated) application. During the additional two-year period of market exclusivity, a generic marketing authorization application can be submitted, and the innovator’sinnovator's data may be referenced, but no generic medicinal product can be marketed until the expiration of the market exclusivity. The overall ten-year period will be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder, or MAH, obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. Even if a compound is considered to be a new chemical entity and the innovator is able to gain the period of data exclusivity, another company nevertheless could also market another version of the drug if such company obtained marketing authorization based on an MAA with a complete independent data package of pharmaceutical test, preclinical tests and clinical trials.
Transparency
There is an increasing trend in the E.U. towards greater transparency and, while the manufacturing or quality information in marketing authorization dossiers is currently generally protected as confidential information, the EMA and national regulatory authorities are now liable to disclose much of the non-clinical and clinical information, including the full clinical study reports, in response to freedom of information requests after the marketing authorization has been granted. In October 2014, the EMA adopted a policy under which clinical study reports would be posted on the agency’sagency's website following the grant, denial or withdrawal of a marketing authorization application, subject to procedures for limited redactions and protection against unfair commercial use. Additional
transparency provisions are contained in the new Clinical Trials Regulation (EU) No 536/2014 that will take effect in May 2016 at the earliest.
Regulatory Requirements After a Marketing Authorization has been Obtained
If we obtain authorization for a medicinal product in the European Union, we will be required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of medicinal products:
Pharmacovigilance and Other Requirements
We will, for example, have to comply with the E.U.’s's stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. E.U. regulators may conduct inspections to verify our compliance with applicable requirements, and we will have to continue to expend time, money and effort to remain compliant. Non-compliance with E.U. requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties in the European Union. Similarly, failure to comply with the European Union’sUnion's requirements regarding the protection of individual personal data can also lead to significant penalties and sanctions. Individual E.U. member states may also impose various sanctions and penalties in case we do not comply with locally applicable requirements.
Manufacturing
The manufacturing of authorized drugs, for which a separate manufacturer’smanufacturer's license is mandatory, must be conducted in strict compliance with the EMA’sEMA's GMP requirements and comparable requirements of other regulatory bodies in the European Union, which mandate the methods, facilities and controls used in manufacturing, processing and packing of drugs to assure their safety and identity. The EMA enforces its GMP requirements through mandatory registration of facilities and inspections of those facilities. The EMA may have a coordinating role for these inspections while the responsibility for carrying them out rests with the member states competent authority under whose responsibility the manufacturer falls. Failure to comply with these requirements could interrupt supply and result in delays, unanticipated costs and lost revenues, and could subject
the applicant to potential legal or regulatory action, including but not limited to warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil and criminal penalties.
Marketing and Promotion
The marketing and promotion of authorized drugs, including industry-sponsored continuing medical education and advertising directed toward the prescribers of drugs and/or the general public, are strictly regulated in the European Union under Directive 2001/83EC, as amended. The applicable regulations aim to ensure that information provided by holders of marketing authorizations regarding their products is truthful, balanced and accurately reflects the safety and efficacy claims authorized by the EMA or by the competent authority of the authorizing member state. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties.
Patent Term Extension
To compensate the patentee for delays in obtaining a marketing authorization for a patented product, a supplementary certificate, or SPC, may be granted extending the exclusivity period for that specific product by up to five years. Applications for SPCs must be made to the relevant patent office in each E.U. member state and the granted certificates are valid only in the member state of grant. An
application has to be made by the patent owner within six months of the first marketing authorization being granted in the European Union (assuming the patent in question has not expired, lapsed or been revoked) or within six months of the grant of the patent (if the marketing authorization is granted first). In the context of SPCs, the term “product”"product" means the active ingredient or combination of active ingredients for a medicinal product and the term “patent”"patent" means a patent protecting such a product or a new manufacturing process or application for it. The duration of an SPC is calculated as the difference between the patent’spatent's filing date and the date of the first marketing authorization, minus five years, subject to a maximum term of five years.
A six-month pediatric extension of an SPC may be obtained where the patentee has carried out an agreed pediatric investigation plan, the authorized product information includes information on the results of the studies and the product is authorized in all member states of the European Union.
Brexit and the Regulatory Framework in the United Kingdom
On June 23, 2016, the electorate in the United Kingdom voted in favor of leaving the European UnionE.U. (commonly referred to as “Brexit”"Brexit"). Thereafter, on March 29, 2017, the country formally notified the E.U. of its intention to withdraw pursuant to Article 50 of the Lisbon Treaty. The withdrawal of the U.K. from the European Union will take effect either on the effective date of the withdrawal agreement or, in the absence of agreement, two years after the U.K. provides a notice of withdrawal pursuant to the E.U. Treaty. The U.K. Prime Minister has stated that notice of withdrawal will be given by the end of March 2017. AsSince the regulatory framework for pharmaceutical products in the U.K. covering quality, safety and efficacy of pharmaceutical products, clinical trials, marketing authorization, commercial sales and distribution of pharmaceutical products is derived from European UnionE.U. directives and regulations, Brexit could materially impact the future regulatory regime which applies to products and the approval of product candidates in the U.K. It remains to be seen how, if at all, Brexit will impact regulatory requirements for product candidates and products in the U.KU.K.
The United Kingdom has a period of a maximum of two years from the date of its formal notification to negotiate the terms of its withdrawal from, and future relationship with, the E.U. If no formal withdrawal agreement is reached between the United Kingdom and the European Union.E.U., then it is expected the United Kingdom's membership of the E.U. will automatically terminate two years after the submission of the notification of the United Kingdom's intention to withdraw from the E.U. Discussions between the United Kingdom and the E.U. focused on finalizing withdrawal issues and transition agreements are ongoing. However, limited progress to date in these negotiations and ongoing uncertainty within the UK Government and Parliament sustains the possibility of the United Kingdom leaving the E.U. on March 29, 2019 without a withdrawal agreement and associated transition period in place, which is likely to cause significant market and economic disruption.
General Data Protection Regulation
The collection, use, disclosure, transfer, or other processing of personal data regarding individuals in the E.U., including personal health data, is subject to the E.U. General Data Protection Regulation, or the GDPR, which became effective on May 25, 2018. The GDPR is wide-ranging in scope and imposes numerous requirements on companies that process personal data, including requirements relating to processing health and other sensitive data, obtaining consent of the individuals to whom the personal data relates, providing information to individuals regarding data processing activities, implementing safeguards to protect the security and confidentiality of personal data, providing notification of data breaches, and taking certain measures when engaging third-party processors. The GDPR also imposes strict rules on the transfer of personal data to countries outside the E.U., including the U.S., and permits data protection authorities to impose large penalties for violations of the GDPR, including potential fines of up to €20 million or 4% of annual global revenues, whichever is greater. The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies, and obtain compensation for
damages resulting from violations of the GDPR. Compliance with the GDPR will be a rigorous and time-intensive process that may increase the cost of doing business or require companies to change their business practices to ensure full compliance.
Pharmaceutical Coverage, Pricing and Reimbursement
In the United States and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Significant uncertainty exists as to the coverage and reimbursement status of products approved by the FDA and other government authorities. Thus, even if a product candidate is approved, sales of the product will depend, in part, on the extent to which third-party payors, including government health programs in the United States such as Medicare and Medicaid, commercial health insurers and managed care organizations, provide coverage, and establish adequate reimbursement levels for, the product. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors are increasingly challenging the prices charged, examining the medical necessity, and reviewing the cost-effectiveness of medical products and services and imposing controls to manage costs. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the approved products for a particular indication.
To secure coverage and reimbursement for any product that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable marketing approvals. Nonetheless, product candidates may not be considered medically necessary or cost effective. A decision by a third-party payor not to cover a product candidate could reduce physician utilization once the product is approved and have a material adverse effect on sales, results of operations and financial condition. Additionally, a payor’spayor's decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Further, one payor’spayor's determination to provide coverage for a drug product does not assure that other payors will also provide coverage and reimbursement for the product, and the level of coverage and reimbursement can differ significantly from payor to payor.
The containment of healthcare costs also has become a priority of federal, state and foreign governments and the prices of drugs have been a focus in this effort. Governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit a company’scompany's revenue generated from the sale of any approved products. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which a company or its collaborators receive marketing approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Outside the United States, ensuring adequate coverage and payment for a product also involves challenges. Pricing of prescription pharmaceuticals is subject to governmental control in many countries. Pricing negotiations with governmental authorities can extend well beyond the receipt of regulatory marketing approval for a product and may require a clinical trial that compares the cost effectiveness of a product to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in commercialization.
In the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that products may be marketed only after a reimbursement price has been
agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular drug candidate to currently available therapies or so called health technology assessments, in order to obtain reimbursement or pricing approval. For example, the European Union provides options for its member states to restrict the range of products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. E.U. European Union member states may approve a specific price for a product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the product on the market. Other member states allow companies to fix their own prices for products, but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. Recently, many countries in the European Union have increased the amount of discounts required on pharmaceuticals and these efforts could continue as countries attempt to manage healthcare expenditures, especially in light of the severe fiscal and debt crises experienced by many countries in the European Union. The downward pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various European Union member states, and parallel trade,i.e., arbitrage between low-priced and high-priced member states, can further reduce prices. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any products, if approved in those countries.
Healthcare Law and Regulation
Healthcare providers and third partythird-party payors play a primary role in the recommendation and prescription of drug products that are granted marketing approval. Arrangements with providers, consultants, third party payors and customers are subject to broadly applicable fraud and abuse, anti-kickback, false claims laws, reporting of payments to physicians and teaching physicians and patient privacy laws and regulations and other healthcare laws and regulations that may constrain business and/or financial arrangements. Restrictions under applicable federal and state healthcare laws and regulations, include the following:
- •
·the federal Anti-Kickback Statute prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, paying, receiving or providing remuneration, directlyor indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid;
·- •
- the federal civil and criminal false claims laws, including the civil False Claims Act and civil monetary penalty laws, which prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false, fictitious or fraudulent or knowingly making, using or causing to be made or used a false record or statement to avoid, decrease or conceal an obligation to pay money to the federal government;
· - •
- the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal laws that prohibit, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters;
· - •
- HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and their regulations, including the Final Omnibus Rule published in January 2013, which impose obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
- •
- the federal false statements statute, which prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services;
· - •
- the Foreign Corrupt Practices Act, or FCPA, which prohibits companies and their intermediaries from making, or offering or promising to make improper payments to non-U.S. officials for the purpose of obtaining or retaining business or otherwise seeking favorable treatment;
- •
- the federal transparency requirements known as the federal Physician Payments Sunshine Act, under the Patient Protection and Affordable Care Act, as amended by the Health Care Education Reconciliation Act, or the Affordable Care Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies to report annually to the Centers for Medicare & Medicaid Services, or CMS, within the United States Department of Health and Human Services, information related to payments and other transfers of value made by that entity to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; and
· - •
- analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to healthcare items or services that are reimbursed by non-governmental third-party payors, including private insurers.
Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’sindustry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Healthcare Reform
A primary trend in the United States healthcare industry and elsewhere is cost containment. There have been a number of federal and state proposals during the last few years regarding the pricing of pharmaceutical and biopharmaceutical products, limiting coverage and reimbursement for drugs and other medical products, government control and other changes to the healthcare system in the United States.
In March 2010, the United States Congress enacted the Affordable Care Act, or the ACA, which, among other things, includes changes to the coverage and payment for products under government health care programs. Among the provisions of the Affordable Care Act of importance to potential drug candidates are:
- •
·an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs, although this fee would not apply to sales of certain products approved exclusively for orphan indications;·- •
- expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a
manufacturer’smanufacturer's Medicaid rebate liability;
- •
- expanded
manufacturers’manufacturers' rebate liability under the Medicaid Drug Rebate Program by increasing the minimum rebate for both branded and generic drugs and revising the definition of“average"average manufacturer price,”" or AMP, for calculating and reporting Medicaid drug rebates on outpatient prescription drug prices and extending rebate liability to prescriptions for individuals enrolled in Medicare Advantage plans;· - •
- addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected;
· - •
- expanded the types of entities eligible for the 340B drug discount program;
· - •
- established the Medicare Part D coverage gap discount program by requiring manufacturers to provide a 50% point-of-sale-discount off the negotiated price of applicable brand drugs to eligible beneficiaries during their coverage gap period as a condition for the
manufacturers’manufacturers' outpatient drugs to be covered under Medicare Part D;· - •
- a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research;
· - •
- the Independent Payment Advisory Board, or IPAB, which has authority to recommend certain changes to the Medicare program to reduce expenditures by the program that could result in reduced payments for prescription drugs. However, the IPAB implementation has been not been clearly defined. PPACA provided that under certain circumstances, IPAB recommendations will become law unless Congress enacts legislation that will achieve the same or greater Medicare cost savings; and
· - •
- established the Center for Medicare and Medicaid Innovation within CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending. Funding has been allocated to support the mission of the Center for Medicare and Medicaid Innovation from 2011 to 2019.
Other legislative changes have been proposed and adopted in the United States since the ACA was enacted. For example, in August 2011,These changes include the Budget Control Act of 2011, which, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2012 through 2021, was unableled to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions ofto Medicare payments to providers of up to 2% per fiscal year which went into effectthat started in April 2013 and will remainstay in effect through 2024 unless additional Congressional action is taken. In January 2013, President Obama signed into lawtaken, and the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several types of providers including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding and otherwise affect the prices we may obtain for any of our product candidates for which we may obtain regulatory approval or the frequency with which any such product candidate is prescribed or used. Further, there have been several recent U.S. congressional inquiries and proposed state and federal legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the costs of drugs under Medicare and reform government program reimbursement methodologies for drug products.
WithSince enactment of the new AdministrationACA, there have been numerous legal challenges and Congress, there will likely be additional legislative changes, includingCongressional actions to repeal and replacementreplace provisions of the law. For example, with enactment of the Tax Cuts and Jobs Act of 2017, which was signed by the President on December 22, 2017, Congress repealed the "individual mandate." The repeal of this provision, which requires most Americans to carry a minimal level of health insurance, will become effective in 2019. According to the Congressional Budget Office, the repeal of the individual mandate will cause 13 million fewer Americans to be insured in 2027 and premiums in insurance markets may rise. Additionally, on January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain
ACA-mandated fees, including the so-called "Cadillac" tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices. Further, the Bipartisan Budget Act of 2018, among other things, amends the ACA, effective January 1, 2019, to increase from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and to close the coverage gap in most Medicare drug plans, commonly referred to as the "donut hole".
The Trump Administration has also taken executive actions to undermine or delay implementation of the ACA. Since January 2017, President Trump has signed two Executive Orders designed to delay the implementation of certain provisions of the ACA. In January 2017, Congress voted to adopt a budget resolution for fiscal year 2017,ACA or the Budget Resolution, that authorizes the implementation of legislation that would repeal portionsotherwise circumvent some of the ACA. The Budget Resolution is not a law, however, it is widely viewed as the first step toward the passage of legislation that would repeal certain aspects ofrequirements for health insurance mandated by the ACA. Further, on January 20, 2017, President Trump signed anOne Executive Order directingdirects federal agencies with authorities and responsibilities under the ACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the ACA that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. The second Executive Order terminates the cost-sharing subsidies that reimburse insurers under the ACA. Several state Attorneys General filed suit to stop the administration from terminating the subsidies, but their request for a restraining order was denied by a federal judge in California on October 25, 2017. In March 2017,addition, CMS has recently proposed regulations that would give states greater flexibility in setting benchmarks for insurers in the United States Houseindividual and small group marketplaces, which may have the effect of Representatives introduced a Budget Resolution to replacerelaxing the essential health benefits required under the ACA will a tax credit based system. It is uncertain if these changes will be approved byfor plans sold through such marketplaces. Further, on June 14, 2018, U.S. Court of Appeals for the United States Senate orFederal Circuit ruled that the President.
federal government was not required to pay more than $12 billion in ACA risk corridor payments to third-party payors who argued were owed to them. The President and congressional leaders have expressed interesteffects of this gap in repealing certain ACA provisions and
replacing them with alternatives that may be less costly and provide state Medicaid programs and private health plans more flexibility. It is possible that these repeal and replacement initiatives, if enacted into law, could ultimately result in fewer individuals having health insurance coverage or in individuals having insurance coverage with less generous benefits. The scope of potential future legislation to repeal and replace ACA provisions is highly uncertain in many respects, and it is possible that somereimbursement on third-party payors, the viability of the ACA provisions that generallymarketplace, providers, and potentially our business, are not favorableyet known.
Further, there have been several recent U.S. congressional inquiries and proposed federal and proposed and enacted state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the costs of drugs under Medicare and reform government program reimbursement methodologies for drug products. For example, there have been several recent U.S. congressional inquiries and proposed federal and proposed and enacted state legislation designed to, among other things, bring more transparency to drug pricing, review the research-based pharmaceutical industryrelationship between pricing and manufacturer patient programs, reduce the costs of drugs under Medicare and reform government program reimbursement methodologies for drug products. At the federal level, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. For example, on May 11, 2018, the Administration issued a plan to lower drug prices. Under this blueprint for action, the Administration indicated that the Department of Health and Human Services (HHS) will: take steps to end the gaming of regulatory and patent processes by drug makers to unfairly protect monopolies; advance biosimilars and generics to boost price competition; evaluate the inclusion of prices in drug makers' ads to enhance price competition; speed access to and lower the cost of new drugs by clarifying policies for sharing information between insurers and drug makers; avoid excessive pricing by relying more on value-based pricing by expanding outcome-based payments in Medicare and Medicaid; work to give Part D plan sponsors more negotiation power with drug makers; examine which Medicare Part B drugs could also be repealed along with ACA coverage expansion provisions. However, at this timenegotiated for a lower price by Part D plans, and improving the coverage expansion provisionsdesign of the ACA appear most likelyPart B Competitive Acquisition Program; update Medicare's drug-pricing dashboard to increase transparency; prohibit Part D contracts that include "gag rules" that prevent pharmacists from informing patients when they could pay less out-of-pocket by not using insurance; and require that Part D plan members be repealedprovided with an annual statement of plan payments, out-of-pocket spending, and replaced.drug price increases.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional health care authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other health care programs. These measures could reduce the ultimate demand for our products, once approved, or put pressure on our product pricing. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
Employees
As of March 15, 2017,January 31, 2019, we had 59110 employees, 381 employee was based in Dublin, Ireland, 39 of our employees are located in Vienna, Austria and 2170 of our employees werelocated in the U.S., with 47 located in King of Prussia, Pennsylvania.Pennsylvania and the remaining 23 employees in the field.
Our employees in Austria are subject to the collective bargaining agreement of the chemical industry. This is an annual agreement between the employer representatives and the trade union of an industry. It defines conditions of employment, such as minimum wages, working hours and conditions, overtime payments, vacations and other matters. We do not have a works council, which would require employee representatives on our supervisory board.
We consider our relations with our employees to be good.
Our Corporate Information
We wereOn March 1, 2017, Nabriva Ireland was incorporated in Ireland under the name Hyacintho 2 plc, and was renamed to Nabriva Therapeutics plc on April 10, 2017, in order to effectuate the change of the jurisdiction of incorporation of the ultimate parent company of the Nabriva Group from Austria to Ireland. Nabriva Ireland replaced Nabriva Austria as the ultimate parent company on June 23, 2017, following the conclusion of a tender offer in which holders of the outstanding share capital of Nabriva Austria exchanged their holdings for ordinary shares, $0.01 nominal value per share, of Nabriva Ireland. The ordinary shares of Nabriva Ireland were issued on a one-for-ten basis to the holders of the Nabriva Austria common shares and on a one-for-one basis to the holders of the Nabriva Austria American Depositary Shares, or Nabriva Austria ADSs. On June 26, 2017, the ordinary shares of Nabriva Ireland began trading on the Nasdaq Global Market under the symbol "NBRV," the same symbol under which the Nabriva Austria ADSs were previously traded. This transaction was accounted for as a merger between entities under common control; accordingly, the historical financial statements of Nabriva Austria for periods prior to this transaction are considered to be the historical financial statements of Nabriva Ireland. Our executive offices are located at 25-28 North Wall Quay IFSC, Dublin 1, Ireland, and our telephone number is +353 1 649 2000.
The predecessor of Nabriva Ireland, Nabriva Austria, was incorporated in Austria as a spin-off from Sandoz GmbH in October 2005 in Austria under the name Nabriva Therapeutics Forschungs GmbH, a limited liability company organized under Austrian law as a spin-off from Sandoz GmbH and commenced operations in February 2006. In 2007, weNabriva Austria transformed into a stock corporation ((Aktiengesellschaft) under the name Nabriva Therapeutics AG. We are incorporatedOn October 19, 2017, Nabriva Austria was converted into a private limited liability company under the laws of the Republic of AustriaAustrian law and registered at the Commercial Register of the Commercial Court of Vienna. Our executive offices are located at Leberstrasse 20, 1110 Vienna, Austria, and our telephone number is +43 (0)1 740 930.renamed Nabriva Therapeutics GmbH.
Our U.S. operations are conducted by our wholly-owned subsidiary Nabriva Therapeutics US, Inc., a Delaware corporation established in August 2014 and located at 1000 Continental Drive, Suite 600, King of Prussia, PA 19406.
Our website address is www.nabriva.com. The information contained on, or that can be accessed from, our website does not form part of this Annual Report. Our agent for service of process in the United States is CT Corporation System, 111 Eighth Avenue, New York, New York 10011.
On March 24, 2017, our supervisory board authorized management to pursue a plan for the redomiciliation of our ultimate parent company from Austria to Ireland. We are working with our advisors to structure and implement such a redomiciliation plan. We currently anticipate that the plan, which will require further approval of the supervisory board, will be presented to our shareholders for their consideration during 2017. We may abandon our redomiciliation plan at any time and there can be no assurance that we will redomicile from Austria to Ireland during 2017, or at all.
Available Information
We make available free of charge through our website our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Previously, as a foreign private issuer, we filed our Annual Report on Form 20-F and furnished information on Form 6-K. We make these reports available through our website as soon as reasonably practicable after we electronically file such reports with, or furnish such reports to, the SEC. You can find, copyPreviously, as a foreign private issuer, we filed our Annual Report on Form 20-F and inspectfurnished information we file at the SEC’s public reference room, which is located at 100 F Street, N.E., Room 1580, Washington, DC 20549. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the SEC’s public reference room.on Form 6-K. You can review our electronically filed reports and other information that we file with the SEC on the SEC’sSEC's web site at http://www.sec.gov. We also make available, free of charge on our website, the reports filed with the SEC by our supervisory board members, management board member, senior managersexecutive officers, directors and 10% shareholders pursuant to Section 16 under the Exchange Act as soon as reasonably practicable after copies of those filings are provided to us by those persons. The information contained on, or that can be accessaccessed through, our website is not a part of or incorporated by reference in this Annual Report on Form 10-K.
You should consider carefully the risks and uncertainties described below, together with all of the other information in this Annual Report on Form 10-K. If any of the following risks are realized, our business, financial condition, results of operations and prospects could be materially and adversely affected. The risks described below are not the only risks facing us. Risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, financial condition, results of operations and/or prospects.
Risks Related to Our Financial Position and Need for Additional Capital
We have incurred significant losses since our inception. We expect to incur losses for at least the next several years and may never generate profits from operations or maintain profitability.
Since inception, we have incurred significant operating losses. Our net losses were $114.8 million for the year ended December 31, 2018, $74.4 million for the year ended December 31, 2017 and $54.9 million for the year ended December 31, 2016, $47.0 million for the year ended December 31, 2015 and $14.2 million for the year ended December 31, 2014.2016. As of December 31, 2016,2018, we had an accumulated lossesdeficit of $204.8$394.0 million. To date, we have financed our operations primarily through the sale of our equity securities, loans, including our American Depositary Shares, or ADSs, and private placements of our common shares, convertible loans, and research and development support from governmental grants and loans. We have devoted substantially allmost of our efforts to research and development, including clinical trials. We have not completed development of any drugs. We expect to continue to incur significant expenses and increasing operating losses for at least the next several years.years, including in connection with our regulatory approval efforts and commercialization of lefamulin and CONTEPO. The net losses we incur may fluctuate significantly from quarter-to-quarter and year-to-year.
We expect to continue to invest in critical pre-commercialization activities prior to potentially receiving marketing approval and making lefamulin and CONTEPO available to patients.
We anticipate thatinitiated our expenses will increase substantially as we progress our two internationalfirst Phase 3 global, pivotal clinical trialstrial of lefamulin, which we refer to as Lefamulin Evaluation Against Pneumonia 1, or LEAP 1, in September 2015, and we initiated our lead product candidate,second Phase 3 global, pivotal clinical trial of lefamulin, which we refer to as Lefamulin Evaluation Against Pneumonia 2, or LEAP 2, in April 2016. In September 2017, we announced positive topline results for LEAP 1. In May 2018, we announced positive topline results from LEAP 2. LEAP 2 evaluated the treatmentsafety and efficacy of five days of oral lefamulin compared to 7 days of oral moxifloxacin in adult patients with moderate community-acquired bacterial pneumonia, or CABP. We initiatedsubmitted two
NDAs for marketing approval of lefamulin for the firsttreatment of these clinical trials, which we refer to as LEAP 1,CABP in September 2015 and initiatedadults in the second trial, which we refer to as LEAP 2,United States in April 2016. If the results of these two trials are favorable, including achievement of the primary efficacy endpoints of the trials, weDecember 2018. We also expect to submit applications fora marketing approvalauthorization application, or MAA, for lefamulin for the treatment of CABP in both the United States andadults in Europe in 2018.the first half of 2019. We also continue to characterize the clinical pharmacology of lefamulin. If we obtain marketing approval of lefamulin for CABP or another indication, we also expect to incur significant additional sales, marketing, distribution and manufacturing expenses.
In June 2016, the first patient was enrolled by Zavante in its pivotal ZTI-01 Efficacy and Safety Study of CONTEPO, which we refer to as the ZEUS Study. In April 2017, Zavante announced positive topline results of the ZEUS Study. The ZEUS Study was a multicenter, randomized, parallel-group, double-blind Phase 2/3, pivotal clinical trial designed to evaluate safety, tolerability, efficacy and pharmacokinetics of seven days of treatment, or up to 14 days of treatment for patients with concurrent bacteremia, with CONTEPO compared to piperacillin-tazobactam, or PIP-TAZ, in the treatment of hospitalized adults with cUTI or acute pyelonephritis, or AP. We submitted an NDA for marketing approval of CONTEPO for the treatment of cUTI in adults in the United States, utilizing the FDA's 505(b)(2) pathway, in October 2018. In June 2018, Zavante initiated a Phase 1, non-comparative, open-label study of the pharmacokinetics and safety of a single dose of CONTEPO in pediatric subjects less than 12 years of age receiving standard-of-care antibiotic therapy for proven or suspected infection or peri-operative prophylaxis. We anticipate completing enrollment in this study in late 2020. We also intend to continue to characterize the clinical pharmacology of CONTEPO. If we obtain marketing approval of CONTEPO for cUTI, including AP, or another indication, we also expect to incur significant additional sales, marketing, distribution and manufacturing expenses.
On July 24, 2018, we completed our Acquisition of Zavante. Upfront consideration in connection with the Acquisition was 8,152,092 of our ordinary shares, including the 815,186 ordinary shares that are issuable upon release of the Holdback Shares subject to the terms of the Merger Agreement. Pursuant to the Merger Agreement, former Zavante stockholders are also entitled to receive from us up to $97.5 million in contingent consideration, consisting of the Approval Milestone Payment and the Net Sales Milestone Payment, subject to the terms and conditions of the Merger Agreement. In connection with the Acquisition, we assumed certain payment obligations under the Stock Purchase Agreement and Zavante manufacturing agreements acquired in the Acquisition. See "—Risks Related to Our Acquisition of Zavante—We may fail to realize the anticipated benefits of our Acquisition of Zavante, those benefits may take longer to realize than expected, and we may encounter significant integration difficulties."
In addition, our expenses will increase if and as we:
- •
·initiate or continue the research and development of lefamulin and CONTEPO for additional indications and of our other product candidates;·- •
- seek to
discover anddevelop additional product candidates;· - •
- seek marketing approval for any product candidates that successfully complete clinical development;
·ultimately establish - •
- are required by the FDA, EMA or other regulators to conduct additional clinical trials prior to or after approval;
- •
- continue to build a medical affairs, sales, marketing and distribution infrastructure and scale up manufacturing capabilities to commercialize any product candidates for which we receive marketing approval;
· - •
- in-license or acquire other products, product candidates or technologies;
· - •
- maintain, expand and protect our intellectual property portfolio;
- •
- expand our physical presence in the United
States;States andIreland;
· - •
- incur additional debt;
- •
- establish and expand manufacturing arrangements with third parties; and
- •
- add operational, financial and management information systems and personnel, including personnel to support our product development, our operations as a larger company following the Acquisition and our operations as a public company
andin addition to our planned future commercialization efforts.
Our ability to generate profits from operations, and to become and remain profitable, depends on our ability to successfully develop and commercialize drugs that generate significant revenue. Based on our current plans, we do not expect to generate significant revenue unless and until we obtain marketing approval for, and commercialize,
lefamulin. lefamulin and CONTEPO. We do not expect to obtain marketing approval for CONTEPO before April 2019 or for lefamulin before August 2019, if at all. This will require us to be successful in a range of challenging activities, including:
- •
·completing enrollment for our Phase 3 clinical trials of lefamulin for the treatment of CABP and completing both trials as and when we expect;·obtaining favorable results from our Phase 3 clinical trials of lefamulin for the treatment of CABP;·subject to obtaining favorable results from our Phase 3 clinical trials, applying for andobtaining marketing approval forlefamulin;lefamulin and CONTEPO;
·establishing- •
- expanding medical affairs, sales, marketing and distribution capabilities to effectively market and sell lefamulin and CONTEPO in the United States;
· - •
- establishing and maintaining collaboration, distribution or other marketing arrangements with third parties to commercialize lefamulin in markets outside the United States;
· - •
- protecting our rights to our intellectual property portfolio related to
lefamulin;lefamulin and CONTEPO;
·contracting - •
- establishing and maintaining arrangements for the manufacture of and obtaining commercial quantities of
lefamulin;lefamulin andCONTEPO; and
· - •
- negotiating and securing adequate reimbursement from third-party payors for
lefamulin.lefamulin and CONTEPO.
We may never succeed in these activities and, even if we do, may never generate revenues that are significant enough to generate profits from operations. Even if we do generate profits from operations, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to generate profits from operations, and to become and remain profitable, would decrease the value of our company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or continue our operations. A decline in the value of our company could also cause our shareholders to lose all or part of their investment.
We will need substantial additional funding. If we are unable to raise capital when needed or on acceptable terms, we could be forced to delay, reduce or eliminate our product development programs or future commercialization efforts.
We expect our research and development, commercialization and other expenses to increase substantiallycontinue to incur substantial costs in connection with our ongoing activities, particularly as we continue development of and potentially seek marketing approval for lefamulin, CONTEPO and, possibly, other product candidates and continue our research activities. Our expenses will increase if we suffer any regulatory delays in our Phase 3or are required to conduct additional clinical program for lefamulin for CABP, including delays in enrollment of patients.trials to satisfy regulatory requirements. If we obtain marketing approval for lefamulin, CONTEPO or any other product candidate that we develop, in-license or acquire, we expect to incur significant commercialization expenses related to product sales, marketing, distribution and manufacturing.
Furthermore, we expect to continue to incur additional costs to service our current debt and any potential future draws on the Loan Agreement (as defined below) and costs associated with operating as a public company.company and as a company with a commercial rather than a research and development
focus. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts.
We expect that our existing cash, cash equivalents and short-term investments as of December 31, 2018, proceeds from the sale of ordinary shares under the our ATM Agreement (as defined below) of $5.9 million in the first quarter of 2019, as well as a $1.5 million milestone payment received in February 2019 under our license agreement with Sinovant Sciences, Ltd., and research premiums from the Austrian government for our qualified research and development expenditures, will be sufficient to enable us to fund our operating expenses, debt service obligations and capital expenditure requirements at least into the second quarter of 2018 and to obtain top-line data for both our Phase 3 clinical trials of lefamulin.2020. We have based these estimatesthis estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect. These estimates assume,This estimate assumes, among other things, that we do not obtain any additional funding through grants and clinical trial support, collaboration agreements, equity or debt financings.financings, including additional advances under the Loan Agreement with Hercules. We may be eligible to borrow up an additional $50.0 million under our Loan Agreement with Hercules if we achieve specified regulatory and product revenue milestones, including $10.0 million that we will be eligible to borrow upon the approval by the FDA of an NDA for lefamulin and $5.0 million that we will be eligible to borrow upon the approval by the FDA of an NDA for CONTEPO.
We expect to seek additional funding in future periods for purposes of investment in our commercial and medical affairs organization, as well as investing in our supply chain in an effort to enhance the potential commercial launch of lefamulin and CONTEPO.
Our future capital requirements will depend on many factors, including:
- •
·the progress, costs and results of our ongoing Phase 3 clinical trials of lefamulin;·the costs and timing of process development and manufacturing scale-up activities associated withlefamulin;lefamulin and CONTEPO;
·- •
- the costs, timing and outcome of regulatory review of
lefamulin;lefamulin and CONTEPO;
· - •
- the costs of commercialization activities for lefamulin and CONTEPO if we receive, or expect to receive, marketing approval, including the costs and timing of establishing product sales, marketing, distribution and outsourced manufacturing
capabilities;capabilities, including the costs of building finished product inventory and its components in preparation of initial marketing of lefamulin and CONTEPO;
· - •
- subject to receipt of marketing approval, revenue received from commercial sales of
lefamulin;lefamulin and CONTEPO;
· - •
- the costs of developing lefamulin and CONTEPO for the treatment of additional indications;
· - •
- our ability to establish collaborations on favorable terms, if at all;
· - •
- the scope, progress, results and costs of product development of
BC-7013 andany other product candidates that we may develop;· - •
- the extent to which we in-license or acquire rights to other products, product candidates or technologies;
· - •
- the costs of preparing, filing and prosecuting patent applications, maintaining and protecting our intellectual property rights and defending against intellectual property-related claims;
· - •
- the continued availability of Austrian governmental grants;
· - •
- the rate of the expansion of our physical presence in the United
States;States and Ireland;
- •
- interest expense on our debt and the eventual repayment of our debt obligations;
- •
- the costs of operating as a company with a commercial rather than a research and development focus; and
- •
- the costs of operating as a public company in the United States.
Conducting clinical trials is a time consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales. Our commercial revenues, if any, will be derived from sales of lefamulin, CONTEPO or any other products that we successfully develop, in-license or acquire, none of which we expect to be commercially available for several years,before April 2019, if at all. In addition, if approved, lefamulin, CONTEPO or any other product candidate that we develop, in-license or acquire may not achieve commercial success. Accordingly, we will need to obtain substantial additional financing to achieve our business objectives.
Adequate additional financing may not be available to us on acceptable terms, or at all. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe that we have sufficient funds for our current or future operating plans.
Raising additional capital may cause dilution to our security holders, restrict our operations or require us to relinquish certain rights to our technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of equity offerings, debt financings, collaborations, and funding from local and international government entities and non-government organizations in the disease areas addressed by our product candidates and marketing, distribution or licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a security holder. DebtAdditional debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. In addition, debt service obligations under any debt financings may limit the availability of our cash for other purposes, and we may be unable to make interest payments or repay the principal of such debt financings when due.
On March 16, 2018, we entered into a Controlled Equity OfferingSM Sales Agreement with Cantor Fitzgerald & Co., or Cantor Fitzgerald, as agent, pursuant to which we may offer and sell ordinary shares, nominal value $0.01 per share, for aggregate gross sale proceeds of up to $50,000,000 from time to time through Cantor Fitzgerald under an "at-the-market" offering program. As of December 31, 2018, $25.8 million remained available for sale under our ATM Agreement. As of March 1, 2019, $20.2 million remained available for sale under the ATM Agreement. If a large number of our ordinary shares is sold in the public market after they become eligible for sale or if we make additional sales under our "at-the-market" offering program, the sales could cause dilution to our security holders, reduce the trading price of our ordinary shares and impede our ability to raise future capital.
In addition, in connection with the closing of the Acquisition, we issued 7,336,906 of our ordinary shares to former Zavante stockholders as initial upfront consideration. An additional 815,186 of ordinary shares will be issued to former Zavante Stockholders upon release of the Holdback Shares, subject to reduction in respect of certain indemnification and other obligations pursuant to the Merger Agreement. The shares that were issued in connection with the closing of the Acquisition are able to be freely sold in the public market, subject to any requirements and restrictions, including any applicable volume limitations, imposed by Rule 144 under the Securities Act. While the Holdback Shares will be restricted as a result of securities laws at the time of issuance, following expiration of applicable holding periods, the Holdback Shares will also be able to be freely sold in the public market, subject to any requirements and restrictions, including any applicable volume limitations, imposed by Rule 144 under the Securities Act. In addition, the Merger Agreement provides that we may issue up
to an additional $97.5 million in our ordinary shares to former Zavante stockholders upon the achievement of specified regulatory and commercial milestones in the future and obligates us to provide registration rights with respect to the registration for resale of such additional ordinary shares that may become issuable upon the achievement of such milestones. The issuance of our ordinary shares to satisfy the milestone payments will cause dilution to our equity holders, and the sale or resale of these shares in the public market, or the market's expectation of such sales, may result in an immediate and substantial decline in our stock price. Such a decline would adversely affect our investors and also might make it difficult for us to sell equity securities in the future at a time and at a price that we deem appropriate.
If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Our limited operating history may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
Our operations to date have been limited to organizing and staffing our company, developing and securing our technology, raising capital, and undertaking preclinical studies and clinical trials of our product candidates, and preparing and filing NDAs for our product candidates. We have not yet demonstrated our ability to successfully complete development of any product candidates, obtain marketing approvals, manufacture a commercial scale product, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history.
In addition, as a new business, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors. WeAlso, we may encounter delays or difficulties in our efforts to, or fail to, successfully integrate the operations of Zavante into our business and CONTEPO into our business strategy. Moreover, we will need to transition from a company with a research and development focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
Our level of indebtedness and debt service obligations could adversely affect our financial condition and may make it more difficult for us to fund our operations.
On December 20, 2018, we entered into a Loan and Security Agreement, or the Loan Agreement, with Hercules Capital, Inc., as administrative agent, collateral agent and lender, pursuant to which an aggregate principal amount of up to $75.0 million is available to us. As of December 31, 2018, we have drawn down on the initial term loan advance under the Loan Agreement of $25.0 million.
All obligations under the Loan Agreement are secured by substantially all of our personal property, intellectual property and other assets owned or later acquired. This indebtedness may create additional financing risk for us, particularly if our business or prevailing financial market conditions are not conducive to paying off or refinancing our outstanding debt obligations at maturity. This indebtedness could also have important negative consequences, including:
- •
- the need to repay our indebtedness by making payment of interest only initially and then interest and principal, which will reduce the amount of funds available to finance our operations, our research and development efforts and our general corporate activities; and
- •
- our failure to comply with the restrictive covenants in the Loan Agreement or the occurrence of an event that has a material adverse effect on our business, operations, properties, assets, condition, our ability to pay any amounts due, the collateral securing our obligations under the Loan Agreement or the ability of Hercules to enforce any of its rights under the Loan Agreement could result in an event of default that, if not cured or waived, would accelerate our obligation to repay this indebtedness, and the lenders could seek to enforce its security interest in the assets securing such indebtedness.
To the extent additional debt is added to our current debt levels, the risks described above could increase.
We may not have cash available to us in an amount sufficient to enable us to make interest or principal payments on our indebtedness when due.
Failure to satisfy our current and future debt obligations under the Loan Agreement could result in an event of default and, as a result, the lenders under the Loan Agreement could accelerate all of the amounts due. In the event of an acceleration of amounts due under the Loan Agreement as a result of an event of default, we may not have sufficient funds or may be unable to arrange for additional financing to repay our indebtedness. In addition, the lenders could seek to enforce their security interests in the assets securing such indebtedness.
We are subject to certain restrictive covenants which, if breached, could have a material adverse effect on our business and prospects.
The Loan Agreement imposes operating and other restrictions on us. Such restrictions will affect, and in many respects limit or prohibit, our ability and the ability of any future subsidiary to, among other things:
- •
- declare dividends or redeem or repurchase equity interests;
- •
- incur additional indebtedness and liens;
- •
- make loans and investments;
- •
- engage in mergers, acquisitions and asset sales;
- •
- certain transactions with affiliates
- •
- undergo a change in control;
- •
- add or change business locations; and
- •
- settle in cash potential milestone payment obligations that may become payable by us in the future to former security holders of Zavante.
We are also required to satisfy certain financial covenants, including an obligation to maintain specified minimum amounts of cash and cash equivalents in accounts pledged to Hercules. These restrictive covenants may prevent us from undertaking an action that we feel is in the best interests of our business. In addition, if we were to breach any of these restrictive covenants, Hercules could accelerate our indebtedness under the Loan Agreement or enforce its security interest against our assets, either of which would materially adversely affect our ability to continue to operate our business.
We have relied on, and expect to continue to rely on, certain government grants and funding from the Austrian government. Should these funds cease to be available, or our eligibility be reduced, or if we are required to repay any of these funds, this could impact our ongoing need for funding and the timeframes within which we currently expect additional funding will be required.
As a company that carriescarried out extensive research and development activities, we benefithave benefited from the Austrian research and development support regime, under which we arewere eligible to receive a research premium from the Austrian government equal to 12% (10%14% (12% for the fiscal years 2016 and 2017 and 10%, in the case of fiscal years prior to 2016) of a specified research and development cost base. Qualifying expenditures largely comprisecomprised research and development activities conducted in Austria, however, the research premium iswas also available for certain related third-party expenses with additional limitations. We received research premiums of $4.3$4.7 million for the year ended December 31, 20152017 and $1.4$5.9 million for the year ended December 31, 2014.2016. We also expect to receive ahave not received any research premium for our qualified 2016 expenditures. However,2018 expenditures as of December 31, 2018. As we increase our personnel and expand our business outside of Austria, we may not be able to continue to claim research premiums to the same extent as we have in previous years or at all, as some research and development activities may no longer be considered to occur in Austria. As research premiums that have been paid out already may be audited by the tax authorities, there is a risk that parts of the submitted cost base may not be considered as eligible and therefore repayments may have to be made.
The intended efficiency of our corporate structure depends on the application of the tax laws and regulations in the countries where we operate, and we may have exposure to additional tax liabilities or our effective tax rate could change, which could have a material impact on our results of operations and financial position.
As a company with international operations, we are subject to income taxes, as well as non-income based taxes, in both the United States and various foreign jurisdictions. We have designed our corporate structure, the manner in which we develop and use our intellectual property, and our intercompany transactions between our subsidiaries in a way that is intended to enhance our operational and financial efficiency. The application of the tax laws and regulations of various countries in which we operate and to our global operations is subject to interpretation. We also must operate our business in a manner consistent with our corporate structure to realize such efficiencies. The tax authorities of the countries in which we operate may challenge our methodologies for valuing developed technology or for transfer pricing. If, for one or more of these reasons, tax authorities determine that the manner in which we operate results in our business not achieving the intended tax consequences, our effective tax rate could increase and harm our financial position and results of operations.
A change in the tax law in the jurisdictions in which we do business, including an increase in tax rates, an adverse change in the treatment of an item of income or expense, a decrease in tax rates in a jurisdiction in which we have significant deferred tax assets, or a new or different interpretation of applicable tax law, could result in a material increase in tax expense.
Risks Related to Product Development and Commercialization
We depend heavily on the success of, our lead product candidate, lefamulin, which we are developing for CABP and potentially other indications.indications, and CONTEPO, which we are developing for cUTI, including AP. If we are unable to complete our Phase 3 clinical program for lefamulin for CABP as and when expected and obtain marketing approvals for lefamulin or CONTEPO, or if thereafter we fail to commercialize lefamulin or CONTEPO or experience significant delays in doing so, our business will be materially harmed.
We have invested a significant portion of our efforts and financial resources in the development of lefamulin.lefamulin and, more recently, in CONTEPO. There remains a significant risk that we will fail to
successfully develop lefamulin for CABP or any other indication or CONTEPO for cUTI or any other indication. Based on our estimates regarding patient enrollment,
In September 2017, we announced positive topline results for LEAP 1. In May 2018, we announced positive topline results from LEAP 2. We submitted two new drug applications, or NDAs, for marketing approval of lefamulin for the treatment of CABP in adults in the United States in December 2018. We also expect to have top-line data from LEAP 1submit a marketing authorization application, or MAA, for lefamulin for the treatment of CABP in the third quarter of 2017. With respect to LEAP 2, based on current projections, we expect to complete patient enrollmentadults in the fourth quarter of 2017, and we anticipate receiving top-line data for LEAP 2Europe in the first quarterhalf of 2019. In mid-2018, we initiated a Phase 1, non-comparative, open-label study of the pharmacokinetics and safety of a single dose of IV lefamulin in pediatric subjects 12 to 18 years of age.
In June 2016, Zavante initiated the ZEUS Study. In April 2017, Zavante announced positive topline results of the ZEUS Study. We submitted an NDA for marketing approval of CONTEPO for the treatment of cUTI in adults in the United States, utilizing the FDA's 505(b)(2) pathway, in October 2018. Our abilityIn June 2018, Zavante initiated a phase 1, non-comparative, open-label study of the pharmacokinetics and safety of a single dose of CONTEPO in pediatric subjects less than 12 years of age receiving standard-of-care antibiotic therapy for proven or suspected infection or peri-operative prophylaxis. We anticipate completing enrollment in this study in late 2020. We also intend to meet our target timing will depend on our enrollment rates. A significant delay in enrollment would result in delayscontinue to our development timeline and additional development costs beyond what we have budgeted.characterize the clinical pharmacology of CONTEPO.
If we ultimately obtain favorable results from our Phase 3 clinical program formarketing approval of lefamulin for CABP, or any other indication, and CONTEPO for cUTI, including AP, or any other indication, we do notalso expect to submit applications forincur significant additional sales, marketing, approval for lefamulin for this indication until 2018.distribution and manufacturing expenses.
Our ability to generate product revenues which may not occur for several years, if ever, will depend heavily on our obtaining marketing approval for and commercializing lefamulin.lefamulin and CONTEPO when and as we expect, and our ability to successfully integrate Zavante into our business and CONTEPO into our business strategy. The success of lefamulin and CONTEPO will depend on a number of factors, including the following:
- •
·completing our ongoing Phase 3 clinical trials asestablishing andwhen expected;·obtaining favorable results from clinical trials;·makingmaintaining arrangements with third-party manufacturers for commercial supply and receiving regulatory approval of our manufacturing processes and our third-partymanufacturers’manufacturers' facilities from applicable regulatory authorities;·- •
- receipt of marketing approvals from applicable regulatory authorities for lefamulin for the treatment of
CABP;CABP and CONTEPO for the treatment of cUTI, including AP;
· - •
- launching commercial sales of lefamulin and CONTEPO, if and when approved,
whether alone orin collaboration with third parties;· - •
- acceptance of lefamulin and CONTEPO, if and when approved, by patients, the medical community and
third- partythird-party payors;· - •
- effectively competing with other therapies;
· - •
- maintaining a continued acceptable safety profile of lefamulin and CONTEPO following approval;
· - •
- obtaining and maintaining patent and trade secret protection and regulatory exclusivity; and
· - •
- protecting our rights in our intellectual property portfolio.
Successful development of lefamulin and CONTEPO for the treatment of additional indications, if any, or for use in other patient populations and our ability, if it is approved, to broaden the labellabels for lefamulin and CONTEPO will depend on similar factors.
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize lefamulin for CABP or for
any additional indications,other indication or CONTEPO for cUTI, including AP or for any other indication, which would materially harm our business.
If clinical trials of lefamulin, CONTEPO or any of our other product candidates fail to demonstrate safety and efficacy to the satisfaction of the U.S. Food and Drug Administration, or FDA, regulatory authorities in the European Union, or other regulatory authorities or do not otherwise produce favorable results, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of lefamulin, CONTEPO or any other product candidate.
Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must complete preclinical development and early clinical trials, including Phase 1 clinical trials, in addition to extensive later-stage Phase 3 clinical trials, to demonstrate the safety and efficacy of our product candidates in humans. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. The outcome of preclinical testing and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. The design of a clinical trial can determine whether its results will support approval of a product, and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced or completed. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products.
We In connection with the ZEUS Study in which CONTEPO met the primary endpoint of statistical non-inferiority versus piperacillin/tazobactam, Zavante conducted a post-hoc primary efficacy analysis of CONTEPO using results of blinded pulsed-field gel electrophoresis molecular typing of urinary tract pathogens. Regulatory authorities typically give greater weight to results from pre-specified analyses and less weight to results from post-hoc, retrospective analyses. While we believe this post-hoc analysis is illustrative information, the FDA may ultimately have not completed any clinical trials of lefamulin specifically for CABP. Our completed Phase 2 clinical trial evaluated lefamulin in patients with acute bacterial skin and skin structure infections, or ABSSSI. Our Phase 1 clinical trials evaluated lefamulin in healthy subjects to obtain tolerance data and to understand the absorption and distribution of lefamulin in the blood and target tissues, evaluate the metabolism and elimination route of lefamulin and obtain safety and tolerability data to help predict safe and effective doses of lefamulin for the treatment of patients. In addition, we are using a different intravenous, or IV, formulationinterpretation of lefamulin for our Phase 3 clinical trials for CABP than we used in our Phase 2 clinical trial for ABSSSI. We have only evaluated this new IV formulation of lefamulin, a sterile saline solution buffered by a citrate salt, in Phase 1 clinical trials. Because of these and other factors, the resultsany of our completed clinical trialsdata that may not predict
success in our Phase 3 clinical trials of lefamulin for CABP. Although we believe that the collective data from prior trials and our preclinical studies provide support for concluding that lefamulin is well suited for treatment of CABP, we may fail to obtain favorable results in our Phase 3 clinical trials of lefamulin for CABP. If the results of our Phase 3 clinical trials are not favorable, including failure to achieve the primary efficacy endpoints of the trials, we may need to conduct additional clinical trials at significant cost or altogether abandon development of lefamulin for CABP and potentially other indications.be based on such post-hoc analysis.
If we are required to conduct additional clinical trials or other testing or studies of lefamulin, CONTEPO or any other product candidate that we develop beyond those that we contemplate,contemplate; if we are unable to successfully complete our clinical trials or other testing or studies; if the results of these trials, tests or testsstudies are not positive or are only modestly positive orpositive; if there are safety concerns,concerns; or if they are otherwise not acceptable to the FDA, we may:
- •
·be delayed in obtaining marketing approval for our product candidates;·- •
- not obtain marketing approval at all;
· - •
- obtain approval for indications or patient populations that are not as broad as intended or desired;
· - •
- obtain approval with labeling that includes significant use or distribution restrictions or safety warnings, including boxed warnings;
· - •
- be subject to additional post-marketing testing requirements or restrictions;
or· - •
- have the product removed from the market after obtaining marketing
approval.approval; or - •
- need to raise capital before we otherwise would or on terms less favorable to us.
The occurrence of any of the developments listed above could materially harm our business, financial condition, results of operations and prospects.
If we experience any of a number of possible unforeseen events in connection with our Phase 3 clinical trials of lefamulin for CABP or other clinical trials, the potential marketing approval or commercialization of lefamulin, CONTEPO or other product candidates could be delayed or prevented.
