UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the Fiscal Year Ended December 31, 20172021
Commission File Number 000-55473001-38659
BIOSIG TECHNOLOGIES, INC.
(Exact name of registrant as specified in its charter)
Delaware | 26-4333375 | |
(State or other jurisdiction of incorporation or organization) | (IRS Employer Identification No.) | |
55 Greens Farms Road, 1st Floor Westport, CT | 06880 | (203) 409-5444 |
(Address of principal executive office) | (Zip Code) | (Registrant’s telephone number, Including area code) |
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Trading Symbol(s) | Name of each exchange on whichregistered |
Common Stock, par value $0.001 per share | BSGM | The NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Act: Common Stock, $0.001 par value per shareNone
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined by Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b–2 of the Exchange Act.
(Check one):
Large accelerated filer | ☐ | Accelerated filer | ☐ | |
Non-accelerated filer | ☒ | Smaller reporting company | ☒ | |
Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal controls over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates as of June 30, 2017,2021, based on the price at which the common stock was last sold on such date, is $24,577,543.$108,511,450. For purposes of this computation, all officers, directors, and 5 percent beneficial owners of the registrant are deemed to be affiliates. Such determination should not be deemed an admission that such directors, officers, or 5 percent beneficial owners are, in fact, affiliates of the registrant.
As of February 27, 2018,March 30, 2022, there were 29,998,46638,424,059 shares of the registrant’s common stock outstanding.
Documents Incorporated by Reference:
The registrant incorporates by reference in Part III (Items 10, 11, 12, 13 and 14) of this Form 10-K portions of its Definitive Proxy Statement for the 2022 Annual Meeting of Stockholders, which shall be filed with the Securities and Exchange Commission within 120 days after the end of the fiscal year.
TABLE OF CONTENTS
PAGE | |||||
PART I | |||||
Item 1. | 4 | ||||
Item 1A. | 24 | ||||
Item 1B. | 46 | ||||
Item 2. | 46 | ||||
Item 3. | 46 | ||||
Item 4. | 46 | ||||
PART II | |||||
Item 5. | 47 | ||||
Item 6. | 47 | ||||
Item 7. | 47 | ||||
Item 7A. | 55 | ||||
Item 8. | F-1 – | ||||
Item 9. | 56 | ||||
Item 9A. | 56 | ||||
Item 9B. | 57 | ||||
Item 9C. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections | 57 | |||
PART III | |||||
Item 10. | 58 | ||||
Item 11. | 58 | ||||
Item 12. | 58 | ||||
Item 13. | 58 | ||||
Item 14. | 58 | ||||
PART IV | |||||
Item 15. | 59 | ||||
Item 16. | 61 | ||||
62 | |||||
PART I
Note on Forward-Looking Statements
This Annual Report on Form 10-K (including the section regarding Management’s Discussion and Analysis of Financial Condition and Results of Operations) contains forward-looking statements regarding our business, financial condition, results of operations and prospects. Words such as “expects,” “anticipates,” “intends,” “plans,” “believes,” “seeks,” “estimates” and similar expressions or variations of such words are intended to identify forward-looking statements, but are not deemed to represent an all-inclusive means of identifying forward-looking statements as denoted in this Annual Report on Form 10-K. Additionally, statements concerning future matters are forward-looking statements.
Although forward-looking statements in this Annual Report on Form 10-K reflect the good faith judgment of our management, such statements can only be based on facts and factors currently known by us. Consequently, forward-looking statements are inherently subject to risks and uncertainties and actual results and outcomes may differ materially from the results and outcomes discussed in or anticipated by the forward-looking statements. Factors that could cause or contribute to such differences in results and outcomes include, without limitation, those specifically addressed under the heading “Risks“Risk Factors” below, as well as those discussed elsewhere in this Annual Report on Form 10-K. Readers are urged not to place undue reliance on these forward-looking statements, which speak only as of the date of this Annual Report on Form 10-K. We file reports with the Securities and Exchange Commission (“SEC”). You can read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. You can obtain additional information about the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. In addition, theThe SEC maintains an Internet site (www.sec.gov) that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC, including us.
We undertake no obligation to revise or update any forward-looking statements in order to reflect any event or circumstance that may arise after the date of this Annual Report on Form 10-K. Readers are urged to carefully review and consider the various disclosures made throughout the entirety of this Annual Report on Form 10-K, which attempt to advise interested parties of the risks and factors that may affect our business, financial condition, results of operations and prospects.
Unless the context indicates otherwise, references in this Annual Report to “BioSig,” the “Company,” “we,” “our” and “us” mean BioSig Technologies, Inc., and its predecessor entities.
ITEM 1 – BUSINESS
Corporate Structure
We were formed as BioSig Technologies, Inc., a Nevada corporation, in February 2009 and in April 2011 we merged with our wholly-ownedwholly owned subsidiary, BioSig Technologies, Inc., a Delaware corporation, with the Delaware corporation continuing as the surviving entity. We areBioSig is principally devoted to improving biomedicalthe standard of care in electrophysiology, or EP, with our PURE EP™ System’s enhanced signal acquisition, digital signal processing, to extract information from physiologic signals. Our initial focus is on providing intracardiac signal information to electrophysiologistsand analysis during electrophysiology (EP) studies and catheter ablation for complex arrhythmias, atrial fibrillation (AF) and ventricular tachycardia (VT). We have notof cardiac arrhythmias. The Company has generated anyminimal revenue to date and consequently ourits operations are subject to all risks inherent in business enterprise in early commercialization stage.
On November 7, 2018, we formed a subsidiary under the establishmentlaws of a new business enterprise.
On July 2, 2020, the Company formed an additional subsidiary, NeuroClear Technologies, Inc. (“NeuroClear”), a Delaware corporation, to pursue additional applications of the PURE EP™ signal processing technology outside of cardiac electrophysiology. We own 100% of the outstanding shares of common stock as of March 30, 2022 and the subsidiary is currently dormant. NeuroClear’s Business Overview can be found on page 19.
Business Overview
We are a development stage medical devicetechnology company developing a proprietary biomedicalthat is commercializing our PURE EP™ System which is an advanced signal acquisition and processing technology platform designed to extract information from physiologic signals. Our initial emphasisprovide essential diagnostic signals with high clinical value in all types of cardiac catheter ablations. PURE EP™ is on providing intracardiac signal informationdesigned to electrophysiologists during EP studiesaddress long-standing limitations that slow and disrupt cardiac catheter ablation procedures, such as environmental lab noise, signal saturation, slow signal recovery, and inaccurate display of AFfractionated potentials.
Cardiac catheter ablation is a procedure that involves delivery of energy through the tip of a catheter that scars or destroys heart tissue to correct heart rhythm disturbances (arrhythmias). In August 2018, we received 510(k) clearance from the U.S. Food and VT. Our first product is theDrug Administration (the “FDA”) to market our PURE (Precise Uninterrupted Real-time evaluation of Electrograms) EP™ System,System.
PURE EP™ is a surface electrocardiogramsignal processing platform that combines advanced hardware and intracardiac multichannel recordingsoftware to address known challenges associated to signal acquisition, to enable electrophysiologists to see more signals and analysis system that acquires, processes and displays electrocardiograms and electrograms required during electrophysiology studies and catheter ablation procedures.
PURE EP™’s initial focus is on improving intracardiac signal processing tools withinacquisition and enhancing diagnostic information for catheter ablation procedures for complex arrhythmias like ventricular tachycardia (“VT”), a potentially life-threatening arrhythmia, and atrial fibrillation (“AF”), the most common cardiac arrhythmia associated with a fivefold risk of stroke.
Clinical data acquired by the PURE EP System. We believe that these will assist electrophysiologistsEP™ System in further differentiating true signals from noise and will provide guidance in identifying ablation targets.
We continue to install PURE EP™ Systems at centers of excellence for clinical evaluation under our market development plan. The physicians had provided us with digital recordings obtained with conventional electrophysiology recording systems during different stages of electrophysiology studies. Using our proprietary signal processing tools that are part of the PURE EPEP™ System we analyzed those recordingshas been utilized at numerous institutions, including Mayo Clinic campuses in Arizona, Florida and successfully removed baseline wander, noise and artifacts from the data thereby providing better diagnostic quality signals.
To date, more than 2,160 patient procedures have been conducted with the PURE EPEP™ System prototype.
In addition to clinical evaluation, we have collaboratedconducted pre-clinical evaluation with Dr. Asirvatham and other physicians affiliated withthe PURE EP™ System under several protocols. At Mayo Clinic in Rochester, Minnesota, and Jacksonville, Florida. Wewe have performed pre-clinical studies at Mayo Clinic since 2015 to validate technology within the PURE EP System prototype. These studies have been designed to determine clinical effectiveness for features within the PURE EP System that aretwenty-seven experiments (including novel research programs such as Artificial Intelligence, or AI, and repolarization) in development. Since March 2016,various animal models; we have published seven manuscripts in collaboration with the physicians from Mayo Clinic evidencing our pre-clinical findings. The publications cover a variety of subjects pertaining to the PURE EP System as an enhanced electrophysiology recording system with signal acquisition and differentiation and having specific visualization of different electrophysiology signals.
In September 2021, we announced that we entered into a manufacturing and professional services agreement with Plexus Corp (“Plexus”) (Nasdaq: PLXS). Under the terms of the agreement, Plexus will manufacture the PURE EP™ System and develop a new product pipeline for our subsidiary, ViralClear.
We intendhave made progress towards obtaining a European CE marking certificate for medical devices. In Q1 2022, we completed the quality management system audit for the International Organization for Standardization (“ISO”) 13485:2016 with the expectation to conduct further pre-clinical studies,obtain the ISO 13485:2016 certification in the first half of 2022 and research studies. The main objectiveproceed to the application for the European CE Marking clearance in the first half of these studies is2023, subject to demonstrate the clinical potentialguidance and availability from the European Notified Body.
In January 2022, U.S. patent claims for our PURE EP™ noise-filtering technology which address computer-implemented systems and methods for filtering noise from input cardiac signals were approved, and the resulting patent issued on March 1, 2022. We now have 48 issued or allowed worldwide patents covering our novel technology for arrhythmia care.
In December 2020, we announced that three PURE EP™ Systems were contracted for purchase by St. David’s Healthcare in Austin, Texas and were subsequently sold in February 2021. We also sold three PURE EP™ Systems to Mayo Foundation for Medical Education and Research in 2021 for use in Mayo Clinic campuses in Rochester, Minnesota, Jacksonville, Florida and Phoenix, Arizona. We are in active discussions with several accounts about the acquisition of the PURE EP SystemEP™ System.
1Evaluation of a medical technology and innovation company, and engaged Quintain Project Solutions LLC as the manufacturing project management leadernovel cardiac signal processing system for theelectrophysiology procedures: The PURE EP System – implementing steps for obtaining 510(k) clearance from the U.S. Food and Drug Administration for the PURE EP System.2.0 study - Al‐Ahmad - 2021 - Journal of Cardiovascular Electrophysiology - Wiley Online Library
Recent Developments
Technion Research & Development Foundation (TRDF) Ltd. Feasibility Study Agreement
On November 16, 2021, we announced the launch of a new Artificial Intelligence development program with Technion – Israel Institute of Technology. Based in Haifa, Israel, Technion – Israel Institute of Technology is a public research university offering degrees in science, engineering, and related fields, such as medicine, industrial management, and education. Over the years, Technion established itself as a leading academic institution in Artificial Intelligence (AI).
The research program is led by Asst. Prof. Joachim Behar, Head of the Artificial Intelligence in Medicine Laboratory (AIMLab) at the Technion. Under the terms of the program, the ECG signals supplied by the conclusionPURE EP™ System are being analyzed in the context of 2018,developing AI-powered algorithms for atrial fibrillation ablation procedures.
Mayo Clinic Artificial Intelligence (AI) Research Agreement
In January 2021, we will have obtained 510(k) marketing clearance fromentered into a research agreement with Mayo Clinic regarding a Novel AI Program for our Novel Signal Recording System. The program is a strategic collaboration with Mayo to develop a next-generation AI- and machine learning-powered software for our PURE EP™ System. The new collaboration includes an R&D program that is expected to expand our proprietary hardware and software with advanced signal processing capabilities and aim to develop novel technological solutions by combining the FDAelectrophysiological signals delivered by PURE EP™ and will be ableother data sources.
The development program is under the leadership of Samuel J. Asirvatham, M.D., Mayo Clinic’s Vice-Chair of Innovation and Medical Director, Electrophysiology Laboratory. We entered into a 10-year collaboration agreement with Mayo Clinic in March 2017 and in November 2019, we signed a patent and know-how license agreement with Mayo Foundation for Medical Education and Research in which such terms apply to commence marketingthis program. On April 9, 2021 and commercializationOctober 22, 2021 we conducted first pre-clinical data collection studies to advance our AI program at Mayo Clinic.
Appointment of Chief Operating Officer
Effective March 21, 2022, we appointed John Sieckhaus as our chief operating officer. Mr. Sieckhaus brings to the PURE EP System. Our abilityCompany 30 years in the healthcare industry, including 21 years at St. Jude Medical and Abbott Laboratories. Mr. Sieckhaus’s annual base salary is $280,000, less applicable payroll deductions and tax withholdings. In addition, Mr. Sieckhaus is eligible to achieve the aforementioned milestones will be principallyreceive an annual discretionary bonus as determined by the Compensation Committee of our ability to obtain necessary financing and regulatory approvals, among other factors.
Our Industry
Pharmacological, or medicine-based, therapies have traditionally been used as initial treatments for cardiac arrhythmias, but they often fail to adequately control the arrhythmia and may have significant side effects. Catheter ablation is now often recommended for an arrhythmia that medicine cannot control. Catheter ablation involves advancing several flexible catheters into the patient’s blood vessels, usually either in the femoral vein, internal jugular vein or subclavian vein. The catheters are then advanced towards the heart. Electrical impulses are then used to induce the arrhythmia and local heating or freezing is used to ablate (destroy) the abnormal tissue that is causing it. Catheter ablation for most of most arrhythmias has a high success raterate. For patients with complex arrhythmias like AF and VT, it is often necessary to perform multiple procedures per patient have been found to be more successful.achieve success.
Catheter ablation is performed by an electrophysiologist (a specially trained cardiologist) in a catheterization lab or a specialized electrophysiologyroom in an EP lab. ItAccording to Health Research International, it is estimated that there are about 3,000 dedicated electrophysiology labs in the U.S. and 1,500 labs outside the U.S.,8,163 global EP lab rooms performing catheter ablations, each typically with an electrophysiologyEP recording system costing an average of $250,000. We believe that the current$160,000. According to Global Market Insights, global cardiac ablation market value of the electrophysiology recording device market in the U.S. is approximately $750 million, based uponprojected to exceed $8.4 billion by 2028. The growing geriatric population is more susceptible to cardiovascular diseases and is expected to contribute to the number of electrophysiology labsablation procedures in U.S. and the average cost of the recording system in each lab. With the potential of 12 million atrial fibrillation patients by the year 2050 (accordingforthcoming years. According to the Atrial Fibrillation Fact Sheet, last updated in August 2017 by the Centers for Disease Control and Prevention) and improvements in technology for atrial fibrillation ablation therapy, significant growth is predicted forWorld Health Organization, the number of hospitals building electrophysiology labs. Accordingindividuals aged 65 years and over is projected to increase from 524 million in 2010 to 1.5 billion by 2050. Aging typically leads to several changes in heart and blood vessels, which result in an increased risk of cardiac disorders. Accordingly, as cardiac ablation is a safe and highly effective treatment for irregular heart rhythm, we believe population aging will drive the 2016 HRI Global Opportunitiesproduct demand in Medical Devices & Diagnostics report, analysts forecastfuture. Along with the globalexpected increased disease burden, we believe that product advancements will significantly drive the industry expansion. Industry players operating in the market for electrophysiology devices will grow at a 10.3 percent compound annual growth rate, from $3.68 billionare continuously developing newer technologies to offer more successful outcomes, and the expected significant investment in 2015research and development activities by these players is anticipated to $6.015 billion in 2020; in addition, global ablation procedure numbers are predictedlead to grow from 865,000 to 1,350,000 per year.new product launches, thereby expanding the product availability.
Catheter Ablation of Atrial FibrillationAF and Ventricular TachycardiaVT
Accurate recording of electrograms is critical to efficient mapping and ablation of complex arrhythmias. We believe that the clearer recordings and additional information the very small amplitude of intracardiac signals--high frequency, small amplitude components in midst of large physiologic signals; signals important to characterize critical substrate, such as fractionated atrial and ventricular electrograms; and high-frequency, low-amplitude signals such as the Purkinje potentials—provided by the PURE EPEP™ System may improve outcomes during electrophysiologyEP studies and ablation procedures for a variety of arrhythmias.
For patients who are candidates for ablation, an electrophysiologyEP study is necessary to define the targeted sites for the ablation procedure. Two common, yet complex, conditions for which ablation procedures are performed are atrial fibrillationAF and ventricular tachycardia. We believe that in the near future, the PURE EP System may have a meaningful impact on assisting ablation strategies for these conditions.
AF is the most common heart rhythm disorder in the world and increases the risk for stroke 5-fold. In 2010,2017, there waswere a reported global37.57 million prevalent cases and 3.05 million incident cases of AF globally, contributing to over 287,000 deaths worldwide (Global, regional, and national prevalence, incidence, mortality, and risk factors for atrial fibrillation, 1990–2017: results from the Global Burden of 33.5 million (20.9 million men and 12.6 million women)Disease Study 2017). In 2017,2020, the Centers for Disease Control and Prevention stated that there are anit is estimated 2.7-6.1that 12.1 million Americans suffering withpeople in the United States will have AF in 2030, more than 750,000454,000 patients hospitalized annually foras the condition,primary diagnosis, and AF contributes to an estimated 130,000158,000 deaths each year. Despite the fact that physicians have been performing radiofrequency ablations since the 1990s, catheter-based treatment is offered to less than 3% of the AF patient population in the U.S. and Europe. An increasing proportion of diagnosed atrial fibrillationAF cases are now being treated via ablation, as both physician confidence and the devices used in these procedures improve. A growing amount of positive clinical data has demonstrated the efficacy of AF ablation when compared to the traditional first-line treatment of anti-arrhythmic drugs. As
Recent studies suggest that COVID-19 may increase the risk of certain arrhythmias. In a result,meta-analysis of 19 observational studies with 21,653 patients hospitalized with COVID-19, the prevalence of AF was 11%. According to the studies, AF was higher in patients with severe versus non-severe COVID-19 (19% versus 3%)2.
In 2021, a meta-analysis of 6 randomized clinical trials involved 1,212 patients with AF (609 were randomized to AF ablation is becomingand 603 to drug therapy (AADs); mean age, 56 years). Compared with AADs, catheter ablation use was associated with reductions in recurrent atrial arrhythmia (32.3% vs 53%; risk ratio (RR), 0.62; 95% CI, 0.51-0.74; P < .001; I2 = 40%), with a number needed to treat with ablation to prevent 1 arrhythmia of 5. Use of ablation was also associated with reduced symptomatic atrial arrhythmia (11.8% vs 26.4%; RR, 0.44; 95% CI, 0.27-0.72; P = .001; I2 = 54%) and hospitalization (5.6% vs 18.7%; RR, 0.32; 95% CI, 0.19-0.53; P < .001) with no significant difference in serious adverse events between the fastest growing procedure typegroups (4.2% vs 2.8%; RR, 1.52; 95% CI, 0.81-2.85; P = .19). In this meta-analysis of randomized clinical trials including first-line therapy of patients with paroxysmal AF, catheter ablation compared with antiarrhythmic drugs was associated with reductions in this market. The American College of Cardiology Foundation/American Heart Association Task Force reported that catheter-directed ablationrecurrence of atrial fibrillation representsarrhythmias and hospitalizations, with no difference in major adverse events.
The AF Ablation Long Term Registry is an international registry of 3,630 patients who underwent AF ablation between 2012 and 2015 – the study reported a substantial achievement that promises better therapy for a large number41% rate of repeat ablation at 3 years post ablation. At 12-month follow-up, the outcome was judged to have been successful in 74% of patients. However, almost 50% of the patients presently resistant to pharmacologicalwere still taking an antiarrhythmic drug. AF recurrences were less common in patients with paroxysmal (31%) than with persistent (40%) or electrical conversion to sinus rhythm (“2014 ACCF/AHA/HRS Focused Update on the Management of Patients With Atrial Fibrillation (Updating the 2011 Guideline)”long-standing persistent (44%). However, rates of success and complications may vary, sometimes considerably. AF.
According to the Heart Rhythm Society, ventricular tachycardiaVT is the most dangerous arrhythmia since it may result in ventricular fibrillation, a rapid chaotic heartbeat in the lower chambers of the heart which can often result in sudden cardiac death. Because the fibrillating muscle cannot contract and pump blood to the brain and vital organs, ventricular fibrillation is the number one cause of sudden cardiac death accountingwhich accounts for more than 350,000approximately 300,000 deaths in the U.S. each year. Ventricular tachycardiaVT is typically treated with implantable cardioverter defibrillators, or ICDs, or a combination of ablation along with an ICD.
2https://www.uptodate.com/contents/covid-19-arrhythmias-and-conduction-system-disease
Catheter ablation of VT has historically been used primarily for drug refractory ventricular arrhythmias in patients with ICDs. However, advances in electro-anatomical mapping systems, techniques to identify ablation sites during sinus rhythm, and the use of hemodynamic support devices has broadened the applicability of catheter ablation for ventricular arrhythmias. When performed in centers with high procedural volumes, the rates of complications remain relatively low. However, success rates
have historically been quite variable and highly dependent on the specific ablation approach adopted.We believe that ablation will continue to becomebe a preferred treatment for ventricular tachycardia, especially considering the challenges presented by ICD therapies; thisAF and VT. This increase in demand for ablation procedures will likelyhas also increaseincreased the demand for technological advances in medical devices essential to ablation procedures. Improvements are needed to help reduce the periprocedural complications and decrease costly lengths of stay in patients undergoing catheter ablation procedures, including electrophysiology recorders, in orderadding focus to better supportimproving outcomes at low volume hospitals and among patients at high risk due to comorbidities. We believe that the PURE EP™ System may have a meaningful impact on assisting ablation procedures.
EP Lab Environment and ElectrophysiologyEP Recording Systems
The electrophysiologyEP lab environment and recording systems create significant amounts of noise and artifacts during electrophysiologyEP procedures. Current surface and intracardiac recording systems typically consist of large workstations interconnected by a complex set of cables that contribute to significant amounts of noise during signal acquisition. Additional noise and artifacts generated from the electrophysiologyEP lab equipment further hamper recordings of small electrophysiological potentials. Preserving spaciotemporal (space and time) characteristics of the signal in a very challenging electrophysiologyEP recording environment is a difficult task. To remove noise and artifacts, recorders that are currently on the market offer a family of low pass, high pass and notch filters, but these filters alter signal information context.
The shape and amplitude of electrocardiograms, unipolar and bipolar electrograms, and, consequently, reconstructed endocardial and epicardial maps, are influenced not only by electrophysiological and structural characteristics of the myocardial tissue involved, but with characteristics of the recording system. Amplitude and morphology of electrocardiogram and intracardiac signals are significantly affected by filters used to remove noise. Because of the number of amplitude and interval measurements made during an electrophysiologyEP study, it is imperative that the recording system faithfully acquires surface electrocardiogram and intracardiac electrograms. We believe that the recording systems that are currently available on the market are ineffective in preserving the optimal amount of original information contained in the cardiac signals.
In addition, the electrophysiologyEP lab consists of sophisticated equipment that requires an electrophysiologist to mentally integrate information from a number of sources during procedures. There are numerous monitors in an electrophysiologyEP lab that provide and display this variety of information. An electrophysiologist needs to evaluate the acquired cardiac signals and the patient’s responses to any induced arrhythmias during the procedure. However, it iscan be difficult for an electrophysiologist to synthesize the disparate information produced by the numerous monitors in the lab and calculate the real-time, three-dimensional orientation of the anatomy and the location of the recording and ablation catheters. As the number of electrophysiologyEP procedures increase, a variety of diagnostic, therapeutic and therapeutichighly specialized ablation catheters are becoming more widely available and new highly specialized catheters are beingcontinue to be developed. In addition, remote robotic and magnetic navigation systems are beinghave been developed to address limitations of dexterity in controlling the catheter tip, especially during complex arrhythmia ablation procedures. We believe that, considering the improvements being made with respect to other equipment used in the electrophysiologyEP lab and the continual increase of ablation procedures, the electrophysiologyEP recorders currently available on the market are not sufficiently advanced with respect to the quality of their recordings to deliver adequate results. We believe that the PURE EPEP™ System will be able to deliver superior quality of recordings that will allow it to successfully integrate with the other advanced equipment found in the EP lab.
Generally, some current electrophysiology lab.recording systems can effectively support the treatment of arrhythmias such as atrial flutter and supraventricular tachycardia, which show up as large-amplitude, low-frequency signals. However, more complex and prevalent arrhythmias, such as AF and VT, which are characterized by low-amplitude, high-frequency signals, have not found an effective evaluation of all relevant signals. This signal detection, acquisition, and isolation can be further complicated by equipment line noise and pacing signals. Current EP recorders use low-pass, high-pass, and notch filters to remove noise and artifacts from the various electrical signal information. Unfortunately, conventional filtering techniques can alter signals and make it difficult or impossible to see low-amplitude, high-frequency signals that can be inherent in cardiac monitoring, the visualization of which signals could help treat atrial fibrillation and ventricular tachycardia. It has been recently recognized that the assurance of waveform integrity, such as for the noise-free acquisition of intracardiac and ECG signals in an EP environment, had not been previously accomplished due to contamination of various signals by artifacts and noise.
The requirement for optimal signal integrity is further amplified during ablation treatments of atrial fibrillationAF and ventricular tachycardia. Presently, oneVT. One of the main objectives of the atrial fibrillationAF ablation procedure is to precisely identify, ablate and eliminate pulmonary vein potentials;potentials and one of the main objectives of the ventricular tachycardiaVT procedure is to map the arrhythmia substrate and precisely identify, ablate and eliminate small abnormal potentials. The information provided by recorders is essential for an electrophysiologist to determine ablation strategy during termination of both pulmonary vein potentials and ventricular tachycardia.VT. Therefore, it is important that the recording system’s noise removal technique does not alt
Our Product
The patented PURE EP™ System is designed to address long-standing limitations that slow and disrupt cardiac catheter ablation procedures, such as environmental lab noise, signal saturation, slow signal recovery, and inaccurate display of fractionated potentials. PURE EP™ is a signal processing platform that combines advanced hardware and software to address known challenges associated to signal acquisition, to enable electrophysiologists to see more signals and analyze them in real-time.The device aims to minimize noise and artifacts from cardiac recordings and acquire high-fidelity cardiac signals. Improving fidelity of acquired cardiac signals may potentially increase the diagnostic value of these signals, thereby possibly improving accuracy and efficiency of the EP studies and ablation procedures.
Cardiac catheter ablation is a procedure that involves delivery of energy through the tip of a catheter that scars or destroys heart tissue to correct heart rhythm disturbances. In August 2018, we received 510(k) clearance from the FDA to market our PURE EP™ System.
Our PURE EP™ System can record raw (unaltered) cardiac and other physiologic signals with multiple display options, low noise, and a large input signal dynamic range. This is achieved using a low-noise amplifier topology with minimal filtering to band-limit the signal and a high-resolution A/D converter. In addition, the PURE EP™ System can provide large-signal (e.g., from a defibrillator) input protection and radio frequency (RF) signal (e.g., from ablation) noise suppression. There is no need for gain switching in this architecture, and the full range of input signals is digitized with high resolution.
Our PURE EP™ System was designed to be useful in arrhythmia diagnosis. For example, in atrioventricular reentrant tachycardia (AVRT) & AV nodal reentrant tachycardia (AVNRT), EP physicians often look for a slow pathway potential or accessory pathway potentials that are not easy to detect. Furthermore, during pacing maneuvers, important diagnostic signals may be buried inside the saturation artifact from the pacing electrode. The wide dynamic range of the PURE EP™ System may allow for better differentiation of those signals, as there is no system saturation and a quicker recovery to baseline.
We are focused on improving intracardiac signal acquisition and enhancing diagnostic information for catheter ablation procedures for all arrhythmias, especially complex types like VT and AF. VT is a fast, abnormal heart rate in the heart’s lower chambers. VT does not give your heart enough time to fill with blood before it contracts again. This can affect blood flow to the rest of your body and is potentially life-threatening. AF is the most common cardiac arrhythmia associated with a fivefold risk of stroke. AF occurs when the upper chambers of the heartbeat irregularly, and do not pump all of the blood to the lower chambers, causing some blood to pool and potentially form clots. If a clot breaks loose, it can travel through the bloodstream to the brain and lead to a stroke. Strokes related to AF are often more severe compared to strokes with other underlying causes.
We intendbelieve that the PURE EP™ System and its advanced signal processing tools may contribute to bringimprovements in patient outcomes in connection with catheter ablation due to the electrophysiology marketfollowing advantages over currently available devices on the market:
● | Less noise: PURE EP™’s low-noise proprietary architecture was engineered to enable acquisition of high-fidelity signals in the original, unfiltered format. PURE EP’s Main System Unit (MSU) topology incorporates advanced shielding and very low noise front-end components. |
● | Wider range: PURE EP™’s wide dynamic range was developed to retain cardiac signal details and reduce saturation. PURE EP™ combines a low-noise signal architecture with a fixed range up to 500mV, so signals are rarely clipped or limited by quantization noise. |
● | Higher fidelity: PURE EP™’s large frequency bandwidth and linear signal acquisition helps to accurately display complex fractionated signals, even at lower amplitudes and higher frequencies. This unique system capability minimizes signal attenuation and maintains original signal amplitude – especially critical for identifying and interpreting complex arrhythmogenic substrates. |
● | Clear, stable unipolar signals: The PURE EP™ System uses an innovative approach to acquiring unipolar signals. The Wilson Central Terminal (WCT+™) relies on a common front-end circuitry similar to how bi-polar intracardiac signals are acquired. This enables clear, stable unipolar signals, without the need for an internal reference catheter. |
● | Customizable software and filters: PURE EP™ offers software modules and specialty digital filters, so electrophysiologists can customize their interface and optimize signals for mapping, signal interpretation and during therapy delivery. |
● | Seamless integration: PURE EP™ integrates with existing EP labs and workflows. It is compatible and complementary with EP recording systems, mapping systems, robotic equipment, and multi-display panels. |
In April 2021, we released PURE EP™ Software Version 4. The latest release builds on the main system capabilities of the PURE EP System, an electrocardiogram/intracardiac recorder that will be coupled with an array ofTM while improving the overall user experience. The software tools intended for electrophysiology studies and procedures ranging from simple diagnostic testsupgrade has been rolled out to ablation forall existing customers. The latest software represents the most complex casesadvanced software version of arrhythmias. the PURE EPTM System. We believe the update adds valuable tools to shorten system set up time and bring innovative features for faster real-time signal analysis, potentially improving the efficiency and accuracy of EP procedures.
Advances in the new PURE EPTM Software Version 4 include user interface enhancements for a more compelling assessment of arrhythmia morphologies, clinical template management for an efficient case setup process, and other software functionalities for real-time signal visualization, such as the “Differential Analysis” allowing the simultaneous display of a channel using various filter settings to assess specific characteristics of a signal or the enhanced “Digital Zoom” permitting to instantly focus on important physiologic details while preserving a high signal-to-noise ratio.
We believe that this system will provide unique recording capabilities because we are developing itPURE EP™’s features may allow physicians to better determine precise ablation targets, strategy, and end point of procedures with the objective of reducing the need for patients to undergo multiple procedures, and to allow precise, uninterrupted, real-time evaluations of electrocardiograms and electrograms, and allow electrophysiologistsfor less experienced EP physicians to obtain data that cannot be acquired from present day recorders.
Initial Analysis
According to S. J. Asirvatham, MD, et. al. (“Signals and Signal Processing for the Electrophysiologist,” Circ Arrhythm Electrophysiol. (2011) 4:965-973), recording environments in a typical electrophysiology laboratory presents challenging situations. S. J. Asirvatham, MD, et. al., state, “Successful mapping and ablation in the electrophysiology laboratory is critically dependent on acquiring multiple, low-amplitude, intracardiac signals in the presence of numerous sources of electric noise and interference and displaying these signals in an uncomplicated and clinically relevant fashion, with minimal artifacts. This represents a significant engineering challenge and, in real-life electrophysiology laboratory, is not always successful.”
To determine and validate the state of present electrophysiology recording technology in the field, we completed a detailed analysis of the effect of filters used by existing EP recorders to reduce noise on spaciotemporal characteristics of electrocardiograms and intracardiac electrograms. We used a custom-built electrocardiogram/intracardiac simulator with a database of various electrocardiogram signals combined with electrophysiology signals, along with waveforms from publicly available databases. The ability to faithfully reproduce database waveforms generated by an electrocardiogram/intracardiac simulator was tested using the PURE EP System and conventional electrophysiology recorders, the GE CardioLab and St. Jude EP-WorkMate.
Proof of Concept Testing
In the second and third quarters of 2013, we performed and finalized testing of our proof of concept unit by initially using an electrocardiogram/intracardiac simulator at our lab, and subsequently by obtaining pre-clinical recordings from the lab at the University of California at Los Angeles. As part of the testing, we simultaneously recorded electrocardiogram and intracardiac signals on our proof of concept unit and GE’s CardioLab recording system. An identical signal was applied to the input of both systems and the monitor of our proof of concept unit was positioned next to the monitor of GE’s CardioLab recording system to allow for visual comparison. We believe that our proof of concept unit performed well as compared to GE’s CardioLab recording system, in that the electrocardiogram and intracardiac signals displayed on our proof of concept unit showed less baseline wander, noise and artifacts compared to signals displayed on GE’s CardioLab recording system. However, because this was a proof of concept test, without any clearly established protocols,Subsequently, we cannot present this data for publication and we do not have any independent verification or peer review of these findings.
