
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-KUNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the fiscal year ended December 31 |
or
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the transition period from ____ to ____ |
For the transition period from ____ to ____
Commission File Number: 001-36357
LIPOCINE INC.
(Exact name of registrant as specified in its charter)
| Delaware | 99-0370688 | |
(State or Other Jurisdiction of Incorporation or Organization) | (IRS Employer Identification No.) | |
675 Arapeen Drive, Suite 202, Salt Lake City, Utah | 84108 | |
| (Address of Principal Executive Offices) | (Zip Code) |
801-994-7383801-994-7383
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Trading Symbol(s) | Name of Each Exchange on Which Registered | ||
| Common Stock, par value $0.0001 per share | LPCN | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrantregistrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o☐ Nox ☒
Indicate by check mark if the Registrantregistrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Securities Exchange Act. Yes o☐ Nox ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes: xYes: ☒ No ¨☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§220.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yesx ☒ No ¨☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act (Check one):Act:
| Large accelerated filer | |
| Accelerated filer | |
| Non-accelerated filer | |
| Smaller reporting company | |
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.¨ ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨☐ Nox ☒
Outstanding Shares
The aggregate market value of the common stock held by non-affiliates of the Registrantregistrant was $78$25.6 million as of June 30, 2017.2023. For purposes of calculating the aggregate market value of shares of our common stock held by non-affiliates as set forth on the cover page of this Annual Report on Form 10-K, we have assumed that all outstanding shares are held by non-affiliates, except for shares held by each of our executive officers, directors and 10% or greater stockholders. However, this assumption should not be deemed to constitute an admission that all executive officers, directors and 10% or greater stockholders are, in fact, affiliates of our company, or that there are not other persons who may be deemed to be affiliates of our company. Further information concerning shareholdings of our officers, directors and principal stockholders is included or incorporated by reference in Part III, Item 12 of this Annual Report on Form 10-K.
As of March 9, 2018,5, 2024, the registrant had 21,264,539 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE:
Portions of the Registrant's definitive Proxy Statement for its 2018 Annual Meeting of Stockholders are incorporated by reference into Part III of this Form 10-K.None
TABLE OF CONTENTS
TABLE OF CONTENTS
| 2 |
FORWARD-LOOKING STATEMENTS
THIS ANNUAL REPORT ON FORM 10-K, IN PARTICULAR “ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION,” AND “ITEM 1. BUSINESS,” CONTAINS FORWARD-LOOKING STATEMENTS WITHIN THE MEANING OF SECTION 27A OF THE SECURITIES ACT OF 1933, AS AMENDED, AND SECTION 21E OF THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED, that involve risks and uncertainties. Forward-looking statements provide current expectations of future events based on certain assumptions and include any statement that does not directly relate to any historical or current fact. Forward-looking statements may refer to such matters as products, product benefits, pre-clinical and clinical development timelines, clinical and regulatory expectations and plans, REGULATORY DEVELOPMENTS AND REQUIREMENTS, THE RECEIPT OF REGULATORY APPROVALS, THE EXPECTATIONS FOR AND RESULTS OF CLINICAL TRIALS, PATIENT ACCEPTANCE OF LIPOCINE’S PRODUCTS, MANUFACTURING AND COMMERCIALIZATION OF LIPOCINE’S PRODUCTS, anticipated financial performance, future revenues or earnings, business prospects, projected ventures, new products and services, anticipated market performance, future expectations for liquidity and capital resources needs and similar matters. Such words as “may”, “will”, “expect”, “continue”, “estimate”, “project”, “intend”, and “potential” and similar terms and expressions are intended to identify forward looking statements. Forward-looking statements are not guarantees of future performance and our actual results may differ significantly from the results discussed in the forward-looking statements. Factors that might cause such differences include, but are not limited to, those discussed in Part I, Item 1A (Risk Factors)“Risk Factors” of this Form 10-K. Except as required by applicable law, we assume no obligation to revise or update any forward-looking statements for any reason.
There are a number of risks, uncertainties and other important factors that could cause our actual results to differ materially from the forward-looking statements contained in this Annual Report on Form 10-K. Such risks, uncertainties and other important factors include, among others, the risks, uncertainties and factors set forth in “Risk Factors,” and the following risks, uncertainties and factors:
| ● | our and our licensee’s plans to develop and commercialize any products or future product candidates; | |
| ● | our ongoing and planned clinical trials; | |
| ● | the timing of and our ability to obtain regulatory approvals or fast track or orphan drug designation, breakthrough designation or IND clearance for any future product candidates; | |
| ● | our ability to monetize non-core product candidates; | |
| ● | our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; | |
| ● | the rate and degree of market acceptance and clinical utility of any products or future product candidates, if approved; | |
| ● | significant competition in our industry; | |
| ● | our intellectual property position; | |
| ● | loss of key members of management; | |
| ● | failure to successfully execute our strategy; and | |
| ● | our failure to maintain effective internal controls. |
There may be other factors that may cause our actual results to differ materially from the forward-looking statements, including factors disclosed in “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” You should evaluate all forward-looking statements made in this Annual Report on Form 10-K in the context of these risks and uncertainties.
We caution you that the risks, uncertainties and other factors referred to above may not contain all of the risks, uncertainties and other factors that are important to you. In addition, we cannot assure you that we will realize the results, benefits or developments that we expect or anticipate or, even if substantially realized, that they will result in the consequences or affect us or our business in the way expected. All forward-looking statements in this Annual Report on Form 10-K apply only as of the date made and are expressly qualified in their entirety by the cautionary statements included in this Annual Report on Form 10-K. We undertake no obligation to publicly update or revise any forward-looking statements to reflect subsequent events or circumstances.
| 3 |
PART I
Organizational BackgroundITEM 1.BUSINESS
Marathon Bar Corp.General
Lipocine Inc. (“Marathon Bar”Lipocine” or the “Company”) wasis incorporated on October 13, 2011, inunder the laws of the State of Delaware. On July 24, 2013, Marathon Bar and MBAR Acquisition Corp. (“Merger Sub”), a wholly owned subsidiary of Marathon Bar, and Lipocine Operating Inc. (“Lipocine Operating”), a privately held company incorporated in Delaware, executed an Agreement and Plan of Merger (“Merger Agreement”). Pursuant to the Merger Agreement, Merger Sub merged with and into Lipocine Operating and Lipocine Operating was the surviving entity. Additionally, pursuant to the Merger Agreement, Marathon Bar changed its name to Lipocine Inc. The Merger is accounted for as a reverse-merger and recapitalization.
General
We are a specialty pharmaceuticalbiopharmaceutical company focused on applyingleveraging our proprietary Lip’ral platform to develop differentiated products through the oral drug delivery technology for the development of pharmaceutical products in the area of men’s and women’s health.previously difficult to deliver molecules, focused on treating Central Nervous System (“CNS”) disorders. Our proprietary delivery technologies are designed to improve patient compliance and safety through orally available treatment options. Our primary development programs are based on oral delivery solutions for poorly bioavailable drugs. We have a portfolio of proprietarydifferentiated innovative product candidates designed to produce favorable pharmacokinetic (“PK”) characteristicsthat target high unmet needs for neurological and facilitate lower dosing requirements, bypass first-pass metabolism in certain cases, reduce side effects,psychiatric CNS disorders, liver diseases, and eliminate gastrointestinal interactions that limit bioavailability. Our leadhormone supplementation for men and women.
We entered into a license agreement for the development and commercialization of our product candidate, TLANDO™TLANDO®, is an oral treatment indicated for testosterone replacement therapy (“TRT”) in adult males for conditions associated with a deficiency or absence of endogenous testosterone (primary or hypogonadotropic hypogonadism) comprised of testosterone undecanoate (“TU”). On January 12, 2024, we entered into a license agreement (the “Verity License Agreement”) with Gordon Silver Limited (“GSL”) and is currently under reviewVerity Pharmaceuticals, Inc. (“Verity” or our “Licensee”), pursuant to which we granted to Verity an exclusive, royalty-bearing, sublicensable right and license to commercialize TLANDO for TRT in the U.S. and Canada. Any required post-marketing studies by the United States Food and Drug Administration (“FDA”) will also be the responsibility of our Licensee, Verity. On January 31, 2024, our license agreement with former licensee, Antares Pharma, Inc. (“Antares”), was terminated and the transition of the U.S. commercial rights for TLANDO from Antares to Verity was completed on February 1, 2024, for the distribution, marketing and sale of TLANDO. The Verity License Agreement also provides Verity with a Prescription Drug User Fee Actlicense to develop and commercialize LPCN 1111 (also referred to as TLANDO XR), the Company’s potential next generation, once daily oral product candidate for testosterone replacement therapy comprised of testosterone tridecanoate (“PDUFA”TT”) action goal date, in the U.S. and Canada.
Additional clinical development pipeline candidates include: LPCN 1154 for postpartum depression (“PPD”); LPCN 2101 for epilepsy; LPCN 2203 for essential tremor; and LPCN 1148 comprising a novel prodrug of May 8, 2018. The FDA has deemed the resubmission a complete response to its June 2016 Complete Response Lettertestosterone, testosterone laurate (“CRL”TL”) that requested additional information related to the dosing algorithm, for the proposed label. Themanagement of decompensated cirrhosis. In addition to our CNS product candidates, we have non-core assets for which we expect to seek partnerships to enable further development for which we do not plan to devote significant resources to developing in the future. These non-core assets include TLANDO New Drug Application (“NDA”) is based on the resultsfor territories outside of North America, LPCN 1148 which we intend to explore partnering upon completion of the Dosing Validation (“DV”)study. The DV study confirmed the efficacy of TLANDO with a fixed dose regimen without need for dose adjustment. TLANDO was well tolerated upon 52-week exposure with no reports of drug related Serious Adverse Events (“SAEs”). On January 10, 2018, the Bone, Reproductive and Urologic Drugs Advisory Committee (“BRUDAC”) of the FDA voted six in favor and thirteen against the acceptability of the overall benefit/risk profile to support approval of TLANDO as a TRT. We continue to work with the FDA in addressing topics discussed by BRUDAC. We may receive another CRL. This would cause delays and added expense to the process of seeking approval of TLANDO. Additional pipeline candidates include LPCN 1111, a next generation oral testosterone therapy product with the potential for once daily dosing, that is currently in Phase 2 testing,study, LPCN 1144, an oral prodrug of androgen receptor modulator for the treatment of non-cirrhotic non-alcoholic steatohepatitis (“NASH”) which has completed Phase 2 testing; and LPCN 1107, which has the potential to becomepotentially the first oral hydroxyprogesteronehydroxy progesterone caproate (“HPC”) product indicated for the prevention of recurrent preterm birth (“PTB”), which has completed a dose finding clinical study in pregnant women and has completed an End-of-Phase 2 meeting withbeen granted orphan drug designation by the FDA.
| 4 |
The following charts summarize the status of our product candidate development programs:

IndustryCorporate Strategy
Our goal is to become a leading biopharmaceutical company focused on leveraging our proprietary Lip’ral drug delivery technology platform to develop differentiated products through oral delivery of previously difficult to deliver molecules for CNS disorders. The key components of our strategy are to:
Testosterone Background
Testosterone, or T, is the primary circulating sex hormone in malesAdvance LPCN 1154 and is criticalother CNS product candidates. We intend to the development and maturation of reproductive tissues as well as other secondary male characteristics such as muscle growth and bone density. Synthesized in the gonads of both males (testis) and females (ovaries), testosterone circulates bound to sex hormone binding globulin (“SHBG”, ~60%), loosely bound to albumin, a protein in the blood that binds to testosterone (~40%), or as a free molecule (~1%). Once circulating, testosterone enters cells directly and activates a network of proteins that ultimately result in metabolic conversions, which in turn produce observable effects. The concentration of circulating testosterone can vary drastically over time or between individuals and can be dependentfocus on genetic factors, other medical conditions, lifestyle behaviors, and/or concurrent medication administration. Although large variability exists, the effects of testosterone are also determined by a number of factors including the amount of steroid penetration, sensitivity of enzymes and cellular proteins to the hormone, and the action of genomic receptors at the cellular level. As a result, assessing clinically low, or potentially high, levels of naturally occurring testosterone often requires a number of quantitative tests in conjunction with clinical evaluations.
Hypogonadism Overview
Low serum testosterone causes significant clinical impact and can result in erectile dysfunction, low libido, decreased muscle mass and strength, increased body fat, decreased bone density, decreased vitality and depressed mood. Furthermore, low serum testosterone concentrations have been found to be an independent predictor of a number of cardiovascular risk factors including obesity, abnormal lipid levels, hypertension, type 2 diabetes, and systemic inflammation. Well-designed, prospective clinical trials have determined that low testosterone levels are also independently associated with mortality risk. These findings have generated interest amongst the medical community and general public regarding the importance of maintaining appropriate serum testosterone levels, which has stimulated growth of the testosterone replacement therapy market which peaked in 2013. The testosterone therapy market has contracted modestly since 2013 due to a number of factors including the withdrawal of direct to consumer advertising mid-2014.
Hypogonadism typically refers to a permanent deficiency of sex hormones rather than a temporary deficiency that may be related to acute/chronic illnesses or other medical, personal, or environmental factors. Primary hypogonadism describes disease states that intrinsically affect the gonads. Examples of these include the genetic disorders, Turner syndrome and Kleinfelter syndrome. Secondary hypogonadism refers to disease states that affect gonadal-related structures such as the hypothalamus and pituitary gland that directly impact the development of gonadsendogenous neuroactive steroids (“NAS”) which have broad applicability in treating various CNS conditions where we can leverage our technology platform to develop highly differentiated oral therapeutics. Our priority is on the development of LPCN 1154, a fast-acting oral antidepressant for postpartum depression (“PPD”) with potential for outpatient use.
Support our Licensee in commercialization of our licensed oral TRT option. We believe the TRT market needs a differentiated, convenient oral option. We have exclusively licensed rights to TLANDO to Verity for commercialization of TLANDO in the U.S. and as suchCanada and have also exclusively licensed development and commercialization rights for LPCN 1111 in the releaseU.S. and Canada to Verity. We plan to support our Licensee’s efforts to effectively enable the availability of testosteroneTLANDO to patients in a timely manner, in addition to receiving milestone and other sexual hormones. Kallmann syndrome, in which patients fail to undergo all of the changesroyalty payments associated with puberty, is a type of secondary hypogonadism. Although a number of inherited diseases are knownTLANDO commercialization as agreed to affect the gonads either directly or indirectly, it is generally believed that the majority of individuals with hypogonadism develop the condition as a result of age-related declines in testosterone or other acquired conditions.
Diagnosis and Treatment of Hypogonadism
Epidemiological studies have determined that total testosterone follows an age-related decline with mean serum concentration at the age of 75 years approximately two thirds that at 25 years. Because naturally occurring testosterone exists at low concentrations, with normal testosterone levels in the rangeVerity License Agreement.
Develop partnership(s) to continue the advancement of 300non-core pipeline assets. We continuously strive to 1080 ng/dL automated platform-based assays have been found to lack specificityprioritize our resources in seeking partnerships of our pipeline assets. We are currently exploring partnering (i) LPCN 1144, our candidate for treatment of non-cirrhotic NASH, (ii) LPCN 1148, for the management of decompensated cirrhosis, and (iii) LPCN 1107, our candidate for prevention of pre-term birth. We are prone to inter-lab variability. The lackalso exploring the possibility of reliable laboratory tests is complicated further by the inter-individual variability seen in an unaffected population. Thus, in order to accurately diagnose hypogonadism in a male, at least two morning serum testosterone levels are performed in conjunction with a clinical assessment of patient symptoms. Patients can only be diagnosed when they present with symptoms that are directly related to low morning serum testosterone level.
Treatment for male hypogonadism (both primary and secondary) is testosterone replacement therapy, or TRT. Some of the reported benefits of TRT include improved libido and sexual function; increased bone density, muscle development, and cognition;licensing LPCN 1021 (known as well as a reduction in other risk factors caused by low testosterone.
Testosterone Replacement Market
Due to the wide variability in therapeutic range and other medical conditions that may confound an accurate diagnosis, there is a consensus that male hypogonadism is significantly undertreated. A large study of 2,162 men over the age of 45 visiting primary care practicesTLANDO in the United States revealed that the prevalence of hypogonadism is about 39%. Based on this prevalence rate and the U.S. Census Bureau’s 2014 estimate that there are 45 million men between 45 and 75 years old, approximately 19 million men in the U.S. may have low testosterone. In the study, fewer than 4% of patients were receiving treatment for hypogonadism.
Testosterone replacement therapies have been commercially available inStates) to third parties outside the United States and Canada, although no licensing agreement has been entered into by the Company.
Our Development Pipeline Product Candidates
Our pipeline of clinical development candidates includes LPCN 1154 for over 70 yearspostpartum depression (“PPD”), LPCN 2101 for epilepsy, LPCN 1148, an androgen therapy for the management of cirrhosis and have followedLPCN 2203 for essential tremor. We will continue to explore other product development candidates targeting CNS indications with a progressionsignificant unmet need. We will also continue efforts to enter into partnership arrangements for the continued development and/or marketing of delivery systems that included subcutaneous, or under-the-skin, intramuscular, transdermal patch,LPCN 1144, LPCN 1148, and finally topical gels, which initially surfaced in 2000,LPCN 1107 and creams. In 2014, a long acting intramuscular injectionfor TRT assets (TLANDO and an intranasal delivery system for testosterone were approved. The difficulty in creating an easy to use/administer and clinically effective testosterone therapy is related to the molecule’s complex pharmacokinetics. Pharmacokinetics, or PK, describe how the body affects a specific drug after administration through the mechanismLPCN 1111) outside of absorption and distribution, as well as the chemical changes of the substance in the body. For example, oral therapies, which would ideally be the most popular route of delivery, require multiple, high daily doses due to low bioavailability. Bioavailability is the fraction of a drug dose that is actually absorbed into the bloodstream. Additionally, the few oral therapies that have been used in the United States previously quickly went out of favor after significant side effects were revealed, most notably liver toxicity.and Canada.
Currently, the U.S. TRT market consists of therapies that exist in four forms:
Although transdermal patches were previously the most desirable application type, gel-based TRT has gained increasing popularity due to improved skin tolerability. Despite becoming a popular approach to male hypogonadism treatment, topical gels are not without limitations. Topical gels place women and children at risk of testosterone transference (secondary exposure to gels), which has prompted the FDA to add black box warnings relating to testosterone transference in the label of approved topical products. Despite these limitations, gels have continued to demonstrate significant market penetration.
The male testosterone market was $1.7 billion in 2017 according to IMS Health data. Additionally testosterone replacement prescriptions were approximately 7.0 million in 2017 according to IMS Health data. Injectables are the predominant dosage form in this market in terms of annual prescriptions written although topical gels garner the majority of the dollar sales and also have a significant share of total annual prescriptions. The historical growth in the market was driven by increasing recognition by both patients and providers of the prevalence of hypogonadism and its far-reaching medical consequences. Top treatments are marketed by AbbVie and Endo.
Product Candidates
Our current portfolio, shown below, includes our lead product candidate, TLANDO, an oral testosterone replacement therapy that is currently under review by the FDA with a PDUFA goal date of May 8, 2018. Additionally, we are in the process of establishing our pipeline of other clinical candidates including a next generation potential once daily oral testosterone replacement therapy, LPCN 1111, and an oral therapy for the prevention of preterm birth, LPCN 1107.
These products are based on our proprietary Lip’ral drug delivery technology platform. Lip’ral-based TLANDO was approved by the FDA in March 2022. Lip’ral technology is a patented technology based on lipidic compositions which form an optimal dispersed phase in the gastrointestinal environment for improved absorption of insoluble drugs. The drug loaded dispersed phase presents the solubilized drug efficiently at the absorption site (gastrointestinal tract membrane) thus improving the absorption process and making the drug less dependent on physiological variables such as dilution, gastro-intestinal pH and food effects for absorption. Lip’ral basedLip’ral-based formulation enables improved solubilization and higher drug-loading capacity, which can lead to improved bioavailability, reduced dose, faster and more consistent absorption, reduced variability, reduced sensitivity to food effects, improved patient compliance, and targeted lymphatic delivery where appropriate.
Oral Programs for CNS Disorders
Some preferred endogenous or naturally occurring NAS present in central nervous system act as positive allosteric modulators (“PAM”) of the GABAA receptor, the major biological target of the inhibitory neurotransmitter γ-aminobutyric acid (“GABAA”). To improve oral delivery of these modulators, several synthetic NAS derivatives of endogenous GABAA receptor PAMs, have been developed for therapeutic use in the past few decades.
We believe through utilization of our proprietary technology we may have the ability to enable effective oral delivery of endogenous GABAA receptor PAMs which historically had been deemed to be not orally bioavailable. As a novel drug class, NAS have received considerable attention because of their potential to treat various neuropsychiatric conditions including depression, movement disorders, epilepsy, anxiety, and neurodegenerative diseases. We have conducted Phase 1 pharmacokinetic (“PK”) studies for each of our three lead NAS candidates which have demonstrated promising PK results, safety, and tolerability, and we will continue to evaluate additional undisclosed CNS-focused candidates.
LPCN 1154: Product Candidate for PPD
Our most advanced NAS candidate is LPCN 1154, a non-invasive, oral formulation of the neuroactive steroid brexanolone which we are developing for the treatment of PPD. The FDA agreed with our proposal for establishing the efficacy of LPCN 1154 through a pivotal PK bridge to an approved IV infusion brexanolone via a 505(b)(2) NDA filing. The company has completed an oral PK study, a food effect study, and a pilot PK bridge study of LPCN 1154, as a prelude to a pivotal study using the scaled up “to be marketed” formulation required for NDA filing.
| 6 |
PPD
PPD (“Postpartum depression”), a type of major depressive disorder with onset either during pregnancy or within four weeks of delivery, refers to depression persisting up to 12 months after childbirth. PPD can be clinically segmented by the severity of symptoms and presence of a comorbidity, including epilepsy. Approximately 1 in 8 mothers suffers from PPD in the United States alone; this equates to approximately 500,000 women being affected by PPD annually.
Disease Overview - PPD
| ● | PPD is distinct from the “baby blues,” a condition that up to 70% of all new mother’s experience; “baby blues” tend to be short-lived emotional conditions that do not interfere with daily activities. | |
| ● | Symptoms of PPD include hallmarks of major depression, including, but not limited to, sadness, depressed mood, loss of interest, change in appetite, insomnia, sleeping too much, fatigue, difficulty thinking/concentrating, excessive crying, fear of harming the baby/oneself, and/or thoughts of death or suicide. | |
| ● | During pregnancy, levels of endogenous NAS increase considerably along with levels of progesterone; however, they drop sharply postpartum. It has been hypothesized that the rapid perinatal decrease in circulating levels of endogenous NASs may be involved in the development of PPD. The first and only approved treatment option for PPD is an injectable containing endogenous NAS. | |
| ● | Depression may persist long after child delivery. Additionally, approximately 40% of women relapse in subsequent pregnancies or on other occasions. | |
| ● | Psychiatric comorbidities are common in patients with epilepsy. Patients with epilepsy are at high risk for major depressive disorders and PPD. Reported PPD rates are higher among women with epilepsy than the general population. |
Associated Risk Factors
| ● | Genetic: family history and/or previous experience of depression or other mood disorders | |
| ● | Physiological: rapid changes in sex hormones, stress hormones, and thyroid hormone levels during and after delivery | |
| ● | Environmental: stressful life events, changes in relationships at home and at work, and/or lack of familial support |
Unmet Medical Need
We believe there is considerable unmet need within women with PPD due to lack of convenient and fast-acting oral therapies. Selective Serotonin Reuptake Inhibitors (“SSRIs”) have been the traditional first-line choice for women with severe PPD requiring weeks for onset of efficacy; therefore, a need for an oral treatment option with a faster onset of action remains a significant unmet need in treating PPD, especially in mothers with moderate to severe depression prone to harmful actions.
Injectable brexanolone (Zulresso™, SAGE Therapeutics) became the first FDA-approved treatment for postpartum depression. However, numerous factors limit the utilization of injectable brexanolone such as method of administration, cost, and safety concerns. In addition to Zulresso, SAGE Therapeutics received FDA approval for zuranolone (brand name ZURZUVAE™) in August, 2023 and Zurzuvae was launched commercially in December 2023. Zuranolone, a synthetic neuroactive steroid derivative, is an oral, once daily 14-day treatment for postpartum depression and is the first oral medication approved by the FDA for the treatment of postpartum depression. Per label, besides long terminal half-life of approximately 19.7 to 24.6 hours and dosage modifications needed for concomitant use with CYP3A4 modulators, warnings and precautions include CNS depressant effects, impaired ability to drive or engage in other potentially hazardous activities and embryo-fetal toxicity.
We believe LPCN 1154 targets the unmet need for robust rapid relief with 48-hour treatment duration through a convenient oral treatment candidate comprising bioidentical NAS with good tolerability.
| 7 |
LPCN 2101: NAS for Epilepsy
We are currently evaluating an additional NAS candidate, LPCN 2101, for women with epilepsy (“WWE”). We have completed pre-clinical and Phase 1 studies for LPCN 2101 which demonstrated promising PK results, safety and tolerability. In July 2022 our IND was accepted by the FDA for LPCN 2101 for adults with epilepsy and we plan to initiate a Phase 2 IND opening proof-of-concept study to evaluate the safety, tolerability, and efficacy of LPCN 2101 in 2024 subject to resource prioritization.
Disease Overview – Epilepsy
Epilepsy is defined by the 1) occurrence of at least two unprovoked seizures more than 24 hours apart, 2) occurrence of one unprovoked seizure and a probability of further seizures occurring over the next 10 years, and/or 3) diagnosis of an epilepsy syndrome. Patients with epilepsy are more likely to be comorbid with other conditions, including depression and anxiety. Patients with epilepsy have increased risk of mortality due to direct effects of seizures (e.g., status epilepticus, car accidents) and indirect effects of seizures (e.g., suicide, cardiovascular effects.)
Epilepsy is a disorder of the brain that causes seizures, affecting the physical, mental, and social well-being of persons, and is associated with a 2 to 3 times greater mortality rate compared with the general population. About 60-65% of epilepsy is idiopathic and about 30% of patients are refractory (i.e., epilepsy not well managed with currently available Anti-Seizure Medications (“ASMs”). Epilepsy is the most common neurological disorder during pregnancy.
It is estimated that approximately 900,000 child-bearing (“CB”) age women suffer from active epilepsy in the U.S. Women of CB age with epilepsy face many additional challenges due to hormonal influences on seizure activity and endocrine function throughout the different phases of their reproductive cycles. Elevated estrogen or decreased progesterone levels can exacerbate seizure frequency. Often, these women experience hormonal and endogenous NAS imbalances, coupled with fluctuations in the blood levels of ASMs that impact control of seizures, efficacy of oral contraceptives, any coexisting anxiety and/or depression and any associated sleep impairment. Epileptic patients are 5-20 times more likely to develop depression.
Clinical segmentation can be categorized by epilepsy type, comorbidities and patient subgroups. Categorization of focal epilepsy, generalized epilepsy, combined focal and generalized epilepsy, and unknown epilepsy can guide the choice of ASM. Special patient subgroups, including WWE of CB age and elderly patients, require special care and management of epilepsy. Comorbidities such as depression and anxiety may be co-treated with therapies that do not aggravate seizures and have no drug interaction with the ASM used for epilepsy. While lowest effective dose and monotherapy are preferred, management of patients with epilepsy is focused on controlling seizures, avoiding adverse events, and maintaining quality of life. Despite a wide range of ASMs available, about 30 % of all people with epilepsy still fail to respond to treatment effectively. Women with epilepsy face specific challenges throughout their lifespan because of seizures, ASMs, and hormonal fluctuations.
Women with epilepsy were once counseled to avoid pregnancy, but epilepsy is no longer considered a contraindication to pregnancy. Caregivers for WWE in the preconception phase either intending to start a family (planning pregnancy) or using contraception to prevent an unplanned pregnancy face significant challenges to balance seizure control efficacy with the selection and dosage of ASMs and ASM-related risks such as, among other risks, fetal-neonatal toxicity, contraception failure, and psychiatric side effects.
Several ASMs are known to have teratogenic effects on the developing fetus (converging evidence from registry studies indicates that teratogenic risks are highest with valproate, followed by carbamazepine and topiramate). Other commonly prescribed ASMs, including older generation agents, such as phenobarbital and phenytoin, have been associated with higher risks as compared with lamotrigine, levetiracetam, clonazepam and gabapentin (Vajda et al., 2014; Voinescu and Pennell, 2015). Moreover, risks associated with ASMs are considerable early in pregnancy; therefore, it is necessary that WWE of CB age undergo counseling, monitoring, and adjustment to the most appropriate ASM prior to becoming pregnant. It is preferable that WWE of CB age discuss seizure control with their doctor for at least 6 months before conception and, if possible, cease ASM therapy or use the lowest effective dose of a single anticonvulsant according to the type of epilepsy and the fetal toxicity of the ASM. Anxiety, depression, lack of adherence to ASM, and/or contraception failure may be experienced by women who are worried about unplanned pregnancy or are late in confirming pregnancy, planned or unplanned. ASMs can reduce the efficacy of oral contraceptives, compounding this problem.
| 8 |
Complex, multidirectional interactions between female hormones, seizures, and ASMs exist. Most hormones act as NAS and can thus modulate brain excitability. Any changes in endogenous or exogenous hormone levels can affect the occurrence of seizures, either directly or via PK interactions that modify the plasma levels of ASMs (Harden, 2008). The PK interactions between oral contraceptives and ASMs are bidirectional (Johnston and Crawford, 2014). The efficacy of hormonal contraception may be diminished for women taking CYP-P450 enzyme inducing ASMs. Epilepsy is not a medical condition in which contraceptives are contraindicated. Contraceptive failure, possibly related to ASMs, may be responsible for up to 1 in 4 unplanned pregnancies in WWE (~12.5% of all WWE pregnancies), versus a rate of 1% in healthy women.
Unmet need to treat WWE in CB age
It is estimated that approximately 900,000 CB age women suffer from active epilepsy in the U.S. Women of CB age with epilepsy face many additional challenges such as hormonal influences on seizure activity and endocrine function throughout the different phases of their reproductive cycles, and approximately 30% of patients with epilepsy cannot be efficiently controlled with available ASMs making consideration of newer pharmacological treatment development options important.
Managing uncontrolled seizures in WWE of CB age is the primary aim during preconception, pregnancy, and postpartum phases. Therefore, uncompromised ASM efficacy with acceptable variability and less or no drug-drug interactions achieved with lowest possible monotherapy dose to address fetal toxicity concerns remain highly unmet needs. Moreover, control of seizures including prevention of breakthrough seizures is critical when planning for pregnancy and also during pregnancy, as it can also lead to undesired falls or auto-accidents and compromise freedom to drive.
Select ASMs have the potential to induce contraception failures, reproductive hormone imbalance, anxiety, and depression. There remains an unmet need for an ASM without the aforementioned downsides, with no to low fetal-neonatal toxicity and without any breast-feeding concerns as well as potential to treat associated comorbidities.
While over 30 molecules have been approved for the treatment of epilepsy in the U.S., no epilepsy drug has been specifically approved for WWE of CB age. We believe our endogenous NASs as GABAA PAMs, while targeting the goal of seizure control, also have the potential for additional benefits in psychiatric disorders comorbidities (e.g., anxiety and/or depression) and sleep impairment. Moreover, these oral endogenous NAS could potentially address some of the fetal toxicity concerns related to unplanned or planned pregnancy in WWE. (1)
(1) Ref: S.Bangar et al. Functional Neurology 2016; 31(3): 127-134; Reimers et al. Seizure. 2015 May; 28: 66-70.
LPCN 1148: Oral Product Candidate for the Management of Decompensated Cirrhosis
We are currently evaluating LPCN 1148 comprising testosterone laurate (“TL”) for the management of decompensated cirrhosis. We believe LPCN 1148 targets unmet needs for subjects with cirrhosis including improvement in the quality of life of patients while on the liver transplant waiting list, prevention or reduction in the occurrence of new decompensation events such as hepatic encephalopathy (“HE”), and improvement in post liver transplant survival, including outcomes and costs.
We recently completed a Phase 2 proof of concept (“POC”) study (NCT04874350) in male cirrhotic subjects to evaluate the therapeutic potential of LPCN 1148 for the management of sarcopenia. The Phase 2 POC study was a prospective, multi-center, randomized, placebo-controlled study in male sarcopenic cirrhotic patients. Subjects were initially randomized 1:1 to 1 of 2 arms. The treatment arm was an oral dose of LPCN 1148, and the second arm was a matching placebo. There were no restrictions on patients with respect to background therapies, including current standard of care, diet or exercise. The primary endpoint was a change in skeletal muscle index at week 24 with key secondary endpoints including change in liver frailty index, rates of breakthrough HE, and number of waitlist events, including all-cause mortality. Total treatment was 52 weeks, with 24-week placebo-controlled treatment subjects receiving LPCN 1148 in the 28-week open-label extension (“OLE”) phase of the study for the duration of the study through week 52. In July 2023, we announced that the Phase 2 study met the study primary endpoint, increased skeletal muscle index (L3-SMI) relative to placebo (P<.01), in patients with cirrhosis. The study also demonstrated improvements in clinical outcomes such as prevention of new decomposition events including HE, rates of hospitalizations, and patient reported outcomes (“PROs”). LPCN 1148 was well-tolerated, with adverse event (“AE”) rates and severities similar to placebo and no mortality was noted in the LPCN 1148 treatment group, nor were there any cases of drug-induced liver injury.
End of study results are expected by the end of the first quarter of 2024. Possible outcomes of interest from the Phase 2 study include continuation and/or extension of clinical outcomes observed after 24 weeks such as overall survival and new decompensation events (including HE and/or ascites occurrences), rates of survival to transplant, rates of hospitalizations, infections, etc., muscle changes such as muscle mass, body composition, myosteatosis (muscle fat), functional capacity changes such as liver frailty index (“LFI”), PROs, and biochemical markers including hematocrit for anemia status, albumin, creatinine/kidney function, etc. We plan to request a Type C meeting with the FDA to discuss the clinical development plan for LPCN 1148 in mid-2024.
| 9 |
Disease Overview – Cirrhosis
There are over 2 million cases of cirrhosis worldwide, with over 500,000 people living with decompensated cirrhosis in the U.S. and nonalcoholic fatty liver disease is the most rapidly increasing indication for liver transplant. 62% of those on the liver transplant (“LT”) waitlist are male and the economic burden (approximately $812,500/transplant) is high and continues to increase. Each year about half of the approximately 17,000 people in U.S. on the LT waitlist undergo transplant, while nearly 3,000 patients either die or are removed from the list because they were “too sick to transplant.”
Liver cirrhosis is defined as the histological development of regenerative nodules surrounded by fibrous bands. Cirrhotic patients typically have a years-long silent, asymptomatic phase (compensated cirrhosis) until decreasing liver function and increasing portal pressure move the patient into the symptomatic phase (decompensated cirrhosis). Transition to decompensated cirrhosis is marked by clinical events including ascites, encephalopathy, jaundice, and/or variceal hemorrhage. Decompensated subjects survive on average less than 2 years. Common causes of liver cirrhosis include alcoholic liver disease, nonalcoholic fatty liver disease (“NAFLD”), chronic hepatitis B and C, primary biliary cirrhosis (“PBC”), primary sclerosing cholangitis (“PSC”) and cryptogenic.
Common complications in cirrhotic patients may include: compromised liver function, portal hypertension, varices in GI tract with internal bleeding, edema, ascites, hepatic encephalopathy, compromised immunity with post-transplant acute rejection risk, high sodium levels, increased bilirubin, low albumin level, insulin resistance with impaired peripheral uptake of glucose, depression, accelerated muscle disorder in the form of sarcopenia, myosteotosis, and frailty with compromised energetics, bone diseases (e.g., osteoporosis), high alkaline phosphatase (“ALP”), cachexia, malnutrition, weight loss (>5%), symptoms of hypogonadism such as abnormal hair distribution, anemia, sexual dysfunction, testicular atrophy, muscle wasting, fatigue, osteoporosis, gynecomastia, inflammation with elevated cytokines, and infection risk leading to hospital admissions and possibly death.
HE, a significant decompensation event in patients with cirrhosis, is a brain dysfunction caused by liver insufficiency and/or portal systemic shunting. Because the damaged liver cannot function normally (as in cirrhosis), neurotoxins such as ammonia are inadequately removed from systemic circulation and travel to the brain, where they affect neurotransmission. This can cause episodes of HE, which may present as alterations in consciousness, cognition, and behavior that range from minimal to severe. Overt HE occurs in 30% to 40% of patients with cirrhosis at some point during the clinical course of their disease. As the burden of chronic liver disease and cirrhosis is increasing, the frequency of HE is also increasing.
LPCN 2203: Oral Product for Management of Essential Tremor
About Essential Tremor:
Essential Tremor (“ET”) is one of the most common movement disorders in the United States, affecting an estimated 7 million in the U.S. For ET patients, uncontrollable shaking of the hands, head, voice, or legs creates difficulty eating, dressing, writing, and pursuing other day-to-day tasks. The etiology of ET is largely unknown, but reduced GABAA receptor levels and decreased GABAergic activity have been observed in ET.
While ET is often associated with aging populations, ET can begin much earlier in life, with a progressive disease course that can eventually necessitate a care partner. Social anxiety and depressive symptoms can manifest in patients with ET as tremor severity increases and may negatively impact a patient’s ability to work and engage in hobbies. In an interview study of ET patients and care partners, the most common impacts on activities of daily living are pouring liquids and writing/typing (100%), and grooming/hygiene, drinking, dressing, eating, and reading (80-85%). Overall, 90% of participants noted the emotional impact of ET, with 75% reporting tremor-related worry or anxiety.
The only FDA approved pharmacological treatment for ET was approved more than 50 years ago, and the majority of patients with ET experience a sub-optimal response with standard-of-care treatments, highlighting numerous and compelling unmet needs in care such as daytime efficacy and improved tolerability, PRN or “as needed use” option, and a superior benefit-to-risk profile.
LPCN 2203 is an oral candidate for management of essential tremor comprising a bioidentical GABA modulating NAS. We have successfully completed oral pharmacokinetics with bioidentical GABA Modulating NAS and are planning to submit a protocol for a proof-of-concept phase 2 study for ET in the first half of 2024.(1) (2)
| (1) | Ref: Louis ED, Ottman R. Tremor Other Kyperkinet Mov (NY). 2014;4:259. | |
| (2) | Ref: Gerbasi et.al. Patient experiences in essential tremor: Mapping functional impacts to existing measures using qualitative research. MDS 2023. |
Our Partnership Pipeline Product Candidates
We continue to pursue opportunities for partnering arrangements for the continued development and/or marketing of LPCN 1144, LPCN 1148, LPCN 1107, in addition to TLANDO and LPCN 1111 outside of the U.S. and Canada. We do not currently anticipate conducting any further significant development activities with respect to these products and product candidates, without the participation of a partner. There can be no guarantee that we will be able to identify or enter into partnering arrangements on terms that are beneficial to us or at all. Even if we do enter into partnering arrangements, such arrangements may not be sufficient to successfully develop and commercialize these products.
TLANDO: An Oral Product Candidate for Testosterone Replacement Therapy
Our leadAs previously described, under the Verity License Agreement, in January 2024 we granted to Verity an exclusive, royalty-bearing, sublicensable right and license to develop and commercialize TLANDO, our product TLANDO, is an oral formulation of the chemical, testosterone undecanoate (“TU”), an eleven carbon side chain attached to testosterone. TU is an ester prodrug of testosterone. An ester is chemically formed by bonding an acid and an alcohol. Upon the cleavage, or breaking, of the ester bond, testosterone is formed. TU has been approved for use outside the United States for many years for delivery via intra-muscular injection and in oral dosage form and recently TU has received regulatory approvalTRT, in the United StatesU.S. and Canada effective February 1, 2024. TLANDO received FDA approval on March 28, 2022. Any FDA requirement to conduct certain post-marketing studies will be the responsibility of our Licensee, Verity.
Proof-of-concept for delivery via intra-muscular injection. We are using our proprietary technology to facilitate steady gastrointestinal solubilization and absorption of TU. Proof of conceptTLANDO was initially established in 2006, and subsequently TLANDO was licensed in 2009 to Solvay Pharmaceuticals, Inc., which was then acquired by Abbott Products, Inc. ("Abbott"(“Abbott”). Following a portfolio review associated with the spin-off of AbbVie Inc. by Abbott in 2011, the rights to TLANDO were reacquired by us. All obligations under the prior license agreement have been completed except that Lipocine will owe Abbott a perpetual 1% royalty on net sales.sales of TLANDO. Such royalties are limited to $1 million in the first two2 calendar years following product launch, after which period there is not ano cap on royalties and no maximum aggregate amount. If generic versions of any such product are introduced, then royalties are reduced by 50%. TLANDO was commercially launched on June 7, 2022. During the years ended December 31, 2023 and December 31, 2022, we incurred royalty expense of approximately $34,000 and $12,000, respectively, resulting from net sales of TLANDO.
| 10 |
Under the Pediatric Research Equity Act (“PREA”), since TLANDO received full FDA approval, under the Verity Licensing Agreement, Verity will need to address the PREA requirement to assess the safety and effectiveness of TLANDO in pediatric patients. The FDA may also require certain post-marketing studies to be conducted which will also be the responsibility of our Licensee, Verity.
Upon execution of the Verity License Agreement, Verity paid us an initial payment of $2.5 million which was received on signing of the License Agreement and $5 million received on February 1, 2024. Verity is also required to make an additional payment of $2.5 million to us before January 1, 2025 and an additional payment of $1 million to us before January 1, 2026. We are also eligible to receive milestone payments of up to $259 million in the aggregate, depending on the achievement of certain sales milestones in a single calendar year and/or development milestones with respect to products licensed by Verity under the Verity License Agreement. In addition, we will receive tiered royalty payments at rates ranging from 12% to up to 18% of net sales of TLANDO in the United States and Canada.
We are exploring the possibility of licensing LPCN 1021 (known as TLANDO in the United States) to third parties outside the United States and Canada, although no licensing agreement has been entered into by the Company. If and when an agreement is made with a partner, such arrangement would likely be partially contingent upon obtaining local regulatory approval. No assurance can be given that any license agreement will be completed, or, if an agreement is completed, that such an agreement would be on terms favorable to us.
LPCN 1144: An Oral Prodrug of Bioidentical Testosterone Product Candidate for the Treatment of NASH
We are exploring the possibility of partnering LPCN 1144 to a third party, although no partnering agreement has been entered into by the Company. No assurance can be given that any license agreement will be completed, or, if an agreement is completed, that such an agreement would be on terms favorable to us.
Disease Overview – NASH
NASH is a more advanced state of non-alcoholic fatty liver disease (“NAFLD”) and can progress to a cirrhotic liver or liver failure, require liver transplant, and can result in hepatocellular carcinoma/ liver cancer, and death. Progression of NASH to end stage liver disease will soon surpass all other causes of liver failure requiring liver transplantation. Importantly, beyond these critical conditions, NASH and NAFLD patients additionally suffer heightened cardiovascular risk and, in fact, die more frequently from cardiovascular events than from liver disease. NAFLD/NASH is becoming more common due to its strong correlation with obesity and metabolic syndrome, including components of metabolic syndrome such as diabetes, cardiovascular disease and high blood pressure. In the U.S., 20% to 30% of the population is estimated to suffer from NAFLD and 15% to 20% percent of this group progress to NASH, which is a substantially large population that lacks effective therapy. NASH is a silent killer that affects millions in the U.S. Diagnoses have been on the rise and are expected to increase dramatically in the next decade. Approximately 50% of NASH patients are in adult males. In men, especially with comorbidities associated with NAFLD/NASH, testosterone deficiency has been associated with an increased accumulation of visceral adipose tissue and insulin resistance, which could be factors contributing to NAFLD/NASH. There is currently no approved therapy for the treatment of NASH although there are several drug candidates currently under development with many having clinical failures to date.
The critical pathophysiologic mechanisms underlying the development and progression of NASH include reduced ability to handle lipids, increased insulin resistance, injury to hepatocytes and liver fibrosis in response to hepatocyte injury. NASH patients have an excessive accumulation of fat in the liver resulting primarily from a caloric intake above and beyond energy needs. A healthy liver contains less than 5% fat, but a liver in someone with NASH can contain more than 20% fat. This abnormal liver fat contributes to the progression to NASH, a liver necro-inflammatory state that can lead to scarring, also known as fibrosis, and, for some, can progress to cirrhosis and liver failure.
Current Status
We have completed the LiFT Phase 2 clinical study in biopsy-confirmed non-cirrhotic NASH subjects. The LiFT clinical study was a prospective, multi-center, randomized, double-blind, placebo-controlled multiple-arm study in biopsy-confirmed hypogonadal and eugonadal male NASH subjects with grade F1-F3 fibrosis and a target NAFLD Activity Score ≥ 4 with a 36-week treatment period. The LiFT clinical study enrolled 56 biopsy confirmed NASH male subjects. Subjects were randomized 1:1:1 to one of three arms (Treatment A is a twice daily oral dose of 142 mg testosterone equivalent, Treatment B is a twice daily oral dose of 142 mg testosterone equivalent formulated with 217 mg of d-alpha tocopherol equivalent, and the third arm is twice daily matching placebo).
The primary endpoint of the NDA ResubmissionLiFT clinical study was change in hepatic fat fraction via MRI-PDFF and exploratory liver fat/marker end points post 12 weeks of treatment. Additionally, key secondary endpoints post 36 weeks of treatment included assessment of histological change for NASH resolution and/or fibrosis improvement (biopsy) as well as liver fat data (MRI-PDFF). The LiFT clinical study was not powered to assess statistical significance of any of the secondary endpoints. Other important endpoints included the following: change in liver injury markers, anthropomorphic measurements, lipids, insulin resistance and inflammatory/fibrosis markers; as well as patient reported outcomes.
We resubmitted our NDA toTreatments with LPCN 1144 post 12 weeks of treatment in the FDALiFT study resulted in August 2017robust liver fat reduction, assessed by MRI-PDFF, and showed improvement of liver injury markers with no observed tolerability issues.
Liver biopsies were performed at baseline (“BL”) and after 36 weeks of treatment (“EOS”). Prespecified biopsy analyses included NASH Clinical Research Network (“CRN”) scoring as well as a continuous paired (“Paired Technique”) and digital technique (“Digital Technique-Fibronest”). All biopsy analyses were performed on the same slides and the reads for the 3 techniques were done independently. Analysis sets included the NASH Resolution Set (all subjects that have BL and EOS biopsy with NASH at BL [NAS ≥4 with lobular inflammation score ≥ 1 and hepatocyte ballooning score ≥1 at BL] (n=37)), the Biopsy Set (all subjects with baseline and EOS biopsies (n=44)), and the Safety Set (all randomized subjects (n=56)).
Both LPCN 1144 treatment arms met with statistical significance the pre-specified accelerated approval regulatory endpoint of NASH resolution with no worsening of fibrosis based on the resultsNASH CRN scoring. Additionally, both treatment arms showed substantial improvement of the DV study. The DV study confirmedobserved NASH activity in steatosis, inflammation, and ballooning.
During the efficacy36 weeks of TLANDO with a fixed dose regimen without need for dose adjustment. TLANDOtreatment, LPCN 1144 was well tolerated upon 52-week exposure with no reportsan overall safety profile comparable to placebo. Additionally, subjects were given the option to have access to LPCN 1144 through an open label extension (“OLE”) study. The extension study enabled the collection of drug related Serious Adverse Events (“SAEs”). Theadditional data on LPCN 1144 for up to a total of 72 weeks of therapy, as well as data for 36 weeks of therapy for those subjects on placebo in the LiFT study. Key results from the OLE study are as follows:
| ● | LPCN 1144 was well tolerated over 72-week exposure with no observed safety signals; | |
| ● | Liver injury markers were reduced and maintained with extended LPCN 1144 treatment; and | |
| ● | Observed liver histology improvements support further development |
In November 2021, the FDA accepted our NDAgranted Fast Track Designation to LPCN 1144 as a completetreatment for non-cirrhotic NASH. The Fast Track program is designed to accelerate the development and expedite the review of products, such as LPCN 1144, which are intended to treat serious diseases and for which there is an unmet medical need.
We had a written only response to their CRL and assignedfrom FDA for a PDUFA goal date of May 8, 2018 for completion of the review. On January 10, 2018, the BRUDAC of the FDA voted six in favor and thirteen against the benefit/risk profile of TLANDO. Discussion topics during the BRUDAC included whether the safety of TLANDO is adequately characterized and whether additional data is need pre-approval or post approval, including the need for an ambulatory blood pressure (“ABPM”) study. Additional areas discussed included the potential to increase cardiovascular risk, lipid changes, hematocrit increase, levels of dihydro-testosterone and estradiol, cosynotropin stimulation results, Cmax excursions, the stopping criteria for use in clinical practice, and whether testosterone concentrations measured in serum tubes are reliable in patients treated with TLANDO. Particularly the FDA may be concerned with TLANDO’s potential for increased adverse cardiovascular outcomes in the population that will likely use the drug, if approved, and observed treatment emergent adverse reactions. We continue to workLPCN 1144 Type C meeting with the FDA in addressing topics discussed by BRUDAC.January 2022 to discuss the development path forward with LPCN 1144. The FDA may or may not view BRUDAC’s TLANDO advice/recommendation similarly. We have submitted two protocolsacknowledged that the NDA submission of LPCN 1144 would be via 505(b)2 regulatory pathway and agreed that no additional non-clinical studies are needed to support an NDA submission. The FDA acknowledged that in the LiFT study subjects achieved improvements in key components associated with NASH histopathology after 36-weeks of treatment with LPCN 1144 in adult males and agreed that the proposed multicomponent primary surrogate endpoint is acceptable for seeking approval under the accelerated approval pathway. The FDA agreed that the proposed primary multicomponent surrogate endpoint, NASH resolution with no worsening of fibrosis, is acceptable for seeking approval under the accelerated approval pathway and the FDA under our TLANDO investigational new drug (“IND”) application. The first protocol is for the conduct of an ABPM clinical study and the second protocol is forrecommended a phlebotomy clinical study to confirm no ex-vivo conversion of TU to T. We plan to initiate both clinical studies ahead of our PDUFA date. Although there is no guarantee of FDA approval of TLANDO, we believe the results from the DV study confirm the validity of a fixed dose approach without the need for dose titration to orally administering TLANDO. However, approval of our NDA on our PDUFA date is not guaranteed and we may receive another CRL. Receipt of another CRL would result in substantial delays which may include additional studies and expense before we would be in a position to re-submit an NDA responsive to such additional CRL.
Previously on June 28, 2016, we received a CRL from the FDA on our original NDA submission. A CRL is a communication from the FDA that informs companies that an application cannot be approved in its present form. The CRL identified deficiencies related to the dosing algorithm for the label. Specifically, the proposed titration scheme for clinical practice was significantly different from the titration scheme used in the Phase 3 trial leadingwith a study duration of 72 weeks. In July 2022, Lipocine held an End of Phase 2 meeting with FDA for LPCN 1144 in NASH. The FDA recommended a Phase 2 dose ranging study be conducted to discordance in titration decisions betweenidentify the Phase 3 trial and real-world clinical practice. In responseoptimal dose prior to conducting a pivotal study. The FDA agreed to the CRL, we met with the FDA in a Post Action meeting and proposed a dosing regimen to the FDA based on analyses of existing data. The FDA noted that while the proposed dosing regimen might be acceptable, validation in aunique testosterone ester, testosterone laurate, for future clinical trial would be needed prior to resubmission. The DV study was in response to the FDA’s request. We also initiated the Dosing Flexibility (“DF”) study to assess TLANDO in hypogonadal males on a fixed daily dose of 450 mg divided into three equal doses.studies.
Results from DV and DF Studies
The DV and DF studies were both an open-label, fixed dose (no titration), single treatment clinical study of oral TRT in hypogonadal males with low testosterone (T) (< 300 ng/dL) that assessed TLANDO in hypogonadal males on a fixed daily dose of 450 mg divided into two equal doses (“BID”) in the DV study and into three equal doses (“TID”) in the DF study. In total, 95 and 100 subjects were enrolled into DV and DF studies, respectively, with 94 and 98 subjects completing the DV and DF studies, respectively.
On June 19, 2017, we announced top-line results of the DV and DF studies. Although there is no guarantee of FDA approval of TLANDO, we believe the results from the DV study confirm the validity of a fixed dose approach without the need for dose titration to orally administering TLANDO. The DV study will be considered our pivotal efficacy clinical study for the NDA resubmission. TLANDO successfully met the FDA primary efficacy guidelines in the DV study safety statistical analysis set (“SS”) where 80% of the subjects achieved average testosterone levels (“Cavg”) within the normal range with a lower bound confidence interval (“CI”) of 72%. The DF study restored 70% of the subjects’ average testosterone levels within the normal range (Cavg) confirming that twice daily (“BID”) dosing is the appropriate dosing regimen for TLANDO and was the basis for resubmission. The safety set is defined as any subject that was randomized into the study and took at least one dose (N=95 subjects in the DV study and N=100 in the DF study). A baseline carried forward approach was used to account for missing data as a result of subject discontinuation.
The primary efficacy endpoint is the percentage of subjects with Cavg within the normal range, which is defined as 300-1080 ng/dL. The FDA guidelines for primary efficacy success is that at least 75% of the subjects on active treatment achieve a testosterone Cavg within the normal range; and the lower bound of the 95% CI must be greater than or equal to 65%.
The adverse event profile of TLANDO in both the DV and DF studies was consistent with the previously conducted 52-week Phase 3 Study of Androgen Replacement (“SOAR”) clinical trial. All drug related adverse events (“AEs”) were either mild or moderate in intensity and none were severe. To date, the safety database of TLANDO includes ~525 unique hypogonadal men demonstrating a profile consistent with other TRT products.
The secondary endpoints assessed the maximum total testosterone concentration (“Cmax”) post dosing using predetermined limits developed by the FDA for transdermals. The FDA guidelines for secondary efficacy success is that at least 85% of the subjects achieve Cmax less than 1500 ng/dL; no greater than 5% of the subjects have Cmax between 1800 ng/dl and 2500 ng/dL; and zero percent of the subjects have Cmax greater than 2500 ng/dL. Consistent with the definition of Cmax and the pharmacokinetic profile of multiple times a day dosing, two pre-specified analyses were performed, Cmax per dose and Cmax per day.
In the DV study SS Cmax per dose analysis, the percentage of subjects with Cmax less than 1500 ng/dL and between 1800 ng/dL and 2500 ng/dL were 85% and 7%, respectively. Deviations from the predetermined limits in the DV study were observed in the Cmax per day dose analysis for these thresholds. Only one subject, who was a major protocol violator, exceeded the 2500 ng/dL limit independent of per dose or per day dose analyses.
The DF study SS met all Cmax thresholds in per dose and per day dose analyses.
Prior to conducting the DV study and the DF study, we completed our SOAR pivotal Phase 3 clinical study evaluating efficacy and 52-week safety of TLANDO. The SOAR study is considered our pivotal safety clinical study for the NDA resubmission.
