VALLON PHARMACEUTICALS,
| Page No. | |||||||||
ITEM 5. | |||||||||
ITEM 9A. | |||||||||
ITEM 10. | |||||||||
ITEM | |||||||||
ITEM 12. | |||||||||
ITEM | |||||||||
ITEM 14. | |||||||||
ITEM 15. | |||||||||
SPECIAL
Although•our history of losses and need for additional capital to fund our operations, our inability to obtain additional capital on acceptable terms, or at all, our ability to continue as a going concern, and our need to liquidate if we believe that we havefail to obtain adequate funding, which could result in our stockholders receiving no value for their investment;
Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified, you
This Annual Report includes trademarks and registered trademarks of Vallon Pharmaceuticals, Inc. Products or service names of other companies mentioneddocuments that we reference in this Annual Report and have filed with the U.S. Securities and Exchange Commisstion (the SEC) as exhibits to the Annual Report with the understanding that our actual future results, levels of activity, performance, and events and circumstances may be trademarks or registered trademarks of their respective owners.
materially different from what we expect.
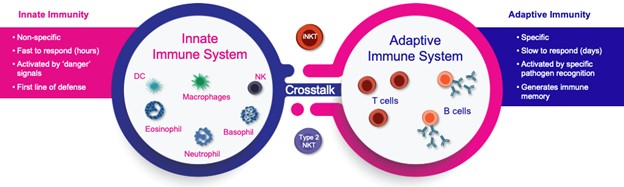
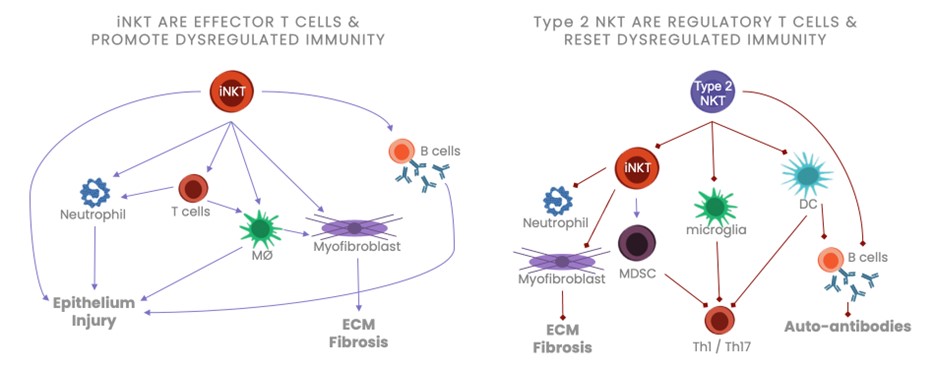
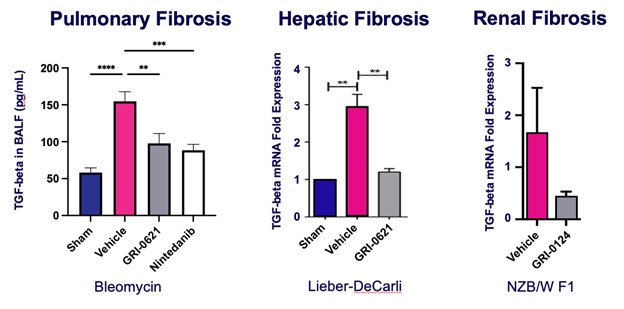
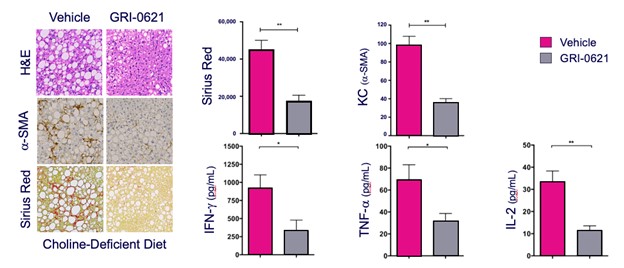
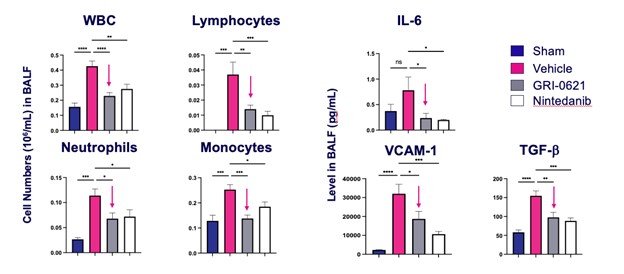
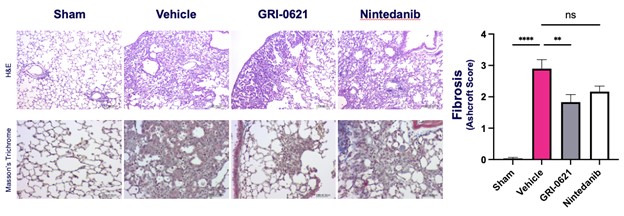
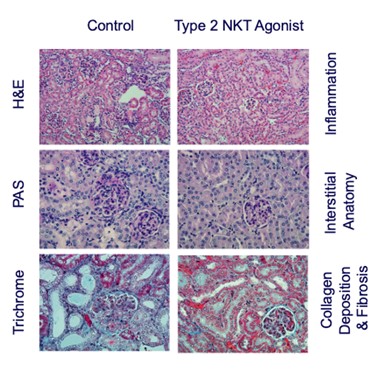
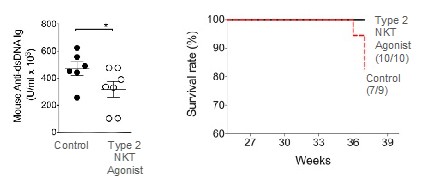
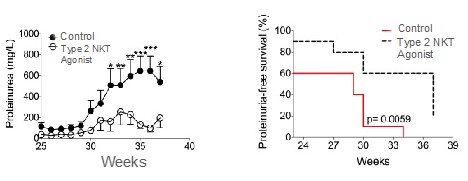
records conducted by researchers at the Mayo Clinic and presented in 2019 found that the adoption of pirfenidone and nintedanib by IPF patients was approximately 10% for each therapy, supporting the earlier observation that the majority of IPF patients are not actively being treated. Despite this, total worldwide sales of pirfenidone and nintedanib in 2019 were over $1.2 billion and $1.6 billion, respectively. proprietary technology and information commercially or strategically important to our business. We Brand patent. The penalties.primarily focused on discovering, developing, and commercializing innovative therapies that target serious diseases associated with dysregulated immune responses leading to inflammatory, fibrotic, and autoimmune disorders. Our goal is to be an industry leader in developing therapies to treat these diseases and to improve the developmentlives of patients suffering from such diseases.commercializationgamma selective agonist, that is approved in the United States for topical treatment of proprietary biopharmaceutical products.psoriasis and acne. As of December 31, 2023, it has been evaluated in over 1,700 patients as an oral product for up to 52-weeks. We are developing prescription drugs for central nervous system (“CNS”) disorders and our current focus is the development of drugs with lower potential for abuse than currently available drugs. Our clinical-stage product currently under development is Abuse-Deterrent Amphetamine Immediate-Release (“ADAIR”), a proprietary, abuse-deterrent oral formulation of immediate-release (short-acting) dextroamphetamineGRI-0621 for the treatment of attention-deficit/hyperactivity disorder (“ADHD”)severe fibrotic lung diseases such as idiopathic pulmonary fibrosis (IPF), a life-threatening progressive fibrotic disease of the lung that affects approximately 140,000 people in the United States, with up to 40,000 new cases per year in the United States. Some estimate that IPF affects 3 million people globally. While there are currently two approved therapies for the treatment of lung fibrosis, neither has been associated with improvements in overall survival, and narcolepsy.both therapies have been associated with significant side effects leading to poor therapeutic adherence. In preliminary data from our trials to date with GRI-0621, and earlier trials with oral tazarotene, we have observed GRI-0621 to be well-tolerated and to inhibit iNKT cell activity in subjects. We and others have shown that activated iNKT are upregulated in IPF, primary sclerosing cholangitis (PSC), non-alcoholic steatohepatitis (NASH), alcoholic liver disease (ALD), systemic lupus erythematosus disease (SLE), multiple sclerosis (MS), ulcerative colitis (UC) patients, as well as other indications. In these patients activated iNKT cells are correlated with more severe disease. The U.S. Food and Drug Administration (FDA) has cleared our IND application for GRI-0621 for the treatment of IPF and we plan to evaluate GRI-0621 in a randomized, double-blind, multi-center Phase 2a biomarker study, for which we commenced enrollment in December 2023. We expect topline results from this trial to be available in the second half of 2024.
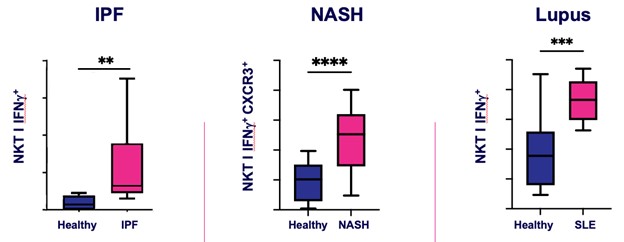
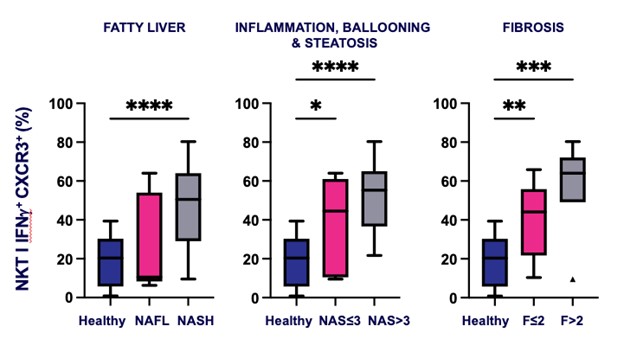
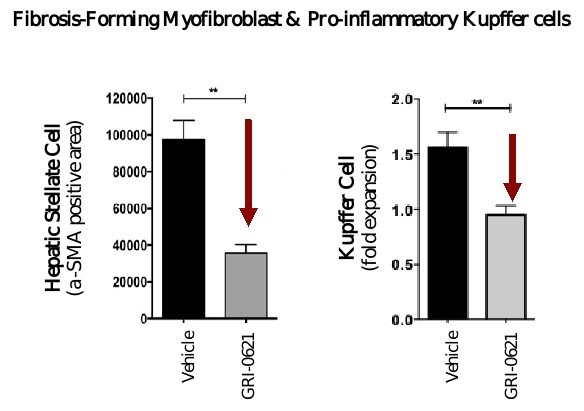
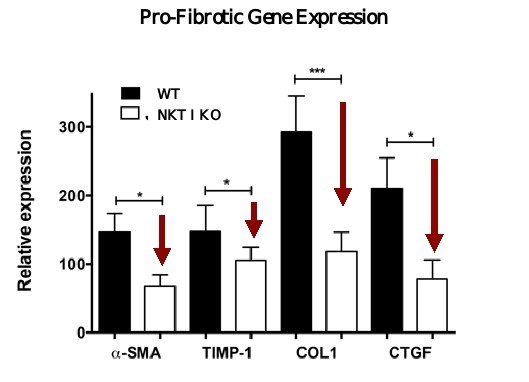
5 million Americans abuse60% of patients dosed with nintedanib have diarrhea and approximately 14% experience elevated levels of liver enzymes. Approximately 30% of patients treated with pirfenidone have skin rash, and approximately 9% experience photosensitivity, both of which can lead to dose reductions or discontinuations. Both agents have some efficacy in patients with more advanced disease, but high rates of discontinuations due to adverse events in these frailer patients limit their use. A survey of 290 physicians published by a third party in 2017 found that over half of IPF patients are not being treated with either agent for multiple reasons, including physicians not having sufficient confidence in clinical benefit and concerns about safety. A retrospective cohort analysis of prescription ADHD stimulants annually.intendare developing GRI-0621 as an oral gel capsule formulation to develop ADAIR for registration throughtreat IPF patients. GRI-0621 is differentiated from current IPF therapies because it is designed to reset the Section 505(b)(2) approval pathway, which we expectdysfunctional immune response driving disease by inhibiting the activity of iNKT cells, as opposed to obviate the need for large Phase 2 and Phase 3 efficacy and safety studies. See the sections entitled “Business — Section 505(b)(2) Pathway” and “Business — Clinical Development” in this Annual Report for more information regarding Section 505(b)(2)targeting a symptom of the FDCA. Althoughdisease that is downstream of the dysregulated immune response. GRI-0621 has been evaluated as an oral formulation in approximately 1,700 psoriasis, acne, and liver disease patients and in those patient populations and studies, the molecule was well tolerated with typical reported adverse events associated with hypervitaminosis A (headache, back pain, foot pain, cheilitis, hyperglycemia, arthralgia, myalgia, joint disorder, nasal dryness, dry skin, rash, and dermatitis).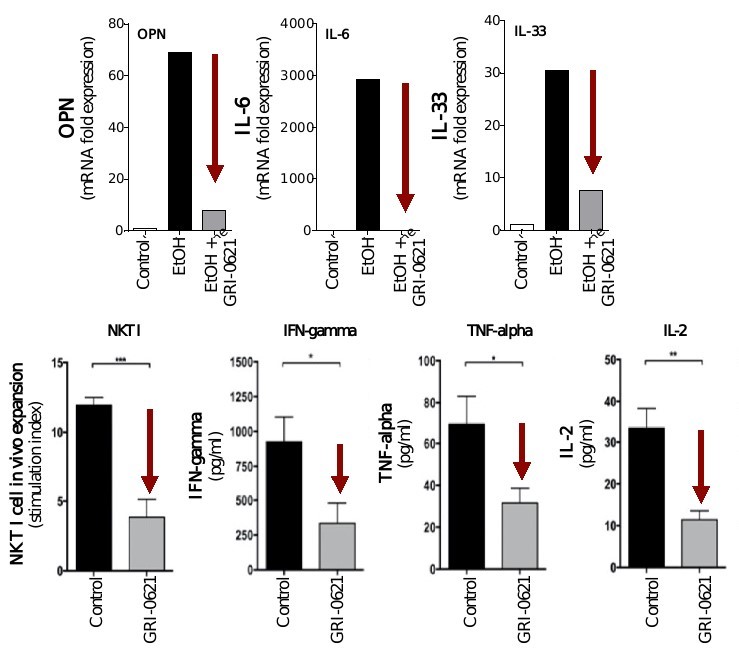
doesregarding the design of NASH clinical studies. In this limited number of patients, GRI-0621 was observed to be well tolerated and showed improvements in liver function tests, serum CK-18, and in iNKT cell activity, however, the study was underpowered to meet its endpoints with statistical significance. Adverse events were generally mild and consistent with RARb g agonism (see table below).ALL-CAUSE PLACEBO (n=4) GRI-0621 4.5mg (n=4) GRI-0621 6.0mg (n=5) SERIOUS TEAEs 0 0 0 GRADE 1 TEAEs 0 0 0 GRADE 2 TEAEs 0 0 0 GRADE 3/4/5 TEAEs 0 0 0 TREATMENT RELATED CHELITIS 0 0 0 NASEAU 0 0 0 DRY SKIN 0 0 0 PURITIS 0 0 0 HEADACHE 0 0 0 MYLAGIA 0 0 0 HYPERTENSION 0 0 1* GASTROENTERITIS 0 0 0 TONSILITIS 0 0 1* CREATINE PHOSPHOKINASE 0 0 0 LACTATE DEHYDROGENASE 0 0 0 POTASSIUM 0 0 0 approveown manufacturing facilities for producing any preclinical study or clinical trial product supplies. We rely on a limited number of suppliers for drug product and engage a single manufacturer to produce our formulated GRI-0621 drug usingproduct for clinical studies, as is standard industry practice in early to mid-stage clinical development. If these suppliers are unable to supply to us in the Section 505(b)(2) pathway until submissionquantities we require, or at all, or otherwise default on their supply obligations to us, we may not be able to obtain alternative supplies from other suppliers on acceptable terms, in a timely manner, or at all. In addition, if we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and acceptanceprocedures that comply with quality standards and with all applicable regulations and guidelines. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturer or manufacturing process will produce our product candidate according to the specifications previously submitted to the FDA or another regulatory authority. We may be unsuccessful in demonstrating the comparability of clinical supplies which could require the conduct of additional clinical trials. The delays associated with the verification of a new drug application (“NDA”), based on discussions heldmanufacturer could negatively affect our ability to develop product candidates in a timely manner or within budget.FDA at a pre-IND meeting in January 2017 and the minutes from such meeting,IPFbelieve the Section 505(b)(2) regulatory pathway is appropriate andcommenced enrollment for our Phase 2a trial. This trial will be acceptablea twelve-week, multicenter, multinational, randomized, placebo-controlled trial in approximately 36 patients with IPF. A 4.5 mg dose will be compared to the FDA. We expectplacebo over twelve weeks of treatment in subjects with a confirmed diagnosis of IPF on background therapy. Subjects will complete a screening visit to request additional labeling basedevaluate their medical history, present condition, laboratory assessments, comorbidities, and concomitant medications. Based on studies that demonstrate the abuse-deterrent characteristicsthese findings, subjects will be randomly assigned to one of the producttwo treatment arms: 4.5 mg of GRI-0621 or placebo in a 2:1 randomization. Weekly visits out to twelve weeks will evaluate safety, pharmacokinetics, and efficacy/mechanism of action of GRI-0621 as they relate to snorting, and possibly IV injection. While dextroamphetamine is approvedassessed by the activation of iNKT cells from both blood at weeks 6 and 12 and bronchi-alveolar lavage fluid at week 12. As a secondary endpoint, various biomarkers will also be evaluated to support theour reformulation, ADAIR,in designing the registration program moving forward.not. Prescription drug abuse is a large and growing problemthe most common type of lupus, affecting between 160,000 - 200,000 patients in the United States, and globally.We filed our Investigational New Drug (“IND”as many as 24,000 people in the United States are diagnosed with the disease each year. SLE predominantly affects women and often starts between the ages of 15 and 44. SLE is an autoimmune disease in which the immune system attacks its own tissues, causing widespread inflammation and tissue damage in the affected organs. It can affect the joints, skin, brain, lungs, kidneys, and blood vessels. There is no cure for lupus, but medical interventions and lifestyle changes can help control it. While people of all races can have the disease, African American women have a three-times higher number of new cases than white, non-Hispanic women. African American women tend to develop the disease at a younger age than white, non-Hispanic women and develop more serious and life-threatening complications. It is also more common in women of Hispanic, Asian and Native American descent. Adherence to treatment regimens is often a problem, especially among young women of childbearing age. Because SLE treatment may require the use of strong immunosuppressive medications that can have serious side effects, female patients must stop taking the medication before and during pregnancy to protect unborn children from harm.applicationan antibody that targets B lymphocyte stimulator, for ADAIRthe treatment of mild to moderate SLE in June 2018combination with standard therapy, providing additional clinical validation of the therapeutic benefit of B cell-targeted therapy for autoimmune diseases. However, the modest therapeutic benefit of Benlysta® and delayed onset of disease intervention indicate the INDneed for additional therapeutic strategies to inhibit overactive B cells. In 2021, the first-in-class type 1 interferon receptor antibody, anifrolumab, the first new drug for the disease in a decade, was clearedapproved for adults with moderate to severe disease who are receiving standard therapy.July 2018. Subsequently,conjunction with complement-mediated injury owing to pathogenic antibodies.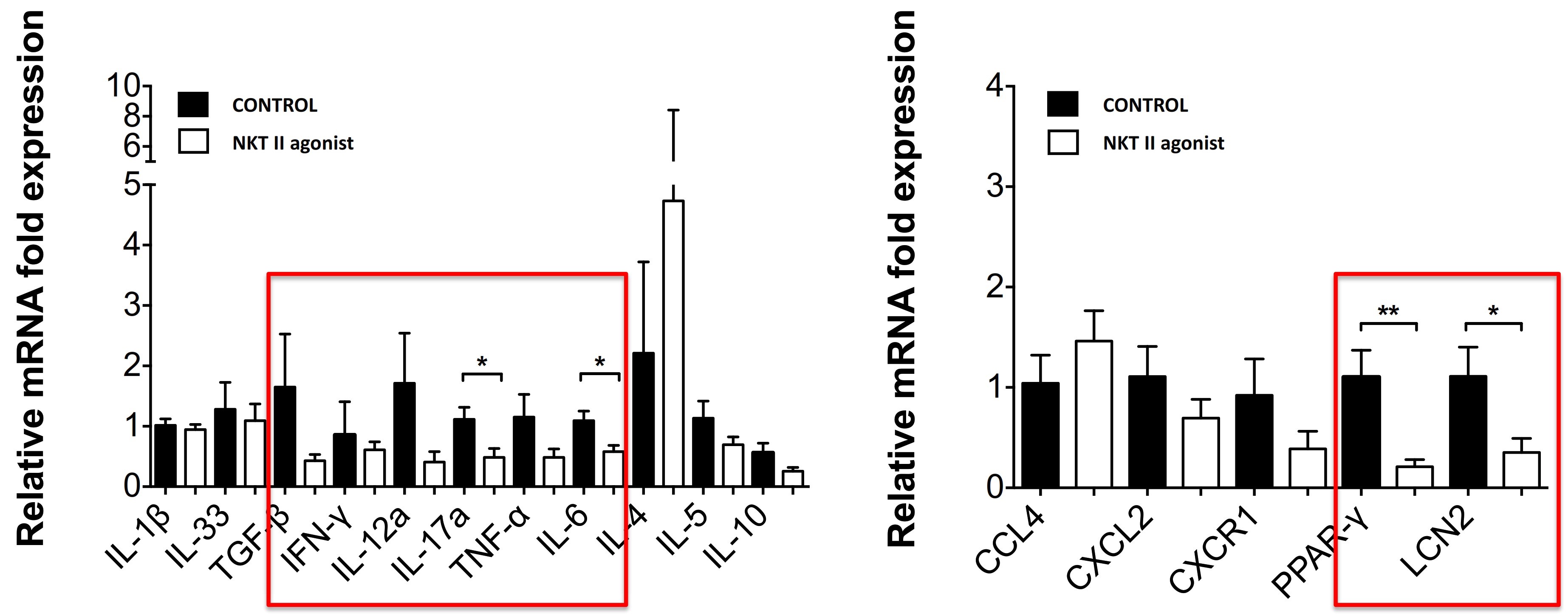
have successfully completedrequire, or at all, or otherwise default on their supply obligations to us, we may not be able to obtain alternative supplies from other suppliers on acceptable terms, in a timely manner, or at all. In addition, if we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturer or manufacturing process will produce our product candidate according to the specifications previously submitted to the FDA or another regulatory authority. We may be unsuccessful in demonstrating the comparability of clinical supplies which could require the conduct of additional clinical trials. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop product candidates in a timely manner or within budget.pivotal bioequivalence studytrial upon completion of ADAIRthe toxicology program for GRI-0803. Assuming positive results, we anticipate filing an IND in the first half of 2024. The Single Ascending Dose (SAD) trial will be run in healthy volunteers. Up to six doses will be evaluated in cohorts of 12 subjects with 10 receiving a dose of GRI-0803 and a Phase 1 food effect study.two receiving placebo. The bioequivalence study enrolled 24safety in each cohort will be evaluated with an Independent Safety Review Board (ISRB) along with the GRI clinical management. After completion of the first cohort, subsequent cohorts will begin within two weeks of dosing the previous cohort. Pharmacokinetics and safety will be the primary endpoint of the SAD trial. The completion of this trial should take approximately three months from when the first cohort is dosed.food effect study enrolled 22 subjects. Both studies were conducted by Altasciences, a contract research organization (“CRO”).In 2019, we conducted a Phase 1 proof-of-concept intranasal human abuse potential study designedtwo highest doses will be completed in patients with SLE. Safety and multi-dose pharmacokinetics will be the primary endpoint of thecompare ADAIR when insufflated (snorted) as compared to the reference comparator, crushed immediate release dextroamphetamine sulfate tablets. The study enrolled 16 subjects and was conducted at a single site by BioPharma Services, a CROcomplete with experience conducting similar trials. The study measured the pharmacokinetic levels of dextroamphetamine of the two compounds when snorted, the subjective “drug-liking” of the two drugs, and the willingness of recreational drug users to take each product again. Thetopline results of this study demonstrated that as compared to standard dextroamphetamine, ADAIR, when snorted, demonstrated an attenuated pharmacokinetic profile and lower drug liking and other abuse liability scores, using standard measures for human abuse potential studies. We have used the results of this proof of concept abuse study to design a larger intranasal abuse study that we will conduct prior to seeking approval of ADAIR. We designed the study to follow the model usedavailable late in intranasal abuse studies that have been conducted for abuse deterrent opioids and following guidance issued by the FDA for such studies. We began enrollment of subjects in this pivotal abuse study during the fourth quarter of 2020.We recently completed2024.preclinical embryofetal study which showed no evidencestrong emphasis on proprietary products. While we believe that our technology, the expertise of developmental effectsour management team, clinical capabilities, research and no clinical observations other than those associateddevelopment experience and scientific knowledge provide us with competitive advantages, we face increasing competition from many different sources, including biotechnology and biopharmaceutical companies, academic institutions, governmental agencies, and public and private research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the pharmacological effects of dextroamphetamine. Wefuture.conducting a 13-week preclinical toxicology study on the final formulation of ADAIR. We also plan to conduct additional preclinical studies of unintended routes of administration such as IV and intranasal administration.On January 6, 2020, Vallon entered into a license agreement with Medice, who is affiliated with one of our principal stockholders, Salmon Pharma, and represented by one member of our board of directors, which grants Medice an exclusive license, with the right to grant sublicenses, to develop, use, manufacture, market and sell ADAIR throughout Europe. Medice currently markets several ADHD products in Europe and is the ADHD market leader in Europe based on branded prescription market share. Medice is responsible for obtaining regulatory approval of ADAIR in the licensed territory. Under the license agreement, Medice paid Vallon a minimal upfront payment and will pay milestone payments of up to $6.3 million in the aggregate upon first obtaining regulatory approval to market and sell ADAIR in any country, territory or region in the licensed territory and upon achieving certain annual net sales thresholds. Medice will also pay tiered royalties on annual net sales of ADAIR at rates in the low double-digits. The initial term of the license agreement will expire five years after the date on which Medice first obtains regulatory approval in any country, territory or region in the licensed territory.We plan to develop other abuse-deterrent products that have potential for abuse in their current forms, beginning withpursuing the development of an abuse deterrent formulation of Ritalin® (“ADMIR”), for which we are conducting formulation development work.The U.S. market for ADHD treatment was estimated to be approximately $9 billion annually, which accounted for over 80% of the global ADHD market in 2019, and the European Union (“EU”) market for ADHD treatment was estimated to be approximately $700 million annually. We plan to target the U.S. ADHD market once we receive FDA approval of ADAIR, followed by the EU market for ADHD with our partner, Medice, who is affiliated with one of our principal stockholders, Salmon Pharma, and represented by one member of our board of directors, once regulatory approval has been granted in the EU.The ADAIR assets were acquired by us on June 22, 2018 pursuant to the terms and conditions of the Amended and Restated Asset Purchase Agreement with Arcturus Therapeutics, Ltd. (“Arcturus”), and Amiservice Development Ltd., dated as of June 22, 2018 (the “Asset Purchase Agreement”). In exchangeproducts for the ADAIR assets, we issued 843,750 sharestreatment of our common stock to Arcturus, which comprised 30% of our then-outstanding common stock on a fully diluted basis.Reverse SplitOn February 10, 2021, the Company filed a certificate of amendment to its amended and restated certificate of incorporation with the Secretary of State of the State of Delaware, which effected a one-for-40 reverse stock split (the “reverse split”) of its issued and outstanding shares of common stock at 11:59 PM Eastern Time on that date. As a result of the reverse split, every 40 shares of common stock issued and outstanding were reclassified into one share of common stock. No fractional shares were issued in connection with the reverse split and any fractional shares were rounded up to the nearest whole share.The reverse split did not change the par value of the common stockconditions GRI is also targeting, or the authorized number of shares of common stock. The reverse split affected all stockholders uniformly and did not alter any stockholder’s percentage interest in equity. All outstanding options and other securities entitling their holders to purchase or otherwise receive shares of common stock have been adjusted as a result of the reverse split, as required by the terms of each security. The number of shares available to be awarded under the Company’s 2018 Equity Incentive Plan have also been appropriately adjusted.All share and per share amounts contained in this Annual Report on Form 10-K give retroactive effect to the reverse split.Our Strategy and PipelineStimulant abuse is a large and growing public health challenge, yet the immediate-release segment of the ADHD market is entirely devoid of any abuse-deterrent products. We intend to address this need by through our abuse-deterrent pharmaceutical products, such as ADAIR and other products we opt to pursuemay target in the future, including ADMIR. IPF, SLE, MS, UC, PSC and NASH. While we know of no other companies currently in clinical development targeting NKT cells as a method of treating any of the above conditions, companies that we are aware of that are targeting the treatment of these diseases include large companies with significant financial resources such as:following table summarizeskey competitive factors affecting the success of our current product candidate portfolio:
Our near-term strategic milestones include:· seeking the necessary regulatory approvals to complete the clinical development of ADAIRcandidates are likely to be efficacy, safety, cost, and convenience. Many of our competitors, either alone or with their collaborators, have significantly greater resources, established presence in the market, expertise in research and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and reimbursement and marketing approved products than we do. These competitors also compete with us in recruiting and retaining qualified scientific, sales, marketing, and management personnel, establishing clinical trial sites and patient registration for the treatment of ADHD and, if successful, file for marketing approval in the United States and other territories;·preparing to commercialize ADAIR by establishing independent distribution capabilities or in conjunction with other biopharmaceutical companies in the United States and other key markets, such as the license agreement with Medice;· commencing development of other abuse-deterrent products such as ADMIR; and·continuing our business development activities and seek partnering, licensing, merger and acquisition opportunities or other transactions to further develop our pipeline and drug-development capabilities and take advantage of our financial resources for the benefit of increasing stockholder value.Section 505(b)(2) PathwayNDAs for most new drug products are based on two adequate and well-controlled clinical trials, which must contain substantial evidence of the safety and efficacy of the proposed new product. These applications are submitted under Section 505(b)(1) of the FDCA. The FDA is, however, authorized to approve an alternative type of NDA under Section 505(b)(2) of the FDCA. This type of application allows the applicant to rely, in part, on the FDA’s previous findings of safety and efficacy for a similar product, or published literature. Specifically, Section 505(b)(2) applies to an NDA for a drug for which the investigations to show whether the drug is safe and effective and relied upon by the applicant for approval of the application “were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted.”Thus, Section 505(b)(2) authorizes the FDA to approve an NDA based in part on safety and effectiveness data that were not developed by the applicant. Section 505(b)(2) may provide an alternate and potentially more expeditious pathway to FDA approval for new or improved formulations or new uses of previously approved products. If the Section 505(b)(2) applicant can establish that reliance on the FDA’s previous approval is scientifically appropriate, the applicant may eliminate the need to conduct certain preclinical studies or clinical trials of the new product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new drug candidate for all or some of the label indications for which the referenced product has been approved, as well as in acquiring technologies complementary to, or necessary for, any new indication sought byour programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Additional mergers and acquisitions may result in even more resources being concentrated in our competitors.Section 505(b)(2) applicant.expect thatseek to obtain and maintain, patent rights intended to cover the technologies incorporated into, or used to produce, our clinical trials described undertherapeutic candidates, the section entitled “Business — Clinical Development” will provide sufficient data to support an NDA filing with the FDA.Prescription Stimulant Abusecompositions of matter of our therapeutic candidates and MisuseAbusetheir methods of use and MisuseStimulants are among the most widely abused substances. This classmanufacture, asdrugs includes amphetamines and methylphenidate. Both of these substances are placed in Schedule II of the U.S. Controlled Substances Act (“CSA”) and the rules and regulation of the U.S. Drug Enforcement Administration (“DEA”), which is reserved for drugs that carry the highest risk of abuse and dependence that have been approved for medicinal use.While the most severe public health and societal problems related to stimulants result from abuse of illicitly manufactured stimulants including methamphetamine, and various synthetic stimulants, prescription stimulants are also widely misused and abused for non-medical uses.·Abuse — means the harmful or hazardous use of psychoactive substances, including alcohol and illicit drugs, and may include misusing a prescribed drug, through snorting, smoking or injecting, that is meant to be administered orally, to “get high” or produce “euphoria.”·Misuse — means the use of a substance or drug for a purpose not consistent with legal or medical guidelines. For example, ADHD medication may be misused through taking high dosages of the drug to enhance alertness and counteract fatigue and sleepiness in order to meet occupational demands, increase alertness while driving, or improve academic performance. Misuse and/or abuse can produce severe adverse consequences, and on rare occasions also death, and contributes to diversion of medicines from prescribed users, as illicit marketing. Furthermore, nonmedical use of prescription stimulants, even for the intent of occasional enhancement of alertness and performance, canother inventions that are important to our business. We also leadseek to more harmful patterns of use of stimulants and other addictive substances.PrevalenceAccording to a 2017 report by the National Survey on Drug Use and Health (the “NSDUH”) over 5 million individuals ages 12 yearsobtain strategic or oldercommercially valuable patent rights in the United States misused prescription stimulants in the previous year. This figure has been rising over time and this represents approximately 2%other jurisdictions.the U.S. population in that age group. Rates of misuse of prescription stimulants increase from age 12proprietary products and peak at age 21, where an estimated 10% of the population reported misuse of prescription stimulants, before declining in older adults.Harmful Effects of Stimulant AbuseAcuteAcute stimulant intoxication may result in a number of cardiovascular-related adverse events, including chest pain, myocardial infarction, palpitations, arrhythmias, thromboembolism, tachycardia, sinus bradycardia, ventricular premature depolarization, ventricular tachycardia degeneration (resulting in the need for defibrillation), asystole, peripheral vascular abnormalities, and/or sudden death from respiratory or cardiac arrest, as well as other adverse eventsrelated methods, such as strokes, seizures, pneumothorax, headaches,methods of use, we have filed patent applications representing six patent families. As of February 15, 2024, our patent estate included 12 issued United States patents, 2 United States pending non-provisional patent applications, 65 issued foreign patents and tinnitus. Acute stimulant intoxication is also associated16 foreign patent applications currently pending in various foreign jurisdictions.several psychiatric symptoms, including rambling speech, transient ideas, paranoid thoughts, auditory hallucinations, tactile hallucinations,claims directed to GRI-0621, and psychosis.High dosagesrelated methods of amphetamines and other stimulants can lead to aggressive or violent behavior (which may lead to self-harm or harm to others), intense temporary anxiety resembling panic disorder, or generalized anxiety disorder or mania, as well as paranoid thoughts and psychotic episodes that resemble schizophrenia. Taking extremely high doses of stimulants may also result in dangerously high body temperatures, irregular heartbeat, cardiovascular problems, and seizures.ChronicExtended abuse of stimulants can lead to psychological symptoms, such as hostility or paranoid psychosis. In addition to health status, the consequences of such substance use impact the individuals using drugs, their families and society at large, with severe repercussions possible at both the individual and public health level resulting from the chronic abuse and/or misuse of stimulants, such as teenage pregnancy, domestic violence, motor vehicle accidents, crime, poor work performance, and impaired personal relationships. Stimulant misuse is also correlated with a higher risk for substance use, with some evidence suggesting greater severity relative to controls, although it remains unclear whether the misuse of controlled medications precedes other substance use behaviors.Long-term stimulant abuse can lead to stimulant use disorder, which may be characterized by chaotic behavior, social isolation, aggressive behavior, and sexual dysfunction. Individuals exposed to amphetamine-type stimulants have been reported to develop stimulant use disorder in as rapidly as one week.Furthermore, individuals may increase their stimulant use in an effort to increase the euphoria, energy, and social and vocational interactions that they feel while using the medications. Individuals may crushsame to treat diseases, e.g. inflammatory conditions. Three United States and snort20 foreign patents (Australia, Brazil, Canada, China, Europe (validated in nine countries), Hong Kong, Japan, South Korea, Mexico, and Russia) were granted in this family. Patent applications in this family are pending in multiple jurisdictions, including, for example, the European Patent Organization, China, Japan, and Korea. Patents in this patent family are expected to expire in 2032, absent any patent term adjustments or inject the stimulants in orderextensions.produce even greater effects. Tolerance will develop with repeated use,GRI-0803 and individuals often increase the frequency and amountrelated methods of use in order to achieve a similar sense of euphoria.Once tolerance has developed, individuals may experience withdrawal symptoms (hypersomnia, increased appetite, and dysphoria) if they try to stop using the medication. Stimulant withdrawal can leadsame to depression, suicidal thoughts, irritability, anhedonia, emotional lability, and disturbances in attention and concentration. There may be temporary depressive symptoms that may meet the criteria for major depressive episode. The effects of withdrawal often lead individuals to abuse the medications again.About ADHD and Existing Treatment OptionsADHD Condition and ImpactADHD is defined as a persistent pattern of inattention and/or hyperactivity-impulsivity that interferes with functioning or development. ADHD causes significant impairment during a patient’s childhood, and throughout the patient’s lifespan, as well as increased morbidity, mortality and psychosocial adversity.Once believed to only affect children, ADHD is now known to persist into adolescence and adulthood in a sizeable number of cases. The following table illustrates how the nature of ADHD symptoms changes with age:ChildrenAdolescentsAdultsHyperactiveEasily distractedShifts activitiesAggressiveInattentiveEasily boredLow frustration toleranceImpatientImpulsiveApproximately 50-60% of adults who suffered from ADHD as children continue to have symptoms of the disorder as adults, with over 90% experiencing inattention symptoms and about 35% experiencing hyperactivity-impulsivity symptoms. As the majority of sufferers of ADHD age, their symptoms tend toward impatience, restlessness, boredom, and low concentration levels away from the more aggressive hyperactivity and impulsive behavior evident in children.Although the definitive causes of ADHD are still unclear, current research suggests that ADHD is caused by an interaction between environmental factors and genetic predispositions. Biologic factors that reportedly increase the risk of having ADHD include maternal smoking, drug or alcohol abuse during pregnancy, brain injury, and exposure to toxins.ADHD is believed to be one of the most under-diagnosed and under-treated mental health conditions facing children and adults. ADHD increases health risks, adverse social externalities and economic costs as illustrated in the following table. Despite the disorder being highly treatable, most adults with ADHD remain undiagnosed and untreated.The following table illustrates the effects on society when ADHD remains untreated:Healthcare SystemPatientFamilyIncreased ER visitsIncreased criminal activityIncreased divorce/separationIncreased car accidentsIncreased incarcerationMore sibling fightsSchool and OccupationSocietyEmployerHigh rates of expulsionSubstance use disorders:Increased parental absenteeism andHigh drop-out ratesHigher risk and earlier onsetlower productivityLower occupational statusLess likely to quit in adulthoodExisting Treatment OptionsCurrent management of ADHD frequently includes a combination of educational support, behavioral interventions, and pharmacotherapy. The current standard of care is the stimulant class of medications including immediate- and extended-release methylphenidate and amphetamine. Amphetamine products comprise the majority of the U.S. ADHD market and immediate-release amphetamines are the fastest growing segment of such market.Stimulant products represent more than 90% of prescriptions of ADHD products in the United States.MethylphenidateAmphetamine(Approx. 30%)(Approx. 60%)Immediate-ReleaseRitalin®Dexedrine®(Approx. 40%)Adderall®Extended-ReleaseConcerta®Adderall XR®(Approx. 50%)Ritalin LA® Vyvanse® Immediate-release (or short-acting) tablets and capsules release the active ingredient within a short period of time, such as 30 minutes and demonstrate efficacy that lasts for four to six hours. The patents covering most of these formulations have expired and most of these medications are now available in generic forms.Extended-release (or long acting) tablets and capsules release the active ingredient at a sustained and controlled release rate over the course of the day, typically demonstrating efficacy for 10 to 14 hours. Some of these are currently covered by patents and are not available in generic form.The four highest-selling drugs for the treatment of ADHD in 2019 on a worldwide basis are shown below: 2019 Global Sales
(in millions)Vyvanse® $ 2,514 Concerta® $ 696 Strattera® $ 243 Adderall XR® $ 223 As of 2019, the two best-selling medications were Vyvanse® and Concerta®, which are both extended-release stimulants. These and other extended-release stimulants are prescribed for both adults and children. For children in particular, the long-acting formulation is preferred because it eliminates the need for the child to take several doses during the school day.Despite the popularity of the long-acting drugs, we believe there is a growing market opportunity for immediate-release treatments among children and adults with ADHD. For instance, some patients taking the extended-release drugs benefit from the addition of a short-acting stimulant taken in the evening to supplement the medication given earlier in the day. This allows the patient to alleviate their ADHD symptoms for an evening meeting or class, without keeping the patient awake all night. In addition, the immediate-release products can be useful when evaluating whether an individual will be able to tolerate a particular stimulant or respond to a dosage titration.Finally, some individuals with ADHD prefer to manage their symptoms with medication only on an as-needed basis, and the immediate-release formulations give the patient more flexibility with the dosing frequency. For instance, many patients experience varying degrees of side effects to stimulant medication, including headaches, jitteriness, irritability, sleep problems, and decreased appetite, and some report that stimulants decrease their creativity and spontaneity. For these reasons, many adults — who now comprise more than 50% of the U.S. prescriptions for ADHD medication — prefer the short-acting formulations. Therefore, although short-acting stimulants are only approved for use in children and adolescents, part of our long-term plan involves seeking approval for use of short-acting stimulants in adults as well.ADHD MarketAccording to IQVIA (formerly, IMS Health), the U.S. market for ADHD treatment was estimated to be approximately $9 billion annually, which accounted for over 80% of the global ADHD market in 2019. The difference in market sizes between the U.S. and other countries is driven by different rates of diagnosis and treatment, different pricing, and the number of available brand name medications (non-U.S. markets are dominated by generic drugs). Global prevalence rates of the disease are estimated to be approximately 8-10% of school-aged children and approximately 4-5% of the adult population. Adult diagnosis and treatment, which has grown at approximately 10% annually over the last few years, is forecasted to grow in the near future due to increased disease awareness and less sociological stigmatization towards the condition. In thetreat diseases. Three United States the rate of treatment with prescription medications is approximately 70%and nine foreign patents (Canada, Europe (validated in childrenseven countries), and 45%Hong Kong) were granted in adults. In 2019, over 75 million prescriptions were filledthis family. Patent applications in this family are pending in the United States and European Patent Organization. Patents in this patent family are expected to expire in 2032, absent any patent term adjustment or extension.approved ADHD medications, whereas less than 44 million prescriptions were filled in 2009. The U.S. market is projected to continue to grow in mid-single digits, driven by an increased prescription rate for adult ADHD. The growth of immediate-release amphetamines averaged over 7% annually from 2014-19 and is projected to continue to grow faster than the overall ADHD market in the foreseeable future. In 2019, 28 million prescriptions were filledexample, in the United States, for immediate-release stimulants, suchUnited Arab Emirates, Brazil, China, Japan, Russia, Canada, Hong Kong and South Korea. Patents in this patent family are expected to expire in 2035, absent any patent term adjustment or extension.Adderallwe develop new technologies and Ritalin. Immediate release amphetamine stimulants,therapeutic candidates. As our business evolves, we may, among other activities, file additional patent applications in pursuit of our intellectual property strategy, to adapt to competition or to seize potential opportunities.segmentlaws of the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing of a non-provisional patent application. However, the term of United States patents may be extended for delays incurred due to compliance with the FDA requirements or by delays encountered during prosecution that are primarily targeting, currently represent approximately 30%caused by the United States Patent and Trademark Office (USPTO). For example, the Hatch-Waxman Act, permits a patent term extension for FDA-approved drugs of up to five years beyond the expiration of the ADHD medications market (prescriptions and patients) and continue to gain market share.international ADHD marketlength of the patent term extension is projected to grow at a faster rate than the U.S. market in part because disease recognition and acceptance is expected to increase in both Japan and Europe. The estimated growth rate for the non-U.S. markets is also higher duerelated to the recent launcheslength of major ADHD drugstime the drug is under regulatory review. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our therapeutic candidates receive FDA approval, we expect to apply for patent term extensions on patents covering those therapeutic candidates. We intend to seek patent term extensions in any jurisdiction where these are available and where we also have already been marketeda patent that may be eligible; however there is no guarantee that the applicable authorities, including the USPTO and FDA, will agree with our assessment of whether such extensions should be granted, and even if granted, the length of such extensions.Vyvanse and Intuniv.Potential for AbuseStimulant abuse is unique and challenging because the abuse and addiction risks of stimulants are not restricted to those who are prescribed the medications. Published data reports that stimulants are almost twice as likely to be diverted (i.e., given away or sold) as other scheduled medications, such as opioid, sleep or anxiety medications. It has been reported that between 25-60% of teenagers and college students with ADHD have been approached at some point to give away or sell their prescription stimulants and over 60% of college students with ADHD admit to having diverted their ADHD prescription medication.Approximately 90% of those who misuse/abuse stimulants do so with prescription amphetamines based on data from the NSDUH.The number of emergency room visits associated with non-medical use of prescription stimulants increased more than four-fold from 2004 to 2011, according to the Drug Abuse Warning Network (“DAWN”), and most of this increase was associated with amphetamine-based prescription stimulants.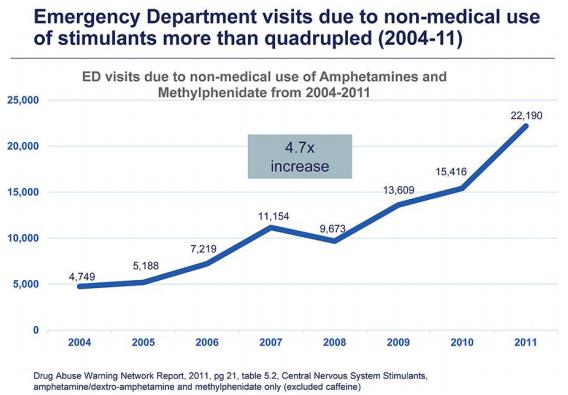
Speed of onset and route of administration has been accepted as being important in evaluating the potential for abuse of certain medications, such as stimulants.In general, the oral route is associated with lower abuse liability because of slower absorption rates and slower onset of effects compared to other routes of administration. Inhalation, snorting, and intravenous (“IV”) injection of drugs are associated with far more rapid absorption and faster onset of effects when compared to oral ingestion. In general, oral use of stimulants results in the slowest rate of absorption, while snorting is relatively faster; smoking and IV injection of stimulants evoke even more rapid absorption and more intense and rapid physiological and subjective responses. Published studies report that 40% or more of people who misuse or abuse prescription stimulants, do so by IV injection or snorting. These methods of abuse drive a more rapid increase in dopamine levels that drive the subjective, or re-enforcing effects of these drugs. Consequently, these abuse routes are thought to bring the abuser one step closer to addiction and dependence. In addition, the quick entry of the drug into the bloodstream increases the risk of chest pain, rapid / irregular heartbeat, heart attack, seizures, hallucinations, hostile/aggressive behavior, suicidal thoughts and behaviors, and stroke.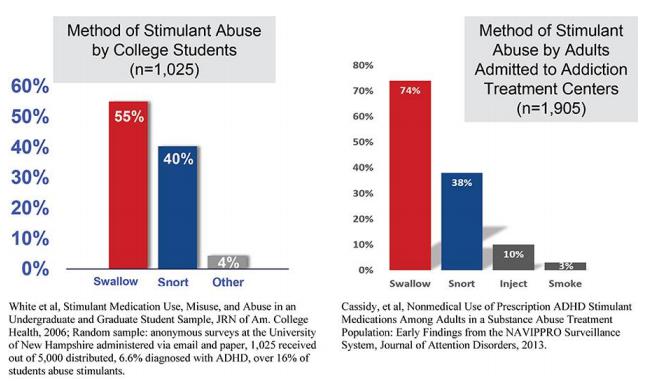
