UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark one)One)
| ☒ | ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
☒ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31 2020., 2023
| ☐ | TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
☐TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE TRANSITION PERIOD FROM TO
Commission file number 001-3200File Number 001-32001
APTOSE BIOSCIENCES INC.Aptose Biosciences Inc.
(Exact name of registrantRegistrant as specified in its charter)Charter)
| |
Canada
| 98-1136802 |
(State or other jurisdiction of incorporation or organization) | 98-1136802
(I.R.S. Employer Identification No.) |
251 Consumers Road, Suite 1105 Toronto, Ontario, CanadaM2J 4R3
| M2J 4R3 |
(Address of principal executive offices) |
647-479-9828
(Registrant’s telephone number, including area code)Zip Code) |
Registrant’s telephone number, including area code: (647) 479-9828
Securities registered pursuant to Section 12(b) of the Act:
| | | | |
Title of each class | Trading Symbol(s)
| Trading Symbol(s) |
| Name of each exchange on which registered |
Common Shares, no par value | APTO
| APTO |
| Nasdaq Capital Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrantRegistrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES Yes ☐ NONo☒
Indicate by check mark if the registrantRegistrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES Yes ☐ NO No ☒
Indicate by check mark whetherwhether the registrantRegistrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrantRegistrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES Yes☒ NO No ☐
Indicate by check mark whether the registrantRegistrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§229.405232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrantRegistrant was required to submit and post such files). YES Yes☒ NO No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | |
Large accelerated filer ☐ |
| ☐ |
| Accelerated filer |
| ☐ |
|
|
|
|
Non-accelerated filer ☒ |
| ☒ |
| Smaller reporting company |
| ☒ |
|
|
|
|
|
|
|
Emerging growth company ☐ |
| ☐ |
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b 212b-2 of the Exchange Act). YES Yes ☐ NO No ☒
The aggregate market value of the voting stock and nonvoting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, or the average bid and asked prices of such common equity, as of June 30, 20202023 was $478,045,833.$29,584,561.
As of March 23, 2021,26, 2024, the registrant had 88,885,238 common shares15,717,701 Common Shares outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of our Proxy Statement for our 20212023 Annual Meeting of Stockholders (the “Proxy Statement”), are incorporated by reference in Part III.
TABLE OF CONTENTS
This Annual Report on Form 10-K contains certain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and is subject to the safe harbor created by those sections. For more information, see “Part I. Item 1. Business — Cautionary Note Regarding Forward-Looking Statements.”
As used in this report, the terms “Aptose,” “Aptose Biosciences,” the “Company,” “we,” “us,” “our” and similar references refer to Aptose Biosciences Inc. (formerly known as Lorus Therapeutics Inc.) and our consolidated subsidiaries, and the term “Common Shares” refers to our common shares, no par value.
Aptose hashad historically qualified as a “foreign private issuer” for purposes of reporting under the Exchange Act, and filing registration statements under the Securities Act of 1933, as amended. Effective December 31, 2018, however, Aptose ceased qualifying as a foreign private issuer and began filing reports with the SECUnited States Securities and Exchange Commission ("SEC") as a “domestic issuer”.issuer.” As a result, Aptose changed the accounting standards by which it prepares its financial statements from International Financial Reporting Standards or “IFRS”, to generally accepted accounting principles in the United States, or “US GAAP”. “U.S. GAAP.” All financial statements contained in this Annual Report are presented on the basis ofin accordance with U.S. GAAP. This report contains the following trademarks,trademark, trade namesname and service marksmark of ours: Aptose. This report also contains trademarks, trade names and service marks that are owned by other persons or entities.
PART I.
Item 1. Business
Overview
Overview
Aptose Biosciences Inc. is a science-driven, clinical-stage biotechnology company committed to precision medicines addressing unmet clinical needs in oncology, with an initial focus on hematology. The Company's small molecule cancer therapeutics pipeline includes products designed to provide single agent efficacy and to enhance the efficacy of other anti-cancer therapies and regimens without overlapping toxicities. The Company’s executive office is located in San Diego, California.
Our Programs
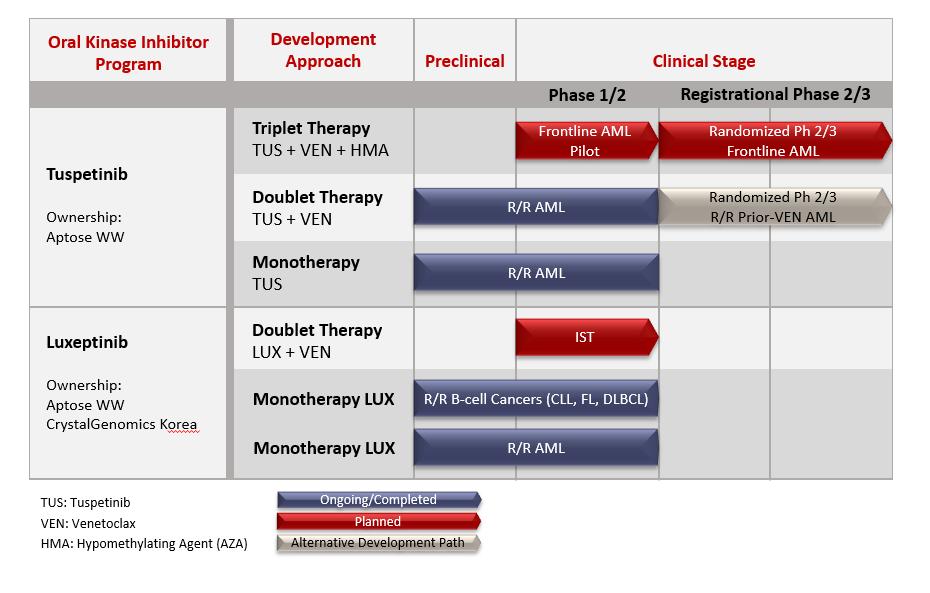
We are advancing highly differentiatedoral targeted agents to treat unmet medical needslife-threatening hematologic cancers that require immediate treatment. We have two clinical-stage oral kinase inhibitors under active development for the treatment of hematologic malignancies: tuspetinib (HM43239) and luxeptinib (CG-806). Tuspetinib and luxeptinib are being evaluated for safety, tolerability, pharmacokinetics and efficacy in life-threatening cancers,Phase 1/2 clinical trials, and each molecule is described below. A third molecule (APTO-253) is not undergoing active clinical development and will not be discussed further.
Tuspetinib, Aptose’s lead asset, is being developed for frontline combination therapy in newly diagnosed AML patients to unlock the most significant patient impact and greatest commercial opportunity. Tuspetinib is a once-daily oral kinase inhibitor, targeting a select group of kinases operative in myeloid malignancies, such as acute myeloid leukemia (“AML”), certain B-cell malignancies, high-risk myelodysplastic syndrome (“MDS”("AML") and the higher risk myelodysplastic syndromes ("hr-MDS"), and known to be involved in tumor proliferation, resistance to therapy, and differentiation. However, tuspetinib avoids kinases that typically cause toxicities associated with other hematologic malignancies. Aptosekinase inhibitors and is consequently a publicly listed company incorporated underwell-tolerated antileukemic agent. The clinical development path for triplet combination therapy in newly diagnosed AML patients with tuspetinib-based triplet combination therapy (tuspetinib + the lawsBCL-2 inhibitor venetoclax + hypomethylating agent; TUS+VEN+HMA) begins with a demonstration of Canada. The Company’s Common Shares are listed on the Nasdaq Capital Marketsafety and the Toronto Stock Exchange. The Company was incorporated on September 5, 1986, under the name RML Medical Laboratories (“RML”activity of tuspetinib as a single agent ("TUS") pursuant to the Business Corporations Act (Ontario) and then continued pursuant towith the Canada Business Corporations Act (“CBCA”TUS+VEN doublet combination therapy in relapsed or refractory ("R/R"). Between 1986 AML patients.
Tuspetinib monotherapy dose escalation and 2014, the Company operated under the names of RML, IMUTEC Corporation and Lorus Therapeutics Inc. On August 28, 2014, the Company changed its name from Lorus Therapeutics Inc. to Aptose Biosciences Inc. and, on October 1, 2014, we consolidated our outstanding Common Shares on the basis of one post-consolidation Common Share for each twelve pre-consolidation Common Shares.
Based on insights into the genetic and epigenetic profiles of certain cancers and patient populations, Aptose is building a pipeline of novel and targeted oncology therapies directed at dysregulated processes and signaling pathways in cancer cells, and this strategy is intended to optimize efficacy through simultaneous targeting of key drivers of disease in cancer cells, while preserving quality of life in patients by minimizing the side effects associated with conventional therapies. Our product pipeline includes cancer drug candidates that exert potent activity as stand-alone agents and that enhance thedose exploration activities of other anticancer agents without causing overlapping toxicities. Indeed, we believe our targeted products can emerge as first-in-class or best-in-class agents that deliver single agent benefit and may servehave been completed as part of an international Phase 1/2 clinical trial designed to assess the safety, tolerability, pharmacokinetics, pharmacodynamic responses, and efficacy as a single agent in patients with R/R AML. Complete responses (“CRs”) without dose limiting toxicities were achieved at four dose levels across a broad diversity of mutationally-defined AML populations and with a highly favorable safety profile. Tuspetinib to date has demonstrated a favorable safety profile and has caused no drug-related QTc prolongations, liver or kidney toxicities, muscle damage, differentiation syndrome, and no
myelosuppression with continuous dosing of patients in remission. A recommended phase 2 dose ("RP2D") of 80 mg tuspetinib once daily as an oral tablet was selected and approved by the U.S. FDA for use as a single agent in patients with R/R AML. At the RP2D, tuspetinib demonstrated notable response rates in R/R AML patients that had never been treated with venetoclax (VEN-naive AML): CR/CRh=36% among all-comers, CR/CRh=50% among patients with mutated FLT3, and CR/CRh=25% in patients with wildtype FLT3.
Following completion of the single agent dose escalation and exploration trial, tuspetinib advanced into the APTIVATE expansion trial of the Phase 1/2 program in R/R AML patient populations treated with tuspetinib combined with the BCL-2 inhibitor venetoclax (TUS+VEN doublet), with the intent to position tuspetinib for triple combination therapeutic strategystudies in frontline therapy for specific populationsnewly diagnosed AML patients. The TUS+VEN doublet combination therapy (with both 40mg and 80mg TUS) maintained a favorable safety profile: no new or unexpected safety signals were observed, and there were no reported drug-related adverse events of cancerQTc prolongation, differentiation syndrome, or deaths. Also, the TUS/VEN doublet combination (with 80mg TUS) achieved responses in heavily pretreated R/R AML patients, including those with wildtype or mutated FLT3, and those who failed prior therapy with venetoclax (Prior-VEN) or FLT3 inhibitors (Prior-FLT3i). Based on the safety and efficacy profile of tuspetinib, we believe that tuspetinib, if approved, can reach annual sales greater than $3 billion by 2035 because we believe tuspetinib could 1) become the preferred kinase inhibitor for inclusion in triplet combination for front line AML patients with FLT3 mutations and for patients with wild type FLT3, 2) become the preferred kinase inhibitor for inclusion in combination with venetoclax for second line AML patients, 3) serve as an effective agent for maintenance therapy to prevent relapse in patients who achieved a complete remission through a stem cell transplant or through drug-based therapy, 4) serve as an effective agent for the treatment of third line FLT3 mutated patients failed by prior therapy with other FLT3 inhibitors and 5) serve in front line triplet combinations, second line doublet combinations, and maintenance therapy for hr-MDS patients. These beliefs related to the potential commercial opportunity are based on management’s current assumptions and estimates, which are subject to change, and there can be no assurance that tuspetinib will ever be approved or successfully commercialized and, if approved and commercialized, that it will ever generate significant revenues. See our “Risk Factors – “We are an early-stage development company with no revenues from product sales.” and “We have a history of operating losses. We expect to incur net losses and we may never achieve or maintain profitability.” in this Annual Report on Form 10-K.
We believeLuxeptinib, Aptose's second agent, is an oral, highly potent kinase inhibitor that selectively targets defined kinases operative in myeloid and lymphoid hematologic malignancies. This small molecule has been evaluated in a Phase 1a/b study for the future of cancer treatment and management lies in the prospective selection and treatment of patients having malignancies that are genetically or epigenetically predisposed to response based onR/R B-cell leukemias and lymphomas (dose escalation from 150mg-900mg BID) and in a drug’s unique mechanism of action. We are of the view that many drugs currently approvedPhase 1a/b study for the treatment of patients with R/R AML or hr-MDS (dose escalation from 450 mg-900mg BID). These clinical studies demonstrated tumor shrinkage among B-cell cancer patients, including a complete response ("CR") in a DLBCL patient who received the original G1 formulation. Likewise, an MRD-negative CR in one R/R AML patient occurred with 450mg BID dosing of the original G1 formulation. Because absorption of the original G1 formulation hampered effectiveness of luxeptinib, a new G3 formulation was developed. Enrollment of patients in the B-cell malignancy trial and managementthe AML trial have been completed, and the initial clinical evaluation of cancerthe G3 formulation with continuous BID dosing has been completed. The G3 formulation delivered superior plasma exposure levels relative to the original G1 formulation. Regarding potential next steps with luxeptinib, a molecularly defined subgroup of hematologic malignancy patients was recently identified that may benefit from treatment with luxeptinib in combination with venetoclax. An investigator-sponsored trial is being considered while non-clinical studies are not selectiveunderway to support the use of LUX+VEN for the specific genetic alterations (targets) and pathways that cause the patient’s tumor and hence allow for disease progression and /or significant toxicities duetreatment of these patients. In parallel, efforts are underway to off-target effects. Aptose’s strategy isidentify sources of capital to develop agents that target underlying disease-promoting mutations or altered pathways withinsupport such a patient population, and we intend to apply this strategy across several therapeutic indications in oncology, including hematologic malignancies and solid tumor indications.trial.
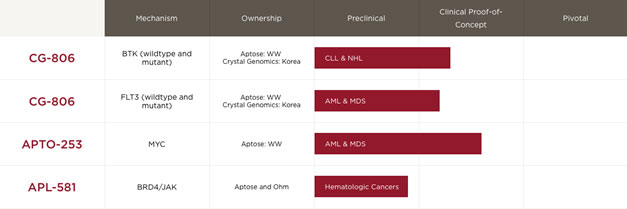
Aptose Programs
Aptose has two clinical-stage assets, and a third program that is discovery-stage and partnered with another company.
| · | Luxeptinib (CG026806, CG-806) is a clinical stage asset with two separate clinical programs: luxeptinib, Aptose’s FMS-like tyrosine kinase 3 (“FLT3”) / Bruton’s tyrosine kinase (“BTK”) inhibitor is being evaluated currently in two separate Phase 1a/b dose escalation studies. The first trial, supported by an investigational new drug (“IND”) application submitted to the U.S. Food and Drug Administration (“FDA”) in February 2019 for luxeptinib, is being conducted in patients with certain B-cell malignancies, including chronic lymphocytic leukemia (“CLL”), small lymphocytic lymphoma (“SLL”) and certain non-Hodgkin’s lymphomas (“NHL”) that are resistant/refractory/intolerant to other therapies. The second trial, supported by an IND application submitted in June 2020 to the FDA for luxeptinib, is being conducted in patients with relapsed/refractory acute myeloid leukemia (“R/R AML”), including the emerging populations resistant to FLT3 inhibitors. |
| · | APTO-253, our second clinical-stage asset: APTO-253 is a small molecule MYC oncogene inhibitor and we are currently enrolling patients in a Phase 1a/b clinical trial for the treatment of patients with relapsed or refractory (“R/R”) blood cancers, including AML and high-risk MDS. |
| · | APL-581, our partnered program, is a dual bromodomain and extra-terminal domain motif (“BET”) protein and kinase inhibitor program which we partnered with Ohm Inc. (“OHM”) on March 7, 2018. |
Aptose is committed to the development of anticancer drugs that target aberrant oncologic signaling that underlies a particular life-threatening malignancy. This targeted approach is intended to impact the disease-causing events in cancer cells without affecting normal processes within cells. Such an approach requires that we first identify critical underlying oncogenic mechanisms in cancer cells and then develop a therapeutic asset that selectively impacts such oncogenic mechanisms.
| · | As a cluster-selective kinase inhibitor, luxeptinib targets the wild type and mutant forms of the FLT3 and BTK validated cancer targets, as well as multiple critical pathways that overlap to lead to the proliferation of cancer cells, including the B-cell receptor signaling pathway and FLT3 receptor pathways, as well as other receptor kinases and signaling cascades that drive dysregulated survival of the cancer cells. |
| · | Further, Aptose created the APTO-253 small molecule targeted drug that inhibits expression of the MYC oncogene and is under development as a novel therapy for AML and related MDS. Dysregulation of the MYC oncogene reprograms signaling of cancer cells to allow for malignant transformation and resistance to typical anticancer drugs. APTO-253 directly targets the MYC gene and inhibits production of MYC mRNA and protein, thereby leading to cancer cell death. |
The following table sets forth various product conditionsfigure identifies the clinical stage agents in our pipeline and their respective stages of development.
Tuspetinib Program
Licensing Overview
On November 4, 2021, we entered into a licensing agreement (the "Tuspetinib Licensing Agreement") with the South Korean company Hanmi Pharmaceutical Co Ltd. (“Hanmi”) for the clinical and commercial development of tuspetinib (formerly HM43239). Under the terms of the Tuspetinib Licensing Agreement, Hanmi granted us exclusive worldwide rights to tuspetinib for all indications. Hanmi received an upfront payment of $12.5 million, including $5 million in cash and $7.5 million in Common Shares. Hanmi will also receive up to $407.5 million in future milestone payments contingent upon achieving certain clinical, regulatory and sales milestones across several potential indications, as well as tiered royalties on net sales. The term of the agreement will continue on a product-by-product and country-by-country basis until the expiration of the royalty period for such product in such country. The licenses to us will survive and become non-exclusive, perpetual, irrevocable and fully paid-up on a product-by-product and country-by-country basis, upon their natural expiration under the terms of the agreement.
Preclinical Profile
Tuspetinib is an oral, once-daily, highly potent myeloid kinome inhibitor designed to target key kinases operative in myeloid malignancies. In preclinical studies, tuspetinib demonstrated potent in vitro and in vivo activity against FLT3 ITD mutated as well as D835 and gatekeeper (F691) tyrosine kinase domain ("TKD") mutated AML that confer resistance to other agents. Additionally, tuspetinib inhibited phosphorylation of the SYK kinase, known to be highly activated in AML and associated with resistance to FLT3 targeted therapy. Tuspetinib also was designed to inhibit several kinases involved in tumor cell proliferation and/or differentiation, including mutant forms of c-KIT, JAK1, JAK2, and RSK, all with half maximal inhibitory concentration ("IC50") values < 10 nM.
Tuspetinib induced in vitro cytotoxicity in AML and Ba/F3 cell lines expressing FLT3 WT, ITD, and/or TKD point mutations. Tuspetinib showed greater inhibitory activity compared to quizartinib on Ba/F3 cells expressing
resistance-conferring ITD/TKD double mutations (ITD/F691L and ITD/D835Y). Thus, Tuspetinib may overcome clinically relevant ITD/TKD double mutations, which may result from sustained FLT3 inhibition. Moreover, target modulation was shown as tuspetinib inhibited FLT3 phosphorylation and downstream signaling molecules such as phospho-ERK and phospho-STAT5.
The in vivo anti-tumor efficacy of tuspetinib was demonstrated in murine xenograft models using MV-4-11 and MOLM-13 human AML cells having the ITD mutant form of FLT3 and using the MOLM-14 model having the ITD and F691L dual mutations of FLT3 with dosing regimens that match those currently under investigation. Tuspetinib exhibited dose-dependent tumor growth inhibition of models of FLT3 ITD mutant AML with complete tumor regression observed in some groups, and no change in body weight. Of note, tuspetinib produced greater tumor growth inhibition in the MOLM-14 FLT3-ITD/F691L model compared to gilteritinib, or entospletinib (SYK inhibitor) as single agents, and comparable activity to the gilteritinib plus entospletinib combination.
Latest Clinical Update and Program Status
On December 9, 2023, Aptose featured tuspetinib in an oral presentation at the 65th American Society of Hematology ("ASH") Annual Meeting and Exposition and announced that a growing body of clinical data for Aptose’s lead compound tuspetinib, demonstrates significant benefit as a single agent and in combination with venetoclax (VEN) in patients with R/R AML in the ongoing APTIVATE Phase 1/2 study. Data were presented in an oral presentation by lead investigator Naval G. Daver, M.D., Professor, Director Leukemia Research Alliance Program, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX.
Dr. Daver reported data from more than 100 relapsed/refractory patients from multiple international clinical sites, who had failed prior therapy and then were treated with TUS as a single agent or the TUS+VEN doublet. Both TUS and the TUS+VEN doublet delivered multiple composite complete remissions ("CRc") in this very ill AML population, while maintaining a favorable safety profile across all treated patients. The data demonstrated tuspetinib as a single agent (TUS) is active and well tolerated in one of the most challenging and heterogeneous disease settings in oncology – relapsed and refractory AML. Tuspetinib demonstrated broad activity, including activity in patients with FLT3 wild-type AML (accounting for more than 70% of the AML population), FLT3 mutated AML, NPM1 mutated AML, as well as in patients with mutations historically associated with resistance to targeted therapy. Most notably, TUS targets VEN resistance mechanisms, enabling the TUS+VEN combination therapy to uniquely treat the very ill prior-VEN AML population, including both FLT3 mutant and FLT3 wildtype disease. From a broader perspective, the growing body of antileukemic activity, and continued favorable safety profile, support advancement of tuspetinib as the TUS+VEN+HMA triplet combination therapy for the treatment of frontline newly diagnosed AML patients.
Dr. Daver also pointed out that while patients on the TUS+VEN therapy are early in their treatment cycles, most patients achieving a response remained on treatment and that responses have begun to mature as dosing continues. Highlights of Dr. Daver’s ASH oral presentation include:
| | | |
| • |
| As a single agent at therapeutic doses of 80-160 mg in 68 evaluable patients, TUS was more active in VEN-naive patients, with an overall CRc rate of 29% (8/28). This included a 42% CRc rate (5/12) in FLT3-mutated patients and a 19% CRc rate (3/16) in FLT3-unmutated, or wildtype, AML patients. Responses and blood counts improved with continuous dosing, many patients bridged to an allogeneic stem cell transplant (HSCT), durability was observed when HSCT was not performed, and 80 mg was selected as the RP2D. Overall, tuspetinib showed a favorable safety profile with only mild adverse events (AEs) and no dose-limiting toxicities (DLTs) up to 160 mg per day, and no drug discontinuations from drug-related toxicity. |
| | | |
| • |
| In the TUS+VEN doublet study, 49 patients were dosed with 80 mg of tuspetinib and 200 mg of venetoclax, with 36 evaluable (and 13 patients too early to assess). Patients were heavily exposed to Prior-VEN and Prior-FLT3 inhibitor treatment. TUS+VEN was active in both VEN-naive and prior Prior-VEN R/R AML patients. TUS demonstrated compelling composite complete remission (CRc) rates. Among all evaluable patients, TUS+VEN demonstrated a CRc rate of 25% (9/36); 43% (3/7) in VEN-naive patients, and 21% (6/29) in Prior-VEN patients. Among FLT3 wildtype patients, TUS+VEN demonstrated an overall CRc rate of 20% (5/25); 33% (2/6) in VEN-naive patients, and 16% (3/19) in |
| | | |
|
|
| Prior-VEN patients. Among FLT3 mutant patients, TUS+VEN demonstrated an overall CRc rate of 36% (4/11); a complete response in a VEN-naive patient (1/1); a 30% (3/10) in Prior-VEN patients; and 44% (4/9) in patients treated prior with a FLT3 inhibitor. |
Clinical data from tuspetinib in AML were presented at the ASH Annual Meeting in December 2022 and during a Corporate Comprehensive Clinical Update Call held December 11, 2022. Data presented demonstrated that tuspetinib delivers single agent responses without prolonged myelosuppression or life-threatening toxicities in these very ill and heavily pretreated relapsed or refractory AML patients. Responses were observed in a broad range of mutationally-defined populations, including those with mutated forms of NPM1, MLL, TP53, NRAS, KRAS, DNMT3A, RUNX1, wild-type FLT3, ITD or TKD mutated FLT3, various splicing factors, and other genes As of October 6, 2022, 60 heavily pretreated R/R AML patients were enrolled at multiple centers and treated at doses escalating from 20 mg to 200 mg, with further dose exploration at the 40 mg, 80 mg, 120 mg and 160 mg dose levels. Tuspetinib delivered multiple CRs at 40 mg, 80 mg, 120 mg and 160 mg dose levels in which no DLTs were observed. Tuspetinib demonstrated clinically meaningful benefit in all responders, by either bridging successfully to HSCT or leading to a durable response, as well as a favorable safety profile. In addition to 5 CRcs and 1 PR reported at ASH 2021, 4 new CRcs and 3 new PR had been generated during 2022. New responses during 2022 were achieved with 160 mg, 120 mg, 80 mg, and 40 mg. Among efficacy-evaluable patients treated with 80 mg, 120 mg, or 160mg, response rates ranging from 19% to 75% were achieved in specific genotypic subpopulations of R/R AML patients. Significant bone marrow leukemic blast reductions were observed broadly in FLT3+ and FLT3 wildtype patients across multiple dose levels, comparable to reported gilteritinib data, except that the patients treated with tuspetinib were more heavily pre-treated relapsed and refractory AML patients than those treated with gilteritinib. Vignettes of patient experiences highlight the potency and breadth of tuspetinib to deliver complete remissions among several mutationally-defined populations with a diversity of adverse mutations. Tuspetinib continued to show a favorable safety profile with only mild AEs and no DLTs up to 160 mg per day, and no drug discontinuations from drug-related toxicity. No drug-related SAE, drug-related deaths, differentiation syndrome, AE of QT prolongation or DLT were observed through the 160 mg level. Tuspetinib avoids many of the typical toxicities observed with other tyrosine kinase inhibitors. We identified a safe therapeutic range with a broad therapeutic window, spanning the dose levels of 40, 80, 120 and 160 milligrams. We also announced that enrollment had been initiated in the APTIVATE expansion trial for monotherapy and drug combination therapy with tuspetinib. For the APTIVATE expansion trial, we selected 120 mg as the initiating single agent expansion dose and 80 mg as the initiating dose selected for combination with venetoclax.
As of January 30, 2023, we announced the dosing of patients in the APTIVATE Phase 1/2 clinical trial of tuspetinib, and that another clinical response has been achieved by a R/R AML patient receiving 40 mg tuspetinib once daily orally in the original dose exploration trial, the second response at the recently launched low-dose 40 mg cohort. In addition, we elucidated a rationale for the superior safety profile of tuspetinib. While several kinase inhibitors require high exposures that exert near complete suppression of a single target to elicit responses, those agents often cause additional toxicity because they also cause extensive inhibition of that target in normal cells. In contrast, tuspetinib simultaneously suppresses a small suite of kinase-driven pathways critical for leukemogenesis. Consequently, tuspetinib achieves clinical responses at lower exposures with less overall suppression of each pathway, thereby avoiding many toxicities observed with competing agents.
Concurrent with the European Hematology Association (EHA) Annual Congress held June 8-11, 2023, Aptose held an interim clinical update webcast on June 10, 2023, to present highlights from the ongoing clinical development of tuspetinib. Aptose reported completion of the tuspetinib dose escalation and dose exploration Phase 1/2 trial in 77 R/R AML patients, tuspetinib demonstrated a favorable safety profile, and tuspetinib delivered monotherapy responses across four dose levels with no dose-limiting toxicity in mutationally diverse and difficult to treat R/R AML populations, including patients with highly adverse mutations that typically do not respond to monotherapy or combination therapy: TP53-mutated patients with a CR/CRh = 20% and RAS-mutated patients with a CR/CRh = 22%. Aptose also reported completion of a successful End of Phase 1 Meeting with the US FDA for tuspetinib, that a monotherapy RP2D was selected as 80mg daily, and that all development paths remain open, including the single arm accelerated path. Following completion of the dose escalation and dose exploration phases of the Phase 1/2 clinical program, Aptose focused attention on the tuspetinib APTIVATE expansion trial. The APTIVATE trial sought to identify patient populations that may serve as development paths in R/R AML patients sensitive to the TUS+VEN doublet and can serve as development paths for accelerated and full approvals. We reported that patient enrollment in
the APTIVATE expansion trial has been brisk and preliminary CR activity had already been reported in patients receiving the TUS+VEN doublet who previously failed therapy with venetoclax.
On October 29, 2023, Aptose presented two posters related to the clinical and preclinical activity of tuspetinib at the European School of Haematology 6th International Conference: Acute Myeloid Leukemia "Molecular and Translational": Advances in Biology and Treatment, held October 29-31, 2023, in Estoril, Portugal. Clinical findings included 1) data from the APTO-TUS-HV01 clinical trial (the "Food Effect Study") evaluating the pharmacokinetic (PK) properties of tuspetinib in healthy human volunteers in which tuspetinib was administered with our without food, and 2) from an international Phase 1/2 study of tuspetinib as a single agent and in combination with venetoclax in patients with R/R AML from across clinical centers in the United States, South Korea, Spain, Australia and other sites. Data from the Food Effect Study in healthy human volunteers demonstrated tuspetinib can be administered with or without food and foresee no clinically meaningful difference in exposure. This is an important finding for patient convenience, as venetoclax is dosed with food and tuspetinib can now be simultaneously administered with the venetoclax rather than require staggered dosing. Findings from the Phase 1/2 clinical trial demonstrated tuspetinib as a single agent was well-tolerated and highly active among R/R AML patients with a diversity of adverse genotypes and delivered a 42% CR/CRh cross-evaluable venetoclax-naive patients at the 80mg daily RP2D. The TUS+VEN doublet has been well tolerated in the APTIVATE international Phase 1/2 expansion trial in R/R AML patients and achieved multiple responses in patients who previously failed venetoclax ("Prior-VEN failure AML"), including Prior-VEN failure patients who also previously failed FLT3 inhibitors, all of whom represent emerging populations of high unmet medical need. Notably, tuspetinib targets venetoclax resistance mechanisms that may re-sensitize Prior-VEN failure patients to venetoclax.
Separate from the clinical studies, the preclinical study (entitled: “Tuspetinib Oral Myeloid Kinase Inhibitor Creates Synthetic Lethal Vulnerability to Venetoclax”) presented by Aptose during the ESH Conference investigated the effects of tuspetinib on key elements of the phosphokinome and apoptotic proteome in both parental and TUS-resistant AML cells. In parental cells, tuspetinib inhibits key oncogenic signaling pathways and shifts the balance of pro- and anti-apoptotic proteins in favor of apoptosis, suggesting that it may generate vulnerability to venetoclax. Indeed, acquired resistance in the AML cells to tuspetinib generated a synthetic lethal vulnerability to venetoclax of unusually high magnitude. Concurrent administration of TUS+VEN therefore may discourage the emergence of resistance to tuspetinib during treatment. In conjunction with poster presentations at the ESH Conference, on October 30, 2023, Aptose held a “Clinical Update and KOL Data Review of AML Drug Tuspetinib” that was webcast and featured Dr. Naval Daver, MD, Professor, Director Leukemia Research Alliance Program, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, Texas. Dr. Daver is the lead investigator on Aptose’s APTIVATE trial and is recognized for significant achievements in the development of novel AML treatments, including several combination therapies. Aptose presented data in 49 patients who received the TUS+VEN doublet, showing an overall response rate (ORR) of 48% among all patients that had achieved an evaluable stage, as well as a 44% ORR among Prior-VEN failure AML patients, including FLT3-unmutated (wildtype) patients (43% ORR) and FLT3-mutated patients (60% ORR), some of whom also had failed prior therapy with FLT3 inhibitors. The TUS+VEN doublet was well tolerated with no unexpected safety signals. The TUS/VEN doublet may serve the Prior-VEN failure R/R AML patients that represent a rapidly growing population that is highly refractory to any salvage therapy with response rates in the 4-15% range. The compelling data with the TUS+VEN doublet in R/R AML patients suggest a TUS+VEN+HMA triplet may serve the needs of frontline (1L) newly diagnosed AML patients.
On December 9, 2023, Aptose featured tuspetinib in an oral presentation at the 65th ASH Annual Meeting and Exposition and announced that a growing body of clinical data for Aptose’s lead compound tuspetinib, demonstrates significant benefit as a single agent and in combination with venetoclax in patients with R/R AML in the ongoing APTIVATE Phase 1/2 study. Data were presented in an oral presentation by lead investigator Naval G. Daver, M.D., Professor, Director Leukemia Research Alliance Program, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX.
Luxeptinib (CG-806) Program
Licensing Overview
Overview
On May 7, 2018, we exercised an option by paying $2.0 million in cash to the South Korean company CrystalGenomics Invites Co. Ltd, formerly Crystal Genomics, Inc. (“CG”), in order to purchase an exclusive license to
research, develop and commercialize luxeptinib in all countries of the world except the Republic of Korea and China, for all fields of use (collectively, the “Rights”). Subsequently, on June 14, 2018, we announced that we entered into a license agreement with CG for Aptoseus to gain a license for Rightsrights to CG-806 in China (including the People’s Republic of China, Hong Kong, and Macau) ) (the “China Rights”). Under the license agreement, Aptosewe made an upfront payment to CG of $3.0 million for the China Rights.rights. CG is eligible for development, regulatory and commercial-based milestones, as well as single-digit royalties on product sales in China. The total deal value for the China Rights, including the upfront payment, is up to $125 million. Aptose now owns worldwide (excluding Korea) Rights, including an issued patent in China,rights to luxeptinib, a first-in-class, highly potent oral small molecule being developed for AML, B-cell malignancies, and other hematologic malignancies. Future possible royalties that might be paid under these agreements are determined on a country-by-country and product-by-product basis, on net sales during the period of time beginning on the first commercial sale of such product in such country and continuing until the later of: (i) the expiration of the last-to-expire valid claim of the CG Patents in such country covering such product; and (ii) ten (10) years after the first commercial sale of such product in such country.
Preclinical Profile
Luxeptinib exhibits a picomolar half maximal inhibitory concentration (“IC50”)IC50 toward FLT3 with the Internal Tandem Duplication (“FLT3-ITD”), potency against the wild type FLT3 and a host of mutant forms of FLT3, as well as single-digit nanomolar IC50’sIC50’s against BTK and its C481S mutant (“BTK-C481S”). Consequently, luxeptinib is characterized as a mutation-agnostic FLT3/ BTK inhibitor. Further, luxeptinib suppresses a small group of other relevant oncogenic kinases/pathways (including CSF1R, PDGFRα, TRK, and the ERK, MYC, AKT/mTOR/S6K and AURK/H3S10 pathways) that are operative in AML and certain B cell malignancies, but does not inhibit the TEC, EGFRepidermal growth factor receptor (EGFR) and ErbB2/4 kinases that are responsible for safety concerns with certain other kinase inhibitors.
As a potent inhibitor of FLT3-ITD, luxeptinib may become an effective therapy in a high-risk subset of AML patients. This is because the FLT3-ITD mutation occurs in approximately 30% of patients with AML and is associated with a poor prognosis. In murine xenograft studies of human AML (FLT3-ITD), CG-806 administered orally resulted in tumor elimination (“cures”) without measurable toxicity. Importantly, luxeptinib targets other oncogenic kinases which may also be operative in FLT3-ITD AML, thereby potentially allowing the agent to become an important therapeutic option for a broader group of this difficult-to-treat AML patient population. The findings that luxeptinib targets all forms of FLT3 and several other key oncogenic pathways, and that luxeptinib was well tolerated from a safety perspective during efficacy and formal Good Laboratory Practice (“GLP”) toxicology studies, suggest that luxeptinib may also have applicability in treating patients, particularly those over the age of 65, who cannot tolerate other therapies.
Separate from the AML and FLT3 story,applications, luxeptinib may be a therapeutic option for patients with B cell malignancies. Overexpression of the BTK enzyme can drive oncogenic signaling of certain B cell malignancies, including CLL and certain NHL such as mantle cell lymphoma, (“MCL”), follicular lymphoma, (“FL”), diffuse large cell B cell lymphoma, (“DLBCL”) and others. Therapy of these patients with covalent, irreversible BTK inhibitors, such as ibrutinib, that target the active site cysteine (“Cys”) residue of BTK can be beneficial in many patients. However, therapy with covalent BTK inhibitors can select for BTK with a C481S mutation, thereby conferring resistance to covalent BTK inhibitors. Furthermore, approximately half of CLL patients have discontinued treatment with ibrutinib after 3.4 years of therapy. Discontinuation of ibrutinib is due to the development of drug resistance (in particular, patients have malignancies that developed the BTK-C481S mutation), or due to refractory disease (patient tumors did not respond to ibrutinib) or intolerance (side effects led to discontinuation of ibrutinib), according to a study performed at The Ohio State University. The C481S mutation is observed in 5-10% of the patients, while 40-45% of the patients were intolerant or refractory to ibrutinib. As a non-covalent, reversible inhibitor of BTK, luxeptinib does not rely on the Cysteine 481 residue (“C481”) for inhibition of the BTK enzyme. Indeed, recent X-ray crystallographic studies (with wild type and C481S BTK) demonstrated that luxeptinib binds productively to the BTK active site in a manner that is indifferent to the presence or absence of mutations at the 481 residue. Moreover, in vitro studies demonstrated that luxeptinib kills B cell malignancy cell lines on average approximately 1000 times more potently than ibrutinib and kills ibrutinib-resistance cancer cells, and that luxeptinib more potently killed primary malignant cells taken from the bone marrow of CLL and ALL B-cell cancer patients. Yet, luxeptinib demonstrated a high degree of safety in animal efficacy and GLP toxicology studies. Consequently, patients who are resistant, refractory or intolerant to ibrutinib or other commercially approved or development-stage BTK inhibitors with B cell malignancies may continue to be sensitive to luxeptinib therapy. This is particularly true since luxeptinib inhibits the wild type and mutant forms of BTK, as well as other kinases/pathways that drive the survival and proliferation of B cell malignancies.
Latest Clinical Update and Program Status
Role of BTK in B-cell signaling
BTK, a memberDuring 2023 and early 2024, clinical evaluation of the TEC family kinase, is an essential element of B-cell receptor (“BCR”) signaling, which is required for B-cell maturation, survival and proliferation. It is an upstream activator of multiple pro-survival / anti-apoptotic pathways, including the NF-KB, mTOR-AKT, RAS, ERK and MAPK pathways. BTK is overexpressed in malignant cells from patients with various B-cell malignancies, such as CLL, MCL, FL, and DLBCL. Disruption of BCR signaling via inhibition of BTK, has been shown to lead to clinical remissions in these patients.
Luxeptinib as a Non-covalent, Reversible Kinase Inhibitor
Binding studiesnew G3 formulation of luxeptinib have confirmed non-covalent, reversible inhibition of BTK, FLT3-ITDwas performed and Aurora Kinase A. Ibrutinib,has now been completed. The G3 formulation was tested in a commercially-approved, covalent BTK inhibitor, possesses a Michael acceptorsingle dose bioavailability study in 20 patients, including both B-cell cancer and AML patients, and across 5 dose levels (10mg to react with C481200mg). The G3 formulation then was evaluated in BTK and irreversibly inactivates the BTK enzyme. In contrast, luxeptinib does not require reactivity with the C481 residue for inhibition of the BTK enzyme, thereby allowing luxeptinib to inhibit the wild type and C481 mutant form of the BTK enzyme.
Preclinical In Vitro Evaluation of Luxeptinib
Luxeptinib is a potent inhibitor of BTK and FLT3 wild types, as well as the BTK C481S and FLT3-ITD mutants, which are strongly associated with clinical relapse or are negative prognostic factors in patients. In enzymatic assays, luxeptinib has demonstrated potency against the BTK C481S mutant with a half maximal IC50 of 2.5 nanomolar (nM). CG-806 also has potent activity against the FLT-ITD mutation, occurring in 30-35% ofR/R AML patients with an IC50 against the purified enzyme of 0.8nM (800pM). Likewise, luxeptinib exerts low nM IC50 values against the FLT3 enzyme having various mutations in the tyrosine kinase domain (TKD) and the Gatekeeper region, and luxeptinib has the ability to potently suppress the CSF1R, PDGFRα, SYK, AKT/mTOR/S6K, ERK, MAPK, MYC and AURK/H3S10 pathways. Finally, luxeptinib does not exhibit any inhibition of epidermal growth factor receptor (“EGFR”), TEC or ErbB2/4 kinases. Inhibition of one or more of these kinases has been speculated to contribute to the toxicity observed from the commercially approved BTK inhibitor.
Luxeptinib Xenograft Studies
In vivo subcutaneous AML tumor models of anti-cancer efficacy revealed luxeptinib induced rapid and sustained tumor eradication (Figure 1a). Luxeptinib was administered orally once daily, for 14 days. Moreover, luxeptinib exhibited the sustained tumor elimination post therapy, while demonstrating no impact to murine body weight, no impacts to hematology cell counts or visible organ toxicities – necropsy and clinical pathology findings did not reveal any abnormal observations. A maximum tolerated dose has not yet been identified with murine xenograft studies.

Figure 1a. Efficacy of luxeptinib (CG-806) in MV4-11 xenograft model.
MV4-11 tumor bearing mice were administered an oral suspension once daily for 14 days of luxeptinib (CG-806) at 2 mg/kg (blue line), 10 mg/kg (green line) or 100 mg/kg (red line), Ibrutinib at 12 mg/kg (turquoise line), or vehicle (Control; black line) with 7-day post-treatment follow-up. Tumor volumes and body weights were measured 3 times weekly.
In a separate MV4-11 xenograft study (Figure 1b), the antitumor efficacy of luxeptinib and mouse survival over 120 days were evaluated when mice were treated orally for 28 consecutive days with luxeptinib atcontinuous dosing using two different dose levels of 0 (vehicle only, red), 10 (olive), 30 (green), 100 (blue), or 300 mg/kg (magenta). In this study, the clinical formulation (luxeptinib co-micronized with 2.5% sodium lauryl sulfate (SLS))(50mg BID and the dosing schedule (“BID”) was utilized. Luxeptinib produced slower tumor growth and extended survival at the 10 and 30mg/kg dose levels, while 100% cure rates were achieved at the 100 and 300 mg/kg dose levels (11/11 mice survived in the latter two groups). Moreover, no signs of toxicity were noted at any dose level.
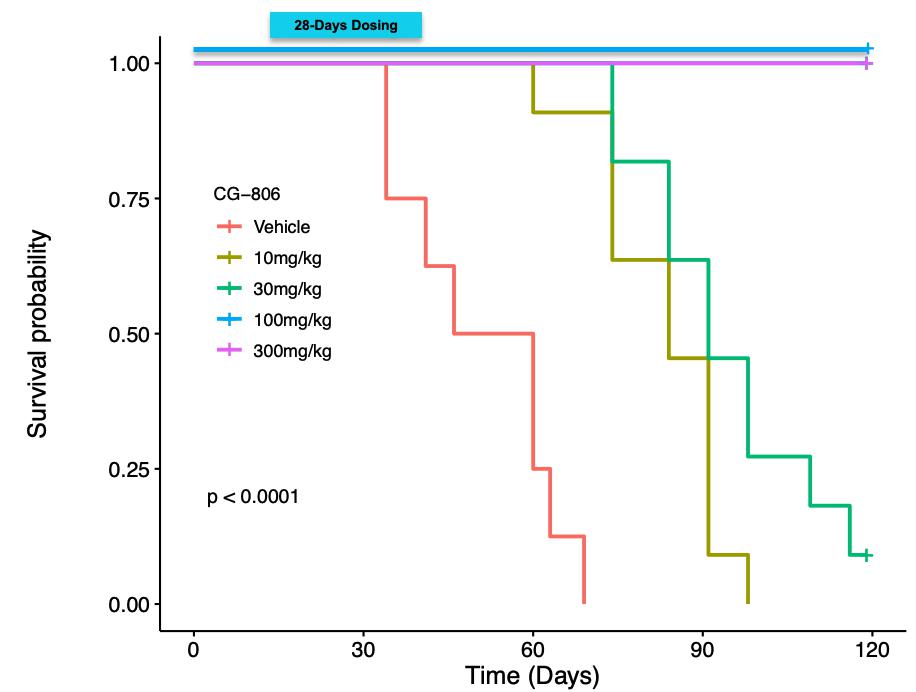
Figure 1b. Luxeptinib (CG-806) Extends Survival200mg BID) in a Dose-Dependent Waytotal of 11 patients. Data show the G3 formulation dosed at 200mg twice daily can achieve 2-3uM steady state plasma levels, with approximately 10-fold better absorption, and interestingly even better tolerability, than the original G1 formulation. Thus, the G3 formulation achieved the desired plasma exposure benchmark and can serve as the formulation of choice for future studies with LUX. Aptose is exploring alternative development paths and collaborations to advance LUX as a single agent or in MV4-11 AML Xenograft Mouse Model Following Oral BID Dosing for 28 Consecutive Days.combination with VEN to treat defined R/R patient populations of high unmet need.
Although the above murine xenograft models demonstrate potent antitumor activity with no observed toxicity, the models utilize an AML cell line rather than cells derived from an AML patient. In a study performed at the University of Texas MD Anderson Cancer Center (“MDACC”), the efficacy of luxeptinibLuxeptinib was evaluated in a patient derived xenograft (“PDX”) model (Figure 1c). Bone marrow cells were collectedPhase 1 a/b trial in patients with relapsed or refractory B cell malignancies who have failed or are intolerant to standard therapies, and in a separate Phase 1 a/b trial in patients with relapsed or refractory AML or high-risk MDS. During 2022, a new G3 formulation was tested as a single dose in 20 patients during the ongoing Phase 1 a/b clinical program. Modeling of the PK properties of G3 predicted steady-state plasma exposure from ancontinuous dosing with 50 mg of G3 (every 12 hours, Q12h) should be comparable to that of 900 mg of the original G1 formulation Q12h, representing a significant improvement in bioavailability with G3. On November 14, 2022, we announced dosing of the first AML patient that had relapsed onto receive a clinical trial. The patient entered the trial with FLT3-ITD AML and was placed on sorafenib and azacytidine. After one cycle, the patient had a complete response but then relapsed after cycle 3. Genetic analysis demonstrated that the AML cells had acquired a second mutation in AML, and this was the D835 mutation, making the patient dual mutant FLT3-ITD/D835. Bone marrow cells from the patient (AML FLT3-ITD/D835) were implanted in mice to establish a PDX model. Expansioncontinuous dosing regimen of the human AML cellsG3 formulation (50 mg G3 Q12h), with the protocol allowing for further dose escalation of G3 in subsequent patients. Clinical data from both studies were presented during a Corporate Comprehensive Clinical Update Call held December 11, 2022. During the bone marrow and peripheral bloodCorporate Update Call, we announced a CR was achieved with a diffuse large B-cell lymphoma patient at the end of Cycle 22 with 900mg BID of the mice tookoriginal G1 formulation. Previously, an MRD-negative CR was reported with a R/R AML patient receiving 450mg BID of the original G1 formulation.
Concurrent with the EHA Annual Congress held June 8-11, 2023, Aptose held an interim clinical update webcast on June 10, 2023. During the update, Aptose reviewed clinical findings with the new G3 formulation of luxeptinib. Aptose confirmed that continuous dosing with 50mg Q12h of the G3 formulation in multiple patients achieves roughly an equivalent pharmacokinetic profile as 900mg original G1 formulation, and that dose escalation with the G3 formulation was anticipated. Since completion of the 50mg G3 Q12h dose exploration, R/R AML patients have been dosed with 200mg Q12h G3.
Safety and PK data with continuous dosing of the G3 formulation have been completed and the 200mg dose of G3 luxeptinib achieved steady state exposure plasma levels of approximately one month.2uM. The amalgam of clinical safety, PK and activity data with all formulations of luxeptinib in B-cell cancer and AML patients are being collected and evaluated, and we plan to disclose the findings at a scientific presentation. In the vehicle (15% Transcutol HP/85% PEG-400) treated mice, the leukemic burdenaddition, a molecularly defined subgroup of CLL patients (harboring mutations in the peripheral blood increased from day 31 through day 50 and beyond. However,FLT3 receptor) has been identified as a potential target population for treatment with 100 mg/kg luxeptinib (orally daily for 5 consecutive days followed by 2 days off every week), resulted in significant reduction in the leukemic burden and reductions in splenomegaly at 52 days post-implantation. These data suggest that luxeptinib may be used to treat patients whose disease has become resistant to other FLT3 inhibitors.