We may experience numerous unforeseen events during, or as a result of, our Phase 3 clinical trials of lefamulin for CABPand CONTEPO or other clinical trials we conductproduct candidates that could delay or prevent our ability to receive marketing approval or commercialize lefamulin, CONTEPO or our other product candidates, including:
- •
·clinical trialsof lefamulin or our other product candidatesmay produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs;·- •
- the number of patients required for clinical trials
of lefamulin for CABP, lefamulin for other indications or our other product candidatesmay be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials at a higher rate than we anticipate;·we may be unable to enroll a sufficient number of patients in our Phase 3 clinical trials of lefamulin for CABP or other clinical trials we conduct to ensure adequate statistical power to detect any statistically significant treatment effects; - •
·our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all;·- •
- regulators, institutional review boards or independent ethics committees may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site or may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks;
· - •
- we may experience delays in reaching or fail to reach agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites;
· - •
- we may have to suspend or terminate
our Phase 3 clinical trials of lefamulin for CABP or otherclinical trials of our product candidates for various reasons, including a finding that the participants are being exposed to unacceptable health or safety risks;· - •
- the cost of clinical trials of our product candidates may be greater than we anticipate;
· - •
- the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; and
· - •
- our product candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators, institutional review boards or independent ethics committees to suspend or terminate the trials.
Our product development costs will increase if we experience delays in enrollment in our clinical development program or our non-clinical development program or in obtaining marketing approvals. We do not know whether any additional non-clinical tests or clinical trials will be required, or if they will begin as planned, or if they will need to be restructured or will be completed on schedule, or at all. Significant non-clinical development program delays, including chemistry, manufacturing and control activities, or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
If we experience delays or difficulties in the enrollment of patients in our clinical trials, our receipt of necessary marketing approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials offor our product candidates, including with respect to lefamulin, CONTEPO or any other product candidate that we develop, if we are unable to locate and enroll a sufficient number of eligible patients to participate in these clinical trials. In particular, we may experience enrollment challenges at trial sites in the United States, where it is a common practice to place patients with potential moderate to severe CABP on antibiotics very shortly after examination. This practice could prevent potential trial patients in the United States from being enrolled in our clinical trials based on our eligibility criteria. In addition, someSome of our competitors have ongoing clinical trials for product candidates that could be competitive with
lefamulin and CONTEPO, and patients who would otherwise be eligible for our clinical trials may instead enroll in clinical trials of our competitors’competitors' product candidates.
Patient enrollment is affected by other factors including:
- •
·severity of the disease under investigation;·- •
- eligibility criteria for the clinical trial in question;
· - •
- perceived risks and benefits of the product candidate under study;
· - •
- approval of other therapies to treat the disease under investigation;
· - •
- efforts to facilitate timely enrollment in clinical trials;
· - •
- patient referral practices of physicians;
· - •
- the time of year in which the trial is initiated or conducted;
· - •
- the geographic distribution of global trial sites, given the timing of pneumonia season globally, and the seasonal variation in the number of patients suffering from pneumonia, including a decline in the number of patients with CABP during the summer months;
· - •
- the ability to monitor patients adequately during and after treatment;
· - •
- proximity and availability of clinical trial sites for prospective patients;
· - •
- delays in the receipt of required regulatory approvals, or the failure to receive required regulatory
approvals, in the jurisdictions in which clinical trials are expected to be conducted; and
· - •
- delays in the receipt of approvals, or the failure to receive approvals, from the relevant institutional review board or ethics committee at clinical trial sites.
For example, in each of our Phase 3 clinical trials of lefamulin, patients who have previously taken no more than one dose of a short acting, potentially effective antibiotic for the treatment of the current CABP episode within 24 hours of receiving the first dose of study medication will be allowed to participate in the trial but will comprise only up to 25% of the total intent to treat populations. Depending upon a region’s or a clinical trial site’s standard of care for the administration of antibiotics, this could affect our ability to enroll patients in these clinical trials in a timely fashion. Also, enrollment for our Phase 3 clinical trials may be negatively impacted by delays in opening clinical trial sites or the duration and/or severity of the influenza season. Moreover, our estimates regarding patient enrollment for our Phase 3 clinical trials of lefamulin depend on increasing enrollment rates as each such trial progresses, making it more difficult to precisely estimate the time of completion of such trials during its earlier stages. Enrollment delays in our clinical trials may result in increased development costs for our product candidates, which would cause the value of the company to decline and limit our ability to obtain additional financing. Our inability to enroll a sufficient number of patients in our Phase 3 clinical trials of lefamulin for CABP or any of our other clinical trials would result in significant delays or may require us to abandon one or more clinical trials altogether.
If serious adverse or undesirable side effects are identified during the development of lefamulin, CONTEPO or any other product candidate that we develop, we may need to abandon or limit our development of that product candidate.
All of our product candidates are in clinical or preclinical development and their risk of failure is high. It is impossible to predict when or if the FDA, EMA or other regulators will view any of our product candidates will proveas effective orand safe in humans or if we will receive marketing approval.marking approval for any of our product candidates. If our product candidates are associated with undesirable side effects or have characteristics that are unexpected, we may need to abandon their development or limit development to certain uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Many compounds that initially showed promise in clinical or earlier stage testing have later been found to cause side effects or other safety issues that prevented further development of the compound.
In LEAP 1, lefamulin was generally well tolerated and exhibited a similar rate of treatment-emergent adverse events to the comparator drug. However, 104 patients in the lefamulin arm of the trial reported at least one treatment-emergent adverse event and eight patients withdrew from the trial following an adverse event. Furthermore, at least 2.0% of patients in LEAP 1 who were dosed with lefamulin reported the following adverse events: hypokalemia, nausea, insomnia, infusion site pain and infusion site phlebitis. Fewer than 2.0% of trial patients dosed with lefamulin also experienced
Lefamulinhypertension and an increase in alanine aminotransaminase, although no patients met Hy's Law criteria, which is an indicator for severe liver damage.
In LEAP 2, lefamulin was again generally well tolerated. However, 120 patients in the lefamulin arm of the trial reported at least one treatment-emergent adverse event and eleven patients withdrew from the trial following an adverse event. Furthermore, at least 2.0% of patients in LEAP 2 who were dosed with lefamulin reported the following adverse events: diarrhea, nausea and vomiting. Fewer than 2.0% of trial patients dosed with lefamulin also experienced hypertension and an increase in alanine aminotransaminase, although no patients met Hy's Law criteria. In addition, one patient who had a positive clinical response to lefamulin in LEAP 2 was later diagnosed with a C. difficile infection during an extended hospital stay.
In addition, lefamulin was well tolerated in our Phase 2 clinical trial for acute bacterial skin and skin structure infection, or ABSSSI. No patient in the trial suffered any serious adverse events that were found to be related to lefamulin, and no patient in the trial died. Some patients experienced adverse events that were assessed by the investigator as possibly or probably related to study medication. The majority of their symptoms were mild in severity. Four patients discontinued study medication following a drug-related event, three of whom were in a lefamulin treatment group: one patient experienced events of hyperhidrosis, vomiting and headache; one patient experienced infusion site pain; and one patient experienced dyspnea.
Because the potential for mild effect on electrocardiogram, or ECG, measurements was observed in preclinical studies of lefamulin, we have continued to assess this potential in all human clinical trials of lefamulin we have conducted. In the Phase 2 clinical trial, no change in ECG measurements was considered to be of clinical significance, and no drug-related cardiovascular adverse event was reported. Both lefamulin and vancomycin treatment were associated with a small increase in the QT interval. The QT interval is a measure of the heart’sheart's electrical cycle, withand a lengthenedprolonged QT interval representingis a markerrisk factor for a potential ventricular arrhythmia. We are continuing to evaluate the effectIn each of LEAP 1 and LEAP 2, while changes in QT that were of potential clinical concern were uncommon, two patients treated with lefamulin on thehad an increase in absolute QT interval in our Phase 3 clinical trialsto greater than 500 msec, regardless of lefamulinbaseline value, which may indicate a higher risk for CABP.ventricular arrhythmias.
There were no systemic adverse events of clinical concern and no drug-related serious adverse events observed in any of our completed Phase 1 clinical trials of lefamulin. In these trials, the most commonly observed adverse effects with oral administration of lefamulin were related to the gastrointestinal tract, including nausea and abdominal discomfort, while the most commonly observed adverse effects related to IV administration were related to irritation at the infusion site. In addition, lefamulin produced a transient, predictable and reproducible prolongation of the QT interval based on the maximum concentration of the drug in the blood plasma. At therapeutic doses, we expect that the drug will not produce large effects on the QT interval that would be of clinical relevance. We did not observe any drug-related cardiac adverse events, such as increase in ectopic ventricular activity or other cardiac arrhythmia, or clinically relevant ECG findings during the conduct of any of our completed Phase 1 clinical trials.
In the ZEUS Study, the incidence of premature discontinuation from study drug was low and similar between treatment groups (6.0% in the CONTEPO treatment group compared to 3.9% in the PIP-TAZ treatment group), and the incidence of not completing the study through the last follow-up visit, which occurred on the 24th through 28th day after completion of seven days of treatment with the study drug, or after up to 14 days of treatment for patients with concurrent bacteremia, was 5.2% in the CONTEPO group compared to 0.9% in the PIP-TAZ group. A total of 42.1% CONTEPO patients and 32.0% PIP-TAZ patients experienced at least one treatment-emergent adverse event. Most treatment-emergent adverse events were mild or moderate in severity, and severe treatment-emergent adverse events were uncommon (2.1% of CONTEPO patients and 1.7% of PIP-TAZ patients). The most common treatment-emergent adverse events in both treatment groups were transient, asymptomatic laboratory abnormalities and gastrointestinal events. Treatment-emergent serious adverse
events were uncommon in both treatment groups (2.1% of CONTEPO patients and 2.6% of PIP-TAZ patients). There were no deaths in the study and one treatment-emergent serious adverse event in each treatment group was deemed related to study drug (hypokalemia in a CONTEPO patient and renal impairment in a PIP-TAZ patient), leading to study drug discontinuation in the PIP-TAZ patient. Study drug discontinuations due to the treatment-emergent adverse events were infrequent and similar between treatment groups (3.0% of CONTEPO patients and 2.6% of PIP-TAZ patients).
The most common laboratory abnormality treatment-emergent adverse events in the ZEUS Study were increases in the levels of alanine aminotransferase, or ALT, (8.6% of CONTEPO patients and 2.6% of PIP-TAZ patients) and aspartate transaminase, or AST, (7.3% of CONTEPO patients and 2.6% of PIP-TAZ patients). None of the ECG stoppingALT or AST elevations were symptomatic or treatment-limiting, and none of the patients met the criteria definedfor Hy's Law. Outside the United States, elevated liver aminotransferases are listed among undesirable effects in the trial protocolslabeling for IV fosfomycin.
In the ZEUS Study, hypokalemia occurred in 71 of 232 (30.6%) CONTEPO patients and 29 of 230 (12.6%) PIP-TAZ patients. Most decreases in potassium levels were mild to moderate in severity. Shifts in potassium levels from normal at baseline to hypokalemia, as determined by worst post-baseline hypokalemia values, were more frequent in the CONTEPO group than the PIP-TAZ group for mild (17.7% compared to 11.3%), moderate (11.2% compared to 0.9%), and severe (1.7% compared to 0.4%) categories of hypokalemia. Hypokalemia was reacheddeemed a treatment-emergent adverse event in any clinical trial. However, if we observe clinically relevant effects on6.4% of patients receiving CONTEPO and 1.3% of patients receiving PIP-TAZ, and all cases were transient and asymptomatic.
While no significant cardiac adverse events were observed in the ZEUS Study, post-baseline QT interval
intervals calculated using Fridericia's formula, or QTcF, of greater than 450 to less than or equal to 480 msec (baseline QTcF of less than or equal to 450 msec) occurred at a higher frequency in our Phase 3 clinical trialsCONTEPO patients (7.3%) compared to PIP-TAZ patients (2.5%). In the CONTEPO arm, these results appeared to be associated with the hypokalemia associated with the salt load of lefamulin for CABPthe IV formulation. Only one patient in the PIP-TAZ arm had a baseline QTcF of less than or in any other clinical trialequal to 500 msec and a post-baseline QTcF of lefamulin, our ability to successfully develop lefamulin for CABP or any other indication may be significantly delayed or prevented.greater than 500 msec.
If we elect or are forced to suspend or terminate any clinical trial of lefamulin, CONTEPO or any other product candidates that we are developing, the commercial prospects of lefamulin, CONTEPO or such other product candidates will be harmed and our ability to generate product revenues, if at all, from lefamulin, CONTEPO or any of these other product candidates will be delayed or eliminated. In addition, a higher rate of adverse events in lefamulin or CONTEPO as compared to the standard of care, even if slight, could negatively impact commercial adoption of lefamulin or CONTEPO by physicians. Any of these occurrences could materially harm our business, financial condition, results of operations and prospects.
Even if lefamulin, CONTEPO or any other product candidate receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success and the market opportunity for lefamulin, CONTEPO or any other product candidate may be smaller than we estimate.
If lefamulin, CONTEPO or any of our other product candidates receive marketing approval, it or they may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. For example, current treatments for pneumonia, including generic options, are well established in the medical community, and doctors may continue to rely on these treatments without lefamulin.lefamulin, CONTEPO or any of our other product candidates. In addition, our efforts to effectively communicate lefamulin’sthe differentiating characteristics and key attributes of lefamulin, CONTEPO or any of our other product candidates to clinicians and hospital pharmacies with the goal of establishing favorable formulary status for lefamulin, CONTEPO or any of our other product
candidates may fail or may be less successful than we expect. If lefamulin, CONTEPO or any of our other product candidates does not achieve an adequate level of acceptance, we may not generate significant product revenues or any profits from operations. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, including:
- •
·the efficacy and potential advantages compared to alternative treatments;·lefamulin’s- •
- the ability of lefamulin, CONTEPO or any other anti-infective product candidate to limit the development of bacterial resistance in the pathogens it targets;
· - •
- the prevalence and severity of any side effects;
· - •
- the ability to offer our product candidates for sale at competitive prices, including in comparison to generic competition;
· - •
- convenience and ease of administration compared to alternative treatments;
· - •
- the willingness of the target patient population to try new therapies, physicians to prescribe these therapies and hospitals to approve the cost and use by their physicians of these therapies;
· - •
- our investment in and the strength of marketing, patient access and distribution support;
· - •
- the availability of third-party coverage and adequate reimbursement; and
· - •
- the timing of any marketing approval in relation to other product approvals.
Although we believe that the mechanism of action for pleuromutilin antibiotics may result in a low propensity for development of bacterial resistance to lefamulin or our other pleuromutilin product candidates, bacteriaBacteria might nevertheless develop resistance to lefamulin, CONTEPO or our other pleuromutilinany future product candidates more rapidly or to a greater degree than we anticipate. If bacteria develop such resistance or if lefamulin, CONTEPO or any future product candidates is not effective against drug-resistant bacteria, the efficacy of these product candidates would decline, which would negatively affect our potential to generate revenues from these product candidates.
Our ability to negotiate, secure and maintain third-party coverage and reimbursement may be affected by political, economic and regulatory developments in the United States, the European Union and other jurisdictions. Governments continue to impose cost containment measures, and third-party payors are increasingly challenging prices charged for medicines and examining their cost effectiveness, in addition to their safety and efficacy. If the level of reimbursement is below our expectations, our revenue and gross margins would be adversely affected. Obtaining formulary approval from third-party payors can be an expensive and time-consuming process. We cannot be certain if and when we will obtain formulary approval to allow us to sell lefamulin, CONTEPO or any future product candidates into our target markets. Even if we do obtain formulary approval, third-party payors, such as government or private health care insurers, carefully review and increasingly question the coverage of, and challenge the prices charged for, drugs. These and other similar developments could significantly limit the degree of market acceptance of lefamulin, CONTEPO or any of our other product candidates
that receive marketing approval.
If we are unable to establish or maintain sales, marketing and distribution capabilities or enter into or maintain sales, marketing and distribution agreements with third parties, we may not be successful in commercializing lefamulin, CONTEPO or any other product candidate if and when they are approved.
We do not have only a very limited sales, marketing, orpatient access and distribution infrastructure, and as a company we have no experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any approved product, we must either develop aestablish an adequate sales, marketing, patient access and distribution organization or outsource these functions to third parties. If lefamulin or CONTEPO receives marketing approval, we plan to commercialize it in the United States with our own targeted hospital sales and marketing organization that we plan to establish.expand beyond its current levels, subject to our ability to access additional capital. In addition, we expect to utilize a
variety of types of collaboration, distribution and other marketing arrangements with one or more third parties to commercialize lefamulin in markets outside the United States. We do not intend to seek approval to commercialize CONTEPO in any markets outside the United States.
There are risks involved with establishing our own sales, marketing, patient access and distribution capabilities and entering into arrangements with third parties to perform these services. For example, recruitingIf we do not establish adequate sales, marketing, patient access and training a sales force is expensivedistribution capabilities prior to or in connection with the commercial launch of any of our products, such products may fail to gain sufficient market acceptance by physicians, patients, third-party payors and time consumingothers in the medical community and could delay any product launch.may fail commercially or be less successful than we expect. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing and distribution capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to commercialize our products on our own include:
- •
·our inability to recruit, train and retain adequate numbers of effective sales, patient access and marketing personnel;·- •
- the inability of sales personnel to obtain access to or persuade adequate numbers of physicians to prescribe any future products;
· - •
- the lack of complementary products to be offered by sales personnel, which may put our sales representatives at a competitive disadvantage relative to sales representatives from companies with more extensive product lines; and
· - •
- unforeseen costs and expenses associated with creating an independent sales, marketing, patient access and distribution organization.
If we enter into arrangements with third parties to perform sales, marketing, patient access and distribution services, our product revenues or the profitability of these product revenues to us are likely to be lower than if we were to market, sell and distribute ourselves any products that we develop. In addition, we may not be successful in entering into arrangements with third parties to sell, market and distribute our product candidates or may be unable to do so on terms that are favorable to us. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish sales, marketing and distribution capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates.
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
The development and commercialization of new drug products is highly competitive. We face competition with respect to lefamulin, CONTEPO and any other products we may seek to develop or commercialize in the future from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization.
There are a variety of available therapies marketed for the treatment of CABP.CABP and cUTI. Currently the treatment of CABP and cUTI is dominated by generic products. For hospitalized patients, combination therapy is frequently used.used in both CABP and cUTI. Many currently approved drugs are well-established therapies and are widely accepted by physicians, patients and third-party payors.
payors for the treatment of CABP. We also are aware of various drugs under development or recently approved by the FDA for the treatment of CABP, including solithromycin, (New Drug Application, or NDA, filed by Cempra Inc. and a complete response letter issuedomadacycline (recently approved by the FDA in December 2016), omadacycline (under Phase 3 clinical development byon behalf of Paratek Pharmaceuticals Inc.), delafloxacin (under Phase(recently approved by the FDA for ABSSSI on behalf of Melinta Therapeutics Inc. with a completed phase 3 clinical development by Melinta Therapeutics Inc.)trial for CABP) and oral nafithromycin (under Phase 2 clinical development by Wockhardt Ltd.). If approved, we expect CONTEPO will face competition from commercially available branded antibiotics such as ceftazidime-avibactam, meropenem-vaborbactam, tigecycline and plazomicin, from other products currently in development for the treatment of cUTI, including AP, such as imipenem-relebactam (under Phase 3 clinical development by Merck), cefiderocol (under Phase 3 clinical development by Shionogi), ETX0282-cefpodoxime proxetil (under Phase 1 clinical development by Entasis Therapeutics), and LYS228 (under development by Novartis), as well as generically available agents including piperacillin-tazobactam, carbapenems, aminoglycosides, and polymyxins.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are approved for broader indications or patient populations, are more convenient or are less expensive than any products that we may develop. Our competitors may also obtain marketing approvals for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, our ability to compete may be affected because in some cases insurers or other third-party payors seek to encourage the use of generic products. This may have the effect of making branded products less attractive, from a cost perspective, to buyers. We expect that if lefamulin is approved for CABP itand CONTEPO is approved for cUTI, including AP, they will be priced at a significant premium over competitive generic products. This pricing difference may make it difficult for us to replace existing therapies with lefamulin.lefamulin and CONTEPO. The key competitive factors affecting the success of our product candidates are likely to be their efficacy, safety, convenience, price and the availability of coverage and reimbursement from government and other third-party payors.
Many of our competitors may have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining approvals from regulatory authorities and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to or necessary for our programs.
Even if we are able to commercialize lefamulin, CONTEPO or any other product candidate that we develop, the product may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which would harm our business.
The regulations that govern marketing approvals, pricing, coverage and reimbursement for new drug products vary widely from country to country. Current and future legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenues we are able to generate from the sale of the product in that country. Adverse pricing
limitations may hinder our ability to recoup our investment in one or more product candidates, even if our product candidates obtain marketing approval.
Our ability to commercialize lefamulin, CONTEPO or any other product candidate successfully also will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and other third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels. A major trend in the healthcare industries in the European Union and the United States and elsewhere is cost containment. Government authorities and other third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that coverage and reimbursement will be available for lefamulin, CONTEPO or any other product that we commercialize and, if coverage and reimbursement are available, the level of reimbursement. Reimbursement may impact the demand for, or the price of, any product candidate for which we
obtain marketing approval. Obtaining and maintaining adequate reimbursement for lefamulin and CONTEPO may be particularly difficult because of the number of generic drugs, which are typically available at lower prices, that are available to treat CABP.CABP and cUTI. In addition, third-party payors are likely to impose strict requirements for reimbursement of a higher priced drug, such as lefamulin.lefamulin and CONTEPO. If reimbursement is not available or is available only to limited levels, we may not be able to successfully commercialize lefamulin, CONTEPO or other product candidates for which we obtain marketing approval.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the applicable regulatory authority. Moreover, eligibility for coverage and reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs, and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. In the United States, third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. In the European Union, reference pricing systems and other measures may lead to cost containment and reduced prices. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Product liability lawsuits against us could divert our resources, cause us to incur substantial liabilities and to limit commercialization of any products that we may develop or in-license.
We face an inherent risk of product liability exposure related to the testing of lefamulin, CONTEPO and any other product candidate that we develop in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop or in-license. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
·
- •
- reduced resources of our management to pursue our business strategy;
- •
- decreased demand for any product candidates or products that we may develop;
· - •
- injury to our reputation and significant negative media attention;
· - •
- withdrawal of clinical trial participants;
· - •
- significant costs to defend the related litigation;
· - •
- substantial monetary awards to trial participants or patients;
· - •
- loss of revenue; and
· - •
- the inability to commercialize any products that we may develop.
We maintain productclinical trial liability insurance that covers bodily injury to patients participating in our clinical trials up to a $10.0 million annual aggregate limit and subject to a per event deductible. This amount of insurance may not be adequate to cover all liabilities that we may incur. We will need to increase our insurance coverage when and if we begin commercializing lefamulin, CONTEPO or any other product candidate that receives marketing approval. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
Risks Related to Our Dependence on Third Parties
Use of third parties to manufacture our product candidates may increase the risk that we will not have sufficient quantities of our product candidates or products or such quantities at an acceptable quality or cost, which
could delay, prevent or impair our development or commercialization efforts.
We do not own or operate manufacturing facilities for the production of lefamulin or CONTEPO that could be used in product candidate development, including clinical trial supply, or for commercial supply, or for the supply of any other compound that we are developing or evaluating in our research program. We have limited personnel with experience in drug manufacturing and lack the resources and the capabilities to manufacture any of our product candidates on a clinical or commercial scale. We currently rely on third parties for supply of lefamulin and CONTEPO, and our strategy is to outsource all manufacturing, packaging, testing, serialization and distribution of our product candidates and products to third parties.
We do not currently have anyentered into agreements, and expect to enter into additional agreements, with third-party manufacturers for the long-term commercial supply of any of our product candidates.lefamulin and CONTEPO. We obtained the pleuromutilin starting material for the clinical trial supply of lefamulin from a single third-party manufacturer, Sandoz GmbH, or Sandoz, a division of Novartis AG, or Novartis. Novartis stopped manufacturing pleuromutilin for us in June 2015 and will not be a commercial supplier of pleuromutilin for us. We have identified and entered into a commercial supply agreement with an alternative supplier that we believe will be able to provide pleuromutilin starting material for the commercial supply of lefamulin.
Another third-party manufacturer synthesizes lefamulin starting from the pleuromutilin starting materialand a readily accessible chiral building block, and provides our supply of the active pharmaceutical ingredient. We may engage a secondary supplier to synthesize lefamulin. However, our current operating plans do not include a secondary supplier unless we obtain additional funding. We engage separate manufacturers to provide filltablets, sterile vials, and finish services for the finished productsterile diluent that we are using in our clinical trials of lefamulin. We have entered into commercial supply agreements with these same manufacturers to support the future commercialization of lefamulin, if approved. We also entered into a long-term commercial supply agreement with Arran Chemical Company Limited, or Arran, for the supply of the chiral acid starting material required in the synthesis of lefamulin API and a commercial packaging and supply agreement with Sharp Corporation for the commercial packaging and of lefamulin.
In addition, we have entered into a manufacturing and supply agreement with Ercros, S.A., pursuant to which Ercros, S.A. supplies to us, on an exclusive basis, the API mixture for CONTEPO in support of our NDA filing and, if CONTEPO is approved, will supply the commercial API Mixture for CONTEPO in the United State. We have also entered into a manufacturing and exclusive supply agreement with Laboratorios ERN, S.A., pursuant to which Laboratorios ERN, S.A. has agreed to supply us with certain technical documentation and data as required for our NDA filing for CONTEPO and certain regulatory support in connection with the commercial sale and use of CONTEPO in the United States. We entered into a commercial packaging agreement with AndersonBrecon, Inc. for the commercial packaging and serialization of CONTEPO in addition to a manufacturing and supply agreement with Fisiopharma S.r.l. for the supply, on a minimum commitment basis, of a percentage of our commercial requirements of CONTEPO in bulk drug vials for the United States as well as the supply of bulk drug vials of CONTEPO in connection with the submission of an NDA.
We may be unable to maintain our current arrangements for commercial supply, or conclude agreements for commercial supply with additional third-party manufacturers, or we may be unable to do so on acceptable terms.
Even if we are able to establish and maintain arrangements with third-party manufacturers, reliance on third-party manufacturers entails additional risks, including:
- •
·reliance on the third party for regulatory compliance and quality assurance;·- •
- an event at one of our manufacturers or suppliers causing an unforeseen disruption of the manufacture or supply of our product candidates;
- •
- the possible breach of the manufacturing agreement by the third party;
· - •
- the possible misappropriation of our proprietary information, including our trade secrets and know-how; and
· - •
- the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us.
Third-party manufacturers may not be able to comply with current good manufacturing practice, or cGMP, regulations or similar regulatory requirements outside the United States. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our product candidates.candidates and products.
Our product candidates and any products that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us.
If the third parties that we engage to supply any materials or manufacture product for our non-clinical testing and clinical trials should cease to continue to do so for any reason, we likely would experience delays in advancing these trials while we identify and qualify replacement suppliers, and we may be unable to obtain replacement supplies on terms that are favorable to us. For example, there are only a limited number of known manufacturers that produce the pleuromutilin starting material used in the synthesis of lefamulin. In early 2015, Novartis announcedcompleted the sale of its animal health division, including its veterinary products, to a third party. As a result, we have identifiedwere required to identify an alternative supplier that currently manufacturesfor pleuromutilin starting material for veterinary products, that we believe will be able to provide pleuromutilin starting material for the commercial scale manufacture of lefamulin. In addition, ifIf we are not able to obtain adequate supplies of our product candidates or the drug substances used to manufacture them, it will be more difficult for us to develop our product candidates and compete effectively.
Our current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our future profit margins and our ability to develop product candidates and commercialize any products that receive marketing approval on a timely and competitive basis.
We rely on third parties to conduct our clinical trials and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials.
We do not independently conduct clinical trials for our product candidates. We rely on third parties, such as contract research organizations, clinical data management organizations, medical institutions and clinical investigators, to perform this function. We expect to continue to rely on such third parties in conducting our clinical trials of lefamulin and CONTEPO, and expect to rely on these third parties to conduct clinical trials of any other product candidate that we develop. Any of these third parties may terminate their engagements with us at any time. If we need to enter into alternative arrangements, it would delay our product development activities.
Our reliance on these third parties for clinical development activities reduces our control over these activities but does not relieve us of our responsibilities. For example, we remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to as Good Clinical Practices, or GCP, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a U.S. government-sponsored database, ClinicalTrials.gov, within certain timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions. Similar GCP and transparency requirements apply in the European Union. Failure to comply with such requirements, including with respect to clinical trials conducted outside the European Union, can also lead regulatory authorities to refuse to take into account clinical trial data submitted as part of a marketing authorization application, oran MAA.
Furthermore, third parties that we rely on for our clinical development activities may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates. Our product development costs will increase if we experience delays in testing or obtaining marketing approvals.
We also rely on other third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidates or commercialization of our products, producing additional losses and depriving us of potential product revenue.
We have entered into and may enter into additional collaborations with third parties for the development or commercialization of lefamulin, CONTEPO and our other product candidates. If those collaborations are not successful, we may not be able to capitalize on the market potential of these product candidates.
If lefamulin receivesand CONTEPO receive marketing approval, we plan to commercialize itthem in the United States with our own targeted hospital sales and marketing organization. Outside the United States, we expect to utilize a variety of types of collaboration, distribution and other marketing arrangements with one or more third parties to commercialize lefamulin. For example, we have entered into a license agreement with Sinovant pursuant to which we granted Sinovant certain rights to manufacture and commercialize lefamulin in the People's Republic of China, Hong Kong, Macau and
Taiwan. We also may seek third-party collaborators for development and commercialization of other product candidates or for lefamulin for indications other than CABP.
Our likely future collaborators for any sales, marketing, distribution, development, licensing or broader collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies and biotechnology companies. We are not currently party toUnder our license agreement with Sinovant, we have, and under any such arrangement. However, ifarrangements we do enter into any such arrangements with any third parties in the future we will likely have, limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidates. Our ability to generate revenues from these arrangements will depend on our collaborators’collaborators' abilities and efforts to successfully perform the functions assigned to them in these arrangements.
CollaborationsOur current collaborations involving our product candidates wouldpose, and any future collaborations likely will pose, numerous risks to us, including the following:
- •
·collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations and may not perform their obligations as expected;·- •
- collaborators may deemphasize or not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on
clinical trial results, changes in the
collaborators’collaborators' strategic focus, product and product candidate priorities, available funding, or external factors such as an acquisition that diverts resources or creates competing priorities;· - •
- collaborators may need to conduct clinical trials, and these clinical trials may not be successful;
- •
- collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing;
· - •
- collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours;
· - •
- a collaborator with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products;
· - •
- collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation;
· - •
- collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability;
· - •
- disputes may arise between the collaborator and us as to the ownership of intellectual property arising during the collaboration;
· - •
- we may grant exclusive rights to our collaborators, which would prevent us from collaborating with others;
- •
- collaborators may be unable to enforce our intellectual property rights in territories where we have licensed, or may license, them such rights, which may expose us to material adverse tax and other consequences;
- •
- disputes may arise between the collaborators and us that result in the delay or termination of the
research,development or commercialization of our products or product candidates or that result in costly litigation or arbitration that diverts management attention and resources; and· - •
- collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates.
For example, in 2012, we entered into a stock purchaseunder our license agreement with ForestSinovant, if any court, tribunal or governmental agency in the People's Republic of China, Hong Kong, Macau or Taiwan determines that the exclusive license granted to Sinovant pursuant to the license agreement is not sufficiently exclusive such that Sinovant does not have sufficient rights to enforce the licensed patent rights in such territories, we and our subsidiary, Nabriva Therapeutics GmbH, have agreed to take such commercially reasonable steps as Sinovant reasonably requests to grant Sinovant such rights. If a court in such jurisdictions were to determine that our license to Sinovant was not sufficiently exclusive and that Sinovant did not have the rights to enforce the licensed patent rights in the licensed territories, Sinovant may require us to take such actions that it deems reasonable but that we do not and which Forest reimbursed us for certain external research and development costs and provided us with a $25.0 million loan in exchange for an exclusive right to acquire 100% of our outstanding shares for a one-year period. However, in 2013, Forest decided not to exercise its right to acquire us and terminated the stock purchase agreement. In connection with this termination, we repurchased the $25.0 million loan for €1.00. We no longermay have a commercial relationship with Forest,material adverse effect on our business, including requiring us to make changes to our organizational structure that may result in adverse tax and no rightsother consequences, or obligations remain outstanding under the stock purchase agreement.to conduct other activities that may cause us to incur significant expenses.
Collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all. If a collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
If we are not able to establish additional collaborations, we may have to alter our development and commercialization plans.
The potential commercialization of lefamulin and CONTEPO and the development and potential commercialization of other product candidates will require substantial additional cash to fund expenses. For some of our product candidates, we may decide to further collaborate with pharmaceutical and biotechnology companies for the development and potential commercialization of those product candidates. For example, we intend to seek to commercialize lefamulin through a variety of types of additional collaboration arrangements outside the United States. These collaborations will help fund the potential commercialization of our product candidates.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaborationadditional collaborations outside greater China will depend, among other things, upon our assessment of the collaborator’s
collaborator's resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’scollaborator's evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge, and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidate. We may also be restricted under future license agreements from entering into agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
If we are unable to reach agreements with suitable collaborators on a timely basis, on acceptable terms, or at all, we may have to curtail the development of a product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to fund and undertake development or commercialization activities on our own, we may need to obtain additional expertise and additional capital, which may not be available to us on acceptable terms or at all. If we fail to enter into additional collaborations and do not have sufficient funds or expertise to undertake the necessary development and commercialization activities, we may not be able to further develop our product candidates or bring them to market and generate product revenue.
Risks Related to Our Intellectual Property
If we are unable to obtain and maintain patent protection for our technology and products, or if the scope of the patent protection is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be adversely affected.
Our success depends in large part on our ability to obtain and maintain patent protection in the United States and other countries with respect to our proprietary technology and products. We seek to protect our proprietary position by filing patent applications in the United States, Europe and in certain additional foreign jurisdictions related to our novel technologies and product candidates that are important to our business. This process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. Moreover, if we license technology or product candidates from third parties in the future, these license agreements may not permit us to control the preparation, filing and prosecution of patent applications, or to maintain or enforce the patents, covering this intellectual property. These agreements could also give our licensors the right to enforce the licensed patents without our involvement, or to decide not to enforce the patents at all. Therefore, in these circumstances, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued which protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
The laws of foreign countries may not protect our rights to the same extent as the laws of the United States. For example, European patent law restricts the patentability of methods of treatment of the human body more than U.S. law does. We also may not pursue or obtain patent protection in all major markets or may not obtain protection that enables us to prevent the entry of third parties onto the market. Assuming the other requirements for patentability are met, currently, the first to file a patent application is generally entitled to the patent. However, prior to March 16, 2013, in the United States, the first to invent was entitled to the patent. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent
applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our U.S. patents or pending U.S. patent applications, or that we were the first to file for patent protection of such inventions.
Moreover, we may be subject to a third partythird-party pre-issuance submission of prior art to the U.S. Patent and Trademark Office, or USPTO, or become involved in opposition, derivation, reexamination,inter partes review, post grant review, interference proceedings or other patent office proceedings or litigation, in the United States or elsewhere, challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.
Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our owned or any future licensed patents by developing similar or alternative technologies or products in a non-infringing manner. In addition, other companies may attempt to circumvent any regulatory data protection or market exclusivity that we obtain under applicable legislation, which may require us to allocate significant resources to preventing such circumvention. Legal and regulatory developments in the European Union and elsewhere may also result in clinical trial data submitted as part of an MAA becoming publicly available. Such developments could enable other companies to circumvent our intellectual property rights and use our clinical trial data to obtain marketing authorizations in the European Union and in other jurisdictions. Such developments may also require us to allocate significant resources to prevent other companies from circumventing or violating our intellectual property rights. Our attempts to prevent third parties from circumventing our intellectual property and other rights may ultimately be unsuccessful. We may also fail to take the required actions or pay the necessary fees to maintain our patents.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and licensed patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents, trademarks, copyrights or other intellectual property. To counter such infringement or unauthorized use, we may be required to file claims, which can be expensive and time consuming. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their intellectual property. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, in whole or in part, construe the patent’spatent's claims narrowly or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability and the ability of our collaborators to develop, manufacture, market and sell our product candidates and use our proprietary technologies without infringing the intellectual property and other proprietary rights of third parties. There is considerable intellectual property litigation in the biotechnology and pharmaceutical industries, and we may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our products and
technology, including interference, derivation,inter partes review or post-grant review proceedings before the USPTO. The risks of being involved in such litigation and proceedings may increase as our product candidates approach commercialization, and as we gain greater visibility as a public company. Third parties may assert infringement claims against us based on existing or future intellectual property rights. We may not be aware of all such intellectual property rights potentially relating to our product candidates. Any freedom-to-operate search or analysis previously conducted may not have uncovered all relevant patents and patent applications, and there may be pending or future patent applications that, if issued, would block us from commercializing lefamulin.lefamulin or CONTEPO. Thus, we do not know with certainty whether lefamulin, CONTEPO or any other product candidate, or our commercialization thereof, does not and will not infringe any third party’sparty's intellectual property.
If we are found to infringe a third party’sparty's intellectual property rights, or to avoid or settle litigation, we could be required to obtain a license to continue developing and marketing our products and technology. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us, and could require us to make substantial payments. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, including treble damages and attorneys’attorneys' fees if we are found to have willfully infringed a patent or other intellectual property right. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
We may be subject to claims by third parties asserting that we or our employees have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these employees have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’semployee's former employer. Litigation may be necessary to defend against these claims.
In addition, while we typically require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own. Our pleuromutilin business was founded as a spin-off from Sandoz. Although all patent applications are fully owned by us and were either filed by Sandoz with all rights fully transferred to us, or filed in our sole name, because we acquired certain of our patents from Sandoz, we must rely on their prior practices, with regard to the assignment of such intellectual property. Similarly, for any patent applications we acquired from Zavante in connection with the Acquisition, we must rely on Zavante's prior practices with regard to the assignment of
intellectual property. Our and their assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to management.
Intellectual property litigation could cause us to spend substantial resources and could distract our personnel from their normal responsibilities.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses, and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of the ADSs.our ordinary shares. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development, sales, marketing or distribution activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and products, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants. However, we cannot guarantee that we have executed these agreements with each party that may have or have had access to our trade secrets or that the agreements we have executed will provide adequate protection. Any party with whom we have executed such an agreement may breach that agreement and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be obtained or independently developed by a competitor, our competitive position would be harmed.
We have not yet registered our trademarks in all of our potential markets, and failure to secure those registrations could adversely affect our business.
Our trademark applications may not be allowed for registration, and our registered trademarks may not be maintained or enforced. During trademark registration proceedings, we may receive rejections. Although we are given an opportunity to respond to those rejections, we may be unable to
overcome such rejections. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. If we do not secure registrations for our trademarks, we may encounter more difficulty in enforcing them against third parties than we otherwise would.
Risks Related to Regulatory Approval and Marketing of Our Product Candidates and Other Legal Compliance Matters
Even if we complete the necessary non-clinical studies and clinical trials, the marketing approval process is expensive, time-consuming and uncertain and may prevent us from obtaining approvals for the commercialization of some or all of our product candidates. If we are not able to obtain, or if there are delays in obtaining, required regulatory approvals, in particular in the United States or the European Union, we will not be able to commercialize our product candidates in those markets, and our ability to generate revenue will be materially impaired.
Our product candidates, including lefamulin and CONTEPO, and the activities associated with their development and commercialization, including their design, testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA and, in the case of lefamulin, by comparable authorities in other countries. Failure to obtain marketing approval for a product candidate will prevent us from commercializing the product candidate. We have not received approval to market lefamulin, CONTEPO or any of our other product candidates from regulatory authorities in any jurisdiction.jurisdiction and we do not intend to seek approval to market CONTEPO outside the United States.
We have nolittle experience in filing and supporting the applications necessary to obtain marketing approvals for product candidates and we have and expect to continue to rely on third-party contract research organizationsthird-parties to assist us in this process. Securing marketing approval requires the submission of extensive non-clinical and clinical data and supporting information to various regulatory authorities for each therapeutic indication to establish the product candidate’scandidate's safety and efficacy. Securing marketing approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the regulatory authorities. Regulatory authorities may determine that lefamulin, CONTEPO or any of our other product candidates are not effective or only moderately effective, or have undesirable or unintended side effects, toxicities, safety profiles or other characteristics that preclude us from obtaining marketing approval or that prevent or limit commercial use.
The process of obtaining marketing approvals is expensive, may take many years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Changes in marketing approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may cause delays in the approval or rejection of an application. For example, on June 23, 2016, eligible members of the electorate in the United Kingdom decided by referendum to leave the European Union, commonly referred to as ‘‘Brexit’’.Brexit. On March 29, 2017, the United Kingdom formally notified the European Union of its intention to withdraw pursuant to Article 50 of the Lisbon Treaty. Because a significant proportion of the regulatory framework in the United Kingdom is derived from European Union directives and regulations, the referendum could materially change the regulatory regime applicable to the approval of any of our product candidates in the United Kingdom. In addition, sincebecause the European Medicines Agency, or EMA, is currently located in the United Kingdom but expected to move to the Netherlands as a result of the Brexit, the implications for the regulatory review process in the European Union has not been fully clarified and could result in relocation of the EMA or a disruption into the EMA review process.
The United Kingdom has a period of a maximum of two years from the date of its formal notification to negotiate the terms of its withdrawal from, and future relationship with, the E.U. If no formal withdrawal agreement is reached between the United Kingdom and the E.U., then it is expected the United Kingdom's membership of the E.U. will automatically terminate two years after the submission of the notification of the United Kingdom's intention to withdraw from the E.U. Discussions between the United Kingdom and the E.U. focused on finalizing withdrawal issues and transition agreements are ongoing. However, limited progress to date in these negotiations and ongoing uncertainty within the UK Government and Parliament sustains the possibility of the United Kingdom leaving the E.U. on March 29, 2019 without a withdrawal agreement and associated transition period in place, which is likely to cause significant market and economic disruption.
The FDA and comparable regulatory authorities in other countries have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies. In addition, varying interpretations of the data obtained from non-clinical and clinical testing could delay, limit or prevent marketing approval of a product candidate. Any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable.
Accordingly, if we or our collaborators experience delays in obtaining approval or if we or they fail to obtain approval of our product candidates, the commercial prospects for our product candidates may be harmed and our ability to generate revenues will be materially impaired.
Our failure to obtain marketing approval in jurisdictions other than the United States and Europe would prevent our product candidates from being marketed in these other jurisdictions, and any approval we are granted for our product candidates in the United States and Europe would not assure approval of product candidates in other jurisdictions.
In order to market and sell lefamulin and our other product candidates in jurisdictions other than the United States and Europe, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval process varies among countries and can involve additional testing. The time required to obtain approval may differ from that required to obtain FDA approval or approvals from regulatory authorities in the European Union. The regulatory approval process outside the United States and Europe generally includes all of the risks associated with obtaining FDA approval or approvals from regulatory authorities in the European Union. In addition, some countries outside the United States and Europe require approval of the sales price of a drug before it can be marketed. In many countries, separate procedures must be followed to obtain reimbursement and a product may not be approved for sale in the country until it is also approved for reimbursement. We may not obtain marketing, pricing or reimbursement approvals outside the United States and Europe on a timely basis, if at all. Approval by the FDA or regulatory authorities in the European Union does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one regulatory authority outside the United States and Europe does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA or regulatory authorities in the European Union. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our products in any market. Marketing approvals in countries outside the United States and Europe do not ensure pricing approvals in those countries or in any other countries, and marketing approvals and pricing approvals do not ensure that reimbursement will be obtained.
Even if we obtain marketing approvals for our product candidates, the terms of approvals and ongoing regulation of our products may limit how we manufacture and market our products and compliance with such requirements may involve substantial resources, which could materially impair our ability to generate revenue.
Even if marketing approval of a product candidate is granted, an approved product and its manufacturer and marketer are subject to ongoing review and extensive regulation, including the potential requirements to implement a risk evaluation and mitigation strategy or to conduct costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of the product. We must also comply with requirements concerning advertising and promotion for any of our product candidates for which we obtain marketing approval. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’sproduct's approved labeling. Thus, we will not be able to promote any products we develop for indications or uses for which they are not approved. In
addition, manufacturers of approved products and those manufacturers’manufacturers' facilities are required to comply with extensive FDA requirements including ensuring that quality control and manufacturing procedures conform to cGMP, which include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation and reporting requirements. We and our contract manufacturers could be subject to periodic unannounced inspections by the FDA to monitor and ensure compliance with cGMP.
Accordingly, assuming we receive marketing approval for one or more of our product candidates, we and our contract manufacturers will continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production, product surveillance and quality control. If we are not able to comply with post-approval regulatory requirements, we could have the marketing approvals for our products withdrawn by regulatory authorities and our ability to market any future products could be limited, which could adversely affect our ability to achieve or sustain profitability. Thus, the cost of compliance with post-approval regulations may have a negative effect on our operating results and financial condition.
Any product candidate for which we obtain marketing approval will be subject to strict enforcement of post-marketing requirements and we could be subject to substantial penalties, including withdrawal of our product from the market, if we fail to comply with all regulatory requirements or if we experience unanticipated problems with our product candidates, when and if any of them are approved.
Any product candidate for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to continual requirements of and review by the FDA and other regulatory authorities. These requirements include, but are not limited to, restrictions governing promotion of an approved product, submissions of safety and other post-marketing information and reports, registration and listing requirements, cGMP requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, and requirements regarding the distribution of samples to physicians and recordkeeping. In addition, even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval.
The FDA and other federal and state agencies, including the U.S. Department of Justice, or DOJ, closely regulate compliance with all requirements governing prescription drug products, including requirements pertaining to marketing and promotion of drugs in accordance with the provisions of the approved labeling and manufacturing of products in accordance with cGMP requirements. The FDA and DOJ impose stringent restrictions on manufacturers’manufacturers' communications regarding off-label use and if we do not market our products for their approved indications, we may be subject to enforcement action for off-label marketing. Violations of such requirements may lead to investigations alleging violations of
the Food, Drug and Cosmetic Act and other statutes, including the False Claims Act and other federal and state health care fraud and abuse laws as well as state consumer protection laws.
Our failure to comply with all regulatory requirements, and later discovery of previously unknown adverse events or other problems with our products, manufacturers or manufacturing processes, may yield various results, including:
- •
·litigation involving patients taking our products;·- •
- restrictions on such products, manufacturers or manufacturing processes;
· - •
- restrictions on the labeling or marketing of a product;
· - •
- restrictions on product distribution or use;
· - •
- requirements to conduct post-marketing studies or clinical trials;
· - •
- warning or untitled letters;
· - •
- withdrawal of the products from the market;
· - •
- refusal to approve pending applications or supplements to approved applications that we submit;
· - •
- recall of products;
· - •
- fines, restitution or disgorgement of profits or revenues;
· - •
- suspension or withdrawal of marketing approvals;
· - •
- damage to relationships with any potential collaborators;
· - •
- unfavorable press coverage and damage to our reputation;
· - •
- refusal to permit the import or export of our products;
· - •
- product seizure;
- •
- exclusion from participation in federal healthcare reimbursement programs or
debarment or the imposition of Corporate Integrity Agreements; or
· - •
- injunctions or the imposition of civil or criminal penalties.
Non-compliance by us or any future collaborator with regulatory requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties. Similarly, failure to comply with regulatory requirements regarding the protection of personal information can also lead to significant penalties and sanctions.
Non-compliance with European Union requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, also can result in significant financial penalties. Similarly, failure to comply with the European Union’sUnion's requirements regarding the protection of personal information can also lead to significant penalties and sanctions.
Inadequate funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the
payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough critical FDA, SEC and other government employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
Governments outside the United States tend to impose strict price controls, which may adversely affect our revenues, if any.
In some countries, particularly the member states of the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. Also, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various European Union member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices. In some countries, we may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our product candidate to other available therapies to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on prices or reimbursement levels within the country of publication and other countries. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely affected.
The FDA’sFDA's agreement to a Special Protocol Assessment, or SPA, with respect to the study design of our first Phase 3 clinical trial of lefamulin for CABP does not guarantee any particular outcome from regulatory review, including ultimate approval, and may not lead to a faster development or regulatory review or approval process.
We reached agreement with the FDA in September 2015 on a SPA, which was later amended in April 2016, regarding the study design of our first Phase 3 clinical trial of lefamulin for the treatment of CABP. The SPA process is designed to facilitate the FDA’sFDA's review and approval of drugs by allowing the FDA to evaluate the proposed design and size of Phase 3 clinical trials that are intended to form the primary basis for determining a product candidate’scandidate's efficacy and safety. Upon specific request by a clinical trial sponsor, the FDA will evaluate the protocol and respond to a sponsor’ssponsor's questions regarding, among other things, primary efficacy endpoints, trial conduct and data analysis, within 45 days of receipt of the request. The FDA ultimately assesses whether the protocol design and planned analysis of the trial are acceptable to support regulatory approval of the product candidate with respect to the effectiveness in the indication studied.
Our agreement with the FDA regarding the SPA may not lead to faster development, regulatory review or approval for lefamulin. Once the FDA and an applicant reach an agreement under the special protocol assessment process regarding the design and size of a clinical trial, the agreement generally cannot be changed after the clinical trial begins. Nevertheless, the FDA may revoke or alter a
SPA under defined circumstances, such as changes in the relevant data or assumptions provided by the sponsor or the emergence of new public health concerns. A revocation or alteration in our SPA could significantly delay or prevent approval of any marketing applications we submit for lefamulin. In addition, any significant change to the protocols for our clinical trial subject to the SPA would require prior FDA approval, which could delay implementation of such a change and the conduct of the trial.
Fast track designation by the FDA may not actually lead to a faster development or regulatory review or approval process and does not assure FDA approval of our product candidate.
If a drug is intended for the treatment of a serious or life threateninglife-threatening condition and the drug demonstrates the potential to address unmet medical need for this condition, the drug sponsor may apply for FDA fast track designation. The FDA has designated each of the IV and oral formulations of lefamulin and the IV formulation of CONTEPO as a qualified infectious disease product, or QIDP, and granted fast track designations to each of these formulations of lefamulin.lefamulin and CONTEPO. However, neither the QIDP nor the fast track designation ensures that lefamulin or CONTEPO will receive marketing approval or that approval will be granted within any particular timeframe. We may also seekhave sought fast track designation for our other product candidates.oral and IV lefamulin, as well as IV CONTEPO. We may, however, not experience a faster development process, review or approval compared to conventional FDA procedures. In addition, the FDA may withdraw fast track designation if it believes that the designation is no longer supported by data from our clinical development program. Fast track designation alone does not guarantee qualification for the FDA’sFDA's priority review procedures.
Priority review designation by the FDA may not lead to a faster regulatory review or approval process and, in any event, does not assure FDA approval of our product candidate.
If the FDA determines that a product candidate offers major advances in treatment or provides a treatment where no adequate therapy exists, the FDA may designate the product candidate for priority review. A priority review designation means that the FDA’sFDA's goal to review an application is six months, rather than the standard review period of ten months. We were notified by the FDA that CONTEPO IV and the IV and oral formulations of lefamulin were granted priority review and have been given a Prescription Drug User Fee Act, or PDUFA, target action date of April 30, 2019 and August 19, 2019, respectively. Because the FDA designated each of the IV and oral formulations of lefamulin as a QIDP, lefamulin also will receive priority review. We may also request priority review for other product candidates. The FDA has broad discretion with respect to whether or not to grant priority review status to a product candidate, so even if we believe a particular product candidate is eligible for such designation or status, the FDA may decide not to grant it. Moreover, a priority review designation does not necessarily mean a faster regulatory review process or necessarily confer any advantage with respect to approval compared to conventional FDA procedures. Receiving priority review from the FDA does not guarantee approval within the six-month review cycle or thereafter.
Designation of our product candidate,each of lefamulin and CONTEPO as a Qualified Infectious Disease Product does not assure FDA approval of thisthese product candidate.candidates.