Results from SOAR
SOAR was a randomized, open-label, parallel-group, active-controlled, Phase 3 clinical study of TLANDO in hypogonadal males with low testosterone (< 300 ng/dL). In total, 315 subjects at 40 active sites were assigned, such that 210 were randomized to TLANDO and 105 were randomized to the active control, AndroGel 1.62%®, for 52 weeks of treatment. The active control is included for safety assessment. TLANDO subjects were started at 225 mg TU (equivalent to ~ 142 mg of T) twice daily (“BID”) with a standard meal and then dose titrated, if needed, based on average T levels during the day, Cavg, and peak serumT levels, Cmax, up to 300 mg TU BID or down to 150 mg TU BID based on serum testosterone measured at weeks 3 and 7 based on PK profile with multiple blood samples drawn at each time period. The mean age of the subjects in the trial was ~53 years with ~91% of the patients < 65 years of age. The discontinuation rate for TLANDO was 38% compared 32% for AndroGel 1.62%®.
Primary statistical analysis was conducted using the Efficacy Population Set ("EPS"). The EPS is defined as subjects randomized into the study with at least one PK profile and no significant protocol deviations and includes imputed missing data by last observation carried forward, N=151. Further analysis was performed using the full analysis set ("FAS") (any subject randomized into the study with at least one post-baseline efficacy variable response, N=193) and the SS (any subject that was randomized into the study and took at least one dose, N=210).
Safety
The safety component of the SOAR trial was completed the last week of April 2015. The safety extension phase was designed to assess safety based on information such as metabolites, biomarkers, laboratory values, serious adverse events ("SAEs") and AEs, with subjects on their stable dose regimen in both the treatment arm and the active control arm. TLANDO treatment was well tolerated in that there were no hepatic, cardiac or drug related SAEs.
TLANDO safety highlights include:
Food Effect Study
We also completed our labeling "food effect" study in May 2015. Results from the labeling "food effect" study indicate that bioavailability of testosterone from TLANDO is not affected by changes in meal fat content. The results demonstrate comparable testosterone levels between the standard fat meal (similar to the meal instruction provided in the Phase 3 clinical study) and both the low and high fat meals. The labeling “food effect” study was conducted per the FDA requirement and we submitted preliminary results from this study to the FDA in the second quarter of 2015 prior to submitting the NDA.
Other Safety Requirements
Based on our meetings with the FDA, we do not expect to be required to conduct a heart attack and stroke risk study or any additional safety studies prior to the potential approval of our NDA for TLANDO. We may, however, be required to conduct a heart attack and stroke risk study on our own or with a consortium of sponsors that have an approved TRT product subsequent to the potential approval of TLANDO.
LPCN 1111: A Next-Generation Long-Acting Oral Product Candidate for TRT
As previously described, under the terms of the Verity License Agreement, we have licensed the development and commercialization rights to LPCN 1111 (TLANDO XR) in the U.S. and Canada to Verity. We will continue to explore the possibility of partnering LPCN 1111 with third parties outside the United States and Canada, although no partnering agreement has been entered into by the Company. No assurance can be given that any license agreement outside North America will be completed, or, if an agreement is completed, that such an agreement would be on terms favorable to us.
| 12 |
LPCN 1111 is a next-generation, novel ester prodrug of testosterone comprised of testosterone tridecanoate (“TT”) which uses the Lip’ralour proprietary delivery technology to enhance solubility and improve systemic absorption. We completed a Phase 2b dose finding study in hypogonadal men in the third quarter of 2016. The primary objectives of the Phase 2b clinical study were to determine the starting Phase 3 dose of LPCN 1111 along with safety and tolerability of LPCN 1111 and its metabolites following oral administration of single and multiple doses in hypogonadal men. The Phase 2b clinical trial was a randomized, open label, two-period, multi-dose PK study that enrolled hypogonadal males into five treatment groups. Each of the 12 subjects in a group received treatment for 14 days. Results of the Phase 2b study suggest that the primary objectives were met, including identifying the dose expected to be tested in a Phase 3 study. Good dose-response relationship was observed over the tested dose range in the Phase 2b study. Additionally, the target Phase 3 dose met primary and secondary end points. Overall, LPCN 1111 was well tolerated with no drug-related severe or serious adverse events reported in the Phase 2b study.
Additionally in October 2014, we completed a Phase 2a proof-of-concept study in hypogonadal men. The Phase 2a open-label, dose-escalating single All future development and multiple dose study enrolled 12 males. Results from the Phase 2a clinical study demonstrated the feasibilitycommercialization of a once daily dosing with LPCN 1111 in hypogonadal menthe U.S. and a good dose response. Additionally,Canada will be the study confirmed that steady state is achieved by day 14 with consistent inter-day performance observed on day 14, 21 and 28. No subjects exceeded Cmax of 1500 ng/dL at any time during the 28-day dosing period on multi-dose exposure. Overall, LPCN 1111 was well tolerated with no serious AE’s reported.
We have also completed a preclinical toxicology study with LPCN 1111 in dogs. In February 2018 we had a meeting with the FDA to discuss these preclinical results and to discuss the Phase 3 clinical study and path forward for LPCN 1111. Based on the results of the FDA meeting, additional pre-clinical or clinical trials may be required before a Phase 3 clinical study can be initiated. Additionally the FDA requested that an ABPM clinical study be conducted. Based on our capital resources and the clinical statusresponsibility of our product candidates, we will primarily focus our efforts in 2018 on TLANDO. We do not anticipate the initiation of a Phase 3 study with LPCN 1111 to occur in 2018 unless and until additional capital is secured or the product candidate is out-licensed.Licensee, Verity.
LPCN 1107: An Oral Product Candidate for the Prevention of Preterm Birth
We are exploring the possibility of partnering LPCN 1107 to a third party, although no partnering agreement has been entered into by the Company. No assurance can be given that any partnership agreement will be completed, or, if an agreement is completed, that such an agreement would be on terms favorable to us.
We believe LPCN 1107 has the potential to become the first oral hydroxyprogesterone caproate (“HPC”) product indicated for the reduction of risk of preterm birth (“PTB”)PTB (delivery less than 37 weeks) in women with singleton pregnancy who have a history of singleton spontaneous PTB. Prevention of PTB is a significant unmet need as ~11.7%approximately 11% of all U.S. pregnancies result in PTB, (delivery less than 37 weeks), a leading cause of neonatal mortality and morbidity.
Current Status
We have completed a multi-dose PK dose selection study in pregnant women. The objective of the multi-dose PK selection study was to assess HPC blood levels in order to identify the appropriate LPCN 1107 Phase 3 dose. The multi-dose PK dose selection study was an open-label, four-period, four-treatment,4-period, 4-treatment, randomized, single and multiple dose PK study in pregnant women of threewith 3 dose levels of LPCN 1107 and the injectable intramuscular ("IM")IM HPC (Makena®). The study enrolled 12 healthy pregnant women (average age of 27 years) with a gestational age of approximately 16 to 19 weeks. Subjects received three dose levels of LPCN 1107 (400 mg BID, 600 mg BID, or 800 mg BID) in a randomized, crossover manner during the first three3 treatment periods and then received five5 weekly injections of HPC during the fourth treatment period. During each of the LPCN 1107 treatment periods, subjects received a single dose of LPCN 1107 on Day 1 followed by twice daily administration from Day 2 to Day 8. Following completion of the three3 LPCN 1107 treatment periods and a washout period, all subjects received five5 weekly injections of HPC. Results from this study demonstrated that average steady state HPC levels (Cavg0-24)(Cavg0-24) were comparable or higher for all three3 LPCN 1107 doses than for injectable HPC. Additionally, HPC levels as a function of daily dose were linear for the three3 LPCN 1107 doses. Also, unlike the injectable HPC, steady state exposure was achieved for all three3 LPCN 1107 doses within seven7 days. We have also completed a proof-of-concept Phase 1b clinical study of LPCN 1107 in healthy pregnant women in January 2015 and a proof-of-concept Phase 1a clinical study of LPCN 1107 in healthy non-pregnant women in May 2014. These studies were designed to determine the PK and bioavailability of LPCN 1107 relative to an IM HPC, as well as safety and tolerability.
| 13 |
A traditional pharmacokinetics/pharmacodynamics (“PK/PD”)PD based Phase 2 clinical study in the intended patient population is not expected to be required prior to entering into Phase 3. Therefore, based on the results of our multi-dose PK study we had an End-of-Phase 2 meeting with the FDA as well as otherand subsequent guidance meetings with the FDA to define a pivotal Phase 2b/3 development plan for LPCN 1107. During the End-of-Phase 2 meeting and subsequent guidance meetings, the FDA agreed toHowever, these discussions may be updated based on recent developments with Covis’ Makena® as described below. We have completed a randomized, open-label, two-arm clinicalfood effect study to include a LPCN 1107 arm and a comparator IM arm with treatment up to 23 weeks. The FDA also provided preliminary feedback on other critical Phase 3 study design considerations including: positive feedback oncharacterize the proposed 800 mg BID Phase 3 dose and dosing regimen; confirmation of the use of a surrogate primary endpoint focusing on rate of delivery less than 37 weeks gestation rather on clinical infant outcomes; acknowledged that the use of a gestational age endpoint would likely lead to any FDA approval, if granted, being a Subpart H approval; and, recommended a non-inferiority study margin of 7% with interim analyses. A standard statistical design for a NI study based on the FDA feedback, a NI margin of 7%regimen for the primary endpoint may require ~1,100 subjects per treatment arm withpivotal study. We plan to submit a 90% power. However, based on the FDA’s suggestion of including an interim analysis in the NI design, an adaptivepivotal clinical study design is under consideration that may allow for fewer subjects. We submitted the initial LPCN 1107 Phase 3 protocol to the FDA via a SPA in June 2017 and have received multiple rounds of FDA’s feedback. Agreement with the FDA on the Phase 3 protocol via SPA has not occurred and will not occur until results from a planned food-effect study with LPCN 1107 are reviewed by the FDA. Final agreement with the FDA on the Phase 3 protocol, if reached, may or may not confirm the FDA’s preliminary feedback on the Phase 3 design. Additionally, manufacturing scale-up work for LPCN 1107 has been completed. Based on our capital resources and the clinical status of our product candidates, we plan to primarily focus our efforts in 2018 on TLANDO. We are not anticipating the initiation of a Phase 3 study with LPCN 1107 to occur in 2018 unless and until additional capital is secured or the product candidate is out-licensed.
The FDA has granted orphan drug designation to LPCN 1107 based on a major contribution to patient care. Orphan designation qualifies Lipocine for various development incentives, including tax credits for qualified clinical testing, and a waiver of the prescription drug user fee when we file our NDA.
Recent Competition Update
On October 5, 2020, the FDA’s Center for Drug Evaluation and Research (“CDER”) proposed that Makena be withdrawn from the market because the PROLONG trial failed to verify the clinical benefit of Makena and concluded that the available evidence does not show Makena is effective for its approved use.
The CDER issued AMAG Pharmaceuticals, the NDA holder at the time, a Notice of Opportunity for Hearing (“NOOH”) to withdraw approval of Makena, for which AMAG Pharmaceuticals responded by requesting a hearing and providing detail on the company’s position, recognizing clinicians’ decade-long use of treatment with Makena and the public health implications of withdrawing approval. The FDA Commissioner held a public hearing with Covis October 17 through 19, 2022, which resulted in a 14-1 vote recommending removal of the product from the market. On October 31, 2022, Covis approached the CDER and outlined a plan of orderly withdrawal which would set a withdrawal timeframe sufficient for current patients to complete their courses of treatment. The CDER declined this proposal. On March 6, 2023, Covis announced its plan to voluntarily withdraw Makena from the market and submitted a request to the CDER for a minimum 21-week wind-down. On April 6, 2023, the FDA withdrew its approval of Makena and ordered the immediate withdrawal of Makena and several approved generic versions of the drug, making it unlawful for the drug to be distributed in the US. The FDA stated that in light of the unmet need for a treatment for preventing preterm birth and improving neonatal outcomes, it is imperative that the medical and scientific communities increase their efforts to find effective treatments and stated their hope that the decision to withdraw Makena will help galvanize further research. The FDA further stated their commitment to working together with patients, researchers, and drug developers to advance the development of safe and effective therapies that are urgently needed as a treatment for the prevention of preterm birth.
Research and Development
We currently have three productsAs disclosed in our development pipeline, (TLANDO, LPCN 1111 and LPCN 1107) and we continue to conceptualizebuild a diversified multi-asset pipeline of novel therapies. In 2023 and discuss new indications for current products as well as new development opportunities. In 2017 and 2016,2022, we spent $11.0$10.2 million and $8.1$8.6 million, respectively, on research and development.
Competition
TestosteroneNeuroactive Steroids Market overview
The unique potential mechanism of action (“MOA”) of NAS presents an opportunity to treat a variety of CNS disorders. Accordingly, multiple NAS as GABAA receptor PAMs are in active development for varied indications. Some companies engaged in development include SAGE Therapeutics, Inc., Marinus Pharmaceuticals, Praxis Precision Medicines, and Eliem Therapeutics.
Postpartum Depression
SAGE Therapeutics is currently marketing an injectable version of an endogenous neuroactive steroid, brexanolone, under the tradename of ZULRESSO™, for treatment in postpartum depression (“PPD”). In August 2023, the FDA approved ZURZUVAE (zuranolone) as an oral, once-daily, 14-day treatment for PPD. ZURZUVAE became commercially available in December 2023.
| 14 |
Cirrhosis Market Overview
Decompensated cirrhosis patients with sarcopenia exhibit significantly shorter overall survival than those without sarcopenia. There are no therapies specifically approved for sarcopenia or decompensated cirrhosis. Currently, the only curative therapy for decompensated cirrhosis is liver transplant; however, liver transplantation is very costly, limited by the supply of available donors, and has a high risk of post-operative complications.
Xifaxan® (rifaximin) is the only FDA-approved medicine indicated for the reduction in risk of overt hepatic encephalopathy (HE) recurrence in adults, a decompensation event typically associated with liver cirrhosis.
Currently, there are no FDA approved drugs to treat secondary sarcopenia in decompensated cirrhosis beyond treatment of the underlying conditions. Lipocine is a leader in pursuing treatment for subjects with decompensated cirrhosis with sarcopenia, however, there are candidates known to be under development for cirrhosis related indication(s).
GB 1211 (by Galecto), an oral galectin-3 inhibitor for advanced liver cirrhosis targeted for directly addressing fibrosis, targeted for initiation of a long-term cirrhosis trial in the first half of 2024, is in development being assessed in patients with moderate-to-severe cirrhosis (Child-Pugh classes B and C).
Reformulated Rifaximin SSD (by BAUSCH health) is in a Phase 3 study for Reduction of Early Decompensation in Cirrhosis with time to first occurrence of hepatic encephalopathy as the primary endpoint. Reportedly, a new drug application (“NDA”) is planned to be submitted 2026.
Testosterone Replacement Therapy Market Overview
The gel-based testosterone replacement products that are currently available include AbbVie’s AndroGel®, marketed by AbbVie,Lilly’s Axiron® Topical Solution and Endo’s Testim® and Fortesta® along with their respective authorized generics.generics as well as the equivalent generic versions of each. Transdermal patches include Allergan’s Androderm®. Intramuscular forms of testosterone also exist although commercialized mostly in generic forms by multiple companies and in branded form as Aveed® by Endo. Additionally, Endo markets the buccal testosterone replacement therapy Striant® and the Testopel® implantable testosterone pellets, which it acquired from Auxillium in 2015. Also, Aytu BioScienceAntares Pharma, Inc. markets a sub-cutaneous weekly auto-injector testosterone therapy, Xyosted®. Acerus Pharmaceuticals markets an intranasal testosterone therapy, Natesto®NATESTO®. Finally, Tolmar Pharmaceuticals markets an oral TRT, JATENZO®, which it licensed from Acerusreceived FDA approval in March 2019 and Marius Pharmaceuticals in 2016.markets an oral TU, KYZATREX®, which received FDA approval for treatment of those with Klinefelter’s Syndrome on August 22, 2022.
Testosterone gels dominate the testosterone replacement therapy market in terms of sales dollars whileCurrently, intramuscular injections have the highest market share in the testosterone replacement market in terms of annual prescriptions. While gels are also a widely-usedwidely used form of testosterone replacement therapy,TRT, there is a risk of transference; additionally, the gels are messy to apply and have significant compliance issues leading to high rates of discontinuance among patients. Additionally, certain intramuscular injections have the potential to cause pulmonary embolisms as well as cause injection site reactions, scarring, pain and risk of infection in patients. We believe a safe and effective oral therapy could potentially increase patient convenience and compliance, while eliminating the testosterone transference risk associated with gels and injection site reaction of injectables. .
The FDA has granted a therapeutic equivalence ("TE") rating of AB to “generic” versions of approved products which have been approved via a 505(b)(2) NDA. In July 2014, the FDA granted the AB rating to Perrigo’s 1% testosterone gel drug product (NDA 203098) approved in January 2013, and a BX rating to Teva’s 1% gel drug product (NDA 202763) approved in February 2012. Each are versions of AbbVie’s AndroGel 1.0% and employed 505(b)(2) submissions citing AndroGel as their reference listed drugs ("RLD").drugs. Teva’s version was found to be bioinequivalent to AndroGel, hence the BX rating. Upsher-Smith Laboratories also received approval for a version of Auxilium’sEndo’s Testim (Vogelxo™; NDA 204399) in June 2014 using the same pathway. In January of 2015, the FDA determined that Vogelxo™ is therapeutically equivalent to Testim and received an AB rating. In August 2015, the FDA granted AB rating to Perrigo’s 1.62% testosterone gel drug product (NDA 204268) which also received FDA approval in August 2015. Eli Lilly and Acrux’s Axiron had patent expiry in February 2017. OnJuly 6, 2017, Acrux confirmed that a generic version of Axiron® Topical Solution, 30 mg/1.5 mL (Testosterone Topical Solution, 30 mg/1.5 mL). has been launched in the United States by Perrigo Company plc. Acrux also confirmed the availability of an authorized generic version of Axiron in the United States, through a marketing and distribution agreement between Eli Lilly and Company and a leading authorized generics company.
| 15 |
Other TRT Therapies in Development
Recently there has been increased interest in developing oral testosterone replacement therapiesTRT’s as well as testosterone therapies which are not considered testosterone replacement and as such will need to achieve efficacy endpoints in addition to endpoints related to serum testosterone levels that are required for testosterone replacement therapies.
Clarus Therapeutics, Inc. has completed three Phase 3 clinical studies. Clarus originally filed an NDA
We believe there remains a significant unmet need in early 2014 with Jatenzo® (formerly Rextoro® and CLR-610),TRT for a twice-dailyonce-a-day convenient oral softgel capsule of TU, as a testosterone replacement therapyoption. LPCN 1111 is targeted to meet this unmet need.
NASH Market Overview
There are currently no medications approved for the treatment of hypogonadism in men. On September 18, 2014, Clarus and the FDA had an Advisory Committee meeting to evaluate the safety and efficacy of Jatenzo. 18 of the 21 members of the Advisory Committee voted that the overall benefit/risk profile of Jatenzo is not acceptable to support approval for T-replacement therapy. The PDUFA date for the Jatenzo NDA was November 2014 with the FDA issuing a CRL. Subsequent to receiving the CRL, Clarus completed another Phase 3 clinical study and subsequently resubmitted its NDA for Jatenzo in June 2017. An Advisory Committee meeting was held for Jatenzo on January 9, 2018 in which the BRUDAC voted nine in favor and ten against the acceptability of overall benefit/risk profile to support approval of Jatenzo as a TRT. The FDA concluded that Jatenzo increased blood pressure in a clinically significant manner compared to Axiron, despite more Jatenzo -treated subjects having escalation of antihypertensive therapies. Jatenzo also increased heart rate, which may amplify the blood pressure effects on cardiovascular risk.
Antares Pharma, Inc. is developing a testosterone enanthate auto-injector administered subcutaneously once each week. The product candidate completed a double-blind, multiple-dose, 12-week efficacy and 52-week safety Phase 3 study in October 2015 and completed a dose-blinded, multiple-dose, concentration controlled 28-week safety and pharmacokinetic study in June 2016. Antares filed an NDA with the FDA for XYOSTED™ (testosterone enanthate) injection, their testosterone auto-injector, in December 2016. On October 20, 2017, Antares announced that it had received a CRL from the FDA regarding its NDA for XYOSTED™ injection. The CRL indicated that the FDA cannot approve the Antares NDA in its present form and identified two deficiencies related to clinical data. Based on findings in two clinical studies, the FDA is concerned XYOSTED could cause a clinically meaningful increase in blood pressure. Additionally, the CRL also raised a concern regarding the occurrence of depression and suicidality.
Marius Pharmaceuticals is developing an oral testosterone undecanoate as a testosterone replacement therapyNASH. However, various therapeutics are used off-label for the treatment of hypogonadismNASH, including vitamin E (an antioxidant), insulin sensitizers (e.g., metformin, pioglitazone), antihyperlipidemic agents (e.g., gemfibrozil), pentoxifylline and ursodeoxycholic acid. There are several product candidates in men and in the treatment of Constitutional Delay of Growth and Puberty in adolescent boys (14-17 years of age). The product candidate has completed Phase 2 clinical trials in hypogonadal males and an end of Phase 2 meeting has been requested of the FDA. Marinus is also currently conducting a 52-week Phase 3 study with an active comparator for the TRT indication.
Novartis is currently developing BGS649, an aromatase inhibitor, as a testosterone therapyor earlier clinical or preclinical development for the treatment of obese, hypogonadotropic hypogonadal men.NASH, including FGF21 stimulants such as BIO89-100 (89bio), Efruxifermin (EFX; Akero Therapeutics), FGF19 Analog:Aldafermin (NGM Biopharmaceuticals); FXR Agonists: PXL065 (Pxel) ;Vonafexor (EYP001) (Enyo Pharma:) Glucagon-like Peptide-1 (GLP-1) Agonist: Peroxisome Proliferator-activated Receptor (PPAR) Regulator: Lanifibranor (Inventiva);THR-β Agonis:t VK2809 (Viking Therapeutics), and Resmetirom (Madrigal Pharmaceuticals).
TesoRx Pharma LLC is developing a potential once-daily oral bio-identical testosterone, TSX-002,Additional pharmaceutical and biotechnology companies with product candidates in the treatment of Constitutional Delay of Growth and Puberty. Phase 2 clinical studies have been completed. TesoRx is also developing a next generation potential once-daily, oral testosterone undecanoate product candidate, THG-1001, as a testosterone replacement therapydevelopment for the treatment of hypogonadism in men. An INDNASH include, Boehringer Ingelheim GmbH, Bristol-Myers Squibb Company, Durect Corporation, Galectin Therapeutics Inc., Galmed Pharmaceuticals Ltd., Immuron Ltd., Ionis Pharmaceuticals, Inc., Islet Sciences, Inc., MediciNova, Inc., MiNA Therapeutics, NGM Biopharmaceuticals, Inc., Novo Nordisk A/S, NuSirt Sciences Inc., Viking Therapeutics, Inc. and Zydus Pharmaceuticals (USA) Inc. In December 2022, Madrigal announced achievement of liver histological improvement endoints that the FDA proposed as reasonably likely to predict clinical benefit from the pivotal Phase 3 MAESTRO-NASH clinical trial of resmetirom for the treatment of NASH and liver fibrosis. Madrigal has submitted an NDA to the FDA and was granted Priority Review for Resmetirom for the treatment of non-cirrhotic NASH with liver fibrosis and has been filed for THG-1001 and TesoRx has commenced enrollment forassigned a Phase1/2a clinical trial in patients with hypogonadism.PDUFA date of March, 14, 2024.
Hydroxyprogesterone caproate, or HPC, Preterm Birth, or PTB, Market Overview
PTB is defined as delivery before 37 weeks of gestation. The only approved therapy for prevention of PTB in women with a prior history of at least one preterm birth (~180,000(approximately 145,000 pregnancies annually) iswhich was a weekly intramuscular injection of hydroxyprogesterone caproate,HPC, marketed by AMAG Pharmaceuticals, Inc.Covis under the brand name Makena®., was pulled from the market effective April 6, 2023 because the PROLONG trial failed to verify the clinical benefit of Makena. The FDA granted a 7-year orphanconcluded that the available evidence does not show Makena is effective for its approved use and withdrew its approval of Makena and ordered the immediate withdrawal of Makena and several approved generic versions of the drug, exclusivitymaking it unlawful for the drug to Makena in February 2011 because the product is intended to treat “rare diseases or conditions” defined as a condition that affects fewer than 200,000 personsbe distributed in the United States which expired in February 2018. AMAG is also developing a potential subcutaneous autoinjector for Makena that received FDA approval on February 14, 2018. Treatment with Makena is initiated in pregnant women between week 16 and week 20 of pregnancy and is continued until up to delivery or week 37, whichever is earlier. The intramuscular injection is administered by a healthcare provider using a 21-gauge needle into the gluteus muscle, alternating sides each week. The intramuscular injections are associated with significant pain, discomfort and associated injection site reactions.US.
AMAG Pharmaceuticals acquired Makena from Lumara Health Inc. in November 2014 for an upfront consideration of $675.0 million ($600.0 million in cash and $75.0 million in AMAG Pharmaceuticals stock) and additional contingent consideration of up to $350.0 million based on achievement of certain sales milestones. Net sales of Makena in 2017 were estimated to be $387.2 million.
Manufacturing Agreement
On March 3, 2016, we entered into a Commercial Manufacturing Services and Supply Agreement (the “Manufacturing Agreement”) with M.W. Encap Ltd. (“Encap”), a United Kingdom based contract manufacturer, a division of Capsugel Dosage Form Solutions which is a subsidiary of Lonza. Pursuant to the Manufacturing Agreement, Encap has agreed to manufacture and supply bulk commercial quantities of TLANDO. From the effective date of the Manufacturing Agreement through the fifth anniversary of the date that FDA approval is obtained for the sale and marketing of TLANDO in the United States unless earlier terminated, we have agreed to purchase a minimum of TLANDO on an annual basis from Encap once we receive commercial approval on the basis of a 12-month rolling commercial forecast in which the first 3 months of each rolling forecast are binding on us. Such forecast may be subsequently increased or decreased by us pursuant to the terms of the Manufacturing Agreement.
In general, we may terminate the Manufacturing Agreement without incurring any fees or costs upon 90 days written notice or immediately if Encap is not able to meet our reasonable requirements of TLANDO. We and Encap may each terminate the Manufacturing Agreement upon a material breach of the Manufacturing Agreement by the other party, so long as the other party has not cured such breach within a defined period after written notice of the breach by the non-breaching party or in the event the other party becomes insolvent or goes into bankruptcy, liquidation or receivership. Encap may terminate the Manufacturing Agreement if we have not placed a firm order for TLANDO within a defined period of time from the date of FDA approval of TLANDO. Additionally, Encap may terminate the Manufacturing Agreement without cause upon the provision of written notice within a defined period of time advance written notice.
Additionally, we entered into an Agreement for the Manufacture of Testosterone Undecanoate Liquid Fill Capsules and the Conduct of an ICH Stability Study in Support of Product Registration with Encap pursuant to which Encap manufactured and supplied to us a total of six lots of TLANDO capsules under current good manufacturing practices. These lots were used in Lipocine’s Phase 3 study for TLANDO. Under the agreement, Encap is also conducting an International Conference on Harmonisation stability program on all six capsule lots in support of our resubmitted NDA filing for TLANDO. If Encap is unable to produce sufficient capsules for our future clinical trials or to support demand for TLANDO if it becomes commercially available, our revenue and profitability would be adversely affected.
Intellectual Property
Drug Delivery Technologies for Lipophilic Drug Substances
TLANDO is an oral formulation of the lipophilic prodrug testosterone undecanoate, utilizing our proprietary technology for improved delivery of lipophilic therapeutic agents. Our patent portfolio is directed to various types of compositions and methods for delivery of lipophilic drugs, which are drugs that are soluble in lipids. Our FDA approved product, TLANDO, is an oral formulation of the lipophilic prodrug TU, utilizing our proprietary technology for improved delivery of lipophilic therapeutic agents.
As of March 6, 2018, we otherwise own or control 207, 2024, our intellectual property patent portfolio consists of various issued U.S. patents 31 pending U.S.and patent applications 20 issued foreign patents, 50 pending foreign patent applicationsrelated to Oral TU, LPCN 1111, LPCN 1107, LPCN 1144/1148, and 3 pending Patent Cooperation Treaty (“PCT”) applications. OfNeurosteroids/CNS treatments both in the above, we have 12 issued U.S. patents, 25 pendingand in multiple countries outside of the U.S. patent applications, 20 issued foreign patents, 6 pending foreign patent applications and 1 PCT applications relating to various aspects of TLANDO.
| 16 |
We also hold license rights in fields other than cough and cold, to 2 U.S. patents and 1 U.S. applicationsapplication (and related foreign patents and applications) that we previously assigned to Spriaso LLC, which could be possibly used with future product candidates.
Our issued U.S. Patent No. 6,267,985 covers pharmaceutical compositions comprising a therapeutic agent solubilized in a triglyceride, and it is expected to expire in 2019. WeAdditionally, we have corresponding patents in Australia, Canada, and New Zealand. These corresponding foreign patents are all expected to expire in 2020. Our issued U.S. Patents No. 6,569,463 and 6,923,988 cover various aspects of pharmaceutical compositions comprising a hydrophobic active ingredient admixed with a hydrophilic surfactant and other components (for example, a lipophilic additive). These issued patents are expected to expire in 2019 and 2020, respectively, and if pending U.S. applications were to issue as patents, their expected expiration would be in 2019. We have corresponding patents in Canada which are expected to expire in 2020.
We have 2 issued US patents (U.S. Patent No. 8,865,695 and U.S. Patent No. 8,778,922), 5 pending U.S. patent applications, one issued patent in each of Canada, Japan, Australia and Mexico and 4 corresponding foreign patent applications (one each in Europe, Hong Kong, Brazil, and India) directed to oral pharmaceutical compositions comprising a testosterone ester and methods of their use. These patents and applications, if they issue, are expected to expire in 2029 in the U.S. and 2030 in foreign jurisdictions.
We have pending U.S. patent applications, 3 foreign patents (one each in Australia, Canada and New Zealand) directed to oral dosage forms comprising a drug, a solubilizer, and a release modulator. The pending U.S. patent applications, if they issue, are expected to expire as early as 2023, and the foreign patents are expected to expire in 2026.
We have pending U.S. patent applications directed to pharmaceutical compositions comprising a sex hormone with corresponding foreign patents in Australia, Canada and Japan. These applications, if they issue, are expected to expire in 2019, while the foreign patents are expected to expire in 2024.
We have 5 issued patents and 6 pending U.S. applications directed to high strength capsule formulations of testosterone undecanoate and methods of their use. These patents and applications, if they issue, are expected to expire in 2030.
We have 2 pending U.S. patent application related to solid dosage forms that have testosterone undecanoate. This application, if it issues, is expected to expire in 2030.
We have pending US applications related to fixed dose dosing regimens, food effects, methods of treatment and high-loading formulations. The high-loading formulations application is also pending in China and Russia.
We currently do not have patent protection for TLANDO in many countries, including territories such as India, Russia, and China, and we will be unable to prevent patent infringement in those countries unless we can file patent applications and obtain patents in those countries that cover TLANDO. We currently have 1 PCT application pending which can be entered into the national phase of such countries to protect TLANDO. Additionally, the 1012 U.S. patents that could be listedwe plan to list in the FDA Orange Book for TLANDO that are expected to expire in 2019, 2020,between 2029 and 2030.2041. If we or our Licensee are marketing the TLANDO product at the time the patents expire and have no other issued U.S. patents covering the product, then we will lose certain advantages that come with FDA Orange Book listing of patents and will no longer be able to prevent others in the U.S. from practicing the inventions claimed by the 1012 patents.
US Patent No. 8,951,996 (along with 4 additional related US issued patents) and 2 pending U.S. patent applications with corresponding counterpart applications filed in Australia (granted), Brazil, Canada (granted), China, Europe, India, Israel (granted), Japan, Mexico (granted), New Zealand (granted), Russia (granted), South Africa (granted) and South Korea are related to our LPCN 1107 product candidate. We have an additional PCT application which was entered into Australia, Brazil, China, Europe, Indonesia, Israel, Japan, Mexico, New Zealand, Philippines, and South Africa, as well as a corresponding US application. These U.S. patents and pending U.S. patent applications, if they issue, are expected to expire as early as 2031, and the foreign patent applications if they issue, are expected to expire in 2032.
US applications with corresponding counterparts filed in Argentina, Australia, Brazil, China, Canada, Europe, India, Israel, Japan, Mexico, New Zealand, Paraguay, South Africa, South Korea, Taiwan, Uruguay and Venezuela and 1 PCT applications as well which can be filed into other foreign jurisdictions at the appropriate time, are being prosecuted to protect our LPCN 1111 product candidate. We have 2 issued US patent related to LPCN 1111 that are expected to expire in 2035. The U.S. patent applications, if they issue, are expected to expire as early as 2029, and the foreign patent applications if they issue, are expected to expire as early as 2034.
We expect to file new patent applications in the future in an attempt to further cover to various aspects of our products and product development.
See Item 3 – Legal Proceedings, for a discussion of intellectual property related legal proceedings.
Government Regulation
The Regulatory Process for Drug Development
The production and manufacture of our product candidates and our research and development activities are subject to regulation by various governmental authorities around the world. In the United States, drugs and products are subject to regulation by the FDA. There are other comparable agencies in Europe and other parts of the world. Regulations govern, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products. Applicable law requires licensing and registration of manufacturing and contract research facilities, carefully controlled research and testing of products, governmental review and/or approval of results prior to marketing therapeutic products. Additionally, adherence to good laboratory practices, or GLP, good clinical practices, or GCP, during clinical testing and current good manufacturing practices, or cGMP, during production is required. The system of new drug approval in the United States is generally considered to be the most rigorous in the world and is described in further detail below under “United States Pharmaceutical Product Development Process.”
United States Pharmaceutical Product Development Process
In the United States, the FDA regulates pharmaceutical products under the Federal Food, Drug and Cosmetic Act and implementing regulations.the regulations it implements. The testing, production, sale, and promotion of pharmaceutical products are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
It takes many years for a typical experimental drug to go from concept to approval. The process required by the FDA before a pharmaceutical product may be marketed in the United States generally includes the following:
| ● | Completion of preclinical laboratory tests and animal studies. The latter often conducted according to GLPs or other applicable regulations, as well as synthesis and drug formulation development leading ultimately to clinical drug supplies manufactured according to cGMPs; | |
| ● | Submission to the FDA of an IND, which must be submitted to the FDA and become effective before human clinical trials may begin in the United States; | |
| ● | Performance of adequate and well-controlled human clinical trials according to the FDA’s current GCPs, to establish the safety and efficacy of the proposed pharmaceutical product for its intended use; | |
| ● | Submission to the FDA of an NDA for a new pharmaceutical product; | |
| ● | Satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the pharmaceutical product is produced to assess compliance with the FDA’s cGMP to assure that the facilities, methods and controls are adequate to preserve the pharmaceutical product’s identity, strength, quality and purity; | |
| ● | Potential FDA audit of the preclinical and clinical trial sites that generated the data in support of the NDA; and | |
| ● | FDA review and approval of the NDA. |
The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources and FDA approval is inherently uncertain.
| 17 |
Preclinical Studies: Prior to preclinical studies, a research phase takes place which involves demonstration of target and function, design, screening and synthesis of agonists or antagonists. Preclinical studies include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to evaluate efficacy and activity, toxic effects, pharmacokineticsPKs and metabolism of the pharmaceutical product candidate and to provide evidence of the safety, bioavailability and activity of the pharmaceutical product candidate in animals. The conduct of the preclinical safety evaluations must comply with federal regulations and requirements including GLPs. The results of the formal IND-enabling preclinical studies, together with manufacturing information, analytical data, any available clinical data or literature as well as the comprehensive descriptions of proposed human clinical studies, are then submitted as part of the IND application to the FDA.
The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the IND on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a pharmaceutical product candidate at any time before or during clinical trials due to safety concerns or non-compliance. Accordingly, we cannot be certain that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such clinical trial.
Clinical Trials: Clinical trials involve the administration of the pharmaceutical product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by the sponsor. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety. Each protocol must be submitted to the FDA if conducted under a U.S. IND. Clinical trials must be conducted in accordance with the FDA’s GCP requirements. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, or ethics committee at or servicing each institution at which the clinical trial will be conducted. An IRB or ethics committee is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB or ethics committee also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
Phase 1 Clinical Trials: Phase 1 clinical trials are usually first-in-man trials, take approximately one1 to two2 years to complete and are generally conducted on a small number of healthy human subjects to evaluate the drug’s activity, schedule and dose, pharmacokineticsPKs and pharmacodynamics. However, in the case of life-threatening diseases, such as cancer, the initial Phase 1 testing may be done in patients with the disease. These trials typically take longer to complete and may provide insights into drug activity.
Phase 2 Clinical Trials: Phase 2 clinical trials can take approximately one1 to three3 years to complete and are carried out on a relatively small to moderate number of patients (as compared to Phase 3) in a specific indication. The pharmaceutical product is evaluated to preliminarily assess efficacy, to identify possible adverse effects and safety risks, and to determine optimal dose, regimens, pharmacokinetics,PKs, pharmacodynamics and dose response relationships. This phase also provides additional safety data and serves to identify possible common short-term side effects and risks in a larger group of patients. Phase 2 clinical trials sometimes include randomization of patients.
Phase 3 Clinical Trials: Phase 3 clinical trials take approximately two2 to five5 years to complete and involve tests on a much larger population of patients (several hundred to several thousand patients) suffering from the targeted condition or disease. These studies usually include randomization of patients and blinding of both patients and investigators at geographically dispersed test sites (multi-center trials). These trials are undertaken to further evaluate dosage, clinical efficacy and safety and are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. Generally, two2 adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA or foreign authorities for approval of marketing applications.NDAs.
Post-approval studies, or Phase 4 clinical trials, may be conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication and may be required by the FDA as a condition of approval.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events or for any finding from tests in laboratory animals that suggests a significant risk for human subjects. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or, if used, its data safety and monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB or ethics committee can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s or ethics committee’s requirements or if the pharmaceutical product has been associated with unexpected serious harm to patients.
| 18 |
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the pharmaceutical product, as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the pharmaceutical product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final pharmaceutical product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the pharmaceutical product candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Pharmaceutical Review and Approval Process
New Drug Application: Upon completion of pivotal Phase 3 clinical studies, the sponsor assembles all the product development, preclinical and clinical data along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the pharmaceutical product, proposed labeling and other relevant information, and submits it to the FDA as part of an NDA. The submission or application is then reviewed by the regulatory body for approval to market the product. This process typically takes eight8 months to one1 year to complete. The FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. If a product receives regulatory approval, the approval may be limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that drug. Orphan drug designation must be requested before submitting an NDA. AfterIf the FDA grants orphan drug designation, the FDA then discloses publicly the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.use. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for seven7 years. Orphan drug exclusivity, however, could also block the approval of one of our products for seven years if a competitor obtains approval of the same drug as defined by the FDA or if our drug candidate is determined to be contained within the competitor’s product for the same indication or disease.
Priority Review
Priority Review is a designation for an NDA after it has been submitted to the FDA for review. Reviews for NDAs are designated as either “Standard” or “Priority.” A Standard designation sets the target date for completing all aspects of a review and the FDA taking an action on 90% of applications (i.e., approve or not approve) at 12 months after the date it was submitted for drugs considered new molecular entities and at 10 months after the date it was submitted for drugs considered non-new molecular entities. A Priority designation sets the target date for the FDA action on 90% of applications at eight months after submission for drugs considered new molecular entities and at 6 months after submission for drugs considered non-new molecular entities. A Priority designation is intended for those products that address unmet medical needs.
Post-Approval RequirementsAccelerated Approval
Accelerated Approval or Subpart H Approval is a program described in the NDA regulations that is intended to make promising products for life threatening diseases available on the basis of evidence of effect on a surrogate endpoint prior to formal demonstration of patient benefit. A surrogate marker is a measurement intended to substitute for the clinical measurement of interest, usually prolongation of survival in oncology that is considered likely to predict patient benefit. The approval that is granted may be considered a provisional approval with a written commitment to complete clinical studies that formally demonstrate patient benefit.
Post-Approval Requirements
Any pharmaceutical products for which we may receive FDA approvalsapproval are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with the FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, prohibitions on promoting pharmaceutical products for uses or in patient populations that are not described in the pharmaceutical product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities and promotional activities involving the internet. Failure to comply with the FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors and civil or criminal penalties.
The FDA also may require post-marketing testing, known as Phase 4 testing, risk evaluation and mitigation strategies and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
21stCentury Cures Act
The 21st Century Cures Act (Public Law No. 144-255) was enacted on December 13, 2016. This sweeping legislation makes significant changes to the way that FDA approves new drugs and medical devices. Among other things, the legislation calls on the FDA to consider new types of data, such as patient experience data, in its drug approval process. The legislation also permits drug manufacturers to utilize new types of clinical trial designs in order to collect data in the drug approval process. The intent of many of the statute’s provisions are to speed the approval of new drugs and medical devices. Whether the 21st Century Cures Act realizes these goals will depend on the adoption of new FDA regulations, policy guidance and FDA approval practices.practices, many of which the agency has not yet proposed or issued.
| 19 |
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including, but not limited, to the Centers for Medicare and Medicaid Services and other divisions of the United States government, including the U.S. Federal Communications Commission, the Department of Health and Human Services, the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, if a drug product is reimbursed by Medicare, Medicaid, or other federal or state healthcare programs, our company,Company, including our sales, marketing and scientific/educational grant programs, among others, must comply with federal healthcare laws, including, but not limited to, the federal Anti-Kickback Statute, false claims laws, civil monetary penalties laws, healthcare fraud and false statement provisions and data privacy and security provisions under the Health Insurance Portability and Accountability Act, or HIPAA, the Physician Payment Sunshine Act, and any analogous state laws. If a drug product is reimbursed by Medicare or Medicaid, pricing and rebate programs must comply with, as applicable, the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 or OBRA,(“OBRA”), and the Medicare Prescription Drug Improvement and Modernization Act of 2003, or MMA.2003. Among other things, OBRA requires drug manufacturers to pay rebates on prescription drugs to state Medicaid programs and empowers states to negotiate rebates on pharmaceutical prices, which may result in prices for our future products that will likely be lower than the prices we might otherwise obtain. Additionally, the Patient Protection and Affordable Care Act as amended by the Health Care and Education Reconciliation Act of 2010 collectively, ACA,(collectively, “ACA”) substantially changes the way healthcare is financed by both governmental and private insurers. Among other cost containment measures, ACA establishes: an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents; a new Medicare Part D coverage gap discount program; and a new formula that increases the rebates a manufacturer must pay under the Medicaid Drug Rebate Program. There may continue to be additional proposals relating to the reform of the U.S. healthcare system, in the future, some of which could further limit coverage and reimbursement of drug products. If drug products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements may apply.
Additionally, to the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including fraud and abuse, privacy and transparency laws.
Pharmaceutical Coverage, Pricing and Reimbursement
In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of coverage and adequate reimbursement from third-party payers, including government health administrative authorities, managed care providers, private health insurers and other organizations. In the United States, private health insurers and other third-party payers often provide reimbursement for products and services based on the level at which the government (through the Medicare or Medicaid programs) provides reimbursement for such treatments. Third-party payers are increasingly examining the medical necessity and cost-effectiveness of medical products and services in addition to their safety and efficacy and, accordingly, significant uncertainty exists as to the coverage and reimbursement status of newly approved therapeutics. In particular, in the United States, the European Union and other potentially significant markets for our product candidates, government authorities and third-party payers are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies, which has resulted in lower average selling prices. Further, the increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the European Union will put additional pressure on product pricing, reimbursement and usage, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of insurers and managed care organizations, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general. As a result, coverage and adequate third-party reimbursement may not be available for our products to enable us to realize an appropriate return on our investment in research and product development.
The Inflation Reduction Act of 2022 (Pub. L. No. 117-169) was signed into law on August 16, 2022, and includes a number of provisions aimed at lowering prescription drug costs and reducing government spending on drugs. This includes a requirement that the Department of Health and Human Services negotiate a “maximum fair price” with drug manufacturers for certain single-source brand drugs or biologics without generic or biosimilar competitors that are covered under Medicare Part D and Part B. This pricing will begin in 2026 for Medicare Part D and 2028 for Medicare Part B. An excise tax is imposed on drug manufacturers that fail to comply with the required negotiation process. In addition, the law requires drug manufacturers to pay a rebate to the federal government if the price for almost all drugs covered under Medicare Part D (starting in 2022), and single-source drug or biologics covered under Medicare B (starting in 2023), increase greater than the inflation rate. The rebate amount equals the number of drug units sold in Medicare multiplied by the amount the drug’s price exceeds the inflation-adjusted price. The law also modifies the Medicare Part D benefit structure to cap the amount beneficiaries must spend on drug costs and increase the discounts manufacturers are required to pay. The Inflation Reduction Act of 2022 signals an increased desire to control the prices and costs associated with pharmaceutical products. In recent years, state laws have been enacted to lower prescription drug costs and prices. In some states, such as Colorado and Maryland, a drug affordability board was created to identify specific drugs that are particularly costly or otherwise create affordability challenges. These drug affordability boards have authority to either implement or recommend upper payment limits for these drugs. This legislation, as well as any future statutes or regulations at the federal or state level, may impact reimbursement for our product candidates and may challenge our ability to realize an appropriate return on our investment in research and product development.
| 20 |
The market for our product candidates for which we may receive regulatory approval will depend significantly on access to third-party payers’ drug formularies or lists of medications for which third-party payers provide coverage and reimbursement. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payers may refuse to include a particular branded drug in their formularies or may otherwise restrict patient access to a branded drug when a less-costly generic equivalent or other alternative is available. In addition, because each third-party payer individually approves coverage and reimbursement levels, obtaining coverage and adequate reimbursement is a time-consuming and costly process. We would be required to provide scientific and clinical support for the use of any product to each third-party payer separately with no assurance that approval would be obtained, and we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. This process could delay the market acceptance of any of our product candidates for which we may receive approval and could have a negative effect on our future revenues and operating results. We cannot be certain that our product candidates will be considered cost-effective. If we are unable to obtain coverage and adequate payment levels for our product candidates from third-party payers, physicians may limit how much or under what circumstances they will prescribe or administer them, and patients may decline to purchase them. This in turn could affect our ability to successfully commercialize our products and impact our profitability, results of operations, financial condition, and future success.
The United States Orphan Drug Act encourages the development of orphan drugs, which are intended to treat “rare diseases or conditions” within the meaning of this Act (i.e., those that affect fewer than 200,000 persons in the United States). The provisions of the Act are intended to stimulate the research, development and approval of products that treat rare diseases. Orphan Drug Designation provides a sponsor with several potential benefits: (1) sponsors may be granted seven years of marketing exclusivity after approval of the orphan-designated indication for the drug product; (2) sponsors are granted U.S. tax incentives for clinical research; (3) the FDA’s office of orphan products development co-ordinates research study design assistance for sponsors of drugs for rare diseases; and (4) grant funding can be obtained to defray costs of qualified clinical testing.
Priority Review
Priority Review is a designation for an NDA after it has been submitted to the FDA for review. Reviews for NDAs are designated as either “Standard” or “Priority.” A Standard designation sets the target date for completing all aspects of a review and the FDA taking an action on 90% of applications (i.e., approve or not approve) at 12 months after the date it was submitted for drugs considered new molecular entities and at 10 months after the date it was submitted for drugs considered non-new molecular entities. A Priority designation sets the target date for the FDA action on 90% of applications at eight months after submission submitted for drugs considered new molecular entities and at 6 months after submission for drugs considered non-new molecular entities. A Priority designation is intended for those products that address unmet medical needs.
Accelerated Approval
Accelerated Approval or Subpart H Approval is a program described in the NDA regulations that is intended to make promising products for life threatening diseases available on the basis of evidence of effect on a surrogate endpoint prior to formal demonstration of patient benefit. A surrogate marker is a measurement intended to substitute for the clinical measurement of interest, usually prolongation of survival in oncology that is considered likely to predict patient benefit. The approval that is granted may be considered a provisional approval with a written commitment to complete clinical studies that formally demonstrate patient benefit.
Related Party Transaction
On July 23, 2013, we entered into assignment/license and services agreements with Spriaso, LLC, an entity that is majority-owned by Mahesh V. Patel, Gordhan Patel, John W. Higuchi, Dr. William I. Higuchi, and their affiliates. Mahesh V. Patel is our President and Chief Executive Officer and a Chairman of our Board of Directors.Officer. Mr. Higuchi is a member of our Board of Directors and Gordhan Patel and Dr. Higuchi, former Board members, were each members of our Board of Directors at the date the license and agreements were entered into.
Under the assignment agreement, we assigned and transferred to Spriaso all of our rights, title and interest in our intellectual property for the cough and cold field. In addition, Spriaso was assigned all rights and obligations under our product development agreement with a co-development partner. In exchange, we would be entitled to receive a potential cash royalty of 20% of the net proceeds received by Spriaso, up to a maximum of $10 million. Spriaso also granted back to us an exclusive license to such intellectual property to develop products outside of the cough and cold field. The assignment agreement will expire upon the expiration of all of Spriaso’s payment obligations thereunder and the expiration of all of the licensed patents thereunder. Spriaso has the right to terminate the assignment agreement with 30 days written notice. We have the right to terminate the assignment agreement upon the complete liquidation or dissolution of Spriaso, unless the assignment agreement is assigned to an affiliate or successor of Spriaso.
Under the services agreement, we willagreed to provide facilities and up to 10% of the services of certain employees to Spriaso for a period of up to 18 months which expired January 23, 2015. Effective January 23, 2015, we entered into an amended services agreement with Spriaso in which we agreed to continue providing up to 10% of the services of certain employees to Spriaso at a rate of $230/hour for a period of six months.time. The agreement was further amended on July 23, 2015, on January 23, 2016, on July 23, 2016 on January 23, 2017, on July 23, 2017 and again on January 23, 2018 to extend the term of the agreement for an additional six months. The agreementprovide services expired in 2021; however, it may be extended upon written agreement of Spriaso and us. Additionally, Spriaso filed its first NDA in 2014, and as an affiliated entity of Lipocine, it used up the one-time waiver of user fees for a small business submitting its first human drug application to FDA.
| 21 |
Employees
As of December 31, 2017,2023, we had 1417 full time employees and we also utilize the services of consultants on a regular basis. EightTwelve employees are engaged in drug development activities and sixfive are in general administration. marketing and sales functions.administration functions and the majority of our employees work out of our Salt Lake City facility. The Company continually evaluates the business need and opportunity and balances in house expertise and capacity with outsourced expertise and capacity. Currently, we outsource substantial clinical trial work to clinical research organizations and certain drug manufacturing to contract manufacturers. None of our employees are represented by labor unions or covered by collective bargaining agreements.agreements and we consider our relations with our employees to be good.
We strive toward having a diverse team of employees and are committed to equality, inclusion and workplace diversity.
Available InformationReverse Stock Split
On May 10, 2023, our Board approved a reverse stock split of 1-for-17. We filed the Amendment to its Certificate of Incorporation with the Secretary of the State of Delaware on May 10, 2023, and the Amendment became effective at 5:00 pm Eastern Time on May 11, 2023. Our shares began trading on a split-adjusted basis on the Nasdaq Capital Market commencing upon market open on May 12, 2021. The par value of the common stock and preferred stock was not adjusted as a result of the reverse stock split. All common stock and per share amounts in the financial statements have been retroactively adjusted for all periods presented to give effect to the reverse stock splits.
Available Information
Our website address is www.lipocine.com. We make available free of charge on the Investor Relations portion of our website, ir.lipocine.com, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission. The SEC maintains an internet website that contains reports, proxy and information statements, and other information that we file electronically, which can be found at http://www.sec.gov.
ITEM 1A. RISK FACTORS
We have identified the following risks and uncertainties that may have a material adverse effect on our business, financial condition, results of operations and future growth prospects. Our business could be harmed by any of these risks. The risks and uncertainties described below are not the only ones we face. The trading price of our common stock could decline due to any of these risks, and you may lose all or part of your investment. In assessing these risks, you should also refer to the other information contained in this Annual Report on Form 10-K, including our consolidated financial statements and related notes.
Risk Factors Summary
Our business operations are subject to numerous risks, factors and uncertainties, including those outside of our control, that could cause our actual results to be harmed, including risks regarding the following:
Risks Relating to Our Business and Industry
| ● | the timelines of our clinical trials; | |
| ● | the early stage of development of LPCN 1154, LPCN 2101, LPCN 2203, LPCN 1148, LPCN 1144, LPCN 1111 and LPCN 1107; | |
| ● | the early stage of development of our research and development programs and processes and the risk of competition; | |
| ● | there can be no assurance as to our ability to file an NDA for LPCN 1154 for postpartum depression or our ability to utilize the streamlined approval pathway under 505(b)(2); | |
| ● | the regulatory requirements for our product candidates and the possibility that regulatory approval will not be received; | |
| ● | the commercial success of our licensed product candidate, TLANDO; | |
| ● | the possibility that T-replacement therapies could be found to create, or could be perceived to create, health risks; | |
| ● | any possible failure to obtain adequate healthcare reimbursement for our products; | |
| ● | competition in the TRT market, including the entrance of generic TRTs into the market; | |
| ● | our Licensee’s ability to commercialize TLANDO may be limited; | |
| ● | successful commercialization of our product candidates internally or through collaborators; | |
| ● | the possibility that we may never receive regulatory approval to market our products outside the United States; | |
| ● | the stringent government regulations concerning the clinical testing of our products; | |
| ● | the market’s acceptance of our products; | |
| ● | physicians and patients using other products may not switch to our product; | |
| ● | the possibility that regulatory agencies could find that we have improperly promoted off-label uses; | |
| ● | any possible failure to comply with federal and state healthcare laws; | |
| ● | our ability to retain our chief executive officer and other key executives and to attract, retain and motivate qualified personnel; | |
| ● | difficulties in managing the growth of the Company; | |
| ● | re-importation of drugs from foreign countries into the United States by our competitors; | |
| ● | any product liability claims; | |
| ● | any failure to comply with the Controlled Substances Act; | |
| ● | the defense and resolution of any litigation; | |
| ● | cyber security risks; |
| 22 |
Risks Related to Our Dependence on Third Parties
| ● | our reliance on third-party contractors and service providers for the execution of some aspects of our development programs; | |
| ● | our reliance on contract research organizations or other third parties to assist us in conducting clinical trials; | |
| ● | our reliance on suppliers for the active and inactive ingredients for our products; | |
| ● | our ability to establish successful collaborations for our products; |
Risks Related to Ownership of Our Common Stock
| ● | our stock price’s reaction to the results and timing of clinical trials, regulatory and other decisions; | |
| ● | the effectiveness of our internal control over financial reporting; | |
| ● | the cost and expense to comply with the requirements of being a public company; | |
| ● | the volatility of our share price; | |
| ● | fluctuations in the value of our warrants outstanding from the November 2019 Offering; | |
| ● | the possibility of delisting of our securities from the Nasdaq Capital Market; | |
| ● | anti-takeover provisions in our amended and restated certificate of incorporation and our amended and restated bylaws, as well as provisions of Delaware law and our stockholder rights plan; | |
| ● | the right of the holders of the common warrants issued in the November 2019 Offering to receive the Black Scholes value of the warrants in the event of a fundamental transaction; | |
| ● | our decision not to pay dividends on our common stock; | |
| ● | our management and directors’ ability to exert influence over our affairs; | |
| ● | volatility in the trading price of our common stock; | |
| ● | any failure of securities or industry analysts to publish accurate research about our business; |
Risks Relating to Our Financial Position and Capital Requirements
| ● | our need for and ability to obtain substantial additional capital in the future; | |
| ● | potential dilution to our existing stockholders from raising any additional capital; | |
| ● | our inability to predict when we will generate product revenues or achieve profitability; | |
| ● | our incurrence of significant operating losses; | |
| ● | any fluctuation in our operating results; |
Risks Relating to Our Intellectual Property
| ● | our ability to protect our intellectual property; | |
| ● | our ability to obtain additional protection under the Drug Price Competition and Patent Term Restoration Act; | |
| ● | the possibility of incurring substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights, or our inability to protect our rights to our products and technology; | |
| ● | the cost and expense, and any unfavorable outcomes, resulting from any claims for infringing intellectual property rights of third parties; | |
| ● | the fact that we do not have patent protection for our product candidates in a significant number of countries; | |
| ● | our ability to comply with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies; and | |
| ● | the possibility that we may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers. |
| 23 |
Risks Relating to Our Business and Industry
The timelines and costs of our clinical trials may be impacted by numerous factors and any delays may adversely affect our ability to execute our current business strategy.