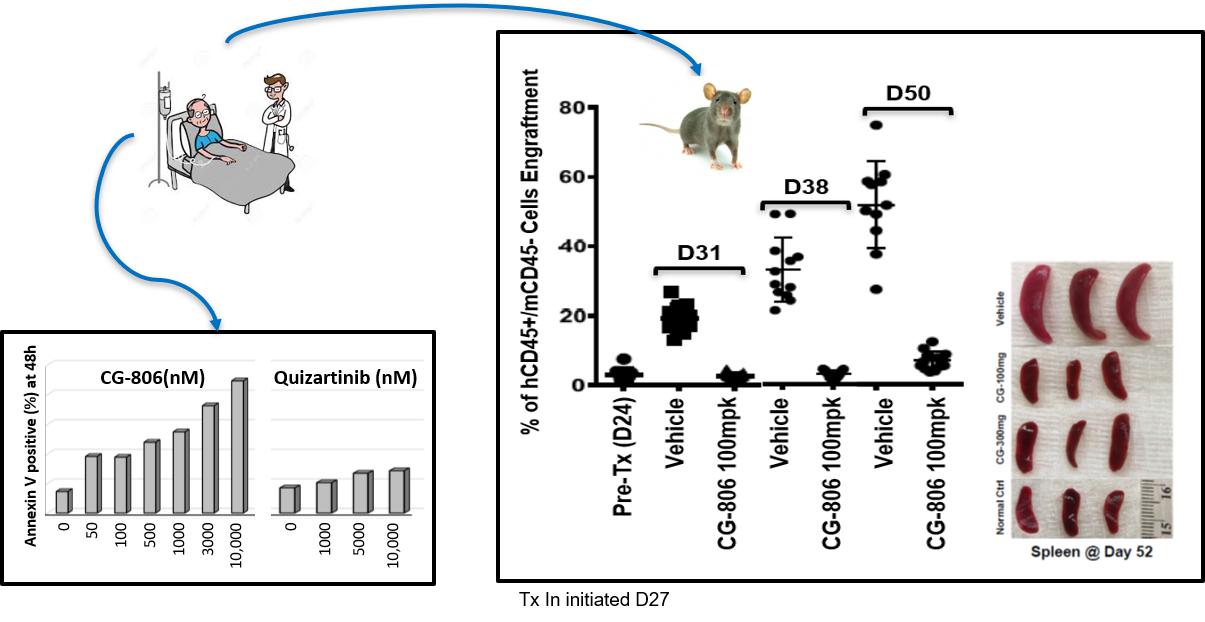
Figure 1c. Luxeptinib (CG-806) Efficacy in PDX Model Against AML Patient Cells with FLT3-ITD+D835Y Mutations
APTO-253 Program
Overview
APTO-253, our second clinical-stage program, is a novel small molecule therapeutic agent that inhibits expression of the MYC oncogene, leading to cell cycle arrest and programmed cell death (apoptosis) in human-derived solid tumor and hematologic cancer cells, without causing general myelosuppression of the healthy bone marrow. The MYC oncogene is overexpressed in hematologic cancers, including AML. MYC is a transcription factor that regulates cell growth, proliferation, differentiation and apoptosis, and overexpression amplifies new sets of genes to promote oncogenesis. APTO-253 dramatically down-regulates expression of the MYC oncogene in AML cells and depletes those cells of the MYC oncoprotein, leading to apoptotic cell death in AML cells. Thus APTO-253 may serve as a safe and effective MYC inhibitor for AML that combines wellcombination with other agents, and does not impact the normal bone marrow.
During 2015, we were evaluating APTO-253 in a Phase 1a/b clinical trial in patients with R/R hematologic malignancies, particularly AML and MDS, before being placed on clinical hold by the FDA in November 2015. The Phase 1a/b trial was placed on clinical hold in order to solve a chemistry-based formulation issue, and the chemistry of the active pharmaceutical ingredient (“API”) and the formulation underwent minor modifications to deliver a stable and soluble drug product for return to the clinical setting. In December 2016, we announced that we had successfully manufactured multiple non-GMP batches of a new drug product formulation for APTO-253, including a batch that had been stable and soluble for over six months. However, the 40L batch that was the intended clinical supply encountered an unanticipated mishap during the filling process that compromised the stability of that batch of drug product. On January 23, 2017, we announced that the root cause and corrective action studies would take longer than originally expected and that we would temporarily delay clinical activities with APTO-253 in order to elucidate the cause of manufacturing mishap, with the intention of restoring the molecule to a state supporting clinical development and partnering. Formal root cause analyses studies were completed to identify the reasonfeasibility analysis for the drug product stability failure, and a correction action was implemented. We then manufactured a new GMP clinical supplypotential development of drug product and performed the studies required to demonstrate the fitness of the drug product for clinical usage, and presented the findings to the FDA in the second quarter of 2018. On June 28, 2018, the FDA notified us that it had lifted the clinical hold on APTO-253. This was followed by resubmission of the revised clinical protocol to Institutional Review Boards (“IRB”) at multiple clinical sites.
On November 28, 2018, we announced that we dosed the first patient in the re-initiation of the Phase 1a/b Clinical Study of APTO-253. Since then, we have completed the first four dose cohorts (20mg/m2, 40mg/m2, 66mg/m2 and 100mg/m2) and are currently dosing patients in the fifth dose cohort at 150mg/m2 dose level. In the patients we have dosed at the first four dose levels, we observed meaningful reductions in MYC expression in the patient PBMC samples and noted that the drug product is well tolerated to date.
APTO-253 Studies on Solid Tumors
In January 2011, Aptose announced the first patient enrollment in a Phase 1 dose-escalation study for APTO-253 in patients with advanced or metastatic solid tumors who are unresponsive to conventional therapy or for whom no effective therapy is available. The study was initially being conducted at Memorial Sloan-Kettering Cancer Center in New York. Objectives of the study included determination or characterization of the safety profile, maximum tolerated dose, and antitumor activity of APTO-253, as well as pharmacokinetics and a recommended Phase 2 dose for subsequent clinical trials.
In June 2012, MDACC in Houston was added as a second site under the direction of Dr. Jennifer Wheler as the principal investigator. In addition, Aptose announced that the study had successfully completed the accelerated drug dose escalation stage (Stage 1), with further escalation under way in the non-accelerated dose escalation stage (Stage 2)luxeptinib for the purpose of determining the maximal tolerated dose level and recommended Phase 2 dose. The addition of a second site expanded patient availability for enrollment.target population is underway.
In January 2013, Aptose announced that Phase 1 clinical study of APTO-253 had successfully escalated to the target dose level based on predicted and observed clinical effects without limitation by toxicity. The success of this study allowed Aptose to initiate a biomarker clinical investigation to further explore the effects of the drug at relevant doses determined in the clinical trial.
In April 2013, Aptose announced that studies demonstrated the antitumor activity of APTO-253 in animal models of human non-small cell lung cancer (“NSCLC”) with a dose-response effect in NSCLC.
In July 2013, Aptose announced the results of the Phase 1 clinical trial of APTO-253. In this first-in-man dose-escalation clinical study, APTO-253 demonstrated a favorable safety profile, as well as encouraging signs of antitumor activity in patients with solid tumors. The design of this trial consisted of APTO-253 as a single agent in patients with advanced solid tumors resistant to multiple standard therapies. The study enrolled 27 patients, all of which had failed a median of four prior chemotherapies. Although this was primarily a dose-escalation safety study, efficacy and pharmacokinetics were also explored.
The clinical trial enrolled patients at seven dose levels ranging from 20 to 229 mg/m2. Of the 27 patients enrolled, 17 were evaluable for efficacy. Of these 17 patients, seven (41%) achieved stable disease by Response Evaluation Criteria In Solid Tumors (“RECIST”). This included patients with colorectal, lung, appendiceal, liver and uterine cancers. Dose related activity was demonstrated at the higher dose levels (176 and 229 mg/m2). At these two highest dose levels, four of five evaluable patients (80%) achieved sustained stable disease by RECIST ranging from 5.6 months to 8 months, representative of disease control. Of these, a patient with NSCLC at the highest dose level additionally demonstrated non-index tumor shrinkage.
The safety assessment indicated that APTO-253 was well tolerated at all dose levels tested in this trial. The dose escalation was not limited by toxicity. The most common adverse event was Grade 1 or 2 fatigue seen in three patients. There was one Grade 3 toxicity, asymptomatic low blood phosphate level that was reversible by supplementation with phosphates. The pharmacokinetic profile was consistent with the predictive profile seen preclinically, and the elimination profile and half-life in patients were suggestive of a very rapid distribution phase and prolonged retention. No further studies were performed after late 2013.
APL-581 Program
In November 2015, Aptose announced an exclusive drug discovery partnership with Laxai Avanti Life Sciences (“LALS”) for their expertise in next generation epigenetic-based therapies. Under the agreement, LALS was to be responsible for developing multiple clinical candidates, including optimizing candidates that exert dual BRD4 / kinase inhibitory activity. Based on available resources, Aptose halted further investment in the collaboration with LALS in late 2016. However, the program delivered novel intellectual property and hit molecules (such as APL-581). Consequently, Aptose chose to out-license the program.
On March 7, 2018, Aptose entered into an exclusive global license agreement with OHM, an affiliate of LALS that was formed in 2016 to advance the clinical development of compelling molecules derived from the LALS initiative, for the development, manufacture and commercialization of APL-581, as well as related molecules, from Aptose’s dual BET protein and kinase inhibitor program. Under the agreement, Aptose retained reacquisition rights to certain molecules, while OHM/LALS has the rights to develop and sublicense all other molecules. Aptose received a nominal upfront cash payment and is eligible to receive up to $125 million of additional payments based on the achievement of certain developmental, regulatory and sales milestones, as well as significant royalties on future sales generated from the program, if any. We have not received any milestone or royalty payments pursuant to this agreement. Future possible royalties that might be paid by OHM to Aptose under these agreements are determined on a country-by-country and product-by-product basis, on net sales during the period of time beginning on the first commercial sale of such product in such country and continuing until the later of: (i) the expiration of the last-to-expire valid claim of the patents in such country covering such product; and (ii) ten (10) years after the first commercial sale of such product in such country.
Competitive Conditions
The biotechnology and pharmaceutical industries are characterized by rapidly evolving technology and intense competition. There are numerous companies in these industries that are focusing their efforts on activities similar to ours. Some of these are companies with established positions in the pharmaceutical industry and may have substantially more financial and technical resources, more extensive research and development capabilities, and greater marketing, distribution, production, and human resources than Aptose.us. In addition, we face competition from other companies for opportunities to enter into partnerships with biotechnology and pharmaceutical companies and academic institutions.
Competition with our potential products may include chemotherapeutic agents, monoclonal antibodies, antisense therapies, small molecules, immunotherapies, vaccines, and other biologics with novel mechanisms of action. These drugs may kill cancer cells indiscriminately, or through a targeted approach, and some have the potential to be used in non-cancer indications. We also expect that we will experience competition from established and
emerging pharmaceutical and biotechnology companies that have other forms of treatment for the cancers that we target, including drugs currently in development for the treatment of cancer that employ a number of novel approaches for attacking these cancer targets. Cancer is a complex disease with more than 100 indications requiring drugs for treatment. The drugs in competition with our potential drugs have specific targets for attacking the disease, targets which are not necessarily the same as ours. These competitive drugs, however, could potentially also be used together in combination therapies with our drugs to manage the disease. Other factors that could render our potential products less competitive may include the stage of development, where competitors’ products may achieve earlier commercialization, as well as superior patent protection, better safety profiles, or a preferred cost-benefit profile.
Luxeptinib Treatment for B Cell Malignancies
We are aware of a number of companies that have developed and are pursuing different approaches to BTK inhibition, both for the wild type and to the C481S-mutant forms. Companies that have developed approved or are currently developing inhibitors that directly target the wild type include AbbVie (IMBRUVICA) and AstraZeneca (CALQUENCE) and Beigene Co., Ltd. (Zanubrutinib).
Others that are developing inhibitors that target the C481S-mutant BTK include Merck (MK-1026), Roche, and Eli Lilly (LY3527727) among others.
Luxeptinib and APTO-253 for AML
We also face intense competition in AML as there is a wide range of therapies that have been approved and are under development for the treatment of AML. Companies that have developed approved or are currently developing non-targeted therapies include Jazz (VYXEOS), Pfizer (MYLOTARG) and AbbVie (VENCLEXTA), among others. Others that have developed or are developing highly targeted therapies such as FLT3 inhibitors include Novartis (RYDAPT), Astellas (XOSPATA), Daiichi Sankyo (quizartinib), Arog (crenolanib), and IDH1/2 inhibitors include Agios/Servier (TIBSOVO) and Celgene/BMS (IDHIFA) among others.
Manufacturers, Suppliers and Other Third Party Contractors
Contract manufacturing organizations (“CMOs”) manufacture our product candidates for all preclinical studies and clinical trials. We rely on CMOs for manufacturing, filling, packaging, storing and shipping of drug product in compliance with Current Good Manufacturing Practice (“cGMP”) regulations applicable to our products. The FDA ensures the quality of drug products by carefully monitoring drug manufacturers’ compliance with cGMP regulations. The cGMP regulations for drugs contain minimum requirements for the methods, facilities and controls used in manufacturing, processing and packing of a drug product. These CMOs are reputable companies active in the biotechnology industry. Pricing is predictable as there are many alternatives of such supplies that are readily available.
We rely and will continue to rely on third party contract research organizations (“CROs”) to conduct a significant portion of our preclinical and clinical development activities. Preclinical activities include in vivo studies providing access to specific disease models, pharmacology and toxicology studies, and assay development. Clinical development activities include trial design, regulatory submissions, clinical patient recruitment, clinical trial monitoring, clinical data management and analysis, safety monitoring and project management, contract manufacturing and quality assurance.
Intellectual Property
We believe that our issued patents and pending applications are important in establishing and maintaining a competitive position with respect to our products and technology.
CG-806Tuspetinib (HM43239)
A Patent Cooperation Treaty (“PCT”In November 2021, we licensed the exclusive rights to research, develop and commercialize tuspetinib (the "Tuspetinib Licensing Agreement") application providing. Under the terms of the Tuspetinib Licensing Agreement, Hanmi granted Aptose exclusive worldwide rights to tuspetinib for all indications.Aptose is now the exclusive licensee of composition of matter and use patents covering tuspetinib, and tuspetinib analogs. Aptose believes that it now owns rights to a strong and defensive intellectual property position.
As of December 31, 2023, Aptose owned rights in 46 issued patents, including 4 issued U.S. patents, and 23 patents validated in countries in Europe, that are in force and cover the tuspetinib compound, or analog compounds. These patents are expected to provide protection until 2038 through 2039. Patent applications are also pending in the United States and in contracting states to the Patent Cooperation Treaty for CG-806 was filed in late 2013,coverage of tuspetinib and analog compounds, with a potentialexpected expiry in 2033 before extension opportunities, across all major geographiesdates between 2038 and 2042.
Luxeptinib (CG-806)
In May 2018 and June 2018, we paid $2.0 million in cash and licensed the Rights to CG-806, for all fields of use, in all territories outside of the Republic of Korea and China, by exercising an option we obtained through a June 2016 option-license agreement with CG that had granted us an exclusive option to research, develop and commercialize CG-806.
In June 2018, we entered into a separate license agreement with CG for Aptose to gain a license for the China Rights. This license agreement was formally executed by Aptose through an upfront payment to CG of $3.0 million for the China Rights. CG is eligible for payments upon the achievement of developmental, regulatory and commercial-based milestones, as well as single-digit royalties on product sales in China. Aptose now owns worldwide Rights to CG-806, including an issued patent in China but excluding any Rights in Korea.
US Patent No. 9,758,508
On September 12, 2017, we announcedAs of December 31, 2023, Aptose owned rights to 49 issued patents, including 3 issued U.S. patents, and 30 patents validated in countries in Europe, that United States Patentare in force and Trademark Office (“USPTO”) issued patent number 9,758,508, entitled “2,3-dihydro-isoindole-1-on derivative as BTK kinase suppressant, and pharmaceutical composition including same”, which claimscover numerous compounds, including the CG-806 compound, pharmaceutical compositions comprising the CG-806 compound, and methods of treating various diseases. The patent is expected to provide protection until December of 2033.
US Patent No. 10,604,508
The issued patent, issued on March 21, 2020, claims a genus that covers the CG-806 compound, pharmaceutical compositions comprising a compound from the genus, and methods ofuse for treating various diseases caused by abnormal or uncontrolled activation of protein kinases, including lymphoma and leukemia. This US patent is expected to provide protection until December 2033.
European Patent No. EP2940014B1
The granted patent claims the CG-806 compound, pharmaceutical compositions comprising the CG-806 compound, and uses for treating diseases caused by abnormal or uncontrolled activation of protein kinases, such as cancer. This European patent will be nationalized in, and cover, approximately twenty European countries including the United Kingdom, France, Germany, Italy, Netherlands and Spain. The patent is expected to provide protection until December of 2033.
Australian Patent Nos. 2013371146 and 2018214134
The granted patents claim numerousadministering various compounds, including the CG-806 compound, pharmaceutical compositions comprising the CG-806 compound, and methods of treating various diseases, including treating cancers such as lymphoma and leukemia. The patent is expected to provide protection until December of 2033.
Chinese Patent No. CN 104995184 B
The granted patent claims numerous compounds, including the CG-806 compound, pharmaceutical compositions comprising the CG-806 compound, and the use of such a compound for the manufacture of a pharmaceutical composition for treating a disease caused by an abnormal or uncontrolled protein kinase. The patent is expected to provide protection until December of 2033.
Japanese Patent Nos. 6325573 and 6596537
The granted patents claim numerous compounds, including the CG-806 compound, pharmaceutical compositions comprising the CG-806 compound, and the use of such a compound for the manufacture of a pharmaceutical composition for treating a disease caused by an abnormal or uncontrolled protein kinase, and pharmaceutical compositions for treating various diseases, including treating cancers such as lymphoma and leukemia. Thecompound. These patents are expected to provide protection until December of 2033.
Canadian2033-2038. Patent No. 2896711
The granted patent claims numerous compounds, including the CG-806 compound, pharmaceutical compositions comprising the CG-806 compound, and the use of such a compound for the manufacture of a pharmaceutical composition for treating a disease caused by an abnormal or uncontrolled protein kinase. The patent is expected to provide protection until December of 2033.
Russian Patent No. 2671847
The granted patent claims various compounds, including the CG-806 compound, pharmaceutical compositions comprising the CG-806 compound, and methods for treating diseases caused by abnormal or uncontrolled activation of protein kinases, and uses for the treatment, relief or prevention of cancer. The patent is expected to provide protection until December 2033.
APTO-253
As of March 23, 2021, weapplications are the owner of record of five issued U.S. patents, which together provide coverage for the APTO-253 compound, its pharmaceutical composition and methods of treating various cancers with APTO-253, including solid tumors and leukemia. The APTO-253 composition of matter has patent protection until February, 2028also pending in the United States and May, 2026 in other countries. We also hold 23 international (non-U.S.) granted patents which together providecontracting states to the Patent Cooperation Treaty for coverage for APTO-253, three of which are granted European patents, validated in at least eight countries in Europe. Our patents also include several compounds that are similar to APTO-253, which provide protection from competitors seeking to develop anticancer products that are related in chemical structure to APTO-253.CG-806, with expected expiry dates between 2038‑2039.
Environmental Protection
The Company’s research and development activities involve the controlled use of hazardous and radioactive materials and, accordingly, the Company is subject to federal, provincial and local laws and regulations in the United States and Canada governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. To the knowledge of the Company, compliance with such environmental laws and regulations does not and will not have any significant impact on its capital spending, profits or competitive position within the normal course of its operating activities. There can be no assurance, however, that the Company will not be required to incur significant costs to comply with environmental laws and regulations in the future or that its operations, business or assets will not be materially adversely affected by current or future environmental laws or regulations.
Employees
Employees
As atof December 31, 2020,2023, we employed 3935 full-time persons and twoone part-time personsperson in research and drug development and administration activities. SixEleven of our employees hold Ph.D.s, two hold M.D.s, and numerous others hold degrees and designations such as MD, MSc, BSc, CPA (CA)M.Sc., CPA (California)B.Sc., C.P.A., C.M.A., M.Acc. and MBA.M.B.A. To encourage a focus on achieving long-term performance, employees and members of the board of directors of the Company (the “Board”) have the ability to acquire an ownership interest in the Company through Aptose’s share option and alternate compensation plans. Of note, in January of 2020, Aptose hired a Chief Medical Officer holding an MD and a Ph.D.
The business of the Company requires personnel with specialized skills and knowledge in oncology. Researchers must be able to design and implement studies to assess the efficacy of anticancer drugs. Specialized knowledge and skills relating to chemistry and formulation process development are also needed. Such knowledge and skills are needed to develop product specific analytical methods and formulation processes. The Company’s business also requires clinical and regulatory expertise and knowledge. The Company has trained scientists and personnel with broad experience in these fields.
None of our employees are unionized, and we consider our relations with our employees to be good.
Government Regulation
Overview
Overview
Our overall regulatory strategy is to work with the appropriate government departments which regulate the use and sale of therapeutic drug products. This includes the FDA in the United States, Health Canada in Canada, the European Medicines Agency (“EMA”) in Europe, and other local regulatory agencies with oversight of preclinical studies, clinical trials and marketing of therapeutic products. Where possible, we intend to take advantage of opportunities for accelerated development of drugs designed to treat rare and serious or life-threatening diseases. We also intend to pursue priority evaluation of any application for marketing approval filed in Canada, the United States or the European Union and to file additional drug applications in other markets where commercial opportunities exist. We may not be able to pursue these opportunities successfully.
Regulation(s) by government authorities in the United States, Canada, and the European Union are significant factors in guiding our current research and drug development activities. To clinically test, manufacture and market drug products for therapeutic use, we must be in compliance with guidance and regulations established by the regulatory agencies in the countries in which we currently operate or intend to operate.
The laws of most of these countries require the licensing of manufacturing facilities, carefully controlled research and the extensive testing of products. Biotechnology companies must establish the safety and efficacy of their new products in clinical trials; they must establish and comply with cGMPscurrent good manufacturing practices ("cGMPs") for the manufacturing of the product and control over marketing activities before being allowed to market a product. The safety and efficacy of a new drug must be shown through human clinical trials of the drug carried out in accordance with the guidance and regulations established by local and federal regulatory agencies.
The process of completing clinical trials and obtaining regulatory approval for a new drug takes a number of years and requires the expenditure of substantial resources. Once a new drug or product license application is submitted, regulatory agencies may not review the application in a timely manner and may not approve the product. Even after a New Drug Application (“NDA”) submission has occurred and/or approval has been obtained, further studies, including post-marketing studies, may be required to provide additional data on the efficacy and safety necessary to confirm the approved indication or to gain approval for the use of the new drug as a treatment for clinical indications other than those for which the new drug was initially tested. Also, regulatoryRegulatory agencies also require post-marketing surveillance programs to monitor a new drug’s side effects, safety and long-term effects of the product. A serious safety or effectiveness problem involving an approved new drug may result in a regulatory agency mandating a withdrawal of the new drug from the market and possible civil action. It is possible that we could encounter such difficulties or excessive costs in our efforts to secure necessary approvals, which could delay or prevent us from manufacturing or marketing our products.
In addition to the regulatory product approval framework, biotechnology companies, including Aptose, are subject to regulation under local, provincial, state and federal law, including requirements regarding occupational safety, laboratory practices, environmental protection and hazardous substance control, and may be subject to other present and future local, provincial, state, federal and foreign regulation, including possible future regulation of the biotechnology industry.
Approval of New Drugs in Canada
In Canada, the manufacture and sale of new drugs are controlled by Health Canada. New drugs must pass through a number of testing stages, including pre-clinical testing and human clinical trials. Pre-clinical testing involves testing the new drug’s chemistry, pharmacology and toxicology in vitro and in vivo.vivo. Successful results (that is, potentially valuable pharmacological activity combined with an acceptable low level of toxicity) enable the developer of the new drug to file a clinical trial application to begin clinical trials involving humans.
To study a drug in Canadian patients, a clinical trial application submission must be filed with Health Canada. The clinical trial application submission must contain specified information, including the results of the pre-clinical tests completed at the time of the submission and any available information regarding use of the drug in humans. In addition, since the method of manufacture may affect the efficacy and safety of a new drug, information on manufacturing methods and standards and the stability of the drug substance and dosage form must be presented. Production methods and quality control procedures must be in place to ensure an acceptably pure product, essentially free of contamination, and to ensure uniformity with respect to all quality aspects.
In addition, all federally regulated trials must be approved and monitored by an independent committee of doctors, scientists, advocates and others to ensure safety and ethical standards, IRBsInstitutional Review Boards (“IRBs”) or Ethics Review Boards (“ERBs”). The review boards study and approve all study-related documents before a clinical trial begins and also carefully monitor data to detect benefit or harm, and validity of results.
Provided Health Canada does not reject a clinical trial application submission and IRB or ERB approval has been obtained, clinical trials can begin. Clinical trials for product candidates in Canada, as in the United States, are generally carried out in three phases. Phase 1 involves studies to evaluate toxicity and ideal dose levels in healthy humans. The new drug is administered to human patients who have met the clinical trial entry criteria to determine pharmacokinetics, human tolerance and prevalence of any adverse side effects. Phases 2 and 3 involve therapeutic studies. In Phase 2, efficacy, dosage, side effects and safety are established in a small number of patients who have the disease or disorder that the new drug is intended to treat. In Phase 3, there are controlled clinical trials in which the new drug is administered to a large number of patients who are likely to receive benefit from the new drug. In Phase 3, the effectiveness of the new drug in patients is compared to that of standard accepted methods of treatment in order to provide sufficient data for the statistical proof of safety and efficacy for the new drug.
If clinical studies establish that a new drug has value, the manufacturer submits a new drug submission application to Health Canada for marketing approval. The new drug submission contains all known information known about the new drug, including the results of pre-clinical testing and clinical trials. Information about a substance contained in new drug submission includes its proper name, its chemical name, and details on its method of manufacturing and purification, and its biological, pharmacological and toxicological properties. The new drug submission also provides information about the dosage form of the new drug, including a quantitative listing of all ingredients used in its formulation, its method of manufacture, manufacturing facility information, packaging and labelling,labeling, the results of stability tests, and its diagnostic or therapeutic claims and side effects, as well as details of the clinical trials to support the safety and efficacy of the new drug. Furthermore, for biological products, an on-site evaluation is completed to assess the production process and manufacturing facility. It is required prior to the issuance of a notice of compliance. All aspects of the new drug submission are critically reviewed by Health Canada. If a new drug submission is found satisfactory, a notice of compliance is issued permitting the new drug to be sold for the approved use. In Canada, an establishment license must be obtained prior to marketing the product.
Health Canada has a policy of priority evaluation of new drug submissions for all drugs intended for serious or life-threatening diseases for which no drug product has received regulatory approval in Canada and for which there is reasonable scientific evidence to indicate that the proposed new drug is safe and may provide effective treatment.
An exception to the foregoing requirements relating to the manufacture and sale of a new drug is the limited authorization that may be available in respect of the sale of new drugs for emergency treatment. Under the special access program, Health Canada may authorize the sale of a quantity of a new drug for human use to a specific practitioner for the emergency treatment of a patient under the practitioner’s care. Prior to authorization, the practitioner must supply Health Canada with information concerning the medical emergency for which the new drug is required, such data as is in the possession of the practitioner with respect to the use, safety and efficacy of the new drug, the names of the institutions at which the new drug is to be used and such other information as may be requested by Health Canada. In addition, the practitioner must agree to report to both the drug manufacturer and Health Canada the results of the new drug’s use in the medical emergency, including information concerning adverse reactions, and must account to Health Canada for all quantities of the new drug made available.
The Canadian regulatory approval requirements for new drugs outlined above are similar to those of other major pharmaceutical markets. While the testing carried out in Canada is often acceptable for the purposes of regulatory submissions in other countries, individual regulatory authorities may request supplementary testing during their assessment of any submission. Therefore, the clinical testing conducted under Health Canada authorization or the approval of regulatory authorities of other countries may not be accepted by regulatory authorities outside Canada or other countries.
Approval of New Drugs in the United States
In the United States, the FDA controls and investigates the investigation, manufacturing, and sale of new drugs. New drugs require FDA approval of an NDA prior to commercial sale. In the case of certain biological products, a Biological License Application (“BLA”) must be obtained prior to marketing and batch releasing. As in Canada, to obtain marketing approval, data from adequate and well-controlled human clinical trials, demonstrating to the FDA’s satisfaction a new drug’s safety and effectiveness for its intended use, are required. Data are generated in studies conducted pursuant to an INDinvestigational new drug (“IND”) submission, similar to that required for a clinical trial application in Canada. Clinical trials with human subjects are characterized as Phase 1, Phase 2 and Phase 3 trials or a combination thereof. In a marketing application, the manufacturer must also demonstrate the identity, potency, quality and purity of the active ingredients of the new drug involved, and the stability of those ingredients. Further, the manufacturing facilities, equipment, processes and quality controls for the new drug must comply with the FDA’s current cGMP regulations for drugs both in a pre-licensing inspection before product licensing and in subsequent periodic inspections after licensing. An establishment license grants the sponsor permission to fabricate, package, label, distribute, import, wholesale or test the newly approved drug.
Federally regulated trials must be approved and monitored by an independent committee of doctors, scientists, advocates, and others to ensure safety and ethical standards, IRBs or ERBs. The review boards study and approve all study-related documents before a clinical trial begins and also carefully monitor data to detect benefit or harm, and validity of results.
Post-Approval Regulation
The monitoring of a new drug does not cease once it is on the market. For example, a manufacturer of a new drug must report any new information received concerning serious side effects, as well as the failure of the new drug to produce desired effects. If Health Canada determines it to be in the interest of public health, a notice of compliance for a new drug may be suspended and the new drug may be removed from the market.
A post surveillance program involves clinical trials conducted after a drug is marketed (referred to as Phase 4 studies in the United States) and is an important source of information on as yet undetected adverse outcomes, especially in populations that may not have been involved in the premarketing trials (e.g., children, the elderly, pregnant women) and the drug’s long-term morbidity and mortality profile. Regulatory authorities may require companies to conduct Phase 4 studies as a condition of market approval. Companies often conduct post-marketing studies in the absence of a regulatory mandate.
The foregoing description is a brief summary of the requirements for a new drug to be approved for marketing in North America. The EMA and Japanese Pharmaceuticals and Medical Devices Agency are also important regulatory authorities in drug development. Together with the FDA, they are the three International Conference on Harmonization parties which oversee the three largest markets for drug sales.
Information About Our Executive Officers
Aptose’s leadership team comprises accomplished industry, financial and clinical research professionals who are dedicated to building a comprehensive anticancer drug pipeline and clinical development programs focused on targeted therapeutics directed against dysregulated oncogenic processes in patients with life. The team includes our President, Chairman and Chief Executive Officer, our Chief Financial Officer, and Chief Operating Officer, our Chief Business and Strategy Officer and our Chief Medical Officer.
Dr. William G. Rice, Ph.D., age 62,64, joined Aptose as Chairman and Chief Executive Officer in October 2013. Dr. Rice brings 25 years of C-level experience in the biotech industry to Aptose. Prior to joining Aptose, Dr. Rice served as the President, Chief Executive Officer and Chairman of the board of Cylene Pharmaceuticals, Inc. (“Cylene”), a private biotechnology company, from 2003 to 2013. Prior to Cylene, Dr. Rice was the founder, President, Chief Executive Officer and Director of Achillion Pharmaceuticals, Inc. from 1998 to 2003. He also served as Senior Scientist and Head of the Drug Mechanism Laboratory at the National Cancer Institute-Frederick Cancer Research and Development Center from 1992 to 1998 and served as a faculty member in the division of Pediatric Hematology and Oncology at Emory University School of Medicine from 1989 to 1992. Dr. Rice received his Ph.D. from Emory University Department of Biochemistry. He continues to serve as the Chairman of the board of Cylene and is a member of the board of directors of Oncolytics Biotech Inc. since 2015.
Gregory K. Chow, M.B.A.Fletcher Payne, age 48,age 61, joined Aptose as Senior Vice President and Chief Financial Officer (“CFO”) in December 2013,June 2022. Mr. Payne was then appointed as Executive Vice President and Chief Financial Officer in 2019, and then Chief Operating Officer and Chief Financial Officer in 2021. Previously, Mr. Chow served as Managing Director, Director of Private Placements at Wedbush Securities from 2012 to 2013, where he led the private placement capital activities within the Life Sciences Investment Banking Group. Prior to joining Wedbush, he was a Director in the Private Placements / Equity Capital Markets Group at RBC Capital Markets from 2006 to 2011, where he led life science private capital activities. From 2003 to 2006, he led the Private Capital Group at Wells Fargo Securities and was a Senior Auditor at BDO Seidman, LLP in their Century City, CA office. Mr. Chow is a Certified Public Accountant (inactive) in the State of California. Mr. Chow received his MBA in Finance from The Wharton School at the University of Pennsylvania, and his BA in Business Economics with an emphasis in Accounting from the University of California, Santa Barbara.
Dr. Jotin Marango, M.D, Ph.D., age 42, joined Aptose as Senior Vice President and Chief Business Officer in June 2019October 2023. With a healthcare tenure of more than 25 years, Mr. Payne most recently served as CFO of Syapse, where he completed several financing transactions and Chief Businessoversaw accounting, finance, corporate development, and Strategy Officerlegal functions. Prior, he served as CFO at Catalyst Bioscience, a publicly traded biotech company. He served in January 2021. Prior to joining Aptose, from 2017 to 2019, Dr. Marango was a managing directorCFO capacity and senior research analystfinancial positions at Roth Capital Partners covering biotechnologyCytomX Therapeutics, Plexxikon Inc., Rinat Neuroscience Corporation, Dynavax Technologies Corporation, and therapeutics. Dr. Marango joined Roth from H.C. Wainwright & Co., where he worked from 2015 to 2017 and covered hematology, oncology, and pulmonary therapeutics, withCell Genesys, among others. Mr. Payne holds a focus on epigenetic and molecularly targeted therapies. Dr. Marango began his careerB.S. in equity research with Collins Stewart/Canaccord Genuity in 2010. Previously, Dr. Marango also served as Chief Operating Officer at the Samuel Waxman Cancer Research Foundation from 2012 to 2015, where he oversaw academic collaborations in translational therapeutics, as well as venture philanthropy initiatives in drug development. Dr. Marango studied theoretical chemistry and classical literature at Harvard University and later received his M.D. and Ph.D. degreesFinance from the Mount SinaiHaas School of Medicine in New York.Business, University of California, Berkeley.
Dr. Rafael Bejar, M.D, Ph.D.Ph.D., age 49,52, joined Aptose as Senior Vice President and Chief Medical Officer in January 2020. Dr. Bejar is an internationally recognized physician scientist with extensive research and clinical experience in the area of hematologic malignancies. Dr. Bejar joined Aptose from UC San Diego (“UCSD”) where he began working in 2012. He continues to serve at UCSD as an Associate Professor of Clinical Medicine, caring for patients and maintaining a research laboratory focused on translational studies of myeloid malignancies and also serves and is an independent consultant as a member of the Independent Data Monitoring Committee for other pharmaceutical companies. At UCSD, he founded the MDS Center of Excellence and led the Hematology Disease Team from 2017 to 2019. There he has directed several clinical studies and served as an advisor for numerous companies including Celgene, Takeda, AbbVie, Astex, Genoptix, Forty Seven, PersImmune, and Daiichi-Sankyo. Outside UCSD, Dr. Bejar sits on the Scientific Advisory Board for the MDS Foundation, is a prior member of the National Comprehensive Cancer Network Guidelines Committee, and has led projects for the International Working Group for MDS. He is frequently invited to speak at national and international meetings and has published articles in a variety of journals including The New England Journal of Medicine,, Journal of Clinical Oncology,, Leukemia, Blood, and Blood Advances.Advances. Dr. Bejar completed his fellowship at the Dana-Farber Cancer Institute and has been board certified in Hematology and Oncology. He completed his internship in Internal Medicine at the University of Chicago followed by his residency at the Brigham and Women’s Hospital in Boston where he later served a Medical Chief Resident and an Instructor in Hematology. He holds an MDM.D. degree and a Neuroscience PhDPh.D. from UCSD and a BSB.S. in Physics from MIT.
Corporate Information
Aptose is a publicly traded company governed by the CBCA.Canada Business Corporations Act ("CBCA"). Our headquarters are located at 251 Consumers Road, Suite 1105 Toronto, Ontario, Canada M2J 4R3 (telephone: 647-479-9828), and our executive offices are located at 12770 High Bluff Drive, Suite 120, San Diego, CA 92130 (telephone: 858-926-2730).
We file annual, quarterly, current reports, proxy statements and other information with the Securities and Exchange Commission (the “SEC”).SEC. The SEC maintains an Internet site that contains our public filings and other information regarding the Company, at www.sec.gov. We make these reports available free of charge at our website http://www.aptose.com (under the “Investors — Financial Information” caption).
Prior to December 31, 2018, Aptose was a foreign private issuer, and in compliance with SEC regulations, filed its Quarterly reports on Form 6-Ks, and its Annual Reports on either Forms F-20 or F-40. These reports were made available on our website as soon as reasonably practicable after their filing with, or furnishing to, the SEC.
We are also a reporting issuer under the securities laws of every province of Canada.
Cautionary Note Regarding Forward-Looking Statements and Risk Factor Summary
This Annual Report on Form 10-K contains forward-looking statements within the meaning of the United States Private Securities Litigation Reform Act of 1995 and “forward-looking information” within the meaning of applicable Canadian securities law. We refer to such forward-looking statements and forward-looking information collectively as “forward-looking statements”. These statements relate to future events or future performance and reflect our expectations and assumptions regarding our growth, results of operations, performance and business prospects and opportunities. Such forward-looking statements reflect our current beliefs and are based on information currently available to us. In some cases, forward-looking statements can be identified by terminology such as “may”, “would”, “could”, “will”, “should”, “expect”, “plan”, “intend”, “anticipate”, “believe”, “estimate”, “predict”, “potential”, “continue” or the negative of these terms or other similar expressions concerning matters that are not historical facts. The forward-looking statements in this Annual Report on Form 10-K include, among others, statements regarding our future operating results, economic performance and product development efforts and statements in respect of:
| · | our ability to obtain the substantial capital we require to fund research and operations; |
•our ability to obtain the substantial capital we require to fund research and operations and to continue as a going concern;
•our clinical development plans;
•our plans to conduct clinical trials and preclinical programs;
•our ability to accrue appropriate numbers and types of patients;
•our reliance on external contract research/manufacturing organizations for certain activities;
•our plans to secure and maintain strategic partnerships to assist in the further development of our product candidates and to build our pipeline;
•our ability to file and maintain intellectual property to protect our pharmaceutical assets;
•potential exposure to legal actions and potential need to take action against other entities;
•our expectations regarding the progress and the successful and timely completion of the various stages of our drug discovery, drug synthesis and formulation, preclinical and clinical studies and the regulatory approval process;
•our plans, objectives, expectations, and intentions; and
•other statements including words such as “anticipate,” “contemplate,” “continue,” “believe,” “plan,” “estimate,” “expect,” “intend,” “will,” “should,” “may,” and other similar expressions.
| · | our clinical development plans; |
| · | our plans to conduct clinical trials and preclinical programs; |
| · | our ability to accrue appropriate numbers and types of patients; |
| · | our reliance on external contract research/manufacturing organizations for certain activities; |
| · | our plans to secure and maintain strategic partnerships to assist in the further development of our product candidates and to build our pipeline; |
| · | our ability to file and maintain intellectual property to protect our pharmaceutical assets; |
| · | potential exposure to legal actions and potential need to take action against other entities; |
| · | our expectations regarding the progress and the successful and timely completion of the various stages of our drug discovery, drug synthesis and formulation, preclinical and clinical studies and the regulatory approval process; |
| · | our plans, objectives, expectations and intentions; and |
| · | other statements including words such as “anticipate”, “contemplate”, “continue”, “believe”, “plan”, “estimate”, “expect”, “intend”, “will”, “should”, “may”, and other similar expressions. |
The forward-looking statements contained in this Annual Report on Form 10-K reflect our current views with respect to future events, are subject to significant risks and uncertainties, and are based upon a number of estimates and assumptions that, while considered reasonable by us, are inherently subject to significant business, economic, competitive, political and social uncertainties and contingencies. Forward-looking statements contained in this Annual Report on Form 10-K are made as of the date of this Annual Report on Form 10-K.
Except as required under applicable securities legislation, we undertake no obligation to publicly update or revise forward-looking statements, whether as a result of new information, future events or otherwise.
Risk Factor Summary
Many factors could cause our actual results, performance or achievements to be materially different from any future results, performance, or achievements that may be expressed or implied by such forward-looking statements, including, among others:
| · | our lack of product revenues and net losses and a history of operating losses; |
| · | our early stage of development, particularly the inherent risks and uncertainties associated with (i) developing new drug candidates generally, (ii) demonstrating the safety and efficacy of these drug candidates in clinical studies in humans, and (iii) obtaining regulatory approval to commercialize these drug candidates; |
| · | our need to raise substantial additional capital in the future and that we may be unable to raise such funds when needed and on acceptable terms; |
| · | further equity financing, which may substantially dilute the interests of our existing shareholders; |
| · | clinical studies and regulatory approvals of our drug candidates are subject to delays, and may not be completed or granted on expected timetables, if at all, and such delays may increase our costs and could substantially harm our business; |
| · | our reliance on external contract research/manufacturing organizations for certain activities and if we are subject to quality, cost, or delivery issues with the preclinical and clinical grade materials supplied by contract manufacturers, our business operations could suffer significant harm; |
| · | clinical studies are long, expensive and uncertain processes and the FDA, or other similar foreign regulatory agencies that we are required to report to, may ultimately not approve any of our product candidates; |
| · | Our operations could be adversely affected by events outside of our control, such as natural disasters, wars or health crises such as the COVID-19 pandemic. |
| · | our ability to comply with applicable governmental regulations and standards; |
| · | our inability to achieve our projected development goals in the time frames we announce and expect; |
•our ability to continue as a going concern
•our lack of product revenues and net losses and a history of operating losses;
•our early stage of development, particularly the inherent risks and uncertainties associated with (i) developing new drug candidates generally, (ii) demonstrating the safety and efficacy of these drug candidates in clinical studies in humans, and (iii) obtaining regulatory approval to commercialize these drug candidates;
•our need to raise substantial additional capital in the future and that we may be unable to raise such funds when needed and on acceptable terms;
•further equity financing, which may substantially dilute the interests of our existing shareholders;
•clinical studies and regulatory approvals of our drug candidates are subject to delays, and may not be completed or granted on expected timetables, if at all, and such delays may increase our costs and could substantially harm our business;
•our reliance on external contract research/manufacturing organizations for certain activities and if we are subject to quality, cost, or delivery issues with the preclinical and clinical grade materials supplied by contract manufacturers, our business operations could suffer significant harm;
•clinical studies are long, expensive and uncertain processes and the FDA, or other similar foreign regulatory agencies that we are required to report to, may ultimately not approve any of our product candidates;
•our operations could be adversely affected by events outside of our control, such as natural disasters, wars or health crises;
•our ability to comply with applicable governmental regulations and standards;
•our inability to achieve our projected development goals in the time frames we announce and expect;
•difficulties in enrolling patients for clinical trials may lead to delays or cancellations of our clinical trials;
•our reliance on third parties to conduct and monitor our preclinical studies;
•our ability to attract and retain key personnel, including key executives and scientists;
•any misconduct or improper activities by our employees;
•our exposure to exchange rate risk;
•our ability to commercialize our business attributed to negative results from clinical trials;
•the marketplace may not accept our products or product candidates due to the intense competition and technological change in the biotechnical and pharmaceuticals industries, and we may not be able to compete successfully against other companies in our industries and achieve profitability;
•our ability to obtain and maintain patent protection;
•our ability to afford substantial costs incurred with defending our intellectual property;
•our ability to protect our intellectual property rights and not infringe on the intellectual property rights of others;
•our business is subject to potential product liability and other claims;
•potential exposure to legal actions and potential need to take action against other entities;
| · | difficulties in enrolling patients for clinical trials may lead to delays or cancellations of our clinical trials; |
| · | our reliance on third-parties to conduct and monitor our preclinical studies; |
•commercialization limitations imposed by intellectual property rights owned or controlled by third parties;
•our ability to maintain adequate insurance at acceptable costs;
•our ability to find and enter into agreements with potential partners;
•extensive government regulation;
•data security incidents and privacy breaches could result in increased costs and reputational harm;
•our share price has been and is likely to continue to be volatile;
•future sales of our Common Shares by us or by our existing shareholders could cause our share price to drop;
•changing global market and financial conditions;
•changes in an active trading market in our Common Shares;
•our ability to maintain listing of our Common Shares on the Nasdaq and / or TSX
•changing regulations may result in additional expenses
•difficulties by non-Canadian investors to obtain and enforce judgments against us because of our Canadian incorporation and presence;
•potential adverse U.S. federal tax consequences for U.S. shareholders because we are a “passive foreign investment company”;
•our “smaller reporting company” status;
•any failures to maintain an effective system of internal controls may result in material misstatements of our financial statements, or cause us to fail to meet our reporting obligations or fail to prevent fraud;
•our broad discretion in how we use the proceeds of the sale of Common Shares;
•our ability to expand our business through the acquisition of companies or businesses; and
•other risks detailed from time-to-time in our on-going filings with the SEC and Canadian securities regulators, and those which are discussed in Item 1A. Risk Factors in this Annual Report on Form 10-K.
| · | our ability to attract and retain key personnel, including key executives and scientists; |
| · | any misconduct or improper activities by our employees; |
| · | our exposure to exchange rate risk; |
| · | our ability to commercialize our business attributed to negative results from clinical trials; |
| · | the marketplace may not accept our products or product candidates due to the intense competition and technological change in the biotechnical and pharmaceuticals, and we may not be able to compete successfully against other companies in our industries and achieve profitability; |
| · | our ability to obtain and maintain patent protection; |
| · | our ability to afford substantial costs incurred with defending our intellectual property; |
| · | our ability to protect our intellectual property rights and not infringe on the intellectual property rights of others; |
| · | our business is subject to potential product liability and other claims; |
| · | potential exposure to legal actions and potential need to take action against other entities; |
| · | commercialization limitations imposed by intellectual property rights owned or controlled by third parties; |
| · | our ability to maintain adequate insurance at acceptable costs; |
| · | our ability to find and enter into agreements with potential partners; |
| · | extensive government regulation; |
| · | data security incidents and privacy breaches could result in increased costs and reputational harm; |
| · | our share price has been and is likely to continue to be volatile; |
| · | future sales of our Common Shares by us or by our existing shareholders could cause our share price to drop; |
| · | changing global market and financial conditions; |
| · | changes in an active trading market in our Common Shares; |
| · | difficulties by non-Canadian investors to obtain and enforce judgments against us because of our Canadian incorporation and presence; |
| · | potential adverse U.S. federal tax consequences for U.S. shareholders because we are a “passive foreign investment company”; |
| · | our “smaller reporting company” status; |
| · | any failures to maintain an effective system of internal controls may result in material misstatements of our financial statements, or cause us to fail to meet our reporting obligations or fail to prevent fraud; |
| · | our broad discretion in how we use the proceeds of the sale of Common Shares; |
| · | our ability to expand our business through the acquisition of companies or businesses; and |
| · | other risks detailed from time-to-time in our on-going filings with the SEC and Canadian securities regulators, and those which are discussed in Item 1A. Risk Factors in this Annual Report on Form 10-K. |
Should one or more of these risks or uncertainties materialize, or should the assumptions described in the Item 1A. Risk Factors in this Annual Report on Form 10-K underlying those forward-looking statements prove incorrect, actual results may vary materially from those described in the forward-looking statements.