A QIDP is an antibacterial or antifungal drug intended to treat serious or life-threatening infections, including those caused by an antibacterial or antifungal resistant pathogen, including novel or emerging infectious pathogens or certain ‘‘qualifying"qualifying pathogens.’’" Upon the approval of an NDA for a drug product designated by the FDA as a QIDP, the product is granted an additional period of five years of regulatory exclusivity. Even though we have received QIDP designation for the IV and oral formulations of lefamulin and IV formulation of CONTEPO, there is no assurance that thisthese product candidatecandidates will be approved by the FDA.
If the FDA does not conclude that our product candidates satisfy the requirements under Section 505(b)(2) of the Federal Food Drug and Cosmetics Act, or if the requirements for such product candidates under Section 505(b)(2) are not as we expect, the approval pathway for those product candidates may take longer, cost more and entail greater complications and risks than anticipated, and may not be successful.
We submitted an NDA for marketing approval of CONTEPO for the treatment of cUTI in adults in the United States in October 2018 utilizing Section 505(b)(2) of the Food, Drug and Cosmetic Act, or the FDCA, which was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Act. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference.
For NDAs submitted under Section 505(b)(2) of the FDCA, the patent certification and related provisions of the Hatch-Waxman Act apply. In accordance with the Hatch-Waxman Act, such NDAs may be required to include certifications, known as Paragraph IV certifications, that certify any patents listed in the Orange Book publication in respect to any product referenced in the 505(b)(2) application are invalid, unenforceable and/or will not be infringed by the manufacture, use or sale of the product that is the subject of the 505(b)(2) application. Under the Hatch-Waxman Act, the holder of the NDA which the 505(b)(2) application references may file a patent infringement lawsuit after receiving notice of the Paragraph IV certification. Filing of a patent infringement lawsuit triggers a one-time automatic 30-month stay of the FDA's ability to approve the 505(b)(2) application. We have not conducted a comprehensive freedom-to-operate review with regard to CONTEPO.
Accordingly, we may invest a significant amount of time and expense in the development of CONTEPO or any other product candidate we may develop and experience significant delays and patent litigation before such products may be commercialized, if at all. A Section 505(b)(2) application also may not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired. The FDA may also require us to perform one or more additional clinical studies or measurements to support the change from the approved product. The FDA may then approve the new formulation for all or only some of the indications sought by us. The FDA may also reject our future Section 505(b)(2) submissions and may require us to file such submissions under Section 501(b)(1) of the FDCA, which could be considerably more expensive and time consuming.
In addition, notwithstanding the approval of a number of products by the FDA under Section 505(b)(2) over the last few years, certain competitors and others have objected to the FDA's interpretation of Section 505(b)(2). If the FDA's interpretation of Section 505(b)(2) is successfully challenged, the FDA may be required to change its 505(b)(2) policies and practices, which could delay or even prevent the FDA from approving any NDA that we submit under Section 505(b)(2). It is not uncommon for a manufacturer of an approved product to file a citizen petition with the FDA seeking to delay approval of, or impose additional approval requirements for, pending competing products. If successful, such petitions can significantly delay, or even prevent, the approval of the new product. However, even if the FDA ultimately denies such a petition, the FDA may substantially delay approval while it considers and responds to the petition. Thus, even if we are able to utilize the Section 505(b)(2) regulatory pathway, there is no guarantee this would ultimately lead to faster product development or earlier approval.
If the FDA does not conclude that CONTEPO, or any of our other product candidates for which we may utilize the 505(b)(2) pathway, satisfies the requirements for the 505(b)(2) regulatory approval pathway, or if the requirements for approval of any of our product candidates, including CONTEPO, under Section 505(b)(2) are not as we expect, the approval pathway for CONTEPO and any of our other product candidates for which we may utilize the 505(b)(2) pathway will likely take significantly
longer, cost significantly more and encounter significantly greater complications and risks than anticipated, and in any case may not be successful.
Under the CURES Act and the Trump Administration's regulatory reform initiatives, the FDA's policies, regulations and guidance may be revised or revoked and that could prevent, limit or delay regulatory approval of our product candidates, which would impact our ability to generate revenue.
In December 2016, the Cures Act was signed into law. The Cures Act, among other things, is intended to modernize the regulation of drugs and spur innovation, but its ultimate implementation is unclear. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would adversely affect our business, prospects, financial condition and results of operations.
We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad. For example, certain policies of the Trump administration may impact our business and industry. Namely, the Trump administration has taken several executive actions, including the issuance of a number of Executive Orders, that could impose significant burdens on, or otherwise materially delay, the FDA's ability to engage in routine regulatory and oversight activities such as implementing statutes through rulemaking, issuance of guidance, and review and approval of marketing applications. An under-staffed FDA could result in delays in the FDA's responsiveness or in its ability to review submissions or applications, issue regulations or guidance, or implement or enforce regulatory requirements in a timely fashion or at all. Moreover, on January 30, 2017, President Trump issued an Executive Order, applicable to all executive agencies, including the FDA, which requires that for each notice of proposed rulemaking or final regulation to be issued in fiscal year 2017, the agency shall identify at least two existing regulations to be repealed, unless prohibited by law. These requirements are referred to as the "two-for-one" provisions. For fiscal years 2018 and beyond, the Executive Order requires agencies to identify regulations to offset any incremental cost of a new regulation and approximate the total costs or savings associated with each new regulation or repealed regulation. In interim guidance issued by the Office of Information and Regulatory Affairs within the Office of Management and Budget on February 2, 2017, the administration indicates that the "two-for-one" provisions may apply not only to agency regulations, but also to significant agency guidance documents. In addition, on February 24, 2017, President Trump issued an executive order directing each affected agency to designate an agency official as a "Regulatory Reform Officer" and establish a "Regulatory Reform Task Force" to implement the two-for-one provisions and other previously issued executive orders relating to the review of federal regulations, however it is difficult to predict how these requirements will be implemented, and the extent to which they will impact the FDA's ability to exercise its regulatory authority. If these executive actions impose constraints on the FDA's ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
Our relationships with healthcare providers, physicians and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which in the event of a violation could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors will play a primary role in the recommendation and prescription of any product candidates, including lefamulin and CONTEPO, for which we obtain marketing approval. Our future arrangements with healthcare providers, physicians and third-party payors may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through
which we market, sell and distribute any products for which we obtain
marketing approval. Restrictions under applicable federal and state healthcare laws and regulations, include the following:
- •
·the federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation or arranging of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid;·- •
- the federal False Claims Act imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, false or fraudulent claims for payment by a federal healthcare program or making a false statement or record material to payment of a false claim or avoiding, decreasing or concealing an obligation to pay money to the federal government, with potential liability including mandatory treble damages and significant per-claim penalties, currently set at $5,500 to $11,000 per false claim;
· - •
- the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters;
·HIPPA, - •
- HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
· - •
- the federal Physician Payments Sunshine Act requires applicable manufacturers of covered products to report payments and other transfers of value to physicians and teaching hospitals, with data collection beginning in August 2013; and
· - •
- analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws and transparency statutes, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third party payors, including private insurers.
Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’sindustry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government and may require product manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not pre-empted by HIPAA, thus complicating compliance efforts.
If our operations are found to be in violation of any of the laws described above or any governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines and the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment or restructuring of our operations could adversely affect our financial results. We are developing and implementing a corporate compliance program designed to ensure that we will market and sell any future products that we successfully develop from our product candidates in compliance with all applicable laws and regulations, but we cannot guarantee that this program will protect us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion of products from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we
expect to do business is found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Current and future legislation may increase the difficulty and cost for us and any future collaborators to obtain marketing approval of and commercialize our product candidates and affect the prices we, or they, may obtain.
In the United States and a number ofsome foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could, among other things, prevent or delay marketing approval of lefamulin, CONTEPO or any of our other product candidates, restrict or regulate post-approval activities and affect our ability, or the ability of any collaborators, to profitably sell any product candidates, including lefamulin and CONTEPO, for which we, or they, obtain marketing approval. We expect that current laws, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we, or any future collaborators, may receive for any approved products.
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the ACA. Among the provisions of the ACA of potential importance to our business and our product candidates are the following:
- •
·an annual, non-deductible fee on any entity that manufactures or imports specified branded prescription products and biologic agents;·- •
- an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program;
·a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for products that are inhaled, infused, instilled, implanted or injected; - •
·expansion of healthcare fraud and abuse laws, including the civil False Claims Act and the federal Anti-Kickback Statute, new government investigative powers and enhanced penalties for noncompliance;·- •
- a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer
50%70% point-of-sale discounts off negotiatedprices of applicable brand products to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient products to be covered under Medicare Part D;prices;
· - •
- extension of
manufacturers’manufacturers' Medicaid rebateliability to individuals enrolled in Medicaid managed care organizations;liability;
· - •
- expansion of eligibility criteria for Medicaid programs;
· - •
- expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program;
· - •
- new requirements to report certain financial arrangements with physicians and teaching hospitals;
· - •
- a new requirement to annually report product samples that manufacturers and distributors provide to physicians; and
- •
- a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such
research;research.
· a new Independent Payment Advisory Board, or IPAB, which has authority to recommend certain changes to the Medicare program to reduce expenditures by the program that could result in reduced payments for prescription products; and
· established the Center for Medicare and Medicaid Innovation within CMS to test innovative payment and service delivery models.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. These changes include the Budget Control Act of 2011, which among other things, led to aggregate reductions to Medicare payments to providers of up to 2% per fiscal year that started in April 2013 and, due to subsequent legislative amendments to the statute,statutes, will stay in effect through 20252027 unless additional Congressional action is taken, and the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding and otherwise affect the prices we may obtain for any of our product candidates for which we may obtain regulatory approval or the frequency with which any such product candidate is prescribed or used. Further, there have been several recent U.S. Congressionalcongressional inquiries and proposed billsstate and federal legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the costs of drugs under Medicare and reform government program reimbursement methodologies for drug products.
We expect that the ACA,these healthcare reforms, as well as other healthcare reform measures that may be adopted in the future, may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, new payment methodologies and additional downward pressure on the price that we receive for any approved product and/or the level of reimbursement physicians receive for administering any approved product we might bring to market. Reductions in reimbursement levels may negatively impact the prices we receive or the frequency with which our products are prescribed or administered. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors.
In addition, with Since enactment of the new AdministrationACA, there have been numerous legal challenges and Congress, there will likely be additional legislative changes, includingCongressional actions to repeal and replacementreplace provisions of the law.
Congress has repeatedly tried to repeal, replace and amend the ACA in recent years. With enactment of the legislation commonly referred to as the Tax Cuts and Jobs Act of 2017, which was signed by the President on December 22, 2017, Congress repealed the "individual mandate" for the ACA. The repeal of this provision, which requires most Americans to carry a minimal level of health insurance, became effective in 2019. According to the Congressional Budget Office, the repeal of the individual mandate will cause 13 million fewer Americans to be insured in 2027 and premiums in insurance markets may rise. Additionally, on January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain ACA-mandated fees, including the so-called "Cadillac" tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices. Further, the Bipartisan Budget Act of 2018, among other things, amends the ACA, effective January 1, 2019, to increase from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and to close the coverage gap in most Medicare drug plans, commonly referred to as the "donut hole." Further, each chamber of the Congress has put forth multiple bills designed to repeal or repeal and replace portions of the ACA. Although none of these measures has been enacted by Congress to date, Congress may consider other legislation to repeal and replace elements of the ACA.
The Trump Administration has also taken executive actions to undermine or delay implementation of the ACA. Since January 2017, President Trump has signed two Executive Orders designed to delay the implementation of certain provisions of the ACA or otherwise circumvent some of the requirements for health insurance mandated by the ACA. It remainsOne Executive Order directs federal agencies with authorities and responsibilities under the ACA to be seen, however, precisely whatwaive, defer, grant exemptions from, or delay the new legislationimplementation of any provision of the ACA that would impose a fiscal or regulatory burden on
states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. The second Executive Order terminates the cost-sharing subsidies that reimburse insurers under the ACA. Several state Attorneys Generals filed suit to stop the administration from terminating the subsidies, but their request for a restraining order was denied by a federal judge in California on October 25, 2017. In addition, CMS has recently proposed regulations that would give states greater flexibility in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing the essential health benefits required under the ACA for plans sold through such marketplaces. Further, on June 14, 2018, U.S. Court of Appeals for the Federal Circuit ruled that the federal government was not required to pay more than $12 billion in ACA risk corridor payments to third-party payors who argued were owed to them. The effects of this gap in reimbursement on third-party payors, the viability of the ACA marketplace, providers, and potentially our business, are not yet known.
We will provide, when it will be enactedcontinue to evaluate the effect that the ACA and what impact it willits possible repeal and replacement could have on our business. It is possible that repeal and replacement initiatives, if enacted into law, could ultimately result in fewer individuals having health insurance coverage or in individuals having insurance coverage with less generous benefits. While the availabilitytiming and scope of healthcareany potential future legislation to repeal and containing or loweringreplace ACA provisions is highly uncertain in many respects, it is also possible that some of the cost of healthcare. SuchACA provisions that generally are not favorable for the research-based pharmaceutical industry could also be repealed along with ACA coverage expansion provisions. Accordingly, such reforms, if enacted, could have an adverse effect on anticipated revenue from product candidates that we may successfully develop and for which we may obtain marketing approval and may affect our overall financial condition and ability to develop or commercialize product candidates. For example,
Further, there have been several recent U.S. congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the Presidentrelationship between pricing and congressional leaders have expressed interest in repealing certain ACA provisionsmanufacturer patient programs, reduce the costs of drugs under Medicare and replacing them with alternativesreform government program reimbursement methodologies for drug products. At the federal level, the Trump administration's budget proposal for fiscal year 2019 contains further drug price control measures that maycould be less costly and provide state Medicaid programs and private health plans more flexibility. It is possible that these repeal and replacement initiatives, if enacted into law, could ultimately result in fewer individuals having health insurance coverageduring the 2019 budget process or in individuals having insurance coverage with less generous benefits. The scope of potentialother future legislation, including, for example, measures to repealpermit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid, and replace ACA provisions is highly uncertainto eliminate cost sharing for generic drugs for low-income patients. While any proposed measures will require authorization through additional legislation to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. At the state level, individual states are increasingly aggressive in many respects,passing legislation and it is possibleimplementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional health care authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other health care programs. These measures could reduce the ultimate demand for our products, once approved, or put pressure on our product pricing.
In addition, on May 11, 2018, the Administration issued a plan to lower drug prices. Under this blueprint for action, the Administration indicated that somethe Department of Health and Human Services, or HHS, will take steps to end the gaming of regulatory and patent processes by drug makers to unfairly protect monopolies; advance biosimilars and generics to boost price competition; evaluate the inclusion of prices in drug makers' advertisements to enhance price competition; speed access to and lower the cost of new drugs by clarifying policies for sharing information between insurers and drug makers; avoid excessive pricing by relying more on value-based pricing by expanding outcome-based
payments in Medicare and Medicaid; work to give Part D plan sponsors more negotiation power with drug makers; examine which Medicare Part B drugs could be negotiated for a lower price by Part D plans, and improving the design of the ACA provisionsPart B Competitive Acquisition Program; update Medicare's drug-pricing dashboard to increase transparency; prohibit Part D contracts that generallyinclude "gag rules" that prevent pharmacists from informing patients when they could pay less out-of-pocket by not using insurance; and require that Part D plan members be provided with an annual statement of plan payments, out-of-pocket spending, and drug price increases.
At the state level, individual states are not favorableincreasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional health care authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other health care programs. These measures could reduce the ultimate demand for the research-based pharmaceutical industry could also be repealed along with ACA coverage expansion provisions.our products, once approved, or put pressure on our product pricing.
LegislativeMoreover, legislative and regulatory proposals have also been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products.drugs. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our productdrug candidates, if any, may be. In addition, increased scrutiny by the United States Congress of the FDA’sFDA's approval process may significantly delay or prevent marketing approval, as well as subject us and any future collaborators to more stringent productdrug labeling and post-marketing testing and other requirements.
We are subject to anti-corruption laws, as well as export control laws, customs laws, sanctions laws and other laws governing our operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures and legal expenses, which could adversely affect our business, results of operations and financial condition.
Our operations are subject to anti-corruption laws, including the U.S. Foreign Corrupt Practices Act, or FCPA, the Irish Criminal Justice (Corruption Offenses) Act, and other anti-corruption laws that apply in countries where we do business and may do business in the future. The FCPA and these other laws generally prohibit us, our officers, and our employees and intermediaries from bribing, being bribed or making other prohibited payments to government officials or other persons to obtain or retain business or gain some other business advantage. We may in the future operate in jurisdictions that pose a high risk of potential FCPA violations, and we may participate in collaborations and relationships with third parties whose actions could potentially subject us to liability under the FCPA or local anti-corruption laws. In addition, we cannot predict the nature, scope or effect of future regulatory requirements to which our international operations might be subject or the manner in that existing laws might be administered or interpreted.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical trials and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
We are also subject to other laws and regulations governing our international operations, including regulations administered by the governments of the United States, and authorities in the European
Union, including applicable export control regulations, economic sanctions on countries and persons, customs requirements and currency exchange regulations, collectively referred to as the trade control laws.
There is no assurance that we will be effective in ensuring our compliance with all applicable anti-corruption laws, including the FCPA or other legal requirements, including trade control laws. If we are not in compliance with the FCPA and other anti-corruption laws or trade control laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, and legal expenses, which could have an adverse impact on our business, financial condition, results of operations and liquidity. Likewise, any investigation of any potential violations of the FCPA, other anti-corruption laws or trade control laws by U.S. or other authorities could also have an adverse impact on our reputation, our business, results of operations and financial condition.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations currently, and may in the future, involve the use of hazardous and flammable materials, including chemicals and medical and biological materials, and produce hazardous waste products. Even if we contract with third parties for the disposal of these materials and wastes, we cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials or disposal of hazardous wastes, we could be held liable for any resulting damages, and any liability could exceed our resources.
Although we maintain workers’workers' compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We also maintain a general liability program for some of the risks, but our insurance program includes limited environmental damage coverage, which has an annual aggregate coverage limit of $2.0 million. Although we maintain an umbrella policy with an annual aggregate coverage limit of $10.0 million, which may provide some environmental coverage, we do not maintain a separate policy covering environmental damages.
In addition, we may incur substantial costs to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Our employees may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of employee fraud or other misconduct, including intentional failures to comply with FDA regulations or similar regulations of comparable non-U.S. regulatory authorities, provide accurate information to the FDA or comparable non-U.S. regulatory authorities, comply with manufacturing standards we have established, comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable non-U.S. regulatory authorities, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements.
Employee misconduct could also involve the improper use of information obtained during clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws, standards or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
We rely significantly on information technology and any failure, inadequacy, interruption or security lapse of that technology, including any cyber security incidents, could harm our ability to operate our business effectively.
Despite the implementation of security measures, our internal computer systems and those of third parties with which we contract are vulnerable to damage from cyber-attacks, computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. System failures, accidents or security breaches could cause interruptions in our operations, and could result in a material disruption of our clinicaldrug development programs and commercialization activities and business operations, in addition to possibly requiring substantial expenditures of resources to remedy. The loss of clinical trial data could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and our product research, development and commercialization efforts could be delayed.
We are subject to various laws protecting the confidentiality of certain patient health information, and our failure to comply could result in penalties and reputational damage.
Certain countries in which we operate have, or are developing, laws protecting the confidentiality of certain patient health information. European Union member states and other jurisdictions have adopted data protection laws and regulations, which impose significant compliance obligations.
For example, the European Union General Data Protection Regulation, or the GDPR, which came into force on May 25, 2018, introduced new data protection requirements in the European Union and substantial fines for breaches of the data protection rules. The GDPR imposes strict obligations and restrictions on controllers and processors of personal data including, for example, expanded disclosures about how personal data is to be used, increased requirements pertaining to health data and pseudonymised (i.e., key-coded) data, mandatory data breach notification requirements and expanded rights for individuals over their personal data. This could affect our ability to collect, analyze and transfer personal data, including health data from clinical trials and adverse event reporting, or could cause our costs to increase, and harm our business and financial condition.
While the GDPR, as a directly effective regulation, was designed to harmonize data protection law across the European Union, it does permit member states to legislate in many areas (particularly with regard to the processing of genetic, biometric or health data), meaning that inconsistencies between different member states will still arise. European Union member states have their own regimes on medical confidentiality and national and European Union-level guidance on implementation and compliance practices is often updated or otherwise revised, which adds to the complexity of processing personal data in the European Union.
Risks Related to Our Acquisition of Zavante
We may fail to realize the anticipated benefits of our Acquisition of Zavante, those benefits may take longer to realize than expected, and we may encounter significant integration difficulties.
On July 24, 2018, we completed our acquisition, or the Acquisition, of Zavante Therapeutics, Inc., or Zavante, pursuant to an Agreement and Plan of Merger, or the Merger Agreement, dated July 23, 2018. Our ability to realize the anticipated benefits of the Acquisition will depend, to a large extent, on our ability to integrate Zavante and CONTEPO into our business and business strategy and realize anticipated growth opportunities and synergies. We expect that the integration process will be complex, costly and time-consuming. As a result, we will be required to devote significant management attention and resources to integrating Zavante into our business and CONTEPO into our business strategy. The integration process may be disruptive to our business and the expected benefits may not be achieved within the anticipated time frame, or at all. The failure to meet the challenges involved and to realize the anticipated benefits of the Acquisition could cause an interruption of, or a loss of momentum in, our development and commercialization efforts, including with respect to lefamulin and CONTEPO, and could adversely affect our business, financial condition and results of operations.
Our ability to realize the anticipated benefits of the Acquisition is expected to entail numerous additional material potential difficulties, including, among others:
- •
- any delay or failure in obtaining marketing approvals for CONTEPO, or any delay or failure to commercialize CONTEPO in the United States thereafter;
- •
- increased scrutiny from third parties, including regulators, legislative bodies and enforcement agencies, including with respect to product pricing and commercialization matters;
- •
- changes in laws or regulations that adversely impact the anticipated benefits of the Acquisition;
- •
- challenges related to the perception by patients, the medical community and third-party payors of CONTEPO for the treatment of complicated urinary tract infections, or cUTIs;
- •
- disruptions to our manufacturing arrangements with third-party manufacturers, including our manufacturing and supply arrangements with respect to CONTEPO and disruptions to our third-party distribution channel;
- •
- difficulties in managing the expanded operations of a larger and more complex company following the Acquisition;
- •
- the diversion of management attention to integration matters;
- •
- difficulties in achieving the anticipated business opportunities and growth prospects from the Acquisition;
- •
- the size of the treatable patient population for CONTEPO may be smaller than we believe it is;
- •
- difficulties in assimilating Zavante employees and in attracting and retaining key personnel; and
- •
- potential unknown liabilities, adverse consequences, unforeseen increased expenses or other unanticipated problems associated with the Acquisition.
Many of these factors are outside of our control, and any one of them could result in increased costs, decreased expected revenues and further diversion of management time and energy, which could materially adversely impact our business, financial condition and results of operations.
Upfront consideration for the Acquisition was comprised of 8,152,092 of our ordinary shares, including the 815,186 ordinary shares that are issuable upon release of the Holdback Shares subject to the terms of the Merger Agreement. Pursuant to the Merger Agreement, former Zavante stockholders are also entitled to receive from us, subject to the terms and conditions of the Merger Agreement, up
to $97.5 million in contingent consideration, of which $25.0 million would become payable upon the first approval of a new drug application from the U.S. Food and Drug Administration, or the FDA, for CONTEPO for injection for any indication, or the Approval Milestone Payment, and an aggregate of up to $72.5 million would become payable upon the achievement of specified net sales milestones, or the Net Sales Milestone Payments. At our Extraordinary General Meeting of Shareholders held in October 2018, our shareholders approved the issuance of ordinary shares in settlement of potential milestone payment obligations that may become payable in the future to former Zavante stockholders, including the Approval Milestone Payment which will be settled in our ordinary shares. We also now have the right to settle the Net Sales Milestone Payments in ordinary shares, except as otherwise provided in the Merger Agreement. The issuance of our ordinary shares in connection with the closing of the Acquisition was dilutive to our existing shareholders, and the future issuance of our ordinary shares to satisfy our milestone payment obligations would be further dilutive to our then existing shareholders.
Also, following the Acquisition, we now possess certain liabilities and obligations, including contractual liabilities and obligations, that were assumed by us upon closing of the Acquisition. Prior to the Acquisition, former Zavante stockholders and SG Pharmaceuticals, Inc. entered into a stock purchase agreement, dated as of May 5, 2015, or the Stock Purchase Agreement, pursuant to which SG Pharmaceuticals, Inc. acquired all of the outstanding capital stock of Zavante from the Zavante selling stockholders and SG Pharmaceuticals, Inc., subsequently merged with and into Zavante, with Zavante as the surviving entity. Pursuant to the Stock Purchase Agreement, Zavante (as successor to SG Pharmaceuticals, Inc.) is obligated to make milestone payments to the selling stockholders of $3.0 million upon marketing approval by the FDA with respect to any oral, intravenous or other form of fosfomycin, or the Zavante Products, and milestone payments of up to $26.0 million in the aggregate upon the occurrence of various specified levels of net sales with respect to the Zavante Products. In addition, Zavante is obligated to make annual royalty payments to the selling stockholders of a mid-single-digit percentage of net sales of Zavante Products, subject to adjustment based on net sales thresholds and with such percentage reduced to low single-digits if generic fosfomycin products account for half of the applicable market on a product-by-product and country-by-country basis. The Stock Purchase Agreement also provides that Zavante will pay to the selling stockholders a mid-single-digit percentage of transaction revenue in connection with the consummation of the grant, sale, license or transfer of market exclusivity rights for a qualified infectious disease product (within the meaning of the 21st Century Cures Act, or the Cures Act) related to a Zavante Product.
In addition, we expect to incur expenses related to the continued development, regulatory approval process and commercialization with respect to CONTEPO. Zavante has entered into a manufacturing and supply agreement with Fisiopharma, S.r.l. pursuant to which Zavante has an obligation to purchase a minimum percentage of its commercial requirements of CONTEPO in the United States. Zavante has also entered into a manufacturing and exclusive supply agreement with Laboratorios ERN, S.A., pursuant to which Laboratorios ERN, S.A. has agreed to supply Zavante with certain technical documentation and data as required for submission of an NDA, or ANDA for CONTEPO and certain regulatory support in connection with the commercial sale and use of CONTEPO in the United States, and which provides for payments to Laboratorios ERN, S.A. of a one-time cash payment upon the first commercial sale of CONTEPO and subsequent quarterly payments thereafter based on the number of vials of CONTEPO sold in the United States during each quarter.
Because we have limited financial resources, by investing in the Acquisition, we may forgo or delay pursuit of other opportunities that may have proven to have greater commercial potential. Further, it is possible that undisclosed, contingent, or other liabilities or problems may arise in the future of which we were previously unaware. These undisclosed liabilities could have an adverse effect on our business, financial condition and results of operations.
All of these factors could decrease or delay the expected accretive effect of the Acquisition and negatively impact our stock price. As a result, it cannot be assured that the Acquisition will result in the full realization of the benefits anticipated from the Acquisition or in the anticipated time frames or at all.
Risks Related to Employee Matters and Managing Growth
Our future success depends on our ability to retain our chief executive officer and other key executives and to attract, retain and motivate key executives and qualified personnel.
We are highly dependent on Dr. Colin Broom, our Chief Executive Officer, and the other principal members of our management and scientificcommercial teams. Although we have formal employment agreements with each of our executive officers, these agreements do not prevent our executives from terminating their employment with us at any time. We do not maintain ‘‘key person’’"key person" insurance on any of our executive officers. The unplanned loss of the services of any of these persons might impede the achievement of our research, development and commercialization objectives.
Recruiting and retaining qualified scientific, clinical, manufacturing and sales and marketing personnel, including in the United States and Ireland where we plan to expand our physical presence, will also be critical to our success. We may not be able to attract and retain these personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we cannot recruit and retain qualified personnel, we may be unable to successfully develop our product candidates, conduct our clinical trials and commercialize our product candidates.
We expect to expand the number of our development, regulatoryemployees and sales and marketing capabilities,the scope of our operations in the certain areas, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of drug development, regulatory affairs, technical operations, supply chain, medical affairs and, subject to obtaining marketing approval of lefamulin and CONTEPO, sales and marketing. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. In addition, our growth in connection with the Acquisition, including expansion of our business operations and employees who joined us in connection with the Acquisition, has imposed added responsibilities on members of our management, including the need to recruit, hire, retain, motivate and integrate additional employees and business operations.
Due to our limited financial resources and the limited experience of our management team in managing a company of our current size, and with such anticipated growth, we may not be able to effectively manage the future expansion of our operations or recruit and train additional qualified personnel. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
Risks Related to Ownership of American DepositaryOur Ordinary Shares
An active trading market for the ADSsour ordinary shares may not be sustained.
Our ADSsordinary shares began trading on the NASDAQNasdaq Global Market on September 18, 2015.June 26, 2017. Given the limited trading history of the ADSs,our ordinary shares, there is a risk that an active trading market for the ADSsour
ordinary shares will not be sustained, which could put downward pressure on the market price of the ADSsour ordinary shares and thereby affect the ability of our security holders to sell their ADSs.shares.
The price of the ADSsour ordinary shares may be volatile and fluctuate substantially.
The trading price of the ADSsour ordinary shares has been and is likely to continue to be volatile. The stock market in general and the market for smaller biopharmaceutical companies in particular have experienced significant volatility that has often been unrelated to the operating performance of particular companies. The market price for the ADSsour ordinary shares may be influenced by many factors, including:
- •
·our ability to successfully implement our proposed business strategy;- •
- the success of competitive products or technologies;
· - •
- results of clinical trials of our product candidates or those of our competitors;
· - •
- regulatory delays and greater government regulation of potential products due to adverse events;
· - •
- regulatory or legal developments in the United States, the European Union and other countries;
· - •
- developments or disputes concerning patent applications, issued patents or other proprietary rights;
· - •
- the recruitment or departure of key scientific or management personnel;
· - •
- the level of expenses related to any of our product candidates or clinical development programs;
· - •
- the results of our efforts to discover, develop, acquire or in-license additional product candidates or products;
· - •
- one of our manufacturers or suppliers could have an event which causes an unforeseen disruption of the manufacture or supply of our product candidates;
- •
- actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts;
· - •
- variations in our financial results or those of companies that are perceived to be similar to us;
· - •
- perception and market performance of companies that are perceived to be similar to us:
- •
- changes in the structure of healthcare payment systems;
· - •
- market conditions in the pharmaceutical and biotechnology sectors;
· - •
- general economic, industry and market conditions; and
· - •
- the other factors described in this
‘‘Risk Factors’’"Risk Factors" section.
In the past, following periods of volatility in the market price of a company’scompany's securities, securities class-action litigation has often been instituted against that company. We also may face securities class-action litigation if we cannot obtain regulatory approvals for or if we otherwise fail to commercialize lefamulin, CONTEPO or any of our other product candidates.candidates or if our securities experience volatility for any reason. Such litigation, if instituted against us, could cause us to incur substantial costs to defend such claims and divert management’smanagement's attention and resources.
Our senior managers, supervisory board membersexecutive officers, directors and principal shareholders, if they choose to act together, have the ability to controlsignificantly influence most matters submitted to shareholders for approval.
Our senior managersexecutive officers and supervisory board members,directors, combined with our shareholders, and their respective affiliates who owned more than 5% of our outstanding commonordinary shares as of DecemberJanuary 31, 20162019 in the aggregate,
beneficially owned approximately 76.9%38.4% of our share capital.capital, assuming the issuance of all Holdback Shares under the Merger Agreement. As a result, if these shareholders were to choose to act together, they would be able to controlsignificantly influence most matters submitted to our shareholders for approval, as well as our management and affairs. For example, these persons, if they choose to act together, would control
significantly influence the election of supervisory board membersdirectors and could, depending on the structure of the particular transaction, significantly influence the approval of anya merger, consolidation or sale of all or substantially all of our assets.
ADSs representing only a relatively small percentage of our common shares are publicly traded, which may limit the liquidity of the ADS and may have a material adverse effect on the price of the ADSs.
As of December 31, 2016 only 23.1% of our common shares were beneficially owned by parties other than our supervisory board members, senior management, shareholders holding 5% or more of our common shares, and their respective affiliates. As a result, ADSs representing only a relatively small number of our common shares are actively traded in the public market. Limited liquidity may increase the volatility of the price of the ADSs.
The ADSs and our common Our ordinary shares do not trade on any exchange outside of the United States.
Our ADSsordinary shares are listed only in the United States on The NASDAQNasdaq Global Market, and we have no plans to list the ADSs or our commonordinary shares in any other jurisdiction. As a result, a holder of ADSs or commonordinary shares outside of the United States may not be able to effect transactions in the ADSsour ordinary shares as readily as the holder may if the ADSsour ordinary shares were listed on an exchange in that holder’sholder's home jurisdiction. Additionally, a holder
Substantial future sales of commonour ordinary shares may not be able to effect transactions in our common shares without depositing such common shares with our depositary in exchange for the issuance of ADSs representing such common shares.
The sale of a substantial number of ADSs maypublic market, or the perception that these sales could occur, could cause the market price of the ADSsour ordinary shares to decline.decline significantly, even if our business is doing well.
Sales of a substantial number of our commonordinary shares, or ADS, or the perception in the market that these sales could occur, could reduce the market price of the ADSs. Each ADS represents one tenth (1/10) of a common share and weour ordinary shares. We had 2,719,695 common67,019,094 ordinary shares outstanding as of December 31, 2016, of which 2,242,302 shares are represented by 22,423,020 American Depositary Shares. Moreover, holders of an aggregate of 951,306 common shares have rights, subject to specified conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other shareholders. Once we register these shares, they can be freely sold in the public market upon issuance, subject to volume limitations applicable to affiliates.2018. To the extent any of these shares are sold into the market, particularly in substantial quantities, the market price of the ADSsour ordinary shares could decline.
Future issuances of commonordinary shares pursuant to our equity incentive plans could also result in additional dilution ofa reduction in the percentage ownershipmarket price of our shareholders.ordinary shares. We have filed a registration statementstatements on Form S-8 on November 18, 2015registering all of the ordinary shares that covers an aggregate of 201,632 common shares reserved for issuance pursuant towe may issue under our equity incentivecompensation plans. Additionally,These shares can be freely sold in the public market upon issuance and once vested, subject to volume, notice and manner of sale limitations applicable to affiliates. The majority of commonordinary shares that may be issued under our equity compensation plans also remain subject to vesting in tranches over a four-year period. As of December 31, 2016,2018, an aggregate of 53,3111,963,877 options to purchase our commonordinary shares had vested and become exercisable. No restricted stock units have vested as of December 31, 2018.
In addition, in March 2018, we entered into a Controlled Equity OfferingSM Sales Agreement, or the ATM Agreement, with Cantor Fitzgerald & Co., or Cantor, pursuant to which, from time to time, we may offer and sell our ordinary shares having an aggregate offering price of up to $50.0 million through Cantor. As of December 31, 2018, we had issued and sold an aggregate of 4,778,031 ordinary shares under the ATM Agreement. From December 31, 2018 through March 1, 2019 , we issued and sold an aggregate of 2,748,048 ordinary shares under the ATM Agreement.
If a large number of the ADSsour ordinary shares are sold in the public market after they become eligible for sale, the sales could reduce the trading price of the ADSsour ordinary shares and impede our ability to raise future capital.
Upfront consideration for the Acquisition is comprised of 8,152,092 of our ordinary shares, including 815,186 ordinary shares that are issuable upon release of the Holdback Shares subject to the terms of the Merger Agreement. The shares that were issued in connection with the closing of the Acquisition are able to be freely sold in the public market, subject to any requirements and restrictions, including any applicable volume limitations, imposed by Rule 144 under the Securities Act. While the Holdback Shares will be restricted as a result of securities laws at the time of issuance, following expiration of applicable holding periods, the Holdback Shares will also be able to be freely sold in the public market, subject to any requirements and restrictions, including any applicable volume limitations, imposed by Rule 144 under the Securities Act. In addition, the Merger Agreement provides that we
may issue up to an additional $97.5 million in our ordinary shares to former Zavante stockholders upon the achievement of specified regulatory and commercial milestones in the future and obligates us to provide registration rights with respect to the registration for resale of such additional ordinary shares that may become issuable upon the achievement of such milestones.
The sale or resale of these shares in the public market, or the market's expectation of such sales, may result in an immediate and substantial decline in our stock price. Such a decline will adversely affect our investors and also might make it difficult for us to sell equity securities in the future at a time and at a price that we deem appropriate.
We are an ‘‘emerging"emerging growth company’’company", and the reduced disclosure requirements applicable to emerging growth companies may make the ADSsour ordinary shares less attractive to investors.
We are an ‘‘emerging"emerging growth company,’’" as that term is used in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and may remain an emerging growth company until December 31, 2020 or such earlier time that we are no longer an emerging growth company. For so long as we remain an emerging growth company, we are permitted and may take advantage of specified reduced reporting and other burdens that are otherwise applicable generally to public companies. These provisions include:
- •
·an exemption from compliance with the auditor attestation requirement of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, on the design and effectiveness of our internal controls over financial reporting;·- •
- an exemption from compliance with any requirement that the Public Company Accounting Oversight Board may adopt regarding mandatory audit firm rotation or a supplement to the
auditor’sauditor's report providing additional information about the audit and the financial statements;· - •
- reduced disclosure about the
company’scompany's executive compensation arrangements; and· - •
- exemptions from the requirements to obtain a non-binding advisory vote on executive compensation or a shareholder approval of any golden parachute arrangements.
We may choose to take advantage of some, but not all, of the available exemptions. We may take advantage of these provisions until December 31, 2020 or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company upon the earlier to occur of: the last day of the fiscal year in which we have more than $1 billion (as may be inflation-adjusted by the SEC from time-to-time) in annual revenues; the date we qualify as a ‘‘large"large accelerated filer,’’" with more than $700 million in market value of our share capital held by non-affiliates; or the issuance by us of more than $1 billion of non-convertible debt over a three-year period.
We may choose to take advantage of some, but not all, of the available benefits under the JOBS Act. We cannot predict whether investors will find the ADSs or commonour ordinary shares less attractive if we rely on such exemptions. If some investors find the ADSsour ordinary shares less attractive as a result, there may be a less active trading market for the ADSsour ordinary shares and the market price of the ADSsour ordinary shares may be more volatile.
In addition, the JOBS Act also provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this exemption and, therefore, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies that are not emerging growth companies.
We lost our foreign private issuer status on January 1, 2017, which requires us to comply with the Exchange Act’s domestic reporting regime, as well as NASDAQ’s domestic company corporate governance requirements, which we expect will cause us to incur significant legal, accounting and other expenses.
We determined that, as of June 30, 2016, we no longer qualified as a ‘‘foreign private issuer’’ under the rules and regulations of the SEC. As a result, beginning January 1, 2017, our future annual filings with the SEC will be made on Form 10-K (including our annual report for the year ending December 31, 2016) rather than on Form 20-F. In addition, commencing on January 1, 2017, we also expanded our reporting to be consistent with that of a domestic filer in the United States, including filing quarterly reports on Form 10-Q and current reports on Form 8-K. In addition, we will prepare our financial statements in accordance with U.S. GAAP, rather than IFRS, and we will adopt new or revised U.S. GAAP accounting standards on the relevant dates on which adoption of such standards is required for other public companies that are not emerging growth companies. We also are now subject to SEC rules governing the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; the provisions of Regulation Fair Disclosure, which regulate the selective disclosure of material information; and the sections of the Exchange Act requiring insiders to file public reports of their share ownership and trading activities and establishing insider liability for profits realized from any ‘‘short-swing’’ transactions in our equity securities. In addition, we are now subject to the NASDAQ Stock Market listing requirements applicable to domestic issuers.
We expect the regulatory and compliance costs to comply with the reporting and corporate governance requirements applicable to a domestic issuer will be significantly higher than the costs we have historically incurred as a foreign private issuer. As a result, we expect that the loss of our foreign private issuer status will increase our legal and financial compliance costs and may make some activities highly time consuming and costly. We also expect that our loss of foreign private issuer status may make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our supervisory board.
We have broad discretion in the use of our funds and may not use them effectively.
Our management hasWe have broad discretion in the application of our available funds and could spend the funds in ways that do not improve our results of operations or enhance the value of the ADSs. Theour ordinary shares. Our failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business, cause the price of the ADSsour ordinary shares to decline and delay the development of our product candidates. Pending their use, we may invest funds in a manner that does not produce income or that loses value.
We incur increased costs as a result of operating as a public company, and our management is required to devote substantial time to new compliance initiatives and corporate governance practices.
As a public company we incur, and particularly after we are no longer an emerging growth company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of The NASDAQNasdaq Global Market and other applicable securities rules and regulations impose various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations have increased our legal and financial compliance costs and make some activities more time-consuming and costly. For example, these rules and regulations have made it more expensive for us to obtain director and officer liability insurance, which in turnand if such insurance becomes prohibitively expensive, this could make it more difficult for us to attract and retain qualified members of our supervisory board.
For as long as we remain an emerging growth company, we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies as described elsewhere in this ‘‘Risk Factors’’ section. We may remain an emerging growth company until December 31, 2020, although if the market value of our share capital that is held by non-affiliates exceeds $700 million as of any June 30 before that time or if we have annual gross revenues of $1 billion or more in any fiscal year, we would cease to be an emerging growth company as of December 31 of the applicable year. We also would cease to be an emerging growth company if we issue more than $1 billion of non-convertible debt over a three-year period.
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, security holders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of the ADSs.our ordinary shares.
Effective internal control over financial reporting is necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, is designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us, as and when required, conducted in connection with Section 404 of the Sarbanes-Oxley Act, or Section 404, or any subsequent testing by our independent registered public accounting firm, as and when required, may reveal deficiencies in our internal control over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement. As a growing company, implementing and maintaining effective controls may require more resources, and we may encounter internal control integration difficulties. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of the ADSs.our ordinary shares.
Pursuant to Section 404, we will be required to furnish a report by our management on our internal control over financial reporting. However, as an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm until we are no longer an emerging growth company. To achieve compliance with Section 404 within the prescribed period, we are engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate
through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
United States investors may have difficulty enforcing civil liabilitiesjudgments against us, our supervisory board members or senior managementdirectors and the experts named in this Annual Report.executive officers.
We are incorporated under the laws of Austria,Ireland, and our registered offices and a substantial portion of our assets are located outside of the United States. In addition, a member of our supervisory board is a resident of a jurisdiction other than the United States. As a result, it may not be possible to effect service of process on such
person or us in the United States or to enforce judgments obtained in courts in the United States against such person or us based on civil liability provisions of the securities laws of the United States. In addition, it
There is questionable whether a court in Austria would accept jurisdictionno treaty between Ireland and impose civil liability if proceedings were commenced in such court predicated solely upon U.S. federal securities laws. As the United States and Austria do not currently have a treaty providing for the reciprocal recognition and enforcement of judgments obtained in civilthe other jurisdiction and commercial matters (other than arbitration awardsIrish common law rules govern the process by which a U.S. judgment may be enforced in such matters),Ireland. The following requirements must be met as a precondition before a U.S. judgment will be eligible for enforcement in Ireland:
- •
- the judgment must be for a definite sum;
- •
- the judgment must be final and conclusive, and the decree must be final and enforceable in the court which pronounces it;
- •
- the judgment
for payment of money renderedmust be provided by afederalcourt of competent jurisdiction, and the procedural rules of the court giving the foreign judgment must have been observed; - •
- the U.S. court must have had jurisdiction in relation to the particular defendant according to Irish conflict of law rules; and
- •
- jurisdiction must be obtained by the Irish courts over judgment debtors in enforcement proceedings by service in Ireland or
stateoutside Ireland in accordance with the applicable court rules in Ireland.
Even if the United States based on civil liability, whetherabove requirements have been met, an Irish court may exercise its right to refuse to enforce the U.S. judgment if the Irish court is satisfied that the judgment (1) was obtained by fraud; (2) is in contravention of Irish public policy; (3) is in breach of natural justice; or not(4) is irreconcilable with an earlier judgment. By way of example, a judgment of a U.S. court of liabilities predicated solely upon U.S. federal securities laws will not be enforceable, either in whole or in part, in Austria. However, if the party in whose favor such final judgment is rendered brings a new suit in a competent court in Austria, such party may submit to the Austrian court the final judgment rendered in the United States. Under such circumstances, a judgment by a federal or state court of the United States against the company will be regarded by an Austrian court only as evidence of the outcome of the dispute to which such judgment relates, and an Austrian court may choose to re-hear the dispute. In addition, awards of punitive damages in actions brought in the United States or elsewhere may be unenforceable in Austria. An award for monetary damages under the securities laws of the United States would be considered punitive if it does not seek to compensate the claimant for loss or damage suffered and is intended to punish the defendant.
Holders of ADSs may not have the same voting rights as the holders of our common shares and may not receive voting materials in time to be able to exercise their right to vote.
Holders of the ADSs may not be able to exercise voting rights attaching to the common shares evidencedenforced by the ADSs. Holders of the ADSs will have the right to instruct the depositary with respect to the voting of the common shares represented by the ADSs. If we tell the depositary to solicit your voting instructions, the depositary is required to endeavor to carry out your instructions. If we do not tell the depositary to solicit your voting instructions (and we are not required to do so), you can still send instructions, and, in that case, the depositary may, but is not required to, carry out those instructions. You may not receive voting materials in time to instruct the depositary to vote, and it is possible that you, or persons who hold their ADSs through brokers, dealers or other third parties, will not have the opportunity to exercise a right to vote.
Holders of ADSs may not have the same rights to participate in subscription rights offering as holders of our common shares.
Under Austrian law, whenever we issue new common shares, we are required by law, subject to certain limited exceptions, to grant subscription rights to all holders of our common shares, giving them the right to purchase a sufficient number of new common shares to maintain their existing ownership percentage. Although we may take steps to offer common shares (in the form of ADSs) to holders of ADSs in connection with any future rights offering, we are not required to do so. We also are not required to ensure that holders of ADSs have an opportunity to participate in any rights offeringIrish courts on the same terms as holdersgrounds of our common shares.public policy if that U.S. judgment includes an award of punitive damages. Further, an Irish court may stay proceedings if concurrent proceedings are being brought elsewhere.
Holders of ADSs may not receive distributions on our common shares represented by the ADSs or any value for them if it is illegal or impractical to make them available to such holders.
The depositary for the ADSs has agreed to pay to holders of ADSs or distribute the cash dividends or other distributions it or the custodian receives on our common shares or other deposited securities after deducting its fees and expenses. Holders of ADSs will receive these distributions in proportion to the number of our common shares their ADSs represent. However, in accordance with the limitations set forth in the deposit agreement, it may be unlawful or impractical to make a distribution available to holders of ADSs. We have no obligation to take any other action to permit the distribution of the ADSs, common shares, rights or anything else to holders of the ADSs. This means that holders of ADSs may not receive the distributions we make on our common shares or any value from them if it is unlawful or impractical to make them available to such holders. These restrictions may have a material adverse effect on the value of the ADSs.
We do not expect to pay dividends in the foreseeable future.
We have not paid any dividends on our commonordinary shares since our incorporation. Even if future operations lead to significant levels of distributable profits, we currently intend that earnings, if any, will be reinvested in our business and that dividends will not be paid until we have an established revenue stream to support continuing dividends. If we propose to pay dividends in the future, we must do so in accordance with Irish law, which provides that distributions including dividend payments, share repurchases and redemptions be funded from "distributable reserves." In addition, the terms of the Loan Agreement with Hercules currently, and the terms of any future debt agreements may in the future, preclude us from paying dividends. Payment of future dividends to security holders will be at the discretion of the management board, subject to the approval of the supervisoryour board, after taking into account various factors including our business prospects, cash requirements, financial performance, debt covenant limitations and new
product development. In addition, Austrian law imposes limitations onAs
a result, capital appreciation, if any, of our ability to pay dividends. Under Austrian law, a company may only pay dividends ifordinary shares will be the distributionsole source of dividends is proposed bygain for holders of our ordinary shares for the management board and the supervisory board and resolved by the company’s shareholders at a general meeting. Our ability to pay dividends is assessed by our management board based primarily on our unconsolidated financial statements prepared in accordance with the Austrian Commercial Code (Unternehmensgesetzbuch). Dividends may be paid only after the relevant balance sheet date from the net profit (Bilanzgewinn) recorded in our unconsolidated annual financial statements as approved by our supervisory board or by our shareholders at a general meeting. In determining the amount available for distribution, the annual net income must be adjusted to account for any accumulated undistributed net profit or loss from previous years as well as for withdrawals from or allocations to reserves. Certain reserves must be established by law, and allocation to such reserves must therefore be deducted from the annual net income to calculate the annual net profit.foreseeable future.
We are exposed to risks related to currency exchange rates.
A significant portion of our expenses are denominated in currencies other than the U.S. dollar. Because our consolidated financial statements are presented in U.S. dollars, changes in currency exchange rates have had and could have a significant effect on our operating results. Exchange rate fluctuations between foreign currencies and the U.S. dollar create risk in several ways, including the following:
- •
·weakening of the U.S. dollar may increase the U.S. dollar cost of overseas research and development expenses;·- •
- strengthening of the U.S. dollar may decrease the value of our revenues denominated in other currencies;
· - •
- the exchange rates on non-U.S. dollar transactions and cash deposits can distort our financial results; and
As a holding company, our operating results, financial condition and ability to pay dividends or other distributions are entirely dependent on funding, dividends and other distributions received from our subsidiaries, which may be subject to restrictions.
Our ability to pay dividends or other distributions and to pay our obligations in the future will depend on the level of funding, dividends and other distributions, if any, received from our subsidiaries and any new subsidiaries we establish in the future. The ability of our subsidiaries to make loans or distributions (directly or indirectly) to us may be restricted as a result of several factors, including restrictions in financing agreements and the requirements of applicable law and regulatory and fiscal or other restrictions. In particular, our subsidiaries and any new subsidiaries may be subject to laws that restrict dividend payments, authorize regulatory bodies to block or reduce the flow of funds from those subsidiaries to us, or limit or prohibit transactions with affiliates. Restrictions and regulatory action of this kind could impede access to funds that we may need to make dividend payments or to fund our own obligations.
Furthermore, we may guarantee some of the payment obligations of certain of our subsidiaries from time to time. These guarantees may require us to provide substantial funds or assets to our subsidiaries or their creditors or counterparties at a time when we are in need of liquidity to fund our own obligations.
· The ownership percentage of our shareholders may be diluted in the future which could dilute the voting power or reduce the value our outstanding ordinary shares. commercial pricing
As with any publicly traded company, the ownership percentage of our shareholders may be diluted in the future because of equity issuances for acquisitions, capital market transactions or otherwise, including equity awards that we intend to continue to grant to our directors, officers and profit marginsemployees. From time to time, we may issue additional options or other share awards to our directors, officers and employees under our benefits plans. Our employees are affected by currency fluctuations.also entitled, subject to certain conditions, to purchase our ordinary shares at a discount pursuant to our Employee Share Purchase Plan.
In addition, our articles of association authorize us to issue, without the approval of our shareholders, one or more classes or series of preferred shares having such designation, powers, preferences and relative, participating, optional and other special rights, including preferences over our ordinary shares respecting dividends and distributions, as our board of directors generally may determine. The terms of one or more classes or series of preferred shares could dilute the voting power
or reduce the value of our ordinary shares. Similarly, the repurchase or redemption rights or liquidation preferences we could assign to holders of preferred shares could affect the residual value of the ordinary shares. Additionally, we may issue and sell our ordinary shares under our ATM Agreement from time to time, and we may issue additional ordinary shares as contingent consideration upon the achievement of certain regulatory and commercialization milestones, subject to the terms and conditions of the Merger Agreement. See"—Risks Related to Ownership of our Ordinary Shares—Substantial future sales of our ordinary shares in the public market, or the perception that these sales could occur, could cause the price of our ordinary shares to decline significantly, even if our business is doing well.".