Our expectations regarding the success of our product candidates, including our clinical candidates and lead compounds, and our business are based on projections which may not be realized for many scientific, business or other reasons. We therefore cannot assure investors that we will be able to adhere to our current schedule. We set goals that forecast the accomplishment of objectives material to our success: selecting clinical candidates, product candidates, failures in research, the inability to identify or advance lead compounds, identifying target patient groups or clinical candidates, the timing and completion of clinical trials, and anticipated regulatory approval. The actual timing of these events can vary dramatically due to factors such as available capital resources, slow enrollment of patientssubjects in studies, uncertainties in scale-up, manufacturing and formulation of our compounds, failures in research, the inability to identify clinical candidates, failures in our clinical trials, requirements for additional clinical trials and uncertainties inherent in the regulatory approval process and regulatory submissions. Decisions by our partners or collaborators may also affect our timelines and delays in achieving manufacturing capacity and marketing infrastructure sufficient to commercialize our products.capacity. The length of time necessary to complete clinical trials and to submit an application for marketing approval by applicable regulatory authorities may also vary significantly based on the type, complexity and novelty of the product candidate involved, as well as other factors. In addition, the development of our product candidates will require significant capital resources and we may not be able to raise sufficient additional capital to fund continued development. Further, we may choose to allocate available capital resources to development of product candidates that do not ultimately achieve commercialization.
LPCN 1154 is still in development and may not be further developed or achieve commercialization for a variety of reasons.
LPCN 1154 is still in development and we plan to conduct a pivotal trial in 2024. There can be no assurance as to the results of the pivotal trial or the timing or completion of any NDA filing relating to LPCN 1154 for postpartum depression. In particular, there can be no assurance that we are able to complete required trials in a timely manner or demonstrate efficacy or meet the requirements of a 505(b)(2) pathway and we may be required to undertake additional clinical studies prior to filing an NDA for LPCN 1154. We are exploring the possibility of partnering LPCN 1154 to a third party for commercialization, however we may not be able to identify potential partners or successfully enter into partnership arrangements on terms favorable to us, if at all. If we are unable to successfully partner or otherwise develop LPCN 1154, LPCN 1154 may never be successfully commercialized.
LPCN 2101 is in a very early stage of development and may not be further developed for a variety of reasons.
Our oral NAS comprising program LPCN 2101 is in a very early stage of development and consequently the risk that we may fail to commercialize LPCN 2101 and related products is high. We have only conducted Phase 1 clinical studies of LPCN 2101 and the ultimate regulatory or technical success of the neuroactive steroids under investigation in these programs is uncertain. The current limited pre-clinical and phase 1 results we have observed may not be replicated in larger studies, future PK, Phase 2, or pivotal studies with a potential “to be marketed formulation”. We may not be able to further test in-clinic in a timely manner or at all due to other regulatory hurdles.
In addition, our oral NAS product candidate LPCN 2101 may not be effective in treating WWE or any other indications or may not have differentiation from competitive products on the market or in development. We may expend significant resources before determining that these programs are not viable candidates for regulatory approval and commercialization.
LPCN 2203 is in an early stage of development and may not be further developed for a variety of reasons.
Our oral NAS comprising programs (including LPCN 2203) are in a very early stage of development and consequently the risk that we may fail to commercialize LPCN 2203 and related products is high. We have only conducted Phase 1 clinical studies with the active pharmaceutical ingredient in LPCN 2203 and the ultimate regulatory or technical success of the neuroactive steroids under investigation in these programs is uncertain. The current limited pre-clinical and Phase 1 results we have observed may not be replicated in larger studies, future PK, Phase 2, or pivotal studies. We may not be able to further test in-clinic in a timely manner or at all due to other regulatory hurdles.
In addition, our oral NAS product candidate LPCN 2203 may not be effective in treating ET or may not have differentiation from competitive products on the market or in development. We may expend significant resources before determining that these programs are not viable candidates for regulatory approval and commercialization.
LPCN 1148 is in a very early stage of development for management of liver cirrhosis in male patients and while there are no therapies specifically approved by the FDA for secondary sarcopenia or cirrhosis beyond treatment of underlying conditions, there are candidates known to be under development for cirrhosis related indication(s).
LPCN 1148 is in a very early stage of development and consequently the risk that we may fail to commercialize or partner LPCN 1148 and related products is high. This development program is susceptible to technical failures in future clinical studies and regulatory hurdles for further testing and/or meeting the FDA’s needs for NDA filing or approval. The result of the Phase 2 study may not be indicative of ultimate success in a larger Phase 2/3 clinical study and, although we are exploring the possibility of partnering LPCN 1148 to a third party for further development and commercialization, we may not be able to identify potential partners or successfully enter into partnership arrangements on terms favorable to us, if at all. While we believe there is a potential to gain Orphan Drug Designation for an indication or condition in male liver cirrhosis, the FDA may not grant such designation which could adversely impact development or the commercial potential of LPCN 1148.
LPCN 1111 is in a very early stage of development and may not be further developed for a variety of reasons.
LPCN 1111 is in a very early stage of development. We have completed a Phase 2a and Phase 2b study in hypogonadal men. Future studies may not have clinical results that support continued development and/or a path towards regulatory approval and commercialization.
In addition, the active ingredient in LPCN 1111 has only been manufactured on a small scale. Further, our Licensee may choose not to engage in further development of LPCN 1111 or, if developed, to effectively commercialize LPCN 1111 in the U.S. or Canada. In addition, the anticipated Phase 3 program for an NDA filing for LPCN 1111 could be very long and expensive, and LPCN 1111 may never be successfully further developed or commercialized.
| 24 |
LPCN 1107 is in a very early stage of development and may not be further developed for a variety of reasons.
LPCN 1107 is in a very early stage of development and consequently, although we are exploring the possibility of partnering LPCN 1107 to a third party for further development and commercialization, we may not be able to identify potential partners or successfully enter into partnership arrangements on terms favorable to us, if at all. If we are unable to successfully partner LPCN 1107, LPCN 1107 may never be successfully commercialized. In particular, we have only conducted three Phase 1 clinical studies with this product candidate. Our completed Phase 1 clinical studies may not be predictive of safety concerns that may arise in pregnant women or demonstrate that LPCN 1107 has an adequate safety profile to warrant further development. These factors can impact the timing of and our ability to continue development or partner LPCN 1107.
In addition, a number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials, even after achieving positive results in early-stage development. Accordingly, our results from our Phase 1a, our Phase 1b and our multi-dose PK dose selection studies may not be predictive of the results we may obtain from further studies and trials.
The FDA has concluded that Makena, based on Makena’s failed definitive PROLONG study, a competing product with the same active ingredient and similar target indication, is ineffective and has withdrawn Makena from the market. It is entirely possible that any pivotal study on LPCN 1107 may require a placebo-controlled trial design. Therefore, we and/or our partner may face significant challenges in patient recruitment for a placebo-controlled trial, be faced with significant resource investment to conduct additional trials, and face potential perceived risk of efficacy failure in a pivotal study resulting in no further development of LPCN 1107.
LPCN 1144 is in a very early stage of development and may not be further developed for a variety of reasons.
LPCN 1144 is in a very early stage of development and consequently the risk that we fail to commercialize LPCN 1144 and related products is high. In particular, we have announced topline primary and key secondary endpoint results from our Phase 2 LiFT and open label extension clinical studies.
Although our results from the LiFT and open label extension clinical study results were positive for NASH resolution with no worsening of fibrosis, these results may not be indicative of ultimate success in a larger Phase 2/3 clinical study with required FDA endpoints and populations needed for regulatory approval of LPCN 1144 for the treatment of NASH.
In addition, a number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials, even after achieving positive results in early-stage development. The FDA currently insists on histopathology endpoints for diagnosis and assessment of efficacy of treatment for NASH with LPCN 1144 in a pivotal trial. Accordingly, our results from our LiFT study may not be predictive of the results we may obtain from further studies and trials.
Several factors could significantly affect the prospects for LPCN 1144, including factors relating to the regulatory approval, competitive landscape and clinical development challenges for LPCN 1144. The anticipated Phase 3 programs for an NDA filing for LPCN 1144 will be very long and resource intensive. Although we are exploring the possibility of partnering LPCN 1144 to a third party for further development and commercialization, we may not be able to identify potential partners or successfully enter into partnership arrangements on terms favorable to us, if at all. If we are unable to successfully partner LPCN 1144, LPCN 1144 may never be successfully commercialized.
Our research and development programs and processes are at an early stage of development, which makes it difficult to evaluate our business and prospects or predict if or when we will successfully commercialize or partner our product candidates.
Our operations to date have primarily been limited to conducting research and development activities under license and collaboration agreements. Our current portfolio consists of product candidates at various clinical stages of development in addition to our out-licensed product TLANDO. We have never marketed or commercialized a drug product. Consequently, any predictions about our future performance may not be as accurate as they could be if we were further along our commercialization path. In addition, as a pre-commercial stage business, we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors.
Our clinical product candidates are at an early stage of development and will require significant further investment and regulatory approvals prior to marketing and commercialization. As such, our product development processes for LPCN 1154, LPCN 2101, LPCN 2203, and LPCN 1148, in addition to LPCN 1111, LPCN 1144, and LPCN 1107 are very risky and uncertain, and our product candidates may fail to advance beyond the current study. Even if we obtain required financing, we cannot ensure successful product development or that we will obtain regulatory approval or successfully commercialize or partner any of our product candidates and generate product revenues.
| 25 |
All of our clinical candidates will be subject to extensive regulation which can be costly and time consuming, cause delays or prevent approval of the products for commercialization.
Our clinical development of LPCN 1154, LPCN 2101, LPCN 2203, LPCN 1148, LPCN 1144, and LPCN 1107 and any future product candidates is subject to extensive regulations by the FDA. Product development is a very lengthy and expensive process and can vary significantly based upon the product candidate’s novelty and complexity. Regulations are subject to change and regulatory agencies have significant discretion in the approval process.
Numerous statutes and regulations govern human testing and the manufacture and sale of human therapeutic products in the United States. Such legislation and regulation bears upon, among other things, the approval of protocols and human testing, the approval of manufacturing facilities, safety of the product candidates, testing procedures and controlled research, review and approval of manufacturing, preclinical and clinical data prior to marketing approval including adherence to cGMP during production and storage as well as regulation of marketing activities including advertising and labeling.
In order to obtain regulatory clearance for the commercial sale of any of our product candidates, we must demonstrate through preclinical studies and clinical trials that the potential product is safe and efficacious for use in humans for each target indication. Obtaining approval of any of our product candidates is an extensive, lengthy, expensive and uncertain process, and the FDA may delay, limit or deny approval for many reasons, including:
| ● | we may not be able to demonstrate that the product candidate is safe and effective to the satisfaction of the FDA; | |
| ● | the results of our clinical trials may not meet the level of statistical or clinical significance required by the FDA for marketing approval; | |
| ● | the FDA may disagree with the number, design, size, conduct or implementation of our clinical trials; | |
| ● | the contract research organization that we retain to manage our clinical trials may take actions outside of our control that materially adversely impact our clinical trials; | |
| ● | the FDA may not find the data from preclinical studies and clinical trials sufficient to demonstrate that a particular product candidate’s clinical and other benefits outweigh its safety risks; | |
| ● | the FDA may disagree with our interpretation of data from our preclinical studies and/or clinical trials or may require that we conduct additional trials; | |
| ● | the FDA may not accept data generated at our clinical trial sites; | |
| ● | if an NDA, once submitted, is reviewed by an Advisory Committee, the FDA may have difficulties scheduling an Advisory Committee meeting in a timely manner or the Advisory Committee may recommend against approval of our application or may recommend that the FDA require, as a condition of approval, additional preclinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; | |
| ● | the FDA may require development of a REMS as a condition of approval; | |
| ● | the FDA may require longer or additional duration of stability data on the clinical lots prior to initiation of further clinical trials; and | |
| ● | the FDA may identify deficiencies in the formulation or stability of our product candidates or products, or relating to our manufacturing processes or facilities, or in the processes and facilities of the contract manufacturing organization (“CMO”), our suppliers, or other third parties that may be utilized in the production supply chain of our products. |
Preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain FDA approval for their products.
| 26 |
No assurance can be given that current regulations relating to regulatory approval will not change or become more stringent. The FDA may also require that we amend clinical trial protocols and/or run additional trials in order to provide additional information regarding the safety, efficacy or equivalency of any compound for which we seek regulatory approval. Moreover, any regulatory approval of a drug which is eventually obtained may entail limitations on the indicated uses for which that drug may be marketed. Furthermore, product approvals may be withdrawn or limited in some way if problems occur following initial marketing or if compliance with regulatory standards is not maintained. The FDA could become more risk averse to any side effects or set higher standards of safety and efficacy prior to reviewing or approving a product. This could result in a product not being approved.
We depend primarilyare dependent on the commercial success of our leadlicensed product, candidate, TLANDO, for which we recently received a negative vote from the BRUDAC against acceptability of the overall benefit/risk profile to support approval of TLANDO as a TRT as well as a previous Complete Response Letter from the FDAroyalty revenue and which may not receive regulatory approval or be successfully commercialized.potential milestone payments.
TLANDO is currently our only product candidate that has completed Phase 3 clinical trials,trials. On February 1, 2024, we transitioned the commercialization of TLANDO to Verity from our previous licensee Antares. In January 2024, we entered into the Verity License Agreement with Verity, pursuant to which we granted Verity an exclusive, royalty-bearing, sublicensable right and license to develop and commercialize our TLANDO product with respect to TRT in the U.S. and Canada. None of our other products have been approved for sale. Therefore, at this stage, our ability to realize revenue depends on TLANDO’s successful commercialization. The commercial success of TLANDO depends almost entirely on Verity’s commercialization efforts and we have very limited ability to influence Verity’s efforts, including the amount and timing of resources they devote, if any, to the commercialization of TLANDO. On March 29, 2022, the FDA granted approval to TLANDO for testosterone replacement therapy in adult males indicated for conditions associated with a deficiency or absence of endogenous testosterone: primary hypogonadism (congenital or acquired) and hypogonadotropic hypogonadism (congenital or acquired). Our ability to realize royalty revenue, will depend on the commercialization efforts of Verity. If Verity is not able to successfully commercialize TLANDO, we may not realize any royalty revenue under the Verity License Agreement and our business currently depends primarily on its successful development,could be adversely affected. Additionally, regulatory approval and commercialization, if approved. We submitted an NDA to the FDA but have not submitted comparable applications to other regulatory authorities. We do not control whether or when we may receive approval of TLANDO may be withdrawn and the failure to maintain regulatory approvals would prevent TLANDO from the FDA. However, we do have a PDUFA goal date of May 8, 2018. We are not permitted to market TLANDO in the United States until we receive approval of an NDA from the FDA, or in any foreign countries until we receive the requisite approval from such countries.
Although we have completed Phase 3 efficacy trials with TLANDO, approval from the FDA is not guaranteed. We resubmitted our NDA to the FDA in August 2017 based on the results of the DV study. The DV study confirmed the efficacy of TLANDO with a fixed dose regimen without need for dose adjustment. TLANDO was well tolerated upon 52-week exposure with no reports of drug related Serious Adverse Events (“SAEs”). The FDA accepted our NDA as a complete response to their CRLbeing marketed and assigned a PDUFA goal date of May 8, 2018 for completion of the review. Previously, on June 28, 2016, we received a CRL from the FDA on our original NDA submission. A CRL is a communication from the FDA that informs companies that an application cannot be approved in its present form. The CRL identified deficiencies related to the dosing algorithm for the label. Specifically, the proposed titration scheme for clinical practice was significantly different from the titration scheme used in the Phase 3 trial leading to discordance in titration decisions between the Phase 3 trial and real-world clinical practice. In response to the CRL, we met with the FDA in a Post Action meeting, and proposed a dosing regimen to the FDA based on analyses of existing data. The FDA noted that while the proposed dosing regimen might be acceptable, validation in a clinical trial would be needed prior to resubmission. If the FDA denies or further delays approval of TLANDO, our business would be materially and adversely harmed. If the FDA does approve TLANDO, but we are unsuccessful in commercializing TLANDO, our business will be materially and adversely harmed.
The FDA may also require the addition of labeling statements or other warnings or contraindications, require us to perform additional clinical trials or studies or provide additional information in order to secure approval. Any such requirement would increase our costs and delay approval and commercialization of TLANDO and wouldcould have a material adverse effect on our businessbusiness.
Under the PREA, our licensing partner, Verity, will need to address the PREA requirement to assess the safety and financial condition.
effectiveness of TLANDO in pediatric patients. The FDA also convened a BRUDAC meeting on January 10, 2018 to seek input fromrequired certain post-marketing studies including: (i) conducting an expert panel regarding TLANDO prior to the PDUFA dateappropriately designed label comprehension and knowledge study that assesses patient understanding of May 8, 2018. The BRUDAC voted six in favor and thirteen against acceptability of overall benefit/key risk profile to support approval of TLANDO as a TRT. As a result, the FDA may not agree with the adequacy of clinical development conducted or clinical data presented. Particularly the FDA may be concerned with TLANDO’s potential for increased adverse cardiovascular outcomesmessages in the population that will likely use the drug, if approved,Medication Guide for TLANDO and observed treatment emergent adverse reactions and/or(ii) conducting an appropriately designed one-year trial to evaluate development of adrenal insufficiency with the risk associated with (associated with but not limited to) the following clinical endpointschronic TLANDO therapy. Verity is responsible for an oral T product; levels of various analytes observed including testosterone, DHT, TU, DHTU, Estradiol; Cmax secondary endpoint excursions; adequacy of long-term safety database; appropriateness and validation of laboratory assays; data collected and tests performed on clinical subjects including vitals such as blood pressure or blood pressure measurement methodology and relating laboratory parameters including levels of hemoglobin, PSA, hematocrit, prolactin, HDL, LDL, Cholesterol, TG, SHGB, alkaline phosphatase, etc.; discontinuation rates experienced in studies; concerns on laboratory normal ranges for our analyte analysis; reliability of serum T measurements, cosynotropin stimulation results, TLANDO stopping criteria, restrictions on food or meal administered to subjects; and data analysis and statistical approaches. Based on the voting outcomeconducting these post-marketing studies. The ramifications of the BRUDAC meeting,results of these studies conducted by Verity, or the FDA mayramifications of Verity’s inability or may not view BRUDAC’s TLANDO advice/recommendation similarly which may result in the receipt of a CRLunwillingness to our NDA resubmission.
We may receive another CRL from the FDA which would result in substantial delaysconduct these studies, are unknown to us and additional studies and expense before we would be in a position to resubmit an NDA responsive to such additional CRL. Our ability to raise capital may also be impaired. If we proceed with any study, such as an ABPM study, we face the risk thatbetween Verity and the FDA would not agree with the design or results of the study or the study may find that blood pressure effects of TLANDO are clinically meaningful and approval of TLANDO may be denied.FDA.
Even if TLANDO is approved, the FDA may limit the indications for which it may be used, include extensive warnings on the product labeling, or require costly ongoing requirements for post-marketing clinical studies including participation in a long-term TRT consortium cardiovascular study and surveillance or other risk management measures to monitor the safety or efficacy of TLANDO. Further, inIn the event that we seek regulatory approval of TLANDO outside the United States and Canada, such markets also have requirements for approval of drug candidates with which we must comply prior to marketing. Obtaining regulatory approval for marketing of TLANDO in one country does not ensure we will be able to obtain regulatory approval in other countries but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in other countries.
Any regulatory approval of TLANDO, once obtained, may be withdrawn. Ultimately, the failure to obtain and maintain regulatory approvals would prevent TLANDO from being marketed and would have a material adverse effect on our business.
If the FDA clarifies, modifies or restricts the indicated population for T-replacement in the "class" label, the market for T-replacement products may shrink and our ability to sell and be reimbursed for TLANDO and LPCN 1111 could be materially adversely affected and our business could be harmed.
On September 17, 2014, the FDA held a T-class Advisory Committee meeting. The Advisory Committee discussed (i) the identification of the appropriate patient population for whom T-replacement therapy should be indicated and (ii) the potential risk of major adverse cardiovascular events, defined as non-fatal stroke, non-fatal myocardial infarction and cardiovascular death associated with T-replacement therapy. At the meeting, 20 of the 21 members of the Advisory Committee voted that the FDA should revise the currently indicated population for T-replacement therapy and recommended changing the label language to restrict the intended uses of the products, particularly in relation to age-related low testosterone. The Committee also supported adding language to the label to guide physicians in better diagnosis of eligible patients for treatment. On March 3, 2015, the FDA issued a safety announcement addressing the Advisory Committee’s recommendations.
The FDA's safety assessment recommended the following label modifications/restrictions in the indicated population for T-replacement therapy:
The actual TRT label revisions have been finalized between the FDA and sponsors with approved T-replacement therapy products. The revised labels are consistent with the FDA's recommendations on March 3, 2015.
Additionally, the FDA stated that they will require manufacturers of approved T-replacement products to conduct a well-designed clinical trial to more clearly address the question of whether an increased risk of heart attack or stroke exists among users of T-replacement products. The FDA encouraged manufacturers to work together on conducting a clinical trial, although the FDA will allow manufacturers to work separately if they so choose. The FDA did not address whether it would require sponsors without an approved T-replacement product to conduct a cardiovascular trial prior to being able to file an NDA. However, on March 19, 2015 we had a pre-NDA meeting with the FDA concerning our pivotal Phase 3 trial for TLANDO. Based on this meeting with the FDA, we do not expect to be required to conduct a heart attack and stroke risk study or any additional safety studies prior to approval of the NDA for TLANDO. The FDA may determine that an ABPM study may be required to better assess any blood pressure effects of TLANDO, a surrogate marker of predicting cardiovascular outcome. If the FDA requires an ABPM study or changes its position, however, and concludes that a cardiovascular trial is required prior to approving our NDA for TLANDO, such trial would require substantial financial resources, and would delay the regulatory process for TLANDO and our entry into the marketplace, all of which would have a material adverse impact on our business. Further, if TLANDO receives FDA approval, it is unclear what our post-approval obligations may be, if any, in relation to a heart attack and stroke risk study. We may be required to contribute to an on-going industry-led heart attack and stroke risk study or to conduct our own long-term heart attack and stroke risk study, either of which would require substantial financial resources and would have a material adverse impact on our business. Regulatory actions related to T-replacement therapy have contributed to a contraction in the market for T-replacement products. If the market for T-replacement products continues to decline, for whatever reason, our business will be materially and adversely harmed.
If T-replacement therapies are found, or are perceived, to create health risks, our ability to sellrealize any revenue from TLANDO and LPCN 1111 could be materially adversely affected, and our business could be harmed. Even if our TLANDO and our LPCN 1111 are approved, physiciansPhysicians and patients may be deterred from prescribing and using T-replacement therapies, which could depress demand for TLANDO and LPCN 1111 and compromise our ability to successfully commercialize TLANDO and LPCN 1111.the successful commercialization of TLANDO.
RecentCertain publications have suggested potential health risks associated with T-replacement therapy, such as increased cardiovascular disease risk, including increased risk of heart attack or stroke, fluid retention, sleep apnea, breast tenderness or enlargement, increased red blood cells, development of clinical prostate disease, including prostate cancer, and the suppression of sperm production. These potential health risks are described in various articles, including the following publications:
| a 2014 publication in PLOS ONE, which found that, compared to the one year prior to beginning T-replacement therapy, the risk of heart attack doubled 90 days after the start of T deficiency treatment in older men regardless of their history of heart disease and was two to three times higher in men younger than 65 with a history of heart disease; |
| ● | a 2013 publication in theJournal of the American Medical Association, which reported that hypogonadal men receiving T-replacement therapy developed a 30% increase in the risk of stroke, heart attack and death; and |
| ● | a 2013 publication in BMC Medicine, which concluded that exogenous T increased the risk of cardiovascular-related events, particularly in trials not funded by the pharmaceutical industry. |
| 27 |
Prompted by these events, the FDA announced on January 31, 2014, that it will investigate the risk of stroke, heart attack, and death in men taking FDA-approved testosterone products and that the FDA would hold a T-class Advisory Committee meeting on September 17, 2014, to discuss this topic further. The FDA has also asked health care professionals and patients to report side effects involving prescription testosterone products to the agency.
Following the FDA'sFDA’s announcement, the Endocrine Society, a professional medical organization, released a statement in February 2014 in support of further studies regarding the risks and benefits of FDA-approved T-replacement products for men with age-related T deficiency. Specifically, the Endocrine Society noted that large-scale randomized controlled trials are needed to determine the risks and benefits of T-replacement therapy in older men. In addition, the Endocrine Society recommended that patients should be informed of the potential cardiovascular risks in middle-aged and older men associated with T-replacement therapies. Also following the FDA'sFDA’s announcement, Public Citizen, a consumer advocacy organization, petitioned the FDA to add a "black box"“black box” warning about the increased risks of heart attacks and other cardiovascular dangers to the product labels of all T-replacement therapies. In addition, this petition urged the FDA to delay its decision date on approving Aveed, a long-acting T-injectable developed by Endo, which was subsequently approved by the FDA in March 2014. In July 2014, the FDA responded to the Public Citizen petition and denied the petition. Additionally, in June 2014 the FDA announced that it would require the manufacturers of testosterone drugs to update the warning label to include blood clots including deep vein thrombosis ("DVT") and pulmonary embolism ("PE").embolism.
At the T-class Advisory Committee meeting held on September 17, 2014, the Advisory Committee discussed (i) the identification of the appropriate patient population for whom T-replacement therapy should be indicated and (ii) the potential risk of major adverse cardiovascular events, defined as non-fatal stroke, non-fatal myocardial infarction and cardiovascular death associated with T-replacement therapy. At the meeting, 16 of the 21 members of the Advisory Committee voted that the FDA should require sponsors of testosterone products to conduct a post marketing study (e.g. observational study or controlled clinical trial) to further assess the potential cardiovascular risk. Further, 12 of these voted that such post marketing study be required only if the T-replacement therapy is also approved for age-related hypogonadism.
The Advisory Committee also held a meeting on September 18, 2014, to evaluate the safety and efficacy of JatenzoJATENZO® (previously Rextoro), an oral TU submitted to the FDA by Clarus Therapeutics for the proposed indication of T-replacement therapy. 18 of the 21 members of the Advisory Committee voted that the overall benefit/risk profile of JatenzoJATENZO® was not acceptable to support approval for T-replacement therapy. The Advisory Committee agreed that an oral TU as a T-replacement therapy is promising and that it would be of great value to patients to have an oral treatment option, but they did not believe the current JatenzoJATENZO® data supported approval.
On March 3, 2015, the FDA issued a safety announcement addressing the Advisory Committee’s recommendations and communicated its expectations related to label revisions and additional clinical requirements.
The FDA'sFDA’s safety assessment recommended the following label modifications/restrictions in the indicated population for T-replacement therapy:
| limiting use of T-replacement products to men who have low testosterone caused by certain medical conditions; |
| ● | prior to initiating use of T-replacement products, confirm diagnosis of hypogonadism by ensuring that serum testosterone has been measured in the morning on at least two separate days and that these concentrations are below the normal range; |
| ● | adding cautionary language stating that the safety and efficacy of TRT products with age-related hypogonadism have not been established; and |
| ● | adding cautionary language stating that some studies have shown an increased risk of myocardial infarction and stroke associated with use of T-replacement products. |
Additionally,On March 29, 2022, the FDA stated that they will require manufacturersapproved TLANDO. As part of approved T-replacement products to conduct a well-designed clinical trial to more clearly address the question of whether an increased risk of heart attack or stroke exists among users of T-replacement products. The FDA encouraged manufacturers to work together on conducting a clinical trial, althoughtheir approval, the FDA will allow manufacturers to work separately if they so choose.
Also, on October 20, 2017, Antares Pharma, Inc. (“Antares”) announced that it had received a CRL fromrequired the FDA regarding its NDA for XYOSTED™ (testosterone enanthate) injection. The CRL indicated that the FDA cannot approve the XYOSTED NDAinclusion of certain warnings and precautions in its present form and identified two deficiencies related to clinical data. Based on findings in two clinical studies, the FDA is concerned XYOSTED could cause a clinically meaningful increase in blood pressure. Additionally, the CRL also raised a concern regarding the occurrence of depression and suicidality. Additionally on January 9, 2018, a BRUDAC meeting was held for Jatenzo, Clarus Therapeutics’ testosterone replacement therapy product candidate. The BRUDAC voted nine in favor and ten against the acceptability of the overall benefit/risk profile to support approval of Jatenzo as a TRT.
It is possible that the FDA's evaluation of this topic and further studies on the effects of T-replacement therapies could demonstrate the risk of major adverse cardiovascular events or other health risks or could impose additional requirements that could delay our approval for TLANDO. During our SOAR trial, we collected safety datalabeling for TLANDO, including a “black box warning,” including warnings relating to blood pressure increases and a control group, the leading approved T-gel product, but we did not compare safety data from TLANDO to a placebo control group or the control group. If, following its evaluation, the FDA concludesan indication that men using FDA-approved T-replacement therapies face serious cardiovascular risks, it may take actions against T-replacement products generally, which could impact us adversely in a variety of ways, including that the FDA could:
Additionally, the FDA convened a BRUDAC meeting on January 10, 2018 to evaluate the safety and efficacy of TLANDO as a T-replacement product. in males less than 18 years has not been established. These warnings may deter physicians and patients from using TLANDO, which could adversely affect our business.
The BRUDAC voted six in favorFDA has also required that certain post-marketing studies be conducted to (i) assess patient understanding of key risks relating to TLANDO and thirteen against(ii) evaluate development of adrenal insufficiency with chronic TLANDO therapy. Verity is responsible for conducting these post-marketing studies. Negative outcomes from such studies could adversely affect the acceptabilityability of the overall benefit/risk profile to support approval of TLANDO as a TRT.
Demonstrated T-replacement therapy safety risks, as well as negative publicity about the risks of hormone replacement therapy, including T-replacement, could hurt sales of and impair our abilityVerity to successfully commercialize TLANDO, and LPCN 1111, if approved. On March 19, 2015, we had a pre-NDA meeting with the FDA concerning our pivotal Phase 3 trial for TLANDO. Based on this meeting with the FDA, we do not expect to be required to conduct a heart attack and stroke risk study or any additional safety studies prior to approval of the NDA for TLANDO. The FDA may determine that an ABPM study may be required to better assess any blood pressure effects of TLANDO, a surrogate marker of predicting cardiovascular outcome. If the FDA requires a ABPM study or changes its position, however, and concludes that a cardiovascular trial is required prior to approving our NDA for TLANDO, such trial would require substantial financial resources, would delay the regulatory process for TLANDO and our entry into the marketplace, all of which would have a material adverse impact onadversely affect our business.ability to realize royalty revenue under the Verity License Agreement.
| 28 |
If we fail to obtain adequate healthcare reimbursement for our products, our revenue-generating ability will be diminished and there is no assurance that the anticipated market for our products will be sustained.
We believe that there could be many different applications for products successfully derived from our technologies and that the anticipated market for products under development could continue to expand. However, due to competition from existing or new products, potential changes to the class TRT label by the FDA and the yet to be established commercial viability of our products, no assurance can be given that these beliefs will prove to be correct. Physicians, patients, formularies, payors or the medical community in general may not accept or utilize any products that we or our collaborative partners may develop. Other drugs may be approved during our clinical testing which could change the accepted treatments for the disease targeted and make our compound obsolete.
Our ability to commercialize our products with success may depend, in part, on the extent to which coverage and adequate reimbursement to patients for the cost of such products and related treatment will be available from governmental health administration authorities, private health coverage insurers and other organizations, as well as the ability of private payors to pay for or afford our drugs. Adequate third-party coverage may not be available to patients to allow us to maintain price levels sufficient for us to realize an appropriate return on our investment in product development.
Coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payers can be critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available. Additionally, current manufacturers of drug products may have agreements with payors that may limit the ability of new products to get on formulary or require a step edit with an existing product before reimbursement orof a new product will occur. Even if we obtain coverage for our products, the resulting reimbursement payment rates might not be adequate or may require co-payments that patients find unacceptably high. Patients are less likely to use our products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products. Payers may require a more arduous prior authorization process as a condition to payment for TRT therapy. This could adversely affect the market for TRT products.
In the United States and in many other countries, pricing and/or profitability of some or all prescription pharmaceuticals and biopharmaceuticals are subject to varying degrees of government control. Healthcare reform and controls on healthcare spending may limit the price we charge for any products and the amounts thereof that we can sell. In particular, in the United States, the federal government and private insurers have changed and have considered ways to change, the manner in which healthcare services are provided. In March 2010, the Patient Protection and Affordable Care Act (“ACA”), as amended by the Healthcare and Education Affordability Reconciliation Act,ACA became law in the United States. ACA substantially changes the way healthcare is financed by both governmental and private insurers and significantly affects the healthcare industry. The provisions of ACA of importance to our potential product candidates include the following:
| an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents; |
| ● | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; |
| ● | expansion of healthcare fraud and abuse laws, including the False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance; |
| ● | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| ● | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| ● | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals beginning in 2014 and by adding new mandatory eligibility categories for certain individuals with specified income levels, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| ● | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
| ● | new requirements to report annually certain financial arrangements with physicians, certain other healthcare professionals, and teaching hospitals; |
| ● | a new requirement to annually report drug samples that manufacturers and distributors provide to licensed practitioners, pharmacies of hospitals and other healthcare entities; and |
| ● | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
| 29 |
In addition, other legislative changes have been proposed and adopted since ACA was enacted. On August 2, 2011, the Budget Control Act of 2011, created, among other things, measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. The Medicare Access and CHIP Reauthorization Act of 2015 was signed into law on April 16, 2015 and implemented the most significant change in Medicare reimbursement since the ACA was enacted. This 2015 law authorizes a new Medicare pay –for-performance reimbursement system for physicians, which will reward physicians for performance on metrics related to quality of care, resource use, meaningful use of electronic medical records, and clinical practice improvement activities. The Bipartisan Budget Act was enacted on November 2, 2015, and among provisions, restricts the types of facilities that may receive hospital reimbursement under Medicare. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our customers and accordingly, our financial operations.
We anticipate that ACA will result in additional downward pressure on the reimbursement we may receive for any approved and covered product and could seriously harm our business. Any reduction in reimbursement from Medicare and other government programs may result in a similar reduction in payments from private payers. In the future, the U.S. government may institute further controls and different reimbursement schemes and limits on Medicare and Medicaid spending or reimbursement that may affect the payments we could collect from sales of any products in the United States.
Moreover, President Trump has indicated a desireThe Department of Health and Human Services Office of Inspector General issued final regulations on November 30, 2020 to repeal ACA. The implementation of further cost containment measures oreliminate safe harbor protection under the repeal of ACA or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our products.
On his first day in office, US President Donald Trump issued an Executive Order to begin the process of dismantling the ACA. The Order affirmatively states the new administration’s intent to seek repeal of the ACA. Although these efforts have not resulted in a wholesale repeal of the ACA, the President and Republicans in Congress have communicatedanti-kickback statute for drug price reductions that they will soon unveil plans to repeal and replace the ACA. Approximately 20 million previously uninsured Americans now have coverage because of the ACA. The potential overhaul of the ACA is creating uncertainty for patients, providers, payers and pharmaceutical manufacturers. In the event that millions of people lose their health insurance with repeal and replace, there will be less spending on pharmaceuticals which could have a material adverse effect on our financial operations.
Furthermore, some Congressional leaders have indicated that further significant changesmanufacturers pay to Medicare and Medicaid benefitsplan sponsors and reimbursement must be made in ordertheir pharmacy benefit managers. The proposal reflects a clear intent to manage the federal budget deficit, particularly in lightsubstantially alter many of the Tax Cutscurrent drug discount and services compensation practices among pharmaceutical manufacturers and Medicare and Medicaid managed care organizations and their pharmacy benefit managers. The proposal also reflects a skepticism that current drug discount and compensation practices among manufacturers and pharmacy benefit managers are sufficiently transparent to health plans to ensure that all appropriate cost reductions and value is passed through to health plans and reflected in lower health plans costs and lower premiums for beneficiaries. The Infrastructure Investment and Jobs Act enacted in 2021 delayed the potential effective date of 2017.the proposal until January 1, 2026, and the Inflation Reduction Act of 2022 further delayed potential implementation of the rule until 2032. If the regulation becomes effective, it could result in lower prices for pharmaceutical products in general.
The Centers for Medicare and Medicaid Services issued an interim final rule on November 20, 2020, that would tie prices for certain drugs under Medicare Part B to the lowest price for those drugs available in certain countries that are members of the Organization for Economic Co-operation and Development. This rule was rescinded in December 2021. If resurrected, any similar proposal could result in lower prices for pharmaceutical products in general.
The Inflation Reduction Act of 2022 (Pub. L. No. 117-169) was signed into law on August 16, 2022 and includes a number of provisions aimed at lowering prescription drug costs and reducing government spending on drugs. This includes a requirement that the Department of Health and Human Services negotiate a “maximum fair price” with drug manufacturers for certain single-source brand drugs or biologics without generic or biosimilar competitors that are covered under Medicare Part D and Part B. This pricing will begin in 2026 for Medicare Part D and 2028 for Medicare Part B. An excise tax is imposed on drug manufacturers that fail to comply with the required negotiation process. In August 2023 the Biden Administration released the first round of drugs subject to this new Medicare Drug Pricing Negotiation Program. In addition, the law requires drug manufacturers to pay a rebate to the federal government if the price for almost all drugs covered under Medicare Part D (starting in 2022), and single-source drug or biologics covered under Medicare B (starting in 2023), increase greater than the inflation rate. The rebate amount equals the number of drug units sold in Medicare multiplied by the amount the drug’s price exceeds the inflation-adjusted price. The law also modifies the Medicare Part D benefit structure to cap the amount beneficiaries must spend on drug costs and increase the discounts manufacturers are required to pay. The Inflation Reduction Act of 2022 signals an increased desire to control the prices and costs associated with pharmaceutical products. A number of states have adopted drug affordability legislation which permits a drug affordability board to implement or recommend upper payment limits for drugs identified as posing affordability challenges. This legislation, as well as any future statutes or regulations at the federal or state level, may impact reimbursement for our product candidates and may challenge our ability to realize an appropriate return on our investment in research and product development. Any further legislative or administrative action to reduce reimbursement or health benefits to beneficiaries under the Medicare or Medicaid program could affect the payment we could collect from sale of any product in the United States.
| 30 |
We faceThere is substantial competition in the TRT market, which may result in others discovering, developing or commercializing products before or more successfully than we do.us or our licensing partner.
We expect to face significant competition for any of our product candidates, if approved. In particular, if approved, TLANDO would competecompetes in the T-replacement therapies market, which is highly competitive and currently dominated by the sale of T-gels in termsand T-injectables. Receipt of sales dollars, which accounted for approximately 72% of U.S. sales in the T-replacement therapies market in 2017 according to IMS Health data. Our successfuture potential payments under our licensing agreement will depend, in large part, on our licensing partner’s ability to obtain an adequate share of the market. Potential competitors in North America, Europe and elsewhere include major pharmaceutical companies, specialty pharmaceutical companies, biotechnology firms, universities and other research institutions and government agencies. Other pharmaceutical companies may develop oral T-replacement therapies that compete with TLANDO. For example, because TU is not a patented compound and is commercially available to third parties, it is possible that competitors may design methods of TU administration that would be outside the scope of the claims of either our issued patents or our patent applications. This would enable their products to effectively compete with TLANDO, which could have a negative effect on potential payments under our business.licensing agreement.
The following T-replacement therapies currently on the market in the United States would compete with TLANDO:
| Oral-T, such as Jatenzo and Kyzatrek; | ||
| ● | T-gels, such as AndroGel (marketed by Abbvie) and | |
| ● | T-injectables, including a |
| Branded, longer-acting injectables, such as Aveed (marketed by Endo); |
| ● | T-nasals, such as Natesto (marketed by |
| ● | methyl-T, such as Methitest (marketed by Impax) and Testred (marketed by Valeant); |
| ● | transdermal patches, such |
| ● | buccal patches, such as Striant (marketed by Endo); |
| ● | generic testosterone enanthate intra-muscular injectables; |
| ● | authorized generic and generic T-gels; and |
| ● | subcutaneous injectable pellets, such as Testopel (marketed by Endo). |
On March 27, 2019, Clarus’ product JATENZO®, an oral TU product, was approved by the FDA and also received three years of marketing exclusivity. On February 10, 2020, Clarus announced that JATENZO® has been launched and is commercially available. The FDA approved TLANDO on March 29, 2022, following the expiration of the exclusivity period granted to Clarus with respect to JATENZO®. In October 2022, Clarus’ assets, including JATENZO®, were purchased by Tolmar Pharmaceuticals Inc. in bankruptcy proceedings.
We are also aware of other pharmaceutical companies that have T-replacement therapies or testosterone therapies in development that may be approved for marketing in the United States or outside of the United States.
Based on publicly available information, we believe that several other T-replacement therapies that would be competitive with TLANDO are in varying stages of development, some of which may be approved, marketed and/or commercialized prior to TLANDO.development. These therapies include T-gels, oral-T, an aromatase inhibitor, a new class of drugs called Selective Androgen Receptor Modulators and hydroalcoholic gel formulations of DHT.dihydrotestosterone (“DHT”).
In light of the competitive landscape above, TLANDO maywill not be the firstonly oral testosterone replacement therapyTRT to market, which may significantly affect the market acceptance and commercial success of TLANDO.
Furthermore, many of our potential competitors have substantially greater financial, technical, and human resources than we do and significantly greater experience in the discovery and development of drug candidates, obtaining FDA and other marketing approvals of products and the commercialization of those products. These competitors have the economic power to acquire and maintain market share, limiting our ability to penetrate the TRT market with our TLANDO product. Accordingly, our competitors may be more successful than we may be in obtaining FDA approval for drugs and achieving widespread market acceptance. Our competitors’ drugs may be more effective, or more effectively marketed and sold, than our products and may render our products obsolete or non-competitive before we can recover the expenses of developing and commercializing them. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. Failure to successfully compete in this market wouldcould materially and negatively impact our business and operations.
| 31 |
The entrance of generic T-gels into the market would likely create downward pricing pressure on all T-replacement therapies and therefore have a negative effect on our business and financial results.Our Licensee’s ability to commercialize TLANDO may be limited.
Several companies have filed Abbreviated New Drug Applications, or ANDAs, seeking approval for generic versions of existing T-gels. For example, in April 2012 Perrigo Company filed an ANDA with the FDA seeking approval for a generic version of AndroGel 1.62%. In responseOur Licensee partner’s ability to this ANDA, the marketer of AndroGel 1.62% filed patent infringement lawsuits against Perrigo Company to block the approval and marketing of the generic product. In August 2015, the FDA approved the ANDA submitted by Perrigo Company and Perrigo’s generic product was also granted 180 days of generic drug exclusivity. The marketer of AndroGel 1.62% may enter into an agreement with Perrigo Company to delay the introduction of a generic AndroGel 1.62%. Additionally, in July 2003, Actavis and Par Pharmaceutical, or Par, filed ANDAs with the FDA seeking approval for generic versions of AndroGel 1%. In response to these ANDAs, the marketer of AndroGel 1% filed patent infringement lawsuits against these two companies to block the approval and marketing of the generic products. In 2006, all the subject companies reached an agreement pursuant to which Actavis agreed not to bring a generic version of AndroGel 1% to the market until August 2015, and Par agreed not to bring a generic version to market until February 2016. The U.S. Federal Trade Commission has questioned the legality of such “pay-to-delay” agreements, and the Supreme Court ruled in June 2013 that such agreements may not be valid. The impact of this ruling on the agreements between the marketer of AndroGel 1% and Actavis and Par, as well as the timing and eventual marketing of generic versions of their respective products,commercialize TLANDO is uncertain at this point.
Additionally, there are several other ANDAs for generic T-gels that have been filed and there is ongoing litigation with each of these ANDAs. Finally, in 2014, two authorized generic T-gels were launched at a lower price than the branded version of the same T-gel. If a generic version of T-gel were to become available in the market, governmental and other pressures to reduce pharmaceutical costs may result in physicians writing prescriptions for generic T-gels as opposed to branded T-gels. The entrance of any generic T-gel into the market would likely cause downward pressure on the pricing of all T-replacement therapies and could materially and adversely affect the level of sales and price at which we could sell TLANDO, and ultimately materially and adversely impact our revenues and financial results.
The introduction of generic T-gel, may also affect the reimbursement policies of government authorities and third-party payors, such as private health insurers and health maintenance organizations. These organizations determine which medications they will pay for and establish reimbursement levels. Cost containment is a primary concern in the U.S. healthcare industry and elsewhere. Government authorities and these third-party payers have attempted to control costs by limiting coverage and the amount of reimbursement for branded medications when there is a generic available. If generic T-gel is available in the market, that may create an additional obstacle to the availability of reimbursement for TLANDO. Even if reimbursement is available, the level of such reimbursement could be reduced or limited. Reimbursement may impact the demand for, or the price of, TLANDO. If reimbursement is not available or is available only at limited levels, we may not be ableuncertain. Our Licensee’s ability to successfully commercialize TLANDO and/oris contingent upon numerous factors including, among other things, the completion of post-marketing studies, the availability of supplies, commercial acceptance by patients, the medical community, and third-party payors, and the resources that our financial results fromLicensee devotes to the salecommercialization of related productsTLANDO. If our Licensee is unable to successfully commercialize TLANDO at scale, our business and operations could be negatively and materially impacted. Rebates and other pricing strategies of generics may erode our revenue and harm our financial performance.adversely affected.
Additionally, TLANDO may not be the first oral testosterone replacement therapy product to market. In this event, if the generic version of a competing oral testosterone replacement therapy product enters the market before our product, then the commercial prospects of TLANDO could be materially and negatively impacted.
We will not be able to successfully commercialize our product candidates without establishing sales, marketing and market access capabilities internally or through collaborators.
We currently do not have a limited sales, marketing and market access staff. This staff was reduced in July 2016 following the receipt of the CRL for TLANDO and further reduced in October 2016. If and when any of our product candidates are commercialized, we may not be able to find suitable sales and marketing staff and collaborators for TLANDO or our other product candidates. The existing sales, marketing and market access staff and outside collaborators we work with, including Verity under the Verity License Agreement with respect to TLANDO, may not be adequate or successful and any collaborators could terminate or materially reduce the effort they direct to our products. The development of collaborations or an internal sales force and marketing, market access and sales capability will require significant capital, management resources and time. The cost of establishing such a sales force may exceed any potential product revenues and our marketing, market access and sales efforts may be unsuccessful. If we are unable to develop an internal marketing, market access and sales capability or if we are unable to enter into a marketing and sales arrangement with a third party on acceptable terms, we may be unable to successfully commercialize TLANDO or develop and seek regulatory approval for our other product candidates.
LPCN 1107 is in a very early stage of development and may not be further developed for a variety of reasons.
LPCN 1107 is in a very early stage of development and consequently the risk that we fail to commercialize LPCN 1107 and related products is high. In particular, we have only conducted three Phase 1 clinical studies with this product candidate. Two of the studies were in healthy pregnant women and one was in healthy women. Although these studies demonstrated oral absorption of LPCN 1107 is possible, we may not be able to match or exceed Cavg blood levels shown with the intramuscular injection comparator product over a longer duration. Furthermore, our completed Phase 1 clinical studies may not be predictive of safety concerns that may arise in pregnant women or demonstrate that LPCN 1107 has an adequate safety profile to warrant further development. The FDA may also require further preclinical studies. All of these factors can impact the timing of and our ability to continue development of LPCN 1107.
In addition, a number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late stage clinical trials, even after achieving positive results in early stage development. Accordingly, our results from our Phase 1a, our Phase 1b and our multi-dose PK dose selection studies may not be predictive of the results we may obtain from further studies and trials.
A traditional pharmacokinetics/pharmacodynamics (“PK/PD”) based Phase 2 clinical study in the intended patient population may not be required prior to entering into Phase 3. Therefore, based on the results of our multi-dose PK study results, we had an End-of-Phase 2 meeting with the FDA in the second quarter of 2016, as well as subsequent guidance meetings to agree on a Phase 3 development plan for LPCN 1107. During the End-of-Phase 2 meeting and subsequent guidance meetings, the FDA agreed to a randomized, open-label, two-arm clinical study to include a LPCN 1107 arm and a comparator IM arm with treatment up to 23 weeks. The FDA also provided feedback on other critical Phase 3 study design considerations including: positive feedback on the proposed 800 mg BID Phase 3 dose and dosing regimen; confirmation of the use of a surrogate primary endpoint focusing on rate of delivery less than 37 weeks gestation rather on clinical infant outcomes; acknowledged that the use of a gestational age endpoint would likely lead to FDA approval, if granted, being a Subpart H approval as opposed to a full approval; and, recommended a non-inferiority study margin of 7% with interim analyses. A standard statistical design for a NI study based on the FDA suggested NI margin of 7% for the primary end point may require ~1,100 subjects per treatment arm with a 90% power. However, based on the FDA’s feedback of including an interim analysis in the NI design, an adaptive study design is under consideration that may allow for fewer subjects. We submitted the initial LPCN 1107 Phase 3 protocol to the FDA via a SPA in June 2017 and have received multiple rounds of FDA feedback. Agreement with the FDA on the Phase 3 protocol via SPA has not occurred and will not occur until results from a planned food-effect study with LPCN 1107 are reviewed by the FDA. Final agreement with the FDA on the Phase 3 protocol, if reached, may or may not confirm the FDA’s preliminary feedback on the Phase 3 design. Additionally, manufacturing scale-up work for LPCN 1107 has been complete. Once the Phase 3 clinical trial is started, the anticipated Phase 3 program for an NDA filing for LPCN 1107 will be very long and expensive.
LPCN 1111 is in a very early stage of development and may not be further developed for a variety of reasons.
LPCN 1111 is in a very early stage of development. We have completed a Phase 2a and Phase 2b study in hypogonadal men. Results from the Phase 2a clinical study demonstrated the feasibility of a once daily dosing with LPCN 1111 in hypogonadal men and a good dose response. Results of the Phase 2b study suggest that the primary objectives were met, including identifying the dose expected to be tested in a Phase 3 study. Future studies may not have similar clinical results. Additionally, we have preliminary data demonstrating absorption of LPCN 1111 in dogs and in postmenopausal females.
In addition, the active ingredient in LPCN 1111 has only been manufactured on a small scale. Scaling up into larger batches could be challenging and our ability to procure adequate material in a timely manner to further develop LPCN 1111 is uncertain. We also may not be able to engage a manufacturer who can supply adequate quantities of the drug substance in compliance with Current Good Manufacturing Practices ("cGMP").
Several factors could significantly affect the prospects for LPCN 1111, including factors relating to the regulatory approval and clinical development challenges for LPCN 1111 discussed above. The anticipated Phase 3 program for an NDA filing for LPCN 1111, however, could be very long and expensive.
Our research and development programs and processes are at an early stage of development, which makes it difficult to evaluate our business and prospects, or predict if or when we will successfully commercialize our product candidates.
Our operations to date have primarily been limited to conducting research and development activities under license and collaboration agreements. Our current portfolio consists of our lead product candidate TLANDO as well as two additional earlier stage clinical candidates, LPCN 1111 and LPCN 1107. We have never marketed or commercialized a drug product. Consequently, any predictions about our future performance may not be as accurate as they could be if we were further along our commercialization path. In addition, as a pre-commercial stage business, we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors.
Our clinical product candidates are at an early stage of development and will require significant further investment and regulatory approvals prior to marketing and commercialization. As such, our product development processes for TLANDO, LPCN 1111 and LPCN 1107 are very risky and uncertain, and our product candidates may fail to advance beyond the current study. Even if we obtain required financing, we cannot ensure successful product development or that we will obtain regulatory approval or successfully commercialize any of our product candidates and generate product revenues.
All of our clinical candidates will be subject to extensive regulation which can be costly and time consuming, cause delays or prevent approval of the products for commercialization.
Our clinical development of TLANDO, LPCN 1111, LPCN 1107 and any future product candidates, is subject to extensive regulations by the FDA. Product development is a very lengthy and expensive process and can vary significantly based upon the product candidate’s novelty and complexity. Regulations are subject to change and regulatory agencies have significant discretion in the approval process.
Numerous statutes and regulations govern human testing and the manufacture and sale of human therapeutic products in the United States. Such legislation and regulation bears upon, among other things, the approval of protocols and human testing, the approval of manufacturing facilities, safety of the product candidates, testing procedures and controlled research, review and approval of manufacturing, preclinical and clinical data prior to marketing approval including adherence to cGMP during production and storage as well as regulation of marketing activities including advertising and labeling.
In order to obtain regulatory clearance for the commercial sale of any of our product candidates, we must demonstrate through preclinical studies and clinical trials that the potential product is safe and efficacious for use in humans for each target indication. Obtaining approval of any of our product candidates is an extensive, lengthy, expensive and uncertain process, and the FDA may delay, limit or deny approval for many reasons, including:
Preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain FDA approval for their products.
No assurance can be given that current regulations relating to regulatory approval will not change or become more stringent. The FDA may also require that we amend clinical trial protocols and/or run additional trials in order to provide additional information regarding the safety, efficacy or equivalency of any compound for which we seek regulatory approval. Moreover, any regulatory approval of a drug which is eventually obtained may entail limitations on the indicated uses for which that drug may be marketed. Furthermore, product approvals may be withdrawn or limited in some way if problems occur following initial marketing or if compliance with regulatory standards is not maintained. FDA could become more risk averse to any side effects or set higher standards of safety and efficacy prior to reviewing or approving a product. This could result in a product not being approved.
Even if we receive marketing approval in the United States, we may never receive regulatory approval to market our products outside the United States, which could reduce the size of our potential markets and have a material adverse impact on our business.
In order to market any products outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy.