Immediate-release stimulants, including amphetamines, are more prone to abuse than extended-release stimulants and are the fastest growing market in the ADHD market in recent years. Amphetamine tablets are easy to crush into a powder suitable for snorting, or mixing with water and injecting. On the other hand, long-acting stimulant capsules are abused less frequently because they contain a combination of immediate-release and extended-release beads with different release profiles that are difficult to crush into a form that can be snorted, smoked, or mixed with water and injected.The rate of amphetamine use disorders doubled between 2010 and 2016: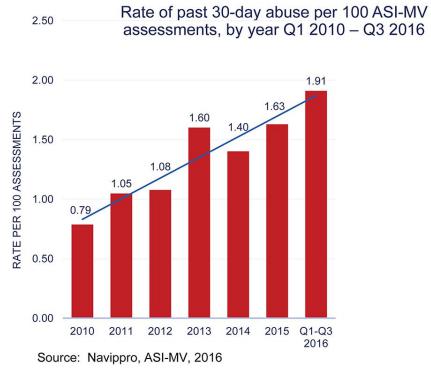
Amphetamine forms molecules that are highly soluble in lipids, which are then rapidly transported to the brain through the blood-brain barrier. Routes of administration that deliver the drug directly into the bloodstream and bypass the digestive system (i.e., snorting, smoking, and IV injection) would be expected to cause faster onset of psychoactive effects. Therefore, reducing the risk of abuse via snorting, smoking, and injection is a potential public health goal because the speed of the absorption of stimulants is an important determinant of a product’s abuse potential, as is the case for opioids, and is also related to the overall potential risks of the drug product.Many people who use amphetamines and other stimulants for recreational use prefer routes of administration that provide rapid onset of effects. In order to achieve its maximum pharmacologic effect, the largest quantity of drug must be delivered into the CNS in the shortest possible time. For instance, a published study found that the reinforcing properties of methylphenidate occur when the drug elicits a large and fast dopamine increase but has only therapeutic properties when there is a slow, steady-state increase in dopamine caused by the drug. This leads drug abusers to progress from relatively safe methods of self-administration, such as oral ingestion of marketed doses of stimulants, to increasingly higher dosages and more dangerous routes of administration, such as smoking, snorting, and injecting.Our SolutionsADAIRStimulant abuse is large and growing public health challenge, yet the immediate-release segment of the ADHD market is entirely devoid of any abuse-deterrent products. This unmet need led to the design of ADAIR as an oral formulation of an immediate-release dextroamphetamine. This included the development and in vitro testing of multiple formulations, followed by the selection of the optimal, proprietary formulation of ADAIR that is intended to introduce certain barriers to abuse of immediate-release dextroamphetamine.ADAIR is an oral, semi-solid, liquid-filled, hard gelatin capsule of dextroamphetamine sulfate, the active ingredient. This formulation resists manipulation and preparation for snorting, and provides meaningful barriers to injection — demonstrated through a set of abuse-deterrence studies conducted in collaboration with M.W. Encap Limited, an affiliate of Lonza Group AG.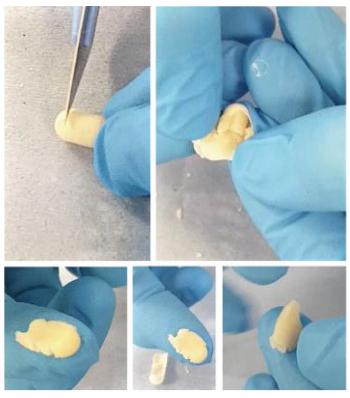
After subjecting ADAIR to grinding, crushing, or cutting the capsule following thermal pre-treatment, minimal quantities of particles could appreciably pass through a 500 micrometer (“µm”) filter (a particle size deemed suitable for snorting). In contrast, 42-47% of the physically manipulated immediate-release dextroamphetamine reference tablet could pass through a 500 µm filter, suggesting that it could be readily crushed and snorted.
We also subjected ADAIR to multiple forms of manipulation, but none of those yielded ADAIR particles that could be easily expelled from a syringe. ADAIR mixed in water yielded a viscous, cloudy material, which was usually impossible, and at other times difficult, to syringe. Texture analysis demonstrated that the force required to push the plunger with a manipulated ADAIR-filled syringe is far greater than that with manipulated immediate-release dextroamphetamine reference tablet.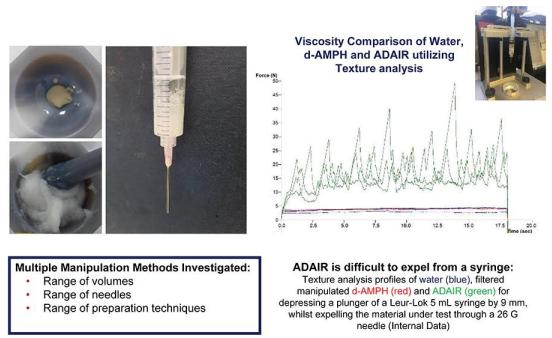
In comparison with the immediate-release dextroamphetamine reference tablet, ADAIR demonstrated reduced syringe-ability across a range of volumes of water (2, 5, and 10 milliliters), needle gauges (26, 23, 20, and 18 gauge), in ambient or hot water, and when passed through a cigarette filter.We believe these studies demonstrate that, as compared to the immediate-release dextroamphetamine reference tablet, ADAIR could display deterrence properties against abuse through snorting or IV injection.Our abuse-deterrent formulation may not meaningfully discourage oral ingestion to enhance occupational or academic performance, or misuse; however, depending on the properties of the formulation, an abuse-deterrent formulation could reduce the risks of adverse effects by anyone who would attempt to abuse it by snorting, smoking, or injecting, and reduce the contribution of prescription stimulants to problems associated with stimulant abuse.In addition, the general pharmacologic rationale for abuse-deterrent stimulants is similar to the rationale of abuse-deterrent opioids used to treat and manage pain as described in the FDA 2015 Guidance on Abuse-Deterrent Opioid. The FDA clearly articulated the rationale for the development of abuse-deterrent technologies, as well as cited its limitations, in its 2015 Guidance, pp. 1-2:Prescription opioid products are an important component of modern pain management. However, abuse and misuse of these products have created a serious and growing public health problem. One potentially important step towards the goal of creating safer opioid analgesics has been the development of opioids that are formulated to deter abuse. FDA considers the development of these products a high public health priority. Because opioid products are often manipulated for purposes of abuse by different routes of administration or to defeat extended-release (ER) properties, most abuse-deterrent technologies developed to date are intended to make manipulation more difficult or to make abuse of the manipulated product less attractive or less rewarding. It should be noted that these technologies have not yet proven successful at deterring the most common form of abuse — swallowing a number of intact capsules or tablets to achieve a feeling of euphoria. Moreover, the fact that a product has abuse-deterrent properties does not mean that there is no risk of abuse. It means, rather, that the risk of abuse is lower than it would be without such properties. Because opioid products must in the end be able to deliver the opioid to the patient, there may always be some abuse of these products.Although ADAIR is very difficult to manipulate into a form that can be snorted, it is not impossible to do so. In order to conduct human abuse studies, Vallon hired a third-party drug laboratory that was able to develop a time- consuming and laborious process to convert ADAIR into a form that could be insufflated. The medical literature reports that recreational abusers of prescription medications are typically not willing to spend more than a few minutes preparing a drug for misuse or abuse and our own research with recreational stimulant users documented that they would not be willing to spend more than 10-12 minutes preparing a drug like ADAIR for snorting. In addition, although ADAIR is difficult to solubilize into a form that can be injected, sophisticated drug abusers may be able to develop methods to manipulate ADAIR into a form that can be injected.Regulatory communications regarding the application of abuse-deterrent technologies for prescription stimulants continue to emerge. In 2014, Janet Woodcock, M.D., Director, Center for Drug Evaluation and Research, stated that the FDA encourages the development of abuse-deterrent formulations for controlled substances, while also noting that the science surrounding abuse-deterrent technology is relatively new. In public meetings, FDA officials have made comments related to interest in the application of abuse-deterrent technologies for stimulants, as well as other drugs of abuse. In practice, the FDA engages with sponsors of abuse-deterrent formulations on a product-by-product basis, sometimes requiring an abuse-deterrent assessment as part of the development to inform approval and labeling processes by the FDA. In September 2019, the FDA issued a Federal Register notice to seek public comment on the development and evaluation of abuse deterrent formulations (ADF) of ADHD stimulants and whether such products could play a role in addressing public health concerns related to prescription stimulant misuse and abuse signaling their interest in this field.Lastly, based on market research conducted by us in conjunction with U.S. health insurers, who collectively manage over 100 million covered lives, a strong majority of insurers are receptive of the ADAIR product concept and indicate that they would be willing to have ADAIR, if approved, placed on their prescription drug formulary and to reimburse the costs for ADAIR through their respective health insurance plans. In addition, the continuing and heightened publicity surrounding the national opioid epidemic continues to result in heightened sensitivity by many health care professionals to prescribe, and pharmacies to dispense, medications with the potential for abuse.Development of ADMIRADMIR is an abuse deterrent formulation of Ritalin for which we are conducting formulation development work. We have developed several prototype formulations that we are continuing to refine. If our formulation development work is successful, we anticipate requesting a pre-IND meeting with the FDA and filing an IND in 2021. ADMIR is designed to have abuse deterrent properties that are similar to ADAIR.Clinical DevelopmentWe aim to be the first company to introduce a proprietary abuse-deterrent immediate-release dextroamphetamine drug to the market and leverage our agility, flexibility, and know-how to utilize such a positiondeveloping. The processes for the benefit of patients, physicians, and our community.We filed our Investigational New Drug (“IND”) application for ADAIR in June 2018 and the IND was cleared in July 2018. Subsequently, we have successfully completed three Phase 1 clinical studies.Phase 1 Bioequivalence StudyIn December 2018, we completed a Phase 1 pivotal bioequivalence study of ADAIR, which was conducted by Altasciences. The study enrolled 24 subjects, who were dosed with 10 mg of ADAIR and reference dextroamphetamine orally on a single occasion for each study drug. There were no serious adverse events (“SAEs”) during the study. The primary objective of the study was to evaluate and compare the pharmacokinetics (“PK”) of ADAIR capsules to dextroamphetamine tablets under fasting conditions. The secondary objectives of the study were to evaluate the safety and tolerability of the test and reference formulations in healthy subjects. The study met the primary endpoint demonstrating bioequivalence and met the secondary endpoints.Food Effect StudyIn December 2018, we completed a Phase 1 food effect study of ADAIR, which was conducted by Altasciences. The study enrolled 22 subjects who were dosed with 10 mg of ADAIR orally twice, once when subjects were fasting and once when they had been fed. One SAE (miscarriage) was reported during the study. A 33-year-old African American female subject had an unplanned pregnancy reported one week after the last study drug dose. The miscarriage occurred at pregnancy day 35. The investigator considered the SAE possibly related to drug treatment. However, because of the timing, background incidence, and risk factors (including age and race), we concluded that this SAE was unlikely to be related to the study drug.The primary objective of this study was to evaluate and compare the PK of d-amphetamine from an abuse-deterrent capsule formulation of dextroamphetamine sulfate when dosed under fasting and fed conditions. The secondary objective was to evaluate the safety and tolerability of the investigational product in healthy subjects. The study met both the primary and secondary endpoints.Human Abuse Proof of Concept StudyI n November 2019, we completed a Phase 1 proof-of-concept intranasal human abuse potential study designed to compare ADAIR when insufflated (snorted) as compared to the reference comparator, crushed immediate release dextroamphetamine sulfate tablets. The study was conducted by BioPharma Services and enrolled 16 subject who received one dose of ADAIR and reference dextroamphetamine administered intranasally each at a dose of 30 mg. The primary objective was to assess safety and tolerability of manipulated ADAIR and crushed dextroamphetamine sulfate IR (“DEX”), when administered intranasally to non-dependent, recreational stimulant users. The secondary objectives were to evaluate and compare the PK profiles of ADAIR and DEX when administered intranasally to non-dependent, recreational stimulant users. The exploratory objectives of this study were to assess and compare abuse liability of ADAIR and DEX when administered intranasally to non- dependent, recreational stimulant to users. There were no SAEs in connection with this trial. The study met the primary, secondary and exploratory endpoints.The results of this study demonstrate that as compared to DEX, ADAIR, when snorted, demonstrated an attenuated pharmacokinetic profile and lower drug liking and other abuse liability scores, using standard measures for human abuse potential studies.We have used the results of this proof of concept abuse study to design a larger intranasal abuse study that we will conduct prior to seeking approval of ADAIR. We designed the study to follow the model used in intranasal abuse studies that have been conducted for abuse deterrent opioids and following guidance issued by the FDA for such studies.Preclinical Studies and Other Clinical Development PlansWe recently completed a preclinical embryofetal study which showed no evidence of developmental effects and no clinical observations other than those associated with the pharmacological effects of dextroamphetamine. We are currently conducting a 13-week preclinical toxicology study on the final formulation of ADAIR. We also plan to conduct additional preclinical studies of unintended routes of administration such as IV and intranasal administration.We plan to develop other abuse-deterrent products that have potential for abuse in their current forms, beginning with the development of an abuse deterrent formulation of Ritalin® (“ADMIR”), for which we are conducting formulation development work.We expect that our clinical trials described above will provide sufficient data to support an NDA filing with the FDA.Government Regulation and Product ApprovalClinical trials, the drug approval process, and the marketing of drugs are intensively regulatedobtaining regulatory approvals in the United States and in all major foreign countries. countries, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.(“FDCA”),(FDCA) and related regulations. Drugs are also subject to other federal, state, and local statutes andits implementing regulations. Failure to comply with the applicable U.S. regulatoryUnited States requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions. These sanctions could include the impositionbrought by the FDA Institutional Review Board (“IRB”)and the Department of a clinical hold on trials,Justice (DOJ), or other governmental entities, such as the FDA’s refusal to approve pending applications or supplements,NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil penalties and/or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, manufacture and marketing of biopharmaceutical products. These agencies and other federal, state, and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, distribution, record keeping, approval, advertising, and promotion of ADAIR or any other product we develop in the future.The FDA’s policies may change, and additional government regulations may be enacted that could prevent or delay regulatory approval of any candidate drug product or approval of new disease indications or label changes. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or abroad.Marketing Approvaldrugsdrug may be marketed in the United States generally involves the following:· laboratory and animal tests;·submission of an IND application, which must become effective before clinical trials may begin;·adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use or uses;·pre-approval inspection of manufacturing facilities and clinical trial sites; and·FDA approval of an NDA which must occur before a drug can be marketed or sold.��The testing and approval process requires substantial timepreclinical studies, such as laboratory tests, potentially animal studies and financial resources,formulation studies, in compliance with FDA regulations for Good Laboratory Practices (GLPs) and we cannot be certain that any approvals will be granted on a timely basis if at all.We will need to successfully complete additional clinical trials in order to be in a position to submit an NDA to the FDA. Future trials may not begin or be completed on schedule, if at all. Trials can be delayed for a variety of reasons, including delays in:·obtaining regulatory approval to commence a study;
other applicable regulations;·reaching agreement with third-party clinical trial sites and vendors and their subsequent performance in conducting accurate and reliable studies on a timely basis;·obtaining institutional review board approval to conduct a study at a prospective site;
We must reach an agreement with the FDA on the proposed protocols for our future clinical trials in the United States. A separate •submission to the FDA of an IND, which must become effective before human clinical trials may begin;
ADAIR
ADAIR was specifically designed Similarly, an IRB can suspend or terminate approval of a clinical trial if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product candidate has been associated with unexpected serious harm to limit abusepatients.
manufacturing facilities for the new product to determine whether the manufacturing processes and facilities comply with cGMPs. The development plan for ADAIR isFDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMPs and are adequate to conductassure consistent production of the product within required specifications.
An NDA would be filed to the FDA only after achieving successnevertheless may accept the data in eachsupport of an NDA if the above milestonesstudy was conducted in accordance with GCPs and any additional milestonesthe FDA is able to validate the data through an on-site inspection, if deemed necessary. Although the FDA generally requests that marketing applications be supported by some data from domestic clinical trials, the FDA may request.
Asaccept foreign data as the sole basis for marketing approval if (1) the foreign data are applicable to the United States population and United States medical practice, (2) the studies were performed by clinical investigators with similarrecognized competence, and (3) the data may be considered valid without the need for an on-site inspection or, if the FDA considers the inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means.
15
Finally, the FDA may designate a product for priority review if it is a drug that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. The FDA determines at the time that the marketing application is submitted, on a case-by-case basis, whether the proposed drug represents a significant improvement in treatment, prevention or diagnosis of disease when compared with other available therapies. Significant improvement may be illustrated by evidence of increased effectiveness in the treatment of a condition, elimination or substantial reduction of a treatment-limiting drug reaction, documented enhancement of patient compliance that may lead to improvement in serious outcomes, or evidence of safety and effectiveness in a new subpopulation. A priority review designation is intended to direct overall attention and resources to the evaluation of such applications, and to shorten the FDA’s goal for taking action on a marketing application from ten months to six months for an NDA for a new molecular entity from the date of filing.
Any products
The FDA may request, or we may propose, to implement a risk management program to educate physicians and parents or patients of appropriate use of ADAIR, and to monitor the real-world use and reports of abuse of ADAIR following its approval. Such risk management programs are common with many medications with abuse potential including many approved ADHD products.
Labeling, Marketing and Promotion
The FDA closely regulates the labeling, marketing, and promotion of drugs. While doctors are free to prescribe any drug approved by the FDA for any use, a company can only make claims relating to theproduct’s safety and efficacy of a drug that are consistent with FDA approval and may only actively market a drug only for the particular use and treatment approved by the FDA. effectiveness after commercialization.