Although we have attempted to identify factors that could cause actual actions, events or results to differ materially from those described in forward-looking statements, there may be other factors that cause actions, events or results not to be as anticipated, estimated or intended. Forward-looking statements are based upon our beliefs, estimates and opinions at the time they are made and we undertake no obligation to update forward-looking statements if these beliefs, estimates and opinions or circumstances should change, except as required by applicable law. There can be no assurance that forward-looking statements will prove to be accurate, as actual results and future events could differ materially from those anticipated in such statements. Accordingly, readers should not place undue reliance on forward-looking statements.
We qualify all the forward-looking statements contained in this Annual Report on Form 10-K by the foregoing cautionary statements.
ITEM 1A. RISK FACTORS
Risk Factors and Uncertainties
Any of the risks and uncertainties described below could significantly and negatively affect our business, prospects, financial condition, operating results, or credit ratings, which could cause the trading price of our Common Shares to decline. Additional risks and uncertainties not presently known to us, or risks that we currently consider immaterial, could also impair our business operations or financial condition. The following discussion of risk factors contains “forward-looking” statements, as discussed above.
Risks Related to our Business
There is substantial doubt that we can remain a going concern over the next twelve months.
As of the filing date, we have sufficient liquidity to support the Company's operations until August 2024. In order for the Company to meet its capital requirements, and continue to operate, additional financing will be necessary. The Company is evaluating strategies to obtain the required additional funding for future operations. These strategies may include, but are not limited to, obtaining equity financing, and restructuring of operations to decrease expenses. However, given the challenges in the U.S. and global financial markets, and the matter in Note 17 to the Financial Statements, Subsequent events, that may impact the Company's ability to raise financing in the capital markets, the Company may be unable to access further equity or when needed, if at all. As the Company is primarily pursuing one compound that is licensed from a related party with significant licensing payments who will have influence on the Company, other investors may not be willing to invest in the Company. As such, there can be no assurance that the Company will be able to obtain additional liquidity when needed or under acceptable terms, if at all. The consolidated financial statements do not reflect any adjustments to the carrying amounts and classification of assets, liabilities, and reported expenses that may be necessary if the Company were unable to continue as a going concern. Such adjustments may be material. See Note 2(a) to the Financial Statements, Going concern.
We are an early stageearly-stage development company with no revenues from product sales.
We are at an early stage of development. In the past five years, noneNone of our potential products has obtained regulatory approval for commercial use and sale in any country and as such, no revenues have resulted from product sales. Significant additional investment will be necessary to complete the development of any of our product candidates. Preclinical and clinical trial work must be completed before our potential products could be ready for use within the markets that we have identified. We may fail to develop any products, obtain regulatory approvals, enter or complete clinical trials or commercialize any products. We do not know whether any of our potential product development efforts will prove to be effective, meet applicable regulatory standards, obtain the requisite regulatory approvals, be capable of being manufactured at a reasonable cost or be accepted in the marketplace. We also do not know whether sales, license fees or related royalties will allow us to recoup any investment we make in the commercialization of our products.
The product candidates we are currently developing are not expected to be commercially viable for at least the next several years and we may encounter unforeseen difficulties or delays in commercializing our product candidates. In addition, our potential products may not be effective or may cause undesirable side effects.
Our product candidates require significant funding to reach regulatory approval assuming positive clinical results. For example, our product candidate APTO-253 began enrollment in aWe are currently conducting Phase 1a/b1 clinical trial in patients with relapsed or refractory AML and high risk MDS and was placed on clinical hold by the FDA following a voluntary suspension of dosing by us. That hold has been lifted, but significant additional funding will be necessary to complete the restarted Phase 1a/b clinical and, if required, Phase 2 or Phase 3 clinical trials. Similarly, we have received FDA approval to initiate a Phase 1a/b clinical trialtrials with our product candidate luxeptinib for patients with B-cell malignancies.candidates tuspetinib and luxeptinib. Significant additional capital will be necessary to complete the Phase 1 clinical trial,trials, and if required, Phase 2 or Phase 3 clinical trials. Such funding for our product candidates may be difficult, or impossible to raise in the public or private markets or through partnerships. If funding or partnerships are not readily attainable, the development of our product candidates may be significantly delayed or stopped altogether. The announcement of a delay or discontinuation of development would likelyof any of our product candidates could have a negative impact on our share price.
We need to raise additional capital.
We have an ongoing need to raise additional capital. As of the filing date, we have sufficient liquidity to support the Company's operations until August 2024. To obtain the necessary capital, we must rely on some or all of the following: additional share issues, debt issuances (including promissory notes), collaboration agreements or corporate partnerships and grants and tax credits to provide full or partial funding for our activities. Additional funding may not be available on terms that are acceptable to us or in amounts that will enable us to carry out our business plan. Although, as of the date of this report, the COVID-19 pandemic did not have and we do not expect that it will have a significant impact on our liquidity and capital resources, the extent to which COVID-19 impacts our business will depend on future developments, which are highly uncertain and cannot be predicted, including the scope, severity and duration of the pandemic, the actions taken to contain the pandemic or mitigate its impact, and the direct and indirect economic effects of the pandemic and containment measures, among other future developments. As such, our ability to raise additional funds could be affected by adverse market conditions resulting from the COVID-19 pandemic and delays related to COVID-19 in enrollment in our trial.
Our need for capital may require us to:
| · | engage in equity financings that could result in significant dilution to existing investors; |
•engage in equity financings that could result in significant dilution to existing investors;
•delay or reduce the scope of or eliminate one or more of our development programs;
•obtain funds through arrangements with collaborators or others that may require us to relinquish rights to technologies, product candidates or products that we would otherwise seek to develop or commercialize ourselves;
•license rights to technologies, product candidates or products on terms that are less favorable to us than might otherwise be available;
•considerably reduce operations; or
| · | delay or reduce the scope of or eliminate one or more of our development programs; |
| · | obtain funds through arrangements with collaborators or others that may require us to relinquish rights to technologies, product candidates or products that we would otherwise seek to develop or commercialize ourselves; |
| · | license rights to technologies, product candidates or products on terms that are less favorable to us than might otherwise be available; |
| · | considerably reduce operations; or |
In addition, sales of a substantial number of our Common Shares in the public markets, or the perception that such sales could occur, could depress the market price of our Common Shares and impair our ability to raise capital through the sale of additional equity securities.
Our operations could be adversely affected by events outside of our control, such as natural disasters, wars or health crises such as the COVID-19 pandemic.crises.
We may be impacted by business interruptions resulting from pandemics and public health emergencies, including those related to COVID-19, geopolitical actions, including war and terrorism or natural disasters including earthquakes, typhoons, floods and fires. An outbreak of infectious disease, a pandemicAny such event, or a similar public health threat, such as the COVID-19 pandemic, or a fear of any of the foregoing, could adversely impact us by causing operating, manufacturing, supply chain, clinical trial and project development delays and disruptions, labourlabor shortages, travel and shipping disruption and shutdowns (including as a result of government regulation and prevention measures). Although, as of the date of this report, we do not expect that COVID-19 will have a significant impact on our liquidity and capital resources, the extent to which COVID-19 impacts our business will depend on future developments, which are highly uncertain and cannot be predicted, including the scope, severity and duration of the pandemic, the actions taken to contain the pandemic or mitigate its impact, and the direct and indirect economic effects of the pandemic and containment measures, among other future developments.shutdowns. We may incur expenses or delays relating to such events outside of our control, which could have a material adverse impact on our business, operating results and financial condition.
We have a history of operating losses. We expect to incur net losses and we may never achieve or maintain profitability.
We have not been profitable since our inception in 1986. We reported net losses of $51.2 million in the fiscal year ended December 31, 2023, $41.8 million in the fiscal year ended December 31, 2022, $65.4 million in the fiscal year ended December 31, 2021, $55.2 million in the fiscal year ended December 31, 2020 $26.3 million in the fiscal year ended December 31, 2019 and $28.8 million in the fiscal year ended December 31, 2018,, and as of December 31, 2020,2023, we had an accumulated deficit of $357.2$515.5 million. We had negative shareholder's equity of $2.9 million as of December 31, 2023 (December 31, 2022, positive shareholder's equity of $37.7 million).
We have not generated any significant revenue to date and it is possible that we will never have sufficient product sales revenue (if any) to achieve profitability. We expect to continue to incur losses for at least the next several years as we or our collaborators and licensees pursue clinical trials, and research and development efforts. To become profitable, we, either alone or with our collaborators and licensees, must successfully develop, manufacture and market our current product candidates APTO-253tuspetinib or luxeptinib, as well as continue to identify, develop, manufacture and market new product candidates. It is possible that we will never have significant product sales revenue or receive royalties on our licensed product candidates. If funding is insufficient at any time in the future, we may not be able to develop or commercialize our products, take advantage of business opportunities or respond to competitive pressures.
We currently do not earn any revenues from our drug candidates and are therefore considered to be in the development stage. The continuation of our research and development activities and the commercialization of the targeted therapeutic products are dependent upon our ability to successfully finance and complete our research and development programs through a combination of equity financing and payments from strategic partners. We have no current sources of significant payments from strategic partners. As of the filing date, we have sufficient liquidity to support the Company's operations until August 2024.
We heavily rely on the capabilities and experience of our key executives and scientists and the loss of any of them could affect our ability to develop our products.
The loss of our executive officers could harm our operations and our ability to achieve strategic objectives. While we have employment agreements with our executive officers, such employment agreements do not guarantee their retention. We also depend on our scientific and clinical collaborators and advisors, all of whom have outside commitments that may limit their availability to us. In addition, we believe that our future success will depend in large part upon our ability to attract and retain highly skilled scientific, managerial, medical, clinical and regulatory personnel, particularly as we expand our activities and seek regulatory approvals for clinical trials. We routinely enter into consulting agreements with our scientific and clinical collaborators and advisors, key opinion leaders and academic partners in the ordinary course of our business. We also enter into contractual agreements with physicians and institutions who will recruit patients into our clinical trials on our behalf in the ordinary course of our business. Notwithstanding these arrangements, we face significant competition for these types of personnel from other companies, research and academic institutions, government entities and other organizations. We cannot predict our success in hiring or retaining the personnel we require for continued growth. The loss of the services of any of our executive officers or other key personnel could potentially harm our business, operating results or financial condition.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include failures to comply with FDA/Health Canada regulations, provide accurate information to the FDA/Health Canada, comply with manufacturing standards we have established, comply with federal, state and provincial health-care fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a substantial impact on our business and results of operations, including the imposition of substantial fines or other sanctions.
We have no sales, marketing or distribution experience and would have to invest significant financial and management resources to establish these capabilities.
We have no sales, marketing or distribution experience. We currently expect to rely heavily on third parties to launch and market our products, if they are approved. However, if we elect to develop internal sales, distribution and marketing capabilities, we will need to invest significant financial and management resources. For products where we decide to perform sales, marketing and distribution functions ourselves, we could face a number of additional risks, including:
•we may not be able to attract and build a significant marketing or sales force;
•the cost of establishing a marketing or sales force may not be justifiable in light of the revenues generated by any particular product; and
•our direct sales and marketing efforts may not be successful.
20
| · | we may not be able to attract and build a significant marketing or sales force; |
| · | the cost of establishing a marketing or sales force may not be justifiable in light of the revenues generated by any particular product; and |
| · | our direct sales and marketing efforts may not be successful. |
If we are unable to develop our own sales, marketing and distribution capabilities, we will not be able to successfully commercialize our products without reliance on third parties.
We may expand our business through the acquisition of companies or businesses or by entering into collaborations or by in-licensing product candidates, each of which could disrupt our business and harm our financial condition.
We may in the future seek to expand our pipeline and capabilities by acquiring one or more companies or businesses, entering into collaborations or in-licensing one or more product candidates. For example, in June 2016, we entered into a definitive agreement with CG, granting Aptose an exclusive option to research, develop and commercialize CG-806 in all countries of the world except the Republic of Korea, for all fields of use.use, and in November 2021 we entered into the Tuspetinib Licensing Agreement with Hanmi granting Aptose exclusive worldwide rights to develop and commercialize tuspetinib.
Acquisitions, collaborations and in-licenses involve numerous risks, including, but not limited to:
| · | substantial cash expenditures; |
| · | technology development risks; |
| · | potentially dilutive issuances of equity securities; |
| · | incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition; |
| · | difficulties in assimilating the operations of the acquired companies; |
| · | potential disputes regarding contingent consideration; |
| · | diverting our management’s attention away from other business concerns; |
•substantial cash expenditures;
•technology development risks;
•potentially dilutive issuances of equity securities;
•incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition;
•difficulties in assimilating the operations of the acquired companies;
•potential disputes regarding contingent consideration;
•diverting our management’s attention away from other business concerns;
•entering markets in which we have limited or no direct experience;
•potential loss of our key employees or key employees of the acquired companies or businesses; and
•failure of the in-licenses agents or technologies to deliver the desired activities or functions.
| · | entering markets in which we have limited or no direct experience; |
| · | potential loss of our key employees or key employees of the acquired companies or businesses; and |
| · | failure of the in-licenses agents or technologies to deliver the desired activities or functions. |
We have experience in entering collaborations and in-licensing product candidates; however, we cannot provide assurance that any acquisition, collaboration or in-license will result in short-term or long-term benefitsany benefit to us. We may incorrectly judge the value or worth of an acquired company or business or in-licensed product candidate. In addition, our future success wouldcould depend in part on our ability to manage the rapid growth associated with some of these acquisitions, collaborations and in-licenses. We cannot assure you that we would be able to successfully combine our business with that of acquired businesses, manage a collaboration or integrate in-licensed product candidates. Furthermore, the development or expansion of our business may require a substantial capital investment by us.
Fluctuations in exchange rates can cause us to incur losses.
We may be exposed to fluctuations of the United StatesU.S. dollar against certain other currencies because we hold most of our cash and cash equivalents in United StatesU.S. dollars, while we incur some of our expenses in foreign currencies, primarily the Canadian dollar. Fluctuations in the value of currencies could cause us to incur currency exchange losses, and we do not currently employ a hedging strategy against exchange rate risk. As a result, changes in the exchange rate between the Canadian dollar and the U.S. dollar could materially impact our reported results of operations and distort period to period comparisons. In particular, to the extent that foreign currency-denominated (i.e., non-U.S. dollar) monetary assets do not equal the amount of our foreign currency denominated monetary liabilities, foreign currency gains or losses could arise and materially impact our financial statements. As a result of such foreign currency fluctuations, it could be more difficult to detect underlying trends in our business and results of operations. In addition, to the extent that fluctuations in currency exchange rates cause our results of operations to differ from our expectations or the expectations of our investors, the trading price of our Common Shares could be adversely affected.
Risks Related to Development, Clinical Testing and Regulatory Approval of Our Product Candidates
Fast Track Designation by the FDA may not lead to a faster development or regulatory review or approval process.
We have obtained Fast Track Designation for tuspetinib for the treatment of patients with R/R AML and FLT3 mutation. We may seek Fast Track Designation for one or more of our other product candidates. If a drug is intended for the treatment of a serious or life-threatening condition and the drug demonstrates the potential to address unmet medical needs for this condition, the product sponsor may apply for FDA Fast Track Designation. The FDA has broad discretion whether or not to grant this designation, so even if we believe a particular product candidate is eligible for this designation, we cannot assure you that the FDA would decide to grant it. Even if we do receive Fast Track Designation, we may not experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may withdraw Fast Track Designation if it believes that the designation is no longer supported by data from our clinical development program.
Clinical trials are long, expensive and uncertain processes and the FDA or Health Canada may ultimately not approve any of our product candidates. We may never develop any commercial drugs or other products that generate revenues.
In the past five years, noneNone of our product candidates has received regulatory approval for commercial use and sale in North America. We cannot market a pharmaceutical product in any jurisdiction until it has completed thorough preclinical testing and clinical trials in addition to that jurisdiction’s extensive regulatory approval process. Approval in one country does not assure approval in another country. In general, significant research and development and clinical studies are required to demonstrate the safety and effectivenessefficacy of our product candidates before we can submit any applications for regulatory approval.
Clinical trials are long, expensive and uncertain processes. Clinical trials may not start or be on schedule and the FDA, or Health Canada or any other regulatory body may not ultimately approve our product candidates for commercial sale in the relevant territory. The clinical trials of any of our drug candidates could be unsuccessful, which would prevent us from advancing, commercializing or partnering the drug.
Even if the results of our preclinical studies or clinical trials are initially positive, it is possible that we will obtain different results in the later stages of drug development or that results seen in clinical trials will not continue with longer term treatment. Positive results in Phase 1 clinical trials may not necessarily repeat in larger Phase 2 or Phase 3 clinical trials.
Our preclinical studies and clinical trials may not generate positivenegative results that will not allow us to move towards the commercial use and sale of our product candidates. Furthermore, negative preclinical or clinical trial results may cause our business, financial condition, or results of operations to be materially adversely affected. For example, our Phase 1a/b clinical trial of APTO-253 in patients with relapsed or refractory AMLOur tuspetinib and high risk MDS was placed on clinical hold by the FDA in November 2015. Those short comings of the drug product were addressed and the clinical hold was lifted. However, there can be no assurance that the Company will have the resources, or that we will decide, to continue the development of APTO-253. There is a long development path ahead that will take many years to complete the development and is prone to the risks of failure or delays inherent in drug development. Likewise, our luxeptinib product candidate iscandidates are currently being evaluated in a Phase 1a/b study for patients having B-cell malignancies,1 studies, and it isare expected to undergo many years of testing and regulatory examinations prior to any potential regulatory approvals.
Preparing, submitting and advancing applications for regulatory approval of products is complex, expensive and time intensive and entails significant uncertainty. A commitment of substantial resources to conduct time-consuming research, preclinical studies and clinical trials is required if we are to complete development of our products.
Clinical trials of our products require that we identify and enroll a large number of patients with the illness under investigation. We may not be able to enroll a sufficient number of appropriate patients to complete our clinical trials in a timely manner, particularly in smaller indications and indications where there is significant competition for patients. If we experience difficulty in enrolling a sufficient number of patients to conduct our clinical trials, we may need to delay or terminate ongoing clinical trials and will not accomplish objectives material to our success. Delays in planned patient enrollment or lower than anticipated event rates in our current clinical trials or future clinical trials also may result in increased costs, program delays, or both.
In addition, unacceptable toxicities or adverse side effects may occur at any time in the course of preclinical studies or human clinical trials or, if any product candidates are successfully developed and approved for marketing, during commercial use of any approved products. The appearance of any unacceptable toxicities or adverse side effects could interrupt, limit, delay or abort the development of any of our product candidates or, if previously approved, necessitate their withdrawal from the market. Furthermore, disease resistance or other unforeseen factors may limit the effectiveness of our potential products.
Our failure to develop safe and commercially viable drugs would substantially impair our ability to generate revenues and sustain our operations and would materially harm our business and adversely affect our share price.
We may choose to expend our limited resources on programs that do not yield successful product candidates as opposed to indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited resources and access to capital to fund our operations, our management must make strategic decisions as to which product candidates and indications to pursue and how much of our resources to allocate to each. Our management must also evaluate the benefits of developing in‑licensed or jointly owned technologies, which in some circumstances we may be contractually obligated to pursue, relative to developing other product candidates, indications or programs. Our management has broad discretion to suspend, scale down, or discontinue any or all of these development efforts, or to initiate new programs to treat other diseases. If we select and commit resources to opportunities that we are unable to successfully develop, or we forego more promising opportunities, our business, financial condition and results of operations will be adversely affected.
We may not achieve our projected development goals in the time frames we announce and expect.
We set goals for, and make public statements regarding, the expected timing of the accomplishment of objectives material to our success, such as the commencement and completion of clinical trials, the submission of a drug-regulatory application, and the expected costs to develop our product candidates. The actual timing and costs of these events can vary dramatically due to factors within and beyond our control, such as delays or failures in our IND submissions or clinical trials, issues related to the manufacturing of drug supply, uncertainties inherent in the regulatory approval process, market conditions and interest by partners in our product candidates, among other things. Our clinical trials may not be completed, we may not make regulatory submissions or receive regulatory approvals as planned; or we may not secure partnerships for any of our product candidates. Any failure to achieve one or more of these milestones as planned would have a material adverse effect on our business, financial condition and results of operations.
Although, as of the date of this report, we do not foresee material delays to the enrollment of patients or timelines for our trials due to COVID-19, the extent to which COVID-19 will impact the projected development goals will depend on future developments, which are highly uncertain and cannot be predicted. In the beginning of April 2020 we learned that certain of our larger sites would not be able to enroll new patients on the fourth dose level of luxeptinib due to the current environment caused by COVID-19 and we therefore expect a slowdown in enrollment at these sites. While it is difficult to estimate the duration and impact of COVID-19 on the larger clinical sites and regional cancer care sites, as of the date of this report, we have not experienced and do not foresee material delays to the enrollment of patients or timelines for the luxeptinib Phase 1a/b B-cell malignancy trial due to the variety of clinical sites that we have actively recruited for this trial. While as of the date of this report we have not experienced any material delays in initiating our luxeptinib Phase 1 clinical study in AML due to COVID-19, we are conducting site initiation visits remotely which could result in delays in site activations and negatively impact this trial. Additionally, COVID-19 could negatively impact patient enrolment if our clinical sites are unable to enroll patients due to either a lack of administrative resources at their sites or decisions made at the clinical sites to limit patient exposure to COVID-19.
Future enrollment of patients on the APTO-253 trial is likely to be negatively impacted as a result of the current environment, as it is administered to patients intravenously, which requires the need for hospital / clinical site resources to assist and monitor patients during each infusion.
Delays in clinical testing could result in delays in commercializing our product candidates and our business may be substantially harmed.
We cannot predict whether any clinical trials will begin as planned, will need to be restructured or will be completed on schedule, if at all. Our product development costs will increase if we experience delays in clinical testing. Significant clinical trial delays could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before us, which would impair our ability to successfully commercialize our product candidates and may harm our financial condition, results of operations and prospects. The completion of clinical trials for our products, including the APTO-253 Phase 1a/btuspetinib and luxeptinib clinical trial, the Phase 1a/b clinical trial for the luxeptinib study for the treatment of patients having B-cell malignancies, and the IND acceptance of our planned Phase 1a/b study for the development of luxeptinib for the treatment of patients with R/R AMLtrials may be delayed for a number of reasons, including delays related, but not limited, to:
| · | failure by regulatory authorities to grant permission to proceed with a clinical trial; |
| · | a regulatory decision to place or placing the clinical trial on hold; |
| · | patients failing to enroll or remain in our trials at the rate we expect; |
•failure by regulatory authorities to grant permission to proceed with a clinical trial;
•a regulatory decision to place or placing the clinical trial on hold;
•patients failing to enroll or remain in our trials at the rate we expect;
•suspension or termination of clinical trials by regulators for many reasons, including concerns about patient safety or failure of our contract manufacturers to comply with cGMP requirements;
•any changes to our manufacturing process that may be necessary or desired;
| · | suspension or termination of clinical trials by regulators for many reasons, including concerns about patient safety or failure of our contract manufacturers to comply with cGMP requirements; |
| · | any changes to our manufacturing process that may be necessary or desired; |
•delays or failure to obtain GMP-grade clinical supply from contract manufacturers of our products necessary to conduct clinical trials;
•product candidates demonstrating a lack of safety or efficacy during clinical trials;
•patients choosing an alternative treatment for the indications for which we are developing any of our product candidates or participating in competing clinical trials;
•patients failing to complete clinical trials due to dissatisfaction with the treatment, side effects or other reasons;
•reports of clinical testing on similar technologies and products raising safety and/or efficacy concerns;
•competing clinical trials and scheduling conflicts with participating clinicians;
•clinical investigators not performing our clinical trials on their anticipated schedule, dropping out of a trial, or employing methods not consistent with the clinical trial protocol, regulatory requirements or other third parties not performing data collection and analysis in a timely or accurate manner;
•failure of our contract research organizations, or CROs, to satisfy their contractual duties or meet expected deadlines;
•inspections of clinical trial sites by regulatory authorities or IRBs, or ethics committees or boards finding regulatory violations that require us to undertake corrective action, resulting in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study;
•one or more IRBs or ethics committees or boards rejecting, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; or
•failure to reach agreement on acceptable terms with prospective clinical trial sites.
| · | delays or failure to obtain GMP-grade clinical supply from contract manufacturers of our products necessary to conduct clinical trials; |
| · | product candidates demonstrating a lack of safety or efficacy during clinical trials; |
| · | patients choosing an alternative treatment for the indications for which we are developing any of our product candidates or participating in competing clinical trials; |
| · | patients failing to complete clinical trials due to dissatisfaction with the treatment, side effects or other reasons; |
| · | reports of clinical testing on similar technologies and products raising safety and/or efficacy concerns; |
| · | competing clinical trials and scheduling conflicts with participating clinicians; |
| · | clinical investigators not performing our clinical trials on their anticipated schedule, dropping out of a trial, or employing methods not consistent with the clinical trial protocol, regulatory requirements or other third parties not performing data collection and analysis in a timely or accurate manner; |
| · | failure of our contract research organizations, or CROs, to satisfy their contractual duties or meet expected deadlines; |
| · | inspections of clinical trial sites by regulatory authorities or IRBs, or ethics committees or boards finding regulatory violations that require us to undertake corrective action, resulting in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study; |
| · | one or more IRBs or ethics committees or boards rejecting, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; or |
| · | failure to reach agreement on acceptable terms with prospective clinical trial sites. |
Our product development costs will increase if we experience delays in testing or approval or if we need to perform more or larger clinical trials than planned. Additionally, changes in regulatory requirements and policies may occur, and we may need to amend study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to regulatory authorities or IRBs or ethics committees or boards for re-examination, which may impact the cost, timing or successful completion of a trial. Delays or increased product development costs may have a material adverse effect on our business, financial condition and prospects.
We rely on contract manufacturers over whom we have limited control. If we are subject to quality, cost or delivery issues with the preclinical and clinical grade materials supplied by contract manufacturers, our business operations could suffer significant harm.
We rely on CMOscontract manufacturing organizations ("CMOs") to manufacture our product candidates for some preclinical studies and clinical trials. We rely on CMOs for manufacturing, filling, packaging, storing and shipping of drug product in compliance with cGMP regulations applicable to our products. The FDA and other regulatory agencies ensure the quality of drug products by carefully monitoring drug manufacturers’ compliance with cGMP regulations. The cGMP regulations for drugs contain minimum requirements for the methods, facilities and controls used in manufacturing, processing and packing of a drug product.
We contracted with multiple CMOs for the manufacture of APTO-253tuspetinib and CG-806luxeptinib to supply the active ingredient and then drug product for our clinical trials. The synthesis of CG-806luxeptinib is challenging from a scale-up synthetic chemistry perspective. The formulation and manufacture of APTO-253 is a complex process with many variables involved. We pre-qualified CMOs to have the capacity, the systems and the experience to supply CG-806tuspetinib and APTO-253luxeptinib for our clinical trials. We have qualified the manufacturing facilities and the FDA has also performed site audits for our selected CMOs. In spite ofDespite the efforts to prequalify CMOs, delays and errors may occur, and any such manufacturing failures, delays or compliance issues could cause delays in the completion of our clinical trial programs.
There can be no assurances that CMOs will be able to meet our timetable and requirements. We have contracted with alternate suppliers in the event our current CMOs are unable to scale up production, or if our current CMOs otherwise experience any other significant problems in the manufacture of CG-806tuspetinib and APTO-253.luxeptinib. However, it is possible that all third-party manufacturing sources may experience failure or delays and may demand commercially unreasonable terms, which may lead to further delays in the development of our product candidates. Further, contract manufacturers must operate in compliance with cGMP and failure to do so could result in, among other things, the disruption of product supplies. Our dependence upon third parties for the manufacture of our products may adversely affect our profit margins and our ability to develop and deliver products on a timely and competitive basis.
Although, as of the date of this report, we have not experienced any material delays in the manufacturing of luxeptinib and APTO-253 due to COVID-19, the extent to which it will impact the manufacturing of our products will depend on future developments, which are highly uncertain and cannot be predicted. Should our suppliers involved in the manufacture of luxeptinib be required to shut down their facilities due to COVID-19 either due to lack of materials or personnel, our trials would be negatively impacted. We are mitigating this risk by continuing to manufacture drug supply, but there is no guarantee that we will have enough drug to supply the trial if any of our manufacturers have a sustained shut down in their operations. COVID-19 may also affect the timing and delivery of labeled and packaged drug product for APTO-253 since it is an intravenous formulation which, compared to orally administered therapies, involves a more complex process. Factors related to COVID-19 caused a delay in the labeling and packaging of the APTO-253 drug product; however, going forward we do not anticipate this to materially affect the patient accrual for the ongoing Phase 1b trial.
Some components of our products are manufactured by third parties outside of the United States, and our business may be harmed by legal, regulatory, economic, political and public health risks associated with international trade and those markets.
We have third-party manufacturing partners in South Korea, Germany and the United Kingdom; in addition, some materials used by our third-party manufacturers are supplied by companies located in other countries, including China. Our reliance on suppliers and manufacturers in foreign markets creates risks inherent in doing business in foreign jurisdictions, including: (a) the burdens of complying with a variety of foreign laws and regulations, including laws relating to the importation and taxation of goods (b) public health crises, such as pandemics and epidemics, in the countries where our suppliers and manufacturers are located; (c) transportation interruptions or increases in transportation costs; and (d) foreign intellectual property infringement risks. For example, the ongoing COVID-19 pandemic has resulted in the extended shutdown of certain businesses and markets in many regions causing reduced availability for certain pharmaceutical ingredients. The current public health crisis or any further political developments or health concerns in markets in which our products are manufactured or from which we obtain necessary pharmaceutical ingredients could adversely affect the supply of our drug products and, in turn, our business, financial condition, and results of operations.
If we have difficulty enrolling patients in clinical trials, the completion of the trials may be delayed or cancelled.canceled.
As our product candidates advance from preclinical testing to clinical testing, and then through progressively larger and more complex clinical trials, we will need to enroll an increasing number of patients that meet our eligibility criteria. There is significant competition for recruiting cancer patients in clinical trials, and we may be unable to enroll the patients we need to complete clinical trials for cancer indications on a timely basis or at all. Certain factors that affect enrollment of patients in our clinical trials are impacted by external forces that may be beyond our control. Such factors include, but are not limited to, the following:
| · | •size and nature of the patient population; |
| · | eligibility and exclusion criteria for the trial; |
| · | design of the study protocol; |
| · | competition with other companies for clinical sites or patients; |
| · | the perceived risks and benefits of the product candidate under study; |
| · | the patient referral practices of physicians; and |
| · | the number, availability, location and accessibility of clinical trial sites. |
Although, as of the datepatient population;
•eligibility and exclusion criteria for the trial;
•design of this report, we do not foresee material delays to the enrollmentstudy protocol;
•competition with other companies for clinical sites or patients;
•the perceived risks and benefits of patients or timelines for our trials due to COVID-19, the extent to which COVID-19 will impact product candidate under study;
•the projected development goals will depend on future developments, which are highly uncertainpatient referral practices of physicians; and cannot be predicted.
•the number, availability, location and accessibility of clinical trial sites.
If we are unable to successfully develop companion diagnostics for our therapeutic product candidates, or experience significant delays in doing so, we may not achieve marketing approval or realize the full commercial potential of our therapeutic product candidates.
We plan to develop companion diagnostics for our therapeutic product candidates. We expect that, at least in some cases, regulatory authorities may require the development and regulatory approval of a companion diagnostic as a condition to approving our therapeutic product candidates. We have limited experience and capabilities in developing or commercializing diagnostics and plan to rely in large part on third parties to perform these functions. We do not currently have any agreement in place with any third party to develop or commercialize companion diagnostics for any of our therapeutic product candidates.
Companion diagnostics are subject to regulation by the FDA, Health Canada and comparable foreign regulatory authorities as medical devices and may require separate regulatory approval or clearance prior to commercialization. If we, or any third parties that we engage to assist us, are unable to successfully develop companion diagnostics for our therapeutic product candidates, or experience delays in doing so, our business may be substantially harmed.
We rely and will continue to rely on third parties to conduct and monitor many of our preclinical studies and our clinical trials, and their failure to perform as required could cause substantial harm to our business.
We rely and will continue to rely on third parties to conduct a significant portion of our preclinical and clinical development activities. Preclinical activities include in vivo studies providing access to specific disease models, pharmacology and toxicology studies, and assay development. Clinical development activities include trial design, regulatory submissions, clinical patient recruitment, clinical trial monitoring, clinical data management and analysis, safety monitoring and project management, contract manufacturing and quality assurance. If there is any dispute or disruption in our relationship with third parties, or if they are unable to provide quality services in a timely manner and at a feasible cost, our active development programs will face delays. Further, if any of these third parties fails to perform as we expect or if their work fails to meet regulatory requirements, our testing could be delayed, cancelledcanceled or rendered ineffective.
Negative results from clinical trials or studies of others and adverse safety events involving the targets of our products may have an adverse impact on our future commercialization efforts.
From time to time, studies or clinical trials on various aspects of biopharmaceutical products are conducted by academic researchers, competitors or others. The results of these studies or trials, when published, may have a significant effect on the market for the biopharmaceutical product that is the subject of the study. The publication of negative results of studies or clinical trials or adverse safety events related to our product candidates, or the therapeutic areas in which our product candidates compete, could adversely affect our share price and our ability to finance future development of our product candidates, and our business and financial results could be materially and adversely affected.
The design or our execution of clinical trials may not support regulatory approval.
The design or execution of a clinical trial can determine whether its results will support regulatory approval and flaws in the design or execution of a clinical trial may not become apparent until the clinical trial is well advanced. In some instances, there can be significant variability in safety or efficacy results between different trials of the same product candidate due to numerous factors, including changes in trial protocols, differences in size and type of the patient populations, adherence to the dosing regimen and other trial protocols and the rate of dropout among clinical trial participants. We do not know whether any Phase 2, Phase 3 or other clinical trials that we may conduct will demonstrate consistent or adequate efficacy and safety to obtain regulatory approval to market our product candidates.
Further, the FDA, Health Canada and comparable foreign regulatory authorities will have some discretion in the approval process and in determining when or whether regulatory approval will be obtained for any of our product candidates. Our product candidates may not be approved even if they achieve their primary endpoints in future Phase 3 clinical trials or registration trials. The FDA, Health Canada or other regulatory authorities may disagree with our trial design and our interpretation of data from preclinical studies and clinical trials. In addition, any of these regulatory authorities may change requirements for the approval of a product candidate even after reviewing and providing comments or advice on a protocol for a pivotal Phase 3 clinical trial that has the potential to result in approval by the FDA, Health Canada or another regulatory agency. In addition, any of these regulatory authorities may also approve a product candidate for fewer or more limited indications than we request or may grant approval contingent on the performance of costly post-marketing clinical trials. The FDA, Health Canada or other regulatory authorities may not approve the labeling claims that we believe would be necessary or desirable for the successful commercialization of our product candidates.
As a result of intense competition and technological change in the biotechnical and pharmaceutical industries, the marketplace may not accept our products or product candidates, and we may not be able to compete successfully against other companies in our industry and achieve profitability.
Many of our competitors have:
| · | drug products that have already been approved or are in development; |
| · | large, well-funded research and development programs in the biotechnical and pharmaceutical fields; |
| · | substantially greater financial, technical and management resources, stronger intellectual property positions and greater manufacturing, marketing and sales capabilities, areas in which we have limited or no experience; and |
| · | significantly greater experience than we do in undertaking preclinical testing and clinical trials of new or improved pharmaceutical products and obtaining required regulatory approvals. |
•drug products that have already been approved or are in development;
•large, well-funded research and development programs in the biotechnical and pharmaceutical fields;
•substantially greater financial, technical and management resources, stronger intellectual property positions and greater manufacturing, marketing and sales capabilities, areas in which we have limited or no experience; and / or
•significantly greater experience in undertaking preclinical testing and clinical trials of new or improved pharmaceutical products and obtaining required regulatory approvals.
Consequently, our competitors may obtain FDA, Health Canada and other regulatory approvals for product candidates sooner and may be more successful in manufacturing and marketing their products than we or our collaborators are.
Our competitors’ existing and future products, therapies and technological approaches will compete directly with the products we seek to develop. Current and prospective competing products may be more effective than our existing and future products insofar as they may provide greater therapeutic benefits for a specific problem or may offer easier delivery or comparable performance at a lower cost.
For luxeptinibtuspetinib and APTO-253luxeptinib in AML, examples of potential competitors include companies that have developed approved or are currently developing inhibitors that directly target the wild type include AbbVie (IMBRUVICA) and AstraZeneca (CALQUENCE) and Beigene Co., Ltd. (Zanubrutinib).
Others that are developing inhibitors that target the C481S-mutant BTK include Merck (MK-1026), Roche, and Eli Lilly (LY3527727) among others.
For luxeptinib and APTO-253 in AML, examples of potential competitors include companies that have developed, approved or are currently developing non-targetedpursuing different therapies include Jazz (VYXEOS), Pfizer (MYLOTARG), Novartis (RYDAPT), Astellas (XOSPATA), AbbVie (VENCLEXTA), Daiichi Sankyo (quizartinib), Arog (crenolanib), Agios/Servier (TIBSOVO), Rigel (REZLIDHA), Celgene/BMS (IDHIFA), Kronos Bio (lanraplenib), Curis (emavusertib), Syndax (revumenib, SNDX-5613), and Roche (VENCLEXTA)Kura (KO-539), among others. Others
For luxeptinib in B cell malignancies, examples of companies that have developed or are developing highly targeted therapies such as FLT-3pursuing different approaches to BTK inhibition, both for the wild type and C481S-mutant forms, include Novartis (RYDAPT)AbbVie (IMBRUVICA), Astellas (XOSPATA)AstraZeneca (CALQUENCE), Daiichi Sankyo (QUIZARTINIB)Beigene Co., Arog (CRENOLANIB)Ltd. (Zanubrutinib), Merck (nemtabrutinib), and IDH1 include Agios (TIBSOVO) and Celgene/BMS (IDHIFA)Eli Lilly (pirtobrutinib), among others.
Any product candidate that we develop and that obtains regulatory approval must then compete for market acceptance and market share. Our products may not gain market acceptance among physicians, patients, healthcare payers, insurers, the medical community and other stakeholders. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, including:
•efficacy and potential advantages compared to alternative treatments;
•the ability to offer our product candidates for sale at competitive prices;
•convenience and ease of administration compared to alternative treatments;
•the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
•the strength of marketing and distribution support;
•sufficient third-party coverage or reimbursement; and
•the prevalence and severity of any side effects.
| · | efficacy and potential advantages compared to alternative treatments; |
| · | the ability to offer our product candidates for sale at competitive prices; |
| · | convenience and ease of administration compared to alternative treatments; |
| · | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
| · | the strength of marketing and distribution support; |
| · | sufficient third-party coverage or reimbursement; and |
| · | the prevalence and severity of any side effects. |
Further, any products we develop may become obsolete or face generic entry before we recover any expenses we incurred in connection with the development of these products. As a result, we may never achieve profitability.
Risks Related to our Intellectual Property
We may be unable to obtain patents to protect our technologies from other companies with competitive products, and patents of other companies could prevent us from manufacturing, developing or marketing our products.
Patent protection
The patent positions of pharmaceutical and biotechnology companies are uncertain and involve complex legal and factual questions. The USPTOUnited States Patent and Trademark Office (“USPTO”) and many other patent offices in the world have not established a consistent policy regarding the breadth of claims that they will allow in biotechnology patents.
Our pending patent applications may not result in issued patents and our issued patents may not be held valid and enforceable if challenged. Competitors may be able to circumvent any such issued patents by adoption of a competitive, though non-infringing product or process. Interpretation and evaluation of pharmaceutical or biotechnology patent claims present complex and often novel legal and factual questions. Our business could be adversely affected by increased competition in the event that any patent granted to it is held to be invalid or unenforceable or is inadequate in scope to protect our operations.
Allowable patentable subject matter and the scope of patent protection obtainable may differ between jurisdictions. If a patent office allows broad claims, the number and cost of patent interference proceedings in the United States, or analogous proceedings in other jurisdictions and the risk of infringement litigation may increase. If it allows narrow claims, the risk of infringement may decrease, but the value of our rights under our patents, licenses and patent applications may also decrease.
The scope of the claims in a patent application can be significantly modified during prosecution before the patent is issued. Consequently, we cannot know whether our pending applications will result in the issuance of patents or, if any patents are issued, whether they will provide us with significant proprietary protection or will be circumvented, invalidated or found to be unenforceable.
Publication of discoveries in scientific or patent literature often lags behind actual discoveries. Patent applications filed in the United States generally will be published 18 months after the filing date unless the applicant certifies that the invention will not be the subject of a foreign patent application. In many other jurisdictions, such as Canada, patent applications are published 18 months from the priority date. We may not be aware of such literature. Accordingly, we cannot be certain that the named inventors of our products and processes were the first to invent that product or process or that we were the first to pursue patent coverage for our inventions.
In addition, United States patent laws may change which could prevent or limit us from filing patent applications or patent claims in the United States to protect our products and technologies or limit the exclusivity periods that are available to patent holders for United States patents. For example, the Leahy-Smith America Invents Act, (the “Leahy-Smith Act”) was signed into law in 2011 and includes a number of significant changes to United States patent law. These include changes to transition from a “first-to-invent” system to a “first-to-file” system and to the way issued patents are challenged. These changes may favor larger and more established companies that have more resources to devote to patent application filing and prosecution. It is not clear what, if any, impact the Leahy-Smith Act will ultimately have on the cost of prosecuting our patent applications in the United States, our ability to obtain patents in the United States based on our discoveries and our ability to enforce or defend our United States issued patents.
Until such time, if ever, that further patents are issued to us, we will rely upon the law of trade secrets to the extent possible given the publication requirements under international patent treaty laws and/or requirements under foreign patent laws to protect our technology and our products incorporating the technology. In this regard, we have adopted certain confidentiality procedures. These include: limiting access to confidential information to certain key personnel; requiring all directors, officers, employees and consultants and others who may have access to our intellectual property to enter into confidentiality agreements which prohibit the use of or disclosure of confidential information to third parties; and implementing physical security measures designed to restrict access to such confidential information and products. Our ability to maintain the confidentiality of our technology is crucial to our ultimate possible commercial success. The procedures adopted by us to protect the confidentiality of our technology may not be effective, third parties may gain access to our trade secrets or our trade secrets or those of our collaborators may be independently discovered by others. Our collaborators, employees and consultants and other parties may not comply with the terms of their agreements with us, and we might be unable to adequately enforce our rights or obtain adequate compensation for the damages caused by unauthorized disclosure or use of our trade secrets or know how. Further, by seeking patent protection in various countries, it is inevitable that a substantial portion of our technology will become available to our competitors, through publication of such patent applications.
Enforcement of intellectual property rights
Protection of the rights revealed in published patent applications can be complex, costly and uncertain. Our commercial success depends in part on our ability to maintain and enforce our proprietary rights. If third parties engage in activities that infringe our proprietary rights, our management’s focus will be diverted and we may incur significant costs in asserting our rights. We may not be successful in asserting our proprietary rights, which could result in our patents being held invalid or a court holding that the third party is not infringing, either of which would harm our competitive position.
Others may design around our patented technology. We may have to participate in interference proceedings declared by the USPTO, European opposition proceedings, or other analogous proceedings in other parts of the world to determine priority of invention and the validity of patent rights granted or applied for, which could result in substantial cost and delay, even if the eventual outcome is favorable to us. Our pending patent applications, even if issued, may not be held valid or enforceable.
Our products and product candidates may infringe the intellectual property rights of others, or others may infringe on our intellectual property rights which could increase our costs.
Our success also depends on avoiding infringement of the proprietary technologies of others. In particular, there may be certain issued patents and patent applications claiming subject matter which we or our collaborators may be required to license in order to research, develop or commercialize APTO-253tuspetinib or luxeptinib. In addition, third parties may assert infringement or other intellectual property claims against us. An adverse outcome in these proceedings could subject us to significant liabilities to third-parties,third parties, require disputed rights to be licensed from third-parties or require us to cease or modify our use of the technology. If we are required to license third-party technology, a license under such patents and patent applications may not be available on acceptable terms or at all. Further, we may incur substantial costs defending ourselves in lawsuits against charges of patent infringement or other unlawful use of another’s proprietary technology. We may also need to bring claims against others who we believe are infringing our rights in order to become or remain competitive and successful. Any such claims can be time consuming and expensive to pursue.
We may incur substantial cost in defending our intellectual property.
While we believe that our products and technology do not infringe proprietary rights of others, third parties may assert infringement claims in the future and such claims could be successful. Even if challenges are unsuccessful, we could incur substantial costs in defending ourselves against patent infringement claims brought by others or in prosecuting suits against others. In addition, others may obtain patents that we would need to license, which may not be available to us on reasonable terms. Whether we are able to obtain a necessary license would depend on the terms offered, the degree of risk of infringement and the need for the patent.
We have licensed important portions of our intellectual property from Hanmi and CG, and are subject to significant obligations under thatthose license agreement.agreements.
The Rightsrights we hold under our license agreementagreements with Hanmi and CG are critical to our business.
Our tuspetinib program is built around patents exclusively in-licensed from Hanmi, which permit us to research, develop and commercialize tuspetinib worldwide. Under the Tuspetinib Licensing Agreement, we are subject to significant obligations, including diligence obligations with respect to development and commercialization activities, payment obligations upon achievement of certain milestones and royalties on product sales, as well as other material obligations. Hanmi is eligible for payments upon the achievement of developmental, regulatory and commercial-based milestones, as well as tiered royalties on product sales.
Our luxeptinib program is built around patents exclusively in-licensed from CG, which permit us to research, develop and commercialize CG-806luxeptinib worldwide except for the Republic of Korea. Under our agreement with CG, we are subject to significant obligations, including diligence obligations with respect to development and commercialization activities, payment obligations upon achievement of certain milestones and royalties on product sales, as well as other material obligations. CG is eligible for payments upon the achievement of developmental, regulatory and commercial-based milestones, as well as low single-digit royalties on product sales in all territories outside of the Republic of Korea.sales.
If there is any conflict, dispute, disagreement or issue of non-performance between us and Hanmi or CG regarding our rights or obligations under the respective license agreements, including any conflict, dispute or disagreement arising from our failure to satisfy diligence or payment obligations under such agreements, Hanmi or CG may have a right to terminate the respective license. The loss of this license agreement could materially and adversely affect our ability to use intellectual property that could be critical to our drug discovery and development efforts, as well as our ability to enter into future collaboration, licensing and/or marketing agreements for one or more affected drug candidates or development programs.
Our business depends, in part, on our ability to use technology that we have licensed or will in the future license from third parties, including CG, and, if these licenses were terminated or if we were unable to license additional technology we may need in the future, our business will be adversely affected.
We currently hold licenses for certain technologies that are or may be critical to our current and subsequent product candidates. These include our exclusive license to research, develop and commercialize luxeptinib worldwide except for the Republic of Korea. TheKorea, and our exclusive license from CG isto develop and commercialize tuspetinib worldwide. Both licenses are subject to termination in the event of a breach by us of the license, if we fail to cure the breach following notice and the passage of a cure period. We may need to acquire additional licenses in the future to technologies developed by others. Furthermore, future license agreements may require us to make substantial milestone payments. We may also be obligated to make royalty payments on the sales, if any, of products resulting from the license. The termination of a license or the inability to license future technologies on acceptable terms may adversely affect our ability to develop or sell our products.
Legal and Regulatory Risk
Our ability to develop, produce and market our products is subject to extensive government regulation.
Government regulation is a significant factor in the development, production and marketing of our products. Research and development, testing, manufacture, marketing and sales of pharmaceutical products or related products are subject to extensive regulatory oversight, often in multiple jurisdictions, which may cause significant additional costs and/or delays in bringing products to market, and in turn, may cause significant losses to investors. The regulations applicable to our product candidates in a given jurisdiction may change. Even if granted, regulatory approvals may include significant limitations on the uses for which products can be marketed or may be conditioned on the conduct of post-marketing surveillance studies. Failure to comply with applicable regulatory requirements can, among other things, result in delay in approving or refusal to approve a product candidate, interruptions of clinical trials or manufacturing, suspension or withdrawal of regulatory approval, warning letters, the imposition of civil penalties or other monetary payments, product recall or seizure, operating restrictions, injunctions or criminal prosecution. In addition, regulatory agencies many not approve the labeling claims that are necessary or desirable for the successful commercialization of our product candidates.