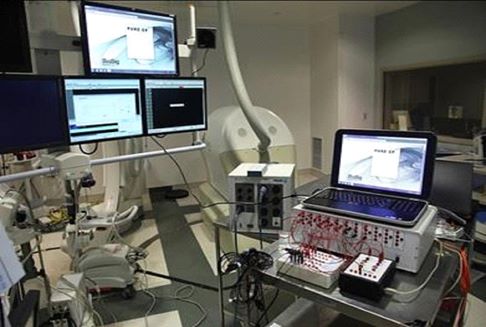
Prototype Testing
After conducting research of peer-reviewed EP publications (see Initial Analysis in Our Products section below)above), we contacted Samuel J. Asirvatham. M.D. (who we believed to be an expert in the field of signal-based catheter ablation), at Mayo Clinic in Rochester, Minnesota. Since the end of 2014, we have collaborated with Dr. Asirvatham and other physicians affiliated with Mayo Clinic in Rochester, Minnesota and Jacksonville, Florida. We have performed pre-clinical studies at Mayo Clinic since 2015 to validate technology within the PURE EP System prototype. These studies have been designed to determine clinical effectiveness for features within the PURE EP System that are in development.System. Since March 2016, we have published sevennine manuscripts in collaboration with the physicians from Mayo Clinic evidencing our pre-clinical findings. The publications coverTo date, we have conducted a varietytotal of subjects pertaining totwenty-four pre-clinical studies with the PURE EP System, twenty-one of which were conducted at Mayo Clinic in Rochester, Minnesota. We also conducted a pre-clinical study at the Mount Sinai Hospital in New York, NY with emphasis on the VT model; and two pre-clinical studies at the University of Pennsylvania in preparation for clinical studies to be conducted there.
Clinical Evaluations
In February 2019, we conducted the first clinical cases with our PURE EP™ System. The observational patient cases were performed by Andrea Natale, M.D., F.A.C.C., F.H.R.S., F.E.S.C., Executive Medical Director, Texas Cardiac Arrhythmia Institute at St. David’s Medical Center in Austin, Texas. In April 2019, we announced the completion of our second set of observational patient cases, which were performed at Prisma Health at Greenville Health System in South Carolina by Andrew Brenyo, MD, FHRS. Dr. Brenyo used the PURE EP™ System during procedures on patients with ischemic ventricular tachycardias, AF, PVC, and atypical flutters.
In May 2019, we announced the completion of our third set of observational patient cases at Indiana University under the leadership of Prof. John M. Miller, M.D., and Dr. Mithilesh K. Das, MBBS. Drs. Miller and Das used the PURE EP™ System during procedures on patients with atypical flutter, atrioventricular nodal reentry tachycardia (AVNRT), AF, supraventricular tachycardia, premature ventricular contractions, and a rare case of dual septal pathway. In August 2019, observational patient cases at Santa Barbara Cottage Hospital in California were performed by Brett Andrew Gidney, M.D. The initial experience across these early evaluation centers showed the PURE EP™ System functions as an enhanced electrophysiologydesigned with positive feedback from EP users about the improved signal detection and fidelity.
In November 2019, we commenced our first clinical study for the PURE EP™ System titled, “Novel Cardiac Signal Processing System for Electrophysiology Procedures (PURE EP 2.0 Study).” The PURE EP 2.0 Study was conducted at three U.S. hospitals: Texas Cardiac Arrhythmia Institute at St. David’s Medical Center in Austin, Texas, Mayo Clinic in Jacksonville, Florida and Massachusetts General Hospital in Boston, Massachusetts.
In April 2021, we announced the completion of the enrollment in the PURE EP 2.0 Study. Intracardiac signal data of clinical interest were collected during 51 cardiac ablation procedures using the PURE EP™ System, the signal recording system, and the 3D mapping system at the same time stamps. The samples were randomized and subjected to blinded, head-to-head evaluation by three independent electrophysiologists to determine the overall quality and clinical utility of PURE EP™ signals when compared to conventional sources. Each reviewer responded to the same 235 signal comparisons using a 10-point rating scale.
Results showed 93% consensus across the blinded reviewers with a 75% overall improvement in intracardiac signal acquisitionquality and differentiation and having specificconfidence in interpreting PURE EP signals over the signals from conventional sources. Further analysis of the responses from the blinded reviewers showed an 83% (p-value <0.001) improved confidence when interpreting complex multi-component signals, leading to a better understanding of the catheter position in relation to the ablation target. Additionally, there was a 73% (p-value <0.001) improved visualization of small, fractionated potentials increasing the proper analysis of scar and abnormal conduction tissue characteristics.
The study manuscript, “Evaluation of a novel cardiac signal processing system for electrophysiology procedures: the PURE EP 2.0 study" has been published in the Journal of Cardiovascular Electrophysiology and is available electronically with open access via the Wiley Online Library. The manuscript is co-authored by Amin Al-Ahmad, M.D., FHRS, Bradley Knight, M.D., FHRS, Wendy Tzou, M.D., FHRS, Robert Schaller, D.O., FHRS, Omar Yasin, M.D, Deepak Padmanabhan, M.D., Jason Zagrodsky, M.D., FHRS, Mohammed Bassiouny, M.D., J David Burkhardt, M.D., FHRS, Joseph Gallinghouse Jr., M.D., FHRS, Moussa Mansour, M.D., FHRS, Christopher McLeod, MBChB, Ph.D., FHRS and Andrea Natale, M.D., FHRS, the Principal Investigator of the study. The independent, blinded reviewers were Bradley P. Knight, M.D. (Northwestern University), Wendy Tzou, M.D. (University of Colorado), and Robert Schaller, M.D. (University of Pennsylvania).
In July, we discussed the completion of enrollment in the Re-Do Atrial Fibrillation Ablation Study. This study enrolled 20 patients undergoing repeat atrial fibrillation ablation at Texas Cardiac Arrhythmia Institute in Austin, TX. The study aims to determine if the PURE EP™ signals can demonstrate different electrophysiology signals.ablation targets and improve procedural efficiency. The results of the study are expected to be announced in early 2022.
We continue to install PURE EP™ Systems at centers of excellence for clinical evaluation under our market development plan. The PURE EP™ System has been utilized at numerous institutions, including Mayo Clinic campuses in Arizona, Florida and Minnesota; the University of Pennsylvania Hospital in Philadelphia, Pennsylvania; Overland Park Regional Medical System in Overland Park, Kansas; Deborah Heart and Lung Center in Browns Mills, New Jersey; St. Elizabeth’s Medical Center in Boston, Massachusetts; Medical City Heart Hospital in Dallas, Texas; Beth Israel Deaconess Medical Center (BIDMC) in Boston, Massachusetts, a teaching hospital of Harvard Medical School; Methodist Hospital in San Antonio, Texas; and Westside Regional Medical Center in Plantation, Florida.
To date, more than 2,160 patient procedures have been conducted with the PURE EP™ System by more than 76 electrophysiologists across seventeen different clinical sites in the United States. Our initial focus is on a targeted commercial launch of the PURE EP™ System in the Northeast, Texas, and Florida. The technology is regularly used in some of the states’ highest-ranked hospitals, including St. David’s Medical Center in Austin, Houston Methodist Hospital, Medical City North Hills in North Richland Hills in Texas and Mayo Clinic Florida Campus in Jacksonville, Florida.
In addition to clinical evaluation, we have conducted pre-clinical evaluation with the PURE EP™ System under several protocols. At Mayo Clinic in Rochester, Minnesota, we have performed twenty-seven experiments (including novel research program such as AI and repolarization) in various animal models; we also conducted a pre-clinical study at the Mount Sinai Hospital in New York, New York, with an emphasis on the VT model; and six experiments to date during a study at the University of Pennsylvania. We intend to continue additional research and development studies with our technology at Mayo Clinic, the University of Pennsylvania and other national centers of excellence.
The current PURE EP System prototype

Examples of PURE EP Signals

Commercialization of the PURE EP™ System
We have developed a marketing strategy to introduce and support our PURE EP™ System. The strategy includes our presence (in-person and virtually) at leading industry events and scientific sessions, both nationally and internationally, for the purposes of physician education, PURE EP System’s demonstrations and select presentations of advanced R&D product pipeline.
We have begun implementing a market development program to commercially launch our PURE EP System. We have installed PURE EP™ Systems at several medical centers of excellence throughout the U.S. during 2021 and will continue to do so in 2022 for clinical evaluation - whereby these systems are installed on a trial basis for system evaluations; data collection for our clinical trials; to gather and publish data in peer-reviewed journals and for presentations at cardiology conferences; and for potential demonstrations to other physicians to observe the technology.
Health systems, facilities, and physicians that have conducted or observed cases performed with our technology may potentially acquire the system. Sales of our systems would potentially consist of hardware, software, and a recurring revenue feature through a technical service contract, including software upgrades, and down the line, include the AI-driven algorithms and applications. In December 2020, we announced that three PURE EP™ Systems were contracted for purchase by St. David’s Healthcare in Austin, Texas and were subsequently sold in February 2021. These units were our first commercial sale. We also sold three PURE EP™ Systems to Mayo Foundation for Medical Education and Research in 2021, and we are in active discussions with several accounts about the acquisition of the PURE EP™ System. We anticipate our following customers will be medical centers of excellence and other healthcare facilities that operate EP labs within our targeted commercial launch markets in the Northeast, Florida, and Texas.
We intend to support our commercial activities by growing clinical validation and educational and training programs, including establishing training hubs at our early hospital partners’ facilities. With the increased commercialization activity planned, we also plan to continue to grow our clinical account management team to support the initial use of the system and assist with ongoing product training and education, and plan to develop an agile regional sales team to escalate our commercialization efforts along with a technical support team.
Our commercial and clinical activities are led by our Chief Commercial Officer, Gray Fleming, an experienced EP sales professional who previously spent 17 years at Abbott Laboratories and St. Jude Medical; Zachary Koch, CCDS, CEPS Clinical Director who spent 16 years at St. Jude Medical and Abbott EP, holding numerous positions across the company’s clinical, sales, training, and commercial teams; Olivier Chaudoir, Senior Director of Marketing from Biosense Webster and DePuy Synthes, Johnson & Johnson companies with 15 year of electrophysiology marketing and sales/clinical support experience. Our team is further complemented by Access Strategy Partners, Inc. (ASPI), a Boston-based consulting firm with a deep expertise in commercialization, contract management, execution, and value proposition optimization. The ASPI team is led by co-founder and president, Jim Walker, a healthcare executive with more than 30 years of experience in sales, marketing, sales operations, and national accounts management in some of the leading companies in the medical device sector, including Boston Scientific Corporation (BSC) and Johnson & Johnson. His experience spans domestic and international responsibilities, focusing on strategic market development and key customer management.
We believe we will have ample inventory to meet planned commercial placement requirements in 2022. We have made progress towards obtaining a European CE marking certificate for medical devices. In Q1 2022, we completed the quality management system audit for the International Organization for Standardization (“ISO”) 13485:2016 with the expectation to obtain the ISO 13485:2016 certification in the first half of 2022 and proceed to the application for the CE Marking clearance in the first half of 2023, subject to the guidance and availability from the European Notified Body.
Technology and Development Plan
Our technology team consists of six engineers and a consulting firmconsultants with expertise in digital signal processing, low power analog and digital circuit design, software development, embedded system development, electromechanical design, testing and system integration, and the regulatory requirements for medical devices. We have also entered into collaboration agreements with advisors and medical institutions in the fields of cardiology and electrophysiology, including Mayo Clinic Mount Sinai Hospital in New York, NY and the Texas Cardiac Arrhythmia Institute in Austin, TX (see “–Strategic Alliances”). We envision outsourcingTexas. Currently, we are transitioning contract manufacturing of the complete PURE EP System.
We intend to continue additional research studies with our first three pre-clinical studies in 2015technology at Mayo Clinic in Rochester, Minnesota. We have continued additional pre-clinical studies as part of an advanced research program since June 2016 atClinic. On November 20, 2019, we entered into licensing agreements with Mayo Clinic in Rochester, Minnesota withunder newly reached terms to establish a new product pipeline to complement the PURE EP System prototype. We also conducted a pre-clinical study at the Mount Sinai Hospital in New York, NY with emphasis on the VT model.
In January 2021, we entered into a research agreement with Mayo Clinic regarding a new AI research Program for our Novel Signal Recording System.
On November 16, 2021, we announced the launch of a new Artificial Intelligence development program with Technion – Israel Institute of Technology. Based in Haifa, Israel, Technion – Israel Institute of Technology is a public research university offering degrees in science, engineering, and we intend to beginrelated fields, such as medicine, industrial management, and education. Over the years, the Technion established itself as a pre-clinical studyleading academic institution in Artificial Intelligence (AI). It is currently ranked as number one in AI in Europe and 15th in the world, with 100 faculty members engaged in areas across the AI spectrum.
The research program is led by Asst. Prof. Joachim Behar, Head of the Artificial Intelligence in Medicine Laboratory (AIMLab) at the Cardiac Arrhythmia Center atTechnion. Under the University of California at Los Angeles. We intend to conduct further pre-clinical studies, and research studies. The main objective of these studies is to demonstrate the clinical potentialterms of the program, the ECG signals supplied by the PURE EP System.EP(tm) System are being analyzed in the context of developing AI-powered algorithms for atrial fibrillation ablation procedures.
Competition
We have initiated technology development with Minnetronix, a medical technology and innovation company, and engaged Quintain Project Solutions LLCare marketing the PURE EP™ System as the manufacturing project management leaderan additional information system for the PURE EP System – implementing steps for obtaining 510(k) clearance fromlab. In general, the U.S. Food and Drug Administration for the PURE EP System.
● | GE Healthcare’s family of CardioLab Recording Systems were initially developed in the early 1990s by Prucka Engineering, which was acquired by General Electric Company in 1999. |
● | The LabSystem PRO EP Recording System was originally designed in the late 1980s by C.R. Bard. C.R. Bard’s electrophysiology business was acquired by Boston Scientific Corporation in 2013. |
● | HeNan HuaNan Medical Science and Technology Co., LTD. offers the |
● | St. Jude Medical, Inc.’s EP-WorkMate Recording System was acquired from EP MedSystems, Inc. in 2008, which had received clearance for the product from the FDA in 2003. In January 2017, Abbott Laboratories acquired St Jude Medical, Inc. |
● | CathVision is developing an EP recording system, ECGenius System™ which is not yet cleared for sale in the US and not authorized for sale in Europe. |
Based upon our analysis of data taken from patent applications filed with the U.S. Patent and Trademark Office (“USPTO”) and 510(k) approval applications filed with the FDA, and various publications, we believe that the above recording systems are built on relatively old technologies and all use the identicalsimilar approach in applying hardware and digital filters to remove noise and artifacts. We are of the opinionreasonably believe that such an approach sacrifices cardiac signal fidelity, and in the case of ablation; the filters haveablation, has a direct impact on the ablation strategy of an electrophysiologist. The imprecise method to remove noise and artifacts used by the oldconventional recorders could be a contributing factor to the multiple (or repeated) ablation procedures that are frequently required in order to completely cure patients from atrial fibrillation and ventricular tachycardia.complex arrhythmias. We are not currently aware of any other companies that are developing new recording technologysimilar signal processing technologies for electrophysiology recorders.laboratories.
Customers
In December 2020, we announced that three PURE EP™ Systems were contracted for purchase by St. David’s Healthcare in Austin, Texas and were subsequently sold in February 2021. These units were our first commercial sales. We also sold three PURE EP™ Systems to Mayo Foundation for Medical Education and Research in 2021 and we are in active discussions with several accounts about the acquisition of the PURE EP™ System. We anticipate our following customers will be medical centers of excellence and other healthcare facilities that operate EP labs within our targeted commercial launch markets in the Northeast, Florida, and Texas.
Suppliers
The PURE EPEP™ System contains proprietary hardware and software modules that are assembled into the system. Hardware boards contain components that are available from different distributors. The parts used to manufacture analog and digital boards are readily available from a number ofseveral distributors or manufacturers. We obtained components from various suppliers and have assembledPlexus is our first prototype in-house. We envision outsourcing manufacturing ofpartner for the complete PURE EP System.
Research and Development Expenses
Research and development expenses for the fiscal years ended December 31, 2017, 20162021, and 2020 were $4,756,468$5,601,508 and $2,654,501,$18,135,862, respectively.
ViralClear Business Overview
ViralClear Pharmaceuticals, Inc.
ViralClear Pharmaceuticals, Inc. (“ViralClear”) is a majority-owned subsidiary of the Company originally known as NeuroClear Technologies, Inc. The subsidiary was established November 2018 to pursue additional applications of the PURE EP™ signal processing technology outside of EP. In March 2020, it was renamed ViralClear in connection with its prior objective to develop merimepodib, a broad-spectrum anti-viral agent that showed potential to treat COVID-19. We currently do not intend to further develop merimepodib and Customer Servicehave discontinued our pharmaceutical operations. Since late 2020, ViralClear has been realigned with its original objective of pursuing additional applications of the PURE EP™ signal processing technology outside of cardiac electrophysiology with an initial emphasis on developing a novel nerve recording system. As of March 30, 2022, the Company retains 60.22% ownership of ViralClear.
Currently, ViralClear is an early stage medical device company that is developing N-SENSE™, a novel sensing technology platform for high-speed electroneurogram (ENG) recordings. The specifications for this new product were based on the core competencies of the PURE EP™ signal processing technology, such as broad dynamic range of recorded signals and low signal-to-noise ratio and adapted to address disorders of the autonomic nervous systems through recordings and analysis of action potentials, the impulses along the membrane of a muscle cell or a nerve cell. These impulses are considered to carry valuable clinical information but may be difficult to detect through conventional recording platforms.
ViralClear aims to address what we believe to be the two main challenges of bioelectronic medicine devices: achieving accurate and targeted stimulation of specific nerves in a nerve bundle and implementing an effective feedback loop that can self-adjust for the optimal amount and timing of stimulation. We believe that advancements in overcoming these challenges will improve the safety and efficacy of current treatments and contribute to the developments of new therapy lines.
On December 18, 2020, we signed a research agreement with the University of Minnesota launching a program to develop novel therapies to treat sympathetic nervous system disease. The program studies are expected to form a foundation for developing a new platform technology to address disorders of the autonomic nervous system. We intend to develop new intellectual properties and products, including new hardware, software, and algorithmic solutions, with the support of Plexus, a tier 1 US-based manufacturing partner and take it through FDA approval, manufacturing, and commercialization. The R&D program is led by Richard W. Bianco, Ph.D., Professor, Director of Experimental Surgical Services (ESS), Department of Surgery in the University of Minnesota Medical School, John W. Osborn, Ph.D., Professor, Department of Surgery and Director of the Minnesota Consortium for Autonomic Neuromodulation (MCAN) in the University of Minnesota Medical School.
In February 2021, we conducted our first preclinical experiment at the University of Minnesota. Further studies to record and evaluate relevant nerve activity were conducted in April and November 2021.
We have partnered with Plexus to design, develop, and manufacture N-SENSE™, a novel sensing and stimulation platform technology.
Our new product pipeline will focus on improving therapies through clearer ENG recordings – methods used to visualize directly recorded electrical activities of neurons in the central nervous system (brain, spinal cord) and/or the peripheral nervous system (nerves, ganglions). ENGs are usually obtained by placing an electrode directly in the neural tissue. ENGs consist of small, high frequency, low amplitude signals, which have been proven hard to detect with conventional signal recording systems.
Our business strategy is to utilize our core signal processing technology to develop superior ENG recording and processing systems and includes the following:
• Develop N-SENSE™, a novel nerve sensing and stimulation platform technology to be used in product candidates which qualify for a nerve mapping and stimulation treatments including, but not limited to, renal denervation, deep brain stimulation and vagus nerve stimulation.
o | The N-SENSE™ is intended to be used as a value add-on to the existing neurostimulation technologies or act as a standalone platform. |
• Pursue licensing opportunities and partnerships to leverage our expertise in high-fidelity signal processing for feedback loop systems for development of products for commercial success.
We believe that the following clinical areas may benefit the most through the advancements in achieving accurate and targeted stimulation and implementation of an effective self-adjusting feedback loop:
• Renal denervation (“RDN”): RDN has been shown to reduce blood pressure and can be an effective treatment for resistant hypertension sufferers who have failed drug therapy. The technique has proven to be effective, but clinical endpoints are still suboptimal. RDN device market is expected to reach $7B by 2027 (CAGR 23.7%).1
o | Potential Application: A device that can measure sympathetic nerve activity will inform the need and potential benefit for performing a procedure. Additionally, a device that can stimulate and elicit a sympathetic response, such as blood pressure, will aid in the assessment of nerve denervation success, and help determine if additional ablation is necessary. Therefore, a device that can perform stimulation on a number of channels, and record nerve activity is needed. |
• Deep Brain Stimulation (“DBS”): DBS is a treatment that involves implanting electrodes (leads) within certain areas of the brain to deliver electrical pulses, which has demonstrated improvements in the treatment of movement disorders, such as the Parkinson’s disease, tremors and dystonia.
o | Potential Application: a new high-speed board-based platform for improved accuracy in lead implantation. Precise positioning of the electrodes during the surgical procedure is important in the success of lead implantation, and highly accurate signal readers can aid in the prediction of the activation of axons surrounding the implanted lead. |
o | We believe that DBS may also be applicable to a substantial number of neurological and psychiatric disorders correlated with dysfunctional circuitry; comparable to a heart pacemaker that uses electric pulses to ultimately regulate brain activity. |
o | Other applications under our investigation include chronic pain management, ADHD, eating disorders, Alzheimer’s, addiction, epilepsy. Alzheimer’s as an application for DBS is currently undergoing clinical trials at several national and international institutions that target the hippocampal outflow pathways by increasing ACh availability, influencing the limbic system, and improving lead placements. |
We may seek additional research collaborations with other academic centers active in one or more fields of clinical interests described above.
Industry and Market Overview
The global neurostimulation devices market is predicted to grow at 15.23% CAGR during the forecast period with the market size reaching $18.667 billion by 2025 from $7.974 billion in 2019. North America is dominating the neurostimulation devices market with highest market share due to robust healthcare infrastructure, growing R&D activity and presence of major healthcare players. The neurostimulation market is primarily driven by deep brain and spinal cord stimulation. The overall neurostimulation market is expected to grow due to societal factors such as an increase in the geriatric population, as well as the associated increase in the prevalence of chronic diseases.
The segment of the neurostimulation market for central nervous system (CNS), which include nVNS and DBS, is projected to exceed $14.5 billion in 2029 from a market value of $5 billion in 2019.2
Non-invasive Vagus Nerve Stimulation
We believe there is a significant opportunity for nVNS based on the potential market size for the treatments for the diseases that nVNS may be applicable. Currently, approximately 1,500 million people worldwide suffer from chronic pain while 1,100 million people worldwide suffer from migraines.
1Source: iHealthcareAnalyst, Inc. Feb. 2020
2Source: Bioelectronic Medicine2019 – 2029. IDTechEx report, Dr. Nadia Tsao.
Most of the currently available VNS products have achieved limited commercial success to date. LivaNova currently sells VNS devices that operate in 3 modes, including a non-rechargeable implantable pulse generator (IPG), SenTiva, which uses a limited closed-loop technology and comes with a wrist-worn magnet and a wireless programming wand. Cerbomed has commercialized a transcutaneous auricular VNS device, NEMOS, which consists of a handheld stimulation unit and an ear electrode worn as an earphone. Cerbomed received the European clearance (CE mark) for the VNS treatment of epilepsies and depression in 2010 and for the treatment of pain in 2012. NEMOS has been commercially available in Germany and Austria since 2013 and has expanded to Great Britain, France, and Spain.
The VNS patent domain is currently dominated by U.S. companies such as Medtronic, LivaNova, and Boston Scientific. Medtronic holds certain patents in closed-loop DBS technology, Medtronic currently markets IPGs such as RestoreSensor SureScan MRI, which is indicted for spinal cord stimulation as an aid in the management of chronic, intractable pain of the trunk and/or limbs and which automatically adjusts stimulation based on the patient's needs and preferences in different body positions, and Activa PC, which is a deep brain stimulator, for investigational loop.
We believe that digital health wearable markets present potential opportunities for our technology. We plan to implementdevelop technology that can provide a signaling feedback loop designed to deliver appropriate stimulation to the vagus nerve through audio and to seek licensing opportunities with consumer electronic market players.
Deep Brain Stimulation:
Deep brain stimulator market is one of the fastest growing sectors in the neurostimulation market worldwide, growing at 9.3% annually and expected to reach $2.3 billion in worldwide market size by 2028. According to the World Health Organization, globally, 264 million people suffer from depression while 50 million people suffer from epilepsy. Parkinson’s disease and essential tremor are FDA-approved indications for DBS, and the deep brain stimulator market is largely dominated by Medtronic, Abbott, and Boston Scientific. These companies have been working on innovations in their electrodes to avoid stimulation of adjacent structures (electric field shaping) which are the root cause of unwanted side effects of DBS. The industry is working on decreasing the size of the implant of the DBS device, which may lead to a skull-mounted implant. Medtronic’s Activa systems consist of dual-channel or single channel IPGs. Abbott sells two devices known as the Infinity DBS IPG and Brio Rechargeable IPG. The Infinity DBS IPG is designated to manage movement disorders including Parkinson’s disease, essential tremor, and dystonia. It utilizes the Bluetooth technology to communicate with a controller and can receive updates through an application. The system allows for currents to be steered towards target areas while avoiding peripheral stimulation. The Brio Rechargeable IPG delivers constant currents to maintain the desired stimulation level. It has shown clinical efficacy in Parkinson’s disease and dystonia. Boston Scientific offers the Vercise directional lead in unison with their Neural Navigator systems ranging from 8 to 16 electrode leads and a directional system. Medtronic’s Percept PC Deep Brain Stimulation (“DBS”) system includes their BrainSense technology making it the first and only DBS neurostimulation system that has the ability to chronically capture and record brain signals while providing therapy to patients with neurologic disorders associated with Parkinson’s Disease (“PD”), among others.
According to the National Institute of Health, future technical innovation in deep brain stimulators will focus on improving the practicability the device, including extension of battery life, reduced size of the devices and development program priorof a device for delivering more tailored and adaptive stimulation and the integration of wireless technology. Clinically, the main challenge will be meeting the needs of an ageing population worldwide and expanding indications for DBS to launch of our PURE EP System. As the product progresses through developmentcircuitopathies other than Parkinson’s disease, including depression and testing, we intend to gather the data produced by the PURE EP System’s processingAlzheimer disease. Even within established indications such as Parkinson’s disease, key questions remain unanswered because biomarkers that predict clinical responses and presenting electrocardiogramaid in patient selection and intracardiac signals and use such data for posters, presentations at cardiology conferences, and, if appropriate, submissions to scientific journals. stimulation parameter settings are still largely lacking.
We believe that asour technology may help advance clinical response to DBS due to more precise stimulation and improve overall safety of the DBS procedures.
On March 5, 2021, we gather additional dataannounced that the U.S. Patent Office had allowed a utility patent which has been exclusively licensed from our existing proofthe Mayo Foundation for Medical Education and Research. The patent application number 16/805,017 entitled, "Systems and Methods for Electroporation" was filed on February 28, 2020. The patent describes and claims methods and materials for improving the treatment of concept tests and our planned pre-clinical and clinical studies and user preference studies, we will be able to better determine the focushypertension via electroporation of our marketing efforts. We also plan to leverage our relationships with cardiac research and treatment centers to gain early product evaluation and validation. We believe that through these efforts, we may be able to gain preliminary acceptance of our PURE EP product by experienced professionals and academicsnerves in the electrophysiology field.
NeuroClear Business Overview
NeuroClear Technologies, Inc.
On July 2, 2020, the Company formed an additional subsidiary, NeuroClear Technologies, Inc. (“NeuroClear”), a Delaware corporation, to begin targeted commercial salespursue additional applications of the PURE EP System inEP™ signal processing technology outside of cardiac electrophysiology. We own 100% of the second halfoutstanding shares of 2018.common stock as of March 30, 2022 and the subsidiary is currently dormant.
Our intention is to move the neurotech assets from ViralClear into NeuroClear where the current and future neurotech assets would be housed. We intend to further develop our nerve recording system and ultimately bring the technology to market under NeuroClear Technologies, Inc.
ViralClear will continue to have cash and a shareholder base. Given its corporate history and almost four years of segregated operations, we believe that this entity can be of great value to the shareholders as we evaluate emerging growth businesses across various industry segments that aim for a Nasdaq listing.
Intellectual Property
Patents
Our success depends in large part on our ability to establish and maintain the proprietary nature of our technology. Our co-founder and former chief technology officer, Budimir S. Drakulic, Ph.D., conceived of the proprietary elements of the PURE EP System in 2009 and 2010. We filed a patent application with the USPTO in December 2013, directed at systems“Systems and methodsMethods for the evaluationEvaluation of electrophysiology systems. In March 2014, the inventors listed on the patent application filed in December 2013 assigned all of their rights to the patent application to us.Electrophysiology Systems.” In December 2014, we filed this patent application under the Patent Cooperation Treaty (PCT) with the U.S. Receiving Office. OurThis patent application filed in December 2013 represents a significant portion of our core proprietary intellectual property. Our patent application filed in December 2013 describes a system that can show comparative output of any two cardiac signal systems—such as the PURE EP System as compared to a competitor system, thus showingsystem. We received notice of allowance on June 5, 2019, and on October 29, 2019, U.S. Patent No. 10,456,057 was issued.
In November 2017, we engaged 3LP Advisors LLC, now Sherpa Technology Group LLC as our intellectual property advisor. We have also retained Sterne Kessler Goldstein & Fox P.L.L.C., a patent firm based in Washington DC, to help develop and execute a strategy for the valuedevelopment of the PURE EP System.
Our owned patent portfolio now includes sixteen allowed/issued patents. Seventeen additional worldwide utility patent applications we believe that the novelare pending covering various aspects of our PURE EP System should be subjectfor recording, measuring, calculating and displaying of electrocardiograms during cardiac ablation procedures. We also have two pending U.S. patent applications directed to pendingartificial intelligence (AI). We also have 30 allowed/issued worldwide design patents, which cover various features of our display screens and graphical user interface for enhanced visualization of biomedical signals. Finally, we have licenses to 3 patents and 14 additional worldwide utility patent application; however, we cannot be assuredapplications from Mayo Foundation for Medical Education and Research that allare pending. These patents and applications are generally directed to electroporation and stimulation.
BioSig and ViralClear signed three patent and know-how license agreements with Mayo Foundation for Medical Education and Research in November 2019. Under the terms of such agreements, BioSig exclusively licensed additional patents and applications of the patentsMayo Clinic related to ournovel ways for ablation therapy and to treat autonomic nervous system disease including hardware, software and algorithmic solutions to be integrated into the PURE EP platform technology. BioSig intends to take the licensed intellectual properties and products, which have been developed by Mayo Clinic over the last decade, through FDA approval, manufacturing, and commercialization. The development program is run under the leadership of Dr. Asirvatham. On March 5, 2021, we announced that the U.S. Patent Office had allowed a utility patent applications, if any, will be granted.that ViralClear has exclusively licensed from the Mayo Foundation for Medical Education and Research. The patent application number 16/805,017 entitled, "Systems and Methods for Electroporation" was filed on February 28, 2020. The patent describes and claims methods and materials for improving the treatment of hypertension via electroporation of nerves in the renal area. Electroporation is an emerging technique that has demonstrated efficacy in treatments for several critical conditions and is currently being evaluated for the treatments of autonomic nervous disorders, including hyper- and hypotension / syncope.
Trademarks
Our trademark for “BIOSIG TECHNOLOGIES”; was registered on April 25, 2017. Our trademark for “PURE EP”; was registered on January 26, 2016. Our trademark for the standard mark, “BIOSIG” was registered January 1, 2019, and our stylized/design trademark mark for the BioSig Technologies’ logo was registered February 12, 2019.
On October 7, 2019, we filed a standard mark trademark application for “SEE MORE, CLEARLY” and was published for opposition on May 26, 2020.
On April 22, 2020, we filed a standard mark trademark application for “DECIBEL” and was published for opposition on July 21, 2020.
On September 20, 2020, we filed a standard mark trademark application for “SMARTFINDER” and was published for opposition on March 16, 2021.
On September 20, 2020, we filed a standard mark trademark application for “COMBIO” and was published for opposition on March 16, 2021.
On October 22, 2020, we filed a standard mark trademark application for “WCT+” and was published for opposition on March 16, 2021.
On October 22, 2020, we filed a standard mark trademark application for “ACCUVIZ.”
On November 5, 2018, we filed a standard mark trademark application for “NEUROCLEAR”,and was registered on September 7, 2021. On January 29, 2019, NeuroClear filed a stylized/design trademark application for the NeuroClear logo and was registered on January 25, 2022.
On October 4, 2019, we filed a stylized/design trademark application for “ALLIANCE FOR ADVANCING BIOELECTRONIC MEDICINE” and was published for opposition on March 16, 2021.
On May 26, 2020, we filed a standard mark trademark application for “N-SENSE” ” and received a notice of allowance on June 2, 2020.
On May 26, 2020, we filed a standard mark trademark application for “N-SENSE TECHNOLOGIES” and received a notice of allowance on November 24, 2020.
In July 2021, we received EU certificates of registration for the following trademarks: ACCUVIZ, WCT+, and COMBIO.
In July 2021, we received UK certificates of registration for the following trademarks: SMARTFINDER, ACCUVIZ, WCT+, and COMBIO.
On May 27, 2021, we filed a standard mark trademark application for “SHOWING THE WAY TO BETTER.”
Government Regulation
The U.S. government regulates healthcare and related products through various agencies, including but not limited to the following: (i) the U.S. Food and Drug Administration (FDA), which enforces the federal Food, Drug and Cosmetic Act (FDCA) and related laws; (ii) the Centers for Medicare & Medicaid Services (CMS), which administers the Medicare and Medicaid programs; (iii) the Office of Inspector General (OIG), which enforces various laws aimed at curtailing fraudulent or abusive practices, including by way of example, the Anti-Kickback Statute, the Physician Self-Referral Law, commonly referred to as the Stark law, the Civil Monetary Penalty Law (including the beneficiary inducement prohibition) (CMP), and the laws that authorize the OIG to exclude healthcare providers and others from participating in federal healthcare programs; and (iv) the Office of Civil Rights (OCR), which administers the privacy aspects of the Health Insurance Portability and Accountability Act of 1996 (HIPAA). All of the aforementioned are agencies within the Department of Health and Human Services (HHS). Healthcare is also provided or regulated, as the case may be, by the Department of Defense through its TRICARE program, the Department of Veterans Affairs, especially through the Veterans Health Care Act of 1992, the Public Health Service within HHS under Public Health Service Act § 340B (42 U.S.C. § 256b), the Department of Justice through the Federal False Claims Act and various criminal statutes, and state governments under the Medicaid and other state sponsored or funded programs. Various states also have state laws equivalent to certain healthcare fraud and abuse laws, including but not limited to state equivalents of the Anti-Kickback Statute and the Stark law, as well as more general state laws regulating all healthcare activities and certain healthcare products, including medical devices.