The rights of our shareholders may differ from the rights typically offered to shareholders of a U.S. corporation. We are organizedincorporated as a stock corporation (Aktiengesellschaft) and incorporatedpublic limited company under AustrianIrish law.
The rights of holdersour shareholders are governed by our memorandum and articles of association and Irish law. The rights associated with our commonordinary shares and, therefore, certain ofare different to the rights generally associated with shares held in a U.S. corporation. Material differences between the rights of holdersshareholders of a U.S. corporation and the ADSs, are governed by Austrianrights of our shareholders include differences with respect to, among other things, distributions, dividends, repurchases and redemptions, bonus issues, the election of directors, the removal of directors, the fiduciary and statutory duties of directors, conflicts of interests of directors, the indemnification of directors and officers, limitations on director liability, the convening of annual meetings of shareholders and special shareholder meetings, notice provisions for meetings, the quorum for shareholder meetings, the adjournment of shareholder meetings, the exercise of voting rights, shareholder suits, rights of dissenting shareholders, anti-takeover measures and provisions relating to the ability to amend the articles of association.
As an Irish public limited company, certain capital structure decisions require shareholder approval, which may limit our flexibility to manage our capital structure.
Under Irish law, includingour board of directors may increase our authorized share capital and issue new ordinary or preferred shares up to a maximum amount equal to the provisions of the Austrian Stock Corporation Act, andauthorized but unissued share capital, without shareholder approval, once authorized to do so by our articles of association. Theseassociation or by an ordinary resolution of our shareholders. Additionally, subject to specified exceptions, Irish law grants statutory preemption rights differto existing shareholders where shares are being issued for cash consideration but allows shareholders to disapply such statutory preemption rights either in important respectsour articles of association or by way of special resolution. Such disapplication can either be generally applicable or be in respect of a particular allotment of shares. Accordingly, our articles of association contain, as permitted by Irish company law, provisions authorizing our board of directors to issue new shares, and to disapply statutory preemption rights. The authorization of our board of directors to issue shares and the disapplication of statutory preemption rights must both be renewed by the shareholders at least every five years, and we cannot provide any assurance that these authorizations will always be approved, which could limit our ability to issue equity and thereby adversely affect the holders of our ordinary shares.
Irish law differs from the laws in effect in the U.S. with respect to defending unwanted takeover proposals and may give our board less ability to control negotiations with hostile offerors.
We are subject to the Irish Takeover Panel Act, 1997, Takeover Rules, 2013. Under those Irish Takeover Rules, the board is not permitted to take any action that might frustrate an offer for our ordinary shares once the board has received an approach that may lead to an offer or has reason to believe that such an offer is or may be imminent, subject to certain exceptions. Potentially frustrating actions such as (i) the issue of ordinary shares, options or convertible securities, (ii) material acquisitions or disposals, (iii) entering into contracts other than in the ordinary course of business or
(iv) any action, other than seeking alternative offers, which may result in frustration of an offer, are prohibited during the course of an offer or at any earlier time during which the board has reason to believe an offer is or may be imminent. These provisions may give the board less ability to control negotiations with hostile offerors and protect the interests of holders of ordinary shares than would be the case for a corporation incorporated in a jurisdiction of the United States.
The operation of the Irish Takeover Rules may affect the ability of certain parties to acquire our ordinary shares.
Under the Irish Takeover Rules, if an acquisition of ordinary shares were to increase the aggregate holding of the acquirer and its concert parties to ordinary shares that represent 30% or more of the voting rights of shareholdersa company, the acquirer and, in typical U.S. corporations. These differences include, in particular:
· Under Austrian law, certain important resolutions, including, for example, capital decreases, mergers, conversions and spin-offs,circumstances, its concert parties would be required (except with the issuance of convertible bonds or bonds with warrants attached and the dissolutionconsent of the stock corporation (apart from insolvencyIrish Takeover Panel) to make an offer for the outstanding ordinary shares at a price not less than the highest price paid for the ordinary shares by the acquirer or its concert parties during the previous 12 months. This requirement would also be triggered by an acquisition of ordinary shares by a person holding (together with its concert parties) ordinary shares that represent between 30% and certain other proceedings), require the vote of a 75% majority (and, in some cases, as high as a 90% majority)50% of the capital present or represented atvoting rights in the relevant general meetingcompany if the effect of shareholders. Therefore, the holder or holders of a blocking minority of 25% or, depending on the attendance level at the general meeting, the holder or holders of a smallersuch acquisition were to increase that person's percentage of the voting rights by 0.05% within a 12-month period. The Irish Takeover Rules could therefore discourage an investor from acquiring 30% or more of our outstanding ordinary shares, unless such investor was prepared to make a bid to acquire all outstanding ordinary shares.
Certain separate concert parties will also be presumed to be acting in concert. Our board of directors and their relevant family members, related trusts and "controlled companies" are presumed to be acting in concert with any corporate shareholder who holds 20% or more of the company. The application of these presumptions may result in restrictions upon the ability of any of the concert parties and members of our board of directors to acquire more of our securities, including under the terms of any executive incentive arrangements. We may consult with the Irish Takeover Panel with respect to the application of this presumption and the restrictions on the ability to acquire further securities if necessary, although we are unable to provide any assurance as to whether the Irish Takeover Panel would overrule this presumption.
We will be exposed to the risk of future changes in law, which could materially adversely affect us.
We are subject to Irish law. As a result, we are subject to the risk of future adverse changes in Irish law (including Irish corporate and tax law). In addition, we and our subsidiaries are also subject to the risk of future adverse changes in Austrian and U.S. law, as well as changes of law in other countries in which we and our subsidiaries operate.
Future adverse changes in law could result in our not being able to maintain a worldwide effective corporate tax rate that is competitive in our industry.
While we believe that being incorporated in Ireland should not affect our ability to maintain a worldwide effective corporate tax rate that is competitive in our industry, we cannot give any assurance as to what our effective tax rate will be because of, among other things, uncertainty regarding the tax policies of the jurisdictions where we will operate. The tax laws of Ireland, Austria, the United States, and other jurisdictions could change in the future, and such changes could cause a material change in our worldwide effective corporate tax rate. In particular, legislative action could be taken by Ireland, Austria, the United States or other jurisdictions which could override tax treaties upon which we expect to rely and adversely affect our effective tax rate. As a result, our actual effective tax rate may be materially different from our expectation.
Comprehensive tax reform legislation could adversely affect our business and financial condition.
The Tax Cuts and Jobs Act of 2017, or the Tax Act, introduced significant changes to the United States Internal Revenue Code of 1986, as amended, or Code.
The Tax Act, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, limitation of the tax deduction for interest expense, limitation of the deduction for net operating losses, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, and modifying or repealing many business deductions and credits.
We continue to examine the impact the Tax Act may have on our business. Notwithstanding the reduction in the federal corporate income tax rate, the overall impact of the Tax Act is uncertain and our business and financial condition could be adversely affected.
After tax reform, U.S. persons who own 10 percent or more of our shares may be subject to U.S. federal income taxation on certain of our foreign subsidiaries' income even if such income is not distributed to such U.S. persons.
A foreign corporation is treated as a "controlled foreign corporation", or CFC, for U.S. federal income tax purposes if, on any day during a taxable year, "United States shareholders" (as defined below) own (directly, indirectly or constructively within the meaning of Section 958 of the Code) more than 50% of the total combined voting power of all classes of our voting shares or more than 50% of the total value of all of our shares. A "United States shareholder" of a foreign corporation is a U.S. person who owns (directly, indirectly or constructively within the meaning of Section 958 of the Code) at least 10% of the total combined voting power of voting shares of such non-U.S. corporation or at least 10% of the total value of shares of all classes of stock of such non-U.S. corporation.
As a result of the Tax Act, all of our non-U.S. subsidiaries will be treated as CFCs. The legislative history under the Tax Act indicates that this change was not intended to cause these non-U.S. subsidiaries to be treated as CFCs with respect to a United States shareholder that is not related to the U.S. subsidiary of the Company. However, it is not clear whether the IRS or a court would interpret the change made by the Tax Act in a manner consistent with such indicated intent.
Any United States shareholder who owns our shares (directly or indirectly within the meaning of Section 958(a) of the Code) on the last day in such taxable year must include in its gross income for U.S. federal income tax purposes its pro rata share (based on direct or indirect ownership of value) of the non-U.S. subsidiaries' "subpart F income," regardless of whether that income was actually distributed to such U.S. person (with certain adjustments). "Subpart F income" of a CFC generally includes among other items passive income, such as dividends, interest, annuities, net gains from sales of property that do not generate active income, net commodities gains, net foreign currency gains, passive rents and royalties.
The Tax Act also requires such United States shareholders to include in their gross income for U.S. federal income tax purposes their pro rata share of a CFC's "global intangible low tax income", or GILTI." In general terms, GILTI is the net income of the CFCs (other than income already included in United States shareholders' taxable income) that exceeds 10% of the CFCs' bases in depreciable tangible assets. GILTI is treated in a manner similar to subpart F income.
In addition, if a U.S. person disposes of shares in an Austrian stocka non-U.S. corporation and the U.S. person was a United States shareholder at any time when the corporation was a CFC during the five-year period ending on the date of disposition, any gain from the disposition will generally be treated as a dividend to the extent of the U.S. person's share of the corporation's undistributed earnings and profits that were accumulated during the period or periods that the U.S. person owned the shares while the
corporation was a CFC (with certain adjustments). Also, a U.S. person may be ablerequired to blockcomply with specified reporting requirements, regardless of the number of shares owned.
A transfer of our ordinary shares, other than a transfer effected by means of the transfer of book-entry interests in the Depository Trust Company, may be subject to Irish stamp duty.
Transfers of our ordinary shares effected by means of the transfer of book entry interests in the Depository Trust Company, or DTC, will not be subject to Irish stamp duty. However, if you hold our ordinary shares directly rather than beneficially through DTC, any such votes, possiblytransfer of your ordinary shares could be subject to our detrimentIrish stamp duty (currently at the rate of 1% of the higher of the price paid or the detrimentmarket value of the shares acquired). Payment of Irish stamp duty is generally a legal obligation of the transferee. The potential for stamp duty could adversely affect the price of our other shareholders.ordinary shares.
Our ordinary shares received by means of a gift or inheritance could be subject to Irish capital acquisitions tax.
· AsIrish capital acquisitions tax, or CAT, could apply to a general rule under Austrian law, a shareholder has no direct recourse against the membersgift or inheritance of our ordinary shares irrespective of the management boardplace of residence, ordinary residence or supervisory board of an Austrian stock corporation in the event that it is alleged that any of them have breached their duty of loyalty or duty of care to the Austrian stock corporation. Apart from insolvency or other special circumstances, only the Austrian stock corporation itself has the right to claim damages from membersdomicile of the managementparties. This is because our ordinary shares will be regarded as property situated in Ireland. The person who receives the gift or supervisory board. An Austrian stock corporation may waiveinheritance has primary liability for CAT. Gifts and inheritances passing between spouses are exempt from CAT. Children have a tax-free threshold of €320,000 in respect of taxable gifts or settle these damages claims only after five years, if the shareholders approve the waiver or settlement at the general meeting with a simple majority of the votes cast and no group of shareholders holding, in the aggregate, at least 20% (and in some cases, 5%) of the Austrian stock corporation’s share capital objects to such waiver or settlement and has its opposition formally noted in the minutes of the general meeting. However, Austrian courts acknowledge a waiver or settlement of claims for damages earlier if all shareholders consent to such waiver.inheritances received from their parents.
We may be classified as a passive foreign investment company for our tax year ending December 31, 2017,2018, which may result in adverse U.S. federal income tax consequence to U.S. holders.
Based on our estimated gross income and average value of our gross assets and the nature of our business, we do not believe that we were a ‘‘passive"passive foreign investment company,’’" or PFIC, for U.S. federal income tax purposes for ourany tax years ended December 31, 2014, 2015 or 2016, although forfrom our taxinitial public offering through the year ended December 31, 2016, our calculations indicate that we were close to being so classified, and to the extent of any differences in its own calculations, the U.S. taxing authority might conclude that we were in fact a PFIC for that year.2018. A corporation organized outside the United States generally will be classified as a PFIC for U.S. federal income tax purposes (1) in any taxable year in which at least 75% of its gross income is passive income or on average at least 50% of the gross value of its assets is attributable to assets that produce passive income or are held for the production of passive income and (2) as to a given holder who was a holder in such year and regardless of such corporation’scorporation's income or asset composition, in any subsequent taxable year, unless certain elections are made by that holder that can discontinue that classification as to that holder, certain elections are made that can entailat the risk of imposing substantial tax costs to that holder. Passive income for this purpose generally includes dividends, interest, royalties, rents and gains from commodities and securities transactions. Our status in any taxable year will depend on our assets and activities in each year, and because this is a factual determination made annually after the end of each taxable year, there can be no assurance that we will not be considered a PFIC for the current taxable year or any future taxable year. The marketgross value of our assets may be determined in large part by reference to the market price of the ADSs,our ordinary shares, which may fluctuate considerably given that market prices of biotechnology companies have been especially volatile. If we were to be treated as a PFIC for the tax year ending December 31, 2017,2018, or any other future taxable year during which a U.S. holder held the ADSs,our ordinary shares, however, certain adverse U.S. federal income tax consequences could apply to the U.S. holder. We currently intend to make available the information necessary to permit a U.S. holder to make a valid QEF election, which may mitigate some of the adverse U.S. federal income tax consequences that could apply to a U.S. holder of ADSs.ordinary shares if we determine that we were a PFIC for any taxable year. However, we may choose not to provide such information at a future date.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
Facilities
Facilities
Our facilities consist of approximately 3,100 square meters of leased laboratory and office space in Vienna, Austria. This space serves as our corporate headquarters. We also lease approximately 18,20015,000 square feet of office space in King of Prussia, Pennsylvania. We believe that our existing facilities are adequate to meet our current needs. However, wealso lease office space in Dublin, Ireland and San Diego, California. We may seek to negotiate new leases or evaluate additional or alternate space as we plan for the growth of our commercial operations in the United States.States and Ireland. We believe that suitable additional or alternative space will be available in the future on commercially reasonable terms.
None
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
ITEM 5. MARKET FOR REGISTRANT’SREGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our American Depositary Shares, or ADSs,ordinary shares have been tradinglisted on the Nasdaq Global Market since June 26, 2017 following the Redomiciliation and trade on the Nasdaq Global Market under the symbol "NBRV". Prior to the Redomiciliation, the ADSs, representing the common shares of our predecessor, Nabriva Therapeutics AG, or Nabriva Austria, had traded on the NASDAQ Global Market under the same symbol “NBRV” since September 18, 2015. Prior to that date, there was no public trading market for ADSs or ourEach ADS represented one tenth (1/10) of a common shares. Our initial public offering was priced at $10.25 per ADS on September 17, 2015. The following table sets forth, for the periods indicated, the reported high and low closing sale pricesshare of our ADSs on the NASDAQ Global Market in U.S. dollars.Nabriva Austria.
Stockholders
|
| Price Per ADS |
| ||
|
| $ |
| ||
|
| High |
| Low |
|
Fiscal Year Ended December 31, 2016 |
|
|
|
|
|
|
|
|
|
|
|
Quarterly: |
|
|
|
|
|
Fourth Quarter 2016 |
| 7.50 |
| 3.52 |
|
Third Quarter 2016 |
| 7.40 |
| 7.01 |
|
Second Quarter 2016 |
| 9.70 |
| 6.85 |
|
First Quarter 2016 |
| 9.80 |
| 7.05 |
|
|
|
|
|
|
|
Fiscal Year Ended December 31, 2015 |
|
|
|
|
|
|
|
|
|
|
|
Quarterly: |
|
|
|
|
|
Fourth Quarter 2015 |
| 10.34 |
| 8.66 |
|
Third Quarter 2015 (from September 18, 2015 through September 30, 2015) |
| 13.24 |
| 9.30 |
|
Stockholders
As of March 1, 2017,January 31, 2019, there were 11sixty-eight holders of record of our common shares and seven holders of record of the ADSs.ordinary shares. The number of record holders may not be representative of the number of beneficial owners because many of our commonordinary shares and ADSs are held by depositories, brokers or other nominees, including the Bank of New York Mellon, which serves as the depositary for our common shares under our American Depositary Receipt program.nominees.
Securities Authorized for Issuance Under Equity Compensation Plans
Information regarding securities authorized for issuance under our equity compensation plans is contained in Part III, Item 12 of this Annual Report.Report on Form 10-K.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings to support our operations and finance the growth and development of our business. In addition, the terms of the Loan Agreement with Hercules preclude us from paying dividends. We do not intend to pay cash dividends on our common stockordinary shares for the foreseeable future.
Performance Graph
The performance graph below compares the cumulative total ADS holder return on the ADSs beginning on September 18, 2015, the date our ADS’s began trading on the NASDAQ Global Market, and for each subsequent quarter period end through and including December 31, 2016, with the cumulative return of the NASDAQ Composite Index and NASDAQ Biotechnology Index.
The performance graph comparison assumes $100 was invested in the ADSs and in each of the other indices described above on September 18, 2015. The stock performance shown on the graph below is not necessarily indicative of future price performance.
Cumulative Total Return
Assumes $100 Initial Investment
December 31, 2016
The performance graph above is being furnished solely to accompany this Annual Report on Form 10-K pursuant to Item 201(e) of Regulation S-K, is not being filed for purposes of Section 18 of the Exchange Act, shall not be deemed to be “soliciting material” or subject to Rule 14A of the Exchange Act and is not to be incorporated by reference into any of our filings, whether made before or after the date hereof, except to the extent that we specifically incorporate this information by reference into such filing.
Recent Sales of Unregistered Securities
We did not sell anyOn July 25, 2018, we granted a non-statutory option to purchase 850,000 of our equity securities or any options, warrants, or rightsordinary shares and 150,000 performance-based restricted share units to purchaseTheodore Schroeder, our equity securities during the year ended December 31, 2016 that were not registered under the Securities Act of 1933, as amended,newly appointed Chief Executive Officer, or the Securities Act.CEO. These equity awards were made as an inducement material to the CEO entering into employment with the Company in accordance with Nasdaq Listing Rule 5635(c)(4). The exercise price per share for the share option is $3.53 per share, and the option award has a ten-year term and will vest over a four-year period, with 25% of the shares underlying the award vesting on the first anniversary of the grant date and the remaining 75% of the shares underlying the option award to vest monthly over the subsequent 36-month period. The performance-based restricted share units are subject to vesting as follows: 50% will vest upon certification by the board of directors of the receipt of approval by the FDA of an NDA for each of lefamulin and CONTEPO for any indication, and 50% will vest on the first anniversary of such certification by the board of directors, provided, in each case, the CEO is performing services to us on the applicable vesting dates. If the FDA does not approve an NDA for both lefamulin and CONTEPO by January 31, 2020, the performance-based restricted share units will terminate in full. The shares underlying these option awards and the performance-based restricted share units have been registered on a Form S-8 registration statement.
Purchase of Equity Securities
We did not purchase any of our registered equity securities during the period covered by this Annual Report on Form 10-K.
UseTable of Proceeds from Registered SecuritiesContents
We effected the initial public offering of our American Depositary Shares, or ADSs, each representing one tenth (1/10) of a common share, through a Registration Statement on Form F-1 (File No. 333-205073) that was declared effective by the Securities and Exchange Commission on September 17, 2015. On September 23, 2015, we completed the sale of 9,000,000 ADSs, representing 900,000 of our common shares, at a public offering price of $10.25 per ADS, before underwriting discounts. In addition, we granted the underwriters a 30-day option to purchase up to 1,350,000 additional ADSs to cover over allotments, if any. On September 30, 2015, we completed the additional sale of 1,350,000 ADSs under this option at a price to the public of $10.25 per ADS, resulting in aggregate net proceeds to us of approximately $92.4 million after deducting underwriting discounts and commissions of $7.4 million and offering expenses of $6.3 million. None of the underwriting discounts and commissions or other offering expenses were paid to directors or officers of ours or their
associates or to persons owning 10% or more of any class of our equity securities or to any affiliates of ours. Leerink Partner LLC, RBC Capital Markets, LLC, Needham & Company, LLC and Wedbush PacGrow Inc. were the underwriters for our initial public offering.
There has been no material change in our planned use of the net proceeds from our initial public offering as described in our final prospectus filed with the SEC pursuant to Rule 424(b)(4) on September 21, 2015.
Our management board retains broad discretion in the allocation and use of the net proceeds of our initial public offering.
ITEM 6. SELECTED FINANCIAL DATA
The selected financial data set forth below for the years ended December 31, 2016, 2015,2017 and 20142018 and as of December 31, 20162017 and 20152018 has been derived from our audited consolidated financial statements which have been prepared in accordance with generally accepted accounting practices in the United States and included elsewhere in this Annual Report. Financial data set forth below for the yearyears ended December 31, 20132014 and 2015 and as of December 31, 2014, 2015 and 20132016 has been derived from the audited consolidated financial statements with retrospective adjustment for change in reporting currency to U.S. dollar effective January 1, 2016 and not included in this Annual Report. The following selected consolidated financial data should be read in conjunction with Item 7, “Management’s"Management's Discussion and Analysis of Financial Condition and Results of Operations”Operations" and the consolidated financial statements and the notes thereto included elsewhere in this Annual Report.Report on Form 10-K. The selected financial data in this section are not intended to replace our consolidated financial statements and the related notes. Our historical results are not necessarily indicative of the results that may be expected in the future.
|
| Year ended December 31, |
| ||||||||||
(in thousands) |
| 2013 |
| 2014 |
| 2015 |
| 2016 |
| ||||
Consolidated Operations Data: |
|
|
|
|
|
|
|
|
| ||||
Revenues |
| $ | 3,194 |
| $ | 2,398 |
| $ | 3,767 |
| $ | 6,482 |
|
Costs and Expenses: |
|
|
|
|
|
|
|
|
| ||||
Research and development |
| (10,471 | ) | (9,355 | ) | (23,604 | ) | (47,994 | ) | ||||
General and administrative |
| (3,582 | ) | (3,739 | ) | (7,921 | ) | (13,535 | ) | ||||
Total operating expenses |
| (14,053 | ) | (13,094 | ) | (31,525 | ) | (61,529 | ) | ||||
Loss from operations |
| (10,859 | ) | (10,696 | ) | (27,758 | ) | (55,047 | ) | ||||
Other income (expense): |
|
|
|
|
|
|
|
|
| ||||
Other income (expense), net |
| 31,805 |
| (524 | ) | 2,427 |
| (783 | ) | ||||
Interest income |
| 5 |
| 2 |
| 14 |
| 343 |
| ||||
Interest expense |
| (2,856 | ) | (2,910 | ) | (22,092 | ) | (75 | ) | ||||
Income (loss) before income taxes |
| 18,095 |
| (14,128 | ) | (47,409 | ) | (55,562 | ) | ||||
Income tax (expense) benefit |
| (1,030 | ) | (94 | ) | 445 |
| 672 |
| ||||
Net income (loss) |
| $ | 17,065 |
| $ | (14,222 | ) | $ | (46,964 | ) | $ | (54,890 | ) |
|
| As of December 31, |
| ||||||||||
(in thousands) |
| 2013 |
| 2014 |
| 2015 |
| 2016 |
| ||||
Consolidated Balance Sheet Data: |
|
|
|
|
|
|
|
|
| ||||
Cash and cash equivalents and short- term investments |
| $ | 4,539 |
| $ | 2,150 |
| $ | 36,446 |
| $ | 32,778 |
|
Total assets |
| 7,343 |
| 4,812 |
| 117,711 |
| 93,240 |
| ||||
Long term liabilities |
| 2,856 |
| 5,741 |
| 84 |
| 107 |
| ||||
Total liabilities |
| 25,969 |
| 33,192 |
| 9,005 |
| 15,984 |
| ||||
Mezzanine equity |
| — |
| 634 |
| — |
| — |
| ||||
Accumulated deficit |
| (106,173 | ) | (120,587 | ) | (171,426 | ) | (204,842 | ) | ||||
Total stockholder’s equity (deficit) |
| (18,626 | ) | (29,014 | ) | 108,706 |
| 77,256 |
| ||||
| | Year ended December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2014 | 2015 | 2016 | 2017 | 2018(a) | |||||||||||
Consolidated Operations Data: | ||||||||||||||||
Revenues | $ | 2,398 | $ | 3,767 | $ | 6,482 | $ | 5,319 | $ | 9,656 | ||||||
Operating Expenses: | ||||||||||||||||
Research and development | (9,355 | ) | (23,604 | ) | (47,994 | ) | (49,615 | ) | (82,288 | ) | ||||||
General and administrative | (3,739 | ) | (7,921 | ) | (13,535 | ) | (29,472 | ) | (41,743 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Total operating expenses | (13,094 | ) | (31,525 | ) | (61,529 | ) | (79,087 | ) | (124,031 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Loss from operations | (10,696 | ) | (27,758 | ) | (55,047 | ) | (73,768 | )) | (114,375 | ) | ||||||
Other income (expense): | ||||||||||||||||
Other income (expense), net | (524 | ) | 2,427 | (783 | ) | 492 | (272 | ) | ||||||||
Interest income | 2 | 14 | 343 | 318 | 49 | |||||||||||
Interest expense | (2,910 | ) | (22,092 | ) | (75 | ) | (43 | ) | (133 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Loss before income taxes | (14,128 | ) | (47,409 | ) | (55,562 | ) | (73,001 | ) | (114,731 | ) | ||||||
Income tax (expense) benefit | (94 | ) | 445 | 672 | (1,355 | ) | (49 | ) | ||||||||
| | | | | | | | | | | | | | | | | |
Net Lossoss | $ | (14,222 | ) | $ | (46,964 | ) | $ | (54,890 | ) | $ | (74,356 | ) | $ | (114,780 | ) | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Loss per share | ||||||||||||||||
Basic and Diluted | $ | (4.44 | ) | $ | (4.80 | ) | $ | (2.56 | ) | $ | (2.49 | ) | $ | (2.26 | ) | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Weighted average number of shares: | ||||||||||||||||
Basic and Diluted | 3,247,030 | 10,583,950 | 21,478,320 | 29,830,669 | 50,795,768 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | As of December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2014 | 2015 | 2016 | 2017 | 2018(a) | |||||||||||
Consolidated Balance Sheet Data: | ||||||||||||||||
Cash and cash equivalents and short-term investments | $ | 2,150 | $ | 111,440 | $ | 83,884 | $ | 86,879 | $ | 102,228 | ||||||
Total assets | 4,812 | 117,711 | 93,240 | 95,763 | 110,418 | |||||||||||
Long-term liabilities | 5,741 | 84 | 107 | 435 | 23,982 | |||||||||||
Total liabilities | 33,192 | 9,005 | 15,984 | 13,695 | 41,788 | |||||||||||
Mezzanine equity | 634 | — | — | — | — | |||||||||||
Accumulated deficit | (120,587 | ) | (171,426 | ) | (204,842 | ) | (279,198 | ) | (393,978 | ) | ||||||
Total stockholder's equity (deficit) | (29,014 | ) | 108,706 | 77,256 | 82,068 | 68,630 | ||||||||||
- (a)
- On July 24, 2018, we completed the acquisition of Zavante Therapeutics, Inc., or Zavante, a privately-held late clinical-stage biopharmaceutical company. Upon the closing of the acquisition, we issued 7,336,906 of our ordinary shares to former Zavante stockholders, which together with the 815,186 ordinary shares that are issuable upon release of the Holdback Shares constitute approximately 19.9% of our ordinary shares outstanding as of immediately prior to the Closing. See Item 7 below for a further description of the transaction.
ITEM 7. MANAGEMENT’SMANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our historical consolidated financial statements and the related notes thereto appearing elsewhere in this Annual Report. Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the “Risk Factors”"Risk Factors" section of this Annual Report, our actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
Overview
We are a clinical stage biopharmaceutical company engaged in the research and development of novel anti-infective agents to treat serious infections, with a focus oninfections. We have two product candidates that have been submitted to the pleuromutilin class of antibiotics. We are developing our lead product candidate,U.S. Food and Drug Administration, or the FDA, for approval: lefamulin, to bepotentially the first pleuromutilin antibiotic available for systemicoral and intravenous (or IV) administration in humans. We are developing both intravenous, or IV, and oral formulations of lefamulinhumans, for the treatment of community-acquired bacterial pneumonia, or CABP and intend toCONTEPO, a potentially first-in-class epoxide antibiotic for intravenous (or IV) use in the United States for complicated urinary tract infections, or cUTI. We may potentially develop lefamulin and CONTEPO for additional indicationsindications. Both lefamulin formulations and CONTEPO were granted Qualified Infectious Disease Product, or QIDP, and Fast Track designation by the FDA, enabling Priority Review of the New Drug Applications, or the NDAs by the FDA.
Lefamulin is a semi-synthetic pleuromutilin antibiotic discovered and developed by our team with the potential to be first-in-class for IV and oral in humans. It inhibits the synthesis of bacterial protein, which is required for bacteria to grow by binding with high affinity, high specificity and at molecular targets that are different than other than pneumonia. We have initiatedantibiotic classes. Based on results from two pivotal, internationalglobal, Phase 3 clinical trials, ofwe believe that lefamulin is well-positioned for use as a first-line monotherapy for the treatment of moderateCABP due to severe CABP. Theseits novel mechanism of action, targeted spectrum of activity, resistance profile, achievement of substantial drug concentration in lung tissue and fluid, availability of oral and IV formulations and a
generally well-tolerated safety profile. We believe lefamulin represents a potentially important new treatment option for the five to six million adults in the United States diagnosed with CABP each year.
On July 24, 2018, we completed the acquisition of Zavante Therapeutics, Inc., or Zavante, a privately-held late clinical-stage biopharmaceutical company focused on developing novel therapies to improve the outcomes of hospitalized patients. Zavante's lead product candidate is CONTEPO™ (fosfomycin for injection, previously referred to as ZTI-01 and ZOLYD).
CONTEPO is a novel, potentially first-in-class investigational intravenous antibiotic in the United States with a broad spectrum of Gram-negative and Gram-positive activity, including activity against most contemporary multi-drug resistant, or MDR, strains such as extended-spectrum b-lactamase-, or ESBL, producing Enterobacteriaceae. IV fosfomycin has been approved for a number of indications and utilized for over 45 years in Europe to treat a variety of serious bacterial infections, including cUTIs. CONTEPO utilizes a new dosing regimen that optimize its pharmacokinetics and pharmacodynamics. We believe these attributes, the extensive clinical experience worldwide and the positive efficacy and safety results from the Phase 2/3 clinical trial support CONTEPO as a first-line treatment for cUTIs, including acute pyelonephritis, or AP, suspected to be caused by MDR pathogens. At least 20% of cUTIs are caused by MDR bacteria and limited treatment options are available in the first clinical trials weU.S. In addition, non-clinical data have conductedshown that CONTEPO acts in combination with lefamulincertain other antibiotics to improve bacterial killing.
We submitted an NDA, for marketing approval of CONTEPO for the treatment of CABP. We initiated the first of these trials, which we refer to as LEAP 1,cUTI in September 2015 and initiated the second trial, which we refer to as LEAP 2, in April 2016. Based on our estimates regarding patient enrollment, we expect to have top-line data available from LEAP1adults in the third quarterUnited States, to the FDA in October 2018. The NDA submission is utilizing the 505(b)(2) regulatory pathway and is supported by a robust data package, including a pivotal Phase 2/3 clinical trial (known as ZEUS™), which met its primary endpoint of 2017statistical non-inferiority to piperacillin/tazobactam in patients with cUTI, including AP. We submitted two NDAs to the FDA for the oral and top-line data available from LEAP 2 in the first quarterIV formulations of 2018. If the results of these trials are favorable, including achievement of the primary efficacy endpoints of the trials, we expect to submit applications for marketing approval for lefamulin for the treatment of CABP in December 2018. The FDA has granted us a Prescription Drug User Fee Act, or PDUFA, target action date of April 30, 2019 for CONTEPO and a PDUFA target action date of August 19, 2019 for lefamulin. We plan to submit a marketing authorization application for lefamulin in Europe in the first half of 2019. The two NDAs are supported by two pivotal, Phase 3 clinical trials (known as LEAP 1 and LEAP 2) that evaluated the safety and efficacy of IV and oral lefamulin compared to moxifloxacin in the treatment of adults with CABP, including the option to switch from IV to oral administration and a short course oral treatment with lefamulin. In both LEAP 1 and LEAP 2, lefamulin was demonstrated to be non-inferior to moxifloxacin, and met both the FDA and European Medicines Agency, or EMA, primary and secondary efficacy endpoints for the treatment of CABP. Lefamulin was also shown to be generally well-tolerated when administered either orally or intravenously.
Both lefamulin formulations and CONTEPO were granted QIDP and Fast Track designation by the FDA, enabling potential Priority Review of the NDAs by the FDA. We were notified by the FDA that CONTEPO IV and both formulations of lefamulin were granted Priority Review following acceptance of the NDAs. We currently have a team of regional business directors and medical science liaisons in the field performing educational and market development activities. We plan to use a targeted hospital-based sales force to promote both lefamulin formulations and CONTEPO, if approved.
We believe that pleuromutilin antibiotics can help address the major public health threat posed by bacterial resistance, which the World Health Organization, or WHO, characterized in 2017 as one of the biggest threats to human health. Increasing resistance to antibiotics used to treat CABP is a growing concern and has become an issue in selecting the appropriate initial antibiotic treatment prior to determining the specific microbiological cause of the infection, referred to as empiric treatment. For example, the U.S. Centers for Disease Control and Prevention, or CDC, has classifiedStreptococcus pneumoniae, the most common respiratory pathogen, as a serious threat to human health as a result of
increasing resistance to currently available antibiotics. In addition, the CDC recently reported on the growing evidence of widespread resistance to macrolides, widely used antibiotics that disrupt bacterial protein synthesis, inMycoplasma pneumoniae, a common cause of CABP that is associated with significant morbidity and mortality. Furthermore,Staphylococcus aureus, including methicillin-resistantS. aureus, or MRSA, which has also been designated as a serious threat to human health by the CDC, has emerged as a more common cause of CABP in some regions of the world, and a possible pathogen to be covered with empiric therapy.
As a result of increasing resistance to antibiotics and the wide array of potential pathogens that cause CABP, the current standard of care for hospitalized patients with CABP whose treatment is initiated in the hospital usually involves first-line empiric treatment with a combination of antibiotics (beta-lactams and macrolides) to address all likely bacterial pathogens or monotherapy with a fluoroquinolone. Combination therapy presents the logistical challenge of administering multiple drugs with different dosing regimens, with some drugs available only as IV, and increases the risk of drug-drug interactions and the potential for serious side effects. Fluoroquinolones are associated with safety and tolerability concerns, including a relatively high risk for developingClostridium difficile infection and increasing rates of resistance for uropathogens. We believe these concerns have resulted in a decreasing use of fluoroquinolones and restriction of their use within a growing number of hospitals. In addition, in May 2016, the FDA announced that an FDA safety review has shown that fluoroquinolones, when used systemically, in the form of tablets, capsules and injectable, are associated with disabling and potentially permanent serious side effects that can occur together. These side effects can involve the tendons, muscles, joints, nerves, and central nervous system. Fluoroquinolones are typically administered in combination with other antibiotics, if community-acquired MRSA is suspected. In addition, many currently available antibiotic therapies are only available for IV administration and are prescribed for seven to fourteen days, meaning continued treatment requires prolonged hospitalization or a switch to a different antibiotic administered orally, with the attendant risk that the patient might respond differently.
Effective January 1, 2017, the Joint Commission & Center for Medicare and Medicaid Services, or CMS, began requiring all U.S. hospitals to have Antibiotic Management guidelines, also known as "Stewardship" Committees, in place to identify antibiotics most appropriate and targeted to each individual patient's infection. Past efforts to "cast the widest net possible" with broad-spectrum antibiotics that affect many types of bacteria have caused problems, such asC. difficile infections, by killing good bacteria or increased antibiotic resistance in other bacteria in different areas of the body. Additionally, in 2016, the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America, or IDSA/SHEA, updated their Antibiotic Stewardship guidelines for antibiotic use. We believe that three key goals from these guidelines are applicable to the treatment of CABP:
- •
- Reduce the risk of antibiotics associated with a high risk ofC. difficile infections;
- •
- Increase use of oral antibiotics as a strategy to improve outcomes or decrease costs; and
- •
- Reduce antibiotic therapy to the shortest effective duration.
Consistent with the Antimicrobial Stewardship principles, we believe that lefamulin could be well suited as either a first-line or second-line empiric monotherapy for the treatment of CABP patients in the hospital setting, outpatient-transition of care or in the community setting, because of its novel mechanism of action, complete spectrum of activity for CABP pathogens, including against multidrug resistant strains, achievement of substantial drug concentrations in lung fluids and lung immune cells, and flexibility as step down oral agent with both the IV and oral formulations and favorable safety and tolerability profile.
CONTEPO (fosfomycin for injection) is a novel, potentially first-in-class investigational intravenous antibiotic in the United States with a broad spectrum of Gram-negative and EuropeGram-positive activity, including activity against most contemporary MDR strains such as ESBL-producing Enterobacteriaceae. CONTEPO inhibits the bacteria's ability to form a normal cell wall, which is critical for the cell's survival and growth. It works at an earlier and different stage of cell wall synthesis than other injectable antibiotics, differentiating its mechanism of action from approved injectable antibiotics. CONTEPO utilizes a dosing approach developed by Zavante for the United States that is designed to optimize its pharmacokinetics and pharmacodynamics in 2018.order to improve treatment outcomes.
The CONTEPO development program has focused on obtaining marketing approval in the United States for the treatment of cUTIs, including AP. In April 2017, Zavante announced the results of the pivotal ZEUS Study, in which CONTEPO met the primary endpoint of statistical non-inferiority versus PIP-TAZ. Zavante then met with the FDA in the second half of 2017 to discuss the filing of an NDA for CONTEPO. We submitted an NDA for marketing approval of CONTEPO for the treatment of cUTI in adults in the United States, to the FDA in October 2018. The FDA has granted us PDUFA target action date of April 30, 2019 for CONTEPO.
We believe that the ZEUS Study results, along with extensive clinical experience with IV fosfomycin for over 45 years outside the United States, support CONTEPO as a potential first-line treatment for cUTI suspected to be caused by MDR pathogens in the United States. A number of studies report that at least 20% of cUTIs are caused by MDR bacteria and limited treatment options are available in the United States. In addition, non-clinical data have completedshown that CONTEPO acts in combination with certain other antibiotics to improve bacterial killing.
The FDA has designated CONTEPO as a Phase 2 clinical trial of lefamulin for QIDP and granted Fast Track designations to CONTEPO under the GAIN Act for:
- •
- complicated urinary tract infections, or cUTI
- •
- complicated intra-abdominal infections, or cIAI
- •
- hospital-acquired bacterial pneumonia, or HABP
- •
- ventilator-associated bacterial pneumonia, or VABP
- •
- acute bacterial skin and skin structure infections, or ABSSSI
Although we have no current plans to finance development of CONTEPO for indications other than cUTI, including AP, these designations make CONTEPO eligible for Fast Track and seventeen Phase 1Generating Antibiotic Incentives Now Act, or the GAIN Act, incentives. We may in the future consider additional non-dilutive financing options to advance these programs in the clinic.
In recognition of the growing need for the development of new antibiotics, recent regulatory changes, including Priority Review and regulatory guidance enabling smaller clinical trials, have led to renewed interest from the pharmaceutical industry in anti-infective development. For example, the FDA Safety and Innovation Act became law in 2012 and included the GAIN Act, which provides incentives, including access to expedited FDA review for approval, Fast Track designation and five years of lefamulinpotential data exclusivity extension for the development of new QIDPs.
On March 1, 2017, Nabriva Therapeutics plc, or Nabriva Ireland, was incorporated in Ireland under the name Hyacintho 2 plc, and was renamed to Nabriva Therapeutics plc on April 10, 2017, in order to effectuate the change of the jurisdiction of incorporation of the ultimate company of the group from Austria to Ireland. Nabriva Ireland replaced Nabriva Therapeutics AG, or Nabriva Austria, as the ultimate parent company on June 23, 2017, following the conclusion of a tender offer, or the Exchange Offer, in which holders of 98.6% of the outstanding share capital of Nabriva Austria exchanged their holdings for ordinary shares, $0.01 nominal value per share, of Nabriva Ireland, which
we exposed healthy subjectsrefer to singleas the Redomiciliation Transaction. The ordinary shares of Nabriva Ireland were issued on a one-for-ten basis to the holders of the Nabriva Austria common shares and on a one-for-one basis to the holders of the Nabriva Austria American Depositary Shares, or multiple dosesNabriva Austria ADSs, participating in the Exchange Offer. On June 26, 2017, the ordinary shares of IV or oral lefamulin. We planNabriva Ireland began trading on the Nasdaq Global Market under the symbol "NBRV," the same symbol under which the Nabriva Austria ADSs were previously traded. This transaction was accounted for as a merger between entities under common control; accordingly, the historical financial statements of Nabriva Austria for periods prior to pursue additional opportunitiesthis transaction are considered to be the historical financial statements of Nabriva Ireland. As of August 18, 2017, 100% of Nabriva Austria share capital had been exchanged for lefamulin, including a development program for use in pediatric patients and potentially for the treatmentordinary shares of ABSSSI. In addition, as an antibiotic with potent activity against a wide variety of multi-drug resistant pathogens, including MRSA, we may explore development of lefamulin in further indications, including ventilator-associated bacterial pneumonia, or VABP and hospital-acquired bacterial pneumonia, or HABP, sexually transmitted infections, or STIs, osteomyelitis, prosthetic joint infections. Through our research and development efforts, we have also identified a topical pleuromutilin product candidate, BC-7013, which has completed a Phase 1 clinical trial.
Nabriva Ireland.
We were Nabriva Austria was incorporated in October 2005 in Austria under the name Nabriva Therapeutics Forschungs GmbH, a limited liability company organized under Austrian law, as a spin-off from Sandoz GmbH and commenced operations in February 2006. In 2007, weNabriva Austria transformed into a stock corporation (Aktiengesellschaft) under the name Nabriva Therapeutics AG. On October 19, 2017, Nabriva Austria was converted into a limited liability company under Austrian law and renamed Nabriva Therapeutics GmbH. In 2014, we established our wholly owned U.S. subsidiary, which began operations in August 2014.
Since inception, we have incurred significant operating losses. As of December 31, 2016,2018, we had an accumulated deficit of $204.8$394.0 million. To date, we have financed our operations primarily through our 2018 equity offering, our term loan, our "at-the-market" offering facility, our 2017 equity offering, our 2016 rights offering, our 2015 initial public offering, private placements of our common shares,equity securities, convertible loans and research and development support from governmental grants and loans. We have devoted substantially all of our efforts to research and development, including clinical trials. Our ability to generate profits from operations and remain profitable depends on our ability to successfully develop and commercialize drugs that generate significant revenue.
We expect to continue to incur significant expenses and increasing operating losses for at least the next several years. We expect our expenses to increase substantiallycontinue to invest in connection with our ongoingcritical pre-commercialization and supply chain activities particularly as we continue the development of andprior to potentially seekreceiving marketing approval for lefamulin and possibly, other product candidatesCONTEPO and continue our research activities.making them available to patients.
Our expenses will increase if we suffer any regulatory delays in our Phase 3or are required to conduct additional clinical program for lefamulin for CABP, including delays in enrollment of patients.trials to satisfy regulatory requirements. If we obtain marketing approval for lefamulin, CONTEPO or any other product candidate that we develop, in-license or acquire, we expect to incur significant commercialization expenses related to product sales, marketing, distribution and manufacturing. Furthermore,
In December 2018, we expectannounced the closing of up to incura $75.0 million term loan with Hercules Capital, Inc., or Hercules, $25.0 million of which was funded on the day of closing. Under the terms of the loan, in addition to the $25.0 million received at closing, we are eligible to receive up to an aggregate of $15.0 million in two tranches upon the approval by the FDA of the NDAs recently submitted for lefamulin and CONTEPO. We will also be eligible to receive an additional costs associated$30.0 million of aggregate term loan advances in three separate tranches upon the achievement of specified product revenue milestones. These additional tranches are at our discretion and may only be drawn upon during specified periods of time. The final $5.0 million tranche is available through December 31, 2021, subject to Hercules' sole discretion. We are entitled to make interest-only payments through July 1, 2020, with operating asincremental extensions through January 1, 2022 upon the achievement of specified performance milestones. We are required to repay the term loan after the interest only period based on a public company.monthly amortization schedule, with a final maturity date occurring on June 1, 2023.
Based on our current plans, we do not expect to generate significant revenue unless and until we obtain marketing approval for, and commercialize, lefamulin.lefamulin and CONTEPO. We do not expect to
obtain marketing approval for CONTEPO before April 2019 or for lefamulin before August 2019, if at all. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. Adequate additional financing may not be available to us on acceptable terms, or at all. If we are unable to raise additional capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization effort.
December 2016 FinancingAcquisition of Zavante
On July 24, 2018, we acquired Zavante, or the Acquisition,, a biopharmaceutical company focused on developing CONTEPO (fosfomycin for injection) to improve the outcomes of hospitalized patients pursuant to the Agreement and Plan of Merger dated July 23, 2018, or the Merger Agreement.
On December 19, 2016, Upon the closing of the Acquisition, or the Closing, we completed a rights offeringissued 7,336,906 of our ordinary shares to former Zavante stockholders, which together with the 815,186 ordinary shares that are issuable upon release of the Holdback Shares (as defined below) constitute approximately 19.9% of our ordinary shares outstanding as of immediately prior to the Closing, or the Upfront Shares.
Pursuant to the Merger Agreement, former Zavante stockholders and a related underwritten offeringother equity holders, in the aggregate and subject to the terms and conditions of the Merger Agreement, will also be entitled to receive from us up to $97.5 million in contingent consideration, of which $25.0 million would become payable upon the first approval of an NDA from the the FDA for fosfomycin for injection for any indication, or the sale ofApproval Milestone Payment, and an aggregate of 588,127 commonup to $72.5 million would become payable upon the achievement of specified sales milestones, or the Net Sales Milestone Payments.
At our Extraordinary General Meeting of Shareholders held in October 2018, our shareholders approved the issuance of ordinary shares resulting in aggregate gross proceedssettlement of approximately $24.8 million and net proceedspotential milestone payment obligations that may become payable in the future to us of approximately $20.6 million, after deducting underwriting fees and offering expenses.former Zavante stockholders, including the Approval Milestone Payment which will be settled in our ordinary shares. We also now have the right to settle the Net Sales Milestone Payments in our ordinary shares, except as otherwise provided in the Merger Agreement.
InSubject to the rights offering, holdersterms of American Depositarythe Merger Agreement, 10% of the Upfront Shares, or ADSs, received 0.276 ADSthe Holdback Shares, will serve as a source for the satisfaction of indemnification and other obligations of the former Zavante stockholders and, subject to reduction in respect of these obligations, will be issued to the former Zavante stockholders following the first anniversary of the Closing.
In addition, we assumed certain liabilities and obligations, including contractual liabilities and obligations, that were assumed by us upon the Closing. Prior to the Acquisition, Zavante was obligated to make milestone payments to the former stockholders of $3.0 million upon marketing approval by the FDA with respect to any oral, intravenous or other form of fosfomycin, or the Zavante Products, and milestone payments of up to $26.0 million in the aggregate upon the occurrence of various specified levels of net sales with respect to the Zavante Products. In addition, Zavante is obligated to make annual royalty payments of a mid-single-digit percentage of net sales of Zavante Products, subject to adjustment based on net sales thresholds and with such percentage reduced to low single-digits if generic fosfomycin products account for half of the applicable market on a product-by-product and country-by-country basis. Zavante will also pay a mid-single-digit percentage of transaction revenue in connection with the consummation of the grant, sale, license or transfer of market exclusivity rights for each ADS owneda qualified infectious disease product (within the meaning of record on November 29, 2016. One ADS right entitled an ADS holderthe 21st Century Cures Act, or the Cures Act) related to subscribe for and purchase one new ADS at the subscription price of $4.32 per ADS, the U.S. dollar equivalent of €4.014 per ADS. An aggregate of 1,592,750 ADSs, representing 159,275 common shares, were subscribed for by holders of ADSs. Each ADS represents one tenth of a common share.Zavante Product.
In the rights offering, holders of common shares received the common share right to subscribe for and purchase 0.276 new common shares, at a subscription price of €40.14 per new common share for each common share owned of record on November 29, 2016. An aggregate of 102,077 new common shares were subscribed for by holders of common shares.
Pursuant to an underwriting agreement that weZavante had entered into a manufacturing and supply agreement with Cantor Fitzgerald & Co.Ercros, S.A., dated December 14, 2016, Cantor Fitzgerald & Co.pursuant to which Ercros, S.A. supplies to Zavante, on an exclusive basis, a blend of fosfomycin disodium and succinic acid, or API Mixture, for CONTEPO and, if CONTEPO is approved, will supply the commercial API Mixture for CONTEPO in the United State. In addition, Zavante had entered into a
manufacturing and supply agreement with Fisiopharma, S.r.l. pursuant to which Zavante has an obligation to purchase a minimum percentage of its commercial requirements of CONTEPO in the United States. Zavante had also entered into a manufacturing and exclusive supply agreement with Laboratorios ERN, S.A., pursuant to which Laboratorios ERN, S.A. has agreed to purchase 326,775 common shares, representing all of the unsubscribed common shares in the rights offering, at a purchase price of €40.14 per common share for purposes of resale of ADSs representing such unsubscribed common shares.
2015 Initial Public Offering
On September 23, 2015 we completed our initial public offering on the NASDAQ Global Market issuing 9,000,000 ADSs at a price to the public of $10.25 per ADS, representing 900,000 of our common shares. On September 30, 2015 the underwriters of our initial public offering exercised in full their over-allotment option to purchase an additional 1,350,000 ADSs, representing 135,000 common shares, at the initial public offering price of $10.25 per ADS, less underwriting discounts. Including the over-allotment ADSs we sold an aggregate of 10,350,000 ADSs representing 1,035,000 common shares, in our initial public offering, which resulted in gross proceeds of approximately $106.1 million and net proceeds to us of approximately $92.4 million, after deducting underwriting discounts and offering expenses.
April 2015 Financing
In March 2015, we entered into an agreementsupply Zavante with certain existingtechnical documentation and data as required for submission of an NDA or an abbreviated new investors to issuedrug application for CONTEPO and sell common shares with contractual preference rights under a shareholders agreement. We refer to this transaction as our April 2015 financing. Incertain regulatory support in connection with our April 2015 financing, we agreed to sell common shares with contractual preference rights under the shareholders agreement in two tranches. In April 2015, we closed thecommercial sale and use of the first tranche of 730,162 common shares, including the sale of 511,188 common shares at a price per share of €82.35 ($87.71) for €42.1 million ($44.8 million) in cash consideration and the sale of 218,974 common shares in exchange for certain contributions in kind consisting of the conversion of outstanding convertible loans and silent partnership interests. We also agreed to sell a second tranche of common shares with contractual preference rights under the shareholders agreement to these investors at their option for an aggregate purchase price of $70.0 million if we did not complete a public offeringCONTEPO in the United States, within specified parameters orand which provides for payments by Zavante to Laboratorios ERN, S.A. of a specified date. Uponone-time cash payment upon the first commercial sale of CONTEPO and subsequent quarterly payments thereafter based on the number of vials of CONTEPO sold in the United States during each quarter.
In connection with the closing of our initial public offeringthe Acquisition, we have assumed other agreements entered into by Zavante, including, among others, an R&D office lease, a collaboration agreement governing the supply and issuancemanufacturing agreements described above and a commercial packaging agreement.
We accounted for the Acquisition as an asset acquisition as the arrangement did not meet the definition of a business pursuant to the guidance prescribed in Accounting Standards Codification, or ASC, Topic 805, Business Combinations. We concluded the Acquisition did not meet the definition of a business because the transaction principally resulted in the acquisition of the sharesexclusive rights to IV fosfomycin in the U.S. which is a single identifiable asset and represents substantially all the fair value of the assets acquired.