Pediatric Research Equity Act
The Pediatric Research Equity Act (“PREA”) amended the FDCA to authorize the FDA to require certain research into drugs used in pediatric patients. The intent of the PREA is to compel sponsors whose drugs have pediatric applicability to study those drugs in pediatric populations, rather than ignoring pediatric indications for adult indications that could be more economically desirable. The Secretary of Health and Human Services may defer or waive these requirements under specified circumstances. The FDA may decide that an NDA will bespecific approved only following completion of additional pediatric studies.
Anti-Kickback and False Claims Laws
In the United States, the research, manufacturing, distribution, sale and promotion of drug products and medical devices are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare & Medicaid Services, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice, state Attorneys General, and other state and local government agencies. For example, sales, marketing, and scientific/educational grant programs must comply with the Anti-Kickback Statute, the False Claims Act, as amended, the privacy regulations promulgated under HIPAA, and similar state laws. Pricing and rebate programs must comply with the Medicaid Drug Rebate Program requirements of the Omnibus Budget Reconciliation Act of 1990, as amended, and the Veterans Health Care Act of 1992, as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws.
In the United States, we are subject to complex laws and regulations pertaining to healthcare “fraud and abuse,” including, but not limited to, the Anti-Kickback Statute, the federal False Claims Act, and other state and federal laws and regulations. The Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid.
The federal civil False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to or approval by the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. Violations of the False Claims Act can result in very significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multi-billion-dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. In addition, the federal civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), also created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the Anti-Kickback Statute a person or entity does not need to have actual knowledge of these statutes or specific intent to violate them in order to have committed a violation.
There are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. In addition, a similar federal requirement Section 6002 of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (the “Affordable Care Act”) commonly referred to as the “Physician Payments Sunshine Act” requires manufacturers to track and report to the federal government certain payments and “transfers of value” made to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members, made in the previous calendar year. There are a number of states that have various types of reporting requirements as well. These laws may affect our sales, marketing, and other promotional activities by imposing administrative and compliance burdens on us. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state, and soon federal, authorities.
Patient Protection and Affordable Health Care Act
In March 2010, the Affordable Care Act was enacted, which includes measures that have or will significantly change the way health care is financed by both governmental and private insurers. The fees, discounts, and other provisions of this law are expected to have a significant negative effect on the profitability of pharmaceuticals.
This legislation is expected to impact the scope of healthcare insurance, the insurance refunds from the insurance companies and possibly also on the costs of medical products.
Other Regulations
We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with such laws and regulations now or in the future.
Manufacturing
We do not currently own or operate any manufacturing facilities and we do not have any experiencein accordance with commercial-scale manufacturing. We currently rely, and expect to continue to rely for the foreseeable future, on a third-party manufacturer to produce our product candidates for preclinical and clinical testing, as well as for commercial manufacture if our product candidates receive marketing approval.
Although we do not have a long-term commercial supply arrangement in place with any of our contract manufacturers, it is our goal to contract with at least one manufacturer in the United States for the commercial supply of ADAIR for the U.S. market.
Our third-party manufacturers, their facilities, and all pharmaceutical products used in our clinical trials are required to comply with cGMP.cGMPs. The cGMP regulationscGMPs include requirements relating to the organization of personnel, buildings and facilities, equipment, control of components and drug product containers and closures, production and process controls, packaging and labeling controls, holding and distribution, laboratory controls, records and reports and returned or salvaged products. TheDrug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and some state agencies and are subject to periodic unannounced inspections by the FDA for compliance with cGMPs and other laws. Changes to the manufacturing facilities for our productsprocess are strictly regulated and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMPs and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers. Accordingly, manufacturers must meet, and continue to meet, cGMP requirementsexpend time, money, and FDA satisfaction before any product is approvedeffort in production and we can manufacture commercial products. Contract manufacturers often encounter difficulties involving production yields,quality control to maintain compliance with cGMPs and other aspects of quality control and quality assurance,assurance.
Salessuch applications, a generic manufacturer must rely on the preclinical and Marketing
We retain advisorsclinical testing previously conducted for a drug product previously approved under an NDA, known as the reference listed drug (RLD).
Medice License Agreement
Onsales and distribution of our products, if approved, either directly or through distribution partners. Whether or not we obtain FDA approval for a product candidate, we must obtain the requisite approvals from regulatory authorities in foreign countries or economic areas, such as the EU, Canada, and the United Kingdom, among other foreign countries, before we may commence clinical trials or market products in those countries or areas. The foreign regulatory approval process includes all of the risks associated with the FDA approval described above, and the time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Some foreign jurisdictions have a drug product approval process similar to that in the United States, which requires the submission of a clinical trial application much like the IND prior to the commencement of clinical studies. In Europe, for example, a clinical trial application (CTA) must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed. To obtain regulatory approval of a medicinal product candidate under EU regulatory systems, we would be required to submit a Marketing Authorisation Application (MAA), which is similar to the NDA, except that, among other things, there are country-specific document requirements. For countries outside of the EU, such as countries in Eastern Europe, Latin America or Asia, and recently the United Kingdom, the requirements governing the conduct of clinical trials, product approval, pricing and reimbursement vary from country to country. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others. Moreover, some nations may not accept clinical studies performed for United States approval to support approval in their countries or require that additional studies be performed on natives of their countries. In addition, in certain foreign markets, the pricing of drug products is subject to government control and reimbursement may in some cases be unavailable or insufficient. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions, and criminal prosecution.
Intellectual Property
We strive to pursue, maintain and defend patent rights developed internally and to protect the technology, inventions and improvements that are commercially important to the development of our business. We currently have two issued U.S. patents directed to specific ADAIR formulations (i.e., composition of matter) and one pending patent application for ADAIR that is under examination with the U.S. PTO. The U.S. patents will expire in 2037. Our international PCT application has entered national phase is under examination in several foreign countries and territories, including the EU, Canada, Japan and China. We also rely on know-how relating to our proprietary technology and product candidates and continuing innovation to develop, strengthen and maintain our proprietary position. We also plan to rely on data exclusivity, market exclusivity and patent term extensions when available.
We cannot be sure that any additional patents will be granted with respect to any of our pending patent applications or with respect to any patent applications we may file in the future. There is also a significant risk that any issued patents will have substantially narrower claims than those that are currently sought. Even with respect to any patents that may be issued to us, we cannot be sure that any such patents will be commercially useful in protecting our technology. Our first two issued patents with respect to ADAIR expire in 2037. Our commercial success will depend in part onimpair our ability to obtain and maintain patentregulatory approval for, and, if approved, commercialize, our products and product candidates in Europe.
With respect to our product candidatesadditional post-market surveillance and processes we intend to develop and commercializereporting requirements.
Our success also depends in part on our ability to preserve trade secrets; prevent third parties from infringing upon our proprietary rights; and operate our business without infringing the patents and proprietary rights of third parties, both in the United States and internationally. We also protect our proprietary technology and processes, in part, by confidentiality and invention assignment agreements with our employees, consultants, scientific advisors and other contractors. These agreements may be breached, and we may not have adequate remediesproducts, if approved for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants, scientific advisors or other contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
The patent positions of companies like ours are generally uncertain and involve complex legal and factual questions. No consistent policy regarding the scope of claims allowable in patents in the field of biopharmaceuticals has emerged in the United States. The relevant patent laws and their interpretation outside of the United States is also uncertain. Changes in either the patent laws or their interpretation in the United States and other countries may diminish our ability to protect our technology or product candidates and could affect the value of such intellectual property. In particular, our ability to stop third parties from making, using, selling, offering to sell or importing products that infringe our intellectual propertymarketing, will depend, in part, on our success in obtainingthe availability and enforcing patent claims that cover our technology, inventions and improvements.
Competition
No immediate-release prescription stimulant product with abuse-deterrent labeling currently exists on the market in the United States or internationally, however, we face competition from established biopharmaceutical companies that currently market a wide range of drugs to treat ADHD. All of these competitors have far greater marketing and research capabilities than we do. We also face potential competition from academic institutions, government agencies, and private and public research institutions, among others, which may in the future develop products to treat ADHD. Any of these companies and institutions may have products in development that are superior to ADAIR.
In addition, the biotechnology and pharmaceutical industries are characterized by rapid technological advancement, significant competition and an emphasis on intellectual property. Any product candidates that we successfully develop and commercialize will compete with current therapies and new therapies that may become available in the future. Our commercial opportunity would be reduced significantly if our competitors develop and commercialize products that are safer, more effective and convenient, have fewer side effects and/or are less expensive than the expected price of ADAIR. Public announcements regarding the development of competing drugs could adversely affect the price of our stock and the commercial potential of ADAIR.
Neither the FDA nor any other regulatory agency has approved any abuse-deterrent immediate-release stimulant. One company has submitted an application to the FDA for the approval of an immediate release stimulant designed to resist physical manipulation. An FDA advisory committee recently recommended against the approval of this product because it did not meet the primary endpoint of its human abuse liability study and based on safety concerns that are specific to the formulation in the other product that do not apply to ADAIR. The formulation technology and active ingredient in that product is distinct from that in ADAIR. While we are aware of several abuse-deterrent long-acting stimulants under development, we do not believe that ADAIR will compete directly with those products because they are designed to last longer and compete in a different segment of the ADHD market for a different patient population than ADAIR.
Third-Party Reimbursement
Sales of biopharmaceutical products depend in significant part on the availabilityextent of coverage and adequate reimbursement by third-party payors, such as state and federal governments,government health programs, including Medicare and Medicaid, commercial insurance and managed care providers,healthcare organizations. These third-party payors are increasingly challenging the price and private insurance plans. Decisions regardinglimiting the extent of coverage and amountreimbursement amounts for medical products and services. There may be significant delays in obtaining coverage and reimbursement for approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or regulatory authorities in other countries. It is time-consuming and expensive to seek reimbursement from third-party payors. Moreover, eligibility for reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim payments for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Payment rates may vary according to the use of the product and the clinical setting in which it is used, may be based on payments allowed for lower-cost products that are already reimbursed and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by third-party payors and by any future relaxation of laws that
Withindrug product basis and the amount of the rebate owed to the federal government is directly dependent on the volume of a drug product that is paid for by Medicare Parts B or D. Additionally, starting for payment year 2026, CMS will negotiate drug prices annually for a select number of single source Part D drugs without generic or biosimilar competition. CMS will also negotiate drug prices for a select number of Part B drugs starting for payment year 2028. If a drug product is selected by CMS for negotiation, it is expected that the revenue generated from such drug will decrease. CMS has begun to implement these new authorities and entered into agreements to conduct price negotiations with pharmaceutical manufacturers in October 2023. However, the impact of this program as a self-administered drug, ADAIR would be reimbursed underon the expanded prescription drug benefit known as Medicare Part D. Thisbiopharmaceutical industry in the United States remains uncertain, in part because multiple large pharmaceutical companies and other stakeholders (e.g., the U.S. Chamber of Commerce) have initiated federal lawsuits against CMS arguing the program is unconstitutional for a voluntary Medicare benefit administered by private plans that operate under contracts withvariety of reasons, among other complaints. Those lawsuits are currently ongoing.
An ongoing trend has been for third-party payors, including the U.S. government, to apply downward pressuredo not hinder, prevent or distort competition on the market. The Price Transparency Directive does not provide any guidance concerning the specific criteria on the basis of which pricing and reimbursement decisions are to be made in the individual EU Member States, nor does it have any direct consequence for pricing or reimbursement levels in the individual EU Member States. The EU Member States are free to restrict the range of medicinal products for which their national health insurance systems provide reimbursement, and to control the prices and/or
There is an emerging trenddemonstrate that they have obtained valid consent or have another legal basis in state legislation requiringplace to justify their data processing activities; the addition of abuse-deterrent formulations of opioid painkillersobligation to appoint data protection officers in certain circumstances; new rights for individuals to be added“forgotten” and rights to managed care formularies. Because theredata portability, as well as enhanced current rights (e.g. access requests); the principal of accountability and demonstrating compliance through policies, procedures, training and audit; and a new mandatory data breach regime. In particular, medical or health data, genetic data and biometric data where the latter is used to uniquely identify an individual are no stimulants currentlyall classified as “special category” data under the GDPR and afforded greater protection and require additional compliance obligations. Further, EU member states have a broad right to impose additional conditions – including restrictions – on these data categories. This is because the GDPR allows EU member states to derogate from the requirements of the GDPR mainly in regard to specific processing situations (including special category data and processing for scientific or statistical purposes). As the EU states continue to reframe their national legislation to harmonize with the GDPR, we will need to monitor compliance with all relevant EU member states’ laws and regulations, including where permitted derogations from the GDPR are introduced. The GDPR also prohibits the international transfer of personal data from the EU to countries outside of the EU unless made to a country deemed to have adequate data privacy laws by the European Commission or made through an approved with similar abuse-deterrent labeling, such legislation has not had an impact on stimulants; however, this could favorably impactdata transfer mechanism. On July 16, 2020, the reimbursementCourt of Justice of the European Union (CJEU), issued a product like ADAIRlandmark opinion in the future.
Asset Purchase Agreement
As described above,case Maximilian Schrems vs. Facebook (Case C-311/18), called Schrems II. This decision (a) calls into question commonly relied upon data transfer mechanisms as between the ADAIR assets were acquired by us pursuantEU Member States and the United States (such as the Standard Contractual Clauses) and (b) invalidates the EU-U.S. Privacy Shield on which many companies had relied as an acceptable mechanism for transferring such data from the EU to the terms and conditions ofUnited States.
the United States – the EU-US Data Privacy Framework (the Framework). The Asset Purchase Agreement also gives ArcturusFramework provides individuals in the EU with several new rights, including the right to appoint one directorobtain access to serve as a membertheir data, or obtain correction or deletion of our boardincorrect or unlawfully handled data. The adequacy decision followed the signing of directors, which was effective immediately uponan executive order introducing new binding safeguards to address the closingpoints raised in the Schrems II decision. Notably, the new obligations were geared to ensure that data can be accessed by US intelligence agencies only to the extent necessary and proportionate and to establish an independent and impartial redress mechanism to handle complaints from Europeans concerning the collection of their data for national security purposes. The European Commission will continually review developments in the United States along with its adequacy decision. Adequacy decisions can be adapted or even withdrawn in the event of developments affecting the level of protection in the applicable jurisdiction. Future actions of EU data protection authorities are difficult to predict. Some customers or other service providers may respond to these evolving laws and regulations by asking us to make certain privacy or data-related contractual commitments that we are unable or unwilling to make. This could lead to the loss of current or prospective customers or other business relationships.
Employees and Human Capital Resources
We recognize that attracting, motivating and retaining talent is vitalUnited Kingdom , or to our continued success. We aimmonitor their behavior will be subject to create an equitable, inclusive and empowering environment in which our employees can grow and advance their careers, with the overall goal of developing, expanding and retaining our workforce to support our current pipeline and future business goals. We value innovation, passion, data-driven decision making, persistence and honesty, and are building a diverse environment where our employees and consultants can thrive and be inspired to make exceptional contributions.
Our current management team, board of directors, and scientific advisors have significant experience in development and marketing of pharmaceutical product candidates from early stage discovery to clinical trials, regulatory approval and commercialization. As of March 15, 2021, we had two full-time employees. We also regularly work with several independent consultants and other contract organizations to support our business and we regularly evaluate additional talent to help support our product development, financial, and other capabilities.
Our human capital resources objectives include identifying, recruiting, retaining, and incentivizing our existing and new employees. We maintain an equity incentive plan,UK GDPR – the principal purposesrequirements of which are (at this time) largely aligned with those under the GDPR and as such, may lead to attract, retainsimilar compliance and reward personnel throughoperational costs with potential fines of up to £17.5 million or 4% of global turnover.
In addition, as a result of the COVID-19 pandemic, we have taken steps to protect the health and safety of our employees in line with directives from state and the applicable local governments, as well as guidance from the CDC.
Facilities
Our executive offices are located at 100 N. 18th Street, Suite 300, Philadelphia, PA 19103. We believe that our current office space will be adequate for the next 12 months. We have no plans to lease additional space in the next twelve months. Should we be required to obtain additional spaceproduct candidates receive foreign regulatory approval in the future, we must have policies and procedures in place sufficient to prevent us and agents acting on our behalf from providing any bribe, gift or gratuity, including excessive or lavish meals, travel or entertainment in connection with marketing our products and services or securing required permits and approvals. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring us to maintain books and records that accurately and fairly reflect all transactions
intellectual capital of our current and future employees and consultants is an impactful driver of our business and is key to our future prospects.
Report.