In addition to being regulated by the FDA, advertising and promotion of certain types of medical devices in the United States is also regulated by the Federal Trade Commission (FTC) and by state regulatory and enforcement authorities. Recently, promotional activities for FDA-regulated products of other companies have been the subject of enforcement action brought under healthcare laws and consumer protection statutes. Further, competitors can initiate litigation relating to advertising claims under the federal Lanham Act and similar state laws.
FDA Regulation
Our solutions include software and hardware which will be used for patient diagnosis and, accordingly, are subject to regulation by the U.S. Food and Drug AdministrationFDA and other regulatory agencies. U.S. Food and Drug AdministrationFDA regulations govern, among other things, the following activities that we perform and will continue to perform in connection with:
● | Product design and development; |
● | Product testing; |
● | Product manufacturing; |
● | Product labeling and packaging; |
● | Product handling, storage, and installation; |
● | Pre-market clearance or approval; |
● | Advertising and promotion; and |
● | Product sales, distribution, and servicing. |
FDA Pre-market Clearance and Approval RequirementsProcesses
The U.S. Food and Drug AdministrationFDA classifies all medical devices into one of three classes. Devicesclasses based on the risks associated with the medical device and the controls deemed necessary to pose lower risksreasonably ensure the device’s safety and effectiveness. Those three classes are:
● | Class I devices present a low risk and are not life-sustaining or life-supporting. The majority of Class I devices are subject only to “general controls” (e.g., prohibition against adulteration and misbranding, registration and listing, good manufacturing practices, labeling, and adverse event reporting. General controls are baseline requirements that apply to all classes of medical devices.) |
● | Class II devices present a moderate risk and are devices for which general controls alone are not sufficient to provide a reasonable assurance of safety and effectiveness. Devices in Class II are subject to both general controls and “special controls” (e.g., special labeling, compliance with performance standards, and post market surveillance. Unless exempted, Class II devices typically require FDA clearance before marketing, through the premarket notification (510(k)) process). |
● | Class III devices present the highest risk. These devices generally are implantable, life-sustaining, life-supporting, or for a use that is of substantial importance in preventing impairment of human health, and/or they present a potential unreasonable risk of illness or injury. Class III devices are devices for which general controls, by themselves, are insufficient and for which there is insufficient information to determine that application of special controls would provide a reasonable assurance of safety and effectiveness. Class III devices are subject to general controls and typically require FDA approval of a premarket approval (“PMA”) application before marketing. |
Unless it is exempt from premarket review requirements, a medical device must receive marketing authorization from the FDA prior to being commercially marketed, distributed, or sold in interstate commerce in the United States. The most common pathways for obtaining marketing authorizations are placed in either Class I or II, which requires510(k) and PMA. With the manufacturer to submit toenactment of the U.S. Food and Drug Administration Safety and Innovation Act (FDASIA), the de novo pathway was made available for certain low-to-moderate risk devices that do not qualify for 510(k) clearance due to the absence of a pre-market notification, knownpredicate device.
510(k) Clearance Process
The 510(k) review process compares a new device to an existing legally marketed device (or, “predicate device”). “Substantial equivalence” means that the proposed new device: (i) has the same intended use as a PMN,the predicate device; (ii) has the same or similar technological characteristics as the predicate device; (iii) is as safe and a 510(k) approval, requesting clearance ofeffective as the predicate device, for commercial distribution in the U.S. Class III devices are devices which must be approvedas shown by the pre-market approval process. These tend to be devices that are permanently implanted into a human body or that may be necessary to sustain life. For example, an artificial heart meets both these criteria. Based on analysissupporting information submitted within the 510(k); and (iv) does not raise different questions of safety and effectiveness than the predicate devices, we believe that our products will be classified as Class II. Pursuant to U.S. Food and Drug Administration guidelines, Class II devices include a programmable diagnostic computer, which is a device that can be programmed to compute various physiologic or blood flow parameters based on the output from one or more electrodes, transducers, or measuring devices; this device includes any associated commercially supplied programs. Because the PURE EP System is a surface electrocardiogram and intracardiac multichannel recording and analysis system that acquires, processes and displays electrocardiogram and electrograms, we believe it will be classified as a Class II device. We must, therefore, first receive a
To obtain 510(k) clearance, from the U.S. Food and Drug Administration for our PURE EP System before we can commercially distribute it in the U.S. In the event that our PURE EP System is classified as a Class III device, which we believe is unlikely to occur, the U.S. Food and Drug Administration regulatory approval process and the subsequent commercialization of our product will require significantly greater time and resources than if it is classified as a Class II device, which would require us to reassess our strategic business plan of operations.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, will require a new 510(k) clearance or could require a pre-market approval, which requires more data and is generally a significantly longer process than the 510(k) clearance process. The U.S. Food and Drug AdministrationFDA requires each manufacturer to make this determination initially, but the U.S. Food and Drug AdministrationFDA can review any such decision and can disagree with a manufacturer’s determination. If the U.S. Food and Drug AdministrationFDA disagrees with a manufacturer’s determination, the U.S. Food and Drug Administrationit can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or a pre-market approval is obtained.
A device that reaches market through the 510(k) process is not considered to be “approved” by the U.S. Food and Drug AdministrationAdministration. They are generally referred to as “cleared” or “510(k) cleared” devices. Nevertheless, it can be marketed and sold in the U.S.
The Premarket Approval Pathway
The PMA process is the most stringent type of device marketing application required by the FDA. Whether PMA is granted is based on a determination by the FDA that the PMA application contains sufficient valid scientific evidence to ensure that the device is safe and effective for its intended use(s). A PMA application generally includes extensive information about the device including the results of clinical testing conducted on the device and a detailed description of the manufacturing process.
After a PMA application is accepted for review, the FDA begins an in-depth review of the submitted information. FDA regulations provide 180 days to review the PMA application and make a determination; however, in practice, the review time is typically longer (e.g., 1-3 years). During this review period, the FDA may request additional information or clarification of information already provided. Also, during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the data supporting the application and provide recommendations as to whether the data provide a reasonable assurance that the device is safe and effective for its intended use. In addition, the FDA generally will conduct a preapproval inspection of the manufacturing facility to ensure compliance with the quality system regulation (QSR), which imposes comprehensive development, testing, control, documentation and other quality assurance requirements for the design and manufacturing of a medical device.
Based on its review, the FDA may (i) issue an order approving the PMA, (ii) issue a letter stating the PMA is “approvable” (e.g., minor additional information is needed), (iii) issue a letter stating the PMA is “not approvable,” or (iv) issue an order denying PMA. A company may not market a device subject to PMA review until the FDA issues an order approving the PMA application. As a condition to approval, the FDA may impose post-approval requirements intended to ensure the continued safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution, and requiring the collection of additional clinical data. Failure to comply with the conditions of approval can result in materially adverse enforcement action, including withdrawal of the approval.
Most modifications to a PMA approved device, including changes to the design, labeling, or manufacturing process, require prior approval before being implemented. Prior approval is obtained through submission of a PMA supplement. The type of information required to support a PMA supplement and the FDA’s time for review of a PMA supplement vary depending on the nature of the modification.
We obtained FDA clearance related to the PURE EP System via the 510(k) process in 2018 and we do not anticipate a PMA for it or other devices at this time.
Pervasive and continuing FDA regulation
After a medical device is placed on the market, numerous U.S. Food and Drug AdministrationFDA regulatory requirements apply, including, but not limited to, the following:
● | Quality System |
● | Establishment Registration, which requires establishments involved in the production and distribution of medical devices intended for commercial distribution in the U.S. to register with the |
● | Medical Device Listing, which requires manufacturers to list the devices they have in commercial distribution with the |
● | Labeling regulations, which prohibit “misbranded” devices from entering the market, as well as mandate the inclusion of certain content in device labels and labeling and prohibit the promotion of products for unapproved or “off-label” uses and impose other restrictions on labeling; and |
● | Medical Device Reporting regulations, which require that manufacturers report to the |
Failure to comply with applicable regulatory requirements can result in enforcement action by the U.S. Food and Drug Administration,FDA, which may include one or more of the following sanctions:
● | Fines, injunctions, and civil penalties; |
● | Mandatory recall or seizure of our products; |
● | Administrative detention or banning of our products; |
● | Operating restrictions, partial suspension or total shutdown of production; |
● | Refusing our request for 510(k) clearance or pre-market approval of new product versions; |
● | Revocation of 510(k) clearance or pre-market approvals previously granted; and |
● | Criminal penalties. |
We are subject to unannounced device inspections by the FDA, as well as other regulatory agencies overseeing the implementation of, and compliance with, applicable state public health regulations. These inspections may include our suppliers’ facilities.
U.S. Healthcare Laws and Regulations
In the United States, there are various healthcare fraud and abuse laws that apply to medical device manufacturers, such as us, with respect to our financial relationships with hospitals, physicians, patients, marketers and sales agents, and other potential purchasers or acquirers of our products or those who are in a position to refer or recommend our products. Federal and state anti-kickback laws prohibit the payment or receipt of kickbacks, bribes or other remuneration intended to induce the purchase or recommendation of healthcare products and services. The U.S. government has published regulations that identify exemptions or “safe harbors,” which describe various payment and business practices that, although they potentially implicate the federal Anti-Kickback Statute, are not treated as offenses under the statute, and thereby, protected from enforcement actions under the federal Anti-Kickback Statute. To qualify, the activity must fit squarely within the safe harbor. Arrangements that do not meet a safe harbor are not necessarily illegal but will be evaluated on a case-by-case basis, and the federal safe harbors may not apply to state anti-kickback laws. Other provisions of state and federal law impose civil and criminal penalties for presenting, or causing to be presented, to third-party payors (including the government) for reimbursement claims that are false or fraudulent, or for items or services that were not provided as claimed. False claims allegations under federal, and some state, laws may be brought on behalf of the government by private persons, or “whistleblowers,” who could then receive a share of any recovery. In addition, the federal Health Insurance Portability and Accountability Act of 1996 (HIPAA) imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services. The Physician Self-Referral Law, commonly referred to as the Stark law, is a strict liability statute that prohibits physicians from referring patients to receive certain services defined as “designated health services” payable by Medicare or Medicaid from entities with which the physician or an immediate family member has a financial relationship, unless a specific exception applies. Violations of these laws can lead to civil and criminal penalties, including but not limited to punitive sanctions, damage assessments, money penalties, imprisonment, denial of payment, exclusion from participation in federal healthcare programs, or some combination thereof.
International Regulation
International sales of medical devices are subject to foreign government regulations, which vary substantially from country to country. The time required to obtain approval by a foreign country may be longer or shorter than that required for U.S. Food and Drug AdministrationFDA approval, and the requirements may differ significantly.
The European Union has adopted legislation, in the form of directives to be implemented in each member state, concerning the regulation of medical devices within the European Union. The directives include, among others, the European Union Medical DevicesDevice Directive (Council Directive 93/42/EEC of 14 June 1993 concerning medical devices, as amended) (the “Medical Device Directive”) that establishes standards for regulating the design, manufacture, clinical trials, labeling, and vigilance reporting for medical devices. Our PURE EP system may be affected by this legislation. Under the European Union Medical Device Directive, medical devices are classified into four classes, I, IIa, IIb, and III, with class I being the lowest risk and class III being the highest risk. Under the Medical Device Directive, a competent authority is nominated by the government of each member state to monitor and ensure compliance with the Medical Device Directive. The competent authority of each member state then designates a notified body to oversee the conformity assessment procedures set forth in the Medical Device Directive, whereby manufacturers demonstrate that their devices comply with the requirements of the Medical Device Directive and are entitled to bear the CE mark. CE is an abbreviation for Conformité Européenne (or European Conformity) and the CE mark, when placed on a product, indicates compliance with the requirements of the applicable directive. Medical devices properly bearing the CE mark may be commercially distributed throughout the European Union. Failure to obtain the CE mark will preclude us from selling the PURE EP System and related products in the European Union.
Employees
As of February 27, 2018,March 30, 2022, we had 1450 full-time employees. Additionally, we use consultants as needed to perform various specialized services. None of our employees are represented under a collective bargaining agreement.
ITEM 1A – RISK FACTORS
RISK FACTORS
There are numerous and varied risks, known and unknown, that may prevent us from achieving our goals. You should carefully consider the risks described below and the other information included in this Annual Report on Form 10-K, including the consolidated financial statements and related notes. If any of the following risks, or any other risks not described below, actually occur, it is likely that our business, financial condition, and/or operating results could be materially adversely affected.The risks and uncertainties described below include forward-looking statements and our actual results may differ from those discussed in these forward-looking statements.
Risk Factor Summary
Below is a summary of the principal factors that make an investment in our common stock speculative or risky. This summary does not address all of the risks that we face. Additional discussion of risks summarized in this risk factor summary, and other risks that we face, can be found below under the heading “Risk Factors” and should be carefully considered, together with other information in this Annual Report on Form 10-K and our other filings with the SEC before making investment decisions regarding our common stock.
● | There is substantial doubt about our ability to continue as a going concern. | |
● | Because we are an early commercialization stage company with one product in commercialization process, we expect to incur substantial additional operating losses. |
● | Our PURE EP System and other product candidates are in continued development and may not be successfully developed or commercialized. |
● | We expect to derive our revenue from sales of our PURE EP System and other products we may develop. If we fail to generate revenue from these sources, our results of operations and the value of our business will be materially and adversely affected. |
● | We may need to finance our future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. Any additional funds that we obtain may not be on terms favorable to us or our stockholders and may require us to relinquish valuable rights. |
● | We may be unable to develop our existing or future technology. |
● | We may experience delays in any phase of the preclinical or clinical development of a product, including during its research and development. |
● | We have not completed a clinical trial of our product. The results of additional clinical studies may not support the usefulness of our technology. |
● | The medical device industry is subject to stringent regulation and failure to obtain regulatory approval will prevent commercialization of our products. |
● | We, and our third-party manufacturer(s), are, and will be, subject to extensive regulation by the FDA. |
● | The market for our technology and revenue generation avenues for our products may be slow to develop, if at all. |
● | Our estimate of the size of our addressable market may prove to be inaccurate. |
● | The EP market is highly competitive. |
● | If we do not effectively manage changes in our business, these changes could place a significant strain on our management and operations. |
● | Our strategic business plan may not produce the intended growth in revenue and operating income. |
● | We currently have limited sales, marketing or distribution operations and will need to expand our expertise in these areas. |
● | Our product development program depends upon third-party researchers, including Mayo, who are outside our control and whose negative performance could materially hinder or delay our pre-clinical testing or clinical trials. |
● | We may face risks associated with future litigation and claims. |
● | If we do not obtain protection for our intellectual property rights, our competitors may be able to take advantage of our research and development efforts to develop competing products. |
● | If we infringe upon the rights of third parties, we could be prevented from selling products and forced to pay damages and defend against litigation. |
● | We depend on our collaboration with Mayo Clinic for the research and development of additional advanced features of PURE EP™ System. If this collaboration is not successful, we may not be able to realize the market potential of such features and may not have rights to use any such developed advanced features. | |
| ● | The market price for our common stock may fluctuate significantly, which could result in substantial losses by our investors. | |
| ● | Although our shares of common stock are now listed on The Nasdaq Capital Market, we currently have a limited trading volume, which results in higher price volatility for, and reduced liquidity of, our common stock. |
● | If we cannot continue to satisfy the continuing listing criteria of the Nasdaq Capital Market, the exchange may subsequently delist our common stock. |
● | Future sales of our common stock in the public market or other financings could cause our stock price to fall. |
● | If we sell additional equity or debt securities to fund our operations, it may impose restrictions on our business. |
Risks Related to Our Business and Industry
There is substantial doubt about our conditionability to continue as agoingconcern.
Our independent registered public accounting firm has issued an opinion on our consolidated financial statements included in this Annual Report on Form 10-K that states that the consolidated financial statements were prepared assuming we will continue as a going concern. Our consolidated financial statements have been prepared using accounting principles generally accepted in the United States of America applicable for a going concern, is in doubt,which assume that we will be forced to ceaserealize our business operations unless we can raise sufficient funds to satisfyassets and discharge our working capital needs.
To date, we have experienced negative cash flow from development of our technology, as well as from the costs associated with building a reasonable period of time.
Because of the numerous risks and there can be no assurance that our efforts will be successful. There is no assurance that can be given that our actions will result in profitable operations or the resolutionuncertainties associated with further development and commercialization of our liquidity problems.
Because we are an early developmentcommercialization stage company with no products nearone product in commercialization process, we expect to incur significantsubstantial additional operating losses.
We are an early developmentcommercialization stage company and we expect to incur substantial additional operating expenses over the next several years as our marketing, commercialization, and customer development along with additional research and development pre-clinical testing, regulatory approvalincrease for our PURE EP System and clinical trial activities increase.other product candidates. The amount of our future losses and when, if ever, we will achieve profitability are uncertain. We have noOur products that have generated anyminimal commercial revenue, and, do notalthough we expect to generate revenues this year from the commercial sale of our products in the near future,PURE EP System, may not be able to generate sufficient revenues to fund our operating expenses, if ever.any. Our ability to generate revenue and achieve profitability will depend on, among other things, the following:
● | successful completion of the pre-clinical and clinical development of our products; |
● | obtaining necessary regulatory approvals from the |
● | establishing manufacturing, sales, and marketing arrangements, either alone or with third parties; and |
● | raising sufficient funds to finance our activities. |
We might not succeed at all, or at any, of these undertakings. If we are unsuccessful at some or all of these undertakings, our business, prospects, and results of operations may be materially adversely affected.
Our PURE EP System and other product candidates are at an early stage ofin continued development and may not be successfully developed or commercialized.
Although our main product candidate, the PURE EP System, is in the early stage of developmentreceived FDA 510(k) clearance from FDA, we are currently conducting clinical trials and willmay conduct additional clinical trials, which may require substantial further capital expenditures,expenditure, to establish the safety and efficacy data needed to obtain acceptance by the medical community and coverage by third-party payors. The continued development testing, and regulatory clearances prior to commercialization, especially given that we have not yet completed pre-clinical testing on this product. The development and regulatory approval process takes several years, and it is not likely thatof the PURE EP System, even if successfully developed and approved by the U.S. Food and Drug Administration,and/or any other product candidates we may not be commercially available for a number of years. In addition, due to budgetary constraints, we have not been able to devote the level of resources that we desired to our research and development efforts. The continued development of our product candidatesdevelop, is dependent upon our ability to obtain sufficient additional financing. However, even if we are able to obtain the requisite financing to fund our development program, we cannot assure you that our current or future product candidates will be successfully developed or commercialized. Our failure to develop, manufacture, or receive regulatory approval for, or successfully commercialize any of our product candidates could result in the failure of our business and a loss of all of your investment in our company.
We expect to derive our revenue from sales of our PURE EP System and other products we may develop. If we fail to generate revenue from these sources, our results of operations and the value of our business will be materially and adversely affected.
As of December 31, 2021, our cash and cash equivalents were approximately $11.7 million. Based on our currently expected level of operating expenditures, we expect that our existing cash and cash equivalents will be sufficient to fund our operations through at least the next 7 months, or July 2022. Our revenue to beis generated from sales of our PURE EP System, for which we made first commercial sale in February 2021, and other products we may develop. Future sales of these products, if any, will be subject to, among other things, the receipt of regulatory approvals and commercial and market uncertainties that may be outside our control. If we fail to generate our intended revenues from these products, our results of operations and the value of our business and securities would be materially and adversely affected.
We may need to finance our future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. Any additional funds that we obtain may not be on terms favorable to us or our stockholders and may require us to relinquish valuable rights.
Until and unless we receive approval from the U.S. Food and Drug Administration and other regulatory authorities for our products, we will not generate revenues from our products. Therefore, for the foreseeable future,PURE EP System or another product of ours become commercially viable, we will have to fund all of our operations and capital expenditures from cash on hand, public or private equity offerings, debt financings, bank credit facilities or corporate collaboration and licensing arrangements. We believe that our existing cash on hand will be sufficient to enable us to fund our projected operating requirements for approximately the next five months.. However, we may need to raise additional funds more quickly if one or more of our assumptions prove to be incorrect or if we choose to expand our product development efforts more rapidly than we presently anticipate. We also may decide to raise additional funds before we require them if we are presented with favorable terms for raising capital.
If we seek to sell additional equity or debt securities, obtain a bank credit facility or enter into a corporate collaboration or licensing arrangement, we may not obtain favorable terms for us and/or our stockholders or be able to raise any capital at all, all of which could result in a material adverse effect on our business and results of operations. The sale of additional equity or debt securities, if convertible, could result in dilution to our stockholders. The incurrence of indebtedness would result in increased fixed obligations and could also result in covenants that would restrict our operations. Raising additional funds through collaboration or licensing arrangements with third parties may require us to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates, or to grant licenses on terms that may not be favorable to us or our stockholders. In addition, we could be forced to discontinue product development, reduce or forego sales and marketing efforts and forego attractive business opportunities, all of which could have an adverse impact on our business and results of operations.
We may be unable to develop our existing or future technology.
Our product, the PURE EP System, may not deliver the levels of accuracy and reliability needed to make it a successful product in the marketplace, and the development of such accuracy and reliability may be indefinitely delayed or may never be achieved. In addition, we may experience delays in the development of our technology for other reasons, including failure to obtain necessary funding and failure to obtain all necessary regulatory approvals. Failure to develop this or other technology could have an adverse material effect on our business, financial condition, results of operations and future prospects.
We may experience delays in any phase of the preclinical or clinical development of a product, including during its research and development.
We may experience delays in any phase of the preclinical or clinical development of a product, including during its research and development. The completion of any of these studies may be delayed or halted for numerous reasons, including, but not limited to, the following:
● | successful completion of the pre-clinical and clinical development of our products; |
● | the FDA or other regulatory authorities do not approve a clinical study protocol or place a clinical study on hold; |
● | patients do not enroll in a clinical study or results from patients are not received at the expected rate; |
● | patients discontinue participation in a clinical study prior to the scheduled endpoint at a higher than expected rate; |
● | patients experience adverse events from a product we develop; |
● | third-party clinical investigators do not perform the studies in accordance with the anticipated schedule or consistent with the study protocol and good clinical practices or other third-party organizations do not perform data collection and analysis in a timely or accurate manner; |
● | third-party clinical investigators engage in activities that, even if not directly associated with our studies, result in their debarment, loss of licensure, or other legal or regulatory sanction; |
● | regulatory inspections of manufacturing facilities, which may, among other things, require us to undertake corrective action or suspend the preclinical or clinical studies; |
● | changes in governmental regulations or administrative actions; |
● | the interim results of the preclinical or clinical study, if any, are inconclusive or negative; and |
● | the study design, although approved and completed, is inadequate to demonstrate effectiveness and safety. |
If the preclinical and clinical studies that we are required to conduct to gain regulatory approval are delayed or unsuccessful, we may not be able to market any product that we develop in the future. Preclinical studies and clinical trials are expensive and difficult to design and implement and any delays or prolongment in our preclinical and clinical studies will require additional capital. There is no assurance that we will be able to acquire additional capital to support our studies. The failure to obtain additional capital would have a material adverse effect on the Company.
We have completed one clinical trial of our product. The results of additional clinical studies may not support the usefulness of our technology.
In November 2019, we commenced our first clinical study with PURE EP System and completed the clinical trial as of September 2021. Conducting clinical trials is a long, expensive, and uncertain process that is subject to delays and failure at any stage. Clinical trials can take months or years. The commencement or completion of any of our subsequent clinical trials may be delayed or halted for numerous reasons, including:
● | the |
● | subjects may not enroll in clinical trials at the rate we expect, or we may not follow up on subjects at the rate we expect; |
● | subjects may experience |
● | third-party clinical investigators may not perform our clinical trials consistent with our anticipated schedule or the clinical trial |
● | interim results of any of our clinical trials may be inconclusive or negative; |
● | regulatory inspections of our clinical trials may require us to undertake corrective action or suspend or terminate the clinical trials if investigators find us |
● | governmental regulations or administrative actions may change and impose new requirements, particularly with respect to reimbursement. |
Results of pre-clinical studies do not necessarily predict future clinical trial results and previous clinical trial results may not be repeated in subsequent medicalclinical trials. We may experience delays, cost overruns and project terminations despite achieving promising results in pre-clinical testing or early clinical testing. In addition, the data obtained from clinical trials may be inadequate to support a device’s approval or clearance, of a submission.or to demonstrate safety and efficacy to the extent required to obtain third-party coverage and/or reimbursement. The U.S. Food and Drug AdministrationFDA may disagree with our interpretation of the data from our clinical trials, or may find the clinical trial design, conduct, or results inadequate to demonstrate the safety and effectiveness of the product candidate. The U.S. Food and Drug AdministrationFDA may also require us to conduct additional pre-clinical studies or clinical trials that could further delay clearance or approval of our products.any product candidates we may develop in the future and/or the PURE EP System to the extent we seek clearance/approval for different indications than that for which it is currently cleared. If we are unsuccessful in receiving U.S. Food and Drug AdministrationFDA clearance approval of a future product candidate, or a product’s clearance or approval is withdrawn, we would not be able to commercialize the productproduct(s) in the U.S., which could seriously harm our business. Moreover, we face similar risks in other jurisdictions in which we may sell or propose to sell our products.
The medical device industry is subject to stringent regulation and failure to obtain regulatory approval will prevent commercialization of our products.
Medical devices are subject to extensive and rigorous regulation by the U.S. Food and Drug AdministrationFDA pursuant to the Federal Food, Drug, and Cosmetic Act, by comparable agencies in foreign countries and by other regulatory agencies and governing bodies. Under the Federal Food, Drug, and Cosmetic Act and associated regulations, manufacturers of medical devices must comply with certain regulations that cover the composition, labeling, testing, clinical study, manufacturing, packaging and distribution of medical devices. In addition, medical devices must receive U.S. Food and Drug AdministrationFDA clearance or approval before they can be commercially marketed in the U.S., and the U.S. Food and Drug AdministrationFDA may require testing and surveillance programs to monitor the effects of approved products that have been commercialized and can prevent or limit further marketing of a product based on the results of these post-market evaluation programs. The process of obtaining marketing clearance or approval from the U.S. Food and Drug AdministrationFDA for new products could take a significant period of time, require the expenditure of substantial resources, involve rigorous pre-clinical and clinical testing, require changes to the products and result in limitations on the indicated uses of the product. In addition, if we seek regulatory approval in non-U.S. markets, we will be subject to further regulatory approvals that may require additional costs and resources. There is no assurance that we will obtain necessary regulatory approvals in a timely manner, or at all.
To obtain 510(k) clearance for a medical device, a pre-market notification must be submitted to the FDA demonstrating that the device is “substantially equivalent” to a previously cleared “predicate” device. A new device is substantially equivalent to a predicate device “at least as safe and effective” as the predicate. The FDA considers a device substantially equivalent to a predicate if it has the same intended use as the predicate and has either: (i) the same technological characteristics as the predicate or (ii) different technological characteristics from the predicate, but the information submitted to the FDA does not raise new questions of safety or effectiveness or demonstrates that the device is at least as safe and effective as the predicate.
We received 510(k) clearance to market our current lead product, the PURE EP System will need to receive 510(k) marketing clearance from the U.S. Food and Drug Administration in order permit us to market this product in the U.S. In addition,However, if we intend to market our productthe PURE EP System for additional medical uses or indications, we willmay need to submit additional 510(k) applications to the U.S. Food and Drug AdministrationFDA that are supported by satisfactory clinical trial results specifically for the additional indication. Clinical trials necessary to support 510(k) clearance or PMA approval for any future product candidates, or any new indications for use for our PURE EP System, would be expensive and could require the enrollment of large numbers of suitable patients who could be difficult to identify and recruit. Delays or failures in any necessary clinical trials could prevent us from commercializing any modified product or new product candidate and could adversely affect our business, operating results and prospects.
The results of our initial clinical trials may not provide sufficient evidence to allow the U.S. Food and Drug AdministrationFDA to grant us such additional marketing clearances and even additional trials requested by the U.S. Food and Drug AdministrationFDA may not result in our obtaining 510(k) marketing clearance for our product. The failure to obtain U.S. Food and Drug AdministrationFDA marketing clearance for the PURE EP System, any additional indications for the PURE EP System or any other of our future products would have a material adverse effect on our business.
We, and our productsthird-party manufacturer(s), are, and will be, subject to extensive post-approval regulation.regulation by the FDA.
In addition to the pre-market regulations, once a productdevice is approved byor cleared for the relevant regulatory bodyapplicable indications for our targeted commercialization market,use, numerous post-approval requirementsFDA regulations apply, including but not limited to requirementsthose relating to manufacturing, labeling, packaging, advertising, and record keeping. Notably, these regulations apply to us, as well as our contract manufacturer(s). Even if regulatory approval or clearance of a product is obtained, the approval or clearance may be subject to limitations on the uses for which the product may be marketed or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Any such post-approval requirementrequirements could reduce our revenues, increase our expenses, and render the approved product candidate not commercially viable. If we fail to comply with the regulatory requirements of the applicable regulatory authorities,requirements, or if previously unknown problems with any approved commercial products, manufacturers, or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other negative consequences, including:
● | restrictions on our products, manufacturers or manufacturing processes; |
● | warning letters and untitled letters; |
● | civil penalties and criminal prosecutions and penalties; |
● | fines; |
● | injunctions; |
● | product seizures or detentions; |
● | import or export bans or restrictions; |
● | voluntary or mandatory product recalls and related publicity requirements; |
● | suspension or withdrawal of regulatory approvals; |
● | total or partial suspension of production; and |
● | refusal to approve pending applications for marketing approval of new products or of supplements to approved applications. |
Regulations are constantly changing, and in the future our business may be subject to additional regulations that increase our compliance costs.
We believe we understand the current laws and regulations to which our products will be subject in the future. However, federal, state and foreign laws and regulations relating to the sale of our products are subject to future changes, as are administrative interpretations of regulatory agencies. If we fail to comply with such federal, state or foreign laws or regulations, we may fail to obtain regulatory approval for our products and, if we have already obtained regulatory approval, we could be subject to enforcement actions, including injunctions preventing us from conducting our business, withdrawal of clearances or approvals and civil and criminal penalties. In the event that federal, state, and foreign laws and regulations change, we may incur additional costs to seek government approvals, in addition to the clearance we intend to seek from the U.S. Food and Drug AdministrationFDA in order to sell or market our products. If we are slow or unable to adapt to changes in existing regulatory requirements or the promulgation of new regulatory requirements or policies, we or our licensees may, following approval, lose marketing approval for our products which will impact our ability to conduct business in the future.
The market for our technology and revenue generation avenues for our products may be slow to develop, if at all.
The market for our products may be slower to develop or smaller than estimated or it may be more difficult to build the market than anticipated. The medical community may resist our products or be slower to accept them than we anticipate. Revenues from our products may be delayed or costs may be higher than anticipated which may result in our need for additional funding. We anticipate that our principal route to market will be through commercial distribution partners. These arrangements are generally non-exclusive and have no guaranteed sales volumes or commitments. The partners may be slower to sell our products than anticipated. Any financial, operational or regulatory risks that affect our partners could also affect the sales of our products. In the current economic environment, hospitals and clinical purchasing budgets may exercise greater restraint with respect to purchases, which may result in purchasing decisions being delayed or denied. If any of these situations were to occur this could have a material adverse effect on our business, financial condition, results of operations and future prospects.
Our estimate of the size of our addressable market may prove to be inaccurate.
While our addressable market size estimate for the EP market was made in good faith and is based on assumptions and estimates we believe to be reasonable, this estimate may not be accurate. If our estimates of the size of our addressable market are not accurate, our potential for future growth may be less than we currently anticipate, which could have a material adverse effect on our business, financial condition, and results of operations.
If we seek to market our products in foreign jurisdictions, we may need to obtain regulatory approval in these jurisdictions.
In order to market our products in the European Union and many other foreign jurisdictions, we may need to obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. Approval procedures vary among countries (except with respect to the countries that are part of the European Economic Area) and can involve additional clinical testing. The time required to obtain approval may differ from that required to obtain U.S. Food and Drug AdministrationFDA approval. Should we decide to market our products abroad, we may fail to obtain foreign regulatory approvals on a timely basis, if at all. Approval by the U.S. Food and Drug AdministrationFDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority, including obtaining CE Mark approval, does not ensure approval by regulatory authorities in other foreign countries or by the U.S. Food and Drug Administration.FDA. We may be unable to file for, and may not receive, necessary regulatory approvals to commercialize our products in any foreign market, which could adversely affect our business prospects. In addition, a new Medical Device Regulation was published in 2017, which includes additional premarket and post-market requirements, as well as potential product reclassifications or more stringent commercialization requirements that could delay or otherwise adversely affect our clearances and approvals.
The electrophysiologyEP market is highly competitive.
There are a number of groups and organizations, such as healthcare, medical device and software companies in the electrophysiologyEP market that may develop a competitive offering to our products. The largest companies in the electrophysiologyEP market are GE, Johnson & Johnson, Boston Scientific, Siemens, Medtronic, and St. Jude Medical.Abbott. All of these companies have significantly greater resources, experience and name recognition than we possess. There is no assurance that they will not attempt to develop similar or superior products, that they will not be successful in developing such products or that any products they may develop will not have a competitive advantage over our products. IfMoreover, our product may not be viewed as superior to existing technology or new technology from our competitors and as a result we may not be able to justify expected selling price our product, which may have a material adverse effect on market acceptance of our product. In addition, if we experience delayed regulatory approvals or disputed clinical claims, we may not have a commercial or clinical advantage over competitors’ products that we believe we currently possess. Should a superior offering come to market, this could have a material adverse effect on our business, financial condition, results of operations and future prospects.
We rely on key officers, consultants and scientific and medical advisors, and their knowledge of our business and technical expertise would be difficult to replace.