We expensed the acquired intellectual property as of the acquisition date as in-process research and development with no alternative future uses. We recorded an in-process research and development expense of $32.0 million which represents $26.9 million for nominalthe fair value in satisfaction of preferred dividends, all contractual preference rights under the shareholders agreement terminated.Upfront Shares, $4.9 million of transaction costs and $0.2 million of net liabilities assumed.
License Agreement with Sinovant Sciences, Ltd.
In March 2018, we entered into a license agreement, or the Sinovant License Agreement, with Sinovant Sciences, Ltd. or Sinovant, an affiliate of Roivant Sciences, Ltd., to develop and commercialize lefamulin in the greater China region. As part of the Sinovant License Agreement, Nabriva Therapeutics Ireland DAC and Nabriva Therapeutics GmbH, our wholly owned subsidiaries, granted Sinovant an exclusive license to develop and commercialize, and a non-exclusive license to manufacture, certain products containing lefamulin, or the Licensed Products, in the People's Republic of China, Hong Kong, Macau, and Taiwan, which we refer to as the Territory. We retain development and commercialization rights in the rest of the world.
Under the Sinovant License Agreement, Sinovant and our subsidiaries have established a joint development committee, or the JDC, to review and oversee development and commercialization plans in the Territory. We received a $5.0 million upfront payment pursuant to the terms of the Sinovant License Agreement and will be eligible for up to an additional $91.5 million in milestone payments upon the achievement of certain regulatory and commercial milestone events related to lefamulin for CABP, plus an additional $4.0 million in milestone payments if any Licensed Product receives a second or any subsequent regulatory approval in the People's Republic of China. The first milestone was a $1.5 million payment for the submission of a Clinical Trial Application, or CTA, by Sinovant to the Chinese Food and Drug Administration that was received in February 2019. The remaining milestone payments are tied to additional regulatory approvals and annual sales targets. In addition, we will be eligible to receive low double-digit royalties on sales, if any, of Licensed Products in the Territory.
Sinovant will be solely responsible for all costs related to developing, obtaining regulatory approval of and commercializing Licensed Products in the Territory and is obligated to use commercially reasonable efforts to develop, obtain regulatory approval for, and commercialize Licensed Product in the Territory. We are obligated to use commercially reasonable efforts to supply, pursuant to supply agreements to be negotiated by the parties, to Sinovant sufficient supply of lefamulin for Sinovant to
manufacture finished drug products for development and commercialization of the Licensed Products in the Territory.
Unless earlier terminated, the Sinovant License Agreement will expire upon the expiration of the last royalty term for the last Licensed Product in the Territory, which we expect will occur in 2033. Following the expiration of the last royalty term, the license granted to Sinovant will become non-exclusive, fully-paid, royalty-free and irrevocable. The Sinovant License Agreement may be terminated in its entirety by Sinovant upon 180 days' prior written notice at any time. Either party may, subject to specified cure periods, terminate the Sinovant License Agreement in the event of the other party's uncured material breach. Either party may also terminate the Sinovant License Agreement under specified circumstances relating to the other party's insolvency. We have the right to terminate the Sinovant License Agreement immediately if Sinovant does not reach certain development milestones by certain specified dates (subject to specified cure periods). The Sinovant License Agreement contemplates that the we will enter into ancillary agreements with Sinovant, including clinical and commercial supply agreements and a pharmacovigilance agreement.
We have identified two performance obligations at inception: (1) the delivery of the licenses to Sinovant; and, (2) the participation in the JDC. The $5.0 million non-refundable upfront payment was allocated entirely to the delivery of the licenses as the JDC deliverable was deemed to be de minimis. In addition, since the first $1.5 million milestone payment is related to a submission of the CTA that is in the control of the parties', we recorded such milestone as variable consideration allocated to the licenses at the inception of the arrangement as we believe it is probable to be met and received. The future regulatory and commercial milestone payments will be recorded during the period the milestone is probable of achievement.
Financial Operations Overview
Revenue
To date, we have not generated any revenues from product sales. Based on our current plans, we do not expect to generate significant revenue unless and until we obtain marketing approval for, and commercialize, lefamulin and CONTEPO. We do not expect to obtain marketing approval for CONTEPO before April 2019 or for lefamulin before August 2019, if at all. If our development efforts result in clinical success and regulatory approval, we may also enter into collaboration agreements with third parties and we may generate revenue from those agreements.
Our revenue consists principally of the collaboration revenues recorded from the Sinovant License Agreement, and includes a $5.0 million non-refundable upfront payment received as consideration for entering into the Sinovant License Agreement, as well as $1.5 million of variable consideration related to a milestone payment that we believe was probable to be met and was received in February of 2019. Revenue also includes governmental research premiums, non-refundable government grants and the benefit of government loans at below-market interest rates, which are more fully described below under "Critical Accounting Policies."
Research and Development Expenses
Research and development expenses represented 78.0%, 62.7% and 66.3% of our total operating expenses for the years ended December 31, 2016, 2017 and 2018, respectively.
For each of our research and development programs, we incur both direct and indirect expenses. Direct expenses include third party expenses related to these programs such as expenses for manufacturing services, non-clinical and clinical studies and other third party development services. Indirect expenses include salaries and related costs, including stock-based compensation, for personnel in research and development functions, infrastructure costs allocated to research and development
operations, costs associated with obtaining and maintaining intellectual property associated with our research and development operations, laboratory consumables, consulting fees related to research and development activities and other overhead costs. We utilize our research and development staff and infrastructure resources across multiple programs, and many of our indirect costs historically have not been specifically attributable to a single program. Accordingly, we cannot state precisely our total indirect costs incurred on a program-by-program basis.
The following table summarizes our direct research and development expenses by program and our indirect costs.
| | Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2016 | 2017 | 2018 | |||||||
Direct costs | ||||||||||
Lefamulin | $ | 36,003 | $ | 34,538 | $ | 20,685 | ||||
CONTEPO | — | — | 3,423 | |||||||
In-process research and development | — | — | 32,048 | |||||||
Other programs and initiatives | 71 | 223 | 53 | |||||||
Indirect costs | 11,920 | 14,854 | 26,079 | |||||||
| | | | | | | | | | | |
Total | $ | 47,994 | $ | 49,615 | $ | 82,288 | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
We expect to continue to incur research and development expenses in connection with required regulatory activities, our activities related to our ongoing pediatric studies of lefamulin for the treatment of CABP and of CONTEPO for the treatment of cUTI, the pursuit of the clinical development of lefamulin and CONTEPO for additional indications and engagement in earlier stage research and development activities. It is difficult to estimate the duration and completion costs of our research and development programs.
The successful development and commercialization of our product candidates is highly uncertain. This is due to the numerous risks and uncertainties associated with product development and commercialization, including the uncertainty of:
- •
- the scope, progress, costs and results of clinical trials and other research and development activities;
- •
- the costs, timing and outcome of regulatory review of our product candidates;
- •
- the efficacy and potential advantages of our product candidates compared to alternative treatments, including any standard of care, and our ability to achieve market acceptance for any of our product candidates that receive marketing approval;
- •
- the costs and timing of commercialization activities, including product sales, marketing, distribution and manufacturing, for any of our product candidates that receive marketing approval; and
- •
- the costs and timing of preparing, filing and prosecuting patent applications, maintaining, enforcing and protecting our intellectual property rights and defending against any intellectual property-related claims.
A change in the outcome of any of these variables with respect to the development of our product candidates could result in a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA or another regulatory authority were to require us to conduct clinical trials or other testing beyond those that we have completed or currently contemplate will be required for the completion of clinical development of any product candidate, we could be required to expend significant additional resources and time on the completion of clinical development of that product candidate.
General and Administrative Expenses
General and administrative expenses represented 22.0%, 37.3% and 33.7% of our total operating expenses for the years ended December 31, 2016, 2017 and 2018, respectively.
General and administrative expenses consist primarily of salaries and related costs, including stock-based compensation not related to research and development activities for personnel in our finance, information technology, commercial, medical affairs and administrative functions. General and administrative expenses also include costs related to professional fees for auditors, lawyers and tax advisors and consulting fees not related to research and development operations, as well as functions that are partly or fully outsourced by us, such as accounting, payroll processing and information technology.
We expect general and general administrative expenses to increase with the expansion of our staff and management team in anticipation of the commercialization of lefamulin and CONTEPO, particularly commercial, medical affairs, technical operations and business development functions
Critical Accounting Policies
Our management’smanagement's discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which we have prepared in accordance with generally accepted accounting principles in the United States, or U.S. GAAP. The preparation of our consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the end of the reporting period, as well as the reported revenues and expenses during the reporting periods. We base our estimates on our limited historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are described in more detail in the notes to our consolidated financial statements appearing at the end of this filing. However, we believe that the following accounting policies are the most critical to aid you in fully understanding and evaluating our financial condition and results of operations.
Research Premium and Grant Revenue
Grant revenue comprises (1)As a company that carries out research and development activities, we benefit from the Austrian research and development support regime, under which we are eligible to receive a research premium from the Austrian government (2) grants received from the Vienna Center for Innovation and Technology (Zentrum für Innovation, or ZIT) and the Vienna Business
Promotion Fund (Wiener Wirtschaftsförderungsfonds, or WWFF) and (3) the benefit of government loans at below-market interest rates. Please referequal to note 3 of our audited consolidated financial statements included elsewhere in this Annual Report for further details on our grant revenue.
The research premium we received from the Austrian government was calculated as 10% of a specified research and development cost base for the years ended December 31, 2015 and December 31, 2014. For the year ended December 31, 2016, the research premium was calculated as 12% of a specified research and development cost base. Qualifying expenditures largely comprise research and development activities conducted in Austria, however, the research premium is also available for certain related third-party expenses with additional limitations. We recorded research premiums of $6.2 million, $4.8 million and $2.6 million for the years ended December 31, 2016, 2017, and 2018, respectively. We have not received any research premium for our qualified 2018 expenditures as of December 31, 2018. We recognize the research premium, as long as we have incurred research and development expenses. The WWFF grantSignificant judgment is paid out through the landlordrequired in the form of a monthly reduction in lease payments and is recognized over the period from grant date in March 2010 until end of the lease termination waiver term in December 2017. The ZIT grantsdetermining which expenditures are provided to support specific research projects and are recognized according to the progress of the respective project. All grants are non-refundable as long as the conditions of the grant are met. We are and have been in full compliance with the conditions of the grants and all related regulations.
The benefit of a government loan at a below-market rate of interest is treated as a government grant. The benefit due to the difference between the market rate of interest and the rate of interest charged by the governmental organization is measured as the difference between the initial carrying value of the loan and the proceeds received. This benefit is deferred, and recognized through profit and loss over the term of the corresponding liabilities.
Convertible Loans and Additional Call Options
Between July 2011 and January 2015, we entered into five convertible loans with certain of our shareholders. Under the loans, the lenders had the right to convert their entire claim for repayment of the loans into common shares with contractual preference rights under a shareholders agreement. Loans were payable in cash on the respective repayment date if not previously converted. In conjunction with the convertible loan agreements we entered into in 2011 and 2012, we also granted the lenders additional call options to acquire common shares with contractual preference rights under the shareholders agreement. No transaction costs were incurred in conjunction with our entry into the convertible loan agreements.
In connection with our April 2015 financing, all of the lenders under our convertible loan agreements waived all rights and claims they had in connection with the convertible loan agreements. In particular, all call option rights as well as claims on payments of accrued interest were waived. All claims for repayment, excluding accrued interest, under all convertible loan agreements, were converted into common shares with contractual preference rights under the shareholders agreement. Any accrued interest, as well as the additional call option rights were forfeited.
Prior to their conversion in April 2015, we presented the convertible loans as a liability in the consolidated balance sheet. We also evaluated the requirement to bifurcate embedded options within the convertible loans in accordance with ASC 815, Derivatives and Hedging, or ASC 815. ASC 815 provides criteria that, if met, require companies to bifurcate conversion options from their host instruments. These criteria include circumstances in which (1) the economic characteristics and risks of the embedded option are not clearly and closely related to the economic characteristics and risks of the host contract, (2) the hybrid instrument is not remeasured at fair value under otherwise applicable generally accepted accounting principles with changes reported in fair value as they occur, and (3) a separate instrument with the same terms as the embedded option would be considered a derivative instrument. Discounts associated with convertible loans were amortized over the term of the related debt using the effective yield method.
We also accounted for the additional call options issued with the convertible loans in 2011 and 2012 as well as our loan from Kreos Capital IV (UK) Limited, or Kreos, in July 2014, pursuant to ASC 815, which provides a two-step modeleligible to be appliedincluded in determining whether a financial instrument or an embedded feature is indexed to an issuer’s own stock. Due to the circumstances that the additional call options did not meet the “fixed for fixed” criteria under ASC 815-40, Contracts in Entity’s Own Equity, and did not meet the definition of a derivative, the additional call options were classified as a liability. The call options were accounted for at fair value by use of an option pricing model, or OPM, at inception and in subsequent periods, with changes in fair value recognized in the other income (expense), net line item within the consolidated statement of comprehensive income (loss).
Silent Partnership
In June 2014 and January 2015, we entered into silent partnership agreements with certain of our
shareholders, which entitled each of the silent partners to a proportionate share in the fair value of the company, similar to a shareholder, including a share in profit or loss, according to an agreed participation rate, were classified as mezzanine equity pursuant to ASC 480, Distinguishing Liabilities from Equity, and ASC 815. The silent partnership interests were evaluated for equity or mezzanine classification based upon the nature of the silent partnership settlement provisions, which provided us the unilateral option to settle the obligation in cash or a variable number of shares. However, when a settlement in shares cannot always be presumed, irrespective of probability of the event occurring, a classification outside of stockholders’ equity is required. Mezzanine equity was initially measured at fair value and subsequently at the redemption value at each reporting period, which represented the anticipated proceeds resulting from an exit event (trade sale or an initial public offering). Such amounts were recognized in retained earnings.
In connection with our April 2015 financing, all of the claims for repayment of the silent partnership interests, including interest accrued thereon, converted into common shares with contractual preference rights under a shareholders agreement.
April 2015 Financing
In March and April 2015, we entered into agreements with our existing shareholders and certain new investors to issue and sell common shares with contractual preference rights under a shareholders agreement.
The April 2015 financing agreements resulted in the following effects with respect to our existing financial instruments:
· all existing convertible loan agreements and silent partnership interests were converted to common shares with contractual reference rights under a shareholders agreement;
· the lenders under our convertible loan agreements irrevocably waived their claims for payment of interest accrued on the loan amounts;
· the lenders under our convertible loan agreements irrevocably waived the call option rights granted under our convertible loan agreements; and
· the silent partners irrevocably agreed to the forfeiture of their claims for payment of interest accrued on their silent partnership investments.
Pursuant to our shareholders agreement, signed on April 2, 2015, the holders of the shares issued in our April 2015 financing were granted certain preferential rights. These rights include the right of certain shareholders to acquire additional common shares against payment of the nominal amount of €1.00 per share following an appropriate resolution of all of our shareholders, which we refer to as the preferred dividend. The preferred dividend accrued at a rate per annum of 8%, based on the number of days that have elapsed from the issuance of such shares until the occurrence of certain triggering events, including an initial public offering, a sale of the company, voluntary conversion to preference shares by the shareholders, and a liquidation. The preferred dividend was cumulative and perpetual, and could be paid in cash or shares based on a shareholders vote. Upon the closing of our initial public offering and the issuance of the shares for nominal value in satisfaction of the preferred dividends, all contractual preference rights terminated in December 2015.
The shares from the April 2015 financing were recorded upon registration of the capital increase in the Austrian commercial register in May 2015. As a result of the preferred dividend rights, which were not legally separable, we were deemed to have issued common shares accompanied by preferred dividends that may be settled for cash or shares. Accordingly, the proceeds from the April 2015 financing, including the consideration from conversion of the convertible loan agreements and silent partnership interests, were recorded as mezzanine equity. A mezzanine equity classification arises as a result of the dividend provision in our shareholders agreement, under which our then-current shareholders covenanted to vote in favor of the requisite shareholder resolutions to allow us to satisfy the preferred dividend rights. As a result, (1) we could not avoid fulfilling the preferred dividend rights if a triggering event occurred that was outside our control, and (2) we could not always presume a settlement in shares. Therefore, when a settlement in shares cannot always be presumed for an event not solely within the control of the issuer, irrespective of probability of the event occurring, a classification outside of stockholders’ equity is required. Mezzanine equity was initially measured at fair value and subsequently at the redemption value at each reporting period, which represented the anticipated proceeds resulting from an exit event (trade sale or an initial public offering). Such amounts were recognized in retained earnings.
The April 2015 financing and the related conversion of our outstanding convertible loan agreements and silent partnership interests resulted in total consideration to us of $77.3 million which was recorded in mezzanine equity. Upon the closing of our initial public offering in September 2015, a triggering event occurred as described above, and the holders of the preferred dividend right received 17,149 additional shares against payment of the nominal amount of €1.00 per share, effectively removing the mezzanine equity classification.
Share-based Payments
Stock Option Plan 2007
Our shareholders adopted our Stock Option Plan 2007 on September 12, 2007 and subsequently approved amendments to the Stock Option Plan 2007 on September, 17, 2009, May 9, 2010 and June 30, 2015. References to our Stock Option Plan 2007 in this Annual Report refer to the plan as amended. No additional awards will be granted under the Stock Option Plan 2007. All employees (including members of the management board), selected members of the supervisory board and further participants were eligible to participate in the Stock Option Plan 2007. Options granted under the Stock Option Plan 2007 give beneficiaries the right to acquire our shares. Options granted under the Stock Option Plan 2007 generally vest over four years from the date of participation. Typically, 25% of the options subject to a particular grant vest on the last day of the last calendar month of the first year of the vesting period, a further 25% of the options vests on the last day of the last calendar month of the second year of the vesting period, and the remaining 50% vests on a monthly pro-rata basis over the third and fourth years of the vesting period (i.e., 2.083% per month). However, alternative vesting schedules applied for beneficiaries who had worked for us prior to the date of the adoption of our Stock Option Plan 2007. All options granted under such alternative vesting schedules have fully vested.
The Stock Option Plan 2007 provides that 50% of any then-unvested options shall automatically vest upon a liquidity event, which refers to an exclusive license of or the sale or other disposal of 50% or more of our assets, a sale or other disposal (but not a pledge) of 50% or more of our shares, a merger of ours with any third party, or a consolidation, liquidation, winding up or other form of dissolution). If a beneficiary has an unjustified termination or a justified premature termination (as such terms are used in the Stock Option Plan 2007) within one year of the liquidity event, all remaining unvested options held by the beneficiary shall automatically vest in full.
Unless otherwise specifically permitted in an option agreement or resolved upon by the management board with the approval of the supervisory board, the exercise of vested options is permitted under the Stock Option Plan 2007 only during specified periods and on specified terms in the case of a liquidity event or following an initial public offering of our shares occurring during the term of the option, regardless of whether or not the beneficiary is then providing services to us. A beneficiary is entitled to exercise vested options at any time during the remaining term of the option. No options may be exercised under the Stock Option Plan 2007 after September 27, 2017. Any options not exercised by September 27, 2017 automatically terminate and are forfeited.
Beneficiaries are not entitled to transfer vested options, except to individuals by way of inheritance or bequest. Options do not entitle beneficiaries to exercise any shareholder rights. Beneficiaries may exercise shareholder rights only with respect to any shares they hold.
Stock Option Plan 2015
Our shareholders, management board and supervisory board adopted our Stock Option Plan 2015 on April 2, 2015 and our shareholders approved an amended and restated version of the Stock Option Plan 2015 on June 30, 2015. An amendment to the amended and restated Stock Option Plan 2015 was approved by our shareholders on July 22, 2015. References to our Stock Option Plan 2015 in this Annual Report refer to the amended and restated version of the Stock Option Plan 2015, as amended. The Stock Option Plan 2015 became effective on July 3, 2015 upon the registration with the commercial register in Austria of our conditional capital increase approved by our shareholders on June 30, 2015. The Stock Option Plan 2015 initially provided for the grant of options for up to 95,000 common shares to our employees, including members of our management board, and to members of our supervisory board. Following approval by our shareholders at our 2016 annual general meeting, the number of shares available for issuance under the Stock Option Plan 2015 was increased to
346,235 common shares. Grants of stock options for 201,568 common shares under this plan to members of the management board, selected members of the supervisory board and certain employees were made as of December 31, 2016. Options granted under the Stock Option Plan 2015 generally have a term of 10 years and vest over four years. Typically 25% of the options subject to a particular grant vest on the last day of the of the last calendar month of the first year of the vesting period, and the remaining 75% vests on a monthly pro-rata basis over the second, third and fourth years of the vesting period (i.e., 2.083% per month). Any alternative vesting period determined by us is subject to approval by our management board, supervisory board or shareholders, in accordance with applicable voting requirements.
The Stock Option Plan 2015 provides that, if a liquidity event (as defined below) occurs, all options outstanding under the Stock Option Plan 2015 will be assumed (or substantially equivalent awards will be substituted by an acquiring or succeeding corporation (or an affiliate of the acquiring or succeeding corporation)), and any then-unvested options shall continue to vest in accordance with the beneficiary’s original vesting schedule. If a beneficiary is terminated due to a good leaver event (within the meaning of the Stock Option Plan 2015), on or prior to the first anniversary of the date of the liquidity event, the beneficiary’s options will be immediately exercisable in full as of the date of such termination. If the acquiring or succeeding corporation (or an affiliate of the acquiring or succeeding corporation) refuses to assume the options outstanding under the Stock Option Plan 2015 or to substitute substantially equivalent options therefore, all then-unvested options under the Stock Option Plan 2015 will automatically vest in full upon the liquidity event. For purposes of the Stock Option Plan 2015, a liquidity event generally refers to an exclusive license of or the sale, lease or other disposal of all or substantially all of our assets, a sale or other disposal (but not a pledge) of 50% or more of our shares, a merger or consolidation of us with or into any third party, or our liquidation, winding up or other form of dissolution of us.
Unless otherwise specifically permitted in an option agreement or resolved upon by the management board with the approval of the supervisory board, the exercise of vested options is permitted under the Stock Option Plan 2015 only during specified periods and on specified terms in the case of a liquidity event or following an initial public offering occurring during the term of the option. A beneficiary is entitled to exercise vested options at any time during the remaining term of the option while the beneficiary is providing services to us, and within the three-month period following a termination of the beneficiary’s services due to a good leaver event. Options granted under the Stock Option Plan 2015 will have a term of no more than ten years from the beneficiary’s date of participation.
Beneficiaries of options granted under the Stock Option Plan 2015 are not entitled to transfer vested options, except to individuals by way of inheritance or bequest. Options do not entitle beneficiaries to exercise any shareholder rights. Beneficiaries may exercise shareholder rights only with respect to any shares they hold.
We measure the options under our equity incentive plans at fair value at their grant date in accordance with ASC 718, Compensation — Stock Compensation,” using the Black-Scholes model. All options under the Stock Option Plan 2007 have an exercise price of €6.72 ($7.32) per share and a maturity of September 27, 2017. The fair value of such share-based compensation is recognized as an expense over the respective vesting period. Share-based compensation expense under the Stock Option Plan 2007 was $96,000, $78,000 and $95,000 for the years ended December 31, 2014, 2015 and 2016, respectively. No options were granted under the Stock Option Plan 2007 during 2015 or 2016. The weighted average fair value of the 1,088 options granted in 2014 was $209.11 per share. Options granted during the year ended December 31, 2016 under the Stock Option Plan 2015 have a weighted-average exercise price of €71.64 ($80.24) and a weighted average fair value of €27.31 ($30.58). We recognized stock-based compensation expense of approximately $2.5 million during the year ending December 31, 2016 related to the options granted under the Stock Option Plan 2015.
We account for related social security contributions as liabilities which are recognized on the date of the event triggering the measurement and payment of the tax to the taxing authority; or the date the options are exercised.
We expect to grant additional stock options that will result in additional share-based compensation expense. Following the consummation of our initial public offering, we determined stock option values based on the market price of our common shares.
Fair Value Estimation
We use valuation techniques that include inputs that are not based on observable market data to estimate
the fair value of certain types of financial instruments, including stock options under our equity incentive plans.
Valuation of Total Equity and Certain Financial Instruments prior to our Initial Public Offering
Prior to our initial public offering, the fair value of the total equity was determined by management and took into account the most recently available valuation of the company and the assessment of additional objective and subjective factors we believed were relevant. We considered numerous objective and subjective factors to determine the best estimate of the fair value of the equity and certain financial instruments that represented potential interests in the equity, including the following:
· the progress of the research and development programs;
· achievement of enterprise milestones, including the entering into collaborationcosts base and licensing agreements;
· contemporaneous third-party valuations of the common shares;
· the forecasted performance and operating results;
· the estimatedsuch costs of capitalare subject to fund operations;
· the rights and preferences of the financial instruments, e.g. liquidation preference of common shares with contractual preference rights relative to other common shares, conversion rights of the convertible loan agreements, etc.;
· the likelihood of achieving a discrete liquidity event, such as a sale of the company or an initial public offering given prevailing market conditions; and
· external market and economic conditions impacting the industry sector.
In determining the fair values of the equity, we considered three generally accepted approaches: the income approach, market approach and cost approach. Based on the stage of development and information available, we determined that the income approach was the most appropriate method.
Discounted cash flow, or DCF, a form of the income approach, is an estimate of the present value of the future monetary benefits expected to flow to the owners of a business. It requires a projection of the cash flows that the business is expected to generate. These cash flows are converted to present value by means of discounting, using a rate of return that accounts for the time value of money and the appropriate degree of risks inherent in the business. The discount rate in the DCF analysis was based upon a weighted average cost of capital, or WACC. The WACC was derived by using the Capital Asset Pricing Model and inputs such as the risk-free rate, beta coefficient, equity risk premiums and the size of the company.
After determining the fair value of total equity for each valuation date, the option pricing method, or OPM, was used to estimate the fair value for all financial instruments that represented claims on the company’s assets, including the different share classes as well as the following instruments:
· options under the Stock Option Plan 2007 and Stock Option Plan 2015;
· the investment from the silent partnership;
· options related to our loan from Kreos, and
· options and conversion rights related to the convertible loans agreements.
Under this approach, each class of securities was modeled as a combination of call options with a unique claim on the assets of the company. The characteristics of each security’s class defined these claims. This reflected differences in value allocation at different company value levels that result from differences in security classes, for example from liquidation preference rights, dividend accrual, etc. The OPM used the Black-Scholes option-pricing model to price the call options. This model defines the securities’ fair values as functions of the current fair value of the company and uses assumptions such as the anticipated timing of a potential liquidity
event and the estimated volatility of the entire equity. Volatility was estimated based on the observed daily share price returns of peer companies over a historic period closely matching the period for which expected volatility is estimated. Volatility is defined as the annualized standard deviation of share price returns. In the allocation of equity, the company also considered valuation outcomes through a sale of the company compared to an initial public offering, and considered the probabilities of each at each valuation date, since the treatment of the liquidation rights were different for these two events. The aggregate value per security class was then dividedreview by the number of securities outstanding to arrive at the value per security.
Our valuations relied on DCF models to derive the total enterprise value. The cash flow projections were based on probability-weighted scenarios which considered estimates of time to market, market share and pricing of lefamulin in the target indications. The cash flow projections were estimated over a period equal to the expected patent life, and a terminal value period was not applied. The expected sales were estimated using a detailed market model that comprises historical and expected number of therapies as well as prices of relevant drugs per indication and region, based on market reports, surveys and estimates by management. Production and research and development costs were estimated at the indication level with general and administrative costs and selling and marketing costs estimated at the overall company level. A WACC of 16.0% was applied for each valuation date. The OPM relied on the anticipated timing and probability of a liquidity event based on then current plans and estimates of the management as per each valuation date. As of July 4, 2014 and December 31, 2014 the probability of an initial public offering was estimated at 60% (2013 and earlier: 10%) and of a sale at 40% (2013 and earlier: 90%). As of December 31, 2014 the estimated volatility was 65% (2013: 80%) based on historical trading volatility for the publicly traded peer companies and a time to liquidity of 0.5 years for the initial public offering scenario and 2.5 years for the trade sales scenario (2013: 1.2 years and 4.4 years, respectively).
In the course of the April 2015 financing the investment from the silent partnership as well as the convertible loans were converted to common shares with contractual preference rights and the lenders under the convertible loan agreements irrevocably waived and acknowledged the termination of their call option rights granted thereunder. The options related to our loan from Kreos were exercised in May 2015. Hence, only the options under the Stock Option Plan 2007 and Stock Option Plan 2015 remained as instruments that required the determination of a fair value.
Valuation of Stock Options Following our Initial Public Offering
Austrian government.
Upon the closing of our initial public offering, the preference rights of certain common shares terminated and the fair market value for all shares equals the market price per share. In accordance with ASC 718, Compensation — Stock Compensation, the grant date fair value of stock options is determined based on the price of the American Depositary Shares on the date of grant by use of a Black Scholes model.
|
| 2015 |
| 2016 |
|
Risk-free interest rate |
| -0.223 | % | -0.605 | % |
Expected term of options (in years) |
| 2.0 |
| 2.2 |
|
Expected volatility |
| 57.39 | % | 81.46 | % |
Dividend yield |
| — |
| — |
|
Research and Development Expenses
Research expenses are defined as costs incurred for current or planned investigations undertaken with the prospect of gaining new scientific or technical knowledge and understanding. Development
expenses are defined as costs incurred for the application of research findings or specialist knowledge to production, production methods, services or goods prior to the commencement of commercial production or use. We expense all research and development costs as incurred.
The following costs, in particular by their nature, constituteAs part of the process of preparing our consolidated financial statements, we are required to estimate our accrued research and development expenses:expenses. This process involves reviewing open contracts and purchase orders, communicating with our applicable personnel to identify services that have been performed on our behalf and estimating the appropriate proportionslevel of direct personnelservice performed and material costs, related overhead for internal or external technology, engineering and other departments that provide services; costs for experimental and pilot facilities (including depreciation of buildings or parts of buildings used for research or development purposes); costs for clinical research; regular coststhe associated cost incurred for the utilizationservice when we have not yet been invoiced or otherwise notified of third parties’ patents foractual costs. We make estimates of our accrued expenses as of each balance sheet date in the consolidated financial statements based on facts and circumstances known to us at that time. Examples of estimated accrued research and development purposes; other taxes related to research facilities;expenses include fees paid to:
- •
- vendors in connection with preclinical development activities;
- •
- the production of preclinical and
fees for the filingclinical trial materials; - •
- CROs in connection with clinical trials; and,
registration of self-generated patents that are not capitalized.Our projects are currently - •
- investigative sites in
the research and development phase and marketing approval by U.S., European and other foreign regulatory authorities is not, nor will be, available for any product in the near future.TaxesWe are subject to income tax in Austria and the United States. Significant judgments and estimates are required in determining the consolidated income tax expense, including a determination of whether and how much of a tax benefit taken by us in our tax filings or positions is more likely than not to be realized. Changes in tax laws and rates could also affect recorded deferred tax assets and liabilities in the future. We are not aware of any such changes that would be expected to have a material effect on our results of operations, cash flows or financial position. There are many transactions and calculations for which the ultimate tax determination is uncertain. Where the final tax outcome of these matters is different from the amounts that were initially recorded, such differences will impact the current and deferred income tax assets and liabilities in the period in which such determination is made.Our U.S. subsidiary is subject to income taxes due to the fact that it provides to us certain management and other services related to research and development activities. These services are rendered on terms that were negotiated at arm’s length pursuant to a services agreementconnection withus.Deferred tax assets have only been recognized to the extent that we believe those assets are more likely than not to be realized. In making such a determination we consider all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax-planning strategies, and results of recent operations. If we determined that we would be able to realize our deferred tax assets in the future in excess of their net recorded amount, we would make an adjustment to the deferred tax asset valuation allowance, which would reduce the provision for income taxes. As of December 31, 2016, we had cumulative net operating loss carryforwards in respect of losses of $216.9 million. The tax loss carry-forwards will not expire in Austria.Change in Functional Currency
trials.We have significantly expanded our presence and operations in the United States, and have begun to, and will continue to, incur a majority of the expenses for ourclinicaltrials in U.S. dollars. Also, the majority of the funds raised from our initial public offering in September 2015 and our rights offering and the related underwritten offering in December 2016, as well as other financing activities, are currently invested, and are expected to remain invested, in U.S. dollar denominated instruments to fund our U.S. operations. As a result, as of January 1, 2016, we determined that our functional and reporting currency had changed from euro to the U.S. dollar.
The change in functional currency is accounted for prospectively from January 1, 2016 and prior year financial statements have not been restated for the change in functional currency. For all relevant periods, foreign currency revenue and expense transactions were recorded using the exchange rates prevailing at the dates of the transactions. However, the change in reporting currency represents an accounting policy change, and accordingly, the change has been applied retrospectively. Therefore, financial information included in our audited consolidated financial statements for the years ended December 31, 2014 and 2015 previously reported in euro has been restated into the new reporting currency U.S. dollars.
For periods prior to January 1, 2016, the effects of exchange rate fluctuations on translating foreign currency monetary assets and liabilities into U.S. dollars were included in the statement of operations and comprehensive income (loss) as foreign exchange gain (loss). Revenue and expense transactions were translated into the U.S. dollar reporting currency at the balance sheet date at average yearly exchange rates prevailing for the relevant period, and assets and liabilities were translated at end of period exchange rates, except for equity transactions, which were translated at historical exchange rates. Translation gains and losses from the application of the U.S. dollar as the reporting currency while the euro was the functional currency are included as part of the cumulative foreign currency translation adjustment, which is reported as a component of shareholders’ equity under accumulated other comprehensive income (loss). The differences between items of equity translated at historical exchange rates as of December 31, 2015 and their value as of January 1, 2016 translated at the exchange rate as of December 31, 2015 are shown in the line item “Change in functional currency” within the condensed consolidated statement of changes in stockholders’ equity.
For periods commencing on and after January 1, 2016, monetary assets and liabilities denominated in foreign currencies are translated into U.S. dollars using exchange rates in effect at the balance sheet date. Opening balances related to non-monetary assets and liabilities are based on prior period translated amounts, and nonmonetary assets and nonmonetary liabilities incurred after January 1, 2016 are translated at the approximate exchange rate prevailing at the date of the transaction. Revenue and expense transactions are translated at the approximate exchange rate in effect at the time of the transaction. Foreign exchange gains and losses are included in the statement of operations and comprehensive income (loss) as foreign exchange gain (loss).
Implications of Being an Emerging Growth Company
As a company with less than $1 billion in revenue during our last fiscal year, weWe qualify as an “emerging"emerging growth company”company" as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. An emerging growth company may take advantage of specified reduced reporting and other burdens that are otherwise applicable generally to public companies. These provisions include:
- •
·an exemption from compliance with the auditor attestation requirement of Section 404 of the Sarbanes-Oxley Act of 2002 on the design and effectiveness of our internal controls over financial reporting;·- •
- an exemption from compliance with any requirement that the Public Company Accounting Oversight Board may adopt regarding mandatory audit firm rotation or a supplement to the
auditor’sauditor's report providing additional information about the audit and the financial statements;· - •
- reduced disclosure about the
company’scompany's executive compensation arrangements; and· - •
- exemptions from the requirements to obtain a non-binding advisory vote on executive compensation or a shareholder approval of any golden parachute arrangements.
We may take advantage of these provisions until December 31, 2020 or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company upon the earlier to occur of: the last day of the fiscal year in which we have more than $1 billion in annual revenues;revenues (as may be inflation adjusted by the SEC from time-to-time); the date we qualify as a “large"large accelerated filer,”" with at least more than $700 million in market value of our share capital held by nonaffiliates; or the issuance by us of more than $1 billion of non-convertible debt over a three-year period. We may choose to take advantage of some, but not all, of the available benefits under the JOBS Act. We have taken advantage of some reduced reporting burdens in this Annual Report. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold stock.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This provision allows an emerging growth company to delay the adoption of some accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of delayed adoption of new or revised accounting standards and, therefore, we will adopt new
or revised accounting standards on the relevant dates on which adoption of such standards is required by for non-emerging growth companies.
Results of Operations
Comparison of Years Ended December 31, 20152017 and 20162018
| | Year ended December 31, | | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2017 | 2018 | Change | |||||||
Consolidated Operations Data: | ||||||||||
Revenues | $ | 5,319 | $ | 9,656 | $ | 4,337 | ||||
Operating Expenses: | ||||||||||
Research and development | (49,615 | ) | (82,288 | ) | (32,673 | ) | ||||
General and administrative | (29,472 | ) | (41,743 | ) | (12,271 | ) | ||||
| | | | | | | | | | | |
Total operating expenses | (79,087 | ) | (124,031 | ) | (44,944 | ) | ||||
| | | | | | | | | | | |
Loss from operations | (73,768 | ) | (114,375 | ) | (40,607 | ) | ||||
Other income (expense): | ||||||||||
Other income (expense), net | 492 | (272 | ) | (764 | ) | |||||
Interest income (expense), net | 275 | (84 | ) | (359 | ) | |||||
| | | | | | | | | | | |
Loss before income taxes | (73,001 | ) | (114,731 | ) | (41,730 | ) | ||||
Income tax (expense) benefit | (1,355 | ) | (49 | ) | 1,306 | |||||
| | | | | | | | | | | |
Net loss | $ | (74,356 | ) | $ | (114,780 | ) | $ | (40,424 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Revenues
Revenues increased by $4.3 million from $5.3 million for the year ended December 30, 2017 to $9.7 million for the year ended December 31, 2018, primarily due to the $5.0 million upfront payment received from our Sinovant License Agreement, as well as a $1.5 million of variable consideration related to a future milestone payment pursuant to the Sinovant License Agreement that we believe is probable to be met. Grant revenue from research premiums provided to us by the Austrian government decreased by $2.2 million as a result of a decrease in our research and development expenses for which we can receive grant revenue.
|
| Year ended December 31, |
|
|
| |||||
(in thousands) |
| 2015 |
| 2016 |
| Change |
| |||
Consolidated Operations Data: |
|
|
|
|
|
|
| |||
Revenues |
| $ | 3,767 |
| $ | 6,482 |
| $ | 2,715 |
|
Costs and Expenses: |
|
|
|
|
|
|
| |||
Research and development |
| (23,604 | ) | (47,994 | ) | (24,390 | ) | |||
General and administrative |
| (7,921 | ) | (13,535 | ) | (5,614 | ) | |||
Total operating expenses |
| (31,525 | ) | (61,529 | ) | (30,004 | ) | |||
Loss from operations |
| (27,758 | ) | (55,047 | ) | (27,289 | ) | |||
Other income (expense): |
|
|
|
|
|
|
| |||
Other income (expense), net |
| 2,427 |
| (783 | ) | (3,210 | ) | |||
Interest income (expense), net |
| (22,078 | ) | 268 |
| (22,346 | ) | |||
Loss before income taxes |
| (47,409 | ) | (55,562 | ) | (8,153 | ) | |||
Income tax (expense) benefit |
| 445 |
| 672 |
| 227 |
| |||
Net loss |
| $ | (46,964 | ) | $ | (54,890 | ) | (7,926 | ) | |
Research and Development Expenses
Research and development expenses increased by $32.7 million from $49.6 million for the year ended December 31, 2017 to $82.3 million for the year ended December 31, 2018. The increase was primarily due to a $32.0 million increase for in-process research and development expenses associated with the acquisition of Zavante assets, a $6.5 million increase in research consulting fees, a $6.5 million increase associated with the payment of the NDA fees to the FDA for lefamulin and CONTEPO, a $1.9 million increase in staff costs due to the addition of employees, a $0.3 million increase in travel and other research and development costs, partly offset by a $13.9 million decrease in research materials and purchased services related to the development of lefamulin and a $0.7 million decrease in stock-based compensation expense.
General and Administrative Expenses
General and administrative expense increased by $12.3 million from $29.5 million for the year ended December 31, 2017 to $41.7 million for the year ended December 31, 2018. The increase was primarily due to a $2.6 million increase of advisory and external consultancy expenses primarily related to pre-commercialization activities and professional service fees, a $9.6 million increase in staff costs
due to the addition of employees, a $0.1 million increase in stock-based compensation expense, a $1.0 million increase in other corporate costs, a $0.7 million increase in travel expenses, and a $0.4 million increase in infrastructure costs, partly offset by a $2.2 million decrease in legal fees.
RevenuesOther Income (Expense), net
Other income (expense), net decreased by $0.8 million from $0.5 million income for the year ended December 31, 2017, to $0.3 million expense for the year ended December 31, 2018 primarily due to re-measurements of our foreign currency account balances.
Interest income (expense), net
During the year ended December 31, 2018, net interest expense increased over the prior year due to lower interest earned on cash and cash equivalents.
Income Tax Expense
Our income tax expense was $1.4 million for the year ended December 31, 2017 compared to $49,000 for the year ended December 31, 2018. The change to income tax expense was primarily due to the recognition of a valuation allowance on deferred tax assets in 2017.
Comparison of Years Ended December 31, 2016 and 2017
| | Year ended December 31, | | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2016 | 2017 | Change | |||||||
Consolidated Operations Data: | ||||||||||
Revenues | $ | 6,482 | $ | 5,319 | $ | (1,163 | ) | |||
Costs and Expenses: | ||||||||||
Research and development | (47,994 | ) | (49,615 | ) | (1,621 | ) | ||||
General and administrative | (13,535 | ) | (29,472 | ) | (15,937 | ) | ||||
| | | | | | | | | | | |
Total operating expenses | (61,529 | ) | (79,087 | ) | (17,558 | ) | ||||
| | | | | | | | | | | |
Loss from operations | (55,047 | ) | (73,768 | ) | (18,721 | ) | ||||
Other income (expense): | ||||||||||
Other income (expense), net | (783 | ) | 492 | 1,275 | ||||||
Interest income (expense), net | 268 | 275 | 7 | |||||||
| | | | | | | | | | | |
Loss before income taxes | (55,562 | ) | (73,001 | ) | (17,439 | ) | ||||
Income tax (expense) benefit | 672 | (1,355 | ) | (2,027 | ) | |||||
Net loss | $ | (54,890 | ) | $ | (74,356 | ) | $ | (19,466 | ) | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Revenues
Revenues, consisting primarily of research premium and grant revenue, increaseddecreased by $2.7$1.2 million from $3.8 million from the year ended December 31, 2015 to $6.5 million for the year ended December 31, 2016.2016 to $5.3 million for the year ended December 31, 2017. The change was primarily due to a $2.6$1.4 million increasedecrease in anticipated grant revenue from research premiums provided to us by the Austrian government as a result of increases in ourlower applicable research and development expenses.expenses, which was offset by a $0.2 million increase in grant income.
Research and Development Expenses
Research and development expenses increased by $24.4$1.6 million from $23.6$48.0 million for the year ended December 31, 20152016 to $48.0$49.6 million for the year ended December 31, 2017. The increase was
primarily due to a $2.1 million increase in staff costs due to the addition of employees and a $1.2 million increase in share-based compensation expense also due to the inclusion of additional employees in our share-based compensation plan, partially offset by a $1.5 million decrease in direct costs for purchased services related to the development of lefamulin and a $0.2 million decrease of advisory and external consultancy, travel and other expenses.
General and Administrative Expenses
General and administrative expense increased by $16.0 million from $13.5 million for the year ended December 31, 2016 to $29.5 million for the year ended December 31, 2017. The increase was primarily due to a $4.1 million increase in legal fees mainly related to the redomiciliation of our parent company from Austria to Ireland, a $6.1 million increase of advisory and external consultancy expenses primarily related to pre-commercialization activities and professional service fees, a $2.0 million increase in share-based compensation expense due to the inclusion of additional employees in our share-based compensation plan, a $2.8 million increase in staff costs due to the addition of employees, a $0.7 million increase in VAT tax expenses, and a $0.3 million increase in support, infrastructure and other corporate costs.
Other income (expense), net
Other income (expense), net increased by $1.3 million from a net expense of $0.8 million for the year ended December 31, 2016 to net income of $0.5 million for the year ended December 31, 2017. The increase was primarily due to re-measurements of our foreign currency account balances.
Interest expense, net
During the year ended December 31, 2017, net interest income was relatively flat compared to the same period in 2016.
Income tax (Expense) Benefit
Our income tax expense was $1.4 million for the year ended December 31, 2017 compared to an income tax benefit of $0.7 million for the year ended December 31, 2016. The increase was primarily due to higher costs related to our Phase 3 clinical trials of lefamulin. Research materials and purchased services for our other programs and initiatives were relatively limited during both periods. Staff costs related to research and development increased for the year ended December 31, 2016 compared to theover year ended December 31, 2015, primarily due to the addition of employees and increased clinical development costs.
General and Administrative Expenses
General and administrative expense increased by $5.6 million from $7.9 million for the year ended December 31, 2015 to $13.5 million for the year ended December 31, 2016. The increase was primarily due to increased staff costs related to the hiring of additional employees and increased professional service fees related to operating as a public company.
Other income (expense), net
Other income (expense), net decreased by $3.2 million to a $0.8 million loss during the year ended December 31, 2016 compared to the same period in 2015. The change was primarily due to an increase in losses from the re-measurementrecognition of foreign currency balances as a result of the changevaluation allowance against deferred tax assets in our functional currency.
Interest expense, net
During the year ended December 31, 2016, net interest expenses decreased by $22.3 million compared to the same period in 2015 primarily due to the decrease in the effective interest accrued under the convertible loan agreements, which were converted into equity securities in connection with our April 2015 financing,foreign subsidiaries. Our income tax (expense) benefit includes Irish, Austrian and U.S. income taxes at statutory rates and the decrease in interest expense on the Kreos loan, which was fully repaid in November 2015.effects of various permanent differences.
Comparison of Years Ended December 31, 2014 and 2015
|
| Year ended December 31, |
|
|
| |||||
(in thousands) |
| 2014 |
| 2015 |
| Change |
| |||
Consolidated Operations Data: |
|
|
|
|
|
|
| |||
Revenues |
| $ | 2,398 |
| $ | 3,767 |
| $ | 1,369 |
|
Costs and Expenses: |
|
|
|
|
|
|
| |||
Research and development |
| (9,355 | ) | (23,604 | ) | (14,249 | ) | |||
General and administrative |
| (3,739 | ) | (7,921 | ) | (4,182 | ) | |||
Total operating expenses |
| (13,094 | ) | (31,525 | ) | (18,431 | ) | |||
Loss from operations |
| (10,696 | ) | (27,758 | ) | (17,062 | ) | |||
Other income (expense): |
|
|
|
|
|
|
| |||
Other income (expense), net |
| (524 | ) | 2,427 |
| (2,951 | ) | |||
Interest income (expense), net |
| (2,908 | ) | (22,078 | ) | (19,170 | ) | |||
Loss before income taxes |
| (14,128 | ) | (47,409 | ) | (33,281 | ) | |||
Income tax (expense) benefit |
| (94 | ) | 445 |
| 539 |
| |||
Net loss |
| (14,222 | ) | $ | (46,964 | ) | (32,742 | ) | ||
Revenues
Revenues, consisting primarily of research premium and grant revenue, increased by $1.4 million from $2.4 million for the year ended December 31, 2014 to $3.8 million for the year ended December 31, 2015. The increase was primarily due to a $2.2 million increase in anticipated research premiums as a result of a higher applicable research and development cost base, offset by a $0.8 million decrease in grant revenue in the year ended December 31, 2015.
Research and Development Expenses
Research and development expenses increased by $14.2 million from $9.4 million for the year ended December 31, 2014 to $23.6 million for the year ended December 31, 2015. The increase was primarily due to higher costs related to preparation for our Phase 3 clinical trials of lefamulin and a $1.1 million increase in staff costs for the year ended December 31, 2015 compared to year ended December 31, 2014 primarily due to the addition of employees during 2015.
General and Administrative Expenses
General and administrative expenses increased by $4.2 million from $3.7 million for the year ended December 31, 2014 to $7.9 million for the year ended December 31, 2015. The increase was primarily due to a $2.2 million increase in professional service fees related to preparing for a potential offering and operating as a public company, and a $2.0 million increase in staff costs related to additional employees, which includes higher stock-based compensation expense of approximately $0.6 million related to the options granted under the Stock Option Plan 2015.
Other income (expense), net
Other income (expense), net increased by $2.9 million to $2.4 million during the year ended December 31, 2015 compared to the same period in 2014. The change was primarily due to higher gains from the re-measurement of foreign currency balances.
Interest expense, net
During the year ended December 31, 2015, interest expense, net increased by $19.2 million compared to the year ended December 31, 2014, primarily due to a $20.5 million increase of effective interest accrued under the convertible loan agreements, which was largely due to the contingent beneficial conversion feature recognized as part of the April 2015 financing. The increase in interest expense was partially offset by the prepayment of the $6.6 million loan from Kreos in November 2015 that included payment of discounted future interest on the loan.
Liquidity and Capital Resources
Since our inception, we have incurred net losses and generated negative cash flows from our operations. To date, we have financed our operations through the sale of equity securities, including our initial public offering, of ADSspublic follow-on offerings and private placements of our common shares,equity securities, convertible and term debt financings, payments from a collaboration partner and research and development support from governmental grants and loans. As
In December 2018, we announced the closing of up to a $75.0 million term loan with Hercules, or the Loan Agreement, $25.0 million of which was funded on the day of closing. Under the terms of the loan, in addition to the $25.0 million received at closing, we are eligible to receive up to an aggregate of $15.0 million in two tranches upon the approval by the FDA of the NDAs recently submitted for lefamulin and CONTEPO. We will also be eligible to receive an additional $30.0 million of aggregate term loan advances in three separate tranches upon the achievement of specified product revenue milestones. These additional tranches are at our discretion and may only be drawn upon during specified periods of time. The final $5.0 million tranche is available through December 31, 2016,2021, subject to the lenders' sole discretion. We are entitled to make interest-only payments through July 1,
2020, with incremental extensions through January 1, 2022 upon the achievement of specified performance milestones. We are required to repay the term loan after the interest only period based on a monthly amortization schedule, with a final maturity date occurring on June 1, 2023. The term loan bears interest at an annual rate equal to the greater of 9.80% and 9.80% plus the prime rate of interest minus 5.50%. The Loan Agreement provides for interest-only payments through July 1, 2020, which may be incrementally extended from time to time upon the occurrence of certain conditions through January 1, 2022, and repayment of the aggregate outstanding principal balance of the term loan thereafter in monthly installments through June 1, 2023. In addition, we hadare required to pay a fee of 6.95% of the aggregate amount of advances under the Loan Agreement at maturity. At our option, we may elect to prepay any portion of the outstanding term loan that is greater than or equal to $5.0 million by paying such portion of the principal balance and all accrued and unpaid interest thereon plus a prepayment charge equal to the following percentage of the principal amount being prepaid: (i) 3.0% if the term loan is prepaid during the first 12 months following the initial closing, (ii) 2.0% if the term loan is prepaid after 12 months following the initial closing but before 24 months following the initial closing and (iii) 1.0% if the term loan is prepaid any time thereafter but prior to the final maturity date. We are also required to satisfy certain financial covenants, including an obligation to maintain specified minimum amounts of cash and cash equivalents and short term investments of $83.9 million.in accounts pledged to Hercules.
In July 2018, we completed an underwritten public offering of 18,181,818 ordinary shares at a public offering price of $2.75 per share, resulting in gross proceeds of $50.0 million and net proceeds to us of $46.3 million, after deducting underwriting discounts and commissions and offering expenses.
On In March 2018, we entered into a sales agreement, or the ATM Agreement, with Cantor Fitzgerald & Co., or Cantor, pursuant to which, from time to time, we may offer and sell our ordinary shares having an aggregate offering price of up to $50.0 million through Cantor pursuant to an effective universal shelf registration statement. Sales of our ordinary shares, if any, under the agreement with Cantor may be made in sales deemed to be an "at-the-market offering" as defined in Rule 415(a)(4) under the Securities Act of 1933, as amended.