Approval procedures vary among countries and can involve additional product candidate testing and additional administrative review periods. The time required to obtain approvals in other countries might differ from that required to obtain FDA approval. The marketing approval process in other countries may include all of the risks detailed above regarding FDA approval in the United States as well as other risks. In particular, in many countries outside of the United States, products must receive pricing and reimbursement approval before the product can be commercialized. This can result in substantial delays in such countries. Marketing approval in one country does not ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one country may have a negative effect on the regulatory process in others. Failure to obtain marketing approval in other countries or any delay or setback in obtaining such approval would impair our ability to market our products in such foreign markets. Any such impairment would reduce the size of our potential markets, which could have a materialan adverse impact on our business, results of operations and prospects.
We are subject to stringent government regulations concerning the clinical testing of our products and will continue to be subject to government regulation of any product that receives regulatory approval.
Numerous statutes and regulations govern human testing and the manufacture and sale of human therapeutic products in the United States and other countries where we intend to market our products. Such legislation and regulation bears upon, among other things, the approval of clinical study protocols and human testing of our products, the approval of manufacturing facilities, testing procedures and controlled research, the review and approval of manufacturing, preclinical and clinical data prior to marketing approval, including adherence to cGMP during production and storage, and marketing activities including advertising and labeling.
Clinical trials may be delayed or suspended at any time by us or by the FDA or by other similar regulatory authorities if it is determined at any time that patients may be or are being exposed to unacceptable health risks, including the risk of death, or if compounds are not manufactured under acceptable cGMP conditions or with acceptable quality. Current regulations relating to regulatory approval may change or become more stringent. The agencies may also require additional clinical trials to be run in order to provide additional information regarding the safety, efficacy or equivalency of any compound for which we seek regulatory approval. Moreover, any regulatory approval of a drug which is eventually obtained may entail limitations on the indicated uses for which that drug may be marketed. Furthermore, product approvals may be withdrawn or limited in some way if problems occur following initial marketing or if compliance with regulatory standards is not maintained. Regulatory agencies could become more risk adverse to any side effects or set higher standards of safety and efficacy prior to reviewing or approving a product. This could result in a product not being approved.
| 32 |
If we, or any future marketing collaborators or CMOs, fail to comply with applicable regulatory requirements, we may be subject to sanctions including fines, product recalls or seizures and related publicity requirements, injunctions, total or partial suspension of production, civil penalties, suspension or withdrawals of previously granted regulatory approvals, warning or untitled letters, refusal to approve pending applications for marketing approval of new products or of supplements to approved applications, import or export bans or restrictions, and criminal prosecution and penalties. Any of these penalties could delay or prevent the promotion, marketing or sale of our products.
The successful commercialization of our product candidates and ability to generate significant revenue will depend on achieving market acceptance.
Even if our product candidates are successfully developed and receive regulatory approval, they may not gain market acceptance among physicians, patients, healthcare payers such as private insurers or governments and other funding parties and the medical community. The degree of market acceptance for our products, if approved, will depend on a number of factors, including:
| ● | the relative convenience and ease of administration, including as compared to alternative treatments and competitive therapies; | |
| ● | the prevalence and severity of any adverse side effects; | |
| ● | limitations or warnings contained in the labeling approved by the FDA; | |
| ● | availability of alternative treatments, including a number of competitive therapies already approved or expected to be commercially launched in the near future; | |
| ● | distribution and use restrictions imposed by the FDA or agreed to by us as part of a mandatory REMS or voluntary risk management plan; | |
| ● | pricing and cost effectiveness; | |
| ● | the effectiveness of our or any future collaborators’ sales and marketing strategies; | |
| ● | our ability to increase awareness of our products through marketing efforts; | |
| ● | our ability to obtain sufficient third-party coverage or reimbursement; and | |
| ● | the willingness of patients to pay out-of-pocket in the absence of third-party coverage. |
If our product candidates are approved but do not achieve an adequate level of acceptance by physicians, healthcare payors and patients, we may not generate sufficient revenue from our products and we may never become or remain profitable. In addition, our efforts to educate the medical community and third-party payors on the benefits of our products may require significant resources and may never be successful.
Even if we obtain marketing approval for our products, physicians and patients using existing products may choose not to switch to our products.
Physicians often show a reluctance to switch their patients from existing drug products even when new and potentially more effective and convenient treatments enter the market. Also, physicians may be reluctant to switch patients if adequate reimbursement for new products is not available. In addition, patients often acclimate to the brand or type of drug product that they are currently taking and do not want to switch unless their physicians recommend switching products or they are required to switch drug treatments due to lack of reimbursement for existing drug treatments and only if the new product has adequate reimbursement. The existence of either or both of physician or patient reluctance in switching to our products would have a materialan adverse effect on our operating results and financial condition.
The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. If we are found to have improperly promoted off-label uses, we may become subject to significant liability.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products, such as our product candidates. In particular, a product may not be promoted for uses that are not approved by the FDA or such other regulatory agencies as reflected in the product’s approved labeling. The FDA may impose further requirements or restrictions on the distribution or use of our product candidates as part of a REMS plan, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. If we receive marketing approval for our product candidates, physicians may nevertheless prescribe our products to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability, including potential liability under federal civil and criminal false claims acts. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed.
| 33 |
If we fail to comply with federal and state healthcare laws, including fraud and abuse and health information privacy and security laws, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected.
As a pharmaceutical company, even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payers, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. We could be subject to healthcare fraud and abuse and patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| ● | the federal Anti-Kickback Statute, which constrains our marketing practices, educational programs, pricing policies, and relationships with healthcare providers or other entities, by prohibiting, among other things, soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, either the referral of an individual or the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; | |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent; | |
| ● | HIPAA, which among other things created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; | |
| ● | the federal Physician Payments Sunshine Act, which, among other things, requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under certain federal healthcare programs to report annually information related to “payments or other transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and ownership and investment interests held by certain healthcare professionals and their immediate family members; | |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and its implementing regulations, which imposes certain requirements relating to the privacy, security, breach notification, and transmission of individually identifiable health information; and | |
| ● | state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers, and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Because of the breadth of these laws and the narrowness of available statutory and regulatory exceptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. To the extent that any of our product candidates is ultimately sold in countries other than the United States, we may be subject to similar laws and regulations in those countries. If we or our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil, criminal and administrative penalties, damages, fines, disgorgement, exclusion from participating in government healthcare programs, contractual damages, reputational harm and the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment or restructuring of our operations could materially adversely affect our ability to operate our business and our financial results. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security and fraud laws may prove costly.
The Department of Health and Human Services Office of Inspector General proposed new regulations on February 6, 2019 to eliminate safe harbor protection under the anti-kickback statute for drug price reductions that pharmaceutical manufacturers pay to Medicare and Medicaid plan sponsors and their pharmacy benefit managers. The proposal reflects a clear intent to substantially alter many of the current drug discount and services compensation practices among pharmaceutical manufacturers and Medicare and Medicaid managed care organizations and their pharmacy benefit managers. The proposal also reflects a skepticism that current drug discount and compensation practices among manufacturers and pharmacy benefit managers are sufficiently transparent to health plans to ensure that all appropriate cost reductions and value is passed through to health plans and reflected in lower health plans costs and lower premiums for beneficiaries. If the proposal is finalized, it could result in lower prices for pharmaceutical products in general. The Infrastructure Investment and Jobs Act enacted in 2021 delayed the potential effective date of the proposal until January 1, 2026, and the Inflation Reduction Act of 2022 further delayed potential implementation of the rule until 2032. If the regulation becomes effective, it could result in lower prices for pharmaceutical products in general.
| 34 |
Any further legislative or administrative action to reduce reimbursement or health benefits to beneficiaries under the Medicare or Medicaid program could affect the payment we could collect from sale of any product in the United States.
Our future success depends on our ability to retain our chief executive officer and other key executives and to attract, retain and motivate qualified personnel.
We are highly dependent on Dr. Mahesh V. Patel and the other principal members of our executive team. Employment with our executives and other employees are “at will”, meaning that there is no mandatory fixed term and their employment with us may be terminated by us or by them for any or no reason. The loss of the services of any of our executives or other key employees might impede the achievement of our research, development and commercialization objectives. Following the receipt of the CRL for LPCN, we reduced our workforce by 8 positions, constituting 33% of our workforce. The reduction in workforce involved all functional disciplines. Further, subsequent to our Post Action meeting with the FDA for TLANDO, we reduced our workforce by two positions in October 2016. Recruiting and retaining qualified scientific personnel accounting personnel and sales and marketingaccounting personnel will also be critical to our success. We may not be able to attract and retain qualified personnel on acceptable terms, or at all, given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific personnel from universities and research institutions. Failure to succeed in clinical trials may make it more challenging to recruit and retain qualified scientific personnel.
In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
We will need to grow our company, and we may encounter difficulties in managing this growth, which could disrupt our operations.
As of December 31, 2017, we had only 14 employees. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Also, our management may need to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. Due to our limited resources, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. This may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. The physical expansion of our operations may lead to significant costs and may divert financial resources from other projects, such as the development of TLANDO. If our management is unable to effectively manage our future growth, our expenses may increase more than expected, our ability to generate revenue could be reduced and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize our product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth.
Recent federalFederal legislation and actions by state and local governments may permit re-importation of drugs from foreign countries into the United States, including foreign countries where the drugs are sold at lower prices than in the United States, which could materially adversely affect our operating results.
WeOur licensing partner may face competition for TLANDO if approved, from lower priced T-replacement therapies from foreign countries that have placed price controls on pharmaceutical products. The Medicare Prescription Drug Improvement and Modernization Act of 2003 (“MMA”) contains provisions that may change U.S. importation laws and expand pharmacists’ and wholesalers’ ability to import lower priced versions of an approved drug and competing products from Canada, where there are government price controls. These changes to U.S. importation laws will not take effect unless and until the Secretary of Health and Human Services certifies that the changes will pose no additional risk to the public’s health and safety and will result in a significant reduction in the cost of products to consumers. The Secretary of Health and Human Services has not yet announced any plans to make this required certification.
A number of federal legislative proposals have been made to implement the changes to the U.S. importation laws without any certification and to broaden permissible imports in other ways. Even if the changes do not take effect, and other changes are not enacted, imports from Canada and elsewhere may continue to increase due to market and political forces, and the limited enforcement resources of the FDA, U.S. Customs and Border Protection and other government agencies. For example, Pub. L. No. 111-83, which was signed into law in October 2009, which provides appropriations for the Department of Homeland Security for the 2010 fiscal year, expressly prohibits U.S. Customs and Border Protection from using funds to prevent individuals from importing from Canada less than a 90-day supply of a prescription drug for personal use, when the drug otherwise complies with the Federal Food, Drug, and Cosmetic Act. Further, several states and local governments have implemented importation schemes for their citizens, and, in the absence of federal action to curtail such activities, we expect other states and local governments to launch importation efforts.
The importation of foreign products that compete with our products could have a materialan adverse effect on our revenue and profitability.
We may become subject to the risk of product liability claims.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize anyon commercialized products. Human therapeutic products involve the risk of product liability claims and associated adverse publicity. Currently, the principal risks we face relate to patients in our clinical trials, who may suffer unintended consequences. Claims might be made by patients, healthcare providers or pharmaceutical companies or others. We may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale.
For example, to our knowledge, hydroxyprogesterone caproate (“HPC”)HPC has not been administered orally in a published clinical trial in any pregnant woman for the prevention of preterm birth.PTB. We cannot be certain of the safety profile upon single oral or multiple oral administration of LPCN 1107 to the patient or the fetus and its long term side effects on the mother as well as the child because (i) oral performance of LPCN 1107 may be substantially different from efficacy and/or safety standpoint compared to FDA approved andpreviously commercialized intramuscular HPC, Makena, and (ii) oral delivery of HPC could have a very different pharmokineticPK and/or pharmacodynamic profile that has never been experienced with non-oral administration of HPC, thus having its own significant liability exposure independent of known safety of non-oral HPC in humans.
| 35 |
Any product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates, if approved. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our product candidates; | |
| ● | injury to our reputation; | |
| ● | withdrawal of clinical trial participants; | |
| ● | initiation of investigations by regulators; | |
| ● | costs to defend the related litigation; | |
| ● | a diversion of management’s time and our resources; | |
| ● | substantial monetary awards to trial participants or patients; | |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; | |
| ● | loss of revenues from product sales; and | |
| ● | the inability to commercialize any of our product candidates, if approved. |
We may not have or be able to obtain or maintain sufficient and affordable insurance coverage, and without sufficient coverage any claim brought against us could have a materially adverse effect on our business, financial condition or results of operations. We run clinical trials through investigators that could be negligent through no fault of our own and which could affect patients, cause potential liability claims against us and result in delayed or stopped clinical trials. We are required in many cases by contractual obligations, to indemnify collaborators, partners, third party contractors, clinical investigators and institutions. These indemnifications could result in a material impact due to product liability claims against us and/or these groups. We currently carry $3$3.0 million in product liability insurance, which we believe is appropriate for our clinical trials. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
Testosterone is a Schedule III substance under the Controlled Substances Act and any failure to comply with this Act or its state equivalents would have a negative impact on our business.
Testosterone is listed by the U.S. Drug Enforcement Agency, or DEA, as a Schedule III substance under the Controlled Substances Act of 1970. The DEA classifies substances as Schedule I, II, III, IV or V substances, with Schedule I substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. Scheduled substances are subject to DEA regulations relating to manufacturing, storage, distribution and physician prescription procedures. For example, all regular Schedule III drug prescriptions must be signed by a physician and may not be refilled more than six months after the date of the original prescription or more than five times unless renewed by the physician.
Entities must register annually with the DEA to manufacture, distribute, dispense, import, export and conduct research using controlled substances. In addition, the DEA requires entities handling controlled substances to maintain records and file reports, follow specific labeling and packaging requirements, and provide appropriate security measures to control against diversion of controlled substances. Failure to follow these requirements can lead to significant civil and/or criminal penalties and possibly even lead to a revocation of a DEA registration. Individual states also have controlled substances laws. State controlled substances laws often mirror federal law, however because the states are separate jurisdictions, they may schedule products separately. While some states automatically schedule a drug when the DEA does so, in other states there must be rulemaking or legislative action, which could delay commercialization.
Our clinical lots of TLANDO for the Phase 3 trials were manufactured in the United Kingdom, or UK. This entailed obtaining additional permits from regulatory authorities in the United States and UK relating to exportation of our active TU, a controlled substance from the United States and importation of the same into the UK, and exportation of finished product from the UK and importation of the same into the United States. Although we were able to manufacture clinical supplies and import these supplies into the United States, these additional requirements could significantly delay the manufacture of the commercial supplies.
Products containing controlled substances may generate public controversy. As a result, these products may have their marketing approvals withdrawn. Political pressuresState and adverse publicity could leadFederal legislatures and administrative agencies may take additional action to delays in, and increased expenses for, and limitcombat a perceived misuse or restrict, the introduction and marketingoveruse of TLANDO.such products.
| 36 |
We may have to dedicate resources to the defense and resolution of litigation.
Securities legislation in the United States makes it relatively easy for stockholders to sue companies. This can lead to frivolous lawsuits which take substantial time, money, resources and attention or force us to settle such claims rather than seek adequate judicial remedy or dismissal of such claims. Historically, securities class action litigation has often been brought against a company following a decline in the market price of its securities. Biotechnology and pharmaceutical companies, including us, have experienced significant stock price volatility in recent years, increasing the risk of such litigation. As we defend class action lawsuits or future patent infringement actions should they be filed, or if we are required to defend future actions brought by shareholders, we may be required to pay substantial litigation costs and managerial attention and financial resources may be diverted from business operations even if the outcome is in our favor. In addition, while our insurance carrier may cover the costs of settling claims, the Company’s capital resources are critical to its continued operations, and the payment of litigation settlements and associated legal fees diverts these capital resources away from our operations, even if such amounts do not have a material impact on our financial statements.
On July 1, 2016,November 14, 2019, the Company and certain of its officers were named as defendants in a purported shareholder class action lawsuit, David LewisSolomon Abady v. Lipocine Inc., et al., 2:19-cv-00906-PMW, filed in the United States District Court for the District of New Jersey. This initial action was followed by additional lawsuits also filed in the District of New Jersey.Utah. The lawsuits contain substantially identical allegations and allegecomplaint alleges that the defendants made false and/or misleading statements and/or failed to disclose that our filing of the NDA for TLANDO to the FDA contained deficiencies and as a result the defendants’ statements about our business and operations were false and misleading and/or lacked a reasonable basis in violation of federal securities laws. The lawsuits seeklawsuit seeks certification as a class action (for a purported class of purchasers of the Company’s securities from March 27, 2019 through November 8, 2019), compensatory damages in an unspecified amount, and unspecified equitable or injunctive relief. On February 15, 2018,We have insurance that covers claims of this nature. The Company filed a motion to dismiss the class action lawsuit on July 24, 2020. In response, the plaintiffs filed their response to the motion to dismiss the class action lawsuit on September 22, 2020 and the Company filed its reply to its motion to dismiss on October 22, 2020. A hearing on the motion to dismiss occurred on January 12, 2022. On April 14, 2023, a judgment was issued ordering the case dismissed with prejudice and closure of the other defendants entered into a memorandumaction. Although this outcome was in favor of understanding to settleour current and former officers and directors, we incurred litigation costs and expended managerial resources defending ourselves against these allegations. In addition, there can be no assurance that we will not experience similar claims in the purported securities class action litigation. future.
Additionally on NovemberApril 2, 2015, Clarus Therapeutics2019, we filed a complaintlawsuit against usClarus in the United States District Court for the District ofin Delaware alleging that TLANDO will infringeClarus’s JATENZO® product infringes six of Lipocine’s issued U.S. patents: 9,034,858; 9,205,057; 9,480,690; 9,757,390; 6,569,463; and 6,923,988. Clarus answered the complaint and asserted counterclaims of non-infringement and invalidity. We answered Clarus’s counterclaims on April 29, 2019. On February 11, 2020, we voluntarily dismissed allegations of patent infringement for expired U.S. Patent Nos. 6,569,463 and 6,923,988 in an effort to streamline the issues and associated costs for dispute. The Court held a scheduling conference on August 15, 2019, a claim construction hearing on February 11, 2020 and a summary judgment hearing on January 15, 2021. In May 2021, the Court granted Clarus’ 428 patent. Themotion for Summary Judgment, finding the asserted claims of Lipocine’s U.S. patents 9,034,858; 9,205,057; 9,480,690; and 9,757,390 invalid for failure to satisfy the written description requirement of 35 U.S.C. § 112. Clarus complaint was dismissed bystill had remaining claims before the District CourtCourt. On July 13, 2021, we entered into a Global Agreement with Clarus which resolved all outstanding claims of this litigation. Under the terms of the settlement, we agreed to pay Clarus $4.0 million, payable as follows: $2.5 million immediately, $1.0 million on July 13, 2022 and $500,000 on July 13, 2023. In April 2022, we amended the Global Agreement with Clarus in 2016, because at the time there was no actionable infringement on Clarus’ 428 patent. While the Clarus complaint has been dismissed, the possibility remains that Clarus could submit its claim again if TLANDO is approved by the FDAan Amended Settlement Agreement and enters the marketplace.
Securities legislation in the United States makes it relatively easy for stockholders to sue. This can lead to frivolous law suits which take substantial time, money, resources and attention or force uswe agreed to settle such claimsthe payments due in July 2022 and 2023 for $1,250,000 rather than seek adequate judicial remedy or dismissalthe $1,500,000 total future payments due under the terms of such claims. Historically, securities class action litigation has often been brought against a company following a declinethe Global Agreement agreed to in the market price2021. The payment of its securities. Biotechnologythis and pharmaceutical companies, including the Company, have experienced significant stock price volatility in recent years, increasing the risk of such litigation. As we defend the class action lawsuits or future patent infringement actions should they be filed, or if we are required to defend additional actions brought by other shareholders, wesettlement payments divert capital resources away from our operations, which may be required to pay substantial litigation costs and managerial attention and financial resources may be diverted from business operations even if the outcome is inadversely affect our favor.business.
Cyber security risks and the failure to maintain the integrity of company, employee or guest data could expose us to data loss, litigation and liability, and our reputation could be significantly harmed.
We collect and third parties collaborating on our clinical trials collect and retain large volumes of data, including personally identifiable information regarding clinical trial participants and others, for business purposes, including for regulatory, research and development and commercialization purposes, and our collaborators’ various information technology systems enter, process, summarize and report such data. We also maintain personally identifiable information about our employees. The integrity and protection of our company,Company, employee and clinical data is critical to our business. We are subject to significant security and privacy regulations, as well as requirements imposed by government regulation. Maintaining compliance with these evolving regulations and requirements could be difficult and may increase our expenses. In addition, a penetrated or compromised data system or the intentional, inadvertent or negligent release or disclosure of data could result in theft, loss or fraudulent or unlawful use of company, employee or clinical data which could harm our reputation, disrupt our operations, or result in remedial and other costs, fines or lawsuits.
| 37 |
Risks Related to Our Dependence on Third Parties
We may enter into license agreements and/or collaborations with third parties for the development and commercialization of our drug candidates. If those collaborations, including, without limitation, our license arrangement with Verity for the development and commercialization of TLANDO, are not successful, we may not be able to capitalize on the market potential of these drug candidates and may have to alter our development and commercialization plans for our products.
Our drug development programs for our product candidates will require substantial additional cash to fund expenses. We have not yet established any collaborative arrangements relating to the development or commercialization of LPCN 1154, LPCN 2101, LPCN 2203, LPCN 1144, LPCN 1148, or LPCN 1107. We have entered into the Verity License Agreement for TLANDO and LPCN 1111 with respect to TRT in the U.S. We intend to continue to develop our product candidates in the United States with or without a partner although our ability to advance these product candidates will depend on our capital resources and/or our ability to find a suitable partner to further develop our product candidates. In order to commercialize our TLANDO product candidates in the United States and Canada, we have partnered with Verity with respect to TLANDO and LPCN 1111 and we will likely look to establish partnership arrangements with respect to the development of some of our other product candidates. We may also seek to enter into collaborative arrangements to develop and commercialize our product candidates outside the United States. We will face significant competition in seeking appropriate collaborators and these collaborations are complex and time-consuming to negotiate and document. We may not be able to negotiate collaborations on acceptable terms or in a timely manner, or at all. If that were to occur, we may have to curtail the development or delay commercialization of our product candidates in certain geographies, reduce the scope of our sales or marketing activities, reduce the scope of our development plans, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities either inside or outside of the United States on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms, or at all.
To the extent we have, and if we do enter into any further such arrangements with any third parties, we will likely have limited control over the amount and timing of resources that our partners dedicate to the development or commercialization of our product candidates. On January 12, 2024, we entered into the Verity License Agreement with Verity, pursuant to which we granted to Verity an exclusive, royalty-bearing, sublicensable right and license to develop and commercialize our TLANDO and LPCN 1111 products with respect to TRT in the U.S. and Canada. Consequently, our ability to generate any revenues from TLANDO with respect to TRT in the U.S. and Canada depends on the efforts of Verity to commercialize TLANDO. We have very limited control over the amount and timing of resources that Verity dedicates to these efforts.
Our ability to generate revenues from this and other collaborative arrangements will depend on our collaborators’ abilities and efforts to successfully perform the functions agreed to with them in these arrangements. License agreements and/or collaborations involving our drug candidates, such as our agreement with Verity, pose numerous risks to us, including the following:
| ● | partners have significant discretion in determining the efforts and resources that they will apply to these efforts and may not perform their obligations as expected; | |
| ● | partners may de-emphasize or not pursue development and commercialization of our drug candidates or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the partners’ strategic focus, including as a result of a sale or disposition of a business unit or development function, or available funding or external factors such as an acquisition that diverts resources or creates competing priorities; | |
| ● | partners may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a drug candidate, repeat or conduct new clinical trials or require a new formulation of a drug candidate for clinical testing; | |
| ● | partners could independently develop, or develop with third parties, products that compete directly or indirectly with our products or drug candidates if the partners believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; | |
| ● | partners may not be able to acquire and maintain supplier and manufacturer relationships necessary to successfully commercialize our products; | |
| ● | a partner with marketing and distribution rights to multiple products may not commit sufficient resources to the marketing and distribution of our product relative to other products; | |
| ● | partners may not properly obtain, maintain, defend or enforce our intellectual property rights or may use our proprietary information and intellectual property in such a way as to invite litigation or other intellectual property related proceedings that could jeopardize or invalidate our proprietary information and intellectual property or expose us to potential litigation or other intellectual property related proceedings; |
| 38 |
| ● | disputes may arise between our partners and us that result in the delay or termination of the research, development or commercialization of our products or drug candidates or that result in costly litigation or arbitration that diverts management attention and resources; | |
| ● | agreements may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable drug candidates; | |
| ● | agreements may not lead to development or commercialization of drug candidates in the most efficient manner or at all; and | |
| ● | if a partner of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated. |
If our license arrangements with Verity, or any future license or collaboration we may enter into, if any, are not successful, our business, financial condition, results of operations, prospects and development and commercialization efforts may be adversely affected. Any termination or expiration of the Verity License Agreement, or any future license or collaboration we may enter into, if any, could adversely affect us financially or harm our business reputation, development and commercialization efforts.
We rely upon third-party contractors and service providers for the execution of some aspects of our development programs. Failure of these collaborators to provide services of a suitable quality and within acceptable timeframes may cause the delay or failure of our development programs.
We outsource certain functions, tests and services to CROs,contract research organizations (“CROs”), medical institutions and collaborators as well as outsourcingcollaborators; and also outsource manufacturing to collaborators and/or contract manufacturers.manufacturers (“CMOs”). We also rely on third parties for quality assurance, clinical monitoring, clinical data management and regulatory expertise. We may also engage a CRO to run all aspects of a clinical trial on our behalf. There is no assurance that such individuals or organizations will be able to provide the functions, tests, drug supply or services as agreed upon or in a quality fashion. Any failure to do so could cause us to suffer significant delays in the development of our products or processes.
Due to our reliance on CROs or other third parties to assist us or who have historically assisted us in conducting clinical trials, we will be unable to directly control all aspects of our clinical trials.
We engaged a CRO to conduct our pivotalSOAR, DV and DF Phase 3 trialclinical studies for TLANDO, as well as the on-going DVABPM study for TLANDO. Additionally, we utilized a CRO for the Phase 2 LiFT clinical study for LPCN 1144, the Phase 2 clinical study for LPCN 1148 and DFthe pilot and future pivotal studies for TLANDO.LPCN 1154. As a result, we have less direct control over the conduct of our clinical trials, the timing and completion of the trials and the management of data developed through the trials than if we were relying entirely upon our own staff. Communicating with outside parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. Outside parties, including CROs, may:
| ● | have staffing difficulties or disruptions; | |
| ● | fail to comply with contractual obligations; | |
| ● | experience regulatory compliance issues; | |
| ● | undergo changes in priorities or may become financially distressed; | |
| ● | form relationships with other entities, some of which may be our competitors; or | |
| ● | manufacturing capacity limitations. |
These factors may materially adversely affect their willingness or ability to conduct our trials in a manner acceptable to us. We may experience unexpected cost increases that are beyond our control.
Moreover, the FDA requires us to comply with standards, commonly referred to as Good Clinical Practices, or GCP,GCP’s for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements.
Problems with the timeliness or quality of the work of a CRO may lead us to seek to terminate the relationship and use an alternative service provider. However, making this change may be costly and may delay our trials, and contractual restrictions may make such a change difficult or impossible. If we must replace any CRO that is conducting our clinical trials, our trials may have to be suspended until we find another CRO that offers comparable services. The time that it takes us to find alternative organizations may cause a delay in the commercialization of our product candidates or may cause us to incur significant expenses to replicate data that may be lost. Although we do not believe that any CRO on which we may rely will offer services that are not available elsewhere, it may be difficult to find a replacement organization that can conduct our trials in an acceptable manner and at an acceptable cost. Any delay in or inability to complete our clinical trials could significantly compromise our ability to secure regulatory approval of our product candidates and preclude our ability to commercialize them, thereby limiting or preventing our ability to generate revenue from their sales.
| 39 |
We and our Licensee rely on a single supplier for our supply of TU,testosterone esters, the active pharmaceutical ingredient of TLANDO, LPCN 1111, LPCN 1148, and LPCN 1144, and the loss of this supplier could harm our business.
We and our Licensee rely on a single third-party supplier for our supply of TU,testosterone esters, the active pharmaceutical ingredient of TLANDO. We have a supply agreement in place with this supplier. We have purchased sufficient quantities of TU for our on-going DVTLANDO, LPCN 1111, LPCN 1148, and DF studies as well as for early commercial launch supplies should TLANDO get approved by the FDA. We plan on using this same supplier for our commercialization needs if TLANDO is approved.LPCN 1144. Since there are only a limited number of TUtestosterone esters suppliers in the world, if this supplier ceases to provide us with TU,testosterone esters, we or our Licensee may be unable to procure TUtestosterone esters on commercially favorable terms and/or may not be able to obtain ittestosterone esters in a timely manner, or may not be able to qualify a new supplier timely post FDA approval, if that occurs.manner. Furthermore, the limited number of suppliers of TUtestosterone esters may provide such companies with greater opportunity to raise their prices. AnyIf we or our Licensee are unable to obtain testosterone esters in a timely manner and/or in sufficient quantities, our ability to develop, and potentially commercialize, LPCN 1111, LPCN 1148, and LPCN 1144 may be adversely affected. In addition, any increase in price for TUtestosterone esters will likely reduce our potential gross margins.margins for LPCN 1148 and LPCN 1144.
We rely on limited suppliers for our supply of inactiveNAS, the active pharmaceutical ingredients of LPCN 1154, LPCN 2101, and LPCN 2203 and the loss of these limited suppliers could harm our business.
We rely on a limited qualified third-party raw material supplierssupplier for our supply of inactiveNAS, the active pharmaceutical ingredients of TLANDO. We do not have supply agreementsLPCN 1154, LPCN 2101, and LPCN 2203. Since there are only a limited number of NAS suppliers in placethe world, if a supplier ceases to provide us with these suppliers. We purchased sufficient quantities of these inactives for our on-going DV and DF studies as well as for early commercial launch of TLANDO if it is approved. We plan on using these same suppliers for our commercialization needs if TLANDO is approved. WeNAS, we may be unable to procure inactivesNAS on developmental or commercially favorable terms, orterms. Furthermore, the limited number of suppliers of NAS may not be ableprovide such suppliers with a greater opportunity to raise their prices. If we are unable to obtain themNAS in a timely manner. Any increasemanner and/or in price for inactives will likely reducesufficient quantities, our gross margins. ability to develop LPCN 1154, LPCN 2101, and LPCN 2203 may be adversely affected.
We depend on M.W. Encap Ltd. for the supply of the TLANDO capsules and the loss of this supplier would significantly harm our business.
We have entered into a Commercial Manufacturing Services and Supply Agreement with M.W. Encap Ltd. (“Encap”), a United Kingdom based contract manufacturer, a division of Capsugel Dosage Form Solutions. Pursuant to the Agreement, Encap has agreed to manufacture and supply bulk commercial quantities of TLANDO. Encap is currently our sole contract manufacturer and is our sole supplier of TLANDO for our clinical trials on a worldwide basis. If Encap is unable to produce sufficient capsules, for whatever reason, to support demand for TLANDO if it becomes commercially available, our revenue and profitability would be materially and adversely harmed. Also, we may not be able to engage an alternative supplier to meet our needs.
Reliance on a third-party manufacturer involves risks, such as capacity and capabilities of the manufacturer to which we would not be subject if we manufactured TLANDO ourselves. We also face risks related to reliance on the third party for regulatory compliance and quality assurance, the possibility of breach of the manufacturing agreement by the third party because of factors beyond our control and the possibility of termination or non-renewal of the agreement by the third party, based on its own business priorities, at a time that is costly or damaging to us. The FDA and other regulatory authorities require that TLANDO be manufactured according to cGMP. Any failure by any third-party manufacturers to comply with cGMP could be the basis for action by the FDA to withdraw approvals previously granted to us and for other regulatory action against us.
If we do not establish successful collaborations, we may have to alter our development and commercialization plans for our products.
Our drug development programs for our product candidates will require substantial additional cash to fund expenses. We have not yet established any collaborative arrangements relating to the development or commercialization of TLANDO, LPCN 11111148, LPCN 1144, or LPCN 1107. We intend to continue to develop some of our product candidates in the United States without a partner.partner although our ability to advance these product candidates will depend on our capital resources. However, in order to commercialize our product candidates in the United States, we will likely look to establish a partnership or co-promotion arrangement with an established pharmaceutical company that has a sales force, collaborate on the establishment of an internal sales force or build an internal sales force on our own. We may also seek to enter into collaborative arrangements to develop and commercialize our product candidates outside the United States. We will face significant competition in seeking appropriate collaborators and these collaborations are complex and time-consuming to negotiate and document. We may not be able to negotiate collaborations on acceptable terms or in a timely manner, or at all. If that were to occur, we may have to curtail the development or delay commercialization of our product candidates in certain geographies, reduce the scope of our sales or marketing activities, reduce the scope of our commercialization plans, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities either inside or outside of the United States on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms, or at all.
If we are successful in entering into collaborative arrangements and any of our collaborative partners doesdo not devote sufficient time and resources to a collaboration arrangement with us, we may not realize the potential commercial benefits of the arrangement, and our results of operations may be materially adversely affected. In addition, if any future collaboration partner were to breach or terminate its arrangements with us, the development and commercialization of our product candidates could be delayed, curtailed or terminated because we may not have sufficient financial resources or capabilities to continue development and commercialization of our product candidates on our own in such locations.
Risks Related to Ownership of Our Common Stock
Our stock price could decline significantly based on the results and timing of clinical trials, and/or regulatory and other decisions affecting our product candidates.
Results of clinical trials and preclinical studies of our current and potential product candidates may not be viewed favorably by us or third parties, including the FDA or other regulatory authorities, investors, analysts and potential collaborators. The same may be true of how we design the clinical trials of our product candidates and regulatory decisions affecting those clinical trials. Pharmaceutical company stock prices have declined significantly when such results and decisions were unfavorable or perceived negatively or when a product candidate did not otherwise meet expectations. The final results from our clinical development programs may be negative, may not meet expectations or may be perceived negatively. The designs of our clinical trials (which may change significantly and be more expensive than currently anticipated depending on our clinical results and regulatory decisions) may also be viewed negatively by third parties. We may not be successful in completing these clinical trials on our projected timetable, if at all. In addition, we may never achieve FDA approval for any of our product candidates other than TLANDO, which could cause our stock price to decline significantly and have other significant adverse effects on our business.
| 40 |
If we do not maintain effective internal controlcontrols over financial reporting in the future, the accuracy and timeliness of our financial reporting may be adversely affected.
The Sarbanes-Oxley Act requires, among other things, that we assess the effectiveness of our internal control over financial reporting annually and disclosure controls and procedures quarterly. In particular, we must perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on the effectiveness of our internal control over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. Additionally, our accounting firm is required to provide an opinion regarding our internal controls over financial reporting on an annual basis. If material weaknesses are identified in the future or we are not able to comply with the requirements of Section 404 in a timely manner, our reported financial results could be materially misstated, we could receive an adverse opinion regarding our internal controls over financial reporting from our accounting firm, and we could be subject to investigations or sanctions by regulatory authorities, which would require additional financial and management resources, and the market price of our stock could decline.
We will incur additionalsignificant expenses in order to comply with the requirements of being a public company in the United States.
As a public company, we incur significantly more legal, accounting and other expenses than as a private company. In addition, the Sarbanes-Oxley Act of 2002 and rules subsequently implemented by the SEC and U.S. stock exchanges impose numerous requirements on public companies, including requiring changes in corporate governance practices. Also, the Exchange Act requires, among other things, that we file annual, quarterly and current reports with respect to our business and operating results. Our management and other personnel will need to devote a substantial amount of time to compliance with these laws and regulations. These requirements have increased and will continue to increase our legal, accounting, and financial compliance costs and have made and will continue to make some activities more time consuming and costly.
Our share price is expected to be volatile and may be influenced by numerous factors that are beyond our control.
A low share price and low market valuation may make it difficult to raise sufficient additional cash due to the significant dilution to current stockholders. Market prices for shares of biotechnology and biopharmaceutical companies such as ours are often volatile. The market price of our common stock may fluctuate significantly in response to a number of factors, most of which we cannot control, including:
| ● | plans for, costs of, progress of and results from clinical trials of our product candidates; |
| ● | the failure of |
| ● | regulatory uncertainty in the TRT class; |
| ● | FDA Advisory Committee meetings and related recommendations including meetings convened on the TRT class or on similar companies; |
| ● | announcements by the FDA that may impact on-going clinical studies related to safety or efficacy of TRT products; |
| ● | product approval and potential FDA required labeling language and/or Phase 4 study commitments; | |
| ● | announcements of new products, technologies, commercial relationships, acquisitions or other events by us or our competitors; |
| ● | our ability to license our products to third parties; | |
| ● | failure to engage with collaborators or build an internal sales force to commercialize our |
| ● | the success or failure of other TRT products or non-testosterone based testosterone therapy products; |
| 41 |
| failure of our products, if approved, to achieve commercial success; |
| ● | fluctuations in stock market prices and trading volumes of similar companies; |
| ● | general market conditions and overall fluctuations in U.S. equity markets; |
| ● | variations in our quarterly operating results; |
| changes in our financial guidance or securities analysts’ estimates of our financial performance; |
| ● | changes in accounting principles; |
| ● | sales of large blocks of our common stock, including sales by our executive officers, directors and significant stockholders; |
| ● | additions or departures of key personnel; |
| ● | discussion of us or our stock price by the press and by online investor communities; |
| ● | our cash balance; and |
| ● | other risks and uncertainties described in these risk factors. |
In recent years, the stock of other biotechnology and biopharmaceutical companies has experienced extreme price fluctuations that have been unrelated to the operating performance of the affected companies. There can be no assurance that the market price of our shares of common stock will not experience significant fluctuations in the future, including fluctuations that are unrelated to our performance. These fluctuations may result due to macroeconomic and world events, national or local events, general perception of the biotechnology industry or to a lack of liquidity. In addition, other biotechnology companies or our competitors’ programs could have positive or negative results that impact their stock prices and their results, or stock fluctuations could have a positive or negative impact on our stock price regardless of whether such impact is direct or not.
Stockholders may not agree with our business, scientific, clinical, commercial, or financial strategy, including additional dilutive financings, and may decide to sell their shares or vote against suchshareholder proposals. Such actions could materially impact our stock price. In addition, portfolio managers of funds or large investors can change or change their view on us and decide to sell our shares. These actions could have a material impact on our stock price. In order to complete a financing, or for other business reasons, we may elect to consolidate our shares of common stock. Investors may not agree with these actions and may sell our shares. We may have little or no ability to impact or alter such decisions.
The stock prices of many companies in the biotechnology industry have experienced wide fluctuations that have often been unrelated to the operating performance of thesethe companies. Following periods of volatility in the market price of a company’s securities, securities class action litigation often has been initiated against a company. For example, on July 1, 2016, the Company and certain of its officers were named as defendants in a purported shareholder class action lawsuit, David Lewis v. Lipocine Inc., et al., filed in the United States District Court for the District of New Jersey. This initial action was followed by additional lawsuits also filed in the District of New Jersey. Due to thisDavid Lewis v Lipocine Inc., et al. was ultimately settled. Additionally on November 14, 2019, the Company and certain of its officers were named as defendants in a purported shareholder class action litigation initiated against us or anylawsuit, Solomon Abady v. Lipocine Inc. et al., 2:19-cv-00906-PMW, filed in the United District Court for the District of Utah. This initial action was followed by additional lawsuits also filed in the United States District Court for the District of Utah. Any future class action litigation that may be initiated against us we may incurresult in us incurring substantial costs and our management’s attention may be diverted from our operations, which could significantly harm our business. In addition, thissuch litigation could lead to increased volatility in our share price.
The value of our warrants outstanding from the November 2019 Offering is subject to potentially material increases and decreases based on fluctuations in the price of our common stock.
In November 2019, we completed a public offering of common stock and warrants to purchase common stock (the “November 2019 Offering”). Gross proceeds from the November 2019 Offering were approximately $6.0 million. In the November 2019 Offering, the Company sold (i) 614,706 Class A Units, with each Class A Unit consisting of one share of common stock and a common stock warrant to purchase one share of common stock, and (ii) 91,177 Class B Units, with each Class B Unit consisting of one pre-funded warrant to purchase one share of a common stock and one common stock warrant to purchase one share of common stock at a price of $8.50 per Class A Unit and $8.4998 per Class B Unit. The pre-funded warrants were issued in lieu of common stock in order to ensure the purchaser did not exceed certain beneficial ownership limitations. The pre-funded warrants were immediately exercisable at an exercise price of $0.0017 per share, subject to adjustment. Additionally, the common stock warrants were immediately exercisable at an exercise price of $8.50 per share and expire on November 17, 2024. As of December 31, 2023, there were 64,362 warrants from the November 2019 offering outstanding.
| 42 |
We account for the common stock warrants as a derivative instrument, and changes in the fair value of the warrants are included under other income (expense) in the Company’s statements of operations for each reporting period. As of December 31, 2023, the aggregate fair value of the warrant liability included in the Company’s consolidated balance sheet was approximately $17,000. We use the Black-Scholes option pricing model to determine the fair value of the warrants. As a result, the option-pricing model requires the input of several assumptions, including the stock price volatility, share price and risk-free interest rate. Changes in these assumptions can materially affect the fair value estimate. While the liability may only result from a change of control at a point in time, we ultimately may incur amounts significantly different than the carrying value of the liability.
We may not be able to maintain our listing on the NASDAQ Capital Market, which would adversely affect the price and liquidity of our common stock.
As a small capitalization pharmaceutical company, the price of our common shares has been, and is likely to continue to be, highly volatile. Any announcements concerning us or our competitors, clinical trial results, quarterly variations in operating results, introduction of new products, delays in the introduction of new products or changes in product pricing policies by us or our competitors, acquisition or loss of significant customers, partners and suppliers, changes in earnings estimates or our ratings by analysts, regulatory developments, or fluctuations in the economy or general market conditions, among other factors, could cause the market price of our common shares to fluctuate substantially. There can be no assurance that the market price of our common shares will not decline below its current price or that it will not experience significant fluctuations in the future, including fluctuations that are unrelated to our performance.
Currently our common stock is quoted on the NASDAQ Capital Market under the symbol “LPCN”. We must satisfy certain minimum listing maintenance requirements to maintain the NASDAQ Capital Market quotation, including certain governance requirements and a series of financial tests relating to stockholders’ equity or net income or market value, public float, number of market makers and stockholders, market capitalization, and maintaining a minimum bid price of $1.00 per share.
If Nasdaq delists our common stock from trading on its exchange and we are not able to list our securities on another national securities exchange, we expect our securities could be quoted on an over-the-counter market. If this were to occur, we could face significant material adverse consequences, including:
| ● | a limited availability of market quotations for our securities; | |
| ● | reduced liquidity for our securities; | |
| ● | a determination that our common stock is a “penny stock” which will require brokers trading in our common stock to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for our securities; | |
| ● | a limited amount of news and analyst coverage; and | |
| ● | a decreased ability to issue additional securities or obtain additional financing in the future. |
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” If our common stock continues to be listed on NASDAQ, our common stock will be a covered security. Although the states are preempted from regulating the sale of our securities, the federal statute does allow the states to investigate companies if there is a suspicion of fraud, and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case.
Anti-takeover provisions in our amended and restated certificate of incorporation and our amended and restated bylaws, as well as provisions of Delaware law and our stockholder rights plan, might discourage, delay or prevent a change in control of our companyCompany or changes in our Board of Directors or management and, therefore, depress the trading price of our common stock.
Our amended and restated certificate of incorporation, amended and restated bylaws and Delaware law contain provisions that may depress the market price of our common stock by acting to discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares of our common stock. These provisions may also prevent or frustrate attempts by our stockholders to replace or remove members of our Board of Directors or our management. Our corporate governance documents include provisions:
| ● | limiting the ability of our stockholders to call and bring business before special meetings and to take action by written consent in lieu of a meeting; |
| 43 |
| ● | requiring advance notice of stockholder proposals for business to be conducted at meetings of our stockholders and for nominations of candidates for election to our Board of Directors; | |
| ● | authorizing blank check preferred stock, which could be issued with voting, liquidation, dividend and other rights superior to our common stock; and | |
| ● | limiting the liability of, and providing indemnification to, our directors and officers. |
As a Delaware corporation, we are also subject to provisions of Delaware law, including Section 203 of the Delaware General Corporation Law, which limits the ability of stockholders owning in excess of 15% of our outstanding voting stock from engaging in certain business combinations with us. Any provision of our amended and restated certificate of incorporation, amended and restated bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
Additionally, on November 12, 2015,5, 2021, we adopted aan amended and restated stockholder rights plan that would cause substantial dilution to, and substantially increase the costs paid by, a stockholder who attempts to acquire us on terms not approved by our board. The intent of the stockholder rights plan is to protect our stockholders’ interests by encouraging anyone seeking control of our companyCompany to negotiate with our board. However, our stockholder rights plan could make it more difficult for a third party to acquire us without the consent of our board, even if doing so may be beneficial to our stockholders. This plan may discourage, delay or prevent a tender offer or takeover attempt, including offers or attempts that could result in a premium over the market price of our common stock. This plan could reduce the price that stockholders might be willing to pay for shares of our common stock in the future. Furthermore, the anti-takeover provisions of our stockholder rights plan may entrench management and make it more difficult to replace management even if the stockholders consider it beneficial to do so.
The common warrants issued in the November 2019 Offering include a right to receive the Black Scholes value of the warrants in the event of a fundamental transaction, which payment would be senior to our common stock.
The common warrants issued in the November 2019 Offering provide that, in the event of a “fundamental transaction,” including, among other things, a merger or consolidation of the Company or sale of all or substantially all of the Company’s assets, the holders of such warrants have the option to require the Company to pay to such holders an amount of cash equal to the Black Scholes value of the warrants. Such amount would be payable prior to any payments to holders of our common stock. The payment of such amount could result in common stockholders and other warrant holders not receiving any consideration if we were to liquidate, dissolve or wind up, either voluntarily or involuntarily. In addition, the existence of such right may reduce the value of our common stock, make it harder for us to sell shares of common stock in offerings in the future, or prevent or delay a change of control.
We have no current plans to pay dividends on our common stock and investors must look solely to stock appreciation for a return on their investment in us.
Although the board of directors of Marathon Bar declared a cash dividend to its stockholders of record in connection with the Merger, weWe do not anticipate paying any further cash dividends on our common stock in the foreseeable future. We currently intend to retain all future earnings to fund the development and growth of our business. Any payment of future dividends will be at the discretion of our board of directors and will depend on, among other things, our earnings, financial condition, capital requirements, level of indebtedness, statutory and contractual restrictions applying to the payment of dividends and other considerations that the board of directors deems relevant. Investors may need to rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize a return on their investment. Investors seeking cash dividends should not purchase our common stock.
Our management and directors will be able to exert influence over our affairs.
As of December 31, 2017,2023, our executive officers and directors beneficially owned approximately 10.7%5.9% of our common stock. These stockholders, if they act together, may be able to influence our management and affairs and all matters requiring stockholder approval, including significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control and might affect the market price of our common stock.
OurThe market price of our common stock is thinly traded,has been volatile over the past year and may continue to be thinlyvolatile.
The market price and trading volume of our common stock has been volatile over the past year, and it may continue to be volatile. During 2023, our common stock has traded as low as $2.36 and as high as $9.86 per share. We cannot predict the price at which our common stock will trade in the future, and our stockholdersit may be unable to selldecline. The price at or near asking prices or at all if they need to sell their shares.
To date, we have a low volume of daily trades inwhich our common stock on NASDAQ. For example,trades may fluctuate significantly and may be influenced by many factors, including our financial results; developments generally affecting our industry; general economic, industry and market conditions; the average daily trading volumedepth and liquidity of the market for our common stock; investor perceptions of our business; reports by industry analysts; announcements by other market participants, including, among others, investors, our competitors, and our customers; regulatory action affecting our business; and the impact of other “Risk Factors” discussed herein and in our common stock on NASDAQ duringAnnual Report. In addition, changes in the fourth quartertrading price of 2017 was approximately 197,000 shares per day with volumes continuing to decrease in 2018. Our stockholders may be unable to sell their common stock at or near their asking prices or at all, which may result in substantial losses to our stockholders.
The market for our common stock may be characterized by significantinconsistent with our operating results and outlook. The volatility of the market price volatility when compared to seasoned issuers, and we expect that our share price will be more volatile than a seasoned issuer for the indefinite future. As noted above,of our common stock may be sporadically and/adversely affect investors’ ability to purchase or thinly traded. As a consequence of this lack of liquidity, the trading of relatively small quantities of shares by our stockholders may disproportionately influence the price of those shares in either direction. The price for our shares could, for example, decline significantly in the event that a large number ofsell shares of our common stock are sold on the market without commensurate demand, as compared to a seasoned issuer that could better absorb those sales without adverse impact on its share price.stock.
| 44 |
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. We currently only have limited securities and industry analysts providing research coverage of our companyCompany and may never obtain additional research coverage by securities and industry analysts. If no additional securities or industry analysts commence coverage of our companyCompany or if current securities analyst coverage of our companyCompany ceases, the trading price for our stock could be negatively impacted. If the analysts downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. If analysts cease coverage of us or failsfail to publish reports on us regularly, demand for our stock could decrease, which could cause our stock price and trading volume to decline.
Risks Relating to Our Financial Position and Capital Requirements
We will need substantial additional capital in the future. If additional capital is not available, we will have to delay, reduce or cease operations.
We will need to raise additional capital to continue to fund our operations. Our future capital requirements may be substantial and will depend on many factors including:
| ● | market conditions for raising capital, particularly for life science companies; | |
| ● | current and future clinical trials for our product candidates, including for LPCN 1154, LPCN 2101, LPCN 2203 and LPCN 1148; | |
| ● | regulatory actions of the FDA; | |
| ● | the scope, size, rate of progress, results and costs of completing ongoing clinical trials and development plans with our product candidates; | |
| ● | the cost, timing and outcomes of our efforts to obtain marketing approval for our product candidates in the United States; | |
| ● | payments received under any current or future license agreements, strategic partnerships or collaborations; | |
| ● | the cost of filing, prosecuting and enforcing patent claims; | |
| ● | the costs associated with commercializing our product candidates if we receive marketing approval for product candidates other than TLANDO, including the cost and timing of developing internal sales and marketing capabilities or entering into strategic collaborations to market and sell our products; | |
| ● | the costs of on-going and future litigation; and | |
| ● | funding additional product line expansions. |
We believe that our existing capital resources, together with interest thereon, will be sufficient to meet our projected operating requirements through at least March 31, 2025. We have based this estimate on assumptions that may prove to be wrong, and future clinical trials forwe could utilize our product candidates, including for TLANDO, and the receipt of any additional CRLs related to TLANDO;
Changing circumstances may cause us to consumeavailable capital significantly fasterresources sooner than we currently anticipate. Also, financial markets may not be conducive to raising the capital we need, and we may not be able to raise capital through partnering arrangements. Additional financing may not be available when we need it or may not be available on terms that are favorable to us. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even ifexpect. While we believe we have sufficient funds forliquidity and capital resources to fund our currentprojected operating requirements through at least March 31, 2025, we will need to raise additional capital at some point through the equity or future operating plans. If adequate funds are not availabledebt markets or through out-licensing activities, either before or after March 31, 2025, to us on a timely basis, or at all, we may be unable to continuesupport our operations, the on-going clinical development of our product candidates, or to commercialize our product, if approved, unless we find a partner that providesand compliance with regulatory requirements. If the Company is unsuccessful in raising additional capital, or reducesits ability to continue as a going concern will become a risk. Further, our operating plan may change, and we may need additional funds to meet operational needs and capital requirements for product development, regulatory compliance, and clinical trial activities sooner than planned. In addition, our capital needs.resources may be consumed more rapidly if we pursue additional clinical studies for LPCN 1154, LPCN 2101, LPCN 2203, LPCN 1148, LPCN 1144, and LPCN 1107. Conversely, our capital resources could last longer if we reduce expenses, reduce the number of activities currently contemplated under our operating plan or if we terminate or suspend on-going clinical studies.
| 45 |
Funding may not be available to us on favorable terms, or at all. Also, market conditions may prevent us from accessing the debt and equity capital markets, including sales of our common stock through the ATM Offering (as defined below). If we are unable to obtain adequate financing when needed, we may have to delay, reduce the scope of or suspend one or more of our clinical studies, research and development programs or, if any of our product candidates other than TLANDO receive approval from the FDA, commercialization efforts. We may seek to raise any necessary additional capital through a combination of public or private equity offerings, including the ATM Offering, debt financings, collaborations, strategic alliances, licensing arrangements and other marketing and distribution arrangements. These arrangements may not be available to us or available on terms favorable to us. To the extent that we raise additional capital through marketing and distribution arrangements, other collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our product candidates, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we do raise additional capital through public or private equity offerings, the ownership interest of our existing stockholders will be diluted, and the terms of these securities may include liquidation or other preferences, warrants or other terms that adversely affect our stockholders’ rights or further complicate raising additional capital in the future. If we raise additional capital through debt financing, we may be subject to covenants limiting or restricting our ability to take thesespecific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we are unable, for any reason, to raise needed capital, we will have to reduce costs, delay research and development programs, liquidate assets, dispose of rights, commercialize products or product candidates earlier than planned or on less favorable terms than desired, or reduce or cease operations.
Our loan agreement contains covenants which may adversely impact our business; the failure to comply with such covenants could cause our outstanding debt to become immediately payable.
On January 5, 2018, we entered into a Loan and Security Agreement (the “Loan and Security Agreement”) with Silicon Valley Bank (“SVB”) pursuant to which SVB has agreed to lend us $10.0 million. The principal borrowed under the Loan and Security Agreement bears a fixed interest rate equal to the Prime Rate plus one percent per annum, which interest is payable monthly. The loan matures on December 1, 2021. In addition, if TLANDO is not approved by the FDA on or prior to May 31, 2018, we will be required to maintain $5.0 million of cash collateral at SVB until such time as TLANDO is approved by the FDA. The Loan Agreement includes a number of restrictive covenants, including restrictions on incurring additional debt, transactions with affiliates, disposing of property, business combinations or acquisitions, paying dividends and making other distributions or payments on our capital stock, subject to limited exceptions. Collectively, these covenants could constrain our ability to grow our business through acquisitions or engage in other transactions. In addition, the Loan Agreement includes covenants requiring, among other things, that we provide financial statements, comply with all laws, pay all taxes and maintain insurance. If we are not able to comply with these covenants, the loans under the Loan Agreement could become immediately due and payable and would have a material adverse effect on our liquidity, financial condition, operating results, business, and prospects and cause the price of our common stock to decline.
We may be unable to generate sufficient cash flow to satisfy our significant debt service obligations, which would adversely affect our financial condition and results of operations.
Our ability to make principal and interest payments on and to refinance our indebtedness will depend on our ability to generate cash in the future. This, to a certain extent, is subject to general economic, financial, competitive, legislative, regulatory and other factors that are beyond our control. If our business does not generate sufficient cash flow from operations, in the amounts projected or at all, or if future borrowings are not available to us under our variable funding notes in amounts sufficient to fund our other liquidity needs, our financial condition and results of operations may be adversely affected. If we cannot generate sufficient cash flow from operations to make scheduled principal amortization and interest payments on our debt obligations in the future, we may need to refinance all or a portion of our indebtedness on or before maturity, sell assets, delay capital expenditures or seek additional equity. If we are unable to refinance any of our indebtedness on commercially reasonable terms, or at all, or to affect any other action relating to our indebtedness on satisfactory terms, or at all, our business may be harmed.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations, or require us to relinquish rights.