We are highly dependent on our officers, consultants and scientific and medical advisors because of their expertise and experience in medical device development. We do not have “key person” life insurance policies for any of our officers. Moreover, if we are unable to obtain additional funding, we will be unable to meet our current and future compensation obligations to such employees and consultants. In light of the foregoing, we are at risk that one or more of our consultants or employees may leave our company for other opportunities where there is no concern about such employers fulfilling their compensation obligations, or for other reasons. The loss of the technical knowledge and management and industry expertise of any of our key personnel could result in delays in product development, loss of customers and sales and diversion of management resources, which could adversely affect our results of operations.
We may fail to attract and retain qualified personnel.
We expect to rapidly expand our operations and grow our sales, research and development and administrative operations. This expansion is expected to place a significant strain on our management and will require hiring a significant number of qualified personnel. Accordingly, recruiting and retaining such personnel in the future will be critical to our success. There is intense competition from other companies, research and academic institutions, government entities and other organizations for qualified personnel in the areas of our activities. Many of these companies, institutions and organizations have greater resources than we do, along with more prestige associated with their names. If we fail to identify, attract, retain and motivate these highly skilled personnel, we may be unable to continue our marketing and development activities, and this could have a material adverse effect on our business, financial condition, results of operations and future prospects.
If we do not effectively manage changes in our business, these changes could place a significant strain on our management and operations.
Our ability to grow successfully requires an effective planning and management process. The expansion and growth of our business could place a significant strain on our management systems, infrastructure and other resources. To manage our growth successfully, we must continue to improve and expand our systems and infrastructure in a timely and efficient manner. Our controls, systems, procedures and resources may not be adequate to support a changing and growing company. If our management fails to respond effectively to changes and growth in our business, including acquisitions, there could be a material adverse effect on our business, financial condition, results of operations and future prospects.
Our strategic business plan may not produce the intended growth in revenue and operating income.
Our strategies ultimately include making significant investments in sales and marketing programs to achieve revenue growth and margin improvement targets. If we do not achieve the expected benefits from these investments or otherwise fail to execute on our strategic initiatives, we may not achieve the growth improvement we are targeting and our results of operations may be adversely affected. We may also fail to secure the capital necessary to make these investments, which will hinder our growth.
In addition, as part of our strategy for growth, we may make acquisitions and enter into strategic alliances such as joint ventures and joint development agreements. However, we may not be able to identify suitable acquisition candidates, complete acquisitions or integrate acquisitions successfully, and our strategic alliances may not prove to be successful. In this regard, acquisitions involve numerous risks, including difficulties in the integration of the operations, technologies, services and products of the acquired companies and the diversion of management’s attention from other business concerns. Although we will endeavor to evaluate the risks inherent in any particular transaction, there can be no assurance that we will properly ascertain all such risks. In addition, acquisitions could result in the incurrence of substantial additional indebtedness and other expenses or in potentially dilutive issuances of equity securities. There can be no assurance that difficulties encountered with acquisitions will not have a material adverse effect on our business, financial condition and results of operations.
We currently have nolimited sales, marketing or distribution operations and will need to expand our expertise in these areas.
We currently have nolimited sales, marketing or distribution operations. We have begun implementing a market development program and are in the process of building such operations and, in connection with the expected commercialization of our planned products, will need to expandPURE EP System, and we are expanding our expertise in these areas.sales, marketing and distribution operations for commercial growth. To increase internal sales, distribution and marketing expertise and be able to conduct these operations, we wouldhave begun to invest in and will have to invest significant amounts of financial and management resources. In developing these functions ourselves, we could face a number of risks, including:
● | we may not be able to attract and build an effective marketing or sales force; |
● | the cost of establishing, training and providing regulatory oversight for a marketing or sales force may be substantial; and |
● | there are significant legal and regulatory risks in medical device marketing and sales that we have never faced, and any failure to comply with applicable legal and regulatory requirements for sales, marketing and distribution could result in an enforcement action by the |
Our product development program depends upon third-party researchers, including Mayo, who are outside our control and whose negative performance could materially hinder or delay our pre-clinical testing or clinical trials.
We do not have the ability to conduct all aspects of pre-clinical testing or clinical trials ourselves. We depend upon independent investigators and collaborators, such as commercial third-parties, government, universities and medical institutions, to conduct our pre-clinical and clinical trials under agreements with us. For our first clinical trial for the PURE EP System, titled “Novel Cardiac Signal Processing System for Electrophysiology Procedures (PURE EP 2.0 Study)” which commenced in November 2019, we rely on third parties, including TCARF and Mayo Clinic to conduct the patient cases. In addition, we are party to various license agreements with Mayo, pursuant to which we rely on research and development information, materials, technical data, unpatented inventions, trade secrets, know-how and supportive information of Mayo to develop, make, have made, use, offer for sale, sell, and import licensed products. These collaborators are not our employees and we cannot control the amount or timing of resources that they devote to our programs. These investigators may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking such programs ourselves. The failure of any of these outside collaborators to perform in an acceptable and timely manner in the future, including in accordance with any applicable regulatory requirements, such as good clinical and laboratory practices, or pre-clinical testing or clinical trial protocols, could cause a delay or otherwise adversely affect our pre-clinical testing or clinical trials, our success in obtaining regulatory approvals and, ultimately, the timely advancement of our development programs. In addition, these collaborators may also have relationships with other commercial entities, some of whom may compete with us. If our collaborators assist our competitors at our expense, our competitive position would be harmed.
If healthcare providers are unable to obtain sufficient reimbursement or other financial incentives from third-party healthcare payers related to the use of our products, their adoption and our future product sales will be materially adversely affected.
Widespread adoption of the PURE EP System, and any other products we may develop in the future, by the medical community is unlikely to occur without a financial incentive from third-party payors for the use of these products. Third-party payors include but are not limited to governmental programs such as Medicare and Medicaid, commercial health insurers and private payors, workers’ compensation programs, and other organizations. Currently, the PURE EP System does not receive separate reimbursement from any third-party payor. Instead, healthcare providers typically receive reimbursement for the procedure in which our product is used. Future regulatory action by CMS or other governmental agencies, or unfavorable mediaclinical data, among other things, may impact coverage could damageand/or reimbursement policies for procedures performed using our reputation and harm our operations.
We may face risks associated with future litigation and claims.
We may, in the future, be involved in one or more lawsuits, claims or other proceedings. These suits could concern issues including contract disputes, employment actions, employee benefits, taxes, environmental, health and safety, personal injury and product liability matters. Due to the uncertainties of litigation, we can give no assurance that we will prevail on any claims made against us in any such lawsuit. Also, we can give no assurance that any other lawsuits or claims brought in the future will not have an adverse effect on our financial condition, liquidity or operating results.
Our business is subject to cybersecurity risks.
Our operations are increasingly dependent on information technologies and services. Threats to information technology systems associated with cybersecurity risks and cyber incidents or attacks continue to grow, and include, among other things, storms and natural disasters, terrorist attacks, utility outages, theft, viruses, phishing, malware, design defects, human error, and complications encountered as existing systems are maintained, repaired, replaced, or upgraded. Risks associated with these threats include, among other things:
● | theft or misappropriation of funds; |
● | loss, corruption, or misappropriation of intellectual property, or other proprietary, confidential or personally identifiable information (including supplier, or employee data); |
● | disruption or impairment of our and our business operations and safety procedures; |
● | damage to our reputation with our potential customers and the market; |
● | exposure to litigation; |
● | increased costs to prevent, respond to or mitigate cybersecurity events. |
Although we utilize various procedures and controls to mitigate our exposure to such risk, cybersecurity attacks and other cyber events are evolving and unpredictable. Moreover, we have no control over the information technology systems of our suppliers, and others with which our systems may connect and communicate. As a result, the occurrence of a cyber incident could go unnoticed for a period time.
We presently maintain insurance coverage to protect against cybersecurity risks. However, we cannot ensure that it will be sufficient to cover any particular losses we may experience as a result of such cyberattacks. Any cyber incident could have a material adverse effect on our business, financial condition and results of operations.
We may be subject, directly or indirectly, to U.S. federal and state health carehealthcare laws, including fraud and abuse, and false claims, and privacy laws and regulations. Prosecutions under such laws have increased in recent years and we may become subject to such litigation.litigation and enforcement. If we are unable to, or have not fully complied with such laws, we could face substantial penalties.
We are successful in achieving regulatory approval to market our PURE EP System, our operations will besubject, directly or indirectly, through our customers and health care professionals, subject to various U.S. federal and state healthcare laws and regulations. These laws include fraud and abuse laws, including, without limitation,such as the federal Anti-Kickback Statute, federal False Claims Act, and federal Foreign Corrupt Practices Act. These laws may impact, among other things, our proposed sales, and marketing and education programs.
● | The federal Anti-Kickback Statute, which prohibits persons from knowingly and willfully soliciting, offering, receiving, or providing remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid programs. |
● | The federal physician self-referral law, commonly referred to as the Stark Law, which prohibits a physician from making a referral for certain designated health services covered by the Medicare program, if the physician or an immediate family member has a financial relationship with the entity providing the designated health services, unless the financial relationship falls within an applicable exception to the prohibition. |
● | Federal civil and criminal false claims laws and civil monetary penalty laws, including the False Claims Act, which prohibits persons from knowingly filing, or causing to be filed, a false claim to, or the knowing use of false statements to obtain payment from, the federal government. Suits may be filed under the federal False Claims Act by the government or by an individual on behalf of the government (known as “qui tam” actions). Such individuals, commonly known as “relators” or “whistleblowers,” may share in any amounts paid by the entity to the government in fines or settlement. |
● | The federal transparency requirements under the Patient Protection and Affordable Care Act and the Health Care and Education Reconciliation Act, including the provision known as the Physician Payments Sunshine Act, which requires manufacturers of drugs, biologics, devices and medical supplies covered under Medicare, Medicaid, or the Children’s Health Insurance Program (CHIP) to record any information related to payments and other transfers of value to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members, and to report this data annually to CMS for subsequent public disclosure. Manufacturers must also disclose investment interests held by physicians and their family members. |
● | The federal Civil Monetary Penalties Law, which prohibits, among other things, the offering or transfer of remuneration to a Medicare or state healthcare program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state healthcare program, unless an exception applies. |
● | Federal criminal statutes created through the Health Insurance Portability and Accountability Act of 1996 (HIPAA), which prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters. |
● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and their respective implementing regulations, which imposes requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform services for them that involve the use, or disclosure of, individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information. |
● | Other federal and state fraud and abuse laws, prohibitions on self-referral and kickbacks, fee-splitting restrictions, prohibitions on the provision of products at no or discounted cost to induce physician or patient adoption, and false claims acts, transparency, reporting, and disclosure requirements, which may extend to services reimbursable by any third-party payer, including private insurers. |
● | State and federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that could potentially harm consumers. |
Additionally, we may be made under a federal health care program such as the Medicare and Medicaid programs. Several courts have interpreted the statute’s intent requirementsubject to mean that if any one purposestate equivalents of an arrangement involving remuneration is to induce referrals of federal health care covered business, the statute has been violated. The federal Anti-Kickback Statute is broad and, despite a series of narrow safe harbors, prohibits many arrangements and practices that are lawful in businesses outsideeach of the health care industry. Penalties for violationshealthcare laws described above, among others, some of which may be broader in scope and may apply regardless of the federal Anti-Kickback Statute include criminal penalties and civil and administrative sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal health care programs. An alleged violation of the federal Anti-Kickback Statute may be used as a predicate offense to establish liability pursuant to other federal laws and regulations such as the federal False Claims Act.payor. Many U.S. states have also adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for health care items orhealthcare services reimbursed by any source, not only the Medicare and Medicaid programs.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our future business activities could be subject to challenge under one or more of such laws. In addition, healthcare reform legislation has strengthened these laws. For example, the Affordable Care Act, among other things, amended the intent requirement of the federal Anti-Kickback and criminal healthcare fraud statutes. As a result of such amendment, a person or entity no longer needs to have actual knowledge of these statutes or specific intent to violate them in order to have committed a violation. Moreover, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including penalties, fines and/or exclusion or suspension from federal and state healthcare programs such as Medicare and Medicaid and debarment from contracting with the U.S. government. In addition, private individuals have the ability to bring actions on behalf of the U.S. government under the False Claims Act as well as under the false claims laws of several states.
Efforts to ensure that our business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our existing or future business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. Any such actions instituted against us could have a significant adverse impact on our business, including the imposition of civil, criminal and administrative penalties, damages, disgorgement, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations. Even if we are successful in defending against such actions, we may nonetheless be subject to substantial costs, reputational harm and adverse effects on our ability to operate our business. In addition, the approval and commercialization of any of our products outside the United States will also likely subject us to non-U.S. equivalents of the healthcare laws mentioned above, among other non-U.S. laws.
If any of our employees, agents, or the impact of such actions. Ifphysicians or other providers or entities with whom we do business are found to be in violation of any of the laws described above and otherhave violated applicable state and federal fraud and abuse laws, we may be subject to penalties,criminal, civil or administrative sanctions, including civil and criminal penalties, damages, fines, or an administrative action of suspension or exclusionexclusions from government health care reimbursementfunded healthcare programs, or, if we are not subject to such actions, we may suffer reputational harm for conducting business with persons or entities found, or accused of being, in violation of such laws. Any such events could adversely affect our ability to operate our business and the curtailment or restructuringour results of our operations.
In addition, to the extent we commence commercial operations overseas, we will be subject to the federal Foreign Corrupt Practices Act and other countries’ anti-corruption/anti-bribery regimes, such as the U.K. Bribery Act. The federal Foreign Corrupt Practices Act prohibits improper payments or offers of payments to foreign governments and their officials for the purpose of obtaining or retaining business. Safeguards we implement to discourage improper payments or offers of payments by our employees, consultants, sales agents or distributors may be ineffective, and violations of the federal Foreign Corrupt Practices Act and similar laws may result in severe criminal or civil sanctions, or other liabilities or proceedings against us, any of which would likely harm our reputation, business, financial condition and results of operations.
We have identified a material weaknesscould be adversely affected if healthcare legislation or reform measures substantially change the market for medical care or healthcare coverage in the U.S., negatively affecting our internal control over financial reporting which, if not remediated, could adversely affect our reputation, business or stock price.revenue for PURE EP or future products.
The Patient Protection and Procedures,Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, commonly referred to as the “Healthcare Reform Law,” we have identifiedincludes a material weakness in our internal control over financial reporting relatednumber of rules regarding health insurance, the provision of healthcare, conditions to reimbursement for healthcare services provided to Medicare and Medicaid patients, and other healthcare policy reforms. Through the segregation of duties in the initiating and recording of transactions.
These structural changes could entail further modifications to the existing system of private payors and government programs (such as Medicare, Medicaid, and the State Children’s Health Insurance Program), creation of government-sponsored healthcare insurance sources, or some combination of both, as well as other changes. Restructuring the coverage of medical care in the U.S. could impact the reimbursement for medical devices, including our current commercial products, those we cannot assure youand our development or commercialization partners are currently developing or those that we will not identify additional material weaknesses in our internal control over financial reportingmay commercialize or promote in the future. If reimbursement for our approved medical devices, products we currently commercialize or promote, or any product we may commercialize or promote is substantially reduced or otherwise adversely affected in the future, or rebate obligations associated with them are unablesubstantially increased, it could have a material adverse effect on our reputation, business, financial condition or results of operations.
Extending medical benefits to remediatethose who currently lack coverage will likely result in substantial costs to the U.S. federal government, which may force significant additional changes to the healthcare system in the U.S. Much of the funding for expanded healthcare coverage may be sought through cost savings. While some of these savings may come from realizing greater efficiencies in delivering care, improving the effectiveness of preventive care and enhancing the overall quality of care, much of the cost savings may come from reducing the cost of care and increased enforcement activities. Cost of care could be reduced further by decreasing the level of reimbursement for medical services or products (including those products currently being developed by us or our development or commercialization partners or any product we may commercialize or promote, including our current commercial products), or by restricting coverage (and, thereby, utilization) of medical services or products. In either case, a reduction in the utilization of, or reimbursement for, any medical device or any product we may commercialize or promote, including our current commercial products, or for which we receive marketing approval in the future, could have a material weakness,adverse effect on our reputation, business, financial condition or results of operations.
Further, the healthcare regulatory environment has seen significant changes in recent years and is still in flux. Legislative initiatives to modify, limit, replace, or repeal the Healthcare Reform Law and judicial challenges continue. Congress has enacted legislation that repeals certain portions of the Healthcare Reform Law, including but not limited to the Tax Cuts and Jobs Act, passed in December 2017, which included a provision that eliminates the penalty under the Healthcare Reform Law’s individual mandate, effective January 1, 2019, as well as the Bipartisan Budget Act of 2018, passed in February 2018, which, among other things, repealed the Independent Payment Advisory Board (which was established by the Healthcare Reform Law and was intended to reduce the rate of growth in Medicare spending). Additionally, in December 2018, a district court in Texas held that the individual mandate is unconstitutional and that the rest of the Healthcare Reform Law is, therefore, invalid. On appeal, the Fifth Circuit Court of Appeals affirmed the holding on the individual mandate but remanded the case back to the lower court to reassess whether and how such holding affects the validity of the rest of the Healthcare Reform Law. The Fifth Circuit’s holding has been appealed to the U.S. Supreme Court, and a decision on the case is pending. Substantial uncertainty remains as to the future of the Healthcare Reform Law. We cannot predict the impact on our business of future legislative and legal challenges to the Healthcare Reform Law or other changes to the current laws and regulations. However, it is possible that such initiatives could have an adverse effect on our ability to record, processobtain approval and/or successfully commercialize products in the U.S. in the future. For example, any changes that reduce, or impede the ability of healthcare providers to obtain reimbursement for medical procedures in which the products we currently, or intend to, commercialize are used, or that reduce medical procedure volumes, could adversely affect our operations and/or future business plans. The financial impact of U.S. healthcare reform legislation over the next few years will depend on a number of factors, including the policies reflected in implementing regulations and report financial information accurately,guidance and to prepare financial statements within the time periods specifiedchanges in sales volumes for medical devices affected by the ruleslegislation. From time to time, legislation is drafted, introduced and formspassed in the U.S. Congress that could significantly change the statutory provisions governing coverage, reimbursement, pricing, and marketing of the Securitiesmedical device products. In addition, third-party payor coverage and Exchange Commission, could be adversely affected. reimbursement policies are often revised or interpreted in ways that may significantly affect our business and our products.
The occurrence of or failure to remediate the material weaknessongoing COVID-19 pandemic may adversely affect our reputationbusiness.
In an effort to contain and mitigate the spread of COVID-19, many countries, including the United States, have imposed unprecedented restrictions on travel, quarantines, and other public health safety measures. Such government-imposed precautionary measures may have been relaxed in certain countries or states, but there is no assurance that more strict measures will be put in place again due to a resurgence in COVID-19 cases. The COVID-19 pandemic may adversely impact our business plan as our clinical studies may be delayed as hospitals in the impacted regions may shift their resources to patients affected by the disease. The rapidly evolving nature of the circumstances is such that it is impossible, at this stage, to determine the full and overall impact the COVID-19 pandemic may have, but it could disrupt production and cause delays in the supply and delivery of products used in our research and development efforts, adversely affect our employees, and disrupt our operations, all of which may have a material adverse effect on our business. In addition, the pandemic may have an adverse effect on the ability of regulatory bodies to review submissions in a timely manner, grant approvals or supervise our candidates and products, and may further divert the attention and efforts of the medical community to coping with the coronavirus and disrupt the marketplace in which we operate and may have a material adverse effects on our operations. Patient enrollment in future clinical trials could be slowed, delayed, or suspended due to the pandemic as well.
Moreover, the COVID-19 pandemic has created significant economic uncertainty and volatility in the credit and capital markets. Management plans to secure the necessary financing through the issue of new equity and/or the entering into of strategic partnership arrangements; however, there is no assurance that our management will be able to obtain such financing on reasonable terms or at all. A continuation or worsening of the levels of market disruption and volatility seen in the recent past could have an adverse effect on our ability to access capital and on the market price of our common stock, and any other securities we may issue.not be able to successfully raise capital through the sale of our securities. If we are unsuccessful in commercializing our products or raising capital, we may need to reduce activities, curtail or cease operations.
In addition, a significant resurgence of COVID-19 or other infectious diseases could result in a widespread health crisis that could adversely affect the economies and financial markets worldwide, resulting in an economic downturn that could impact our business, financial condition and results of operations.
As a smaller reporting company, we are subject to scaled disclosure requirements that may make it more challenging for investors to analyze our results of operations and financial prospects.
Currently, we are a “smaller reporting company,” as defined by Rule 12b-2 of the Exchange Act. As a “smaller reporting company,” we are able to provide simplified executive compensation disclosures in our filings and have certain other decreased disclosure obligations in our filings with the SEC, including being required to provide only two years of audited financial statements in annual reports. Consequently, it may be more challenging for investors to analyze our results of operations and financial prospects.
Furthermore, we are a non-accelerated filer as defined by Rule 12b-2 of the Exchange Act, and, as such, are not required to provide an auditor attestation of management’s assessment of internal control over financial reporting, which is generally required for SEC reporting companies under Section 404(b) of the Sarbanes-Oxley Act. Because we are not required to, and have not, had our auditor’s provide an attestation of our management’s assessment of internal control over financial reporting, a material weakness in internal controls may remain undetected for a longer period.
There are inherent limitations in all control systems, and misstatements due to error or fraud may occur and not be detected.
The ongoing internal control provisions of Section 404 of the Sarbanes-Oxley Act of 2002 require us to identify material weaknesses in internal control over financial reporting, which is a process to provide reasonable assurance regarding the reliability of financial reporting for external purposes in accordance with accounting principles generally accepted in the United States. Our management, including our chief executive officer and chief financial officer, does not expect that our internal controls and disclosure controls will prevent all errors and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. In addition, the design of a control system must reflect the fact that there are resource constraints and the benefit of controls must be relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, in our company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple errors or mistakes. Further, controls can be circumvented by individual acts of some persons, by collusion of two or more persons, or by management override of the controls. The design of any system of controls is also based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, a control may be inadequate because of changes in conditions, such as growth of the company or increased transaction volume, or the degree of compliance with the policies or procedures may deteriorate. Because of inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
In addition, discovery and disclosure of a material weakness, by definition, could have a material adverse impact on our financial statements. Such an occurrence could discourage certain customers or suppliers from doing business with us and adversely affect how our stock trades. This could in turn negatively affect our ability to access equity markets for capital.
Risks Related to Our Intellectual Property
If we do not obtain protection for our intellectual property rights, our competitors may be able to take advantage of our research and development efforts to develop competing products.
We intend to rely on a combination of patents, trade secrets, and nondisclosure and non-competition agreements to protect our proprietary intellectual property. Our owned patent portfolio now includes sixteen allowed/issued patents. Seventeen additional worldwide utility patent applications are pending covering various aspects of our PURE EP System for recording, measuring, calculating and displaying of electrocardiograms during cardiac ablation procedures. We also have filed atwo pending U.S. patent application with the U.S. Patentapplications directed to artificial intelligence (AI). We also have 30 allowed/issued worldwide design patents, which cover various features of our display screens and Trademark Office, andgraphical user interface for enhanced visualization of biomedical signals. Finally, we have filed thislicenses to 3 patents and 14 additional worldwide utility patent application under the Patent Cooperation Treaty (PCT) with the U.S. Receiving Office. applications from Mayo Foundation for Medical Education and Research that are pending. These patents and applications are generally directed to electroporation and stimulation.
We plan to file additional patent applications in the U.S. and in other countries as we deem appropriate for our products. Our applications have and will include claims intended to provide market exclusivity for certain commercial aspects of the products, including the methods of production, the methods of usage and the commercial packaging of the products. However, we cannot predict:
● | the degree and range of protection any patents will afford us against competitors, including whether third parties will find ways to invalidate or otherwise circumvent our patents; |
● | if and when such patents will be issued, and, if granted, whether patents will be challenged and held invalid or unenforceable; |
● | whether or not others will obtain patents claiming aspects similar to those covered by our patents and patent applications; or |
● | whether we will need to initiate litigation or administrative proceedings which may be costly regardless of outcome. |
Our success also depends upon the skills, knowledge and experience of our scientific and technical personnel, our consultants and advisors as well as our licensors and contractors. To help protect our proprietary know-how and our inventions for which patents may be unobtainable or difficult to obtain, we rely on trade secret protection and confidentiality agreements. To this end, it is our policy to require all of our employees, consultants, advisors and contractors to enter into agreements which prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business. These agreements may not provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information. If any of our trade secrets, know-how or other proprietary information is disclosed, the value of our trade secrets, know-how and other proprietary rights would be significantly impaired and our business and competitive position would suffer.
Given the fact that we may pose a competitive threat, competitors, especially large and well-capitalized companies that own or control patents relating to electrophysiology recording systems, may successfully challenge our current and planned patent applications, produce similar products or products that do not infringe our future patents, or produce products in countries where we have not applied for patent protection or that do not respect our patents.
If any of these events occurs, or we otherwise lose protection for our trade secrets or proprietary know-how, the value of our intellectual property may be greatly reduced. Patent protection and other intellectual property protection are important to the success of our business and prospects, and there is a substantial risk that such protections will prove inadequate.
If we infringe upon the rights of third parties, we could be prevented from selling products and forced to pay damages and defend against litigation.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may be required to:
● | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
● | abandon an infringing product candidate; |
● | redesign our product candidates or processes to avoid infringement; |
● | cease usage of the subject matter claimed in the patents held by others; |
● | pay damages; and/or |
● | defend litigation or administrative proceedings which may be costly regardless of outcome, and which could result in a substantial diversion of our financial and management resources. |
Any of these events could substantially harm our earnings, financial condition and operations.
We depend on our collaboration with Mayo Clinic for the research and development of additional advanced features of PURE EP™ System. If this collaboration is not successful, we may not be able to realize the market potential of such features and may not have rights to use any such developed advanced features.
On March 15, 2017, we entered into a know-how license agreement with Mayo Foundation for Medical Education and Research (“Mayo Clinic”), effective December 2, 2016, and as amended whereby we were granted an exclusive license, with the right to sublicense, certain know how and patent applications in the fields of signal processing, physiologic recording, electrophysiology recording, electrophysiology software and autonomics to develop, make and offer for sale. The agreement expires ten years from the effective date. In furtherance of this collaboration, we subsequently entered into four additional agreements whereby we were granted exclusive licenses, with the right to sublicense additional Mayo Clinic patents and know-how. Pursuant to these agreements, Mayo Clinic retains ownership of the licensed intellectual property and any developed intellectual property. Mayo Clinic also retains the right to prosecute and enforce the developed intellectual property. If our agreements with Mayo Clinic terminate, our access to technology and intellectual property licensed to us by Mayo Clinic may be restricted or terminate entirely, which may delay our continued development of such advanced features utilizing the Mayo Clinic’s technology or intellectual property or require us to stop development of those product candidates completely. Additional risks posed by this collaboration include:
● | Mayo Clinic may not properly obtain, maintain, enforce, or defend intellectual property or proprietary rights relating to our advanced features or may use our proprietary information in such a way as to expose us to potential litigation or other intellectual property related proceedings, including proceedings challenging the scope, ownership, validity, and enforceability of our intellectual property; |
● | Mayo Clinic may own or co-own intellectual property covering our advanced features that results from our collaboration with them, and in such cases, we may not have the exclusive right or any right to commercialize such intellectual property or such product candidates or research programs; or |
● | We may be prevented from enforcing or defending any intellectual property that we contribute to or that arises out of the collaboration, if Mayo Clinic refuses to cooperate with such action. |
Our collaboration with Mayo Clinic is made subject to the rights of the U.S. government to the extent that the technology covered by the licensed intellectual property was developed under a funding agreement between Mayo Clinic and the U.S. government. Additionally, to the extent there is any conflict between our agreements with Mayo Clinic and applicable laws or regulations, applicable laws and regulations will prevail. Some, and possibly all, of the developed intellectual property rights relating to our advanced features may have been developed in the course of research funded by the U.S. government. As a result, the U.S. government may have certain rights to intellectual property embodied in our current or future products pursuant to the Bayh-Dole Act of 1980. Government rights in certain inventions developed under a government-funded program include a nonexclusive, non-transferable, irrevocable worldwide license to use inventions for any governmental purpose. In addition, the U.S. government has the right to require us, or an assignee or exclusive licensee to such inventions, to grant licenses to any of these inventions to a third party if the U.S. government determines that adequate steps have not been taken to commercialize the invention, that government action is necessary to meet public health or safety needs, that government action is necessary to meet requirements for public use under federal regulations, or that the right to use or sell such inventions is exclusively licensed to an entity within the U.S. and substantially manufactured outside the U.S. without the U.S. government’s prior approval. Additionally, we may be restricted from granting exclusive licenses for the right to use or sell our inventions created pursuant to such agreements unless the licensee agrees to additional restrictions (e.g., manufacturing substantially all of the invention in the U.S.). The U.S. government also has the right to take title to these inventions if we fail to disclose the invention to the government and fail to file an application to register the intellectual property within specified time limits. In addition, the U.S. government may acquire title in any country in which a patent application is not filed within specified time limits. Additionally, certain inventions are subject to transfer restrictions during the term of these agreements and for a period, thereafter, including sales of products or components, transfers to foreign subsidiaries for the purpose of the relevant agreements, and transfers to certain foreign third parties. If any of our intellectual property becomes subject to any of the rights or remedies available to the U.S. government or third parties pursuant to the Bayh-Dole Act of 1980, this could impair the value of our intellectual property and could adversely affect our business. The U.S. government has not exercised any of these rights or provided us with any notice of its intent to exercise any of these rights with respect to any of the intellectual property licensed to us by Mayo Clinic. We are not aware of any instance in which the U.S. government has ever exercised any such rights with respect to any technologies or other intellectual property developed under funding agreements with the U.S. government.
Risks Related to our Common Stock
The market price for our common stock may fluctuate significantly, which could result in substantial losses by our investors.
The stock market in general, and Nasdaq in particular, as well as biotechnology companies, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of small companies. The market price of our common stock may fluctuate significantly in response to numerous factors, some of which are beyond our control, such as:
announcements of technological innovations, new products or product enhancements by us or others;
actual or anticipated quarterly increases or decreases in revenue, gross margin or earnings, and changes in our business, operations or prospects;
announcements of significant strategic partnerships, out-licensing, in-licensing, joint ventures, acquisitions or capital commitments by us or our competitors;
conditions or trends in the biotechnology industry;
changes in the economic performance or market valuations of other biotechnology companies;
general market conditions or domestic or international macroeconomic and geopolitical factors unrelated to our performance or financial condition;
purchase or sale of our common stock by stockholders, including executives and directors;
volatility and limitations in trading volumes of our common stock;
changes in our capital structure or dividend policy, future issuances of securities, sales or distributions of large blocks of our common stock by stockholders;
our cash position;
announcements and events surrounding financing efforts, including debt and equity securities;
changes in earnings estimates or recommendations by security analysts, if our common stock is covered by analysts;
the addition or departure of key personnel;
disputes and litigation related to intellectual property rights, proprietary rights, and contractual obligations;
changes in applicable laws, rules, regulations, or accounting practices and other dynamics; and
other events or factors, many of which may be out of our control.
These factors and any corresponding price fluctuations may materially and adversely affect the market price of our common stock and result in substantial losses by our investors.
Further, the stock market in general, and the market for technology companies in particular, has experienced extreme price and volume fluctuations in the past. Continued market fluctuations could result in extreme volatility in the price of our common stock, which could cause a decline in the value of our common stock.
Moreover, the COVID-19 pandemic has resulted in significant financial market volatility and uncertainty in recent months. A continuation or worsening of the levels of market disruption and volatility seen in the recent past could have an adverse effect on our ability to access capital, on our business, results of operations and financial condition, and on the market price of our common stock.
Price volatility of our common stock might be worse if the trading volume of our common stock is low. In the past, following periods of market volatility, stockholders have often instituted securities class action litigation. If we were involved in securities litigation, it could have a substantial cost and divert resources and attention of management from our business, even if we are successful. Future sales of our common stock could also reduce the market price of such stock.
Moreover, the liquidity of our common stock is limited, not only in terms of the number of shares that can be bought and sold at a given price, but by delays in the timing of transactions and reduction in security analysts’ and the media’s coverage of us, if any. These factors may result in lower prices for our common stock than might otherwise be obtained and could also result in a larger spread between the bid and ask prices for our common stock. In addition, without a large float, our common stock is less liquid than the stock of companies with broader public ownership and, as a result, the trading prices of our common stock may be more volatile. In the absence of an active public trading market, an investor may be unable to liquidate its investment in our common stock. Trading of a relatively small volume of our common stock may have a greater impact on the trading price of our stock than would be the case if our public float were larger. We cannot predict the prices at which our common stock will trade in the future.
Although our shares of Contents
Although our shares of common stock are now listed on The Nasdaq Capital Market under the symbol “BSGM,” trading volume in our common stock has been limited and an active trading market for our shares of common stock may never develop or be maintained. The absence of an active trading market increases price volatility and reduces the liquidity of our common stock. As long as this condition continues, the sale of a significant number of shares of common stock at any particular time could be difficult to achieve at the market prices prevailing immediately before such shares are offered.
If we cannot continue to satisfy the continuing listing criteria of the Nasdaq Capital Market, the exchange may subsequently delist our common stock.
Nasdaq requires us to meet certain financial, public float, bid price and liquidity standards on an ongoing basis in order to continue the listing of our common stock. Generally, we must maintain a minimum amount of stockholders’ equity and a minimum number of holders of our securities. If we fail to meet any of the continuing listing requirements, our common stock may be subject to delisting. If our common stock is a “penny stock,” which makes it more difficult fordelisted and we are not able to list our investors to sell their shares.
Future sales of our securities.
Sales of a substantial number of shares of our common stock may cause the price of our common stock to decline.
If we sell additional equity or equity-relateddebt securities to fund our operations, it may impose restrictions on our business.
In order to raise additional funds to support our operations, we may sell additional equity or debt securities, which may impose restrictive covenants that adversely impact our business. The incurrence of indebtedness would result in the future at a timeincreased fixed payment obligations and pricecould also result in restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. If we deem reasonableare unable to expand our operations or appropriate.otherwise capitalize on our business opportunities due to such restrictions, our business, financial condition and results of operations could be materially adversely affected.
Our stockholders may experience substantial dilution as a result of the conversionexercise of outstanding convertible preferred stockoptions or the exercise of options and warrants to purchase shares of our common stock, or upon conversion of our Series C preferred stock into shares of our common stock.