Requirements for regulatory approval vary widely from country to country. Whether or not approved in Canada or the United States, regulatoryRegulatory authorities in other countries must approve a product prior to the commencement of marketing the product in those countries.product. The time required to obtain any such approval may be longer or shorter than in Canada or the United States. Approved drugs, as well as their manufacturers, are subject to continuing and ongoing review, and discovery of problems with these products or the failure to adhere to manufacturing or quality control requirements may result in regulatory restrictions being imposed.
Current and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and may adversely affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could, among other things, prevent or delay marketing approval of our product candidates, restrict or regulate post approval activities and affect our ability to profitably sell any products for which we obtain marketing approval.
For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care Education Reconciliation Act, or collectively the Affordable Care Act, was enacted to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for health care and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Additionally, the Drug Supply Chain Security Act, enacted in 2013, imposed new obligations on manufacturers of pharmaceutical products related to product tracking and tracing.
Members of CongressSince its enactment, there have been judicial, executive and the Trump Administration have considered legislationCongressional challenges to fundamentally change or repeal the Affordable Care Act. While Congress has not passed repeal legislation to date, the Tax Cuts and Jobs Act (“TCJA”) includes a provision repealing the individual insurance coverage mandate included in the Affordable Care Act, effective January 1, 2019. Further, President Trump signed an Executive Order directing federal agencies with authorities and responsibilities under the Affordable Care Act to waive, defer, grant exemptions from, or delay the implementation of any provisioncertain aspects of the Affordable Care Act, that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. On October 13, 2017, the President signed an Executive Order terminating the cost-sharing subsidies that reimburse insurers under the Affordable Care Act. Several state Attorneys General filed suitand we expect there will be additional challenges and amendments to stop the administration from terminating the subsidies, but their request for a restraining order was denied by a federal judge in California on October 25, 2017. In addition, the Centers for Medicare and Medicaid Services has recently proposed regulations that would give states greater flexibility in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing the essential health benefits required under the Affordable Care Act for plans sold through such marketplaces. Congress may consider other legislationin the future. On June 17, 2021, the United States Supreme Court dismissed the most recent judicial challenge to replace elementsthe Affordable Care Act without specifically ruling on the constitutionality of the Affordable Care Act. The implicationsPrior to the Supreme Court’s decision, President Biden issued an executive order initiating a special enrollment period from February 15, 2021 through August 15, 2021 for purposes of obtaining health insurance coverage through the Affordable Care Act itsmarketplace. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the Affordable Care Act. It is possible repeal, any legislation that may be proposed to replace the Affordable Care Act will be subject to judicial or Congressional challenges in the political uncertainty surroundingfuture. It is unclear how any repeal or replacement legislation forsuch challenges and the healthcare reform measures of the Biden administration will impact the Affordable Care Act and our business and financial condition, if any, are not yet clear.business.
We expect ongoing initiatives in the United States and internationally to increase pressure on drug pricing. Regulations that mandate price controls and limitations on patient access to products or establish prices paid by government entities or programs may impact product candidates that we may successfully develop. Pharmaceutical product pricing is subject to enhanced government and public scrutiny and calls for reform. Some U.S. states have implemented, and other U.S. states are considering, pharmaceutical price controls or patient access constraints under the Medicaid program, and some U.S. states are considering price-control regimes that would apply to broader segments of their populations that are not Medicaid eligible. Efforts by government officials or legislators to implement measures to regulate prices or payments for pharmaceutical products, including legislation on drug importation, could have an adverse effect on anticipated revenues from product candidates that we may successfully develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop drug candidates.
Legislative and regulatory proposals have also been made to expand post approval requirements and restrict sales and promotional activities for pharmaceutical products in the US. Any healthcare reforms enacted in the future may, like the Affordable Care Act, be phased in over a number of years but, if enacted, could reduce our revenue, increase our costs, or require us to revise the ways in which we conduct business or put us at risk for loss of business. We areIt is not sureclear whether additional legislative changes will be enacted, or whether the current regulations, guidance or interpretations will be changed, or what the impact of such changes on our business, if any, may be.
In Canada, the Patented Medicine Prices Review Board (“PMPRB”(the “PMPRB”) has jurisdiction to control prices of patented medicines that are considered excessive. Recent changes to the regulations governing the PMPRB are intended to lower the prices of patented medicines even further. The PMPRB’s jurisdiction could extend to any of our drug products that are approved in Canada and protected under Canadian patents, with an adverse effect on the prices that we would otherwise obtain for these drugs in the relevant market.
Coverage and adequate reimbursement may not be available for our product candidates, which could make it difficult for us to sell our products profitably.
Market acceptance and sales of any drug candidates that we develop will depend in part on the extent to which reimbursement for these products and related treatments will be available from third party payors, including government health administration authorities and private health insurers. Third party payors decide which drugs they will pay for and establish reimbursement levels. Third party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. However, decisions regarding the extent of coverage and amount of reimbursement to be provided for each of our drug candidates will be made on a plan by planplan-by-plan basis. One payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage, and adequate reimbursement, for the product. Additionally, a third partythird-party payor’s decision to provide coverage for a drug does not imply that an adequate reimbursement rate will be approved. Each plan determines whether or not it will provide coverage for a drug, what amount it will pay the manufacturer for the drug, and on what tier of its formulary the drug will be placed. The position of a drug on a formulary generally determines the copayment that a patient will need to make to obtain the drug and can strongly influence the adoption of a drug by patients and physicians. Patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products.
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Third party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. We cannot be sure that coverage and reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be. Inadequate coverage and reimbursement may impact the demand for, or the price of, any product for which we obtain marketing approval. If coverage and adequate reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize any drug candidates that we develop.
Additionally, there have been a number of legislative and regulatory proposals to change the healthcare system in the United States and in some foreign jurisdictions, including Canada, that could affect our ability to sell any future drugs profitably. These legislative and regulatory changes may negatively impact the reimbursement for any future drugs, following approval.
We are subject to U.S. and Canadian healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, fines, disgorgement, exclusion from participation in government healthcare programs, curtailment or restricting of our operations and diminished profits and future earnings.
Healthcare providers, physicians and others will play a primary role in the recommendation and prescription of any products for which we obtain marketing approval. Our future arrangements with healthcare providers, patients and third partythird-party payors could expose us to broadly applicable U.S. and Canadian laws and regulations relating to fraud abuse and healthcare more generally that may constrain the business or financial arrangements and collaborative partners through which we market, sell and distribute any products for which we obtain marketing approval.
Efforts to ensure that our collaborations with third parties, and our business generally, will comply with applicable U.S. and Canadian healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental laws and regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion of products from government funded healthcare programs, contractual damages, reputational harm, disgorgement, curtailment or restricting of our operations, any of which could substantially disrupt our operations and diminish our profits and future earnings. If any of the physicians or other providers or entities with whom we expect to do business isare found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. The risk of ourthe Company being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations.
If product liability, clinical trial liability or environmental liability claims are brought against us or we are unable to obtain or maintain product liability, clinical trial or environmental liability insurance, we may incur substantial liabilities that could reduce our financial resources.
The clinical testing and commercial use of pharmaceutical products involves significant exposure to product liability, clinical trial liability, environmental liability and other risks that are inherent in the testing, manufacturing and marketing of our products. These liabilities, if realized, could have a material adverse effect on our business, results of operations and financial condition.
We have obtained limited product liability insurance coverage for our clinical trials on humans; however, our insurance coverage may be insufficient to protect us against all product liability damages. Regardless of merit or eventual outcome, liability claims may result in decreased demand for a future product, injury to reputation, withdrawal of clinical trial volunteers, loss of revenue, costs of litigation, distraction of management and substantial monetary awards to plaintiffs. Additionally, if we are required to pay a product liability claim, we may not have sufficient financial resources to complete development or commercialization of any of our product candidates and our business and results of operations will be adversely affected. In general, insurance will not protect us against some of our own actions, such as negligence.
As our development activities progress towards the commercialization of product candidates, our liability coverage may not be adequate, and we may not be able to obtain adequate product liability insurance coverage at a reasonable cost, if at all. Even if we obtain product liability insurance, our financial position may be materially adversely affected by a product liability claim. A product liability claim could also significantly harm our reputation and delay market acceptance of our product candidates. Additionally, product recalls may be issued at the direction of the FDA, other government agencies or other companies having regulatory control for pharmaceutical sales. If a product recall occurs in the future, such a recall could adversely affect our business, financial condition or reputation.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and radioactive and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
We may be unable to obtain partnerships for our product candidates, which could curtail future development and negatively affect our share price. In addition, our partners might not satisfy their contractual responsibilities or devote sufficient resources to our partnership.
Our strategy for the research, development and commercialization of our products requires entering into various arrangements with corporate collaborators, licensors, licensees and others, and our commercial success is dependent upon these outside parties performing their respective contractual responsibilities. The amount and timing of resources that such third parties will devote to these activities may not be within our control. These third parties may not perform their obligations as expected and our collaborators may not devote adequate resources to our programs. In addition, we could become involved in disputes with our collaborators, which could result in a delay or termination of the related development programs or result in litigation. We intend to seek additional collaborative arrangements to develop and commercialize some of our products. We may not be able to negotiate collaborative arrangements on favorable terms, or at all, in the future, and our current or future collaborative arrangements may not be successful.
If we cannot negotiate collaboration, license or partnering agreements, we may never achieve profitability and we may not be able to continue to develop our product candidates. Continuing Phase 1a/b,1, and commencing Phase 2 and Phase 3 clinical trials for luxeptinibtuspetinib and APTO-253luxeptinib would require significant amounts of funding and such funding may not be available to us.
Risks Related to Our Common Shares
Our share price has been and is likely to continue to be volatile and an investment in our Common Shares could suffer a decline in value.
You should consider an investment in our Common Shares as risky and invest only if you can withstand a significant loss and wide fluctuations in the market value of your investment. The market price of our Common Shares has been highly volatile and is likely to continue to be volatile. This leads to a heightened risk of securities litigation pertaining to such volatility. Factors affecting our Common Share price include but are not limited to:
| · | the progress of our pre-clinical and clinical trials; |
| · | our ability to obtain partners and collaborators to assist with the future development of our products; |
| · | general market conditions; |
| · | announcements of technological innovations or new product candidates by us, our collaborators or our competitors; |
•the progress of our pre-clinical and clinical trials;
•our ability to obtain partners and collaborators to assist with the future development of our products;
•general market conditions;
•announcements of technological innovations or new product candidates by us, our collaborators or our competitors;
•published reports by securities analysts;
•developments in patent or other intellectual property rights;
•the cash and investments held by us and our ability to secure future financing;
•our ability to raise additional capital;
•public concern as to the safety and efficacy of drugs that we and our competitors develop;
•shareholder interest in our Common Shares;
•low liquidity in the daily trading volume of our Common Shares; and
•our ability to continue as a going concern.
| · | published reports by securities analysts; |
| · | developments in patent or other intellectual property rights; |
| · | the cash and investments held by us and our ability to secure future financing; |
| · | our ability to raise additional capital; |
| · | public concern as to the safety and efficacy of drugs that we and our competitors develop; |
| · | shareholder interest in our Common Shares; |
| · | low liquidity in the daily trading volume of our Common Shares; and |
| · | our ability to continue as a going concern. |
Future sales of our Common Shares by us or by our existing shareholders could cause our share price to fall.
The issuance of Common Shares by usthe Company could result in significant dilution in the equity interest of existing shareholders and adversely affect the market price of our Common Shares. Sales by existing shareholders of a large number of our Common Shares in the public market and the issuance of Common Shares in connection with strategic alliances, or the perception that such additional sales could occur, could cause the market price of our Common Shares to decline and have an undesirable impact on our ability to raise capital.
We are susceptible to stress in the global economy and therefore, our business may be affected by the current and future global financial conditions.
If the increased level of volatility and market turmoil that have marked recent years continue, our operations, business, financial condition and the trading price of our Common Shares could be materially adversely affected. Furthermore, general economic conditions may have a great impact on us, including our ability to raise capital, our commercialization opportunities and our ability to establish and maintain arrangements with others for research, manufacturing, product development and sales.
An active trading marketFailure to meet the TSX's and the Nasdaq’s continued listing requirements could result in the delisting of our Common Shares, may not be sustained.
Ournegatively impact the price of our Common Shares are listed for trading onand negatively impact our ability to raise additional capital.
If we fail to satisfy the continued listing requirements of the Nasdaq Capital Market, andsuch as the Toronto Stock Exchange. However, an active trading market incorporate governance requirements or the minimum closing bid price requirement, the exchange may take steps to delist our Common Shares. A delisting would likely have a negative effect on the price of our Common Shares and would impair your ability to sell or purchase our Common Shares when you wish to do so. In the event of a delisting notification, we anticipate that we would take actions to restore our compliance with applicable exchange requirements, such as stabilize our market price, improve the liquidity of our Common Shares, prevent our Common Shares from dropping below such exchange’s minimum bid price requirement, or prevent future non-compliance with such exchange’s listing requirements.
On February 29, 2024, the Company received a deficiency letter (the “2024 Deficiency Letter”) from the Nasdaq Listing Qualifications Department of The Nasdaq Stock Market LLC (“Nasdaq”) notifying the Company that the Company’s January 2024 private placement (the "Private Placement") of securities to Hanmi violated rule 5635(d) because the Company did not obtain shareholder approval prior to such issuance. Nasdaq stated that the Private Placement involved the issuance of greater than 20% of the issued and outstanding Common Shares of the Company at a discount to the Nasdaq official closing price on January 25, 2024, the date of the subscription agreement between the Company and Hanmi. The 2024 Deficiency Letter has no immediate effect on the stock exchanges may notlisting of the Company’s Common Shares. In accordance with the Nasdaq Listing Rules, the Company has been given forty-five (45) calendar days, or until April 14, 2024, to submit a plan to regain compliance. If Nasdaq accepts the Company’s plan to regain compliance, Nasdaq can grant an extension of up to 180 calendar days from the date of the Deficiency Letter to evidence compliance. Although the Company believes that the Private Placement was completed in accordance with the Nasdaq Listing Rules, the Company respects Nasdaq’s query and intends to work with Nasdaq to resolve Nasdaq’s concerns and will consider available options to regain compliance. However, there can be sustained and we may notno assurance that the Company will be able to maintainregain compliance with the applicable Nasdaq Listing Rules.
Compliance with changing regulation of corporate governance and public disclosure may result in additional expenses.
Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Dodd-Frank Wall Street Reform and Consumer Protection Act, new SEC regulations and Nasdaq rules, are creating uncertainty for companies such as ours. These laws, regulations and standards are subject to varying interpretations in some cases due to their lack of specificity, and as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies, which could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We are committed to maintaining high standards of corporate governance and public disclosure. As a result, our listings.efforts to comply with evolving laws, regulations and standards have resulted in, and are likely to continue to result in, increased general and administrative expenses and management time related to compliance
activities. If we fail to comply with these laws, regulations and standards, our reputation may be harmed and we might be subject to sanctions or investigation by regulatory authorities, such as the SEC. Any such action could adversely affect our financial results and the market price of our common stock.
Certain Canadian laws could delay or deter a change of control.
Limitations on the ability to acquire and hold our Common Shares may be imposed by the Competition Act in Canada. This legislation permits the Commissioner of Competition of Canada to review any acquisition of a significant interest in us.the Company. This legislation grants the Commissioner jurisdiction to challenge such an acquisition before the Canadian Competition Tribunal if the Commissioner believes that it would, or would be likely to, result in a substantial lessening or prevention of competition in any market in Canada. The Investment Canada Act subjects an acquisition of control of a company by a non-Canadian to government review if the value of our assets, as calculated pursuant to the legislation, exceeds a threshold amount. A reviewable acquisition may not proceed unless the relevant minister is satisfied that the investment is likely to result in a net benefit to Canada. Any of the foregoing could prevent or delay a change of control and may deprive or limit strategic opportunities for our shareholders to sell their shares.
The exercise of all or any number of outstanding stock options, the award of any additional options, restricted stock units or other stock-based awards or any issuance of shares to raise funds or acquire a business may dilute your Common Shares.
We have in the past and may in the future grant to some or all of our directors, officers and employees options to purchase our Common Shares and other stock-based awards as non-cash incentives to those persons. The issuance of any equity securities could, and the issuance of any additional shares will, cause our existing shareholders to experience dilution of their ownership interests.
Any additional issuance of shares or a decision to acquire other businesses through the sale of equity securities may dilute our investors’ interests, and investors may suffer dilution in their net book value per share depending on the price at which such securities are sold. Such issuance may cause a reduction inreduce the proportionate ownership and voting power of all other shareholders. The dilution may result in a decline in the price of our Common Shares or a change in control.
We do not expect to pay dividends for the foreseeable future.
We have not paid any cash dividends to date and we do not intend to declare dividends for the foreseeable future, as we anticipate that we will reinvest future earnings, if any, in the development and growth of our business. Therefore, investors will not receive any funds unless they sell their Common Shares, and shareholders may be unable to sell their shares on favorable terms or at all. We cannot assure you of a positive return on investment or that you will not lose the entire amount of your investment in our Common Shares. Prospective investors seeking or needing dividend income or liquidity should not purchase our Common Shares.
General Risks
It may be difficult for non-Canadian investors to obtain and enforce judgments against us because of our Canadian incorporation and presence.
We are a corporation existing under the laws of Canada. Some of our directors and officers, and manysome of the experts named or unnamed in this Annual Report on Form 10-K, are residents of Canada, and all or a substantial portion of their assets, and a substantial portion of our assets, are located outside the United States. Consequently, although we have appointed an agent for service of process in the United States, it may be difficult for holders of our shares who reside in the United States to effect service within the United States upon our directors and officers and experts who are not residents of the United States. It may also be difficult for holders of our shares who reside in the United States to realize in the United States upon judgments of courts of the United States predicated upon our civil liability and the civil liability of our directors, officers and experts under the United States federal securities laws. Investors should not assume that Canadian courts (i) would enforce judgments of United States courts obtained in actions against us or our directors, officers or experts predicated upon the civil liability provisions of the United States federal securities laws or the securities or “blue sky” laws of any state within the United States or (ii) would enforce, in original actions, liabilities against us or our directors, officers or experts predicated upon the United States federal securities laws or any such state securities or “blue sky” laws. In addition, we have been advised by our Canadian counsel that in normal circumstances, only civil judgments and not other rights arising from United States securities legislation are enforceable in Canada and that the protections afforded by Canadian securities laws may not be available to investors in the United States.
We are likely a “passive foreign investment company” which may have adverse United States federal income tax consequences for United States shareholders.
United States investors in our Common Shares should be aware that we believe we are classified as a passive foreign investment company (“PFIC”) during the tax year ended December 31, 2020,2022, and based on the nature of our business, the projected composition of our gross income and the projected composition and estimated fair market value of our assets, we expect to be a PFIC for the year endingended December 31, 2021,2023, and may be a PFIC in subsequent tax years. If the Company is a PFIC for any year during a United States shareholder’s holding period, then such United States shareholder generally will be required to treat any gain realized upon a disposition of Common Shares, or any so-called “excess distribution” received on its Common Shares, as ordinary income, and to pay an interest charge on a portion of such gain or distributions, unless the shareholder makes a timely and effective “qualified electing fund” election (“QEF election”) or a “mark-to-market” election with respect to the Common Shares. A United States shareholder who makes a QEF election generally must report on a current basis its share of the Company’s net capital gain and ordinary earnings for any year in which the Company is a PFIC, whether or not the Company distributes any amounts to its shareholders. However, United States shareholders should be aware that we do not intend to satisfy record keeping requirements that apply to a qualified electing fund, and we do not intend to supply United States shareholders with information that such United States shareholders require to report under the QEF election rules, in the event that we are a PFIC and a United States shareholder wishes to make a QEF election. Thus, United States shareholders should assume that they will not be able to make a QEF election with respect to their Common Shares. A United States shareholder who makes the mark-to-market election generally must include as ordinary income each year the excess of the fair market value of the Common Shares over the taxpayer’s basis therein. Each United States shareholder should consult its own tax advisor regarding the United States federal, United States local, and foreign tax consequences of the PFIC rules and the acquisition, ownership, and disposition of our Common Shares.
Any failure to maintain an effective system of internal controls may result in material misstatements of our consolidated financial statements or cause us to fail to meet our reporting obligations or fail to prevent fraud; and in that case, our shareholders could lose confidence in our financial reporting, which would harm our business and could negatively impact the price of our Common Shares.
Section 404(a) of the Sarbanes-Oxley Act of 2002 requires that our management assess and report annually on the effectiveness of our internal control over financial reporting and identify any material weaknesses in our internal control over financial reporting.
Effective internal controls are necessary for us to provide reliable financial reports and prevent fraud. If we fail to maintain an effective system of internal controls, we might not be able to report our financial results accurately or prevent fraud; and in that case, our shareholders could lose confidence in our financial reporting, which would harm our business and could negatively impact the price of our Common Shares. While we believe that we have sufficient personnel and review procedures to allow us to maintain an effective system of internal controls, we cannot assure you that we will not experience potential material weaknesses in our internal control. Even if we conclude that our internal control over financial reporting provides reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with US GAAP, because of its inherent limitations, internal control over financial reporting may not prevent or detect fraud or misstatements. Failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm our results of operations or cause us to fail to meet our future reporting obligations.
If we fail to timely achieve and maintain the adequacy of our internal control over financial reporting, we may not be able to produce reliable financial reports or help prevent fraud. Our failure to achieve and maintain effective internal control over financial reporting could prevent us from complying with our reporting obligations on a timely basis, which could result in the loss of investor confidence in the reliability of our consolidated financial statements, harm our business and negatively impact the trading price of our Common Shares.
Prior to December 31, 2018, we were a foreign private issuer and were therefore not subject to certain United States securities law disclosure requirements that apply to a domestic United States issuer, which may limit the historical information publicly available to our shareholders.
As a foreign private issuer prior to December 31, 2018, we were exempt from certain rules under the Exchange Act that impose disclosure requirements as well as procedural requirements for proxy solicitations under Section 14 of the Exchange Act. In addition, our officers, directors and principal shareholders were exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act. Moreover, we were not required to file periodic reports and financial statements with the SEC as frequently or as promptly as a company that files as a domestic issuer whose securities are registered under the Exchange Act, nor were we generally required to comply with the SEC’s Regulation Fair Disclosure, which restricts the selective disclosure of material non-public information. For as long as we were a “foreign private issuer” or an eligible Canadian issuer under the Multijurisdictional Disclosure System, we filed our annual financial statements on Form 20-F, or on Form 40-F, respectively, and furnished our quarterly updates on Form 6-K to the SEC. However, the information we filed or furnished was not the same as the information required in annual and quarterly reports on Form 10-K or Form 10-Q for United States domestic issuers. Accordingly, there may be less historical information publicly available concerning us than there is for a company that has filed as a domestic issuer for longer.
Data security incidents and privacy breaches could result in important remediation costs, increased cyber security costs, litigation and reputational harm.
Cyber security incidents can result from deliberate attacks or unintentional events. Cyber-attacks and security breaches could include unauthorized attempts to access, disable, improperly modify or degrade the Company’s information, systems and networks, the introduction of computer viruses and other malicious codes and fraudulent “phishing” emails that seek to misappropriate data and information or install malware onto users’ computers. Cyber-attacks in particular vary in technique and sources, are persistent, frequently change and are increasingly more targeted and difficult to detect and prevent against. Our network security and data recovery measures and those of third parties with which we contract, may not be adequate to protect against cyber-attacks.
Disruptions due to cyber security incidents could adversely affect our business. In particular, a cyber security incident could result in the loss or corruption of data from our research and development activities, including clinical trials, which may cause significant delays to some or all of our clinical programs. Also, our trade secrets, including unpatented know how, technology and other proprietary information could be disclosed to competitors further to a breach, which would harm our business and competitive position. We expect that risks and exposures related to cyber security attacks will remain high for the foreseeable future due to the rapidly evolving nature and sophistication of these threats. While we have invested in the protection of data and information technology, there can be no assurance that our efforts to implement adequate security measures would be sufficient to protect us against cyber-attacks.
We must successfully upgrade and maintain our information technology systems.
We rely on various information technology systems to manage our operations. There are inherent costs and risks associated with maintaining, modifying and/or changing these systems and implementing new systems, including potential disruption of our internal control structure, substantial capital expenditures, additional administration and operating expenses, retention of sufficiently skilled personnel to implement and operate its systems, demands on management time and other risks and costs of delays or difficulties in transitioning to new systems or of integrating new systems into our current systems. In addition, our information technology system implementations may not result in productivity improvements at a level that outweighs the costs of implementation, or at all. The implementation of new information technology systems may also cause disruptions in our business operations and have an adverse effect on our business, prospects, financial condition and operating results.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 1C. CYBERSECURITY
None.Cybersecurity Risk Management and Strategy
We have developed and maintain a cybersecurity program designed to assess, identify, and manage risks from cybersecurity threats. As part of this program, we conduct periodic assessments of our IT Systems to evaluate the effectiveness of applicable security controls. These assessments follow industry-standard frameworks and include a review of our information security controls to assess cybersecurity capabilities and maturity. The results of these assessments are reported to the Audit Committee of the Board of Directors..
In general, we seek to address cybersecurity risks through a cross-functional approach that is focused on preserving the confidentiality, integrity, and availability of the information that we collect and store by identifying, preventing, and mitigating cybersecurity threats and effectively responding to cybersecurity incidents when they occur.
We have established a cybersecurity policy that outlines the governance processes for identifying and managing material risks to privacy and cybersecurity. In addition, our cybersecurity policy describes our capabilities and processes for the detection, response, analysis, mitigation, recovery, and reporting of cybersecurity incidents. We also manage and maintain business continuity and disaster recovery capabilities to help ensure the availability of business-critical technology resources.
Governance Related to Cybersecurity Risks
Management is responsible for the day-to-day management of risks we face, while our board of directors, as a whole and through committees, has responsibility for the oversight of risk management. Our Audit Committee oversees the management of risks from cybersecurity threats. In addition, the full board reviews our major risk exposures, their potential impact on us, and the steps we take to manage them.
Our Chief Information Officer ("CIO") is responsible for developing, implementing, and maintaining our cybersecurity risk management policies and procedures. The individual currently serving in the role of CIO has over thirty-five years of experience in cybersecurity, information security, data protection, regulatory compliance, and risk management within complex and international business verticals such as pharmaceutical/biotech, technology, and logistics. The CIO provides regular cybersecurity updates to our board of directors.
Our Information Technology Steering Committee ("ITSC") oversees matters regarding the Company’s Information Technology strategy, priorities, and governance, including cybersecurity threats and risk assessments, through periodic meetings and frequent communications. ITSC members include representatives from the Finance, Regulatory Affairs, Operations, and Information Technology departments. The ITSC has a charter that is reviewed internally to ensure it is aligned with our business strategy. As outlined in its charter, and relative to cybersecurity, the ITSC is responsible for identifying and assessing material cybersecurity risks across the Company, including escalating to our Audit Committee and Executive Management where appropriate.
ITEM 2. PROPERTIES
We lease approximately 7,3097,556 square feet of office space and 2,168 square feet of lab space in San Diego, California. The lease for the office space expires on March 31, 2023,California and can be extended for an additional 5 year period. The lease for our lab space expires on February 28, 2022. We lease approximately 2,078 square feet of office space in Toronto, Ontario, Canada. The lease for the San Diego office space is scheduled to expire on May 31, 2026. Aptose previously leased 2,618 square feet of laboratory space in San Diego. We exited this location expireslaboratory space prior to the expiration of the lease on February 28, 2023. The lease for the Toronto office space is scheduled to expire on June 30, 2023, with an option to renew for another five-year period.2024. We believe that our facilities are sufficient to meet our needs and that suitable additional space will be available as and when needed.
ITEM 3. LEGAL PROCEEDINGS
We know of no material pending legal proceedings to which our company or subsidiaries is a party or of which any of our properties, or the properties of our subsidiaries, is the subject. However, from time to time, we may be subject to various pending or threatened legal actions and proceedings, including those that arise in the ordinary course of our business. Such matters are subject to many uncertainties and to outcomes that are not predictable with assurance and that may not be known for extended periods of time.
ITEM 4. MINE SAFETY DISCLOSURES
None.
PART II.
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our Common Shares are currently traded on The Nasdaq Capital Market under the symbol “APTO” and the Toronto Stock Exchange under the symbol “APS”.“APS.”
As of March 23, 2021,26, 2024, there were approximately 3230 shareholders of record of our Common Shares, which included Cede & Co., a nominee for Depository Trust Company, or DTC, and CDS & Co., a nominee for The Canadian Depository for Securities Ltd., or CDS.("CDS"). Common shares that are held by financial institutions as nominees for beneficial owners are deposited into participant accounts at either DTC or CDS, and are considered to be held of record by Cede & Co. or CDS & Co., each as one shareholder.
We currently intend to retain all future earnings, if any, for the operation and expansion of our business and, therefore, do not anticipate declaring or paying cash dividends on our Common Shares in the foreseeable future.
Repurchases of Equity Securities
There were no repurchases of equity securities during the fourth quarter of 2020.2023.
ITEM 6. SELECTED financial dataRESERVED
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and have therefore omitted the information required by this Item 6.
ITEM 7 - MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
This discussion contains forward-looking statements that involve risks and uncertainties. When reviewing the discussion below, you should keep in mind the substantial risks and uncertainties that impact our business. In particular, we encourage you to review the risks and uncertainties described in “Risk Factors” in Part I, Item 1A in this Annual Report on Form 10-K. These risks and uncertainties could cause actual results to differ materially from those projected or implied by our forward-looking statements contained in this report. These forward-looking statements are made as of the date of this management’s discussion and analysis, and we do not intend, and do not assume any obligation, to update these forward-looking statements, except as required by law.
All amounts are expressed in United States dollars unless otherwise stated.
OVERVIEW
Aptose Biosciences Inc. is a science-driven clinical stage biotechnology company committed to precision medicines addressing the unmet clinical needs in oncology, with an initial focus on hematology. The Company's small molecule cancer therapeutics pipeline includes products designed to provide single agent efficacy and to enhance the efficacy of other anti-cancer therapies and regimens without overlapping toxicities. The Company’s executive office is in San Diego, California, and our head office is in Toronto, Canada.
Aptose Programs
We are advancing first-in-classoral targeted agents to treat life-threatening hematologic cancers such as AML, high-risk MDS, CLL and other hematologic malignancies. Based on insights intothat require immediate treatment. We have two clinical-stage oral kinase inhibitors under active development for the genetic and epigenetic profiles of certain cancers and patient populations, Aptose is building a pipeline of novel oncology therapies directed at dysregulated processes and signaling pathways. Aptose is developing targeted medicines for precision treatment of these diseases to optimize efficacyhematologic malignancies: tuspetinib ("HM43239" or "TUS") and quality of life by minimizing the side effects associated with conventional therapies. We currently have in development two molecules: luxeptinib (CG-806)("CG-806" or "LUX"). Tuspetinib and APTO-253, bothluxeptinib are being evaluated for safety, tolerability, pharmacokinetics and signals of efficacy in Phase 11/2 clinical trials. Eachtrials, and each molecule is described below. A third molecule (APTO-253) is not under clinical development and will not be discussed further.
Tuspetinib, Aptose’s lead asset, is being developed for frontline combination therapy in newly diagnosed AML patients to unlock the most significant patient impact and greatest commercial opportunity. Tuspetinib is a once-daily oral kinase inhibitor, targeting a select group of kinases operative in myeloid malignancies, such as acute myeloid leukemia ("AML") and the higher risk myelodysplastic syndromes ("hr-MDS"), and known to be involved in tumor proliferation, resistance to therapy, and differentiation. However, tuspetinib avoids kinases that typically cause toxicities associated with other kinase inhibitors and is consequently a well-tolerated antileukemic agent. The clinical development path for triplet combination therapy in newly diagnosed AML patients with tuspetinib-based triplet combination therapy (TUS + VEN + hypomethylating agent; (TUS+VEN+HMA), the TUS+VEN doublet in relapsed or refractory R/R AML patients.
Tuspetinib has completed the dose escalation and dose exploration stages of an international Phase 1/2 clinical trial designed to assess the safety, tolerability, pharmacokinetics, pharmacodynamic responses, and efficacy as a single agent in patients with R/R AML. Complete responses (“CRs”) without dose limiting toxicities were achieved at four dose levels across a broad diversity of mutationally-defined AML populations and with a highly favorable safety profile. Tuspetinib to date has demonstrated a favorable safety profile and has caused no drug-related QTc prolongations, liver or kidney toxicities, muscle damage, differentiation syndrome, and no myelosuppression with continuous dosing of patients in remission. A recommended phase 2 dose (RP2D) of 80 mg tuspetinib once daily as an oral tablet was selected and approved by the U.S. FDA for use as a single agent in patients with R/R AML. At the RP2D, tuspetinib demonstrated notable response rates in R/R AML patients that had never been treated with venetoclax (VEN-naive AML): CR/CRh=36% among all-comers, CR/CRh=50% among patients with mutated FLT3, and CR/CRh=25% in patients with wildtype FLT3.
Following completion of the single agent dose escalation and exploration trial, tuspetinib advanced into the APTIVATE expansion trial of the Phase 1/2 program in R/R AML patient populations treated with the TUS+VEN doublet, with the intent to position tuspetinib for triple combination studies in frontline therapy. The TUS+VEN doublet combination therapy maintained a favorable safety profile: no new or unexpected safety signals were observed, and there were no reported drug-related adverse events of QTc prolongation, differentiation syndrome, or deaths. The TUS+VEN doublet combination also achieved responses in heavily pretreated R/R AML patients, including those with wildtype or mutated FLT3, and those who failed prior therapy with venetoclax ("Prior-VEN") or FLT3 inhibitors ("Prior-FLT3i"). Based on the safety and efficacy profile of tuspetinib, we believe that tuspetinib, if
approved, can reach annual sales greater than $3 billion by 2035 because we believe tuspetinib could 1) become the preferred kinase inhibitor for inclusion in triplet combination for front line AML patients with FLT3 mutations and for patients with wild type FLT3, 2) become the preferred kinase inhibitor for inclusion in combination with venetoclax for second line AML patients, 3) serve as an effective agent for maintenance therapy to prevent relapse in patients who achieved a complete remission through a stem cell transplant or through drug-based therapy, 4) serve as an effective agent for the treatment of third line FLT3 mutated patients failed by prior therapy with other FLT3 inhibitors and 5) serve in front line triplet combinations, second line doublet combinations, and maintenance therapy for hr-MDS patients. These beliefs related to the potential commercial opportunity are based on management’s current assumptions and estimates, which are subject to change, and there can be no assurance that tuspetinib will ever be approved or successfully commercialized and, if approved and commercialized, that it will ever generate significant revenues. See our “Risk Factors – “We are an early-stage development company with no revenues from product sales.” and “We have a history of operating losses. We expect to incur net losses and we may never achieve or maintain profitability.” in this Annual Report on Form 10-K.
Luxeptinib is an orally administered,oral, highly potent first-in-class FLT3/BTKkinase inhibitor that selectively targets defined clusters of kinases that are operative in myeloid and lymphoid hematologic malignancies. This mutationally agnostic small molecule anticancer agent is currently beinghas been evaluated in a Phase 1a/b study for the treatment of patients having B-cell malignancies including classic CLL, SLL and certain NHL that are resistant/refractory/intolerant to other therapies. In addition, Aptose received IND allowance and initiated patient dosing in a Phase 1a/b study to develop luxeptinib for the treatment of patients with R/R AML, including the emerging populations resistant to FLT3 inhibitors. In this trial of patients with R/R AML, the luxeptinib starting dose of 450 mg BID was selected because the plasma from B-cell cancer patients at that dose completely inhibited phospho-FLT3, suggesting that this starting dose might be active in the AML patient population,leukemias and that dose escalation is planned to identify an optimal dose for treating a breadth of AML patients. It is important to note that luxeptinib now is undergoing formal clinical development in both lymphoidlymphomas and myeloid hematologic malignancies.
APTO-253 is a first-in-class small molecule therapeutic agent that clinically inhibits expression of the MYC oncogene without causing, to date, general myelosuppression of the bone marrow. The MYC oncogene is overexpressed across many hematologic cancers, including AML and certain B cell malignancies, as well as certain solid tumor indications. MYC acts as a transcription factor that regulates cell growth, proliferation, differentiation and apoptosis, and overexpression of MYC amplifies new sets of genes to promote survival of cancer cells. APTO-253 suppresses expression of the MYC oncogene in AML cells and depletes those cells of the MYC oncoprotein, leading to apoptotic cell death. APTO-253 is currently being evaluated in a Phase 1a/b study for the treatment of patients with R/R AML and high-risk MDS. APTO-253 may serve asor hr-MDS. These clinical studies demonstrated tumor shrinkage among B-cell cancer patients, including a safe and effective MYC inhibitor for AML/MDS patientsCR in a diffuse large B-cell lymphoma patient that combines wellwas determined via biopsy analysis at the end of Cycle 22 with other agents and does not significantly impact900mg BID dosing of the normal bone marrow.
Impactoriginal G1 formulation. Likewise, an MRD-negative CR in one R/R AML patient occurred with 450mg BID dosing of COVID 19 on our Research Programs:
We are advancing first-in-class targeted agents to treat life-threatening cancers that, in most cases, are not elective for patients and require immediate treatment. However, COVID-19 has caused global economic and social disruptions that could adversely affect our ongoing or planned research and development and clinical trial activities including enrollmentthe original G1 formulation. Because absorption of the original G1 formulation hampered effectiveness of luxeptinib, a new G3 formulation was developed. Enrollment of patients in our ongoing clinical trials, collection and analysis of patient data and eventually, the reporting of top-line results from our trials.
Our team proactively addressed these new challenges swiftly and appropriately, implementing safeguards and procedures to ensure both the safety of our employees and stakeholders, and accommodate the potential challenges due to COVID-19. Aptose was early in directing its employees to work-from-home and provided the tools to minimize productivity disruptions. Our clinical operations team reached out to active and future clinical sites to determine their needs and challenges and assist where possible, including virtual monitoring of patients, which reduces patients’ visits. We also have contacted our drug manufacturers to identify any potential supply chain disruptions and are adjusting accordingly. During the early part of the first quarter of 2020, we began to carefully monitor the potential impact of COVID-19, and on a regular basis, we communicated with investigators at our clinical sites to gain an evolving understanding of competing COVID-19 related activities and clinical trial related activities.
In the beginning of April 2020, we learned that some of our larger clinical sites that are impacted by COVID-19 may either postpone or face delays in the enrollment of patients on all on-going clinical trials due to a number of factors, including the re-allocation of resources and to avoid clinical trial patients being exposed to COVID-19. Such measures taken at the clinical sites could lead to a slowdown in the enrollment of patients on our trials at these sites. To minimize the impact of COVID-19, we focused efforts on our other larger clinical sites and regional cancer care sites that are not/less impacted by COVID-19 to recruit patients into the fourth cohort. While it is difficult to estimate the duration and impact of COVID-19 on the larger clinical sites and regional cancer care sites, as of the date of this report, we have not experienced and do not foresee material delays to the enrollment of patients or timelines for the luxeptinib Phase 1a/b B-cell malignancy trial dueand the AML trial have been completed, and clinical evaluation of the G3 formulation has been completed. The G3 formulation was determined to deliver superior plasma exposure levels relative to the varietyoriginal G1 formulation, and any future trial with luxeptinib should use the G3 formulation. Regarding potential next steps with luxeptinib, recent therapeutic strategies with CLL B-cell cancer patients typically involve therapy with certain BTK inhibitors in combination with venetoclax (VEN). Drug resistance has begun to emerge in a molecularly defined subgroup of clinical sites that we have actively recruited forthese patients, and the drug resistance has been correlated with mutations in the FLT3 receptor. Although FLT3 mutations are typically associated with AML patients, these R/R CLL prior-BTKi/Prior-VEN/FLT3-mutated patients are difficult to treat and represent a potential commercial market of approximately $200 million by 2039. The Dana Farber Cancer Institute identified this trial. APTO-253, which is administered intravenously, requiresemerging patient population and has requested luxeptinib be tested as part of an investigator sponsored trial in combination with VEN in the need for hospital / clinical site resourcesR/R CLL prior-BTKi/Prior-VEN/FLT3-mutated patients. Non-clinical studies are underway to assist and monitor patients during each infusion and based on the current conditions caused by COVID-19, future enrollment of patients on this trial is likely to be negatively impacted.
PROGRAM UPDATES
Luxeptinib (CG-806)
Indication and Clinical Trials:
Luxeptinib is being developed with the intent to deliver the agent as an oral therapeuticposition LUX+VEN for the treatment of R/R AMLthese patients, and efforts are underway to identify sources of capital to support such a trial to develop LUX for the treatmenta molecularly defined CLL subpopulation with a high unmet medical need.
PROGRAM UPDATES
Tuspetinib
Tuspetinib is an oral, highly potent, small molecule inhibitor of a spectrum of B cellkinases operative in myeloid malignancies (including but not limitedand known to CLL, SLLbe involved in tumor proliferation, resistance to therapy and NHL).
On March 25, 2019, we announceddifferentiation. Preclinical in vitro and in vivo studies suggest that the FDA granted Aptose IND allowance to initiate its Phase 1a/b clinical trial for luxeptinib. The clinical trial is a multicenter, open label, dose-escalation study with additional optional expansion cohorts to assess the safety, tolerability, pharmacokineticsTuspetinib may be an effective monotherapy and pharmacodynamic effects, and prelimnary efficacy of luxeptinibcombination therapy in patients with CLL, SLL or NHL. In this study, luxeptinib is administered in gelatin capsules twice daily (BID during a 28-day cycle.
As of the date of this report, we have initiated thirtyhematologic malignancies including AML. An international Phase 1/2 clinical sites for the Phase 1a/b trial in patients with CLL/SLLrelapsed or NHL which include specialty regional cancer care centers as well as large hospitals and key academic institutions. As of the daterefractory AML is ongoing. The dose escalation portion of this report, we have completed the first, second, third and fourth dose levels (150 mg, 300 mg, 450 mg and 600 mg BID, respectively). Cohort 5 (750mg) enrollment is ongoing. Under an FDA-approved accelerated titration protocol, only one patient was required at eachstudy to date has observed evidence of the first two dose levels, followed by three patients at each dose level thereafter. Intra-patient dose escalation is allowed if the higher dose is safe in three or more patients, and additional patients may be enrolled at dose levels previously declared safe. To date, we have reported that among enrolled patients with an array of B-cell malignancies, three classic CLL patients have received luxeptinib and all three demonstrated inhibition of phospho-BTK and “on-target” lymphocytosis and modest tumor reductions in different tumor types, indicating target engagement and pharmacologicrobust clinical activity, of luxeptinib. As luxeptinib moves from low/intermediate dose levels and into the higher dose levels, it is hoped that an optimal dose can be selected that demonstrates formal clinicalincluding multiple complete responses without excessive toxicity.
We are also advancing luxeptinib into myeloid malignancies, with an initial focus on AML, in a separate Phase 1a/b trial. On June 29, 2020, we announced that we had received allowance from the FDA to proceed into a study in R/R AML with a starting dose of 450 mg BID, and subsequently on October 19, 2020, announced that we had initiated dosing of the first patient with AML. As of the date of this report we have initiated six clinical sites for the Phase 1a/b trial and dosing continues at the 600 mg dose cohort.
The clinical trial is a multicenter, open label, dose-escalation study with additional optional expansion cohorts to assess the safety, tolerability, pharmacokinetics and pharmacodynamic effects, and preliminary efficacy of luxeptinib in patients with R/R AML. In this study, luxeptinib is administered in gelatin capsules BID during a 28-day cycle. Our strategy was to identify a startingvarious disease genotypes, and no toxicity trends that should prevent further dose of luxeptinib that we believe could be therapeutically active in critically ill patients with R/R AML. In our ongoing Phase 1a/b study in patients with CLL and other B-cell malignancies, 450 mg BID luxeptinib delivered plasma levels potently inhibited phospho-FLT3 in a plasma inhibitory activity (PIA) reporter cell assay, suggesting that the 450 mg BID dose may be active in patients with AML. Aptose plans to dose escalate beyond the 450 mg BID dose level, provided the 450 mg BID dose level is safe and well tolerated in R/R AML patients. Based on strong preclinical evidence of luxeptinib’s activity against AML – including demonstration of mutation-agnostic and genotype-agnostic potency, particularly compared against other FLT3 inhibitors, and its ability to safely cure AML in murine leukemia models – we believe that luxeptinib may offer hope to the fragile and difficult-to-treat AML patient population. escalation.
The FDA has granted orphan drug designation to luxeptinibtuspetinib for the treatment of patients with AML.AML in October 2018. Orphan drug designation is granted by the FDA to encourage companies to develop therapies for the treatment of diseases that affect fewer than 200,000 individuals in the United States. Orphan drug status provides research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees and other benefits. The orphan drug designation also provides us with seven additional years of marketing exclusivity in this indication.
Manufacturing:
Following the Tuspetinib licensing agreement between Aptose and Hanmi on November 4, 2021 (the "Tuspetinib Licensing Agreement"), Aptose received from Hanmi an existing inventory of drug product expected to support continuation of the current Phase 1/2 study. The Company and Hanmi also entered into a separate supply agreement in 2022 for additional production of new drug substance and drug product to support further clinical development. Additional batches of API and drug product have been produced by other companies during 2022 and 2023.
Manufacturing:Program Updates at Recent Scientific Forums:
On December 9, 2023, Aptose featured tuspetinib in an oral presentation at the 65th American Society of Hematology (ASH) Annual Meeting and Exposition and announced that a growing body of clinical data for Aptose’s lead compound tuspetinib, demonstrates significant benefit as a single agent and in combination with venetoclax in patients with R/R AML in the ongoing APTIVATE Phase 1/2 study. Data were presented in an oral presentation by lead investigator Naval G. Daver, M.D., Professor, Director Leukemia Research Alliance Program, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX.
Dr. Daver reported data from more than 100 relapsed/refractory patients from multiple international clinical sites, who had failed prior therapy and then were treated with TUS as a single agent or TUS+VEN. Both TUS and TUS+VEN delivered multiple composite complete remissions (CRc) in this very ill AML population, while maintaining a favorable safety profile across all treated patients. The data demonstrated tuspetinib is active and well tolerated in one of the most challenging and heterogeneous disease settings in oncology – relapsed and refractory AML. Tuspetinib demonstrated broad activity, including activity in patients with FLT3 wild-type AML (accounting for more than 70% of the AML population), FLT3 mutated AML, NPM1 mutated AML, as well as in patients with mutations historically associated with resistance to targeted therapy. Most notably, TUS targets VEN resistance mechanisms, enabling TUS+VEN uniquely to treat the very ill prior-VEN AML population, including both FLT3 mutant and FLT3 wildtype disease. From a broader perspective, the growing body of antileukemic activity, and continued favorable safety profile, support advancement of tuspetinib in a TUS+VEN+HMA triplet for the treatment of frontline newly diagnosed AML patients.”