We may seek additional capital through a combination of private and public equity offerings, debt financings, collaborations, and strategic and licensing arrangements. To the extent that we raise additional capital through the sale of common stock or securities convertible or exchangeable into common stock, current stockholders’ ownership interest in the companyCompany will be diluted. In addition, the terms may include liquidation or other preferences that materially adversely affect their rights as a stockholder. Debt financing, if available, would increase our fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration, strategic alliance and licensing arrangements with third parties, we may have to relinquish valuable rights to our product candidates, our intellectual property, future revenue streams or grant licenses on terms that are not favorable to us.
We cannot predict when we will generate product revenues and may never achieve or maintain profitability.
Our ability to become profitable depends upon our ability to generate revenue from product sales.sales and/or licensing agreements. To date, we have not generated any significant revenue from product sales of TLANDO or our other drug candidates in the current pipeline, and we do not know when, or if, we will generate anysignificant revenue from product sales. We do not expect to generate significant revenue unless or until we obtain marketing approval of, and begin to sell, TLANDO. Our ability to generate revenue depends on a number of factors, including, but not limited to, our ability to:
| ● | other than for TLANDO in the U.S., obtain U.S. and foreign marketing approval for our product candidates; | |
| ● | commercialize our product candidates by developing a sales force and/or entering into licensing agreements or collaborations with partners/third parties, either before or after obtaining marketing approval for our product candidates; and | |
| ● | achieve market acceptance of our product candidates in the medical community and with third-party payors. |
Even if our product candidates other than TLANDO isare approved for commercial sale, we expect to incur significant costs as we prepare to commercialize TLANDO.them. Even if we receive FDA approval for TLANDO, TLANDOour product candidates, they may not be a commercially successful drug.drugs. We may not achieve profitability soon after generating product sales, if ever. If we are unable to generate product revenue, we will not become profitable and may be unable to continue operations without continued funding.
Accordingly, the likelihood of our success must be evaluated in light of many potential challenges and variables associated with an early-stage drug development company, many of which are outside of our control, and past operating or financial results should not be relied on as an indication of future results. If one or more of our product candidates is approved for commercial sale and we retain commercial rights, we anticipate incurring significant costs associated with commercializing any such approved product candidate. Therefore, even if we are able to generate revenues from the sale of any approved product, we may never become profitable. Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to predict the timing or amount of expenses and when we will be able to achieve or maintain profitability, if ever.
We have incurred significant operating losses in most years since our inception and anticipate that we will incur continued losses for the foreseeable future.
We have focused a significant portion of our efforts on developing TLANDO.TLANDO and more recently on our oral neuroactive steroids LPCN 1154 and LPCN 2101, LPCN 1148, and LPCN 1144. We have funded our operations to date through proceeds from sales of common stock, preferred stock and convertibleour equity securities, debt, and from license and milestone revenues and research revenue frompayments received under our license and collaboration agreements with corporate partners.arrangements. We have incurred losses in most years since our inception. As of December 31, 2017,2023, we had an accumulated deficit of $126.4$199.8 million. Substantially all of our operating losses resulted from costs incurred in connection with our research and development programs and from general and administrative costs associated with our operations. These losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ equity and working capital. We expect our research and development expenses to significantly increase in connection with clinical trials associated with TLANDO,our oral neuroactive steroids LPCN 11111154, LPCN 2101, LPCN 2203, and LPCN 1148, and LPCN 1144 and LPCN 1107, if further clinical trials are initiated. In addition, if we obtain marketing approval for TLANDO, we will incur significant sales, marketing and commercialization expenses. As a result, we expect to continue to incur significant operating losses for the foreseeable future as we advance our lead product candidate, TLANDO, andevaluate further clinical development of LPCN 1111,1154, LPCN 2101, LPCN 2203, LPCN 1148, LPCN 1144, and LPCN 1107 and our other programs and continued research efforts. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or when we will become profitable, if at all.
| 46 |
Our operating results may fluctuate significantly, and any failure to meet financial expectations may disappoint securities analysts or investors and result in a decline in the price of our securities.
We have a history of operating losses. Our operating results have fluctuated in the past and are likely to do so in the future. These fluctuations could cause our share price to decline. Due to fluctuations in our operating results, we believe that period-to-period comparisons of our results are not indicative of our future performance. It is possible that in some future quarter or quarters, our operating results will be above or below the expectations of securities analysts or investors. In this case, the price of our securities could decline.
We cannot predict the impact of new tax legislation on our operating results or the impact on holders of our common stock.
On December 22, 2017, the Tax Cuts and Jobs Act (“TCJA”) was signed into law, significantly reforming the U.S. Internal Revenue Code. The TCJA, among other things, includes changes to U.S. federal tax rates, imposes significant additional limitations on the deductibility of interest, allows for the expensing of capital expenditures, puts into effect the migration from a “worldwide” system of taxation to a territorial system and modifies or repeals many business deductions and credits. We continue to examine the impact the TCJA may have on our business. We will evaluate the effect of the TCJA on our projection of minimal cash taxes or to our net operating losses. The estimated impact of the TCJA is based on our management’s current knowledge and assumptions and recognized impacts could be materially different from current estimates based on our actual results and our further analysis of the new law. Our net deferred tax assets and liabilities will be revalued at the newly enacted U.S. corporate rate, and the impact will be recognized in our tax expense, if any, in the year of enactment. The impact of the TCJA on holders of common shares is uncertain and could be adverse. This Form 10-K does not discuss any such tax legislation or the manner in which it might affect purchasers of common shares. We urge our stockholders, including purchasers of common shares, to consult with their own legal and tax advisors with respect to such legislation and the potential tax consequences of investing in common shares.
Risks Relating to Our Intellectual Property
Our success depends in part on our ability to protect our intellectual property. It is difficult and costly to protect our proprietary rights and technology, and we may not be able to ensure their protection.
Our commercial success will depend in large part on obtaining and maintaining patent, trademark and trade secret protection of our product candidates, their respective formulations, methods used to manufacture them and methods of treatment, as well as successfully defending these patents against third party challenges. Our ability to stop unauthorized third parties from making, using, selling, offering to sell, or importing our product candidates, once commercialized, is dependent upon the extent to which we have rights under valid and enforceable patents or trade secrets that cover these products and activities.
The patent positions of pharmaceutical, biopharmaceutical and related companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in patents in these fields has emerged to date in the United States. There have been recent changes regarding how patent laws are interpreted, and both the United States Patent and Trademark Office (“PTO”USPTO”) and Congress have enacted radical changes to the patent system. We cannot accurately predict future changes in the interpretation of patent laws or changes to patent laws which might be enacted into law. Those changes may materially affect our patents, our ability to obtain patents and/or the patents and applications of our collaborators and licensors. The patent situation in these fields outside the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in the patents we own or which we license or third-party patents.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep a competitive advantage. For example:
| ● | others may be able to make or use compounds that are the same or similar to the pharmaceutical compounds used in our product candidates but that are not covered by the claims of our patents; | |
| ● | the Active Pharmaceutical Ingredients (“APIs”) in our licensed product TLANDO and current product candidates LPCN 1154, LPCN 1148, LPCN 1144, LPCN 1111, and LPCN 1107 are, or may soon become, commercially available in generic drug products, and no patent protection may be available without regard to formulation or method of use; | |
| ● | we may not be able to detect infringement against our owned or licensed patents, which may be especially difficult for manufacturing processes or formulation patents; | |
| ● | we might not have been the first to make the inventions covered by our issued patents or pending patent applications or those we license; | |
| ● | we might not have been the first to file patent applications for these inventions; | |
| ● | others may independently develop similar or alternative technologies or duplicate any of our technologies; | |
| ● | it is possible that our pending patent applications or those of our licensor will not result in issued patents; | |
| ● | it is possible that there are dominating patents to any of our product candidates of which we are not aware; | |
| ● | it is possible that there are prior public disclosures that could invalidate our patents, or parts of our patents, of which we are not aware; | |
| ● | it is possible that others may circumvent our owned or licensed patents; |
| 47 |
| ● | it is possible that there are unpublished applications or other patent applications maintained in secrecy that may issue later than our patents/applications but may have priority dates that are earlier than our priority dates and may have claims covering our products or technology similar to ours; | |
| ● | the laws of foreign countries may not protect our proprietary rights to the same extent as the laws of the United States; | |
| ● | the claims of our owned or licensed issued patents or patent applications, if and when issued, may not cover our product candidates; | |
| ● | our issued patents or those of our licensor may not provide us with any competitive advantages, or may be narrowed in scope, be held invalid or unenforceable as a result of legal challenges by third parties; | |
| ● | our licensor or licensees as the case may be, who have access to our patents, may attempt to enforce our owned or licensed patents, which if unsuccessful, may result in narrower scope of protection of our owned or licensed patents or our owned or licensed patents becoming invalid or unenforceable; | |
| ● | we may not develop additional proprietary technologies for which we can obtain patent protection; or | |
| ● | the patents of others may have an adverse effect on our business. |
We also may rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect, and we have limited control over the protection of trade secrets used by our collaborators and suppliers. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using for any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods, and know-how. If our confidential or proprietary information is divulged to or acquired by third parties, including our competitors, our competitive position in the marketplace will be harmed and our ability to successfully penetrate our target markets could be severely compromised.
If any of our owned or licensed patents are found to be invalid or unenforceable, or if we are otherwise unable to adequately protect our rights, it could have a material adverse impact on our business and our ability to commercialize or license our technology and products. Additionally, we currently do not have patent protection for TLANDOsome of our product candidates in many countries, including large territories such as India, Russia, and China, and we will be unable to prevent patent infringementunauthorized third parties from using our intellectual property in those countries unless we can file patent applications and obtain patents in those countries that cover TLANDO.our product candidates. Likewise, our United States patents covering certain technology used in our product candidates, including TLANDO, are expected to expire on various dates from November 23, 2019 through November 2030.2041. Upon the expiration of these patents, we will lose the right to exclude others from practicing these inventions to the extent that at those times we have no additional issued patents to protect our product candidates, including TLANDO. Additionally, if these are our only patents listed in the FDA Orange Book, should we have aan FDA-approved and marketed product at that time, their expiration will mean that we lose certain advantages that come with Orange Book listing of patents. The expiration of these patents could also have a similar material adverse effect on our business, results of operations, financial condition and prospects. Moreover, if we are unable to commence or continue any action relating to the defense of our patents, we may be unable to protect our product candidates.
If we do not obtain additional protection under the Drug Price Competition and Patent Term Restoration Act and similar foreign legislation by extending the patent terms and obtaining data exclusivity for our product candidates, our business may be materially harmed.
Depending upon the timing, duration and specifics of FDA marketing approval of our product candidates, one or more of our U.S. patents may be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permitpermits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, we may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or competitor’s prior product launch or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or restoration or the term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and our ability to generate revenues could be materially adversely affected.
| 48 |
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights, and we may be unable to protect our rights to our products and technology.
If we or our collaborators choose to go to court to stop a third party from using the inventions claimed in our owned or licensed patents, that third party may ask a court to rule that the patents are invalid and should not be enforced against that third party. These lawsuits are expensive and would consume time and other resources, including financial resources, even if we were successful in stopping the infringement of these patents. In addition, there is a risk that a court will decide that these patents are not valid or not enforceable and that we do not have the right to stop others from using the inventions.
There is also the risk that, even if the validity of these patents is not challenged or is upheld, the court will refuse to stop the third party on the ground that such third-party’s activities do not infringe on our owned or licensed patents. In addition, the U.S. Supreme Court has recently changed and continues to change some standards relating to the granting of patents and assessing the validity of patents. As a consequence, issued patents may be found to contain invalid claims according to the newly revised standards. Some of our owned or licensed patents may be subject to challenge and subsequent invalidation or significant narrowing of claim scope in a reexamination or other proceeding before the U.S. Patent and Trademark Office ("PTO"),USPTO, or during litigation, under the revised criteria which make it more difficult to obtain or maintain patents.
While our in-licensed patents and applications are not currently used in our product candidates, should we develop other product candidates that are covered by this intellectual property, we willmay rely on our licensor to file and prosecute patent applications and maintain patents and otherwise protect the intellectual property we license from them. Our licensor has retained the first right, but not the obligation, to initiate an infringement proceeding against a third-party infringer of the intellectual property licensed to us, and enforcement of our in-licensed patents or defense of any claims asserting the invalidity or unenforceability of these patents would also be subject to the control or cooperation of our licensor. It is possible that our licensor’s defense activities may be less vigorous than had we conducted the defense ourselves.
We also license our patent portfolio, including U.S. and foreign patents and patent applications that cover our TLANDO and our other product candidates, to third parties for their respective products and product candidates. Under our agreements with our licensees, we have the right, but not the obligation, to enforce our current and future licensed patents against infringers of our licensees. In certain cases, our licensees may have primary enforcement rights and we have the obligation to cooperate. In the event of an enforcement action against infringers of our licensees, our licensees might not have the interest or resources to successfully preserve the patents, the infringers may countersue, and as a result our patents may be found invalid or unenforceable or of a narrower scope of coverage and leave us with no patent protection for TLANDO and our other product candidates.
In addition, on May 15, 2015 we filed a patent application with the PTO, and our application requests that the PTO declare an interference between our patent application and Clarus Therapeutics’ (“Clarus”) U.S. Patent No. 8,828,428 (“the Clarus 428 Patent”). Pursuant to Lipocine's request, on December 4, 2015, the Patent Trial and Appeal Board (“PTAB”) declared an interference between the Clarus 428 Patent and Lipocine's application to determine, as between Clarus and Lipocine, who was the first to invent the subject matter of the claimed invention. Lipocine was declared the senior party in the interference. On September 20, 2017 the PTAB issued a Motions Decision based on each party’s motions in the interference case. The PTAB granted Lipocine’s motion to deny Clarus’ previously accorded priority date for the Clarus 428 Patent. Therefore, Clarus has a new priority date of April 16, 2014 for the Clarus 428 patent. The PTAB also granted Clarus’ motion to deny Lipocine’s accorded priority date. Therefore, Lipocine has an accorded priority date of May 15, 2015 on its application. As a consequence of this decision, the PTAB has redeclared the interference and named Clarus as the senior party and Lipocine as the junior party. All other motions were denied. A conference call with the PTAB was held on October 4, 2017 to discuss the next steps, including priority schedule. As a result of the conference call, the PTAB has issued an order setting priority times. The order indicated that since Lipocine is the only party that filed a priority statement, only Lipocine shall be permitted to put on a priority case. The priority statement filed by Lipocine included a invention date prior to Clarus’ accorded benefit date. Interference proceedings may fail and could require us to cease using the related technology or to attempt to license rights to it from the prevailing party; our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms, if any license is offered at all. Even if we are successful in such interference, it may result in substantial costs to us and distraction to our management.
This interference proceeding will consume a portion of our capital resources. Moreover, weWe may be subject to a third party preissuancethird-party pre-issuance submission of prior art to the PTO,USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our owned or licensed patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our owned or licensed patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates and impair our ability to raise needed capital.
If we are required to defend patent infringement actions brought by other third parties, or if we sue to protect our own patent rights or otherwise to protect our proprietary information and to prevent its disclosure, we may be required to pay substantial litigation costs and managerial attention and financial resources may be diverted from business operations even if the outcome is in our favor.
If we are sued for infringing intellectual property rights of third parties, it will be costly and time consuming, and an unfavorable outcome in that litigation would have a material adverse effect on our business.
Our commercial success depends upon our ability and the ability of our collaborators to develop, manufacture, market and sell our product candidates and use our proprietary technologies without infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields relating to our product candidates. As the biotechnology, pharmaceutical, and related industries expand and more patents are issued, the risk increases that others may assert that our product or product candidates infringe the patent rights of others. Moreover, it is not always clear to industry participants, including us, which patents cover various types of drugs, products or their formulations or methods of use. Thus, because of the large number of patents issued and patent applications filed in our fields, there may be a risk that third parties may allege they have patent rights encompassing our product, product candidates, technology, or methods. For example, on November 2, 2015, Clarus Therapeutics Holdings, Inc. (Clarus) filed a complaint against us in the United States District Court for the District of Delaware alleging that TLANDO willwould infringe the Clarus 428 Patent, and the complaint sought damages, declaratory and injunctive relief. On October 6, 2016, United States District Court of the District of Delaware granted our motion to dismiss the lawsuit filed by Clarus, because at the time there was no actionable infringement on Clarus’ 428 patent. While the Clarus complaint has been dismissed, the possibility remains that Clarus could submit its claim again if TLANDO is approved by the FDA and enters the marketplace.
| 49 |
In addition, there may be issued patents of third parties of which we are currently unaware, that are infringed or are alleged to be infringed by our product candidates or proprietary technologies. Because some patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in the United States and many foreign jurisdictions are typically not published until eighteen months after filing, and because publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our or our licensor’s issued patents or our pending applications, or that we were the first to invent the technology. Our competitors may have filed, and may in the future file, patent applications covering our products or technology similar to ours. Any such patent application may have priority over our owned or licensed patent applications or patents, which could further require us to obtain rights to issued patents covering such technologies. If another party has filed a U.S. patent application on inventions similar to those owned or licensed by us, we may have to participate in an interference proceeding declared by the PTOUSPTO to determine priority of invention in the United States. If another party has an allowed reason to question the validity of our owned or licensed U.S. patents, the third party can request that the PTOUSPTO reexamine the patent claims, which may result in a loss of scope of some claims or a loss of the entire patent. In addition to potential infringement claims, interference and reexamination proceedings, we may become a party to patent opposition proceedings in the European Patent Office or post-grant proceedings in the United States where either our patents are challenged, or we are challenging the patents of others. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful, for example if the other party had independently arrived at the same or similar invention prior to our invention, resulting in a loss of our U.S. patent position with respect to such inventions. We may be exposed to, or threatened with, future litigation by third parties having patent or other intellectual property rights alleging that our product candidates and/or proprietary technologies infringe their intellectual property rights. These lawsuits are costly and could adversely affect our results of operations and divert the attention of managerial and technical personnel. There is a risk that a court would decide that we or our commercialization partners are infringing the third party’s patents and would order us or our partners to stop the activities covered by the patents. In addition, there is a risk that a court will order us or our partners to pay the other party damages for having violated the other party’s patents.
If a third-party’s patent was found to cover our product candidates, proprietary technologies or their uses, we or our collaborators could be enjoined by a court and required to pay damages and could be unable to commercialize any one or more of our product candidates or use our proprietary technologies unless we or they obtainedobtain a license to the patent. A license may not be available to us or our collaborators on acceptable terms, if at all. In addition, during litigation, the patent holder could obtain a preliminary injunction or other equitable relief which could prohibit us from making, using or selling our products, technologies or methods pending a trial on the merits, which could be years away.
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology, pharmaceutical, and related industries generally. If a third-party claims that we or our collaborators infringe its intellectual property rights, we may face a number of issues, including, but not limited to:
| ● | infringement and other intellectual property claims which, regardless of merit, may be expensive and time-consuming to litigate and may divert our management’s attention from our core business; | |
| ● | substantial damages for infringement, which we may have to pay if a court decides that the product at issue infringes on or violates the third party’s rights, and if the court finds that the infringement was willful, we could be ordered to pay treble damages and the patent owner’s attorneys’ fees; | |
| ● | a court prohibiting us from selling or licensing the product unless the third-party licenses its product rights to us, which it is not required to do; | |
| ● | if a license is available from a third party, we may have to pay substantial royalties, upfront fees and/or grant cross-licenses to intellectual property rights for our products; and | |
| ● | redesigning our products or processes so they do not infringe, which may not be possible or may require substantial monetary expenditures and time. |
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations or otherwise have a material adverse effect on our business, results of operations, financial condition, and prospects.
Although we own worldwide rights to our product candidates, we do not have patent protection for the product candidates in a significant number of countries, and we will be unable to prevent infringement in those countries.
Our patent portfolio related to our product candidates includes patents in the United States and other foreign countries. The covered technology and the scope of coverage varies from country to country. For those countries where we do not have granted patents, we have no ability to prevent the unauthorized use of our intellectual property, and third parties in those countries may be able to make, use, or sell products identical to, or substantially similar to our product candidates.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenancemaintenance/annuity fees on our owned or licensed patents and patent applications are due to be paid to the PTOrespective patent offices in several stages over the lifetime of the patents. Future maintenance fees will also need to be paid on other patents which may be issued to us. We have systems in place to remind us to pay these fees, and we employ outside firms to remind us to pay annuity fees due to foreign patent agencies on our pending foreign patent applications. We have even less control over our in-licensed patents and applications, for which our licensor retains responsibility. The PTOapplications. In addition, the USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, thereThere are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business.
| 50 |
We also may rely on trade secrets and confidentiality agreements to protect our technology and know-how, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect, and we have limited control over the protection of trade secrets used by our collaborators and suppliers. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators, and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods, and know-how. If our confidential or proprietary information is divulged to or acquired by third parties, including our competitors, our competitive position in the marketplace will be harmed and our ability to successfully generate revenues from our product candidates, and if approved by the FDA or other regulatory authorities, our product candidates could be adversely affected.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology, pharmaceutical and related industries, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management, which would adversely affect our financial condition.
ITEM 1B.UNRESOLVED STAFF COMMENTS
None.
ITEM 1C.CYBERSECURITY
Risk Management and Strategy
We regularly assess risks from cybersecurity threats, monitor our information systems for potential vulnerabilities, and test those systems pursuant to our cybersecurity policies, processes, and practices, which are integrated into our overall risk management program. To protect our information systems from cybersecurity threats, we use various security tools that are designed to help identify, escalate, investigate, resolve, and recover from security incidents in a timely manner. Our board of directors assesses risks based on probability and potential impact to key business systems and processes as part of our overall risk management program overseen by the board of directors. Risks that are considered high are incorporated into our overall risk management program. We collaborate with third parties to assess the effectiveness of our cybersecurity prevention and response systems and processes and to assist in the identification, verification, and validation of cybersecurity risks, as well as to support associated mitigation plans when necessary. We have also developed a third-party cybersecurity risk management process to conduct due diligence on external entities, including those that perform cybersecurity services. Cybersecurity threats, including those resulting from any previous cybersecurity incidents, have not materially affected our Company, including our business strategy, results of operations, or financial condition. Refer to the risk factor captioned “Cyber security risks and the failure to maintain the integrity of company, employee or guest data could expose us to data loss, litigation and liability, and our reputation could be significantly harmed” in Part I, Item 1A. “Risk Factors” for additional details regarding cybersecurity risks and potential impacts on our business.
Governance
Our board of directors oversees our risk management process, including as it pertains to cybersecurity risks, which focuses on the most significant risks we face in the short-, intermediate-, and long-term timeframe. Management is responsible for the operational oversight of company-wide cybersecurity strategy, policy, and standards across relevant departments to assess and help prepare us to address cybersecurity risks. Meetings of our board of directors include discussions and presentations from management regarding specific risk areas throughout the year, including, among others, those relating to cybersecurity threats, and reports from management on our enterprise risk profile on an annual basis. The board of directors reviews our cybersecurity risk profile with management on a periodic basis using key performance and/or risk indicators. These key performance indicators are metrics and measurements designed to assess the effectiveness of our cybersecurity program in the prevention, detection, mitigation, and remediation of cybersecurity incidents. We take a risk-based approach to cybersecurity and have implemented cybersecurity policies throughout our operations that are designed to address cybersecurity threats and incidents.
ITEM 2. PROPERTIES
Our corporate headquarters are located in a leased facility in Salt Lake City, Utah. Our lease expires on February 28, 2019. Additionally, we had a satellite office located in a leased facility in Lawrenceville, New Jersey. Our lease for this facility expired on January 31, 2018 and we did not renew this lease.2025. We believe that our existing facility is suitable and adequate and that we have sufficient capacity to meet our current anticipated needs.
ITEM 3. LEGAL PROCEEDINGS
On May 15, 2015,April 2, 2019, we filed a patent application with the PTO, and our application requested that the PTO declare an interference between our patent application and thelawsuit against Clarus 428 Patent. Pursuant to Lipocine's request, on December 4, 2015, the Patent Trial and Appeal Board (“PTAB”) declared an interference between the Clarus 428 Patent and Lipocine's application to determine, as between Clarus and Lipocine, who was the first to invent the subject matter of the claimed invention. Lipocine was declared the Senior Party in the interference. On September 20, 2017 the PTAB issued a Decisions on Motions. The PTAB granted Lipocine’s motion to deny Clarus’ previously accorded priority dateUnited States District Court for the District of Delaware alleging that Clarus’s JATENZO® product infringes six of Lipocine’s issued U.S. patents: 9,034,858; 9,205,057; 9,480,690; 9,757,390; 6,569,463; and 6,923,988. However, on February 11, 2020, we voluntarily dismissed allegations of patent infringement for expired U.S. Patent Nos. 6,569,463 and 6,923,988 in an effort to streamline the issues and associated costs for dispute. Clarus 428 Patentanswered the complaint and deniedasserted counterclaims of non-infringement and invalidity. We answered Clarus’s counterclaims on April 29, 2019. The Court held a scheduling conference on August 15, 2019, a claim construction hearing on February 11, 2020, and a summary judgment hearing on January 15, 2021. In May 2021, the Court granted Clarus’ motion for an earlier priority date based uponsummary judgment, finding the filingasserted claims of its provisional applications. Therefore,Lipocine’s U.S. patents 9,034,858; 9,205,057; 9,480,690; and 9,757,390 invalid for failure to satisfy the written description requirement of 35 U.S.C. § 112. Clarus has a new priority date of April 16, 2014 forstill had remaining claims before the Court. On July 13, 2021, we entered into the Global Agreement with Clarus 428 patent. The PTAB also granted Clarus’ motion to deny Lipocine’s accorded priority date. Therefore, Lipocine has an accorded priority date of May 15, 2015 on its application. As a consequencewhich resolved all outstanding claims of this decision, the PTAB has redeclared the interference and named Claruslitigation as well as the senior partyon-going United States Patent and Trademark Office (“USPTO”) Interference No. 106,128 between the parties (as described below). Under the terms of the Global Agreement, Lipocine agreed to pay Clarus $4.0 million payable as the junior party. All other motions were denied. A conference call with the PTAB was heldfollows: $2.5 million immediately, $1.0 million on October 4, 2017 to discuss the next steps, including a priority schedule. After the conference call, the PTAB issued an order setting times in the priority phase. The order indicated that since Lipocine is the only party that filed a priority statement, only Lipocine shall be permitted to putJuly 13, 2022, and $500,000 on a priority case. The priority statement filed by Lipocine included a claimed date of invention well prior to Clarus’ accorded benefit date. Lipocine has filed its initial motion in the priority phase.
July 13, 2023. On July 1, 2016,15, 2021, the CompanyCourt dismissed with prejudice Lipocine’s claims and Clarus’ counterclaims. In April 2022, we agreed to an amendment to Section 3.1 of the Global Agreement with Clarus pursuant to which we agreed to settle the payments due in July 2022 and 2023 for $1,250,000 rather than the $1,500,000 total future payments due. No future royalties are owing from either party.
On November 14, 2019, we and certain of itsour officers were named as defendants in a purported shareholder class action lawsuit, David LewisSolomon Abady v. Lipocine Inc., et al., 3:16-cv-04009-BRM-LHG,2:19-cv-00906-PMW, filed in the United States District Court for the District of New Jersey. This initial action was followed by additional lawsuits also filed in the District of New Jersey. On December 2, 2016, the court granted plaintiff’s motion to consolidate the various lawsuits and appointed Pomerantz LLP as lead counsel and Lipocine Investor Group as lead plaintiff. The court also stated that all filings shall bear the captionIn re Lipocine Inc. Securities Litigation. On March 14, 2017, the court granted our motion to transfer the action to the United States District Court for the District of Utah. On April 27, 2017, Plaintiff filed an Amended Complaint against the Company and certain of its officers and/or directors in the United States District Court for the District of Utah. This is a purported class action seeking relief for violations of Section 10(b) and 20(a) of the Securities Exchange Act of 1934 and Rule 10b-5 promulgated thereunder. The Amended Complaintcomplaint alleges that the defendants made false and/or misleading statements and/or failed to disclose that our filing of the NDA for TLANDO to the FDA contained deficiencies and as a result the defendants’ statements about our business and operations were false and misleading and/or lacked a reasonable basis in violation of federal securities laws. PlaintiffThe lawsuit seeks certification as a class action (for a purported class of purchasers of the Company’s securities from March 27, 2019, through November 8, 2019), compensatory damages ofin an unspecified amount, pre-judgment and post-judgment interest, reasonable attorneys’ fees, expert fees, and unspecified other costs, as well as any further relief the court deems just and proper.equitable or injunctive relief. We filed a motion to dismiss the Amended Complaint on June 12, 2017, in compliance with the scheduling order entered by the court on December 20, 2016. Oral arguments on the motion to dismiss were held on October 24, 2017, and the judge denied our motion to dismiss. On February 15, 2018 we and the other defendants entered into a memorandum of understanding to settle the purported securities class action litigation. The memorandum of understanding contemplateshave insurance that the parties will enter into a settlement agreement, which, if entered into, will be subject to customary conditions including court approval following notice to our stockholders, and a hearing at which time the court will consider the fairness, reasonableness and adequacy of the settlement. If a settlement is finally approved by the court, it will resolve all of the claims that were or could have been brought in the action being settled. We continue to believe that the claims in the lawsuits are without merit and, to the extent the parties do not enter into a settlement agreement or the court does not approve a settlement, will defend against them vigorously. We maintain insurance forcovers claims of this nature, which management believesnature. The retention amount payable by us under our policy is adequate. Moreover, we believe, based on information currently available, that$1.25 million. On April 14, 2023, a judgment was issued ordering the filingcase dismissed with prejudice and ultimate outcomeclosure of the lawsuits will not have a material impact on our financial position, although we will have to pay up to the insurance retention amount in connection with the lawsuit.action.
ITEM 4. MINE SAFETY DISCLOSURES
Not Applicable.
Not Applicable.
ITEM 5. MARKET FOR THE REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is quoted on The NASDAQ Capital Market.Market under the symbol “LPCN”.
The following table sets forth, for the periods indicated, the high and low sales prices for our common stock, as reported on The NASDAQ Capital Market.Holders
| High | Low | |||||||
| 2016 | ||||||||
| First Quarter | $ | 12.99 | $ | 7.90 | ||||
| Second Quarter | 12.66 | 2.51 | ||||||
| Third Quarter | 4.93 | 2.96 | ||||||
| Fourth Quarter | 5.90 | 3.03 | ||||||
| 2017 | ||||||||
| First Quarter | $ | 4.86 | $ | 3.25 | ||||
| Second Quarter | 5.14 | 3.33 | ||||||
| Third Quarter | 5.33 | 3.52 | ||||||
| Fourth Quarter | 4.38 | 3.34 | ||||||
Holders
As of March 9, 2018,5, 2024, there were approximately 10987 holders of record of our common stock. This number does not include an undetermined number of stockholders whose stock is held in “street” or “nominee” name.
Performance Graph and TableDividends
The following graph shows a comparison from October 21, 2013 (the date of Lipocine Inc.’s Exchange Act registration through the filing of a Form 8-A) through December 31, 2017 of the cumulative total return for (i) our ordinary shares, (ii) the NASDAQ Composite Index, (iii) the NASDAQ Biotechnology Index and (iv) the NYSE Pharmaceutical Index.
The graph assumes an initial investment of $100 on October 21, 2013. The comparisons in the graph are required by the Securities and Exchange Commission and are not intended to forecast or be indicative of possible future performance of our ordinary shares.
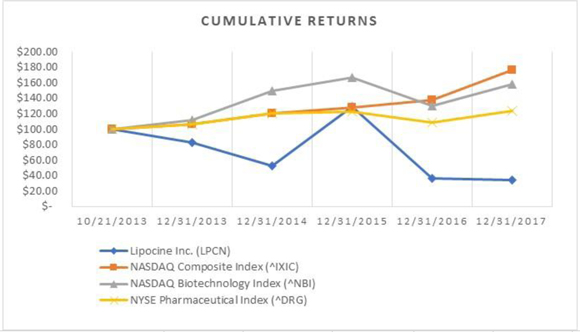
| Cumulative Returns | ||||||||||||||||||||||||
| 10/21/2013 | 12/31/2013 | 12/31/2014 | 12/31/2015 | 12/31/2016 | 12/31/2017 | |||||||||||||||||||
| Lipocine Inc. (LPCN) | $ | 100.00 | $ | 82.50 | $ | 52.60 | $ | 129.30 | $ | 36.80 | $ | 34.40 | ||||||||||||
| NASDAQ Composite Index (^IXIC) | 100.00 | 106.54 | 120.82 | 127.74 | 137.32 | 176.10 | ||||||||||||||||||
| NASDAQ Biotechnology Index (^NBI) | 100.00 | 111.40 | 149.38 | 166.45 | 130.35 | 157.80 | ||||||||||||||||||
| NYSE Pharmaceutical Index (^DRG) | 100.00 | 105.94 | 120.59 | 122.55 | 108.99 | 123.34 | ||||||||||||||||||
The foregoing graph and table are furnished solely with this report, and are not filed with this report, and shall not be deemed incorporated by reference into any other filing under the Securities Act of 1933, as amended, or the Securities Act, or the Securities Exchange Act of 1934, as amended, whether made by us before or after the date hereof, regardless of any general incorporation language in any such filing, except to the extent we specifically incorporate this material by reference into any such filing.
Dividends
Although the board of directors of Marathon Bar declared a cash dividend to its stockholders of record in connection with the Merger, weWe do not anticipate paying any further cash dividends on our common stock in the foreseeable future. We intend to retain any future earnings to finance growth and development and therefore do not anticipate paying cash dividends in the foreseeable future.
The selected statement of operations comprehensive loss data for the years ended December 31, 2017, 2016 and 2015, and the balance sheet data as of December 31, 2017 and 2016 have been derived from our audited financial statements included elsewhere in this Annual Report on Form 10-K. The selected statement of operations and comprehensive income (loss) data for the years ended December 31, 2014 and 2013, and the balance sheet data as of December 31, 2015, 2014 and 2013 have been derived from audited financial statements which are not included in this Annual Report on Form 10-K.ITEM 6.RESERVED
Our historical results are not necessarily indicative of future results. The selected financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes included elsewhere in this Annual Report on Form 10-K.
| Years Ended December 31, | ||||||||||||||||||||
| 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||||
| Selected Balance Sheet Data | ||||||||||||||||||||
| Cash, cash equivalents and marketable investment securities | $ | 21,468,070 | $ | 26,840,286 | $ | 44,382,827 | $ | 27,666,055 | $ | 45,263,698 | ||||||||||
| Total assets | 25,325,114 | 27,342,970 | 45,377,278 | 27,993,502 | 46,107,522 | |||||||||||||||
| Total liabilities | 6,345,126 | 1,326,169 | 3,391,861 | 1,633,532 | 1,283,775 | |||||||||||||||
| Accumulated deficit | (126,399,823 | ) | (105,416,963 | ) | (86,445,455 | ) | (68,237,077 | ) | (47,864,401 | ) | ||||||||||
| Shareholders' equity | 18,979,988 | 26,016,801 | 41,985,417 | 26,539,970 | 44,823,747 | |||||||||||||||
| Years Ended December 31, | ||||||||||||||||||||
| 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||||
| Selected Statement of Operations and Loss Data | ||||||||||||||||||||
| Operating loss | $ | (21,217,976 | ) | $ | (19,186,834 | ) | $ | (18,382,068 | ) | $ | (20,480,814 | ) | $ | (10,683,630 | ) | |||||
| Net loss | (20,982,860 | ) | (18,971,508 | ) | (18,208,378 | ) | (20,372,676 | ) | (10,590,106 | ) | ||||||||||
| Net loss per share - basic | (1.05 | ) | (1.04 | ) | (1.11 | ) | (1.60 | ) | (1.44 | ) | ||||||||||
| Net loss per share - diluted | (1.05 | ) | (1.04 | ) | (1.11 | ) | (1.60 | ) | (1.44 | ) | ||||||||||
ITEM 7.MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and the related notes thereto and other financial information included elsewhere in this report.
As used in the discussion below, “we,” “our,” and “us” refers to the historical financial results of Lipocine.
Forward Looking Statements
This section and other parts of this report contain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, that involve risks and uncertainties. Forward-looking statements provide current expectations of future events based on certain assumptions and include any statement that does not directly relate to any historical or current fact. Forward-looking statements may refer to such matters as products, product benefits, pre-clinical and clinical development timelines, clinical and regulatory expectations and plans, anticipated financial performance, future revenues or earnings, business prospects, projected ventures, new products and services, anticipated market performance, future expectations for liquidity and capital resources needs and similar matters. Such words as “may”, “will”, “expect”, “continue”, “estimate”, “project”, and “intend” and similar terms and expressions are intended to identify forward looking statements. Forward-looking statements are not guarantees of future performance and our actual results may differ significantly from the results discussed in the forward-looking statements. Factors that might cause such differences include, but are not limited to, those discussed in Part I, Item 1A (Risk Factors) of this Form 10-K. Except as required by applicable law, we assume no obligation to revise or update any forward-looking statements for any reason.
Overview of Our Business
We are a specialty pharmaceuticalbiopharmaceutical company focused on applyingleveraging our proprietary Lip’ral platform to develop differentiated products through the oral drug delivery technology for the development of pharmaceutical products in the area of men’s and women’s health.previously difficult to deliver molecules, focused on treating Central Nervous System (“CNS”) disorders. Our proprietary delivery technologies are designed to improve patient compliance and safety through orally available treatment options. Our primary development programs are based on oral delivery solutions for poorly bioavailable drugs. We have a portfolio of proprietarydifferentiated innovative product candidates designed to produce favorable pharmacokinetic (“PK”) characteristicsthat target high unmet needs for neurological and facilitate lower dosing requirements, bypass first-pass metabolism in certain cases, reduce side effects,psychiatric CNS disorders, liver diseases, and eliminate gastrointestinal interactions that limit bioavailability. Our leadhormone supplementation for men and women.
We entered into a license agreement for the development and commercialization our product candidate, TLANDO™TLANDO®, is an oral testosterone replacement therapy (“TRT”) and is currently under review by the United States Food and Drug Administrationcomprised of testosterone undecanoate (“FDA”) with a Prescription Drug User Fee Act (“PDUFA”) action goal date of May 8, 2018. The FDA has deemed the resubmission a complete response to its June 2016 Complete Response Letter (“CRL”) that requested additional information related to the dosing algorithm for the proposed label. The TLANDO New Drug Application (“NDA”) is based on the results of the Dosing Validation (“DV”)study. The DV study confirmed the efficacy of TLANDO with a fixed dose regimen without need for dose adjustment. TLANDO was well tolerated upon 52-week exposure with no reports of drug related Serious Adverse Events (“SAEs”TU”). On January 10, 2018,12, 2024, we entered into a license agreement (the “Verity License Agreement”) with Verity Pharmaceuticals, Inc. (“Verity” or our “Licensee”), pursuant to which we granted to Verity an exclusive, royalty-bearing, sublicensable right and license to develop and commercialize the Bone, ReproductiveTLANDO product for TRT in the U.S. and Urologic Drugs Advisory Committee (“BRUDAC”)Canada. Any FDA required post-marketing studies will also be the responsibility of our Licensee, Verity. On March 28, 2022, the FDA voted six in favor and thirteen against the acceptability of the overall benefit/risk profile to support approval ofapproved TLANDO as a TRT. We continue to workTRT in adult males for conditions associated with a deficiency of endogenous testosterone, also known as hypogonadism. On June 7, 2022, our former commercial partner Antares (a wholly owned subsidiary of Halozyme) announced the FDAcommercial launch of TLANDO, an oral treatment indicated for testosterone replacement therapy in addressing topics discussed by BRUDAC. We may receive another CRL. This would cause delays and added expense to the processadult males for conditions associated with a deficiency or absence of seeking approval of TLANDO. endogenous testosterone (primary or hypogonadotropic hypogonadism).
| 53 |
Additional clinical development pipeline candidates includeinclude: LPCN 1111,1154 for postpartum depression (“PPD”); LPCN 2101 for epilepsy; and LPCN 2203 for essential tremor. In addition to our CNS product candidates, we have assets for which we expect to seek partnerships to enable further development including TLANDO for territories outside of North America, LPCN 1148 comprising a next generationnovel prodrug of testosterone, and testosterone laurate (“TL”), for the management of decompensated cirrhosis, LPCN 1144, an oral testosterone therapy product withprodrug of androgen receptor modulator for the potential for once daily dosing, that is currently intreatment of non-cirrhotic non-alcoholic steatohepatitis (“NASH”) which has completed Phase 2 testing,testing; and LPCN 1107, which has the potential to becomepotentially the first oral hydroxyprogesteronehydroxy progesterone caproate (“HPC”) product indicated for the prevention of recurrent preterm birth (“PTB”), which has completed a dose finding clinical study in pregnant women and has completed an End-of-Phase 2 meeting withbeen granted orphan drug designation by the FDA.
To date, we have funded our operations primarily through the sale of equity securities, debt and convertible debt and through up-front payments, research funding and royalty and milestone payments from our license and collaboration arrangements. We have not generated any revenues from product sales and while we expect to generate royalties from our Licensee’s sales of TLANDO, we do not expect to generate revenue from product sales from our other product candidates unless and until we obtain regulatory approval of TLANDO or other products.approval.
We have incurred losses in most years since our inception. As of December 31, 2017,2023, we had an accumulated deficit of $126.4$199.8 million. Income and losses fluctuate year to year, primarily depending on the nature and timing of recognition of revenues fromresearch and development occurring on our license and collaboration agreements.product candidates. Our net loss was $21.0$16.4 million for the year ended December 31, 2017,2023, compared to $19.0$10.8 million for the year ended December 31, 2016.2022. Substantially all of our operating losses resulted from expenses incurred in connection with our product candidate development programs, our research activities and general and administrative costs associated with our operations.
We expect to continue to incur significant expenses and operating losses for the foreseeable future as we:
| ● | subject to resource availability, conduct further development of our other product candidates, including LPCN 1154, LPCN 2101, LPCN 2203, and LPCN 1148; | |
| ● | continue our research efforts; | |
| ● | research new products or new uses for our existing products; | |
| ● | maintain, expand and protect our intellectual property portfolio; and | |
| ● | provide general and administrative support for our operations. |
To fund future long-term operations, including the potential commercialization of any of our product candidates, we will need to raise additional capital. The amount and timing of future funding requirements will depend on many factors, including capital market conditions, regulatory requirements and outcomes related tocommercial success of TLANDO, including our ABPM clinical study, regulatory requirements related to our other product development programs, the timing and results of our ongoing development efforts, the potential expansion of our current development programs, potential new development programs, our ability to license and/or partner our products to third parties, the pursuit of various potential commercial activities and strategies associated with our development programs and related general and administrative support. We anticipate that we will seek to fund our operations through public or private equity or debt financings or other sources, such as potential license, partnering and collaboration agreements. We cannot be certain that anticipated additional financing will be available to us on favorable terms, in amounts sufficient to fund our operations, or at all. Although we have previously been successful in obtaining financing through public and private equity securities offerings and our license and collaboration agreements, there can be no assurance that we will be able to do so in the future.
Corporate Strategy
Our Product Candidatesgoal is to become a leading biopharmaceutical company focused on leveraging our proprietary Lip’ral drug delivery technology platform to develop differentiated products through oral delivery of previously difficult to deliver molecules for CNS disorders. The key components of our strategy are to:
Advance LPCN 1154 and other CNS product candidates. We intend to focus on the development of endogenous neuroactive steroids (“NAS”) which have broad applicability in treating various CNS conditions where we can leverage our technology platform to develop highly differentiated oral therapeutics. Our current portfolio, shown below, includes our lead product candidate, TLANDO, anpriority is on the development of LPCN 1154, a fast-acting oral testosterone replacement therapy that is currently under review by the FDAantidepressant for postpartum depression (“PPD”) with a PDUFA goal date of May 8, 2018. Additionally, we are in the process of establishing our pipeline of other clinical candidates including a next generation potential once daily oral testosterone replacement therapy, LPCN 1111, and an oral therapy for the prevention of preterm birth, LPCN 1107.outpatient use.
| 54 |
Our Development PipelineSupport our Licensee in commercialization of our licensed oral TRT option. We believe the TRT market needs a differentiated, convenient oral option. We have exclusively licensed rights to TLANDO to Verity for commercialization of TLANDO in the U.S. and Canada. We plan to support our Licensee’s efforts to effectively enable the availability of TLANDO to patients in a timely manner, in addition to receiving milestone and royalty payments associated with TLANDO commercialization as agreed to in the Verity License Agreement.
TLANDO: An Oral Product CandidateDevelop partnership(s) to continue the advancement of non-core pipeline assets. We continuously strive to prioritize our resources in seeking partnerships of our pipeline assets. We are currently exploring partnering of our liver programs LPCN 1144, our candidate for Testosterone Replacement Therapy
Our lead product,treatment of non-cirrhotic NASH and LPCN 1148 for the management of decompensated cirrhosis, and LPCN 1107, our candidate for prevention of pre-term birth. We are exploring the possibility of licensing LPCN 1021 (known as TLANDO is an oral formulation ofin the chemical, testosterone undecanoate (“TU”), an eleven carbon side chain attachedUnited States) and LPCN 1111 to testosterone. TU is an ester prodrug of testosterone. An ester is chemically formed by bonding an acid and an alcohol. Upon the cleavage, or breaking, of the ester bond, testosterone is formed. TU has been approved for usethird parties outside the United States for many years for delivery via intra-muscular injection and in oral dosage form and recently TUCanada, although no licensing agreement has received regulatory approval in the United States for delivery via intra-muscular injection. We are using our proprietary technology to facilitate steady gastrointestinal solubilization and absorption of TU. Proof of concept was initially established in 2006, and subsequently TLANDO was licensed in 2009 to Solvay Pharmaceuticals, Inc. which was then acquired by Abbott Products, Inc. ("Abbott"). Following a portfolio review associated with the spin-off of AbbVie by Abbott in 2011, the rights to TLANDO were reacquired by us. All obligations under the prior license agreement have been completed except that Lipocine will owe Abbott a perpetual 1% royalty on net sales. Such royalties are limited to $1 million in the first two calendar years following product launch, after which period there is not a cap on royalties and no maximum aggregate amount. If generic versions of any such product are introduced, then royalties are reduced by 50%.
NDA Resubmission
We resubmitted our NDA to the FDA in August 2017 based on the results of the DV study. The DV study confirmed the efficacy of TLANDO with a fixed dose regimen without need for dose adjustment. TLANDO was well tolerated upon 52-week exposure with no reports of drug related Serious Adverse Events (“SAEs”). The FDA accepted our NDA as a complete response to their CRL and assigned a PDUFA goal date of May 8, 2018 for completion of the review. On January 10, 2018, the BRUDAC of the FDA voted six in favor and thirteen against the benefit/risk profile of TLANDO. Discussion topics during the BRUDAC included whether the safety of TLANDO is adequately characterized and whether additional data is need pre-approval or post approval, including the need for an ambulatory blood pressure (“ABPM”) study. Additional areas discussed included the potential to increase cardiovascular risk, lipid changes, hematocrit increase, levels of dihydro-testosterone and estradiol, cosynotropin stimulation results, Cmax excursions, the stopping criteria for use in clinical practice, and whether testosterone concentrations measured in serum tubes are reliable in patients treated with TLANDO. Particularly the FDA may be concerned with TLANDO’s potential for increased adverse cardiovascular outcomes in the population that will likely use the drug, if approved, and observed treatment emergent adverse reactions. We continue to work with the FDA in addressing topics discussed by BRUDAC. The FDA may or may not view BRUDAC’s TLANDO advice/recommendation similarly. We have submitted two protocols to the FDA under our TLANDO investigational new drug (“IND”) application. The first protocol is for the conduct of an ABPM clinical study and the second protocol is for a phlebotomy clinical study to confirm no ex-vivo conversion of TU to T. We plan to initiate both clinical studies ahead of our PDUFA date. Although there is no guarantee of FDA approval of TLANDO, we believe the results from the DV study confirm the validity of a fixed dose approach without the need for dose titration to orally administering TLANDO. However, approval of our NDA on our PDUFA date is not guaranteed and we may receive another CRL. Receipt of another CRL would result in substantial delays which may include additional studies and expense before we would be in a position to re-submit an NDA responsive to such additional CRL.
Previously on June 28, 2016, we received a CRL from the FDA on our original NDA submission. A CRL is a communication from the FDA that informs companies that an application cannot be approved in its present form. The CRL identified deficiencies related to the dosing algorithm for the label. Specifically, the proposed titration scheme for clinical practice was significantly different from the titration scheme used in the Phase 3 trial leading to discordance in titration decisions between the Phase 3 trial and real-world clinical practice. In response to the CRL, we met with the FDA in a Post Action meeting and proposed a dosing regimen to the FDA based on analyses of existing data. The FDA noted that while the proposed dosing regimen might be acceptable, validation in a clinical trial would be needed prior to resubmission. The DV study was in response to the FDA’s request. We also initiated the Dosing Flexibility (“DF”) study to assess TLANDO in hypogonadal males on a fixed daily dose of 450 mg dividedentered into three equal doses.
Results from DV and DF Studies
The DV and DF studies were both an open-label, fixed dose (no titration), single treatment clinical study of oral TRT in hypogonadal males with low testosterone (T) (< 300 ng/dL) that assessed TLANDO in hypogonadal males on a fixed daily dose of 450 mg divided into two equal doses (“BID”) in the DV study and into three equal doses (“TID”) in the DF study. In total, 95 and 100 subjects were enrolled into DV and DF studies, respectively, with 94 and 98 subjects completing the DV and DF studies, respectively.
On June 19, 2017, we announced top-line results of the DV and DF studies. Although there is no guarantee of FDA approval of TLANDO, we believe the results from the DV study confirm the validity of a fixed dose approach without the need for dose titration to orally administering TLANDO. The DV study will be considered our pivotal efficacy clinical study for the NDA resubmission. TLANDO successfully met the FDA primary efficacy guidelines in the DV study safety statistical analysis set (“SS”) where 80% of the subjects achieved average testosterone levels (“Cavg”) within the normal range with a lower bound confidence interval (“CI”) of 72%. The DF study restored 70% of the subjects’ average testosterone levels within the normal range (Cavg) confirming that twice daily (“BID”) dosing is the appropriate dosing regimen for TLANDO and was the basis for resubmission. The safety set is defined as any subject that was randomized into the study and took at least one dose (N=95 subjects in the DV study and N=100 in the DF study). A baseline carried forward approach was used to account for missing data as a result of subject discontinuation.
The primary efficacy endpoint is the percentage of subjects with Cavg within the normal range, which is defined as 300-1080 ng/dL. The FDA guidelines for primary efficacy success is that at least 75% of the subjects on active treatment achieve a testosterone Cavg within the normal range; and the lower bound of the 95% CI must be greater than or equal to 65%.
The adverse event profile of TLANDO in both the DV and DF studies was consistent with the previously conducted 52-week Phase 3 Study of Androgen Replacement (“SOAR”) clinical trial. All drug related adverse events (“AEs”) were either mild or moderate in intensity and none were severe. To date, the safety database of TLANDO includes ~525 unique hypogonadal men demonstrating a profile consistent with other TRT products.
The secondary endpoints assessed the maximum total testosterone concentration (“Cmax”) post dosing using predetermined limits developed by the FDA for transdermals. The FDA guidelines for secondary efficacy success is that at least 85% of the subjects achieve Cmax less than 1500 ng/dL; no greater than 5% of the subjects have Cmax between 1800 ng/dl and 2500 ng/dL; and zero percent of the subjects have Cmax greater than 2500 ng/dL. Consistent with the definition of Cmax and the pharmacokinetic profile of multiple times a day dosing, two pre-specified analyses were performed, Cmax per dose and Cmax per day.Company.
In the DV study SS Cmax per dose analysis, the percentage of subjects with Cmax less than 1500 ng/dL and between 1800 ng/dL and 2500 ng/dL were 85% and 7%, respectively. Deviations from the predetermined limits in the DV study were observed in the Cmax per day dose analysis for these thresholds. Only one subject, who was a major protocol violator, exceeded the 2500 ng/dL limit independent of per dose or per day dose analyses.
The DF study SS met all Cmax thresholds in per dose and per day dose analyses.
Prior to conducting the DV study and the DF study, we completed our SOAR pivotal Phase 3 clinical study evaluating efficacy and 52-week safety of TLANDO. The SOAR study is considered our pivotal safety clinical study for the NDA resubmission.
Results from SOAR
SOAR was a randomized, open-label, parallel-group, active-controlled, Phase 3 clinical study of TLANDO in hypogonadal males with low testosterone (< 300 ng/dL). In total, 315 subjects at 40 active sites were assigned, such that 210 were randomized to TLANDO and 105 were randomized to the active control, AndroGel 1.62%®, for 52 weeks of treatment. The active control is included for safety assessment. TLANDO subjects were started at 225 mg TU (equivalent to ~ 142 mg of T) twice daily (“BID”) with a standard meal and then dose titrated, if needed, based on average T levels during the day, Cavg, and peak serumT levels, Cmax, up to 300 mg TU BID or down to 150 mg TU BID based on serum testosterone measured at weeks 3 and 7 based on PK profile with multiple blood samples drawn at each time period. The mean age of the subjects in the trial was ~53 years with ~91% of the patients < 65 years of age. The discontinuation rate for TLANDO was 38% compared to 32% for AndroGel 1.62%.
Primary statistical analysis was conducted using the Efficacy Population Set ("EPS"). The EPS is defined as subjects randomized into the study with at least one PK profile and no significant protocol deviations and includes imputed missing data by last observation carried forward, N=151. Further analysis was performed using the full analysis set ("FAS") (any subject randomized into the study with at least one post-baseline efficacy variable response, N=193) and the SS (any subject that was randomized into the study and took at least one dose, N=210).
Safety
The safety component of the SOAR trial was completed the last week of April 2015. The safety extension phase was designed to assess safety based on information such as metabolites, biomarkers, laboratory values, serious adverse events ("SAEs") and AEs, with subjects on their stable dose regimen in both the treatment arm and the active control arm. TLANDO treatment was well tolerated in that there were no hepatic, cardiac or drug related SAEs.
TLANDO safety highlights include:
Food Effect Study
We also completed our labeling "food effect" study in May 2015. Results from the labeling "food effect" study indicate that bioavailability of testosterone from TLANDO is not affected by changes in meal fat content. The results demonstrate comparable testosterone levels between the standard fat meal (similar to the meal instruction provided in the Phase 3 clinical study) and both the low and high fat meals. The labeling “food effect” study was conducted per the FDA requirement and we submitted preliminary results from this study to the FDA in the second quarter of 2015 prior to submitting the NDA.
Other Safety Requirements
Based on our meetings with the FDA, we do not expect to be required to conduct a heart attack and stroke risk study or any additional safety studies prior to the potential approval of our NDA for TLANDO. We may, however, be required to conduct a heart attack and stroke risk study on our own or with a consortium of sponsors that have an approved TRT product subsequent to the potential approval of TLANDO.