In September 2017 we completed an underwritten public offering of 9,411,765 ordinary shares at a public offering price of $8.50 per share, resulting in gross proceeds of $80.0 million and net proceeds to us of $73.3 million, after deducting underwriting discounts and commissions and offering expenses.
In December 19, 2016, we completed a rights offering and a related underwritten offering for the sale of an aggregate of 588,127 common shares resulting in aggregate gross proceeds of approximately $24.8 million and net proceeds to us of approximately $20.6 million, after deducting underwriting fees and offering expenses.
On September 23, 2015 we completed our initial public offering on the NASDAQ Global Market issuing 9,000,000 ADSs at a price to the public of $10.25 per ADS, representing 900,000 of our common shares. Each ADS represents one tenth of a common share. On September 30, 2015 the underwriters of our initial public offering exercised in full their over-allotment option to purchase an additional 1,350,000 ADSs, representing 135,000 common shares, at the initial public offering price of $10.25 per ADS, less underwriting discounts. Including the over-allotment ADSs, we sold an aggregate of 10,350,000 ADSs representing 1,035,000 common shares, in our initial public offering, which resulted in gross proceeds of approximately $106.1 million and net proceeds to us of approximately $92.4 million, after deducting underwriting discounts and offering expenses.
In connection with our April 2015 financing, we sold 730,162 common shares with contractual preference rights under a shareholders agreement, including the sale of 511,188 common shares at a price per share of €82.35 ($87.71) for €42.1 million ($44.8 million) in cash consideration and the sale of 218,974 common shares in exchange for certain contributions in-kind consisting of the conversion of outstanding convertible loans and silent partnership interests. We also agreed to sell a second tranche of common shares with contractual preference rights under the shareholders agreement to the investors in our April 2015 financing at their option for an aggregate purchase price of $70.0 million if we did not complete a public offering in the United States within specified parameters or by a specified date. Upon the closing of our initial public offering and the issuance of the shares for nominal value in satisfaction of the preferred dividend rights, all contractual preference rights under the shareholders agreement terminated.
Between 2011 and 2015 we entered into five convertible loan agreements with certain of our shareholders for proceeds in the aggregate amount of $18.2 million. All outstanding convertible loans converted into common shares with contractual preference rights under the shareholders agreement in connection with our April 2015 financing.
We entered into silent partnership agreements with certain of our shareholders for aggregate proceeds of $0.5 million in the second quarter of 2014 and $0.9 million in the first quarter of 2015. These agreements have terminated and the related claims for repayment were converted into common shares with contractual preference rights under the shareholders agreement in connection with our April 2015 financing.
Also during the second quarter of 2014, we entered into a €5.0 million ($6.6 million) loan agreement with Kreos that resulted in net proceeds of $6.2 million after deduction of the initial interest and principal payments and transaction costs at closing. In connection with the loan agreement, we granted Kreos Capital IV (Expert Fund) Limited a warrant to purchase our common shares with contractual preference rights under the shareholders agreement, which Kreos Capital IV (Expert Fund) Limited has exercised in full. As collateral for the loan, we pledged our intellectual property, fixed assets exceeding a book value of €1,000, the receivables related to the research premium and our bank accounts. In July 2015, Kreos Capital IV (UK) Limited agreed to release us from the pledge of our intellectual property upon the closing of our initial public offering. We prepaid the Kreos loan in accordance with the terms of the loan agreement in November 2015.
Cash Flows
Comparison of Years Ended December 31, 20152017 and 20162018
The following table summarizes our cash flows for the years ended December 31, 20152017 and 2016:2018:
| | Year ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
(in thousands) | 2017 | 2018 | |||||
Net cash (used in) provided by: | |||||||
Operating activities | $ | (69,348 | ) | $ | (72,723 | ) | |
Investing activities | 49,749 | (4,604 | ) | ||||
Financing activities | 72,219 | 92,923 | |||||
Effect of foreign currency translation on cash | 1,371 | (362 | ) | ||||
| | | | | | | | |
Net increase (decrease) in cash and cash equivalents | $ | 53,991 | $ | 15,234 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Operating Activities
|
| Year ended December 31, |
| ||||
(in thousands) |
| 2015 |
| 2016 |
| ||
Net cash (used in) provided by: |
|
|
|
|
| ||
Operating activities |
| $ | (21,858 | ) | $ | (49,321 | ) |
Investing activities |
| (76,704 | ) | 23,352 |
| ||
Financing activities |
| 133,018 |
| 22,301 |
| ||
Effect of foreign currency translation on cash |
| (160 | ) | — |
| ||
Net increase (decrease) in cash |
| 34,296 |
| (3,668 | ) | ||
Operating Activities
Cash flow utilized byused in operating activities increased by $27.4$3.4 million from $21.9$69.3 million for the year ended December 31, 20152017 to $49.3$72.7 million for the year ended December 31, 20162018 primarily due to a $28.4$8.7 million increase in net loss, after adjustments for non-cash amounts included in net income partially offset by improvedloss and higher working capital of $1.0$5.3 million primarily from higher trade payablesdue to changes in accrued expenses and other current liabilities.
Investing Activities
Cash flow fromprovided by investing activities changeddecreased by $100.1$54.4 million from $76.7$49.7 million of cash outflow inprovided for the year ended December 31, 20152017 to $23.4$4.6 million cash inflow inused for the year ended December 31, 20162018 primarily due to the redemptiona $51.0 million decrease in proceeds from sale of
term deposits. Other investing activities were relatively insignificant available-for-sale financial assets to fund operational cash outflows and $4.3 million in both periods andtransaction costs related primarily to the acquisition of equipment in support of our research and development activities.Zavante assets.
Financing Activities
Financing Activities
Cash flow generated fromprovided by financing activities decreasedincreased by $110.7$20.7 million from $133.0$72.2 million for the year ended December 31, 20152017 to $22.3$92.9 million for the year ended December 31, 20162018 primarily due to net proceeds of $44.8$24.2 million related to sales of our ordinary shares under our ATM Agreement, net proceeds of $46.3 million from our April 2015 financing and proceeds of $106.1 million from our initialJuly 2018 public offering in September 2015, $3.4of ordinary shares and the $25.0 million from the issuance of an additional convertible loan in January 2015 and proceeds of $0.9 million from a silent partnership agreement entered into in January 2015. The year over year decrease in financing cash inflows was partially offset by proceeds of $24.8 million fromadvance under our December 2016 rights offering, a $7.4 million decrease of cash outflows for repayments of long-term borrowings and a $12.1 million decrease in equity transaction costs.2018 Loan Agreement with Hercules.
Comparison of Years Ended December 31, 20142016 and 20152017
The following table summarizes our cash flows for the years ended December 31, 20142016 and 2015:2017:
| | Year ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
(in thousands) | 2016 | 2017 | |||||
Net cash (used in) provided by: | |||||||
Operating activities | $ | (48,325 | ) | $ | (69,348 | ) | |
Investing activities | 23,352 | 49,749 | |||||
Financing activities | 22,301 | 72,219 | |||||
Effect of foreign currency translation on cash | (996 | ) | 1,371 | ||||
| | | | | | | | |
Net increase (decrease) in cash and cash equivalents | $ | (3,668 | ) | $ | 53,991 | ||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Operating Activities
|
| Year ended December 31, |
| ||||
(in thousands) |
| 2014 |
| 2015 |
| ||
Net cash (used in) provided by: |
|
|
|
|
| ||
Operating activities |
| $ | (11,407 | ) | $ | (21,858 | ) |
Investing activities |
| (87 | ) | (76,704 | ) | ||
Financing activities |
| 9,098 |
| 133,018 |
| ||
Effect of foreign currency translation on cash |
| (411 | ) | (160 | ) | ||
Net increase (decrease) in cash |
| (2,807 | ) | 34,296 |
| ||
Operating Activities
Cash flow utilized by operating activities increased by $10.5$21.0 million from $11.4$48.3 million for the year ended December 31, 20142016 to $21.9$69.3 million for the year ended December 31, 20152017 primarily due to a higher$16.1 million increase in net loss, of $13.1 million during the year ended December 31, 2015, after adjustments for non-cash amounts included in net income, partly offset by improvedloss and higher working capital of $2.6$4.9 million primarily from higher accounts payabledue to changes in accrued expenses and accrued expenses.other current liabilities.
Investing Activities
Investing Activities
Cash flow utilizedprovided by investing activities increased by $76.6$26.4 million from $0.1$23.4 million cash inflow in the year ended December 31, 20142016 to $76.7$49.7 million cash inflow in the year ended December 31, 20152017 primarily due to thean increase of $15.0 million in proceeds from sale of available-for-sale securities to fund operational cash out flows and a decrease of $57.0 million in purchases of marketable securities and investmentsavailable-for
sale-securities in term deposits. Other2017. The year over year increase in investing activities were relatively insignificantwas partially offset by a $45.0 million decrease in both years and related primarily to the acquisitionproceeds from maturities of equipment in support of our research and development activities.term deposits.
Financing Activities
Financing Activities
Cash flow generated fromprovided by financing activities increased by $123.9$49.9 million from $9.1$22.3 million for the year ended December 31, 20142016 to $133.0$72.2 million for the year ended December 31, 20152017 primarily due to the gross proceeds of $106.1$80.0 million from our initial public offering in September 2015, cash2017 equity financing, compared to the proceeds of $44.8$24.8 million from our April 2015rights offering and related underwritten offering in December 2016. The year-over-year increase in financing $3.4 million from the issuance of an additional convertible loan in January 2015 and proceeds of $0.9 million from a new silent partnership agreement in January 2015, compared with net proceeds from our loan from Kreos of $6.2 million, additional convertible loans in the aggregate principle amount of $4.7 million and proceeds of $0.5 million from a new silent partnership agreement in the year ended December 31, 2014. The year over year increasecash inflows was partially offset by a $5.3 million increase in equity transaction costs of $14.9 million and a $5.1 million increase of cash outflows for repayments of long-term borrowings, including $5.4 million from the prepayment of the Kreos loan in 2015.costs.
Operating and Capital Expenditure Requirements
We anticipate that our expenses will increase substantially as we continue the development of and potentially seekto invest in critical pre-commercialization activities prior to obtaining marketing approval for lefamulin and possibly, other product candidates and continue our research activities. Our expenses will increase if we suffer any delays in our Phase 3 clinical program for lefamulin for the treatment of CABP, including delays in enrollment of patients.CONTEPO. If we obtain marketing approval for lefamulin, CONTEPO or any other product candidate that we develop, in-license or acquire, we expect to incur significant additional commercialization expenses related to product sales, marketing, distribution and manufacturing. Furthermore,In addition, our expenses will increase if we expectsuffer any regulatory delays or are required to incurconduct additional costs associated with operating as a public company.clinical trials to satisfy regulatory requirements.
In addition, our expenses will increase if and as we:
- •
·initiate or continue the research and development of lefamulin and CONTEPO for additional indications and of our other product candidates;·- •
- seek to discover and develop additional product candidates;
· - •
- seek marketing approval for any product candidates that successfully complete clinical development;
·ultimately - •
- continue to establish a medical affairs, sales, marketing and distribution infrastructure and build up inventory of finished product and its components to commercialize any product candidates for which we receive marketing approval;
· - •
- in-license or acquire other products, product candidates or technologies;
· - •
- maintain, expand and protect our intellectual property portfolio;
· - •
- establish and expand manufacturing arrangements with third parties;
- •
- Draw additional funds under the Loan Agreement with Hercules;
- •
- expand our physical presence in the United
States;States andIreland; and,
· - •
- add operational, financial and management information systems and personnel, including personnel to support our product development, our operations as a public company and our planned
futurecommercialization efforts.
We expect that our existing cash, cash equivalents and short-term investments as of December 31, 2018, proceeds from the sale of ordinary shares under the our ATM Agreement of $5.9 million in the first quarter of 2019, as well as a $1.5 million milestone payment received in February 2019 under our license agreement with Sinovant Sciences, Ltd., and research premiums from the Austrian government for our qualified research and development expenditures, will be sufficient to enable us to fund our operating expenses, debt service obligations and capital expenditure requirements at least into the second quarter of 2018 and to obtain top-line data for both our Phase 3 clinical trials of lefamulin.2020. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect. This estimate assumes, among other
things, that we do not obtain any additional funding through grants and clinical trial support, collaboration agreements, equity or debt financings.financings, including additional advances under the Loan Agreement with Hercules. We may be eligible to borrow up an additional $50.0 million under our Loan Agreement with Hercules if we achieve specified regulatory and product revenue milestones, including $10.0 million that we will be eligible to borrow upon the approval by the FDA of an NDA for lefamulin and $5.0 million that we will be eligible to borrow upon the approval by the FDA of an NDA for CONTEPO.
We expect to seek additional funding in future periods for purposes of investment in our commercial and medical affairs organization, as well as investing in our supply chain, in an effort to enhance the potential commercial launch of lefamulin and CONTEPO.
Our future capital requirements will depend on many factors, including:
- •
·the progress, costs and results of our Phase 3 clinical trials for lefamulin;·the costs and timing of process development and manufacturing scale-up activities associated withlefamulin;lefamulin and CONTEPO;
·- •
- the costs, timing and outcome of regulatory review of
lefamulin;lefamulin and CONTEPO;
· - •
- the costs of commercialization activities for lefamulin and CONTEPO if we receive, or expect to receive, marketing approval, including the costs and timing of establishing product sales, marketing, distribution and outsourced manufacturing capabilities, including the costs of building finished product inventory and its components in preparation of initial marketing of
lefamulin;lefamulin and CONTEPO;
· - •
- subject to receipt of marketing approval, revenue received from commercial sales of
lefamulin;lefamulin and CONTEPO;
· - •
- the costs of developing lefamulin and CONTEPO for the treatment of additional indications;
· - •
- our ability to establish collaborations on favorable terms, if at all;
· - •
- the scope, progress, results and costs of product development of any other product candidates that we may develop;
· - •
- the extent to which we in-license or acquire rights to other products, product candidates or technologies;
· - •
- the costs of preparing, filing and prosecuting patent applications, maintaining and protecting our intellectual property rights and defending against intellectual property-related claims;
· - •
- the continued availability of Austrian governmental grants;
· - •
- the need to satisfy interest and principal obligations in our debt facility;
- •
- the rate of the expansion of our physical presence in the United
States;States andIreland; and
· - •
- the costs of operating as a public company in the United States.
Conducting clinical trials is a time-consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales. Our commercial revenues, if any, will be derived from sales of lefamulin, CONTEPO or any other products that we successfully develop, in-license or acquire, none of which we expect to beare yet commercially available for several years, if at all.available. In addition, if approved, lefamulin, CONTEPO or any other product candidate that we develop, in-license or acquire may not achieve commercial success. Accordingly, we will need to obtain substantial additional financing to achieve our business objectives. Adequate additional financing may not be available to us on acceptable terms, or at all. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe that we have sufficient funds for our current or future operating plans.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of equity offerings, debt financings, collaborations, and funding from local and international government entities and non-government organizations in the disease areas addressed by our product candidates and marketing, distribution or licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, yourthe ownership interest of our shareholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect yourthe rights as a security holder. Debtof our shareholders. Additional debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends.
If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Contractual Obligations and Commitments
The table below sets forth our contractual obligations and commercial commitments as of December 31, 2016 that are expected to have an impact on liquidity and cash flow in future periods. The amounts disclosed are the contractual undiscounted cash flow values.
|
| Payments Due by Period |
| |||||||||
(in thousands) |
| Less than |
| Between 1 |
| Between 3 |
| More than 5 |
| Total |
| |
Operating lease obligations |
| $ | 1,484 |
| 1,499 |
| 1,037 |
| 507 |
| 4,527 |
|
|
| Payments Due by Period |
| |||||||||
(in thousands) |
| Less than |
| Between 1 |
| Between 3 |
| More than 5 |
| Total |
| |
Other contractual commitments |
| 51,685 |
| 6,241 |
| — |
| — |
| 57,926 |
| |
Total |
| $ | 53,169 |
| 7,740 |
| 1,037 |
| 507 |
| 62,453 |
|
Operating lease obligations include rental agreements for our facilities in Austria and the United States.
Other contractual commitments relate to contracts entered into with contract research organizations and contract manufacturing organizations in connection with the conduct of clinical trials and other research and development activities. Some of these commitments include early termination clauses exercisable at our discretion. The amounts shown above are estimated based on the assumptions that all remaining services will be performed as agreed and all milestones and other conditions of the respective contracts are met.
Capital Expenditures
Our total purchases related to capitalCapital expenditures were $216,000$1.2 million and $603,000$0.2 million for the years ended December 31, 20152017 and 2016,2018, respectively. We made no significant investments in intangible assets during the years ended December 31, 20152017 and 2016.2018. Currently, there are no material capital projects planned in 2017. However, we expect our capital expenditures may increase over the next 12 to 18 months due to the expansion of our U.S. presence, our two Phase 3 clinical trials for lefamulin and the continued enhancements of our information technology infrastructure.2019.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balanceoff-balance sheet arrangements, as defined under SEC rules, other than our operating lease obligations for our facilities in Austria, Ireland and the United States.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to a variety of financial risks in the ordinary course of our business: market risk, credit risk and liquidity risk. Our overall risk management program focuses on preservation of capital given the unpredictability of financial markets. These market risks are principally limited to interest rate and foreign currency fluctuations.
Market Risk
We do not have any significant credit risk exposure to any single counterparty or any group of counterparties having similar characteristics. The credit risk on liquid funds (bank accounts, cash balances, marketable securities and term deposits) is limited because the counterparties are banks with high credit ratings from international credit rating agencies. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes.
We are exposed to foreign exchange risk arising from various currency exposures, primarily with respect to the euro and the British pound. Our functional currency is the U.S. dollar, but we receive payments from several of our collaborators, and acquire materials, in each of these other currencies. We have not established any formal practice to manage the foreign exchange risk against our functional currency. However, we attempt to minimize our net exposure by buying or selling foreign currencies at spot rates upon receipt of new funds to facilitate committed or anticipated foreign currency transactions.
Interest rate risk may arise from short-term or long-term borrowings. Asdebt. In December 2018, we announced the closing of December 31, 2016, we had no borrowings that exposed usup to a $75.0 million term loan with Hercules, $25.0 million of which was funded on the day of closing. This initial draw and subsequent draws will have interest rates set based on US Prime Interest Rate at the time of the draw. If the US Prime Interest Rate increases, our interest rate risk. As of December 31, 2016, we had neither significant long-term interest-bearing assets nor significant long-term interest-bearing liabilities.will increase.
Due to the short-term nature of our investment portfolio, we do not believe an immediate 10% increase in interest rates would have a material effect on the fair market value of our portfolio, and accordingly we do not expect our operating results or cash flows to be materially affected by a sudden change in market interest rates.
Liquidity Risk
Since our inception, we have incurred net losses and generated negative cash flows from our operations. Based on our current operating plans, we believeexpect that our existing cash, cash equivalents and short-term investments as of December 31, 2018, proceeds from the sale of ordinary shares under the our ATM Agreement of $5.9 million in the first quarter of 2019, as well as a $1.5 million milestone payment received in February 2019 under our license agreement with Sinovant Sciences, Ltd., and research premiums from the Austrian government for our qualified research and development expenditures, will be sufficient to enable us to fund our operationsoperating expenses, debt service obligations and capital expenditure requirements at least into the second quarter of 2018.2020. We have based these estimatesthis estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect. This estimate assumes, among other things, that we do not obtain any additional funding through grants and clinical trial support, collaboration agreements, equity or debt financings, including additional advances under the Loan Agreement with Hercules. We may be eligible to borrow up an additional $50.0 million under our Loan Agreement with Hercules if we achieve specified regulatory and product revenue milestones, including $10.0 million that we will be eligible to borrow upon the approval by the FDA of an NDA for lefamulin and $5.0 million that we will be eligible to borrow upon the approval by the FDA of an NDA for CONTEPO.
We anticipate that our expenses will increase substantially as we continue to seek marketing approval for and prepare for the commercialization of lefamulin and CONTEPO, and, possibly, other product candidates. If we obtain marketing approval for lefamulin and CONTEPO or any other product candidate that we develop, in-license or acquire, we expect to incur significant commercialization expenses related to product sales, marketing, distribution and manufacturing. We have developed plans to mitigate this risk, which primarily consist of raising additional capital through a combination of equity or debt financings, new collaborations, and reducing cash expenditures.
However, there can be no assurance that we will be successful in acquiring additional capital at level sufficient to fund our operations or on terms favorable to us.
If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce to eliminate our research and development programs or any future commercialization effort.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our consolidated financial statements, together with the report of our independent registered public accounting firm, appear on pages F-1 through F-33F-31 of this Annual Report on Form 10-K.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Conclusions Regarding the Effectiveness of Disclosure Controls and Procedures
We have carried out an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) under the supervision and the participation of the company’scompany's management, which is responsible for the management of the internal controls, and which includes our Chief Executive Officer and Chief Financial Officer (our principal executive officer and principal financial officer, respectively). The term “disclosure"disclosure controls and procedures,”" as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’sCommission's rules and forms.
Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Securities Exchange Act of 1934 is accumulated and communicated to the company’scompany's management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. There are inherent limitations to the effectiveness of any system of disclosure controls and procedures, including the possibility of human error and the circumvention or overriding of the controls and procedures. Accordingly, even effective disclosure controls and procedures can only provide reasonable assurance of achieving their control objectives. Based upon our evaluation of our disclosure controls and procedures as of December 31, 2016,2018, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at a reasonable level of assurance.
Management’sManagement's Annual Report on Internal Control Over Financial Reporting
Internal control over financial reporting refers to the process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer, and effected by our supervisory board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that: (1) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and supervisory board;board of directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’scompany's assets that could have a material effect on the financial statements.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human
diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Management is responsible for establishing and maintaining adequate internal control over our financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(e) under the Exchange Act. Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting. Management has used the framework set forth in the report entitled “"Internal Control—Integrated Framework (2013)”" published by the Committee of Sponsoring Organizations of the Treadway Commission to evaluate the effectiveness of our internal control over financial reporting. Based on its evaluation, management has concluded that our internal control over financial reporting was effective as of December 31, 2016,2018, the end of our most recent fiscal year.
Our independent registered public accounting firm has not performed an evaluation of our internal control over financial reporting during any period in accordance with the provisions of the Sarbanes-Oxley Act. For as long as we remain an “emerging"emerging growth company”company" as defined in the JOBS Act, we intend to take advantage of the exemption permitting us not to comply with the requirement that our independent registered public accounting firm provide an attestation on the effectiveness of our internal control over financial reporting.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting (as defined in Exchange Act Rule 13a-15(f)) that occurred during the three months ended December 31, 20162018 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
None.Table of Contents
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
General
We have a two tier board structure consistingBoard of our management board (Vorstand) and a separate supervisory board (Aufsichtsrat). The management board is responsible for managing the business and represents the company in dealings with third parties. The supervisory board is responsible for appointing and removing the members of the management board and supervising the business conducted by the management board. Although the supervisory board does not actively manage the company, both the Austrian Stock Corporation Act (Aktiengesetz) and our articles of association (Satzung), together with the management board’s internal rules of procedure (Geschäftsordnung), require that the prior approval of the supervisory board is obtained before the management board takes certain actions. Below is a summary of relevant information concerning our supervisory board, management board and senior management, as well as a summary of certain significant provisions of Austrian corporate law, the articles of association and the Austrian Stock Corporation Act in respect of our management board and supervisory board.Directors
Supervisory Board
Members of the Supervisory Board
Set forth below are the names and certain biographical information about each member of our supervisory board membersof directors as of December 31, 2016.March 1, 2019. The information presented includes each supervisory board member’sdirector's principal occupation and business experience for at least the past five years and the names of other public companies of which he or she has served as a director during the past five years. The business address
Name | Age | Position | |||
|---|---|---|---|---|---|
| Daniel Burgess(1)(3) | 57 | Director, Chairman of the Board | |||
| Theodore Schroeder | 63 | Director, Chief Executive Officer | |||
| Colin Broom, MD | 63 | Director | |||
| Carrie Bourdow(2) | 56 | Director | |||
| Mark Corrigan, MD(2)(3) | 61 | Director | |||
| Charles A. Rowland, Jr.(1)(2) | 60 | Director | |||
| George H. Talbot, MD(3) | 70 | Director | |||
| Stephen Webster(1)(3) | 57 | Director | |||
- (1)
- Member of
our supervisory board members is our registered office address at Leberstrasse 20, 1110 Vienna, Austria.the audit committee. - (2)
- Member of the compensation committee.
- (3)
- Member of the nominating and corporate governance committee.
Name |
| Age |
| Position |
| Initial Year of |
| Year of |
Daniel Burgess |
| 55 |
| Member of the Supervisory Board (Chairman) |
| 2016 |
| 2019 |
Axel Bolte |
| 45 |
| Member of the Supervisory Board (Deputy Chairman) |
| 2007 |
| 2017 |
Chau Khuong |
| 41 |
| Member of the Supervisory Board |
| 2015 |
| 2018 |
Stephen Webster |
| 55 |
| Member of the Supervisory Board |
| 2016 |
| 2019 |
Mark Corrigan, MD |
| 59 |
| Member of the Supervisory Board |
| 2016 |
| 2019 |
George H. Talbot, MD |
| 68 |
| Member of the Supervisory Board |
| 2009 |
| 2017 |
Charles A. Rowland, Jr. |
| 58 |
| Member of the Supervisory Board |
| 2015 |
| 2018 |
Daniel Burgesshas served on our board of directors since June 23, 2017. Mr. Burgess was a member of the supervisory board of Nabriva Austria and served as its chairman since August 2016.from October 2016 until the Redomiciliation. Mr. Burgess has been a venture partner at SV Life Sciences since 2014. He was previously president and chief executive officer of Rempex Pharmaceuticals, an antibiotics company he co-founded in 2011 and that was subsequently sold to The Medicines Company in 2013. Prior to this, Mr. Burgess was president and chief executive officer of Mpex Pharmaceuticals from 2007 until its acquisition by Aptalis Inc. in 2011. He also served as chief operating officer and chief financial officer of Hollis-Eden Pharmaceuticals from 1999 to 2007 and Chief Financial Officerchief financial officer at Nanogen IncInc. from 1998 to 1999. Prior to this, Mr. Burgess spent 10 years at Gensia Sicor, Inc. (acquired by Teva Pharmaceutical Industries Ltd), where he held a variety of executive-level positions with responsibility for overall finance for the company. He began his career at Castle & Cooke, and Smith Barney, Harris Upham and Company. Mr. Burgess also is chairman of the board of directors of Atox Bio, a private biotechnology company, and of Pulmocide Ltd., a private drug discovery company; and executive chairman of Qpex Biopharma, a drug discovery company. In addition, he serves as a member of the boards of directors of Cidara Therapeutics, Inc., Arsanisa public biotechnology company; Arbutus Biopharma Corp., a public biotechnology company; and Leiter's Inc., and Genoaa private compounding pharmacy. Mr. Burgess was a member of the board of directors of Santarus, Inc., from 2004 until its acquisition in 2014 by Salix Pharmeceuticals Inc. (now Valeant Pharmaceuticals International, Inc.). He received his B.A. in economics from Stanford University and an M.B.A. from Harvard University. We believe Mr. Burgess is qualified to serve on our supervisory boardas a director because of his expertise and experience as an executive in the pharmaceutical industry and his educational background.
Axel BolteTheodore Schroeder has served on our board of directors and as deputy chairman of our supervisory boardchief executive officer since 2013 andJuly 24, 2018. During the last 30 years, Mr. Schroeder has been focused on our supervisory board since 2007. Since 2003, Mr. Bolte has been an investment advisor at HBM Partners AG, a provider of investment advisory servicesdrug development and commercialization in the life sciences industry. Previously,both large and small pharmaceutical companies. Most recently, he was an investment manager at NMT New Medical
Technologies AG from 2001 to 2003, and prior to that, Mr. Bolte served as a scientistpresident, chief executive officer and director of Zavante Therapeutics from June 2015 until its acquisition by Nabriva Therapeutics in July 2018. Mr. Schroeder co-founded Cadence Pharmaceuticals
in 2004 and previously held leadership roles at Serono SA.Elan Pharmaceuticals, Dura Pharmaceuticals and earlier in his career, Bristol-Myers Squibb. He currently serves on the board of Cidara Therapeutics, Otonomy and Collegium Pharmaceutical. He is a former chair of BIOCOM, the California life sciences trade association and in 2014, he was named the EY Entrepreneur of the Year for the San Diego region and was listed as a national finalist. He received a bachelor's degree in management from Rutgers University. We believe Mr. Shroeder is qualified to serve as a director because of his expertise and experience as an executive in the pharmaceutical industry, his service on other boards of directors and his educational background.
Colin Broom has served on our board of Ophthotech Corporationdirectors since June 23, 2017 and as our chief executive officer since April 12, 2017 until July 24, 2018. Dr. Broom was previously chief executive officer of Nabriva Austria from August 2014 until the Redomiciliation. Prior to joining Nabriva Austria, he served as chief scientific officer at ViroPharma Incorporated from 2004 until it was acquired by Shire plc in 2014. Dr. Broom served as vice president of clinical development and medical affairs in Europe for Amgen Inc. from 2000 to 2003 and previously held several leadership positions with Hoechst Marion Roussel (now Sanofi), SmithKline Beecham and Glaxo (now GlaxoSmithKline). Dr. Broom served onas a member of the board of directors of PTC Therapeutics,NPS Pharmaceuticals, Inc. Mr. Boltefrom 2009 until its acquisition by Shire in 2015. He is a member of the U.K. Royal College of Physicians and a fellow of the Faculty of Pharmaceutical Medicine. Dr. Broom received a degreehis B.Sc. from the Swiss Federal Institute of TechnologyUniversity College London and an M.B.A.M.B.B.S. from the University of St. Gallen.George's Hospital Medical School. We believe that Mr. BolteDr. Broom is qualified to serve on our supervisory board because of his many years of service on our supervisory board, his extensive experience as a venture capital investor in the life sciences industry and his service on the boards of directors of other life sciences companies.
Chau Khuong has served on our supervisory board since April 2015. Mr. Khuong is a private equity partner at OrbiMed Advisors LLC, which he joined in 2003. Previously, he served as a manager at Veritas Medicine, Inc. from 2000 to 2001. Mr. Khuong serves on the boards of directors of Otonomy, Inc. and Pieris Pharmaceuticals, Inc. He received both his B.S. and M.P.H. from Yale University. We believe that Mr. Khuong is qualified to serve on our supervisory board due to his extensive experience as a venture capital investor in the life sciences industry and his service on the boards of directors of other life sciences companies.
Stephen Webster has served on our supervisory board since August 2016. Mr. Webster has been chief financial officer of Spark Therapeutics since July 2014. He was previously senior vice president and chief financial officer of Optimer Pharmaceuticals, Inc. from June 2012 until its acquisition by Cubist Pharmaceuticals in November 2013. Prior to this, Mr. Webster served as senior vice president and chief financial officer of Adolor Corporation, also acquired by Cubist, from 2008 to 2011. Previously, Mr. Webster served as managing director Investment Banking Division, Health Care Group for Broadpoint Capital Inc. (formerly First Albany Capital). He also was a co-founder and served as president and chief executive officer of Neuronyx, Inc. Prior to this, Mr. Webster held positions of increasing responsibility, including as director, Investment Banking Division, Health Care Group, for PaineWebber Incorporated. Mr. Webster holds an A.B. in economics from Dartmouth College and an M.B.A. from the University of Pennsylvania. We believe that Mr. Webster is qualified to serve on our supervisory board due to his extensive experience in all stages of drug development and commercialization.
Carrie Bourdow has served on our board of directors since June 23, 2017. Ms. Bourdow has been the President, Chief Executive Officer, and member of the Trevena Board of Directors since October 2018. She has served in various senior positions at Trevena since May 2015. She joined Trevena as Chief Commercial Officer and was appointed Executive Vice President and Chief Operating Officer in January 2018. Prior to joining Trevena, Ms. Bourdow was Vice President of Marketing at Cubist Pharmaceuticals, Inc., from 2013 until its acquisition by Merck & Co., Inc. in January 2015. At Cubist, Ms. Bourdow led launch strategy, marketing, reimbursement, and operations for five acute care hospital pharmaceuticals. Prior to Cubist, Ms. Bourdow served for more than 20 years at Merck & Co., Inc., where she held positions of increasing responsibility across multiple therapeutic Ms. Bourdow holds a B.A. degree from Hendrix College and an M.B.A. from Southern Illinois University. We believe Ms. Bourdow is qualified to serve as a director due to her extensive experience in the biopharmaceutical industry, particularly his prior service as a chief financial officerincluding her experience with anti-infectives and in other executive management roles.with the commercialization of new drugs.
Mark Corrigan has served on our board of directors since June 23, 2017. Dr. Corrigan previously served on the supervisory board since August 2016.of Nabriva Austria from October 2016 until the Redomiciliation. Since January 2015, Dr. Corrigan has been executive chairman of BlackThorn Therapeutics. Dr. Corrigan served as president and chief executive officer of Zalicus, Inc. from January 2010 until July 2014. Previously, Dr. Corrigan was executive vice president of research and development at the specialty pharmaceutical company Sepracor Inc., and prior to this, he spent 10 years with Pharmacia & Upjohn, most recently as Group Vice President of Global Clinical Research and Experimental Medicine. Before entering the healthcare industry, Dr. Corrigan was in academic research at the University of North Carolina at Chapel Hill School of Medicine, where he maintains a faculty appointment as Adjunct Professor in the Psychiatry Department. Dr. Corrigan hascurrently serves on the boards of directors of BlackThorn Therapeutics, Inc., a private clinical-stage biopharmaceutical company, Novelin Therapeutics, Inc., a public biopharmaceutical company, Correvio Pharma Corporation (formerly Cardiome Pharma), a public biopharmaceutical company, Quartet Medicine, a private biotechnology company, and Accele BioPharma Inc., a private biopharmaceutical company, and previously served on the boardboards of directors of numerous companies, includingCoLucid Pharmaceuticals, Inc., Cubist Pharmaceuticals, Inc., Avanair Pharmaceuticals, Inc., and Avanir Pharmaceuticals prior to their acquisitions by Merck and Otsuka Holdings, respectively, andEPIRUS Biopharmaceuticals, Inc., where he served as chairman of EPIRUS Biopharmaceuticals’ the
board of directors. Dr. Corrigan holds an M.D. from the University of Virginia and received specialty training in psychiatry at Maine Medical Center and Cornell University. We believe Dr. Corrigan is qualified to serve on our supervisory boardas a director due to his extensive experience in the biopharmaceutical industry as both an executive and a board member and because of his education and training.
George H. Talbot has served on our board of directors since June 23, 2017. Dr. Talbot previously served on the supervisory board since 2009.of Nabriva Austria from 2009 until the Redomiciliation. Dr. Talbot has been the principal at Talbot Advisors LLC, a biopharmaceutical company consultancy, since 2007 and prior to that, from 2000 to 2006. From 2006 to 2007, he served as chief medical officer and executive vice president of Cerexa, Inc. prior to its acquisition by Forest Laboratories, Inc. Dr. Talbot also worked closely with Calixa Therapeutics, Inc. and Durata Therapeutics, Inc., prior to their acquisitions by Cubist Pharmaceuticals, Inc. and Actavis plc, respectively. He was an initial member of the Infectious Diseases Society of America's Antimicrobial Availability Task Force ("Bad Bugs, No Drugs") and recently completed a seven-year tenure as co-chair of the Foundation for the National Institutes of Health (FNIH) Biomarkers Consortium Projects for Endpoint Development in Acute Bacterial Skin and Skin Structure Infections, Community-acquired Bacterial Pneumonia, and Hospital-acquired Bacterial Pneumonia/Ventilator-associated Bacterial Pneumonia, which made evidence-based recommendations to the Food and Drug Administration for its Guidance development in these indications. Dr. Talbot received his B.A. from Wesleyan University, his M.D. from the Yale University School of Medicine, and his Infectious Diseases fellowship training at the University of Pennsylvania. After serving as a faculty member of the Infectious Diseases Section at the University of Pennsylvania, he joined the anti-infectives group at Rhone-Poulenc-Rorer in 1990. We believe that Dr. Talbot is qualified to serve on our supervisory boardas a director due to his education, training and extensive experience in the biopharmaceutical industry.
Charles A. Rowland, Jr. has served on our board of directors since June 23, 2017. Mr. Rowland previously served on the supervisory board sinceof Nabriva Austria from January 2015.2015 until the Redomiciliation. Mr. Rowland served as chief executive officer of Aurinia Pharmaceuticals Inc. from April 2016 to January 2017. Mr. Rowland previously served as vice president and chief financial officer of ViroPharma Incorporated from 2008 until it was acquired by Shire plc in 2014. Prior to joining ViroPharma, Mr. Rowland served as executive vice president and chief financial officer, as well as interim co-chief executive officer, for Endo Pharmaceuticals Inc. from 2006 to 2008 and chief financial officer at Biovail Corporation from 2004 to 2006. He previously held finance and operational positions of increasing responsibility at Breakaway Technologies, Inc., Pharmacia, Novartis International AG and Bristol-Myers Squibb Company. Mr. Rowland currently serves onas a member of the board of directors offor Blueprint Medicines Corporation, a public biopharmaceutical company, Viking Therapeutics, a public, clinical-stage biopharmaceutical company, and previouslyOrchard Terapeutics, a public, clinical-stage biopharmaceutical company. In addition, Mr. Rowland serves as a member of the board of directors for Generation Bio and PsiOxus Therapeutics Ltd., both of which are privately held biopharmaceutical companies. Previously, he served on the board of directors at Idenix Pharmaceuticals, Inc., Vitae Pharmaceuticals, Inc., Bind Therapeutics Inc. and Aurinia Pharmaceuticals Inc. Mr. Rowland received his B.S. from Saint Joseph’sJoseph's University and M.B.A. from Rutgers University. We believe that Mr. Rowland is qualified to serve on our supervisory boardas a director due to his extensive experience in pharmaceutical operations and all areas of finance and accounting.
Supervisory Board Responsibilities
Our supervisoryStephen Webster has served on our board is responsible for the supervision of the activities of our management board and our company’s general affairs and business but does not actively engage in the management of the company. Supervision is exercised by the examination of regular reports which must be provided by the management board. The supervisory board must also approve certain transactions prior to their implementation. Our supervisory board may also,directors since June 23, 2017. Mr. Webster previously served on its own initiative, provide the management board with advice and may request any information from the management board that it deems appropriate. In performing its duties, the supervisory board is required to act in the interests of our company and its associated business as a whole. The supervisory board must convene a general meeting of shareholders if it is in the best interest of the company. The members of the supervisory board are not authorized to represent us in dealings with third parties, except for legal transactions concluded by the company with members of the management board and legal proceedings which have been approved at a general meeting of shareholders against such members.
Members of the supervisory board are appointed by the general meeting of shareholders. Pursuant to our articles of association, the supervisory board consists of a minimum of three and a maximum of ten supervisory board members. Supervisory board members are appointed at the general meeting of shareholders and may, if not appointed for a shorter period of time, serveNabriva Austria from October 2016 until the annual meeting occurringRedomiciliation. Mr. Webster has been chief financial officer of Spark Therapeutics since July 2014. He was previously senior vice president and chief financial officer of Optimer Pharmaceuticals, Inc. from June 2012 until its acquisition by Cubist Pharmaceuticals in the fifth calendar year after such board members’ initial appointment.November 2013. Prior to this, Mr. Webster served as senior vice president and chief financial officer of Adolor Corporation, also acquired by Cubist, from 2008 to 2011. Previously, Mr. Webster served as managing director, Investment Banking Division, Health Care
MembersGroup for Broadpoint Capital Inc. (formerly First Albany Capital). He also was a co-founder and served as president and chief executive officer of the supervisory board may be re-elected. They may also be removed by the voteNeuronyx, Inc. Prior to this, Mr. Webster held positions of three-quarters of the votes cast at the relevant general meeting of shareholders or resign by written notice to the company. Resignation upon written noticeincreasing responsibility, including as director, Investment Banking Division, Health Care Group, for PaineWebber Incorporated. Mr. Webster is subject tocurrently a four-week notice period unless otherwise agreed. In the event an elected member resigns from the supervisory board before the expiration of his or her term, the next general meeting may elect a replacement. The term of office of the replacement member runs until the expiration of the original term of the resigning member. In case the number of supervisory board members falls below three (the statutory minimum), an extraordinary general meeting of shareholders must be convened to elect a replacement. The supervisory board appoints a chairman and a deputy chairman from among its members for the entire period of their respective appointments. The supervisory board adopts its own rules of procedure.
The supervisory board meets at least quarterly. At least half of the members of the supervisory board including either the chairman or the deputy chairman must be present at a supervisory board meeting to constitute a quorum, in each case however at least three members need to be present. Except where a different majority is required by law or the articles of association, the supervisory board acts by a simple majority of the votes cast. In case of a split vote, the chairman casts the deciding vote. A member of the supervisory board may authorizeof directors of Viking Therapeutics, Inc. He holds an A.B. in writing another membereconomics from Dartmouth College and an M.B.A. from the University of Pennsylvania. We believe that Mr. Webster is qualified to serve as a director due to his extensive experience in the supervisory board or any third party to represent him or herbiopharmaceutical industry, particularly his service as a chief financial officer and exercise his or her voting rights. Such representative is not taken into account in determining a quorum. The right to chair a supervisory board meeting cannot be transferred.other executive management roles.
Board Committees
The supervisoryOur board may also adopt resolutions outside a meeting, provided that such resolutions are adopted in writing and submitted to all members of the supervisory board and provided that no supervisory board member objects to adopting resolutions without conducting a meeting. Each supervisory board member is entitled to cast one vote.
Committees of the Supervisory Board
We havedirectors has established an audit committee, a compensation committee and a nominating and corporate governance committee, and have adoptedeach of which operates under a charter for eachthat has been approved by our board. Copies of these committees.the committee charters are posted under the heading "Corporate Governance" on the Investor section of our website, which is located at http://investors.nabriva.com.
Audit Committee
Our audit committee consists of Charles A. Rowland, Jr., Daniel Burgess and Stephen Webster, andWebster. Charles A. Rowland, Jr. iswas the chair of the audit committee.committee until August 1, 2018, and Stephen Webster has been chair since August 1, 2018. The audit committee oversees our accounting and financial reporting processes and the audits of our consolidated financial statements. The audit committee is responsible for, among other things:
- •
·making recommendations to oursupervisoryboard regarding theappointmentratification by the annual general meeting ofshareholders of our independent auditors;
·- •
- overseeing the work of the independent auditors, including resolving disagreements between management and the independent auditors relating to financial reporting;
· - •
- pre-approving all audit and non-audit services permitted to be performed by the independent auditors;
· - •
- reviewing the independence and quality control procedures of the independent auditors;
·discussing material off-balance sheet transactions, arrangements and obligations with management and the independent auditors; - •
·reviewing and approving all proposed related-party transactions;·- •
- discussing the annual audited consolidated and statutory financial statements with management;
· - •
- annually reviewing and reassessing the adequacy of our audit committee charter;
· - •
- meeting separately with the independent auditors to discuss critical accounting policies, recommendations on internal controls, the
auditor’sauditor's engagement letter and independence letter and other material written communications between the independent auditors and the management; and· - •
- attending to such other matters as are specifically delegated to our audit committee by our
supervisoryboard from time to time.
Our supervisory boardBoard has determined that Charles A. Rowland, Jr. is an “auditand Stephen Webster are "audit committee financial expert”experts" as defined in the applicable SEC rules.
To satisfy the independence criteria for audit committee members set forth in Rule 10A-3 under the Exchange Act, each member of an audit committee of a listed company may not, other than in his capacity as a member of the audit committee, the board of directors, or any other board committee, accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries or otherwise be an affiliated person of the listed company or any of its subsidiaries. We believe that the composition of our audit committee meets the requirements for independence under current NASDAQ and SEC rules and regulations.
The audit committee met five times in 2016.
Compensation Committee
Our compensation committee consists of Axel Bolte, Charles A. Rowland, Jr., Mark Corrigan and Chau Khuong, and Axel BolteCarrie Bourdow. Charles A. Rowland, Jr. is the chair of the compensation committee. The compensation committee assists the supervisory board in reviewing and approving or recommending our compensation structure,
including all forms of compensation relating to our supervisory board membersdirectors and management. The compensation committee is responsible for, among other things:
- •
·reviewing and making recommendations to thesupervisoryboard with respect to compensation of ourmanagementboard of directors andsupervisory board members;management;
·- •
- reviewing and approving the compensation, including equity compensation, change-of-control benefits and severance arrangements, of our chief executive officer, chief financial officer and such other members of our management as it deems appropriate;
· - •
- overseeing the evaluation of our management;
· - •
- reviewing periodically and making recommendations to our
supervisoryboard with respect to any incentive compensation and equity plans, programs or similar arrangements;· - •
- exercising the rights of our
supervisoryboard under any equity plans, except for the right to amend any suchplans unless otherwise expressly authorized to do so; and
· - •
- attending to such other matters as are specifically delegated to our compensation committee by our
supervisoryboard from time to time.
To satisfy the independence criteria for compensation committee members set forth in Rule 10C-1 under the Exchange Act, all factors specifically relevant to determining whether a member of a compensation committee has a relationship to such company which is material to that member’s ability to be independent from management in connection with the duties of a compensation committee member must be considered, including, but not limited to: (1) the source of compensation of the committee member, including any consulting advisory or other compensatory fee paid by such company to the member; and (2) whether the member is affiliated with the company or any of its subsidiaries or affiliates. We believe the composition of our compensation committee meets the requirements for independence under current NASDAQ and SEC rules and regulations. In determining the independence of the members of our compensation committee, our supervisory board considered that Mr. Khuong is a private equity partner at OrbiMed Advisors, which was the beneficial owner of approximately 14.4% of our outstanding common shares as of December 31, 2016.
The compensation committee met three times in 2016.
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Daniel Burgess, Mark Corrigan and Stephen Webster, andWebster. Daniel Burgess is the chair of the nominating and corporate governance committee. The nominating and corporate governance committee assists the supervisory board in selecting individuals qualified to become our supervisory board membersdirectors and in determining the composition of the supervisory board and its committees. The nominating and corporate governance committee is responsible for, among other things:
- •
·recommending to thesupervisoryboard persons to be nominated for election or re-election to thesupervisoryboard at any meeting oftheshareholders;·- •
- overseeing the
supervisory board’sboard's annual review of its own performance and the performance of its committees; and· - •
- considering, preparing and recommending to the
supervisoryboard a set of corporate governance guidelines.
Compensation Committee Interlocks and Insider Participation
The nominating and corporate governance committee met twice in 2016.
Board Meetings and Attendance
Our supervisory board met 14 times during 2016 and took action by written consent four times. During 2016, each supervisory board member attended at least 75% ofFor the aggregate offiscal year ended December 31, 2018, the number of supervisory board meetings held during his or her term, and of the meetings held by all committees of the supervisory board on which he or she then served.
Our supervisory board members are expected to attend our general meeting of shareholders. In August 2016, all of our then-current supervisory board members attendedcompensation committee were Axel Bolte, Carrie Bourdow, Mark Corrigan and Charles Rowland, Jr. No member of our general meetingcompensation committee is, or has been, an officer or employee of shareholders either in personours or by telephone.any subsidiary of ours. None of our executive officers served as a director or a member of a compensation committee (or other committee serving an equivalent function) of any other entity that had one or more executive officers serving as a director or member of our compensation committee during the year ended December 31, 2018.
Executive Officers
Management Board
Member of the Management Board
The following table sets forth information with respect toregarding our management board member, his age and positionexecutive officers as of March 1, 2019:
Name | Age | Position | |||
|---|---|---|---|---|---|
| Theodore Schroeder | 63 | Chief Executive Officer | |||
| Robert Crotty | 45 | General Counsel and Secretary | |||
| Steven Gelone | 51 | President and Chief Operating Officer | |||
| Francesco Maria Lavino | 45 | Chief Commercial Officer | |||
| Jennifer Schranz | 54 | Chief Medical Officer | |||
| Gary Sender | 56 | Chief Financial Officer | |||
In addition to the datebiographical information for Mr. Schroeder, which is set forth above under "Board of this Annual ReportDirectors," set forth below is certain biographical information about Drs. Gelone and the year our supervisory board appointed him. The business address for Colin Broom is c/o Nabriva Therapeutics AG, 1000 Continental Drive, Suite 600, King of Prussia, Pennsylvania.Schranz, and Messrs. Crotty, Lavino and Sender:
Name |
| Age |
| Position |
| Initial Year |
Colin Broom |
| 61 |
| Chief Executive Officer |
| 2014 |
Colin BroomRobert Crotty has served as our chief executive officergeneral counsel and secretary since 2014. PriorJune 23, 2017. Mr. Crotty joined Nabriva Austria as general counsel and secretary prior to joining our company, he served as chief scientific officer at ViroPharma Incorporated from 2004 until it was acquired by Shire plc in 2014. Dr. Broom alsothe Redomiciliation on June 14, 2017. Previously, Mr. Crotty served as vice president, general counsel, chief compliance officer and secretary of clinical development and medical affairs at AmgenVernalis Therapeutics, Inc. from 2000January 2016 to 2003. He is a member of the U.K. Royal College of PhysiciansJune 2017. Prior to joining Vernalis, Mr. Crotty held several positions at Dendreon Corporation from April 2012 to July 2015, including president, general counsel and a fellow of the Faculty of Pharmaceutical Medicine. Dr. Broomsecretary from February 2015 to July 2015, executive vice president, general counsel and secretary from March 2014 to February 2015, and vice president, assistant general counsel and assistant secretary from April 2012 to February 2014. Before Dendreon, Mr. Crotty was senior counsel at NPS Pharmaceuticals from 2009 until 2012 and at ImClone Systems, Inc. from 2006 to 2009. Prior to going in-house, Mr. Crotty was an associate at Morgan, Lewis & Bockius and Norton Rose Fulbright. Mr. Crotty received his B.Sc.B.A. from Princeton University and J.D. from University College London and M.B.B.S. from St. George’s Hospital Medical School. We believe that Dr. Broom is qualified to serve on our management board due to his extensive experience in all stages of drug development and commercialization.
Management Board Responsibilities
Our management board is responsible for the day-to-day management of our operations under the supervision of the supervisory board. The management board is required to:
· keep the supervisory board informed in a timely manner in order to allow the supervisory board to carry out its responsibilities;
· consult with the supervisory board on important matters; and
· submit certain important decisions to the supervisory board for its approval, as more fully described below.
Our management board may perform all acts necessary or useful for achieving our corporate purposes, other than those acts that are prohibited by law or by our articles of association, as more fully discussed below. Members of the management board of an Austrian stock corporation are appointed by the supervisory board for a maximum period of five years and may be re-appointed. The supervisory board may remove a member of the management board prior to the expiration of his term only for a significant cause, such as a material breach of duty, the inability to manage the business properly or a vote of no-confidence at a shareholders’ meeting (Vertrauensentzug). The shareholders themselves are not entitled to appoint or dismiss the members of the management board.Pennsylvania.
The management board manages the business and represents the company in dealings with third parties and is responsible for the financial books and records of the company. The management board is required to report to the supervisory board at least annually regarding fundamental questions of future business policy and the future development of the assets and financial situation of the company (annual report; Lagebericht). The management board is also required to report to the supervisory board regularly, at least quarterly, on the progress of business and the results of the company against the annual forecast results and considering future developments to the extent determined by Section 81 of the Austrian Stock Corporation Act (quarterly reports; Quartalsberichte). In addition, the management board is required to promptly inform the supervisory board of any matter that may be of significance to the company’s business operations, in particular with respect to any circumstances relating to the company’s profitability and liquidity (special report; Sonderbericht). The annual report and the quarterly report have to be in writing and must be explained to the supervisory board on demand. Each member of the supervisory board has to be provided with a copy of these reports. The special reports may be oral reports or in writing.