Dr. Daver also pointed out that while patients on the TUS+VEN therapy are early in their treatment cycles, most achieving a response remained on treatment and that responses have begun to mature as dosing continues. Highlights of Dr. Daver’s ASH oral presentation include:
| | | |
| • |
| As a single agent at therapeutic doses of 80-160 mg in 68 evaluable patients, TUS was more active in VEN-naive patients, with an overall CRc rate of 29% (8/28). This included a 42% CRc rate (5/12) in FLT3-mutated patients and a 19% CRc rate (3/16) in FLT3-unmutated, or wildtype, AML patients. Responses and blood counts improved with continuous dosing, many patients bridged to an allogeneic stem cell transplant ("HSCT"), durability was observed when HSCT was not performed, and 80 mg was selected as the RP2D. Overall, tuspetinib showed a favorable safety profile with only mild adverse events ("AEs") and no dose-limiting toxicities ("DLTs") up to 160 mg per day, and no drug discontinuations from drug-related toxicity. |
| | | |
| • |
| In the TUS+VEN doublet study, 49 patients were dosed with 80 mg of tuspetinib and 200 mg of venetoclax, with 36 evaluable (and 13 patients too early to assess). Patients were heavily exposed to Prior-VEN and Prior-FLT3 inhibitor treatment. TUS+VEN was active in both VEN-naive and prior Prior-VEN R/R AML patients. TUS demonstrated compelling composite complete remission (CRc) rates. Among all evaluable patients, TUS+VEN demonstrated a CRc rate of 25% (9/36); 43% (3/7) in VEN-naive patients, and 21% (6/29) in Prior-VEN patients. Among FLT3 wildtype patients, TUS+VEN demonstrated an overall CRc rate of 20% (5/25); 33% (2/6) in VEN-naive patients, and 16% (3/19) in Prior-VEN patients. Among FLT3 mutant patients, TUS+VEN demonstrated an overall CRc rate of 36% (4/11); a complete response in a VEN-naive patient (1/1); a 30% (3/10) in Prior-VEN patients; and 44% (4/9) in patients treated prior with a FLT3 inhibitor. |
On October 29, 2023, Aptose presented two posters related to the clinical and preclinical activity of tuspetinib at the European School of Haematology 6th International Conference: Acute Myeloid Leukemia "Molecular and Translational": Advances in Biology and Treatment, held October 29-31, 2023, in Estoril, Portugal. Clinical findings included 1) data from the APTO-TUS-HV01 clinical trial (the "Food Effect Study") evaluating the pharmacokinetic (PK) properties of tuspetinib in healthy human volunteers in which tuspetinib was administered with our without food, and 2) from an international Phase 1/2 study of tuspetinib as a single agent (TUS) and in combination with venetoclax in patients with R/R AML from across clinical centers in the United States, South Korea, Spain, Australia and other sites. Data from the Food Effect Study in healthy human volunteers demonstrated tuspetinib can be administered with or without food and foresee no clinically meaningful difference in exposure. This is an important finding for patient convenience, as venetoclax is dosed with food and tuspetinib can now be co-administered with venetoclax rather than in staggered dosing. Findings from the Phase 1/2 clinical trial demonstrated tuspetinib as a single agent was well-tolerated and highly active among R/R AML patients with a diversity of adverse genotypes and delivered a 42% CR/CRh cross-evaluable venetoclax (VEN) naive patients at the 80mg daily RP2D. The TUS+VEN doublet has been well tolerated in the APTIVATE international Phase 1/2 expansion trial in R/R AML patients and achieved multiple responses in patients who previously failed venetoclax ("Prior-VEN failure AML"), including Prior-VEN failure patients who also previously failed FLT3 inhibitors, all of whom represent emerging populations of high unmet medical need. Notably, tuspetinib targets venetoclax resistance mechanisms that may re-sensitize Prior-VEN failure patients to venetoclax.
Separate from the clinical studies, the preclinical study (entitled: “Tuspetinib Oral Myeloid Kinase Inhibitor Creates Synthetic Lethal Vulnerability to Venetoclax”) presented by Aptose during the ESH Conference investigated the effects of tuspetinib on key elements of the phosphokinome and apoptotic proteome in both parental and TUS-resistant AML cells. In parental cells, tuspetinib inhibits key oncogenic signaling pathways and shifts the balance of pro- and anti-apoptotic proteins in favor of apoptosis, suggesting that it may generate vulnerability to venetoclax. In addition, acquired resistance in the AML cells to tuspetinib generated a synthetic lethal vulnerability to venetoclax of unusually high magnitude. Concurrent administration of TUS+VEN therefore may discourage the emergence of resistance to tuspetinib during treatment.
In conjunction with poster presentations at the ESH Conference, on October 30, 2023, Aptose held a “Clinical Update and KOL Data Review of AML Drug Tuspetinib” that was webcast and featured Dr. Naval Daver, MD, Professor, Director Leukemia Research Alliance Program, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, Texas. Dr. Daver is the lead investigator on Aptose’s APTIVATE trial and is recognized for significant achievements in the development of novel AML treatments, including several combination therapies. Aptose presented data in 49 patients who received the TUS+VEN doublet, showing an overall response rate ("ORR") of 48% among all patients that had achieved an evaluable stage, as well as a 44% ORR among Prior-VEN failure AML patients, including FLT3-unmutated ("wildtype") patients (43% ORR) and FLT3-mutated patients (60% ORR), some of whom also had failed prior therapy with FLT3 inhibitors. The TUS+VEN doublet was well tolerated with no unexpected safety signals. The TUS+VEN doublet may serve the Prior-VEN failure R/R AML patients that represent a rapidly growing population that is highly refractory to any salvage therapy. The compelling data with the TUS+VEN doublet in R/R AML patients suggest a TUS+VEN+HMA triplet may also serve the needs of frontline (1L) newly diagnosed AML patients.
Concurrent with the European Hematology Association (EHA) Annual Congress held June 8-11, 2023, Aptose held an interim clinical update webcast on June 10, 2023, to present highlights from the ongoing clinical development of tuspetinib. Aptose reported completion of the tuspetinib dose escalation and dose exploration Phase 1/2 trial in 77 R/R AML patients, tuspetinib demonstrated a favorable safety profile, and tuspetinib delivered monotherapy responses across four dose levels with no dose-limiting toxicity in mutationally diverse and difficult to treat R/R AML populations, including patients with highly adverse mutations that typically do not respond to monotherapy or combination therapy: TP53-mutated patients with a CR/CRh = 20% and RAS-mutated patients with a CR/CRh = 22%. Aptose also reported completion of a successful End of Phase 1 Meeting with the US FDA for tuspetinib, that a monotherapy RP2D was selected as 80mg daily, and that all development paths remain open, including the single arm accelerated path. Following completion of the dose escalation and dose exploration phases of the Phase 1/2 clinical program, Aptose focused attention on the tuspetinib APTIVATE expansion trial. The APTIVATE trial is designed to identify patient populations sensitive to tuspetinib monotherapy that may serve as development paths for single arm accelerated approval and to use the TUS+VEN doublet in R/R AML patients and identify patient populations of unmet need that are sensitive to the TUS+VEN doublet and can serve as development paths for accelerated and full approvals.
We reported that patient enrollment in the APTIVATE expansion trial has been brisk and preliminary CR activity had already been reported in patients receiving the TUS+VEN doublet who previously failed therapy with venetoclax. During the interim clinical update webcast Aptose also reviewed clinical findings with the new G3 formulation of luxeptinib. Aptose disclosed that continuous dosing with 50mg of the G3 formulation achieves roughly an equivalent pharmacokinetic profile as 900mg original G1 formulation, and that dose escalation with the G3 formulation was anticipated.
On March 23, 2023, Aptose announced the APTIVATE Phase 1/2 expansion trial with tuspetinib had been initiated and already had treated several R/R AML patients in the monotherapy arm, and that patient enrollment had been initiated in the doublet combination treatment arm of the APTIVATE trial with the TUS+VEN doublet. Since then, patients have continued to enroll and receive tuspetinib on the monotherapy arm. Plus, enrollment and dosing of patients on the TUS+VEN doublet arm have been brisk. Clinical investigator interest for tuspetinib is evident, and early signs of antileukemic activity during the APTIVATE trial have fueled the level of excitement for the trial.
Clinical responses to monotherapy with tuspetinib have been observed in a broad range of mutationally defined populations, including those with mutated forms of NPM1, MLL, TP53, DNMT3A, RUNX1, wild-type FLT3, ITD or TKD mutated FLT3, various splicing factors, and other genes. In the March 23, 2023, announcement, Aptose also highlighted an unexpected observation of a 29% CR/CRh response rate with tuspetinib monotherapy in R/R AML patients having mutations in the RAS gene or other genes in the RAS pathway. Responses in RAS-mutated patients are important because the RAS pathway is often mutated in response to therapy by other agents as the AML cells mutate toward resistance to those other agents. Collectively, these observations of broad clinical activity of tuspetinib, along with its favorable safety profile, position tuspetinib for potential accelerated development paths, as well as for doublet, triplet and maintenance therapy indications.
On January 30, 2023, Aptose announced dosing of patients in the APTIVATE Phase 1/2 clinical trial of tuspetinib, and that another clinical response has been achieved by a R/R AML patient receiving 40 mg tuspetinib once daily orally in the original dose exploration trial, the second response at the recently launched low-dose 40 mg cohort. In addition, Aptose elucidated a rationale for the superior safety profile of tuspetinib. While several kinase inhibitors require high exposures that exert near complete suppression of a single target to elicit responses, those agents often cause additional toxicity because they also cause extensive inhibition of that target in normal cells. In contrast, tuspetinib simultaneously suppresses a small suite of kinase-driven pathways critical for leukemogenesis. Consequently, tuspetinib achieves clinical responses at lower exposures with less overall suppression of each pathway, thereby avoiding many of the toxicities observed with competing agents.
Tuspetinib has completed the dose escalation and dose exploration phases of an international Phase 1/2 study in patients with relapsed or refractory AML across clinical centers in the United States and South Korea. Clinical data from tuspetinib in AML were presented at the ASH Annual Meeting in December 2022 and presented during a Corporate Comprehensive Clinical Update Call held December 11, 2022. Data presented demonstrated that tuspetinib delivers single agent responses without prolonged myelosuppression or life-threatening toxicities in these very ill and heavily pretreated relapsed or refractory AML patients. Responses were observed in a broad range of mutationally-defined populations, including those with mutated forms of NPM1, MLL, TP53, NRAS, KRAS, DNMT3A, RUNX1, wild-type FLT3, ITD or TKD mutated FLT3, various splicing factors, and other genes As of October 6, 2022, 60 heavily pretreated R/R AML patients were enrolled at multiple centers and treated at doses escalating from 20 mg to 200 mg, with further dose exploration at the 40 mg, 80 mg, 120 mg and 160 mg dose levels. Tuspetinib delivered multiple CRs at 40 mg, 80 mg, 120 mg and 160 mg dose levels in which no DLTs were observed. Tuspetinib demonstrated clinically meaningful benefit in all responders, by either bridging successfully to HSCT or leading to a durable response, as well as a favorable safety profile. In addition to 5 CRc and 1 PR reported at ASH 2021, 4 new CRc and 3 new PR had been generated during 2022. New responses during 2022 were achieved with 160 mg,120 mg, 80 mg, and 40 mg. Among efficacy-evaluable patients treated with 80 mg, 120 mg, or 160mg, the following response rates ranging from 19% to 75% were achieved in specific genotypic subpopulations of r/r AML patients. Significant bone marrow leukemic blast reductions were observed broadly in FLT3+ and FLT3 wildtype patients across multiple dose levels, comparable to reported gilteritinib data, but in more heavily pre-treated relapsed and refractory AML patients. Vignettes of patient experiences highlight the potency and breadth of tuspetinib to deliver complete remissions among several mutationally-defined populations with a diversity of adverse mutations. Tuspetinib continued to show a favorable safety profile with only mild AEs and no DLTs up to 160 mg per day, and no drug discontinuations from drug-related toxicity. No drug-related SAE, drug-related deaths, differentiation syndrome, AE of QT prolongation or DLT were observed through the 160 mg level. Tuspetinib avoids many of the typical toxicities observed with other tyrosine kinase inhibitors. Aptose identified a safe therapeutic range with a broad therapeutic window, spanning the dose levels of 40, 80, 120 and 160 milligrams. Aptose also announced that enrollment had been initiated in the APTIVATE expansion trial for monotherapy and drug combination therapy with tuspetinib. For the APTIVATE expansion trial, Aptose selected 120 mg as the initiating single agent expansion dose and 80 mg as the initiating dose selected for combination with venetoclax.
At the European Hematology Association Annual Congress 2022 held June 9-12, 2022, Aptose presented preclinical data from tuspetinib in a poster entitled “Myeloid Kinome Inhibitor HM43239 Overcomes Acquired Resistance in Acute Myeloid Leukemia Models.” Oral HM43239 potently inhibits kinases that drive AML, including SYK, diverse forms of the FLT3, JAK1 and JAK2, and mutant forms of the c-KIT kinases. The SYK and JAK1/2 intracellular kinases and the FLT3 (mutated and wildtype) and cKIT (mutated) receptor kinases mediate oncogenic signaling pathways in AML that can drive malignant proliferation and promote drug resistance to certain drugs. Tuspetinib was developed to overcome shortcomings of other drugs, such as simple SYK inhibitors and approved inhibitors of FLT3. These preclinical findings support the continued clinical development of tuspetinib for the treatment of multiple AML populations, particularly those who have failed other therapies.
Major conclusions include
•tuspetinib inhibits wild type and mutant forms of FLT3 at low nM concentrations
•tuspetinib HM43239 inhibits SYK, JAK1, JAK2 and mutant forms of c-KIT at low nM concentrations
•tuspetinib inhibits phospho-FLT3, phospho-SYK, phospho-EKR1/2 and phospho-JAK/STAT5 that participate in signaling and rescue pathways
•tuspetinib has potential to kill cells and tumors resistant to other FLT3 inhibitors
•tuspetinib, at doses that are well tolerated, demonstrates in vivo efficacy on tumors resistant to other FLT3 inhibitors
Luxeptinib
Indication and Clinical Trials:
Luxeptinib is currently being evaluated in a Phase 1 a/b trial in patients with relapsed or refractory B cell malignancies who have failed or are intolerant to standard therapies, and in a separate Phase 1 a/b trial in patients with relapsed or refractory AML or high-risk MDS. During 2022, a new G3 formulation was tested as a single dose in 20 patients during the ongoing Phase 1 a/b clinical program. Modeling of the PK properties of G3 predicted steady-state plasma exposure from continuous dosing with 50 mg of G3 (every 12 hours, Q12h) should be comparable to that of 900 mg of the original G1 formulation Q12h, representing a significant improvement in bioavailability with G3. On November 14, 2022, Aptose announced dosing of the first AML patient to receive a continuous dosing regimen of the G3 formulation (50 mg G3 Q12h), with the protocol allowing for further dose escalation of G3 in subsequent patients. Clinical data from both studies were presented during a Corporate Comprehensive Clinical Update Call held December 11, 2022. During the Corporate Update Call, Aptose announced a CR was achieved with a DLBCL patient at the end of Cycle 22 with 900mg BID of the original G1 formulation. Previously, an MRD-negative CR was reported with a R/R AML patient receiving 450mg BID of the original G1 formulation. Aptose expects that 9-15 patients will determine if G3 is safe and achieves desired exposures to deliver clinical responses.
Manufacturing:
During fiscal years 2017 and 2018, we created a scalable chemical synthetic route for the manufacture of luxeptinib drug substance and have scaled the manufacture of API (active pharmaceutical ingredient, or drug substance) to multi-kg levels, we completed the manufacture of a multi-kg batch of API under GMP conditions as our API supply for our first-in-human clinical trials, and we manufactured under GMP conditions two dosage strengths of capsules to serve as our clinical supply in those human studies. During fiscal 2019 and 2020, we completed successful manufacture of multiple batches of API and drug product, and planned numerous GMP production campaigns to supply the ongoing trial and planned trials into the future. To date we have been able to manufacture API and capsules to support clinical supplies under GMP conditions. We are continuingIn fiscal 2021 and 2022 we continued our manufacturing campaigns in the current 2021 fiscal period and continue scale-up and tech transfer activities to support additional manufacturing capacity for the ongoing and planned clinical trials of luxeptinib. Additional research and development funds are beingwere utilized to support exploratory formulation studies in an ongoing effort to craft a superior formulation for later stagethe development of the G3 formulation of luxeptinib. Now that the G3 formulation has been successfully manufactured and has demonstrated encouraging PK properties, the manufacture of additional batches of the first and second generation formulation of luxeptinib are discontinued, and the amount of drug substance manufacturing is reduced.
Publication of Peer-Reviewed Research Articles Related to Luxeptinib:
During the first quarter of 2023, Aptose and its scientific collaborators published a peer reviewed research article, entitled “Luxeptinib interferes with LYN-mediated activation of SYK and modulates BCR signaling in lymphoma,” in the online journal PLOS ONE. The article elucidates the mechanism by which luxeptinib suppresses the B-cell receptor and compares it to BTK inhibitor ibrutinib. Results showed that both luxeptinib and ibrutinib are potent inhibitors of recombinant forms of BTK but have different efficacy profiles and effects on BTK activity. Luxeptinib was more effective than ibrutinib at reducing both steady state and anti-IgM-induced phosphorylation of the LYN and SYK kinases upstream of BTK where IB has little or no effect.
During the first quarter of 2022, three separate peer-reviewed research articles were published that presented preclinical data related to the application of luxeptinib to the treatment of AML, certain B-cell lymphomas and inflammation. These publications contribute to the body of preclinical data demonstrating luxeptinib’s activity as a lymphoid and myeloid kinome inhibitor, and now as an inflammation kinome inhibitor, and support its continued clinical development in several therapeutic areas.
Program Updates at Recent Scientific Forums:
On December 11, 2021, we presented clinical updates from luxeptinib in patients with R/R B-cell malignancies and R/R AML in two virtual poster presentations at the 63rd ASH Annual Meeting (A Phase 1 a/b Dose Escalation Study of the Mutation Agnostic BTK/FLT3 Inhibitor Luxeptinib (CG-806) in Patients with Relapsed or Refractory B-Cell Malignancies; A Phase 1 a/b Dose Escalation Study of the Mutation Agnostic FLT3/BTK Inhibitor Luxeptinib
(CG-806) in Patients with Relapsed or Refractory Acute Myeloid Leukemia. The presentations highlighted that in both of these Phase 1/2 studies luxeptinib has been generally well tolerated at dose levels of 450, 600 and 750 mg BID over multiple cycles, and that patients already were being dosed at the 900 mg level. Target engagement of BTK and FLT3, and anti-tumor activity, including dose- and exposure-dependent tumor reductions, have been observed in multiple patients collectively between the studies, including in patients with follicular lymphoma, diffuse large B-cell lymphoma, CLL/SLL, and AML.
Preclinical Program Updates:
We have completed several non-clinical and clinical studies that demonstrate the highly differentiated profile of luxeptinib. Key studies that have been presented at scientific forums are as follows:
| · | On April 15, 2018, at the 2018 Annual Meeting of the American Association for Cancer Research (“AACR”), we presented with the OHSU Knight Cancer Institute preclinical data demonstrating that luxeptinib, a pan-FLT3/pan-BTK inhibitor, demonstrates broader activity and superior potency to other FLT3 and BTK inhibitors against primary bone marrow samples from patients with hematologic malignancies. We also presented preclinical data demonstrating that luxeptinib targets multiple pathways to kill diverse subtypes of AML and B-cell malignancies in vitro. |
| · | On June 15, 2018, at the 23rd Congress of the European Hematology Association (“EHA”), we presented, during a poster presentation, preclinical data demonstrating a unique binding mode of luxeptinib to wild type and C481S mutant BTK. Further, we presented that luxeptinib suppresses the BCR, AKT/PI3K, ERK and NFkB signaling pathways and exerts broader and far greater potency of direct cancer cell killing that ibrutinib against malignant bone marrow cells from patients with CLL, ALL and a host of other hematologic malignancies. |
| · | On December 3, 2018, we announced two separate poster presentations at the American Society of Hematology (“ASH”) Annual Meeting. The OHSU Knight Cancer Institute and Aptose presented data in one poster and the team at The University of MDACC presented data in a separate poster. These presentations highlighted several key findings. First, in collaboration with the MDACC, orally administered luxeptinib demonstrated efficacy in a PDX study in which the bone marrow cells from a patient with AML having dual ITD and D835 mutations in FLT3 were implanted into a mouse. The dual FLT3 mutant form of AML represents a very difficult-to-treat population that has shown resistance to other FLT3 inhibitors, and data from the PDX model suggest that luxeptinib may be useful in treating such patients. Secondly, Aptose presented high level data from preclinical GLP toxicology studies that demonstrate orally administered luxeptinib is a well-tolerated targeted molecule. Finally, in collaboration with the OHSU Knight Cancer Center, studies of luxeptinib on 124 samples of freshly isolated bone marrow from CLL patients demonstrated both broader and greater cell killing potency for luxeptinib than Ibrutinib. |
| · | On April 1, 2019, at the 2019 Annual Meeting of the AACR, Aptose, along with our collaborators at OHSU Knight Cancer Institute, presented data highlighting luxeptinib was more potent in killing AML patient-derived samples than other FLT3 inhibitors including midostaurin, sorafenib, sunitinib, dovitinib, quizartinib, crenolanib and gilteritinib. Luxeptinib was equally potent against cells from patients in the adverse, intermediate and favorable risk groups (2017 ELN risk stratification), and cells from patients with relapsed or transformed AML (World Health Organization classification) were as sensitive as those from patients with de novo AML. The data demonstrated potency on primary AML patient samples across all AML subgroups including relapsed/refractory/transformed AML and those with genetic abnormalities related to poor prognosis. While patient samples with FLT3-ITD mutations were expected to have greater sensitivity to luxeptinib, the most surprising correlation was the sensitivity of patient samples with IDH1 R132 mutations. The enhanced sensitivity of IDH-1 mutant AML to luxeptinib warrants investigation in the clinical setting. Moreover, in studies of luxeptinib on AML patient bone marrow samples, we demonstrated that mutations in p53, ASXL1 and NPM1 do not hinder the potency of luxeptinib. |
| · | On June 14, 2019, we presented new preclinical data for luxeptinib in a poster presentation at the 24th Congress of the EHA in Amsterdam, the Netherlands. The poster, CG-806, preclinical in vivo efficacy and safety profile as a pan-FLT3 / pan-BTK inhibitor, highlights the in vivo anti-leukemic efficacy of luxeptinib and its GLP toxicology and toxicokinetic profile. In a preclinical MV4-11 FLT3-ITD AML xenograft mouse model, luxeptinib suppressed leukemia growth at all doses tested throughout the 28-day period of dosing. In the mice treated with 100 mg/kg, 5 of 11 (45%) were cured through day 120, and in the 300 mg/kg group, 10 of 11 (91%) of the mice were cured. Retreating the “uncured’ mice in these two dose groups for an additional 28 days beginning on day 88 led to rapid and robust antitumor response in all retreated mice through day 120. In the “re-treated” mice, no drug resistance and no toxicities were observed. GLP 28-day toxicology and TK studies mice and dogs showed no adverse luxeptinib-related effects on body weight, ophthalmic, respiratory or neurological examinations, clinical pathology (coagulation, clinical chemistry, or urinalysis), organ weight or macroscopic evaluations. No luxeptinib-related cardiovascular effects were noted in the 28-day GLP toxicology study or in a separate preclinical cardiovascular safety study. |
| · | On October 24, 2019, we presented preclinical data in a poster presentation at the 5th International Conference on Acute Myeloid Leukemia “Molecular and Translational” Advances in Biology and Treatment in Estoril, Portugal. The poster, CG-806 Pan-FLT3/Pan-BTK Inhibitor Simultaneously Suppresses Multiple Oncogenic Signaling Pathways to Treat AML, highlighted that luxeptinib acts on large xenograft tumors with no evidence of drug resistance and with no observed toxicity, enhances killing of patient-derived AML and B-cell cancer cells when combined with venetoclax, and retains activity in patient-derived AML cells even when cells harbor mutations of FLT3, IDH-1, NPM1, ASXL1 or p53. |
• | · | On December 8 and 9, 2019, we presented new preclinical data in two separate poster presentations at the 61st ASH Annual Meeting. On December 8, 2019, the poster CG-806, a First-in-Class Pan-FLT3/Pan-BTK Inhibitor, Exhibits Broad Signaling Inhibition in Chronic Lymphocytic Leukemia Cells compared luxeptinib and ibrutinib, the standard of care, on primary patient cells of CLL highlighting that CG-806 broadly inhibits B-cell receptor signaling in CLL cells, resulting in CLL cell apoptosis and reduced proliferation, luxeptinib is more potent than ibrutinib in inducing apoptosis of MEC1 CLL cells and, finally, luxeptinib targets elements of the CLL microenvironment, and thereby potentially targets pro-survival signals from the microenvironment. The poster presented on December 9, 2019 titled Synergistic Targeting of BTK and E-Selectin/CXCR4 in the Microenvironment of Mantle Cell Lymphomas,explored the effects of luxeptinib on cells of MCL, a rare subtype of aggressive B cell non Hodgkin lymphoma that is incurable with standard therapy, and investigated the molecular mechanisms of acquired resistance to treatment, highlighted that luxeptinib demonstrated superior anti-lymphoma effects compared with ibrutinib, exerting potent cell growth inhibitory effects on ibrutinib-resistant MCL cells, luxeptinib suppresses phospho-BTK, -Stat3, -AKT, -ERK, -Src, NF-kB, and the anti-apoptotic protein Mcl1, while upregulating p53, luxeptinib increased autophagy in MCL cells, which may be associated with resistance to luxeptinib-mediated apoptosis. Inhibition of autophagy re-sensitizes MCL cells to luxeptinib-induced apoptosis, luxeptinib treatment upregulates CXCR4/E-selectin levels in MCL cells and finally, combination of CXCR4/E-selectin antagonists with luxeptinib enhances luxeptinib-induced apoptotic killing of MCL cells in the presence of the tumor microenvironment. On December 7, 2019, Aptose also hosted a corporate event and clinical update, where the company’s management and invited Key Opinion Leaders highlighted some early clinical observations on safety, tolerability, pharmacokinetics, and activity, including. The discussion focused on key findings from dose levels one and two of luxeptinib in heavily pretreated R/R CLL patients, including: the clean safety profile to date, with no myelosuppression, drug-related adverse events or dose-limiting toxicity observed; meaningful oral absorption and predictable pharmacokinetic (“PK”) profile; evidence of target engagement manifesting as inhibition of Phospho-BTK, Phospho-SYK and Phospho-ERK in a plasma inhibitory assay (“PIA”) using plasma from the CLL patient on dose level two, and early evidence of clinical activity in the same patient manifesting as increase in peripheral blood lymphocytes (lymphocytosis), typically associated with BTK inhibition. |
| · | On April 27, 2020, we presented the early clinical data on luxeptinib at the AACR Virtual Annual Meeting I in lieu of the live oral presentation originally planned. A video summary of Abstract # 9967 - Early clinical findings from a Phase 1a/b dose escalation trial to evaluate the safety and tolerability of CG-806 in patients with relapsed or refractory CLL/SLL or non-Hodgkin’s lymphomasOn April 27, 2020, we presented the early clinical data on luxeptinib at the AACR Virtual Annual Meeting I in lieu of the live oral presentation originally planned. A video summary of Abstract # 9967 - (Early clinical findings from a Phase 1a/b dose escalation trial to evaluate the safety and tolerability of CG-806 in patients with relapsed or refractory CLL/SLL or non-Hodgkin’s lymphomas) described the first-in-human tests of luxeptinib which are being carried out in a Phase 1a/b clinical study in patients with significant unmet needs including patients with relapsed or refractory CLL, SLL or NHL who had been failed by or been intolerant to two lines of established therapy. We noted that the second patient, treated at the 300 mg BID dose level, represented a classic CLL patient that developed a brisk lymphocytosis (evidence of BTK target engagement and evidence of pharmacologic activity), and that enrollment was continuing. |
| · | On June 12, 2020, we presented new clinical data on luxeptinib in a poster presentation at the 25th Congress of the EHA. The poster, Early Clinical Findings from a Phase 1 a/b Dose Escalation Trial to Evaluate the Safety and Tolerability of CG-806 in Patients with Relapsed or Refractory CLL/SLL or Non-Hodgkin’s Lymphomas (EHA2020 Abstract# EP711), reviewed luxeptinib data for eight patients (as of the data cut-off date on May 5, 2020) with relapsed or refractory CLL, SLL or NHL in the first in-human Phase 1a/b, open-label, single arm, multicenter dose-escalation clinical study. Data from the ongoing trial demonstrated that luxeptinib was well-tolerated in patients treated at 150 mg, 300 mg, 450 mg BID over multiple cycles, with no dose-limiting toxicities or serious adverse events observed, supporting continued dose escalation. Luxeptinib treatment achieved human steady state PK levels known to be effective in murine tumor models and led to complete inhibition of phospho-BTK and multiple CLL survival pathways. Luxeptinib treatment also led to lymphocytosis in both classic CLL patients entering study with elevated lymphocyte counts and led to complete inhibition of phospho-FLT3, suggesting that dose levels evaluated in this study may be therapeutic in patients with AML. |
| · | On June 22, 2020, we presented new preclinical data on luxeptinib in a poster presentation at the AACR Virtual Annual II 2020. The poster, CG-806, a First-in-Class FLT3/BTK Inhibitor, and Venetoclax Synergize to Inhibit Cell Proliferation and to Induce Apoptosis and Aggressive B-cell Lymphomas, illustrated how luxeptinib simultaneously inhibits the driver BCR pathway and PI3K/AKT, NFᴋB and MAPK-mediated rescue pathways to kill aggressive double-hit and double-expressor B-cell lymphoma cells. Overall, the presented work provided additional mechanistic evidence to support the clinical development of CG-806 as a single agent or in combination with venetoclax in patients with aggressive B-cell lymphomas harboring unfavorable BCL2/MYC/BCL6 translocations and / or overexpression. |
| · | On December 6, 2020, we presented new clinical data in a virtual poster presentation at the 62nd ASH Annual Meeting. The poster, A Phase 1 a/b Dose Escalation Study of the Mutation Agnostic BTK/FLT3 Inhibitor CG-806 in Patients with Relapsed or Refractory CLL/SLL or Non-Hodgkin’s Lymphomasreviewed luxeptinib data for fourteen patients (as of the cutoff date of November 2, 2020) with relapsed or refractory CLL, SLL or NHL in the first in-human Phase 1a/b, open-label, single arm, multicenter dose-escalation clinical study. Data from the ongoing trial demonstrated that luxeptinib was generally well-tolerated in patients treated at 150 mg, 300 mg, 450 mg, and 600 mg BID over multiple cycles, supporting continued dose escalation. At the ongoing 750 mg dose, luxeptinib achieved steady state plasma concentration greater than 2 micromolar at the of Cycle 1. Luxeptinib treatment also led to modest reductions in patients with different B-cell malignancies. On December 6, 2020, Aptose also hosted a corporate event and clinical update, where the company’s management and invited Key Opinion Leaders highlighted some early clinical observations on safety, tolerability, pharmacokinetics, and activity, including. The discussion focused on key findings from dose levels one and two of luxeptinib in heavily pretreated R/R CLL patients, including: the clean safety profile to date, with no myelosuppression, drug-related adverse events or dose-limiting toxicity observed; meaningful oral absorption and predictable PK profile; evidence of target engagement manifesting as inhibition of Phospho-BTK, Phospho-SYK and Phospho-ERK in a PIA using plasma from the CLL patient on dose level two, and early evidence of clinical activity in the same patient manifesting as increase in peripheral blood lymphocytes (lymphocytosis), typically associated with BTK inhibition. |
APTO-253
APTO-253, a small molecule inhibitor of MYC gene expression, is being evaluated by Aptose in a Phase 1a/b clinical trialstudy in patients with R/R hematologic malignancies, particularly R/R AMLsignificant unmet needs including patients with relapsed or refractory CLL, SLL or NHL who had been failed by or been intolerant to two lines of established therapy. We noted that the second patient, treated at the 300 mg BID dose level, represented a classic CLL patient that developed a brisk lymphocytosis (evidence of BTK target engagement and high-risk MDS.evidence of pharmacologic activity), and that enrollment was continuing.
•On June 12, 2020, we presented new clinical data on luxeptinib in a poster presentation at the 25th Congress of the EHA. The poster, Early Clinical Findings from a Phase 1b,1 a/b Dose Escalation Trial to Evaluate the Safety and Tolerability of CG-806 in Patients with Relapsed or Refractory CLL/SLL or Non-Hodgkin’s Lymphomas (EHA2020 Abstract# EP711), reviewed luxeptinib data for eight patients (as of the data cut-off date on May 5, 2020) with relapsed or refractory CLL, SLL or NHL in the first in-human Phase 1a/b, open-label, single arm, multicenter open-label, dose-escalation clinical study. Data from the ongoing trial demonstrated that luxeptinib was well-tolerated in patients treated at 150 mg, 300 mg, 450 mg BID over multiple cycles, with no dose-limiting toxicities or serious adverse events observed, supporting continued dose escalation. Luxeptinib treatment achieved human steady state PK levels known to be effective in murine tumor models and led to complete inhibition of APTO-253 is designedphospho-BTK and multiple CLL survival pathways. Luxeptinib treatment also led to assesslymphocytosis in both classic CLL patients entering study with elevated lymphocyte counts and led to complete inhibition of phospho-FLT3, suggesting that dose levels evaluated in this study may be therapeutic in patients with AML.
•On December 6, 2020, we presented new clinical data in a virtual poster presentation at the 62nd ASH Annual Meeting. The poster, A Phase 1 a/b Dose Escalation Study of the Mutation Agnostic BTK/FLT3 Inhibitor CG-806 in Patients with Relapsed or Refractory CLL/SLL or Non-Hodgkin’s Lymphomas reviewed luxeptinib data for fourteen patients (as of the cutoff date of November 2, 2020) with R/R CLL, SLL or NHL in the first in-human Phase 1a/b, open-label, single arm, multicenter dose-escalation clinical study. Data from the ongoing trial demonstrated that luxeptinib was generally well-tolerated in patients treated at 150 mg, 300 mg, 450 mg, and 600 mg BID over multiple cycles, supporting continued dose escalation. At the ongoing 750 mg dose, luxeptinib achieved steady state plasma concentration greater than 2 micromolar at the of Cycle 1. Luxeptinib treatment also led to modest reductions in patients with different B-cell malignancies. On December 6, 2020, Aptose also hosted a corporate event and clinical update, where the company’s management highlighted some early clinical observations on safety, tolerability, pharmacokinetics and pharmacodynamic responsesactivity from the Phase 1a/b study in B-cell malignancies as well as from the recently initiated Phase 1a/b study in AML. On June 11, 2021, we presented clinical data on luxeptinib in a poster presentation at the EHA June 2021 Congress. With luxeptinib in heavily pretreated B-cell cancer patients we presented that many of the patients rapidly progressed immediately before luxeptinib treatment was initiated, resulting in a trend of tumor growth early in treatment, often followed by tumor reductions. We observed dose-dependent anti-leukemic activity to luxeptinib in patients who received dose escalation, including one follicular lymphoma patient who experienced tumor growth while on 450mg BID and efficacy of APTO-253 as aupon dose escalation to 600mg BID the patient experienced 43% tumor reduction from peak (12% from baseline). In that patient, luxeptinib was well-tolerated with single agent activity for the duration of 16+ cycles of therapy. In addition, one CLL patient and determineone WM patient reported >25% tumor volume reduction.
•On June 11, 2021, during a virtual corporate update event we provided updated clinical findings with luxeptinib for the recommendedtreatment of patients with relapsed or refractory AML. We presented dose-dependent inhibition of phospho-FLT3, -BTK, -SYK, and -PDGFRα signaling and that all three R/R-AML patients with FLT3-ITD mutations who received 450mg BID luxeptinib (the lowest dose) for 28 days experienced blast reductions. Two patients experienced blast reduction of 67-90% but later experienced disease progression. However, one patient who failed chemotherapy twice, failed prior FLT3 inhibitor therapy, failed venetoclax and decitabine treatment and failed AHSC transplants twice, achieved a MRD-negative CR with monotherapy of 450mg BID luxeptinib.
•On December 11, 2021, we presented clinical updates from luxeptinib in patients with relapsed or refractory B-cell malignancies and relapsed or refractory AML in two virtual poster presentations at the 63rd ASH Annual Meeting (A Phase 2 dose. APTO-253 is being administered once weekly, over a 28-day cycle. The dose escalation stage1 a/b Dose Escalation Study of the study could potentially enroll up to 20Mutation Agnostic BTK/FLT3 Inhibitor Luxeptinib (CG-806) in Patients with Relapsed or Refractory B-Cell Malignancies; A Phase 1 a/b Dose Escalation Study of the Mutation Agnostic FLT3/BTK Inhibitor Luxeptinib (CG-806) in Patients with Relapsed or Refractory Acute Myeloid Leukemia). The presentations highlighted that in both of these Phase 1/2 studies luxeptinib has been generally well tolerated at dose levels of 450, 600 and 750 mg BID over multiple cycles, and is currently being dosed in 900 mg BID cohorts in parallel. Target engagement of BTK and FLT3, and anti-tumor activity, including dose- and exposure-dependent tumor reductions, have been observed in multiple patients collectively between the studies, including in patients with R/R AML or high-risk MDS.FL, DLBCL, CLL/SLL, and AML.
Other corporate matters
Nasdaq Notice and Reverse Stock Split
On July 18, 2022, we received a letter from the Nasdaq Stock Market, LLC (“Nasdaq”) indicating that, for the last 30 consecutive business days, the bid price for our Common Shares had closed below the minimum $1.00 per share required for continued inclusion on the Nasdaq Capital Market under the Nasdaq Listing Rules. The study is designednotice had no effect on the listing of our Common Shares.
On May 23, 2023, our shareholders voted to approve special resolutions providing for an amendment to our articles of incorporation to effect the Reverse Stock Split at a ratio in the range of 1-for-10 to 1-for-20. Our Board of Directors then transition, as appropriate,approved a ratio of 1-for-15 on May 23, 2023. On May 24, 2023, we filed articles of amendment under the CBCA to single-agent expansion cohortsgive effect to the Reverse Stock Split (consolidation) of our Common Shares on the basis of one post-consolidation Common Share for each 15 pre-consolidation Common Shares. The Common Shares commenced trading on a post-Reverse Stock Split basis at market open on Tuesday, June 6, 2023. The Reverse Share Split was primarily intended to bring us into compliance with the minimum bid price requirement for maintaining the listing of the Common Shares on Nasdaq and to make the bid price more attractive to investors. On June 21, 2023, Nasdaq confirmed that we had regained compliance with the minimum bid price requirement. All references in R/R AML and/or high-risk MDS.this report to historical Common Share prices, numbers of Common Shares, and earnings per share calculations have been presented to reflect the effect of the Reverse Stock Split.
AsOn February 29, 2024, we received a deficiency letter (the “2024 Deficiency Letter”) from the Nasdaq Listing Qualifications Department of The Nasdaq Stock Market LLC (“Nasdaq”) notifying the Company that the Company’s January 2024 Private Placement of securities to Hanmi violated 5635(d) because the Company did not obtain shareholder approval prior to such issuance. Nasdaq stated that the Private Placement involved the issuance of greater than 20% of the issued and outstanding Common Shares of the Company at a discount to the Nasdaq official closing price on January 25, 2024, the date of this report, we have multiple active sites recruiting patients in the dose escalation stagesubscription agreement between the Company and Hanmi. The 2024 Deficiency Letter has no immediate effect on the listing of the trial. AsCompany’s Common Shares. In accordance with the Nasdaq Listing Rules, the Company has been given forty-five (45) calendar days, or until April 14, 2024, to submit a plan to regain compliance. If Nasdaq accepts the Company’s plan to regain compliance, Nasdaq can grant an extension of up to 180 calendar days from the date of this report, we havethe Deficiency Letter to evidence compliance. Although the Company believes that the Private Placement was completed enrollmentin accordance with the Nasdaq Listing Rules, the Company respects Nasdaq’s query and treatment of patients onintends to work with Nasdaq to resolve Nasdaq’s concerns and will consider available options to regain compliance. However, there can be no assurance that the first, second, third and fourth dose levels (20, 40, 66, and 100 mg/m2, respectively). Under an FDA-approved accelerated titration protocol, only one patient was required at each ofCompany will be able to regain compliance with the first two dose levels, followed by three patients at each dose level thereafter. Aptose is currently enrolling and treating patients in the fifth dose level (150 mg/m2) of APTO-253. During the second quarter of 2020, the FDA allowed an amendment for Aptose to initiate more aggressive dose escalations with APTO-253, provided the tolerability profile remains favorable. The first four dosing cohorts have enrolled a mix of patients with AML and MDS. To date, we have observed meaningful reductions in MYC expression in peripheral blood mononuclear cells (PBMCs) from treated patients with AML and MDS, demonstrating MYC target engagement and mechanistic proof of concept in different indications.applicable Nasdaq Listing Rules.
Manufacturing:
We are continuing to manufacture additional drug substance and drug product for use in the ongoing trial.
We are exploring additional drug delivery methods for APTO-253 and plan to initiate additional non-clinical studies for solid tumor and hematologic cancer development. As preparing, submitting, and advancing applications for regulatory approval, developing drugs and drug product and clinical trials are sometimes complex, costly, and time-consuming processes, an estimate of the future costs is not reasonable at this time.
Preclinical data presented at scientific forums are as follows:
| · | On April 17, 2018, at the 2018 Annual Meeting of the AACR, we presented preclinical data demonstrating that APTO-253 is a new addition to the repertoire of drugs that can exploit DNA BRCA1/2 deficiency, broadening the potential applicability of APTO-253 towards solid cancer indications. |
| · | On June 4, 2018, we announced that preclinical data elucidating the mechanism of action of APTO-253 were published in two separate articles in the June 2018 issue (Volume 17, Number 6) of Molecular Cancer Therapeutics, a peer-reviewed journal of the AACR. The most important finding disclosed in the published articles is the ability of the APTO-253 small molecule to bind to and stabilize a G-quadruplex DNA motif found in the promoter regulatory region of the MYC oncogene and to inhibit expression of the MYC gene, thereby depleting the cells of the MYC oncoprotein and leading to cancer cell death. These findings make APTO-253 the only clinical stage molecule that can directly target the MYC gene and inhibit its expression. |
| · | On April 1, 2019, at the 2019 Annual Meeting of the AACR, we presented in vitro studies that further define the mechanism of action of APTO-253. Researchers found that APTO-253 targets a G-quadruplex motif in the P1/P2 promoter region of the MYC gene and inhibits MYC gene expression to induce apoptosis, resulting in its ability to potently kill hematologic malignant cell lines and primary samples from AML and CLL patients. In this study, researchers performed long-term in vitro studies to determine if and how cells might develop resistance to APTO-253. MYC driven Raji cells required three years in increasing concentrations of APTO-253 in order to adopt multiple modifications and develop high level resistance to APTO-253. These modifications include up-regulation of the ABCG2 transporter, acquisition of a more stable MYC protein lacking the conserved core sequence of MYC Box III generated by deletion of an internal region of the MYC gene exon 2, and utilization of alternate P3 promoter not inhibited by G4 binding and stabilization. Importantly, these studies confirmed the MYC gene as a target of APTO-253. |
| · | On December 6, 2020, we presented new clinical data in a virtual poster presentation at the 62nd ASH Annual Meeting. The poster, A Phase 1a/b Dose Escalation Study of the MYC Repressor APTO-253 in Patients with Relapsed or Refractory AML or Higher-risk MDS reviewed APTO-253 data for 10 patients with relapsed or refractory AML and MDS at 20 mg/m2, 40 mg/m2, 66 mg/m2 and 100 mg/m2 once weekly over multiple cycles. APTO-253 demonstrated MYC reduction in 5 out of 6 patients 24 hours after dosing C1D1 providing proof of concept that APTO-253 is a MYC repressor. APTO-253 was well tolerated with no dose-limiting toxicities or serious adverse events observed, supporting continued dose escalation. |
While as of the date of this report we have not experienced any material delays in initiating our luxeptinib Phase 1 clinical study in AML due to COVID-19, we are conducting site initiation visits remotely which could result in delays in site activations and negatively impact this trial. Additionally, COVID-19 could negatively impact patient enrollment if our clinical sites are unable to enroll patients due to either a lack of administrative resources at their sites or decisions made at the clinical sites to limit patient exposure to COVID-19.
As of the date of this report, we have not experienced material delays in the manufacturing of luxeptinib or APTO-253 related to COVID-19. Should our manufacturers experience shortages in staffing or be required to shut down their facilities due to COVID-19 for an extended period of time, our trials may be negatively impacted.
LIQUIDITY AND CAPITAL RESOURCES
We are an early stageearly-stage development company and we currently do not earn any revenues from our drug candidates. The continuation of our research and development activities and the commercialization of the targeted therapeutic products are dependent upon our ability to successfully finance and complete our research and development programs through a combination of equity financing and payments from strategic partners. We have no current sources of significant payments from strategic partners. As of the filing date, we have sufficient liquidity to support the Company's operations until August 2024.
Sources of liquidity:
The following table presents our cash and cash equivalents, investments and working capital as atof December 31, 20202023 and 2019.2022.
| | (in thousands) | | Balances at
December 31, 2020 | | Balances at
December 31, 2019 | | Balances at December 31, 2023 | | | Balances at December 31, 2022 | |
| Cash and cash equivalents | | $ | 117,393 | | | $ | 79,842 | | | $ | 9,252 | | | $ | 36,970 | |
| Investments | | | 5,000 | | | | 17,758 | | | | — | | | | 9,989 | |
| Total | | $ | 122,393 | | | $ | 97,600 | | | $ | 9,252 | | | $ | 46,959 | |
| | | | | | | | | |
| Working capital | | | 118,264 | | | | 93,227 | | | $ | (3,375 | ) | | $ | 37,235 | |
Working capital is a non-GAAP measure and represents primarily cash, cash equivalents, investments, prepaid expenses and other current assets less current liabilities.
We believe that our cash, cash equivalents and investments on hand atAs of December 31, 20202023, we reported negative shareholder's equity of $2.9 million (December 31, 2022, positive shareholder's equity of $37.7 million). In order for the Company to meet its capital requirements, and continue to operate, additional financing will be sufficientnecessary. The Company is evaluating strategies to financeobtain the required additional funding for future operations. These strategies may include, but are not limited to, obtaining equity financing, and restructuring of operations to decrease expenses. However, given the challenges in the U.S. and global financial markets, and the matter in Note 17 to the Financial Statements, Subsequent events, that may impact the Company's ability to raise financing in the capital markets, the Company may be unable to access further equity or when needed, if at all. As the Company is primarily pursuing one compound that is licensed from a related party with significant licensing payments who will have influence on the Company, other investors may not be willing to invest in the Company. As such, there can be no assurance that the Company will be able to obtain additional liquidity when needed or under acceptable terms, if at all. The consolidated financial statements do not reflect any adjustments to the carrying amounts and classification of assets, liabilities, and reported expenses that may be necessary if the Company were unable to continue as a going concern. Such adjustments may be material. If necessary funds are not available, we may have to delay, reduce the scope of, or eliminate some of our operationsdevelopment programs, potentially delaying the time to market for at least 12 months from the issuance dateany of these financial statements. our product candidates.
Our cash needs for the next twelve months include estimates of the number of patients and rate of enrollment of our clinical trials, the amount of drug product that we will require to support our clinical trials, and our general corporate overhead costs to support our operations, and our reliance on our manufacturers. We have based these estimates on assumptions and plans which may change and which could impact the magnitude and/or timing of operating expenses and our cash runway.
Since our inception, we have financed our operations and technology acquisitions primarily from equity financing, proceeds from the exercise of warrants and stock options, and interest income on funds held for future investment.
Hanmi Investment September 2023
On July 20, 2020 and August 10, 2020,2023, the Company completedentered into a confidentially marketed public offering (“CMPO”),binding term sheet with Piper Sandler & Co. (“Piper Sandler”) asHanmi whereby Hanmi agreed to invest, at their sole discretion, up to a maximum of $7 million in Aptose up to a total ownership of 19.99 percent of Aptose by Hanmi. On September 6, 2023, the representative ofCompany entered into a subscription agreement with Hanmi, pursuant to which the underwriters, through the issuance of, in the aggregate, 11,854,472Company sold 668,449 Common Shares to Hanmi for gross proceeds of $62.2$3 million. Hanmi held 884,152 Common Shares of Aptose as of December 31, 2023. Hanmi subsequently invested $4 million (approximately $58.2in Aptose as noted below. See below, "January 2024 Public Offering Financing and Concurrent Hanmi Investment." In addition, see Note 17 to the Financial Statements, Subsequent events.