As of February 27, 2018,March 30, 2022, we have grantedoutstanding options to purchase 8,230,3194,869,484 shares of common stock, 82,500 restricted stock units and have reserved 1,7402,582,522 shares of our common stock for further issuances pursuant to our 2012 Equity Incentive Plan (the “2012 Plan”).Plan. In addition, as of February 27, 2018,March 30, 2022, we may be required to issue 3,713,101159,822 shares of our common stock for issuance upon conversion of outstanding convertible Series C preferred stock which includes accrued dividends as of March 30, 2022, and 12,583,1293,432,040 shares of our common stock for issuance upon exercise of outstanding warrants. Should all of these shares be issued, you would experience dilution in ownership of our common stock and the price of our common stock will decrease unless the value of our company increases by a corresponding amount.
The interests of our controlling stockholders may not coincide with yours and such controlling stockholders may make decisions with which you may disagree.
As of February 27, 2018, twoMarch 30, 2022, five of our stockholders beneficially owned over 25.52%17.4% of our common stock. As a result, these stockholders may be able to influence the outcome of matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. In addition, this concentration of ownership may delay or prevent a change in control of our company and make some future transactions more difficult or impossible without the support of our controlling stockholders. The interests of our controlling stockholders may not coincide with our interests or the interests of other stockholders.
Delaware law and our Amended and Restated Certificate of Incorporation and By-laws contain anti-takeover provisions that could delay or discourage takeover attempts that stockholders may consider favorable.
Our board of directors is authorized to issue shares of preferred stock in one or more series and to fix the voting powers, preferences and other rights and limitations of the preferred stock. Accordingly, we may issue shares of preferred stock with a preference over our common stock with respect to dividends or distributions on liquidation or dissolution, or that may otherwise adversely affect the voting or other rights of the holders of common stock. Issuances of preferred stock, depending upon the rights, preferences and designations of the preferred stock, may have the effect of delaying, deterring or preventing a change of control, even if that change of control might benefit our stockholders. In addition, we are subject to Section 203 of the Delaware General Corporation Law. Section 203 generally prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless (i) prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; (ii) the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding (a) shares owned by persons who are directors and also officers and (b) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or (iii) on or subsequent to the date of the transaction, the business combination is approved by the board and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the interested stockholder.
Section 203 could delay or prohibit mergers or other takeover or change in control attempts with respect to us and, accordingly, may discourage attempts to acquire us even though such a transaction may offer our stockholders the opportunity to sell their stock at a price above the prevailing market price.
Risks Related to our Series C Preferred Stock
Our Series C Preferred Stock contains covenants that could limit our financing options and liquidity position, which would limit our ability to grow our business.
Covenants in the certificate of designation for our Series C Preferred Stock impose operating and financial restrictions on us. These restrictions prohibit or limit our ability to, among other things:
● | incur additional indebtedness; |
● | permit liens on assets; |
● | repay, repurchase or otherwise acquire more than a de minimis number of shares of capital stock; |
● | pay cash dividends to our stockholders; and |
● | engage in transactions with affiliates. |
These restrictions may limit our ability to obtain financing, withstand downturns in our business or take advantage of business opportunities. Moreover, debt financing we may seek may contain terms that include more restrictive covenants, may require repayment on an accelerated schedule or may impose other obligations that limit our ability to grow our business, acquire needed assets, or take other actions we might otherwise consider appropriate or desirable.
In addition, the certificate of designation for our Series C Preferred Stock requires us to redeem shares of our Series C Preferred Stock, at each holder’s option and for an amount greater than their stated value, upon the occurrence of certain events, including our being subject to a judgment of greater than $100,000 or our initiation of bankruptcy proceedings.
The holders of our Series C Preferred Stock are entitled to receive a dividend, which may be increased if we do not comply with certain covenants.
The holders of the Series C Preferred Stock are entitled to a 9% annual dividend on the $1,000 per share stated value of our Series C Preferred Stock, which is payable in cash or, subject to the satisfaction of certain conditions, in pay-in-kind shares. The dividend may be increased to a 18% annual dividend if we fail to comply with certain covenants, including our being subject to a judgment of greater than $100,000 or our initiation of bankruptcy proceedings. As a result of the payment of dividends related to our Series C Preferred Stock, we may be obligated to pay significant sums of money or issue a significant number of shares of our common stock, which could negatively affect our operations or result in the dilution of the holders of our common stock, respectively.
The terms of our Series C Preferred Stock and certain of our warrants contain anti-dilution provisions that may result in the reduction of theirthe conversion prices or exercise prices in the future.
The terms of our Series C Preferred Stock and such warrants, respectively, we will be required to further reduce the relevant conversion or exercise prices, and the number of shares underlying such warrants will be increased. We may find it more difficult to raise additional equity capital while our Series C Preferred Stock and such warrants are outstanding.
The terms of our Series DC Preferred Stock is outstanding.prohibit us from paying dividends in the future on our common stock. As a result, any return on investment may be limited to the value of our common stock.
The terms of our Series C Preferred Stock prohibit us from paying dividends in the future on our common stock, absent consent from the holders representing a super-majority of the outstanding shares of our Series C Preferred Stock and a certain investor. Because we will likely not pay dividends, our common stock may be less valuable because a return on an investment in our common stock will only occur if our stock price appreciates.
General Risk Factors
The holdersliability of our Series E Preferred Stockdirectors and officers is limited.
The applicable provisions of the Delaware General Corporation Law and our Amended and Restated Certificate of Incorporation and By-laws limit the liability of our directors to us and our stockholders for monetary damages for breaches of their fiduciary duties, with certain exceptions, and for other specified acts or omissions of such persons. In addition, the applicable provisions of the Delaware General Corporation Law and of our Amended and Restated Certificate of Incorporation and By-laws provide for indemnification of such persons under certain circumstances. In the event we are entitledrequired to indemnify any of our directors or any other person, our financial strength may be harmed.
Negative publicity or unfavorable media coverage could damage our reputation and harm our operations.
In the event that the marketplace perceives our products as not offering the benefits which we believe they offer, we may receive negative publicity. This publicity may result in litigation and increased regulation and governmental review. If we were to receive a dividend.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. We currently have new research coverage by securities and industry analysts. If one or more of the analysts who covers us downgrades our stock or publishes inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of us or fails to publish reports on us regularly, demand for our stock could decrease, which could negatively affectcause our operations.
We are subject to financial reporting and other requirements that place significant demands on our resources.
We are subject to reporting and other obligations under the Securities Exchange Act of 1934, as amended, including the requirements of Section 404 of the Sarbanes-Oxley Act of 2002. Section 404 requires us to conduct an annual management assessment of the effectiveness of our warrants contain anti-dilution provisions thatinternal controls over financial reporting. These reporting and other obligations place significant demands on our management, administrative, operational, internal audit and accounting resources. Any failure to maintain effective internal controls could have a material adverse effect on our business, operating results and stock price. Moreover, effective internal control is necessary for us to provide reliable financial reports and prevent fraud. If we cannot provide reliable financial reports or prevent fraud, we may result in the reductionnot be able to manage our business as effectively as we would if an effective control environment existed, and our business and reputation with investors may be harmed.
ITEM 1B – UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 2 – PROPERTIES
We maintain our principal executive and engineering office at 12424 Wilshire Boulevard, Suite 745, Los Angeles, California which is55 Greens Farms Road, Westport, Connecticut, where we sublease approximately 2,2006,590 square feet in size. On February 8, 2017, we extended ourof office space. This lease for office space in Los Angeles, California to Augustruns until December 31, 2019,2024, with monthly payments of $8,139 beginning September$14,828 from January 1, 2017 until August2022 through December 31, 20182022, $15,377 per month from January 1, 2023 through December 31, 2023 and $8,423 until August$15,926 from January 1, 2024 through December 31, 2019.2024 plus any additional utility expenses. In connection with the lease, we paid a security deposit of our office space, we are obligated$14,828. There is no option to extend the lease parking spaces at an aggregate approximate cost of $978 per month. past its initial term.
In addition, the Company entered into a lease for storage space with thewe maintain our engineering offices at 12424 Wilshire Boulevard, Los Angeles, California, buildingwhere we lease approximately 4,000 square feet of office space. This lease runs until June 30, 2022, with monthly payments of $13,702. In connection with the lease, we paid a security deposit of $32,852. Although we do not have an option to extend past its lease term, we are currently in negotiations to replace our expiring existing lease.
In October 2021, we exercised our option to extend our lease agreement for approximately 1,400 square feet of office space in Rochester Minnesota commencing on DecemberNovember 1, 20172021, and expiring on AugustOctober 31, 20192023, at a rate of $3,513 per month through October 31, 2023. This lease agreement includes an option to extend the lease for approximately $200 per month. In February 2018,one additional period of two years each its existing term.
We believe we opened an administrative office located in Austin, Texas. We believemay need to expand our current facilities are sufficient to meet our future needs.
Year Ending December 31, | ||||
| 2018 | $ | 112,994 | ||
| 2019 | 76,995 | |||
| $ | 189,989 | |||
ITEM 3 – LEGAL PROCEEDINGS
Aurigene Pharmaceutical Services LTD vs. ViralClear Pharmaceuticals Inc. and BioSig Technologies, Inc.
On January 8, 2021, Aurigene Pharmaceutical Services, LTD (“Aurigene”) filed a complaint with the United States District Court for the District of Connecticut claiming the Company is in default of certain milestone payments for manufacturing and services under contracts dated June 23, 2020 and July 16, 2020 in aggregate amount of $1,530,000.
On September 23, 2021, we entered into a settlement agreement with Aurigene for a sum of $1,000,000 payable in three installments of $400,000, $300,000, and $300,000 on September 30, 2021, December 31, 2021, and March 31, 2022, respectively, with no admission or concession by either party. Balance due under the settlement is $300,000 as of March 30, 2022.
From time to time, we may become involved in other various lawsuits and legal proceedings which arise in the ordinary course of business. However, litigation is subject to inherent uncertainties, and an adverse result in these or other matters may arise from time to time that may harm our business. We are currently not aware of any such legal proceedings or claims that we believe will have, individually or in the aggregate, a material adverse effect on our business, financial condition or operating results.
There are no material proceedings in which any of our directors, officers or affiliates or any registered or beneficial shareholder of more than 5% of our common stock is an adverse party or has a material interest adverse to our interest.
ITEM 4 – MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5 – MARKET FOR REGISTRANT’SREGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market for Common Stock
On October 29, 2014, our common stock commenced trading on OTCQB under the symbol “BSGM.”“BSGM” and on September 21, 2018 we commenced trading on the Nasdaq Capital Market exchange under the same ticker symbol. Prior to October 29, 2014, there was no established trading price for our common stock. The following table sets forth, for the periods indicated, the high and low bid prices per sharelast reported sales price of our common stock as reported byon the OTCQB. The quotations reflect inter-dealer prices, without retail markup, markdown or commissions, and may not represent actual transactions.Nasdaq Capital Market on March 29, 2022, was $1.30 per share.
| Fiscal Year 2017 | ||||||||
| High | Low | |||||||
| First Quarter | $ | 2.00 | $ | 1.20 | ||||
| Second Quarter | $ | 1.76 | $ | 1.23 | ||||
| Third Quarter | $ | 1.60 | $ | 1.25 | ||||
| Fourth Quarter | $ | 1.75 | $ | 1.29 | ||||
| Fiscal Year 2016 | ||||||||
| High | Low | |||||||
| First Quarter | $ | 1.59 | $ | 0.90 | ||||
| Second Quarter | $ | 2.15 | $ | 1.33 | ||||
| Third Quarter | $ | 1.60 | $ | 1.05 | ||||
| Fourth Quarter | $ | 1.59 | $ | 1.25 | ||||
Holders of Record
As of February 27, 2018,March 30, 2022, there were approximately 285290 holders of our common stock, as determined by counting our record holders and the number of participants reflected in a security position listing provided to us by the Depository Trust Company. Because the “DTC participants” are brokers and other institutions holding shares of our common stock on behalf of their customers, we do not know the actual number of unique shareholders represented by these record holders.
Dividends
We have never paid cash dividends on our common stock and do not anticipate paying any cash dividends in the foreseeable future but intend to retain our capital resources for reinvestment in our business. In addition, the terms of our Series C, Series D, and Series E Preferred Stock prohibit us from paying dividends in the future on our common stock. We may not pay dividends on our common stock: (i) so long as at 25% of the originally issued shares of Series D Preferred Stock remain outstanding; (ii) absent consent from the holders representing a super-majority of the outstanding shares of our Series C Preferred Stock and a certain investor; and (iii) absent consent from a majority of the outstanding shares of our Series E Preferred Stock, which majority must include a certain investor
ITEM 6 – SELECTED FINANCIAL DATA
ITEM 7 – MANAGEMENT’S MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Management’s Discussion and Analysis of Financial Condition and Results of Operations is intended to provide a reader of our financial statements with a narrative from the perspective of our management on our financial condition, results of operations, liquidity, and certain other factors that may affect our future results. You should read the following discussion and analysis of our financial condition and results of operations in conjunction with our financial statements and the related notes thereto that are included in this Form 10-K.In addition to historical information, the following discussion and analysis includes forward-looking statements that reflect our plans, estimates and beliefs.Our actual results could differ materially from those discussed in the forward-looking statements. Factors that could cause or contribute to these differences include those discussed below and elsewhere in this prospectus, particularly in the section entitled “Risk“Risk Factors.” See “Special“Special Note Regarding Forward-Looking Statements.”
Overview
We are a development stage medical devicetechnology company developing a proprietary biomedicalthat is commercializing our PURE EP™ System which is an advanced signal acquisition and processing technology platform designed to extract information from physiologic signals. Our initial emphasisprovide essential diagnostic signals with high clinical value in all types of cardiac catheter ablations. PURE EP™ is on providing intracardiac signal informationdesigned to electrophysiologists during EP studiesaddress long-standing limitations that slow and disrupt cardiac catheter ablation procedures, such as environmental lab noise, signal saturation, slow signal recovery, and inaccurate display of AFfractionated potentials.
Cardiac catheter ablation is a procedure that involves delivery of energy through the tip of a catheter that scars or destroys heart tissue to correct heart rhythm disturbances (arrhythmias). In August 2018, we received 510(k) clearance from the U.S. Food and VT. Our first product is theDrug Administration (the “FDA”) to market our PURE (Precise Uninterrupted Real-time evaluation of Electrograms) EP™ System,System.
PURE EP™ is a surface electrocardiogramsignal processing platform that combines advanced hardware and intracardiac multichannel recordingsoftware to address known challenges associated to signal acquisition, to enable electrophysiologists to see more signals and analysis system that acquires, processesanalyze them in real-time.The device aims to minimize noise and displays electrocardiogramsartifacts from cardiac recordings and electrograms required during electrophysiologyacquire high-fidelity cardiac signals. Improving fidelity of acquired cardiac signals may potentially increase the diagnostic value of these signals, thereby possibly improving accuracy and efficiency of the EP studies and ablation procedures.
PURE EP™’s initial focus is on improving intracardiac signal acquisition and enhancing diagnostic information for catheter ablation procedures.procedures for complex arrhythmias like ventricular tachycardia (“VT”), a potentially life-threatening arrhythmia, and atrial fibrillation (“AF”), the most common cardiac arrhythmia associated with a fivefold risk of stroke.
Clinical data acquired by the PURE EP™ System in a multi-center study at Texas Cardiac Arrhythmia Institute at St. David’s Medical Center in Austin, Texas, Mayo Clinic in Jacksonville, Florida, and Massachusetts General Hospital in Boston, Massachusetts was published in September 2021 in the Journal of Cardiovascular Electrophysiology and is available electronically with open access via the Wiley Online Library. Study results showed 93% consensus across the blinded reviewers with a 75% overall improvement in intracardiac signal quality and confidence in interpreting PURE EP™ signals over conventional sources. AF accounted for over 40% of enrollments.
We continue to install PURE EP™ Systems at centers of excellence for clinical evaluation under our market development plan. The PURE EP™ System has been utilized at numerous institutions, including Mayo Clinic campuses in Arizona, Florida and Minnesota; the University of Pennsylvania Hospital in Philadelphia, Pennsylvania; Overland Park Regional Medical System in Overland Park, Kansas; Deborah Heart and Lung Center in Browns Mills, New Jersey; St. Elizabeth’s Medical Center in Boston, Massachusetts; Medical City Heart Hospital in Dallas, Texas; Beth Israel Deaconess Medical Center (BIDMC) in Boston, Massachusetts, a teaching hospital of Harvard Medical School; Methodist Hospital in San Antonio, Texas; Houston Methodist Hospital; Medical City North Hills in North Richland Hills; and Westside Regional Medical Center in Plantation, Florida.
To date, more than 2,160 patient procedures have been conducted with the PURE EP™ System by more than 76 electrophysiologists across seventeen different clinical sites in the United States.
In addition to clinical evaluation, we have conducted pre-clinical evaluation with the PURE EP™ System under several protocols. At Mayo Clinic in Rochester, Minnesota, we have performed twenty-seven experiments (including novel research programs such as Artificial Intelligence, or AI, and repolarization) in various animal models; we also conducted a pre-clinical study at the Mount Sinai Hospital in New York, New York, with an emphasis on the VT model; and six experiments to date during a study at the University of Pennsylvania. We intend to continue additional research and development studies with our technology at Mayo Clinic, the University of Pennsylvania and other national centers.
In September 2021, we announced that we entered into a manufacturing and professional services agreement with Plexus Corp (“Plexus”) (Nasdaq: PLXS). Under the terms of the agreement, Plexus will manufacture the PURE EP™ System and develop a new product pipeline for our subsidiary, ViralClear.
We have not generated any revenuemade progress towards obtaining a European CE marking certificate for medical devices. In Q1 2022, we completed the quality management system audit for the International Organization for Standardization (“ISO”) 13485:2016 with the expectation to dateobtain the ISO 13485:2016 certification in the first half of 2022 and consequently our operations areproceed to the application for the European CE Marking clearance in the first half of 2023, subject to all risks inherentthe guidance and availability from the European Notified Body.
In January 2022, we were awarded U.S. patent claims for our PURE EP™ noise-filtering technology which address computer-implemented systems and methods for filtering noise from input cardiac signals. We now have 49 issued or allowed worldwide patents covering our novel technology for arrhythmia care.
In December 2020, we announced that three PURE EP™ Systems were contracted for purchase by St. David’s Healthcare in Austin, Texas and were subsequently sold in February 2021. We also sold three PURE EP™ Systems to Mayo Foundation for Medical Education and Research in 2021 for use in Mayo Clinic campuses in Rochester, Minnesota, Jacksonville, Florida and Phoenix, Arizona. We are in active discussions with several accounts about the establishmentacquisition of a new business enterprise.the PURE EP™ System.
Critical Accounting Policies and Estimates
The following discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements, which have been prepared in accordance with generally accepted accounting principles in the U.S. The preparation of consolidated financial statements in accordance with generally accepted accounting principles in the U.S. requires us to make estimates and assumptions that affect the amounts reported in our consolidated financial statements. The consolidated financial statements include estimates based on currently available information and our judgment as to the outcome of future conditions and circumstances. Significant estimates in these financial statements include allowance for doubtful accounts and accruals for inventory claims. Changes in the status of certain facts or circumstances could result in material changes to the estimates used in the preparation of the financial statements and actual results could differ from the estimates and assumptions.
Among the significant judgments made by management in the preparation of our financial statements are the following:
We believe the following critical accounting estimates affect our more significant judgments and estimates used in the preparation of our financial statements.
Revenue Recognition
We derive our revenue primarily from the sale of our medical device, the PURE EP™ System, as well as related support and maintenance services and software upgrades in connection with the system.
We recognize revenue in accordance with Accounting Standards Codification (ASC) 606, Revenue from Contracts with Customers (“ASC 606”). The core principle of ASC 606 is that an entity recognizes revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services.
We determine revenue recognition through the following five steps:
● | Identify the contract with the customer; |
● | Identify the performance obligations in the contract; |
● | Determine the transaction price; | |
● | Allocate the transaction price to the performance obligation in the contract; and |
● | Recognize revenue when, or as, the performance obligations are satisfied. |
Performance obligations are the unit of accounting for revenue recognition and generally represent the distinct goods or services that are promised to the customer. If we determine that it has not satisfied a performance obligation, it will defer recognition of the revenue until the performance obligation is deemed to be satisfied. Support, maintenance, and software upgrades are performance obligations over a defined period and are recognized ratably over the contractual service period. Customers typically purchase these services with the initial sale of the PURE EP System and do not have the right to terminate their contracts unless we fail to perform material obligations.
We may execute more than one contract with a single customer. If so, it is evaluated whether the agreements were negotiated as a package with a single objective, whether the amount of consideration to be paid in one agreement depends on the price and/or performance of another agreement, or whether the goods or services promised in the agreements represent a single performance obligation. The conclusions reached can impact the allocation of the transaction price to each performance obligation and the timing of revenue recognition related to those arrangements.
We estimate the transaction price based on the amount of consideration we expect to receive for transferring the promised goods or services in the contract. The consideration may include both fixed consideration and variable consideration. At the inception of each arrangement that includes variable consideration, we evaluate the amount of the potential payments and the likelihood that the payments will be received. If it is probable that a significant revenue reversal would not occur, the variable consideration is included in the transaction price.
We record accounts receivable for amounts invoiced to customers for which the Company has an unconditional right to consideration as provided under the contractual arrangement. Unbilled receivables, if any, include amounts related to our contractual right to consideration for completed performance obligations not yet invoiced. Deferred revenue includes payments received in advance of performance under the contract. Our unbilled receivables and deferred revenue are reported on an individual contract basis at the end of each reporting period. Unbilled receivables are classified as current or noncurrent based on the timing of when we expect to bill the customer. Deferred revenue is classified as current or noncurrent based on the timing of when we expect to recognize revenue.
Research and Development
We account for research and development costs in accordance with the Accounting Standards Codification subtopic 730-10, Research and Development (“ASC 730-10”). Under ASC 730-10, all research and development costs must be charged to expense as incurred. Accordingly, internal research and development costs are expensed as incurred. Third-party research and developmentsdevelopment costs are expensed when the contracted work has been performed or as milestone results have been achieved. Company-sponsored research and development costs related to both present and future products are expensed in the period incurred.
Stock Based Compensation
All stock-based payments to employees and to nonemployee directors for their services as directors consisted of grants of restricted stock and stock options, which are measured at fair value on the grant date and recognized in the statements of operations as compensation expense over the relevant vesting period. Restricted stock payments and stock-based payments to nonemployees are recognized as an expense over the period of performance.
Such payments are measured at fair value at the earlier of the date a performance commitment is reached, or the date performance is completed. In addition, for awards that vest immediately and are non-forfeitable, the measurement date is the date the award is issued.
Use of Estimates
The preparation of our common stock onconsolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the award.
Acquisition of Intellectual Property
Intellectual property acquired are accounted for changes inunder the acquisition method of accounting. This method requires the recording of acquired assets, including separately identifiable intangible assets, and assumed liabilities at their acquisition date fair values. Any excess consideration transferred over fair value is allocated on a relative fair value basis to the identifiable net assets.
The acquired intellectual property from the Trek acquisition was considered unproven compounds, the success of which was uncertain at the time of the acquisition. Accordingly, the fair value of the derivative instruments depends on whether the derivatives qualifyconsideration paid was charged as hedge relationshipsacquired research and the types of relationships designated are based on the exposures hedged. At December 31, 2017 and 2016, the Company did not have any derivative instruments that were designated as hedges.
Results of Operations (000’s)
We anticipate that our results of operations will fluctuate for the foreseeable future due to several factors, such as the progress of our research and development efforts and the timing and outcome of regulatory submissions. Due to these uncertainties, accurate predictions of future operations are difficult or impossible to make.
Twelve Months Ended December 31, 20172021, Compared to Twelve Months Ended December 31, 20162020
Revenues and Cost of Goods Sold
.We derive our revenue primarily from the sale of Contents
Cost of sales for the year ended December 31, 2021, was $199 comprised of the delivered product and cost of services as compared to nil for the year ended December 31, 2020.
Gross profit from the year ended December 31, 2021, was $242 or 54.9% as compared to nil for the year ended December 31, 2020.
Research and Development Expenses. Research and development expenses for the twelve months ended December 31, 20172021, were $4,756,468, an increase$5,602, a decrease of $2,101,967$12,534 or 79%69.1%, from $2,654,501$18,136 for the twelve months ended December 31, 2016.2020. This increasedecrease is primarily due additional personnelto ceasing development of merimepodib in 2020, which led to a reduction of $13,116 from 2020 to 2021 in the ViralClear segment, net with a $614 increase in the BioSig segment research and outside design costs as we develop our proprietary technology platform. development from $4,399 for the twelve months ended December 31, 2020, to $5,013 for the twelve months ended December 31, 2021.
Research and development expenses were comprised of the following:
2021 | 2020 | |||||||
Salaries and equity compensation | $ | 2,833 | $ | 3,030 | ||||
Consulting expenses | 725 | 2,374 | ||||||
Research, clinical studies, and design work | 1,159 | 2,068 | ||||||
Regulatory | 142 | 68 | ||||||
Data/AI development | 307 | 505 | ||||||
Product development and formulation | 15 | 4,910 | ||||||
Acquired research and development | 150 | 4,883 | ||||||
Travel, supplies, other | 271 | 298 | ||||||
Total | $ | 5,602 | $ | 18,136 | ||||
| 2017 | 2016 | |||||||
| Salaries and equity compensation | $ | 1,481,421 | $ | 1,744,780 | ||||
| Consulting expenses | 500,628 | 322,520 | ||||||
| Clinical studies and design work | 2,066,028 | 388,447 | ||||||
| Acquired research and development | 543,927 | - | ||||||
| Travel, supplies, other | 164,464 | 198,754 | ||||||
| Total | $ | 4,756,468 | $ | 2,654,501 | ||||
Stock-based compensation for research and development personnel was $432,929$759 and $760,732$1,253 for the yeartwelve months ended December 31, 20172021, and 2016,2020, respectively.
On March 15, 2017, we24, 2020, ViralClear entered into an asset purchase agreement (the “Asset Purchase Agreement”) with Trek Therapeutics, PBC (“Trek”). Pursuant to the Asset Purchase Agreement, Trek sold to ViralClear all right, title and interest of Trek and its affiliates to certain assets (the “Purchased Assets”). As consideration for the Purchased Assets, ViralClear agreed to pay Trek in upfront and milestone payments a combination of cash, shares of ViralClear’s common stock, which common stock may equal up to 10% of ViralClear’s outstanding equity, and sublicense fees in the event ViralClear sublicenses the Purchased Assets. On March 30, 2020, pursuant to the Asset Purchase Agreement, ViralClear paid $350,000 in cash and issued 634,910 shares of ViralClear’s common stock to Trek at a fair of $3,174,550. The Purchased Assets were recorded as acquired research and development.
On April 8, 2020, ViralClear entered into a know-how license agreement (the “Agreement”) with Mayo Foundation for Medical Education and Research whereby we were grantedMayo. The Agreement grants to ViralClear (i) an exclusive worldwide license, with the right to sublicense, certain know how and patent applications inwithin the field of signal processing, physiologic recording, electrophysiology recording, electrophysiology softwareanti-viral agents to target COVID-19 (the “Field”) to certain patent rights for the development and autonomicscommercialization of products, methods, and processes for public use and benefit (the “Licensed Products”) and (ii) a non-exclusive worldwide license, with the right to sublicense, within the Field, to use the know-how of Mayo that is necessary to develop make and offerthe Licensed Products.
The Agreement will expire upon the later of either (a) the expiration of the licensed patent rights or (b) the 7th anniversary of the date of the first commercial sale of a Licensed Product, unless earlier terminated by Mayo for sale. The agreement expires in ten years fromViralClear’s failure to cure a material breach of the effective date. As such, we are obligated to payAgreement, ViralClear’s or a sublicensee’s commencement of any action or proceedings against Mayo or its affiliates other than for an uncured material breach of the Agreement by Mayo, or insolvency ViralClear.
In connection with the Agreement, ViralClear issued to Mayo Foundation a 1% or 2%259,959 shares of ViralClear’s common stock. ViralClear also agreed to make earned royalty payment on netpayments to Mayo in connection with ViralClear’s sales of licensed products, as defined.
General and Administrative Expenses
. General and administrative expenses for the twelve months ended December 31,Payroll related expenses (including equity compensation) decreased to $5,579,117$17,360 in the twelve months ended December 31, 20172021, from $6,381,610$31,080 for the twelve months ended December 31, 2016,2020, a decrease of $802,493,$13,720, or 13%44.1%. This decrease is due to the value of the stock-based compensation decreasing to $4,316,542$9,062 in 2017,2021, as a result of the vesting of stock and stock options issued to board members, officers, and employees, as compared to $5,233,818$23,911 of stock-based compensation in 2016,2020, net with added additional personnel.personnel in 2021, a decrease of $14,849 or 62.1%.
Professional services for the twelve months ended December 31, 20172021, totaled $362,663, an increase$1,261, a decrease of $2,968,$687, or 1%35.3%, over the $359,695$1,948 recognized for the twelve months ended December 31, 2016.2020. Of professional services, legal fees totaled $273,663$943 for the twelve months ended December 31, 2017,2021, a decrease of $12,532,$560, or 4%37.3%, from $286,195$1,503 incurred for the twelve months ended December 31, 2016.2020. The significant decrease in legal fees in 2021 is due to reduction in legal work in asset acquisitions, financing and in developing and registering patents. Accounting fees incurred in the twelve months ended December 31, 20172021, amounted to $89,000, an increase$179, a decrease of $15,500,$79 or 21%30.6%, from $73,500$258 incurred for the same period in 2016.2020. The increasessignificant decrease is due to reduction in professional fees was primarily related2021 work relating to an increase in legalinternal control audit, design and monitoring, audit requirements in 2017 as comparedwork relating to 2016 as we continue to develop our operations, including legal fees associated with our capital raising transactions and the filingViralClear segment.
Consulting fees and marketing totaled $1,453,005$4,763 for the twelve months ended December 31, 2017, an increase2021, a decrease of $285,585$2,106 or 24%30.7%, from $1,167,420$6,869 for the twelve months ended December 31, 2016.2020. The increasedecrease primarily relates to ourreductions in fund raising and investor relations to support our increased efforts in market research and potential investor identification.
Travel, meals and entertainment costs for the twelve months ended December 31, 20172021, were $379,970,$1,010, an increase of $105,008,$637, or 38%170.8%, from $274,962$373 incurred during the twelve months ended December 31, 2016. During 2017, additional travel2020. The significant increase in 2021 was required than in 2016 due to our marketing and fund-raising efforts. lifting of various restrictions imposed by the COVID-19 pandemic-related measures as compared to 2020.
Rent for the twelve months ended December 31, 20172021, totaled $142,975, an increase$466, a decrease of $14,419,$18, or 11%3.7%, from $128,556$484 incurred during the same period in 2016.2020. In 2017,2021, we incurred a rent reduction with our significant increase was the resultrelocation of our corporate offices in Connecticut and our lease renewalextension in California along with final settlement with closing our Minneapolis, Minnesota office in late 2017.Los Angeles facility.
Depreciation and Amortization Expense
. Depreciation and amortization expense for the twelve months endedInterest Income (expense)
Gain on Settlement of Debt. On September 23, 2021, we negotiated a lawsuit settlement with Aurigene relating to certain milestone payments for manufacturing and services under a contract with our ViralClear subsidiary. In connection with the settlement, we recognized a gain on settlement of debt of $553 during the twelve months ended December 31, 2021, as compared to nil for the twelve months ended December 31, 2020.
Preferred Stock Dividend. Our preferredPreferred stock dividend for the twelve months ended December 31, 20172021, totaled $119,877, an increase$9, a decrease of $9,854,$5, or 9%35.7% from $110,023$14 incurred during the twelve months ended December 31, 2016. The increase in2020. Preferred stock dividends is a result addedare related to the issuance of our Series DC Preferred Stock issuedfrom 2013 through 2015. The significant decrease in 2017, net2021 as compared to 2020 is the result of 2020 conversions of the Series C Preferred StockStock.
Noncontrolling Interest. In 2019 and 2020, ViralClear sold shares of its common stock to common reducingfund its initial and ongoing operations. As of December 31, 2021, we had a majority interest in ViralClear of 68.4%. The proportionate loss attributed to noncontrolling interests for the number of Series C Preferred shares outstanding. Preferred stock dividends are relatedtwelve months ended December 31, 2021, was $939 as compared to our Series C and D Preferred Stock issued in 2013, 2015 and 2017.$6,922 for 2020.
Net Loss Available to Common Stockholders
Segment Results
The Company reports segment information based on the “management” approach. The management approach designates the internal reporting used by management for making decisions and Capital Resources
Summary Statement of current assets) of $2,300,644, comprised of cash of $1,547,579 and prepaid expenses of $116,938, which was offset by $473,098 of accounts payable and accrued expenses, accrued dividends on preferred stock issuances of $447,901, warrant liability of $2,358,240 and derivative liability of $685,922. ExcludingOperations for the derivative and warrant liabilities, our working capital would have been $743,518. For the twelve monthsyear ended December 31, 2017, cash provided by financing activities totaled $7,971,174, comprised of proceeds from the sale of our common stock and subscriptions of $6,041,214 and sale of our Series D Preferred stock of $1,929,960. In the comparable period in 2016, $5,226,368 was raised through the sale of our common stock. At December 31, 2017, we had cash of $1,547,5792021, as compared to $1,055,895 at December 31, 2016. Our cash is held in bank deposit accounts. At December 31, 2017 and 2016, we had no convertible debentures outstanding.
COVID-19
The full public-health impact of the ongoing COVID-19 pandemic is currently indeterminable and 2016 was $7,470,054rapidly evolving, and $5,107,452, respectively, which represent cash outlays for researchthe related health crisis has adversely affected and development and general and administrative expensesmay continue to adversely affect the global economy, resulting in such periods. Increase in cash outlays principally resulted from increased research and development and general and administrative expensespossibly delaying our commercialization objectives of the PURE EP Systems due to limited resources and accessibility of hospitals as they cope with the continued development of our operations.pandemic.
Liquidity and Capital Resources
We had an accumulated deficit as of December 31, 2017 was $9,436, compared to $16,255 for the twelve months ended December 31, 2016. During both the twelve months ended December 31, 20172021, of approximately $189 million, as well as a net loss of approximately $32 million and the twelve months ended December 31, 2016, we purchased office furniture and computer equipment.