SummaryRisk FactorsThe followingDecember 31, 2023, we had an accumulated deficit of $31.5 million. We have devoted substantially all of our resources and efforts to research and development, and we expect that it will be several years, if ever, before we generate revenue from product sales. Even if we receive marketing approval for and commercialize one or more of our product candidates, we expect that we will continue to incur substantial research and development and other expenses in order to develop and, if approved, market additional potential product candidates.summaryvery time-consuming, expensive, and uncertain process that takes years to complete. Our operations have consumed substantial amounts of the principal risks described belowcash since inception. We expect our expenses to increase in Part I, Item 1A “Risk Factors” in this Annual Report on Form 10-K. We believe that the risks described in the “Risk Factors” section are material to investors, butconnection with our ongoing activities, particularly as we conduct our planned clinical trials of GRI-0621, GRI-0803 and any other factors not presently known to us orproduct candidates that we currently believe are immaterial may also adversely affect us. The following summary should not be considered an exhaustive summary of the material risks facing us,develop or seek regulatory approvals for and, it should be read in conjunction with the “Risk Factors” sectionif approved, launch and the other information contained in this Annual Report on Form 10-K.·We anticipate future losses and negative cash flow, and it is uncertain if or whencommercialize. In particular, we will become profitable.·We are a clinical-stage company with no approved products and a lack of operating history, which makes it difficult to assess our future viability.·As a result of our limited operating history, we may not be able to correctly estimate our future revenues, operating expenses, need for investment capital, or stability of operations, which could lead to cash shortfalls.·We do not currently have any drug products for sale, and only one clinical stage product under development, ADAIR. Our prospects currently depend largely on the success of ADAIR, which is still in clinical development, and we may not be able to generate revenues from ADAIR.·If serious adverse or unacceptable side effects are identified during the development of ADAIR or any potential future products, such as ADMIR, we may need to abandon or limit our development of some of such products.·If ADAIR does not achieve broad market acceptance, the revenues that we generate from its sales will be limited.·If we obtain approval to commercialize ADAIR, or any other future product, such as ADMIR, outside of the U.S., a variety of risks associated with international operations could materially adversely affect our business.·If the government or third-party payors fail to provide adequate coverage and payment rates for ADAIR or any future products, such as ADMIR, we may develop, license or acquire, if any, our revenue and prospects for profitability will be limited.·If we are unable to establish sales, marketing, and distribution capabilities or to enter into agreements with third parties to market and sell ADAIR or any other future product, such as ADMIR, we may not be successful in commercializing ADAIR or any other future product if and when they are approved.·We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations.·We will continue to incur significant increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.·We may not receive regulatory approval for ADAIR or any future product, such as ADMIR, or its or their approvals may be delayed, which would have a material adverse effect on our business and financial condition.·Even if ADAIR or any other proposed product that we develop, such as ADMIR, receives marketing approval, we will continue to face extensive regulatory requirements and the product may still face future development and regulatory difficulties.·The abuse, misuse or off-label use of our products may harm our image in the marketplace, result in injuries that lead to product liability suits or result in costly investigations, fines or sanctions by regulatory bodies if we are deemed to have engaged in the promotion of these uses, any of which could be costly to our business.·We have filed multiple patent applications and have two issued patents by the U.S. PTO. These or any other patent applications may not result in issued patents, and as a result we may have limited protection of our proprietary technology in the marketplace.·If we are unable to obtain and maintain patent protection for our technology and products or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired.·Because it is difficult and costly to protect our proprietary rights, we may not be able to ensure their protection.·If we cannot continue to satisfy the listing requirements of The Nasdaq Capital Market and other rules, including the director independence requirements, our securities may be delisted, which could negatively impact the price of our securities and your ability to sell them.·Our stock may be subject to substantial price and volume fluctuations due to a number of factors, many of which are beyond our control and may prevent our stockholders from reselling our common stock at a profit.Risks Relating to Our Business and IndustryWe anticipate future losses and negative cash flow, and it is uncertain if or when we will become profitable.We do not expect to generate any significant revenues untilbe able to continue our clinical trials or development efforts without raising additional funds. We also expect to incur additional costs associated with operating as a public company. Accordingly, we successfully complete developmentwill need to obtain substantial additional funding in order to maintain our continuing operations. If we are unable to raise capital when needed or on acceptable terms, we may be forced to delay, reduce, or eliminate one or more of our firstresearch and drug development programs or future commercialization efforts.including obtaining all required regulatory approvals,development efforts. We are also party to a registration rights agreement that requires us to, among other things, obtain effectiveness of a registration statement and to maintain the effectiveness of the registration statements for resale of the shares underlying the Exchange Warrants (as defined below) and the Equity Warrants (as defined below). If we are ablefail to successfully commercialize the product through salescomply with these obligations, we would be subject to substantial cash penalties and, licensing. Asas of the date of this Annual Report, would likely not have the resources to make such payments and continue our operations.still inunable to estimate the actual funds we will require for development, and haveassuming approval, marketing and commercialization activities.been approved bybe available on acceptable terms, if at all. Until we can generate sufficient revenue to finance our cash requirements, which we may never do, we expect to finance our future cash needs through a combination of public or private equity offerings, debt financings, collaborations, strategic alliances, licensing arrangements and other marketing or distribution arrangements. If weFDA.We have not yet demonstratedterms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders. Further, to the extent that we raise additional capital through the sale of Common Stock or securities convertible or exchangeable into Common Stock, our stockholders’ ownership interest will be diluted. In addition, any debt financing may subject us to fixed payment obligations and covenants limiting or restricting our ability to generate revenue,take specific actions, such as incurring additional debt, making capital expenditures, or declaring dividends. If we raise additional capital through marketing and distribution arrangements or collaborations, strategic alliances, or licensing arrangements with third parties, we may never be ablehave to produce revenuesrelinquish certain valuable intellectual property or operate on a profitable basis. We have incurred losses since our inception (January 11, 2018) and expect to experience operating losses and negative cash flow for the foreseeable future. ADAIR or any other future product, such as ADMIR, may never be approved or become commercially viable. Even if we and any collaborators are able to commercialize our technology, which may include licensing, we may never recover our research and development expenses.We are a clinical-stage company with no approved products and a lack of operating history, which makes it difficult to assess our future viability.We were incorporated on January 11, 2018, and our operations to date have been limited to organizing and staffing our company, acquiring rights to ADAIR, preparing and filingour product candidates, technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable to us. We also may be required to seek collaborators for any of our product candidates at an IND application for ADAIR, conducting four Phase I studies and working on the commercial formulation and manufacturing of ADAIR. We have not yet demonstrated anearlier stage than otherwise would be desirable or relinquish our rights to product candidates or technologies that we otherwise would seek to develop or commercialize ourselves. Market volatility resulting from inflation, pandemics, geopolitical events or other financial markets factors could also adversely impact our ability to successfully overcome many of the risksaccess capital as and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical area. For example,when needed. If we are unable to execute our business plan,secure adequate additional funding, we will need to successfully:·obtain adequate financing on terms that are acceptable to us;·execute product development activities;·obtain required regulatory approvals for the development and commercialization of ADAIR or any other future product, such as ADMIR;·maintain, leverage, and expand our intellectual property portfolio;·build and maintain robust sales, distribution, and marketing capabilities, either on our own or in collaboration with strategic partners;·gain market acceptance for ADAIR or any other future product, such as ADMIR;·develop and maintain any strategic relationships we elect to enter into; and·manage our spending as costs and expenses increase due to clinical trials, regulatory approvals, and commercialization. Ifreevaluate our operating plans and may be forced to make reductions in spending, extend payment terms with suppliers, liquidate assets where possible, delay, scale back or eliminate some or all of our development programs, relinquish rights to our technology on less favorable terms than we are unsuccessful in accomplishing these objectives, we may not be able to develop ADAIRwould otherwise choose or any other future product, such as ADMIR, raise capital, expandcease operations entirely. These actions could materially impact our business, or continueresults of operations, our operations.Further, our lackfuture prospects and the value of operating history makes it difficult to evaluate our business and prospects. Our prospects must be considered in light of the risks, uncertainties, expenses, and difficulties frequently encountered by companies in their early stages of development, particularly companies in the biopharmaceutical industry. As a young business, we may encounter unforeseen expenses, difficulties, complications, delays, and other known and unknown factors. We will need to expand our capabilities to support commercial activities. We may not be successful in adding such capabilities. The historical information in this Annual Report may not be indicativeshares of our future financial conditionCommon Stock, and future performance. We expect our financial condition and operating results to continue to fluctuate significantly from quarter to quarter and year to year due to a variety of factors, many of which are beyond our control. Accordingly, you should not rely upon our past results of as an indication of future operating performance.As a result of our limited operating history, we may not be able to correctly estimate our future revenues, operating expenses, need for investment capital, or stability of operations, which could lead to cash shortfalls.We have a limited operating history from which to evaluate our business. As a result, our historical financial data is of limitedstockholders may receive no value in estimating future operating expenses.for their investment. In addition, although we are a clinical-stage company, weattempting to secure additional financing diverts the time and attention of management from day-to-day activities and distract from our discovery and product development efforts.not yet completed all of the non-clinical safety studies for ADAIR or other pivotal clinical trials. We also have not obtained regulatory approvals for any ofexpressed substantial doubt about our products, manufactured a commercial scale product, arranged for a third party to do so on our behalf, or conducted sales and marketing activities necessary for successful product commercialization. Therefore, our budgeted operating expense levels are based in part on our expectations concerning the FDA approval process and expenses related to development of other product candidates. Failing to reach our short-term developmental milestones within anticipated timelines due to delays caused by the COVID-19 outbreak, serious adverse or unacceptable side effects caused by our product candidates, or other events, many of which may be beyond our control, may cause our financial condition and operating resultsability to continue to fluctuate significantly from quarter to quarter and year to year.We do not currently have any drug products for sale, and only one clinical stage product under development, ADAIR. Our prospects currently depend largely on the success of ADAIR, which is still in clinical development,as a going concern, and we may not be able to generate revenues from ADAIR.Wecontinue as a going concern if we do not obtain additional financing.any prescriptionincurred losses since inception and, to date, have financed our operations by issuing equity and debt securities. We anticipate that we will continue to incur losses and generate negative operating cash flows in the foreseeable future as we continue to develop our drug productscandidates and that have been approved bywe will require additional funding to support our planned operating activities. The report of our independent registered public accounting firm on our financial statements as of and for the FDA, or any similar regulatory authority. Our business success depends onyear ended December 31, 2023 includes an explanatory paragraph indicating that there is substantial doubt about our ability to obtaincontinue as a going concern. Until such time, if ever, in which we can generate substantial product revenue, we expect we may continue to fund our operations and capital funding needs through equity offerings, debt financings or other capital sources, including strategic licensing, collaboration or other similar agreements. As stated above, if we are unable to secure adequate additional funding, we will need to reevaluate our operating plans and may be forced to make reductions in spending, extend payment terms with suppliers, liquidate assets where possible, delay, scale back or eliminate some or all of our development programs, or relinquish rights to our technology on less favorable terms than it would otherwise choose. These actions could materially impact our business, results of operations, our future prospects and the value of shares of our Common Stock, and, as a result, our stockholders may receive no value for their investment.successfully commercializemay never be able to develop marketable products. Because GRI-0621 is our onlylead product currently undercandidate, if GRI-0621 encounters safety or efficacy problems, development ADAIR, which will require substantial investment, access to sufficient commercial manufacturing capacity,delays, regulatory issues or other problems, our development plans and significant marketing efforts beforebusiness would be significantly harmed. Before we can generate any revenue from sales of ADAIR, if it is ever approved for commercialization.We have only oneour lead product candidate, GRI-0621, GRI-0803 or any of our other prescription drug candidate in early development today and so our business prospects currently depend primarily on the successfulproduct candidates, we must undergo additional clinical development, regulatory review, and approval and commercialization of ADAIR, which may never occur. Even though ADAIR is in the clinical development stage, it has an uncertain chance of successfully completing clinical developmentone or more jurisdictions. These efforts will require substantial investment, and gaining regulatory approval. Any significant delays in obtaining approval for and commercializing ADAIR will have a substantial adverse impact on our business and financial condition.Even if approved, we may be unable to successfully commercialize ADAIR and we may never generate meaningful revenues. Our ability to generate revenues from ADAIR will depend on our ability to:·hire, train, deploy, and support our sales force, including any contract sales force engaged;·create market demand for ADAIR through our own marketing and sales activities, and any other promotional arrangements we may later establish;·obtain sufficient quantities of ADAIR from our third-party manufacturers as required to meet commercial demand at launch and thereafter;·establish and maintain agreements with wholesalers, distributors and group purchasing organizations on commercially reasonable terms; and·maintain patent protection and regulatory exclusivity for ADAIR.In addition, if the market for ADAIR develops less rapidly than we anticipate, we may not have the financial resources to continue development of our product candidates.shiftcommercialize, our resources to the development of alternative products.24 If serious adverse•negative or unacceptable side effects are identified during the development of ADAIR or any potential future products, such as ADMIR, we may need to abandon or limitinconclusive results from our development of some of such products.If ADAIR or potential future products, such as ADMIR, are associated with undesirable side effects in preclinical studies or clinical trials or have characteristics that are unexpected, we may needthe clinical trials of others for product candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon theira program;limitto support market authorization;more narrow usesour intellectual property rights and our manufacturing, marketing, distribution and sales efforts or subpopulationsthat of any future collaborator. Delays in whichregulatory approvals or our failure to obtain regulatory approvals would harm our business, prospects and results of operations.undesirableresults of preclinical studies and early-stage clinical trials of our product candidates may not be predictive of the results of later-stage clinical trials.other characteristics are less prevalent, less severe, or more acceptable from a risk-benefit perspective. In our industry, many compoundstoxicities;initially showed promise in early stage testing have later been found to cause side effects that prevented further applicable regulatory authorities would consider clinically meaningful;compound. same disease state;eventstandards that the FDA and comparable foreign regulatory authorities use when regulating our product candidates require judgment and can change, which makes it difficult to predict with certainty how they will be applied. Any analysis we perform of data from preclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. We may also encounter unexpected delays or increased costs due to new government regulations. Examples of such regulations include future legislation or administrative action, or changes in FDA policy during the period of product development and FDA regulatory review. We cannot predict whether legislative changes will be enacted, or whether FDA or foreign regulations, guidance or interpretations will be changed, or what the impact of such changes, if any, may be. For example, in April 2023 the European Commission issued a proposal for a new directive and a new regulation, which will revise and replace the existing general pharmaceutical legislation. If adopted and implemented as currently proposed, these revisions will significantly change several aspects of drug development and approval in the EU.revealour clinical development activities could be delayed or otherwise adversely affected.highsufficient number of eligible patients to participate in these trials as required by the FDA.unacceptable severityexclusion criteria defined in the protocol;prevalencethe process for identifying patients;side effects,patients to participate in our trials;suspendedviewed with caution until the final, complete data are available.terminated,more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects. There can be no guarantee that a favorable interim analysis will result in a favorable final result at the completion of the clinical trial.could order usand IRBs for review and approval, which may increase the cost or delay the timing or successful completion of clinical studies. Similarly, amendments to cease further developmentour preclinical studies may increase the cost or deny approvaldelay the timing or successful completion of ADAIRthose preclinical studies. If we experience delays completing, or potential future productsif we terminate, any of our preclinical or clinical studies, or if we are required to conduct additional preclinical or clinical studies, the commercial prospects for any or all targeted indications. The FDA could also issue a letter requesting additional data or information prior to making a final decision regarding whether or not to approve a product. The number of requests for additional data or information issued by the FDA in recent years has increased, and resulted in substantial delays in the approval of several new drugs. Undesirable side effects caused by ADAIR or potential future products could also result in the inclusion of unfavorable information in our product labeling, denial of regulatory approval by the FDA,candidates may be harmed and our ability to recognize product revenue will be delayed.regulatory authoritiesliabilities and may be required to limit commercialization of our product candidates.any or all targetedtopical administration, in particular, GRI-0621 may be subject to the identification of new serious adverse events as it is administered to larger numbers of research subjects in order to evaluate its safety/effectiveness in chronic use indications and in turn prevent us from commercializingnew patient populations. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even a successful defense of these claims would require significant financial and generating revenues frommanagement resources. Regardless of the salemerits or eventual outcome, liability claims may result in:ADAIRclinical trial participants and inability to continue clinical trials;potential future products. Drug-related side effects could affect patient recruitmentcriminal penalties;abilityinability to commercialize any product candidate, if approved; andcomplete the trial and could result inobtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims.Additionally,claims could prevent or inhibit the commercialization of products we develop. We may be unable to obtain, or may obtain on unfavorable terms, clinical trial insurance in amounts adequate to cover any liabilities from any of our clinical trials. Our insurance policies may also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Even if our agreements with any future corporate collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.currentcandidates, there is no guarantee this would ultimately lead to faster product development or earlier approval.future products, including ADAIR or ADMIR,to address an unmet medical need for this condition, a product sponsor may apply for FDA Fast Track designation. If we seek Fast Track designation for a product candidate, we may not receive it from the FDA. However, even if we receive Fast Track designation, Fast Track designation does not ensure that we will receive marketing approval and we or others later identify undesirable side effects causedthat approval will be granted within any particular time frame. We may not experience a faster development or regulatory review or approval process with Fast Track designation compared to conventional FDA procedures. In addition, the FDA may withdraw Fast Track designation if the designation is no longer supported by this product, a number of potentially significant negative consequences could result, including:·regulatory authorities may require the addition of unfavorable labeling statements, specific warnings, or a contraindication;·regulatory authorities may suspend or withdraw their approval of the product, or require it to be removed from the market;·we may be required to change the way the product is administered, conduct additional clinical trials, or change the labeling of the product; or·our reputation may suffer. Any of these events could prevent usdata from achieving or maintaining market acceptance of ADAIR or potential future products, such as ADMIR, or could substantially increase our commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from its sale.If ADAIRclinical development program. Fast Track designation alone does not achieve broad market acceptance,guarantee qualification for the revenues thatFDA’s priority review procedures.generate from its sales will be limited.The commercial success of ADAIR, if approved by the FDA, will depend upon its acceptance by the medical community, our ability to ensure that the drug is included in hospital formularies, and coverage and reimbursement for ADAIR by third-party payors, including government payors. The degree of market acceptance of ADAIR or any other product we may license or acquire will depend on a number of factors, including, but not necessarily limited to:·the efficacy and safety as demonstrated in clinical trials;·the timing of market introduction of such proposed product as well as competitive products;·the clinical indications for which the drug is approved;·acceptance by physicians and patients of the drug as a safe and effective treatment;·the safety of such proposed product seen in a broader patient group, including its use outside the approved indications;·the availability, cost, and potential advantages of alternative treatments, including less expensive generic drugs;·the availability of adequate reimbursement and pricing by third-party payors and government authorities;·the relative convenience and ease of administration of the proposed product for clinical practices;·the product labeling or product insert required by the FDA orreceive regulatory authority in other countries;·the approval availability, market acceptance, and reimbursement for a companion diagnostic, if any;·the prevalence and severity of adverse side effects;·the effectiveness of our sales and marketing efforts;·limitations or warnings contained in the product’s FDA-approved labeling;·changes in the standard of care for the targeted indications for ADAIR or any future product, such as ADMIR, which could reduce the marketing impact of any superiority claims that we could make following FDA approval; and·potential advantages over, and availability of, alternative treatments. If any proposed product that we develop does not provide a treatment regimen that is as beneficial as, or is not perceived as being as beneficial as, the current standard of care or otherwise does not provide patient benefit, that proposed product, if approved for commercial sale by the FDA or other regulatory authorities, likely will not achieve market acceptance. Our ability to effectively promote and sell ADAIR and any other product we may license or acquire will also depend on pricing and cost effectiveness, including our ability to produce a product at a competitive price and achieve acceptance of the product onto hospital formularies, as well as our ability to obtain sufficient third-party coverage or reimbursement. Since many hospitals are members of group purchasing organizations, which leverage the purchasing power of a group of entities to obtain discounts based on the collective buying power of the group, our ability to attract customers will also depend on our ability to effectively promote ADAIR or any other future product to group purchasing organizations. We will also need to demonstrate acceptable evidence of safety and efficacy, as well as relative convenience and ease of administration. Market acceptance could be further limited depending on the prevalence and severity of any expected or unexpected adverse side effects associated with ADAIR or any other future product such as ADMIR. If ADAIR or any other future product are approved but do not achieve an adequate level of acceptance by physicians, health care payors, and patients, we may not generate sufficient revenue from these products, and we may not become or remain profitable. In addition, our efforts to educate the medical community and third-party payors on the benefits of ADAIR or any other future product may require significant resources and may never be successful.If we obtain approval to commercialize ADAIR, or any other future product, such as ADMIR, outside of the U.S., a variety of risks associated with international operations could materially adversely affect our business.If ADAIR, or any other future product, such as ADMIR, is approved for commercialization outside the U.S., such as pursuant to the license agreement with Medice, who is affiliated with one of our principal stockholders, Salmon Pharma, and represented by one member of our board of directors, we will likely enter into agreements with third parties to market such product outside the U.S. We expect thatcandidates, we will be subject to ongoing regulatory obligations and continued regulatory review. Maintaining compliance with ongoing regulatory requirements may result in significant additional risksexpense to us, and any failure to maintain such compliance could subject us to penalties and cause our business to suffer.entering intopermit the import or maintaining international business relationships, including:·different regulatory requirements for drug approvals in foreign countries;·differing U.S. and foreign drug import and export rules, particularly regarding controlled substances and scheduled products, such as ADAIR;
export of our product candidates; and·reduced protection for intellectual property rights in foreign countries;
•injunctions, consent decrees, or the imposition of civil or criminal penalties.·unexpected changes in tariffs, trade barriers, and regulatory requirements;
In addition, the policies of the FDA and of other regulatory authorities may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the governmentadoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
Medicaid programs and managed care organizations, and others in the medical community. In both domestic and foreign markets, our sales of any future products, such as ADMIR, will depend in part uponaddition, the availability of coverage and reimbursement from third-party payors. Suchby third-party payors include governmentmay be affected by existing and future health programs such as Medicare, Medicaid, managed care providers, privatereform measures designed to reduce the cost of health insurers and other organizations. Accordingly, ADAIR or any other futurecare. If the product thatcandidates we develop do not achieve an adequate level of acceptance, we may develop, in-license or acquire,not generate significant product revenues and we may not become profitable.
candidates approved for sale in any jurisdiction, including international markets, and we do not have experience in obtaining regulatory
Other than the license agreement with Medice, we currently do not have a marketing or sales organization for the marketing, sales and distribution of biopharmaceutical products. In ordermaintaining necessary rights to commercialize any product that receives marketing approval, we would need to build marketing, sales, distribution, managerial, and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. In the event of successful development and regulatory approval of ADAIR or another future product, such as ADMIR, we expect to build a targeted specialist sales force to market or co-promote the product. There are risks involved with establishing our own sales, marketing, and distribution capabilities. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to commercialize our products, if any, on our own include, but are not necessarily limited to:
As an alternative to establishing our own sales force, we may choose to partner with third parties that have well-established direct sales forces to sell, market, and distribute our products.
We rely on a limited number of suppliers and manufacturers for ADAIR and expect to continue to do so fordevelop any future product candidates on acceptable terms.
The manufacture of biopharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls andrequire the use of specialized processing equipment. We have entered into an agreement to complete precommercial manufacturing development activities with one contract manufacturer and expect to contract with them forproprietary rights held by third parties, the commercial manufacturegrowth of ADAIR. However, we have not yet negotiated and signed a commercial supply agreement. The inability of a third-party manufacturer to successfully manufacture ADAIR may materially harm our business may depend in part on our ability to acquire, in-license or use these proprietary rights.
We do not have any of our own manufacturing facilities or personnel. We rely,pre-existing biotechnology and expect to continue to rely, on third parties for the manufacture of ADAIR or any other future product, such as ADMIR, for clinical testing, as well as for commercial manufacture if any of ADAIR or any other future product receive marketing approval. This reliance on third parties increases the risk that we will not have sufficient quantities of ADAIR or any other future product or such quantities at an acceptable cost or quality, which could delay, prevent, or impair our development or commercialization efforts.
We also expect to rely on third-party manufacturers or third-party collaborators for the manufacture of commercial supply of any product for which our collaborators or we obtain marketing approval.biopharmaceutical compounds. We may be unable to establishacquire or in-license any agreements withcompositions, methods of use, processes, or other third-party manufacturersintellectual property rights from third parties that we identify as necessary or important to our business operations. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all, which would harm our business. We may need to cease use of the compositions or methods covered by such third-party intellectual property rights and may need to seek to develop alternative approaches that do sonot infringe on acceptable terms.such intellectual property rights which may entail additional costs and development delays, even if we were able to develop such alternatives, which may not be feasible. Even if we are able to establish agreements with third-party manufacturers, reliance on third-party manufacturers entails additional risks, including, but not necessarily limited to:
All ofobtain a license, it may be non-exclusive, thereby giving our contract manufacturers must comply with strictly enforced federal, state, and foreign regulations, including current Good Manufacturing Practice (“cGMP”) requirements enforced by the FDA through its facilities inspection program. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturers for compliance with cGMP regulations for manufacture of ADAIR or any other future product, such as ADMIR. Our failure, or the failure of our third-party manufacturers, to comply with applicable regulations or similar regulatory requirements outside the U.S. could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, suspension of production, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our products. Any manufacturing defect or error discovered after products have been produced and distributed could result in even more significant consequences, including costly recall procedures, re-stocking costs, damage to our reputation, and potential for product liability claims.
ADAIR and any products that we may develop may compete with other products forcompetitors access to manufacturing facilities. There are a limited number of manufacturersthe same technologies licensed to us. In that operate under cGMP regulations and that might be capable of manufacturing for us. Any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval. We do not currently have arrangements in place for redundant supply or a second source for bulk drug substance. If our current contract manufacturers cannot perform as agreed,event, we may be required to replaceexpend significant time and resources to develop or license replacement technology.
Our current and anticipated future dependence upon others for the manufacture of ADAIR or any other future products, such as ADMIR, may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis.
We also expect to rely on other third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of ADAIR or any other future product, such as ADMIR, or commercialization of any of our products, producing additional losses and depriving us of potential product revenue.
Public health crises such as pandemics or similar outbreaks could materially and adversely affect our preclinical and clinical trials, business, financial condition and results of operations.
In March 2020, the World Health Organization declared COVID-19 a global pandemic and the United States declared a national emergency with respect to COVID-19. In response to the COVID-19 pandemic, “shelterpatents are issued, patent applications in place” orders and other public health guidance measures have been implemented across much of the United States and Europe, includingmany foreign jurisdictions are typically not published until 18 months after filing, and publications in the locations ofscientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our principal place of business, clinical trial sites,owned and locations ofin-licensed issued patents or our key vendors and partners.pending applications, or that we or, if applicable, a licensor were the first to invent or first to file a patent application covering the technology. Our clinical development program and preclinical study timelinescompetitors may be negatively affected by COVID-19, which could materially and adversely affect our business, financial condition and results of operations. Further, due to “shelter in place” orders and other public health guidance measures, we and our third-party vendors and suppliers have implemented remote working policies. Our third-party vendors’ and suppliers’, and our increased reliance on personnel working from home may negatively impact productivity, or disrupt, delay or otherwise adversely impact our business. For example, the COVID-19 pandemic caused some delays at our clinical trial sites, contract research organizations (“CROs”), and third-party manufacturers, which in turn resulted in some delays in the FDA approval process; however, these delays have been largely remediated.