2023 Committed Equity Facility
On May 25, 2023, the Company and Keystone Capital Partners, LLC ("Keystone") entered into a committed equity facility, (the "2023 Committed Equity Facility"), which provides that subject to the terms and conditions set forth therein, we may sell to Keystone up to the lesser of (i) $25.0 million of the Common Shares and (ii) a number of Common Shares equal to 19.99% of the Common Shares outstanding immediately prior to the execution of the 2023 Committed Equity Facility Agreement with Keystone which respect to the 2023 Committed Equity Facility (subject to certain exceptions) (the “Total Commitment”), from time to time during the 24-month term of the 2023 Committed Equity Facility. Additionally, on May 25, 2023, the Company entered into a Registration Rights Agreement with Keystone, pursuant to which the Company agreed to file a registration statement with the SEC covering the resale of Common Shares that are issued to Keystone under the 2023 Committed Equity Facility. This registration statement became effective on June 30, 2023 and the 2023 Committed Equity Facility commencement date was July 12, 2023 (the "Commencement Date").
Upon entering into the 2023 Committed Equity Facility, the Company agreed to issue to Keystone an aggregate of 25,156 Common Shares (the “Commitment Shares”) as consideration for Keystone’s commitment to purchase Common Shares upon the Company’s direction under the 2023 Committed Equity Facility. The Company issued 7,547 Common Shares, or 30% of the Commitment Shares, on the date of the 2023 Committed Equity Facility Agreement (the “Initial Commitment Shares”). An additional 7,547 Common Shares, or 30% of the Commitment Shares, were issued to Keystone 90 days following the Commencement Date (the “First Back-End Commitment Shares”). The remaining 10,062 Common Shares, or 40% of the Commitment Shares, were issued to Keystone subsequent to December 31, 2023 (the “Second Back-End Commitment Shares”). See Note 17 to the Financial Statements, Subsequent events.
In the year ended December 31, 2023, the Company's sales of Common Shares to Keystone comprised 720,494 Common shares sold to Keystone at an average price of $2.91 per Common share for gross cash proceeds of $2.1 million. In addition, 15,094 Commitment Shares were issued in 2023 and the remaining 10,062 Commitment Shares were issued in January 2024.
January 2024 Public Offering Financing and Concurrent Hanmi Investment
On January 31, 2024, the Company announced the closing of a $9.7 million public offering (the “Public Offering”) and a $4 million private placement (the “Private Placement”) with Hanmi Pharmaceutical. The Public Offering comprised 5,649,122 Common Shares and warrants at a combined offering price of US $1.71 per share. This included 736,842 Common Shares and warrants pursuant to a full exercise by the underwriter of its over-allotment option (the “Over-Allotment Option”). The Private Placement with Hanmi comprised 2,105,263 Common Shares of the Company sold at a price of $1.90, representing an 11% premium over the price of the Common Shares issued as part of the Public Offering. Financing costs included underwriting costs of 7% and approximately $400,000 in professional fees. The Company also issued Hanmi warrants to purchase Common Shares at an exercise price of $1.71 per Warrant Share. The total number of Common Shares outstanding after the closing of the Public Offering, including the Over-Allotment Option, and Private Placement was 15,706,810 and warrants outstanding were 8,332,163. As of the filing date, the total Common Share ownership by Hanmi was 2,989,414, representing ownership of 19.03% of the outstanding Common Shares of the Company. Hanmi also owns 2,339,181 warrants to purchase Common Shares at an exchange price of $1.71 per Warrant Share.
At-The-Market Facilities
On December 9, 2022, the Company entered into an equity distribution agreement pursuant to which the Company may, from time to time, sell Common Shares having an aggregate offering value of up to $50 million through Jones Trading Institutional Services LLC ("Jones Trading") on Nasdaq (the "2022 ATM Facility"). During the year ended December 31, 2022, the Company issued 4,836 Common Shares under this 2022 ATM Facility at an average price of $10.81 per Common Share for gross proceeds of $52 thousand ($51 thousand net of share issuance costs). During the year ended December 31, 2023, the Company issued 336,690 Common Shares under this 2022 ATM Facility at an average price of $5.62 for gross proceeds of $1.9 million ($1.8 million net of share issueissuance costs). As of December 31, 2023, the Company had raised a total of $1.9 million gross proceeds ($1.9 million net of share issuance costs) under the 2022 ATM Facility. Costs associated with the proceeds consisted of 3% cash commission.
On May 5, 2020, the Company entered an “at-the-market” equity distribution agreement with Piper Sandler & Co. (“Piper Sandler”) and Canaccord Genuity LLC (“Canaccord Genuity”) acting as co-agents (the “2020 ATM Facility”). Under the terms of the 2020 ATM Facility, the Company may,could, from time to time, sell Common Shares having an aggregate offering value of up to $75 million through Piper Sandler and Canaccord Genuity on Nasdaq. During the Nasdaq Capital Market.period from January 1, 2022 to October 31, 2022, the date the Agreement was terminated, the company issued During the year ended December 31, 2020,2022, the Company did not issue any sharesissued 3,646 Common Shares under the 2020 ATM Facility.
During the year ended December 31, 2019, the Company completed two CMPOs, with RBC Capital Markets, LLC (“RBC Capital Markets”) and Canaccord Genuity, as representativesFacility at an average price of the underwriters, and Piper Jaffray & Co (now Piper Sandler) as the representative of the underwriters, respectively, through the issuance of, in the aggregate, 30,043,750$14.25 per Common SharesShare for aggregate gross proceeds of $95.45 million (approximately $88.18 million$52 thousand ($50 thousand net of share issueissuance costs). TheAs of October 31, 2022, the date the 2020 ATM Facility was terminated, the Company alsohad raised capital pursuant to two separate share purchase agreements with Aspire Capital Fund, LLC (“Aspire Capital”) through the issuancea total of an aggregate of 7,302,433 Common Shares for aggregate$89 thousand gross proceeds ($86 thousand net of $14.4 million. We do not expect that COVID-19 will haveshare issuance costs) under the 2020 ATM Facility. Costs associated with the proceeds consisted of a significant impact on our liquidity and capital resources and we are not incurring significant additional costs to support our ongoing operations during this time. We have not entered into long term manufacturing contracts and should there be a delay in our trials we have flexibility to reduce future planned manufacturing campaigns without incurring additional costs.3% cash commission.
2022 Base Shelf
We expect that we will need to raise additional capital or incur indebtedness to continue to fund our operations in the future. In December 2019,October 2022, we filed a short form base shelf prospectus (the 2022 “Base Shelf”) that allows us to distribute, upon the filing of prospectus supplements, up to $200,000,000 of Common Shares, warrants, or units comprising any combination of Common Shares and warrants. The Base Shelf was declared effective by the SEC on January 9, 2020October 21, 2022 and expires on January 9, 2023.October 7, 2025.
Our ability to raise additional funds could be affected by adverse market conditions, the status of our product pipeline, possible delays in enrollment in our trial related to COVID-19, and various other factors and we may be unable to raise capital when needed, or on terms favorable to us. If the necessary funds are not available, we may need to delay, reduce the scope of, or eliminate some of our development programs, potentially delaying the time to market for any of our product candidates. As of the filing date, we have sufficient liquidity to support the Company's operations until August 2024.
Cash flows:
The following table presents a summary of our cash flows for the years ended December 31, 20202023 and 2019:2022:
| | | | For the Years Ended, | | For the Years Ended, | |
| (in thousands) | | December 31,2020 | | December 31, 2019 | | December 31, 2023 | | | December 31, 2022 | |
| | | | | |
| | | | | |
| Net cash provided by (used in): | | | | | | | | | | | | | |
| Operating activities | | $ | (33,891 | ) | | $ | (21,558 | ) | | $ | (44,590 | ) | | $ | (32,322 | ) |
| Investing activities | | | 12,628 | | | | (17,370 | ) | | | 9,960 | | | | 30,066 | |
| Financing activities | | | 58,807 | | | | 103,448 | | | | 6,910 | | | | 116 | |
| Effect of exchange rates changes on cash and cash equivalents | | | 7 | | | | 23 | | | | 2 | | | | (4 | ) |
| Net increase in cash and cash equivalents | | $ | 37,551 | | | $ | 64,543 | |
Net (decrease)/increase in cash and
cash equivalents | | | $ | (27,718 | ) | | $ | (2,144 | ) |
Cash used in operating activities:
Our cash used fromin operating activities for the years ended December 31, 20202023 and 20192022 was approximately $33.9$44.6 million and $21.6$32.3 million, respectively. Net cash used in operating activities was higher in the year ended December 31, 2020 as compared with the year ended December 31, 2019 resulting mostly from a higher net loss in the current year. See “Results of Operations”. Our uses of cash for operating activities for both years consisted primarily of salaries and wages for our employees, facility and facility-related costs for our offices and laboratories, fees paid in connection with preclinical and clinical studies, drug manufacturing costs, laboratory supplies and materials, and professional fees. Net cash used in operating activities was higher in the year ended December 31, 2023 as compared with the year ended December 31, 2022 resulting mostly from higher operating expenses, as discussed further below. See “Results of Operations.”
We do not expect to generate positive cash flow from operations for the foreseeable future due to additional research and development costs, including costs related to drug discovery, preclinical testing, clinical trials, and manufacturing, as well as operating expenses associated with supporting these activities, and potential milestone payment to our collaborators. It is expected that negative cash flow will continue until such time, if ever, that we receive regulatory approval to commercialize any of our products under development and/or royalty or milestone revenue from any such products exceeds expenses.
Cash flow from investing activities:
Our cash provided by investing activities for the yearyears ended December 31, 20202023 and December 31, 2022 was $12.6$10.0 million and consisted$30.1 million, consisting mainly of maturitiesproceeds from the maturity of investments of $12.7 million and purchases of property and equipment of $79 thousand. Our cash used by investing activities for the year ended December 31, 2019 was $17.4 million, and consisted of net purchases of investments of $17.3 million and purchases of property and equipment of $102 thousand.
investments.
The composition and mix of cash, cash equivalents and investments is based on our evaluation of conditions in financial markets and our near-term liquidity needs. We have exposure to credit risk, liquidity risk and market risk related to our investments. The Company manages credit risk associated with its cash and cash equivalents and investments by maintaining minimum standards of R1-lowR1‑low or A-lowA‑low investments. The Company invests only in highly rated corporations and treasury bills which are capable of prompt liquidation. The Company manages its liquidity risk by continuously monitoring forecasts and actual cash flows. The Company is subject to interest rate risk on its cash and cash equivalents and investments. The Company does not believe that the results of operations or cash flows would be affected to any significant degree by a sudden change in market interest rates relative to interest rates on the investments, owing to the relative short-termshort‑term nature of the investments.
Cash flow from financing activities:
Our cash flow from financing activities for the year ended December 31, 20202023 was approximately $58.8$6.9 million, and consisted mostly of $3.0 million in proceeds from shares issued to Hanmi, $2.1 million in proceeds from the CMPO we completed in JulyCommitted Equity Facility, $1.8 million from shares issued from the 2022 ATM facility, and August$29 thousand from the issuance of shares under the ESPP plan.
Our cash flow from financing activities for the year ended December 31, 2022 was approximately $116 thousand, and consisted of proceeds from the from shares issued from the 2022 ATM Facility of approximately $51 thousand, proceeds from shares issued from the 2020 as described aboveATM Facility of $50 thousand, and of proceeds from the exercise of stock options of $573$15 thousand. Net cash provided by financing activities in the year ended December 31, 2019 reflects mostly:
| i) | 18,543,750 Common Shares issued pursuant to the CMPO we completed in December 2019 with Piper Jaffray & Co. (now Piper Sandler) as representatives of the underwriters, for net proceeds of approximately $68.6 million, |
| ii) | 11,500,000 Common Shares issued pursuant to the CMPO we completed in June 2019 with RBC Capital Markets and Canaccord Genuity, as representatives of the underwriters, for net proceeds of approximately $19.6 million, |
| iii) | 1,800,000 shares issued to Aspire Capital pursuant to the 2019 Aspire Purchase Agreement, as described below, for net proceeds of approximately $4.4 million, |
| iv) | 5,502,433 shares issued to Aspire Capital pursuant to the 2018 Aspire Purchase Agreement, as described below, for net proceeds of approximately $10 million, |
| v) | 77,349 shares issued pursuant to the 2018 ATM Facility with Cantor Fitzgerald, as described below, for net proceeds of approximately $178 thousand, and |
| vi) | proceeds from the exercise of stock options of $718 thousand. |
At-The-Market Facilities
On May 5, 2020, the Company entered into an equity distribution agreement with Piper Sandler and Canaccord Genuity acting as co-agents in connection with the 2020 ATM Facility. Under the terms of the 2020 ATM Facility, the Company may, from time to time, sell Common Shares having an aggregate offering value of up to $75 million through Piper Sandler and Canaccord Genuity on the Nasdaq Capital Market. During the year ended December 31, 2020, the Company did not issue any shares under the 2020 ATM Facility.
On May 24, 2019, we entered an at-the-market equity facility (the “2019 ATM Facility”) with Piper Jaffray & Co. (now Piper Sandler) and Canaccord Genuity, acting as co-agents. The 2019 ATM Facility replaced the previous facility that we had entered into with Cantor Fitzgerald (the “2018 ATM Facility”). The 2019 ATM Facility allowed us to instruct our co-agents to offer up to approximately 20.2 million Common Shares, having an aggregate offering value of up to $40.0 million, at the prevailing market price from time to time. The Company did not issue any shares under the 2019 ATM Facility, and on December 16, 2019, the Company terminated the 2019 ATM Facility.
On March 27, 2018, the Company entered into an equity distribution agreement with Cantor Fitzgerald acting as sole agent in connection with the 2018 ATM Facility. Under the terms of the 2018 ATM Facility, the Company was allowed, from time to time, to sell Common Shares having an aggregate offering value of up to $30 million through Cantor Fitzgerald on the Nasdaq Capital Market. During the year ended December 31, 2019, the Company issued 77,349 shares under the 2018 ATM Facility at an average price of $2.37 for gross proceeds of $183 thousand ($178 thousand net of share issue costs). On a cumulative basis to December 31, 2019, the Company had raised a total of $11.2 million gross proceeds ($10.9 million net of share issue costs) under the 2018 ATM Facility. The Company terminated this agreement on May 24, 2019.
Common Share Purchase Agreements
On May 30, 2018, the Company entered into a common share purchase agreement to sell up to $20.0 million of Common Shares to Aspire Capital over approximately 30 months (the “2018 Aspire Purchase Agreement”). Pursuant to the terms of this agreement, on June 8, 2018, the Company issued 170,261 Common Shares to Aspire Capital in consideration for entering into the 2018 Aspire Purchase Agreement for a total cost of $600 thousand. During the year ended December 31, 2019, the Company issued 5,502,433 Common Shares to Aspire Capital pursuant to the 2018 Aspire Purchase Agreement at an average price of $1.82 per Common Share for gross and net proceeds of $10 million. On a cumulative basis, the Company raised a total of approximately $11.9 million gross and net proceeds under the 2018 Aspire Purchase Agreement. As of May 24, 2019 the Company had issued 6,409,980 Common Shares, the maximum number of Common Shares issuable under this facility without shareholder approval, and the 2018 Aspire Purchase Agreement was accordingly terminated.
On May 7, 2019, the Company entered into the a common share purchase agreement with Aspire Capital (the “2019 Aspire Purchase Agreement”), which provided that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital was committed to purchase up to an aggregate of $20 million of Common Shares over approximately 30 months. Pursuant to the terms of this agreement, on May 13, 2019, the Company issued 171,428 Common Shares to Aspire Capital in consideration for entering into the 2019 Aspire Purchase Agreement for a total cost of $360 thousand. During the period from May 7, 2019 up to December 16, 2019, the date the 2019 Aspire Purchase Agreement was terminated, the Company issued 1,800,000 Common Shares under the agreement at an average price of $2.43 per Common Share for gross and net proceeds of $4.4 million.
Contractual Obligations and Off-Balance Sheet Financing
As atof December 31, 2020,2023, we have not entered into any off-balance sheet arrangements.
The Company enters into research, development and license agreements in the ordinary course of business where the Company receives research services and rights to proprietary technologies. Milestone and royalty payments that may become due under various agreements are dependent on, among other factors, clinical trials, regulatory approvals and ultimately the successful development of a new drug, the outcome and timing of which is uncertain.
On November 4, 2021, the Company entered into the Tuspetinib Licensing Agreement with Hanmi for exclusive global rights to its compound named tuspetinib. Under the Tuspetinib Licensing Agreement, the Company has maximum obligations for clinical development and global regulatory milestones totaling $64.5 million for the first potential clinical indication of tuspetinib, $34 million for the second indication, and $29 million for the third indication. The Company has maximum obligations for tiered global sales-based milestones totaling $280 million. The Company also has an obligation for tiered royalty payments on global sales of commercialized product. The timing of any milestone or royalty payments that may become due is not yet determinable.
Under the license agreement with CG with regards to the Rights (other than the China Rights), the Company has obligations for development milestones of $16 million related to the initiation of Phase 2 and pivotal clinical trials, and regulatory milestones totaling $44 million. The Company also has an obligation to pay royalty payments on sales of commercialized product. The timing of any milestone or royalty payments that may become due is not yet determinable.
Under the license agreement with CG with regards to the China Rights, we entered into a license agreement with CG to gain an exclusive license to CG-806luxeptinib in China (including the People’s Republic of China, Hong Kong and Macau). The Company has future obligations of development milestones of $6 million related to approval of an IND and to the initiation of Phase 2 and pivotal clinical trials, and regulatory milestones totaling $20 million. The Company also has an obligation to pay sales milestones and royalty payments on sales of commercialized product. The timing of any milestone or royalty payments that may become due is not yet determinable.
RESULTS OF OPERATIONS
A summary of the results of operations for the years ended December 31, 20202023 and 20192022 is presented below:
| | | | Year ended December 31, | | Year ended December 31, | |
| (in thousands except per Common Share data) | | 2020 | | 2019 | | 2023 | | | 2022 | |
| | | | | | | | | | |
| Revenues | | $ | — | | | $ | — | | | $ | - | | | $ | - | |
| Research and development expenses | | | 29,288 | | | | 16,835 | | | | 36,765 | | | | 28,088 | |
| General and administrative expenses | | | 26,480 | | | | 10,022 | | | | 15,591 | | | | 14,514 | |
| Net finance income | | | 530 | | | | 580 | | | | 1,149 | | | | 779 | |
| Net loss | | $ | (55,238 | ) | | $ | (26,277 | ) | | $ | (51,207 | ) | | $ | (41,823 | ) |
| Unrealized gain/(loss) on securities available-for-sale | | | (18 | ) | | | 18 | | | | 2 | | | | (2 | ) |
| Total comprehensive loss | | $ | (55,256 | ) | | $ | (26,259 | ) | | $ | (51,205 | ) | | $ | (41,825 | ) |
| Basic and diluted loss per Common Share | | $ | (0.67 | ) | | $ | (0.52 | ) | | $ | (7.58 | ) | | $ | (6.80 | ) |
Net loss of $55.2$51.2 million for the year ended December 31, 20202023 increased by approximately $28.9$9.4 million as compared with $26.3$41.8 million for the year ended December 31, 2019,2022, primarily as of a result of an increase of $19.1 million in stock-based compensation in the current period, a combined increase inresearch and development program costs and related labor costspersonnel expenses of approximately $9.8$8.7 million, on our luxeptinib development program and higher cash-baseda $1.1 million increase in general and administrative expenses of approximately $549 thousand. These expenses were partially offset by lower costs of $545 thousand on our APTO-253 development programs.
costs.
Research and Development Expenses
Research and development expenses consist primarily of costs incurred related to the research and development of our product candidates. Costs include the following:
•External research and development expenses incurred under agreements with third parties, such as CROs, consultants, members of our scientific advisory boards, external labs and CMOs;
•Employee-related expenses, including salaries, benefits, travel, and stock-based compensation for personnel directly supporting our clinical trials and manufacturing, and development activities;
We have ongoing Phase 1 clinical trials for our product candidates tuspetinib and luxeptinib. Tuspetinib was licensed into Aptose in November 2021 and we assumed sponsorship, and the related costs, of the tuspetinib study effective January 1, 2022. In December 2021, we discontinued the APTO-253 program.
We expect our research and development expenses to be higher for the foreseeable future as we continue to advance tuspetinib into larger clinical trials.
The research and development (“R&D”) expenses for the years ended December 31, 20202023 and 20192022 were as follows:
| | | | Year ended December 31, | | Year ended December 31, | |
| (in thousands) | | 2020 | | 2019 | | 2023 | | | 2022 | |
| | | | | |
| Program costs – luxeptinib | | $ | 16,329 | | | $ | 8,475 | |
Program costs – Tuspetinib | | | $ | 24,925 | | | $ | 10,083 | |
Program costs – Luxeptinib | | | | 3,510 | | | | 8,426 | |
| Program costs – APTO-253 | | | 3,632 | | | | 4,177 | | | | 40 | | | | 141 | |
| Personnel expenses | | | 5,590 | | | | 3,679 | | | | 6,878 | | | | 7,181 | |
| Stock-based compensation | | | 3,720 | | | | 474 | | | | 1,373 | | | | 2,218 | |
| Depreciation of equipment | | | 17 | | | | 30 | | | | 39 | | | | 39 | |
| | | $ | 29,288 | | | $ | 16,835 | | | $ | 36,765 | | | $ | 28,088 | |
R&D expenses increased by $12.5$8.7 million to $29.3$36.8 million for the year ended December 31, 20202023 as compared with $16.8$28.1 million for the comparative period in 2019.2022. Changes to the components of our R&D expenses presented in the table above are primarily as a result of the following activities:
•Program costs for tuspetinib increased by $14.8 million. The higher program costs for tuspetinib in 2023 represent the enrollment of patients in our APTIVATE clinical trial, our healthy volunteer trial, manufacturing activities to support clinical development, and related expenses.
•Program costs for luxeptinib decreased by approximately $4.9 million, primarily due to lower clinical trial costs and lower manufacturing costs as a result of the current formulation requiring less API than the prior formulation.
•Program costs for APTO-253 decreased by approximately $101 thousand due to the Company's decision on December 20, 2021 to discontinue further development of APTO-253.
•Personnel-related expenses decreased by $0.3 million, due to lower headcount in 2023.
•Stock-based compensation decreased by approximately $845 thousand in the year ended December 31, 2023, compared with the year ended December 31, 2022, primarily due to stock options granted with lower grant date fair values in the current period.
| · | Program costs for luxeptinib increased by approximately $7.9 million, mostly as a result of higher manufacturing costs, including costs to scale up manufacturing and research costs associated with optimizing the formulation, higher costs associated with the luxeptinib Phase 1a/b trial and the costs associated with the luxeptinib AML trial. |
| · | Program costs for APTO-253 decreased by approximately $545 thousand, mostly as a result of lower manufacturing costs and lower clinical trial costs related to the APTO-253 Phase 1a/b trial. |
| · | Personnel-related expenses increased by $1.9 million, mostly related to new positions hired since the second quarter of 2019 to support the luxeptinib Phase 1a/b and APTO-253 Phase 1a/b clinical trials and the luxeptinib AML Phase 1a/b clinical trial. |
| · | Stock-based compensation increased by approximately $3.2 million in the year ended December 31, 2020, compared with the year ended December 31, 2019, mostly related to an increase in the number of options granted during the year ended December 31, 2020 and a higher grant date fair value of options as compared with the year ended December 31, 2019, and a higher rate of forfeitures in the comparative period in 2019. |
General and Administrative Expenses
General and administrative expenses consist primarily of salaries, benefits and travel, including stock-based compensation for our executive, finance, business development, human resource, and support functions. Other general and administrative expenses and professional fees for auditing, and legal services, investor relations and other consultants, insurance and facility related expenses.
We expect that our general and administrative expenses will increase for the foreseeable future as we incur additional costs associated with being a publicly traded company and to support our expanding pipeline of activities. We also expect our intellectual property related legal expenses to increase as our intellectual property portfolio expands.
The general and administrative expenses for the years ended December 31, 20202023 and 20192022 are as follows:
| | | | Year ended December 31, | | Year ended December 31, | |
| (in thousands) | | 2020 | | 2019 | | 2023 | | | 2022 | |
| | | | | |
| General and administrative, excluding items below: | | $ | 8,627 | | | $ | 8,078 | | | $ | 13,262 | | | $ | 11,444 | |
| Stock-based compensation | | | 17,718 | | | | 1,822 | | | | 2,280 | | | | 2,989 | |
| Depreciation of equipment | | | 135 | | | | 122 | | | | 49 | | | | 81 | |
| | | $ | 26,480 | | | $ | 10,022 | | | $ | 15,591 | | | $ | 14,514 | |
General and administrative expenses for the year ended December 31, 20202023 were approximately $26.5$15.6 million as compared with $10.0$14.5 million for the comparative period in 2019,2022, an increase of approximately $16.5$1.1 million. The increase was primarily as a result of the following:higher salaries expenses, higher travel expenses, and higher professional fees, partly offset by a decrease in stock-based compensation costs of $709 thousand.
| · | General and administrative expenses, other than stock-based compensation and depreciation of equipment, increased by approximately $549Stock-based compensation decreased by approximately $709 thousand mostly as a result of a lower number of options granted in the year ended December 31, 2020 primarily as a result of higher personnel related costs, higher insurance costs and higher office administrative costs offset by lower financing costs and lower travel expenses. |
| · | Stock-based compensation increased by approximately $15.9 million in the year ended December 31, 2020, compared with the year ended December 31, 2019 mostly related to an increase in the number of restricted share units and options granted during the year ended December 31, 2020, and a higher grant date fair value of options as compared with December 30, 2019. |
COVID-19 did not have a significant impact on our results of operations for the year ended December 31, 2020. We have not experienced and do not foresee material delays to2023, with those options having a lower grant date fair value as compared with the enrollment of patients or timelines for the luxeptinib Phase 1a/b trial due to the variety of clinical sites that we have actively recruited for this trial. Similarly, we do not expect our enrollment of the luxeptinib AML trial to be negatively impacted by COVID-19 as we plan to use a variety of clinical sites for this trial as well. APTO-253, which is administered intravenously, requires the need for hospital / clinical site resources to assist and monitor patients during each infusion and, based on the current conditions caused by COVID-19, future enrollment of patients on this trial is likely to be negatively impacted. As of the date of this report, we have not experienced material delaysoptions granted in the manufacturingcomparative period, and additional compensation recognized in the comparative period for modifications made to then vested and unvested stock options for one former company officer, as part of luxeptinib or APTO-253 related to COVID-19. Should our manufacturers be required to shut down their facilities due to COVID-19 for an extended period of time, our trials may be negatively impacted.a separation and release agreement.
CRITICAL ACCOUNTING POLICIES
Critical Accounting Policies and Estimates
We periodically review our financial reporting and disclosure practices and accounting policies to ensure that they provide accurate and transparent information relative to the current economic and business environment. As part of this process, we have reviewed our selection, application and communication of critical accounting policies and financial disclosures. A “critical accounting policy” is one which is both important to the portrayal of our financial condition and results and requires management’s most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. Management has discussed the development and selection of the critical accounting policies with the Audit Committee of the Board, and the Audit Committee has reviewed the disclosure relating to critical accounting policies in this MD&A. For additional information, please see the discussion of our significant accounting policies in Note 2 to the Financial Statements.
Significant accounting judgments and estimates
Management’s assessment of our ability to continue as a going concern involves making a judgment, at a particular point in time, about inherently uncertain future outcomes and events or conditions. Please see the “Liquidity and Capital Resources” section in this document for a discussion of the factors considered by management in arriving at its assessment.
Other important The critical accounting policies, judgments and estimates made by management are the estimates related to prepaid and accrued R&D activities, the valuation of contingent liabilities, the valuation of tax accounts, and the assumptions used in determining the valuation of share-based compensation.activities.
Research and Development Activities:
R&D costs are expensed as incurred. R&D costs consist primarily of salaries and benefits, stock-based compensation, manufacturing, contract services, clinical trials, and research related overhead. Non-refundable advance payments for goods and services that will be used in future research are recorded in prepaid and other assets and are expensed when the services are performed.
The Company records expenses for research and development activities based on Management’s estimates of services received and efforts expended pursuant to contracts with vendors that conduct research and development on Aptose’sthe Company’s behalf. The financial terms vary from contract to contract and may result in uneven payment flows as compared with services performed or products delivered. As a result, the Company is required to estimate research and development expenses incurred during the period, which impacts the amount of accrued expenses and prepaid balances related to such costs as of each balance sheet date. Management estimates the amount of work completed through discussions with internal personnel and external service providersthe contract research and contract manufacturing organizations as to the progress or stage of completion of the services. The Company’s estimates are based on a number of factors, including the Company’s knowledge of the status of each of the research and development project milestones, and contract terms together with related executed change orders. Management makes significant judgments and estimates in determining the accrued balance inat the end of each reporting period. As actual costs become known, the Company adjusts the accrued estimates.
Although Management does not expect our estimates to be materially different from amounts actually incurred, if the estimates of the status and timing of services performed differ from the actual status and timing of services performed, it could result in the Company reporting amounts that are too high or too low in any particular period. As of December 31, 2023, the Company has recorded approximately $0.7 million in prepaid expenses and approximately $6.5 million in accrued liabilities related to its research and development activities. If the estimates are too high or too low by a factor of 10% this would mean that prepaid expenses would be over or understated by approximately $70 thousand, and accrued liabilities would be over or understated by approximately $650 thousand. On a combined basis, this could mean an increase or decrease in research and development expenses by approximately $720 thousand. To date, there have been no material differences between the estimates of such expenses and the amounts actually incurred.
Valuation of contingent liabilities:
The Company utilizes considerable judgment in the measurement and recognition of provisions and the Company’s exposure to contingent liabilities. Judgment is required to assess and determine the likelihood that any potential or pending litigation or any and all potential claims against the Company may be successful. The Company must estimate if an obligation is probable, as well as quantify the possible economic cost of any claim or contingent liability. Such judgments and assumptions are inherently uncertain. The increase or decrease of one of these assumptions could materially increase or decrease the fair value of the liability and the associated expense.
Valuation of tax accounts:
Uncertainties exist with respect to the interpretation of complex tax regulations and the amount and timing of future taxable income. Currently, the Company has deductible temporary differences which would create a deferred tax asset. Deferred tax assets are recognized for all deductible temporary differences to the extent that it is probable that future taxable profit will be available against which the deductible temporary differences can be utilized. Management judgment is required to determine the amount of deferred tax assets that can be recognized, based upon the likely timing and the level of future taxable profits together with future tax planning strategies. To date, the Company has determined that none of its deferred tax assets should be recognized. The Company’s deferred tax assets are mainly comprised of its net operating losses from prior years and prior year research and development expenses not yet deducted for income tax purposes. These tax pools relate to entities that have a history of losses, have varying expiry dates, and may not be used to offset taxable income. As well, there are no taxable temporary differences or any tax planning opportunities available that could partly support the recognition of these losses as deferred tax assets. The generation of future taxable income could result in the recognition of some portion or all of the remaining benefits, which could result in an improvement in the Company’s results of operations through the recovery of future income taxes.
Valuation of share based compensation:
Management measures the costs for share based payments using market-based option valuation techniques. Assumptions are made and judgment is used in applying valuation techniques. These assumptions and judgments include estimating the future volatility of the share price, expected dividend yield, and expected life of the options. The Company uses historical data to estimate the expected dividend yield and expected volatility of its Common Shares in determining the fair value of stock options. The expected life of the options represents the estimated length of time the options are expected to remain outstanding. Such judgments and assumptions are inherently uncertain. The increase or decrease of one of these assumptions could materially increase or decrease the fair value of share based payments and share purchase warrants issued and the associated expense.
The weighted average assumptions that were used in the Black Scholes option pricing model to determine the fair value of stock options granted during the periods ended December 31, 2020 and 2019, respectively, are presented in Note 12 to the consolidated financial statements.
Leases:
Effective January 1, 2019, the Company adopted Financial Accounting Standards Board, or FASB, standard ASU No. 2016-02, “Leases (Topic 842)”. The Company’s operating leases of tangible property with terms greater than twelve months are recognized as right of use assets, which represents the lessee’s right to use, or control the use of, a specified asset for the lease term, and a corresponding lease liability, which represents the lessee’s obligation to make lease payments under a lease, measured on a discounted basis. The Company adopted the new standard using the alternative transition method, which permits a company to use its effective date as the date of initial application without restating comparative period financial statements. Landlord inducements in the form of free rent periods are netted against lease payments to the landlord in measuring right-of-use assets and lease liabilities.
Impact of adoption:
As a result of adopting Topic 842, we recorded as of January 1, 2019, a right of use asset of approximately $1.570 million, and a lease liability of approximately $1.647 million. Upon adoption, landlord inducements of approximately $78 thousand were de-recognized, and a corresponding adjustment was made to right-of-use assets.
Updated share information
As atof March 23, 2021,26, 2024, we had 88,885,23815,717,701 Common Shares issued and outstanding. In addition, there were 14,643,0531,504,636 Common Shares issuable upon the exercise of outstanding stock options.
ITEM 7A. QUALITATIVE AND QUANTITATIVE DISCLOSURES ABOUT MARKET RISK
Under SEC rules and regulations, as a smaller reporting company, we are not required to provide this information.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements and supplementary data required bypursuant to this itemitems are included in the Exhibits toItem 15 of this Annual Report and are presented beginning on page F-1 of this Annual Report on Form 10-K.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
None.
ITEM 9A. CONTROLS AND PROCEDURES
As of the end of our fiscal year ended December 31, 2020,2023, an evaluation of the effectiveness of our “disclosure controls and procedures” (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) was carried out by our management, with the participation of our principal executive officer and principal financial officer. Based upon that evaluation, our principal executive officer and principal financial officer have concluded that as of the end of that fiscal year, our disclosure controls and procedures are effective to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is (i) recorded, processed, summarized and reported within the time periods specified in SEC rules and forms and (ii) accumulated and communicated to our management, including our principal executive officer and principal financial officers, to allow timely decisions regarding required disclosure.
It should be noted that while our principal executive officer and principal financial officer believe that our disclosure controls and procedures provide a reasonable level of assurance that they are effective, they do not expect that our disclosure controls and procedures or internal control over financial reporting will prevent all errors or fraud. A control system, no matter how well conceived or operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as such term is defined in Exchange Act Rule 13a-15(f). Internal control over financial reporting is a process designed under the supervision and with the participation of our management, including our principal executive and financial officer, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America.
As of December 31, 2020,2023, our management assessed the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 Framework). Based on this assessment, our management concluded that, as of December 31, 2020,2023, our internal control over financial reporting was effective based on those criteria. We are a “smaller reporting company” as defined in Item 10(f)(1) of Regulation S-K under the Securities Act. For as long as we continue to be a smaller reporting company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not smaller reporting companies.
CHANGES IN INTERNAL CONTROL OVER FINANCIAL REPORTING
There were no changes in our internal control over financial reporting (as defined in Rule 13a-15(f) under the 1934 Act) during our fiscal quarter ended December 31, 2020,2023, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
None.ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
PART III.
Certain information required by Part III of this Annual Report on Form 10-K is omitted from this report because we are incorporating by reference to the definitive Proxy Statement for our 20212024 Annual Meeting of Shareholders, referred to as the Proxy Statement, which will be filed with the SEC within 120 days of the 20202023 fiscal year-end.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item is incorporated herein by reference to the information from the Proxy Statement under the sections entitled “Election of Directors,” “Nomination of Directors,” and “Corporate Governance – Board Committees” andCommittees,” except for the information required with respect to our executive officers, which has been included under the heading “Executive Officers” in Item 1, Part I of this Form 10-K, and is incorporated herein by reference, and except for information on our code of ethics:ethics.
We have adopted a code of ethics for directors, officers (including our principal executive officer, principal financial officer and principal accounting officer) and employees, known as the Code of Business Conduct and Ethics. The Code of Business Conduct and Ethics is available on our website at http://www.aptose.com under the Corporate Governance section of our Investor Relations page. We will promptly disclose on our website (i) the nature of any amendment to the policy that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and (ii) the nature of any waiver, including an implicit waiver, from a provision of the policy that is granted to one of these specified individuals that is required to be disclosed pursuant to SEC rules and regulations, the name of such person who is granted the waiver and the date of the waiver.
Item 11. EXECUTIVE COMPENSATION
The information required by this item is incorporated herein by reference to the information from the Proxy Statement under the sections entitled “Executive Compensation,” and “Director Compensation.”
Item 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is incorporated herein by reference to the information from the Proxy Statement under the sections entitled “Share Ownership of Certain Beneficial Owners, Management and Directors” and “Equity Compensation Plan Information.”
Item 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AN DIRECTOR INDEPENDENCE
The information required by this item is incorporated herein by reference to the information from the Proxy Statement under the sections entitled “Corporate Governance - Independence of the Board” and “Interest of Related Persons in Transactions.”
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this item is incorporated herein by reference to the information from the Proxy Statement under the section entitled “Audit, Audit-Related, Tax and Other Fees” and “Pre-Approval Policies and Procedures.”
PART IV.
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
(a)Documents filed as part of this report.
1.Financial Statements. We have filed the following documents as part of this Annual Report:
2.2. Financial Statement Schedules.
All schedules are omitted because they are not applicable or the required information is shown in the Financial Statements or notes thereto.
(b)Exhibits
The following exhibits are filed as part of, or incorporated by reference into, this report:
| | |
Exhibit Number |
| Description of Document |
3.1
|
|
|
3.1 |
| Articles of Incorporation, Arrangement and Amendment (incorporated herein by reference to Exhibit 99.3 to the Company’s Current Report on Form 6-K filed with the SEC on June 12, 2015) |
3.2
|
|
|
3.2 |
| By-law #2 of the Company (incorporated herein by reference to Exhibit 99.2 to the Company’s Current Report on Form 6-K filed with the SEC on June 12, 2015) |
4.1*
|
|
|
3.3 |
| Certificate of Amendment (incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K filed with the SEC on June 5, 2023) |
|
|
|
4.1* |
| Description of Securities (incorporated by reference to Exhibit 4.1 to the Company's Annual report on Form 10-K filed with the SEC on March 22, 2022) |
10.1
|
|
|
10.1 |
| Indemnification Agreement dated July 10, 2007 between Lorus Therapeutics Inc. and the Company (incorporated herein by reference to Exhibit 99.1 to the Company’s Current Report on Form 6-K filed with the SEC on September 4, 2007) |
10.2+
|
|
|
10.2+ |
| Amended and Restated Executive Employment Agreement between the Company and Dr. William G. Rice dated August 19, 2014 (incorporated herein by reference to Exhibit 4.9A to the Company’s Annual Report on Form 20-F filed with the SEC on March 4, 2015) |
10.3+
| Executive Employment Agreement between the Company and Gregory K. Chow dated November 29, 2013 (incorporated herein by reference to Exhibit 4.9.1 to the Company’s Annual Report on Form 20-F filed with the SEC on May 16, 2014)
|
|
10.4+10.3+ |
| Share Option Plan as amended May 5, 2015 (incorporated herein by reference to Exhibit 99.2 to the Company’s Current Report on Form 6-K filed with the SEC on June 12, 2015) |
10.5+
|
|
|
10.4+ |
| Stock Incentive Plan as adopted May 5, 2015 (incorporated herein by reference to Exhibit 99.1 to the Company’s Current Report on Form 6-K filed with the SEC on June 12, 2015) |
10.6+
| Form of Executive Employment Agreement, dated June 3, 2019, between the Company and Dr. Jotin Marango (incorporated herein by reference to Exhibit 10.4 to the Company’s Quarterly Report filed on Form 10-Q on August 6, 2019)
|
|
10.7+10.5+ |
| Form of Executive Employment Agreement, dated December 4, 2019, between the Company and Dr. Rafael Bejar (incorporated herein by reference to Exhibit 10.7 to the Company’s Annual Report filed on Form 10-K filed with the SEC on March 10, 2020) |
10.8^
|
|
|
10.6^ |
| License agreement dated June 13, 2018 by and between the Company and CrystalGenomics, Inc. (incorporated herein by reference to Exhibit 1.1 to the Company’s Current Report on Form 6-K filed with the SEC filed on June 22, 2018) |
10.9^
|
|
|
| | |
10.7^ |
| Option and License Agreement between the Company and CrystalGenomics, Inc. dated March 21, 2016 (incorporated herein by reference on Form 10-KA/3 filed with the SEC on April 22, 2019) |
10.10^
|
|
|
10.8^ |
| Amendment to Option and License Agreement between the Company and CrystalGenomics, Inc., dated April 26, 2016 (incorporated herein by reference to Exhibit 99.2 to the Company’s Current Report on Form 6-K filed with the SEC on June 8, 2016) |
10.11^
|
|
|
10.9^ |
| Second Amendment to Option and License Agreement between the Company and CrystalGenomics, Inc., dated May 13, 2016 (incorporated herein by reference to Exhibit 99.3 to the Company’s Current Report on Form 6-K filed with the SEC on June 8, 2016) |
Exhibit Number
| Description of Document
|
10.12^
|
|
|
10.10^ |
| Third Amendment to Option and License Agreement between the Company and CrystalGenomics, Inc., dated May 19, 2016 (incorporated herein by reference to Exhibit 99.4 to the Company’s Current Report on Form 6-K filed with the SEC on June 8, 2016) |
10.13^
|
|
|
10.11^ |
| Fourth Amendment to Option and License Agreement between the Company and CrystalGenomics, Inc., dated June 1, 2016 (incorporated herein by reference to Exhibit 99.5 to the Company’s Current Report on Form 6-K filed with the SEC on June 8, 2016) |
10.14^
|
|
|
10.12^ |
| License Agreement dated as of March 6, 2018 by and between the Company and Ohm Oncology Inc. (incorporated herein by reference to Exhibit 99.2 on Form 6-K filed with the SEC filed on March 8, 2018) |
10.15^
|
|
|
10.13+ |
| Underwriting Agreement, dated May 30, 2019, between Aptose Biosciences Inc. 2021 Employee Stock Purchase Plan ( incorporated by reference to the Definitive Proxy statement on Schedule 14A filed with the SEC on April 1, 2021)(File no. 1-32001)
|
|
|
|
10.14+ |
| Aptose Biosciences Inc. 2021 Employee Stock Incentive Plan ( incorporated by reference to the Definitive Proxy statement on Schedule 14A filed with the SEC on April 1, 2021)(File no. 1-32001) |
|
|
|
10.15^ |
| Exclusive License Agreement, dated November 4, 2021, by and RBC Capital Markets, LLCbetween Hanmi Pharmaceutical Co. Ltd. and Canaccord Genuity LLC (incorporatedAptose Biosciences Inc. ( incorporated herein by reference to Exhibit 1.110.1 to the Company’s Current Report filed on Form 8-K on May 30, 2019).November 4, 2021) |
10.16^
|
|
|
10.16 |
| UnderwritingEmployment Agreement dated December 16,June 3, 2019 between Aptose Biosciences Inc. and Piper Sandler & Co. (incorporatedPhilippe Ledru ( incorporated herein by reference to Exhibit 1.110.1 to the Company’s Current Report filed on Form 8-K on April 11, 2022)
|
|
|
|
10.17 |
| Employment Agreement, dated June 27, 2022, between Aptose Biosciences Inc. and Fletcher Payne ( incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report filed on Form 8-K on June 28, 2022) |
|
|
|
10.18 |
| Equity Distribution Agreement, dated December 9, 2022, among Aptose Biosciences Inc. and JonesTrading Institutional Services LLC( incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report filed on Form 8-K on December 17, 2020).12, 2022) |
10.17^
|
|
|
10.19 |
| Equity DistributionRegistration Rights Agreement, dated as of May 5, 202025, 2023, by and between the Company and Piper Sandler & Co. and Cannacord GenuityKeystone Capital Partners, LLC (incorporated herein by reference to Exhibit 10.110.2 to the Company’s Current Report on Form 8K8-K filed with the SEC on May 5, 2020).26, 2023)
|
10.18^
|
|
|
10.20 |
| UnderwritingCommon Share Purchase Agreement, dated July 15, 2020,as of May 25, 2023, by and between Aptose Biosciences Inc.the Company and Piper Sandler & Co.Keystone Capital Partners, LLC (incorporated herein by reference to Exhibit 1.110.1 to the Company’s Current Report filed on Form 8-K filed with the SEC on July 16, 2020).May 26, 2023)
|
21.1*
|
|
|
10.21 |
| Subscription Agreement, dated September 6, 2023, by and between the Company and Hanmi Pharmaceutical Co., Ltd. (incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the SEC on September 12, 2023) |
|
|
|
10.22 |
| Investor Rights Agreement, dated September 6, 2023, by and between the Company and Hanmi Pharmaceutical Co., Ltd. (incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed with the SEC on September 12, 2023) |
| | |
|
|
|
21.1* |
| List of Subsidiaries |
23.1*
|
|
|
23.1* |
| Consent of Independent Registered Public Accounting Firm (KPMG) |
24.1*
|
|
|
24.1* |
| Powers of Attorney (included on signature page) |
31.1*
|
|
|
31.1* |
| Certification of Principal Executive Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
31.2*
|
|
|
31.2* |
| Certification of Principal Financial Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
32.1*
|
|
|
32.1* |
| Certification of Principal Executive Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
32.2*
|
|
|
32.2* |
| Certification of Principal Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
101**
| The following consolidated financial statements from the Aptose Biosciences Inc. Annual Report on Form 10-K for the year ended December 31, 2020, formatted in Extensible Business Reporting Language (XBRL): (i) statements of operations and comprehensive loss, (ii) balance sheets, (iii) statements of changes of shareholders’ equity, (iv) statements of cash flows, and (v) the notes to the financial statements.
|
|
97.1* |
| Aptose Bioscience Inc. Clawback Policy |
|
|
|
101.INS* |
| Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document. |
101.SCH |
| Inline XBRL Taxonomy Extension Schema With Embedded Linkbase Documents |
104 |
| Cover Page Interactive Data File (embedded within the Inline XBRL document) |
+ | Indicates management contract or compensatory plan. |
* | Filed herewith. |
^ | Confidential treatment has been sought with respect to certain portions of this exhibit. |
^^ | Confidential treatment has been granted with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission. |
** | In accordance with Rule 406T of Regulation S-T, the XBRL related information in Exhibit 101 to this Annual Report on Form 10-K is deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act, is deemed not filed for purposes of Section 18 of the Exchange Act, and otherwise is not subject to liability under these sections. |
| | |
+ Indicates management contract or compensatory plan.
Confidential treatment has been sought with respect to certain portions of this exhibit.
Confidential treatment has been granted with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission.
** In accordance with Rule 406T of Regulation S-T, the Inline XBRL related information in Exhibit 101 to this Annual Report on Form 10-K is deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act, is deemed not filed for purposes of Section 18 of the Exchange Act, and otherwise is not subject to liability under these sections.
ITEM 16. form 10-k summary
None.
SIGNATURES
SIGNATURES
Pursuant to the requirements of the Securities Act, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of San Diego, State of California, on the 23rd26thday of March, 2021.
2024.
| |
Aptose Biosciences Inc. |
| |
| |
| By: | /s/ William G. Rice |
By: | | William G. Rice, Ph.D.
|
| President, Chief Executive Officer and Chairman of the Board of Directors |
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Dr. William G. Rice and Mr. Gregory K. Chow,Fletcher Payne, and each of them, his or her true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments (including post-effective amendments) to this report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or either of them, or their or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | |
Signature |
| Title |
|
|
|
/s/ William G. Rice |
|
|
William G. Rice, Ph.D. |
| President, Chief Executive Officer and Chairman of the Board of Directors (Principal Executive Officer) |
|
|
|
/s/ Gregory K. Chow | Fletcher Payne | Executive
|
|
Fletcher Payne |
| Senior Vice PresidentandChief Financial Officer (Principal Financial Officer and Accounting Officer) and Chief Operating Officer
|
|
|
|
/s/ Denis R. Burger |
|
|
Denis R. Burger, Ph.D. |
| Director, Lead Independent |
|
|
|
/s/ Carol G. Ashe |
| Director
|
Carol G. Ashe |
| Director |
/s/ Caroline Loewy
|
| Director
|
/s/ Erich M. Platzer |
|
|
/s/ Erich M. Platzer, M.D., Ph.D. |
| Director |
|
|
|
/s/ Bernd R. Seizinger |
|
|
Bernd R. Seizinger, M.D., Ph.D. |
| Director |
|
|
|
/s/ Mark D.Vincent, M.D.D. Vincent |
| Director
|
Mark D.Vincent, M.D. |
| Director |
|
|
|
/s/ Warren Whitehead |
|
|
Warren Whitehead |
| Director |


Consolidated Financial Statements
APTOSE BIOSCIENCES INC.