Under the articles of association, if the management board consists of one member only, this member may, to the extent permitted by law, represent the company solely. If the management board consists of more than one member, the company shall be represented by two members of the management board acting jointly or by a one member of the management board together with the holder of a general commercial power of attorney (Prokurist). The supervisory board may grant individual members of the management board the power to independently represent the company. Currently, our sole management board member is empowered with independent signing authority.
The management board has no obligation to obey orders or directives originating from the general meeting of shareholders or the supervisory board. However, both the Austrian Stock Corporation Act and our articles of association, together with the by-laws of our management board, require the prior approval of the supervisory board or one of its committees before the management board may take certain actions. A failure by the management board to obtain such
approval does not affect the validity of transactions with respect to third parties, but may render the management board personally liable for any damages resulting therefrom. Pursuant to our articles of association, as well as pursuant to Section 95 paragraph 5 of the Austrian Stock Corporation Act, the following transactions require the prior approval of our supervisory board:
· the acquisition and sale of shareholdings in terms of Section 228 of the Austrian Commercial Code (UGB) as well as the acquisition, disposal and closing down of companies and businesses;
· the acquisition, disposal and encumbrance of real estate outside of the ordinary course of business;
· the establishment and closing of branch offices;
· investments in excess of €500,000;
· the issuance of bonds or entering into loans or credits in excess of €500,000;
· the granting of loans outside of the ordinary course of business;
· the introduction and termination of lines of business or product lines;
· the determination of general principles of business policy;
· the determination of general policies for the granting of participations in profit or revenues and pension promises to executive staff in accordance with Section 80 paragraph 1 of the Austrian Stock Corporation Act;
· the determination of general principles for the granting of options to receive shares in the company to employees and executive staff (leitende Angestellte) of the company or its affiliates, or to members of the management board and of the supervisory board of our affiliates;
· the granting of special power of attorney (Prokura);
· the entry into contracts with members of the supervisory board pursuant to which such members commit themselves to render services outside of their activities on the supervisory board for the company or a subsidiary for a remuneration not of minor value (although this shall also apply to contracts with companies in which the supervisory board member has a material economic interest);
· the acceptance of an executive position (leitende stellung) within the company within two years after issuance of an audit opinion, by the auditor, by the group auditor, by the auditor of an affiliated major company, or by the certified accountant who signed the audit opinion or a person working for him or her, who has had a significant position in the audit, to the extent not prohibited pursuant to Section 271c of the Austrian Commercial Code (UGB); and
· measures pursuant to which the management board makes use of an authorization pursuant to Section 102 paragraph 3 or 4 of the Austrian Stock Corporation Act.
Our supervisory board may also require that additional actions, beyond those listed above, by the management board be conditioned upon the supervisory board’s approval. Such actions must be clearly specified to the management board in writing. The absence of approval of the supervisory board does not affect the authority of the management board or its members to represent us in dealings with third parties.
Senior Management
Our management board is supported by our senior management team. The following table sets forth information with respect to each of the members of our senior management team, their respective ages, their positions as of the date of this Annual Report and the year our management board appointed them. The business address for Steven Gelone, Elyse Seltzer and Gary Sender is c/o Nabriva Therapeutics AG, 1000 Continental Drive, Suite 600, King of Prussia, Pennsylvania.
Name |
| Age |
| Position |
| Year of |
Steven Gelone |
| 49 |
| Chief Development Officer |
| 2014 |
Elyse Seltzer |
| 52 |
| Chief Medical Officer |
| 2015 |
Gary Sender |
| 55 |
| Chief Financial Officer |
| 2016 |
Steven Gelone has served as our president and chief operating officer since July 24, 2018. Dr. Gelone previously served as Nabriva Austria's chief development officer since 2014.and head of business development from 2014 until the Redomiciliation, our chief development officer from the Redomiciliation until June 30, 2017 and our chief scientific officer from June 30, 2017 until July 24, 2018. Prior to joining our company,Nabriva Austria, he served as head of clinical research and development at Spark Therapeutics, Inc. in 2014 and vice president of clinical and preclinical development at ViroPharma Incorporated from 2005 to 2014. Dr. Gelone also served as director of medical affairs at Vicuron Pharmaceuticals from 2002 to 2003 and director of clinical pharmacology and experimental medicine at GlaxoSmithKline Pharmaceuticals from 2000 to 2002. Dr. Gelone received his B.S. Pharm. and Pharm.D. from Temple University.
Francesco Maria Lavino has served as our chief commercial officer since July 10, 2017. Previously, Mr. Lavino served as associate vice president and global brand leader for the anti-infective portfolio at Merck & Co. from September 2015 to July 2017. Prior to Merck, Mr. Lavino was vice president of international marketing for Cubist Pharmaceuticals from December 2013 until September 2015. Before joining Cubist, Mr. Lavino spent 10 years with Merck & Co. in various roles, including serving as executive director and global brand leader for Merck's anti-fungal portfolio from January 2011 to November 2013. Mr. Lavino began his career in pharmaceutical sales at UCB S.A. and 3M Company in Italy. He has a B.A. in Pharmacy from the Federico II University of Napoli, Italy and an M.B.A. from SDA Bocconi School of Management in Milan, Italy.
Elyse G. SeltzerJennifer Schranz has served as our chief medical officer since May 2015.March 21, 2018. Previously, Dr. Schranz served as vice president, clinical research and development, global development team lead, for hereditary angioedema at Shire plc from January 2014 until March 2018. Prior to joining our company, she held several positions at GlaxoSmithKline from 2009 to 2015, includingShire, Dr. Schranz served as vice president of global clinical sciences and operationsdevelopment for ViroPharma, Inc. from 2014 to 2015,March 2011 until January
2014. Before joining ViroPharma, Dr. Schranz was vice president, of therapeutic area delivery from 2012 to 2013clinical research at Cempra, Inc., where she was responsible for clinical and vice president of cardiovascular metabolic operations and clinical head of cardiovascular metabolic from 2009 to 2011. She also served as chief medical officer and vice president ofregulatory strategy. Earlier in her career, Dr. Schranz worked in clinical development and medical affairs at Tengion,several pharmaceutical companies, including Wyeth (now Pfizer), Vicuron Pharmaceuticals, Inc. from 2006(now Pfizer), GlaxoSmithKline plc, and Merck & Co. Inc. Dr. Schranz completed two years of biology and psychology at McMaster University prior to 2009. Prior to working in the pharmaceutical industry, Dr. Seltzer practiced clinical infectious diseases medicine. Dr. Seltzer received her B.A.acceptance and subsequent completion of an M.D. from the University of PennsylvaniaToronto, where she completed her internal medicine training and her M.D. from the New York University School of Medicine.was a fellow in infectious diseases.
Gary Sender has served as our chief financial officer since April 12, 2017. Mr. Sender previously served as our chief financial officer from May 2016.2016 until the Redomiciliation. Prior to joining our company,Nabriva Austria, he served as chief financial officer and executive vice president at Synergy Pharmaceuticals from 2015 to 2016. From 2009 until 2015, Mr. Sender served as senior vice president, Finance at Shire plc., supporting its Specialty Pharmaceuticals business and subsequently its Global Commercial businesses. HeAt Shire he was responsible for financial management and support of all commercial areas of Shire’sShire's Specialty Pharmaceutical and Rare Disease businesses, with an emphasis on resource allocation, financial forecasting, business cases and mergers and acquisitions. Prior to joining Shire, Mr. Sender was the founding CFO of Tengion, Inc. Mr. Sender also spent 15 years in a number of leadership roles within Merck. Mr. Sender received his B.S. from Boston University and an M.B.A from Carnegie-Mellon University.
Code of Business Conduct and Ethics
Our Code of Business Conduct and Ethics is applicable to all of our employees, management board memberdirectors, officers and supervisory board membersemployees and is available on our website at http://www.nabriva.com.investors.nabriva.com. Our Code of Business Conduct and Ethics provides that our employees, management board memberdirectors, officers and supervisory board membersemployees are expected to avoid any action, position or interest that conflicts with the interests of our company or gives the appearance of a conflict. We expect that any amendment to this code, or any waivers of its requirements, will be disclosed on our website. Information contained on, or that can be accessed through, our website is not incorporated by reference into this document, and you should not consider information on our website to be part of this document.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our supervisory board members, management board member, senior managersdirectors, executive officers and holders of more than 10% of our commonordinary shares to file with the SEC initial reports of ownership of our commonordinary shares and other equity securities on a Form 3 and reports of changes in such ownership on a Form 4 or Form 5. Our supervisory board members, management board member and senior managersdirectors, executive officers and holders of more than 10% of our commonordinary shares are required by SEC regulations to furnish us with copies of all Section 16(a) forms they file. Prior to the loss of our “foreign private issuer” status on January 1, 2017, our supervisory board members, management board
member, senior managers and holders of more than 10% of our common shares were not required to file Section 16(a) reports. However, with the loss our “foreign private issuer” status, our supervisory board members, management board member, senior managers and holders of more than 10% of our common shares are now required to comply with such filing requirements. Based solely on a review of our records and representations made by the persons required to file these reports, we believe that, during the year ended December 31, 2016,2018, our supervisory board members, management board member, senior managersdirectors, executive officers and holders of more than 10% of our common sharesany class of equity securities complied with all Section 16(a) filing requirements applicable to them.
ITEM 11. EXECUTIVE COMPENSATION
The following discussion provides the amount of compensation paid, and benefits in-kind granted, by us and our subsidiaries to the members of our supervisory board of directors and certain executives (including members of our management board and senior management) for services provided in all capacities to us and our subsidiaries for the year ended December 31, 2016.2018.
Executive and Director Compensation Processes
Our executive compensation program is administered by the compensation committee of our supervisory board of directors, subject to the oversight and approval of our full supervisory board.board of directors. Our compensation
committee reviews our executive compensation practices on an annual basis and based on this review approves, or, as appropriate, makes recommendations to our supervisory board of directors for approval of our executive compensation program.
In designing our executive compensation program, our compensation committee considers publicly available compensation data for national and regional companies in the biotechnology/pharmaceutical industry to help guide its executive compensation decisions at the time of hiring and for subsequent adjustments in compensation. Since 2016, our compensation committee has retained Radford, a part of Aon Hewitt, a business unit of Aon plc, as its independent compensation consultant, to provide comparative data on executive compensation practices in our industry and to advise on our executive compensation program generally. The committee also has retained Radford for guidelines and review of non-employee director compensation. Although our compensation committee considers the advice and guidelines of Radford as to our executive compensation program, our compensation committee ultimately makes its own decisions about these matters. In the future, we expect that our compensation committee will continue to engage independent compensation consultants to provide additional guidance on our executive compensation programs and to conduct further competitive benchmarking against a peer group of publicly traded companies.
The compensation committee reviewed information regarding the independence and potential conflicts of interest of Radford, taking into account, among other things, the factors set forth in the NASDAQ listing standards. Based on such review, the committee concluded that the engagement of Radford did not raise any conflict of interest. Outside of services provided for the compensation committee, the compensation consultant provided nominal additional services to the company in 2016 related to benchmarking data with respect to certain non-executive positions in an effort to ensure that our compensation practices are competitive so that we can attract, reward, motivate and retain all employees. The total amount paid to Radford in connection with these additional engagements was less than $120,000 in 2016.
Our director compensation program is administered by our supervisory board of directors with the assistance of the compensation committee. The compensation committee conducts an annual review of director compensation and makes recommendations to the supervisory board of directors with respect thereto.
Summary Compensation Table
Our “named"named executive officers”officers" for the year ended December 31, 20162018 were as follows: Dr. Broom, our Chief Executive Officerformer chief executive officer, Mr. Schroeder, our chief executive officer, Dr. Gelone, our president and member of the management board; Dr. Seltzer, our Chief Medical Officer;chief operating officer, and Mr. Wolf,Lavino, our former General Counsel.chief commercial officer. The following table sets forth information regarding compensation awarded to, earned by or paid to our named executive officers duringfor the years ended December 31, 2016 and December 31, 2015.
|
|
|
|
|
|
| |||||||
|
|
|
|
|
|
| |||||||
|
|
|
|
|
|
| |||||||
|
|
|
|
|
|
|
periods presented.
Name and principal position | Year | Salary($) | Bonus ($) | Share Awards ($)(1) | Option Awards ($)(1) | Non-Equity Incentive Plan Compensation ($)(2) | All Other Compensation ($)(3) | Total ($) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Colin Broom | 2018 | 275,137 | — | 436,725 | 754,400 | 355,594 | 223,131 | 2,044,987 | |||||||||||||||||
Chief Executive | 2017 | 457,387 | — | — | 1,235,835 | 183,501 | 40,158 | 1,916,881 | |||||||||||||||||
Officer(4) | 2016 | 410,354 | — | — | 769,798 | 175,776 | 43,043 | 1,398,971 | |||||||||||||||||
Theodore Schroeder | 2018 | 220,360 | — | 529,500 | 1,739,100 | — | 20,380 | 2,509,340 | |||||||||||||||||
Chief Executive Officer | |||||||||||||||||||||||||
Francesco Maria Lavino | 2018 | 360,298 | 50,000 | (5) | 145,575 | 377,200 | 119,438 | 36,253 | 1,088,764 | ||||||||||||||||
Chief Commercial | |||||||||||||||||||||||||
Officer | |||||||||||||||||||||||||
Steven Gelone | 2018 | 426,104 | — | 225,255 | 546,738 | 126,822 | 15,116 | 1,340,035 | |||||||||||||||||
President and Chief | 2017 | 370,278 | — | — | 535,055 | 93,766 | 15,942 | 1,015,041 | |||||||||||||||||
Operating Officer | |||||||||||||||||||||||||
Narrative Disclosure to Summary Compensation Table Base Salary In In None of our named executive officers is currently party to an employment agreement or other agreement or arrangement that provides for automatic or scheduled increases in base salary. Annual Performance-Based Compensation Our “Non-Equity Incentive Plan Compensation” column represent awards to our named executive officers under our annual cash bonus program.(2) The amounts reported in the “Option Awards” column"Share Awards" and "Option Awards" columns reflect the aggregate grant-date fair value of share-based compensation awarded during the year computed in accordance with the provisions of ASC Topic 718. See Note 11 to ourthe consolidated audited financial statements regarding assumptions underlying the valuation of equity awards.“All"All Other Compensation”Compensation" column consists of amounts we contributed to our 401(k) plan and medical insurance premiums paid by us on behalf of such individual.(4) This column also includes severance benefits for Dr. Seltzer was appointed as our Chief Medical Officer on May 4, 2015.(5) Mr. Wolf was appointed as our General Counsel on September 28, 2015. Mr. WolfBroom of $195,868.company, effective February 7, 2017, to pursue other opportunities.first anniversary of his start date.2016,2018, we paid annualized base salaries of $436,000$471,534 to Dr. Broom; $368,740$530,000 to Mr. Schroeder; $360,500 to Mr. Lavino; and $450,000 to Dr. Seltzer; and $323,200 to Mr. Wolf.Gelone. In 2015,2017, we paid annualized base salaries of $414,000$457,800 to Dr. Broom; $358,000$350,000 to Mr. Lavino; and $390,000 to Dr. Seltzer; and $320,000 to Mr. Wolf.Gelone.February 2017,January 2019, our supervisory board of directors, following approval and recommendation from the compensation committee and consistent with the recommendations of the compensation committee’scommittee's independent compensation consultant, approved an increase to the base salaries of our named executive officers for 20172019 as follows: $457,800$560,000 for Mr. Schroeder, $371,800 for Mr. Lavino and $472,100 for Dr. Bloom and $394,552 for Dr. Seltzer. As Mr. Wolf had already announced his resignation from the company, which became effective on February 7, 2017, he did not receive a base salary increase for 2017.Gelone. The supervisory board also approved 20172019 base salaries for Mr. Crotty, our general counsel and secretary, of Mr. Gelone,$371,100, Dr. Schranz, our Chief Development Officer,chief medical officer, of $353,280$436,200 and Mr. Sender, our Chief Financial Officer,chief financial officer, of $360,500,$382,500, which also were consistent with the recommendation of the compensation committee’scommittee's independent consultant.management board and senior management team,executive officers, which includesinclude the named executive officers, participate in our performance-based bonus program. All annual cash bonuses for our executives under the performance-based bonus program are tied to the achievement of strategic and operational corporate goals for the company, which are set by the compensation committee and approved by the supervisory board. There are no discretionary individual goals under the bonus program. The 20162018 strategic and operational goals for the companyNabriva related to the following objectives:·corporate strategy and financial reach, including executing on financing plans, grant opportunities and development program objectives;
strategy;·
filing the NDA for lefamulin in the fourth quarter of 2018; and,
· discovery programs, specifically identifying feasibility and intellectual property differentiation of additional compounds; and
· communications, including enhancing communications with key stakeholders.
Under their respective employment agreements, the annual target bonus for Dr. BroomMr. Schroeder is 50% of his current base salary, the annual target bonus for Dr. Gelone is 40% of his current base salary and the annual target bonus for each of Drs. SeltzerDr. Schranz, Mr. Sender, Mr. Crotty and Gelone and Messrs. Sender and WolfMr. Lavino is 35% of their respective current base salaries. The annual target bonus for Dr. Broom was 50% of his then-current base salary. In October 2018, the compensation committee recommended, and the board of directors approved, an annual target bonus for each of Dr. Schranz, Mr. Sender, Mr. Crotty and Mr. Lavino at 40% of their respective salaries.
At a meeting held in January 2017,October 2018, our compensation committee reviewed the accomplishments of the senior management teamnamed executive officers as measured against the aforementioned 20162018 goals. The compensation committee reviewed whether each goal had been obtained and the weight such goals should be given in determining the bonus payout for 20162018 performance. Based on its review, the compensation committee recommended an 85%108% payout of the target bonuses for 2016,2018, which was approved by the supervisory board on February 7, 2016.were paid in January 2019. Accordingly, the 20162018 bonus payouts were $183,501$119,250 for Mr. Schroeder, $136,269 for Mr. Lavino and $181,420 for Dr. Gelone. Dr. Broom $108,635 for Dr. Seltzer and $96,152 for Mr. Wolf, who remained employed with us through such time.received a lump sum cash payment of $132,417 in full satisfaction of any bonus as part of his separation agreement.
Equity Incentive Awards
We believe that equity grants provide our executivesexecutive officers with a strong link to our long-term performance, create an ownership culture and help to align the interests of our executivesexecutive officers and our shareholders. In addition, we believe that equity grants with a time-based vesting feature promote executive retention because this feature incents our management board and senior management teamexecutive officers to remain in our employment during the vesting period. Accordingly, our supervisory board of directors periodically reviews the equity incentive compensation of our management board and senior management team,executive officers, which includes the named executive officers, and from time to time may grant equity incentive awards to them in the form of stock options. We also generally make stock option grants to new management team membersexecutive officers in connection with the commencement of their employment.
We have historically granted stock options with exercise prices that are set at no less than the fair market value of our common sharesthe underlying award on the date of grant, as determined by contemporaneous valuations and reviewed, and approved by our compensation committee or our supervisory board.
On February 7, 2017,January 31, 2019, our supervisory board of directors granted stock options under our Stockthe 2017 Share Incentive Plan 2015 to Drs. BroomMr. Schroeder, Mr. Lavino and Seltzer. Mr. Wolf did not receiveDr. Gelone. The options will vest over a grant as he had resigned from the company, effective February 7, 2017. Vesting of the options beganfour-year period beginning on February 28, 2017 and will end on February 28, 2021.January 31, 2019. Twenty-five percent (25%) of the option will vest on February 28, 2018,January 31, 2020, and the remaining seventy-five percent (75%) will vest on a monthly pro-rata basis over the remaining vesting period. Each of the option awards hashad an exercise price of €79.63 ($85.00)$1.90 per share, which was the equivalent of the closing sale price of the commonour ordinary shares underlying our American Depositary Shares on the NASDAQNasdaq Global Market on the grant date. Mr. Schroeder, Mr. Lavino and Dr. Gelone were also granted Restricted Stock Units, or RSUs. The options also hadRSUs will vest over a grant date fair valuefour-year period beginning on January 31, 2019. Twenty-five percent (25%) of €30.25 ($32.29) per share, as determined in accordance with ASC Topic 718.the RSUs will vest on January 31, 2020, and the remaining seventy-five percent (75%) will vest on a monthly pro-rata basis over the remaining vesting period.
The following table sets forth the number of our commonordinary shares issuable upon exercise of the share awards granted to our named executive officers in 2019:
Name | Option Award (#) | RSU Award (#) | |||||
|---|---|---|---|---|---|---|---|
Theodore Schroeder | 429,800 | 163,250 | |||||
Colin Broom | — | — | |||||
Steven Gelone | 278,400 | 105,750 | |||||
Francesco Maria Lavino | 138,200 | 52,500 | |||||
On July 23, 2018, Nabriva Therapeutics US, Inc. ("Nabriva US") entered into an Employment Agreement with Theodore Schroeder (the "Schroeder Agreement") providing for Mr. Schroeder, age 63, to serve as chief executive officer of the Company and president and chief executive officer of Nabriva US, contingent on and effective as of the Closing of the Zavante Acquisition (the "Commencement Date). Pursuant to the Schroeder Agreement, the Board also approved the grant to Mr. Schroeder, effective as of the first business day immediately following the Commencement Date (the "Grant Date"), of a non-statutory stock options grantedoption to purchase 850,000 ordinary shares of the Company at an exercise price per share equal to the closing price per share of the Company's ordinary
shares on the Nasdaq Global Select Market on the Grant Date. The option award will have a ten-year term and will vest over a four-year period, with 25% of the shares underlying the award vesting on the first anniversary of the Commencement Date and the remaining 75% of the shares underlying the option award to vest monthly over the subsequent 36-month period. In addition, the Board approved the grant to Mr. Schroeder, effective as of the Grant Date, of 150,000 performance-based restricted share units (the "Schroeder PRSUs"). The Schroeder PRSUs will vest as follows: 50% of the Schroeder PRSUs will vest upon Board certification of the receipt of FDA approval of a new drug application for each of (x) lefamulin and (y) CONTEPO for any indication, and 50% of the Schroeder PRSUs will vest on the first anniversary of such Board certification, provided, in 2017:each case, that Mr. Schroeder is performing services to Nabriva US on the applicable vesting dates. If the FDA does not approve an NDA for both lefamulin and CONTEPO by January 31, 2020, the Schroeder PRSUs award will terminate in full.
|
| ||
|
| ||
|
|
On February 5, 2016,January 31, 2018, our supervisory board of directors granted stock options under our Stockthe 2017 Share Incentive Plan 2015 to each of our named executive officers. Vesting beganDr. Broom, Mr. Sender and Dr. Gelone. The options will vest over a four-year period beginning on February 29, 2016 and will end on February 29, 2020.January 31, 2018. Twenty-five percent (25%) of the option vestedwill vest on February 28, 2017,January 31, 2019, and the remaining seventy-five percent (75%) vestswill vest on a
monthly pro-rata basis over the remaining vesting period. Each of the option awards hashad an exercise price of €74.45 ($83.40)$6.47 per share, which was the equivalent of the closing sale price of the commonour ordinary shares underlying our American Depositary Shares on the NASDAQNasdaq Global Market on the grant date. The optionsDr. Broom, Mr. Lavino and Dr. Gelone were also had agranted Restricted Stock Units, or RSUs. Vesting of the RSUs is subject to FDA approval of an NDA, for lefamulin. Fifty percent (50%) of each RSU award will vest upon FDA approval of an NDA for lefamulin, and the remaining fifty percent (50%) will vest on the one-year anniversary of such approval. If the FDA does not approve an NDA for lefamulin within two years of the grant date, fair value of €29.96 ($33.56) per share, as determinedthe RSU award will terminate in accordance with ASC Topic 718.full.
The following table sets forth the number of our commonordinary shares issuable upon exercise of the stock optionsshare awards granted to our named executive officers in 2016:2018:
Name | Option Award (#) | RSU Award (#) | |||||
|---|---|---|---|---|---|---|---|
Theodore Schroeder | 850,000 | 150,000 | |||||
Colin Broom | 200,000 | 67,500 | |||||
Steven Gelone | 185,000 | 54,500 | |||||
Francesco Maria Lavino | 100,000 | 22,500 | |||||
|
| ||
|
| ||
|
| ||
|
|
Outstanding Equity Awards as of December 31, 20162018
The following table sets forth information regarding outstanding stock options and RSUs held by our named executive officers as of December 31, 2016:
|
| Option Awards |
| |||||||
Name |
| Number of |
| Number of |
| Option |
| Option |
| |
Colin Broom |
| 9,233 |
| 6,594 | (1) | 66.18 |
| 7/5/2025 |
| |
|
| 6,974 |
| 12,716 | (2) | 66.18 |
| 7/5/2025 |
| |
|
|
|
| 15,100 | (3) | 74.45 |
| 2/4/2026 |
| |
Elyse Seltzer |
| 3,515 |
| 5,364 | (4) | 66.18 |
| 7/5/2025 |
| |
|
|
|
| 5,590 | (5) | 74.45 |
| 2/4/2026 |
| |
Peter Wolf(8) |
| 1,719 |
| 3,781 | (6) | 84.80 |
| 9/29/2025 |
| |
|
|
|
| 5,590 | (7) | 74.45 |
| 2/4/2026 |
| |
2018:
| | | | | | Share Awards | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Option Awards | | | | Equity incentive plan awards: Market or payout value of unearned shares, units or other rights that have not vested ($) | ||||||||||||||||||||
| | | Market value of shares of units of stock that have not vested ($) | Equity incentive plan awards: Number of unearned shares, units or other rights that have not vested (#) | ||||||||||||||||||||||
Name | Number of securities underlying unexercised options (#) exercisable | Number of securities underlying unexercised options (#) unexercisable | Option exercise price ($) | Option expiration date | Number of shares or units of stock that have not vested (#) | ||||||||||||||||||||
Colin Broom | 158,270 | — | (1) | 7.21 | 7/5/2025 | — | — | 67,500 | (14) | 98,550 | |||||||||||||||
| 168,185 | 28,715 | (2) | 7.21 | 7/5/2025 | — | — | — | — | |||||||||||||||||
| 106,958 | 44,042 | (3) | 8.34 | 2/4/2026 | — | — | — | — | |||||||||||||||||
| 119,625 | 141,375 | (4) | 8.50 | 2/7/2027 | — | — | — | — | |||||||||||||||||
| — | 200,000 | (5) | 6.47 | 1/31/2028 | — | — | — | — | |||||||||||||||||
Theodore Schroeder | — | 850,000 | (6) | 3.53 | 7/25/2028 | 150,000 | (15) | 219,000 | — | — | |||||||||||||||
Francesco Maria Lavino | 53,125 | 96,875 | (7) | 10.48 | 7/31/2027 | — | — | 22,500 | (14) | 32,850 | |||||||||||||||
| — | 100,000 | (8) | 6.47 | 1/31/2028 | — | — | — | — | |||||||||||||||||
Steven Gelone | 81,997 | 6,793 | (9) | 7.21 | 7/5/2025 | — | — | 54,500 | (14) | 79,570 | |||||||||||||||
| 39,596 | 16,304 | (10) | 8.34 | 2/4/2026 | — | — | — | — | |||||||||||||||||
| 51,792 | 61,208 | (11) | 8.50 | 2/7/2027 | — | — | — | — | |||||||||||||||||
| — | 100,000 | (8) | 6.47 | 1/31/2028 | — | — | — | — | |||||||||||||||||
| — | 77,500 | (12) | 3.53 | 7/25/2028 | — | — | — | — | |||||||||||||||||
| — | 7,500 | (13) | 2.49 | 2/8/2028 | — | — | — | — | |||||||||||||||||
Employment Agreements with Agreement with Pursuant to his employment agreement, Mr. Schroeder receives an annual base salary of $530,000 and is eligible to receive an annual performance bonus targeted at 50% of his annual base salary, with the actual amount of such bonus, if any, to be determined by the Board. For 2018, Mr. Schroeder's bonus was pro-rated to reflect his 2018 service to us. Mr. Schroeder is also eligible to receive equity awards at such times and on such terms and conditions as the Board may determine and is also entitled to participate in any and all benefit programs that we make available to our executive officers, for which he may be eligible, under the plan documents governing such programs. Pursuant to his employment agreement, the Board also approved the grant to Mr. Schroeder, effective as of the first business day immediately following his employment commencement date (the "Grant Date"), of a non-statutory stock option to purchase 850,000 ordinary shares at an exercise price per share equal to the closing price per share of the Company's ordinary shares on the Nasdaq Global Select Market on the Grant Date. The option award has a ten-year term and vests over a four-year period, with 25% of the shares underlying the award vesting on the first anniversary of his employment commencement date and the remaining 75% of the shares underlying the option award to vest monthly over the subsequent 36-month period. In addition, the Board approved the grant to Mr. Schroeder, effective as of the Grant Date, of 150,000 performance-based restricted share units (the "Schroeder PRSUs"). The Schroeder PRSUs vest as follows: 50% of the Schroeder PRSUs will vest upon Board certification of the receipt of FDA approval of a new drug application for each of (x) lefamulin and (y) CONTEPO for any indication, and 50% of the Schroeder PRSUs will vest on the first anniversary of such Board certification, provided, in each case, that Mr. Schroeder is performing services for us on the applicable vesting dates. If the FDA does not approve an NDA for both lefamulin and CONTEPO by January 31, 2020, the Schroeder PRSUs award will terminate in full. The option being granted to Mr. Schroeder and the Schroeder PRSUs were awarded outside of our 2017 Share Incentive Plan as an inducement material to Mr. Schroeder's entering into employment with us in accordance with Nasdaq Stock Market Listing Rule 5635(c)(4). The employment agreement, and In the event of the termination of sum payment equal to a prorated annual bonus for the year in which In the event of the termination of If As a condition of his employment, Agreements with other Mr. Sender was appointed chief financial officer of Nabriva Austria and entered into an employment agreement dated and effective as of May 2, 2016. The employment agreements and the employment of each of Mr. Sender, Dr. In the event of the termination of such In the event of the termination of the If such accrued and to which he or she is entitled as of the termination date and, solely if such Pursuant to their respective employment agreements, each of these As a condition to their employment, each of Mr. Sender, Dr. Under the Transition Agreement, Dr. Broom will be entitled to compensation for such services consistent with the Company's Non-Employee Director Compensation Policy as in effect from time to time (the "Policy"). However, Dr. Broom was not eligible to receive an "initial grant" of share options under such Policy or an "annual grant" of share options under such Policy in 2018. Pursuant to the Transition Agreement, in return for Dr. Broom's execution of the Transition Agreement and timely execution and nonrevocation of an additional release of claims at the time of the Transition Date, Dr. Broom received the "severance benefits" described in his amended and restated employment agreement dated as of June 17, 2016 that he In addition, pursuant to the Transition Agreement, Dr. Broom will continue to vest in any options he holds to purchase the Company's ordinary shares that were outstanding as of the Transition Date (the "Outstanding Options") for the award will accelerate and vest in full at the time he The Transition Agreement also provided for, among other things, a release of claims by Dr. Broom in favor of us and our affiliates, continuing confidentiality, non-solicitation and non-competition obligations applicable to Dr. Broom under his existing Proprietary Rights, Non-Disclosure and Developments Agreement, and a mutual non-disparagement obligation applicable to us and Dr. Broom. Pursuant to the Consulting Agreement, Dr. Broom receives $500.00 per hour in consulting fees for his performance of the Services under the Consulting Agreement. The term of the Consulting Agreement will be from the Transition Date until January 31, 2020, and may be extended for additional periods upon mutual written agreement of both us and Dr. Broom. In addition, the Consulting Agreement may be terminated by us if Dr. Broom materially breaches the Consulting Agreement or the Transition Agreement; by Dr. Broom, if we materially breach the Consulting Agreement or the Transition Agreement; at any time by either us or Dr. Broom upon not less than 15 days prior written notice to the other party; at any time upon the mutual written consent of the parties; or automatically upon the death, physical incapacitation or mental incompetence of Dr. Broom. Equity In this section, we describe our 2017 Share Incentive Plan The 2017 Share Incentive Plan permits the award of share options, share appreciation rights, or SARs, restricted shares, restricted share units or RSUs, and other share-based awards to our employees, officers, directors, consultants and advisers. Our board of directors will administer the 2017 Share Incentive Plan. As of January 31, 2019, under our 2017 Share Incentive Plan, there were options to purchase an aggregate of 2,398,425 of our ordinary shares at a weighted average exercise price of $5.41 per share, 1,222,100 restricted stock units outstanding with a weighted average grant date fair value of $3.33 per share, and 1,443,853 ordinary shares available for future issuance under the plan. Shares covered by awards under the 2017 Share Incentive Plan that expire or are terminated, surrendered, or cancelled without having been fully exercised or are forfeited in whole or in part or that result in any shares not being issued will again be available for the grant of awards under the 2017 Share Incentive Plan (subject, in the case of incentive share options, to any limitations under the Internal Revenue Code, or the Code). Options and SARs granted under the 2017 Share Incentive Plan may not have an exercise price or measurement price, respectively, that is less than 100% of the fair market value of our ordinary shares on the date of grant; provided, however, that if our board of directors approves the grant of an option or SAR with an exercise price or measurement price to be determined on a future date, such price may not be less than 100% of the fair market value of our ordinary shares on such future date. Such options and SARs will be exercisable at such times and subject to such terms and conditions as the board may specify in the applicable option or SAR agreement. No option or SAR will be granted with a term in excess of ten years. Restricted shares and RSUs granted under the 2017 Share Incentive Plan will determine the terms and conditions of restricted shares and RSUs, including the conditions for vesting and repurchase (or forfeiture) and the issue price, if any. If, during the term of the 2017 Share Incentive Plan, there is a change in our capital or a restructuring measure which has an effect on our capital, such as a share split or reverse share split, which change or measure results in a change in the value of the share-based awards outstanding under the 2017 Share Incentive Plan, the board will make appropriate adjustments to the price or the amount of such outstanding awards. The 2017 Share Incentive Plan also contains provisions addressing the consequences of any reorganization event. A reorganization event is defined as (a) any merger or consolidation of us with or into another entity as a result of which all of our ordinary shares are converted into or exchanged for the right to receive cash, securities or other property, or are cancelled, (b) any transfer or disposition of all of our ordinary shares for cash, securities or other property pursuant to a share exchange or other transaction or (c) our liquidation or dissolution; any one of which, (a), (b) or (c), may be effected pursuant to the laws of the Republic of Ireland. The 2017 Share Incentive Plan provides that, if a reorganization event occurs, the board of directors may take one or more of the following actions to all or any outstanding awards other than restricted shares on such terms as the board of directors determines: (1) provide that such awards will be assumed, or substantially equivalent awards will be substituted, by the acquiring or succeeding corporation (or an affiliate thereof), (2) upon written notice to a participant, provide that all of the participant's unvested awards will be forfeited immediately prior to the consummation of such reorganization event and/or that all of the participant's unexercised awards will terminate immediately prior to the consummation of such reorganization event unless exercised by the participant (to the extent then exercisable) within a specified period following the date of such notice, (3) provide that outstanding awards will become exercisable, realizable, or deliverable, or restrictions applicable to an award will lapse, in whole or in part prior to or upon such reorganization event, (4) in the event of a reorganization event under the terms of which holders of our ordinary shares will receive, upon consummation thereof, a cash payment for each share surrendered in the reorganization event, which we refer to as the Acquisition Price, make or provide for a cash payment to participants with respect to each award held by a participant equal to (A) the number of ordinary shares subject to the vested portion of the award (after giving effect to any acceleration of vesting that occurs upon or immediately prior to such reorganization event) multiplied by (B) the excess, if any, of (I) the Acquisition Price over (II) the exercise, measurement or purchase price of such award and any applicable tax withholdings, in exchange for the termination of such award, (5) provide that, in connection with our liquidation or dissolution, awards will convert into the right to receive liquidation proceeds (if applicable, net of the exercise, measurement or purchase price thereof and any applicable tax withholdings) and (6) any combination of the foregoing. Our board is not obligated to treat all awards, all awards held by a participant, or all awards of the same type, identically. No award may be granted under the 2017 Share Incentive Plan after September 14, 2027. The board of directors may, at any time, amend, suspend or terminate the 2017 Share Incentive Plan or any portion thereof. However, if shareholder approval is required, including by application of Irish law, the board may not effect such modification or amendment without such approval. Stock Option Plan 2015 under the plan. Following the approval of the 2017 Share Incentive Plan by our shareholders Options granted under the Stock Option Plan 2015 entitle beneficiaries thereof to purchase our requirements. The Stock Option Plan 2015 provides that, if a liquidity event (as defined below) occurs, all options outstanding under the Stock Option Plan 2015 will be assumed (or substantially equivalent awards will be substituted by an acquiring or succeeding corporation (or an affiliate of the acquiring or succeeding corporation)), and any then-unvested options shall continue to vest in accordance with the Unless otherwise specifically permitted in an option agreement or resolved upon by the If, during the term of the Stock Option Plan 2015, there is a change in our capital or a restructuring measure which has an effect on our capital, such as a stock split or reverse stock split, which change or measure results in a change in the value of the options outstanding under the Stock Option Plan 2015, the 401(k) Plan We maintain a defined contribution employee retirement plan for our U.S.-based employees. Our 401(k) plan is intended to qualify as a tax-qualified plan under Section 401 of the Internal Revenue Code, so that contributions to our 401(k) plan, and income earned on such contributions, are not taxable to participants until withdrawn or distributed from the 401(k) plan. Our 401(k) plan provides that each participant may contribute up to 90% of his or her pre-tax compensation, up to a statutory limit, which is $18,000 for 2017. Participants who are at least 50 years old can also make Name Fees Earned or Option Awards Total ($) Daniel Burgess (3) 26,301 58,873 (5) 85,174 Axel Bolte (4) — — — Chau Khuong 41,740 19,624 (6) 61,364 George Talbot(7) 39,500 19,624 (6) 59,124 Charles Rowland 49,369 19,624 (6) 68,993 Stephen Webster (3) 16,132 58,873 (5) 75,005 Mark Corrigan (3) 13,501 58,873 (5) 72,374 Denise Pollard-Knight (8) 48,904 — 48,904 Chen Yu (8) 28,365 — 28,365 David Chiswell (8) 30,706 — 30,706 Risk Considerations in Our Compensation Program Our compensation committee has reviewed and evaluated the philosophy and standards on which our compensation plans have been developed and implemented across our company. It is our belief that our compensation programs do not encourage inappropriate actions or risk taking by our executive officers. We do not believe that any risks arising from our employee compensation policies and practices are reasonably likely to have a material adverse effect on our company. In addition, we do not believe that the mix and design of the components of our executive compensation program encourage management to assume excessive risks. Summary Compensation Table The following table sets forth a summary of the compensation earned by the non-employee members of the board of directors for the year ended December 31, 2018. Daniel Burgess Axel Bolte(3) Colin Broom George Talbot Charles Rowland, Jr. Stephen Webster Mark Corrigan Carrie Bourdow Director Compensation Arrangements Following the Redomiciliation, we adopted a non-employee director compensation policy for our directors, which provided for the following: On March 12, 2018, our board of directors approved an amendment to our non-employee director compensation policy. Effective as of March 12, 2018, the amendment increased the initial grant of an option to purchase our ordinary shares to new non-employee directors upon their initial election to the board of directors to 58,000 ordinary shares and increased the annual grant of an option to purchase our ordinary shares for each non-employee director to 29,000 ordinary shares. The board of directors approved the annual grants to non-employee directors for the 2018 fiscal year on May 2, 2018. On October 31, 2018, our board of directors approved an amendment to our non-employee director compensation policy. Effective as of October 31, 2018, we adopted a non-employee director compensation policy for our directors, which provides for the following: The stock options to be granted to our non-employee directors under our non-employee director compensation policy have an exercise price equal to the fair market value of our ordinary shares on the date of grant and will expire ten years after the date of grant. The initial stock options granted to newly elected director vest, subject to such director's continued service on the board, over a three-year period on a monthly pro-rata basis at the end of each successive month following the date of the initial grant. The annual stock options granted to directors will vest, subject to such director's continued service on the board, fully on the last date of the month of the first anniversary of the grant date. Under our non-employee director compensation policy, the annual cash fees are payable in arrears in four equal quarterly installments payable the week following the end of each quarter. Each non-employee director is also entitled to reimbursement for reasonable travel and other expenses incurred in connection with attending meetings of the board of directors and any committee on which he or she serves or otherwise in direct service of the company. ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS The following table sets forth information with respect to the beneficial ownership of our Broom’sBroom's option to purchase 15,827 common158,270 of our ordinary shares vests over four years, with 25% of the options vesting on August 31, 2015, and the remaining 75% of the option vesting on a monthly pro-rata basis over the remaining three years of the vesting period.Broom’sBroom's option to purchase 19,690 common196,900 of our ordinary shares vests over four years, with 25% of the options vesting on July 31, 2016, and the remaining 75% of the option vesting on a monthly pro-rata basis over the remaining three years of the vesting period.Broom’sBroom's option to purchase 15,100 common151,000 of our ordinary shares vests over four years, with 25% of the options vesting on February 28, 2017, and the remaining 75% of the option vesting on a monthly pro-rata basis over the remaining three years of the vesting period.Seltzer’sBroom's option to purchase 8,879 common261,000 of our ordinary shares vests over four years, with 25% of the options vesting on February 28, 2018, and the remaining 75% of the option vesting on a monthly pro-rata basis over the remaining three years of the vesting period. (5)Seltzer’sGelone's option to purchase 5,590 common55,900 of our ordinary shares vests over four years, with 25% of the options vesting on February 28, 2017, and the remaining 75% of the option vesting on a monthly pro-rata basis over the remaining three years of the vesting period.(6) Mr. Wolf’s5,500 common113,000 of our ordinary shares vests over four years, with 25% of the options vesting on September 30, 2016,February 28, 2018, and the remaining 75% of the option vesting on a monthly pro-rata basis over the remaining three years of the vesting period. (7) Mr. Wolf’s5,590 common77,500 of our ordinary shares vests over four years, with 25% of the options vesting on February 28, 2017,July 25, 2019, and the remaining 75% of the option vesting on a monthly pro-rata basis over the remaining three years of the vesting period.(8) Mr. Wolf’s unvested stock options were forfeited upon his resignation from the company effective February 7, 2016.Management Board Member and Senior ManagementExecutive OfficersColin Broom,Theodore Schroeder, Chief Executive Officer and Management Board MemberDirectorDr. BroomMr. Schroeder was appointed our chief executive officer and entered into an employment agreement dated and effective as of July 23, 2018. He was appointed to our board on August 28, 2014, which was amended and restated as of June 17, 2016. He also serves on our management board.1, 2018. His employment agreement automatically renews each August 28 for an additional one-year term, unless either we or Dr. Broom timely provide a notice of non-renewal,continues until terminated in accordance with its terms, as described below.Dr. Broom’sMr. Schroeder's employment, may be terminated as follows: (1) either party may notify the other, in writing and not less than 90 days prior to the applicable term’s expiration date, of its intention not to renew the term of employment; (2) upon Dr. Broom’sMr. Schroeder's death or “disability”"disability" (as disability is defined in his employment agreement); (3)(2) at our election, with or without “cause”"cause" (as cause is defined in his employment agreement); and (4)(3) at Dr. Broom’sMr. Schroeder's election, with or without “good reason”"good reason" (as good reason is defined in his employment agreement).Dr. Broom’sMr. Schroeder's employment by us without cause, including as a result of a termination of his employment following our delivery to Dr. Broom of a notice of non-renewal, or by him for good reason prior to, or more than twelve months following, a “change"change in control”control" (as change in control is defined in his employment agreement), Dr. BroomMr. Schroeder will be entitled to his base salary that has accrued and to which he is entitled as of the termination date. In addition, subject to his execution and nonrevocation of a release of claims in our favor and his continued compliance with his proprietary rights, non-disclosure and developments agreement with us, he is entitled to (1) continued payment of his base salary, in accordance with our regular payroll procedures, for a period of 18 months (2) provided he is eligible for and timely elects to continue receiving group medical insurance under COBRA and the payments would not result in the violation of nondiscrimination requirements of applicable law, payment by us of the portion of health coverage premiums we pay for similarly-situated, active employees who receive the same type of coverage, for a period of up to 18 months following his date of termination, (3) a lump sum payment equal to any earned but unpaid annual bonus for a previously completed calendar year and (4) a lumpDr. Broom’sMr. Schroeder's employment is terminated based on the number of days he provided services to us during the year in which his employment is terminated.Dr. Broom’sMr. Schroeder's employment by us without cause, including as a result of a termination of his employment following our delivery to Dr. Broom of a notice of non-renewal, or by him for good reason prior to, or by him for good reason within twelve months following a change in control, subject (as described above with respect to certain payments), to his execution and nonrevocation of a release of claims in our favor and his continued compliance with his proprietary rights, non-disclosure and developments agreement with us, Dr. BroomMr. Schroeder would be entitled to the same payments and benefits as described in the preceding paragraph, except that, in lieu of a pro-rated annual bonus payment, he would be entitled to receive a lump sum payment equal to 100% of his target bonus for the year in which his employment is terminated and he shall also be entitled to full vesting acceleration of his then-unvested equity awards, whether granted under the Stock Option2017 Share Incentive Plan 2015 or any successor equity incentive plan or as an inducement to his employment, such that his equity awards become fully exercisable and non-forfeitable as of the termination date.Dr. Broom’sMr. Schroeder's employment is terminated for any other reason, including as a result of his death or disability, for cause, or voluntarily by Dr. BroomMr. Schroeder without good reason, our obligations under the employment agreement cease immediately, and Dr. BroomMr. Schroeder is only entitled to his base salary that has accrued and to which he is entitled as of the termination date and solely if his employment is terminated as a result of his death or disability, subject to his execution and nonrevocation of a release of claims in our favor and his continued compliance with his proprietary rights, non-disclosure and developments agreement with us, he or his estate, as applicable, is entitled to any earned but unpaid annual bonus from a previously completed calendar year.Pursuant to his amended and restated employment agreement, Dr. Broom is entitled to receive an annual base salary of $435,999. His base salary is reviewed by our compensation committee and supervisory board in the first quarter of each fiscal year and any adjustment to his base salary is retroactively effective to the first day of such fiscal year. In addition, Dr. Broom is eligible for an annual discretionary bonus of 50% of his current base salary. He is also eligible to receive equity awards at such times and on such terms and conditions as the supervisory board may determine and is also entitled to participate in any and all benefit programs that we make available to our senior managers, for which he may be eligible, under the plan documents governing such programs.Dr. BroomMr. Schroeder signed a proprietary rights, non-disclosure and developments agreement.Senior ManagersExecutive OfficersDr. SeltzerHe was appointed our chief medicalfinancial officer and entered into an employment agreement dated and effective as ofon April 14, 2015, which was amended and restated as of May 26, 2016. Mr.12, 2017. Dr. Gelone was appointed chief development officer and entered into an employment agreement dated and effective as of December 1, 2014, which was amended and restated as of May 26, 2016.2016 and further amended on restated on July 24, 2018. Mr. Crotty was appointed general counsel and secretary of Nabriva Austria and entered into an employment agreement dated and effective as of June 14, 2017. Dr. Gelone was appointed as our chief development officer and Mr. Crotty was appointed our general counsel and secretary effective as of June 23, 2017. Dr. Gelone was subsequently appointed our chief scientific officer on June 30, 2017, and our president and chief operating officer on July 24, 2018. Dr. Schranz was appointed our chief medical officer and entered into an employment agreement dated and effective as of March 21, 2018. Mr. Lavino was appointed our chief commercial officer and entered into an employment agreement dated and effective as of July 10, 2017. Each of these employment agreements provides that such senior managerexecutive officer is an at-will employee, and his or her employment with us can be terminated by the respective senior managerexecutive officer or us at any time and for any reason.SeltzerGelone, Mr. Crotty, Dr. Schranz and Mr. GeloneLavino may be terminated in one of three ways: (1) upon the death or “disability”"disability" (as disability is defined in the applicable employment agreement) of such senior manager;executive officer; (2) at our election, with or without “cause”"cause" (as cause is defined in the applicable employment agreement); and (3) at such senior manager’sexecutive officer's election, with or without “good reason”"good reason" (as good reason is defined in the applicable employment agreement).senior manager’sexecutive officer's employment by us without cause or by him or her for good reason prior to, or more than twelve months following, a “change"change in control”control" (as change in control is defined in the applicable employment agreement), such senior managerexecutive officer will be entitled to his or her base salary that has accrued and to which he or she is entitled as of the termination date. In addition, subject to such senior manager’sexecutive officer's execution and nonrevocation of a release of claims in our favor and his or her continued compliance with his or her proprietary rights, non-disclosure and developments agreement with us, such senior managerexecutive officer is entitled to (1) continued payment of such senior manager’sexecutive officer's base salary, in accordance with our regular payroll procedures, for a period of 12 months, (2) provided he or she is eligible for and timely elects to continue receiving group medical insurance under COBRA and the payments would not result in the violation of nondiscrimination requirements of applicable law, payment by us of the portion of health coverage premiums we pay for similarly-situated, active employees, who receive the same type of coverage, for a period of up to 12 months following the date of termination, (3) a lump sum payment equal to any earned but unpaid annual bonus for a previously completed calendar year and (4) a lump sum payment equal to a prorated annual bonus for the year in which such senior manager’sexecutive officer's employment is terminated based on the number of days such senior managerexecutive officer provided services to us during the year in which such senior manager’sexecutive officer's employment is terminated.senior manager’sexecutive officer's employment by us without cause or by him or her for good reason within twelve months following a change in control, subject (as describedescribed above with respect to certain payments) to such senior manager’sexecutive officer's execution and nonrevocation of a release of claims in our favor and his or her continued compliance with his or her proprietary rights, non-disclosure and developments agreement with us, such senior managerexecutive officer will be entitled to the same payments and benefits as described in the preceding paragraph, except that, in lieu of a pro-rated annual bonus payment, such senior managerexecutive officer will be entitled to receive a lump sum payment equal to 100% of such senior manager’sexecutive officer's target bonus for the year in which his or her employment is terminated, and such senior managerexecutive officer shall also be entitled to full vesting acceleration of his or her then-unvested equity awards, whether granted under the Stock Option2017 Share Incentive Plan 2015 or any successor equity incentive plan, such that his or her equity awards