LPCN 1111: A Next-Generation Oral Product Candidate for TRT
LPCN 1111 is a next-generation, novel ester prodrug of testosterone which uses the Lip’ral technology to enhance solubility and improve systemic absorption. We completed a Phase 2b dose finding study in hypogonadal men in the third quarter of 2016. The primary objectives of the Phase 2b clinical study were to determine the starting Phase 3 dose of LPCN 1111 along with safety and tolerability of LPCN 1111 and its metabolites following oral administration of single and multiple doses in hypogonadal men. The Phase 2b clinical trial was a randomized, open label, two-period, multi-dose PK study that enrolled hypogonadal males into five treatment groups. Each of the 12 subjects in a group received treatment for 14 days. Results of the Phase 2b study suggest that the primary objectives were met, including identifying the dose expected to be tested in a Phase 3 study. Good dose-response relationship was observed over the tested dose range in the Phase 2b study. Additionally, the target Phase 3 dose met primary and secondary end points. Overall, LPCN 1111 was well tolerated with no drug-related severe or serious adverse events reported in the Phase 2b study.
Additionally in October 2014, we completed a Phase 2a proof-of-concept study in hypogonadal men. The Phase 2a open-label, dose-escalating single and multiple dose study enrolled 12 males. Results from the Phase 2a clinical study demonstrated the feasibility of a once daily dosing with LPCN 1111 in hypogonadal men and a good dose response. Additionally, the study confirmed that steady state is achieved by day 14 with consistent inter-day performance observed on day 14, 21 and 28. No subjects exceeded Cmax of 1500 ng/dL at any time during the 28-day dosing period on multi-dose exposure. Overall, LPCN 1111 was well tolerated with no serious AE’s reported.
We have also completed a preclinical toxicology study with LPCN 1111 in dogs. In February 2018 we had a meeting with the FDA to discuss these preclinical results and to discuss the Phase 3 clinical study and path forward for LPCN 1111. Based on the results of the FDA meeting, additional pre-clinical or clinical trials may be required before a Phase 3 clinical study can be initiated. Additionally the FDA requested that an ABPM clinical study be conducted. Based on our capital resources and the clinical status of our product candidates, we will primarily focus our efforts in 2018 on TLANDO. We do not anticipate the initiation of a Phase 3 study with LPCN 1111 to occur in 2018 unless and until additional capital is secured or the product candidate is out-licensed.
LPCN 1107: An Oral Product Candidate for the Prevention of Preterm Birth
We believe LPCN 1107 has the potential to become the first oral hydroxyprogesterone caproate (“HPC”) product indicated for the reduction of risk of preterm birth (“PTB”) in women with singleton pregnancy who have a history of singleton spontaneous PTB. Prevention of PTB is a significant unmet need as ~11.7% of all U.S. pregnancies result in PTB (delivery less than 37 weeks), a leading cause of neonatal mortality and morbidity.
We have completed a multi-dose PK dose selection study in pregnant women. The objective of the multi-dose PK selection study was to assess HPC blood levels in order to identify the appropriate LPCN 1107 Phase 3 dose. The multi-dose PK dose selection study was an open-label, four-period, four-treatment, randomized, single and multiple dose, PK study in pregnant women of three dose levels of LPCN 1107 and the injectable intramuscular ("IM") HPC (Makena®). The study enrolled 12 healthy pregnant women (average age of 27 years) with a gestational age of approximately 16 to 19 weeks. Subjects received three dose levels of LPCN 1107 (400 mg BID, 600 mg BID, or 800 mg BID) in a randomized, crossover manner during the first three treatment periods and then received five weekly injections of HPC during the fourth treatment period. During each of the LPCN 1107 treatment periods, subjects received a single dose of LPCN 1107 on Day 1 followed by twice daily administration from Day 2 to Day 8. Following completion of the three LPCN 1107 treatment periods and a washout period, all subjects received five weekly injections of HPC. Results from this study demonstrated that average steady state HPC levels (Cavg0-24) were comparable or higher for all three LPCN 1107 doses than for injectable HPC. Additionally, HPC levels as a function of daily dose were linear for the three LPCN 1107 doses. Also unlike the injectable HPC, steady state exposure was achieved for all three LPCN 1107 doses within seven days. We have also completed a proof-of-concept Phase 1b clinical study of LPCN 1107 in healthy pregnant women in January 2015 and a proof-of-concept Phase 1a clinical study of LPCN 1107 in healthy non-pregnant women in May 2014. These studies were designed to determine the PK and bioavailability of LPCN 1107 relative to an IM HPC, as well as safety and tolerability.
A traditional pharmacokinetics/pharmacodynamics (“PK/PD”) based Phase 2 clinical study in the intended patient population is not expected to be required prior to entering into Phase 3. Therefore, based on the results of our multi-dose PK study we had an End-of-Phase 2 meeting with the FDA as well as other guidance meetings with the FDA to define a Phase 3 development plan for LPCN 1107. During the End-of-Phase 2 meeting and subsequent guidance meetings, the FDA agreed to a randomized, open-label, two-arm clinical study to include a LPCN 1107 arm and a comparator IM arm with treatment up to 23 weeks. The FDA also provided preliminary feedback on other critical Phase 3 study design considerations including: positive feedback on the proposed 800 mg BID Phase 3 dose and dosing regimen; confirmation of the use of a surrogate primary endpoint focusing on rate of delivery less than 37 weeks gestation rather on clinical infant outcomes; acknowledged that the use of a gestational age endpoint would likely lead to any FDA approval, if granted, being a Subpart H approval; and, recommended a non-inferiority study margin of 7% with interim analyses. A standard statistical design for a NI study based on the FDA feedback, a NI margin of 7% for the primary endpoint may require ~1,100 subjects per treatment arm with a 90% power. However, based on the FDA’s suggestion of including an interim analysis in the NI design, an adaptive study design is under consideration that may allow for fewer subjects. We submitted the initial LPCN 1107 Phase 3 protocol to the FDA via a SPA in June 2017 and have received multiple rounds of FDA’s feedback. Agreement with the FDA on the Phase 3 protocol via SPA has not occurred and will not occur until results from a planned food-effect study with LPCN 1107 are reviewed by the FDA. Final agreement with the FDA on the Phase 3 protocol, if reached, may or may not confirm the FDA’s preliminary feedback on the Phase 3 design. Additionally, manufacturing scale-up work for LPCN 1107 has been completed. Based on our capital resources and the clinical status of our product candidates, we plan to primarily focus our efforts in 2018 on TLANDO. We do not anticipate the initiation of a Phase 3 study with LPCN 1107 to occur in 2018 unless and until additional capital is secured or the product candidate is out-licensed.
The FDA has granted orphan drug designation to LPCN 1107 based on a major contribution to patient care. Orphan designation qualifies Lipocine for various development incentives, including tax credits for qualified clinical testing, and a waiver of the prescription drug user fee when we file our NDA.
Financial Operations Overview
Revenue
Revenue
To date, we have not generated any revenues from product sales and do not expect to do so until one of our product candidates receives approval from the FDA. Revenues to date have been generated substantially from license fees, royalty and milestone payments and research support from our licensees. Since our inception through December 31, 2017,2023, we have generated $27.5$41.9 million in revenue under our various license and collaboration arrangements and from government grants. We have entered into the Verity License Agreement with the potential for revenue from future milestones and royalties, but we may never generate revenues from TLANDO or any of our other clinical or preclinical development programs or licensed products as we may never succeed in obtaining regulatory approval or commercializing any of these product candidates.
Research and Development Expenses
Research and development expenses consist primarily of salaries, benefits, stock-based compensation and related personnel costs, fees paid to external service providers such as contract research organizations and contract manufacturing organizations, contractual obligations for clinical development, clinical sites, manufacturing and scale-up for late-stage clinical trials, formulation of clinical drug supplies, and expenses associated with regulatory submissions. Research and development expenses also include an allocation of indirect costs, such as those for facilities, office expense travel, and depreciation of equipment based on the ratio of direct labor hours for research and development personnel to total direct labor hours for all personnel. We expense research and development expenses as incurred. Since our inception, we have spent approximately $97.2$147.2 million in research and development expenses through December 31, 2017.2023.
We expectOn January 12, 2024, we entered into the Verity License Agreement, pursuant to incur approximately $3.9 millionwhich we granted to Verity an exclusive, royalty-bearing, sublicensable right and license to develop and commercialize our TLANDO product for TRT in additional researchthe U.S. and developments costs forCanada. Any FDA required post-marketing studies will also be the responsibility of our Licensee, Verity. On March 28, 2022, TLANDO as we complete the ABPM and phlebotomy clinical studies. However, these expenditures are subject to numerous uncertainties regarding timing and cost to completion.
Approval, if ever, of TLANDO will require approvalwas approved by the FDA as a TRT in adult males for conditions associated with a deficiency of endogenous testosterone, also known as hypogonadism. On January 31, 2024, our license agreement with former licensee, Antares Pharma, Inc. (“Antares”), was terminated and the transition of the resubmitted NDA,U.S. commercial rights for TLANDO from Antares to Verity was completed on February 1, 2024, for the distribution, marketing and wesale of TLANDO. The Verity License Agreement also provides Verity with a license to develop and commercialize TLANDO XR (LPCN 1111), the Company’s potential next generation, once daily oral product candidate for testosterone replacement therapy comprised of testosterone tridecanoate (“TT”), in the U.S. and Canada.
We expect to continue to incur significant costs as we seek approvaldevelop our other product candidates, including our CNS product candidates as well as the development of TLANDO.any future pipeline product candidates.
In general, the cost of clinical trials may vary significantly over the life of a project as a result of uncertainties in clinical development, including, among others:
| ● | the number of sites included in the trials; | |
| ● | the length of time required to enroll suitable subjects; |
| 55 |
| ● | the duration of subject follow-ups; | |
| ● | the length of time required to collect, analyze and report trial results; | |
| ● | the cost, timing and outcome of regulatory review; and | |
| ● | potential changes by the FDA in clinical trial and NDA filing requirements. |
Future research and report trial results;
We also incurred significant manufacturing costs to prepare launch supplies for TLANDO and expect to incur additional manufacturing costs related to TLANDO. However, thesedevelopment expenditures are subject to numerous uncertainties regarding timing and cost to completion, including, among others:
| ● | the timing and outcome of regulatory filings and FDA reviews and actions for product candidates; | |
| ● | our dependence on third-party manufacturers for the production of satisfactory finished product for registration and launch should regulatory approval be obtained on any of our product candidates; | |
| ● | the potential for future license or co-promote arrangements for our product candidates, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our future plans and capital requirements; and | |
| ● | the effect on our product development activities of actions taken by the FDA or other regulatory authorities. |
A change of outcome for any of these variables with respect to the development of TLANDOour product development candidates could mean a substantial change in the costs and timing associated with these efforts, willcould require us to raise additional capital, and may require us to reduce operations.
Given the stage of clinical development and the significant risks and uncertainties inherent in the clinical development, manufacturing and regulatory approval process, we are unable to estimate with any certainty the time or cost to complete the development of LPCN 1154, LPCN 2101, LPCN 2203, LPCN 1148, LPCN 1144, LPCN 1111, LPCN 1107 and other product candidates. Clinical development timelines, the probability of success and development costs can differ materially from expectations and results from our clinical trials may not be favorable. If we are successful in progressing LPCN 1111,1154, LPCN 11072101, LPCN 2203 or other future product candidates into later stage development, we will require additional capital. The amount and timing of our future research and development expenses for these product candidates will depend on the preclinicalpre-clinical and clinical success of both our current development activities and potential development of new product candidates, as well as ongoing assessments of the commercial potential of such activities. We will continue efforts to enter into partnership arrangements for the continued development and/or marketing of LPCN 1144, LPCN 1148, LPCN 1107 and for TLANDO and LPCN 1111 outside of the U.S. and Canada.
Summary of Research and Development Expense
We are conducting on-going clinical and regulatory activities with all three of our product candidates. Additionally, we incur costs for our other research programs. The following table summarizes our research and development expenses:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| External service provider costs: | ||||||||||||
| TLANDO | $ | 7,354,321 | $ | 3,492,422 | $ | 8,265,031 | ||||||
| LPCN 1111 | 326,602 | 1,706,637 | 848,743 | |||||||||
| LPCN 1107 | 803,679 | 268,470 | 861,847 | |||||||||
| Other product candidates | - | 30,000 | 54,716 | |||||||||
| Total external service provider costs | 8,484,602 | 5,497,529 | 10,030,337 | |||||||||
| Internal personnel costs | 2,092,217 | 2,036,773 | 2,032,501 | |||||||||
| Other research and development costs | 427,462 | 541,751 | 517,407 | |||||||||
| Total research and development | $ | 11,004,281 | $ | 8,076,053 | $ | 12,580,245 | ||||||
We expect research and development expenses to decreaseincrease in 2018the future as cost of conducting the ABPM and phlebotomywe complete on-going clinical studies, including the studies for TLANDO in 2018 are less than the cost of conducting the DVour CNS product candidates and DFas we conduct future clinical studies, for TLANDOincluding when and if we conduct Phase 2 clinical studies with our development product candidates and when and if we conduct Phase 3 clinical studies with LPCN 1144, LPCN 1148, and LPCN 1107. We are exploring the possibility of licensing LPCN 1144, LPCN 1148, and LPCN 1107, although we have not entered into a licensing agreement and no assurance can be given that occurredany license agreement will be completed, or, if an agreement is completed, that such an agreement would be on terms favorable to us. If we are unable to raise additional capital or obtain non-dilutive financing, we may need to reduce research and development expenses in 2017.order to extend our ability to continue as a going concern.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related benefits, including stock-based compensation related to our executive, finance, business development, marketing, sales and support functions. Other general and administrative expenses include rent and utilities, travel expenses, professional fees for auditing, tax, legal and legal services, litigation settlementvarious other services.
General and market research and market analytics.
Theyadministrative expenses also include expenses for the cost of preparing, filling and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims.
We expect that general and administrative expenses will continue to increase in the future as we maturecontinue as a public company. These increases will likely includecompany, including legal and consulting fees, accounting and audit fees, director fees, increased directors’ and officers’ insurance premiums, fees for investor relations services and enhanced business and accounting systems, litigation costs, professional fees and other costs. However, outside spend on sales and marketing pre-commercialization activities will be consistent with spend during 2017 untilif we receive clarity on the regulatory path forward for TLANDO. If the FDA approves TLANDO, we will increase our outside spend on pre-commercialization and commercialization activities substantially and will needare unable to raise additional capital, we may need to fund these expenses.
Restructuring Charges
Restructuring charges relate to our initiative to restructure operations which was approved by the board of directors on July 13, 2016. Under the July 2016 restructuring, we reduced our workforce by eight positions, constituting 33% of our workforce. The reduction in workforce involved all functional disciplines includingreduce general and administrative employees, sales and marketing and research and development personnel. Additionally, the Board approvedexpenses in order to extend our ability to continue as a further restructuring in October 2016 whereby we reduced our workforce by an additional two positions in the sales and marketing functions. The restructurings that occurred in 2016 are jointly referred to as the 2016 Restructuring Plan.going concern.
| 56 |
Other Income,Expense (Income), Net
Other income,expense (income), net consists primarily of interest income earned on our cash, cash equivalents and cash equivalents.marketable investment securities, imputed interest on minimum royalties under the license agreement we had with Antares, which was terminated January 31, 2024, gains on our warrant liability and gains on our litigation liability.
Results of Operations
Comparison of the Years Ended December 31, 20172023 and 20162022
The following table summarizes our results of operations for the years ended December 31, 20172023 and 2016:2022:
| Years Ended December 31, | Years Ended December 31, | |||||||||||||||||||||||
| 2017 | 2016 | Variance | 2023 | 2022 | Variance | |||||||||||||||||||
| Revenue | $ | (2,850,818 | ) | $ | 500,000 | $ | (3,350,818 | ) | ||||||||||||||||
| Research and development expenses | $ | 11,004,281 | $ | 8,076,053 | 2,928,228 | 10,175,251 | 8,556,888 | 1,618,363 | ||||||||||||||||
| General and administrative expenses | 10,213,695 | 10,382,146 | (168,451 | ) | 4,904,888 | 4,062,487 | 842,401 | |||||||||||||||||
| Restructuring costs | - | 728,635 | (728,635 | ) | ||||||||||||||||||||
| Other income, net | 235,816 | 216,078 | 19,738 | |||||||||||||||||||||
| Interest and investment income | 1,366,940 | 572,578 | 794,362 | |||||||||||||||||||||
| Interest expense | - | (27,098 | ) | 27,098 | ||||||||||||||||||||
| Unrealized gain on warrant liability | 212,690 | 565,940 | (353,250 | ) | ||||||||||||||||||||
| Gain on litigation settlement | - | 250,000 | (250,000 | ) | ||||||||||||||||||||
| Income tax expense | 700 | 752 | (52 | ) | (755 | ) | (681 | ) | (74 | ) | ||||||||||||||
Revenue
We recognized a net reversal of variable consideration revenue of $2.9 million during the year ended December 31, 2023, compared to revenue of $500,000 during the year ended December 31, 2022. Net reversal of variable consideration revenue in 2023 was mainly attributable to the reversal of variable consideration revenue recognized for minimum guaranteed royalties in 2021 under the license agreement with Antares, offset by $110,000 in license revenue payments received from Spriaso under a licensing agreement in the cough and cold field. License revenue in 2022 was related to a non-refundable cash fee of $500,000 received from Antares for consideration of a 90-day extension to exercise its option to license LPCN 1111. On June 30, 2022, Antares’ option to license TLANDO XR expired and was not exercised. On October 2, 2023, the Company received notice from Antares of Antares’ termination of the Antares License Agreement which stated that the Antares License Agreement will terminate effective January 31, 2024. On January 12, 2024, the Company entered into the Verity License Agreement with Verity. Upon termination of the Antares License Agreement, all rights and licenses granted by the Company to Antares under the Antares License Agreement terminated and all rights in TLANDO were transferred to the Company’s new licensing partner, Verity.
Research and Development Expenses
We recorded research and development expenses of $10.2 million and $8.6 million, respectively, for the years ended December 31, 2023 and 2022. The increase in research and development expenses during the year ended December 31, 20172023 was primarily due to increaseda $1.2 million increase in contract research organization and consultant costs of $5.0 million for the DV and DF studiesexpense related to our LPCN 1154 clinical studies, a $591,000 increase in TLANDO increased contract manufacturing related costs, for LPCN 1107 of $725,000, and increased outside services of $666,000a $560,000 increase related to the Advisory Committee meeting for TLANDO.our Phase 2 POC study in male patients with cirrhosis with LPCN 1148, and a $537,000 increase in personnel expense primarily resulting from additional headcount. These increases wereare offset by decreased contract research organization costs fora $600,000 decrease related to LPCN 1111 of $1.4 million,scale up costs in 2022, a decrease in validation and commercial batch manufacturing costs for TLANDO of $1.9 million, and a$416,000 decrease in contract research organization expense and outside consulting costs forrelated to the LPCN 1144 LiFT Phase 2 clinical study in NASH subjects, a $144,000 decrease in our LPCN 1107 of $153,000.clinical study, and a $123,000 decrease in other research and development activities.
General and Administrative Expenses
The decrease inWe recorded general and administrative expenses during the year ended December 31, 2017 was primarily due to a decrease of $534,000 for pre-commercialization marketing$4.9 million and sales activities related to TLANDO and a decrease of $558,000 in legal costs for patent interference offset by an increase of $930,000 related to class-action litigation settlement fees.
Restructuring Charges
The decrease in restructuring charges in the year ended December 31, 2017 was the result of our 2016 Restructuring Plan during the year ended December 31, 2016. The charge related to restructuring during the year ended December 31, 2016 was $729,000 and was comprised of $678,000 in severance related expenses and $51,000 for extending the exercise period of certain options under an existing employee severance agreement. We did not incur similar charges in the year ended December 31, 2017.
Other Income, Net
The increase in other income, net, primarily reflects increased interest income earned as a result of increased interest rates and corresponding earnings on average balances in cash, cash equivalents and marketable investment securities in 2017 as compared to 2016.
Comparison of the Years Ended December 31, 2016 and 2015
The following table summarizes our results of operations$4.1 million, respectively, for the years ended December 31, 20162023 and 2015:
| Years Ended December 31, | ||||||||||||
| 2016 | 2015 | Variance | ||||||||||
| Research and development expenses | $ | 8,076,053 | $ | 12,580,245 | (4,504,192 | ) | ||||||
| General and administrative expenses | 10,382,146 | 5,801,823 | 4,580,323 | |||||||||
| Restructuring costs | 728,635 | - | 728,635 | |||||||||
| Other income, net | 216,078 | 173,890 | 42,188 | |||||||||
| Income tax expense | 752 | 200 | 552 | |||||||||
Research and Development Expenses
The decrease in research and development expenses during the year ended December 31, 2016 was primarily due to decreased contract research organization and consultant costs of $3.8 million due to the completion of our Phase 3 SOAR clinical study of TLANDO during 2015. In addition, during the year ended December 31, 2015, we incurred a $2.3 million fee to file our NDA for TLANDO with the FDA. These decreases were offset by an increase in validation and commercial batch manufacturing costs during 2016 for TLANDO of $1.7 million.
General and Administrative Expenses
2022. The increase in general and administrative expenses during the year ended December 31, 20162023 was primarily due to ana $374,000 increase of $1.2 million for pre-commercialization marketingin business development and sales activitiesstrategic advisory services related to TLANDO, anexpenses, a $217,000 increase of $750,000 in legal costs for class action defensefees relating to corporate matters such as our reverse stock split, potential licensing partnerships, and patent interference and ancorporate strategy, a $219,000 increase of $2.0 million in personnel related costs, due primarily to increased salariesa $188,000 increase in franchise taxes resulting from our increase in authorized shares and stock-based compensation related to higher average headcount. The remainingreverse stock split, a $133,000 increase relates to several other items including increasedin director fees, increased audit fees, increased D&O insurance costs, increased rent cost related to our New Jersey location and overall higher allocated overhead costs toa $41,000 increase in other general and administrative expense.expenses and a $22,000 increase in royalty expense relating to the net sales of TLANDO. These increases were offset by a $222,000 decrease in corporate insurance expense, a $138,000 decrease related to the recruitment of two additional directors, and a $8,000 decrease in our various consulting and professional fees.
| 57 |
Restructuring ChargesInterest and Investment Income
The increase in restructuring charges in the year ended December 31, 2016 was the result of our 2016 Restructuring Plan. The charge related to restructuringinterest and investment income during the year ended December 31, 20162023 was $729,000due to higher interest rates in 2023 compared to 2022, despite declining cash and was comprised of $678,000marketable investment securities balances quarter over quarter in severance related expenses2023.
Interest Expense
The decrease in interest expense during the year ended December 31, 2023 is due to a decrease in interest expense on our Loan and $51,000 for extending the exercise period of certain options under an existing employee severance agreement.
Other Income, Net
The increase in other income, net, primarily reflects increased interest income earnedSecurity Agreement with SVB, mainly as a result of increased interest rates and corresponding earnings on averagelower principal balances in cash, cash equivalents2023 compared to 2022 as the SVB loan matured and marketable investment securitieswas paid in 2016full in June of 2022.
Unrealized Loss (Gain) on Warrant Liability
We recorded gains of $213,000 and $566,000, respectively, on warrant liability during the years ended December 31, 2023 and 2022 related to the change in the fair value of outstanding common stock warrants issued in the November 2019 Offering. The gains in 2023 and 2022 were attributable to a decrease in the value of warrants outstanding as of December 31, 2023 as compared to 2015.December 31, 2022, and a decrease in the value of warrants outstanding as of December 31, 2022 as compared to December 31, 2021. These decreases in value were due to a decrease in our stock price and a shorter term remaining on the outstanding warrants. There were no common stock warrants from the November 2019 Offering exercised during 2023 or 2022. The warrants are classified as a liability due to a provision contained within the warrant agreement which allows the warrant holder the option to elect to receive an amount of cash equal to the value of the warrants as determined in accordance with the Black-Scholes option pricing model with certain defined assumptions upon a change of control. The warrant liability will continue to fluctuate in the future based on inputs to the Black-Scholes model including our current stock price, the remaining life of the warrants, the volatility of our stock price, the risk-free interest rate and the number of common stock warrants outstanding.
Litigation Settlement
During the year ended December 31, 2022, we recorded a gain on the settlement of litigation liability of $250,000 as a result of the April 2022 Amendment to Global Agreement with Clarus (“Amended Settlement Agreement”). The Amended Settlement Agreement settled the payments due in July 2022 and 2023 for $1,250,000 rather than the $1,500,000 total future payments due under the terms of the Global Agreement agreed to in 2021. Under the terms of the Global Agreement we entered into in 2021, we had agreed to pay Clarus $4.0 million payable as follows: $2.5 million which was paid in July 2021, $1.0 million which was to be paid on July 13, 2022 and $500,000 which was to be paid on July 13, 2023. No future royalties are owing from either party. On July 15, 2021, the Court dismissed with prejudice the Company’s claims and Clarus’ counterclaims.
Liquidity and Capital Resources
Since our inception, our operations have been primarily financed through sales of our equity securities, debt and payments received under our license and collaboration arrangements. In 2018, we secured a term loan with Silicon Valley Bank for $10.0 million. We have devoted our resources to funding research and development programs, including discovery research, preclinical and clinical development activities. We have incurred operating losses in most years since our inception and we expect to continue to incur operating losses into the foreseeable future as we seek to advance our lead product candidate, TLANDO, and further clinical development of LPCN 1111,1154, LPCN 11072101, LPCN 2203 and ourany other programs andproduct candidate, including continued research efforts.
As of December 31, 2017,2023, we had $21.5$22.0 million of unrestricted cash, cash equivalents and marketable investment securities compared to $26.8$32.5 million at December 31, 2016.2022.
On January 5, 2018,12, 2024, we entered into a Loan and Securitythe Verity License Agreement (the “Loan and Security Agreement”) with Silicon Valley Bank (“SVB”)Verity, pursuant to which SVB has agreed to lend us $10.0 million. The principal borrowed under the Loan and Security Agreement bears a fixed interest rate equal to the Prime Rate plus one percent per annum, which interest is payable monthly. The loan matures on December 1, 2021. We are only required to make monthly interest payments until December 31, 2018, following which we will be required to also make equal monthly payments of principal for the remainder of the term; provided, however, that if on or prior to May 31, 2018, we receive evidence reasonably satisfactory to SVB that we have received FDA approval for TLANDOTM, the interest-only payment period will be extended to June 30, 2019. We will also be required to pay an additional final payment at maturity equal to $650,000 (the “Final Payment Charge”). At our option, we may prepay all amounts owed under the Loan and Security Agreement (including all accrued and unpaid interest and the Final Payment Charge), subject to a prepayment charge if the loan has been outstanding for less than two years, which prepayment charge is determined based on the date the loan is prepaid. In connection with the Loan and Security Agreement, we granted to SVBVerity an exclusive, royalty-bearing, sublicensable right and license to develop and commercialize our TLANDO product with respect to TRT in the U.S. and Canada. Upon execution of the Verity License Agreement in January 2024 and upon transition of the commercialization of TLANDO from Antares to Verity in February 2024, Verity paid to us initial payments of $2.5 million and $5 million, respectively. Verity has also agreed to make additional payments to us of $2.5 million before January 1, 2025 and $1 million before January 1, 2026. The Verity License Agreement also provides Verity with a security interestlicense to develop and commercialize TLANDO XR (LPCN 1111), the Company’s potential next generation, once daily oral product candidate for testosterone replacement therapy comprised of testosterone tridecanoate (“TT”), in substantiallythe U.S. and Canada. We are also eligible to receive milestone payments of up to $259 million in the aggregate, depending on the achievement of certain development milestones and sales milestones in a single calendar year with respect to all of our assets now owned or hereafter acquired, excluding intellectual property and certain other assets.products licensed by Verity under the Verity License Agreement. In addition, if TLANDOwe receive tiered royalty payments at rates ranging from percentages of 12% up to 18% of net sales of all products licensed to Verity in the United States and Canada. Our ability to realize benefits from the Verity License Agreement, including milestone and royalty payments, is not approved by the FDA on or prior to May 31, 2018, we will be required to maintain $5.0 million of cash collateral at SVB until such time as TLANDO is approved by the FDA. While any amounts are outstanding under the Loan and Security Agreement, we are subject to a number of affirmative and negative covenants, including covenants regarding dispositions of property, business combinationsrisks. We may not realize milestone or acquisitions, incurrence of additional indebtedness and transactions with affiliates, among other customary covenants.royalty payments in anticipated amounts, or at all.
| 58 |
On March 6, 2017, we entered into the a sales agreement (“Sales AgreementAgreement”) with Cantor Fitzgerald & Co. (“Cantor”) pursuant to which we may issue and sell, from time to time, shares of our common stock having an aggregate offering price of up to $20.0the amount we have registered on an effective registration statement pursuant to which the offering is being made. We currently have registered up to $50.0 million for sale under the Sales Agreement, pursuant to our Registration Statement on Form S-3 (the “Form S-3”), through Cantor as our sales agent. Cantor may sell our common stock by any method permitted by law deemed to be an “at the market offering” as defined in Rule 415(a)(4) of the Securities Act of 1933, as amended, including sales made directly on or through the NasdaqNASDAQ Capital Market or any other existing trade market for our common stock, in negotiated transactions at market prices prevailing at the time of sale or at prices related to prevailing market prices, or any other method permitted by law.
The shares of our common stock to be sold under the Sales Agreement will be sold and issued pursuant to the Company’s Registration Statement on Form S-3 (File No. 333-199093) (the “Existing Form S-3”), which was previously declared effective by the Securities and Exchange Commission, and the related prospectus and one or more prospectus supplements. Cantor will useuses its commercially reasonable efforts consistent with its normal trading and sales practices and applicable law and regulations to sell these shares. We will pay Cantor 3.0% of the aggregate gross proceeds from each sale of shares under the Sales Agreement. We have also provided Cantor with customary indemnification rights.
On October 13, 2017, we filed a Form S-3 (File No. 333-220942) (the “New Form S-3”) to replace the Existing Form S-3. The New Form S-3 has been declared effective by the Securities and Exchange Commission. The New Form S-3 registered the sale of up to $150,000,000 of any combination of common stock, preferred stock, debt securities, warrants and units pursuant to a shelf registration statement. The New Form S-3 also contains a prospectus pursuant to which we may sell, from time to time, shares of our common stock having an aggregate offering price of up to $25 million through Cantor as our sales agent, pursuant to the Sales Agreement that we currently have in place with Cantor. The other terms of the Sales Agreement that are described above will apply to the up to $25 million “at the market offering” anticipated to be made pursuant to the prospectus in the New Form S-3. Pursuant to Rule 415(a)(6) of the Securities Act of 1933, as amended, the offering of securities on the Existing Form S-3 were deemed terminated as of the date of effectiveness of the New Form S-3.
We are not obligated to make any sales of our common stock under the Sales Agreement. The offering of our common stock pursuant to the Sales Agreement will terminate upon the termination of the Sales Agreement as permitted therein. We and Cantor may each terminate the Sales Agreement at any time upon ten days’ prior notice.
As of December 31, 2017,2023, we havehad sold 2,518,109an aggregate of 964,711 shares at a weighted-average sales price of $34.52 per share under the At the Market Offering (the “ATM Offering”) for aggregate gross proceeds of $33.3 million and net proceeds of $32.1 million, after deducting sales agent commission and discounts and our other offering costs. During the year ended December 31, 2023, we sold 81,000 shares of our common stock pursuant to the ATM Offering at a weighted-average sales price of $5.36 per share, resulting in net proceeds of approximately $10.6 million$405,000, under the Sales Agreement which is net of $260,000 commissions paidapproximately $24,000 in expenses. During the year ended December 31, 2022, we did not sell any shares of our common stock pursuant to Cantor in connection with these sales.the ATM Offering. As of December 31, 2023, we had $40.8 million available for sale under the Sales Agreement. However, as of November 22, 2023, we are subject to General Instruction I.B.6 of Form S-3 which limits the amounts that we may sell under the registration statement. As a result of such limitations, we have currently registered the offer and sale of shares of common stock pursuant to the Sales Agreement having an aggregate offering price of up to $5.3 million.
We believe that our existing capital resources, together with interest thereon, will be sufficient to meet our projected operating requirements through at least March 31, 2019. While we believe we have sufficient liquidity2025 which include on-going clinical studies for LPCN 1154 and/or LPCN 2101 or LPCN 2203 and capital resources to fund our projected operating requirements through March 31, 2019, we will need to raise additional capital at some point, either before or after March 31, 2019, to support our operations, ongoing clinical studies,research and development activities and compliance with regulatory requirements, long-term research and development and commercialization of TLANDO, if we receive approval of TLANDO from the FDA.requirements. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. Additionalexpect if additional activities are performed by us including new clinical studies mayfor LPCN 1148, LPCN 1144, and/or LPCN 1107. While we believe we have sufficient liquidity and capital resources to fund our projected operating requirements through at least March 31, 2025, we will need to raise additional capital at some point through the equity or debt markets or through additional out-licensing activities, either before or after March 31, 2025, to support our operations. If we are unsuccessful in raising additional capital as necessary, our ability to continue as a going concern will be required to obtain approval of TLANDO and these studies would put additional demands on our limited capital resources.limited. Further, our operating plan may change, and we may need additional funds to meet operational needs and capital requirements for product development, regulatory compliance and clinical trials and pre-commercializationtrial activities sooner than planned. We may consumeIn addition, our capital resources may be consumed more rapidly if the FDA approvalwe pursue additional clinical studies for TLANDO is delayed LPCN 1154, LPCN 2101, LPCN 2203, LPCN 1148, LPCN 1144, and/or denied, or if we elect to pursue the build out of an internal sales force as part of our commercialization launch plan if our product candidates receive approval from the FDA.LPCN 1107. Conversely, our capital resources could last longer if we reduce expenses, andreduce the number of activities currently contemplated under our operating plan.
We currently have no credit facility but did secure $10.0 million in term debt in January 2018.plan or if we terminate, modify or suspend on-going clinical studies. We can raise capital pursuant to the Sales Agreement in the ATM Offering but may choose not to issue common stock if our market price is too low to justify such sales in our discretion. There are numerous risks and uncertainties associated with the development and, subject to approval by the FDA, commercialization of our product candidates. There are numerous risks and uncertainties impacting our ability to enter into collaborations with third parties to participate in the development and potential commercialization of our product candidates. We are unable to precisely estimate the amounts of increased capital outlays and operating expenditures associated with our anticipated or unanticipated clinical studies and ongoing development and pre-commercialization efforts. All of these factors affect our need for additional capital resources. To fund future operations, we will need to ultimately raise additional capital and our requirements will depend on many factors, including the following:
| ● | the scope, rate of progress, results and cost of our clinical studies, preclinical testing and other related activities for all of our product candidates, including LPCN 1154, LPCN 2101 and LPCN 2203, LPCN 1148, LPCN 1144, LPCN 1107; |
| 59 |
| ● | the cost of manufacturing clinical supplies, and establishing commercial supplies, of our product candidates and any products that we may develop; | |
| ● | the cost and timing of establishing sales, marketing and distribution capabilities, if any; | |
| ● | the terms and timing of any collaborative, licensing, settlement and other arrangements that we may establish; | |
| ● | the number and characteristics of product candidates that we pursue; | |
| ● | the cost, timing and outcomes of regulatory approvals; | |
| ● | the timing, receipt and amount of sales, profit sharing or royalties, if any, from our potential products; | |
| ● | the cost of preparing, filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; | |
| ● | the extent to which we acquire or invest in businesses, products or technologies, although we currently have no commitments or agreements relating to any of these types of transactions; and | |
| ● | the extent to which we grow significantly in the number of employees or the scope of our operations. |
Funding may not be available to us on acceptablefavorable terms, or at all. Also, market conditions may prevent us from accessing the debt and equity capital markets, including sales of our common stock through the ATM Offering.Sales Agreement. If we are unable to obtain adequate financing when needed, we may have to delay, reduce the scope of or suspend one or more of our clinical studies, research and development programs or, if any of our product candidates receive approval from the FDA, commercialization efforts. We may seek to raise any necessary additional capital through a combination of public or private equity offerings, including the ATM Offering,Sales Agreement, debt financings, collaborations, strategic alliances, licensing arrangements and other marketing and distribution arrangements. These arrangements may not be available to us or available on terms favorable to us. To the extent that we raise additional capital through marketing and distribution arrangements, other collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our product candidates, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we do raise additional capital through public or private equity offerings, the ownership interest of our existing stockholders will be diluted, and the terms of these securities may include liquidation or other preferences, warrants or other terms that adversely affect our stockholders’ rights or further complicate raising additional capital in the future. If we raise additional capital through debt financing, we may be subject to covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we are unable, for any reason, to raise needed capital, we will have to reduce costs, delay research and development programs, liquidate assets, dispose of rights, commercialize products or product candidates earlier than planned or on less favorable terms than desired or reduce or cease operations.
Sources and Uses of Cash
The following table provides a summary of our cash flows for the years ended December 31, 2017, 20162023 and 2015:2022:
| Years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Cash used in operating activities | $ | (16,702,015 | ) | $ | (18,281,938 | ) | $ | (15,356,304 | ) | |||
| Cash provided by (used in) investing activities | 3,126,020 | 3,229,125 | (25,018,399 | ) | ||||||||
| Cash provided by financing activities | 11,226,028 | 605,870 | 32,716,307 | |||||||||
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Cash used in operating activities | $ | (11,865,991 | ) | $ | (11,968,819 | ) | ||
| Cash provided by investing activities | 13,084,686 | 14,293,707 | ||||||
| Cash provided by (used in) financing activities | 404,567 | (2,126,944 | ) | |||||
Net Cash Used in Operating Activities
During each of the years ended December 31, 2017, 20162023 and 2015,2022, net cash used in operating activities was $16.7$11.9 and $12.0 million, $18.3 million and $15.4 million, respectively.
Net cash used in operating activities during the years ended December 31, 2017, 20162023 and 20152022 was primarily attributable to cash outlays to support on-going operations, including research and development expenses and general and administrative expenses. During 2017,2023, we were conductingperforming activities mainly related to our DV and DFLPCN 1154 clinical studies and our LPCN 1148 Phase 2 POC Study in male subjects with TLANDO, we resubmitted our NDA with the FDA andcirrhosis. During 2022, we were transferringperforming activities related to our manufacturing technology to a contract manufacturer for LPCN 1107. During 2016, we had our TLANDO NDA under reviewPhase 2 POC study in male subjects with the FDA, we were conducting a Phase 2b clinical studycirrhosis with LPCN 11111148, our PK and we were building out our commercial infrastructurefood effect studies with LPCN 1154 and capabilities leading up to our PDUFA date of June 28, 2016 with TLANDO. During 2015,LPCN 1107, and the SOAR trial for TLANDO was being conducted and significant cash outlays were made to support that clinical study and related NDA submission. Additionally, clinical studies for LPCN 1111 and 1107 were also being conducted in 2015.manufacturing scale up.
| 60 |
Net Cash Provided by (Used in) Investing Activities
During the yearsyear ended December 31, 2017, 2016 and 2015,2023, net cash provided by (used in) investing activities was $3.1 million, $3.2$13.1 million and $(25.0) million, respectively.during the year ended December 31, 2022, net cash used in investing activities was $14.3 million.
Net cash provided by investing activities during 20172023 and 20162022 was primarily the result of selling marketable investment securities, net $3.1 million and $3.2 million, respectively, to fund current operations. Net cash used in investing activities during 2015 was primarily the resultmaturity of investing excess cash not currently required to fund operations to purchase marketable investment securities, net of $25.0 million. Capital$36.0 million and $59.5 million, respectively. There were $13,000 and $134,000 in capital expenditures for 2017, 2016the years ended December 31, 2023 and 2015 were zero, $60,000 and $29,000,2022, respectively.
Net Cash Provided by (Used In) Financing Activities
During the yearsyear ended December 31, 2017, 2016 and 2015,2023, net cash provided by financing activities was $11.2 million, $606,000$405,000 and $32.7 million, respectively.during the year ended December 31, 2022 net used in financing activities was $2.1 million.
Net cash provided by financing activities during 2017the year ended December 31, 2023, was primarily attributablerelated to net proceeds from the sale of common stock of $10.6 million and proceeds from the exercise of stock options.
Net cash provided by financing activities during 2016 was primarily attributable to proceeds from the exercise of stock options.
Net cash provided by financing activities during 2015 was primarily attributable to net proceeds from the sale of common stock in April 2015 of $32.4 million and proceeds from the exercise of stock options.
Employee stock option exercises provided approximately $581,000, $606,000, and $277,000 of cash during 2017, 2016 and 2015, respectively. Proceeds from the exercise of employee stock options vary from period to period based upon, among other factors, fluctuations in the market price81,000 shares of our common stock relativeunder our ATM Offering, less associated costs. Net cash used in financing activities during the year ended December 31, 2022 was mainly due to loan repayments of $1.7 million and payment of the Final Payment Charge of $650,000 related to the exercise price of such options.SVB Loan and Security Agreement, offset by $211,000 in cash provided by proceeds from stock option exercises.
Contractual Commitments and Contingencies
Long-Term Debt Obligations and Interest on Debt
On January 5, 2018, we entered into a Loan and Security Agreement with SVB pursuant to which SVB agreed to lend us $10.0 million. The following table represents our contractual obligations as of December 31, 2017:principal borrowed under the Loan and Security Agreement bore interest at a rate equal to the Prime Rate plus one percent per annum, which interest was payable monthly. The loan matured on June 1, 2022 and the outstanding principal, interest and Final Payment Charge were paid in full.
| Less Than | 1-3 | 4-5 | More Than | |||||||||||||||||
| Total | 1 Year | Years | Years | 5 Years | ||||||||||||||||
| Purchase obligations | $ | 682,433 | $ | 682,433 | $ | - | $ | - | $ | - | ||||||||||
| Operating leases | 58,903 | 58,903 | - | - | - | |||||||||||||||
| Total | $ | 741,336 | $ | 741,336 | $ | - | $ | - | $ | - | ||||||||||
Purchase Obligations
We enter into contracts and issue purchase orders in the normal course of business with clinical research organizations for clinical trials and clinical and commercial supply manufacturing and with vendors for preclinical research studies, research supplies and other services and products for operating purposes. These contracts generally provide for termination on notice and are cancellable obligations.
Operating Leases
In August 2004, we entered into an agreement to lease our facility in Salt Lake City, Utah consisting of office and laboratory space which serves as our corporate headquarters. On February 9, 2018,January 24, 2024, we modified and extended the lease through February 28, 2019. Additionally, on December 28, 2015, we entered into an agreement to lease office space in Lawrenceville, New Jersey which has an occupancy date of February 1, 2016 and an end date of January 31, 2018.2025.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements which we have prepared in accordance with U.S. generally accepted accounting principles.principles (“GAAP”). In preparing our financial statements, we are required to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. We have identified the following accounting policies that we believe require application of management’s most subjective judgments, often requiring the need to make estimates about the effect of matters that are inherently uncertain and may change in subsequent periods. Our actual results could differ from these estimates and such differences could be material.
While our significant accounting policies are described in more detail in Note 2 of our annual financial statements included in this filing, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our financial statements.
| 61 |
Revenue Recognition
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (Topic 606) with amendments in2015 (ASU 2015-14) and 2016 (ASU 2016-8, ASU 2016-10, ASU 2016-12 and ASU 2016-20). The updated standard is a new comprehensive revenue recognition model that requires revenue to be recognized in a manner that depicts the transfer of goods or services to a customer at an amount that reflects the consideration expected to be received in exchange for those goods or services. The guidance also requires disclosures regarding the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers. We adopted this pronouncement effective January 1, 2017, and it did not have any effect on our financial position or results2017. We recognized net reversal of operations forvariable consideration revenue of $2.9 million during the years endingyear ended December 31, 2017, 20162023, and 2015 as nolicense revenue of $500,000 during the year ended December 31, 2022. Net reversal of variable consideration revenue in 2023 was mainly attributable to the reversal of variable consideration revenue recognized during these years.for minimum guaranteed royalties in 2021 under the license agreement with Antares, offset by $110,000 in license revenue payments received from Spriaso under a licensing agreement in the cough and cold field. License revenue in 2022 was related to a non-refundable cash fee of $500,000 received from Antares for consideration of a 90-day extension to exercise its option to license LPCN 1111. On June 30, 2022, Antares’ option to license TLANDO XR expired and was not exercised.
We may provide research and development services under collaboration arrangements to advance the development of jointly owned products. We record the expenses incurred and reimbursed on a net basis in research and development expense.
As of December 31, 2017,2023, we do not have any active collaboration agreements except for an agreement to provide joint research and development services which was assigned to Spriaso LLC as described in Note 12 of Lipocine Inc.’s annual financial statements included in this filing.agreements.
Accrued Research and Development Expenses
We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on the facts and circumstances known to us at that time. Our expense accruals for contract research, contract manufacturing and other contract services are based on estimates of the fees associated with services provided by the contracting organizations. Payments under some of the contracts we have with such parties depend on factors such as successful enrollment of patients, site initiation and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If possible, we obtain information regarding unbilled services directly from these service providers. However, we may be required to estimate these services based on other information available to us. If we underestimate or overestimate the activity or fees associated with a study or service at a given point in time, adjustments to research and development expenses may be necessary in future periods. Subsequent changes in estimates may result in a material change in our accruals.
Stock-Based Compensation
We recognize stock-based compensation expense for grants of stock option awards, restricted stock units and restricted stock under our Incentive Plan to employees, nonemployees and nonemployee members of our board of directors based on the grant-date fair value of those awards. The grant-date fair value of an award is generally recognized as compensation expense over the award’s requisite service period. In addition, in the past we have granted performance-based stock option awards and restricted stock grants, which vest based upon our satisfying certain performance conditions. Potential compensation cost, measured on the grant date, related to these performance options will be recognized only if, and when, we estimate that these options will vest, which is based on whether we consider the options’ performance conditions to be probable of attainment. Our estimates of the number of performance-based options that will vest will be revised, if necessary, in subsequent periods.
In addition, we grant stock options to nonemployee consultants from time to time in exchange for services performed for us. Equity instruments granted to nonemployees are subject to periodic revaluation over their vesting terms.
We use the Black-Scholes model to compute the estimated fair value of stock option awards. Using this model, fair value is calculated based on assumptions with respect to (i) expected volatility of our Common Stockcommon stock price, (ii) the periods of time over which employees and members of the board of directors are expected to hold their options prior to exercise (expected term), (iii) expected dividend yield on the Common Stock,common stock, and (iv) risk-free interest rates. Stock-based compensation expense also includes an estimate, which is made at the time of grant, of the number of awards that are expected to be forfeited. This estimate is revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
As of December 31, 2017,2023, there was $2.8 million$455,000 of total unrecognized compensation cost related to unvested share-based compensation arrangements granted under our Incentive Plan. Additionallythe Company’s stock option plan.
Warrant Liability
In connection with the November 2019 public offering, we issued warrants to purchase common stock. The warrants require us to pay such holders an amount of cash in the event of a fundamental transaction, as defined in the warrant agreement. As the cash payment is at the option of the warrant holder, we account for the common stock warrants as a liability, which is adjusted to fair value each reporting period as well as upon exercise of such warrants. The Company estimates the fair value of the warrant liability based on a hypothetical payout associated with a fundamental transaction. The fair value estimate utilizes a pricing model and unobservable inputs. Unlike the fair value of other assets and liabilities which are readily observable and therefore more easily independently corroborated, the warrants are not actively traded, and fair value is determined based on significant judgments regarding models, unobservable inputs and valuation methodologies.
As of December 31, 2017, there2023 and 2022, the warrant liability was $736,000 of total unrecognized compensation cost related to unvested restricted stock units that have performance vesting.$17,000 and $230,000, respectively.
Accounting Standards Issued Not Adopted
Refer to Note 13 in “Notes to Consolidated Financial Statements” for a discussion of new accounting standards.None.
Off-Balance Sheet Arrangements
None.
ITEM 7A.QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
We are exposed to various market risks, which include potential losses arising from adverse changes in market rates and prices, such as interest rates. We do not enter into derivatives or other financial instruments for trading or speculative purposes.
Interest Rate Risk.Our interest rate risk exposure results from our investment portfolio. Our primary objectives in managing our investment portfolio are to preserve principal, maintain proper liquidity to meet operating needs and maximize yields. The securities we hold in our investment portfolio are subject to interest rate risk. At any time, sharp changes in interest rates can affect the fair value of the investment portfolio and its interest earnings. After a review of our marketable investment securities, we believe that in the event of a hypothetical ten percent increase in interest rates, the resulting decrease in fair value of our marketable investment securities would be insignificant to the consolidated financial statements. Currently, we do not hedge these interest rate exposures. We have established policies and procedures to manage exposure to fluctuations in interest rates. We place our investments with high quality issuers and limit the amount of credit exposure to any one issuer and do not use derivative financial instruments in our investment portfolio. We invest in highly liquid, investment-grade securities and money market funds of various issues, types and maturities. These securities are classified as available-for-sale and, consequently, are recorded on the balance sheet at fair value with unrealized gains or losses reported as accumulated other comprehensive income as a separate component in stockholders' deficit unless a loss is deemed other than temporary, in which case the loss is recognized in earnings.
Subsequent to December 31, 2017, we entered into the Loan and Security Agreement with SVB for $10.0 million. A one percent increase in the prime rate would result in a $263,000 increase in interest expense, while a one percent decrease in the prime rate would result in a $262,000 decrease in interest expense.
ITEM 8.FINANCIAL STATEMENTS AND SUPPLEMENTAL DATA
LIPOCINE INC.
INDEX TO FINANCIAL STATEMENTS
| Page | ||
| Audited Financial Statements of Lipocine Inc. for the Years ended December 31, | ||
| Report of Independent Registered Public Accounting Firm (PCAOB ID No. 270) | 64 | |
| Consolidated Balance Sheets | 65 | |
| Consolidated Statements of Operations and Comprehensive Loss | 66 | |
| Consolidated Statements of Changes in Stockholders’ Equity | 67 | |
| Consolidated Statements of Cash Flows | 68 | |
| Notes to Consolidated Financial Statements | 69 |
| 63 |
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors
and Stockholders
Lipocine Inc.:
OpinionsOpinion on the Consolidated Financial Statements and Internal Control Over Financial Reporting
We have audited the accompanying consolidated balance sheets of Lipocine Inc. and subsidiaries (the Company) as of December 31, 20172023 and 2016,2022, the related consolidated statements of operations and comprehensive loss, changes in stockholders’ equity, and cash flows for each of the years in the three-year periodthen ended, December 31, 2017, and the related notes (collectively, the consolidated financial statements). We also have audited the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established inInternal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of the Company as of December 31, 20172023 and 2016,2022, and the consolidated results of its operations and its cash flows for each of the years in the three-year periodthen ended, December 31, 2017, in conformity with U.S.accounting principles generally accepted accounting principles. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting asUnited States of December 31, 2017, based on criteria established inInternal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.America.
Basis for OpinionsOpinion
The Company’s management is responsible for theseThese consolidated financial statements for maintaining effective internal control over financial reporting, and for its assessmentare the responsibility of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting.Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements and an opinion on the Company’s internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the auditsaudit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud, and whether effectivefraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, was maintained in all material respects.
Our auditsbut not for the purpose of expressing an opinion on the effectiveness of the consolidatedCompany’s internal control over financial statementsreporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.opinion.
Definition and Limitations of Internal Control Over Financial ReportingCritical Audit Matter
A company’s internal control over financial reportingThe critical audit matter communicated below is a process designed to provide reasonable assurance regardingmatter arising from the reliabilitycurrent period audit of financial reporting and the preparation ofconsolidated financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertainwas communicated or required to be communicated to the maintenanceaudit committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of recordscritical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which is relates.
Revenue Recognition
The Company entered into a license agreement during 2021 that includes a license fee, guaranteed minimum royalties, ongoing sales royalties, milestone payments and transfer of materials. On October 2, 2023, the licensee provided notice that the contract would be terminated effective January 31, 2024, resulting in reasonable detail, accurately and fairly reflect the transactions and dispositionsa reversal of a portion of the assetsvariable consideration revenue that had been previously recognized.
Management is required to determine the transaction price and allocate the transaction price to the performance obligations in the license agreement. Management is also required to make estimates of when achievement of a particular milestone becomes probable. Milestone payments are included in the transaction price when it becomes probable that such inclusion would not result in a significant revenue reversal. A reversal of variable consideration is recorded in the period that the change becomes known.
We identified revenue recognition as a critical audit matter because of the company; (2) provide reasonable assurance that transactions are recordedsignificant judgment by management in determining the transaction price and allocating the transaction price to the performance obligations as necessary to permit preparation of financial statements in accordance with generally acceptedwell as the accounting principles, and that receipts and expenditurestreatment of the company are being made only in accordance with authorizations of management and directorsconstrained revenue resulting from the termination of the company;license agreement. This in turn led to a high degree of auditor judgment and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subjecteffort in performing procedures and evaluating audit evidence related to the risk that controls may become inadequate because of changes in conditions, or thatjudgments made by management.