We have incurred net losses and negative cash flows from operations since inception and our expectation is that these conditions will continue for the foreseeable future. In addition, we will require additional financing to fund future operations. Further,Although we do not have any commercial products available for sale, andwe have not generated significant revenues to date, and there is no assurance that if approval of our products is received, we will be able to generate cash flow to fund operations. In addition, there can be no assurance that our research and development will be successfully completed or that any productadditional products will be approved or commercially viable. Our ability to continue as a going concern is subject to our ability to obtain necessary funding from outside sources, including obtaining additional funding from the sale of our securities, obtaining loans from various financial institutions or being awarded grants from government agencies, where possible. Our continued net operating losses increase the difficulty in meeting such goals and there can be no assurances that such methods will prove successful.
Our plans include the continued commercialization of the PURE EP System and other applications of our core technology and raising capital through the sale of additional equity securities, debt or capital inflows from strategic partnerships. Our shift from a focus on technology development to commercialization has allowed us to reduce our annual expenses in a meaningful way. As a result of this transition, we have been able to achieve savings through reductions in executive and management compensation and a reduction of our utilization of external consultants and professional service providers. We believe these cost-saving measures combined with our expectations of positive trends in commercial activity create the potential for us to achieve a lower cash flow breakeven rate. There are no assurances, however, that we will be successful in obtaining the level of financing needed for our operations. The ongoing COVID-19 pandemic has resulted and continues to result in significant financial market volatility and uncertainty in recent months. In addition, U.S. and global markets are experiencing volatility and disruption following the escalation of geopolitical tensions and the start of the military conflict between Russia and Ukraine.
A continuation or worsening of the levels of market disruption and volatility seen in the recent past could have an adverse effect on our ability to access capital and on the market price of our common stock, and we may not be able to successfully raise capital through the sale of our securities.
Our Series C Preferred Stock contains triggering events which would, among other things, require redemption (i) in cash, at the greater of (a) 120% of the stated value of $1 or (b) the product of (I) the variable weighted average price of our common stock on the trading day immediately preceding the date of the triggering event and (II) the stated value divided by the then conversion price or (ii) in shares of our common stock, equal to a number of shares equal to the amount set forth in (i) above divided by 75%. As of June 30, 2021, the aggregate stated value of our Series C Preferred Stock was $105. The triggering events include our being subject to a judgment of greater than $100 or our initiation of bankruptcy proceedings. If any of the triggering events contained in our Series C Preferred Stock occur, the holders of our Series C Preferred Stock may demand redemption, an obligation we may not have the ability to meet at the time of such demand. We will be required to pay interest on any amounts remaining unpaid after the required redemption of our Series C Preferred Stock, at a rate equal to the lesser of 18% per annum or the maximum rate permitted by applicable law.
We expect to incur losses from operations for the near future. We expect to incur increasing marketing and commercialization expenses related to our PURE EP system in addition to additional research and development costs relating to the PURE EP and other product candidates, including expenses related to clinical trials. We expect that our general and administrative expenses will increase in the future as we expand our business development, add infrastructure and incur additional costs related to being a public company, including incremental audit fees, investor relations programs and increased professional services.
Our future capital requirements will depend on a number of factors, including the progress of our research and development of product candidates, the timing and outcome of regulatory approvals, the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims and other intellectual property rights, the status of competitive products, the availability of financing and our success in developing markets for our product candidates.
Future financing may include the issuance of equity or debt securities, obtaining credit facilities, or other financing mechanisms. Even if we are able to raise the funds required, it is possible that we could incur unexpected costs and expenses or experience unexpected cash requirements that would force us to seek alternative financing. Furthermore, if we issue additional equity or debt securities, existing holders of our securities may experience additional dilution or the new equity securities may have rights, preferences or privileges senior to those of existing holders of our securities.
If additional financing is not available or is not available on acceptable terms, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our commercialization efforts or obtain funds through arrangements with collaborative partners or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently.
Equity Financing
On July 2, 2021, we entered into an underwriting agreement (the “Underwriting Agreement”) with Laidlaw & Company (UK) Ltd. (the “Underwriter”), relating to an underwritten public offering of 2,500,000 shares of the Company’s common stock, $0.001 par value per share. The public offering price of the shares was $4.00 per share, and the Underwriter agreed to purchase the shares from us pursuant to the Underwriting Agreement at a price of $3.68 per share. After the underwriting discount, offering and other related expenses, we received net proceeds from the offering of approximately $9.0 million. Pursuant to the Underwriting Agreement, we also granted the Underwriter an option to purchase up to 375,000 additional shares of common stock, or 15% of the number of Shares sold in the offering, at a price of $3.68 per share, for a period of 30 days from the date of the Underwriting Agreement, of which none were exercised.
Pursuant to the Underwriting Agreement, we issued to the Underwriter or its designees warrants to purchase up to an aggregate 125,000 shares of common stock, or 5% of the number of shares sold in the offering (the “Underwriter Warrants”). The Underwriter Warrants are exercisable following the date of issuance, July 7, 2021, and ending five years from the date of the execution of the Underwriting Agreement, July 2, 2026, at a price per share equal to $4.80 per share (120% of the public offering price per share) and are exercisable on a “cashless” basis.
The shares were sold and issued pursuant to our shelf registration statement on Form S-3 (Registration Statement No. 333-251859) previously filed with the Securities and Exchange Commission and declared effective by the Securities and Exchange Commission on January 12, 2021. A preliminary prospectus supplement and prospectus supplement and the accompanying prospectus relating to the offering were filed with the Securities and Exchange Commission. The offering closed on July 7, 2021.
On March 22, 2022, we closed a registered direct offering (the “Offering”) of an aggregate of 2,613,130 shares of our common stock, at an offering price of $1.15 per share and (ii) warrants to purchase up to 2,613,130 shares of our common stock, at an exercise price of $1.40 per share, that will become exercisable six months after the date of issuance and will expire three and one-half years following the date of issuance, for gross proceeds of approximately $3.0 million before the deduction of fees and offering expenses.
The common stock and warrants were offered by us pursuant to a shelf registration statement on Form S-3 (File No. 333-251859) (the “Shelf Registration Statement”), previously filed with the SEC on December 31, 2020, and declared effective by the SEC on January 12, 2021, and a prospectus supplement, dated March 21, 2022, to the Shelf Registration Statement, filed with the SEC on March 22, 2022.
At-the-Market Offering
On August 28, 2020, we entered into an Open Market Sale Agreement (the “Sales Agreement”) with Jefferies LLC to act as our sales agent and/or principal (“Jefferies” or the “Agent”), with respect to the issuance and sale of up to $45,000,000 of our shares of common stock, par value $0.001 per share (the “Shares”), from time to time in an at-the-market offering (the “Offering”).
Jefferies sold the Shares by an “at the market offering” as defined in Rule 415(a)(4) promulgated under the Securities Act of 1933, as amended (the “Securities Act”). We sold the Shares in amounts from time to time subject to the terms and conditions of the Sales Agreement, and we had no obligation to sell any of the Shares under the Sales Agreement. We or Jefferies were able to suspend or terminate the offering of Shares upon notice to the other party and subject to other conditions. Jefferies acted as sales agent on a commercially reasonable efforts basis consistent with its normal trading and sales practices and applicable state and federal law, rules and regulations and the rules of Nasdaq.
We paid the Agent a commission equal to 3.0% of the gross proceeds from the sale of the Shares pursuant to the Sales Agreement. The Company has also agreed to provide Jefferies with customary indemnification and contribution rights.
From January 15, 2021, through February 16, 2021, the Company sold 251,720 shares of its common stock through the Open Market Sales Agreement for net proceeds of $1,300,135, after transactional costs of $40,365.
The Shares were sold and issued pursuant the Company’s shelf registration statement on Form S-3 (File No. 333-230448), which was previously declared effective by the Securities and Exchange Commission, and a related prospectus. On March 25, 2021, the Sales Agreement was terminated with approximately $41M remaining available under the Shelf registration.
On March 25, 2021, the Company delivered written notice to Jefferies to terminate the Sales Agreement effective as of April 8, 2021, pursuant to Section 7(b)(i) thereof. The Company was not subject to any termination penalties related to the termination of the Sales Agreement.
Twelve Months Ended December 31, 2021, Compared to Twelve Months Ended December 31, 2020
As of December 31, 2021, we had a working capital of $11,318, comprised of cash of $11,659, inventory of $1,881 and prepaid expenses of $354, which was offset by $2,179 of accounts payable and accrued expenses, accrued dividends on preferred stock issuances of $82, short term deferred revenue of $32 and short term lease liabilities of $283. For the twelve months ended December 31, 2021, cash provided by financing activities totaled $10,332, comprised of proceeds from the sale of our common stock of $9,004, proceeds from At-the-market sale of our common stock of $1,300 and proceeds from the exercise of options of $28. In the comparable period in 2020, $25,215 was raised through the sale of our common stock, sale of subsidiary stock to non-controlling interests of $10,592, proceeds from At-the-market sale of our common stock of $2,228 and proceeds of $4,812 from the exercise of options and warrants. At December 31, 2021, we had cash of $11,659 compared to $28,268 at December 31, 2020. Our cash is held in bank deposit accounts. At December 31, 2021 and 2020, we had no convertible debentures outstanding.
Cash used in operations for the twelve months ended December 31, 2021, and 2020 was $26,399 and $26,601, respectively, which represent cash outlays for research and development and general and administrative expenses in such periods. The decrease in cash outlays principally resulted from reduced research and development and general and administrative expenses from 2020 to 2021.
Cash used in investing activities for the twelve months ended December 31, 2021, was $542, compared to $87 for the twelve months ended December 31, 2020. During the twelve months ended December 31, 2021, we purchased office furniture, manufacturing equipment, computer equipment and leasehold improvements. For the twelve months ended December 31, 2020, we incurred $87 purchases of office furniture and computer equipment.
Our Series C Preferred Stock contains triggering events which would, among other things, require redemption (i) in cash, at the greater of (a) 120% of the stated value of $1,000 or (b) the product of (I) the variable weighted average price of our common stock on the trading day immediately preceding the date of the triggering event and (II) the stated value divided by the then conversion price or (ii) in shares of our common stock, equal to a number of shares equal to the amount set forth in (i) above divided by 75%. As of December 31, 2017,2020, the aggregate stated value of our Series C Preferred Stock was $985,000.$105,000. The triggering events include our being subject to a judgment of greater than $100,000 or our initiation of bankruptcy proceedings. If any of the triggering events contained in our Series C Preferred Stock occur, the holders of our Series C Preferred Stock may demand redemption, an obligation we may not have the ability to meet at the time of such demand. We will be required to pay interest on any amounts remaining unpaid after the required redemption of our Series C Preferred Stock, at a rate equal to the lesser of 18% per annum or the maximum rate permitted by applicable law.
Recent Accounting Pronouncements
There were various updates recently issued, most of which represented technical corrections to the accounting literature or application to specific industries and are not expected to a have a material impact on the Company’s consolidated financial position, results of operations or cash flows.
ITEM 7A – QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
ITEM 8 – FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
BIOSIG TECHNOLOGIES, INC.
CONSOLIDATED FINANCIAL STATEMENTS
TABLE OF CONTENTS
| (PCAOB ID 711) | F-2 |
| 2020 | F-4 |
| 2020 | F-5 |
| 2021 and 2020 | F-6 |
| 2020 | F-8 |
F-9 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheetsheets of BioSig Technologies.Technologies, Inc. (“the Company”and subsidiaries (the “Company”) as of December 31, 20172021 and 2016,2020, and the related consolidated statements of operations, stockholders’ deficit,changes in equity, and cash flows for each of the years then ended. in the two-year period ended December 31, 2021, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the years in the two year period ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
The Company’s Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company’s accumulated deficit and operating losses raise a substantial doubt about its ability to continue as a going concern. Management’s evaluation of the events and conditions and management’s plans regarding those matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty. Our opinion is not modified with respect to that matter.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on thesethe Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States).PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement.misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included considerationAs part of our audits, we are required to obtain an understanding of internal control over financial reporting, as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company’sCompany’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence supportingregarding the amounts and disclosures in the consolidated financial statements, assessingstatements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statement presentation.statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matter does not alter in any way our opinion on the consolidated financial statements, referredtaken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to above present fairly,which it relates.
Recognition and Disclosure of Research and Development Costs
Description of the Matter
As described in allNote 3 of the consolidated financial statements, research and development costs are expensed as incurred. Significant estimates include assessment of work in progress of multiple third-party contracts and evaluation whether milestones and contingent payments are probable in estimating the research and development costs to accrue or disclose in the reporting period. Recognition and disclosure of research and development costs were considered a critical audit matter due their material respects,impact on disclosures in the consolidated financial positionstatements and the nature and extend of audit effort required to evaluate the results of audit procedures.
How We Addressed the Matter in Our Audits
We reviewed third-party contracts, statements of work and purchase orders, discussed with personnel and obtained confirmations with external service providers as to the progress or stage of completion of services, the agreed-upon fee to be paid for such services, and probability of milestones and contingent payments.
/s/ Friedman LLP | |
We have served as the Company’s auditor since 2020. | |
Marlton, New Jersey | |
March 31, 2022 | |
BIOSIG TECHNOLOGIES, INC. | ||||||||
CONSOLIDATED BALANCE SHEETS | ||||||||
(In Thousands, Except Par Value and Share Amounts) | ||||||||
December 31, | ||||||||
2021 | 2020 | |||||||
ASSETS | ||||||||
Current assets: | ||||||||
Cash | $ | 11,659 | $ | 28,268 | ||||
Inventory | 1,881 | 768 | ||||||
Prepaid expenses and vendor deposits | 354 | 301 | ||||||
Total current assets | 13,894 | 29,337 | ||||||
Property and equipment, net | 652 | 289 | ||||||
Right-to-use assets, net | 604 | 306 | ||||||
Other assets: | ||||||||
Patents, net | 326 | 346 | ||||||
Trademarks | 1 | 1 | ||||||
Prepaid expenses, long term | - | 5 | ||||||
Deposits | 42 | 102 | ||||||
Total assets | $ | 15,519 | $ | 30,386 | ||||
LIABILITIES AND EQUITY | ||||||||
Current liabilities: | ||||||||
Accounts payable and accrued expenses, including $86 and $317 to related parties as of December 31, 2021 and 2020, respectively | $ | 2,179 | $ | 4,722 | ||||
Deferred revenue, short term | 32 | - | ||||||
Dividends payable | 82 | 73 | ||||||
Lease liability, short term | 283 | 313 | ||||||
Total current liabilities | 2,576 | 5,108 | ||||||
Deferred revenue, long term | 5 | - | ||||||
Lease liability, long term | 373 | 1 | ||||||
Total long-term liabilities | 378 | 1 | ||||||
Total liabilities | 2,954 | 5,109 | ||||||
Commitments and contingencies (Note 11) | ||||||||
Series C 9% Convertible Preferred Stock, $0.001 par value, $1,000 stated value, authorized 4,200 shares, 105 shares issued and outstanding: liquidation preference of $105 as of December 31, 2021 and 2020 | 105 | 105 | ||||||
Equity: | ||||||||
Preferred stock, $0.001 par value, authorized 1,000,000 shares, designated 200 shares of Series A, 600 shares of Series B, 4,200 shares of Series C, 1,400 shares of Series D, 1,000 shares of Series E, 200,000 shares of Series F Preferred Stock, none issued | - | - | ||||||
Common stock, $0.001 par value, authorized 200,000,000 shares, 35,567,180 and 30,764,792 issued and outstanding as of December 31, 2021 and 2020, respectively | 36 | 31 | ||||||
Additional paid in capital | 201,127 | 181,344 | ||||||
Accumulated deficit | (188,922 | ) | (157,005 | ) | ||||
Total stockholders' equity attributable to BioSig Technologies, Inc. | 12,241 | 24,370 | ||||||
Non-controlling interest | 219 | 802 | ||||||
Total equity | 12,460 | 25,172 | ||||||
Total liabilities and equity | $ | 15,519 | $ | 30,386 | ||||
See the accompanying notes to the consolidated financial statements.
BIOSIG TECHNOLOGIES, INC. | ||||||||
CONSOLIDATED STATEMENTS OF OPERATIONS | ||||||||
(In Thousands, Except Par Value and Share Amounts) | ||||||||
Year ended December 31, | ||||||||
2021 | 2020 | |||||||
Revenue: | ||||||||
Product sales | $ | 414 | $ | - | ||||
Service | 27 | - | ||||||
Total revenue | 441 | - | ||||||
Cost of goods sold | 199 | - | ||||||
Gross profit | 242 | - | ||||||
Operating expenses: | ||||||||
Research and development | 5,602 | 18,136 | ||||||
General and administrative | 27,853 | 40,954 | ||||||
Depreciation and amortization | 198 | 94 | ||||||
Total operating expenses | 33,653 | 59,184 | ||||||
Loss from operations | (33,411 | ) | (59,184 | ) | ||||
Other income (expense): | ||||||||
Interest income, net | 2 | 45 | ||||||
Gain on settlement of debt | 553 | - | ||||||
Loss on foreign currency translation | - | (1 | ) | |||||
Loss before income taxes | (32,856 | ) | (59,140 | ) | ||||
Income taxes (benefit) | - | - | ||||||
Net loss | (32,856 | ) | (59,140 | ) | ||||
Non-controlling interest | 939 | 6,922 | ||||||
Net loss attributable to BioSig Technologies, Inc. | (31,917 | ) | (52,218 | ) | ||||
Preferred stock dividend | (9 | ) | (14 | ) | ||||
NET LOSS ATTRIBUTABLE TO COMMON SHAREHOLDERS | $ | (31,926 | ) | $ | (52,232 | ) | ||
Net loss per common share, basic and diluted | $ | (0.95 | ) | $ | (1.87 | ) | ||
Weighted average number of common shares outstanding, basic and diluted | 33,511,941 | 27,906,584 | ||||||
See the accompanying notes to the consolidated financial statements.
BIOSIG TECHNOLOGIES, INC. | ||||||||||||||||||||||||
CONSOLIDATED STATEMENT OF CHANGES IN EQUITY | ||||||||||||||||||||||||
YEARS ENDED DECEMBER 31, 2021 AND 2020 | ||||||||||||||||||||||||
(In Thousands, Except Par Value and Share Amounts) | ||||||||||||||||||||||||
Additional | Non- | |||||||||||||||||||||||
| Common stock | Paid in | Accumulated | controlling | |||||||||||||||||||||
Shares | Amount | Capital | Deficit | Interest | Total | |||||||||||||||||||
Balance, December 31, 2019 | 23,323,087 | $ | 23 | $ | 115,910 | $ | (104,787 | ) | $ | 515 | $ | 11,661 | ||||||||||||
Sale of common stock, net transactional costs | 4,687,500 | 5 | 25,210 | - | - | 25,215 | ||||||||||||||||||
Sale of common stock under At-the-market offering, net of transaction expenses of $222 | 424,357 | * | 2,228 | - | - | 2,228 | ||||||||||||||||||
Sale of subsidiary shares to non-controlling interest | - | - | 7,124 | - | 3,468 | 10,592 | ||||||||||||||||||
Common stock issued for services | 679,555 | 1 | 4,399 | - | - | 4,400 | ||||||||||||||||||
Fair value of subsidiary shares issued to acquire research and development | - | - | 1,051 | - | 248 | 1,299 | ||||||||||||||||||
Common stock issued upon conversion of Series C Preferred Stock at $3.75 per share | 29,334 | * | 110 | - | - | 110 | ||||||||||||||||||
Common stock issued settlement of Series C Preferred Stock accrued dividends at $4.53 per share | 15,516 | * | 70 | - | - | 70 | ||||||||||||||||||
Common stock issued upon cashless exercise of warrants | 12,840 | * | - | - | - | - | ||||||||||||||||||
Common stock issued upon cashless exercise of options | 160,743 | * | - | - | - | - | ||||||||||||||||||
Common stock issued upon exercise of options at an average of $4.64 per share | 586,825 | 1 | 2,721 | - | - | 2,722 | ||||||||||||||||||
Common stock issued upon exercise of warrants at an average of $3.88 per share | 542,646 | 1 | 2,089 | - | - | 2,090 | ||||||||||||||||||
Common stock issued in exchange for subsidiary shares | 83,055 | * | 24 | - | (24 | ) | - | |||||||||||||||||
Fair value of subsidiary shares issued to acquire research and development from Trek Therapeutics, PBC | - | - | 2,439 | - | 735 | 3,174 | ||||||||||||||||||
Stock based compensation | 219,334 | * | 17,983 | - | 2,782 | 20,765 | ||||||||||||||||||
Preferred stock dividend | - | - | (14 | ) | - | - | (14 | ) | ||||||||||||||||
Net loss | - | - | - | (52,218 | ) | (6,922 | ) | (59,140 | ) | |||||||||||||||
Balance, December 31, 2020 | 30,764,792 | $ | 31 | $ | 181,344 | $ | (157,005 | ) | $ | 802 | $ | 25,172 | ||||||||||||
*- less than $1
BIOSIG TECHNOLOGIES, INC. | ||||||||||||||||||||||||
CONSOLIDATED STATEMENT OF CHANGES IN EQUITY | ||||||||||||||||||||||||
YEARS ENDED DECEMBER 31, 2021 AND 2020 | ||||||||||||||||||||||||
(In Thousands, Except Par Value and Share Amounts) | ||||||||||||||||||||||||
Additional | Non- | |||||||||||||||||||||||
Common stock | Paid in | Accumulated | controlling | |||||||||||||||||||||
Shares | Amount | Capital | Deficit | Interest | Total | |||||||||||||||||||
Balance, December 31, 2020 | 30,764,792 | $ | 31 | $ | 181,344 | $ | (157,005 | ) | $ | 802 | $ | 25,172 | ||||||||||||
Common stock issued for services | 1,124,341 | 1 | 3,974 | - | - | 3,975 | ||||||||||||||||||
Common stock issued upon exercise of options at $2.96 per share | 9,375 | * | 28 | - | - | 28 | ||||||||||||||||||
Sale of common stock, net transactional costs of $995 | 2,500,000 | 3 | 9,001 | - | - | 9,004 | ||||||||||||||||||
Sale of common stock under At-the-market offering, net of transaction expenses of $40 | 251,720 | * | 1,300 | - | - | 1,300 | ||||||||||||||||||
Change in fair value of modified options | - | - | 313 | - | 8 | 321 | ||||||||||||||||||
Stock based compensation | 916,952 | 1 | 5,176 | - | 348 | 5,525 | ||||||||||||||||||
Preferred stock dividend | - | - | (9 | ) | - | - | (9 | ) | ||||||||||||||||
Net loss | - | - | - | (31,917 | ) | (939 | ) | (32,856 | ) | |||||||||||||||
Balance, December 31, 2021 | 35,567,180 | 36 | 201,127 | (188,922 | ) | 219 | 12,460 | |||||||||||||||||
*- less than $1
See the accompanying notes to the consolidated financial statements.
BIOSIG TECHNOLOGIES, INC. | ||||||||
CONSOLIDATED STATEMENTS OF CASH FLOWS | ||||||||
(In Thousands, Except Par Value and Share Amounts) | ||||||||
Year ended December 31, | ||||||||
2021 | 2020 | |||||||
CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
Net loss | $ | (32,856 | ) | $ | (59,140 | ) | ||
Adjustments to reconcile net loss to cash used in operating activities: | ||||||||
Depreciation and amortization | 198 | 94 | ||||||
Non-cash lease expense | 441 | 456 | ||||||
Equity based compensation | 9,500 | 25,165 | ||||||
Gain on settlement of debt | (553 | ) | - | |||||
Change in fair value of modified options | 321 | - | ||||||
Fair value of subsidiary stock issued to acquire research and development from Trek Therapeutics, PBC | - | 3,174 | ||||||
Fair value of subsidiary stock issued to acquire research and development | - | 1,299 | ||||||
Changes in operating assets and liabilities: | ||||||||
Inventory | (1,114 | ) | (287 | ) | ||||
Prepaid expenses and other | (50 | ) | (119 | ) | ||||
Deferred revenue | 38 | - | ||||||
Deposits | 60 | (18 | ) | |||||
Accounts payable and accrued expenses | (1,988 | ) | 3,233 | |||||
Operating lease liabilities | (396 | ) | (458 | ) | ||||
Net cash used in operating activities | (26,399 | ) | (26,601 | ) | ||||
CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
Purchase of property and equipment | (542 | ) | (87 | ) | ||||
Net cash used in investing activity | (542 | ) | (87 | ) | ||||
CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
Proceeds from sale of common stock, net of issuance costs | 9,004 | 25,215 | ||||||
Proceeds from sale subsidiary stock to non-controlling interest, net of issuance costs | - | 10,592 | ||||||
Proceeds from sale of common stock under a At-the-market offering, net of issuance costs | 1,300 | 2,228 | ||||||
Proceeds from exercise of options | 28 | 2,722 | ||||||
Proceeds from exercise of warrants | - | 2,090 | ||||||
Net cash provided by financing activities | 10,332 | 42,847 | ||||||
Net (decrease) increase in cash and cash equivalents | (16,609 | ) | 16,159 | |||||
Cash, beginning of the year | 28,268 | 12,109 | ||||||
Cash, end of the year | $ | 11,659 | $ | 28,268 | ||||
Supplemental disclosures of cash flow information: | ||||||||
Cash paid during the period for interest | $ | - | $ | - | ||||
Cash paid during the period for income taxes | $ | - | $ | - | ||||
Noncash investing and financing activities: | ||||||||
Common stock issued upon conversion of Series C Preferred Stock and accrued dividends | $ | - | $ | 180 | ||||
Dividend payable on preferred stock charged to additional paid in capital | $ | 9 | $ | 14 | ||||
Record right-to-use assets and related lease liability | $ | 800 | $ | 2 | ||||
See the accompanying notes to the consolidated financial statements.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
NOTE 1 – NATURE OF OPERATIONS AND BASIS OF PRESENTATION
Business and organization
BioSig Technologies, Inc. aswas initially incorporated on February 24, 2009 under the laws of the State of Nevada and subsequently re-incorporated in the state of Delaware in 2011. The Company is principally devoted to improving the standard care in electrophysiology with our PURE EP System’s enhanced signal acquisition, digital signal processing, and analysis during ablation of cardiac arrhythmias. The Company has generated minimal revenue to date and consequently its operations are subject to all risks inherent in business enterprises in early commercialization stage.
On November 7, 2018, the Company formed a subsidiary under the laws of the State of Delaware originally under the name of NeuroClear Technologies, Inc. which was renamed to ViralClear Pharmaceuticals, Inc. (“ViralClear”) in March 2020. The subsidiary was established to pursue additional applications of the PURE EP™ signal processing technology outside of cardiac electrophysiology, and subsequently in 2020, was repurposed to develop merimepodib, a broad-spectrum anti-viral agent that showed potential for the treatment of COVID-19. Since late 2020, ViralClear has been realigned with its original objective of pursuing additional applications of the PURE EP™ signal processing technology outside of cardiac electrophysiology.
In 2019 and 2020, ViralClear sold an aggregate of 1,965,240 shares of its common stock to investors for net proceeds of $15.6 million and issued an aggregate of 894,869 shares of its common stock in connection with acquiring assets and with know-how agreements. As of December 31, 20172021, the Company had a majority interest in ViralClear of 68.44%.
On July 2, 2020, the Company formed an additional subsidiary, NeuroClear Technologies, Inc., a Delaware corporation.
COVID-19
On March 11, 2020, the World Health Organization declared a pandemic related to the rapidly spreading coronavirus (COVID-19) outbreak, which has led to a global health emergency. The full public-health impact of the ongoing pandemic is currently indeterminable and 2016,rapidly evolving, and the resultsrelated health crisis has adversely affected and may continue to adversely affect the global economy, resulting in delaying to our commercialization objectives of its operations and its cash flows for the years then ended in conformity with accounting principles generally accepted in the United StatesPURE EP Systems into 2022.
NOTE 2 – GOING CONCERN AND MANAGEMENT’S LIQUIDITY PLANS
As of America.
The Company’s primary source of operating funds since inception has been cash proceeds from sale of of common and preferred stock. The Company has experienced net losses and negative cash flows from operations since inception and expects these conditions to continue for the foreseeable future.
The Company’s plans include the continued commercialization of the PURE EP System and other applications of our core technology and raising capital through the sale of additional equity securities, debt or capital inflows from strategic partnerships. The Company’s strategic shift from a focus on technology development to commercialization will allow the Company to significantly reduce operating expenses.
The Company will require additional financing to fund future operations. Further, although the Company began commercial operations; there is no assurance that the Company will be able to generate sufficient cash flow to fund operations. In addition, there can be no assurance that the Company’s continuing research and development will be successfully completed or that any additional products will be commercially viable.
Accordingly, the accompanying consolidated financial statements have been prepared in regardconformity with U.S. GAAP, which contemplates continuation of the Company as a going concern and the realization of assets and satisfaction of liabilities in the normal course of business. The carrying amounts of assets and liabilities presented in the consolidated financial statements do not necessarily purport to these matters are also described in Note 2.represent realizable or settlement values. The consolidated financial statements do not include any adjustmentsadjustment that might result from the outcome of this uncertainty.