become fully exercisable and non-forfeitable as of the termination date.senior manager’sexecutive officer's employment is terminated for any other reason, including as a result of his or her death or disability, for cause, or voluntarily by such senior managerexecutive officer without good reason, our obligations under the applicable employment agreement cease immediately, and such senior managerexecutive officer is only entitled to his or her base salary that hassenior manager’sexecutive officer's employment is terminated as a result of his or her death or disability and subject to his or her execution and nonrevocation of a release of claims in our favor and his or her continued compliance with his or her proprietary rights, non-disclosure and developments agreement with us, such senior managerexecutive officer or the estate of such senior manager,executive officer, as applicable, is entitled to any earned but unpaid annual bonus from a previously completed calendar year.senior managersexecutive officers is entitled to an annual base salary, as follows: Mr. Sender: $350,000; Ms. Seltzer: $368,740$382,500; Dr. Gelone: $472,100; Mr. Crotty; $371,100; Dr. Schranz: $436,200 and Mr. Gelone: $318,270.Lavino: $371,800. Such base salary is reviewed by our compensation committee and supervisorythe board of directors in the first quarter of each fiscal year and any adjustment to such base salary is retroactively effective to the first day of such fiscal year. In addition, such senior managersexecutive officers are eligible for an annual discretionary bonus of 35% of their current base salary. Each senior managerexecutive officer is also eligible to receive equity awards at such times and on such terms and conditions as the supervisory boardBoard may determine and is also entitled to participate in any and all benefit programs that we make available to our senior managers,executive officers, for which he or she may be eligible, under the plan documents governing such programs.SeltzerGelone, Mr. Crotty, Dr. Schranz and Mr. GeloneLavino signed a proprietary rights, non-disclosure and developments agreement.AgreementAgreements with Peter Wolf, former General CounselColin Broom, Director and Former Chief Executive OfficerMr. WolfDr. Broom was appointed general counselchief executive officer of Nabriva Austria and entered into an employment agreement dated and effective as of September 24, 2015,August 28, 2014, which was amended and restated as of May 26,June 17, 2016. Mr. WolfHe was appointed our chief executive officer on April 12, 2017. Dr. Broom resigned as general counsel,chief executive officer, effective February 7, 2017.July 24, 2018 (the "Transition Date"). We entered into a Transition, Separation and Release of Claims Agreement, or the Transition Agreement, with Dr. Broom on July 23, 2018. We also entered into a Consulting Agreement with Dr. Broom, effective as of the Transition Date, pursuant to which Dr. Broom provides to us, upon the request of the Board, consulting and advisory services relating to the preparation and submission of a new drug application for the Company's lefamulin product candidate to the FDA (collectively, the "Services"). Dr. Broom continues to serve as a director on the Board.waswould be entitled to anreceive upon a "qualifying termination" occurring prior to or more than 12 months following a "change in control" (as such terms are defined in the Employment Agreement). Specifically, he received continued payment of his current annual base salary of $323,200. In addition,$471,534 for 2016, Mr. Wolf was eligiblea period of 18 months, or $707,301 in the aggregate; subject to certain conditions, payment of a portion of health coverage premiums for an annual discretionary bonusa period of up to 35%18 months following the Transition Date; and a lump sum payment of $132,417 which is equal to the prorated portion of Dr. Broom's 2018 annual target bonus of $235,767, representing 50.0% of his current2018 annual base salary, based on the number of days of his employment in 2018.whicha period of 12 months following the Transition Date provided he was awardedremains on the Board during such 12-month period. In the event his service as a bonusdirector on the Board terminates before the 12-month anniversary of $96,152. Upon his resignation, Mr. Wolf was notthe Transition Date, the portion of the Outstanding Options that would have vested had Dr. Broom remained on the Board through the 12-month anniversary of the Transition Date will be accelerated at that time. Any of Dr. Broom's options that are unvested at the 12-month anniversary of the Transition Date will terminate and Dr. Broom will no longer have any rights with respect to them. In addition, the post-termination exercise period of the vested Outstanding Options will be extended such that Dr. Broom is entitled to exercise any additional compensation, othervested Outstanding Options until the later of the date that is 24 months following the Transition Date and the date that is three months following his cessation of service on the Board (but in no event later than his base salarythe final exercise date applicable to such option). The Transition Agreement also provides that, had accrued as ofif Dr. Broom continues to serve on the Board or to provide Services under the Consulting Agreement on the effective date of his resignation,grant, he shall continue to be entitled to receive the grant of performance-based restricted share units (the "Broom PRSUs") previously approved by the Board, which will vest in accordance with, and otherwise be subject to the terms and conditions of, the previously-approved award agreement. However, if Dr. Broom ceases to serve on the Board and ceases to provide Services under the Consulting Agreement following achievement of the applicable performance metric under the Broom PRSUs but before the first anniversary of the achievement of the performance vesting metric,forfeitedceases to provide all of his unvested stock options.services on the Board and as a consultant.Compensation ArrangementsIncentive PlansStock Option2017 Share Incentive Plan 2007 and our Stock Option Plan 2015. Prior to our initial public offering, wethe Redomiciliation, Nabriva Austria granted awards to eligible recipients under both the Stock Option Plan 2007 and the Stock Option Plan 2015. In connection with the Redomiciliation, both plans were amended to take account of certain requirements under Irish law and assumed by us, with each option to acquire one Nabriva Austria common share becoming an option to acquire ten of our ordinary shares on the same terms and conditions. We currently make option grantsshare awards to eligible recipients solely under the Stock Optionour 2017 Share Incentive Plan. 2015.Our shareholders, management boardThe Stock Option Plan 2015 provided for the grant of options to purchase our ordinary shares to our employees, including executive officers, and supervisory board adoptedto directors. As of January 31, 2019, under our Stock Option Plan 2015, on April 2, 2015,there were options to purchase an aggregate of 2,842,913 of our ordinary shares at a weighted average exercise price of $8.34 per share and no ordinary shares were available for issuanceapproved an amended and restated version ofon September 15, 2017, we ceased granting awards under the Stock Option Plan 2015, on June 30, 2015. An amendment toUnless the amended and restated Stock Option Plan 2015 was approved by our shareholders on July 22, 2015. Referencescontext specifically indicates otherwise, references to our Stock Option Plan 2015 in this Annual Report on Form 10-K refer to the amended and restated version of the Stock Option Plan 2015, as amended. The Stock Option Plan 2015 became effective on July 3, 2015 upon the registration with the commercial register in Austria of our conditional capital increase approvedamended and adopted by our shareholders on June 30, 2015. The Stock Option Plan 2015 provides for the grant of options to purchase common shares to our employees, including members of our management board, and to members of our supervisory board. Following approval by our shareholders at our 2016 annual general meeting, the number of shares available for issuance under the Stock Option Plan 2015 was increased to 346,235 common shares. The grant of stock options for 201,568 common shares under this plan to members of the management board, certain members of the supervisory board and certain employees had been made as of December 31, 2016 at a weighted-average exercise price of €70.63 ($77.99) per share.us.commonordinary shares at an exercise price equal to 100% of the fair market value per share on the beneficiary’sbeneficiary's date of participation, which following our initial public offeringthe Redomiciliation was derived from the closing sale price of our ADSsordinary shares on the NASDAQNasdaq Global Market. Options granted under the Stock Option Plan 2015 generally vest over four years from the beneficiary’sbeneficiary's date of participation. Typically, 25% of the options subject to a particular grant vest on the last day of the last calendar month of the first year of the vesting period, and the remaining 75% vests on a monthly pro-rata basis over the second, third and fourth years of the vesting period (i.e., 2.083% per month). Any alternative vesting period determined by us is subject to approval by our managementexecutive officers, board supervisory boardof directors or shareholders, in accordance with any applicable votingbeneficiary’sbeneficiary's original vesting schedule. If a beneficiary is terminated due to a good leaver event (within the meaning of the Stock Option Plan 2015), on or prior to the first anniversary of the date of the liquidity event, the beneficiary’sbeneficiary's options will be immediately exercisable in full as of the date of such termination. If the acquiring or succeeding corporation (or an affiliate of the acquiring or succeeding corporation) refuses to assume the options outstanding under the Stock Option Plan 2015 or to substitute substantially equivalent options therefor, all then-unvested options under the Stock Option Plan 2015 will automatically vest in full upon the liquidity event. For purposes of the Stock Option Plan 2015, a liquidity event generally refers to an exclusive license of or the sale, lease or other disposal of all or substantially all of our assets, a sale or other disposal (but not a pledge) of 50% or more of our shares, a merger or consolidation of us with or into any third party, or our liquidation, winding up or other form of dissolution of us.management board with the approval of the supervisory board,directors, the exercise of vested options is permitted under the Stock Option Plan 2015 only during specified periods and on specified terms in the case of a liquidity event or following an initial public offering occurring during the term of the option. A beneficiary is entitled to exercise vested options at any time during the remaining term of the option while the beneficiary is providing services to us, and within the three-month period following a termination of the beneficiary’sbeneficiary's services due to a good leaver event. Options granted under the Stock Option Plan 2015 will have a term of no more than ten years from the beneficiary’sbeneficiary's date of participation.supervisory board, upon a recommendation from the management board may make appropriate adjustments to the price or the amount of such outstanding options.No options may be granted under the Stock Option Plan 2015 after July 22, 2025, but options previously granted to a beneficiary may extend beyond that date. The supervisory board of directors may, at any time, amend, suspend or terminate the Stock Option Plan 2015 in whole or in part. However, if shareholder approval is required, including by application of AustrianIrish law, the supervisory board may not effect such modification or amendment without such approval.Stock Option Plan 2007Our shareholders adopted our Stock Option Plan 2007 on September 12, 2007 and subsequently approved amendments to the Stock Option Plan 2007 on September, 17, 2009, May 9, 2010 and June 30, 2015. References to our Stock Option Plan 2007 in this Annual Report refer to the plan as amended. No additional awards will be granted under the Stock Option Plan 2007. The Stock Option Plan 2007 provided for the grant of up to 29,889 options to certain of our employees, including members of our management board and certain members of our supervisory board, and other beneficiaries. As of December 31, 2016, a total of 10,996 options were outstanding at a weighted-average exercise price of €6.72 ($7.32) per share. The options provide for the right to purchase our common shares at an exercise price determined by us with the assistance of an Austrian Independent Certified Public Accountant as of August 24, 2007.Options granted under the Stock Option Plan 2007 generally vest over four years from the date of participation. Typically, 25% of the options subject to a particular grant vest on the last day of the last calendar month of the first year of the vesting period, a further 25% of the options vests on the last day of the last calendar month of the second year of the vesting period, and the remaining 50% vests on a monthly pro-rata basis over the third and fourth years of the vesting period (i.e., 2.083% per month). However, alternative vesting schedules applied for beneficiaries who had worked for us prior to the date of the adoption of our Stock Option Plan 2007. All options granted under such alternative vesting schedules have fully vested.The Stock Option Plan 2007 provides that 50% of any then-unvested options shall automatically vest upon a liquidity event, which refers to an exclusive license of or the sale or other disposal of 50% or more of our assets, a sale or other disposal (but not a pledge) of 50% or more of our shares, a merger of ours with any third party, or a consolidation, liquidation, winding up or other form of dissolution. If a beneficiary has an unjustified termination or a justified prematuretermination (as such terms are used in the Stock Option Plan 2007) within one yearTable of the liquidity event, all remaining unvested options held by the beneficiary shall automatically vest in full.ContentsUnless otherwise specifically permitted in an option agreement or resolved upon by the management board with the approval of the supervisory board, the exercise of vested options is permitted under the Stock Option Plan 2007 only during specified periods and on specified terms in the case of a liquidity event or following an initial public offering of our shares occurring during the term of the option, regardless of whether or not the beneficiary is then providing services to us. A beneficiary is entitled to exercise vested options at any time during the remaining term of the option. No options may be exercised under the Stock Option Plan 2007 after September 27, 2017. Any options not exercised by September 27, 2017 automatically terminate and are forfeited.If, during the term of the Stock Option Plan 2007, there is a change in our capital or a restructuring measure which has an effect on our capital, such as a stock split or reverse stock split, which change or measure results in a change in the value of the options outstanding under the Stock Option Plan 2007, the supervisory board, upon a recommendation from the management board, may make appropriate adjustments to the price or the amount of such outstanding options.To date, 22,979 options have been exercised under the Stock Option Plan 2007.Founders Program 2007In November 2007, we granted common shares and stock options to Gerd Ascher and Rodger Novak as compensation and in recognition of their status as founders of our company. We refer to these grants as our Founders Program 2007. Under the Founders Program 2007, a total of 4,982 common shares were granted, including 623 common shares granted in the form of stock options to Dr. Novak. The 623 options granted under the Founders Program 2007 have an exercise price of €1.00 per share and all of the options became fully vested in February 2010. No other options have been granted under the program, and none of the options have been exercised.“catch-up”"catch-up" contributions, which in 2017 may be up to an additional $6,000 above the statutory limit. Under our 401(k) plan, each employee is fully vested in his or her deferred salary contributions. Employee contributions are held and invested by the plan’splan's trustee, subject to participants’participants' ability to give investment directions by following certain procedures. We match 100.0% of the first 3.0% of the employee’semployee's voluntary contribution to the 401(k) plan and 50.0% of the next 2.0% contributed by the employee.SUPERVISORY BOARD COMPENSATIONSummary Compensation TableThe following table sets forth a summary of the compensation earned by the members of our supervisory board for the year ended December 31, 2016:
Paid in Cash
($) (1)
($) (2)(1) Fees earned or paid in cash consist of retainer fees paid in cash or accrued for the supervisory board member.(2) The amounts reported in the “Option Awards” column reflect the aggregate fair value of share-based compensation awarded during the year computed in accordance with the provisions of ASC Topic 718.(3) Fees paid in cash from the date of election to our supervisory board in August 2016 through the end of the year.(4) Mr. Bolte has declined to accept either cash or equity compensation for his services on our supervisory board.(5) Consists of (i) option to purchase 1,010 shares of common stock vesting with respect to all of the shares on the last date of the month of the first anniversary of the grant date, and (ii) option to purchase 2,020 shares of common stock vesting over a three-year period on a monthly pro-rata basis at the end of each successive month following the date of the initial grant.(6) Consists of option to purchase 1,010 shares of common stock vesting with respect to all of the shares on the last date of the month of the first anniversary of the grant date.(7) Includes approximately $4,500 paid to Talbot Advisors LLC, a single-member limited liability company of which George H. Talbot is the principal, for Dr. Talbot’s service as chairman of our Clinical Advisory Board and for consulting services related to our clinical development strategy, engagement with strategic partners and related travel expenses.(8) Fees paid in cash in a lump sum following our annual general meeting of shareholders in August 2016 for such board member’s service from Jaunary 1, 2016 through the annual general meeting of shareholders at which such board member did not stand for re-election to our supervisory board. Such fees are the pro-rata amount of the annual cash retainer fees approved by our shareholders at our annual general meeting of shareholders.Director Compensation ArrangementsAt our annual general meeting of shareholders in August 2016, the shareholders approved a compensation plan for our non-employee supervisory board members, effective as of the date of such meeting. Our supervisory board compensation policy provides for the following:· each new non-employee supervisory board member will receive an initial grant of an option under our Stock Option Plan 2015 to purchase 2,020 common shares upon his or her initial election to our supervisory board;· each non-employee supervisory board member will receive an annual grant of an option under our Stock Option Plan 2015 to purchase 1,010 common shares on the date of our annual general meeting of shareholders;· each non-employee supervisory board member will receive an annual cash fee of $35,000;· the chairman of our supervisory board will receive an additional annual cash fee of $25,000;· each non-employee supervisory board member who is a member of the audit committee will receive an additional annual cash fee of $7,500 ($15,000 for the audit committee chair);· each non-employee supervisory board member who is a member of the compensation committee will receive an additional annual cash fee of $5,000 ($10,000 for the compensation committee chair); and· each non-employee supervisory board member who is a member of the nominating and corporate governance committee will receive an additional annual cash fee of $3,500 ($7,500 for the nominating and corporate governance committee chair).The stock options granted to our non-employee supervisory board members will have an exercise price equal to the fair market value of our common shares on the date of grant and will expire ten years after the date of grant. The initial stock options granted to our newly elected non-employee supervisory board members will, subject to such member’s continued service on our supervisory board, vest over a three-year period on a monthly pro-rata basis at the end of each successive month following the date of the initial grant. The annual stock options granted to our non-employee supervisory board members will, subject to such member’s continued service on our supervisory board, vest fully on the last date of the month of the first anniversary of the grant date.Upon approval of the non-employee supervisory board compensation plan by our shareholders in August 2016, we paid the members who had served on the supervisory board prior to the approval date, the cash compensation owed to them under the approved plan for their service in 2016 in a lump sum payment. Thereafter, the annual cash fee will be payable in arrears in four equal quarterly installments payable the week following the end of each quarter. The amount of each payment will be prorated for any portion of a quarter that a member is not serving on our supervisory board. Each non-employee supervisory board member will also be entitled to reimbursement for reasonable travel and other expenses incurred in connection with attending meetings of the supervisory board and any committee on which he or she serves or otherwise in direct service of our company. These amounts are excluded from the table above.Compensation Committee Interlocks and Insider ParticipationFor 2016, the members of our compensation committee were Axel Bolte (chair), Chau Khuong and Charles Rowland. No member of our compensation committee is, or has been, an officer or employee of ours or any subsidiary of ours. None of our executive officers served as a director or a member of a compensation committee (or other committee serving an equivalent function) of any other entity that had one or more executive officers serving as a supervisory board member or member of our compensation committee during the year ended December 31, 2016. Fees Earned or
Paid in Cash
($)(1) Option Awards
($)(2) Total ($) 82,500 101,113 (4) 183,613 — — — 16,982 — 16,982 40,114 79,663 (6) 119,777 62,542 93,963 (5) 156,505 54,058 79,663 (6) 133,721 44,630 79,663 (6) 124,293 44,000 79,663 (6) 123,663 Security Ownership of Certain Beneficial Owners and Managementcommonordinary shares as of March 1, 2017January 31, 2019 by:· each of the members of our supervisory board;
·each of our “nameddirectors and director nominees;
The percentages in the columns entitled “Percentage"Percentage of Shares Beneficially Owned”Owned" are based on a total of 2,719,851 common67,645,598 ordinary shares outstanding as of March 1, 2017.January 31, 2019.
Beneficial ownership is determined in accordance with the rules and regulations of the SEC and includes voting or investment power with respect to our commonordinary shares. Our commonordinary shares subject to options that are currently exercisable or exercisable within 60 days of March 1, 2017January 31, 2019 are considered outstanding and beneficially owned by the person holding the options for the purpose of calculating the percentage ownership of that person but not for the purpose of calculating the percentage ownership of any other person. Except as otherwise noted, the persons and entities in this table have sole voting and investing power with respect to all of the commonordinary shares beneficially owned by them, subject to
community property laws, where applicable. Except as otherwise set forth below, the address of the beneficial owner is c/o Nabriva Therapeutics AG, Leberstrasse 20, 1110 Vienna, Austria.
Name and Address of Beneficial Owner |
| Number of |
| Percentage of |
|
Supervisory Board Members and Named Executive Officers: |
|
|
|
|
|
Chau Khoung (1) |
| 391,960 |
| 14.41 | % |
Daniel Burgess (2) |
| 449 |
| — |
|
Axel Bolte (3) |
| — |
| — |
|
George H. Talbot (4) |
| 4,650 |
| * |
|
Mark Corrigan (2) |
| 449 |
| — |
|
Stephen Webster (2) |
| 449 |
| — |
|
Charles A. Rowland, Jr. (5) |
| 700 |
| * |
|
Colin Broom (6) |
| 39,140 |
| 1.40 | % |
Elyse Seltzer (7) |
| 6,125 |
| * |
|
Peter Wolf (8)(9) |
| 2,103 |
| * |
|
All current senior managers, management board member and supervisory board members as a group (10 individuals) |
| 446,025 |
| 15.81 | % |
|
|
|
|
|
|
5% Shareholders: | �� |
|
|
|
|
Entities affiliated with Vivo Capital (10) |
| 402,156 |
| 14.79 | % |
OrbiMed Private Investments V, L.P. (11) |
| 391,960 |
| 14.41 | % |
HBM Healthcare Investments (Cayman) Ltd. and an affiliated entity (12) |
| 315,153 |
| 11.59 | % |
Novo A/S (13) |
| 232,655 |
| 8.55 | % |
venBio Global Strategic Fund II, L.P. (14) |
| 222,296 |
| 8.17 | % |
Wellington Management Group LLP (15) |
| 196,031 |
| 7.21 | % |
Phase4 Ventures III L.P. (16) |
| 159,882 |
| 5.88 | % |
plc, 25-28 North Wall Quay, Dublin 1, Ireland.
Name and Address of Beneficial Owner | Number of Shares Beneficially Owned | Percentage of Shares Beneficially Owned | |||||
|---|---|---|---|---|---|---|---|
Directors, Director Nominees and Named Executive Officers: | |||||||
Daniel Burgess(1) | 73,594 | * | % | ||||
George H. Talbot(2) | 104,507 | * | % | ||||
Mark Corrigan(3) | 37,594 | * | % | ||||
Stephen Webster(4) | 45,594 | * | % | ||||
Charles A. Rowland, Jr.(5) | 87,867 | * | % | ||||
Carrie Bourdow(6) | 21,322 | * | % | ||||
Colin Broom(7) | 824,429 | 1.21 | % | ||||
Steven Gelone(8) | 238,759 | * | % | ||||
Theodore Schroeder(9) | 60,741 | * | % | ||||
Francesco Lavino(10) | 95,386 | * | % | ||||
All current directors and executive officers as a group (13 individuals)(11) | 1,868,456 | 2.70 | % | ||||
5% Shareholders: | |||||||
Entities affiliated with Vivo(12) | 6,619,190 | 9.79 | % | ||||
Novo Holdings A/S(13) | 4,703,720 | 6.95 | % | ||||
Longitude Capital Management Co., LLC(14) | 4,012,375 | 5.93 | % | ||||
Frazier Healthcare(15) | 4,012,374 | 5.93 | % | ||||
OrbiMed Private Investments V, L.P.(16) | 3,919,600 | 5.79 | % | ||||
January 31, 2019. OrbiMed Private Investments V, L.P., or OPI V. OrbiMed Private Investments V Cooperatief U.A., or Cooperatief, is the sole Securities Authorized for Issuance under Equity Compensation Plans The following table contains information about our equity compensation plans as of December 31, Equity compensation plans approved by security holders Equity compensation plans not approved by security holders Total Plan category Number of Weighted-average Number of (a) (b) (c) Equity compensation plans approved by security holders 191,055 $ 73.92 (1) 164,663 Equity compensation plans not approved by security holders — — — Total 191,055 $ 73.92 (1) 164,663 ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE Board Determination of Independence Applicable In March Board Policies Our If a related person proposes to enter into such a transaction, arrangement or relationship, which we refer to as a person transaction to our A related person transaction reviewed under the policy will be considered approved or ratified if it is authorized by the audit committee after full disclosure of the related Based solely upon the Form 3 filed on December 30, 2016, which sets forth beneficial ownership as of December 30, 2015. the(i) 36,000 ordinary shares listed in footnote (11) below, which are held by OrbiMed Private Investments V-NB B.V and OrbiMed Private Investments V, L.P. Mr. Khuong, one(ii) 37,594 ordinary shares issuable upon exercise of our supervisory board members, is a Private Equity Partner at OrbiMed Advisors, the managing memberstock options exercisable within 60 days of the general partner of the sole shareholder of OrbiMed Private Investments V, L.P. and OrbiMed Private Investments V-NB B.V. Mr. Khuong disclaims beneficial ownership of these shares except to the extent of any pecuniary interest therein.449 common(i) 47,640 ordinary shares and (ii) 56,867 ordinary shares issuable upon exercise of stock options exercisable within 60 days of January 31, 2019.March 1, 2017.(3) Based solely upon the Form 3 filed on December 30, 2016, which sets forth beneficial ownership as of December 30, 2015. Mr. Bolte, a member of our supervisory board, is an advisor to HBM Partners AG. HBM Partners AG provides investment management services to HBM Healthcare Investments (Cayman) Ltd. and HBM BioCapital Invest Ltd. Mr. Bolte has no voting or investment power over the shares held by HBM Healthcare Investments (Cayman) Ltd. or HBM BioCapital Invest Ltd. and disclaims beneficial ownership of such shares.(4) Consists of (i) 2,849 common shares and (ii) 1,801 common shares issuable upon exercise of stock options within 60 days of March 1, 2017.(5) Consists of 700 common shares issuable upon exercise of stock options within 60 days of March 1, 2017.(6) Consists of (i) 3,009 common shares directly owned by Dr. Broom, (ii) 12,572 common shares held by the Colin Broom Grantor Trusts I and II, and (iii) 23,569 common shares issuable upon exercise of stock options within 60 days of March 1, 2017.(7) Consists of (i) 240 common shares and (ii) 5,885 common shares issuable upon exercise of stock options within 60 days of March 1, 2017.(8) Consists of (i) 270 common shares and (ii) 1,833 common shares issuable upon exercise of stock options that had vested as of February 7, 2017.(9) Mr. Wolf resigned as from the company effective February 7, 2017.(10)January 31, 2019.13, 2017,14, 2019, which sets forth beneficial ownership as of December 31, 2016.2018. Consists of (i) 170,611 common4,218,777 ordinary shares and 1,827,506 ADSs held by Vivo Hong Kong VIII Co,Co., Limited, wholly owned subsidiary of Vivo Capital Fund VIII, L.P. and, (ii) 23,559 common582,556 ordinary shares and 252,353 ADSs held by Vivo Hong Kong VIII Surplus Co., Limited, wholly owned subsidiary of Vivo Capital Surplus Fund VIII, L.P. and (iii) 1,817,857 ordinary shares and held by Vivo Opportunity, LLC. Vivo Capital VIII, LLC is the general partner of both Vivo Capital Fund VIII, L.P. and Vivo Capital Surplus Fund VIII, L.P. The voting members of Vivo Capital VIII, LLC are Frank Kung, Albert Cha, Edgar Engleman, Chen Yu and Shan Fu, none of whom has individual voting or investment power with respect to these shares and each of whom disclaims beneficial ownership of such shares of Vivo Hong Kong VIII Co., Limited and Vivo Hong Kong VIII Surplus Co., Limited. Vivo Opportunity, LLC is the general partner of Vivo Opportunity Fund, L.P. The voting members of Vivo Opportunity, LLC are Albert Cha, Gaurav Aggarwal, Shan Fu, Frank Kung and Michael Chang, none of whom has individual voting or investment power with respect to these shares of Vivo Opportunity, LLC and each of whom disclaims beneficial ownership of such shares. The address for Vivo Capital VIII, LLC and Vivo Opportunity, LLC is 505 Hamilton Avenue, Suite 207, Palo Alto, California 94301.(11)the Form 3Schedule 13G filed on December 30, 2016,February 1, 2019, which sets forth beneficial ownership as of December 30, 2015. Consists31, 2018. Novo Holdings A/S, a Danish limited liability company, is wholly owned by Novo Nordisk Fonden (the "Foundation"), a Danish commercial foundation. Novo Holdings A/S is the holding company in the group of 175,679 commonNovo companies (currently comprised of Novo Nordisk A/S, Novozymes A/S and NNIT A/S) and is responsible for managing the Foundation's assets, including its financial assets. Based on the governance structure of Novo Holdings A/S and the Foundation, the Foundation disclaims any beneficial ownership of the shares held by Novo Holdings A/S. Novo Holdings A/S, through its board of directors (the "Novo Board"), has the sole power to vote and dispose of the shares. Lars Rebien Sorensen, Steen Riisgaard, Francis Cuss, Jean-Luc Butel, Jeppe Christiansen and Vivian Monges serve on the Novo Board and may exercise voting and dispositive control over the shares only with the support of a majority of the Novo Board. As such, no individual member of the Novo Board is deemed to hold any beneficial ownership or reportable pecuniary interest in the shares. The business address of Novo Holdings A/S is Tuborg Havnevej 19, 2900 Hellerup, Denmark. 131,500 common shares held by shareholderstockholder of OPI V-NB. OPI V is the majority member of Cooperatief, and OrbiMed Capital GP V LLC, or GP V, is the sole general partner of OPI V. OrbiMed Advisors LLC, or OrbiMed Advisors, is the managing member of GP V. GP V and OrbiMed Advisors may be deemed to have beneficial ownership of the shares held by OPI V. Samule D. Islay is the managing member ofV and owner of a controlling interest inOPI V-NB. OrbiMed Advisors exercises its investment and as such may be deemedvoting power with respect to have beneficial ownership of the shares held by OPI V. Chau Khuong, oneV-NB and OPI V through a management committee comprised of our supervisory board members, is employed as a Private Equity Partner at OrbiMed Advisors.Carl L. Gordon, Sven H. Borho and Jonathan T. Silverstein, each of whom disclaims beneficial ownership of such shares. Each of GP V and OrbiMed Advisors Mr. Islay and Mr. Khuong disclaims beneficial ownership of the shares held by OPI V and OPI V-NB except to the extent of its or his pecuniary interest therein, if any. The address for these entities is 601 Lexington Avenue, 54th floor, New York, New York 10022.(12) Based solely upon the Form 3 filed on December 30, 2016, which sets forth beneficial ownership as of December 30, 2015. Consists of (i) 275,988 common shares held by HBM Healthcare Investments (Cayman) Ltd., and (ii) 39,165 common shares held by HBM BioCapital Invest Ltd. The board of directors of HBM Healthcare Investments (Cayman) Ltd. has sole voting and investment power with respect to the shares held by such entity. The board of directors of HBM Healthcare Investments (Cayman) Ltd. is comprised of Jean-Marc Lesieur, Richard Coles, Sophia Harris, Dr. Andreas Wicki, Paul Woodhouse and John Urquhart, none of whom has individual voting or investment power with respect to these shares and each of whom disclaims beneficial ownership of the shares held by HBM Healthcare Investments (Cayman) Ltd., except to the extent of any pecuniary interest therein. The board of directors of HBM BioCapital Invest Ltd. has sole voting and investment power with respect to the shares held by such entity. The board of directors of HBM BioCapital Invest Ltd. is comprised of Jean-Marc LeSieur and Dr. Andreas Wicki, none of whom has individual voting or investment power with respect to these shares and each of whom disclaims beneficial ownership of the shares held by HBM BioCapital Invest Ltd., except to the extent of any pecuniary interest therein. The address for HBM Healthcare Investments (Cayman) Ltd. and HBM BioCapital Invest Ltd. is Governor’s Square, Suite # 4-212-2, 23 Lime Tree Bay Avenue, West Bay, Grand Cayman, Cayman Islands.(13) Based solely upon Schedule 13G filed on February 8, 2017, which sets forth beneficial ownership as of December 31, 2016. Novo A/S, a Danish limited liability company, is wholly owned by Novo Nordisk Fonden (the “Foundation”), a Danish commercial foundation. Novo A/S is the holding company in the group of Novo companies (currently comprised of Novo Nordisk A/S, Novozymes A/S and NNIT A/S) and is responsible for managing the Foundation’s assets, including its financial assets. Based on the governance structure of Novo A/S and the Foundation, the Foundation disclaims any beneficial ownership of the shares held by Novo A/S. Novo A/S, through its board of directors (the “Novo Board”), has the sole power to vote and dispose of the shares. Sten Scheibye, Goran Ando, Jeppe Christiansen, Steen Riisgaard and Per Wold-Olsen serve on the Novo Board and may exercise voting and dispositive control over the shares only with the support of a majority of the Novo Board. As such, no individual member of the Novo Board is deemed to hold any beneficial ownership or reportable pecuniary interest in the shares. The business address of Novo A/S is Tuborg Havnevej 19, 2900 Hellerup, Denmark.(14) Based solely upon Schedule 13G filed on February 13, 2017, which sets forth beneficial ownership as of December 31, 2016. Consists of 222,296 common shares held by venBio Global Strategic Fund II, L.P. (the “Fund”). venBio Global Strategic GP II, L.P. (the “General Partner”) is the sole general partner of the Fund, and venBio Global Strategic GP II, Ltd. (the “GP Ltd.”) is the sole general partner of the General Partner. The General Partner and GP Ltd., as well as Robert Adelman and Corey Goodman, as directors of GP Ltd. may be deemed beneficially own the shares The business address of venBio Global Strategic Fund II, L.P.is c/o venBio Partners, LLC, 1700 Owens Street, Suite 595, San Francisco, California 94158.(15) Based solely upon Schedule 13G filed on February 9, 2017, which sets forth beneficial ownership as of December 31, 2016. Consists of common shares reported as being beneficially owned by Wellington Management Group LLP, Wellington Group Holdings LLP, Wellington Investment Advisors Holdings LLP and Wellington Management Company LLP. The shares are owned of record by clients of the one or more investment advisors (the “Wellington Investment Advisors”). Wellington Investment Advisors Holdings LLP controls directly or indirectly, through Wellington Management Global Holdings, Ltd., the Wellington Investment Advisors. Wellington Investment Advisors Holdings LLP is owned by Wellington Group Holdings LLP. Wellington Group Holdings LLP is owned by Wellington Management Group LLP. The address of Wellington Management Group LLP is c/o Wellington Management Company LLP, 280 Congress Street, Boston, Massachusetts 02210.(16) Based solely upon Schedule 13G filed on February 10, 2017, which sets forth beneficial ownership as of December 31, 2016. Consists of 159,882 common shares held by Phase4 Ventures III General Partner Limited (“Phase4 GP”), Phase4 Partners Limited (“Phase4 Partners”), Phase4 Ventures III LP (“Phase4”) and Phase4 Ventures III GP LP (“Phase4 GPLP”, and together with Phase4 GP, Phase4 Partners, and Phase 4, the “Phase4 Reporting Persons”). The general partner of Phase4 is Phase4 GPLP. The general partner of Phase4 GPLP is Phase4 GP. Phase4 GP has appointed Phase4 Partners to act as the manager of Phase4. The Phase4 Reporting Persons may be deemed a “group” for purposes of Section 13 of the Exchange Act and expressly disclaim status as a “group” for purposes of this Schedule 13G. Each of the Phase4 Reporting Persons is deemed to beneficially own shares held by Phase4.2016.2018, as adjusted to reflect our assumption of the option granted under the Stock Option Plan 2015 in connection with the Redomiciliation. As of December 31, 2016, we2018, Nabriva had three equity compensation plans, the 2017 Share Incentive Plan, the Stock Option Plan 2015 and the 2018 Employee Share Purchase Plan, each of which were approved by our shareholders:shareholders. In addition, from time to time, the 2015compensation committee grants inducement equity awards to individuals as an inducement material to the individual's entry into employment with us within the meaning of Nasdaq Listing Rules. Number of
securities to be
issued upon
exercise
of outstanding
options, warrants
and rights Weighted-average
exercise price of
outstanding
options, warrants
and rights Number of
securities remaining
available for future
issuance under
equity
compensation plans
(excluding
securities reflected
in column(a)) (a) (b) (c) 6,463,438 (1) $ 6.52 (2) 1,543,853 (3) 1,000,000 (4) 3.53 (2) — 7,463,438 $ 6.52 (2) 1,543,853 2007 Stock Optionweighted-average exercise price.2007 Founders Program.company in accordance with Nasdaq Listing Rule 5635(c)(4).
securities to be
issued upon
exercise
of outstanding
options, warrants
and rights
exercise price of
outstanding
options, warrants
and��rights
securities remaining
available for future
issuance under
equity
compensation plans
(excluding
securities reflected
in column(a))(1) The U.S. dollar equivalentTable of €66.93.ContentsNASDAQNasdaq rules require a majority of a listed company’scompany's board of directors to be comprised of independent directors within one year of listing. In addition, the NASDAQNasdaq rules require that, subject to specified exceptions, each member of a listed company’scompany's audit, compensation and nominating and corporate governance committees be independent under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act, and compensation committee members must also satisfy the independence criteria set forth in Rule 10C-1 under the Exchange Act. Under applicable NASDAQNasdaq rules, a director will only qualify as an “independent director”"independent director" if, in the opinion of the listed company’scompany's board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. In order toTo be considered independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee, accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries or otherwise be an affiliated person of the listed company or any of its subsidiaries. In order to be considered independent for purposes of Rule 10C-1, the board must consider, for each member of a compensation committee of a listed company, all factors specifically relevant to determining whether a director has a relationship to such company which is material to that director’sdirector's ability to be independent from management in connection with the duties of a compensation committee member, including, but not limited to: (1) the source of compensation of the director, including any consulting, advisory or other compensatory fee paid by such company to the director; and (2) whether the director is affiliated with the company or any of its subsidiaries or affiliates.2017,2019, our supervisory board of directors undertook its annuala review of the independence of each supervisory board member.director. Based upon information requested from and provided by each supervisory board memberdirector concerning his or her background, employment and affiliations, including family relationships, our supervisory board has determined that each of our supervisory board members,directors, with the exception of George H. Talbot,Colin Broom and Theodore Schroeder, is an “independent director”"independent director" as defined under applicable NASDAQNasdaq rules, including, in the case of all the members of our audit committee, the independence criteria set forth in Rule 10A-3 under the Exchange Act, and in the case of all the members of our compensation committee, the independence criteria set forth in Rule 10C-1 under the Exchange Act. In making such determination, our supervisory board considered the relationships that each such supervisory board memberdirector has with Nabriva Therapeutics,us, including each of the transactions described below in “—"—Board Policies—Related Person Transactions—Certain Relationships and Related Transactions,”" and all other facts and circumstances that our supervisory board deemed relevant in make such independence determination.Policies and Procedures for Related Person Transactionssupervisory board of directors has adopted written policies and procedures for the review of any transaction, arrangement or relationship in which Nabriva Therapeuticsthe company is a participant, the amount involved exceeds the lesser of $120,000 and one percent of the average of the our total assets at year-end for the last two completed fiscal years and one of our members of senior management, member of the supervisory board, nominee for the supervisory boardexecutive officers, directors, director nominees or 5% shareholder,shareholders, or their immediate family members, each of whom we refer to as a “related"related person,”" has a direct or indirect material interest.“related"related person transaction,”" the related person must report the proposed relatedChief Financial Officerchief financial officer or General Counsel.general counsel. The policy calls for the proposed related person transaction to be reviewed and, if deemed appropriate, approved by our audit committee. Whenever practicable, the reporting, review and approval will occur prior to entry into the transaction. If advance review and approval is not practicable, the committee will review, and, in its discretion, may ratify the related person transaction. The policy also permits the chairmanchair of the audit committee to review and, if deemed appropriate, approve proposed related person transactions that arise between committee meetings, subject to ratification by the committee at its next meeting. Any related person transactions that are ongoing in nature will be reviewed annually.person’sperson's interest in the transaction. As appropriate for the circumstances, the audit committee will review and consider:·the related person’sperson's interest in the related person transaction;
·
Our audit committee may approve or ratify the transaction only if it determines that, under all of the circumstances, the transaction is in our best interests. Our audit committee may impose any conditions on the related person transaction that it deems appropriate.
In addition to the transactions that are excluded by the instructions to the SEC’sSEC's related person transaction disclosure rule, our supervisory board of directors has determined that the following transactions do not create a material direct or indirect interest on behalf of related persons and, therefore, are not related person transactions for purposes of this policy:
- •
·interests arising solely from the relatedperson’sperson's position as an executive officer of another entity, whether or not the person is also a director of the entity, that is a participant in the transaction where the related person and all other related persons own in the aggregate less than a 10% equity interest in such entity, the related person and his or her immediate family members are not involved in the negotiation of the terms of the transaction and do not receive any special benefits as a result of the transaction and the amount involved in the transaction is less than the greater of $200,000 or 5% of the annual gross revenues of the company receiving payment under the transaction; and·- •
- a transaction that is specifically contemplated by provisions of our memorandum and articles of
association or by-laws.association.
The policy provides that transactions involving compensation of our management board or senior managersexecutive officers shall be reviewed and approved by our compensation committee in the manner specified in the compensation committee’scommittee's charter.
In addition, under our Code of Business Conduct and Ethics, our employees, management board memberdirectors, executive officers and supervisory board membersemployees have an affirmative responsibility to disclose any transaction or relationship that reasonably could be expected to give rise to a conflict of interest.
Certain Relationships and Related Transactions
Since January 1, 2016,2018, we have engaged in the following transactions with our senior managers, management board member, supervisory board membersexecutive officers, directors and holders of more than 5% of our voting securities, and affiliates of our senior managers, management board member, supervisory board membersexecutive officers, directors and 5% shareholders. We believe that all of the transactions described below were made on terms no less favorable to us than could have been obtained from unaffiliated third parties:
December 2016July 2018 Financing
In December 2016,July 2018, we completed a rights offering and a relatedan underwritten offering for the sale of an aggregate of 588,127 of our common shares (including common shares represented by American Depositary Shares, or ADSs).18,181,818 ordinary shares. In connection with such offerings, our chief executive officer and our principal5% shareholders, and their affiliated entities purchased an aggregate of 102,077 common8,715,000 ordinary shares at a purchase price of €40.14$2.75 per common share andordinary share.
5% Shareholders: | Number of Shares Acquired | |||
|---|---|---|---|---|
Entities affiliated with Vivo Capital | 2,545,000 | |||
Novo A/S | 1,815,000 | |||
Longitude Ventures Partners | 1,815,000 | |||
Frazier Healthcare Partners | 1,815,000 | |||
OrbiMed Private Investments V, L.P. | 725,000 | |||
At-the-Market Offering
In March 2018, Wellington Management Group LLP, purchased an aggregate of 2,865,277 ADSs (representing approximately 286,528 common shares)3,414,100 of our ordinary shares at a purchase price of $4.32$5.50 per ADS. The following table sets forth the aggregate number of common shares and ADSs thatshare under our chief executive officer and our principal shareholders and their affiliated entities purchased.
Beneficial Owner |
| Number of Common Shares Purchased |
| Number of ADSs Purchased |
|
OrbiMed Private Investments V, L.P |
| 48,487 |
| 362,940 |
|
venBio |
| — |
| 1,045,092 |
|
Entities affiliated with Vivo Capital |
| 53,590 |
| 333,959 |
|
Novo A/S |
| — |
| 1,089,244 |
|
Colin Broom |
| — |
| 34,042 |
|
Relationship with George Talbot
We paid Talbot Advisors LLC, a single-member limited liability company of which George H. Talbot is the principal, approximately $4,500, $17,400 and $163,900 during the years ended December 31, 2016, 2015, and 2014, for Dr. Talbot’s service as chairman"at-the-market" offering program. Following such purchase, Wellington Management Group LLP beneficially owned more than 5% of our Clinical Advisory Board and for consulting services related to our clinical development strategy, engagement with strategic partners and related travel expenses.outstanding share capital.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The following table sets forth, for each of the years indicated, the aggregate fees billed or expected to be billed to us for services rendered by KPMG LLP, or KPMG, and PwC Wirtschaftsprüfung GmbH, or PwC, our independent registered public accounting firm.
|
| Year Ended December 31, |
| ||||
(in thousands) |
| 2016 |
| 2015 |
| ||
|
|
|
|
|
| ||
Audit Fees |
| $ | 417 |
| $ | 382 |
|
Audit-Related Fees(1) |
| 253 |
| 133 |
| ||
Tax Fees(2) |
| 30 |
| — |
| ||
All Other Fees(3) |
| 989 |
| 1,857 |
| ||
|
|
|
|
|
| ||
Total |
| $ | 1,689 |
| $ | 2,372 |
|
firms during the periods indicated. Effective June 2017, we engaged KPMG as our independent registered public accounting firm replacing PwC, as further described in Part II, Item 9 "Changes in and Disagreements with Accountants on Accounting and Financial Disclosure".
| | Year Ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
(in thousands) | 2018 | 2017 | |||||
Audit Fees | $ | 654 | $ | 467 | |||
Audit-Related Fees(1) | — | — | |||||
Tax Fees(2) | 23 | 24 | |||||
All Other Fees(3) | 235 | 1,210 | |||||
| | | | | | | | |
Total | $ | 912 | $ | 1,701 | |||
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Our All of the services ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES None Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized. Date: March 12, 2019 By: /s/ THEODORE SCHROEDER Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated: /s/ Director, Chief Executive Officer March /s/ Incorporated by Exhibit Description of Exhibit Form File Date of Exhibit Filed 3.1 Articles of Association of Nabriva Therapeutics AG 6-K 001-37558 10-17-16 99.1 3.2 By-Laws of the Supervisory Board of Nabriva Therapeutics AG 20-F 001-37558 04/28/16 1.2 3.3 By-Laws of the Management Board of Nabriva Therapeutics AG 20-F 001-37558 04/28/16 1.3 4.1 Deposit Agreement, dated September 17, 2015, among Nabriva Therapeutics AG, The Bank of New York Mellon, as depositary, and all owners and holders of ADSs issued thereunder 6-K 001-37558 09/17/15 99.3 4.2 Form of American Depositary Receit 6-K 001-37558 09/17/15 99.3 4.3 Registration Rights Agreement, dated September 4, 2015, among Nabriva Therapeutics AG and the parties listed therein F-1 333-205073 09/08/15 4.4 10.1 # Stock Option Plan 2007, as amended F-1 333-205073 07/7/15 10.1 10.2 # Stock Option Plan 2015, as amended F-1 333-205073 8/24/15 10.2 10.3 Lease Agreement, dated December 1, 2014, by and between Nabriva Therapeutics AG and EOS at 1000 Continental, LLC F-1 333-205073 06/18/15 10.3 10.4 Lease Agreement, dated March 16, 2007, by and between Nabriva Therapeutics AG and CONTRA Liegenschaftsverwaltung GmbH F-1 333-205073 06/18/15 10.4 10.5 Sublease Agreement, dated July 7, 2015, by and between Nabriva Therapeutics AG and Card Connect, LLC F-1 333-205073 8/24/15 10.11 10.6 Consultancy Service Agreement, dated January 1, 2014, between Nabriva Therapeutics AG and Talbot Advisors LLC F-1 333-205073 06/18/15 10.10 10.7 # Amended and Restated Employment Agreement dated June 17, 2016 by and between Nabriva Therapeutics US, Inc. and Colin Broom 6-K 001-37558 08/09/16 10.1 10.8 # Employment Agreement dated May 2, 2016 by and between Nabriva Therapeutics US, Inc. and Gary Sender 6-K 001-37558 08/09/16 10.2 10.9 # Amended and Restated Employment Agreement dated May 26, 2016 by and between Nabriva Therapeutics US, Inc. and Elyse Seltzer 6-K 001-37558 08/09/16 10.3 10.10 # Amended and Restated Employment Agreement dated May 26, 2016 by and between Nabriva Therapeutics US, Inc. and Steven Gelone 6-K 001-37558 08/09/16 10.4 10.11 # Amended and Restated Employment Agreement dated May 26, 2016 by and between Nabriva Therapeutics US, Inc.and Peter Wolf 6-K 001-37558 08/09/16 10.5 10.12 Loan Agreement, dated July 4, 2014, between Nabriva Therapeutics AG and Kreos Capital IV (UK) Limited F-1 333-205073 06/18/15 10.9 10.13 Loan Prepayment Agreement, dated November 20, 2015, between Nabriva Therapeutics AG and Kreos Capital IV 20-F 001-37558 04/28/16 4.12 21.1 Subsidiaries of Nabriva Therapeutics AG X 23.1 Consent of PwC Wirtschaftsprüfung GmbH X 31.1 Certification of principal executive officer pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934, as amended. X 31.2 Certification of principal financial officer pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934, as amended. X 32.1 Certification of principal executive officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 X 32.2 Certification of principal financial officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 X 101.INS XBRL Instance Document X 101.SCH XBRL Taxonomy Extension Schema Document X 101.CAL XBRL Taxonomy Calculation Linkbase Document X 101.DEF XBRL Taxonomy Extension Definition Linkbase Document X Table of Nabriva Therapeutics plc Audited Consolidated Financial Statements Report of KPMG LLP, Independent Registered Public Accounting Firm Consolidated balance sheets as of December 31, Consolidated statements of cash flows for the years ended December 31, included in our initial public offering registration statement that was filed with the Securities and Exchange Commission in 2015 and other audit related assurance services.Audit Committee Pre-Approval Policies and ProceduresAudit Committee reviewsaudit committee has adopted policies and pre-approvesprocedures relating to the scopeapproval of all audit and the cost of audit services and permissible non-audit services that are to be performed by our independent registered public accounting firm. This policy generally provides that we will not engage our independent registered public accounting firm to render audit or non-audit services unless the independent auditors, other than those forservice is specifically approved in advance by our audit committee or the engagement is entered into pursuant to a de minimis services which are approved by the Audit Committee prior to the completion of the audit.exception in accordance with applicable SEC rules.relatedprovided to our company providedus by KPMG and PwC Wirtschaftsprüfung GmbH during the last two fiscal year have beenyears were approved by the Audit Committee.audit committee.(a)(1)(a) (1) Financial Statements: See Index to Consolidated Financial Statements on page F-1. (2)
(2)
No financial statement schedules have been filed as part of this Annual Report on Form 10-K because they are not applicable, not required or because the information is otherwise included in our financial statements or notes thereto.(3)
(3)
The exhibits listed on the Exhibit Index set forth immediately following Item 16 are filed or furnished as part of this Annual Report on Form 10-K are set forth on the Exhibit Index immediately following our financial statements.Report. The Exhibit Index is incorporated herein by reference. Incorporated by Reference Exhibit
Number Description of Exhibit Form File Number Date of
Filing Exhibit
Number Filed
Herewith 2.1 * Agreement and Plan of Merger dated as of July 23, 2018, by and among Nabriva Therapeutics plc, Zuperbug Merger Sub I, Inc., Zuperbug Merger Sub II, Inc., Zavante Therapeutics, Inc. and Cam Gallagher, solely in his capacity as Stockholder Representative 8-K 001-37558 07/25/2018 2.1
3.1
Memorandum and Articles of Association of Nabriva Therapeutics plc.
8-K
001-38132
06/26/2017
3.1
10.1
Form of Indemnification Agreement.
8-K
001-38132
06/26/2017
10.1
10.2
#
2017 Share Incentive Plan, as Amended.
10-Q
001-37558
11/09/2017
10.2
10.3
#
Stock Option Plan 2007, as Amended.
8-K
001-38132
06/26/2017
10.2
10.4
#
Stock Option Plan 2015, as Amended.
8-K
001-38132
6/26/2017
10.3
10.5
Lease Agreement, dated March 16, 2007, by and between Nabriva Therapeutics AG and CONTRA Liegenschaftsverwaltung GmbH
F-1
333-205073
06/18/15
10.4
Incorporated by Reference Exhibit
NumberDescription of Exhibit Form File Number Date of
FilingExhibit
NumberFiled
Herewith101.CAL XBRL Taxonomy Calculation Linkbase Document X
101.DEF
XBRL Taxonomy Extension Definition Linkbase Document
X
101.LAB
XBRL Taxonomy Label Linkbase Document
X
101.PRE
XBRL Taxonomy Presentation Linkbase Document
X
#
Management contract or compensatory plan or arrangement filed in response to Item 15(a)(3) of the Instructions to the Annual Report on Form 10-K.
*
Confidential treatment was granted for certain portions that are omitted from this exhibit. The omitted information has been filed separately with the U.S. Securities and Exchange Commission (the "SEC") pursuant to the registrant's application for confidential treatment. In addition, schedules have been omitted from this exhibit pursuant to Item 601(b)(2) of Regulation S-K. A copy of any omitted schedule will be furnished supplementally to the SEC upon request; provided, however, that the registrant may request confidential treatment for any document so furnished.
**
Confidential treatment has been requested as to certain portions, which portions have been omitted and separately filed with the Securities and Exchange Commission.Not Applicable.SIGNATURESNABRIVA THERAPEUTICS PLC NABRIVA THERAPEUTICS AG Date: March 24, 2017Colin BroomColin Broom
Theodore Schroeder
Chief Executive OfficerColin Broom
Theodore SchroederColin Broom24, 2017(Principal Executive Officer)
/s/ GARY SENDER
Gary SenderGary Sender
Chief Financial Officer(Principal (Principal Financial and Accounting Officer)
March 24, 2017
/s/ DANIEL BURGESS
Daniel BurgessDaniel Burgess
Chairman of the Supervisory Board
March 24, 2017
/s/ COLIN BROOM
Colin Broom
Director
March 12, 2019
/s/ Axel Bolte
Carrie Bourdow
Director
March 12, 2019Axel Bolte
/s/ GEORGE TALBOT
George TalbotDeputy Chairman of the Supervisory Board
Director
March 24, 2017
/s/ CHARLES A. ROWLAND JR.
Charles A. Rowland Jr.
Director
March 12, 2019Chau Khuong
Stephen WebsterDirector March 12, 2019 Chau KhuongSupervisory Board MemberMarch 24, 2017
/s/ George TalbotGeorge TalbotSupervisory Board MemberMarch 24, 2017/s/ Charles RowlandCharles RowlandSupervisory Board MemberMarch 24, 2017/s/ Stephen WebsterStephen WebsterSupervisory Board MemberMarch 24, 2017/s/ MARK CORRIGAN
Mark Corrigan
DirectorMark CorriganSupervisory Board Member
March 24, 2017EXHIBIT INDEX
Number
Number
Filing
Number
Herewith101.LABXBRL Taxonomy Label Linkbase DocumentX101.PREXBRL Taxonomy Presentation Linkbase DocumentX# Management contract or compensatory plan or arrangement filed in response to Item 15(a)(3)the Instructions to theAnnual Report on Form 10-K.ContentsNabriva Therapeutics AGConsolidated Audited Financial StatementsF-2 20152017 and 2016 2018F-3 F-4 F-5 2014, 20152016, 2017 and 2016 2018F-6 F-7