Our audit procedures for auditing revenue included the degree of compliance with the policies orfollowing procedures, may deteriorate.among others:
| ● | We obtained and read the material license and royalty agreements. | |
| ● | We tested management’s determination of the transaction price and the allocation of the transaction price to the performance obligations. | |
| ● | We evaluated the reasonableness of management’s judgments and estimates. |
/s/ Tanner LLC
We have served as the Company’s auditor since 2011.2018
Salt Lake City, Utah
March 12, 20187, 2024
| 64 |
LIPOCINE INC. AND SUBSIDIARIES
Consolidated Balance Sheets
December 31, 2017 and 2016
| December 31, | December 31, | |||||||
| 2023 | 2022 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 4,771,758 | $ | 3,148,496 | ||||
| Marketable investment securities | 17,263,788 | 29,381,410 | ||||||
| Accrued interest income | 52,254 | 80,427 | ||||||
| Contract asset - current portion | - | 579,428 | ||||||
| Prepaid and other current assets | 773,424 | 945,319 | ||||||
| Total current assets | 22,861,224 | 34,135,080 | ||||||
| Contract asset - non-current portion | - | 3,252,500 | ||||||
| Property and equipment, net of accumulated depreciation of $1,182,191 and $1,153,530 respectively | 116,095 | 131,589 | ||||||
| Other assets | 23,753 | 23,753 | ||||||
| Total assets | $ | 23,001,072 | $ | 37,542,922 | ||||
| Liabilities and Stockholders’ Equity | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 1,395,977 | $ | 600,388 | ||||
| Accrued expenses | 1,218,486 | 1,077,738 | ||||||
| Warrant liability - current portion | 17,166 | - | ||||||
| Total current liabilities | 2,631,629 | 1,678,126 | ||||||
| Warrant liability | - | 229,856 | ||||||
| Total liabilities | 2,631,629 | 1,907,982 | ||||||
| Commitments and contingencies (notes 8, 9 and 11) | - | - | ||||||
| Stockholders’ equity: | ||||||||
| Common stock, par value $ per share, shares authorized; and issued and and outstanding | 8,860 | 8,852 | ||||||
| Additional paid-in capital | 220,171,250 | 219,112,164 | ||||||
| Treasury stock at cost, shares | (40,712 | ) | (40,712 | ) | ||||
| Accumulated other comprehensive gain (loss) | 7,259 | (20,321 | ) | |||||
| Accumulated deficit | (199,777,214 | ) | (183,425,043 | ) | ||||
| Total stockholders’ equity | 20,369,443 | 35,634,940 | ||||||
| Total liabilities and stockholders’ equity | $ | 23,001,072 | $ | 37,542,922 | ||||
| 2017 | 2016 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 3,210,749 | $ | 5,560,716 | ||||
| Marketable investment securities | 18,257,321 | 21,279,570 | ||||||
| Accrued interest income | 23,067 | 38,943 | ||||||
| Litigation insurance recovery | 3,319,927 | - | ||||||
| Prepaid and other current assets | 408,227 | 329,548 | ||||||
| Total current assets | 25,219,291 | 27,208,777 | ||||||
| Property and equipment, net of accumulated depreciation of $1,121,080 and $1,092,710, respectively | 75,070 | 103,440 | ||||||
| Other assets | 30,753 | 30,753 | ||||||
| Total assets | $ | 25,325,114 | $ | 27,342,970 | ||||
| Liabilities and Stockholders' Equity | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 598,070 | $ | 245,915 | ||||
| Litigation settlement payable | 4,250,000 | - | ||||||
| Accrued expenses | 1,497,056 | 1,080,254 | ||||||
| Total current liabilities | 6,345,126 | 1,326,169 | ||||||
| Total liabilities | 6,345,126 | 1,326,169 | ||||||
| Commitments and contingencies (notes 8 and 11) | ||||||||
| Stockholders' equity: | ||||||||
| Preferred stock, par value $0.0001 per share, 10,000,000 shares authorized; zero issued and outstanding | - | - | ||||||
| Common stock, par value $0.0001 per share, 100,000,000 shares authorized; 21,270,249 and 18,462,325 issued and 21,264,539 and 18,456,615 outstanding | 2,127 | 1,846 | ||||||
| Additional paid-in capital | 145,423,012 | 131,481,123 | ||||||
| Treasury stock at cost, 5,710 shares | (40,712 | ) | (40,712 | ) | ||||
| Accumulated other comprehensive loss | (4,616 | ) | (8,493 | ) | ||||
| Accumulated deficit | (126,399,823 | ) | (105,416,963 | ) | ||||
| Total stockholders' equity | 18,979,988 | 26,016,801 | ||||||
| Total liabilities and stockholders' equity | $ | 25,325,114 | $ | 27,342,970 | ||||
See accompanying notes to consolidated financial statements
| 65 |
LIPOCINE INC. AND SUBSIDIARIES
Consolidated Statements of Operations and Comprehensive Loss
Years Ending December 31, 2017, 2016, and 2015
| 2023 | 2022 | |||||||
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Revenues: | ||||||||
| License revenue | $ | 109,987 | $ | 500,000 | ||||
| Minimum guaranteed royalties revenue (reversal of variable consideration) | (2,960,805 | ) | - | |||||
| Total revenues (reversal of variable consideration), net | (2,850,818 | ) | 500,000 | |||||
| Operating expenses: | ||||||||
| Research and development | 10,175,251 | 8,556,888 | ||||||
| General and administrative | 4,904,888 | 4,062,487 | ||||||
| Total operating expenses | 15,080,139 | 12,619,375 | ||||||
| Operating loss | (17,930,957 | ) | (12,119,375 | ) | ||||
| Other income (expense): | ||||||||
| Interest and investment income | 1,366,940 | 572,578 | ||||||
| Interest expense | - | (27,098 | ) | |||||
| Unrealized gain on warrant liability | 212,690 | 565,940 | ||||||
| Gain on litigation settlement liability | - | 250,000 | ||||||
| Total other income, net | 1,579,630 | 1,361,420 | ||||||
| Loss before income tax expense | (16,351,327 | ) | (10,757,955 | ) | ||||
| Income tax expense | (755 | ) | (681 | ) | ||||
| Net loss | (16,352,082 | ) | (10,758,636 | ) | ||||
| Issuance of Series B preferred stock dividend | (89 | ) | - | |||||
| Net loss attributable to common shareholders Net loss attributable to common shareholders | $ | (16,352,171 | ) | $ | (10,758,636 | ) | ||
| Basic loss per share attributable to common stock | $ | (3.10 | ) | $ | (2.06 | ) | ||
| Weighted average common shares outstanding, basic | 5,269,671 | 5,231,681 | ||||||
| Diluted loss per share attributable to common stock | $ | (3.14 | ) | $ | (2.15 | ) | ||
| Weighted average common shares outstanding, diluted | 5,269,671 | 5,256,169 | ||||||
| Comprehensive loss: | ||||||||
| Net loss | $ | (16,352,082 | ) | $ | (10,758,636 | ) | ||
| Net unrealized gain (loss) on available-for-sale securities | 27,580 | (2,305 | ) | |||||
| Comprehensive loss | $ | (16,324,502 | ) | $ | (10,760,941 | ) | ||
| 2017 | 2016 | 2015 | ||||||||||
| Operating expenses: | ||||||||||||
| Research and development | $ | 11,004,281 | $ | 8,076,053 | $ | 12,580,245 | ||||||
| General and administrative | 10,213,695 | 10,382,146 | 5,801,823 | |||||||||
| Restructuring costs | - | 728,635 | - | |||||||||
| Total operating expenses | 21,217,976 | 19,186,834 | 18,382,068 | |||||||||
| Operating loss | (21,217,976 | ) | (19,186,834 | ) | (18,382,068 | ) | ||||||
| Other income, net | 235,816 | 216,078 | 173,890 | |||||||||
| Loss before income tax expense | (20,982,160 | ) | (18,970,756 | ) | (18,208,178 | ) | ||||||
| Income tax expense | (700 | ) | (752 | ) | (200 | ) | ||||||
| Net loss | $ | (20,982,860 | ) | $ | (18,971,508 | ) | $ | (18,208,378 | ) | |||
| Basic loss per share attributable to common stock | $ | (1.05 | ) | $ | (1.04 | ) | $ | (1.11 | ) | |||
| Weighted average common shares outstanding, basic | 20,051,934 | 18,258,149 | 16,470,814 | |||||||||
| Diluted loss per share attributable to common stock | $ | (1.05 | ) | $ | (1.04 | ) | $ | (1.11 | ) | |||
| Weighted average common shares outstanding, diluted | 20,051,934 | 18,258,149 | 16,470,814 | |||||||||
| Comprehensive loss: | ||||||||||||
| Net loss | $ | (20,982,860 | ) | $ | (18,971,508 | ) | $ | (18,208,378 | ) | |||
| Unrealized net gain (loss) on available-for-sale securities | 3,877 | 24,407 | (32,900 | ) | ||||||||
| Comprehensive loss | $ | (20,978,983 | ) | $ | (18,947,101 | ) | $ | (18,241,278 | ) | |||
See accompanying notes to consolidated financial statements
| 66 |
LIPOCINE INC. AND SUBSIDIARIES
Consolidated Statements of Changes in Stockholders’ Equity
For the Years EndingEnded December 31, 2017, 20162023 and 20152022
| Common Stock | Treasury Stock | |||||||||||||||||||||||||||||||
| Number of Shares | Amount | Number of Shares | Amount | Additional Paid-In Capital | Accumulated Other Comprehensive Loss | Accumulated Deficit | Total Stockholders' Equity | |||||||||||||||||||||||||
| Balances at December 31, 2014 | 12,794,672 | $ | 1,280 | 5,710 | $ | (40,712 | ) | $ | 94,636,479 | $ | - | $ | (68,237,077 | ) | $ | 26,359,970 | ||||||||||||||||
| Net loss | - | - | - | - | - | - | (18,208,378 | ) | (18,208,378 | ) | ||||||||||||||||||||||
| Unrealized net loss on marketable investment securities | - | - | - | - | - | (32,900 | ) | - | (32,900 | ) | ||||||||||||||||||||||
| Stock-based compensation | - | - | - | - | 1,150,418 | - | - | 1,150,418 | ||||||||||||||||||||||||
| Option exercises | 98,574 | 10 | - | - | 276,984 | - | - | 276,994 | ||||||||||||||||||||||||
| Vesting of restricted stock awards | 4,000 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
| Issuance of common stock in offering | 5,347,500 | 535 | - | - | 32,438,778 | - | - | 32,439,313 | ||||||||||||||||||||||||
| Balances at December 31, 2015 | 18,244,746 | $ | 1,825 | 5,710 | $ | (40,712 | ) | $ | 128,502,659 | $ | (32,900 | ) | $ | (86,445,455 | ) | $ | 41,985,417 | |||||||||||||||
| Net loss | - | - | - | - | - | - | (18,971,508 | ) | (18,971,508 | ) | ||||||||||||||||||||||
| Unrealized net gain on marketable investment securities | - | - | - | - | - | 24,407 | - | 24,407 | ||||||||||||||||||||||||
| Stock-based compensation | - | - | - | - | 2,372,615 | - | - | 2,372,615 | ||||||||||||||||||||||||
| Option exercises | 208,869 | 21 | - | - | 605,849 | - | - | 605,870 | ||||||||||||||||||||||||
| Vesting of restricted stock awards | 3,000 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
| Balances at December 31, 2016 | 18,456,615 | $ | 1,846 | 5,710 | $ | (40,712 | ) | $ | 131,481,123 | $ | (8,493 | ) | $ | (105,416,963 | ) | $ | 26,016,801 | |||||||||||||||
| Net loss | - | - | - | - | - | - | (20,982,860 | ) | (20,982,860 | ) | ||||||||||||||||||||||
| Unrealized net gain on marketable investment securities | - | - | - | - | - | 3,877 | - | 3,877 | ||||||||||||||||||||||||
| Stock-based compensation | - | - | - | - | 2,716,142 | - | - | 2,716,142 | ||||||||||||||||||||||||
| Option exercises | 206,813 | 21 | - | - | 581,124 | - | - | 581,145 | ||||||||||||||||||||||||
| Vesting of restricted stock units | 83,002 | 8 | - | - | (8 | ) | - | - | - | |||||||||||||||||||||||
| Common stock sold through ATM offering | 2,518,109 | 252 | - | - | 10,644,631 | - | - | 10,644,883 | ||||||||||||||||||||||||
| Balances at December 31, 2017 | 21,264,539 | $ | 2,127 | 5,710 | $ | (40,712 | ) | $ | 145,423,012 | $ | (4,616 | ) | $ | (126,399,823 | ) | $ | 18,979,988 | |||||||||||||||
| Number of Shares | Amount | Amount | Number of Shares | Amount | Number of Shares | Amount | Paid-In Capital | Comprehensive Loss | Accumulated Deficit | Stockholders’ Equity | |||||||||||||||||||||||||||||||
| Mezzanine Equity | Stockholder’s Equity | ||||||||||||||||||||||||||||||||||||||||
| Series B Preferred Stock | Common Stock | Treasury Stock | Additional | Accumulated Other | Total | ||||||||||||||||||||||||||||||||||||
| Number of Shares | Amount | Number of Shares | Amount | Number of Shares | Amount | Paid-In Capital | Comprehensive Loss | Accumulated Deficit | Stockholders’ Equity | ||||||||||||||||||||||||||||||||
| Balances at December 31, 2021 | - | $ | - | - | 5,221,883 | $ | 8,830 | 336 | $ | (40,712 | ) | $ | 218,286,323 | $ | (18,016 | ) | $ | (172,666,407 | ) | $ | 45,570,018 | ||||||||||||||||||||
| Mezzanine equity, balance, shares | - | ||||||||||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | - | - | - | (10,758,636 | ) | (10,758,636 | ) | ||||||||||||||||||||||||||||
| - | |||||||||||||||||||||||||||||||||||||||||
| Unrealized net loss on marketable investment - securities | - | - | - | - | - | - | - | (2,305 | ) | - | (2,305 | ) | |||||||||||||||||||||||||||||
| - | |||||||||||||||||||||||||||||||||||||||||
| Stock-based compensation | - | - | - | - | - | - | 636,140 | - | - | 636,140 | |||||||||||||||||||||||||||||||
| Mezzanine equity, Issuance of Series B preferred stock dividend, value | |||||||||||||||||||||||||||||||||||||||||
| Mezzanine equity, Issuance of Series B preferred stock dividend, share | |||||||||||||||||||||||||||||||||||||||||
| Mezzanine equity, Redemption of Series B preferred stock, value | |||||||||||||||||||||||||||||||||||||||||
| Mezzanine equity, Redemption of Series B preferred stock, share | |||||||||||||||||||||||||||||||||||||||||
| - | |||||||||||||||||||||||||||||||||||||||||
| Option exercises | - | - | 12,947 | 22 | - | - | 211,401 | - | - | 211,423 | |||||||||||||||||||||||||||||||
| - | |||||||||||||||||||||||||||||||||||||||||
| Costs associated with ATM Offering | - | - | - | - | - | - | (21,700 | ) | - | - | (21,700 | ) | |||||||||||||||||||||||||||||
| Balances at December 31, 2022 | - | $ | - | - | 5,234,830 | $ | 8,852 | 336 | $ | (40,712 | ) | $ | 219,112,164 | $ | (20,321 | ) | $ | (183,425,043 | ) | $ | 35,634,940 | ||||||||||||||||||||
| Mezzanine equity, balance, shares | - | ||||||||||||||||||||||||||||||||||||||||
| Number of Shares | Amount | Amount | Number of Shares | Amount | Number of Shares | Amount | Paid-In Capital | Comprehensive Loss | Accumulated Deficit | Stockholders’ Equity | |||||||||||||||||||||||||||||||
| Mezzanine Equity | Stockholder’s Equity | ||||||||||||||||||||||||||||||||||||||||
| Series B Preferred Stock | Common Stock | Treasury Stock | Additional | Accumulated Other | Total | ||||||||||||||||||||||||||||||||||||
| Number of Shares | Amount | Number of Shares | Amount | Number of Shares | Amount | Paid-In Capital | Comprehensive Loss | Accumulated Deficit | Stockholders’ Equity | ||||||||||||||||||||||||||||||||
| Balances at December 31, 2022 | - | $ | - | - | 5,234,830 | $ | 8,852 | 336 | $ | (40,712 | ) | $ | 219,112,164 | $ | (20,321 | ) | $ | (183,425,043 | ) | $ | 35,634,940 | ||||||||||||||||||||
| Balances, value | - | $ | - | - | 5,234,830 | $ | 8,852 | 336 | $ | (40,712 | ) | $ | 219,112,164 | $ | (20,321 | ) | $ | (183,425,043 | ) | $ | 35,634,940 | ||||||||||||||||||||
| Net loss | - | - | - | - | - | - | - | - | - | (16,352,082 | ) | (16,352,082 | ) | ||||||||||||||||||||||||||||
| Unrealized net gain on marketable investment securities | - | - | - | - | - | - | - | 27,580 | - | 27,580 | |||||||||||||||||||||||||||||||
| Stock-based compensation | - | - | - | - | - | - | 654,438 | - | - | 654,438 | |||||||||||||||||||||||||||||||
| Issuance of Series B preferred stock dividend | 88,511 | 9 | - | - | - | - | 80 | - | 80 | ||||||||||||||||||||||||||||||||
| Redemption of Series B preferred stock | (88,511 | ) | (9 | ) | - | - | - | - | 9 | - | (89 | ) | (80 | ) | |||||||||||||||||||||||||||
| Common stock sold through ATM offering | - | - | 81,000 | 8 | - | - | 404,559 | - | - | 404,567 | |||||||||||||||||||||||||||||||
| Balances at December 31, 2023 | - | $ | - | - | 5,315,830 | $ | 8,860 | 336 | $ | (40,712 | ) | $ | 220,171,250 | $ | 7,259 | $ | (199,777,214 | ) | $ | 20,369,443 | |||||||||||||||||||||
| Balances, value | - | $ | - | - | 5,315,830 | $ | 8,860 | 336 | $ | (40,712 | ) | $ | 220,171,250 | $ | 7,259 | $ | (199,777,214 | ) | $ | 20,369,443 | |||||||||||||||||||||
See accompanying notes to consolidated financial statements
| 67 |
LIPOCINE INC. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
Years Ending December 31, 2017, 2016 and 2015
| 2023 | 2022 | |||||||
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (16,352,082 | ) | $ | (10,758,636 | ) | ||
| Adjustments to reconcile net loss to cash used in operating activities: | ||||||||
| Depreciation expense | 28,661 | 9,453 | ||||||
| Stock-based compensation expense | 654,438 | 636,140 | ||||||
| Non-cash interest expense | - | 5,842 | ||||||
| Non-cash gain on change in fair value of warrant liability | (212,690 | ) | (565,940 | ) | ||||
| Gain on settlement of litigation liability | - | (250,000 | ) | |||||
| Amortization of discounts on marketable investment securities | (952,651 | ) | (122,048 | ) | ||||
| Write off of contract asset due to variable consideration revenue reversal | 2,960,805 | - | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Accrued interest income | 28,173 | 166,826 | ||||||
| Contract asset | 709,933 | 218,072 | ||||||
| Prepaid and other current assets | 333,085 | 569,146 | ||||||
| Accounts payable | 795,589 | (688,954 | ) | |||||
| Accrued expenses | 140,748 | 61,280 | ||||||
| Litigation settlement liability | - | (1,250,000 | ) | |||||
| Cash used in operating activities | (11,865,991 | ) | (11,968,819 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Purchase of property and equipment | (13,167 | ) | (133,831 | ) | ||||
| Purchases of marketable investment securities | (22,902,147 | ) | (45,074,462 | ) | ||||
| Maturities of marketable investment securities | 36,000,000 | 59,502,000 | ||||||
| Cash provided by investing activities | 13,084,686 | 14,293,707 | ||||||
| Cash flows from financing activities: | ||||||||
| Debt repayments | - | (1,666,667 | ) | |||||
| End of loan payment | - | (650,000 | ) | |||||
| Net proceeds from sale of common stock through ATM | 404,567 | (21,700 | ) | |||||
| Proceeds from stock option exercises | - | 211,423 | ||||||
| Cash provided by (used in) financing activities | 404,567 | (2,126,944 | ) | |||||
| Net increase in cash and cash equivalents | 1,623,262 | 197,944 | ||||||
| Cash and cash equivalents at beginning of period | 3,148,496 | 2,950,552 | ||||||
| Cash and cash equivalents at end of period | $ | 4,771,758 | $ | 3,148,496 | ||||
| Supplemental disclosure of cash flow information: | ||||||||
| Interest paid | $ | - | $ | 21,256 | ||||
| Income taxes paid | $ | 225 | 200 | |||||
| Supplemental disclosure of non-cash investing and financing activity: | ||||||||
| Net unrealized gain (loss) on available-for-sale securities | $ | 27,580 | $ | (2,305 | ) | |||
| Accrued final payment charge on debt | $ | - | $ | 5,842 | ||||
| Issuance of Series B preferred stock | $ | 89 | $ | - | ||||
| 2017 | 2016 | 2015 | ||||||||||
| Cash flows from operating activities: | ||||||||||||
| Net loss | $ | (20,982,860 | ) | $ | (18,971,508 | ) | $ | (18,208,378 | ) | |||
| Adjustments to reconcile net loss to cash used in operating activities: | ||||||||||||
| Depreciation expense | 28,370 | 31,960 | 26,721 | |||||||||
| Stock-based compensation expense | 2,716,142 | 2,372,615 | 1,150,418 | |||||||||
| Accretion (amortization) of premium/discount on marketable investment securities | (99,894 | ) | 224,482 | 181,390 | ||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Accrued interest income | 15,876 | 105,593 | (144,536 | ) | ||||||||
| Litigation insurance recovery | (3,319,927 | ) | - | - | ||||||||
| Prepaid and other current assets | (78,679 | ) | 20,612 | (120,248 | ) | |||||||
| Accounts payable | 352,155 | (261,152 | ) | 200,791 | ||||||||
| Litigation settlement payable | 4,250,000 | - | - | |||||||||
| Accrued expenses | 416,802 | (1,804,540 | ) | 1,557,538 | ||||||||
| Cash used in operating activities | (16,702,015 | ) | (18,281,938 | ) | (15,356,304 | ) | ||||||
| Cash flows from investing activities: | ||||||||||||
| Payment of rental deposit | - | (7,000 | ) | - | ||||||||
| Purchases of property and equipment | - | (59,650 | ) | (28,689 | ) | |||||||
| Purchases of marketable investment securities | (33,055,980 | ) | (25,272,225 | ) | (25,789,710 | ) | ||||||
| Maturities of marketable investment securities | 36,182,000 | 28,568,000 | 800,000 | |||||||||
| Cash provided by (used in) investing activities | 3,126,020 | 3,229,125 | (25,018,399 | ) | ||||||||
| Cash flows from financing activities: | ||||||||||||
| Proceeds from stock option exercises | 581,145 | 605,870 | 276,994 | |||||||||
| Net proceeds from sale of common stock through ATM | 10,644,883 | - | - | |||||||||
| Net proceeds from common stock offering | - | - | 32,439,313 | |||||||||
| Cash provided by financing activities | 11,226,028 | 605,870 | 32,716,307 | |||||||||
| Net decrease in cash and cash equivalents | (2,349,967 | ) | (14,446,943 | ) | (7,658,396 | ) | ||||||
| Cash and cash equivalents at beginning of period | 5,560,716 | 20,007,659 | 27,666,055 | |||||||||
| Cash and cash equivalents at end of period | $ | 3,210,749 | $ | 5,560,716 | $ | 20,007,659 | ||||||
| Supplemental disclosure of cash flow information: | ||||||||||||
| Unrealized net gain (loss) on marketable investment securities | 3,877 | 24,407 | (32,900 | ) | ||||||||
| Cash paid for income taxes | 700 | 752 | 200 | |||||||||
See accompanying notes to consolidated financial statements
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 20162023 and 20152022
(1) Description of Business
Lipocine Inc. (“Lipocine” or the “Company”), a clinical-stage biopharmaceutical company focused on central nervous system (“CNS”) disorders, is engaged in research and development for the delivery of drugs using its proprietary delivery technology. The Company’s principal operation is to provide oral delivery solutions for existing drugs. Lipocine develops its own drug candidates or it develops drug candidates on behalf of or in collaboration with corporate partners. The Company has funded operating costs primarily through collaborative license, milestone and research arrangements, through federal grants, and through the sale of equity securities.securities and through debt. The Company is incorporated under the laws of the State of Delaware.
(2) Summary of Significant Accounting Policies
(a) Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles (“US GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant items subject to such estimates and assumptions include those related to the timing and amount of revenue recognized from licensing agreements, stock-based compensation; income tax uncertainties; the fair value of the warrant liability and the useful lives of property and equipment.
(b) Cash and Cash Equivalents
The Company considers all highly liquid investments with original maturities to the Company of three months or less to be cash equivalents. Although the Company may deposit its cash and cash equivalents with multiple financial institutions, its deposits, at times, may exceed federally insured limits. Cash and cash equivalents were $2.2$4.8 million and $2.9$3.1 million at December 31, 20172023 and 2016.2022.
(c) Receivables
Accounts receivable are recorded at the invoiced amount and do not bear interest.
The Company maintains an allowance for doubtful accounts for estimated losses. In establishing the allowance, management considers historical losses adjusted to take into account current market conditions and their customers’ financial condition, the amount of receivables in dispute, and the current receivables aging and current payment patterns. The Company had no write-offs in 2017, 20162023 and 20152022 and the Company did not record an allowance for doubtful accounts as of December 31, 20172023 and 20162022 as there were no accounts receivable outstanding. The Company does not have any off-balance-sheet credit exposure related to its customers.
(d)Revenue Recognition
The Company generates most of its revenue from license and royalty arrangements. At the inception of each contract, the Company identifies the goods and services that have been promised to the customer and each of those that represent a distinct performance obligation, determines the transaction price including any variable consideration, allocates the transaction price to the distinct performance obligations and determines whether control transfers to the customer at a point in time or over time. Variable consideration is included in the transaction price to the extent that it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur or when the uncertainty associated with the variable consideration is subsequently resolved. The Company reassesses its reserves for variable consideration at each reporting date and makes adjustments, if necessary, which may affect revenue and earnings in periods in which any such changes become known.
| 69 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 20162023 and 20152022
In May 2014, the Financial(2) Summary of Significant Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (Topic 606) with amendments inPolicies – (continued)
2015 (ASU 2015-14)
Disaggregation of Revenue. In the following tables, revenues reported for the years ended December 31, 2023 and 2016 (ASU 2016-8, ASU 2016-10, ASU 2016-122022, under Topic 606, are disaggregated by type of revenue.
Schedule of Disaggregation of Revenue
| Type of Revenue | 2023 | 2022 | ||||||
| License | $ | 109,987 | $ | 500,000 | ||||
| Minimum guaranteed royalties revenue (reversal of variable consideration) | (2,960,805 | ) | - | |||||
| Revenue | $ | (2,850,818 | ) | $ | 500,000 | |||
Under Topic 606, all revenue has been recognized as point in time for the years ended December 31, 2023 and ASU 2016-20)2022.
See Note 4 for a description of the license agreement with Antares Pharma, Inc. (the “Antares License Agreement”). See Note 12 for a description of the agreement with Spriaso.
The updated standard
License Fees. For distinct license performance obligations, upfront license fees are recognized when the Company satisfies the underlying performance obligation. This generally occurs upon transfer of the right to use the Company’s licensed technology to the customer. In addition, license arrangements may include contingent milestone payments, which are due following achievement by our licensee of specified sales or regulatory milestones and for which the licensee and/or Company must fulfill its performance obligation prior to achievement of these milestones. Because of the uncertainty of the milestone achievement, and/or the dependence on sales of our licensee, variable consideration for contingent milestones is fully constrained and is not recognized as revenue until the milestone is achieved by our licensee, to the extent collectability is reasonably certain.
Royalties. Royalties revenue consists of sales-based and minimum royalties earned under licenses agreements for our products. Performance obligations under these licenses, which consist of the right to use the Company’s proprietary technology, are satisfied at a new comprehensivepoint in time corresponding with delivery of the underlying technology rights to the licensee, which is generally upon transfer of the licensed technology/product to the customer. Sales-based royalties revenue recognition model that requires revenue to berepresents variable consideration under the license agreements and is recognized in a manner that depicts the transfer of goods or services toperiod a customer sells products incorporating the Company’s licensed technologies/products. The Company estimates sales-based royalties revenue earned but unpaid at an amounteach reporting period using information provided by the licensee. The Company’s license arrangements may also provide for minimum royalties, which the Company recognizes upon the satisfaction of the underlying performance obligation, which generally occurs with delivery of the underlying technology rights to the licensee. Sales-based and minimum royalties are generally due within 45 days after the end of each quarter in which they are earned.
Contract Assets
Contract assets consist of minimum royalty revenue earned in relation to the license agreement but not yet payable based on the terms of the contract. On October 2, 2023, the Company received notice from Antares of Antares’ termination of the Antares License Agreement which stated that reflects the Antares License Agreement will terminate effective January 31, 2024. The Company received approximately $902,000 from Antares during the year ended December 31, 2023 under the terms of our license agreement, of which approximately $710,000 was applied to the contract asset and $192,000 was applied to imputed interest receivable. Based on the termination notice, the Company recorded a non-cash revenue reversal of variable consideration relating to the minimum guaranteed royalties recorded as part of the Antares License Agreement of approximately $3.0 million for the balance of the contract asset that is not expected to be received in exchange for those goods or services.received. The guidance also requires disclosures regardingcontract asset as of December 31, 2023 and 2022, was zero and $3,832,000, respectively and is related to the nature, amount, timing and uncertaintyAntares License Agreement.
Revenue Concentration
A major partner is considered to be one that comprises more than 10% of the Company’s total revenues. The Company recognized a reversal of revenue relating to variable consideration of the Antares License Agreement of $3.0 million and cash flows arising from contracts with customers. The Company adopted this pronouncement effective January 1, 2017 and it did not have any effect on the Company's financial position or resultsrevenue of operations$500,000 for the years endingended December 31, 2017, 20162023, and 2015 as no2022, respectively. The reversal of revenue in 2023 is due to the write off of the contract asset resulting in a reversal of variable consideration recognized in 2021 for minimum guaranteed royalties which resulted from termination of the Antares License Agreement. License revenue recognized in 2022 was recognized during these years.100% from one major customer, Antares.
| 70 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
(2) Summary of Significant Accounting Policies – (continued)
(e) Property and Equipment
Property and equipment are recorded at cost, less accumulated depreciation. Maintenance and repairs that do not extend the life or improve the asset are expensed in the year incurred.
Depreciation is computed using the straight-line method over the estimated useful lives of the assets, which are five years for laboratory and office equipment, three years for computer equipment and software, and seven years for furniture and fixtures.
(f) Accounting for Impairment of Long-Lived Assets
Long-lived assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets held and used is measured by a comparison of the carrying amount of an asset to future net cash flows (undiscounted) expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured as the amount by which the carrying amount of the assets exceeds the fair value of the assets. Assets held for sale are reported at the lower of the carrying amount, or fair value, less costs to sell.
(g) Income Taxes
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 and 2015
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is provided against net deferred tax assets if, based upon the available evidence, it is more likely than not that some or all of the net deferred tax assets will not be realized.
The Company recognizes the effect of income tax positions only if those positions are more likely than not of being sustained. Recognized income tax positions are measured at the largest amount that is greater than 50 percent likely of being realized. Changes in recognition or measurement are reflected in the period in which the change in judgment occurs. The Company records interest and penalties related to unrecognized tax benefits as a component of its income tax expense.
(h)
Share Based Payments
The Company recognizes stock-based compensation expense for grants of stock option awards, restricted stock units and restricted stock under the Company’s Incentive Plan to employees, nonemployees and nonemployee members of the Company’s board of directors based on the grant-date fair value of those awards. The grant-date fair value of an award is generally recognized as compensation expense over the award’s requisite service period. In addition, in the past the Company has granted performance-based stock option awards and restricted stock grants,units, which vest based upon the Company satisfying certain performance conditions. Potential compensation cost, measured on the grant date, related to these performance options will be recognized only if, and when, the Company estimates that these options or units will vest, which is based on whether the Company considers the options’ performance conditions to be probable of attainment. The Company’s estimates of the number of performance-based options or units that will vest will be revised, if necessary, in subsequent periods. In addition, the Company grants stock options to nonemployee consultants from time to time in exchange for services performed for the Company. Equity instruments granted to nonemployees are subject to periodic revaluation over their vesting terms.
| 71 |
During July 2017, the Company modified 11,250 existing performance-vesting restricted stock unitsLIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
(2) Summary of a terminated employee by accelerating the vesting of these restricted stock units under the terms of the employee’s respective employment and severance agreement. Compensation expense of $46,000 was recorded as a result of the modification and recorded as general and administrative expense. Additionally, during August 2016 and in conjunction with the 2016 Restructuring Plan (see note 5), the Company modified 61,487 existing time-vested options of a terminated employee by extending the exercise period to three years from the date of modification under the terms of the employee’s respective employment and severance agreement. Compensation expense of $51,000 was recorded as a result of the modification and recorded as a restructuring charge.Significant Accounting Policies – (continued)
The Company uses the Black-Scholes model to compute the estimated fair value of stock option awards. Using this model, fair value is calculated based on assumptions with respect to (i) expected volatility of the Company’s Common Stockcommon stock price, (ii) the periods of time over which employees, nonemployees and members of the board of directors are expected to hold their options prior to exercise (expected term), (iii) expected dividend yield on the Common Stock,common stock, and (iv) risk-free interest rates. Stock-based compensation expense also includes an estimate, which is made at the time of grant, of the number of awards that are expected to be forfeited. This estimate is revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Stock-based compensation cost that has been expensed in the statements of operations amounted to $2.7 million, $2.4 million $and $1.2 million $for the years ended December 31, 2017, 20162023 and 2015,2022, allocated as follows:
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Research and development | $ | 358,352 | $ | 338,018 | ||||
| General and administrative | 296,086 | 298,122 | ||||||
| $ | 654,438 | $ | 636,140 | |||||
DECEMBER 31, 2017, 2016 and 2015
| Year Ended | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Research and development | $ | 794,367 | $ | 629,400 | $ | 287,491 | ||||||
| General and administrative | 1,921,775 | 1,691,949 | 862,927 | |||||||||
| Restructuring costs | - | 51,266 | - | |||||||||
| $ | 2,716,142 | $ | 2,372,615 | $ | 1,150,418 | |||||||
The Company issued 543,000 stock options, 990,000 stock options and 301,500 stock options respectively, during the years ended December 31, 2017, 20162023 and 2015. Additionally, the Company issued 287,000 restricted stock units during the year ended December 31, 2017 and did not issue any restricted stock units during the years ended December 31, 2016 and 2015.2022, respectively.
Key assumptions used in the determination of the fair value of stock options granted are as follows:
Expected Term: The expected term represents the period that the stock-based awards are expected to be outstanding. Due to limited historical experience of similar awards, the expected term was estimated using the simplified method in accordance with the provisions of Staff Accounting Bulletin (“SAB”) No. 107,Share-Based Payment, for awards with stated or implied service periods. The simplified method defines the expected term as the average of the contractual term and the vesting period of the stock option. For awards with performance conditions, and that have the contractual term to satisfy the performance condition, the contractual term was used.
Risk-Free Interest Rate: The risk-free interest rate used was based on the implied yield currently available on U.S. Treasury issues with an equivalent remaining term.
Expected Dividend: The expected dividend assumption is based on management’s current expectation about the Company’s anticipated dividend policy. The Company does not anticipate declaring dividends in the foreseeable future.
Expected Volatility: Since the Company did not have sufficient trading history, the volatility factor was based on the average of similar public companies through August 2014. When selecting similar companies, the Company considered the industry, stage of life cycle, size, and financial leverage. Beginning in August 2014, the volatility factor is based on a combination of the Company's trading history since March 2014 and the average of similar public companies. Beginning in July 2017, theThe volatility factor is based solely on the Company’s trading history since March 2014.history.
| 72 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
(2) Summary of Significant Accounting Policies – (continued)
Schedule of Key Assumption of Fair Value of Stock Options Granted
| 2023 | 2022 | |||||||
| Expected term | years | years | ||||||
| Risk-free interest rate | 3.73 | % | 3.06 | % | ||||
| Expected dividend yield | — | — | ||||||
| Expected volatility | 98.97 | % | 99.31 | % | ||||
| 2017 | 2016 | 2015 | ||||||||||
| Expected term | 5.85 | years | 5.84 | years | 5.75 | years | ||||||
| Risk-free interest rate | 2.13 | % | 1.76 | % | 1.63 | % | ||||||
| Expected dividend yield | — | — | — | |||||||||
| Expected volatility | 80.54 | % | 84.26 | % | 81.39 | % | ||||||
FASB Accounting Standards Codification (“ASC”) 718,Stock Compensation, requires the Company to recognize compensation expense for the portion of options that are expected to vest. Therefore, the Company applied estimated forfeiture rates that were derived from historical employee termination behavior. If the actual number of forfeitures differs from those estimated by management, additional adjustments to compensation expense may be required in future periods.
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 and 2015
As of December 31, 2017,2023, there was $2.8 million$ of total unrecognized compensation cost related to unvested share-based compensation arrangements granted under the Company’s stock option plan. That cost is expected to be recognized over a weighted average period of 2.04 years and will be adjusted for subsequent changes in estimated forfeitures. The weighted average fair value of share-based compensation awards granted during the years ended December 31, 2017, 20162023 and 20152022 was approximately $2.45 per share, $6.06$ per share and $6.60$ per share, respectively. Additionally as of December 31, 2017, there was $736,000 of total unrecognized compensation cost related to unvested restricted stock units that have performance vesting.
(i) Fair Value
The Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible. The Company determines fair value based on assumptions that market participants would use in pricing an asset or liability in the principal or most advantageous market. When considering market participant assumptions in fair value measurements, the following fair value hierarchy distinguishes between observable and unobservable inputs, which are categorized in one of the following levels:
| ● | Level 1 Inputs: Quoted prices for identical instruments in active markets. | |
| ● | Level 2 Inputs: Quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active, and model-derived valuation in which all significant inputs and significant value drivers are observable in active markets. | |
| ● | Level 3 Inputs: Valuations derived from valuation techniques in which one or more significant inputs or significant value drivers are unobservable. |
| 73 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022model-derived valuation in which all significant inputs and significant value drivers are observable in active markets.Level 3 Inputs: Valuations derived from valuation techniques in which one or more significant inputs or significant value drivers are unobservable.
(2) Summary of Significant Accounting Policies – (continued)
All of the Company’s financial instruments are valued using quoted prices in active markets or based on other observable inputs. For accrued interest income, prepaid and other current assets, accounts payable, and accrued expenses, the carrying amounts approximate fair value because of the short maturity of these instruments. The following table presents the placement in the fair value hierarchy of assets and liabilities that are measured at fair value on a recurring basis at December 31, 20172023 and 2016:2022:
Schedule of Fair Value, Assets and Liabilities Measured on Recurring Basis
| Fair value measurements at reporting date using | Fair value measurements at reporting date using | |||||||||||||||||||||||||||||||
| December 31, 2017 | Level 1 inputs | Level 2 inputs | Level 3 inputs | December 31, 2023 | Level 1 inputs | Level 2 inputs | Level 3 inputs | |||||||||||||||||||||||||
| Assets: | ||||||||||||||||||||||||||||||||
| Cash equivalents - money market funds | $ | 2,171,814 | $ | 2,171,814 | $ | - | $ | - | $ | 4,695,491 | $ | 4,695,491 | $ | - | $ | - | ||||||||||||||||
| Government bonds and notes | 4,741,690 | 4,741,690 | - | - | ||||||||||||||||||||||||||||
| Corporate bonds, notes and commercial paper | 13,515,631 | - | 13,515,631 | - | ||||||||||||||||||||||||||||
| Government treasury bills | 14,281,104 | 14,281,104 | - | - | ||||||||||||||||||||||||||||
| US. Government agency securities | 2,982,684 | - | 2,982,684 | - | ||||||||||||||||||||||||||||
| $ | 20,429,135 | $ | 6,913,504 | $ | 13,515,631 | $ | - | |||||||||||||||||||||||||
| $ | 21,959,279 | $ | 18,976,595 | $ | 2,982,684 | $ | - | |||||||||||||||||||||||||
| Liabilities: | ||||||||||||||||||||||||||||||||
| Warrant liability | $ | 17,166 | $ | - | $ | - | $ | 17,166 | ||||||||||||||||||||||||
| $ | 21,976,445 | $ | 18,976,595 | $ | 2,982,684 | $ | 17,166 | |||||||||||||||||||||||||
| Fair value measurements at reporting date using | ||||||||||||||||
| Deember 31, 2022 | Level 1 inputs | Level 2 inputs | Level 3 inputs | |||||||||||||
| Assets: | ||||||||||||||||
| Cash equivalents - money market funds | $ | 2,694,434 | $ | 2,694,434 | $ | - | $ | - | ||||||||
| Government treasury bills | 5,959,000 | 5,959,000 | - | - | ||||||||||||
| Commercial paper | 14,586,930 | - | 14,586,930 | - | ||||||||||||
| Corporate bonds and notes | 5,454,690 | - | 5,454,690 | - | ||||||||||||
| U.S. government agency securities | 3,380,790 | - | 3,380,790 | - | ||||||||||||
| $ | 32,075,844 | $ | 8,653,434 | $ | 23,422,410 | $ | - | |||||||||
| Liabilities: | ||||||||||||||||
| Warrant liability | $ | 229,856 | $ | - | $ | - | $ | 229,856 | ||||||||
| $ | 32,305,700 | $ | 8,653,434 | $ | 23,422,410 | $ | 229,856 | |||||||||
| 74 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 20162023 and 20152022
| Fair value measurements at reporting date using | ||||||||||||||||
| December 31, 2016 | Level 1 inputs | Level 2 inputs | Level 3 inputs | |||||||||||||
| Assets: | ||||||||||||||||
| Cash equivalents - money market funds | $ | 2,920,577 | $ | 2,920,577 | $ | - | $ | - | ||||||||
| Government bonds and notes | 7,471,054 | 3,752,350 | 3,718,704 | - | ||||||||||||
| Corporate bonds, notes and commercial paper | 13,808,516 | - | 13,808,516 | - | ||||||||||||
| $ | 24,200,147 | $ | 6,672,927 | $ | 17,527,220 | $ | - | |||||||||
(2) Summary of Significant Accounting Policies – (continued)
The following methods and assumptions were used to determine the fair value of each class of assets and liabilities recorded at fair value in the balance sheets:
Cash equivalents: Cash equivalents primarily consist of highly-ratedhighly rated money market funds and commercial papertreasury bills with original maturities to the Company of three months or less and are purchased daily at par value with specified yield rates. Cash equivalents related to money market funds and treasury bills are classified within Level 1 of the fair value hierarchy because they are valued using quoted market prices or broker or dealer quotations for similar assets. Cash equivalents related
Government bonds and notes: The Company uses a third-party pricing service to value these investments. United States bonds and notes are classified within Level 1 of the fair value hierarchy because they are valued using quoted market prices for identical assets and reportable trades.
Corporate bonds, notes, and commercial paper: The Company uses a third-party pricing service to value these investments. Corporate bonds, notes and commercial paper are classified within Level 2 of the fair value hierarchy because they are valued using broker/dealer quotes, bids and offers, benchmark yields and credit spreads and other observable inputs.
Government bondsWarrant liability: The warrant liability (which relates to warrants to purchase shares of common stock) is marked-to-market each reporting period with the change in fair value recorded to other income (expense) in the accompanying statements of operations until the warrants are exercised, expire or other facts and notes:circumstances lead the warrant liability to be reclassified to stockholders’ equity. The Company uses a third-party pricing service tofair value these investments. United States Treasury bonds and notes are classified within Level 1 of the warrant liability is estimated using a Black-Scholes option-pricing model. The significant assumptions used in preparing the option pricing model for valuing the warrant liability as of December 31, 2023, include (i) volatility of %, (ii) risk free interest rate of %, (iii) strike price of $, (iv) fair value hierarchy because they are valued using quoted market pricesof common stock of $, and (v) expected life of years. The significant assumptions used in active marketspreparing the option pricing model for identical assets and reportable trades. Other United States government agency bonds are classified within Level 2valuing the warrant liability as of theDecember 31, 2022, include (i) volatility of %, (ii) risk free interest rate of %, (iii) strike price of $, (iv) fair value hierarchy because they are valued using broker/dealer quotes, bidsof common stock of $, and offers, benchmark yields and credit spreads and other observable inputs.(v) expected life of years.
Corporate bonds, notes, and commercial paper: The Company uses a third-party pricing service to value these investments. The pricing service utilizes broker/dealer quotes, bids and offers, benchmark yields and credit spreads and other observable inputs.
The Company’s accounting policy is to recognize transfers between levels of the fair value hierarchy on the date of the event or change in circumstances that caused the transfer. There were no transfers into or out of Level 1, Level 2 or Level 23 for the years ended December 31, 20172023 and 2016.December 31, 2022.
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 and 2015
Basic earnings (loss) per share is calculated by dividing net income (loss) available to common shareholders by the weighted average number of common shares outstanding during the period. Net income (loss) available to common shareholders for the year ended December 31, 2017, 2016 and 2015 was calculated using the two-class method, which is an earnings (loss) allocation method for computing earnings (loss) per share when an entity’s capital structure includes common stock and participating securities. The two-class method determines earnings (losses) per share based on dividends declared on common stock and participating securities (i.e., distributed earnings) and participation rights of participating securities in any undistributed earnings (loss). The application of the two-class method was required since the Company’s unvested restricted stock contains non-forfeitable rights to dividends or dividend equivalents. However, unvested restricted stock grants are not included in computing basic earnings (loss) per share for periods where the Company has losses as these securities are not contractually obligated to share in losses of the Company.
Diluted earnings (loss) per share is based on the weighted average number of common shares outstanding plus, where applicable, the additional potential common shares that would have been outstanding related to dilutive options, warrants, and unvested restricted stock units to the extent such shares are dilutive.
| 75 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
(2)Summary of Significant Accounting Policies – (continued)
Schedule of Computation of Basic and Diluted Earnings (Loss) Per Share of Common Stock
| 2023 | 2022 | |||||||||||||||||||
| Year Ended December 31, | Years Ended December 31, | |||||||||||||||||||
| 2017 | 2016 | 2015 | 2023 | 2022 | ||||||||||||||||
| Basic loss per share attributable to common stock: | ||||||||||||||||||||
| Numerator | ||||||||||||||||||||
| Net loss | $ | (20,982,860 | ) | $ | (18,971,508 | ) | $ | (18,208,378 | ) | $ | (16,352,171 | ) | $ | (10,758,636 | ) | |||||
| Denominator | ||||||||||||||||||||
| Weighted avg. common shares outstanding | 20,051,934 | 18,258,149 | 16,470,814 | 5,269,671 | 5,231,681 | |||||||||||||||
| Basic loss per share attributable to common stock | $ | (1.05 | ) | $ | (1.04 | ) | $ | (1.11 | ) | $ | (3.10 | ) | $ | (2.06 | ) | |||||
| Diluted loss per share attributable to common stock: | ||||||||||||||||||||
| Numerator | ||||||||||||||||||||
| Net loss | $ | (20,982,860 | ) | $ | (18,971,508 | ) | $ | (18,208,378 | ) | $ | (16,352,171 | ) | $ | (10,758,636 | ) | |||||
| Effect of dilutive securities on net loss: | ||||||||||||||||||||
| Common stock warrants | 212,690 | 565,940 | ||||||||||||||||||
| Total net loss for purpose of calculating diluted net loss per common share | $ | (16,564,861 | ) | $ | (11,324,576 | ) | ||||||||||||||
| Denominator | ||||||||||||||||||||
| Weighted avg. common shares outstanding | 20,051,934 | 18,258,149 | 16,470,814 | 5,269,671 | 5,231,681 | |||||||||||||||
| Weighted average effect of dilutive securities: | ||||||||||||||||||||
| Common stock warrants | - | 24,488 | ||||||||||||||||||
| Total shares for purpose of calculating diluted net loss per common share | 5,269,671 | 5,256,169 | ||||||||||||||||||
| Diluted loss per share attributable to common stock | $ | (1.05 | ) | $ | (1.04 | ) | $ | (1.11 | ) | $ | (3.14 | ) | $ | (2.15 | ) | |||||
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 and 2015
The computation of diluted earnings per share for the years ended December 31, 2017, 20162023 and 20152022 does not include the following stock options unvested restricted stock awards andor warrants to purchase shares in the computation of diluted earnings per share because these instruments were antidilutive:
Schedule of Anti-dilutive Securities Excluded from Computation of Earnings Per Share
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Stock options | 262,247 | 277,225 | ||||||
| Warrants | 49,433 | 49,433 | ||||||
| 76 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
(2) Summary of Significant Accounting Policies – (continued)
| December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Stock options | 2,374,449 | 2,225,850 | 1,722,552 | |||||||||
| Unvested restricted stock awards | - | - | 3,000 | |||||||||
| Unvested restricted stock units | 203,998 | - | - | |||||||||
(k) Segment Information
The Company is a single reportable segment engaged in research and development for the delivery of drugs using its proprietary delivery technology. Operating segments are identified as components of an enterprise for which separate discrete financial information is available for evaluation by the chief operating decision maker in making decisions regarding resource allocation and assessing performance. The chief operating decision maker made such decisions and assessed performance at the company level, as one segment.
(l) Principles of Consolidation
The consolidated financial statements include the accounts of the Company and all subsidiaries. The Company eliminates all intercompany accounts and transactions in consolidation.
(3) Marketable Investment Securities
The Company has classified its marketable investment securities as available-for-sale securities, all of which are debt securities. These securities are carried at fair value with unrealized holding gains and losses, net of the related tax effect, included in accumulated other comprehensive lossincome (loss) in stockholders’ equity until realized. Gains and losses on investment security transactions are reported on the specific-identification method. Dividend income is recognized on the ex-dividend date and interest income is recognized on an accrual basis. The amortized cost, gross unrealized holding gains, gross unrealized holding losses, and fair value for available-for-sale securities by major security type and class of security at December 31, 20172023 and 20162022 were as follows:
| December 31, 2017 | Amortized Cost | Gross unrealized holding gains | Gross unrealized holding losses | Aggregate fair value | ||||||||||||
| Government bonds and notes | $ | 4,744,566 | $ | - | $ | (2,876 | ) | $ | 4,741,690 | |||||||
| Corporate bonds, notes and commercial paper | 13,517,371 | - | (1,740 | ) | 13,515,631 | |||||||||||
| $ | 18,261,937 | $ | - | $ | (4,616 | ) | $ | 18,257,321 | ||||||||
Schedule of Available for Sale Securities
| December 31, 2023 | Amortized Cost | Gross unrealized holding gains | Gross unrealized holding losses | Aggregate fair value | ||||||||||||
| Government treasury bills | $ | 14,272,530 | $ | 8,574 | $ | - | $ | 14,281,104 | ||||||||
| U.S. government agency securities | 2,983,999 | - | (1,315 | ) | 2,982,684 | |||||||||||
| $ | 17,256,529 | $ | 8,574 | $ | (1,315 | ) | $ | 17,263,788 | ||||||||
| December 31, 2022 | Amortized Cost | Gross unrealized holding gains | Gross unrealized holding losses | Aggregate fair value | ||||||||||||
| Government treasury bills | $ | 5,973,087 | $ | - | $ | (14,087 | ) | $ | 5,959,000 | |||||||
| Commercial paper | 20,052,505 | - | (10,885 | ) | 20,041,620 | |||||||||||
| U.S. government agency securities | 3,376,139 | 4,651 | - | 3,380,790 | ||||||||||||
| $ | 29,401,731 | $ | 4,651 | $ | (24,972 | ) | $ | 29,381,410 | ||||||||
| 77 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 20162023 and 20152022
| (3) | Marketable Investment Securities - (continued) |
| December 31, 2016 | Amortized Cost | Gross unrealized holding gains | Gross unrealized holding losses | Aggregate fair value | ||||||||||||
| Government bonds and notes | $ | 7,473,273 | $ | - | $ | (2,219 | ) | $ | 7,471,054 | |||||||
| Corporate bonds, notes and commercial paper | 13,814,790 | - | (6,274 | ) | 13,808,516 | |||||||||||
| $ | 21,288,063 | $ | - | $ | (8,493 | ) | $ | 21,279,570 | ||||||||
Maturities of debt securities classified as available-for-sale securities at December 31, 20172023 are as follows:
| Amortized Cost | Aggregate fair value | |||||||
| Due within one year | $ | 18,261,937 | $ | 18,257,321 | ||||
| $ | 18,261,937 | $ | 18,257,321 | |||||
Schedule of Maturities of Debt Securities Classified as Available-for-Sale Securities
| December 31, 2023 | Amortized Cost | Aggregate fair value | ||||||
| Due within one year | $ | 17,256,529 | $ | 17,263,788 | ||||
| $ | 17,256,529 | $ | 17,263,788 | |||||
There were no sales of marketable investment securities during the years ended December 31, 2017, 20162023 and 20152022 and therefore no realized gains or losses. Additionally, $36.2 million, $28.6$36.0 million and $800,000$59.5 million of marketable investment securities matured during the years ended December 31, 2017, 20162023 and 2015,2022, respectively. The Company determined there were no other-than-temporary impairments for the years ended December 31, 2017, 20162023 and 2015.2022.
(4) Contractual Agreements
| (a) | Abbott Products, Inc. |
On March 29, 2012, the Company terminated its collaborative agreement with Solvay Pharmaceuticals, Inc. (later acquired by Abbott Products, Inc.) for TLANDO. As part of the termination, the Company reacquired the rights to the intellectual property from Abbott. All obligations under the prior license agreement have been completed except that Lipocine will owe Abbott a perpetual 1% royalty on net sales. Such royalties are limited to $1.0$1.0 million in the first two calendar years following product launch, after which period there is not a cap on royalties and no maximum aggregate amount. If generic versions of any such product are introduced, then royalties are reduced by 50%. The Company didincurred royalty expense of $34,000 and $12,000 in the years ended December 31, 2023 and 2022, respectively.
| (b) | Antares Pharma, Inc. |
On October 14, 2021, the Company entered into a license agreement (“License Agreement”) with Antares Pharma, Inc. (“Antares”) pursuant to which the Company granted to Antares an exclusive, royalty-bearing, sublicensable right and license to develop and commercialize, upon final approval of TLANDO® from the U.S. Food and Drug Administration (“FDA”), the Company’s TLANDO product with respect to testosterone replacement therapy in males for conditions associated with a deficiency or absence of endogenous testosterone, as indicated in NDA No. 208088, treatment of Klinefelter syndrome, and pediatric indications relating to testosterone replacement therapy in males for conditions associated with a deficiency or absence of endogenous testosterone (the “Field”), in each case within the United States. TLANDO received FDA approval on March 29, 2022.
Upon execution of the Antares License Agreement, Antares paid to the Company an initial payment of $11.0 million. Antares agreed to make additional payments of $5.0 million to the Company on each of January 1, 2025, and January 1, 2026, provided that certain conditions were satisfied. The Company was also eligible to receive milestone payments of up to $160.0 million in the aggregate, depending on the achievement of certain sales milestones in a single calendar year with respect to all products licensed by Antares under the Antares License Agreement. In addition, upon commercialization, the Company was to receive tiered royalty payments at rates ranging from percentages in the mid-teens to up to 20% of net sales of TLANDO in the United States, subject to certain minimum royalty obligations. On October 2, 2023, the Company received notice from Antares of Antares’ termination of the License Agreement. In accordance with the terms of the License Agreement, the License Agreement will terminate effective January 31, 2024. On January 12, 2024, the Company entered into a license agreement (the “Verity License Agreement”) with Verity
| 78 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
| (4) | Contractual Agreements – (continued) |
Pharmaceuticals Inc. (Verity). See Note 4(c) for a description of the Verity License Agreement. Upon termination of the Antares License Agreement, all rights and licenses granted by the Company to Antares under the Antares License Agreement will terminate and all rights in TLANDO will be transferred to the Company’s new licensing partner, Verity.
Under the Antares License Agreement, the Company retained development and commercialization rights in the rest of the world, and with respect to applications outside of the Field inside or outside the United States. The License Agreement also provided Antares with an option, exercisable on or before March 31, 2022, to license TLANDO XR (LPCN 1111), the Company’s potential once-daily oral product candidate for testosterone replacement therapy. On April 1, 2022, the Company entered into the First Amendment to the License Agreement (the “Amendment”), pursuant to which the License Agreement was amended to extend the deadline by which Antares was to exercise its option to license TLANDO XR to June 30, 2022. As consideration for the Company agreeing to enter into the Amendment, in April 2022 Antares paid the Company a non-refundable cash fee of $500,000. On June 30, 2022, Antares’ option to license TLANDO XR expired and was not incur any royaltiesexercised. Lipocine retains all development and commercialization rights to TLANDO XR. The Company recognized a reversal of variable consideration revenue under the Antares License Agreement of $3.0 million during the year ended December 31, 2023 and license revenue $500,000 during the years ended December 31, 2017, 2016 and 2015.2022.
| Verity Pharmaceuticals, Inc. |
On January 12, 2024, the Company entered into a License Agreement (the “License Agreement”) with Gordon Silver Limited (“GSL”) and Verity Pharmaceuticals, Inc. (“Verity Pharma”), pursuant to which the Company granted to GSL (an affiliate of Verity Pharma) an exclusive, royalty-bearing, sublicensable right and license to commercialize the Company’s TLANDO product with respect to testosterone replacement therapy in males for conditions associated with a deficiency or absence of endogenous testosterone, as indicated in NDA No. 208088, treatment of Klinefelter syndrome, and pediatric indications relating to testosterone replacement therapy in males for conditions associated with a deficiency or absence of endogenous testosterone (the “Field”), in each case within the United States and Canada. The License Agreement also provides GSL with a license to develop and commercialize TLANDO XR, the Company’s potential once-daily oral product candidate for testosterone replacement therapy. The Company retains development and commercialization rights outside of the United States and Canada, and with respect to applications outside of the Field inside or outside the United States and Canada.