As a result of the COVID-19 pandemic, or similar pandemics, and related “shelter in place” orders and other public health guidance measures, we havefiled, and may in the future experience disruptions that could materiallyfile, patent applications covering our products or technology similar to ours. Any such patent application may have priority over our owned and adversely impact our clinical trials, business, financial condition and results of operations. Potential disruptions include but are not limited to:
These and other factors arising from the COVID-19 global pandemic could worsen in countries that are already afflicted with COVID-19, could continue to spread to additional countriesin-licensed patent applications or could return to countries where the pandemic has been partially contained, each ofpatents, which could further adversely impact our abilityrequire us to conduct clinical trials and our business generally, and could materially and adversely affect our business, financial condition and resultsobtain rights to issued patents covering such technologies. If another party has filed a U.S. patent application on inventions similar to those owned by or in-licensed to us, we or, in the case of operations.
The COVID-19 global pandemic continuesin-licensed technology, the licensor may have to rapidly evolve. The extentparticipate in an interference or derivation proceeding declared by the USPTO to which the outbreak may affect our clinical trials, business, financial condition and resultsdetermine priority of operations will depend on future developments, which are highly uncertain and cannot be predicted at this time, such as the ultimate geographic spread of the disease, the duration of the outbreak, travel restrictions and actions to contain the outbreak or treat its impact, such as social distancing and quarantines or lock-downsinvention in the United StatesStates. If we or one of our licensors is a party to an interference or derivation proceeding involving a U.S. patent application on inventions owned by or in-licensed to us, we may incur substantial costs, divert management’s time and expend other countries, business closuresresources, even if we are successful.
Our future growthour competitors gain access to the same technology. Litigation or interference proceedings may depend onresult in a decision adverse to our ability to identifyinterests and, acquire or in-license products andeven if we do not successfully identifyare successful, may result in substantial costs and acquire or in-license related product candidates and products or integrate them intodistract our operations, we may have limited growth opportunities.
An important part of our business strategy is to continue to develop a pipeline of product candidates and products by acquiring or in-licensing products, businesses or technologies that we believe are a strategic fit with our business. Future in-licenses or acquisitions, however, may entail numerous operational and financial risks, including:
We have limited resources to identify and execute the acquisition or in-licensing of third-party products, businesses and technologies and integrate them into our current infrastructure. In particular, we may compete with larger biopharmaceutical companies and other competitors in our efforts to establish new collaborations and in-licensing opportunities. These competitors likely will have access to greater financial resources than us and may have greater expertise in identifying and evaluating new opportunities. Moreover, we may devote resources to potential acquisitions or in-licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts.
Expanding our product offerings may not be profitable.
We may choose to develop new products to offer. We are currently developing an abuse deterrent formulation of Ritalin, ADMIR, another commonly prescribed product for the treatment of ADHD. Developing new products involves inherent risks, including our inability to estimate demand for the new offerings, competition from more established market participants, and a lack of market understanding. In addition, expanding into new geographic areas and/or expanding product offerings will be challenging and may require integrating new employees into our culture as well as assessing the demand in the applicable market.
We may expend our limited resources to pursue a particular proposed product or indication, and fail to capitalize on a different proposed product or indication that may have been more profitable or for which there would have been a greater likelihood of success.
Because we have limited financial and managerial resources, we focus on research programs and proposed products that we identify for specific indications. As a result, we may forego or delay pursuit of opportunities with other proposed products, or for other indications, that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and proposed products for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular proposed product, we may relinquish valuable rights to that proposed product through collaboration, licensing, or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such proposed product.
If we fail to effectively manage our growth, our business and reputation, results of operations, and financial condition may be adversely affected.
We may experience a rapid growth in operations, which may place significant demands on our management team and our operational and financial infrastructure. As we continue to grow, we must effectively identify, integrate, develop and motivate new employees, and maintain the beneficial aspects of our corporate culture. To attract top talent, we believe we will have to offer attractive compensation packages. The risks of over-hiring or overcompensating and the challenges of integrating a rapidly growing employee base may impact profitability.
Additionally, if we do not effectively manage our growth, the quality of our services could suffer, which could adversely affect our business and reputation, results of operations, and financial condition. If operational, technology, and infrastructure improvements are not implemented successfully, our ability to manage our growth will be impaired and we may have to make significant additional expenditures to address these issues. To effectively manage our growth, we will need to continue to improve our operational, financial, and management controls and our reporting systems and procedures. This will require that we refine our information technology systems to maintain effective online services and enhance information and communication systems to ensure that our employees effectively communicate with each other and our growing base of customers. These system enhancements and improvements will require significant incremental and ongoing capital expenditures and allocation of valuable management and employee resources. If we fail to implement these improvements and maintenance programs effectively, our ability to manage our expected growth and comply with the rules and regulations that are applicable to publicly reporting companies will be impaired and we may incur additional expenses.
employees. We may not be able to manageprevent, alone or with our business effectivelylicensors, misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
Our key employees currently include David Baker, our President and Chief Executive Officer, and Ms. Penny Toren, our Senior Vice President, Regulatory Affairs & Program Management, who is responsible for regulatory affairs, and consulting arrangements with individuals such as our Chief Medical Officer, Dr. Timothy Whitaker, who is responsible for overseeing clinical developmentperhaps all, of the patent protection on our product candidates. Our future growthSuch a loss of patent protection could have a material adverse impact on our business and success depend on our ability to recruit, retain, manage,commercialize or license our technology and motivateproduct candidates.
Because of the specialized scientificexisting patent portfolio and managerial nature of our business, we rely heavily on our ability to attractprotect and retain qualified scientificenforce our intellectual property in the future.
manner or do so on commercially reasonable terms. In addition, our CROs may not prioritize our clinical trials relative to those of other customers, and any turnover in personnel or delays in the allocation of CRO employees by the CRO may negatively affect our clinical trials. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, our clinical trials may be delayed or terminated, and we may not be able to meet our current plans with respect to our product candidates. CROs also may involve higher costs than anticipated, which could negatively affect our financial condition and operations.
Ifour product candidates and our current costs to manufacture our product candidates may not be commercially feasible. As a result, we are notmay never be able to attract and retain necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
Our directors, consultants and advisors are not obligated to commit their time and attention exclusively to our business and therefore they may encounter conflicts of interest with respect to the allocation of time and business opportunities between our operations and those of other businesses.
Our directors are not obligated to commit their time and attention exclusively to our business and, accordingly, they may encounter conflicts of interest in allocating their own time, or any business opportunities which they may encounter, between our operations and those of other businesses.
Currently, our full-time employees consist of David Baker, who is our President and Chief Executive Officer, and Penny Toren, who is Senior Vice President, Regulatory Affairs, and our key consultants consist of Dr. Timothy Whitaker, our Chief Medical Officer, as well as consultants for finance, bookkeeping, pre-clinical and formulation development, chemistry manufacturing and controls (CMC), and clinical operations. Currently, consulting arrangements with individuals, such as Dr. Whitaker, only require them to devote an average of approximately 10 to 20 hours per week to our business. develop a commercially viable product.
Additionally, all of our officers and directors,limited. Following NDA approval, a change in the course of their other business activities, may become aware of investments, business opportunities,manufacturing site could require additional approval from the FDA. This approval would require new testing and compliance inspections;
•our third-party manufacturers could breach or terminate their agreements with us.
requirements.requirements, which could have a material adverse effect on our business.of employee fraudthat our employees, independent contractors, principal investigators, CROs, consultants or vendors may engage in fraudulent or other misconduct.illegal activity. Misconduct by employeesthese parties could include intentional, failuresreckless and/or negligent conduct or disclosure of unauthorized activities to comply withus that violates: FDA regulations, provideincluding those laws requiring the reporting of true, complete and accurate information to the FDA, comply withFDA; manufacturing standards we have established, comply withstandards; federal and state health-carehealthcare fraud and abuse laws and regulations, reportregulations; or laws that require the true, complete and accurate reporting of financial information or data accurately or disclose unauthorized activities to us under the Federal Physician Payments Sunshine Act and similar state laws. In particular, sales, marketing, and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing, making or contributing to the making of a false claim for reimbursement to federal, state or private payors, and other abusive practices.data. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct couldActivities subject to these laws also involve the improper use or misrepresentation of information obtained in the course of clinical trials or creating fraudulent data in our preclinical studies or clinical trials, which could result in regulatory sanctions and serious harm to our reputation. Thecomplybe in compliance with such laws or regulations. Additionally, we are subject to the risk that a person could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, and results of operations, including the imposition of significantcivil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished potential profits and future earnings, and curtailment of our operations, any of which could adversely affect our business, financial condition, results of operations or prospects.sanctions.Our management hasmaterials becomes limited experience in managing the day to day operations of a public company and, as a result,or interrupted for other reasons, we may incur additional expenses associatedbe forced to manufacture the materials ourselves, for which we currently do not have the capabilities or resources, or enter into an agreement with another third party, which we may not be able to do on reasonable terms, if at all. In either scenario, our clinical trials could be delayed significantly as we establish alternative supply sources. In some cases, the managementtechnical skills or technology required to manufacture our product candidates may be unique or proprietary to the original manufacturer and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills or technology to a back-up or alternative supplier, or we may not be able to transfer such skills or technology at all. Furthermore, a manufacturer may possess technology related to the manufacture of our company.Our Chief Executive Officer is responsible for the operations and reporting ofproduct candidate that such manufacturer owns independently. These factors would increase our company. The requirements of operating as a small public company are new to our management. This mayreliance on such manufacturer or require us to obtain outside assistancea license from such manufacturer in order to have another third party manufacture our product candidates.accounting, investor relations, or other professionals that couldremedies associated with such a breach may be more costly than planned. Weinsufficient to compensate us for any damages we may alsosuffer. In addition, if we are required to change manufacturers for any reason, we will be required to hire additional staff toverify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturer or manufacturing process will produce our product candidate according to the specifications previously submitted to the FDA or another regulatory authority. We may be unsuccessful in demonstrating the comparability of clinical supplies which could require the conduct of additional SEC reporting requirements. These compliance costs will make some activities significantly more time-consuming and costly. If we lack cash resources to cover these costs inclinical trials. The delays associated with the future, our failure to comply with reporting requirements and other provisionsverification of securities lawsa new manufacturer could negatively affect our stock priceability to develop product candidates in a timely manner or within budget.adversely affect our potential results of operations, cash flow,regulatory requirements, including those related to quality control and financial condition after we commence operations.Our market is subject to intense competition.assurance. If we are unable to compete effectively, ADAIRobtain or maintain third-party manufacturing for product candidates, or to do so on commercially reasonable terms, we may not be able to develop and commercialize our product candidates successfully.proposed product thatcompanies to provide us with important technologies and funding for our programs and technology. Any future collaborations we developenter into may be rendered noncompetitive or obsolete.There arepose a number of existing treatments for ADHD currently onrisks, including that collaborators have significant discretion in determining the market, allefforts and resources that they will apply and may not perform their obligations as expected, collaborators may not provide us with timely and accurate information regarding development progress and activity under any future license agreement, which could adversely impact our ability to report progress to our investors and otherwise plan development of which are marketed by pharmaceutical companies that are far larger and more experienced than we are. Patients and doctors are often unwilling to change medications, and this factor will make it difficult for ADAIR or any other proposedour product to penetrate the market. Further, our industry is highly competitive and subject to rapid and significant technological change. Our potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and research institutions. All of these competitors currently engage in, have engaged in or may engage in the future in the development, manufacturing, marketing and commercialization of new pharmaceuticals, some of which may compete with ADAIR or other proposed productcandidates, we may have disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or terminations of the research, development or commercialization of product candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive and collaborations may be terminated by the future. Smallercollaborator, and, if terminated, we could be required to raise additional capital to pursue further development or early stage companies may also provecommercialization of the applicable product candidates.significant competitors, particularly through collaborative arrangementsattractive in light of the terms that we are seeking and other available products for licensing by other companies. Collaborations are complex and time-consuming to negotiate and document. If we are unable to reach agreements with large, established companies. These companiessuitable collaborators on a timely basis, on acceptable terms, or at all, we may have products in development that are superior to ADAIR or any other future product, such as ADMIR. Key competitive factors affecting the commercial success of ADAIR and any other proposed product that we may develop in the future are likely to be efficacy, time of onset, safety and tolerability profile, reliability, convenience of dosing, price, and reimbursement.Many of our potential competitors have substantially greater financial, technical, and human resources than we do and significantly greater experience in the discovery and development of drug candidates, obtaining FDA and other regulatory approvals of products, and the commercialization of those products. Established competitors may invest heavily to quickly discover and develop novel compounds that could make ADAIR or other proposed product we may develop obsolete. Other companies may be developing abuse deterrent formulations of ADHD treatments that may be approved and marketed before ADAIR, limiting the commercial potential of ADAIR. Accordingly, our competitors may be more successful than us in obtaining FDA and other marketing approvals, or more favorable language in the prescription information, for their drugs, and achieving widespread market acceptance. Our competitors’ drugs may be more effective, or more effectively marketed and sold, than any drug we may commercialize and may render ADAIR or any other proposed product that we develop obsolete or non-competitive before we can recover the expenses of developing and commercializing the product. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. Finally,curtail the development of new treatment methods fora product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the diseasesscope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we are targeting could render ADAIRelect to increase our expenditures to fund development or any other proposed product that we develop non-competitive or obsolete.We face potential product liability exposure, and if successful claims are brought against us,commercialization activities on our own, we may incur substantial liability for ADAIR or other proposed product we may license or acquireneed to obtain additional expertise and may have to limit their commercialization.The use of ADAIR and any other proposed product we may license or acquire in clinical trials and the sale of any products foradditional capital, which we obtain marketing approval expose us to the risk of product liability claims. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during clinical testing, manufacturing, marketing, or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability, or a breach of warranties. Product liability claims might be brought against us by consumers, health care providers, or others using, administering, or selling our products. If we cannot successfully defend ourselves against these claims, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:·withdrawal of clinical trial participants;·termination of clinical trial sites or entire trial programs;·decreased demand for any proposed product or products that we may develop;·initiation of investigations by regulators;·impairment of our business reputation;·costs of related litigation;·substantial monetary awards to patients or other claimants;·loss of revenues;·reduced resources of our management to pursue our business strategy; and·the inability to commercialize ADAIR or any future product, such as ADMIR.We will obtain limited product liability insurance coverage for any and all of our clinical trials. However, our insurance coverage may not reimburse us or may not be available to us on acceptable terms, or at all. If we fail to enter into future collaborations or do not have sufficient funds or expertise to reimburse us for any expenses or lossesundertake the necessary development and commercialization activities, we may suffer. Moreover, insurance coverage is becoming increasingly expensive,not be able to further develop our product candidates, bring them to market and generate revenue from sales of drugs or continue to develop our technology, and our business may be materially and adversely affected. Even if we are successful in our efforts to establish new strategic collaborations, the future,terms that we agree upon may not be favorable to us, and we may not be able to maintain insurance coverage atsuch strategic collaborations if, for example, development or approval of a reasonable costproduct candidate is delayed or sales of an approved product are disappointing. Any delay in sufficient amountsentering into new strategic collaboration agreements related to protect us against losses dueour product candidates could delay the development and commercialization of our product candidates and reduce their competitiveness even if they reach the market.liability. When needed,Managing Our Business and Operationsintend to expand our insurance coverage to include the sale of commercial productslose key management personnel, or if we obtain marketing approvalfail to recruit additional highly skilled personnel, our ability to develop current product candidates or identify and develop new product candidates will be impaired, could result in loss of markets or market share and could make us less competitive.ADAIRskilled personnel in our market is intense and may limit our ability to hire and retain highly qualified personnel on acceptable terms or at all.future product incompanies. Despite our efforts to retain valuable employees, members of our management, scientific and development such as ADMIR, butteams may terminate their employment with us on short notice. Our key employees are at-will employees, which means that any of our employees could leave our employment at any time, with or without notice. There is no guarantee that any “key person” insurance policy we have or may enter into would adequately compensate us for the loss of any key employee. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior, mid-level and senior scientific and medical personnel.obtain commercially reasonablesuccessfully develop or commercialize our product liability insurancecandidates or otherwise implement our business plan.any product approveda limited number of qualified personnel among biopharmaceutical companies. Many of the other biopharmaceutical companies against which we will compete have greater financial and other resources, different risk profiles, and a longer history in the industry. Our competitors may provide higher compensation, more diverse opportunities, and/or better opportunities for marketing. On occasion, large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claimcareer advancement. Any or seriesall of claims brought against usthese competing factors may limit our ability to continue to attract and retain high quality personnel, which could cause our stock price to fall and, if judgments exceed our insurance coverage, could decrease our cash and adverselynegatively affect our business.Our internal computerability to successfully develop and commercialize our product candidates and to grow our business and operations as currently contemplated.those used by third-party CROs, manufacturers, or other contractors or consultants, may fail or suffer security breaches.Despite the implementation of security measures,proprietary information, including personal data, damage our reputation, and subject us to significant financial and legal exposure.ourany future CROs, manufacturers,collaborators and other contractors andor consultants are vulnerable to damage from computer viruses, phishing or other unauthorized access, natural disasters, terrorism, war and unauthorized access. Although to our knowledge we have not experienced any such material system failure or security breach to date, iftelecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations.operations, financial loss, a loss of our trade secrets or other proprietary information and damage to our reputation and otherwise negatively impact us. For example, the loss of clinical trial data from future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, (such as individually identifiable health information), we could incur significant liabilitiesliability, our competitive position could be harmed and the further development and commercialization of ADAIR or any other futureour product including ADMIR,candidates could be delayed.
MediceWe rely on information technology systems that we or our third-party providers operate to process, transmit and store electronic information in our day-to-day operations. In connection with our product discovery efforts, we may not successfully developcollect and use a variety of personal data, such as names, mailing addresses, email addresses, phone numbers and clinical trial information. A successful cyberattack could result in the theft or commercialize ADAIRdestruction of intellectual property, data, or other misappropriation of assets, or otherwise compromise our confidential or proprietary information and disrupt our operations. Cyberattacks are increasing in Europe.
On January 6, 2020, Vallon entered intotheir frequency, sophistication and intensity, and have become increasingly difficult to detect. Cyberattacks could include wrongful
Risks Relating to Finances and Capital Requirements
We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations.
Our operations have consumed substantial amounts of cash since our inception. As of December 31, 2020, we had an accumulated deficit of $12.6 milliondisputes with physicians, patients and our net loss was $4.8 million for the year ended December 31, 2020. We expect to continue to incur significantpartners, regulatory sanctions or penalties, increases in operating expenses, and increasing operating losses for the foreseeable future. Our business will require additional capital for implementation of our long-term business plan and product development and commercialization.
Our ability to raise additional funds may be adversely impacted by potential worsening global economic conditions and the recent disruptions to, and volatility in, the credit and financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic. As we require additional funds, we may seek to fund our operations through the sale of additional equity securities, debt financing and/expenses or strategic collaboration agreements. We cannot be sure that additional financing fromlost revenues, civil liability or other adverse consequences, any of these sources will be available when needed or that, if available, the additional financing will be obtained on favorable terms.
Our future funding requirements will depend on many factors, including, but not limited to:
If we raise additional funds by selling shares of our common stock or other equity-linked securities, the ownership interest of our current stockholders will be diluted. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time. If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or products or to grant licenses on terms that may not be acceptable to us. If we raise additional funds through debt financing, we may have to grant a security interest on our assets to the future lenders, our debt service costs may be substantial, and the lenders may have a preferential position in connection with any future bankruptcy or liquidation involving the company.
If we are unable to raise additional capital when needed, we may be required to curtail the development of our technology or materially curtail or reduce our operations. We could be forced to sell or dispose of our rights or assets. Any inability to raise adequate funds on commercially reasonable terms could have a material adverse effect on our business, results of operations, financial condition, prospects and cash flows. Any failure by such third parties to prevent or mitigate security breaches or improper access to or disclosure of such information could have similarly adverse consequences for us. If we are unable to prevent or mitigate the impact of such security or data privacy breaches, we could be exposed to litigation and governmental investigations, which could lead to a potential disruption to our business. By way of example, the CCPA, which went into effect on January 1, 2020, creates individual privacy rights for California consumers and increases the privacy and security obligations of entities handling certain personal data. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches increasing the potential for data breach litigation. The CPRA became effective on January 1, 2023 to strengthen elements of the CCPA. While there are exceptions for information that is subject to HIPAA and clinical trial regulations, the CCPA, as applicable, would still impact our business, and may increase our compliance costs and potential liability, and many similar laws have been proposed at the federal level and actually implemented in other states. By way of example regarding foreign laws and regulations with respect to data privacy and security, the GDPR went into effect in the EU in May 2018 introducing strict requirements for processing the personal data of EU data subjects. Companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater.
the extent exercised on a cashless basis. The holders of these securities or their affiliates have and may sell large amounts of our Common Stock in the open market or in privately negotiated transactions, which, in the past and again may result in a lower trading price of our Common Stock and substantial dilution to our stockholders. Additionally, the registration and availability of such a significant number of shares of Common Stock for trading in the public market has and may increase the volatility in our stock price or put significant downward pressure on the price of our stock.
In addition, if we fail to maintain proper and effective internal controls, our ability to produce accurate financial statements on a timely basis could be impaired.
The Sarbanes-Oxley Act of 2002
We are an “emerging growth company” and we cannot be certain if the reduced disclosuregovernance requirements applicable to emerging growth companies, will makewhich could result in our securitiesCommon Stock being less attractive to investors.
After we become a reporting company under the Exchange Act, we
Our ability to utilize our net operating loss (“NOL”) carryforwards may be limited.
As of December 31, 2020, we had NOL carryforwards available to reduce future taxable income, if any, for federal and state income tax purposes of $11.6 million.