Years ended December 31, 20202023 and 20192022
| |

| F-1 |
|

KPMG LLP
100 New Park Place, Suite 1400
Vaughan, ON L4K 0J3
Tel 905-265 5900
Fax 905-265 6390
www.kpmg.ca
Report of Independent Registered Public Accounting Firm
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors
Aptose Biosciences Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated statements of financial position of Aptose Biosciences Inc. and subsidiaries (the Company) as of December 31, 20202023 and 2019,2022, the related consolidated statements of loss and comprehensive loss, changes in shareholders’ equity, and cash flows for each of the years in the two-year periodthen ended, December 31, 2020, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 20202023 and 2019,2022, and the results of its operations and its cash flows for each of the years in the two-year periodthen ended, December 31, 2020, in conformity with U.S. generally accepted accounting principles.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Notes 1 and 2(a) to the consolidated financial statements, the Company has suffered recurring losses from operations and has a net capital deficiency that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Notes 1 and 2(a). The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but
|
KPMG LLP, an Ontario limited liability partnership and member firm of the KPMG global organization of independent member firms affiliated with KPMG International Limited, a private English company limited by guarantee. KPMG Canada provides services to KPMG LLP. |
F-2
| |
| Aptose Biosciences Inc. March 26, 2024 |
not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
KPMG LLP, an Ontario limited liability partnership and member firm of the KPMG global organization of independent member firms affiliated with KPMG International Limited, a private English company limited by guarantee. KPMG Canada provides services to KPMG LLP.
| Aptose Biosciences Inc.
|
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Evaluation of Research and Development Prepaid and Accrued Costs
As discussed in Notes 2(i), 4 and 9 to the consolidated financial statements, the Company records expenses for research and development activities based on Management’s estimates of services received and efforts expended pursuant to contracts with vendors that conduct research and development on the Company’s behalf. The financial terms vary from contract to contract and may result in uneven payment flows as compared with services performed or products delivered. As a result, the Company is required to estimate research and development expenses incurred during the period, which impacts the amount of accrued expenses and prepaid balances related to such costs as of each balance sheet date. Management estimates the amount of work completed through discussions with internal personnel and external service providersthe contract research and contract manufacturing organizations as to the progress or stage of completion of the services. Management makes significant judgments and estimates in determining the accrued balance at the end of each reporting period.
We identified the evaluation of research and development prepaid and accrued costs as a critical audit matter. The Company’s estimates are based on a number of factors, including the Company’s knowledge of the status of each of the research and development project milestones, and contract terms together with related executed change orders. Management makes significant judgments and estimates in determining the accrued balance at the end of each reporting period.
We identified the evaluation of research and development prepaid and accrued costs as a critical audit matter. Higher degree of auditor judgment was required in evaluating the results of our audit procedures regarding the Company’s estimates, because of the subjectivity and estimation uncertainty in the significant assumptions used in the calculation.associated with this estimate.
| Aptose Biosciences Inc.Inc. March 26, 2024 |
The following are the primary procedures we performed to address this critical audit matter. We evaluated the design of certain internal controls related to the critical audit matter. This included controls over the development of the estimated amountFor a selection of prepaid and accrued costs incurred by the contract research organizations and contract manufacturing organizations during the period. For a selection ofamounts for research and development projects, we assessed the Company’s estimates of a selection ofby:
•inquiring with Company personnel responsible for overseeing the research and development activities completed to date by:understand progress of the activities including project milestones, and contract terms together with related executed change orders
· | inquiring with Company personnel responsible for overseeing the research and development activities to understand progress of the activities; |
· | inspecting the terms of the contracts between the Company and the respective contract research and contract manufacturing organizations, the correspondence between the Company and these organizations as to the completion status, arriving at an estimate of the prepaid or accrual amounts based thereon and comparing it to the amounts recorded by the Company; and |
· | performing a lookback analysis by comparing the estimated accrual balances at prior annual reporting date to the actual amounts that were ultimately invoiced. |
•inspecting the terms of the contracts, including related executed change orders, between the Company and the respective contract research and contract manufacturing organizations, the correspondence between the Company and these organizations as to the completion status, invoices received by the Company subsequent to period end, and using this information to arrive at an independent estimate of the prepaid or accrual amounts and comparing it to the amounts recorded by the Company
We have served as the Company’s auditor since 2003.1994.
Chartered Professional Accountants, Licensed Public Accountants
Vaughan, Canada
March 23, 202126, 2024
APTOSE BIOSCIENCES INC.
Consolidated Statements of Financial Position
(Expressed in thousands of US dollars)
| | | | December 31, 2020 | | | | December 31, 2019 | |
| | | | | | | | | | | December 31,
2023 | | | December 31,
2022 | |
| Assets | | | | | | | | | | | | | |
| Current assets: | | | | | | | | | | | | | |
| Cash and cash equivalents | | $ | 117,393 | | | $ | 79,842 | | | $ | 9,252 | | | $ | 36,970 | |
| Investments | | | 5,000 | | | | 17,758 | | | | — | | | | 9,989 | |
| Prepaid expenses | | | 2,554 | | | | 1,025 | | | | 2,042 | | | | 2,303 | |
| Other current assets | | | 129 | | | | 141 | | | | 600 | | | | 257 | |
| Total current assets | | | 125,076 | | | | 98,766 | | | | 11,894 | | | | 49,519 | |
| Non-current assets: | | | | | | | | | | | | | |
| Property and equipment | | | 261 | | | | 334 | | | | 152 | | | | 211 | |
| Right-of-use assets, operating leases | | | 925 | | | | 1,376 | | | | 943 | | | | 1,297 | |
| Total non-current assets | | | 1,186 | | | | 1,710 | | | | 1,095 | | | | 1,508 | |
| Total assets | | $ | 126,262 | | | $ | 100,476 | | | $ | 12,989 | | | $ | 51,027 | |
| Liabilities and Shareholders’ Equity | | | | | | | | | | | | | |
| Current liabilities: | | | | | | | | | | | | | |
Accounts payable to related party | | | $ | 2,554 | | | $ | 2,984 | |
| Accounts payable | | $ | 2,171 | | | $ | 1,960 | | | | 3,492 | | | | 3,342 | |
Accrued liabilities to related party | | | | — | | | | 572 | |
| Accrued liabilities | | | 4,102 | | | | 3,058 | | | | 8,829 | | | | 5,085 | |
| Current portion of lease liability, operating leases | | | 539 | | | | 521 | | | | 394 | | | | 301 | |
Total current liabilities
| | | 6,812 | | | | 5,539 | | | | 15,269 | | | | 12,284 | |
| Non-current liabilities: | | | | | | | | | | | | | |
| Lease liability, operating leases | | | 535 | | | | 1,011 | | | | 621 | | | | 1,002 | |
| Total liabilities | | | 7,347 | | | | 6,550 | | | | 15,890 | | | | 13,286 | |
| | | | | | | | | |
| Shareholders’ equity: | | | | | | | | | | | | | |
| Share capital: | | | | | | | | | | | | | |
| Common shares, no par value, unlimited authorized shares, 88,881,737 and 76,108,031 shares issued and outstanding at December 31, 2020 and December 31, 2019 | | | 429,523 | | | | 365,490 | |
Common shares, no par value, unlimited authorized shares,
7,942,363 and 6,157,749 shares issued and outstanding at
December 31, 2023 and December 31, 2022, respectively | | | | 444,806 | | | | 437,520 | |
| Additional paid-in capital | | | 50,861 | | | | 34,649 | | | | 72,146 | | | | 68,869 | |
| Accumulated other comprehensive loss | | | (4,316 | ) | | | (4,298 | ) | | | (4,316 | ) | | | (4,318 | ) |
| Deficit | | | (357,153 | ) | | | (301,915 | ) | | | (515,537 | ) | | | (464,330 | ) |
| Total shareholders’ equity | | | 118,915 | | | | 93,926 | | | | (2,901 | ) | | | 37,741 | |
| | | | | | | | | |
| Total liabilities and shareholders’ equity | | $ | 126,262 | | | $ | 100,476 | | | $ | 12,989 | | | $ | 51,027 | |
See accompanying notes to consolidated financial statementsstatements.
Going concern, see Note 2(a).
Commitments, see Note 10.
Subsequent events, (note 17)see Note 17.
APTOSE BIOSCIENCES INC.
Consolidated Statements of Loss and Comprehensive Loss
(Expressed in thousands of US dollars, except for per common share data)
| | | | | | | | |
| | Year ended December 31, 2023 | | | Year ended December 31, 2022 | |
Revenue | | $ | — | | | $ | — | |
Expenses: | | | | | | |
Research and development, related party | | | 3,492 | | | | 3,556 | |
Research and development | | | 33,273 | | | | 24,532 | |
General and administrative | | | 15,591 | | | | 14,514 | |
Operating expenses | | | 52,356 | | | | 42,602 | |
Other income: | | | | | | |
Interest income | | | 1,151 | | | | 788 | |
Foreign exchange loss | | | (2 | ) | | | (9 | ) |
Total other income | | | 1,149 | | | | 779 | |
Net loss | | | (51,207 | ) | | | (41,823 | ) |
Other comprehensive loss: | | | | | | |
Unrealized gain (loss) on securities available-for-sale securities | | | 2 | | | | (2 | ) |
Total comprehensive loss | | $ | (51,205 | ) | | $ | (41,825 | ) |
Basic and diluted loss per common share | | $ | (7.58 | ) | | $ | (6.80 | ) |
Weighted average number of common shares outstanding used in the
calculation of basic and diluted loss per share | | | 6,755 | | | | 6,151 | |
| | | | Year ended
December 31, 2020 | | | | Year ended
December 31, 2019 | |
| | | | | | | | | |
| | | | | | | | | |
| Revenue | | $ | - | | | $ | - | |
| | | | | | | | | |
| Expenses: | | | | | | | | |
| Research and development | | | 29,288 | | | | 16,835 | |
| General and administrative | | | 26,480 | | | | 10,022 | |
| Operating expenses | | | 55,768 | | | | 26,857 | |
| Other income : | | | | | | | | |
| Interest income | | | 522 | | | | 574 | |
| Foreign exchange gain | | | 8 | | | | 6 | |
| Total other income | | | 530 | | | | 580 | |
| | | | | | | | | |
| Net loss | | | (55,238 | ) | | | (26,277 | ) |
| | | | | | | | | |
| Other comprehensive loss: | | | | | | | | |
| Unrealized gain/(loss) on securities available-for-sale | | | (18 | ) | | | 18 | |
| Total comprehensive loss | | $ | (55,256 | ) | | $ | (26,259 | ) |
| | | | | | | | | |
| Basic and diluted loss per common share | | $ | (0.67 | ) | | $ | (0.52 | ) |
| | | | | | | | | |
| Weighted average number of common shares outstanding used in the calculation of (in thousands) | | | | | | | | |
| Basic and diluted loss per common share | | | 81,837 | | | | 50,160 | |
| | | | | | | | | |
See accompanying notes to consolidated financial statements.
APTOSE BIOSCIENCES INC.
Consolidated Statements of Changes in Shareholders’ Equity
(Expressed in thousands of US dollars)dollars, except for per common share data)
| | | | Common Shares | | | | | | | | | | | | | | | | | |
| | | | Shares (thousands) | | | | Amount | | | | Additional paid-in capital | | | | Accumulated other comprehensive loss | | | | Deficit | | | | Total | |
| Balance, December 31, 2019 | | | 76,108 | | | $ | 365,490 | | | $ | 34,649 | | | $ | (4,298 | ) | | $ | (301,915 | ) | | $ | 93,926 | |
| Common shares issued pursuant to public offering | | | 11,854 | | | | 58,234 | | | | - | | | | - | | | | - | | | | 58,234 | |
| Common shares issued on redemption of restricted share units | | | 685 | | | | 4,801 | | | | (4,801 | ) | | | - | | | | - | | | | - | |
| Common shares issued upon exercise of stock options | | | 235 | | | | 998 | | | | (425 | ) | | | - | | | | - | | | | 573 | |
| Stock-based compensation | | | - | | | | - | | | | 21,438 | | | | - | | | | - | | | | 21,438 | |
| Other comprehensive gain | | | - | | | | - | | | | - | | | | (18 | ) | | | - | | | | (18 | ) |
| Net loss | | | - | | | | - | | | | - | | | | - | | | | (55,238 | ) | | | (55,238 | ) |
| Balance, December 30, 2020 | | | 88,882 | | | $ | 429,523 | | | $ | 50,861 | | | $ | (4,316 | ) | | $ | (357,153 | ) | | $ | 118,915 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance, December 31, 2018 | | | 38,162 | | | $ | 261,072 | | | $ | 32,963 | | | $ | (4,316 | ) | | $ | (275,638 | ) | | $ | 14,081 | |
| Common shares issued pursuant to the December 2019 public offering | | | 18,544 | | | | 68,588 | | | | - | | | | - | | | | - | | | | 68,588 | |
| Common shares issued pursuant to the June 2019 public offering | | | 11,500 | | | | 19,594 | | | | - | | | | - | | | | - | | | | 19,594 | |
| Common shares issued pursuant to 2019 share purchase agreement | | | 1,971 | | | | 4,730 | | | | - | | | | - | | | | - | | | | 4,730 | |
| Common shares issued under the 2018 ATM | | | 77 | | | | 178 | | | | - | | | | - | | | | - | | | | 178 | |
| Common shares issued pursuant to 2018 share purchase agreement | | | 5,502 | | | | 10,000 | | | | - | | | | - | | | | - | | | | 10,000 | |
| Common shares issued upon exercise of stock options | | | 312 | | | | 1,248 | | | | (530 | ) | | | - | | | | - | | | | 718 | |
| Common shares issued on redemption of restricted share units | | | 40 | | | | 80 | | | | (80 | ) | | | - | | | | - | | | | - | |
| Stock-based compensation | | | - | | | | - | | | | 2,296 | | | | - | | | | - | | | | 2,296 | |
| Other comprehensive gain | | | - | | | | - | | | | - | | | | 18 | | | | - | | | | 18 | |
| Net loss | | | - | | | | - | | | | - | | | | - | | | | (26,277 | ) | | | (26,277 | ) |
| Balance, December 30, 2019 | | | 76,108 | | | $ | 365,490 | | | $ | 34,649 | | | $ | (4,298 | ) | | $ | (301,915 | ) | | $ | 93,926 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
| | Common Shares | | | | | | | | | | | | | |
| | Shares
(thousands) | | | Amount | | | Additional
paid-in capital | | | Accumulated other
comprehensive loss | | | Deficit | | | Total | |
Balance, December 31, 2022 | | | 6,158 | | | $ | 437,520 | | | $ | 68,869 | | | $ | (4,318 | ) | | $ | (464,330 | ) | | $ | 37,741 | |
Common shares issued under
the Hanmi Subscription Agreement | | | 668 | | | | 2,989 | | | | — | | | | — | | | | — | | | | 2,989 | |
Common shares issued in exchange for RSUs | | | 38 | | | | 376 | | | | (376 | ) | | | — | | | | — | | | | — | |
Common shares issued under
the October 2022 ATM | | | 337 | | | | 1,809 | | | | — | | | | — | | | | — | | | | 1,809 | |
Common shares issued under
the 2023 Committed Equity Facility | | | 735 | | | | 2,083 | | | | — | | | | — | | | | — | | | | 2,083 | |
Stock-based compensation | | | — | | | | — | | | | 3,653 | | | | — | | | | — | | | | 3,653 | |
Common shares issued under the ESPP plan | | | 6 | | | | 29 | | | | — | | | | — | | | | — | | | | 29 | |
Other comprehensive gain / loss | | | — | | | | — | | | | — | | | | 2 | | | | — | | | | 2 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | (51,207 | ) | | | (51,207 | ) |
Balance, December 31, 2023 | | | 7,942 | | | $ | 444,806 | | | $ | 72,146 | | | $ | (4,316 | ) | | $ | (515,537 | ) | | $ | (2,901 | ) |
Balance, December 31, 2021 | | | 6,147 | | | $ | 437,386 | | | $ | 63,673 | | | $ | (4,316 | ) | | $ | (422,507 | ) | | $ | 74,236 | |
Common shares issued under
the October 2022 ATM | | | 5 | | | | 51 | | | | — | | | | — | | | | — | | | | 51 | |
Common shares issued under the May 2020 ATM | | | 4 | | | | 50 | | | | — | | | | — | | | | — | | | | 50 | |
Common shares issued upon exercise of stock options | | | 1 | | | | 26 | | | | (11 | ) | | | — | | | | — | | | | 15 | |
Stock-based compensation | | | — | | | | — | | | | 5,207 | | | | — | | | | — | | | | 5,207 | |
Common shares issued under the ESPP plan | | | 1 | | | | 7 | | | | — | | | | — | | | | — | | | | 7 | |
Other comprehensive loss | | | — | | | | — | | | | — | | | | (2 | ) | | | — | | | | (2 | ) |
Net loss | | | — | | | | — | | | | — | | | | — | | | | (41,823 | ) | | | (41,823 | ) |
Balance, December 31, 2022 | | | 6,158 | | | $ | 437,520 | | | $ | 68,869 | | | $ | (4,318 | ) | | $ | (464,330 | ) | | $ | 37,741 | |
See accompanying notes to consolidated financial statements.
APTOSE BIOSCIENCES INC.
Consolidated Statements of Cash Flows
(Expressed in thousands of US dollars)
| | | | Year ended
December 31, 2020 | | | | Year ended
December 31, 2019 | |
| | | | | | | | | |
| | | | | | | | | |
| | | | | | | | | | | Year ended December 31, 2023 | | | Year ended December 31, 2022 | |
| Cash flows from operating activities: | | | | | | | | | | | | | | |
| Net loss for the year | | $ | (55,238 | ) | | $ | (26,277 | ) | | $ | (51,207 | ) | | $ | (41,823 | ) |
| Items not involving cash: | | | | | | | | | | | | | | |
| Stock-based compensation | | | 21,438 | | | | 2,296 | | | | 3,653 | | | | 5,207 | |
| Shares issued to Aspire Capital as commitment fees | | | - | | | | 360 | |
| Depreciation and amortization | | | 152 | | | | 152 | | | | 88 | | | | 120 | |
Disposal of property and equipment | | | | — | | | | 16 | |
| Amortization of right-of-use assets | | | 462 | | | | 461 | | | | 378 | | | | 408 | |
| Interest on lease liabilities | | | 69 | | | | 90 | | | | 93 | | | | 35 | |
| Unrealized foreign exchange gain/(loss) | | | (8 | ) | | | (23 | ) | | | — | | | | 4 | |
| Accrued interest on investments | | | 33 | | | | (34 | ) | | | — | | | | (60 | ) |
| Change in operating working capital: | | | | | | | | | | | | | | |
| Prepaid expenses | | | (1,529 | ) | | | (379 | ) | | | 261 | | | | 173 | |
| Operating lease payments | | | (537 | ) | | | (471 | ) | | | (405 | ) | | | (546 | ) |
| Other assets | | | 12 | | | | (40 | ) | | | (343 | ) | | | (124 | ) |
Accounts payable, related party | | | | (430 | ) | | | 2,984 | |
| Accounts payable | | | 211 | | | | 645 | | | | 150 | | | | 4,627 | |
Accrued liabilities, related party | | | | (572 | ) | | | 572 | |
| Accrued liabilities | | | 1,044 | | | | 1,662 | | | | 3,744 | | | | (3,915 | ) |
| Cash used in operating activities | | | (33,891 | ) | | | (21,558 | ) | | | (44,590 | ) | | | (32,322 | ) |
| | | | | | | | | |
| Cash flows from financing activities: | | | | | | | | | | | | | | |
| Issuance of common shares pursuant to July/August 2020 Public Offering, net of broker commission and agent legal fees | | | 58,402 | | | | - | |
| Issuance of common shares pursuant to December 2019 Public Offering, net of broker commission and agent legal fees | | | - | | | | 68,883 | |
| Issuance of common shares pursuant to June 2019 Public Offering, net of broker commission and agent legal fees | | | - | | | | 19,736 | |
| Issuance of common shares under the 2018 ATM, net of broker commission | | | - | | | | 178 | |
| Issuance of common shares under 2019 share purchase agreement | | | - | | | | 4,370 | |
| Issuance of common shares under 2018 share purchase agreement | | | - | | | | 10,000 | |
| Offering costs paid | | | (168 | ) | | | (437 | ) |
Issuance of common shares to Hanmi | | | | 2,989 | | | | — | |
Issuance of common shares under Committed Equity Facility | | | | 2,083 | | | | — | |
Issuance of common shares under 2022 ATM | | | | 1,809 | | | | 51 | |
Issuance of common shares under 2020 ATM | | | | — | | | | 50 | |
Issuance of commons shares under the ESPP share purchase | | | | 29 | | | | — | |
| Issuance of common shares pursuant to exercise of stock options | | | 573 | | | | 718 | | | | — | | | | 15 | |
| Cash provided by financing activities | | | 58,807 | | | | 103,448 | | | | 6,910 | | | | 116 | |
| | | | | | | | | |
| Cash flows from (used in) investing activities: | | | | | | | | | | | | | | |
| Maturity (acquisition) of investments, net | | | 12,707 | | | | (17,268 | ) | | | 9,989 | | | | 30,090 | |
| Purchase of property and equipment | | | (79 | ) | | | (102 | ) | | | (29 | ) | | | (24 | ) |
| Cash provided by (used in) investing activities | | | 12,628 | | | | (17,370 | ) | | | 9,960 | | | | 30,066 | |
| | | | | | | | | |
| Effect of exchange rate fluctuations on cash and cash equivalents held | | | 7 | | | | 23 | | | | 2 | | | | (4 | ) |
| | | | | | | | | |
| Increase in cash and cash equivalents | | | 37,551 | | | | 64,543 | |
| | | | | | | | | |
Decrease in cash and cash equivalents | | | | (27,718 | ) | | | (2,144 | ) |
| Cash and cash equivalents, beginning of year | | | 79,842 | | | | 15,299 | | | | 36,970 | | | | 39,114 | |
| Cash and cash equivalents, end of year | | $ | 117,393 | | | $ | 79,842 | | | $ | 9,252 | | | $ | 36,970 | |
See accompanying notes to consolidated financial statements.
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
1. Reporting entity:
Aptose Biosciences Inc. (“Aptose”Aptose,” “the Company,” “we,” “us,” or the “Company”“our”) is a science-driven, clinical-stage biotechnology company committed to discoveringthe development and developing personalized therapiescommercialization of precision medicines addressing unmet medicalclinical needs in oncology.oncology, with an initial focus on hematology. The Company's small molecule cancer therapeutics pipeline includes products designed to provide single agent efficacy and to enhance the efficacy of other anti-cancer therapies and regimens without overlapping toxicities. The Company’s executive offices areoffice is located in San Diego, California, and itsour head office is located in Toronto, Canada.
Aptose has two clinical-stage programs and a second program that is discovery-stage and partnered with another company. CG026806 (“CG-806”), Aptose’s pan-FMS-like tyrosine kinase 3 / pan-Bruton’s tyrosine kinase inhibitor, is currently enrolling patients in a Phase 1, multicenter, open label, dose-escalation study with expansions to assess the safety, tolerability, PK, and preliminary efficacy of CG-806 in patients with chronic lymphocytic leukemia (CLL/SLL) or non-Hodgkin lymphomas (NHL). Aptose was granted IND allowance from the U.S Food and Drug Administration (FDA) to initiate a separate Phase 1 trial in patients with relapse or refractory acute myeloid leukemia (AML) in June 2020, and this trial is also enrolling patients. APTO-253, Aptose’s second program, is a small molecule MYC inhibitor and is currently enrolling patients in a Phase 1b clinical trial for the treatment of patients with R/R blood cancers, including AML and high-risk Myelodysplastic Syndrome.
We are advancing first-in-class targeted agents to treat life-threatening hematologic cancers that, in most cases, are not elective for patients and require immediate treatment. However, COVID-19 has caused global economicWe have two clinical-stage investigational products for hematological malignancies: tuspetinib, an oral, potent myeloid kinase inhibitor, and social disruptions that could adversely affect our ongoingluxeptinib, an oral, dual lymphoid and planned research and development of our clinical-stage programs including but not limited to drug manufacturing campaigns, clinical trial activities including enrollment of patients in our ongoing and planned clinical trials, collection and analysis of patient data and eventually, the reporting of results from our trials.myeloid kinase inhibitor.
Since our inception, we have financed our operations and technology acquisitions primarily from equity financing, proceeds from the exercise of warrants and stock options, and interest income on funds held for future investment. Our uses of cash for operating activities have primarily consisted of salaries and wages for our employees, facility and facility-related costs for our offices and laboratories, fees paid in connection with preclinical and clinical studies, licensing fees, drug manufacturing costs, laboratory supplies and materials, and professional fees.
Management recognizes that in order for us to meet our capital requirements, and continue to operate, additional financing will be necessary. We plan to raise additional funds to fund our business operations but there is no assurance that such additional funds will be available for us to finance our operations on acceptable terms, if at all. These conditions raise substantial doubt about the Company’s ability to continue as a going concern, see Note 2(a), Going concern. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
In order for the Company to meet its capital requirements, and continue to operate, additional financing will be necessary. The Company is evaluating strategies to obtain the required additional funding for future operations. These strategies may include, but are not limited to, obtaining equity financing, and restructuring of operations to decrease expenses. However, given the impact of the economic downturn on the U.S. and global financial markets, the Company may be unable to access further equity or when needed. As such, there can be no assurance that the Company will be able to obtain additional liquidity when needed or under acceptable terms, if at all. The consolidated financial statements do not reflect any adjustments to the carrying amounts and classification of assets, liabilities, and reported expenses that may be necessary if the Company were unable to continue as a going concern. Such adjustments may be material.
Our ability to raise additional funds could be affected by adverse market conditions, the status of our product pipeline, possible delays in enrollment in our trial, and various other factors and we may be unable to raise capital when needed, or on terms favorable to us. If necessary funds are not available, we may have to delay, reduce the scope of, or eliminate some of our development programs, potentially delaying the time to market for any of our product candidates.
We do not expect to generate positive cash flow from operations for the foreseeable future due to the early stage of our clinical trials. It is expected that negative cash flow will continue until such time, if ever, that we receive regulatory approval to commercialize any of our products under development and/or royalty or milestone revenue from any such products exceeds expenses.
We believe that our cash, cash equivalents and investments on hand at December 31, 2020 will be sufficient to finance our operations for at least 12 months from the issuance date of these financial statements. Our cash needs for the next twelve months include estimates of the number of patients and rate of enrollment of our clinical trials, the amount of drug product that we will require to support our clinical trials, and our general corporate overhead costs to support our operations, and our reliance on our manufacturers. We have based these estimates on assumptions and plans which may change and which could impact the magnitude and/or timing of operating expenses and our cash runway.runway, See Note 2(a), Going concern.
APTOSE BIOSCIENCES INC.
Our abilityNotes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
The Company's financial statements are prepared using generally accepted accounting principles in the United States of America applicable to a going concern which contemplates the realization of assets and liquidation of liabilities in the normal course of business. As of December 31, 2023, the Company had an accumulated deficit of approximately $515.5 million (December 31, 2022, $464.3 million); cash and cash equivalents and investment balances of approximately $9.3 million (December 31, 2022, $47.0 million); negative working capital of approximately $3.4 million (December 31, 2022, positive working capital of $37.2 million); and negative shareholder's equity of $2.9 million (December 31, 2022, positive shareholder's equity of $37.7 million). Management recognizes that in order to meet the capital requirements, and continue to operate, additional financing will be necessary. The Company plans to raise additional funds could be affected by adverse market conditions,to fund our business operations through equity financing under the status2022 ATM Facility, the 2023 Committed Equity Facility and other facilities, as further described in Note 12. Management continues considering other options for raising capital including debt, equity, collaborations, and reorganization to reduce operational expenses. However, given the impact of our product pipeline, possible delaysthe financial markets, and the matter in enrollmentNote 17, Subsequent events, that may impact the Company's ability to raise financing in our trial related to COVID-19, and various other factors and wethe capital markets, the Company may be unable to raise capitalaccess financing when needed, if at all. As such, there can be no assurance that the Company will be able to obtain additional liquidity when needed or onunder acceptable terms, favorableif at all. These conditions raise substantial doubt about the Company’s ability to us. If necessary funds arecontinue as a going concern. The financial statements do not available, weinclude any adjustments that might result from the outcome of this uncertainty. For information about financing events and the regulatory matter that may haveimpact the Company's ability to delay, reduceraise financing in the scopecapital markets which arose subsequent to December 31, 2023, see Note 17, Subsequent events.
On May 23, 2023, during the Aptose Annual and Special Meeting of or eliminate someShareholders, our shareholders voted to approve special resolutions providing for an amendment to our articles of incorporation to effect a reverse share split of our development programs, potentially delayingoutstanding Common Shares, at a ratio in the timerange of 1-for-10 to market for any1-for-20. Our Board of Directors then approved a ratio of 1-for-15 on May 23, 2023. On May 24, 2023, we filed articles of amendment under the Canada Business Corporations Act ("CBCA") to give effect to the reverse stock split (consolidation) of our product candidates.Common Shares on the basis of one post-consolidation Common Share for each 15 pre-consolidation Common Shares (the “Reverse Stock Split”). The Common Shares commenced trading on a post-Reverse Stock Split basis at market open on Tuesday, June 6, 2023. All references in this report to historical Common Share prices, numbers of Common Shares, and earnings per share calculations have been presented to reflect the effect of the Reverse Stock Split.
2. | Significant accounting policies |
| (a) | 2.Significant accounting policies (a)Basis of consolidation: |
These consolidated financial statements include the accounts of its subsidiaries. All intercompany transactions, balances, revenue and expenses are eliminated on consolidation.
presentation - Going concern | (b) | Basis of presentation: |
These consolidated financial statements have been prepared in conformity with generally accepted accounting principles in the United States, or GAAP and the rules and regulations of the Securities and Exchange Commission, or SEC, related to annual reports filed on Form 10-K. 10-K, assuming the Company will continue as a going concern. The going concern assumption contemplates the realization of assets and satisfaction of liabilities in the normal course of business. However, substantial doubt about the Company's ability to continue as a going concern exists. As of the filing date, we have sufficient liquidity to support the Company's operations until August 2024.
In order for the Company to meet its capital requirements, and continue to operate, additional financing will be necessary. The Company is evaluating strategies to obtain the required additional funding for future operations. These strategies may include, but are not limited to, obtaining equity financing, and restructuring of operations to decrease expenses. However, given the challenges in the U.S. and global financial markets, and the matter in Note 17, Subsequent events, that may impact the Company's ability to raise financing in the capital markets, the Company may be unable to access further equity or when needed, if at all. As the Company is primarily pursuing one compound that is licensed from a related party with significant licensing payments who will have influence on the Company, other investors may not be willing to invest in the Company. As such, there can be no assurance that the Company will be able to obtain additional liquidity when needed or under acceptable terms, if at all. The consolidated financial statements do not reflect any adjustments to the carrying amounts and classification of assets, liabilities, and reported expenses that may be necessary if the Company were unable to continue as a going concern. Such adjustments may be material.
The functional and presentation currency of the Company is the US dollar.
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
F-10
APTOSE BIOSCIENCES INC.
| (c) | Significant accounting policies, estimates and judgments: |
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
(b)Basis of consolidation:
These consolidated financial statements include the accounts of its subsidiaries. All intercompany transactions, balances, revenue, and expenses are eliminated on consolidation.
(c)Significant accounting policies, estimates and judgments
The preparation of the consolidated financial statements requires management to make judgments, estimates and assumptions that affect the application of accounting policies and reported amounts of assets and liabilities at the date of the consolidated financial statements and reported amounts of revenue and expenses during the reporting period. Actual outcomes could differ from those estimates. The consolidated financial statements includecontain estimates, which by their nature, are uncertain.
The impacts of such estimates are pervasive throughout the consolidated financial statements and mayrequire accounting adjustments based on future occurrences.
The estimates and underlying assumptions are reviewed on a regular basis. Revisions to accounting estimates are recognized in the period in which the estimate is revised and in any future periods affected.
Effective January 1, 2019, the Company adopted Financial Accounting Standards Board, or FASB, standard ASU No. 2016-02, “Leases (Topic 842)”. The Company’s operating leases of tangible property with terms greater than twelve months are recognized as right of use assets, which represents the lessee’s right to use, or control the use of, a specified asset for the lease term, and a corresponding lease liability, which represents the lessee’s obligation to make lease payments under a lease, measured on a discounted basis. The Company adopted the new standard using the alternative transition method, which permits a company to use its effective date as the date of initial application without restating comparative period financial statements. Landlord inducements in the form of free rent periods are netted against lease payments to the landlord in measuring right-of-use assets and lease liabilities.
(e) Cash and cash equivalents:
Impact of adoption:
As a result of adopting Topic 842, we recorded as of January 1, 2019, a right of use asset of approximately $1.570 million, and a lease liability of approximately $1.647 million. Upon adoption, landlord inducements of approximately $78 thousand were de-recognized, and a corresponding adjustment was made to right-of-use assets.
| (e) | Cash and cash equivalents: |
Cash and cash equivalents are short-term highly liquid investments with original maturities of 90 days or less as atof the date of purchase. Cash equivalents are accounted for onan amortized cost basis, which approximates itstheir fair value due to their short-term maturities.
Investments consist of timeterm deposits with original maturities greater than 90 days and are classified by management as securities available-for-sale. These available-for-sale securities are recorded at estimated fair values. Unrealized gains and losses on these investments are recorded in accumulated other comprehensive income (AOCI) in shareholder’s equity. Realized gains and losses and declines in value that are judged to be other than temporary are included in interest income.
(g)Concentration of risk:
| (g) | Concentration of risk: |
The Company is subject to credit risk from the Company’s cash and cash equivalents and investments. The carrying amount of the financial assets represents the maximum credit exposure. The Company manages credit risk associated with its cash and cash equivalents and investments by maintaining minimum standards of R1-lowR1‑low or A-lowA‑low investments and the Company invests only in highly rated Canadian corporations and treasury bills, which are capable of prompt liquidation.
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
The Company has cash accounts in Canada and the US. The Canada Deposit Insurance Corporation (CDIC) and the US Federal Deposit Insurance Corporation (FDIC) provide insurance to protect depositors against the loss of their deposits in case of a bank failure. However, the maximum amount of coverage varies by jurisdiction and account type. In Canada, the CDIC insures eligible deposits up to $100,000 (CAD) per depositor, per insured category, per member institution. In the United States, the FDIC insures deposits up to $250,000 per depositor, per insured bank, for each account ownership category. It is important to note that not all deposits are eligible for insurance coverage. For example, deposits in foreign currency, deposits held in trust, and investments such as mutual funds, stocks, and bonds are not insured by either the FDIC or CDIC.
The Company is subject to intermediary risk associated with the actions of financial intermediaries, such as banks or investment managers, who act on behalf of clients to buy and sell assets. The Company has diversified the investments with two large financial institutions to reduce the concentration of risk in any one institution, and spread the risk. This measure reduces the likelihood of being significantly impacted by the failure of a single financial institution.
The Company has reduced the exposure to individual investment vehicles to minimize the risk of loss in case of adverse events. The Company has diversified the investment portfolio across different asset classes and investment vehicles to achieve this goal.
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
(h) Property and equipment:
| (h) | Property and equipment: |
Property and equipment is measured at cost less accumulated depreciation and accumulated impairment losses. Cost includes expenditures that are directly attributable to the acquisition of the asset. The Company records depreciation at rates that charge operations with the cost of the assets over their estimated useful lives on a straight-linestraight‑line basis as follows:
| | |
Office furniture |
| | | 5 years |
Office furniture (years)Laboratory equipment |
| | 5 | years |
Laboratory equipment (years)Computer hardware |
| | 5 | 3 years |
Computer hardware (years)software |
| | 3 | years |
Computer software (years) | | | 3 | |
Leasehold improvements | | |
| Life of lease | |
| | | | |
The residual value, useful life and methods of depreciation of the assets are reviewed at each reporting period and adjusted prospectively if appropriate.
(i)Research and development:
| (i) | Research and development: |
Research and development (R&D) costs are expensed as incurred. R&D costs consist primarily of salaries and benefits, stock-based compensation, manufacturing, contract services, clinical trials and research related overhead. Non-refundable advance payments for goods and services that will be used in future research are recorded in prepaid and other assets and are expensed when the services are performed.
The Company records expenses for research and development activities based on Management’s estimates of services received and efforts expended pursuant to contracts with vendors that conduct research and development on Aptose’sthe Company’s behalf. The financial terms vary from contract to contract and may result in uneven payment flows as compared with services performed or products delivered. As a result, the Company is required to estimate research and development expenses incurred during the period, which impacts the amount of accrued expenses and prepaid balances related to such costs as of each balance sheet date. Management estimates the amount of work completed through discussions with internal personnel and external service providersthe contract research and contract manufacturing organizations as to the progress or stage of completion of the services. The Company’s estimates are based on a number of factors, including the Company’s knowledge of the status of each of the research and development project milestones, and contract terms together with related executed change orders. Management makes significant judgments and estimates in determining the accrued balance inat the end of each reporting period. As actual costs become known, the Company adjusts the accrued estimates.
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
The Company measures its financial assets and liabilities at fair value. The carrying amounts for the Company’s financial instruments, including cash and cash equivalents, accounts payable and accrued liabilities approximate their fair value due to their short maturities. Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date.
(k)Stock-based compensation:
| (k) | Stock-based compensation: |
The Company has a stock-basedstock‑based compensation plan (the “Plan”) available to officers, directors, employees, and consultants with grants under the Plan approved by the Company’s Board of Directors. Under the Plan, the exercise price of each option equals the closing trading price of the Company’s stock on the day prior to the grant if the grant is made during the trading day or the closing trading price on the day of the grant if the grant is issued after markets have closed. Vesting is provided for at the discretion of the Board of Directors and the expiration of options is to be no greater than 10 years from the date of grant.
The Company uses the fair value based method of accounting for employee awards granted under the Plan. The Company calculates the fair value of each stock option grant using the Black-ScholesBlack‑Scholes option pricing model at the grant date. The stock-basedstock‑based compensation cost of the options is recognized as stock-basedstock‑based compensation expense over the relevant vesting period of the stock options using an estimate of the number of options that will eventually vest.
Stock options awarded to non-employeesnon‑employees are measured at the grant-date fair value of the equity instruments issued in accordance with FASB issued accounting standards update No 2018-07, Topic 718.
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
The Company has a stock incentive plan pursuant to which the Board maygrant equity settled stock-basedstock‑based awards comprised of restricted stock units or dividend equivalents to employees, officers, consultants, independent contractors, advisors and non-employeenon‑employee directors of the Company. Compensation cost for restricted share units is measured at fair value at the date of grant, which is the market price of the underlying security, and is expensed over the award’s vesting period on a straight-line basis using an estimate of the number of awards that will eventually vest.
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker, or CODM. The Company’s Chief Executive Officer serves as its CODM. The Company views its operations and manages its business as one segment, which is the discovery and development of personalized therapies addressing unmet medical needs in oncology. The Company operates primarily in the US.U.S.
Basic loss per share is computed by dividing the net loss available to common shareholders by the weighted average number of shares outstanding during the year. Diluted loss per share is computed similarly to basic loss per share except that the weighted average share outstanding is increased to include additional shares for the assumed exercise of stock options and warrants, if dilutive. The number of additional shares is calculated by assuming that outstanding stock options and warrants were exercised and that the proceeds from such exercises were used to acquire common stock at the average market price during the year. The inclusion of the Company’s stock options and warrants in the computation of diluted loss per share has an anti-dilutiveanti‑dilutive effect on the loss per share and, therefore, they have been excluded from the calculation of diluted loss per share.
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
The Company accounts for income taxes under the asset and liability method. Under this method, deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted rates in effect for the year in which these temporary differences are expected to be recovered or settled. Valuation allowances are provided if, based on the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
The Company provides reserves for potential payments of tax to various tax authorities related to uncertain tax positions and other issues. Reserves are based on a determination of whether and how much of a tax benefit taken by the Company in its tax filing is more likely than not to be realized following resolution of any potential contingencies present related to the tax benefit. Potential interest and penalties associated with such uncertain tax positions are recorded as components of income tax expense. As atof December 31, 20202023 and December 31, 2019,2022, the Company has not recorded any reserves for potential payments as the Company has a history of losses and does not have any revenue from operations.
(o) Recent Accounting Pronouncements
There were various accounting standards and interpretations issued recently, none of which are expected to have a material impact on our financial position, operations, or cash flows.
3.Cash and cash equivalents:
3. | Cash and cash equivalents: |
Cash and cash equivalents consistconsists of cash of $329$2,764 thousand (December 31, 2019 - $1.640 million)2022 ‑ $596 thousand) and deposits in high interest savings accounts, money market funds and accounts and other term deposits with original maturities of less than 90 days totaling $117.064 million$6,488 thousand (December 31, 2019 - $78.202 million)2022 ‑ $36,374 thousand).
| | | | | | | | |
| | December 31,
2023 | | | December 31,
2022 | |
Prepaid research and development expenses | | $ | 720 | | | $ | 1,271 | |
Prepaid insurance | | | 882 | | | | 893 | |
Other prepaid expenses | | | 440 | | | | 139 | |
Total | | $ | 2,042 | | | $ | 2,303 | |
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
5.Property and equipment:
| | | | | | | | | | | | |
December 31, 2023 | | Cost | | | Accumulated
depreciation | | | Net book value | |
Laboratory equipment | | $ | 197 | | | $ | 87 | | | $ | 110 | |
Computer hardware | | | 29 | | | | 25 | | | | 4 | |
Computer software | | | 222 | | | | 222 | | | | 0 | |
Office furniture | | | 140 | | | | 132 | | | | 8 | |
Leasehold improvements | | | 213 | | | | 183 | | | | 30 | |
Total | | $ | 801 | | | $ | 649 | | | $ | 152 | |
| | | | | | | | | |
December 31, 2022 | | Cost | | | Accumulated
depreciation | | | Net book value | |
Laboratory equipment | | $ | 197 | | | $ | 48 | | | $ | 149 | |
Computer hardware | | | 198 | | | | 177 | | | | 21 | |
Computer software | | | 222 | | | | 222 | | | | — | |
Office furniture | | | 140 | | | | 117 | | | | 23 | |
Leasehold improvements | | | 184 | | | | 166 | | | | 18 | |
Total | | $ | 941 | | | $ | 730 | | | $ | 211 | |
In the year ended December 31, 2022, the company recorded a loss on disposition of fixed assets of $16 thousand, with these assets having had an original cost of $196 thousand and accumulated depreciation of $180 thousand. There was no loss on disposition of fixed assets in the year ended December 31, 2023. In the years ended December 31, 2023 and December 31, 2022, the Company had additions to fixed assets of $29 thousand and $24 thousand, respectively.
| | | | December 31, | | | | December 31, | |
| | | | 2020 | | | | 2019 | |
| | | | | | | | | |
| Prepaid research and development expenses | | $ | 622 | | | $ | 451 | |
| Other prepaid expenses | | | 1,932 | | | | 574 | |
| | | $ | 2,554 | | | $ | 1,025 | |
| | | | | | | | | |
Right-of-use assets, operating leases:
| | | | | | | | |
| | Year ended December 31, 2023 | | | Year ended December 31, 2022 | |
| | | | | | |
Right-of-use assets, beginning of year | | $ | 3,100 | | | $ | 1,860 | |
Additions to right-of-use assets | | | 24 | | | | 1,240 | |
Right-of-use assets, end of year | | | 3,124 | | | | 3,100 | |
Accumulated amortization | | | (2,181 | ) | | | (1,803 | ) |
Right-of use assets, NBV | | $ | 943 | | | $ | 1,297 | |
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
5.
| Property and equipment: |
| December 31, 2020 | | | Cost | | | | Accumulated depreciation | | | | Net book value | |
| | | | | | | | | | | | | |
| Laboratory equipment | | $ | 185 | | | $ | 177 | | | $ | 8 | |
| Computer hardware | | | 170 | | | | 99 | | | | 71 | |
| Computer software | | | 222 | | | | 174 | | | | 48 | |
| Office furniture | | | 140 | | | | 72 | | | | 68 | |
| Leasehold improvements | | | 184 | | | | 118 | | | | 66 | |
| | | $ | 901 | | | $ | 640 | | | $ | 261 | |
| | | | | | | | | | | | | |
| December 31, 2019 | | | Cost | | | | Accumulated depreciation | | | | Net book value | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Laboratory equipment | | $ | 185 | | | $ | 160 | | | $ | 25 | |
| Computer hardware | | | 122 | | | | 60 | | | | 62 | |
| Computer software | | | 222 | | | | 128 | | | | 94 | |
| Office furniture | | | 116 | | | | 51 | | | | 65 | |
| Leasehold improvements | | | 177 | | | | 89 | | | | 88 | |
| | | $ | 822 | | | $ | 488 | | | $ | 334 | |
| 6. | Right of-use assets, operating leases: |
| | | | Year ended December 31, 2020 | | | | Year ended
December 31, 2019 | |
| | | | | | | | | |
| Right-of-use assets, beginning of year | $ | 1,837 | | | $ | 1,570 | |
| Additions to right-of-use assets | | | 11 | | | | 267 | |
| Right-of-use assets, end of year | | | 1,848 | | | | 1,837 | |
| Accumulated amortization | | | (923 | ) | | | (461 | ) |
| Right-of use assets, NBV | | | 925 | | | | 1,376 | |
| | | | | | | | | |
Investments consisted of the following as of December 31, 20202023 and December 31, 2019:2022:
| | | | December 31, 2020 | |
| | | | Cost | | | | Unrealized gain/(loss) | | | | Market value | |
| | | | | | | | | | | | | |
| United States Treasury Bills | | | 5,000 | | | | - | | | | 5,000 | |
| | | | 5,000 | | | | - | | | | 5,000 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | |
APTOSE BIOSCIENCES INC.
|
| December 31, 2023 |
|
Notes to Consolidated Financial Statements
|
| Cost |
|
| Unrealized loss |
|
| Market value |
|
Year ended December 31, 2020 and 2019United States Treasury Bills
|
| $ | — |
|
| $ | — |
|
| $ | — |
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)Total
|
| $ | — |
|
| $ | — |
|
| $ | — |
|
| | | | | | | | | | | | |
| | December 31, 2022 | |
| | Cost | | | Unrealized gain | | | Market value | |
United States Treasury Bills | | $ | 9,991 | | | $ | (2 | ) | | $ | 9,989 | |
Total | | $ | 9,991 | | | $ | (2 | ) | | $ | 9,989 | |
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
| | | | December 31, 2019 | |
| | | | Cost | | | | Unrealized gain | | | | Market value | |
| | | | | | | | | | | | | |
| Guaranteed investment certificates, issued by a Canadian financial institution | | | 12,008 | | | | 18 | | | | 12,026 | |
| Commercial notes | | | 3,736 | | | | - | | | | 3,736 | |
| Canadian provincial promissory note | | | 1,996 | | | | - | | | | 1,996 | |
| | | | 17,740 | | | | 18 | | | | 17,758 | |
| | | | | | | | | | | | | |
8.Fair value measurements and financial instruments:
8. | Fair value measurements and financial instruments: |
The fair value hierarchy establishes three levels to classify the inputs to valuation techniques used to measure fair value.
Level 1 -‑ inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities;
Level 2 -‑ inputs are quoted prices in markets that are not active, quoted prices for similar assets or liabilities in active markets, inputs other than quoted prices that are observable for the asset or liability, or inputs that are derived principally from or corroborated by observable market data or other means; and
Level 3 -‑ inputs are unobservable (supported by little or no market activity).
The fair value hierarchy gives the highest priority to Level 1 inputs and the lowest priority to Level 3 inputs.