| BIOSIG TECHNOLOGIES, INC. | ||||||||
| DECEMBER 31, 2017 AND 2016 | ||||||||
| 2017 | 2016 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 1,547,579 | $ | 1,055,895 | ||||
| Prepaid expenses | 116,938 | 134,263 | ||||||
| Total current assets | 1,664,517 | 1,190,158 | ||||||
| Property and equipment, net | 18,716 | 24,188 | ||||||
| Other assets: | ||||||||
| Deposits | 17,084 | 27,612 | ||||||
| Total assets | $ | 1,700,317 | $ | 1,241,958 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses, including $27,375 and $15,755 to related parties as of December 31, 2017 and 2016, respectively | $ | 473,098 | $ | 373,103 | ||||
| Dividends payable | 447,901 | 359,891 | ||||||
| Warrant liability | 2,358,240 | 1,937,234 | ||||||
| Derivative liability | 685,922 | 288,934 | ||||||
| Total current liabilities | 3,965,161 | 2,959,162 | ||||||
| Series C Preferred Stock, 985 and 1,070 shares issued and outstanding; liquidation preference of $985,000 and $1,070,000 as of December 31, 2017 and 2016, respectively | 985,000 | 1,070,000 | ||||||
| Stockholders’ deficit | ||||||||
| Preferred stock, $0.001 par value, authorized 1,000,000 shares, designated 200 shares of Series A, 600 shares of Series B, 4,200 shares of Series C and 1,400 shares of Series D Preferred Stock | ||||||||
| Series D Preferred Stock, $0.001 par value, 1,334 and 0 shares issued and outstanding; liquidation preference of $2,001,000 and $0 as of December 31, 2017 and 2016, respectively | 1 | - | ||||||
| Common stock, $0.001 par value, authorized 200,000,000 shares, 29,321,204 and 22,588,184 issued and outstanding as of December 31, 2017 and 2016, respectively | 29,321 | 22,588 | ||||||
| Additional paid in capital | 53,215,635 | 41,019,251 | ||||||
| Common stock subscription | 29,985 | - | ||||||
| Accumulated deficit | (56,524,786 | ) | (43,829,043 | ) | ||||
| Total stockholders’ deficit | (3,249,844 | ) | (2,787,204 | ) | ||||
| Total liabilities and stockholders’ deficit | $ | 1,700,317 | $ | 1,241,958 | ||||
| BIOSIG TECHNOLOGIES, INC. | ||||||||
| Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Operating expenses: | ||||||||
| Research and development | $ | 4,756,468 | $ | 2,654,501 | ||||
| General and administrative | 8,138,117 | 8,499,304 | ||||||
| Depreciation | 11,698 | 10,475 | ||||||
| Total operating expenses | 12,906,283 | 11,164,280 | ||||||
| Loss from operations | (12,906,283 | ) | (11,164,280 | ) | ||||
| Other income (expense): | ||||||||
| Gain (loss) on change in fair value of derivatives | 210,465 | (422,908 | ) | |||||
| Interest income | 75 | 1 | ||||||
| Loss before income taxes | (12,695,743 | ) | (11,587,187 | ) | ||||
| Income taxes (benefit) | - | - | ||||||
| Net loss | (12,695,743 | ) | (11,587,187 | ) | ||||
| Preferred stock dividend | (119,877 | ) | (110,023 | ) | ||||
| NET LOSS AVAILABLE TO COMMON STOCKHOLDERS | $ | (12,815,620 | ) | $ | (11,697,210 | ) | ||
| Net loss per common share, basic and diluted | $ | (0.50 | ) | $ | (0.60 | ) | ||
| Weighted average number of common shares outstanding, basic and diluted | 25,550,686 | 19,490,767 | ||||||
| BIOSIG TECHNOLOGIES, INC. | ||||||||||||||||||||||||||||||||
| TWO YEARS ENDED DECEMBER 31, 2017 | ||||||||||||||||||||||||||||||||
| Additional | Common | |||||||||||||||||||||||||||||||
| Series D Preferred stock | Common stock | Paid in | stock | Accumulated | ||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Subscription | Deficit | Total | |||||||||||||||||||||||||
| Balance, January 1, 2016 | - | $ | - | 16,825,703 | $ | 16,826 | $ | 29,314,399 | $ | - | $ | (32,241,856 | ) | $ | (2,910,631 | ) | ||||||||||||||||
| Sale of common stock | - | - | 3,798,417 | 3,798 | 5,222,570 | - | - | 5,226,368 | ||||||||||||||||||||||||
| Common stock issued for services | - | - | 1,335,000 | 1,335 | 2,469,715 | - | - | 2,471,050 | ||||||||||||||||||||||||
| Common stock issued upon conversion of Series C Preferred Stock at $1.50 per share | - | - | 267,334 | 267 | 400,733 | - | - | 401,000 | ||||||||||||||||||||||||
| Common stock issued settlement of Series C Preferred Stock accrued dividends at $1.55 per share | - | - | 58,185 | 58 | 90,365 | - | - | 90,423 | ||||||||||||||||||||||||
| Reclassify fair value of derivative liability to equity upon conversion of Series C Preferred Stock to common shares | - | - | - | - | 103,096 | - | - | 103,096 | ||||||||||||||||||||||||
| Stock based compensation | - | - | 303,545 | 304 | 3,528,396 | - | - | 3,528,700 | ||||||||||||||||||||||||
| Preferred Stock dividend | - | - | - | - | (110,023 | ) | - | - | (110,023 | ) | ||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (11,587,187 | ) | (11,587,187 | ) | ||||||||||||||||||||||
| Balance, December 31, 2016 | - | $ | - | 22,588,184 | $ | 22,588 | $ | 41,019,251 | $ | - | $ | (43,829,043 | ) | $ | (2,787,204 | ) | ||||||||||||||||
| BIOSIG TECHNOLOGIES, INC. | ||||||||||||||||||||||||||||||||
| CONDENSED STATEMENT OF STOCKHOLDERS’ DEFICIT | ||||||||||||||||||||||||||||||||
| TWO YEARS ENDED DECEMBER 31, 2017 | ||||||||||||||||||||||||||||||||
| Additional | Common | |||||||||||||||||||||||||||||||
| Series D Preferred stock | Common stock | Paid in | stock | Accumulated | ||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Subscription | Deficit | Total | |||||||||||||||||||||||||
| Balance, December 31, 2016 | - | $ | - | 22,588,184 | $ | 22,588 | $ | 41,019,251 | $ | - | $ | (43,829,043 | ) | $ | (2,787,204 | ) | ||||||||||||||||
| Sale of common stock | - | - | 4,131,536 | 4,131 | 6,007,098 | - | - | 6,011,229 | ||||||||||||||||||||||||
| Sale of Series D preferred stock | 1,334 | 1 | - | - | 1,929,959 | - | - | 1,929,960 | ||||||||||||||||||||||||
| Common stock issued for services | - | - | 2,271,788 | 2,272 | 3,384,345 | - | - | 3,386,617 | ||||||||||||||||||||||||
| Common stock issued upon conversion of Series C Preferred Stock at $1.50 per share | - | - | 56,669 | 57 | 84,943 | - | - | 85,000 | ||||||||||||||||||||||||
| Common stock issued settlement of Series C Preferred Stock accrued dividends at $1.37 per share | - | - | 24,021 | 24 | 31,844 | - | - | 31,868 | ||||||||||||||||||||||||
| Common stock received and canceled in connection with short term swing profit reimbursement | - | - | (10,744 | ) | (11 | ) | 11 | - | - | - | ||||||||||||||||||||||
| Common stock subscription received | - | - | - | - | - | 29,985 | - | 29,985 | ||||||||||||||||||||||||
| Reclassify initial fair value of derivative and warrant liability of Series D preferred stock and warrants at issuance | - | - | - | - | (1,049,216 | ) | - | - | (1,049,216 | ) | ||||||||||||||||||||||
| Reclassify fair value of derivative liability to equity upon conversion of Series C Preferred Stock to common shares | - | - | - | - | 20,757 | - | - | 20,757 | ||||||||||||||||||||||||
| Fair value of warrant issued to acquire research and development | - | - | - | - | 543,927 | - | - | 543,927 | ||||||||||||||||||||||||
| Stock based compensation | - | - | 259,750 | 260 | 1,362,593 | - | - | 1,362,853 | ||||||||||||||||||||||||
| Preferred stock dividend | - | - | - | - | (119,877 | ) | - | - | (119,877 | ) | ||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (12,695,743 | ) | (12,695,743 | ) | ||||||||||||||||||||||
| Balance, December 31, 2017 | 1,334 | $ | 1 | 29,321,204 | $ | 29,321 | $ | 53,215,635 | $ | 29,985 | $ | (56,524,786 | ) | $ | (3,249,844 | ) | ||||||||||||||||
| BIOSIG TECHNOLOGIES, INC. | ||||||||
| Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (12,695,743 | ) | $ | (11,587,187 | ) | ||
| Adjustments to reconcile net loss to cash used in operating activities: | ||||||||
| Depreciation | 11,698 | 10,475 | ||||||
| Equipment distribution as officer compensation | 3,210 | - | ||||||
| Change in derivative liabilities | (210,465 | ) | 422,908 | |||||
| Equity based compensation | 4,749,470 | 5,999,750 | ||||||
| Fair value of issued warrant to acquire research and development | 543,927 | - | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses | 17,325 | (102,955 | ) | |||||
| Security deposit | 10,528 | - | ||||||
| Accounts payable | 102,338 | 149,661 | ||||||
| Deferred rent payable | (2,342 | ) | (104 | ) | ||||
| Net cash used in operating activities | (7,470,054 | ) | (5,107,452 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||
| Purchase of property and equipment | (9,436 | ) | (16,255 | ) | ||||
| Net cash used in investing activity | (9,436 | ) | (16,255 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Proceeds from sale of common stock | 6,011,229 | 5,226,368 | ||||||
| Proceeds from sale of Series D preferred stock | 1,929,960 | - | ||||||
| Proceeds from common stock subscription | 29,985 | - | ||||||
| Net cash provided by financing activities | 7,971,174 | 5,226,368 | ||||||
| Net increase in cash and cash equivalents | 491,684 | 102,661 | ||||||
| Cash and cash equivalents, beginning of the period | 1,055,895 | 953,234 | ||||||
| Cash and cash equivalents, end of the period | $ | 1,547,579 | $ | 1,055,895 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid during the period for interest | $ | - | $ | - | ||||
| Cash paid during the period for income taxes | $ | - | $ | - | ||||
| Non cash investing and financing activities: | ||||||||
| Common stock issued upon conversion of Series C Preferred Stock and accrued dividends | $ | 116,868 | $ | 491,423 | ||||
| Reclassify initial fair value of derivative and warrant liabilities from equity upon issuance of Series D preferred stock | $ | 1,049,216 | $ | - | ||||
| Reclassify fair value of derivative liability to equity | $ | 20,757 | $ | 103,096 | ||||
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 20172021
NOTE 13 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
A summary of the significant accounting policies applied in the preparation of the accompanying consolidated financial statements follows.
Principals of consolidation
The accompanying consolidated financial statements include the accounts of BioSig Technologies, Inc. (the “Company”) was initially incorporated on February 24, 2009 under the laws of the State of Nevada and subsequently re-incorporated in the state of Delaware in 2011. The Company and its efforts are principally devotedmajority owned subsidiary, ViralClear Pharmaceuticals, Inc., and wholly owned subsidiary, NeuroClear Technologies, Inc. herein referred to improvingas the quality of cardiac recordings obtained during ablation of atrial fibrillation (AF)“Company” or “BioSig”. All significant intercompany accounts and ventricular tachycardia (VT). The Company has not generated any revenue to date and consequently its operations are subject to all risks inherenttransactions have been eliminated in the establishment of a new business enterprise.consolidation.
Use of estimatesEstimates
The preparation of these consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Significant estimates include the recoverability and useful lives of long-lived assets, the fair value of the Company’s stock,acquired assets, stock-based compensation fair values relating to warrant and other derivative liabilities and the valuation allowance related to deferred tax assets. Actual results may differ from these estimates.
Revenue Recognition
The Company placesderives its cash and temporary cash investments with credit quality institutions. At times, such amounts may be in excess of the FDIC insurance limit. At December 31, 2017 and 2016, deposits in excess of FDIC limits were $1,297,579 and $805,895, respectively.
The Company followsrecognizes revenue in accordance with Accounting Standards Codification 360-10-15-3, “Impairment(ASC) 606, Revenue from Contracts with Customers (“ASC 606”). The core principle of ASC 606 is that an entity recognizes revenue to depict
the transfer of promised goods or Disposal of Long-lived Assets,”services to customers in an amount that reflects the consideration to which established a “primary asset” approachthe entity expects to determinebe entitled in exchange for those goods or services.
The Company determines revenue recognition through the cash flow estimation period for a group of assets and liabilities that representsfollowing five steps:
● | Identify the contract with the customer; |
● | Identify the performance obligations in the contract; |
● | Determine the transaction price; |
● | Allocate the transaction price to the performance obligation in the contract; and |
● | Recognize revenue when, or as, the performance obligations are satisfied. |
Performance obligations are the unit of accounting for revenue recognition and generally represent the distinct goods or services that are promised to the customer. If the Company determines that it has not satisfied a long-lived assetperformance obligation, it will defer recognition of the revenue until the performance obligation is deemed to be heldsatisfied. Once the PURE EP system is delivered, installed, and used. Long-lived assetsaccepted by the customer, our performance obligation is recognized. Support, maintenance, and software upgrades are performance obligations over a defined period and are recognized ratably over the contractual service period. Customers typically purchase these services with the initial sale of the PURE EP System and do not have the right to terminate their contracts unless we fail to perform material obligations.
The Company may execute more than one contract with a single customer. If so, it is evaluated whether the agreements were negotiated as a package with a single objective, whether the amount of consideration to be held and used are reviewed for impairment whenever events paid in one agreement depends on the price and/or changesperformance of another agreement, or whether the goods or services promised in circumstances indicate that the carrying amount of an asset may not be recoverable.agreements represent a single performance obligation. The carrying amount of a long-lived asset is not recoverable if it exceedsconclusions reached can impact the sumallocation of the undiscounted cash flows expectedtransaction price to result fromeach performance obligation and the usetiming of revenue recognition related to those arrangements.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
The Company records accounts receivable for amounts invoiced to customers for which the Company has an unconditional right to consideration as provided under the contractual arrangement. Unbilled receivables, if any, include amounts related to the Company’s contractual right to consideration for completed performance obligations not yet invoiced. Deferred revenue includes payments received in advance of performance under the contract. Our unbilled receivables and eventual dispositiondeferred revenue are reported on an individual contract basis at the end of each reporting period. Unbilled receivables are classified as current or noncurrent based on the timing of when we expect to bill the customer. Deferred revenue is classified as current or noncurrent based on the timing of when we expect to recognize revenue.
The Company’s unconditional right to consideration for goods and services transferred to the customer is included in accounts receivable, net (if any) in the Company’s consolidated balance sheet.
There are no contract liabilities for the year ended December 31, 2020.
A reconciliation of contract liabilities with customers for the year ended December 31, 2021, are presented below:
Balance at December 31, 2020 (000’s) | Consideration Received (000’s) | Recognized in Revenue (000’s) | Balance at December 31, 2021 (000’s) | |||||||||||||
Product revenue | $ | - | $ | 414 | $ | (414 | ) | $ | - | |||||||
Service revenue | - | 64 | (27 | ) | 37 | |||||||||||
Total | $ | - | $ | 478 | $ | (441 | ) | $ | 37 | |||||||
The table below summarizes our deferred revenue as of December 31, 2021 and 2020:
December 31, 2021 (000’s) | December 31, 2020 (000’s) | |||||||
Deferred revenue-current | $ | 32 | $ | - | ||||
Deferred revenue-noncurrent | 5 | - | ||||||
Total deferred revenue | $ | 37 | $ | - | ||||
The Company had two customers which accounted for approximately 68% and 32% of their revenue in the year ended December 31, 2021.
The Company utilized one contract manufacturer for the manufacture and supply of the asset. Long-lived assetsPure EP system for the years ended December 31, 2021 and 2020.
Cost of Goods Sold
Cost of goods sold consists primarily of the delivered cost of our medical device(s) sold.
Allowance for Doubtful Accounts
The Company adjusts accounts receivable down to be disposednet realizable value with its allowance methodology. In determining the allowance for doubtful accounts for estimated losses, aged receivables are analyzed periodically by management. Each identified receivable is reviewed based upon historical collection experience, financial condition of are reportedthe client and the status of any open or unresolved issues with the client preventing the payment thereof. Corrective action, if necessary, is taken by the Company to resolve open issues related to unpaid receivables. The allowance for doubtful accounts was $0 at December 31, 2021 and 2020. The Company believes that its reserve is adequate, however results may differ in future periods. For the loweryear ended December 31, 2021 and 2020, bad debt expense totaled $0.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
Fair Value of Financial Instruments
Accounting Standards Codification subtopic 825-10, Financial Instruments (“ASC 825-10”) requires disclosure of the fair value of certain financial instruments. The carrying value of cash, and cash equivalents, accounts payable and accrued liabilities as reflected in the balance sheets, approximate fair value because of the short-term maturity of these instruments. All other significant financial assets, financial liabilities and equity instruments of the Company are either recognized or disclosed in the financial statements together with other information relevant for making a reasonable assessment of future cash flows, interest rate risk and credit risk. Where practicable the fair values of financial assets and financial liabilities have been determined and disclosed; otherwise only available information pertinent to fair value has been disclosed.
The Company follows Accounting Standards Codification subtopic 820-10, Fair Value Measurements and Disclosures (“ASC 820-10”) and Accounting Standards Codification subtopicASC 825-10, Financial Instruments (“ASC 825-10”), which permits entities to choose to measure many financial instruments and certain other items at fair value.
Concentrations of Credit Risk
Financial instruments and related items, which potentially subject the Company to concentrations of credit risk, consist primarily of cash and cash equivalents. The Company accountsplaces its cash and temporary cash investments with credit quality institutions. At times, such amounts may be in excess of the FDIC insurance limit. At December 31, 2021 and 2020, deposits in excess of FDIC limits were $11.2 million and $27.8 million, respectively.
Inventory
The inventory is comprised of work in process and finished goods available for derivative instrumentssale and are stated at the lower of cost or net realizable value using specific identification method for serial numbered inventory and first-in, first-out method for all other inventory for valuation. The inventory at December 31, 2021 and 2020 were $1.9 million and $0.8 million, respectively, comprised of finished goods.
Prepaid Expenses and Vendor Deposits
Prepaid expenses and vendor deposits are comprised of prepaid insurance, operating expenses and other prepayments.
Leases
The Company determines if a contractual arrangement is a lease at inception. Operating leases are included in accordance with ASC 815, which establishes accountingoperating lease right-of-use (“ROU”) assets, current operating lease liabilities, and reporting standards for derivative instruments and hedging activities, including certain derivative instruments embedded in other financial instruments or contracts and requires recognition of all derivativesnoncurrent operating lease liabilities on the Company’s consolidated balance sheetsheet. The Company evaluates and classifies leases as operating or finance leases for financial reporting purposes. The classification evaluation begins at fair value, regardless of hedging relationship designation. Accountingthe commencement date and the lease term used in the evaluation includes the non-cancellable period for changes in fair valuewhich the Company has the right to use the underlying asset, together with renewal option periods when the exercise of the derivative instruments depends on whetherrenewal option is reasonably certain and failure to exercise such option which result in an economic penalty. All the derivatives qualifyCompany’s real estate leases are classified as hedge relationshipsoperating leases. ROU assets represent the Company’s right to use an underlying asset for the lease term and lease liabilities represent the typesCompany’s obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at the commencement date of relationships designated arethe lease based on the exposures hedged. At Decemberpresent value of lease payments over the lease term.
The lease payments included in the present value are fixed lease payments. As most of the Company’s leases do not provide an implicit rate, the Company estimates its collateralized incremental borrowing rate, based on information available at the commencement date, in determining the present value of lease payments. The Company applies the portfolio approach in applying discount rates to its classes of leases. The operating lease ROU assets include any payments made before the commencement date. Lease expense for lease payments is recognized on a straight-line basis over the lease term. The Company does not currently have subleases. The Company does not currently have residual value guarantees or restrictive covenants in its leases.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 20172021
Property and 2016,Equipment
Property and equipment are stated at cost and depreciated using the straight-line method over their estimated useful lives of 3 to 5 years. When retired or otherwise disposed, the related carrying value and accumulated depreciation are removed from the respective accounts and the net difference less any amount realized from disposition, is reflected in earnings.
Impairment of Long-lived Assets
The Company recognizes an impairment of long-lived assets used in operations, other than goodwill, when events or circumstances indicate that the asset might be impaired and the estimated undiscounted cash flows to be generated by those assets over their remaining lives are less than the carrying amount of those items. The net carrying value of assets not recoverable is reduced to fair value, which is typically calculated using the discounted cash flow method. The Company did not haverecognize and record any derivative instruments that were designated as hedges.
Research and development costsDevelopment Costs
The Company accounts for research and development costs in accordance with the Accounting Standards Codification subtopic 730-10, Research and Development (“ASC 730-10”). Under ASC 730-10, all research and development costs must be charged to expense as incurred. Accordingly, internal research and development costs are expensed as incurred. Third-party research and developments costs are expensed when the contracted work has been performed or as milestone results have been achieved. Company-sponsored research and development costs related to both present and future products are expensed in the period incurred. The Company incurred research and development expenses of $4,756,468$5.6 million and $2,654,501$18.1 million for the year ended December 31, 20172021 and 2016,2020, respectively.
Acquisition of Intellectual Property
Intellectual property acquired are accounted for under the acquisition method of accounting. This method requires the recording of acquired assets, including separately identifiable intangible assets, and assumed liabilities at their acquisition date fair values. Any excess consideration transferred over fair value is allocated on a relative fair value basis to the identifiable net assets.
The acquired intellectual property from the Trek acquisition was considered unproven compounds, the success of which was uncertain at the time of the acquisition. Accordingly, the fair value of the consideration paid was charged as acquired research and development to current period operations.
Net Income (loss) Per Common Share
The Company computes earnings (loss) per share under Accounting Standards Codification subtopic 260-10, Earnings Per Share (“ASC 260-10”). Net loss per common share is computed by dividing net loss by the weighted average number of shares of common stock outstanding during the period. Diluted earnings per share, if presented, would include the dilution that would occur upon the exercise or conversion of all potentially dilutive securities into common stock using the “treasury stock” and/or “if converted” methods as applicable.
The computation of basic and diluted loss per share as of December 31, 2021 and 2020 excludes potentially dilutive securities when their inclusion would be anti-dilutive, or if their exercise prices were greater than the average market price of the common stock during the period.
Potentially dilutive securities excluded from the computation of basic and diluted net income (loss) per share are as follows:
|
| December 31, 2021 |
|
| December 31, 2020 |
| ||
Series C convertible preferred stock |
|
| 83,468 |
|
|
| 47,578 |
|
Options to purchase common stock |
|
| 4,568,484 |
|
|
| 3,568,497 |
|
Warrants to purchase common stock |
|
| 818,910 |
|
|
| 1,446,200 |
|
Restricted stock units to acquire common stock |
|
| 141,250 |
|
|
| 218,334 |
|
Totals |
|
| 5,612,112 |
|
|
| 5,280,609 |
|
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 20172021
Stock Based Compensation
The Company measures the cost of services received in exchange for an award of equity instruments based on the fair value of the award as measured on the grant date. The fair value amount is then recognized over the period during which services are required to be provided in exchange for the award, usually the vesting period.
Income Taxes
The Company follows Accounting Standards Codification subtopic 740-10, Income Taxes (“ASC 740-10”) for recording the provision for income taxes. Deferred tax assets and liabilities are computed based upon the difference between the financial statement and income tax basis of assets and liabilities using the enacted marginal tax rate applicable when the related asset or liability is expected to be realized or settled. Deferred income tax expenses or benefits are based on the changes in the asset or liability during each period. If available evidence suggests that it is more likely than not that some portion or all of the deferred tax assets will not be realized, a valuation allowance is required to reduce the deferred tax assets to the amount that is more likely than not to be realized. Future changes in such valuation allowance are included in the provision for deferred income taxes in the period of change. Deferred income taxes may arise from temporary differences resulting from income and expense items reported for financial accounting and tax purposes in different periods.
Patents, Net
The Company computes earnings (loss) per share under Accounting Standards Codification subtopic 260-10, Earnings Per Sharecapitalizes certain initial asset costs in connection with patent applications including registration, documentation and other professional fees associated with the application. Patent costs incurred prior to the Company’s U.S. Food and Drug Administration (“ASC 260-10”FDA”). Net loss per common share 510(k) application on March 28, 2018 were charged to research and development expense as incurred. Commencing upon first in-man trials on February 18 and 19, 2019, capitalized costs are amortized to expense using the straight-line method over the lesser of the legal patent term or the estimated life of the product of 20 years. During the year ended December 31, 2021 and 2020, the Company recorded amortization of $19,006 and $19,005 to current period operations, respectively.
Warranty
The Company generally warrants its products to be free from material defects and to conform to material specifications for a period of up to two (2) years. Warranty expense is computed by dividingestimated based primarily on historical experience and is reflected in the consolidated financial statements.
Non-controlling Interest
The Company’s non-controlling interest represents the non-controlling shareholders ownership interests related to the Company’s subsidiary, ViralClear. The Company reports its non-controlling interest in subsidiaries as a separate component of equity in the consolidated balance sheets and reports both net loss byattributable to the weighted average numbernon-controlling interest and net loss attributable to the Company’s common shareholders on the face of sharesthe consolidated statements of common stock outstanding duringoperations. The Company’s equity interest in ViralClear is 68.44% and the year. Diluted earnings per share, if presented, would include the dilution that would occur upon the exercise or conversion of all potentially dilutive securities into common stock using the “treasury stock” and/or “if converted” methods as applicable.
Segment Information
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker, or decision-making group, in making decisions how to allocate resources and 2016 excludes potentially dilutive securities when their inclusion would be anti-dilutive, or if their exercise prices were greater than the average market priceassess performance. The information disclosed herein represents all of the common stock duringmaterial financial information related to the period.Company’s principal operating segments. (See Note 12 – Segment Reporting).
| 2017 | 2016 | |||||||
| Series C convertible preferred stock | 656,667 | 713,333 | ||||||
| Series D convertible preferred stock | 1,334,000 | - | ||||||
| Options to purchase common stock | 8,510,319 | 8,245,190 | ||||||
| Warrants to purchase common stock | 12,789,086 | 9,128,189 | ||||||
| Totals | 23,290,072 | 18,086,712 | ||||||
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2017
Recent Accounting Pronouncements
In June 2016, the FASB issued ASU 2016-13, Financial Instruments-Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments (“ASU 2016-13”), which requires the measurement and recognition of expected credit losses for financial assets held at amortized cost. ASU 2016-13 replaces the existing incurred loss impairment model with an expected loss model that requires the use of forward-looking information to calculate credit loss estimates. It also eliminates the concept of other-than-temporary impairment and requires credit losses on available-for-sale debt securities to be recorded through an allowance for credit losses instead of as a reduction in the amortized cost basis of the securities. ASU 2016-13 was effective for annual periods, and interim periods within those annual periods, beginning after December 15, 2019. Early adoption was permitted, including adoption in any interim period.
In February 2020, the FASB issued ASU 2020-02, Financial Instruments-Credit Losses (Topic 326) and Leases (Topic 842) - Amendments to SEC Paragraphs Pursuant to SEC Staff Accounting Bulletin No. 119 and Update to SEC Section on Effective Date Related to Accounting Standards Update No. 2016-02, Leases (Topic 842), which amended the effective date of the original pronouncement for smaller reporting companies. ASC 2016-13 and its amendments will be effective for annual and interim periods beginning after December 15, 2022 for smaller reporting companies. The Company is currently assessing the impact the adoption of this new standard will have on its consolidated financial statements and related disclosures.
There were other various updates recently issued, most of which represented technical corrections to the accounting literature or application to specific industries and are not expected to a have a material impact on the Company’s financial position, results of operations or cash flows.
NOTE 4 – PROPERTY AND EQUIPMENT
Property and equipment as of December 31, 20172021 and 20162020 is summarized as follows:
| 2017 | 2016 | December 31, 2021 (000’s) | December 31, 2020 (000’s) | |||||||||||||
| Computer equipment | $ | 87,059 | $ | 84,704 | $ | 383 | $ | 234 | ||||||||
| Furniture and fixtures | 12,975 | 10,117 | 88 | 75 | ||||||||||||
| Subtotal | 100,034 | 94,821 | ||||||||||||||
Manufacturing equipment | 286 | 34 | ||||||||||||||
Testing/Demo equipment | 145 | 96 | ||||||||||||||
Leasehold improvements | 79 | - | ||||||||||||||
Total | 981 | 439 | ||||||||||||||
| Less accumulated depreciation | (81,318 | ) | (70,633 | ) | (329 | ) | (150 | ) | ||||||||
| Property and equipment, net | $ | 18,716 | $ | 24,188 | $ | 652 | $ | 289 | ||||||||
Property and equipment are stated at cost and depreciated using the straight-line method over their estimated useful lives of 3 to 5 years. Leasehold improvements are depreciated over the related expected lease term. When retired or otherwise disposed, the related carrying value and accumulated depreciation are removed from the respective accounts and the net difference less any amount realized from disposition, is reflected in earnings.
Depreciation expense was $11,698$179,136 and $10,475$74,527 for years ended December 31, 2021 and 2020, respectively.
NOTE 5 – RIGHT TO USE ASSETS AND LEASE LIABILITY
Operating leases:
On February 10, 2021 the Company entered into a Sixth Amendment to the Office Lease at 12424 Wilshire Blvd in Los Angeles dated August 9, 2011 – it is the Fourth Extended Term with respect to Suite 745 and the Expansion Term with respect to Suite 740 which is from July 1, 2021 until June 30, 2022 with a fixed monthly rent equal to $13,702 (down from $16,289); and the security deposit will be reduced by $5,448 so that the balance remaining shall be $27,404.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
The Company determined that the Sixth Amendment was a lease modification and accordingly reassessed the lease classification, remeasured the lease liability and adjusted the right-to-use asset. At February 10, 2021 the Company removed the remaining right-to-use net assets of $60,881 and related lease liability of $63,076 and recorded right-to-use assets and related lease liability of $217,903.
On August 2, 2021, the Company exercised its option to extend its Rochester, Minnesota lease of approximately 1,400 square feet of office space for two additional years expiring on October 31, 2023 with a fixed monthly rate of $3,513, increasing to $3,618 for the second year.
The Company determined that the lease option exercised was a lease modification and accordingly reassessed the lease classification, remeasured the lease liability and adjusted the right-to-use asset. On August 2, 2021 the Company removed the remaining right-to-use net assets of $10,247 and related lease liability of $10,400 and recorded right-to-use assets and related lease liability of $89,629. At the lease modification date, the Company estimated the lease liability and the right of use assets at present value using the Company’s estimated incremental borrowing rate of 6.5%.
On August 3, 2021, the Company entered into a sublease agreement whereby the Company leased approximately 6,590 square feet of office space at 55 Greens Farms Road, Westport, Connecticut commencing September 1, 2021 and expiring December 31, 2024 (40 months) at the initial rate beginning January 1, 2022 of $14,828 with escalating payments. In connection with the lease, the Company paid a security deposit of $14,232. There is no option to extend the lease past its initial term. At the lease commencement date, the Company estimated the lease liability and right-to-use assets at present value using the Company’s incremental borrowing rate of 6.5% and determined their initial present values, at inception, of $492,876. In conjunction with the lease, the Company terminated, without penalty, the sublease at 54 Wilton Road, Westport, CT effective September 4, 2021 and removed the remaining right-to-use assets of $36,756 and related lease liability of $37,625 with a credit to rent expense of $868 relating to the lease termination.
As of December 31, 2021, the Company had outstanding 5 leases with aggregate payments of $32,143 per month, expiring through December 31, 2024.
Right to use assets is summarized below:
December 31, 2021 (000’s) | December 31, 2020 (000’s) | |||||||
Right to use assets, net | $ | 803 | $ | 1,087 | ||||
Less accumulated amortization | (199 | ) | (781 | ) | ||||
Right to use assets, net | $ | 604 | $ | 306 | ||||
During the years ended December 31, 20172021 and 2016,2020, the Company recorded $479,746 and $492,844 as lease expense to current period operations, respectively.
Lease liability is summarized below:
December 31, 2021 (000’s) | December 31, 2020 (000’s) | |||||||
Total lease liability | $ | 656 | $ | 314 | ||||
Less: short term portion | (283 | ) | (313 | ) | ||||
Long term portion | $ | 373 | $ | 1 | ||||
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
Maturity analysis under these lease agreements are as follows (000’s):
Year ended December 31, 2022 | $ | 304 | ||
Year ended December 31, 2023 | 221 | |||
Year ended December 31, 2024 | 191 | |||
Total | 716 | |||
Less: Present value discount | (60 | ) | ||
Lease liability | $ | 656 |
Lease expense for the year ended December 31, 2021 and 2020 was comprised of the following:
December 31, 2021 (000’s) | December 31, 2020 (000’s) | |||||||
Operating lease expense | $ | 441 | $ | 456 | ||||
Short-term lease expense | 39 | 35 | ||||||
Variable lease expense | - | 2 | ||||||
Total | $ | 480 | $ | 493 | ||||
NOTE 56 – ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Accounts payable and accrued expenses at December 31, 20172021 and 20162020 consist of the following:
| 2017 | 2016 | December 31, 2021 (000’s) | December 31, 2020 (000’s) | |||||||||||||
| Accrued accounting and legal | $ | 93,595 | $ | 120,464 | $ | 204 | $ | 177 | ||||||||
| Accrued reimbursements | 2,600 | 43,116 | ||||||||||||||
Accrued reimbursements and travel | 56 | 56 | ||||||||||||||
| Accrued consulting | 109,059 | 1,192 | 264 | 256 | ||||||||||||
| Accrued research and development expenses | 246,030 | 181,884 | 367 | 3,127 | ||||||||||||
Accrued product purchases | 1 | 30 | ||||||||||||||
Accrued marketing | 38 | - | ||||||||||||||
| Accrued office and other | 7,912 | 10,202 | 84 | 127 | ||||||||||||
| Deferred rent | 569 | 2,912 | ||||||||||||||
Accrued payroll | 552 | 936 | ||||||||||||||
| Accrued settlement related to arbitration | 13,333 | 13,333 | 613 | 13 | ||||||||||||
| $ | 473,098 | $ | 373,103 | $ | 2,179 | $ | 4,722 | |||||||||
NOTE 67 – SERIES C 9% CONVERTIBLE PREFERRED STOCK
Series C 9% Convertible Preferred Stock
On January 9, 2013, the Board of Directors authorized the issuance of up to 4,200 shares of 9% Series C Convertible Preferred Stock (the “Series C Preferred Stock”).
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 20172021
The Series C Preferred Stock is entitled to preference over holders of junior stock upon liquidation in the amount of $1,000 plus any accrued and unpaid dividends; entitled to dividends as a preference to holders of junior stock at a rate of 9% per annum of the stated value of $1,000 per share, payable quarterly beginning on September 30, 2013 and are cumulative. The holders of the Series C Preferred Stock vote together with the holders of our common stock on an as-converted basis but may not vote the Series C Preferred Stock in excess of the beneficial ownership limitation of the Series C Preferred Stock. The beneficial ownership limitation is 4.99% of our then outstanding shares of common stock following such conversion or exercise, which may be increased to up to 9.99% of our then outstanding shares of common stock following such conversion or exercise upon the request of an individual holder. The beneficial ownership limitation is determined on an individual holder basis, such that the as-converted number of shares of one holder is not included in the shares outstanding when calculating the limitation for a different holder.
As a result of an amendment to the conversion price of our Series C Preferred Stock, the full-ratchet anti-dilution protection provisionconversion price effective as of December 31, 2020 was $3.75 per share, subject to certain reset provisions. On December 12, 2021, the conversion price was reset to $2.27 per share. The effect was de minimis.
The Series C Preferred Stock contains triggering events which would, among other things, require redemption (i) in cash, at the greater of (a) 120% of the warrants decreasedstated value of $1,000 or (b) the exerciseproduct of (I) the variable weighted average price of our common stock on the warrants from $2.61 per sharetrading day immediately preceding the date of the triggering event and (II) the stated value divided by the then conversion price or (ii) in shares of our common stock, equal to $1.50 per share and increased the aggregatea number of shares issuable underequal to the warrantsamount set forth in (i) above divided by 75%. As of December 31, 2021 and 2020, the aggregate stated value of our Series C Preferred Stock was $105,000. The triggering events include our being subject to 2,315,301.
The Company issued an aggregate of 54,85944,850 shares of its common stock in exchange for 75110 shares of the Company’s Series C Preferred Stockstock (stated value of $110,000) and $70,341 accrued dividends.
Series C Preferred Stock issued and outstanding totaled 985 and 1,070105 as of December 31, 20172021 and 2016, respectively.2020. As of December 31, 20172021 and 2016,2020, the Company has accrued $419,283$81,667 and $359,891$72,517 dividends payable on the Series C Preferred Stock.
NOTE 8 – STOCKHOLDER EQUITY
Shareholder rights plan
On July 14, 2020, our board of directors adopted a stockholder rights plan (the “Rights Plan”) and declared a dividend of one preferred share purchase right for each outstanding share of BioSig’s common stock to stockholders of record on July 27, 2020, and one right will be issued for each new share of common stock issued thereafter. Each right will initially trade with common stock, and will allow its holder to purchase from BioSig one one-thousandth of a share of Series F Junior Participating Preferred stock, par value $0.001 per share, for an exercise price of $50.00, once the rights become exercisable. In the event that a person or group acquires beneficial ownership of 12% or more of BioSig’s then outstanding common stock, subject to certain exceptions, each right would entitle its holder (other than such person or members of such group) to purchase additional shares of BioSig’s common stock having a market value of two times the exercise price of the right. In addition, at any time after a person or group acquires 12% or more of BioSig’s outstanding common stock (unless such person or group acquires 50% or more), the Board may exchange one share of BioSig’s common stock for each outstanding right (other than rights owned by such person or group, which would have become void). The Rights Plan could make it more difficult for a third party to acquire control of BioSig or a large block of our common stock without the approval of our board of directors. The rights expired on July 13, 2021.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
Preferred stock
The Company is authorized to issue 1,000,000 shares of $0.001 par value preferred stock. As of December 31, 20172021 and 2016,2020, the Company has authorizeddesignated 200 shares of Series A preferred stock, 600 shares of Series B preferred stock, 4,200 shares of Series C Preferred Stock, and (in 2017) 1,400 shares of Series D Preferred Stock, 1,000 shares of Series E Preferred Stock and 200,000 shares of Series F Preferred Stock. As of December 31, 20172021, and 2016,2020, there were no outstanding shares of Series A, Series B, Series D, Series E and Series BF preferred stock.
Common stock
BioSig Technologies, Inc.
The Company is authorized to issue 200,000,000 shares of $0.001 par value common stock. As of December 31, 2021 and 2020, the Company had 35,567,180 and 30,764,792 shares issued 54,859 shares of its common stock in exchange for 75 shares ofand outstanding, respectively.
During the Company’s Series C Preferred Stock and accrued dividends.