Upon execution of the License Agreement, GSL agreed to pay the Company a license fee of $11.0 million with an initial payment of $2.5 million which was received on signing of the License Agreement, $5.0 million received on February 1, 2024, $2.5 million to be paid no later than January 1, 2025, and $1.0 million to be paid no later than January 1, 2026. The Company is also eligible to receive development and sales milestone payments of up to $259.0 million in the aggregate, depending primarily on the achievement of certain sales milestones in a single calendar year with respect to all products licensed by GSL under the License Agreement. In addition, the Company is eligible to receive tiered royalty payments at rates ranging from 12% up to 18% of net sales of licensed products in the United States and Canada.
Pursuant to the terms of the License Agreement, GSL is generally responsible for expenses relating to the development (including the conduct of any clinical trials) and commercialization of licensed products in the Field in the United States and Canada, while the Company is generally responsible for expenses relating to development activities outside of the Field and/or the United States and Canada.
| (d) | Contract Research and Development |
The Company has entered into agreements with various contract organizations that conduct preclinical, clinical, analytical and manufacturing development work on behalf of the Company as well as a number of independent contractors, primarily clinical researchers, who serve as advisors to the Company. The Company incurred expenses of $8.5 million, $5.5$6.7 million and $10.0$5.6 million under these agreements in 2017, 20162023 and 20152022 and has recorded these expenses in research and development expenses.
| 79 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 20162023 and 20152022
Restructuring charges relate(5) Loan and Security Agreement
Silicon Valley Bank Loan
On January 5, 2018, the Company entered into a Loan and Security Agreement (the “Loan and Security Agreement”) with Silicon Valley Bank (“SVB”) pursuant to our initiativewhich SVB agreed to restructure operationslend the Company $10.0 million. The principal borrowed under the Loan and Security Agreement bore interest at a rate equal to the Prime Rate, as reported in the money rates section of The Wall Street Journal or any successor publication representing the rate of interest per annum then in effect, plus one percent per annum, which interest was approved bypayable monthly. Additionally on April 1, 2020, the board of directors on July 13, 2016.Company entered into a Deferral Agreement with SVB. Under the July 2016 restructuring,Deferral Agreement, principal repayments were deferred by six months and the Company reduced its workforce by eight positions, constituting 33%was only required to make monthly interest payments. The loan matured on June 1, 2022. The Company made a final payment at maturity equal to $650,000 (the “Final Payment Charge”) at the time the loan matured. The expense of the Company’s workforce. The reduction in workforce involved all functional disciplines including generalfinal payment charge had been recognized over the term of the facility using the effective interest method.
(6) Property and administrative employees, sales and marketing and research and development personnel. Additionally, the Board approved a further restructuring in October 2016 whereby the Company reduced its workforce by an additional two positions in the sales and marketing functions. The restructurings that occurred in 2016 are jointly referred to as the 2016 Restructuring Plan.Equipment
The charge related to the 2016 Restructuring Plan during the year ended December 31, 2016 was $729,000 and was comprised of $678,000 in severance related expenses and $51,000 for extending the exercise period of certain options under an existing employee severance agreement. The activity for the year ended December 31, 2017 for restructuring charges is as follows:
| December 31, | ||||
| 2017 | ||||
| Accrued restructuring charges payable at January 1, 2017 | $ | 239,573 | ||
| Restructuring expenses during 2017 | - | |||
| Restructuring payments during 2017 | (239,573 | ) | ||
| Accrued restructuring charges payable at December 31, 2017 | $ | - | ||
Property and equipment consisted of the following:
| December 31, 2017 | December 31, 2016 | |||||||
| Computer equipment and software | $ | 43,361 | $ | 43,361 | ||||
| Lab and office equipment | 1,048,932 | 1,048,932 | ||||||
| Furniture and fixtures | 103,857 | 103,857 | ||||||
| 1,196,150 | 1,196,150 | |||||||
| Less accumulated depreciation | (1,121,080 | ) | (1,092,710 | ) | ||||
| $ | 75,070 | $ | 103,440 | |||||
Schedule of Property and Equipment
| 2023 | 2022 | |||||||
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Computer equipment and software | $ | 66,830 | $ | 53,663 | ||||
| Lab and office equipment | 1,180,052 | 1,180,052 | ||||||
| Furniture and fixtures | 51,404 | 51,404 | ||||||
| Property and equipment, gross | 1,298,286 | 1,285,119 | ||||||
| Less accumulated depreciation | (1,182,191 | ) | (1,153,530 | ) | ||||
| Property and equipment, net | $ | 116,095 | $ | 131,589 | ||||
Depreciation expense for the years ended December 31, 20172023 and 20162022 was $28,000approximately $29,000 and $32,000.$9,500, respectively.
| 80 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 20162023 and 20152022
(7) Income Taxes
| (a) | Income Tax Expense |
Income tax expense consists of:
Schedule of Income Tax Expense
| 2023 | 2022 | |||||||||||||||||||
| December 31, | December 31, | |||||||||||||||||||
| 2017 | 2016 | 2015 | 2023 | 2022 | ||||||||||||||||
| U.S. federal | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||
| State and local | 700 | 752 | 200 | 755 | 681 | |||||||||||||||
| Deferred | - | - | - | - | - | |||||||||||||||
| Total | $ | 700 | $ | 752 | $ | 200 | $ | 755 | $ | 681 | ||||||||||
| (b) | Tax Rate Reconciliation |
Income tax expense was $700, $752$755 and $200,$681, respectively, for the years ended December 31, 2017, 20162023 and 20152022 and differed from the amounts computed by applying the U.S. federal income tax rate of 34%21% for 2023 and 2022, respectively, to pretax income from continuing operations as a result of the following:
Schedule of Pretax Income from Continuing Operations
| December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Computed "expected" tax expense (benefit) | $ | (7,133,934 | ) | $ | (6,450,075 | ) | $ | (6,190,781 | ) | |||
| Increase (reduction) in income taxes resulting from: | ||||||||||||
| Change in valuation allowance | (6,173,708 | ) | 6,709,591 | 6,691,992 | ||||||||
| Difference associated with federal rate change | 13,240,342 | - | - | |||||||||
| State and local income taxes, net of federal income tax benefit | 462 | 496 | 132 | |||||||||
| Stock expense | 684,854 | 200,002 | 102,221 | |||||||||
| Research and development tax credits | (394,470 | ) | (337,968 | ) | (452,609 | ) | ||||||
| Orphan drug tax credit | (227,109 | ) | (127,641 | ) | (155,452 | ) | ||||||
| Other, net | 4,263 | 6,347 | 4,697 | |||||||||
| $ | 700 | $ | 752 | $ | 200 | |||||||
| 2023 | 2022 | |||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Computed “expected” tax expense (benefit) | $ | (3,433,893 | ) | $ | (2,259,272 | ) | ||
| Increase (reduction) in income taxes resulting from: | ||||||||
| Change in valuation allowance | 3,619,564 | 2,529,547 | ||||||
| State and local income taxes, net of federal income tax benefit | 596 | 538 | ||||||
| Stock expense | 319,214 | 314,186 | ||||||
| Research and development tax credits | (434,858 | ) | (422,495 | ) | ||||
| Orphan drug tax credit | (26,245 | ) | (42,976 | ) | ||||
| Warrant Liability | (44,665 | ) | (118,847 | ) | ||||
| Other, net | 1,042 | - | ||||||
| Total | $ | 755 | $ | 681 | ||||
| 81 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 20162023 and 20152022
| (7) | Income Taxes – (continued) |
| (c) | Significant Components of Deferred Taxes |
The tax effects of temporary differences that give rise to significant portions of the deferred tax assets and deferred tax liabilities at December 31, 20172023 and 20162022 are presented below.below:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Deferred tax assets: | ||||||||
| Stock-based compensation | $ | 1,330,706 | $ | 1,924,260 | ||||
| Net operating loss carryforwards | 25,395,518 | 30,700,358 | ||||||
| Employee benefits | 54,729 | 69,112 | ||||||
| Research and development tax credits | 3,012,081 | 2,465,893 | ||||||
| Orphan drug tax credits | 1,023,290 | 679,185 | ||||||
| Other deductible tempory differences | 104 | 70,231 | ||||||
| Total gross deferred tax assets | 30,816,428 | 35,909,039 | ||||||
| Less valuation allowance | (30,813,270 | ) | (35,904,927 | ) | ||||
| Net deferred tax assets | 3,158 | 4,112 | ||||||
| Deferred tax liabilities: | ||||||||
| Plant and equipment | (3,158 | ) | (4,112 | ) | ||||
| Total gross deferred tax liabilities | (3,158 | ) | (4,112 | ) | ||||
| Net deferred tax liabilities | $ | - | $ | - | ||||
Schedule of Deferred Tax Assets and Liabilities
On December 22, 2017, the Tax Cuts and Jobs Act of 2017 (“Tax Act”) was signed into law. The new law includes, among other items, a permanent reduction to the U.S. corporate income tax rate from 34% to 21% effective January 1, 2018. As a result, at December 31, 2017, we recognized a tax expense of $13.2 million from revaluing U.S net deferred tax assets which was offset by a corresponding change in the Company’s valuation allowance.
| 2023 | 2022 | |||||||
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Deferred tax assets: | ||||||||
| Stock-based compensation | $ | 1,188,535 | $ | 1,428,167 | ||||
| Net operating loss carryforwards | 35,108,766 | 35,595,940 | ||||||
| Employee benefits | 45,592 | 44,602 | ||||||
| Research and development tax credits | 6,032,559 | 5,491,805 | ||||||
| Orphan drug tax credits | 1,274,204 | 1,240,982 | ||||||
| Plant and equipment | - | - | ||||||
| Sec. 174 Expenses | 3,945,862 | 1,997,787 | ||||||
| Other deductible temporary differences | 167,371 | 69,273 | ||||||
| Total gross deferred tax assets | 47,762,889 | 45,868,556 | ||||||
| Net deferred tax assets | $ | 47,762,889 | $ | 45,868,556 | ||||
| Deferred tax liabilities: | ||||||||
| Plant and equipment | (7,227 | ) | (9,052 | ) | ||||
| Total gross deferred tax liabilities | (7,227 | ) | (9,052 | ) | ||||
| Net deferred tax liabilities | $ | (7,227 | ) | $ | (9,052 | ) | ||
| Deferred tax asset/deferred tax liability | 47,755,662 | 45,859,504 | ||||||
| Valuation allowance | (47,755,662 | ) | (45,859,504 | ) | ||||
| Net deferred tax asset | $ | - | $ | - | ||||
The valuation allowance for deferred tax assets as of December 31, 20172023 and 20162022 was $30.8$47.8 million and $35.9 million.$45.9 million, respectively. The net change in the valuation allowance was a decreasean increase of $5.1$1.9 million in 20172023 and an increase $7.7of $2.9 million in 2016.2022. A valuation allowance has been provided for the full amount of the Company’s net deferred tax assets as the Company believes it is more likely than not that these benefits will not be realized. In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities (including the impact of available carryback and carryforward periods), projected future taxable income, and tax planning strategies in making this assessment.
| 82 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 AND 20152023 and 2022
| (7) | Income Taxes – (continued) |
During the year ended December 31, 2013, the Company experienced a change in ownership, as defined by the Internal Revenue Code, as amended (the “Code”) under Section 382. A change of ownership occurs when ownership of a company increases by more than 50 percentage points over a three-year testing period of certain stockholders. As a result of this ownership change, we determined that our annual limitation on the utilization of our federal net operating loss (“NOL”) and credit carryforwards is approximately $1.1$1.1 million per year. We will only be able to utilize $20.2$9.8 million of our pre-ownership change NOL carryforwards and will forgo utilizing $5.5$3.3 million of our pre-ownership change NOL carryforwards and $1.2$1.2 million of our pre-change credit carryforwards as a result of this ownership change. We do not account for forgone NOL and credit carryovers in our deferred tax assets and only account for the NOL and credit carryforwards that will not expire unutilized as a result of the restrictions of Code Section 382.
As of December 31, 2017,2023, we had NOL and research and development credit carryforwards for U.S. federal income tax reporting purposes of approximately $96.5$138.6 million and $2.0$4.4 million, respectively. Approximately $25.7$33.7 million of the NOL will expire between 20232024 and 20332034 and $70.8$52.4 million of the NOL will expire 20342035 through 2037.2037. Pursuant to the Tax Cuts and Jobs Act of 2017, NOL’s generated in 2018 and subsequent years have an unlimited carryforward therefore the 2023, 2022, 2020, 2019 and 2018 NOL of $52.5 million can be carried forward indefinitely. The research and development credits will begin to expire in 2033 through 2037.2043. We have orphan drug credit carry forwards of approximately $1.0$1.3 million which will expire if unused through 2037.2043.
We also have state NOL and research and development credit carry-forwardscarry forwards of approximately $102.5$128.9 million and $971,000,$1.7 million, respectively. Approximately $12.4 million of the Company'sThe Company’s state NOL expires in 2018, $37.7of $31.2 million expireswill expire between 20192024 and 2029, and $52.4$97.7 million will expire in 2030 through 2033.2037. The state research and development credits expire in 20232024 through 2030.2038.
The Company'sCompany’s federal and state income tax returns for December 31, 20142020 through 20172023 are open tax years.
A reconciliation of the beginning and ending amount of total unrecognized tax contingencies, excluding interest and penalties, for the years ended December 31, 20172023 and 20162022 are as follows:
Schedule of Reconciliation of the Beginning and Ending Amount of Total Unrecognized Tax Contingencies, Excluding Interest and Penalties
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Balance, beginning of year | $ | - | $ | - | ||||
| Balance, end of year | $ | - | $ | - | ||||
| 83 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Balance, beginning of year | $ | - | $ | - | ||||
| Balance, end of year | $ | - | $ | - | ||||
(8) Leases
On August 6, 2004, the
The Company assumedhas a non-cancelable operating lease for office space and laboratory facilities in Salt Lake City, Utah. On May 6, 2014,January 24, 2024, the Company modified and extendedterm of the lease has been extended through February 28, 2018. On February 8, 2018, the Company extended the lease through February 28, 2019. Additionally, on December 28, 2015, the Company entered into an operating lease for office space in Lawrenceville, New Jersey through January 31, 2018.2025.
Future minimum lease payments under the non-cancelable operating leaseslease as of December 31, 20172023 are:
| Operating | ||||
| leases | ||||
| Year ending December 31: | ||||
| 2018 | 58,903 | |||
| Total minimum lease payments | $ | 58,903 | ||
Schedule of Future Minimum Rental Payments for Operating Leases
| Operating | ||||
| leases | ||||
| Year ending December 31: | ||||
| 2024 | $ | 366,290 | ||
| 2025 | 61,346 | |||
| Total minimum lease payments | $ | 427,636 | ||
The Company’s rent expense was $379,000, $374,000$355,000 and $295,000$341,000 for the years ended December 31, 2017, 20162023 and 2015.2022, respectively.
(9) Stockholders’ Equity
On May 1, 2023, at the 2023 annual meeting of the stockholders, the Company’s stockholders approved an amendment to the Company’s Amended and Restated Certificate of Incorporation to effect a reverse stock split at a ratio not less than 1-for-5 and not more than 1-for-20, with the exact ration to be set within that range at the discretion of the Board without further approval or authorization from stockholders.
On May 10, 2023, the Company’s Board approved a reverse stock split of 1-for-17. The Company filed the Amendment to its Certificate of Incorporation with the Secretary of the State of Delaware on May 10, 2023, and the Amendment became effective at 5:00 pm Eastern Time on May 11, 2023. The Company’s shares began trading on a split-adjusted basis on the Nasdaq Capital Market commencing upon market open on May 12, 2023.
All common stock share data and per share price data of the Company reflect the reverse stock split effective May 11, 2023.
On June 8, 2022, at the 2022 annual meeting of the stockholders, the Company’s stockholders approved an amendment to the Company’s Amended and Restated Certificate of Incorporation to increase the number of authorized shares of the Company’s common stock, par value $, from shares to shares. The Company filed the amendment to the Restated Certificate with the Secretary of State of the State of Delaware on June 28, 2022. The amendment to the Restated Certificate became effective upon filing with the Secretary of State of the State of Delaware.
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 AND 2015
| (a) | Issuance of Common Stock |
InOn March 6, 2017, the Company entered into a Controlled Equity Offering sales agreement (“Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co. (“Cantor”), pursuant to which the Company may issue and sell, shares of our common stock, with aggregate gross sales proceeds of up to $20.0 million, from time to time, through an “at the market” (“ATM”), equity offering program, under which Cantor acts as sales agent. The shares of its common stock having an aggregate offering price of up to be soldthe amount the Company registered on an effective registration statement pursuant to which the offering is being made. The Company currently has registered up to $50.0 million for sale under the Sales Agreement, will be sold and issued pursuant to the Company’s Registration Statement on Form S-3 (File No. 333-199093) (the “Existing Form S-3”), which was previously declared effective333-250072) through Cantor as the Company’s sales agent. Cantor may sell the Company’s common stock by any method permitted by law deemed to be an “at the market offering” as defined in Rule 415(a)(4) of the Securities Act, including sales made directly on or through the NASDAQ Capital Market or any other existing trade market for our common stock, in negotiated transactions at market prices prevailing at the time of sale or at prices related to prevailing market prices, or any other method permitted by law. Cantor uses its commercially reasonable efforts consistent with its normal trading and Exchange Commission,sales practices and applicable law and regulations to sell these shares. The Company pays Cantor of the related prospectusaggregate gross proceeds from each sale of shares under the Sales Agreement. In addition, the Company has also provided Cantor with customary indemnification rights.
The Company is not obligated to make any sales of its common stock under the Sales Agreement. The offering of common stock pursuant to the Sales Agreement will terminate upon the termination of the Sales Agreement as permitted therein. The Company and one or more prospectus supplements. Cantor may each terminate the Sales Agreement at any time upon ten days’ prior notice.
| 84 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
| (9) | Stockholders’ Equity – (continued) |
As of December 31, 2017,2023, we had sold an aggregate of 2,518,109 shares at a weighted-average sales price of $4.40$ per share under the ATMAt the Market Offering (the “ATM Offering”) for aggregate gross proceeds of $11.1$33.3 million and net proceeds of $10.6$32.1 million, after deducting sales agent commission and discounts and our other offering costs.
On October 13, 2017, During the year ended December 31, 2023, the Company filedsold shares of its common stock pursuant to the ATM offering at a weighted-average sales price of $ per share, resulting in net proceeds of approximately $405,000 under the Sales Agreement which is net of approximately $24,000 in expenses. During the year ended December 31, 2022, the Company did not sell any shares of its common stock pursuant to the ATM Offering. As of December 31, 2023, the Company had $40.8 million available for sale under the Sales Agreement. However, as of November 22, 2023, the Company is now subject to General Instruction I.B.6 of Form S-3 (File No. 333-220942) (the “New Form S-3”) to replacewhich limits the Existing Form S-3. The New Form S-3 registered the sale of up to $150.0 million of any combination of common stock, preferred stock, debt securities, warrants and units pursuant to a shelf registration statement. The New Form S-3 also contains a prospectus pursuant to whichamounts that we may sell from time to time,under the registration statement. As a result of such limitations, the Company has currently registered the offer and sale of shares of ourthe Company’s common stock pursuant to the Sales Agreement having an aggregate offering price of up to $25.0 million through Cantor as our sales agent, pursuant to$5.3 million.
| (b) | Series B Preferred Stock |
On March 7, 2023, the Sales Agreement. Pursuant to Rule 415(a)(6)Board of the Securities ActCompany declared a dividend of 1933, as amended,one one-thousandth (1/1,000th) of a share of Series B Preferred Stock, par value $ per share (“Series B Preferred Stock”), for each outstanding share of common stock of the offeringCompany, to stockholders of securitiesrecord on March 24, 2023. The Certificate of Designation of Series B Preferred Stock (the “Certificate of Designation”) was filed with the Delaware Secretary of State and became effective on March 10, 2023.
The dividend was based on the Existing Form S-3 are deemed terminated asnumber of shares of outstanding common stock on March 24, 2023, and resulted in Series B Preferred shares being issued. Each whole share of Series B Preferred Stock entitled the dateholder thereof to 1,000,000 votes per share, and each fraction of effectivenessa share of Series B Preferred Stock had a ratable number of votes. Thus, each one-thousandth of a share of Series B Preferred Stock was entitled to 1,000 votes. The outstanding shares of Series B Preferred Stock were entitled to vote together with the New Form S-3.
On April 29, 2015, the Company sold 5,347,500outstanding shares of common stock inas a single class exclusively with respect to any proposal to adopt an underwritten offering. Net proceedsamendment to the Company from the sale totaled approximately $32.4 million, after deducting the directCompany’s Amended and incremental expensesRestated Certificate of Incorporation, as amended (the “Certificate of Incorporation”), to effect a reverse stock split of the offering and the commissionsoutstanding shares of Common Stock at a ratio determined in connectionaccordance with the offering paidterms of such amendment (the “Reverse Stock Split”), and (ii) any proposal to adjourn any meeting of stockholders called for the purpose of voting on the Reverse Stock Split (the “Adjournment Proposal”) in conjunction with the Company’s 2023 annual meeting of stockholders.
All shares of Series B Preferred Stock that were not present in person or by proxy at the 2023 annual meeting as of immediately prior to the opening of the polls (the “Initial Redemption Time”) were automatically redeemed by the Company without further action on the part of the Company or the holder of shares of Series B Preferred Stock (the “Initial Redemption”). The remaining shares of Series B Preferred Stock that were not redeemed pursuant to the Initial Redemption were redeemed automatically upon the effectiveness of the amendment to the Certificate of Incorporation implementing the Reverse Stock Split (the “Subsequent Redemption”).
Each “beneficial owner” (as such terms are defined in the Certificate of Designation with respect to the Series B Preferred Stock) of shares of Series B Preferred Stock redeemed in the redemptions described above has the right to receive an amount equal to $0.01 in cash for each ten whole shares of Series B Preferred Stock that were “beneficially owned” by the beneficial owner as of immediately prior to the applicable redemption time and redeemed pursuant to such redemption, payable upon receipt by the Company of $2.3 million.a written request submitted by the applicable beneficial owner to the corporate secretary of the Company following the applicable redemption time.
The Series B Preferred Stock was not convertible into, or exchangeable for, shares of any other class or series of stock or other securities of the Company. The Series B Preferred Stock had no stated maturity and was not subject to any sinking fund. The Series B Preferred Stock was not subject to any restriction on the redemption or repurchase of shares by the Company while there is any arrearage in the payment of dividends or sinking fund installments.
| 85 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
| (9) | Stockholders’ Equity – (continued) |
The Company was not solely in control of the redemption of the shares of Series B Preferred Stock prior to the annual meeting of stockholders since the holders had the option of deciding whether to vote in respect of the above-described Reverse Stock Split, which determined whether a given holder’s shares of Series B Preferred Stock was redeemed in the Initial Redemption or the Subsequent Redemption. Since the redemption of the Series B Preferred Stock was not solely in the control of the Company, the shares of Series B Preferred Stock were classified within the mezzanine equity in the Company’s unaudited consolidated statement of stockholder’s equity. Upon issuance, the shares of Series B Preferred Stock were measured at redemption value. As of June 30, 2023, all shares of Series B Preferred Stock had been redeemed by the Company.
The foregoing description of the Series B Preferred Stock does not purport to be complete and is qualified in its entirety by reference to the Certificate of Designation, which is filed as Exhibit 3.2 to the Form 8-K filed with the SEC on March 10, 2023.
| Rights Agreement |
On November 13, 2015, the Company and American Stock Transfer & Trust Company, LLC, as Rights Agent, entered into a Rights Agreement. Also on November 12, 2015, the Boardboard of Directorsdirectors of the Company authorized and the Company declared a dividend of one preferred stock purchase right (each a “Right” and collectively, the “Rights”) for each outstanding share of common stock of the Company. The dividend was payable to stockholders of record as of the close of business on November 30, 2015 and entitles the registered holder to purchase from the Company one one-thousandth of a fully paid non-assessable share of Series A Junior Participating Preferred Stock of the Company at a price of $63.96$ per one-thousandth share (the “Purchase Price”). The Rights will generally become exercisable upon the earlier to occur of (i) 10 business days following a public announcement that a person or group of affiliated or associated persons has become an Acquiring Person (as defined below) or (ii) 10 business days (or such later date as may be determined by action of the board of directors prior to such time as any person or group of affiliated or associated persons becomes an
Acquiring Person) following the commencement of, or announcement of an intention to make, a tender offer or exchange offer the consummation of which would result in the beneficial ownership by a person or group of 15% or more of the outstanding common stock of the Company. Except in certain situations, a person or group of affiliated or associated persons becomes an “Acquiring Person” upon acquiring beneficial ownership of 15% or more of the outstanding shares of common stock of the Company.
In general, in the event a person becomes an Acquiring Person, then each Right not owned by such Acquiring Person will entitle its holder to purchase from the Company, at the Right’s then current exercise price, in lieu of shares of Series A Junior Participating Preferred Stock, common stock of the Company with a market value of twice the Purchase Price.
In addition, if after any person has become an Acquiring Person, (a) the Company is acquired in a merger or other business combination, or (b) 50% or more of the Company’s assets, or assets accounting for 50% or more of its earning power, are sold, leased, exchanged or otherwise transferred (in one or more transactions), proper provision shall be made so that each holder of a Right (other than the Acquiring Person, its affiliates and associates and certain transferees thereof, whose Rights became void) shall thereafter have the right to purchase from the acquiring corporation, for the Purchase Price, that number of shares of common stock of the acquiring corporation which at the time of such transaction would have a market value of twice the Purchase Price.
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 AND 2015
The Company will be entitled to redeem the Rights at $0.001$ per Right at any time prior to the time an Acquiring Person becomes such. The terms of the Rights are set forth in the Rights Agreement, which is summarized in the Company'sCompany’s Current Report on Form 8-K dated November 13, 2015. The rights plan willwas originally set to expire on November 12, 2018; however, on November 5, 2018 our Board of Directors approved an Amended and Restated Rights Agreement pursuant to which the expiration date was extended to November 5, 2021 and again on November 1, 2021, the Company adopted a Second Amended and Restated Rights Agreement pursuant to which the expiration date was extended to November 1, 2024, unless the rights are earlier redeemed or exchanged by the Company.
| 86 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
| (9) | Stockholders’ Equity – (continued) |
| Stock Option Plan |
In April 2014, the board of directors adopted the 2014 Stock and Incentive Plan ("(“2014 Plan"Plan”) subject to shareholder approval which was received in June 2014. The 2014 Plan provides for the granting of nonqualified and incentive stock options, stock appreciation rights, restricted stock units, restricted stock and dividend equivalents. An aggregate of 1,000,000 shares arewere authorized for issuance under the 2014 Plan. Additionally, 271,906 remaining authorized shares under the 2011 Equity Incentive Plan ("(“2011 Plan"Plan”) were issuable under the 2014 Plan at the time of the 2014 Plan adoption. Upon receiving shareholder approval in June 2016, the 2014 Plan was amended and restated to increase the authorized number of shares of common stock of the Company issuable under all awards granted under the 2014 Plan from 1,271,906 to 2,471,906.. Additionally, upon receiving shareholder approval in June 2018, the 2014 Plan was further amended and restated to increase the authorized number of shares of common stock of the Company issuable under all awards granted under the 2014 Plan from to . Finally, upon receiving shareholder approval in June 2020, the 2014 Plan was further amended and restated to increase the authorized number of shares of common stock of the Company issuable under all awards granted under the 2014 Plan from to . The board of directors, on an option-by-option basis, determines the number of shares, exercise price, term, and vesting period.period for options granted. Options granted generally have a ten-year-year contractual life. The Company issues shares of common stock upon the exercise of options with the source of those shares of common stock being either newly issued shares or shares held in treasury. An aggregate of 2,471,906 shares are authorized for issuance under the 2014 Plan, with 573,108 shares remaining available for grant as of December 31, 2017.2023.
| Outstanding stock options | ||||||||
| Number of shares | Weighted average exercise price | |||||||
| Balance at December 31, 2016 | 2,225,850 | $ | 6.12 | |||||
| Options granted | 543,000 | 3.55 | ||||||
| Options exercised | (206,813 | ) | 2.81 | |||||
| Options forfeited | - | - | ||||||
| Options cancelled | (187,588 | ) | 8.47 | |||||
| Balance at December 31, 2017 | 2,374,449 | 5.64 | ||||||
| Options exercisable at December 31, 2017 | 1,462,333 | 6.12 | ||||||
Schedule of Stock Option Activity
| Outstanding stock options | ||||||||
Number of shares | Weighted average exercise price | |||||||
| Balance at December 31, 2022 | 277,225 | $ | 38.44 | |||||
| Options granted | 26,467 | 6.19 | ||||||
| Options exercised | - | - | ||||||
| Options forfeited | (7,352 | ) | 6.91 | |||||
| Options cancelled | (34,093 | ) | 52.72 | |||||
| Balance at December 31, 2023 | 262,247 | 34.21 | ||||||
| Options exercisable at December 31, 2023 | 194,228 | 42.72 | ||||||
| 87 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 AND 20152023 and 2022
| (9) | Stockholders’ Equity – (continued) |
| Options outstanding | Options exercisable | |||||||||||||||||||||||||||||
| Number outstanding | Weighted average remaining contractual life (Years) | Weighted average exercise price | Aggregate intrinsic value | Number exerciseable | Weighted average remaining contractual life (Years) | Weighted average exercise price | Aggregate intrinsic value | |||||||||||||||||||||||
| 2,374,449 | 7.45 | $ | 5.64 | $ | 381,294 | 1,462,333 | 6.26 | $ | 6.12 | $ | 381,294 | |||||||||||||||||||
Schedule of Share-based Compensation of Stock Options Outstanding and Exercisable
| Options outstanding | Options exercisable | |||||||||||||||||||||||||||||
| Number outstanding | Weighted average remaining contractual life (Years) | Weighted average exercise price | Aggregate intrinsic value | Number exerciseable | Weighted average remaining contractual life (Years) | Weighted average exercise price | Aggregate intrinsic value | |||||||||||||||||||||||
| 262,247 | $ | 34.21 | $ | - | 194,228 | $ | 42.72 | $ | - | |||||||||||||||||||||
The intrinsic value for stock options is defined as the difference between the current market value and the exercise price. The total intrinsic value of stock options exercised during the years ended December 31, 2017, 20162023 and 20152022, was $223,000, $216,000$ and $658,000.$, respectively. There were 206,813, 208,869 and 98,574 stock options exercised during the years ended December 31, 2017, 20162023 and 2015.2022, respectively.
A summary of restricted common stock activity is as follows:
| (e) | Common Stock Warrants |
For charitable purposes,The Company accounts for its common stock warrants under ASC 480, Distinguishing Liabilities from Equity, which requires any financial instrument, other than an outstanding share, that, at inception, embodies an obligation to repurchase the issuer’s equity shares, or is indexed to such an obligation, and requires or may require the issuer to settle the obligation by transferring assets, to be classified as a liability. In accordance with ASC 480, the Company’s outstanding warrants from the November 2019 Offering are classified as a liability. The liability is adjusted to fair value at each reporting period, with the changes in fair value recognized as gain (loss) on change in fair value of warranty liability in the Company’s consolidated statements of operations. The warrants issued in the November 2019 Offering allow the warrant holder, if certain change in control events occur, the option to receive an amount of cash equal to the value of the warrants as determined in accordance with the Black-Scholes option pricing model with certain defined assumptions upon a fundamental transaction.
As of December 23, 2003,31, 2023 and 2022, the Company grantedhad warrants outstanding from the November 2019 Offering to a local university for 20,467purchase an equal number of shares of common stock at a pricestock. The fair value of $12.21 per share with an original expirationthese warrants on November 18, 2019 (closing date of November 2019 Offering) and December 31, 2010. In January 2011,2023 and 2022 was determined using the Company extendedBlack-Scholes option pricing model with the term to December 31, 2015 atfollowing Level 3 inputs (as defined in the same price. The warrants were not exercised by December 31, 2015 and were cancelled.November 2019 Offering):
Schedule of Fair Value of Warrants
| December 31, 2023 | December 31, 2022 | November 18, 2019 | ||||||||||
| Expected life in years | ||||||||||||
| Risk-free interest rate | 4.79 | % | 4.41 | % | 1.63 | % | ||||||
| Dividend yield | — | — | — | |||||||||
| Volatility | 100.00 | % | 100.00 | % | 224.47 | % | ||||||
| Stock price | $ | 2.79 | $ | 6.77 | $ | 6.89 | ||||||
| 88 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 AND 20152023 and 2022
| (9) |
During the years ended December 31, 2023 and 2022, the Company recorded a non-cash gain of $213,000 and $566,000, respectively, from the change in fair value of the November 2019 Offering warrants. The following table is a reconciliation of the warrant liability measured at fair value using level 3 inputs:
Schedule of Reconciliation of Warrant Liability
| Warrant Liability | ||||
| Balance at December 31, 2022 | $ | 229,856 | ||
| Change in fair value of common stock warrants | (212,690 | ) | ||
| Balance at December 31, 2023 | $ | 17,166 | ||
Additionally, in the February 2020 Offering, the Company issued common stock warrants. However, because these warrants do not provide the warrant holder the option to put the warrant back to the Company, the warrants are classified as equity. As of December 31, 2023, there were warrants outstanding that were issued in the February 2020 Offering.
The following table summarizes the number of common stock warrants outstanding and the weighted average exercise price:
Schedule of Number of Warrants Outstanding and the Weighted Average Exercise Price
| Warrants | Weighted Average Exercise Price | |||||||
| Outstanding at December 31, 2022 | 113,795 | $ | 8.72 | |||||
| Issued | - | - | ||||||
| Exercised | - | - | ||||||
| Expired | - | - | ||||||
| Cancelled | - | - | ||||||
| Forfeited | - | - | ||||||
| Balance at December 31, 2023 | 113,795 | $ | 8.72 | |||||
During the years ended December 31, 2023 and 2022, common stock warrants to purchase one share of our common stock were exercised.
The following table summarizes information about common stock warrants outstanding at December 31, 2023:
Schedule of Common Stock Warrants Outstanding
| Warrants outstanding | ||||||||||||||
| Number exercisable | Weighted average remaining contractual life (Years) | Weighted average exercise price | Aggregate intrinsic value | |||||||||||
| 113,795 | $ | $ | - | |||||||||||
| 89 |
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
(10) 401(k) Plan
On January 1, 2002, the Company adopted a tax qualified employee savings and retirement plan (the “401(k) Plan”) covering eligible employees. Pursuant to the 401(k) Plan, employees may elect to reduce current compensation by a percentage of eligible compensation, not to exceed legal limits, and contribute the amount of such reduction to the 401(k) Plan. Beginning April 1, 2014, the 401(k) Plan was amended to require matching contributions to the 401(k) Plan by the Company on behalf of the participants of 100 percent Company match on up to four percent of an employee'semployee’s compensation computed on a per pay period basis. Prior to April 1, 2014, the 401(k) Plan permitted but did not require additional matching and profit sharing contributions to the 401(k) Plan by the Company on behalf of the participants. The Company contributed $82,000, $128,000$102,000 and $74,000,$94,000, respectively, to the 401(k) Plan during the years ended December 31, 2017, 20162023 and 2015.2022.
(11) Commitments and Contingencies
Litigation
The Company is involved in various lawsuits, claims and other legal matters from time to time that arise in the ordinary course of conducting business. The Company records a liability when a particular contingency is probable and estimable.
On April 2, 2019, the Company filed a lawsuit against Clarus in the United States District Court for the District of Delaware alleging that Clarus’s JATENZO® product infringes six of Lipocine’s issued U.S. patents: 9,034,858; 9,205,057; 9,480,690; 9,757,390; 6,569,463; and 6,923,988. However, on February 11, 2020, the Company voluntarily dismissed allegations of patent infringement for expired U.S. Patent Nos. 6,569,463 and 6,923,988 in an effort to streamline the issues and associated costs for dispute. Clarus answered the complaint and asserted counterclaims of non-infringement and invalidity. The Company answered Clarus’s counterclaims on April 29, 2019. The Court held a scheduling conference on August 15, 2018,2019, a claim construction hearing on February 11, 2020, and a summary judgment hearing on January 15, 2021. In May 2021, the Court granted Clarus’ motion for Summary Judgment, finding the asserted claims of Lipocine’s U.S. patents 9,034,858; 9,205,057; 9,480,690; and 9,757,390 invalid for failure to satisfy the written description requirement of 35 U.S.C. § 112. Clarus still had remaining claims before the Court. On July 13, 2021, the Company entered into the Global Agreement with Clarus which resolved all outstanding claims of this litigation as well as the on-going United States Patent and Trademark Office (“USPTO”) Interference No. 106,128 between the parties. Under the terms of the Global Agreement, the Company agreed to pay Clarus $4.0 million payable as follows: $2.5 million immediately, $1.0 million on July 13, 2022 and $500,000 on July 13, 2023. On April 29, 2022, the Company agreed to an amendment to Section 3.1 of the Global Agreement, pursuant to which the Company agreed to pay Clarus $1,250,000 in May 2022, with no additional payments required thereafter. No future royalties are owing from either party. On July 15, 2021, the Court dismissed with prejudice the Company’s claims and Clarus’ counterclaims.
On November 14, 2019, the Company and the othercertain of our officers were named as defendants entered intoin a memorandum of understanding to settle the purported securitiesshareholder class action litigation captionedlawsuit, In reSolomon Abady v. Lipocine Inc. Securities Litigation,et al., 2:17CV00182 DB (D. Utah) which was originally19-cv-00906-PMW, filed againstin the Company on July 1, 2016.United District Court for the District of Utah. The memorandum of understanding contemplatescomplaint alleges that the parties will enter into a settlement agreement, which, if entered into, will be subjectdefendants made false and/or misleading statements and/or failed to customary conditions including court approval following notice to our stockholders, and a hearing at which timedisclose that the court will consider the fairness, reasonableness and adequacyCompany’s filing of the settlement. IfNDA for TLANDO to the FDA contained deficiencies and as a settlement is finally approved byresult the court, it will resolve alldefendants’ statements about our business and operations were false and misleading and/or lacked a reasonable basis in violation of federal securities laws. The lawsuit seeks certification as a class action (for a purported class of purchasers of the claims that wereCompany’s securities from March 27, 2019 through November 8, 2019), compensatory damages in an unspecified amount, and unspecified equitable or could have been brought in the action being settled.injunctive relief. The Company continues to believehas insurance that the claims in the lawsuits are without merit and, to the extent the parties do not enter into a settlement agreement or the court does not approve a settlement, the Company will defend against them vigorously. The Company maintains insurance forcovers claims of this nature. AsOn April 14, 2023, a result of signingjudgment was issued ordering the memorandum of understandingcase dismissed with prejudice and the potential liability becoming probable and estimable, the Company has recorded a litigation settlement liability for $4.3 million as of December 31, 2017. Additionally, the Company has recorded a litigation insurance settlement recovery receivable of $3.3 million as of December 31, 2017 which represents the estimated insurance claims proceeds from our insurance carrier in excessclosure of the Company’s retention.action.
| 90 |
BeyondIn re Lipocine Inc. Securities Litigation, 2:17CV00182 DB (D. Utah), managementLIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2023 and 2022
| (11) | Commitments and Contingencies (continued) |
Management does not currently believe that any other matter, individually or in the aggregate, will have a material adverse effect on our financial condition, liquidity or results of operations.
Guarantees and Indemnifications
In the ordinary course of business, the Company enters into agreements, such as lease agreements, licensing agreements, clinical trial agreements, and certain services agreements, containing standard guarantee and / or indemnifications provisions. Additionally, the Company has indemnified its directors and officers to the maximum extent permitted under the laws of the State of Delaware.
LIPOCINE INC. AND SUBSIDIARIES(12) Agreement with Spriaso, LLC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 AND 2015
On July 23, 2013, theThe Company entered into an assignment/has a license and a services agreement with Spriaso, a related-party that is majority-owned by certain current and former directors of Lipocine Inc. and their affiliates. Under the license agreement, the Company assigned and transferred to Spriaso all of the Company’s rights, title and interest in its intellectual property to develop products for the cough and cold field. In addition, Spriaso received all rights and obligations under the Company’s product development agreement with a third-party. In exchange, the Company will receive a royalty of 20 percent of the net proceeds received by Spriaso, up to a maximum of $10.0$10.0 million. Spriaso also granted back to the Company an exclusive license to such intellectual property to develop products outside of the cough and cold field. Under the service agreement, theThe Company provided facilities andalso agreed to continue providing up to 10 percent of the services of certain employees to Spriaso for a period of 18 months which expired January 23, 2015. Effective January 23, 2015, the Company entered into an amended services agreement with Spriaso in which the Company agreed to continue providing up to 10 percent of the services of certain employees to Spriaso at a rate of $230/hour for a period of six months.time. The agreement was further amended on July 23, 2015, on January 23, 2016, on July 23, 2016, on January 23, 2017, on July 23, 2017 and again on January 23, 2018 to extend the term of the agreement for an additional six months. The agreementprovide services expired in 2021; however, it may be extended upon written agreement of Spriaso and the Company. The Company receiveddid not receive any reimbursements of $31,000, $3,000 and $61,000from Spriaso for the years ended December 31, 2017, 20162023 and 2015,2022, respectively. Additionally, during the years ended December 31, 2023 and 2022, the Company received $110,000 and $0, respectively, in royalty revenue from Spriaso. Spriaso filed its first NDA and as an affiliated entity of the Company, it used up the one-time waiver for user fees for a small business submitting its first human drug application to the FDA. Spriaso is considered a variable interest entity under the FASB ASC Topic 810-10,Consolidations, however the Company is not the primary beneficiary and has therefore not consolidated Spriaso.
In February 2018, the FASB issued ASU No. 2018-02 “Reclassification of Certain Tax Effects from Accumulated Other Comprehensive Income”. ASU 2018-02 allows for a reclassification from accumulated other comprehensive income or loss to retained earnings or accumulated deficit for stranded tax effects resulting from the Tax Cuts and Jobs Act of 2017 (“TCJA”). ASU 2018-02 also requires certain related disclosures. ASU 2018-02 is effective for annual periods, and interim periods within those annual periods, beginning after December 15, 2018 and should be applied either in the period of adoption or retrospectively to each period in which the effect of the change in the U.S. federal corporate income tax rate in the TCJA is recognized. Early adoption is permitted. The Company is currently evaluating the impact of ASU 2018-02 but does not believe it will have a material effect on the Company's financial position or results of operations. In May 2017, the FASB issued ASU 2017-09,Compensation-Stock Compensation (Topic 718): Scope Modification Accounting. This update provides guidance about which changes to the terms or conditions of a share-based payment awards require an entity to apply modification accounting. This update is effective for annual and interim reporting periods beginning after December 15, 2017. Early adoption is permitted. The Company plans to adopt this pronouncement effective January 1, 2018 and does not believe it will have a material effect on the Company's financial position or results of operations.(13) Subsequent Events
In August 2016, the FASB issued ASU 2016-15,Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments. This update addresses how certain cash inflows and outflows are classified in the statement of cash flows to eliminate existing diversity in practice. This update is effective for annual and interim reporting periods beginning after December 15, 2017. Early adoption is permitted. The Company plans to adopt this pronouncement effective January 1, 2018 and does not believe it will have a material effect on the Company's financial position or results of operations.
In June 2016, the FASB issued ASU 2016-13,Financial Instruments - Credit Losses. The new standard amends guidance on reporting credit losses for assets held at amortized cost basis and available-for-sale debt securities.ASU 2016-13 is effective for interim and annual reporting periods beginning after December 15, 2019; early adoption is permitted for interim and annual reporting periods beginning after December 15, 2018. Entities will apply the standard’s provisions as a cumulative-effect adjustment to retained earnings as of the beginning of the first reporting period in which the guidance is effective (i.e., modified retrospective approach). The Company plans to adopt this pronouncement effective January 1, 2019 and does not believe it will have a material effect on the Company's financial position or results of operations.
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 AND 2015
In February 2016, FASB issued ASU 2016-02,Leases,which provides new guidance for lease accounting including recognizing most leases on-balance sheet. The standard becomes effective for annual and interim periods in fiscal years beginning after December 15, 2018. ASU 2016-02 mandates a modified retrospective transition method for all entities.The Company does not have any lease that extends beyond December 31, 2018 other that its facility lease that was extended in February 2018 for a period of one-year until February 28, 2019. The Company plans to adopt this pronouncement effective January 1, 2019 and does not believe it will have a material effect on the Company's financial position or results of operations.
In January 2016, FASB issuedASU 2016-01,Financial Instruments, Recognition and Measurement of Financial Assets and Financial Liabilities, which provides new guidance for the recognition, measurement, presentation, and disclosure of financial assets and liabilities. The standard becomes effective for the Company beginning in the first quarter of our fiscal year ended December 31, 2018 and early adoption is permitted.The Company plans to adopt this pronouncement effective January 1, 2018 and does not believe it will have a material effect on the Company's financial position or results of operations.
The following table provides a summary of selected financial results of operations by quarter for the years ended December 31, 2017 and 2016:
| 2017 | First | Second | Third | Fourth | ||||||||||||
| Operating loss | $ | (4,908,915 | ) | $ | (6,140,618 | ) | $ | (4,766,059 | ) | $ | (5,402,384 | ) | ||||
| Net loss | (4,861,260 | ) | (6,089,766 | ) | (4,700,248 | ) | (5,331,586 | ) | ||||||||
| Basic loss per share attributable to common stock | (0.26 | ) | (0.31 | ) | (0.22 | ) | (0.25 | ) | ||||||||
| Diluted loss per share attributable to common stock | (0.26 | ) | (0.31 | ) | (0.22 | ) | (0.25 | ) | ||||||||
| 2016 | First | Second | Third | Fourth | ||||||||||||
| Operating loss | $ | (7,070,404 | ) | $ | (5,815,120 | ) | $ | (3,286,220 | ) | $ | (3,015,090 | ) | ||||
| Net loss | (7,009,445 | ) | (5,760,111 | ) | (3,235,485 | ) | (2,966,467 | ) | ||||||||
| Basic loss per share attributable to common stock | (0.38 | ) | (0.32 | ) | (0.18 | ) | (0.16 | ) | ||||||||
| Diluted loss per share attributable to common stock | (0.38 | ) | (0.32 | ) | (0.18 | ) | (0.16 | ) | ||||||||
LIPOCINE INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017, 2016 AND 2015
On January 5, 2018,12, 2024, the Company entered into a Loan and SecurityLicense Agreement (the “Loan and Security Agreement”) with Silicon Valley Bank (“SVB”) pursuant to which SVB has agreed to lend the Company $10.0 million. The principal borrowed under the Loan and Security Agreement bears a fixed interest rate equal to the Prime Rate plus one percent per annum, which interest is payable monthly. The loan matures on December 1, 2021. The Company is only required to make monthly interest payments until December 31, 2018, following which the Company will be required to also make equal monthly payments of principalVerity Pharmaceuticals, Inc. for the remaindercommercialization of TLANDO® in the term; provided, however, that if on or priorU.S. and for the development and commercialization rights to May 31, 2018, the Company receives evidence reasonably satisfactory to SVB that the Company has received FDA approval for TLANDOTM, the interest-only payment period will be extended to June 30, 2019. The Company will also be required to pay an additional final payment at maturity equal to $650,000 (the “Final Payment Charge”). At its option, the Company may prepay all amounts owed under the Loan and Security Agreement (including all accrued and unpaid interest and the Final Payment Charge), subject to a prepayment charge if the loan has been outstanding for less than two years, which prepayment charge is determined based on the date the loan is prepaid. In connection with the Loan and Security Agreement, the Company and the Subsidiary granted to SVB a security interest XR in substantially all of the Company’s assets now owned or hereafter acquired, excluding intellectual property and certain other assets. In addition, if TLANDO is not approved by the FDA on or prior to May 31, 2018, the Company will be required to maintain $5.0 million of cash collateral at SVB until such timeCanada as TLANDO is approved by the FDA. While any amounts are outstanding under the Loan and Security Agreement, the Company is subject to a number of affirmative and negative covenants, including covenants regarding dispositions of property, business combinations or acquisitions, incurrence of additional indebtedness and transactions with affiliates, among other customary covenants.described in Note 4(c).
| 91 |
None.ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
We maintain "disclosure“disclosure controls and procedures"procedures” within the meaning of Rule 13a-15(e) of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Our disclosure controls and procedures or (“Disclosure Controls,Controls”) are designed to ensure that information required to be disclosed by us in the reports we file or submit under the Exchange Act, such as this Annual Report on Form 10-K, is recorded, processed, summarized and reported within the time periods specified in the U.S. Securities and Exchange Commission'sSEC’s rules and forms. Our Disclosure Controls include, without limitation, controls and procedures designed to ensure that such information is accumulated and communicated to our management, including our Chief Executive Officer and ChiefPrincipal Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
As of the end of the period covered by this Annual Report on Form 10-K, we evaluated the effectiveness of the design and operation of our Disclosure Controls, which was done under the supervision and with the participation of our management, including our Chief Executive Officer and our ChiefPrincipal Financial Officer. Based on the controls evaluation, our Chief Executive Officer and ChiefPrincipal Financial Officer have concluded that, as of the date of their evaluation, our Disclosure Controls were effective as of December 31, 2017.2023.
Management'sManagement’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control system was designed to provide our management and board of directors reasonable assurance regarding the reliability of financial reporting and preparation of financial statements for external purposes in accordance with GAAP. Internal control over financial reporting has inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements will not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Our management has assessed the effectiveness of internal control over financial reporting as of December 31, 2017.2023. In making this assessment, we used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission or COSO, inInternal Control-Integrated Framework (2013). Based on our assessment we believe that, as of December 31, 2017,2023, our internal control over financial reporting is effective based on those criteria.
Change in Internal Control over Financial Reporting
During the quarteryear ended December 31, 2017,2023 there have been no changes in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
During the three months ended December 31, 2023, none of our directors or officers adopted or terminated a “Rule 10-b5-1 trading arrangement” or “non-Rule 10-b5-1 trading arrangement” as each term is identified in Item 408 of Regulation S-K.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Certain of the information required by this item will be contained in our definitive Proxy Statement with respect to our 20182024 Annual Meeting of Stockholders, under the captions "Election“Election of Directors,"” and "Compliance“Compliance with Section 16(a) of the Exchange Act"Act” and is incorporated into this item by reference.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item will be contained in our definitive Proxy Statement with respect to our 20182024 Annual Meeting of Stockholders, under the captions "Executive Compensation"“Executive Compensation”, "Compensation“Compensation Committee Interlocks and Insider Participation"Participation”, and "Compensation“Compensation Committee Report"Report” and is incorporated into this item by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT, AND RELATED STOCKHOLDER MATTERS
The information required by this item will be contained in our definitive Proxy Statement with respect to our 20182024 Annual Meeting of Stockholders, under the captions "Security“Security Ownership of Certain Beneficial Owners and Management"Management” and "Equity“Equity Compensation Plan Information"Information” and is incorporated into this item by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item will be contained in our definitive Proxy Statement with respect to our 20182024 Annual Meeting of Stockholders under the captions "Certain“Certain Relationships and Related Transactions"Transactions” and "Independence“Independence of the Board"Board” and is incorporated into this item by reference.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item will be contained in our definitive Proxy Statement with respect to our 20182024 Annual Meeting of Stockholders, under the caption "Principal“Principal Accountant Fees and Services"Services” and is incorporated into this item by reference.
ITEM 15.EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) The following documents are filed as part of this Annual Report on Form 10-K.
1. Financial Statements. The financial statements listed on the accompanying Index to Consolidated Financial Statements are filed as part of this report.
2. Financial statement schedules. There are no financial statements schedules included because they are either not applicable or the required information is shown in the consolidated financial statements or the notes thereto.
3. Exhibits. The following exhibits are filed or incorporated by reference as part of this Form 10-K.
INDEX TO EXHIBITS
| Exhibit | Incorporation By Reference | |||||||||
| Number | Exhibit Description | Form | SEC File No. | Exhibit | Filing Date | |||||
10.28*, *** | ||||||||||
| 21.1* | Subsidiaries | |||||||||
| 23.1* | Consent of Tanner LLC | |||||||||
| 31.1* | Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |||||||||
| 31.2* | Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |||||||||
| 32.1* | Certification of Principal Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, 18 U.S.C. 1350. | |||||||||
| 32.2* | Certification of Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, 18 U.S.C. 1350. | |||||||||
| 97* | ||||||||||
| XBRL Instance Document – the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | ||||||||||
| 101.SCH* | Inline XBRL Taxonomy Extension Schema Document | |||||||||
| 101.CAL* | Inline XBRL Taxonomy Extension Calculation Linkbase Document | |||||||||
| 101.DEF* | Inline XBRL Taxonomy Extension Definition Linkbase Document | |||||||||
| 101.LAB* | Inline XBRL Taxonomy Extension Labels Linkbase Document | |||||||||
| 101.PRE* | Inline XBRL Taxonomy Extension Presentation Linkbase Document | |||||||||
| 104* | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101) | |||||||||
| * | Filed herewith |
| ** | Management contract or compensation plan or arrangement |
| + | Confidential treatment has been granted with respect to certain portions of this exhibit. Omitted portions have been submitted separately with the Securities and Exchange Commission. | |
| *** | Certain portions of this exhibit have been omitted pursuant to Item 601(b)(10) of Regulation S-K. The Registrant hereby undertakes to furnish to the SEC, upon request, copies of any such instruments. |
ITEM 16.FORM 10-K SUMMARY
None
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| (Registrant) | |
| Dated: March | /s/ Mahesh V. Patel |
Mahesh V. Patel, President and Chief Executive Officer (Principal Executive Officer and Principal Financial Officer) | |
| Dated: March | /s/ |
(Principal |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the date indicated.
| Signature | Title | Date | ||
| /s/ Mahesh V. Patel | President and Chief Executive Officer (Principal Executive | March 7, 2024 | ||
| Mahesh V. Patel | Officer and Principal Financial Officer) | |||
| /s/ Krista Fogarty | Corporate Controller (Principal Accounting Officer) | March 7, 2024 | ||
| Krista Fogarty | ||||
/s/
| March | |||
| Jeffrey Fink | ||||
/s/
| March | |||
| Jill M. Jene |
| /s/ John Higuchi | Director | March 7, 2024 | ||
| John Higuchi | ||||
/s/
| Director | March | ||
| R. Dana Ono | ||||
/s/
| Independent Lead Director and Chairman of the Board | March | ||
| Spyros Papapetropoulos | ||||
| ||||
|
| 97 |