The following table presents the Company’s assets that are measured at fair value on a recurring basisof the Company’s financial instruments for the periods presented:
| | | | | | | | | | | | | | | | |
| | December 31,
2023 | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets | | | | | | | | | | | | |
High interest savings accounts | | $ | 2,002 | | | $ | — | | | $ | 2,002 | | | $ | — | |
United States Treasury Bills | | | 4,486 | | | | — | | | | 4,486 | | | | — | |
Total | | $ | 6,488 | | | $ | — | | | $ | 6,488 | | | $ | — | |
| | | | | | | | | | | | | | | | |
| | December 31,
2022 | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets | | | | | | | | | | | | |
Money Market accounts | | $ | 165 | | | $ | — | | | $ | 165 | | | $ | — | |
Money Market Funds | | | 22,343 | | | | — | | | | 22,343 | | | | — | |
High interest savings accounts | | | 13,866 | | | | — | | | | 13,866 | | | | — | |
United States Treasury Bills | | | 9,989 | | | | — | | | | 9,989 | | | | — | |
Total | | $ | 46,363 | | | $ | — | | | $ | 46,363 | | | $ | — | |
| | | | December 31, 2020 | | | | Level 1 | | | | Level 2 | | | | Level 3 | |
| Assets | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Money Market accounts | | $ | 668 | | | | - | | | $ | 668 | | | | - | |
| Money Market Funds | | | 44,000 | | | | - | | | | 44,000 | | | | - | |
| High interest savings accounts | | | 48,397 | | | $ | - | | | | 48,397 | | | | - | |
| United States Treasury Bill | | | 5,000 | | | | - | | | | 5,000 | | | | - | |
| Government of Canada Treasury Bill | | | 23,999 | | | | - | | | | 23,999 | | | | - | |
| | | $ | 122,064 | | | $ | - | | | $ | 122,064 | | | $ | - | |
9.Accrued liabilities:
| | | | December 31, 2019 | | | | Level 1 | | | | Level 2 | | | | Level 3 | |
| Assets | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| High interest savings account | | $ | 2,989 | | | $ | - | | | $ | 2,989 | | | | - | |
| Commercial notes | | | 6,235 | | | | - | | | | 6,235 | | | | - | |
| Canadian provincial promissory notes | | | 5,493 | | | | - | | | | 5,493 | | | | - | |
| Guaranteed investment certificates, issued by a Canadian financial institution | | | 81,243 | | | | - | | | | 81,243 | | | | - | |
| | | | | | | | | | | | | | | | | |
| | | $ | 95,960 | | | $ | - | | | $ | 95,960 | | | $ | - | |
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
Accrued liabilities as of December 31, 20202023 and December 31, 20192022 consisted of the following:
| | | | | | | | |
| | December 31,
2023 | | | December 31,
2022 | |
Accrued personnel-related costs | | $ | 1,989 | | | $ | 2,302 | |
Accrued research and development expenses | | | 6,527 | | | | 2,550 | |
Other accrued expenses | | | 313 | | | | 233 | |
Total | | $ | 8,829 | | | $ | 5,085 | |
| | | | December 31, | | | | December 31, | |
| | | | 2020 | | | | 2019 | |
| | | | | | | | | |
| Accrued personnel related costs | | $ | 1,917 | | | $ | 1,739 | |
| Accrued research and development expenses | | | 1,932 | | | | 1,062 | |
| Other accrued expenses | | | 253 | | | | 257 | |
| | | $ | 4,102 | | | $ | 3,058 | |
Aptose leases office space in San Diego, California and Toronto, Canada. The lease for the San Diego office space is scheduled to expire in May 31, 2026. Aptose previously leased lab space in San Diego, California. Thewhich we exited prior to the expiration of the lease for the office space expires on March 31, 2023 and can be extended for an additional 5 year period. The lease for our lab space expires on February 28, 2022.2023. The costs incurred in exiting this laboratory space were not material. We lease office space in Toronto, Ontario, Canada, and thewith this lease for this location expiresscheduled to expire on June 30, 2023 with an option to renew for another 5-year period.2024. The Company has not included any extension periods in calculating its right-to-use assets and lease liabilities. The Company also enters into leases for small office equipment.
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
Minimum payments, undiscounted, under our operating leases are as follows:
| Years ending December 31, | | | | |
| 2021 | | $ | 552 | |
| 2022 | | | 464 | |
| 2023 | | | 119 | |
| Thereafter | | | - | |
| | | $ | 1,135 | |
| | | | |
Years ending December 31, | | | |
2024 | | | 459 | |
2025 | | | 462 | |
2026 | | | 198 | |
Total | | $ | 1,119 | |
To calculate the lease liability, the lease payments in the table above were discounted over the remaining term of the leases using the Company’s incremental borrowing rate as atof January 1, 2019 for existing leases at the time of adopting the TopicASC 842, and for new leases after the date adoption, as atof the date of the execution date of the new lease. The following table presents the weighted average remaining term of the leases and the weighted average discount rate:
| | | | Year ended December 31, 2020 | | | | Year ended December 31, 2019 | |
| Weighted-average remaining term – operating leases (years) | | | 2.1 | | | | 3.3 | |
| Weighted-average discount rate – operating leases | | | 5.40 | % | | | 5.43 | % |
| | | | | | | | | |
| Lease liability, current portion | | | 539 | | | | 521 | |
| Lease liability, long term portion | | | 535 | | | | 1,011 | |
| Lease liability, total | | | 1,074 | | | | 1,532 | |
| | | | | | | | |
| | December 31,
2023 | | | December 31,
2022 | |
Weighted-average remaining term – operating
leases (years) | | | 2.4 | | | | 3.3 | |
Weighted-average discount rate – operating
leases | | | 7.38 | % | | | 6.62 | % |
Lease liability, current portion | | $ | 394 | | | $ | 301 | |
Lease liability, long term portion | | | 621 | | | | 1,002 | |
Lease liability, total | | $ | 1,015 | | | $ | 1,303 | |
Right-of-use assets obtained in exchange for new operating lease liabilities are as follows:
| | | | Year ended December 31, 2020 | | | | Year ended December 31, 2019 | |
| | | | | | | | | |
| Right-of-use assets recorded upon adoption of Topic 842, beginning of period | | | - | | | $ | 1,570 | |
| Right-of-use assets obtained in exchange for new operating lease liabilities in the period | | $ | 11 | | | $ | 267 | |
| | | | | | | | | |
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
Operating lease costs and operating cash flows from our operating leases are as follows:
| | | | Year ended December 31, 2020 | | | | Year ended December 31, 2019 | |
| | | | | | | | | |
| Operating lease cost | | $ | 530 | | | $ | 551 | |
| | | | | | | | | |
| Operating cash flows from operating leases | | $ | 537 | | | $ | 471 | |
| | | | | | | | | |
| | | | | | | | |
| | Year ended
December 31, 2023 | | | Year ended
December 31, 2022 | |
| | | | | | |
Operating lease cost | | $ | 471 | | | $ | 443 | |
Operating cash flows from operating leases | | $ | 405 | | | $ | 546 | |
11.Related party transactions
Hanmi Pharmaceutical Co. Ltd.
On November 4, 2021, Aptose entered into a licensing agreement (the "Tuspetinib Licensing Agreement") with the South Korean company Hanmi. Under the terms of the Tuspetinib Licensing Agreement, Hanmi granted Aptose exclusive worldwide rights to tuspetinib for all indications. Hanmi received an upfront payment of $12.5 million, including $5 million in cash and $7.5 million in Common Shares. Aptose issued Hanmi 215,703 Common Shares in this upfront licensing payment. Hanmi will also receive up to $407.5 million in future milestone payments contingent upon achieving certain clinical, regulatory and sales milestones across several potential indications, as well as tiered royalties on net sales. The term of the agreement will continue on a product-by-product and country-by-country basis until the expiration of the royalty period for such product in such country. The licenses to Aptose will survive and become non-exclusive, perpetual, irrevocable and fully paid-up on a product-by-product and country-by-country basis, upon their natural expiration under the terms of the agreement.
In 2022, the Company and Hanmi also entered into a separate supply agreement for additional production of new drug substance ("API") and drug product to support further tuspetinib clinical development, for which the Company pays Hanmi per batch of production. Expenses related to this supply agreement have been recognized by the Company, amounting to $3.5 million and approximately $3.6 million, for the years ended December 31, 2023 and 2022, respectively. Since inception to December 31, 2023, $7.1 million has been recognized for the period under the supply agreement.
The Company paid supply costs to Hanmi of $4.5 million and nil in the years ended December 31, 2023 and 2022, respectively. Since inception to December 31, 2023, payments of $4.5 million have been made under the supply
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
agreement. At December 31, 2023, there was $2.6 million in accounts payable and nil in accrued liabilities to Hanmi related to the supply agreement. (At December 31, 2022, there was $3.0 million in accounts payable and $0.6 million in accrued liabilities). Transactions involving related parties cannot be presumed to be carried out on an arm's-length basis, as the requisite conditions of competitive, free-market dealings may not exist.
On August 10, 2023, the Company entered into a binding term sheet with Hanmi whereby Hanmi agreed at their sole discretion to invest, up to a maximum of $7 million in Aptose up to a total ownership of 19.99 percent of Aptose by Hanmi. On September 6, 2023, the Company entered into a subscription agreement with Hanmi, pursuant to which the Company sold 668,449 Common Shares to Hanmi for gross proceeds of $3 million. The issuance of shares, in exchange for gross proceeds of $3 million, is recorded in Common Shares on the balance sheet as of December 31, 2023. For information regarding Hanmi's investment subsequent to December 31, 2023, see Note 17, Subsequent events.
Hanmi held 884,152 Common Shares of Aptose as of December 31, 2023. As of the filing date, the total common share ownership by Hanmi was 2,989,414, representing ownership of 19.03% of the outstanding Common Shares of the Company. Hanmi also owns 2,339,181 warrants to purchase Common Shares at an exchange price of $1.71 per Warrant Share. See Note 17, Subsequent events for information about shares sold to Hanmi subsequent to December 31, 2023 and the Nasdaq Deficiency Letter.
The Company has authorized share capital of an unlimited number of common voting shares.
| (i) | July/August 2020 Confidentially Marketed Public Offering (CMPO) |
(i) Hanmi
On July 20, 2020 and August 10, 2020, the Company completed a confidentially marketed public offering through the issuance of 11,854,472 common shares at a price of $5.25 per share for gross proceeds of $62.236 million (approximately $58.234 million net of share issue costs). Costs associated with the proceeds consisted of a 6% cash commissions and share issue costs, which consisted of agent commission, legal and professional fees and listing fees.
| (ii) | December 2019 Confidentially Marketed Public Offering (CMPO) |
On December 19, 2019, the Company completed a confidentially marketed public offering through the issuance of 18,543,750 common shares at a price of $4.00 per share for gross proceeds of $74.175 million (approximately $68.588 million net of share issue costs). Costs associated with the proceeds consisted of a 7% cash commissions and share issue costs, which consisted of agent commission, legal and professional fees and listing fees.
| (iii) | June 2019 Confidentially Marketed Public Offering (CMPO) |
On June 3, 2019, the Company completed a confidentially marketed public offering through the issuance of 11,500,000 common shares at a price of $1.85 per share for gross proceeds of $21.275 (approximately $19.594 million net of share issue costs). Costs associated with the proceeds consisted of a 7% cash commissions and share issue costs, which consisted of agent commission, legal and professional fees and listing fees.
| (iv) | 2019 Share Purchase agreement |
On May 7, 2019,2023, the Company entered into a binding term sheet with Hanmi whereby Hanmi agreed at their sole discretion to invest, up to a maximum of $7 million in Aptose up to a total ownership of 19.99 percent of Aptose by Hanmi. On September 6, 2023, the 2019 Aspire Purchase Agreement,Company entered into a subscription agreement with Hanmi, pursuant to which providedthe Company sold 668,449 Common Shares to Hanmi for proceeds of $3 million.
Hanmi held 884,152 Common Shares of Aptose as of December 31, 2023 See Note 17, Subsequent events.
(ii) 2023 Committed Equity Facility
On May 25, 2023, the Company and Keystone Capital Partners, LLC ("Keystone") entered into a committed equity facility, (the "2023 Committed Equity Facility"), which provides that uponsubject to the terms and subject to the conditions and limitations set forth therein, Aspire Capitalwe may sell to Keystone up to the lesser of (i) $25.0 million of the Common Shares and (ii) a number of Common Shares equal to 19.99% of the Common Shares outstanding immediately prior to the execution of the 2023 Committed Equity Facility Agreement with Keystone which respect to the 2023 Committed Equity Facility (subject to certain exceptions) (the “Total Commitment”), from time to time during the 24-month term of the 2023 Committed Equity Facility. Additionally, on May 25, 2023, the Company entered into a Registration Rights Agreement with Keystone, pursuant to which the Company agreed to file a registration statement with the SEC covering the resale of Common Shares that are issued to Keystone under the 2023 Committed Equity Facility. This registration statement became effective on June 30, 2023 and the 2023 Committed Equity Facility commencement date was committedJuly 12, 2023 (the "Commencement Date").
Upon entering into the 2023 Committed Equity Facility, the Company agreed to purchase upissue to Keystone an aggregate of $20 million25,156 Common Shares (the “Commitment Shares”) as consideration for Keystone’s commitment to purchase Common Shares upon the Company’s direction under the 2023 Committed Equity Facility. The Company issued 7,547 Common Shares, or 30% of the Commitment Shares, on the date of the 2023 Committed Equity Facility Agreement (the “Initial Commitment Shares”). An additional 7,547
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
Common Shares, or 30% of the Commitment Shares, were issued to Keystone 90 days following the Commencement Date (the “First Back-End Commitment Shares”). The remaining 10,062 Common Shares, or 40% of the Commitment Shares, shall be issued to Keystone 180 days following the Commencement Date (the “Second Back-End Commitment Shares”, together with the First Back-End Commitment Shares, the “Back-End Commitment Shares”). See Note 17, Subsequent events.
In the year ended December 31, 2023, the Company's issuance of Common Shares over approximately 30 months. Pursuant to the terms of this agreement, on May 13, 2019, the Company issued 171,428Keystone comprised 720,494 Common Shares (“Commitment Shares”)shares sold to Aspire Capital in consideration for entering into the 2019 Aspire Purchase Agreement for a total cost of $360 thousand. During the period from May 7, 2019 up to December 16, 2019, the date the 2019 Aspire Purchase Agreement was terminated, the Company issued 1,800,000 common shares under the agreementKeystone at an average price of $2.43$2.91 per Common share for gross and netcash proceeds of $4.37 million.$2.1 million and the 15,094 Commitment Shares. See Note 17, Subsequent events.
| (v) | 2018 Share Purchase agreement |
(iii) 2022 At-The-Market Facility
On May 30, 2018, the Company entered into the 2018 Aspire Purchase Agreement, which provided that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital was committed to purchase up to an aggregate of $20 million of Common Shares over approximately 30 months. Pursuant to the terms of this agreement, on June 8, 2018, the Company issued 170,261 Common Shares (“Commitment Shares”) to Aspire Capital in consideration for entering into the 2018 Aspire Purchase Agreement for a total cost of $600 thousand. During the period from January 1, 2019 up to May 24, 2019, the date the 2018 Aspire Purchase Agreement was terminated, the Company issued 5,502,433 common shares under the agreement at an average price of $1.82 per share for gross and net proceeds of $10 million. On a cumulative basis up to May 24, 2019, the Company raised a total of approximately $11.9 million gross and net proceeds under the 2018 Aspire Purchase Agreement. As of May 24, 2019, the Company has issued 6,409,980, the maximum number of shares issuable under this facility without shareholder approval.
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
| (vi) | 2020 At-The-Market (“ATM”) Facility |
On May 5, 2020,December 9, 2022, the Company entered into an equity distribution agreement with Piper Sandler and Canaccord Genuity acting as co-agents in connection with the 2020 ATM Facility. Under the terms of the 2020 ATM Facility,pursuant to which the Company may, from time to time, sell Common Shares having an aggregate offering value of up to $75$50 million through Jones Trading Institutional Services LLC ("Jones Trading") on Nasdaq (the "2022 ATM Facility"). During the year ended December 31, 2022, the Company issued 4,836 Common Shares under this 2022 ATM Facility at an average price of $10.81 per Common Share for gross proceeds of $52 thousand ($51 thousand net of share issuance costs). During the year ended December 31, 2023, the Company issued 336,690 Common Shares under this 2022 ATM Facility at an average price of $5.62 for gross proceeds of $1.9 million ($1.8 million net of share issuance costs). Since inception to December 31, 2023, the Company had raised a total of $1.9 million gross proceeds ($1.9 million net of share issuance costs) under the 2022 ATM Facility. Costs associated with the proceeds consisted of 3% cash commission.
(iv) 2020 At-The-Market Facility
On May 5, 2020, the Company entered an “at-the-market” equity distribution agreement with Piper Sandler & Co. (“Piper Sandler”) and Canaccord Genuity LLC (“Canaccord Genuity”) acting as co-agents (the “2020 ATM Facility”). Under the terms of the 2020 ATM Facility, the Company could, from time to time, sell Common Shares having an aggregate offering value of up to $75 million through Piper Sandler and Canaccord Genuity on the Nasdaq Capital Market. During the year ended December 31, 2020, the Company did not issue any shares under the 2020 ATM Facility.
| (vii) | 2019 At-The-Market (“ATM”) Facility |
On May 24, 2019, the Company entered into an “At-The-Market” Facility (“ATM”) equity distribution agreement with Piper Jaffray and Canaccord Genuity acting as co-agents. Under the terms of this facility, the Company may, from time to time, sell shares of our common stock having an aggregate offering value of up to $40 million through Piper Jaffray and Canaccord Genuity on the Nasdaq Capital Market. During the period from May 24, 2019 to December 16, 2019, the date the “ATM” Facility was terminated, the Company did not issue any shares under this ATM equity.
| (viii) | 2018 At-The-Market (“ATM”) Facility |
On March 27, 2018, the Company entered into an “At-The-Market” Facility (“ATM”) equity distribution agreement with Cantor Fitzgerald acting as sole agent. Under the terms of this facility, the Company may, from time to time, sell shares of our common stock having an aggregate offering value of up to $30 million through Cantor Fitzgerald on the Nasdaq Capital Market.Nasdaq. During the period from January 1, 20192022 to May 24, 2019,October 31, 2022, the date the Agreement was terminated, the company issued During the year ended December 31, 2022, the Company issued 77,349 shares3,646 Common Shares under thisthe 2020 ATM equity facilityFacility at an average price of $2.37$14.25 per Common Share for gross proceeds of $183$52 thousand ($17850 thousand net of share issueissuance costs). As of October 31, 2022, the date the 2020 ATM Facility was terminated, the Company had raised a total of $89 thousand gross proceeds ($86 thousand net of share issuance costs) under the 2020 ATM Facility. Costs associated with the proceeds consisted of a 3%3% cash commission. As of May 24, 2019, the Company has raised a total of $11.2 million gross proceeds ($10.9 million net of share issue costs) under the ATM Facility.
For information regarding recent equity issuances subsequent to December 31, 2023, see Note 17, Subsequent events.
Loss per common share is calculated using the weighted average number of common sharesCommon Shares outstanding and is presented in the table below:
| (in thousands) | | | Year ended
December 31, 2020 | | | | Year ended
December 31, 2019 | |
| | | | | | | | | |
| Net loss | | $ | (55,238 | ) | | $ | (26,277 | ) |
| Weighted-average common shares – basic and diluted | | | 81,837 | | | | 50,160 | |
| Net loss per share – basic and diluted | | $ | (0.67 | ) | | $ | (0.52 | ) |
| | | | | | | | |
(in thousands) | | Year ended December 31, 2023 | | | Year ended December 31, 2022 | |
Net loss | | $ | (51,207 | ) | | $ | (41,823 | ) |
Weighted-average common shares – basic
and diluted | | | 6,755 | | | | 6,151 | |
Net loss per share – basic and diluted | | $ | (7.58 | ) | | $ | (6.80 | ) |
The effect of any potential exercise of the Company’s stock options outstanding during the yearyears ended December 31, 2023 and December 31, 2022 has been excluded from the calculation of diluted loss per common share as it would be anti-dilutive.anti‑dilutive.
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
F-19
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
| 12. | Stock-based compensation: |
| (a) | 13.Stock‑based compensation: |
Under the Company’s stock option plan and employee stock purchase plan
Effective June 1, 2021,the Company adopted a new stock incentive plan (New Incentive Plan) and an employee stock purchase plan (ESPP).
The New Incentive Plan authorizes the Board of Directors to administer the New Incentive Plan to provide equity‑based compensation in the form of stock options, rightsstock appreciation rights., restricted stock, restricted stock units and other entitlementsDividend Equivalents.
The Corporation currently maintains its existing Share Option Plan and 2015 Stock Incentive Plan (2015 SIP). Effective June 1, 2021 no further grants will be made under the Share Option Plan or 2015 SIP, though existing grants under the Share Option Plan will remain in effect in accordance with their terms.
The aggregate number of our common shares, no par value, that may be grantedissued under all awards under the New Incentive Plan is (i) 691,400, plus (ii) any of our Common Shares subject to directors, officers, employees and consultantsany outstanding award under our prior plans that, after June 1, 2021, are not purchased or are forfeited or reacquired by us, or otherwise not delivered to the participant due to termination, cancellation or cash settlement of such award subject to the share counting provisions of the Company to purchase up to a maximumNew Incentive Plan.
Under both the Share Option Plan and the New Incentive Plan, the exercise price of 17.5% of the total number of outstanding common shares, estimated at 15.6 million options, rights and other entitlements as at December 31, 2020. Options are granted at the fair market value of the common shares oneach option equals the closing trading price of the Company’s stock on the day prior to the grant if the grant is made during the trading day or the closing trading price on the day of grant if the grant is issued after markets have closed. Options vestVesting is provided for at various rates (immediatethe discretion of the Board of Directors and the expiration of options is to four years)be no greater than 10 years from the date of grant.
The Company uses the fair value-based method of accounting for employee awards granted under both plans. The Company calculates the fair value of each stock option grant using the Black‑Scholes option pricing model at the grant date. The stock‑based compensation cost of the options is recognized as stock‑based compensation expense over the relevant vesting period of the stock options using an estimate of the number of options that will eventually vest.
The ESPP, which is administered by the Board of Directors, allows eligible employees of the Company with an opportunity to purchase Common Shares through accumulated payroll deductions up to a maximum 15% of eligible compensation. The ESPP will be implemented by consecutive offering periods with a new offering period commencing on the first trading day on or after February 1 and haveAugust 1 each year, or on such other date as the Board of Directors will determine, and continuing thereafter until terminated in accordance with the Plan. Unless the Board of Directors provides otherwise, the purchase price will be equal to eighty‑five percent (85%) of the fair market value of a termCommon Share on the offering date or the exercise date, whichever is lower.
The maximum number of 10 years.Common Shares which will be made available for sale under the ESPP will be 113,333 Common Shares.
The first six-month offering period began on February 1, 2022 and ended on August 1, 2022. The second six-month period began on August 1, 2022 and ended on February 1, 2023. The third six-month period began on February 2, 2023, and ended on August 1, 2023. There were 5,891 and 724 Common Shares issued under the ESPP in the years ended December 31, 2023 and 2022, respectively.
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
Stock option transactions for the yearsyear ended December 31, 20192023 and 2020December 31, 2022, are summarized as follows:
| Option numbers are in (000’s) | | | | | | | | |
| | | | | | | Weighted average | | Aggregate |
| | | Options | | Weighted average
exercise price | | remaining contractual
life (years) | | Intrinsic
Value |
| | | | | | | | | |
| Outstanding, December 31, 2018 | | | 4,489 | | | | 3.11 | | | | | | | | | |
| Granted | | | 2,160 | | | | 2.00 | | | | | | | | | |
| Exercised | | | (312 | ) | | | 2.32 | | | | | | | | | |
| Expired | | | (67 | ) | | | 4.61 | | | | | | | | | |
| Forfeited | | | (329 | ) | | | 2.36 | | | | | | | | | |
| Outstanding, December 31, 2019 | | | 5,941 | | | $ | 2.84 | | | | | | | | | |
| Granted | | | 6,362 | | | | 6.81 | | | | | | | | | |
| Exercised | | | (235 | ) | | | 2.45 | | | | | | | | | |
| Forfeited | | | (126 | ) | | | 4.50 | | | | | | | | | |
| Outstanding, December 31, 2020 | | | 11,942 | | | | 4.97 | | | | 7.9 | | | $ | 9,142,263 | |
| Exercisable, December 31, 2020 | | | 4,363 | | | | 3.06 | | | | 6.2 | | | $ | 6,470,708 | |
Vested and expected to vest, December 31, 2020 | | | 10,804 | | | | 4.86 | | | | 7.8 | | | $ | 8,741,530 | |
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | |
| | Options | | | Weighted average
exercise price | | | Weighted
average
remaining
contractual
life (years) | | | Aggregate
Intrinsic
Value | |
| | (in thousands) | | | | | | | | | | |
Outstanding, December 31, 2021 | | | 1,007 | | | $ | 69.15 | | | | | | | |
Granted | | | 420 | | | | 17.85 | | | | | | | |
Exercised | | | (1 | ) | | | 16.20 | | | | | | | |
Forfeited | | | (326 | ) | | | 57.60 | | | | | | | |
Outstanding, December 31, 2022 | | | 1,100 | | | $ | 52.20 | | | | | | | |
Granted | | | 217 | | | | 9.87 | | | | | | | |
Exercised | | | 0 | | | | | | | | | | |
Forfeited | | | (133 | ) | | | 50.87 | | | | | | | |
Outstanding, December 31, 2023 | | | 1,184 | | | $ | 44.78 | | | | 6.9 | | | $ | — | |
Exercisable, December 31, 2023 | | | 714 | | | $ | 59.00 | | | | 5.9 | | | $ | — | |
Vested and expected to vest, December 31, 2023 | | | 1,099 | | | $ | 46.46 | | | | 6.8 | | | $ | — | |
Aggregate intrinsic value represents the excess of the value of the closing stock price on the previous trading day of the respective balance sheet dates over the exercise price of the stock options. Total intrinsic value of options exercised was $850 thousandnil for 2020 (20192023 (2022 – $259$3 thousand).
As of December 31, 2020, 2023, there was $9.52$1.2 million of total unrecognized compensation cost related to non-vested stock options, which is expected to be recognized over an estimated weighted-average period of 1.831.7 years.
The following table presents the weighted average assumptions that were used in the Black-ScholesBlack‑Scholes option pricing model to determine the fair value of stock options granted during the year,period, and the resultant weighted average fair values:
| | | | Year ended December 31, 2020 | | | | Year ended December 31, 2019 | |
| | | | | | | | | | | Year ended December 31, 2023 | | | Year ended December 31, 2022 | |
| Risk-free interest rate | | | 1.27 | % | | | 2.18 | % | | | 3.42 | % | | | 2.17 | % |
| Expected dividend yield | | | - | | | | - | | | | — | | | | — | |
| Expected volatility | | | 85.9 | % | | | 83.9 | % | | | 80.3 | % | | | 82.7 | % |
| Expected life of options (years) | | | 5 | | | | 5 | | | | 5 | | | | 5 | |
| Grant date fair value | | $ | 4.59 | | | $ | 1.34 | | | $ | 6.53 | | | $ | 11.85 | |
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
The Company uses historical data to estimate the expected dividend yield and expected volatility of its common sharesCommon Shares in determining the fair value of stock options. The expected life of the options represents the estimated length of time the options are expected to remain outstanding.
The following table presents the vesting terms of options granted in the period:
| | | | | | | | |
| | Year ended December 31, 2023 | | | Year ended December 31, 2022 | |
| | Options | | | Options | |
| | (in thousands) | | | (in thousands) | |
3-year vesting (50%-25%-25%) | | | 48 | | | | 28 | |
4-year vesting (50%-16 2/3%-16 2/3%-16 2/3%) | | | 169 | | | | 352 | |
Earlier of performance criteria or 4 years | | | — | | | | 40 | |
Total stock options granted in the year | | | 217 | | | | 420 | |
| Option numbers are in (000’s) | | | Year ended
December 31, 2020 | | | | Year ended
December 31, 2019 | |
| | | | Number of options | | | | Number of options | |
| | | | | | | | | |
| Cliff vesting after one year anniversary | | | 300 | | | | 335 | |
| 3 year vesting (50%-25%-25%) | | | 862 | | | | 160 | |
| 4 year vesting (50%-16 2/3%-16 2/3%-16 2/3%) | | | 5,200 | | | | 1,665 | |
| Total stock options granted in the period | | | 6,362 | | | | 2,160 | |
F-21
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
During the year ended December 31, 2022, the option agreements of one Company officer were modified as part of a separation and release agreement. Vested options of 56,737, with exercise prices ranging from $20.1 to $103.65, were allowed to continue to be exercisable for an additional 12-month period, and also 31,810 options that would have expired unvested, were allowed to continue to vest for a 12-month period. As there was no service requirement, during the year ended December 31, 2022, the company recorded $67 thousand in additional compensation related to these modifications.
| (b) | Restricted share units |
The Company has a stock incentive plan (SIP) pursuant to which the Board may grant stock-based awards comprised of restricted stock units or dividend equivalents to employees, officers, consultants, independent contractors, advisors and non-employee directors of the Company. Each restricted stock unit ("RSU") is automatically redeemed for one common shareCommon Share of the Company upon vesting. During the year ended December 31, 2023, the Company granted 38,000 RSUs with immediate vesting and an exercise price of $9.90 (the "2023 RSU Grant"). On February 6, 2023, all of these RSUs were redeemed for 38,000 Common Shares. The following table presents the activity undervesting and redemption of the SIP plan forRSUs granted in the year ended December 31, 2020 and 2019 the units outstanding.
| | | Year ended,
December 31, 2020 | | Year ended,
December 31, 2019 |
| | | | Number (in thousands) | | | | Weighted average grant date fair value | | | | Number (in thousands) | | | | Weighted average grant date fair value | |
| Outstanding, beginning of year | | | 40 | | | $ | 2.00 | | | | - | | | $ | - | |
| Granted | | | 645 | | | | 7.32 | | | | 80 | | | | 2.00 | |
| Redeemed | | | (685 | ) | | | 7.01 | | | | (40 | ) | | | 2.00 | |
| Outstanding, end of year | | | - | | | $ | - | | | | 40 | | | $ | 2.00 | |
On March 10, 2020, the Company granted 645,000 restricted share units (RSUs) with a vesting term of three months. On May 5, 2020, the vesting term on the RSUs was extended from three months to four months. On July 10, 2020, all of these restricted share units were vested and were redeemed for 645,000 common shares.
On June 3, 2019, the Company granted 80,000 restricted share units (RSUs), 40,000 restricted share units of which have a vesting term of three months and the balance having a vesting term of one year. On September 3, 2019, 50% of these restricted share units were vested and were redeemed for 40,000 common shares. On May 5, 2020, the vesting term on the balance was extended from one year to one year and one month. On July 2, 2020, the remaining of these restricted share units were vested and were redeemed for 40,000 common shares.
The grant date fair value of the March 10, 2020 and June 3, 20192023. No RSUs were determined asgranted in the closing value of the common shares of the Company on the Nasdaq Stock Market on the date prior to the date of grant.year ended December 31, 2022.
| | | | | | | | | | | | |
| Year ended
December 31, 2023 |
| Year ended
December 31, 2022 |
|
| Number of options
(in thousands) |
| Weighted average grant date fair value |
| Number of options
(in thousands) |
| Weighted average grant date fair value |
|
Outstanding, beginning of period |
| - |
| $ | - |
|
| - |
| $ | - |
|
Granted |
| 38 |
|
| 9.90 |
|
| - |
|
| - |
|
Vested and redeemed |
| (38 | ) |
| (9.90 | ) |
| - |
|
| - |
|
Outstanding, ending of period |
| - |
| $ | - |
|
| - |
| $ | - |
|
(b)Share-based payment expense
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
| (c) | Share-based payment expense |
The Company recorded share-based payment expense related to stock options and RSUs as follows:
| | | | | | | | |
| | Year ended December 31, 2023 | | | Year ended December 31, 2022 | |
Research and development | | $ | 1,373 | | | $ | 2,218 | |
General and administrative | | | 2,280 | | | | 2,989 | |
Total | | $ | 3,653 | | | $ | 5,207 | |
| | | | Year ended
December 31, 2020 | | | | Year ended
December 31, 2019 | |
| | | | | | | | | |
| Research and development | | $ | 3,720 | | | $ | 474 | |
| General and administrative | | | 17,718 | | | | 1,822 | |
| Total | | $ | 21,438 | | | $ | 2,296 | |
14.Collaborative agreements:
13. | Related party transactions: |
The Company uses Moores Cancer Center at the University of California San Diego (UCSD) to provide pharmacology lab services to the Company. Dr. Stephen Howell is a member of our Scientific Advisory Board and former Acting Chief Medical Officer of Aptose up to January 1, 2020 and is also a Professor of Medicine at UCSD and oversees the laboratory work. The work is completed under the terms of research services agreements executed in March 2015 and has been extended annually. These transactions are in the normal course of business and are measured at the amount of consideration established and agreed to by the related parties.
During the comparative year ended December 31, 2019, while Dr. Howell was Acting Chief Medical Officer, the Company recorded $223 thousand in research and development expenses related to the agreement.
14. | Collaborative agreements: |
The Company enters into research, development and license agreements in the ordinary course of business where the Company receives research services and rights to proprietary technologies. Milestone and royalty payments that may become due under various agreements are dependent on, among other factors, clinical trials, regulatory approvals and ultimately the successful development of a new drug, the outcome and timing of which is uncertain.
On November 4, 2021, the Effective Date, the Company entered into the Tuspetinib Licensing Agreement with Hanmi for global rights to tuspetinib. In consideration of the license and other rights granted, Aptose made an upfront payment to Hanmi in the amount of $12.5 million, including $5.0 million in cash and $7.5 million in Aptose Common Shares (the “Shares”). The number of Shares issued was determined using the average market closing price of the Common Shares on the NASDAQ stock market over the five (5) trading day period ending on the Effective Date. Accordingly, Aptose issued 215,703 shares to Hanmi.
APTOSE BIOSCIENCES INC.
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
Under the Company’s license agreement with Hanmi, the Company has maximum obligations for clinical development and global regulatory milestones totaling $64.5 million for the first potential clinical indication of tuspetinib, $34 million for the second indication, and $29 million for the third indication. The Company has maximum obligations for tiered global sales-based milestones totaling $280 million. The Company also has an obligation for tiered royalty payments on global sales of commercialized product. The timing of any milestone or royalty payments that may become due is not yet determinable.
On March 21, 2016, the Company entered into a license agreement with CrystalGenomics Invites Co. Ltd. ("CG"), formerly CrystalGenomics, Inc.) for rights to CG-806,luxeptinib, in all territories outside of the Republic of Korea and China, the Company has obligations for development milestones of $16$16 million related to the initiation of Phase 2 and pivotal clinical trials, and regulatory milestones totaling $44$44 million. The Company also has an obligation to pay royalty payments on sales of commercialized product. The timing of any milestone or royalty payments that may become due is not yet determinable.
On June 13, 2018, the Company entered into a license agreement with CrystalGenomicsCG to gain an exclusive license to CG-806luxeptinib in China. The Company has potential future obligations of development milestones of $6$6 million related to approval of an Investigational New Drug (“IND”) and to the initiation of Phase 2 and pivotal clinical trials, and regulatory milestones totaling $20$20 million. The Company also has an obligation to pay sales milestones and royalty payments on sales of commercialized product. The timing or likelihood of any milestone or royalty payments that may become due is not yet determinable.
On March 7, 2018, we entered into an exclusive global license agreement with Ohm Oncology (OHM), for the development, manufacture and commercialization of APL-581, as well as related molecules from our dual bromodomain and extra-terminal domain motif (BET) protein and kinase inhibitor program. Under the agreement, we will retain reacquisition rights to certain molecules, while OHM/LALS will have the rights to develop and sublicense all other molecules. We have received two nominal upfront cash payments and are eligible to receive up to $125 million of additional payments based on the achievement of certain development, regulatory and sales milestones, as well as significant royalties on future sales generated from the program, if any.
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
| (a) | Recent tax legislation |
In December 2017 the U.S. government enacted comprehensive tax legislation, the Tax Cuts and Jobs Act (the "Tax Act"), which significantly revises the U.S. tax code, generally effective January 1, 2018, by lowering the U.S. federal corporate income tax rate from 35% to 21%, implementing a territorial tax system and setting limitations on the deductibility of certain costs (e.g. Interest expenses) among other things. As a Canadian entity, we generally would be classified as a foreign entity (and, therefore, a non-U.S. tax resident) under general rules of U.S. federal income taxation. However, we have a branch and U.S. subsidiary subject to U.S. federal income taxation.
For the years ended December 31, 20192023 and 2020,2022, the total comprehensive loss is as follows:
| | | | | | | | |
| | December 31,
2023 | | | December 31,
2022 | |
Loss attributed to US foreign operations | | $ | (45,652 | ) | | $ | (36,615 | ) |
Loss attributed to Canadian operations | | | (5,555 | ) | | | (5,208 | ) |
Loss before income taxes | | $ | (51,207 | ) | | $ | (41,823 | ) |
| | | | December 31, 2020 | | | | December 31, 2019 | |
| | | | | | | | | |
| | | | | | | | | |
| Loss attributed to US foreign operations | | $ | (39,757 | ) | | $ | (20,470 | ) |
| Loss attributed to Canadian operations | | | (15,481 | ) | | | (5,807 | ) |
| Income (loss) before income taxes | | $ | (55,238 | ) | | $ | (26,277 | ) |
Tax rate reconciliation
| (c) | Tax rate reconciliation |
Major items causing the Company’s income tax rate to differ from the statutory rate of approximately 26.5%26.5% (December 31, 20192022 – 26.5%26.5%) are as follows:
| | | | | | | | |
| | Year ended December 31, 2023 | | | Year ended December 31, 2022 | |
Net loss | | $ | (51,207 | ) | | $ | (41,823 | ) |
Statutory Canadian corporate tax rate | | | 26.5 | % | | | 26.5 | % |
Computed expected tax recovery | | $ | (13,570 | ) | | $ | (11,083 | ) |
Non-deductible permanent differences | | | 873 | | | | 1,376 | |
Change in valuation allowance | | | 13,059 | | | | 10,821 | |
Foreign tax rate differential | | | (677 | ) | | | (466 | ) |
Prior year true-up adjustments | | | 355 | | | | (703 | ) |
Other | | | (40 | ) | | | 55 | |
| | $ | — | | | $ | — | |
| | | | Year ended
December 31, 2020 | | | | Year ended
December 31, 2019 | |
| | | | | | | | | |
| Net loss | | $ | (55,238 | ) | | $ | (26,277 | ) |
| Statutory Canadian corporate tax rate | | | 26.5 | % | | | 26.5 | % |
| | | | | | | | | |
| Computed expected tax recovery | | $ | (14,638 | ) | | $ | (6,963 | ) |
| Non-deductible permanent differences | | | 4,959 | | | | (1,305 | ) |
| Change in valuation allowance | | | 10,383 | | | | 12,146 | |
| Foreign tax rate differential | | | (428 | ) | | | (286 | ) |
| Foreign exchange differences | | | - | | | | - | |
| Prior year true-up adjustments | | | (230 | ) | | | (3,563 | ) |
| Other | | | (46 | ) | | | (29 | ) |
| | | $ | - | | | $ | - | |
F-23
APTOSE BIOSCIENCES INC.
| (d) | Significant components of deferred taxes |
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
(c)Significant components of deferred taxes
The tax effects of temporary differences that give rise to significant portions of the unrecognized deferred tax assets are presented below:
| | | | December 31, 2020 | | | | December 31, 2019 | |
| | | | | | | | | |
| | | | | | | | | |
| Net operating losses carried forward | | $ | 37,362 | | | $ | 26,786 | |
| Research and development expenditures | | | 5,032 | | | | 5,031 | |
| Property, equipment, and other intangible assets | | | 3,760 | | | | 4,191 | |
| Research and development tax credits | | | 3,597 | | | | 3,685 | |
| Financing costs | | | 2,336 | | | | 2,010 | |
| Right-of-use assets | | | 40 | | | | 41 | |
| Total deferred tax assets | | | 52,127 | | | | 41,744 | |
| Valuation allowance | | | (52,127 | ) | | | (41,744 | ) |
| Net deferred tax asset | | $ | - | | | $ | - | |
| | | | | | | | |
| | December 31,
2023 | | | December 31,
2022 | |
Net operating losses carried forward | | $ | 73,552 | | | $ | 60,092 | |
Research and development expenditures | | | 5,025 | | | | 5,023 | |
Property, equipment, and other intangible assets | | | 7,321 | | | | 7,264 | |
Research and development tax credits | | | 4,930 | | | | 4,968 | |
Financing costs | | | 431 | | | | 873 | |
Right-of-use assets | | | 19 | | | | 2 | |
Total deferred tax assets | | | 91,278 | | | | 78,222 | |
Valuation allowance | | | (91,278 | ) | | | (78,222 | ) |
Net deferred tax asset | | $ | — | | | $ | — | |
APTOSE BIOSCIENCES INC.
|
Notes to Consolidated Financial Statements
|
Year ended December 31, 2020 and 2019
|
(Tabular amounts in thousands of US dollars, unless otherwise noted)
|
|
The valuation allowance at December 31, 20202023 was primarily related to net operating loss carryforwards that, in the judgment of management, are not more-likely than-not to be realized. In assessing the realizability of deferred tax assets, management considers whether it is more-likely than-notmore-likely-than-not that all or some portion of the deferred assets will not be realized. This ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the period in which those deductible temporary difference become deductible. Based on the history of losses and projections for future taxable income, management believes that it is not more-likely than-not that the Company will realize the benefits of these deductible temporary differences (e.g. deferred tax assets).
The Company has certain deductible Canadian undeducted research and development expenditures that have not been deducted for tax purposes, totaling $19.0$19.0 million, that can be carried forward indefinitely. The Company also has Canadian non-refundablenon‑refundable federal and provincial investment tax credits of approximately $3.6$2.4 million which are available to reduce future federal taxes payable and begin to expire in 2021,2024, as well as non-refundablenon‑refundable US research and development tax credits of approximately $0.9$3.1 million which are available to reduce future US taxes payable and begin to expire in 2038.2038.
In addition, the Company has Canadian non-capital loss carryforwards of $135.2$266.2 million. To the extent that the non-capital loss carryforwards are not used, they begin to expire in 2026. The Company also has a US non-capital loss carryforward of $0.7 million,$1.1 million. To the extent that the non-capital loss carryforwards are not used, they begin to expire in 2034.2034.
The Company files income tax returns with Canada and its provinces and territories. Generally, we are subject to routine examinations by the Canada Revenue Agency ("CRA"). Income tax returns filed with various provincial jurisdictions are generally open to examination for periods of four to five years subsequent to the filing of the respective return.
The Company also files income tax returns for our U.S. operations and subsidiary with the U.S. federal and state tax jurisdictions. Generally, we are subject to routine examination by taxing authorities in the U.S. jurisdictions. There are presently no examination of our U.S. federal and U.S. state returns. We believe that our tax positions comply with the applicable tax law.
16.Selected quarterly financial data (unaudited):
16. | Selected quarterly financial data (unaudited): |
Selected financial data (unaudited) for the periods presented was as follows:
| | | | | | | | | | | | | | | | |
| | March 31,
2023 | | | June 30,
2023 | | | September 30,
2023 | | | December 31,
2023 | |
Revenue | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
Net loss | | $ | (13,676 | ) | | $ | (14,129 | ) | | $ | (11,447 | ) | | $ | (11,955 | ) |
Basic and diluted loss per common share | | $ | (2.25 | ) | | $ | (2.27 | ) | | $ | (1.76 | ) | | $ | (1.57 | ) |
| | | | March 31, 2020 | | | | June 30, 2020 | | | | September 30, 2020 | | | | December 31, 2020 | |
| | | | | | | | | | | | | | | | | |
| Revenue | | $ | - | | | $ | - | | | $ | - | | | $ | - | |
| Net loss | | | (11,526 | ) | | | (15,750 | ) | | | (13,249 | ) | | | (14,713 | ) |
| Basic and diluted loss per common share | | | (0.15 | ) | | | (0.21 | ) | | | (0.15 | ) | | | (0.17 | ) |
F-24
APTOSE BIOSCIENCES INC.
| | | | March 31, 2019 | | | | June 30, 2019 | | | | September 30, 2019 | | | | December 31, 2019 | |
| | | | | | | | | | | | | | | | | |
| Revenue | | $ | - | | | $ | - | | | $ | - | | | $ | - | |
| Net loss | | | (5,506 | ) | | | (6,218 | ) | | | (6,844 | ) | | | (7,709 | ) |
| Basic and diluted loss per common share | | | (0.14 | ) | | | (0.13 | ) | | | (0.12 | ) | | | (0.13 | ) |
Notes to Consolidated Financial Statements Years ended December 31, 2023 and 2022
(Tabular amounts in thousands of United States dollars, except as otherwise noted)
| | | | | | | | | | | | | | | | |
| | March 31,
2022 | | | June 30,
2022 | | | September 30,
2022 | | | December 31,
2022 | |
Revenue | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
Net loss | | $ | (11,481 | ) | | $ | (10,565 | ) | | $ | (9,777 | ) | | $ | (10,000 | ) |
Basic and diluted loss per common share | | $ | (1.81 | ) | | $ | (1.66 | ) | | $ | (1.66 | ) | | $ | (1.67 | ) |
Subsequent to the year end, the Company issued 2,746,750408,168 stock options to directors, officers, employees and consultants with an average exercise price of $4.36. The$2.00. 388,170 stock options vest 50%50% after one year and 16.67%16.67% on each of the next three anniversaries, except for 430,000and 19,998 options which vest 50%50% after one year and 25%25% on each of the next two anniversaries. The future expense associated with this option grant is $402 thousand.
In January 2024, the Company issued 10,062 Common Shares to Keystone under the 2023 Committed Equity Facility, as the Second Back-End Commitment Shares.
On January 31, 2024, the Company announced the closing of a $9.7 million public offering (the “Public Offering”) and a $4 million private placement (the “Private Placement”) with Hanmi Pharmaceutical. The Public Offering comprised 5,649,122 common shares of the Company (the “Common Shares”) and warrants at a combined offering price of US $1.71 per share. This included 736,842 Common Shares and warrants pursuant to a full exercise by the underwriter of its over-allotment option (the “Over-Allotment Option”). The Private Placement comprised 2,105,263 Common Shares of the Company sold at a price of $1.90, representing an 11% premium over the price of the Common Shares issued as part of the Public Offering. Nasdaq subsequently issued a letter to the Company regarding the value and the date of the Private Placement, as discussed in this note, below. Financing costs of approximately $1.4 million included underwriting costs of 7% and approximately $0.4 million in professional fees. The Company also issued Hanmi warrants to purchase Common Shares at an exercise price of US $1.71 per Warrant Share. The total number of Common Shares outstanding after the closing of the Public Offering, including the Over-Allotment Option, and Private Placement was 15,706,810 and warrants outstanding were 8,332,163. As of the date of this report, the total common share ownership by Hanmi was 2,989,414. Hanmi also owns 2,339,181 warrants to purchase Common Shares at an exchange price of $1.71 per Warrant Share.
On February 29, 2024, the Company received a deficiency letter (the “2024 Deficiency Letter”) from the Nasdaq Listing Qualifications Department of The Nasdaq Stock Market LLC (“Nasdaq”) notifying the Company that the Company’s January 2024 Private Placement of securities to Hanmi violated 5635(d) because the Company did not obtain shareholder approval prior to such issuance. Nasdaq stated that the Private Placement involved the issuance of greater than 20% of the issued and outstanding Common Shares of the Company at a discount to the Nasdaq official closing price on January 25, 2024, the date of the subscription agreement between the Company and Hanmi. The 2024 Deficiency Letter has no immediate effect on the listing of the Company’s Common Shares. In accordance with the Nasdaq Listing Rules, the Company has been given forty-five (45) calendar days, or until April 14, 2024, to submit a plan to regain compliance. If Nasdaq accepts the Company’s plan to regain compliance, Nasdaq can grant an extension of up to 180 calendar days from the date of the Deficiency Letter to evidence compliance. Although the Company believes that the Private Placement was completed in accordance with the Nasdaq Listing Rules, the Company respects Nasdaq’s query and intends to work with Nasdaq to resolve Nasdaq’s concerns and will consider available options to regain compliance. However, there can be no assurance that the Company will be able to regain compliance with the applicable Nasdaq Listing Rules.
On February 5, 2024, the Company issued 10,891 Common Shares under the ESPP.