On November 3, 2017, the Board of Directors authorized the issuance of up to 1,400 shares of Series D Convertible Preferred Stock (the “Series D Preferred Stock”) and accordingly, the Company filed the Certificate of Designations for the Series D Preferred Stock with the Secretary of State of the State of Delaware. Pursuant to such Certificate of Designations, in the event of the Company’s liquidation or winding up of its affairs, the holders of Preferred Shares will be entitled to a liquidation preference of the stated value per Preferred Share of $1,500 (the “Stated Value”) plus any accrued but unpaid dividends or any other fees due the holder.
On June 24, 2020, the Company entered into securities purchase agreements with investors pursuant to which the Company issued 4,131,5362,187,500 shares of common stock and 2,246,300 warrants for aggregate proceeds of $6,011,229,$16,161,980, net of $186,075$1,338,020 in expenses.
During the year ended December 31, 2020, the Company received and canceled 10,744issued 679,555 shares of common stock for services at a fair value of $4,399,533 ($6.47 per share).
During the year ended December 31, 2020, the Company issued 542,646 shares of common stock in exchange for proceeds of $2,088,997 from the exercise of warrants.
During the year ended December 31, 2020, the Company issued 586,825 shares of common stock in exchange for proceeds of $2,722,012 from the exercise of options.
During the year ended December 31, 2020, the Company issued 12,840 shares of common stock in exchange for the exercise of 37,841 cashless exercises of warrants.
During the year ended December 31, 2020, the Company issued 160,743 shares of common stock in exchange for the exercise of 616,398 cashless exercises of options.
During the year ended December 31, 2020, the Company issued 83,055 shares of common stock in exchange for 80,958 previously issued ViralClear shares (see below).
In January 2021, the Company issued an aggregate of 658,868 shares of its common stock as payment for short-swing profit pursuant to Section 16(b)services at a fair value previously recorded in 2020 of the U.S. Securities Exchange Act of 1934, as amended from an officer and member of the Company’s Board of Directors.$2,658,224.
On July 2, 2021, the Company entered into registration rightssecurities purchase agreements with the purchasers in such private placementsinvestors pursuant to which the Company agreedissued 2,500,000 shares of common stock for aggregate proceeds of $9,004,033, net of $995,966 in expenses.
During the year ended December 31, 2021, the Company issued 1,124,341 shares of common stock for services at a fair value of $3,975,451.
During the year ended December 31, 2021, the Company issued 9,375 shares of common stock in exchange for proceeds of $27,750 from the exercise of options.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
During the year ended December 31, 2021, the Company issued an aggregate of 258,084 shares of its common stock for vested restricted stock units.
At-The-Market Sale Agreement
August 28, 2020, the Company entered into an Open Market Sale Agreement (the “Sales Agreement”) with Jefferies LLC to provide certain registration rightsact as the Company’s sales agent and/or principal (“Jefferies” or the “Agent”), with respect to the common stock issuedissuance and sale of up to the investors participating in such private placements and the common stock issuable upon exercise$45.0 million of the related warrants issued such investors. Specifically, the Company agreed to file a registration statement with the Securities and Exchange Commission covering the resale of theCompany’s shares of common stock issuedfrom time to time in an at-the-market offering.
Upon delivery of a placement notice and subject to the terms and conditions of the Sales Agreement, Jefferies may sell the Shares by any method permitted by law deemed to be an “at the market offering” as defined in Rule 415(a)(4) promulgated under the Securities Act of 1933, as amended. The Company may sell the common stock in amounts and at times to be determined by the Company from time to time subject to the terms and conditions of the Sales Agreement, but it has no obligation to sell any of the shares under the Sales Agreement. The Company or Jefferies may suspend or terminate the offering of shares upon notice to the other party and subject to other conditions. Jefferies will act as sales agent on a commercially reasonable efforts basis consistent with its normal trading and sales practices and applicable state and federal law, rules and regulations and the rules of Nasdaq.
The Company paid Agent a commission equal to 3.0% of the gross proceeds from the sale of the shares pursuant to the private placementSales Agreement. The Company has also agreed to provide Jefferies with customary indemnification and issuablecontribution rights.
The offering of shares pursuant to the Sales Agreement will terminate upon the exerciseearlier of (i) the sale of all common stock subject to the Sales Agreement or (ii) termination of the warrants within 45 days ofSales Agreement in accordance with its terms.
The common stock was sold and issued pursuant the termination date of such private placement and to cause suchCompany’s shelf registration statement to beon Form S-3, which was previously declared effective by the Securities and Exchange Commission, and a related prospectus.
From August 28, 2020 through December 31, 2020, the Company sold 424,357 shares of its common stock through the Sales Agreement for net proceeds of $2,228,000, after transactional costs of $222,397.
From January 15, 2021 through February 16, 2021, the Company sold 251,720 shares of its common stock through the Open Market Sales Agreement for net proceeds of $1,300,135, after transactional costs of $40,365.
On March 25, 2021, the Company delivered written notice to Jefferies to terminate the Sales Agreement effective as of April 8, 2021, pursuant to Section 7(b)(i) thereof. The Company was not subject to any termination penalties related to the termination of the Sales Agreement.
ViralClear Pharmaceuticals, Inc.
On May 20, 2020, ViralClear and the Company entered into a securities purchase agreement, pursuant to which ViralClear agreed to sell in a private placement transaction an aggregate of 1,068,550 shares of ViralClear’s common stock at $10.00 per share, for an aggregate consideration of $10,592,075. This private placement closed on May 20, 2020.
The Company was party to certain 2019 purchase agreements between ViralClear and the private placement investors with respect to a provision in each securities purchase agreement which provides that in the event that the registration statement(i) ViralClear common stock is not reviewedlisted on a national securities exchange by October 31, 2020, or (ii) a change of control (as defined in each securities purchase agreement) of ViralClear occurs, whichever is earlier, at the Securitiesoption of the holder of ViralClear common stock, each share of ViralClear common stock may be exchanged into 0.9 of a share our common stock if the ViralClear common stock subject to the share exchange was purchased in the August or September 2019 private placements, or 1.1 shares of our common stock if the ViralClear common stock subject to the share exchange was purchased in the private placement closed in October 2019 through December 2019. In November and Exchange Commission, within 30 calendar days afterDecember 2020, the Company is notified that registration statement is not being reviewed by the Securities and Exchange Commission, and within 180 calendar daysissued an aggregate of the initial filing date83,055 shares of the registration statementits common stock in the event that the registration statement is reviewed by the Securities and Exchange Commission and the Securities and Exchange Commission issues comments.exchange for 80,958 previously issued shares of ViralClear pursuant with 2019 purchase agreements.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 20172021
NOTE 9 – OPTIONS, RESTRICTED STOCK UNITS AND WARRANTS
BioSig Technologies, Inc.
2012 Equity Incentive Plan
On October 19, 2012, the Company’s Board of Directors of BioSig Technologies, Inc. approved the 2012 Equity Incentive Plan (“the “Plan)“Plan”) and terminated the Long-Term Incentive Plan (the “2011 Plan”). The Plan provides for the issuance of options, stock appreciation rights, restricted stock and restricted stock units to purchase up to 15,186,12314,474,450 (as amended) shares of the Company’s common stock to officers, directors, employees and consultants of the Company (as amended).Company. Under the terms of the Plan the Company may issue Incentive Stock Options as defined by the Internal Revenue Code to employees of the Company only and nonstatutory options. The Board of Directors of the Company or a committee thereof administers the Plan and determines the exercise price, vesting and expiration period of the grants under the Plan.
However, the exercise price of an Incentive Stock Option should not be less than 110% of fair value of the common stock at the date of the grant for a 10% or more stockholder and 100% of fair value for a grantee who is not 10% stockholder. The fair value of the common stock is determined based on the quoted market price or in absence of such quoted market price, by the administrator in good faith.
Additionally, the vesting period of the grants under the Plan will be determined by the administrator, in its sole discretion, with an expiration period of not more than ten years. The Company reserved 227,388There are 3,048,522 shares of its common stockremaining available for future issuance of awards under the terms of the Plan.Plan as of December 31, 2021.
During the yearyears ended December 31, 2016,2021 and 2020, the Company granted an aggregate of 750,000, net1,818,000 and 1,070,000 (net of 100,000 canceled,50,000 canceled) options to officers, directors, and key consultants.
During the yearyears ended December 31, 2016,2021 and 2020, the Company grantedissued an aggregate of 723,5451,185,872 and 634,517 stock grants to officers, employees and key consultants under the plan. See Note 8.
| Options Outstanding | Options Exercisable | |||||||||||||
| Weighted | ||||||||||||||
| Average | Exercisable | |||||||||||||
| Exercise | Number of | Remaining Life | Number of | |||||||||||
| Price | Options | In Years | Options | |||||||||||
| $ | 1.01-2.00 | 3,825,540 | 6.8 | $ | 2,662,707 | |||||||||
| 2.01-3.00 | 4,384,779 | 3.7 | 4,384,779 | |||||||||||
| 3.01-4.00 | 300,000 | 7.3 | 300,000 | |||||||||||
| 8,510,319 | 5.2 | 7,347,486 | ||||||||||||
| Weighted-Average | ||||||||||||||||
| Weighted-Average | Remaining | Aggregate | ||||||||||||||
| Shares | Exercise Price | Contractual Term | Intrinsic Value | |||||||||||||
| Outstanding at January 1, 2016 | 7,780,190 | $ | 2.30 | 6.4 | $ | - | ||||||||||
| Grants | 905,000 | 1.71 | 10.0 | $ | - | |||||||||||
| Exercised | - | - | - | |||||||||||||
| Canceled | (440,000 | ) | 2.24 | - | ||||||||||||
| Outstanding at December 31, 2016 | 8,245,190 | $ | 2.24 | 5.7 | $ | - | ||||||||||
| Grants | 1,680,898 | 1.50 | 10.0 | $ | - | |||||||||||
| Exercised | - | |||||||||||||||
| Canceled | (1,415,769 | ) | $ | 2.17 | ||||||||||||
| Outstanding at December 31, 2017 | 8,510,319 | $ | 2.11 | 5.2 | $ | 27,045 | ||||||||||
| Exercisable at December 31, 2017 | 7,347,486 | $ | 2.19 | 4.8 | $ | 25,394 | ||||||||||
Option valuation models require the input of highly subjective assumptions. The fair value of stock-based payment awards was estimated using the Black-Scholes option model with a volatility figure derived from historical stock prices of the Company. The Company accounts for the expected life of options using the based on the contractual life of options for non-employees.
For employees, the Company accounts for the expected life of options in accordance with the “simplified” method, which is used for “plain-vanilla” options, as defined in the accounting standards codification.
The following table presents information related to stock options at December 31, 2021:
| Options Outstanding |
|
| Options Exercisable |
| ||||||||||
|
|
|
|
|
|
|
|
| Weighted |
|
|
|
|
| |
|
|
|
|
|
|
|
|
| Average |
|
| Exercisable |
| ||
| Exercise |
|
| Number of |
|
| Remaining Life |
|
| Number of |
| ||||
| Price |
|
| Options |
|
| In Years |
|
| Options |
| ||||
| $ | Under 3.00 |
|
|
| 1,035,375 |
|
|
| 9.5 |
|
|
| 560,000 |
|
|
| 3.00-3.99 |
|
|
| 587,466 |
|
|
| 6.5 |
|
|
| 387,466 |
|
4.00-4.99 | 1,762,916 | 6.2 | 1,151,545 | ||||||||||||
5.00-5.99 | 156,132 | 7.1 | 119,464 | ||||||||||||
6.00-6.99 | 591,542 | 5.0 | 478,846 | ||||||||||||
|
| 7.00-7.99 |
|
|
| 191,720 |
|
|
| 6.9 |
|
|
| 177,138 |
|
|
| Over 8.00 |
|
|
| 243,333 |
|
|
| 6.3 |
|
|
| 197,351 |
|
|
|
|
|
|
| 4,568,484 |
|
|
| 6.9 |
|
|
| 3,071,810 |
|
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
A summary of the stock option activity and related information for the Plan for the two years ended December 31, 2021 is as follows:
Weighted-Average | ||||||||||||||||
Weighted-Average | Remaining | Aggregate | ||||||||||||||
Shares | Exercise Price | Contractual Term | Intrinsic Value | |||||||||||||
Outstanding at January 1, 2020 | 3,980,804 | $ | 5.58 | 6.3 | $ | 3,130,791 | ||||||||||
Grants | 1,120,000 | $ | 4.98 | 10.0 | - | |||||||||||
Exercised | (1,203,223 | ) | $ | 5.08 | ||||||||||||
Forfeited/expired | (329,084 | ) | $ | 5.19 | ||||||||||||
Outstanding at December 31, 2020 | 3,568,497 | $ | 5.59 | 7.0 | $ | 110,961 | ||||||||||
Grants | 1,818,000 | $ | 3.69 | 10.0 | $ | - | ||||||||||
Exercised | (9,375 | ) | $ | 2.96 | ||||||||||||
Forfeited/expired | (808,638 | ) | $ | 6.19 | ||||||||||||
Outstanding at December 31, 2021 | 4,568,484 | $ | 4.57 | 6.9 | $ | - | ||||||||||
Exercisable at December 31, 2021 | 3,071,810 | $ | 4.83 | 6.0 | $ | - | ||||||||||
The aggregate intrinsic value in the preceding tables represents the total pretax intrinsic value, based on options with an exercise price less than the stock price of BioSig Technologies, Inc. of $2.23 as of December 31, 2021, which would have been received by the option holders had those option holders exercised their options as of that date.
On January 10, 2020, BioSig Technologies, Inc. granted 60,000 options to purchase the company stock in connection with the services rendered at the exercise price of $6.00 per share for a term of ten years with quarterly vesting beginning March 31, 2020 for three years.
On March 24, 2020, BioSig Technologies, Inc. granted 100,000 options to purchase the company stock in connection with the services rendered at the exercise price of $2.96 per share for a term of ten years with 25,000 vesting immediately and 75,000 quarterly vesting beginning June 30, 2020 for two years.
On March 31, 2020, BioSig Technologies, Inc. granted 50,000 options to purchase the company stock in connection with the services rendered at the exercise price of $3.73 per share for a term of ten years with vesting quarterly vesting beginning June 30, 2020 for three years. On August 12, 2020, this option was cancelled and a was replaced for a restricted stock award for 50,000 shares.
On April 14, 2020, BioSig Technologies, Inc. granted an aggregate of 625,000 options to purchase the company stock to directors and an employee. The options are exercisable at $4.66 per share for ten years and fully vested and exercisable at the date of grant. On April 14, 2020, BioSig Technologies, Inc. granted an aggregate of 90,000 options to purchase shares of its common stock to employees. The options are exercisable at $4.66 per share for ten years and vest quarterly over three years.
On May 20, 2020, BioSig Technologies, Inc. granted an aggregate of 65,000 options to purchase the company stock to consultants and an employee. The options are exercisable at $10.49 per share for ten years with 40,000 fully vested and exercisable at the date of grant and 25,000 options vesting quarterly over three years.
On August 26, 2020, BioSig Technologies, Inc. granted an aggregate of 25,000 options to purchase the company stock to 3 employees at the exercise price of $7.57 per share for a term of ten years with one-third vesting on the one year anniversary and two-thirds vesting quarterly thereafter beginning November 26, 2021 for two years.
On October 9, 2020, BioSig Technologies, Inc. granted an aggregate of 105,000 options to purchase the company stock to 3 employees at the exercise price of $5.03 per share for a term of ten years with one-third vesting on the one year anniversary and two-thirds vesting quarterly thereafter beginning January 9, 2022 for two years.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
On January 12, 2021, BioSig Technologies, Inc. granted 387,500 options to purchase the company stock in connection with the services rendered at the exercise price of $4.23 per share for a term of ten years with one-third vesting on the one-year anniversary and two-thirds vesting quarterly thereafter beginning January 12, 2022 for two years.
On February 16, 2021, BioSig Technologies, Inc. granted 102,000 options to purchase the company stock in connection with the services rendered at the exercise price of $4.97 per share for a term of ten years with one-third vesting on the one year anniversary and two-thirds vesting quarterly thereafter beginning February 16, 2022 for two years.
On April 9, 2021, BioSig Technologies, Inc. granted 90,000 options to purchase the company stock in connection with the services rendered at the exercise price of $4.38 per share for a term of ten years with one-third vesting on the one-year anniversary and two-thirds vesting quarterly thereafter beginning April 9, 2022 for two years.
On April 13, 2021, BioSig Technologies, Inc. granted 25,000 options to purchase the company stock in connection with the services rendered at the exercise price of $4.42 per share for a term of ten years with one-third vesting on the one-year anniversary and two-thirds vesting quarterly thereafter beginning April 13, 2022 for two years.
On May 18, 2021, BioSig Technologies, Inc. granted 150,000 options to purchase the company stock in connection with the services rendered at the exercise price of $3.20 per share for a term of ten years with one-third vesting on the one-year anniversary and two-thirds vesting quarterly thereafter beginning May 18, 2022 for two years.
On August 3, 2021, BioSig Technologies, Inc. granted an aggregated of 75,000 options to purchase shares of its common stock to 3 employees. The options are exercisable at $3.61 per share for ten years with one-third vesting on the first anniversary of the date of grant, and the remaining two-thirds vesting in substantially equal quarterly installments over the following two years.
On August 31, 2021, BioSig Technologies, Inc. granted an aggregated of 47,500 options to purchase shares of its common stock to 3 employees. The options are exercisable at $2.98 per share for ten years with immediate vesting.
On September 17, 2021, BioSig Technologies, Inc. granted an aggregated of 40,000 options to purchase shares of its common stock to 2 employees. The options are exercisable at $2.99 per share for ten years with one-third vesting on the first anniversary of the date of grant, and the remaining two-thirds vesting in substantially equal quarterly installments over the following two years.
On October 4, 2021, BioSig Technologies, Inc. granted 50,000 options to purchase shares of its common stock to a newly appointed Board member. The options are exercisable at $2.89 per share for ten years with half immediate vesting and half vesting on September 20, 2022.
On December 15, 2021, BioSig Technologies, Inc. granted an aggregate of 351,000 options to purchase shares of its common stock to 2 employees. The options are exercisable at $2.58 per share for ten years with one-third vesting on the first anniversary of the date of grant, and the remaining two-thirds vesting in substantially equal quarterly installments over the following two years.
On December 28, 2021, BioSig Technologies, Inc. granted an aggregate of 425,000 options to purchase shares of its common stock with as compensation to the Company’s Board of Directors. The options are exercisable at $2.44 per share with immediate vesting. Also, on December 28, 2021, BioSig Technologies issued 75,000 options to purchase shares of its common stock to a consultant. The options are exercisable at $2.44 per share with 25,000 options vested immediately and 50,000 options vesting on the one-year anniversary.
The following assumptions were used in determining the fair value of options during the years ended December 31, 2021 and 2020:
2021 | 2020 | |||||||
Risk-free interest rate | 0.77% - 1.49 | % | 0.42% to 1.83 | % | ||||
Dividend yield | 0 | % | 0 | % | ||||
Stock price volatility | 82.50% to 95.98 | % | 86.51% to 93.43 | % | ||||
Expected life | 5 – 10 years | 5-10 years | ||||||
Weighted average grant date fair value | $ | 2.55 | $ | 4.03 | ||||
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
On June 28, 2021, in connection with the exit of two members of the Company’s board of directors, the Company extended the life of 145,000 previously issued director options from the contractual 90 days from termination of service to the earlier of the initial life or June 28, 2023. The change in estimated fair value of the modified options of $182,514 was charged to current period operations.
The following assumptions were used in determining the change in fair value of the modified options at June 28, 2021:
Risk-free interest rate | 0.05% - 0.25 | % | ||
Dividend yield | 0 | % | ||
Stock price volatility | 88.57 | % | ||
Expected life | 0.25 – 2 years | |||
On June 30, 2021, in connection with the resignation of a member of the Company’s board of directors, the Company entered into a one-year consulting contract and extended the life of 221,240 previously issued director options from the contractual 90 days from termination of service to the earlier of the initial life or two years after service contract completion. The change in estimated fair value of the modified options of $111,402 was charged to current period operations.
The following assumptions were used in determining the change in fair value of the modified options on June 30, 2021:
Risk-free interest rate | 0.06% - 0.46 | % | ||
Dividend yield | 0 | % | ||
Stock price volatility | 88.59 | % | ||
Expected life | 0.59 – 3 years | |||
The fair value of all options vesting during the year ended December 31, 2021 and 2020 of $3,357,274 and $5,217,761, respectively, was charged to current period operations. Unrecognized compensation expense of $3,655,519 at December 31, 2021 will be expensed in future periods.
Warrants
The following table summarizes information with respect to outstanding warrants to purchase common stock of BioSig Technologies, Inc. at December 31, 2021:
| Exercise |
|
| Number |
| Expiration | ||
| Price |
|
| Outstanding |
| Date | ||
| $ | 4.80 |
|
|
| 250,000 |
| February 2025 to July 2026 |
| $ | 6.16 |
|
|
| 568,910 |
| November 2027 |
|
|
|
|
|
| 818,910 |
|
|
On February 25, 2020, BioSig Technologies, Inc. issued warrants to purchase 125,000 shares of its common stock at $4.80 per share, expiring on February 21, 2025, for placement agent services in connection with the sale of the company’s common stock.
On July 7, 2021, BioSig Technologies, Inc. issued warrants to purchase 125,000 shares of its common stock at $4.80 per share, expiring on July 2, 2026, for placement agent services in connection with the sale of the company’s common stock.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
A summary of the warrant activity for the two years ended December 31, 2021 is as follows:
Weighted-Average | ||||||||||||||||
Weighted-Average | Remaining | Aggregate | ||||||||||||||
Shares | Exercise Price | Contractual Term | Intrinsic Value | |||||||||||||
Outstanding at January 1, 2020 | 2,744,718 | $ | 5.40 | 2.2 | $ | 3,410,763 | ||||||||||
Issued | 125,000 | $ | 4.80 | 4.2 | - | |||||||||||
Exercised | (580,487 | ) | $ | 3.89 | ||||||||||||
Expired | (843,031 | ) | $ | 6.29 | ||||||||||||
Outstanding at December 31, 2020 | 1,446,200 | $ | 5.44 | 3.3 | $ | 1,500 | ||||||||||
Issued | 125,000 | $ | 4.80 | 5.0 | ||||||||||||
Expired | (752,290 | ) | $ | 5.00 | - | - | ||||||||||
Outstanding at December 31, 2021 | 818,910 | $ | 5.74 | 5.3 | $ | - | ||||||||||
Vested and expected to vest at December 31, 2021 | 818,910 | $ | 5.74 | 5.3 | $ | - | ||||||||||
Exercisable at December 31, 2021 | 818,910 | $ | 5.74 | 5.3 | $ | - | ||||||||||
The aggregate intrinsic value in the preceding tables represents the total pretax intrinsic value, based on options with an exercise price less than the company’s stock price of $2.23 of December 31, 2021, which would have been received by the option holders had those option holders exercised their options as of that date.
Restricted Stock Units
The following table summarizes the restricted stock activity for the two years ended December 31, 2021:
Restricted shares issued as of January 1, 2020 | 262,668 | |||
Granted | 175,000 | |||
Vested and issued | (219,334 | ) | ||
Restricted shares issued as of December 31, 2020 | 218,334 | |||
Granted | 301,000 | |||
Vested and issued | (258,084 | ) | ||
Forfeited | (120,000 | ) | ||
Vested restricted shares as of December 31, 2021 | - | |||
Unvested restricted shares as of December 31, 2021 | 141,250 |
In 2020, the Company granted an aggregate of 175,000 restricted stock grants for services with vesting from one year to three years from grant date.
On January 4, 2021, the Company granted 220,000 restricted stock units for services with 105,000 vesting one-third on the one-year anniversary and two-thirds vesting quarterly thereafter beginning January 4, 2022 for two years and with 115,000 vesting quarterly for one year.
On March 8, 2021 the Company granted 31,000 restricted stock units for services vesting on August 31, 2021.
On June 1, 2021, in connection with the termination of an employee, the Company accelerated vesting of 30,000 previously granted restricted stock units from a three-year period to fully vested. The change in vesting of the modified restricted stock unit resulted in a $109,725 charge to current period operations.
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
On June 30, 2021, in connection with the resignation of a member of the Company’s board of directors, the Company accelerated vesting of 50,000 previously granted restricted stock units from a three-year period to fully vested. The change in vesting of the modified restricted stock unit resulted in a $232,375 charge to current period operations.
On August 14, 2021 the Company granted 50,000 restricted stock units for services vesting quarterly for one year.
Stock based compensation expense related to restricted stock grants was $950,281 and $1,151,676 for the year ended December 31, 2021 and 2020, respectively. As of December 31, 2021, the stock-based compensation relating to restricted stock of $286,417 remains unamortized.
ViralClear Pharmaceuticals, Inc.
2019 Long-Term Incentive Plan
On September 24, 2019, ViralClear’s Board of Directors approved the 2019 Long-Term Incentive Plan (as subsequently amended, the “ViralClear Plan”). The ViralClear Plan was approved by BioSig as ViralClear’s majority stockholder. The ViralClear Plan provides for the issuance of options, stock appreciation rights, restricted stock and restricted stock units to purchase up to 4,000,000 shares of ViralClear’s common stock to officers, directors, employees and consultants of the ViralClear. Under the terms of the ViralClear Plan, ViralClear may issue Incentive Stock Options as defined by the Internal Revenue Code to employees of ViralClear only and nonstatutory options. The Board of Directors of ViralClear or a committee thereof administers the ViralClear Plan and determines the exercise price, vesting and expiration period of the grants under the ViralClear Plan.
However, the exercise price of an Incentive Stock Option should not be less than 110% of fair market value of the common stock at the date of the grant for a 10% or more stockholder and 100% of fair market value for a grantee who is not 10% stockholder. The fair market value of the common stock is determined based on the quoted market price or in absence of such quoted market price, by the administrator in good faith.
Additionally, the vesting period of the grants under the ViralClear Plan will be determined by the administrator, in its sole discretion, with an expiration period of not more than ten years. There are 2,330,750 shares remaining available for future issuance of awards under the terms of the ViralClear Plan.
ViralClear Options
A summary of the stock option activity and related information for the ViralClear Plan for the two years ended December 31, 2021 is as follows:
Weighted-Average | ||||||||||||
Weighted-Average | Remaining | |||||||||||
Shares | Exercise Price | Contractual Term | ||||||||||
Outstanding at January 1, 2020 | 575,000 | $ | 5.00 | 9.3 | ||||||||
Grants | 1,599,173 | $ | 5.31 | 9.6 | ||||||||
Forfeited/expired | (646,507 | ) | $ | 5.77 | ||||||||
Outstanding at December 31, 2020 | 1,527,666 | $ | 5.00 | 4.0 | ||||||||
Exercised | (550,000 | ) | $ | 5.00 | ||||||||
Forfeited/expired | (852,666 | ) | $ | 5.00 | ||||||||
Outstanding at December 31, 2021 | 125,000 | $ | 5.00 | 7.2 | ||||||||
Exercisable at December 31, 2021 | 83,331 | $ | 5.00 | 6.6 | ||||||||
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
The following table presents information related to stock options at December 31, 2021:
Options Outstanding |
|
| Options Exercisable |
| ||||||||||
|
|
|
|
|
|
|
| Weighted |
|
|
|
|
| |
|
|
|
|
|
|
|
| Average |
|
| Exercisable |
| ||
Exercise |
|
| Number of |
|
| Remaining Life |
|
| Number of |
| ||||
Price |
|
| Options |
|
| In Years |
|
| Options |
| ||||
$ | 5.00 |
|
|
| 125,000 |
|
|
| 7.2 |
|
|
| 83,331 |
|
The fair value of the stock-based payment awards was estimated using the Black-Scholes option model with a volatility figure derived from an index of historical stock prices of comparable entities until sufficient data exists to estimatewith the volatility using the Company’s own historicalmarket value of stock prices. Management determined this assumption to be a more accurate indicator of value. The Company accounts for the expected life of optionsprice based on the contractual life of options for non-employees.
In 2020, ViralClear granted an aggregate of 685,0001,599,173 options to purchase the Company stock in connectionshares with the services rendered at thean exercise price of $1.84 per share$5.00 to $10.00 for a term of ten years vesting immediately. In September 2016,with 1,278,999 vested immediately, 120,174 quarterly over one year and 200,000 quarterly over two years.
The following assumptions were used in determining the Companychange in fair value of the ViralClear options for the year ended December 31, 2020:
Risk-free interest rate | 0.36% to 0.52 | % | ||
Dividend yield | 0 | % | ||
Stock price volatility | 125.16% to 126.03 | % | ||
Expected life | 5 – 6 years | |||
Weighted average grant date fair value | $ | 4.51 | ||
On July 1, 2021, ViralClear issued 83,545206,250 shares of its common stock in exchange for 100,000 common stockthe cashless exercise of 550,000 options previously issuedgranted on October 16, 2019.
On June 30, 2021, in May 2016 underconnection with the termsresignation of its 2012 Equity Plan. The equalitya member of the fair value was determined usingCompany’s board of directors, the Black Scholes option pricing model withCompany entered into a one-year consulting contract and extended the following assumptions: dividend yield: 0%; volatility: 122.82%; risk free rate: 1.08%, term: 5life of 25,000 previously issued director options from the contractual 90 days from termination of service to the earlier of the initial life or two years andafter service contract completion. The change in estimated fair value of the Company’s common stock: $1.84.modified options of $26,577 was charged to current period operations.
The following assumptions were used in determining the change in fair value of employee and vesting non-employeethe modified options during the year ended December 31, 2016:
| Risk-free interest rate | 1.08% - 2.04 | % | ||
| Dividend yield | 0 | % | ||
| Stock price volatility | 109.3% to 122.82 | % | ||
| Expected life | 5 – 10 years | |||
| Weighted average grant date fair value | $ | 1.47 | ||
Risk-free interest rate | 0.07% - 0.46 | % | ||
Dividend yield | 0 | % | ||
Stock price volatility | 88.59 | % | ||
Expected life | 1.25 - 3 years | |||
| Risk-free interest rate | 2.01% - 2.34 | % | ||
| Dividend yield | 0 | % | ||
| Stock price volatility | 95.72% to 107.17 | % | ||
| Expected life | 5 – 10 years | |||
| Weighted average grant date fair value | $ | 1.17 | ||
The fair value of all options vesting during the yearyears ended December 31, 20172021 and 20162020 of $1,269,591$146,083 and $2,801,948,$5,873,376, respectively, was charged to current period operations. Unrecognized compensation expense of $979,812 and $310,817$182,604 at December 31, 2017 and 2016, respectively,2021 will be expensed in future periods.
Warrants (ViralClear)
The following table presents information related to warrants (ViralClear) at December 31, 2021:
| Exercise |
|
| Number |
| Expiration | ||
| Price |
|
| Outstanding |
| Date | ||
| $ | 5.00 |
|
|
| 473,772 |
| November 2027 |
|
| 10.00 |
|
|
| 6,575 |
| May 2025 |
|
|
|
|
|
| 480,347 |
|
|
BIOSIG TECHNOLOGIES INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
On May 20, 2020, ViralClear issued warrants to purchase 6,575 shares of its common stock at $10.00 per share, expiring on May 20, 2025, for placement agent services in connection with the sale of ViralClear’s common stock.
Restricted Stockstock units (ViralClear)
The following table summarizes the restricted stock activity for the two years ended December 31, 2017:2021:
Restricted shares | ||||
Granted | ||||
Restricted shares outstanding at December 31, 2020: | 1,420,716 | |||
Issued | ( | ) | ||
Forfeited | ( | ) | ||
Total restricted shares outstanding at December 31, 2021: | 1,318,679 | |||
Comprised of: | ||||
Vested restricted shares as of December 31, | ||||
Unvested restricted shares as of December 31, | ||||
Total | 1,318,679 |
On September 7, 2016, the CompanyMarch 25, 2020, ViralClear granted 180,000an aggregate of 338,000 restricted stock units (“RSU”)to two ViralClear board members for services vesting immediately.
On March 30, 2020, ViralClear granted an aggregate of 960,000 restricted stock units to ViralClear board members and employees for services with 320,000 vesting immediately, and 640,000 vesting upon ViralClear meeting certain milestones.
On July 13, 2020, ViralClear granted 82,716 restricted stock units to a consultant for services with vesting monthly over one year beginning October 7, 2016.from date of grant.
Stock based compensation expense related to restricted stock unit grants of ViralClear was $93,261$904,112 and $213,174$5,893,320 for the years ended December 31, 20172021 and 2016,2020, respectively. As of December 31, 2017,2021, the stock-based compensation relating to restricted stock of $-0-$186,047 remains unamortized.
NOTE 10 – NON-CONTROLLING INTEREST
On November 7, 2018, the Company formed a subsidiary, now known as ViralClear, to pursue additional applications of the PURE EP™ signal processing technology outside of cardiac electrophysiology, and subsequently in 2020, was repurposed to develop merimepodib, a broad-spectrum anti-viral agent that showed potential for the treatment of COVID-19. Since late 2020, ViralClear has been realigned with its original objective of pursuing additional applications of the PURE EP™ signal processing technology outside of cardiac electrophysiology.
On March 24, 2020, ViralClear entered into an asset purchase agreement (the “Asset Purchase Agreement”) with Trek Therapeutics, PBC (“Trek”), a related party; an entity controlled by a member of the Company’s board of directors. Pursuant to the Asset Purchase Agreement, Trek sold to ViralClear all right, title and interest of Trek and its affiliates to certain assets (the “Purchased Assets”). As consideration for the Purchased Assets, ViralClear agreed to pay Trek in upfront and milestone payments a combination of cash, shares of ViralClear’s common stock, which common stock may equal up to 10% of ViralClear’s outstanding equity, and sublicense fees in the event ViralClear sublicenses the Purchased Assets. On March 30, 2020, pursuant to the Asset Purchase Agreement, ViralClear paid $350,000 in cash and issued 634,910 shares of ViralClear’s common stock valued at $3,174,550 to Trek. In addition, in the event of sublicensing, sale, transfer, assignment or similar transaction, ViralClear agreed to pay Trek 10% of the consideration received.
As part of the Purchased Assets, ViralClear received an assignment and licensing rights agreement from Trek with a third-party vendor regarding certain formulas and compounds usage. The agreement, as amended on September 2, 2020, calls for milestone payments upon marketing authorization (as defined) in any first and second country of $10 million and $5 million, respectively, in addition to 6% royalty payments.