The report of our independent registered public accounting firm included a “going concern” explanatory paragraph.
The report of our independent registered public accounting firm on our financial statements as of and for the year ended December 31, 2020 included an explanatory paragraph indicating that there is substantial doubt about our ability to continue as a going concern. We have financed our working capital requirements to date by raising capital through private placementslisting of shares of our common stock, issuingCommon Stock does not currently comply with the rules of short-term and convertible notes, and the proceedsThe Nasdaq Capital Market or any other Nasdaq Market tier. A delisting of our Common Stock from our initial public offering completed in February 2021. If we are unable to raise sufficient capital as and when needed, our business, financial condition and results of operations will be materially andNasdaq could adversely affected, and we will need to significantly modify our operational plans to continue as a going concern. If we are unable to continue as a going concern, we may have to liquidate our assets, and the values we receive for our assets in liquidation or dissolution could be significantly lower than the values reflected in our financial statements. The inclusion of a going concern explanatory paragraph by our independent registered public accounting firm, our lack of cash resources and our potential inability to continue as a going concern may materially adversely affect our share price and our ability to raise newadditional capital enter into critical contractual relations with third partiesthrough the public or private sale of equity securities and otherwise execute our development strategy.
Risks Relatinginvestors’ ability to Regulatory Matters
We may not receive regulatory approvaldispose of, or obtain accurate quotations as to the market value of, our Common Stock.
ADAIR and any other future product, such as ADMIR, as well as the activities associated with their development and commercialization, including their design, testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA and other regulatory agencies in the United States and by the European Medicines Agency, and similar regulatory authorities outside the U.S. Failure to obtain marketing approval for ADAIR or any future product will prevent us from commercializing such products. We have not received approval to market ADAIR from regulatory authorities in any jurisdiction. We have only limited experience in filing and supporting the applications necessary to gain marketing approvals and expect to rely on third-party contract research organizations and regulatory consultants to assist us in this process. Securing marketing approval requires the submission of extensive preclinical and clinical data and supporting information to regulatory authorities for each therapeutic indication to establish the proposed product’s safety and efficacy. Securing marketing approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the regulatory authorities. ADAIR or any future product may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit commercial use. If ADAIR or any future product receives marketing approval, the accompanying label may limit the approved use of our drug, which could limit sales of the product. We may never succeed in convincing regulatory agencies to allow us to promote the abuse deterrent properties of ADAIR, even if we are successful in demonstrating these characteristics in studies. The FDA and other regulatory agencies may never allow us to study ADAIR in human abuse liability studies, thereby limiting our ability to generate differentiating data and claims.
The process of obtaining marketing approvals, both in the United States and abroad, is expensive, may take many years if approval is granted at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the proposed product involved. Changes in marketing approval policiesdays during the development180-calendar day period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may cause delays in the approval or rejection of an application. Regulatory authorities have substantialunless Nasdaq exercises its discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional preclinical studies and/or clinical trials. In addition, varying interpretations of the data obtained from preclinical studies and/or clinical testing could delay, limit, or prevent marketing approval of a proposed product. Any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable.
If we experience delays in obtaining approval or if we fail to obtain approval of our proposed product or any future product, the commercial prospects for our proposed product may be harmed and our ability to generate revenue will be materially impaired.
In addition, even if we obtain approval, regulatory authorities may approve our proposed product or any future product for fewer or more limited indications than we request, may not approve the price we intend to charge for our product, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a proposed product with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product. Any of these scenarios could compromise the commercial prospects for our proposed product or any future product.
Separately, in response to the global pandemic of COVID-19, on March 10, 2020 the FDA announced its intention to postpone most foreign inspections of manufacturing facilities and products, and subsequently, on March 18, 2020, the FDA announced its intention to temporarily postpone routine surveillance inspections of domestic manufacturing facilities. The FDA reiterated its stance to Congress on June 2, 2020 stating, “only inspections deemed mission-critical will be considered on a case-by-case basis as this outbreak continues to unfold.” On August 20, 2020, and subsequently updated on January 29, 2021, the FDA’s Guidance for Industry against stated, “foreign pre-approval and for-cause inspection assignments that are not deemed mission-critical remain temporarily postponed, while those deemed mission-critical will still be considered for inspection on a case-by-case basis.”
Regulatory authorities outside the United States may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic. If a prolonged government shutdown occurs, or if global health concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process our regulatory submissions, which could have a material adverse effect on our business. If the FDA becomes unable to continue its current level of performance, we could experience delays and setbacks for our product candidates, including ADAIR and ADMIR, and for any approvals we may seek which could adversely affect our business.
Even if ADAIR or any other proposed product that we develop,extend such as ADMIR, receives marketing approval, we will continue to face extensive regulatory requirements and the product may still face future development and regulatory difficulties.
ADAIR and any other proposed product we may develop, such as ADMIR, or license or acquire, will also be subject to ongoing requirements and review of the FDA and other regulatory authorities. These requirements include labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information and reports, registration and listing requirements, cGMP requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, and requirements regarding the distribution of samples to physicians and recordkeeping of the drug.
The FDA may also impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of the product. The FDA closely regulates the post-approval marketing and promotion of drugs to ensure drugs are marketed only for the approved indications and in accordance with the provisions of the approved labeling. The FDA imposes stringent restrictions on manufacturers’ communications regarding off-label use and if10‑day period. If we do not market ADAIR or any other future product for only their approved indications,regain compliance by the Compliance Date, we may be eligible for an additional 180-calendar day period, subject to enforcement action for off-label marketing. Violations ofsatisfying the FDCA, relating to the promotion of prescription drugs may lead to investigations alleging violations of federal and state health care fraud and abuse laws, false claims acts, as well as state consumer protection laws.
In addition, later discovery of previously unknown adverse events or other problems with ADAIR or any other future product, manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may yield various results, including:
In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP and other regulations.
The FDA’s policies may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our proposed product. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained.
The abuse, misuse or off-label use of our products may harm our imageconditions in the marketplace, result in injuries that lead to product liability suitsapplicable Nasdaq Listing Rules. If, before the Compliance Date, our Common Stock has a closing bid price of $0.10 per share or result in costly investigations, fines or sanctions by regulatory bodies if we are deemed to have engaged inless for ten consecutive trading days, the promotion of these uses, any of which could be costly to our business.
The products we marketStaff (as defined below) will be approved by the FDA for specific treatments. We will train our marketing and direct sales force to not promote our products for uses outside of the FDA-approved indications for use, known as “off-label uses.” We cannot, however, preventissue a physician from using our products off-label, when in the physician’s independent professional medical judgement, he or she deems it appropriate. There may be increased risk of injury to patients if physicians attempt to use our products off-label. Furthermore, the use of our products for indications other than those approved by the FDA or approved by any foreign regulatory body may not effectively treat such conditions, which could harm our reputation in the marketplace among physicians and patients. In addition, although ADAIR is very difficult to manipulate into a form that can be snorted, it is not impossible to do so, as was shown by a third-party drug laboratory, hired by us in preparation for human abuse studies, that was able to develop a time-consuming and laborious process to convert ADAIR into a form that could be insufflated. In addition, although ADAIR is difficult to solubilize into a form that can be injected, sophisticated drug abusers may be able to develop methods to manipulate ADAIR into a form that can be injected. We cannot assure you that users of ADAIR or such other products we may develop in the future will not manipulate our products with the intention of abusing our products. Such abuse of our products may harm our image in the marketplace and damage our reputation.
If the FDA or any foreign regulatory body determines that our promotional materials or training constitute promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our business activities to constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs, and the curtailment of our operations.
In addition, physicians may misuse our products if they are not adequately trained, potentially leading to injury and an increased risk of product liability. If our products are misused or used with improper technique, we may become subject to costly litigation by our customers or their patients. As described elsewhere, product liability claims could divert management’s attention from our core business, be expensive to defend and result in sizeable damage awards against us that may not be covered by insurance.
Our current and future relationships with customers and third-party payors in the United States and elsewhere may be subject, directly or indirectly, to applicable anti-kickback, fraud and abuse, false claims, transparency, health information privacy and security, and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, administrative burdens, and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors in the United States and elsewhere will play a primary role in the recommendation and prescription of any product for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations, including, without limitation, the federal Anti-Kickback Statute and the federal False Claims Act, which may constrain the business or financial arrangements and relationships through which we sell, market, and distribute any product for which we obtain marketing approval. In addition, we may be subject to transparency laws and patient privacy regulation by U.S. federal and state governments and by governments in foreign jurisdictions in which we conduct our business. The applicable federal, state, and foreign healthcare laws and regulations that may affect our ability to operate include, but are not necessarily limited to:
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations may involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, including, without limitation, damages, fines, imprisonment, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations, which could have a material adverse effect on our business. If any of the physicians or other healthcare providers or entities with whom we expect to do business, including our collaborators, is found not to be in compliance with applicable laws, it may be subject to criminal, civil or administrative sanctions, including exclusions from participation in government healthcare programs, which could also materially affect our business.
Regulatory approval for any approved product is limited by the FDA to those specific indications and conditions for which clinical safety and efficacy have been demonstrated.
Any regulatory approval is limited to those specific diseases and indications for which a product is deemed to be safe and effective by the FDA. In addition to the FDA approval required for new formulations, any new indication for an approved product also requires FDA approval. If we are not able to obtain FDA approval for any desired future indications for ADAIR or any other future product, such as ADMIR, our ability to effectively market and sell ADAIR or any other future product may be reduced, and our business may be adversely affected.
While physicians may choose to prescribe drugs for uses that are not described in the product’s labeling and for uses that differ from those tested in clinical studies and approved by the regulatory authorities, our ability to promote the products is limited to those indications that are specifically approved by the FDA. These “off-label” uses are common across medical specialties and may constitute an appropriate treatment for some patients in varied circumstances. Regulatory authorities in the United States generally do not regulate the behavior of physicians in their choice of treatments. Regulatory authorities do, however, restrict communications by pharmaceutical companies on the subject of off-label use. If our promotional activities fail to comply with these regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, our failure to follow FDA rules and guidelines relating to promotion and advertising may cause the FDA to send a Warning Letter to the Company (which becomes public) alleging violations of the FDCA and subjecting the Company to other potential enforcement such as to suspend or withdraw an approved product from the market, require a recall or institute fines, or could diminish our reputation and result in disgorgement of money, operating restrictions, injunctions, or criminal prosecution, any of which could harm our business.
Public concern regarding the safety of drug products such as ADAIR could delay or limit our ability to obtain regulatory approval, result in the inclusion of unfavorable information in our labeling, or require us to undertake other activities that may entail additional costs.
In light of widely publicized events concerning the safety risk of certain drug products, the FDA, members of Congress, the Government Accountability Office, medical professionals and the general public have raised concerns about potential drug safety issues. These events have resulted in the withdrawal of drug products, revisions to drug labeling that further limit use of the drug products and the establishment of risk management programs. The Food and Drug Administration Amendments Act of 2007 (“FDAAA”) grants significant expanded authority to the FDA, much of which is aimed at improving the safety of drug products before and after approval. In particular, this law authorizes the FDA to, among other things, require post-approval studies and clinical trials, mandate changes to drug labeling to reflect new safety information and require risk evaluation and mitigation strategies for certain drugs, including certain currently approved drugs. It also significantly expands the federal government’s clinical trial registry and results databank, which we expect will result in significantly increased government oversight of clinical trials. Under the FDAAA, companies that violate these and other provisions of this law are subject to substantial civil monetary penalties, among other regulatory, civil and criminal penalties. The increased attention to drug safety issues may result in a more cautious approach by the FDA in its review of data from our preclinical studies and clinical trials. Data from preclinical studies and clinical trials may receive greater scrutiny, particularlyStaff Delisting Determination under Nasdaq Listing Rule 5810 with respect to safety, which may makeour Common Stock. On January 29, 2024, we filed an amendment to our Charter to implement the FDA or other regulatory authorities more likelyJanuary 2024 Reverse Stock Split to require additional preclinical studies and/or clinical trials. Ifattempt to regain compliance with the FDA requires usMinimum Bid Price Rule. Nasdaq subsequently reported that our Common Stock had a closing bid price above $1.00 per share for 10 consecutive trading days during the 180-day period, but Nasdaq exercised its discretion to conduct additional preclinical studies or clinical trials prior to approving ADAIR, our ability to obtain approval of this proposed product will be delayed. Ifextend the FDA requires us to provide additional preclinical studies and/or clinical data following the approval of ADAIR, the indications for which this proposed product is approved may be limited or there may be specific warnings or limitations on dosing,10-day period and our efforts to commercialize ADAIR may be otherwise adversely impacted.
If we experience delays or difficulties in the enrollment of patients in clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented.
We maydid not be able to continue existing clinical trials, or initiate new clinical trials, for our proposed product if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory authorities outside the U.S. Some of our competitors have ongoing clinical trials for proposed products that treat the same indications as our proposed product, and patients who would otherwise be eligible for our clinical trials may instead enroll in clinical trials of our competitors’ proposed products. Available therapies for the indications we are pursuing can also affect enrollment in our clinical trials. Patient enrollment is affected by other factors including, but not necessarily limited to:
Our inability to enroll a sufficient number of patients for our clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for ADAIR or any future product, such as ADMIR, which would cause the value of our company to decline and limit our ability to obtain additional financing.
Current and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize ADAIR or any other proposed productdeem that we develop and affect the prices we may set.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for ADAIR or any other proposed product that we develop, restrict or regulate post-approval activities and affect our ability to profitably sell ADAIR or any other proposed product for which we obtain marketing approval.
Legislative and regulatory proposals have been made to expand post-approval requirements, restrict sales and promotional activities for pharmaceutical products, and regulate pricing. We are not sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our proposed product, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
Recently enacted legislation, future legislation and healthcare reform measures may increase the difficulty and cost for us to obtain marketing approval for and commercialize ADAIR and for any future product, such as ADMIR, and may affect the prices we may set.
In the United States and some foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system, including cost-containment measures that may reduce or limit coverage and reimbursement for newly approved drugs and affect our ability to profitably sell ADAIR and for any future product, such as ADMIR, for which we obtain marketing approval. In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs, restrict or regulate post-approval activities and improve the quality of healthcare.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (the “MMA”) changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, this legislation authorized Medicare Part D prescription drug plans to limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for any approved products and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
For example, in March 2010, the Affordable Care Act, was enacted in the United States. Among the provisions of the Affordable Care Act of importance to ADAIR and for any future product, the Affordable Care Act: establishes an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents; extends manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; expands eligibility criteria for Medicaid programs; expands the entities eligible for discounts under the Public Health program; increases the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; creates a new Medicare Part D coverage gap discount program; establishes a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical effectiveness research, along with funding for such research; and establishes a Center for Medicare Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending.
At this time, we are unsure of the full impact that the Affordable Care Act will have on our business. There have been judicial and political challenges to certain aspects of the Affordable Care Act. For example, since January 2017, President Trump has signed two executive orders and other directives designed to delay, circumvent, or loosen certain requirements of the Affordable Care Act. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the Affordable Care Act. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the Affordable Care Act have been signed into law. The Tax Cuts and Jobs Act of 2017 (the “Tax Act”) included a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” On January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain Affordable Care Act-mandated fees, including the so-called “Cadillac” tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share. The Bipartisan Budget Act of 2018 (the “BBA”) among other things, amends the Affordable Care Act, effective January 1, 2019, to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole,” by increasing from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D. In July 2018, CMS published a final rule permitting further collections and payments to and from certain Affordable Care Act qualified health plans and health insurance issuers under the Affordable Care Act risk adjustment program in response to the outcome of federal district court litigation regarding the method CMS uses to determine this risk adjustment. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas (“Texas District Court Judge”) ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the Tax Act, the remaining provisions of the ACA are invalid as well. While the Texas District Court Judge, as well as the Trump Administration and CMS, have stated that the ruling will have no immediate effect, it is unclear how this decision, subsequent appeals, and other efforts to repeal and replace the ACA will impact the ACA and our business.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. The Coronavirus Aid, Relief, and Economic Security Act, which was signed into law on March 27, 2020, designed to provide financial support and resources to individuals and businesses affected by the COVID-19 pandemic, suspended these reductions from May 1, 2020 through December 31, 2020, and extended the sequester by one year, through 2030. In addition, on January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Further, there has been heightened governmental scrutiny in the United States of pharmaceutical pricing practices in light of the rising cost of prescription drugs. Such scrutiny has resulted in several recent congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products.
At the federal level, the Trump administration’s budget proposal for fiscal year 2019 contains further drug price control measures that could be enacted during the 2019 budget process or in other future legislation, including, for example, measures to permit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid and to eliminate cost sharing for generic drugs for low-income patients. Additionally, the Trump administration released a “Blueprint” to lower drug prices through proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. The U.S. Department of Health and Human Services has begun the process of soliciting feedback on some of these measures and, at the same time, is implementing others under its existing authority. Although some of these, and other, proposals will require authorization through additional legislation to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for ADAIR and for any future product, such as ADMIR, if approved, or put pressure on our product pricing, which could negatively affect our business, results of operations, financial condition and prospects.
We expect that the Affordable Care Act, these new laws and other healthcare reform measures that may be adopted in the future may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, new payment methodologies and additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize ADAIR and for any future product, such as ADMIR, if approved.
We rely, and expect to continue to rely, on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such trials or complying with applicable regulatory requirements.
We rely on third-party contract research organizations and clinical research organizations to conduct our clinical trials for ADAIR and for any future product, such as ADMIR. We expect to continue to rely on third parties, such as contract research organizations, clinical research organizations, clinical data management organizations, medical institutions and clinical investigators, to conduct all of our clinical trials. The agreements with these third parties might terminate for a variety of reasons, including a failure to perform by the third parties. If we need to enter into alternative arrangements, that could delay our product development activities.
Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we will remain responsible for ensuring that each of our clinical trials are conducted in accordanceregained compliance with the general investigational plan and protocols for the trial and in accordance with good laboratory practice, as appropriate. Moreover, the FDA requires us to comply with standards, commonly referred to as good clinical practices (“GCPs”) for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Regulatory authorities enforce these requirements through periodic inspections of trial sponsors, clinical investigators, and trial sites. If we or any of our clinical research organizations fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure investors that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials complies with GCP regulations. In addition, our clinical trials must be conducted with product produced under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within specified timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
The third parties with whom we have contracted to help perform our clinical trials may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines, or conduct our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our proposed product and will not be able to, or may be delayed in our efforts to, successfully commercialize our proposed product.
If any of our relationships with these third-party contract research organizations or clinical research organizations terminates, we may not be able to enter into arrangements with alternative contract research organizations or clinical research organizations or to do so on commercially reasonable terms. Switching or adding additional contract research organizations or clinical research organizations involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new contract research organization or clinical research organization commences work. As a result, delays could occur, which could compromise our ability to meet our desired development timelines. Though we carefully manage our relationships with our contract research organizations or clinical research organizations, thereMinimum Bid Price Rule. There can be no assurance that we will not encounter similar challenges or delays in the future.
We rely on clinical data and results obtained by third parties that could ultimately prove to be inaccurate or unreliable.
As part of our strategy to mitigate development risk, we seek to develop proposed products with validated mechanisms of action, and we utilize biomarkers to assess potential clinical efficacy early in the development process. This strategy necessarily relies upon clinical data and other results obtained by third parties that may ultimately prove to be inaccurate or unreliable. Further, such clinical data and results may be based on products or proposed products that are significantly different from ADAIR or any future product, such as ADMIR. If the third-party data and results we rely upon prove to be inaccurate, unreliable or not applicable to ADAIR or any future product, we could make inaccurate assumptions and conclusions about our proposed product and our research and development efforts could be compromised.
If we fail to comply with environmental, health, and safety laws and regulations, we could become subject to fines or penalties or incur costs that could harm our business.
We are subject to numerous environmental, health, and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment, and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. Although we believe that the safety procedures for handling and disposing of these materials comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Our failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
If the FDA does not conclude that ADAIR is sufficiently bioequivalent, or demonstrate comparable bioavailability to its reference listed drug (“RLD “), or if the FDA otherwise does not conclude that ADAIR satisfies the requirements of Section 505(b)(2) of the FDCA, approval pathway, the approval pathway for this proposed product will likely take significantly longer, cost significantly more and entail significantly greater complications and risks than anticipated, and the FDA may not approve this proposed product.
A key element of our strategy is to seek FDA approval for ADAIR through Section 505(b)(2) approval pathway. Section 505(b)(2) of the FDCA permits the filing of an NDA that contains full safety and efficacy reports but where at least some of the information required for approval comes from studies not conducted by or for the applicant. Such information could include the FDA’s findings of safety and efficacy in the approval of a similar drug, and for which the applicant has not obtained a right of reference and/or published literature. Such reliance is typically predicated on a showing of bioequivalence or comparable bioavailability to an approved drug.
If the FDA does not allow us to pursue the Section 505(b)(2) approval pathway for ADAIR, or if we cannot demonstrate bioequivalence or comparable bioavailability of ADAIR to an approved product, we may need to conduct additional clinical trials, provide additional data and information, and meet additional standards for regulatory approval. If this were to occur, the time and financial resources required to obtain FDA approval for ADAIR would increase. Moreover, our inability to pursue the Section 505(b)(2) approval pathway could result in new competitive products reaching the market sooner than ADAIR, which could have a material adverse effect on our competitive position and our business prospects. Even if we are allowed to pursue the Section 505(b)(2) approval pathway, we cannot assure investors that ADAIR will receive the requisite approval for commercialization on a timely basis, if at all.
In addition, notwithstanding the approval of a number of products by the FDA under the Section 505(b)(2) approval pathway over the last few years, pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). If the FDA’s interpretation of Section 505(b)(2) is successfully challenged, the FDA may change its policies and practices with respect to the Section 505(b)(2) approval pathway, which could delay or even prevent the FDA from approving any NDA that we submit under Section 505(b)(2).
Even if ADAIR is approved under Section 505(b)(2), the approval may be subject to limitations on the indicated uses for which the product may be marketed or to other conditions of approval, or may contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product, including additional clinical trials.
ADAIR contains a controlled substance, the manufacture, use, sale, importation, exportation, and distribution of which are subject to regulation by state, federal, and foreign law enforcement and other regulatory agencies.
ADAIR contains, and any future product, such as ADMIR, if any, may contain, controlled substances which are subject to state, federal, and foreign laws and other regulations regarding their manufacturing, use, sale, importation, exportation, and distribution. ADAIR’s active ingredient, dextroamphetamine, is classified as a controlled substance under the CSA, and regulations of the DEA. A number of states also independently regulate these drugs as controlled substances. Controlled substances are classified by the DEA as Schedule I, II, III, IV, or V substances, with Schedule I substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. The active ingredient in ADAIR, dextroamphetamine, is listed by the DEA as a Schedule II controlled substance under the CSA. For our current proposed product, and any potential future product, such as ADMIR, containing a controlled substance, we and our suppliers, manufacturers, contractors, customers, and distributors are required to obtain and maintain applicable registrations from state, federal, and foreign law enforcement and regulatory agencies and comply with their laws and regulations regarding the manufacturing, use, sale, importation, exportation, and distribution of controlled substances. An example of such practice is that all Schedule II drug prescriptions must be signed by a physician, physically presented to a pharmacist and may not be refilled without a new prescription. Furthermore, the amount of Schedule II substances that can be obtained for clinical trials and commercial distribution is limited by the CSA and DEA regulations. We may not be able to obtain sufficient quantities of these controlled substances in order to complete our clinical trialsregain compliance or meet commercial demand, even if ADAIR is approved for marketing.
In addition, controlled substances are subject to regulations governing manufacturing, labeling, packaging, testing, dispensing, production and procurement quotas, recordkeeping, reporting, handling, shipment, and disposal. These regulations increasethat the personnel needs and the expense associated with development and commercialization of proposed products that include controlled substances. The DEA and some states conduct periodic inspections of registered establishments that handle controlled substances.
Failure to obtain and maintain required registrations or to comply with any applicable regulations, including quotas imposed by the DEA, could delay or preclude us from developing and commercializing our proposed product that contains a controlled substance and subject us to enforcement action. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to revoke those registrations. In some circumstances, violations could lead to criminal proceedings. Because of their restrictive nature, these regulations could limit commercializationbid price of our proposed product containing controlled substances.
If we,Common Stock will remain above the minimum $1.00 bid price required for any post-split Nasdaq monitoring period or otherwise. We will likely need to effect an additional reverse stock split of our drug products or the manufacturing facilities for our drug products failCommon Stock in an effort to comply with applicable regulatory requirements, a regulatory agency may:
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatoryregain compliance we may lose any marketing approval that we may have been obtained and we may not achieve or sustain profitability, which would adversely affect our business.
The FDA may not approve product labeling for ADAIR that would permit us to market and promote our product in the United States by describing its abuse-deterrent features.
We have invested resources conducting Laboratory Manipulation and Extraction Studies (Category 1), and may spend substantial additional resources conducting Pharmacokinetic Studies (Category 2) and Clinical Abuse Potential Studies (Category 3) to support ADAIR’s abuse-deterrence characteristics, and we believe such studies are consistent with the April 2015 final FDA guidance on opioid medications (the “2015 FDA Guidance”), as no guidance exists on stimulants. However, we have not yet received regulatory approval to market ADAIR and additional data may emerge that could change the FDA’s position on the proposed product label, andMinimum Bid Price Rule, but there can be no assurance that ADAIRsuch a split may be approved or completed.
Even if any other proposed products are approved for marketing with certain abuse-deterrence claims, the 2015 FDA Guidance may not apply to such proposed product, as the said guidance is not binding law and may be superseded or modified at any time. Also, if the FDA determines that our post-marketing data do not demonstrate that the abuse-deterrent properties result in reduction of abuse, or demonstrate a shift to routes of abuse that present a greater risk, the FDA may find that product labeling revisions are needed, and may require the removal of our abuse-deterrence claims. Although ADAIR is very difficult to manipulate into a form that can be snorted, it is not impossible to do so, as was shown by a third-party drug laboratory, hired by us in preparation for human abuse studies, that was able to develop a time-consuming and laborious process to convert ADAIR into a form that could be insufflated. In addition, although ADAIR is difficult to solubilize into a form that can be injected, sophisticated drug abusers may be able to develop methods to manipulate ADAIR into a form that can be injected.
Further, in October 2020, an FDA advisory committee reviewed another abuse deterrent stimulant which relied on different delivery technology and a different active molecule than ADAIR. The advisory committee recommended against the approval of the other product, voting that the benefits did not outweigh the risks. In particular, the advisory committee determined that the other product had not established that it could deter the risk of intranasal abuse based on the results of its human abuse liability study because it failed the study’s primary endpoint. Further the advisory committee voted that the safety of the other product had not been adequately characterized because of safety concerns with two inactive ingredients contained in the other product. ADAIR does not contain either of these inactive ingredients. Nevertheless, we may not be able to conduct a successful pivotal human abuse trial and an FDA advisory committee could vote that the benefits of ADAIR or ADMIR do not outweigh the risks.
We may need to comply with the requirements of the Drug Supply Chain Security Act, which outlines critical steps required to build an electronic, interoperable system to identify and trace certain prescription drugs as they are distributed in the United States.
We may need to comply with the requirements of the Drug Supply Chain Security Act, including those related to product tracing, verification, and authorized trading partners. Signed into law in 2013, the Drug Supply Chain Security Act, amended the FDCA, and is being implemented over a 10-year period. The law’s requirements include the ability to quarantine and promptly investigate a suspect product,national securities exchange, such as a potentially counterfeit, diverted or stolen product, to determine if it is illegitimate, and notifyThe Nasdaq Capital Market, our trading partners and the FDA of any illegitimate product. Such compliance may be time consuming and expensive to implement. Also, in the event that we fail to comply with these requirements, we may face regulatory actions that could affect our ability to commercialize ADAIR.
Even if ADAIR or any other future product, such as ADMIR, we may develop receives marketing approval, there could be adverse effects not discovered during development.
Even if any of our proposed products receive marketing approval, we or others may later identify undesirable side-effects caused by the product or problems with our third-party manufacturers or manufacturing processes, and in either event a number of potentially significant negative consequences could result, including:
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product and could substantially increase commercialization costs or even force us to cease operations.
Our products may cause or contribute to adverse medical events that we are required to report to the FDA and if we fail to do so, we would be subject to sanctions that would materially harm our business.
Post-marketing safety data collection and adverse event reporting are critical elements of FDA’s oversight of drugs and therapeutic biologics available to the American public. The testing that helps to establish the safety of products, such as drugs and therapeutic biologics, is typically conducted on small groups before FDA approves the products for sale. Some problems can remain unknown, only to be discovered when a product is used by a large number of people. An adverse event is any unanticipated experience or side effect associated with the use of a drug or therapeutic biologic in humans, whether or not it is considered related to the product. An adverse event could occur:
During inspections, FDA investigators will review a company’s post-marketing adverse event information to ensure compliance with federal laws and regulations.
Our marketed products are subject to adverse event reporting (ADR) which require that we report to the FDA events related to our products. The timing of our obligation to report under the ADR regulations is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the FDA could take action, including warning letters, untitled letters, administrative actions, criminal prosecution, imposition of civil monetary penalties, revocation of our device clearances or approvals, seizure of our products, or delay in clearance or approval of future products.
Our products may in the future be subject to product recalls. A recall of our products, either voluntarily or at the direction of the FDA or another governmental authority, or the discovery of serious safety issues with our products, could have a significant adverse impact on us.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in their design or manufacture. A government-mandated or voluntary recall could occur as a result of an unacceptable risk to health, contaminated product, manufacturing errors, or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our reputation, results of operations and financial condition, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. WeCommon Stock may also be subject to liability claims,the regulations and restrictions regarding trading in “penny stocks,” which are those securities trading for less than $5.00 per share, and that are not otherwise exempted from the definition of a penny stock under other exemptions provided for in the applicable regulations. These requirements and regulations could severely limit the liquidity of securities in the secondary market because fewer brokers or dealers would likely to be requiredwilling to bearundertake related compliance activities. If our Common Stock is not listed on a national securities exchange, the rules and restrictions regarding penny stock transactions may limit an investor’s ability to sell to a third party and our trading activity in the secondary market may be reduced.
Changes in regulatory requirementsraise capital, adversely affect our business and guidance or unanticipated events during our clinical trials may occur, which may result in necessary changes to clinical trial protocols, which could result in increased costs to us, delay our development timeline or reduce the likelihood of successful completionprice of our clinical trials.
ChangesCommon Stock.
Our amended and restated certificate of incorporation designate specific courts as the exclusive forum for certain litigation that may be initiated by the Company’s stockholders, which could limit its stockholders’ ability to obtain a favorable judicial forum for disputes with us.
Pursuant to our amended and restated certificate of incorporation, unless we consent in writing to the selection of an alternative forum, the Court of ChanceryApril 2023 Reverse Stock Split. The liquidity of the State of Delaware is the sole and exclusive forum for any state law claims for (1) any derivative action or proceeding brought on our behalf; (2) any action asserting a claim of or based on a breach of a fiduciary duty owed by any director, officer or other employee of ours to us or our stockholders; (3) any action asserting a claim pursuant to any provision of the Delaware General Corporation Law, our amended and restated certificate of incorporation; or (4) any action asserting a claim governed by the internal affairs doctrine (the “Delaware Forum Provision”). The Delaware Forum Provision will not apply to any causes of action arising under the Securities Act or the Exchange Act. Our amended and restated certificate of incorporation further provide that unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America shall be the sole and exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act (the “Federal Forum Provision”). In addition, our amended and restated certificate of incorporation provide that any person or entity purchasing or otherwise acquiring any interest in shares of our capitalCommon Stock may be affected adversely by the January 2024 Reverse Stock Split given the reduced number of shares outstanding following the January 2024 Reverse Stock Split, especially if the market price of our Common Stock does not increase as a result of the January 2024 Reverse Stock Split. In addition, the January 2024 Reverse Stock Split may have increased the number of stockholders who own odd lots (less than 100 shares) of our Common Stock, creating the potential for such stockholders to experience an increase in the cost of selling their shares and greater difficulty effecting such sales. We expect to need to complete an additional reverse stock is deemed to have noticesplit of and consented to the Delaware Forum Provision and the Federal Forum Provision; provided, however,our Common Stock given that stockholders cannot and willour current bid price does not be deemed to have waived our compliancecomply with the federal securities lawsMinimum Bid Price Rule, but there can be no assurance that such a split will be approved or completed.
We recognizeinvesting requirements of those investors. Consequently, the trading liquidity of our Common Stock may not improve.
Risks Relating to Intellectual Property
We have filed multiple patent applications and have two issued patents by the U.S. PTO. These or any other patent applications mayJanuary 2024 Reverse Stock Split did not result in issued patents, and asthe price of our Common Stock meeting the Minimum Bid Price Rule. As a result, we may have limited protectionwill likely need to effect an additional reverse stock split of our proprietary technology in the marketplace.
We have had two patents granted and one additional patent application directed to ADAIR for ADHD and narcolepsy filed in the United States, and we are seeking patent protection for ADAIR internationally in several foreign countries and territories, including the EU, Canada, Japan and China. The U.S. patents will expire in 2037.Common Stock, which may not be approved or completed. It is impossible to predict whether or how our PCT applicationcannot be assured that a reverse stock split will result in any issued patent. Even if the pending application issues, it may issue with claims significantly narrower than those we currently seek.
The patent position of biotechnology and biopharmaceutical companies is generally uncertain because it involves complex legal and factual considerations. The standards applied by the U.S. PTO and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in biotechnology and biopharmaceutical patents. Consequently, a patent may not issue from our pending patent applications. Therefore, we do not know the degree of future protection that we will have on any proprietary product or technology that we have or may develop.
If we are unable to obtain and maintain patent protection for our technology and products or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection in the United States and other countries with respect to ADAIR or any other future product, such as ADMIR, that we may license or acquire and the methods we use to manufacture them, as well as successfully defending these patents and trade secrets against third-party challenges. We seek to protect our proprietary position by filing patent applications in the United States and abroad related to our proposed products. We will only be able to protect our technologies from unauthorized use by third parties to the extent that valid and enforceable patents or trade secrets cover them.
The patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. If our licensors or we fail to obtain or maintain patent protection or trade secret protection for ADAIR or any other future product, such as ADMIR, we may license or acquire, third parties could use our proprietary information, which could impair our ability to competesustained proportionate increase in the market price of our Common Stock, which is dependent upon many factors, including our business and financial performance, general market conditions, and prospects for future success, which are unrelated to the number of shares of our Common Stock outstanding. In fact, the January 2024 Reverse Stock Split did not result in a sustained proportionate increase in the market price of our Common Stock, in part due to the dilutive effects of the Equity Offering and subsequent warrant exercises. It is not uncommon for the market price of a company’s common stock to decline in the period following a reverse stock split or following an offering.
The patent position of biotechnology and biopharmaceutical companies generally is highly uncertain, involves complex legal and factual questions, and has inCommon Stock. In recent years, many such changes have been the subject of much litigation. In addition, no consistent policy regarding the breadth of claims allowed in biopharmaceutical or biotechnology patents has emergedmade and changes are likely to datecontinue to occur in the United States. The patent situation outside the U.S. is even more uncertain. Thefuture. Future changes in tax laws of foreign countries may not protect our rights to the same extent as the laws of the U.S. For example, European patent law restricts the patentability of methods of treatment of the human body more than U.S. law does. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until eighteen (18) months after a first filing, or in some cases at all. Therefore, we cannot know with certainty whether we or our licensors were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or whether we were the first to file for patent protection of such inventions. In the event that a third party has also filed a U.S. patent application relating to our proposed products or a similar invention, we may have to participate in interference proceedings declared by the U.S. PTO to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that our efforts would be unsuccessful, resulting in a material adverse effect on our U.S. patent position. As a result, the issuance, scope, validity, enforceability, and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued which protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. On September 16, 2011, the Leahy-Smith America Invents Act (the “Leahy-Smith Act”) was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted and may also affect patent litigation. The U.S. PTO recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, andcash flow, financial condition.
Moreover, we may be subject to a third-party pre-issuance submissioncondition, or results of prior art to the U.S. PTO, or become involved in opposition, derivation, reexamination, inter parties review, post-grant review or interference proceedings challengingoperations.
Even if our patent applications issue as patents, theyCommon Stock may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumventdevelop and our owned or licensed patents by developing similar or alternative technologies or products in a non-infringing manner.
The issuance of a patent does not foreclose challenges to its inventorship, scope, validity or enforceability. Therefore, our owned and licensed patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing, and regulatory review of new proposed products, patents protecting such proposed products might expire before or shortly after such proposed products are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
Because it is difficult and costly to protect our proprietary rights, westockholders may not be able to ensureresell their protection.
The degreeshares of future protectionCommon Stock for our proprietary rights is uncertain, because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our issued patents or other intellectual property. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their patents. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, in whole or in part, construe the patent’s claims narrowly or refuse to stop the other party from using the technologyprofit, if at issue on the grounds that our patents do not cover the technology in question. all.
If we are sued for infringing intellectual property rights of third parties, it will be costly and time consuming, and an unfavorable outcome in any litigation would harm our business.
Our ability to develop, manufacture, market and sell ADAIR or any other future product, such as ADMIR, that we may license or acquire depends upon our ability to avoid infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the general fields of ADHD and cover the use of numerous compounds and formulations in our targeted markets. Because of the uncertainty inherent in any patent or other litigation involving proprietary rights, we and our licensors may not be successful in defending intellectual property claims by third parties, which could have a material adverse effect on our results of operations. Regardless of the outcome of any litigation, defending the litigation may be expensive, time-consuming and distracting to management. In addition, because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that ADAIR may infringe. There could also be existing patents of which we are not aware that ADAIR may inadvertently infringe
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology and biopharmaceutical industries generally. If a third party claims that we infringe on their patents or misappropriated their technology, we could face a number of issues, including:
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses, and could distract our technical and management personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could compromise our ability to compete in the marketplace.
We may need to license certain intellectual property from third parties, and such licenses may not be available or may not be available on commercially reasonable terms.
A third party may hold intellectual property, including patent rights that are important or necessary to the development and commercialization of ADAIR or any other future product, such as ADMIR. It may be necessary for us to use the patented or proprietary technology of third parties to commercialize ADAIR or any other future product. If we are unable to obtain a license from these third parties, or unable to obtain a license, on commercially reasonable terms, our business could be harmed.
If we fail to comply with our obligations in our intellectual property licenses and funding arrangements with third parties, we could lose rights that are important to our business.
In the future, we may become party to licenses that are important for product development and commercialization. If we fail to comply with our obligations under current or future license and funding agreements, our counterparties may have the right to terminate these agreements, in which event we might not be able to develop, manufacture or market any product or utilize any technology that is covered by these agreements or may face other penalties under the agreements. Such an occurrence could materially and adversely affect the value of a proposed product being developed under any such agreement or could restrict our drug discovery activities. Termination of these agreements or reduction or elimination of our rights under these agreements may result in our having to negotiate new or reinstated agreements with less favorable terms, or cause us to lose our rights under these agreements, including our rights to important intellectual property or technology.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and biopharmaceutical industry, we employ individuals who were previously employed at other biotechnology or biopharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patent protection for ADAIR or any future product, such as ADMIR, we also rely on trade secrets, including unpatented know-how, technology, and other proprietary information, to maintain our competitive position, particularly where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We limit disclosure of such trade secrets where possible, but we also seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who do have access to them, such as our employees, our licensors, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors, and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants. Despite these efforts, any of these parties may breach the agreements and may unintentionally or willfully disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the U.S. are less willing or unwilling to protect trade secrets. Moreover, if any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
Risks Relating to Securities Markets and Our Common Stock
An active, liquid and orderly market for our common stock may not be maintained.
Prior to our February 2021 initial public offering, there had been no public market for our common stock. Our common stock only recently began trading on the Nasdaq Capital Market, but we can provide no assurance that we will be able to develop and sustain an active trading market for our common stock. Even if an active trading market is developed, itshares of Common Stock may notnever develop or be sustained. The lack ofIf an active market for our Common Stock does not develop or is not sustained, it may impair your abilitybe difficult for our stockholders to sell yourtheir shares at the time you wish to sell theman attractive price or at a price that you consider reasonable. An inactive market may also impair our ability to raise capital by selling shares and may impair our ability to acquire other businesses or technologies using our shares as consideration, which, in turn, could materially adversely affect our business.
all.
Although common stock is listed on The Nasdaq Capital Market, we may be unable to continue to satisfy the listing requirements and rules, including the director independence requirements and certain financial metrics for our stockholders’ equity and market value of listed securities or net income from continuing operations. If we are unable to satisfy The Nasdaq Capital Market criteria for maintaining our listing, our securities could be subject to delisting. If The Nasdaq Capital Market delists our securities, we could face significant consequences, including:
In addition, we would no longer be subject to The Nasdaq Capital Market rules, including rules requiring us to have a certain number of independent directors and to meet other corporate governance standards.
Our stock may be subject to substantial price and volume fluctuations due to a number of factors, many of which are beyond our control and may prevent our stockholders from reselling our common stock at a profit.
The market prices for securities of biotechnology and biopharmaceutical companies have historically been highly volatile, and the market has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. In particular, the trading prices for pharmaceutical, biopharmaceutical and biotechnology companies have been highly volatile as a result of the COVID-19 pandemic.
The market price of our common stock is likely to be highly volatile and may fluctuate substantially due to many factors, including:
In addition, the stock markets have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many biopharmaceutical companies. If we were to become involved in securities litigation, it could subject us to substantial costs, divert resources and the attention of management from our business and adversely affect, our business, operating results, financial condition and cash flows.
If securities or industryresearch analysts do not publish research or cease publishingreports, or publish unfavorable research or reports, about us, our business, or our market, or if they adversely change their recommendations or publish negative reports regarding our business or our common stock a liquid trading market, if any, for our common stock may not develop, and if it does, our share price and trading volume could decline.
Because
We have never declared or paid cash dividends on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our businesscash flow and, do not anticipate declaring or paying any cash dividends inconsequently, could negatively impact the foreseeable future.
Our significant stockholders may exert a substantial influence on actions requiring a stockholder vote, potentially in a manner that you do not support.
As of March 15, 2021, Medice, through its affiliated entity, Salmon Pharma owns approximately 22.4% of our issued and outstanding shares of common stock, and accordingly controls approximately 22.4% of our voting power. In addition, Arcturus owns approximately 12.4% of our issued and outstanding shares of common stock, and accordingly controls approximately 12.4% of our voting power. Members of our board of directors and management beneficially own approximately 39.4% of our issued and outstanding shares of common stock, and accordingly will control approximately 39.4% of our voting power, assuming exercise of all stock options exercisable within 60 days of March 15, 2021. If Salmon Pharma, Arcturus or any member of our board or management acquires additional shares of common stock in the aftermarket or in privately negotiated transactions, this would increase their control. Factors that would be considered in making such additional purchases would include consideration of the current trading price of our common stock.
Salmon Pharma and Arcturus’s large ownership stake may allow it to exert a substantial influence on actions requiring a stockholder vote, potentially in a manner that you do not support, including amendments to our amended and restated certificate of incorporation, election of our board of directors, removal of any of our directors, adoption of measures that could delay or prevent a change in control or impede a merger, takeover, or other business combination involving us, and approval of other major corporate transactions. In addition, Salmon Pharma and Arcturus’s stock ownership may discourage a potential acquirer from making a tender offer or otherwise attempting to obtain control of us, which in turn could reduce our stock price or prevent our stockholders from realizing a premium over our stock price. Accordingly, our stockholders other than Salmon Pharma and Arcturus may be unable to influence management and exercise control over our business.
Our charter documents and Delaware law may inhibit a takeover that stockholders consider favorable.